Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - NEKTAR THERAPEUTICS | nktr-ex321_10.htm |
| EX-31.2 - EX-31.2 - NEKTAR THERAPEUTICS | nktr-ex312_6.htm |
| EX-31.1 - EX-31.1 - NEKTAR THERAPEUTICS | nktr-ex311_9.htm |
| EX-23.1 - EX-23.1 - NEKTAR THERAPEUTICS | nktr-ex231_8.htm |
| EX-21.1 - EX-21.1 - NEKTAR THERAPEUTICS | nktr-ex211_7.htm |
| EX-10.18 - EX-10.18 - NEKTAR THERAPEUTICS | nktr-ex1018_184.htm |
| EX-10.9 - EX-10.9 - NEKTAR THERAPEUTICS | nktr-ex109_195.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
Form 10-K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the fiscal year ended December 31, 2017
or
|
☐ |
TRANSITION REPORTS PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the transition period from to
Commission File Number: 0-24006
NEKTAR THERAPEUTICS
(Exact name of registrant as specified in its charter)
|
Delaware |
|
94-3134940 |
|
(State or other jurisdiction of incorporation or organization) |
|
(IRS Employer Identification No.) |
455 Mission Bay Boulevard South
San Francisco, California 94158
(Address of principal executive offices and zip code)
415-482-5300
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Each Exchange on Which Registered |
|
Common Stock, $0.0001 par value |
|
NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days) Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. :
|
Large accelerated filer |
|
☒ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☐ (Do not check if a smaller reporting company) |
|
Smaller reporting company |
|
☐ |
|
Emerging growth company |
|
☐ |
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2) Yes ☐ No ☒
The approximate aggregate market value of voting stock held by non-affiliates of the registrant, based upon the last sale price of the registrant’s common stock on the last business day of the registrant’s most recently completed second fiscal quarter, June 30, 2017, as reported on the NASDAQ Global Select Market, was approximately $3,028,667,056. This calculation excludes approximately 941,915 shares held by directors and executive officers of the registrant. Exclusion of these shares does not constitute a determination that each such person is an affiliate of the registrant.
As of February 22, 2018, the number of outstanding shares of the registrant’s common stock was 160,922,921.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of registrant’s definitive Proxy Statement to be filed for its 2018 Annual Meeting of Stockholders are incorporated by reference into Part III hereof. Such Proxy Statement will be filed with the Securities and Exchange Commission within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
NEKTAR THERAPEUTICS
2017 ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
|
|
|
Page |
|
|
|
|
|
Item 1. |
4 |
|
|
Item 1A. |
32 |
|
|
Item 1B. |
46 |
|
|
Item 2. |
46 |
|
|
Item 3. |
47 |
|
|
Item 4. |
47 |
|
|
|
|
|
|
Item 5. |
48 |
|
|
Item 6. |
50 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
51 |
|
Item 7A. |
63 |
|
|
Item 8. |
64 |
|
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
98 |
|
Item 9A. |
98 |
|
|
Item 9B. |
99 |
|
|
|
|
|
|
Item 10. |
100 |
|
|
Item 11. |
100 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
100 |
|
Item 13. |
Certain Relationships and Related Transactions and Director Independence |
100 |
|
Item 14. |
100 |
|
|
|
|
|
|
Item 15. |
101 |
|
|
106 |
||
2
This report includes “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical fact are “forward-looking statements” for purposes of this annual report on Form 10-K, including any projections of market size, earnings, revenue, milestone payments, royalties, sales or other financial items, any statements of the plans and objectives of management for future operations (including, but not limited to, preclinical development, clinical trials and manufacturing), any statements related to our financial condition and future working capital needs, any statements regarding potential future financing alternatives, any statements concerning proposed drug candidates, any statements regarding the timing for the start or end of clinical trials or submission of regulatory approval filings, any statements regarding future economic conditions or performance, any statements regarding the success of our collaboration arrangements, timing of commercial launches and product sales levels by our collaboration partners and future payments that may come due to us under these arrangements, any statements regarding our plans and objectives to initiate or continue clinical trials, and any statements of assumptions underlying any of the foregoing. In some cases, forward-looking statements can be identified by the use of terminology such as “may,” “will,” “expects,” “plans,” “anticipates,” “estimates,” “potential” or “continue,” or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements contained herein are reasonable, such expectations or any of the forward-looking statements may prove to be incorrect and actual results could differ materially from those projected or assumed in the forward-looking statements. Our future financial condition and results of operations, as well as any forward-looking statements, are subject to inherent risks and uncertainties, including, but not limited to, the risk factors set forth in Part I, Item 1A “Risk Factors” below and for the reasons described elsewhere in this annual report on Form 10-K. All forward-looking statements and reasons why results may differ included in this report are made as of the date hereof and we do not intend to update any forward-looking statements except as required by law or applicable regulations. Except where the context otherwise requires, in this annual report on Form 10-K, the “Company,” “Nektar,” “we,” “us,” and “our” refer to Nektar Therapeutics, a Delaware corporation, and, where appropriate, its subsidiaries.
Trademarks
The Nektar brand and product names, including but not limited to Nektar®, contained in this document are trademarks and registered trademarks of Nektar Therapeutics in the United States (U.S.) and certain other countries. This document also contains references to trademarks and service marks of other companies that are the property of their respective owners.
3
Nektar Therapeutics is a research-based biopharmaceutical company that discovers and develops innovative medicines in areas of high unmet medical need. Our research and development pipeline of new investigational drugs includes treatments for cancer, autoimmune disease and chronic pain. We leverage our proprietary and proven chemistry platform to discover and design new drug candidates. These drug candidates utilize our advanced polymer conjugate technology platforms, which are designed to enable the development of new molecular entities that target known mechanisms of action. We refer to our drug candidates where we retain at least U.S. commercial rights as “proprietary programs” and our other drug candidate programs that we have licensed U.S. and potentially other commercial rights to collaboration partners as “collaboration partner programs.”
Our Proprietary Programs
Immuno-oncology (I-O)
In the area of I-O, we are developing medicines that target biological pathways, which stimulate and sustain the body’s immune response in order to fight cancer. We are developing medicines designed to directly or indirectly modulate the activity of key immune cells, such as cytotoxic T cells and natural killer (NK) cells, to increase their numbers and improve their function to recognize and attack cancer cells.
NKTR-214, our lead I-O candidate, is a biologic with biased signaling through one of the IL-2 receptor subunits (CD 122) that can stimulate proliferation and growth of tumor-killing immune cells in the tumor micro-environment and increase expression of PD-1 on these immune cells.
On September 21, 2016, we entered into a Clinical Trial Collaboration Agreement (BMS Agreement) with Bristol-Myers Squibb Company (BMS), pursuant to which we and BMS are collaborating to conduct Phase 1/2 clinical trials evaluating NKTR-214 and BMS’ human monoclonal antibody that binds PD-1, known as Opdivo® (nivolumab), as a potential combination treatment regimen in at least five tumor types and eight indications, and such other clinical trials evaluating the combined therapy as may be mutually agreed upon by the parties (each, a Combined Therapy Trial). Under the BMS Agreement, BMS is responsible for 50% of all out-of-pocket costs reasonably incurred by us in connection with third party contract research organizations, laboratories, clinical sites and institutional review boards. Each party is otherwise responsible for its own internal costs, including internal personnel costs, incurred in connection with each Combination Therapy Trial. Interim data from the dose-escalation phase of the trial was presented at the 2017 Society for Immunotherapy of Cancer (SITC) meeting in November 2017. We identified the Phase 2 dose for NKTR-214 and we are currently enrolling subjects in the expansion phase of the study.
On February 13, 2018, we entered into a Strategic Collaboration Agreement (the BMS Collaboration Agreement) with BMS, pursuant to which we and BMS will jointly develop NKTR-214, including, without limitation, in combination with BMS’s Opdivo® (nivolumab) and Opdivo® plus Yervoy® (ipilimumab), and other compounds of BMS, us or any third party. The parties have agreed to jointly commercialize NKTR-214 on a worldwide basis. BMS will pay us a non-refundable upfront cash payment of $1.0 billion and purchase $850.0 million of shares of our common stock at a purchase price of $102.60 per share pursuant to a Share Purchase Agreement (Purchase Agreement).
We are eligible to receive additional cash payments of a total of up to $1.43 billion upon achievement of certain development and regulatory milestones and a total of up to $350.0 million upon achievement of certain sales milestones. We will book all worldwide sales and revenue for NKTR-214. We will share global commercialization profits and losses with BMS for NKTR-214, with Nektar sharing 65% and BMS sharing 35% of the net profits and losses. BMS will lead commercialization for combinations of NKTR-214 with BMS proprietary medicines, and we will lead all other commercialization efforts for NKTR-214. We will have the final decision-making authority regarding the pricing for NKTR-214. NKTR-214 will be sold on a stand-alone basis and there will be no fixed-dose combinations or co-packaging without the consent of both parties.
Under the BMS Collaboration Agreement, we and BMS will collaborate to develop and conduct clinical studies of NKTR-214 pursuant to a joint development plan, which initially includes a series of registration-enabling trials in more than 20 indications in nine tumor types and may be updated and expanded only upon mutual agreement of the parties. The parties will share the development costs for NKTR-214 in combination regimens based on each party’s relative ownership interest in the compounds included in the regimens. Nektar’s share of such development costs are limited to an annual cap of $125.0 million. Neither party will develop a therapy using an IL-2 agonist in combination with a small or large molecule that binds to the PD(L)-1 target (and in certain indications the anti-CTLA4 target), in indications included in the joint development plan (each, a Competing Combination), whether alone or in collaboration with any third party, during a limited exclusivity period from the closing date under the BMS Collaboration Agreement until the later of (i) the first commercial sale of NKTR-214 or (ii) the third anniversary of the closing date, but each party may develop a Competing Combination on its own (but not in collaboration with any third party) during the three years after the end of the
4
foregoing limited exclusivity period. If a registration-enabling study included in the joint development plan does not have the first patient enrolled prior to the date which is 14 months from the closing date, subject to allowable delays, the indication covered by that study is no longer subject to the above exclusivity. Other than as described above, Nektar may independently develop and commercialize NKTR-214 either alone or in combination with other Nektar proprietary compounds or third party compounds.
The closing date under the BMS Collaboration Agreement will occur simultaneously with the closing under the Purchase Agreement, which is subject to the expiration or early termination of the waiting period (and any extension thereof) under the Hart-Scott-Rodino Antitrust Improvements Act of 1976 and other customary closing conditions. The closings are expected to occur during the second quarter of 2018, upon which the BMS Collaboration Agreement will supersede and replace the existing BMS Agreement.
On May 22, 2017, we announced a research collaboration that is currently underway bewteen Nektar and Takeda Pharmaceutical Company Limited (Takeda) to evaluate NKTR-214 with five compounds from Takeda’s cancer portfolio.
On September 12, 2017, we announced that we had begun dosing in a clinical study evaluating the efficacy and safety of NKTR-214 in combination with approved checkpoint inhibitors, TECENTRIQ® (atezolizumab) and KEYTRUDA® (pembrolizumab). This study is planned to enroll patients into two separate arms concurrently with the first arm evaluating NKTR-214 in combination with atezolizumab in up to 30 patients, and the second arm evaluating NKTR-214 in combination with pembrolizumab in up to 30 patients. This study is not impacted by the BMS Collaboration Agreement and we plan to continue this study through completion.
In addition to the existing clinical programs, we plan to continue to advance a broad clinical development program, both on our own or in collaboration with other potential partners, to explore the potential of combining NKTR-214 with therapies such as cancer vaccines, adoptive cell therapy, small molecules, and other biological agents in order to generate novel immune-oncology approaches.
NKTR-262 is a small molecule agonist that targets toll-like receptors (TLRs) found on innate immune cells in the body. NKTR-262 is designed to stimulate the innate immune system and promote maturation and activation of antigen-presenting cells (APC), such as dendritic cells, which are critical to induce the body’s adaptive immunity and create antigen-specific cytotoxic T cells. NKTR-262 is being developed as an intra-tumoral injection in combination with systemic NKTR-214 in order to induce an abscopal response and achieve the goal of complete tumor regression in cancer patients treated with both therapies. We filed an IND for NKTR-262 in December 2017 and a Phase 1 clinical trial is expected to start before the end of the first quarter of 2018.
NKTR-255 is a biologic that targets the interleukin-15 pathway in order to activate the body’s innate and adaptive immunity. Signaling of the IL-15 pathway induces the proliferation and growth of CD8 memory T cells so the body’s immune system retains the ability to identify cancer cells if they re-grow in the body. NKTR-255 has also been shown to stimulate NK cell development. NKTR-255 is currently advancing through preclinical development.
Immunology
In February 2017 we submitted an investigational new drug application (IND) for a new biologic that we are developing, NKTR-358, which is designed to correct the underlying immune system imbalance in the body that occurs in patients with autoimmune disease. The breakdown of mechanisms assuring recognition of self and non-self is what underlies all autoimmune diseases. A failure of the body's self-tolerance mechanisms is known to result from pathogenic auto reactive T lymphocytes. By increasing the number of regulatory T cells (which are specific immune cells in the body that modulate the immune system and prevent autoimmune disease by maintaining self-tolerance), these pathogenic auto reactive T cells can be reduced and the proper balance of effector and regulatory T cells can be achieved to restore the body's self-tolerance mechanisms. There is consistent evidence that suboptimal regulatory T cell numbers and their lack of activity play a significant role in a myriad of autoimmune diseases. NKTR-358 is designed to optimally target the IL-2 receptor complex in order to stimulate proliferation and growth of regulatory T cells. NKTR-358 is being developed as a once or twice monthly self-administered injection for a number of autoimmune diseases.
On July 23, 2017, we entered into a worldwide license agreement with Eli Lilly and Company (Lilly) to co-develop NKTR-358. We received an initial payment of $150.0 million in September 2017 and are eligible for up to an additional $250.0 million for development and regulatory milestones. We will be responsible for completing Phase 1 clinical development and certain drug product development and supply activities. We will also share Phase 2 development costs with Lilly, with 75% of those costs borne by Lilly and 25% of the costs borne by us. We will have the option to contribute funding to Phase 3 development on an indication-by-indication basis, ranging from zero to 25% of the global Phase 3 development costs. We are eligible for tiered royalties on global sales up to the low twenties that escalate based upon our level of our contribution to Phase 3 development costs and the level of global product annual sales. Lilly will be responsible for all costs of global commercialization and we will have an option to co-promote in the U.S. under certain conditions. In March 2017, we began the first Phase 1 dose-finding trial of NKTR-358 to evaluate single-ascending doses of NKTR-358 in approximately 50 healthy patients. The study is still ongoing. The Phase 1 multiple-ascending dose
5
trial to evaluate NKTR-358 in patients with systemic lupus erythematosus (SLE) is expected to be initiated in the second quarter of 2018.
Pain - NKTR-181
NKTR-181 is a novel mu-opioid analgesic drug candidate for chronic pain conditions. NKTR-181 is currently in Phase 3 clinical development, which we call the SUMMIT Phase 3 program, which includes the SUMMIT-07 efficacy study, a human abuse potential (HAP) study, and the SUMMIT-LTS long-term safety study.
On March 20, 2017, we announced that NKTR-181 met its primary and secondary endpoints in the SUMMIT-07 Phase 3 efficacy study. We enrolled the first patient in the first SUMMIT-07 study in February 2015, and we completed enrollment in the study in late 2016. The SUMMIT-07 study compared twice-daily dosing of NKTR-181 tablets to placebo in the treatment of over 600 patients with moderate to severe chronic low back pain who were new to opioid therapy (opioid-naïve). SUMMIT-07 evaluated four analgesic doses of NKTR-181 (100 mg, 200 mg, 300 mg and 400 mg). Patients in the trial achieved an average pain score reduction of over 65% (from 6.73 at screening to 2.32 at randomization) during the dose titration period. The primary efficacy endpoint of the study demonstrated significantly improved chronic back pain relief with NKTR-181 compared to placebo (p=0.0019). Key secondary endpoints of the study also achieved high statistical significance. The study demonstrated that NKTR-181 had a favorable safety profile and was well tolerated.
On July 18, 2017, we announced positive top-line data for our pivotal human abuse potential study (HAP) for NKTR-181. The HAP study was designed to confirm and assess the relative oral abuse potential of NKTR-181, at its maximum analgesic therapeutic, dose (400 mg) studied in the SUMMIT-07 trial and at a supratherapeutic dose (3 times to 12 times greater than the analgesic dose range of 100 mg to 400 mg used in the SUMMIT-07 trial), compared to common therapeutic doses of oxycodone (40 mg and 60 mg) in 54 healthy non-dependent recreational drug users. For the primary endpoint of Drug Liking, NKTR-181 (400 mg and 600 mg) rated less likable compared to oxycodone 40 mg and 60 mg (p<0.0001), and a supratherapeutic dose of NKTR-181 (1200 mg) rated less likable than oxycodone 60 mg (p=0.0071). Key secondary endpoints of Area Under Effect for Drug Liking (0-1 hours, 0-2 hours, 0-3 hours), Drug High and Take Drug Again scores met statistical significance for all doses of NKTR-181 (1200 mg, 600 mg, 400 mg) compared to oxycodone (60 mg).
The SUMMIT Phase 3 program also includes a 52-week long-term safety study, which we call SUMMIT-LTS. SUMMIT-LTS is evaluating the long-term safety and tolerability of NKTR-181 in 638 patients (opioid-naïve and opioid-experienced) with moderate to severe chronic low pain or chronic non-cancer pain. The study was completed in December 2017.
Following our success of the SUMMIT-07 Phase 3 efficacy study and the HAP study, we are seeking a partner to support future development and commercialization activities for NKTR-181. We have completed pre-NDA meetings with FDA and we are currently planning to file an NDA for NKTR-181 in the first half of 2018.
Oncology - ONZEALDTM
ONZEALDTM (also known as NKTR-102, etirinotecan pegol) is our next-generation topoisomerase I inhibitor proprietary drug candidate. On March 17, 2015, we announced top-line data from a Phase 3 clinical study for ONZEALDTM, which we call the BEACON study (BrEAst Cancer Outcomes with NKTR-102), as a single-agent therapy for women with advanced metastatic breast cancer. The BEACON study compared ONZEALDTM to an active control arm comprised of a single chemotherapy agent of physician’s choice (TPC) in patients who were heavily pre-treated with a median of three prior therapies for metastatic disease. In a top-line analysis of 852 patients from the trial, ONZEALDTM provided a 2.1 month improvement in median overall survival over TPC (12.4 months for patients receiving ONZEALDTM compared to 10.3 months for patients receiving TPC). Based on a stratified log-rank analysis, the primary endpoint measuring the Hazard Ratio (HR) for survival in the ONZEALDTM group compared to the active control arm was 0.87 with a p-value of 0.08, which did not achieve statistical significance. Secondary endpoints in the BEACON study included objective response rate and progression-free survival, which did not achieve statistical significance in the study. We also announced that we observed a significant overall survival benefit in two pre-specified subgroup populations—patients with a history of brain metastases and patients with baseline liver metastases at study entry.
We have explored future regulatory and development paths forward for ONZEALDTM with the European Union (EU) and U.S. health authorities. In Europe, we met with the National Authorities in Sweden and the United Kingdom, as well as the European Medicines Agency (EMA) to discuss the BEACON data. In June 2016, we filed a Marketing Authorization Application (MAA) for conditional approval of ONZEALDTM for adult patients with advanced breast cancer who have brain metastases and began enrolling in the ATTAIN study, which compares the overall survival in patients with breast cancer and brain metastases treated with ONZEALDTM versus physicians treatment of choice. On July 21, 2017, we were informed by the EMA’s Committee for Medicinal Products for Human Use (CHMP) that it had adopted a negative opinion for the conditional marketing authorization application for ONZEALDTM in the European Union. On July 26, 2017, we filed a request for re-examination of the opinion adopted by the CHMP (the “CHMP Appeal”) but such request was not successful. On December 6, 2017, we received a notice from Daiichi Sankyo Europe
6
GmbH (Daiichi), exercising its right to terminate the Collaboration and License Agreement between us and Daiichi (the Daiichi Collaboration Agreement), which termination became effective on February 4, 2018. As a result of the termination, we made a $12.5 million termination payment to Daiichi in December 2017, and all rights and licenses granted to Daiichi under the Daiichi Collaboration Agreement reverted exclusively to Nektar. The Phase 3 study (ATTAIN) in breast cancer patients having brain metastases is ongoing.
Collaboration Partner Programs
In 2014, we achieved the first approval of one of our proprietary drug candidates, MOVANTIK® (naloxegol), under a global license agreement with AstraZeneca AB (AstraZeneca). MOVANTIK® is an oral peripherally-acting opioid antagonist, for the treatment of opioid-induced constipation, or OIC, a side effect caused by chronic administration of prescription opioid pain medicines. AstraZeneca markets and sells MOVANTIK® in the United States in collaboration with Daiichi Sankyo, Inc. (Daiichi Inc.). Kyowa Hakko Kirin Co. Ltd. (Kirin) has exclusive marketing rights to MOVENTIG® (the naloxegol brand name in the EU) in the EU, Iceland, Liechtenstein, Norway and Switzerland.
We have a collaboration with Baxalta Incorporated, (a wholly-owned subsidiary of Shire plc) to develop and commercialize PEGylated drug candidates with the objective of providing new long-acting therapies for hemophilia patients. Under this collaboration, we worked with Baxalta to develop ADYNOVATE® (previously referred to as BAX 855), an extended half-life recombinant factor VIII (rFVIII) treatment for Hemophilia A based on ADVATE® [Antihemophilic Factor (Recombinant)]. ADYNOVATE®, was first approved by the United States Food and Drug Administration (FDA) in late 2015 for use in adults and adolescents, aged 12 years and older, who have Hemophilia A. In December 2016 the FDA expanded the approval of ADYNOVATE® for the treatment of Hemophilia A in pediatric patients under 12 years of age, and for the use in surgical settings for both adult and pediatric patients. In March 2016, ADYNOVATE® was approved in Japan for patients aged 12 years and older with Hemophilia A and in November 2017 that approval was extended to include pediatric patients under 12 years of age, and those undergoing surgery. In late 2016, ADYNOVATE® (marketed as ADYNOVI) was approved in Switzerland for use in Hemophilia A patients aged 12 years and older and in March 2017, ADYNOVI was approved for use in pediatric patients. In November 2016, ADYNOVATE® was approved in Canada for patients 12 years of age and older for the treatment of Hemophilia A, including for perioperative management. In March 2017, ADYNOVATE® was approved in Australia for use in adult and pediatric patients with Hemophilia A. In September 2017, ADYNOVATE® was approved in Colombia for use in children and adults with Hemophilia A In January 2018, ADYNOVATE® (marketed as ADYNOVI) was approved in the European Union for on-demand and prophylactic use in patients 12 years and older with Hemophilia A. In January 2018, ADYNOVATE® was also approved in S. Korea for use in children and adults with Hemophilia A.
We retained our right to receive royalties on net sales of the Cipro DPI (Cipro Dry Powder Inhaler, previously called Cipro Inhale), pursuant to a drug development program with Bayer Schering Pharma AG (Bayer Schering) that we transferred to Novartis as part of the 2008 pulmonary asset divestiture transaction. In August 2012, Bayer Schering initiated a global Phase 3 program called RESPIRE for the Cipro DPI product candidate in patients with non-cystic fibrosis bronchiectasis (NCFB). In September 2016, Bayer Schering presented data from the first Phase 3 trail (RESPIRE 1) at the 2016 European Respiratory Society Annual Meeting which showed in the 14 days on/off regimen, the RESPIRE 1 trial met its primary endpoints of significantly prolonged time to first exacerbation and significantly reduced frequency of exacerbations versus placebo. The top-line results of the second Phase 3 trial (RESPIRE 2) were presented at the American Thoracic Society 2017 International Conference. A pooled analysis of the primary efficacy results from RESPIRE 1 and RESPIRE 2 is positive and the data from both RESPIRE 1 and RESPIRE 2 indicate that Cipro DPI has a positive safety profile. On November 16, 2017, the Antimicrobial Drugs Advisory Committee of the FDA voted not to recommend Cipro DPI to be approved for the treatment of non-cystic fibrosis. Bayer Schering has informed us that they are discontinuing development of Cipro DPI in NCFB for the time being as they evaluate possible future options for this asset.
We have a license, manufacturing and supply agreement with UCB Pharma (UCB) for dapirolizumab pegol, a monovalent pegylated Fab antibody fragment against the CD40 ligand (CD40L), being developed for the treatment of autoimmune diseases, including systemic lupus erythematosus (SLE) for which the candidate is entering Phase 2 development with UCB partner Biogen Inc. We also have a number of license, manufacturing and supply agreements with other leading biotechnology and pharmaceutical companies, including Amgen, Inc., Allergan, Inc., Merck & Co., Inc. and Pfizer, Inc. More than 10 products using our PEGylation technology have received regulatory approval in the U.S. or the EU. There are also a number of other products in clinical development that incorporate our advanced polymer conjugate technologies.
Corporate Information
We were incorporated in California in 1990 and reincorporated in Delaware in 1998. We maintain our executive offices at 455 Mission Bay Boulevard South, San Francisco, California 94158, and our main telephone number is (415) 482-5300. Our website is located at www.nektar.com. The information contained in, or that can be accessed through, our website is not part of, and is not incorporated in, this Annual Report on Form 10-K.
7
Our Technology Platform
As a leader in the polymer conjugation field, we have advanced our technology platform to include new advanced polymer technologies that can be tailored in specific and customized ways with the objective of optimizing and significantly improving the profile of a wide range of molecules, including many classes of drugs targeting numerous disease areas. Polymer conjugation or PEGylation has been a highly effective technology platform for the development of therapeutics with significant commercial success, such as Amgen’s Neulasta® (pegfilgrastim) and Roche’s PEGASYS® (PEG-interferon alfa-2a). Nearly all of the PEGylated drugs approved over the last fifteen years were enabled with our PEGylation technology through our collaborations and licensing partnerships with a number of well-known biotechnology and pharmaceutical companies. PEGylation is a versatile technology as a result of polyethylene glycol (PEG) being a water soluble, amphiphilic, non-toxic, non-immunogenic compound that has been shown to safely clear from the body. Its primary use to date has been in currently approved biologic drugs to favorably alter their pharmacokinetic or pharmacodynamic properties. However, in spite of its widespread success in commercial drugs, there are some limitations with the first-generation PEGylation approaches that have been used with biologics. For example, these techniques cannot be used successfully to create small molecule drugs which could potentially benefit from the application of the technology. Other limitations of the early applications of PEGylation technology include sub-optimal bioavailability and bioactivity, and its limited ability to be used to fine-tune properties of the drug, as well as its inability to be used to create oral drugs.
With our expertise and proprietary technology in polymer conjugation, we have created the next generation of PEGylation technology. Our advanced polymer conjugation technology platform is designed to overcome the limitations of the first generation of the technology platform and to allow the platform to be utilized with a broader range of molecules across many therapeutic areas. We have also developed robust manufacturing processes for generating second generation PEGylation reagents that allow us to utilize the full potential of these newer approaches.
Our advanced polymer conjugate technology platforms have the potential to offer one or more of the following benefits:
|
|
• |
improve efficacy or safety of a drug as a result of better pharmacokinetics, pharmacodynamics, longer half-life and sustained exposure of the drug; |
|
|
• |
improve targeting or binding affinity of a drug to its target receptors with the potential to improve efficacy and reduce toxicity or drug resistance; |
|
|
• |
improve solubility of a drug; |
|
|
• |
enable oral administration of parenterally-administered drugs, or drugs that must be administered intravenously or subcutaneously, and increase oral bioavailability of small molecules; |
|
|
• |
prevent drugs from crossing the blood-brain barrier, or reduce their rate of passage into the brain, thereby limiting undesirable central nervous system effects; |
|
|
• |
reduce first-pass metabolism effects of certain drug classes with the potential to improve efficacy, which could reduce the need for other medicines and reduce toxicity; |
|
|
• |
reduce the rates of drug absorption and of elimination or metabolism by improving stability of the drug in the body and providing it with more time to act on its target; |
|
|
• |
differentially alter binding affinity of a drug for multiple receptors, improving its selectivity for one receptor over another; and |
|
|
• |
reduce immune response to certain macromolecules with the potential to prolong their effectiveness with repeated doses. |
We have a broad range of approaches that we may use when designing our own drug candidates, some of which are further described below.
Small Molecule Stable Polymer Conjugates
Our customized approach for small molecule polymer conjugates allows for the fine-tuning of the physicochemical and pharmacological properties of small molecule oral drugs to potentially increase their therapeutic benefit. In addition, this approach can enable oral administration of subcutaneously or intravenously delivered small molecule drugs that have low bioavailability when delivered orally. The benefits of this approach can also include: improved potency, modified biodistribution with enhanced pharmacodynamics, and reduced transport across specific membrane barriers in the body, such as the blood-brain barrier. An example of reducing transport across the blood-brain barrier is MOVANTIK®, an orally-available peripherally-acting opioid antagonist that is approved in the United States and the EU. An additional example of the application of membrane transport, specifically slowing transport across the blood-brain barrier is NKTR-181, an orally-available mu-opioid analgesic molecule that is currently in Phase 3 clinical development.
8
Small Molecule Pro-Drug Releasable Polymer Conjugates
The pro-drug polymer conjugation approach can be used to optimize the pharmacokinetics and pharmacodynamics of a small molecule drug to substantially increase its efficacy and improve its side effect profile. We are currently using this platform with oncolytics, which typically have sub-optimal half-lives that can limit their therapeutic efficacy. With our releasable polymer conjugate technology platform, we believe that these drugs can be modulated for programmed release within the body, optimized bioactivity and increased sustained exposure of active drug to tumor cells in the body.
Large Molecule Polymer Conjugates (Proteins and Peptides)
Our customized approaches with large molecule polymer conjugates have enabled numerous successful PEGylated biologics on the market today. Based on our knowledge of the technology and biologics, our scientists have designed novel hydrolyzable linkers that in many cases can be used to optimize bioactivity. Through rational drug design, a protein or peptide’s pharmacokinetics and pharmacodynamics can be substantially improved and its half-life can be significantly extended. An example of this is Baxalta’s ADYNOVATE®, a longer-acting (PEGylated) form of a full-length recombinant factor VIII (rFVIII) protein, which was approved by the FDA in November 2015 for use in adults and adolescents, aged 12 years and older, who have Hemophilia A. In December 2016, Shire plc announced the FDA approved ADYNOVATE® for use in surgical settings for both adults and pediatric patients, and that the FDA also approved ADYNOVATE® for the treatment of Hemophilia A in pediatric patients under 12 years of age.
More recently, our scientists have shown that we can also optimize relative receptor binding characteristics of large molecule conjugates. For instance, the cytokine Interleukin-2 (IL-2) has two different receptor complexes in the body that cause opposing effects on the immune system. We have engineered different novel conjugates of IL-2 with optimized differential receptor binding to the IL-2 receptor categories in the immune system. By biasing the receptor binding of these molecules in complementary ways, we have made two different drug candidates: NKTR-214, which selectively activates effector T cells, which kill tumors; and NKTR-358, which selectively activates regulatory T cells, which can reduce the pathological immune activation that underlies many autoimmune diseases.
Large Molecule Pro-Drug Releasable Polymer Conjugates (Cytokines)
Our customized approaches with large molecule polymer conjugates have expanded to include a new approach with biologics, in particular cytokines, which utilizes the polymer as a means to bias action to a certain receptor or receptor sub-type. In addition, a cytokine’s pharmacokinetics and pharmacodynamics can be substantially improved and its half-life can be significantly extended. An example of this is NKTR-214, which is a CD122-biased immune-stimulatory cytokine with an every two or every three-week dosing schedule.
Antibody Fragment Polymer Conjugates
This approach uses a large molecular weight PEG conjugated to antibody fragments in order to potentially improve their toxicity profile, extend their half-life and allow for ease of synthesis with the antibody. The specially designed PEG replaces the function of the fragment crystallizable (Fc) domain of full length antibodies with a branched architecture PEG with either stable or degradable linkage. This approach can be used to reduce antigenicity, reduce glomerular filtration rate, enhance uptake by inflamed tissues, and retain antigen-binding affinity and recognition. There is currently one approved product on the market that utilizes our technology with an antibody fragment, CIMZIA® (certoluzimab pegol), which was developed by our partner UCB Pharma and is approved for the treatment of Crohn’s Disease, psoriatic arthritis and ankylosing spondylitis in the U.S. and rheumatoid arthritis in the U.S. and EU.
Our Strategy
The key elements of our business strategy are described below:
Advance Our Proprietary Clinical Pipeline of Drug Candidates that Leverage Our Advanced Polymer Conjugate Platform
Our objective is to create value by advancing our lead drug candidates through various stages of clinical development. To support this strategy, we have significantly expanded and added expertise to our internal preclinical, clinical development and regulatory departments. A key component of our development strategy is to potentially reduce the risks and time associated with drug development by capitalizing on the known safety and efficacy of existing drugs and drug candidates as well as established pharmacologic targets and drugs directed to those targets. For many of our novel drug candidates, we may seek to study the drug candidates in indications for which the parent drugs have not been studied or approved. We believe that the improved characteristics of our drug candidates will provide meaningful benefit to patients compared to the existing therapies. In addition, in certain instances we have the opportunity to develop new treatments for patients for which the parent drugs are not currently approved.
9
Ensure Future Growth of our Proprietary Pipeline through Internal Research Efforts and Advancement of our Preclinical Drug Candidates into Clinical Trials
We believe it is important to maintain a diverse pipeline of new drug candidates to continue to build on the value of our business. Our discovery research organization is continuing to identify new drug candidates by applying our technology platform to a wide range of molecule classes, including small molecules and large proteins, peptides and antibodies, across multiple therapeutic areas. We continue to advance our most promising research drug candidates into preclinical development with the objective of advancing these early-stage research programs to human clinical studies over the next several years.
Enter into Strategic and High-Value Partnerships to Bring Certain of Our Drug Candidates to Market
We decide on a drug candidate-by-drug candidate basis, how far to advance clinical development (e.g. Phase 1, 2 or 3) and whether to commercialize products on our own, or seek a partner, or pursue a combination of these approaches. When we determine to seek a partner, our strategy is to enter into collaborations with leading pharmaceutical and biotechnology companies to fund further clinical development, manage the global regulatory filing process, and market and sell drugs in one or more geographies. The options for future collaboration arrangements range from comprehensive licensing and commercialization arrangements to co-promotion and co-development agreements with the structure of the collaboration depending on factors such as the structure of economic risk sharing, the cost and complexity of development, marketing and commercialization needs, therapeutic area and geographic capabilities.
Continue to Build a Leading Intellectual Property Estate in the Field of Polymer Conjugate Chemistry across Therapeutic Modalities
We are committed to continuing to build on our intellectual property position in the field of polymer conjugate chemistry. To that end, we have a comprehensive patent strategy with the objective of developing a patent estate covering a wide range of novel inventions, including among others, polymer materials, conjugates, formulations, synthesis, therapeutic areas, methods of treatment and methods of manufacture.
Nektar Proprietary Programs
The following table summarizes our proprietary drugs and drug candidates that have either received regulatory approval or are being developed by us or in collaboration with other pharmaceutical companies or independent investigators. The table includes the type of molecule or drug, the target indications for the drug candidate, and the status of the clinical development program.
|
Drug Candidate |
|
Target Indication |
|
Status(1) |
|
|
|
|
|
|
|
ONZEALDTM (next-generation topoisomerase I inhibitor) |
|
Advanced metastatic breast cancer in patients with brain metastases |
|
Phase 3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
NKTR-181 (orally-available mu-opioid analgesic molecule) |
|
Moderate to severe chronic pain |
|
Phase 3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
NKTR-214 (CD122-biased immune-stimulatory cytokine) |
|
Oncology |
|
Phase 1/2 in at least eight potential solid tumor indications |
|
|
|
|
|
|
|
NKTR-358 |
|
Autoimmune Disease |
|
Phase 1 |
|
|
|
|
|
|
|
NKTR-262 |
|
Solid Tumors |
|
IND Filed |
|
|
|
|
|
|
|
NKTR-255 |
|
Immuno-Oncology |
|
Research/Preclinical |
|
(1) |
Status definitions are: |
Phase 3 or Pivotal — product in large-scale clinical trials conducted to obtain regulatory approval to market and sell the drug (these trials are typically initiated following encouraging Phase 2 trial results).
Phase 2 — a drug candidate in clinical trials to establish dosing and efficacy in patients.
Phase 1 — a drug candidate in clinical trials, typically in healthy subjects, to test safety.
Research/Preclinical — a drug candidate is being studied in research by way of in vitro studies and/or animal studies
10
Overview of Nektar Proprietary Programs
Immuno-Oncology (I/O)
NKTR-214 (cytokine immunostimulatory therapy)
NKTR-214 is a CD122-biased immune-stimulatory cytokine designed to preferentially activate the IL-2 beta sub-receptors and gamma sub-units of the IL-2 receptor in order to proliferate tumor-killing T cells within the body (CD8-positive effector T cells and natural killer T cells) without stimulating regulatory T cells (CD4-positive T cells). This receptor selectivity is intended to increase efficacy and improve safety over existing immunostimulatory cytokine drugs.
Under a research collaboration with The University of Texas MD Anderson Cancer Center, in December 2015 we commenced a Phase 1 study to evaluate NKTR-214 as a monotherapy in a variety of tumor types to evaluate safety and efficacy, and define the recommended Phase 2 dose of NKTR-214 in patients with solid tumors. In addition, the study also assessed the safety profile of NKTR-214, the immunologic effect of NKTR-214 on tumor-infiltrating lymphocytes and other immune activation markers in both blood and tumor tissue, the pharmacokinetic and pharmacodynamic profile of NKTR-214. Interim data from this dose-escalation monotherapy trial was presented at the 2016 Society for Immunotherapy of Cancer (SITC) meeting in November 2016.
The development program for NKTR-214 includes combinations with a number of therapeutic approaches where we believe there is a strong biologic rationale for complimentary mechanisms of action. On September 21, 2016, we entered into a Clinical Trial Collaboration Agreement with BMS, pursuant to which we and BMS are collaborating to conduct Phase 1/2 clinical trials evaluating NKTR-214 and BMS’ human monoclonal antibody that binds PD-1, known as Opdivo® (nivolumab), as a potential combination treatment regimen in five tumor types and eight potential indications, and such other clinical trials evaluating the combined therapy as may be mutually agreed upon by the parties (each, a Combined Therapy Trial). The dose expansion portion of the Phase 1/2 program evaluating NKTR-214 in combination with Opdivo® is currently enrolling patients at multiple clinical sites. In the first phase of the PIVOT-02 study, we evaluated the clinical benefit, safety, and tolerability of combining NKTR-214 with Opdivo® in thirty-eight patients. Interim data from the dose-escalation phase of the trial was presented at the 2017 SITC meeting in November 2017. We identified the recommended Phase 2 dose for NKTR-214 in combination with Opdivo® and are currently enrolling subjects in the expansion phase of the study. The second phase of the expansion cohorts is evaluating the safety and efficacy of combining NKTR-214 with Opdivo®. Under the Clinical Trial Collaboration Agreement, BMS is responsible for 50% of all out-of-pocket costs reasonably incurred in connection with third party contract research organization, laboratories, clinical sites and institutional review boards. Each party will otherwise be responsible for its own internal costs, including internal personnel costs, incurred in connection with each Combined Therapy Trial. Nektar and BMS will use commercially reasonable efforts to manufacture and supply its compound for each Combined Therapy Trial and will bear the costs related thereto.
On February 13, 2018, we entered into the BMS Collaboration Agreement with BMS, pursuant to which we and BMS will jointly develop NKTR-214, including, without limitation, in combination with BMS’s Opdivo® (nivolumab) and Opdivo® plus Yervoy® (ipilimumab), and other compounds of BMS, us or any third party. The parties have agreed to jointly commercialize NKTR-214 on a worldwide basis. BMS will pay us a non-refundable upfront cash payment of $1 billion and purchase $850.0 million of shares of our common stock at a purchase price of $102.60 per share pursuant to a Share Purchase Agreement (Purchase Agreement). We are eligible to receive additional cash payments of a total of up to $1.43 billion upon achievement of certain development and regulatory milestones and a total of up to $350.0 million upon achievement of certain sales milestones. We will book all worldwide sales and revenue for NKTR-214. We will share global commercialization profits and losses with BMS for NKTR-214, with Nektar sharing 65% and BMS sharing 35% of the net profits and losses. BMS will lead commercialization for combinations of NKTR-214 with BMS proprietary medicines, and we will lead all other commercialization efforts for NKTR-214. We will have the final decision-making authority regarding the pricing for NKTR-214. NKTR-214 will be sold on a stand-alone basis and there will be no fixed-dose combinations or co-packaging without the consent of both parties.
Under the BMS Collaboration Agreement, we and BMS will collaborate to develop and conduct clinical studies of NKTR-214 pursuant to a joint development plan, which initially includes a series of registration-enabling trials in more than 20 indications in nine tumor types and may be updated and expanded only upon mutual agreement of the parties. The parties will share the development costs for NKTR-214 in combination regimens based on each party’s relative ownership interest in the compounds included in the regimens. Nektar’s share of such development costs are limited to an annual cap of $125.0 million. Neither party will develop any Competing Combination, whether alone or in collaboration with any third party, during a limited exclusivity period from the closing date under the BMS Collaboration Agreement until the later of (i) the first commercial sale of NKTR-214 or (ii) the third anniversary of the closing date, but each party may develop a Competing Combination on its own (but not in collaboration with any third party) during the three years after the end of the foregoing limited exclusivity period. If a registration-enabling study included in the joint development plan does not have the first patient enrolled prior to the date which is 14 months from the closing date, subject to allowable delays, the indication covered by that study is no longer subject to the above exclusivity. Other than as described above, Nektar may independently develop and commercialize NKTR-214 either alone or in combination with other Nektar proprietary compounds or third party compounds.
11
The closing under the BMS Collaboration Agreement will occur simultaneously with the closing under a Share Purchase Agreement entered into between us and BMS on February 13, 2018, which is subject to the expiration or early termination of the waiting period (and any extension thereof) under the Hart-Scott-Rodino Antitrust Improvements Act of 1976 and other customary closing conditions. The closings are expected to occur during the second quarter of 2018, upon which the BMS Collaboration Agreement will supersede and replace the existing Clinical Trial Collaboration Agreement.
In addition to the clinical trials in collaboration with BMS, we also plan to initiate a broad clinical development program, both on our own or in collaboration with other potential partners, to explore the potential of combining NKTR-214 with therapies such as cancer vaccines, adoptive cell therapy, small molecules, and other biological agents in order to generate novel immune-oncology approaches.
NKTR-262
NKTR-262 is a small molecule agonist that targets toll-like receptors (TLRs) found on innate immune cells in the body. NKTR-262 is designed to overcome the body’s dysfunction of antigen-presenting cells (APC), such as dendritic cells, which are critical to induce the body’s adaptive immunity and create antigen-specific cytotoxic T cells. NKTR-262 is being developed as a single intra-tumoral injection to be administered at the start of therapy with NKTR-214 in order to induce an abscopal response and achieve the goal of complete tumor regression in cancer patients treated with both therapies. We filed an IND for NKTR-262 in December 2017 and expect to begin enrolling patients in the initial Phase 1 study before the end of the first quarter of 2018.
NKTR-255
NKTR-255 is a memory T cell stimulating cytokine designed to engage the IL-15 pathway to induce long-term T cell activation and improve the quality of T cell memory response to treat cancer. Through optimal engagement of the IL-15Rα/IL-2Rγ receptor complex, NKTR-255 stimulates proliferation and survival of CD8+ T cells, natural killer (NK) cells and enhances formation of long-term immunological memory which may lead to sustained anti-tumor immune response. Native rhIL-15 is rapidly cleared from the body and must be administered frequently and in high doses limiting its utility due to toxicity. NKTR-255 is designed with IL-15 receptor alpha specificity to optimize biological activity and is uniquely engineered to provide optimal exposure and an improved safety profile.
Immunology
NKTR-358 is designed to correct the underlying immune system imbalance in the body which occurs in patients with autoimmune disease. Current systemic treatments for autoimmune disease, including corticosteroids and anti-TNF agents, suppress the immune system broadly and come with severe side effects. NKTR-358 targets the CD25 sub-receptor in the interleukin-2 pathway in order to stimulate proliferation and growth of regulatory T cells, which are specific immune cells in the body that modulate the immune system and prevent autoimmune disease by maintaining self-tolerance.
On July 23, 2017, we entered into the Lilly Agreement, pursuant to which we and Lilly will co‑develop NKTR-358. Under the terms of the Lilly Agreement, we received an initial payment of $150.0 million in September 2017 and are eligible for up to $250.0 million in additional development and regulatory milestones. We will be responsible for completing Phase 1 clinical development and certain drug product development and supply activities. We will also share Phase 2 development costs with Lilly, with 75% of those costs borne by Lilly and 25% of the costs borne by us. We will have the option to contribute funding to Phase 3 development on an indication-by-indication basis, ranging from zero to 25% of the global Phase 3 development costs. We are eligible to receive up to double-digit sales royalty rates that escalate based upon our contribution to Phase 3 development cost and the level of global product annual sales. Lilly will be responsible for all costs of global commercialization and we will have an option to co-promote in the U.S. under certain conditions.
In February 2017 we filed an IND for NKTR-358 and we are currently enrolling patients in the initial Phase 1 dose-finding trial of NKTR-358 to evaluate single-ascending doses of NKTR‑358 in approximately 50 healthy patients. The Phase 1 multiple-ascending dose trial is expected to be initiated in the second quarter of 2018 to evaluate NKTR‑358 in patients with systemic lupus erythematosus and other indications.
