Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-31326
ELOXX PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 84-1368850 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) |
950 Winter Street
Waltham, Massachusetts 02451
(Address of Principal Executive Offices and Zip Code)
(781) 577-5300
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.01 par value | The OTCQB Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ☐ NO ☒
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. YES ☐ NO ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of “accelerated filer”, “large accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
| Non-accelerated filer | ☐ | Smaller reporting company | ☒ | |||
| Emerging growth company | ☐ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, based on the closing price for such stock as reported on the OTCQB Market on June 30, 2017, the last business day of the registrant’s most recently completed second quarter, was: $7,092,700.
As of December 31, 2017, there were 27,527,738 shares of the Registrant’s common stock, par value $0.01 per share, outstanding.
Table of Contents
ELOXX PHARMACEUTICALS INC.
i
Table of Contents
Special Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K, or this report and the other documents we have filed with the SEC that are incorporated herein by reference, contains forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, are forward-looking statements. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “target,” “potential,” “will,” “would,” “could,” “should,” “continue,” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. In particular, you should consider the numerous risks described in the “Risk Factors” section in this Report on Form 10-K.
Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. You should not rely upon forward-looking statements as predictions of future events. Unless required by law, we will not undertake and we specifically disclaim any obligation to release publicly the result of any revisions which may be made to any forward-looking statements to reflect events or circumstances after the date of such statements or to reflect the occurrence of events, whether or not anticipated. In that respect, we wish to caution readers not to place undue reliance on any such forward-looking statements, which speak only as of the date they are made.
This report and the other documents incorporated by reference herein includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third party research, surveys and studies are reliable, we have not independently verified such data.
The following are some risks and uncertainties, among others, that could cause actual results to differ materially from those expressed or implied by forward looking statements in this prospectus:
| • | risks related to the reverse merger and potentially significant, unexpected costs and liabilities arising with respect to the historic Sevion business and operations; |
| • | risks related to our ability to obtain adequate financing in the future through product licensing, public or private equity or debt financing or otherwise; general business conditions; competition; business abilities and judgment of personnel; and the availability of qualified personnel; |
| • | risks related to the ability to obtain the capital necessary to find our operations; |
| • | risks related to our ability to progress any product candidates in preclinical or clinical trials; |
| • | risks related to the scope, rate and progress of our preclinical studies and clinical trials and other research and development activities; |
| • | the uncertainty of clinical trial results and the fact that positive results from preclinical studies are not always indicative of positive clinical results; |
| • | risks that our product candidates may not prove to be safe and efficacious; |
| • | risks relating to the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and |
| • | risks related to the competition for patient enrollment from drug candidates in development. |
ii
Table of Contents
Merger of Sevion Therapeutics, Inc. and Eloxx Pharmaceuticals, Limited
On December 19, 2017, the Sevion Therapeutics, Inc. (“Sevion”) acquired Eloxx Pharmaceuticals, Limited (“Private Eloxx”) pursuant to a merger between the companies (the “Transaction”). Upon consummation of the Transaction (the “Closing”), Sevion adopted the business plan of Private Eloxx and discontinued the pursuit of Sevion’s business plan pre-Closing. In connection with the Transaction, Sevion agreed to acquire all of the outstanding capital stock of Private Eloxx in exchange for the issuance of an aggregate 20,316,656 shares of the Sevion’s common stock, par value $0.01 per share (the “Common Stock”), after giving effect to a 1-for-20 reverse split effected immediately prior to the Transaction. As a result of the Transaction, Private Eloxx became a wholly-owned subsidiary of Sevion. While Sevion was the legal acquirer in the transaction, Private Eloxx was deemed the accounting acquirer. Immediately after giving effect to the Transaction, on December 19, 2017, Sevion changed its name to Eloxx Pharmaceuticals, Inc. (“Eloxx” or the “Company”). Our current trading symbol is “ELOX.” Our principal executive offices are located in Waltham, Massachusetts and we have a research and development center in Rehovot, Israel. Our telephone number is (781) 577-5300.
Company Overview
We are a global biopharmaceutical company focused on discovering and developing novel therapeutics for the treatment of rare and ultra-rare premature stop codon diseases. We are harnessing the science of genetic read-through to develop novel drug product candidates that interact with the ribosome to overcome these premature stop codons. Our revolutionary small molecule approach is designed to unleash the potential to restore production of full length functional proteins with the goal of enabling a return toward normal cellular function. We believe there is a broad application of this approach to the over 1800 rare and ultra-rare diseases where nonsense mutation has been implicated in the cause or pathway of human disease.
Our research and development strategy is to target rare or ultra-rare diseases where a high unmet medical need, nonsense mutation bearing patient population has been identified. We focus on clinical indications where there is a high unmet medical need, established preclinical read-through or personalized medicine experiments that are predictive of clinical activity, and a definable path for Orphan Drug development, regulatory approval, patient access and commercialization. We believe patient advocacy to be an important element of patient focused drug development and seek opportunities to collaborate with patient advocacy groups throughout the discovery and development process. Our current clinical focus is on cystic fibrosis (or “CF”) and cystinosis where we are advancing our lead drug product candidate, ELX-02.
We intend to be the global leader in the application of the science of translational read through and the associated pathway of nonsense mediated messenger ribonucleic acid (“mRNA”) decay. We believe that expanding our expertise across these basic science areas of mRNA regulation, ribosomal function, and protein translation forms a solid foundation to support our discovery and development activities. Our compounds modulate the activity of the ribosome, the organelle within living cells responsible for protein production, a process also known as translation. These novel small molecule compounds are designed to allow the ribosome to read-through a nonsense mutation in mRNA (which is transcribed from the DNA sequence), to restore the translation process to produce full length, functional proteins and increase the amount of mRNA that would otherwise be degraded as part of a phenomenon called nonsense mediated mRNA decay. As our compounds target the general mechanism for protein production in the cell, we believe they have the potential to treat hundreds of genetic diseases where nonsense mutations have impaired gene function. Our subcutaneously injected small molecules have the potential to be self-administered and to be active at most tissue locations across the body.
We believe that our library of related novel small molecules hold the potential to be disease-modifying therapies that may change the course of hundreds of genetic diseases and improve the lives of patients. Our early
1
Table of Contents
preclinical data in animal models of nonsense mutations suggests that drug product candidates from our read through compound library may have potential beneficial effects for each of the following diseases: cystic fibrosis, cystinosis, mucopolysaccharidosis type 1, Duchenne muscular dystrophy and Rett syndrome, and have demonstrated the potential for beneficial effects in multiple organs such as the brain, kidney, muscles and others. We intend to advance one or more additional molecules from our drug product candidate library toward clinical development by initiating the required investigational new drug (“IND”)-enabling studies in 2018.
Currently our lead program ELX-02 is focused on development for cystic fibrosis and cystinosis patients with diagnosed nonsense mutations. To advance the program, we have held pre-IND pre-clinical trial application (“CTA”) discussions with the Federal Agency for Medicines and Health Products (the “FAMHP”) in Brussels Belgium and pre-IND discussions with the U.S. Food & Drug Administration (the “FDA”) for cystic fibrosis and cystinosis, respectively. We are on-track for an expected mid-2018 submission of our IND and CTA. Approval of these submissions will be required for initiation of Phase 2 studies in cystic fibrosis and cystinosis in 2018.
As part of our clinical program, we have completed a Phase 1 single ascending dose (“SAD”) study in a total of 60 healthy volunteers at sites in Israel (ClinicalTrials.gov Identifier: NCT02807961) and Belgium (ClinicalTrials.gov Identifier: NCT03292302). Currently ongoing is the Phase 1 multiple ascending dose (“MAD”) study in 45 healthy volunteers in Belgium (ClinicalTrials.gov Identifier: NCT03309605). We anticipate that the Phase 1 MAD study will be completed in 2018. The results from the completed Phase 1 study will be included in the planned IND and CTA submissions.
We believe there is a significant unmet medical need in the treatment of cystic fibrosis patients carrying nonsense mutations on one or both alleles of the Cystic Fibrosis Transmembrane Conductance Regulator (“CFTR”) gene. Cystic fibrosis is the most prevalent genetic disease in the western world and there are no currently approved therapies that target the impairment associated with Class 1 CFTR mutations. We believe that nonsense mutations may impact a similar proportion of patients diagnosed with cystinosis. There are no currently approved therapeutics that target the nonsense mutation mediated impairment of cystinosin the cystine-selective transport channel in the lysosomal membrane that is attributed as the cause for the accumulation of cystine in this disease state. Given the high proportion of pediatric patients in each of these rare orphan diseases we intend to apply for relevant Orphan Drug incentives in the US and Europe, including the Rare Pediatric Disease Priority Review Voucher in the U.S.
Currently, the European Medicines Agency (the “EMA”) has designated ELX-02 as an orphan medicine for the treatment of mucopolysaccharidosis type I (“MPS I”), and the FDA has granted orphan drug designation to ELX-02 for the treatment of MPS I and for the treatment of Rett Syndrome.
We hold worldwide development and commercialization rights to ELX-02 and novel compounds in our read-through library, for all indications, in all territories, under a license from the Technion Research and Development Foundation Ltd. Professor Timor Baasov, the inventor of our compounds, has served as our senior consultant since our incorporation.
Our Technology
Nonsense mutations, also known as premature termination or stop codons, are single point mutations within the DNA sequence which are either inherited or acquired that result in premature termination of the translational process leading to truncated or absent proteins. Nonsense mutations are the cause of a large number of genetic diseases such as cystic fibrosis, cystinosis, mucopolysaccharidosis type 1 (“nmMPS-1”), Duchenne muscular dystrophy (“nmDMD”), Rett syndrome, and a variety of cancers. According to the human gene mutation database (http://www.hgmd.cf.ac.uk/ac/index.php), nonsense mutations account for approximately twelve percent (12%) of patients with a given genetic disease. The disease phenotypes caused by nonsense mutations are frequently more severe than those caused by other kinds of mutations because these mutations often lead to a complete loss of protein production or function. In general, these diseases do not have specific therapies beyond symptomatic and palliative interventions.
2
Table of Contents
In eukaryotic cells, the cytoplasmic ribosome is responsible for the production of proteins by a process called translation. As part of the translation process, the genetic information is transcribed to the mRNA arranged as codons that specify the corresponding amino acid, the building block of a protein. The ribosome pairs a specific mRNA codon with an aminoacyl transfer RNA (aa-tRNA) containing an anticodon sequence causing elongation of the nascent protein.
Normal translation termination in eukaryotic cells occurs when a natural (canonical) termination codon enters the ribosomal A site, the protein production site within the ribosome, and no complementary aa-tRNA is found. Termination codon recognition is not carried out by codon-anticodon interactions, since no tRNA anticodon is complementary to any of the mRNA termination codons. Rather, a complex of releasing factors recognize the termination codons and interact with the ribosome to release the completed protein, resulting in termination of the translation process.
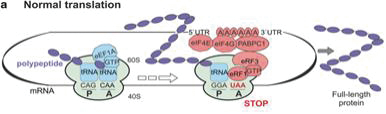
Translation terminates efficiently when the termination codon (“TC”) is in physical proximity to the 3’ poly(A) tail (AAAAAAAA) and/or the 5’ 7-methylguanosine (m7G) cap of the mRNA. Efficient translation termination prevents nonsense-mediated delay (“NMD”) of the mRNA.
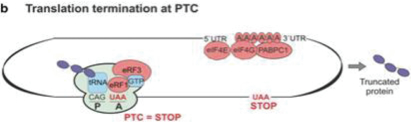
In the presence of a nonsense mutation the ribosome cannot pair the mRNA with a corresponding aa-tRNA and protein elongation stops and terminates, giving rise to a truncated protein.
3
Table of Contents
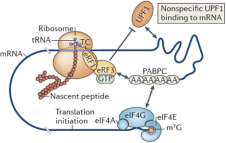
When the ribosome stalls after finding a premature termination codon, upstream protein factor 1 (“UPF1”), UPF2 and UPF3 are recruited. UPF1 binds nonspecifically to the mRNA, ribosome-associated eRF3 interacts with UPF1, thereby recruiting UPF2 and/or UPF3 (assisted by an exon–junction complex (“EJC”) bound to the 3’ untranslated region (“UTR”) or independently) and thus enables NMD. Some mRNAs may escape NMD for one or more rounds of translation, due to the inefficient recruitment of UPF1, UPF2 and/or UPF3 to the terminating ribosome.
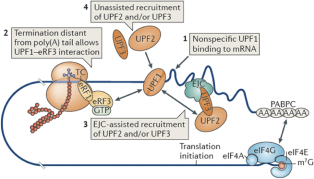
The assembly of a protein complex including UPF1, UPF2, UPF3, suppressor of morphogenetic effect on genitalia 1 (SMG1), SMG8, SMG9, DEAH box polypeptide 34 (DHX34) and the EJC signals that the TC is a PTC. At this point, translation may terminate, ultimately leading to the dissociation of the individual ribosomal subunits, the release factors and the nascent protein.
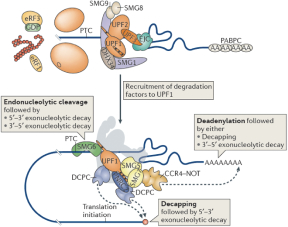
4
Table of Contents
Translation read-through across a premature termination codon (nonsense mutation) is a process in which the ribosome inserts related (near cognate) tRNAs which compete with the releasing factor complex and enable the insertion of a near cognate amino acid in the protein leading to translation of the full protein. Translation read-through across a nonsense mutation is a natural process that occurs at the rate of 1%. In such instances, the ribosome will not terminate the translational process prematurely regardless of a premature termination. ELX-02 is designed to enhance this natural process by increasing the read-through activity and the frequency of near cognate aa-tRNA binding within the A site of the ribosome. ELX-02 enables the production of sufficient amounts of full-length protein to restore activity of the mutated protein.
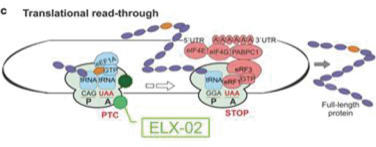
Current Data Indicating the Mechanism of Action of ELX-02
ELX-02 is an advanced aminoglycoside with poor antibiotic activity and markedly decreased affinity for the prokaryotic and mitochondrial ribosomes. Aminoglycosides, such as gentamicin, are potent antibiotics that bind to the decoding site in the prokaryotic ribosome and prevent protein translation in bacteria. In eukaryotic cells, aminoglycosides induce a conformational change that reduces the codon-anticodon recognition, enhancing the ability of an aa-tRNA to compete with the release factor complex for binding to the premature termination codon and increasing the probability that translational read-through of premature termination codons occurs. Despite promising results, aminoglycoside use as a read through therapy is restricted since they cause damage to the kidney and ear after prolonged administration. In addition, prolonged administration of antibiotic aminoglycosides may cause antibiotic resistance and may damage the natural microflora. Because it stabilizes the ribosomal RNA (or “rRNA”), ELX-02 prevents the assembly of the NMD factors required to initiate decay of mRNA. In this manner the PTC is not recognized and the insertion of the near cognate amino acid to the nascent polypeptide drives translation to produce a full-length, functional protein.
5
Table of Contents
ELX-02 is an investigational new chemical entity (NCE) advanced aminoglycoside optimized by successive rounds of medicinal chemistry to separate the sections of the molecule interacting with the prokaryotic ribosome responsible for the antibiotic activity from those portions of the molecule inducing translational read-through. ELX-02 has poor antibiotic activity and binds preferentially to the eukaryotic ribosome and is thereby designed to improve translational read-through. ELX-02’s low affinity for the bacterial ribosome decouples the antibacterial activity from the read-through activity. When compared in laboratory tests to gentamicin, a classic aminoglycoside, ELX-02 thus far has shown a 100-fold lower antibacterial activity and nine-fold higher read-through activity for nonsense mutations; this has been attributed to higher selectivity towards the cytoplasmic eukaryotic ribosome. Consequently, ELX-02 could potentially be used to treat hundreds of genetic diseases caused by nonsense mutations.
Our Disease Focus
We believe that the segment of cystic fibrosis and cystinosis patients with diagnosed nonsense mutations on one or both alleles represents a high unmet medical need as there are currently no approved therapeutics targeting the impairment caused by these mutations. There are existing in vitro assays, animal models and/or biomarker screens that have been demonstrated to be useful in assessing the potential therapeutic benefit of development compounds for these disease states. The design of clinical trials and the endpoints for measuring clinical benefit have been established for the currently approved therapeutics for these disorders. We believe these to be attractive development targets based on the potential use of these precedents to de-risk the program.
We believe that our library of related novel small molecules hold the potential to be disease-modifying therapies that may change the course of hundreds of genetic diseases and improve the lives of patients. Our early preclinical data in animal models of nonsense mutations suggest that drug product candidates from our read through compound library may have potential beneficial effects for each of the following diseases: cystic fibrosis, cystinosis, mucopolysaccharidosis type 1, Duchenne muscular dystrophy and Rett syndrome, and have demonstrated the potential for beneficial effects in multiple organs such as the brain, kidney, muscles and others. We intend to advance one or more additional molecules from our drug product candidate library toward clinical development by initiating the required investigational new drug (IND)-enabling studies in 2018.
Nonsense Mutation Cystic Fibrosis
Cystic fibrosis (CF) is the most prevalent genetic disease in the western world and affects an estimated 70,000 to 100,000 patients worldwide, with the vast majority of affected individuals in the United States, Canada, Europe and Australia. CF is the most common fatal inherited disease in Caucasians. The incidence of CF varies across the globe. CF affects one out of 3,500 births in the United States, one out of 2,000 to 3,000 in Europe, and one out of 2,500 in Australia.
Approximately 13% of the CF patients carry a nonsense mutation on the CFTR gene. CF is a progressive disease caused by a deficiency in CFTR activity with insufficient ionic transconductance in the cell membrane, which, in turn, leads to the accumulation of thick mucus in vital organs, particularly the lungs, pancreas and gastrointestinal tract. As a result, CF patients experience respiratory infections, chronic lung inflammation, and poor absorption of nutrients as well as many other conditions, and, in most cases, progressive respiratory failure. Although the life expectancy of CF patients has improved, the median age of death in the United States in 2014 was only 29 years, with a vast majority of such deaths resulting from respiratory failure.
The disease occurs at a rate of 1 in 2,500–6,000 newborns, depending on the region and ethnic origin. Patients with CF caused by nonsense mutations have some of the most severe forms of the disease and, other than palliative therapies, no treatment currently exists for them.
Mutations in the gene that encodes CFTR protein, which play a critical role in regulating the viscosity of the mucus layer that lines human organs, cause CF. The CFTR protein forms an ion channel that regulates the flow of ions in and out of the cells of vital organs such as the lungs, pancreas and gastrointestinal tract. We refer to
6
Table of Contents
this as ion flow. When CFTR protein expels the ions, osmosis draws water out of the cell and hydrates the cell surface. Through regulation of the location of the ions across the cell membrane, the amount of salts in the fluid both inside and outside the cell remains balanced.
In CF patients, the CFTR gene is defective, and as a result, CF patients lack the functional CFTR protein ion channel necessary to regulate ion flow. An altered ion concentration gradient between the inside and the outside of the cell reduces the amount of water molecules outside the cell, causing the accumulation of thick mucus on the epithelial surface as shown in Figure 1.
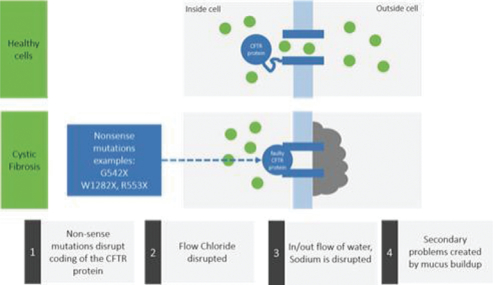
Figure 1: Ion Flow in Normal CFTR Protein Compared to Mutant CFTR Protein
The deficiency in CFTR protein activity in CF patients is particularly problematic in the lungs, where the build-up of thick mucus obstructs airflow and impairs proper immune response, which leads to chronic infection and persistent inflammation. In the pancreas and the gastrointestinal tract, the build-up of mucus prevents the release of digestive enzymes that help the body break down food and impairs the absorption of nutrients, resulting in poor growth and development.
Nonsense mutation Cystinosis
Cystinosis is an ultra-rare autosomal recessive lysosomal storage disease. Mutations in the CTNS gene (cystinosin), on the short arm of chromosome 17 (17p13), cause the primary defect in the disease. Cystinosin is a ubiquitous cystine-selective transport channel in the lysosomal membrane. Loss-of-function mutations prevent cystine efflux from the lysosome, causing massive accumulation of intra-lysosomal cystine in tissues throughout the body, and lead to apoptotic cell death, impaired physiology and end organ damage.
Affected children may appear fairly well until the age of 4-6 months, when progressive dysfunction and atrophy of the proximal renal tubule cause Fanconi syndrome and failure to thrive. By 10-12 years of age, dialysis or kidney transplantation is required to treat end-stage renal disease. Although the renal allograft is spared, lifespan is diminished by the inexorable dysfunction of other organs.
The most common nonsense mutation in the CTNS gene is W138X which has an overall incidence rate of 1 in every 62,500 live births in Quebec, Canada.
Current treatment includes cysteamine bitartrate (Cystagon® or Procysbi®). Cystagon was approved in the USA and Europe in 1994 and Procysbi was approved in the USA and Europe in 2013. Both therapies delay but
7
Table of Contents
do not cure the condition and despite treatment, patients eventually require dialysis and renal transplantation and experience significant morbidity in other organ systems.
Nonsense mutation Duchenne muscular dystrophy (nmDMD)
Muscular dystrophies are genetic disorders involving progressive muscle wasting and weakness. DMD is the most common and one of the most severe types of muscular dystrophy. DMD occurs when a mutation in the dystrophin gene prevents the cell from making a functional dystrophin protein. Dystrophin is a muscle membrane associated protein and is critical to the structural and membrane stability of muscle fibers in skeletal, diaphragm and heart muscle. The absence of normally functioning dystrophin results in muscle fragility, such that muscle injury occurs when muscles contract or stretch during normal use. As muscle damage progresses, connective tissue and fat replace muscle fibers, resulting in inexorable muscle weakness.
Because the dystrophin gene is located on the X chromosome, DMD occurs almost exclusively in young boys. According to Parent Project Muscular Dystrophy, DMD occurs in approximately 1 in 3,500 live male births, while information from Moat, et al. (2013) in the European Journal of Human Genetics indicate prevalence of approximately 1 in 5,000 live male births. Genetic tests are available to determine if a patient’s DMD is caused by a nonsense mutation. Based on information from Prior, et al. (1995) in the American Journal of Human Genetics, we estimate that a nonsense mutation is the cause of DMD in approximately 13% of patients. Overall, we estimate that there are approximately 7,000 nmDMD patients worldwide, with approximately 85% of such patients outside of the United States, including in Europe, Latin America, Asia Pacific, Middle East and Northern Africa regions. nmDMD is an ultra-rare, life threatening disorder. Without treatment, patients with DMD typically lose walking ability by their early teens, require ventilation support in their late teens and, eventually, experience premature death due to heart and lung failure. The average age of death for DMD patients is in their mid-twenties.
Two main treatments have received approval for DMD, Translarna™ (ataluren), which has received approval in the European Union (“EU”) for the treatment of underlying cause of nmDMD, and received a complete response letter from the FDA and is not approved in the US. Another marketed product is EXONDYS 51® (eteplirsen) Injection, approved in the US for the treatment of DMD patients who are amenable for exon 51 skipping.
Nonsense mutation Mucopolysaccharidosis type I (nmMPS I)
Mucopolysaccharidosis type I (MPS I) is a chronic, progressive genetic disorder caused by a deficiency of the enzyme alpha-L-iduronidase (IDUA). The deficiency of this enzyme leads to the accumulation of a class of molecules called glycosaminoglycans (GAGs). The accumulation of GAGs causes disruption in the movement of molecules inside the cell and leads to the subsequent dysfunction of cells, tissues and organs. Globally, MPS I occurs in about 1 in every 100,000 births for the severe form and 1 in 500,000 for the attenuated form. About 70% of MPS I patients carry one of two nonsense mutations, Q70X and W402X. Estimates suggest that 50%- 80% of all MPS I patients present with the severe form.
MPS I is broadly classified in two groups; severe MPS I and the attenuated MPS I. The symptoms of the severe form of MPS I develop after birth and progress rapidly, causing progressive respiratory, cardiac and musculoskeletal manifestations along with coarse facies, hepatosplenomegaly, hernias, deafness, and a shortened life expectancy. Lack of reabsorption of cerebrospinal fluid (CSF) in the severe phenotype leads to communicating hydrocephalus, delayed neuromotor and impaired cognitive development. Patients usually have increased intracranial pressure due to accumulation of macromolecules, which causes optic atrophy, corneal clouding, glaucoma and vision problems including corneal opacity, acute blindness and corneal thickening. Children with severe MPS I often die in the first decade of life due to respiratory failure, cardiac valvulopathy, and cardiorespiratory problems. The attenuated form of MPS I progresses slowly and usually manifests in early childhood. Patients with the attenuated phenotype have valvular, left ventricular diastolic and systolic abnormalities. Patients typically face cervical spinal cord injury, carpal tunnel syndrome and joint stiffness along with other deformities like kyphosis, scoliosis and spondylolisthesis. Children with attenuated MPS I have
8
Table of Contents
decreased intelligence quotient and language skills as compared to healthy children. Patients also suffer from recurrent headaches and optic nerve compression due to increased levels of CSF.
Treatment of severe and attenuated forms of MPS I is aimed at slowing the progression of the disease and improving the quality of life. Treatment can be broken into two classifications: supportive, symptom-based treatment and disease-specific treatment. The symptom-based treatment is coordinated by a specialized team to maintain patients’ health and prevent the comorbidity which may arise due to the progression of the disease. The disease-specific treatments include enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT). HSCT is considered the standard of care for children with severe MPS I. HSCT therapy is based on the principle that donor-derived hematopoietic stem cells (HSC) engraft in the recipient and can differentiate into numerous cell types, thus providing enzyme to deficient cells via metabolic cross-correction and clearing GAG storage material from host tissues. Recombinant α-L-iduronidase is used for the ERT treatment in the form of Laronidase and is currently licensed in the US, Europe, and Canada for treating non-CNS manifestations of MPS I. In this treatment, drugs are administered exogenously by weekly intravenous infusion. At this time, more effective and affordable strategies are being developed as an alternative approach to treat patients with MPS disorders.
Nonsense mutation Rett Syndrome
Rett syndrome is a X-linked neurodevelopmental disorder that predominantly affects girls and has a worldwide incidence of 1 in every 10,000-15,000 female births. The condition is characterized by normal development for the first 6-18 months of age, followed by a period of regression in which the girls lose language and motor skills and purposeful hand use is replaced by repetitive stereotyped hand movements. Decelerating head growth and autistic features such as diminished eye contact and emotional withdrawal also occur. Additional characteristics include anxiety, respiratory dysfunctions, impairment of sleeping patterns, cardiac abnormalities, seizures, loss of locomotion, and bone density deficits. Furthermore, girls with Rett syndrome tend to be growth-retarded and have a reduced life-span. Currently, no treatment exists for the underlying cause of the disease. Treatment is symptomatic and palliative. Thus, a high unmet medical need exists for patients with Rett syndrome.
Loss-of-function mutations in the gene encoding the transcriptional regulator methyl-CpG binding protein 2 (“Mecp2”) account for most cases of Rett syndrome. Mecp2 is a transcriptional repressor that binds to methylated promoters and recruits the histone deacetylases (“HDACs”) machinery to induce chromatin condensation. In neurons, Mecp2 has been implicated in the modulation of specific neuronal target genes in an activity-dependent manner, such as brain-derived neurotrophic factor (“BDNF”), but also has been implicated in both repression and activation of a large number of genes, in modulation of RNA splicing, and most recently has been suggested to affect global chromatin structure impacting the entire neuronal genome.
Recent work in mouse models of Rett syndrome suggests that the clinical condition may be reversible, insofar as the reintroduction of functional Mecp2, either ubiquitously or selectively, in the brain of Mecp2-deficient mice significantly improved at least some of their Rett-like behavioral deficits. Collectively, these results indicate that the neurological defects seen in Rett syndrome are amenable to rescue, either by gene or protein reintroduction or by the reactivation of a silenced or dysfunctional Mecp2 allele.
9
Table of Contents
Nonsense mutations in the Mecp2 gene account for approximately 30% of Rett syndrome cases. The most prominent nonsense mutations found in Rett syndrome, R168X, R255X, R270X and R294X, are all caused by a change of arginine to the stop codon, UGA.
Currently, no cure for Rett Syndrome exists. Treatment of Rett syndrome focuses on the management of symptoms, e.g., physical, occupational and speech-language therapy. Medicines can be used for seizure control and movement disorders along with treatments for breathing and gastrointestinal symptoms. The long-term prognosis of Rett patients is unknown. Patients have numerous comorbidities that are thought to contribute to a shortened lifespan.
Status of Clinical Programs
We are conducting a Phase 1 program in healthy volunteers that is designed to support studies of ELX-02 in patient populations in any indication caused by nonsense mutations and assess the safety of ELX-02. This initial phase of testing includes a small number of healthy volunteers. The studies assess the effects of ELX-02 on humans and measure bioavailability, excretion, safety and side effects, as well as the pharmacokinetics (what the body does to the drug) with increasing doses. Phase 1 studies include single ascending dose SAD, or Phase 1a, and multiple ascending dose MAD, or Phase 1b, studies.
We conducted a SAD study at the Tel Aviv Sourasky Medical Center in Israel (“TASMC”) between July 12, 2016 and March 15, 2017 and between November 2017 and December 2017 at SGS in Antwerp, Belgium. The study was designed as a Phase 1a, randomized, double-blinded, placebo-controlled, single dose escalation study to evaluate the safety, tolerability and pharmacokinetics of ELX-02 in healthy adult volunteers. The study was designed and executed in compliance with the International Conference of Harmonisation Good Clinical Practices E6 guideline and in compliance with applicable regulatory requirements in Israel, the United States and the European Union. Subjects were allocated to one of seven cohorts and received doses of ELX-02 ranging between 0.3 mg/kg and 7.5 mg/kg injected either IV (only in the 0.3 mg/kg) or SC. A total of 60 subjects participated in the study. The study did not show acute or chronic changes in vital signs, chemistry, hematology, biomarkers of early tubular injury, changes in serum creatinine, evidence of aberrant translational read-through of housekeeping genes or impact in auditory function using a battery of tests that included pure tone audiometry (“PTA”), high frequency audiometry (“HFA”), tympanometry, and Speech Reception Threshold (“SRT”), or vestibular function, using electronystagmography (“ENG”), the Dizziness Handicap Inventory (“DHI”) and the Tinnitus handicap Inventory (“THI”). No significant adverse events (“SAEs”), or serious adverse events of interest (“AEOIs”) or deaths occurred in the study. We did report an AEOI of unclear physiological significance when we observed high frequency pure tone fluctuations outside the normal hearing range in a single subject at 5 mg/kg in the Israeli cohort.
