Attached files
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended December 31, 2017 |
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to |
Commission file number: 001-34263
Impax Laboratories, Inc. | ||
(Exact name of registrant as specified in its charter) | ||
Delaware | 65-0403311 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
30831 Huntwood Avenue, Hayward, CA | 94544 | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code:
(510) 476-2000
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered: | |
Common Stock, par value $0.01 per share | The NASDAQ Stock Market LLC | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation of S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and "emerging growth company" in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ☒ | Accelerated filer ☐ | Non-accelerated filer ☐ |
Smaller reporting company ☐ | Emerging growth company ☐ | |
(Do not check if a smaller reporting company)
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ☐ No ☒
The aggregate market value of the registrant’s outstanding shares of common stock, other than shares held by persons who may be deemed affiliates of the registrant, computed by reference to the price at which the registrant’s common stock was last sold on The NASDAQ Stock Market LLC as of the last business day of the registrant’s most recently completed second fiscal quarter (June 30, 2017), was approximately $1,043,981,075.
As of February 26, 2018, there were 73,936,490 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the definitive proxy statement to be filed pursuant to Regulation 14A promulgated under the Securities Exchange Act of 1934, as amended, in connection with the 2018 Annual Meeting of Stockholders of the registrant (the “Annual Meeting Proxy Statement”) have been incorporated by reference into Part III of this Annual Report on Form 10-K. In the event the registrant does not file the Annual Meeting Proxy Statement, the information will be provided instead by an amendment to this report not later than 120 days after the end of the registrant’s fiscal year.
1
TABLE OF CONTENTS
Forward-Looking Statements | |||
PART I. | |||
Item 1. | Business | ||
Item 1A. | Risk Factors | ||
Item 1B. | Unresolved Staff Comments | ||
Item 2. | Properties | ||
Item 3. | Legal Proceedings | ||
Item 4. | Mine Safety Disclosures | ||
PART II. | |||
Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | ||
Item 6. | Selected Financial Data | ||
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | ||
Item 7A. | Quantitative and Qualitative Disclosures about Market Risk | ||
Item 8. | Financial Statements and Supplementary Data | ||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | ||
Item 9A. | Controls and Procedures | ||
Item 9B. | Other Information | ||
PART III. | |||
Item 10. | Directors, Executive Officers and Corporate Governance | ||
Item 11. | Executive Compensation | ||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | ||
Item 13. | Certain Relationships and Related Transactions, and Director Independence | ||
Item 14. | Principal Accounting Fees and Services | ||
PART IV. | |||
Item 15. | Exhibits and Financial Statement Schedules | ||
SIGNATURES | |||
EXHIBIT INDEX | |||
2
Forward-Looking Statements
Statements included in this Annual Report on Form 10-K that do not relate to present or historical conditions are “forward-looking statements.” Such forward-looking statements involve risks and uncertainties that could cause results or outcomes to differ materially from those expressed in the forward-looking statements. Forward-looking statements may include statements relating to our plans, strategies, objectives, expectations and intentions. Words such as “believes,” “forecasts,” “intends,” “possible,” “estimates,” “anticipates,” and “plans” and similar expressions are intended to identify forward-looking statements. Our ability to predict results or the effect of events on our operating results is inherently uncertain. Forward-looking statements involve a number of risks, uncertainties and other factors that could cause actual results to differ materially from those discussed in this Annual Report on Form 10-K. Such risks and uncertainties include, but are not limited to, fluctuations in our operating results and financial condition, the volatility of the market price of our common stock, our ability to successfully develop and commercialize pharmaceutical products in a timely manner, the impact of competition, the effect of any manufacturing or quality control problems, our ability to manage our growth, risks related to acquisitions of or investments in technologies, products or businesses, risks relating to goodwill and intangibles, the reduction or loss of business with any significant customer, the substantial portion of our total revenues derived from sales of a limited number of products, the impact of continuing consolidation of our customer base, our ability to sustain profitability and positive cash flows, the impact of any valuation allowance on our deferred tax assets, the restrictions imposed by our credit facility and indenture, our level of indebtedness and liabilities and the potential impact on cash flow available for operations, the availability of additional funds in the future, any delays or unanticipated expenses in connection with the operation of our manufacturing facilities or at our third party suppliers, the effect of foreign economic, political, legal and other risks on our operations abroad, the uncertainty of patent litigation and other legal proceedings, the increased government scrutiny on our agreements to settle patent litigations, product development risks and the difficulty of predicting FDA filings and approvals, consumer acceptance and demand for new pharmaceutical products, the impact of market perceptions of us and the safety and quality of our products, our determinations to discontinue the manufacture and distribution of certain products, our ability to achieve returns on our investments in research and development activities, changes to FDA approval requirements, our ability to successfully conduct clinical trials, our reliance on third parties to conduct clinical trials and testing, our lack of a license partner for commercialization of Numient® (IPX066) outside of the United States and Taiwan, impact of illegal distribution and sale by third parties of counterfeits or stolen products, the availability of raw materials and impact of interruptions in our supply chain, our policies regarding returns, rebates, allowances and chargebacks, the use of controlled substances in our products, the effect of global economic conditions on our industry, business, results of operations and financial condition, disruptions or failures in our information technology systems and network infrastructure caused by cyberattacks or other third party breaches or other events, our reliance on alliance and collaboration agreements, our reliance on licenses to proprietary technologies, our dependence on certain employees, our ability to comply with legal and regulatory requirements governing the healthcare industry, the regulatory environment, the effect of certain provisions in our government contracts, our ability to protect our intellectual property, exposure to product liability claims, changes in tax regulations, uncertainties involved in the preparation of our financial statements, our ability to maintain an effective system of internal control over financial reporting, the effect of terrorist attacks on our business, the location of our manufacturing and research and development facilities near earthquake fault lines, expansion of social media platforms, risks related to our proposed business combination with Amneal Pharmaceuticals, Inc. (“Amneal”), including whether the transactions (the “Combination”) contemplated by the Business Combination Agreement dated as of October 17, 2017 by and among us, Amneal, Atlas Holdings, Inc., and K2 Merger Sub Corporation as amended by Amendment No. 1, dated November 21, 2017 and Amendment No. 2 dated December 16, 2017 (the “Business Combination Agreement”) will be completed on the terms or timeline contemplated, if at all, the risk that governmental entities could take actions under antitrust laws to enjoin the completion of the Combination, business uncertainties and contractual restrictions while the Combination is pending, challenges related to our integration with Amneal after the closing, the fact that ownership interests will not be adjusted if there is a change in value of our company or Amneal, provisions in the Business Combination Agreement that may discourage other companies from acquiring us, transaction related costs related to the Combination and integration, the lower ownership and voting interests that our stockholders will have in New Amneal after the closing, the pending litigation related to the Combination and other risks described below in “Item 1A. Risk Factors.” You should not place undue reliance on forward-looking statements. Such statements speak only as to the date on which they are made, and we undertake no obligation to update or revise any forward-looking statement, regardless of future developments or availability of new information.
Rytary® and Emverm® are registered trademarks of Impax Laboratories, Inc. Other names are for informational purposes only and are used to identify companies and products and may be trademarks of their respective owners.
3
PART I.
Item 1. Business
Overview
We are a specialty pharmaceutical company applying formulation and development expertise, as well as our drug delivery technology, to the development, manufacture and marketing of bioequivalent pharmaceutical products, commonly referred to as “generics,” in addition to the development, manufacture and marketing of branded products. We operate in two segments, referred to as “Impax Generics” and “Impax Specialty Pharma.” The Impax Generics division includes our legacy Global Pharmaceuticals business as well as the generic businesses from our acquisition of Tower Holdings, Inc. ("Tower") and its subsidiaries on March 9, 2015 (the "Tower Acquisition"). The Impax Specialty Pharma division includes our legacy Impax Pharmaceuticals business as well as the acquired business of Amedra Pharmaceuticals, LLC ("Amedra") from the Tower Acquisition. Impax Generics concentrates its efforts on generic products, which are the pharmaceutical and therapeutic equivalents of brand-name drug products and are usually marketed under their established nonproprietary drug names rather than by a brand name. Impax Specialty Pharma utilizes its specialty sales force to market proprietary branded pharmaceutical products for the treatment of central nervous system (“CNS”) disorders and other select specialty segments. See “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements— Note 20. Segment Information,” for financial information about our segments for the years ended December 31, 2017, 2016 and 2015.
Business Combination with Amneal Pharmaceuticals LLC
On October 17, 2017, we entered into a Business Combination Agreement (the “Business Combination Agreement”) with Atlas Holdings, Inc., a Delaware corporation and our wholly-owned subsidiary (“Holdco”), K2 Merger Sub Corporation, a Delaware corporation and a wholly-owned subsidiary of Holdco (“Merger Sub”), and Amneal Pharmaceuticals LLC (“Amneal”). The Business Combination Agreement was unanimously approved by our board of directors on October 16, 2017.
At the closing (the “Closing”) of the transactions contemplated by the Business Combination Agreement (the “Transactions”), (i) Merger Sub will merge with and into our company (the “Impax Merger”), with our company surviving the Impax Merger as a direct wholly-owned subsidiary of Holdco, (ii) each share of our common stock, par value $0.01 per share (“Impax Common Stock”), issued and outstanding immediately prior to the Impax Merger, other than Impax Common Stock held by us in treasury, by Amneal or by any of their respective subsidiaries, will be converted into the right to receive one fully paid and nonassessable share of Class A common stock of Holdco, par value $0.01 per share (“Holdco Class A Common Stock”), (iii) we will convert to a limited liability company pursuant to the General Corporation Law of the State of Delaware and the Delaware Limited Liability Company Act, (iv) Holdco will contribute to Amneal all of Holdco’s equity interests in our company to Amneal, in exchange for common units of Amneal, (v) Holdco will issue an aggregate number of shares of Class B common stock of Holdco, par value $0.01 per share (“Holdco Class B Common Stock”, and together with Holdco Class A Common Stock, “Holdco Common Stock”) to the existing members of Amneal (the “Existing Amneal Members”) and (vi) Holdco will become the managing member of Amneal. In connection with the Closing, Holdco will be renamed Amneal Pharmaceuticals, Inc. (“New Amneal”).
Immediately following the Closing, (i) the Existing Amneal Members will hold approximately 75% of the voting power and economic interests in New Amneal, and (ii) our stockholders immediately prior to the Closing will hold approximately 25% of the voting power and economic interests in New Amneal. Following the Closing and the PIPE Investment (as described below), it is expected that the Existing Amneal Members will hold approximately 60% of the voting power of the outstanding shares of New Amneal common stock (the “New Amneal Shares”) and TPG Improv Holdings, L.P. (“TPG”) and other institutional investors will hold approximately 15% of the voting power of the outstanding New Amneal Shares.
4
In connection with the Combination and the investment (the “PIPE Investment”) by certain institutional investors including TPG and funds affiliated with Fidelity Management & Research Company (“Fidelity”), the Existing Amneal Members (together with their affiliates, successors and permitted assigns, the “Amneal Group”) entered into a definitive purchase agreement (the “PIPE Purchase Agreement”) with select institutional investors, including TPG and funds affiliated with Fidelity (the “PIPE Investors”). Pursuant to the PIPE Purchase Agreement, upon the Closing, members of the Amneal Group will exercise their right to cause Amneal to redeem certain of the Amneal common units (the “Redeemed Units”) held by such members pursuant to the Third Amended and Restated Limited Liability Company Agreement of Amneal (the “LLC Agreement”). In connection with such redemption, such members of the Amneal Group will receive shares of Class A Common Stock or shares of Class B-1 Common Stock in exchange for such Redeemed Units, in each case pursuant to the LLC Agreement (such redemption and issuance of Class A Common Stock and Class B-1 Common Stock to the members of the Amneal Group, the “Redemption”). Following the Redemption, the members of the Amneal Group will sell such shares of Class A Common Stock and Class B-1 Common Stock to the PIPE Investors at a per share purchase price of $18.25 for gross proceeds of approximately $855.0 million. Following the PIPE Investment, the PIPE Investors will own collectively approximately 15% of the New Amneal Shares on a fully diluted and as converted basis. The PIPE Investors other than TPG will receive shares of Class A Common Stock only, with approximately 30.4 million shares of Class A Common Stock issued to such investors in connection with the PIPE Investment. TPG will receive up to approximately 16.4 million shares of Class A Common Stock or Class B-1 Common Stock in connection with the PIPE Investment, with the exact split between Class A Common Stock and Class B-1 Common Stock to be determined prior to the Closing upon written notice by TPG to the Amneal Group in accordance with the PIPE Purchase Agreement. Following the PIPE Investment, TPG will own all outstanding shares of Class B-1 Common Stock, to the extent they elect to receive shares of Class B-1 Common Stock. The Class B-1 Common Stock held by TPG following the PIPE Investment will not be entitled to any voting rights, will have economic rights that are identical to those of the Class A Common Stock, and will be convertible into shares of Class A Common Stock. The Class A Common Stock and Class B-1 Common Stock held by the PIPE Investors following the PIPE Investment will represent approximately 15% of the voting shares and 37.5% of the economic interest of New Amneal (assuming the conversion of all such shares of Class B-1 Common Stock into shares of Class A Common Stock).
Consummation of the Transactions is subject to customary closing conditions, including, among other things, (i) the approval of our stockholders holding a majority of the outstanding Impax Common Stock entitled to vote (the “Requisite Stockholder Approval”), (ii) expiration of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and (iii) NYSE listing approval for Holdco Class A Common Stock. The obligation to consummate the Transactions is also conditioned upon each party’s representations and warranties being true and correct (subject to certain materiality exceptions) and each party having performed in all material respects its obligations under the Business Combination Agreement.
The Business Combination Agreement may be terminated by each of us and Amneal under certain circumstances, including if the Closing does not occur on or before July 17, 2018 (the “Outside Date”). Amneal also has certain additional termination rights, including in connection with a change of our Board’s recommendation that our stockholders adopt and approve the Business Combination Agreement. We are required to pay Amneal a termination fee of $45.0 million (the “Termination Fee”) in connection with such a termination by Amneal, as well as under certain other circumstances, including if the Business Combination Agreement is terminated by us in connection with a Superior Proposal (as defined in the Business Combination Agreement). Additionally, Amneal will be entitled to reimbursement for up to $15.0 million of its reasonable out-of-pocket expenses incurred in connection with the Business Combination Agreement and the Transactions if the Business Combination Agreement is terminated due to the failure to obtain the Requisite Stockholder Approval.
5
On October 17, 2017, Holdco and the Amneal Members also entered into a Stockholders Agreement, which was subsequently amended and restated on December 16, 2017 (the “Stockholders Agreement”). The Stockholders Agreement provides, among other things, that:
• | Board Representation. Following the Closing, the board of directors of New Amneal (the “New Amneal Board”) will consist of no more than 13 directors, subject to increase for a Qualifying Investor (as defined below). If an Executive Event (as defined below) has occurred, the New Amneal Board will consist of 11 members, subject to increase for a Qualifying Investor. Immediately following the Closing, the New Amneal Board will consist of (i) seven directors (the “Amneal Directors”) designated by Amneal Holdings, LLC (the “Amneal Group Representative”), including the co-chairmen of the New Amneal Board, Chirag Patel and Chintu Patel, unless an Executive Event has occurred, in which case the number of Amneal Directors will be six and (ii) five directors (the “Non-Amneal Directors”) designated by us, including Paul M. Bisaro, our current President and Chief Executive Officer, four directors from our board that meet the NYSE independence requirements (including Robert L. Burr, our current Chairman of our board who will serve as Lead Independent Director) and unless and Executive Event has occurred, Robert A. Stewart, Amneal’s current President and Chief Executive Officer, who will also serve as President and Chief Executive Officer of New Amneal after the Closing. “Executive Event” means the failure of Robert A. Stewart to be serving as the President of Amneal as of immediately prior to the Closing, including by reason of his death, resignation, retirement, disqualification, removal from office or other cause. |
In the event that the Amneal Group transfers more than 4% of the outstanding New Amneal Shares to an investor (a “Qualifying Investor”) and, following such transfer, the Amneal Group Members continue to beneficially own more than 50% of the outstanding New Amneal Shares, then the Amneal Group will have a one-time right to increase the size of the New Amneal Board by two directors and to fill the resulting vacancies with one new director designated by the Amneal Group Representative and one new director designated by the Qualifying Investor. Such Qualifying Investor may designate a board observer if it has not appointed a director. Immediately following the Closing and the PIPE Investment, TPG, as a Qualifying Investor, will have the right to designate a director for appointment to the New Amneal Board and the New Amneal Board may be expanded from 13 directors to 15 directors (or, if an Executive Event has occurred, from 11 directors to 13 directors) to accommodate TPG’s exercise of such right and the appointment of such designated director. In the event that the Amneal Group Members transfer more than 5% of the outstanding New Amneal Shares to a Qualifying Investor and, immediately prior to or following such transfer, the Amneal Group Members beneficially own less than 50% of the outstanding New Amneal Shares, then the Amneal Group Representative will have a one-time right to replace any existing Amneal Director with a director designated by such a Qualifying Investor.
For so long as the Amneal Group Members beneficially own more than 50% of the New Amneal Shares, (i) the Amneal Directors will have the right to designate the Co-Chairmen of the New Amneal Board, and the Non-Amneal Directors will have the right to designate the Lead Independent Director of the New Amneal Board and (ii) the Amneal Group Representative will have the right to designate a number of directors for nomination by the New Amneal Board for election to the New Amneal Board a number of designees that constitutes a majority of the total number of directors comprising the New Amneal Board. If the Amneal Group Members own less than 50% but more than 10% of the outstanding New Amneal Shares, the Amneal Group Representative will have the right to designate a number of directors proportionate to the beneficial ownership of outstanding New Amneal Shares by the Amneal Group Members (rounded up to the nearest whole number).
Nominating Committee and Compensation Committee. The Amneal Group Representative will have the right to nominate two of the four directors serving on each of the Nominating Committee and Compensation Committee for so long as the Amneal Group Members beneficially own more than 50% of the outstanding New Amneal Shares. The remaining directors on the Nominating Committee and Compensation Committee will be designated by a majority of the independent directors of the New Amneal Board.
• | Conflicts Committee. Until the Amneal Group Members beneficially own less than 10% of the New Amneal Shares, the New Amneal Board will have a Conflicts Committee comprised solely of independent directors to provide leadership and guidance to the New Amneal Board and New Amneal regarding potential conflicts of interest between New Amneal and any Amneal Group Member, including with respect to related party transactions. |
• | Integration Committee. For at least two years following the Closing, the New Amneal Board will have an Integration Committee comprised of Chirag Patel, Chintu Patel, the current Co-Chief Executive Officers and Co-Chairmen of Amneal, and Paul M. Bisaro, our current President and Chief Executive Officer, which will serve as an advisory committee to management to provide input in connection with the integration of our company and Amneal. |
6
• | Chief Executive Officer. Robert A. Stewart will be the Chief Executive Officer of New Amneal, unless an Executive Event has occurred. If an Executive Event has occurred, Paul M. Bisaro will be the Chief Executive Officer of New Amneal following the Closing. |
• | Executive Chairman. Paul M. Bisaro, our current President and Chief Executive Officer, will be the Executive Chairman of New Amneal following the Closing. For 18 months following the Closing, the removal of Mr. Bisaro as the Executive Chairman (or as CEO if an Executive Event has occurred) will require the approval of (i) a majority of the New Amneal Board and a majority of the Non-Amneal Directors (other than Mr. Bisaro). |
• | Standstill Provisions. The Amneal Group Members will be subject to customary standstill provisions, subject to certain exceptions, until the earlier of (i) the third anniversary of the Closing Date and (ii) such time when the Amneal Group Members beneficially own less than 20% of the outstanding shares of the New Amneal Shares. |
• | Amneal Buyout Transactions. Any proposal by an Amneal Group Member to acquire all outstanding New Amneal Shares held by all other stockholders (other than other Amneal Group Members) must be approved by the Conflicts Committee and, as long as the Amneal Group Members beneficially own 37.5% of the outstanding shares of Holdco Common Stock, be subject to a non-waivable condition that a majority of the voting power of the outstanding shares of Holdco Common Stock held by such other stockholders approve the transaction. |
• | Transfer Restrictions. At any time, an Amneal Group Member may transfer shares of New Amneal Shares to an affiliate. For the period of 180 days following the closing (the “Lock-Up Period”), no Amneal Group Member may transfer any shares of New Amneal Shares, unless with the prior written consent of the Conflicts Committee, subject to certain exceptions. Following the expiration of the Lock-Up Period, Amneal Group Members may transfer New Amneal Shares pursuant to an effective registration statement, or in transactions exempt from or not subject to registration requirements, subject to certain customary restrictions; provided, without the approval of the Conflicts Committee, Amneal Group Members will be prohibited from making transfers of New Amneal Shares (i) if after such transfer, such transferee or group of transferees would own more than 15% of the voting power of the outstanding New Amneal Shares, or (ii) to any person or group who, prior to such transfer, beneficially owned 15% or more of the outstanding New Amneal Shares. |
The Stockholders Agreement will terminate when the Amneal Group Members cease to own 10% of the outstanding shares of Holdco Common Stock.
The Closing is currently expected to occur during the first half of 2018, however, we cannot assure that the Closing will be completed on the terms or timeline currently contemplated, or at all. See “Item IA. Risk Factors - Risks Related to Our Proposed Business Combination with Amneal Pharmaceuticals LLC” below for additional information regarding the risks related to the Transactions.
Teva Transaction
On August 3, 2016, we completed our previously announced acquisition of (A) certain assets related to (i) 15 then currently marketed generic pharmaceutical products, (ii) one then approved generic product and two then tentatively approved strengths of a then currently marketed product, which at the time of the closing had not yet launched, (iii) one pipeline generic product and one pipeline strength of a then currently marketed product, which at the time of the closing were pending approval by the FDA and (iv) one generic product then under development, and (B) the return to us of our full commercial rights to our then pending ANDA for the generic equivalent to Concerta® (methylphenidate hydrochloride), a product we had previously partnered with Teva Pharmaceuticals USA, Inc. (“Teva USA”) (collectively, the products and pipeline products and the assets related thereto in (A) and (B), the “Acquired Product Lines” and the transactions related thereto the “Teva Transaction”), pursuant to (x) an Asset Purchase Agreement, dated as of June 20, 2016, as amended on June 30, 2016, with Teva Pharmaceutical Industries Ltd. (“Teva”), acting directly or through its affiliates (the “Teva APA”), (y) an Asset Purchase Agreement, dated as of June 20, 2016, as amended on June 30, 2016, with affiliates of Allergan plc (“Allergan”), (the “Allergan APA” and collectively with the Teva APA, the "APAs"), and (z) a Termination Agreement, dated as of June 20, 2016, between our company and Teva USA, terminating each party’s rights and obligations with respect to methylphenidate hydrochloride under the Strategic Alliance Agreement, dated June 27, 2001, as amended between our company and Teva USA. The aggregate purchase price for the Acquired Product Lines pursuant to the terms of the Teva APA and the Allergan APA, including the upfront payment to Teva in accordance with the Termination Agreement, was $585.8 million in cash at closing. We are also obligated to make future payments to Teva of up to $40.0 million under the terms of the Termination Agreement, payable upon the achievement of specified commercialization events related to methylphenidate hydrochloride.
7
Our Strategy
We plan to continue to expand our Impax Generics division by targeting complex solid oral and alternative dosage form Abbreviated New Drug Applications (“ANDAs”) with high revenue potential, including products with the potential to be first-to-file or first-to-market. Our products and product candidates are generally difficult to formulate and manufacture, providing certain competitive advantages. In addition to our product pipeline of 17 pending applications at the FDA as of December 31, 2017, we are continuing to evaluate and pursue external growth initiatives including acquisitions and partnerships.
The following information summarizes our generic pharmaceutical product development activities since inception through December 31, 2017:
• | 107 ANDAs approved by the U.S. Food and Drug Administration (“FDA”), including two tentatively approved (i.e., satisfying substantive FDA requirements but remaining subject to statutory restrictions). In addition, we have rights to market and/or share in profits to 18 approved ANDAs held by our third party alliance partners. The approved ANDAs (including those held by our partners) include generic versions of brand name pharmaceuticals such as Adderall XR®, Lofibra®, Opana ER® (NDA 021610), Pulmicort Respules® and Solaraze®. |
• | 17 applications pending at the FDA that represent approximately $14 billion in 2017 U.S. product sales. |
• | A number of products in various stages of development for which applications have not yet been filed. |
A core component of our strategy includes an ongoing focus in our Impax Specialty Pharma division on proprietary brand-name pharmaceutical products to treat CNS disorders and other specialty segments. We believe that we have the research, development and formulation expertise to develop branded products that will deliver significant improvements over existing therapies. We plan to continue investing in our development pipeline, both internally and through acquisitions and partnerships primarily focused on late-stage and next generation product opportunities.
Impax Generics Division
In the generic pharmaceutical market, we focus our efforts on developing, manufacturing, selling and distributing complex solid dose and alternative dosage form products covering a broad range of therapeutic areas and having technically challenging drug-delivery mechanisms or unique product development formulations. We employ our technologies and formulation expertise to develop generic products that reproduce brand-name products’ physiological characteristics but do not infringe any valid patents relating to such brand-name products. Generic products contain the same active ingredient and are of the same route of administration, dosage form, strength and indication(s) as brand-name products already approved for use in the United States by the FDA. We generally focus our generic product development on brand-name products as to which the patents covering the active pharmaceutical ingredient have expired or are near expiration, and we employ our experience to develop bioequivalent versions of such brand-name products. We also develop, manufacture, sell and distribute specialty generic pharmaceuticals that we believe present certain competitive advantages, such as difficulty in raw materials sourcing, complex formulation or development characteristics or special handling requirements. We have generally obtained rights to our alternative dosage form products through third party alliance and collaboration agreements, such as through our partnership agreement with Tolmar, Inc. (“Tolmar”).
We sell and distribute generic pharmaceutical products primarily through four sales channels:
• | the “Impax Generics sales channel" for sales of generic prescription products we sell directly to wholesalers, large retail drug chains, and others; |
• | the “Private Label sales channel" for generic pharmaceutical over-the-counter (“OTC”) and prescription products we sell to unrelated third party customers who in-turn sell the product to third parties under their own label; |
• | the “Rx Partner sales channel" for generic prescription products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements; and |
• | the “OTC Partner sales channel" for sales of generic pharmaceutical OTC products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements. |
8
As of December 31, 2017, we marketed 225 generic pharmaceutical products representing dosage variations of 77 different pharmaceutical compounds through our Impax Generics division, and five other generic pharmaceutical products, representing dosage variations of two different pharmaceutical compounds, through our alliance and collaboration agreement partners. As of December 31, 2017, our significant marketed generic products were Epinephrine Auto-Injector (generic Adrenaclick®), oxymorphone hydrochloride extended release tablets (AB rated to original OPANA® ER), Budesonide Inhalation Suspension (gPulmicort Respules®), Mixed Amphetamine Salts ER Capsules (generic Adderall XR®), and fenofibrate (generic Lofibra®).
As of December 31, 2017, we had 17 applications pending at the FDA. The following table lists our publicly identified product applications pending at the FDA as of December 31, 2017:
Product | Generic of | |
Apixaban Tablets 2.5, 5 mg | Eliquis® | |
Carvedilol Phosphate ER Capsules 10, 20, 40, 80 mg | Coreg CR® | |
Colesevelam Tablets 625 mg | Welchol® | |
Dimethyl Fumarate DR Capsules 120, 240mg | Tecfidera® | |
Fentanyl Buccal Tablet 100, 200, 400, 600, 800 mcg | Fentora® | |
Mixed Amphetamine Salts ER Capsules 5, 10, 15, 20, 25, 30 mg | Adderall XR® | |
Oxycodone ER Tablets (new formulation) 10, 15, 20, 30, 40, 60, 80 mg | Oxycontin® | |
Risedronate Sodium DR Tablets 35 mg | Atelvia® | |
Teriflunomide Tablets 14 mg | Aubagio® | |
Impax Specialty Pharma
Impax Specialty Pharma is engaged in the development, sale and distribution of proprietary branded pharmaceutical products that we believe represent improvements to already-approved pharmaceutical products addressing CNS disorders and other select specialty segments. We estimate there are approximately 16,000 neurologists in the United States. Historically, a concentrated number of these neurologists are responsible for writing the majority of neurology prescriptions. CNS is the largest therapeutic category in the United States with 2017 sales of about $66.7 billion, or 14.2% of the $470 billion U.S. prescription drug market. CNS product sales contracted (5.2%) in 2017, compared to 1.5% growth for the overall pharmaceutical market, while total CNS prescriptions declined 1.1%, compared to a 0.2% reduction in the overall pharmaceutical industry prescriptions. (Source: IQVIA).
Our branded pharmaceutical product portfolio consists of commercial CNS and other select specialty products, as well as development stage projects. In February 2012, we licensed from AstraZeneca UK Limited ("AstraZeneca") the exclusive U.S. commercial rights to Zomig® (zolmitriptan) tablet, orally disintegrating tablet and nasal spray formulations pursuant to the terms of a Distribution, License, Development and Supply Agreement with AstraZeneca, which was subsequently amended (the "AZ Agreement") and began sales of the Zomig® products under our label during the year ended December 31, 2012 through our specialty sales force. In May 2013, our exclusivity period for branded Zomig® tablets and orally disintegrating tablets expired and we launched authorized generic versions of those products in the United States. In June 2015, the FDA approved the Zomig® nasal spray for use in pediatric patients 12 years of age or older for the acute treatment of migraine with or without aura. In addition to the Zomig® products and our internally developed pharmaceutical product, Rytary® for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication and/or manganese intoxication, which was approved by the FDA on January 7, 2015, we are currently engaged in the sales and marketing of Emverm® (mebendazole) 100 mg chewable tablets, indicated for the treatment of pinworm, whipworm, common roundworm, common hookworm, and two other products, all acquired in our acquisition of Tower and Lineage which closed in March 2015. In November 2015, the European Commission granted marketing authorization for Numient® (referred to as Rytary® in the United States). The review of the Numient® application was conducted under the centralized licensing procedure as a therapeutic innovation, and the authorization is applicable in all 28 member states of the European Union, as well as Iceland, Liechtenstein and Norway.
We have a couple of product candidates that are in varying stages of development and we currently intend to expand our portfolio of branded pharmaceutical products primarily through internal development and through licensing and acquisitions, with a focus on late-stage product opportunities.
9
Alliance and Collaboration Agreements
We have entered into several alliance, collaboration or license and distribution agreements with respect to certain of our products and services and may enter into similar agreements in the future. These agreements typically obligate us to deliver multiple goods and/or services over extended periods. Such deliverables include manufactured pharmaceutical products, exclusive and semi-exclusive marketing rights, distribution licenses, and research and development services. Our alliance and collaboration agreements often include milestones and provide for payments upon achievement of these milestones. For more information about the types of milestone events in our agreements and how we categorize them, see “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements — Note 17. Alliance and Collaboration Agreements.”
Impax Generics Division – Alliance and Collaboration Agreements
License and Distribution Agreement with Shire
In January 2006, we entered into a License and Distribution Agreement with an affiliate of Shire Laboratories, Inc., which was subsequently amended (“Prior Shire Agreement”), under which we received a non-exclusive license to market and sell an authorized generic of Shire’s Adderall XR® product (“AG Product”) subject to certain conditions, but in any event by no later than January 1, 2010. We commenced sales of the AG Product in October 2009. On February 7, 2013, we entered into an Amended and Restated License and Distribution Agreement with Shire (the “Amended and Restated Shire Agreement”), which amended and restated the Prior Shire Agreement. Pursuant to the terms of the Amended and Restated Shire Agreement, we are required to pay to Shire a specified profit share based on sales of the AG Product and a specified profit share based on sales of our generic Adderall XR® product. We began selling our generic Adderall XR® product during the second quarter of 2016. We accrued a profit share payable to Shire of $2.2 million during the year ended December 31, 2017, based on sales of our generic Adderall XR® product and reflecting adjustments for returns and government rebates from our previous sales of the AG Product and of $7.5 million and $19.5 million during the years ended December 31, 2016 and 2015, respectively, based on sales of the AG Product and our generic Adderall XR® product, in each case with a corresponding charge included in the cost of revenues line in the consolidated statements of operations.
Development, Supply and Distribution Agreement with Tolmar, Inc.
In June 2012, we entered into the Tolmar Agreement with Tolmar. Under the terms of the Tolmar Agreement, Tolmar granted us an exclusive license to commercialize up to 11 generic topical prescription drug products, including 10 currently approved products in the United States and its territories; the parties agreed in 2015 to terminate development efforts of one product under the Tolmar Agreement that had been pending approval at the FDA. Under the terms of the Tolmar Agreement, Tolmar is responsible for developing and manufacturing the products, and we are responsible for marketing and sale of the products. As of December 31, 2017, we were currently marketing and selling four approved products. We are required to pay a profit share to Tolmar on sales of each product commercialized pursuant to the terms of the Tolmar Agreement.
We paid Tolmar a $21.0 million upfront payment upon signing of the agreement and, pursuant to the terms of the agreement, are also required to make payments to Tolmar up to an aggregate amount of $25.0 million upon the achievement of certain specified milestone events. As of December 31, 2017, we had paid a total of $20.0 million to Tolmar upon the achievement of certain specified milestone events, including $12.0 million upon the achievement of a regulatory milestone event and $5.0 million upon the achievement of a commercialization event, and do not currently expect to make any additional milestone payments to Tolmar under the agreement. We are also required to pay a profit share to Tolmar on sales of the topical products, of which we accrued a profit share payable to Tolmar of $10.0 million, $36.4 million and $77.7 million during the years ended December 31, 2017, 2016 and 2015, respectively, with a corresponding charge included in the cost of revenues line in our consolidated statements of operations.
Mebendazole Product Acquisition Agreement with Teva Pharmaceuticals USA, Inc.
In August 2013, we, through our Amedra Pharmaceuticals subsidiary, entered into a product acquisition agreement (the “Mebendazole Product Acquisition Agreement”) with Teva pursuant to which we acquired the assets (including the ANDA and other regulatory materials) and related liabilities related to Teva’s mebendazole tablet product in all dosage forms. Pursuant to the Mebendazole Product Acquisition Agreement, we were required to pay certain milestone payments up to an aggregate amount of $3.5 million upon the approval and launch of the mebendazole tablet product; we paid the $3.5 million to Teva during the quarter ended March 31, 2016 upon the FDA's approval and our subsequent launch of Emverm® (mebendazole) 100 mg chewable tablets. We are also obligated to pay Teva a royalty payment based on net sales of Emverm®, including a specified annual minimum royalty payment, subject to customary reductions and the other terms and conditions set forth in the Mebendazole Product Acquisition Agreement.
10
Rx Partner and OTC Partner Alliance Agreements
We have entered into alliance agreements with unrelated third-party pharmaceutical companies pursuant to which our partner distributes a specified product or products which we developed and, in some cases manufacture. Pursuant to these alliance agreements we typically receive payment on delivery of the product, and share in the resulting profits, or receive a royalty or other payments from our partners. Our alliance agreements are separated into two sales channels, the “Rx Partner” sales channel, for generic prescription products sold through our partners under their own label, and the “OTC Partner” sales channel, for sales of generic pharmaceutical OTC products sold through our partner under their own label. The revenue recognized and the percentage of gross revenue for each of the periods noted, for the Rx Partner and the OTC Partner alliance agreements, was as follows:
Year Ended December 31, | ||||||||||||||||||||
2017 | 2016 | 2015 | ||||||||||||||||||
(in thousands) | ||||||||||||||||||||
Gross Revenue and % Gross Revenue | ||||||||||||||||||||
Rx Partner | $ | 22,674 | 1 | % | $ | 14,339 | 1 | % | $ | 9,307 | 1 | % | ||||||||
OTC Partner | $ | 196 | * | $ | 225 | * | $ | 1,744 | 1 | % | ||||||||||
* Not material
Strategic Alliance Agreement with Teva
We are a party to a Strategic Alliance Agreement dated as of June 27, 2001 with Teva Pharmaceutical USA, Inc. ("Teva USA"), an affiliate of Teva, which was subsequently amended (“Teva Agreement”). The Teva Agreement commits us to develop and manufacture, and Teva to distribute, a specified number of controlled release generic pharmaceutical products (“generic products”), each for a 10-year period. As of December 31, 2017, we were supplying Teva with oxybutynin extended release tablets (Ditropan XL® 5 mg, 10 mg and 15 mg extended release tablets); the other products under the Teva Agreement have either been returned to us, are being manufactured by Teva at its election, were voluntarily withdrawn from the market or our obligations to supply such product had expired or were terminated in accordance with the Teva Agreement.
Impax Specialty Pharma – Alliance and Collaboration Agreements
Distribution, License, Development and Supply Agreement with AstraZeneca UK Limited
In January 2012, we entered into the AZ Agreement with AstraZeneca and the parties subsequently entered into a First Amendment to the AZ Agreement dated May 31, 2016. Under the terms of the AZ Agreement, AstraZeneca granted us an exclusive license to commercialize the tablet, orally disintegrating tablet and nasal spray formulations of Zomig® (zolmitriptan) products for the treatment of migraine headaches in the United States and in certain U.S. territories, except during an initial transition period when AstraZeneca fulfilled all orders of Zomig® products on our behalf and AstraZeneca paid us the gross profit on such Zomig® product sales. Under the terms of the AZ Amendment, under certain conditions and depending on the nature and terms of the study agreed to with the FDA, we agreed to conduct, at our own expense, the juvenile toxicity study and pediatric study required by the FDA under the Pediatric Research Equity Act (“PREA”) for approval of the nasal formulation of Zomig® for the acute treatment of migraine in pediatric patients ages six through eleven years old, as further described in the study protocol mutually agreed to by the parties (the “PREA Study”). In consideration for us conducting the PREA Study at our own expense, the AZ Amendment provides for the total royalty payments payable by us to AstraZeneca on net sales of Zomig® products under the AZ Agreement to be reduced by certain specified amounts beginning from the quarter ended June 30, 2016 and through the quarter ended December 31, 2020, with such reduced royalty amounts totaling an aggregate amount of $30.0 million. In the event the royalty reduction amounts exceed the royalty payments payable by us to AstraZeneca pursuant to the AZ Agreement in any given quarter, AstraZeneca will be required to pay us an amount equal to the difference between the royalty reduction amount and the royalty payment payable by us to AstraZeneca. Our commitment to perform the PREA Study may be terminated, without penalty, under certain circumstances as set forth in the AZ Amendment.
11
In May 2013, our exclusivity period for branded Zomig® tablets and orally disintegrating tablets expired and we launched authorized generic versions of those products in the United States. As discussed above, pursuant to the AZ Amendment, the total royalty payments payable by us to AstraZeneca on net sales of Zomig® products under the AZ Agreement is reduced by certain specified amounts beginning from the quarter ended June 30, 2016 and through the quarter ended December 31, 2020, with such reduced royalty amounts totaling an aggregate amount of $30.0 million. We accrued a royalty payable to AstraZeneca of $17.8 million, $17.2 million and $16.8 million for the years ended December 31, 2017, 2016 and 2015, respectively, with a corresponding charge included in the cost of revenues line on our consolidated statements of operations.
Our Controlled-Release Technology
We have developed a number of different controlled-release delivery technologies which may be utilized with a variety of oral dosage forms and drugs. Controlled-release drug delivery technologies are designed to release drug dosages at specific times and in specific locations in the body and generally provide more consistent and appropriate drug levels in the bloodstream than immediate-release dosage forms. Controlled-release pharmaceuticals may improve drug efficacy, ensure greater patient compliance with the treatment regimen, reduce side effects or increase drug stability and be more patient friendly by reducing the number of times a drug must be taken.
We believe our controlled-release drug delivery technologies are flexible and can be applied to develop a variety of pharmaceutical products, both generic and branded. Our technologies utilize a variety of polymers and other materials to encapsulate or entrap the active pharmaceutical ingredients and to release them at varying rates or at predetermined locations in the gastrointestinal tract.
Competition
The pharmaceutical industry is highly competitive and is affected by new technologies, new developments, government regulations, health care legislation, availability of financing, and other factors. Many of our competitors have longer operating histories and substantially greater financial, research and development, marketing, and other resources than we have. We compete with numerous other companies that currently operate, or intend to operate, in the pharmaceutical industry, including companies that are engaged in the development of controlled-release drug delivery technologies and products, and other manufacturers that may decide to undertake development of such products.
Due to our focus on relatively hard to replicate controlled-release products, competition in the generic pharmaceutical market is sometimes limited to those competitors who possess the appropriate drug delivery technology. The principal competitive factors in the generic pharmaceutical market are:
• | the ability to introduce generic versions of products promptly after a patent expires; |
• | price; |
• | product quality; |
• | customer service (including maintenance of inventories for timely delivery); and |
• | the ability to identify and market niche products. |
In the brand-name pharmaceutical market, our principal competitors are pharmaceutical companies that are focused on Parkinson’s disease and other CNS disorders. In addition, with respect to products that we are developing internally and/or any additional products we may in-license from third parties, we expect that we will face increased competition from large pharmaceutical companies, drug delivery companies and other specialty pharmaceutical companies that have focused on the same disorders as our branded products. Our principal competitors in the generic pharmaceutical products market are Teva Pharmaceutical Industries Ltd., Mylan N.V., Cipla Inc., Lannett Company, Inc., and Sandoz.
A description of the competition we face from brand-name and generic pharmaceutical companies is included in “Item 1A. Risk Factors.”
12
Sales and Marketing
We market and sell our generic pharmaceutical prescription drug products within the continental United States and the Commonwealth of Puerto Rico. We have not made sales in any other jurisdictions over the last three fiscal years. We derive a substantial portion of our revenue from sales to a limited number of customers. The customer base for our products consists primarily of drug wholesalers, warehousing chain drug stores, mass merchandisers, and mail-order pharmacies. We market our products both directly, through our Impax Generics and Impax Specialty Pharma divisions, and indirectly through our Rx Partner and OTC Partner alliance and collaboration agreements. Together, our three major customers, Cardinal Health, McKesson Corporation, and Amerisource-Bergen accounted for 88% of our gross revenue for the year ended December 31, 2017. These three customers individually accounted for 33%, 30%, and 25%, respectively, of our total gross revenue for the year ended December 31, 2017. We do not have long-term contracts in effect with our three major customers. A reduction in or loss of business with any one of these customers, or any failure of a customer to pay us on a timely basis, would adversely affect our business.
Manufacturing and Distribution
We source our finished dosage form products from our own facility in Hayward, California. We also use several contract manufacturers for this purpose. During 2015, we restructured our packaging and distribution operations. As a result, we closed our Philadelphia packaging site and all of our company-wide distribution operations were outsourced to United Parcel Services (UPS). During 2017, we closed our Middlesex, New Jersey manufacturing facility and in early 2018, we sold CorePharma, LLC, our wholly owned subsidiary that held the leases to the site. We additionally announced during 2017 that we entered into a stock and asset purchase agreement with Bora Pharmaceuticals Co., Ltd., pursuant to which we agreed to sell Impax Laboratories (Taiwan), Inc. (“Impax Taiwan”), our wholly owned subsidiary which owns our manufacturing facility in Taiwan, R.O.C. The sale of Impax Taiwan subsequently closed in February 2018.
We maintain an inventory of our products in connection with our obligations under our alliance and collaboration agreements. In addition, for products pending approval, we may produce batches for inventory in anticipation of the launch of the products. In the event that FDA approval is denied or delayed, we could be exposed to the risk of this inventory becoming obsolete.
Raw Materials
The raw materials we use in the production of our products consist of pharmaceutical chemicals in various forms that are generally available from several sources in the United States and throughout the world. In some cases, however, the raw materials, such as the active pharmaceutical ingredients (“API”) used to manufacture our products, are available only from a single supplier. Further, even if more than one supplier exists, we may choose, and have done so in the case of our API suppliers for a majority of our products, to list only one supplier in our product applications submitted to the FDA. The FDA requires identification of raw material suppliers in applications for approval of drug products. If raw materials were unavailable from a specified supplier or the supplier was not in compliance with FDA or other applicable requirements, the FDA approval of a new supplier could delay the manufacture of the drug involved. As a result, there is no guarantee we will always have timely and sufficient access to a required raw material or other product. Generally, we would need as long as 18 months to find and qualify a new sole-source supplier. If we receive less than one year’s termination notice from a sole-source supplier that it intends to cease supplying raw materials, it could result in disruption of our ability to produce the drug involved. We currently do not have long-term supply agreements with the majority of our API suppliers and although to date we have only experienced occasional interruptions in supplies, no assurance can be given that we will continue to receive uninterrupted or adequate supplies of such raw materials. Any inability to obtain raw materials on a timely basis, or any significant price increases not passed on to customers, could have a material adverse effect on us.
13
Quality Control
Regulatory agencies such as the FDA regularly inspect our manufacturing facilities and the facilities of our third party suppliers. The failure of one of our facilities, or a facility of one of our third party suppliers, to comply with applicable laws and regulations may lead to breach of representations made to our customers or to regulatory or government action against us related to products made in that facility. We have in the past received a warning letter from the FDA regarding certain operations within our manufacturing network at our Hayward manufacturing facility, which we subsequently resolved in 2015. We remain committed to continuing to improve our quality control and manufacturing practices, however, we cannot be assured that the FDA will continue to be satisfied with our corrective actions and with our quality control and manufacturing systems and standards. Failure to comply strictly with these regulations and requirements may damage our reputation and lead to financial penalties, compliance expenditures, the recall or seizure of products, total or partial suspension of production and/or distribution, withdrawal or suspension of the applicable regulator’s review of our submissions, enforcement actions, injunctions and criminal prosecution. Further, other federal agencies, our customers and partners in our alliance, development, collaboration and other partnership agreements with respect to our products and services may take any such FDA observations or warning letters into account when considering the award of contracts or the continuation or extension of such partnership agreements. Because regulatory approval to manufacture a drug is site-specific, the delay and cost of remedial actions, or obtaining approval to manufacture at a different facility, could negatively impact our business. Any failure by us to comply with applicable laws and regulations and/or any actions by the FDA and other agencies as described above could have a material adverse effect on our business, financial position and results of operations.
Research and Development
We conduct most of our research and development activities at our facility in Hayward, California with a staff of 153 employees as of December 31, 2017. In addition, we have outsourced a number of research and development projects to third-party laboratories.
We spent $80.8 million, $80.5 million and $70.6 million on research and development activities during the years ended December 31, 2017, 2016 and 2015, respectively, as more fully set out in the tables below (in millions).
Year Ended December 31, 2017 | Impax Generics | Impax Specialty Pharma | Total Impax | ||||||||
Clinical study expenses | $ | 9.0 | $ | 1.7 | $ | 10.7 | |||||
Personnel expenses | 24.8 | 8.0 | 32.8 | ||||||||
Experimental materials | 4.7 | 0.1 | 4.8 | ||||||||
Outside services | 7.6 | 4.2 | 11.8 | ||||||||
Facility expenses | 3.6 | 0.5 | 4.1 | ||||||||
Legal expenses | 0.5 | 0.2 | 0.7 | ||||||||
Other | 13.1 | 2.8 | 15.9 | ||||||||
Total | $ | 63.3 | $ | 17.5 | $ | 80.8 | |||||
Year Ended December 31, 2016 | Impax Generics | Impax Specialty Pharma | Total Impax | ||||||||
Clinical study expenses | $ | 11.1 | $ | 2.4 | $ | 13.5 | |||||
Personnel expenses | 25.0 | 10.8 | 35.8 | ||||||||
Experimental materials | 6.1 | — | 6.1 | ||||||||
Outside services | 7.1 | 3.3 | 10.4 | ||||||||
Facility expenses | 3.7 | 0.5 | 4.2 | ||||||||
Legal expenses | 0.1 | 0.2 | 0.3 | ||||||||
Other | 9.1 | 1.1 | 10.2 | ||||||||
Total | $ | 62.2 | $ | 18.3 | $ | 80.5 | |||||
14
Year Ended December 31, 2015 | Impax Generics | Impax Specialty Pharma | Total Impax | ||||||||
Clinical study expenses | $ | 4.6 | $ | 0.8 | $ | 5.4 | |||||
Personnel expenses | 28.6 | 10.0 | 38.6 | ||||||||
Experimental materials | 4.3 | — | 4.3 | ||||||||
Outside services | 5.8 | 4.5 | 10.3 | ||||||||
Facility expenses | 4.2 | 0.4 | 4.6 | ||||||||
Legal expenses | 0.4 | 0.2 | 0.6 | ||||||||
Other | 4.6 | 2.2 | 6.8 | ||||||||
Total | $ | 52.5 | $ | 18.1 | $ | 70.6 | |||||
We do not generally track research and development expense by individual product in either the Impax Generics division or the Impax Specialty Pharma division.
In the Impax Generics division, we focus our research and development efforts based on drug-delivery technology and on products that we believe may have certain competitive advantages, rather than on any particular therapeutic area. As of December 31, 2017, the Impax Generics division had 17 product applications pending with the FDA and another 20 products in development. Accordingly, we believe that our generic pipeline products will, in the aggregate, generate a significant amount of revenue for us in the future. However, while a generic product is still in development, we are unable to predict the level of commercial success that the product may ultimately achieve given the uncertainties relating to the successful and timely completion of bioequivalence studies, ANDA filing, receipt of marketing approval and resolution of any related patent litigation, as well as the amount of competition in the market at the time of product launch and thereafter and other factors detailed in “Item 1A. Risk Factors.” Additionally, we do not believe that any individual generic pipeline product is currently significant in terms of accrued or anticipated research and development expense given the large volume of products under development in the Impax Generics division, as detailed above. Further, on a per product basis, development costs for generic products tend to be significantly lower than for branded products, as the process for establishing bioequivalence is significantly less extensive than the standard clinical trial process. The regulatory approval process is significantly less onerous as well compared to the process for branded products.
In the Impax Specialty Pharma division, we currently market one internally developed branded pharmaceutical product, Rytary® (IPX066) for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication and/or manganese intoxication, which was approved by the FDA on January 7, 2015 and which we launched in the United States in April 2015. In addition to Rytary®, Impax Specialty Pharma is also currently engaged in the sale and distribution of four other branded products; the more significant include Zomig® (zolmitriptan) products, indicated for the treatment of migraine headaches, under the terms the AZ Agreement, and Emverm® (mebendazole) 100 mg chewable tablets, indicated for the treatment of pinworm, whipworm, common roundworm, common hookworm, and American hookworm in single or mixed infection. We also have a number of product candidates that are in varying stages of development. While we believe the pipeline products in this division are potentially viable, profitable product candidates for us, given the uncertainties relating to the successful completion of clinical trials, the FDA approval process for branded products, reimbursement levels, the amount of competition at the time of product launch and thereafter and other factors detailed in “Item 1A. Risk Factors,” such pipeline products are too early in the development process to be considered significant at this point in time.
Regulation
The manufacturing and distribution of pharmaceutical products are subject to extensive regulation by the federal government, primarily through the FDA and the Drug Enforcement Administration (“DEA”), and to a lesser extent by state and local governments. The Food, Drug, and Cosmetic Act, Controlled Substances Act and other federal statutes and regulations govern or influence the manufacture, labeling, testing, storage, record keeping, approval, advertising and promotion of our products. As described above under "Quality Control", facilities used in the manufacture, packaging, labeling and repackaging of pharmaceutical products must be registered with the FDA and are subject to FDA inspection to ensure that drug products are manufactured in accordance with current Good Manufacturing Practices. Noncompliance with applicable requirements can result in product recalls, seizure of products, injunctions, suspension of production and/or distribution, refusal of the government or third parties to enter into contracts with us, withdrawal or suspension of the applicable regulator's review of our drug applications, civil penalties and criminal fines, and disgorgement of profits.
15
FDA approval is required before any “new drug” may be marketed, including new formulations, strengths, dosage forms and generic versions of previously approved drugs. Generally, the following two types of applications are used to obtain FDA approval of a “new drug.”
New Drug Application (“NDA”). For a drug product containing an active ingredient not previously approved by the FDA, a prospective manufacturer must submit a complete application containing the results of clinical studies supporting the drug product’s safety and efficacy. A NDA is also required for a drug with a previously approved active ingredient if the drug will be used to treat an indication for which the drug was not previously approved or if the dosage form, strength or method of delivery is changed. The process required by the FDA before a pharmaceutical product may be approved for marketing in the U.S. generally involves the steps listed below, which could take from approximately three to more than ten years to complete.
• | Laboratory and clinical tests; |
• | Submission of an Investigational New Drug (“IND”) application, which must become effective before clinical studies may begin; |
• | Adequate and well-controlled human clinical studies to establish the safety and efficacy of the proposed product for its intended use; |
• | Submission of a NDA containing the results of the preclinical tests and clinical studies establishing the safety and efficacy of the proposed product for its intended use, as well as extensive data addressing such matters such as manufacturing and quality assurance; |
• | Scale-up to commercial manufacturing; and |
• | FDA approval of a NDA. |
As noted above, the submission of a NDA is not a guarantee that the FDA will find it complete and accept it for filing. The FDA reviews all NDAs submitted before it accepts them for filing. It may refuse to file the application and instead request additional information, in which case, the application must be resubmitted with the supplemental information. After the application is deemed filed by the FDA, FDA staff will review a NDA to determine, among other things, whether a product is safe and efficacious for its intended use.
If, after reviewing the NDA, the FDA determines that the application cannot be approved in its current form, the FDA sends the NDA applicant a Complete Response Letter identifying all outstanding deficiencies that preclude final approval. The FDA then halts its review until the applicant resubmits the NDA with new information designed to address the deficiencies. An applicant receiving a Complete Response Letter may resubmit the application with data and information addressing the FDA’s concerns or requirements, withdraw the application without prejudice to a subsequent submission of a related application or request a hearing on whether there are grounds for denying approval of the application. If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may require an applicant to conduct Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval, and may require surveillance programs to monitor the safety of approved products which have been commercialized. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety or efficacy questions are raised after the product reaches the market. The agency may also impose requirements that the NDA holder conduct new studies, make labeling changes, implement Risk Evaluation and Mitigation Strategies, and take other corrective measures.
16
Abbreviated New Drug Application (“ANDA”). For a generic version of an approved drug — a drug product that contains the same active ingredient as a drug previously approved by the FDA and is in the same dosage form and strength, utilizes the same method of delivery and will be used to treat the same indications as the approved product — the FDA requires only an abbreviated new drug application that ordinarily need not include clinical studies demonstrating safety and efficacy. An ANDA typically requires only data demonstrating that the generic formulation is bioequivalent to the previously approved “reference listed drug,” indicating that the rate of absorption and levels of concentration of the generic drug in the body do not show a significant difference from those of the reference listed drug. In July 2012, the Generic Drug Fee User Amendments of 2012 (“GDUFA”) was enacted into law. The GDUFA legislation implemented fees for new ANDA applications, Drug Master Files, product and establishment fees and a one-time fee for back-logged ANDA applications pending approval as of October 1, 2012. In return, the program was intended to provide faster and more predictable ANDA reviews by the FDA and increased inspections of drug facilities. Under GDUFA, generic product companies face significant penalties for failure to pay the new user fees, including rendering an ANDA application not “substantially complete” until the fee is paid. Prior to the implementation of GDUFA, the FDA took an average of approximately 30 months to approve an ANDA. Following the implementation of GDUFA, the FDA’s stated internal goal for ANDAs submitted in fiscal year 2016 was to have a “first-action” goal date within 15 months of submission on 75% of submitted ANDAs. The “first-action” goal date is referred to by the FDA as the date in which the FDA takes a first action on an application by either granting approval or tentative approval or in the event of deficiencies, identifying those deficiencies in a complete response letter or in a refusal to receive the application.
Under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the “Hatch-Waxman Act,” which established the procedures for obtaining approval of generic drugs, an ANDA filer must make certain patent certifications that can result in significant delays in obtaining FDA approval. If the applicant intends to challenge the validity or enforceability of an existing patent covering the reference listed drug or asserts that its drug does not infringe such patent, the applicant files a so called “Paragraph IV” certification and notifies the patent holder that it has done so, explaining the basis for its belief that the patent is not infringed or is invalid or unenforceable. If the patent holder initiates a patent infringement suit within 45 days after receipt of the Paragraph IV Certification, the FDA is automatically prevented from approving an ANDA until the earlier of 30 months after the date the Paragraph IV Certification is given to the patent holder, expiration of the patents involved in the certification, or when the infringement case is decided in the ANDA applicant’s favor. In addition, the first company to file an ANDA for a given drug containing a Paragraph IV certification can be awarded 180 days of market exclusivity following approval of its ANDA, during which the FDA may not approve any other ANDAs for that drug product.
During any period in which the FDA is required to withhold its approval of an ANDA due to a statutorily imposed non-approval period, the FDA may grant tentative approval to an applicant’s ANDA. A tentative approval reflects the FDA’s preliminary determination that a generic product satisfies the substantive requirements for approval, subject to the expiration of all statutorily imposed non-approval periods. A tentative approval does not allow the applicant to market the generic drug product.
The Hatch-Waxman Act contains additional provisions that can delay the launch of generic products. A five year marketing exclusivity period is provided for new chemical compounds, and a three year marketing exclusivity period is provided for approved applications containing new clinical investigations essential to an approval, such as a new indication for use, or new delivery technologies, or new dosage forms. The three year marketing exclusivity period applies to, among other things, the development of a novel drug delivery system, as well as a new use. In addition, companies can obtain six additional months of exclusivity if they perform pediatric studies of a reference listed drug product. The marketing exclusivity provisions apply to both patented and non-patented drug products. The Act also provides for patent term extensions to compensate for patent protection lost due to time taken in conducting FDA required clinical studies and during FDA review of NDAs.
The Generic Drug Enforcement Act of 1992 establishes penalties for wrongdoing in connection with the development or submission of an ANDA. In general, the FDA is authorized to temporarily bar companies, or temporarily or permanently bar individuals, from submitting or assisting in the submission of an ANDA, and to temporarily deny approval and suspend applications to market generic drugs under certain circumstances. In addition to debarment, the FDA has numerous discretionary disciplinary powers, including the authority to withdraw approval of an ANDA or to approve an ANDA under certain circumstances and to suspend the distribution of all drugs approved or developed in connection with certain wrongful conduct. The FDA may also withdraw product approval or take other correct measures if ongoing regulatory requirements are not met or if safety or efficacy questions are raised after the product reaches the market.
17
Other Regulatory Requirements
We are subject to the Maximum Allowable Cost Regulations, which limit reimbursements for certain generic prescription drugs under Medicare, Medicaid, and other programs to the lowest price at which these drugs are generally available. In many instances, only generic prescription drugs fall within the regulations’ limits. Generally, the pricing and promotion of, method of reimbursement and fixing of reimbursement levels for, and the reporting to federal and state agencies relating to drug products is under active review by federal, state and local governmental entities, as well as by private third-party reimbursers and individuals under whistleblower statutes. At present, the Justice Department and U.S. Attorneys Offices and State Attorneys General have initiated investigations, reviews, and litigation into industry-wide pharmaceutical pricing and promotional practices, and whistleblowers have filed qui tam suits. We cannot predict the results of those reviews, investigations, and litigation, or their impact on our business.
Virtually every state, as well as the District of Columbia, has enacted legislation permitting the substitution of equivalent generic prescription drugs for brand-name drugs where authorized or not prohibited by the prescribing physician, and some states mandate generic substitution in Medicaid programs.
In addition, numerous state and federal requirements exist for a variety of controlled substances, such as narcotics, that may be part of our product formulations. The DEA, which has authority similar to the FDA’s and may also pursue monetary penalties, and other federal and state regulatory agencies have far reaching authority.
The State of California requires that any manufacturer, wholesaler, retailer or other entity in California that sells, transfers, or otherwise furnishes certain so called precursor substances must have a permit issued by the California Department of Justice, Bureau of Narcotic Enforcement. The substances covered by this requirement include ephedrine, pseudoephedrine, norpseudoephedrine, and phenylpropanolamine, among others. The Bureau has authority to issue, suspend and revoke precursor permits, and a permit may be denied, revoked or suspended for various reasons, including (i) failure to maintain effective controls against diversion of precursors to unauthorized persons or entities; (ii) failure to comply with the Health and Safety Code provisions relating to precursor substances, or any regulations adopted thereunder; (iii) commission of any act which would demonstrate actual or potential unfitness to hold a permit in light of the public safety and welfare, which act is substantially related to the qualifications, functions or duties of the permit holder; or (iv) if any individual owner, manager, agent, representative or employee of the permit applicant/permit holder willfully violates any federal, state or local criminal statute, rule, or ordinance relating to the manufacture, maintenance, disposal, sale, transfer or furnishing of any precursor substances.
Patents, Trademarks and Licenses
We own or license a number of patents in the U.S. and other countries covering certain products and product candidates and have also developed brand names and trademarks for other products and product candidates. Generally, the brand pharmaceutical business relies upon patent protection to ensure market exclusivity for the life of the patent. We consider the overall protection of our patents, trademarks and license rights to be of material value and act to protect these rights from infringement. However, our business is not dependent upon any single patent, trademark or license.
In the branded pharmaceutical industry, the majority of an innovative product’s commercial value is usually realized during the period in which the product has market exclusivity. In the U.S. and some other countries, when market exclusivity expires and generic versions of a product are approved and marketed, there can often be very substantial and rapid declines in the branded product’s sales. The rate of this decline varies by country and by therapeutic category; however, following patent expiration, branded products often continue to have market viability based upon the goodwill of the product name, which typically benefits from trademark protection.
An innovator product’s market exclusivity is generally determined by two forms of intellectual property: patent rights held by the innovator company and any regulatory forms of exclusivity to which the innovator is entitled.
Patents are a key determinant of market exclusivity for most branded pharmaceuticals. Patents provide the innovator with the right to exclude others from practicing an invention related to the medicine. Patents may cover, among other things, the active ingredient(s), various uses of a drug product, pharmaceutical formulations, drug delivery mechanisms and processes for (or intermediates useful in) the manufacture of products. Protection for individual products extends for varying periods in accordance with the expiration dates of patents in the various countries. The protection afforded, which may also vary from country to country, depends upon the type of patent, its scope of coverage and the availability of meaningful legal remedies in the country.
18
Market exclusivity is also sometimes influenced by regulatory exclusivity rights. Many developed countries provide certain non-patent incentives for the development of medicines. For example, the U.S., the EU and Japan each provide for a minimum period of time after the approval of a new drug during which the regulatory agency may not rely upon the innovator’s data to approve a competitor’s generic copy. Regulatory exclusivity rights are also available in certain markets as incentives for research on new indications, on orphan drugs and on medicines useful in treating pediatric patients. Regulatory exclusivity rights are independent of any patent rights and can be particularly important when a drug lacks broad patent protection. However, most regulatory forms of exclusivity do not prevent a competitor from gaining regulatory approval prior to the expiration of regulatory data exclusivity on the basis of the competitor’s own safety and efficacy data on its drug, even when that drug is identical to that marketed by the innovator.
We estimate the likely market exclusivity period for each of our branded products on a case-by-case basis. It is not possible to predict the length of market exclusivity for any of our branded products with certainty because of the complex interaction between patent and regulatory forms of exclusivity, and inherent uncertainties concerning patent litigation. There can be no assurance that a particular product will enjoy market exclusivity for the full period of time that we currently estimate or that the exclusivity will be limited to the estimate.
In addition to patents and regulatory forms of exclusivity, we also market products with trademarks. Trademarks have no effect on market exclusivity for a product, but are considered to have marketing value. Trademark protection continues in some countries as long as used; in other countries, as long as registered. Registration is for fixed terms and may be renewed indefinitely.
Environmental Laws
We are subject to comprehensive federal, state and local environmental laws and regulations that govern, among other things, air polluting emissions, waste water discharges, solid and hazardous waste disposal and the remediation of contamination associated with current or past generation handling and disposal activities. We are subject periodically to environmental compliance reviews by various environmental regulatory agencies. While it is impossible to predict accurately the future costs associated with environmental compliance and potential remediation activities, compliance with environmental laws is not expected to require significant capital expenditures and has not had, and is not expected to have, a material adverse effect on our business, operations or financial condition.
