Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Invuity, Inc. | ivty-20171231ex32186e624.htm |
| EX-31.2 - EX-31.2 - Invuity, Inc. | ivty-20171231ex31234ab13.htm |
| EX-31.1 - EX-31.1 - Invuity, Inc. | ivty-20171231ex311ac9356.htm |
| EX-23.1 - EX-23.1 - Invuity, Inc. | ivty-20171231ex2317163fc.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
FOR THE TRANSITION PERIOD FROM ________________ TO ________________
Commission File Number 001-37417
Invuity, Inc.
(Exact name of Registrant as specified in its Charter)
|
Delaware (State or other jurisdiction of incorporation or organization) |
04-3803169 (I.R.S. Employer Identification No.) |
|
|
|
|
444 De Haro Street San Francisco, CA (Address of principal executive offices) |
94107 (Zip Code) |
Registrant’s telephone number, including area code: (415) 665-2100
Securities registered pursuant to Section 12(b) of the Act: Common Stock, par value $0.001 per share; Common Stock traded on the NASDAQ Stock Market
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ☐ NO ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. YES ☐ NO ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES ☒ NO ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES ☒ NO ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act.
|
x |
|
|
|
|
|
Large accelerated filer |
☐ |
|
Accelerated filer |
☒ |
|
|
|
|
|
|
|
Non-accelerated filer |
☐ |
(Do not check if a small reporting company) |
Small reporting company |
☐ |
|
|
|
|
|
|
|
|
|
|
Emerging growth company |
☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ☐ NO ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, based on the closing price of a share of common stock on June 30, 2017 (the last business day of the registrant’s most recently completed second fiscal quarter) as reported by the NASDAQ Stock Market on such date was $119,135,219.
The number of shares of registrant’s Common Stock outstanding as of February 28, 2018 was 17,204,000.
Portions of the registrant’s Definitive Proxy Statement relating to the Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10K where indicated. Such Definitive Proxy Statement will be filed with the Securities and Exchange Commission within 120 days after the end of the registrant’s fiscal year ended December 31, 2017.
i
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which statements involve substantial risks and uncertainties. Some of the statements under the captions “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business” and elsewhere in this Annual Report on Form 10-K are forward-looking statements. These statements are based on our current expectations, assumptions, estimates and projections about our business and our industry and involve known and unknown risks, uncertainties and other factors that may cause our company’s or our industry’s results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied in, or contemplated by, the forward-looking statements. Words such as “believe,” “anticipate,” “expect,” “intend,” “plan,” “focus,” “assume,” “goal,” “objective,” “will,” “may” “should,” “would,” “could,” “estimate,” “predict,” “potential,” “continue,” “encouraging” or the negative of such terms or other similar expressions identify forward-looking statements. Our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Factors that might cause such a difference include those discussed in Item 1A. “Risk Factors” as well as those discussed elsewhere in this Annual Report on Form 10-K. These and many other factors could affect our future financial and operating results. We undertake no obligation to update any forward-looking statement to reflect events after the date of this report.
Unless the context otherwise requires, references to “Invuity,” “we,” “us,” “our” or “the Company” in this Annual Report on Form 10-K refer to Invuity, Inc.
Overview of Our Company
We are a leading medical technology company focused on developing and marketing advanced surgical devices to improve the ability of physicians to perform minimal access surgery through smaller and hidden incisions. Our patented Intelligent Photonics® technology delivers enhanced visualization which facilitates surgical precision, efficiency and safety. In addition, we utilize comprehensive strategic marketing programs to create stronger institutional partnerships. Clinical applications include women's health, encompassing breast cancer and breast reconstruction surgery, gynecology and thyroid surgery. Additional applications include procedures for electrophysiology, spine, orthopedic, cardiothoracic, and general surgery.
We channel our development through two broad categories of innovation. First, we integrate our Intelligent Photonics® technology platform into our single-use and reusable advanced surgical devices to address some of the critical intracavity illumination and visualization challenges facing surgeons today. We utilize this proprietary technology to develop optical waveguides that direct and shape thermally cool, brilliant light into broad, uniform and volumetric illumination of the surgical target. We believe that improving a surgeon’s ability to see critical anatomical structures can lead to better clinical and aesthetic outcomes, improved patient safety and reduced surgical time and healthcare costs.
Our second broad category of innovations for minimally invasive and minimal access surgical procedures is in the development and commercialization of a novel advanced energy platform. In September 2016, we received U.S. Food and Drug Administration, or FDA, 510(k) clearance of PhotonBlade® , a dynamic precision illuminator with enhanced energy delivery. PhotonBlade® is a first-of-its-kind device, delivering directed, thermally cool illumination at the precise point of surgical treatment in conjunction with a novel advanced energy platform allowing for precise tissue cutting and coagulation with minimal tissue damage. As such, PhotonBlade® represents a new category of Intelligent Photonics® and strategically expands our current product portfolio. After a preliminary market launch in March 2017, we fully launched the PhotonBlade® in September 2017.
Finally, in the third quarter of 2017, through a distribution arrangement with our manufacturing partner, Fluoptics Imaging Inc., we began a limited market launch of a fluorescence imaging system, called PhotonVue, which is used for the visual assessment of blood flow in adults as an adjunctive method for the evaluation of tissue perfusion
2
We sold our devices to approximately 870 hospitals in the fourth quarter of 2017, as compared to approximately 745 hospitals in the fourth quarter of 2016. Based on the number of single-use units we have shipped as of December 31, 2017, we estimate that our devices have been used in approximately 367,000 surgical procedures. We are also using our advanced photonics technology to develop new devices and modalities to broaden the application and adoption of minimal access procedures and enable new advanced surgical techniques.
Recent Developments
On February 27, 2018, Philip Sawyer informed the board of directors that he will resign from his role as President, Chief Executive Officer, and a member of our board of directors, effective as of February 28, 2018. Scott Flora, currently serving as a member of our board of directors, assumed the additional role of Interim President and Chief Executive Officer, and as such became the Company’s principal executive officer effective on March 1, 2018.
Illumination and Visualization
Photonics is the science and technological applications of light. We have applied advanced principles of photonics to develop our Intelligent Photonics® technology platform, which enables the transmission, management and manipulation of light in surgical procedures. Our initial application of this technology is integrated into our family of proprietary optical waveguides, which we call Photonguides. Photonguides are sophisticated devices that rely on the principles of optics to shape and direct light. They are coupled to a modified fiber optic cable and are designed to work with the standard xenon or light-emitting diode, or LED, light sources typically found and utilized in the operating room. Photonguides are incorporated into surgical devices, including our customized line of illuminated surgical retractors, handheld illuminated aspiration devices, drop-in intracavity illuminators and, most recently, PhotonBlade®. Our handheld illuminated aspiration devices, drop-in intracavity illuminator, and PhotonBlade® are single-use products. Our retractors are reusable, but utilize a single-use optical Photonguide with each procedure.
The fundamental attributes of Photonguides include a solid core optical-grade polymer, total internal reflection of light waves, light mixing and extraction by a complex geometry of refractive microstructures or microlenses. The solid core optical-grade polymer Photonguide is coupled to a fiber optic cable in order to facilitate the efficient transfer of light. This unique coupling results in Photonguides capturing maximum light with minimal heat build-up. Photonguides use critical angles and the properties of total internal reflection to retain and transmit maximum light as it travels through the device. In addition, each Photonguide utilizes various novel optical methods to mix light during the total internal reflection transmission process to enable more uniform light extraction across its output surface. The output surface consists of a complex geometry of refractive microstructures or microlenses that extract, direct and shape volumetric illumination into the surgical cavity while virtually eliminating shadows and glare. This complex geometric structure extracts and directs light at numerous different angles to enable illumination of the surgical target, even if blood or debris accumulates on the surface of the Photonguide. The uniform distribution of light extraction from the microstructures or microlenses throughout the entire output surface of the Photonguide, as well as the proprietary solid core optical-grade polymer and patented design of Photonguides, results in thermally cool illumination.
Advances in medical technology have resulted in growing adoption of minimally invasive and minimal access surgical procedures. Minimally invasive surgery refers to surgeries performed through one or more small incisions, which offer several benefits over traditional invasive open surgery, such as fewer surgical target complications and infections, overall reduced trauma to the anatomy, less bleeding, shorter hospitalization time, less postoperative pain, faster recovery time and improved aesthetic outcomes. Some minimally invasive procedures, such as endoscopic, laparoscopic and arthroscopic procedures, use small tubes, tiny cameras and surgical instruments to access, visualize and perform the surgery. Other procedures also use smaller incisions than conventional open surgery, but still provide the surgeon with direct visualization of the surgical target and the ability to use traditional surgical instruments. We refer to these procedures as minimal access procedures. We estimate that approximately 40% of all surgical procedures in the United States are open or minimal access, representing a sizable clinical and commercial opportunity. We are targeting our sales and marketing efforts to surgeons for the following women’s health clinical applications: breast cancer and breast reconstruction surgery, gynecology and thyroid surgery. Additional applications include procedures for electrophysiology, spine, orthopedic, cardiothoracic, and general surgery. Furthermore, our current illuminated surgical devices have a broader indication for use and can be marketed to other specialties with limited or no additional regulatory clearance. We currently estimate the annual total addressable market for our devices in these surgical specialties in the United States to be approximately $2.0 billion, based on the estimate of our average revenue per procedure.
3
In the last several years, we have transitioned from a focus on research and development to the commercialization of our device portfolio. As of December 31, 2017, we market six categories of illuminated surgical devices, consisting of over 40 devices. Our goal is to develop advanced surgical devices to improve the ability of physicians to perform minimal access surgery through smaller and hidden incisions. The company's patented Intelligent Photonics™ technology delivers enhanced visualization, which facilitates surgical precision, efficiency and safety. We market and sell our devices in the United States primarily through a direct sales force, which has changed from 67 representatives as of December 31, 2016 to 58 representatives as of December 31, 2017. Throughout the year, we have continued to work on optimizing the sales force, and we believe that a sales force in the low 60s strikes the right balance between sales force investment and revenue growth.
In June 2017, we initiated commercial activity in Japan, our first market outside of the U.S. We partnered with MC Medical, a division of Mitsubishi Corp., for distribution. MC Medical is an established entity in the Japanese surgical market and is involved in multiple specialties that pertain to the Invuity portfolio of products.
Traditional Illumination Devices and Their Limitations
Lighting is a critical element of every open surgical procedure. Traditional surgical lighting options in the operating room include overhead lighting systems, surgical headlights and on-field fiber optic lighting systems. The most common illumination method in the operating room setting today is overhead lighting systems. When used in open and minimal access procedures, overhead lighting systems can present numerous challenges. It can be difficult to maintain the required direct path of illumination given changes of patient and surgeon positioning. Adjustments to the lighting position may be required which may inconvenience the surgeon, disrupt the surgical flow, increase operating procedure time, and increase risk of contamination given that overhead lighting systems are not sterile. Moreover, overhead lighting systems may be inadequate for surgery in deeper cavities due to the creation of shadows and the inability of the light to reach the depth of the incision.
Surgical headlights overcome some of the shortcomings of overhead lighting systems by bringing the light source closer to the surgical cavity, eliminating shadows caused by the surgeons head, and reducing the need to adjust the overhead lighting system. Despite these benefits, we believe headlights still present limitations. Use of headlights can cause head, neck and shoulder fatigue from the prolonged improper posture during their use. Because the source of light is still above the surgical cavity, we believe the use of headlights can create shadows and glare caused by hands, instruments and anatomy, which may limit visualization in deep surgical cavities. Headlights can also generate considerable amounts of heat during use, which can further limit comfort and can cause burns if an operator accidently mishandles the device.
Due to the limitations of overhead lighting systems and surgical headlights, on-field fiber optic lighting systems have been developed in an effort to provide intracavity lighting of the surgical field. On-field fiber optic lighting systems consist of a fiber optic cable attached to a fiber optic retractor. While traditional on-field fiber optic lighting systems can be effective at bringing the light source closer to the surgical field, we believe they also have inherent limitations and risks. Traditional on-field fiber optic lighting systems represent a thermal hazard in the operating room, creating the risk of burns to patients, surgeons and hospital staff, and operating room fires. The light generated by the xenon or LED light source is extremely powerful and can create temperatures exceeding 100°C. Thermal heat builds up whenever light is obstructed. We believe another limitation of traditional on-field fiber optic lighting systems is the potential for hot spots and glare. The general light output shape emanating from a fiber optic cable that resembles a cone and is circular, with a common center around the mechanical axis of the fiber bundle. Since this bright, narrow outlet of light exits the fiber in a straight line in the direction and orientation of the fiber, the light may be reflected back in the same direction, and can create glare in the line of vision of the surgeon. In an attempt to minimize this glare, the surgeon may be required to constantly reposition the fiber optic retractor during the procedure.
Market Need for Advanced Intracavity Illumination and Visualization Devices
Given the limitations of traditional surgical lighting options in the operating room, we believe there is a significant opportunity to enhance intracavity illumination and visualization during open and minimal access procedures. In addition, we believe that an advanced illumination and visualization technology could broaden the application and adoption of less invasive surgical techniques.
4
Our Illumination and Visualization Solution
We utilize our Intelligent Photonics technology platform to develop surgical devices designed to overcome the significant limitations of traditional surgical lighting options in the operating room. Based on surgeon feedback, surgeon observation and bench testing, we believe our technology may provide the following benefits:
|
· |
Enhanced illumination and visualization of the surgical field. Our devices are designed to provide enhanced intracavity illumination and visualization of the surgical field during open and minimal access surgeries. The proprietary complex geometry of refractive microstructures or microlenses along the surface of the Photonguides allow for the extraction of light in a manner that distributes light at different angles in a broad, uniform and volumetric pattern that is intended to reduce shadows, glare and excessive heat that are commonly associated with traditional surgical lighting options. In bench testing comparing light distribution and thermal profile of our Eikon retractor to a traditional fiber optic retractor, we found our Eikon retractor system had approximately five times the illumination area with a thermal profile that is below the risk of burn. |
|
· |
Improved surgical precision during open and minimal access procedures. Our technology is designed to improve intracavity visualization to allow surgeons to identify, differentiate and avoid vital anatomical structures. We believe this enables surgeons to dissect with great precision, while also allowing them to differentiate tissue planes, identify and avoid nerves and blood vessels, and quickly locate and control bleeding vessels to achieve rapid hemostasis. With this precise visualization, we believe surgeons may be able to use smaller, and in some cases fewer, incisions. |
|
· |
Reduced risks to patients and surgeons. Our technology is developed with design elements to help create thermally cool illumination as well as ergonomics to improve ease of use while performing a procedure. Our advanced surgical devices incorporate a solid core optical-grade polymer that facilitates efficient coupling to the surgical instrument to offer significantly improved light transfer while concurrently reducing heat transfer. We believe this is an important advancement over traditional on-field fiber optic lighting systems that do not efficiently transfer light through the fiber-to-fiber coupling, resulting in the generation of excess heat, which can increase the risk of burn to patients and surgical staff and create the potential for operating room fires. In addition, by improving visualization, our devices may also decrease risk of unintended retained foreign objects by improving the surgeon’s ability to see and dispose of such objects that might have otherwise been left in the surgical cavity inadvertently. Finally, by being directly incorporated into a variety of illuminated surgical retractors, handheld illuminated aspiration devices, and drop-in intracavity illuminators, we believe our technology may help to decrease surgeon fatigue by reducing or eliminating the need for surgical headlights, thereby helping to reduce some of the associated head, neck and shoulder fatigue, frequent headaches, neck pain and injury to the cervical spine. In addition, our non-conductive Eikon LT retractors virtually eliminate the potential for burns due to arcing from electrosurgical devices coming in contact with traditional stainless steel retractors. |
|
· |
Enhanced operating room efficiency. We believe our technology improves operating room workflow by reducing the need for perioperative repositioning of traditional surgical lighting options. Overhead lighting systems and headlights require frequent readjustment, which may interrupt operating room workflow and extend surgical procedure time. Many open and minimal access procedures are time sensitive and the treatment area requires constant attention of the surgeon and operating team. Because the Photonguides are directly connected to the surgical instrument that is used to access the deep surgical cavity, surgeons are able to clearly illuminate the surgical target and effectively focus on performing the procedure. As an example, in a survey we conducted with 12 surgeons that use our devices, each of whom is considered a leading breast surgeon, 11 of these surgeons reported that procedure time during nipple-sparing mastectomy procedures when using our devices was reduced by an average of 24%. |
|
· |
Economic value proposition to healthcare systems. We believe our devices have the potential to substantially reduce procedure costs as well as create incremental revenue opportunities. We believe the improved efficiency of the operating room workflow and the related reduced procedure and anesthesia time can translate to meaningful cost savings for the hospital. In addition, we believe the reduction in procedure times also creates additional capacity in the operating room for surgeons to perform more procedures, which we believe can create incremental revenue for the hospital. |
5
Our Intelligent Photonics Technology
Photonics is the science and technical application of light. We have applied advanced principles of photonics to develop our Intelligent Photonics technology platform, which enables the transmission, management and manipulation of light in surgical procedures. Our initial application of this technology is our family of proprietary optical waveguides, called Photonguides. The fundamental attributes of Photonguides include a solid core optical-grade polymer, total internal reflection of light waves, light mixing and extraction by a complex geometry of refractive microstructures or microlenses.
Fundamental Attributes of Photonguides

Solid Core Optical-Grade Polymer
Photonguides are fabricated from a proprietary solid core optical-grade polymer, specifically selected for its key optical and mechanical characteristics, which enable the efficient transmission and management of light. These optical characteristics include the ability to mold the material into various complex geometries, which is of particular importance when molding ultra-precise structures. Certain mechanical properties of the polymer, such as structural integrity, hydrophobicity and thermal stability, are critical to its use during surgical procedures. In addition, our solid core design facilitates the coupling of the Photonguide to the modified fiber optic cable in order to allow the efficient transfer of light into the solid core Photonguide, while remaining thermally cool. All these characteristics are critical in order for the Photonguide to function as an advanced illuminated surgical device.
Total Internal Reflection of Light Waves
One of the key aspects of the Photonguide technology is the ability to transmit light in a highly efficient manner prior to its extraction. Light travels in waves. As a wave travels through a medium it will reach a boundary where there is a different medium on the other side of the boundary. At the point where the wave meets the boundary, three phenomena can occur: reflection, refraction or some combination of both. Reflection occurs when light bounces off the boundary and refraction occurs when waves pass through a boundary and change direction. The angle at which the wave hits the boundary is referred to as the angle of incidence. That angle is usually referenced to the line that is perpendicular to the boundary. A zero incidence angle means that the wave is traveling perpendicular to the boundary. At that angle most of the light will pass most of its energy through the boundary and will not refract as long as the index of refraction is less on the other side of the boundary than in the medium the light is traveling. As the angle of incidence increases, the wave will get split into two components: one portion will pass the boundary and refract and the other portion will reflect back into the medium in which the wave was originally traveling. As the angle increases, the amount of refraction will decrease and reflection will increase. The smallest angle where the light is completely reflected and not refracted is called the critical angle. At any angle of incidence greater than the critical angle, all of the light is reflected off the boundary with no refraction. This is referred to as total internal reflection. We designed the structural and material properties of our devices to maximize locations of total internal reflection as the light propagates along the central axis of the Photonguide.
6
Light Mixing
Photonguides utilize various novel optical methods to mix light, or randomize its reflections, during the total internal reflection transmission process. The design and shape of the optical stem, or area of the Photonguide that is between the input of the Photonguide and the array of refractive microstructures or microlenses, enhance the mixing of light waves, while maintaining total internal reflection. Photonguides utilize light mixing before extraction to significantly reduce glare and bright spots, leading to a more uniform illumination profile across the surgical target while remaining thermally cool.
Complex Geometry of Refractive Microstructures and Microlenses
We designed a proprietary complex geometry of refractive microstructures and microlenses that are placed on the surface of the optical waveguide to extract light from the device in a manner that distributes light over the surgical target. This distribution of light from the Photonguide also reduces the energy density in the device, thus reducing heat. Without the microstructures to extract the light uniformly on the surgical target, the Photonguide would dissipate an energy density across its surface that is in excess of the amount that the tissue could absorb without causing thermal injury. The surface of the Photonguide contains a complex geometry of zones with corresponding refractive microstructures or microlenses at varying angles. These extraction zones allow the Photonguide to direct the extracted light onto the surgical target and shape it into a broad, uniform and volumetric pattern. The ability to direct light is especially important when the Photonguide is mounted on surgical retractors, because our device is able to push the light away from the retractor, thus maintaining its efficiency on the surgical target. We believe this is a significant advantage over traditional on-field fiber optic lighting systems, which lack the microstructures to direct light and instead direct light in a straight line in the shape of a cone from the end of the fiber. As a result, a portion of the illumination is obstructed and absorbed by the surgical retractors when the fiber is adjacent to the surgical instrument. The ability to shape light is also critical, as it reduces the focal intensity of light. With traditional fiber optic retractors, light is directed in a narrow beam, with intensity at a maximum in the center of the spot of light, and dropping off exponentially toward the edges. As a result, it typically does not illuminate the entire surgical cavity and heat builds up significantly in that focal zone. In contrast, Photonguides broaden this intensity of distribution, which allows the pattern of light to have uniform brightness across the surface of the surgical target, while minimizing the thermal profile.
Photonguides are also designed to extract light from multiple zones, allowing the surgical target to be illuminated from various angles. As light is extracted across the Photonguide at numerous different points along the surface at slightly different angles, if any of the features on the surface become blocked by an instrument, blood or tissue, there are multiple other microstructures from which light is extracted to provide illumination. This proprietary complex geometry also provides off-axis illumination on the surgical target, meaning that the light originates from a different angle than in direct orientation to the Photonguide. As such, when light reflects off the tissue of the surgical target, instead of reflecting upwards towards the surgeon, the light is generally reflected onto the surface opposite the retractor. This feature of the Photonguide is important because it allows the surgeon and operating staff much better visual perception of the surgical target with less shadows and glare.
Advanced/Enhanced Energy
In September 2016, we received FDA 510(k) clearance of PhotonBlade®, a dynamic precision illuminator with enhanced energy delivery. The PhotonBlade® is a first-of-its-kind device, delivering directed, thermally cool illumination at the precise point of surgical treatment in conjunction with a novel energy platform allowing for precise tissue cutting and coagulation with minimal tissue damage. As such, PhotonBlade® represents a new category of Intelligent Photonics®and strategically expands our current product portfolio. After a preliminary market launch in March 2017, we fully launched the PhotonBlade® in September 2017.
7
Market Need for Improved Advanced/Enhanced Energy Devices
We estimate at least 16 million surgical procedures in the U.S. use some form of electrical energy for cutting of tissue and/or coagulation of blood. While effective and standard for surgery, there are applications where more precise tissue cutting is needed and minimal impact on collateral tissue is preferred. For these cases, enhanced energy devices were introduced to address those needs. Further, while surgeons operate in dark cavities or difficult to see areas, illumination is needed to provide a greater measure of safety and efficiency. PhotonBlade® is a precision illuminator that adds the attributes of Advanced/Enhanced energy, giving surgeons the ability to see better while addressing tissue in a more precise fashion as compared to standard electrosurgical instruments.
Our Advanced Energy Solution
PhotonBlade® has the following benefits as compared to other energy devices:
|
· |
Intelligent Photonics: Dynamic, precision illumination ~2cm from the point of treatment; |
|
· |
Enhanced Energy Delivery: Proprietary insulated tip design for the lowest thermal spread of all competitive devices; |
|
· |
Universal Compatibility: Compatible with any 510k cleared electrosurgical unit, eliminating the need to manage dedicated generators; and |
|
· |
Adjustable Illumination and Energy: 2”-5” adjustable shaft length. |
Our Market Opportunity
Advances in medical technology have resulted in growing adoption of minimal access surgical procedures. The increased utilization of these procedures by surgeons is primarily driven by their significant benefits compared to conventional open surgery including:
|
· |
smaller incisions resulting in less scarring and fewer complications; |
|
· |
less trauma to the organs, muscles, nerves, and tissue; |
|
· |
less bleeding and reduced need for blood transfusions; |
|
· |
fewer surgical infections; |
|
· |
shortened hospital stays, potentially reducing hospital costs; |
|
· |
less postoperative pain and reduced need for associated narcotics; |
|
· |
faster recovery time; and |
|
· |
improved aesthetic outcomes. |
Though some minimally invasive procedures, such as endoscopic, laparoscopic and arthroscopic procedures, have several of the benefits described above, surgeons are only able to view the surgical target through a tiny camera, which can cause reduced depth perception and field of vision, diminished hand-eye coordination, limited mobility of the surgical instruments, and reduced tactile feedback. These limitations can increase the cognitive and physical load on the surgeon and, consequently, increase the possibility of surgical error. We believe that minimal access procedures provide many of the benefits described above. However, the small incisions used in these procedures inherently reduce a surgeon’s ability to directly see the surgical target, particularly deep within the surgical cavity, which can impact surgical precision, procedural efficiency and patient safety.
8
Our Products
Our advanced photonics technology has allowed us to design multiple variations of Photonguides in order to target different illumination patterns for different shapes of surgical cavities. Because we can mold our solid core optical-grade polymer into different shapes, we are able to design Photonguides that either directs the light narrowly for deep cavities, or broadly for larger blade cavities. Photonguides also come in narrow or wide configurations to accommodate various retractor widths that are designed for varying patient anatomies. Our versatile design and manufacturing capabilities allow us to develop Photonguides with a variety of extraction patterns. For example, our current retractor based Photonguides utilize a complex geometry of refractive microstructures and microlenses, whereas as our handheld illuminated aspiration devices have integrated microlens arrays. Using advanced ray-trace software modeling programs, we are able to perform three-dimensional optical performance modeling of Photonguides, as well as an entire assembly including the retractor. We are capable of analyzing the entire optical performance of the assembly as we monitor various characteristics such as extracted light direction, uniformity on the target, glare to the user, as well as thermal profile. This ray-trace modeling process helps us develop illuminated surgical devices that are designed to provide optimal intracavity illumination.
We currently market six categories of illuminated surgical devices, consisting of over 40 devices. Our advanced photonics technology is integrated into each of these device families. Our device portfolio includes reusable illuminated surgical retractors that include a single-use Photonguide, single-use handheld illuminated aspiration devices and single-use drop-in intracavity illuminators. Photonguides are integrated into these customized devices to deliver improved visualization of the surgical cavity without generating excessive heat. Our accessories include sterilization trays and light cables.
|
Product Family |
|
Image |
|
Description |
|
Surgical Specialties |
|
|
|
|
|
|
|
|
|
Eikon LT Illuminated Retractor System |
|
|
|
Illuminated surgical retractor with a low-profile design. Lightweight, radiolucent, non-conductive retractors provide electrical insulation from electrosurgical device preventing inadvertent thermal damage. Atraumatic and elevated tip for easy maneuverability, dissection and retraction. Available in multiple blade sizes, with or without teeth, for varying patient anatomies, surgeon preferences, and surgical specialties. |
|
Breast / Oncoplastic / Surgery / Gynecology / EP / Plastic / Endocrine |
|
|
|
|
|
|
|
|
|
PhotonBlade® |
Dynamic recision illuminator with enhanced energy. Precise tissue cutting and coagulation with minimal collateral damage. No need for propriety generator. |
Breast / Plastics / EP / Orthopedics |
||||
|
|
|
|
|
|
|
|
|
Eiberg Illuminated Retractor System |
|
|
|
Illuminated hohmann-style retractor with a low-profile design. Made of stainless steel, with an ergonomic design to be comfortable to hold. Designed with subtle curvatures and smooth edges to provide atraumatic, secure retraction. |
|
Orthopedic |



9
|
|
|
|
|
|
|
|
|
PhotonSaber Y |
|
|
|
Handheld illuminator incorporated in a traditional Yankauer aspiration platform. Provides on-field illumination, aspiration, smoke evacuation, soft tissue retraction and blunt dissection in one device. Low-profile design enables surgeons to work efficiently in deep, dark cavities through smaller incisions. Available in multiple tip configurations (taper, metal, and bulb) for various surgical needs and in an optional pistol grip handle for improved ergonomics and visualization. |
|
Orthopedic / Spine / Cardiothoracic / Breast / General / Gynecology / Plastic |
|
|
|
|
|
|
|
|
|
PhotonSaber F |
|
|
|
Handheld illuminator incorporated in a traditional Frazier aspiration platform. Provides on-field illumination, aspiration, smoke evacuation, and soft tissue retraction in one device. Low-profile design enables surgeons to work efficiently in deep, dark cavities through smaller incisions. Available in multiple tip configurations (8 Fr and 12 Fr). |
|
Spine / Orthopedic / Neurosurgery |
|
|
|
|
|
|
|
|
|
Breiten Illuminated Retractor System |
|
|
|
Illuminated surgical retractor with a low-profile design. Radiolucent to enable visibility during fluoroscopy. Color-coded for easy identification. Provides an offset hub for blade positioning. Available in multiple blade sizes and blade tips for varying patient anatomies and surgeon preferences. |
|
Spine / Orthopedics |
|
|
|
|
|
|
|
|
|
Photonguide XT System |
Drop-in intracavity illuminator with a low-profile design. Anchors to the incision wall providing a stand-alone, hands-free device. Minimal profile design is compatible with existing retractors and instrumentation and accommodates preferred surgical exposure techniques. |
Spine |
||||
|
Eika Illuminated Retractor System |
Illuminated surgical retractor with a low-profile design. Self-retracting handle design enables either hands-free or manual retraction. Includes a handle slot for ideal cable management and placement. Available in multiple blade sizes for varying patient anatomies and surgeon preferences. Designed for anterior neck approaches, including thyroid and cervical spine surgeries. |
Endocrine / Spine / Orthopedics |
||||
|
PhotonVue®* |
System used in conjunction with IC Indocyanine for identifying and verifying blood flow in tissue. Small, portable system with attractive economic options. |
Breast / Plastics / Colorectal |





* By way of distribution and manufacturing agreement with Fluoptics Imaging Inc.
10
Selected Surgical Applications
Our commercial strategy is initially focused on targeting open and minimal access procedures where there is a significant need for improved illumination and direct visualization. These procedures span a broad spectrum of women’s health clinical applications: breast cancer and breast reconstruction surgery, gynecology and thyroid surgery. Additional applications include procedures for electrophysiology, spine, orthopedic, cardiothoracic, and general surgery. We believe our technology has enabled surgeons to perform procedures that were previously difficult to perform due to visualization and illumination challenges. The selected procedures discussed below illustrate some of the benefits of our technology.
Breast: Nipple Sparing Mastectomy
Surgical management of breast cancer has evolved dramatically over the past several decades. Surgeons have continuously looked for ways to improve oncologic outcomes while combining the techniques of oncoplastic surgery to maximize both the treatment of cancer and the aesthetic outcome with the optimal goal of preserving the nipple areola complex. Skin and nipple preservation during breast cancer surgery is essential to attain ideal aesthetic results.
A nipple sparing mastectomy, or NSM, is a procedure in which the cancerous breast tissue is removed but the breast skin and nipple are left intact. We believe the relatively limited adoption to date of the NSM procedure is attributed to a number of surgical limitations. Some of these limitations include limited access and visualization through smaller and distant incision location, and difficulty in maintaining consistent breast flap thickness and viability. We believe our advanced photonics technology can facilitate a surgeon’s ability to:
|
· |
use a single infra-mammary fold incision in NSM to access and visualize deep into the surgical cavity; |
|
· |
access and visualize the lymphatic tree without a second axillary incision in most cases; and |
|
· |
assess the breast flap thickness and viability via trans-illumination. |
Electrophysiology: Pacemaker and ICD changeouts
In the U.S. there are approximately 200,000 procedures to remove or replace pacemakers or implantable cardiac defibrillators (“ICDs”). When accessing pacemakers/ICDs, surgeons must make an incision in the chest and work around leads that run into the chambers of the heart. In many of these procedures, complications may occur such as:
|
· |
damage to the leads from the use of mechanical or electrosurgical devices; |
|
· |
untreated bleeding vessel on the chest wall due to poor visualization, which can result in a post-operative hematoma; and |
|
· |
post-operative infection, which is highly correlated with post-operative hematoma and patient mortality. |
PhotonBlade® allows the surgeon to effectively see in a dark chest pocket during the procedure. The illumination which enhances the ability to see may allow the surgeon to address bleeding during the procedure and avoid a post-operative hematoma and subsequent complications.
GYN: Vaginal Hysterectomy
Hysterectomy, or removal of the uterus, is one of the most common types of surgery for women in the United States. Depending on the condition, many women will be presented with several surgical procedural options. Such surgical approaches may be an abdominal, robotic, vaginal, laparoscopic or laparoscopically assisted hysterectomy. All options, other than vaginal hysterectomy will result in some form of visible scarring and a potentially greater degree of postoperative pain.
A vaginal hysterectomy allows the surgeon to perform a total or radical hysterectomy through the vagina, a natural opening in the body. We believe that our advanced photonics technology can facilitate the surgeon’s ability to:
|
· |
effectively perform a vaginal hysterectomy through enhanced visualization of key structures and anatomy |
11
|
· |
optimally treat pelvic organ prolapse, concomitant in approximately 40% of vaginal or laparoscopically assisted hysterectomies; and |
|
· |
provide for a safe and effective operation through the use of illuminated non-conductive retractors. |
Orthopedics: Total Hip Arthroplasty including the Anterior Approach
The growth of minimally invasive surgery in orthopedics has been dramatic worldwide, as clinical results indicate that patients who undergo these procedures typically experience improved clinical outcomes, shorter hospital stays, faster rehabilitation and improved aesthetic outcomes. Our technology has been used in a range of procedures including, among others, hip arthroplasty, within which the use of our technology has enabled a less invasive approach.