We are also conducting a multiple ascending dose MAD study in healthy volunteers. The study has been designed as a Phase 1b, randomized, double-blinded, placebo-controlled, multiple dose escalating study in healthy male and female subjects. The study consists of 5 cohorts of 9 subjects each. Subjects will be randomized to receive nine doses of ELX-02 or placebo at a ratio of 2:1 in each cohort. The study has been reviewed and approved by the Federal Agency for Medicines and Health Products (FAMHP) in Belgium, and by the Institutional Review Board in August 2017 in Antwerp, Belgium. The screening began in October 2017 and the study commenced in November 2017.
In November 2017, we submitted a Pre-IND package to the FDA to initiate regulatory discussions around our submission of an IND supporting our Phase 2 study of cystinosis in the U.S. In December 2017, we received FDA’s very productive written response, and we are on track for a mid-2018 IND submission in the U.S., and, subject to regulatory review of the IND and the IND becoming effective, we are targeting the 4th quarter 2018 for the first FPFV for our phase 2 cystinosis study in the U.S.
In January 2018, we held a Pre-CTA regulatory meeting with the FAMHP to discuss our submission of a CTA supporting our Phase 2 study of cystic fibrosis in Belgium. Based upon our very productive regulatory
10
Table of Contents
dialogue with FAMHP, we are on track for a mid-2018 CTA submission in Belgium, and, subject to regulatory review and approval of the CTA, we are targeting the 4th quarter 2018 for the first patient first visit (FPFV) for our phase 2 cystic fibrosis study in Belgium.
Status of Preclinical Programs
We have completed a comprehensive series of preclinical studies to assess the safety, pharmacokinetics and pharmacology of ELX-02.
Safety and Pharmacokinetic Studies of ELX-02
A comprehensive toxicology program in accordance with the ICH guideline M3 (R2) was completed for ELX-02 to support clinical studies.
We conducted repeated subcutaneous-dose toxicity studies in rats and beagle dogs for up to 28 days at dose levels significantly higher than those intended for humans. Both of these species are routinely selected for toxicology testing. Both species exhibited renal toxicities that were monitorable and reversible at doses higher than those intended for humans. The toxicology data generated thus far in these species suggest the kidney and urinary bladder may be a target organ at higher exposures. In addition, local injection site reactions were observed at all dose levels in both animal species. These injection site reactions are likely due to the unique anatomy of the cutaneous musculature in animals compared to humans and available literature suggests that injection site reactions in animals bear a poor concordance between animal and humans. Based on the 28-day rat study, the expected safety margin is more than 50X at the starting dose in the MAD study (0.1 mg/kg/dose) and 30X times the starting dose to be tested in subjects with CF (0.3 mg/kg/dose). At the anticipated efficacious clinical doses of 1 or 2.5 mg/kg the safety margin based on steady state plasma AUC values in the rat study are anticipated to be approximately 10 or 4X, respectively. The rat 28-day data is used to define the safety margin since the rat was determined to be the most sensitive species. We believe these data provide support for human clinical trials with durations up to 4 weeks, but we plan to complete long-term toxicity studies prior to initiation of our Phase 3 clinical trials. In definitive repeat-dose toxicity studies in rats and dogs, ELX-02 given as intermittent (twice weekly) SC doses over a 28-day period had little or no effect on body weight, food consumption, clinical signs of toxicity, ophthalmology, cardiovascular parameters, hematology or coagulation parameters. ELX-02 has no cochlear toxicity as evidenced in anatomic and functional hearing studies in 28-day rat studies at exposures where renal toxicity was noted (240 mg/kg/day). We are currently conducting 3-month toxicology studies in juvenile rats and in young dogs, as well as chronic toxicology studies in these 2 species for 6- and 9-months, respectively. The 3-month studies have both completed the in-life phase with no mortality and no significant in-life toxicity noted. Both studies are in reporting phase and pathology review. ELX-02 was not genotoxic in the core battery of in vitro and in vivo genotoxicity assays. As an aminoglycoside, ELX-02 has poor oral bioavailability but is 100% bioavailable following SC administration. In rats and dogs, ELX-02’s pharmacokinetic profile is comparable to that of conventional aminoglycosides. Additionally, ELX-02 does not undergo metabolization and is excreted unchanged almost exclusively via the urine.
Pharmacology Studies of ELX-02
We have conducted a series of preliminary studies to demonstrate the primary pharmacodynamics of ELX-02 in several genetic disease indications. We have tested the translational read-through capabilities of ELX-02 in vitro and in vivo, in cells and in animal models of nonsense mutations.
We have shown the in vitro read-through activity of ELX-02 in an array of plasmids engineered to contain nonsense mutations of genetic diseases and in cell-based models of CF, cystinosis, DMD, MPS 1, and Rett syndrome.
In CF, ELX-02 induced about 30% of wild type CFTR levels after 48 h in heterozygous G542/F508del human bronchial epithelial cells. In the G542X transgenic mouse, ELX-02 showed a ~5-fold increase in CFTR activity compared to control after twice weekly treatment for four weeks with 60 mg/kg.
11
Table of Contents
In DMD, ELX-02 induced a 35-fold increase in read-through in the R3381X mutation in the dystrophin gene in vitro, and in a preliminary study in the mdx mouse increased muscle force (forelimb grip strength tests) and motor activity (rotarod performance) and showing a trend of decreased serum creatine kinase (a measure of muscle injury).
In MPS 1, ELX-02 induced a 48-fold and a 98-fold increase in read-through of the W392X and Q70X mutations, respectively, in the in vitro assay of the Idua gene. In primary mouse embryonic fibroblasts carrying the Idua W392X mutation, ELX-02 led to a dose-dependent increase in α-L-iduronidase activity up to 24-fold and a concomitant reduction in stored GAGs to control levels. In Idua-W392X (Iduatm1Kmke) mice, ELX-02 treatment for 4-week resulted in elevated levels of α-L-iduronidase activity and reduced GAG storage in the brain, spleen, heart, liver, kidneys, lungs, and femoral bone in a dose-dependent manner. In brain and spleen tissues of the Idua-W392X mouse model, ELX-02 treatment reduced the compensatory increases seen in the activity of the lysosomal enzymes ß-glucuronidase and ß-hexosaminidase.
In Rett syndrome, ELX-02 increased translational read-through of multiple nonsense mutations of the MECP2 gene, R168X (14-fold), R255X (32-fold), R270X (83-fold), and R294X (25-fold) in vitro. In fibroblasts derived from a human male Rett syndrome patient carrying the R294X mutation, ELX-02 increased Mecp2 protein translation and expression levels in nuclei. In neurons and glial cells derived from stem cells overexpressing Mecp2 R168X-GFP and Mecp2 R255X-GFP, ELX-02 induced a dose-dependent increase in Mecp2-GFP protein. In Mecp2R168X cells, ELX-02 increased BDNF mRNA levels by ~ 4-fold, suggesting a downstream effect of the increased Mecp2 protein. In female Mecp2R168X/x mice, ELX-02 was measurable in and increased Mecp2 in the brain and lengthened the latency period of time to fall and in distance traveled on a rotarod test.
In cystinosis, ELX-02 increased read-through of the W138X mutation in the CTNS gene by 30-fold in vitro. In primary homozygous W138X fibroblasts, ELX-02 led to a dose-dependent increase in normalized CTNS mRNA levels, suggesting a decrease in nonsense mediated mRNA decay, and a corresponding reduction in cystine levels to wild-type levels, suggesting translation of a functional CTNS channel.
Intellectual Property
Patents and Trade Secrets
Our licensed and owned patents and patent applications relate to our lead compounds that exhibit read-through properties and include patent applications directed to new compositions of matter and to methods of treating genetic diseases such as cystic fibrosis, cystinosis, Duchenne’s muscular dystrophy, ataxia-telengiectasia, Hurler syndrome, hemophilia A and B, Usher syndrome, Tay-Sachs and Rett syndrome, including combination therapies with existing treatments for these indications, such as CFTR modulators for the CF indication.
As of August, 29 2013, we licensed two pending U.S. provisional patent applications and subsequent Patent Cooperation Treaty (“PCT”) applications claiming priority from these, from which we have so far gained patent protection in the United States and in Europe, Japan, Canada and Israel for composition of matter, methods of use, and combination therapies relating to our lead compound, ELX-02 (formerly known as NB124) and other compounds (e.g. ELX-03; formerly known as NB84). Additional patent applications are pending in India, as are divisional applications in Europe, Israel and Japan. If we continue to pursue protection, and if any patents issue based on these applications, we expect such patents to expire between 2027 and 2031, depending on any extensions of term for which we may be eligible that we may be granted.
As of June 04, 2015, we own a PCT application for methods of use relating to our lead compound, ELX-02, and other related compounds for treatment of Rett Syndrome and we intend to seek patent protection in the United States and in selected jurisdictions (Canada, Europe, Hong Kong, India, Israel, and Japan) for such
12
Table of Contents
methods. If any patents are issued in connection with this application, we expect such patents to expire in 2036, depending on any extensions of term for which we may be eligible that we may be granted.
In addition, we have four pending PCT applications, filed on September 2, 2016, all of which generally relate to new compositions of matter and to methods of treating genetic diseases.
As of March 15, 2018, we have a pending patent application in India related to the large-scale synthesis of our compound, ELX-02, and other related new compounds, and we intend to seek similar patent protection in the United States and in selected jurisdictions worldwide. If any patents are issued based on this application, we expect such patents to expire in 2037, depending on any extensions of term for which we may be eligible that we may be granted.
With respect to our synthetic-aminoglycosides-based technology platform, we primarily rely on trade secrets and know-how to protect the proprietary nature of our platform. However, trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data, know-how and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
License Agreements
Research and License Agreement with Technion Research and Development Foundation Ltd.
On August 29, 2013, we entered into a license agreement with the Technion Research and Development Foundation Ltd., or TRDF, which was further amended and addended to reflect, inter alia, the assignment of patents and extension of research periods, with respect to certain technology relating to aminoglycosides and the redesign of aminoglycosides for the treatment of human genetic diseases caused by premature stop mutations and further results of the research of the technology, in order to develop and commercialize products based on such technology. The license agreement provides us with an exclusive, worldwide, non-transferrable license, with a right to grant sublicenses, and royalty-bearing licenses to the TRDF inventions, TRDF patent rights, TRDF’s interest in the joint inventions and joint patent rights, and certain materials and research results owned by TRDF, solely with respect to products in the field of prevention, diagnosis or treatment of any human disease or condition therefor. In return for the license we will pay TRDF (i) milestone payments with respect to each licensed product upon the achievement of certain pre-defined goals by us or one of our sublicensees as follows: $100,000 upon first dosing of a patient in Phase II clinical study; $1,000,000 upon first dosing of a patient in pivotal study; $1,000,000 upon first filing on a new drug application (NDA); (ii) certain royalties on a low- to mid- single-digit percentage of all net sales (subject to change in the case of (a) sublicensing to a big pharmaceutical or biotechnology company, or (a) payment of royalties to third parties, or (c) commercialization by a third party of an authorized generic to a licensed product); (iii) a low- to mid- double-digit percentage of any non-royalty sublicense income; (iv) an exit fee in the amount of a one digit percentage of any consideration paid upon an exit event (as defined in the agreement); and (v) in the case of an initial public offering for a number of ordinary shares equal to 3% of our outstanding shares on a fully diluted basis (as defined in the agreement) immediately prior to the closing of such initial public offering. If we distribute any dividends prior to an exit event, TRDF will be entitled to dividends as if it was holding 3% of our outstanding shares. In addition to the milestone payments, we undertook to annually fund the research activities under the license, currently in the amount of $0.1 million per year. The license agreement further provides TRDF with an additional pre-emptive right, in force until the first exit event, to invest an amount equal to up to 5% of the amount contemplated to be
13
Table of Contents
raised in a proposed investment. TRDF is also entitled, until the closing of an exit event, to appoint an observer to the board under certain restrictions such as confidentiality or conflict of interest. In addition, we will reimburse TRDF for all patent filing expenses as of the effective date of the license agreement and for past patent filing expenses in the amount of several hundred thousand New Israeli Shekels upon the occurrence of certain conditions.
Under the license agreement, TRDF reserved the right, for itself, the Technion and other not-for-profit research organizations to utilize the technology solely for educational purposes. Furthermore, Professor Bassov, the principal investigator, had ongoing research programs involving covered compounds (as defined in the agreement) that are being funded by the National Institute of Health in the U.S., or the NIH, under sub-awards from the University of Alabama and the University of Michigan and it is possible that such research programs will overlap with the research conducted according to the terms of the agreement. In the case of any such overlap, the work product of such research will be subject to the terms and conditions of such sub-awards, including certain obligations under 35 U.S.C. §§ 200-212 or 37 C.F.R § 401 et seq. in the case of any TRDF inventions that are also “subject invention” as defined in 35 U.S.C. §201.
The license agreement shall continue in full force and effect on a product-by-product and country-by-country basis until the expiration of all payment obligations for any such licensed product as described above. Upon the expiration, we will have a fully-paid up, worldwide non-exclusive, perpetual, irrevocable license (with the right to grant sublicenses) to use certain materials and the research results, solely with respect to products in the field of prevention, diagnosis or treatment of any human disease or condition.
Manufacturing
ELX-02 is manufactured under current Good Manufacturing Practice (“cGMP”) conditions and is formulated as a sterile frozen liquid in glass vials for parenteral subcutaneous (SC injection) administration.
We do not own or operate manufacturing or distribution facilities for the production of clinical quantities of ELX-02 or for our other preclinical product candidates. We currently rely, and expect to continue to rely, on third parties for the manufacture, packaging, labeling and distribution of clinical supplies of ELX-02 as well as any other candidate that we may develop.
We engage separate manufacturers for drug substance and drug product. We have a relationship with a manufacturer that is capable of providing fill and finish services for our clinical product at the current scale. To support later clinical trials, transfer of the manufacturing and release to a manufacturer with higher lot scale capacity will be needed for our clinical product.
All of our current drug candidates are organic compounds of low molecular weight. We have selected our lead compounds not only on the basis of their potential efficacy and safety but also for their ease of synthesis and reasonable cost of their starting materials. ELX-02 is manufactured in reliable and reproducible synthetic processes. We currently rely on a single third-party manufacturing source for the production of a key raw material, produced by bacterial fermentation. We do not currently have any agreements with third-party manufacturers for the long-term commercial supply of ELX-02 or the fermentation-derived starting material, although we may seek to establish such arrangements in the future.
We currently obtain supplies of ELX-02 from third-party manufacturers pursuant to agreements that include specific supply timelines and volume expectations. If a manufacturer should become unavailable to us for any reason, we would seek to obtain supply from another manufacturer engaged by us for the applicable product or service. In the event that we were unable to procure the applicable supply from a currently qualified manufacturer, we believe that there are a number of potential replacements for each of our outsourced services, however we would likely experience delays in our ability to supply ELX-02 in advancing our clinical trials while we identify and qualify replacement suppliers.
14
Table of Contents
Government Regulation
Drug Development and Approval in the United States
The preclinical studies and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, export, and marketing, among other things, of our product candidates are subject to extensive regulation by governmental authorities in the United States, the European Union and other territories. In the United States, pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and other laws, including, in the case of biologics, the Public Health Service Act. Failure to comply with FDA requirements, both before and after product approval, may subject us and/or our partners, contract manufacturers, and suppliers to administrative or judicial sanctions, including FDA refusal to approve applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, fines and/or criminal prosecution.
The process for obtaining regulatory approval to market a medicine is expensive, often takes many years, and can vary substantially based on the type, complexity, and novelty of the product candidates involved. The steps required before a drug may be approved for marketing of an indication in the United States generally include:
| (a) | preclinical laboratory tests and animal tests; |
| (b) | submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may commence; |
| (c) | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended use; |
| (d) | submission to the FDA of a NDA; |
| (e) | FDA pre-approval inspection of the manufacturing and clinical study sites identified in the NDA; and |
| (f) | FDA review and approval of the NDA. |
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological animal studies to assess the potential safety and efficacy of the product candidates. Preclinical safety tests intended for submission to FDA must be conducted in compliance with FDA’s Good Laboratory Practice (“GLP”) regulations and the U.S. Department of Agriculture’s Animal Welfare Act. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application that must become effective before human clinical trials may be commenced. The IND will automatically become effective 30 days after receipt by the FDA, unless the FDA before that time raises concerns about the drug candidate or the conduct of the trials as outlined in the IND. The IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. We cannot assure you that submission of an IND will result in FDA authorization to commence clinical trials or that once commenced, other concerns will not arise. FDA may stop the clinical trials by placing them on “clinical hold” because of concerns about the safety of the product being tested, or for other reasons.
Clinical trials involve the administration of the investigational product to healthy volunteers or to patients, under the supervision of qualified principal investigators. The conduct of clinical trials is subject to extensive regulation, including compliance with the FDA’s bioresearch monitoring regulations and Good Clinical Practice (“GCP”) requirements, which establish standards for conducting, recording data from, and reporting the results of clinical trials, and are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected.
Clinical trials must be conducted in accordance with protocols that detail the objectives of the study, the criteria for determining subject eligibility, the dosing plan, patient monitoring requirements, timely reporting of adverse events, and other elements necessary to ensure patient safety, and any efficacy criteria to be evaluated.
15
Table of Contents
Each protocol must be submitted to FDA as part of the IND; further, each clinical study at each clinical site must be reviewed and approved by an independent institutional review board, prior to the recruitment of subjects. The institutional review board’s role is to protect the rights and welfare of human subjects involved in clinical studies by evaluating, among other things, the potential risks and benefits to subjects, processes for obtaining informed consent, monitoring of data to ensure subject safety, and provisions to protect the subjects’ privacy. Foreign studies conducted under an IND application must meet the same requirements that apply to studies being conducted in the United States. Data from a foreign study not conducted under an IND may be submitted in support of a NDA if the study was conducted in accordance with GCP and FDA is able to validate the data.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap, and different trials may be initiated with the same drug candidate within the same phase of development in similar or differing patient populations. Phase I studies may be conducted in a limited number of patients but are usually conducted in healthy volunteer subjects. The drug is usually tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmacokinetics and pharmacodynamics. Phase II usually involves studies in a larger, but still limited patient population to evaluate preliminarily the efficacy of the drug candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible short-term adverse effects and safety risks. Phase III trials are undertaken to gather additional information to evaluate the product’s overall risk-benefit profile, and to provide a basis for physician labeling. Phase III trials evaluate clinical efficacy of a specific endpoint and test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase I, Phase II or Phase III testing might not be completed successfully within any specific time period, if at all, with respect to any of our product candidates. Results from one trial are not necessarily predictive of results from later trials. Furthermore, the FDA, sponsor or institutional review board may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
We must register each controlled clinical trial, other than Phase I trials, on a website administered by the NIH (http://clinicaltrials.gov). Registration must occur not later than 21 days after the first patient is enrolled, and the submission must include descriptive information (e.g., a summary in lay terms of the study design, type and desired outcome), recruitment information (e.g., target number of participants and whether healthy volunteers are accepted), location and contact information, and other administrative data (e.g., FDA identification numbers). Within one year of a trial’s completion, information about the trial including characteristics of the patient sample, primary and secondary outcomes, trial results written in lay and technical terms, and the full trial protocol must be submitted to the FDA. The results information is posted to the website unless the drug has not yet been approved, in which case the FDA posts the information shortly after approval. A NDA, and certain other submissions to the FDA require certification of compliance with these clinical trials database requirements. There are proposals to expand these registration requirements to additional studies.
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture and composition of the product and proposed labeling for the product, are submitted to the FDA as part of a NDA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act, as amended, the fees payable to the FDA for reviewing a NDA, as well as annual fees for commercial manufacturing establishments and for approved products, can be substantial. The NDA review fee alone can exceed $2.4 million subject to certain limited deferrals, waivers and reductions that may be available. Each NDA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within sixty days following submission of the application. If the FDA finds the NDA sufficiently complete, the FDA will “file” the NDA, thus triggering a full review of the application. The FDA may refuse to file any NDA that it deems incomplete or not properly reviewable at the time of submission. Current FDA performance goals provide for action on an application within 12 months of submission. The FDA, however, may not approve a drug within these established timeline goals and its review clock for a particular NDA is subject to change from time to time because the review process is often significantly extended by FDA requests for additional information or clarification. As part of its review, the FDA may refer the NDA to an advisory committee composed of outside experts for evaluation and a recommendation
16
Table of Contents
as to whether the application should be approved. Although the FDA is not bound by the recommendation of an advisory committee, the agency usually has followed such recommendations.
Further, the outcome of the review, even if generally favorable, may not be an actual approval but instead a “complete response letter” communicating the FDA’s decision not to approve the application at that time, outlining the deficiencies in the NDA that need to be addressed in order to be eligible for approval, and identifying what information and/or data (including additional preclinical or clinical data) is required before the application can be approved. Even if such additional information and data are submitted, the FDA may decide that the NDA still does not meet the standards for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we do.
The FDA will typically inspect one or more clinical sites to assure compliance with GCP before approving an NDA. The FDA also will inspect the facility or the facilities at which the product is manufactured before the NDA is approved to assure compliance with cGMP. The FDA will not approve the product unless GCP and cGMP compliance is satisfactory. The FDA may also take into account results of inspections performed by certain counterpart foreign regulatory agencies in assessing compliance with GCP or cGMP. The FDA has entered into international agreements with foreign agencies, including the EMA, in order to facilitate this type of information sharing. If the FDA determines the application, clinical sites, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information.
The FDA may deny approval of a NDA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, and may require that additional studies be conducted following approval as a condition of the approval. FDA also may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a Risk Evaluation Mitigation Strategy (“REMS”), or otherwise limit the scope of any approval. A REMS may include various elements, ranging from a medication guide to limitations on who may prescribe or dispense the drug, depending on what the FDA considers necessary for the safe use of the drug. To market a product for other indicated uses, or to make certain manufacturing or other changes, requires FDA review and approval of a NDA Supplement or new NDA and the payment of applicable review fees. Further post-marketing testing and surveillance to monitor the safety or efficacy of a product may be required. In addition, new government requirements may be established that could delay or prevent regulatory approval of our product candidates under development.
Under the Pediatric Research Equity Act of 2003 (“PREA”), NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is determined by the FDA to be safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted. As the FDA has not issued regulations applying PREA to orphan-designated indications, submission of a pediatric assessment is not presently required for an application to market a product for an orphan-designated indication. However, PREA compliance may be required if approval is sought for other indications for which the drug has not received orphan designation.
Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take years to complete. Data obtained from clinical trials are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all.
17
Table of Contents
We may encounter difficulties or unanticipated costs in our efforts to secure necessary FDA approvals, which could delay or preclude us from marketing our products. The FDA may refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. The advisory committee process may cause delays in the approval timeline. The FDA is not bound by the recommendation of an advisory committee, but it considers such recommendations carefully, particularly any negative recommendations or limitations, when making drug approval decisions.
The FDA may limit the indications for use, approve narrow labeling relegating a drug to second- line or later-line use, add limitations of use to the labeling or place other conditions on approvals, which could restrict the marketing of an approved product. Further, FDA may require that certain contraindications, warnings or precautions be included in the product labeling. After approval, some types of changes to the approved product, such as adding new indications, which may themselves require further clinical testing, or changing the manufacturing process are subject to further FDA review and approval.
Post-approval Requirements
After FDA approval of a product is obtained, we may be required to comply with a number of post-approval requirements, including, among other things, establishment registration and product listing, record-keeping requirements, reporting certain adverse reactions and production problems to the FDA, providing updated safety and efficacy information, and complying with requirements concerning advertising and promotional labeling. As a condition of approval of an NDA, the FDA may require the applicant to conduct additional clinical trials or other post-market testing and surveillance to further monitor and assess the drug’s safety and efficacy.
The FDA also has the authority to require a REMS to ensure the safe use of the drug. In determining whether a REMS is necessary, the FDA must consider the size of the population likely to use the drug, the seriousness of the disease or condition to be treated, the expected benefit of the drug, the duration of treatment, the seriousness of known or potential adverse events, and whether the drug is a new molecular entity. A REMS may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate health care providers of the drug’s risks, limitations on who may prescribe or dispense the drug, or other measures that the FDA deems necessary to assure the safe use of the drug. In addition, the REMS must include a timetable to assess the strategy at 18 months, three years, and seven years after the strategy’s approval. The FDA may also impose a REMS requirement on an approved drug if the FDA determines, based on new safety information, that a REMS is necessary to ensure that the drug’s benefits outweigh its risks.
The FDA regulates strictly the marketing, labeling, advertising and promotion of drug products that are placed on the market. Although physicians may prescribe a drug for off-label uses, manufacturers may only promote for the approved indications and in accordance with the approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with the laws and regulations governing advertising and promotion can have negative consequences, including adverse publicity, warning and untitled letters from the FDA, requests for corrective advertising or communications with doctors, and civil penalties or criminal prosecution.
In addition, the distribution of approved prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (“PDMA”), which regulates the distribution of drugs and drug samples at the federal level and sets minimum standards for the registration and regulation of drug distributors by the states. Similarly, the Drug Supply Chain Security Act (“DSCSA”), regulates the distribution of prescription pharmaceutical drugs, requiring passage of a pedigree to track and trace each prescription drug at the saleable unit level through the distribution system. The DSCSA also imposes obligations on drug manufacturers related to suspect product identification/removal, verification, dealing only with authorized trading partners, and other elements. The DSCSA will be effective incrementally over a 10-year period, with serialization of prescription drug products
18
Table of Contents
distributed in the United States effective November 27, 2017 for drug manufacturers. The PDMA, DSCSA, and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes certain procedural and recordkeeping requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of process and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and any future commercial quantities of our product candidates. Future FDA inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution or require substantial resources to correct. In addition, discovery of problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing.
Once approval is granted, FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if issues bearing on the product’s safety or efficacy are discovered. Newly discovered or developed safety or effectiveness data or other information may also require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. New government requirements, including those resulting from new legislation, may be established that could delay or prevent FDA approval of our products under development or negatively impact the marketing of any future approved products.
Orphan Drug Designation
We have received orphan drug designation from the FDA for ELX-02 for the treatment of MPS I for the treatment of Rett syndrome. The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which is defined as a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Orphan drug designation can provide opportunities for grant funding towards clinical trial costs, tax advantages and FDA user-fee benefits. In addition, if a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity.
Rare Pediatric Disease Designation and Priority Review Voucher
Some orphan drugs may also qualify for designation as a “rare pediatric disease” under Section 529 of the FDCA. Section 529 is similar to the Orphan Drug Act, as both require that the “rare disease or condition” affect fewer than 200,000 persons in the United States. In the Advancing Hope Act of 2016, Section 529 was changed so that the “rare pediatric disease” must also meet the additional criteria of being a serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents. Under Section 529 of the
19
Table of Contents
FDCA, FDA will award priority review vouchers to sponsors of rare pediatric disease product applications that meet these criteria. Under this program, a sponsor who receives an approval for a drug or biologic for a “rare pediatric disease” may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product.
Hatch-Waxman Exclusivity
Market and data exclusivity provisions under the FDCA can delay the submission or the approval of certain applications for competing products. The FDCA provides a five-year period of non-patent data exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety. During the exclusivity period, the FDA generally may not accept for review an abbreviated new drug application (“ANDA”) or a 505(b)(2) NDA submitted by another company that references the previously approved drug. An ANDA or 505(b)(2) NDA may be submitted after four years if it contains a certification of patent invalidity or non-infringement.
For some applications that do not qualify for five-year exclusivity, the FDCA provides a shorter three-year period of market exclusivity. Three-year exclusivity applies to an NDA, 505(b)(2) NDA, or supplement to an existing NDA or 505(b)(2) NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant, are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, strengths or dosage forms of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for generic versions of the original, unmodified drug product. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric Exclusivity
Pediatric exclusivity is another type of non-patent market exclusivity in the United States and, if granted, provides for the attachment of an additional six months of market protection to the term of any existing Orange Book-listed patents or regulatory exclusivity, including the non-patent exclusivity periods described above. This six-month exclusivity may be granted based on the voluntary completion of a pediatric study or studies in accordance with an FDA-issued “Written Request” for such a study or studies.
Regulation Outside the United States
In order to market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of any future approved products. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Regulation in the European Union
We have obtained an orphan medicinal product designation from the European Commission, following an evaluation by the EMA’s Committee for Orphan Medicinal Products, for ELX-02 for the treatment of nmMPS I.
20
Table of Contents
The European Commission can grant orphan medicinal product designation to products for which the sponsor can establish that it is intended for the diagnosis, prevention, or treatment of (1) a life-threatening or chronically debilitating condition affecting not more than five in 10,000 people in the European Union, or (2) a life threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that sales of the drug in the European Union would generate a sufficient return to justify the necessary investment. In addition, the sponsor must establish that there is no other satisfactory method approved in the European Union of diagnosing, preventing or treating the condition, or if such a method exists, the proposed orphan drug will be of significant benefit to patients. Orphan drug designation is not a marketing authorization. It is a designation that provides a number of benefits, including fee reductions, regulatory assistance, and the possibility to apply for a centralized EU marketing authorization, as well as 10 years of market exclusivity following a marketing authorization. During this market exclusivity period, neither the European Medicines Agency, nor the European Commission nor the Member States can accept an application or grant a marketing authorization for a “similar medicinal product.” A ‘similar medicinal product’ is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may be reduced to six years if, at the end of the fifth year, it is established that the orphan designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. In addition, a competing similar medicinal product may in limited circumstances be authorized prior to the expiration of the market exclusivity period, including if it is shown to be safer, more effective or otherwise clinically superior to our product. Our product can lose orphan designation, and the related benefits, prior to us obtaining a marketing authorization if it is demonstrated that the orphan designation criteria are no longer met.