Available Information
We maintain an Internet website at the following address: www.impaxlabs.com. We make available on or through our Internet website certain reports and amendments to those reports, as applicable, that we file with or furnish to the Securities and Exchange Commission (the “SEC”) in accordance with the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These include our Annual Report on Form 10-K, our Quarterly Reports on Form 10-Q and our Current Reports on Form 8-K. Our website also includes our Code of Conduct and the charters of our Audit Committee, Nominating Committee, Compensation Committee and Compliance Committee of our Board of Directors. We make this information available on our website free of charge, as soon as reasonably practicable after we electronically file the information with, or furnish it to, the SEC. The contents of our website are not incorporated by reference in this Annual Report on Form 10-K and shall not be deemed “filed” under the Exchange Act.
Corporate and Other Information
We were incorporated in the State of Delaware in 1995. Our corporate headquarters are located at 30831 Huntwood Avenue, Hayward, California, 94544. We were formerly known as Global Pharmaceutical Corporation until December 14, 1999, when Impax Pharmaceuticals, Inc., a privately held drug delivery company, merged into Global Pharmaceutical Corporation and the name of the resulting entity was changed to Impax Laboratories, Inc.
Unless otherwise indicated, all product sales data and U.S. market size data in this Annual Report on Form 10-K are based on information obtained from IQVA, formerly known as IMS Health, unrelated third-party providers of prescription market data. We did not independently engage IQVA to provide this information.
Employees
As of December 31, 2017, we had 1,257 full-time employees, of which 409 were in operations, 153 in research and development, 320 in the quality area, 210 in legal and administration, and 165 in sales and marketing. None of our employees are subject to collective bargaining agreements with labor unions, and we believe our employee relations are good.
19
Item 1A. Risk Factors
An investment in our common stock involves a high degree of risk. In deciding whether to invest in our common stock, you should consider carefully the following risk factors, as well as the other information included in this Annual Report on Form 10-K. The materialization of any of these risks could have a material adverse effect on our business, results of operations and financial condition. This Annual Report on Form 10-K contains forward looking statements that involve risks and uncertainties. Our actual results could differ materially from the results discussed in the forward looking statements. Factors that could cause or contribute to these differences include those discussed in this “Risk Factors” section. See “Forward-Looking Statements” on page 1 of this Annual Report on Form 10-K.
Risks Related to Our Business
Our operating results and financial condition could fluctuate significantly.
Our operating results and financial condition may vary significantly from year to year and quarter to quarter as well as in comparison to the corresponding year or quarter of the preceding year, as the case may be, for a number of reasons, including all the risks described in this section. We also cannot predict with any certainty the timing or level of sales of our products in the future. Our operating results and profitability are also dependent upon the costs for us to purchase products from third parties and our ability to manufacture our products in a cost effective manner. If we are unable to reduce our operating expenses to offset a decline in revenue of our products for a particular fiscal period due to existing or new competition, product supply or any other reasons, we could experience a material and adverse effect on our business, results of operations and financial condition in such periods.
Due to the fluctuations in our operating results and financial condition, we believe that period-to-period comparisons of our operating results are not necessarily meaningful and should not be relied upon as indications of our future performance and any full-year financial forecast should not be relied upon as a guarantee of future performance for that year or for any given quarter within that year. If our operating results fall below the expectations of investors or securities analysts, the value of our securities could decline substantially and our business, results of operations and financial condition could be materially and adversely affected, as further described below under the risk factor, “The market price of our common stock has been volatile and may continue to be volatile in the future, and the value of any investment in our common stock could decline significantly.”
The market price of our common stock has been volatile and may continue to be volatile in the future, and the value of any investment in our common stock could decline significantly.
The market price for our shares of common stock listed on the Nasdaq Stock Market has fluctuated significantly from time to time, for example, varying between a high of $25.70 on September 21, 2017 to a low of $7.75 on March 7, 2017 during the year ended December 31, 2017. The market price of our common stock is likely to continue to be volatile and subject to significant price and volume fluctuations in response to market, industry and other factors, including the risks described in this section. Further, the stock market for pharmaceutical companies has recently experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. In particular, recent negative publicity regarding pricing and price increases by pharmaceutical companies has negatively impacted, and may continue to negatively impact, the market for pharmaceutical companies. These broad market and industry factors have negatively impacted, and in the future may seriously negatively impact, the market price of our common stock, regardless of our operating performance.
Our stock market price may also be dependent upon the valuations and recommendations of the analysts who cover our business. If our results do not meet these analysts’ forecasts, the expectations of our investors or the financial guidance we provide to investors in any period, the market price of our common stock could decline. In the past, following periods of volatility in the market or significant price decline, securities class-action litigation has often been instituted against companies and we have in the past been subject to such suits. Such suits could result in substantial costs and diversion of management’s attention and resources, which could materially and adversely affect our business, results of operations and financial condition.
20
Our continued growth is dependent on our ability to continue to successfully develop and commercialize new products in a timely manner.
Our financial results depend upon our ability to introduce and commercialize additional generic and branded products in a timely manner. As of December 31, 2017, we had 17 product applications pending at the FDA and 20 product candidates under development for generic versions of brand-name pharmaceuticals. In our branded products division, we have a few product candidates in various stages of development, including IPX203, a new extended-release oral capsule formulation of carbidopa and levodopa, as a potential treatment for symptoms of Parkinson’s disease.
The future profitability of our Impax Generics Division depends, to a significant extent, upon our ability to introduce, on a timely basis, new generic products that are either the first-to-market (or among the first-to-market) or that otherwise can gain significant share during the 180-day marketing period as permitted by the Hatch-Waxman Act. The Hatch-Waxman amendments to the Federal Food, Drug, and Cosmetic Act provide for a period of 180 days of marketing exclusivity for a generic version of a previously approved drug for any applicant that is first-to-file an ANDA containing a certification of invalidity, non-infringement or unenforceability related to a patent listed with respect to the corresponding brand-name drug (commonly referred to as a “Paragraph IV certification”). ANDAs that contain Paragraph IV certifications challenging patents, however, generally become the subject of patent litigation that can be both lengthy and costly. We cannot assure that we will prevail in any such patent litigation, that we will be the first-to-file and be granted the 180-day marketing exclusivity period, or, if we are granted the 180-day marketing exclusivity period, that we will not forfeit such period. Even in the event where our ANDA is awarded marketing exclusivity, we may be required to share our exclusivity period with other ANDA applicants who submit Paragraph IV certifications. Further, branded pharmaceutical companies often authorize a generic version of the corresponding brand name drug to be sold during any period of marketing exclusivity that is awarded. Furthermore, timely commencement of the litigation by the patent owner imposes an automatic stay of ANDA approval by the FDA for 30 months, unless the case is decided in the ANDA applicant’s favor during that period. Finally, if the court decision is adverse to the ANDA applicant, the ANDA approval will be delayed until the challenged patent expires, and the applicant will not be granted the 180-day marketing exclusivity. Our ability to timely bring our products to market is thus dependent upon, among other things, the timing of regulatory approval of our products, which to a large extent is outside of our control, as well as the timing of competing products. Our revenues and future profitability are also dependent, in large part, upon our ability or the ability of our development partners to file, timely and effectively, ANDAs with the FDA or to enter into collaboration agreements or other contractual relationships with third parties that have obtained marketing exclusivity.
The development and commercialization process for our branded products in the Impax Specialty Pharma Division is time-consuming, costly and involves processes and expertise that are different from those used in the development of our generic products, thus creating a higher degree of risk of failure. For example, the time from discovery to commercial launch of a branded specialty product can be 15 years or even longer, and involves multiple stages from intensive preclinical and clinical testing to a highly complex, lengthy and expensive approval process with the FDA. The longer it takes to develop a product, the less time there will be for us to recover our development costs and generate profits. The FDA and the regulatory authorities may not approve our products submitted to them or our other products under development. Additionally, we may not successfully complete our development efforts. Even if the FDA approves our products, we may not be able to market them successfully or profitably or our future results of operations will depend significantly upon our ability to timely develop, receive FDA approval for, and market new pharmaceutical products or otherwise acquire new products.
No assurances can be given that we will be able to develop and introduce commercially successful products in the future within the time constraints necessary to be successful. If we or our development partners are unable to continue to timely and effectively file ANDAs with the FDA or to partner with other parties that have obtained marketing exclusivity or if we are unable to timely develop and receive approval of our branded pipeline products, our revenues and operating results may decline significantly and our business, results of operations and financial condition could be materially and adversely affected.
We face intense competition from both brand-name and generic pharmaceutical companies.
The pharmaceutical industry is highly competitive and many of our competitors have longer operating histories and substantially greater financial, research and development, marketing, and other resources than we have. Further, the pharmaceutical industry has in recent years seen increased consolidation, resulting in larger competitors and placing further pressure on prices, development activities and customer retention. In addition, pharmaceutical manufacturers’ customer base consists of an increasingly limited number of large pharmaceutical wholesalers, chain drug stores that warehouse products, mass merchandisers and mail order pharmacies. Our competitors may be able to develop products competitive with or more effective or less expensive than our own for many reasons, including that they may have:
• | proprietary processes or delivery systems; |
• | greater resources in the area of research and development and marketing; |
21
• | larger or more efficient production capabilities; |
• | more expertise in a particular therapeutic area; |
• | more expertise in preclinical testing and human clinical trials; |
• | more experience in obtaining required regulatory approvals, including FDA approval; |
• | more breadth of products; or |
• | more experience in developing new drugs and financial resources, particularly with regard to brand manufacturers. |
In the generic products market, we face competition from other generic pharmaceutical companies, which may impact our selling price and revenues from such products. The FDA approval process often results in the FDA granting final approval to a number of ANDAs for a given product at the time a patent for a corresponding brand product or other market exclusivity expires. This often forces us to face immediate competition when we introduce a generic product into the market. As competition from other generic pharmaceutical companies intensifies, selling prices and gross profit margins often decline, which has been our experience with our existing products. Moreover, with respect to products for which we file a Paragraph IV certification, if we are not the first ANDA filer challenging a listed patent for a product, we are at a significant disadvantage to the competitor that first filed an ANDA for that product containing such a challenge, which is awarded 180 days of market exclusivity for the product. Conversely, in some cases when we are the first ANDA filer to challenge a listed patent, we may forfeit our 180 days of market exclusivity under certain circumstances. In that case, a competitor may obtain ANDA approval earlier than we obtain ANDA approval, in which case we will be at a disadvantage to such competitor. Accordingly, the level of market share, revenue and gross profit attributable to a particular generic product that we develop is generally related to the number of competitors in that product’s market and the timing of that product’s regulatory approval and launch, in relation to competing approvals and launches. Although we cannot assure, we strive to develop and introduce new products in a timely and cost effective manner to be competitive in our industry (see “Item 1. Business — Regulation”). Additionally, ANDA approvals often continue to be granted for a given product subsequent to the initial launch of the generic product. These circumstances generally result in significantly lower prices and reduced margins for generic products compared to brand products. New generic market entrants generally cause continued price and margin erosion over the generic product life cycle.
In addition to the competition we face from other generic pharmaceutical companies related to our generic products, we also face competition from brand-name pharmaceutical companies that may try to prevent, discourage or delay the use of generic versions through various measures, including introduction of new branded products, legislative initiatives, changing dosage forms or dosing regimens, regulatory processes, filing new patents or patent extensions, lawsuits, citizens’ petitions, and negative publicity prior to introduction of a generic product. In addition, brand-name competitors may lower their prices to compete with generic products, increase advertising, or launch, either through an affiliate or licensing arrangements with another company, an authorized generic at or near the time the first generic product is launched, reducing the generic product market exclusivity provided by the Hatch-Waxman Act.
Our principal competitors in the generic pharmaceutical products market are Teva Pharmaceutical Industries Ltd., Mylan N.V., Cipla, Inc, Lannett Company, Inc., Lupin Pharmaceuticals, Inc., and Sandoz.
In the brand-name pharmaceutical market, our principal competitors are pharmaceutical companies that are focused on Parkinson’s disease and other CNS disorders, many of whom have substantially greater resources and longer operating histories and experience with the development, marketing and/or acquisition of branded products. Such companies have greater resources to devote to the marketing of new products and as such, we face increased pressure to demonstrate to our physicians, patients and third party payors the benefits of our branded products compared to competing or comparable products, whose benefits may be more familiar or better established. Further, our branded products also face competition from generic companies who may develop and receive approval on generic versions of our products, resulting in decreased selling price and reduced revenue from the sale of such products. For instance, we have filed suits against Actavis Laboratories, Inc., Sandoz Inc. and Zydus Pharmaceuticals USA relating to their respective ANDAs for carbidopa and levodopa extended release capsules, generic to Rytary® (see “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 19. Legal and Regulatory Matters” for a description of the suits). If such litigation is resolved unfavorably for us such that we are unable to rely on our patent protection for Rytary® as a barrier to entry of potential generic versions of the product, we may experience significant decreased revenue from sales of Rytary® which could materially and adversely affect our business, results of operations and financial condition.
22
Manufacturing or quality control problems may damage our reputation for quality production, demand costly remedial activities and negatively impact our business, results of operations and financial condition.
As a pharmaceutical company, we are subject to substantial regulation by various governmental authorities. For instance, we must comply with requirements of the U.S. Food and Drug Administration (“FDA”) and other healthcare regulators with respect to the manufacture, labeling, sale, distribution, marketing, advertising, promotion and development of pharmaceutical products. We must register our facilities, whether located in the United States or elsewhere, with the FDA as well as regulators outside the United States, and our products must be made in a manner consistent with current good manufacturing practices (“cGMP”), or similar standards in each territory in which we manufacture. The failure of one of our facilities, or a facility of one of our third party suppliers, to comply with applicable laws and regulations may lead to breach of representations made to our customers or to regulatory or government action against us related to products made in that facility.
In addition, the FDA and other agencies periodically inspect our manufacturing facilities. Following an inspection, an agency may issue a notice listing conditions that are believed to violate cGMP or other regulations, or a warning letter for violations of “regulatory significance” that may result in enforcement action if not promptly and adequately corrected. We have in the past received a warning letter from the FDA regarding certain operations within our manufacturing network at our Hayward manufacturing facility, which we subsequently resolved in 2015. We remain committed to continuing to improve our quality control and manufacturing practices; however, we cannot be assured that the FDA will continue to be satisfied with our corrective actions and with our quality control and manufacturing systems and standards. Failure to comply strictly with these regulations and requirements may damage our reputation and lead to financial penalties, compliance expenditures, the recall or seizure of products, total or partial suspension of production and/or distribution, withdrawal or suspension of the applicable regulator’s review of our submissions, enforcement actions, injunctions and criminal prosecution. Further, other federal agencies, our customers and partners in our alliance, development, collaboration and other partnership agreements with respect to our products and services may take any such FDA observations or warning letters into account when considering the award of contracts or the continuation or extension of such partnership agreements. Because regulatory approval to manufacture a drug is site-specific, the delay and cost of remedial actions, or obtaining approval to manufacture at a different facility, could negatively impact our business. Any failure by us to comply with applicable laws and regulations and/or any actions by the FDA and other agencies as described above could have a material adverse effect on our business, financial position and results of operations.
If we are unable to manage our growth, our business will suffer.
We have experienced rapid growth in the past several years, including through acquisitions such as the Teva Transaction in 2016, and we anticipate continued rapid expansion in the future, including through our proposed business combination with Amneal as described above under “Item 1. Business - Business Combination with Amneal Pharmaceuticals LLC”. This growth has required us to expand, upgrade, and improve our administrative, operational, and management systems, internal controls and resources. Although we cannot assure you that we will, in fact, grow as we expect, if we fail to manage growth effectively or to develop a successful marketing approach, our business and financial results will be materially harmed. We may also seek to expand our business through complementary or strategic acquisitions of other businesses, products or assets, or through joint ventures, strategic agreements or other arrangements. Any such acquisitions, joint ventures or other business combinations may involve significant integration challenges, operational complexities and time consumption and require substantial resources and effort. It may also disrupt our ongoing businesses, which may adversely affect our relationships with customers, employees, regulators and others with whom we have business or other dealings. Further, if we are unable to realize synergies or other benefits expected to result from any acquisitions, joint ventures or other business combinations, or to generate additional revenue to offset any unanticipated inability to realize these expected synergies or benefits, our growth and ability to compete may be impaired, which would require us to focus additional resources on the integration of operations rather than other profitable areas of our business, and may otherwise cause a material adverse effect on our business, results of operations and financial condition.
23
We may make acquisitions of, or investments in, complementary technologies, businesses or products, which may be on terms that are not commercially advantageous, may require additional debt or equity financing, and may involve numerous risks, including the risks that we may be unable to integrate the acquired business successfully and that we may assume liabilities that adversely affect us.
We regularly review the potential acquisition of technologies, products, product rights and complementary businesses. We may choose to enter into such transactions at any time. Nonetheless, we cannot provide assurance that we will be able to identify suitable acquisition or investment candidates. To the extent that we do identify candidates that we believe to be suitable, we cannot provide assurance that we will be able to make such acquisitions or investments on commercially advantageous terms or at all. Further, there are a number of risks and uncertainties relating to closing such transactions. If such transactions are not completed for any reason, we will be subject to several risks, including the following: (i) the market price of shares of our common stock may reflect a market assumption that such transactions will occur, and a failure to complete such transactions could result in a negative perception by the market of us generally and a decline in the market price of our common stock; and (ii) many costs relating to the such transactions may be payable by us whether or not such transactions are completed.
If we make any acquisitions or investments, we may finance such acquisitions or investments through our cash reserves, debt financing, or by issuing additional equity securities, which could dilute the holdings of our then-existing stockholders. If we require financing, we cannot provide assurance that we will be able to obtain required financing when needed on acceptable terms or at all. Any such acquisitions or investments could also result in an increase in goodwill, intangible assets and amortization expenses that could ultimately negatively impact our profitability. As further described below under the risk factor “Our significant balances of intangible assets, including product rights and goodwill acquired, are subject to impairment testing,” if the fair value of our goodwill or intangible assets is determined at some future date to be less than its recorded value, a charge to earnings may be required. Further, our consolidated financial statements may also be impacted in future periods based on the accuracy of our valuations of any businesses or assets we acquire. Such a charge to earnings or impact on our consolidated financial statements could be in amounts that are material to our business, results of operations and financial condition.
Additionally, acquisitions involve numerous risks, including difficulties in assimilating the personnel, operations and products of the acquired companies, the diversion of management’s attention from other business concerns, risks of entering markets in which we have limited or no prior experience, and the potential loss of key employees of the acquired company. There may be overlap between our products or customers and those of an acquired entity that may create conflicts in relationships or other commitments detrimental to the integrated businesses. If we are unable to successfully or timely integrate the operations of acquired companies with our business, we may incur unanticipated liabilities and be unable to realize the revenue growth, synergies and other anticipated benefits resulting from the acquisition, and our business, results of operations and financial condition could be materially and adversely affected.
As a result of acquiring businesses, we may incur significant transaction costs, including substantial fees for investment bankers, attorneys, accountants and financial printing. Any acquisition could result in our assumption of unknown and/or unexpected, perhaps material liabilities. Additionally, in any acquisition agreement, the negotiated representations, warranties and agreements of the selling parties may not entirely protect us, and liabilities resulting from any breaches could exceed negotiated indemnity limitations.
Our significant balances of intangible assets, including product rights and goodwill acquired, are subject to impairment testing and may result in impairment charges. Impairment charges, and the factors contributing to the incurrence of such impairment charges, could have a material and adverse effect on our business, financial position and results of operations.
A significant amount of our total assets is related to acquired intangible assets and goodwill. At December 31, 2017, the carrying value of our goodwill, which includes goodwill generated as a result of the Tower Acquisition, in addition to goodwill generated as a result of the December 1999 merger of Global Pharmaceuticals Corporation and Impax Pharmaceuticals, Inc., was $207.3 million, or approximately 15% of our total assets. At December 31, 2017, the carrying value of our acquired intangible assets, composed of currently marketed product rights, in-process research and development product rights, and future royalties was $262.5 million, or approximately 19% of our total assets.
24
We have in recent years seen significant balances of intangible assets on our consolidated balance sheet. For instance, the carrying value of our intangible assets was $262.5 million and $620.5 million during the years ended December 31, 2017 and December 31, 2016, respectively. We regularly evaluate and will continue to regularly evaluate whether events or circumstances have occurred to indicate all, or a portion, of the carrying amount of intangible assets or goodwill may no longer be recoverable, in which case an impairment charge to earnings would become necessary. As part of our regular evaluation, we test indefinite-lived intangible assets and goodwill for impairment at least annually during the fourth quarter of our fiscal year, in accordance with Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 350, Intangibles - Goodwill and Other.
We may never fully realize the value of our intangibles assets and goodwill on our consolidated balance sheet, and we have in recent years incurred a significant amount of intangible asset impairment charges as a result of our recent acquisitions, such as the Teva Transaction. For instance, during the year ended December 31, 2017, we recognized a total of $289.7 million of intangible asset impairment charges, with a significant portion of such charges attributable to products acquired in the Teva Transaction as a result of manufacturing issues, product launch delays, increased competition or significant price or volume erosion without an offsetting increase in customer demand.
There was no impairment charge related to goodwill as a result of our annual testing in 2017.
Any future acquisitions or investments in businesses could also result in an increase in goodwill, intangible assets and amortization expenses that could have a negative impact on our profitability. If the fair value of our goodwill or intangible assets is determined at some future date to be less than its recorded value, a charge to earnings may be required. Any such charge or future determination requiring the write-off of a significant portion of the carrying value of our goodwill or intangible assets, and the factors leading to the incurrence of such charges or write-off, could have a material adverse effect on our business, results of operations and financial condition.
A substantial portion of our total revenues is derived from sales to a limited number of customers.
We derive a substantial portion of our revenue from sales to a limited number of customers. In 2017, our three major customers, Cardinal Health, McKesson Corporation, and Amerisource-Bergen, accounted for 33%, 30%, and 25%, respectively, or an aggregate of 88%, of our gross revenue.
Our net sales and quarterly growth comparisons may also be affected by fluctuations in the buying patterns of our customers, whether resulting from pricing, wholesaler buying decisions or other factors. Since such a significant portion of our revenues is derived from relatively few customers, any financial difficulties experienced by a single customer, any delay in receiving payments from a single customer, or any reduction or loss of business with one of these customers, could have a material adverse effect on our business, results of operations and financial condition.
A substantial portion of our total revenues is derived from sales of a limited number of products.
We derive a substantial portion of our revenue from sales of a limited number of products. In 2017, our significant products accounted for 15%, 12%, 9%, 7% and 7%, or an aggregate of 50%, of our product sales, net. The sale of our products can be significantly influenced by market conditions, as well as regulatory actions. We may experience decreases in the sale of our products in the future as a result of actions taken by our competitors, such as price reductions, or as a result of regulatory actions related to our products or to competing products, which could have a material impact on our results of operations. Actions which could be taken by our competitors, which may materially and adversely affect our business, results of operations and financial condition, may include, without limitation, pricing changes and entering or exiting the market for specific products.
25
Sales volume and prices of our products may be adversely affected by the continuing trend of consolidation of certain customer groups, which could have a material and adverse effect on our business, results of operations and financial condition.
A significant proportion of our sales is made to relatively few retail drug chains, wholesalers, and managed care organizations. These customers are continuing to undergo significant consolidation. Such consolidation has provided and may continue to provide them with additional purchasing leverage, and consequently may increase the pricing pressures that we face. Additionally, the emergence of large buying groups representing independent retail pharmacies, and the prevalence and influence of managed care organizations and similar institutions, enable those groups to extract price discounts on our products. For example, there has been a recent trend of large wholesalers and retailer customers forming partnerships, such as the alliance between Walgreens and AmerisourceBergen Corporation, the alliance between Rite Aid and McKesson Drug Company, and the alliance between CVS Caremark and Cardinal Health. Increased pricing pressure and other adverse effects from the continued consolidation of our customers and the creation of partnerships between wholesalers and retailer customers could have a material and adverse effect on our business, results of operations and financial condition.
We have experienced operating losses and negative cash flow from operations in the past, and our future profitability is uncertain.
We experienced net losses during the years ended December 31, 2017 and 2016 and cannot assure that our business will return to profitability or generate positive cash flow in the future. To remain operational and profitable, we must, among other things:
• | obtain FDA approval of our products; |
• | successfully launch and market new products; |
• | prevail in patent infringement litigation in which we are involved; |
• | continue to generate or obtain sufficient capital on acceptable terms to fund our operations; and |
• | comply with the many complex governmental regulations that deal with virtually every aspect of our business activities. |
We have recorded a valuation allowance against our deferred tax assets, which could reduce our earnings and have a material adverse effect on our business, results of operations and financial condition.
Our inability to realize deferred tax assets may have a material and adverse effect on our business, results of operations and financial condition. Deferred tax assets are recorded for net operating losses and temporary differences between the book and tax basis of assets and liabilities expected to produce tax deductions in future periods. The ultimate realization of the deferred tax assets is dependent upon the generation of future taxable income during the periods in which those deferred tax assets would be deductible. We assess the realizability of the deferred tax assets each period by considering whether it is more likely than not that all or a portion of our deferred tax assets will not be realized. If we conclude that it is more likely than not that the deferred tax assets will not be realized, we record a valuation allowance against the net deferred tax asset. For instance, when we experienced an operating loss for the year ended December 31, 2016, we recorded a valuation allowance against a significant portion of our net deferred tax assets we subsequently recorded a valuation allowance against all of our net deferred tax assets for the year ended December 31, 2017 for the reasons outlined below. We incurred operating losses for the years ended December 31, 2016 and December 31, 2017 largely due in part to impairment charges we incurred during each of the years. As part of our evaluations for the years ended December 31, 2016 and December 31, 2017, we weighed both the positive and negative evidence available, such as scheduled reversal of deferred tax liabilities, prior earnings history, projected future earnings, carry-back and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income exclusive of reversing taxable temporary differences and carryforwards to outweigh objective negative evidence of financial reporting losses. In addition, due to the elimination of the net operating loss carryback pursuant to the Tax Cuts and Jobs Act (the "2017 Tax Reform Act"), we excluded 2016 taxable income as a source of taxable income to realize the deferred tax asset. Based on an evaluation of both the positive and negative evidence available we determined that it was necessary to continue to record a valuation allowance against all of our net deferred tax assets that we recorded during the year ended December 31, 2016 and for the year ended December 31, 2017 as well. The valuation allowance reduces earnings and our shareholders’ equity and increases the balance sheet leverage as measured by debt-to-total capitalization. The valuation allowance will remain until such time, if ever, that we can determine that the net deferred tax assets are more likely than not to be realized.
26
The terms of our revolving credit facility, term loan facility, and the indenture governing our 2.00% Convertible Senior Notes Due June 2022 impose financial and operating restrictions on us.
We have a $200.0 million senior secured revolving credit facility (the “Revolving Credit Facility”) and a $400.0 million term loan (the "Term Loan Facility", and together with the Revolving Credit Facility, the "RBC Credit Facilities") pursuant to an amended credit agreement, dated as of August 3, 2016, by and among us, the lenders party thereto from time to time and Royal Bank of Canada, as administrative agent and collateral agent. We are also party to an indenture dated June 30, 2015 between us and Wilmington Trust, National Association (the “Indenture”) governing our 2.00% Convertible Senior Notes due 2022 (the “Notes”). Refer to “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 10. Debt” for a detailed description of our outstanding indebtedness.
Our RBC Credit Facilities and Indenture contain a number of negative covenants that limit our ability to engage in activities. These covenants limit or restrict, among other things, our ability to:
• | incur additional debt, guarantee other obligations or grant liens on our assets; |
• | make certain loans or investments; |
• | undertake certain acquisitions, mergers or consolidations, or dispose of assets; |
• | make optional payments or modify certain debt instruments; |
• | pay dividends or other payments on our capital stock, enter into arrangements that restrict our and our restricted subsidiaries’ ability to pay dividends or grant liens; or |
• | engage in certain transactions with our affiliates. |
The terms of our RBC Credit Facilities also include a financial covenant which requires us to maintain a certain total net leverage ratio. These limitations and restrictions may adversely affect our ability to finance our future operations or capital needs or engage in other business activities that may be in our best interests. If we breach any of the covenants in our RBC Credit Facilities or Indenture, we may be in default and our borrowings under the facilities and the Notes could be declared due and payable, including accrued interest and other fees, which could have a material adverse effect on our business, results of operations and financial condition.
Our level of indebtedness and liabilities could limit cash flow available for our operations, expose us to risks that could adversely affect our business, results of operations and financial condition and impair our ability to satisfy our obligations under our convertible notes and other debt instruments.
At December 31, 2017, our total consolidated liabilities were $1.2 billion, including $600.0 million of outstanding convertible notes and a $325.0 million term loan. Refer to “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 10. Debt” for a detailed description of our outstanding indebtedness. We may also incur additional indebtedness to meet future financing needs. Our indebtedness could have significant negative consequences for our business, results of operations and financial condition, including:
• | increasing our vulnerability to adverse economic and industry conditions; |
• | limiting our ability to obtain additional financing; |
• | requiring the dedication of a substantial portion of our cash flow from operations to service our indebtedness, thereby reducing the amount of our cash flow available for other purposes; |
• | limiting our flexibility in planning for, or reacting to, changes in our business; |
• | dilution experienced by our existing stockholders as a result of the conversion of the convertible notes into shares of common stock; and |
• | placing us at a possible competitive disadvantage with less leveraged competitors and competitors that may have better access to capital resources. |
We cannot assure you that we will be able to continue to maintain sufficient cash reserves or continue to generate cash flow from operations at levels sufficient to permit us to pay principal, premium, if any, and interest on our indebtedness, or that our cash needs will not increase. If we are unable to generate sufficient cash flow or otherwise obtain funds necessary to make required payments, or if we fail to comply with the various requirements of our indebtedness then outstanding, we would be in default, which would permit the holders of the affected indebtedness to accelerate the maturity of such indebtedness and could cause defaults under our other indebtedness. Any default under any indebtedness could have a material adverse effect on our business, results of operations and financial condition.
27
We may need to raise additional funds in the future which may not be available on acceptable terms or at all.
We may consider issuing additional debt or equity securities in the future to fund potential acquisitions or investments, to refinance existing debt, or for general corporate purposes. If we issue equity, convertible preferred equity or convertible debt securities to raise additional funds, our existing stockholders may experience dilution, and the new equity or debt securities may have rights, preferences and privileges senior to those of our existing shareholders. If we incur additional debt, it may increase our leverage relative to our earnings or to our equity capitalization, requiring us to pay additional interest expenses and potentially lowering our credit ratings. We may not be able to market such issuances on favorable terms, or at all, in which case, we may not be able to develop or enhance our products, execute our business plan, take advantage of future opportunities, or respond to competitive pressures or unanticipated customer requirements.
Any delays or unanticipated expenses in connection with the operation of our facility or at one of our third party suppliers could have a material adverse effect on our business.
During the year ended December 31, 2017, we closed our Middlesex, New Jersey manufacturing facility and announced that we have entered into a stock and purchase agreement with a third party pursuant to which we subsequently sold Impax Laboratories (Taiwan) Inc. ("Impax Taiwan"), our wholly owned subsidiary which owns our manufacturing facility in Taiwan, R.O.C.; the sale subsequently closed in February 2018. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 15. Restructurings." As such, we are increasingly dependent on our manufacturing facility located in Hayward, California and on our third party suppliers to manufacture our products. A significant disruption at our Hayward facility or at any of our third party suppliers, even on a short-term basis, whether due to an adverse quality or compliance observation, including a total or partial suspension of production and/or distribution by regulatory authorities, an act of God, terrorism, civil or political unrest, or other events could impair our ability to produce and ship products to the market on a timely basis and could, among other consequences, subject us to exposure to claims from customers. Any of these events could have a material adverse effect on our business, results of operations and financial condition.
Our business is subject to the economic, political, legal and other risks of maintaining facilities and conducting clinical trials in foreign countries.
We conduct certain operations in foreign countries, primarily related to clinical trials for our product candidates at various sites in Europe. These foreign operations are subject to risks inherent in maintaining operations and doing business abroad, such as economic and political destabilization, international conflicts, restrictive actions by foreign governments, expropriation or nationalization of property, changes in laws and regulations, changes in regulatory requirements, the difficulty of effectively managing diverse global operations, adverse foreign tax or tariff laws, more limited intellectual property protection in certain foreign jurisdictions, and the threat posed by potential international disease pandemics in countries that do not have the resources necessary to deal with such outbreaks. Further, as our global operations require compliance with a complex set of foreign and U.S. laws and regulations, including data privacy requirements, labor relations laws, tax laws, anti-competition regulations, import and trade restrictions, export requirements, U.S. laws such as the Foreign Corrupt Practices Act of 1977, as amended, and local laws which also prohibit payments to governmental officials or certain payments or remunerations to customers, there is a risk that some provisions may be inadvertently breached. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers or our employees, and prohibitions on the conduct of our business. These foreign economic, political, legal and other risks could impact our operations and have an adverse effect on our business, results of operations and financial condition.
We are involved in various legal proceedings, including patent litigation that can delay or prevent our commercialization of generic products or accelerate generic competition for our branded products, all of which are uncertain, force us to incur substantial expense to defend and/or expose us to substantial liability.
Patent infringement litigation involves many complex technical and legal issues and its outcome is often difficult to predict, and the risk involved in doing so can be substantial. For generic product manufacturers, the potential consequences to such generic companies in the event of an unfavorable outcome include delaying generic launch until patent expiration and potential damages measured by the profits lost by the branded product manufacturer rather than the profits earned by the generic pharmaceutical company. For brand drug manufacturers, an unfavorable outcome may significantly accelerate generic competition ahead of patent expiration. Such litigation usually involves significant expense and can delay or prevent introduction or sale of our products. Our generic products division is routinely subject to patent infringement litigation brought by branded pharmaceutical manufacturers seeking to delay FDA approval to manufacture and market generic forms of their branded products. Likewise, our branded products division is currently involved in patent infringement litigation against generic drug manufacturers seeking FDA approval to market their generic drugs prior to expiration of patents covering our branded products.
28
We and/or our third party partners are routinely subject to patent infringement suits related to our Generics Division products, including as of December 31, 2017, suits related to our oxymorphone hydrochloride tablets, apixaban tablets and dimethyl furarate capsules. If this or any of our future patent litigation matters involving generic products are resolved unfavorably, we or our alliance or collaboration partners may be enjoined from manufacturing, developing or selling the generic product that is the subject of such litigation without a license from the other party. In addition, if we decide to market and sell generic products prior to the resolution of patent infringement suits, we could be held liable for lost profits if we are found to have infringed a valid patent, or liable for treble damages if we are found to have willfully infringed a valid patent. In our branded products division, as of December 31, 2017, we were involved in two patent infringement suits related to Zomig® nasal spray and three patent infringement suits related to Rytary®. If these patent litigation matters involving our branded products are resolved unfavorably, our Zomig® nasal spray product and/or Rytary® may face generic competition significantly earlier than the date of patent expirations for the products. We have incurred substantial expense to defend the foregoing patent litigation suits; during fiscal year 2017, we incurred costs of $5.1 million in connection with our participation in the patent litigation matters described above, as well as for other matters that were resolved in 2017. Although it is not currently possible to quantify the liability we could incur if any of the above referenced patent litigation suits are decided against us, any unfavorable outcome on such matters could have a material adverse effect on our business, results of operations and financial condition.
In addition to patent infringement litigation claims, we are or may become a party to other litigation in the ordinary course of our business, including, among others, matters alleging product liability, other intellectual property rights infringement, violations of securities laws, employment discrimination or breach of commercial contract. A detailed description of our significant legal proceedings are described in “Item 15. Exhibits and Financial Statement Schedules – Notes to Consolidated Financial Statements - Note 19. Legal and Regulatory Matters.” In general, litigation claims can be expensive and time consuming to bring or defend against and could result in settlements or damages that could have a material adverse effect on our business, results of operations and financial condition.
Our agreements to settle patent litigations, which are important to our business, are facing increased government scrutiny in the United States, which may result in increased government actions and private litigation suits.
We are involved in numerous patent litigations in which we challenge the validity or enforceability of innovator companies’ listed patents and/or their applicability to our generic pharmaceutical products, as well as patent infringement litigation in which generic companies challenge the validity or enforceability of our patents and/or their applicability to their generic pharmaceutical products, and therefore settling patent litigations has been and is likely to continue to be an important part of our business. Parties to such settlement agreements in the United States, including us, are required by law to file them with the Federal Trade Commission (“FTC”) and the Antitrust Division of the Department of Justice for review. The FTC has publicly stated that, in its view, some of the brand - generic settlement agreements violate the antitrust laws and has brought actions against some brand and generic companies that have entered into such agreements. In June 2013, the U.S. Supreme Court in its decision in FTC v. Actavis determined that “reverse payment” settlement agreements between brand and generic companies could violate antitrust laws. The Supreme Court held that such settlement agreements are neither immune from antitrust attack nor presumptively illegal but rather should be analyzed under the “Rule of Reason.” It is currently uncertain the effect the Supreme Court’s decision will have on our existing settlement agreements or its impact on our ability to enter into such settlement agreements in the future or the terms thereof. The Supreme Court’s decision may result in heightened scrutiny from the FTC of such settlement agreements and we may become subject to increased FTC investigations or enforcement actions arising from such settlement agreements. Further, private plaintiffs, including direct and indirect purchasers of our products, may also become more active in bringing private litigation claims against us and other brand and generic pharmaceutical companies alleging that such settlement agreements violate antitrust laws.
Our approved products may not achieve expected levels of market acceptance.
Even if we are able to obtain regulatory approvals for our new products, the success of those products is dependent upon market acceptance. Levels of market acceptance for our new products could be affected by several factors, including:
• | the availability of alternative products from our competitors; |
• | the prices of our products relative to those of our competitors; |
• | the timing of our market entry; |
• | the ability to market our products effectively at the retail level; |
• | the perception of patients and the healthcare community, including third-party payors, regarding the safety, efficacy and benefits of our drug products compared to those of competing products; and |
• | the acceptance of our products by government and private formularies. |
29
Some of these factors are not within our control, and our products may not achieve expected levels of market acceptance. Additionally, continuing and increasingly sophisticated studies of the proper utilization, safety and efficacy of pharmaceutical products are being conducted by the industry, government agencies and others which can call into question the utilization, safety and efficacy of previously marketed products. In some cases, studies have resulted, and may in the future result, in the discontinuance of product marketing or other risk management programs such as the need for a patient registry.
Our business is highly dependent on market perceptions of us and the safety and quality of our products. Our business, products or product pricing could be subject to negative publicity, which could have a material adverse effect on our business, results of operations and financial condition.
Market perceptions of our business are very important to us, especially market perceptions of the safety and quality of our products. If any of our products or similar products that other companies distribute are subject to market withdrawal or recall or are proven to be, or are claimed to be, harmful to consumers, then this could have a material adverse effect on our business, results of operations and financial condition. Also, because our business is dependent on market perceptions, negative publicity associated with product quality, illness or other adverse effects resulting from, or perceived to be resulting from, our products could have a material adverse impact on our business, results of operations and financial condition.
The generic pharmaceutical industry has also in recent years been the subject of significant publicity regarding the pricing of pharmaceutical products more generally, including publicity and pressure resulting from prices charged by competitors and peer companies for new products as well as price increases by competitors and peer companies on older products that the public has deemed excessive. Any downward pricing pressure on the price of certain of our products arising from social or political pressure to lower the cost of pharmaceutical products could have a material adverse impact on our business, results of operations and financial condition.
Accompanying the press and media coverage of pharmaceutical pricing practices and public complaints about the same, there has been increasing U.S. federal and state legislative and enforcement interest with respect to drug pricing. For instance, the United States Department of Justice issued subpoenas to pharmaceutical companies, including us, seeking information about the sales, marketing and pricing of certain generic drugs. A detailed description of the United States Department of Justice’s investigation is described in “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 19. Legal and Regulatory Matters”. In addition to the effects of any investigations or claims brought against us, our business, results of operations and financial condition could also be adversely affected if any such inquiries, of us or of other pharmaceutical companies or the industry more generally, were to result in legislative or regulatory proposals that limit our ability to increase the prices of our products.
We may discontinue the manufacture and distribution of certain existing products, which may adversely impact our business, results of operations and financial condition.
We continually evaluate the performance of our products, and may determine that it is in our best interest to discontinue the manufacture and distribution of certain of our products. We cannot guarantee that we have correctly forecasted, or will correctly forecast in the future, the appropriate products to discontinue or that our decision to discontinue various products is prudent if market conditions change. In addition, we cannot assure you that the discontinuance of products will reduce our operating expenses or will not cause us to incur material charges associated with such a decision. Furthermore, the discontinuance of existing products entails various risks, including, in the event that we decide to sell the discontinued product, the risk that we will not be able to find a purchaser for such products or that the purchase price obtained will not be equal to at least the book value of the net assets for such products. Other risks include managing the expectations of, and maintaining good relations with, our customers who previously purchased products from our discontinued products, which could prevent us from selling other products to them in the future. Moreover, we may incur other significant liabilities and costs associated with our discontinuance of products, which could have a material adverse effect on our business, results of operations and financial condition.
30
We expend a significant amount of resources on research and development efforts that may not lead to successful product introductions or the recovery of our research and development expenditures.
We conduct research and development primarily to enable us to manufacture and market pharmaceuticals in accordance with FDA regulations. We spent $80.8 million, $80.5 million and $70.6 million on research and development activities during the years ended December 31, 2017, 2016 and 2015, respectively. We are required to obtain FDA approval before marketing our drug products. The FDA approval process is costly and time consuming. Typically, research expenses related to the development of innovative products and the filing of NDAs are significantly greater than those expenses associated with ANDAs. As we continue to develop new products, our research expenses will likely increase. Because of the inherent risk associated with research and development efforts in our industry, our research and development expenditures may not result in the successful introduction of FDA-approved pharmaceuticals.
Our bioequivalence studies, other clinical studies and/or other data may not result in FDA approval to market our new drug products. While we believe that the FDA’s ANDA procedures will apply to our bioequivalent versions of branded drugs, these drugs may not be suitable for, or approved as part of, these abbreviated applications. In addition, even if our drug products are suitable for FDA approval by filing an ANDA, the abbreviated applications are costly and time consuming to complete. After we submit a NDA or ANDA, the FDA may require that we conduct additional studies, and as a result, we may be unable to reasonably determine the total research and development costs to develop a particular product. Also, for products pending approval, we may obtain raw materials or produce batches of inventory to be used in anticipation of the product’s launch. In the event that FDA approval is denied or delayed, we could be exposed to the risk of this inventory becoming obsolete. Finally, we cannot be certain that any investment made in developing products or product-delivery technologies will be recovered, even if we are successful in commercialization. To the extent that we expend significant resources on research and development efforts and are not able, ultimately, to introduce successful new products or new delivery technologies as a result of those efforts, we will be unable to recover those expenditures.
The time necessary to develop generic drugs may adversely affect whether, and the extent to which, we receive a return on our capital.
We generally begin our development activities for a new generic drug product several years in advance of the patent expiration date of the brand-name drug equivalent. The development process, including drug formulation, testing, and FDA review and approval, often takes three or more years. This process requires that we expend considerable capital to pursue activities that do not yield an immediate or near-term return. Also, because of the significant time necessary to develop a product, the actual market for a product at the time it is available for sale may be significantly less than the originally projected market for the product. If this were to occur, our potential return on our investment in developing the product, if approved for marketing by the FDA, would be adversely affected and we may never receive a return on our investment in the product. It is also possible for the manufacturer of the brand-name product for which we are developing a generic drug to obtain approvals from the FDA to switch the brand-name drug from the prescription market to the OTC market. If this were to occur, we would be prohibited from marketing our product other than as an OTC drug, in which case revenues could be substantially less than we anticipated.
Research and development efforts invested in our branded pharmaceutical products may not achieve expected results.
We invest increasingly significant resources to develop our branded products, both through our own efforts and through collaborations, in-licensing and acquisition of products from or with third parties. The development of proprietary branded drugs involves processes and expertise different from those used in the development of generic products, which increases the risks of failure that we face. For example, the time from discovery to commercial launch of a branded product can be 15 years or even longer, and involves multiple stages: not only intensive preclinical and clinical testing, but also highly complex, lengthy and expensive approval processes which can vary from country to country. The longer it takes to develop a product, the longer time it may take for us to recover our development costs and generate profits, if at all.
During each development stage, we may encounter obstacles that delay the process or approval and increase expenses, leading to significant risks that we will not achieve our goals and may be forced to abandon a potential product in which we have invested substantial amounts of time and money. These obstacles may include: preclinical failures; difficulty enrolling patients in clinical trials; delays in completing formulation and other work needed to support an application for approval; adverse reactions or other safety concerns arising during clinical testing; insufficient clinical trial data to support the safety or efficacy of the product candidate; and failure to obtain, or delays in obtaining, the required regulatory approvals for the product candidate or the facilities in which it is manufactured. As a result of the obstacles noted above, our investment in research and development of branded products can involve significant costs with no assurances of future revenues or profits.
31
Approvals for our new generic drug products may be delayed or become more difficult to obtain if the FDA institutes changes to its approval requirements.
The FDA may institute changes to its ANDA approval requirements, such as implementing new or additional fees similar to the fees imposed by the Generic Drug Fee User Amendments of 2012 (“GDUFA”) and its second iteration (GDUFA II), which may make it more difficult or expensive for us to obtain approval for our new generic products. The FDA may also implement other changes that may directly affect some of our ANDA filings pending approval from the FDA, such as changes to guidance from the FDA regarding bioequivalency requirements for particular drugs. Such changes may cause our development of such generic drugs to be significantly more difficult or result in delays in FDA approval or result in our decision to abandon or terminate certain projects. Any changes in FDA requirements may make it more difficult for us to file ANDAs or obtain approval of our ANDAs and generate revenues and thus have a material adverse effect on our business, results of operations and financial condition.
The risks and uncertainties inherent in conducting clinical trials could delay or prevent the development and commercialization of our own branded products, which could have a material adverse effect on our business, results of operations and financial condition.
With respect to our branded products which do not qualify for the FDA’s abbreviated application procedures, we must demonstrate through clinical trials that these products are safe and effective for use. We have only limited experience in conducting and supervising clinical trials. The process of completing clinical trials and preparing a NDA may take several years and requires substantial resources. Our studies and filings may not result in FDA approval to market our new drug products and, if the FDA grants approval, we cannot predict the timing of any approval. There are substantial filing fees for NDAs that are not refundable if FDA approval is not obtained.
There are a number of risks and uncertainties associated with clinical trials. The results of clinical trials may not be indicative of results that would be obtained from large scale testing. Clinical trials are often conducted with patients having advanced stages of disease and, as a result, during the course of treatment these patients can die or suffer adverse medical effects for reasons that may not be related to the pharmaceutical agents being tested, but which nevertheless affect the clinical trial results. In addition, side effects experienced by the patients may cause delay of approval or limit the profile of an approved product. Moreover, our clinical trials may not demonstrate sufficient safety and efficacy to obtain approval from the FDA or foreign regulatory authorities. The FDA or foreign regulatory authorities may not agree with our assessment of the clinical data or they may interpret it differently. Such regulatory authorities may require additional or expanded clinical trials. Even if the FDA or foreign regulatory authorities approve certain products developed by us, there is no assurance that such regulatory authorities will not subject marketing of such products to certain limits on indicated use.
Failure can occur at any time during the clinical trial process and, in addition, the results from early clinical trials may not be predictive of results obtained in later and larger clinical trials, and product candidates in later clinical trials may fail to show the desired safety or efficacy despite having progressed successfully through earlier clinical testing. A number of companies in the pharmaceutical industry, including us, have suffered significant setbacks in clinical trials, even in advanced clinical trials after showing positive results in earlier clinical trials. The completion of clinical trials for our product candidates may be delayed or halted for the reasons noted above in addition to many other reasons, including:
• | delays in patient enrollment, and variability in the number and types of patients available for clinical trials; |
• | regulators or institutional review boards may not allow us to commence or continue a clinical trial; |
• | our inability, or the inability of our partners, to manufacture or obtain from third parties materials sufficient to complete our clinical trials; |
• | delays or failure in reaching agreement on acceptable clinical trial contracts or clinical trial protocols with prospective clinical trial sites; |
• | risks associated with trial design, which may result in a failure of the trial to show statistically significant results even if the product candidate is effective; |
• | difficulty in maintaining contact with patients after treatment commences, resulting in incomplete data; |
• | poor effectiveness of product candidates during clinical trials; |
• | safety issues, including adverse events associated with product candidates; |
• | the failure of patients to complete clinical trials due to adverse side effects, dissatisfaction with the product candidate, or other reasons; |
• | governmental or regulatory delays or changes in regulatory requirements, policy and guidelines; and |
• | varying interpretation of data by the FDA or foreign regulatory authorities. |
In addition, our product candidates could be subject to competition for clinical study sites and patients from other therapies under development which may delay the enrollment in or initiation of our clinical trials.
32
The FDA or foreign regulatory authorities may require us to conduct unanticipated additional clinical trials, which could result in additional expense and delays in bringing our product candidates to market. Any failure or delay in completing clinical trials for our product candidates would prevent or delay the commercialization of our product candidates. We cannot assure that our expenses related to clinical trials will lead to the development of brand-name drugs that will generate revenues in the near future. Delays or failure in the development and commercialization of our own branded products could have a material adverse effect on our business, results of operations and financial condition.
We rely on third parties to conduct clinical trials and testing for our product candidates, and if they do not properly and successfully perform their legal and regulatory obligations, as well as their contractual obligations to us, we may not be able to obtain regulatory approvals for our product candidates.
We design the clinical trials for our product candidates, but rely on contract research organizations and other third parties to assist us in managing, monitoring and otherwise carrying out these trials, including with respect to site selection, contract negotiation, analytical testing and data management. We do not control these third parties and, as a result, they may not treat our clinical studies as their highest priority, or in the manner in which we would prefer, which could result in delays.
Although we rely on third parties to conduct our clinical trials and related activities, we are responsible for confirming that each of our clinical trials is conducted in accordance with our general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices and good laboratory practices, for conducting, recording and reporting the results of clinical trials to ensure that the data and results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. The FDA enforces good clinical practices and good laboratory practices through periodic inspections of trial sponsors, principal investigators and trial sites. If we, our contract research organizations or our study sites fail to comply with applicable good clinical practices and good laboratory practices, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with good clinical practices and good laboratory practices. In addition, our clinical trials must be conducted with product manufactured under the FDA’s cGMP regulations. Our failure or the failure of our contract manufacturers if any are involved in the process, to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
If third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to failure to adhere to our clinical protocols or regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, our clinical trials may not meet regulatory requirements. If our clinical trials do not meet regulatory requirements or if these third parties need to be replaced, our clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates, which could have a material adverse effect on our business, results of operations and financial condition.
We currently do not have a license partner for commercialization of Numient® outside of the United States and Taiwan.
In November 2015, the European Commission granted marketing authorization for Numient® (IPX066) (referred to as Rytary® in the United States). The review of the Numient® application was conducted under the centralized licensing procedure as a therapeutic innovation, and the authorization is applicable in all 28 member states of the European Union, as well as Iceland, Liechtenstein and Norway. We have no experience marketing or selling our pharmaceutical products outside of the United States and Puerto Rico and to date, we have not launched commercialization activities for Numient® outside of the United States. In connection with our sale of Impax Taiwan in February 2018, we entered into a license agreement for the third party buyer to manufacture and commercialize Numient® in Taiwan, R.O.C., for a period of up to ten years. If we are unsuccessful in entering into third party collaboration arrangements for commercialization activities of Numient® outside of the United States or Taiwan , R.O.C., such failure could have a material adverse effect on our business, results of operations and financial condition.
The illegal distribution and sale by third parties of counterfeit versions of our products or of stolen products could have a negative impact on our reputation and a material adverse effect on our business, results of operations and financial condition.
Third parties could illegally distribute and sell counterfeit versions of our products, which do not meet the rigorous manufacturing and testing standards that our products undergo. Counterfeit products are frequently unsafe or ineffective, and can be life-threatening. Counterfeit medicines may contain harmful substances, the wrong dose of the active pharmaceutical ingredient or no active pharmaceutical ingredients at all. However, to distributors and users, counterfeit products may be visually indistinguishable from the authentic version.
33
Reports of adverse reactions to counterfeit drugs or increased levels of counterfeiting could materially affect patient confidence in the authentic product. It is possible that adverse events caused by unsafe counterfeit products will mistakenly be attributed to the authentic product. In addition, thefts of inventory at warehouses, plants or while in-transit, which are not properly stored and which are sold through unauthorized channels could adversely impact patient safety, our reputation and our business.
Public loss of confidence in the integrity of pharmaceutical products as a result of counterfeiting or theft could have a material adverse effect on our business, results of operations and financial condition.
We are dependent on a small number of suppliers for our raw materials that we use to manufacture our products and interruptions in our supply chain could materially and adversely affect our business.
The raw materials we use in the production of our products consist of pharmaceutical chemicals in various forms that are generally available from several sources in the United States and throughout the world. In some cases, however, the raw materials, such as the active pharmaceutical ingredients (“API”) used to manufacture our products, are available only from a single supplier. Further, even if more than one supplier exists, we may choose, and have done so in the case of our API suppliers for a majority of our products, to list only one supplier in our product applications submitted to the FDA. The FDA requires identification of raw material suppliers in applications for approval of drug products. If raw materials were unavailable from a specified supplier or the supplier was not in compliance with FDA or other applicable requirements, the FDA approval of a new supplier could delay the manufacture of the drug involved. As a result, there is no guarantee we will always have timely and sufficient access to a required raw material or other product. Generally, we would need as long as 18 months to find and qualify a new sole-source supplier. If we receive less than one year’s termination notice from a sole-source supplier that it intends to cease supplying raw materials, it could result in disruption of our ability to produce the drug involved. We currently do not have long-term supply agreements with the majority of our API suppliers and although to date we have only experienced occasional interruptions in supplies, no assurance can be given that we will continue to receive uninterrupted or adequate supplies of such raw materials.
A significant portion of our raw materials may also be available only from foreign sources. Foreign sources can be subject to the special risks of doing business abroad, including:
• | greater possibility for disruption due to transportation or communication problems; |
• | the relative instability of some foreign governments and economies; |
• | interim price volatility based on labor unrest, materials or equipment shortages, export duties, restrictions on the transfer of funds, or fluctuations in currency exchange rates; and |
• | uncertainty regarding recourse to a dependable legal system for the enforcement of contracts and other rights. |
Many third-party suppliers are subject to governmental regulation and, accordingly, we are dependent on the regulatory compliance of these third parties. We also depend on the strength, enforceability and terms of our various contracts with these third-party suppliers. We also rely on complex shipping arrangements throughout the various facilities of our supply chain spectrum. Customs clearance and shipping by land, air or sea routes rely on and may be affected by factors that are not in our full control or are hard to predict.
Any inability to obtain raw materials on a timely basis, or any significant price increases which cannot be passed on to customers, could have a material adverse effect on our business, results of operations and financial condition.
34
Our policies regarding returns, rebates, allowances and chargebacks, and marketing programs adopted by wholesalers may reduce our revenues in future fiscal periods.
Based on industry practice, many generic drug manufacturers’ policies give customers post-sale inventory allowances on returns. Under these arrangements, from time to time, we give our customers credits on our generic products that our customers hold in inventory after we have decreased the market prices of the same generic products due to competitive pricing. Therefore, if new competitors enter the marketplace and significantly lower the prices of any of their competing products, we would likely reduce the price of our product. As a result, we would be obligated to provide credits to our customers who are then holding inventories of such products, which could reduce sales revenue and gross margin for the period the credit is provided. Like our competitors, we also give credits for chargebacks to wholesalers that have contracts with us for their sales to hospitals, group purchasing organizations, pharmacies or other customers. A chargeback is the difference between the price the wholesaler pays and the price that the wholesaler’s end-customer, who is covered under a contract with the manufacturer allowing it to purchase at a lower price, pays for a product. Although we establish reserves based on our prior experience and our best estimates of the impact that these policies may have in subsequent periods, we cannot ensure that our reserves are adequate or that actual product returns, rebates, allowances and chargebacks will not exceed our estimates. Any failure to establish adequate reserves with respect to returns, rebates, allowances and chargebacks may result in a material adverse effect on our business, results of operations and financial condition.
Certain of our products use controlled substances, the availability of which may be limited by the DEA and other regulatory agencies.
We utilize controlled substances in certain of our current products and products in development and therefore must meet the requirements of the Controlled Substances Act of 1970 and the related regulations administered by the DEA in the United States. These laws relate to the manufacture, shipment, storage, sale and use of controlled substances. The DEA and other regulatory agencies limit the availability of the active ingredients used in certain of our current products and products in development and, as a result, our procurement quota of these active ingredients may not be sufficient to meet commercial demand or complete clinical trials. We must annually apply to the DEA for procurement quota in order to obtain these substances. Any delay or refusal by the DEA in establishing our procurement quota for controlled substances could delay or stop our clinical trials or product launches, or could cause trade inventory disruptions for those products that have already been launched, which could have a material adverse effect on our business, results of operations and financial condition.
Global economic conditions may adversely affect our industry, business, results of operations and financial condition.
Although global economic conditions have been fairly stable as a whole in recent years, continued concerns regarding the systemic impact of potential geopolitical issues and economic policy uncertainty could potentially cause economic and market instability in the future and could adversely affect our business, results of operations and financial condition. We have exposure to many different industries and counterparties, including our partners under our alliance and collaboration agreements, suppliers of raw chemical materials, drug wholesalers and other customers that may be affected by an unstable economic environment. Any economic instability or challenging economic conditions could result in tighter credit conditions and thus affect these parties’ ability to fulfill their respective contractual obligations to us, cause them to limit or place burdensome conditions upon future transactions with us or drive us and our competitors to decrease prices, each of which could materially and adversely affect our business, results of operations and financial condition.
We may be subject to disruptions or failures in our information technology systems and network infrastructures, including through cyber-attacks or other third party breaches, that could have a material adverse effect on our business.
We rely on the efficient and uninterrupted operation of complex information technology systems and network infrastructures to operate our business. We also hold data in various data center facilities upon which our business depends. A disruption, infiltration or failure of our information technology systems or any of our data centers as a result of software or hardware malfunctions, system implementations or upgrades, computer viruses, third-party security breaches, employee error, theft or misuse, malfeasance, power disruptions, natural disasters or accidents could cause breaches of data security, loss of intellectual property and critical data and the release and misappropriation of sensitive competitive information.
Further, data maintained in digital form is subject to risk of cyber-attacks, which are increasing in frequency and sophistication. Cyber-attacks could include the deployment of harmful malware and other means to affect service reliability and threaten data confidentiality, integrity and availability. Despite our efforts to monitor and safeguard our systems to prevent data compromise, the possibility of a future data compromise cannot be eliminated entirely, and risks associated with intrusion, tampering, and theft remain. In addition, we do not have insurance coverage with respect to system failures or cyber-attacks.
35
While we have implemented a number of protective measures, including firewalls, antivirus, patches, data encryption, log monitors, routine back-ups with offsite retention of storage media, system audits, data partitioning, routine password modifications and disaster recovery procedures, such measures may not be adequate or implemented properly to prevent or fully address the adverse effect of such events. If our systems were to fail or we are unable to successfully expand the capacity of these systems, or we are unable to integrate new technologies into our existing systems, our operations and financial results could suffer.
We have also outsourced significant elements of our information technology infrastructure and as a result we depend on third parties who are responsible for maintaining significant elements of our information technology systems and infrastructure and who may or could have access to our confidential information. The size and complexity of our information technology systems, and those of our third party vendors, make such systems potentially vulnerable to service interruptions and security breaches from inadvertent or intentional actions by its employees, partners or vendors. These systems are also vulnerable to attacks by malicious third parties, and may be susceptible to intentional or accidental physical damage to the infrastructure maintained by us or by third parties. A breach of our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination, misappropriation or misuse of trade secrets, proprietary information, or other confidential information, whether as a result of theft, hacking, fraud, trickery or other forms of deception, or for any other cause, could enable others to produce competing products, use our proprietary technology or information, and/or adversely affect our business. Further, any such interruption, security breach, loss or disclosure of confidential information could result in financial, legal, business, and reputational harm to us and could have a material adverse effect on our business, results of operations and financial condition.
We may be adversely affected by alliance, collaboration, supply, or license and distribution agreements we enter into with other companies.
We have entered into several alliance, collaboration, supply or license and distribution agreements with respect to certain of our products and services and may enter into similar agreements in the future. These arrangements may require us to relinquish rights to certain of our technologies or product candidates, or to grant licenses on terms that ultimately may prove to be unfavorable to us. Relationships with alliance partners may also include risks due to regulatory requirements, incomplete marketplace information, inventories, and commercial strategies of our partners, and our agreements may be the subject of contractual disputes. If we or our partners are not successful in commercializing the products covered by the agreements, such commercial failure could adversely affect our business.
Pursuant to license and distribution agreements with unrelated third party pharmaceutical companies, we are dependent on such companies to supply us with product that we market and sell, and we may enter into similar agreements in the future. Any delay or interruption in the supply of product under such agreements could curtail or delay our product shipment and adversely affect our revenues, as well as jeopardize our relationships with our customers.
From time to time we may need to rely on licenses to proprietary technologies, which may be difficult or expensive to obtain.
We may need to obtain licenses to patents and other proprietary rights held by third parties to develop, manufacture and market products. If we are unable to timely obtain these licenses on commercially reasonable terms, our ability to commercially market our products may be inhibited or prevented, which could have a material adverse effect on our business, results of operations and financial condition.
We depend on qualified scientific and technical employees and are increasingly dependent on our direct sales force, and our limited resources may make it more difficult to attract and retain these personnel.
Because of the specialized scientific nature of our business, we are highly dependent upon our ability to continue to attract and retain qualified scientific and technical personnel. We are not aware of any pending, significant losses of scientific or technical personnel. Loss of the services of, or failure to recruit, key scientific and technical personnel, however, would be significantly detrimental to our product-development programs. As a result of our small size and limited financial and other resources, it may be difficult for us to attract and retain qualified officers and qualified scientific and technical personnel.
In addition, marketing of our branded products, such as the Zomig® products pursuant to our AZ Agreement with AstraZeneca and Rytary®, requires much greater use of a direct sales force compared to marketing of our generic products. Our ability to realize significant revenues from marketing and sales activities depends on our ability to attract and retain qualified sales personnel. Competition for qualified sales personnel is intense. Any failure to attract or retain qualified sales personnel could negatively impact our sales revenue and have a material adverse effect on our business, results of operations and financial condition.
36
We have entered into employment agreements with our executive officers and certain other key employees that provide that the executive officer may terminate his or her employment upon 60 days prior written notice to us. All of our other key personnel are employed on an at-will basis with no formal employment agreements. We do not maintain “Key Man” life insurance on any executives.
We are subject to significant costs and uncertainties related to compliance with the extensive regulations that govern the manufacturing, labeling, distribution, promotion and sale of pharmaceutical products as well as environmental, safety and health regulations.
The manufacturing, distribution, processing, formulation, packaging, labeling, promotion and sale of our products are subject to extensive regulation by federal agencies, including the FDA, DEA, FTC, Consumer Product Safety Commission and Environmental Protection Agency, among others. We are also subject to state and local laws, regulations and agencies in California, New Jersey, Pennsylvania and elsewhere. Such regulations are also subject to change by the relevant federal, state and international agencies. For instance, beginning from January 1, 2015, we have been required to comply with certain requirements under the Drug Quality and Security Act (“DQSA”), specifically Title II of the DQSA, referred to as the Drug Supply Chain Security Act, which requires companies in certain prescription drugs’ chain of distribution to build electronic, interoperable systems to identify and trace the products as they are distributed in the United States. Compliance with the Drug Supply Chain Security Act has resulted in increased expenses for us and has imposed greater administrative burdens on our organization. Compliance with further requirements of the DQSA or any future federal or state electronic pedigree requirements may result in further expenditures.