Pain - NKTR-181 (mu-opioid analgesic molecule for chronic pain)
NKTR-181 is an orally-available novel mu-opioid analgesic molecule in development as a long-acting analgesic to treat chronic pain. NKTR-181 is designed with the objective to address the abuse liability and serious central nervous system (CNS) side effects associated with current opioid therapies. NKTR-181 was created using Nektar’s proprietary polymer conjugate technology, which provides it with a long-acting profile and slows its entry into the CNS. The abuse deterrent properties of NKTR-181 are inherent to its novel molecular structure and do not rely on a formulation approach to prevent its conversion into a more abusable form of an opioid. In May 2012, the FDA granted Fast Track designation for the NKTR-181 development program.
12
In June 2012, we initiated a Phase 2 clinical study to evaluate the efficacy, safety and tolerability of NKTR-181 in patients with moderate to severe chronic pain from osteoarthritis of the knee. The Phase 2 clinical study utilized a double-blind, placebo-controlled, randomized withdrawal, enriched enrollment study design. The study enrolled 295 opioid-naïve patients with osteoarthritis of the knee who were not getting adequate pain relief from their current non-opioid pain medication. Patients who qualified during the baseline period entered a titration phase, during which they were titrated on NKTR-181 tablets administered orally twice-daily until a dose was reached that provided a reduction of at least 20% in the patient’s pain score as compared to the patient’s own baseline. Patients that achieved this level of analgesia were then randomized on a 1:1 basis to either continue to receive their analgesic dose of NKTR-181 or to receive placebo for up to 25 days. The primary endpoint of the study was the average change in a patient’s pain score from baseline to the end of the double-blind, randomized treatment period.
In the first half of 2013, we conducted a human abuse liability study, or HAL study, for NKTR-181. In this study, NKTR-181 had highly statistically significant lower "drug liking" scores and reduced "feeling high" scores as compared to oxycodone at all doses tested (p < 0.0001). On June 19, 2013, we presented data from the HAL study at the 2013 Annual Meeting of The College on Problems of Drug Dependence in San Diego, California.
On September 26, 2013, we announced results from this Phase 2 efficacy study. Of the 295 patients that entered the study, only 9 patients (representing 3% of the patient population) were unable to achieve meaningful pain relief with NKTR-181. A total of 213 patients achieved an average 40% reduction in pain and entered the randomized phase of the study. NKTR-181 performed as expected as an opioid analgesic throughout the study with patients continuing to show a reduction in pain scores throughout the randomized phase of the study. However, patients who were randomized to placebo did not show the expected increase in pain scores observed in similar enriched enrollment, randomized withdrawal studies. This unusual lack of a placebo rebound caused the Phase 2 study to miss the primary endpoint in the study.
In October 2014, we engaged in an end-of-Phase 2 meeting for NKTR-181 with the FDA, which included discussions of the design of the Phase 3 clinical study program. We enrolled the first patient in the first Phase 3 efficacy study, which we call SUMMIT-07 in February 2015 and we completed enrollment in the study in the fourth quarter of 2016. In this study, we randomized patients with chronic low back pain in an enriched enrollment randomized withdrawal design which included a qualifying screening period, an open-label titration period where NKTR-181 was given to all patients, followed by a 12-week double-blind randomized period where subjects were randomized on a 1:1 basis to receive either NKTR-181 or placebo. On February 29, 2016, we increased the sample size of this trial by approximately 200 patients following a pre-specified sample size assessment by the independent analysis center (IAC) after approximately fifty percent of the initially planned 416 patients completed the study. The protocol for the NKTR-181 study defined only two possible outcomes for this pre-planned blinded interim sample size assessment: (1) if the conditional powering at the midpoint of the trial fell between 50-85%, the sample size was to be increased by approximately 200 patients; or (2) if the conditional powering fell below 50%, or above 85%, the sample size was not to be changed. The IAC’s determination was nondiscretionary and was based upon our determination of pre-defined acceptable power to detect a statistically significant difference between NKTR-181 and placebo based on the primary efficacy endpoint. In January 2017, we started enrollment in a second human abuse liability study where we are assessing abuse liability of supra-therapeutic doses of NKTR-181 in order to further evaluate NKTR-181 for labeling and scheduling purposes. On March 20, 2017, we announced that NKTR-181 met its primary and secondary endpoints in the SUMMIT-07 Phase 3 efficacy Study. On July 18, 2017, we announced positive top-line data for our pivotal human abuse potential study for NKTR-181, which we call the HAP study.
The SUMMIT Phase 3 program also includes a 52-week long-term safety study, which we call SUMMIT-LTS. SUMMIT-LTS is evaluating the long-term safety and tolerability of NKTR-181 in over 600 patients (opioid-naïve and opioid-experienced) with moderate to severe chronic low pain or chronic non-cancer pain. The study was completed in December 2017.
Following our success of the SUMMIT-07 Phase 3 efficacy study and the HAP study, we are seeking a partner to support future development and commercialization activities for NKTR-181. We have completed pre-NDA meetings with the FDA to discuss plans for an NDA submission and we are currently planning to file an NDA for NKTR-181 in the first half of 2018.
According to a 2011 report from the National Academy of Sciences, chronic pain conditions, such as osteoarthritis, back pain and cancer pain, affect at least 100 million adults in the U.S. annually and contribute to over $300 billion a year in lost productivity. Opioids are considered to be the most effective therapeutic option for pain. However, opioids cause significant problems for physicians and patients because of their serious side effects such as respiratory depression and sedation, as well as the risks they pose for addiction, abuse, misuse, and diversion. The FDA has cited prescription opioid analgesics as being at the center of a major public health crisis of addiction, misuse, abuse, overdose and death. According to the American Society of Addiction Medicine 2016 report, there are 1.9 million Americans which have a substance use disorder involving prescription pain relievers. This same report attributes 18,893 overdose deaths in 2015 were related to prescription pain relievers.
13
Oncology - ONZEALDTM (next generation, long-acting topoisomerase I inhibitor)
ONZEALDTM (previously known as NKTR-102 or etirinotecan pegol) is our next-generation topoisomerase I inhibitor proprietary drug candidate. In 2015, we announced top-line data from a Phase 3 clinical study for ONZEALDTM, which we call the BEACON study (BrEAst Cancer Outcomes with ONZEALDTM), as a single-agent therapy for women with advanced metastatic breast cancer. The BEACON study compared ONZEALDTM to an active control arm comprised of a single chemotherapy agent of physician’s choice (TPC) in patients who were heavily pre-treated with a median of three prior therapies for metastatic disease. In a top-line analysis of 852 patients from the trial, ONZEALDTM provided a 2.1 month improvement in median overall survival over TPC (12.4 months for patients receiving ONZEALDTM compared to 10.3 months for patients receiving TPC). Based on a stratified log-rank analysis, the primary endpoint measuring the hazard ratio for survival in the ONZEALDTM group compared to the active control arm was 0.87 with a p-value of 0.08, which did not achieve statistical significance. Secondary endpoints in the BEACON study included objective response rate and progression-free survival, which did not achieve statistical significance in the study. We also announced that we observed a significant overall survival benefit in two pre-specified subgroups—patients with a history of brain metastases and patients with baseline liver metastases at study entry.
We have also conducted clinical studies for ONZEALDTM in other solid tumor settings. In 2013, we completed a Phase 2 clinical study for ONZEALDTM in approximately 170 patients with platinum-resistant/refractory ovarian cancer. We also initiated a Phase 2 clinical study of ONZEALDTM monotherapy versus irinotecan in second-line metastatic colorectal cancer patients with the KRAS mutant gene. The Phase 2 clinical study was designed to enroll 174 patients with metastatic colorectal cancer. In February 2014, we decided to close enrollment in this study after 80 patients were randomized due to challenges in recruiting new patients because the comparator arm of this study, single-agent irinotecan, is not the common standard of care for second line metastatic colorectal therapy in the U.S. or EU.
We also conducted a Phase 1 dose-escalation clinical study which enrolled 26 patients to evaluate ONZEALDTM in combination with 5-Fluorouracil (5-FU)/leucovorin in refractory solid tumor cancers. The chemotherapy agent 5-FU is currently used as a part of a combination treatment regimen for colorectal cancer in combination with irinotecan, which is also known as the FOLFIRI regimen. On January 18, 2014, we presented data from this study at the 2014 Gastrointestinal Cancers Symposium in San Francisco, California. In addition to the clinical study of ONZEALDTM being conducted by us, we have also provided support for four investigator-initiated Phase 2 studies being conducted for ONZEALDTM. On August 7, 2012, we announced a Phase 2 investigator-initiated clinical study of ONZEALDTM in patients with bevacizumab (Avastin)-resistant high-grade glioma being conducted at the Stanford Cancer Institute. In May 2013, the study completed enrollment of 20 patients with high-grade glioma who had received a median of three prior lines of therapy before enrolling in the study. A separate investigator-initiated clinical study is also being conducted at Stanford to evaluate ONZEALDTM in patients with brain metastasis from primary lung cancer. On February 5, 2013, we announced a Phase 2 investigator-initiated clinical study of ONZEALDTM in patients with metastatic and recurrent non-small cell lung cancer being conducted at the Abramson Cancer Center of the University of Pennsylvania. On October 24, 2013, we announced a Phase 2 investigator-initiated clinical study of ONZEALDTM in patients with relapsed or refractory small-cell lung cancer at the Roswell Park Cancer Institute.
We have explored future regulatory and development paths forward for ONZEALDTM with the EU and U.S. health authorities. In Europe, we met with the National Authorities in Sweden and the United Kingdom, as well as the EMA to discuss the BEACON data. In June 2016, we filed an MAA for conditional approval of ONZEALDTM for adult patients with advanced breast cancer who have brain metastases treated with ONZEALDTM versus physicians treatment of choice in the ATTAIN trial. On July 21, 2017, we were informed by the EMA’s Committee for Medicinal Products for Human Use (CHMP) that it had adopted a negative opinion for the conditional marketing authorization application for ONZEALD™ in the EU. On July 26, 2017, we filed a request for re-examination of the opinion adopted by the CHMP (the “CHMP Appeal”) but we were informed by the CHMP in November 2017 that our request was not successful. On December 6, 2017 we received notice of termination from Daiichi exercising its right to terminate the Daiichi Collaboration Agreement, which termination became effective on February 4, 2018. As a result of the termination, we made a $12.5 million termination payment to Daiichi in December 2017, and all rights and licenses granted to Daiichi under the Daiichi Collaboration Agreement reverted exclusively to Nektar. The Phase 3 study (ATTAIN) in breast cancer patients having brain metastases is ongoing.
According to the American Cancer Society and World Health Organization, more than 1.4 million women worldwide are diagnosed with breast cancer globally every year. The chance of developing invasive breast cancer at some time in a woman’s life is a little less than one in eight (approximately 12%). In 2017, the American Cancer Society estimates there will be 252,710 new cases of invasive breast cancer diagnosed in the U.S. and about 40,610 women will die from breast cancer. Anthracyclines and taxanes are the among the most active and widely used chemotherapeutic agents for breast cancer, but the increased use of these agents at an early stage of disease often renders tumors resistant to these drugs by the time the disease recurs, thereby reducing the number of treatment options for metastatic disease. There are currently no FDA-approved topoisomerase I inhibitors indicated to treat breast cancer.
14
Collaboration Partner Programs
The following table outlines our collaborations with a number of pharmaceutical companies that currently license our intellectual property and, in some cases, purchase our proprietary PEGylation materials for their drug products. More than ten products using our PEGylation technology have received regulatory approval in the U.S. or Europe. There are also a number of other candidates that have been filed for approval or are in various stages of clinical development. These collaborations generally contain one or more elements including a license to our intellectual property rights and manufacturing and supply agreements under which we may receive manufacturing revenue, milestone payments, and/or royalties on commercial sales of drug products.
|
Drug |
|
Primary or Target Indications |
|
Drug Marketer/Partner |
|
Status(1) |
|
|
|
|
|
|
|
|
|
MOVANTIK® (naloxegol tablets) |
|
Opioid-induced constipation in adult patients with chronic non-cancer pain |
|
AstraZeneca AB |
|
Approved |
|
|
|
|
|
|
|
|
|
MOVENTIG® (brand name for MOVANTIK® in Europe) |
|
Opioid-induced constipation in adult patients who have an inadequate response to laxatives |
|
AstraZeneca AB |
|
Approved |
|
|
|
|
|
|
|
|
|
ADYNOVATE® (previously referred to as BAX 855, PEGylated rFVIII) |
|
Hemophilia A |
|
Shire plc |
|
Approved |
|
|
|
|
|
|
|
|
|
ADYNOVI® (brand name for ADYNOVATE® in Europe) |
|
Hemophilia A |
|
Shire plc |
|
Approved |
|
|
|
|
|
|
|
|
|
Somavert® (pegvisomant) |
|
Acromegaly |
|
Pfizer Inc. |
|
Approved |
|
|
|
|
|
|
|
|
|
Neulasta® (pegfilgrastim) |
|
Neutropenia |
|
Amgen Inc. |
|
Approved |
|
PEG-INTRON® (peginterferon alfa-2b) |
|
Hepatitis-C |
|
Merck (through its acquisition of Schering-Plough Corporation) |
|
Approved |
|
|
|
|
|
|
|
|
|
Macugen® (pegaptanib sodium injection) |
|
Age-related macular degeneration |
|
Valeant Pharmaceuticals International, Inc. |
|
Approved |
|
|
|
|
|
|
|
|
|
CIMZIA® (certolizumab pegol) |
|
Rheumatoid arthritis |
|
UCB Pharma |
|
Approved * |
|
|
|
|
|
|
|
|
|
CIMZIA® (certolizumab pegol) |
|
Crohn’s disease |
|
UCB Pharma |
|
Approved * |
|
|
|
|
|
|
|
|
|
CIMZIA® (certolizimab pegol) |
|
Psoriasis/Ankylosing Spondylitis |
|
UCB Pharma |
|
Approved * |
|
|
|
|
|
|
|
|
|
MIRCERA® (C.E.R.A.) (Continuous Erythropoietin Receptor Activator) |
|
Anemia associated with chronic kidney disease in patients on dialysis and patients not on dialysis |
|
F. Hoffmann-La Roche Ltd |
|
Approved ** |
|
SEMPRANA® |
|
Migraine |
|
Allergan, Inc. |
|
Filed for approval in U.S. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dapirolizumab Pegol |
|
Systemic Lupus Erythematosus |
|
UCB Pharma (Biogen) |
|
Phase 2 |
|
|
|
|
|
|
|
|
|
PEGPH20 |
|
Pancreatic, Non-Small Cell Lung Cancer, and other multiple tumor types |
|
Halozyme Therapeutics, Inc. |
|
Phase 1, 2, and 3 |
|
|
|
|
|
|
|
|
|
Longer-acting blood clotting proteins |
|
Hemophilia |
|
Baxalta |
|
Research/Preclinical |
|
|
|
|
|
|
|
|
|
(1) |
Status definitions are: |
Approved — regulatory approval to market and sell product obtained in one or more of the U.S., EU or other countries.
Filed — an application for approval and marketing has been filed with the applicable government health authority.
15
Phase 3 or Pivotal — product in large-scale clinical trials conducted to obtain regulatory approval to market and sell the drug
(these trials are typically initiated following encouraging Phase 2 trial results).
Phase 2 — a drug candidate in clinical trials to establish dosing and efficacy in patients.
Phase 1 — a drug candidate in clinical trials, typically in healthy subjects, to test safety.
Research/Preclinical — a drug candidate is being studied in research by way of in vitro studies and/or animal studies
|
* |
In February 2012, we sold our rights to receive royalties on future worldwide net sales of CIMZIA® effective as of January 1, 2012. |
|
** |
In February 2012, we sold our rights to receive royalties on future worldwide net sales of MIRCERA® effective as of January 1, 2012 until the agreement with Roche is terminated or expires. |
With respect to all of our collaboration and license agreements with third parties, please refer to Item 1A, Risk Factors, including without limitation, “We are a party to numerous collaboration agreements and other significant agreements which contain complex commercial terms that could result in disputes, litigation or indemnification liability that could adversely affect our business, results of operations and financial condition.”
Overview of Collaboration Partner Programs
We have a number of product candidates in clinical development and approved products in collaboration with our partners where we invented the drug candidate or where our collaboration partners have licensed our proprietary intellectual property to enable one of their drug candidates. Our agreements with collaboration partners may involve several elements including a technology license as well as the development, commercialization, and manufacturing and supply obligations. We typically receive consideration from our collaboration partners in the form of upfront payments, or milestone payments and royalties on sales. In certain cases, we also manufacture and supply our proprietary polymer materials to our partners.
MOVANTIK® and MOVANTIK® Fixed-Dose Combination Products (MOVANTIK® previously referred to as naloxegol or NKTR-118 and MOVANTIK® Fixed-Dose Combination Products previously referred to as NKTR-119), License Agreement with AstraZeneca AB
In September 2009, we entered into a global license agreement with AstraZeneca AB (AstraZeneca) pursuant to which we granted AstraZeneca a worldwide, exclusive, perpetual, royalty-bearing license under our patents and other intellectual property to develop, market and sell MOVANTIK® and MOVANTIK® fixed-dose combination products. MOVANTIK® was developed using our oral small molecule polymer conjugate technology and we advanced this drug through the completion of Phase 2 clinical studies prior to licensing it to AstraZeneca. MOVANTIK® is an orally-available peripherally-acting mu-opioid antagonist which is a medication for the treatment of opioid-induced constipation (OIC), which is a common side effect of prescription opioid medications. Opioids attach to specific proteins called opioid receptors. When the opioids attach to certain opioid receptors in the gastrointestinal tract, constipation may occur. OIC is a result of decreased fluid absorption and lower gastrointestinal motility due to opioid receptor binding in the gastrointestinal tract.
On September 16, 2014, the FDA approved MOVANTIK® as the first once-daily oral peripherally-acting mu-opioid receptor antagonist (PAMORA) medication for the treatment of OIC in adult patients with chronic, non-cancer pain. On December 9, 2014, the European Commission, or EC, granted Marketing Authorisation to MOVENTIG® (the naloxegol brand name in the EU) as the first once-daily oral PAMORA to be approved in the EU for the treatment of OIC in adult patients who have had an inadequate response to laxative(s). The EC’s approval applies to all 28 EU member countries plus Iceland and Norway. AstraZeneca launched the commercial sales of MOVANTIK® in the United States in March 2015 and MOVENTIG® in Germany, the first EU member country, in August 2015. Under the terms of our license agreement with AstraZeneca, AstraZeneca made an initial license payment of $125.0 million to us and has responsibility for all activities and bears all costs associated with research, development and commercialization for MOVANTIK® and MOVANTIK® fixed-dose combination products. We received milestone payments of $70.0 million and $25.0 million upon the acceptance of regulatory approval applications of MOVANTIK® by the FDA and the EMA, respectively, in 2013. We received an additional developmental milestone payment of $35.0 million upon the FDA’s approval of MOVANTIK® in 2014 and a total of $140.0 million upon commercial launches in 2015, including $100.0 million for MOVANTIK® in the U.S. and $40.0 million for MOVENTIG® in Germany. We are also entitled to up to $375.0 million in sales milestones for MOVANTIK® if the program achieves certain annual commercial sales levels. For the MOVANTIK® fixed-dose combination products, we are also eligible to receive significant development milestones as well as significant sales milestone payments if the program achieves certain annual commercial sales levels. For both MOVANTIK® and the fixed-dose combination products, we are also entitled to significant double-digit royalty payments starting at 20% of net sales in the U.S. and 18% of net sales in the EU and rest of world, varying by country of sale and level of annual net sales. On March 1, 2016, AstraZeneca announced that it had entered into an agreement with Kyowa Hakko Kirin Co. Ltd. (Kirin), granting Kirin exclusive marketing rights to MOVENTIG® in the EU, Iceland, Liechtenstein, Norway and Switzerland. Nektar receives a 40% share of royalty payments made by Kirin to AstraZeneca will be financially equivalent to Nektar receiving high single-digit to low double-digit royalties depending on Kirin’s annual net sales levels. Our right to receive royalties (subject to certain adjustments) in any particular country will expire upon the later of (a) a
16
specified period of time after the first commercial sale of the product in that country or (b) the expiration of patent rights in that particular country. AstraZeneca has agreed to use commercially reasonable efforts to develop one MOVANTIK® fixed-dose combination product and has the right to develop multiple products which combine MOVANTIK® with opioids.
There are a number of patents relevant to MOVANTIK®, some of which are listed in the FDA’s “Orange Book.” The “Orange Book” currently lists six patents for MOVANTIK®. Four patents (i.e., U.S. Patent Nos. 7,056,500, 7,662,365, 7,786,133 and 9,012,469) are “composition of matter patents” - one of which has a patent expiry extending into 2032. In addition, two patents (i.e., U.S. Patent Nos. 8,067,431 and 8,617,530) are directed to methods of treatment.
ADYNOVATE® (previously referred to as BAX 855) and Long-Acting Therapies for Hemophilia A, Agreement with Subsidiaries of Baxalta Incorporated
In September 2005, we entered into an exclusive research, development, license, manufacturing and supply agreement (Baxalta License Agreement) with certain subsidiaries of Baxalta (which has been acquired by Shire plc), formerly Baxter before the separation of Baxalta from Baxter in July 2015, to develop products with an extended half-life for the treatment and prophylaxis of Hemophilia A patients using our proprietary PEGylation technology. The first product in this collaboration, ADYNOVATE® (previously referred to as BAX 855), is a longer-acting (PEGylated) form of a full-length recombinant factor VIII (rFVIII) protein. ADYNOVATE® is a full-length PEGylated longer-acting recombinant factor VIII (rFVIII) that was developed to increase the half-life of ADVATE® (Antihemophilic Factor (Recombinant) Plasma/Albumin-Free Method). We are entitled to up to $55.0 million in total development and sales milestone payments, as well as royalties on net sales varying by product and country of sale. The royalties start in the mid-single digits for net sales of ADYNOVATE® up to $1.2 billion and then in the low teens for net sales exceeding $1.2 billion. Our right to receive these royalties in any particular country will expire upon the later of ten years after the first commercial sale of the product in that country or the expiration of patent rights in certain designated countries or in that particular country.
In November 2015, ADYNOVATE® was approved by the FDA for use in adults and adolescents, aged 12 years and older, who have Hemophilia A. On November 30, 2015, Baxalta announced that it had initiated sales of ADYNOVATE® in the U.S. In December 2016 the FDA expanded the approval of ADYNOVATE® for the treatment of Hemophilia A in pediatric patients under 12 years of age, and for the use in surgical settings for both adult and pediatric patients. In March 2016, ADYNOVATE® was approved in Japan for patients aged 12 years and older with Hemophilia A and in November 2017 that approval was extended to include pediatric patients under 12 years of age, and those undergoing surgery. In late 2016, ADYNOVATE® (marketed as ADYNOVI) was approved in Switzerland for use in Hemophilia A patients aged 12 years and older and in March 2017, ADYNOVI was approved for use in pediatric patients. In November 2016, ADYNOVATE® was approved in Canada for patients 12 years of age and older for the treatment of Hemophilia A, including for perioperative management. In March 2017, ADYNOVATE® was approved in Australia for use in adult and pediatric patients with Hemophilia A. In September 2017, ADYNOVATE® was approved in Colombia for use in children and adults with Hemophilia A. In January 2018 ADYNOVATE® (marketed as ADYNOVI) was approved in the European Union for on-demand and prophylactic use in patients 12 years and older with Hemophilia A. In January 2018, ADYNOVATE® was also approved in S. Korea for use in children and adults with Hemophilia A.
In October 2017, we entered into a right to sublicense agreement with Baxalta, under which we granted to Baxalta the right to grant a nonexclusive sublicense to certain patents to a third party that were previously exclusively licensed to Baxalta under the Baxalta License Agreement. Under the right to sublicense agreement, Baxalta paid us $12.0 million in November 2017 and agreed to pay us single digit royalty payments based upon net sales of the third party products covered under the sublicense throughout the term of the right to sublicense agreement.
Hemophilia A, also called factor VIII (FVIII) deficiency or classic hemophilia, is a genetic disorder caused by missing or defective factor VIII, a clotting protein. According to the US Centers for Disease Control and Prevention, hemophilia occurs in approximately one in 5,000 live births and there are about 20,000 people with hemophilia in the US. All races and ethnic groups are affected. Hemophilia A is four times as common as Hemophilia B while more than half of patients with Hemophilia A have the severe form of hemophilia. In 2014, according to the Evaluate Group, sales of FVIII replacement products exceeded $6.0 billion globally.
Amikacin Inhale (BAY41-6551, formerly NKTR-061), Agreement with Bayer Healthcare LLC
In August 2007, we entered into a co-development, license and co-promotion agreement (Bayer Agreement) with Bayer Healthcare LLC (Bayer) to develop a specially-formulated Amikacin (BAY41-6551, Amikacin Inhale, formerly called NKTR-061) for the treatment of gram-negative pneumonias. Under the terms of the agreement, Bayer was responsible for most future clinical development and commercialization costs, all activities to support worldwide regulatory filings, approvals and related activities, further development of formulated Amikacin and final product packaging for Amikacin Inhale. We were responsible for all future development, manufacturing and supply of the nebulizer device for clinical and commercial use.
17
In April 2013, Bayer initiated enrollment in a global Phase 3 clinical study, which it calls INHALE, to evaluate the efficacy and safety of Amikacin Inhale versus aerosolized placebo in the treatment of intubated and mechanically ventilated patients with Gram-negative pneumonia receiving standard of care intravenous antibiotics. On November 24, 2017, Bayer announced that the Phase 3 Amikacin Inhale clinical program did not meet its primary endpoint or key secondary endpoints. On December 14, 2017, we received notice of termination from Bayer of the Bayer Agreement effective as of January 13, 2018. No payment obligations to Bayer arose as a result of the termination of the Bayer Agreement.
Cipro DPI (formerly known as Cipro Inhale), Bayer Schering Pharma AG
We were a party to a collaborative research, development and commercialization agreement with Bayer Schering Pharma AG (Bayer Schering), related to the development of an inhaled powder formulation of ciprofloxacin delivered by way of a dry powder inhaler, Cipro DPI (formerly known as Cipro Inhale) for the treatment of chronic lung infections caused by Pseudomonas aeruginosa in cystic fibrosis patients. On December 31, 2008, we assigned the agreement to Novartis Pharma AG in connection with the completion of the pulmonary asset sale transaction. However, we retained our economic interest in the future potential net sales royalties if Cipro DPI is approved by health authorities and is successfully commercialized by Bayer Schering. In August 2012, Bayer Schering initiated a Phase 3 clinical development program which it calls RESPIRE for Cipro DPI in patients with non-cystic fibrosis bronchiectasis (NCFB). In these patients with NCFB, the bronchial tubes are enlarged, allowing mucus to pool and making the area prone to infection. In September 2016, Bayer Schering presented data from the first Phase 3 trail (RESPIRE 1) at the 2016 European Respiratory Society Annual Meeting which showed that the study met its co-primary endpoints for the every 14-day dosing arm of Cipro DPI. The second Phase 3 trial (RESPIRE 2) completed recruitment in September 2016. On April 5, 2017, an abstract with data from the RESPIRE 2 trial was published by the American Thoracic Society in connection with the American Thoracic Society 2017 International Conference. A pooled analysis of the primary efficacy results of RESPIRE 1 and RESPIRE 2 is positive and the data of both RESPIRE 1 and RESPIRE 2 indicated that Cipro DPI has a positive safety profile. On November 16, 2017, the Antimicrobial Drugs Advisory Committee of the FDA voted not to recommend Cipro DPI to be approved for the treatment of non-cystic fibrosis. Bayer Schering has informed us that they are discontinuing development of Cipro DPI in NCFB for the time being as they evaluate possible future options for this asset.
FOVISTA® (Anti-PDGF Therapy), Agreement with Ophthotech Corporation
On May 19, 2014, Ophthotech entered into a Licensing and Commercialization Agreement with Novartis Pharma AG to develop and commercialize Fovista® and related combination products in all countries outside of the U.S. Under the original license and supply agreement entered into between us and Ophthotech, we received a $19.8 million payment in June 2014 in connection with this licensing agreement. In December 2016 Ophthotech announced that two pivotal Phase 3 clinical trials for Fovista® failed to meet their primary endpoints. In August 2017, Opthotech announced that the third Fovista® Phase 3 trial also failed to meet its primary endpoint. On October 27, 2017, we agreed to terminate our license and supply agreement with Ophthotech.
PEGPH20, Agreement with Halozyme Therapeutics, Inc.
In December 2006, we entered into a license agreement with Halozyme pursuant to which we granted Halozyme a worldwide, limited exclusive license to certain of our proprietary PEGylation technology to develop, manufacture and commercialize particular products that use our proprietary PEGylation materials linked only with certain qualifying hyaluronidase protein molecules including PEGPH20. According to Halozyme, certain cancers, including pancreatic, breast, colon and prostate, have been shown to accumulate high levels of hyaluronan (HA). Halozyme’s FDA-approved, HYLENEX® recombinant human hyaluronidase, rHuPH20, is administered subcutaneously and temporarily and reversibly degrades HA to facilitate the absorption and dispersion of other injected drugs or fluids and for subcutaneous fluid administration. However, rHuPH20 acts only locally at the injection site, is rapidly inactivated in the body, and does not survive in the blood. PEGPH20 is an investigational PEGylated form of rHuPH20, under development by Halozyme to increase the half-life of the compound in the blood and allow for intravenous administration. Halozyme is currently evaluating PEGPH20 in a Phase 3 clinical study combining PEGPH20 with ABRAXANE® (nab-paclitaxel) and gemcitabine in stage IV pancreatic ductal adenocarcinoma (PDA) (HALO 109-301), in Phase 1b clinical testing for PEGPH20 with KEYTRUDA® (pembrolizumab) in non-small cell lung cancer and gastric cancer (HALO 107-101), in Phase 1b/2 clinical testing for PEGPH20 with HALAVEN® (eribulin) in patients treated with up to two lines of prior therapy for HER2-negative metastatic breast cancer, in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq® (atezolizumab) in patients with previously treated metastatic PDA, in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq in patients with gastric cancer and in Phase 1b/2 clinical testing for PEGPH20 with Tecentriq in patients with cholangiocarcinoma and gall bladder cancer (HALO 110-101/MATRIX). We are entitled to future development milestones and royalties on net sales subject to reduction in the absence of patent coverage. Our right to receive royalties in any particular country will expire upon the later of twelve years after first commercial sale of the product or expiration of patent rights in the particular country. We also manufacture and supply Halozyme with clinical and future commercial supply of our proprietary PEGylation materials used in the manufacture of PEGPH20.
18
SEMPRANA®, Agreement with MAP Pharmaceuticals, Inc. (a wholly-owned subsidiary of Allergan, Inc.)
In June 2004, we entered into a license agreement with MAP Pharmaceuticals, Inc. (MAP), which includes a worldwide, exclusive license, to certain of our patents and other intellectual property rights to develop and commercialize a formulation of dihydroergotamine (DHE) for administration to patients via the pulmonary or nasal delivery route, which resulted in the development of SEMPRANA®, formerly known as LEVADEX®. In 2006, we amended and restated this agreement. Under the terms of the agreement, we have the right to receive certain milestone payments based on development criteria that are solely the responsibility of MAP and royalties based on net sales of SEMPRANA®. Our right to receive royalties for the net sales of SEMPRANA® under the license agreement in any particular country will expire upon the later of (i) 10 years after first commercial sale in that country, (ii) the date upon which the licensed know-how becomes known to the general public, and (iii) expiration of certain patent claims, each on a country-by-country basis. Either party may terminate the agreement upon a material, uncured default of the other party.
SEMPRANA® is a self-administered formulation of DHE using an inhaler device that is currently under review by the FDA. On May 26, 2011, MAP submitted an NDA to the FDA for SEMPRANA®. In March of 2012, the FDA issued a complete response letter to MAP identifying issues relating to chemistry, manufacturing and controls deficiencies of the product at a contracted third party manufacturer. On April 17, 2013, the FDA issued a second complete response letter identifying issues related to a supplier that provided the canister filling unit for SEMPRANA®. In June 2014, Allergan which acquired MAP as described below, announced that it had received a third complete response letter from the FDA related to specifications around content uniformity on the improved canister filling process and on standards for device actuation. Allergan has responded to the FDA’s latest complete response letter and has stated that it continues to work with the FDA to resolve outstanding Chemistry, Manufacturing and Controls (CMC) issues.
On January 28, 2011, MAP entered into a Collaboration Agreement with Allergan, Inc. (Allergan), pursuant to which Allergan received a co-exclusive license to market and promote SEMPRANA® to neurologists and pain specialists in the U.S. Under this arrangement, Allergan paid MAP an upfront payment of $60.0 million. MAP was also entitled to receive up to an additional $97.0 million in the form of regulatory milestones, which includes milestones for the FDA’s acceptance of filing of the SEMPRANA® NDA and first commercial sale associated with the initial acute migraine indication. On March 1, 2013, Allergan completed a merger and acquisition transaction with MAP pursuant to which MAP become a wholly-owned subsidiary of Allergan. In December 2017, we, MAP and Allergan entered into a Settlement Agreement and Release. Pursuant to this agreement, Allergan paid Nektar $15.0 million in December 2017 in exchange for relief of any claims related to our share of payments received by MAP from Allergan as well as a specified development milestone payment due to Nektar.
Dapirolizumab Pegol
In 2010, we entered into a license, manufacturing and supply agreement with UCB Pharma S.A., (UCB) under which we granted UCB a worldwide, exclusive license to certain of our proprietary PEGylation technology to develop, manufacture and commercialize an anti-CD40L PEGylated Fab being developed by UCB and their partner Biogen Idec, for the treatment of autoimmune disorders, including systemic lupus erythemastosus (SLE). In 2014, UCB and Biogen completed a Phase 1b randomized, double-blind, placebo-controlled clinical study in approximately 24 patients with SLE. Data from the study was published in September 2015 at the Annual American College of Rheumatology Meeting and showed that multiple administrations of dapirolizumab pegol given over 12 weeks were well-tolerated and the safety profile supported further development of the compound. Exploratory analyses from the same study showed greater improvement in clinical measures of disease activity in the dapriolizumab pegol group versus placebo. In 2016, UCB initiated a multi-center, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging Phase 2 clinical study followed by an observational period to evaluate the efficacy and safety of patients with moderately to severely active SLE receiving stable standard of care medications. This Phase 2 clinical trial is currently recruiting patients.
Neulasta®, Agreement with Amgen, Inc.
In July 1995, we entered into a non-exclusive supply and license agreement (the 1995 Agreement) with Amgen, Inc., pursuant to which we licensed our proprietary PEGylation technology to be used in the development and manufacture of Neulasta®. Neulasta® selectively stimulates the production of neutrophils that are depleted by cytotoxic chemotherapy, a condition called neutropenia that makes it more difficult for the body to fight infections. On October 29, 2010, we amended and restated the 1995 Agreement by entering into a supply, dedicated suite and manufacturing guarantee agreement (the 2010 Agreement) and an amended and restated license agreement with Amgen Inc. and Amgen Manufacturing, Limited (together referred to as Amgen). Under the terms of the 2010 Agreement, we guarantee the manufacture and supply of our proprietary PEGylation materials (Polymer Materials) to Amgen in an existing manufacturing suite to be used exclusively for the manufacture of Polymer Materials for Amgen in our manufacturing facility in Huntsville, Alabama. This supply arrangement is on a non-exclusive basis (other than the use of the manufacturing suite and certain equipment) whereby we are free to manufacture and supply the Polymer Materials to any other third party and Amgen is free to procure the Polymer Materials from any other third party. Under the terms of the 2010 Agreement, we received a $50.0 million upfront payment in return for guaranteeing supply of certain quantities of Polymer Materials to Amgen and the Additional Rights
19
described below, and Amgen will pay manufacturing fees calculated based on fixed and variable components applicable to the Polymer Materials ordered by Amgen and delivered by us. Amgen has no minimum purchase commitments. If quantities of the Polymer Materials ordered by Amgen exceed specified quantities (with each specified quantity representing a small portion of the quantity that we historically supplied to Amgen), significant additional payments become payable to us in return for guaranteeing supply of additional quantities of the Polymer Materials.
The term of the 2010 Agreement runs through October 29, 2020. In the event we become subject to a bankruptcy or insolvency proceeding, we cease to own or control the manufacturing facility in Huntsville, Alabama, we fail to manufacture and supply the Polymer Materials or certain other events occur, Amgen or its designated third party will have the right to elect, among certain other options, to take title to the dedicated equipment and access the manufacturing facility to operate the manufacturing suite solely for the purpose of manufacturing the Polymer Materials (Additional Rights). Amgen may terminate the 2010 Agreement for convenience or due to an uncured material default by us. Either party may terminate the 2010 Agreement in the event of insolvency or bankruptcy of the other party.
PEGASYS®, Agreement with F. Hoffmann-La Roche Ltd
In February 1997, we entered into a license, manufacturing and supply agreement with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (Roche), under which we granted Roche a worldwide, exclusive license to use certain intellectual property related to our PEGylation materials to manufacture and commercialize a certain class of products, of which PEGASYS® is the only product currently commercialized. PEGASYS® is approved in the U.S., EU and other countries for the treatment of Hepatitis C and is designed to help the patient’s immune system fight the Hepatitis C virus. As a result of Roche exercising a license extension option in December 2009, beginning in 2010 Roche has the right to manufacture all of its requirements for our proprietary PEGylation materials for PEGASYS® and we supplied raw materials or performed additional manufacturing, if any, only on a back-up basis. In connection with Roche’s exercise of the license extension option in December 2009, we received a payment of $31.0 million. In 2013, we delivered additional quantities of PEGylation materials used by Roche to produce PEGASYS® and MIRCERA® for total consideration of approximately $18.6 million. The agreement expires on the expiration of our last relevant patent containing a valid claim. As of December 31, 2015, we no longer had any continuing manufacturing or supply obligations under this PEGASYS® agreement.
Somavert®, Agreement with Pfizer, Inc.
In January 2000, we entered into a license, manufacturing and supply agreement with Sensus Drug Development Corporation (subsequently acquired by Pharmacia Corp. in 2001 and then acquired by Pfizer, Inc. in 2003), for the PEGylation of Somavert® (pegvisomant), a human growth hormone receptor antagonist for the treatment of acromegaly. We currently manufacture our proprietary PEGylation reagent for Pfizer, Inc. on a price per gram basis.
PEG-Intron®, Agreement with Merck (through its acquisition of Schering-Plough Corporation)
In February 2000, we entered into a manufacturing and supply agreement with Schering-Plough Corporation (Schering) for the manufacture and supply of our proprietary PEGylation materials to be used by Schering in production of a PEGylated recombinant human interferon-alpha (PEG-Intron). PEG-Intron is a treatment for patients with Hepatitis C. Schering was acquired by, and became a wholly-owned subsidiary of, Merck & Co., Inc. We manufactured our proprietary PEGylation materials for Schering on a price per gram basis. In December 2010, the parties amended the manufacturing and supply agreement to provide for a transition plan to an alternative manufacturer and extension of the term through December 31, 2017. The amended agreement provided for a one-time payment and milestone payments as well as increased pricing for any future manufacturing performed by us. As of December 31, 2017, we no longer have any continuing manufacturing or supply obligations under this agreement.
Macugen®, Agreement with Valeant Pharmaceuticals International, Inc.
In 2002, we entered into a license, manufacturing and supply agreement with Eyetech, Inc. (subsequently acquired by Valeant Pharmaceuticals International, Inc. or Valeant), pursuant to which we license certain intellectual property related to our proprietary PEGylation technology for the development and commercialization of Macugen®, a PEGylated anti-vascular endothelial growth factor aptamer currently approved in the U.S. and EU for age-related macular degeneration. We currently manufacture our proprietary PEGylation materials for Valeant on a price per gram basis. Under the terms of the agreement, we will receive royalties on net product sales in any particular country for the longer of ten years from the date of the first commercial sale of the product in that country or the duration of patent coverage. Our agreement with Valeant expires upon the expiration of our last relevant patent containing a valid claim. In addition, Valeant may terminate the agreement if marketing authorization is withdrawn or marketing is no longer feasible due to certain circumstances, and either party may terminate for cause if certain conditions are met.
20
CIMZIA®, Agreement with UCB Pharma
In December 2000, we entered into a license, manufacturing and supply agreement covering our proprietary PEGylation materials for use in CIMZIA® (certolizumab pegol) with Celltech Chiroscience Ltd., which was acquired by UCB Pharma (UCB) in 2004. Under the terms of the agreement, UCB is responsible for all clinical development, regulatory, and commercialization expenses. We also manufacture and supply UCB with our proprietary PEGylation reagent used in the manufacture of CIMZIA® on a fixed price per gram. We were also entitled to receive royalties on net sales of the CIMZIA® product for the longer of ten years from the first commercial sale of the product anywhere in the world or the expiration of patent rights in a particular country. In February 2012, we sold our rights to receive royalties on future worldwide net sales of CIMZIA® effective as of January 1, 2012 until the agreement with UCB is terminated or expires. This sale is further discussed in Note 7 of Item 8, Financial Statements and Supplementary Data. Our agreement with UCB Pharma expires upon the expiration of all of UCB’s royalty obligations, provided that the agreement can be extended for successive two year renewal periods upon mutual agreement of the parties. In addition, UCB may terminate the agreement should it cease the development and marketing of CIMZIA® and either party may terminate for cause under certain conditions.
MIRCERA® (C.E.R.A.) (Continuous Erythropoietin Receptor Activator), Agreement with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc.
In December 2000, we entered into a license, manufacturing and supply agreement with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (Roche), which was amended and restated in its entirety in December 2005. Pursuant to the agreement, we license our intellectual property related to our proprietary PEGylation materials for the manufacture and commercialization of Roche’s MIRCERA® product. MIRCERA® is a novel continuous erythropoietin receptor activator indicated for the treatment of anemia associated with chronic kidney disease in patients on dialysis and patients not on dialysis. As of the end of 2006, we were no longer required to manufacture and supply our proprietary PEGylation materials for MIRCERA® under our original agreement. In February 2012, we entered into a toll-manufacturing agreement with Roche under which we manufactured our proprietary PEGylation material for MIRCERA®. Roche entered into the toll-manufacturing agreement with the objective of establishing us as a secondary back-up source on a non-exclusive basis through December 31, 2016. Under the terms of this agreement, Roche paid us an up-front payment of $5.0 million plus a total of $22.0 million in performance-based milestone payments upon our achievement of certain manufacturing readiness, validation and production milestones, including the delivery of specified quantities of PEGylation materials, all of which were successfully completed by the end of January 2013. In 2013, we delivered additional quantities of PEGylation materials used by Roche to produce PEGASYS® and MIRCERA® for total consideration of approximately $18.6 million. We were also entitled to receive royalties on net sales of the MIRCERA® product. In February 2012, we sold all of our future rights to receive royalties on future worldwide net sales of MIRCERA® effective as of January 1, 2012. This sale is further discussed in Note 7 of Item 8, Financial Statements and Supplementary Data. As of December 31, 2016, we no longer had any continuing manufacturing or supply obligations under this MIRCERA® agreement.
Government Regulation
Product Development and Approval Process
The research and development, clinical testing, manufacture and marketing of products using our technologies are subject to regulation by the FDA and by comparable regulatory agencies in other countries. These national agencies and other federal, state and local entities regulate, among other things, research and development activities and the testing (in vitro, in animals, and in human clinical trials), manufacture, labeling, storage, recordkeeping, approval, marketing, advertising and promotion of our products.
The approval process required by the FDA before a product using any of our technologies may be marketed in the U.S. depends on whether the chemical composition of the product has previously been approved for use in other dosage forms. If the product is a new chemical entity that has not been previously approved, the process includes the following:
|
|
• |
extensive preclinical laboratory and animal testing; |
|
|
• |
submission of an IND prior to commencing clinical trials; |
|
|
• |
adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for the intended indication; |
|
|
• |
extensive pharmaceutical development for the characterization of the chemistry, manufacturing process and controls for the active ingredient and drug product; and |
|
|
• |
submission to the FDA of an NDA for approval of a drug, a Biological License Application (BLA) for approval of a biological product or a Premarket Approval Application (PMA) or Premarket Notification 510(k) for a medical device product (a 510(k)). |
21
If the active chemical ingredient has been previously approved by the FDA, the approval process is similar, except that certain preclinical tests, including those relating to systemic toxicity normally required for the IND and NDA or BLA, and clinical trials, may not be necessary if the company has a right of reference to existing preclinical or clinical data under section 505(j) of the Federal Food, Drug, and Cosmetic Act (FDCA) or is eligible for approval under Section 505(b)(2) of the FDCA or the biosimilars provisions of the Public Health Services Act.
Preclinical tests include laboratory evaluation of product chemistry and animal studies to assess the safety and efficacy of the product and its chosen formulation. Preclinical safety tests must be conducted by laboratories that comply with FDA good laboratory practices (GLP) regulations. The results of the preclinical tests for drugs, biological products and combination products subject to the primary jurisdiction of the FDA’s Center for Drug Evaluation and Research (CDER) or Center for Biologics Evaluation and Research (CBER) are submitted to the FDA as part of the IND and are reviewed by the FDA before clinical trials can begin. Clinical trials may begin 30 days after receipt of the IND by the FDA, unless the FDA raises objections or requires clarification within that period. Clinical trials involve the administration of the drug to healthy volunteers or patients under the supervision of a qualified, identified medical investigator according to a protocol submitted in the IND for FDA review. Drug products to be used in clinical trials must be manufactured according to current good manufacturing practices (cGMP). Clinical trials are conducted in accordance with protocols that detail the objectives of the study and the parameters to be used to monitor participant safety and product efficacy as well as other criteria to be evaluated in the study. Each protocol is submitted to the FDA in the IND.