Traditional hip replacement, also known as hip arthroplasty, involves operating from the side or the back of the hip, which can create a significant disturbance of the muscles and tendons and an incision approximately eight to twelve inches long. In comparison, the direct frontal, or anterior, approach requires an incision that is only three to four inches long and located at the front of the hip. In this position, the surgeon does not need to detach any of the muscles or tendons, but rather can move them aside along their natural tissue planes. This approach often results in faster recovery, less pain and more normal function after hip replacement. In addition, there is a lower risk of dislocating the new prosthesis when placed via the anterior approach, as the strength and integrity of the adjacent tendons and muscles surrounding the hip are maintained.
To date, we believe the less invasive anterior approach has been underutilized due, in part, to the visualization challenges associated with the procedure. More specifically, because the acetabulum and femoral canal are difficult to visualize using this approach, component positioning, sizing, and stability are more likely to be compromised, all of which are critical factors to yielding a successful and durable clinical outcome.
We believe the visualization provided by our devices can facilitate the surgeon’s ability to:
|
· |
expose, prepare and seat the acetabular shell and liner within the acetabulum; |
|
· |
place the acetabular screw; |
|
· |
evaluate stability and impingement of the ball against the socket; |
|
· |
prepare and mobilize the femur; and |
|
· |
internally inspect the femoral canal. |
Additional Applications
Our existing portfolio of devices is also eligible for use in, and could potentially improve the viability of, a multitude of additional surgical procedures. Importantly, our devices could be marketed and sold for a broad spectrum of surgical specialties without the need for any additional regulatory clearance. We believe our technology could help address the illumination and visualization challenges associated with various general surgery procedures, including appendectomy and herniorrhaphy; hysterectomy and other urological, gynecological and colorectal procedures; thyroidectomy and parathyroidectomy and other ear, nose and throat procedures; cardiac, cardiothoracic and cardiovascular procedures; craniomaxillofacial procedures and aesthetic plastic surgery.
We also continue to research and develop new devices, as well as pursue new clinical applications.
Sales and Marketing
We began selling our first FDA-cleared Photonguide-based device in March 2009. As a result, we have limited experience marketing and selling our devices. We currently sell our devices through our direct sales representatives only in the United States. While we primarily sell directly to hospitals, surgeons typically drive the purchasing decision. We sold our devices to approximately 870 hospitals in the fourth quarter of 2017. As of December 31, 2017, we had a sales and marketing team of 92 employees. Our sales team consisted of 58 direct sales representatives and 18 independent
12
sales agents or agencies, whom we refer to as independent sales agents, all of whom had significant sales experience before joining our sales team. The direct sales representatives are supported by 28 employees in sales management, training and customer support. Additionally, we have six employees in marketing. Over the past two years, we have significantly expanded our direct sales force to help facilitate further adoption among existing hospital accounts, as well as to broaden awareness of our advanced photonics technology to new hospitals. Using our expanded direct sales force, we intend to continue to educate and train surgeons on the advantages of our advanced photonics technology compared to traditional operating room lighting options. We believe the benefits of our technology should also enable the broader application and adoption of minimal access surgical procedures by more surgeons. Our operating results are directly dependent upon the sales and marketing efforts of our employees.
Our marketing efforts are focused on developing a strong reputation with major teaching institutions and hospitals, as well as surgeons that we have identified as key opinion leaders based on their knowledge of our devices, clinical expertise and reputation. We developed the Invuity Hidden ScarTM Surgery program to train and certify surgeons on minimal access surgical approaches and designate Centers of Excellence in Hidden Scar Surgery at hospitals and medical centers. Breast cancer surgery is our initial focus with the Hidden Scar Breast Surgery program and we are expanding into other specialties to include Hidden Scar Breast Reconstruction and Hidden Scar Hysterectomy to form a broader women’s health initiative.
We also sell and market to third-party medical device manufacturers. There were no sales to any customer in excess of 10% of our total revenue for any of the years ended December 31, 2017, 2016, and 2015.
Coverage and Reimbursement
Payment for patient care in the United States is generally made by third-party payors, including private insurers and government insurance programs. The reimbursement to the facility from third-party payors is intended to cover the overall cost of treatment, including the cost of our devices used during the procedure as well as the overhead cost associated with the facility where the procedure is performed. We do not directly bill any third-party payors; instead, we receive payment from the hospital or surgical center for our devices. Failure by physicians, hospitals, ambulatory surgery centers and other users of our devices to obtain sufficient coverage and reimbursement from healthcare payors for procedures in which our devices are used, or adverse changes in government and private third-party payors’ policies could have a material adverse effect on our business, financial condition, results of operations and future growth prospects.
In addition, there are periodic changes to reimbursement. Third-party payors regularly update reimbursement amounts and also from time to time revise the methodologies used to determine reimbursement amounts. This includes annual updates to payments to physicians, hospitals and ambulatory surgery centers for procedures during which our devices are used. Because the cost of our devices generally is recovered by the healthcare provider as part of the payment for performing a procedure and not separately reimbursed, these updates could directly impact the demand for our devices. An example of such payment updates is the Medicare program’s updates to hospital and physician payments, which are done on an annual basis using a prescribed statutory formula. In the past, with respect to reimbursement for physician services under the Medicare Physician Fee Schedule, when the application of the formula resulted in lower payment, Congress has passed interim legislation to prevent the reductions.
Any changes in coverage and reimbursement that lowers reimbursement for procedures using our devices could materially affect our business.
Competition
The medical device industry is highly competitive. We believe that the primary competitive factors in the surgical illumination and visualization and advanced energy market segments are clinical safety and effectiveness, price, surgeon experience and comfort with use of particular systems, reliability and durability, ease of use, device support and service, sales force experience and relationships. We face significant competition from competitors that are based in the United States and internationally in these market segments, and we expect the intensity of competition will increase over time. Many of the companies developing or marketing competing products enjoy several competitive advantages, including:
|
· |
more established sales and marketing programs and distribution networks; |
13
|
· |
long established relationships with surgeons and hospitals; |
|
· |
contractual relationships with customers; |
|
· |
products that have already received approval from the relevant value analysis committees, or VACs,; |
|
· |
greater financial and human resources for product development, sales and marketing; |
|
· |
greater name recognition; |
|
· |
the ability to offer rebates or bundle multiple product offerings to offer greater discounts or incentives; and |
|
· |
greater experience in and resources for conducting research and development, clinical studies, manufacturing, preparing regulatory submissions, obtaining regulatory clearance or approval for products and marketing approved products. |
Any device we develop must compete for market acceptance and market share. Our success in selling our devices to hospitals is dependent on our ability to demonstrate that the clinical, qualitative and economic value delivered by our products outweighs their increase to the cost per procedure. Our ability to compete on price depends on our ability to demonstrate to surgeons, hospitals and surgery centers that the potential benefits of improved clinical outcomes and reduced procedure costs from the increased efficiency in the operating room workflow and related reduced procedure and anesthesia time using our medical devices outweigh the price of our devices compared to our competitors’ products.
Intellectual Property
In order to remain competitive, we must protect the proprietary technology that we believe is important to our business, including seeking and, if granted, maintaining patents intended to cover our products and inventions that are commercially important to the development of our business. We also rely on trademarks, trade secret laws and confidentiality and invention assignment agreements to protect our intellectual property rights.
It is our policy to require our employees, consultants, contractors, outside scientific collaborators and other advisors to execute non-disclosure and assignment of invention agreements on commencement of their employment or engagement. Agreements with our employees also forbid them from using the proprietary rights of third parties in their work for us. We also require confidentiality agreements from third parties that receive our confidential data or materials.
Our success will depend on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. For more information, please see “Risk Factors—Risks Related to Our Intellectual Property.”
As of December 31, 2017, we held 97 issued U.S. and foreign patents and had 100 U.S. and foreign utility patent applications, and 14 Patent Cooperation Treaty, or PCT, applications pending. As we continue to research and develop our Intelligent Photonics technology, we intend to file additional U.S. and foreign patent applications related to the design, manufacture and clinical uses of our illuminated devices and other products. Our issued patents expire between the years 2026 and 2035. Our pending patent applications and issued patents include claims directed to coupling of an illumination device with a light source or an instrument, as well as efficient and safe transmission of light through the illumination device.
As of December 31, 2017, we held 55 U.S., foreign and international trademark registrations and had nine U.S. and foreign trademark applications.
Manufacturing, Raw Materials and Quality Assurance
Our manufacturing involves the combined utilization of our internal manufacturing resources and expertise, approved suppliers and contract manufacturers. Our internal manufacturing activities, located in San Francisco, California, include the inspection, assembly and packaging of the Photonguides, retractor systems, aspiration devices and accessories associated with each of our device families. We outsource the manufacture of components, subassemblies and certain
14
finished devices that are produced to our specifications and shipped to our facilities for final assembly or inspection, and certification. Finished products are stored at and distributed from our facility. Quality control, risk management, efficiency and the ability to respond quickly to changing requirements are the primary goals of our manufacturing operations. Unlike our other products, PhotonVue® is manufactured by Fluoptics Imaging Inc.
We have arrangements with our suppliers that allow us to adjust the delivery quantities of components, subassemblies and finished products, as well as delivery schedules, to match our changing requirements. The forecasts we use are based on historical trends, current utilization patterns and sales forecasts of future demand. Lead times for components, subassemblies and finished products may vary significantly depending on the size of the order, specific supplier requirements and current market demand for the components and subassemblies. Most of our suppliers have no contractual obligations to supply us with, and we are not contractually obligated to purchase from them, the components used in our devices.
We obtain the optical polymer used in the manufacture of Photonguides and certain accessories from single suppliers, for which we attempt to mitigate risks through inventory management and purchase order commitments. While we believe alternate sources exist for the optical polymer, we have not qualified an alternate provider. Other products and components come from single suppliers, but alternate suppliers have been qualified or, we believe, can be readily identified and qualified. In addition, we rely on a single provider for sterilization of our devices that require sterilization. While we believe replacement suppliers exist for all components, materials and services we obtain from single sources, establishing additional or replacement suppliers for any of these components, materials or services, if required, may not be accomplished quickly. Even if we are able to find a replacement supplier, the replacement supplier may need to be qualified and may require additional regulatory authority approval, which could result in further delay. While we seek to maintain adequate inventory of the single-source components and materials used in our products, in the event of disruption, those inventories may not be sufficient. To date, we have not experienced material delays in obtaining any of our components, subassemblies or finished products, nor has the ready supply of finished products to our customers been adversely affected. To date, we have not experienced any material delays by our sterilization provider and will continue to evaluate the cost and benefit of qualifying a second sterilization provider.
We have implemented a quality management system designed to comply with FDA regulations and International Standards Organization, or ISO, standards governing medical device products. These regulations govern the design, manufacture, testing and release of diagnostic products as well as raw material receipt and control. We have received ISO 13485 certification as well as an European Counsel, or EC, Certificate under Directive 93/42/EEC on Medical Devices, Annex II, excluding section 4. Our key outsourcing partners are ISO-certified.
We use small quantities of common cleaning products in our manufacturing operations, which are lawfully disposed of through a normal waste management program. We do not forecast any material costs due to compliance with environmental laws or regulations.
Segment Reporting
We manage our operations as a single operating segment for the purposes of assessing performance and making operating decisions. All of our assets are maintained in the United States. We derive our revenue primarily from sales to customers in the United States, based upon the billing address of the customer. In 2017, we started selling into Asia.
Government Regulation
Our products are medical devices and are therefore subject to extensive regulation by the FDA under the authority of the Federal Food, Drug and Cosmetic Act, or FDCA, and the regulations promulgated thereunder, as well as by corresponding state and international regulatory authorities. The regulations govern the following activities that we and our suppliers, licensors and partners engage in:
|
· |
product design and development; |
|
· |
pre-clinical and clinical testing; |
|
· |
establishment registration and product listing; |
15
|
· |
product manufacturing; |
|
· |
labeling and storage; |
|
· |
pre-market clearance or approval; advertising and promotion; |
|
· |
product sales and distribution; |
|
· |
recalls and field safety corrective actions; and |
|
· |
servicing and post-market surveillance. |
Regulatory Clearances and Approvals. Unless an exemption applies, each medical device that we wish to commercially distribute in the United States will require either prior 510(k) clearance or Premarket Approval Application, or PMA, approval from the FDA. The FDA classifies medical devices into one of three classes. Devices requiring fewer controls because they are deemed to pose low or moderate risk are placed in Classes I or II, which, unless subject to an exemption, requires the manufacturer to submit to FDA a 510(k) premarket notification requesting clearance for commercial distribution. Exempt Class I and II devices do not require submission of a 510(k) but are otherwise subject to general controls such as labeling, pre-market notification and adherence to the FDA’s Quality System Regulation, or QSR, which cover manufacturers’ methods and documentation of the design, testing, production, control quality assurance, labeling, packaging, sterilization, storage and shipping of products. Certain Class II devices are also subject to special controls such as performance standards, post-market surveillance, FDA guidelines, or particularized labeling. The Photonguides, retractor and aspiration devices are marketed as Class I exempt devices. The fiber optic cables we supply as part of our illuminated retractor and aspiration systems are marketed as Class II exempt devices. The metal and plastic sterilization trays used by the customer to sterilize our reusable retractors and fiber optic cables are Class II 510(k) products.
To obtain 510(k) clearance, we must submit a premarket notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976 and for which the FDA has not yet called for the submission of PMAs. The FDA’s 510(k) clearance pathway usually takes between three and twelve months from the date the notification is submitted, but can take considerably longer, depending on the extent of requests for additional information from the FDA and the amount of time a sponsor takes to fulfill them. FDA requests for additional information can include clinical data that the FDA determines is necessary to make a determination regarding substantial equivalence. We obtained 510(k) clearance for the BriteField McCulloch Retractor System, in April of 2009 and for the Eigr Surgical Illumination System in February of 2012. All of our other commercial products to date have been commercialized as Class I, Class II exempt or Class II devices.
After a device receives 510(k) clearance or is commercialized as a Class I or II exempt device, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k) clearance or could require premarket approval. The FDA requires each manufacturer to make this decision initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or premarket approval is obtained. We have made, and plan to continue to make, product enhancements that we believe do not require new 510(k) clearances. If the FDA requires us to seek 510(k) clearance or premarket approval for any such modifications to previously commercialized products, we may be required to cease marketing or recall the modified device until we obtain this clearance or approval, and we could be subject to significant regulatory fines or penalties.
A PMA must be submitted if a device cannot be cleared through the 510(k) clearance process. A PMA application must be supported by extensive data, including, but not limited to, technical information, preclinical data, clinical trial data, manufacturing data and labeling to demonstrate to the FDA’s satisfaction the safety and efficacy of the device for its intended use. None of our existing products are currently approved under a PMA, and we have no plans to develop products that would require a PMA.
16
Continuing FDA Regulation. Even after a device receives clearance or approval and is placed in commercial distribution, numerous regulatory requirements apply. These include:
|
· |
establishment registration and device listing with FDA; |
|
· |
Quality System Regulation, or QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, production, control, supplier/contractor selection, complaint handling, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
|
· |
labeling regulations that prohibit the promotion of products for uncleared, unapproved or “off-label” uses, and impose other restrictions on labeling, advertising and promotional activities; |
|
· |
Medical Device Reporting, or MDR, regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
|
· |
voluntary and mandatory device recalls to address problems when a device is defective and could be a risk to health; and |
|
· |
corrections and removals reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health. |
We and our contract manufacturers, specification developers and some suppliers of components or device accessories, also are required to manufacture our products in compliance with current Good Manufacturing Practice, or GMP, requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and it includes extensive requirements with respect to quality management and organization, device design, buildings, equipment, purchase and handling of components or services, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing, and record keeping. The FDA and the California Food and Drug Branch evaluate compliance with the QSR through periodic unannounced inspections that may include the manufacturing facilities of our subcontractors. If the FDA or the California Food and Drug Branch believes that we or any of our contract manufacturers or regulated suppliers are not in compliance with these requirements, it can shut down our manufacturing operations, require recall of our products, refuse to clear new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations or assess civil and criminal penalties against us or our officers or other employees.
Failure to comply with applicable regulatory requirements can result in enforcement actions by the FDA and other regulatory agencies. These may include any of the following sanctions or consequences:
|
· |
warning letters or untitled letters that require corrective action; |
|
· |
fines and civil penalties; |
|
· |
delays in clearing or refusal to clear future products; |
|
· |
FDA refusal to issue certificates to foreign governments needed to export products for sale in other countries; |
|
· |
suspension or withdrawal of FDA clearances; |
|
· |
product recall or seizure; |
|
· |
interruption or total shutdown of production; |
|
· |
operating restrictions; |
17
|
· |
injunctions; and |
|
· |
criminal prosecution. |
Fraud and Abuse Laws. There are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback, false claims, physician payment and privacy and security laws. Our relationships with healthcare providers and other third parties are subject to scrutiny under these laws. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state healthcare programs, including the Medicare, Medicaid and Veterans Administration health programs.
Federal Anti-Kickback Laws. The Federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, receiving, offering or providing remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging of a good or service, for which payment may be made under a federal healthcare program, such as Medicare and Medicaid. The definition of “remuneration” has been broadly interpreted to include anything of value, including such items as gifts, discounts, the furnishing of supplies or equipment, credit arrangements, waiver of payments and providing anything at less than its fair market value. The Department of Health and Human Services, or HHS, has issued regulations, commonly known as safe harbors, that set forth certain provisions which, if fully met, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the HHS Office of Inspector General.
The penalties for violating the Anti-Kickback Statute include imprisonment for up to ten years, fines of up to $100,000 per violation and possible exclusion from federal healthcare programs such as Medicare and Medicaid. Many states have adopted prohibitions similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items and services reimbursed by any source, not only by the Medicare and Medicaid programs. Further, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or PPACA, amends the intent requirement of the Anti-Kickback Statute and criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. The PPACA also provides that the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes.
Federal False Claims Act. The Federal False Claims Act provides, in part, that the federal government may bring a lawsuit against any person whom it believes has knowingly presented, or caused to be presented, a false or fraudulent request for payment from the federal government, or who has made a false statement or used a false record to get a claim approved. In addition, amendments in 1986 to the Federal False Claims Act have made it easier for private parties to bring “qui tam” whistleblower lawsuits against companies under the Federal False Claims Act. Penalties include fines ranging from $11,181 to $22,363 for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person. Qui tam actions have increased significantly in recent years, causing greater numbers of healthcare companies to have to defend a false claim action, pay fines or be excluded from Medicare, Medicaid or other federal or state healthcare programs as a result of an investigation arising out of such action.
Civil Monetary Penalties Law. The Federal Civil Monetary Penalties Law prohibits the offering or transferring of remuneration to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection of a particular supplier of Medicare or Medicaid payable items or services. Noncompliance can result in civil money penalties of up to $15,270 for each wrongful act, assessment of three times the amount claimed for each item or service and exclusion from the federal healthcare programs.
State Fraud and Abuse Provisions. Many states have also adopted some form of anti-kickback and anti-referral laws and a false claims act. A determination of liability under such laws could result in fines and penalties and restrictions on our ability to operate in these jurisdictions.
Health Insurance Portability and Accountability Act of 1996. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, created two new federal crimes: healthcare fraud and false statements relating to healthcare
18
matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines or imprisonment. In addition, similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation.
Physician Payment Transparency Laws. There has also been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. PPACA, among other things, imposes new reporting requirements on device manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Device manufacturers are required to submit reports to the government by the 90th day of each calendar year. Failure to submit the required information may result in civil monetary penalties up to an aggregate of $165,786 per year (and up to an aggregate of $1.105 million per year for “knowing failures”) for all payments, transfers of value or ownership or investment interests not reported in an annual submission, and may result in liability under other federal laws or regulations. Certain states also mandate implementation of compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians.
Data Privacy and Security Laws. We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, impose specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
U.S. Foreign Corrupt Practices Act. The U.S. Foreign Corrupt Practices Act, or FCPA, prohibits U.S. corporations and their representatives from offering, promising, authorizing or making corrupt payments, gifts or transfers to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA would include interactions with certain healthcare professionals in many countries.
International Regulation
We may evaluate international expansion opportunities in the future. International sales of medical devices are subject to local government regulations, which may vary substantially from country to country. The time required to obtain approval in another country may be longer or shorter than that required for FDA approval, and the requirements may differ. There is a trend towards harmonization of quality system standards among the European Union, United States, Canada and various other industrialized countries.
The primary regulatory body in Europe is that of the European Union, which includes most of the major countries in Europe. Other countries, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the European Union with respect to medical devices. The European Union has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices. Devices that comply with the requirements of a relevant directive will be entitled to bear the conformity CE marking, indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout Europe. The method of assessing conformity varies depending on the class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a “Notified Body.” This third-party assessment may consist of an audit of the manufacturer’s quality system and specific testing of the manufacturer’s product. An assessment by a Notified Body of one country within the European Union is required in order for a manufacturer to commercially distribute the product throughout the European Union. Additional local
19
requirements may apply on a country-by-country basis. Outside of the European Union, regulatory approval would need to be sought on a country-by-country basis in order for us to market our devices.
Research and Development
We have an experienced research and development team with the scientific, engineering and process talent that we believe is necessary to grow our business. As of December 31, 2017, we had 11 employees engaged in research and development. Our research and development team has the technical and engineering knowledge that we believe is necessary to develop next-generation technology, as well as the advanced educational backgrounds in physics, optics, biomedical and mechanical engineering to support innovation in these areas.
We expect to commit significant resources to developing new technologies and devices, improving product performance and reducing costs. We continually seek to enhance and iterate our existing devices. Our research and development expenses totaled $9.0 million, $9.9 million and $7.9 million in the years ended December 31, 2017, 2016, and 2015, respectively. We also expect to expand our technology to create next generation devices and new platform offerings. In addition, we are engaged in advanced research related to inclusion of illumination in other medical devices, as well as further improvements in visualization and tissue differentiation.
Seasonality
Traditionally, we have experienced seasonality during the year. Revenue tends to be the lowest in the first quarter as the result of the resetting of annual patient healthcare insurance plan deductibles and by hospitals and military facilities working off their inventories of products purchased in the fourth quarter. The third quarter is similarly negatively impacted by lower procedure rates typically associated with vacation plans of both patients and surgeons. Revenue in the fourth quarter tends to be the highest as demand may be impacted by the desire of patients to spend their remaining balances in their flexible spending accounts or because they have met their annual deductibles under their health insurance plans. In addition, in the fourth quarter, our results can be impacted by the budgeting and buying patterns of hospitals and military facilities.
Employees
As of December 31, 2017, we had 161 full-time employees, which included 92 employees engaged in sales and marketing, 11 employees engaged in research and development, 43 employees engaged in manufacturing and quality assurance and 15 general and administrative employees. None of our employees are represented by collective bargaining agreement and we have never experienced any work stoppages. We believe that we have good relations with our employees.
Corporate Information
We were incorporated in California in 2004 as Spotlight Surgical, Inc. We changed our name to Invuity, Inc. in 2007, and reincorporated in Delaware in May 2015. Our principal executive offices are located at 444 De Haro Street, San Francisco, California 94107, and our telephone number is (415) 665-2100. In June 2015, we closed our initial public offering, or IPO, and our common stock is listed on the NASDAQ Global Market under the symbol “IVTY”.
Invuity, Inc. and our logo, as well as Intelligent Photonics, are our trademarks and are used in this Annual Report on Form 10-K. This Annual Report on Form 10-K also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, our trademarks and tradenames referred to in this Annual Report on Form 10-K appear without the ® and ™ symbols, but those references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights, or the right of the applicable licensor to these trademarks and tradenames. Additionally, we do not intend for our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, these other companies.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, and, as such, we have elected to comply with certain reduced public company reporting requirements. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year following the fifth anniversary of the completion of our initial public offering, (ii) the last day of the first fiscal year in which our annual gross revenue is $1 billion or more, (iii) the date on which we have, during the previous rolling three-year period, issued more than $1 billion in non-convertible debt securities or (iv) the date on which we are deemed to be a “large accelerated filer” as defined in the
20
Exchange Act. We refer to the Jumpstart Our Business Startups Act of 2012 herein as the “JOBS Act,” and references herein to “emerging growth company” are intended to have the meaning associated with it in the JOBS Act.
Available Information
Our website is located at www.invuity.com, and our investor relations website is located at http://investors.invuity.com/. We also use our investor relations website as a channel of distribution for important company information. Important information, including press releases, analyst presentations and financial information regarding us, as well as corporate information, is routinely posted and accessible on our investor relation website for complying with our disclosure obligations under Regulation FD. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to reports filed pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act, as amended, or the Exchange Act, are filed with the Securities and Exchange Commission, the SEC. We are subject to the informational requirements of the Exchange Act and file or furnish reports, proxy statements and other information with the SEC. Such reports and other information filed by us with the SEC are available free of charge on our website at www.investors.invuity.com when such reports are made available on the SEC’s website. The public may read and copy any materials filed by us with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Room 1580, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at www.sec.gov. The contents of these
websites are not incorporated into this filing. Further, our references to the URLs for these websites are intended to be inactive textual references only.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information contained in this Annual Report on Form 10-K, including our financial statements and the related notes thereto, before making a decision to invest in our common stock. The realization of any of the following risks could materially and adversely affect our business, financial condition, operating results and prospects. In that event, the price of our common stock could decline, and you could lose part or all of your investment.
Risks Related to Our Business and Industry
We currently are required to obtain additional funds to continue as a going concern. If we are unable to obtain sufficient funds, we may be unable to invest in our sales and marketing efforts and research and development activities in order to growg our business. Additionally, we may not be able to secure these funds on commercially reasonable terms, or at all.
We have experienced significant operating losses, and we expect to continue to incur operating losses for the next several years as we implement additional initiatives designed to grow our business, including, among other things, increasing sales and developing new devices. We incurred net losses of $39.9 million, $40.6 million and $37.6 million for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, our accumulated deficit was $186.1 million. Our prior losses, combined with expected future losses, have had and will continue to have, for the foreseeable future, an adverse effect on our stockholders’ deficit and working capital. These factors raise substantial doubt about our ability to continue as a going concern for the one year period from the date of issuance of the financial statements, included elsewhere in this Annual Report on Form 10-K. To date, we have financed our operations primarily through the sale of equity securities, certain debt-related financing arrangements and from sales of our approved devices. We have devoted substantially all of our resources to research and development of our devices, sales and marketing activities and certain clinical and quality assurance initiatives. Our ability to generate sufficient revenue from our existing devices or from any of our device candidates in development, and to transition to profitability and generate consistent positive cash flows, is uncertain. We will need to generate significant sales to achieve profitability, and we might not be able to do so. If our revenue grows more slowly than we anticipate or if we fail to continue to grow our revenue, or if our operating expenses are higher than we expect, we may not be able to achieve profitability as anticipated, or ever, our financial condition will suffer and our stock price could decline. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
21
Substantially all of our revenue to date has been generated from devices incorporating our Intelligent Photonics technology, and any decline in the sales of these devices or failure to gain market acceptance of these devices will negatively impact our business.
We have focused heavily on the development and commercialization of devices using our photonics technology platform for the illumination of certain open and minimal access surgeries. For the years ended December 31, 2017, 2016 and 2015, our revenues of $39.6 million, $32.5 million, and $21.0 million, respectively, were derived entirely from sales of devices incorporating our Intelligent Photonics® technology. Because we expect our revenue to be derived substantially from sales of these devices for the foreseeable future, our ability to execute our growth strategy and become profitable will depend not only upon an increase in the number of hospitals using our devices, but also an increase in the number of specialties using our devices within those hospitals in which our devices are utilized. If our advanced photonics technology, and the devices that incorporate it, fail to achieve and maintain wide market acceptance for any reason, our business may be adversely affected, as we will be severely constrained in our ability to fund our operations and develop and commercialize improvements to existing and new product lines.
We introduced our PhotonBlade® device in 2017 and there can be no assurance on the level of market acceptance or revenue generated by this product.
In the third quarter of 2016, we announced that we had received FDA 510(k) clearance for PhotonBlade®, a new device that integrates our Intelligent Photonics® technology into an advanced energy device used for cutting and coagulation of soft-tissue during surgical procedures. We initiated a limited launch of the PhotonBlade® device in the first quarter of 2017 to obtain feedback from initial users with a goal of launching the product more broadly in the middle of 2017. Shortly after the limited launch, we received a small number of customer complaints which related to possible unintended energy discharge from the device with the potential to cause tissue damage to patients. In June 2017, we initiated a voluntary recall of the device. We redesigned the device to correct the performance issues that prompted the recall and relaunched the product in September of 2017. We cannot be assured that we have successfully redesigned the PhotonBlade® so as to eliminate the performance issues that prompted the recall. Furthermore, in light of the recall, we cannot be assured that the redesigned PhotonBlade® will achieve market acceptance. We will be required to devote significant resources to relaunch and market the PhotonBlade® and cannot be assured that these activities will generate revenue as anticipated. If our redesign of the device fails to successfully eliminate the performance issue or if our revenue grows more slowly than we expect, our business and financials will be adversely affected.
Failure to achieve revenue targets under our credit facility with MidCap would constitute an event of default, which may require us to repay the amounts outstanding under the facility earlier than expected.
Our credit facility with MidCap requires us to comply with a financial covenant relating to certain quarterly minimum Net Revenue requirements on a trailing twelve month basis. The minimum Net Revenue requirements on a trailing twelve month basis for 2018 is $40,488,151 for the period ending March 31, 2018, $42,248,214 for the period ending June 30, 2018, $46,574,986 for the period ending September 30, 2018, and $50,000,000 for the period ending December 31, 2018. If we do not achieve these quarterly minimum Net Revenue requirements, it would constitute a breach of the credit agreements, may cause all of the outstanding indebtedness under our credit agreements, as amended, to become immediately due and payable and, if the Company was unable to obtain a waiver for this default, would permit the lenders to terminate all commitments to extend further credit and permit the agent, on behalf of the lenders, to proceed against the collateral in which we granted the agent and the lenders a security interest. As of December 31, 2017, we had borrowed $30.0 million in term loans and drawn down $5.9 million under our revolving credit facility.
If we are unable to convince hospital facilities to approve the use of our devices, our sales may decrease.
In the United States, in order for surgeons to use our devices, the hospital facilities in which surgeons treat patients will typically require us to receive approval from the facility’s Value Analysis Committee (“VAC”). VACs typically review the comparative effectiveness and cost of medical devices used in the facility. The makeup and evaluation processes for VACs vary considerably, and it can be a lengthy, costly and time-consuming effort to obtain approval by the relevant VAC. For example, even if we have an agreement with a hospital system for the purchase of our devices, in most cases, we must obtain VAC approval by each hospital within the system to sell at that particular hospital. Additionally, hospitals typically require separate VAC approval for each specialty in which our device is used, which may result in multiple VAC approval processes within the same hospital even if such device has already been approved for use by a different specialty group. We often need VAC approval for each different device to be used by surgeons in each discrete
22
specialty. In addition, hospital facilities and group purchasing organizations, or GPOs, which manage purchasing for multiple facilities, may also require us to enter into a purchasing agreement and satisfy numerous elements of their administrative procurement process, which can also be a lengthy, costly, and time-consuming effort. If we do not receive access to hospital facilities in a timely manner, or at all, via these VAC and purchasing contract processes, or otherwise, or if we are unable to secure contracts in a timely manner, or at all, our operating costs will increase, our sales may decrease, and our operating results may be harmed. Furthermore, we may expend significant effort and still be unable to obtain VAC approval or a purchase contract from hospitals or GPOs.
We must demonstrate to surgeons and hospitals the merits of our devices in order to facilitate greater adoption of our devices.
Surgeons play a significant role in determining the devices used in the operating room and assisting in obtaining approval by the relevant VAC. Educating surgeons on the benefits of our devices requires a significant commitment by our marketing team and sales organization. Surgeons and hospitals may be slow to change their practices because of perceived risks arising from the use of new devices, lack of experience using new devices, lack of clinical data supporting the benefits of such devices or the cost of new devices. We cannot predict when, or if ever, there will be widespread adoption of our devices by surgeons and hospitals. If we are unable to educate surgeons and hospitals about the advantages of our devices as compared to alternatives, we may face challenges in obtaining approval by the relevant VAC, and we will not achieve significantly greater market acceptance of our devices, gain momentum in our sales activities, significantly grow our market share or grow our revenue, and our business and financial condition will be adversely affected.
If we fail to develop and retain our direct sales force and independent sales agents, our business could suffer.
We currently sell our devices through our direct sales representatives in the United States. Our direct sales force works with independent sales agents or agencies, who assist us in educating targeted surgeons. Our operating results are dependent upon the sales and marketing efforts of our direct sales representatives. If our direct sales force fails to adequately promote, market and sell our devices, our sales may suffer.