Overview of Application Process
To obtain regulatory approval of a drug under the European Union’s regulatory systems and authorization procedures, an applicant may submit a Marketing Authorization Application (“MAA”) under a centralized, decentralized, or national procedure. The centralized procedure is compulsory for certain medicinal products, including orphan medicinal products, like ELX-02 and medicinal products produced by certain biotechnological processes, and optional for certain other innovative products. The centralized procedure enables applicants to obtain a marketing authorization that is valid in all EU member states based on a single application. Under the centralized procedure, the EMA’s Committee for Human Medicinal Products (“CHMP”), is required to adopt an opinion on a valid application within 210 days, excluding clock stops, during which additional written or oral information is to be provided by the applicant in response to questions. More specifically, on day 120 of the procedure, once the CHMP has received the preliminary assessment reports and opinions from the rapporteur and co- rapporteur, the CHMP prepares a list of potential outstanding issues, referred to as “other concerns” or “major objections.” These are sent to the applicant together with CHMP’s recommendation. The CHMP can make one of two recommendations: (1) the marketing authorization could be granted provided that satisfactory answers are given to the “other concerns” and/or “major objections” identified and that all conditions outlined in the list of outstanding issues are implemented and complied with; or (2) the product is not approvable since there are “major objections.”
Applicants have three months from the date of receiving the potential outstanding issues to respond to the CHMP, and can request a three-month extension if necessary. The granting of a marketing authorization will depend on the recommendations and potential major objections identified by the CHMP as well as the ability of the applicant to adequately respond to these findings. An accelerated assessment can be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of major public health interest, in particular from the viewpoint of therapeutic innovation. In this circumstance, the EMA ensures that the opinion of the CHMP is given within 150 days. After the adoption of the CHMP opinion, a decision on the MAA must be adopted by the European Commission, after consulting the EU member states, which in total should be completed in 67 days.
An applicant for an MAA may request a re-examination in the event of a negative opinion, in connection with which CHMP appoints new rapporteurs. Within 60 days of receipt of the negative opinion, the applicant
21
Table of Contents
must submit a document explaining the basis for its request for re-examination. The CHMP has 60 days to consider the applicant’s request for re-examination. The applicant may request an oral explanation before the CHMP, which is routinely granted, following which CHMP will adopt a final opinion. The final opinion, whether positive or negative, is published by the CHMP shortly following the CHMP meeting at which the oral explanation takes place.
Conditional Marketing Authorizations
In specific circumstances, EU legislation enables applicants to obtain a marketing authorization on a conditional basis prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for products designated as orphan medicinal products, if (1) the risk-benefit balance of the product is positive, (2) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (3) the product fulfills unmet medical needs, and (4) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions and/or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization. The granting of a conditional marketing authorization will depend on the applicant’s ability to fulfill the conditions imposed within the agreed upon deadline.
Variations to Conditional Marketing Authorizations
After the granting of a conditional marketing authorization, the marketing authorization holder may submit an application to vary the conditional marketing authorization under a variation procedure. In the case of the introduction of an additional therapeutic indication, the timeframe for the variation procedure for the initial assessment of the dossier is generally 90 days (plus up to 20 days for validation).
In the framework of a variation application assessment procedure, however, the EMA may send one or more requests for supplementary information to the marketing authorization holder, requiring that additional information be provided by the marketing authorization holder to support its variation application. Such supplementary requests will be sent together with a timetable stating the date by when the marketing authorization holder must submit the requested data and, where appropriate, the extended evaluation period to be applied to such variation procedure. The 90-day variation procedure may be suspended for up to three months for the marketing authorization holder to submit its responses to such supplementary requests. The marketing authorization holder will be notified of the outcome of the CHMP’s assessment of the variation procedure within 15 days from the adoption of the CHMP opinion. If unfavorable, the CHMP opinion may be subject to a re-examination procedure upon the marketing authorization holder’s request. This may imply an additional minimum two-month procedure. If the CHMP opinion is favorable, the European Commission will vary the marketing authorization to introduce the additional therapeutic indication within approximately two months from the receipt of the final CHMP opinion.
Additional Requirements and Considerations
Prior to obtaining a marketing authorization in the European Union, applicants have to demonstrate compliance with all measures included in an EMA-approved Pediatric Investigation Plan (“PIP”), covering all subsets of the pediatric population, unless the EMA has granted (1) a product-specific waiver, (2) a class waiver, or (3) a deferral for one or more of the measures included in the PIP. In the case of orphan medicinal products, completion of an approved PIP can result in an extension of the aforementioned market exclusivity period from ten to twelve years.
22
Table of Contents
In the European Union, independently generated data submitted as part of a full marketing authorization application dossier are protected by regulatory data protection (‘data exclusivity’) for a period of eight years from the granting of a marketing authorization for a ‘reference product’. This means that for a period of eight years, competent authorities may not accept marketing authorization applications that rely on the independently generated data in the marketing authorization dossier of the reference product. Generic medicinal products that rely on the independently generated data of the reference product may not be placed on the market for 10 years from the granting of the initial marketing authorization for the reference medicinal product. These periods of data exclusivity and market exclusivity do not prevent other companies from obtaining a marketing authorization based on their own independently generated data.
Were we able to obtain a marketing authorization for ELX-02 for any indication in the European Union, we would be required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products. We must, for example, comply with the EU’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. Other requirements relate to, for example, the manufacturing of products and active pharmaceutical ingredients in accordance with good manufacturing practice standards. Competent authorities of EU member states may conduct inspections to verify our compliance with applicable requirements, and we will have to continue to expend time, money and effort to remain compliant. Non-compliance with EU requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties in the EU Similarly, failure to comply with the EU’s requirements regarding the protection of individual personal data can also lead to significant penalties and sanctions. Individual EU member states may also impose various sanctions and penalties in case we do not comply with locally applicable requirements.
Off-label promotion of medicinal products is prohibited in the European Union. The applicable laws at EU level and in the individual EU member states also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the European Union could be penalized by administrative measures, fines and imprisonment. These laws may further limit or restrict our promotional activities with health care professionals. In addition, legislation adopted at the EU level and by individual EU member states require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics (“SmPC”), as approved by the competent authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. Promotion of indications not covered by the SmPC is specifically prohibited.
The EMA is responsible for coordinating inspections to verify compliance with the principles of GCP, cGMP, GLP, and good pharmacovigilance practice (“GVP”). These inspections are also intended to verify compliance with other aspects of the supervision of authorized medicinal products in use in the European Union. The EMA coordinates any inspection requested by the CHMP in connection with the assessment of MAAs or matters referred to these committees. Inspections may be routine or triggered by issues arising during the assessment of the dossier or by other information, such as previous inspection experience. Inspections usually are requested during the initial review of an MAA but could arise post-authorization.
Inspectors are drawn from member states of the European Union and the European Economic Area. Following an inspection, the inspectors provide a written inspection report to the inspected site or applicant and provide an opportunity for response. Some inspection reports require follow-up and may result in additional adverse consequences due to critical or major findings. The inspectors and the CHMP will comment on any response from an inspected site or applicant and may monitor future compliance with any proposed corrective action plan.
23
Table of Contents
In the GCP area, inspectors grade their findings according to the following scale:
| • | Critical: Conditions, practices or processes that adversely affect the rights, safety or well-being of the subjects or the quality and integrity of data. Observations classified as critical may include a pattern of deviations classified as major. |
| • | Major: Conditions, practices or processes that might adversely affect the rights, safety or well-being of the subjects and/or the quality and integrity of data. Observations classified as major may include a pattern of deviations or numerous minor observations. |
| • | Minor: Conditions, practices or processes that would not be expected to adversely affect the rights, safety or wellbeing of the subjects or the quality and integrity of data. Minor observations indicate the need for improvement of conditions, practices and processes. |
| • | Comments: Suggestions on how to improve quality or reduce the potential for a deviation to occur in the future. |
Possible consequences of critical and major findings include rejection of clinical trial data, causing significant delays in obtaining final marketing authorization, or other direct action by national regulatory authorities.
Early Access Programs
Many jurisdictions allow the supply of unauthorized medicinal products in the context of strictly regulated and exceptional early access programs, and some countries may provide reimbursement for drugs provided in the context of such programs. In the European Union, the legal basis for early access programs, also referred to as named-patient and compassionate use programs, is set out in the EU legislation regulating the authorization, manufacture, distribution and marketing of medicinal products. Detailed regulatory requirements applicable to early access programs have been adopted and implemented by EU member states in their national laws. The promotion, advertising and marketing of unauthorized medicinal products is generally prohibited, and authorization for early access programs must generally be obtained from national competent authorities, which might not grant such authorization. Obtaining authorization for an early access program in one country does not ensure that authorization will be obtained in another country. U.S. law permits “expanded access” (also known as compassionate use and treatment use) for certain patients with serious diseases who have no comparable alternative treatment options. To provide expanded access, sponsors must submit detailed regulatory information to the FDA. FDA authorization depends on several different factors, including whether expanded access will interfere with related clinical trials or drug development. Sponsors may not promote products as safe or effective for expanded-access uses.
Pharmaceutical Pricing and Reimbursement
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of pharmaceuticals have been a focus of this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. For example, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 expanded Medicare coverage for drug purchases by the elderly and changed the way Medicare covers and pays for pharmaceutical products. Cost reduction initiatives and other provisions of this law may decrease the coverage and reimbursement rate that we may receive for any approved products. Likewise, healthcare reform measures under the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, referred to together as the Affordable Care Act, contain provisions that may reduce the profitability of drug products by increasing the minimum level of Medicaid rebates payable by manufacturers of brand-name drugs from 15.1% to 23.1%, effective 2011, extending the Medicaid rebate to Medicaid managed care plans, changing the Medicaid rebate rates for line
24
Table of Contents
extensions or new formulations of oral solid dosage form, mandating discounts for certain Medicare Part D beneficiaries, and imposing a non-deductible annual fee on pharmaceutical manufacturers or importers who sell “branded prescription drugs,” effective 2011, expanding the types of entities eligible for the “Section 340B discounts” for outpatient drugs, requiring manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50% point-of-sale discounts off negotiated prices of applicable branded drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D and creating a process for approval of biologic therapies that are similar or identical to approved biologics. There are numerous steps required to implement the Affordable Care Act, and implementation remains ongoing. Congress also has enacted, and may continue to seek, legislative changes that alter, delay, or eliminate some of its provisions. On February 1, 2016, the Centers for Medicare and Medicaid Services released a long-awaited new rule, the Medicaid Program Covered Outpatient Drug Final Rule, effective April 1, 2016, implementing various provisions of the Affordable Care Act related to “covered outpatient drugs,” including revising the calculation of “average manufacturer price” and addressing other issues relating to Medicaid price reporting and reimbursement. These and other changes contribute to the uncertainty of the ongoing implementation and impact of the Affordable Care Act; they also underscore the potential for additional reform going forward. Certain provisions of enacted or proposed legislative changes may negatively impact coverage and reimbursement of healthcare items and services.
Increasing pricing pressure continues from managed care organizations, government agencies and programs, particularly for new and innovative therapies, that could negatively affect the company’s sales and profit margins. In the United States, these include practices of managed care groups, federal and state exchanges, and institutional and governmental purchasers. Changes to the health care system enacted as part of health care reform in the United States, as well as increased purchasing power of entities that negotiate on behalf of Medicare, Medicaid, and private sector beneficiaries, could negatively impact the company’s sales and profit margins. Such pressures may also increase the risk of litigation or investigations by the government regarding pricing calculations. There has also been recent negative publicity and Congressional scrutiny around pharmaceutical drug pricing in the United States. These dynamics may give rise to negative reactions to pricing decisions for products for which we may receive regulatory approval in the future, possibly limiting our ability to generate revenue and attain profitability. Moreover, the pharmaceutical industry will likely face greater regulation and political and legal action in the future. In this healthcare regulatory climate, there may be significant delays in and impediments to obtaining coverage and reimbursement for newly approved drugs. Any regulatory approval of our products is limited to specific diseases and indications for which our products have been deemed safe and effective by the FDA. Coverage by federal healthcare programs may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities’ coverage of the same products. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the extent to which the costs of the products will be covered and reimbursed by third-party payors, including government healthcare programs such as Medicare and Medicaid, private health insurers and other organizations. Obtaining reimbursement for orphan drugs may be particularly difficult because of the higher prices typically associated with drugs directed at smaller populations of patients. In addition, third-party payors are likely to impose strict requirements for reimbursement in the use of a higher priced drug. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States.
The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the FDA approved products for a particular indication. Third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. In the future, we may need to conduct
25
Table of Contents
direct head-to-head studies to demonstrate clinical superiority and cost-effectiveness. Our product candidates may not be considered clinically superior and cost-effective to competitor products.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and other third-party payors fail to provide adequate coverage and reimbursement. In addition, there is an increasing emphasis on managed care in the United States that may negatively impact pharmaceutical pricing.
In some countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing and reimbursement negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. In some countries, governments can set conditions that must be satisfied for prices to be set at a certain value. Political, economic and regulatory developments may further complicate pricing and reimbursement negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU member states, and parallel distribution (arbitrage between low-priced and high-priced member states), can further reduce prices. In some countries we may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidate to other available therapies in order to obtain reimbursement or pricing approval.
Freedom of Information Requests
We are also subject, in the United States and many other countries, to various regulatory schemes that require disclosure of clinical trial data or allow access to our data via freedom of information requests. We have been and may, from time to time, be notified by regulators, such as the EMA or the competent authorities of EU member states that they have received a freedom of information request for documents that they hold relating to our company, including information related to our product or our product candidates.
Fraud and Abuse Laws
Any present or future arrangements with third-party payors, healthcare providers and professionals and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may restrict certain marketing and contracting practices. These laws include, and are not limited to, anti-kickback and false claims statutes.
Both the federal Foreign Corrupt Practices Act (“FCPA”), and the UK Bribery Act of 2010 (“Bribery Act”), are broad in scope and will require companies to make and keep books and records that accurately and fairly reflect the transactions of the company and to devise and maintain an adequate system of internal accounting controls. The FCPA prohibits the offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to improperly influence any act or decision, secure any other improper advantage, or obtain or retain business. Under the Bribery Act, companies which carry on a business or part of a business in the United Kingdom may be held liable for bribes given, offered or promised to any person, including non-UK government officials and private persons, by employees and persons associated with the company in order to obtain or retain business or a business advantage for the company.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, in cash or kind, to induce or reward either the referral of an individual for, or the purchase, or order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. This statute has been broadly interpreted to apply to manufacturer arrangements with prescribers, purchasers and formulary managers, among others. Although a number of statutory exemptions and regulatory safe harbors exist to protect certain
26
Table of Contents
common activities from prosecution, the exemptions and safe harbors for this statute are narrow, and practices that involve compensation intended to induce prescriptions, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not always meet all of the criteria for safe harbor protection. Further, the Affordable Care Act amended the intent requirement of the federal anti-kickback and criminal health care fraud statutes. This amendment provides that a person or entity no longer needs to have knowledge of these statutes or specific intent to violate them. In addition, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. Several other countries, including the United Kingdom, have enacted similar anti-kickback, fraud and abuse laws and regulations.
The federal False Claims Act imposes civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. Several pharmaceutical and health care companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the free product. Other companies have been prosecuted for causing false claims to be submitted because of these companies’ marketing of a product for unapproved, and thus non-reimbursable, uses. Potential liability under the federal False Claims Act includes mandatory treble damages and significant per claim penalties, currently set at $5,500 to $11,000 per false claim. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs; furthermore, in several states, these statutes and regulations apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s product from reimbursement under government programs, debarment, criminal fines, and imprisonment.
The Affordable Care Act included a provision requiring certain providers and suppliers of items and services to Federal Health Care Programs to report and return overpayments within sixty days after they are “identified,” or the Overpayment Statute. In February 2016, the Centers for Medicare and Medicaid Services (“CMS”) released long-awaited regulatory guidance (in the form of a final rule) to Medicare Part A and Part B providers and suppliers regarding how to comply with the Overpayment Statute. CMS had previously released a final rule addressing overpayments involving Medicare Part C and Part D providers in May 2014. Although Medicare Part A/B/C/D providers and suppliers have faced federal False Claims Act liability since 2010 for failures to comply with the Overpayment Statute, these final rules interpreting the Overpayment Statute provide guidance to providers and suppliers regarding how to comply appropriately with applicable obligations, and guidance to government regulators and enforcement authorities regarding monitoring and prosecuting suspected violations. This final rule is not directly applicable to manufacturers, but may impact their customers and potential customers who are Medicare providers and suppliers.
The federal Physician Payments Sunshine Act, enacted as part of the Affordable Care Act, and its implementing regulations, require manufacturers of drugs, devices, biologics and medical supplies to report to the Department of Health and Human Services information related to payments and other transfers of value made to covered recipients, such as physicians and teaching hospitals, as well as physician ownership and investment interests. Payments made to physicians and certain research institutions for clinical trials are included within the ambit of this law. Pharmaceutical manufacturers are required to report and disclose payments and ownership and investment interests held by physicians and their immediate family members during the preceding calendar year. Manufacturers were required to make these first reports for information collected in 2013 by March 31, 2014. Such information is publicly available from the Secretary of Health and Human Services in a searchable format, with data collected in each calendar year published the following June. Failure to submit required information may result in civil monetary penalties of up to $150,000 per year (and up to $1.0 million per year for “knowing failures”) for all payments, transfers of value or ownership or investment interests not reported in an annual submission. If not preempted by this federal law, several states currently require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and
27
Table of Contents
payments to individual physicians in those states. Depending on the state, legislation may prohibit various other marketing related activities, or require the posting of information relating to clinical studies and their outcomes. In addition, certain states, such as California, Nevada, Connecticut and Massachusetts, require pharmaceutical companies to implement compliance programs or marketing codes and several other states are considering similar proposals. Manufacturers that fail to comply with these state laws can face civil penalties.
Statutory requirements to disclose publicly payments made to healthcare professionals and healthcare organizations have also been enacted in certain EU member states. In addition, self-regulatory bodies of the pharmaceuticals industry, such as the European Federation of Pharmaceutical Industries and Associations (EFPIA”) have published codes of conduct to which its members have agreed to abide by, that require the public disclosure of payments made to healthcare professionals and healthcare organizations.
The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal liability for executing a scheme to defraud any healthcare benefit program and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services. HIPAA also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information, and imposes criminal and civil liability for violations of these obligations. Recently, the U.S. federal government criminally prosecuted an employee of a pharmaceutical company for an alleged violation of the privacy requirements under HIPAA. Furthermore, certain privacy laws and genetic testing laws may apply directly to our operations and/or those of our collaborators and may impose restrictions on our use and dissemination of individuals’ health information.
The foregoing discussion should be read in conjunction with the information appearing under “Risk Factors—Our relationships with customers, healthcare providers and professionals and third-party payors are or will be subject to applicable anti-kickback, fraud and abuse, transparency and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings” which contains important information regarding some of the risks to our business arising as a result fraud and abuse laws.
Competition
Our industry is highly competitive and subject to rapid and significant technological change. New therapies and treatments based on innovative discoveries emerge frequently.
Our potential competitors are public and private companies, pharmaceutical companies and biotechnology companies who may be engaged in targeting the same biological processes that our compounds impact and who may be developing products for the same indications as our investigational drug candidates. Potential competitors could also include academic institutions, government agencies, other public and private research organizations and charitable venture philanthropic organizations that conduct research, seek patent protection and/or establish collaborative arrangements for research, development, manufacturing and commercialization.
Many of our competitors have substantially greater financial resources, technical resources, expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials. As a result, our competitors may commercialize products more rapidly or effectively than we do, which would affect our competitive position, the likelihood that our drug candidates, if approved, would achieve and maintain market acceptance and our ability to generate meaningful revenues from our products.
28
Table of Contents
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects, are more convenient or are more affordable than any products that we may develop. The key competitive factors affecting the success of ELX-02 and our other product candidates are their impact on the targeted diseases, superiority over competing products, long-term safety, convenience, price and the availability of coverage and reimbursement from government and other third-party payors.
Several companies are involved in researching and developing molecules targeting suppression of nonsense mutations and enhancement of translational read-through. PTC Therapeutics is developing ataluren (Translarna®) as translational read-through inducing drug. PTC has gained approval of ataluren for Duchenne muscular dystrophy under exceptional circumstances in the European Union. In January 2017, the European Commission renewed the conditional marketing authorization for ataluren to treat certain nonsense mutations of dystrophin. The renewal of the conditional marketing authorization is subject to a requirement to conduct an 18-month, randomized, placebo-controlled study of ataluren in nmDMD patients followed by an 18-month, open-label extension period with results expected by early 2021. Ataluren has not been approved by the FDA for any indication. Ten out of 11 members from a Peripheral and Central Nervous Systems Drugs Advisory Committee on September 28, 2017 stated more data are needed to prove the drug’s efficacy. In a Complete Response Letter, the FDA’s Office of Drug Evaluation I stated that it is unable to approve the ataluren application in its current form. Specifically, the letter indicated that evidence of effectiveness from one or more additional adequate and well-controlled clinical trials will be necessary to provide substantial evidence of effectiveness.
We believe that ELX-02 is the only drug candidate in clinical development designed to treat nonsense mutations in CFTR the underlying cause of cystic fibrosis and cystinosis, our lead indications. La Jolla Pharmaceuticals is testing a sub-fraction of gentamicin at a preclinical stage and PTC Therapeutics discontinued its CF program as ataluren did not show efficacy in a Phase 3 CF study.
Additional competition to ELX-02 may arise from other programs that do not target a specific CFTR mutation class but work via other mechanisms. Proteostasis Therapeutics is developing PTI-428, a CFTR amplifier in Phase 2; and Apteeus is developing TEE786 (Amlexanox), a NMD manipulator, in Phase 1. Other companies are developing RNA based therapeutics, gene therapy and cell therapy. Most of these products are in preclinical stages and these platforms face great technological challenges.
Employees
We currently have fifteen full-time employees. Of these employees, ten are located at our Rehovot, Israel research and development facility and five, including some executive officers, are located at our Waltham, Massachusetts facility. None of our employees are covered by a collective bargaining agreement and we have never experienced any work stoppage. We consider our relations with our employees to be good.
Additional Information
Our website address is http://www.eloxxpharma.com. Information on our website is not incorporated by reference herein. Copies of our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, are available on our website as soon as reasonably practicable after we electronically file those reports with, or furnish them to the SEC. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling 1-800-SEC-0330. The SEC also maintains a website at http://www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically. (This website address is not intended to function as a hyperlink.)
29
Table of Contents
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below and all information contained in this report before you decide to purchase our common stock. If any of the possible adverse events described below actually occurs, we may be unable to conduct our business as currently planned and our financial condition and operating results could be harmed. In addition, the trading price of our common stock could decline due to the occurrence of any of the events described below, and you may lose all or part of your investment.
Risks Related to the Reverse Merger
The risks arising with respect to the historic Sevion business and operations may be different from what we anticipate, which could lead to significant, unexpected costs and liabilities and could materially and adversely affect our business going forward.
We may not have appreciated, understood or fully anticipated the extent of the risks associated with the recent reverse merger between Sevion and Ellox Limited. After the reverse merger, Sevion’s historic business was discontinued, but prior to the transaction Sevion had a long operating history. As a consequence, we may be subject to claims, demands for payment, regulatory issues, costs and liabilities that were not and are not currently expected or anticipated. Notwithstanding our exercise of due diligence pre-transaction and risk mitigation strategies post-transaction, the risks involved with taking over a business with a long operating history and the costs and liabilities associated with these risks may be greater than we anticipate. Further, we do not have rights of indemnification against the pre-transaction stockholders of Sevion. We may not be able to contain or control the costs or liabilities associated with Sevion’s historic business, which could materially and adversely affect our business, liquidity, capital resources or results of operation.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant operating losses since our inception and anticipate that we will continue to incur substantial operating losses for the foreseeable future. We may never achieve or maintain profitability.
Since our inception, we have incurred significant operating losses. Our net loss was $21.2 million and $9.8 million for 2017 and 2016, respectively. As of December 31, 2017, we had an accumulated deficit of $39.0 million. To date, we have financed our operations primarily through equity capital investments, and to a lesser extent from loans and grants from Israeli Innovation Authority of the Ministry of Economy and Industry, or the IIA. We have devoted substantially all of our financial resources and efforts to research and development. We expect that it will be many years, if ever, before we receive regulatory approval and have a product candidate ready for commercialization. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. Our net losses may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially if and as we:
| • | advance ELX-02 further into clinical trials; |
| • | continue the preclinical development of our research programs and advance candidates into clinical trials; |
| • | identify additional product candidates and advance them into preclinical development; |
| • | pursue regulatory approval of product candidates; |
| • | seek marketing approvals for our product candidates that successfully complete clinical trials; |
| • | establish a sales, marketing and distribution infrastructure to commercialize any product candidates for which we obtain marketing approval; |
30
Table of Contents
| • | maintain, expand and protect our intellectual property portfolio; |
| • | hire additional clinical, regulatory and scientific personnel; |
| • | add operational, financial and management information systems and personnel, including personnel to support product development; |
| • | acquire or in-license other product candidates and technologies; and |
| • | operate as a public company. |
We have never generated any revenue from product sales and may never be profitable. To become and remain profitable, we and our collaborators must develop and eventually commercialize one or more products with significant market potential. This will require us to be successful in a range of challenging activities, including completing preclinical studies and clinical trials of our product candidates, obtaining marketing approval for these product candidates, manufacturing, marketing and selling those product candidates for which we may obtain marketing approval, securing coverage and reimbursement for those product candidates, and satisfying any post-marketing requirements. We may never succeed in these activities and, even if we do, may never generate revenue that is significant or large enough to achieve profitability. Our failure to become and remain profitable would decrease the value of the company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We will need substantial additional funding. If we are unable to raise capital when needed, we would be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research and development of, continue and initiate clinical trials of and seek marketing approval for ELX-02, and as we become obligated to make milestone payments pursuant to our outstanding license agreements. In addition, if we obtain marketing approval for any of our current or future product candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution of the approved product.
Our future capital requirements will depend on many factors, including:
| • | the scope, progress, results and costs of drug discovery, clinical development, laboratory testing and clinical trials for ELX-02; |
| • | the costs, timing and outcome of any regulatory review of ELX-02; |
| • | the cost of any other product candidate programs we pursue; |
| • | the costs and timing of commercialization activities, including manufacturing, marketing, sales and distribution, and securing coverage and reimbursement for any product candidates that receive marketing approval; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| • | our ability to establish and maintain collaborations on favorable terms, if at all; and |
| • | the extent to which we acquire or in-license other product candidates and technologies. |
Identifying potential product candidates and conducting preclinical studies and clinical trials are time consuming, expensive and uncertain processes that take years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales for any of our current or future product candidates. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenue, if any, will be derived from sales of products that we do not expect to be commercially available for many years, if at all.
31
Table of Contents
Accordingly, we will need substantial additional funding in connection with our continuing operations and to achieve our goals. We expect that our existing cash and cash equivalents will be sufficient to enable us to meet our current operating plan at least through the end of the first quarter of 2019. However, our existing cash and cash equivalents may prove to be insufficient for these activities. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs, product portfolio expansion or future commercialization efforts. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional financing due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our operating plans.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity and debt financings, as well as entering into new collaborations, strategic alliances and licensing arrangements. We do not have any committed external source of funds, other than our collaborations, which are limited in scope and duration. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends, and may be secured by all or a portion of our assets. If we raise funds by entering into new collaborations, strategic alliances or licensing arrangements in the future with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or through collaborations, strategic alliances or licensing arrangements when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Risks Related to Drug Discovery, Development, Regulatory Approval and Commercialization
We depend heavily on the success of our lead product candidate, ELX-02. If ELX-02 fails during development or suffers any material delays, it may adversely impact the commercial viability of ELX-02 and our business.
We currently have no products approved for sale. To date, we have invested substantially all of our efforts and financial resources in the research and development of ELX-02, which is currently our only product candidate in clinical development. Our ability to achieve and sustain profitability depends on obtaining regulatory approvals, and successfully commercializing (if ever), ELX-02 and any future product candidates, either alone or with third parties. Before obtaining regulatory approval for the commercial distribution of our therapeutic product candidates, we or a collaborator must conduct extensive preclinical studies and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. The clinical trials, manufacturing and marketing of ELX-02, and any future product candidates, will be subject to extensive and rigorous review and regulation by numerous government authorities in the United States, the European Union and other jurisdictions where we intend to test and, if approved, market our current and future product candidates. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through preclinical studies and clinical trials that the product candidate is safe and effective for use in each target indication, and potentially in specific patient populations, including the pediatric population. This process can take many years and may include post-marketing studies and surveillance, which would require the expenditure of substantial resources. Of the large number of drugs in development for approval in the United States and the European Union, only a small percentage successfully complete the FDA or EMA regulatory approval processes, as applicable, and are commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our research, development and clinical programs, we cannot assure you that ELX-02 or any of our future product candidates will be successfully developed or commercialized.
32
Table of Contents
Preclinical studies and clinical trials are expensive, difficult to design and implement, can take many years to complete and are uncertain as to outcome. The start or end of a clinical trial is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative therapeutic or required prior or combination therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, the age and condition of the patients, the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments for the relevant disease.
We and our collaborating partners may be subject, directly or indirectly, to federal and state healthcare fraud and abuse and false claims laws and regulations. If we or our collaborating partners are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
All marketing activities associated with product candidates that are approved for sale in the United States, if any, will be, directly or indirectly through our customers, subject to numerous federal and state laws governing the marketing and promotion of pharmaceutical products in the United States, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act and HIPAA. These laws may adversely impact, among other things, our proposed sales, marketing and education programs.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging for a good or service, for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value, including for example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of co-payments and deductibles, ownership interests and providing anything at less than its fair market value. The reach of the Anti-Kickback Statute was also broadened by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the PPACA, which, among other things, amends the intent requirement of the federal Anti-Kickback Statute and the applicable criminal healthcare fraud statutes contained within 42 U.S.C. § 1320a-7b, effective March 23, 2010. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below) or the civil monetary penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. The federal Anti-Kickback Statute is broad, and despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Penalties for violations of the federal Anti-Kickback Statute include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other state or federal healthcare programs. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs, and do not contain identical safe harbors.