Apart from the IND process described above, each clinical study must be reviewed by an independent Institutional Review Board (IRB) and the IRB must be kept current with respect to the status of the clinical study. The IRB considers, among other things, ethical factors, the potential risks to subjects participating in the trial and the possible liability to the institution where the trial is conducted. The IRB also reviews and approves the informed consent form to be signed by the trial participants and any significant changes in the clinical study.
Clinical trials are typically conducted in three sequential phases. Phase 1 involves the initial introduction of the drug into healthy human subjects (in most cases) and the product generally is tested for tolerability, pharmacokinetics, absorption, metabolism and excretion. Phase 2 involves studies in a limited patient population to:
|
|
• |
determine the preliminary efficacy of the product for specific targeted indications; |
|
|
• |
determine dosage and regimen of administration; and |
|
|
• |
identify possible adverse effects and safety risks. |
If Phase 2 trials demonstrate that a product appears to be effective and to have an acceptable safety profile, Phase 3 trials are undertaken to evaluate the further clinical efficacy and safety of the drug and formulation within an expanded patient population at geographically dispersed clinical study sites and in large enough trials to provide statistical proof of efficacy and tolerability. The FDA, the clinical trial sponsor, the investigators or the IRB may suspend clinical trials at any time if any one of them believes that study participants are being subjected to an unacceptable health risk. In some cases, the FDA and the drug sponsor may determine that Phase 2 trials are not needed prior to entering Phase 3 trials.
Following a series of formal meetings and communications between the drug sponsor and the regulatory agencies, the results of product development, preclinical studies and clinical studies are submitted to the FDA as an NDA or BLA for approval of the marketing and commercial shipment of the drug product. The FDA may deny approval if applicable regulatory criteria are not satisfied or may require additional clinical or pharmaceutical testing or requirements. Even if such data are submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy all of the criteria for approval. Additionally, the approved labeling may narrowly limit the conditions of use of the product, including the intended uses, or impose warnings, precautions or contraindications which could significantly limit the potential market for the product. Further, as a condition of approval, the FDA may impose post-market surveillance, or Phase 4, studies or risk evaluation and mitigation strategies. Product approvals, once obtained, may be withdrawn if compliance with regulatory standards is not maintained or if safety concerns arise after the product reaches the market. The FDA may require additional post-marketing clinical testing and pharmacovigilance programs to monitor the effect of drug products that have been commercialized and has the power to prevent or limit future marketing of the product based on the results of such programs. After approval, there are ongoing reporting obligations concerning adverse reactions associated with the product, including expedited reports for serious and unexpected adverse events.
Each manufacturing establishment producing the active pharmaceutical ingredient and finished drug product for the U.S. market must be registered with the FDA and typically is inspected by the FDA prior to NDA or BLA approval of a drug product manufactured by such establishment. Such inspections are also held periodically after the commercialization. Establishments handling controlled substances must also be licensed by the U.S. Drug Enforcement Administration. Manufacturing establishments of U.S. marketed products are subject to inspections by the FDA for compliance with cGMP and other U.S. regulatory requirements.
22
They are also subject to U.S. federal, state, and local regulations regarding workplace safety, environmental protection and hazardous and controlled substance controls, among others.
For product candidates currently under development utilizing pulmonary technology, the pulmonary inhaler devices are considered to be part of a drug and device combination for deep lung delivery of each specific molecule. The FDA will make a determination as to the most appropriate center and division within the agency that will assume primary responsibility for the review of the applicable applications, which would consist of an IND and an NDA or BLA where CDER or CBER are determined to have primary jurisdiction or an investigational device exemption application and PMA or 510(k) where the Center for Devices and Radiological Health (CDRH) is determined to have primary jurisdiction. In the case of our product candidates, CDER in consultation with CDRH could be involved in the review. The assessment of jurisdiction within the FDA is based upon the primary mode of action of the drug or the location of the specific expertise in one of the centers.
Where CDRH is determined to have primary jurisdiction over a product, 510(k) clearance or PMA approval is required. Medical devices are classified into one of three classes — Class I, Class II, or Class III — depending on the degree of risk associated with each medical device and the extent of control needed to ensure safety and effectiveness. Devices deemed to pose lower risks are placed in either Class I or II, which requires the manufacturer to submit to the FDA a Premarket Notification requesting permission to commercially distribute the device. This process is known as 510(k) clearance. Some low risk devices are exempted from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device are placed in Class III, requiring PMA approval.
In situations where our partners are responsible for clinical and regulatory approval procedures, we may still participate in this process by submitting to the FDA a drug master file developed and maintained by us which contains data concerning the manufacturing processes for the inhaler device, polymer conjugation materials or drug. For our proprietary products, we prepare and submit an IND and are responsible for additional clinical and regulatory procedures for product candidates being developed under an IND. The clinical and manufacturing, development and regulatory review and approval process generally takes a number of years and requires the expenditure of substantial resources. Our ability to manufacture and market products, whether developed by us or under collaboration agreements, ultimately depends upon the completion of satisfactory clinical trials and success in obtaining marketing approvals from the FDA and equivalent foreign health authorities.
Sales of our products outside the U.S. are subject to local regulatory requirements governing clinical trials and marketing approval for drugs. Such requirements vary widely from country to country.
In the U.S., under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the U.S. The company that obtains the first FDA approval for a designated orphan drug for a rare disease receives marketing exclusivity for use of that drug for the designated condition for a period of seven years. In addition, the Orphan Drug Act provides for protocol assistance, tax credits, research grants, and exclusions from user fees for sponsors of orphan products. Once a product receives orphan drug exclusivity, a second product that is considered to be the same drug for the same indication generally may be approved during the exclusivity period only if the second product is shown to be “clinically superior” to the original orphan drug in that it is more effective, safer or otherwise makes a “major contribution to patient care” or the holder of exclusive approval cannot assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Similar incentives also are available for orphan drugs in the EU.
In the U.S., the FDA may grant Fast Track or Breakthrough Therapy designation to a product candidate, which allows the FDA to expedite the review of new drugs that are intended for serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs. Important features of Fast Track or Breakthrough Therapy designation include a potentially reduced clinical program and close, early communication between the FDA and the sponsor company to improve the efficiency of product development.
Coverage, Reimbursement, and Pricing
Sales of any products for which we may obtain regulatory approval depends, in part, on the coverage and reimbursement status of those products. In the U.S., sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payors. Third-party payors include government programs such as Medicare, Medicaid, TRICARE and the Veterans Administration, as well as managed care providers, private health insurers and other organizations.
The process for determining whether a payor will provide coverage for a product is typically separate from the process for setting the reimbursement rate that the payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list or formulary which might not include all of the FDA-approved products for a particular indication. Third-party payors
23
may also refuse to include a particular branded drug on their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available. Further, private payors often follow the coverage and payment policies established by certain government programs, such as Medicare and Medicaid, which require manufacturers to comply with certain rebate, price reporting, and other obligations. For example, the Medicaid Drug Rebate Program, which is part of the Medicaid program (a program for financially needy patients, among others), requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services under which the manufacturer agrees to report certain prices to the government and pay rebates to state Medicaid programs on outpatient drugs furnished to Medicaid patients, as a condition for receiving federal reimbursement for the manufacturer’s outpatient drugs furnished to Medicaid patients. Further, in order for a pharmaceutical product to receive federal reimbursement under Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the Public Health Service’s 340B drug pricing program.
Third-party payors are increasingly challenging the prices charged for medical products and services, and examining the medical necessity and cost- effectiveness of medical products and services, in addition to their safety and efficacy. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the price of therapeutics have been a focus in this effort. The U.S. government and state legislatures have shown significant interest in implementing cost-containment programs, including price controls and restrictions on reimbursement, among other controls. Adoption of price controls or other cost-containment measures could limit coverage for or the amounts that federal and state governments or private payors will pay for health care products and services, which could also result in reduced demand for our drug candidates or additional pricing pressures and affect our ultimate profitability. If third-party payors do not consider a product to be cost-effective compared to other available therapies, they may not cover an approved product or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Regulations
If we obtain regulatory approval of our products, we may be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales and marketing programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration (a term interpreted broadly to include anything of value, including, for example, gifts, discounts, and credits), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order, or recommendation of, an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
|
|
• |
federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to Medicare, Medicaid, or other third-party payors that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money owed to the federal government; |
|
|
• |
provisions of the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes, referred to as the “HIPAA All-Payor Fraud Prohibition,” that prohibit knowingly and willfully executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
|
|
• |
federal transparency laws, including the federal Physician Payment Sunshine Act, which require manufacturers of certain drugs and biologics to track and disclose payments and other transfers of value they make to U.S. physicians and teaching hospitals as well as physician ownership and investment interests in the manufacturer, and that such information is subsequently made publicly available in a searchable format on a CMS website; |
24
|
|
• |
provisions of HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; and |
|
|
• |
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state transparency reporting and compliance laws; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and which may not have the same effect, thus complicating compliance efforts. |
The Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act (collectively, the “Affordable Care Act”), enacted in 2010, expanded the reach of the fraud and abuse laws by, among other things, amending the intent requirement of the federal Anti-Kickback Statute and the applicable criminal fraud statutes contained within 42 U.S.C. § 1320a-7b. Pursuant to the Affordable Care Act, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act or the civil monetary penalties statute. Many states have adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The federal False Claims Act prohibits anyone from, among other things, knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services that are false or fraudulent. Although we would not submit claims directly to payors, manufacturers can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. In addition, our future activities relating to the reporting of wholesaler or estimated retail prices for our products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state, and third-party reimbursement for our products, and the sale and marketing of our products, are subject to scrutiny under this law. For example, pharmaceutical companies have been prosecuted under the federal False Claims Act in connection with their alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal health care programs for the product. Penalties for a False Claims Act violation include three times the actual damages sustained by the government, plus mandatory civil penalties of between $10,781 and $21,916 for each separate false claim, the potential for exclusion from participation in federal healthcare programs, and, although the federal False Claims Act is a civil statute, conduct that results in a False Claims Act violation may also implicate various federal criminal statutes. In addition, private individuals have the ability to bring actions under the federal False Claims Act and certain states have enacted laws modeled after the federal False Claims Act.
Legislative and Regulatory Landscape
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing, marketing, coverage and reimbursement of products regulated by the FDA or other government agencies. In addition to new legislation, FDA and healthcare fraud and abuse and coverage and reimbursement regulations and policies are often revised or interpreted by the agency in ways that may significantly affect our business and our products. Further, the 2016 Presidential and Congressional elections and subsequent developments have caused the future state of many core aspects of the current health care marketplace to be uncertain, as the new Presidential Administration and Congress have repeatedly expressed a desire to repeal all or portions of the Affordable Care Act. While specific changes and their timing are not yet apparent, there may be significant changes to the healthcare environment in the future that could have an adverse effect on anticipated revenues from therapeutic candidates that we may successfully develop and for which we may obtain regulatory approval. Furthermore, federal agencies, Congress, state legislatures, and the privacy sector have shown significant interest in implementing cost containment programs to limit the growth of health care costs, including price controls, restrictions on reimbursement and other fundamental changes to the healthcare delivery system. Any proposed or actual changes could limit coverage for or the amounts that federal and state governments will pay for health care products and services, which could also result in reduced demand for our products or additional pricing pressures and affect our ultimate profitability.
Patents and Proprietary Rights
We own more than 250 U.S. and 800 foreign patents and a number of pending patent applications that cover various aspects of our technologies. We have filed patent applications, and plan to file additional patent applications, covering various aspects of our advanced polymer conjugate technologies and our proprietary product candidates. More specifically, our patents and patent applications cover polymer architecture, drug conjugates, formulations, methods of making polymers and polymer conjugates,
25
methods of administering polymer conjugates, and methods of manufacturing polymers and polymer conjugates. Our patent portfolio contains patents and patent applications that encompass our advanced polymer conjugate technology platforms, some of which we acquired in our acquisition of Shearwater Corporation in June 2001. Our patent strategy is to file patent applications on innovations and improvements to cover a significant majority of the major pharmaceutical markets in the world. Generally, patents have a term of twenty years from the earliest priority date (assuming all maintenance fees are paid). In some instances, patent terms can be increased or decreased, depending on the laws and regulations of the country or jurisdiction that issued the patent.
In January 2002, we entered into a Cross-License and Option Agreement with Enzon, pursuant to which we and Enzon provided certain licenses to selected portions of each party’s PEGylation patent portfolio.
On December 31, 2008, we completed the sale of certain assets related to our pulmonary business, associated technology and intellectual property to Novartis Pharma AG and Novartis Pharmaceuticals Corporation (together referred to as Novartis) for a purchase price of $115.0 million in cash (Novartis Pulmonary Asset Sale). In connection with the Novartis Pulmonary Asset Sale, as of December 31, 2008, we entered into an exclusive license agreement with Novartis Pharma AG. Pursuant to the exclusive license agreement, Novartis Pharma AG grants back to us an exclusive, irrevocable, perpetual, royalty-free and worldwide license under certain specific patent rights and other related intellectual property rights acquired by Novartis from us in the Novartis Pulmonary Asset Sale, as well as certain improvements or modifications thereto that are made by Novartis. Certain of such patent rights and other related intellectual property rights relate to our development program for inhaled vancomycin or are necessary for us to satisfy certain continuing contractual obligations to third parties, including in connection with development, manufacture, sale, and commercialization activities related to BAY41-6551 partnered with Bayer Healthcare LLC.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to, or disclose, our trade secrets. Please refer to Item 1A, Risk Factors, including but not limited to “We rely on trade secret protection and other unpatented proprietary rights for important proprietary technologies, and any loss of such rights could harm our business, results of operations and financial condition.” In certain situations in which we work with drugs covered by one or more patents, our ability to develop and commercialize our technologies may be affected by limitations in our access to these proprietary drugs. Even if we believe we are free to work with a proprietary drug, we cannot guarantee that we will not be accused of, or determined to be, infringing a third party’s rights and be prohibited from working with the drug or found liable for damages. Any such restriction on access or liability for damages would have a material adverse effect on our business, results of operations and financial condition.
The patent positions of pharmaceutical and biotechnology companies, such as ours, are uncertain and involve complex legal and factual issues. There can be no assurance that patents that have issued will be held valid and enforceable in a court of law. Even for patents that are held valid and enforceable, the legal process associated with obtaining such a judgment is time consuming and costly. Additionally, issued patents can be subject to opposition or other proceedings that can result in the revocation of the patent or maintenance of the patent in amended form (and potentially in a form that renders the patent without commercially relevant or broad coverage). Further, our competitors may be able to circumvent and otherwise design around our patents. Even if a patent is issued and enforceable, because development and commercialization of pharmaceutical products can be subject to substantial delays, patents may expire early and provide only a short period of protection, if any, following the commercialization of products encompassed by our patent. We may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office, which could result in a loss of the patent and/or substantial cost to us. Please refer to Item 1A, Risk Factors, including without limitation, “If any of our pending patent applications do not issue, or are deemed invalid following issuance, we may lose valuable intellectual property protection.”
U.S. and foreign patent rights and other proprietary rights exist that are owned by third parties and relate to pharmaceutical compositions and reagents, medical devices and equipment and methods for preparation, packaging and delivery of pharmaceutical compositions. We cannot predict with any certainty which, if any, of these rights will be considered relevant to our technology by authorities in the various jurisdictions where such rights exist, nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. We could incur substantial costs in defending ourselves and our partners against any such claims. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from third parties. There can be no assurance that we can obtain a license to any technology that we determine we need on reasonable terms, if at all, or that we could develop or otherwise obtain alternative technology. The failure to obtain licenses if needed may have a material adverse effect on our business, results of operations and financial condition. Please refer to Item 1A, Risk Factors, including without limitation, “We may not be able to obtain intellectual property licenses related to the development of our drug candidates on a commercially reasonable basis, if at all.”
26
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee shall be our property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Customer Concentrations
Our revenue is derived from our collaboration agreements with partners, under which we may receive contract research payments, milestone payments based on clinical progress, regulatory progress or net sales achievements, royalties or product sales revenue. The revenue generated under the Lilly Agreement and the license, manufacturing and supply agreement with UCB represented 42% and 12% of our revenue, respectively, for the year ended December 31, 2017. No other collaboration partner accounted for more than 10% of our total revenue during the year ended December 31, 2017.
Backlog
Pursuant to our collaboration agreements, we manufacture and supply our proprietary polymer conjugation materials. Inventory is produced and sales are made pursuant to customer purchase orders for delivery. The volume of our proprietary polymer conjugation materials actually ordered by our customers, as well as shipment schedules, are subject to frequent revisions that reflect changes in both the customers’ needs and our manufacturing capacity. In our partnered programs where we provide contract research services, those services are typically provided under a work plan that is subject to frequent revisions that change based on the development needs and status of the program. The backlog at a particular time is affected by a number of factors, including scheduled date of manufacture and delivery and development program status. In light of industry practice and our own experience, we do not believe that backlog as of any particular date is indicative of future results.
Competition
Competition in the pharmaceutical and biotechnology industry is intense and characterized by aggressive research and development and rapidly-evolving science, technology, and standards of medical care throughout the world. We frequently compete with pharmaceutical companies and other institutions with greater financial, research and development, marketing and sales, manufacturing and managerial capabilities. We face competition from these companies not just in product development but also in areas such as recruiting employees, acquiring technologies that might enhance our ability to commercialize products, establishing relationships with certain research and academic institutions, enrolling patients in clinical trials and seeking program partnerships and collaborations with larger pharmaceutical companies.
Science and Technology Competition
We believe that our proprietary and partnered products will compete with others in the market on the basis of one or more of the following parameters: efficacy, safety, ease of use and cost. We face intense science and technology competition from a multitude of technologies seeking to enhance the efficacy, safety and ease of use of approved drugs and new drug molecule candidates. A number of the drug candidates in our pipeline have direct and indirect competition from large pharmaceutical companies and biopharmaceutical companies. With our advanced polymer conjugate technologies, we believe we have competitive advantages relating to factors such as efficacy, safety, ease of use and cost for certain applications and molecules. We constantly monitor scientific and medical developments in order to improve our current technologies, seek licensing opportunities where appropriate, and determine the best applications for our technology platforms.
In the fields of advanced polymer conjugate technologies, our competitors include Biogen Idec Inc., Savient Pharmaceuticals, Inc., Dr. Reddy’s Laboratories, Ltd., Mountain View Pharmaceuticals, Inc., SunBio Corporation, NOF Corporation, and Novo Nordisk A/S (assets formerly held by Neose Technologies, Inc.). Several other chemical, biotechnology and pharmaceutical companies may also be developing advanced polymer conjugate technology or technologies intended to deliver similar scientific and medical benefits. Some of these companies license intellectual property or PEGylation materials to other companies, while others apply the technology to create their own drug candidates.
27
Product and Program Specific Competition
NKTR-214 (immunostimulatory CD122-biased cytokine)
There are numerous companies engaged in developing immunotherapies to be used alone, or in combination, to treat a wide range of oncology indications targeting both solid and liquid tumors. In particular, we expect to compete with therapies with tumor infiltrating lymphocytes, or TILS, chimeric antigen receptor-expressing T cells, or CAR-T, cytokine-based therapies, and checkpoint inhibitors. Potential competitors in the TIL and CAR-T space include Kite Pharma/NCI, Adaptimmune LLC, Celgene Corporation, Juno Therapeutics, and Novartis, Alkermes, Altor, and Armo in the cytokine-based therapies space, and Tesaro, Macrogenics, Merck, BMS, and Roche in the checkpoint inhibitor space.
NKTR-358
There are a number of competitors in various stages of clinical development that are working on programs which are designed to correct the underlying immune system imbalance in the body due to autoimmune disease. In particular, we expect to compete with therapies that could be cytokine-based therapies (Symbiotix, LLC and Tizona Therapeutics), regulatory T cell therapies (Targazyme, Inc., Juno Therapeutics and Tract Therapeutics, Inc.), or IL-2-based and toleragen-based therapies (Celgene Corporation, Amgen Inc., Tolera Therapeutics, Inc., and Caladrius BioSciences, Inc.).
NKTR-181 (mu-opioid analgesic molecule for chronic pain)
There are numerous companies developing pain therapies designed to have less abuse potential primarily through formulation technologies and techniques applied to existing pain therapies. Potential competitors include Acura Pharmaceuticals, Inc., Cara Therapeutics, Inc., Collegium Pharmaceutical, Inc., Egalet Ltd, Elite Pharmaceuticals, Inc., Endo Health Solutions Inc., KemPharm, Inc., Pfizer, Inc., Purdue Pharma L.P., and Teva Pharmaceutical Industries Ltd.
MOVANTIK® (previously referred to as naloxegol and NKTR-118) (orally-available peripheral opioid antagonist)
There are no other once-daily oral drugs that act specifically to block or reverse the action of opioids on receptors in the gastrointestinal tract which are approved specifically for the treatment of opioid-induced constipation (OIC) or opioid bowel dysfunction (OBD) in patients with chronic, non-cancer pain. The only approved oral treatment for opioid-induced constipation in adults with chronic, non-cancer pain is a twice daily oral therapy called AMITIZA® (lubiprostone), which acts by specifically activating CIC-2 chloride channels in the gastrointestinal tract to increase secretions. AMITIZA® is marketed by Sucampo Pharmaceuticals and Takeda. There is also a subcutaneous treatment and an oral treatment known as RELISTOR® which is marketed by Valeant Pharmaceuticals, Ltd (formerly Salix) under a license from Progenics Pharmaceuticals, Inc. In 2014, RELISTOR® Subjectaneous Injection was approved by the FDA for adult patients with chronic non-cancer pain. On July 22, 2016, Relistor (methylnaltrexone bromide) oral tablets for the treatment of OCI in adult patients with chronic non-cancer pain was approved by FDA. Other therapies used to treat OIC and OBD include over-the-counter laxatives and stool softeners, such as docusate sodium, senna, and milk of magnesia. These therapies do not address the underlying cause of constipation as a result of opioid use and are generally viewed as ineffective or only partially effective to treat the symptoms of OIC and OBD.
There are a number of companies developing potential products which are in various stages of clinical development and are being evaluated for the treatment of OIC and OBD in different patient populations. Potential competitors include Merck & Co., Inc., GlaxoSmithKline plc, Ironwood Pharmaceuticals, Inc. in collaboration with Actavis plc, Purdue Pharma L.P. in collaboration with Shionogi & Co., Ltd., Mundipharma Int. Limited, Theravance, Inc., Develco Pharma, Sucampo Pharmaceuticals, Inc., and Takeda Pharmaceutical Company Limited.
ADYNOVATE® (previously referred to as BAX 855, PEGylated rFVIII)
On June 6, 2014, the FDA approved Biogen Idec ’s ELOCTATETM [Antihemophilic Factor (Recombinant), Fc Fusion Protein] for the control and prevention of bleeding episodes, perioperative (surgical) management and routine prophylaxis in adults and children with Hemophilia A. ELOCTATETM is intended to be an extended half-life Factor VIII therapy with prolonged circulation in the body with the potential to extend the interval between prophylactic infusions. Prior to its 2014 approval, the fusion protein in ELOCTATETM was not used outside of the clinical trial setting for Hemophilia A patients. There are other long-acting Factor VIII programs in late-stage development for Hemophilia A patients. Bayer Healthcare and Novo Nordisk have ongoing Phase 3 clinical development programs for longer acting Factor VIII proteins based on PEGylation technology approaches. These programs, if developed successfully and approved by health authorities, would be competitors in the longer acting Factor VIII market.
28
ONZEALDTM (next-generation, long acting topoisomerase I inhibitor)
There are a number of chemotherapies and cancer therapies approved today and in various stages of clinical development for advanced breast cancer. These therapies are only partially effective in treating advanced or metastatic breast cancer and none of them have a specific indication in either the U.S. or Europe for treatment of patients with advanced breast cancer and co-existing brain metastases. These therapies include but are not limited to: Abraxane® (paclitaxel protein-bound particles for injectable suspension (albumin bound)), Afinitor® (everolimus), Ellence® (epirubicin), Gemzar® (gemcitabine), Halaven® (eribulin), Herceptin® (trastuzumab), Ixempra® (ixabepilone), Navelbine® (vinolrebine), Xeloda® (capecitabine) and Taxotere® (docetaxel). Major pharmaceutical or biotechnology companies with approved drugs or drugs in development for these cancers include Bristol-Myers Squibb Company, Eisai, Inc., Roche Holding Group (including its Genentech subsidiary), GlaxoSmithKline plc, Pfizer, Inc., Eli Lilly & Co., Sanofi Aventis S.A., and others.
Research and Development
Our total research and development expenditures can be disaggregated into the following significant types of expenses (in millions):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Third party and direct materials costs |
|
$ |
125.4 |
|
|
$ |
98.2 |
|
|
$ |
89.3 |
|
|
Personnel, overhead and other costs |
|
|
113.5 |
|
|
|
84.6 |
|
|
|
77.8 |
|
|
Stock-based compensation and depreciation |
|
|
29.6 |
|
|
|
21.0 |
|
|
|
15.7 |
|
|
Research and development expense |
|
$ |
268.5 |
|
|
$ |
203.8 |
|
|
$ |
182.8 |
|
Manufacturing and Supply
We have a manufacturing facility located in Huntsville, Alabama that is capable of manufacturing our proprietary PEGylation materials for active pharmaceutical ingredients (APIs). The facility is also used to produce APIs to support the early phases of clinical development of our proprietary drug candidates. The facility and associated equipment are designed and operated to be consistent with all applicable laws and regulations.
As we do not maintain the capability to manufacture APIs (including biologics) nor finished drug products for all of our development programs, we primarily utilize contract manufacturers to manufacture active pharmaceutical ingredients and finished drug product for us. We source drug starting materials for our manufacturing activities from one or more suppliers. For the drug starting materials necessary for our proprietary drug candidate development, we have agreements for the supply of such drug components with drug manufacturers or suppliers that we believe have sufficient capacity to meet our demands. However, from time to time, we source critical raw materials and services from one or a limited number of suppliers and there is a risk that if such supply or services were interrupted, it would materially harm our business. In addition, we typically order raw materials and services on a purchase order basis and do not enter into long-term dedicated capacity or minimum supply arrangements. We also utilize the services of contract manufacturers to manufacture APIs required for later phases of clinical development and eventual commercialization for us under all applicable laws and regulations.
Environment
As a manufacturer of PEG reagents for the U.S. market, we are subject to inspections by the FDA for compliance with cGMP and other U.S. regulatory requirements, including U.S. federal, state and local regulations regarding environmental protection and hazardous and controlled substance controls, among others. Environmental laws and regulations are complex, change frequently and have tended to become more stringent over time. We have incurred, and may continue to incur, significant expenditures to ensure we are in compliance with these laws and regulations. We would be subject to significant penalties for failure to comply with these laws and regulations.
Employees and Consultants
As of December 31, 2017 we had 509 employees, of which 396 employees were engaged in research and development, commercial operations and quality activities and 113 employees were engaged in general administration and business development. Of the 509 employees, 432 were located in the U.S. and 77 were located in India. We have a number of employees who hold advanced degrees, such as Ph.D. None of our employees are covered by a collective bargaining agreement, and we have experienced no work stoppages. We believe that we maintain good relations with our employees.
29
To complement our own expert professional staff, we utilize specialists in regulatory affairs, pharmacovigilance, process engineering, manufacturing, quality assurance, clinical development and business development. These individuals include scientific advisors as well as independent consultants.
Available Information
Our website address is http://www.nektar.com. The information in, or that can be accessed through, our website is not part of this annual report on Form 10-K. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports are available, free of charge, on or through our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities Exchange Commission (SEC). The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding our filings at www.sec.gov.
EXECUTIVE OFFICERS OF THE REGISTRANT
The following table sets forth the names, ages and positions of our executive officers as of February 22, 2018:
|
Name |
|
Age |
|
|
Position |
|
|
Howard W. Robin |
|
|
65 |
|
|
Director, President and Chief Executive Officer |
|
Stephen K. Doberstein, Ph.D. |
|
|
59 |
|
|
Senior Vice President, Research & Development and Chief Research & Development Officer |
|
Maninder Hora, Ph.D |
|
|
64 |
|
|
Senior Vice President, Pharmaceutical Development and Chief Technical Operations Officer |
|
Gil M. Labrucherie, J.D. |
|
|
46 |
|
|
Senior Vice President and Chief Financial Officer |
|
John Nicholson |
|
|
66 |
|
|
Senior Vice President and Chief Operating Officer |
|
Jillian B. Thomsen |
|
|
52 |
|
|
Senior Vice President, Finance and Chief Accounting Officer |
Howard W. Robin has served as our President and Chief Executive Officer since January 2007 and has served as a member of our board of directors since February 2007. Mr. Robin served as Chief Executive Officer, President and a director of Sirna Therapeutics, Inc., a biotechnology company, from July 2001 to November 2006 and from January 2001 to June 2001, served as their Chief Operating Officer, President and as a director. From 1991 to 2001, Mr. Robin was Corporate Vice President and General Manager at Berlex Laboratories, Inc. (Berlex), a pharmaceutical products company that is a subsidiary of Schering, AG, and from 1987 to 1991 he served as Vice President of Finance and Business Development and Chief Financial Officer of Berlex. From 1984 to 1987, Mr. Robin was Director of Business Planning and Development at Berlex. He was a Senior Associate with Arthur Andersen & Co. prior to joining Berlex. Mr. Robin serves as a director of the Biotechnology Industry Organization, the world’s largest biotechnology industry trade organization, and also serves as a director of BayBio, a non-profit trade association serving the Northern California life sciences community. He received his B.S. in Accounting and Finance from Fairleigh Dickinson University in 1974.
Stephen K. Doberstein, Ph.D. has served as our Senior Vice President, Research & Development and Chief Research & Development Officer since November 2017. Dr. Doberstein served as Senior Vice President and Chief Scientific Officer from January 2010 to November 2017 when he was promoted to Senior Vice President, Research & Development and Chief Research & Development Officer. From October 2008 through December 2009, Dr. Doberstein served as Vice President, Research at Xoma (US) LLC, a publicly traded clinical stage biotechnology company. From July 2004 until August 2008, he served as Vice President, Research at privately held Five Prime Therapeutics, Inc., a clinical stage biotechnology company. From September 2001 until July 2004, Dr. Doberstein was Vice President, Research at privately held Xencor, Inc., a clinical stage biotechnology company. From 1997 to 2000, he held various pharmaceutical research positions at Exelixis, Inc. (Exelixis), a publicly traded clinical stage biotechnology company. Prior to working at Exelixis, Dr. Doberstein was a Howard Hughes Postdoctoral Fellow and a Muscular Dystrophy Association Senior Postdoctoral Fellow at the University of California, Berkeley. Dr. Doberstein received his Ph.D. in Biochemistry, Cell and Molecular Biology from the Johns Hopkins University School of Medicine and received a B.S. in Chemical Engineering from the University of Delaware.
Maninder Hora, Ph.D. has served as our Senior Vice President, Pharmaceutical Development and Chief Technical Operations Officer since August 2010. From July 2006 to July 2010, he held various executive positions most recently as Vice President, Product and Quality Operations at Facet Biotech Corporation (now Abbvie Biotherapeutics), a clinical stage biotechnology company, which was acquired in 2010 by Abbvie Biotherapeutics (formerly Abbot). From 1986 to 2006, Dr. Hora held positions of increasing responsibility with Chiron Corporation (acquired in 2005 by Novartis), a pharmaceutical company, serving most recently at Chiron as Vice President of Process and Product Development. Dr. Hora has also held positions at Wyeth Pharmaceuticals and GlaxoSmithKline plc prior to joining Chiron. Dr. Hora served as a key member of various teams that successfully registered ten drugs
30
or vaccines in the U.S. and Europe during his professional career. Dr. Hora completed his Ph.D. in Bioengineering from the Indian Institute of Technology, Delhi, India, and was a Fulbright Scholar at the University of Washington, and received his B.S. in chemistry from the University of Jabalpur.
Gil M. Labrucherie has served as our Senior Vice President and Chief Financial Officer since June 2016. Mr. Labrucherie served as our Vice President, Corporate Legal from October 2005 through April 2007 and served as our Senior Vice President, General Counsel and Secretary from April 2007 through June 2016 when he was promoted to Senior Vice President and Chief Financial Officer. From October 2000 to September 2005, Mr. Labrucherie was Vice President of Corporate Development at E2open. While at E2open, Mr. Labrucherie was responsible for global corporate alliances and merger and acquisitions. Prior to E2open, he was the Senior Director of Corporate Development at AltaVista Company, an Internet search company, where he was responsible for strategic partnerships and mergers and acquisitions. Mr. Labrucherie began his career as an associate in the corporate practice of the law firm of Wilson Sonsini Goodrich & Rosati, P.C. Mr. Labrucherie received his J.D. from the Berkeley Law School and a B.A. from the University of California Davis.
John Nicholson has served as our Senior Vice President and Chief Operating Officer since June 2016. Mr. Nicholson joined the Company as Senior Vice President of Corporate Development and Business Operations in October 2007 and was appointed Senior Vice President and Chief Financial Officer in December 2007 and served as our Chief Financial Officer until June 2016 when he was promoted to Senior Vice President and Chief Operating Officer. Before joining Nektar, Mr. Nicholson spent 18 years in various executive roles at Schering Berlin, Inc., the U.S. management holding company of Bayer Schering Pharma AG, a pharmaceutical company. From 1997 to September 2007, Mr. Nicholson served as Schering Berlin Inc.’s Vice President of Corporate Development and Treasurer. From 2001 to September 2007, he concurrently served as President of Schering Berlin Insurance Co., and from February 2007 through September 2007, he also concurrently served as President of Bayer Pharma Chemicals and Schering Berlin Capital Corp. Mr. Nicholson holds a B.B.A. from the University of Toledo.
Jillian B. Thomsen has served as our Senior Vice President, Finance and Chief Accounting Officer since February 2010. From March 2006 through March 2008, Ms. Thomsen served as our Vice President Finance and Corporate Controller and from April 2008 through January 2010 she served as our Vice President Finance and Chief Accounting Officer. Before joining Nektar, Ms. Thomsen was Vice President Finance and Deputy Corporate Controller of Calpine Corporation from September 2002 to February 2006. Ms. Thomsen is a certified public accountant and previously was a senior manager at Arthur Andersen LLP, where she worked from 1990 to 2002, and specialized in audits of multinational consumer products, life sciences, manufacturing and energy companies. Ms. Thomsen holds a Masters of Accountancy from the University of Denver and a B.A. in Business Economics from Colorado College.
31
We are providing the following cautionary discussion of risk factors, uncertainties and assumptions that we believe are relevant to our business. These are factors that, individually or in the aggregate, we think could cause our actual results to differ materially from expected and historical results and our forward-looking statements. We note these factors for investors as permitted by Section 21E of the Exchange Act and Section 27A of the Securities Act. You should understand that it is not possible to predict or identify all such factors. Consequently, you should not consider this section to be a complete discussion of all potential risks or uncertainties that may substantially impact our business. Moreover, we operate in a competitive and rapidly changing environment. New factors emerge from time to time and it is not possible to predict the impact of all of these factors on our business, financial condition or results of operations.
Risks Related to Our Business
We are highly dependent on the success of NKTR-214, our lead I-O candidate. We are executing a broad development program for NKTR-214 and clinical and regulatory outcomes for NKTR-214, if not successful, will significantly harm our business.
Our future success is highly dependent on our ability to successfully develop, obtain regulatory approval for, and commercialize NKTR-214. In general, most early stage investigatory drugs, including oncology drug candidates such as NKTR-214, do not become approved drugs. Accordingly, there is a very meaningful risk that NKTR-214 will not succeed in one or more clinical trials sufficient to support one or more regulatory approvals. To date, clinical outcomes from NKTR-214 have had a significant impact on our market valuation, financial position, and business prospects and we expect this to continue in future periods. If one or more clinical studies of NKTR-214 are not successful, it would materially harm our market valuation, prospects, financial condition and results of operations. For example, upon the effective date of the strategic collaboration agreement with Bristol-Myers Squibb, we would be entitled to up to $1.43 billion in development milestones that are based upon clinical and regulatory successes from the NKTR-214 development program. Failures in NKTR-214 studies could jeopardize such milestone payments.
Drug development is a long and inherently uncertain process with a high risk of failure at every stage of development.
We have a number of proprietary drug candidates and partnered drug candidates in research and development ranging from the early discovery research phase through preclinical testing and clinical trials. Preclinical testing and clinical studies are long, expensive, difficult to design and implement and highly uncertain as to outcome. It will take us, or our collaborative partners, many years to conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparator drug or required prior therapy, clinical outcomes, or our and our partners’ financial constraints.
Drug development is a highly uncertain scientific and medical endeavor, and failure can unexpectedly occur at any stage of preclinical and clinical development. Typically, there is a high rate of attrition for drug candidates in preclinical and clinical trials due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The risk of failure increases for our drug candidates that are based on new technologies, such as the application of our advanced polymer conjugate technology to ONZEALDTM, NKTR-181, NKTR-214, NKTR-358, NKTR-262, NKTR-255, and other drug candidates currently in discovery research or preclinical development. The failure of one or more of our drug candidates could have a material adverse effect on our business, financial condition and results of operations.
The risk of clinical failure for any drug candidate remains high prior to regulatory approval.
A number of companies have suffered significant unforeseen failures in clinical studies due to factors such as inconclusive efficacy or safety, even after achieving preclinical proof-of-concept or positive results from earlier clinical studies that were satisfactory both to them and to reviewing regulatory authorities. Clinical study outcomes remain very unpredictable and it is possible that one or more of our clinical studies could fail at any time due to efficacy, safety or other important clinical findings or regulatory requirements. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA, an independent Institutional Review Board (IRB), an independent ethics committee, or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects. Similarly, an IRB or ethics committee may suspend a clinical trial at a particular trial site. If one or more of our drug candidates fail in clinical studies, it could have a material adverse effect on our business, financial condition and results of operations.
32
Our results of operations and financial condition depend significantly on the ability of our collaboration partners to successfully develop and market drugs and they may fail to do so.
Under our collaboration agreements with various pharmaceutical or biotechnology companies (other than the BMS Collaboration Agreement), our collaboration partner is generally solely responsible for:
|
|
• |
designing and conducting large scale clinical studies; |
|
|
• |
preparing and filing documents necessary to obtain government approvals to sell a given drug candidate; and/or |
|
|
• |
marketing and selling the drugs when and if they are approved. |
Our reliance on collaboration partners poses a number of significant risks to our business, including risks that:
|
|
• |
we have very little control over the timing and level of resources that our collaboration partners dedicate to commercial marketing efforts such as the amount of investment in sales and marketing personnel, general marketing campaigns, direct-to-consumer advertising, product sampling, pricing agreements and rebate strategies with government and private payers, manufacturing and supply of drug product, and other marketing and selling activities that need to be undertaken and well executed for a drug to have the potential to achieve commercial success; |
|
|
• |
collaboration partners with commercial rights may choose to devote fewer resources to the marketing of our partnered drugs than they devote to their own drugs or other drugs that they have in-licensed; |
|
|
• |
we have very little control over the timing and amount of resources our partners devote to development programs in one or more major markets; |
|
|
• |
disagreements with partners could lead to delays in, or termination of, the research, development or commercialization of product candidates or to litigation or arbitration proceedings; |
|
|
• |
disputes may arise or escalate in the future with respect to the ownership of rights to technology or intellectual property developed with partners; |
|
|
• |
we do not have the ability to unilaterally terminate agreements (or partners may have extension or renewal rights) that we believe are not on commercially reasonable terms or consistent with our current business strategy; |
|
|
• |
partners may be unable to pay us as expected; and |
|
|
• |
partners may terminate their agreements with us unilaterally for any or no reason, in some cases with the payment of a termination fee penalty and in other cases with no termination fee penalty. |
Given these risks, the success of our current and future collaboration partnerships is highly unpredictable and can have a substantial negative or positive impact on our business. If the approved drugs fail to achieve commercial success or the drugs in development fail to have positive late stage clinical outcomes sufficient to support regulatory approval in major markets, it could significantly impair our access to capital necessary to fund our research and development efforts for our proprietary drug candidates. If we are unable to obtain sufficient capital resources to advance our drug candidate pipeline, it would negatively impact the value of our business, results of operations and financial condition.
We are a party to numerous collaboration agreements and other significant agreements which contain complex commercial terms that could result in disputes, litigation or indemnification liability that could adversely affect our business, results of operations and financial condition.
We currently derive, and expect to derive in the foreseeable future, all of our revenue from collaboration agreements with biotechnology and pharmaceutical companies. These collaboration agreements contain complex commercial terms, including:
|
|
• |
clinical development and commercialization obligations that are based on certain commercial reasonableness performance standards that can often be difficult to enforce if disputes arise as to adequacy of our partner’s performance; |
|
|
• |
research and development performance and reimbursement obligations for our personnel and other resources allocated to partnered drug candidate development programs; |
|
|
• |
clinical and commercial manufacturing agreements, some of which are priced on an actual cost basis for products supplied by us to our partners with complicated cost allocation formulas and methodologies; |
|
|
• |
intellectual property ownership allocation between us and our partners for improvements and new inventions developed during the course of the collaboration; |
33
|
|
• |
royalties on drug sales based on a number of complex variables, including net sales calculations, geography, scope of patent claim coverage, patent life, generic competitors, bundled pricing and other factors; and |
|
|
• |
indemnity obligations for intellectual property infringement, product liability and certain other claims. |
We are a party to numerous significant collaboration agreements and other strategic transaction agreements (e.g., financings and asset divestitures) that contain complex representations and warranties, covenants and indemnification obligations. If we are found to have materially breached such agreements, it could subject us to substantial liabilities and harm our financial condition.
From time to time, we are involved in litigation matters involving the interpretation and application of complex terms and conditions of our agreements. For example, in February 2015, we filed a claim against Allergan and MAP seeking monetary damages related to a dispute over the economic sharing provisions of our collaboration agreement with MAP. On December 12, 2017, Nektar, MAP and Allergan entered into a Settlement Agreement and Release, for which MAP paid us $15.0 million in December 2017. On August 14, 2015, Enzon, Inc. filed a breach of contract claim for alleged unpaid licensing fees (the “Enzon Litigation”). On June 26, 2017, we entered into a Second Amendment to our Cross-License and Option Agreement (“Cross-License Agreement”) with Enzon in which we agreed to pay Enzon a sum of $7.0 million to satisfy all past and future obligations of royalty payments pursuant to the Cross-License Agreement and to have the Enzon Litigation dismissed. The Enzon Litigation was dismissed with prejudice on June 30, 2017. In 2013, we settled a breach of contract litigation matter with the Research Foundation of the State University of New York (SUNY) pursuant to which we paid an aggregate of $12.0 million. One or more disputes may arise or escalate in the future regarding our collaboration agreements, transaction documents, or third-party license agreements that may ultimately result in costly litigation and unfavorable interpretation of contract terms, which would have a material adverse effect on our business, financial condition and results of operations.
If we or our partners do not obtain regulatory approval for our drug candidates on a timely basis, or at all, or if the terms of any approval impose significant restrictions or limitations on use, our business, results of operations and financial condition will be negatively affected.
We or our partners may not obtain regulatory approval for drug candidates on a timely basis, or at all, or the terms of any approval (which in some countries includes pricing approval) may impose significant restrictions or limitations on use. Drug candidates must undergo rigorous animal and human testing and an extensive review process for safety and efficacy by the FDA and equivalent foreign regulatory authorities. The time required for obtaining regulatory decisions is uncertain and difficult to predict. The FDA and other U.S. and foreign regulatory authorities have substantial discretion, at any phase of development, to terminate clinical studies, require additional clinical development or other testing, delay or withhold registration and marketing approval and mandate product withdrawals, including recalls. For example, while data from certain pre-specified subgroups in our BEACON study for etirinotecan pegol (NKTR-102) in 2015 was positive, the study did not achieve statistical significance for its primary endpoint and the FDA and European Medicines Agency rarely approve drugs on the basis of studies that do not achieve statistical significance on the primary endpoint. Further, while the results from the Phase 3 study of NKTR-181 were positive, NKTR-181 has Fast Track designation and we intend to file an NDA following our pre-NDA meetings with FDA, the regulatory pathway for NKTR-181 remains subject to substantial uncertainty including the amount of data required to support an approval of NKTR-181. In particular, regulations concerning and controlling the access to opioid-based pharmaceuticals are strict and there is no guarantee which scheduling category will apply to NKTR-181 if regulatory approval is achieved. Further, regulatory authorities have the discretion to analyze data using their own methodologies that may differ from those used by us or our partners, which could lead such authorities to arrive at different conclusions regarding the safety or efficacy of a drug candidate. In addition, undesirable side effects caused by our drug candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restricted label or the delay or denial of regulatory approval by regulatory authorities. For example, AstraZeneca will be conducting a post-marketing, observational epidemiological study comparing MOVANTIK® to other treatments of OIC in patients with chronic, non-cancer pain and the results of this study could at some point in the future negatively impact the labeling, regulatory status, and commercial potential of MOVANTIK®.
Even if we or our partners receive regulatory approval of a product, the approval may limit the indicated uses for which the drug may be marketed. Our partnered drugs that have obtained regulatory approval, and the manufacturing processes for these products, are subject to continued review and periodic inspections by the FDA and other regulatory authorities. Discovery from such review and inspection of previously unknown problems may result in restrictions on marketed products or on us, including withdrawal or recall of such products from the market, suspension of related manufacturing operations or a more restricted label. The failure to obtain timely regulatory approval of product candidates, any product marketing limitations or a product withdrawal would negatively impact our business, results of operations and financial condition.