As we launch new devices and increase our current marketing efforts with respect to existing devices and expand into new geographies, our future success will depend largely on our ability to continue to hire, train, retain and motivate skilled sales personnel with significant technical knowledge of our devices. We have made, and intend to continue to make, a significant investment in recruiting and training sales representatives. There is significant competition for sales personnel who are experienced in relevant medical device sales. Once hired, the training process is lengthy because of the significant education required to achieve the level of competency that surgeons expect from sales representatives with respect to understanding our devices. Upon completion of the training, our sales representatives typically require lead time in the field to grow their network of accounts and achieve the productivity levels we expect them to reach. If we are unable to attract, motivate, develop and retain a sufficient number of qualified sales personnel, or if our sales representatives do not achieve the productivity levels we expect them to reach, our revenue will not grow at the rate we expect and our financial performance will suffer. Also, to the extent we hire personnel from our competitors, we may have to wait until applicable non-competition provisions expire before deploying such personnel in restricted territories, or else incur costs to relocate personnel outside of such territories, and we may be subject to allegations that such new hires have been improperly solicited, or that they have divulged to us proprietary or other confidential information.
We operate in a highly competitive market segment. If our competitors are better able to market and develop devices than we are able to market or develop devices, our business will be adversely impacted.
The medical device industry is highly competitive. Any device we develop will have to compete for market acceptance and market share. We believe that the primary competitive factors in the surgical illumination and visualization, advanced energy and fluorescent imaging market segments are clinical safety and effectiveness, price, surgeon experience and comfort with use of particular illumination systems, reliability and durability, ease of use, device support and service, sales force experience and relationships. We face significant competition from competitors based in the United States and internationally in these market segments, and we expect the intensity of competition will increase over time. Many of the companies developing or marketing competing products enjoy several competitive advantages over us, including:
|
· |
more established sales and marketing programs and distribution networks; |
23
|
· |
long established relationships with surgeons and hospitals; |
|
· |
contractual relationships with customers; |
|
· |
products that have already received approval from the relevant VACs; |
|
· |
greater financial and human resources for product development, sales and marketing; |
|
· |
greater name recognition; |
|
· |
the ability to offer rebates or bundle multiple product offerings to offer greater discounts or incentives; and |
|
· |
greater experience in and resources for conducting research and development, clinical studies, manufacturing, preparing regulatory submissions, obtaining regulatory clearance or approval for products and marketing approved products. |
Our competitors may develop and patent processes or devices earlier than us, obtain regulatory clearance or approvals for competing devices more rapidly than us or develop more effective or less expensive devices or technologies that render our technology or devices obsolete or less competitive. We also face fierce competition in recruiting and retaining qualified sales, scientific, and management personnel. If our competitors are more successful than us in these matters, our business may be harmed.
Our ability to sell our devices at prices necessary to support our current business strategies depends on demonstrating that the benefits of devices incorporating our technology outweigh the increased cost of such devices compared to alternatives.
Hospital and other healthcare provider customers that purchase our devices typically bill various third-party payors to cover all or a portion of the costs and fees associated with the surgical procedures in which our devices are used and bill patients for any deductibles or copayments. Supplies used in surgery, such as our devices, are typically not separately reimbursed by third-party payors, but are rather included in the overall reimbursement for the procedure involved. Because there is no separate reimbursement for medical devices and supplies used in surgical procedures, the additional cost associated with the use of our devices can impact the profit margin of the hospital or surgery center where the surgery is performed. If reimbursement is inadequate, hospitals may choose to use less expensive instruments. Some of our target customers may be unwilling to adopt our devices in light of the additional associated cost. Our success depends on our ability to convince such cost-restricted customers that the potential benefits of using our devices, such as reduced surgery time, reduced surgery blood transfusion, and reduced post-surgery complications, outweigh the additional cost of such devices.
It is difficult to forecast future performance and our financial results may vary from forecasts and may fluctuate from quarter to quarter.
Our limited operating history and commercial experience make it difficult for us to predict future performance and growth as such forecasts are limited and subject to a number of uncertainties, including our ability to market our devices successfully, our ability to maintain or obtain regulatory clearances, unexpected or serious complications related to our devices or other factors discussed in these risk factors. A number of factors over which we have limited control may contribute to fluctuations in our financial results. These factors include, without limitation:
|
· |
surgeon and hospital acceptance of our devices; |
|
· |
the productivity of our sales representatives; |
|
· |
the introduction of new devices and technologies or acquisitions by us or our competitors; |
|
· |
fluctuations in our expenses associated with expanding our operations and operating as a public company; |
24
|
· |
the timing, expense and results of research and development activities and obtaining future regulatory clearances and approvals; |
|
· |
supplier, manufacturing or quality problems with our devices; |
|
· |
the impact and timing of taxes or changes in tax law; and |
|
· |
changes in our pricing policies or in the pricing policies of our competitors or suppliers. |
Additionally, we may experience seasonal variations in revenue. For example, our revenue tends to be the lowest in the first quarter as the result of the resetting of annual patient healthcare insurance plan deductibles and by hospitals and military facilities working off their inventories of products purchased in the fourth quarter. Revenue in the third quarter can be impacted by summer vacation season. Revenue in the fourth quarter tends to be the highest as demand may be impacted by the desire of patients to spend their remaining balances in their flexible spending accounts or because they have met their annual deductibles under their health insurance plans. In addition, in the fourth quarter, our results can be impacted by the budgeting and buying patterns of hospitals and military facilities.
The loss of one or more of our key customers could slow our revenue growth or cause our revenue to decline.
A material portion of our total revenue in any given period may come from a relatively small number of customers. We do not expect sales to these customers to increase significantly in the future, and as our revenue increases, we expect sales to these customers to decrease as a percent of revenue. However, the loss of any of our key customers for any reason, or a change in our relationship with any of our key customers may cause a significant decrease in our total revenue.
Our manufacturing operations are dependent upon third-party suppliers, making us vulnerable to supply problems and price fluctuations, which could harm our business.
We rely on a number of suppliers who manufacture certain components of our devices, including specialty machining for our retractors and molding for Photonguides and handheld components. We do not have long-term supply agreements with most of our suppliers, and, in many cases, we purchase components on a purchase order basis. Our suppliers may encounter problems during manufacturing for a variety of reasons, including failure to follow specific protocols and procedures, failure to comply with applicable regulations, equipment malfunction and environmental factors, any of which could delay or impede their ability to meet our demand. Our reliance on these third-party suppliers also subjects us to other risks that could harm our business, including:
|
· |
we are not a major customer of many of our suppliers, and these suppliers may therefore give other customers’ needs higher priority than ours; |
|
· |
we may not be able to obtain an adequate supply in a timely manner or on commercially reasonable terms; |
|
· |
price fluctuations due to a lack of long-term supply arrangements with our suppliers for components; |
|
· |
our suppliers, especially new suppliers, may make errors in manufacturing that could negatively affect the efficacy or safety of our devices or cause delays in shipment; |
|
· |
we may have difficulty locating and qualifying alternative suppliers; |
|
· |
switching components or suppliers may require device redesign and possibly new premarket submissions to the FDA; |
|
· |
the failure of our suppliers to comply with strictly enforced regulatory requirements, which could result in disruption of supply and/or increased expenses; |
|
· |
the occurrence of a fire, natural disaster or other catastrophe impacting one or more of our suppliers may affect the supplier’s ability to deliver components to us in a timely manner; |
25
|
· |
additional political, business, logistical and regulatory risks affecting our relationships with international suppliers in China and South Korea; |
|
· |
our suppliers may encounter financial hardships unrelated to our demand, which could inhibit their ability to fulfill our orders and meet our requirements; and |
|
· |
we may encounter undesirable issues, such as the inability to exercise the same degree of quality control as we can our own facilities, with our overseas suppliers that may force us to find another supplier, which would create a delay in service. |
In addition, we rely on single- and limited-source suppliers for several of our components and sub-assemblies. For example, the optical molding for Photonguides is provided by one supplier. These components are critical to our devices and there are relatively few alternative sources of supply. We do not carry a significant inventory of these components. Identifying and qualifying additional or replacement suppliers for any of these components or sub-assemblies used in our devices could involve significant time and cost.
Although we could temporarily assemble some of these components internally, we may incur greater costs, delay production or divert attention from other critical projects until we find an alternate source. Any interruption or delay in obtaining components from our third-party suppliers, or our inability to obtain components from qualified alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to switch to competing devices.
We are required to maintain high levels of inventory, which could consume a significant amount of our resources and reduce our cash flows.
We need to maintain substantial levels of inventory to protect ourselves from supply interruptions. In addition, because of the broad choice of devices we offer the many surgeon specialists who use our devices, we must maintain sufficient inventory on hand to ensure each order is filled when received, and we provide our sales representatives with trunk stock inventory to allow them to demonstrate the breath of our offering. As a result of our substantial inventory levels, we are subject to the risk that a substantial portion of our inventory could become obsolete, which could have a material adverse effect on our earnings and cash flows due to the resulting costs associated with the inventory impairment charges and costs required to replace such inventory. For example, we introduced our Eikon LT retractor series at the beginning of 2016 which quickly displaced its predecessor the Eikon Classic retractor series. As a result, we recorded a 100% reserve against finished good inventory on hand and trunk stock inventory held by our sales representatives for the Eikon Classic retractor series for the year ended December 31, 2016. In the year ended December 31, 2017, we recorded a charge of $456,000 related to on hand and trunk stock inventory that was obsolete due to a design modification to PhotonBlade®. We may need to write off inventory for other reasons as well.
We have limited clinical data to support the clinical and cost benefits of use of our devices, which could be a barrier to further surgeon adoption of our devices
For FDA purposes, our devices are classified as Class I, Class II exempt or Class II devices. Class I and Class II exempt devices do not require a 510(k) premarket notification. Our Class II devices, which require a 510(k) premarket notification, do not require clinical data or completion of clinical studies to obtain clearance for marketing. As a result, the FDA has not required, and we have not developed, clinical data supporting the cost effectiveness of our devices. Therefore, we currently lack clinical data supporting the benefits and cost effectiveness of our devices compared to other surgical solutions. As a result, surgeons may be slow to adopt or recommend our devices, and we may encounter difficulty obtaining approval from VACs. Further, any clinical studies that we initiate or the clinical experience of surgeons may indicate that our devices do not provide cost advantages over our competitors’ surgical devices or that our devices do not deliver sufficient benefits to justify their cost. Such results could slow the adoption of our devices and significantly reduce our sales, which could harm our business and reputation.
We may need to conduct clinical studies in the future to support new device regulatory clearances or approvals, gain acceptance of our products in hospitals or to secure approval of the use of our devices in some foreign countries. Clinical testing is time-consuming and expensive and carries uncertain outcomes. The initiation and completion of any of these studies may be prevented, delayed or halted for numerous reasons. Moreover, we cannot assure you that the results of any clinical trials would support the promoted benefits of our devices. Failure or perceived failures in any clinical trials
26
will delay and may prevent our device development and regulatory clearance or approval processes, damage our business prospects and negatively affect our reputation and competitive position.
Our long-term growth depends on our ability to develop and commercialize additional devices.
The medical device industry is highly competitive and subject to rapid change and technological advancements. Therefore, it is important to our business that we continue to enhance our device offerings and introduce new devices. Developing new devices is expensive and time-consuming and could divert management’s attention away from our core business. Even if we are successful in developing additional devices, the success of any new device offering or enhancements to existing devices will depend on several factors, including our ability to:
|
· |
properly identify and anticipate surgeon and patient needs; |
|
· |
develop and introduce new devices or device enhancements in a timely manner; |
|
· |
develop an effective and dedicated sales and marketing team; |
|
· |
avoid infringing upon the intellectual property rights of third-parties by developing our own intellectual property to protect our inventions; |
|
· |
demonstrate, if required, the safety and efficacy of new devices with data from preclinical studies and clinical trials; |
|
· |
obtain the necessary regulatory clearances or approvals for new devices or device enhancements; |
|
· |
comply with FDA regulations regarding the advertising and promotion of new devices or modified devices; |
|
· |
provide adequate training to potential users of our devices; and |
|
· |
receive adequate coverage and reimbursement for procedures performed with our devices. |
If we are unsuccessful in developing and commercializing additional devices in other areas, our ability to increase our revenue may be impaired.
We may face product liability claims that could result in costly litigation and significant liabilities, and we may not be able to maintain adequate product liability insurance.
Our business exposes us to the risk of product liability claims that are inherent in the testing, manufacturing and marketing of medical devices. This risk exists even if a device is cleared or approved for commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority. Manufacturing and marketing of our commercial devices, and clinical testing of our devices under development, may expose us to product liability and other tort claims. Additionally, regardless of the merit or eventual outcome, product liability claims may result in:
|
· |
litigation costs; |
|
· |
distraction of management’s attention from our primary business; |
|
· |
impairment of our business reputation; |
|
· |
the inability to commercialize our devices; |
|
· |
decreased demand for our devices or devices in development, if cleared or approved; |
|
· |
device recall or withdrawal from the market; |
27
|
· |
withdrawal of clinical trial participants; |
|
· |
substantial monetary awards to patients or other claimants; or |
|
· |
loss of revenue. |
Although we have, and intend to maintain, liability insurance, the coverage limits of our insurance policies may not be adequate, and one or more successful claims brought against us may have a material adverse effect on our business and results of operations. If we are unable to obtain insurance in the future at an acceptable cost or on acceptable terms with adequate coverage, we will be exposed to significant liabilities.
Adverse litigation or settlements resulting from legal proceedings in which we may be involved in the normal course of our business could negatively impact our business, financial condition and results of operations.
From time to time, we are subject to allegations, claims and legal actions arising in the ordinary course of our business, which may include claims by third parties, including customers, competitors, employees or regulators. In this regard, we are currently subject to a securities litigation. In February 2017, a purported stockholder class action was filed against us, alleging that we made false or misleading statements about our business to investors who purchased our common stock between July 19, 2016 and November 3, 2016 in violation of Sections 10(b) and 20(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and the SEC Rule 10b‑5. This securities litigation is discussed in greater detail under Item 3, “Legal Proceedings” and Note 7, “Commitments and Contingencies” to our financial statements included elsewhere in this Annual Report on Form 10‑K.
Additionally, one of our employees has received a letter from Medtronic PLC, or Medtronic, indicating a concern about “inevitable disclosure” of trade secrets, which did not relate to specific accusations of actual misconduct. We may be subject to litigation as a result of this letter or others that we or our employees may receive in the future. In December 2016, Medtronic, which competes with us across multiple product lines, filed a civil complaint in the State Court of Minnesota against us, alleging tortious interference with contract, and two of our sales employees, alleging breach of contract, misappropriation of trade secrets and breach of duty of loyalty. The complaint seeks unspecified monetary damages. Additionally, in connection with the civil complaint, a temporary restraining order was filed against us and the two named employees to stop the employees from selling PhotonBlade®. In January 2017, the State Court of Minnesota entered into a temporary restraining order against us, relating to sales by the two employees of PhotonBlade®. A temporary injunction hearing was held in May 2017, and the court entered an order in July 2017, granting in part and denying in part the motion for temporary injunction. This lawsuit is scheduled for trial in April 2018. We are incurring and expect to continue incurring significant costs related to defending this action.
The outcome of these and other proceedings cannot be predicted. We intend to defend any proceedings vigorously. If any proceedings were to be determined adversely against us or resulted in legal actions, claims, regulatory proceedings, enforcement actions, or judgments, fines, or settlements involving a payment of material sums of money, or if injunctive relief were issued against us, our business, financial condition and results of operations could be materially adversely affected. Even the successful defense of legal proceedings may cause us to incur substantial legal costs and may divert management’s attention and resources.
Our ability to maintain our competitive position depends on our ability to attract, integrate and retain highly qualified personnel.
We believe that our continued success depends to a significant extent upon the efforts and abilities of our executive officers and other key personnel. Our executive officers and other key personnel are critical to the strategic direction and overall management of our company as well as our research and development process. All of our executive officers and other employees are at-will employees and, therefore, may terminate employment with us at any time with no advance notice. The loss of any of our executive officers and other key personnel could adversely affect our business, financial condition, and operating results. Our productivity may be adversely affected if we do not integrate and train our new employees quickly and effectively.
We invest significant time and expense in training our employees, which increases their value to competitors who may seek to recruit them. Many of our competitors have greater resources than we have. We do not carry any “key person”
28
insurance policies. The replacement of any of our key personnel likely would involve significant time and costs and may significantly delay or prevent the achievement of our business objectives and would harm our business.
In addition, our employees may be more likely to leave us if the shares they own or the shares underlying their vested options have significantly appreciated in value relative to the original purchase prices of the shares or the exercise prices of the options, or if the exercise prices of the options that they hold are significantly below the market price of our common stock.
On February 27, 2018, Philip Sawyer informed the board of directors that he will resign from his role as President, Chief Executive Officer, and a member of our board of directors, effective as of February 28, 2018. Scott Flora, currently serving as a member of our board of directors, assumed the additional role of Interim President and Chief Executive Officer, and as such became the Company’s principal executive officer effective on March 1, 2018.
The board of directors will commence a search to recruit a permanent successor with the assistance of a leading executive search firm. These changes in the Company’s management team and to the board of directors, may be disruptive to, or cause uncertainty in, the Company’s business, and any additional changes to the management team or the board of directors could have a negative impact on the Company’s ability to manage and grow its business effectively. In addition, if the Company is not effective in its succession planning, it may have a negative impact on the Company’s ability to successfully recruit for its management team. Any such disruption or uncertainty or difficulty in efficiently and effectively filling key management roles could have a material adverse impact on the Company’s business, results of operations and/or the price of the Company’s common stock.
If we fail to properly manage our anticipated growth, our business could suffer.
We have been growing in recent periods and have a relatively short history of operating as a commercial company. We intend to continue to grow and may experience periods of rapid growth and expansion. Future growth will impose significant added responsibilities on management, including the need to identify, recruit, train and integrate additional employees. In addition, rapid and significant growth will place a strain on our administrative personnel, information technology systems and other operational infrastructure. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals. To achieve our revenue goals, we must continue to hire, train, retain and motivate skilled personnel.
In order to manage our operations and growth, we will need to continue to improve our operational and management controls, reporting and information technology systems and financial internal control procedures. If we are unable to manage our growth effectively, it may be difficult for us to execute our business strategy and our operating results and business could suffer.
We must also successfully increase production output to meet expected customer demand. In the future, we may experience difficulties with production yields and quality control, component supply, and shortages of qualified personnel, among other problems. These problems could result in delays in product availability and increases in expenses. Any such delay or increased expense could adversely affect our ability to generate revenues.
Our ability to achieve profitability will depend, in part, on our ability to reduce the per unit manufacturing cost of our current devices and attain a low per unit manufacturing cost for our future devices.
Currently, the gross profit generated from the sale of our devices is not sufficient to cover our operating expenses. To achieve profitability, we need to, among other things, reduce the per unit manufacturing cost of our current devices and attain low per unit manufacturing costs for our future devices, including the PhotonBlade®. This cannot be achieved without improving manufacturing efficiency and increasing our manufacturing volume to leverage manufacturing overhead costs. If we are unable to improve manufacturing efficiency and reduce manufacturing overhead costs per unit, our ability to achieve profitability will be constrained. Any increase in manufacturing volumes is dependent upon a corresponding increase in sales. The occurrence of one or more factors that negatively impact the manufacturing or sales of our devices or reduce our manufacturing efficiency may prevent us from achieving our desired decrease in manufacturing costs, which would prevent us from attaining profitability.
29
If our facilities are damaged or become inoperable, we will be unable to continue to research, develop and manufacture our devices and, as a result, there will be an adverse impact on our business until we are able to secure a new facility.
We do all of our internal manufacturing, development and management activities in a single location in San Francisco, California. Our facility and equipment would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including, but not limited to, earthquakes, flooding, fire, vandalism and power outages, which may render it difficult or impossible for us to perform our research, development, manufacturing and commercialization activities for some period of time. While we have taken precautions to safeguard our facilities, including through insurance and health and safety protocols, the inability to perform those activities may result in the inability to continue manufacturing our devices during such periods and the loss of customers or harm to our reputation. We also possess insurance for damage to our property and the disruption of our business, but this insurance may not be sufficient to cover all of our potential losses and this insurance may not continue to be available to us on acceptable terms, or at all.
We have limited prior experience selling devices outside of the United States. If we commercialize any devices outside of the United States, a variety of risks associated with international operations could adversely impact our net sales, results of operations and financial condition.
We currently sell our devices in the United States and began selling in Asia in June 2017. We also expect to expand sales to Europe and other regions, both directly and through distributors, which will require us to identify and develop relationships with distributors who will focus on marketing our devices.
The sale and shipment of our devices across international borders, as well as the purchase of components from international sources, such as from suppliers in China and South Korea, subject us to United States and foreign governmental trade, import and export, and customs regulations and laws. Compliance with these regulations and laws is costly and exposes us to penalties for non-compliance. Other laws and regulations that can significantly impact us include various anti-bribery laws, including the FCPA and anti-boycott laws, as well as export controls laws. Any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, seizure of shipments, restrictions on certain business activities and exclusion or debarment from government contracting. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our shipping and sales activities.
Additionally, the countries into which we expand our sales in the future may have different practices than the United States regarding the use of disposable medical devices. In the United States, our single-use optical waveguides, called Photonguides, for use with reusable retractors, single-use handheld illuminated aspiration devices, PhotonBlade®, and single-use drop-in intracavity illuminators are not reused, whereas surgeons in some countries may reuse our single-use devices. Customers in these countries may be less willing to purchase our single-use devices as they were not designed to be reusable, or they may purchase fewer of our single-use devices than United States-based customers purchase, because they choose to reuse our devices rather than purchasing additional single-use devices from us.
International operations will expose us and our distributors to risks inherent in operating in foreign jurisdictions. These risks include:
|
· |
difficulties in obtaining, enforcing or defending intellectual property rights; |
|
· |
pricing pressure that we may experience internationally; |
|
· |
a shortage of high-quality sales people and distributors; |
|
· |
third-party reimbursement policies that may require some of the patients who receive our devices to directly absorb medical costs or that may necessitate the reduction of the selling prices of our devices; |
|
· |
competitive disadvantage with established businesses and customer relationships; |
|
· |
the imposition of additional United States and foreign governmental controls or regulations; |
30
|
· |
economic instability; |
|
· |
changes in duties and tariffs, license obligations and other non-tariff barriers to trade; |
|
· |
the imposition of restrictions on the activities of foreign agents, representatives and distributors; |
|
· |
scrutiny of foreign tax authorities which could result in significant fines, penalties and additional taxes being imposed on us; |
|
· |
laws and business practices favoring local companies; |
|
· |
longer payment cycles; |
|
· |
foreign currency exchange rate fluctuations; |
|
· |
difficulties in maintaining consistency with our internal guidelines; |
|
· |
difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
|
· |
the imposition of costly and lengthy new export licensing requirements; |
|
· |
the imposition of U.S. or international sanctions against a country, company, person or entity with whom we do business that would restrict or prohibit continued business with the sanctioned country, company, person or entity; and |
|
· |
the imposition of new trade restrictions. |
If we experience any of these risks, our sales in international countries may be harmed and our results of operations would suffer.
Our operations involve the use of hazardous and toxic materials, and we must comply with environmental, health and safety laws and regulations, which can be expensive, and could have an adverse impact on our business.
Our operations use or generate small volumes of hazardous or toxic materials. We are, therefore, subject to a variety of federal, state and local regulations relating to the use, handling, storage, disposal and human exposure to hazardous and toxic materials. Liability under environmental laws can be joint and several and without regard to comparative fault, and environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations, which could have an adverse impact on our business. Although we believe that our activities conform in all material respects with environmental, health and safety laws, there can be no assurance that violations of environmental, health and safety laws will not occur in the future as a result of human error, accident, equipment failure or other causes. The failure to comply with past, present or future laws could result in the imposition of fines, third-party property damage and personal injury claims, investigation and remediation costs, the suspension of production or a cessation of operations. We also expect that our operations will be affected by other new environmental and health and safety laws and regulations on an ongoing basis. Although we cannot predict the ultimate impact of any such new laws and regulations, they will likely result in additional costs, and may require us to change how we manufacture our devices, which could have an adverse impact on our business. We cannot predict what impact the new administration will have on the political and regulatory environment in the United States, the timing of any such changes, or the impact of any such changes on our business.
We may acquire other companies or technologies, which could divert our management’s attention, result in additional dilution to our stockholders and otherwise disrupt our operations and harm our operating results.
We may in the future seek to acquire or invest in companies or technologies that we believe could complement or expand our platform, enhance our technical capabilities or otherwise offer growth opportunities. The pursuit of potential acquisitions may divert the attention of management and cause us to incur various costs and expenses in identifying, investigating and pursuing suitable acquisitions, whether or not they are consummated. We may not be able to identify
31
desirable acquisition targets, be successful in entering into an agreement with any particular target, or obtain the expected benefits of any acquisition or investment.
To date, the growth in our business has been organic, and we have no experience in acquiring other businesses. In any acquisition, we may not be able to successfully integrate acquired personnel, operations and technologies, or effectively manage the combined business following the acquisition. Acquisitions could also result in dilutive issuances of equity securities, the use of our available cash or the incurrence of debt, which could harm our operating results. In addition, if an acquired company or technology fails to meet our expectations, or if we are unable to integrate any acquired company or technology, our operating results, business and financial condition may suffer.
Failure to protect our information technology infrastructure against cyber-based attacks, network security breaches, service interruptions, or data corruption could significantly disrupt our operations and adversely affect our business and operating results.
We rely on information technology and telephone networks and systems, including the Internet, to process and transmit sensitive electronic information and to manage or support a variety of business processes and activities, including sales, billing, customer service, procurement and supply chain, manufacturing, and distribution. We use enterprise information technology systems to record, process, and summarize financial information and results of operations for internal reporting purposes and to comply with regulatory financial reporting, legal, and tax requirements. Our information technology systems, some of which are managed by third-parties, may be susceptible to damage, disruptions or shutdowns due to computer viruses, attacks by computer hackers, failures during the process of upgrading or replacing software, databases or components thereof, power outages, hardware failures, telecommunication failures, user errors or catastrophic events. We are not aware of any breaches of our information technology infrastructure. Despite the precautionary measures we have taken to prevent breakdowns in our information technology and telephone systems, if our systems suffer severe damage or disruption or shutdown and we are unable to effectively resolve the issues in a timely manner, our business and operating results may suffer.
Risks Related to Our Intellectual Property
If our intellectual property rights are not adequately protected, our business will be negatively affected.
Our success depends in large part on our intellectual property rights, including patents, trademarks, trade secrets, copyrights and know-how. The steps we have taken and may take in the future to protect our intellectual property may not adequately prevent misappropriation or ensure that others will not develop competitive technologies or devices. We cannot assure you that our competitors will not successfully challenge the validity or ownership of our patents or design products that avoid infringement of our proprietary rights with respect to our technology. There can be no assurance that other companies are not investigating or developing other similar technologies, that any patents will be issued from any application pending or filed by us, or that, if patents are issued, the issued claims will be sufficiently broad to deter or prohibit others from marketing similar devices. We may also not be able to detect infringement of our patents by third parties. In addition, we cannot assure you that any patents issued to us will not be challenged, invalidated or circumvented, or that the rights under those patents will provide a competitive advantage to us or that our devices and technology will be adequately covered by our patents and other intellectual property. Additionally, as our patents expire, we may be unsuccessful in extending their protection through adjustments in patent term. The expiration of, or the failure to maintain or extend our patents, could have a material adverse effect on us.
Furthermore, we do not have any patent rights in certain foreign countries in which a market may exist in the future, and the laws of many foreign countries may not protect our intellectual property rights to the same extent as the laws of the U.S. The scope of our patent claims may vary between countries, as individual countries have distinctive patent laws. Thus, we may not be able to stop a competitor from marketing and selling in certain foreign countries devices that are the same as or similar to our devices.
We also own trade secrets and confidential information that we try to protect by entering into invention assignment and confidentiality agreements with our employees and other parties. However, these agreements may not be honored or, if breached, we may not have sufficient remedies to protect our confidential or proprietary information. Further, our competitors may independently learn our trade secrets and develop similar or superior technologies. To the extent that our consultants, key employees or others apply technological information to our projects that they develop independently or others develop, disputes may arise regarding the ownership of proprietary rights to such information, and such
32
disputes may not be resolved in our favor. If we are unable to protect our intellectual property adequately, our business and commercial prospects will suffer.
The medical device industry is characterized by extensive patent litigation, and we could become subject to patent or other proprietary rights litigation that could be costly to defend or settle, result in the diversion of management’s attention, require us to pay significant damages or royalty payments or prevent us from marketing and selling our existing or future devices, and, if adversely adjudicated, could harm our business.
Our success depends, in part, on not infringing the patents or violating the proprietary rights of others. Significant litigation regarding patent rights occurs in the medical device industry. It is possible that U.S. and foreign patents and pending patent applications controlled by third parties may be alleged to cover our devices. Our competitors in both the United States and abroad, many of which have substantially greater resources and have made substantial investments in patent portfolios and competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with our ability to make, use, sell, or offer to sell our devices. We or our employees may receive in the future and have received in the past, particularly as a public company, communications from patent holders or other third parties, including non-practicing entities, alleging infringement of patents or other intellectual property rights, actual or potential future misappropriation of trade secrets, or offering licenses to such intellectual property. In this regard, one of our employees has received a letter from Medtronic indicating a concern about “inevitable” disclosure of trade secrets, which did not relate to specific accusations of actual misconduct. We may be subject to litigation as a result of this letter or others that we or our employees may receive in the future.
In December 2016, Medtronic, which competes with us across multiple product lines, filed a civil complaint in the State Court of Minnesota against us, alleging tortious interference with contract, and two of our sales employees, alleging breach of contract, misappropriation of trade secrets and breach of duty of loyalty. The complaint seeks unspecified monetary damages. Additionally, in connection with the civil complaint, a temporary restraining order was filed against us and the two named employees to stop the employees from selling PhotonBlade®. In January 2017, the State Court of Minnesota entered into a temporary restraining order against us, relating to sales by the two employees of PhotonBlade®. A temporary injunction hearing was held in May 2017, and the court entered an order in July 2017, granting in part and denying in part the motion for temporary injunction. This lawsuit is scheduled for trial in April 2018. We are incurring and could continue to incur significant costs related to defending this action.
At any given time, we may be involved as either a plaintiff or a defendant in a number of patent infringement actions related to other proprietary rights, the outcomes of which may not be known for prolonged periods of time. Such intellectual property litigation is typically costly and time-consuming. Litigation proceedings, if instituted against us, could divert our management’s and technical team’s attention and resources. Adverse determinations in any such litigation could result in significant liabilities to third parties or injunctions, or could require us to seek licenses from third parties and, if such licenses are not available on commercially reasonable terms, prevent us from manufacturing, selling or using certain devices, any one of which could have a material adverse effect on us. In addition, some licenses may be nonexclusive, which could provide our competitors access to the same technologies. Third parties could also obtain patents that may require us to either redesign products or, if possible, negotiate licenses from such third parties. Such licenses may materially increase our expenses.
The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technologies involved and the uncertainty of litigation significantly increase the risks related to patents or other proprietary rights. Any potential intellectual property litigation also could force us to do one or more of the following:
|
· |
stop selling, offering to sell, making, or using devices that use the disputed intellectual property; |
|
· |
obtain a license from the intellectual property owner to continue selling, offering to sell, making, licensing, or using devices, which license may require substantial royalty payments and may not be available on reasonable terms, or at all; |
|
· |
incur significant legal expenses; |
|
· |
pay substantial damages, treble damages, or royalties to the party whose intellectual property rights we may be found to be infringing; |
33
|
· |
pay the attorney fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; and |
|
· |
redesign those devices that contain the allegedly infringing intellectual property, which could be costly, disruptive and/or infeasible. |
If any of the foregoing occurs, we may have to withdraw existing devices from the market or may be unable to commercialize one or more of our devices, all of which could have a material adverse effect on our business, results of operations and financial condition. Any litigation or claim against us, even those without merit, may cause us to incur substantial costs and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. Further, as the number of participants in the industry grows, the possibility of intellectual property infringement claims against us increases.
In addition, we may be required to indemnify our customers, distributors and OEM partners with respect to infringement by our devices of the proprietary rights of third parties. Third parties may assert infringement claims against our customers or distributors which may require us to initiate or defend protracted and costly litigation on behalf of our customers or distributors, regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of our customers or distributors or may be required to obtain licenses for the devices they use. If we cannot obtain all necessary licenses on commercially reasonable terms, our customers may be forced to stop using our devices.
Risks Related to Our Capital Structure
We currently are required to obtain additional funds and may also be required to obtain additional funds in the future, and these funds may not be available on acceptable terms at all.