As further described above under the risk factor, “Manufacturing or quality control problems may damage our reputation for quality production, demand costly remedial activities and negatively impact our business, results of operations and consolidated financial condition,” we are required to comply with the requirements of the FDA and our products are required to be manufactured in a manner consistent with cGMP and other similar standards. If we, or one of our third party suppliers, fail to comply with the FDA requirements and other applicable laws and regulations, we may breach our representations made to our customers or the FDA or other governmental may take action against us or our products, which could have a material adverse effect on our business, results of operations and financial condition.
The FDA may also require labeling revisions, formulation or manufacturing changes and/or product modifications or additional safety data for our new or existing products. If the FDA imposes more stringent requirements or requires any additional safety, testing or remedial measures on our products or product candidates, we could incur increased costs for, or delays in, obtaining approval of such products or be required to remove such products from the market. For instance, we have derived a portion of our revenue from the sale of generic oxymorphone hydrochloride ER, a pharmaceutical product in the opioid class of drugs. The U.S. Department of Health and Human Services has declared the wide spread addiction to and abuse of opioid products a public health emergency in the United States. Changing public and clinical perceptions of opioid products and the risks relating to their use may result in the imposition of even stricter regulation, or even the removal of, such products by the FDA or other governmental agencies, any of which could have a material adverse effect on our business, results of operations and financial condition.
With respect to environmental, safety and health laws and regulations, we cannot accurately predict the outcome or timing of future expenditures that we may be required to make in order to comply with such laws as they apply to our operations and facilities. We are also subject to potential liability for the remediation of contamination associated with both present and past hazardous waste generation, handling, and disposal activities. We are subject periodically to environmental compliance reviews by environmental, safety, and health regulatory agencies. Environmental laws are subject to change and we may become subject to stricter environmental standards in the future and face larger capital expenditures in order to comply with environmental laws.
Compliance with federal and state and local law regulations, including compliance with any newly enacted regulations, requires substantial expenditures of time, money and effort to ensure full technical compliance. Failure to comply with applicable governmental regulations can result in fines, disgorgement, unanticipated compliance expenditures, recall or seizure of products, exposure to product liability claims, total or partial suspension of production or distribution, suspension of the FDA’s review of NDAs or ANDAs, enforcement actions, injunctions and civil or criminal prosecution, any of which could have a material and adverse effect on our business, results of operations and financial condition.
37
We may experience reductions in the levels of reimbursement for pharmaceutical products by governmental authorities, HMOs or other third-party payors. Any such reductions could have a material adverse effect on our business, results of operations and financial condition.
Various governmental authorities and private health insurers and other organizations, such as HMOs, provide reimbursement to consumers for the cost of certain pharmaceutical products. Demand for our products depends in part on the extent to which such reimbursement is available. In addition, third-party payors are attempting to control costs by limiting the level of reimbursement for medical products, including pharmaceuticals, and increasingly challenge the pricing of these products which may adversely affect the pricing of our products. Moreover, health care reform has been, and is expected to continue to be, an area of national and state focus, which could result in the adoption of measures that could adversely affect the pricing of pharmaceuticals or the amount of reimbursement available from third-party payors for our products.
Reporting and payment obligations under the Medicaid rebate program and other government programs are complex, and failure to comply could result in sanctions and penalties or we could be required to reimburse the government for underpayments, which could have a material adverse effect on our business.
Medicaid and other government reporting and payment obligations are highly complex and somewhat ambiguous. State attorneys general and the U.S. Department of Justice have brought suits or instituted investigations against a number of other pharmaceutical companies for failure to comply with Medicaid and other government reporting obligations. Our methodologies for making these calculations are complex and the judgments involved require us to make subjective decisions, such that these calculations are subject to the risk of errors. Government agencies may impose civil or criminal sanctions, including fines, penalties and possible exclusion from federal health care programs, including Medicaid and Medicare. Any such penalties or sanctions could have a material adverse effect on our business, results of operations and financial condition.
Legislative or regulatory programs that may influence prices of prescription drugs could have a material adverse effect on our business.
Current or future federal or state laws and regulations may influence the prices of drugs and, therefore, could adversely affect the prices that we receive for our products. Programs in existence in certain states seek to set prices of all drugs sold within those states through the regulation and administration of the sale of prescription drugs. Expansion of these programs, in particular, state Medicaid programs, or changes required in the way in which Medicaid rebates are calculated under such programs, could adversely affect the price we receive for our products and could have a material adverse effect on our business, results of operations and financial condition. Further, as described in detail above under the risk factor “Our business is highly dependent on market perceptions of us and the safety and quality of our products. Our business, products or pricing could be subject to negative publicity, which could have a material adverse effect on our business, results of operations and financial condition” pharmaceutical product prices have been the focus of increased scrutiny by the government, including certain state attorneys general, members of congress and the United States Department of Justice. Decreases in health care reimbursements or prices of our prescription drugs could limit our ability to sell our products or decrease our revenues, which could have a material adverse effect on our business, results of operations and financial condition.
Our failure to comply with the legal and regulatory requirements governing sales, marketing and pricing of our products may result in substantial fines, sanctions and restrictions on our business activities.
Our practices and activities related to the sales and marketing of our products, as well as the pricing of our products, are subject to extensive regulation under U.S. federal and state healthcare statutes and regulations intended to combat fraud and abuse to federal and state healthcare payment programs, such as Medicare and Medicaid, Tri-Care, CHAMPUS, and Department of Defense programs. These laws include the federal Anti-Kickback Statute, the federal False Claims Act, and similar state laws and implementing regulations. For example, the payment of any incentive to a healthcare provider to induce the recommendation of our product or the purchase of our products reimbursable under a federal or state program is prohibited under these laws. Likewise, knowingly presenting or causing to be presented a false claim for payment to a federal or state health care program would expose a company to sanctions and penalties. Similarly, the inaccurate reporting of prices leading to inflated reimbursement rates would also be considered a violation of these laws. The Physician Payment Sunshine Act enacted in 2010 imposes reporting and disclosure requirements on drug manufacturers for any “transfer of value” made or distributed to prescribers and other healthcare providers. Failure to submit this required information may result in significant civil monetary penalties. These laws and regulations are enforced by the U.S. Department of Justice, the U.S. Department of Health and Human Services, Office of Inspector General, state Medicaid Fraud Units and other state enforcement agencies.
38
Violations of the laws and regulations described above are punishable by criminal and civil sanctions, including substantial fines and penal sanctions, such as imprisonment. It is common for enforcement agencies to initiate investigations into sales and marketing practices, as well as pricing practices, regardless of merit. These types of investigations and any related litigation can result in: (i) large expenditures of cash for legal fees, payment for penalties, and compliance activities; (ii) limitations on operations; (iii) diversion of management resources; (iv) injury to our reputation; and (v) decreased demand for our products.
While we have developed corporate compliance programs based on what we believe to be current best practices, we cannot assure you that we or our employees or agents are or will be in compliance with all applicable federal or state regulations and laws. Further, the criteria for determining compliance are often complex and subject to change and interpretation. If we are in violation of any of these requirements or any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could include the imposition of significant criminal and civil fines and penalties, exclusion from federal healthcare programs or other sanctions, any which could have a material and adverse effect on our business, results of operations and financial condition.
We have entered into, and anticipate entering into, contracts with various U.S. government agencies. Unfavorable provisions in government contracts, some of which may be customary, may harm our business, results of operations and financial condition.
Government contracts customarily contain provisions that give the government substantial rights and remedies, many of which are not typically found in commercial contracts, including provisions that allow the government to:
• | suspend or debar the contractor from doing business with the government or a specific government agency; |
• | terminate existing contracts, in whole or in part, for any reason or no reason; |
• | reduce the scope and value of contracts; |
• | change certain terms and conditions in contracts; |
• | claim rights to products, including intellectual property, developed under the contract; |
• | take actions that result in a longer development timeline than expected; |
• | direct the course of a development program in a manner not chosen by the government contractor; |
• | audit and object to the contractor’s contract-related costs and fees, including allocated indirect costs; and |
• | control and potentially prohibit the export of the contractor’s products. |
Generally, government contracts contain provisions permitting unilateral termination or modification, in whole or in part, at the government’s convenience. Under general principles of government contracting law, if the government terminates a contract for convenience, the terminated company may recover only its incurred or committed costs, settlement expenses and profit on work completed prior to the termination.
If the government terminates a contract for default, the defaulting company is entitled to recover costs incurred and associated profits on accepted items only and may be liable for excess costs incurred by the government in procuring undelivered items from another source. Some government contracts grant the government the right to use, for or on behalf of the U.S. government, any technologies developed by the contractor under the government contract. If we were to develop technology under a contract with such a provision, we might not be able to prohibit third parties, including our competitors, from using that technology in providing products and services to the government.
As a government contractor, we may also become subject to periodic audits and reviews. As part of any such audit or review, the government may review the adequacy of, and our compliance with, our internal control systems and policies, including those relating to our purchasing, property, compensation and/or management information systems. In addition, if an audit or review uncovers any improper or illegal activity, we may be subject to civil and criminal penalties and administrative sanctions, including termination of our contracts, forfeiture of profits, suspension of payments, fines and suspension or prohibition from doing business with the government. We could also suffer serious harm to our reputation if allegations of impropriety were made against us.
Legislative or regulatory reform of the healthcare system in the United States may harm our future business.
In recent years, there have been numerous initiatives on the federal and state levels for comprehensive reforms affecting the payment for, the availability of and reimbursement for, healthcare services in the United States, and it is likely that Congress and state legislatures and health agencies will continue to focus on healthcare reform in the future. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively commonly referred to as the “Healthcare Reform Act” became effective on January 1, 2010 and amongst other changes, increased the minimum Medicaid drug rebates for pharmaceutical companies and revised the definition of “average manufacturer price” for reporting purposes, which affected the amount of our Medicaid drug rebates to states and imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
39
We are unable to predict the future course of federal or state healthcare legislation. If significant additional reforms are made to the United States healthcare system, those reforms could have a material adverse effect on our business, results of operations and financial condition.
We depend on our intellectual property, and our future success is dependent on our ability to protect our intellectual property and not infringe on the rights of others.
We believe intellectual property protection is important to our business and that our future success will depend, in part, on our ability to obtain patent protection, maintain trade secret protection and operate without infringing on the rights of others. We cannot assure you that:
• | any of our future processes or products will be patentable; |
• | our processes or products will not infringe upon the patents of third parties; or |
• | we will have the resources to defend against charges of patent infringement by third parties or to protect our own rights against infringement by third parties. |
We rely on trade secrets and proprietary knowledge related to our products and technology which we generally seek to protect by confidentiality and non-disclosure agreements with employees, consultants, licensees and pharmaceutical companies. If these agreements are breached, we may not have adequate remedies for any breach, and our trade secrets may otherwise become known by our competitors.
We are subject to potential product liability claims that can result in substantial litigation costs and liability.
The design, development and manufacture of pharmaceutical products involve an inherent risk of product liability claims and associated adverse publicity. Product liability insurance coverage is expensive, difficult to obtain, and may not be available in the future on acceptable terms, or at all. Although we currently carry $50.0 million of such insurance, we believe that no reasonable amount of insurance can fully protect against all such risks because of the potential liability inherent in the business of producing pharmaceutical products for human consumption.
Changes in tax regulations, including the U.S. federal tax reform, and varying application and interpretations of these regulations could result in an increase in our existing and future tax liabilities.
We have potential tax exposures resulting from the varying application of statutes, regulations and interpretations, including exposures with respect to manufacturing, research and development, marketing, sales and distribution functions. For instance, on December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act (H.R. 1) (the “2017 Tax Reform Act”). The 2017 Tax Reform Act contains significant changes to U.S. federal income tax laws, including reduction of the federal corporate tax rate from 35% to 21%. The reduction in the corporate income tax rate does not currently materially impact the value of our net deferred tax assets, because we are currently in a full valuation allowance. However, the limitations on the deductibility of interest expense and executive compensation under the terms of the 2017 Tax Reform Act may have a material and adverse effect on our business, financial condition and results of operations. Moreover, the limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks under the 2017 Tax Reform Act may also have a material effect on our cash flows.
There are inherent uncertainties involved in estimates, judgments and assumptions used in the preparation of financial statements in accordance with GAAP. Any future changes in estimates, judgments and assumptions used or necessary revisions to prior estimates, judgments or assumptions could lead to a restatement of our results.
The consolidated financial statements included in this Annual Report on Form 10-K are prepared in accordance with GAAP. This involves making estimates, judgments and assumptions that affect reported amounts of assets (including intangible assets), liabilities, revenues, expenses and income. Estimates, judgments and assumptions are inherently subject to change in the future and any necessary revisions to prior estimates, judgments or assumptions could lead to a restatement. Any such changes could result in corresponding changes to the amounts of assets (including goodwill and other intangible assets), liabilities, revenues, expenses and income.
40
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results, timely file our periodic reports, maintain our reporting status or prevent fraud.
Our management or our independent registered public accounting firm may identify material weaknesses in our internal control over financial reporting in the future. The existence of internal control material weaknesses may result in current and potential stockholders and alliance and collaboration agreements’ partners losing confidence in our financial reporting, which could harm our business, the market price of our common stock, and our ability to retain our current, or obtain new, alliance and collaboration agreements’ partners.
In addition, the existence of material weaknesses in our internal control over financial reporting may affect our ability to timely file periodic reports under the Exchange Act. Although we remedied any past accounting issues and do not believe similar accounting problems are likely to recur, an internal control material weakness may develop in the future and affect our ability to timely file our periodic reports. The inability to timely file periodic reports under the Exchange Act could result in the SEC revoking the registration of our common stock, which would prohibit us from listing or having our stock quoted on any public market. This would have an adverse effect on our business and stock price by limiting the publicly available information regarding us and greatly reducing the ability of our stockholders to sell or trade our common stock.
Terrorist attacks and other acts of violence or war may adversely affect our business.
Terrorist attacks at or nearby our facility in Hayward, California may negatively affect our operations. While we do not believe that we are more susceptible to such attacks than other companies, such attacks could directly affect our physical facilities or those of our suppliers or customers and could make the transportation of our products more difficult and more expensive and ultimately affect our sales.
We carry insurance coverage on our facilities of types and in amounts that we believe are in line with coverage customarily obtained by owners of similar properties. We continue to monitor the state of the insurance market in general and the scope and cost of coverage for acts of terrorism in particular, but we cannot anticipate what coverage will be available on commercially reasonable terms in future policy years. Currently, we carry terrorism insurance as part of our property and casualty and business interruption coverage. If we experience a loss that is uninsured or that exceeds policy limits, we could lose the capital invested in the damaged facilities, as well as the anticipated future net sales from those facilities.
We are party to various agreements as part of our normal course of business which periodically incorporate provisions whereby we indemnify the other party to the agreement.
In the normal course of business, we periodically enter into commercial, employment, legal settlement and other agreements which incorporate indemnification provisions. In some but not all cases, we maintain insurance coverage which we believe will effectively mitigate our obligations under certain of these indemnification provisions. However, should our obligation under an indemnification provision exceed any applicable coverage or should coverage be denied, we could experience a material and adverse effect on our business, financial condition and results of operations.
Because of the location of our manufacturing and research and development facilities, our operations could be interrupted by an earthquake or be susceptible to climate changes.
Our corporate headquarters, manufacturing operations, and research and development activities related to process technologies located in California are located near major earthquake fault lines. Although we use third party manufacturers to produce a number of our products, we produce a substantial portion of our products at our California facility. A disruption at these California facilities due to an earthquake, other natural disaster, or due to climate changes, even on a short-term basis, could impair our ability to produce and ship products to the market on a timely basis. In addition, we could experience a destruction of facilities which would be costly to rebuild, or loss of life, all of which could materially adversely affect our business and results of operations.
We presently carry $100.0 million and $25.0 million of earthquake coverage related to our facilities and property (including inventory) located in Hayward, California and Memphis, TN, respectively. For all other worldwide locations where we have facilities and/or property, we presently carry $100.0 million of earthquake coverage. We believe the aggregate amount of earthquake coverage we currently carry is appropriate in light of the risks; however, the amount of our earthquake insurance coverage may not be sufficient to cover losses from earthquakes. We may discontinue some or all of this insurance coverage in the future if the cost of premiums exceeds the value of the coverage discounted for the risk of loss. If we experience a loss that is uninsured or that exceeds policy limits, we could lose the capital invested in the damaged facilities, as well as the anticipated future net sales from those facilities.
41
The expansion of social media platforms present new risks and challenges, which could cause a material adverse effect on our business, results of operations and financial condition.
The inappropriate use of certain media vehicles could cause brand damage or information leakage or could lead to legal implications from the improper collection and/or dissemination of personally identifiable information. In addition, negative posts or comments about us on any social networking website could seriously damage our reputation. Further, the disclosure of non-public company sensitive information through external media channels could lead to information loss as there might not be structured processes in place to secure and protect information. If our non-public sensitive information is disclosed or if our reputation is seriously damaged through social media, it could have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Our Proposed Business Combination with Amneal Pharmaceuticals LLC
The transactions contemplated by the Business Combination Agreement with Amneal may not be completed on the terms or timeline currently contemplated, or at all, and failure to complete the transactions may result in material adverse consequences to our business, results of operations and financial condition.
As discussed under “Item 1. Business - “Business Combination with Amneal Pharmaceuticals”, we entered into the Business Combination Agreement whereby we have agreed with Amneal to combine our generics and specialty pharmaceutical businesses with the generic drug development and manufacturing business of Amneal.
The Combination is subject to several closing conditions, including the adoption of the Business Combination Agreement by our stockholders, the approval of the listing of the Class A Common Stock to be issued in connection with the issuance of shares by Holdco, and the expiration or termination of any applicable waiting period under (“HSR Act”). If any one of these conditions is not satisfied or waived, the Combination may not be completed. We cannot assure that we will complete the Combination on the terms or timeline currently contemplated, or at all.
The parties have not yet obtained all regulatory clearances, consents and approvals required to complete the Combination. Governmental or regulatory agencies could still seek to block or challenge the Combination or could impose restrictions they deem necessary or desirable in the public interest as a condition to approving the Combination. These restrictions could include a requirement that we sell certain specified assets to obtain such regulatory approvals, which could have a material and adverse effect on our business, results of operations and financial condition. If these approvals are not received, then we will not be obligated to complete the Combination.
If our stockholders do not approve and adopt the Business Combination Agreement and thereby approve the Combination or if the Combination is not completed for any other reason, we would be subject to a number of risks, including the following:
• | we and are our stockholders would not realize the anticipated benefits of the Combination, including any anticipated synergies from combining the two companies; |
• | we may be required to pay a termination fee of $45.0 million if the Business Combination Agreement is terminated in accordance with the specified terms thereof; |
• | we may be required to reimburse Amneal for all reasonable out-of-pocket fees and expenses incurred by Amneal in connection with the Business Combination Agreement and the Combination up to a maximum of $15.0 million in the event that we fail to receive the required approval from our stockholders; and |
• | the trading price of our shares may experience increased volatility to the extent that the current market prices reflect a market assumption that the Combination will be completed. |
We are also exposed to general competitive pressures and risks, which may be increased if the Combination is not completed. The occurrence of any of these events individually or in combination could have a material adverse effect on our business, results of operation and financial condition.
42
Governmental entities or private parties could take actions under antitrust laws to enjoin the completion of the Combination or to seek the divestiture of substantial assets of Impax.
At any time before or after consummation of the Combination, notwithstanding the termination of the waiting period under the HSR Act, the DOJ or the FTC or other state governmental entity could take such action under the antitrust laws as it deems necessary or desirable in the public interest including seeking to enjoin the completion of the Combination or seeking divestiture of a substantial amount of our assets. A divestiture of a substantial amount of our assets could have a material adverse effect on our business, results of operations and financial condition or prospects before and after Closing, diminish the anticipated benefits of the Combination in full or in part, or cause the benefits to take longer to be realized than expected. Further, private parties may also seek to take legal action under the antitrust laws under certain circumstances.
We are subject to business uncertainties and contractual restrictions while the Combination is pending that could have a material and adverse effect on our business, results of operations and financial condition.
Uncertainty about the effect of the Combination on our employees and customers may have a material and adverse effect on our business, regardless of whether the Combination is eventually completed. These uncertainties may impair our ability to attract, retain and motivate key personnel until the Combination is completed, or the Business Combination Agreement is terminated, and for a period of time thereafter, and could cause customers, suppliers and others that deal with us to seek to change our existing business relationships.
Employee retention and recruitment may be particularly challenging for us during the pendency of the Combination, as employees and prospective employees may experience uncertainty about their future roles with us or with New Amneal. The departure of existing key employees or the failure of potential key employees to accept or maintain employment with us despite our retention and recruiting efforts, could have a material adverse impact on our business, results of operation and financial condition, regardless of whether the Combination is eventually completed.
The pursuit of the Combination and the preparation for the integration of the two companies have placed, and will continue to place, a significant burden on our management and internal resources. There is a significant degree of difficulty and management distraction inherent in the process of closing the Combination and planning for the integration of the two companies, which could cause an interruption of, or loss of momentum in, the activities of our existing businesses, regardless of whether the Combination is eventually completed. Before and immediately following Closing, our management team will be required to devote considerable amounts of time to this integration planning process, which will decrease the time they will have to manage their respective existing businesses, service existing customers, attract new customers and develop new products, services or strategies. One potential consequence of such distractions could be the failure of management to realize other opportunities that could be beneficial to us. If our senior management is not able to effectively manage the process leading up to and immediately following Closing, or if any significant business activities are interrupted as a result of the integration planning process, our business, results of operations and financial condition could be materially and adversely affected.
In addition, the Business Combination Agreement restricts us from making certain acquisitions and taking other specified actions without the consent of Amneal until the Combination is consummated or the Business Combination Agreement is terminated. These restrictions may prevent us from pursuing otherwise attractive business opportunities and making other changes to our businesses before completion of the Combination or termination of the Business Combination Agreement, which could have a material and adverse effect on our business, results of operations and financial condition.
The integration of Impax and Amneal following the Closing will present challenges that may result in a decline in the anticipated benefits of the Combination.
The Combination involves the integration of two businesses that currently operate as independent businesses. We and Amneal will be required to devote management attention and resources to integrating our business practices and operations following the Closing, and prior to the Combination, management attention and resources will be required to plan for such integration. Potential difficulties we, Amneal or New Amneal may encounter in the integration process include the following:
• | the inability to successfully integrate the two businesses, including operations, technologies, products and services, in a manner that permits us, Amneal or New Amneal to achieve the cost savings and operating synergies anticipated to result from the Combination, which could result in the anticipated benefits of the Combination not being realized partly or wholly in the time frame currently anticipated or at all; |
43
• | the loss of sales and customers as a result of certain customers of either or both of the two businesses deciding not to continue to do business with us or Amneal, or deciding to decrease their amount of business in order to reduce their reliance on a single company; |
• | the necessity of coordinating geographically separated organizations, systems and facilities; |
• | potential unknown liabilities and unforeseen expenses, delays or regulatory conditions associated with the Combination; |
• | the integration of personnel with diverse business backgrounds and business cultures, while maintaining focus on providing consistent, high-quality products and services; |
• | the consolidation and rationalization of information technology platforms and administrative infrastructures as well as accounting systems and related financial reporting activities; |
• | the potential weakening of established relationships with regulators; and |
• | the challenge of preserving important relationships of both us and Amneal and resolving potential conflicts that may arise. |
Furthermore, it is possible that the integration process could result in the loss of our talented employees or skilled workers. The loss of talented employees and skilled workers could adversely affect our, Amneal’s or New Amneal’s ability to successfully conduct their respective businesses because of such employees’ experience and knowledge of our business and Amneal’s business. In addition, we, Amneal or New Amneal could be adversely affected by the diversion of management’s attention and any delays or difficulties encountered in connection with the integration of the two businesses. The process of integrating operations could cause an interruption of, or loss of momentum in, the activities of one or more of our or Amneal’s segments. If we, Amneal or New Amneal experience difficulties with the integration process, the anticipated benefits of the Combination may not be realized fully or at all, or may take longer to realize than expected. These integration matters could have a material and adverse effect on the business, results of operations, financial condition or prospects of us, Amneal or New Amneal during this transition period and for an undetermined period after completion of the Combination.
Ownership interests will not be adjusted if there is a change in the value of our company or Amneal and their respective assets before the Combination is completed.
The interests of Class A Common Stock received by our stockholders in connection with the Combination will not be adjusted if there is a change in the value or assets of Amneal, Impax or New Amneal prior to the consummation of the Combination. No termination rights will arise in connection with an adverse change at Amneal or our company unless there has been an Amneal Material Adverse Effect or an Impax Material Adverse Effect as applicable (in each case as defined in the Business Combination Agreement).
The Business Combination Agreement contains provisions that may discourage other companies from trying to acquire us.
The Business Combination Agreement contains provisions that may discourage third parties from submitting business combination proposals to us that might result in greater value to our stockholders than the Combination. The Business Combination Agreement generally prohibits us from soliciting any competing acquisition proposal. In addition, if the Business Combination Agreement is terminated by us or Amneal in circumstances that obligate us to pay a termination fee of $45.0 million or to reimburse transaction expenses of up to $15.0 million incurred by Amneal, our financial condition may be adversely affected as a result of the payment of the termination fee and transaction expenses, which might deter third parties from proposing alternative business combination proposals.
After the Combination is complete, our stockholders will have a significantly lower ownership and voting interest in New Amneal than the Existing Amneal Members and will exercise less influence over New Amneal than they do over our company.
Upon the completion of the Combination, each holder of Impax shares of common stock will have a percentage ownership of New Amneal that is smaller than such holder’s percentage ownership of our company immediately prior to the Combination. At the Closing, approximately 75% of the total New Amneal Shares will not be held by our stockholders. Following the Closing, Existing Amneal Members are expected to hold approximately 60% of the voting power of the outstanding New Amneal Shares and following the PIPE Investment, the PIPE investors will hold approximately 15% of the voting power of the outstanding New Amneal Shares.
In addition, the Class A Common Stock to be received by our stockholders as consideration if the Combination is completed will have different rights than shares in Impax common stock.
44
We will incur transaction-related costs in connection with the Combination and the integration of our business with Amneal’s business.
We will incur transaction-related costs in connection with the Combination and in connection with the integration of our business with Amneal’s business following the Closing. There are many systems that must be integrated, including information management, purchasing, accounting and finance, sales, billing, payroll and benefits, and regulatory compliance. We and Amneal are in the early stages of assessing the magnitude of these costs and are therefore unable to provide estimates of these costs. Moreover, many of the expenses that will be incurred, by their nature, are difficult to estimate accurately at the present time. Such expenses could, particularly in the near term, reduce the cost synergies that we expect to achieve from the elimination of duplicative expenses and the realization of economies of scale and cost synergies related to the integration of the businesses following the completion of the Combination. Accordingly, any net synergies may not be achieved in the near term or at all. These integration expenses may result in us, Amneal or New Amneal taking significant charges against earnings following the completion of the Combination. Some of these costs and expenses will be incurred even if the Combination is not consummated, which could have a material and adverse effect on our business, results of operations and financial condition.
Pending litigation against us or Amneal could result in an injunction preventing the completion of the Combination or a judgment resulting in the payment of damages.
We, Amneal and members of our respective boards are currently and may in the future be parties, among others, to various claims and litigation related to the Business Combination Agreement and the Combination, including putative shareholder class actions - see “Note 19. Legal and Regulatory Matters.” for additional details regarding the shareholder class actions. Among other remedies, the plaintiffs in such matters are seeking to enjoin the Combination. The results of complex legal proceedings are difficult to predict, and could delay or prevent the Combination from becoming effective in a timely manner. The existence of litigation relating to the Combination could impact the likelihood of obtaining the required approval from our stockholders in order to complete the Combination. Moreover, the pending litigation is, and any future additional litigation could be, time consuming and expensive, could divert the attention of our management away from our regular business, and, if any one of these lawsuits is adversely resolved against either us or Amneal, could have a material adverse effect on our respective businesses, results of operation and financial condition.
One of the conditions to the Closing is that no governmental entity will have issued an order or injunction or taken any other action enjoining or otherwise prohibiting the consummation of the Combination, and that no law will have been enacted or promulgated by any governmental authority of competent jurisdiction which prohibits or makes illegal the consummation of the Combination. Consequently, if a settlement or other resolution is not reached in the litigation referenced above and the plaintiffs secure injunctive or other relief prohibiting, delaying or otherwise adversely affecting our and/or Amneal’s ability to complete the Combination on the terms contemplated by the Business Combination Agreement, then such injunctive or other relief may prevent the Combination from being completed in a timely manner or at all.
Item 1B. Unresolved Staff Comments
Not applicable.
45
Item 2. Properties
Our primary properties consist of various owned and leased facilities in California, Pennsylvania and New Jersey. As of December 31, 2017, we also owned a significant manufacturing facility in Taiwan, R.O.C. classified as held for sale as discussed in "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 15. Restructuring." The expiration dates of the lease agreements for our leased facilities range between January 14, 2018 and December 31, 2027. Our properties are generally used to support the operations of both the Impax Generics division and the Impax Specialty Pharma division. The table below shows the square feet owned or leased by function at each location.
Location | Owned | Leased | Total | Function | |||||||
Hayward, CA | 35,000 | — | 35,000 | Research & development | |||||||
Hayward, CA | 50,000 | — | 50,000 | Manufacturing | |||||||
Hayward, CA | 19,000 | — | 19,000 | Administration & lab | |||||||
Hayward, CA | 13,300 | — | 13,300 | Manufacturing support | |||||||
Hayward, CA | — | 76,180 | 76,180 | Warehouse & lab | |||||||
Hayward, CA | — | 45,000 | 45,000 | Corporate offices | |||||||
Hayward, CA | — | 88,677 | 88,677 | Manufacturing & lab | |||||||
California Properties | 117,300 | 209,857 | 327,157 | ||||||||
Fort Washington, PA | — | 47,379 | 47,379 | Administration | |||||||
Middlesex, NJ | — | 37,500 | 37,500 | Manufacturing ** | |||||||
Middlesex, NJ | — | 18,593 | 18,593 | Packaging ** | |||||||
Middlesex, NJ | — | 816 | 816 | Research & development ** | |||||||
Middlesex, NJ | — | 32,516 | 32,516 | Administration ** | |||||||
Bridgewater, NJ | — | 32,806 | 32,806 | Administration | |||||||
New Jersey Properties | — | 122,231 | 122,231 | ||||||||
Taiwan | — | 397,917 | 397,917 | Manufacturing * | |||||||
Totals | 117,300 | 777,384 | 894,684 | ||||||||
* This facility is on land that is leased from the state. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 15 . Restructurings" for additional description of our sale of our Taiwan facility.
**Our leases in Middlesex, NJ expire on March 31, 2018.
In our various facilities we maintain an extensive equipment base that includes new or recently reconditioned equipment for the manufacturing and packaging of compressed tablets, coated tablets and capsules. The manufacturing and research and development equipment includes mixers and blenders for capsules and tablets, automated capsule fillers, tablet presses, particle reduction, sifting equipment, and tablet coaters. The packaging equipment includes fillers, cottoners, cappers, and labelers. We also maintain two well equipped, modern laboratories used to perform all the required physical and chemical testing of our products. We also maintain a broad variety of material handling and cleaning, maintenance, and support equipment. We own substantially all of our manufacturing equipment and believe it is well maintained and suitable for its requirements.
We maintain property and casualty and business interruption insurance in amounts we believe are sufficient and consistent with practices for companies of comparable size and business.
Item 3. Legal Proceedings
Information pertaining to legal proceedings can be found in “Item 15. Exhibits and Financial Statement Schedules -Notes to Consolidated Financial Statements – Note 19. Legal and Regulatory Matters” and is incorporated by reference herein.
Item 4. Mine Safety Disclosures
Not applicable.
46
PART II.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Stock Price
Our common stock is traded on the NASDAQ Global Market under the symbol "IPXL". The following table sets forth the high and low sales prices for our common stock as reported by the NASDAQ Global Market, as follows:
Price Range per Share | |||||||
High | Low | ||||||
Year Ended December 31, 2017 | |||||||
First Quarter | $ | 15.05 | $ | 7.75 | |||
Second Quarter | $ | 17.95 | $ | 11.85 | |||
Third Quarter | $ | 25.70 | $ | 14.65 | |||
Fourth Quarter | $ | 22.45 | $ | 15.60 | |||
Year Ended December 31, 2016 | |||||||
First Quarter | $ | 43.16 | $ | 29.66 | |||
Second Quarter | $ | 37.20 | $ | 27.62 | |||
Third Quarter | $ | 32.20 | $ | 20.97 | |||
Fourth Quarter | $ | 24.47 | $ | 12.28 | |||
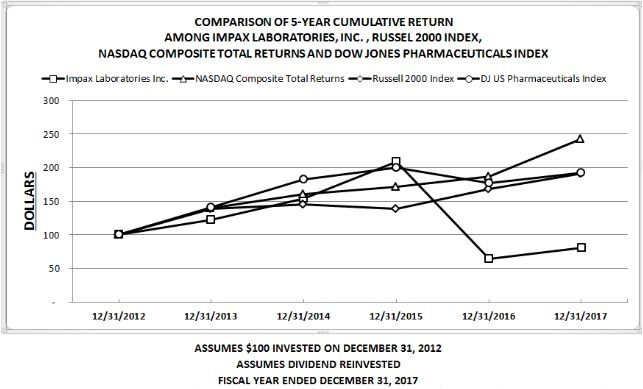
This performance graph shall not be deemed "soliciting material" or to be "filed" with the Securities and Exchange Commission for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any filing of Impax Laboratories, Inc. under the Securities Act of 1933, as amended, or the Exchange Act.
47
Holders
As of December 31, 2017, there were approximately 1,128 holders of record of our common stock, solely based upon the count our transfer agent provided us as of that date.
Dividends
We have never paid cash dividends on our common stock and have no present plans to do so. Our current policy is to retain all earnings, if any, for use in the operation of our business. The payment of future cash dividends, if any, will be at the discretion of our Board of Directors and will be dependent upon our earnings, financial condition, capital requirements and other factors as our Board of Directors may deem relevant.
Unregistered Sales of Securities
There were no sales of unregistered securities during the year ended December 31, 2017.
Purchases of Equity Securities by the Issuer
The following table provides information regarding the purchases of our equity securities by us during the quarter ended December 31, 2017.
Period | Total Number of Shares (or Units) Purchased(1) | Average Price Paid Per Share (or Unit) | Total Number of Shares (or Units) Purchased as Part of Publicly Announced Plans or Programs | Maximum Number (or Approximate Dollar Value) of Shares (or Units) that May Yet Be Purchased Under the Plans or Programs | |||||||||
October 1, 2017 to October 31, 2017 | 78,790 | $ | 19.74 | — | — | ||||||||
November 1, 2017 to November 30, 2017 | — | — | — | — | |||||||||
December 1, 2017 to December 31, 2017 | 597 | $ | 12.15 | — | — | ||||||||
Total | 79,387 | $ | 19.69 | — | — | ||||||||
(1) | Represents shares of our common stock that were repurchased to settle employee tax withholding obligations upon the vesting of shares of restricted stock and/or exercise of stock options pursuant to the terms of our Fourth Amended and Restated 2002 Equity Incentive Plan (the “2002 Plan”). |
48
Equity Compensation Plans
The following table details information regarding our existing equity compensation plans as of December 31, 2017:
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | Weighted Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities reflected in Column (a) | |||||||||
Plan Category | (a) | (b) | (c) | ||||||||
Equity compensation plans approved by security holders | 2,324,997 | (1) | $ | 20.43 | 1,932,375 | (1) | |||||
Equity compensation plans not approved by security holders | 850,000 | (2) | 12.70 | — | |||||||
Total: | 3,174,997 | $ | 18.36 | 1,932,375 | |||||||
(1) | Represents options issued pursuant to the 2002 Plan. There were 296,921 available for issuance under the 1999 Plan, however, we have ceased granting equity awards under this plan. |
(2) | Represents 850,000 options issued to Paul Bisaro, our President and Chief Executive Officer appointed in March 2017 in accordance with NASDAQ's employment inducement grant exemption. |
See “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements — Note 14. Employee Benefit Plans and Note 13. Share-Based Compensation” for information concerning our employee benefit plans and equity compensation plans.
Item 6. Selected Financial Data
The following selected financial data should be read together with our consolidated financial statements and accompanying consolidated financial statement footnotes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” appearing elsewhere in this Annual Report on Form 10-K. The selected consolidated financial statement data in this section are not intended to replace our consolidated financial statements and the accompanying consolidated financial statement footnotes. Our historical consolidated financial results are not necessarily indicative of our future consolidated financial results.
The selected financial data set forth below are derived from our consolidated financial statements. The consolidated statements of operations data for the years ended December 31, 2017, 2016 and 2015 and the consolidated balance sheet data as of December 31, 2017 and 2016 are derived from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K. The data set forth below with respect to our consolidated statements of operations for the years ended December 31, 2014 and 2013 and the consolidated balance sheet data as of December 31, 2015, 2014 and 2013 are derived from our consolidated financial statements, which are not included in this Annual Report on Form 10-K.
49
(In thousands, except per share data) | Years Ended December 31, | ||||||||||||||||||
Statements of Operations Data: | 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||
Total revenues | $ | 775,787 | $ | 824,429 | $ | 860,469 | $ | 596,049 | $ | 511,502 | |||||||||
Research and development | 80,846 | 80,466 | 70,622 | 78,642 | 68,854 | ||||||||||||||
Total operating expenses | 546,491 | 343,080 | 282,836 | 223,837 | 205,687 | ||||||||||||||
(Loss) income from operations | (402,692 | ) | (494,182 | ) | 69,568 | 88,816 | (6,387 | ) | |||||||||||
Net (loss) income | (469,287 | ) | (472,031 | ) | 38,997 | 57,353 | 101,259 | ||||||||||||
Net (loss) income per share — basic | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.56 | $ | 0.84 | $ | 1.51 | |||||||
Net (loss) income per share — diluted | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.54 | $ | 0.81 | $ | 1.47 | |||||||
(In thousands) | As of December 31, | ||||||||||||||||||
Balance Sheet Data: | 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||
Cash, cash equivalents and short-term investments | $ | 181,778 | $ | 180,133 | $ | 340,351 | $ | 414,856 | $ | 413,133 | |||||||||
Working capital | 341,317 | 309,817 | 495,312 | 516,927 | 505,852 | ||||||||||||||
Total assets | 1,351,300 | 1,823,018 | 1,922,487 | 1,079,197 | 996,923 | ||||||||||||||
Long-term debt | 769,524 | 813,545 | 424,595 | — | — | ||||||||||||||
Total liabilities | 1,164,099 | 1,199,044 | 860,078 | 191,320 | 186,720 | ||||||||||||||
Retained (deficit) earnings | (372,445 | ) | 98,192 | 570,223 | 531,226 | 473,873 | |||||||||||||
Total stockholders’ equity | 187,201 | 623,974 | 1,062,409 | 887,877 | 810,203 | ||||||||||||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis, as well as other sections in this report, should be read in conjunction with the consolidated financial statements and related Notes to Consolidated Financial Statements included elsewhere herein. All references to years mean the relevant 12-month period ended December 31.
Overview
We are a specialty pharmaceutical company applying formulation and development expertise, as well as our drug delivery technology, to the development, manufacture and marketing of bioequivalent pharmaceutical products, commonly referred to as “generics,” in addition to the development, manufacture and marketing of branded products. We operate in two segments, referred to as “Impax Generics” and “Impax Specialty Pharma.” Impax Generics concentrates its efforts on generic products, which are the pharmaceutical and therapeutic equivalents of brand-name drug products and are usually marketed under their established nonproprietary drug names rather than by a brand name. Impax Specialty Pharma utilizes its specialty sales force to market proprietary branded pharmaceutical products for the treatment of CNS disorders and other select specialty segments. We sell our Impax Generics division products within the continental United States and the Commonwealth of Puerto Rico. We have no sales in foreign countries.
50
We plan to continue to expand Impax Generics through targeted ANDAs and a first-to-file and first-to-market strategy and to continue to evaluate and pursue external growth initiatives, including acquisitions and partnerships. We focus our efforts on a broad range of therapeutic areas including products that have technically challenging drug-delivery mechanisms or unique product formulations. We employ our technologies and formulation expertise to develop generic products that reproduce brand-name products’ physiological characteristics but do not infringe any valid patents relating to such brand-name products. We generally focus our generic product development on brand-name products as to which the patents covering the active pharmaceutical ingredient have expired or are near expiration, and we employ our proprietary formulation expertise to develop controlled-release technologies that do not infringe patents covering the brand-name products’ controlled-release technologies. We also develop, manufacture, sell and distribute specialty generic pharmaceuticals that we believe present one or more competitive advantages, such as difficulty in raw materials sourcing, complex formulation or development characteristics or special handling requirements. In addition to our focus on solid oral dosage products, we have expanded our generic pharmaceutical products portfolio to include alternative dosage form products, primarily through alliance and collaboration agreements with third parties. As of December 31, 2017, we marketed 225 generic pharmaceuticals, which represent dosage variations of 77 different pharmaceutical compounds through our Impax Generics division; another five of our generic pharmaceuticals representing dosage variations of two different pharmaceutical compounds are marketed by our alliance and collaboration agreement partners. As of December 31, 2017, in our Impax Generics Division, we had 17 applications pending at the FDA and 20 other products in various stages of development for which applications have not yet been filed.
The Impax Generics division develops, manufactures, sells, and distributes generic pharmaceutical products primarily through the following sales channels:
• | the “Impax Generics sales channel” for sales of generic prescription products we sell directly to wholesalers, large retail drug chains, and others; |
• | the “Private Label Product sales channel” for generic pharmaceutical over-the-counter and prescription products we sell to unrelated third-party customers who in-turn sell the product to third parties under their own label; |
• | the “Rx Partner sales channel” for generic prescription products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements; and |
• | the “OTC Partner sales channel” for sales of generic pharmaceutical over-the-counter products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements. |
Revenues from generic products are reported under the caption "Impax Generics, net."
Impax Specialty Pharma is engaged in the development, sale and distribution of proprietary branded pharmaceutical products that we believe represent improvements to already-approved pharmaceutical products addressing CNS disorders, including migraine, multiple sclerosis, Parkinson's disease and post-herpetic neuralgia, and other select specialty segments. We believe that we have the research, development and formulation expertise to develop branded products that will deliver significant improvements over existing therapies.
Our branded pharmaceutical product portfolio consists of commercial CNS and other select specialty products, as well as development stage projects. In February 2012, we licensed from AZ the exclusive U.S. commercial rights to Zomig® (zolmitriptan) tablet, orally disintegrating tablet and nasal spray formulations pursuant to the terms of the AZ Agreement (which was subsequently amended) and began sales of the Zomig® products under our label during the year ended December 31, 2012 through our specialty sales force. In May 2013, our exclusivity period for branded Zomig® tablets and orally disintegrating tablets expired and we launched authorized generic versions of those products in the United States. In June 2015, the FDA approved the Zomig® nasal spray for use in pediatric patients 12 years of age or older for the acute treatment of migraine with or without aura. In addition to the Zomig® products and our internally developed pharmaceutical product, Rytary® for the treatment of Parkinson's disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication and/or manganese intoxication, which was approved by the FDA on January 7, 2015, we are currently engaged in the sales and marketing of Emverm® (mebendazole) 100 mg chewable tablets, indicated for the treatment of pinworm, whipworm, common roundworm, common hookworm, and two other products, all acquired in the Tower Acquisition which closed in March 2015 as described further below. In November 2015, the European Commission granted marketing authorization for Numient® (referred to as Rytary® in the United States). The review of the Numient® application was conducted under the centralized licensing procedure as a therapeutic innovation, and the authorization is applicable in all 28 member states of the European Union, as well as Iceland, Liechtenstein and Norway.
51
Business Combination with Amneal Pharmaceuticals LLC
As discussed above under "Item 1. Business - Business Combination with Amneal Pharmaceuticals LLC", on October 17, 2017, we entered into a Business Combination Agreement (the “Business Combination Agreement”) with Atlas Holdings, Inc., a Delaware corporation and our wholly-owned subsidiary (“Holdco”), K2 Merger Sub Corporation, a Delaware corporation and a wholly-owned subsidiary of Holdco (“Merger Sub”), and Amneal Pharmaceuticals LLC (“Amneal”).
At the closing (the “Closing”) of the transactions contemplated by the Business Combination Agreement (the “Transactions”), (i) Merger Sub will merge with and into our company (the “Impax Merger”), with our company surviving the Impax Merger as a direct wholly-owned subsidiary of Holdco, (ii) each share of our common stock, par value $0.01 per share (“Impax Common Stock”), issued and outstanding immediately prior to the Impax Merger, other than Impax Common Stock held by us in treasury, by Amneal or by any of their respective subsidiaries, will be converted into the right to receive one fully paid and nonassessable share of Class A common stock of Holdco, par value $0.01 per share (“Holdco Class A Common Stock”), (iii) we will convert to a limited liability company pursuant to the General Corporation Law of the State of Delaware and the Delaware Limited Liability Company Act, (iv) Holdco will contribute to Amneal all of Holdco’s equity interests in the Company to Amneal, in exchange for common units of Amneal, (v) Holdco will issue an aggregate number of shares of Class B common stock of Holdco, par value $0.01 per share (“Holdco Class B Common Stock”, and together with Holdco Class A Common Stock, “Holdco Common Stock”) to the existing members of Amneal (the “Existing Amneal Members”) and (vi) Holdco will become the managing member of Amneal. In connection with the Closing, Holdco will be renamed Amneal Pharmaceuticals, Inc. (“New Amneal”).
Immediately following the Closing, (i) the Existing Amneal Members will hold approximately 75% of the voting power and economic interests in New Amneal, and (ii) our stockholders immediately prior to the Closing will hold approximately 25% of the voting power and economic interests in New Amneal. Following the Closing and the PIPE Investment (as described above under “Item 1. Business - Business Combination with Amneal Pharmaceuticals LLC”), it is expected that the Existing Amneal Members will hold approximately 60% of the voting power of the outstanding shares of New Amneal common stock (the “New Amneal Shares”) and TPG Improv Holdings, L.P. (“TPG”) and other institutional investors will hold approximately 15% of the voting power of the outstanding New Amneal Shares.
Consummation of the Transactions is subject to customary closing conditions, including, among other things, (i) the approval of our stockholders holding a majority of the outstanding Impax Common Stock entitled to vote (the “Requisite Stockholder Approval”), (ii) expiration of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and (iii) NYSE listing approval for Holdco Class A Common Stock. The obligation to consummate the Transactions is also conditioned upon each party’s representations and warranties being true and correct (subject to certain materiality exceptions) and each party having performed in all material respects its obligations under the Business Combination Agreement. The Closing is currently expected to occur during the first half of 2018, however, we cannot assure that the Closing will be completed on the terms or timeline currently contemplated, or at all. See “Item IA. Risk Factors - Risks Related to Our Proposed Business Combination with Amneal Pharmaceuticals LLC” above for additional information regarding the risks related to the Transactions.
52
Results of Operations
Year Ended December 31, 2017 Compared to Year Ended December 31, 2016
Overview
The following table sets forth our summarized, consolidated results of operations for the years ended December 31, 2017 and 2016 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2017 | 2016 | Dollars | Percentage | |||||||||||
Total revenues | $ | 775,787 | $ | 824,429 | $ | (48,642 | ) | (6 | )% | |||||
Gross profit (loss) | 143,799 | (151,102 | ) | 294,901 | * | |||||||||
(Loss) income from operations | (402,692 | ) | (494,182 | ) | 91,490 | (19 | )% | |||||||
(Loss) income before income taxes | (450,961 | ) | (576,325 | ) | 125,364 | (22 | )% | |||||||
Provision for (benefit from) income taxes | 18,326 | (104,294 | ) | 122,620 | * | |||||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 2,744 | (1 | )% | ||||
* Percentage exceeds 100%
Consolidated total revenues for the year ended December 31, 2017 decreased by 6%, or $48.6 million, to $775.8 million compared to $824.4 million for the year ended December 31, 2016. The decrease was primarily attributable to lower Impax Generics division product sales. Selling price for existing products decreased consolidated total revenues by 22%, while volumes for existing products increased consolidated total revenues by 14%, in each case compared to the prior year. The decrease in selling price was primarily the result of additional competition during the year ended December 31, 2017 in generic Adderall XR®, fenofibrate, diclofenac sodium gel, metaxalone and lower prices on epinephrine auto injector, partially offset by volume increases in epinephrine auto injector and Rytary®. New product launches increased consolidated total revenues by 2% compared to the prior year. We currently expect pricing pressures on generic products to continue in the industry at least in the near term. We are closely monitoring these developments as they related to our products, customers and end users.
Revenues from our Impax Generics division for the year ended December 31, 2017 were $549.1 million, a decrease of $57.2 million or 9%, over the prior year. The decrease compared to the prior year period was primarily due to increased competition on diclofenac sodium gel, metaxalone, generic Adderall XR® and fenofibrate. These decreases were partially offset by increased sales of our epinephrine auto-injector, budesonide and other products we acquired as part of the Teva Transaction compared to the prior year period.
Revenues from our Impax Specialty Pharma division for the year ended December 31, 2017 were $226.7 million, an increase of $8.6 million or 4% over the prior year. The increase from the prior year period was primarily due to higher sales of Rytary®, partially offset by lower sales of our anthelmintic products franchise and Zomig®.
Net loss for the year ended December 31, 2017 was $469.3 million, a decrease in our loss of $2.7 million compared to a net loss of $472.0 million for the year ended December 31, 2016. The net loss for the year ended December 31, 2017 was due to $289.7 million in intangible asset impairment charges and an approximate $74.1 million fixed assets impairment charge of our Taiwan manufacturing facility associated with our announced sale of the Taiwan operations. Additionally, during the year ended December 31, 2017, revenue from our generic products decreased due to increased competition and an approximate $48.2 million increase in cost of revenues caused by under-utilization of our plants associated with our restructuring initiatives. Our fiscal year 2016 net loss was driven largely by our $541.6 million asset impairment charges and a $40.3 million reserve recorded as a result of the uncertainty of collection of the receivable due from Turing for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities.
Of the $289.7 million intangible asset impairment charges we incurred during the year ended December 31, 2017, we recognized $96.9 million in cost of revenues impairment charges and $192.8 million in in-process research and development impairment charges on our consolidated statement of operations. The impairment charge was attributable to eight currently marketed products and four in-process research and development ("IPR&D") product rights, the majority of which were acquired as part of the Teva Transaction. For the currently marketed products, the impairment charge was the result of continued significant price and volume erosion during the year ended December 31, 2017, resulting in significantly lower expected future cash flows. The IPR&D impairment was the result of delays in the anticipated product launch and related competition in the market.
53
Impax Generics
The following table sets forth results of operations for the Impax Generics division for the years ended December 31, 2017 and 2016 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2017 | 2016 | Dollars | Percentage | |||||||||||
Revenues: | ||||||||||||||
Impax Generics sales, net | $ | 549,077 | $ | 606,320 | $ | (57,243 | ) | (9 | )% | |||||
Cost of revenues | 454,911 | 417,316 | 37,595 | 9 | % | |||||||||
Cost of revenues impairment charges | 96,865 | 464,319 | (367,454 | ) | (79 | )% | ||||||||
Gross loss | (2,699 | ) | (275,315 | ) | 272,616 | (99 | )% | |||||||
Operating expenses: | ||||||||||||||
Selling, general and administrative | 28,294 | 20,508 | 7,786 | 38 | % | |||||||||
Research and development | 63,245 | 61,980 | 1,265 | 2 | % | |||||||||
In-process research and development impairment charges | 192,809 | 27,765 | 165,044 | * | ||||||||||
Patent litigation expense | 827 | 829 | (2 | ) | — | % | ||||||||
Change in fair value of contingent consideration | (31,048 | ) | — | (31,048 | ) | * | ||||||||
Fixed assets impairment charges | 8,380 | — | 8,380 | * | ||||||||||
Total operating expenses | 262,507 | 111,082 | 151,425 | * | ||||||||||
Loss from operations | $ | (265,206 | ) | $ | (386,397 | ) | $ | 121,191 | (31 | )% | ||||
* Percentage exceeds 100%
Revenues
Total revenues for the Impax Generics division for the year ended December 31, 2017 were $549.1 million, a decrease of $57.2 million or 9%, compared to the prior year. The decrease compared to the prior year period was primarily due to increased competition on diclofenac sodium gel, metaxalone, generic Adderall XR® and fenofibrate. The decreases in revenue related to these products were partially offset by increased sales of our epinephrine auto-injector, budesonide and other products we acquired as part of the Teva Transaction compared to the prior year period.
Cost of Revenues
Cost of revenues was $454.9 million for the year ended December 31, 2017, an increase of $37.6 million from the prior year. The increase was due to $22.4 million of higher intangible asset amortization expenses resulting from the Teva Transaction, an increase of $14.4 million of inventory reserves primarily for bad batches and short-dated product, $11.6 million of additional restructuring costs incurred in connection with the closure of our Middlesex, New Jersey facility and the reduction-in-force of our technical operations group. The additional costs are offset by lower production costs due to increased absorption primarily as a result of restocking of new product launch inventory.
Cost of Revenues Impairment Charges
Cost of revenues impairment charges was $96.9 million for the year ended December 31, 2017, as compared to $464.3 million for the year ended December 31, 2016. The $96.9 million of impairment charges for the year ended December 31, 2017 were mostly due to continued price and volume erosion on eight currently marketed products, of which six were acquired in the Teva Transaction without an offsetting increase in customer demand, resulting in significantly lower expected future cash flows. The $464.3 million of impairment charges for the year ended December 31, 2016 were primarily due to price reductions taken on certain products acquired as part of the Teva Transaction in order to retain key customers.
54
Gross Loss
Gross loss for the year ended December 31, 2017 was $2.7 million as compared to gross loss of $275.3 million for the prior year. The decrease in gross loss was due primarily to $367.5 million of lower intangible asset impairment charges offset by an increase of $22.4 million intangible asset amortization expenses both relating to assets acquired in the Teva Transaction. The gross loss decrease was also partially offset by continued price erosion due to competition and customer mix along with an increase of $14.4 million of inventory reserves, $11.6 million of additional restructuring costs incurred with the closure of our Middlesex, New Jersey facility and the reduction-in-force of our technical operations group.
Selling, General and Administrative Expenses
Selling, general, and administrative expenses for the year ended December 31, 2017 were $28.3 million, as compared to $20.5 million for the year ended December 31, 2016. The $7.8 million increase from the prior year was primarily due to $2.9 million of additional freight costs, $2.8 million of higher supply claims from our wholesale customers and $2.2 million higher marketing costs.
Research and Development Expenses
Research and development expenses for the year ended December 31, 2017 were $63.2 million, as compared to $62.0 million for the year ended December 31, 2016. The $1.2 million increase from the prior year period was primarily due to $3.3 million of higher internal project costs and $0.8 million of employee termination benefits from the closure of our Generic Division's research and development site in Middlesex, New Jersey partially offset by $3.4 million of lower external development.
In-process Research and Development Impairment Charges
In-process research and development impairment charges were $192.8 million for the year ended December 31, 2017, as compared to $27.8 million for the year ended December 31, 2016. The $192.8 million of impairment charges for the year ended December 31, 2017 were due to delays in the anticipated launch of products and marketing rights acquired in the Teva Transaction and associated competition in the market. The $27.8 million of impairment charges for the year ended December 31, 2016 were due to delays in the expected start of commercialization and/or lower pricing amid highly competitive market conditions of certain pipeline products, resulting in lower expected future cash flows.
Change in Fair Value of Contingent Consideration
During the year ended December 31, 2017, we recognized $31.0 million of income on the change in the fair value of contingent consideration, compared to a minimal change in fair value of contingent consideration recognized during the prior year. We are required under the Termination Agreement with Teva to make certain milestone payments to Teva associated with our methylphenidate hydrochloride (generic Concerta®) product. The Company conducted a review of the underlying inputs and assumptions at December 31, 2017, and based on timing and probability of the product launch, and corresponding number of competitors expected to be in the market at both launch and the one-year anniversary of launch, the Company concluded that the fair value of its contingent consideration was $0. Refer to “Item 15. Financial Information - Notes to the Consolidated Financial Statements - Note 4. Fair Value Measurement and Financial Instruments” for additional information related to the contingent consideration associated with our methylphenidate hydrochloride product.
Fixed Assets Impairment Charges
The fixed assets impairment charges recognized during the year ended December 31, 2017 were primarily due to the closure of our Middlesex, New Jersey manufacturing facility; we sold the entity which held the leases to the site to a third party in early 2018. In addition, we recognized fixed impairment charges associated with abandoned software. Refer to “Item 15. Financial Information - Notes to the Consolidated Financial Statements - Note 15. Restructurings” for additional information related to the sale of Middlesex, New Jersey assets. There was no comparable loss in 2016.
55
Impax Specialty Pharma
The following table sets forth results of operations for the Impax Specialty Pharma division for the years ended December 31, 2017 and 2016 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2017 | 2016 | Dollars | Percentage | |||||||||||
Revenues: | ||||||||||||||
Rytary®, net | $ | 91,637 | $ | 73,834 | $ | 17,803 | 24 | % | ||||||
Zomig®, net | 51,115 | 53,539 | (2,424 | ) | (5 | )% | ||||||||
All other Specialty Pharma Products, net | 83,958 | 90,736 | (6,778 | ) | (7 | )% | ||||||||
Total revenues | 226,710 | 218,109 | 8,601 | 4 | % | |||||||||
Cost of revenues | 80,212 | 69,583 | 10,629 | 15 | % | |||||||||
Cost of revenues impairment charges | — | 24,313 | (24,313 | ) | (100 | )% | ||||||||
Gross profit | 146,498 | 124,213 | 22,285 | 18 | % | |||||||||
Operating expenses: | ||||||||||||||
Selling, general and administrative | 67,949 | 61,448 | 6,501 | 11 | % | |||||||||
Research and development | 17,602 | 18,486 | (884 | ) | (5 | )% | ||||||||
In-process research and development impairment charges | — | 25,200 | (25,200 | ) | (100 | )% | ||||||||
Fixed assets impairment charges | 74,128 | — | 74,128 | * | ||||||||||
Patent litigation expense | 4,278 | 6,990 | (2,712 | ) | (39 | )% | ||||||||
Total operating expenses | 163,957 | 112,124 | 51,833 | 46 | % | |||||||||
(Loss) income from operations | $ | (17,459 | ) | $ | 12,089 | $ | (29,548 | ) | * | |||||
* Percentage exceeds 100%
Revenues
Total revenues for the Impax Specialty Pharma division for the year ended December 31, 2017 were $226.7 million, an increase of $8.6 million or 4% over the prior year. The increase from the prior year period was primarily due to higher sales of Rytary®, partially offset by lower sales of our anthelmintic products franchise and Zomig®.
Cost of Revenues
Cost of revenues was $80.2 million for the year ended December 31, 2017, a $10.6 million increase over the prior year. The increase is primarily due to higher sales of Rytary, increase in inventory reserve of $4.6 million and increase in accelerated depreciation expenses of $9.1 million related to our manufacturing facility located in Taiwan, which we sold in February of 2018. The cost of revenues increase was partially offset by a reduction in amortization of $10.6 million due to impairment of Emverm® intangible asset in 2016.
Cost of Revenues Impairment Charges
Cost of revenues impairment charges were $24.3 million for the year ended December 31, 2016, primarily as a result of lower than expected script volume for Emverm®. There were no comparable charges during 2017.
Gross Profit
Gross profit for the year ended December 31, 2017 was $146.5 million, or 65% of total revenues, as compared to $124.2 million, or 57% of total revenues, in the prior year. The increase in gross profit was primarily due to higher product sales of Rytary® during the year ended December 31, 2017 and a reduction in intangible asset impairment charges compared to 2016.
56
Selling, General and Administrative Expenses
Selling, general and administrative expenses for the year ended December 31, 2017 were $67.9 million, as compared to $61.4 million for the year ended December 31, 2016. The $6.5 million increase compared to the prior year was primarily due to certain employee termination benefits and higher advertising and promotion costs related to Emverm® and Zomig®.
Research and Development Expenses
Research and development expenses for the year ended December 31, 2017 were $17.6 million, as compared to $18.5 million for the year ended December 31, 2016. The $0.9 million decrease compared to the prior year was primarily due to a $2.6 million AstraZeneca reimbursement to us related to the juvenile toxicity study and pediatric study required by the FDA under the Pediatric Research Equity Act for approval of the nasal formulation of Zomig® for the acute treatment of migraine in pediatric patients ages six through eleven years old pursuant to the terms of the AZ Agreement, as well as reduced expenses related to our branded initiatives, partially offset by higher spend for our Drug Safety/Pharmacovigilance group of $2.5 million. See “Item 1. Alliance and Collaboration Agreements - Impax Specialty Pharma - Alliance and Collaboration Agreements” for details regarding the AZ Agreement.
In-process Research and Development Impairment Charges
In-process research and development impairment charges were $25.2 million for the year ended December 31, 2016. The impairment charges resulted from management’s decision during the fourth quarter of 2016 to cease development on our next generation Albenza® product due to continued difficulties in sourcing the active pharmaceutical ingredient for the product. There were no comparable charges during 2017.
Fixed Assets Impairment Charges
The fixed assets impairment charges recorded during the year ended December 31, 2017 were primarily due to a $74.1 million loss associated with a stock and asset purchase agreement we entered into with a third party during the year ended December 31, 2017 pursuant to which we agreed to sell Impax Taiwan, including our Taiwan facility. Refer to “Item 15. Exhibits and Financial Statement Schedules - Notes to the Consolidated Financial Statements - Note 15. Restructurings” for additional information related to the sale of our Taiwan operations. There was no comparable fixed asset impairment charges recorded in 2016.
Patent Litigation Expenses
Patent litigation expenses for the year ended December 31, 2017 were $4.3 million, as compared to $7.0 million for the year ended December 31, 2016. The $2.7 million higher cost during the prior year was primarily due to patent litigation activity related to Zomig® trial during the third quarter of 2016. Refer to “Item 15. Financial Information - Notes to the Consolidated Financial Statements - Note 19. Legal and Regulatory Matters” for more information.
57
Corporate and Other
The following table sets forth corporate general and administrative expenses, as well as other items of income and expense presented below income or loss from operations for the years ended December 31, 2017 and 2016 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2017 | 2016 | Dollars | Percentage | |||||||||||
General and administrative expenses | $ | 120,027 | $ | 119,874 | $ | 153 | — | % | ||||||
Interest expense, net | (53,412 | ) | (40,419 | ) | (12,993 | ) | 32 | % | ||||||
Reserve for Turing receivable | (3,999 | ) | (40,312 | ) | 36,313 | (90 | )% | |||||||
Gain on sale of assets | 17,236 | 175 | 17,061 | * | ||||||||||
Loss on debt extinguishment | (1,215 | ) | — | (1,215 | ) | * | ||||||||
Other expense, net | (6,879 | ) | (1,587 | ) | (5,292 | ) | * | |||||||
Loss before income taxes | (168,296 | ) | (202,017 | ) | 33,721 | (17 | )% | |||||||
Provision for (benefit from) income taxes | $ | 18,326 | $ | (104,294 | ) | $ | 122,620 | * | ||||||
* Percentage exceeds 100%
General and Administrative Expenses
General and administrative expenses for the year ended December 31, 2017 were $120.0 million, as compared to $119.9 million for the year ended December 31, 2016. The $0.1 million increase during 2017 compared to the prior year was primarily due to $8.6 million higher legal expenses compared to the prior year period and $11.7 million integration costs. These higher expenses were largely offset primarily by $5.4 million of lower executive costs, $4.8 million lower share-based compensation costs, $4.3 million of reduction in IT spending, and $1.5 million lower business development spending. The expenses in 2016 also included $3.7 million related to the Teva Transaction, of which there were no comparable charges during year ended December 31, 2017.