34
We have substantial future capital requirements and there is a risk we may not have access to sufficient capital to meet our current business plan. If we do not receive substantial milestone or royalty payments from our existing collaboration agreements, execute new high value collaborations or other arrangements, or are unable to raise additional capital in one or more financing transactions, we would be unable to continue our current level of investment in research and development.
As of December 31, 2017, we had cash and investments in marketable securities valued at approximately $353.2 million and had debt of $250.0 million in principal of senior secured notes. In addition, as described above and in Note 14 to our Consolidated Financial Statements, in February 2018, we entered into a collaboration agreement with BMS under which BMS agreed to pay us a non-refundable upfront cash payment of $1.0 billion and a share purchase agreement under which BMS agreed to purchase $850.0 million of shares of our common stock. The closings under the two agreements, subject to satisfaction of certain closing conditions, are expected to occur during the second quarter of 2018. While we believe that our cash position will be sufficient to meet our liquidity requirements through at least the next 12 months, our future capital requirements will depend upon numerous unpredictable factors, including:
|
|
• |
the cost, timing and outcomes of clinical studies and regulatory reviews of our proprietary drug candidates that we have licensed to our collaboration partners —important examples include NKTR-214 in collaboration with BMS, Cipro DPI licensed to Bayer and NKTR-358 licensed to Lilly; |
|
|
• |
the commercial launch and sales levels of products marketed by our collaboration partners for which we are entitled to royalties and sales milestones—importantly, the level of success in marketing and selling MOVANTIK® by AstraZeneca in the U.S. and ADYNOVATE® by Baxalta globally, as well as MOVENTIG® (the naloxegol brand name in the EU) by Kirin in the EU; |
|
|
• |
if and when we receive potential milestone payments and royalties from our existing collaborations if the drug candidates subject to those collaborations achieve clinical, regulatory or commercial success; |
|
|
• |
the progress, timing, cost and results of our clinical development programs; |
|
|
• |
the success, progress, timing and costs of our efforts to implement new collaborations, licenses and other transactions that increase our current net cash, such as the sale of additional royalty interests held by us, term loan or other debt arrangements, and the issuance of securities; |
|
|
• |
the number of patients, enrollment criteria, primary and secondary endpoints, and the number of clinical studies required by the regulatory authorities in order to consider for approval our drug candidates and those of our collaboration partners; |
|
|
• |
our general and administrative expenses, capital expenditures and other uses of cash; and |
|
|
• |
disputes concerning patents, proprietary rights, or license and collaboration agreements that negatively impact our receipt of milestone payments or royalties or require us to make significant payments arising from licenses, settlements, adverse judgments or ongoing royalties. |
A significant multi-year capital commitment is required to advance our drug candidates through the various stages of research and development in order to generate sufficient data to enable high value collaboration partnerships with significant upfront payments or to successfully achieve regulatory approval. In the event we do not enter into any new collaboration partnerships with significant upfront payments and we choose to continue our later stage research and development programs, we may need to pursue financing alternatives, including dilutive equity-based financings, such as an offering of convertible debt or common stock, which would dilute the percentage ownership of our current common stockholders and could significantly lower the market value of our common stock. If sufficient capital is not available to us or is not available on commercially reasonable terms, it could require us to delay or reduce one or more of our research and development programs. If we are unable to sufficiently advance our research and development programs, it could substantially impair the value of such programs and result in a material adverse effect on our business, financial condition and results of operations.
While we have conducted numerous experiments using laboratory and home-based chemistry techniques that have not been able to convert NKTR-181 into a rapid-acting and more abusable opioid, there is a risk that a technique could be discovered in the future to convert NKTR-181 into a rapid-acting and more abusable opioid, which would significantly diminish the value of this drug candidate.
An important objective of our NKTR-181 drug development program is to create a unique opioid molecule that does not rapidly enter a patient’s central nervous system and therefore has the potential to be less susceptible to abuse than alternative opioid therapies. To date, we have conducted numerous experiments using laboratory and home-based chemistry techniques that have been unable to convert NKTR-181 into a rapidly-acting, more abusable form of opioid. In the future, an alternative chemistry technique, process or method of administration, or combination thereof, may be discovered to enable the conversion of NKTR-181 into a more abusable opioid, which could significantly and negatively impact the commercial potential or diminish the value of NKTR-181.
35
The commercial potential of a drug candidate in development is difficult to predict. If the market size for a new drug is significantly smaller than we anticipate, it could significantly and negatively impact our revenue, results of operations and financial condition.
It is very difficult to estimate the commercial potential of product candidates due to important factors such as safety and efficacy compared to other available treatments, including potential generic drug alternatives with similar efficacy profiles, changing standards of care, third party payer reimbursement standards, patient and physician preferences, drug scheduling status, the availability of competitive alternatives that may emerge either during the long drug development process or after commercial introduction, and the availability of generic versions of our product candidates following approval by regulatory authorities based on the expiration of regulatory exclusivity or our inability to prevent generic versions from coming to market by asserting our patents. In particular, regulations concerning and controlling the access to opioid-based pharmaceuticals are strict and there is no guarantee which scheduling category will apply to NKTR-181 if regulatory approval is achieved. If due to one or more of these risks the market potential for a drug candidate is lower than we anticipated, it could significantly and negatively impact the commercial terms of any collaboration partnership potential for such drug candidate or, if we have already entered into a collaboration for such drug candidate, the revenue potential from royalty and milestone payments could be significantly diminished and this would negatively impact our business, financial condition and results of operations. We also depend on our relationships with other companies for sales and marketing performance and the commercialization of product candidates. Poor performance by these companies, or disputes with these companies, could negatively impact our revenue and financial condition.
If we are unable to establish and maintain collaboration partnerships on attractive commercial terms, our business, results of operations and financial condition could suffer.
We intend to continue to seek partnerships with pharmaceutical and biotechnology partners to fund a portion of our research and development capital requirements. The timing of new collaboration partnerships is difficult to predict due to availability of clinical data, the outcomes from our clinical studies, the number of potential partners that need to complete due diligence and approval processes, the definitive agreement negotiation process and numerous other unpredictable factors that can delay, impede or prevent significant transactions. If we are unable to find suitable partners or negotiate collaboration arrangements with favorable commercial terms with respect to our existing and future drug candidates or the licensing of our intellectual property, or if any arrangements we negotiate, or have negotiated, are terminated, it could have a material adverse effect on our business, financial condition and results of operations.
Preliminary and interim data from our clinical studies that we announce or publish from time to time are subject to audit and verification procedures that could result in material changes in the final data and may change as more patient data become available.
From time to time, we publish preliminary or interim data from our clinical studies. Preliminary data remain subject to audit confirmation and verification procedures that may result in the final data being materially different from the preliminary data we previously published. Interim data are also subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. As a result, preliminary and interim data should be viewed with caution until the final data are available. Material adverse changes in the final data could significantly harm our business prospects.
Delays in clinical studies are common and have many causes, and any significant delay in clinical studies being conducted by us or our partners could result in delay in regulatory approvals and jeopardize the ability to proceed to commercialization.
We or our partners may experience delays in clinical trials of drug candidates. We have ongoing trials evaluating NKTR-214 including a trial evaluating NKTR-214 as a potential combination treatment with BMS’s Opdivo® (nivolumab) as well as other ongoing and planned combination trials. We also have an ongoing Phase 1 dose-escalation study for NKTR-358 under our collaboration with Lilly, including an on-going dose-finding trial of NKTR-358 to evaluate single-ascending doses of NKTR-358 in healthy subjects, and a multiple-ascending dose trial expected to be initiated in the first half of 2018 to evaluate NKTR-358 in patients with systemic lupus erythematosus. We also have ongoing trials with our partners for the following: Halozyme has trials in Pancreatic, Non-Small Cell Lung Cancer and other multiple tumor types in Phase 1, 2, and 3 development. These and other clinical studies may not begin on time, enroll a sufficient number of patients or be completed on schedule, if at all. Clinical trials for any of our product candidates could be delayed for a variety of reasons, including:
|
|
• |
delays in obtaining regulatory authorization to commence a clinical study; |
|
|
• |
delays in reaching agreement with applicable regulatory authorities on a clinical study design; |
|
|
• |
imposition of a clinical hold by the FDA or other health authorities, which may occur at any time including after any inspection of clinical trial operations or trial sites; |
36
|
|
• |
suspension or termination of a clinical study by us, our partners, the FDA or foreign regulatory authorities due to adverse side effects of a drug on subjects in the trial; |
|
|
• |
delays in recruiting suitable patients to participate in a trial; |
|
|
• |
delays in having patients complete participation in a trial or return for post-treatment follow-up; |
|
|
• |
clinical sites dropping out of a trial to the detriment of enrollment rates; |
|
|
• |
delays in manufacturing and delivery of sufficient supply of clinical trial materials; and |
|
|
• |
changes in regulatory authorities policies or guidance applicable to our drug candidates. |
If the initiation or completion of any of the planned clinical studies for our drug candidates is delayed for any of the above or other reasons, the regulatory approval process would be delayed and the ability to commercialize and commence sales of these drug candidates could be materially harmed, which could have a material adverse effect on our business, financial condition and results of operations. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
We may not be able to obtain intellectual property licenses related to the development of our drug candidates on a commercially reasonable basis, if at all.
Numerous pending and issued U.S. and foreign patent rights and other proprietary rights owned by third parties relate to pharmaceutical compositions, methods of preparation and manufacturing, and methods of use and administration. We cannot predict with any certainty which, if any, patent references will be considered relevant to our or our collaboration partners’ technology or drug candidates by authorities in the various jurisdictions where such rights exist, nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. In certain cases, we have existing licenses or cross-licenses with third parties; however, the scope and adequacy of these licenses is very uncertain and can change substantially during long development and commercialization cycles for biotechnology and pharmaceutical products. There can be no assurance that we can obtain a license to any technology that we determine we need on reasonable terms, if at all, or that we could develop or otherwise obtain alternate technology. If we are required to enter into a license with a third party, our potential economic benefit for the products subject to the license will be diminished. If a license is not available on commercially reasonable terms or at all, we may be prevented from developing and commercializing the drug, which could significantly harm our business, results of operations, and financial condition.
If any of our pending patent applications do not issue, or are deemed invalid following issuance, we may lose valuable intellectual property protection.
The patent positions of pharmaceutical and biotechnology companies, such as ours, are uncertain and involve complex legal and factual issues. We own more than 250 U.S. and 800 foreign patents and have a number of pending patent applications that cover various aspects of our technologies. There can be no assurance that patents that have issued will be held valid and enforceable in a court of law. Even for patents that are held valid and enforceable, the legal process associated with obtaining such a judgment is time consuming and costly. Additionally, issued patents can be subject to opposition or other proceedings that can result in the revocation of the patent or maintenance of the patent in amended form (and potentially in a form that renders the patent without commercially relevant and/or broad coverage). Further, our competitors may be able to circumvent and otherwise design around our patents. Even if a patent is issued and enforceable, because development and commercialization of pharmaceutical products can be subject to substantial delays, patents may expire early and provide only a short period of protection, if any, following the commercialization of products encompassed by our patents. We may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office, which could result in a loss of the patent and/or substantial cost to us.
We have filed patent applications, and plan to file additional patent applications, covering various aspects of our PEGylation and advanced polymer conjugate technologies and our proprietary product candidates. There can be no assurance that the patent applications for which we apply would actually issue as patents, or do so with commercially relevant and/or broad coverage. The coverage claimed in a patent application can be significantly reduced before the patent is issued. The scope of our claim coverage can be critical to our ability to enter into licensing transactions with third parties and our right to receive royalties from our collaboration partnerships. Since publication of discoveries in scientific or patent literature often lags behind the date of such discoveries, we cannot be certain that we were the first inventor of inventions covered by our patents or patent applications. In addition, there is no guarantee that we will be the first to file a patent application directed to an invention.
An adverse outcome in any judicial proceeding involving intellectual property, including patents, could subject us to significant liabilities to third parties, require disputed rights to be licensed from or to third parties or require us to cease using the technology in
37
dispute. In those instances where we seek an intellectual property license from another, we may not be able to obtain the license on a commercially reasonable basis, if at all, thereby raising concerns on our ability to freely commercialize our technologies or products.
We are involved in legal proceedings and may incur substantial litigation costs and liabilities that will adversely affect our business, financial condition and results of operations.
From time to time, third parties have asserted, and may in the future assert, that we or our partners infringe their proprietary rights, such as patents and trade secrets, or have otherwise breached our obligations to them. A third party often bases its assertions on a claim that its patents cover our technology platform or drug candidates or that we have misappropriated its confidential or proprietary information. Similar assertions of infringement could be based on future patents that may issue to third parties. In certain of our agreements with our partners, we are obligated to indemnify and hold harmless our collaboration partners from intellectual property infringement, product liability and certain other claims, which could cause us to incur substantial costs and liability if we are called upon to defend ourselves and our partners against any claims. If a third party obtains injunctive or other equitable relief against us or our partners, they could effectively prevent us, or our partners, from developing or commercializing, or deriving revenue from, certain drugs or drug candidates in the U.S. and abroad. Costs associated with litigation, substantial damage claims, indemnification claims or royalties paid for licenses from third parties could have a material adverse effect on our business, financial condition and results of operations.
We are involved in legal proceedings where we or other third parties are enforcing or seeking intellectual property rights, invalidating or limiting patent rights that have already been allowed or issued, or otherwise asserting proprietary rights through one or more potential legal remedies. For example, we are currently involved in a German litigation proceeding whereby Bayer is seeking co-ownership rights in certain of our patent filings pending at the European Patent Office covering, among other things, PEGylated Factor VIII which we have exclusively licensed to Baxalta. The subject matter of our patent filings in this proceeding relates to Bayer’s PEGylated recombinant Factor VIII compound, BAY 94-9027. We believe that Bayer’s claim to an ownership interest in these patent filings is without merit and are vigorously defending sole and exclusive ownership rights to this intellectual property. In addition, Bayer has filed claims in the U.S. against Baxalta and Nektar. In one U.S. proceeding, Bayer alleges ADYNOVATE® infringes a Bayer patent. In another U.S. proceeding, Bayer is seeking a declaratory judgement that BAY 94-9027 does not infringe specified Nektar patents or in the alternative that the specified patents are invalid. As part of its intellectual property litigation strategy relating to PEGylated Factor VIII products, Nektar has also filed claims against Bayer. We are also regularly involved in opposition proceedings at the European Patent Office where third parties seek to invalidate or limit the scope of our allowed European patent applications covering (among other things) our drugs and platform technologies. The cost to us in initiating or defending any litigation or other proceeding, even if resolved in our favor, could be substantial, and litigation would divert our management’s attention. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could delay our research and development efforts or result in financial implications either in terms of seeking license arrangements or payment of damages or royalties.
Our manufacturing operations and those of our contract manufacturers are subject to laws and other governmental regulatory requirements, which, if not met, would have a material adverse effect on our business, results of operations and financial condition.
We and our contract manufacturers are required in certain cases to maintain compliance with current good manufacturing practices (cGMP), including cGMP guidelines applicable to active pharmaceutical ingredients, and with laws and regulations governing manufacture and distribution of controlled substances, and are subject to inspections by the FDA, the Drug Enforcement Administration or comparable agencies in other jurisdictions administering such requirements. We anticipate periodic regulatory inspections of our drug manufacturing facilities and the manufacturing facilities of our contract manufacturers for compliance with applicable regulatory requirements. Any failure to follow and document our or our contract manufacturers’ adherence to such cGMP and other laws and governmental regulations or satisfy other manufacturing and product release regulatory requirements may disrupt our ability to meet our manufacturing obligations to our customers, lead to significant delays in the availability of products for commercial use or clinical study, result in the termination or hold on a clinical study or delay or prevent filing or approval of marketing applications for our products. Failure to comply with applicable laws and regulations may also result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delays, suspension or withdrawal of approvals, license revocation, seizures, administrative detention, or recalls of products, operating restrictions and criminal prosecutions, any of which could harm our business. Regulatory inspections could result in costly manufacturing changes or facility or capital equipment upgrades to satisfy the FDA that our manufacturing and quality control procedures are in substantial compliance with cGMP. Manufacturing delays, for us or our contract manufacturers, pending resolution of regulatory deficiencies or suspensions could have a material adverse effect on our business, results of operations and financial condition.
38
If we or current or future collaborators or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions and civil or criminal penalties.
Although we do not currently have any products on the market, once we begin commercializing our drug candidates, we will be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal and state governments of the jurisdictions in which we conduct our business. Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any drug candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our therapeutic candidates for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration (a term interpreted broadly to include anything of value, including, for example, gifts, discounts, and credits), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order, or recommendation of, an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
|
|
• |
federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to Medicare, Medicaid, or other third-party payors that are false or fraudulent, or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money owed to the federal government; |
|
|
• |
provisions of the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes, referred to as the “HIPAA All-Payor Fraud Prohibition,” that prohibit knowingly and willfully executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
|
|
• |
federal transparency laws, including the federal Physician Payment Sunshine Act, which require manufacturers of certain drugs and biologics to track and disclose payments and other transfers of value they make to U.S. physicians and teaching hospitals as well as physician ownership and investment interests in the manufacturer, and that such information is subsequently made publicly available in a searchable format on a CMS website; |
|
|
• |
provisions of HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; and |
|
|
• |
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, state transparency reporting and compliance laws; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and which may not have the same effect, thus complicating compliance efforts. |
Ensuring that our future business arrangements with third-parties comply with applicable healthcare laws and regulations could involve substantial costs. If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, monetary damages, the curtailment or restructuring of our operations, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely affect financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
If we or our contract manufacturers are not able to manufacture drugs or drug substances in sufficient quantities that meet applicable quality standards, it could delay clinical studies, result in reduced sales or constitute a breach of our contractual obligations, any of which could significantly harm our business, financial condition and results of operations.
If we or our contract manufacturers are not able to manufacture and supply sufficient drug quantities meeting applicable quality standards required to support large clinical studies or commercial manufacturing in a timely manner, it could delay our or our collaboration partners’ clinical studies or result in a breach of our contractual obligations, which could in turn reduce the potential
39
commercial sales of our or our collaboration partners’ products. As a result, we could incur substantial costs and damages and any product sales or royalty revenue that we would otherwise be entitled to receive could be reduced, delayed or eliminated. In some cases, we rely on contract manufacturing organizations to manufacture and supply drug product for our clinical studies and those of our collaboration partners. Pharmaceutical manufacturing of drugs and devices involves significant risks and uncertainties related to the demonstration of adequate stability, sufficient purification of the drug substance and drug product, the identification and elimination of impurities, optimal formulations, process and analytical methods validations, device performance and challenges in controlling for all of these variables. We have faced and may in the future face significant difficulties, delays and unexpected expenses as we validate third party contract manufacturers required for drug and device supply to support our clinical studies and the clinical studies and products of our collaboration partners. Failure by us or our contract manufacturers to supply drug product or devices in sufficient quantities that meet all applicable quality requirements could result in supply shortages for our clinical studies or the clinical studies and commercial activities of our collaboration partners. Such failures could significantly and materially delay clinical trials and regulatory submissions or result in reduced sales, any of which could significantly harm our business prospects, results of operations and financial condition.
Building and validating large scale clinical or commercial-scale manufacturing facilities and processes, recruiting and training qualified personnel and obtaining necessary regulatory approvals is complex, expensive and time consuming. In the past, we have encountered challenges in scaling up manufacturing to meet the requirements of large scale clinical trials without making modifications to the drug formulation, which may cause significant delays in clinical development. Drug and device combination products are particularly complex, expensive and time-consuming to develop due to the number of variables involved in the final product design, including ease of patient and doctor use, maintenance of clinical efficacy, reliability and cost of manufacturing, regulatory approval requirements and standards and other important factors. There continues to be substantial and unpredictable risk and uncertainty related to manufacturing and supply until such time as the commercial supply chain is validated and proven.
We purchase some of the starting material for drugs and drug candidates from a single source or a limited number of suppliers, and the partial or complete loss of one of these suppliers could cause production delays, clinical trial delays, substantial loss of revenue and contract liability to third parties.
We often face very limited supply of a critical raw material that can only be obtained from a single, or a limited number of, suppliers, which could cause production delays, clinical trial delays, substantial lost revenue opportunities or contract liabilities to third parties. For example, there are only a limited number of qualified suppliers, and in some cases single source suppliers, for the raw materials included in our PEGylation and advanced polymer conjugate drug formulations. Any interruption in supply or failure to procure such raw materials on commercially feasible terms could harm our business by delaying our clinical trials, impeding commercialization of approved drugs or increasing our costs.
Our revenue is exclusively derived from our collaboration agreements, which can result in significant fluctuation in our revenue from period to period, and our past revenue is therefore not necessarily indicative of our future revenue.
Our revenue is exclusively derived from our collaboration agreements, from which we receive upfront fees, contract research payments, milestone and other contingent payments based on clinical progress, regulatory progress or net sales achievements, royalties and manufacturing revenue. Significant variations in the timing of receipt of cash payments and our recognition of revenue can result from significant payments based on the execution of new collaboration agreements, the timing of clinical outcomes, regulatory approval, commercial launch or the achievement of certain annual sales thresholds. The amount of our revenue derived from collaboration agreements in any given period will depend on a number of unpredictable factors, including our ability to find and maintain suitable collaboration partners, the timing of the negotiation and conclusion of collaboration agreements with such partners, whether and when we or our collaboration partners achieve clinical, regulatory and sales milestones, the timing of regulatory approvals in one or more major markets, reimbursement levels by private and government payers, and the market introduction of new drugs or generic versions of the approved drug, as well as other factors. The application of the Accounting Standards Codification (ASC) 606, Revenue Recognition - Revenue from Contracts with Customers, which will apply beginning in the first quarter of 2018, may have a material impact on revenue recognition under our collaboration agreements. Our past revenue generated from collaboration agreements is not necessarily indicative of our future revenue. If any of our existing or future collaboration partners fails to develop, obtain regulatory approval for, manufacture or ultimately commercialize any product candidate under our collaboration agreement, our business, financial condition, and results of operations could be materially and adversely affected.
40
If we are unable either to create sales, marketing and distribution capabilities or to enter into agreements with third parties to perform these functions, we will be unable to commercialize our product candidates successfully.
We currently have no sales, marketing or distribution capabilities. To commercialize any of our drugs that receive regulatory approval for commercialization, we must either develop internal sales, marketing and distribution capabilities, which would be expensive and time consuming, or enter into collaboration arrangements with third parties to perform these services. If we decide to market our products directly, we must commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and with supporting distribution, administration and compliance capabilities. Factors that may inhibit our efforts to commercialize our products directly or indirectly with our partners include:
|
|
• |
our inability to recruit and retain adequate numbers of effective sales and marketing personnel; |
|
|
• |
the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to use or prescribe our products; |
|
|
• |
the lack of complementary products or multiple product pricing arrangements may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
|
|
• |
unforeseen costs and expenses associated with creating and sustaining an independent sales and marketing organization. |
If we, or our partners through our collaborations, are not successful in recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty commercializing our products, which would adversely affect our business, results of operations and financial condition.
To the extent we rely on other pharmaceutical or biotechnology companies with established sales, marketing and distribution systems to market our products, we will need to establish and maintain partnership arrangements, and we may not be able to enter into these arrangements on acceptable terms or at all. To the extent that we enter into co-promotion or other arrangements, any revenue we receive will depend upon the efforts of third parties, which may not be successful and over which we have little or no control—important examples of this risk include MOVANTIK® partnered with AstraZeneca and ADYNOVATE® (previously referred to as BAX 855) partnered with Baxalta. In the event that we market our products without a partner, we would be required to build a sales and marketing organization and infrastructure, which would require a significant investment, and we may not be successful in building this organization and infrastructure in a timely or efficient manner.
We rely on trade secret protection and other unpatented proprietary rights for important proprietary technologies, and any loss of such rights could harm our business, results of operations and financial condition.
We rely on trade secret protection for our confidential and proprietary information. No assurance can be given that others will not independently develop substantially equivalent confidential and proprietary information or otherwise gain access to our trade secrets or disclose such technology, or that we can meaningfully protect our trade secrets. In addition, unpatented proprietary rights, including trade secrets and know-how, can be difficult to protect and may lose their value if they are independently developed by a third party or if their secrecy is lost. Any loss of trade secret protection or other unpatented proprietary rights could harm our business, results of operations and financial condition.
We expect to continue to incur substantial losses and negative cash flow from operations and may not achieve or sustain profitability in the future.
For the year ended December 31, 2017, we reported a net loss of $96.7 million. If and when we achieve profitability depends upon a number of factors, including the timing and recognition of milestone and other contingent payments and royalties received, the timing of revenue under our collaboration agreements, the amount of investments we make in our proprietary product candidates and the regulatory approval and market success of our product candidates. We may not be able to achieve and sustain profitability.
Other factors that will affect whether we achieve and sustain profitability include our ability, alone or together with our partners, to:
|
|
• |
develop drugs utilizing our technologies, either independently or in collaboration with other pharmaceutical or biotechnology companies; |
|
|
• |
effectively estimate and manage clinical development costs, particularly the cost of the clinical studies for NKTR-214, NKTR-358, NKTR-262, and NKTR-255; |
|
|
• |
receive necessary regulatory and marketing approvals; |
|
|
• |
maintain or expand manufacturing at necessary levels; |
41
|
|
• |
achieve market acceptance of our partnered products; |
|
|
• |
receive royalties on products that have been approved, marketed or submitted for marketing approval with regulatory authorities; and |
|
|
• |
maintain sufficient funds to finance our activities. |
If government and private insurance programs do not provide payment or reimbursement for our partnered products or proprietary products, those products will not be widely accepted, which would have a negative impact on our business, results of operations and financial condition.
In both domestic and foreign markets, sales of our partnered and proprietary products that have received regulatory approval will depend in part on market acceptance among physicians and patients, pricing approvals by government authorities and the availability of coverage and payment or reimbursement from third-party payers, such as government programs, including Medicare and Medicaid, managed care providers, private health insurers and other organizations. However, eligibility for coverage does not necessarily signify that a drug candidate will be adequately reimbursed in all cases or at a rate that covers costs related to research, development, manufacture, sale, and distribution. Third-party payers are increasingly challenging the price and cost effectiveness of medical products and services. Therefore, significant uncertainty exists as to the coverage and pricing approvals for, and the payment or reimbursement status of, newly approved healthcare products.
Moreover, legislation and regulations affecting the pricing of pharmaceuticals may change before regulatory agencies approve our proposed products for marketing and could further limit coverage or pricing approvals for, and reimbursement of, our products from government authorities and third-party payers. For example, Congress passed the Affordable Care Act in 2010 which enacted a number of reforms to expand access to health insurance while also reducing or constraining the growth of healthcare spending, enhancing remedies against fraud and abuse, adding new transparency requirements for healthcare industries, and imposing new taxes on fees on healthcare industry participants, among other policy reforms. Federal agencies, Congress and state legislatures have continued to show interest in implementing cost containment programs to limit the growth of health care costs, including price controls, restrictions on reimbursement and other fundamental changes to the healthcare delivery system. In addition, in recent years, Congress has enacted various laws seeking to reduce the federal debt level and contain healthcare expenditures, and the Medicare and other healthcare programs are frequently identified as potential targets for spending cuts. New government legislation or regulations related to pricing or other fundamental changes to the healthcare delivery system as well as a government or third-party payer decision not to approve pricing for, or provide adequate coverage or reimbursement of, our products hold the potential to severely limit market opportunities of such products.
We depend on third parties to conduct the clinical trials for our proprietary product candidates and any failure of those parties to fulfill their obligations could harm our development and commercialization plans.
We depend on independent clinical investigators, contract research organizations and other third-party service providers to conduct clinical trials for our proprietary product candidates. We rely heavily on these parties for the successful execution of our clinical trials. Though we are ultimately responsible for the results of their activities, many aspects of their activities are beyond our control. For example, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trials, but the independent clinical investigators may prioritize other projects over ours or communicate issues regarding our products to us in an untimely manner. Third parties may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or our stated protocols. The early termination of any of our clinical trial arrangements, the failure of third parties to comply with the regulations and requirements governing clinical trials or the failure of third parties to properly conduct our clinical trials could hinder or delay the development, approval and commercialization of our product candidates and would adversely affect our business, results of operations and financial condition.
Significant competition for our polymer conjugate chemistry technology platforms and our partnered and proprietary products and product candidates could make our technologies, products or product candidates obsolete or uncompetitive, which would negatively impact our business, results of operations and financial condition.
Our advanced polymer conjugate chemistry platforms and our partnered and proprietary products and product candidates compete with various pharmaceutical and biotechnology companies. Competitors of our polymer conjugate chemistry technologies include Biogen Inc., Savient Pharmaceuticals, Inc., Dr. Reddy’s Laboratories Ltd., SunBio Corporation, Mountain View Pharmaceuticals, Inc., Novo Nordisk A/S (formerly assets held by Neose Technologies, Inc.), and NOF Corporation. Several other chemical, biotechnology and pharmaceutical companies may also be developing polymer conjugation technologies or technologies that have similar impact on target drug molecules. Some of these companies license or provide the technology to other companies, while others are developing the technology for internal use.
42
There are many competitors for our proprietary product candidates currently in development. For MOVANTIK®, there are currently several alternative therapies used to address opioid-induced constipation (OIC) and opioid-induced bowel dysfunction (OBD), including Symproic® (naldemedine) from Shionogi and Purdue Pharma L.P., RELISTOR® Subcutaneous Injection (methylnaltrexone bromide), oral therapy Amitizia (lubiprostone), and oral and rectal over-the-counter laxatives and stool softeners such as docusate sodium, senna and milk of magnesia. In addition, there are a number of companies developing potential products which are in various stages of clinical development and are being evaluated for the treatment of OIC and OBD in different patient populations, including Merck & Co., Inc., Progenics Pharmaceuticals, Inc. in collaboration with Salix Pharmaceuticals, Ltd., Purdue Pharma L.P. in collaboration with Shionogi & Co., Ltd., Mundipharma Int. Limited, Sucampo Pharmaceuticals, Inc., Develco Pharma GmbH, Alkermes plc, GlaxoSmithKline plc, Theravance, Inc., and Takeda Pharmaceutical Company Limited. For ADYNOVATE®, on June 6, 2014, the FDA approved Biogen Idec’s Fc fusion protein ELOCTATETM for the control and prevention of bleeding episodes, perioperative (surgical) management and routine prophylaxis in adults and children with Hemophilia A. Longer acting Factor VIII proteins based on polymer conjugation technology approaches are being pursued by Bayer Healthcare LLC (which has filed for regulatory approval in the U.S.) and Novo Nordisk (which has an ongoing Phase 3 clinical development program). In addition, technologies other than those based on Fc fusion and polymer conjugation approaches (such as gene therapy) are being pursued to treat patients with Hemophilia A. For NKTR-181, there are numerous companies developing pain therapies designed to have less abuse potential primarily through formulation technologies and techniques applied to existing pain therapies. Potential competitors include Acura Pharmaceuticals, Inc., Cara Therapeutics, Inc., Collegium Pharmaceutical, Inc., Egalet Ltd, Elite Pharmaceuticals, Inc., Endo Health Solutions Inc., KemPharm, Inc., Pfizer, Inc., Purdue Pharma L.P., and Teva Pharmaceutical Industries Ltd. For ONZEALDTM there are a number of chemotherapies and cancer therapies approved today and in various stages of clinical development for breast cancer, including, but not limited to: Abraxane® (paclitaxel protein-bound particles for injectable suspension (albumin bound)), Xeloda® (capecitabine), Afinitor® (everolimus), Doxil® (doxorubicin HCl), Ellence® (epirubicin), Gemzar® (gemcitabine), Halaven® (eribulin), Herceptin® (trastuzumab), Hycamtin® (topotecan), Ibrance® (palbociclib), Ixempra® (ixabepilone), Navelbine® (vinolrebine), Iniparib, Paraplatin® (carboplatin), Taxol® (paclitaxel) and Taxotere® (docetaxel). Major pharmaceutical or biotechnology companies with approved drugs or drugs in development for breast cancers include, but are not limited to, Bristol-Myers Squibb Company, Eli Lilly & Co., Roche, GlaxoSmithKline plc, Johnson and Johnson, Pfizer Inc., Eisai Inc., and Sanofi Aventis S.A. For NKTR-214, there are numerous companies engaged in developing immunotherapies to be used alone, or in combination, to treat a wide range of oncology indications targeting both solid and liquid tumors. In particular, we expect to compete with therapies with tumor infiltrating lymphocytes, or TILS, chimeric antigen receptor-expressing T cells, or CAR-T, cytokine-based therapies, and checkpoint inhibitors. Potential competitors in the TIL and CAR-T space include Kite Pharma/NCI, Adaptimmune LLC, Celgene Corporation, Juno Therapeutics, and Novartis, Alkermes, Altor, and Armo in the cytokine-based therapies space, and Tesaro, Macrogenics, Merck, BMS, and Roche in the checkpoint inhibitor space. For NKTR‑358, there are a number of competitors in various stages of clinical development that are working on programs which are designed to correct the underlying immune system imbalance in the body due to autoimmune disease. In particular, we expect to compete with therapies that could be cytokine-based therapies (Symbiotix, LLC and Tizona Therapeutics), regulatory T cell therapies (Targazyme, Inc., Juno Therapeutics and Tract Therapeutics, Inc.), or IL-2-based and toleragen-based therapies (Celgene Corporation, Amgen Inc., Tolera Therapeutics, Inc., and Caladrius BioSciences, Inc.).
There can be no assurance that we or our partners will successfully develop, obtain regulatory approvals for and commercialize next-generation or new products that will successfully compete with those of our competitors. Many of our competitors have greater financial, research and development, marketing and sales, manufacturing and managerial capabilities. We face competition from these companies not just in product development but also in areas such as recruiting employees, acquiring technologies that might enhance our ability to commercialize products, establishing relationships with certain research and academic institutions, enrolling patients in clinical trials and seeking program partnerships and collaborations with larger pharmaceutical companies. As a result, our competitors may succeed in developing competing technologies, obtaining regulatory approval or gaining market acceptance for products before we do. These developments could make our products or technologies uncompetitive or obsolete.
If product liability lawsuits are brought against us, we may incur substantial liabilities.
The manufacture, clinical testing, marketing and sale of medical products involve inherent product liability risks. If product liability costs exceed our product liability insurance coverage, we may incur substantial liabilities that could have a severe negative impact on our financial position. Whether or not we are ultimately successful in any product liability litigation, such litigation would consume substantial amounts of our financial and managerial resources and might result in adverse publicity, all of which would impair our business. Additionally, we may not be able to maintain our clinical trial insurance or product liability insurance at an acceptable cost, if at all, and this insurance may not provide adequate coverage against potential claims or losses.
43
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs or the theft of our confidential information or patient confidential information.
Despite the implementation of security measures, our internal computer systems and those of our contract research organizations (CROs) and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such events could cause interruptions of our operations. For instance, the loss of preclinical data or data from any future clinical trial involving our product candidates could result in delays in our development and regulatory filing efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential or proprietary information of our company or clinical patients, we could suffer or be subject to reputational harm, monetary fines, civil suits, civil penalties or criminal sanctions and requirements to disclose the breach, and other forms of liability, and the development of our product candidates could be delayed.
Our future depends on the proper management of our current and future business operations and their associated expenses.
Our business strategy requires us to manage our business to provide for the continued development and potential commercialization of our proprietary and partnered drug candidates. Our strategy also calls for us to undertake increased research and development activities and to manage an increasing number of relationships with partners and other third parties, while simultaneously managing the capital necessary to support this strategy. If we are unable to manage effectively our current operations and any growth we may experience, our business, financial condition and results of operations may be adversely affected. If we are unable to effectively manage our expenses, we may find it necessary to reduce our personnel-related costs through reductions in our workforce, which could harm our operations, employee morale and impair our ability to retain and recruit talent. Furthermore, if adequate funds are not available, we may be required to obtain funds through arrangements with partners or other sources that may require us to relinquish rights to certain of our technologies, products or future economic rights that we would not otherwise relinquish or require us to enter into other financing arrangements on unfavorable terms.
We are dependent on our management team and key technical personnel, and the loss of any key manager or employee may impair our ability to develop our products effectively and may harm our business, operating results and financial condition.
Our success largely depends on the continued services of our executive officers and other key personnel. The loss of one or more members of our management team or other key employees could seriously harm our business, operating results and financial condition. The relationships that our key managers have cultivated within our industry make us particularly dependent upon their continued employment with us. We are also dependent on the continued services of our technical personnel because of the highly technical nature of our products and the regulatory approval process. Because our executive officers and key employees are not obligated to provide us with continued services, they could terminate their employment with us at any time without penalty. We do not have any post-employment noncompetition agreements with any of our employees and do not maintain key person life insurance policies on any of our executive officers or key employees.
Because competition for highly qualified technical personnel is intense, we may not be able to attract and retain the personnel we need to support our operations and growth.
We must attract and retain experts in the areas of clinical testing, manufacturing, research, regulatory and finance, and may need to attract and retain marketing and distribution experts and develop additional expertise in our existing personnel. We face intense competition from other biopharmaceutical companies, research and academic institutions and other organizations for qualified personnel. Many of the organizations with which we compete for qualified personnel have greater resources than we have. Because competition for skilled personnel in our industry is intense, companies such as ours sometimes experience high attrition rates with regard to their skilled employees. Further, in making employment decisions, job candidates often consider the value of the stock options they are to receive in connection with their employment. Our equity incentive plan and employee benefit plans may not be effective in motivating or retaining our employees or attracting new employees, and significant volatility in the price of our stock may adversely affect our ability to attract or retain qualified personnel. If we fail to attract new personnel or to retain and motivate our current personnel, our business and future growth prospects could be severely harmed.
If earthquakes or other catastrophic events strike, our business may be harmed.
Our corporate headquarters, including a substantial portion of our research and development operations, are located in the San Francisco Bay Area, a region known for seismic activity and a potential terrorist target. In addition, we own facilities for the manufacture of products using our advanced polymer conjugate technologies in Huntsville, Alabama and own and lease offices in Hyderabad, India. There are no backup facilities for our manufacturing operations located in Huntsville, Alabama. In the event of an earthquake or other natural disaster, political instability, or terrorist event in any of these locations, our ability to manufacture and supply materials for drug candidates in development and our ability to meet our manufacturing obligations to our customers would be
44
significantly disrupted and our business, results of operations and financial condition would be harmed. Our collaborative partners may also be subject to catastrophic events, such as earthquakes, floods, hurricanes and tornadoes, any of which could harm our business, results of operations and financial condition. We have not undertaken a systematic analysis of the potential consequences to our business, results of operations and financial condition from a major earthquake or other catastrophic event, such as a fire, sustained loss of power, terrorist activity or other disaster, and do not have a recovery plan for such disasters. In addition, our insurance coverage may not be sufficient to compensate us for actual losses from any interruption of our business that may occur.
We have implemented certain anti-takeover measures, which make it more difficult to acquire us, even though such acquisitions may be beneficial to our stockholders.
Provisions of our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even though such acquisitions may be beneficial to our stockholders. These anti-takeover provisions include:
|
|
• |
establishment of a classified board of directors such that not all members of the board may be elected at one time; |
|
|
• |
lack of a provision for cumulative voting in the election of directors, which would otherwise allow less than a majority of stockholders to elect director candidates; |
|
|
• |
the ability of our board to authorize the issuance of “blank check” preferred stock to increase the number of outstanding shares and thwart a takeover attempt; |
|
|
• |
prohibition on stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of stockholders; |
|
|
• |
establishment of advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon by stockholders at stockholder meetings; and |
|
|
• |
limitations on who may call a special meeting of stockholders. |
Further, provisions of Delaware law relating to business combinations with interested stockholders may discourage, delay or prevent a third party from acquiring us. These provisions may also discourage, delay or prevent a third party from acquiring a large portion of our securities or initiating a tender offer or proxy contest, even if our stockholders might receive a premium for their shares in the acquisition over the then-current market prices. We also have a change of control severance benefit plan, which provides for certain cash severance, stock award acceleration and other benefits in the event our employees are terminated (or, in some cases, resign for specified reasons) following an acquisition. This severance plan could discourage a third party from acquiring us.
The price of our common stock is expected to remain volatile.
Our stock price is volatile. During the year ended December 31, 2017, based on closing prices on The NASDAQ Global Select Market, the closing price of our common stock ranged from $11.75 to $60.50 per share. We expect our stock price to remain volatile. A variety of factors may have a significant effect on the market price of our common stock, including the risks described in this section titled “Risk Factors” and the following:
|
|
• |
announcements of data from, or material developments in, our clinical studies and those of our collaboration partners, including data regarding efficacy and safety, delays in clinical development, regulatory approval or commercial launch – in particular, data from clinical studies of NKTR-214 has had a significant impact on our stock price; |
|
|
• |
announcements by collaboration partners as to their plans or expectations related to drug candidates and approved drugs in which we have a substantial economic interest; |
|
|
• |
announcements regarding terminations or disputes under our collaboration agreements; |
|
|
• |
fluctuations in our results of operations; |
|
|
• |
developments in patent or other proprietary rights, including intellectual property litigation or entering into intellectual property license agreements and the costs associated with those arrangements; |
|
|
• |
announcements of technological innovations or new therapeutic products that may compete with our approved products or products under development; |
|
|
• |
announcements of changes in governmental regulation affecting us or our competitors; |
|
|
• |
litigation brought against us or third parties to whom we have indemnification obligations; |
|
|
• |
public concern as to the safety of drug formulations developed by us or others; |
45
|
|
• |
our financing needs and activities; and |
|
|
• |
general market conditions. |
At times, our stock price has been volatile even in the absence of significant news or developments. The stock prices of biotechnology companies and securities markets generally have been subject to dramatic price swings in recent years.
The indenture governing our 7.75% senior secured notes imposes significant operating and financial restrictions on us and our subsidiaries that may prevent us from pursuing certain business opportunities and restrict our ability to operate our business.
On October 5, 2015, we issued $250.0 million in aggregate principal amount of 7.75% senior secured notes due October 2020. The indenture governing the senior secured notes contains covenants that restrict our and our subsidiaries’ ability to take various actions, including, among other things:
|
|
• |
incur or guarantee additional indebtedness or issue disqualified capital stock or cause certain of our subsidiaries to issue preferred stock; |
|
|
• |
pay dividends or distributions, redeem equity interests or subordinated indebtedness or make certain types of investments; |
|
|
• |
create or incur liens; |
|
|
• |
transfer, sell, lease or otherwise dispose of assets and issue or sell equity interests in certain of our subsidiaries; |
|
|
• |
incur restrictions on certain of our subsidiaries’ ability to pay dividends or other distributions to the Company or to make intercompany loans, advances or asset transfers; |
|
|
• |
enter into transactions with affiliates; |
|
|
• |
engage in any business other than businesses which are the same, similar, ancillary or reasonably related to our business as of the date of the indenture; and |
|
|
• |
consummate a merger, consolidation, reorganization or business combination, sell, lease, convey or otherwise dispose of all or substantially all of our assets or other change of control transaction. |
This indenture also requires us to maintain a minimum cash and investments in marketable securities balance of $60.0 million. We have certain reporting obligations under the indenture regarding cash position and royalty revenue. The indenture specifies a number of events of default, some of which are subject to applicable grace or cure periods, including, among other things, non-payment defaults, covenant defaults, cross-defaults to other material indebtedness, bankruptcy and insolvency defaults, non-payment of material judgments, loss of any material business license, criminal indictment of the Company, and certain civil forfeiture proceedings involving material assets of the Company. Our ability to comply with these covenants will likely be affected by many factors, including events beyond our control, and we may not satisfy those requirements. Our failure to comply with our obligations could result in an event of default under our other indebtedness and the acceleration of our other indebtedness, in whole or in part, could result in an event of default under the indenture governing the senior secured notes.
The restrictions contained in the indenture governing the senior secured notes could also limit our ability to plan for or react to market conditions, meet capital needs or otherwise restrict our activities or business plans and adversely affect our ability to finance our operations, enter into acquisitions or to engage in other business activities that would be in our interest.
None.
California
We lease a 129,732 square foot facility in the Mission Bay Area of San Francisco, California (Mission Bay Facility), under an operating lease which expires in January 2030. The Mission Bay Facility is our corporate headquarters and also includes our research and development operations.
46
Alabama
We currently own five facilities consisting of approximately 175,000 square feet in Huntsville, Alabama, which house laboratories as well as administrative, clinical and commercial manufacturing facilities for our PEGylation and advanced polymer conjugate technology operations as well as manufacturing of APIs for early clinical studies.
In July 2012, we consolidated our U.S.-based research activities into our Mission Bay Facility and ceased use of one of our buildings located in Huntsville that was dedicated to research activities. We are currently seeking a buyer for the land and building.
India
We own a research and development facility consisting of approximately 88,000 square feet, near Hyderabad, India. In addition, we lease approximately 1,600 square feet of office space in Hyderabad, India, under a three-year operating lease that will expire in 2018.
From time to time, we are subject to legal proceedings. We are not currently a party to or aware of any proceedings that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition or results of operations.
Not applicable.
47
|
Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock trades on The NASDAQ Global Select Market under the symbol “NKTR.” The table below sets forth the high and low closing sales prices for our common stock as reported on The NASDAQ Global Select Market during the periods indicated.
|
|
|
High |
|
|
Low |
|
||
|
Year Ended December 31, 2016: |
|
|
|
|
|
|
|
|
|
1st Quarter |
|
$ |
16.20 |
|
|
$ |
11.00 |
|
|
2nd Quarter |
|
|
16.28 |
|
|
|
13.09 |
|
|
3rd Quarter |
|
|
19.68 |
|
|
|
14.09 |
|
|
4th Quarter |
|
|
17.48 |
|
|
|
11.85 |
|
|
Year Ended December 31, 2017: |
|
|
|
|
|
|
|
|
|
1st Quarter |
|
$ |
24.20 |
|
|
$ |
11.75 |
|
|
2nd Quarter |
|
|
22.57 |
|
|
|
17.54 |
|
|
3rd Quarter |
|
|
24.00 |
|
|
|
17.79 |
|
|
4th Quarter |
|
|
60.50 |
|
|
|
23.02 |
|
Holders of Record
As of February 22, 2018, there were approximately 177 holders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently expect to retain any future earnings for use in the operation and expansion of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future.