Our operations have consumed substantial amounts of cash since inception, and we anticipate our expenses will increase as we seek to continue to grow our business and transition to operating as a public company. We believe that our growth will depend, in part, on our ability to fund our commercialization efforts and our efforts to develop new technologies, as well as technology complementary to our current devices. Our existing resources may not allow us to conduct all of these activities that we believe would be beneficial for our future growth. As a result, we intend to obtain additional funding through public or private financing, collaborative arrangements with strategic partners, or through additional credit lines or other debt financing sources to increase the funds available to fund operations. If we are unable to raise funds on favorable terms, or at all, we may not be able to support our commercialization efforts or increase our research and development activities and the growth of our business may be negatively impacted. As a result, we may be unable to compete effectively. For the year ended December 31, 2017, 2016 and 2015, our net cash used in operating activities was $38.9 million, $36.6 million, and $31.2 million, respectively. As of December 31, 2017, we had working capital of $22.7 million, which included $21.0 million in cash and cash equivalents and short-term investments. Our cash requirements in the future may be significantly different from our current estimates and depend on many factors, including:
|
· |
the results of our commercialization efforts for our existing and future devices, including international expansion; |
|
· |
the rate at which we continue to grow our direct sales force and increase our marketing activities; |
|
· |
the establishment of high volume manufacturing; |
|
· |
the need for additional capital to fund future development programs; |
|
· |
the costs involved in obtaining and enforcing patents or any litigation by third parties regarding intellectual property; and |
|
· |
our success in entering into collaborative relationships with other parties. |
34
To finance these activities, we may seek funds through borrowings or through additional rounds of financing, including private or public equity or debt offerings and collaborative arrangements with corporate partners. In July 2016, in order to facilitate the raising of additional funds, we filed a shelf registration statement on Form S‑3 (Registration No. 333‑212395) that allows us to sell up to an aggregate of $100,000,000 of our common stock, preferred stock, warrants, depository shares and/or units. In July 2016, we also filed a prospectus supplement for an at-the-market, or ATM, program of up to $25.0 million. In August 2016, we sold 3,220,000 shares in a secondary offering pursuant to this shelf registration and a prospectus supplement. If we raise additional funds by issuing equity securities, our stockholders may experience dilution. In March 2017, we also entered into a debt agreement with MidCap (as defined below), as amended in September 2017, for up to $30.0 million in term loans and up to $20.0 million under a revolving credit facility. As of December 31, 2017, we had borrowed $30.0 million in term loans and drawn down $5.9 million under our revolving credit facility. This debt arrangement imposes upon us, and any future debt financing into which we enter may impose upon us covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our common stock, make certain investments and engage in certain merger, consolidation or asset sale transactions. See below for more information. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third-parties, it may be necessary to relinquish some rights to our technologies or our devices, or grant licenses on terms that are not favorable to us.
We cannot be certain that additional funding will be available on acceptable terms, if at all. Additionally, we cannot be certain that we will have access to the full amount available under our revolving credit agreement with MidCap if we fail to achieve certain revenue thresholds or meet other conditions, or that we will be able to renegotiate the terms of our credit facility to access further funding. We may not be able to issue equity securities under our shelf registration statement or ATM program due to unacceptable terms and conditions to us in the capital markets. If we do not have, or are not able to obtain, sufficient funds, we may have to reduce our cash burn rate, delay hiring of new staff, delay research and development projects, delay development or commercialization of our devices in development, license to third parties the rights to commercialize devices or technologies that we would otherwise seek to commercialize, and reduce discretionary operating expenses. If we are unable to generate sufficient cash flows or to raise adequate funds to finance our forecasted expenditures, we may have to liquidate some or all of our assets, or delay, reduce the scope of or eliminate some or all of our development programs. We also may have to reduce sales, marketing, engineering, customer support or other resources devoted to our existing products, or cease operations. Any of these actions could impede our ability to achieve our business objectives and harm our operating results. As a result of our obligations and lack of immediately available financial resources, there is uncertainty regarding our ability to maintain liquidity sufficient to operate our business effectively, which raises substantial doubt about our ability to continue as a going concern. The report of our independent registered public accounting firm, included in this Annual Report on Form 10-K, includes a going concern explanatory paragraph stating that our recurring losses from operations and net capital deficiency raise substantial doubt about our ability to continue as a going concern.
We may not be able to generate sufficient cash to service our indebtedness, which currently consists of our credit agreements with MidCap.
On March 10, 2017, we entered into a credit and security agreement with MidCap Financial Trust and their affiliates, or MidCap, that provides for up to $30.0 million in term loans. On September 26, 2017, we amended this credit and security agreement. Under the terms of the agreement, we borrowed the first term loan of $20.0 million on March 10, 2017 and an additional term loan of $10.0 million on September 26, 2017. The term loans accrue interest at a floating rate equal to 6.50% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. Principal is payable in 36 equal monthly installments beginning April 1, 2019, subject to extension to October 1, 2019 if the Company achieves a certain revenue target, until paid in full on March 1, 2022. In connection with entry into the credit and security agreement with MidCap, we terminated the HCRP loan agreement and used a portion of the term loan proceeds to repay all amounts outstanding under such agreement.
Also on March 10, 2017, we entered into a separate credit and security agreement with MidCap that provides for a revolving credit facility of up to $10.0 million based on the eligible accounts receivable and inventory balances. On September 26, 2017, we amended this credit and security agreement. As of December 31, 2017, we have outstanding borrowings of $5.9 million under this revolving credit facility. We may increase the total commitments under the revolving credit facility by up to an additional $10.0 million, subject to certain conditions. Loans under the revolving credit facility accrue interest at a floating rate equal to 3.25% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. The facility terminates in full on March 1, 2022 unless terminated earlier. This new revolving credit facility
35
replaces our $7.5 million accounts receivable credit facility that existed with SVB, under which no principal or interest was outstanding.
Our ability to make scheduled payments or to refinance our debt obligations depends on numerous factors, including the amount of our cash balances and our actual and projected financial and operating performance. These amounts and our performance are subject to certain financial and business factors, as well as prevailing economic and competitive conditions, some of which may be beyond our control. We may be unable to maintain a level of cash balances or cash flows from operating activities sufficient to permit us to pay the principal, premium, if any, and interest on our existing or future indebtedness. If our cash flows and capital resources are insufficient to fund our debt service obligations, we may be forced to reduce or delay capital expenditures, sell assets or operations, seek additional capital or restructure or refinance our indebtedness. Our future working capital, borrowings or equity financing could be unavailable to repay or refinance the amounts outstanding under the loan agreements, and even if they were, these actions may be insufficient to permit us to meet our scheduled debt service obligations. In addition, in the event of our breach of the credit agreements with MidCap, we may not be allowed to draw additional amounts and we may be required to repay any outstanding amounts earlier than anticipated. In the event of a liquidation, MidCap would be repaid all outstanding principal, premium, if any, and interest prior to distribution of assets to unsecured creditors, and the holders of our common stock would receive a portion of any liquidation proceeds only if all of our creditors, including MidCap, were first repaid in full.
The credit agreements, as amended, with MidCap contain restrictive covenants that may limit our operating flexibility.
Borrowings under the credit agreements, as amended, with MidCap are secured by substantially all of our assets, including our intellectual property. The credit agreements, as amended, with MidCap contain customary restrictive covenants that, among other things, limit our ability to dispose of assets, undergo a change in control, merge or consolidate, make acquisitions, incur debt, incur liens, pay dividends, repurchase stock and make investments, in each case subject to certain exceptions. We, therefore, may not be able to engage in any of the foregoing transactions unless we obtain the consent of our lenders. We must also comply with a financial covenant relating to certain quarterly minimum Net Revenue requirements on a trailing twelve month basis. The minimum Net Revenue requirements on a trailing twelve month basis for 2018 is $40,488,151 for the period ending March 31, 2018, $42,248,214 for the period ending June 30, 2018, $46,574,986 for the period ending September 30, 2018, and $50,000,000 for the period ending December 31, 2018.
The operating and financial restrictions and covenants in the credit agreements, as amended, as well as any future financing agreements that we may enter into, could restrict our ability to finance our operations and to engage in, expand or otherwise pursue business activities and strategies that we or our stockholders may consider beneficial. Our ability to comply with these covenants may be affected by events beyond our control, and future breaches of any of these covenants could result in a default under the credit agreements, as amended. Future defaults, if not waived, could cause all of the outstanding indebtedness under our credit agreements, as amended, to become immediately due and payable and would permit the lenders to terminate all commitments to extend further credit and permit the agent, on behalf of the lenders, to proceed against the collateral in which we granted the agent and the lenders a security interest.
Our ability to use our net operating losses to offset future taxable income may be subject to certain limitations; in addition, we may be unable to use a substantial part of our net operating losses if we do not attain profitability in an amount necessary to offset such losses.
As of December 31, 2017, we had net operating loss, or NOLs, carryforwards for federal and state income tax purposes of approximately $171.0 million and $124.8 million, respectively. In general, under Section 382 of the Internal Revenue Code of 1986, as amended, or Section 382, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change NOLs to offset future taxable income. Our existing NOLs may be subject to limitations arising from previous ownership changes, and if we undergo an ownership change in the future, our ability to utilize NOLs could be further limited by Section 382. Future changes in our stock ownership, some of which are outside of our control, could also result in an ownership change under Section 382. Furthermore, we may be unable to use a substantial part of our NOLs if we do not attain profitability in an amount sufficient to offset such losses. Any limitation on using NOLs could result in our retaining less cash after payment of U.S. federal and state income taxes during any year in which we have taxable income than we would be entitled to retain if such NOLs were available as an offset
36
against such income for U.S. federal income and state tax reporting purposes, which could materially and adversely affect our results of operations.
The Tax Cuts and Jobs Act (the “Tax Reform Act”) was enacted on December 22, 2017 and provides for significant changes to U.S. tax law. Among other provisions, the Tax Reform Act reduces the U.S. corporate income tax rate to 21%, effective in 2018. As a result, the Company has remeasured its U.S. deferred tax assets and liabilities as of December 31, 2017 to reflect the lower rate expected to apply when these temporary differences reverse. The Company estimates that the remeasurement resulted in a decrease in deferred tax assets of $22.5 million, which was fully offset by a corresponding change to the Company’s valuation allowance. The impact will likely be subject to ongoing technical guidance and accounting interpretation, which the Company will continue to monitor and assess.
Risks Related to Government Regulation
Our business is subject to extensive governmental regulation that could make it more expensive and time consuming for us to introduce new or improved devices.
Our devices are medical devices and must comply with regulatory requirements imposed by the FDA in the United States and similar agencies in foreign jurisdictions. While our current devices are classified as Class I, Class II 510(k) exempt, or Class II medical devices in the United States and, with respect to our Class I and Class II exempt devices, are not subject to premarket clearance or approval by the FDA, these requirements could change and new devices may be subject to more extensive regulation. Premarket clearance or approval has become more stringent over time and can involve lengthy and detailed laboratory and clinical testing procedures and an extensive agency review process, and other costly and time-consuming procedures. It often takes several years to satisfy these requirements depending on the complexity and novelty of the device. We also are subject to numerous additional licensing and regulatory requirements relating to safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances.
Government regulation may impede our ability to develop and manufacture our existing and future devices. Government regulation also could delay our marketing of new devices for a considerable period of time and impose costly procedures on our activities. The FDA and other regulatory agencies may not approve or clear any of our future devices on a timely basis, if at all. Any delay in obtaining, or failure to obtain, such approvals or clearances could negatively impact our marketing of any future devices and reduce our device revenues.
Our devices remain subject to strict regulatory controls on manufacturing, advertising and promotion. We may be forced to modify or recall a device after release in response to regulatory action or unanticipated difficulties encountered in general use. Any such action could have a material adverse effect on the reputation of our devices and on our business and financial position.
Further, regulations may change, and any additional regulation could limit or restrict our ability to use any of our technologies, which could harm our business. We could also be subject to new international, federal, state or local regulations that could affect our research and development programs and harm our business in unforeseen ways. If this happens, we may have to incur significant costs to comply with such laws and regulations, which will harm our results of operations.
If we fail to obtain and maintain necessary regulatory clearances or approvals for our devices, or if clearances or approvals for future devices and indications are delayed or not issued, our commercial operations would be harmed.
Our devices are medical devices that are subject to extensive regulation by the FDA in the United States and by regulatory agencies in other countries where we plan to do business. Government regulations specific to medical devices are wide-ranging and govern, among other things:
|
· |
device design, development and manufacture; |
|
· |
laboratory, preclinical and clinical testing, labeling, packaging, storage and distribution; |
|
· |
premarketing clearance and approval; |
37
|
· |
record keeping; |
|
· |
device marketing, promotion and advertising, sales and distribution; and |
|
· |
post-marketing surveillance, including reporting of deaths and serious injuries and recalls and correction and removals. |
Before a new medical device, or a new intended use for, an existing device can be marketed in the United States, a company must first submit and receive either 510(k) clearance or premarketing approval, or PMA from the FDA, unless an exemption applies. In the 510(k) clearance process, the FDA will determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based on extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. Products that are approved through a PMA application generally need FDA approval before they can be modified. Similarly, some modifications made to products cleared through a 510(k) may require a new 510(k). Either process can be expensive, lengthy and unpredictable. We may not be able to obtain the necessary clearances or approvals or may be unduly delayed in doing so, which could harm our business. Furthermore, even if we are granted regulatory clearances or approvals, they may include significant limitations on the indicated uses for the device, which may limit the market for the device. Although we have obtained 510(k) clearance to market our sterilization trays, our clearance can be revoked if safety or efficacy problems develop.
In addition, we are required to timely file various reports with the FDA, including reports required by the medical device reporting regulations, or MDRs, that require we report to the regulatory authorities if our devices may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur. If these reports are not filed in a timely manner, regulators may impose sanctions and sales of our devices may suffer, and we may be subject to product liability or regulatory enforcement actions, all of which could harm our business. If we initiate a correction or removal for one of our devices to reduce a risk to health posed by the device, we would be required to submit a publically available Correction and Removal report to the FDA and in many cases, similar reports to other regulatory agencies. This report could be classified by the FDA as a device recall which could lead to increased scrutiny by the FDA, other international regulatory agencies and our customers regarding the quality and safety of our devices. Furthermore, the submission of these reports has been and could be used by competitors against us in competitive situations and cause customers to delay purchase decisions or cancel orders and would harm our reputation. In June 2017, we submitted a Correction and Removal report to the FDA in connection with the voluntary recall of our PhotonBlade® product, and had not received additional information from the FDA that would increase our exposure to additional risks.
The FDA and the Federal Trade Commission, or FTC, also regulate the advertising and promotion of our devices to ensure that the claims we make are consistent with our regulatory clearances, that there are adequate and reasonable data to substantiate the claims and that our promotional labeling and advertising is neither false nor misleading in any respect. If the FDA or FTC determines that any of our advertising or promotional claims are misleading, not substantiated or not permissible, we may be subject to enforcement actions, including FDA warning letters, and we may be required to revise our promotional claims and make other corrections or restitutions.
FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA or state agencies, which may include any of the following sanctions:
|
· |
adverse publicity, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
· |
repair, replacement, refunds, recall or seizure of our devices; |
|
· |
operating restrictions, partial suspension or total shutdown of production; |
|
· |
refusing our requests for 510(k) clearance or premarket approval of new devices, new intended uses or modifications to existing devices; |
38
|
· |
withdrawing 510(k) clearance or premarket approvals that have already been granted; and |
|
· |
criminal prosecution. |
If any of these events were to occur, our business and financial condition would be harmed.
The misuse of our devices may harm our image in the marketplace, result in injuries that lead to product liability lawsuits, which could be costly to our business, or result in costly investigations and FDA sanctions if we are deemed to have engaged in such promotion.
Surgeons may misuse our devices or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our devices are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Product liability claims could divert management’s attention from our core business, be expensive to defend, and result in sizable damage awards against us that may not be covered by insurance. Any of these events could significantly harm our business and results of operations and cause our stock price to decline.
Our devices may, in the future, be subject to recalls or voluntary market withdrawals that could harm our reputation, business and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized devices in the event of material deficiencies or defects in the design, manufacture or labeling of the device that could affect patient safety or in the event that a product poses an unacceptable risk to health. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious adverse health consequences or death. Further, under the FDA’s MDR regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Manufacturers may, under their own initiative, conduct a field action notice or correction to inform surgeons of changes to instructions for use or of a deficiency, or of a suspected deficiency, found in a device. A government-mandated recall or voluntary recall by us or one of our distributors could occur as a result of component failures, manufacturing errors, design or labeling defects or other issues.
Repeated product malfunctions may result in a voluntary or involuntary product recall, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner and have an adverse effect on our reputation, financial condition and operating results. For example, shortly after the limited launch of PhotonBlade® we received a small number of customer complaints which related to possible unintended energy discharge from the device with the potential to cause tissue damage to patients. In June 2017, we initiated a voluntary recall of the device. We will be required to devote significant resources to relaunch and market the PhotonBlade® and cannot be assured that these activities will generate revenue as anticipated. If our redesign of the device fails to successfully eliminate the performance issue or if our revenue grows more slowly than we expect, our business and financials will be adversely affected.
Depending on the corrective action we take to address a product’s deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new approvals or clearances for the device before we may market or distribute the corrected device. Seeking such approvals or clearances may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties, or civil or criminal fines. We may also be required to bear other costs or take other actions that may have a negative impact on our sales as well as face significant adverse publicity or regulatory consequences, which could harm our business, including our ability to market our products in the future.
Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or regulatory agency action, which could include inspection, mandatory recall or other enforcement action. Recalls, which include certain notifications and corrections as well as removals, of any of our devices, could divert managerial and financial resources and could have an adverse effect on our financial condition, harm our reputation with customers, and reduce our ability to achieve expected revenues.
39
Material modifications to our devices may require new 510(k) clearances or premarket approvals or may require us to recall or cease marketing our devices until clearances or approvals are obtained.
Material modifications to the intended use or technological characteristics of our devices will require new 510(k) clearances or premarket approvals, or require us to recall or cease marketing the modified devices until these clearances or approvals are obtained. The FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement, or clearance; however, the FDA can review a manufacturer’s decision. Any modification to an FDA-cleared device that could significantly affect its safety or efficacy or that would constitute a major change in its intended use would constitute a material modification and would require a new 510(k) clearance or possibly a premarket approval. We may not be able to obtain additional 510(k) clearances or premarket approvals for new devices or for modifications to, additional indications for, our devices in a timely fashion, or at all. Delays in obtaining required future clearances would harm our ability to introduce new or enhanced devices in a timely manner, which in turn would harm our future growth. We have made modifications to our devices in the past and will make additional modifications in the future that we believe do not or will not require additional clearances or approvals. If the FDA disagrees and requires new clearances or approvals for the modifications, we may be required to recall and to stop selling or marketing our devices as modified, which could harm our operating results and require us to redesign our platform devices. In these circumstances, we may also be subject to significant enforcement actions such as significant regulatory fines or penalties.
If we or our suppliers fail to comply with the FDA’s Quality System Regulation, our manufacturing operations could be delayed or shut down and our sales could suffer.
Our manufacturing processes and those of our third-party suppliers are required to comply with the FDA’s Quality System Regulation, or QSR, which covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our devices. We are also subject to similar state requirements and licenses. In addition, we must engage in extensive recordkeeping and reporting and must make available our manufacturing facilities and records for periodic announced and unannounced inspections by governmental agencies, including the FDA, state authorities and comparable agencies in other countries. If we fail a Quality System inspection, our operations could be disrupted and our manufacturing interrupted. Failure to take adequate and prompt corrective action in response to an adverse Quality System inspection could result in, among other things, a partial or total shut-down of our manufacturing operations, significant fines, consent decrees, injunctions, untitled letters, warning letters, injunctions, customer notifications or repair, replacement, refunds, recall, detention or seizure of our products, suspension of marketing clearances and approvals, seizures or recalls of our devices, operating restrictions, refusal to grant export approval for our products, refusing or delaying our requests for 510(k) clearance or pre-market approval of new products or modified products, withdrawing 510(k) clearances or pre-market approvals that have already been granted, and criminal prosecutions, any of which would cause our business to suffer. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements, which may result in manufacturing delays for our devices and cause revenues to decline.
We have registered with the FDA as a medical device manufacturer and have obtained a manufacturing license from the California Department of Public Health, or CDPH. The FDA has broad post-market and regulatory enforcement powers. We are subject to announced and unannounced inspections by FDA and the Food and Drug Branch of CDPH to determine our compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of our suppliers. We passed the most recent audit by the Food and Drug Branch of CDPH in February 2015, and the inspection revealed no minor or major issues. We were also subjected to an FDA inspection in April 2016 resulting in two observations. The FDA will evaluate and validate whether our corrective actions have been effective during its next inspection, which has not be scheduled. However, we cannot assure you that we will pass future inspections or audits by the FDA or other regulatory bodies or that the FDA will consider the observations it previously identified to be closed out.
40
We may be subject to federal, state and foreign healthcare laws and regulations, and a finding of failure to comply with such laws and regulations could have a material adverse effect on our business.
Our operations are, and will continue to be, directly and indirectly affected by various federal, state or foreign healthcare laws, including, but not limited to, those described below. These laws include:
|
· |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the Anti-Kickback Statute or specific intent to violate it to have committed a violation; in addition, the government may assert that a claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
|
· |
federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other federal third-party payors that are false or fraudulent. Lawsuits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals, commonly known as “whistleblowers”, may share in any amounts paid by the entity to the government in fines or settlement. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim; |
|
· |
the federal Civil Monetary Penalties Law, which prohibits, among other things, offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier; |
|
· |
federal criminal laws that prohibit executing a scheme to defraud any federal healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
|
· |
the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; |
|
· |
the federal Physician Payment Sunshine Act, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the Centers for Medicare and Medicaid Services, or CMS, information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and requires applicable manufacturers and group purchasing organizations to report annually to the government ownership and investment interests held by the physicians described above and their immediate family members and payments or other “transfers of value” to such physician owners. Manufacturers are required to submit reports to CMS by the 90th day of each calendar year. Failure to submit the required information may result in civil monetary penalties up to an aggregate of $165,786 per year (and up to an aggregate of $1.105 million per year for “knowing failures”) for all payments, transfers of value or ownership or investment interests not reported in an annual submission, and may result in liability under other federal laws or regulations; |
|
· |
the U.S. Foreign Corrupt Practices Act, or the FCPA, which prohibits corporations and individuals from paying, offering to pay or authorizing the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity; the United Kingdom Bribery Act, which prohibits both domestic and international bribery, as well as bribery across both public and private sectors; |
41
and bribery provisions contained in the German Criminal Code, which, pursuant to draft legislation being prepared by the German government, may make the corruption and corruptibility of physicians in private practice and other healthcare professionals a criminal offense; and |
|
· |
analogous state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require device companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or that otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under such laws, it is possible that some of our business activities, including our consulting agreements and other relationships with surgeons and other healthcare providers, some of whom receive stock or stock options as compensation for their services and/or recommend, purchase and/or use our devices, group purchasing organizations and our independent sales agents and distributors, could be subject to challenge under one or more of such laws. We are also exposed to the risk that our employees, independent contractors, principal investigators, consultants, vendors, independent sales agents and distributors may engage in fraudulent or other illegal activity. While we have policies and procedures in place prohibiting such activity, misconduct by these parties could include, among other infractions or violations, intentional, reckless and/or negligent conduct or unauthorized activity that violates FDA regulations, including those laws that require the reporting of true, complete and accurate information to the FDA, manufacturing standards, federal and state healthcare fraud and abuse laws and regulations, laws that require the true, complete and accurate reporting of financial information or data or other commercial or regulatory laws or requirements. It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us now or in the future, we may be subject to penalties, including civil and criminal penalties, damages, fines, disgorgement, exclusion from governmental health care programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
We may fail to obtain foreign regulatory approvals to market our devices in other countries.
In June 2017, we expanded sales to Asia, and we expect to expand sales to Europe. Prior to such date, we did not have any direct sales outside of the United States. Our corporate partners, however, sell certain of our devices outside of the United States and have already obtained the necessary regulatory approvals to sell certain of our devices outside of the United States. Sales of our devices outside the United States are subject to foreign regulatory requirements that vary widely from country to country. In addition, the FDA regulates the exports of medical devices from the United States. Complying with international regulatory requirements can be an expensive and a time-consuming process and clearance or approval is not certain. The time required to obtain clearances or approvals, if required by other countries, may be longer than that required for FDA clearances or approvals, and requirements for such clearances or approvals may significantly differ from FDA requirements. In certain countries we may rely upon third-party or third-party distributors to obtain all required regulatory clearances or approvals, and these distributors may be unable to obtain or maintain such clearances or approvals. Our distributors in these countries may also incur significant costs in attempting to obtain and in maintaining foreign regulatory approvals or qualifications, which could increase the difficulty of attracting and retaining qualified distributors. If these distributors experience delays in receiving necessary qualifications, clearances or approvals to market our devices outside the United States, or if they fail to receive those qualifications, clearances or approvals, we may be unable to market our devices in certain international markets effectively, or at all, which will adversely affect our results of operations and financial condition generally.
42
Healthcare policy changes, including recent federal legislation to reform the U.S. healthcare system, may have a material adverse effect on us.
Our operations are impacted by the PPACA. Compliance with the ACA has imposed significant administrative and financial burdens on us. For example, the PPACA imposed, among other things, a 2.3% federal excise tax, with limited exceptions, on any entity that manufactures or imports Class I, II and III medical devices offered for sale in the United States that began on January 1, 2013. Through a series of legislative amendments, the tax was suspended for 2016 through 2019. Absent further legislative action, the device excise tax will be reinstated on medical device sales starting January 1, 2020. The current administration have expressed an intention to repeal the ACA and replace it with alternative reforms. For example, the Tax Cuts and Jobs Acts was enacted, which, among other things, removes penalties for not complying with the individual mandate to carry health insurance. The details and timing of any further such actions are unknown at this time. However, it is possible that these changes could adversely affect our business.
On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and, due to subsequent legislative amendments to the statute, will remain in effect through 2025 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012, or the ATRA, came into effect, which, among other things, further reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare devices and services, which could result in reduced demand for our devices or additional pricing pressures.
We face uncertainties that might result from modification or repeal of any of the provisions of the PPACA, including as a result of current and future executive orders and legislative actions. The impact of those changes on us and potential effect on the medical device industry as a whole is currently unknown. But, any changes to the PPACA are likely to have an impact on our results of operations, and may have a material adverse effect on our results of operations. We cannot predict what other healthcare programs and regulations will ultimately be implemented at the federal or state level or the effect of any future legislation or regulation in the United States may have on our business.
Risks Related to our Common Stock
We expect that the price of our common stock will fluctuate substantially.
The market price of our common stock is likely to be highly volatile and may fluctuate substantially in response to, among other things, the risk factors described in this Annual Report on Form 10‑K and other factors, many of which are beyond our control, including:
|
· |
variance in our financial performance from the financial projects we may provide to the public, any changes in these projections or our failure to meet these projections; |
|
· |
changes in analysts’ estimates, investors’ perceptions, recommendations by securities analysts or our failure to achieve analysts’ estimates; |
|
· |
announcements of significant new devices or device enhancements by us or our competitors; |
|
· |
actual or anticipated quarterly variations in our or our competitors’ results of operations; |
|
· |
changes in operating performance and stock market valuations of other technology companies generally, or those in the medical device industry in particular; |
|
· |
changes in our pricing policies or the pricing policies of our competitors; |
43
|
· |
legislation or regulatory policies, practices or actions affecting our business; |
|
· |
lawsuits threatened or filed against us; |
|
· |
the sale of our common stock or other securities in the future by us or our stockholders, including upon expiration of market standoff or contractual lock-up agreements; |
|
· |
developments or disputes concerning our intellectual property or other proprietary rights; |
|
· |
announcements related to patents issued to us or our competitors and to litigation; |
|
· |
recruitment or departure of key personnel, including changes in our board of directors and management; |
|
· |
changes in market valuation or earnings of our competitors; |
|
· |
the trading volume of our common stock; |
|
· |
changes in the estimation of the future size and growth rate of our markets; |
|
· |
general market conditions and other factors unrelated to our operating performance or the operating performance of our competitors; and |
|
· |
developments in our industry. |
In addition, the market prices of the stock of many new issuers in the medical device industry and of other companies with smaller market capitalizations like us have been volatile and from time to time have experienced significant share price and trading volume changes unrelated or disproportionate to the operating performance of those companies. As a result, stockholders have filed securities class action litigation following periods of market volatility. Any securities litigation, it could subject us to substantial costs, divert resources and the attention of management from our business, and adversely affect our business, results of operations, financial condition, reputation and cash flows. See Part I, Item 3, “Legal Proceedings” and Note 7, “Commitments and Contingencies” to our financial statements included elsewhere in this Annual Report on Form 10-K for more information. These factors may materially and adversely affect the market price of our common stock.
A substantial number of additional shares may be sold into the public market in the near future, which may cause the market price of our common stock to decline significantly, even if our business is doing well.
Sales of a substantial amount of common stock in the public market, or the perception that these sales may occur, could adversely affect the market price of our common stock. As of December 31, 2017, we have 17,179,258 shares of common stock outstanding. This includes the 4,600,000 shares of our common stock we sold in our IPO in June 2015 and 3,220,000 shares of our common stock we sold in a secondary offering in August 2016, which may be resold in the public market immediately. In July 2016, in order to facilitate the raising of additional funds, we filed a shelf registration statement on Form S‑3 (Registration No. 333‑212395) that allows us to sell up to an aggregate of $100,000,000 of our common stock, preferred stock, warrants, depository shares and/or units. The shares we sold in a secondary offering in August 2016 were sold pursuant to this shelf registration and a prospectus supplement.
A significant portion of the holders of our common stock and warrants have the right, subject to some conditions, to require us to file registration statements under the Securities Act of 1933, as amended, or the Securities Act covering their shares or to include their shares in registration statements that we may file for ourselves or our stockholders pursuant to a stockholders agreement between such holders and us. If such holders, by exercising their registration rights, sell a large number of shares, they could adversely affect the market price for our common stock. If we file a registration statement for the purpose of selling additional shares to raise capital and are required to include shares held by these holders pursuant to the exercise of their registration rights, our ability to raise capital may be impaired. These registration rights terminate in June 2022.
We filed a registration statement under the Securities Act to register all shares subject to options outstanding or reserved for future issuance under our equity incentive plans. Our 2015 Equity Incentive Plan provides for annual automatic increases in the shares reserved for issuance under the plan without stockholder approval, which would result in
44
additional dilution to our stockholders. These shares can be freely sold in the public market upon issuance and vesting, subject to any applicable lock-up period or other restrictions provided under the terms of the applicable plan and/or the option agreements entered into with option holders.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
As of December 31, 2017, our directors and executive officers and stockholders holding more than 5% of our capital stock, and their affiliates, own a significant portion of our outstanding common stock. To the extent our existing stockholders purchase additional shares, this ownership concentration would increase. As a result, if these stockholders were to choose to act together, they would exercise significant influence over most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions, such as a merger or other sale of our company or our assets. This concentration of ownership could limit your ability to influence corporate matters and may have the effect of delaying or preventing a third party from acquiring control over us.
If securities or industry analysts do not publish research or reports about our business, or if they issue a negative opinion regarding our common stock, the price of our common stock and trading volume could decline.
The trading market for our common stock will be influenced by the research reports and opinions that securities or industry analysts publish about our business, our market and our competitors. We are pioneering the use of our technology in minimal access surgery and thus, analysts may be less likely to publish reports and opinions about our industry. Therefore, we may be required to educate analysts on the nature of our industry in order to obtain research coverage, and such efforts may not be successful. We do not have any control over these analysts. Investors have numerous investment opportunities and may limit their investments to publicly traded companies that receive thorough research coverage. If one or more analysts who cover us downgrade our shares, cease to cover us or fail to publish reports in a regular manner, our share price would likely decline, or we could lose visibility in the financial markets, which could cause a significant and prolonged decline in our stock price due to lack of investor awareness.
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members.
As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Act, the listing requirements of the NASDAQ Global Market and other applicable securities laws, rules and regulations. Despite recent reforms made possible by the JOBS Act, compliance with these laws, rules and regulations will nonetheless increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources, particularly after we are no longer an “emerging growth company.” The Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and operating results. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight may be required. As a result, management’s attention may be diverted from other business concerns and our costs and expenses will increase, which could harm our business and operating results. We may need to hire more employees in the future or engage outside consultants to comply with these requirements, which will increase our costs and expenses.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
45
We incur substantial compensation costs as we pay our executive officers cash compensation commensurate with that of executive officers of other similarly sized public medical device companies, which increases our general and administrative expense and could harm our profitability. Any future equity awards will also increase our compensation expense. We also expect that being a public company and compliance with applicable rules and regulations increases the cost of director and officer liability insurance, and, to the extent our risk profile changes, we may be required to accept reduced coverage or incur substantially higher costs to maintain coverage. These factors could also make it more difficult for us to attract and retain qualified executive officers and members of our board of directors, particularly to serve on our audit committee and compensation committee.
As disclosure of information in filings required increases in a public company, our business and financial condition has become more visible, which could be advantageous to our competitors and clients and could result in threatened or actual litigation, including by competitors and other third parties. If such claims are successful, our business and operating results could be harmed, and even if the claims are resolved in our favor, these claims, and the time and resources necessary to resolve them, could divert the resources of our management and harm our business, brand, reputation and operating results.
We are an “emerging growth company” and the reduced disclosure requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we may take, and intend to take, advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not “emerging growth companies.” In particular, while we are an “emerging growth company” (1) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (2) we will be exempt from any rules that could be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements, (3) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (4) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved.
We may remain an “emerging growth company” until as late as December 31, 2020, the fiscal year-end following the fifth anniversary of the completion of our IPO, though we may cease to be an “emerging growth company” earlier under certain circumstances, including if (1) we have more than $1.07 billion in annual revenue in any fiscal year, (2) the market value of our common stock that is held by non-affiliates exceeds $700 million as the last business day of our most recently completed second fiscal quarter or (3) we issue more than $1.0 billion of non-convertible debt over a three-year period.
The exact implications of the JOBS Act are still subject to interpretations and guidance by the SEC and other regulatory agencies, and we cannot assure you that we will be able to take advantage of all of the benefits of the JOBS Act. In addition, investors may find our common stock less attractive to the extent we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline or become more volatile.
In the event that our internal control over financial reporting is perceived as inadequate, or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results and the trading price of our common stock could decline.
We are subject to the periodic reporting requirements of the Exchange Act. As a result, our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. generally accepted accounting principles. As a public company, we are required, under Section 404(a) of the Sarbanes-Oxley Act of 2002, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. We designed our disclosure controls and procedures to provide reasonable assurance that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC.