Interest Expense, net
Interest expense,net was $53.4 million for the year ended December 31, 2017, a $13.0 million increase from the prior year. Interest expense for 2017 reflected interest on our $600.0 million convertible senior notes issued in 2015, interest on our $400.0 million Term Loan with Royal Bank of Canada entered into in the third quarter of 2016 to fund the Teva Transaction, and unused line of credit fees on our Revolving Credit Facility with Royal Bank of Canada entered into in 2016. In contrast, prior year interest expense of $40.4 million reflected interest expense on our Term Loan with Barclays Bank PLC entered into in connection with the financing of the Tower Acquisition, which was repaid in full on June 30, 2016 using proceeds from the issuance of our $600.0 million convertible senior notes. Refer to "Outstanding Debt Obligations" below for additional information related to our outstanding convertible notes and credit facilities. Interest income was $1.0 million for the year ended December 31, 2017, which was relatively consistent with interest income for the year ended December 31, 2016.
Reserve for Turing Receivable
During the year ended December 31, 2016, we recorded a reserve of $40.3 million as a result of the uncertainty of collection of the receivable due from Turing for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities, as compared to a net $4.0 million of such charges during the year ended December 31, 2017. Refer to “Item 15. Exhibits and Financial Statement Schedules - Notes to the Consolidated Financial Statements - Note 5. Accounts Receivable” for additional information related to the Turing receivable.
Gain on Sale of Assets
During the year ended December 31, 2017, we recognized a $12.5 million gain on the sale of 29 ANDAs and one NDA for non-strategic approved generic products, the vast majority of which products were not marketed, and all acquired as part of the Tower Acquisition, and $4.7 million gain from the sale of the our storage warehouse in Hayward, California.
58
Loss on Debt Extinguishment
During the year ended December 31, 2017, we recognized a $1.2 million loss on debt extinguishment related to the voluntary prepayment of $50.0 million on our Term Loan Facility with Royal Bank of Canada. There was no comparable loss in 2016.
Other Expense, Net
Other expense, net was $6.9 million for year ended December 31, 2017, as compared to $1.6 million for the year ended December 31, 2016. The expense for the year ended December 31, 2017 was primarily due to legal settlement costs related to our settlement with Endo Pharmaceuticals Inc. on our marketed oxymorphone hydrochloride tablets, which we settled in August 2017, and the suit related to the Telephone Consumer Protection Act. Refer to "Item 15. Financial Information - Notes to the Consolidated Financial Statements - Note 19. Legal and Regulatory Matters" for more information related to the Telephone Consumer Protection Act suit.
Income Taxes
During the year ended December 31, 2017, we recorded an aggregate tax provision of $18.3 million for U.S. domestic income taxes and foreign income taxes, an increase of $122.6 million compared to an aggregate tax benefit of $104.3 million we recorded during the prior year. The effective tax rate decreased to (4.1)% for the year ended December 31, 2017 compared to 18.1% for the year ended December 31, 2016.
The effective income tax rate was (4.1)% for the fiscal year ended December 31, 2017, and reflected the increase in valuation allowance of $77.1 million against net deferred tax assets. Based on an evaluation of both the positive and negative evidence available, as discussed below, we determined that it was necessary to establish a valuation allowance against all of our net deferred tax assets for the fiscal year ended December 31, 2017.
A valuation allowance, if needed, reduces deferred tax assets to the amount expected to be realized. When determining the amount of net deferred tax assets that are more likely than not to be realized, we assess all available positive and negative evidence. This evidence includes, but is not limited to, scheduled reversal of deferred tax liabilities, prior earnings history, projected future earnings, carry-back and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income (exclusive of reversing taxable temporary differences and carryforwards) to outweigh objective negative evidence of a recent financial reporting loss for the year ended December 31, 2017.
Based on these criteria and the relative weighting of both the positive and negative evidence available, and in particular the activity surrounding our prior earnings history, including the intangible impairments charges recognized during 2017, we determined that it was necessary to establish a valuation allowance against all of our net deferred tax assets as of December 31, 2017. Given the objectively verifiable negative evidence of a three-year cumulative loss which, under the provisions of FASB ASC Topic 740 is a significant element of negative evidence that is difficult to overcome, and the weighting of all available positive evidence, we excluded projected taxable income from the assessment of income that could be used as a source of taxable income to realize the deferred tax assets.
59
Year Ended December 31, 2016 Compared to Year Ended December 31, 2015
Overview
The following table sets forth our summarized, consolidated results of operations for the years ended December 31, 2016 and 2015 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2016 | 2015 | Dollars | Percentage | |||||||||||
Total revenues | $ | 824,429 | $ | 860,469 | $ | (36,040 | ) | (4 | )% | |||||
Gross (loss) profit | (151,102 | ) | 352,404 | (503,506 | ) | * | ||||||||
(Loss) income from operations | (494,182 | ) | 69,568 | (563,750 | ) | * | ||||||||
(Loss) income before income taxes | (576,325 | ) | 59,368 | (635,693 | ) | * | ||||||||
(Benefit from) provision for income taxes | (104,294 | ) | 20,371 | (124,665 | ) | * | ||||||||
Net (loss) income | $ | (472,031 | ) | $ | 38,997 | $ | (511,028 | ) | * | |||||
* Percentage exceeds 100%
Consolidated total revenues for the year ended December 31, 2016 decreased by 4%, or $36.1 million, to $824.4 million compared to $860.5 million for the year ended December 31, 2015. The decrease was primarily attributable to lower Impax Generics division product sales, partially offset by higher Impax Specialty Pharma division product sales. Selling price for existing products decreased consolidated total revenues by 16%, while volumes for existing products increased consolidated total revenues by 2%, in each case compared to the prior year. New product launches, including those resulting from acquisitions, increased consolidated total revenues by 9% compared to the prior year.
Revenues from our Impax Generics division decreased by $104.6 million during the year ended December 31, 2016, as compared to the prior year. This decrease was primarily due to lower selling prices across a majority of the products in the division, partially offset by higher sales volumes, including those resulting from product acquisitions. The products that experienced significant declines in selling price during the year ended December 31, 2016 compared to the prior year included diclofenac sodium gel, metaxalone, generic Adderall XR®, and fenofibrate family products. In connection with the pricing declines, we recorded $15.0 million in shelf-stock adjustments related to diclofenac sodium gel and metaxalone during 2016. Partially offsetting these pricing declines were price and volume increases of certain products compared to 2015 primarily related to our epinephrine auto-injector and oxymorphone products.
Revenues from our Impax Specialty Pharma division increased by $68.6 million during the year ended December 31, 2016, as compared to the prior year. The increase was primarily due to higher selling prices and higher sales volumes across a majority of the products in the division including Zomig®, Rytary®, which launched in April 2015, and our anthelmintic products franchise.
Net loss for the year ended December 31, 2016 was $472.0 million, a decrease of $511.0 million compared to net income of $39.0 million for the year ended December 31, 2015. The net loss for the year ended December 31, 2016 was primarily driven by $541.6 million in intangible asset impairment charges, as compared to $13.7 million of such charges in the prior year, as well as a $40.3 million reserve recorded as a result of the uncertainty of collection of the receivable due from Turing for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities. Included in our 2015 results was a $45.6 million gain related to the sale of Daraprim® to Turing, for which there was no comparable gain in 2016. Refer to "Item 15. Exhibits and Financial Schedules - Note 5. Accounts Receivable" for information related to the Daraprim® sale and Turing reserve.
60
Of the $541.6 million in intangible asset impairment charges we incurred in 2016, $308.4 million of such charges related to certain intangible assets acquired as part of the Teva Transaction. Upon closing the Teva Transaction on August 3, 2016, we initiated the process of transferring and securing Teva’s and Allergan’s customers for the acquired products to our account. We assumed certain price concessions would occur following the closing. However, we elected to take additional price reductions on certain of the acquired products in order to retain key customers. These reductions produced significantly lower than expected operating cash flows from the acquired product lines and triggered an impairment charge of $251.0 million during the third quarter of 2016. We experienced even further price reductions on certain of the products acquired in the Teva Transaction during the fourth quarter of 2016, which resulted in $57.4 million of additional intangible asset impairment charges. In total, our impairment analyses for the products acquired in the Teva Transaction resulted in the recognition of $308.4 million of non-cash impairment charges to earnings, comprised of a $301.7 million charge recorded in cost of revenues impairment charges and a $6.7 million charge recorded in-process research and development impairment charges in our consolidated statement of operations for the year ended December 31, 2016.
During 2016, we also incurred other non-cash impairment charges on certain of our intangible assets, primarily related to the products acquired from the Tower Acquisition, totaling $233.2 million. These impairment charges arose primarily due to increased competition, price degradation, product discontinuations and delays in expected product launches. The largest intangible asset impairment charge related to products acquired in the Tower Acquisition was for our epinephrine auto-injector product, which occurred during the fourth quarter of 2016 and accounted for more than half of the $233.2 million in charges. The impairment charge on the epinephrine auto-injector product was triggered by current and projected price degradation as a result of unexpected changes in the pricing environment and additional competition.
Impax Generics
The following table sets forth results of operations for the Impax Generics division for the years ended December 31, 2016 and 2015 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2016 | 2015 | Dollars | Percentage | |||||||||||
Revenues: | ||||||||||||||
Impax Generics sales, net | $ | 591,744 | $ | 699,844 | $ | (108,100 | ) | (15 | )% | |||||
Rx Partner | 14,339 | 9,307 | 5,032 | 54 | % | |||||||||
Other Revenues | 237 | 1,781 | (1,544 | ) | (87 | )% | ||||||||
Total revenues | 606,320 | 710,932 | (104,612 | ) | (15 | )% | ||||||||
Cost of revenues | 417,316 | 442,742 | (25,426 | ) | (6 | )% | ||||||||
Cost of revenues impairment charges | 464,319 | 7,303 | 457,016 | * | ||||||||||
Gross (loss) profit | (275,315 | ) | 260,887 | (536,202 | ) | * | ||||||||
Operating expenses: | ||||||||||||||
Selling, general and administrative | 20,508 | 29,641 | (9,133 | ) | (31 | )% | ||||||||
Research and development | 61,980 | 52,478 | 9,502 | 18 | % | |||||||||
In-process research and development impairment charges | 27,765 | 6,360 | 21,405 | * | ||||||||||
Patent litigation expense | 829 | 2,942 | (2,113 | ) | (72 | )% | ||||||||
Total operating expenses | 111,082 | 91,421 | 19,661 | 22 | % | |||||||||
(Loss) income from operations | $ | (386,397 | ) | $ | 169,466 | $ | (555,863 | ) | * | |||||
* Percentage exceeds 100%
Revenues
Total revenues for the Impax Generics division for the year ended December 31, 2016 were $606.3 million, a decrease of $104.6 million or 15%, over the prior year. The decrease was primarily due to increased competition on diclofenac sodium gel, metaxalone, and fenofibrate, coupled with lower market share for generic Adderall XR® during the first half of 2016, in each case compared to the prior year. These decreases were partially offset by increased sales of oxymorphone, increased sales of epinephrine auto-injector, which was acquired as part of the Tower Acquisition in March 2015, and sales of the products acquired as part of the Teva Transaction in August 2016, in each case compared to the prior year. In addition, during the year ended December 31, 2016, we recorded a $15.0 million shelf-stock adjustment related to diclofenac sodium gel and metaxalone as a result of declining prices during 2016, for which there was no comparable charge in the prior year.
61
Cost of Revenues
Cost of revenues was $417.3 million for the year ended December 31, 2016, a decrease of $25.4 million from the prior year. The decrease was primarily attributable to lower costs related to decreased product revenue compared to the prior year and the absence of costs related to (i) the step-up to fair value of inventory in connection with the Tower Acquisition, (ii) Hayward remediation activities and (iii) the Philadelphia restructuring, which were all incurred in the prior year but for which we did not incur comparable costs in 2016. The reduced costs during 2016 compared to the prior year were partially offset by higher intangible asset amortization expenses resulting from the Teva Transaction and a full year of amortization expense related to products acquired in the Tower Acquisition, along with higher restructuring costs incurred in conjunction with the previously announced closure of the Middlesex, New Jersey facility.
Cost of Revenues Impairment Charges
Cost of revenues impairment charges was $464.3 million for the year ended December 31, 2016, a $457.0 million increase over the prior year. Of this increase, $301.7 million related to impairments recognized on certain intangible assets acquired as part of the Teva Transaction. As discussed above, we assumed certain price concessions would occur following the closing of the Teva Transaction on August 3, 2016. However, we elected to take additional price reductions on certain of the acquired products in order to retain key customers. These reductions produced significantly lower than expected operating cash flows from the acquired product lines and triggered an impairment charge of $248.0 million during the third quarter of 2016. We experienced even further price reductions on certain of the products acquired in the Teva Transaction during the fourth quarter of 2016, which resulted in $53.7 million of additional intangible asset impairment charges recorded in cost of revenues impairment charges.
During 2016, we also incurred other non-cash impairment charges recorded to cost of revenues impairment charges on certain of our intangible assets, primarily related to the products acquired from the Tower Acquisition, totaling $162.6 million. These impairment charges arose primarily due to increased competition, price degradation, and product discontinuations. The largest intangible asset impairment charge related to the products acquired in the Tower Acquisition was on our epinephrine auto-injector product, which occurred during the fourth quarter of 2016. The impairment charge on the epinephrine auto-injector product was triggered by current and projected price degradation as a result of changes in the pricing environment and additional competition.
Gross (Loss) Profit
Gross (loss) for the year ended December 31, 2016 was ($275.3) million, or 45% of total revenues, as compared to gross profit of $260.9 million, or 37% of total revenues, for the prior year. The decreases in gross profit and gross margin were primarily due to intangible asset impairment charges, lower product sales, higher shelf-stock adjustments, increased intangibles amortization, and increased restructuring costs, as noted above. These decreases were partially offset by the absence of remediation costs related to the Hayward facility and the absence of restructuring costs related to the Philadelphia facility in 2016, both incurred in 2015.
Selling, General and Administrative Expenses
Selling, general, and administrative expenses for the year ended December 31, 2016 were $20.5 million, as compared to $29.6 million for the year ended December 31, 2015. The $9.1 million decrease from the prior year was primarily attributable to a decrease in failure to supply claims during 2016.
Research and Development Expenses
Research and development expenses for the year ended December 31, 2016 were $62.0 million, as compared to $52.5 million for the year ended December 31, 2015. The $9.5 million increase from the prior year was primarily due to an increase in external development costs from increased research and development activities and a full year of research and development expenses from the Tower acquired companies.
62
In-process Research and Development Impairment Charges
In-process research and development impairment charges were $27.8 million for the year ended December 31, 2016, an increase of $21.4 million from the prior year. The 2016 impairment charges included $21.1 million related to products acquired as part of the Tower Acquisition and caused primarily due to delays in the expected start of commercialization and/or lower anticipated pricing of such products amid highly competitive market conditions, resulting in lower forecasted future cash flows. There were $6.4 million of similar charges recorded in the prior year. In addition, the 2016 impairment charges included $6.7 million related to products acquired as part of the Teva Transaction and caused by lower anticipated pricing amid highly competitive market conditions, resulting in lower forecasted future cash flows.
Patent Litigation Expenses
Patent litigation expenses for the year ended December 31, 2016 were $0.8 million, as compared to $2.9 million for the year ended December 31, 2015. The $2.1 million decrease was due to reduced legal activity in 2016 compared to the prior year.
Impax Specialty Pharma
The following table sets forth results of operations for the Impax Specialty Pharma division for the years ended December 31, 2016 and 2015 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2016 | 2015 | Dollars | Percentage | |||||||||||
Revenues: | ||||||||||||||
Rytary®, net | $ | 73,834 | $ | 42,364 | $ | 31,470 | 74 | % | ||||||
Zomig®, net | 53,539 | 49,251 | 4,288 | 9 | % | |||||||||
All other Specialty Pharma Products, net | 90,736 | 57,922 | 32,814 | 57 | % | |||||||||
Total revenues | 218,109 | 149,537 | 68,572 | 46 | % | |||||||||
Cost of revenues | 69,583 | 58,020 | 11,563 | 20 | % | |||||||||
Cost of revenues impairment charges | 24,313 | — | 24,313 | * | ||||||||||
Gross profit | 124,213 | 91,517 | 32,696 | 36 | % | |||||||||
Operating expenses: | ||||||||||||||
Selling, general and administrative | 61,448 | 52,427 | 9,021 | 17 | % | |||||||||
Research and development | 18,486 | 18,144 | 342 | 2 | % | |||||||||
In-process research and development impairment charges | 25,200 | — | 25,200 | * | ||||||||||
Patent litigation expense | 6,990 | 1,625 | 5,365 | * | ||||||||||
Total operating expenses | 112,124 | 72,196 | 39,928 | 55 | % | |||||||||
Income from operations | $ | 12,089 | $ | 19,321 | $ | (7,232 | ) | (37 | )% | |||||
* Percentage exceeds 100%
Revenues
Total revenues for the Impax Specialty Pharma division for the year ended December 31, 2016 were $218.1 million, an increase of $68.6 million or 46% over the prior year. The increase was primarily due to increased sales from Rytary®, which we launched in April 2015, and increased revenues resulting from the Tower Acquisition, including sales from our anthelmintic products franchise.
Cost of Revenues
Cost of revenues was $69.6 million for the year ended December 31, 2016, an $11.6 million increase over the prior year. The increase was primarily due to higher costs related to increased product sales and a full year of amortization expense related to products acquired in the Tower Acquisition. Additionally, cost of revenues for the prior year included a $5.4 million step-up to fair value of inventory charge in connection with the Tower Acquisition, for which there was no comparable charge in 2016.
63
Cost of Revenues Impairment Charges
Cost of revenues impairment charges were $24.3 million for the year ended December 31, 2016. There were no comparable charges during the prior year. The impairment charge was primarily the result of lower than expected script volume for Emverm®.
Gross Profit
Gross profit for the year ended December 31, 2016 was $124.2 million, or 57% of total revenues, as compared to $91.5 million, or 61% of total revenues, in the prior year. The increase in gross profit in 2016 compared to the prior year was primarily due to increased product sales and the absence in 2016 of the $5.4 million step-up to fair value of inventory charge in connection with the Tower Acquisition we incurred in 2015, partially offset by higher impairment charges during 2016. The decrease in gross margin during the year ended December 31, 2016 was primarily due to lower selling prices on certain products compared to the prior year.
Selling, General and Administrative Expenses
Selling, general and administrative expenses for the year ended December 31, 2016 were $61.4 million, as compared to $52.4 million for the year ended December 31, 2015. The $9.0 million increase during the year ended December 31, 2016 was primarily due to expenses related to the sales force expansion to support sales and marketing activities for Rytary® and increased advertising and promotion expenses to support the launch of Emverm® and the new indication of Zomig® nasal spray for pediatric patients approved by the FDA in June 2015. The increase in expenses during 2016 was partially offset by training expenses incurred during the year ended December 31, 2015 to support the launch of Rytary®.
Research and Development Expenses
Research and development expenses for the year ended December 31, 2016 were $18.5 million, as compared to $18.1 million for the year ended December 31, 2015. The $0.4 million increase compared to the prior year was primarily due to increased research and development activities related to our branded initiatives.
In-process Research and Development Impairment Charges
In-process research and development impairment charges were $25.2 million for the year ended December 31, 2016. There were no comparable charges during the prior year. The impairment charges resulted from management’s decision during the fourth quarter of 2016 to cease development on our next generation Albenza® product due to continued difficulties in sourcing the active pharmaceutical ingredient for the product.
Patent Litigation Expenses
Patent litigation expenses for the year ended December 31, 2016 were $7.0 million, as compared to $1.6 million for the year ended December 31, 2015. The $5.4 million increase during 2016 compared to the prior year was due to increased patent litigation activity in 2016.
64
Corporate and Other
The following table sets forth corporate general and administrative expenses, as well as other items of income and expense presented below income from operations for the years ended December 31, 2016 and 2015 (in thousands):
Year Ended December 31, | Increase / (Decrease) | |||||||||||||
2016 | 2015 | Dollars | Percentage | |||||||||||
General and administrative expenses | $ | 119,874 | $ | 119,219 | $ | 655 | 1 | % | ||||||
Interest expense, net | (40,419 | ) | (26,226 | ) | (14,193 | ) | 54 | % | ||||||
Reserve for Turing receivable | (40,312 | ) | — | (40,312 | ) | * | ||||||||
Gain on sale of asset | — | 45,574 | (45,574 | ) | * | |||||||||
Loss on debt extinguishment | — | (16,903 | ) | 16,903 | * | |||||||||
Net change in fair value of derivatives | — | (13,000 | ) | 13,000 | * | |||||||||
Other (expense) income, net | (1,412 | ) | 355 | (1,767 | ) | * | ||||||||
Loss before income taxes | (202,017 | ) | (129,419 | ) | (72,598 | ) | 56 | % | ||||||
(Benefit from) provision for income taxes | $ | (104,294 | ) | $ | 20,371 | $ | (124,665 | ) | * | |||||
* Percentage exceeds 100%
General and Administrative Expenses
General and administrative expenses for the year ended December 31, 2016 were $119.9 million, as compared to $119.2 million for the year ended December 31, 2015. The $0.7 million increase during 2016 compared to the prior year was primarily due to costs recognized in 2016 related to the separation of G. Frederick Wilkinson as our President and Chief Executive Officer in December 2016 and higher legal expenses compared to the prior year, partially offset by lower transaction and integration expenses related to strategic transactions during 2016 as compared to the transaction and integration expenses incurred related to the Tower Acquisition during the prior year.
Interest Expense, net
Interest expense, net was $40.4 million for the year ended December 31, 2016, a $14.2 million increase from the prior year. Interest expense for 2016 reflected interest on our $600.0 million convertible senior notes issued in 2015, interest on our $400.0 million Term Loan with Royal Bank of Canada entered into in 2016 to fund the Teva Transaction, and unused line of credit fees on our Revolving Credit Facility with Royal Bank of Canada entered into in 2016. In contrast, prior year interest expense of $27.3 million reflected interest expense on our Term Loan with Barclays Bank PLC entered into in connection with the financing of the Tower Acquisition, which was repaid in full on June 30, 2016 using proceeds from the issuance of our $600.0 million senior notes. Refer to "Outstanding Debt Obligations" below for additional information related to our outstanding convertible notes and credit facilities. Interest income was $1.0 million for the year ended December 31, 2016, which was relatively consistent with interest income for the year ended December 31, 2015.
Reserve for Turing Receivable
During the year ended December 31, 2016, we recorded a reserve of $40.3 million, representing the amount of the estimated receivable due from Turing for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities. We received $7.7 million in payments from Turing during the fourth quarter of 2016, which reduced the reserve balance of $48.0 million as of September 30, 2016 to the reserve balance of $40.3 million as of December 31, 2016. Refer to “Item 15. Financial Information - Notes to the Consolidated Financial Statements - Note 5. Accounts Receivable” for additional information related to the Turing receivable.
Gain on Sale of Asset
During the year ended December 31, 2015, we recognized a $45.6 million gain on the sale of our right to Daraprim®. There was no comparable gain in 2016.
65
Loss on Debt Extinguishment
During the year ended December 31, 2015, we recognized a $16.9 million loss on debt extinguishment related to the repayment of our $435.0 million term loan with Barclays Bank PLC. There was no comparable loss in 2016.
Net Change in Fair Value of Derivatives
During the year ended December 31, 2015, we recognized a $13.0 million expense as the net change in the fair value of our derivative instruments entered into in conjunction with our convertible senior notes due 2022. This expense resulted from the change in our stock price from June 30, 2015 to December 31, 2015. A third party valuation firm with expertise in valuing financial instruments was engaged to determine the fair value of our bond hedge derivative asset and conversion option derivative liability at each reporting period. There was no comparable change in the fair value of derivatives during 2016.
Other (Expense) Income, Net
Other expense, net was $1.4 million for the year ended December 31, 2016, a $1.8 million increase from the prior year. The increase was primarily due to the change in the fair value of the contingent consideration due to Teva pursuant to the Termination Agreement with Teva whereby Teva returned to us our full commercial rights to our then pending ANDA for methylphenidate hydrochloride and due to an increase in fixed asset impairments over the prior year. Refer to "Item 15. Exhibits and Financial Statement Schedules - Note 3. Business Acquisitions" for more information on the Termination Agreement with Teva.
Income Taxes
During the year ended December 31, 2016, we recorded an aggregate tax benefit of $104.3 million for U.S. domestic income taxes and for foreign income taxes, a decrease of $124.7 million compared to an aggregate tax provision of $20.4 million we recorded during the prior year. The decrease in the tax provision during 2016 compared to the prior year resulted from lower income before taxes in the year ended December 31, 2016. The effective tax rate decreased to 18.1% for the year ended December 31, 2016 compared to 34.3% for the year ended December 31, 2015.
The effective income tax rate was 18.1% for the fiscal year ended December 31, 2016, and reflected the establishment of a valuation allowance of $108.8 million against net deferred tax assets. Based on an evaluation of both the positive and negative evidence available, as discussed below, we determined that it was necessary to establish a valuation allowance against a significant portion of our net deferred tax assets for the fiscal year ended December 31, 2016.
A valuation allowance, if needed, reduces deferred tax assets to the amount expected to be realized. When determining the amount of net deferred tax assets that are more likely than not to be realized, we assess all available positive and negative evidence. This evidence includes, but is not limited to, scheduled reversal of deferred tax liabilities, prior earnings history, projected future earnings, carry-back and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income (exclusive of reversing taxable temporary differences and carryforwards) to outweigh objective negative evidence of a recent financial reporting loss for the year ended December 31, 2016.
Based on these criteria and the relative weighting of both the positive and negative evidence available, and in particular the activity surrounding our prior earnings history, including the intangible impairments charges recognized during 2016, we determined that it was necessary to establish a valuation allowance against a significant portion of our net deferred tax assets as of December 31, 2016. Given the objectively verifiable negative evidence of a three-year cumulative loss which, under the provisions of FASB ASC Topic 740 is a significant element of negative evidence that is difficult to overcome, and the weighting of all available positive evidence, we excluded projected taxable income from the assessment of income that could be used as a source of taxable income to realize the deferred tax assets.
Liquidity and Capital Resources
We generally fund our operations with cash from operating activities, although we have also funded our operations with proceeds from the sale of debt and equity securities. Our cash flows from operating activities consist primarily of the proceeds from sales of our products and services.
66
We expect to incur significant operating expenses, including research and development and patent litigation expenses, for the foreseeable future. In addition, we are generally required to make cash expenditures to manufacture and/or acquire finished product inventory in advance of selling the finished product to our customers and collecting payment, which may result in a significant use of cash. We believe our existing cash and cash equivalents, together with cash expected to be generated from operations and our revolving line of credit facility, will be sufficient to meet our financing requirements through the next 12 months. We may, however, seek additional financing through alliance, collaboration, and licensing agreements, as well as from the debt and equity capital markets, to fund capital expenditures, research and development plans, potential acquisitions, and potential revenue shortfalls due to delays in new product introductions or otherwise. We cannot be assured that such financing will be available on favorable terms, or at all. Refer to "Item 1A. Risk Factors" above for additional information related to the risks related to our ability to raise additional funds.
Cash Flows - Year Ended December 31, 2017 Compared to Year Ended December 31, 2016
Net cash provided by operations increased by $0.4 million to $84.2 million for the year ended December 31, 2017, from $83.8 million for year ended December 31, 2016. Our cash flows are impacted by our underlying results from operations and related timing of cash receipts and cash disbursements. For the year ended December 31, 2017, while we experienced reduced operating results, our working capital management improved in 2017, most notably with inventory and payables.
Net cash used in investing activities for the year ended December 31, 2017 was $9.7 million, a decrease of $617.4 million compared to $627.1 million in the prior year. In 2017, net cash used in investing activities primarily consisted of a $26.7 million for capital expenditures partially offset by proceeds from the sale of intangible assets and property, plant and equipment of $21.5 million. In 2016, net cash used in investing activities primarily consisted of a $585.8 million payment to fund the Teva Transaction. Increased capital expenditures in 2016 were partially offset by proceeds from the repayment by Tolmar of the outstanding $15.0 million balance due to us under our loan and security agreement with Tolmar pursuant to which provided to Tolmar one or more loans in an aggregate amount not to exceed $15.0 million (the "Tolmar Loan Agreement").
Net cash used in financing activities for the year ended December 31, 2017 was $73.7 million, representing a decrease of $456.2 million as compared to $382.5 million net cash provided by financing activities in the prior year. In 2017, $70.0 million of principal payments were made on the $400.0 million of proceeds from the term loan entered into with Royal Bank of Canada to finance the Teva Transaction in 2016. In 2016, net cash provided by financing activities primarily consisted of $400.0 million of proceeds from the term loan entered into with Royal Bank of Canada to finance the Teva Transaction. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 10. Debt" below for additional information regarding our outstanding convertible notes and credit facilities.
Cash Flows - Year Ended December 31, 2016 Compared to Year Ended December 31, 2015
Net cash provided by operating activities for the year ended December 31, 2016 was $83.9 million, a decrease of $8.6 million as compared to the prior year $92.5 million net cash provided by operating activities. While the 2016 cash flows from operations were relatively stable compared to 2015, there were some large variations in the line items. Our lower net income during 2016 was more than offset by higher non-cash items. Significant changes in non-cash items during 2016 included higher depreciation and amortization resulting from acquisition activity, non-cash interest expense, intangible asset impairment charges, and the reserve related to the receivable from Turing. Working capital items also experienced significant changes in 2016 compared to the prior year as increased cash flow from accounts receivable collections were more than offset by higher cash outflows related to profit sharing payments, higher inventory in support of product launches as well as lower cash inflows from accounts payable and accrued expenses largely related to payments made on behalf of Turing. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 5. Accounts Receivable" for additional information related to the Turing receivable and payments.
Net cash used in investing activities for the year ended December 31, 2016 was $627.1 million, an increase of $159.6 million compared to $467.5 million in the prior year. In 2016, net cash used in investing activities primarily consisted of a $585.8 million payment to fund the Teva Transaction. Increased capital expenditures in 2016 were partially offset by proceeds from the repayment by Tolmar of the outstanding $15.0 million balance due to us under the Tolmar Loan Agreement. Net cash used in investing activities for the prior year included a $691.3 million payment to fund the Tower Acquisition, partially offset by $200.1 million from the maturity of investments and $59.5 million in proceeds from the sale to Turing of our rights to Daraprim®, both of which had no similar activity during 2016.
67
Net cash provided by financing activities for the year ended December 31, 2016 was $382.5 million, representing a decrease of $118.4 million as compared to $500.9 million in the prior year. In 2016, net cash provided by financing activities primarily consisted of $400.0 million of proceeds from the term loan entered into with Royal Bank of Canada to finance the Teva Transaction. In contrast, prior year net cash provided by financing activities included $600.0 million from the issuance of convertible notes and $88.3 million from the sale of warrants, offset by the payment of $147.0 million to purchase the bond hedge derivative asset, for which similar activity did not occur during 2016. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 10. Debt" below for additional information regarding our outstanding convertible notes and credit facilities.
Commitments and Contractual Obligations
Our contractual obligations as of December 31, 2017 were as follows (in thousands):
Payments Due by Period | |||||||||||||||||||
Contractual Obligations | Total | Less Than 1 Year | 1-3 Years | 3-5 Years | More Than 5 Years | ||||||||||||||
Open Purchase Order Commitments | $ | 108,071 | $ | 108,071 | $ | — | $ | — | $ | — | |||||||||
Operating Leases (a) | 28,142 | 5,575 | 6,318 | 5,136 | 11,113 | ||||||||||||||
Long-term debt obligations | 925,000 | 20,000 | 305,000 | 600,000 | — | ||||||||||||||
Interest payments on long-term debt obligations (b) | 112,852 | 29,286 | 77,566 | 6,000 | — | ||||||||||||||
Total (c) | $ | 1,174,065 | $ | 162,932 | $ | 388,884 | $ | 611,136 | $ | 11,113 | |||||||||
(a) | We lease office, warehouse, and laboratory facilities under non-cancelable operating leases with expiration dates through December 2027. We also lease certain equipment under various non-cancelable operating leases with various expiration dates through July 2022. |
(b) | Interest on existing debt obligations was calculated based on applicable rates at December 31, 2017. |
(c) | Liabilities for uncertain tax positions FASB ASC Topic 740, Sub-topic 10, were excluded as we are not able to make a reasonably reliable estimate of the amount and period of related future payments. As of December 31, 2017, we had a $3.5 million provision for uncertain tax positions. Refer to "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 16. Income Taxes" for additional information. |
Off-Balance Sheet Arrangements
We did not have any off-balance sheet arrangements as of December 31, 2017 and 2016.
Outstanding Debt Obligations
Royal Bank of Canada Credit Facilities
On August 3, 2016, we entered into a restatement agreement with Royal Bank of Canada, as administrative agent, and the lenders and guarantors party thereto (the "Restatement Agreement"). The Restatement Agreement amends and restates the existing Revolving Credit Facility Agreement (as amended and restated and amended to date, the "Amended and Restated Credit Agreement") to, among other things, (i) add a term loan feature to allow for the borrowing of up to $400.0 million of term loans (the "Term Loan Facility") by us in accordance with the terms of the Amended and Restated Credit Agreement, (ii) increase the aggregate principal amount of revolving loans permitted under the Amended and Restated Credit Agreement (the "Revolving Credit Facility," and, together with the Term Loan Facility, the "RBC Credit Facilities"), from $100.0 million to $200.0 million and (iii) extend the maturity date of the Revolving Credit Facility from August 4, 2020 to August 3, 2021. On March 27, 2017, we entered into Amendment No. 1 by and among us, Royal Bank of Canada, as administrative agent, and the lenders party thereto (the “Amendment”) to the Amended and Restated Credit Agreement.
68
Borrowings under the Amended and Restated Credit Agreement will accrue interest at a rate equal to LIBOR or the base rate, plus an applicable margin. The applicable margin may be increased or reduced by 0.25% based on our total net leverage ratio. Up to $12.5 million of the Revolving Credit Facility is available for issuance of letters of credit and any such letters of credit will reduce the amount available under the Revolving Credit Facility on a dollar-for-dollar basis. We are required to pay a commitment fee to the lenders on the average daily unused portion of the Revolving Credit Facility at 0.50% or 0.375% per annum, depending our total net leverage ratio.
The Amended and Restated Credit Agreement contains certain negative covenants (subject to exceptions, materiality thresholds and other allowances) including, without limitation, negative covenants that limit our and our restricted subsidiaries' ability to incur additional debt, guarantee other obligations, grant liens on assets, make loans, acquisitions or other investments, dispose of assets, make optional payments in connection with or modify certain debt instruments, pay dividends or make other payments on capital stock, engage in mergers or consolidations, enter into arrangements that restrict our and our restricted subsidiaries' ability to pay dividends or grant liens, engage in transactions with affiliates, or change our fiscal year. Prior to the effective date of the Amendment on March 27, 2017 the Amended and Restated Credit Agreement also included a financial maintenance covenant whereby we must not permit our total net leverage ratio in any 12-month period to exceed 5.00:1.00, as tested at the end of each fiscal quarter. Effective as of March 27, 2017 and pursuant to Amendment, the total net leverage ratio financial covenant was replaced with a senior secured net leverage ratio financial covenant. Pursuant to the Amendment, we must not permit our senior secured net leverage ratio to exceed 2.50:1.00 and our interest coverage ratio to be less than 3.00:1.00, in each case in any 12-month period, as tested at the end of each fiscal quarter. We were in compliance with all of our covenants under the Amended and Restated Credit Agreement as of December 31, 2017.
The Amended and Restated Credit Agreement contains events of default, including, without limitation (subject to customary grace periods and materiality thresholds), events of default upon (i) the failure to make payments pursuant to the terms of the Amended and Restated Credit Agreement, (ii) violation of covenants, (iii) incorrectness of representations and warranties, (iv) cross-default and cross-acceleration to other material indebtedness, (v) bankruptcy events, (vi) material monetary judgments (to the extent not covered by insurance), (vii) certain matters arising under the Employee Retirement Income Security Act of 1974, as amended, that could reasonably be expected to result in a material adverse effect, (viii) the actual or asserted invalidity of the documents governing the RBC Credit Facilities, any material guarantees or the security interests (including priority thereof) required under the Amended and Restated Credit Agreement and (ix) the occurrence of a change of control (as defined therein). Upon the occurrence of certain events of default, the obligations under the Amended and Restated Credit Agreement may be accelerated and any remaining commitments thereunder may be terminated.
The full amount of the proceeds from the Term Loan Facility of $400.0 million, along with $196.4 million of cash were used to finance the Teva Transaction, including transaction fees, on its closing date of August 3, 2016. As of December 31, 2017, the full amount of the $200.0 million Revolving Credit Facility remains available to us for working capital and other general corporate purposes.
In connection with the Term Loan Facility, we incurred $11.0 million of debt issuance costs for banking, legal and accounting fees and other expenses during the third quarter of 2016. In connection with the Amendment, we incurred $0.8 million of debt issuance costs for banking fees during the first quarter of 2017. These debt issuance costs were recorded on our consolidated balance sheet as a reduction to the current and long-term portions of debt related to the Term Loan Facility. These deferred debt issuance costs will be accreted to interest expense over the term of the debt using the effective interest method. In connection with the increase in the aggregate principal amount of revolving loans permitted under the Revolving Credit Facility, we incurred $0.8 million of debt issuance costs for banking fees which were recorded as an asset with current and long-term portions on our consolidated balance sheet. These deferred debt issuance costs, in addition to the $0.3 million balance remaining from the initial balance remaining from the initial $100.0 million revolving credit facility, will be amortized to interest expense over the term of the Revolving Credit Facility using the straight-line method.
For the year ended December 31, 2017, we recognized $17.7 million of interest expense related to the Term Loan Facility, of which $15.5 million was cash and $2.2 million was non-cash accretion of the debt discount recorded for deferred debt issuance costs. For the period of August 3, 2016 through December 31, 2016, we recognized $6.9 million of interest expense related to the Term Loan Facility, of which $6.0 million was cash and $0.9 million was non-cash accretion of debt discounts recorded for deferred debt issuance costs. As of December 31, 2017, the Term Loan Facility had a carrying value of $317.5 million, of which $17.8 million is classified as current debt and $299.7 million is classified as long-term debt on our consolidated balance sheet. The Term Loan Facility requires us to make quarterly principal payments of $5.0 million beginning from December 2016 through June 2021, and the remaining principal balance is payable in August 2021. As of December 31, 2017, the outstanding principal amount for the Term Loan Facility was $325.0 million.
69
Loss on Early Extinguishment of Debt - Voluntary Prepayment of $50.0 Million of Principal - RBC Term Loan Facility
On February 28, 2017, we made a voluntary prepayment in the amount of $50.3 million under our Term Loan Facility representing $50.0 million of principal amount and $0.3 million of accrued interest thereon. As a result of the voluntary prepayment, for the quarter ended March 31, 2017, we recorded a loss on early extinguishment of debt of $1.2 million to write-off a pro rated portion of the related unaccreted debt issuance costs.
2% Convertible Senior Notes due June 2022
On June 30, 2015, we issued an aggregate principal amount of $600.0 million of 2.00% Convertible Senior Notes due June 2022 (the “Notes”) in a private placement offering, which are our senior unsecured obligations. The Notes were issued pursuant to an Indenture dated June 30, 2015 (the “Indenture”) between us and Wilmington Trust, N.A., as trustee. The Indenture includes customary covenants and sets forth certain events of default after which the Notes may be due and payable immediately. The Notes will mature on June 15, 2022, unless earlier redeemed, repurchased or converted. The Notes bear interest at a rate of 2.00% per year, and interest is payable semiannually in arrears on June 15 and December 15 of each year, beginning on December 15, 2015.
The conversion rate for the Notes is initially set at 15.7858 shares per $1,000 of principal amount, which is equivalent to an initial conversion price of $63.35 per share of our common stock. If a Make-Whole Fundamental Change (as defined in the Indenture) occurs or becomes effective prior to the maturity date and a holder elects to convert its Notes in connection with the Make-Whole Fundamental Change, we are obligated to increase the conversion rate for the Notes so surrendered by a number of additional shares of our common stock as prescribed in the Indenture. Additionally, the conversion rate is subject to adjustment in the event of an equity restructuring transaction such as a stock dividend, stock split, spinoff, rights offering, or recapitalization through a large, nonrecurring cash dividend (“standard antidilution provisions,” per FASB ASC 815-40, Contracts in Entity’s Own Equity ("ASC 815-40")).
The Notes are convertible at the option of the holders at any time prior to the close of business on the business day immediately preceding December 15, 2021 only under the following circumstances:
(i) | If during any calendar quarter commencing after the quarter ending September 30, 2015 (and only during such calendar quarter) the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than 130% of the conversion price on each applicable trading day; or |
(ii) | If during the five business day period after any 10 consecutive trading day period (the “measurement period”) in which the trading price per $1,000 of principal amount of Notes for each trading day of the measurement period was less than 98% of the product of the last report sale price of our common stock and the conversion rate on each such trading day; or |
(iii) | Upon the occurrence of corporate events specified in the Indenture. |
On or after December 15, 2021 until the close of business on the second scheduled trading day immediately preceding the maturity date, the holders may convert their Notes at any time, regardless of the foregoing circumstances. We may satisfy our conversion obligation by paying or delivering, as the case may be, cash, shares of our common stock, or a combination of cash and shares of our common stock, at our election and in the manner and subject to the terms and conditions provided in the Indenture.
Concurrently with the offering of the Notes and using a portion of the proceeds from the sale of the Notes, we entered into a series of convertible note hedge and warrant transactions (the “Note Hedge Transactions” and “Warrant Transactions”) which are designed to reduce the potential dilution to our stockholders and/or offset the cash payments we are required to make in excess of the principal amount upon conversion of the Notes. The Note Hedge Transactions and Warrant Transactions are separate transactions, in each case, entered into by us with a financial institution and are not part of the terms of the Notes. These transactions will not affect any holder’s rights under the Notes, and the holders of the Notes have no rights with respect to the Note Hedge Transactions and Warrant Transactions. See “Note 10. Debt” and “Note 11. Stockholders’ Equity” for additional information.
70
For the years ended December 31, 2017 and December 31, 2016, we recognized $35.5 million and $33.8 million, respectively, of interest expense related to the Notes, of which $12.0 million and $12.0 million, respectively, was cash and $23.5 million and $21.8 million, respectively, was non-cash accretion of the debt discounts recorded. As the Notes mature in 2022, they have been classified as long-term debt on our consolidated balance sheets, with a carrying value of $469.9 million and $446.4 million as of December 31, 2017 and December 31, 2016, respectively. Accrued interest payable on the Notes of $0.5 million as of both December 31, 2017 and December 31, 2016 is included in accrued expenses on our consolidated balance sheets.
Critical Accounting Policies and Use of Estimates
The preparation of our consolidated financial statements in accordance with accounting principles generally accepted in the United States (“GAAP”) and the rules and regulations of the U.S. Securities & Exchange Commission (“SEC”) require the use of estimates and assumptions, based on complex judgments considered reasonable, and affect the reported amounts of assets and liabilities and disclosure of contingent assets and contingent liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The most significant judgments are employed in estimates used in determining values of tangible and intangible assets, contingent consideration, legal contingencies, tax assets and tax liabilities, fair value of share-based compensation related to equity incentive awards issued to employees and directors, and estimates used in applying our revenue recognition policy including those related to accrued chargebacks, rebates, distribution service fees, product returns, Medicare, Medicaid, and other government rebate programs, shelf-stock adjustments, and the timing and amount of deferred and recognized revenue under our several alliance and collaboration agreements. Actual results may differ from estimated results. Certain prior year amounts have been reclassified to conform to the presentation for the year ended December 31, 2017.
Although we believe our estimates and assumptions are reasonable when made, they are based upon information available to us at the time they are made. We periodically review the factors having an influence on our estimates and, if necessary, adjust such estimates. Due to the risks and uncertainties involved in our business and evolving market conditions, and given the subjective element of the estimates made, actual results may differ from estimated results. This possibility may be greater than normal during times of pronounced economic volatility.
Impax Generics sales, net, and Impax Specialty Pharma sales, net. We recognize revenue from the sale of products when title and risk of loss of the product is transferred to the customer and the sales price is fixed and determinable. Provisions for discounts, early payments, rebates, sales returns and distributor chargebacks under terms customary in the industry are provided for in the same period the related sales are recorded. We record estimated reductions to revenue at the time of the initial sale and these estimates are based on the sales terms, historical experience and trend analysis.
Gross to Net Sales Accruals. Sales returns accruals are based on using a historical lag period, which is the time between when the product is sold and when it is ultimately returned, and estimated return rates which may be adjusted based on various assumptions including: changes to internal policies and procedures, business practices, commercial terms with customers, and the competitive position of each product; the amount of inventory in the wholesale and retail supply chain; the introduction of new products; and changes in market sales information. We also consider other factors, including significant market changes which may impact future expected returns, and actual product returns. We allow our customers to return product if approved by authorized personnel in writing or by telephone with the lot number and expiration date accompanying any request and if such products are returned within six months prior to or until twelve months following, the product’s expiration date. We estimate and recognize an accrued provision for product returns as a percentage of gross sales based upon historical experience. Any changes from the historical trend rates are considered in determining the current sales return allowance. If the historical data we use to calculate these estimates do not properly reflect future returns, then a change in the allowance would be made in the period in which such a determination is made and revenues in that period could be materially affected.
Cash discount accruals are based on payment terms extended to customers which are generally 2% to 3% of the gross selling price, as an incentive for paying within invoice terms, which generally range from 30 to 90 days. An estimate of cash discounts is recorded in the same period when revenue is recognized.
71
Government rebate accruals are based on estimated payments due to governmental agencies for purchases made by third parties under various governmental programs. U.S. Medicaid rebate accruals are generally based on historical payment data and estimates of future Medicaid beneficiary utilization applied to the Medicaid unit rebate formula established by the Center for Medicaid and Medicare Services. The accrual of the rebates associated with Medicaid Managed Care Organizations is calculated based on actual billings received from the states. We adjust the rebate accruals as more information becomes available and to reflect actual claims experience. Effective January 1, 2011, manufacturers of pharmaceutical products are responsible for 50% of the patient’s cost of branded prescription drugs related to the Medicare Part D Coverage Gap. In order to estimate the cost to us of this coverage gap responsibility, we analyze the historical invoices. This expense is recognized throughout the year as costs are incurred. TRICARE is a health care program of the U.S. Department of Defense Military Health System that provides civilian health benefits for military personnel, military retirees and their dependents. TRICARE rebate accruals are based on estimated Department of Defense eligible sales multiplied by the TRICARE rebate formula.
Rebates and administrative fees are offered to certain customers, group purchasing organizations and pharmacy benefit managers, consistent with pharmaceutical industry practices. Settlement of rebates and fees may generally occur from one to 15 months from the date of sale. We provide a provision for rebates and administrative fees at the time of sale based on contracted rates and historical redemption rates. Assumptions used to establish the provision include level of customer inventories, contract sales mix and average contract pricing. We regularly review the information related to these estimates and adjust the provision accordingly.
Chargeback accruals are based on the differentials between product acquisition prices paid by wholesalers and lower contract pricing paid by eligible customers.
Distribution service fee accruals are based on contractual fees to be paid to the wholesale distributor for services provided.
A significant majority of our gross to net accruals are the result of chargebacks and rebates and administrative fees, with the majority of those programs having an accrual to payment cycle of three months. In addition to this relatively short accrual to payment cycle, we receive monthly information from the wholesalers regarding their sales of our products and actual on hand inventory levels of our products. During the year ended December 31, 2017, the three large wholesalers account for 99% of our chargebacks and 66% of our indirect sales rebates. Consistent with the pharmaceutical industry, the accrual to payment cycle for returns is longer and can take several years depending on the expiration of the related products. However, returns represent the smallest gross to net adjustment. We have not experienced any significant changes in our estimates as it relates to our chargebacks, rebates or returns in each of the years in the three-year period ended December 31, 2017.
The following tables are rollforwards of the activity in the reserves for the years ended December 31, 2017, 2016 and 2015 with an explanation for any significant changes in the accrual percentages (in thousands):
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Chargeback reserve | |||||||||||
Beginning balance | $ | 151,978 | $ | 102,630 | $ | 43,125 | |||||
Acquired balances | — | — | 24,532 | ||||||||
Provision recorded during the period | 1,212,039 | 1,011,400 | 833,157 | ||||||||
Credits issued during the period | (1,227,126 | ) | (962,052 | ) | (798,184 | ) | |||||
Ending balance | $ | 136,891 | $ | 151,978 | $ | 102,630 | |||||
Provision as a percent of gross product sales | 42 | % | 36 | % | 34 | % | |||||
As noted in the table above, the provision for chargebacks, as a percent of gross product sales, increased to 42% in 2017 from 36% in 2016 primarily due to the change in products sales mix due to the Teva Transaction, which closed in August 2016 and which products carry a higher chargeback rate, a higher chargeback rate on both Fenofibrate and Budesonide product sales due to increase market competition in 2017 and lower product sales of Diclofenac Sodium Gel, which carried a lower chargeback rate.
72
The aggregate provision for chargebacks, as a percent of gross product sales, increased to 36% in 2016 from 34% in 2015 primarily as a result of product sales mix and inclusion of product sales from the Tower Acquisition and Teva Transaction.
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Rebate reserve | |||||||||||
Beginning balance | $ | 300,647 | $ | 265,229 | $ | 88,812 | |||||
Acquired balances | — | — | 75,447 | ||||||||
Provision recorded during the period | 663,724 | 768,629 | 571,642 | ||||||||
Credits issued during the period | (769,104 | ) | (733,211 | ) | (470,672 | ) | |||||
Ending balance | $ | 195,267 | $ | 300,647 | $ | 265,229 | |||||
Provision as a percent of gross product sales | 23 | % | 27 | % | 23 | % | |||||
As noted in the table above, the provision for rebates, as a percent of gross product sales, decreased from 27% during the year ended December 31, 2016 to 23% during the year ended December 31, 2017 as a result of lower product sales of Diclofenac Sodium Gel, which carried a higher rebate rate, and the discontinuation of the Amphetamine Salts IR products in May 2017, which carried a higher rebate rate.
The provision for rebates, as a percent of gross product sales, increased from 23% during the year ended December 31, 2015 to 27% during the year ended December 31, 2016 as a result of product sales mix, the formation of alliances between certain major wholesalers and major retailers and the inclusion of product sales from the Tower Acquisition, which carry a higher rebate rate.
The table above represents rebates in both the Impax Generics and Impax Specialty Pharma divisions. The payment mechanisms for rebates in the Impax Generics and Impax Specialty Pharma divisions are different, which impacts the location on our balance sheet. Only rebates in the Impax Generics division are shown in "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 5. Accounts Receivable," as Impax Specialty Pharma rebates are classified as Accrued Expenses on our consolidated balance sheets.
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Returns reserve | |||||||||||
Beginning balance | $ | 72,888 | $ | 48,950 | $ | 27,174 | |||||
Acquired balances | — | — | 11,364 | ||||||||
Provision related to sales recorded in the period | 47,709 | 52,383 | 43,967 | ||||||||
Credits issued during the period | (44,304 | ) | (28,445 | ) | (33,555 | ) | |||||
Ending balance | $ | 76,293 | $ | 72,888 | $ | 48,950 | |||||
Provision as a percent of gross product sales | 1.7 | % | 1.9 | % | 2.0 | % | |||||
As noted in the table above, the provision for returns as a percent of gross product sales decreased to 1.7% in 2017 compared to 1.9% in 2016 as a result of slightly lower historical returns experience.
The provision for returns as a percent of gross product sales decreased to 1.9% in 2016 compared to 2.0% in 2015 as a result of slightly lower historical returns experience.
Medicaid and Other Government Pricing Programs. As required by law, we provide a rebate payment on drugs dispensed under the Medicaid, Medicare Part D, TRICARE, and other U.S. government pricing programs. We determine our estimate of the accrued rebate reserve for government programs primarily based on historical experience of claims submitted by the various states, and other jurisdictions, as well as any new information regarding changes in the pricing programs that may impact our estimate of rebates. In determining the appropriate accrual amount, we consider historical payment rates and processing lag for outstanding claims and payments. We record estimates for government rebate payments as a deduction from gross sales, with corresponding adjustments to accrued liabilities. The accrual for payments under government pricing programs totaled $60.3 million, $72.1 million, and $91.7 million as of December 31, 2017, December 31, 2016 and December 31, 2015, respectively.
73
Shelf-Stock Adjustments. Based upon competitive market conditions, we may reduce the selling price of some of our products to customers for certain future product shipments. We may issue a credit against the sales amount to a customer based upon its remaining inventory of the product in question, provided the customer agrees to continue to make future purchases of product from us. This type of customer credit is referred to as a shelf-stock adjustment, which is the difference between the sales price and the revised lower sales price, multiplied by an estimate of the number of product units on hand at a given date. Decreases in selling prices are discretionary decisions made by us in response to market conditions, including estimated launch dates of competing products and estimated declines in market price. The accrued reserve for shelf-stock adjustments totaled $7.5 million, $7.0 million, and $6.6 million as of December 31, 2017, December 31, 2016 and December 31, 2015, respectively.
Rx Partner and OTC Partner. Each of our Rx Partner and OTC Partner agreements contain multiple deliverables in the form of products, services and/or licenses over extended periods. FASB ASC Topic 605-25 supplemented SAB 104 and provides guidance for accounting for such multiple-element revenue arrangements. With respect to our multiple-element revenue arrangements that are material to our financial results, we determine whether any or all of the elements of the arrangement should be separated into individual units of accounting under FASB ASC Topic 605-25. If separation into individual units of accounting is appropriate, we recognize revenue for each deliverable when the revenue recognition criteria specified by SAB 104 are achieved for the deliverable. If separation is not appropriate, we recognize revenue and related direct manufacturing costs over the estimated life of the agreement or our estimated expected period of performance using either the straight-line method or a modified proportional performance method.
The Rx Partners and OTC Partners agreements obligate us to deliver multiple goods and/or services over extended periods. Such deliverables include manufactured pharmaceutical products, exclusive and semi-exclusive marketing rights, distribution licenses, and research and development services. In exchange for these deliverables, we receive payments from our agreement partners for product shipments and research and development services, and may also receive other payments including royalty, profit sharing, upfront payments, and periodic milestone payments. Revenue received from our partners for product shipments under these agreements is generally not subject to deductions for chargebacks, rebates, product returns, and other pricing adjustments. Royalty and profit sharing amounts we receive under these agreements are calculated by the respective agreement partner, with such royalty and profit share amounts generally based upon estimates of net product sales or gross profit which include estimates of deductions for chargebacks, rebates, product returns, and other adjustments the alliance agreement partners may negotiate with their customers. We record the agreement partner's adjustments to such estimated amounts in the period the agreement partner reports the amounts to us.
OTC Partner revenue was previously related to our alliance and collaboration agreement with Pfizer, Inc., formerly Wyeth LLC (“Pfizer”) and our supply agreement with L. Perrigo Company (“Perrigo”) with respect to the supply of over-the-counter pharmaceutical product Loratadine and Pseudoephedrine Sulfate 5 mg/120 mg 12-hour Extended Release Tablets (the "D12 Product"). Following the expiration of our obligation to supply the D12 Product to Pfizer and Perrigo as described below, we do not currently sell any over-the-counter pharmaceutical products through this sales channel. We previously recognized profit share revenue in the period earned.
During the quarter ended September 30, 2016, we sold the ANDAs for both the D12 Product and Loratadine and Pseudoephedrine Sulfate 10 mg/240 mg 24-hour Extended Release Tablets, in addition to other specified assets, to Perrigo pursuant to an asset purchase agreement with Perrigo dated as of March 31, 2016 (the "Perrigo APA"). Under the terms of the Perrigo APA, we were required to continue to supply the D-12 Product to Pfizer and Perrigo until the date that was the earliest of (i) the date that Perrigo's manufacturing facility was approved to manufacture the D-12 Product and (ii) December 31, 2017. On November 30, 2017, we transferred manufacturing of the D12 Product to Perrigo and assigned and transferred our supply agreement with Pfizer in its entirety to Perrigo in accordance with the Perrigo APA.
Research Partner. We have entered into development agreements with unrelated third-party pharmaceutical companies under which we are collaborating in the development of five dermatological products, including four generic products and one branded dermatological product. We are not currently in the process of developing the branded dermatological product. Under each of the development agreements, we received an upfront fee with the potential to receive additional milestone payments upon completion of contractually specified clinical and regulatory milestones. We defer and recognize revenue received from the achievement of contingent research and development milestones in the period such payment is earned. We will recognize royalty fee income, if any, as current period revenue when earned.
74
Estimated Lives of Alliance and Collaboration Agreements. Because we may defer revenue we receive under our alliance agreements, and recognize it over the estimated life of the related agreement, or our expected period of performance, we are required to estimate the recognition period under each such agreement in order to determine the amount of revenue to be recognized in each period. Sometimes this estimate is based on the fixed term of the particular alliance agreement. In other cases the estimate may be based on more subjective factors as noted in the following paragraphs. While changes to the estimated recognition periods have been infrequent, such changes, should they occur, may have a significant impact on our consolidated financial statements.
Third-Party Research Agreements. In addition to our own research and development resources, we may use unrelated third-party vendors, including universities and independent research companies, to assist in our research and development activities. These vendors provide a range of research and development services to us, including clinical and bio-equivalency studies. We generally sign agreements with these vendors which establish the terms of each study performed by them, including, among other things, the technical specifications of the study, the payment schedule, and timing of work to be performed. Third-party researchers generally earn payments either upon the achievement of a milestone, or on a pre-determined date, as specified in each study agreement. We account for third-party research and development expenses as they are incurred according to the terms and conditions of the respective agreement for each study performed, with an accrued expense at each balance sheet date for estimated fees and charges incurred by us, but not yet billed to us. We monitor aggregate actual payments and compare them to the estimated provisions to assess the reasonableness of the accrued expense balance at each quarterly balance sheet date.
Share-Based Compensation. We recognize the grant date fair value of each option and restricted share over its vesting period. Stock options and restricted stock awards granted under the 2002 Plan generally vest over a four year period and, in the case of stock options, have a term of ten years. We estimate the fair value of each stock option award on the grant date using the Black-Scholes-Merton option-pricing model, wherein expected volatility is based on historical volatility of our common stock. We base the expected term calculation on the “simplified” method described in SAB No. 107, Share-Based Payment and SAB No. 110, Share-Based Payment, because it provides a reasonable estimate in comparison to our actual experience. We base the risk-free interest rate on the U.S. Treasury yield in effect at the time of grant for an instrument with a maturity that is commensurate with the expected term of the stock options. The dividend yield is zero as we have never paid cash dividends on our common stock, and have no present intention to pay cash dividends.
Income Taxes. We are subject to U.S. federal, state and local income taxes, Netherlands income tax, Republic of Ireland income tax and Taiwan R.O.C. income taxes. In accordance with U.S. Securities and Exchange Commission Staff Accounting Bulletin No. 118 ("SAB 118") the amounts recorded in the fourth quarter of 2017 related to the 2017 Tax Reform Act represent reasonable estimates based on our analysis to date and are considered to be provisional and subject to revision during 2018. Provisional amounts were recorded for the Transition Tax, and the re-measurement of our 2017 U.S. net deferred tax liabilities. These amounts are considered to be provisional as we continue to assess available tax methods and elections and refine our computations. In addition, further regulatory guidance related to the 2017 Tax Reform Act is expected to be issued in 2018 which may result in changes to our current estimates. Any revisions to the estimated impacts of the 2017 Tax Reform Act will be recorded quarterly until the computations are complete which is expected no later than the fourth quarter of 2018.
Significant management judgment is required in determining the provision for income taxes, deferred tax assets and liabilities, and any valuation allowance recorded against net deferred tax assets. The process involves summarizing temporary differences between the financial statement carrying values (in accordance with U.S. GAAP) and the tax bases of our assets and liabilities. These differences result in a net deferred tax asset or liability, which is included within the consolidated balance sheet. In addition, we are required to assess whether valuation allowances should be established against our deferred tax assets based on consideration of all available evidence using a "more likely than not" standard. To the extent a valuation allowance is established in a period, an expense must generally be recorded within the income tax provision in the statement of operations.
In assessing the realizability of our deferred tax assets, we consider whether it is more likely than not that our deferred tax assets will be realized based upon all available evidence, including, but not limited to, scheduled reversal of deferred tax liabilities, prior earnings history, projected future earnings, carryback and carryforward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight we afford the evidence is commensurate with the extent the evidence may be objectively verified. As such, we did not rely on or project future taxable income (exclusive of reversing taxable temporary differences and carryforwards) to outweigh objective negative evidence of a recent financial reporting loss for the years ended December 31, 2017 or December 31, 2016.
75
In relying on the objectively verifiable negative evidence of the three-year cumulative loss, and in not considering or projecting taxable income under the provisions of FASB ASC Topic 740, “Income Taxes,” we confined our sources of income to realize the deferred tax assets to (1) carryback to recover taxes paid in the current year or prior years and (2) offsetting taxable amounts related to taxable temporary differences within the carryback or carryforward period for which deferred tax liabilities are more likely than not to be realized. The deferred tax liabilities consist of indefinite-lived acquired in-process research and development ("IPR&D") product rights.
Our consolidated net deferred tax asset valuation allowance totaled $184.6 million as of December 31, 2017, such that we realize on a more likely than not basis, a tax-effected net deferred tax liability of $3.2 million. If actual results differ from these estimates or these estimates are adjusted in future periods, the valuation allowance may need to be adjusted, which could materially impact our financial position and results of operations. If sufficient positive evidence arises in the future indicating that all or a portion of the deferred tax assets meet the more likely than not standard for realization, the valuation allowance would be reduced accordingly in the period that such a conclusion is reached.
We recognize the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized. We reevaluate the effect of uncertain income tax positions on a quarterly basis, and any changes in recognition or measurement are reflected in the period in which the change in judgment occurs. This evaluation is based on factors including, but not limited to, changes in facts and circumstances, changes in tax law, effectively settled issues, and new audit activity. Any changes in these factors could result in changes to a tax benefit or tax provision.
Contingencies. In the normal course of business, we are subject to loss contingencies, such as legal proceedings and claims arising out of our business, covering a wide range of matters, including, among others, patent litigation, stockholder lawsuits, and product and clinical trial liability. In accordance with FASB ASC Topic 450, "Contingencies," we record accrued loss contingencies when it is probable a liability will be incurred and the amount of loss can be reasonably estimated. We do not recognize gain contingencies until they have been realized.
Intangible Assets. Our intangible assets include both finite lived and indefinite lived assets. Finite lived intangible assets, consisting of marketed product rights and royalties received from product sales by our third party partners, are amortized over the estimated useful life of the asset based on the pattern in which the economic benefits are expected to be consumed or otherwise used up or, if that pattern is not readily determinable, on a straight-line basis. Indefinite-lived intangible assets consist of acquired IPR&D product rights and acquired future royalty rights to be paid based on other companies’ net sales of products not yet approved. IPR&D assets acquired in a business combination are considered indefinite-lived until the completion or abandonment of the associated research and development efforts. Amortization over the estimated useful life will commence at the time of the respective product’s launch. If FDA approval to market the product is not obtained, we will immediately expense the related capitalized cost.
Finite lived intangible assets are tested for impairment when events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. All of our indefinite-lived intangible assets are tested for impairment at least annually during the fourth quarter of the fiscal year, or more often if indicators of impairment are present. Impairment testing requires management to estimate the future undiscounted cash flows of an intangible asset using assumptions believed to be reasonable, but which are unpredictable and inherently uncertain. Actual future cash flows may differ from the estimates used in the impairment testing. We recognize an impairment loss when and to the extent that the estimated fair value of an intangible asset is less than its carrying value.
Goodwill. In accordance with FASB ASC Topic 350, "Goodwill and Other Intangibles," rather than recording periodic amortization of goodwill, goodwill is subject to an annual assessment for impairment. Under FASB ASC Topic 350, if the fair value of the reporting unit exceeds the reporting unit’s carrying value, including goodwill, then goodwill is considered not impaired, making further analysis not required. We consider each of our Impax Generics division and Impax Specialty Pharma division operating segments to be a reporting unit, as this is the lowest level for each of which discrete financial information is available. We attribute $59.7 million of goodwill to the Impax Specialty Pharma division and $147.6 million of goodwill to the Impax Generics division.