The federal False Claims Act imposes liability on any person who, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The “qui tam” provisions of the False Claims Act allow a private individual to bring civil actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any
33
Table of Contents
monetary recovery. In addition, various states have enacted false claims laws analogous to the False Claims Act. Many of these state laws apply where a claim is submitted to any third-party payer and not merely a federal healthcare program. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of $5,500 to $11,000 for each separate false claim.
HIPAA created several new federal crimes, including health care fraud, and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private third-party payers. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services.
We are unable to predict whether we could be subject to actions under any of these or other fraud and abuse laws, or the impact of such actions. Moreover, to the extent that any of our product candidates, if approved for marketing, will be sold in a foreign country, we and our future collaborators, may be subject to similar foreign laws and regulations. If we or any of our future collaborators are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from government healthcare reimbursement programs and the curtailment or restructuring or our operations, any of which could have a material adverse effect on our business, results of operations and financial condition.
Positive results from preclinical or in vitro and in vivo testing of ELX-02 are not necessarily predictive of the results of future clinical trials of ELX-02. If we cannot achieve positive results in our clinical trials for ELX-02, we may be unable to successfully develop, obtain regulatory approval for and commercialize ELX-02.
Positive results from our preclinical testing of ELX-02 in vitro and in vivo may not necessarily be predictive of the results from our planned clinical trials in humans. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in preclinical and in vitro and in vivo studies, and we, or the third parties whose product candidates we expect to be co-administered with ELX-02, may face similar setbacks. Preclinical and clinical data are often susceptible to varying interpretations and analyses, and the FDA or EMA or other regulatory agencies may require changes to our protocols or other aspects of our clinical trials or require additional studies. Additionally, many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or EMA approval. If we fail to secure positive results from our clinical trials of ELX-02, the development timeline and regulatory approval and commercialization prospects for our lead product candidate, and, correspondingly, our business and financial prospects would be materially adversely affected.
Our product candidates, including ELX-02, may cause adverse effects or have other properties that could delay or prevent their regulatory approval or limit the scope of any approved label or market acceptance.
Undesirable side effects caused by our product candidates, such as ELX-02, could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. It is possible that, during the course of the clinical development of ELX-02, results of our clinical trials could reveal an unacceptable severity and prevalence of this or other side effects. For example, in preclinical testing of ELX-02, we observed renal toxicities in the animals we tested following administration of this compound at doses in excess of the doses we expect to administer in our clinical trials. As a result of this or any other side effects, our clinical trials could be suspended or terminated or not even allowed to commence, and the FDA or comparable foreign regulatory authorities could order us to cease further development, or deny approval, of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims.
34
Table of Contents
Additionally if one or more of our product candidates receive marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw approvals of such product or impose restrictions on its distribution in a form of a modified risk evaluation and mitigation strategy; |
| • | regulatory authorities may require additional labeling, such as additional warnings or contraindications; |
| • | we may be required to change the way the product is administered or to conduct additional clinical studies; |
| • | we could be sued and held liable for harm caused to patients; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Our clinical trials may be costly and lengthy, time-consuming and difficult to design and implement, may result in unforeseen costs and could be delayed or terminated, which may have a material adverse effect on our business, results of operations and financial condition.
For human trials, patients must be recruited and each product candidate must be tested at various doses and formulations for each clinical indication. In addition, to ensure safety and effectiveness, the effect of drugs often must be studied over a long period of time, especially for the chronic genetic diseases that we will be studying. Many of our programs focus on diseases with small patient populations making patient recruitment and enrollment difficult. Insufficient patient enrollment in our clinical trials could delay or cause us to abandon a product development program. We may decide to abandon development of a product candidate or a study at any time due to unfavorable results, or we may have to spend considerable resources repeating clinical trials or conducting additional trials, either of which would increase costs and delay any revenue from those product candidates, if any.
Failure or delay in the commencement or completion of our clinical trials may be caused by several factors, including:
| • | slower than expected rates of patient recruitment, particularly with respect to trials of rare diseases such as nmCF; |
| • | determination of dosing issues; |
| • | unforeseen safety issues; |
| • | lack of effectiveness during clinical trials; |
| • | inability to monitor patients adequately during or after treatment; |
| • | inability or unwillingness of medical investigators and institutional review boards to follow our clinical protocols; and |
| • | lack of sufficient funding to finance the clinical trials. |
We may find it difficult to recruit and enroll patients in our clinical trials, which could cause significant delays in the completion of such trials or may cause us to abandon one or more clinical trials.
Some of the diseases that our product candidates are intended to treat are rare and ultra-rare and we expect only a subset of the patients with these diseases will be eligible for our clinical trials. Because ELX-02 targets small populations and patient numbers have not been determined definitively, we must be able to identify patients in order to complete our development programs and commercialize ELX-02 successfully.
35
Table of Contents
In addition, the protocol for our clinical trials generally mandates that a patient cannot be involved in more than one clinical trial for the same indication. Therefore, subjects that participate in ongoing clinical trials for products that are competitive with our product candidates are not available to participate in our clinical trials. We cannot guarantee that any of our programs will identify a sufficient number of patients to complete clinical development and market our product candidates if approved. The combined number of patients in the United States, Japan and Europe and elsewhere may turn out to be lower than expected, may not be otherwise amenable to treatment with ELX-02, or new patients may become increasingly difficult to identify, all of which would adversely affect our results of operations and our business. An inability to recruit and enroll a sufficient number of patients for any of our current or future clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether, which could impact our ability to develop our product candidates and may have a material adverse effect on our business, results of operations and financial condition.
Because our clinical trials depend upon third-party researchers, scientists and consultants the results of our clinical trials and such research activities are subject to delays and other risks that are, to a certain extent, beyond our control, which could impair our clinical development programs and our competitive position.
We depend on independent investigators, consultants, researchers, medical experts, collaborators, chemists, toxicologist and a small number of medical institutions and third-party contract organizations to assist with our research efforts and conduct our preclinical and clinical trials and related activities. These collaborators, scientists, consultants and other third parties have provided, and we expect that they will continue to provide, valuable advice regarding our clinical development programs and product candidates. These collaborators, scientists, consultants and other third parties are not our employees, may have other commitments that would limit their future availability to us and typically will not enter into non-compete agreements with us. We cannot control the amount or timing of resources that they devote to our preclinical and or clinical development programs and they may not assign as great a priority to our preclinical or clinical development programs or pursue them as diligently as we would if we were undertaking such programs directly. If outside collaborators fail to devote sufficient time and resources to our preclinical and clinical development programs, or if their performance is substandard, the approval of anticipated NDAs and other marketing applications, and our introduction of new drugs, if any, may be delayed, which could impair our clinical development programs and would have a material adverse effect on our business and results of operations. The collaborators may also have relationships with other commercial entities, some of whom may compete with us and we may be unable to prevent them from establishing competing businesses or developing competing products. For example, if a key scientist acting as a principal investigator in any of our clinical trials identifies a potential product or compound that is more scientifically interesting to his or her professional interests, his or her availability to remain involved in our clinical trials could be restricted or eliminated, which may have a material adverse effect on our business, results of operations and financial condition.
We are subject to extensive governmental regulation including the requirement of FDA or comparable foreign regulatory authorities for approval of our product candidates before they can be marketed.
We, our product candidates, our suppliers, our contract manufacturers, our contract testing laboratories and our clinical trial sites and clinical trial researchers are subject to extensive regulation by the FDA and comparable foreign regulatory authorities. Failure to comply with applicable requirements of the FDA or comparable foreign regulatory authorities could result in, among other things, any of the following actions:
| • | warning letters; |
| • | fines and other monetary penalties; |
| • | unanticipated expenditures; |
| • | holds on the initiation of clinical trials; |
| • | delays in the FDA’s or other foreign regulatory authorities’ approving, or the refusal of any regulatory authority to approve, any product candidate; |
36
Table of Contents
| • | product recall or seizure; |
| • | interruption of manufacturing or clinical trials; |
| • | operating restrictions; |
| • | injunctions; and |
| • | criminal prosecutions. |
In addition to the approval requirements, other numerous and pervasive regulatory requirements apply, both before and after approval of our product candidates, to us, our product candidates, and our suppliers, contract manufacturers, and contract laboratories, including requirements related to testing, manufacturing, quality control, labeling, advertising, promotion, distribution, export, reporting to the FDA of certain adverse experiences associated with use of the product candidate, and obtaining additional approvals for certain modifications to the product candidate or its labeling or claims.
We also are subject to inspection by the FDA and comparable foreign regulatory authorities, to determine our compliance with regulatory requirements, as are our suppliers, contract manufacturers, contract testing laboratories, and our clinical trial sites and clinical researchers and there can be no assurance that the FDA or any other comparable foreign regulatory authority, will not identify compliance issues that may disrupt production or distribution, or require substantial resources to correct. We may be required to make modifications to our manufacturing operations in response to these inspections, which may require significant resources and may have a material adverse effect upon our business, results of operations and financial condition.
The approval process for any product candidate may also be delayed by changes in government regulation, future legislation or administrative action or changes in policy of the FDA and comparable foreign regulatory authorities that occur prior to or during their respective regulatory reviews of such product candidate. Delays in obtaining regulatory approvals with respect to any product candidate may:
| • | delay commercialization of, and our ability to derive product revenues from, such product candidate; |
| • | delay any regulatory-related milestone payments payable under outstanding collaboration agreements; |
| • | require us to perform costly procedures with respect to such product candidate; or |
| • | otherwise diminish any competitive advantages that we may have with respect to such product candidate. |
We may not obtain the necessary U.S., EMA or other worldwide regulatory approvals to commercialize our product candidates in a timely manner, if at all, which would have a material adverse effect on our business, results of operations and financial condition.
We need FDA approval to commercialize our product candidates in the United States, EMA approval to commercialize our product candidates in the European Union and approvals from other foreign regulatory authorities to commercialize our product candidates elsewhere in the world. In order to obtain FDA approval of any of our product candidates, we must submit to the FDA an NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as preclinical studies, as well as human tests, which are referred to as clinical trials. In the European Union, we must submit a Marketing Authorization Application, or MAA, to the EMA. Satisfaction of the FDA’s, the EMA’s and foreign regulatory authorities’ regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. Even if we comply with all the requests of regulatory authorities, the authorities may ultimately reject the marketing applications that we file for our product candidates in the future, if any, or we might not obtain regulatory clearance in a timely manner. Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced or late-stage clinical trials, even after
37
Table of Contents
obtaining promising earlier trial results or in preliminary findings or other comparable authorities for such clinical trials. Further, even if favorable testing data is generated during the clinical trials of a product candidate, the applicable regulatory authority may not accept or approve the marketing application filed by a pharmaceutical or biotechnology company for the product candidate. Failure to obtain approval of the FDA, EMA or comparable foreign regulatory authorities of any of our product candidates in a timely manner, if at all, will severely undermine our business, financial condition and results of operation by reducing our potential marketable products and our ability to generate corresponding product revenues.
Our research and clinical efforts may not result in drugs that the FDA, EMA or foreign regulatory authorities consider safe for humans and effective for indicated uses, which would have a material adverse effect on our business, results of operations and financial condition. After clinical trials are completed for any product candidate, if at all, the FDA, EMA and foreign regulatory authorities have substantial discretion in the drug approval process of the product candidate in their respective jurisdictions and may require us to conduct additional clinical testing or perform post-marketing studies, which would cause us to incur additional costs. Incurring such costs may have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Commercialization
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell any of our product candidates that obtain regulatory approval, we may be unable to generate any revenue.
We have no experience selling and marketing our product candidates or any other products. To successfully commercialize any products that may result from our clinical development programs and obtain regulatory approval, we will need to develop these capabilities, either on our own or with the assistance of others. We may seek to enter into collaborations with other entities to utilize their marketing and distribution capabilities, but we may be unable to do so on favorable terms, if at all. If any future collaborative partners do not commit sufficient resources to commercialize our future products, if any, and we are unable to develop the necessary marketing capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies or successfully commercialize any of our product candidates.
Developments by competitors may render our products or technologies obsolete or non-competitive which would have a material adverse effect on our business, results of operations and financial condition.
We compete with fully integrated biopharmaceutical companies and smaller biopharmaceutical companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Our product candidates will have to compete with existing therapies and therapies under development by our competitors. In addition, our commercial opportunities may be reduced or eliminated if our competitors develop and market products that are less expensive, more effective or safer than our product candidates. Other companies have product candidates in various stages of preclinical or clinical development to treat diseases for which we are also seeking to develop product candidates. Some of these potential competing drugs are further advanced in development than our product candidates and may be commercialized earlier. Even if we are successful in developing effective drugs, our products may not compete successfully with products produced by our competitors.
Most of our competitors, either alone or together with their collaborative partners, operate larger research and development programs, staff and facilities and have substantially greater financial resources than we do, as well as significantly greater experience in:
| • | developing drugs; |
38
Table of Contents
| • | undertaking preclinical testing and human clinical trials; |
| • | obtaining marketing approvals from the FDA and other regulatory authorities; |
| • | formulating and manufacturing drugs; and |
| • | launching, marketing and selling drugs. |
These organizations also compete with us to attract qualified personnel, acquisitions and joint ventures candidates and for other collaborations. Efforts to compete and the pursuit of activities of our competitors may impose unanticipated costs on our business, which would have a material adverse effect on our business, results of operations and financial condition.
If we are unable to develop and commercialize our product candidates, our business will be adversely affected.
A key element of our strategy is to develop and commercialize a portfolio of new products. We seek to do so through our internal research programs and strategic collaborations for the development of new products. Research programs to identify new product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for many reasons, including the following:
| • | a product candidate is not capable of being produced in commercial quantities at an acceptable cost, or at all; |
| • | a product candidate that is developed and approved may not be accepted by patients, the medical community or third-party payors; |
| • | competitors may develop alternatives that render our product candidates obsolete; |
| • | the research methodology used may not be successful in identifying potential product candidates; or |
| • | a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory approval requirements. |
Any failure to develop or commercialize any of our product candidates may have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Our Business and Operations
Maintaining and improving our financial controls and the requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified board members.
The trading market for our common stock is influenced by the research and reports that securities or industry analysts publish. As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and OTCQB Market rules. The requirements of these rules and regulations have increased and will continue to significantly increase our legal and financial compliance costs, including costs associated with the hiring of additional personnel, making some activities more difficult, time-consuming or costly, and may also place undue strain on our personnel, systems and resources. The Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and financial condition.
39
Table of Contents
The Sarbanes-Oxley Act requires, among other things, that we maintain disclosure controls and procedures and internal control over financial reporting. Ensuring that we have adequate internal financial and accounting controls and procedures in place, as well as maintaining these controls and procedures, is a costly and time-consuming effort that needs to be re-evaluated frequently. Section 404 of the Sarbanes-Oxley Act, or Section 404, requires that we annually evaluate our internal control over financial reporting to enable management to report on, the effectiveness of those controls. In connection with the Section 404 requirements, we test our internal controls and could, as part of that documentation and testing, identify material weaknesses, significant deficiencies or other areas for further attention or improvement.
Implementing any appropriate changes to our internal controls may require specific compliance training for our directors, officers and employees, require the hiring of additional finance, accounting and other personnel, entail substantial costs to modify our existing accounting systems, and take a significant period of time to complete. These changes may not, however, be effective in maintaining the adequacy of our internal controls, and any failure to maintain that adequacy, or consequent inability to produce accurate financial statements on a timely basis, could increase our operating costs and could materially impair our ability to operate our business. Moreover, adequate internal controls are necessary for us to produce reliable financial reports and are important to help prevent fraud. As a result, our failure to satisfy the requirements of Section 404 on a timely basis could result in the loss of investor confidence in the reliability of our financial statements, which in turn could cause the market value of our common stock to decline.
Various rules and regulations applicable to public companies make it more difficult and more expensive for us to maintain directors’ and officers’ liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to maintain coverage. If we are unable to maintain adequate directors’ and officers’ liability insurance, our ability to recruit and retain qualified officers and directors, especially those directors who may be deemed independent for purposes of the OTCQB Market rules, will be significantly curtailed.
The requirements of being a public company may strain our resources and distract management.
As a public company, we are subject to the reporting requirements of the Exchange Act and the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act. These requirements are extensive. The Exchange Act requires that we file annual, quarterly and current reports with respect to our business and financial condition. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal controls over financial reporting.
We may incur significant costs associated with our public company reporting requirements and costs associated with applicable corporate governance requirements. These applicable rules and regulations significantly increase our legal and financial compliance costs and make some activities more time consuming and costly. This may divert management’s attention from other business concerns, which could have a material adverse effect on our business, financial condition and results of operations. We also expect that these applicable rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as executive officers. We are currently evaluating and monitoring developments with respect to these rules, and we cannot predict or estimate the amount of additional costs we may incur or the timing of such costs.
40
Table of Contents
We are seeking to expand our business through strategic initiatives. Our efforts to identify opportunities or complete transactions that satisfy our strategic criteria may not be successful, and we may not realize the anticipated benefits of any completed acquisition or other strategic transaction.
Our business strategy includes expanding our products and capabilities. We regularly evaluate potential merger, acquisition, partnering and in-license opportunities that we expect will expand our pipeline or product offerings, and enhance our research platforms.
To manage effectively our current and future potential growth, we must continue to enhance and develop our global employee base, and our operational and financial processes. Supporting our growth strategy will require significant capital expenditures and management resources, including investments in research, development, sales and marketing, manufacturing and other areas of our operations. The development or expansion of our business, any acquired business or any acquired or in-licensed products may require a substantial capital investment by us. We may not have these necessary funds or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our capital stock, which could dilute current stockholders’ ownership interest in our company, or securities convertible into our capital stock, which could dilute current stockholders’ ownership interest in our company upon conversion.
Our business could be affected by litigation, government investigations and enforcement actions.
We operate in many jurisdictions in a highly regulated industry and we could be subject to litigation, government investigation and enforcement actions on a variety of matters in the U.S. or foreign jurisdictions, including, without limitation, intellectual property, regulatory, product liability, environmental, whistleblower, Qui Tam, false claims, privacy, anti-kickback, anti-bribery, securities, commercial, employment, and other claims and legal proceedings which may arise from conducting our business.
The intended efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate and we may have exposure to additional tax liabilities or our effective tax rate could change, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the U.S. and various foreign jurisdictions. Significant judgment is required in determining our worldwide tax liabilities. Although we believe our estimates are reasonable, the ultimate outcome with respect to the taxes we owe may differ from the amounts recorded in our financial statements. If the Internal Revenue Service, or other taxing authority, disagrees with the positions we take, we could have additional tax liability, and this could have a material impact on our results of operations and financial position. Our effective tax rate could be adversely affected by changes in the mix of earnings in countries with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in tax laws and regulations, changes in interpretations of tax laws, including pending tax law changes, changes in our manufacturing activities and changes in our future levels of research and development spending.
We have designed our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our affiliates in a way that is intended to enhance our operational and financial efficiency and increase our overall profitability. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing. If tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase and harm our financial position and results of operations.
41
Table of Contents
In addition, the U.S. federal government and other U.S. state and foreign governments are considering and may adopt tax reform measures that significantly increase our worldwide tax liabilities. The U.S. Congress, the Organization for Economic Co-operation and Development and other government agencies in countries where we and our affiliates operate have focused on issues related to the taxation of multinational corporations, including, for example, in the area of “base erosion and profit shifting,” where payments are made between affiliates from a jurisdiction with high tax rates to a jurisdiction with lower tax rates. These changes and other prospective changes in the U.S. and other countries in which we and our affiliates operate could increase our effective tax rate, and harm our financial position and results of operations.
Changes in healthcare laws and implementing regulations, as well as changes in healthcare policy, may affect coverage and reimbursement of our product candidates in ways that we cannot currently predict and these changes could adversely affect our business and financial condition.
In the U.S., a number of legislative and regulatory initiatives have focused on containing the cost of healthcare. The Patient Protection and Affordable Care Act, or PPACA, was enacted in the U.S. in March 2010. This law substantially changes the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, fraud and abuse enforcement and rules governing the approval of biosimilar products. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program. In early 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate Program under PPACA. These regulations became effective on April 1, 2016. Moreover, in the future, Congress could enact legislation that further increases Medicaid drug rebates or other costs and charges associated with participating in the Medicaid Drug Rebate Program. Legislative changes to the PPACA also remain possible and appear likely in the 115th U.S. Congress under the Trump administration. The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program has and will continue to increase our costs and the complexity of compliance, has been and will be time-consuming, and could have a material adverse effect on our results of operations.
Governments in countries where we operate have adopted or have shown significant interest in pursuing legislative initiatives to reduce costs of healthcare. We expect that the implementation of current laws and policies, the amendment of those laws and policies in the future, as well as the adoption of new laws and policies, could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or successfully commercialize our product candidates, or could limit or eliminate our future spending on development projects. In many cases, these government initiatives, even if enacted into law, are subject to future rulemaking by regulatory agencies. Although we have evaluated these government initiatives and the impact on our business, we cannot know with certainty whether any such law, rule or regulation will adversely affect coverage and reimbursement of our product candidates, or to what extent, until such laws, rules and regulations are promulgated, implemented and enforced, which could sometimes take many years. The announcement or adoption of regulatory or legislative proposals could delay or prevent our entry into new markets, affect our reimbursement or sales in the markets where we are already selling our products and materially harm our business, financial condition and results of operations.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our
42
Table of Contents
business. In the U.S., we may be subject to state security breach notification laws, state health information privacy laws and federal and state consumer protections laws which impose requirements for the collection, use, disclosure and transmission of personal information. Each of these laws are subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting the violation of HIPAA.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. EU member states and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. For example, the EC adopted the EU Data Protection Directive, as implemented into national laws by the EU member states, which imposed strict obligations and restrictions on the ability to collect, analyze, and transfer personal data, including health data from clinical trials and adverse event reporting. Data protection authorities from different EU member states have interpreted the privacy laws differently, which adds to the complexity of processing personal data in the European Union, and guidance on implementation and compliance practices are often updated or otherwise revised. Any failure to comply with the rules arising from the EU Data Protection Directive and related national laws of EU member states could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
In May 2016, the European Union formally adopted the General Data Protection Regulation, which will apply to all EU member states from May 25, 2018 and will replace the current EU Data Protection Directive on that date. The regulation introduces new data protection requirements in the European Union and substantial fines for breaches of the data protection rules. It will increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
Security breaches, cyber-attacks, or other disruptions could expose us to liability and affect our business and reputation.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store, and transmit sensitive information including intellectual property, proprietary business information and personal information in connection with business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack by third parties with a wide range of motives and expertise, including organized criminal groups, “hactivists,” patient groups, disgruntled current or former employees, and others. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance. We have implemented information security measures to protect patients’ personal information against the risk of inappropriate and unauthorized external use and disclosure. However, despite these measures, and due to the ever-changing information cyber-threat landscape, we may be subject to data breaches through cyber-attacks. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. If our systems become compromised, we may not promptly discover the intrusion. Like other companies in our industry, we have experienced attacks to our data and systems, including malware and computer viruses. If our systems failed or were breached or disrupted, we could lose product sales, and suffer reputational damage and loss of customer confidence. Such incidents would result in notification obligations to affected individuals and government agencies, legal claims or proceedings, and liability under federal and state laws that protect the privacy and security of personal information. Any one of these events could cause our business to be materially harmed and our results of operations would be adversely impacted.
43
Table of Contents
We expect to rely on third parties to conduct some or all aspects of our product manufacturing, protocol development, research and preclinical and clinical testing, and these third parties may not perform satisfactorily.
We do not expect to independently conduct all aspects of our product manufacturing, protocol development, research and preclinical and clinical testing. We currently rely, and expect to continue to rely, on third parties with respect to these items.
Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it could delay our product development activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations and study protocols. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, we will not be able to complete, or may be delayed in completing, the preclinical studies and clinical trials required to support future NDA submissions and approval of our product candidates.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the product candidates ourselves, including:
| • | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
| • | reduced control as a result of using third-party manufacturers for all aspects of manufacturing activities; |
| • | termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; and |
| • | disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier. |
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize future products. Some of these events could be the basis for FDA action, including injunction, recall, seizure or total or partial suspension of production. Any one of these events could cause our business to be materially harmed and our results of operations would be adversely impacted.
Our future success depends on our ability to retain key employees, consultants and advisors and to attract, retain and motivate qualified personnel.
The success of our business is dependent in large part on our continued ability to attract and retain our senior management, and other highly qualified personnel in our scientific, clinical, manufacturing and commercial organizations. Intense competition exists in the biopharmaceutical industry for these types of personnel. Our business is specialized and global and we must attract and retain highly qualified individuals across many geographies. We may not be able to continue to attract and retain the highly qualified personnel necessary for developing, manufacturing and commercializing our products and product candidates. If we are unsuccessful in our recruitment and retention efforts, or if our recruitment efforts take longer than anticipated, our business may be harmed.
We are highly dependent on principal members of our senior management, including Robert Ward, our Chief Executive Officer. While we have entered into employment agreements or offer letters with each of our executive officers, any of them could leave our employment at any time, as all of our employees are “at will” employees. Recruiting and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will also be critical to our success. As a result, competition for
44
Table of Contents
skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for individuals with similar skill sets. In addition, failure to succeed in preclinical studies or clinical trials may make it more challenging to recruit and retain qualified personnel. The inability to recruit or loss of the services of any executive, key employee, consultant or advisor may impede the progress of our research, development and commercialization objectives. If we fail to attract and retain highly qualified personnel, we may not be able to successfully develop, manufacture or commercialize our products or products candidates.
Risks Related to Intellectual Property
If we fail to adequately protect or enforce our intellectual property rights or secure rights to third party patents, the value of our intellectual property rights would diminish and our business, competitive position and results of operations would suffer.
As of December 31, 2017, we had 25 pending patent applications. However, the filing of a patent application does not mean that we will be issued a patent, or that any patent eventually issued will be as broad as requested in the patent application or sufficient to protect our technology. Any modification required to a current patent application may delay the approval of such patent application which would have a material adverse effect on our business, results of operations and financial condition. In addition, there are a number of factors that could cause our patents, if granted, to become invalid or unenforceable or that could cause our patent applications to not be granted, including known or unknown prior art, deficiencies in the patent application or the lack of originality of the technology. Our competitive position and future revenues will depend in part on our ability and the ability of our licensors and collaborators to obtain and maintain patent protection for our product candidates, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We have filed U.S. and international patent applications for process patents; however, we cannot predict:
| • | the degree and range of protection any patents will afford us against competitors and those who infringe upon our patents, including whether third parties will find ways to invalidate or otherwise circumvent our licensed patents; |
| • | if and when patents will issue; |
| • | whether or not others will obtain patents claiming aspects similar to those covered by our licensed patents and patent applications; or |
| • | whether we will need to initiate litigation or administrative proceedings, which may be costly, and whether we win or lose. |
If patent rights covering our products or technologies are not sufficiently broad, they may not provide us with sufficient proprietary protection or competitive advantages against competitors with similar products and technologies. Furthermore, if the U.S. Patent and Trademark Office or foreign patent offices issue patents to us or our licensors, others may challenge the patents or circumvent the patents, or the patent office or the courts may invalidate the patents. Thus, any patents we own or license from or to third parties may not provide any protection against our competitors and those who infringe upon our patents.
Furthermore, the life of our patents is limited. The patents we hold, and the patents that may be issued in the future based on patent applications from the patent families, relating to our lead product candidate are expected to expire between 2031 and 2037 depending on any extensions of term for which we may be eligible that we may be granted.
If we cannot obtain new patents, maintain our existing patents and protect the confidentiality and proprietary nature of our trade secrets and other intellectual property, our business and competitive position will be harmed.
45
Table of Contents
Our success will depend in part on our ability to obtain and maintain patent and regulatory protections for our products and investigational compounds, to preserve our trade secrets and other proprietary rights, to operate without infringing the proprietary rights of third parties, and to prevent third parties from circumventing our rights. Due to the time and expense of bringing new product candidates through development and regulatory approval to the marketplace, there is particular importance in obtaining patent and trade secret protection for significant new technologies, products and processes.
We have and may in the future obtain patents or the right to practice patents through ownership or license. Our patent applications may not result in the issue of patents in the U.S. or other countries. Our patents may not afford adequate protection for our products. Third parties may challenge our patents. If any of our patents are narrowed, invalidated or become unenforceable, competitors may develop and market products similar to ours that do not conflict with or infringe our patents rights, which could have a material adverse effect on our financial condition. We may also finance and collaborate in research conducted by government organizations, hospitals, universities or other educational or research institutions. Such research partners may be unwilling to grant us exclusive rights to technology or products developed through such collaborations. There is also a risk that disputes may arise as to the rights to technology or products developed in collaboration with other parties. Our products and product candidates are expensive and time-consuming to test and develop. Even if we obtain and maintain patents, our business may be significantly harmed if the patents are not broad enough to protect our products from copycat products.
Significant legal questions exist concerning the extent and scope of patent protection for biopharmaceutical products and processes in the U.S. and elsewhere. Accordingly, there is no certainty that patent applications owned or licensed by us will issue as patents, or that our issued patents will afford meaningful protection against competitors. Once issued, patents are subject to challenge through both administrative and judicial proceedings in the U.S. and other countries. Such proceedings include re-examinations, inter partes reviews, post-grant reviews and interference proceedings before the U.S. Patent and Trademark Office, as well as opposition proceedings before the European Patent Office and other non-U.S. patent offices. Litigation may be required to enforce, defend or obtain our patent and other intellectual property rights. Any administrative proceeding or litigation could require a significant commitment of our resources and, depending on outcome, could adversely affect the scope, validity or enforceability of certain of our patent or other proprietary rights.
In addition, our business requires using sensitive technology, techniques and proprietary compounds that we protect as trade secrets. However, we may also rely heavily on collaboration with, or discuss the potential for collaboration with, suppliers, outside scientists and other biopharmaceutical companies. Collaboration and discussion of potential collaboration present a strong risk of exposing our trade secrets. If our trade secrets were exposed, it would help our competitors and adversely affect our business prospects.
If we are found to be infringing on patents owned by others, we may be forced to pay damages to the patent owner and/or obtain a license to continue the manufacture, sale or development of our products. If we cannot obtain a license, we may be prevented from the manufacture, sale or development of our products, which would adversely affect our business.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages and required to defend against litigation which could result in substantial costs and may have a material adverse effect on our business, results of operations and financial condition.