46
We believe that any disclosure controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur in the future and not be detected.
In addition, once we cease to be an emerging growth company under the federal securities laws, our auditors will be required to express an opinion on the effectiveness of our internal controls under Section 404(b) of the Sarbanes-Oxley Act. While we may remain an “emerging growth company” until as late as December 31, 2020, the fiscal year-end following the fifth anniversary of the completion of our IPO, we may cease to be an “emerging growth company” earlier under certain circumstances and that could accelerate our timeline for complying with Section 404(b).
If we are unable to confirm that our internal control over financial reporting is effective, or if our auditors, when required, are unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, or be delayed in producing these financial reports, both of which could cause the price of our common stock to decline. We could also be subject to, among other things, regulatory or enforcement actions by the SEC and the Nasdaq Global Market and could be subject to securities litigation.
Anti-takeover provisions in our amended and restated certificate of incorporation and amended and restated bylaws and Delaware law could discourage a takeover and may prevent attempts by our stockholders to replace or remove current management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that might discourage, delay or prevent a merger, acquisition or change of control, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions include:
|
· |
a classified board of directors; |
|
· |
advance notice requirements applicable to stockholders for matters to be brought before a meeting of stockholders and requirements as to the form and content of a stockholder’s notice; |
|
· |
a supermajority stockholder vote requirement for amending certain provisions of our certificate of incorporation and bylaws; |
|
· |
the right to issue preferred stock without stockholder approval, which could be used to dilute the stock ownership of a potential hostile acquirer; |
|
· |
allowing stockholders to remove directors only for cause and only with a supermajority stockholder vote; |
|
· |
a requirement that the authorized number of directors may be changed only by resolution of the board of directors; |
|
· |
allowing all vacancies, including newly created directorships, to be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum, except as otherwise required by law; |
|
· |
a requirement that our stockholders may only take action at annual or special meetings of our stockholders and not by written consent; and |
|
· |
limiting the persons that can call special meetings of our stockholders to our board of directors, the chairperson of our board of directors, the chief executive officer or the president (in the absence of a chief executive officer). |
These provisions might discourage, delay or prevent a change in control of our company or a change in our management. The existence of these provisions could adversely affect the voting power of holders of common stock and limit the price that investors might be willing to pay in the future for shares of our common stock. In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation
47
from engaging in any of a broad range of business combinations with any “interested” stockholder for a period of three years following the date on which the stockholder became an “interested” stockholder.
Our issuance of preferred stock could adversely affect holders of our common stock.
Pursuant to our amended and restated certificate of incorporation, our board is authorized to issue up to 10,000,000 shares of preferred stock without any action on the part of our stockholders. Our board also has the power, without stockholder approval, to set the terms of any series of preferred stock that may be issued, including voting rights, except that shares of preferred stock may not have more than one vote per share, dividend rights, preferences over our common stock with respect to dividends or in the event of a dissolution, liquidation or winding up and other terms. In the event that we issue preferred stock in the future that has preference over our common stock with respect to payment of dividends or upon our liquidation, dissolution or winding up, or if we issue preferred stock that is convertible into our common stock at greater than a one-to-one ratio, the voting and other rights of the holders of our common stock or the market price of our common stock could be adversely affected.
We have not paid dividends in the past and do not expect to pay dividends in the future on our common stock, and any return on investment may be limited to the value of our common stock.
We have never paid cash dividends and we currently intend to retain any future earnings and do not anticipate paying cash dividends in the foreseeable future. We are not legally or contractually required to pay dividends and the credit agreements with MidCap contain restrictions on our ability to pay cash dividends The declaration and payment of all future dividends, if any, will be at the sole discretion our board of directors, which retains the right to change our dividend policy at any time, and may be limited by our debt arrangements in place from time to time. The payment of dividends will depend on our earnings, capital requirements, financial condition, prospects and other factors our board of directors may deem relevant. If we do not pay dividends, our common stock may be less valuable because stockholders must rely on sales of their common stock after price appreciation, which may never occur, to realize any future gains on their investment.
ITEM 1B.UNRESOLVED STAFF COMMENTS
Not applicable.
We lease an aggregate of approximately 38,135 square feet of manufacturing, office and research space in San Francisco, California under a lease expiring in 2024. We currently conduct all of our internal manufacturing at this facility. We believe this facility is sufficient to support our operations and that suitable facilities would be available to us should our operations require it.
In April 2015, we entered into a lease termination agreement with our landlord to terminate the lease for our former facility in San Francisco, California, prior to its scheduled expiration in January 2016. We are no longer obligated to make any lease payments subsequent to the lease termination date of April 30, 2015.
On February 27, 2017, a purported stockholder class action titled Paciga v. Invuity, Inc., et al., Case No. 3:17‑cv‑01005, was filed in the United States District Court for the Northern District of California against us, our Chief Executive Officer, and our Chief Financial Officer. The complaint alleges that the defendants made false or misleading statements to investors regarding our business prospects. The complaint purports to assert claims for violation of Sections 10(b) and 20(a) of the Exchange Act of 1934, and SEC Rule 10b‑5 on behalf of a purported class consisting of all purchasers of our common stock between July 19, 2016 and November 3, 2016, and seeks unspecified compensatory damages, attorney fees and costs, and other relief. On May 30, 2017, the Court appointed Mike Paciga as lead plaintiff. The lead plaintiff filed an amended complaint on July 31, 2017. Defendants filed a motion to dismiss on September 14, 2017, the lead plaintiff filed his opposition to the motion on October 30, 2017 and Defendants filed a reply brief on December 4, 2017. The motion to dismiss is currently scheduled for hearing on April 6, 2018. We intend to defend the litigation vigorously. Based on information currently available, we have determined that the amount of any possible loss or range of possible loss is not reasonably estimable.
48
In addition, we are, and from time to time we may become, involved in legal proceedings arising from the ordinary course of our business. Management is currently not aware of any matters that will have a material adverse effect on our financial position, results of operations or cash flows.
ITEM 4.MINE SAFETY DISCLOSURES
Not Applicable.
49
ITEM 5.MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market for Common Equity
Our common stock has traded on the NASDAQ Global Market under the symbol “IVTY” since June 15, 2015. The following table sets forth, for the periods indicated, the high and low intraday sales prices for our common stock as reported by the NASDAQ Global Market:
|
|
|
Common Stock |
|
||||
|
|
|
Price |
|
||||
|
|
|
High |
|
Low |
|
||
|
Year Ended December 31, 2017 |
|
|
|
|
|
|
|
|
Quarter Ended December 31, 2017 |
|
$ |
9.55 |
|
$ |
6.15 |
|
|
Quarter Ended September 30, 2017 |
|
$ |
9.25 |
|
$ |
5.95 |
|
|
Quarter Ended June 30, 2017 |
|
$ |
9.70 |
|
$ |
5.75 |
|
|
Quarter Ended March 31, 2017 |
|
$ |
8.75 |
|
$ |
5.75 |
|
|
Quarter Ended December 31, 2016 |
|
$ |
13.95 |
|
$ |
4.50 |
|
|
Quarter Ended September 30, 2016 |
|
$ |
14.25 |
|
$ |
9.28 |
|
|
Quarter Ended June 30, 2016 |
|
$ |
10.03 |
|
$ |
4.80 |
|
|
Quarter Ended March 31, 2016 |
|
$ |
9.21 |
|
$ |
6.06 |
|
On February 28, 2018, the last reported sale price on the NASDAQ Global Market for our common stock was $4.15 per share.
As of February 28, 2018, we had 32 holders of record of our common stock. The number of record holders does not include beneficial holders who hold their shares in “street name,” meaning that the shares are held for their accounts by a broker or other nominee. In these instances, the brokers or other nominees are included in the number of registered holders, but the underlying holders of the common stock that have shares held in “street name” are not.
Stock Performance Graph
This performance graph shall not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, or the SEC, for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any of our filings under the Securities Act of 1933, as amended, or the Securities Act, or the Exchange Act.
The graph below shows the cumulative total stockholder return assuming the investment of $100.00 on the date specified in each of our common stock, The NASDAQ Global Market Index, the NASDAQ Biotechnology Index and iShares US Medical Devices Index for the period commencing on June 15, 2015 (the first day of trading of our common stock) and ending on December 31, 2017. We have added the iShares US Medical Devices Index to this year’s stock performance graph because it is comprised of companies with operations more similar to ours than those companies in the Nasdaq Biotechnology Index. Accordingly, we believe that including this new index provides a more appropriate comparison of our stock performance with the performance of other companies in our industry. In this transition year, we have retained the Nasdaq Biotechnology Index for comparison purposes, but will not include that index in our stock performance graph going forward. The comparisons in the table are required by the SEC and are not intended to forecast or be indicative of future performance of our common stock. All amounts are shown are based on the closing prices of the respective indices and our stock:
50
Performance Graph
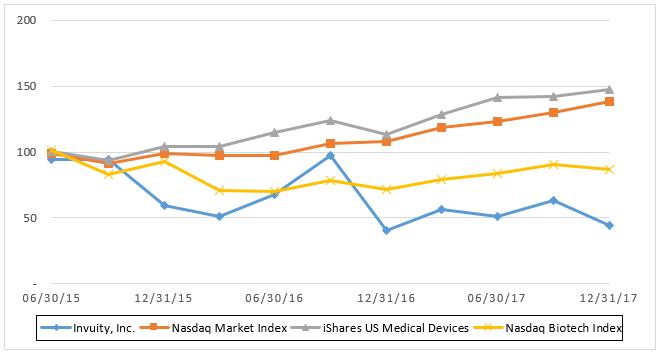
Dividend Policy
We have never declared or paid any dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance our operations and do not anticipate paying any cash dividends on our capital stock in the foreseeable future. Future determination as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors and will depend on then existing conditions, including our operating results, financial conditions, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
Securities Authorized for Issuance under Equity Compensation Plan
The information required by this item will be included in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2017, and is incorporated herein by reference.
Recent Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
None.
ITEM 6.SELECTED FINANCIAL DATA.
The following selected financial data is qualified in its entirety by, and should be read in conjunction with the financial statements and the notes thereto included in Part II, Item 8, “Financial Statements and Supplementary Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Part II, Item 7 of this Annual Report on Form 10-K. The following selected statement of operations data for the years ended December 31, 2017, 2016, and 2015 and the balance sheet data as of December 31, 2017 and 2016 have been derived from our audited financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K. We derived the selected statements of operations data for the year ended December 31, 2014 and the selected balance sheet data as of December 31, 2015 and 2014 from our audited financial statements which are not
51
included in this Annual Report on Form 10-K. Our historical results are not necessarily indicative of the results that may be expected in the future.
|
|
|
December 31, |
|
||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
||||
|
Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
|
$ |
39,619 |
|
$ |
32,461 |
|
$ |
21,031 |
|
$ |
13,103 |
|
|
Cost of goods sold |
|
|
11,741 |
|
|
8,824 |
|
|
7,733 |
|
|
4,630 |
|
|
Gross profit |
|
|
27,878 |
|
|
23,637 |
|
|
13,298 |
|
|
8,473 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
9,018 |
|
|
9,908 |
|
|
7,869 |
|
|
5,181 |
|
|
Selling, general and administrative |
|
|
54,119 |
|
|
52,409 |
|
|
40,636 |
|
|
23,044 |
|
|
Total operating expenses |
|
|
63,137 |
|
|
62,317 |
|
|
48,505 |
|
|
28,225 |
|
|
Loss from operations |
|
|
(35,259) |
|
|
(38,680) |
|
|
(35,207) |
|
|
(19,752) |
|
|
Interest expense |
|
|
(2,370) |
|
|
(2,018) |
|
|
(1,881) |
|
|
(1,402) |
|
|
Interest income |
|
|
222 |
|
|
133 |
|
|
28 |
|
|
51 |
|
|
Other income (expense), net |
|
|
(208) |
|
|
(44) |
|
|
(510) |
|
|
441 |
|
|
Loss on extinguishment of debt |
|
|
(2,303) |
|
|
— |
|
|
— |
|
|
— |
|
|
Net loss and comprehensive loss |
|
$ |
(39,918) |
|
$ |
(40,609) |
|
$ |
(37,570) |
|
$ |
(20,662) |
|
|
Net loss per common share, basic and diluted |
|
$ |
(2.34) |
|
$ |
(2.73) |
|
$ |
(4.94) |
|
$ |
(31.63) |
|
|
Weighted-average shares used to compute net loss per common share, basic and diluted |
|
|
17,051,037 |
|
|
14,868,501 |
|
|
7,606,172 |
|
|
653,195 |
|
|
|
|
December 31, |
|
||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents, and short-term investments |
|
$ |
21,002 |
|
$ |
39,037 |
|
$ |
46,296 |
|
$ |
6,048 |
|
|
Working capital |
|
|
22,678 |
|
|
41,235 |
|
|
49,314 |
|
|
10,366 |
|
|
Total assets |
|
|
45,495 |
|
|
60,335 |
|
|
66,305 |
|
|
25,324 |
|
|
Convertible preferred stock warrant liability |
|
|
— |
|
|
— |
|
|
— |
|
|
136 |
|
|
Total long-term debt |
|
|
29,116 |
|
|
13,261 |
|
|
14,480 |
|
|
9,347 |
|
|
Convertible preferred stock |
|
|
— |
|
|
— |
|
|
— |
|
|
73,755 |
|
|
Accumulated deficit |
|
|
(186,134) |
|
|
(146,216) |
|
|
(105,607) |
|
|
(68,037) |
|
|
Total stockholders’ equity (deficit) |
|
$ |
(862) |
|
$ |
34,448 |
|
$ |
42,343 |
|
$ |
(65,827) |
|
52
ITEM 7.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with “Selected Financial Data” and the financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those discussed in “Risk Factors” and in other parts of this Annual Report on Form 10-K.
Overview
We are a leading medical technology company focused on developing and marketing advanced surgical devices to improve the ability of physicians to perform minimal access surgery through smaller and hidden incisions. Our patented Intelligent Photonics® technology delivers enhanced visualization which facilitates surgical precision, efficiency and safety. In addition, we utilize comprehensive strategic marketing programs to create stronger institutional partnerships. Clinical applications include women's health, encompassing breast cancer and breast reconstruction surgery, gynecology and thyroid surgery. Additional applications include procedures for electrophysiology, spine, orthopedic, cardiothoracic, and general surgery.
We channel our development through three broad categories of innovation. First, we integrate our Intelligent Photonics® technology platform into our single-use and reusable advanced surgical devices to address some of the critical intracavity illumination and visualization challenges facing surgeons today. We utilize this proprietary technology to develop optical waveguides, called Photonguides, that direct and shape thermally cool, brilliant light into broad, uniform and volumetric illumination of the surgical target. We believe that improving a surgeon’s ability to see critical anatomical structures can lead to better clinical and aesthetic outcomes, improved patient safety and reduced surgical time and healthcare costs.
Our second broad category of innovation for minimally invasive and minimal access surgical procedures is in the development and commercialization of a novel advanced energy platform. In September 2016, we received U.S. Food and Drug Administration, or FDA, 510(k) clearance of PhotonBlade®, a dynamic precision illuminator with enhanced energy delivery. PhotonBlade® is a first-of-its-kind device, delivering directed, thermally cool illumination at the precise point of surgical treatment in conjunction with a novel advanced energy platform allowing for precise tissue cutting and coagulation with minimal tissue damage. As such, PhotonBlade® represents a new category of Intelligent Photonics®and strategically expands our current product portfolio. After a preliminary market launch in March 2017, we fully launched the PhotonBlade® in September 2017.
Finally, in the third quarter of 2017, through a distribution arrangement with our manufacturing partner, Fluoptics Imaging Inc., we began a limited market launch of a fluorescence imaging system, called PhotonVue, which is used for the visual assessment of blood flow in adults as an adjunctive method for the evaluation of tissue perfusion.
We sold our devices to approximately 870 hospitals in the fourth quarter of 2017, as compared to approximately 745 hospitals in the fourth quarter of 2016. Based on the number of single-use units we have shipped as of December 31, 2017, we estimate that our devices have been used in approximately 367,000 surgical procedures cumulative from inception.
In addition to marketing and selling our existing products, we are engaged in ongoing research and development. Our research and development efforts are focused on developing new devices and modalities to broaden the application and adoption of minimal access procedures and enable new advanced surgical techniques. Our manufacturing involves the combined utilization of our internal manufacturing resources and expertise, approved suppliers and contract manufacturers. We outsource the manufacture of components, subassemblies and certain finished devices that are produced to our specifications and shipped to our facilities in San Francisco, California for final assembly or inspection, and certification. Finished products are stored at and distributed from our facility. We are in the process of securing additional space for storage and distribution, and believe these facilities are sufficient to support our operations and that suitable facilities would be available to us should our operations require it.
53
Revenue
Substantially all of our revenue is derived from sales of our devices in the United States. In June 2017, we started selling into Asia with sales through December 31, 2017 not being considered material. We earn revenue from the sale of our devices primarily through our direct sales force as complemented by independent sales agents.
We consider revenue derived from our single-use products to be the most important part of our business. Therefore, our focus is on procedure adoption. Recent revenue growth has been driven by the growth of our sales and marketing infrastructure and increased surgeon awareness of the benefits of our advanced photonics technology platform over traditional surgical lighting options in the operating room. We are pursuing a number of strategies that we believe will enable us to continue to grow. Our products have broad applicability to open, smaller incision surgeries which we estimate to be approximately 40% of all surgical procedures in the United States. We are targeting our sales and marketing efforts to surgeons for the following women’s health clinical applications: breast cancer and breast reconstruction surgery, gynecology, and thyroid surgery. Additional applications include procedures for electrophysiology, spine, orthopedic, cardiothoracic, and general surgery. Although we have made progress towards our goals there are still many areas for growth. To achieve these goals, we are focused on the following initiatives:
|
· |
Leveraging our unique marketing programs that align with hospital initiatives including safety, patient outcomes and patient satisfaction, for example our Hidden Scar Surgery Program and Operating Room Safety Program; |
|
· |
Cross selling within our initial target markets and developing adjacent surgical specialties to broaden the three core categories to Women’s health, spine/orthopedic and electrophysiology; and |
|
· |
Introducing new products to support our ability to expand our clinical application, as demonstrated by the launch of the PhotonBlade® and PhotonVue® in 2017. |
We have experienced seasonality during the year. Revenue tends to be the lowest in the first quarter as the result of the resetting of annual patient healthcare insurance plan deductibles and by hospitals and military facilities working off their inventories of products purchased in the fourth quarter. The third quarter is similarly negatively impacted by lower procedure rates typically associated with vacation plans of both patients and surgeons. Revenue in the fourth quarter tends to be the highest as demand may be impacted by the desire of patients to spend their remaining balances in their flexible spending accounts or because they have met their annual deductibles under their health insurance plans. In addition, in the fourth quarter, our results can be impacted by the budgeting and buying patterns of hospitals and military facilities.
Cost of Goods Sold and Gross Margin
Cost of goods sold consists primarily of material costs, manufacturing overhead, direct labor and third-party services, such as sterilization. Manufacturing overhead represents a significant portion of cost of goods sold and includes the cost of quality assurance, material procurement, inventory control, facilities, equipment and operations supervision and management. New product launches typically have higher costs due to new manufacturing processes and lower production volumes. However, we expect overhead costs as a percentage of revenue to decrease as our production volume increases and our production processes become more efficient. Cost of goods sold also includes depreciation expense for production equipment and certain direct costs such as shipping cost
We calculate gross margin as gross profit divided by revenue. Our gross margin has been and will continue to be affected by a variety of factors, primarily production volumes, manufacturing costs and product yields, adjustments to inventory reserves and the implementation of cost-reduction strategies. Gross margin on our current product portfolio may increase over the long term as our production volume increases and we are able to spread the fixed portion of our manufacturing overhead costs over a larger number of units produced, thereby reducing our per unit manufacturing costs; however this positive trend may be offset by new product introductions, notably the PhotonBlade® and PhotonVue®, as new products are produced at low volumes which may result in lower margins as component costs are usually higher at lower volumes and manufacturing processes are less efficient. Therefore, our gross margin will likely fluctuate from quarter to quarter in the near term.
54
Research and Development Expenses
Our research and development, or R&D, expenses consist primarily of product research, engineering, product development, quality assurance and depreciation. These expenses include personnel costs, including salaries, healthcare benefits, bonuses and stock-based compensation expense, consulting services, laboratory materials and supplies and an allocation of related facilities costs. In the near term, we expect our R&D costs to be fairly constant in absolute dollars as we look to increase our operating efficiencies and then over the longer term to increase in absolute dollars as we hire additional personnel to develop new devices and device enhancements. We expense R&D costs as they are incurred.
Selling, General and Administrative Expenses
Our selling, general and administrative, or SG&A, expenses consist primarily of personnel costs including salaries, healthcare benefits, sales commissions, bonuses and stock compensation for sales, executive, and administrative personnel. Other significant SG&A expenses include independent sales agent commissions, conferences, trade shows, promotional activities, including trunk stock expenses associated with the sales team demonstrating our products, professional fees for legal and accounting services, consulting fees, insurance costs and travel expenses.
In the near term, we expect SG&A expenses to be fairly constant in absolute dollars as we look to increase operating efficiencies, and then over the longer term, to increase in absolute dollars as we expect to hire incremental direct sales representatives and expand our commercial infrastructure to both drive and support our planned revenue growth. We also expect to incur additional SG&A expenses as a result of supporting a dynamically growing company, including investments in human resources and other administrative functions.
Interest Expense
Interest expense consists of cash and non-cash components. The cash component of interest expense is attributable to our borrowings under our loan agreements. The non-cash component consists of interest expense recognized from the amortization of debt discounts derived from the issuance of warrants and debt issuance costs capitalized on our balance sheets.
Interest Income and Other Income (Expense), Net
Interest income and other income (expense), net consists primarily of interest income earned on our cash, cash equivalents and marketable securities, offset by tax expenses.
Results of Operations
Revenue, cost of revenue, gross profit and gross margin
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
|
|
(in thousands, except percentages) |
|
|||||||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
Single use devices |
|
$ |
32,389 |
|
$ |
24,548 |
|
$ |
15,269 |
|
|
Reusable retractors |
|
|
4,225 |
|
|
5,401 |
|
|
3,333 |
|
|
Sales to 3rd party medical device manufacturers |
|
|
1,775 |
|
|
1,386 |
|
|
1,422 |
|
|
Accessories |
|
|
1,230 |
|
|
1,126 |
|
|
1,007 |
|
|
Total revenue |
|
|
39,619 |
|
|
32,461 |
|
|
21,031 |
|
|
Cost of Revenue |
|
|
11,741 |
|
|
8,824 |
|
|
7,733 |
|
|
Gross Profit |
|
$ |
27,878 |
|
$ |
23,637 |
|
$ |
13,298 |
|
|
Gross Margin |
|
|
70 |
% |
|
73 |
% |
|
63 |
% |
Twelve Months Ended December 31, 2017 as compared to Twelve Months Ended December 31, 2016
Revenue increased $7.2 million, or 22%, to $39.6 million during the year ended December 31, 2017, compared to $32.5 million during the year ended December 31, 2016. Revenue growth was driven by our single use devices which increased $7.8 million, or 32%. We continue to see an increase in procedural adoption for our single-use devices as a
55
result of selling our products to new customers and also increasing sales to existing customers. We continue to see increased adoption of our devices in breast cancer and breast reconstruction surgery driven by our Hidden Scar marketing programs, advances within certain gynecological procedures and growing interest in certain electrophysiology procedures. The number of customers ordering our devices increased from approximately 745 in the fourth quarter of 2016 to approximately 870 in the fourth quarter of 2017, and procedural volumes increased to 127,000 for 2017 compared to 95,000 for 2016. Compared to the year ended December 31, 2016, revenue from sales of our retractors decreased $1.2 million, or 22%, due to a decrease of the total number of retractors sold, including sales of retractors that were being phased out in 2017. In addition, our retractor devices are reusable, but utilize a single-use optical waveguide, which we sell separately because a new waveguide must be used for each procedure. We use various promotional programs to incentivize customers to purchase our retractors with the aim of increasing the installed base of retractors that subsequently may drive increased waveguide sales.
Cost of goods sold increased $2.9 million, or 33%, to $11.7 million during the year ended December 31, 2017, compared to $8.8 million for the year ended December 31, 2016. This increase in cost of goods sold was primarily due to the increase of number of devices sold as we expanded our sales and marketing efforts and increased the number of units sold.
Gross margin for the year ended December 31, 2017 was 70% compared to 73% for the year ended December 31, 2016. Gross margin declined in the second half of 2017 due to the launch of our new products PhotonBlade® and PhotonVue® that, as is common with new product launches, have yet to benefit from improvements in manufacturing processes and lower costs associated with higher production volumes.
Twelve Months Ended December 31, 2016 as compared to Twelve Months Ended December 31, 2015
Revenue increased $11.4 million, or 54%, to $32.5 million during the year ended December 31, 2016, compared to $21.0 million during the year ended December 31, 2015. The growth in revenue was attributable to an increase in unit sales as a result of selling our products to new customers and also selling more units to existing customers. The increase in units was driven by the expansion of our direct salesforce, which increased the number of customers to whom we sold devices. The number of customers ordering our devices increased from approximately 530 in the fourth quarter of 2015 to approximately 745 in the fourth quarter of 2016.
Cost of goods sold increased $1.1 million, or 14%, to $8.8 million during the year ended December 31, 2016, compared to $7.7 million during the same period in 2015. This increase in cost of goods sold was primarily due to the increase of number of devices sold as we expanded our sales and marketing efforts and increased the number of units sold.
Gross margin for the year ended December 31, 2016 was 73% compared to 63% for the year ended December 31, 2015. The increase was due to the introduction of the Eikon LT retractor series at the beginning of the year, which replaced our Eikon Classic retractor series and had significantly improved margins as a result of being made from a polymer compared to metal which is a lower cost material. The Eikon LT comprised 12% of total sales for 2016.
Operating Expenses
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||||||||
|
|
|
|
|
|
% of |
|
|
|
|
% of |
|
|
|
|
% of |
|
|
|
|
Amount |
|
Revenue |
|
Amount |
|
Revenue |
|
Amount |
|
Revenue |
|
|||
|
|
|
(in thousands, except percentages) |
|
|||||||||||||
|
Operating Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
$ |
9,018 |
|
23 |
% |
$ |
9,908 |
|
31 |
% |
$ |
7,869 |
|
37 |
% |
|
Selling general and administrative |
|
|
54,119 |
|
137 |
|
|
52,409 |
|
161 |
|
|
40,636 |
|
193 |
|
|
Total operating expenses |
|
$ |
63,137 |
|
160 |
% |
$ |
62,317 |
|
192 |
% |
$ |
48,505 |
|
230 |
% |
Twelve Months Ended December 31, 2017 as compared to Twelve Months Ended December 31, 2016
Research and development expenses: expenses decreased $0.9 million, or 9%, to $9.0 million during the year ended December 31, 2017, compared to $9.9 million during the year ended December 31, 2016. Expenses are substantially impacted by the cadence of product releases and the majority of PhotonBlade® expenses were incurred in 2016 with a corresponding reduction of these expenses in 2017 as a result of the PhotonBlade® being launched.
56
Selling, general and administrative expenses: expenses increased $1.7 million, or 3%, to $54.1 million during the year ended December 31, 2017, compared to $52.4 million during the year ended December 31, 2016. Expenses increased $4.0 million due to increase professional fees, which included $3.2 million of litigation costs. This increase was offset by a reduction in selling and marketing expenses of $1.4 million, reduced payroll related costs of $0.6 million, and reduced depreciation, facilities and maintenance costs of $0.3 million. During the year we took advantage of turnover in our direct sales representatives to increase sales productivity by placing replacement hires into major metropolitan markets and consolidating some smaller sales territories.
Twelve Months Ended December 31, 2016 as compared to Twelve Months Ended December 31, 2015
Research and development expenses: expenses increased $2.0 million, or 26%, to $9.9 million during the year ended December 31, 2016, compared to $7.9 million during the year ended December 31, 2015. Expenses increased $1.1 million as a result of increasing our headcount and $1.3 million for consultants, research on new product development, testing and associated regulatory and quality costs, including PhotonBlade® development.
Selling, general and administrative expenses: expenses increased $11.8 million, or 29%, to $52.4 million during the year ended December 31, 2016, compared to $40.6 million during the year ended December 31, 2015. Personnel-related expenses, including salaries and employee benefits, increased $11.5 million, as a result of increasing our direct sales force, marketing and administrative staff. Travel and entertainment expenses increased $1.1 million mainly due to the increased sales force, and sales commissions increased $1.2 million as a result of increased revenues driven by the increased sales force. In addition, in 2016, we incurred $0.8 million in expense as a result of reserves against our trunk stock inventory related to our Eikon Classic retractor that was replaced by our new Eikon LT retractor series. These increases were offset a $1.1 million decrease in professional services, consultants, and marketing expenses.
Interest expense, interest income, and other expense, net
Non-operating items, including interest expense, interest income, and other income (expense), were as follows for the periods presented:
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||||||||
|
|
|
|
|
|
% of |
|
|
|
|
% of |
|
|
|
|
% of |
|
|
|
|
Amount |
|
Revenue |
|
Amount |
|
Revenue |
|
Amount |
|
Revenue |
|
|||
|
|
|
(in thousands, except percentages) |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(2,370) |
|
(6) |
% |
$ |
(2,018) |
|
(6) |
% |
$ |
(1,881) |
|
(9) |
% |
|
Interest income |
|
|
222 |
|
1 |
|
|
133 |
|
0 |
|
|
28 |
|
0 |
|
|
Other expense, net |
|
|
(208) |
|
(1) |
|
|
(44) |
|
(0) |
|
|
(510) |
|
(2) |
|
|
Loss on extinguishment of debt |
|
|
(2,303) |
|
(6) |
|
|
— |
|
— |
|
|
— |
|
— |
|
|
Total |
|
$ |
(4,659) |
|
(12) |
% |
$ |
(1,929) |
|
(6) |
% |
$ |
(2,363) |
|
(11) |
% |
Twelve Months Ended December 31, 2017 as compared to Twelve Months Ended December 31, 2016
Interest expense increased $0.4 million to $2.4 million during the year ended December 31, 2017 from $2.0 million during the year ended December 31, 2016 as a result of increasing our term loan from $15 million to $30 million and utilizing our revolving credit facility during 2017.
Interest income increased $0.1 million to $0.2 million during the year ended December 31, 2017 from $0.1 million during the year ended December 31, 2016, due to additional interest income earned when compared to 2016.
Other expense, net changed $0.2 million to expense of $0.2 million during the year ended December 31, 2017, compared to expense of $44,000 during the year ended December 31, 2016, due to additional tax expenses incurred when compared to 2016.
A loss of extinguishment of debt costs of $2.3 million was incurred in March 2017 when we re-financed our loan with HCRP and our credit facility with SVB and replaced them with our current loan facility with MidCap. These expenses included a $1.8 million prepayment fee, approximately $0.4 million in expenses relating to the immediate recognition of
57
debt discounts that were being amortized over the term of the original loan, and a $150,000 fee for ending our access to our SVB credit facility.
Twelve Months Ended December 31, 2016 as compared to Twelve Months Ended December 31, 2015
Interest expense increased $0.1 million to $2.0 million during the year ended December 31, 2016 from $1.9 million during the year ended December 31, 2015 primarily due to increased interest payments on our loan with HealthCare Royalty Partners, or HCRP.
Interest income increased $0.1 million to $0.1 million during the year ended December 31, 2016, compared to income of $28,000 during the year ended December 31, 2015, due to additional interest income earned on a higher average cash balance held throughout the year.
Other expense, net changed $0.5 million to $44,000 during the year ended December 31, 2016, compared to expense of $0.5 million during the year ended December 31, 2015. In 2015, we incurred an expense related to the fair value re-measurement of the liability related to our outstanding convertible preferred stock warrants prior to our IPO.
Liquidity and Capital Resources
Liquidity is our ability to generate sufficient cash flows from operating activities to meet our obligations and commitments. In addition, liquidity includes the ability to obtain appropriate financing or to raise capital. The accompanying financial statements have been prepared on a basis which assumes we will continue as a going concern and which contemplates the realization of assets and satisfaction of liabilities and commitments in the normal course of business. We have incurred net losses from operations since inception, including $39.9 million for the year ended December 31, 2017, and have an accumulated deficit of $186.1 million as of December 31, 2017. We have $21.0 million in cash and cash equivalents and short-term investments, $35.0 million in debt outstanding and a working capital of $22.7 million at December 31, 2017. We are required to comply with a financial covenant relating to certain quarterly minimum Net Revenue requirements on a trailing twelve-month basis. The minimum Net Revenue requirements on a trailing twelve month basis for 2018 is $40,488,151 for the period ending March 31, 2018, $42,248,214 for the period ending June 30, 2018, $46,574,986 for the period ending September 30, 2018, and $50,000,000 for the period ending December 31, 2018. As of December 31, 2017, we were in compliance with this covenant. We expect to incur additional losses and negative cash flows for the foreseeable future as we continue to invest in our sales and marketing efforts and research and development activities in order to continue to grow our business.
We believe that our cash, cash equivalents and short-term investments as of December 31, 2017, and expected sales from our products together with additional funding available under our revolving credit facility will not provide sufficient funds to enable us to meet our projected operating requirements for the next twelve months from the issuance of the financial statements included in this Annual Report on Form 10-K.