76
We concluded the carrying value of goodwill was not impaired as of December 31, 2017 and 2016, as the fair value of the Impax Specialty Pharma division and the Impax Generics division exceeded their respective carrying values at each date. In the fourth quarter of 2017, we determined that it was not more likely than not that the fair value of goodwill was less than its carrying value. As a result we did not perform a quantitative analysis. In the fourth quarter of 2016, we performed a quantitative analysis and estimated the fair value of the Impax Specialty Pharma division and the Impax Generics division using a discounted cash flow model for both the reporting unit and the enterprise, as well as earnings and revenue multiples per common share outstanding for enterprise fair value. In addition, on a quarterly basis, we perform a review of our business operations to determine whether events or changes in circumstances have occurred that could have a material adverse effect on the estimated fair value of each reporting unit, and thus indicate a potential impairment of the goodwill carrying value. If such events or changes in circumstances were deemed to have occurred, we would perform an interim impairment analysis, which may include the preparation of a discounted cash flow model, or consultation with one or more valuation specialists, to analyze the impact, if any, on our assessment of the reporting unit’s fair value.
Recent Accounting Pronouncements
Recently issued accounting standards are discussed in "Item 15. Exhibits and Financial Statements - Notes to Consolidated Financial Statements - Note 2. Summary of Significant Accounting Policies."
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Our cash is held on deposit in demand accounts at large financial institutions in amounts in excess of the Federal Deposit Insurance Corporation (FDIC) insurance coverage limit of $250,000 per depositor, per FDIC-insured bank, per ownership category. Our cash equivalents are comprised of highly-rated money market funds. We had no short-term investments as of December 31, 2017 or December 31, 2016.
Financial instruments that potentially subject us to concentrations of credit risk consist principally of cash equivalents and accounts receivable. We limit our credit risk associated with cash equivalents by placing investments with high credit quality securities, including U.S. government securities, treasury bills, corporate debt, short-term commercial paper and highly-rated money market funds. As discussed above under “Item 7. Outstanding Debt Obligations,” we are party to a Term Loan facility of $400.0 million (of which $325.0 million is outstanding as of December 31, 2017) and a Revolving Credit Facility, of up to $200.0 million pursuant to the RBC Credit Facilities. The amount under our Revolving Credit Facility is available for working capital and other general corporate purposes. We also issued the Notes in a private placement offering on June 30, 2015, which are our senior unsecured obligations, as described above under “Outstanding Debt Obligations.”
We limit our credit risk with respect to accounts receivable by performing credit evaluations when deemed necessary. We do not require collateral to secure amounts owed to us by our customers. As discussed above under "Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 5. Accounts Receivable” we recorded a reserve in the amount of $48.0 million on our consolidated statement of operations for the period ended March 31, 2016, representing the full amount of the estimated receivable due from Turing for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities as of March 31, 2016. During the fourth quarter of 2016, we received $7.7 million in payments from Turing. During the year ended December 31, 2017, we increased the reserve balance by a net $4.0 million, consisting of a $5.0 million increase in the reserve resulting from additional Medicaid rebate claims received during the period and a $1.0 million reduction in the reserve resulting from payments received from Turing during the period. As of December 31, 2017, the $44.3 million estimated receivable due from Turing was fully reserved.
Prior to June 30, 2015, we had no derivative assets or liabilities and did not engage in any hedging activities. As a result of our June 30, 2015 issuance of the Notes described above under “Item 7. Outstanding Debt Obligations” and in “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements - Note 10. Debt”, we entered into a series of convertible note hedge and warrant transactions (the “Note Hedge Transactions” and “Warrant Transactions”) which are designed to reduce the potential dilution to our stockholders and/or offset the cash payments we are required to make in excess of the principal amount upon conversion of the Notes.
We do not use derivative financial instruments or engage in hedging activities in our ordinary course of business and have no material foreign currency exchange exposure or commodity price risks. See “Item 15. Exhibits and Financial Statement Schedules - Notes to Consolidated Financial Statements – Note 20. Segment Information” for more information regarding the value of our investment in Impax Laboratories (Taiwan), Inc.
We do not believe that inflation has had a significant impact on our revenues or operations to date.
77
Item 8. Financial Statements and Supplementary Data
The consolidated financial statements and schedule listed in the Index to Financial Statements beginning on page F-1 are filed as part of this Annual Report on Form 10-K and incorporated by reference herein.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) that are designed to ensure information required to be disclosed by us in reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based upon this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures, as defined in Rule 13a-15(e) of the Exchange Act, were effective as of December 31, 2017 at the reasonable assurance level.
Management Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Exchange Act Rule 13a-15(f), to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles used in the United States (GAAP). Internal control over financial reporting includes our policies and procedures, such as our Code of Conduct, which (i) require our employees, directors and contingent workers and business partners who perform work on our behalf to adhere to certain ethical standards; (ii) require the maintenance of records, in reasonable detail, to help to ensure that our transactions, assets and liabilities are accurately and fairly recorded; (iii) provide reasonable assurance that transactions are authorized by our management and directors and are recorded as necessary to allow for the accurate preparation of financial statements in accordance with GAAP; and (iv) provide reasonable assurance regarding the safeguarding of our assets and the prevention or timely detection of the unauthorized acquisition, use or disposition of our assets which could have a material effect on the financial statements. Internal control over financial reporting includes the controls themselves, management’s monitoring of those controls, the independent assessment of the design and effectiveness of those controls by our internal audit team, actions taken to correct deficiencies as identified, and oversight of our internal control environment by our Audit Committee of our Board of Directors. Any system of internal control has inherent limitations and therefore may not prevent or detect misstatements. Projections of any evaluation of the effectiveness of internal control over financial reporting to future periods has risks as controls may become inadequate over time because of changes in conditions.
Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017, the end of our fiscal year, and has reviewed the results of this assessment with the Audit Committee of our Board of Directors. Management based its assessment on criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s assessment included evaluation of such elements as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies, and our overall control environment. Based on the assessment, management has concluded our internal control over financial reporting was effective as of the end of the fiscal year 2017 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with GAAP.
The scope of management’s assessment of the effectiveness of its internal control over financial reporting as of December 31, 2017 included our consolidated operations.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by KPMG LLP, an independent registered public accounting firm, as stated in their report which is included immediately below.
78
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Impax Laboratories, Inc.:
Opinion on Internal Control Over Financial Reporting
We have audited Impax Laboratories, Inc. and subsidiaries’ (the Company) internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2017 and 2016, the related consolidated statements of operations, comprehensive (loss) income, changes in stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2017, and the related notes, and the consolidated financial statement schedule, "Schedule II - Valuation and Qualifying Accounts” (collectively, the consolidated financial statements), and our report dated March 1, 2018 expressed an unqualified opinion on those consolidated financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that the receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ KPMG LLP
Philadelphia, Pennsylvania
March 1, 2018
79
Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2017, there were no changes in the Company’s internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act) which materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Item 9B. Other Information
None.
80
PART III.
Item 10. Directors, Executive Officers and Corporate Governance
Code of Conduct
We have adopted a Code of Conduct, which was amended and restated effective February 16, 2016 (“Code of Conduct”). The Code of Conduct applies to all of our directors, employees, including our Chief Executive Officer, Chief Financial Officer and any other accounting officer, controller or persons performing similar functions, and contingent workers and business partners who perform work on our behalf. The Code of Conduct is available on our website (www.impaxlabs.com) and accessible via the “Investor Relations” page. Any amendments to, or waivers of, the Code of Conduct will be disclosed on our website within four business days following the date of such amendment or waiver.
Additional information required by this item is incorporated by reference to our definitive proxy statement for the 2018 Annual Meeting of Stockholders (“the Annual Meeting Proxy Statement”). In the event we do not file the Annual Meeting Proxy Statement, the information will be provided instead by an amendment to this report not later than 120 days after the end of our fiscal year.
Item 11. Executive Compensation
The information required by this item is incorporated by reference to the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item is incorporated by reference to the Proxy Statement, except information concerning the equity compensation plans table which is set forth in “Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities” and which is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated by reference to the Proxy Statement.
Item 14. Principal Accounting Fees and Services
The information required by this item is incorporated by reference to the Proxy Statement.
81
PART IV.
Item 15. Exhibits and Financial Statement Schedules
(a)(1) Consolidated Financial Statements
The consolidated financial statements listed in the Index to Financial Statements beginning on page F-1 are filed as part of this Annual Report on Form 10-K.
(a)(2) Financial Statement Schedules
The financial statement schedule listed in the Index to Financial Statements on page F-1 is filed as part of this Annual Report on Form 10-K.
(a)(3) Exhibits
See the "Exhibit Index" beginning on the next page of this Annual Report on Form 10-K.
Item 16. Form 10-K Summary
Not Applicable.
82
EXHIBIT INDEX
Exhibit No. | Description of Document | ||
Business Combination Agreement, dated as of October 17, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation, and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on October 17, 2017).† | Incorporated by Reference | ||
Amendment No. 1 to the Business Combination Agreement, dated as of November 21, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on November 21, 2017). | Incorporated by Reference | ||
Amendment No. 2 to the Business Combination Agreement, dated as of December 16, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on December 20, 2017). | Incorporated by Reference | ||
Stock Purchase Agreement, dated as of October 8, 2014, by and among the Company, Tower Holdings, Inc. (“Tower”), Lineage Therapeutics Inc. (“Lineage”), Roundtable Healthcare Partners II, L.P., Roundtable Healthcare Investors II, L.P., the other stockholders of Tower and Lineage, the holders of options to purchase shares of Tower common stock and options to purchase shares of Lineage common stock, the holders of warrants to acquire shares of Tower common stock and warrants to acquire shares of Lineage common stock and, solely with respect to Section 8.3, Roundtable Healthcare Management II, LLC. (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on October 10, 2014).† | Incorporated by Reference | ||
Restated Certificate of Incorporation of the Company dated as of August 30, 2004 (incorporated by reference to Exhibit 3.1 to Amendment No. 5 to the Company’s Registration Statement on Form 10 filed on December 23, 2008). | Incorporated by Reference | ||
Certificate of Amendment of the Restated Certificate of Incorporation of the Company dated as of December 9, 2015 (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on December 9, 2015). | Incorporated by Reference | ||
Certificate of Designation of Series A Junior Participating Preferred Stock, as filed with the Secretary of State of Delaware on January 21, 2009 (incorporated by reference to Exhibit 3.3 to the Company’s Current Report on Form 8-K filed on January 22, 2009). | Incorporated by Reference | ||
Amended and Restated Bylaws of the Company, effective as of May 14, 2014. | Filed Herewith | ||
Amendment No. 1 to Amended and Restated Bylaws of the Company, effective as of March 24, 2015. | Filed Herewith | ||
Amendment No. 2 to Amended and Restated Bylaws of the Company, effective as of July 7, 2015. | Filed Herewith | ||
Amendment No. 3 to Amended and Restated Bylaws of the Company, effective as of October 7, 2015. | Filed Herewith | ||
Amendment No. 4 to Amended and Restated Bylaws of the Company, effective as of May 17, 2016. | Filed Herewith | ||
Amendment No. 5 to Amended and Restated Bylaws of the Company, effective as of August 19, 2016. | Filed Herewith | ||
Amendment No. 6 to Amended and Restated Bylaws of the Company, effective as of November 23, 2016. | Filed Herewith | ||
Amendment No. 7 to Amended and Restated Bylaws of the Company, effective as of December 19, 2016. | Filed Herewith | ||
Amendment No. 8 to Amended and Restated Bylaws of the Company, effective as of March 24, 2017. | Filed Herewith | ||
83
Amendment No. 9 to Amended and Restated Bylaws of the Company, effective as of November 10, 2017. | Filed Herewith | ||
Specimen of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form 10 filed on October 10, 2008). | Incorporated by Reference | ||
Preferred Stock Rights Agreement, dated as of January 20, 2009, by and between the Company and StockTrans, Inc., as Rights Agent (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on January 22, 2009). | Incorporated by Reference | ||
Indenture, dated as of June 30, 2015, between the Company and Wilmington Trust, National Association, as trustee (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Supplemental Indenture, dated as of November 6, 2017, between the Company and Wilmington Trust, National Association, as trustee (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on November 7, 2017). | Incorporated by Reference | ||
Letter Agreement, dated as of June 25, 2015, between RBC Capital Markets LLC and the Company regarding the Base Warrants (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 25, 2015, between RBC Capital Markets LLC and the Company regarding the Base Call Option Transaction (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 26, 2015, between RBC Capital Markets LLC and the Company regarding the Additional Warrants (incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 26, 2015, between RBC Capital Markets LLC and the Company regarding the Additional Call Option Transaction (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Credit Agreement, dated as of August 4, 2015, by and among the Company, the lenders party thereto from time to time and Royal Bank of Canada, as administrative agent and collateral agent (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 5, 2015). | Incorporated by Reference | ||
Restatement Agreement, dated as of August 3, 2016, by and among the Company, the guarantors party thereto, Royal Bank of Canada, as administrative agent, and the lenders party thereto (incorporated by reference to Exhibit 10.7 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016). | Incorporated by Reference | ||
Amendment No. 1, dated as of March 27, 2017, to the Credit Agreement, dated as of August 4, 2015, as amended and restated as of August 3, 2016, among the Company, as borrower, Royal Bank of Canada, as administrative agent and collateral agent, the lenders party thereto and the other agents and parties thereto (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017). | Incorporated by Reference | ||
Distribution, License, Development and Supply Agreement, dated as of January 31, 2012, between the Company and AstraZeneca UK Limited (incorporated by reference to Exhibit 10.1 to Amendment No. 1 to the Company’s Current Report on Form 8-K filed on April 2, 2012)**. | Incorporated by Reference | ||
First Amendment, dated as of May 31, 2016, to the Distribution, License, Development and Supply Agreement by and between AstraZeneca UK Limited and the Company dated as of January 31, 2012 (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016).** | Incorporated by Reference | ||
Stock and Asset Purchase Agreement, dated as of December 19, 2017, by and between the Company and Bora Pharmaceuticals Co., Ltd. † | Filed Herewith | ||
Master Supply Agreement, dated as of December 19, 2017, between the Company, Bora Pharmaceuticals Co., Ltd. and Impax Laboratories (Taiwan), Inc.*** | Filed Herewith | ||
Asset Purchase Agreement, dated as of June 20, 2016, between Teva Pharmaceutical Industries Ltd. and the Company (incorporated by reference to Exhibit 10.2.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).†** | Incorporated by Reference | ||
84
Amendment No. 1, dated as of June 30, 2016, to the Asset Purchase Agreement between Teva Pharmaceutical Industries Ltd. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.2.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016). | Incorporated by Reference | ||
Asset Purchase Agreement, dated as of June 20, 2016, by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company (incorporated by reference to Exhibit 10.3.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).†** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Asset Purchase Agreement by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.3.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2106 filed on August 9, 2016).** | Incorporated by Reference | ||
Supply Agreement, dated as of June 20, 2016, between Teva Pharmaceutical Industries Ltd. and the Company (incorporated by reference to Exhibit 10.4.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Supply Agreement between Teva Pharmaceutical Industries Ltd. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.4.2 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Supply Agreement, dated as of June 20, 2016, by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company (incorporated by reference to Exhibit 10.5.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Supply Agreement by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.5.2 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Impax Laboratories, Inc. 1999 Equity Incentive Plan (incorporated by reference to Exhibit 10.4 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2008 filed on March 12, 2009).* | Incorporated by Reference | ||
Form of Stock Option Grant under the Impax Laboratories, Inc. 1999 Equity Incentive Plan (incorporated by reference to Exhibit 10.4.1 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2008 filed on March 12, 2009).* | Incorporated by Reference | ||
Impax Laboratories, Inc. 2001 Non-Qualified Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to the Company’s Registration Statement on Form 10 filed on October 10, 2008).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Amended and Restated Non-Qualified Employee Stock Purchase Plan (incorporated by reference to Exhibit 4.13 to the Company’s Registration Statement on Form S-8 filed on August 29, 2017).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Appendix B to the Company’s definitive proxy statement on Schedule 14A filed on April 5, 2017).* | Incorporated by Reference | ||
Form of Stock Option Agreement under the Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2017 filed on August 9, 2017).* | Incorporated by Reference | ||
85
Form of Restricted Stock (Stock Bonus) Award Agreement under the Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2017 filed on August 9, 2017).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Executive Non-Qualified Deferred Compensation Plan, amended and restated effective January 1, 2008 (incorporated by reference to Exhibit 10.1.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 filed on May 6, 2010).* | Incorporated by Reference | ||
Amendment to Impax Laboratories, Inc. Executive Non-Qualified Deferred Compensation Plan, effective as of January 1, 2009 (incorporated by reference to Exhibit 10.1.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 filed on May 6, 2010).* | Incorporated by Reference | ||
Employment Agreement, dated as of March 24, 2017, between the Company and Paul M. Bisaro (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017).* | Incorporated by Reference | ||
Stock Option Agreement with Paul M. Bisaro (incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of December 16, 2017, by and among Amneal Pharmaceuticals LLC, Atlas Holdings, Inc., a wholly owned subsidiary of the Company, and Robert A. Stewart (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 20, 2017).* | Incorporated by Reference | ||
Memorandum of Understanding, dated as of December 16, 2017, by and among Amneal Pharmaceuticals LLC, Paul M. Bisaro, the Company and Atlas Holdings, Inc., a wholly owned subsidiary of the Company (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on December 20, 2017).* | Incorporated by Reference | ||
Letter Agreement, dated as of December 19, 2016, between the Company and J. Kevin Buchi (incorporated by reference to Exhibit 10.19 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 filed on March 1, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of April 21, 2014, by and between the Company and G. Frederick Wilkinson (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 24, 2014).* | Incorporated by Reference | ||
General Release and Waiver, dated as of December 19, 2016, by and between the Company and G. Frederick Wilkinson (incorporated by reference to Exhibit 10.20.2 of the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 filed on March 1, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of January 1, 2010, between the Company and Michael J. Nestor (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on January 14, 2010).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of January 1, 2014, between the Company and Michael Nestor (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Separation Agreement, dated as of January 8, 2018, between the Company and Michael J. Nestor (incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K filed on January 9, 2018).* | Incorporated by Reference | ||
Employment Agreement, dated as of July 14, 2016, between the Company and Douglas S. Boothe (incorporated by reference to Exhibit 10.6 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016).* | Incorporated by Reference | ||
Offer of Employment Letter, dated as of March 17, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.13.1 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2011 filed on February 28, 2012).* | Incorporated by Reference | ||
Employment Agreement, dated as of May 2, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.13.2 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2011 filed on February 28, 2012).* | Incorporated by Reference | ||
86
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of May 2, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Employment Agreement, dated as of December 12, 2012, between the Company and Bryan M. Reasons (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 13, 2012).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of December 12, 2012 between the Company and Bryan M. Reasons (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Employment Agreement, dated as of November 28, 2011, by and between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of November 28, 2011, by and between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
Letter Agreement, dated as of April 1, 2014, between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.3 of the Company’s Quarterly Report on Form 10-Q to the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
11.1 | Statement re computation of per share earnings (incorporated by reference to Note 12 to the Notes to Consolidated Financial Statements in this Annual Report on Form 10-K). | ||
Subsidiaries of the registrant. | Filed Herewith | ||
Consent of Independent Registered Public Accounting Firm. | Filed Herewith | ||
24.l | Powers of Attorney (included on the signature page of this Annual Report on Form 10-K) | ||
Certification of the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
101 | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2017, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets as of December 31, 2017 and 2016, (ii) Consolidated Statements of Operations for each of the three years in the period ended December 31, 2017, (iii) Consolidated Statements of Comprehensive (Loss) Income for each of the three years in the period ended December 31, 2017, (iv) Consolidated Statements of Changes in Stockholders’ Equity for each of the three years in the period ended December 31, 2017, (v) Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2017 and (vi) Notes to Consolidated Financial Statements for each of the three years in the period ended December 31, 2017. | ||
* | Management contract, compensatory plan or arrangement. | ||
** | Confidential treatment granted for certain portions of this exhibit pursuant to Rule 24b-2 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which portions are omitted and filed separately with the SEC. | ||
*** | Confidential treatment requested for certain portions of the exhibit pursuant to Rule 24b-2 under the Exchange Act, which portions are omitted and filed separately with the SEC. | ||
† | Schedules omitted pursuant to Item 601(b)(2) of Regulation S-K. The Company agrees to furnish a supplemental copy of any omitted schedule to the SEC upon request. | ||
87
Impax Laboratories, Inc.
INDEX TO FINANCIAL STATEMENTS
Consolidated Statements of Operations for the years ended December 31, 2017, 2016, and 2015 | |
Consolidated Statements of Comprehensive (Loss) Income for the years ended December 31, 2017, 2016, and 2015 | |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Impax Laboratories, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Impax Laboratories, Inc. and subsidiaries (the Company) as of December 31, 2017 and 2016, and the related consolidated statements of operations, comprehensive (loss) income, changes in stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2017, and the related notes and the consolidated financial statement schedule, "Schedule II - Valuation and Qualifying Accounts" (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results its operations and its cash flows for each of the years in the three-year period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated March 1, 2018 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KPMG LLP
We have served as the Company's auditor since 2011.
Philadelphia, Pennsylvania
March 1, 2018
F-2
IMPAX LABORATORIES, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
December 31, 2017 | December 31, 2016 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 181,778 | $ | 180,133 | |||
Accounts receivable, net | 240,753 | 257,368 | |||||
Inventory, net | 158,471 | 175,230 | |||||
Prepaid expenses and other current assets | 21,086 | 14,897 | |||||
Income tax receivable | 61,201 | 3,513 | |||||
Assets held for sale | 32,266 | — | |||||
Total current assets | 695,555 | 631,141 | |||||
Property, plant and equipment, net | 124,813 | 233,372 | |||||
Intangible assets, net | 262,467 | 620,466 | |||||
Goodwill | 207,329 | 207,329 | |||||
Deferred income taxes, net | — | 69,866 | |||||
Other non-current assets | 61,136 | 60,844 | |||||
Total assets | $ | 1,351,300 | $ | 1,823,018 | |||
Liabilities and Stockholders’ Equity | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 81,093 | $ | 58,952 | |||
Accrued expenses | 248,127 | 244,653 | |||||
Liabilities held for sale | 7,170 | — | |||||
Current portion of long-term debt, net | 17,848 | 17,719 | |||||
Total current liabilities | 354,238 | 321,324 | |||||
Long-term debt, net | 769,524 | 813,545 | |||||
Deferred income taxes | 3,226 | — | |||||
Other non-current liabilities | 37,111 | 64,175 | |||||
Total liabilities | 1,164,099 | 1,199,044 | |||||
Commitments and contingencies (Note 18) | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $0.01 par value; 2,000,000 shares authorized; No shares issued or outstanding at December 31, 2017 and 2016 | — | — | |||||
Common stock, $0.01 par value; 150,000,000 shares authorized; 74,234,076 issued and 73,990,347 outstanding shares at December 31, 2017; 73,948,340 issued and 73,704,611 outstanding shares at December 31, 2016 | 742 | 739 | |||||
Treasury stock at cost: 243,729 shares at December 31, 2017 and 2016 | (2,157 | ) | (2,157 | ) | |||
Additional paid-in capital | 559,632 | 535,056 | |||||
Retained (deficit) earnings | (372,445 | ) | 98,192 | ||||
Accumulated other comprehensive income (loss) | 1,429 | (7,856 | ) | ||||
Total stockholders’ equity | 187,201 | 623,974 | |||||
Total liabilities and stockholders’ equity | $ | 1,351,300 | $ | 1,823,018 | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
IMPAX LABORATORIES, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except share and per share data)
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Revenues: | |||||||||||
Impax Generics, net | $ | 549,077 | $ | 606,320 | $ | 710,932 | |||||
Impax Specialty Pharma, net | 226,710 | 218,109 | 149,537 | ||||||||
Total revenues, net | 775,787 | 824,429 | 860,469 | ||||||||
Cost of revenues | 535,123 | 486,899 | 500,762 | ||||||||
Cost of revenues impairment charges | 96,865 | 488,632 | 7,303 | ||||||||
Gross profit (loss) | 143,799 | (151,102 | ) | 352,404 | |||||||
Operating expenses: | |||||||||||
Selling, general and administrative | 216,270 | 201,830 | 201,287 | ||||||||
Research and development | 80,847 | 80,466 | 70,622 | ||||||||
In-process research and development impairment charges | 192,809 | 52,965 | 6,360 | ||||||||
Fixed asset impairment charges | 82,508 | — | — | ||||||||
Change in fair value of contingent consideration | (31,048 | ) | — | — | |||||||
Patent litigation | 5,105 | 7,819 | 4,567 | ||||||||
Total operating expenses | 546,491 | 343,080 | 282,836 | ||||||||
(Loss) income from operations | (402,692 | ) | (494,182 | ) | 69,568 | ||||||
Other income (expense): | |||||||||||
Interest expense, net | (53,412 | ) | (40,419 | ) | (26,226 | ) | |||||
Reserve for Turing receivable | (3,999 | ) | (40,312 | ) | — | ||||||
Gain on sale of assets | 17,236 | 175 | 45,574 | ||||||||
Loss on debt extinguishment | (1,215 | ) | — | (16,903 | ) | ||||||
Net change in fair value of derivatives | — | — | (13,000 | ) | |||||||
Other, net | (6,879 | ) | (1,587 | ) | 355 | ||||||
(Loss) income before income taxes | (450,961 | ) | (576,325 | ) | 59,368 | ||||||
Provision for (benefit from) income taxes | 18,326 | (104,294 | ) | 20,371 | |||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | |||
Net (loss) income per common share: | |||||||||||
Basic | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.56 | |||
Diluted | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.54 | |||
Weighted-average common shares outstanding: | |||||||||||
Basic | 71,856,950 | 71,147,397 | 69,640,417 | ||||||||
Diluted | 71,856,950 | 71,147,397 | 72,027,344 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
IMPAX LABORATORIES, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME
(In thousands)
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | |||
Other comprehensive (loss) income component: | |||||||||||
Currency translation adjustments | 9,285 | 2,607 | (4,454 | ) | |||||||
Comprehensive (loss) income | $ | (460,002 | ) | $ | (469,424 | ) | $ | 34,543 | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
IMPAX LABORATORIES, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(In thousands)
Accumulated | ||||||||||||||||||||||||||
Common Stock | Additional | Other | ||||||||||||||||||||||||
Number of Shares | Par Value | Treasury Stock | Paid-in Capital | Retained (deficit) Earnings | Comprehensive Income (Loss) | Total | ||||||||||||||||||||
Balance, December 31, 2014 | 71,228 | $ | 714 | $ | (2,157 | ) | $ | 364,103 | $ | 531,226 | $ | (6,009 | ) | $ | 887,877 | |||||||||||
Net income | — | — | — | — | 38,997 | — | 38,997 | |||||||||||||||||||
Other comprehensive loss: | ||||||||||||||||||||||||||
Currency translation adjustment | — | — | — | — | — | (4,454 | ) | (4,454 | ) | |||||||||||||||||
Exercises of stock options, issuances of restricted stock and sales of common stock under ESPP | 1,698 | 15 | — | (3,533 | ) | — | — | (3,518 | ) | |||||||||||||||||
Share-based compensation | — | — | — | 28,613 | — | — | 28,613 | |||||||||||||||||||
Sale of warrants | — | — | — | 88,320 | — | — | 88,320 | |||||||||||||||||||
Reclassification of derivatives to equity, net of related taxes | — | — | — | 21,038 | — | — | 21,038 | |||||||||||||||||||
Tax benefit related to exercises of stock options and vestings of restricted stock | — | — | — | 5,536 | — | — | 5,536 | |||||||||||||||||||
Balance, December 31, 2015 | 72,926 | 729 | (2,157 | ) | 504,077 | 570,223 | (10,463 | ) | 1,062,409 | |||||||||||||||||
Net loss | — | — | — | — | (472,031 | ) | — | (472,031 | ) | |||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||||
Currency translation adjustment | — | — | — | — | — | 2,607 | 2,607 | |||||||||||||||||||
Exercises of stock options, issuances of restricted stock and sales of common stock under ESPP | 1,022 | 10 | — | (612 | ) | — | — | (602 | ) | |||||||||||||||||
Share-based compensation | — | — | — | 32,180 | — | — | 32,180 | |||||||||||||||||||
Tax benefit related to exercises of stock options and vestings of restricted stock | — | — | — | (589 | ) | — | — | (589 | ) | |||||||||||||||||
Balance, December 31, 2016 | 73,948 | 739 | (2,157 | ) | 535,056 | 98,192 | (7,856 | ) | 623,974 | |||||||||||||||||
Cumulative effect of change in accounting principle (Note 3) | — | — | — | 1,350 | (1,350 | ) | — | — | ||||||||||||||||||
Net loss | — | — | — | — | (469,287 | ) | — | (469,287 | ) | |||||||||||||||||
Other comprehensive income: | ||||||||||||||||||||||||||
Currency translation adjustment | — | — | — | — | — | 9,285 | 9,285 | |||||||||||||||||||
Exercises of stock options, issuances of restricted stock and sales of common stock under ESPP | 286 | 3 | — | (2,855 | ) | — | — | (2,852 | ) | |||||||||||||||||
Share-based compensation | — | — | — | 26,258 | — | — | 26,258 | |||||||||||||||||||
Other | — | — | — | (177 | ) | — | — | (177 | ) | |||||||||||||||||
Balance, December 31, 2017 | 74,234 | $ | 742 | $ | (2,157 | ) | $ | 559,632 | $ | (372,445 | ) | $ | 1,429 | $ | 187,201 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
IMPAX LABORATORIES, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Cash flows from operating activities: | |||||||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | |||
Adjustments to reconcile net (loss) income to net cash provided by operating activities: | |||||||||||
Depreciation and amortization | 109,449 | 88,348 | 68,637 | ||||||||
Non-cash interest expense | 25,950 | 22,845 | 11,230 | ||||||||
Share-based compensation expense | 26,258 | 32,180 | 28,613 | ||||||||
Deferred income taxes, net and uncertain tax positions | 74,873 | (127,405 | ) | (29,558 | ) | ||||||
Intangible asset impairment charges | 289,674 | 541,597 | 13,664 | ||||||||
Reserve for Turing receivable | 3,999 | 40,312 | — | ||||||||
Fixed asset impairment charges | 82,508 | — | — | ||||||||
Gain on sale of assets | (17,236 | ) | (175 | ) | (45,574 | ) | |||||
Loss on debt extinguishment | 1,215 | — | 16,903 | ||||||||
Change in fair value of contingent consideration | (31,048 | ) | — | — | |||||||
Net change in fair value of derivatives | — | — | 13,000 | ||||||||
Recognition of deferred revenue | — | — | (4,310 | ) | |||||||
Other | (1,018 | ) | 2,853 | (81 | ) | ||||||
Changes in certain assets and liabilities: | |||||||||||
Accounts receivable | 12,552 | 26,771 | (121,110 | ) | |||||||
Inventory | 6,650 | (45,561 | ) | (14,035 | ) | ||||||
Prepaid expenses and other assets | (65,576 | ) | (573 | ) | 9,330 | ||||||
Accounts payable and accrued expenses | 32,377 | (27,949 | ) | 107,402 | |||||||
Other liabilities | 2,882 | 2,638 | (656 | ) | |||||||
Net cash provided by operating activities | 84,222 | 83,850 | 92,452 | ||||||||
Cash flows from investing activities: | |||||||||||
Payment for business acquisition (net of cash acquired) | (121 | ) | (585,800 | ) | (691,348 | ) | |||||
Purchases of property, plant and equipment | (26,749 | ) | (49,402 | ) | (25,199 | ) | |||||
Proceeds from sales of property, plant and equipment | 9,111 | 1,360 | — | ||||||||
Payments for licensing agreements | (50 | ) | (3,500 | ) | (5,850 | ) | |||||
Investment in cash surrender value of insurance | (4,750 | ) | (4,750 | ) | (4,750 | ) | |||||
Proceeds from cash surrender value of insurance | 529 | — | — | ||||||||
Proceeds from repayment of Tolmar loan | — | 15,000 | — | ||||||||
Proceeds from sale of intangible assets | 12,350 | — | 59,546 | ||||||||
Maturities of short-term investments | — | — | 200,064 | ||||||||
Net cash used in investing activities | (9,680 | ) | (627,092 | ) | (467,537 | ) | |||||
Cash flows from financing activities: | |||||||||||
Proceeds from sale of convertible notes | — | — | 600,000 | ||||||||
Proceeds from issuance of term loan | — | 400,000 | 435,000 | ||||||||
Repayment of term loan | (70,000 | ) | (5,000 | ) | (435,000 | ) | |||||
Payment of deferred financing fees | (818 | ) | (11,867 | ) | (36,941 | ) | |||||
Purchase of bond hedge derivative asset | — | — | (147,000 | ) | |||||||
Proceeds from sale of warrants | — | — | 88,320 | ||||||||
Payment of withholding taxes related to restricted stock awards | (4,231 | ) | (9,842 | ) | (14,990 | ) | |||||
Proceeds from exercises of stock options and ESPP | 1,379 | 9,239 | 11,472 | ||||||||
Net cash (used in) provided by financing activities | (73,670 | ) | 382,530 | 500,861 | |||||||
Effect of exchange rate changes on cash and cash equivalents | 773 | 494 | (298 | ) | |||||||
Net increase (decrease) in cash and cash equivalents | 1,645 | (160,218 | ) | 125,478 | |||||||
Cash and cash equivalents, beginning of year | 180,133 | 340,351 | 214,873 | ||||||||
Cash and cash equivalents, end of year | $ | 181,778 | $ | 180,133 | $ | 340,351 | |||||
Supplemental disclosure of cash flow information: | |||||||||||
Cash paid for interest | $ | 28,374 | $ | 18,139 | $ | 15,365 | |||||
Cash paid for income taxes, net | $ | 2,928 | $ | 23,053 | $ | 43,223 | |||||
Supplemental disclosure of non-cash investing activity: | |||||||||||
Fair value of contingent consideration issued in business acquisition | $ | — | $ | 30,100 | $ | — | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
IMPAX LABORATORIES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS
Operating and Reporting Structure
Impax Laboratories, Inc. (“Impax” or the “Company”) is a specialty pharmaceutical company that focuses on developing, manufacturing, marketing and distributing generic and branded pharmaceutical products. The Company has two reportable segments, referred to as “Impax Generics” and “Impax Specialty Pharma.” The Impax Generics division includes the Company’s legacy Global Pharmaceuticals business as well as the acquired businesses from the Company's acquisition of Tower Holdings, Inc. ("Tower") and its subsidiaries on March 9, 2015 (the "Tower Acquisition").
The Impax Generics division focuses on a broad range of therapeutic areas, including products having technically challenging drug-delivery mechanisms or unique product formulations. In addition to developing solid oral dosage products, the Impax Generics division’s portfolio includes alternative dosage form products, primarily through alliance and collaboration agreements with third parties. Impax Generics develops, manufactures, sells, and distributes generic pharmaceutical products primarily through the following four sales channels: the “Impax Generics” sales channel, for generic pharmaceutical prescription products the Company sells directly to wholesalers, large retail drug chains, and others; the “Private Label” sales channel, for generic pharmaceutical over-the-counter (“OTC”) and prescription products the Company sells to unrelated third-party customers who, in turn, sell the product to third parties under their own label; the “Rx Partner” sales channel, for generic prescription products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements; and the “OTC Partner” sales channel, for generic pharmaceutical OTC products sold through unrelated third-party pharmaceutical entities under their own labels pursuant to alliance and supply agreements. Revenues from generic products are reported under the caption "Impax Generics, net."
The Impax Specialty Pharma division includes the legacy Impax Pharmaceuticals business as well as the acquired business of Amedra Pharmaceuticals, LLC ("Amedra") from the Tower Acquisition. The Company’s Impax Specialty Pharma division is focused on the development and promotion, through the Company’s specialty sales force, of proprietary branded pharmaceutical products for the treatment of central nervous system (“CNS”) disorders and other select specialty segments. Impax Specialty Pharma currently has one internally developed branded pharmaceutical product, Rytary® (IPX066), an extended release oral capsule formulation of carbidopa-levodopa for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication and/or manganese intoxication, which was approved by the FDA on January 7, 2015 and which the Company began marketing in the United States (“U.S.”) in April 2015. The Company received marketing authorization from the European Commission for Numient® (the brand name of IPX066 outside of the United States) during the fourth quarter of fiscal year 2015. In addition to Rytary®, Impax Specialty Pharma is also currently engaged in the sale and distribution of four other branded products; the more significant include Zomig® (zolmitriptan) products, indicated for the treatment of migraine headaches, under the terms of a Distribution, License, Development and Supply Agreement with AstraZeneca UK Limited (“AstraZeneca”) in the United States and in certain U.S. territories (as amended, the "AZ Agreement"), and Emverm® (mebendazole) 100 mg chewable tablets, indicated for the treatment of pinworm, whipworm, common roundworm, common hookworm, and American hookworm in single or mixed infections. Revenues from branded products are reported under the caption “Impax Specialty Pharma sales, net.” Impax Specialty Pharma also has a number of product candidates that are in varying stages of development. See “Note 20. Segment Information,” for financial information about our segments for the years ended December 31, 2017, 2016 and 2015.
Operating Locations
As of December 31, 2017, the Company owned and/or leased facilities in California, Pennsylvania, New Jersey and Taiwan, Republic of China (“R.O.C.”). In California, the Company utilizes a combination of owned and leased facilities mainly located in Hayward. The Company’s primary properties in California consist of a leased office building used as the Company’s corporate headquarters, in addition to four properties it owns, including a research and development center facility and a manufacturing facility. Additionally, the Company leases two facilities in Hayward, utilized for additional research and development, equipment storage and quality assurance support. In Pennsylvania, the Company leases office space for sales and marketing, finance, and administrative personnel in Fort Washington. In New Jersey, the Company leases manufacturing, packaging, research and development and warehousing facilities in Middlesex and office space in Bridgewater.
During the year ended December 31, 2017, the Company closed its Middlesex, New Jersey manufacturing facility and on February 6, 2018, we completed the sale of Impax Laboratories (Taiwan), Inc. (“Impax Taiwan”) through a stock and purchase agreement.
F-8
Business Combination with Amneal Pharmaceuticals LLC
On October 17, 2017, the Company entered into a Business Combination Agreement (the “Business Combination Agreement”) with Atlas Holdings, Inc., a Delaware corporation and a wholly-owned subsidiary of the Company (“Holdco”), K2 Merger Sub Corporation, a Delaware corporation and a wholly-owned subsidiary of Holdco (“Merger Sub”), and Amneal Pharmaceuticals LLC (“Amneal”). The Business Combination Agreement was unanimously approved by the board of directors of the Company on October 16, 2017. Consummation of the Transactions is subject to customary closing conditions, including, among other things, the approval of the Company’s stockholders holding a majority of the outstanding Company Common Stock entitled to vote. The Company and Amneal expect the merger to close in the first half of 2018. However, the Company cannot predict with certainty when, or if, the merger will be completed because completion of the merger is subject to conditions beyond the control of the Company.
At the closing (the “Closing”) of the transactions contemplated by the Business Combination Agreement (the “Transactions”), (i) Merger Sub will merge with and into the Company (the “Impax Merger”), with the Company surviving the Impax Merger as a direct wholly-owned subsidiary of Holdco, (ii) each share of the Company’s common stock issued and outstanding immediately prior to the Impax Merger, other than Company Common Stock held by the Company in treasury, by Amneal or by any of their respective subsidiaries, will be converted into the right to receive one fully paid and nonassessable share of Class A common stock of Holdco, (“Holdco Class A Common Stock”), (iii) the Company will convert to a limited liability company pursuant to the General Corporation Law of the State of Delaware and the Delaware Limited Liability Company Act, (iv) Holdco will contribute to Amneal all of Holdco’s equity interests in the Company to Amneal, in exchange for common units of Amneal, (v) Holdco will issue an aggregate number of shares of Class B common stock of Holdco, par value $0.01 per share (“Holdco Class B Common Stock”, and together with Holdco Class A Common Stock, “Holdco Common Stock”) to the existing members of Amneal (the Existing “Amneal Members”) and (vi) Holdco will become the managing member of Amneal. Upon closing of the transactions, the combination will be accounted for as a business combination under the acquisition method of accounting in accordance with Financial Accounting Standards Board ("FASB") Accounting Standards Codification ("ASC") Topic 805, "Business Combinations," with Amneal treated as the "acquirer" and Impax treated as the "acquired" company for financial reporting purposes. In connection with the Closing, Holdco will be renamed Amneal Pharmaceuticals, Inc. (“New Amneal”).
Immediately following the Closing, (i) the Existing Amneal Members will hold 100% of Holdco Class B Common Stock, which, together with their common units of Amneal, will represent approximately 75% of the voting power and economic interests in New Amneal, and (ii) the Company’s stockholders immediately prior to the Closing will hold 100% of the Holdco Class A Common Stock, which will represent approximately 25% of the voting power and economic interests in New Amneal.
Consummation of the Transactions is subject to customary closing conditions, including, among other things, (i) the approval of the Company’s stockholders holding a majority of the outstanding Company Common Stock entitled to vote (the “Requisite Stockholder Approval”), (ii) expiration of the applicable waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and (iii) NYSE listing approval for Holdco Class A Common Stock. The obligation to consummate the Transactions is also conditioned upon each party’s representations and warranties being true and correct (subject to certain materiality exceptions) and each party having performed in all material respects its obligations under the Business Combination Agreement.
The Business Combination Agreement contains customary and reciprocal representations and warranties of the Company and Amneal, many of which are subject to and qualified by materiality qualifiers. The Company and Amneal have also made customary covenants in the Business Combination Agreement regarding the operation of their respective businesses and the businesses of their respective subsidiaries in the ordinary course prior to the Closing.
The Business Combination Agreement also contains a customary “no shop” covenant prohibiting the Company from soliciting proposals for alternative proposals to acquire the Company, or providing information or participating in any discussions in connection with any such proposals. However, prior to adoption of the Business Combination Agreement by the Company’s stockholders, the Board may, in the exercise of its fiduciary duties, (i) withhold, withdraw, qualify or modify its recommendation that the Company’s stockholders adopt the Business Combination Agreement in connection with certain intervening events, or (ii) terminate the Business Combination Agreement to enter into an agreement in connection with an alternative proposal to acquire the Company that is more favorable to the Company’s stockholders from a financial point of view than the Transactions (a “Superior Proposal”), in each case, subject to complying with certain notice and other specified requirements, including giving Amneal the opportunity to propose revisions to the terms of the Transactions and the payment of the Termination Fee (as defined below).
F-9
Consummation of the Transactions is not subject to a financing condition. However, Amneal is required to use its reasonable best efforts to obtain financing to (i) fund repayment of the Company’s Notes and refinance the RBC Credit Facilities and (ii) refinance outstanding Amneal debt. The Company is required to use reasonable best efforts to provide cooperation in connection with the financing process.
The Business Combination Agreement may be terminated by each of the Company and Amneal under certain circumstances, including if the Closing does not occur on or before July 17, 2018 (the “Outside Date”). Amneal also has certain additional termination rights, including in connection with a change of the Impax Board’s recommendation that the Company’s stockholders adopt and approve the Business Combination Agreement. The Company is required to pay Amneal a termination fee of $45.0 million (the “Termination Fee”) in connection with such a termination by Amneal, as well as under certain other circumstances, including if the Business Combination Agreement is terminated by the Company in connection with a Superior Proposal (as defined in the Business Combination Agreement). Additionally, Amneal will be entitled to reimbursement for up to $15.0 million of its reasonable out-of-pocket expenses incurred in connection with the Business Combination Agreement and the Transactions if the Business Combination Agreement is terminated due to the failure to obtain the Requisite Stockholder Approval.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation
As of December 31, 2017, the consolidated financial statements of the Company include the accounts of the operating parent company, Impax Laboratories, Inc., its wholly owned subsidiaries, including Impax Laboratories USA, LLC, Impax Laboratories (Taiwan), Inc., ThoRx Laboratories, Inc., Impax International Holdings, Inc., Impax Holdings, LLC, Impax Laboratories (Netherlands) C.V., Impax Laboratories (Netherlands) B.V., Impax Laboratories Ireland Limited, Atlas Holdings, Inc., Lineage and Tower, including operating subsidiaries CorePharma LLC, Amedra Pharmaceuticals LLC, Mountain LLC and Trail Services, Inc., and Prohealth Biotech (Taiwan), Inc. (“Prohealth”). The Company acquired all the issued and outstanding share capital in Prohealth on October 24, 2017 and previously held a 57.54% majority ownership interest in the entity prior to such date. All significant intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”) and the rules and regulations of the U.S. Securities & Exchange Commission (“SEC”) requires the use of estimates and assumptions, based on complex judgments considered reasonable, which affect the reported amounts of assets and liabilities and disclosure of contingent assets and contingent liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The most significant judgments are employed in estimates used in determining values of tangible and intangible assets, contingent consideration, legal contingencies, tax assets and tax liabilities, fair value of share-based compensation related to equity incentive awards issued to employees and directors, and estimates used in applying the Company’s revenue recognition policy, including those related to accrued chargebacks, rebates, product returns, Medicare, Medicaid, and other government rebate programs, shelf-stock adjustments, and the timing and amount of deferred and recognized revenue and deferred and amortized product manufacturing costs related to alliance and collaboration agreements. Actual results may differ from estimated results.
Reclassifications
Certain prior year amounts have been reclassified to conform to the presentation for the year ended December 31, 2017.
Revenue Recognition
The Company recognizes revenue when the earnings process is complete, which under SEC Staff Accounting Bulletin No. 104, Topic No. 13, “Revenue Recognition” (“SAB 104”), is when revenue is realized or realizable and earned, there is persuasive evidence a revenue arrangement exists, delivery of goods or services has occurred, the sales price is fixed or determinable, and collectability is reasonably assured.
The Company accounts for material revenue arrangements which contain multiple deliverables in accordance with FASB ASC Topic 605-25, Revenue Recognition - Multiple Element Arrangements ("ASC 605-25"), which addresses the determination of whether an arrangement involving multiple deliverables contains more than one unit of accounting. A delivered item within an arrangement is considered a separate unit of accounting only if both of the following criteria are met:
• | the delivered item has value to the customer on a stand-alone basis; and |
F-10
• | if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the control of the vendor. |
Under ASC Topic 605-25, if both of the criteria above are not met, then separate accounting for the individual deliverables is not appropriate. Revenue recognition for arrangements with multiple deliverables constituting a single unit of accounting is recognized generally over the greater of the term of the arrangement or the expected period of performance, either on a straight-line basis or on a modified proportional performance method.
The Company accounts for milestones related to research and development activities in accordance with FASB ASC Topic 605-28, Revenue Recognition - Milestone Method ("ASC 605-28"). ASC Topic 605-28 allows for the recognition of consideration, which is contingent on the achievement of a substantive milestone, in its entirety in the period the milestone is achieved. A milestone is considered to be substantive if all of the following criteria are met:
• | the milestone is commensurate with either: (1) the performance required to achieve the milestone, or (2) the enhancement of the value of the delivered items resulting from the performance required to achieve the milestone; |
• | the milestone relates solely to past performance; and |
• | the milestone payment is reasonable relative to all of the deliverables and payment terms within the agreement. |
Impax Generics revenues, net, and Impax Specialty Pharma revenues, net
The Impax Generics revenues, net and Impax Specialty Pharma revenues, net include revenue recognized related to shipments of generic and branded pharmaceutical products to the Company’s customers, primarily drug wholesalers and retail chains. Gross sales revenue is recognized at the time title and risk of loss passes to the customer, which is generally when product is received by the customer. Net revenues may include deductions from the gross sales price related to estimates for chargebacks, rebates and administrative fees, distribution service fees, returns, shelf-stock adjustments, and other pricing adjustments. The Company records an estimate for these deductions in the same period when revenue is recognized. A description of each of these gross-to-net deductions follows.
• | Chargebacks |
The Company has agreements establishing contract prices for certain products with certain indirect customers, such as retail pharmacy chains, group purchasing organizations, managed care organizations, hospitals and government agencies who purchase products from drug wholesalers. The contract prices are lower than the prices the customer would otherwise pay to the wholesaler, and the price difference is referred to as a chargeback, which generally takes the form of a credit memo issued by the Company to reduce the invoiced gross selling price charged to the wholesaler. An estimated accrued provision for chargeback deductions is recognized at the time of product shipment. The primary factors considered when estimating the provision for chargebacks are the average historical chargeback credits given, the mix of products shipped, and the amount of inventory on hand at the major drug wholesalers with whom the Company does business. The Company also monitors actual chargebacks granted and compares them to the estimated provision for chargebacks to assess the reasonableness of the chargeback reserve at each quarterly balance sheet date.
• | Rebates and Administrative Fees |
The Company maintains various rebate and administrative fee programs with its customers in an effort to maintain a competitive position in the marketplace and to promote sales and customer loyalty. The rebates generally take the form of a credit memo to reduce the invoiced gross selling price charged to a customer for products shipped. An estimated accrued provision for rebate deductions is recognized at the time of product shipment. The primary factors the Company considers when estimating the provision for rebates are the average historical experience of aggregate credits issued, the mix of products shipped and the historical relationship of rebates as a percentage of total gross product sales, the contract terms and conditions of the various rebate programs in effect at the time of shipment, and the amount of inventory on hand at the major drug wholesalers with whom the Company does business. The Company also monitors actual rebates granted and compares them to the estimated provision for rebates to assess the reasonableness of the rebate reserve at each quarterly balance sheet date.
F-11
• | Distribution Service Fees |
The Company pays distribution service fees to several of its wholesaler customers related to sales of its Impax Products. The wholesalers are generally obligated to provide the Company with periodic outbound sales information as well as inventory levels of the Company’s Impax Products held in their warehouses. Additionally, the wholesalers have agreed to manage the variability of their purchases and inventory levels within specified days on hand limits. An accrued provision for distribution service fees is recognized at the time products are shipped to wholesalers.
• | Returns |
The Company allows its customers to return product if approved by authorized personnel in writing or by telephone with the lot number and expiration date accompanying any request and if such products are returned within six months prior to or until twelve months following, the product’s expiration date. The Company estimates and recognizes an accrued provision for product returns as a percentage of gross sales based upon historical experience. The product return reserve is estimated using a historical lag period, which is the time between when the product is sold and when it is ultimately returned, and estimated return rates which may be adjusted based on various assumptions including: changes to internal policies and procedures, business practices, commercial terms with customers, and the competitive position of each product; the amount of inventory in the wholesale and retail supply chain; the introduction of new products; and changes in market sales information. The Company also considers other factors, including significant market changes which may impact future expected returns, and actual product returns. The Company monitors actual returns on a quarterly basis and may record specific provisions for returns it believes are not covered by historical percentages.
• | Shelf-Stock Adjustments |
Based upon competitive market conditions, the Company may reduce the selling price of certain Impax Generics division products. The Company may issue a credit against the sales amount to a customer based upon their remaining inventory of the product in question, provided the customer agrees to continue to make future purchases of product from the Company. This type of customer credit is referred to as a shelf-stock adjustment, which is the difference between the initial sales price and the revised lower sales price, multiplied by an estimate of the number of product units on hand at a given date. Decreases in selling prices are discretionary decisions made by the Company in response to market conditions, including estimated launch dates of competing products and declines in market price. The Company records an estimate for shelf-stock adjustments in the period it agrees to grant such a credit memo to a customer.
• | Cash Discounts |
The Company offers cash discounts to its customers, generally 2% to 3% of the gross selling price, as an incentive for paying within invoice terms, which generally range from 30 to 90 days. An estimate of cash discounts is recorded in the same period when revenue is recognized.
• | Medicaid and Other U.S. Government Pricing Programs |
As required by law, the Company provides a rebate on drugs dispensed under the Medicaid program, Medicare Part D, TRICARE, and other U.S. government pricing programs. The Company determines its estimated government rebate accrual primarily based on historical experience of claims submitted by the various states and other jurisdictions and any new information regarding changes in the various programs which may impact the Company’s estimate of government rebates. In determining the appropriate accrual amount, the Company considers historical payment rates and processing lag for outstanding claims and payments. The Company records estimates for government rebates as a deduction from gross sales, with a corresponding adjustment to accrued liabilities.
• | Rx Partner and OTC Partner |
The Rx Partner and OTC Partner contracts include revenue recognized under alliance and collaboration agreements between the Company and unrelated third-party pharmaceutical companies. The Company has entered into these alliance agreements to develop marketing and/or distribution relationships with its partners to fully leverage its technology platform.
F-12
The Rx Partners and OTC Partners alliance agreements obligate the Company to deliver multiple goods and/or services over extended periods. Such deliverables include manufactured pharmaceutical products, exclusive and semi-exclusive marketing rights, distribution licenses, and research and development services. In exchange for these deliverables the Company receives payments from its agreement partners for product shipments and research and development services, and may also receive other payments including royalties, profit sharing payments, and upfront and periodic milestone payments. Revenue received from the alliance agreement partners for product shipments under these agreements is not subject to deductions for chargebacks, rebates, product returns, and other pricing adjustments. Royalty and profit sharing amounts the Company receives under these agreements are calculated by the respective agreement partner, with such royalty and profit share amounts generally based upon estimates of net product sales or gross profit which include estimates of deductions for chargebacks, rebates, product returns, and other adjustments the alliance agreement partners may negotiate with their respective customers. The Company records the agreement partner's adjustments to such estimated amounts in the period the agreement partner reports the amounts to the Company.
The Company applies the updated guidance of FASB ASC Topic 605-25 to the Strategic Alliance Agreement, as amended with Teva Pharmaceuticals USA, Inc., an affiliate of Teva Pharmaceutical Industries Limited (the “Teva Agreement”). The Company looks to the underlying delivery of goods and/or services which give rise to the payment of consideration under the Teva Agreement to determine the appropriate revenue recognition. The Company initially defers consideration received as a result of research and development-related activities performed under the Teva Agreement. The Company recognizes deferred revenue on a straight-line basis over the expected period of performance for such services. Consideration received as a result of the manufacture and delivery of products under the Teva Agreement is recognized at the time title and risk of loss passes to the customer, which is generally when product is received by Teva. The Company recognizes profit share revenue in the period earned.
OTC Partner revenue was previously, related to agreements with Pfizer, Inc., formerly Wyeth LLC (“Pfizer”) and L. Perrigo Company (“Perrigo”) with respect to the supply of the Company's over-the-counter pharmaceutical product Loratadine and Pseudoephedrine Sulfate 5 mg/120 mg 12-hour Extended Release Tablets (the "D12 Product"). Following the expiration of the Company's obligation to supply the D12 Product to Pfizer and Perrigo as described below, the company does not currently sell any over-the-counter pharmaceutical products. The Company previously recognized profit share revenue in the period earned. During the quarter ended September 30, 2016, the Company sold the ANDAs for both the D12 Product and the Loratadine and Pseudoephedrine Sulfate 10 mg/240 mg 24-hour Extended Release Tablets, in addition to other specified assets, to Perrigo pursuant to an asset purchase agreement with Perrigo dated as of March 31, 2016 (the "Perrigo APA"). Under the terms of the Perrigo APA, the Company was required to continue to supply the D-12 Product to Pfizer and Perrigo until the date that was the earliest of (i) the date Perrigo’s manufacturing facility is approved to manufacture the D-12 Product and (ii) December 31, 2017. On November 30, 2017, the Company assigned and transferred its supply agreement with Pfizer in its entirety to Perrigo in accordance with the Perrigo APA.
• | Research Partner |
The Research Partner contract revenue results from development agreements the Company enters into with unrelated third-party pharmaceutical companies. The development agreements generally obligate the Company to provide research and development services over multiple periods. In exchange for this service, the Company generally receives upfront payments upon signing of each development agreement and is eligible to receive contingent milestone payments, payment of which is based upon the achievement of contractually specified events. Additionally, the Company may also receive royalty payments from the sale, if any, of a successfully developed and commercialized product under one of these development agreements. The Company recognizes revenue received from the achievement of contingent research and development milestones in the period such payment is earned. Royalty revenue, if any, will be recognized as current period revenue when earned.
Cash and Cash Equivalents
The Company considers all short-term investments with a maturity of three months or less at the date of purchase to be cash equivalents. Cash and cash equivalents are stated at cost, which, for cash equivalents, approximates fair value due to the short-term nature. The Company is potentially subject to financial instrument concentration of credit risk through its cash and cash equivalents.
F-13
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk are cash, cash equivalents and accounts receivable. Cash is held on deposit in demand accounts at large financial institutions in amounts in excess of the Federal Deposit Insurance Corporation (FDIC) insurance coverage limit of $250,000 per depositor, per FDIC-insured bank, per ownership category. Cash equivalents are comprised of highly-rated money market funds. The Company limits its credit risk with respect to accounts receivable by performing credit evaluations of customers when deemed necessary. The Company does not require collateral to secure amounts due from its customers.
The following tables present the percentage of total accounts receivable and gross revenues represented by the Company’s three largest customers as of and for the years ended December 31, 2017, 2016 and 2015:
Percent of Total Accounts Receivable | 2017 | 2016 | 2015 | |||||
Customer #1 | 44.7 | % | 36.2 | % | 52.4 | % | ||
Customer #2 | 23.6 | % | 35.6 | % | 24.8 | % | ||
Customer #3 | 23.4 | % | 20.5 | % | 14.4 | % | ||
Top three largest customers | 91.7 | % | 92.3 | % | 91.6 | % | ||
Percent of Gross Revenues | 2017 | 2016 | 2015 | |||||
Customer #1 | 32.9 | % | 40.1 | % | 45.6 | % | ||
Customer #2 | 30.0 | % | 28.4 | % | 21.7 | % | ||
Customer #3 | 25.0 | % | 20.1 | % | 18.8 | % | ||
Top three largest customers | 87.9 | % | 88.6 | % | 86.1 | % | ||
Allowance for Doubtful Accounts
The Company maintains allowances for doubtful accounts for estimated losses resulting from amounts deemed to be uncollectible from its customers; these allowances are for specific amounts on certain accounts based on facts and circumstances determined on a case-by-case basis.
Inventory
Inventory is stated at the lower of cost or net realizable value. Cost is determined using a standard cost method, and the cost flow assumption is first in, first out (“FIFO”) flow of goods. Standard costs are revised annually, and significant variances between actual costs and standard costs are apportioned to inventory and cost of goods sold based upon inventory turnover. Costs include materials, labor, quality control, and production overhead. Inventory is adjusted for short-dated, unmarketable inventory equal to the difference between the cost of inventory and the estimated value based upon assumptions about future demand and market conditions. If actual market conditions are less favorable than those projected by the Company, additional inventory write-downs may be required. Consistent with industry practice, the Company may build pre-launch inventories of certain products which are pending required approval from the FDA and/or resolution of patent infringement litigation, when, in the Company’s assessment, such action is appropriate to prepare for the anticipated commercial launch and FDA approval is expected in the near term and /or the related litigation will be resolved in the Company’s favor. The Company accounts for all costs of idle facilities, excess freight and handling costs, and wasted materials (spoilage) as a current period charge in accordance with U.S. GAAP.
F-14
Assets Held for Sale
The Company classifies its long-lived assets to be sold as held for sale in the period (i) it has approved and committed to a plan to sell the asset, (ii) the asset is available for immediate sale in its present condition, (iii) an active program to locate a buyer and other actions required to sell the asset have been initiated, (iv) the sale of the asset is probable, (v) the asset is being actively marketed for sale at a price that is reasonable in relation to its current fair value, and (vi) it is unlikely that significant changes to the plan will be made or that the plan will be withdrawn. The Company initially measures a long-lived asset that is classified as held for sale at the lower of its carrying value or fair value less any costs to sell. Any loss resulting from this measurement is recognized in the period in which the held for sale criteria are met. Conversely, gains are not recognized on the sale of a long-lived asset until the date of sale. Upon designation as an asset held for sale, the Company stops recording depreciation expense on the asset. The Company assesses the fair value of a long-lived asset less any costs to sell at each reporting period and until the asset is no longer classified as held for sale.
Property, Plant and Equipment
Property, plant and equipment are recorded at cost. Maintenance and repairs are charged to expense as incurred and costs of improvements and renewals are capitalized. Costs incurred in connection with the construction or major renovation of facilities, including interest directly related to such projects, are capitalized as construction in progress. Depreciation is recognized using the straight-line method based on the estimated useful lives of the related assets, which are generally 40 years for buildings, 10 to 15 years for building improvements, eight to 10 years for equipment, and four to 10 years for office furniture and equipment. Land and construction-in-progress are not depreciated.
Intangible Assets
The Company’s intangible assets include both finite lived and indefinite-lived assets. Finite lived intangible assets, consisting of marketed product rights and royalties received from product sales by the Company's third party partners, are amortized over the estimated useful life of the asset based on the pattern in which the economic benefits are expected to be consumed or otherwise used up or, if that pattern is not readily determinable, on a straight-line basis. Indefinite-lived intangible assets consist of acquired in process research and development ("IPR&D") product rights and acquired future royalty rights to be paid based on other companies’ net sales of products not yet approved. IPR&D assets acquired in a business combination are considered indefinite-lived until the completion or abandonment of the associated research and development efforts. Amortization over the estimated useful life will commence at the time of the respective product’s launch. If FDA approval to market the product is not obtained, the Company will immediately expense the related capitalized cost.
Finite lived intangible assets are tested for impairment when events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. All of the Company's indefinite-lived intangible assets are tested for impairment at least annually during the fourth quarter of the fiscal year, or more often if indicators of impairment are present. Impairment testing requires management to estimate the future undiscounted cash flows of an intangible asset using assumptions believed to be reasonable, but which are unpredictable and inherently uncertain. Actual future cash flows may differ from the estimates used in the impairment testing. The Company recognizes an impairment loss when and to the extent that the estimated fair value of an intangible asset is less than its carrying value.
Goodwill
In accordance with FASB ASC Topic 350, "Goodwill and Other Intangibles," rather than recording periodic amortization, goodwill is subject to an annual assessment for impairment by applying a fair value based test. If the fair value of the reporting unit exceeds the reporting unit’s carrying value, including goodwill, then goodwill is considered not impaired, making further analysis not required. The Company considers the Impax Generics division and the Impax Specialty Pharma division operating segments to each be a reporting unit. The Company attributes $59.7 million of goodwill to the Impax Specialty Pharma division and $147.6 million of goodwill to the Impax Generics division.
F-15
The Company concluded the carrying value of goodwill was not impaired as of December 31, 2017 and 2016 as the fair value of the Impax Specialty Pharma division and the Impax Generics division exceeded their carrying value at each date. The Company performs its annual impairment test in the fourth quarter of each year. In the fourth quarter of 2017, the Company determined that it was not more likely than not that the fair value of goodwill was less than its carrying value. As a result, the Company did not perform a quantitative analysis. In the fourth quarter of 2016, the Company performed a quantitative analysis and estimated the fair value of the Impax Specialty Pharma division and the Impax Generics division using a discounted cash flow model for both the reporting unit and the enterprise, as well as earnings and revenue multiples per common share outstanding for enterprise fair value. In addition, on a quarterly basis, the Company performs a review of its business operations to determine whether events or changes in circumstances have occurred that could have a material adverse effect on the estimated fair value of each reporting unit, and thus indicate a potential impairment of the goodwill carrying value. If such events or changes in circumstances were deemed to have occurred, the Company would perform an interim impairment analysis, which may include the preparation of a discounted cash flow model, or consultation with one or more valuation specialists, to analyze the impact, if any, on our assessment of the reporting unit’s fair value.