We have not received to date any claims of infringement by any third parties. However, as our product candidates progress into clinical trials and commercialization, if at all, our public profile and that of our product candidates may be raised and generate such claims. Defending against such claims, and occurrence of a judgment adverse to us, could result in unanticipated costs and may have a material adverse effect on our business and competitive position. If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we may incur substantial costs and we may have to:
| • | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
46
Table of Contents
| • | redesign our products or processes to avoid infringement; |
| • | stop using the subject matter claimed in the patents held by others, which could cause us to lose the use of one or more of our product candidates; |
| • | defend litigation or administrative proceedings that may be costly whether we win or lose, and which could result in a substantial diversion of management resources; or |
| • | pay damages. |
Any costs incurred in connection with such events or the inability to sell our products may have a material adverse effect on our business, results of operations and financial condition.
We rely on confidentiality agreements that could be breached and may be difficult to enforce which could have a material adverse effect on our business and competitive position.
Our policy is to enter agreements relating to the non-disclosure of confidential information with third parties, including our contractors, consultants, advisors and research collaborators, as well as agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees and consultants while we employ them. However, these agreements can be difficult and costly to enforce. Moreover, to the extent that our contractors, consultants, advisors and research collaborators apply or independently develop intellectual property in connection with any of our projects, disputes may arise as to the proprietary rights to the intellectual property. If a dispute arises, a court may determine that the rights belongs to a third party, and enforcement of our rights can be costly and unpredictable. In addition, we rely on trade secrets and proprietary know-how that we seek to protect in part by confidentiality agreements with our employees, contractors, consultants, advisors and other third parties. Despite the protective measures we employ, we still face the risk that:
| • | these agreements may be breached; |
| • | these agreements may not provide adequate remedies for the applicable type of breach; or |
| • | our trade secrets or proprietary know-how will otherwise become known. |
Any breach of our confidentiality agreements or our failure to effectively enforce such agreements may have a material adverse effect on our business and competitive position.
If we cannot meet requirements under our license agreement, we could lose the rights to our products, which could have a material adverse effect on our business.
We depend on the license agreement with TRDF to maintain the intellectual property rights to certain of our product candidates. Our license agreement requires us to make payments and satisfy performance obligations in order to maintain our rights under this agreement. This agreement lasts either throughout the life of the patents that are the subject of the agreement, or with respect to other licensed technology, for a number of years after the first commercial sale of the relevant product.
In addition, we are responsible for the cost of filing and prosecuting certain patent applications and maintaining certain issued patents licensed to us. If we do not meet our obligations under our license agreement in a timely manner, we could lose the rights to our proprietary technology, which could have a material adverse effect on our business, results of operations and financial condition.
47
Table of Contents
Risks Related to Our Operations in Israel
Potential political, economic and military instability in the State of Israel, where our research facilities are located, may adversely affect our results of operations.
Our research offices and lab are located in the State of Israel. Accordingly, political, economic and military conditions in Israel and the surrounding region may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its neighboring Arab countries, the Hamas militant group and the Hezbollah. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect our operations and results of operations. Since October 2000, there have been increasing occurrences of terrorist violence. In 2006, a conflict between Israel and the Hezbollah in Lebanon resulted in thousands of rockets being fired from Lebanon into Israel. In 2008, Israel engaged in an armed conflict with Hamas in the Gaza Strip, which involved missile strikes against Israel and negatively affected business conditions in Israel. In 2012, Israel experienced a similar armed conflict, resulting in hundreds of rockets being fired from the Gaza Strip. In 2014, Israel yet again experienced rocket strikes against civilian targets in various parts of Israel, as part of an armed conflict commenced between Israel and Hamas. Ongoing and revived hostilities or other Israeli political or economic factors, such as, an interruption of operations at the Tel Aviv airport, could prevent or delay shipments of our components or products. If continued or resumed, these hostilities may negatively affect business conditions in Israel in general and our business in particular. In the event that hostilities disrupt the ongoing operation of our facilities or the airports and seaports on which we depend to import and export our supplies and product candidates, our operations may be materially adversely affected.
In addition, since 2010 political uprisings and conflicts in various countries in the Middle East, including Egypt and Syria, are affecting the political stability of those countries. It is not clear how this instability will develop and how it will affect the political and security situation in the Middle East. This instability has raised concerns regarding security in the region and the potential for armed conflict. In Syria, a country bordering Israel, a civil war is taking place. In addition, it is widely believed that Iran, which has previously threatened to attack Israel, has been stepping up its efforts to achieve nuclear capability. Iran is also believed to have a strong influence among extremist groups in the region, such as Hamas in Gaza and Hezbollah in Lebanon. Additionally, the Islamic State of Iraq and Levant, or ISIL, a violent jihadist group, is involved in hostilities in Iraq and Syria. Although ISIL’s activities have not directly affected the political and economic conditions in Israel, ISIL’s stated purpose is to take control of the Middle East, including Israel. The tension between Israel and Iran and/or these groups may escalate in the future and turn violent, which could affect the Israeli economy in general and us in particular. Any potential future conflict could also include missile strikes against parts of Israel, including our offices and facilities. Such instability may lead to deterioration in the political and trade relationships that exist between the State of Israel and certain other countries. Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions, could harm our results of operations and could make it more difficult for us to raise capital. Parties with whom we do business may sometimes decline to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary in order to meet our business partners face to face. Several countries, principally in the Middle East, still restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business with Israel and Israeli companies if hostilities in Israel or political instability in the region continues or increases. Similarly, Israeli companies are limited in conducting business with entities from several countries. For instance, in 2008, the Israeli legislature passed a law forbidding any investments in entities that transact business with Iran. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements.
Our insurance does not cover losses that may occur as a result of an event associated with the security situation in the Middle East or for any resulting disruption in our operations. Although the Israeli government has in the past covered the reinstatement value of direct damages that were caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained or, if maintained, will be sufficient to
48
Table of Contents
compensate us fully for damages incurred and the government may cease providing such coverage or the coverage might not suffice to cover potential damages. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions generally and could harm our results of operations.
Furthermore, in the past, the State of Israel and Israeli companies have been subjected to economic boycotts. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial conditions or the expansion of our business.
Our research operations may be disrupted by the obligations of our personnel to perform military service which could have a material adverse effect on our business.
Our employees and consultants in Israel may be obligated to perform one month, and in some cases longer periods, of military reserve duty until they reach the age of 40 (or older, for citizens who hold certain positions in the Israeli armed forces reserves) and, in the event of a military conflict or emergency circumstances, may be called to immediate and unlimited active duty. In the event of severe unrest or other conflict, individuals could be required to serve in the military for extended periods of time. In response to increases in terrorist activity, there have been periods of significant call-ups of military reservists. It is possible that there will be similar large-scale military reserve duty call-ups in the future. Our operations could be disrupted by the absence of a significant number of our officers, directors, employees and consultants related to military service. Such disruption could materially adversely affect our business and operations. Additionally, the absence of a significant number of the employees of our Israeli suppliers and contractors related to military service or the absence for extended periods of one or more of their key employees for military service may disrupt their operations.
Because a certain portion of our expenses are incurred in New Israeli Shekels, or NIS, our results of operations may be seriously harmed by currency fluctuations and inflation.
We report our financial statements in U.S. dollars, our functional currency. Although most of our expenses are incurred in U.S. dollars, we pay a portion of our expenses in New Israeli Shekels, or NIS, and as a result, we are exposed to risk to the extent that the inflation rate in Israel exceeds the rate of devaluation of the NIS in relation to the U.S. dollar or if the timing of these devaluations lags behind inflation in Israel. In that event, the U.S. dollar cost of our operations in Israel will increase and our U.S. dollar-measured results of operations will be adversely affected. To the extent that the value of the NIS increases against the dollar, our expenses on a dollar cost basis increase. Our operations also could be adversely affected if we are unable to guard against currency fluctuations in the future. To date, we have not engaged in hedging transactions. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rate of the U.S. dollar against the NIS. These measures, however, may not adequately protect us from material adverse effects.
We received Israeli government grants for our research and development activities and programs. The terms of such grants may require us, in the future, to pay royalties and to satisfy specific conditions if and to the extent we receive future royalties or in order to complete the sale of such grant-based technologies and programs. We may be required to pay penalties in addition to payment of the royalties.
Our research and development efforts have been financed, in part, through royalty-bearing grants from the IIA. As of December 31, 2017, we have received the aggregate amount of approximately $2.6 million from the IIA for the development of our abovementioned technologies. With respect to such grants we are committed to pay certain royalties (including accrued LIBOR interest) up to $2.7 million. We are required to comply with the requirements of the Israeli Encouragement of Research, Development and Technological Innovation in the Industry Law, 5744-1984, as amended, and related regulations, or the Research Law, with respect to these past grants. If we fail to comply with the Research Law, we may be required to refund certain grants previously received and/or to pay interest and penalties and we may become subject to criminal charges.
49
Table of Contents
We have not commenced the payment obligation of the royalties and have a contingent obligation with respect to royalty-bearing participation received or accrued, to include LIBOR interest, in the amount of $2.7 million.
In the past, we received Israeli government grants for certain of our research and development activities. The terms of those grants may require us, in addition to payment of royalties, to satisfy specified conditions in order to manufacture products and transfer technologies outside of Israel. We may be required to pay penalties in addition to repayment of the grants.
We received Israeli government grants for certain of our research and development activities from the IIA. With respect to such grants we are obligated to pay royalties at a rate of 3% to 6% from the revenues generated from the sale of product (as well as revenue from associated services) developed using the IIA grants up to the total amount of grants received, linked to the U.S. dollar and bearing interest at an annual rate of LIBOR applicable to dollar deposits.
A recent amendment, or Amendment No. 7, to the Law for the Encouragement of Research, Development and Technological Innovation in the Industry, 1984-5744, or the R&D Law, mandated the formation of the IIA to replace the Chief Scientist. Pursuant to Amendment No. 7, the IIA may establish new guidelines and promulgate new regulations under the R&D Law. These changes in the structure of the IIA and the R&D Law may affect our existing or future IIA programs and related obligations. At this stage, we cannot predict what changes, if any, the new authority may make.
The R&D Law and the regulations promulgated thereunder provide that when a company develops know-how, technology or products using IIA grants, the terms of these grants and the R&D Law restrict the transfer of such know-how, and the transfer of manufacturing or manufacturing rights of such products, technologies or know-how outside of Israel, without the prior approval of the IIA. Therefore, if aspects of our technologies are deemed to have been developed with IIA funding according to the R&D Law, the discretionary approval of the IIA may be required for any assignment and/or transfer to third parties inside or outside of Israel of know-how or transfer outside of Israel of manufacturing or manufacturing rights related to those aspects of such technologies, and may result in payment of increased royalties (both increased royalty rates and increased royalties ceilings) and/or payment of additional amounts to the IIA. Such approvals may be subject to conditions and\or may not be received. Furthermore, according to the R&D Law, the IIA may impose certain conditions on any arrangement under which it permits us to transfer technology or development out of Israel (including for the purpose of manufacturing).
The R&D Law and the regulations promulgated thereunder provide that the transfer of IIA-supported technology or know-how outside of Israel may involve the payment of additional amounts depending upon the value of the transferred technology or know-how, the amount of IIA support, the time of completion of the IIA-supported research project and other factors up to a maximum of six times the amount of grants received. These restrictions and requirements for payment may impair our ability to sell our technology assets outside of Israel or to outsource or transfer development or manufacturing activities with respect to any product or technology outside of Israel. Furthermore, the consideration available to our stockholders in a transaction involving the transfer outside of Israel of technology or know-how developed with IIA funding (such as a merger or similar transaction) may be reduced by any amounts that we are required to pay to the IIA. Our obligations and limitations pursuant to the R&D Law are not limited in time and may not be terminated by us at will. As of the date hereof, we have not been required to pay any royalties with respect to the IIA grants.
We may become subject to claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and adversely affect our business.
We enter into agreements with our employees pursuant to which they agree that any inventions created in the scope of their employment or engagement are assigned to us or owned exclusively by us, depending on the jurisdiction, without the employee retaining any rights. A significant portion of our intellectual property has been
50
Table of Contents
developed by our employees in the course of their employment for us. Under the Israeli Patent Law, 5727-1967 (the “Patent Law”), inventions conceived by an employee during the scope of his or her employment with a company are regarded as “service inventions,” which belong to the employer, absent a specific agreement between the employee and employer giving the employee service invention rights. The Patent Law also provides that if there is no such agreement between an employer and an employee, the Israeli Compensation and Royalties Committee (the “Committee”), a body constituted under the Patent Law, shall determine whether the employee is entitled to remuneration for his or her inventions. Recent decisions by the Committee and the Israeli Supreme Court have created uncertainty in this area, as the Israeli Supreme Court held that employees may be entitled to remuneration for their service inventions despite having specifically waived any such rights. Further, the Committee has not yet determined the method for calculating this Committee-enforced remuneration. Although our employees have agreed that any rights related to their inventions are owned exclusively by us, we may face claims demanding remuneration in consideration for such acknowledgement. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current and/or former employees, or be forced to litigate such claims, which could negatively affect our business.
Risks Related to Our Common Stock
Our stock price may be volatile and purchasers of our common stock could incur substantial losses.
The trading price of our common stock has been volatile and may continue to be volatile and subject to wide fluctuations in the future. Many factors could have an impact on our stock price, including fluctuations in our or our competitors’ operating results, clinical trial results or adverse events associated with our product candidates, product development by us or our competitors, changes in laws, including healthcare, tax or intellectual property laws, intellectual property developments, acquisitions or other strategic transactions, changes in financial or operational estimates or projections and the perceptions of our investors that we are not performing or meeting expectations. The trading price of the common stock of many biopharmaceutical companies, including ours, has experienced extreme price and volume fluctuations, which have at times been unrelated to the operating performance of the companies whose stocks were affected.
In addition, the securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of shares of our common stock.
Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that an investor may not consider to be in the best interests of our stockholders.
Our directors, executive officers, principal stockholders and affiliated entities beneficially own, in the aggregate, approximately 70% of our common stock, as of December 31, 2017, giving effect to options, convertible notes and other derivative securities that are held by such persons that are exercisable within such 60 days from such date. As a result, if some or all of them acted together, they would have the ability to exert substantial influence over the election of our board of directors and the outcome of issues requiring approval by our stockholders. This concentration of ownership may have the effect of delaying or preventing a change in control of our company that may be favored by other stockholders. This could prevent the consummation of transactions favorable to other stockholders, such as a transaction in which stockholders might otherwise receive a premium for their shares over current market prices.
Future sales and issuances of our securities or rights to purchase securities, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause the prices of our securities to fall.
Additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell
51
Table of Contents
common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner, we determine from time to time. If we sell common stock, convertible securities or other equity securities in one or more transactions, existing investors may be materially diluted by subsequent sales, and new investors could gain rights superior to our existing stockholders.
Pursuant to the Share Ownership and Option Plan (2013), or the 2013 Plan, and the 2008 Equity Incentive Plan, or the 2008 Plan, our management is authorized to grant share options and other equity-based awards to our employees, directors and consultants. As of December 31, 2017, our employees and officers held share options to purchase an aggregate of 3,252,785 shares of common stock under our 2013 Plan. If our board of directors elects to increase the number of shares available for future grant by the maximum amount each year, our stockholders may experience additional dilution, which could cause our share price to fall.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
Our principal executive offices are currently located at 950 Winter Street, Waltham, Massachusetts, and consist of 3,736 square feet of office space under lease until December 2020, with an option to extend the lease period for additional 3 years. Our research headquarters is located in Park Tamar, Rehovot, Israel and consists of approximately 225 square meters of office space under a lease that expires on April 2020.
From time to time, we may become involved in various lawsuits and legal proceedings, which arise in the ordinary course of business. We are currently unaware of any material pending legal proceedings to which we are party or of which our property is the subject. However, we may at times in the future become involved in litigation in the ordinary course of business, which may include actions related to or based on our intellectual property and its use, customer claims, employment practices and employee complaints and other events arising out of our operations. When appropriate in management’s estimation, we will record adequate reserves in our financial statements for pending litigation. Litigation is subject to inherent uncertainties, and an adverse result in any such matters could adversely impact our reputation, operations, and our financial operating results or overall financial condition. Additionally, any litigation to which we may become subject could also require significant involvement of our senior management and may divert management’s attention from our business and operations.
ITEM 4. MINE SAFETY DISCLOSURES
None.
52
Table of Contents
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information for Common Stock
Our common stock is traded on the OTCQB Market under the symbol “ELOX.” Prior to completion of the Transaction, our common stock was traded under the symbol “SVON.”
The following table sets forth, for the fiscal periods indicated, the high and low bid prices of a share of our common stock as reported by the OTCQB Market. All periods except for the quarter ending December 31, 2017 reflect the periods prior to the completion of the Transaction and do not reflect the reverse stock split effected on December 19, 2017. Such quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
| HIGH | LOW | |||||||
| Quarters Ended |
||||||||
| September 30, 2015 |
$ | 0.99 | $ | 0.55 | ||||
| December 31, 2015 |
$ | 0.72 | $ | 0.32 | ||||
| March 31, 2016 |
$ | 0.40 | $ | 0.19 | ||||
| June 30, 2016 |
$ | 0.28 | $ | 0.15 | ||||
| September 30, 2016 |
$ | 0.22 | $ | 0.08 | ||||
| December 31, 2016 |
$ | 0.20 | $ | 0.11 | ||||
| March 31, 2017 |
$ | 0.30 | $ | 0.16 | ||||
| June 30, 2017 |
$ | 0.38 | $ | 0.16 | ||||
| September 30, 2017 |
$ | 0.36 | $ | 0.18 | ||||
| December 31, 2017* |
$ | 8.80 | $ | 4.00 | ||||
| * | Reflects reverse stock split effected on December 19, 2017. |
The closing price of our common stock as reported by the OTCQB Market on March 14, 2018 was $7.15 per share. As of March 14, 2018 there were approximately 23 holders of record of our common stock. The actual number of holders of our common stock is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers or held by other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Issuer Purchases of Equity Security
There were no repurchases of our common stock during the fourth quarter of 2017.
Dividend Policy
We have not paid dividends on our common stock since inception and we do not intend to pay any dividends to our stockholders in the foreseeable future. We expect that any earnings, which we may realize, will be retained to finance the growth of our company. The declaration of dividends in the future will be at the election of our board of directors and will depend upon our earnings, capital requirements, financial position, general economic conditions, and other factors the board of directors deem relevant.
Recent Sales of Unregistered Securities
None, except as previously disclosed on our Quarterly Reports on Forms 10-Q and Current Reports on Forms 8-K.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable to a “smaller reporting company”, as defined in Item 10(f)(1) of SEC Regulation S-K.
53
Table of Contents
| ITEM 7. | MANAGEMENT’ S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION |
The following information should be read in conjunction with the consolidated financial statements and related notes thereto included in this Annual Report on Form 10-K
Except for the historical information contained herein, the matters discussed in this Annual Report on Form 10-K may be deemed to be forward-looking statements that involve risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. In this Annual Report on Form 10-K, words such as “may,” “expect,” “anticipate,” “estimate,” “intend,” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements.
Our actual results and the timing of certain events may differ materially from the results discussed, projected, anticipated, or indicated in any forward-looking statements. We caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this Annual Report. In addition, even if our results of operations, financial condition and liquidity, and the development of the industry in which we operate are consistent with the forward-looking statements contained in this Annual Report, they may not be predictive of results or developments in future periods.
The following information and any forward-looking statements should be considered in light of factors discussed elsewhere in this Annual Report on Form 10-K, including those risks identified under Item 1A. Risk Factors. In many instances, dollar amounts contained in the narrative descriptions in the following section of this Annual Report are stated in approximate values, pursuant to generally accepted rounding conventions. We caution readers not to place undue reliance on any forward-looking statements made by us, which speak only as of the date they are made. We disclaim any obligation, except as specifically required by law and the rules of the SEC, to publicly update or revise any such statements to reflect any change in our expectations or in events, conditions or circumstances on which any such statements may be based, or that may affect the likelihood that actual results will differ from those set forth in the forward-looking statements.
Reverse Merger
On December 19, 2017, Sevion Therapeutics, Inc. (“Sevion”) acquired Eloxx Pharmaceuticals, Limited (“Private Eloxx” or “Eloxx Limited”) pursuant to a merger between the companies (the “Transaction” or “Reverse Merger”). Upon consummation of the Transaction (the “Closing”), Sevion adopted the business plan of Private Eloxx and discontinued the pursuit of Sevion’s business plan pre-Closing. In connection with the Transaction, Sevion agreed to acquire all of the outstanding capital stock of Private Eloxx in exchange for the issuance of an aggregate 20,316,656 shares of Sevion’s common stock, par value $0.01 per share (the “Common Stock”), after giving effect to a 1-for-20 reverse split effected immediately prior to the Transaction. As a result of the Transaction, Private Eloxx became a wholly-owned subsidiary of Sevion. While Sevion was the legal acquirer in the transaction, Private Eloxx was deemed the accounting acquirer. Immediately after giving effect to the Transaction, on December 19, 2017, Sevion changed its name to Eloxx Pharmaceuticals, Inc. (“Eloxx” or the “Company”).
The annual consolidated financial statements of the Company reflect the operations of Private Eloxx as the acquirer for accounting purposes, together with a deemed issuance of shares, equivalent to the shares held by the stockholders of the legal acquirer, Sevion, prior to the Transaction, and a recapitalization of the equity of the accounting acquirer. The annual consolidated financial statements include the accounts of the Company since the effective date of the Reverse Merger and the accounts of Private Eloxx since inception.
54
Table of Contents
Upon closing of the Reverse Merger, the Company assumed the obligations under outstanding warrants previously issued by Eloxx Limited to purchase its share capital and, in connection therewith, issued warrants to purchase 346,307 shares of the Company’s common stock to certain warrant holders of Eloxx Limited.
In addition, upon closing of the Reverse Merger, the Company assumed all of the outstanding obligations under the Eloxx 2013 Share Ownership and Option Plan (the “2013 Plan”) and, accordingly, the Company has reserved 2,307,738 shares of the Company’s common stock for issuance upon the exercise of such options. As part of the Company’s assumption of the outstanding options under the 2013 Plan, the Company also assumed the 2013 Plan and accordingly reserved 189,751 shares of the Company’s common stock for future grants.
Immediately prior to the closing of the Reverse Merger the Company raised gross proceeds of $13.5 million at a price per share of $0.15 from accredited investors as a private placement. The amount was raised pursuant a share purchase agreement dated May 31, 2017, as amended between Eloxx Limited and a group of accredited investors, (“Eloxx SPA”). Under the Eloxx SPA and the first joinder thereunder executed on June 29, 2017, Eloxx Limited received gross proceeds of $15.0 million from the group of accredited investors. In accordance with the terms of the Eloxx SPA, each of the investors executed a separate subscription agreement with the Company for the total investment of an additional $15.0 million in exchange for the Company’s shares of common stock at a price per share of $0.15 immediately prior to the consummation of the Reverse Merger. With the consent of the parties, an amount of $1.5 million was invested by an accredited investor under the subscription agreement into Sevion.
On August 2, 2017, Eloxx Limited raised under a second joinder to the Eloxx SPA, an additional aggregate amount of $8.0 million, half of the amount was invested in Eloxx Limited on August 2, 2017 and the remainder was invested in Eloxx Limited immediately prior to the consummation of the Reverse Merger but was deemed an investment in the Company’s share capital for the purpose of the exchange ratio under the Agreement.
This private placement was made solely to “accredited investors,” as that term is defined in Regulation D under the Securities Act of 1933, as amended (the “Securities Act”), and was conducted in reliance on the exemption from registration afforded by Section 4(2), Rule 506 of Regulation D and Regulation S under the Securities Act, as amended, and corresponding provisions of state securities laws.
Following the Reverse Merger and reverse stock split, and commencing December 20, 2017, the Company’s Common Stock symbol on OTCQB marketplace changed to “SVOND”, and subsequently changed to “ELOX” on January 19, 2018.
Effective with the Reverse Merger each member of the Board of Directors of Eloxx Limited prior to the Reverse Merger was appointed to the Company’s Board of Directors. In addition, each officer of Eloxx Limited was reappointed as an officer of the Company. Also effective with the Reverse Merger, the Company’s Board affirmed its financial year end as December 31, 2017 to align with the fiscal year end of Eloxx Limited.
Company Overview
We are a global biopharmaceutical company focused on discovering and developing novel therapeutics for the treatment of rare and ultra-rare premature stop codon diseases. We are harnessing the science of genetic read-through to develop novel drug product candidates that interact with the ribosome to overcome these premature stop codons. Our revolutionary small molecule approach is designed to unleash the potential to restore production of full length functional proteins with the goal of enabling a return toward normal cellular function. We believe there is a broad application of this approach to the over 1800 rare and ultra-rare diseases where nonsense mutation has been implicated in the cause or pathway of human disease.
55
Table of Contents
Our research and development strategy is to target rare or ultra-rare diseases where a high unmet medical need, nonsense mutation bearing, patient population has been identified. We focus on clinical indications where there is a high unmet medical need, established preclinical read-through or personalized medicine experiments that are predictive of clinical activity, and a definable path for Orphan Drug development, regulatory approval, patient access and commercialization. We believe patient advocacy to be an important element of patient focused drug development and seek opportunities to collaborate with patient advocacy groups throughout the discovery and development process. Our current clinical focus is on cystic fibrosis (or “CF”) and cystinosis where we are advancing our lead drug product candidate ELX-02.
We intend to be the global leader in the application of the science of translational read through and the associated pathway of nonsense mediated messenger ribonucleic acid (“mRNA”) decay. We believe that expanding our expertise across these basic science areas of mRNA regulation, ribosomal function, and protein translation forms a solid foundation to support our discovery and development activities. Our compounds modulate the activity of the ribosome, the organelle within living cells responsible for protein production, a process also known as translation. These novel small molecule compounds are designed to allow the ribosome to read-through a nonsense mutation in mRNA (which is transcribed from the DNA sequence), to restore the translation process to produce full length, functional proteins and increase the amount of mRNA that would otherwise be degraded as part of a phenomenon called nonsense mediated mRNA decay. As our compounds target the general mechanism for protein production in the cell, we believe they have the potential to treat hundreds of genetic diseases where nonsense mutations have impaired gene function. Our subcutaneously injected small molecules have the potential to be self-administered and to be active at most tissue locations across the body.
We believe that our library of related novel small molecules hold the potential to be a disease-modifying therapies that may change the course of hundreds of genetic diseases and improve the lives of patients. Our early preclinical data in animal models of nonsense mutations suggests that drug product candidates from our read through compound library may have potential beneficial effects for each of the following diseases: cystic fibrosis, cystinosis, mucopolysaccharidosis type 1, Duchenne muscular dystrophy and Rett syndrome, and have demonstrated the potential for beneficial effects in multiple organs such as the brain, kidney, muscles and others. We intend to advance one or more additional molecules from our drug product candidate library toward clinical development by initiating the required investigational new drug (“IND”)-enabling studies in 2018.
Currently our lead program, ELX-02, is focused on development for cystic fibrosis and cystinosis patients with diagnosed nonsense mutations. To advance the program, we have held pre-clinical trial application (CTA) discussions with the Federal Agency for Medicines and Health Products (the “FAMHP”) in Brussels Belgium and pre-IND discussions with the U.S. Food & Drug Administration (the “FDA”) for cystic fibrosis and cystinosis, respectively. We are on-track for mid-2018 submission of our IND and CTA. Approval of these submissions will be required for initiation of Phase 2 studies in cystic fibrosis and cystinosis in 2018.
As part of our clinical program, we have completed a Phase 1 single ascending dose (“SAD”) study in a total of 60 healthy volunteers at sites in Israel (ClinicalTrials.gov Identifier: NCT02807961) and Belgium (ClinicalTrials.gov Identifier: NCT03292302). Currently ongoing is the Phase 1 multiple ascending dose (“MAD”) study in 45 healthy volunteers in Belgium (ClinicalTrials.gov Identifier: NCT03309605). We anticipate that the Phase 1 MAD study will be completed in 2018. The results from the completed Phase 1 study will be included in the planned IND and CTA submissions.
We believe there is a significant unmet medical need in the treatment of Cystic Fibrosis patients carrying nonsense mutations on one or both alleles of the Cystic Fibrosis Transmembrane Conductance Regulator (“CFTR”) gene. Cystic fibrosis is the most prevalent genetic disease in the western world and there are no currently approved therapies that target the impairment associated with Class 1 CFTR mutations. We believe that nonsense mutations may impact a similar proportion of patients diagnosed with cystinosis. There are no currently approved therapeutics that target the nonsense mutation mediated impairment of cystinosin the cystine-selective
56
Table of Contents
transport channel in the lysosomal membrane that is attributed as the cause for the accumulation of cystine in this disease state. Given the high proportion of pediatric patients in each of these rare orphan diseases we intend to apply for relevant Orphan Drug incentives in the US and Europe, including the Rare Pediatric Disease Priority Review Voucher.
Currently, the European Medicines Agency (the “EMA”) has designated ELX-02 as an orphan medicine for the treatment of mucopolysaccharidosis type I (“MPS I”), and the FDA has granted orphan drug designation to ELX-02 for the treatment of MPS I and for the treatment of Rett Syndrome.
We hold worldwide development and commercialization rights to ELX-02 and novel compounds in our read-through library, for all indications, in all territories, under a license from the Technion Research and Development Foundation Ltd. Professor Timor Baasov, the inventor of our compounds, has served as our senior consultant since our incorporation.
As of December 31, 2017, we had cash and cash equivalents of $24.0 million. We expect that our current cash and cash equivalents will be sufficient to fund our current operations at least through the end of the first quarter of 2019.