There were no sales of unregistered securities and there were no common stock repurchases made during the year ended December 31, 2017.
Securities Authorized for Issuance Under Equity Compensation Plans
Information regarding our equity compensation plans as of December 31, 2017 is disclosed in Item 12 “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” of this Annual Report on Form 10-K and is incorporated herein by reference from our proxy statement for our 2018 annual meeting of stockholders to be filed with the SEC pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
48
Performance Measurement Comparison
The material in this section is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act or otherwise subject to the liability of that section, nor shall the material in this section be deemed to be incorporated by reference in any registration statement or other document filed with the SEC under the Securities Act or the Exchange Act, except as otherwise expressly stated in such filing.
The following graph compares, for the five year period ended December 31, 2017, the cumulative total stockholder return (change in stock price plus reinvested dividends) of our common stock with (i) the NASDAQ Composite Index, (ii) the NASDAQ Pharmaceutical Index, (iii) the RDG SmallCap Pharmaceutical Index, (iv) the NASDAQ Biotechnology Index and (v) the RDG SmallCap Biotechnology Index. Measurement points are the last trading day of each of our fiscal years ended December 31, 2013, December 31, 2014, December 31, 2015, December 31, 2016 and December 31, 2017. The graph assumes that $100 was invested on December 31, 2012 in the common stock of the Company, the NASDAQ Composite Index, the Nasdaq Pharmaceutical Index, the RDG SmallCap Pharmaceutical Index, the NASDAQ Biotechnology Index and the RDG SmallCap Biotechnology Index and assumes reinvestment of any dividends. The stock price performance in the graph is not intended to forecast or indicate future stock price performance.
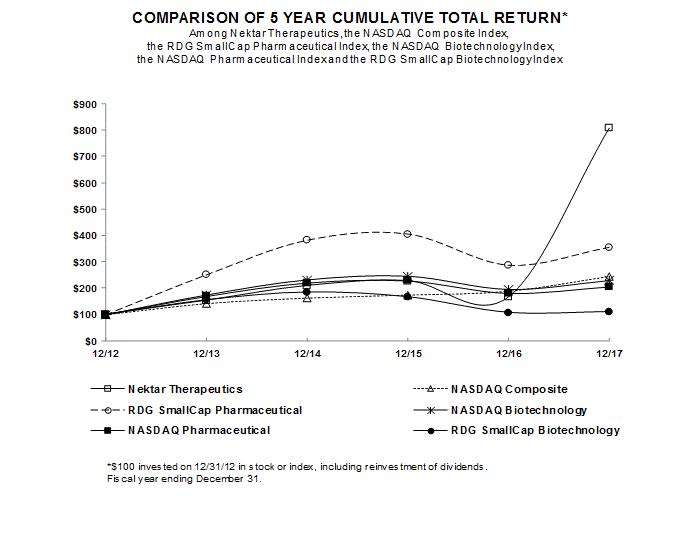
49
SELECTED CONSOLIDATED FINANCIAL INFORMATION
(In thousands, except per share information)
The selected consolidated financial data set forth below should be read together with the consolidated financial statements and related notes, “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the other information contained herein.
|
|
|
Year Ended December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales |
|
$ |
32,688 |
|
|
$ |
55,354 |
|
|
$ |
40,155 |
|
|
$ |
25,152 |
|
|
$ |
44,846 |
|
|
Royalty revenue |
|
|
33,527 |
|
|
|
19,542 |
|
|
|
2,967 |
|
|
|
329 |
|
|
|
1,148 |
|
|
Non-cash royalty revenue related to sale of future royalties(1) |
|
|
30,531 |
|
|
|
30,158 |
|
|
|
22,058 |
|
|
|
21,937 |
|
|
|
22,055 |
|
|
License, collaboration and other revenue |
|
|
210,965 |
|
|
|
60,382 |
|
|
|
165,604 |
|
|
|
153,289 |
|
|
|
80,872 |
|
|
Total revenue |
|
|
307,711 |
|
|
|
165,436 |
|
|
|
230,784 |
|
|
|
200,707 |
|
|
|
148,921 |
|
|
Total operating costs and expenses(2) |
|
|
367,353 |
|
|
|
278,291 |
|
|
|
260,155 |
|
|
|
217,192 |
|
|
|
269,051 |
|
|
Loss from operations |
|
|
(59,642 |
) |
|
|
(112,855 |
) |
|
|
(29,371 |
) |
|
|
(16,485 |
) |
|
|
(120,130 |
) |
|
Non-cash interest expense on liability related to sale of future royalties(1) |
|
|
(18,869 |
) |
|
|
(19,712 |
) |
|
|
(20,619 |
) |
|
|
(20,888 |
) |
|
|
(22,309 |
) |
|
Interest income (expense) and other income (expense), net |
|
|
(17,565 |
) |
|
|
(20,081 |
) |
|
|
(16,602 |
) |
|
|
(17,055 |
) |
|
|
(17,329 |
) |
|
Loss on extinguishment of debt |
|
|
— |
|
|
|
— |
|
|
|
(14,079 |
) |
|
|
— |
|
|
|
— |
|
|
Provision (benefit) for income taxes |
|
|
616 |
|
|
|
876 |
|
|
|
506 |
|
|
|
(512 |
) |
|
|
2,245 |
|
|
Net loss |
|
$ |
(96,692 |
) |
|
$ |
(153,524 |
) |
|
$ |
(81,177 |
) |
|
$ |
(53,916 |
) |
|
$ |
(162,013 |
) |
|
Basic and diluted net loss per share(3) |
|
$ |
(0.62 |
) |
|
$ |
(1.10 |
) |
|
$ |
(0.61 |
) |
|
$ |
(0.42 |
) |
|
$ |
(1.40 |
) |
|
Weighted average shares outstanding used in computing basic and diluted net loss per share(3) |
|
|
155,953 |
|
|
|
139,596 |
|
|
|
132,458 |
|
|
|
126,783 |
|
|
|
115,732 |
|
|
|
|
As of December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and investments in marketable securities |
|
$ |
353,220 |
|
|
$ |
389,102 |
|
|
$ |
308,944 |
|
|
$ |
262,824 |
|
|
$ |
262,026 |
|
|
Working capital |
|
$ |
270,657 |
|
|
$ |
353,730 |
|
|
$ |
288,805 |
|
|
$ |
224,153 |
|
|
$ |
159,661 |
|
|
Total assets |
|
$ |
508,866 |
|
|
$ |
568,871 |
|
|
$ |
498,642 |
|
|
$ |
441,621 |
|
|
$ |
434,527 |
|
|
Deferred revenue |
|
$ |
37,970 |
|
|
$ |
66,239 |
|
|
$ |
83,854 |
|
|
$ |
101,384 |
|
|
$ |
106,048 |
|
|
Senior secured notes, net |
|
$ |
245,207 |
|
|
$ |
243,464 |
|
|
$ |
241,699 |
|
|
$ |
125,000 |
|
|
$ |
125,000 |
|
|
Liability related to the sale of future royalties(1) |
|
$ |
94,655 |
|
|
$ |
105,950 |
|
|
$ |
116,029 |
|
|
$ |
120,471 |
|
|
$ |
128,520 |
|
|
Other long-term liabilities |
|
$ |
5,992 |
|
|
$ |
7,223 |
|
|
$ |
10,813 |
|
|
$ |
18,204 |
|
|
$ |
25,775 |
|
|
Accumulated deficit |
|
$ |
(2,117,941 |
) |
|
$ |
(2,021,010 |
) |
|
$ |
(1,867,486 |
) |
|
$ |
(1,786,309 |
) |
|
$ |
(1,732,393 |
) |
|
Total stockholders’ equity (deficit) |
|
$ |
87,828 |
|
|
$ |
88,125 |
|
|
$ |
6,429 |
|
|
$ |
36,332 |
|
|
$ |
(89,903 |
) |
|
(1) |
In February 2012, we sold all of our rights to receive future royalty payments on net sales of UCB’s CIMZIA® and Roche’s MIRCERA®. As described in Note 7 to our Consolidated Financial Statements, this royalty sale transaction has been recorded as a liability that amortizes over the estimated royalty payment period. As a result of this liability accounting, even though the royalties from UCB and Roche are remitted directly to the purchaser of these royalty interests starting in the second quarter of 2012, we will continue to record non-cash revenue for these royalties and related non-cash interest expense. |
|
(2) |
Operating costs and expenses in 2017 includes $16.0 million for the impairment of equipment and related costs resulting from the termination of the Amikacin Inhale development program. |
|
(3) |
Basic and diluted net loss per share is based upon the weighted average number of common shares outstanding. |
50
The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed here. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in this section as well as factors described in “Part I, Item 1A — Risk Factors.”
Overview
Strategic Direction of Our Business
Nektar Therapeutics is a research-based biopharmaceutical company that discovers and develops innovative new medicines in areas of high unmet medical need. Our research and development pipeline of new investigational drugs includes treatments for cancer, autoimmune disease and chronic pain. We leverage our proprietary and proven chemistry platform to discover and design new drug candidates. These drug candidates utilize our advanced polymer conjugate technology platforms, which are designed to enable the development of new molecular entities that target known mechanisms of action.
We continue to make significant investments in building and advancing our pipeline of proprietary drug candidates as we believe that this is the best strategy to build stockholder value. Described below are certain key events and activities where we are making investments in advancing our research and development pipeline.
|
|
• |
On February 13, 2018, we entered into a Strategic Collaboration Agreement (BMS Collaboration Agreement) with Bristol-Myers Squibb Company (BMS), pursuant to which we and BMS will jointly develop NKTR-214, our lead immuno-oncology drug candidate, in combination with BMS’s Opdivo® (nivolumab) and Opdivo® plus Yervoy® (ipilimumab), in more than 20 indications across nine tumor types, as well as potential combinations with other anti-cancer agents from BMS, us and third parties. We also entered into a Share Purchase Agreement (Purchase Agreement) with BMS, pursuant to which BMS has agreed to purchase $850.0 million of shares of our common stock at a purchase price of $102.60 per share. The closings under the two agreements, subject to satisfaction of certain closing conditions, are expected to occur during the second quarter of 2018. |
|
|
• |
In early 2017, we commenced a broad clinical development program for NKTR-214, our lead immuno-oncology drug candidate in combination with other immuno-oncology agents including Opdivo® (nivolumab) as part of our broad Phase 1/2 clinical collaboration with BMS in five tumor types and eight potential indications, a dose-escalation study with atezolizumab, and numerous preclinical collaboration programs. |
|
|
• |
In the last half of 2017 we completed enrollment in the dose-escalation phase of the NKTR-214 study evaluating NKTR-214 in combination with Opdivo® (nivolumab) in patients with melanoma, renal cell carcinoma and non-small cell lung cancer which we call the PIVOT-02 study. On November 11, 2017 we announced interim data from the dose-escalation phase of the PIVOT-02 Phase 1/2 study. We have identified the Phase 2 dose for NKTR-214 and are currently enrolling subjects in the expansion phase of the study. |
|
|
• |
On May 22, 2017, we announced a research collaboration that will allow Nektar and Takeda Pharmaceutical Company Limited (Takeda) to evaluate NKTR-214 with five compounds from Takeda’s cancer portfolio. |
|
|
• |
On September 12, 2017, we announced that we had begun dosing in a clinical study evaluating the efficacy and safety of NKTR-214 in combination with approved checkpoint inhibitors, TECENTRIQ® (atezolizumab) and KEYTRUDA® (pembrolizumab). |
|
|
• |
In February 2017, we filed an investigational new drug (IND) application for NKTR-358, our autoimmune disease drug candidate. We began the Phase 1 clinical study to evaluate single-ascending doses of NKTR-358 in healthy volunteers in March 2017. This study is designed to establish a range of dose levels and evaluate pharmacokinetics and safety. A Phase 1 multiple-ascending dose trial is expected to be initiated in the second quarter of 2018 in lupus patients. On July 24, 2017, we entered into a license agreement with Lilly to co-develop NKTR-358. |
|
|
• |
On March 20, 2017, we announced that NKTR-181, met its primary and secondary endpoints in the SUMMIT-07 Phase 3 efficacy study. On July 18, 2017, we announced positive top-line data for our pivotal human abuse potential study (the HAP study) for NKTR-181. The HAP study was designed to assess the relative oral abuse potential of NKTR-181 at its highest tested therapeutic dose as well as at the highest dose to which patients have been exposed in our long-term safety study and at a supratherapeutic dose compared to common therapeutic doses of oxycodone, a Schedule II opioid. Following the success of the SUMMIT-07 Phase 3 efficacy study and the HAP study, we are seeking a partner to support future development and commercialization activities for NKTR-181. We are currently planning to file the NDA for NKTR-181 in the first half of 2018. |
51
|
|
• |
We filed the IND for NKTR-262 in December 2017 and expect to begin enrolling patients in the initial Phase 1 study by the end of the first quarter of 2018. We are also completing preclinical research for NKTR-255 with the goal of advancing this program into the clinic in 2019. |
The level of our future research and development investment will depend on a number of trends and uncertainties including clinical outcomes, future studies required to advance programs to regulatory approval, and the economics related to potential future collaborations that may include up-front payments, development funding, milestones, and royalties.
We also have significant milestone and royalty economic interests in approved drugs and drug candidates in late stage development with our collaboration partners. With AstraZeneca, we have a collaboration for MOVANTIK®, an oral peripherally-acting mu-opioid antagonist for the treatment of opioid-induced constipation in adult patients with non-cancer pain. MOVANTIK® is approved by health authorities in the United States, European Union, and many other countries. We have a collaboration with Baxalta (a wholly-owned subsidiary of Shire plc) for ADYNOVATE®, that was approved by the FDA in late 2015 for use in adults and adolescents, aged 12 years and older, who have Hemophilia A. ADYNOVATE® is approved by health authorities in the United States, European Union, and many other countries.
Our business is subject to significant risks, including the risks inherent in our development efforts, the results of our clinical trials, our dependence on the marketing efforts by our collaboration partners, uncertainties associated with obtaining and enforcing patents, the lengthy and expensive regulatory approval process and competition from other products. For a discussion of these and some of the other key risks and uncertainties affecting our business, see Item 1A "Risk Factors" of this Annual Report on Form 10-K.
While the approved drugs and clinical development programs described above are key elements of our future success, we believe it is critically important that we continue to make substantial investments in our earlier-stage drug candidate pipeline. We have several drug candidates in earlier stage clinical development or being explored in research that we are preparing to advance into the clinic in future years. We are also advancing several other drug candidates in preclinical development in the areas of cancer immunotherapy, immunology, and other therapeutic indications. We believe that our substantial investment in research and development has the potential to create significant value if one or more of our drug candidates demonstrates positive clinical results, receives regulatory approval in one or more major markets and achieves commercial success, drug research and development is an inherently uncertain process and there is a high risk of failure at every stage prior to approval and the timing and outcome of clinical trial results are extremely difficult to predict. Clinical development successes and failures can have a disproportionately positive or negative impact on our scientific and medical prospects, financial condition and prospects, results of operations and market value.
Historically, we have entered into a number of license and supply contracts under which we manufactured and supplied our proprietary polymer reagents on a fixed price or cost-plus basis. Our current strategy is to manufacture and supply polymer reagents to support our proprietary drug candidates or our third-party collaborators where we have a strategic development and commercialization relationship or where we derive substantial economic benefit.
Key Developments and Trends in Liquidity and Capital Resources
We estimate that we have working capital to fund our current business plans through at least March 1, 2019. At December 31, 2017, we had approximately $353.2 million in cash and investments in marketable securities and had debt of $250.0 million in principal of senior secured notes due in October 2020. In addition, as described above and in Note 14 to our Consolidated Financial Statements, in February 2018, we entered into a collaboration agreement with BMS under which BMS will pay us a non-refundable upfront cash payment of $1.0 billion and a share purchase agreement under which BMS will purchase $850.0 million of shares of our common stock. The closings under the two agreements, subject to satisfaction of certain closing conditions, are expected to occur during the second quarter of 2018.
52
Results of Operations
Years Ended December 31, 2017, 2016, and 2015
Revenue (in thousands, except percentages)
|
|
|
Year Ended December 31, |
|
|
Increase/ (Decrease) |
|
|
Increase/ (Decrease) |
|
|
Percentage Increase/ (Decrease) |
|
|
Percentage Increase/ (Decrease) |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 vs. 2016 |
|
|
2016 vs. 2015 |
|
|
2017 vs. 2016 |
|
|
2016 vs. 2015 |
|
|||||||
|
Product sales |
|
$ |
32,688 |
|
|
$ |
55,354 |
|
|
$ |
40,155 |
|
|
$ |
(22,666 |
) |
|
$ |
15,199 |
|
|
|
(41 |
)% |
|
|
38 |
% |
|
Royalty revenue |
|
|
33,527 |
|
|
|
19,542 |
|
|
|
2,967 |
|
|
|
13,985 |
|
|
|
16,575 |
|
|
|
72 |
% |
|
> 100 |
% |
|
|
Non cash royalty revenue related to sale of future royalties |
|
|
30,531 |
|
|
|
30,158 |
|
|
|
22,058 |
|
|
|
373 |
|
|
|
8,100 |
|
|
|
1 |
% |
|
|
37 |
% |
|
License, collaboration and other revenue |
|
|
210,965 |
|
|
|
60,382 |
|
|
|
165,604 |
|
|
|
150,583 |
|
|
|
(105,222 |
) |
|
> 100 |
% |
|
|
(64 |
)% |
|
|
Total revenue |
|
$ |
307,711 |
|
|
$ |
165,436 |
|
|
$ |
230,784 |
|
|
$ |
142,275 |
|
|
$ |
(65,348 |
) |
|
|
86 |
% |
|
|
(28 |
)% |
Our revenue is derived from our collaboration agreements, under which we may receive product sales revenue, royalties, license fees, milestone and other contingent payments and/or contract research payments. Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured. The amount of upfront fees received under our license and collaboration agreements allocated to continuing obligations, such as manufacturing and supply commitments, is generally recognized ratably over our expected performance period under the arrangement. As a result, there may be significant variations in the timing of receipt of cash payments and our recognition of revenue. We make our best estimate of the period over which we expect to fulfill our performance obligations. Given the uncertainties in research and development collaborations, significant judgment is required by us to determine the performance periods.
Product sales
Product sales include predominantly fixed price manufacturing and supply agreements with our collaboration partners and are the result of firm purchase orders from those partners. The timing of shipments is based solely on the demand and requirements of our collaboration partners and is not ratable throughout the year.
Product sales decreased for the year ended December 31, 2017 compared to the year ended December 31, 2016 primarily due to decreased product demand from our collaboration partner Ophthotech related to its drug candidate Fovista®. Product sales increased for the year ended December 31, 2016 compared to the year ended December 31, 2015 primarily as a result of increased product demand from Ophthotech. In December 2016, Ophthotech announced that two pivotal Phase 3 clinical trials for Fovista® failed to meet their primary endpoints. In the year ended December 31, 2017, we recognized $10.4 million of product sales to Ophthotech based on prior binding purchase commitments. In August 2017, Ophthotech announced that the third Fovista® Phase 3 trial also failed to meet its primary endpoint and, in October 2017, our agreement with Ophthotech was terminated. We expect product sales in 2018 to decrease compared to 2017 primarily due to the termination of the Ophthotech agreement.
Royalty revenue
We receive royalty revenue from certain of our collaboration partners based on their net sales of commercial products. Royalty revenue received in cash increased for the years ended December 31, 2017 and 2016 compared to the years ended December 31, 2016 and 2015, respectively, due primarily to the commercial launch by AstraZeneca of MOVANTIK® in the U.S. in March 2015 and MOVENTIG® in the EU in August 2015 and the launch of ADYNOVATE® by Baxalta in the U.S. in November 2015. We expect royalty revenue in 2018 will increase as compared to 2017 due to royalties we expect to receive as a result of sales growth of these partnered products.
As part of its approval of MOVANTIK®, the FDA required AstraZeneca to perform a post-marketing, observational epidemiological study comparing MOVANTIK® to other treatments of OIC in patients with chronic, non-cancer pain. As a result, the royalty rate payable to us from net sales of MOVANTIK® in the U.S. by AstraZeneca can be reduced by up to two percentage points to fund 33% of the external costs incurred by AstraZeneca to fund such post approval study, subject to a $35.0 million aggregate cap. As of December 31, 2017, our cumulative share of the post-approval study expenses has been $0.7 million. Any costs incurred by AstraZeneca can only be recovered by the reduction of the royalty paid to us. In no case can amounts be recovered by the reduction of a contingent payment due from AstraZeneca to us or through a payment from us to AstraZeneca.
53
Non-cash royalty revenue related to sale of future royalties
In February 2012, we sold all of our rights to receive future royalty payments on CIMZIA® and MIRCERA®. As described in Note 7 to our Consolidated Financial Statements, this royalty sale transaction has been recorded as a liability that amortizes over the estimated royalty payment period. As a result of this liability accounting, even though the royalties from UCB and Roche are remitted directly to the purchaser of these royalty interests, we will continue to record revenue for these royalties. We expect non-cash royalties from net sales of CIMZIA® and MIRCERA® in 2018 to increase marginally compared to 2017.
License, collaboration and other revenue
License, collaboration and other revenue includes the recognition of upfront payments, milestone and other contingent payments received in connection with our license and collaboration agreements and certain research and development activities. The level of license, collaboration and other revenue depends in part upon the estimated amortization period of the upfront payments, the achievement of milestones and other contingent events, the continuation of existing collaborations, the amount of research and development work, and entering into new collaboration agreements, if any.
License, collaboration and other revenue increased for the year ended December 31, 2017 compared to the year ended December 31, 2016 primarily due to the recognition of $130.1 million of the $150.0 million upfront payment we received in September 2017 from our collaboration agreement with Lilly for NKTR-358 as described in Note 10 to our Consolidated Financial Statements. In addition, in the three months ended December 31, 2017, we recognized $38.0 million related to the termination of our collaboration agreements with Bayer, Ophthotech, and Daiichi as described in Note 10 to our Consolidated Financial Statements. These increases in 2017 were partially offset by the recognition of $28.0 million in March 2016 for our 40% share of the $70.0 million sublicense payment received by AstraZeneca from Kirin.
License, collaboration and other revenue decreased for the year ended December 31, 2016 compared to the year ended December 31, 2015 primarily as a result of the recognition in March 2015 of the $100.0 million milestone payment received from AstraZeneca as a result of the U.S. commercial launch of MOVANTIK® and the $40.0 million milestone payment received from AstraZeneca in August 2015 as a result of the EU commercial launch of MOVENTIG® partially offset by the recognition in 2016 of a $28.0 million payment received from AstraZeneca described above.
We currently expect, subject to our adoption of ASC 606 and completion of our determination of revenue to be recognized related to the BMS Collaboration Agreement executed in February 2018, that our license, collaboration and other revenue from existing collaboration agreements will increase in 2018 compared to 2017.
The timing and future success of our drug development programs and those of our collaboration partners are subject to a number of risks and uncertainties. See “Part I, Item 1A — Risk Factors” for discussion of the risks associated with the complex nature of our collaboration agreements.
Revenue by geography (in thousands)
Revenue by geographic area is based on the locations of our partners. The following table sets forth revenue by geographic area:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
190,810 |
|
|
$ |
39,147 |
|
|
$ |
40,400 |
|
|
Europe |
|
|
116,901 |
|
|
|
126,289 |
|
|
|
190,384 |
|
|
Total revenue |
|
$ |
307,711 |
|
|
$ |
165,436 |
|
|
$ |
230,784 |
|
The increase in revenue attributable to the United States for the year ended December 31, 2017 compared to the year ended December 31, 2016 is primarily attributable to the recognition of $130.1 million of the $150.0 million upfront payment we received in September 2017 from our collaboration agreement with Lilly for NKTR-358. The decrease in revenue attributable to European countries for the year ended December 31, 2016 compared to the year ended December 31, 2015 is primarily attributable to decreased milestone and other contingent payments from our existing European based collaboration partners, including the recognition in 2015 of a total of $140.0 million payments from AstraZeneca in connection with its commercial launches of MOVANTIK® described above.
54
Cost of goods sold (in thousands, except percentages)
|
|
|
Year Ended December 31, |
|
|
Increase/ (Decrease) 2017 vs. |
|
|
Increase/ (Decrease) 2016 vs. |
|
|
Percentage Increase/ (Decrease) 2017 vs. |
|
Percentage Increase/ (Decrease) 2016 vs. |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
2015 |
|
|||||||
|
Cost of goods sold |
|
$ |
30,547 |
|
|
$ |
30,215 |
|
|
$ |
34,102 |
|
|
$ |
332 |
|
|
$ |
(3,887 |
) |
|
|
1 |
% |
|
(11 |
)% |
|
Product gross profit |
|
|
2,141 |
|
|
|
25,139 |
|
|
|
6,053 |
|
|
|
(22,998 |
) |
|
|
19,086 |
|
|
|
(91 |
)% |
> 100 |
% |
|
|
Product gross margin |
|
|
7 |
% |
|
|
45 |
% |
|
|
15 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As noted above, our strategy is to manufacture and supply polymer reagents to support our proprietary drug candidates or our third-party collaborators where we have a strategic development and commercialization relationship or where we derive substantial economic benefit. We have elected to only enter into and maintain those manufacturing relationships associated with long-term collaboration agreements which include multiple sources of revenue, which we view holistically and in aggregate. We have a predominantly fixed cost base associated with our manufacturing activities, which generally results in similar total cost of goods sold amounts each year. As a result, our product gross profit and margin are significantly impacted by the mix and volume of products sold in each period.
Cost of goods sold during the year ended December 31, 2017 was consistent with the year ended December 31, 2016. Cost of goods sold decreased during the year ended December 31, 2016 compared to the year ended December 31, 2015 primarily due to the mix of product sales, which resulted in decreases to cost of goods sold even though product sales increased during the same period.
The decrease in product gross profit and product gross margin during the year ended December 31, 2017 compared to the year ended December 31, 2016 is primarily due to decreased product sales as well as a less favorable product mix in 2017 compared to 2016. In particular, we have a manufacturing arrangement with a partner that includes a fixed price which is less than the fully burdened manufacturing cost for the reagent, and we expect this situation to continue with this partner in future years. There were more shipments to this partner relative to shipments to other customers during the year ended December 31, 2017 compared to the year ended December 31, 2016. The improvement in product gross profit and product gross margin during the year ended December 31, 2016 compared to the year ended December 31, 2015 is primarily due to a more favorable product mix. In particular, the increased demand from our collaboration partners in 2016 resulted in product sales where we have a relatively higher gross margin. This increased margin was partially offset by the manufacturing arrangement mentioned above. In addition to product sales from reagent materials supplied to the partner where our sales are less than our fully burdened manufacturing cost, we also receive royalty revenue from this collaboration. In the years ended December 31, 2017, 2016 and 2015, the royalty revenue from this collaboration exceeded the related negative gross profit.
We expect product gross margin to continue to fluctuate in future periods depending on the level and mix of manufacturing orders from our customers. We currently expect product gross margin to decrease in 2018 as compared to 2017 and gross margin may be negative in 2018 as a result of the anticipated decrease in product sales described above.
Research and development expense (in thousands, except percentages)
|
|
|
Year Ended December 31, |
|
|
Increase/ (Decrease) 2017 vs. |
|
|
Increase/ (Decrease) 2016 vs. |
|
|
Percentage Increase/ (Decrease) 2017 vs. |
|
|
Percentage Increase/ (Decrease) 2016 vs. |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|||||||
|
Research and development expense |
|
$ |
268,461 |
|
|
$ |
203,801 |
|
|
$ |
182,787 |
|
|
$ |
64,660 |
|
|
$ |
21,014 |
|
|
|
32 |
% |
|
|
11 |
% |
Research and development expense consists primarily of clinical study costs, direct costs of outside research, materials, supplies, licenses and fees as well as personnel costs (including salaries, benefits, and stock-based compensation). Research and development expense also includes certain overhead allocations consisting of support and facilities-related costs. Where we perform research and development activities under a clinical joint development collaboration, such as our collaboration with Bristol-Myers Squibb, we record the cost reimbursement from our partner as a reduction to research and development expense when reimbursement amounts are due to us under the agreement.
Research and development expense increased during the year ended December 31, 2017 compared to the year ended December 31, 2016 and includes increased costs for our clinical development of NKTR-214, NKTR-358, NKTR-262, and preclinical activities for NKTR-255. In addition, the increase in research and development expense during the year ended December 31, 2017 compared with the year ended December 31, 2016 includes increased costs related to personnel and outside services. Research and development
55
expense increased during the year ended December 31, 2016 compared to the year ended December 31, 2015 primarily due to costs incurred in our NKTR-214 clinical and NKTR-358 pre-clinical programs as well as increased headcount costs, partially offset by a decrease in our ONZEALDTM program costs.
We utilize our employee and infrastructure resources across multiple development and research programs. The following table shows expenses incurred for clinical and regulatory services, clinical supplies, and preclinical study support provided by third parties as well as direct materials costs for each of our drug candidates. The table also presents other costs and overhead consisting of personnel, facilities and other indirect costs (in thousands):
|
|
|
Clinical |
|
Year Ended December 31, |
|
|||||||||
|
|
|
Study Status(1) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
NKTR-181 (mu-opioid analgesic molecule for chronic pain) |
|
Phase 3 |
|
$ |
41,680 |
|
|
$ |
46,783 |
|
|
$ |
45,307 |
|
|
NKTR-214 (cytokine immunostimulatory therapy)(2) |
|
Phase 1/2 |
|
|
33,834 |
|
|
|
16,118 |
|
|
|
8,637 |
|
|
ONZEALDTM (topoisomerase I inhibitor-polymer conjugate) |
|
Phase 3 |
|
|
15,052 |
|
|
|
14,940 |
|
|
|
19,295 |
|
|
BAY41-6551 (Amikacin Inhale)(3) |
|
Phase 3 |
|
|
10,977 |
|
|
|
10,995 |
|
|
|
9,947 |
|
|
NKTR-358 |
|
Phase 1 |
|
|
10,607 |
|
|
|
5,695 |
|
|
|
132 |
|
|
NKTR-255 |
|
Pre-clinical |
|
|
4,881 |
|
|
|
1,297 |
|
|
|
76 |
|
|
NKTR-262 |
|
Phase 1 |
|
|
4,837 |
|
|
|
11 |
|
|
|
— |
|
|
Other product candidates |
|
Various |
|
|
3,500 |
|
|
|
2,358 |
|
|
|
5,917 |
|
|
Total third party and direct materials costs |
|
|
|
|
125,368 |
|
|
|
98,197 |
|
|
|
89,311 |
|
|
Personnel, overhead and other costs |
|
|
|
|
113,466 |
|
|
|
84,563 |
|
|
|
77,752 |
|
|
Stock-based compensation and depreciation |
|
|
|
|
29,627 |
|
|
|
21,041 |
|
|
|
15,724 |
|
|
Research and development expense |
|
|
|
$ |
268,461 |
|
|
$ |
203,801 |
|
|
$ |
182,787 |
|
|
(1) |
Clinical Study Status definitions are provided in the chart found in Part I, Item 1. Business. |
|
(2) |
The amount for the year ended December 31, 2017 includes $7.8 million of clinical study cost reimbursements from BMS under our collaboration. No reimbursement amounts were recorded in 2016 and 2015. |
|
(3) |
We partnered this program with Bayer Healthcare LLC in August 2007. As part of the Novartis Pulmonary Asset Sale in 2008, we retained an exclusive license to this technology for the development and commercialization of this drug candidate. In November 2017, Bayer announced that its two Phase 3 studies of Amikacin Inhale both failed to meet their primary and secondary endpoints and, in December 2017, Bayer terminated our collaboration to develop Amikacin Inhale. |
We expect research and development expense to increase significantly in 2018 compared to 2017 primarily as a result of the development of NKTR-214 under the BMS Collaboration Agreement. In 2018, we plan to continue and expand a broad clinical development program for NKTR-214. Under the BMS Collaboration Agreement, we and BMS will jointly develop NKTR-214 in combination with BMS’s Opdivo® (nivolumab) and Opdivo® plus Yervoy® (ipilimumab), in more than 20 indications across nine tumor types, as well as potential combinations with other anti-cancer agents from BMS, us and third parties.
In addition, we are collaborating with Lilly to develop NKTR-358 and are continuing its Phase 1 clinical development program in 2018. We have initiated start-up activities on our Phase 1/2 program for NKTR-262 and plan to enroll the initial patients in the second quarter of 2018. We plan to file an IND for NKTR-255 in 2019. The timing and amount of our future clinical investments will vary significantly based upon our evaluation of ongoing clinical results and the structure, timing, and scope of potential collaboration partnerships (if any) for these programs. We plan to file an NDA for NKTR-181 in the first half of 2018 and plan to pursue a collaboration to secure a development and commercialization partner to fund further development for NKTR-181 through direct program funding, up-front payments, milestones, or a combination thereof. In addition, we expect non-cash stock-based compensation expense to increase in 2018 due to the increase in our stock price.
In addition to our drug candidates that we plan to evaluate in clinical development during 2018 and beyond, we believe it is vitally important to continue our substantial investment in a pipeline of new drug candidates to continue to build the value of our drug candidate pipeline and our business. Our discovery research organization is identifying new drug candidates by applying our polymer conjugation technology platform to a wide range of molecule classes, including small molecules and large proteins, peptides and antibodies, across multiple therapeutic areas. We plan to continue to advance our most promising early research drug candidates into preclinical development with the objective to advance these early stage research programs to human clinical studies over the next several years.
Our expenditures on current and future preclinical and clinical development programs are subject to numerous uncertainties in timing and cost to completion. In order to advance our drug candidates through clinical development, each drug candidate must be
56
tested in numerous preclinical safety, toxicology and efficacy studies. We then conduct clinical studies for our drug candidates that take several years to complete. The cost and time required to complete clinical trials may vary significantly over the life of a clinical development program as a result of a variety of factors, including but not limited to:
|
|
• |
the number of patients required for a given clinical study design; |
|
|
• |
the length of time required to enroll clinical study participants; |
|
|
• |
the number and location of sites included in the clinical studies; |
|
|
• |
the clinical study designs required by the health authorities (i.e. primary and secondary endpoints as well as the size of the study needed to demonstrate efficacy and safety outcomes); |
|
|
• |
the potential for changing standards of care for the target patient population; |
|
|
• |
the competition for patient recruitment from competitive drug candidates being studied in the same clinical setting; |
|
|
• |
the costs of producing supplies of the product candidates needed for clinical trials and regulatory submissions; |
|
|
• |
the safety and efficacy profile of the drug candidate; |
|
|
• |
the use of clinical research organizations to assist with the management of the trials; and |
|
|
• |
the costs and timing of, and the ability to secure, approvals from government health authorities. |
Furthermore, our strategy includes the potential of entering into collaborations with third parties to participate in the development and commercialization of some of our drug candidates such as those collaborations that we have already completed for NKTR-214 and MOVANTIK®. In these situations, the clinical development program and process for a drug candidate and the estimated completion date will largely be under the control of that third party and not under our control. We cannot forecast with any degree of certainty which of our drug candidates will be subject to future collaborations or how such arrangements would affect our development plans or capital requirements.
The risks and uncertainties associated with our research and development projects are discussed more fully in Item 1A — Risk Factors. As a result of the uncertainties discussed above, we are unable to determine with any degree of certainty the duration and completion costs of our research and development projects, anticipated completion dates or when and to what extent we will receive cash inflows from a collaboration arrangement or the commercialization of a drug candidate.
General and administrative expense (in thousands, except percentages)
|
|
|
Year Ended December 31, |
|
|
Increase/ (Decrease) 2017 vs. |
|
|
Increase/ (Decrease) 2016 vs. |
|
|
Percentage Increase/ (Decrease) 2017 vs. |
|
|
Percentage Increase/ (Decrease) 2016 vs. |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|||||||
|
General and administrative expense |
|
$ |
52,364 |
|
|
$ |
44,275 |
|
|
$ |
43,266 |
|
|
$ |
8,089 |
|
|
$ |
1,009 |
|
|
|
18 |
% |
|
|
2 |
% |
General and administrative expense includes the cost of administrative staffing, business development, marketing, finance, and legal activities. General and administrative expense increased during the year ended December 31, 2017 compared with the year ended December 31, 2016 primarily due to increased costs related to personnel, facilities and outside services. General and administrative expense increased marginally during the year ended December 31, 2016 compared to the year ended December 31, 2015. In 2018, we expect general and administrative expenses to increase compared to 2017, including an increase in non-cash stock-based compensation expense due to the increase in our stock price.
Interest expense (in thousands, except percentages)
|
|
|
Year Ended December 31, |
|
|
Increase/ (Decrease) 2017 vs. |
|
|
Increase/ (Decrease) 2016 vs. |
|
|
Percentage Increase/ (Decrease) 2017 vs. |
|
|
Percentage Increase/ (Decrease) 2016 vs. |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|
2016 |
|
|
2015 |
|
|||||||
|
Interest expense |
|
$ |
22,085 |
|
|
$ |
22,468 |
|
|
$ |
18,282 |
|
|
$ |
(383 |
) |
|
$ |
4,186 |
|
|
|
(2 |
)% |
|
|
23 |
% |
|
Non-cash interest expense on liability related to sale of future royalties |
|
$ |
18,869 |
|
|
$ |
19,712 |
|
|
$ |
20,619 |
|
|
$ |
(843 |
) |
|
$ |
(907 |
) |
|
|
(4 |
)% |
|
|
(4 |
)% |
57
Interest expense for the year ended December 31, 2017 decreased marginally compared with the year ended December 31, 2016 due to decreased interest expense from our capital leases. Interest expense for the year ended December 31, 2016 increased compared to the year ended December 31, 2015 due primarily to our secured notes transaction completed in October 2015. As is further described in Note 5 to our Consolidated Financial Statements, on October 5, 2015, we issued $250.0 million in aggregate principal amount of 7.75% senior secured notes due October 2020 and used a portion of the proceeds from these notes to redeem the $125.0 million in aggregate principal amount of 12.0% senior secured notes due July 2017. Interest on the 7.75% senior secured notes is calculated based on actual days outstanding over a 360-day year. We expect interest expense in 2018 to decrease marginally compared to 2017.
Non-cash interest expense on the liability related to sale of future royalties for the years ended December 31, 2017 and 2016 decreased marginally as compared to the years ended December 31, 2016 and 2015, respectively, due to the decrease in 2017 and 2016, respectively, of the average balance of the related liability as that liability balance amortizes. On February 24, 2012, we sold all of our rights to receive future royalty payments on CIMZIA® and MIRCERA® in exchange for $124.0 million. As described in Note 7 to our Consolidated Financial Statements, this royalty sale transaction has been recorded as a liability that amortizes over the estimated royalty payment period as CIMZIA® and MIRCERA® royalties are remitted directly to the purchaser. We impute interest on the transaction and record interest expense at the effective interest rate, which we estimated to be approximately 17% from inception to 2017. During the three month period ended December 31, 2017, as a result of increases in the forecasted sales of CIMZIA®, our estimate of the effective annual interest rate over the life of the agreement increased to approximately 17.6%, which results in a prospective interest rate of 21%. There are a number of factors that could materially affect the estimated interest rate, in particular, the amount and timing of royalty payments from future net sales of CIMZIA® and MIRCERA®, and we will assess this estimate on a periodic basis. As a result, future interest rates could differ significantly and any such change in interest rate will be adjusted prospectively. Unless we adjust our estimated interest rate, we expect non-cash interest expense on the liability related to sale of future royalties for 2018 to increase marginally compared with 2017 as a result of the increase of the estimated prospective interest rate noted above.
Impairment of equipment and other costs for terminated program
As described in Note 4 to our Consolidated Financial Statements, in December 2017, Bayer terminated our collaboration to develop Amikacin Inhale. As a result of the termination of the program, in the year ended December 31, 2017, we wrote off program-specific manufacturing equipment with a net book value of $15.1 million. In addition, in the year ended December 31, 2017, we incurred approximately $0.9 million of other program termination costs related to our manufacturing obligations.
Loss on Extinguishment of Debt
As a result of the secured note financing transaction in October 2015 discussed above, in the year ended December 31, 2015, we recognized a $14.1 million loss on extinguishment of our 12% notes, which consists of a $11.3 million redemption premium payment, $1.2 million of incremental interest paid at redemption to the 12% note holders and the write-off of $1.6 million of unamortized issuance costs on the 12% notes.
Liquidity and Capital Resources
We have financed our operations primarily through revenue from product sales, royalties and strategic collaboration agreements, as well as public offering and private placements of debt and equity securities. At December 31, 2017, we had approximately $353.2 million in cash and investments in marketable securities and had debt of $250.0 million in principal of senior secured notes due in October 2020.
We estimate that we have working capital to fund our current business plans through at least March 1, 2019. We expect the clinical development of our proprietary drug candidates including NKTR-214, NKTR-358, NKTR-262, NKTR-255, NKTR-181, and ONZEALDTM will continue to require significant investment in order to continue to advance in clinical development with the objective of entering into a collaboration partnership or obtaining regulatory approval. In the past, we have received a number of significant payments from collaboration agreements and other significant transactions. In July 2017, we entered into a collaboration agreement for NKTR-358 with Lilly, under which we received a $150.0 million upfront payment. Subject to the closing of the BMS Collaboration Agreement and the Purchase Agreement, BMS will pay us a non-refundable upfront cash payment of $1.0 billion and purchase $850.0 million of shares of our common stock at a purchase price of $102.60 per share. We have no credit facility or any other sources of committed capital. In the future, we expect to receive increasing royalties from commercial sales of products such as MOVANTIK®, MOVENTIG® and ADYNOVATE® as they continue to increase sales. We also expect potential substantial payments from future collaboration transactions if drug candidates in our pipeline achieve positive clinical or regulatory outcomes. Our current business plan is also subject to significant uncertainties and risks as a result of, among other factors, clinical and regulatory outcomes for NKTR-214, the sales levels of products for which we are entitled to royalties such as MOVANTIK®, MOVENTIG® and
58
ADYNOVATE®, clinical program outcomes, whether, when and on what terms we are able to enter into new collaboration transactions, expenses being higher than anticipated, unplanned expenses, cash receipts being lower than anticipated, and the need to satisfy contingent liabilities, including litigation matters and indemnification obligations.
The availability and terms of various financing alternatives substantially depend on many factors including the success or failure of drug development programs in our pipeline, including NKTR-214, NKTR-181, NKTR-358, NKTR-262, and NKTR-255 as well as other early stage development programs. The availability and terms of financing alternatives and any future significant payments from existing or new collaborations depend on the positive outcome of ongoing or planned clinical studies, whether we or our partners are successful in obtaining regulatory authority approvals in major markets, and if approved, the commercial success of these drugs, as well as general capital market conditions. We will pursue various financing alternatives as needed to continue to fund our research and development activities and to fund the expansion of our business as appropriate.
Due to the potential for adverse developments in the credit markets, we may experience reduced liquidity with respect to some of our investments in marketable securities. These investments are generally held to maturity, which, in accordance with our investment policy, is less than two years. However, if the need arises to liquidate such securities before maturity, we may experience losses on liquidation. At December 31, 2017, the average time to maturity of the investments held in our portfolio was approximately eight months. To date we have not experienced any liquidity issues with respect to these securities. We believe that, even allowing for potential liquidity issues with respect to these securities, our remaining cash and investments in marketable securities will be sufficient to meet our anticipated cash needs for at least the next twelve months.
Cash flows from operating activities
Cash flows used in operating activities for the year ended December 31, 2017 totaled $80.4 million, which includes $225.3 million of net operating cash uses as well as $19.6 million for interest payments on our senior secured notes and the $12.5 million termination payment to Daiichi in December 2017 related to our ONZEALDTM collaboration arrangement in Europe, partially offset by the receipt of $150.0 million upfront payment in September 2017 from our collaboration agreement with Lilly for NKTR-358, the receipt of $12.0 million in November 2017 related to our collaboration agreement with Baxalta and the receipt of $15.0 million in December 2017 resulting from a settlement agreement related to our collaboration agreement with MAP Pharmaceuticals, Inc.
Cash flows used in operating activities for the year ended December 31, 2016 totaled $117.0 million, which includes $158.3 million of net operating cash uses as well as $19.7 million for interest payments on our senior secured notes, partially offset by the receipt of $31.0 million of payments from AstraZeneca related to its sub-license to Kirin, the receipt of a $20.0 million upfront payment in August 2016 from Daiichi related to our ONZEALDTM collaboration arrangement in Europe, as well as the receipt of a $10.0 million milestone in January 2016 from our Baxalta collaboration agreement, which was recorded in accounts receivable in our Consolidated Balance Sheet at December 31, 2015.
Cash flows used in operating activities for the year ended December 31, 2015 totaled $73.1 million, which includes $196.3 million of net operating cash uses as well as $18.8 million for interest payments on our senior secured notes, partially offset by the receipt of $142.0 million of milestones and other contingent payments from collaboration agreements.
We expect that cash flows used in operating activities, excluding upfront, milestone and other contingent payments received, if any, will increase in the full year of 2018 compared to 2017 primarily as a result of increased research and development expenses.
Cash flows from investing activities
We paid $9.7 million, $6.4 million, and $11.2 million to purchase property, plant and equipment in the years ended December 31, 2017, 2016, and 2015, respectively. We expect our capital expenditures in 2018 to increase compared to 2017.