Derivatives
The Company generally does not use derivative instruments or engage in hedging activities in its ordinary course of business. Prior to June 30, 2015, the Company had no derivative assets or liabilities and did not engage in any hedging activities. As a result of the Company’s June 30, 2015 issuance of the convertible senior notes described in “Note 10. Debt”, the conversion option of the notes temporarily met the criteria for an embedded derivative liability which required bifurcation and separate accounting. Concurrently with the issuance of the notes, the Company entered into a series of convertible note hedge and warrant transactions which in combination are designed to reduce the potential dilution to the Company’s stockholders and/or offset the cash payments the Company is required to make in excess of the principal amount upon conversion of the notes. See “Note 11. Stockholders’ Equity” for additional information regarding the note hedge transactions and warrant transactions. While the warrants sold were classified as equity and recorded in additional paid-in capital, the call options purchased were temporarily classified as a bond hedge derivative asset on the Company’s consolidated balance sheet. The Company engaged a third-party valuation firm with expertise in valuing financial instruments to determine the fair value of the bond hedge derivative asset and conversion option derivative liability at each reporting period. The Company’s consolidated balance sheets reflected the fair value of the derivative asset and liability as of the reporting date, and changes in the fair value were reflected in current period earnings, as appropriate. As result of the amendment to the Company’s Restated Certificate of Incorporation to increase the number of authorized shares of the Company's common stock discussed in “Note 11. Stockholders’ Equity,” both the derivative asset and liability were reclassified to additional paid-in capital. The Company had no derivative assets or liabilities and did not engage in any hedging activities during the years ended December 31, 2017 or 2016.
Contingencies
In the normal course of business, the Company is subject to loss contingencies, such as legal proceedings and claims arising out of its business, covering a wide range of matters, including, among others, patent litigation, stockholder lawsuits, and product and clinical trial liability. The Company records accruals for such loss contingencies when it is probable a liability will have been incurred and the amount of loss can be reasonably estimated. The Company does not recognize gain contingencies until realized. The Company records an accrual for legal costs in the period incurred. A discussion of contingencies is included in “Note 18. Commitments and Contingencies” and “Note 19. Legal and Regulatory Matters”.
Deferred Financing Costs
The Company capitalizes direct costs incurred to obtain debt financing and amortizes these costs to interest expense using the effective interest method over the term of the debt. These costs are recorded as a debt discount and the unamortized costs are netted against the related debt on the Company’s consolidated balance sheets. For line-of-credit arrangements with no outstanding borrowing, the costs incurred to obtain the credit facility are amortized to interest expense using the straight-line method over the term of the line-of-credit arrangement. The unamortized balance is included in other assets on the Company’s consolidated balance sheets.
Shipping and Handling Fees and Costs
Shipping and handling fees related to sales transactions are recorded as selling expense. Shipping costs were $7.0 million, $3.7 million and $2.3 million for the years ended December 31, 2017, 2016 and 2015, respectively.
F-16
Research and Development Expenses
Research and development activities are expensed as incurred and consist of self-funded research and development costs and costs associated with work performed by other participants under collaborative research and development agreements.
Share-Based Compensation
The Company accounts for stock-based employee compensation arrangements in accordance with provisions of FASB ASC Topic 718 “Stock Compensation.” Under FASB ASC Topic 718, the Company recognizes the grant date fair value of stock-based employee compensation as expense on a straight-line basis over the vesting period of the grant. The Company uses the Black Scholes option pricing model to determine the grant date fair value of employee stock options. The fair value of restricted stock awards is equal to the closing price of the Company’s stock on the date such award was granted.
Effective January 1, 2017, the Company adopted Accounting Standards Update ("ASU") 2016-09 "Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting" and elected to eliminate the use of a forfeiture rate estimate in the determination of share-based compensation expense for restricted stock awards using the modified retrospective transition method. Adoption of the new guidance using this method resulted in a $1.4 million charge to opening retained earnings for 2017.
Income Taxes
The Company provides for income taxes using the asset and liability method as required by FASB ASC Topic 740, “Income Taxes.” This approach recognizes the amount of federal, state, local and foreign taxes payable or refundable for the current year, as well as deferred tax assets and liabilities for the future tax consequences of events recognized in the consolidated financial statements and income tax returns. Deferred income tax assets and liabilities are adjusted to recognize the effects of changes in tax laws or enacted tax rates in the period during which they are signed into law. FASB ASC Topic 740 requires an assessment of whether valuation allowances are needed against deferred tax assets based upon consideration of all available evidence using a more likely than not standard. See "Note 16. Income Taxes" for further discussion of the Company’s valuation allowances.
FASB ASC Topic 740, Sub-topic 10 “Tax Positions,” defines the criterion an individual tax position must meet for any part of the benefit of the tax position to be recognized in financial statements prepared in conformity with generally accepted accounting principles. Under FASB ASC Topic 740, Sub-topic 10, the Company may recognize the tax benefit from an uncertain tax position only if it is more likely than not the tax position will be sustained on examination by the taxing authorities, based solely on the technical merits of the tax position. The tax benefits recognized in the financial statements from such a tax position should be measured based on the largest benefit having a greater than 50% likelihood of being realized upon ultimate settlement with the tax authority. Additionally, FASB ASC Topic 740, Sub-topic 10 provides guidance on measurement, de-recognition, classification, interest and penalties, accounting in interim periods, disclosure, and transition. In accordance with the disclosure requirements of FASB ASC Topic 740, Sub-topic 10, the Company’s policy on income statement classification of interest and penalties related to income tax obligations is to include such items as part of total interest expense and other expense, respectively.
Other Comprehensive Income
The Company follows the provisions of FASB ASC Topic 220, ”Comprehensive Income,” which establishes standards for the reporting and display of comprehensive income and its components. Comprehensive income is defined to include all changes in equity during a period except those resulting from investments by owners and distributions to owners. The Company recorded foreign currency translation gains and losses, which are reported as comprehensive income. Foreign currency translation gains (losses) for the years ended December 31, 2017, 2016 and 2015 were $9.3 million, $2.6 million and $(4.5) million, respectively.
Foreign Currency Translation
The Company translates the assets and liabilities of the Taiwan dollar functional currency of Prohealth and its wholly-owned subsidiary Impax Laboratories (Taiwan), Inc. into the U.S. dollar reporting currency using exchange rates in effect at the end of each reporting period. The revenues and expenses of these entities are translated using an average of the rates in effect during the reporting period. Gains and losses from these translations are recorded as currency translation adjustments included in the consolidated statements of comprehensive (loss) income and the consolidated statements of changes in stockholders’ equity.
F-17
Recent Accounting Pronouncements
Accounting Guidance Issued Not Yet Adopted
In May 2014, the FASB issued ASU 2014-09, “Revenue from Contracts with Customers” (Topic 606) regarding the accounting for and disclosures of revenue recognition, with an effective date for annual and interim periods beginning after December 15, 2016. This update provided a single comprehensive model for accounting for revenue from contracts with customers. The model requires that revenue recognized reflect the actual consideration to which the entity expects to be entitled in exchange for the goods or services defined in the contract, including in situations with multiple performance obligations. In July 2015, the FASB issued ASU 2015-14, “Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date,” which deferred the effective date of the previously issued revenue recognition guidance by one year. The guidance is effective for annual and interim periods beginning after December 15, 2017. In April 2016 and May 2016, the FASB issued ASU 2016-10, "Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing" and ASU 2016-12, "Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients," respectively. Both of these updates provide improvements and clarification to the previously issued revenue recognition guidance. The new standard can be adopted using one of two methods: the full retrospective method, which requires the standard to be applied to each prior period presented, or the modified retrospective method, which requires the cumulative effect of adoption to be recognized as an adjustment to opening retained earnings in the period of adoption. The Company adopted the new revenue recognition standard as of January 1, 2018 using the modified retrospective method. The Company has substantially completed its analysis of the impact that adoption will have on its consolidated financial statements. The majority of the Company's revenue relates to the sale of finished products to various customers, and the adoption will not have an impact on revenue recognized from these transactions. The Company has also evaluated the impact on certain less significant transactions involving third-party collaborations and other arrangements, whereby the Company will recognize revenue earlier under the new standard. The Company has estimated that a cumulative effect adjustment of approximately $0.5 million will be recognized as of January 1, 2018 to reflect the recognition of revenue related to the Company' profit sharing agreements. During fiscal year 2018, the Company will disclose the amount by which revenue was affected for each period presented. In addition, the new standard will require changes to the Company’s processes and controls and the Company has identified and designed changes to processes and controls to ensure readiness.
In February 2016, the FASB issued ASU 2016-02, "Leases" (Topic 842), with guidance regarding the accounting for and disclosure of leases. The update requires lessees to recognize all leases, including operating leases, with a term greater than 12 months on the balance sheet. This update also requires lessees and lessors to disclose key information about their leasing transactions. The guidance will be effective for annual and interim periods beginning after December 15, 2018. The Company is currently evaluating the effect that this guidance will have on its consolidated financial statements and related disclosures. The Company's expects the implementation of this standard to have an impact on its consolidated financial statements and related disclosures as it has aggregate future minimum lease payments of $28.1 million as of December 31, 2017 under the current portfolio of non-cancelable leases for land, office space, and manufacturing, warehouse and research and development facilities with various expiration dates between January 2018 and December 2027. The Company anticipates recognition of additional assets and corresponding liabilities related to these leases on its consolidated balance sheet.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): "Classification of Certain Cash Receipts and Cash Payments," with guidance intended to reduce the diversity in practice regarding how certain cash receipts and cash payments are presented and classified within the statement of cash flows. The update addresses eight specific cash flow issues including debt prepayment or debt extinguishment costs, the settlement of zero-coupon debt instruments or other debt instruments with coupon interest rates that are insignificant in relation to the effective interest rate of the borrowing, contingent consideration payments made after a business combination, proceeds from the settlement of insurance claims, proceeds from the settlement of corporate-owned life insurance policies (COLIs) (including bank-owned life insurance policies (BOLIs)), distributions received from equity method investees, beneficial interests in securitization transactions, and separately identifiable cash flows and application of the predominance principle. The guidance is effective for annual and interim periods beginning after December 15, 2017. The adoption of this guidance will not have any impact on the Company's consolidated financial statements.
In October 2016, the FASB issued ASU-2016-16, Income Taxes (Topic 740): "Intra-Entity Transfers of Assets Other Than Inventory," with guidance intended to more faithfully represent the economics of intra-entity asset transfers. The update clarifies that entities must recognize the income tax consequences of intra-entity asset transfers, other than inventory, when the transfer occurs. The guidance is effective for annual and interim periods beginning after December 15, 2017. The adoption of this guidance will not have any impact on the Company's consolidated financial statements.
F-18
In January 2017, the FASB issued ASU-2017-01, Business Combinations (Topic 805): "Clarifying the Definition of a Business," with guidance intended to assist entities in evaluating whether transactions should be accounted for as acquisitions (or disposals) of businesses. The update provides a screen to determine whether an integrated set of assets and activities constitute a business. If the screen is not met, the guidance (1) requires that to be considered a business, a set must include, at a minimum, an input and a substantive process that together significantly contribute to the ability to create output and (2) removes the evaluation of whether a market participant could replace the missing elements. The guidance is effective for annual and interim periods beginning after December 15, 2017 and will be applied prospectively. The Company adopted this guidance as of January 1, 2018 and the guidance will not have any impact on the Company's consolidated financial statements.
In May 2017, the FASB issued ASU 2017-09, Compensation - Stock Compensation (Topic 718): "Scope of Modification Accounting," which provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The guidance is effective for annual and interim periods beginning after December 15, 2017, with early adoption permitted. The amendments in this ASU are applied prospectively to an award modified on or after the adoption date. The Company adopted this guidance as of January 1, 2018 and the guidance will not have any impact on the Company's consolidated financial statements.
Recently Adopted Accounting Guidance
In July 2015, the FASB issued ASU 2015-11, Inventory (Topic 330): “Simplifying the Measurement of Inventory,” with guidance regarding the accounting for and measurement of inventory. The update requires that inventory measured using first-in, first-out ("FIFO") shall be measured at the lower of cost and net realizable value. When there is evidence that the net realizable value of inventory is lower than its cost, the difference shall be recognized as a loss in earnings in the period in which it occurs. The guidance is effective for annual and interim periods beginning after December 15, 2016. The Company adopted this guidance during the first quarter of 2017, and it did not have a material effect on the Company's consolidated financial statements.
In March 2016, the FASB issued ASU 2016-06, Derivatives and Hedging (Topic 915): "Contingent Put and Call Options in Debt Instruments," with guidance regarding the accounting for embedded derivatives related to debt contracts. The update clarifies that determining whether the economic characteristics of a put or call are clearly and closely related to its debt host requires only an assessment of the four-step decision sequence outlined in FASB ASC paragraph 815-15-25-24. The update also indicates that entities are not required to separately assess whether the contingency itself is clearly and closely related. The guidance will be effective for annual and interim periods beginning after December 15, 2016. The Company adopted this guidance during the first quarter of 2017, and it did not have an effect on the Company's consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation - Stock Compensation (Topic 718): "Improvements to Employee Share-Based Payment Accounting," with guidance regarding the simplification of accounting for share-based payment award transactions. The update changes the accounting for such areas as the accounting and cash flow classification for excess tax benefits and deficiencies; forfeitures; and tax withholding requirements and cash flow classification. The guidance is effective for annual and interim periods beginning after December 15, 2016. The Company adopted the new guidance effective January 1, 2017 and elected to eliminate the use of a forfeiture rate estimate in the determination of share-based compensation expense for restricted stock awards using the modified retrospective transition method, which resulted in a $1.4 million charge to opening retained earnings for 2017. In addition, the Company is now presenting the cash paid for tax withholdings on stock options exercised and restricted stock awards vested retrospectively in cash flows from financing activities as opposed to the historical presentation in cash flows from operating activities. The adoption resulted in an increase to net cash from operations and decrease net cash provided by financing of $9.3 million and $20.5 million for the years ended December 31, 2016 and 2015, respectively. Excess tax benefits or deficiencies, historically recorded to additional paid-in capital, are recorded to income tax expense as they occur on a prospective basis.
In January 2017, the FASB issued ASU 2017-03, "Accounting Changes and Error Corrections" (Topic 250) and Investments - Equity Method and Joint Ventures (Topic 323), which add to and amend SEC paragraphs pursuant to the SEC Staff Announcements at the September 22, 2016 and November 17, 2016 Emerging Issues Task Force (EITF) meetings. The guidance provides additional disclosure requirements regarding the impact of recently issued accounting standards on the financial statements of a registrant when such standards are adopted in a future period. The Company adopted this guidance during the first quarter of 2017, and it did not have an effect on the Company’s consolidated financial statements.
F-19
In January 2017, the FASB issued ASU-2017-04, "Intangibles - Goodwill and Other" (Topic 350): "Simplifying the Test for Goodwill Impairment," which removes the second step of the two-step goodwill impairment test. In order to reduce the cost and complexity of testing goodwill for impairment, entities are now only required to perform a one-step quantitative impairment test and to record the amount of goodwill impairment as the excess of a reporting unit's carrying amount over its fair value, not to exceed the total amount of goodwill allocated to the reporting unit. The new guidance does not amend the optional qualitative assessment of a reporting unit to determine if the quantitative impairment test is necessary. Entities should apply the guidance on a prospective basis and disclose the nature of and reason for the change in accounting principle upon transition. The guidance will be effective for annual and interim periods beginning after December 15, 2019, with early adoption permitted for interim or annual goodwill impairment tests performed after January 1, 2017. The Company adopted this guidance during the first quarter of 2017, and it did not have an effect on the Company’s consolidated financial statements.
3. BUSINESS ACQUISITIONS
Teva Transaction
On August 3, 2016, the Company completed its previously announced acquisition of (A) certain assets related to (i) 15 then currently marketed generic pharmaceutical products, (ii) one then approved generic product and two then tentatively approved strengths of a then currently marketed product, which at the time of the closing had not yet launched, (iii) one pipeline generic product and one pipeline strength of a then currently marketed product, which at the time of the closing were pending approval by the FDA and (iv) one generic product then under development, and (B) the return to the Company of its full commercial rights to its then pending ANDA for the generic equivalent to Concerta® (methylphenidate hydrochloride), a product the Company previously partnered with Teva Pharmaceuticals USA, Inc. (“Teva USA”) (collectively, the products and pipeline products and the assets related thereto in (A) and (B), the “Acquired Product Lines” and the transactions related thereto the “Teva Transaction”), pursuant to (x) an Asset Purchase Agreement, dated as of June 20, 2016, as amended on June 30, 2016, with Teva Pharmaceutical Industries Ltd. (“Teva”), acting directly or through its affiliates (the “Teva APA”), (y) an Asset Purchase Agreement, dated as of June 20, 2016, as amended on June 30, 2016, with affiliates of Allergan plc (“Allergan”), (the “Allergan APA” and collectively with the Teva APA, the "APAs"), and (z) a Termination Agreement, dated as of June 20, 2016, between the Company and Teva USA, terminating each party’s rights and obligations with respect to methylphenidate hydrochloride under the Strategic Alliance Agreement, dated June 27, 2001, as amended between the Company and Teva USA. The aggregate purchase price for the Acquired Product Lines pursuant to the terms of the Teva APA and the Allergan APA, including the upfront payment to Teva in accordance with the Termination Agreement, was $585.8 million in cash at closing. The Company is also obligated to make future payments to Teva of up to $40.0 million under the terms of the Termination Agreement, payable upon the achievement of specified commercialization events related to methylphenidate hydrochloride.
The Company financed the Teva Transaction utilizing cash on hand and $400.0 million, the full amount of borrowing available, from its Term Loan Facility with Royal Bank of Canada, as discussed in "Note 11. Debt." The Company incurred acquisition-related costs for the Teva Transaction of $3.1 million and $0.6 million during for the years ended December 31, 2016, and 2015, respectively, which are included in selling, general, and administrative expenses in the Company's consolidated statements of operations.
The acquisition of the foregoing currently marketed and pipeline products fits with the Company’s strategic priorities of maximizing its Generics Division’s platform and optimizing research and development opportunities. Through the Teva Transaction, the Company expanded its portfolio of difficult-to-manufacture or limited-competition products and maximized utilization of its existing manufacturing facilities.
As part of the closing of the Teva Transaction, the Company, Teva and Allergan agreed to certain transition related services pursuant to which the Company agreed to manage the payment process for certain commercial chargebacks and rebates on behalf of Teva and Allergan related to products each of Teva and Allergan sold into the channel prior to the closing date. On August 18, 2016, the Company received a payment totaling $42.4 million from Teva and Allergan, which represented their combined estimate of the amount of commercial chargebacks and rebates to be paid by the Company on their behalf to wholesalers who purchased products from Teva and Allergan prior to the closing. Pursuant to the agreed upon transition services, Teva and Allergan are obligated to reimburse the Company for additional payments related to chargebacks and rebates made on their behalf in excess of the $42.4 million. If the total payments made by the Company on behalf of Teva and Allergan are less than $42.4 million, the Company is obligated to refund the difference to Teva and/or Allergan. As of December 31, 2017, the Company had paid $29.1 million on behalf of Teva and Allergan related to chargebacks and rebates as described above and $13.3 million remained in accrued expenses on the consolidated balance sheet.
F-20
Purchase Accounting and Consideration
FASB ASC Topic 805, Business Combinations ("ASC 805") defines a business as consisting of inputs and processes applied to those inputs that have the ability to create outputs. The Company has determined that the Acquired Product Lines meet the definition of a business and, accordingly, has accounted for the Teva Transaction as a business combination under the acquisition method of accounting.
The following is an estimate of the purchase price for the Teva Transaction as of the closing date of August 3, 2016 (in thousands):
Estimated Fair Value | |||
Purchase price per the APAs | $ | 575,800 | |
Upfront payment pursuant to Termination Agreement | 10,000 | ||
Total cash consideration | 585,800 | ||
Fair value of contingent consideration pursuant to Termination Agreement (1) | 30,100 | ||
Total consideration transferred | $ | 615,900 | |
(1) The contingent consideration arrangement pursuant to the Termination Agreement potentially requires the Company to pay up to $40.0 million of additional consideration to Teva upon the achievement of specified commercialization events related to methylphenidate hydrochloride. The $30.1 million fair value of the potential contingent consideration payments recognized on the acquisition date was estimated by applying a probability-weighted expected return methodology. The Company conducted a review of the underlying inputs and assumptions at December 31, 2017, and based on timing and probability of the product launch, and corresponding number of competitors expected to be in the market at both launch and the one-year anniversary of launch, the Company concluded that the fair value of its contingent consideration was $0.
Recognition and Measurement of Assets Acquired at Fair Value
The Company has allocated the purchase price for the Teva Transaction based upon the estimated fair value of the assets acquired at the date of acquisition.
The following is an estimate of the fair value of the intangible and tangible assets acquired in connection with the Teva Transaction on the closing date of August 3, 2016 (in thousands):
Estimated Fair Value | Weighted-Average Estimated Useful Life | |||
Marketed product rights | $ | 455,529 | 19 years | |
Acquired IPR&D product rights (1) | 157,503 | n/a | ||
Total intangible assets | 613,032 | |||
Inventory - raw materials | 2,868 | |||
Total assets acquired | $ | 615,900 | ||
(1) IPR&D refers to the Company's in-process research and development product rights. Pursuant to the Termination Agreement, Teva returned to the Company its full commercial rights to its then pending ANDA for the generic equivalent to Concerta® (methylphenidate hydrochloride), a product the Company previously partnered with Teva USA under a Strategic Alliance Agreement dated June 27, 2001, as amended. As a result, the Company recognized an intangible asset of $78.9 million related to the reacquired IPR&D. The Company engaged a third-party valuation specialist to measure the value of the reacquired product right using a discounted cash flow analysis. The asset was determined to be indefinite-lived based on the market participant methodology prescribed in ASC 805.
F-21
The estimated fair value of the IPR&D and identifiable intangible assets was determined using the “income approach,” which is a valuation technique that provides an estimate of the fair value of an asset based on market participant expectations of the cash flows an asset would generate over its remaining useful life. The assumptions, including the expected projected cash flows, utilized in the purchase price allocation and in determining the purchase price were based on management's best estimates as of the closing date of the Teva Transaction on August 3, 2016. Some of the more significant assumptions inherent in the development of those asset valuations include the estimated net cash flows for each year for each asset or product (including net revenues, cost of sales, research and development costs, selling and marketing costs and working capital / contributory asset charges), the appropriate discount rate to select in order to measure the risk inherent in each future cash flow stream, the assessment of each asset’s life cycle, the potential regulatory and commercial success risks, competitive trends impacting the asset and each cash flow stream, as well as other factors. The discount rate used to arrive at the present value at the closing date of the intangible assets was 6.7%. No assurances can be given that the underlying assumptions used to prepare the discounted cash flow analysis will not change. For these and other reasons, actual results may vary significantly from estimated results. During the year ended December 31, 2017 and 2016, the Company recognized impairment charges of $213.9 million and $308.4 million, respectively, related to the intangible assets from the Teva Transaction as described in "Note 8. Intangible Assets and Goodwill."
Revenues and Earnings for Acquired Product Lines
Included in the Company's consolidated statement of operations for the year ended December 31, 2016 were revenues of $44.8 million and a net loss of $244.7 million (including $308.4 million of impairment charges - See "Note 8. Intangible Assets and Goodwill"), representing the results of operations for the Acquired Product Lines from the Teva Transaction from the August 3, 2016 closing date through December 31, 2016.
Unaudited Pro Forma Results of Operations
The unaudited pro forma combined results of operations for the years ended December 31, 2016 and 2015 (assuming the closing of the Teva Transaction occurred on January 1, 2015) are as follows (in thousands):
Years Ended December 31, | ||||||||
2016 | 2015 | |||||||
Total revenues | $ | 927,593 | $ | 1,025,598 | ||||
Net (loss) income | (450,190 | ) | 70,057 | |||||
The pro forma adjustments reflected herein include only those adjustments that are directly attributable to the Teva Transaction, factually supportable and expected to have a continuing impact on the Company. The pro forma results have been prepared for comparative purposes only and are not necessarily indicative of the actual results of operations had the closing of the Teva Transaction taken place on January 1, 2015. Furthermore, the pro forma results do not purport to project the future results of operations of the Company.
The unaudited pro forma information reflects primarily the following adjustments:
•Adjustments to amortization expense related to identifiable intangible assets acquired;
•Adjustments to interest expense to reflect the Company's Term Loan Facility (described in “Note 10. Debt”); and
• | Adjustments to selling, general and administrative expense related to transaction costs directly attributable to the transaction include the elimination of $3.1 million of charges in the pro forms results for the year ended December 31, 2016 which have been included in the pro forma results for the year ended December 31, 2015. |
All of the items above were adjusted for the applicable tax impact.
F-22
Tower Acquisition
On March 9, 2015, the Company completed the Tower Acquisition for a purchase price of $691.3 million, net of $41.5 million of cash acquired and including the repayment of indebtedness of Tower and Lineage and post-closing working capital adjustments. The privately-held companies specialized in the development, manufacture and commercialization of complex generic and branded pharmaceutical products. The Tower Acquisition included the Company's acquisition of all of the outstanding shares of common stock of Tower and Lineage, pursuant to the Stock Purchase Agreement dated as of October 8, 2014, by and among the Company, Tower, Lineage, Roundtable Healthcare Partners II, L.P., Roundtable Healthcare Investors II, L.P., and the other parties thereto, including holders of certain options and warrants to acquire the common stock of Tower or Lineage. In connection with the Tower Acquisition, the options and warrants of Tower and Lineage that were outstanding at the time of the acquisition were cancelled. The Company incurred acquisition-related costs of $10.9 million, of which $6.7 million were incurred during the year ended December 31, 2015 and were included in selling, general and administrative expenses in the Company's consolidated statement of operations for that period. In connection with the Tower Acquisition, the Company recorded an accrual for severance and related termination costs of $2.4 million during 2015 related to the elimination of approximately 10 positions at the acquired companies. The Company paid all severance and related termination costs related to the Tower Acquisition as of the end of the second quarter of 2016.
The Tower Acquisition allowed the Company to expand its commercialized generic and branded product portfolios. The Company has also leveraged its sales and marketing organization to promote the marketed products acquired.
Purchase Accounting and Consideration
The Company has accounted for the Tower Acquisition as a business combination under the acquisition method of accounting. The Company allocated the purchase price for the transaction based upon the fair value of net assets acquired and liabilities assumed at the date of acquisition.
Recognition and Measurement of Assets Acquired and Liabilities Assumed at Fair Value
The following tables summarize the final fair values of the tangible and identifiable intangible assets acquired and liabilities assumed in the Tower Acquisition at the closing date, net of cash acquired of $41.5 million (in thousands):
Accounts receivable (1) | $ | 56,851 | |
Inventory | 31,259 | ||
Income tax receivable and other prepaid expenses | 2,407 | ||
Property, plant and equipment | 27,540 | ||
Intangible assets | 632,600 | ||
Intangible assets held for sale | 4,000 | ||
Goodwill | 179,755 | ||
Deferred income taxes | 37,041 | ||
Other non-current assets | 3,844 | ||
Total assets acquired | 975,297 | ||
Current liabilities | 67,584 | ||
Deferred tax liabilities | 210,005 | ||
Other non-current liabilities | 6,360 | ||
Total liabilities assumed | 283,949 | ||
Cash paid, net of cash acquired | $ | 691,348 | |
(1) | The accounts receivable acquired in the Tower Acquisition had a fair value of $56.9 million, including an allowance for doubtful accounts of $9.0 million, which represented the Company’s best estimate on March 9, 2015 (the closing date of the transaction) of the contractual cash flows not expected to be collected by the acquired companies. |
F-23
Intangible Assets
The following table identifies the Company’s allocations, by category, of the Tower Acquisition purchase price to the intangible assets acquired as of the closing date (in thousands):
Estimated Fair Value | Weighted-Average Estimated Useful Life (years) | ||||
Marketed product rights | $ | 381,100 | 13 | ||
Royalty rights | 80,800 | 12 | |||
Acquired IPR&D product rights | 170,700 | n/a | |||
Total intangible assets | $ | 632,600 | |||
The estimated fair value of the IPR&D and identifiable intangible assets was determined using the “income approach,” which is a valuation technique that provides an estimate of the fair value of an asset based on market participant expectations of the cash flows an asset would generate over its remaining useful life. Some of the more significant assumptions inherent in the development of those asset valuations include the estimated net cash flows for each year for each asset or product (including net revenues, cost of sales, research and development costs, selling and marketing costs and working capital / contributory asset charges), the appropriate discount rate to select in order to measure the risk inherent in each future cash flow stream, the assessment of each asset’s life cycle, the potential regulatory and commercial success risks, competitive trends impacting the asset and each cash flow stream as well as other factors. The discount rates used to arrive at the present value at the acquisition date of currently marketed products was 15%. For in-process research and development, the discount rate used was 16% to reflect the internal rate of return and incremental commercial uncertainty in the cash flow projections. No assurances can be given that the underlying assumptions used to prepare the discounted cash flow analysis will not change. For these and other reasons, actual results may vary significantly from estimated results.
Goodwill
The Company recorded approximately $179.8 million of goodwill in connection with the Tower Acquisition, some of which will not be tax-deductible. Goodwill of $59.7 million was assigned to the Impax Specialty Pharma segment and $120.1 million was assigned to the Impax Generics segment. Factors that contributed to the Company’s recognition of goodwill include the Company’s intent to expand its generic and branded pharmaceutical product portfolios and to acquire certain benefits from the Tower and Lineage product pipelines, in addition to the anticipated synergies that the Company expects to generate from the acquisition.
Unaudited Pro Forma Results of Operations
The unaudited pro forma combined results of operations for the year ended December 31, 2015 (assuming the closing of the Tower Acquisition occurred on January 1, 2014) are as follows (in thousands):
Year Ended December 31, 2015 | |||
Total revenues | $ | 892,906 | |
Net income | $ | 54,285 | |
The pro forma adjustments reflected herein include only those adjustments that are directly attributable to the Tower Acquisition, factually supportable and expected to have a continuing impact on the Company. The pro forma results have been prepared for comparative purposes only and are not necessarily indicative of the actual results of operations had the closing of the Tower Acquisition taken place on January 1, 2014. Furthermore, the pro forma results do not purport to project the future results of operations of the Company.
F-24
The unaudited pro forma information reflects primarily the following adjustments:
• | Adjustments to amortization expense related to identifiable intangible assets acquired; |
• | Adjustments to depreciation expense related to property, plant and equipment acquired; |
• | Adjustments to interest expense to reflect the long-term debt held by Tower and Lineage paid out and eliminated at the closing and the Company's Senior Secured Credit Facilities (described in “Note 10. Debt”); |
• | Adjustments to cost of revenues related to the fair value adjustments in inventory sold, including elimination of $6.1 million for the year ended December 31, 2015; |
• | Adjustments to selling, general and administrative expense related to the elimination of severance and retention costs of $3.4 million incurred as part of the transaction; |
• | Adjustments to selling, general and administrative expense related to transaction costs directly attributable to the transaction include the elimination of $12.2 million of charges in the pro forma results for the year ended December 31, 2015; and |
• | Adjustments to reflect the elimination of $2.3 million in commitment fees related to the Company's $435.0 million term loan with Barclays Bank PLC (described in "Note 10. Debt") that were incurred during the year ended December 31, 2015. |
All of the items above were adjusted for the applicable tax impact.
4. FAIR VALUE MEASUREMENT AND FINANCIAL INSTRUMENTS
The carrying values of cash equivalents, accounts receivable, prepaid expenses and other current assets, and accounts payable in the Company’s consolidated balance sheets approximated their fair values as of December 31, 2017 and 2016 due to their short-term nature.
Certain of the Company’s financial instruments are measured at fair value using a three-level hierarchy that prioritizes the inputs used to measure fair value. This hierarchy maximizes the use of observable inputs and minimizes the use of unobservable inputs. The three levels of inputs used to measure fair value are as follows:
• | Level 1 - Inputs are quoted prices for identical instruments in active markets. |
• | Level 2 - Inputs are quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; or model-derived valuations whose inputs are observable or whose significant value drivers are observable. |
• | Level 3 - Inputs are unobservable and reflect the Company's own assumptions, based on the best information available, including the Company's own data. |
F-25
The carrying amounts and fair values of the Company’s financial instruments as of December 31, 2017 and 2016 are indicated below (in thousands):
As of December 31, 2017 | |||||||||||||||||||
Fair Value Measurement Based on | |||||||||||||||||||
Carrying Amount | Fair Value | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||||
Assets | |||||||||||||||||||
Deferred Compensation Plan assets(1) | $ | 43,023 | $ | 43,023 | $ | — | $ | 43,023 | $ | — | |||||||||
Liabilities | |||||||||||||||||||
Term Loan Facility due August 2021, current portion (2) | $ | 20,000 | $ | 20,000 | $ | — | $ | 20,000 | $ | — | |||||||||
Term Loan Facility due August 2021, long-term portion (2) | $ | 305,000 | $ | 305,000 | $ | — | $ | 305,000 | $ | — | |||||||||
2% Convertible Senior Notes due June 2022 (3) | $ | 600,000 | $ | 579,378 | $ | 579,378 | $ | — | $ | — | |||||||||
Deferred Compensation Plan liabilities (1) | $ | 33,413 | $ | 33,413 | $ | — | $ | 33,413 | $ | — | |||||||||
Contingent consideration, long-term portion (4) | $ | — | $ | — | $ | — | $ | — | $ | — | |||||||||
As of December 31, 2016 | |||||||||||||||||||
Fair Value Measurement Based on | |||||||||||||||||||
Carrying Amount | Fair Value | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||||
Assets | |||||||||||||||||||
Deferred Compensation Plan assets(1) | $ | 37,382 | $ | 37,382 | $ | — | $ | 37,382 | $ | — | |||||||||
Liabilities | |||||||||||||||||||
Term Loan Facility due August 2021, current portion (2) | $ | 20,000 | $ | 20,000 | $ | — | $ | 20,000 | $ | — | |||||||||
Term Loan Facility due August 2021, long-term portion (2) | $ | 375,000 | $ | 375,000 | $ | — | $ | 375,000 | $ | — | |||||||||
2% Convertible Senior Notes due June 2022 (3) | $ | 600,000 | $ | 469,800 | $ | 469,800 | $ | — | $ | — | |||||||||
Deferred Compensation Plan liabilities (1) | $ | 28,582 | $ | 28,582 | $ | — | $ | 28,582 | $ | — | |||||||||
Contingent consideration, long-term portion (4) | $ | 31,048 | $ | 31,048 | $ | — | $ | — | $ | 31,048 | |||||||||
(1) | The Deferred Compensation Plan liabilities are non-current liabilities recorded at the value of the amount owed to the plan participants, with changes in value recognized as compensation expense in the Company’s consolidated statements of operations. The calculation of the Deferred Compensation Plan obligation is derived from observable market data by reference to hypothetical investments selected by the participants and is included in the line item captioned “Other non-current liabilities” on the Company’s consolidated balance sheets. The Company invests participant contributions in corporate-owned life insurance (“COLI”) policies, for which the cash surrender value is included in the line item captioned “Other non-current assets” on the Company’s consolidated balance sheets. |
(2) | The difference between the amount shown as the carrying value in the above tables and the amount shown on the Company’s consolidated balance sheets as of December 31, 2017 and 2016 represents the unaccreted discount related to deferred debt issuance costs. |
F-26
(3) | The difference between the amount shown as the carrying value in the above tables and the amount shown on the Company’s consolidated balance sheets at December 31, 2017 and 2016 represents the unaccreted discounts related to deferred debt issuance costs and bifurcation of the conversion feature of the notes. |
(4) | Under the terms of the Termination Agreement related to the Teva Transaction as described in "Note 3. Business Acquisitions.", the Company could be contractually obligated to make payments up to $40.0 million based on the achievement of certain commercial and time-based milestones associated with its methylphenidate hydrochloride product. A discounted cash flow valuation model was used to value the contingent consideration using significant unobservable inputs, including the probability and timing of successful product launch, the expected number of product competitors in the market at the time of launch (as defined in the Termination Agreement) and the expected number of such competitors in the market on the one-year launch anniversary date. The Company conducted a review of the underlying inputs and assumptions at December 31, 2017, and based on timing and probability of the product launch, and corresponding number of competitors expected to be in the market at both launch and the one-year anniversary of launch, the Company concluded that the fair value of its contingent consideration is $0. |
The following table presents the changes in Level 3 instruments measured on a recurring basis for the years ended December 31, 2017 and 2016 (in thousands):
Years Ended December 31, | |||||||
Contingent consideration | 2017 | 2016 | |||||
Beginning balance | $ | 31,048 | $ | — | |||
Completion of Teva Transaction August 3, 2016 | — | 30,100 | |||||
Change in fair value included in earnings | (31,048 | ) | 948 | ||||
Ending balance | $ | — | $ | 31,048 | |||
5. ACCOUNTS RECEIVABLE
The composition of accounts receivable, net is as follows (in thousands):
December 31, 2017 | December 31, 2016 | ||||||
Gross accounts receivable (1) | $ | 634,059 | $ | 794,173 | |||
Less: Rebate reserve | (181,611 | ) | (293,816 | ) | |||
Less: Chargeback reserve | (136,891 | ) | (151,978 | ) | |||
Less: Distribution services reserve | (11,037 | ) | (18,318 | ) | |||
Less: Discount reserve | (14,344 | ) | (17,957 | ) | |||
Less: Uncollectible accounts reserve (2) | (49,423 | ) | (54,736 | ) | |||
Accounts receivable, net | $ | 240,753 | $ | 257,368 | |||
(1) | Includes estimated $44.3 million and $40.3 million as of December 31, 2017 and 2016, respectively, receivable due from Turing Pharmaceuticals AG ("Turing") for reimbursement of Daraprim® chargebacks and Medicaid rebate liabilities pursuant to an Asset Purchase Agreement between the Company and Turing dated August 7, 2015 (the "Turing APA"). In accordance with the terms of the Turing APA and in accordance with federal laws and regulations, the Company receives, and is initially responsible for processing and paying (subject to reimbursement by Turing), all chargebacks and rebates resulting from utilization by Medicaid, Medicare and other federal, state and local government programs, health plans and other health care providers for products sold under the Company's labeler code. Under the terms of the Turing APA, Turing is responsible for liabilities related to chargebacks and rebates that arise as a result of Turing's marketing or selling related activities in connection with Daraprim®. Refer to "Note 19. Legal and Regulatory Matters" for a description of the Company's suit against Turing related to, among other matters, Turing's failure to reimburse the Company for chargebacks and Medicaid rebate liabilities when due. |
F-27
(2) | As a result of the uncertainty of collection from Turing that developed during the first quarter of 2016, the Company recorded a reserve of $48.0 million as of March 31, 2016, which represented the full amount of the estimated receivable due from Turing. During the fourth quarter of 2016, the Company received a $7.7 million payment from Turing. During the year ended December 31, 2017, the Company increased the reserve balance by a net $4.0 million, consisting of a $5.0 million increase in the reserve resulting from additional Medicaid rebate claims received during the period and a $1.0 million reduction in the reserve balance resulting from payments received from Turing during the period. As of December 31, 2017, the $44.3 million estimated receivable due from Turing was fully reserved. |
A roll-forward of the rebate and chargeback reserves activity for the years ended December 31, 2017, 2016 and 2015 is as follows (in thousands):
Years Ended December 31, | |||||||||||
Rebate reserve | 2017 | 2016 | 2015 | ||||||||
Beginning balance | $ | 293,816 | $ | 265,229 | $ | 88,812 | |||||
Acquired balances | — | — | 75,447 | ||||||||
Provision recorded during the period for Impax Generics rebates | 642,447 | 756,774 | 571,642 | ||||||||
Credits issued during the period for Impax Generics rebates | (754,652 | ) | (728,187 | ) | (470,672 | ) | |||||
Ending balance | $ | 181,611 | $ | 293,816 | $ | 265,229 | |||||
The payment mechanisms for rebates in the Impax Generics and Impax Specialty Pharma divisions are different, which impacts the location on the Company's consolidated balance sheets. Impax Generics rebates are classified as "Accounts receivable, net" on the Company's consolidated balance sheets. Impax Specialty Pharma rebates are classified as "Accrued expenses" on the Company's consolidated balance sheets.
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Chargeback reserve | |||||||||||
Beginning balance | $ | 151,978 | $ | 102,630 | $ | 43,125 | |||||
Acquired balances | — | — | 24,532 | ||||||||
Provision recorded during the period | 1,212,039 | 1,011,400 | 833,157 | ||||||||
Credits issued during the period | (1,227,126 | ) | (962,052 | ) | (798,184 | ) | |||||
Ending balance | $ | 136,891 | $ | 151,978 | $ | 102,630 | |||||
6. INVENTORY
Inventory, net of carrying value reserves, as of December 31, 2017 and 2016 consisted of the following (in thousands):
December 31, 2017 | December 31, 2016 | ||||||
Raw materials | $ | 63,732 | $ | 53,808 | |||
Work in-process | 3,046 | 3,280 | |||||
Finished goods | 104,187 | 130,879 | |||||
Total inventory | 170,965 | 187,967 | |||||
Less: Non-current inventory | 12,494 | 12,737 | |||||
Total inventory-current, net | $ | 158,471 | $ | 175,230 | |||
Inventory carrying value reserves were $71.6 million and $38.0 million as of December 31, 2017 and 2016, respectively. Included in the $71.6 million of inventory reserves at December 31, 2017 was a pre-launch product inventory reserve of $20.5 million, primarily related to colesevelam, recognized during the third quarter of 2017.
F-28
The Company recognizes pre-launch inventories at the lower of its cost or the expected net selling price. Cost is determined using a standard cost method, which approximates actual cost, and assumes a FIFO flow of goods. Costs of unapproved products are the same as approved products and include materials, labor, quality control, and production overhead. When the Company concludes FDA approval is expected within approximately six months, the Company will generally begin to schedule manufacturing process validation studies as required by the FDA to demonstrate the production process can be scaled up to manufacture commercial batches. Consistent with industry practice, the Company may build quantities of pre-launch inventories of certain products pending required final FDA approval and/or resolution of patent infringement litigation, when, in the Company’s assessment, such action is appropriate to prepare for the anticipated commercial launch, FDA approval is expected in the near term, and/or the related litigation will be resolved in the Company’s favor. The capitalization of unapproved pre-launch inventory involves risks, including, among other items, FDA approval of product may not occur; approvals may require additional or different testing and/or specifications than used for unapproved inventory; and, in cases where the unapproved inventory is for a product subject to litigation, the litigation may not be resolved or settled in favor of the Company. If any of these risks were to materialize and the launch of the unapproved product delayed or prevented, then the net carrying value of unapproved inventory may be partially or fully reserved. Generally, the selling price of a generic pharmaceutical product is at discount from the corresponding brand product selling price. Typically, a generic drug is easily substituted for the corresponding branded product, and once a generic product is approved, the pre-launch inventory is typically sold within the subsequent three months. If the market prices become lower than the product inventory carrying costs, then the pre-launch inventory value is reduced to such lower market value. If the inventory produced exceeds the estimated market acceptance of the generic product and becomes short-dated, a carrying value reserve will be recorded. In all cases, the carrying value of the Company's pre-launch product inventory is lower than the respective estimated net selling prices. The carrying value of unapproved inventory less reserves was $19.3 million and $29.2 million at December 31, 2017 and 2016, respectively.
To the extent inventory is not scheduled to be utilized in the manufacturing process and/or sold within twelve months of the balance sheet date, it is included as a component of other non-current assets. Amounts classified as non-current inventory consist of raw materials, net of valuation reserves. Raw materials generally have a shelf life of approximately three to five years, while finished goods generally have a shelf life of approximately two years.
7. PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment, net of accumulated depreciation, consisted of the following (in thousands):
December 31, 2017 | December 31, 2016 | ||||||
Land | $ | 3,500 | $ | 5,603 | |||
Buildings and improvements | 96,775 | 174,303 | |||||
Equipment | 82,442 | 143,818 | |||||
Office furniture and equipment | 11,082 | 15,767 | |||||
Construction-in-progress | 46,622 | 50,191 | |||||
Property, plant and equipment, gross | 240,421 | 389,682 | |||||
Less: Accumulated depreciation | (115,608 | ) | (156,310 | ) | |||
Property, plant and equipment, net | $ | 124,813 | $ | 233,372 | |||
Depreciation expense was $38.3 million, $29.1 million and $25.5 million for the years ended December 31, 2017, 2016 and 2015, respectively.
Unpaid vendor invoices relating to purchases of property, plant and equipment of $3.1 million, $4.0 million and $4.5 million, which were accrued as of December 31, 2017, 2016 and 2015, respectively, have been excluded from the purchase of property, plant, and equipment and the change in accounts payable and accrued expenses in the Company’s consolidated statements of cash flows.
During the third quarter of 2017, the Company sold a storage warehouse in Hayward, California for $8.8 million in cash proceeds, representing the gross proceeds of $9.4 million less fees and costs related to the sale of approximately $0.6 million. Prior to the sale, the net book value of the storage warehouse was $4.1 million and was included in the Impax Generics segment. The gain of $4.7 million is included in gain on sale of assets in the Company's consolidated statement of operations.
F-29
During 2017, the Company closed its Middlesex, New Jersey manufacturing facility and in early 2018, the Company sold CorePharma, LLC, its wholly owned subsidiary that held the leases to the site . The Company additionally announced during 2017 that it had entered into a stock and asset purchase agreement with Bora Pharmaceuticals Co., Ltd., pursuant to which the Company agreed to sell Impax Laboratories (Taiwan), Inc., its wholly owned subsidiary which owns the manufacturing facility in Taiwan, R.O.C. The sale of Impax Taiwan subsequently closed in February 2018. Refer to "Note 15. Restructurings" for disclosures relating to these assets.
8. INTANGIBLE ASSETS AND GOODWILL
Intangible Assets
The Company's intangible assets include both finite lived and indefinite-lived assets. Finite lived intangible assets, consisting of marketed product rights and royalties received from product sales by the Company's third party partners, are amortized over the estimated useful life of the asset based on the pattern in which the economic benefits are expected to be consumed or otherwise used up or, if that pattern is not readily determinable, on a straight-line basis. The remaining weighted-average amortization period for the Company's finite lived intangible assets not yet fully amortized is 6.6 years as of December 31, 2017. Indefinite-lived intangible assets consist of acquired IPR&D product rights and acquired future royalty rights to be paid based on other companies’ net sales of products not yet approved. IPR&D assets acquired in a business combination are considered indefinite-lived until the completion or abandonment of the associated research and development efforts. Amortization over the estimated useful life will commence at the time of the respective product’s launch. If FDA approval to market the product is not obtained, the Company will immediately expense the related capitalized cost.
Finite lived intangible assets are tested for impairment when events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. All of the Company's indefinite-lived intangible assets are tested for impairment at least annually during the fourth quarter of the fiscal year, or more often if indicators of impairment are present. Impairment testing requires management to estimate the future undiscounted cash flows of an intangible asset using assumptions believed to be reasonable, but which are unpredictable and inherently uncertain. Actual future cash flows may differ from the estimates used in the impairment testing. The Company recognizes an impairment loss when and to the extent that the estimated fair value of an intangible asset is less than its carrying value.
The following tables show the gross carrying values and accumulated amortization, where applicable, of the Company’s intangible assets by type for the consolidated balance sheets presented (in thousands):
Marketed Product Rights | IPR&D and Royalties | Total Company | ||||||||||||||||
Gross Carrying Value | Accumulated Amortization | Intangible Assets, Net | Non-amortized Value | Intangible Assets, Net | ||||||||||||||
Balance as of December 31, 2015 | $ | 460,875 | $ | (83,095 | ) | $ | 377,780 | $ | 224,240 | $ | 602,020 | |||||||
Additions | (1) | 455,529 | — | 455,529 | 161,003 | 616,532 | ||||||||||||
Amortization | — | (56,489 | ) | (56,489 | ) | — | (56,489 | ) | ||||||||||
Commercial Launch | (2) | 97,300 | — | 97,300 | (97,300 | ) | — | |||||||||||
Impairment Charge | (3) | (488,632 | ) | — | (488,632 | ) | (52,965 | ) | (541,597 | ) | ||||||||
Balance as of December 31, 2016 | 525,072 | (139,584 | ) | 385,488 | 234,978 | 620,466 | ||||||||||||
Additions | — | 50 | 50 | |||||||||||||||
Amortization | — | (68,375 | ) | (68,375 | ) | — | (68,375 | ) | ||||||||||
Commercial Launch | (2) | 4,216 | — | 4,216 | (4,216 | ) | — | |||||||||||
Divestiture | (4) | (2,414 | ) | 2,414 | — | — | — | |||||||||||
Impairment Charge | (3) | (96,865 | ) | — | (96,865 | ) | (192,809 | ) | (289,674 | ) | ||||||||
Balance as of December 31, 2017 | $ | 430,009 | $ | (205,545 | ) | $ | 224,464 | $ | 38,003 | $ | 262,467 | |||||||
F-30
(1) | During the first quarter of 2016, the Company capitalized $3.5 million of milestone payments due to an affiliate of Teva under the terms of the Mebendazole Product Agreement related to the FDA's approval and the Company's subsequent commercial launch of Emverm® (mebendazole) 100 mg chewable tablets. See "Note 17. Alliance and Collaboration Agreements" for additional information related to the Mebendazole Product Agreement. |
During the third quarter of 2016, the Company recorded $613.0 million of intangible asset additions as a result of the Teva Transaction, of which $455.5 million were amortized, finite-lived marketed product rights and $157.5 million were non-amortized, indefinite-lived acquired IPR&D product rights. Refer to "Note 3. Business Acquisitions" for additional information on the Teva Transaction.
Pursuant to the Termination Agreement related to the Teva Transaction, the Company reacquired its full commercial rights to its then pending ANDA for the generic equivalent to Concerta® (methylphenidate hydrochloride), a product candidate the Company had acquired in the Tower Acquisition that the Company had previously partnered with Teva USA, by terminating each party's rights and obligations with respect to such product under the Strategic Alliance Agreement between the Company and Teva, as amended. Pursuant to the terms of the Strategic Alliance Agreement, each party would retain 50% of the gross profit realized upon sales of the product following approval. As such the Company's 50% interest in the product was previously considered a non-amortized, indefinite-lived acquired future royalty right owing to the fact that Teva would sell the product upon receiving FDA approval and pay the Company 50% of the gross profit realized. Upon the Company's reacquisition of the full rights in this product pursuant to the Termination Agreement, the $70.8 million asset value of the Company's 50% interest, determined at the time of the Tower Acquisition, was transferred to non-amortized, indefinite-lived acquired IPR&D products rights, as reflected in the tables above.
(2) | During the year ended December 31, 2017, the Company commercially launched two products acquired as IPR&D as part of the Teva Transaction and Tower Acquisition and, as a result, transferred the $4.2 million asset value from non-amortized, indefinite-lived acquired IPR&D product rights to amortized, finite lived marketed product rights. These assets will be amortized over an estimated useful life ranging from seven to eight years based on the pattern of economic benefit expected to be realized through 2025. |
As of December 31, 2015, the Emverm® acquired IPR&D product right had a carrying value of $82.8 million, which was the fair value assigned by the Company during the purchase price allocation accounting for the Tower Acquisition. As a result of the Company's commercial launch of the product during the first quarter of 2016, the Company transferred the total $86.3 million of asset value from non-amortized, indefinite-lived acquired IPR&D product rights to amortized, finite-lived marketed product rights and began amortization of the asset. The Emverm® marketed product right intangible asset will be amortized over an estimated useful life of nine years based on the pattern of economic benefit expected to be realized through 2024.
In addition to the intangible asset additions resulting from the Teva Transaction as described above, during the third quarter of 2016, the Company also commercially launched two products, resulting in the transfer of $11.0 million of asset value from non-amortized, indefinite-lived acquired IPR&D product rights to amortized, finite-lived marketed products rights.
(3) | For the year ended December 31, 2017 the Company recognized a total of $289.7 million of intangible asset impairment charges, of which $96.9 million were recognized in cost of revenues impairment charges and $192.8 million were recognized in in-process research and development impairment charges on the Company’s consolidated statement of operations. |
The $192.8 million in-process research and development impairment charge was attributable to four products, most of which were acquired in the Teva Transaction. The Company incurred a full impairment charge of $149.7 million during the fourth quarter of 2017 related to the Company's AB-rated methylphenidate hydrochloride (generic equivalent to Concerta) product. The validation efforts for the product, produced by the Company's third party manufacturer, were not immediately successful and will require additional time and effort which is anticipated to result in a delay in the launch of up to 12-15 months. The delayed launch is currently expected to result in reduced volume and lower pricing than originally anticipated due to increased competition, resulting in significantly lower expected future cash flows. The Company also reduced the forecasted market share for another IPR&D product due to the introduction of a similar product by a competitor which administers the same active drug ingredient but with a different mode of delivery resulting in a $37.0 million impairment charge incurred during the fourth quarter of 2017. The remainder of the impairment charges were primarily related to the delayed launch of two products which are currently expected to result in reduced volume after launch due to increased competition.
F-31
The $96.9 million cost of revenue impairment charge for currently marketed products was attributable to eight currently marketed products. The Company experienced even further price and volume erosion throughout the year without an offsetting increase in customer demand, resulting in significantly lower expected future cash flows. The impairment charge was related to six of the products acquired as part of the Teva Transaction and two products acquired as part of the Tower Acquisition.
During the second quarter of 2016, the Company recognized a total of $1.5 million of charges within cost of revenues impairment charges on the Company's consolidated statement of operations related to two currently marketed products, which were acquired as part of the Tower Acquisition, primarily due to active pharmaceutical ingredient ("API") supply issues and minimal sales activity, resulting in immediate discontinuation of one product and rapid phase-out of the other. Additionally, one of the Company's IPR&D generic products, also acquired as part of the Tower Acquisition, was determined to be impaired as a result of the commercial launch of a competitor's generic product, resulting in a $1.0 million charge to in-process research and development impairment charges on the Company's consolidated statement of operations.
Upon closing the Teva Transaction on August 3, 2016, the Company initiated the process of transferring and securing Teva’s and Allergan’s customers for the acquired products to its account. The Company assumed certain price concessions would occur following the closing, however, the Company elected to take additional price reductions on certain of the acquired products in order to retain key customers. These reductions produced significantly lower than expected operating cash flows from the Acquired Product Lines and triggered an impairment analysis. The Company's impairment analysis for the third quarter of 2016 resulted in the recognition of a total $251.0 million non-cash impairment charge to earnings. Of the total $251.0 million impairment charge, $248.0 million was recorded in cost of revenues impairment charges and $3.0 million was recorded in in-process research and development impairment charges, each in the Company’s consolidated statement of operations for the third quarter of 2016.
Certain other non-cash impairment charges unrelated to the Teva Transaction were also recorded in the third quarter of 2016. During the third quarter of 2016, the Company also recognized a total of $34.2 million of intangible asset impairment charges, of which $8.5 million was recognized in cost of revenues impairment charges on the Company's consolidated statement of operations and attributable to the full impairment of three marketed products and one third-party partnered product where the Company received royalties from the sale of such product. The affected products were manufactured in the Company's Middlesex, New Jersey facility, which the Company is in the process of closing as discussed in "Note 15. Restructurings." The products were discontinued for several reasons, including the inability to efficiently transfer technology to another manufacturing site, the inability to continue to secure API from third parties on a timely basis, and/or minimal current and projected sales activity. The remaining $25.7 million of impairment charges recognized by the Company during the third quarter of 2016 were recognized in in-process research and development impairment charges and related to two of the Company's IPR&D product rights acquired in the Tower Acquisition due to delays in expected start of commercialization and lower pricing amid highly competitive market conditions, resulting in lower expected future cash flows.
During the fourth quarter of 2016, the Company recognized a total of $253.9 million of intangible asset impairment charges, of which $230.6 million were recognized in cost of revenues impairment charges and $23.3 million were recognized in in-process research and development impairment charges on the Company's consolidated statement of operations. More than half of the total impairment charges incurred during the fourth quarter of 2016 was attributable to the Company’s epinephrine auto-injector product, which was acquired as part of the Tower Acquisition. The impairment charge on the epinephrine auto-injector product was triggered by current and projected price degradation as a result of changes in the pricing environment and additional competition. The Company also experienced even further price reductions on certain of the products acquired as part of the Teva Transaction during the fourth quarter of 2016, resulting in $57.4 million of additional intangible asset impairment charges, of which $53.7 million was recorded to cost of revenues impairment charges and $3.7 million was recorded to in-process research and development impairment charges. In addition, the Company recognized $36.3 million of intangible asset impairment related to its anthelmintic product franchise, of which $24.3 million was recorded to cost of revenues impairment charges and $12.0 million was recorded to in-process research and development impairment charges. The $24.3 million charge was attributable to lower than expected script volume for Emverm®. The $12.0 million charge recorded to in-process research and development during the fourth quarter of 2016 was attributable to a decision by the Company's management during the fourth quarter of 2016 to cease development on a next-generation version of Albenza® as a result of continued difficulties sourcing the API. The remainder of the fourth quarter of 2016 impairment charges were primarily attributable to the products acquired as part of the Tower Acquisition and resulted from lower current and/or forecasted pricing amid highly competitive market conditions, resulting in lower forecasted future cash flows.
F-32
(4) | During the second quarter of 2017, the Company divested 29 ANDAs and one NDA for non-strategic approved generic products, the vast majority of which were not marketed, and all acquired as part of the Tower Acquisition, for gross proceeds of $12.0 million. These intangible assets had a fully amortized gross carrying value of $2.4 million at the time of the sale. The Company incurred $0.1 million of legal expense in connection with the divestiture, resulting in a net gain on sale of $11.9 million recognized as gain on sale of assets on the Company’s consolidated statement of operations. |
Amortization
The Company recognized amortization expense of $68.4 million, $56.5 million and $40.2 million for the years ended December 31, 2017, 2016 and 2015, respectively, in cost of revenues in the consolidated statements of operations presented.
The following table shows the expected future amortization of the Company’s finite lived intangible assets as of December 31, 2017 (in thousands):
For the years ending December 31, | Amortization Expense | ||
2018 | $ | 56,431 | |
2019 | 46,771 | ||
2020 | 36,140 | ||
2021 | 23,778 | ||
2022 | 19,701 | ||
Thereafter | 41,643 | ||
Total | $ | 224,464 | |
Sale of Daraprim® to Turing
In July 2015, the Company received an unsolicited offer from Turing to purchase the U.S. rights to Daraprim®, one of the marketed products acquired in the Tower Acquisition, as well as the active pharmaceutical ingredient for the product and the finished goods inventory on hand. The sale closed on August 7, 2015, and the Company received proceeds of $55.5 million at closing. The net book value of the Daraprim® product rights at the time of sale was $9.3 million , and the Company recognized a gain on the sale of the intangible asset of $45.6 million , net of expenses. Pursuant to the terms of the Asset Purchase Agreement between the Company and Turing dated August 7, 2015 (the "Turing APA"), the Company also granted a limited license to sell the existing Daraprim® product under the Company’s labeler code with the Company’s trade dress.
In accordance with the terms of the Turing APA and in accordance with federal laws and regulations, the Company received and was initially responsible for processing and paying (subject to reimbursement by Turing), all chargebacks and rebates resulting from utilization by Medicaid, Medicare and other federal, state and local governmental programs, health plans and other health care providers for product sold under the Company’s labeler code. Under the terms of the Turing APA, Turing is responsible for liabilities related to chargebacks and rebates that arise as a result of Turing’s marketing or selling related activities in connection with Daraprim®.
Goodwill
Goodwill had a carrying value on the Company’s consolidated balance sheets of $207.3 million and $207.3 million as of December 31, 2017 and 2016, respectively. As of December 31, 2017, the Company attributed $147.6 million and $59.7 million to the Impax Generics division and the Impax Specialty Pharma division, respectively. The Company concluded based on the results of the annual testing performed that the carrying value of goodwill was not impaired as of December 31, 2017 or 2016.
F-33
9. ACCRUED EXPENSES
The following table sets forth the Company’s accrued expenses (in thousands):
December 31, 2017 | December 31, 2016 | ||||||
Payroll-related expenses | $ | 38,415 | $ | 37,986 | |||
Product returns | 76,293 | 72,888 | |||||
Accrued shelf stock | 7,525 | 7,032 | |||||
Government rebates | 73,970 | 72,063 | |||||
Legal and professional fees | 14,005 | 8,395 | |||||
Estimated Teva and Allergan chargebacks and rebates (1) | 13,277 | 14,813 | |||||
Accrued profit sharing and royalty expenses | 8,373 | 13,642 | |||||
Other | 16,269 | 17,834 | |||||
Total accrued expenses | $ | 248,127 | $ | 244,653 | |||
(1) | As discussed in "Note 3. Business Acquisitions," in connection with the Teva Transaction, the Company, Teva and Allergan agreed to certain transition related services pursuant to which the Company agreed to manage the payment process for certain commercial chargebacks and rebates on behalf of Teva and Allergan related to products each of Teva and Allergan sold into the channel prior to the Company's acquisition of the products. On August 18, 2016, the Company received a payment totaling $42.4 million from Teva and Allergan, which represented their combined estimate of the amount of commercial chargebacks and rebates to be paid by the Company on their behalf to wholesalers who purchased products from Teva and Allergan prior to the closing. Pursuant to the agreed upon transition services, Teva and Allergan are obligated to reimburse the Company for additional payments related to chargebacks and rebates for products they sold into the channel prior to the closing and made on their behalf in excess of the $42.4 million. If the total payments made by the Company on behalf of Teva and Allergan are less than $42.4 million, the Company is obligated to refund the difference to Teva and/or Allergan. As of December 31, 2017, the Company had paid $29.1 million related to chargebacks and rebates as described above and $13.3 million remained in accrued expenses on the Company's consolidated balance sheet. |
Product Returns
The Company maintains a return policy to allow customers to return product within specified guidelines. The Company estimates a provision for product returns as a percentage of gross sales based upon historical experience for sales made through its Impax Generics and Impax Specialty Pharma sales channels. Sales of product under the Private Label, Rx Partner and OTC Partner alliance, collaboration and supply agreements are not subject to returns.
A rollforward of the return reserve activity for the years ended December 31, 2017, 2016 and 2015 is as follows (in thousands):
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Returns reserve | |||||||||||
Beginning balance | $ | 72,888 | $ | 48,950 | $ | 27,174 | |||||
Acquired balances | — | — | 11,364 | ||||||||
Provision related to sales recorded in the period | 47,709 | 52,383 | 43,967 | ||||||||
Credits issued during the period | (44,304 | ) | (28,445 | ) | (33,555 | ) | |||||
Ending balance | $ | 76,293 | $ | 72,888 | $ | 48,950 | |||||
F-34
10. DEBT
Royal Bank of Canada Credit Facilities
On August 3, 2016, the Company entered into a restatement agreement with Royal Bank of Canada, as administrative agent, and the lenders and guarantors party thereto (the "Restatement Agreement"). The Restatement Agreement amends and restates the Company's existing Revolving Credit Facility Agreement (as amended and restated and amended to date, the "Amended and Restated Credit Agreement") to, among other things, (i) add a term loan feature to allow for the borrowing of up to $400.0 million of term loans (the "Term Loan Facility") by the Company in accordance with the terms of the Amended and Restated Credit Agreement, (ii) increase the aggregate principal amount of revolving loans permitted under the Amended and Restated Credit Agreement (the "Revolving Credit Facility," and, together with the Term Loan Facility, the "RBC Credit Facilities"), from $100.0 million to $200.0 million; and (iii) extend the maturity date of the Revolving Credit Facility from August 4, 2020 to August 3, 2021. On March 27, 2017, the Company entered into Amendment No. 1 by and among the Company, Royal Bank of Canada, as administrative agent, and the lenders party thereto (the “Amendment”) to the Amended and Restated Credit Agreement.
Borrowings under the Amended and Restated Credit Agreement will accrue interest at a rate equal to LIBOR or the base rate, plus an applicable margin. The applicable margin may be increased or reduced by 0.25% based on the Company's total net leverage ratio. Up to $12.5 million of the Revolving Credit Facility is available for issuance of letters of credit and any such letters of credit will reduce the amount available under the Revolving Credit Facility on a dollar-for-dollar basis. The Company is required to pay a commitment fee to the lenders on the average daily unused portion of the Revolving Credit Facility at 0.50% or 0.375% per annum, depending on the Company's total net leverage ratio.