We intend to obtain additional funding through public or private financing, collaborative arrangements with strategic partners, or through additional credit lines or other debt financing sources to increase the funds available to support our operating and capital needs. If we are not able to perform according to our 2018 operating plan, our available capital resources may be consumed more rapidly than currently expected, and therefore we may be required to raise additional funds sooner than anticipated. However, we may not be able to secure such financing in a timely manner or on favorable terms, if at all. Without additional funds, we will be forced to delay, scale back or eliminate some of our sales and marketing efforts, research and development activities, or other operations and potentially delay product development in an effort to provide sufficient funds to continue our operations.
The uncertainty around the achievement of our plans to mitigate the risk of going concern raises substantial doubt about our ability to continue as a going concern for a one year period from the issuance of the financial statements included in this Annual Report on Form 10-K. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
58
Cash Flows
The following table summarizes our cash flows for the periods indicated:
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Net cash (used in) provided by: |
|
(in thousands) |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(38,885) |
|
$ |
(36,624) |
|
$ |
(31,165) |
|
|
Investing activities |
|
|
6,870 |
|
|
(11,783) |
|
|
(3,748) |
|
|
Financing activities |
|
|
21,495 |
|
|
30,411 |
|
|
75,126 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
(10,520) |
|
$ |
(17,996) |
|
$ |
40,213 |
|
Twelve Months Ended December 31, 2017
Net cash used in operating activities was $38.9 million, which consisted of a net loss of $39.9 million, adjusted by non-cash charges of $5.6 million and net change of $4.5 million in our net operating assets. The change in our operating assets is primarily due to an increase in our accounts receivable of $1.6 million, and increase in our inventory of $2.4 million and a reduction in our accrued liabilities of $1.3 million, offset by an increase in our accounts payable of $1.4 million. The increase in our accounts receivable is in line with increased sales, increased inventories are due to the launch of PhotonBlade® and the increase in our accounts payable is primarily due to the timing of December payments made in January. Accrued liabilities reduced as a result of lower operating expenses in the fourth quarter.
Net cash used in investing activities consisted of the purchase and sale of marketable securities and capital expenditures to purchase property and equipment.
Net cash from financing activities included $30 million of proceeds from the issuance of long-term debt net of issuance costs under the credit and security agreement the Company entered into with MidCap in March 2017. The Company received $20.0 million upon closing and $10 million in September 2017. The Company used $17.2 million of the initial funding to pay off in full the outstanding $15.0 million loan with HCRP including a $1.8 million prepayment fee and $0.4 million in interest. In addition the Company entered a separate credit and security agreement with MidCap in March 2017 that provides for a revolving credit facility of up to $10.0 million based on the eligible accounts receivable and inventory balances, of which, at December 31, 2017, the Company had drawn down $5.9 million.
Twelve Months Ended December 31, 2016
Net cash used in operating activities was $36.6 million, which consisted of a net loss of $40.6 million, adjusted by non-cash charges of $4.6 million and net change of $0.6 million in our net operating assets. The change in our operating assets is primarily due to an increase in our accounts receivable of $2.3 million and a reduction in our accounts payable of $0.3 million offset by an increase in our accrued liabilities of $2.1 million. Our increase in our accounts receivable is in line with increased sales and our accounts payable and accrued liabilities is primarily due to the timing of December payments made in January.
Net cash used in investing activities consisted of the purchase and sale of marketable securities and capital expenditures to purchase property and equipment in connection with the new facility.
In July 2016, we filed a shelf registration statement on Form S-3 (Registration No. 333-212395) with the SEC to offer for sale up to an aggregate of $100,000,000 of common stock, preferred stock, depositary shares, warrants and/or units in one or more offerings and in any combinations. We also established an ATM program of up to $25 million. The associated costs were fully expensed in 2016.
In August 2016, we sold 3,220,000 shares of our common stock in a secondary offering. The total net proceeds from the offering were approximately $29.7 million.
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements and do not have any holdings in variable interest entities.
59
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2017:
|
|
|
Payments Due by Period |
|
|||||||||||||
|
|
|
Less Than |
|
1 to 3 |
|
3 to 5 |
|
More than |
|
|
|
|
||||
|
(in thousands) |
|
1 Year |
|
Years |
|
Years |
|
5 Years |
|
Total |
|
|||||
|
Debt, including interest |
|
$ |
2,490 |
|
$ |
24,838 |
|
$ |
8,795 |
|
$ |
— |
|
$ |
36,123 |
|
|
Operating leases |
|
|
2,178 |
|
|
4,553 |
|
|
4,831 |
|
|
4,680 |
|
|
16,242 |
|
|
Purchase obligations |
|
|
1,275 |
|
|
— |
|
|
— |
|
|
— |
|
|
1,275 |
|
|
Total contractual obligations (1) |
|
$ |
5,943 |
|
$ |
29,391 |
|
$ |
13,626 |
|
$ |
4,680 |
|
$ |
53,640 |
|
|
(1) |
Our term loans with MidCap accrue interest at a floating rate equal to 6.50% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. Interest shall accrue on the date of the commencement of funding and is payable in arrears on the first day of each month. The debt payments noted are estimates of our principal and interest payments using the floating rate as of December 31, 2017. Actual payments relating to interest may differ. |
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires our management to make judgments and estimates that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue generated and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these judgments and estimates under different assumptions or conditions and any such differences may be material. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgment and estimates.
Revenue Recognition
We earn revenue from the sale of our devices to hospitals through direct sales representatives and independent sales agents. Our revenue is recognized when the following criteria are met:
|
· |
Persuasive evidence of an arrangement exists. We consider this criterion satisfied when we have a purchase order or contract with the customer in place. |
|
· |
Delivery has occurred and title passed to the customer, which is typically upon shipment of the device from our location or when received by the customer based on the shipping terms. |
|
· |
The price is fixed or determinable and collectability is reasonably assured. We determine the satisfaction of these criteria based on our judgment regarding the nature of the fee charged for devices, contractual agreements entered into, and the collectability of those fees under any contract or agreement. |
We do not offer rights of return other than our standard warranty, or price protection. With the exception of PhotonVue®, whereby customers can purchase service contracts for future periods of time, we have no post-delivery obligations other than our standard warranty for our other products.
Stock-based Compensation
We recognize compensation costs related to stock options granted to employees based on the estimated fair value of the awards on the date of grant, net of actual forfeitures. We estimate the grant date fair value, and the resulting stock-based compensation expense, using the Black-Scholes option pricing model. The grant date fair value of stock-based awards is expensed on a straight-line basis over the period during which the employee is required to provide service in exchange for the award (generally the vesting period).
60
We estimate the fair value of our stock-based awards using the Black-Scholes option-pricing model, which requires the input of highly subjective assumptions. Our assumptions are as follows:
|
· |
Expected term. The expected term represents the period that the stock-based awards are expected to be outstanding. We use the simplified method to determine the expected term, which is calculated as the average of the time to vesting and the contractual life of the options. |
|
· |
Expected volatility. The expected volatility is derived from the average historical volatilities of publicly traded companies within our industry that we consider to be comparable to our business over a period approximately equal to the expected term for employee’s options and the remaining contractual life for non-employees options blended with a review of our actual historical volatility since our IPO in June 2015. |
|
· |
Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield with a maturity equal to the expected term of the option in effect at the time of grant. |
|
· |
Dividend yield. The expected dividend is assumed to be zero as we have never paid dividends and have no current plans to pay any dividends on our common stock. |
Prior to our IPO, the fair value of the shares of our common stock underlying the stock options had historically been determined by our board of directors. Because there had been no public market for our common stock, our board of directors had determined the fair value of our common stock at the time of grant of the option by considering a number of objective and subjective factors, including our stage of development, sales of our convertible preferred stock, our operating and financial performance, equity market conditions affecting comparable public companies, the lack of liquidity of our capital stock, and the general and industry-specific economic outlooks.
Since our IPO in June 2015, the fair value of our common stock is based on the closing price of our common stock, as quoted on the NASDAQ Global Market, on the date of grant.
Stock-based compensation expense for options granted to non-employees as consideration for services received is measured on the date of performance at the fair value of the consideration received or the fair value of the equity instruments issued, using the Black-Scholes option-pricing model, whichever can be more reliably measured. Stock-based compensation expense for options granted to non-employees is periodically remeasured as the underlying options vest.
The following table summarizes the assumptions we used to determine the fair value of stock options granted to employees:
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Expected term (in years) |
|
6.0 |
|
5.0-6.0 |
|
5.0-6.0 |
|
|
Expected volatility |
|
60 |
% |
50 |
% |
35-50 |
% |
|
Risk-free interest rate |
|
1.87-2.22 |
% |
1.24-2.26 |
% |
1.31-1.89 |
% |
|
Dividend yield |
|
0 |
% |
0 |
% |
0 |
% |
We recorded total stock-based compensation expense of $3.3 million, $2.3 million and $1.2 million, for the years ended December 31, 2017, 2016, and 2015, respectively. We expect to continue to grant stock options and other equity-based awards in the future, and to the extent that we do, our stock-based compensation expense recognized in future periods will likely increase.
JOBS Act Accounting Election
We are an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will
61
be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Recent Accounting Pronouncements
See Note 2, “Summary of Significant Accounting Policies”, of the Notes to Financial Statements included in Part II, Item 8 of this Annual Report on Form 10-K for a full description of recent accounting pronouncements including the respective expected dates of adoption and estimated effects, if any on our financial statements.
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
Our operations are primarily within the United States. We are exposed to market risks in the ordinary course of our business. These risks primarily relate to interest rate, inflation risks, and changes in the general economic conditions in the countries where we conduct business. We had cash, cash equivalents, and short term investments of $21.0 million and $39.0 million as of December 31, 2017 and December 31, 2016, respectively, which consist of corporate bonds, commercial paper, bank deposits and money market funds.
Our short-term investments primarily consisted of corporate bonds. The cash and cash equivalents are held for working capital purposes. Our investments are made for capital preservation purposes. We do not enter into investments for trading or speculative purposes. Because of the short-term nature of the instruments in our portfolio, a sudden change in market interest rates would not be expected to have a material impact on our financial statements.
As of December 31, 2017, we had drawn down a principal debt balance of $30.0 million with a floating interest equal to 6.50% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. In addition, we had also drawn down $5.9 million against our accounts receivable facility. This facility accrues interest at a floating rate equal to 3.25% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. As of December 31, 2016, we had a principal debt balance of $15.0 million with a fixed interest rate equal to 12.5%. A hypothetical 100 basis point change in interest rates during any of the periods presented would not have had a material impact on our financial statements. See Note 5 and Note 6 of the Notes to Financial Statements included in Part II, Item 8 of this Annual Report on Form 10-K for more details
Foreign Currency Risk
As of December 31, 2017, our business has been almost entirely conducted in U.S. dollars, but we have expanded sales to Asia and expect to expand sales to Europe. As we begin to conduct transactions in foreign currencies, we may become increasingly subject to foreign currency risks related to our revenue and operating expenses denominated in currencies other than the U.S. Dollar. As our international operations grow, our risks associated with fluctuation in currency rates will become greater, and we will continue to reassess our approach to managing this risk.
Inflation Risk
We do not believe that inflation has had a material effect on our business, financial condition or results of operations. Nonetheless, if our costs were to become subject to significant inflationary pressures, we may not be able to fully offset such higher costs through price increases. Our inability or failure to do so could harm our business, financial condition and results of operations.
62
ITEM 8.FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
INVUITY, INC.
INDEX TO FINANCIAL STATEMENTS
| 64 | ||
| 65 | ||
| 66 | ||
|
Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit) |
67 | |
| 68 | ||
| 69 | ||
| 88 | ||
63
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Invuity, Inc.:
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Invuity, Inc. as of December 31, 2017 and 2016, and the related statements of operations and comprehensive loss, of convertible preferred stock and stockholders’ equity (deficit), and of cash flows for each of the three years in the period ended December 31, 2017, including the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017 in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Change in Accounting Principles
As discussed in Note 2 to the financial statements, in 2017, the Company changed the manner in which it accounts for certain elements of its employee share-based payments and the manner in which it presents restricted cash in the statement of cash flows.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
San Jose, California
March 5, 2018
We have served as the Company's auditor since 2010, which includes periods before the Company became subject to SEC reporting requirements.
64
INVUITY, INC.
(in thousands, except share data)
|
|
|
December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
17,962 |
|
$ |
28,300 |
|
|
Short-term investments |
|
|
3,040 |
|
|
10,737 |
|
|
Restricted cash - current |
|
|
181 |
|
|
181 |
|
|
Accounts receivable, net |
|
|
7,421 |
|
|
5,782 |
|
|
Inventory |
|
|
7,436 |
|
|
5,052 |
|
|
Prepaid expenses and other current assets |
|
|
1,274 |
|
|
1,088 |
|
|
Total current assets |
|
|
37,314 |
|
|
51,140 |
|
|
Restricted cash |
|
|
727 |
|
|
909 |
|
|
Property and equipment, net |
|
|
7,169 |
|
|
8,286 |
|
|
Other long-term assets |
|
|
285 |
|
|
— |
|
|
Total assets |
|
$ |
45,495 |
|
$ |
60,335 |
|
|
Liabilities and Stockholders’ Equity (Deficit) |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
3,598 |
|
$ |
2,192 |
|
|
Accrued and other current liabilities |
|
|
5,179 |
|
|
6,351 |
|
|
Short-term debt |
|
|
5,859 |
|
|
1,362 |
|
|
Total current liabilities |
|
|
14,636 |
|
|
9,905 |
|
|
Deferred rent |
|
|
2,569 |
|
|
2,721 |
|
|
Deferred revenue - long term |
|
|
36 |
|
|
— |
|
|
Long-term debt |
|
|
29,116 |
|
|
13,261 |
|
|
Total liabilities |
|
|
46,357 |
|
|
25,887 |
|
|
Commitments and contingencies (Note 7) |
|
|
|
|
|
|
|
|
Stockholders’ equity (deficit): |
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value—10,000,000 shares authorized at December 31, 2017 and December 31, 2016, no shares issued and outstanding at December 31, 2017 and December 31, 2016 |
|
|
— |
|
|
— |
|
|
Common stock, $0.001 par value—100,000,000 shares authorized at December 31, 2017 and December 31, 2016 17,179,258 and 16,950,940 shares issued and outstanding at December 31, 2017 and December 31, 2016 |
|
|
17 |
|
|
17 |
|
|
Additional paid-in capital |
|
|
185,255 |
|
|
180,647 |
|
|
Accumulated deficit |
|
|
(186,134) |
|
|
(146,216) |
|
|
Total stockholders’ equity (deficit) |
|
|
(862) |
|
|
34,448 |
|
|
Total liabilities and stockholders’ equity (deficit) |
|
$ |
45,495 |
|
$ |
60,335 |
|
The accompanying notes are an integral part of these financial statements.
65
INVUITY, INC.
Statements of Operations and Comprehensive Loss
(in thousands, except share and per share data)
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
|
|
|
|
|||||||
|
Revenue |
|
$ |
39,619 |
|
$ |
32,461 |
|
$ |
21,031 |
|
|
Cost of goods sold |
|
|
11,741 |
|
|
8,824 |
|
|
7,733 |
|
|
Gross profit |
|
|
27,878 |
|
|
23,637 |
|
|
13,298 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
9,018 |
|
|
9,908 |
|
|
7,869 |
|
|
Selling, general and administrative |
|
|
54,119 |
|
|
52,409 |
|
|
40,636 |
|
|
Total operating expenses |
|
|
63,137 |
|
|
62,317 |
|
|
48,505 |
|
|
Loss from operations |
|
|
(35,259) |
|
|
(38,680) |
|
|
(35,207) |
|
|
Interest expense |
|
|
(2,370) |
|
|
(2,018) |
|
|
(1,881) |
|
|
Interest income |
|
|
222 |
|
|
133 |
|
|
28 |
|
|
Other expense, net |
|
|
(208) |
|
|
(44) |
|
|
(510) |
|
|
Loss on extinguishment of debt |
|
|
(2,303) |
|
|
— |
|
|
|
|
|
Net loss and comprehensive loss |
|
$ |
(39,918) |
|
$ |
(40,609) |
|
$ |
(37,570) |
|
|
Net loss per common share, basic and diluted |
|
$ |
(2.34) |
|
$ |
(2.73) |
|
$ |
(4.94) |
|
|
Weighted-average shares used to compute net loss per common share, basic and diluted |
|
|
17,051,037 |
|
|
14,868,501 |
|
|
7,606,172 |
|
The accompanying notes are an integral part of these financial statements.
66
INVUITY, INC.
Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(in thousands, except share and per share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
Stockholders' |
|
|||
|
|
|
Convertible Preferred Stock |
|
|
Common Stock |
|
Paid-In |
|
Accumulated |
|
Equity |
|
||||||||||
|
|
|
Shares |
|
Amount |
|
|
Shares |
|
Amount |
|
Capital |
|
Deficit |
|
(Deficit) |
|
||||||
|
Balances at December 31, 2014 |
|
6,056,403 |
|
$ |
73,755 |
|
|
|
711,249 |
|
$ |
1 |
|
$ |
2,209 |
|
$ |
(68,037) |
|
$ |
(65,827) |
|
|
Issuance of Series F convertible preferred stock for cash at $14.3449 per share, net of issuance costs of $128 |
|
1,596,212 |
|
|
22,769 |
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Conversion of Convertible Preferred Stock upon IPO |
|
(7,652,615) |
|
|
(96,524) |
|
|
|
7,979,332 |
|
|
8 |
|
|
96,516 |
|
|
— |
|
|
96,524 |
|
|
Conversion of preferred stock warrants to common stock warrants |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
608 |
|
|
— |
|
|
608 |
|
|
Proceeds from issuance of common stock, net issuance costs of $7,988 |
|
— |
|
|
— |
|
|
|
4,600,000 |
|
|
4 |
|
|
47,215 |
|
|
— |
|
|
47,219 |
|
|
Exercise of common stock options |
|
— |
|
|
— |
|
|
|
102,250 |
|
|
— |
|
|
139 |
|
|
— |
|
|
139 |
|
|
Repurchase of early exercised options |
|
— |
|
|
— |
|
|
|
(473) |
|
|
— |
|
|
(1) |
|
|
— |
|
|
(1) |
|
|
Vesting of early exercise options |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
2 |
|
|
— |
|
|
2 |
|
|
Stock-based compensation expense |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
1,249 |
|
|
— |
|
|
1,249 |
|
|
Net loss |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
(37,570) |
|
|
(37,570) |
|
|
Balances at December 31, 2015 |
|
— |
|
|
— |
|
|
|
13,392,358 |
|
|
13 |
|
|
147,937 |
|
|
(105,607) |
|
|
42,343 |
|
|
Proceeds from secondary offering, net issuance costs of $590 |
|
— |
|
|
— |
|
|
|
3,220,000 |
|
|
3 |
|
|
29,675 |
|
|
— |
|
|
29,678 |
|
|
Exercise of common stock options |
|
— |
|
|
— |
|
|
|
338,582 |
|
|
1 |
|
|
732 |
|
|
— |
|
|
733 |
|
|
Stock-based compensation expense |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
2,303 |
|
|
— |
|
|
2,303 |
|
|
Net loss |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
(40,609) |
|
|
(40,609) |
|
|
Balances at December 31, 2016 |
|
— |
|
|
— |
|
|
|
16,950,940 |
|
|
17 |
|
|
180,647 |
|
|
(146,216) |
|
|
34,448 |
|
|
Exercise of common stock options |
|
— |
|
|
— |
|
|
|
165,687 |
|
|
— |
|
|
526 |
|
|
— |
|
|
526 |
|
|
Issuance of common stock due to vesting of restricted stock units |
|
— |
|
|
— |
|
|
|
27,231 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Issuance of common stock warrants related to term loan facilities |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
556 |
|
|
— |
|
|
556 |
|
|
Proceeds from at-the-market offering, net issuance costs of $125 |
|
— |
|
|
— |
|
|
|
35,400 |
|
|
— |
|
|
183 |
|
|
— |
|
|
183 |
|
|
Stock-based compensation expense |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
3,343 |
|
|
— |
|
|
3,343 |
|
|
Net loss |
|
— |
|
|
— |
|
|
|
— |
|
|
— |
|
|
— |
|
|
(39,918) |
|
|
(39,918) |
|
|
Balances at December 31, 2017 |
|
— |
|
$ |
— |
|
|
|
17,179,258 |
|
$ |
17 |
|
$ |
185,255 |
|
$ |
(186,134) |
|
$ |
(862) |
|
The accompanying notes are an integral part of these financial statements.
67
INVUITY, INC.
(in thousands)
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Cash flows from operating activities |
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(39,918) |
|
$ |
(40,609) |
|
$ |
(37,570) |
|
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
1,992 |
|
|
1,995 |
|
|
1,710 |
|
|
Stock-based compensation |
|
|
3,343 |
|
|
2,303 |
|
|
1,249 |
|
|
Changes in fair value of convertible preferred stock warrant liability |
|
|
— |
|
|
— |
|
|
472 |
|
|
Provision for (recovery of) doubtful accounts |
|
|
(23) |
|
|
136 |
|
|
216 |
|
|
Non-cash portion of extinguishment loss |
|
|
352 |
|
|
— |
|
|
— |
|
|
Payment of original issue discount |
|
|
(210) |
|
|
— |
|
|
— |
|
|
Noncash interest expense |
|
|
122 |
|
|
143 |
|
|
143 |
|
|
Accretion of premium (discount) on marketable securities |
|
|
(17) |
|
|
9 |
|
|
— |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
(1,581) |
|
|
(2,300) |
|
|
(1,033) |
|
|
Inventory |
|
|
(2,384) |
|
|
130 |
|
|
(911) |
|
|
Prepaid expenses and other current assets |
|
|
(186) |
|
|
(165) |
|
|
1,568 |
|
|
Other non-current assets |
|
|
(285) |
|
|
— |
|
|
— |
|
|
Accounts payable |
|
|
1,372 |
|
|
(284) |
|
|
1,349 |
|
|
Accrued and other current liabilities |
|
|
(1,310) |
|
|
2,107 |
|
|
1,537 |
|
|
Deferred rent |
|
|
(152) |
|
|
(89) |
|
|
105 |
|
|
Net cash used in operating activities |
|
|
(38,885) |
|
|
(36,624) |
|
|
(31,165) |
|
|
Cash flows from investing activities |
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(844) |
|
|
(1,037) |
|
|
(3,748) |
|
|
Purchases of marketable securities |
|
|
(12,503) |
|
|
(10,746) |
|
|
— |
|
|
Maturities of marketable securities |
|
|
20,217 |
|
|
— |
|
|
— |
|
|
Net cash used in (provided by) investing activities |
|
|
6,870 |
|
|
(11,783) |
|
|
(3,748) |
|
|
Cash flows from financing activities |
|
|
|
|
|
|
|
|
|
|
|
Proceeds from revolving credit facility, net of payment |
|
|
5,859 |
|
|
— |
|
|
— |
|
|
Proceeds from issuance of common stock upon initial public offering, net of issuance costs |
|
|
— |
|
|
— |
|
|
47,219 |
|
|
Proceeds from common stock offerings, net of offering costs |
|
|
203 |
|
|
29,678 |
|
|
— |
|
|
Proceeds from issuance of long-term debt, net of issuance costs |
|
|
29,672 |
|
|
— |
|
|
— |
|
|
Proceeds from issuance of long-term debt-related party, net of issuance costs |
|
|
— |
|
|
— |
|
|
5,000 |
|
|
Repayments of original proceeds from long-term debt-related party |
|
|
(14,765) |
|
|
— |
|
|
— |
|
|
Proceeds from issuance of common stock upon exercise of stock options |
|
|
526 |
|
|
733 |
|
|
139 |
|
|
Payments to repurchase early exercised common stock |
|
|
— |
|
|
— |
|
|
(1) |
|
|
Proceeds from issuance of convertible preferred stock, net of issuance costs |
|
|
— |
|
|
— |
|
|
22,769 |
|
|
Net cash provided by financing activities |
|
|
21,495 |
|
|
30,411 |
|
|
75,126 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
(10,520) |
|
|
(17,996) |
|
|
40,213 |
|
|
Cash and cash equivalents and restricted cash, beginning of year |
|
|
29,390 |
|
|
47,386 |
|
|
7,173 |
|
|
Cash and cash equivalents and restricted cash, end of year |
|
$ |
18,870 |
|
$ |
29,390 |
|
$ |
47,386 |
|
|
Reconciliation of cash, cash equivalents and restricted cash as shown in the statement of cash flows |
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
|
17,962 |
|
|
28,300 |
|
|
46,296 |
|
|
Restricted cash classified as current assets |
|
|
181 |
|
|
181 |
|
|
— |
|
|
Restricted cash classified as long-term assets |
|
|
727 |
|
|
909 |
|
|
1,090 |
|
|
Total cash, cash equivalents and restricted cash |
|
|
18,870 |
|
|
29,390 |
|
|
47,386 |
|
|
Supplemental disclosures of cash flow information |
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest to related party |
|
$ |
377 |
|
$ |
1,875 |
|
$ |
1,740 |
|
|
Cash paid for interest |
|
$ |
1,536 |
|
$ |
— |
|
$ |
— |
|
|
Non-cash investing and financing activities |
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment in accounts payable and accrued liabilities at period end |
|
$ |
59 |
|
$ |
48 |
|
$ |
78 |
|
|
At-the-market offering in accounts payable and accrued liabilities |
|
$ |
20 |
|
$ |
— |
|
$ |
— |
|
|
Reclassification of convertible preferred stock warrant liability to additional paid-in capital upon conversion of preferred stock warrants into common stock warrants |
|
$ |
— |
|
$ |
— |
|
$ |
608 |
|
|
Conversion of convertible preferred stock into common stock and additional paid in capital |
|
$ |
— |
|
$ |
— |
|
$ |
96,524 |
|
The accompanying notes are an integral part of these financial statements.
68
INVUITY, INC.
1.ORGANIZATION AND DESCRIPTION OF BUSINESS
Invuity, Inc. (the “Company”) was incorporated in California on November 29, 2004 and reincorporated in Delaware in May 2015. The Company is a commercial-stage medical technology company that utilizes its proprietary Intelligent Photonics technology to develop single-use and reusable illuminated surgical devices, which provide surgeons with illumination and direct visualization of surgical cavities during minimal access procedures. The Company’s manufacturing, development and management facilities are located in San Francisco, California.
Liquidity and Going Concern
The Company has incurred net losses from operations since inception, including $39.9 million for the year ended December 31, 2017, and has an accumulated deficit of $186.1 million as of December 31, 2017. The Company has $21.0 million in cash and cash equivalents and short-term investments, $35.0 million in debt outstanding and a working capital of $22.7 million at December 31, 2017. The Company is required to comply with a financial covenant relating to certain quarterly minimum Net Revenue (as defined in the debt agreements) requirements on a trailing twelve-month basis. See Note 6 for further details. As of December 31, 2017, the Company was in compliance with this covenant. The Company expects to incur additional losses and negative cash flows for the foreseeable future as the Company continues to invest in its sales and marketing efforts and research and development activities to continue to grow its business.
The Company believes that its cash, cash equivalents and short-term investments as of December 31, 2017, and expected sales from its products together with additional funding available under the Company’s revolving credit facility will not provide sufficient funds to enable the Company to meet its projected operating requirements for the next twelve months from the issuance of these financial statements.
The Company intends to obtain additional funding through public or private financing, collaborative arrangements with strategic partners, or through additional credit lines or other debt financing sources to increase the funds available to support its operating and capital needs. If the Company is not able to perform according to the Company’s 2018 operating plan, the Company’s available capital resources may be consumed more rapidly than currently expected, and therefore may be required to raise additional funds sooner than anticipated. However, the Company may not be able to secure such financing in a timely manner or on favorable terms, if at all. Without additional funds, the Company will be forced to delay, scale back or eliminate some of its sales and marketing efforts, research and development activities, or other operations and potentially delay product development in an effort to provide sufficient funds to continue its operations.
The uncertainty around the achievement of the Company’s plans to mitigate the risk of going concern raises substantial doubt about the Company’s ability to continue as a going concern for a one year period from the issuance of these financial statements. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
2.SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
These financial statements have been prepared in accordance with accounting principles generally accepted in the U.S. (U.S. GAAP).
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. On an ongoing basis, management evaluates its estimates, including those related to revenue recognition, fair value of assets
69
and liabilities, inventory, income taxes, common stock, and stock-based compensation. Actual results could differ from those estimates and assumptions.
Out-of-period and Other Adjustments
In the twelve-months ended December 31, 2015, the Company recorded an out-of-period adjustment to reverse revenue that the Company had originally recorded in the fourth quarter of 2014 associated with sales to a distributor for military facilities. The correction of this error resulted in an increase to the Company’s net loss of $302,000 for the twelve months ended December 31, 2015 and a corresponding decrease to accounts receivable. The distributor returned the underlying inventory, and the Company terminated the relationship with the distributor involved, and started working with a new distributor for military accounts.
In addition, during the twelve months ended December 31, 2015, the Company recorded an out-of-period adjustment to increase the fair value of the convertible preferred stock warrant liability, which was incorrectly valued at December 31, 2014 due to an error in the expected term assumption. The correction of this error resulted in an increase to the Company’s net loss of $370,000 for the twelve months ended December 31, 2015 and a corresponding increase to the convertible preferred stock warrant liability.
Management assessed the impact of these adjustments and did not believe the amounts were material to any prior period financial statements, and the impact of correcting these errors in the twelve months ended December 31, 2015 was not material to those financial statements. As a result, the Company did not restate any prior period amounts.
Cash Equivalents
Cash equivalents consist of short-term, highly liquid investments with original maturities of three months or less from the date of purchase. Cash equivalents consist primarily of amounts invested in money market funds.
Restricted Cash
Restricted cash represents a letter of credit related to the Company’s facility lease.
Short-Term Investments
All short-term investments are classified as “available-for-sale” and carried at estimated fair value as determined based upon quoted market prices or pricing models for similar securities. Management determines the appropriate classification of its investments in debt securities at the time of purchase and reevaluates such designation as of each balance sheet date. Unrealized gains and losses are excluded from earnings and are reported as a component of comprehensive loss. Realized gains and losses and declines in fair value judged to be other than temporary, if any, on available-for-sale securities are included in interest and other income (expense), net, respectively, and are derived using the specific identification method for determining the cost of securities sold. Interest on available-for-sale securities is included in interest and other income (expense), net. Unrealized gains and losses and realized gains and losses on sale of short-term investments were not material for the years ended December 31, 2017, 2016, and 2015. The Company had total short-term investments of $3.0 million and $10.7 million as of December 31, 2017 and 2016, respectively.
Accounts Receivable
Accounts receivable are recorded at the invoiced amount and do not bear interest. Allowances are provided for individual accounts receivable when the Company becomes aware of a customer’s inability to meet its financial obligations, such as in the case of bankruptcy, deterioration in the customer’s operating results or change in financial position. If circumstances related to customers change, estimates of the recoverability of receivables would be further adjusted. The Company also considers broad factors in evaluating the sufficiency of its allowance for doubtful accounts, including the length of time receivables are past due, significant one-time events, creditworthiness of customers and historical experience. Account balances are charged off against the allowance after all means of collection have been exhausted and the potential for recovery is considered remote.
70
The allowance for doubtful accounts balance was $268,000 and $300,000 as of December 31, 2017 and 2016, respectively. The Company wrote off $0, $6,000 and $165,000 as uncollectible accounts receivables to the allowance for doubtful accounts during the years ended December 31, 2017, 2016, and 2015, respectively.
Fair Value of Financial Instruments
Carrying amounts of the Company’s financial instruments, including cash equivalents, short-term investments, accounts receivable, and accounts payable approximate fair value due to their relatively short maturities. As of December 31, 2017, based on Level 2 inputs and the borrowing rates available to the Company for loans with similar terms and consideration of the Company’s credit risk, the carrying value of the Company’s debt approximates its fair value.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk at December 31, 2017 and 2016 consist primarily of cash and cash equivalents, which are held primarily by one domestic financial institution and exceeds federally insured limits. The Company manages its liquidity risk by investing in a variety of money market funds and corporate debt. This diversification of investments is consistent with the Company’s policy to maintain liquidity and ensure the ability to collect principal. All investments are made pursuant to corporate investment policy guidelines which restrict investments to issuers evaluated as creditworthy.
Significant customers are those which represent 10% or more of the Company’s total revenue for each period presented in the statements of operations and comprehensive loss or 10% or more of the Company’s net accounts receivable balance at each respective balance sheet date. As of December 31, 2017 and 2016 and for the years ended December 31, 2017, 2016, and 2015, the Company had no customers that represented 10% or more of its revenue or accounts receivable balances.
Inventory
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the standard cost method, which approximates the first-in, first out basis. The Company periodically assesses the recoverability of all inventories, including raw materials and finished goods, to determine whether adjustments to the carrying value are required. Inventory that is obsolete or in excess of forecasted usage is written down to its estimated net realizable value based on assumptions about future demand and market conditions. Inventory write-downs are charged to cost of goods.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Depreciation is provided using the straight-line method over the estimated useful lives of the respective assets. The estimated useful lives of the Company’s assets are as follows:
|
Laboratory equipment |
|
5 years |
|
Leasehold improvements |
|
Shorter of lease term or estimated life of the assets |
|
Furniture and fixtures |
|
3 years |
|
Computer equipment and software |
|
2 to 3 years |
|
Manufacturing equipment |
|
5 years |
Maintenance and repairs that do not extend the life or improve the asset are expensed when incurred.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset (or asset group) may not be recoverable. An impairment loss is recognized when the total of estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. Impairment, if any, would be assessed using discounted cash flows or other appropriate measures of fair value. The Company has not recorded impairment charges on long-lived assets for the periods presented in these financial statements.