Restricted cash of $25.0 million was required to be maintained in a separate account until July 1, 2015 under the terms of our 12% senior secured notes due July 2017. This restriction expired on July 1, 2015 and the restricted funds were returned to us.
Cash flows from financing activities
On October 24, 2016, we completed the issuance and sale of 14,950,000 shares of our common stock in an underwritten public offering with total proceeds of approximately $189.7 million after deducting the underwriting commissions and discounts of approximately $12.1 million. In addition, we incurred approximately $0.4 million in legal and accounting fees, filing fees, and other costs in connection with this offering.
59
On October 5, 2015, we issued $250.0 million in aggregate principal amount of 7.75% senior secured notes due 2020. The notes bear interest at a rate of 7.75% per annum payable in cash quarterly in arrears on January 15, April 15, July 15, and October 15 of each year. In connection with the issuance of the notes, we paid $8.7 million of transaction and facility fees paid to the purchasers of the notes and other direct costs, which were capitalized as a debt discount and issuance costs and are recorded as a reduction to the note payable liability. On October 5, 2015, we used a portion of the proceeds from these notes to redeem the $125.0 million in aggregate principal amount of 12.0% senior secured notes due in 2017. In addition, on October 5, 2015, we paid accrued interest of $3.3 million and made a $12.5 million redemption payment, which includes $1.2 million additional interest paid to the 12% note holders at redemption.
We received proceeds from issuance of common stock related to our employee option and stock purchase plans of $59.5 million, $20.3 million, and $32.2 million in the years ended December 31, 2017, 2016, and 2015, respectively.
Contractual Obligations (in thousands)
|
|
|
Payments Due by Period |
|
|||||||||||||||||
|
|
|
Total |
|
|
<=1 Yr 2018 |
|
|
2-3 Yrs 2019-2020 |
|
|
4-5 Yrs 2021-2022 |
|
|
2023+ |
|
|||||
|
Obligations(1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7.75% senior secured notes due October 2020, including interest |
|
$ |
308,394 |
|
|
$ |
19,644 |
|
|
$ |
288,750 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Operating leases(2) |
|
|
106,250 |
|
|
|
5,965 |
|
|
|
11,395 |
|
|
|
17,763 |
|
|
|
71,127 |
|
|
Purchase commitments(3) |
|
|
16,456 |
|
|
|
16,456 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
$ |
431,100 |
|
|
$ |
42,065 |
|
|
$ |
300,145 |
|
|
$ |
17,763 |
|
|
$ |
71,127 |
|
|
(1) |
The above table does not include certain commitments and contingencies which are discussed in Note 8 of Item 8. Financial Statements and Supplementary Data. |
|
(2) |
These amounts primarily result from our Mission Bay Facility lease, which expires in 2030. The lease is discussed in Note 6 of Item 8. Financial Statements and Supplementary Data. |
|
(3) |
Substantially all of this amount was subject to open purchase orders as of December 31, 2017 that were issued under existing contracts. This amount does not represent minimum contract termination liabilities for our existing contracts. |
Off Balance Sheet Arrangements
We do not utilize off-balance sheet financing arrangements as a source of liquidity or financing.
Critical Accounting Policies
The preparation and presentation of financial statements in conformity with U.S. generally accepted accounting principles (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form our basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources, and evaluate our estimates on an ongoing basis. Actual results may differ materially from those estimates under different assumptions or conditions. We have determined that for the periods in this report, the following accounting policies and estimates are critical in understanding our financial condition and the results of our operations.
Revenue Recognition
License, collaboration and other research revenue is recognized based on the facts and circumstances of each contractual agreement and includes amortization of upfront fees. We defer income under contractual agreements when we have further obligations that indicate that a separate earnings process has not been completed. The amount of upfront fees and other payments received under our license and collaboration agreements that are allocated to our continuing obligations are generally recognized ratably over our expected performance period under each arrangement. Management makes its best estimate of the period over which we expect to fulfill our performance obligations, which may include technology transfer assistance, research and development activities, or manufacturing activities through the completion of clinical development or the termination or expiration of the collaboration agreement. Given the complexities and uncertainties of collaboration arrangements, significant judgment is required by management
60
to determine the duration of the performance period. Our original estimates are periodically evaluated to determine if circumstances have caused the estimates to change and if so, amortization of revenue is adjusted prospectively.
In addition, at the inception of each new multiple-element arrangement or the material modification of an existing multiple-element arrangement, we allocate arrangement consideration to all units of accounting based on the relative selling price method, generally based on our best estimate of selling price (ESP). The objective of ESP is to determine the price at which we would transact a sale if the product or service was sold on a stand-alone basis. We determine ESP for the elements in our collaboration arrangements by considering multiple factors including, but not limited to, technical complexity of the performance obligation and similarity of elements to those performed under previous arrangements. Since we apply significant judgment in arriving at the ESPs, any material changes would significantly affect the allocation of the total consideration to the different elements of a multiple element arrangement.
Clinical Trial Accruals
We record accruals for the estimated costs of our clinical study activities performed by third parties. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows to our vendors. Payments under the contracts depend on factors such as the achievement of certain events, successful enrollment of patients and completion of certain clinical trial activities. We generally accrue costs associated with the start-up and reporting phases of the clinical studies ratably over the estimated duration of the start-up and reporting phases. We generally accrue costs associated with the treatment phase of clinical studies based on the total estimated cost of the treatment phase on a per patient basis and we expense the per patient cost ratably over the estimated patient treatment period based on patient enrollment in the studies. In specific circumstances, such as for certain time-based costs, we recognize clinical trial expenses using a methodology that we consider to be more reflective of the timing of costs incurred. Advance payments for goods or services that will be used or rendered for future research and development activities are capitalized as prepaid expenses and recognized as expense as the related goods are delivered or the related services are performed. We base our estimates on the best information available at the time. However, additional information may become available to us which may allow us to make a more accurate estimate in future periods. In this event, we may be required to record adjustments to research and development expenses in future periods when the actual level of activity becomes more certain. Such increases or decreases in cost are generally considered to be changes in estimates and will be reflected in research and development expenses in the period identified.
Stock-Based Compensation
We use the Black-Scholes option pricing model for each respective grant to determine the estimated fair value of stock options on the date of grant (grant date fair value). We expense the estimated fair value of each award ratably over the expected service period of the award and recognize forfeitures as they occur. The Black-Scholes option pricing model requires the input of highly subjective assumptions. These variables include, but are not limited to, our stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors. Because our employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect fair value estimates, in management’s opinion, the existing models may not provide a reliable single measure of the fair value of our employee stock options. Management continually assesses the assumptions and methodologies used to calculate the estimated fair value of stock-based compensation. In addition, for awards that vest upon the achievement of performance milestones, we estimate whether the awards will vest and the vesting period based on our evaluation of the probability of achievement of each respective milestone and the related estimated date of achievement. Circumstances may change and additional data may become available over time, which could result in changes to the assumptions and methodologies, and which could materially impact our fair value determination, as well as our stock-based compensation expense.
Non-cash Interest Expense on Liability Related to Sale of Future Royalties
In February 2012, we sold all of our rights to receive future royalty payments from sales of the CIMZIA® and MIRCERA® drug products marketed by UCB and Roche, respectively. Although we are required to make payments to the purchaser (RPI) only in certain situations, including the event of our breach of a representation, warranty or covenant in the Purchase and Sale Agreement that gives rise to a liability in accordance with the terms and conditions of such agreement, this royalty sale transaction was recorded as a liability (Royalty Obligation) that we will amortize using the interest method over the estimated life of the Purchase and Sale Agreement. As a result, we impute interest on the transaction and record interest expense at the estimated interest rate. Our estimate of the interest rate under the agreement is based on the amount of royalty payments to be received by RPI over the life of the arrangement and payments we are required to make to RPI under the agreement. We will periodically assess the expected royalty payments to RPI from UCB and Roche using a combination of historical results and forecasts from market data sources. To the extent such payments are greater or less than our initial estimates or the timing of such payments is materially different than our original estimates, we will prospectively adjust the amortization of the Royalty Obligation. There are a number of factors that could materially affect the amount and timing of royalty payments from CIMZIA® and MIRCERA®, most of which are not within our control. Such factors include, but are not limited to, changing standards of care, the introduction of competing products, manufacturing or other delays, biosimilar competition, intellectual property matters, adverse events that result in health authority imposed restrictions on the use of the drug products, significant changes in foreign exchange rates as the royalties remitted to RPI are made in U.S. dollars (USD)
61
while significant portions of the underlying sales of CIMZIA® and MIRCERA® are made in currencies other than USD, and other events or circumstances that result in reduced royalty payments from CIMZIA® and MIRCERA®, all of which would result in a reduction of non-cash royalty revenue and non-cash interest expense over the life of the Royalty Obligation. Conversely, if sales of CIMZIA® and MIRCERA® are higher than expected, non-cash royalty revenue and non-cash interest expense would be greater over the term of the Royalty Obligation. During the three month period ended December 31, 2017, as a result of increases in the forecasted sales of CIMZIA®, our estimate of the effective annual interest rate over the life of the agreement increased from 17% to approximately 17.6%, which results in a prospective interest rate of 21%. If we had determined that the interest rate used in 2017 should have been one percentage point higher than our quarterly estimates during 2017, the non-cash interest expense recognized in the year ended December 31, 2017 and the Royalty Obligation balance as of December 31, 2017 would have increased by $1.1 million. If we had determined that the interest rate used in 2017 should have been 21% during 2017, the non-cash interest expense recognized in the year ended December 31, 2017 and the Royalty Obligation balance as of December 31, 2017 would have increased by $3.5 million.
Recent Accounting Pronouncements
In May 2014, the FASB issued guidance codified in Accounting Standards Codification (ASC) 606, Revenue Recognition — Revenue from Contracts with Customers, which supersedes the guidance in ASC 605, Revenue Recognition, and is effective for public companies for annual and interim periods beginning after December 15, 2017. The FASB has issued numerous updates that provide clarification on a number of specific issues as well as requiring additional disclosures. We plan to adopt the standard in the first quarter of 2018 using the modified retrospective method.
The new guidance requires the application of a five-step model to determine the amount and timing of revenue to be recognized and requires that we recognize revenue in a manner that reasonably reflects the delivery of our goods or services to customers in return for expected consideration. To achieve this core principle, the guidance provides the following steps: (1) identify the contract(s) with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation. Under ASC 606, companies may need to use more judgment and make more estimates than under ASC 605 in the application of this five-step process.
We are continuing to assess the impact of the new guidance on our accounting policies and procedures and are evaluating the new requirements as applied to existing collaboration agreements, both in terms of the cumulative adjustment to opening accumulated deficit and our subsequent recognition of revenue. Since each collaboration agreement is unique, we will separately assess each agreement (see Note 10 for a discussion of our existing collaboration agreements) under the new standard. We anticipate that the adoption of ASC 606 will have the following impact to our revenue recognition for a collaboration agreement:
(i) Changes in revenue recognition for distinct licenses of functional intellectual property may result in a timing difference of revenue recognition between the current literature and ASC 606. For certain of our arrangements, the value associated with the licenses and certain other deliverables have been assessed as one unit of accounting and recognized over a period of time in which services are rendered or made available to our customer pursuant to revenue recognition guidance in effect for such arrangements at the time such arrangements commenced. For certain other arrangements, under current ASC 605 guidance, we identified the value of the license as a separate unit of accounting and recognize revenue upon issuance of the license. Under ASC 606, we may continue to recognize revenue for such licenses upon delivery of the license.
(ii) For other consideration, including milestone payments or contingent payments from our collaboration partners, under our current accounting policy, we recognize such payments as revenue in the period that the payment-triggering event occurred or is achieved. The new revenue standard, however, may require us to recognize these payments before the payment-triggering event is completely achieved, subject to management’s assessment of whether it is probable that the triggering event will be achieved and that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved.
(iii) We will recognize revenue for sales-based royalties and commercial sales-based milestones in the period of the related sale based on estimates, rather than recording them as reported by the customer.
We currently estimate that, upon adoption on January 1, 2018, we will record an adjustment to decrease our accumulated deficit by approximately $11.0 million to $13.0 million for amounts that would be recognized as of December 31, 2017 under ASC 606, but are not recognized under the current ASC 605 guidance. This estimated adjustment primarily consists of approximately $11.0 million for the accrual of royalties due to us related to net sales in the three months ended December 31, 2017 that we would have recognized in 2018 under current guidance. The estimated adjustment also includes up to approximately $2.0 million related to the recognition of the unamortized portion of upfront payments under ASC 606 that are required to be deferred as of December 31, 2017 under current guidance. We will finalize our assessments in the first quarter of 2018 as we adopt ASC 606.
62
In February 2016, the FASB issued guidance to amend a number of aspects of lease accounting, including requiring lessees to recognize almost all leases with a term greater than one year as a right-of-use asset and corresponding liability, measured at the present value of the lease payments. The guidance will become effective for us beginning in the first quarter of 2019 and is required to be adopted using a modified retrospective approach. Early adoption is permitted. We are currently evaluating the impact of the adoption of this standard.
Interest Rate and Market Risk
The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in liquid, high quality debt securities. Our investments in debt securities are subject to interest rate risk. To minimize the exposure due to an adverse shift in interest rates, we invest in short-term securities and maintain a weighted average maturity of one year or less.
A hypothetical 50 basis point increase in interest rates would result in an approximate $1.1 million decrease, less than 1%, in the fair value of our available-for-sale securities at December 31, 2017. This potential change is based on sensitivity analyses performed on our investment securities at December 31, 2017. Actual results may differ materially. The same hypothetical 50 basis point increase in interest rates would have resulted in an approximate $0.8 million decrease, less than 1%, in the fair value of our available-for-sale securities at December 31, 2016.
As of December 31, 2017, we held $347.3 million of available-for-sale investments, excluding money market funds, with an average time to maturity of eight months. To date we have not experienced any liquidity issues with respect to these securities, but should such issues arise, we may be required to hold some, or all, of these securities until maturity. We believe that, even allowing for potential liquidity issues with respect to these securities, our remaining cash, cash equivalents, and investments in marketable securities will be sufficient to meet our anticipated cash needs for at least the next twelve months. Based on our available cash and our expected operating cash requirements, we currently do not intend to sell these securities prior to maturity and it is more likely than not that we will not be required to sell these securities before we recover the amortized cost basis. Accordingly, we believe there are no other-than-temporary impairments on these securities and have not recorded any provisions for impairment.
Foreign Currency Risk
The majority of our revenue, expense, and capital purchasing activities are transacted in U.S. dollars. However, since a portion of our operations consists of research and development activities outside the United States, we have entered into transactions in other currencies, primarily the Indian Rupee, and we therefore are subject to foreign exchange risk.
Our international operations are subject to risks typical of international operations, including, but not limited to, differing economic conditions, changes in political climate, differing tax structures, other regulations and restrictions, and foreign exchange rate volatility. We do not utilize derivative financial instruments to manage our exchange rate risks.
63
NEKTAR THERAPEUTICS
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
|
Page |
|
65 |
|
|
67 |
|
|
68 |
|
|
69 |
|
|
70 |
|
|
71 |
|
|
72 |
64
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Nektar Therapeutics
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Nektar Therapeutics (the “Company”) as of December 31, 2017 and 2016, the related consolidated statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes. In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 1993.
Redwood City, California
March 1, 2018
65
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Nektar Therapeutics
Opinion on Internal Control over Financial Reporting
We have audited Nektar Therapeutics’ internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Nektar Therapeutics (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2017 and 2016, the related consolidated statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Redwood City, California
March 1, 2018
66
NEKTAR THERAPEUTICS
(In thousands, except par value information)
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
4,762 |
|
|
$ |
59,640 |
|
|
Short-term investments |
|
|
291,370 |
|
|
|
329,462 |
|
|
Accounts receivable, net of allowance of nil at December 31, 2017 and 2016 |
|
|
5,014 |
|
|
|
15,678 |
|
|
Inventory |
|
|
10,726 |
|
|
|
11,109 |
|
|
Other current assets |
|
|
14,948 |
|
|
|
10,063 |
|
|
Total current assets |
|
|
326,820 |
|
|
|
425,952 |
|
|
Long-term investments |
|
|
57,088 |
|
|
|
— |
|
|
Property, plant and equipment, net |
|
|
47,463 |
|
|
|
65,601 |
|
|
Goodwill |
|
|
76,501 |
|
|
|
76,501 |
|
|
Other assets |
|
|
994 |
|
|
|
817 |
|
|
Total assets |
|
$ |
508,866 |
|
|
$ |
568,871 |
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
4,782 |
|
|
$ |
2,816 |
|
|
Accrued compensation |
|
|
8,263 |
|
|
|
18,280 |
|
|
Accrued clinical trial expenses |
|
|
9,461 |
|
|
|
7,958 |
|
|
Other accrued expenses |
|
|
10,064 |
|
|
|
4,711 |
|
|
Interest payable |
|
|
4,198 |
|
|
|
4,198 |
|
|
Liability related to refundable upfront payment |
|
|
— |
|
|
|
12,500 |
|
|
Deferred revenue, current portion |
|
|
18,949 |
|
|
|
14,352 |
|
|
Other current liabilities |
|
|
446 |
|
|
|
7,407 |
|
|
Total current liabilities |
|
|
56,163 |
|
|
|
72,222 |
|
|
Senior secured notes, net |
|
|
245,207 |
|
|
|
243,464 |
|
|
Liability related to the sale of future royalties, net |
|
|
94,655 |
|
|
|
105,950 |
|
|
Deferred revenue, less current portion |
|
|
19,021 |
|
|
|
51,887 |
|
|
Other long-term liabilities |
|
|
5,992 |
|
|
|
7,223 |
|
|
Total liabilities |
|
|
421,038 |
|
|
|
480,746 |
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Preferred stock, $0.0001 par value; 10,000 shares authorized; no shares designated, issued or outstanding at December 31, 2017 or 2016 |
|
|
— |
|
|
|
— |
|
|
Common stock, $0.0001 par value; 300,000 shares authorized; 159,524 shares and 153,212 shares issued and outstanding at December 31, 2017 and 2016, respectively |
|
|
15 |
|
|
|
15 |
|
|
Capital in excess of par value |
|
|
2,207,865 |
|
|
|
2,111,483 |
|
|
Accumulated other comprehensive loss |
|
|
(2,111 |
) |
|
|
(2,363 |
) |
|
Accumulated deficit |
|
|
(2,117,941 |
) |
|
|
(2,021,010 |
) |
|
Total stockholders’ equity |
|
|
87,828 |
|
|
|
88,125 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
508,866 |
|
|
$ |
568,871 |
|
The accompanying notes are an integral part of these consolidated financial statements.
67
NEKTAR THERAPEUTICS
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share information)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales |
|
$ |
32,688 |
|
|
$ |
55,354 |
|
|
$ |
40,155 |
|
|
Royalty revenue |
|
|
33,527 |
|
|
|
19,542 |
|
|
|
2,967 |
|
|
Non-cash royalty revenue related to sale of future royalties |
|
|
30,531 |
|
|
|
30,158 |
|
|
|
22,058 |
|
|
License, collaboration and other revenue |
|
|
210,965 |
|
|
|
60,382 |
|
|
|
165,604 |
|
|
Total revenue |
|
|
307,711 |
|
|
|
165,436 |
|
|
|
230,784 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of goods sold |
|
|
30,547 |
|
|
|
30,215 |
|
|
|
34,102 |
|
|
Research and development |
|
|
268,461 |
|
|
|
203,801 |
|
|
|
182,787 |
|
|
General and administrative |
|
|
52,364 |
|
|
|
44,275 |
|
|
|
43,266 |
|
|
Impairment of equipment and other costs for terminated program |
|
|
15,981 |
|
|
|
— |
|
|
|
— |
|
|
Total operating costs and expenses |
|
|
367,353 |
|
|
|
278,291 |
|
|
|
260,155 |
|
|
Loss from operations |
|
|
(59,642 |
) |
|
|
(112,855 |
) |
|
|
(29,371 |
) |
|
Non-operating income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
(22,085 |
) |
|
|
(22,468 |
) |
|
|
(18,282 |
) |
|
Non-cash interest expense on liability related to sale of future royalties |
|
|
(18,869 |
) |
|
|
(19,712 |
) |
|
|
(20,619 |
) |
|
Loss on extinguishment of debt |
|
|
— |
|
|
|
— |
|
|
|
(14,079 |
) |
|
Interest income and other income (expense), net |
|
|
4,520 |
|
|
|
2,387 |
|
|
|
1,680 |
|
|
Total non-operating expense, net |
|
|
(36,434 |
) |
|
|
(39,793 |
) |
|
|
(51,300 |
) |
|
Loss before provision for income taxes |
|
|
(96,076 |
) |
|
|
(152,648 |
) |
|
|
(80,671 |
) |
|
Provision for income taxes |
|
|
616 |
|
|
|
876 |
|
|
|
506 |
|
|
Net loss |
|
$ |
(96,692 |
) |
|
$ |
(153,524 |
) |
|
$ |
(81,177 |
) |
|
Basic and diluted net loss per share |
|
$ |
(0.62 |
) |
|
$ |
(1.10 |
) |
|
$ |
(0.61 |
) |
|
Weighted average shares outstanding used in computing basic and diluted net loss per share |
|
|
155,953 |
|
|
|
139,596 |
|
|
|
132,458 |
|
The accompanying notes are an integral part of these consolidated financial statements.
68
NEKTAR THERAPEUTICS
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net loss |
|
$ |
(96,692 |
) |
|
$ |
(153,524 |
) |
|
$ |
(81,177 |
) |
|
Other comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net unrealized gain (loss) on available-for-sale investments, net of tax |
|
|
(533 |
) |
|
|
79 |
|
|
|
(226 |
) |
|
Net foreign currency translation gain (loss) |
|
|
785 |
|
|
|
(272 |
) |
|
|
(377 |
) |
|
Other comprehensive income (loss), net of tax |
|
|
252 |
|
|
|
(193 |
) |
|
|
(603 |
) |
|
Comprehensive loss |
|
$ |
(96,440 |
) |
|
$ |
(153,717 |
) |
|
$ |
(81,780 |
) |
The accompanying notes are an integral part of these consolidated financial statements.
69
NEKTAR THERAPEUTICS
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands)
|
|
|
Common Shares |
|
|
Par Value |
|
|
Capital in Excess of Par Value |
|
|
Accumulated Other Comprehensive Income/(Loss) |
|
|
Accumulated Deficit |
|
|
Total Stockholders’ Equity |
|
||||||
|
Balance at December 31, 2014 |
|
|
131,216 |
|
|
$ |
13 |
|
|
$ |
1,824,195 |
|
|
$ |
(1,567 |
) |
|
$ |
(1,786,309 |
) |
|
$ |
36,332 |
|
|
Shares issued under equity compensation plans |
|
|
4,073 |
|
|
|
— |
|
|
|
32,208 |
|
|
|
— |
|
|
|
— |
|
|
|
32,208 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
19,669 |
|
|
|
— |
|
|
|
— |
|
|
|
19,669 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(603 |
) |
|
|
— |
|
|
|
(603 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(81,177 |
) |
|
|
(81,177 |
) |
|
Balance at December 31, 2015 |
|
|
135,289 |
|
|
|
13 |
|
|
|
1,876,072 |
|
|
|
(2,170 |
) |
|
|
(1,867,486 |
) |
|
|
6,429 |
|
|
Sale of common stock, net of issuance costs of $439 |
|
|
14,950 |
|
|
|
2 |
|
|
|
189,274 |
|
|
|
— |
|
|
|
— |
|
|
|
189,276 |
|
|
Shares issued under equity compensation plans |
|
|
2,973 |
|
|
|
— |
|
|
|
20,287 |
|
|
|
— |
|
|
|
— |
|
|
|
20,287 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
25,850 |
|
|
|
— |
|
|
|
— |
|
|
|
25,850 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(193 |
) |
|
|
— |
|
|
|
(193 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(153,524 |
) |
|
|
(153,524 |
) |
|
Balance at December 31, 2016 |
|
|
153,212 |
|
|
|
15 |
|
|
|
2,111,483 |
|
|
|
(2,363 |
) |
|
|
(2,021,010 |
) |
|
|
88,125 |
|
|
Shares issued under equity compensation plans |
|
|
6,312 |
|
|
|
— |
|
|
|
59,528 |
|
|
|
— |
|
|
|
— |
|
|
|
59,528 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
36,615 |
|
|
|
— |
|
|
|
— |
|
|
|
36,615 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
239 |
|
|
|
252 |
|
|
|
(239 |
) |
|
|
252 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(96,692 |
) |
|
|
(96,692 |
) |
|
Balance at December 31, 2017 |
|
|
159,524 |
|
|
$ |
15 |
|
|
$ |
2,207,865 |
|
|
$ |
(2,111 |
) |
|
$ |
(2,117,941 |
) |
|
$ |
87,828 |
|
The accompanying notes are an integral part of these consolidated financial statements.
70
NEKTAR THERAPEUTICS
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(96,692 |
) |
|
$ |
(153,524 |
) |
|
$ |
(81,177 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash royalty revenue related to sale of future royalties |
|
|
(30,531 |
) |
|
|
(30,158 |
) |
|
|
(22,058 |
) |
|
Non-cash interest expense on liability related to sale of future royalties |
|
|
18,869 |
|
|
|
19,712 |
|
|
|
20,619 |
|
|
Stock-based compensation |
|
|
36,615 |
|
|
|
25,850 |
|
|
|
19,669 |
|
|
Depreciation and amortization |
|
|
14,741 |
|
|
|
15,351 |
|
|
|
12,855 |
|
|
Loss from redemption premium and incremental interest on 12% senior secured notes |
|
|
— |
|
|
|
— |
|
|
|
12,500 |
|
|
Write-off of deferred financing costs on 12% senior secured notes |
|
|
— |
|
|
|
— |
|
|
|
1,579 |
|
|
Impairment of equipment from terminated program |
|
|
15,081 |
|
|
|
— |
|
|
|
— |
|
|
Other non-cash transactions |
|
|
(881 |
) |
|
|
(2,185 |
) |
|
|
(2,365 |
) |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable, net |
|
|
10,664 |
|
|
|
4,269 |
|
|
|
(16,340 |
) |
|
Inventory |
|
|
383 |
|
|
|
237 |
|
|
|
1,606 |
|
|
Other assets |
|
|
(4,800 |
) |
|
|
(312 |
) |
|
|
(825 |
) |
|
Accounts payable |
|
|
2,074 |
|
|
|
518 |
|
|
|
(412 |
) |
|
Accrued compensation |
|
|
(10,017 |
) |
|
|
12,282 |
|
|
|
249 |
|
|
Accrued clinical trial expenses |
|
|
1,503 |
|
|
|
(262 |
) |
|
|
512 |
|
|
Other accrued expenses |
|
|
5,774 |
|
|
|
191 |
|
|
|
(2,278 |
) |
|
Interest payable |
|
|
— |
|
|
|
— |
|
|
|
(2,719 |
) |
|
Liability related to refundable upfront payment |
|
|
(12,500 |
) |
|
|
12,500 |
|
|
|
— |
|
|
Deferred revenue |
|
|
(28,269 |
) |
|
|
(17,615 |
) |
|
|
(17,530 |
) |
|
Other liabilities |
|
|
(2,428 |
) |
|
|
(3,878 |
) |
|
|
3,032 |
|
|
Net cash used in operating activities |
|
|
(80,414 |
) |
|
|
(117,024 |
) |
|
|
(73,083 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of investments |
|
|
(404,425 |
) |
|
|
(334,659 |
) |
|
|
(297,608 |
) |
|
Maturities of investments |
|
|
347,743 |
|
|
|
253,682 |
|
|
|
226,923 |
|
|
Sales of investments |
|
|
37,549 |
|
|
|
4,969 |
|
|
|
42,544 |
|
|
Release of restricted cash |
|
|
— |
|
|
|
— |
|
|
|
25,000 |
|
|
Purchases of property, plant and equipment |
|
|
(9,676 |
) |
|
|
(6,392 |
) |
|
|
(11,195 |
) |
|
Net cash used in investing activities |
|
|
(28,809 |
) |
|
|
(82,400 |
) |
|
|
(14,336 |
) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Payment of capital lease obligations |
|
|
(5,131 |
) |
|
|
(5,945 |
) |
|
|
(5,187 |
) |
|
Proceeds from shares issued under equity compensation plans |
|
|
59,522 |
|
|
|
20,287 |
|
|
|
32,208 |
|
|
Issuance of common stock, net of issuance costs |
|
|
— |
|
|
|
189,276 |
|
|
|
— |
|
|
Proceeds from issuance of 7.75% senior secured notes, net of issuance costs |
|
|
— |
|
|
|
— |
|
|
|
241,262 |
|
|
Repayment of 12% senior secured notes |
|
|
— |
|
|
|
— |
|
|
|
(125,000 |
) |
|
Payment of redemption premium and incremental interest on 12% senior secured notes |
|
|
— |
|
|
|
— |
|
|
|
(12,500 |
) |
|
Net cash provided by financing activities |
|
|
54,391 |
|
|
|
203,618 |
|
|
|
130,783 |
|
|
Effect of exchange rates on cash and cash equivalents |
|
|
(46 |
) |
|
|
(124 |
) |
|
|
(159 |
) |
|
Net increase (decrease) in cash and cash equivalents |
|
|
(54,878 |
) |
|
|
4,070 |
|
|
|
43,205 |
|
|
Cash and cash equivalents at beginning of year |
|
|
59,640 |
|
|
|
55,570 |
|
|
|
12,365 |
|
|
Cash and cash equivalents at end of year |
|
$ |
4,762 |
|
|
$ |
59,640 |
|
|
$ |
55,570 |
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
20,116 |
|
|
$ |
20,589 |
|
|
$ |
20,225 |
|
|
Cash paid for income taxes |
|
$ |
556 |
|
|
$ |
757 |
|
|
$ |
860 |
|
The accompanying notes are an integral part of these consolidated financial statements.
71
NEKTAR THERAPEUTICS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2017
Note 1 — Organization and Summary of Significant Accounting Policies
Organization
We are a research-based biopharmaceutical company headquartered in San Francisco, California and incorporated in Delaware. We are developing a pipeline of drug candidates that utilize our advanced polymer conjugate technology platforms, which are designed to enable the development of new molecular entities that target known mechanisms of action. Our research and development pipeline of new investigational drugs includes treatments for cancer, autoimmune disease and chronic pain.
Our research and development activities have required significant ongoing investment to date and are expected to continue to require significant investment. As a result, we expect to continue to incur substantial losses and negative cash flows from operations in the future. We have financed our operations primarily through cash generated from licensing, collaboration and manufacturing agreements and financing transactions. At December 31, 2017, we had approximately $353.2 million in cash and investments in marketable securities and had debt of $250.0 million in principal of senior secured notes due in October 2020.
Basis of Presentation, Principles of Consolidation and Use of Estimates
Our consolidated financial statements include the financial position, results of operations and cash flows of our wholly-owned subsidiaries: Nektar Therapeutics (India) Private Limited and Nektar Therapeutics UK Limited. All intercompany accounts and transactions have been eliminated in consolidation.
Our Consolidated Financial Statements are denominated in U.S. dollars. Accordingly, changes in exchange rates between the applicable foreign currency and the U.S. dollar will affect the translation of each foreign subsidiary’s financial results into U.S. dollars for purposes of reporting our consolidated financial results. Translation gains and losses are included in accumulated other comprehensive income (loss) in the stockholders’ equity section of the Consolidated Balance Sheets. To date, such cumulative translation adjustments have not been significant to our consolidated financial position. Aggregate gross foreign currency transaction gains (losses) recorded in operations for the years ended December 31, 2017, 2016, and 2015 were not significant.
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Accounting estimates and assumptions are inherently uncertain. Actual results could differ materially from those estimates and assumptions. Our estimates include those related to estimated selling prices of deliverables in collaboration agreements, estimated periods of performance, the net realizable value of inventory, the impairment of investments, the impairment of goodwill and long-lived assets, contingencies, accrued clinical trial expenses, estimated non-cash royalty revenue and non-cash interest expense from our liability related to our sale of future royalties, stock-based compensation, and ongoing litigation, among other estimates. We base our estimates on historical experience and on other assumptions that management believes are reasonable under the circumstances. These estimates form the basis for making judgments about the carrying values of assets and liabilities when these values are not readily apparent from other sources. As appropriate, estimates are assessed each period and updated to reflect current information and any changes in estimates will generally be reflected in the period first identified.
Reclassifications
Certain items previously reported in specific financial statement captions have been reclassified to conform to the current period presentation, including as a result of the adoption of new accounting guidance related to the classification of deferred tax assets described below. Such reclassifications do not materially impact previously reported revenue, operating loss, net loss, total assets, liabilities or stockholders’ equity.
Cash, Cash Equivalents, and Investments, and Fair Value of Financial Instruments
We consider all investments in marketable securities with an original maturity of three months or less when purchased to be cash equivalents. Investments in securities with remaining maturities of less than one year, or where our intent is to use the investments to fund current operations or to make them available for current operations, are classified as short-term investments. Investments in securities with remaining maturities of over one year are classified as long-term investments.
72
Investments are designated as available-for-sale and are carried at fair value, with unrealized gains and losses reported in stockholders’ equity as accumulated other comprehensive income (loss). The disclosed fair value related to our cash equivalents and investments is based on market prices from a variety of industry standard data providers and generally represent quoted prices for similar assets in active markets or have been derived from observable market data.
Interest and dividends on securities classified as available-for-sale, as well as amortization of premiums and accretion of discounts to maturity, are included in interest income. Realized gains and losses and declines in value of available-for-sale securities judged to be other-than-temporary, if any, are included in other income (expense). The cost of securities sold is based on the specific identification method.
Our cash, cash equivalents, short-term investments and long-term investments are exposed to credit risk in the event of default by the third parties that hold or issue such assets. Our cash, cash equivalents, short-term investments and long-term investments are held by financial institutions that management believes are of high credit quality. Our investment policy limits investments to fixed income securities denominated and payable in U.S. dollars such as U.S. government obligations, money market instruments and funds, and corporate bonds and places restrictions on maturities and concentrations by type and issuer.
Accounts Receivable and Significant Customer Concentrations
Our customers are primarily pharmaceutical and biotechnology companies that are located in the U.S. and Europe and with whom we have multi-year arrangements. Our accounts receivable balance contains billed and unbilled trade receivables from product sales, milestones, other contingent payments and royalties, as well as time and materials based billings from collaborative research and development agreements. When appropriate, we provide for an allowance for doubtful accounts by reserving for specifically identified doubtful accounts. We generally do not require collateral from our customers. We perform a regular review of our customers’ credit risk and payment histories, including payments made subsequent to year-end. We have not experienced significant credit losses from our accounts receivable. At December 31, 2017, four different customers represented 43%, 20%, 19% and 14%, respectively, of our accounts receivable. At December 31, 2016, three different customers represented 39%, 32%, and 13%, respectively, of our accounts receivable.
Inventory and Significant Supplier Concentrations
Inventory is generally manufactured upon receipt of firm purchase orders from our collaboration partners. Inventory includes direct materials, direct labor, and manufacturing overhead and cost is determined on a first-in, first-out basis. Inventory is valued at the lower of cost or net realizable value and defective or excess inventory is written down to net realizable value based on historical experience or projected usage. Inventory related to our research and development activities is expensed when purchased.
We are dependent on our suppliers and contract manufacturers to provide raw materials, drugs and devices of appropriate quality and reliability and to meet applicable contract and regulatory requirements. In certain cases, we rely on single sources of supply of one or more critical materials. Consequently, in the event that supplies are delayed or interrupted for any reason, our ability to develop and produce our drug candidates or our ability to meet our supply obligations could be significantly impaired, which could have a material adverse effect on our business, financial condition and results of operations.
Long-Lived Assets
Property, plant and equipment are stated at cost. Major improvements are capitalized, while maintenance and repairs are expensed when incurred. Manufacturing, laboratory and other equipment are depreciated using the straight-line method generally over estimated useful lives of three to ten years. Buildings are depreciated using the straight-line method generally over the estimated useful life of twenty years. Leasehold improvements are amortized using the straight-line method over the shorter of the estimated useful life or the remaining term of the lease.
Goodwill represents the excess of the price paid for another entity over the fair value of the assets acquired and liabilities assumed in a business combination. We are organized in one reporting unit and evaluate the goodwill for the Company as a whole. Goodwill has an indefinite useful life and is not amortized, but instead tested for impairment at least annually in the fourth quarter of each year using an October 1 measurement date.
We assess the impairment of long-lived assets whenever events or changes in business circumstances indicate that the carrying amounts of the assets may not be fully recoverable. When such events occur, we determine whether there has been an impairment in value by comparing the carrying value of the asset with its fair value, as measured by the anticipated undiscounted net cash flows associated with the asset. In the case of goodwill impairment, market capitalization is generally used as the measure of fair value. If an impairment in value exists, the asset is written down to its estimated fair value.
73
Revenue Recognition
Our revenue is derived from our arrangements with pharmaceutical and biotechnology collaboration partners and may result from one or more of the following: upfront and license fees, payments for contract research and development, milestone and other contingent payments, manufacturing and supply payments, and royalties. Our performance obligations under our collaborations may include licensing our intellectual property, manufacturing and supply obligations, and research and development obligations. In order to account for the multiple-element arrangements, we identify the deliverables included within the arrangement and evaluate which deliverables represent separate units of accounting. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver goods or services, a right or license to use an asset, or another performance obligation. Revenue is recognized separately for each identified unit of accounting when the basic revenue recognition criteria are met: there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured.
At the inception of each new multiple-element arrangement or the material modification of an existing multiple-element arrangement, we allocate all consideration received under multiple-element arrangements to all units of accounting based on the relative selling price method, generally based on our best estimate of selling price (ESP). The objective of ESP is to determine the price at which we would transact a sale if the product or service was sold on a stand-alone basis. We determine ESP for the elements in our collaboration arrangements by considering multiple factors including, but not limited to, technical complexity of the performance obligation and similarity of elements to those performed under previous arrangements. Since we apply significant judgment in arriving at the ESPs, any material change in our estimates would significantly affect the allocation of the total consideration to the different elements of a multiple element arrangement.
Product sales
Product sales are primarily derived from fixed price manufacturing and supply agreements with our collaboration partners. Our products are tested for adherence to technical specifications prior to shipment, accordingly, we have not experienced any significant returns from our customers.
Royalty revenue
Generally, we are entitled to royalties from our collaboration partners based on the net sales of their approved drugs that are marketed and sold in one or more countries where we hold royalty rights. We recognize royalty revenue when the cash is received or when the royalty amount to be received is estimable and collection is reasonably assured. With respect to the non-cash royalties related to our sale of future royalties described in Note 7, revenue is recognized when estimable, otherwise, revenue is recognized during the period in which the related royalty report is received, which generally occurs in the quarter after the applicable product sales are made.
License, collaboration and other revenue
The amount of upfront fees and other payments received by us in license and collaboration arrangements that are allocated to continuing performance obligations, such as manufacturing and supply obligations, is deferred and generally recognized ratably over our expected performance period under each respective arrangement. We make our best estimate of the period over which we expect to fulfill our performance obligations, which may include technology transfer assistance, research activities, clinical development activities, and manufacturing activities from research and development through the commercialization of the product. Given the uncertainties of these collaboration arrangements, significant judgment is required to determine the duration of the performance period and this estimate is periodically re-evaluated. Upon the termination of each of our license and collaboration arrangements and if no additional services are required to be performed by us, all non-refundable, non-creditable amounts previously deferred from the arrangement are recognized as revenue.
Contingent consideration received from the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved, which we believe is consistent with the substance of our performance under our various license and collaboration agreements. A milestone is defined as an event (i) that can only be achieved based in whole or in part either on the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved, and (iii) that would result in additional payments being due to the entity. A milestone is substantive if the consideration earned from the achievement of the milestone is consistent with our performance required to achieve the milestone or the increase in value to the collaboration resulting from our performance, relates solely to our past performance, and is reasonable relative to all of the other deliverables and payments within the arrangement.
74
Our license and collaboration agreements with our partners provide for payments to us upon the achievement of development milestones, such as the completion of clinical trials or regulatory submissions, approvals by regulatory authorities, and commercial launches of drugs. Given the challenges inherent in developing and obtaining regulatory approval for drug products and in achieving commercial launches, there was substantial uncertainty whether any such milestones would be achieved at the time of execution of these licensing and collaboration agreements. In addition, we evaluated whether the development milestones met the remaining criteria to be considered substantive. As a result of our analysis, we consider our remaining development milestones under all of our license and collaboration agreements to be substantive and, accordingly, we recognize as revenue payments received from each milestone only if and as such milestone is achieved.
Our license and collaboration agreements with certain partners also provide for contingent payments to us based solely upon the performance of the respective partner. For such contingent amounts, we recognize the payments as revenue when earned under the applicable contract, which is generally upon completion of performance by the respective partner, provided that collection is reasonably assured.
Our license and collaboration agreements with our partners also provide for payments to us upon the achievement of specified sales volumes of approved drugs. We consider these payments to be similar to royalty payments and we recognize such sales-based payments upon achievement of such sales volumes, provided that collection is reasonably assured.
Shipping and Handling Costs
We recognize costs related to shipping and handling of product to customers in cost of goods sold.
Research and Development Expense
Research and development costs are expensed as incurred and include salaries, benefits and other operating costs such as outside services, supplies and allocated overhead costs. We perform research and development for our proprietary drug candidates and technology development and for certain third parties under collaboration agreements. For our proprietary drug candidates and our internal technology development programs, we invest our own funds without reimbursement from a third party. Where we perform research and development activities under a clinical joint development collaboration, such as our collaboration with Bristol-Myers Squibb, we record the cost reimbursement from our partner as a reduction to research and development expense when reimbursement amounts are due to us under the agreement.
We record accruals for the estimated costs of our clinical trial activities performed by third parties. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows to our vendors. Payments under the contracts depend on factors such as the achievement of certain events, successful enrollment of patients, and completion of certain clinical trial activities. We generally accrue costs associated with the start-up and reporting phases of the clinical trials ratably over the estimated duration of the start-up and reporting phases. We generally accrue costs associated with the treatment phase of clinical trials based on the total estimated cost of the treatment phase on a per patient basis and we expense the per patient cost ratably over the estimated patient treatment period based on patient enrollment in the trials. In specific circumstances, such as for certain time-based costs, we recognize clinical trial expenses using a methodology that we consider to be more reflective of the timing of costs incurred. Advance payments for goods or services that will be used or rendered for future research and development activities are capitalized as prepaid expenses and recognized as expense as the related goods are delivered or the related services are performed. We base our estimates on the best information available at the time. However, additional information may become available to us which may allow us to make a more accurate estimate in future periods. In this event, we may be required to record adjustments to research and development expenses in future periods when the actual level of activity becomes more certain. Such increases or decreases in cost are generally considered to be changes in estimates and will be reflected in research and development expenses in the period identified.
Stock-Based Compensation
Stock-based compensation arrangements include stock option grants and restricted stock unit (RSU) awards under our equity incentive plans, as well as shares issued under our Employee Stock Purchase Plan (ESPP), through which employees may purchase our common stock at a discount to the market price.
We use the Black-Scholes option pricing model for the respective grant to determine the estimated fair value of the option on the date of grant (grant date fair value) and the estimated fair value of common stock purchased under the ESPP. The Black-Scholes option pricing model requires the input of highly subjective assumptions. These variables include, but are not limited to, our stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors. Because our employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect the fair value estimate, in management’s opinion, the existing models may not provide a reliable
75
single measure of the fair value of our employee stock options or common stock purchased under the ESPP. The fair value of an RSU is equal to the closing price of our common stock on the grant date. Management will continue to assess the assumptions and methodologies used to calculate the estimated fair value of stock-based compensation. Circumstances may change and additional data may become available over time, which could result in changes to these assumptions and methodologies, and which could materially impact our fair value determination.
We expense the value of the portion of the option or award on a straight line basis over the requisite service periods in our Consolidated Statements of Operations and recognize forfeitures of options and awards as they occur. For options and awards that vest upon the achievement of performance milestones, we estimate the vesting period based on our evaluation of the probability of achievement of each respective milestone and the related estimated date of achievement. Stock-based compensation expense for purchases under the ESPP is recognized over the respective six-month purchase period. Expense amounts are recorded in cost of goods sold, research and development expense, and general and administrative expense based on the function of the applicable employee. Stock-based compensation charges are non-cash charges and as such have no impact on our reported cash flows.
Net Loss Per Share
Basic net loss per share is calculated based on the weighted-average number of common shares outstanding during the periods presented. For all periods presented in the Consolidated Statements of Operations, basic and diluted net loss per share are the same due to our net losses and the requirement to exclude potentially dilutive securities which would have an anti-dilutive effect on net loss per share. During 2017, 2016 and 2015, potentially dilutive securities consisted of common shares underlying outstanding stock options and RSUs. There were weighted average outstanding stock options and RSUs of 20.6 million, 19.9 million and 21.1 million during the years ended December 31, 2017, 2016 and 2015, respectively.
Income Taxes
We account for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on differences between the financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain. We record a valuation allowance against deferred tax assets to reduce their carrying value to an amount that is more likely than not to be realized. When we establish or reduce the valuation allowance related to the deferred tax assets, our provision for income taxes will increase or decrease, respectively, in the period such determination is made.
We utilize a two-step approach to recognize and measure uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained upon tax authority examination, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount of benefit, determined on a cumulative probability basis, that is more than 50% likely of being realized upon ultimate settlement.
Comprehensive loss
Comprehensive loss is the change in stockholders’ equity from transactions and other events and circumstances other than those resulting from investments by stockholders and distributions to stockholders. Our other comprehensive income (loss) is comprised of net loss, gains and losses from the foreign currency translation of the assets and liabilities of our India and UK subsidiaries, and unrealized gains and losses on investments in available-for-sale securities.