The Amended and Restated Credit Agreement contains certain negative covenants (subject to exceptions, materiality thresholds and other allowances) including, without limitation, negative covenants that limit the Company's and its restricted subsidiaries' ability to incur additional debt, guarantee other obligations, grant liens on assets, make loans, acquisitions or other investments, dispose of assets, make optional payments in connection with or modify certain debt instruments, pay dividends or make other payments on capital stock, engage in mergers or consolidations, enter into arrangements that restrict the Company's and its restricted subsidiaries' ability to pay dividends or grant liens, engage in transactions with affiliates, or change its fiscal year. Prior to the effective date of the Amendment on March 27, 2017, the Amended and Restated Credit Agreement also included a financial maintenance covenant whereby the Company must not permit its total net leverage ratio in any 12-month period to exceed 5.00:1.00, as tested at the end of each fiscal quarter. Effective as of March 27, 2017 and pursuant to the Amendment, the total net leverage ratio financial covenant was replaced with a new senior secured net leverage ratio financial covenant. Pursuant to the Amendment, the Company must not permit its senior secured net leverage ratio to exceed 2.50:1.00 and the interest coverage ratio to be less than 3.00:1.00, in each case in any 12-month period, as tested at the end of each fiscal quarter. The Company was in compliance with all of its covenants under the Amended and Restated Credit Agreement as of December 31, 2017.
The Amended and Restated Credit Agreement contains events of default, including, without limitation (subject to customary grace periods and materiality thresholds), events of default upon (i) the failure to make payments pursuant to the terms of the Amended and Restated Credit Agreement, (ii) violation of covenants, (iii) incorrectness of representations and warranties, (iv) cross-default and cross-acceleration to other material indebtedness, (v) bankruptcy events, (vi) material monetary judgments (to the extent not covered by insurance), (vii) certain matters arising under the Employee Retirement Income Security Act of 1974, as amended, that could reasonably be expected to result in a material adverse effect, (viii) the actual or asserted invalidity of the documents governing the RBC Credit Facilities, any material guarantees or the security interests (including priority thereof) required under the Amended and Restated Credit Agreement and (ix) the occurrence of a change of control (as defined therein). Upon the occurrence of certain events of default, the obligations under the Amended and Restated Credit Agreement may be accelerated and any remaining commitments thereunder may be terminated.
The full amount of proceeds from the Term Loan Facility of $400.0 million, along with $196.4 million of cash were used to finance the Teva Transaction (including transaction costs) at closing on August 3, 2016. As of December 31, 2017, $199.7 million Revolving Credit Facility remains available to the Company for working capital and other general corporate purposes.
F-35
In connection with the Term Loan Facility, the Company incurred $11.0 million of debt issuance costs for banking, legal and accounting fees and other expenses during the third quarter of 2016. In connection with the Amendment, the Company incurred $0.8 million of debt issuance costs for banking fees during the first quarter of 2017. These debt issuance costs were recorded on the Company's consolidated balance sheet as a reduction to the current and long-term portions of debt related to the Term Loan Facility. These deferred debt issuance costs will be accreted to interest expense over the term of the debt using the effective interest method. In connection with the increase in the aggregate principal amount of revolving loans permitted under the Revolving Credit Facility, the Company incurred $0.8 million of debt issuance costs for banking fees which were recorded as an asset with current and long-term portions on the Company's consolidated balance sheet. These deferred debt issuance costs, in addition to the $0.3 million balance remaining from the initial $100.0 million revolving credit facility, will be amortized to interest expense over the term of the Revolving Credit Facility using the straight-line method.
For the year ended December 31, 2017, the Company recognized $17.7 million of interest expense related to the Term Loan Facility, of which $15.5 million was cash and $2.2 million was non-cash accretion of the debt discount recorded for deferred debt issuance costs. For the period of August 3, 2016 through December 31, 2016, the Company recognized $6.9 million of interest expense related to the Term Loan Facility, of which $6.0 million was cash and $0.9 million was non-cash accretion of the debt discount recorded for deferred debt issuance costs. As of December 31, 2017, the Term Loan Facility had a carrying value of $317.5 million, of which $17.8 million is classified as current debt and $299.7 million is classified as long-term debt on the Company's consolidated balance sheets. The Term Loan Facility requires the Company to make quarterly principal payments of $5.0 million beginning from December 2016 through June 2021, and the remaining principal balance is payable in August 2021. As of December 31, 2017, the outstanding principal amount for the Term Loan Facility was $325.0 million.
Loss on Early Extinguishment of Debt - Voluntary Prepayment of $50.0 Million of Principal - RBC Term Loan Facility
On February 28, 2017, the Company made a voluntary prepayment in the amount of $50.3 million under its Term Loan Facility, representing $50.0 million of principal amount and $0.3 million of accrued interest thereon. As a result of this voluntary prepayment, for the quarter ended March 31, 2017, the Company recorded a loss on early extinguishment of debt of $1.2 million to write-off a pro rated portion of the related unaccreted debt issuance costs.
2% Convertible Senior Notes due June 2022
On June 30, 2015, the Company issued an aggregate principal amount of $600.0 million of 2.00% Convertible Senior Notes due June 2022 (the “Notes”) in a private placement offering, which are the Company’s senior unsecured obligations. The Notes were issued pursuant to an Indenture dated June 30, 2015 (the “Indenture”) between the Company and Wilmington Trust, N.A., as trustee. The Indenture includes customary covenants and sets forth certain events of default after which the Notes may be due and payable immediately. The Notes will mature on June 15, 2022, unless earlier redeemed, repurchased or converted. The Notes bear interest at a rate of 2.00% per year, and interest is payable semiannually in arrears on June 15 and December 15 of each year, beginning on December 15, 2015.
The conversion rate for the Notes is initially set at 15.7858 shares per $1,000 of principal amount, which is equivalent to an initial conversion price of $63.35 per share of the Company’s common stock. If a Make-Whole Fundamental Change (as defined in the Indenture) occurs or becomes effective prior to the maturity date and a holder elects to convert its Notes in connection with the Make-Whole Fundamental Change, the Company is obligated to increase the conversion rate for the Notes so surrendered by a number of additional shares of the Company’s common stock as prescribed in the Indenture. Additionally, the conversion rate is subject to adjustment in the event of an equity restructuring transaction such as a stock dividend, stock split, spinoff, rights offering, or recapitalization through a large, nonrecurring cash dividend (“standard antidilution provisions,” per FASB ASC 815-40).
Contracts in Entity’s Own Equity ("ASC 815-40")).
The Notes are convertible at the option of the holders at any time prior to the close of business on the business day immediately preceding December 15, 2021 only under the following circumstances:
(i) | If during any calendar quarter commencing after the quarter ending September 30, 2015 (and only during such calendar quarter) the last reported sale price of the Company’s common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than 130% of the conversion price on each applicable trading day; or |
F-36
(ii) | If during the five business day period after any 10 consecutive trading day period (the “measurement period”) in which the trading price per $1,000 of principal amount of Notes for each trading day of the measurement period was less than 98% of the product of the last report sale price of the Company’s common stock and the conversion rate on each such trading day; or |
(iii) | Upon the occurrence of corporate events specified in the Indenture. |
On or after December 15, 2021 until the close of business on the second scheduled trading day immediately preceding the maturity date, the holders may convert their Notes at any time, regardless of the foregoing circumstances. The Company may satisfy its conversion obligation by paying or delivering, as the case may be, cash, shares of the Company’s common stock, or a combination of cash and shares of the Company’s common stock, at the Company’s election and in the manner and subject to the terms and conditions provided in the Indenture.
Concurrently with the offering of the Notes and using a portion of the proceeds from the sale of the Notes, the Company entered into a series of convertible note hedge and warrant transactions (the “Note Hedge Transactions” and “Warrant Transactions”) which are designed to reduce the potential dilution to the Company’s stockholders and/or offset the cash payments the Company is required to make in excess of the principal amount upon conversion of the Notes. The Note Hedge Transactions and Warrant Transactions are separate transactions, in each case, entered into by the Company with a financial institution and are not part of the terms of the Notes. These transactions will not affect any holder’s rights under the Notes, and the holders of the Notes have no rights with respect to the Note Hedge Transactions and Warrant Transactions. See “Note 11. Stockholders’ Equity” for additional information.
At the June 30, 2015 issuance date of the Notes, the Company did not have the necessary number of authorized but unissued shares of its common available to share-settle the conversion option of the Notes. Therefore, in accordance with guidance found in FASB ASC 470-20, Debt with Conversion and Other Options, and FASB Topic ASC 815-15, Embedded Derivatives, the conversion option of the Notes was deemed an embedded derivative requiring bifurcation from the Notes (host contract) and separate accounting as a derivative liability. The fair value of the conversion option derivative liability at June 30, 2015 was $167.0 million, which was recorded as a reduction to the carrying value of the debt and will be accreted to interest expense over the term of the debt using the effective interest method. Although the Company subsequently amended the Company's Restated Certificate of Incorporation to increase the authorized number of shares of the Company's common stock in December 2015, the debt discount remained and continues to be accreted to interest expense. See "Note 11. Stockholders' Equity" for additional information.
In connection with the issuance of the Notes, the Company incurred $18.7 million of debt issuance costs for banking, legal and accounting fees and other expenses. This amount was also recorded on the Company’s balance sheet as a reduction to the carrying value of the debt and is being accreted to interest expense over the term of the debt using the effective interest method.
For the years ended December 31, 2017 and 2016, the Company recognized $35.5 million and $33.8 million, respectively, of interest expense related to the Notes, of which $12.0 million and $12.0 million, respectively, was cash and $23.5 million and $21.8 million, respectively, was non-cash accretion of the debt discounts recorded. As the Notes mature in 2022, they have been classified as long-term debt on the Company’s consolidated balance sheets, with a carrying value of $469.9 million and $446.4 million as of December 31, 2017 and 2016, respectively.
Loss on Early Extinguishment of Debt – Barclays $435.0 million Term Loan
In connection with the Tower Acquisition during the first quarter of 2015, the Company entered into a $435.0 million senior secured term loan facility (the “Barclays Term Loan”) and a $50.0 million senior secured revolving credit facility (the “Barclays Revolver” and collectively with the Barclays Term Loan, the “Barclays Senior Secured Credit Facilities”), pursuant to a credit agreement, dated as of March 9, 2015, by and among the Company, the lenders party thereto from time to time and Barclays Bank PLC ("Barclays"), as administrative and collateral agent (the “Barclays Credit Agreement”). In connection with the Barclays Senior Secured Credit Facilities, the Company incurred debt issuance costs for banking, legal and accounting fees and other expenses of $17.8 million, which were previously reflected as a discount to the carrying value of the debt on the Company's consolidated balance sheet in accordance with ASU 2015-03. Prior to repayment of the Barclays Term Loan on June 30, 2015, this debt discount was accreted to interest expense over the term of the loan using the effective interest rate method.
On June 30, 2015, the Company used $436.4 million of the proceeds from the sale of the Notes to repay the $435.0 million of principal and $1.4 million of accrued interest due on its Barclays Term Loan under the Barclays Credit Agreement. In connection with this repayment of the loan, for the quarter ended June 30, 2015, the Company recorded a loss on early extinguishment of debt of $16.9 million related to the unaccreted portion of the debt discount.
F-37
For the six months ended June 30, 2015, the Company incurred total interest expense related to the Barclays Term Loan of $10.7 million, of which $9.8 million was cash and $0.9 million was non-cash accretion of the debt discount recorded. In addition, included in interest expense for 2015 is a $2.3 million ticking fee paid to Barclays during the first quarter of 2015, prior to the funding of the Barclays Senior Secured Credit Facilities on March 9, 2015, to lock in the financing terms from the lenders’ commitment of the Barclays Term Loan until the actual allocation of the loan occurred at the closing of the Tower Acquisition.
Future principal maturities of December 31, 2017 are as follows (in thousands):
Years ending December 31, | |||
2018 | $ | 20,000 | |
2019 | 20,000 | ||
2020 | 20,000 | ||
2021 | 265,000 | ||
2022 | 600,000 | ||
Total | $ | 925,000 | |
11. STOCKHOLDERS’ EQUITY
Preferred Stock
Pursuant to its Restated Certificate of Incorporation (the “Certificate of Incorporation”), the Company is authorized to issue 2,000,000 shares of “blank check” preferred stock, $0.01 par value per share, which enables the Board of Directors, from time to time, to create one or more new series of preferred stock. Each series of preferred stock issued can have the rights, preferences, privileges and restrictions designated by the Board of Directors. The issuance of any new series of preferred stock could affect, among other things, the dividend, voting, and liquidation rights of the Company’s common stock. The Company had no preferred stock issued or outstanding as of December 31, 2017 or 2016.
Common Stock
Pursuant to its Certificate of Incorporation, the Company is authorized to issue 150,000,000 shares of common stock, $0.01 par value per share, of which 74,234,076 shares have been issued and 73,990,347 shares were outstanding as of December 31, 2017. In addition, the Company had reserved for issuance the following amounts of shares of its common stock for the purposes described below as of December 31, 2017 (in thousands):
Shares issued | 74,234 | |
Stock options outstanding (1) | 3,175 | |
Conversion of Notes payable (2) | 9,471 | |
Warrants outstanding (see below) | 9,471 | |
Total shares of common stock issued and reserved for issuance | 96,351 | |
(1) See “Note 13. Share-Based Compensation”
(2) See “Note 10. Debt”
Warrants
As discussed in “Note 10. Debt”, on June 30, 2015, the Company entered into a series of Note Hedge Transactions and Warrant Transactions with a financial institution which are designed to reduce the potential dilution to the Company’s stockholders and/or offset the cash payments the Company is required to make in excess of the principal amount upon conversion of the Notes. Pursuant to the Warrant Transactions, the Company sold to a financial institution 9.47 million warrants to purchase the Company’s common stock, for which it received proceeds of $88.3 million. The warrants have an exercise price of $81.277 per share (subject to adjustment), are immediately exercisable, and have an expiration date of September 15, 2022.
F-38
Additional Paid-In Capital
Pursuant to the Note Hedge Transactions, the Company purchased from a financial institution 0.6 million call options on the Company's common stock, for which it paid consideration of $147.0 million. Each call option entitles the Company to purchase 15.7858 shares of the Company's common stock at an exercise price of $63.35 per share, is immediately exercisable, and has an expiration date of June 15, 2022, subject to earlier exercise. At the time of the Note Hedge Transactions, because of an insufficient number of authorized but unissued shares of the Company's common stock, these call options did not meet the criteria for equity classification under FASB ASC Topic 815-40, Derivatives and Hedging and were accounted for as a derivative asset.
As of December 8, 2015, pursuant to the Company's amendment to its Certificate of Incorporation to increase the number of authorized shares of common stock, the call options purchased pursuant to the Note Hedge Transactions (formerly a derivative asset) and the conversion option of the Notes (formerly an embedded derivative liability) were reclassified to equity in additional paid-in capital. The net effect of the reclassification of these derivatives was a $21.0 million, net of tax, increase in additional paid-in capital reflected on the Company's December 31, 2015 consolidated balance sheet.
During the year ended December 31, 2015, the Company recognized in its consolidated statement of operations $13.0 million of net expense related to the change in the fair value of the former derivative asset and liability. There was no comparable expense recognized in 2016 or 2017.
12. EARNINGS PER SHARE
The Company's basic earnings per common share (“EPS”) is computed by dividing net (loss) income available to the Company’s common stockholders (as presented on the consolidated statements of operations) by the weighted-average number of shares of the Company’s common stock outstanding during the period. The Company’s restricted stock awards (non-vested shares) are issued and outstanding at the time of grant but are excluded from the Company’s computation of weighted-average shares outstanding in the determination of basic EPS until vesting occurs.
For purposes of calculating diluted EPS, the denominator includes both the weighted-average number of shares of common stock outstanding and the number of common stock equivalents if the inclusion of such common stock equivalents would be dilutive. Dilutive common stock equivalents potentially include warrants, stock options and non-vested restricted stock awards using the treasury stock method and the number of shares of common stock issuable upon conversion of the Company’s outstanding convertible notes payable. In the case of the Company’s outstanding convertible notes payable, the diluted EPS calculation is further affected by an add-back of interest expense, net of tax, to the numerator under the assumption that the interest would not have been incurred if the convertible notes had been converted into common stock.
F-39
The following is a reconciliation of basic and diluted net (loss) income per share of common stock for the three years ended December 31, 2017, 2016 and 2015 (in thousands, except per share amounts):
Years Ended December 31, | ||||||||||||||
2017 | 2016 | 2015 | ||||||||||||
Basic (Loss) Earnings Per Common Share: | ||||||||||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | ||||||
Weighted-average common shares outstanding | 71,857 | 71,147 | 69,640 | |||||||||||
Basic (loss) earnings per share | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.56 | ||||||
Diluted (Loss) Earnings Per Common Share: | ||||||||||||||
Net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | ||||||
Add-back of interest expense on outstanding convertible notes payable, net of tax | — | (1) | — | (1) | — | (2) | ||||||||
Adjusted net (loss) income | $ | (469,287 | ) | $ | (472,031 | ) | $ | 38,997 | ||||||
Weighted-average common shares outstanding | 71,857 | 71,147 | 69,640 | |||||||||||
Weighted-average incremental shares related to assumed exercise of warrants, stock options, vesting of non-vested shares and ESPP share issuance | — | (3) | — | (4) | 2,387 | (5) | ||||||||
Weighted-average incremental shares assuming conversion of outstanding notes payable | — | (1) | — | (1) | — | (2) | ||||||||
Diluted weighted-average common shares outstanding | 71,857 | (3) | 71,147 | (4) | 72,027 | (6) | ||||||||
Diluted net (loss) income per share | $ | (6.53 | ) | $ | (6.63 | ) | $ | 0.54 | ||||||
(1) | For the years ended December 31, 2017 and 2016, the Company incurred a net loss, which cannot be diluted, so basic and diluted loss per common share were the same. Accordingly, there were no numerator or denominator adjustments related to the Company's outstanding Notes. |
(2) | The numerator and denominator adjustments related to the Company’s convertible notes payable were excluded from the computation because the add-back of interest expense, net of tax, to the numerator had a greater effect on the quotient than the inclusion of the incremental shares assuming conversion of the convertible notes payable in the denominator, resulting in anti-dilution. |
(3) | For the year ended December 31, 2017, the Company incurred a net loss, which cannot be diluted, so basic and diluted loss per common share were the same. As of December 31, 2017, shares issuable but not included in the Company's calculation of diluted EPS, which could potentially dilute future earnings, included 9.47 million warrants outstanding, 9.47 million shares for conversion of outstanding Notes payable, 3.2 million stock options outstanding and 1.9 million non-vested restricted stock awards. |
(4) | For the year ended December 31, 2016, the Company incurred a net loss, which cannot be diluted, so basic and diluted loss per common share were the same. As of December 31, 2016, shares issuable but not included in the Company's calculation of diluted EPS, which could potentially dilute future earnings, included 9.47 million warrants outstanding, 9.47 million shares for conversion of outstanding Notes payable, 2.2 million stock options outstanding and 2.2 million non-vested restricted stock awards. |
(5) | As of December 31, 2015, the approximately 9.47 million warrants outstanding have been excluded from the denominator of the diluted EPS computation under the treasury stock method because the exercise price of the warrants exceeds the average market price of the Company’s common stock for the period, so inclusion in the calculation would be anti-dilutive. |
(6) | As of December 31, 2015, shares issuable but not included in the Company’s calculation of diluted EPS, which could potentially dilute future earnings, included 9.47 million for warrants outstanding, 9.47 million shares for conversion of outstanding Notes payable, 1.7 million stock options outstanding and 1.5 million non-vested restricted stock awards. |
F-40
13. SHARE-BASED COMPENSATION
The Company recognizes the grant date fair value of each option and share of restricted stock over its vesting period. Stock options and restricted stock awards are granted under the Company’s Fourth Amended and Restated 2002 Equity Incentive Plan and generally vest over a four year period and, in the case of stock options, have a term of 10 years.
Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan ("2002 Plan")
The aggregate number of shares of common stock authorized for issuance pursuant to the Company's 2002 Plan is 18,050,000 shares. There were 2,324,997, 2,233,393 and 2,394,433 stock options outstanding as of December 31, 2017, 2016 and 2015, respectively, and 1,861,489, 2,160,127 and 2,146,498 non-vested restricted stock awards outstanding as of December 31, 2017, 2016 and 2015, respectively, under the 2002 Plan.
Impax Laboratories, Inc. 1999 Equity Incentive Plan ( "1999 Plan" )
The aggregate number of shares of common stock authorized for issuance pursuant to the Company's 1999 Plan is 5,000,000 shares. There were 0, 938 and 10,938 stock options outstanding as of December 31, 2017, 2016 and 2015, respectively, under the 1999 Plan. The Company has ceased granting equity awards under the 1999 Plan.
Awards Granted Out of Plan - CEO Inducement
On March 27, 2017, the Company granted Paul M. Bisaro, its new President and Chief Executive Officer, an option to purchase 850,000 shares of the Company’s common stock pursuant to the terms of his Employment Agreement dated as of March 24, 2017 with the Company. The grant was made in accordance with NASDAQ’s employment inducement grant exemption and therefore was not granted under a stockholder approved plan. The grant is subject to the terms of an option agreement with Mr. Bisaro to evidence the award. There were 850,000 stock options outstanding related to this grant as of December 31, 2017.
The following table summarizes all of the Company's stock option activity for the years ended December 31, 2017, 2016, and 2015:
Stock Options | Number of Shares Under Option | Weighted- Average Exercise Price per Share | ||||
Outstanding at December 31, 2014 | 3,042,180 | $ | 14.78 | |||
Options granted | 406,950 | 41.27 | ||||
Options exercised | (1,042,198 | ) | 9.87 | |||
Options forfeited | (1,561 | ) | 16.70 | |||
Outstanding at December 31, 2015 | 2,405,371 | 21.39 | ||||
Options granted | 572,625 | 12.27 | ||||
Options exercised | (477,910 | ) | 19.09 | |||
Options forfeited | (265,755 | ) | 35.88 | |||
Outstanding at December 31, 2016 | 2,234,331 | 22.67 | ||||
Options granted | 1,198,726 | 12.21 | ||||
Options exercised | (74,643 | ) | 10.22 | |||
Options forfeited | (183,417 | ) | 33.07 | |||
Outstanding at December 31, 2017 | 3,174,997 | 18.36 | ||||
Options exercisable at December 31, 2017 | 1,634,133 | $ | 19.63 | |||
In May 2016, a retiring member of the Company's Board of Directors exercised vested stock options on a cashless basis, whereby the Company withheld 19,022 shares to cover the $0.6 million of proceeds due to the Company, representing the aggregate exercise price of the options.
F-41
As of December 31, 2017, stock options outstanding and exercisable had average remaining contractual lives of 6.70 years and 5.20 years, respectively. Also, as of December 31, 2017, stock options outstanding and exercisable each had aggregate intrinsic values of $9.9 million and $4.6 million, respectively, and restricted stock awards outstanding had an aggregate intrinsic value of $31.0 million.
The Company grants restricted stock to certain eligible employees as a component of its long-term incentive compensation program. The restricted stock award grants are made in accordance with the Company’s 2002 Plan and are issued and outstanding at the time of grant but are subject to forfeiture if the vesting conditions are not met. A summary of the non-vested restricted stock awards is as follows:
Restricted Stock Awards | Non-Vested Restricted Stock Awards | Weighted- Average Grant Date Fair Value | ||||
Non-vested at December 31, 2014 | 2,327,176 | $ | 23.61 | |||
Granted | 973,742 | 45.40 | ||||
Vested | (930,159 | ) | 22.64 | |||
Forfeited | (224,261 | ) | 29.01 | |||
Non-vested at December 31, 2015 | 2,146,498 | 33.20 | ||||
Granted | 1,245,184 | 31.77 | ||||
Vested | (893,190 | ) | 28.97 | |||
Forfeited | (338,365 | ) | 33.87 | |||
Non-vested at December 31, 2016 | 2,160,127 | 34.02 | ||||
Granted | 980,419 | 13.89 | ||||
Vested | (730,160 | ) | 31.99 | |||
Forfeited | (548,897 | ) | 30.27 | |||
Non-vested at December 31, 2017 | 1,861,489 | $ | 25.36 | |||
Included in the 730,160 shares of restricted stock vested during the year ended December 31, 2017 are 268,512 shares with a weighted-average fair value of $15.77 per share that were withheld for tax withholding obligations upon vesting of such awards from stockholders who elected to net share settle such tax withholding obligation. Included in the 893,190 shares of restricted stock vested during the year ended December 31, 2016 are 335,423 shares with a weighted-average fair value of $27.69 per share that were withheld for tax withholding purposes upon vesting of such awards from stockholders who elected to net share settle such tax withholding obligation. Included in the 930,159 shares of restricted stock vested during the year ended December 31, 2015 are 370,449 shares with a weighted-average fair value of $40.48 per share that were withheld for tax withholding purposes upon vesting of such awards from stockholders who elected to net share settle such tax withholding obligation.
As of December 31, 2017, the Company had 1,932,375 shares available for issuance for either stock options or restricted stock awards under the 2002 Plan. Although there were also 296,921 shares available for issuance under the 1999 Plan, the Company has ceased granting equity awards under this plan. Additionally, the Company had 1,501,351 shares available for issuance under its 2001 Non-Qualified Employee Stock Purchase Plan, as amended (“ESPP”). The Company's Board of Directors has determined that the final purchase period prior to December 31, 2017 would be the final purchase period under the ESPP, and the ESPP was terminated thereafter.
As of December 31, 2017, the Company had total unrecognized share-based compensation expense of $41.8 million related to all of its share-based awards, which is expected to be recognized over a weighted average period of 1.75 years. The intrinsic value of options exercised during the years ended December 31, 2017, 2016 and 2015 was $0.5 million, $5.8 million and $33.0 million, respectively. The total fair value of restricted stock which vested during the years ended December 31, 2017, 2016 and 2015 was $23.4 million, $25.9 million and $21.1 million, respectively.
F-42
The Company estimated the fair value of each stock option award on the grant date using the Black-Scholes option pricing model with the following assumptions:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Volatility (range) | 46.5% | - | 49.2% | 38.1% | - | 40.3% | 39.9% | - | 40.1% | ||
Volatility (weighted average) | 48.1% | 38.3% | 40.0% | ||||||||
Risk-free interest rate (range) | 1.9% | - | 2.2% | 1.2% | - | 1.9% | 0.8% | - | 1.8% | ||
Risk-free interest rate (weighted average) | 2.1% | 1.4% | 1.7% | ||||||||
Dividend yield | —% | —% | —% | ||||||||
Weighted-average expected life (years) | 6.18 | 6.14 | 6.18 | ||||||||
Weighted average grant date fair value | $5.93 | $12.27 | $17.08 | ||||||||
The Company estimated the fair value of each stock option award on the grant date using the Black-Scholes option pricing model, wherein expected volatility is based on historical volatility of the Company’s common stock. The expected term calculation is based on the “simplified” method described in SAB No. 107, Share-Based Payments and SAB No. 110, Share-Based Payment, as the result of the simplified method provides a reasonable estimate in comparison to actual experience. The risk-free interest rate is based on the U.S. Treasury yield at the date of grant for an instrument with a maturity that is commensurate with the expected term of the stock options. The dividend yield of zero is based on the fact that the Company has never paid cash dividends on its common stock and has no present intention to pay cash dividends. Options granted under each of the above plans generally vest over four years and have a term of 10 years.
The amount of share-based compensation expense recognized by the Company is as follows (in thousands):
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Manufacturing expenses | $ | 4,975 | $ | 6,364 | $ | 4,479 | |||||
Research and development | 16,174 | 5,697 | 5,996 | ||||||||
Selling, general and administrative | 5,109 | 20,119 | 18,138 | ||||||||
Total | $ | 26,258 | $ | 32,180 | $ | 28,613 | |||||
The after tax impact of recognizing the share-based compensation expense related to FASB ASC Topic 718 on basic earnings per common share was $0.30, $0.31 and $0.20 for the years ended December 31, 2017, 2016 and 2015, respectively, and diluted earnings per common share was $0.30, $0.31 and $0.20 for the years ended December 31, 2017, 2016 and 2015, respectively. The Company recognized a deferred tax benefit, before consideration of tax valuation allowances, of $4.8 million, $9.6 million and $9.2 million in the years ended December 31, 2017, 2016 and 2015, respectively, related to share-based compensation expense recorded for non-qualified employee stock options and restricted stock awards.
The Company’s policy is to issue new shares to satisfy stock option exercises and to grant restricted stock awards.
Share based Compensation Expense related to Former Executives
In December 2016, the Company announced that G. Frederick Wilkinson and the Company mutually agreed that Mr. Wilkinson would separate from his positions as President and Chief Executive Officer of Impax and resign as a member of the Board of Directors of the Company, effective December 19, 2016. In connection with his separation from the Company, Mr. Wilkinson and the Company entered into a General Release and Waiver dated as of December 19, 2016 (the “General Release and Waiver”). The General Release and Waiver provided for 12 month accelerated vesting of certain of Mr. Wilkinson's stock options and shares of restricted stock in accordance with the terms therein. As a result, during the year ended December 31, 2016, the Company recorded $0.5 million of accelerated expense related to the accelerated vesting of certain of Mr. Wilkinson's outstanding stock options and restricted stock.
F-43
The Company appointed Mr. Wilkinson as its President and Chief Executive Officer effective as of April 29, 2014. In accordance with Mr. Wilkinson’s appointment and pursuant to Mr. Wilkinson’s employment agreement with the Company, the Company granted to Mr. Wilkinson 150,000 shares of the Company’s restricted stock with a grant date fair value of $3.9 million, which vested as to one-third of the underlying shares on each of the six, 12 and 18 month anniversaries of April 29, 2014. Mr. Wilkinson also received pursuant to his employment agreement with the Company an award of 375,000 shares of restricted stock. The performance goals were achieved during fiscal year 2015 and pursuant to the terms of the employment agreement, 50% of Mr. Wilkinson’s performance-based restricted stock vested in 2015 and 50% vested in 2016. The Company valued these restricted stock awards subject to performance-based vesting using a Monte Carlo simulation and recognized the $7.6 million value of these awards over the longer of the derived or explicit service period, which was two years.
14. EMPLOYEE BENEFIT PLANS
401(k) Defined Contribution Plan
The Company sponsors a 401(k) defined contribution plan covering all employees. Participants are permitted to contribute up to 25% of their eligible annual pre-tax compensation up to established federal limits on aggregate participant contributions. The Company matches 100% of the employee contributions up to a maximum of 5% of employee compensation. Discretionary profit-sharing contributions made by the Company, if any, are determined annually by the Board of Directors. Participants are 100% vested in discretionary profit-sharing and matching contributions made by the Company after three years of service, and are 25% and 50% vested after one and two years of service, respectively. There were $6.1 million, $7.4 million and $3.7 million in matching contributions and no discretionary profit-sharing contributions made under this plan for the years ended December 31, 2017, 2016 and 2015, respectively.
Employee Stock Purchase Plan
In February 2001, the Board of Directors approved the 2001 Non-Qualified Employee Stock Purchase Plan (“ESPP”), with a 500,000 share reservation. The purpose of the ESPP was to enhance employee interest in the success and progress of the Company by encouraging employee ownership of common stock of the Company. The ESPP provided the opportunity to purchase the Company’s common stock at a 15% discount to the market price through payroll deductions or lump sum cash investments. Under the ESPP plan, for the years ended December 31, 2017, 2016 and 2015, the Company sold shares of its common stock to its employees in the amount of 50,185, 29,612 and 35,275, respectively, for net proceeds of $0.6 million, $0.7 million and $1.2 million, respectively. The Company's Board of Directors determined that the final purchase period prior to December 31, 2017 would be the final purchase period under the ESPP, and the ESPP was terminated thereafter.
Deferred Compensation Plan
In February 2002, the Board of Directors approved the Executive Non-Qualified Deferred Compensation Plan (“ENQDCP”) effective August 15, 2002 covering executive level employees of the Company as designated by the Board of Directors. Participants can defer up to 75% of their base salary and quarterly sales bonus and up to 100% of their annual performance based bonus. The Company matches 50% of employee deferrals up to 10% of base salary and bonus compensation. The maximum total match by the Company cannot exceed 5% of total base and bonus compensation. Participants are vested in the employer match contribution at 20% each year, with 100% vesting after five years of employment. Participants can earn a return on their deferred compensation based on hypothetical investments in investment funds. Changes in the market value of the participant deferrals and earnings thereon are reflected as an adjustment to the liability for deferred compensation with an offset to compensation expense. There were $1.0 million, $1.0 million and $1.1 million in matching contributions under the ENQDCP for the years ended December 31, 2017, 2016 and 2015, respectively.
The deferred compensation liability is a non-current liability recorded at the value of the amount owed to the ENQDCP participants, with changes in the value of such amounts recognized as compensation expense in the consolidated statements of operations. The calculation of the deferred compensation obligation is derived from observable market data by references to hypothetical investments selected by the participants and is included in the line item captioned “Other liabilities” on the consolidated balance sheets. The Company invests in corporate owned life insurance (“COLI”) policies, of which the cash surrender value is included in the line item captioned “Other assets” on the consolidated balance sheets. As of December 31, 2017 and 2016, the Company had a cash surrender value asset of $43.0 million and $37.4 million, respectively, and a deferred compensation liability of $33.4 million and $28.6 million, respectively, which approximated fair value. The asset representing the cash surrender value of the corporate owned life insurance and the deferred compensation liability are both Level 2 fair value measurements.
F-44
15. RESTRUCTURINGS
Consolidation and Improvement Plan
On May 10, 2017, the Company announced that it initiated a series of actions designed to improve manufacturing and research and development ("R&D") efficiencies, capitalize on growth opportunities, improve profitability and mitigate current challenges. The actions include:
• | Consolidating all of Generic R&D and U.S. manufacturing and packing operations to the Company's Hayward, California facility; |
• | Continuing the previously announced closure of the Middlesex, New Jersey manufacturing site, which will now include the closure of the Middlesex Generic R&D site as further discussed below under "Middlesex, New Jersey Manufacturing and Packaging Operations" and "Middlesex, New Jersey Generic R&D"; |
• | Reorganizing certain functions including quality, engineering and supply chain operations as further described below under "Technical Operations Reduction-in-Force"; |
• | Reviewing strategic alternatives for the Company’s Taiwan facility, including a sale of the facility as further described below under "Sale of Impax Laboratories (Taiwan), Inc." and |
• | Rationalizing the generic portfolio to eliminate low-value products and streamline operations such as the Company's divestment during the second quarter of 2017 of 29 ANDAs and one NDA for approved non-strategic generic products, the vast majority of which were not marketed, and all acquired as part of the Tower Acquisition, as described in "Note 8. Intangible Assets and Goodwill." |
By consolidating activities as outlined above, the Company expects to achieve cost savings and operating efficiency benefits while maintaining the infrastructure and expertise needed to capitalize on product and pipeline strengths. The Company currently expects to incur estimated charges for each initiative as described below. There are no charges currently expected to be incurred related to the rationalization of the generic product portfolio.
Middlesex, New Jersey Manufacturing and Packaging Operations
In March 2016, the Company's Board of Directors approved a plan of restructuring designed to reduce costs, improve operating efficiencies and enhance the Company's long-term competitive position by closing the Company's Middlesex, New Jersey manufacturing and packaging site and transferring the products and the functions performed there to the Company's other facilities or to third-party manufacturers. This restructuring was expected to take up to two years to complete. As a result of the restructuring, 215 positions were eliminated.
The Company incurred aggregate pre-tax charges of $43.4 million in connection with this plan through the year ended 2017 and does not anticipate any significant future charges. The following is a summary of the cumulative charges incurred by major type of cost (in thousands):
Type of Cost | Cumulative Amount Incurred | ||
Employee retention and severance payments | $ | 12,725 | |
Technical transfer of products | 9,544 | ||
Asset impairment and accelerated depreciation charges | 20,900 | ||
Facilities lease terminations and asset retirement obligations | 209 | ||
Legal and professional fees | 12 | ||
Total estimated restructuring charges | $ | 43,390 | |
Employee retention and severance payments are being accrued over the estimated service period. For the years ended December 31, 2017 and 2016, the Company recorded expense of $16.3 million and $27.1 million, respectively, to general and administrative expense in the Corporate and Other segment on the consolidated statements of operations.
F-45
A rollforward of the charges incurred to general and administrative expense for the year ended December 31, 2016 is as follows (in thousands):
Balance as of December 31, 2015 | Expensed /Accrued Expense | Cash Payments | Non-Cash Items | Balance as of December 31, 2016 | ||||||||||||||||
Employee retention and severance payments | $ | — | $ | 6,636 | $ | (691 | ) | $ | — | $ | 5,945 | |||||||||
Technical transfer of products | — | 6,573 | (6,573 | ) | — | — | ||||||||||||||
Asset impairment and accelerated depreciation charges | — | 13,678 | — | (13,678 | ) | — | ||||||||||||||
Facilities lease terminations and asset retirement obligations | — | 209 | — | — | 209 | |||||||||||||||
Legal and professional fees | — | 12 | (12 | ) | — | — | ||||||||||||||
Total | $ | — | $ | 27,108 | $ | (7,276 | ) | $ | (13,678 | ) | $ | 6,154 | ||||||||
A rollforward of the charges incurred to general and administrative expense for the year ended December 31, 2017 is as follows (in thousands):
Balance as of December 31, 2016 | Expensed /Accrued Expense | Cash Payments | Non-Cash Items | Balance as of December 31, 2017 | ||||||||||||||||
Employee retention and severance payments | $ | 5,945 | $ | 6,089 | $ | (4,648 | ) | $ | — | $ | 7,386 | |||||||||
Technical transfer of products | — | 2,671 | (2,671 | ) | — | — | ||||||||||||||
Asset impairment and accelerated depreciation charges | — | 7,525 | — | (7,525 | ) | — | ||||||||||||||
Facilities lease terminations and asset retirement obligations | 209 | — | — | — | 209 | |||||||||||||||
Total | $ | 6,154 | $ | 16,285 | $ | (7,319 | ) | $ | (7,525 | ) | $ | 7,595 | ||||||||
For the years ended December 31, 2017 and 2016, the Company recognized a liability of $7.6 and $6.2 million, respectively, in accrued expenses on the Company's consolidated balance sheet and anticipates remaining payments to be made through early 2018.
Middlesex, New Jersey Generic R&D
In May 2017, as part of its Consolidation and Improvement Plan, the Company announced its plan to close its Middlesex, New Jersey Generic R&D site and consolidate all Generic R&D activities to its Hayward, California facility. As a result, the Company eliminated a total of 31 positions in Middlesex. In connection with this Generic R&D consolidation, the Company incurred aggregate pre-tax charges for employee termination benefits, program termination costs and accelerated depreciation charges of $3.4 million through the end of 2017. For the year ended December 31, 2017, the Company recorded $3.0 million of employee termination benefits and program termination costs and $0.4 million for accelerated depreciation charges, all to research and development on the consolidated statement of operations. As of December 31, 2017, $3.0 million of employee termination benefits and program termination costs had been paid.
Sale of Middlesex, New Jersey Assets
In the fourth quarter of 2017, management completed an evaluation of the assets located at the Company's Middlesex, New Jersey facilities in accordance with ASC 360 - Property, Plant and Equipment (“ASC 360”) to determine whether all of the “held for sale” criteria under subsection 360-10-45-9 had been met. Based upon management's evaluation of the criteria under ASC 360, the Middlesex, New Jersey assets were determined to meet all of the “held for sale” criteria. As a result, the Company completed an impairment assessment related to the net book value of the assets of $5.6 million and based upon the estimated fair value less estimated costs to sell the assets the Company recorded a fixed asset impairment charges of $3.3 million in the Generic segment of its consolidated statement of operations for the year ended December 31, 2017.
F-46
On January 16, 2018, the Company sold all of its outstanding membership interests in CorePharma LLC, its wholly owned subsidiary, including certain specified assets within the entity, to a third party for a purchase price of $2.2 million.
Technical Operations Reduction-in-Force
In March 2017, the Company's management determined that a reduction-in-force was necessary in the Company's technical operations group in order to achieve greater operational efficiencies and to further streamline the organization. The Company identified 48 positions for elimination as of December 31, 2017. In connection with this reduction-in-force, the Company incurred aggregate pre-tax charges for employee termination benefits and other associated costs of $3.7 million through the end of 2017. For the year ended December 31, 2017, the Company recorded $3.7 million of employee termination benefits and other associated costs to cost of revenues in the Impax Generics segment on the consolidated statement of operations. As of December 31, 2017, $2.0 million had been paid and $1.7 million of employee termination benefits were included in accrued expenses on the Company’s consolidated balance sheet and the Company estimates that all payments will be made by early 2018.
Sale of Impax Laboratories (Taiwan), Inc.
In May 2017, as part of its Consolidation and Improvement Plan, the Company announced that it was reviewing strategic alternatives for its Taiwan facility, including a sale of the facility to a qualified buyer capable of reliably producing Rytary® in accordance with FDA requirements as the Company’s third party contract manufacturer (“CMO”) or, in the alternative, a closure of the facility following the completion of the technology transfer process to allow Rytary® to be manufactured either in the Company’s Hayward, California facility or at a CMO. Following this announcement, management completed an evaluation of the Taiwan facility in accordance with ASC 360 to determine whether all of the “held for sale” criteria under subsection 360-10-45-9 had been met. Based upon the evaluation of the criteria, including management's assessment of whether it was probable that a sale to a qualified buyer could be completed within one year, the Taiwan facility was determined to be “held and used” as of May 31, 2017.
Following the “held and used” determination, management next evaluated the Taiwan facility for recoverability. Recoverability of property is evaluated by a comparison of the carrying amount of an asset or asset group to the future net undiscounted cash flows expected to be generated by the asset or asset group. As the activities at the Taiwan facility were primarily focused on manufacturing Rytary®, which product represented the majority of the unit volume produced and cash flows generated, the Taiwan facility was included in the Impax Specialty Pharma asset group. Based upon the cash flows expected to be generated by the Impax Specialty Pharma asset group, management determined that there was no impairment of the asset group which included the Taiwan facility as of May 31, 2017.
As of May 31, 2017, the remaining useful life of the Taiwan facility was estimated to be two years, which was based on the estimated time required to complete the technology transfer process for Rytary® and reflected the new pattern of consumption of the expected benefits of the facility. The Company will recognize accelerated depreciation expense on a straight-line basis through May 31, 2019 to write the building and equipment associated with the Taiwan facility down to their estimated salvage values. For the year ended December 31, 2017 the Company recorded accelerated depreciation of $9.1 million.
After May 31, 2017 the Company continued to assess whether the Taiwan facility met the ASC 360 criteria. In the fourth quarter of 2017 based upon management's valuation of the criteria the Taiwan facility was determined to meet all of the “held for sale” criteria. As a result, excluding assets and liabilities subject to customary working capital adjustment, the Company completed an impairment assessment of the net book value of $91.7 million related to the net assets to be sold, and based upon an estimated fair value less estimated costs to sell for the net assets, the Company recorded an asset impairment charge of $74.1 million in the Company's consolidated statement of operations, of which $73.6 million related to property, plant and equipment. The remaining assets and liabilities associated with the Taiwan entity, which were part of the Impax Specialty Pharma segment, were reclassified as held for sale.
F-47
The following table provides the components of assets and liabilities of the Taiwan operations held for sale as of December 31, 2017 (in thousands):
December 31, 2017 | |||
Current assets | $ | 11,527 | |
Property, plant and equipment | 18,500 | ||
Assets held for sale | $ | 30,027 | |
Current liabilities | $ | 7,170 | |
Liabilities held for sale | $ | 7,170 | |
On December 19, 2017, the Company entered into a stock and asset purchase agreement with Bora Pharmaceuticals Co., Ltd. (“Bora”) pursuant to which Bora agreed to acquire the outstanding shares of Impax Laboratories (Taiwan), Inc. for $18.5 million in cash plus reimbursement for the closing working capital, subject to adjustment as defined in the agreement. The closing of the sale was completed on February 6, 2018.
Hayward, California Technical Operations and R&D
In November 2015, the Company's management assessed the headcount in the technical operations and research and development groups in Hayward, California, primarily as a result of the resolution of the warning letter at the Hayward facility, and determined that a reduction-in-force was necessary to adjust the headcount to the operating conditions of the post-warning letter resolution environment. The Company eliminated 27 positions and recorded an accrual in the Impax Generics segment for severance and related employee termination benefits of $2.5 million during the quarter ended December 31, 2015. As of December 31, 2017, $2.3 million has been paid, and the Company currently expects the remainder of this balance to be paid by early 2018.
Philadelphia, Pennsylvania Packaging and Distribution Operations
On June 30, 2015, the Company committed to a plan of restructuring of its packaging and distribution operations and as a result of this plan, the Company closed its Philadelphia packaging site and all Company-wide distribution operations were outsourced to United Parcel Services during the fiscal year ended December 31, 2015. The Company eliminated 93 positions and recorded an accrual for severance and related employee termination benefits of $2.6 million during the quarter ended June 30, 2015. As of June 30, 2016, the full $2.6 million had been paid.
F-48
16. INCOME TAXES
The Company is subject to federal, state and local income taxes in the United States, and income taxes in Taiwan, R.O.C., the Republic of Ireland and the Netherlands. The provision for (benefit from) income taxes is comprised of the following (in thousands):
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Current: | |||||||||||
Federal taxes | $ | (55,844 | ) | $ | 21,386 | $ | 48,078 | ||||
State taxes | (372 | ) | 266 | 2,286 | |||||||
Foreign taxes | 639 | 1,377 | (442 | ) | |||||||
Total current tax (benefit) expense | (55,577 | ) | 23,029 | 49,922 | |||||||
Deferred: | |||||||||||
Federal taxes | $ | 73,357 | $ | (133,387 | ) | $ | (23,605 | ) | |||
State taxes | (371 | ) | 5,502 | (5,733 | ) | ||||||
Foreign taxes | 917 | 562 | (213 | ) | |||||||
Total deferred tax expense (benefit) | 73,903 | (127,323 | ) | (29,551 | ) | ||||||
Provision for (benefit from) income taxes | $ | 18,326 | $ | (104,294 | ) | $ | 20,371 | ||||
F-49
A reconciliation of the difference between the tax provision (benefit) at the federal statutory rate and actual income taxes on income before income taxes, which includes federal, state, and other income taxes, is as follows (in thousands):
Years Ended December 31, | ||||||||||||||||||||
2017 | 2016 | 2015 | ||||||||||||||||||
(Loss) income before income taxes | $ | (450,961 | ) | $ | (576,325 | ) | $ | 59,368 | ||||||||||||
Tax (benefit) provision at the federal statutory rate | (157,836 | ) | 35.0 | % | (201,714 | ) | 35.0 | % | 20,779 | 35.0 | % | |||||||||
Increase (decrease) in tax rate resulting from: | ||||||||||||||||||||
Tax rate differential and permanent items on foreign income | 662 | (0.2 | )% | 186 | — | % | 412 | 0.7 | % | |||||||||||
State income taxes, net of federal benefit | (8,291 | ) | 1.8 | % | (7,394 | ) | 1.3 | % | 365 | 0.6 | % | |||||||||
State research and development credits | (1,324 | ) | 0.3 | % | (1,767 | ) | 0.3 | % | (2,357 | ) | (4.0 | )% | ||||||||
Federal research and development credits | (1,243 | ) | 0.3 | % | (2,213 | ) | 0.4 | % | (2,672 | ) | (4.5 | )% | ||||||||
Share-based compensation | 5,471 | (1.2 | )% | 1,768 | (0.3 | )% | 968 | 1.6 | % | |||||||||||
Executive compensation | 543 | (0.1 | )% | (761 | ) | 0.1 | % | 3,140 | 5.3 | % | ||||||||||
Domestic manufacturing deduction | — | — | % | (1,286 | ) | 0.2 | % | (1,422 | ) | (2.4 | )% | |||||||||
Other permanent book/tax differences | (1,846 | ) | 0.4 | % | (258 | ) | — | % | 2,003 | 3.4 | % | |||||||||
Provision for uncertain tax positions | (807 | ) | 0.2 | % | 337 | — | % | 184 | 0.3 | % | ||||||||||
Revision of prior years’ estimates | 1,371 | (0.3 | )% | (792 | ) | 0.1 | % | 859 | 1.5 | % | ||||||||||
Taiwan rural area investment tax credit | — | — | % | — | — | % | (2,134 | ) | (3.6 | )% | ||||||||||
Impact on gross deferred net assets from 2017 Tax Reform Act | 100,065 | (22.2 | )% | — | — | % | — | — | % | |||||||||||
Foreign withholding tax | 1,534 | (0.3 | )% | — | — | % | — | — | % | |||||||||||
Other, net | 2,888 | (0.7 | )% | 842 | (0.1 | )% | 246 | 0.4 | % | |||||||||||
Valuation allowance | 77,139 | (17.1 | )% | 108,758 | (18.9 | )% | — | — | % | |||||||||||
Provision for (benefit from) income taxes | $ | 18,326 | (4.1 | )% | $ | (104,294 | ) | 18.1 | % | $ | 20,371 | 34.3 | % | |||||||
Deferred income taxes result from temporary differences between the financial statement carrying values and the tax bases of the Company’s assets and liabilities. Deferred tax assets principally result from certain accruals and reserves currently not deductible for tax purposes, acquired product rights and intangibles, capitalized legal and share based compensation expense. Deferred tax liabilities principally result from acquired product rights and intangibles and the use of accelerated depreciation methods for income tax purposes.
A valuation allowance, if needed, reduces deferred tax assets to the amount expected to be realized. When determining the amount of net deferred tax assets that are more likely than not to be realized, the Company assesses all available positive and negative evidence. This evidence includes, but is not limited to, scheduled reversal of deferred tax liabilities, prior earnings history, projected future earnings, carry-back and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a deferred tax asset. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income (exclusive of reversing taxable temporary differences and carryforwards) to outweigh objective negative evidence of recent financial reporting losses for the years ended December 31, 2017 and 2016.
Based on an evaluation of both the positive and negative evidence available, the Company determined that it was necessary to establish a valuation allowance against all of the net deferred tax assets for the year ended December 31, 2017 and against a significant portion of the net deferred tax assets for the year ended December 31, 2016. Given the objectively verifiable negative evidence of a three-year cumulative loss which, under the provisions of FASB ASC Topic 740 is a significant element of negative evidence that is difficult to overcome, and the weighting of all available positive evidence, the Company excluded projected taxable income from the assessment of income that could be used as a source of taxable income to realize the deferred tax assets. The valuation allowance recorded against the consolidated net deferred tax asset in 2017 and 2016 were $185.9 million and $108.8 million, respectively.
F-50
The components of the Company’s deferred tax assets and liabilities are as follows (in thousands):
December 31, | |||||||
2017 | 2016 | ||||||
Deferred tax assets: | |||||||
Accrued expenses | $ | 60,069 | $ | 114,825 | |||
Inventory reserves | 17,602 | 15,873 | |||||
Net operating loss carryforwards | 2,518 | 2,302 | |||||
Depreciation and amortization | 2,657 | 651 | |||||
Acquired product rights and intangibles | 118,168 | 128,401 | |||||
Capitalized legal fees | 6,695 | 10,231 | |||||
Credit carryforwards | 11,205 | 8,453 | |||||
Share based compensation expense | 3,535 | 6,371 | |||||
Sale of subsidiary | 7,794 | — | |||||
Other | 495 | 525 | |||||
Deferred tax assets | 230,738 | 287,632 | |||||
Deferred tax liabilities: | |||||||
Tax depreciation and amortization in excess of book amounts | 3,808 | 5,428 | |||||
Acquired product rights and intangibles | 35,698 | 95,517 | |||||
Derivative | 3,411 | 6,192 | |||||
Foreign withholding tax | 1,824 | — | |||||
Other | 3,326 | 1,871 | |||||
Deferred tax liabilities | 48,067 | 109,008 | |||||
Deferred tax assets (liabilities), net | 182,671 | 178,624 | |||||
Valuation allowance | (185,897 | ) | (108,758 | ) | |||
Deferred tax assets (liabilities), net after valuation allowance | $ | (3,226 | ) | $ | 69,866 | ||
A rollforward of unrecognized tax benefits for the years ended December 31, 2017, 2016 and 2015 is as follows (in thousands):
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Unrecognized tax benefits beginning of year | $ | 6,425 | $ | 5,680 | $ | 6,517 | |||||
Gross change for current year positions | 328 | 549 | 1,079 | ||||||||
Gross change for prior period positions | (105 | ) | 1,318 | (673 | ) | ||||||
Gross change due to Tower Acquisition | — | — | 1,037 | ||||||||
Decrease due to expiration of statutes of limitations | (972 | ) | — | — | |||||||
Decrease due to settlements and payments | — | (1,122 | ) | (2,280 | ) | ||||||
Unrecognized tax benefits end of year | $ | 5,676 | $ | 6,425 | $ | 5,680 | |||||
F-51
The amount of unrecognized tax benefits at December 31, 2017, 2016 and 2015 was $5.7 million, $6.4 million and $5.7 million, respectively, of which $5.0 million, $5.3 million and $4.3 million would impact the Company’s effective tax rate, respectively, if recognized. The Company currently does not believe that the total amount of unrecognized tax benefits will increase or decrease significantly over the next 12 months. Interest expense related to income taxes is included in “Interest expense, net” on the consolidated statements of operations. Net interest expense related to unrecognized tax benefits for the year ended December 31, 2017 was $(24,000), compared to $125,000 in 2016. Accrued interest expense as of December 31, 2017 and 2016 was $0.3 million and $0.4 million, respectively. Income tax penalties are included in “Other income (expense)” on the consolidated statements of operations. Accrued tax penalties of $0.6 million were booked in 2015 related to the 2010-2011 California audit and were paid in 2016.
Tower Holdings, Inc. (“Tower”) is currently under audit for federal income tax by the U.S. Internal Revenue Service ("IRS") for the tax year ended March 9, 2015, which pre-dates the Company’s acquisition of Tower. The Company and the former stockholders of Tower are currently cooperating with the IRS in connection with the audit. Under the terms of the Stock Purchase Agreement related to the Tower Acquisition, the Company is not responsible for pre-acquisition income tax liabilities. Neither the Company nor any of its other affiliates is currently under audit for federal income tax.
Through March 31, 2017, no provision had been made for U.S. federal deferred income taxes on the excess of the amount for financial reporting over the tax basis of investments in foreign subsidiary since it had been the current intention of management to indefinitely reinvest the undistributed earnings in the foreign subsidiary.
As of June 30, 2017, following management’s announcement in May 2017 that it was reviewing potential options to either sell or close the Taiwan manufacturing facility and dissolve operations at Impax Taiwan, the Company changed its assertion related to the accumulated unremitted foreign earnings of its Taiwan subsidiary. The Company was no longer able to assert under ASC 740-30-25 that the unremitted foreign earnings are indefinitely reinvested outside the United States. Accordingly, the Company has recorded a deferred tax liability associated with remitting these earnings back to the United States.
Effect of 2017 Tax Reform Act
On December 22, 2017, the 2017 Tax Reform Act was signed into law. Among other things, the 2017 Tax Reform Act permanently lowers the corporate tax rate to 21% from the existing maximum rate of 35%, effective for tax years including or commencing January 1, 2018. As a result of the reduction of the corporate tax rate to 21%, U.S. GAAP require companies to re-value their deferred tax assets and liabilities as of the date of enactment, with resulting tax effects accounted for in the reporting period of enactment.
In connection with the Company's initial analysis of the impact of the 2017 Tax Reform Act, the Company recorded a discrete net tax benefit of $0.4 million in the period ending December 31, 2017. This net tax benefit primarily consisted of the corporate rate reduction of $0.5 million and a net expense for the Transition Tax (as described below) of $0.1 million.
Although the Company is able to make a reasonable estimate of the impact of the reduction in its corporate tax rate, due to the 2017 Tax Reform Act, the Company's estimate may be affected by other analyses related to the 2017 Tax Reform Act, including, but not limited to, the Company's calculation of deemed repatriation of deferred foreign income and the state tax effect of adjustments made to federal temporary differences. The deemed repatriation transition tax, also referred to as the "Transition Tax", is a tax on previously untaxed accumulated and current earnings and profits ("E&P") of a company's foreign subsidiaries. To determine the amount of the Transition Tax, the Company determined, in addition to other factors, the amount of post-1986 E&P of the Company's relevant subsidiaries - including Impax Laboratories (Netherlands) CV, Impax (Netherlands) BV, Impax Laboratories Ireland Limited, and Impax Taiwan Inc, - as well as the amount of non-U.S. income taxes paid on such earnings. As such, the Company has made a reasonable estimate of the Transition Tax and recorded a Transition Tax obligation of $0.1 million, however, the Company continues to gather additional information to more precisely compute the amount of the Transition Tax. The Company continues to evaluate legislative changes, regulations, and notices regarding the applicable mechanics of the relevant rules impacting the estimate of the Transition Tax, and, the Company continues to evaluate cash versus non-cash earnings and profits, as the rates differ for the two different categories of earnings and profits.
17. ALLIANCE AND COLLABORATION AGREEMENTS
The Company has entered into several alliance, collaboration, license and distribution agreements, and similar agreements with respect to certain of its products and services, with unrelated third-party pharmaceutical companies. The consolidated statements of operations include revenue recognized under agreements the Company has entered into to develop marketing and/or distribution relationships with its partners to fully leverage the technology platform and revenue recognized under development agreements which generally obligate the Company to provide research and development services over multiple periods.
F-52
The Company’s alliance and collaboration agreements often include milestones and provide for milestone payments upon achievement of these milestones. Generally, the milestone events contained in the Company’s alliance and collaboration agreements coincide with the progression of the Company’s products and technologies from pre-commercialization to commercialization.
The Company groups pre-commercialization milestones in its alliance and collaboration agreements into clinical and regulatory categories, each of which may include the following types of events:
Clinical Milestone Events:
• | Designation of a development candidate. Following the designation of a development candidate, generally, IND-enabling animal studies for a new development candidate take 12 to 18 months to complete. |
• | Initiation of a Phase I clinical trial. Generally, Phase I clinical trials take one to two years to complete. |
• | Initiation or completion of a Phase II clinical trial. Generally, Phase II clinical trials take one to three years to complete. |
• | Initiation or completion of a Phase III clinical trial. Generally, Phase III clinical trials take two to four years to complete. |
• | Completion of a bioequivalence study. Generally, bioequivalence studies take three months to one year to complete. |
Regulatory Milestone Events:
• | Filing or acceptance of regulatory applications for marketing approval such as a New Drug Application in the United States or Marketing Authorization Application in Europe. Generally, it takes six to 12 months to prepare and submit regulatory filings and two months for a regulatory filing to be accepted for substantive review. |
• | Marketing approval in a major market, such as the United States or Europe. Generally it takes one to three years after an application is submitted to obtain approval from the applicable regulatory agency. |
• | Marketing approval in a major market, such as the United States or Europe for a new indication of an already-approved product. Generally it takes one to three years after an application for a new indication is submitted to obtain approval from the applicable regulatory agency. |
Commercialization Milestone Events:
• | First commercial sale in a particular market, such as in the United States or Europe. |
• | Product sales in excess of a pre-specified threshold, such as annual sales exceeding $100 million. The amount of time to achieve this type of milestone depends on several factors including but not limited to the dollar amount of the threshold, the pricing of the product and the pace at which customers begin using the product. |
License and Distribution Agreement with Shire
In January 2006, the Company entered into a License and Distribution Agreement with an affiliate of Shire Laboratories, Inc., which was subsequently amended (“Prior Shire Agreement”), under which the Company received a non-exclusive license to market and sell an authorized generic of Shire’s Adderall XR® product (“AG Product”) subject to certain conditions, but in any event by no later than January 1, 2010. The Company commenced sales of the AG Product in October 2009. On February 7, 2013, the Company entered into an Amended and Restated License and Distribution Agreement with Shire (the “Amended and Restated Shire Agreement”), which amended and restated the Prior Shire Agreement. Pursuant to the terms of the Amended and Restated Shire Agreement, the Company is required to pay to Shire a specified profit share based on sales of the AG Product and a specified profit share based on sales of our generic Adderall XR® product. The Company began selling our generic Adderall XR® product during the second quarter of 2016. The Company accrued a profit share payable to Shire of $2.2 million during the year ended December 31, 2017, based on sales of its generic Adderall XR® product and reflecting adjustments for returns and government rebates from its previous sales of the AG Product and of $7.5 million and $19.5 million during the years ended December 31, 2016 and 2015, respectively, based on sales of the AG Product and the Company's generic Adderall XR® product, in each case with a corresponding charge included in the cost of revenues line in the consolidated statements of operations.
F-53
Development, Supply and Distribution Agreement with Tolmar, Inc.
In June 2012, the Company entered into the Tolmar Agreement with Tolmar. Under the terms of the Tolmar Agreement, Tolmar granted to the Company an exclusive license to commercialize up to 11 generic topical prescription drug products, including 10 currently approved products in the United States and its territories; the parties agreed in 2015 to terminate development efforts of one product under the Tolmar Agreement that had been pending approval at the FDA. Under the terms of the Tolmar Agreement, Tolmar is responsible for developing and manufacturing the products, and the Company is responsible for marketing and sale of the products. As of December 31, 2017, the Company was currently marketing and selling four approved products. The Company is required to pay a profit share to Tolmar on sales of each product commercialized pursuant to the terms of the Tolmar Agreement.
The Company paid Tolmar a $21.0 million upfront payment upon signing of the agreement and, pursuant to the terms of the agreement, is also required to make payments to Tolmar up to an aggregate amount of $25.0 million upon the achievement of certain specified milestone events. As of December 31, 2017, the Company had paid a total of $20.0 million to Tolmar upon the achievement of certain specified milestone events, including $12.0 million upon the achievement of a regulatory milestone event and $5.0 million upon the achievement of a commercialization event, and does not currently expect to make any additional milestone payments under the agreement. The Company is required to pay a profit share to Tolmar on sales of the topical products, of which it accrued a profit share payable to Tolmar of $10.0 million, $36.4 million and $77.7 million during the years ended December 31, 2017, 2016 and 2015, respectively, with a corresponding charge included in the cost of revenues line in the Company’s consolidated statement of operations.
The Company entered into a Loan and Security Agreement with Tolmar in March 2012 (the “Tolmar Loan Agreement”), under which the Company agreed to lend to Tolmar one or more loans through December 31, 2014, in an aggregate amount not to exceed $15.0 million. The outstanding principal amount of, including any accrued and unpaid interest on, the loans under the Tolmar Loan Agreement are payable by Tolmar beginning from March 31, 2017 through March 31, 2020 or the maturity date, in accordance with the terms therein. Pursuant to the Tolmar Loan Agreement, Tolmar could prepay all or any portion of the outstanding balance of the loans prior to the maturity date without penalty or premium. In May 2016, Tolmar repaid in full the $15.0 million due to the Company under the Tolmar Loan Agreement.
Strategic Alliance Agreement with Teva
The Company is a party to a Strategic Alliance Agreement dated as of June 27, 2001 with Teva Pharmaceuticals USA, Inc. ("Teva USA"), an affiliate of Teva, which was subsequently amended (“Teva Agreement”). The Teva Agreement commits the Company to develop and manufacture, and Teva to distribute, a specified number of controlled release generic pharmaceutical products (“generic products”), each for a 10-year period. As of December 31, 2017, the Company was supplying Teva with oxybutynin extended release tablets (Ditropan XL® 5 mg, 10 mg and 15 mg extended release tablets); the other products under the Teva Agreement have either been returned to the Company, are being manufactured by Teva at its election, were voluntarily withdrawn from the market or the Company’s obligations to supply such product had expired or were terminated in accordance with the Teva Agreement.