71
Convertible Preferred Stock Warrant Liability
Freestanding warrants for shares that were contingently redeemable were classified as liabilities on the balance sheet at their estimated fair value because the shares underlying the warrants may obligate the Company to transfer assets to the holders at a future date under certain circumstances such as a deemed liquidation event. The warrants were subject to re-measurement at each balance sheet date and the change in fair value, if any, was recognized as interest and other income (expense), net in the statements of operations. The Company adjusted the liability for changes in fair value until the completion of its IPO, at which time all convertible preferred stock warrants were converted into warrants to purchase common stock and the liability was reclassified to additional paid-in capital.
Other Comprehensive Income (Loss)
Other comprehensive income (loss) represents all changes in stockholders’ equity (deficit) except those resulting from distributions to stockholders. The Company’s unrealized loss on short-term available-for-sale securities represent the components of other comprehensive income (loss) that are excluded from the reported net loss and are presented in the statements of comprehensive loss.
Revenue Recognition
The Company’s revenue is generated from the sale of its products to hospitals and medical centers through direct sales representatives and independent sales agents. The Company recognizes revenue when all of the following criteria are met:
|
· |
persuasive evidence of an arrangement exists; |
|
· |
the sales price is fixed or determinable; |
|
· |
collection of the relevant receivable is reasonably assured at the time of sale; and |
|
· |
delivery has occurred or services have been rendered. |
The Company recognizes revenue when title to the goods and risk of loss transfers to the customer, which is upon shipment of the product under the Company’s standard terms and conditions. Shipping and handling costs billed to the customer are recorded in revenue.
In certain circumstances, the Company enters into arrangements in which multiple deliverables are provided to customers. Under multiple deliverable arrangements, the Company recognizes revenue in accordance with the principles described above and allocates the revenue based on the relative selling price of each deliverable, which is based on stand alone selling price.
Warranty Obligations
The Company does not offer rights of return or price protection and has no post-delivery obligations other than its standard warranty which entitles the customer to return defective products for a period of one year after sale. The warranty liability was $50,000 as of December 31, 2017. Historical warranty costs have been insignificant.
Medical Device Excise Tax
In March 2010, the Patient Protection and Affordable Care Act was signed into law which included a deductible 2.3% excise tax on any entity that manufactures or imports medical devices offered for sale in the United States, with limited exceptions, effective January 1, 2013. Subsequently, this excise tax was suspended effective January 1, 2016 through December 31, 2017 and suspended again under the 2018 short-term spending bill. Under this bill, this tax was suspended through December 31, 2020, unless subsequent legislation changes this provision.
72
Research and Development
The Company’s research and development costs are expensed as incurred. Research and development costs includes but are not limited to, payroll and personnel-related expenses, including stock-based compensation, laboratory supplies, consulting costs, and allocated facilities and information services costs.
Income Taxes
The Company accounts for income taxes under the asset and liability method, whereby deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. A valuation allowance is established when, in management’s estimate, it is more likely than not that the deferred tax asset will not be realized.
The tax effects of the Company’s income tax positions are recognized only if they are more likely than not to be sustained based solely on the technical merits as of the reporting date. The Company considers many factors when evaluating and estimating its tax positions and benefits, which may require periodic adjustments and which may not accurately anticipate actual outcomes.
Stock-based Compensation
The Company measures its stock-based awards made to employees based on the estimated fair values of the awards as of the grant date using the Black-Scholes option-pricing model. Stock-based compensation expense is recognized over the requisite service period using the straight-line method and is based on the value of the portion of stock-based payment awards that ultimately vests. The Company’s stock-based compensation is reduced for actual forfeitures in the period and upon the actual date of forfeiture.
Stock-based compensation expense for options granted to non-employees as consideration for services received is measured on the date of performance at the fair value of the consideration received or the fair value of the equity instruments issued, using the Black-Scholes option-pricing model, whichever can be more reliably measured. Compensation expense for options granted to non-employees is periodically remeasured as the underlying options vest.
Segment Reporting
The Company manages its operations as a single operating segment for the purposes of assessing performance and making operating decisions. The majority of the Company’s assets are maintained in the United States. The Company derives its revenue primarily from sales to customers in the United States, based upon the billing address of the customer. In June 2017, the Company started selling into Asia with sales through December 31, 2017 being immaterial.
Net Loss per Common Share
Basic net loss per common share is calculated by dividing the net loss by the weighted-average number of shares of common stock outstanding during the period, without consideration of potentially dilutive securities. Diluted net loss per common share is the same as basic net loss per common share since the effect of potentially dilutive securities are anti-dilutive. Shares subject to repurchase are excluded from the weighted-average shares.
Recent Accounting Pronouncements
|
· |
In May 2014, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (“ASU”) No. 2014‑09, Revenue from Contracts with Customers (Topic 606), which supersedes the revenue recognition requirements in ASC 605, Revenue Recognition. This ASU is based on the principle that revenue is recognized to depict the transfer of goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The ASU also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. In August 2015, the FASB issued ASU No. 2015‑14, Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date, which |
73
effectively delayed the adoption date by one year, to an effective date for public entities for annual and interim periods beginning after December 15, 2017. |
|
· |
In March 2016, the FASB issued ASU No. 2016‑08, Revenue from Contracts with Customers (Topic 606): Principal Versus Agent Considerations (Reporting Revenue Gross Versus Net), to clarify certain aspects of the principal-versus-agent guidance in its new revenue recognition standard. |
|
· |
In April 2016, the FASB issued ASU No. 2016‑10, Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing to clarify on how to identify the performance obligations and the licensing implementation guidance in its new revenue recognition standard. |
|
· |
In May 2016, the FASB issued ASU No. 2016‑12, Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients, to address certain issues identified by the Transition Resource Group, (the “TRG”) in the guidance on assessing collectability, presentation of sales tax, noncash consideration, and completed contracts and contracts modifications at transition. |
The Company will adopt the new revenue standard on January 1, 2018, using the modified retrospective method. The new revenue standard is principles-based and interpretation of those principles may vary from company to company based on their unique circumstances. It is possible that interpretation, industry practice, and guidance may evolve as companies and the accounting profession work to implement this new standard. The Company has determined that the new guidance will not have a material impact on its financial statements.
|
· |
In July 2015, the FASB issued ASU No. 2015‑11, Inventory (Topic 330): Simplifying the Measurement of Inventory, which permits companies to measure inventory at the lower of cost and realizable value. ASU 2015‑11 applies to all business entities and is effective for public business entities for annual periods, and interim periods within those annual periods, beginning after December 15, 2016. The adoption of this standard in the first quarter of 2017 did not have a material impact on the Company’s financial statements. |
|
· |
In January 2016, the FASB issued ASU No. 2016‑01, Recognition and Measurement of Financial Assets and Financial Liabilities (Topic 825), which addresses certain aspects of recognition, measurement, presentation and disclosure of financial instruments. ASU No. 2016‑01 is effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2016. The adoption of this standard in the first quarter of 2017 did not have a material impact on the Company’s financial statements. |
|
· |
In February 2016, the FASB issued ASU No. 2016‑02—Leases (“ASC 842”), which sets out the principles for the recognition, measurement, presentation and disclosure of leases for both parties to a contract (i.e. lessees and lessors). The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight line basis over the term of the lease, respectively. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than twelve months regardless of their classification. Leases with a term of twelve months or less will be accounted for similar to existing guidance for operating leases today. ASC 842 supersedes the previous leases standard, ASC 840 Leases. The standard is effective on January 1, 2019, with early adoption permitted. The Company is in the process of evaluating the impact of this new guidance on its financial statements. |
|
· |
In March 2016, the FASB issued ASU No. 2016‑09, Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. This update simplifies the accounting for employee share-based payment transactions, including the accounting for income taxes, forfeitures, and statutory tax withholding requirements, as well as classification in the statement of cash flows. ASU No. 2016‑09 is effective for public entities for annual periods beginning after December 15, 2016. |
As a result of adopting ASU No. 2016‑09 in the first quarter of 2017, the Company has made an accounting policy election to account for forfeitures as they occur. This change has been applied on a modified retrospective basis, with no material impact on the Company’s financial statements. The adoption of ASU
74
No. 2016‑09 also requires excess tax benefits and tax deficiencies be recorded in the statement of operations as opposed to additional paid‑in capital when the awards vest or are settled, and has been applied on a prospective basis with no impact on the financial statements as of and for the year ended December 31, 2017. As a result of the adoption, the Company's increased its total NOLs by $1.0 million on January 1, 2017 related to deferred tax assets that arose directly from tax deductions related to equity compensation greater than compensation recognized for financial reporting purposes. This amount is fully offset by a valuation allowance.
The adoption of ASU No. 2016‑09 related to the accounting for minimum statutory withholding tax requirements and cash paid by an employer when directly withholding shares for tax-withholding purposes had no impact on the Company's current financial statements or on any prior period financial statements presented.
|
· |
In June 2016, the FASB issued ASU No. 2016‑13, Measurement of Credit Losses on Financial Statements (Topic 326). This update provides financial statement users with more decision-useful information about the expected credit losses on financial instruments and other commitments to extend credit held by a reporting entity at each reporting date. The update replaces the incurred loss impairment methodology in current GAAP with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. ASU No. 2016‑13 is effective for public entities for annual periods beginning after December 15, 2019. The Company is in the process of evaluating the impact of this new guidance on its financial statements. |
|
· |
In August 2016, the FASB issued ASU No. 2016‑15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments (a consensus of the FASB Emerging Issues Task Force). The new guidance is intended to reduce diversity in practice in how certain transactions are classified in the statement of cash flows. This update addresses the following eight specific cash flow issues: debt prepayment or debt extinguishment costs; settlement of zero-coupon debt instruments or other debt instruments with coupon interest rates that are insignificant in relation to the effective interest rate of the borrowing; contingent consideration payments made after a business combination; proceeds from the settlement of insurance claims; proceeds from the settlement of corporate-owned life insurance policies (“COLIs”) (including bank-owned life insurance policies (“BOLIs”); distributions received from equity method investees; beneficial “interests in securitization transactions; and separately identifiable cash flows and application of the predominance principle. The amendments in ASU No. 2016‑15 should be applied using a retrospective transition method to each period presented. The standard is effective for public entities for annual periods beginning after December 15, 2017. The Company will adopt this standard beginning on January 1, 2018 and as a result, expects to reclassify the $2.0 million debt extinguishment cost paid in cash in the first quarter of 2017 from operating activities to financing activities. |
|
· |
In November 2016, the FASB issued ASU No. 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash. The amendments in this Update require that a statement of cash flows explain the change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. Therefore, amounts generally described as restricted cash and restricted cash equivalents should be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. The amendments in this Update are effective for public business entities for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. The Company early adopted this standard in the fourth quarter of 2017. The adoption is currently reflected in the Company’s financial statements. |
3.FAIR VALUE MEASUREMENTS
The Company discloses and recognizes the fair value of its assets and liabilities using a hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. The hierarchy gives the highest priority to valuations based upon unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to valuations based upon unobservable
75
inputs that are significant to the valuation (Level 3 measurements). The guidance establishes three levels of the fair value hierarchy as follows:
Level 1—Inputs are unadjusted quoted prices in active markets for identical assets or liabilities at the measurement date.
Level 2—Inputs (other than quoted market prices included in Level 1) are either directly or indirectly observable for the asset or liability through correlation with market data at the measurement date and for the duration of the instrument’s anticipated life.
Level 3—Inputs reflect management’s best estimate of what market participants would use in pricing the asset or liability at the measurement date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.
The Company’s financial instruments consist of Level 1 and 2 assets. Level 1 assets consist primarily of highly liquid money market funds that are included in cash, cash equivalents, and restricted cash. Commercial Paper and corporate debt securities are classified in Level 2 of the fair value hierarchy because these valuation inputs are observable or market-corroborated for similar securities.
Prior to the IPO, the Company had a convertible preferred stock warrant liability relating to certain prior year financings. See Note 8 for further discussion. Upon the IPO, in June 2015, all convertible preferred stock warrants were converted into common stock warrants and the company recorded expense of $136,000 relating to the change in fair value of this convertible preferred stock warrant. For the periods ended December 31, 2017 and December 31, 2016 respectively, the Company had no Level 3 assets or liabilities.
The following table sets forth the fair value of the Company’s financial assets and liabilities measured at fair value on a recurring basis based on the three-tier fair value hierarchy (in thousands):
|
|
|
December 31, 2017 |
|
||||||||||
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
13,071 |
|
$ |
— |
|
$ |
— |
|
$ |
13,071 |
|
|
Commercial paper |
|
|
— |
|
|
3,763 |
|
|
— |
|
|
3,763 |
|
|
Corporate debt securities |
|
|
— |
|
|
1,753 |
|
|
— |
|
|
1,753 |
|
|
|
|
$ |
13,071 |
|
$ |
5,516 |
|
$ |
— |
|
$ |
18,587 |
|
|
|
|
December 31, 2016 |
|
||||||||||
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds |
|
$ |
18,755 |
|
$ |
— |
|
$ |
— |
|
$ |
18,755 |
|
|
Commercial paper |
|
|
— |
|
|
9,582 |
|
|
— |
|
|
9,582 |
|
|
Corporate debt securities |
|
|
— |
|
|
9,533 |
|
|
— |
|
|
9,533 |
|
|
|
|
$ |
18,755 |
|
$ |
19,115 |
|
$ |
— |
|
$ |
37,870 |
|
As of December 31, 2017 and 2016, the carrying value of the Company’s short term investments approximates its fair value.
76
4.BALANCE SHEET COMPONENTS
Inventory
Inventory consisted of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Raw materials |
|
$ |
1,606 |
|
$ |
699 |
|
|
Work-in-process |
|
|
1,860 |
|
|
1,144 |
|
|
Finished goods |
|
|
3,970 |
|
|
3,209 |
|
|
Total inventory |
|
$ |
7,436 |
|
$ |
5,052 |
|
Prepaid expenses and other current assets
Prepaid expenses and other current assets consisted of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Prepaid expenses |
|
$ |
1,252 |
|
$ |
1,004 |
|
|
Other |
|
|
22 |
|
|
84 |
|
|
Total prepaid expenses and other assets |
|
$ |
1,274 |
|
$ |
1,088 |
|
Property and Equipment, Net
Property and equipment, net consisted of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Computer equipment and software |
|
$ |
1,368 |
|
$ |
1,330 |
|
|
Laboratory and manufacturing equipment |
|
|
3,008 |
|
|
2,422 |
|
|
Furniture and fixtures |
|
|
1,516 |
|
|
1,442 |
|
|
Leasehold improvements |
|
|
7,155 |
|
|
7,153 |
|
|
Assets in progress |
|
|
167 |
|
|
47 |
|
|
Total property and equipment, gross |
|
|
13,214 |
|
|
12,394 |
|
|
Less: accumulated depreciation and amortization |
|
|
(6,045) |
|
|
(4,108) |
|
|
Total property and equipment, net |
|
$ |
7,169 |
|
$ |
8,286 |
|
Depreciation and amortization expense was $2.0 million during each of the years ended December 31, 2017 and 2016, and $1.7 million during the year ended December 31, 2015.
Accrued and Other Current Liabilities
Accrued and other current liabilities consisted of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Accrued payroll-related expenses |
|
$ |
3,777 |
|
$ |
5,301 |
|
|
Accrued independent sales agent commissions |
|
|
181 |
|
|
193 |
|
|
Accrued professional fees |
|
|
501 |
|
|
246 |
|
|
Deferred rent |
|
|
261 |
|
|
261 |
|
|
Other |
|
|
459 |
|
|
350 |
|
|
Total accrued and other current liabilities |
|
$ |
5,179 |
|
$ |
6,351 |
|
77
5.RELATED PARTY LOAN AGREEMENT
In February 2014, the Company entered into a loan agreement with HealthCare Royalty Partners (“HCRP”), a related party due to its equity ownership interest in the Company, and drew down the first tranche of $10.0 million. The second tranche associated with HCRP, of $5.0 million, was drawn down in March 2015.
Interest was payable quarterly at a fixed rate of 12.5% per annum with interest-only payments to be made from the effective date of the loan until March 31, 2017. The Company was permitted to make a voluntary prepayment in full, but not in part, prior to December 31, 2020, including all accrued and unpaid fixed interest on the amount prepaid and any additional amounts due in respect thereof, including an additional percentage of the aggregate loan amount or outstanding principal amount, depending on the date of prepayment. The Company’s obligations under the loan agreement were secured by a first priority security interest in all of the Company’s assets, other than bank accounts, accounts receivable and inventory. The loan agreement imposed customary affirmative and restrictive covenants, including with respect to fundamental transactions, the incurrence of additional indebtedness or liens and the payment of cash dividends, but did not include any financial covenants. The loan agreement contained a material adverse event clause which provided that an event of default would occur if, among other triggers, there occurred any circumstance that would reasonably expect to result in a material adverse effect on the Company’s business, operations or condition, or on the Company’s ability to perform its obligations under the loan.
In connection with the loan agreement, the Company issued HCRP a warrant to purchase 84,553 shares of Series E convertible preferred stock at $13.3052 per share. The warrant was recorded on the balance sheet on the date of issuance at its fair value of $572,000 and recorded as a reduction in the carrying value of the debt. Upon completion of the IPO in June 2015, this warrant automatically converted into a warrant to purchase 86,891 shares of common stock and the liability was reclassified to additional paid-in capital. The Company also paid $200,000 in debt issuance costs to HCRP, which were recorded as a debt discount. The total debt discount was amortized as interest expense in the straight-line method over the term of the loan.
On March 10, 2017, the Company entered into a new credit and security agreement with MidCap Financial Trust and affiliates (“MidCap”) and used $17.2 million received under the new term loan to terminate the loan agreement with HRCP and payoff in full the loan of $15 million and $2.2 million in associated prepayment fees and interest. The Company recorded interest expense of $0.4 million and $2.0 million on the loan, for the years ended December 31, 2017 and 2016, respectively.
6.DEBT
MidCap credit and security agreement:
On March 10, 2017, the Company entered into a credit and security agreement with MidCap, as agent, for up to $30.0 million in term loans. Under the terms of the agreement, the Company borrowed the first term loan of $20.0 million (“Tranche 1”) at closing. The Tranche 1 term loan accrues interest at a floating rate equal to 6.50% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. Interest shall accrue on the date of the commencement of funding and is payable in arrears on the first day of each month. Principal is payable in 36 equal monthly installments beginning April 1, 2019, subject to extension to October 1, 2019, if the Company achieves a certain revenue target, until paid in full on March 1, 2022. The Company used $17.2 million of the $20.0 million Tranche 1 term loan to pay off in full the outstanding $15.0 million loan with Health Care Royalty Partners. The Company also terminated its accounts receivable credit facility with Silicon Valley Bank which was never drawn down. The transaction was accounted for as a debt extinguishment and a loss of $2.3 million was accounted for as loss on extinguishment of debt in the income statement. The loss amount includes a $1.8 million prepayment fee paid to HCRP, a $150,000 fee paid to Silicon Valley Bank for termination of the accounts receivable facility and a $0.3 million non-cash expense related to unamortized issuance costs.
On September 26, 2017, the Company amended the credit and security agreement with MidCap and borrowed the second term loan of $10.0 million (“Tranche 2”). The Tranche 2 term loan accrues interest at a floating rate equal to 6.50% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. Interest shall accrue on the date of the commencement of funding and is payable in arrears on the first day of each month. Principal is payable in 36 equal monthly installments beginning April 1, 2019, subject to extension to October 1, 2019, if the Company achieves a certain revenue target, until paid in full on March 1, 2022.
78
The Company also entered into a separate credit and security agreement with MidCap on March 10, 2017 that provides for a revolving credit facility of up to $10.0 million based on the eligible accounts receivable and inventory balances, as amended on September 26, 2017. The Company may increase the total commitments under the revolving credit facility by up to an additional $10.0 million, subject to the Company meeting certain conditions. Loans under the revolving credit facility accrue interest at a floating rate equal to 3.25% per annum, plus the greater of (i) 1.5% or (ii) one month LIBOR. Interest is payable in arrears on the first day of each month subsequent to the draw down date. The facility terminates in full on March 1, 2022 unless terminated earlier. As of December 31, 2017, the Company had drawn down $5.9 million under the revolving credit facility.
The term loan facility and the revolving credit facility are secured by substantially all of the Company’s assets, including intellectual property. In addition, under the terms of the agreement, the Company is required to meet certain covenants which if the Company is unable to meet, or if the Company does not make its payments, the Company may be found in default and all obligations may be accelerated and become immediately due and payable upon the sole election of the lenders. The Company must also comply with a financial covenant relating to certain quarterly minimum Net Revenue requirements on a trailing twelve month basis. The minimum Net Revenue requirements on a trailing twelve month basis for 2018 is $40,488,151 for the period ending March 31, 2018, $42,248,214 for the period ending June 30, 2018, $46,574,986 for the period ending September 30, 2018, and $50,000,000 for the period ending December 31, 2018. Additionally, the credit and security agreement with MidCap, includes customary events of default, including failure to pay amounts due, breaches of covenants and warranties, and material adverse effect events . If an event of default occurs, MidCap may require immediate repayment of all amounts due. As of December 31, 2017, the Company was in compliance with all required covenants.
In connection with the term loan facility, the Company agreed to issue to each lender warrants to purchase shares of the Company’s common stock upon the drawdown of each tranche in an aggregate amount equal to 2.0% of the amount drawn, divided by the exercise price per share for that tranche. See Note 8 – Warrants for further details.
Future payments due under the Company’s term loans as of December 31, 2017 are as follow (in thousands):
|
Year ending December 31, |
|
|
|
|
|
2018 |
|
|
2,490 |
|
|
2019 |
|
|
14,644 |
|
|
2020 |
|
|
10,194 |
|
|
2021 |
|
|
7,105 |
|
|
Thereafter |
|
|
1,690 |
|
|
|
|
|
36,123 |
|
|
Less: Amount representing interest |
|
|
(6,122) |
|
|
Less: Amount representing debt discount (1) |
|
|
(885) |
|
|
Total |
|
$ |
29,116 |
|
|
|
|
|
|
|
|
(1) Interest expense is based on a 6.5% base rate plus one month Libor rate |
|
|
|
|
Silicon Valley Bank Accounts Receivable Credit Facility:
In February 2015, the Company entered into an accounts receivable credit facility with Silicon Valley Bank (“SVB”) that allowed the Company to borrow the lesser of $7.5 million or an amount representing up to 80% of eligible accounts receivable. In March 2017, this facility was replaced by the MidCap facility noted above. The Company paid a fee of $150,000 to terminate this facility.
7.COMMITMENTS AND CONTINGENCIES
Operating Leases
The Company leases manufacturing and office space in San Francisco, California, under a non-cancelable operating lease entered into in May 2014. The lease commencement date was November 1, 2014 and the lease expires on October 31, 2024. At the inception of the lease, the Company provided the landlord with a security deposit of $1.1
79
million in the form of an irrevocable letter of credit, which was recorded in restricted cash on the balance sheet at both December 31, 2017 and December 31, 2016.
Rent expense is recognized on a straight-line basis over the term of the leases and accordingly, the Company records the difference between cash rent payments and the recognition of rent expense as a deferred rent liability. Incentives granted under the Company’s facilities leases, including allowances to fund leasehold improvements, are deferred and are recognized as adjustments to rental expense on a straight-line basis over the term of the lease. The Company was entitled to a $2.6 million tenant allowance in connection with the lease entered into in November 2014. The Company utilized the entire $2.6 million allowance in connection with qualified costs as of December 31, 2014 and was fully reimbursed by the landlord as of December 31, 2015. The allowance has been recorded on the balance sheet as a leasehold improvement and is being amortized over the term of the lease as a reduction to rent expense.
The following table summarizes the Company’s future minimum lease payments as of December 31, 2017 (in thousands):
|
Year ending December 31: |
|
|
|
|
|
2018 |
|
$ |
2,178 |
|
|
2019 |
|
|
2,243 |
|
|
2020 |
|
|
2,310 |
|
|
2021 |
|
|
2,380 |
|
|
2022 |
|
|
2,451 |
|
|
Thereafter |
|
|
4,680 |
|
|
Total |
|
$ |
16,242 |
|
The Company’s rent expense was $2.0 million for the years ended December 31, 2017, 2016, and 2015, respectively.
Legal Proceedings
On February 27, 2017, a purported stockholder class action titled Paciga v. Invuity, Inc., et al., Case No. 3:17‑cv‑01005, was filed in the United States District Court for the Northern District of California against us, our Chief Executive Officer, and our Chief Financial Officer. The complaint alleges that the defendants made false or misleading statements to investors regarding our business prospects. The complaint purports to assert claims for violation of Sections 10(b) and 20(a) of the Exchange Act of 1934, and SEC Rule 10b‑5 on behalf of a purported class consisting of all purchasers of our common stock between July 19, 2016 and November 3, 2016, and seeks unspecified compensatory damages, attorney fees and costs, and other relief. On May 30, 2017, the Court appointed Mike Paciga as lead plaintiff. The lead plaintiff filed an amended complaint on July 31, 2017. Defendants filed a motion to dismiss on September 14, 2017, the lead plaintiff filed his opposition to the motion on October 30, 2017 and Defendants filed a reply brief on December 4, 2017. The motion to dismiss is currently scheduled for hearing on April 6, 2018. The Company intends to defend the litigation vigorously. Based on information currently available, the Company has determined that the amount of any possible loss or range of possible loss is not reasonably estimable.
In addition, the Company is, and from time to time may become, involved in legal proceedings arising from the ordinary course of its business. Management is currently not aware of any matters that will have a material adverse effect on the Company’s financial position, results of operations or cash flows.
Indemnifications
In the ordinary course of business, the Company enters into agreements that may include indemnification provisions. Pursuant to such agreements, the Company may indemnify, hold harmless and defend an indemnified party for losses suffered or incurred by the indemnified party. Some of the provisions will limit losses to those arising from third-party actions. In some cases, the indemnification will continue after the termination of the agreement. The maximum potential amount of future payments the Company could be required to make under these provisions is not determinable. The Company has never incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. The Company has also entered into indemnification agreements with its directors and officers that may require the Company to indemnify its directors and officers against liabilities that may arise by reason of their status or service as directors or officers to the fullest extent permitted by Delaware corporate law. The Company currently has directors’ and officers’ insurance. To date, the Company has not paid any claims, and the Company believes that the
80
estimated fair value of these indemnification obligations is minimal and it has not accrued any amounts for these obligations.
8.WARRANTS
Common Stock Warrants
In connection with Tranche 1 of the MidCap term loan facility, the Company issued warrants to purchase an aggregate of 50,618 shares of the Company’s common stock, at an exercise price equal to $7.90 per share. These warrants, which were recorded within stockholders’ equity (deficit), were fair valued at $279,000 upon issuance using a Black-Scholes valuation model. The assumptions used in the Black-Scholes model consisted of a 10 year contractual term, interest free rate of 2.58%, dividend yield of 0.0% and volatility of 60.0%. The fair value was recorded as a discount to the initial $20.0 million term loan and will be amortized as interest expense over the term of the agreement, which is approximately five years. In connection with the Tranche 2 term loan, the Company issued warrants to purchase an aggregate of 47,790 shares of the Company’s common stock, at an exercise price equal to $8.37 per share. These warrants, which were recorded within stockholders’ equity (deficit), were fair valued at $278,000 upon issuance using a Black-Scholes valuation model. The assumptions used in the Black-Scholes model consisted of a 10 year contractual term, interest free rate of 2.24%, dividend yield of 0.0% and volatility of 60.0%. The fair value was recorded as a discount to the initial $10.0 million term loan and will be amortized as interest expense over the term of the agreement, which is approximately five years.
In March 2010, the Company issued a warrant to purchase 3,532 shares of common stock at an exercise price of $1.30 per share to a third party in exchange for recruiting services. The Company recorded the warrants in stockholders’ equity (deficit) at their fair value of $3,000 on the date of issuance using the Black-Scholes option-pricing model. The warrant was fully exercisable upon grant and expired upon the Company’s IPO in June 2015.
Preferred Stock Warrants
In conjunction with various financings between 2008 and 2014, the Company issued warrants to purchase 130,540 shares of convertible preferred stock. The relative fair value of these warrants was determined using the Black-Scholes model and was amortized to interest expense over the term of each loan, unless subsequently modified. All convertible preferred stock warrants were classified as liabilities on the balance sheet at their estimated fair value because the shares underlying the warrants could obligate the Company to transfer assets to the holders at a future date under certain circumstances such as a deemed liquidation event. The warrants were subject to re-measurement at each balance sheet date and the change in fair value, if any, was recognized as interest and other income, net in the statements of operations and comprehensive loss. The Company adjusted the liability for changes in fair value until the completion of its IPO, at which time all convertible preferred stock warrants were converted into 137,007 warrants to purchase common stock and the liability was reclassified to additional paid-in capital.
The Company recorded a loss of $472,000 during the year ended December 31, 2015 relating to the change in fair value of the convertible preferred stock warrant liability.
9.CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIT
Convertible Preferred Stock
In February and March 2015, the Company issued an aggregate of 1,596,212 shares of Series F convertible preferred stock. Upon the closing of the Company’s IPO in June 2015, all 7,652,615 shares of convertible preferred stock then outstanding converted into 7,979,332 shares of common stock, which includes an aggregate of 326,717 additional shares of common stock related to anti-dilution adjustments upon conversion of the convertible preferred stock.
Reverse Stock Split
In May 2015, the Company’s board of directors and its stockholders approved an amendment to the Company’s amended and restated articles of incorporation to effect a reverse split of shares of the Company’s common stock on a 1-for-18.5 basis (the “Reverse Stock Split”). All authorized, issued and outstanding shares of common stock, convertible preferred stock, warrants for common stock and preferred stock, options to purchase common stock and the related per share
81
amounts contained in the financial statements have been retroactively adjusted to reflect this Reverse Stock Split for all periods presented. The Reverse Stock Split was effected on May 27, 2015.
Initial Public Offering
In June 2015, the Company completed an initial public offering (the “IPO”) of its common stock. In connection with its IPO, the Company sold 4,600,000 shares of common stock at $12.00 per share, for aggregate net proceeds of $47.2 million after underwriting discounts and commissions and offering costs incurred by the Company. The net proceeds include the exercise in full by the underwriters of their option to purchase up to 600,000 additional shares of common stock at the same price to cover over-allotments. Upon the closing of the IPO, all shares of convertible preferred stock then outstanding converted into 7,979,332 shares of common stock.
Upon the effectiveness of the Amended and Restated Certificate of Incorporation of the Company on June 18, 2015, the number of shares of capital stock the Company is authorized to issue was increased to 110,000,000 shares, of which 100,000,000 shares are common stock and 10,000,000 shares are preferred stock. Both the common stock and preferred stock have a par value of $0.001 per share. There are no shares of preferred stock outstanding at December 31, 2017.
Secondary Offering
On July 1, 2016, the Company filed a shelf registration statement on Form S-3 (Registration No. 333-212395) with the SEC to offer for sale up to an aggregate of $100.0 million of common stock, preferred stock, depositary shares, warrants and/or units in one or more offerings and in any combinations. On that date, the Company also filed a prospectus supplement for an ATM Offering of up to $25.0 million. On August 2, 2016, the Company completed a secondary offering of 3,220,000 shares of its common stock at a price to the public of $10.00 per share, which included the exercise in full by the underwriters of their option to purchase an additional 420,000 shares of its common stock. The total net proceeds from this offering were $29.7 million, after deducting underwriting discounts and commissions and offering expenses of $0.6 million payable by the Company.
10.STOCK OPTION PLANS
In April 2015, the Company’s board of directors approved the 2015 Equity Incentive plan (the “2015 Plan”), effective June 11, 2015, covering incentive stock options (“ISOs”), nonstatutory stock options (“NSOs”), restricted stock, restricted stock awards (“RSU”), stock appreciation rights and performance units that may be granted to employees, directors and consultants. In connection with the approval of the 2015 Plan, all remaining shares available for future award under the 2005 Stock Option Plan (the “2005 Plan”) were transferred to the 2015 Plan, and the 2005 Plan was terminated. The number of shares initially authorized for issuance under the 2015 Plan was 1,494,272 in addition to 169,529 shares remaining available for future awards under the Company’s 2005 Plan. Any options under the 2005 Plan or 2015 Plan (collectively “the Plans”) that expire or otherwise terminate will revert to the 2015 Plan and again become available for issuance.
The number of shares available for issuance under the 2015 Plan will be increased on the first day of each fiscal year in an amount equal to the lessor of (i) 1,494,272 shares; (ii) five percent of the outstanding shares on the last day of the immediately preceding fiscal year or (iii) such number of shares determined by the Company’s board of directors. ISOs may be granted to employees or directors holding more than 10% of the voting power of all classes of stock of the Company at an exercise price of no less than 110% of the fair value of the common stock on the grant date and to all other employees or directors at an exercise price of no less than 100% of the fair value of the common stock on the grant date. NSOs may be granted to employees, directors and consultants at an exercise price no less than 100% of the fair value of the common stock on the grant date. Employee stock options under the 2015 plan generally vest 25% upon one year of continued service to the Company, with the remainder in monthly increments over three additional years. Options expire no more than ten years after the date of grant.