Adoption of New Accounting Principle
In March 2016, the Financial Accounting Standards Board (FASB) issued guidance to simplify several aspects of employee share-based payment accounting, including forfeitures, income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. This guidance was effective for our interim and annual periods beginning January 1, 2017. As a result of the adoption of this guidance, as of January 1, 2017, we recorded a $0.2 million charge to our accumulated deficit in our Consolidated Balance Sheet related to our election to recognize forfeitures of awards as they occur. In addition, prior to adoption of this guidance, tax attributes related to stock option windfall deductions were not recorded until they resulted in a reduction of cash tax payable. As of December 31, 2016, the excluded windfall deductions for federal and state purposes were $20.6 million and $9.8 million, respectively. Upon adoption, we recognized the excluded windfall deductions as a deferred tax asset on a tax-effected basis with a corresponding increase in the valuation allowance.
In November 2015, the FASB issued guidance to require that deferred tax assets and liabilities be classified as noncurrent on the balance sheet. Previous guidance required deferred tax assets and liabilities to be separated into current and noncurrent amounts on the
76
balance sheet. Accordingly, as of January 1, 2017, we reclassified $0.3 million from other current assets to our other assets balance. This reclassification was applied retrospectively to these balances in our Consolidated Balance Sheet as of December 31, 2016.
Recent Accounting Pronouncements
In May 2014, the FASB issued guidance codified in Accounting Standards Codification (ASC) 606, Revenue Recognition — Revenue from Contracts with Customers, which supersedes the guidance in ASC 605, Revenue Recognition, and is effective for public companies for annual and interim periods beginning after December 15, 2017. The FASB has issued numerous updates that provide clarification on a number of specific issues as well as requiring additional disclosures. We plan to adopt the standard in the first quarter of 2018 using the modified retrospective method.
The new guidance requires the application of a five-step model to determine the amount and timing of revenue to be recognized and requires that we recognize revenue in a manner that reasonably reflects the delivery of our goods or services to customers in return for expected consideration. To achieve this core principle, the guidance provides the following steps: (1) identify the contract(s) with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation. Under ASC 606, companies may need to use more judgment and make more estimates than under ASC 605 in the application of this five-step process.
We are continuing to assess the impact of the new guidance on our accounting policies and procedures and are evaluating the new requirements as applied to existing collaboration agreements, both in terms of the cumulative adjustment to opening accumulated deficit and our subsequent recognition of revenue. Since each collaboration agreement is unique, we will separately assess each agreement (see Note 10 for a discussion of our existing collaboration agreements) under the new standard. We anticipate that the adoption of ASC 606 will have the following impact to our revenue recognition for a collaboration agreement:
(i) Changes in revenue recognition for distinct licenses of functional intellectual property may result in a timing difference of revenue recognition between the current literature and ASC 606. For certain of our arrangements, the value associated with the licenses and certain other deliverables have been assessed as one unit of accounting and recognized over a period of time in which services are rendered or made available to our customer pursuant to revenue recognition guidance in effect for such arrangements at the time such arrangements commenced. For certain other arrangements, under current ASC 605 guidance, we identified the value of the license as a separate unit of accounting and recognize revenue upon issuance of the license. Under ASC 606, we may continue to recognize revenue for such licenses upon delivery of the license.
(ii) For other consideration, including milestone payments or contingent payments from our collaboration partners, under our current accounting policy, we recognize such payments as revenue in the period that the payment-triggering event occurred or is achieved. The new revenue standard, however, may require us to recognize these payments before the payment-triggering event is completely achieved, subject to management’s assessment of whether it is probable that the triggering event will be achieved and that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved.
(iii) We will recognize revenue for sales-based royalties and commercial sales-based milestones in the period of the related sale based on estimates, rather than recording them as reported by the customer.
We currently estimate that, upon adoption on January 1, 2018, we will record an adjustment to decrease our accumulated deficit by approximately $11.0 million to $13.0 million for amounts that would be recognized as of December 31, 2017 under ASC 606, but are not recognized under the current ASC 605 guidance. This estimated adjustment primarily consists of approximately $11.0 million for the accrual of royalties due to us related to net sales in the three months ended December 31, 2017 that we would have recognized in 2018 under current guidance. The estimated adjustment also includes up to approximately $2.0 million related to the recognition of the unamortized portion of upfront payment amounts under ASC 606 that are required to be deferred as of December 31, 2017 under current guidance. We will finalize our assessments in the first quarter of 2018 as we adopt ASC 606.
In February 2016, the FASB issued guidance to amend a number of aspects of lease accounting, including requiring lessees to recognize almost all leases with a term greater than one year as a right-of-use asset and corresponding liability, measured at the present value of the lease payments. The guidance will become effective for us beginning in the first quarter of 2019 and is required to be adopted using a modified retrospective approach. Early adoption is permitted. We are currently evaluating the impact of the adoption of this standard.
77
Note 2 — Cash and Investments in Marketable Securities
Cash and investments in marketable securities, including cash equivalents, are as follows (in thousands):
|
|
|
Estimated Fair Value at |
|
|||||
|
|
|
December 31, 2017 |
|
|
December 31, 2016 |
|
||
|
Cash and cash equivalents |
|
$ |
4,762 |
|
|
$ |
59,640 |
|
|
Short-term investments |
|
|
291,370 |
|
|
|
329,462 |
|
|
Long-term investments |
|
|
57,088 |
|
|
|
— |
|
|
Total cash and investments in marketable securities |
|
$ |
353,220 |
|
|
$ |
389,102 |
|
We invest in liquid, high quality debt securities. Our investments in debt securities are subject to interest rate risk. To minimize the exposure due to an adverse shift in interest rates, we invest in securities with maturities of two years or less and maintain a weighted average maturity of one year or less. As of December 31, 2017, $57.1 million of our total investments had maturities between one and two years and, as of December 31, 2016, all of our investments had maturities of one year or less.
Gross unrealized gains and losses were not significant at either December 31, 2017 or 2016. During the years ended December 31, 2017, 2016 and 2015, we sold available-for-sale securities totaling $37.5 million, $5.0 million, and $42.5 million, respectively, and realized gains and losses were not significant in any of those periods.
Under the terms of our 7.75% senior secured notes due October 2020, we are required to maintain a minimum cash and investments in marketable securities balance of $60.0 million.
Our portfolio of cash and investments in marketable securities includes (in thousands):
|
|
|
|
|
|
|
Estimated Fair Value at |
|
|||||
|
|
|
Fair Value Hierarchy Level |
|
|
December 31, 2017 |
|
|
December 31, 2016 |
|
|||
|
Corporate notes and bonds |
|
|
2 |
|
|
$ |
216,253 |
|
|
$ |
156,044 |
|
|
Corporate commercial paper |
|
|
2 |
|
|
|
128,096 |
|
|
|
160,920 |
|
|
Obligations of U.S. government agencies |
|
|
2 |
|
|
|
2,977 |
|
|
|
13,749 |
|
|
Available-for-sale investments |
|
|
|
|
|
|
347,326 |
|
|
|
330,713 |
|
|
Money market funds |
|
|
1 |
|
|
|
302 |
|
|
|
51,104 |
|
|
Certificate of deposit |
|
N/A |
|
|
|
1,132 |
|
|
|
2,930 |
|
|
|
Cash |
|
N/A |
|
|
|
4,460 |
|
|
|
4,355 |
|
|
|
Total cash and investments in marketable securities |
|
|
|
|
|
$ |
353,220 |
|
|
$ |
389,102 |
|
|
Level 1 — |
Quoted prices in active markets for identical assets or liabilities. |
|
Level 2 — |
Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices for identical or similar assets or liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
|
Level 3 — |
Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
During the years ended December 31, 2017, 2016 and 2015 there were no transfers between Level 1 and Level 2 of the fair value hierarchy.
At December 31, 2017 and 2016, we had letter of credit arrangements in favor of a landlord and certain vendors totaling $1.1 million and $2.4 million, respectively. These letters of credit are secured by investments of similar amounts.
78
Note 3 — Inventory
Inventory consists of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Raw materials |
|
$ |
1,796 |
|
|
$ |
2,055 |
|
|
Work-in-process |
|
|
4,843 |
|
|
|
7,311 |
|
|
Finished goods |
|
|
4,087 |
|
|
|
1,743 |
|
|
Total inventory |
|
$ |
10,726 |
|
|
$ |
11,109 |
|
Note 4 — Property, Plant and Equipment
Property, plant and equipment consists of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Building and leasehold improvements |
|
$ |
81,444 |
|
|
$ |
75,137 |
|
|
Laboratory equipment |
|
|
31,214 |
|
|
|
27,424 |
|
|
Furniture, fixtures and other equipment |
|
|
27,800 |
|
|
|
26,590 |
|
|
Manufacturing equipment |
|
|
20,695 |
|
|
|
44,237 |
|
|
Depreciable property, plant and equipment at cost |
|
|
161,153 |
|
|
|
173,388 |
|
|
Less: accumulated depreciation and amortization |
|
|
(115,090 |
) |
|
|
(111,243 |
) |
|
Depreciable property, plant and equipment, net |
|
|
46,063 |
|
|
|
62,145 |
|
|
Construction-in-progress |
|
|
1,400 |
|
|
|
3,456 |
|
|
Property, plant and equipment, net |
|
$ |
47,463 |
|
|
$ |
65,601 |
|
Building and leasehold improvements include our manufacturing, research and development and administrative facilities and the related improvements to these facilities. Laboratory and manufacturing equipment include assets that support both our manufacturing and research and development efforts. Construction-in-progress includes assets being built to enhance our manufacturing and research and development efforts.
Depreciation and amortization expenses on property, plant and equipment, including depreciation of assets acquired through capital leases, for the years ended December 31, 2017, 2016, and 2015 was $12.6 million, $13.2 million, and $11.4 million, respectively.
In November 2017, Bayer announced that the Phase 3 Amikacin Inhale clinical program did not meet its primary endpoint or key secondary endpoints and, in December 2017, Bayer terminated our related collaboration agreement. Under this collaboration, we were responsible for the development, manufacturing and supply of our proprietary nebulizer device included in the Amikacin product and had acquired specific manufacturing equipment for this purpose. As a result of the termination of the program, in the three months ended December 31, 2017, we wrote off program specific manufacturing equipment with an original cost of $23.4 million and a net book value of $15.1 million. We expect to complete the disposal of this equipment in the first quarter of 2018. In addition, in the three months ended December 31, 2017, we incurred approximately $0.9 million of other program termination costs related to our manufacturing obligations.
Note 5 — Senior Secured Notes
On October 5, 2015, we completed the sale and issuance of $250.0 million in aggregate principal amount of 7.75% senior secured notes due 2020 (the “Notes”). The Notes are secured by a first-priority lien on substantially all of our assets and bear interest at a rate of 7.75% per annum payable in cash quarterly in arrears on January 15, April 15, July 15, and October 15 of each year. Interest is calculated based on actual days outstanding over a 360 day year. The Notes will mature on October 5, 2020, at which time the outstanding principal will be due and payable.
In connection with the issuance of the Notes, we paid fees and expenses of $8.9 million, of which $8.7 million of transaction and facility fees paid directly to the purchasers of the Notes and other direct issuance costs were capitalized as a debt discount and issuance costs and are recorded as a reduction to the senior secured notes, net liability balance in our Consolidated Balance Sheet. The unamortized balance of these costs is $4.8 million at December 31, 2017 and will be amortized to interest expense over the remainder of the five year term of the Notes.
79
On October 5, 2015, we used a portion of the proceeds from the Notes to redeem the $125.0 million in aggregate principal amount of 12.0% senior secured notes due in 2017 (12% Notes). In addition, on October 5, 2015, we paid $3.3 million of accrued interest on the 12% Notes and made a $12.5 million redemption payment, which includes $1.2 million of additional interest paid to the note holders at redemption. As a result, we received $100.3 million in net proceeds in connection with these transactions. In addition, as a result of these transactions, in the year ended December 31, 2015, we recognized a $14.1 million loss on extinguishment of our 12% Notes, which consists of the $11.3 million redemption premium and $1.2 million of incremental interest paid to the note holders and the write-off of $1.6 million of unamortized issuance costs.
The agreement, pursuant to which the Notes were issued, contains customary covenants, including covenants that limit or restrict our ability to incur liens, incur indebtedness, declare or pay dividends, redeem stock, issue preferred stock, make certain investments, merge or consolidate, make dispositions of assets, or enter into certain new businesses or transactions with affiliates, but do not contain covenants related to future financial performance. In particular, the Notes agreement requires us to maintain a minimum cash and investments in marketable securities balance of $60.0 million during the term of the Notes. The Notes agreement provides that, beginning on October 5, 2017, we may redeem some or all of the Notes, subject to certain prepayment premiums and conditions. If we experience certain change of control events, the holders of the Notes will have the right to require us to purchase all or a portion of the Notes at a purchase price in cash equal to 101% of the principal amount thereof, plus accrued and unpaid interest to the date of purchase. In addition, upon certain asset sales, we may be required to offer to use the net proceeds thereof to purchase some of the Notes at 100% of the principal amount thereof, plus accrued and unpaid interest to the date of purchase.
As of December 31, 2017, based on a discounted cash flow analysis using Level 3 inputs including financial discount rates, we believe the $250.0 million in principal amount of the Notes is consistent with its fair value.
Note 6 — Leases
Operating Leases
In August 2017, we entered into a Lease Agreement (the “Lease”) with ARE-San Francisco No. 19, LLC (ARE) and terminated our sublease with Pfizer, Inc., effectively extending our lease term from 2020 to 2030 for our 129,732 square foot corporate office and R&D facility located at 455 Mission Bay Boulevard, San Francisco, California (the “Mission Bay Facility”).
The term of the Lease commenced on September 1, 2017, and will expire January 31, 2030, subject to our right to extend the term of the Lease for two consecutive five-year periods. The monthly base rent for the Mission Bay Facility will escalate over the term of the Lease at various intervals. During the term of the Lease, we are responsible for paying our share of operating expenses specified in the Lease, including insurance costs and taxes. The Lease also obligates Nektar to rent from ARE a total of an additional approximately 23,000 square feet of space at the Mission Bay Facility at specified delivery dates. The Lease includes various covenants, indemnities, defaults, termination rights, security deposits and other provisions customary for lease transactions of this nature. In addition to the Lease, we have entered into other operating leases for locations in San Francisco and India.
We recognize rent expense on a straight-line basis over the lease period. For the year ended December 31, 2017, rent expense was approximately $4.7 million and for the years ended December 31, 2016, and 2015, rent expense was approximately $3.2 million in each year. As of December 31, 2017 and 2016, we had total deferred rent balances of $6.0 million and $6.6 million, respectively, which are recorded in other current and other long-term liabilities in our Consolidated Balance Sheets.
Our future minimum lease payments for our operating leases at December 31, 2017 are as follows (in thousands):
|
Year ending December 31, |
|
|
|
|
|
2018 |
|
$ |
5,965 |
|
|
2019 |
|
|
5,511 |
|
|
2020 |
|
|
5,884 |
|
|
2021 |
|
|
8,739 |
|
|
2022 |
|
|
9,024 |
|
|
2023 and thereafter |
|
|
71,127 |
|
|
Total future minimum lease payments |
|
$ |
106,250 |
|
Capital Leases
During the year ended December 31, 2017, we repaid the $5.1 million due under our capital leases on our Consolidated Balance Sheet as of December 31, 2016 and have no capital lease balances as of December 31, 2017.
80
Note 7 — Liability Related to the Sale of Future Royalties
On February 24, 2012, we entered into a Purchase and Sale Agreement (the Purchase and Sale Agreement) with RPI Finance Trust (RPI), an affiliate of Royalty Pharma, pursuant to which we sold, and RPI purchased, our right to receive royalty payments (the Royalty Entitlement) arising from the worldwide net sales, from and after January 1, 2012, of (a) CIMZIA®, under our license, manufacturing and supply agreement with UCB Pharma (UCB), and (b) MIRCERA®, under our license, manufacturing and supply agreement with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (together referred to as Roche). We received aggregate cash proceeds of $124.0 million for the Royalty Entitlement. As part of this sale, we incurred approximately $4.4 million in transaction costs, which will be amortized to interest expense over the estimated life of the Purchase and Sale Agreement. Although we sold all of our rights to receive royalties from the CIMZIA® and MIRCERA® products, as a result of our ongoing manufacturing and supply obligations related to the generation of these royalties, we will continue to account for these royalties as revenue, and we recorded the $124.0 million in proceeds from this transaction as a liability (Royalty Obligation) that will be amortized using the interest method over the estimated life of the Purchase and Sale Agreement.
The following table shows the activity within the liability account during the year ended December 31, 2017 and for the period from the inception of the royalty transaction on February 24, 2012 (inception) to December 31, 2017 (in thousands):
|
|
|
Year ended December 31, 2017 |
|
|
Period from inception to December 31, 2017 |
|
||
|
Liability related to the sale of future royalties—beginning balance |
|
$ |
108,584 |
|
|
$ |
— |
|
|
Proceeds from sale of future royalties |
|
|
— |
|
|
|
124,000 |
|
|
Payments from Nektar to RPI |
|
|
— |
|
|
|
(10,000 |
) |
|
Non-cash CIMZIA® and MIRCERA® royalty revenue |
|
|
(30,531 |
) |
|
|
(137,531 |
) |
|
Non-cash interest expense recognized |
|
|
18,869 |
|
|
|
120,453 |
|
|
Liability related to the sale of future royalties – ending balance |
|
|
96,922 |
|
|
|
96,922 |
|
|
Less: unamortized transaction costs |
|
|
(2,267 |
) |
|
|
(2,267 |
) |
|
Liability related to the sale of future royalties, net |
|
$ |
94,655 |
|
|
$ |
94,655 |
|
Pursuant to the Purchase and Sale Agreement, in March 2014 and March 2013, we were required to pay RPI $7.0 million and $3.0 million, respectively, as a result of worldwide net sales of MIRCERA® for the 12 month periods ended December 31, 2013 and 2012 not reaching certain minimum thresholds. The Purchase and Sale Agreement does not include any other potential payments related to minimum net sales thresholds and, therefore, we do not expect to make any further payments to RPI related to this agreement.
During the years ended December 31, 2017, 2016 and 2015, we recognized $30.5 million, $30.2 million, and $22.1 million, respectively, in non-cash royalties from net sales of CIMZIA® and MIRCERA®, and we recorded $18.9 million, $19.7 million and $20.6 million, respectively, of related non-cash interest expense.
As royalties are remitted to RPI from Roche and UCB, the balance of the Royalty Obligation will be effectively repaid over the life of the agreement. In order to determine the amortization of the Royalty Obligation, we are required to estimate the total amount of future royalty payments to be received by RPI. The sum of these amounts less the $124.0 million proceeds we received will be recorded as interest expense over the life of the Royalty Obligation. From inception until 2017, our estimate of this total interest expense resulted in an effective annual interest rate of approximately 17%. We periodically assess the estimated royalty payments to RPI from UCB and Roche and to the extent the amount or timing of such payments is materially different than our original estimates, we will prospectively adjust the amortization of the Royalty Obligation. During the three month period ended December 31, 2017, as a result of increases in the forecasted sales of CIMZIA®, our estimate of the effective annual interest rate over the life of the agreement increased to 17.6%, which results in a prospective interest rate of 21%.
There are a number of factors that could materially affect the amount and timing of royalty payments from CIMZIA® and MIRCERA®, most of which are not within our control. Such factors include, but are not limited to, changing standards of care, the introduction of competing products, manufacturing or other delays, biosimilar competition, intellectual property matters, adverse events that result in governmental health authority imposed restrictions on the use of the drug products, significant changes in foreign exchange rates as the royalties remitted to RPI are made in U.S. dollars (USD) while significant portions of the underlying sales of CIMZIA® and MIRCERA® are made in currencies other than USD, and other events or circumstances that could result in reduced royalty payments from CIMZIA® and MIRCERA®, all of which would result in a reduction of non-cash royalty revenues and the non-cash interest expense over the life of the Royalty Obligation. Conversely, if sales of CIMZIA® and MIRCERA® are more than
81
expected, the non-cash royalty revenues and the non-cash interest expense recorded by us would be greater over the term of the Royalty Obligation.
In addition, the Purchase and Sale Agreement grants RPI the right to receive certain reports and other information relating to the Royalty Entitlement and contains other representations and warranties, covenants and indemnification obligations that are customary for a transaction of this nature. To our knowledge, we are currently in compliance with these provisions of the Purchase and Sale Agreement; however, if we were to breach our obligations, we could be required to pay damages to RPI that are not limited to the purchase price we received in the sale transaction.
Note 8 — Commitments and Contingencies
Purchase Commitments
In the normal course of business, we enter into various firm purchase commitments related to contract manufacturing, clinical development and certain other items. As of December 31, 2017, these commitments were approximately $16.5 million, all of which are expected to be paid in 2018.
Legal Matters
From time to time, we are involved in lawsuits, arbitrations, claims, investigations and proceedings, consisting of intellectual property, commercial, employment and other matters, which arise in the ordinary course of business. We make provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are reviewed at least quarterly and adjusted to reflect the impact of settlement negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case. Litigation is inherently unpredictable. If any unfavorable ruling were to occur in any specific period, there exists the possibility of a material adverse impact on the results of our operations of that period and on our cash flows and liquidity.
On August 14, 2015, Enzon, Inc. filed a breach of contract complaint in the Supreme Court of the State of New York (Court) claiming damages of $1.5 million (plus interest) for unpaid licensing fees (the “Enzon Litigation”) through the date of the complaint. Enzon alleged that we failed to pay a post-patent expiration immunity fee related to one of the licenses. On June 26, 2017, we entered into a Second Amendment to the Cross-License and Option Agreement (Cross-License Agreement) with Enzon in which we agreed to pay Enzon a sum of $7.0 million to satisfy all past and future obligations of royalty payments pursuant to the Cross-License Agreement and to have the Enzon Litigation dismissed. The Enzon Litigation was dismissed with prejudice on June 30, 2017. We paid $3.5 million in June 2017 and the remaining $3.5 million in December 2017. Of the total $7.0 million consideration, $1.4 million represents our accrued royalty liability to Enzon related to commercial sales of certain products from January 2017 through June 2017 recorded in cost of goods sold for year ended December 31, 2017. In addition, $2.3 million was recorded as a prepaid royalty asset for estimated future commercial sales of certain products through the term of the applicable underlying Enzon patents expiring in March 2018, which Nektar will amortize over this period. As of December 31, 2017, the unamortized prepaid royalty balance is $0.6 million and is included in other current assets. We recorded the remaining $3.3 million of consideration in general and administrative expense in the year ended December 31, 2017. No liability was recorded for this matter on our Consolidated Balance Sheets as of December 31, 2016.
Royalty Expense
After the termination of our royalty obligations under the Enzon agreement discussed above, we will have no third party licenses that require us to pay royalties based on our sales of certain products and/or on our recognition of royalty revenue under certain of our collaboration agreements. Royalty expense, which is reflected in cost of goods sold in our Consolidated Statements of Operations, was approximately $3.1 million, $3.7 million, and $2.8 million for the years ended December 31, 2017, 2016, and 2015, respectively.
Foreign Operations
We operate in a number of foreign countries. As a result, we are subject to numerous local laws and regulations that can result in claims made by foreign government agencies or other third parties that are often difficult to predict even after the application of good faith compliance efforts.
Indemnification Obligations
During the course of our normal operating activities, we have agreed to certain contingent indemnification obligations as further described below. The term of our indemnification obligations is generally perpetual. There is generally no limitation on the potential amount of future payments we could be required to make under these indemnification obligations. To date, we have not incurred
82
significant costs to defend lawsuits or settle claims based on our indemnification obligations. If any of our indemnification obligations is triggered, we may incur substantial liabilities. Because the aggregate amount of any potential indemnification obligation is not a stated amount, the overall maximum amount of any such obligations cannot be reasonably estimated. No liabilities have been recorded for these obligations on our Consolidated Balance Sheets as of December 31, 2017 or 2016.
Indemnifications in Connection with Commercial Agreements
As part of our collaboration agreements with our partners related to the license, development, manufacture and supply of drugs based on our proprietary technologies and drug candidates, we generally agree to defend, indemnify and hold harmless our partners from and against third party liabilities arising out of the agreement, including product liability (with respect to our activities) and infringement of intellectual property to the extent the intellectual property is developed by us and licensed to our partners.
As part of the sale of our royalty interest in the CIMZIA® and MIRCERA® products, we and RPI made representations and warranties and entered into certain covenants and ancillary agreements which are supported by indemnity obligations. Additionally, as part of our pulmonary asset sale to Novartis in 2008, we and Novartis made representations and warranties and entered into certain covenants and ancillary agreements which are supported by an indemnity obligation. In the event it is determined that we breached certain of the representations and warranties or covenants and agreements made by us in any such agreements, we could incur substantial indemnification liabilities depending on the timing, nature, and amount of any such claims.
Indemnification of Underwriters and Initial Purchasers of our Securities
In connection with our sale of equity and senior secured debt securities, we have agreed to defend, indemnify and hold harmless our underwriters or initial purchasers, as applicable, as well as certain related parties from and against certain liabilities, including liabilities under the Securities Act of 1933, as amended.
Director and Officer Indemnifications
As permitted under Delaware law, and as set forth in our Certificate of Incorporation and our Bylaws, we indemnify our directors, executive officers, other officers, employees, and other agents for certain events or occurrences that may arise while in such capacity. The maximum potential amount of future payments we could be required to make under this indemnification is unlimited; however, we have insurance policies that may limit our exposure and may enable us to recover a portion of any future amounts paid. Assuming the applicability of coverage, the willingness of the insurer to assume coverage, and subject to certain retention, loss limits and other policy provisions, we believe any obligations under this indemnification would not be material, other than an initial $1.8 million per incident for merger and acquisition related claims, $1.8 million per incident for securities related claims and $0.5 million per incident for non-securities related claims retention deductible per our insurance policy. However, no assurances can be given that the covering insurers will not attempt to dispute the validity, applicability, or amount of coverage without expensive litigation against these insurers, in which case we may incur substantial liabilities as a result of these indemnification obligations.
Note 9 — Stockholders’ Equity
Preferred Stock
We have authorized 10,000,000 shares of Preferred Stock with each share having a par value of $0.0001. As of December 31, 2017 and 2016, no preferred shares are designated, issued or outstanding.
Common Stock
On October 24, 2016, we completed the issuance and sale of 14,950,000 shares of our common stock in an underwritten public offering with total proceeds of approximately $189.7 million after deducting the underwriting commissions and discounts of approximately $12.1 million. In addition, we incurred approximately $0.4 million in legal and accounting fees, filing fees, and other costs in connection with this offering.
83
Equity Compensation Plans
At December 31, 2017, we had 26,761,122 reserved shares of common stock, all of which are reserved for issuance under our equity compensation plans as summarized in the following table (share numbers in thousands):
|
Plan Category |
|
Number of Securities to be Issued Upon Exercise of Outstanding Options & Vesting of RSUs (a) |
|
|
Weighted- Average Exercise Price of Outstanding Options (b) |
|
|
Number of Securities Remaining Available for Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column(a) (c) |
|
|||
|
Equity compensation plans approved by security holders(1) |
|
|
20,999 |
|
|
$ |
20.55 |
|
|
|
5,360 |
|
|
Equity compensation plans not approved by security holders |
|
|
402 |
|
|
$ |
11.01 |
|
|
|
— |
|
|
Total |
|
|
21,401 |
|
|
$ |
20.35 |
|
|
|
5,360 |
|
|
(1) |
Includes shares of common stock available for future issuance under our ESPP as of December 31, 2017. |
2017 Performance Incentive Plan
Our 2017 Performance Incentive Plan (2017 Plan) was adopted by the Company’s board of directors (Board of Directors) on March 28, 2017 and was approved by our stockholders on June 14, 2017. On the date of approval, any shares of our common stock that were available for issuance under our 2012 Performance Incentive Plan (2012 Plan) ceased to be available for future grants.
Subject to the terms of the 2017 Plan, 8,300,000 shares of our common stock, reduced by the number of shares of common stock subject to awards granted under the 2012 Plan on or after March 31, 2017 and prior to the adoption of the 2017 Plan, were initially available for awards under the 2017 Plan. Shares issued in respect of any “full-value award” granted under the 2017 Plan will be counted against the share limit described in the preceding sentence as 1.5 shares for every one share actually issued in connection with the award. Shares that are subject to or underlie awards which expire or for any reason are cancelled or terminated, are forfeited, fail to vest, or for any other reason are not paid or delivered under the 2017 Plan or any Prior Plan (as defined below) will again be available for subsequent awards under the 2017 Plan (with any such shares subject to full-value awards increasing the 2017 Plan’s share limit based on the full-value award ratio described above or, in the case of an award granted under a Prior Plan, the full-value award ratio set forth in such Prior Plan). Notwithstanding the foregoing, shares that are exchanged by a participant or withheld by the Company to pay the exercise price of an award granted under the 2017 Plan, as well as any shares exchanged or withheld to satisfy the tax withholding obligations related to any award, will not be available for subsequent awards under the 2017 Plan.
The purpose of the 2017 Plan and our other incentive plans is to promote the success of the Company by providing an additional means for us to attract, motivate, retain and reward directors, officers, employees, and other eligible persons through the grant of awards. Equity-based awards are also intended to further align the interests of award recipients and our stockholders. The 2017 Plan authorizes stock options, stock appreciation rights, stock bonuses, restricted stock, performance stock, stock units, phantom stock or similar rights to purchase or acquire shares, and other forms of awards granted or denominated in our common stock or units of the Company’s common stock, as well as cash bonus awards. Members of the Board of Directors, officers or employees, certain consultants and advisors of the Company and its subsidiaries are eligible to receive awards under the 2017 Plan. Pursuant to the 2017 Plan, we granted or issued non-qualified stock options and RSUs to employees, officers, and non-employee directors during 2017. The requisite service period for stock options granted to our employees under the 2017 Plan as well as our Prior Plans is generally four years; the requisite service period for stock options granted to our directors is generally one year. The requisite service period for RSUs granted under the 2017 Plan and our Prior Plans is generally three years for employees and one year for directors.
The 2017 Plan will terminate on March 27, 2027, unless earlier terminated by the Board of Directors. The maximum term of a stock option or stock appreciation right under the 2017 Plan and our Prior Plans is eight years from the date of grant. The per share exercise price of an option generally may not be less than the fair market value of a share of the Company’s common stock on The NASDAQ Stock Market on the date of grant.
84
Other Equity Incentive Plans
In addition to the 2017 Plan, we have other equity incentive plans under which options and restricted stock units granted remain outstanding but no new options or restricted stock units may be granted either as a result of the effectiveness of the 2017 Plan or the expiration of such other plan. These other equity incentive plans include: (i) the 2012 Plan which was adopted by the Board of Directors on April 4, 2012 and approved by our stockholders on June 28, 2012, amended on June 16, 2015 by approval of our stockholders to make available for award grants 7,000,000 additional shares, and replaced by the 2017 Plan; (ii) the 2008 Equity Incentive Plan (2008 Plan) which was adopted by the Board of Directors on March 20, 2008 and approved by our stockholders on June 6, 2008; (iii) the 2000 Equity Incentive Plan (2000 Plan) which was adopted by the Board of Directors on April 19, 2000 by amending and restating our 1994 Equity Incentive Plan, and which expired on February 9, 2010; and (iv) the 1998 Non-Officer Equity Incentive Plan which was adopted by our Board of Directors on August 18, 1998, and amended and restated in its entirety and renamed the 2000 Non-Officer Equity Incentive Plan on June 6, 2000 (2000 Non-Officer Plan and collectively with the 2012 Plan, the 2008 Plan and the 2000 Plan, the Prior Plans).
Pursuant to the Prior Plans, we previously granted or issued incentive stock options to employees and officers and non-qualified stock options, rights to acquire restricted stock, restricted stock units, and stock bonuses to employees, officers, non-employee directors, and consultants. Pursuant to the 2000 Non-Officer Plan, we previously granted or issued non-qualified stock options, rights to acquire restricted stock and stock bonuses to employees and consultants who are neither officers nor directors of Nektar.
Employee Stock Purchase Plan
In February 1994, our Board of Directors adopted the Employee Stock Purchase Plan (ESPP) pursuant to section 423(b) of the Internal Revenue Code of 1986. Under the ESPP, 2,500,000 shares of our common stock have been authorized for issuance. The terms of the ESPP provide eligible employees with the opportunity to acquire an ownership interest in Nektar through participation in a program of periodic payroll deductions for the purchase of our common stock. Employees may elect to enroll or re-enroll in the ESPP on a semi-annual basis. Stock is purchased at 85% of the lower of the closing price on the first day of the enrollment period or the last day of the enrollment period.
401(k) Retirement Plan
We sponsor a 401(k) retirement plan whereby eligible employees may elect to contribute up to the lesser of 60% of their annual compensation or the statutorily prescribed annual limit allowable under Internal Revenue Service regulations. The 401(k) plan permits us to make matching contributions on behalf of all participants, up to a maximum of $4,000 per participant. For the years ended December 31, 2017, 2016, and 2015, we recognized $1.6 million, $1.1 million, and $0.9 million, respectively, of compensation expense in connection with our 401(k) retirement plan.
Change in Control Severance Plan
On December 6, 2006, our Board of Directors approved a Change of Control Severance Benefit Plan (CIC Plan). This CIC Plan has subsequently been amended a number of times by our Board of Directors with the most recent amendment occurring on April 5, 2011. The CIC Plan is designed to make certain benefits available to our eligible employees in the event of a change of control of Nektar and, following such change of control, an employee’s employment with us or a successor company is terminated in certain specified circumstances. We adopted the CIC Plan to support the continuity of the business in the context of a change of control transaction. The CIC Plan was not adopted in contemplation of any specific change of control transaction.
Under the CIC Plan, in the event of a change of control of Nektar and a subsequent termination of employment initiated by us or a successor company other than for Cause (as defined in the CIC Plan) or initiated by the employee for a Good Reason Resignation (as defined in the CIC Plan) in each case within twelve months following a change of control transaction, (i) the Chief Executive Officer would be entitled to receive cash severance pay equal to 24 months base salary plus annual target incentive pay, the extension of employee benefits over this severance period and the full acceleration of unvested outstanding equity awards, and (ii) our Senior Vice Presidents and Vice Presidents (including Principal Fellows) would each be entitled to receive cash severance pay equal to twelve months base salary plus annual target incentive pay, the extension of employee benefits over this severance period and the full acceleration of unvested outstanding equity awards. In the event of a change of control of Nektar and a subsequent termination of employment initiated by the Company or a successor company other than for Cause within twelve months following a change of control transaction, all other employees would each be entitled to receive cash severance pay equal to 6 months base salary plus a pro-rata portion of annual target incentive pay, the extension of employee benefits over this severance period and the full acceleration of each such employee’s unvested outstanding equity awards. Under the CIC Plan, as amended, non-employee directors would also be entitled to full acceleration of vesting of all outstanding stock awards in the event of a change of control transaction.
85
Note 10 — License and Collaboration Agreements
We have entered into various collaboration agreements including license agreements and collaborative research, development and commercialization agreements with various pharmaceutical and biotechnology companies. Under these collaboration arrangements, we are entitled to receive license fees, upfront payments, milestone and other contingent payments, royalties, sales milestone payments, and payments for the manufacture and supply of our proprietary PEGylation materials and/or for research and development activities. All of our collaboration agreements are generally cancelable by our partners without significant financial penalty. Our costs of performing these services are generally included in research and development expense, except that costs for product sales to our collaboration partners are included in cost of goods sold.
In accordance with our collaboration agreements, we recognized license, collaboration and other revenue as follows (in thousands):
|
|
|
|
|
Year Ended December 31, |
|
|||||||||
|
Partner |
|
Agreement |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Eli Lilly and Company |
|
NKTR-358 |
|
$ |
130,087 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Ophthotech Corporation(1) |
|
Fovista® |
|
|
19,123 |
|
|
|
1,408 |
|
|
|
1,398 |
|
|
Bayer Healthcare LLC(1) |
|
BAY41-6551 (Amikacin Inhale) |
|
|
17,931 |
|
|
|
1,429 |
|
|
|
1,919 |
|
|
Baxalta Incorporated / Shire |
|
ADYNOVATE® |
|
|
11,443 |
|
|
|
650 |
|
|
|
10,694 |
|
|
MAP Pharmaceuticals, Inc. / Allergan |
|
SEMPRANA® |
|
|
11,000 |
|
|
|
— |
|
|
|
— |
|
|
Amgen, Inc. |
|
Neulasta® |
|
|
5,000 |
|
|
|
5,000 |
|
|
|
5,000 |
|
|
AstraZeneca AB |
|
MOVANTIK® and MOVANTIK® fixed-dose combination program |
|
|
4,600 |
|
|
|
33,000 |
|
|
|
130,000 |
|
|
Daiichi Sankyo Europe GmbH(1) |
|
ONZEALDTM (NKTR-102) |
|
|
3,811 |
|
|
|
3,690 |
|
|
|
— |
|
|
Roche(1) |
|
PEGASYS® and MIRCERA® |
|
|
— |
|
|
|
7,685 |
|
|
|
12,816 |
|
|
Other |
|
|
|
|
7,970 |
|
|
|
7,520 |
|
|
|
3,777 |
|
|
License, collaboration and other revenue |
|
|
|
$ |
210,965 |
|
|
$ |
60,382 |
|
|
$ |
165,604 |
|
|
(1) |
These collaboration agreements were completed as of December 31, 2017. |
As of December 31, 2017, our collaboration agreements with partners included potential future payments for development milestones totaling approximately $305.5 million, including amounts from our agreements with Lilly and Baxalta described below. In addition, under our collaboration agreements we are entitled to receive other contingent payments, including contingent sales milestones and royalty payments, as described below.
86
Eli Lilly and Company (Lilly): NKTR-358
Effective August 23, 2017, we entered into a worldwide license agreement with Eli Lilly and Company (Lilly) to co-develop NKTR-358, a novel immunological drug candidate that we invented. Under the terms of the agreement we (i) received an initial payment of $150.0 million in September 2017 and are eligible for up to $250.0 million in additional development milestones, (ii) will co-develop NKTR-358 with Lilly with Nektar responsible for completing Phase 1 clinical development and certain drug product development and supply activities, (iii) will share with Lilly Phase 2 development costs with 75% of those costs borne by Lilly and 25% of the costs borne by Nektar, (iv) will have the option to contribute funding to Phase 3 development on an indication-by-indication basis ranging from zero to 25% of development costs, and (v) will have the opportunity to receive up to double-digit sales royalty rates that escalate based upon our Phase 3 development cost contribution and the level of annual global product sales. Lilly will be responsible for all costs of global commercialization, and we will have an option to co-promote in the U.S. under certain conditions. A portion of the development milestones may be reduced by 50% under certain conditions, related to the final formulation of the approved product and the timing of prior approval (if any) of competitive products with a similar mechanism of action, which could reduce these milestone payments by 75% if both conditions occur.
The agreement will continue until Lilly no longer has any royalty payment obligations to us or, if earlier, the termination of the agreement in accordance with its terms. The agreement may be terminated by Lilly for convenience, and may also be terminated under certain other circumstances, including material breach.
We identified our license grant to Lilly, our ongoing Phase 1 clinical development obligation, our drug product development obligation and our obligation to supply clinical trial materials as the significant, non-contingent deliverables under the agreement and concluded that each of these deliverables represents a separate unit of accounting. The valuation of each unit of accounting involves significant estimates and assumptions, including but not limited to, expected market opportunity and pricing, assumed royalty rates, clinical trial costs, timelines and likelihood of success; in each case these estimates and assumptions covering long time periods. We determined the best estimate of the selling price for the license based on a discounted cash flow analysis of projected revenues from NKTR-358 and development and commercial costs using a discount rate based on a market participant’s weighted average cost of capital adjusted for forecasting risk. We determined the best estimate of selling prices for Phase 1 clinical development, drug product development and clinical supply deliverables based on the nature of the services to be performed and estimates of the associated efforts and third-party rates for similar services.
Based on these estimates at agreement inception, the $150.0 million upfront payment was allocated $125.9 million to the license, $17.6 million to the Phase 1 clinical development and $6.5 million to the drug product development based on our estimate of their relative selling prices. We did not allocate any of the upfront arrangement consideration to the clinical trial material supply obligations as we will receive incremental consideration when we deliver such materials. We recognized license revenue upon the effective date of the arrangement in August 2017. We will recognize revenue related to Phase 1 clinical development and drug product development activities using the proportionate performance method as services are provided over the estimated service period, which we estimate will continue until the end of 2019. As a result, during the year ended December 31, 2017, we recognized $125.9 million of revenue related to the license and $4.2 million of revenue related to Phase 1 clinical development and drug product development activities. We recorded the remaining $19.9 million related to Phase 1 clinical development and drug product development activities as deferred revenue as of December 31, 2017.
Ophthotech Corporation: Fovista®
On October 27, 2017, we terminated our license and supply agreement with Ophthotech Corporation (Ophthotech) dated September 2006, pursuant to which Ophthotech received a worldwide, exclusive license to certain of our proprietary PEGylation technology to develop, manufacture and sell Fovista®. Under the terms of our agreement, we were the exclusive supplier of all of Ophthotech’s clinical and commercial requirements for our proprietary PEGylation reagent used in Fovista®. The termination of our agreement with Ophthotech followed Opthotech’s previous announcements, in December 2016 and August 2017, that their three pivotal Phase 3 studies investigating the superiority of Fovista® therapy in combination with Lucentis® therapy compared to Lucentis® monotherapy and evaluating Fovista® in combination with Eylea® or Avastin® compared to Eylea® or Avastin® monotherapy for the treatment of wet age-related macular degeneration (AMD) did not achieve the pre-specified primary endpoints.
Under our agreement with Ophthotech, in June 2014, we received a $19.8 million payment from Ophthotech in connection with its licensing agreement with Novartis. In addition, in January 2017, we received a $12.7 million advance payment from Ophthotech, which included $10.4 million for reagent shipments recognized in the second quarter of 2017 as well as approximately $2.3 million for 2017 minimum purchase requirements. As a result of the termination of this agreement, we recognized the remaining $18.0 million of deferred revenue from this arrangement in the three months ended December 31, 2017.
87
Bayer Healthcare LLC: BAY41-6551 (Amikacin Inhale)
In December 2017, Bayer Healthcare LLC (Bayer) terminated our co-development, license and co-promotion agreement entered into in August 2007 to develop a specially-formulated inhaled Amikacin. Under this agreement, we were responsible for development, manufacturing and supply of our proprietary nebulizer device included in the Amikacin product. Bayer was responsible for most clinical development and commercialization activities and costs, all activities and costs to support worldwide regulatory filings, approvals and related activities, further development of Amikacin Inhale and final product packaging and distribution. The termination of this agreement followed Bayer’s announcement in November 2017 that the Phase 3 Amikacin Inhale clinical program for the treatment of intubated and mechanically ventilated patients with Gram-negative pneumonia did not meet its primary endpoint or key secondary endpoints.
Under this collaboration, we received an upfront payment of $40.0 million (which was paid to us in 2007) and milestone payments totaling $30.0 million (the last of which was paid to us in 2013). As a result of the termination of the agreement, we recognized the remaining $16.8 million of deferred revenue related to this arrangement in the three months ended December 31, 2017.
Baxalta Incorporated: Hemophilia
We are a party to an exclusive research, development, license and manufacturing and supply agreement with Baxalta Incorporated (Baxalta), a subsidiary of Shire plc, entered into in September 2005 to develop products designed to improve therapies for Hemophilia A patients using our PEGylation technology. Under the terms of the agreement, we are entitled to research and development funding for our active programs, which are now complete for Factor VIII, and are responsible for supplying Baxalta with its requirements of our proprietary materials. Baxalta is responsible for all clinical development, regulatory, and commercialization expenses. The agreement is terminable by the parties under customary conditions.
This Hemophilia A program includes ADYNOVATE®, which was approved by the FDA in November 2015 for use in adults and adolescents, aged 12 years and older, who have Hemophilia A, and is now marketed in the U.S. As a result of the FDA’s approval, we earned a $10.0 million development milestone in November 2015. Under the terms of this agreement, we are also entitled to a $10.0 million development milestone due upon marketing authorization in the EU, which was achieved in January 2018. In addition, we are entitled to sales milestones upon achievement of annual sales targets and royalties based on annual worldwide net sales of products resulting from this agreement.
In October 2017, we entered into a right to sublicense agreement with Baxalta under which we granted to Baxalta the right to grant a nonexclusive sublicense to certain patents that were previously exclusively licensed to Baxalta under our 2005 agreement. Under the right to sublicense agreement, Baxalta paid us $12.0 million in November 2017 and agreed to pay us single digit royalty payments based upon net sales of the products covered under the sublicense throughout the term of the agreement.
We determined that this right to sublicense agreement should be considered a material modification of the existing arrangement described above. We have identified our grant of the right to sublicense and our ongoing obligation to supply PEGylation materials specified in the 2005 agreement as the undelivered units of accounting in the arrangement. We made our best estimates of the selling price of these deliverable and recognized the $11.0 million allocated to the right to sublicense and allocated $1.0 million to our ongoing supply obligation unit of accounting. As of December 31, 2017, we have deferred revenue of approximately $1.0 million related to this agreement, which we expect to recognize through January 2028, the estimated end of our supply obligations under the arrangement.