Since our inception, we have incurred significant operating losses. Our net losses were $21.2 million and $9.8 million for each of the years ended December 31, 2017 and 2016, respectively. As of December 31, 2017, we had an accumulated deficit of $39.0 million. To date, we have financed our operations primarily through equity capital investments, and to a lesser extent from loans and grants from Israeli Innovation Authority of the Ministry of Economy and Industry, or the IIA. We have devoted substantially all of our financial resources and efforts to research and development. We expect that it will be many years, if ever, before we receive regulatory approval and have a product candidate ready for commercialization. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. Our net losses may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially if and as we:
| • | advance ELX-02 further into clinical trials; |
| • | continue the preclinical development of our research programs and advance candidates into clinical trials; |
| • | identify additional product candidates and advance them into preclinical development; |
| • | pursue regulatory approval of product candidates; |
| • | seek marketing approvals for our product candidates that successfully complete clinical trials; |
| • | establish a sales, marketing and distribution infrastructure to commercialize any product candidates for which we obtain marketing approval; |
| • | maintain, expand and protect our intellectual property portfolio; |
| • | hire additional clinical, regulatory and scientific personnel; |
| • | add operational, financial and management information systems and personnel, including personnel to support product development; |
| • | acquire or in-license other product candidates and technologies; and |
| • | operate as a public company. |
57
Table of Contents
Results of Operations
Comparison of the Years Ended December 31, 2017 and 2016
The following table is in thousands:
| Year Ended December 31, |
||||||||
| 2017 | 2016 | |||||||
| Operating expenses: |
||||||||
| Research and development, net |
$ | 16,398 | $ | 8,986 | ||||
| General and administrative |
3,992 | 854 | ||||||
|
|
|
|
|
|||||
| Total operating costs |
20,390 | 9,840 | ||||||
| Financial and other expenses, net |
824 | 7 | ||||||
| Net loss |
$ | 21,214 | $ | 9,847 | ||||
|
|
|
|
|
|||||
Research and development expenses, net.
Research and development expenses were $16.4 million for the year ended December 31, 2017 compared to $9.0 million for the year ended December 31, 2016, an increase of $7.4 million. The increase in research and development expenses was primarily related to the provision recorded related to the exit fee for Technion of $3.4 million, along with fees incurred to subcontractors, consultants and advisors in connection with research and development of our ELX-02 of $3.2 million (including a deduction of research and development grants we received from the IIA) and salaries and other personnel related costs of $0.8 million.
General and administrative expenses.
General and administrative expenses were $4.0 million for the year ended December 31, 2017 compared to $0.8 million for the year ended December 31, 2016, an increase of $3.1 million. The increase in general and administrative expenses was primarily related to salaries, stock-based compensation, and other personnel related costs of $1.0 million, professional services of $0.6 million, and Reverse Merger related costs of $1.3 million.
Financial and other expenses, net.
We recorded $0.8 million in financial and other expenses for the year ended December 31, 2017 compared to $7,000 for the year ended December 31, 2016, an increase of $0.8 million. The increase in other expenses, net was primarily due to $0.6 million of amortization and revaluation of embedded conversion feature in respect to convertible loan and $0.2 million of exchange rate differences.
Net operating loss carryforwards
As of December 31, 2017, we had U.S. federal and state NOL carryforwards of $77.2 million and $27.4 million, respectively, and federal research tax credit carryforwards of $0.7 million. Our U.S. net operating loss carryforwards will begin to expire, if not utilized, beginning in 2019 through 2037, and the research tax credits will expire beginning in 2027 through 2037. These NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the newly enacted Tax Cuts and Jobs Act (“Tax Act”), federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited. It is uncertain if and to what extent various states will conform to the newly enacted federal tax law. See Note 12: Income Taxes.
58
Table of Contents
Comparison of the Years Ended December 31, 2016 and 2015
The following table is in thousands:
| Year Ended December 31, |
||||||||
| 2016 | 2015 | |||||||
| Operating expenses: |
||||||||
| Research and development, net |
$ | 8,986 | $ | 5,842 | ||||
| General and administrative |
854 | 442 | ||||||
|
|
|
|
|
|||||
| Total operating costs |
9,840 | 6,284 | ||||||
| Financial and other expenses, net |
7 | 122 | ||||||
| Net loss |
$ | 9,847 | $ | 6,406 | ||||
|
|
|
|
|
|||||
Research and development expenses, net.
Research and development expenses were $9.0 million for the year ended December 31, 2016, compared to $5.8 million for the year ended December 31, 2015, an increase of $3.2 million. The increase in research and development expenses was primarily related to salaries, stock-based compensation and other personnel related costs of $0.7 million and net fees incurred to subcontractors, consultants and advisors in connection with research and development of ELX-02 of $2.5 million.
General and administrative expenses.
General and administrative expenses were $0.9 million for the year ended December 31, 2016, compared to $0.4 million for the year ended December 31, 2015, an increase of $0.5 million. The increase in general and administrative expenses during these periods is primarily related to patent fees of $0.3 million and professional service fees of $0.1 million.
Financial and other expenses, net.
We recorded $7,000 in financial and other expenses for the year ended December 31, 2016 compared to $0.1 million for the year ended December 31, 2015. The decrease was due to $0.1 million of exchange rate differences in 2015.
Liquidity and Capital Resources
General
Liquidity is the ability of a company to generate funds to support its current and future operations, satisfy its obligations, and otherwise operate on an ongoing basis. Significant factors in the management of liquidity are funds generated by operations, levels of accounts receivable and accounts payable and capital expenditures. Since our inception and through December 31, 2017 we have funded our operations primarily through equity and convertible debt financings in private placements, as described below.
As of December 31, 2017, we had cash and cash equivalents of $24.0 million. We expect that our cash and cash equivalents as of December 31, 2017 will enable us to fund our current operations at least through the end of the first quarter of 2019. Our future viability beyond that point is dependent on our ability to raise additional capital to finance our operations. Although we have been successful in raising capital in the past, there is no assurance that we will be successful in obtaining such additional financing on terms acceptable to us, if at all. If we are unable to obtain funding, we could be forced to delay, reduce or eliminate our research and development programs, product portfolio expansion or commercialization efforts, which could adversely affect our business prospects, or we may be unable to continue operations.
We reported cash of $13.5 million in our September 30, 2017 balance sheet, and our use of cash in operations in Q4 2017 was $6.3 million. Q4 2017 research and development expense totaled $8.4 million which included $3.4 million in non-cash expense related to the Technion Agreement. Q4 2017 general & administrative expense totaled $2.2 million, transaction related costs were $0.7 million, and net loss was $10.6 million. We received net proceeds of $16.8 million in Q4 2017 related to completing our Series C financing.
59
Table of Contents
Principal Financing Activities
In April 2015, Eloxx Limited achieved a clinical milestone in connection with the share purchase agreement signed in 2013, pursuant to which, Eloxx Limited issued to investors 1,073,157 shares of Series A preferred stock with a par value of $0.01 for an aggregate amount of $0.9 million.
In July 2015, Eloxx Limited entered into a Share Purchase Agreement (the “2015 SPA”) whereby Eloxx Limited issued to existing investors 1,002,049 shares of Series B-1 preferred stock with a par value of $0.01 and 1,503,068 warrants to purchase 1,503,068 shares of Series B-1 preferred stock with an exercise price of $3.11 for an aggregate gross amount of $3.1 million, representing a price per unit of $3.11. In connection with the 2015 SPA, Eloxx Limited paid a contractor cash consideration of $0.1 million as a finder fee and granted 30,563 warrants to purchase 30,563 shares of Series B-1 preferred stock with an exercise price of $3.11.
In July 2015, one of Eloxx Limited’s employees exercised 99,829 options with an exercise price of $0.01 per share to purchase 99,829 shares of common stock.
In February 2016, Eloxx Limited entered into Shares Purchase Agreement (the “2016 SPA”) whereby Eloxx Limited issued to existing investors 1,929,676 shares of Series B-1 preferred stock with a par value of $0.01 and 2,894,519 warrants to purchase 2,894,519 shares of Series B-1 preferred stock with an exercise price of $3.11 per share for an aggregate gross amount of $6.0 million.
In connection with the 2016 SPA, Eloxx Limited paid a contractor cash consideration of $0.2 million as a finder fee and granted 48,242 warrants to purchase 48,242 shares of Series B-1 preferred stock with an exercise price of $3.11 per share.
In August 2016, Technion Investment Opportunities Fund L.P (the “TIOF) and TRDF exercised 124,786 and 311,964 warrants, respectively, to purchase shares of Series A preferred stock at an exercise price of $0.80 per share, respectively, for total consideration of $0.4 million.
In September 2016, Eloxx Limited achieved a milestone in connection with the Share Purchase Agreement signed in 2014 (“2014 SPA”), pursuant to which, Eloxx Limited paid a $0.1 million milestone payment and issued to investors 1,174,138 shares of Series B-1 preferred stock with a par value of $0.01 and 587,072 warrants to purchase 587,072 shares of Series B-1 preferred stock for an aggregate amount of $3.7 million.
In connection with the 2014 SPA milestone, Eloxx Limited paid a contractor cash consideration of $0.1 million as a finder fee and granted 36,593 warrants to purchase 36,593 shares of Series B-1 preferred stock with exercise price of $3.11 per share.
On May 22, 2017, Eloxx Limited entered into a Share Purchase Agreement (the “2017 SPA”) (and subsequently on joinder agreements)with certain existing and new investors, whereby, an aggregate gross amount of $21.5 million, which included the conversion of the loan as detailed in Note 7, was received by Eloxx Limited in exchange for the issuance of 7,136,289 shares of Series C preferred stock with par value of $0.01 with the initial closing, of which 39,293 were issued as a result of the anti-dilution effect of the Reverse Merger. The related issuance costs were $0.6 million.
In connection with the 2017 SPA, the Company granted 142,524 warrants to purchase 142,524 shares of Series C preferred stock to certain service providers as finder fee compensation.
Upon the closing of the Reverse Merger the Company issued 6,333,333 shares of common stock related to the closing of the 2017 SPA with a par value of $0.01 for an aggregate gross amount of $17.5 million. Additionally, Sevion raised $1.5 million prior to the Reverse Merger. The related issuance costs for these transactions was $0.5 million.
60
Table of Contents
Cash Flows
The following table presents the major components of net cash flows provided by (used in) operating, investing and financing activities for the periods presented (in thousands):
| Year Ended December 31, 2017 |
Year Ended December 31, 2016 |
Year Ended December 31, 2015 |
||||||||||
| Net cash used in operating activities |
$ | (15,935 | ) | $ | (8,844 | ) | $ | (5,735 | ) | |||
| Net cash (used in) provided by investing activities |
$ | (178 | ) | $ | (50 | ) | $ | 1,486 | ||||
| Net cash provided by financing activities |
$ | 37,950 | $ | 9,736 | $ | 3,882 | ||||||
Operating Activities
During the year ended December 31, 2017, the net cash used in operating activities was $15.9 million, primarily driven by our net loss of $21.2 million, partially offset by non-cash charges of $3.4 million for the provision related to the Technion exit fee of $3.4 million, $1.0 million related to changes in working capital, $0.6 million related to the amortization and revaluation of the discount of our convertible loan, and $0.1 million related to stock-based compensation.
During the year ended December 31, 2016, the net cash used in operating activities was $8.8 million, primarily driven by our net loss of $9.8 million, offset by non-cash charges including $0.9 million related to changes in working capital and $0.1 million related to stock-based compensation.
During the year ended December 31, 2015, the net cash used in operating activities was $5.7 million, primarily driven by our net loss of $6.4 million, offset by non-cash charges including $0.6 million related to changes in working capital and $0.1 million related to stock-based compensation.
Investing Activities
During the year ended December 31, 2017, the net cash used in investing activities was $0.2 million, consisting of the purchase of property and equipment of $0.2 million and restricted cash deposits of $0.1 million, offset by cash acquired in the Merger of $0.1 million.
During the year ended December 31, 2016, the net cash used in investing activities was $0.1 million, primarily consisting of the purchase of property and equipment.
During the year ended December 31, 2015, the net cash provided by investing activities was $1.5 million, primarily consisting of the purchase of the maturity of a restricted bank deposit.
Financing Activities
During the year ended December 31, 2017, the net cash provided by financing activities was $38.0 million, primarily resulting from the net proceeds of $18.4 million from the sale of preferred stock and $17.0 million from the sale of common stock.
During the year ended December 31, 2016, the net cash provided by financing activities was $9.7 million, primarily resulting from the net proceeds of $9.4 million from the sale of preferred stock and warrants.
During the year ended December 31, 2015, the net cash provided by financing activities was $3.9 million, primarily resulting from the net proceeds of $3.9 million from the sale of preferred stock and warrants.
61
Table of Contents
Government Grants from the Israeli Innovation Authority
Under the research and development agreements with the IIA and pursuant to applicable laws, we are required to pay royalties at the rate of 3% on sales to end customers of product candidates developed with funds provided by the IIA, up to an amount equal to 100% of the IIA research and development grants received, linked to the dollar plus interest on the unpaid amount received based on the 12-month LIBOR rate (from the year the grant was approved) applicable to dollar deposits. If we do not generate sales of product candidates developed with funds provided by the IIA, we are not obligated to pay royalties or repay the grants.
We received research and development grants from the IIA in the amounts of $0.9 million, $1.2 million, and $0.3 million for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, we have not commenced the payment obligation of the royalties and have a contingent obligation with respect to royalty-bearing participation received or accrued, amounting to $2.7 million.
Technion Research and Development Foundation Limited Agreement
On August 29, 2013, we entered into an agreement (“Technion Agreement”) with Technion Research and Development Foundation Limited (“TRDF”), with respect to certain technology relating to aminoglycosides and the redesign of aminoglycosides for the treatment of human genetic diseases caused by premature stop mutations and further results of the research of the technology, in order to develop and commercialize products based on such technology. Under the agreement, TRDF shall provide us research services for an estimated annual payment of $0.1 million, to be agreed exactly by the parties prior to the beginning of each year of the research period. During the years ended December 31, 2017, 2016 and 2015, we recorded general and administrative expenses amounting to $7,000, $185,000 and $0, respectively, in relation to the TRDF reimbursement for the preparation, filling, prosecution and maintenance of TRDF patents rights related to Eloxx Limited. In addition, during the years ended December 31, 2017, 2016 and 2015 the Company recorded research and development expenses in connection to the TRDF amounting to $3,465,000, $33,000 and $58,000, respectively. As of December 31, 2017 and 2016, amounts recorded in accrued expenses were $25,000 and $185,000, respectively.
In addition, TRDF shall grant the Company a license to use, market, sell or sub-license the rights of the product developed under the TRDF research results (the “Licensed Product”), as fully defined in the Technion Agreement, for the following considerations: (a) milestone payments, to be transferred upon meeting certain milestones as defined in the Technion Agreement, up to total consideration of $6.1 million; (b) certain royalties on a low- to mid- single-digit percentage of net sales (subject to change in the case of (x) sublicensing to a big pharmaceutical or biotechnology company, or (y) payment of royalties to third parties, or (z) commercialization by a third party of an authorized generic to a licensed product), for a period until the later of (i) the expiration of a valid claim on the Licensed Product in each country the Licensed Product is sold to, or (ii) a certain amount of years from the date of the first commercial sale of the Licensed Product in such country, and (c) a low- to mid- double-digit percentage of any non-royalty sub-license income received by the Company from a sub-licensed entity. In addition, the Company shall pay certain fee to TRDF upon an exit event as described in the Technion Agreement.
Moreover, upon the closing of an Exit Event which is not an Initial Public Offering (“IPO”), as defined in the Technion Agreement, TRDF shall be entitled to an amount equal to 3% of all non-refundable, non-contingent consideration, whether in cash or in kind, actually received by the Company and / or its shareholders. Upon the closing of an exit event which is an IPO, as defined in the Technion Agreement, TRDF shall be entitled to a number of Ordinary Shares of the Company representing 3% of the Company’s outstanding shares on a fully diluted basis immediately prior to the closing of such IPO.
On August 9, 2017 the Company received a legal claims letter from TRDF regarding TRDF’s alleged entitlement to an exit fee in accordance with the Technion Agreement. The parties are in discussion regarding a settlement of the legal claim, whereby the Company would issue shares to TRDF representing approximately 2.1% of the outstanding shares of the Company representing fulfillment of the “exit clause”. Therefore, the
62
Table of Contents
Company has recorded a $3.4 million research and development expense with an offsetting adjustment to additional paid-in capital for the year ended December 31, 2017 related to this legal claim.
Taurus Sublicense Agreement
On December 18, 2017, the Company executed a binding term sheet with Taurus Biosciences Inc. (“Taurus”) pursuant to which the Company grants Taurus a worldwide exclusive, sublicensable, license to the antibody SVN-001 for the development and commercialization of a product in the field of immunology, and Taurus will pay Eloxx 2% royalties on net sales. Taurus will file and prosecute any and all patents and patent applications, and shall pay all related patent expenses, with respect to this license. The Company assumed a license agreement with The Scripps Research Institute through the Reverse Merger pursuant to which the Company is required to pay a 2% royalty of net sales.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations and commitments as of December 31, 2017 and the effects such obligations are expected to have on our liquidity and cash flows in future periods (in thousands):
| Total | Less Than a Year |
2 - 3 Years |
4 - 5 Years |
More Than Five Years |
||||||||||||||||
| Contractual Obligations |
||||||||||||||||||||
| Operating lease obligations(1) |
$ | 676 | $ | 258 | $ | 232 | $ | 186 | $ | — | ||||||||||
| Total contractual cash obligations |
$ | 676 | $ | 258 | $ | 232 | $ | 186 | $ | — | ||||||||||
| (1) | Represents operating lease costs, consisting of leases for our office space in Waltham, Massachusetts that expires in December 2020, with an option to extend the lease term for an additional three years, along with our office space in Rehovot, Israel that expires in April 2020, with an option to extend the lease term for two years and laboratory space in Rehovot, Israel that expires in March 2018, with an option to extend the lease term for one year. |
Off-Balance Sheet Arrangements
As of December 31, 2017 and 2016, we do not have any off-balance sheet arrangements, as such term is defined under Item 303 of Regulation S-K, that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Recently Issued Accounting Pronouncements
For information with respect to recent accounting pronouncements, see Note 2 to the audited consolidated financial statements of Eloxx included elsewhere in this Form 10-K.
Critical Accounting Policies
The preparation of annual consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent liabilities at the date of the annual consolidated financial statements and the reported amounts of expenses during the reporting period. Our significant accounting policies, which include our management’s best estimates and judgments, are included in Note 2 to the annual consolidated financial statements of Eloxx for the year ended December 31, 2017 included in this Form 10-K.
63
Table of Contents
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of its financial condition and results of comprehensive loss is based on our consolidated financial statements, which we have prepared in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”). The preparation of these annual consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the annual consolidated financial statements, as well as the reported expenses during the reporting periods. We evaluate these estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our annual consolidated financial statements appearing elsewhere in this Form 10-K, we believe that the following accounting policies related to the treatment of stock-based compensation and contingencies are the most critical for fully understanding and evaluating our financial condition and results of operations.
Stock-Based Compensation Expense
We account for stock-based compensation granted to employees in accordance with ASC Topic 718, “Compensation-Stock Compensation” which requires the measurement and recognition of compensation expense for all stock-based payment awards based on fair value.
The fair value of each option award is estimated on the grant date using the Binomial Option-Pricing Model (“Binomial Model”). The stock-based compensation expense, is recognized using the straight-line method over the requisite service period of the award.
Key Assumptions
The Binomial Model requires the input of highly subjective assumptions, including the fair value of the underlying Ordinary Shares, the expected volatility of the price of our ordinary shares, the expected term of the option, risk-free interest rates and the expected dividend yield of our ordinary shares. These estimates involve inherent uncertainties and the application of management’s judgment. These assumptions are estimated as follows:
| • | Fair Value of our Ordinary Shares. Because our ordinary shares have not been publicly traded, we estimated the fair value of its ordinary shares, as discussed in “ordinary shares valuations” below. Upon the completion of the Reverse Merger transaction between Eloxx and Sevion, the ordinary shares will be valued by reference to its publicly-traded price. |
| • | Expected Volatility. As we do not have a trading history for its Ordinary Shares, the expected price volatility for our ordinary shares was estimated by taking the average historical price volatility for industry peers based on weekly price observations over a period equivalent to the expected term of the ordinary share option grants. Industry peers consist of several public companies that are similar in size and stage of our life cycle. |
| • | Expected Term. The expected term of options granted is derived from the output of the option valuation model and represents the period of time that options granted are expected to be outstanding. |
| • | Suboptimal exercise factor. The suboptimal exercise factor is estimated using historical option exercise information. The suboptimal exercise factor is the ratio by which the stock price must increase over the exercise price before employees are expected to exercise their stock options. |
| • | Risk-Free Rate. The risk-free interest rate is based on the yields of U.S. Treasury securities with maturities similar to the expected term of the options for each option group. |
| • | Dividend Yield. We have never declared or paid any cash dividends and do not presently plan to pay cash dividends in the foreseeable future. Consequently, we used an expected dividend yield of zero. |
64
Table of Contents
Stock-based compensation expense was $0.1 million for each of the years ended December 31, 2017, 2016 and 2015. See Notes 2 and 11 to our annual consolidated financial statements as of December 31, 2017 included elsewhere in this Form 10-K for information concerning specific assumptions used in applying the Binomial Model to determine the estimated fair value of employee shares options granted. We will continue to use judgment in evaluating the expected volatility and expected terms utilized for our stock-based compensation expense calculations on a prospective basis.
Ordinary Share Valuations
Since inception date and until December 31, 2017, our Board of Directors approved the grant of 3,310,621 options exercisable into its Ordinary Shares at exercise prices which are ranging from $0.002 to $8.00 per share.
Since inception date, the estimated fair value of the ordinary shares underlying our share options was determined at the grant date of each option by our board of directors with input from management and with the assistance of independent third-party valuations. The valuations of our ordinary shares for these dates were determined in accordance with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation (the “Practice Aid”). The methodology used by the third-party valuation specialists to assist in determining the fair value of our Ordinary Shares included estimating the fair value of the equity and then allocating this value to all of the equity interests using the option pricing method. The assumptions used in the valuation model to determine the estimated fair value of our ordinary shares as of the grant date of each option are based on numerous objective and subjective factors, combined with management judgment, including the following:
| • | Our operating and financial performance, including our levels of available capital resources; |
| • | The valuation of publicly-traded companies in the life sciences and biotechnology sectors, as well as recently completed mergers and acquisitions of peer companies; |
| • | Rights and preferences of our ordinary shares compared to the rights and preferences of its other outstanding equity securities; |
| • | Equity market conditions affecting comparable public companies, as reflected in comparable companies’ market multiples, initial public offering valuations and other metrics; |
| • | The achievement of enterprise milestones, including our development, intellectual property and regulatory progress; |
| • | The likelihood of achieving a liquidity event for our ordinary shares, such as an initial public offering or an acquisition of its company given prevailing market and biotechnology sector conditions; |
| • | Sales of our preferred shares in arms-length transactions; |
| • | The illiquidity of our securities by virtue of being a private company; and |
| • | Business risks. |
Ordinary Share Valuation Methodologies
The valuations were performed in accordance with applicable elements of the Practice Aid. The Practice Aid prescribes several valuation approaches for estimating the value of an enterprise, such as the cost, market and income approaches, and various methodologies for allocating the value of an enterprise to its ordinary shares.
The Practice Guide identifies various available methods for allocating enterprise value across classes and series of share capital to determine the estimated fair value of the ordinary shares at each valuation date. In accordance with the Practice Guide, we considered the following methods:
65
Table of Contents
| • | Option Pricing Method. Under the option pricing method (“OPM”), shares are valued by creating a series of call options with exercise prices based on the liquidation preferences and conversion terms of each equity class. The estimated fair values of the Preferred and Ordinary Shares are inferred by analyzing these options. |
| • | Hybrid Method. The hybrid method typically is a combination of the OPM and PWERM. It is appropriate when a company is likely to go through a transformative event (for example, an initial public offering or liquidation) in the near future. Just like the PWERM, the hybrid method is a scenario-based analysis. |
Based on our pre-revenue stage of development and other relevant factors, we determined that the OPM was the most appropriate method for allocating our enterprise value to determine the estimated fair value of its ordinary shares for valuations performed since inception date and until June 30, 2017. Commencing June 30, 2017, we began using the Hybrid Method by combining the OPM and M&A scenario to determine the fair value of our ordinary shares.
Under the OPM methodology, we used the pricing data from the recent rounds of preferred financings to estimate the value of the equity. Under the Hybrid Method, we estimated the probability and timing of the M&A based on management’s best estimate, taking into consideration all available information as of the valuation date, including the stage of development of our product candidates, its expected near-term and long-term funding requirements, an assessment of the current financing and life science industry environment and the economic trends, market conditions at the time of valuation and the assistance of an independent third-party valuation.
Following the closing of the Reverse Merger, the fair value of the Ordinary Shares was determined based on the closing price of our common stock on the OTCOB Market.
Contingencies
We account for our contingent liabilities in accordance with ASC Topic 450, “Contingencies”. A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated.
With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter. Currently, we are not a party to any ligation that could have a material adverse effect on our business, financial position, results of operations or cash flows.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
Not applicable to a “smaller reporting company”, as defined in Item 10(f)(1) of SEC Regulation S-K.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The consolidated financial statements and supplementary data required by this item are included in this report immediately following Part IV and are incorporated herein by reference.
66
Table of Contents
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Effective on December 19, 2017, the Company dismissed RSM US LLP, or RSM, as its independent registered public accounting firm. Effective December 20, 2017, the Board engaged Kost Forer Gabbay & Kasierer, a member of Ernst & Young Global, or EY, as the independent registered public accounting firm to audit the Company’s financial statements for the fiscal year ended December 31, 2017.
RSM audit reports on the Company’s financial statements for the fiscal years ended June 30, 2017 and June 30, 2016 did not contain an adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles, except that the reports contained an explanatory paragraph noting that there was substantial doubt as to the Company’s ability to continue as a going concern. In addition, Sevion management concluded that there was a reportable event pursuant to Item 304(a)(1)(v)(A) of Regulation S-K, due to Sevion management’s determination that material weaknesses existed in Sevion’s internal control over financial reporting as of June 30, 2017 and June 30, 2016 and, as a result, its disclosure controls and procedures were not effective.
During the years ended June 30, 2017 and June 30, 2016 and the subsequent interim period through the date of RSM dismissal, there were no disagreements with RSM on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of RSM would have caused it to make reference to the subject matter thereof in connection with its report.
During the years ended June 30, 2017 and June 30, 2016 and the subsequent interim period through the date of RSM’s dismissal, neither the Company nor anyone acting on its behalf consulted EY regarding the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Disclosure Controls and Procedures
Our management evaluated, with the participation and under the supervision of our Chief Executive Officer and our Chief Financial Officer, the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective in ensuring that information required to be disclosed by us in reports that we file or submit under the Exchange Act (i) is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and (ii) is accumulated and communicated to the Company’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. We maintain a system of internal control that is designed to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles.
67
Table of Contents
Notwithstanding that we do not qualify for the relief afforded by Instruction 1 to Item 308 of Regulation S-K to newly public companies, management has not assessed nor attested to our internal control over financial reporting as is set forth in Item 308 of Regulation S-K promulgated under the Securities Exchange Act 1934, as amended, and Section 404 of the Sarbanes-Oxley Act as of December 31, 2017, the end of our last fiscal year. We will do so initially as of December 31, 2018.
We were unable to conduct the required assessment primarily due to the Transaction that closed on December 19, 2017 and the substantial change in operational focus, management and the internal control environment following the Transaction and due to the fact the internal controls of the legal acquirer were no longer existed as of the required assessment date and during that period. Therefore this annual report does not include a report of management’s assessment regarding internal control over financial reporting.
Since the reverse merger concluded in December 2017, management has been performing a comprehensive post-transaction review of the adequacy of its internal controls over financial reporting. This discovery process has led to a diagnosis of various needs and has begun the process of taking targeted actions that are being implemented immediately, including the hiring of experienced accounting and finance staff, systems implementations, new policies and procedures, IT controls, and other steps planned throughout the 2018 fiscal year. In addition, the company is engaging an external consulting firm to assist in the implementation of ICFR best practices necessary to position management to report on its assessment of internal controls for 2018.
Changes in Internal Control over Financial Reporting
Other than as discussed above, there have not been any changes in our internal controls over financial reporting (as such item is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during our fiscal quarter ended December 31, 2017 that have materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
Inherent Limitations on the Effectiveness of Controls
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the controls are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected. Accordingly, our controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our control system are met.
| ITEM 9B. | OTHER INFORMATION |
None.
68
Table of Contents
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference to our definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
ITEM 11. EXECUTIVE AND DIRECTOR COMPENSATION
The information required by this item is incorporated by reference to our definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference to our definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference to our definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item is incorporated by reference to our definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
69
Table of Contents
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
Item 15(a)
(1) Financial Statements
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this report.
(2) Financial Statement Schedules
Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in the financial statements or notes thereto beginning on page F-1 of this report.
(3) Exhibits:
The exhibits listed in the Exhibit Index at the end of this report are filed or incorporated by reference as part of this report.
| Item 15(b) | Exhibits |
See (a)(3) above.
| Item 15(c) | Financial Statement Schedules |
See (a)(2) above.
| ITEM 16. | FORM 10-K SUMMARY |
Not applicable.
70
Table of Contents
EXHIBIT INDEX
71
Table of Contents
72
Table of Contents
73
Table of Contents
| + | Confidential treatment was granted for portions of such exhibit. |
| * | Confidential treatment requested under 17 C.F.R. §§200.80(b)(4) and 24b-2. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the Securities and Exchange Commission pursuant to the confidential treatment request. |
| ** | Indicates a management contract or compensatory plan or arrangement required to be filed pursuant to Item 15(b) of Form 10-K |
74
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, as amended the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| ELOXX PHARMACEUTICALS INC. (Registrant) | ||||||
| Date: March 16, 2018 |
/s/ Robert E. Ward | |||||
| Robert E. Ward | ||||||
| Chairman of the Board of Directors and Chief Executive Officer (On behalf of the Registrant and as Principal Executive Officer) | ||||||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Robert E. Ward and Gregory Weaver, and each of them, as his or her true and lawful attorneys-in-fact and agents, each with the full power of substitution, for him or her and in his or her name, place or stead, in any and all capacities, to sign any and all amendments to this report, with exhibits thereto and other documents in connection therewith, with the U.S. Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or their, his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Robert E. Ward Robert E. Ward |
Chairman of the Board of Directors and Chief Executive Officer (Principal Executive Officer) | March 16, 2018 | ||
| /s/ Gregory Weaver Gregory Weaver |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | March 16, 2018 | ||
| /s/ Tomer Kariv Tomer Kariv |
Director | March 16, 2018 | ||
| /s/ Ran Nussbaum Ran Nussbaum |
Director | March 16, 2018 | ||
| /s/ Silvia Noiman Silvia Noiman, PhD |
Director | March 16, 2018 | ||
| /s/ Gadi Veinrib Gadi Veinrib |
Director | March 16, 2018 | ||
| /s/ Zafrira Avnur Zafrira Avnur, PhD |
Director | March 16, 2018 | ||
| /s/ Martijn Kleijwegt Martijn Kleijwegt |
Director | March 16, 2018 | ||
75
Table of Contents
| Signature |
Title |
Date | ||
| /s/ Steven D. Rubin Steven D. Rubin |
Director | March 16, 2018 | ||
| /s/ Jasbir Seehra Jasbir Seehra, Ph.D. |
Director | March 16, 2018 | ||
76
Table of Contents
ELOXX PHARMACEUTICAL INC.
CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2017
| Page | ||||
| 77 | ||||
| 79 | ||||
| 80 | ||||
| 81 | ||||
| 82 | ||||
| 83 | ||||

|
Kost Forer Gabbay & Kasierer 144 Menachem Begin Road, Building A Tel-Aviv 6492102, Israel |
Tel: +972-3-6232525 Fax: +972-3-5622555 ey.com |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of Eloxx Pharmaceuticals Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Eloxx Pharmaceuticals, Inc. (the “Company”) as of December 31, 2017 and 2016, the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017 and the related notes (collectively referred to as the “ consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016 and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
77
Table of Contents
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KOST FORER GABBAY & KASIERER
A Member of Ernst & Young Global
We have served as the Company‘s auditor since 2015.
Tel-Aviv, Israel
March 16, 2018
78
Table of Contents
ELOXX PHARMACEUTICALS, INC.
Amounts in thousands, except share and per share data
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 24,049 | $ | 2,212 | ||||
| Restricted bank deposit |
102 | 38 | ||||||
| Prepaids and other current assets |
355 | 837 | ||||||
|
|
|
|
|
|||||
| Total current assets |
24,506 | 3,087 | ||||||
| Property and equipment, net |
278 | 41 | ||||||
| Total assets |
24,784 | 3,128 | ||||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities |
||||||||
| Accounts payables |
1,530 | 1,899 | ||||||
| Accrued expenses |
1,893 | 619 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
3,423 | 2,518 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 9) |
||||||||
| Stockholders’ equity: |
||||||||
| Series A, B-1, B-2 and C Preferred Stock, $0.01 par value 5,000,000 and 19,965,708 shares authorized as of December 31, 2017 and 2016, respectively; 0 and 7,638,263 shares issued and outstanding as of December 31, 2017 and 2016, respectively |
— | 76 | ||||||
| Common stock, $0.01 par value 500,000,000 and 29,948,562 shares authorized as of December 31, 2017 and 2016, respectively; 27,527,738 and 4,205,278 shares issued and outstanding as of December 31, 2017 and 2016, respectively |
274 | 42 | ||||||
| Additional paid-in capital |
60,047 | 18,238 | ||||||
| Accumulated deficit |
(38,960 | ) | (17,746 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
21,361 | 610 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 24,784 | $ | 3,128 | ||||
|
|
|
|
|
|||||
See accompanying notes consolidated financial statements
79
Table of Contents
ELOXX PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
Amounts in thousands, except per share and per share data
| Year ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Operating expenses: |
||||||||||||
| Research and development, net |
$ | 16,398 | $ | 8,986 | $ | 5,842 | ||||||
| General and administrative expenses |
3,992 | 854 | 442 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
20,390 | 9,840 | 6,284 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(20,390 | ) | (9,840 | ) | (6,284 | ) | ||||||
| Financial and other expenses, net |
824 | 7 | 122 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | 21,214 | $ | 9,847 | $ | 6,406 | ||||||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | 4.75 | $ | 2.60 | $ | 1.67 | ||||||
|
|
|
|
|
|
|
|||||||
| Weighted average number of Common Stock used in computing basic and diluted loss per share |
4,976,377 | 4,205,277 | 4,148,389 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes consolidated financial statements
80
Table of Contents
ELOXX PHARMACEUTICALS, INC.
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
Amounts in thousands, except share and per share data
| Additional paid-in capital |
Total Stockholders’ Equity |
|||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Accumulated deficit |
||||||||||||||||||||||||||
| Number | Amount | Number | Amount | |||||||||||||||||||||||||
| Balance as of January 1, 2015 |
2,022,493 | $ | 20 | 4,105,449 | $ | 41 | $ | 4,207 | $ | (1,493 | ) | $ | 2,775 | |||||||||||||||
| Issuance of Series A preferred stock |
1,073,157 | 11 | — | — | 849 | — | 860 | |||||||||||||||||||||
| Issuance of Series B-1 preferred stock and warrants to purchase Series B-1 preferred stock, net of $93 issuance costs |
1,002,049 | 10 | — | — | 3,012 | — | 3,022 | |||||||||||||||||||||
| Exercise of stock options |
— | — | 99,829 | 1 | (1 | ) | — | — | ||||||||||||||||||||
| Receipt on account of shares |
— | — | — | — | 300 | — | 300 | |||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 92 | — | 92 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (6,406 | ) | (6,406 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance as of December 31, 2015 |
4,097,699 | 41 | 4,205,278 | 42 | 8,459 | (7,899 | ) | 643 | ||||||||||||||||||||
| Issuance of Series B-1 preferred stock and warrants to purchase common stock, net of $264 issuance costs |
1,929,676 | 19 | — | — | 5,717 | — | 5,736 | |||||||||||||||||||||
| Exercise of warrants into Series A preferred stock |
436,750 | 4 | — | — | 346 | — | 350 | |||||||||||||||||||||
| Issuance of Series B-1 preferred stock and warrants to purchase Series B-1 preferred stock |
1,174,138 | 12 | — | — | 3,638 | — | 3,650 | |||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 78 | — | 78 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (9,847 | ) | (9,847 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance as of December 31, 2016 |
7,638,263 | 76 | 4,205,278 | 42 | 18,238 | (17,746 | ) | 610 | ||||||||||||||||||||
| Issuance of Series C preferred stock, net of $573 issuance costs |
6,311,076 | 63 | — | — | 18,364 | — | 18,427 | |||||||||||||||||||||
| Conversion of convertible loan into Series C preferred stock of $0.01 par value |
825,213 | 8 | — | — | 3,160 | 3,168 | ||||||||||||||||||||||
| Exercise of stock options |
— | — | 16,699 | — | 17 | — | 17 | |||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 101 | — | 101 | |||||||||||||||||||||
| Issuance of common stock, net of $494 issuance costs |
— | — | 6,333,333 | 63 | 16,943 | — | 17,006 | |||||||||||||||||||||
| Conversion of Series A, B-1, B-2 and C preferred stock into common stock with respect to the Reverse Merger |
(14,774,552 | ) | (147 | ) | 14,774,552 | 147 | — | — | — | |||||||||||||||||||
| Shares issued with respect to the Reverse Merger |
— | — | 2,197,876 | 22 | (192 | ) | — | (170 | ) | |||||||||||||||||||
| Provision related to the Technion exit fee |
— | — | — | — | 3,416 | — | 3,416 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (21,214 | ) | (21,214 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance as of December 31, 2017 |
— | $ | — | 27,527,738 | $ | 274 | $ | 60,047 | $ | (38,960 | ) | $ | 21,361 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes consolidated financial statements.
81
Table of Contents
ELOXX PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
Amounts in thousands
| Year ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (21,214 | ) | $ | (9,847 | ) | $ | (6,406 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation and restricted shares |
101 | 78 | 92 | |||||||||
| Depreciation |
39 | 8 | 5 | |||||||||
| Amortization and revaluation of discount in respect to convertible loan |
668 | — | — | |||||||||
| Provision related to the Technion exit fee |
3,416 | — | — | |||||||||
| Change in operating assets and liabilities: |
||||||||||||
| Prepaid expenses and other current assets |
709 | (482 | ) | (276 | ) | |||||||
| Accounts payable |
(583 | ) | 1,250 | 529 | ||||||||
| Accrued expenses |
929 | 149 | 321 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(15,935 | ) | (8,844 | ) | (5,735 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Investment in restricted bank deposit |
(64 | ) | (14 | ) | — | |||||||
| Maturity of (investment in) restricted bank deposit |
— | — | 1,496 | |||||||||
| Purchase of property and equipment |
(237 | ) | (36 | ) | (10 | ) | ||||||
| Cash received upon the Reverse Merger |
123 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by investing activities |
(178 | ) | (50 | ) | 1,486 | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from finance activities: |
||||||||||||
| Proceeds from exercise of warrants into Series A preferred stock |
— | 350 | — | |||||||||
| Proceeds from exercise of options into common stock |
17 | — | — | |||||||||
| Proceeds from issuance of Series B-1 preferred stock and warrants to purchase Series B-1 preferred stock, net of issuance costs |
— | 9,386 | 3,022 | |||||||||
| Proceeds from issuance of Series A preferred stock |
— | — | 860 | |||||||||
| Proceeds from issuance of Series C preferred stock |
18,427 | — | — | |||||||||
| Proceeds from issuance of common stock |
17,006 | — | — | |||||||||
| Proceeds from convertible loan and related financial derivative into Series C preferred stock |
2,500 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
37,950 | 9,736 | 3,882 | |||||||||
|
|
|
|
|
|
|
|||||||
| Increase (decrease) in cash and cash equivalents |
21,837 | 842 | (367 | ) | ||||||||
| Cash and cash equivalents at the beginning of the year |
2,212 | 1,370 | 1,737 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at the end of the year |
$ | 24,049 | $ | 2,212 | $ | 1,370 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental disclosure of non-cash financing activities: |
||||||||||||
| Conversion of convertible loan into Series C preferred stock |
$ | 3,168 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes consolidated financial statements
82
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of the Business
We are a global biopharmaceutical company focused on discovering and developing novel therapeutics for the treatment of rare and ultra-rare premature stop codon diseases. We are harnessing the science of genetic read-through to develop novel drug product candidates that interact with the ribosome to overcome these premature stop codons. Our revolutionary small molecule approach is designed to unleash the potential to restore production of full length functional proteins with the goal of enabling a return toward normal cellular function. We believe there is a broad application of this approach to the over 1800 rare and ultra-rare diseases where nonsense mutation has been implicated in the cause or pathway of human disease.
Our research and development strategy is to target rare or ultra-rare diseases where a high unmet medical need, nonsense mutation bearing, patient population has been identified. We focus on clinical indications where there is a high unmet medical need, established preclinical read-through or personalized medicine experiments that are predictive of clinical activity, and a definable path for Orphan Drug development, regulatory approval, patient access and commercialization. We believe patient advocacy to be an important element of patient focused drug development and seek opportunities to collaborate with patient advocacy groups throughout the discovery and development process. Our current clinical focus is on cystic fibrosis and cystinosis where we are advancing our lead drug product candidate ELX-02. Eloxx is headquartered in Waltham, MA with research and development operations in Rehovot, Israel.
Eloxx Pharmaceuticals Ltd. (“Eloxx Limited”) was incorporated in Israel on September 17, 2013. The Company focuses its activity on the discovery, development and commercialization of compounds for the treatment of genetic diseases caused by nonsense mutations. In 2013, the Company entered into a license agreement (the “Technion Agreement”) with the Technion Research and Development Foundation Ltd. (“TRDF”).
Reverse Merger
On December 19, 2017, Sevion Therapeutics, Inc. (“Sevion”) acquired Eloxx Pharmaceuticals, Limited (“Private Eloxx” or “Eloxx Limited”) pursuant to a merger between the companies (the “Transaction” or “Reverse Merger”). Upon consummation of the Transaction (the “Closing”), Sevion adopted the business plan of Private Eloxx and discontinued the pursuit of Sevion’s business plan pre-Closing. In connection with the Transaction, Sevion agreed to acquire all of the outstanding capital stock of Private Eloxx in exchange for the issuance of an aggregate 20,316,656 shares of the Sevion’s common stock, par value $0.01 per share (the “Common Stock”), after giving effect to a 1-for-20 reverse split effected immediately prior to the Transaction. As a result of the Transaction, Private Eloxx became a wholly-owned subsidiary of Sevion. While Sevion was the legal acquirer in the transaction, Private Eloxx was deemed the accounting acquirer. Immediately after giving effect to the Transaction, on December 19, 2017, Sevion changed its name to Eloxx Pharmaceuticals, Inc. (“Eloxx” or the “Company”).
The annual consolidated financial statements of the Company reflect the operations of Private Eloxx as the acquirer for accounting purposes, together with a deemed issuance of shares, equivalent to the shares held by the stockholders of the legal acquirer, Sevion, prior to the Transaction, and a recapitalization of the equity of the accounting acquirer. The annual consolidated financial statements include the accounts of the Company since the effective date of the reverse merger and the accounts of Private Eloxx since inception.
Upon closing of the Reverse Merger, the Company assumed the obligations under outstanding warrants previously issued by Eloxx Limited to purchase its share capital and, in connection therewith, issued warrants to purchase 346,307 shares of the Company’s common stock to certain warrant holders of Eloxx Limited.
83
Table of Contents
In addition, upon closing of the Reverse Merger, Eloxx Limited assumed all of the outstanding obligations under the Eloxx 2013 Share Ownership and Option Plan (the “2013 Plan”) and, accordingly, the Company has reserved 2,307,738 shares of the Company’s common stock for issuance upon the exercise of such options. As part of the Company’s assumption of the outstanding options under the 2013 Plan, the Company also assumed the 2013 Plan and accordingly reserved 189,751 shares of the Company’s common stock for future grants.
Immediately prior to the closing of the Reverse Merger the Company raised gross proceeds of $13.5 million at a price per share of $0.15 from accredited investors as a private placement. The amount was raised pursuant a share purchase agreement dated May 31, 2017, as amended between Eloxx Limited and a group of accredited investors, (“Eloxx SPA”). Under the Eloxx SPA and the first joinder thereunder executed on June 29, 2017, Eloxx Limited received gross proceeds of $15.0 million from the group of accredited investors. In accordance with the terms of the Eloxx SPA, each of the investors executed a separate subscription agreement with the Company for the total investment of an additional $15.0 million in exchange for the Company’s shares of common stock at a price per share of $0.15 immediately prior to the consummation of the Reverse Merger. With the consent of the parties, an amount of $1.5 million was invested by an accredited investor under the subscription agreement into Sevion.
On August 2, 2017, Eloxx Limited raised under a second joinder to the Eloxx SPA, an additional aggregate amount of $8.0 million, half of the amount was invested in Eloxx Limited on August 2, 2017 and the remainder was invested in Eloxx Limited immediately prior to the consummation of the Reverse Merger but was deemed an investment in the Company’s share capital for the purpose of the exchange ratio under the Agreement.
This private placement was made solely to “accredited investors,” as that term is defined in Regulation D under the Securities Act of 1933, as amended (the “Securities Act”), and was conducted in reliance on the exemption from registration afforded by Section 4(2), Rule 506 of Regulation D and Regulation S under the Securities Act, as amended, and corresponding provisions of state securities laws.
Following the Reverse Merger and reverse stock split, and commencing December 20, 2017, the Company’s Common Stock symbol on OTCQB marketplace changed to “SVOND”, and subsequently changed to “ELOX” on January 19, 2018.
Effective with the Reverse Merger each member of the Board of Directors of Eloxx Limited prior to the Reverse Merger was appointed to the Company’s Board of Directors. In addition, each officer of Eloxx Limited was reappointed as an officer of the Company. Also effective with the Reverse Merger, the Company’s Board affirmed its financial year end as December 31, 2017 to align with the fiscal year end of Eloxx Limited.
Taurus Sublicense Agreement
On December 18, 2017, the Company executed a binding term sheet with Taurus Biosciences Inc. (“Taurus”) pursuant to which the Company grants Taurus a worldwide exclusive, sublicensable, license to the antibody SVN-001 for the development and commercialization of a product in the Field of immunology and Taurus will pay Eloxx 2% royalties on net sales. Taurus will file and prosecute any and all patents and patent applications, and shall pay all related patent expenses, with respect to this license. The Company assumed a license agreement with The Scripps Research Institute through the Reverse Merger pursuant to which the Company is required to pay a 2% royalty of net sales.
84
Table of Contents
Liquidity
As reflected in the accompanying audited consolidated financial statements, the Company has not generated revenue from the sale of any product and does not expect to generate significant revenue unless and until obtaining marketing approval and commercialization. As of December 31, 2017, the Company had cash and cash equivalents of $24.0 million. The Company expects its cash and cash equivalents will fund operations at least through the end of the first quarter of 2019 based on its current operating plans. The Company incurred a loss for the year ended December 31, 2017 of $21.2 million and had a negative cash flow from operating activities of $15.9 million during the year ended December 31, 2017. The accumulated deficit as of December 31, 2017 was $39.0 million.
If we are unable to obtain funding, we could be forced to delay, reduce or eliminate our research and development programs, product portfolio expansion or commercialization efforts, which could adversely affect our business prospects, or we may be unable to continue operations.
The Company reported cash of $13.5 million in its September 30, 2017 balance sheet, and its use of cash in operations in Q4 2017 was $6.3 million. Q4 2017 research and development expense totaled $8.4 million which included $3.4 million in non-cash expense related to the Technion Agreement. Q4 2017 general & administrative expense totaled $2.2 million, transaction related costs were $0.7 million, and net loss was $10.6 million. The Company received net proceeds of $16.8 million in Q4 2017 related to completing its Series C financing.
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”).
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its subsidiaries. Intercompany balances and transactions have been eliminated upon consolidation.
Use of Estimates
The preparation of the financial statements in conformity with U.S. GAAP requires management to make estimates, judgments and assumptions. The Company’s management believes that the estimates, judgments and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the dates of the financial statements, and the reported amounts of expenses during the reporting period. The Company evaluates estimates on an ongoing basis. Actual results could differ from those estimates.
Foreign Currency Translation
The functional currency of the Company is the U.S. dollar.
Accordingly, monetary accounts maintained in currencies other than the dollar are re-measured into U.S. dollars in accordance with Accounting Standards Codification (“ASC”) Topic 830, “Foreign Currency Matters”. All transaction gains and losses of the re-measurement of monetary balance sheet items are reflected in the statements of operation as other income or expenses, as appropriate.
Cash and Cash Equivalents
Cash equivalents are short-term highly liquid investments that are readily convertible to cash with original maturities of three months or less at the date of purchase to be cash equivalents.
Restricted bank deposits
At December 31, 2017, and 2016, restricted bank deposits consisted of guarantees related to the Company’s credit card and corporate facilities leases.
85
Table of Contents
Concentrations of credit risk
Financial instruments that subject us to significant concentrations of credit risk consist primarily of cash. Substantially all of the Company’s cash is held at financial institutions that management believes to be of high-credit quality. Deposits with these financial institutions may exceed the amount of insurance provided on such deposits; however, these deposits may be redeemed upon demand and, therefore, bear minimal risk.
The Company has no off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other foreign hedging arrangements.
Property and Equipment
Property and equipment are recorded at cost, less accumulated depreciation. Costs associated with maintenance and repairs are expensed as incurred. Depreciation expense is recognized using the straight-line method over the estimated useful lives:
| Useful Life (Years) | ||
| Computers and software |
3 years | |
| Office furniture and equipment |
5 – 12 years | |
| Laboratory equipment |
5 years | |
| Leasehold improvement |
Over the shorter of the expected lease term or estimated useful life |
Impairment of long-lived assets
Property and equipment subject to amortization are reviewed for impairment in accordance with ASC Topic 360, “Accounting for the Impairment or Disposal of Long-Lived Assets,” whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted cash flows expected to be generated by the assets. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the fair value of the assets. The Company has not recorded any impairment losses to date.
Legal and Other Contingencies
The Company accounts for its contingent liabilities in accordance with ASC Topic 450 “Contingencies”. A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter. For the years ended December 31, 2017 and 2016, the Company was not a party to any litigation that could have a material adverse effect on the Company’s business, financial position, results of operations or cash flows (see also Note 9). Legal costs incurred in connection with loss contingencies are expensed as incurred.
Severance Pay
Eloxx Limited’s liability for severance pay is pursuant to Section 14 of the Severance Compensation Act, 1963 (“Section 14”) under Israeli law, pursuant to which all Eloxx Limited’s employees are included under Section 14, and are entitled only to monthly deposits, at a rate of 8.33% of their monthly salary, made in the employee’s name with insurance companies. Under Israeli employment law, payments in accordance with Section 14 release Eloxx Limited from any future severance payments in respect of those employees. The fund is made available to the employee at the time the employer-employee relationship is terminated, regardless of cause of termination. The severance pay liabilities and deposits under Section 14 are not reflected in the consolidated balance sheets as the severance pay risks have been irrevocably transferred to the severance funds. Severance expenses for the years ended December 31, 2017, 2016 and 2015 amounted to $64,000, $44,000 and $35,000, respectively.
86
Table of Contents
Research and Development Costs
Research and development costs are expensed as incurred except royalty-bearing participation from the Israeli Innovation Authority (previously known as Office of the Chief Scientist) of the Ministry of Economy (“IIA”), as described in “Government Grants”. Research and development expenses are comprised of costs incurred in performing research and development activities, including salaries, stock-based compensation and benefits, facilities costs, depreciation, third-party license fees, and external costs of outside vendors engaged to conduct preclinical development activities and clinical trials. Research and development expenses include the Company’s costs of performing services in connection with its collaboration agreements and research grants.
Nonrefundable prepayments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered, or the services rendered.
Government Grants
The Company receives royalty-bearing grants, which represents participation of IIA in approved programs for research and development. These amounts are recognized on the accrual basis as a reduction of research and development expenses as such expenses are incurred.
Fair value of financial instruments:
ASC Topic 820, “Fair Value Measurements and Disclosures” (“ASC 820”), defines fair value as the price that would be received to sell an asset or paid to transfer a liability (i.e., the “exit price”) in an orderly transaction between market participants at the measurement date.
In determining fair value, the Company uses various valuation approaches. ASC 820 establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy is broken down into three levels based on the inputs as follows:
| Level 1 — |
Valuations based on quoted prices in active markets for identical assets that the Company has the ability to access. | |
| Level 2 — |
Valuations based on one or more quoted prices in markets that are not active or for which all significant inputs are observable, either directly or indirectly. | |
| Level 3 — |
Valuations based on inputs that are unobservable and significant to the overall fair value measurement. |
The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
The carrying amounts of cash and cash equivalents, restricted bank deposits, prepaids and other assets, trade payables and accrued expenses approximate their fair value due to the short-term maturities of such instruments.
Stock-Based Compensation
The Company accounts for stock-based compensation in accordance with ASC Topic 718, “Compensation-Stock Compensation”, (“ASC 718”), which requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The Company recognizes compensation expenses for the value of its awards granted based on the straight-line method over the requisite service period of each of the awards or over the implicit service period when a performance condition affects the vesting, and it is considered probable that the performance condition will be achieved.
87
Table of Contents
The Company estimates the fair value of stock options granted using the Binomial Option-Pricing Model (“Binomial Model”) which requires a number of assumptions, of which the most significant are the fair market value of the underlying Ordinary Shares, expected stock price volatility, suboptimal exercise factor and the expected option term. Expected volatility was calculated based upon historical volatilities of similar entities in the related sector index. The expected option term represents the period that the Company’s stock options are expected to be outstanding and is determined based on the simplified method until sufficient historical exercise data will support using expected life assumptions. The suboptimal exercise factor is estimated using historical option exercise information. The suboptimal exercise factor is the ratio by which the stock price must increase over the exercise price before employees are expected to exercise their stock options. The risk-free interest rate is based on the yield from U.S. treasury bonds with an equivalent term. The Company has historically not paid dividends and has no foreseeable plans to pay dividends.
The fair value of ordinary shares underlying the options was determined by the Company’s Board of Directors with the assistance of an independent valuation firm. Because there has been no public market for the ordinary shares, the Board of Directors has determined fair value of the ordinary shares at the time of grant by considering a number of objective and subjective factors including data from other comparable companies, sales of series preferred shares to unrelated third parties, operating and financial performance, the lack of liquidity of capital stock and general and industry specific economic outlook, amongst other factors. The fair value of the underlying ordinary shares shall be determined by management until such time as the ordinary shares are listed on an established stock exchange, national market system or other quotation system. The Company determined that the Option Pricing Method (“OPM”) was the most appropriate method for allocating its enterprise value to determine the estimated fair value of its ordinary shares for valuations performed since its inception date and until June 30, 2017. Commencing June 30, 2017 and until the Closing of the Reverse Merger, the Company began using the Hybrid Method by combining the OPM and M&A scenario to determine the fair value of its ordinary shares.
Following the closing of the Reverse Merger, the fair value of the Company’s common stock is determined based on the closing price on the OTCQB.
Income taxes
The Company account for income taxes in accordance with ASC Topic 740, “Income Taxes” (“ASC 740”) which prescribes the use of the liability method whereby deferred tax assets and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, if necessary, to reduce deferred tax assets to their estimated realizable value if it is more likely than not that a portion or all of the deferred tax assets will be realized.
Based on ASC 740, a two-step approach is used to recognize and measure uncertain tax positions. The first step is to evaluate the tax position taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% likely to be realized upon ultimate settlement. As of December 31, 2017, and 2016, no liability for unrecognized tax positions has been recorded. Accordingly, no interest or penalties related to uncertain tax positions are recorded, either. It is the Company’s policy that any interest or penalties associated with unrecognized tax positions would be reflected in income tax expense.
Net Loss per Share
The Company applies the two-class method as required by ASC Topic 260-10, “Earnings Per Share” (“ASC 260-10”), which requires the income or loss per share for each class of shares (ordinary and preferred shares) to be calculated assuming 100% of the Company’s earnings are distributed as dividends to each class of shares based on their contractual rights.
88
Table of Contents
No dividends were declared or paid during the reported periods. According to the provisions of ASC 260-10, the Company’s preferred shares are not participating securities in losses and, therefore, are not included in the computation of net loss per share.
Basic loss per share is computed by dividing the loss for the period applicable to ordinary shareholders by the weighted average number of ordinary shares outstanding during the period. In computing diluted income per share, basic earnings per share are adjusted to reflect the potential dilution that could occur upon the exercise of share options granted to grantees and upon conversion of shares and warrants issued to investors and service providers using the “treasury stock method”.
For the years ended December 31, 2017, 2016 and 2015, all outstanding preferred stock, stock options, stock warrants and restricted stock have been excluded from the calculation of the diluted net loss per share as all such securities are anti-dilutive for all years presented (see Note 13).
Recent Accounting Pronouncements adopted
On March 30, 2016, the FASB issued ASU 2016-09, “Compensation—Stock Compensation”, which effects all entities that issue share-based payment awards to their employees. The amendments in this update cover such areas as the recognition of excess tax benefits and deficiencies, the classification of those excess tax benefits on the statement of cash flows, an accounting policy election for forfeitures, the amount an employer can withhold to cover income taxes and still qualify for equity classification and the classification of those taxes paid on the statement of cash flows. This update is effective for annual and interim periods beginning after December 15, 2016. This guidance can be applied either prospectively, retrospectively or using a modified retrospective transition method. The Company adopted the new guidance prospectively. This new guidance does not have a material impact on the Company’s consolidated financial statements.
Recent Accounting Pronouncements not adopted yet
In February 2016, the FASB issued ASU No. 2016-02, Leases. This guidance will require that lease arrangements longer than 12 months result in an entity recognizing an asset and liability equal to the present value of the lease payments in the statement of financial position. This guidance is effective for annual periods beginning after December 15, 2018, and interim periods therein. This standard requires a modified retrospective transition approach for all leases existing at, or entered into after, the date of initial application, with an option to use certain transition relief. Early adoption is permitted. The Company is currently evaluating the impact that the adoption of ASU 2016-02 will have on its financial statements and related disclosures.
In November 2016, the FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230): “Restricted Cash” (ASU 2016-18), which requires companies to include amounts generally described as restricted cash and restricted cash equivalents in cash and cash equivalents when reconciling beginning-of-period and end-of-period total amounts shown on the statement of cash flows. The amendments in this update are effective for public business entities for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. Early adoption is permitted, including adoption in an interim period. This new guidance does not have a material impact on the Company’s consolidated financial statements.
In January 2017, the FASB issued ASU 2017-01 Business Combinations (Topic 805): “Clarifying the Definition of a Business”. ASU 2017-01 provides amendments to clarify the definition of a business and affect all companies and other reporting organizations that must determine whether they have acquired or sold a business. The amendments are intended to help companies and other organizations evaluate whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. The guidance is effective for public business entities for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years and should be applied prospectively as of the beginning of the period of adoption. Early adoption is permitted under certain circumstances. The Company adopted ASU 2017-01 on January 1, 2018 and it did not have an impact on its accounting and disclosures.
89
Table of Contents
In July 2017, the FASB issued ASU 2017-11, which allows companies to exclude a down round feature when determining whether a financial instrument is considered indexed to the entity’s own stock. As a result, financial instruments with down round features may no longer be required to be classified as liabilities. A company will recognize the value of a down round feature only when it is triggered, and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, such as warrants, an entity will treat the value of the effect of the down round, when triggered, as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. The guidance in ASU 2017-11 is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted, and the guidance is to be applied using a full or modified retrospective approach. The Company is evaluating the impact of the revised guidance.
3. Reverse Merger
As described in Note 1: “Nature of the Business”, the Reverse Merger was accounted for as a reverse recapitalization which is outside the scope of ASC Topic 805, “Business Combinations” (“ASC 805”). Under reverse capitalization accounting, Eloxx Limited is considered the acquirer for accounting and financial reporting purposes and acquired the assets and assumed the liabilities of the Company. The assets acquired and liabilities assumed are reported at their historical amounts. The annual consolidated financial statements of the Company reflect the operations of the acquirer for accounting purposes together with a deemed issuance of shares, equivalent to the shares held by the former stockholders of the legal acquirer and a recapitalization of the equity of the accounting acquirer. The annual consolidated financial statements include the accounts of the Company since the effective date of the reverse capitalization and the accounts of Eloxx Limited since inception.