The Company’s board of directors and stockholders previously approved the 2005 Plan. Pursuant to the 2005 plan, options and restricted stock may be granted to employees, directors and consultants of the Company. Options granted under the Company’s 2005 plan may be either incentive stock options or nonstatutory stock options. ISOs may be granted to employees with exercise prices of no less than 100% the fair value of the common stock on the grant date and NSOs may be granted to employees, directors or consultants at exercise prices of no less than 85% of the fair value of the common stock on the grant date, as determined by the board of directors. All options granted under the 2005 plan
82
may be exercised before they are vested. Employee stock options granted under the 2005 plan generally vest 25% upon one year of continued service to the Company, with the remainder in monthly increments over three additional years. Stock options granted to consultants generally vest over the performance period of the consultancy agreement, ranging from two to four years. Options expire no more than ten years after the date of grant.
As of December 31, 2017, there were 4,430,892 shares authorized for issuance under the Plans, of which 929,940 were available for grant. In the event of stock splits and stock dividends, the board of directors may increase or decrease proportionately the number of shares and the exercise (purchase) price per share deliverable to the 2015 Plan participants. In the event of a merger in which the Company is not the surviving entity or sale of substantially all the Company’s assets, all outstanding options must be either assumed or substituted by the surviving corporation, or may be required to be exercised or settled.
The following table summarizes stock option and restricted stock unit activities and related information:
|
|
|
|
|
Options Outstanding |
|
RSUs Outstanding |
|||||||||||||||
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
Weighted- |
|
Weighted- |
|
|
|
|||
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
Average |
|
Average |
|
|
|
|||
|
|
|
Shares |
|
|
|
Exercise |
|
Aggregate |
|
|
|
Grant Date |
|
Remaining |
|
Aggregate |
|||||
|
|
|
Available |
|
Number of |
|
Price Per |
|
Intrinsic |
|
Number of |
|
Fair Value Per |
|
Contractual |
|
Intrinsic |
|||||
|
|
|
for Grant |
|
Shares |
|
Share |
|
Value |
|
Shares |
|
Share |
|
Terms |
|
Value |
|||||
|
|
|
|
|
|
|
|
|
|
(in thousands) |
|
|
|
|
|
|
(in years) |
|
(in thousands) |
|||
|
Balances at December 31, 2016 |
|
1,198,856 |
|
2,445,600 |
|
$ |
6.86 |
|
$ |
2,920 |
|
131,807 |
|
$ |
7.82 |
|
|
|
|
|
|
|
Options authorized |
|
847,547 |
|
— |
|
|
|
|
|
. |
|
— |
|
|
|
|
|
|
|
|
|
|
Options granted |
|
(1,227,205) |
|
1,227,205 |
|
$ |
7.29 |
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
RSUs awarded |
|
(183,657) |
|
|
|
|
|
|
|
|
|
183,657 |
|
|
6.61 |
|
|
|
|
|
|
|
Options exercised |
|
— |
|
(165,687) |
|
$ |
3.18 |
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
RSUs released |
|
|
|
|
|
|
|
|
|
|
|
(36,804) |
|
|
4.47 |
|
|
|
|
|
|
|
Options cancelled |
|
274,426 |
|
(274,426) |
|
$ |
9.22 |
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
RSUs forfeited/canceled |
|
19,973 |
|
|
|
|
|
|
|
|
|
(10,400) |
|
|
7.45 |
|
|
|
|
|
|
|
Balances at December 31, 2017 |
|
929,940 |
|
3,232,692 |
|
$ |
7.01 |
|
$ |
2,890 |
|
268,260 |
|
$ |
6.84 |
|
|
1.53 |
|
$ |
1,663 |
|
Options exercisable—December 31, 2017 |
|
|
|
1,489,787 |
|
$ |
6.02 |
|
$ |
2,686 |
|
|
|
|
|
|
|
|
|
|
|
|
Options vested and expected to vest—December 31, 2017 |
|
|
|
3,232,692 |
|
$ |
7.01 |
|
$ |
2,890 |
|
|
|
|
|
|
|
|
|
|
|
|
RSUs vested and expected to vest—December 31, 2017 |
|
|
|
|
|
|
|
|
|
|
|
268,260 |
|
$ |
6.84 |
|
|
1.43 |
|
$ |
1,663 |
The intrinsic value is the difference between the estimated fair value of the Company’s common stock at the date of exercise and the exercise price for in-the-money options. The aggregate intrinsic value of options exercised was $0.7 million, $2.3 million and $0.8 million for the years ended December 31, 2017, 2016, and 2015, respectively. The weighted-average grant-date fair value of options granted during the years ended December 31, 2017, 2016, and 2015 was $7.29, $3.43 and $4.82 per share, respectively.
As of December 31, 2017 and 2016, the weighted-average remaining contractual life of options outstanding was 7.0 and 7.5 years, respectively, and for options vested and expected to vest, was 7.0 and 7.5 years, respectively.
83
The options outstanding, vested and currently exercisable by exercise price under the 2005 and 2015 Plans at December 31, 2017 are as follows:
|
|
|
Options Outstanding |
|
Options Exercisable |
|
||||||||
|
|
|
|
|
Weighted- |
|
Weighted- |
|
|
|
Weighted- |
|
||
|
|
|
|
|
Average |
|
Average |
|
|
|
Average |
|
||
|
|
|
|
|
Remaining |
|
Exercise |
|
|
|
Exercise |
|
||
|
|
|
Number of |
|
Contractual |
|
Price Per |
|
Number of |
|
Price Per |
|
||
|
Exercise Price |
|
Options |
|
Life (years) |
|
Share |
|
Options |
|
Share |
|
||
|
$1.30-2.78 |
|
228,049 |
|
2.8 |
|
$ |
1.43 |
|
228,049 |
|
$ |
1.43 |
|
|
$3.15 |
|
503,113 |
|
5.2 |
|
$ |
3.15 |
|
455,914 |
|
$ |
3.15 |
|
|
$4.81-6.50 |
|
799,835 |
|
7.6 |
|
$ |
6.05 |
|
203,489 |
|
$ |
5.24 |
|
|
$6.75-7.45 |
|
558,200 |
|
7.5 |
|
$ |
7.32 |
|
204,657 |
|
$ |
7.32 |
|
|
$7.60-8.40 |
|
468,713 |
|
8.7 |
|
$ |
8.16 |
|
68,865 |
|
$ |
8.26 |
|
|
$8.95-13.00 |
|
500,920 |
|
7.6 |
|
$ |
10.93 |
|
229,696 |
|
$ |
11.33 |
|
|
$13.47-15.91 |
|
173,862 |
|
7.5 |
|
$ |
14.62 |
|
99,117 |
|
$ |
14.87 |
|
|
|
|
3,232,692 |
|
7.0 |
|
$ |
7.01 |
|
1,489,787 |
|
$ |
6.02 |
|
Early Exercise of Stock Options
The 2005 Plan allowed for the granting of options that may be exercised before the options have vested. Shares issued as a result of early exercise that have not vested are subject to repurchase by the Company upon termination of the purchaser’s employment or services, at the price paid by the purchaser. The Company’s right to repurchase these shares generally lapses 1/48 of the original grant date amount per month over four years. As of December 31, 2017 and 2016, there were zero and 2,661 shares of common stock outstanding, respectively, subject to the Company’s right of repurchase. The weighted-average exercise price for the 2,661 shares of common stock was $2.59 per share.
Employee Stock-Based Compensation
Stock-based compensation expense recognized during the years ended December 31, 2017, 2016, and 2015, includes compensation expense for stock-based awards granted to employees based on the grant date fair value of $3.1 million, $2.2 million, and $1.1 million respectively.
As of December 31, 2017 and 2016, there were total unamortized compensation costs of $6.2 million and $4.3 million, respectively, related to unvested stock options which the Company expects to recognize over a period of approximately 2.8 years and 3.1 years, respectively. As of December 31, 2017 and 2016, there were total unamortized compensation costs of $1.3 million and $0.7 million, respectively related to unvested RSUs which the Company expects to recognize over a period of approximately 2.3 years and 3.8 years, respectively
The Company estimates the fair value of stock options using the Black-Scholes option valuation model. The fair value of employee stock options is being amortized on a straight-line basis over the requisite service period of the awards. Prior to the Company’s IPO, the fair value of the shares of the Company’s common stock underlying the stock options has historically been determined by the Company’s board of directors. Because there had been no public market for the Company’s common stock, its board of directors has determined the fair value of the Company’s common stock at the time of grant of the option by considering a number of objective and subjective factors, including the Company’s stage of development, sales of the Company’s convertible preferred stock, the Company’s operating and financial performance, equity market conditions affecting comparable public companies, the lack of liquidity of the Company’s capital stock, and the general and industry-specific economic outlooks.
Since the Company’s IPO in June 2015, the fair value of the Company’s common stock is based on the closing price of its common stock, as quoted on the NASDAQ Global Market, on the date of grant. In addition to the value determined
84
by the board and closing price on NASDAQ Global Market, the fair value is estimated using the assumptions below. Each of these inputs is subjective and its determination generally requires significant judgment.
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Expected term (in years) |
|
6.0 |
|
5.0–6.0 |
|
5.0–6.0 |
|
|
Expected volatility |
|
60 |
% |
50 |
% |
35–50 |
% |
|
Risk-free interest rate |
|
1.87-2.22 |
% |
1.24–2.26 |
% |
1.31–1.89 |
% |
|
Dividend yield |
|
— |
% |
— |
% |
— |
% |
Expected Term. The expected term of stock-based awards represents the weighted-average period that the stock-based awards are expected to remain outstanding. The Company opted to use the “simplified method” for estimating the expected term of the awards, whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the awards.
Expected Volatility. The Company determined the share price volatility for stock-based awards based on an analysis of the historical volatilities of a peer group of publicly traded medical device companies. In evaluating similarity, the Company considered factors such as industry, stage of life cycle and size.
Risk-Free Interest Rate. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant for zero-coupon U.S. Treasury notes with remaining terms similar to the expected term of the stock-based awards.
Dividend Rate. The expected dividend was assumed to be zero as the Company has never paid dividends and has no current plans to do so.
Expected Forfeiture Rate. As allowed under ASU No. 2016‑09, Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, the Company accounts for forfeitures as they occur.
Non-Employee Stock-Based Compensation
Stock-based compensation expense related to non-employee awards was $256,000, $121,000, and $103,000 during the years ended December 31, 2017, 2016, and 2015, respectively.
Total Stock-Based Compensation
The following table summarizes total stock-based compensation expense for the years ended December 31, 2017, 2016, and 2015, which was included in the statements of operations as follows (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Cost of goods sold |
|
$ |
229 |
|
$ |
133 |
|
$ |
89 |
|
|
Selling, general and administrative |
|
|
2,402 |
|
|
1,751 |
|
|
860 |
|
|
Research and development |
|
|
712 |
|
|
419 |
|
|
300 |
|
|
Total stock-based compensation expense |
|
$ |
3,343 |
|
$ |
2,303 |
|
$ |
1,249 |
|
11.INCOME TAXES
The Company has incurred net operating losses for the years ended December 31, 2017, 2016, and 2015, therefore has no provision for income taxes recorded for such years. For the years ended December 31, 2017, 2016, and 2015, the Company generated losses before taxes in the United States of $39.9 million, $40.6 million and $37.6 million, respectively and no foreign income or losses. The Company’s deferred tax assets are offset by a full valuation allowance.
85
The reconciliation of the statutory federal income tax rate to the Company’s effective tax rate is as follows:
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Tax at statutory federal rate |
|
34.0 |
% |
34.0 |
% |
34.0 |
% |
|
State taxes, net of federal benefit |
|
8.0 |
|
3.5 |
|
0.4 |
|
|
Tax credits |
|
0.8 |
|
1.2 |
|
0.9 |
|
|
Change in valuation allowance |
|
17.9 |
|
(37.7) |
|
(33.9) |
|
|
Other |
|
(0.8) |
|
(1.0) |
|
(1.4) |
|
|
Federal Tax Rate Change |
|
(59.9) |
|
— |
|
— |
|
|
Provision for income taxes |
|
0.0 |
% |
0.0 |
% |
(0.0) |
% |
The tax effects of temporary differences and carryforwards that give rise to significant portions of the deferred tax assets are as follows (in thousands):
|
|
|
December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
43,168 |
|
$ |
49,261 |
|
$ |
35,474 |
|
|
Research and development |
|
|
1,732 |
|
|
1,343 |
|
|
986 |
|
|
Accrued liabilities and other |
|
|
1,282 |
|
|
1,567 |
|
|
935 |
|
|
Stock-based compensation |
|
|
998 |
|
|
823 |
|
|
485 |
|
|
Fixed assets |
|
|
482 |
|
|
477 |
|
|
219 |
|
|
Tenant improvement allowance |
|
|
447 |
|
|
744 |
|
|
836 |
|
|
Total deferred tax assets |
|
|
48,109 |
|
|
54,215 |
|
|
38,935 |
|
|
Valuation allowance |
|
|
(48,109) |
|
|
(54,215) |
|
|
(38,935) |
|
|
Net deferred tax assets |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by $7.1 million in the year ended December 31, 2017, due to an increase of $16.4 million in total deferred tax assets due to the increase in net operating loss carryforwards generated during the year, a decrease in the deferred tax assets related to the reduction of the U.S. corporate income tax rate from the 2017 Tax Act of $22.5 million and a decrease of $1.0 million due to the adoption of ASU No. 2016-09. The valuation allowance increased by $15.3 million and $12.7 million in the years ended December 31, 2016, and 2015, respectively, and there were no releases of the valuation allowance in any of these years.
The adoption of ASU No. 2016‑09 requires excess tax benefits and tax deficiencies be recorded in the statement of operations as opposed to additional paid‑in capital when the awards vest or are settled, and has been applied on a prospective basis with no impact on the financial statements as of December 31, 2017. As a result of the adoption, the Company's increased its total NOLs by $1.0 million on January 1, 2017 related to deferred tax assets that arose directly from tax deductions related to equity compensation greater than compensation recognized for financial reporting purposes. This amount is fully offset by the Company’s valuation allowance.
The 2017 Tax Act was signed into law on December 22, 2017. The 2017 Tax Act significantly revises the U.S. corporate income tax by, among other things, lowering the statutory corporate tax rate from 35% to 21%, eliminating certain deductions, imposing a mandatory one-time tax on accumulated earnings of foreign subsidiaries as of 2017, introducing new tax regimes, and changing how foreign earnings are subject to U.S. tax. The 2017 Tax Act also enhanced and extended through 2026 the option to claim accelerated depreciation deductions on qualified property. The Company has not completed its determination of the accounting implications of the 2017 Tax Act on its tax accruals. However, the Company has reasonably estimated the effects of the 2017 Tax Act to be zero as of December 31, 2017 for the following reasons. Due to the Company not having foreign subsidiaries, the mandatory one-time tax on accumulated foreign earnings is not applicable to the Company. The remeasurement of federal net deferred tax assets resulting from the permanent reduction in the U.S. statutory corporate tax rate to 21% from 35% results in no effect to the Company's provision for income taxes due to the full valuation allowance recorded on deferred tax assets.
86
On December 22, 2017, the SEC staff issued Staff Accounting Bulletin No. 118 (“SAB 118”), which provides guidance for the tax effect of the Tax Act. SAB 118 provides a measurement period that should not extend beyond one year from the Tax Act’s enactment date for companies to complete the accounting under Accounting Standards Codification Topic 740, Income Taxes (“ASC 740”). In accordance with SAB 118, the Company must reflect the income tax effects of those aspects of the Tax Act for which the accounting under ASC 740 is complete. To the extent that its accounting for certain income tax effects of the Tax Act is incomplete, but the Company is able to determine a reasonable estimate, the Company must record a provisional estimate in its consolidated financial statements. If the Company cannot determine a provisional estimate to be included in its consolidated financial statements, the Company should continue to apply ASC 740 on the basis of the provisions of the tax laws that were in effect immediately before the enactment of the Tax Act. Also, it is expected that the U.S. Treasury will issue regulations and other guidance on the application of certain provisions of the Tax Act. In subsequent periods, but within the measurement period, the Company will analyze that guidance and other necessary information to refine its estimates and complete its accounting for the tax effects of the Tax Act as necessary.
As of December 31, 2017, the Company had net operating loss (“NOL”) carryforwards (before tax effects) for federal and state income tax purposes of $171.0 million and $124.8 million, respectively. These federal and state NOL carryforwards will begin to expire in 2026 and 2027, respectively, if not utilized. In addition, the Company has federal and state research and development tax credit carryforwards of $1.1 million and $1.5 million, respectively, to offset future income tax liabilities. The federal research and development tax credits will begin to expire in 2026, if not utilized, while the state research and development tax credit can be carried forward indefinitely.
Federal and California tax laws impose substantial restrictions on the utilization of net operating losses and credit carry-forwards in the event of an “ownership change” for tax purposes, as defined in Section 382 of the Internal Revenue Code. Due to ownership changes since inception, the Company’s net operating losses may be limited as to their usage. In the event the Company has additional changes in ownership, utilization of the carryforwards could be further restricted.
A reconciliation of the Company’s unrecognized tax benefits for the years ended December 31, 2017, 2016, and 2015 is as follows (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Balance at beginning of year |
|
$ |
448 |
|
$ |
329 |
|
$ |
261 |
|
|
Additions for tax positions taken in current year |
|
|
81 |
|
|
124 |
|
|
86 |
|
|
Increases (reductions) for tax positions taken in prior years |
|
|
48 |
|
|
(5) |
|
|
(18) |
|
|
Balance at end of year |
|
$ |
577 |
|
$ |
448 |
|
$ |
329 |
|
The unrecognized tax benefits, if recognized, would not have an impact on the Company’s effective tax rate to the extent that the Company continues to maintain a full valuation allowance against its deferred tax assets. The Company does not expect a material change to its unrecognized tax benefits over the next twelve months.
The Company’s policy is to include interest and penalties related to unrecognized tax benefits within the provision for income taxes. Management determined that no accrual for interest and penalties was required as of December 31, 2017, 2016, and 2015, respectively.
The Company’s tax years 2005-2016 will remain open for examination by the federal and state authorities for three and four years, respectively, from the date of utilization of any NOL or research and development credits.
87
12.NET LOSS PER COMMON SHARE
The following table sets forth the computation of the basic and diluted net loss per share during the years ended December 31, 2017, 2016, and 2015 (in thousands, except share and per share data):
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Numerator: |
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(39,918) |
|
$ |
(40,609) |
|
$ |
(37,570) |
|
|
Denominator: |
|
|
|
|
|
|
|
|
|
|
|
Weighted-average common shares outstanding |
|
|
17,051,629 |
|
|
14,869,691 |
|
|
7,619,696 |
|
|
Less: weighted-average unvested common shares subject to repurchase |
|
|
(592) |
|
|
(1,190) |
|
|
(13,524) |
|
|
Weighted-average shares used to compute net loss per common share, basic and diluted |
|
|
17,051,037 |
|
|
14,868,501 |
|
|
7,606,172 |
|
|
Net loss per common share, basic and diluted |
|
$ |
(2.34) |
|
$ |
(2.73) |
|
$ |
(4.94) |
|
Basic net loss per share attributable to common stockholders is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding for the period. Diluted net loss per share attributable to common stockholders is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares and potentially dilutive securities outstanding for the period, determined using the treasury-stock method and the as-if converted method, for convertible securities, if inclusion of these is dilutive. Because the Company has reported a net loss for all periods presented, diluted net loss per common share is the same as basic net loss per common share for those periods.
The following potentially dilutive securities outstanding at the end of the periods presented have been excluded from the computation of diluted shares outstanding:
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Options to purchase common stock |
|
3,232,692 |
|
2,445,600 |
|
2,038,789 |
|
|
Restricted Stock Units |
|
268,260 |
|
131,807 |
|
— |
|
|
Warrants to purchase common stock |
|
235,415 |
|
137,007 |
|
137,007 |
|
|
Total |
|
3,736,367 |
|
2,714,414 |
|
2,175,796 |
|
13.SUBSEQUENT EVENTS
On February 27, 2018, Philip Sawyer informed the board of directors that he will resign from his role as President, Chief Executive Officer, and a member of the Company’s board of directors, effective as of February 28, 2018. Scott Flora, currently serving as a member of the Company’s board of directors, assumed the additional role of Interim President and Chief Executive Officer, and as such became the Company’s principal executive officer effective on March 1, 2018.
SUPPLEMENTARY FINANCIAL DATA (unaudited)
The following table presents selected unaudited financial data for each of the eight quarters in the two-year period ended December 31, 2017. The selected quarterly financial data should be read in conjunction with the Company's financial statements and the related notes and Item 7 “Management's Discussion and Analysis of Financial Condition and Results of Operations." This information has been derived from the Company's unaudited financial statements that, in management's opinion, reflect all recurring adjustments necessary to fairly state this information when read in conjunction with the Company's financial statements and the related notes appearing in the section entitled " Financial Statements and Supplementary Data," Net loss per share-basic and diluted, for the four quarters of each fiscal year may not sum to the total for the fiscal year because of the different number of shares outstanding during each period. The results of operations for any quarter are not necessarily indicative of the results to be expected for any future period.
88
Statements of Operations Data:
|
|
|
Three Months Ended |
|
||||||||||
|
|
|
December 31, 2017 |
|
September 30, 2017 |
|
June 30, 2017 |
|
March 31, 2017 |
|
||||
|
|
|
(in thousands except share and per share data) |
|
||||||||||
|
Revenue |
|
$ |
11,228 |
|
$ |
9,600 |
|
$ |
9,768 |
|
$ |
9,023 |
|
|
Gross profit |
|
|
7,489 |
|
|
6,712 |
|
|
6,753 |
|
|
6,924 |
|
|
Loss from operations |
|
|
(6,636) |
|
|
(8,404) |
|
|
(9,861) |
|
|
(10,357) |
|
|
Net loss |
|
$ |
(7,407) |
|
$ |
(8,907) |
|
$ |
(10,387) |
|
$ |
(13,217) |
|
|
Net loss per common share, basic and diluted |
|
$ |
(0.43) |
|
$ |
(0.52) |
|
$ |
(0.61) |
|
$ |
(0.78) |
|
|
Weighted-average shares used to compute net loss per common share, basic and diluted |
|
|
17,154,060 |
|
|
17,093,183 |
|
|
16,986,074 |
|
|
16,958,332 |
|
|
|
|
Three Months Ended |
|
||||||||||
|
|
|
December 31, 2016 |
|
September 30, 2016 |
|
June 30, 2016 |
|
March 31, 2016 |
|
||||
|
|
|
(in thousands except share and per share data) |
|
||||||||||
|
Revenue |
|
$ |
9,356 |
|
$ |
8,478 |
|
$ |
8,223 |
|
$ |
6,404 |
|
|
Gross profit |
|
|
6,948 |
|
|
6,259 |
|
|
6,132 |
|
|
4,298 |
|
|
Loss from operations |
|
|
(9,074) |
|
|
(8,346) |
|
|
(9,637) |
|
|
(11,623) |
|
|
Net loss |
|
$ |
(9,549) |
|
$ |
(8,821) |
|
$ |
(10,129) |
|
$ |
(12,110) |
|
|
Net loss per common share, basic and diluted |
|
$ |
(0.56) |
|
$ |
(0.56) |
|
$ |
(0.76) |
|
$ |
(0.90) |
|
|
Weighted-average shares used to compute net loss per common share, basic and diluted |
|
|
14,868,501 |
|
|
15,690,785 |
|
|
13,404,007 |
|
|
13,392,976 |
|
ITEM 9.CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
Not Applicable
ITEM 9A.CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act refers to controls and procedures that are designed to provide reasonable assurance that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to provide reasonable assurance that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its Chief Executive Officer and Chief Financial Officers, as appropriate to allow timely decisions regarding required disclosure.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2017, the end of the period covered by this Annual Report on Form 10-K. Based upon such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of such date. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their control objectives.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting identified in connection with the evaluation required by Rules 13(a)-15(d) and 15d-15(d) under the Exchange Act that occurred during our most recently completed fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
89
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, conducted an evaluation of the effectiveness of our internal control over financial reporting. This evaluation was based on the framework established in “Internal Control-Integrated Framework” (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our assessment under the framework established in “Internal Control-Integrated Framework” (2013), our management concluded that, as of December 31, 2017, our internal control over financial reporting was effective.
Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements will not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
This Annual Report on Form 10-K does not include an attestation report on our internal control over financial reporting of our independent registered public accounting firm. Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal control over financial reporting as long as we are an “emerging growth company” pursuant to the provisions of the JOBS Act.
On February 27, 2018, Philip Sawyer informed the board of directors that he will resign from his role as President, Chief Executive Officer, and a member of the Company’s board of directors, effective as of February 28, 2018. Scott Flora, currently serving as a member of the Company’s board of directors, assumed the additional role of Interim President and Chief Executive Officer, and as such became the Company’s principal executive officer effective on March 1, 2018. The separation and compensation arrangements with Mr. Sawyer and Mr. Flora, respectively, have not yet been finalized. Mr. Flora will continue to serve on the Company’s board of directors.
Mr. Flora, age 62, has been a member of our board of directors since November 2017. Since 2016, Mr. Flora has served as the President of SFlora Consulting LLC, a consulting company. From October 2011 to April 2014, Mr. Flora served as the Director, President, and Chief Executive Officer of OmniGuide, Inc. a medical device company. From November 2006 to June 2011, he served as the Global Business Unit President for the surgical device division of Covidien plc, a publicly traded, global healthcare products company later acquired by Medtronic PLC. Mr. Flora currently serves on the board of directors of Conventus Orthopaedics, a medical device company, and AgNovos Healthcare, a bone health medical device company. Previously, Mr. Flora has served on the board of directors of MAKO Surgical Corp., a medical device company (acquired by Stryker Corporation in December 2013). Mr. Flora received a B.S. in marketing from Millikin University.
There is no arrangement or understanding between Mr. Flora and any other persons pursuant to which Mr. Flora was selected as an officer. There are no family relationships between Mr. Flora and any director or executive officer of the Company and, other than as described above, he has no direct or indirect material interest in any transaction required to be disclosed pursuant to Item 404(a) of Regulation S-K.
90
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The information required by this item will be included in our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission, or the SEC, within 120 days of the fiscal year ended December 31, 2017, and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION.
The information required by this item will be set forth in our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders and is incorporated in this Annual Report on Form 10-K by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The information required by this item will be set forth in our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders and is incorporated in this Annual Report on Form 10-K by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The information required by this item will be set forth in our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders and is incorporated in this Annual Report on Form 10-K by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES.
The information required by this item will be set forth in our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders and is incorporated in this Annual Report on Form 10-K by reference.
With the exception of the information incorporated in Items 10, 11, 12, 13, and 14 of this Annual Report on Form 10-K, our Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the fiscal year ended December 31, 2017 is not deemed “filed” as part of this Annual Report on Form 10-K.
91
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
The following documents are filed as part of this report:
1. Financial Statements
Our Financial Statements are listed in the “Index to Financial Statements” under Part II, Item 8 of this Annual Report on Form 10-K.
2. Financial Statement Schedules
All schedules are omitted because they are not applicable or the required information is shown in the financial statements and the related notes thereto.
3. Exhibits
The documents listed in the Exhibit Index immediately preceding the signature page of this Annual Report on Form 10-K are incorporated by reference or are filed or furnished with this Annual Report on Form 10-K, in each case as indicted therein (numbered in accordance with Item 601 of Regulation S-K).
Not applicable.
92
|
Exhibit |
|
Exhibit |
|
Exhibit Description |
|
(1) |
|
3.1 |
|
|
|
|
|
|
|
|
|
(1) |
|
3.2 |
|
Certificate of Amendment to the Amended and Restated Certificate of Incorporation. |
|
|
|
|
|
|
|
(1) |
|
3.3 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.1 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.2 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.3 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.4 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.5 |
|
|
|
|
|
|
|
|
|
(2) |
|
4.6 |
|
|
|
|
|
|
|
|
|
(3) |
|
4.7 |
|
|
|
|
|
|
|
|
|
(3) |
|
4.8 |
|
|
|
|
|
|
|
|
|
(3) |
|
4.9 |
|
|
|
|
|
|
|
|
|
(4) |
|
4.10 |
|
|
|
|
|
|
|
|
|
(4) |
|
4.11 |
|
|
|
|
|
|
|
|
|
(4) |
|
4.12 |
|
|
|
|
|
|
|
|
|
(2) |
|
10.1 |
|
Form of Indemnification Agreement for directors and executive officers. |
|
|
|
|
|
|
|
(2) |
|
10.2+ |
|
Invuity, Inc. 2005 Stock Incentive Plan and form of agreements thereunder. |
|
|
|
|
|
|
|
(2) |
|
10.3+ |
|
|
|
|
|
|
|
|
|
(2) |
|
10.4+ |
|
2015 Equity Incentive Plan and forms of agreements thereunder. |
|
|
|
|
|
|
|
(5) |
|
10.5+ |
|
Invuity, Inc. Restricted Stock Unit Deferral Program for Outside Directors. |
|
|
|
|
|
|
93
|
(5) |
|
10.6+ |
|
Invuity, Inc. Restricted Stock Unit Deferral Program for Outside Directors Award Notice. |
|
|
|
|
|
|
|
(2) |
|
10.7 |
|
|
|
|
|
|
|
|
|
(3) |
|
10.8 |
|
|
|
|
|
|
|
|
|
(3) |
|
10.9 |
|
|
|
|
|
|
|
|
|
(4) |
|
10.10 |
|
|
|
|
|
|
|
|
|
(4) |
|
10.11 |
|
|
|
(6) |
|
10.12+ |
|
Executive Employment Agreement, dated May 10, 2016, by and between the Registrant and Philip Sawyer. |
|
|
|
|
|
|
|
(6) |
|
10.13+ |
|
|
|
|
|
|
|
|
|
(6) |
|
10.14+ |
|
Executive Severance Agreement, dated May 10, 2016, by and between the Registrant and Philip Sawyer. |
|
|
|
|
|
|
|
(7) |
|
10.15+ |
|
|
|
|
|
|
|
|
|
(6) |
|
10.16+ |
|
|
|
|
|
|
|
|
|
(6) |
|
10.17+ |
|
|
|
|
|
|
|
|
|
|
|
23.1* |
|
Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm. |
|
|
|
|
|
|
|
|
|
24.1* |
|
|
|
|
|
|
|
|
|
|
|
31.1* |
|
|
|
|
|
|
|
|
|
|
|
31.2* |
|
|
|
|
|
|
|
|
|
|
|
32.1† |
|
|
|
|
|
|
|
|
|
|
|
101.INS* |
|
XBRL Instance Document |
|
|
|
|
|
|
|
|
|
101.SCH* |
|
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
94
|
|
|
101.CAL* |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
|
|
|
|
101.DEF* |
|
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
|
|
101.LAB* |
|
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
|
|
|
|
101.PRE* |
|
XBRL Taxonomy Extension Presentation Linkbase Document |
*Filed herewith.
+Indicates management contract or compensatory plan.
†These certification attached as Exhibit 32.1 that accompany this Annual Report on Form 10-K, are deemed “furnished” and not “filed” with the Securities and Exchange Commission and are not incorporated by reference into any filing of Invuity, Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
|
(1) |
Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q filed with the SEC on August 12, 2015 and incorporated herein by reference. |
|
(2) |
Filed as an exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-203505) filed with the SEC and incorporated herein by reference. |
|
(3) |
Filed as an exhibit to the Registrant’s Annual Report on Form 10-K filed with the SEC on March 16, 2017 and incorporated herein by reference. |
|
(4) |
Filed as an exhibit to the Registrant’s Current Report on Form 8-K filed with the SEC on September 27, 2017 and incorporated herein by reference. |
|
(5) |
Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q filed with the SEC on July 26, 2017 and incorporated herein by reference. |
|
(6) |
Filed as an exhibit to the Registrant’s Quarterly Report on Form 10-Q filed with the SEC on July 20, 2016 and incorporated herein by reference. |
|
(7) |
Filed as an exhibit to the Registrant’s Annual Report on Form 10-K filed with the SEC on March 25, 2016 and incorporated herein by reference. |
95
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized
|
|
|
Invuity, Inc. |
|
|
|
|
|
|
|
Date: March 5, 2018 |
|
By: |
/s/ Scott Flora |
|
|
|
|
Scott Flora |
|
|
|
|
Interim President and Chief Executive Officer |
|
|
|
|
|
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Scott Flora and James Mackaness and each of them, any of whom may act without the joinder of the others, as his or her true and lawful attorneys-in-fact and agents with full power of substitution and re-substitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his or her substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
|
Name |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Scott Flora |
|
Director and Interim President and Chief Executive Officer |
|
March 5, 2018 |
|
Scott Flora |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/s/ James Mackaness |
|
Chief Financial Officer |
|
March 5, 2018 |
|
James Mackaness |
|
(Principal Financial Officer and Chief Accounting Officer) |
|
|
|
|
|
|
|
|
|
/s/ Eric Roberts |
|
Director |
|
March 5, 2018 |
|
Eric Roberts |
|
|
|
|
|
|
|
|
|
|
|
/s/ Gregory Lucier |
|
Director |
|
March 5, 2018 |
|
Gregory Lucier |
|
|
|
|
|
|
|
|
|
|
|
/s/ Randall Lipps |
|
Director |
|
March 5, 2018 |
|
Randall Lipps |
|
|
|
|
|
|
|
|
|
|
|
/s/ William Burke |
|
Director |
|
March 5, 2018 |
|
William Burke |
|
|
|
|
|
|
|
|
|
|
|
/s/ Daniel Wolterman |
|
Director |
|
March 5, 2018 |
|
Daniel Wolterman |
|
|
|
|
|
|
|
|
|
|
96