MAP Pharmaceuticals, Inc.: SEMPRANA®
In August 2006, we entered into a restated and amended license agreement with MAP Pharmaceuticals, Inc. (MAP) under which we granted a license to technology used by MAP in the development of its drug candidate SEMPRANA®, a potential treatment for chronic migraine headache. Under our agreement, we are entitled to development milestone payments, royalties and a share of sublicensing payments received by MAP. In January 2011, MAP announced a collaboration with Allergan, Inc. (Allergan) to co-promote SEMPRANA®, under which MAP has received upfront and sublicensing payments totaling $80.0 million from Allergan. In 2013, Allergan acquired MAP and MAP became a wholly-owned subsidiary of Allergan.
In January 2015, we filed a breach of contract action against Allergan and MAP. In December 2017, we, Allergan and MAP entered into a Settlement Agreement and Release. Pursuant to this agreement, Allergan paid Nektar $15.0 million in December 2017 in exchange for relief of any claims related to our share of payments received by MAP from Allergan as well as a specified development milestone payment due to Nektar. We have no continuing performance obligations under the MAP agreement. As a result, in the year ended December 31, 2017, we recognized $11.0 million as license revenue related to our share of payments due to us under the MAP agreement and recorded the remaining $4.0 million, which primarily represents the reimbursement of legal
88
expenses, as a gain on settlement recorded in general and administrative expenses. As of December 31, 2017, we have no deferred revenue related to this agreement.
Amgen, Inc.: Neulasta®
In October 2010, we amended and restated an existing supply and license agreement by entering into a supply, dedicated suite and manufacturing guarantee agreement (the amended and restated agreement) and a license agreement with Amgen Inc. and Amgen Manufacturing, Limited (together referred to as Amgen). Under the terms of the amended and restated agreement, we guarantee the manufacture and supply of our proprietary PEGylation materials (Polymer Materials) to Amgen in an existing manufacturing suite to be used exclusively for the manufacture of Polymer Materials for Amgen (the Manufacturing Suite) in our manufacturing facility in Huntsville, Alabama (the Facility). This supply arrangement is on a non-exclusive basis (other than the use of the Manufacturing Suite and certain equipment) whereby we are free to manufacture and supply the Polymer Materials to any other third party and Amgen is free to procure the Polymer Materials from any other third party. Under the terms of the amended and restated agreement, we received a $50.0 million payment in the fourth quarter of 2010 in return for our guaranteeing the supply of certain quantities of Polymer Materials to Amgen including without limitation the Additional Rights described below and manufacturing fees that are calculated based on fixed and variable components applicable to the Polymer Materials ordered by Amgen and delivered by us. Amgen has no minimum purchase commitments. If quantities of the Polymer Materials ordered by Amgen exceed specified quantities, significant additional payments become payable to us in return for our guaranteeing the supply of additional quantities of the Polymer Materials.
The term of the amended and restated agreement ends on October 29, 2020. In the event we become subject to a bankruptcy or insolvency proceeding, we cease to own or control the Facility, we fail to manufacture and supply or certain other events, Amgen or its designated third party will have the right to elect, among certain other options, to take title to the dedicated equipment and access the Facility to operate the Manufacturing Suite solely for the purpose of manufacturing the Polymer Materials. Amgen may terminate the amended and restated agreement for convenience or due to an uncured material default by us.
As of December 31, 2017, we have deferred revenue of approximately $14.2 million related to this agreement, which we expect to recognize through October 2020, the estimated end of our obligations under this agreement.
AstraZeneca AB: MOVANTIK® (naloxegol oxalate), previously referred to as naloxegol and NKTR-118, and MOVANTIK® fixed-dose combination program, previously referred to as NKTR-119
In September 2009, we entered into an agreement with AstraZeneca AB (AstraZeneca) under which we granted AstraZeneca a worldwide, exclusive license under our patents and other intellectual property to develop, market, and sell MOVANTIK® and MOVANTIK® fixed-dose combination program. AstraZeneca is responsible for all research, development and commercialization and is responsible for all drug development and commercialization decisions for MOVANTIK® and the MOVANTIK® fixed-dose combination program. AstraZeneca paid us an upfront payment of $125.0 million, which we received in the fourth quarter of 2009 and which was fully recognized as of December 31, 2010. In addition, we have received the payments described further below based on development events related to MOVANTIK® completed solely by AstraZeneca. We are entitled to receive up to $75.0 million of commercial launch contingent payments related to the MOVANTIK® fixed-dose combination program, based on development events to be pursued and completed solely by AstraZeneca. In addition, we are entitled to significant and escalating double-digit royalty payments and sales milestone payments based on annual worldwide net sales of MOVANTIK® and MOVANTIK® fixed-dose combination products.
On September 16, 2014, the United States Food and Drug Administration (FDA) approved MOVANTIK® for the treatment of opioid-induced constipation (OIC) in adult patients with chronic, non-cancer pain. On December 9, 2014, AstraZeneca announced that MOVENTIG® (the naloxegol brand name in the European Union or EU) had been granted Marketing Authorisation by the European Commission (EC) for the treatment of opioid-induced constipation (OIC) in adult patients who have had an inadequate response to laxative(s). In March 2015, we received and recognized a $100.0 million non-refundable payment resulting from the U.S. commercial launch of MOVANTIK®. In March 2015, we agreed to pay AstraZeneca a total of $10.0 million to fund U.S. television advertising in consideration for certain additional commercial information rights. We recorded this $10.0 million obligation as a liability and made the initial $5.0 million payment to AstraZeneca in July 2015 and the remaining $5.0 million payment in July 2016. We determined that this $10.0 million obligation should be recorded as a reduction of revenue, which we recorded in 2015. In August 2015, we received and recognized as revenue an additional $40.0 million non-refundable payment triggered by the first commercial sale of MOVENTIG® in Germany.
On March 1, 2016, AstraZeneca announced that it had entered into an agreement with ProStrakan Group plc, a subsidiary of Kyowa Hakko Kirin Co. Ltd. (Kirin), granting Kirin exclusive marketing rights to MOVENTIG® in the EU, Iceland, Liechtenstein, Norway and Switzerland. Under the terms of AstraZeneca’s agreement with Kirin, Kirin made a $70.0 million upfront payment to AstraZeneca and will make additional payments based on achieving market access milestones, tiered net sales royalties, as well as sales milestones. Under our license agreement with AstraZeneca, we will share the upfront payment, market access milestones,
89
royalties and sales milestones from Kirin with AstraZeneca receiving 60% and Nektar receiving 40%. This payment sharing arrangement is in lieu of other royalties payable by AstraZeneca to us and a portion of the sales milestones as described below. Our 40% share of royalty payments made by Kirin to AstraZeneca will be financially equivalent to us receiving high single-digit to low double-digit royalties dependent on the level of Kirin’s net sales. Kirin’s MOVENTIG® net sales will be included for purposes of achieving the annual global sales milestones payable to us by AstraZeneca and will also be included for purposes of determining the applicable ex-U.S. royalty rate, from the tier schedule in our AstraZeneca license agreement, that will be applied to ex-U.S. sales outside of the Kirin territory. The global sales milestones under our license agreement with AstraZeneca will be reduced in relation to the amount of Kirin MOVENTIG® net sales that contribute to any given annual sales milestone target. As a result, in April 2016, we received 40% (or $28.0 million) of the $70.0 million payment received by AstraZeneca from Kirin. In the years ended December 31, 2017 and 2016, we recognized a total of $4.6 million and $33.0 million, respectively, related to our share of such license-related payments made to AstraZeneca. As of December 31, 2017, we do not have deferred revenue related to our agreement with AstraZeneca.
Daiichi Sankyo Europe GmbH: ONZEALDTM (etirinotecan pegol), also referred to as NKTR-102
In December 2017, Daiichi Sankyo Europe GmbH (Daiichi) terminated our collaboration and license agreement made effective in May 2016, under which we granted Daiichi exclusive commercialization rights in the European Economic Area, Switzerland, and Turkey (collectively, the European Territory) to ONZEALDTM (etirinotecan pegol). Daiichi was responsible for all commercialization activities in the European Territory and we were responsible for supplying Daiichi with ONZEALDTM and for funding and conducting a Phase 3 confirmatory trial in patients with advanced breast cancer who have brain metastases, which was initiated in the fourth quarter of 2016. Daiichi terminated the agreement after the European Medicines Agency’s Committee for Medicinal Products for Human Use confirmed in November 2017 that it declined the conditional marketing authorization application for ONZEALDTM in the EU.
Under the terms of the agreement, Daiichi paid us a $20.0 million up-front payment in August 2016, of which $12.5 million was refundable to Daiichi if conditional approval was not achieved and the agreement was terminated. This $12.5 million contingent termination payment from us to Daiichi was recorded in our liability related to refundable upfront payment balance in our Consolidated Balance Sheet at December 31, 2016 and was paid to Daiichi in December 2017.
In 2016, we allocated the $7.5 million non-refundable portion of the $20.0 million upfront payment from Daiichi to the license grant and our development service deliverables based on their relative estimated selling prices. As a result, we recognized $3.7 million of revenue in 2016 from this arrangement, primarily related to the delivery of the license. As a result of the termination of the arrangement, we recognized the remaining $3.2 million of deferred revenue in the three months ended December 31, 2017.
Roche: PEGASYS® and MIRCERA®
In February 2012, we entered into a toll-manufacturing agreement with Roche under which we agreed to manufacture the proprietary PEGylation material used by Roche to produce MIRCERA®. Roche entered into the toll-manufacturing agreement with the objective of establishing us as a secondary back-up supply source on a non-exclusive basis. Under the terms of our toll-manufacturing agreement, Roche paid us a total of $27.0 million, including amounts for the delivery of specified quantities of PEGylation materials, all of which were completed as of January 2013. In addition, in 2013, we delivered additional quantities of PEGylation materials used by Roche to produce PEGASYS® and MIRCERA® for total consideration of $18.6 million. As of December 31, 2016, we no longer had any continuing manufacturing or supply obligations under this MIRCERA® agreement and, therefore, we have no deferred revenue related to this agreement.
In February 1997, we entered into a license, manufacturing and supply agreement with Roche, under which we granted Roche a worldwide, exclusive license to certain intellectual property related to our proprietary PEGylation materials used in the manufacture and commercialization of PEGASYS®. Our performance obligations under this PEGASYS® agreement ended on December 31, 2015.
Bristol-Myers Squibb: NKTR-214
On September 21, 2016, we entered into a Clinical Trial Collaboration Agreement (Clinical Trial Agreement) with Bristol-Myers Squibb Company (BMS), pursuant to which we and BMS collaborated to conduct Phase 1/2 clinical trials evaluating our IL-2-based CD122-biased agonist, known as NKTR-214, and BMS’ human monoclonal antibody that binds PD-1, known as Opdivo® (nivolumab), as a potential combination treatment regimen in five tumor types and eight indications, and such other clinical trials evaluating the combined therapy as may be mutually agreed upon by the parties (each, a Combination Therapy Trial).
We acted as the sponsor of each Combination Therapy Trial. Under the Clinical Trial Agreement, BMS was responsible for 50% of all out-of-pocket costs reasonably incurred in connection with third party contract research organizations, laboratories, clinical
90
sites and institutional review boards. We recorded cost reimbursement payments to us from BMS as a reduction to research and development expense and we have recorded a total of $7.8 million of such reimbursements in the year ended December 31, 2017 and since the inception of our collaboration. Each party was otherwise responsible for its own internal costs, including internal personnel costs, incurred in connection with each Combination Therapy Trial. Nektar and BMS used commercially reasonable efforts to manufacture and supply NKTR-214 and Opdivo® (nivolumab), respectively, for each Combination Therapy Trial with each party bearing its own costs related thereto.
Ownership of, and global commercial rights to, NKTR-214 remain solely with us under the Clinical Trial Agreement. If we wish to license the right to commercialize NKTR-214 in one of certain major market territories prior to September 30, 2018 (Exclusivity Expiration Date), we must first negotiate with BMS, for a period of three months (Negotiation Period), to grant an exclusive license to develop and commercialize NKTR-214 in any of these major market territories. If we do not reach an agreement with BMS for an exclusive license within the Negotiation Period, we will be free to license any right to NKTR-214 to other parties in any territory worldwide except that in the event that we receive a license offer from a third party during a period of 90 calendar days after the end of the Negotiation Period, we will provide BMS ten business days to match the terms of such third-party offer. After the Exclusivity Expiration Date, we are free to license NKTR-214 without any further obligation to BMS. Each party grants to the other party a non-exclusive, worldwide (subject to certain exceptions in the case of the license granted by BMS), non-transferable and royalty-free research and development license to such licensing party’s patent rights, technology and regulatory documentation to use its compound solely to the extent necessary to discharge its obligations under the Clinical Trial Agreement with respect to the conduct of the Combination Therapy Trials.
The Clinical Trial Agreement will be superseded and replaced by the Strategic Collaboration Agreement entered into between us and BMS on February 13, 2018 (BMS Collaboration Agreement), which is summarized in Note 14, if and when the BMS Collaboration Agreement becomes effective.
Other
In addition, as of December 31, 2017, we have a number of other collaboration agreements, including with our collaboration partners UCB and Halozyme, under which we are entitled to up to a total of $45.5 million of development milestones upon achievement of certain development objectives, as well as sales milestones upon achievement of annual sales targets and royalties based on net sales of commercialized products, if any. However, given the current phase of development of the potential products under these collaboration agreements, we cannot estimate the probability or timing of achieving these milestones. As of December 31, 2017, we have deferred revenue of approximately $2.9 million related to these other collaboration agreements, which we expect to recognize through 2020, the estimated end of our obligations under those agreements.
Note 11 — Stock-Based Compensation
We issue stock-based awards from our equity incentive plans, which are more fully described in Note 9. Stock-based compensation expense was recognized as follows (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cost of goods sold |
|
$ |
2,333 |
|
|
$ |
1,620 |
|
|
$ |
1,142 |
|
|
Research and development |
|
|
21,252 |
|
|
|
13,093 |
|
|
|
9,207 |
|
|
General and administrative |
|
|
13,030 |
|
|
|
11,137 |
|
|
|
9,320 |
|
|
Total stock-based compensation |
|
$ |
36,615 |
|
|
$ |
25,850 |
|
|
$ |
19,669 |
|
As of December 31, 2017, total unrecognized compensation costs of $211.8 million related to unvested stock-based compensation arrangements are expected to be recognized as expense over a weighted-average period of 2.1 years.
91
Black-Scholes Assumptions
The following table lists the Black-Scholes option-pricing model assumptions used to calculate the fair value of employee and director stock options:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Average risk-free interest rate |
|
|
2.0 |
% |
|
|
1.4 |
% |
|
|
1.6 |
% |
|
Dividend yield |
|
|
0.0 |
% |
|
|
0.0 |
% |
|
|
0.0 |
% |
|
Average volatility factor |
|
|
54.2 |
% |
|
|
51.7 |
% |
|
|
50.8 |
% |
|
Average weighted average expected life |
|
5.3 years |
|
|
5.3 years |
|
|
5.3 years |
|
|||
The average risk-free interest rate is based on the U.S. treasury yield curve in effect at the time of grant for periods commensurate with the expected life of the stock-based award. We have never paid dividends, nor do we expect to pay dividends in the foreseeable future; therefore, we used a dividend yield of 0.0%. Our estimate of expected volatility is based on the daily historical trading data of our common stock at the time of grant over a historical period commensurate with the expected life of the stock-based award. We estimated the weighted-average expected life based on the contractual and vesting terms of the stock options, as well as historical cancellation and exercise data.
Stock-based compensation expense resulting from our ESPP was not significant in the years ended December 31, 2017, 2016, and 2015.
Summary of Stock Option Activity
The table below presents a summary of stock option activity under our equity incentive plans (in thousands, except for price per share and contractual life information):
|
|
|
Number of Shares |
|
|
Weighted- Average Exercise Price per Share |
|
|
Weighted- Average Remaining Contractual Life (in Years) |
|
|
Aggregate Intrinsic Value(1) |
|
||||
|
Outstanding at December 31, 2016 |
|
|
19,413 |
|
|
$ |
12.01 |
|
|
|
|
|
|
|
|
|
|
Options granted |
|
|
5,322 |
|
|
|
40.56 |
|
|
|
|
|
|
|
|
|
|
Options exercised |
|
|
(5,284 |
) |
|
|
10.88 |
|
|
|
|
|
|
|
|
|
|
Options forfeited & canceled |
|
|
(625 |
) |
|
|
13.69 |
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2017 |
|
|
18,826 |
|
|
$ |
20.35 |
|
|
|
5.15 |
|
|
$ |
741,257 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at December 31, 2017 |
|
|
10,504 |
|
|
$ |
11.95 |
|
|
|
3.57 |
|
|
$ |
501,818 |
|
|
(1) |
Aggregate intrinsic value represents the difference between the exercise price of the option and the closing market price of our common stock on December 31, 2017. |
The weighted-average grant-date fair value per share of options granted during the years ended December 31, 2017, 2016, and 2015 was $20.08, $6.54, and $6.55, respectively. The total intrinsic value of options exercised during the years ended December 31, 2017, 2016, and 2015 was $84.0 million, $17.9 million, and $20.5 million, respectively. The estimated fair value of options vested during the years ended December 31, 2017, 2016, and 2015 was $19.3 million, $16.7 million, and $16.8 million, respectively.
92
Summary of RSU Activity
A summary of RSU award activity is as follows (in thousands except for per share amounts):
|
|
|
Units Issued |
|
|
Weighted- Average Grant Date Fair Value |
|
|
Aggregate Intrinsic Value(1) |
|
|||
|
Balance at December 31, 2016 |
|
|
2,248 |
|
|
$ |
13.53 |
|
|
|
|
|
|
Granted |
|
|
1,405 |
|
|
|
54.14 |
|
|
|
|
|
|
Vested and released |
|
|
(875 |
) |
|
|
14.05 |
|
|
|
|
|
|
Forfeited and canceled |
|
|
(200 |
) |
|
|
13.14 |
|
|
|
|
|
|
Balance at December 31, 2017 |
|
|
2,578 |
|
|
$ |
35.52 |
|
|
$ |
153,941 |
|
|
(1) |
Aggregate intrinsic value represents the difference between the grant price of the award, which is zero, and the closing market price of our common stock on December 31, 2017. |
Note 12 — Income Taxes
Loss before provision for income taxes includes the following components (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Domestic |
|
$ |
(97,938 |
) |
|
$ |
(155,375 |
) |
|
$ |
(81,931 |
) |
|
Foreign |
|
|
1,862 |
|
|
|
2,727 |
|
|
|
1,260 |
|
|
Loss before provision for income taxes |
|
$ |
(96,076 |
) |
|
$ |
(152,648 |
) |
|
$ |
(80,671 |
) |
Provision for Income Taxes
The provision for income taxes consists of the following (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
State |
|
|
1 |
|
|
|
(1 |
) |
|
|
(1 |
) |
|
Foreign |
|
|
580 |
|
|
|
992 |
|
|
|
529 |
|
|
Total Current |
|
|
581 |
|
|
|
991 |
|
|
|
528 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
State |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
35 |
|
|
|
(115 |
) |
|
|
(22 |
) |
|
Total Deferred |
|
|
35 |
|
|
|
(115 |
) |
|
|
(22 |
) |
|
Provision for income taxes |
|
$ |
616 |
|
|
$ |
876 |
|
|
$ |
506 |
|
93
Income tax provision related to continuing operations differs from the amount computed by applying the statutory income tax rate of 35% to pretax loss as follows (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
U.S. federal provision (benefit) |
|
|
|
|
|
|
|
|
|
|
|
|
|
At statutory rate |
|
$ |
(33,627 |
) |
|
$ |
(53,427 |
) |
|
$ |
(28,235 |
) |
|
Tax law changes |
|
|
248,155 |
|
|
|
— |
|
|
|
— |
|
|
Non-cash interest expense on liability related to sale of future royalties |
|
|
6,604 |
|
|
|
6,899 |
|
|
|
7,217 |
|
|
Non-deductible officers' compensation |
|
|
2,547 |
|
|
|
220 |
|
|
|
184 |
|
|
Change in valuation allowance |
|
|
(186,124 |
) |
|
|
51,981 |
|
|
|
22,210 |
|
|
Stock-based compensation |
|
|
(20,665 |
) |
|
|
528 |
|
|
|
674 |
|
|
Sale of future royalties |
|
|
(8,236 |
) |
|
|
— |
|
|
|
— |
|
|
Research credits |
|
|
(8,038 |
) |
|
|
(4,543 |
) |
|
|
(4,529 |
) |
|
Foreign tax deduction |
|
|
— |
|
|
|
— |
|
|
|
1,773 |
|
|
Other |
|
|
— |
|
|
|
(782 |
) |
|
|
1,212 |
|
|
Provision for income taxes |
|
$ |
616 |
|
|
$ |
876 |
|
|
$ |
506 |
|
Tax Law Changes
The U.S. Tax Cuts and Jobs Act (the Tax Act) was enacted on December 22, 2017. The Tax Act reduces the U.S. federal corporate tax rate from 35% in 2017 to 21% in 2018, requires companies to pay a one-time transition tax on earnings of certain foreign subsidiaries that were previously tax deferred and creates new taxes on certain foreign sourced earnings. At December 31, 2017, we have not completed our accounting for the tax effects of enactment of the Tax Act; however, in certain cases, as described below, we have made a reasonable estimate of the effects on our existing deferred tax balances and the one-time transition tax.
Deferred Tax Assets and Liabilities
Deferred income taxes reflect the net tax effects of loss and credit carryforwards and temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. We remeasured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which includes the change in the federal rate to 21%. Significant components of our deferred tax assets for federal and state income taxes are as follows (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
364,864 |
|
|
$ |
508,406 |
|
|
Research and other credits |
|
|
94,103 |
|
|
|
74,917 |
|
|
Stock-based compensation |
|
|
14,552 |
|
|
|
26,357 |
|
|
Property, plant and equipment |
|
|
6,848 |
|
|
|
6,882 |
|
|
Reserves and accruals |
|
|
5,659 |
|
|
|
12,269 |
|
|
Capitalized research expenses |
|
|
5,497 |
|
|
|
11,587 |
|
|
Deferred revenue |
|
|
3,796 |
|
|
|
23,035 |
|
|
Sale of future royalties |
|
|
— |
|
|
|
1,611 |
|
|
Other |
|
|
1,156 |
|
|
|
750 |
|
|
Deferred tax assets before valuation allowance |
|
|
496,475 |
|
|
|
665,814 |
|
|
Valuation allowance for deferred tax assets |
|
|
(496,191 |
) |
|
|
(665,514 |
) |
|
Total deferred tax assets |
|
|
284 |
|
|
|
300 |
|
|
Total deferred tax liabilities |
|
|
— |
|
|
|
— |
|
|
Net deferred tax assets |
|
$ |
284 |
|
|
$ |
300 |
|
Realization of our deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Because of our lack of U.S. earnings history, the net U.S. deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by $169.3 million during the year ended December 31, 2017 and increased by $52.5 million and $17.3 million during the years ended December 31, 2016 and 2015, respectively. The decrease in the valuation allowance for year ended December 31, 2017 is primarily due to the change in the federal rate. The valuation allowance includes approximately
94
$35.6 million of income tax benefit at both December 31, 2017 and December 31, 2016 related to stock-based compensation that will be included in income tax expense in our Consolidated Statement of Operations when realized.
The one-time transition tax is based on our total post-1986 earnings and profits (E&P) that we previously deferred from U.S. income taxes. As of December 31, 2017, we estimate that there is negative E&P on an aggregate basis and we have not recorded a provisional amount for any one-time transition tax triggered by the Tax Act. We have not yet completed our calculation of the total post-1986 E&P for these foreign subsidiaries. Further, the transition tax is based in part on the amount of those earnings held in cash and other specified assets. This amount may change when we finalize the calculation of post-1986 foreign E&P previously deferred from U.S. federal taxation and finalize the amounts held in cash or other specified assets. No additional income taxes have been provided for any remaining undistributed foreign earnings not subject to the transition tax, or any additional outside basis difference inherent in these entities, as these amounts continue to be indefinitely reinvested in foreign operations.
Net Operating Loss and Tax Credit Carryforwards
As of December 31, 2017, we had a net operating loss carryforward for federal income tax purposes of approximately $1,610.3 million, portions of which will begin to expire in 2018. As of December 31, 2017, we had a total state net operating loss carryforward of approximately $474.0 million, portions of which will begin to expire in 2019. Utilization of some of the federal and state net operating loss and credit carryforwards are subject to annual limitations due to the “change in ownership” provisions of the Internal Revenue Code of 1986 and similar state provisions.
We have federal research credits of approximately $64.0 million, which will begin to expire in 2019 and state research credits of approximately $33.3 million which have no expiration date. We have federal orphan drug credits of $17.7 million which will begin to expire in 2026. These tax credits are subject to the same limitations discussed above.
Unrecognized tax benefits
We have incurred net operating losses since inception. Our policy is to include interest and penalties related to unrecognized tax benefits, if any, within the provision for income taxes in the consolidated statements of operations. If we are eventually able to recognize our uncertain positions, our effective tax rate may be reduced. We currently have a full valuation allowance against our U.S. net deferred tax asset which would impact the timing of the effective tax rate benefit should any of these uncertain tax positions be favorably settled in the future. Any adjustments to our uncertain tax positions would result in an adjustment of our net operating loss or tax credit carry forwards rather than resulting in a cash outlay. The decrease in the unrecognized tax benefits in 2015 primarily relates to a California Supreme Court decision relating to the calculation of apportionment of income.
We file income tax returns in the U.S., California, Alabama, and India. Because of net operating losses and research credit carryovers, substantially all of our domestic tax years remain open and subject to examination. We are currently under examination in India for the fiscal years ending 2009 and 2016.
We have the following activity relating to unrecognized tax benefits (in thousands):
|
|
|
December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Beginning balance |
|
$ |
18,413 |
|
|
$ |
17,384 |
|
|
$ |
27,522 |
|
|
Tax positions related to current year |
|
|
|
|
|
|
|
|
|
|
|
|
|
Additions: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
1,206 |
|
|
|
530 |
|
|
|
529 |
|
|
State |
|
|
1,666 |
|
|
|
499 |
|
|
|
443 |
|
|
Reductions |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Tax positions related to prior year |
|
|
|
|
|
|
|
|
|
|
|
|
|
Additions: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
State |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Reductions |
|
|
(802 |
) |
|
|
— |
|
|
|
(11,110 |
) |
|
Settlements |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Lapses in statute of limitations |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Ending balance |
|
$ |
20,483 |
|
|
$ |
18,413 |
|
|
$ |
17,384 |
|
95
Although it is reasonably possible that certain unrecognized tax benefits may increase or decrease within the next twelve months, we do not anticipate any significant changes to unrecognized tax benefits over the next twelve months. During the years ended December 31, 2017, 2016 and 2015, no significant interest or penalties were recognized relating to unrecognized tax benefits.
Note 13 — Segment Reporting
We operate in one business segment which focuses on applying our technology platforms to improve the performance of established drugs and to develop novel drug candidates. Our business offerings have similar economics and other characteristics, including the nature of products and manufacturing processes, types of customers, distribution methods and regulatory environment. We are comprehensively managed as one business segment by our Chief Executive Officer.
Our revenue is derived primarily from clients in the pharmaceutical and biotechnology industries. Lilly and UCB represented 42% and 12% of our revenue, respectively, for the year ended December 31, 2017. Revenue from AstraZeneca, UCB, Ophthotech and Roche represented 29%, 21%, 19% and 11% of our revenue, respectively, for the year ended December 31, 2016. Revenue from AstraZeneca and UCB represented 57% and 13% of our revenue, respectively, for the year ended December 31, 2015 .
Revenue by geographic area is based on the locations of our partners. The following table sets forth revenue by geographic area (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
190,810 |
|
|
$ |
39,147 |
|
|
$ |
40,400 |
|
|
Europe |
|
|
116,901 |
|
|
|
126,289 |
|
|
|
190,384 |
|
|
Total revenue |
|
$ |
307,711 |
|
|
$ |
165,436 |
|
|
$ |
230,784 |
|
At December 31, 2017, $41.7 million, or approximately 88%, of the net book value of our property, plant and equipment was located in the United States and $5.8 million, or approximately 12%, was located in India. At December 31, 2016, $48.8 million, or approximately 74%, of the net book value of our property, plant and equipment was located in the United States, $10.4 million, or approximately 16% was located at contract manufacturers in Germany and the Netherlands, $5.6 million, or approximately 9%, was located in India, and $0.8 million or approximately 1%, was located in other countries.
Note 14 — Subsequent Event
On February 13, 2018, we entered into the BMS Collaboration Agreement with BMS, pursuant to which we and BMS will jointly develop NKTR-214, including, without limitation, in combination with BMS’s Opdivo® (nivolumab) and Opdivo® plus Yervoy® (ipilimumab), and other compounds of BMS, us or any third party. The parties have agreed to jointly commercialize NKTR-214 on a worldwide basis. BMS will pay us a non-refundable upfront cash payment of $1.0 billion. We are eligible to receive additional cash payments of a total of up to $1.43 billion upon achievement of certain development and regulatory milestones and a total of up to $350.0 million upon achievement of certain sales milestones. We will book all worldwide sales and revenue for NKTR-214. We will share global commercialization profits and losses with BMS for NKTR-214, with Nektar sharing 65% and BMS sharing 35% of the net profits and losses. BMS will lead commercialization for combinations of NKTR-214 with BMS proprietary medicines, and we will lead all other commercialization efforts for NKTR-214. We will have the final decision-making authority regarding the pricing for NKTR-214. NKTR-214 will be sold on a stand-alone basis and there will be no fixed-dose combinations or co-packaging without the consent of both parties. Under the BMS Collaboration Agreement, we and BMS will collaborate to develop and conduct clinical studies of NKTR-214 pursuant to a joint development plan, which initially includes a series of registration-enabling trials in more than 20 indications in nine tumor types and may be updated and expanded only upon mutual agreement of the parties. The parties will share the development costs for NKTR-214 in combination regimens based on each party’s relative ownership interest in the compounds included in the regimens.
Pursuant to a Share Purchase Agreement entered into by us and BMS on February 13, 2018 (Purchase Agreement), BMS has also agreed to purchase 8,284,600 shares of our common stock for total cash consideration of $850.0 million, or $102.60 per share. The closing of the share purchase is subject to the expiration or early termination of the waiting period (and any extension thereof) under the Hart-Scott-Rodino Antitrust Improvements Act of 1976 and other customary closing conditions. The closings of the BMS Collaboration Agreement and the Purchase Agreement will be simultaneous and are expected to occur during the second quarter of 2018.
96
Note 15 — Selected Quarterly Financial Data (Unaudited)
The following table sets forth certain unaudited quarterly financial data. In our opinion, the unaudited information set forth below has been prepared on the same basis as our audited information and includes all adjustments necessary to present fairly the information set forth herein. We have experienced fluctuations in our quarterly results and expect these fluctuations to continue in the future. Due to these and other factors, we believe that quarter-to-quarter comparisons of our operating results will not be meaningful, and you should not rely on our results for any one quarter as an indication of our future performance. Certain items previously reported in specific financial statement captions have been reclassified to conform to the current period presentation. Such reclassifications have not materially impacted previously reported total revenues, operating loss or net loss. All data is in thousands except per share information.
|
|
|
Year Ended December 31, 2017 |
|
|
Year Ended December 31, 2016 |
|
||||||||||||||||||||||||||
|
|
|
Q1 |
|
|
Q2 |
|
|
Q3 |
|
|
Q4 |
|
|
Q1 |
|
|
Q2 |
|
|
Q3 |
|
|
Q4 |
|
||||||||
|
Product sales |
|
$ |
4,756 |
|
|
$ |
15,693 |
|
|
$ |
4,448 |
|
|
$ |
7,791 |
|
|
$ |
14,099 |
|
|
$ |
12,867 |
|
|
$ |
14,698 |
|
|
$ |
13,690 |
|
|
Total revenue |
|
$ |
24,728 |
|
|
$ |
34,589 |
|
|
$ |
152,928 |
|
|
$ |
95,466 |
|
|
$ |
58,882 |
|
|
$ |
32,768 |
|
|
$ |
36,336 |
|
|
$ |
37,450 |
|
|
Cost of goods sold |
|
$ |
6,131 |
|
|
$ |
8,989 |
|
|
$ |
5,674 |
|
|
$ |
9,753 |
|
|
$ |
8,870 |
|
|
$ |
7,708 |
|
|
$ |
7,033 |
|
|
$ |
6,604 |
|
|
Research and development expenses |
|
$ |
61,058 |
|
|
$ |
60,260 |
|
|
$ |
65,714 |
|
|
$ |
81,429 |
|
|
$ |
49,268 |
|
|
$ |
52,350 |
|
|
$ |
51,951 |
|
|
$ |
50,232 |
|
|
Operating income (loss) |
|
$ |
(54,437 |
) |
|
$ |
(50,656 |
) |
|
$ |
69,485 |
|
|
$ |
(24,034 |
) |
|
$ |
(9,484 |
) |
|
$ |
(38,325 |
) |
|
$ |
(32,901 |
) |
|
$ |
(32,145 |
) |
|
Net income (loss) |
|
$ |
(63,866 |
) |
|
$ |
(59,871 |
) |
|
$ |
60,871 |
|
|
$ |
(33,826 |
) |
|
$ |
(19,498 |
) |
|
$ |
(48,603 |
) |
|
$ |
(43,224 |
) |
|
$ |
(42,199 |
) |
|
Net income (loss) per share(1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
(0.42 |
) |
|
$ |
(0.39 |
) |
|
$ |
0.39 |
|
|
$ |
(0.21 |
) |
|
$ |
(0.14 |
) |
|
$ |
(0.36 |
) |
|
$ |
(0.32 |
) |
|
$ |
(0.28 |
) |
|
Diluted |
|
$ |
(0.42 |
) |
|
$ |
(0.39 |
) |
|
$ |
0.37 |
|
|
$ |
(0.21 |
) |
|
$ |
(0.14 |
) |
|
$ |
(0.36 |
) |
|
$ |
(0.32 |
) |
|
$ |
(0.28 |
) |
|
(1) |
Quarterly loss per share amounts may not total to the year-to-date loss per share due to rounding. |
97
None.
Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Securities Exchange Act of 1934 (Exchange Act) reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required financial disclosure.
As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including the Chief Executive Officer and the Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15. Based upon, and as of the date of, this evaluation, the Chief Executive Officer and the Chief Financial Officer concluded that our disclosure controls and procedures were effective. Accordingly, management believes that the financial statements included in this report fairly present in all material respects our financial condition, results of operations and cash flows for the periods presented.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP.
Our management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making its assessment of internal control over financial reporting, management used the criteria described in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework).
Based on our evaluation under the framework described in Internal Control — Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2017.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Changes in Internal Control Over Financial Reporting
We continuously seek to improve the efficiency and effectiveness of our internal controls. This results in refinements to processes throughout the Company. There was no change in our internal control over financial reporting during the quarter ended December 31, 2017, which was identified in connection with our management’s evaluation required by Exchange Act Rules 13a-15(f) and 15d-15(f) that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
98
Inherent Limitations on the Effectiveness of Controls
Our management, including the Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the company have been detected. These inherent limitations include the realities that judgments in decision making can be faulty and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
None.
99
Information relating to our executive officers required by this item is set forth in Part I — Item 1 of this report under the caption “Executive Officers of the Registrant” and is incorporated herein by reference. The other information required by this Item is incorporated by reference from the definitive proxy statement for our 2018 Annual Meeting of Stockholders to be filed with the SEC pursuant to Regulation 14A (Proxy Statement) not later than 120 days after the end of the fiscal year covered by this Form 10-K under the captions “Corporate Governance and Board of Directors,” “Proposal 1 — Election of Directors” and “Section 16(a) Beneficial Ownership Reporting Compliance.” Joseph J. Krivulka, who had served as our director since March 2005, passed away on February 17, 2018, resulting in a vacancy on the Board of Directors. The Board of Directors has not yet nominated an individual to fill this vacancy.
Information regarding our audit committee financial expert will be set forth in the Proxy Statement under the caption “Audit Committee,” which information is incorporated herein by reference.
We have a Code of Business Conduct and Ethics applicable to all employees, including the principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions. The Code of Business Conduct and Ethics is posted on our website at www.nektar.com. Amendments to, and waivers from, the Code of Business Conduct and Ethics that apply to any of these officers, or persons performing similar functions, and that relate to any element of the code of ethics definition enumerated in Item 406(b) of Regulation S-K will be disclosed at the website address provided above and, to the extent required by applicable regulations, on a current report on Form 8-K.
As permitted by SEC Rule 10b5-1, certain of our executive officers, directors and other employees have or may set up a predefined, structured stock trading program with their broker to sell our stock. The stock trading program allows a broker acting on behalf of the executive officer, director or other employee to trade our stock during blackout periods or while such executive officer, director or other employee may be aware of material, nonpublic information, if the trade is performed according to a pre-existing contract, instruction or plan that was established with the broker when such executive officer, director or employee was not aware of any material, nonpublic information. Our executive officers, directors and other employees may also trade our stock outside of the stock trading programs set up under Rule 10b5-1 subject to our securities trading policy.
The information required by this Item is included in the Proxy Statement and incorporated herein by reference.
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item is included in the Proxy Statement and incorporated herein by reference.
The information required by this Item is included in the Proxy Statement and incorporated herein by reference.
The information required by this Item is included in the Proxy Statement and incorporated herein by reference.
100
|
|
(a) |
The following documents are filed as part of this report: |
|
|
(1) |
Consolidated Financial Statements: |
The following financial statements are filed as part of this Annual Report on Form 10-K under Item 8 “Financial Statements and Supplementary Data.”
|
|
|
Page |
|
|
65 |
|
|
|
|
|
|
|
67 |
|
|
|
|
|
|
|
68 |
|
|
|
|
|
|
|
69 |
|
|
|
|
|
|
|
70 |
|
|
|
|
|
|
|
71 |
|
|
|
|
|
|
|
72 |
|
|
(2) |
Financial Statement Schedules: |
All financial statement schedules have been omitted because they are not applicable, or the information required is presented in our consolidated financial statements and notes thereto under Item 8 of this Annual Report on Form 10-K.
|
|
(3) |
Exhibits. |
Except as so indicated in Exhibit 32.1, the following exhibits are filed as part of, or incorporated by reference into, this Annual Report on Form 10-K.
|
Exhibit Number |
|
Description of Documents |
|
|
|
|
|
2.1(1) |
|
|
|
|
|
|
|
3.1(2) |
|
Certificate of Incorporation of Inhale Therapeutic Systems (Delaware), Inc. |
|
|
|
|
|
3.2(3) |
|
|
|
|
|
|
|
3.3(4) |
|
|
|
|
|
|
|
3.4(5) |
|
|
|
|
|
|
|
3.5(6) |
|
|
|
|
|
|
|
4.1 |
|
|
|
|
|
|
|
4.2(4) |
|
|
|
|
|
|
|
4.3(7) |
|
|
|
|
|
|
|
10.1(8) |
|
2000 Equity Incentive Employee Stock Purchase Plan, as amended and restated.++ |
|
|
|
|
|
10.2(8) |
|
2000 Non-Officer Equity Incentive Plan, as amended and restated.++ |
|
|
|
|
|
10.3(8) |
|
|
|
|
|
|
|
10.4(8) |
|
|
|
|
|
|
101
|
Exhibit Number |
|
Description of Documents |
|
10.5(8) |
|
Amended and Restated Change of Control Severance Benefit Plan.++
|
|
10.6(9) |
|
2012 Performance Incentive Plan.++
|
|
10.7(10) |
|
|
|
10.8(11) |
|
2017 Performance Incentive Plan.++
|
|
10.9(25) |
|
|
|
|
|
|
|
10.10(12) |
|
|
|
|
|
|
|
10.11(13) |
|
Amended and Restated Compensation Plan for Non-Employee Directors.++ |
|
|
|
|
|
10.12(14) |
|
|
|
|
|
|
|
10.13(15) |
|
Form of Severance Letter for executive officers of the company.++ |
|
|
|
|
|
10.14(1) |
|
|
|
|
|
|
|
10.15(1) |
|
|
|
|
|
|
|
10.16(16) |
|
Letter Agreement, executed effective on December 10, 2009, with Stephen K. Doberstein, Ph.D.++ |
|
|
|
|
|
10.17(17) |
|
|
|
|
|
|
|
10.18(25) |
|
|
|
|
|
|
|
10.19(15) |
|
|
|
|
|
|
|
10. 20(18) |
|
|
|
|
|
|
|
10.21(19) |
|
|
|
|
|
|
|
10.22(1) |
|
|
|
|
|
|
|
10.23(1) |
|
|
|
|
|
|
|
10.24(16) |
|
|
|
|
|
|
|
10.25(20) |
|
License Agreement by and between AstraZeneca AB and Nektar Therapeutics, dated September 20, 2009.+ |
|
|
|
|
|
10.26(21) |
|
|
|
|
|
|
|
10.27(22) |
|
|
|
10.28(18) |
|
|
|
|
|
|
|
10.29(7) |
|
|
|
|
|
|
|
10.30(7) |
|
|
|
|
|
|
102
|
Exhibit Number |
|
Description of Documents |
|
10.31(23) |
|
|
|
|
|
|
|
10.32(24) |
|
|
|
|
|
|
|
21.1(25) |
|
|
|
|
|
|
|
23.1(25) |
|
|
|
|
|
|
|
24 |
|
Power of Attorney (reference is made to the signature page). |
|
|
|
|
|
31.1(25) |
|
|
|
|
|
|
|
31.2(25) |
|
|
|
|
|
|
|
32.1* |
|
|
|
|
|
|
|
101** |
|
The following materials from Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2017, formatted in XBRL (Extensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Comprehensive Loss, (iv) Consolidated Statements of Stockholders’ Equity, (v) Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements. |
|
+ |
Confidential treatment with respect to specific portions of this Exhibit has been requested, and such portions are omitted and have been filed separately with the SEC. |
|
++ |
Management contract or compensatory plan or arrangement. |
|
* |
Exhibit 32.1 is being furnished and shall not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liability of that section, nor shall such exhibit be deemed to be incorporated by reference in any registration statement or other document filed under the Securities Act of 1933, as amended, or the Securities Exchange Act, except as otherwise stated in such filing. |
|
** |
XBRL information is filed herewith. |
|
(1) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2008. |
|
(2) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended June 30, 1998. |
|
(3) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended June 30, 2000. |
|
(4) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on January 23, 2003. |
|
(5) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2009. |
|
(6) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on April 15, 2014. |
|
(7) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on October 6, 2015. |
|
(8) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2011. |
|
(9) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on June 17, 2015. |
|
(10) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K filed on December 17, 2015. |
|
(11) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on June 15, 2017. |
|
(12) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Current Report on Form 8-K, filed on June 27, 2014. |
|
(13) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2012. |
|
(14) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics Quarterly Report on Form 10-Q for the quarter ended June 30, 2004. |
|
(15) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended September 30, 2007. |
|
(16) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2010. |
|
(17) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Annual Report on Form 10-K for the year ended December 31, 2014. |
103
|
(18) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended September 30, 2017. |
|
(19) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended June 30, 2006. |
|
(20) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended September 30, 2009. |
|
(21) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended June 30, 2016. |
|
(22) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended September 30, 2016. |
|
(23) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended March 31, 2012. |
|
(24) |
Incorporated by reference to the indicated exhibit in Nektar Therapeutics’ Quarterly Report on Form 10-Q for the quarter ended September 30, 2013. |
|
(25) |
Filed herewith. |
104
|
Item 16. |
Form 10-K Summary |
None.
105
Pursuant to the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City and County of San Francisco, State of California on March 1, 2018.
|
By: |
/s/ Gil M. Labrucherie |
|
|
Gil M. Labrucherie |
|
|
Senior Vice President and Chief Financial Officer |
|
By: |
/s/ JILLIAN B. THOMSEN |
|
|
Jillian B. Thomsen |
|
|
Senior Vice President, Finance and Chief Accounting Officer |
106
KNOW ALL PERSON BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Gil M. Labrucherie and Jillian B. Thomsen and each of them, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratify and confirming all that said attorneys-in-fact and agents, or any of them, or their or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed by the following persons in the capacities and on the dates indicated:
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Howard W. Robin |
|
Chief Executive Officer, President and Director |
|
March 1, 2018 |
|
Howard W. Robin |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/s/ Gil M. Labrucherie |
|
Senior Vice President and Chief Financial Officer |
|
March 1, 2018 |
|
Gil M. Labrucherie |
|
(Principal Financial Officer) |
|
|
|
|
|
|
|
|
|
/s/ Jillian B. Thomsen |
|
Senior Vice President, Finance and Chief Accounting Officer |
|
March 1, 2018 |
|
Jillian B. Thomsen |
|
(Principal Accounting Officer) |
|
|
|
|
|
|
|
|
|
/s/ Robert B. Chess |
|
Director, Chairman of the Board of Directors |
|
March 1, 2018 |
|
Robert B. Chess
|
|
|
|
|
|
/s/ Jeffrey R. Ajer |
|
Director |
|
March 1, 2018 |
|
Jeffrey R. Ajer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ R. Scott Greer |
|
Director |
|
March 1, 2018 |
|
R. Scott Greer |
|
|
|
|
|
|
|
|
|
|
|
/s/ Christopher A. Kuebler |
|
Director |
|
March 1, 2018 |
|
Christopher A. Kuebler |
|
|
|
|
|
|
|
|
|
|
|
/s/ Lutz Lingnau |
|
Director |
|
March 1, 2018 |
|
Lutz Lingnau |
|
|
|
|
|
|
|
|
|
|
|
/s/ Roy A. Whitfield |
|
Director |
|
March 1, 2018 |
|
Roy A. Whitfield |
|
|
|
|
|
|
|
|
|
|
|
/s/ Dennis L. Winger |
|
Director |
|
March 1, 2018 |
|
Dennis L. Winger |
|
|
|
|
|
|
|
|
|
|
|
|
107