The following summarizes the estimated fair value of the assets and liabilities assumed at the date of the Reverse Merger (in thousands):
| December 19, 2017 | ||||
| Cash and cash equivalents |
$ | 123 | ||
| Prepaid expenses and other current assets |
220 | |||
| Property, plant and equipment, net |
39 | |||
| Restricted bank deposits |
6 | |||
|
|
|
|||
| Total assets acquired |
388 | |||
|
|
|
|||
| Accounts payable |
(215 | ) | ||
| Accrued expenses |
(343 | ) | ||
|
|
|
|||
| Total liabilities acquired |
(558 | ) | ||
|
|
|
|||
| Total net liabilities acquired |
$ | (170 | ) | |
|
|
|
|||
Additionally, the Company incurred approximately $1.3 million in professional fees related to the Reverse Merger.
4. Prepaids and other current assets
Prepaids and other current assets consisted of the following (in thousands):
| December 31 | ||||||||
| 2017 | 2016 | |||||||
| Government grants from IIA |
— | 605 | ||||||
| Other governmental agencies |
88 | 221 | ||||||
| Prepaid expenses |
267 | 11 | ||||||
|
|
|
|
|
|||||
| $ | 355 | $ | 837 | |||||
|
|
|
|
|
|||||
90
Table of Contents
5. Property and Equipment, net
Property and equipment, net consisted of the following (in thousands):
| December 31 | ||||||||
| 2017 | 2016 | |||||||
| Cost: |
||||||||
| Computers and software |
$ | 124 | $ | 54 | ||||
| Office furniture and equipment |
118 | 2 | ||||||
| Laboratory equipment |
37 | — | ||||||
| Leasehold improvement |
53 | — | ||||||
|
|
|
|
|
|||||
| 332 | 56 | |||||||
| Accumulated depreciation |
54 | 15 | ||||||
|
|
|
|
|
|||||
| 54 | 15 | |||||||
| Property and equipment, net |
$ | 278 | $ | 41 | ||||
|
|
|
|
|
|||||
Depreciation expense was $39,000, $8,000 and $5,000 for the years ended December 31, 2017, 2016 and 2015, respectively.
6. Accrued Expenses
Accrued expenses consisted of the following (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Accrued payroll and related expenses |
$ | 402 | $ | 393 | ||||
| Accrued research and development expenses |
704 | 119 | ||||||
| Accrued professional services |
787 | 107 | ||||||
|
|
|
|
|
|||||
| $ | 1,893 | $ | 619 | |||||
|
|
|
|
|
|||||
7. Convertible Loan
On January 26, 2017 (the “Closing Date”), the Company entered into a Convertible Loan Agreement (the “Agreement”) with five of its shareholders (the “Lenders”), pursuant to which the Company raised an aggregate amount of $2.5 million (the “Convertible Loan”). The Convertible Loan shall bear a 5% annual interest rate. According to the Agreement, the outstanding portion of the Convertible Loan (without accrued interest) should be automatically converted upon the consummation of equity investment by a third party of an aggregate amount of at least $5.0 million (the “Qualified Equity Investment”), prior to the lapse of two years from the Closing Date (the “Maturity Date”), into equity securities of the same class issued by the Company in such Qualified Equity Investment, in a conversion price which was equal to 80% of the price per share paid by the third party in such Qualified Equity Investment (the “Automatic Conversion”). In addition, upon the earlier of (i) Maturity Date or (ii) Event of Default as defined in the Agreement, the outstanding portion of the Convertible Loan (including accrued interest) should be converted into an equity investment of the then existing most senior class, at a conversion price per share which was equal to 65% of the original issue price per share applicable to such class. At the end of May 2017, the Lenders signed a waiver agreement pursuant to which they waived the potential discount as described above.
In accordance with ASC Topic 815 “Derivatives and Hedging”, features related to convertible loans qualify as embedded derivative instruments at the date of issuance, since these are considered as stock settled debt. In determining fair value, the Company uses various valuation approaches. ASC 820 establishes a hierarchy for
91
Table of Contents
inputs used in measuring fair value. The embedded conversion feature is classified under level 3 in the hierarchy. The fair value assigned to the embedded conversion feature on the issuance dates amounted to $0.3 million. The embedded instruments are marked to market in each reporting period and changes are recorded in financial expenses. The discount is amortized using the effective interest over the loan period.
On May 31, 2017, the Convertible Loan (without accrued interest) was converted into 825,213 Series C preferred stock, according to the price per share that was paid in the 2017 Share Purchase Agreement (see Note 10). During the year ended December 31, 2017, the Company recorded $0.7 million as financial and other expenses, net, as a result of changes in the embedded instruments. In connection with the conversion, the embedded instrument together with all accrued interest in the amount of $0.7 million and was classified to additional paid in capital.
The following table presents reconciliations for the Company’s liabilities measured and recorded at fair value on a recurring basis, using significant unobservable inputs (in thousands):
| Significant Unobservable Inputs (Level 3) |
||||
| Balance at January 26, 2017 |
$ | (308 | ) | |
| Amortization and revaluation embedded conversion feature |
(317 | ) | ||
| Conversion of convertible loan into Series C preferred stock |
625 | |||
|
|
|
|||
| Balance at December 31, 2017 |
$ | — | ||
|
|
|
|||
8. Related Parties
On August 29, 2013, the Company entered into an agreement (“Technion Agreement”) with TRDF, with respect to certain technology relating to aminoglycosides and the redesign of aminoglycosides for the treatment of human genetic diseases caused by premature stop mutations and further results of the research of the technology, in order to develop and commercialize products based on such technology. Under the agreement, TRDF shall provide the Company research services for an estimated annual payment of $0.1 million, to be agreed exactly by the parties prior to the beginning of each year of the research period. During the years ended December 31, 2017, 2016 and 2015, the Company recorded general and administrative expenses amounting to $7,000, $185,000 and $0, respectively, in relation to the TRDF reimbursement for the preparation, filling, prosecution and maintenance of TRDF patents rights related to Eloxx Limited. In addition, during the years ended December 31, 2017, 2016 and 2015 the Company recorded research and development expenses in relation to the TRDF amounting to $3,465,000, $33,000 and $58,000, respectively. As of December 31, 2017 and 2016, amounts recorded in accrued expenses were $25,000 and $185,000, respectively.
In addition, TRDF shall grant the Company a license to use, market, sell or sub-license the rights of the product developed under the TRDF research results (the “Licensed Product”), as fully defined in the Technion Agreement, for the following considerations: (a) milestone payments, to be transferred upon meeting certain milestones as defined in the Technion Agreement, up to total consideration of $6.1 million; (b) certain royalties on a low- to mid- single-digit percentage of net sales (subject to change in the case of (x) sublicensing to a big pharmaceutical or biotechnology company, or (y) payment of royalties to third parties, or (z) commercialization by a third party of an authorized generic to a licensed product), for a period until the later of (i) the expiration of a valid claim on the Licensed Product in each country the Licensed Product is sold to, or (ii) a certain amount of years from the date of the first commercial sale of the Licensed Product in such country, and (c) a low- to mid- double-digit percentage of any non-royalty sub-license income received by the Company from a sub-licensed entity. In addition, the Company shall pay certain fee to TRDF upon an exit event as described in the Technion Agreement.
92
Table of Contents
Moreover, upon the closing of an Exit Event which is not an Initial Public Offering (“IPO”), as defined in the Technion Agreement, TRDF shall be entitled to an amount equal to 3% of all non-refundable, non-contingent consideration, whether in cash or in kind, actually received by the Company and / or its shareholders. Upon the closing of an exit event which is an IPO, as defined in the Technion Agreement, TRDF shall be entitled to a number of Ordinary Shares of the Company representing 3% of the Company’s outstanding shares on a fully diluted basis immediately prior to the closing of such IPO.
On August 9, 2017 the Company received a legal claims letter from TRDF regarding TRDF’s alleged entitlement to an exit fee in accordance with the Technion Agreement. The parties are in discussion regarding a settlement of the legal claim, whereby the Company would issue shares to TRDF representing approximately 2.1% of the outstanding shares of the Company representing fulfillment of the “exit clause.” Therefore, the Company has recorded a $3.4 million research and development expense with an offsetting adjustment to additional paid-in capital for the year ended December 31, 2017 related to this legal claim.
9. Commitments and Contingencies
Operating Leases
The Company entered into a lease agreement for office space in Rehovot, Israel for a period of three years commencing March 8, 2017 and until April 30, 2020 with annual lease payments in the amount of $0.1 million. The Company has an option to extend the lease for a term of two years.
The Company entered into a lease agreement for laboratory space in Rehovot, Israel for a period of one year commencing March 8, 2017 and until March 8, 2018 with annual lease payments in the amount of $48,000. The Company has an option to extend the lease for a term of one year.
The Company entered into a lease agreement for office space in Waltham, MA for a period of 37 months commencing November 15, 2017 and until December 17, 2020 with annual lease payments in the amount of $0.2 million. The Company has an option to extend the lease for a term of three years.
Future minimum lease commitments as of December 31, 2017 were as follows:
| As of December 31, 2017 |
Total | |||
| 2018 |
$ | 258 | ||
| 2019 |
232 | |||
| 2020 |
186 | |||
|
|
|
|||
| $ | 676 | |||
|
|
|
|||
The Company recorded lease expenses in the amounts of $0.2 million for the year ended December 31, 2017 and $0.1 million for the each of the years ended December 31, 2016 and 2015.
Royalty Commitments to the IIA
Under the research and development agreements with the IIA and pursuant to applicable laws, the Company is required to pay royalties at the rate of 3% on sales to end customers of products developed with funds provided by the IIA, up to an amount equal to 100% of the IIA research and development grants received, linked to the dollar plus interest on the unpaid amount received based on the 12-month LIBOR rate (from the year the grant was approved) applicable to dollar deposits. If the Company does not generate sales of products developed with funds provided by the IIA, the Company is not obligated to pay royalties or repay the grants.
93
Table of Contents
The Company received research and development grants from the IIA in the amounts of $0.9 million, $1.2 million, and $0.3 million for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, the Company has not commenced the payment obligation of the royalties and has a contingent obligation with respect to royalty-bearing participation received or accrued, amounting to $2.7 million.
Commitments to Scripps
The Company assumed a license agreement with Scripps Research Institute through the Reverse Merger pursuant to which the Company is required to pay a 2% royalty of net sales.
Commitments to TRDF
Since August 29, 2013, the Company has had an ongoing agreement with TRDF. Refer to Note 8: Related Parties for further information.
10. Stockholders’ Equity
For accounting purposes, all common stock, preferred stock, warrants, options to purchase common stock and loss per share amounts have been adjusted to give retroactive effect to the exchange ratio and reverse stock split for all periods presented in these consolidated financial statements.
Preferred and Common Stock
In April 2015, Eloxx Limited achieved a clinical milestone in connection with the share purchase agreement signed in 2013, pursuant to which, Eloxx Limited issued to investors 1,073,157 shares of Series A preferred stock with a par value of $0.01 for an aggregate amount of $0.9 million.
In July 2015, Eloxx Limited entered into a Share Purchase Agreement (the “2015 SPA”) whereby Eloxx Limited issued to existing investors 1,002,049 shares of Series B-1 preferred stock with a par value of $0.01 and 1,503,068 warrants to purchase 1,503,068 shares of Series B-1 preferred stock with an exercise price of $3.11 for an aggregate gross amount of $3.1 million, representing a price per unit of $3.11 per share. In connection with the 2015 SPA, Eloxx Limited paid a contractor cash consideration of $0.1 million as a finder fee and granted 30,563 warrants to purchase 30,563 shares of Series B-1 preferred stock with an exercise price of $3.11 per share.
In July 2015, one of Eloxx Limited’s employees exercised their 99,829 options with an exercise price of $0.01 per share to purchase 99,829 shares of common stock.
In February 2016, Eloxx Limited entered into Shares Purchase Agreement (the “2016 SPA”) whereby Eloxx Limited issued to existing investors 1,929,676 shares of Series B-1 preferred stock with a par value of $0.01 and 2,894,519 warrants to purchase 2,894,519 shares of Series B-1 preferred stock with an exercise price of $3.11 per share for an aggregate gross amount of $6.0 million.
In connection with the 2016 SPA, Eloxx Limited paid a contractor cash consideration of $0.2 million as finder fee and granted 48,242 warrants to purchase 48,242 shares of Series B-1 preferred stock with an exercise price of $3.11 per share.
In August 2016, Technion Investment Opportunities Fund L.P (the “TIOF) and TRDF exercised 124,786 and 311,964 warrants, respectively, to purchase shares of Series A preferred stock at an exercise price of $0.80 per share, respectively, for total consideration of $0.4 million.
94
Table of Contents
In September 2016, Eloxx Limited achieved a milestone in connection with the Share Purchase Agreement signed in 2014 (“2014 SPA”), pursuant to which, Eloxx Limited paid a $0.1 million milestone payment and issued to investors 1,174,138 shares of Series B-1 preferred stock with a par value of $0.01 and 587,072 warrants to purchase 587,072 shares of Series B-1 preferred stock for an aggregate amount of $3.7 million.
In connection with the 2014 SPA milestone, Eloxx Limited paid a contractor cash consideration of $0.1 million as a finder fee and granted 36,593 warrants to purchase 36,593 shares of Series B-1 preferred stock with exercise price of $3.11 per share.
On May 22, 2017, Eloxx Limited entered into a Share Purchase Agreement (the “2017 SPA”) (and subsequently on joinder agreements) with certain existing and new investors, whereby, an aggregate gross amount of $21.5 million, which included the conversion of the loan (as detailed in Note 7), was received by Eloxx Limited in exchange for the issuance of 7,136,289 shares of Series C preferred stock with a par value of $0.01 with the initial closing, of which 39,293 were issued as a result of the anti-dilution effect of the Reverse Merger. The related issuance costs were $0.6 million.
In connection with the 2017 SPA, the Company granted 142,524 warrants to purchase 142,524 shares of Series C preferred stock to certain service providers of as finder fee compensation.
Upon the closing of the Reverse Merger the Company issued 6,333,333 shares of common stock related to the closing of the 2017 SPA with a par value of $0.01 for an aggregate gross amount of $17.5. Additionally, Sevion raised $1.5 million prior to the Reverse Merger. The related issuance costs for these transactions was $0.5 million.
Warrants
As of December 31, 2017, the Company had 480,049 outstanding warrants to purchase 480,049 shares of the Company’s common stock.
The table below summarizes the outstanding warrants as of December 31, 2017:
| Amount | Weighted average exercise price |
Expiration Date | ||||||||||
| 64,374 | 0.800 | 12/19/2022 | ||||||||||
| 59,415 | 3.030 | 12/19/2022 | ||||||||||
| 60,832 | 3.000 | 12/19/2022 | ||||||||||
| 22,282 | 3.029 | 12/19/2022 | ||||||||||
| 151,984 | 3.109 | 12/19/2022 | ||||||||||
| 48,252 | 4.664 | 12/19/2022 | ||||||||||
| 68,434 | 8.000 | 1/27/2018 | ||||||||||
| 4,474 | 40.000 | 5/16/2019 | ||||||||||
| 2 | 520.000 | 11/17/2020 | ||||||||||
|
|
|
|||||||||||
| 480,049 | ||||||||||||
|
|
|
|||||||||||
11. Stock-Based Compensation
The Company has two equity compensation plans; the Company has a 2008 Plan and Eloxx Limited has a 2013 plan, both are explained in detail below.
In December 2008, the Company adopted the 2008 Incentive Compensation Plan (the “2008 Plan”), which provides for the grant of stock options, stock grants and stock purchase rights to certain designated employees and certain other persons performing services for the Company, as designated by the Company’s Board of
95
Table of Contents
Directors. Pursuant to the 2008 Plan, an aggregate of 245,884 shares of common stock has been reserved for issuance. On January 1 of each calendar year beginning with the calendar year 2015, the share reserve will automatically increase by 5% of the fully-diluted equity outstanding on the immediately preceding December 31, up to an annual maximum of 75,000 shares of common stock, provided that the aggregate number of shares subject to outstanding awards will not exceed 25% of the fully-diluted equity outstanding. Consequently, the pool of options was increased by additional 126,051 shares of common stock. The terms and vesting schedules for share-based awards vary by type of grant and the employment status of the grantee. Generally, the awards vest based upon time-based conditions or achievement of specified goals and milestones. As of December 31, 2017, 116,466 Common Stock are available for future grant under the 2008 Plan.
In December 2013, Eloxx Limited’s Board of Directors adopted the 2013 Share Ownership and Option Plan in accordance with section 102 and 3(i) of the Israeli Income Tax Ordinance (the “2013 Plan”). Under the 2013 Plan, options to purchase ordinary shares of Eloxx Limited or ordinary shares of Eloxx Limited may be granted to employees, officers, directors, service providers and consultants of Eloxx Limited Each option granted can be exercised for one ordinary share of Eloxx Limited. Options granted generally become fully exercisable after a two to four-year vesting period and expire no later than ten years from the date of grant. Any option forfeited or cancelled before expiration becomes available for future grants under the 2013 Plan until the tenth anniversary of the 2013 Plan, after which no further grants under the 2013 Plan are permissible. On April 21, 2015, Eloxx Limited’s Board of Directors adopted the US Share Ownership and Option Appendix under 2013 Plan pursuant to which Eloxx Limited may grant options to purchase its ordinary shares to U.S. grantees of Eloxx Limited or any of its parent or subsidiary companies. Upon the closing of the Reverse Merger, the Company assumed the 2013 Plan and all outstanding options thereunder and thereafter the 2013 Plan and any options or shares previously granted thereunder were replaced with options to purchase or shares of our common stock. As of December 31, 2017, the of the 2013 Plan amounted to 2,473,255 shares of common stock and 119,762 shares of Common Stock were available for future grant under the 2013 Plan.
Transactions related to the grant of options to employees and directors under the 2008 Plan during the year ended December 31, 2017, were as follows:
| Amount | Weighted average exercise price |
Weighted average remaining contractual life |
Aggregate intrinsic value |
|||||||||||||
| Options outstanding at the closing of the Reverse Merger |
215,723 | $ | 28.93 | 7.80 | $ | 61,358 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Options outstanding at end of year |
215,723 | $ | 28.93 | 7.80 | $ | 338,056 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Options exercisable at end of year |
215,723 | $ | 28.93 | 7.80 | $ | 338,056 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Transactions related to the grant of options to employees and directors under the 2013 Plan during the year ended December 31, 2017, were as follows:
| Amount | Weighted average exercise price |
Weighted average remaining contractual life |
Aggregate intrinsic value |
|||||||||||||
| Options outstanding at beginning of year |
1,948,154 | $ | 0.47 | 7.93 | $ | 1,571,406 | ||||||||||
| Granted |
582,961 | 5.40 | ||||||||||||||
| Exercised |
(16,669 | ) | 0.96 | |||||||||||||
| Forfeited |
(177,720 | ) | 0.98 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Options outstanding at end of year |
2,336,726 | $ | 1.82 | 7.12 | $ | 14,835,970 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Options exercisable at end of year |
1,624,407 | $ | 0.36 | 6.04 | $ | 12,408,281 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
96
Table of Contents
The aggregate intrinsic value represents the total intrinsic value (the difference between the deemed fair value of the Company’s Common Stock on the last day of fiscal year 2017 and the exercise price, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on December 31, 2017. This amount is impacted by the changes in the fair value of the Company’s shares.
The weighted average grant date fair value of the options granted during the years ended December 31, 2017, 2016 and 2015 were $3.68, $0.14 and $0.17, respectively.
The following table presents the assumptions used to estimate the fair values of the options granted in the period presented:
| December 31, | ||||||
| 2017 |
2016 |
2015 | ||||
| Dividend yield |
0% | 0% | 0% | |||
| Volatility |
87.17%-116.69% | 64.46%-105.57% | 73.70%-88.72% | |||
| Risk free interest |
1.22%-2.5% | 0.47%-2.35% | 0.25%-1.87% | |||
| Contractual term (years) |
10 | 10 | 10 | |||
| Forfeiture rate post-vesting |
10% | 10% | 10% | |||
| Suboptimal exercise |
2.3 | 2.3 | 2.3 | |||
As of December 31, 2017, there was $2.1 million of total unrecognized compensation cost related to non-vested stock options granted under the 2013 Plan. This cost is expected to be recognized over a weighted-average period of 2.09 years.
The total equity-based compensation expense related to all of the Company’s equity-based awards were recognized as follows:
| Year ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Research and development |
$ | 39 | $ | 61 | $ | 68 | ||||||
| General and administrative(1) |
62 | 17 | 24 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation expenses |
$ | 101 | $ | 78 | $ | 92 | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | Including expenses of $19,000 in connection with restricted shares that were granted during the year ended December 31, 2017. |
The Company issued an inducement award outside of the 2008 Plan and 2013 Plan to the Company’s Chief Executive Officer in the form of an option to purchase 22,427 shares of the Company’s common stock with an exercise price per share equal to $8.00, and an award of restricted stock units for 22,427 shares of the Company’s common stock (collectively the “Performance Option Awards”). Subject to continued service through the vesting date, the Performance Option Awards will vest and become immediately exercisable upon the date that marks the first successful completion of a Phase-2B study with respect to any indication.
In addition, the Company issued an inducement award outside of the 2008 Plan and 2013 Plan to the Company’s Chief Executive Officer in the form of an option to purchase 640,785 shares of the Company’s common stock with an exercise price per share equal to $8.00, and an award of restricted stock units for 640,785 shares of the Company’s common stock (collectively the “Time-Vesting Awards”). Subject to continued service through the vesting date, 1/3 of the Time-Vesting Awards will vest and become immediately exercisable on the first anniversary of the effective date, with an additional 1/12 of the Time-Vesting Awards vesting on each quarterly anniversary of the effective date, provided that vesting of the Time-Vesting Awards shall be subject to acceleration following the achievement of certain milestones.
97
Table of Contents
12. Income Taxes
The Company is subject to income taxes in the United States and Israel. In general, the U.S. federal and state income tax returns remain open to examination by taxing authorities for tax years beginning in June 30, 2014 to present. The Israeli income tax returns remain open to examination beginning in 2013 to present. However, if and when the Company claims net operating loss (“NOL”) carryforwards from any prior years against future taxable income, those losses may be examined by the taxing authorities.
The United States enacted the Tax Cuts and Jobs Act (“Tax Act”) on December 22, 2017, most provisions of which will take effect starting in years beginning after December 31, 2017. The Tax Act makes substantial changes to U.S. taxation of corporations, including lowering the U.S. federal corporate income tax rate from 34% to 21%. The effect on deferred tax assets and liabilities of a change in law or tax rates is recognized in income in the period that includes the enactment date. The Tax Act also includes a provision designed to currently tax global intangible low-taxed income (“GILTI”). The Company will record the U.S. income tax effect of future GILTI inclusions in the period in which they arise, if ever.
After the enactment of the Tax Act, the SEC issued Staff Accounting Bulletin No. 118 (“SAB 118”) to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Tax Act. We have calculated an estimate of the impact of the Tax Act in our year-end income tax provision in accordance with our understanding of the Tax Act and guidance available as of the date of this filing. The provisional amount related to the remeasurement of our net U.S. deferred tax asset, based on the rate at which they are now expected to reverse in the future, was deferred tax expense of $10.2 million, but which was fully and equally offset by a corresponding reduction in our valuation allowance. The effect of the change in federal corporate tax rate from 34% to 21% is subject to change based on resolution of estimates used in determining the amounts of deferred tax assets and liabilities that were remeasured. The Company will reflect any adjustments to the provisional amounts in the period the accounting is completed and expects to complete this analysis within the one-year measurement period provided by SAB 118.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. A deferred tax liability or asset shall be recognized for all of the Company’s estimated future tax effects attributable to temporary differences and carryforwards, unless an exception applies. A common exception for businesses that operate in multiple jurisdictions is a temporary difference that is essentially permanent in duration related to the excess of the amount for financial reporting over the tax basis of an investment attributable to unremitted earnings in a foreign subsidiary. However, there are no or trivial unremitted earnings in foreign jurisdictions, so no provision for deferred taxes thereupon is required for the Company.
Significant components of the Company’s deferred tax assets are as follows (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: |
||||||||
| Net operation loss carryforward |
$ | 23,689 | $ | 2,811 | ||||
| Stock-based compensation |
1,125 | — | ||||||
| Reserves and allowances |
11 | 17 | ||||||
| U.S. tax credits and other |
714 | — | ||||||
| Research and development |
2,604 | 1,195 | ||||||
|
|
|
|
|
|||||
| Net deferred tax asset before valuation allowance |
28,143 | 4,023 | ||||||
| Valuation allowance |
(28,143 | ) | (4,023 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax asset |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
98
Table of Contents
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that all or some portion of the deferred tax assets will not be realized. The ultimate realization of the deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences are deductible and net operating losses are utilized. Based on consideration of these factors, the Company recorded a full valuation allowance at December 31, 2017 and 2016. As of December 31, 2017 and 2016, we have provided a valuation allowance of approximately $28.1 million and $4.0 million, respectively, on U.S. federal and state and Israeli tax jurisdiction deferred tax assets to reduce the amount of these assets to zero. The net change in our valuation allowance was an increase of $24.1 million for the year ended December 31, 2017 of which $30.2 million was related to the Reverse Merger of Sevion on December 19, 2017, that was partially offset by a $10.2 million decrease related to the reduction in the U.S. federal corporate tax rate on December 22, 2017. The remaining $4.1 million increase was primarily related to losses generated in the current period.
As of December 31, 2017, we had U.S. federal and state NOL carryforwards of $77.2 million and $27.4 million, respectively, and federal research tax credit carryforwards of $0.7 million. Our U.S. net operating loss carryforwards will begin to expire, if not utilized, beginning in 2019 through 2037, and the research tax credits will expire beginning in 2027 through 2037. These NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the newly enacted Tax Act, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited. It is uncertain if and to what extent various states will conform to the newly enacted federal tax law.
In addition, under Section 382 of the Code and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50 percent change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. We may have experienced ownership changes in the past, including the Reverse Merger of Sevion Therapeutics, Inc. on December 19, 2017 at which time our pre-change U.S. federal NOL carryforward was $77.1 million and research tax credit was $0.7 million. We may experience additional ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. Although we have not completed our analysis, it is reasonably possible that our federal NOLs available to offset future taxable income could materially decrease. This reduction will be offset by an adjustment to the existing valuation allowance for an equal and offsetting amount. Additionally, our state NOLs available to offset future state income could similarly decrease which would also be offset by an equal and offsetting adjustment to the existing valuation allowance. Given the offsetting adjustments to the existing valuation allowance, any ownership change is not expected to have an adverse material effect on our Consolidated Financial Statements. Finally, as of December 31, 2017, we had Israeli NOL carryforwards of $24.9 million, which carryforward indefinitely.
The components of income (loss) before taxes on income are as follows (in thousands):
| Year ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| U.S. |
$ | (21 | ) | $ | 44 | $ | 16 | |||||
| Israel |
(21,145 | ) | (9,883 | ) | (6,418 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Income (loss) before taxes on income |
$ | (21,166 | ) | $ | (9,839 | ) | $ | (6,402 | ) | |||
|
|
|
|
|
|
|
|||||||
Taxes on income during the years ended December 31, 2017 and 2016 are comprised from taxes incurred as a result of the implementation of the cost-plus method between the Company and its subsidiary were immaterial.
The main reconciling item between the statutory tax rate of the Company and the effective tax rate is the recognition of valuation allowances in respect to deferred taxes relating to accumulated net operating losses carried forward and temporary differences due to the uncertainty of the realization of such deferred taxes.
99
Table of Contents
13. Net Loss Per Share
The loss and the weighted average number of shares used in computing basic and diluted net loss per share for the years ended December 31, 2017, 2016 and 2015, is as follows (amounts in thousands, except share numbers):
| Year ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | 21,214 | $ | 9,847 | $ | 6,406 | ||||||
| Dividends accumulated for the period(1) |
2,404 | 1,100 | 516 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss available to stockholders of Common Stock |
$ | 23,618 | $ | 10,947 | $ | 6,922 | ||||||
|
|
|
|
|
|
|
|||||||
| Denominator: |
||||||||||||
| Shares used in computing net loss per share of Common Stock, basic and diluted |
4,976,377 | 4,205,277 | 4,148,389 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss per share of Common Stock, basic and diluted |
$ | 4.75 | $ | 2.60 | $ | 1.67 | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | The net loss used for the computation of basic and diluted net loss per share include 8% per share per annum compounded annually which was related to distributions for preferred stockholders of Eloxx Limited. On December 19, 2017 in conjunction with the Reverse Merger all preferred shares were converted to common shares. |
The total weighted average numbers of shares related to outstanding preferred stock, stock options under the 2008 Plan and 2013 Plan, stock warrants and restricted shares that have been excluded from the calculation of the diluted net loss per share due to their anti-dilutive effect was 19,027,306, 12,746,823 and 6,778,980 for the years ended December 31, 2017, 2016 and 2015, respectively.
14. Financial and Other Expenses, net
Financial and other expenses consisted of the following (in thousands):
| Year ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Exchange rate differences |
156 | 7 | 122 | |||||||||
| Amortization and revaluation of embedded conversion feature in respect to convertible loan |
625 | — | — | |||||||||
| Interest expenses in respect to convertible loan |
43 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total financial and other expense, net |
824 | 7 | 122 | |||||||||
|
|
|
|
|
|
|
|||||||
15. Segment and Geographic Information
Operating segments are defined as components of an enterprise (business activity from which it earns revenue and incurs expenses) about which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is the Chief Executive Officer. The chief operating decision maker reviews consolidated operating results to make decisions about allocating resources and assessing performance for the entire company. The Company views its operations and manages its business as one operating segment; however, it operates in two geographic regions: United States (Waltham, MA) and Israel (Rehovot).
100
Table of Contents
16. Subsequent Events
On January 10, 2018, the Company filed a Registration Statement on Form S-8 for the purpose of registering (i) 2,353,493 shares common stock of the Company, $0.01 par value per share (the “Common Stock”) issuable pursuant to outstanding but unexercised option awards previously issued under 2013 Plan, (ii) 119,762 shares of common stock issuable upon the exercise of stock options reserved for issuance pursuant to future awards under the 2013 Plan, and (iii) 663,212 restricted stock units and 663,212 options to purchase common stock granted to Robert E. Ward on December 26, 2017, as Chief Executive Officer and Chairman of the Board of Directors of the Company.
101