F-54
Distribution, License, Development and Supply Agreement with AstraZeneca UK Limited
In January 2012, the Company entered into the AZ Agreement with AstraZeneca and the parties subsequently entered into a First Amendment to the AZ Agreement dated May 31, 2016 (as amended, the "AZ Amendment"). Under the terms of the AZ Agreement, AstraZeneca granted to the Company an exclusive license to commercialize the tablet, orally disintegrating tablet and nasal spray formulations of Zomig® (zolmitriptan) products for the treatment of migraine headaches in the United States and in certain U.S. territories, except during an initial transition period when AstraZeneca fulfilled all orders of Zomig® products on the Company’s behalf and AstraZeneca paid to the Company the gross profit on such Zomig® products. Under the terms of the AZ Amendment, under certain conditions and depending on the nature and terms of the study agreed to with the FDA, the Company agreed to conduct, at its own expense, the juvenile toxicity study and pediatric study required by the FDA under the Pediatric Research Equity Act (“PREA”) for approval of the nasal formulation of Zomig® for the acute treatment of migraine in pediatric patients ages six through eleven years old, as further described in the study protocol mutually agreed to by the parties (the “PREA Study”). In consideration for the Company conducting the PREA Study at its own expense, the AZ Amendment provides for the total royalty payments payable by the Company to AstraZeneca on net sales of Zomig® products under the AZ Agreement to be reduced by certain specified amounts beginning from the quarter ended June 30, 2016 and through the quarter ended December 31, 2020, with such reduced royalty amounts totaling an aggregate amount of $30.0 million. In the event the royalty reduction amounts exceed the royalty payments payable by the Company to AstraZeneca pursuant to the AZ Agreement in any given quarter, AstraZeneca will be required to pay the Company an amount equal to the difference between the royalty reduction amount and the royalty payment payable by the Company to AstraZeneca. The Company’s commitment to perform the PREA Study may be terminated, without penalty, under certain circumstances as set forth in the AZ Amendment.
In May 2013, the Company’s exclusivity period for branded Zomig® tablets and orally disintegrating tablets expired and the Company launched authorized generic versions of those products in the United States. As discussed above, pursuant to the AZ Amendment, the total royalty payments payable by the Company to AstraZeneca on net sales of Zomig® products under the AZ Agreement is reduced by certain specified amounts beginning from the quarter ended June 30, 2016 and through the quarter ended December 31, 2020, with such reduced royalty amounts totaling an aggregate amount of $30.0 million. The Company accrued a royalty payable to AstraZeneca of $17.8 million, $17.2 million and $16.8 million for the years ended December 31, 2017, 2016 and 2015, respectively, with a corresponding charge included in the cost of revenues line on the consolidated statements of operations.
Mebendazole Product Acquisition Agreement with Teva Pharmaceuticals USA, Inc.
In August 2013, the Company, through its Amedra Pharmaceuticals subsidiary, entered into a product acquisition agreement (the “Mebendazole Product Acquisition Agreement”) with Teva pursuant to which the Company acquired the assets (including the ANDA and other regulatory materials) and related liabilities related to Teva’s mebendazole tablet product in all dosage forms. Pursuant to the Mebendazole Product Acquisition Agreement, the Company was required to pay certain milestone payments up to an aggregate amount of $3.5 million upon the approval and launch of the mebendazole tablet product; the Company paid the $3.5 million to Teva during the quarter ended March 31, 2016 upon the FDA's approval and the Company's subsequent launch of Emverm® (mebendazole) 100 mg chewable tablets. The Company is also obligated to pay Teva a royalty payment based on net sales of Emverm®, including a specified annual minimum royalty payment, subject to customary reductions and the other terms and conditions set forth in the Mebendazole Product Acquisition Agreement.
18. COMMITMENTS AND CONTINGENCIES
Executive Employment Agreements
The Company is a party to employment and separation agreements with certain members of its executive management team that provide for severance and other payments following termination of their employment for various reasons.
F-55
Lease Agreements
The Company leases land, office space, manufacturing, warehouse and research and development facilities, and equipment under non-cancelable operating leases expiring between January 2018 and December 2027. Rent expense for the years ended December 31, 2017, 2016 and 2015 was $5.2 million, $4.9 million and $4.1 million, respectively. The Company recognizes rent expense on a straight-line basis over the lease period. The Company also leases certain equipment under various non-cancelable operating leases with various expiration dates between April 2018 and July 2022. Future minimum lease payments under the non-cancelable operating leases are as follows (in thousands):
Years ending December 31, | |||
2018 | $ | 5,575 | |
2019 | 3,740 | ||
2020 | 2,578 | ||
2021 | 2,551 | ||
2022 | 2,585 | ||
Thereafter | 11,113 | ||
Total minimum lease payments | $ | 28,142 | |
Purchase Order Commitments
As of December 31, 2017, the Company had $108.1 million of open purchase order commitments, primarily for raw materials. The terms of these purchase order commitments are generally less than one year in duration.
19. LEGAL AND REGULATORY MATTERS
Patent Litigation
There is substantial litigation in the pharmaceutical, biological, and biotechnology industries with respect to the manufacture, use, and sale of new products which are the subject of conflicting patent and intellectual property claims. One or more patents often cover the brand name products for which the Company is developing generic versions and the Company typically has patent rights covering the Company’s branded products.
Under federal law, when a drug developer files an ANDA for a generic drug seeking approval before expiration of a patent, which has been listed with the FDA as covering the brand name product, the developer must certify its product will not infringe the listed patent(s) and/or the listed patent is invalid or unenforceable (commonly referred to as a “Paragraph IV” certification). Notices of such certification must be provided to the patent holder, who may file a suit for patent infringement within 45 days of the patent holder’s receipt of such notice. If the patent holder files suit within the 45 day period, the FDA can review and approve the ANDA, but is prevented from granting final marketing approval of the product until a final judgment in the action has been rendered in favor of the generic drug developer, or 30 months from the date the notice was received, whichever is sooner. The Company’s generic products division is typically subject to patent infringement litigation brought by branded pharmaceutical manufacturers in connection with the Company’s Paragraph IV certifications seeking an order delaying the approval of the Company’s ANDA until expiration of the patent(s) at issue in the litigation. Likewise, the Company’s branded products division is currently involved in patent infringement litigation against generic drug manufacturers who have filed Paragraph IV certifications to market their generic drugs prior to expiration of the Company’s patents at issue in the litigation.
The uncertainties inherent in patent litigation make the outcome of such litigation difficult to predict. For the Company’s generic products division, the potential consequences in the event of an unfavorable outcome in such litigation include delaying launch of its generic products until patent expiration. If the Company were to launch its generic product prior to successful resolution of a patent litigation, the Company could be liable for potential damages measured by the profits lost by the branded product manufacturer rather than the profits earned by the Company if we are found to infringe a valid, enforceable patent. For the Company’s branded products division, an unfavorable outcome may significantly accelerate generic competition ahead of expiration of the patents covering the Company’s branded products. All such litigation typically involves significant expense.
F-56
The Company is generally responsible for all of the patent litigation fees and costs associated with current and future products not covered by its alliance and collaboration agreements. The Company has agreed to share legal expenses with respect to third-party and Company products under the terms of certain of the alliance and collaboration agreements. The Company records the costs of patent litigation as expense in the period when incurred for products it has developed, as well as for products which are the subject of an alliance or collaboration agreement with a third-party.
Although the outcome and costs of the asserted and unasserted claims is difficult to predict, based on the information presently known to management, the Company does not currently expect the ultimate liability, if any, for such matters to have a material adverse effect on its business, financial condition, results of operations, or cash flows.
Patent Infringement Litigation
Endo Pharmaceuticals Inc. and Grunenthal GmbH v. Impax Laboratories, Inc. and ThoRx Laboratories, Inc. (Oxymorphone hydrochloride); Endo Pharmaceuticals Inc. and Grunenthal GmbH v. Impax Laboratories, Inc. (Oxymorphone hydrochloride)
In November 2012, Endo Pharmaceuticals, Inc. and Grunenthal GmbH filed suit against ThoRx Laboratories, Inc., a wholly owned subsidiary of the Company (“ThoRx”), and the Company in the U.S. District Court for the Southern District of New York alleging patent infringement based on the filing of ThoRx’s ANDA relating to Oxymorphone hydrochloride, Extended Release tablets, 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg, 30 mg and 40 mg, generic to Opana ER®. In January 2013, Endo filed a separate suit against the Company in the U.S. District Court for the Southern District of New York alleging patent infringement based on the filing of the Company’s ANDA relating to the same products. ThoRx and the Company filed an answer and counterclaims to the November 2012 suit and the Company filed an answer and counterclaims with respect to the January 2013 suit. A bench trial was completed in April 2015. In June 2016, the Court entered an amended judgment in both cases that the products described in the Company’s and ThoRx’s ANDAs would, if marketed, infringe certain claims of the patents asserted by Endo and Grunenthal. The Court also found that the asserted claims of patents owned by Endo were not invalid, but that the asserted claims of patents owned by Grunenthal were invalid. As a result, the Court enjoined the Company and ThoRx from marketing their products until expiration of the Endo patents in 2023. The Company and ThoRx are appealing the Court's judgment.
In November 2014, Endo Pharmaceuticals Inc. and Mallinckrodt LLC filed suit against the Company in the U.S. District Court for the District of Delaware making additional allegations of patent infringement based on the filing of the Company’s Oxymorphone hydrochloride ANDA described above. Also in November 2014, Endo and Mallinckrodt filed a separate suit in the U.S. District Court for the District of Delaware making additional allegations of patent infringement based on the filing of ThoRx’s Oxymorphone hydrochloride ANDA described above. ThoRx and the Company filed an answer and counterclaim to those suits in which they are named as a defendant. The cases are currently stayed.
Impax Laboratories Inc., et al. v. Lannett Holdings, Inc. and Lannett Company (Zomig®)
In July 2014, the Company filed suit against Lannett Holdings, Inc. and Lannett Company (collectively, “Lannett”) in the United States District Court for the District of Delaware, alleging patent infringement based on the filing of the Lannett ANDA relating to Zolmitriptan Nasal Spray, 5mg, generic to Zomig® Nasal Spray. The case went to trial in September 2016. On March 29, 2017, the District Court issued a Trial Opinion finding the asserted patents valid and infringed. On April 17, 2017, the District Court entered a Final Judgment and Injunction that, inter alia, bars FDA approval of Lannett’s proposed generic product prior to May 29, 2021. On May 12, 2017, Lannett filed a Notice of Appeal with the United States Court of Appeals for the Federal Circuit. Briefing of Lannett’s appeal has been completed and oral argument is scheduled for April 5, 2018.
Impax Laboratories Inc., et al. v. Par Pharmaceutical, Inc. (Zomig®)
On September 23, 2016, the Company filed suit against Par Pharmaceutical, Inc. (“Par”) in the United States District Court for the District of Delaware, alleging patent infringement based on the filing of the Par ANDA relating to Zolmitriptan Nasal Spray, 2.5 mg and 5 mg, generic to Zomig® Nasal Spray. On October 12, 2016, the parties stipulated to stay the case pending the outcome of the related case, Impax Laboratories Inc., et al. v. Lannett matter described above. On April 24, 2017, the parties stipulated that the stay shall remain in effect until the Impax Laboratories Inc., et al. v. Lannett matter is fully resolved. As such, Par has not yet filed an answer or counterclaims to the Company’s complaint. The 30-month stay of approval for applicable to the Par ANDA has been tolled pending resumption of this case.
F-57
Impax Laboratories Inc., et al. v. Actavis Laboratories, Inc. and Actavis Pharma Inc. (Rytary®)
In September 2015, the Company filed suit against Actavis Laboratories, Inc. and Actavis Pharma Inc. (collectively, “Actavis”) in the United States District Court for the District of New Jersey, alleging patent infringement of U.S. Patent Nos. 7,094,427; 8,377,474; 8,454,998; 8,557,283; 9,089,607; 9,089,608, based on the filing of the Actavis ANDA relating to carbidopa and levodopa extended release capsules, generic to Rytary®. The Company filed related actions alleging infringement of later-issued U.S. Patent No. 9,463,246 in December 2016 and of later-issued U.S. Patent No. 9,533,046 in May 2017. Both related actions were consolidated with the lead action. On December 15, 2017, the Patent and Trademark Office issued an Ex Parte Reexamination Certificate canceling all claims of the '427 patent; the parties subsequently stipulated to dismiss with prejudice all claims and counterclaims relating to the '427 patent. Fact discovery and claim construction briefing have concluded and a claim construction hearing was held on April 26, 2017. On May 9, 2017, the District Court issued a decision interpreting certain claim terms in dispute in the litigation. Subject to reservation of all rights to appeal the Court’s May 9, 2017 decision, the parties stipulated to dismiss without prejudice all claims and counterclaims relating to the ‘474, ‘998, and ‘607 patents, and the Court entered an order recognizing this stipulation on June 8, 2017. The parties have completed expert discovery and Actavis filed a summary judgment motion on October 23, 2017. Briefing on the summary judgment motion is complete and an oral hearing was scheduled for February 27, 2018. On February 20, 2018, the Court issued an order setting trial for March 6, 2018. On February 23, 2018, the parties filed a joint letter requesting a trial date in the first two weeks of May 2018. The Court has not yet responded to the parties’ letter.
Impax Laboratories, Inc. v. Sandoz Inc. (Rytary®)
On March 31, 2017, the Company filed suit against Sandoz Inc. in the United States District Court for the District of New Jersey, alleging infringement of U.S. Patent Nos. 7,094,427; 8,377,474; 8,454,998; 8,557,283; 9,089,607; 9,089,608; 9,463,246; and 9,533,046, based on the filing of Sandoz’s ANDA relating to carbidopa and levodopa extended release capsules, generic to Rytary®. Sandoz has not yet answered or otherwise responded to the Complaint.
Impax Laboratories, Inc. v. Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Ltd. (Rytary®)
On December 21, 2017, the Company filed suit against Zydus Pharmaceuticals USA, Inc. and Cadila Healthcare Ltd. (collectively, “Zydus”) in the United States District Court for the District of New Jersey, alleging infringement of U.S. Patent No. 9,089,608, based on the filing of Zydus’s ANDA relating to carbidopa and levodopa extended release capsules, generic to Rytary®. Zydus has not yet answered or otherwise responded to the Complaint.
Bristol-Myers Squibb Company, et al. v. Impax Laboratories, Inc. (Apixiban)
On April 10, 2017, Bristol-Myers Squibb Company and Pfizer Inc. filed suit against the Company in the United States District Court for the District of Delaware alleging patent infringement based on the filing of the Company’s ANDA related to Apixaban Tablets, 2.5 mg and 5 mg, generic to Eliquis®. The Company responded to the complaint on June 2, 2017 and Plaintiffs further responded on June 22, 2017. On September 22, 2017, the parties jointly filed a proposed schedule with the Court, proposing that the Company’s case and a number of related cases be consolidated. On November 3, 2017, the Court consolidated the related cases and set the case schedule. Trial is scheduled for October 15, 2019.
Biogen MA Inc. v. Impax Laboratories, Inc. (Dimethyl Fumarate)
On June 26, 2017, Biogen MA Inc. filed suit against the Company in the U.S. District Court for the District of Delaware alleging patent infringement based on the filing of the Company’s ANDA relating to Dimethyl Fumarate 120 and 240 mg capsules, generic to Tecfidera®. The Company answered the complaint on October 16, 2017. On February 2, 2017, the Court consolidated the related cases and set the case schedule. Trial is scheduled for December 9, 2019.
Other Litigation Related to the Company’s Business
Solodyn® Antitrust Class Actions
From July 2013 to January 2016, 18 complaints were filed as class actions on behalf of direct and indirect purchasers, as well as by certain direct purchasers, against manufacturers of the brand drug Solodyn® and its generic equivalents, including the Company.
F-58
On July 22, 2013, Plaintiff United Food and Commercial Workers Local 1776 & Participating Employers Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On July 23, 2013, Plaintiff Rochester Drug Co-Operative, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 1, 2013, Plaintiff International Union of Operating Engineers Local 132 Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Northern District of California on behalf of itself and others similarly situated. On August 29, 2013, this Plaintiff withdrew its complaint from the United States District Court for the Northern District of California, and on August 30, 2013, re-filed the same complaint in the United States Court for the Eastern District of Pennsylvania, on behalf of itself and others similarly situated.
On August 9, 2013, Plaintiff Local 274 Health & Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 12, 2013, Plaintiff Sheet Metal Workers Local No. 25 Health & Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 27, 2013, Plaintiff Fraternal Order of Police, Fort Lauderdale Lodge 31, Insurance Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 29, 2013, Plaintiff Heather Morgan, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 30, 2013, Plaintiff Plumbers & Pipefitters Local 178 Health & Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On September 9, 2013, Plaintiff Ahold USA, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the District of Massachusetts on behalf of itself and others similarly situated.
On September 24, 2013, Plaintiff City of Providence, Rhode Island, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Arizona on behalf of itself and others similarly situated.
On October 2, 2013, Plaintiff International Union of Operating Engineers Stationary Engineers Local 39 Health & Welfare Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Massachusetts on behalf of itself and others similarly situated.
On October 7, 2013, Painters District Council No. 30 Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Massachusetts on behalf of itself and others similarly situated.
On October 25, 2013, Plaintiff Man-U Service Contract Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On March 13, 2014, Plaintiff Allied Services Division Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Massachusetts on behalf of itself and others similarly situated.
On March 19, 2014, Plaintiff NECA-IBEW Welfare Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Massachusetts on behalf of itself and others similarly situated.
On February 25, 2014, the United States Judicial Panel on Multidistrict Litigation ordered the pending actions transferred to the District of Massachusetts for coordinated pretrial proceedings, as In Re Solodyn (Minocycline Hydrochloride) Antitrust Litigation.
F-59
On March 26, 2015, Walgreen Co., The Kruger Co., Safeway Inc., HEB Grocery Company L.P., Albertson’s LLC, direct purchasers, filed a separate complaint in the United States District Court for the Middle District of Pennsylvania. On April 8, 2015, the Judicial Panel on Multi-District Litigation ordered the action be transferred to the District of Massachusetts, to be coordinated or consolidated with the coordinated proceedings. The original complaint filed by the plaintiffs asserted claims only against defendant Medicis. On October 5, 2015, the plaintiffs filed an amended complaint asserting claims against the Company and the other generic defendants.
On April 16, 2015, Rite Aid Corporation and Rite Aid Hdqtrs. Corp, direct purchasers, filed a separate complaint in the United States District Court for the Middle District of Pennsylvania. On May 1, 2015, the Judicial Panel on Multi-District Litigation ordered the action be transferred to the District of Massachusetts, to be coordinated or consolidated with the coordinated proceedings. The original complaint filed by the plaintiffs asserted claims only against defendant Medicis. On October 5, 2015, the plaintiffs filed an amended complaint asserting claims against the Company and the other generic defendants.
On January 25, 2016, CVS Pharmacy, Inc., a direct purchaser, filed a separate complaint in the United States District Court for the Middle District of Pennsylvania. On February 11, 2016, the Judicial Panel on Multi-District Litigation ordered the action to be transferred to the District of Massachusetts to be coordinated or consolidated with the coordinated proceedings.
The consolidated amended complaints allege that Medicis engaged in anticompetitive schemes by, among other things, filing frivolous patent litigation lawsuits, submitting frivolous Citizen Petitions, and entering into anticompetitive settlement agreements with several generic manufacturers, including the Company, to delay generic competition of Solodyn® and in violation of state and federal antitrust laws. Plaintiffs seek, among other things, unspecified monetary damages and equitable relief, including disgorgement and restitution. On August 14, 2015, the District Court granted in part and denied in part defendants’ motion to dismiss the consolidated amended complaints. On October 16, 2017, the Court certified the Direct Purchaser Plaintiffs’ and End-Payor Plaintiffs’ classes. On October 30, 2017, the Company filed a petition for interlocutor appeal challenging the Court's certification of the End-Payor Plantiffs' class. On January 25, 2018, the Court denied Plaintiffs' and the Company's summary judgment motions. Trial is currently set for March 12, 2018.
Opana ER® FTC Antitrust Suit
On February 25, 2014, the Company received a Civil Investigative Demand ("CID") from the FTC concerning its investigation into the drug Opana® ER and its generic equivalents. On March 30, 2016, the FTC filed a complaint against the Company, Endo, and others in the United States District Court for the Eastern District of Pennsylvania, alleging that the Company and Endo violated antitrust laws when they entered into a June 2010 co-promotion and development agreement and a June 2010 settlement agreement that resolved patent litigation in connection with the submission of the Company’s ANDA for generic original Opana® ER. In July 2016, the defendants filed a motion to dismiss the complaint, and a motion to sever the claims regarding Opana® ER from claims with respect to a separate settlement agreement that was challenged by the FTC. On October 20, 2016, the Court granted the motion to sever, formally terminating the suit against the Company, with an order that the FTC re-file no later than November 3, 2016 and dismissed the motion to dismiss as moot. On October 25, 2016, the FTC filed a notice of voluntary dismissal. On January 19, 2017, the FTC filed a Part 3 Administrative complaint against the Company with similar allegations regarding the Company’s June 2010 settlement agreement with Endo that resolved patent litigation in connection with the submission of the Company’s ANDA for generic original Opana® ER. The Company filed its answer to the Administrative Complaint on February 7, 2017. Trial concluded on November 15, 2017. Post-trial briefing is complete and closing arguments were held February 15, 2018. A decision is pending.
Opana ER® Antitrust Class Actions
From June 2014 to April 2015, 14 complaints were filed as class actions on behalf of direct and end-payor (indirect) purchasers, as well as by certain direct purchasers, against the manufacturer of the brand drug Opana ER® and the Company.
On June 4, 2014, Plaintiff Fraternal Order of Police, Miami Lodge 20, Insurance Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 4, 2014, Plaintiff Rochester Drug Co-Operative, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
F-60
On June 6, 2014, Plaintiff Value Drug Company, a direct purchaser, filed a class action complaint in the United States District Court for the Northern District of California on behalf of itself and others similarly situated. On June 26, 2014, this Plaintiff withdrew its complaint from the United States District Court for the Northern District of California, and on July 16, 2014, re-filed the same complaint in the United States District Court for the Northern District of Illinois, on behalf of itself and others similarly situated.
On June 19, 2014, Plaintiff Wisconsin Masons’ Health Care Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Northern District of Illinois on behalf of itself and others similarly situated.
On July 17, 2014, Plaintiff Massachusetts Bricklayers, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On August 11, 2014, Plaintiff Pennsylvania Employees Benefit Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Northern District of Illinois on behalf of itself and others similarly situated.
On September 19, 2014, Plaintiff Meijer Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Northern District of Illinois on behalf of itself and others similarly situated.
On October 3, 2014, Plaintiff International Union of Operating Engineers, Local 138 Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Northern District of Illinois on behalf of itself and others similarly situated.
On November 17, 2014, Louisiana Health Service & Indemnity Company d/b/a Blue Cross and Blue Shield of Louisiana, an indirect purchaser, filed a class action complaint in the United States District Court for the Middle District of Louisiana on behalf of itself and others similarly situated.
On December 12, 2014, the United States Judicial Panel on Multidistrict Litigation ordered the pending actions transferred to the Northern District of Illinois for coordinated pretrial proceedings, as In Re Opana ER Antitrust Litigation.
On December 19, 2014, Plaintiff Kim Mahaffay, an indirect purchaser, filed a class action complaint in the Superior Court of the State of California, Alameda County, on behalf of herself and others similarly situated. On January 27, 2015, the Defendants removed the action to the United States District Court for the Northern District of California.
On January 12, 2015, Plaintiff Plumbers & Pipefitters Local 178 Health & Welfare Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Northern District of Illinois on behalf of itself and others similarly situated.
On March 26, 2015 Walgreen Co., The Kruger Co., Safeway Inc., HEB Grocery Company L.P., Albertson’s LLC, direct purchasers, filed a separate complaint in the United States District Court for the Northern District of Illinois.
On April 23, 2015, Rite Aid Corporation and Rite Aid Hdqtrs. Corp, direct purchasers, filed a separate complaint in the United States District Court for the Northern District of Illinois.
In each case, the complaints allege that Endo engaged in an anticompetitive scheme by, among other things, entering into an anticompetitive settlement agreement with the Company to delay generic competition of Opana ER® and in violation of state and federal antitrust laws. Plaintiffs seek, among other things, unspecified monetary damages and equitable relief, including disgorgement and restitution. Consolidated amended complaints were filed on May 4, 2015 by direct purchaser plaintiffs and end-payor (indirect) purchaser plaintiffs.
On July 3, 2015, defendants filed motions to dismiss the consolidated amended complaints, as well as the complaints of the “Opt-Out Plaintiffs” (Walgreen Co., The Kruger Co., Safeway Inc., HEB Grocery Company L.P., Albertson’s LLC, Rite Aid Corporation and Rite Aid Hdqtrs. Corp.).
On February 1, 2016, CVS Pharmacy, Inc. filed a complaint in the United States District Court for the Northern District of Illinois. The parties agreed that CVS Pharmacy, Inc. would be bound by the court’s ruling on the defendants’ motion to dismiss the Opt-Out Plaintiffs’ complaints.
F-61
On February 10, 2016, the court granted in part and denied in part defendants’ motion to dismiss the end-payor purchaser plaintiffs’ consolidated amended complaint, and denied defendants’ motion to dismiss the direct purchaser plaintiffs’ consolidated amended complaint. The end-payor purchaser plaintiffs have filed a second consolidated amended complaint and the Company has moved to dismiss certain state law claims.
On February 25, 2016, the court granted defendants’ motion to dismiss the Opt-Out Plaintiffs’ complaints, with leave to amend. The Opt-Out Plaintiffs and CVS Pharmacy, Inc. have filed amended complaints and the Company has filed its answer.
Discovery is ongoing. No trial date has been scheduled.
United States Department of Justice Investigations
Previously on November 6, 2014, the Company disclosed that one of its sales representatives received a grand jury subpoena from the Antitrust Division of the United States Justice Department (the “Justice Department”). In connection with this same investigation, on March 13, 2015, the Company received a grand jury subpoena from the Justice Department requesting the production of information and documents regarding the sales, marketing, and pricing of certain generic prescription medications. In particular, the Justice Department’s investigation currently focuses on four generic medications: digoxin tablets, terbutaline sulfate tablets, prilocaine/lidocaine cream, and calcipotriene topical solution. The Company has been cooperating and intends to continue cooperating with the investigation. However, no assurance can be given as to the timing or outcome of the investigation.
Attorney General of the State of Connecticut Interrogatories and Subpoena Duces Tecum
On July 14, 2014, the Company received a subpoena and interrogatories (the “Subpoena”) from the State of Connecticut Attorney General (“Connecticut AG”) concerning its investigation into sales of the Company’s generic product, digoxin. According to the Connecticut AG, the investigation is to determine whether anyone engaged in a contract, combination or conspiracy in restraint of trade or commerce which has the effect of (i) fixing, controlling or maintaining prices or (ii) allocating or dividing customers or territories relating to the sale of digoxin in violation of Connecticut state antitrust law. The Company intends to cooperate with the Connecticut AG in producing documents and information in response to the Subpoena. To the knowledge of the Company, no proceedings by the Connecticut AG have been initiated against the Company at this time; however no assurance can be given as to the timing or outcome of this investigation.
In re Generic Pharmaceuticals Pricing Antitrust Litigation
From March 2016 to April 2017, 22 complaints were filed as class actions on behalf of direct and indirect purchasers against manufacturers of generic digoxin and doxycycline and the Company alleging a conspiracy to fix, maintain and/or stabilize prices of these generic products. From January 2017 to April 2017, three complaints were filed on behalf of indirect purchasers against manufacturers of generic lidocaine/prilocaine and the Company alleging a conspiracy to fix, maintain and/or stabilize prices of these generic products.
On March 2, 2016, Plaintiff International Union of Operating Engineers Local 30 Benefits Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated. The plaintiff filed an amended complaint on June 9, 2016.
On March 25, 2016, Plaintiff Tulsa Firefighters Health and Welfare Trust, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On March 25, 2016, Plaintiff NECA-IBEW Welfare Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On April 4, 2016, Plaintiff Pipe Trade Services MN, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On April 25, 2016, Plaintiff Edward Carpinelli, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On April 27, 2016, Plaintiff Fraternal Order of Police, Miami Lodge 20, Insurance Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
F-62
On May 2, 2016, Plaintiff Nina Diamond, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On May 5, 2016, Plaintiff UFCW Local 1500 Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On May 6, 2016, Plaintiff Minnesota Laborers Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On May 12, 2016, Plaintiff the City of Providence, Rhode Island, an indirect purchaser, filed a class action complaint in the United States District Court for the District of Rhode Island on behalf of itself and others similarly situated.
On May 18, 2016, Plaintiff KPH Healthcare Services, Inc. a/k/a Kinney Drugs, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On May 19, 2016, Plaintiff Philadelphia Federation of Teachers Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 8, 2016, Plaintiff United Food & Commercial Workers and Employers Arizona Health and Welfare Trust, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 17, 2016, Plaintiff Ottis McCrary, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 20, 2016, Plaintiff Rochester Drug Co-Operative, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 27, 2016, Plaintiff César Castillo Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On June 29, 2016, Plaintiff Plumbers & Pipefitters Local 33 Health and Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On July 1, 2016, Plaintiff Plumbers & Pipefitters Local 178 Health and Welfare Trust Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On July 15, 2016, Plaintiff Ahold USA, Inc., a direct purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On September 7, 2016, Plaintiff United Here Health, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On September 20, 2016, Plaintiff Valerie Velardi, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated.
On January 13, 2017, Plaintiff International Union of Operating Engineers Local 30 Benefits Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated against manufacturers of generic lidocaine/prilocaine and the Company alleging a conspiracy to fix, maintain and/or stabilize prices of this generic drug.
F-63
On April 17, 2017, Plaintiff UFCW Local 1500 Welfare Fund, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated against manufacturers of generic lidocaine/prilocaine and the Company alleging a conspiracy to fix, maintain and/or stabilize prices of this generic drug.
On April 25, 2017, Plaintiff Louisiana Health Service Indemnity Company, an indirect purchaser, filed a class action complaint in the United States District Court for the Eastern District of Pennsylvania on behalf of itself and others similarly situated against manufacturers of generic lidocaine/prilocaine and the Company alleging a conspiracy to fix, maintain and/or stabilize prices of this generic drug.
On May 19, 2016, several indirect purchaser plaintiffs filed a motion with the Judicial Panel on Multidistrict Litigation to transfer and consolidate the actions in the United States District Court for the Eastern District of Pennsylvania. The Judicial Panel ordered the actions consolidated in the Eastern District of Pennsylvania and ordered that the actions be renamed “In re Generic Digoxin and Doxycycline Antitrust Litigation”. On January 27, 2017, plaintiffs filed two consolidated class action complaints. With respect to doxycycline, the plaintiffs dropped their allegations against the Company. On March 28, 2017, the Company, separately and along with other defendants, filed motions to dismiss the digoxin class action complaint. On April 6, 2017, the Judicial Panel on Multidistrict Litigation ordered the consolidation of all civil actions involving allegations of antitrust conspiracies in the generic pharmaceutical industry regarding 18 generic drugs to the Eastern District of Pennsylvania. The consolidated actions have been renamed In re Generic Pharmaceuticals Pricing Antitrust Litigation. On October 6, 2017, the Company filed a motion to dismiss the digoxin complaint. Briefing on the motion to dismiss is complete and a decision is pending.
On January 19, 2018, Plaintiffs The Kroger Co., Albertsons Companies, LLC, and H.E. Butt Grocery Company L.P., opt-outs, filed a complaint in the United States District Court for the Eastern District of Pennsylvania against 35 companies, including the Company, alleging a conspiracy to fix, maintain and/or stabilize prices of thirty drugs and specifically digoxin and lidocaine/prilocaine with respect to the Company. No schedule has been set.
AWP Litigation
On December 30, 2015, Plumbers’ Local Union No. 690 Health Plan and others similarly situated filed a class action against several generic drug manufacturers, including the Company, in the Court of Common Pleas of Philadelphia County, First Judicial District of Pennsylvania, Civil Trial Division, alleging that the Company and others violated the law, including the Pennsylvania Unfair Trade Practices and Consumer Protection law, by inflating the Average Wholesale Price (“AWP”) of certain generic drugs. The case has since been removed to federal court in the United States District Court for the Eastern District of Pennsylvania. By virtue of an amended complaint filed on March 29, 2016, the suit has been amended to comprise a nationwide class of third party payors that allegedly reimbursed or purchased certain generic drugs based on AWP and to assert causes of action under the laws of other states in addition to Pennsylvania. On May 17, 2016, this case was stayed. On January 18, 2017, the Company, along with the other defendants, filed a joint motion to dismiss the complaint. On September 15, 2017, the Court dismissed the complaint with prejudice. The time period to file an appeal has elapsed.
On February 5, 2016, Delaware Valley Health Care Coalition filed a lawsuit based on substantially similar allegations in the Court of Common Pleas of Philadelphia County, First Judicial District of Pennsylvania, Civil Trial Division that seeks declaratory judgment. On May 20, 2016, this case was stayed pending resolution of the federal court action described above.
F-64
Impax Laboratories, Inc. v. Turing Pharmaceuticals AG
On May 2, 2016, the Company filed suit against Turing Pharmaceuticals AG ("Turing") in the United States District Court for the Southern District of New York alleging breach of the terms of the contract by which Turing purchased from the Company the right to sell the drug Daraprim®, as well as the right to sell certain Daraprim® inventory (the “Purchase Agreement”). Specifically, the Company seeks (i) a declaratory judgment that the Company may revoke Turing’s right to sell Daraprim® under the Company’s labeler code and national drug codes; (ii) specific performance to require Turing to comply with its obligations under the Purchase Agreement for past due reports and for reports going forward; and (iii) money damages to remedy Turing’s failure to reimburse the Company for chargebacks and Medicaid rebate liability when due, currently in excess of $40.9 million, and for future amounts that may be due. Turing has filed its answer and a counterclaim against the Company alleging breach of contract and breach of the duty of good faith and fair dealing. Discovery is closed. On October 14, 2016, the Company filed a motion for summary judgment. The District Court issued its order on September 29, 2017. The Court found that Turing breached the Purchase Agreement by failing to reimburse the Company for Medicaid rebate liability, however, the Court also found that the Company breached the Purchase Agreement by not filing a restatement with the Centers for Medicare and Medicaid Services at Turing’s request. Therefore, the Company was not entitled to damages. On October 13, 2017, the Company filed a Motion for Clarification / Reconsideration of the Summary Judgment Order. Briefing on the motion is complete and a decision is pending.
Telephone Consumer Protection Act Cases
On January 31, 2017, Plaintiff Family Medicine Pharmacy LLC filed a class action complaint in the United States District Court for the Southern District of Alabama on behalf of itself and others similarly situated against the Company alleging violation of the Telephone Consumer Protection Act, as amended by the Junk Fax Prevention Act of 2005 (the "Telephone Consumer Protection Act"). On March 27, 2017, the Company filed a motion to dismiss the complaint and plaintiff filed an amended complaint on April 10, 2017. On July 18, 2017, the parties reached an agreement in principle regarding the class settlement. On September 29, 2017, the District Court preliminarily approved the proposed class settlement. The Court is scheduled to hold a hearing on March 6, 2018 regarding the final approval of the proposed class settlement.
On February 14, 2017, Plaintiff Medicine To Go Pharmacies, Inc. filed a class action complaint in the United States District Court for the District of New Jersey on behalf of itself and others similarly situated against the Company alleging violation of the Telephone Consumer Protection Act. On April 17, 2017, the Company filed a motion to dismiss, transfer, or stay this case in light of the first-filed case described above. This case was transferred to the Southern District of Alabama. On September 15, 2017, the Court stayed this matter pending the final approval of the class settlement described above.
Securities Class Action
On April 17, 2017, Lead Plaintiff New York Hotel Trades Council & Hotel Association of New York City, Inc. Pension Fund filed an amended class action complaint in the United States District Court for the Northern District of California on behalf of itself and others similarly situated against the Company alleging violations of Sections 10(b) and 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5. The Company filed its motion to dismiss the amended complaint on June 1, 2017 and briefing has been completed.
Shareholder Derivative Action
On February 22, 2017, Plaintiff Ed Lippman filed a shareholder derivative complaint in the Superior Court for the State of California in the County of Alameda on behalf of the Company against former executives, a current executive, and certain current members of the board of directors alleging breach of fiduciary duty, unjust enrichment, abuse of control, gross mismanagement, and corporate waste. This matter has been stayed pending the securities class action referenced above.
Teva v. Impax Laboratories, Inc.
On February 15, 2017, Plaintiffs Teva Pharmaceuticals USA, Inc. and Teva Pharmaceuticals Curacao N.V. (“Teva”) filed a Praecipe to Issue Writ of Summons and Writ of Summons (precursor to a complaint) in the Philadelphia County Court of Common Pleas against the Company alleging that the Company breached the Strategic Alliance Agreement between the parties by not indemnifying Teva in its two litigations with GlaxoSmithKline LLC regarding Wellbutrin® XL. The Company filed a Motion to Disqualify Teva’s counsel related to the matter, and on August 23, 2017, the Court denied the Company’s motion. Following the Court’s order, Teva filed its complaint. The Company has filed its appeal regarding the disqualification order, and oral argument will be held on April 10, 2018. The matter is currently stayed.
F-65
California Wage and Hour Class Action
On August 3, 2017, Plaintiff Emielou Williams filed a class action complaint in the Superior Court for the State of California in the County of Alameda on behalf of herself and others similarly situated against the Company alleging violation of California Business and Professions Code section 17200 by violating various California wage and hour laws. On October 10, 2017, the Company filed a Demurrer and Motion to Strike Class Allegations. On December 12, 2017, the Court overruled the Company's Demurrer to Plaintiff's individual claims, however, it struck all of Plaintiff's class allegations. Discovery is ongoing.
Securities Class Actions
On December 12, 2017 and December 14, 2017, Plaintiffs Susan Vana and David Stone, respectively, filed class action complaints in the United States District Court for the Northern District of California on behalf of themselves and others similarly situated against the Company alleging violations of Sections 14(a) and 20(a) of the Securities Exchange Act of 1934 generally alleging that the Registration Statement on Form S-4 related to the proposed business combination with Amneal Pharmaceuticals, LLC (“Amneal”) contains false and misleading statements and/or omissions concerning the financial projections of the Company, Amneal, and New Amneal; Morgan Stanley & Co. LLC’s valuation analyses and Fairness Opinions relating to the Company and Amneal; potential conflicts of interest associated with one of the Company’s financial advisors and the proposed business combination with Amneal; and background information of the proposed business combination, including confidentiality agreements entered into by the Company in connection with the proposed business combination. No schedule has been set.
20. SEGMENT INFORMATION
The Company has two reportable segments, Impax Generics and Impax Specialty Pharma. Impax Generics develops, manufactures, sells, and distributes generic pharmaceutical products, primarily through the following sales channels: the Impax Generics sales channel for sales of generic prescription products directly to wholesalers, large retail drug chains, and others; the Private Label Product sales channel for generic over-the-counter and prescription products sold to unrelated third-party customers who, in turn, sell the products under their own label; the Rx Partner sales channel for generic prescription products sold through unrelated third-party pharmaceutical entities under their own label pursuant to alliance agreements; and the OTC Partner sales channel for over-the-counter products sold through unrelated third-party pharmaceutical entities under their own labels pursuant to alliance and supply agreements. Revenues from generic products are reported under the caption "Impax Generics, net."
Impax Specialty Pharma is engaged in the development, sale and distribution of proprietary brand pharmaceutical products that the Company believes represent improvements to already-approved pharmaceutical products addressing central nervous system (“CNS”) disorders and other select specialty segments. Impax Specialty Pharma currently has one internally developed branded pharmaceutical product, Rytary® (IPX066), an extended release oral capsule formulation of carbidopa-levodopa for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication and/or manganese intoxication, which was approved by the FDA on January 7, 2015 and which the Company launched in April 2015. In November 2015, the European Commission granted marketing authorization for Numient® (IPX066) (referred to as Rytary® in the United States). The review of the Numient® application was conducted under the centralized licensing procedure as a therapeutic innovation, and authorization is applicable in all 28 member states of the European Union, as well as Iceland, Liechtenstein and Norway. Impax Specialty Pharma is also engaged in the sale and distribution of four other branded products including Zomig® (zolmitriptan) products, indicated for the treatment of migraine headaches, under the terms of the AZ Agreement with AstraZeneca in the United States and in certain U.S. territories, and Emverm® (mebendazole) 100 mg chewable tablets, indicated for the treatment of pinworm, whipworm, common roundworm, common hookworm, and American hookworm in single or mixed infections.
Revenues from branded products are reported under the cation "Impax Specialty Pharma, net." Impax Specialty Pharma also has a number of product candidates that are in varying stages of development.
The Company’s chief operating decision maker evaluates the financial performance of the Company’s segments based upon segment income (loss) before income taxes. Items below income (loss) from operations are not reported by segment, since they are excluded from the measure of segment profitability reviewed by the Company’s chief operating decision maker. Additionally, general and administrative expenses, certain selling expenses, certain litigation settlements, and non-operating income and expenses are included in “Corporate and Other.” The Company does not report balance sheet information by segment since it is not reviewed by the Company’s chief operating decision maker. The accounting policies for the Company’s segments are the same as those described above in the discussion of "Revenue Recognition" and in “Note 2. Summary of Significant Accounting Policies.” The Company has no inter-segment revenue.
F-66
The tables below present segment information reconciled to total Company financial results, with segment operating income or loss including gross profit less direct research and development expenses and direct selling expenses as well as any litigation settlements, to the extent specifically identified by segment (in thousands):
Year Ended December 31, 2017 | Impax Generics | Impax Specialty Pharma | Corporate and Other | Total Company | |||||||||||
Revenues, net | $ | 549,077 | $ | 226,710 | $ | — | $ | 775,787 | |||||||
Cost of revenues | 454,911 | 80,212 | — | 535,123 | |||||||||||
Cost of revenues impairment charges | 96,865 | — | — | 96,865 | |||||||||||
Selling, general and administrative | 28,294 | 67,949 | 120,027 | 216,270 | |||||||||||
Research and development | 63,245 | 17,602 | — | 80,847 | |||||||||||
In-process research and development impairment charges | 192,809 | — | — | 192,809 | |||||||||||
Fixed assets impairment charges | 8,380 | 74,128 | — | 82,508 | |||||||||||
Change in fair value of contingent consideration | (31,048 | ) | — | — | (31,048 | ) | |||||||||
Patent litigation | 827 | 4,278 | — | 5,105 | |||||||||||
(Loss) before income taxes | (265,206 | ) | (17,459 | ) | (168,296 | ) | (450,961 | ) | |||||||
Year Ended December 31, 2016 | Impax Generics | Impax Specialty Pharma | Corporate and Other | Total Company | |||||||||||
Revenues, net | $ | 606,320 | $ | 218,109 | $ | — | $ | 824,429 | |||||||
Cost of revenues | 417,316 | 69,583 | — | 486,899 | |||||||||||
Cost of revenues impairment charges | 464,319 | 24,313 | — | 488,632 | |||||||||||
Selling, general and administrative | 20,508 | 61,448 | 119,874 | 201,830 | |||||||||||
Research and development | 61,980 | 18,486 | — | 80,466 | |||||||||||
In-process research and development impairment charges | 27,765 | 25,200 | — | 52,965 | |||||||||||
Patent litigation | 829 | 6,990 | — | 7,819 | |||||||||||
(Loss) income before income taxes | (386,397 | ) | 12,089 | (202,017 | ) | (576,325 | ) | ||||||||
Year Ended December 31, 2015 | Impax Generics | Impax Specialty Pharma | Corporate and Other | Total Company | |||||||||||
Revenues, net | $ | 710,932 | $ | 149,537 | $ | — | $ | 860,469 | |||||||
Cost of revenues | 442,742 | 58,020 | — | 500,762 | |||||||||||
Cost of revenues impairment charges | 7,303 | — | — | 7,303 | |||||||||||
Selling, general and administrative | 29,641 | 52,427 | 119,219 | 201,287 | |||||||||||
Research and development | 52,478 | 18,144 | — | 70,622 | |||||||||||
In-process research and development impairment charges | 6,360 | — | — | 6,360 | |||||||||||
Patent litigation | 2,942 | 1,625 | — | 4,567 | |||||||||||
Income (loss) before income taxes | 169,466 | 19,321 | (129,419 | ) | 59,368 | ||||||||||
F-67
Significant Products
The Company generally consolidates net revenue by “product family,” meaning that it consolidates net revenue from products containing the same active ingredient(s) irrespective of dosage strength, delivery method or packaging size. The Company’s significant product families, as determined based on net revenue, and their percentage of the Company’s consolidated net revenue for each of the years ended December 31, 2017, 2016 and 2015 are set forth in the tables below (in thousands):
Segment | Product Family | 2017 | |||||||
$ | % | ||||||||
Impax Generics | Epinephrine Auto-Injector family (generic Adrenaclick®) | $ | 113,931 | 15 | % | (1) | |||
Impax Specialty Pharma | Rytary® family | $ | 91,637 | 12 | % | (2) | |||
Impax Generics | Oxymorphone HCI ER family | $ | 68,587 | 9 | % | (3) | |||
Impax Generics | Budesonide family | $ | 51,548 | 7 | % | (4) | |||
Impax Generics | Zomig family | $ | 51,115 | 7 | % | (5) | |||
Segment | Product Family | 2016 | |||||||
$ | % | ||||||||
Impax Generics | Epinephrine Auto-Injector family (generic Adrenaclick®) | $ | 91,572 | 11 | % | (1) | |||
Impax Specialty Pharma | Rytary® family | $ | 73,833 | 9 | % | (2) | |||
Impax Generics | Oxymorphone HCI ER family | $ | 72,661 | 9 | % | (3) | |||
Impax Generics | Diclofenac Sodium Gel family (generic Solaraze®) | $ | 69,035 | 8 | % | (6) | |||
Impax Generics | Fenofibrate family | $ | 64,001 | 8 | % | (7) | |||
Segment | Product Family | 2015 | |||||||
$ | % | ||||||||
Impax Generics | Diclofenac Sodium Gel family (generic Solaraze®) | $ | 148,610 | 17 | % | (6) | |||
Impax Generics | Amphetamine Salts ER (CII) family (generic Adderall®) | $ | 106,252 | 12 | % | (8) | |||
Impax Generics | Fenofibrate family | $ | 93,458 | 11 | % | (7) | |||
Impax Generics | Metaxalone family (generic Skelaxin) | $ | 69,876 | 8 | % | (9) | |||
Impax Generics | Oxymorphone HCI ER family | $ | 59,175 | 7 | % | (3) | |||
(1) Epinephrine Auto-Injector (generic Adrenaclick®) product family consists of the injector product in two different strengths and is indicated in the emergency treatment of allergic reactions (Type 1) including anaphylaxis.
(2) Rytary® product family consists of the capsules product in four different strengths and is indicated for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication or manganese intoxication.
(3) Oxymorphone Hydrochloride Extended Release product family consists of the oxymorphone hydrochloride extended release tablet formulation of the product in seven different strengths and is indicated for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate.
(4) Budesonide Inhalation Suspension (generic Pulmicort Respules®) product family consists of two products strengths and is indicated for the maintenance treatment of asthma.
(5) Zomig® product family consists of products in tablet, orally disintegrating tablet, and nasal spray dosage forms in six different strengths and is indicated for the acute treatment of migraine with or without aura in adults and pediatric patients 12 years of age or older.
F-68
(6) Diclofenac Sodium Gel (generic Solaraze®) product family consists of one product strength and is indicated for the topical treatment of actinic keratosis.
(7) Fenofibrate product family consists of products in both capsule and tablet dosage forms in seven different strengths and is indicated as adjunctive therapy to diet to reduce elevated LDL-C, Total-C, Triglycerides and Apo B, and to increase HDL-C in adult patients with primary hypercholesterolemia or mixed dyslipidemia (Fredrickson Types IIa and IIb); and also indicated as adjunctive therapy to diet for treatment of adult patients with hypertriglyceridemia (Fredrickson Types IV and V hyperlipidemia).
(8) Amphetamine Salts extended release capsules, CII (generic Adderall XR®) product family consists of the capsules product in six different strengths and is indicated for the treatment of attention deficit hyperactivity disorder.
(9) Metaxalone (generic Skelaxin®) product family consists of the tablet product in two different strengths and is indicated as an adjunct to rest, physical therapy, and other measures for the relief of discomforts associated with acute, painful musculoskeletal conditions.
Foreign Operations
The Company’s wholly-owned subsidiary, Impax Laboratories (Taiwan), Inc., constructed a manufacturing facility in Taiwan which was utilized for manufacturing, warehouse, and administrative functions, as well as some limited research and development activities. On the Company's consolidated balance sheet as of December 31, 2017, Impax Laboratories (Taiwan), Inc. represented $22.9 million of net carrying value of assets, which are included in assets and liabilities held for sale. See "Note 15. Restructurings" for additional information related to the sale of the Taiwan operations in the first quarter of 2018.
F-69
21. SUPPLEMENTARY FINANCIAL INFORMATION (Unaudited)
Selected financial information for the quarterly periods noted is as follows:
2017 Quarters Ended | ||||||||||||||||
(in thousands, except share and per share amounts) | March 31 | June 30 | September 30 | December 31 | ||||||||||||
Revenue: | ||||||||||||||||
Impax Generics sales, gross | $ | 635,897 | $ | 663,167 | $ | 622,252 | $ | 584,374 | ||||||||
Less: | ||||||||||||||||
Chargebacks | 298,744 | 286,092 | 281,835 | 302,394 | ||||||||||||
Rebates | 164,792 | 170,398 | 162,914 | 144,344 | ||||||||||||
Product returns | 9,733 | 15,210 | 7,003 | 4,657 | ||||||||||||
Other credits | 28,481 | 40,578 | 19,402 | 20,036 | ||||||||||||
Impax Generics sales, net | 134,147 | 150,889 | 151,098 | 112,943 | ||||||||||||
Impax Specialty Pharma sales, gross | 84,133 | 84,238 | 107,407 | 111,918 | ||||||||||||
Less: | ||||||||||||||||
Chargebacks | 9,828 | 8,967 | 14,121 | 10,058 | ||||||||||||
Rebates | 4,483 | 4,682 | 5,914 | 6,198 | ||||||||||||
Product returns | 1,844 | 1,416 | 3,614 | 4,234 | ||||||||||||
Other credits | 17,722 | 17,980 | 28,464 | 21,461 | ||||||||||||
Impax Specialty Pharma revenues, net | 50,256 | 51,193 | 55,294 | 69,967 | ||||||||||||
Total revenues | 184,403 | 202,082 | 206,392 | 182,910 | ||||||||||||
Gross profit | 24,891 | 72,406 | 34,033 | 12,469 | ||||||||||||
Net loss | $ | (98,431 | ) | $ | (20,417 | ) | $ | (49,369 | ) | $ | (301,070 | ) | ||||
Net loss per common share: | ||||||||||||||||
Basic | $ | (1.37 | ) | $ | (0.28 | ) | $ | (0.69 | ) | $ | (4.18 | ) | ||||
Diluted | $ | (1.37 | ) | $ | (0.28 | ) | $ | (0.69 | ) | $ | (4.18 | ) | ||||
Weighted-average common shares outstanding: | ||||||||||||||||
Basic | 71,594,472 | 71,803,920 | 71,924,592 | 72,098,533 | ||||||||||||
Diluted | 71,594,472 | 71,803,920 | 71,924,592 | 72,098,533 | ||||||||||||
F-70
Quarterly computations of net loss per share amounts are made independently for each quarterly reporting period, and the sum of the per share amounts for the quarterly reporting periods may not equal the per share amounts for the year-to-date reporting period.
2016 Quarters Ended | ||||||||||||||||
(in thousands, except share and per share amounts) | March 31 | June 30 | September 30 | December 31 | ||||||||||||
Revenue: | ||||||||||||||||
Impax Generics sales, gross | $ | 614,176 | $ | 532,968 | $ | 658,099 | $ | 690,674 | ||||||||
Less: | ||||||||||||||||
Chargebacks | 217,354 | 197,864 | 252,303 | 308,253 | ||||||||||||
Rebates | 185,476 | 178,097 | 183,347 | 211,359 | ||||||||||||
Product returns | 11,913 | 10,237 | 16,151 | 7,920 | ||||||||||||
Other credits | 29,354 | 25,075 | 30,978 | 23,916 | ||||||||||||
Impax Generics revenues, net | 170,079 | 121,695 | 175,320 | 139,226 | ||||||||||||
Impax Specialty Pharma sales, gross | 82,073 | 81,254 | 77,841 | 108,121 | ||||||||||||
Less: | ||||||||||||||||
Chargebacks | 6,111 | 8,826 | 5,439 | 15,253 | ||||||||||||
Rebates | 2,853 | 2,430 | 3,556 | 3,016 | ||||||||||||
Product returns | 1,508 | 1,279 | 574 | 2,802 | ||||||||||||
Other credits | 16,172 | 17,824 | 15,683 | 27,854 | ||||||||||||
Impax Specialty Pharma revenues, net | 55,429 | 50,895 | 52,589 | 59,196 | ||||||||||||
Total revenues | 225,508 | 172,590 | 227,909 | 198,422 | ||||||||||||
Gross profit (loss) | 102,590 | 72,984 | (165,426 | ) | (161,250 | ) | ||||||||||
Net loss | $ | (10,408 | ) | $ | (2,701 | ) | $ | (179,337 | ) | $ | (279,585 | ) | ||||
Net loss per common share: | ||||||||||||||||
Basic | $ | (0.15 | ) | $ | (0.04 | ) | $ | (2.51 | ) | $ | (3.91 | ) | ||||
Diluted | $ | (0.15 | ) | $ | (0.04 | ) | $ | (2.51 | ) | $ | (3.91 | ) | ||||
Weighted-average common shares outstanding: | ||||||||||||||||
Basic | 70,665,394 | 71,100,123 | 71,331,247 | 71,487,071 | ||||||||||||
Diluted | 70,665,394 | 71,100,123 | 71,331,247 | 71,487,071 | ||||||||||||
Quarterly computations of net loss per share amounts are made independently for each quarterly reporting period, and the sum of the per share amounts for the quarterly reporting periods may not equal the per share amounts for the year-to-date reporting period.
F-71
SCHEDULE II, VALUATION AND QUALIFYING ACCOUNTS
(In thousands)
Column A | Column B | Column C | Column D | Column E | |||||||||||||
Description | Balance at Beginning of Period | Charge to Costs and Expenses | Charge to Other Accounts | Deductions | Balance at End of Period | ||||||||||||
For the Year Ended December 31, 2015: | |||||||||||||||||
Reserve for bad debts | $ | 515 | 5,122 | 9,550 | * | — | $ | 15,187 | |||||||||
For the Year Ended December 31, 2016: | |||||||||||||||||
Reserve for bad debts | $ | 15,187 | 41,213 | — | (1,664 | ) | $ | 54,736 | |||||||||
For the Year Ended December 31, 2017: | |||||||||||||||||
Reserve for bad debts | $ | 54,736 | 3,804 | — | (9,117 | ) | $ | 49,423 | |||||||||
* Represents reserve for bad debts acquired.
S-1
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
IMPAX LABORATORIES, INC. | |||
By: | /s/ Paul M. Bisaro | ||
Name: | Paul M. Bisaro | ||
Title: | President and | ||
Chief Executive Officer | |||
Date: March 1, 2018
Each person whose signature appears below constitutes and appoints each of Paul M. Bisaro and Bryan M. Reasons, acting alone or together with another attorney-in-fact, as his or her true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for such person and in his or her name, place and stead, in any and all capacities, to sign any or all further amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ Paul M. Bisaro | President, Chief Executive Officer | March 1, 2018 | ||
Paul M. Bisaro | (Principal Executive Officer) and Director | |||
/s/ Bryan M. Reasons | Senior Vice President, Finance and | March 1, 2018 | ||
Bryan M. Reasons | Chief Financial Officer | |||
(Principal Financial Officer and | ||||
Principal Accounting Officer) | ||||
/s/ Robert L. Burr | Chairman of the Board | March 1, 2018 | ||
Robert L. Burr | ||||
/s/ Leslie Z. Benet, Ph.D. | Director | March 1, 2018 | ||
Leslie Z. Benet, Ph.D. | ||||
/s/ J. Kevin Buchi | Director | March 1, 2018 | ||
J. Kevin Buchi | ||||
/s/ Allen Chao, Ph.D. | Director | March 1, 2018 | ||
Allen Chao, Ph.D. | ||||
/s/ Mary K. Pendergast | Director | March 1, 2018 | ||
Mary K. Pendergast | ||||
/s/ Peter R. Terreri | Director | March 1, 2018 | ||
Peter R. Terreri | ||||
/s/ Janet S. Vergis | Director | March 1, 2018 | ||
Janet S. Vergis | ||||
EXHIBIT INDEX
Exhibit No. | Description of Document | ||
Business Combination Agreement, dated as of October 17, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation, and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on October 17, 2017).† | Incorporated by Reference | ||
Amendment No. 1 to the Business Combination Agreement, dated as of November 21, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on November 21, 2017). | Incorporated by Reference | ||
Amendment No. 2 to the Business Combination Agreement, dated as of December 16, 2017, by and among the Company, Atlas Holdings, Inc., K2 Merger Sub Corporation and Amneal Pharmaceuticals LLC (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on December 20, 2017). | Incorporated by Reference | ||
Stock Purchase Agreement, dated as of October 8, 2014, by and among the Company, Tower Holdings, Inc. (“Tower”), Lineage Therapeutics Inc. (“Lineage”), Roundtable Healthcare Partners II, L.P., Roundtable Healthcare Investors II, L.P., the other stockholders of Tower and Lineage, the holders of options to purchase shares of Tower common stock and options to purchase shares of Lineage common stock, the holders of warrants to acquire shares of Tower common stock and warrants to acquire shares of Lineage common stock and, solely with respect to Section 8.3, Roundtable Healthcare Management II, LLC. (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on October 10, 2014).† | Incorporated by Reference | ||
Restated Certificate of Incorporation of the Company dated as of August 30, 2004 (incorporated by reference to Exhibit 3.1 to Amendment No. 5 to the Company’s Registration Statement on Form 10 filed on December 23, 2008). | Incorporated by Reference | ||
Certificate of Amendment of the Restated Certificate of Incorporation of the Company dated as of December 9, 2015 (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on December 9, 2015). | Incorporated by Reference | ||
Certificate of Designation of Series A Junior Participating Preferred Stock, as filed with the Secretary of State of Delaware on January 21, 2009 (incorporated by reference to Exhibit 3.3 to the Company’s Current Report on Form 8-K filed on January 22, 2009). | Incorporated by Reference | ||
Amended and Restated Bylaws of the Company, effective as of May 14, 2014. | Filed Herewith | ||
Amendment No. 1 to Amended and Restated Bylaws of the Company, effective as of March 24, 2015. | Filed Herewith | ||
Amendment No. 2 to Amended and Restated Bylaws of the Company, effective as of July 7, 2015. | Filed Herewith | ||
Amendment No. 3 to Amended and Restated Bylaws of the Company, effective as of October 7, 2015. | Filed Herewith | ||
Amendment No. 4 to Amended and Restated Bylaws of the Company, effective as of May 17, 2016. | Filed Herewith | ||
Amendment No. 5 to Amended and Restated Bylaws of the Company, effective as of August 19, 2016. | Filed Herewith | ||
Amendment No. 6 to Amended and Restated Bylaws of the Company, effective as of November 23, 2016. | Filed Herewith | ||
Amendment No. 7 to Amended and Restated Bylaws of the Company, effective as of December 19, 2016. | Filed Herewith | ||
Amendment No. 8 to Amended and Restated Bylaws of the Company, effective as of March 24, 2017. | Filed Herewith | ||
Amendment No. 9 to Amended and Restated Bylaws of the Company, effective as of November 10, 2017. | Filed Herewith | ||
Specimen of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form 10 filed on October 10, 2008). | Incorporated by Reference | ||
Preferred Stock Rights Agreement, dated as of January 20, 2009, by and between the Company and StockTrans, Inc., as Rights Agent (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on January 22, 2009). | Incorporated by Reference | ||
Indenture, dated as of June 30, 2015, between the Company and Wilmington Trust, National Association, as trustee (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Supplemental Indenture, dated as of November 6, 2017, between the Company and Wilmington Trust, National Association, as trustee (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on November 7, 2017). | Incorporated by Reference | ||
Letter Agreement, dated as of June 25, 2015, between RBC Capital Markets LLC and the Company regarding the Base Warrants (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 25, 2015, between RBC Capital Markets LLC and the Company regarding the Base Call Option Transaction (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 26, 2015, between RBC Capital Markets LLC and the Company regarding the Additional Warrants (incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Letter Agreement, dated as of June 26, 2015, between RBC Capital Markets LLC and the Company regarding the Additional Call Option Transaction (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on June 30, 2015). | Incorporated by Reference | ||
Credit Agreement, dated as of August 4, 2015, by and among the Company, the lenders party thereto from time to time and Royal Bank of Canada, as administrative agent and collateral agent (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 5, 2015). | Incorporated by Reference | ||
Restatement Agreement, dated as of August 3, 2016, by and among the Company, the guarantors party thereto, Royal Bank of Canada, as administrative agent, and the lenders party thereto (incorporated by reference to Exhibit 10.7 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016). | Incorporated by Reference | ||
Amendment No. 1, dated as of March 27, 2017, to the Credit Agreement, dated as of August 4, 2015, as amended and restated as of August 3, 2016, among the Company, as borrower, Royal Bank of Canada, as administrative agent and collateral agent, the lenders party thereto and the other agents and parties thereto (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017). | Incorporated by Reference | ||
Distribution, License, Development and Supply Agreement, dated as of January 31, 2012, between the Company and AstraZeneca UK Limited (incorporated by reference to Exhibit 10.1 to Amendment No. 1 to the Company’s Current Report on Form 8-K filed on April 2, 2012)**. | Incorporated by Reference | ||
First Amendment, dated as of May 31, 2016, to the Distribution, License, Development and Supply Agreement by and between AstraZeneca UK Limited and the Company dated as of January 31, 2012 (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016).** | Incorporated by Reference | ||
Stock and Asset Purchase Agreement, dated as of December 19, 2017, by and between the Company and Bora Pharmaceuticals Co., Ltd. † | Filed Herewith | ||
Master Supply Agreement, dated as of December 19, 2017, between the Company, Bora Pharmaceuticals Co., Ltd. and Impax Laboratories (Taiwan), Inc.*** | Filed Herewith | ||
Asset Purchase Agreement, dated as of June 20, 2016, between Teva Pharmaceutical Industries Ltd. and the Company (incorporated by reference to Exhibit 10.2.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).†** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Asset Purchase Agreement between Teva Pharmaceutical Industries Ltd. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.2.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016). | Incorporated by Reference | ||
Asset Purchase Agreement, dated as of June 20, 2016, by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company (incorporated by reference to Exhibit 10.3.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).†** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Asset Purchase Agreement by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.3.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2106 filed on August 9, 2016).** | Incorporated by Reference | ||
Supply Agreement, dated as of June 20, 2016, between Teva Pharmaceutical Industries Ltd. and the Company (incorporated by reference to Exhibit 10.4.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017).** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Supply Agreement between Teva Pharmaceutical Industries Ltd. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.4.2 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Supply Agreement, dated as of June 20, 2016, by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company (incorporated by reference to Exhibit 10.5.1 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Amendment No. 1, dated as of June 30, 2016, to the Supply Agreement by and among Actavis Elizabeth LLC, Actavis Group PTC Ehf., Actavis Holdco US, Inc., Actavis LLC, Actavis Mid Atlantic LLC, Actavis Pharma, Inc., Actavis South Atlantic LLC, Andrx LLC, Breath Ltd., The Rugby Group, Inc., Watson Laboratories, Inc. and the Company dated as of June 20, 2016 (incorporated by reference to Exhibit 10.5.2 to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on January 6, 2017)** | Incorporated by Reference | ||
Impax Laboratories, Inc. 1999 Equity Incentive Plan (incorporated by reference to Exhibit 10.4 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2008 filed on March 12, 2009).* | Incorporated by Reference | ||
Form of Stock Option Grant under the Impax Laboratories, Inc. 1999 Equity Incentive Plan (incorporated by reference to Exhibit 10.4.1 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2008 filed on March 12, 2009).* | Incorporated by Reference | ||
Impax Laboratories, Inc. 2001 Non-Qualified Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to the Company’s Registration Statement on Form 10 filed on October 10, 2008).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Amended and Restated Non-Qualified Employee Stock Purchase Plan (incorporated by reference to Exhibit 4.13 to the Company’s Registration Statement on Form S-8 filed on August 29, 2017).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Appendix B to the Company’s definitive proxy statement on Schedule 14A filed on April 5, 2017).* | Incorporated by Reference | ||
Form of Stock Option Agreement under the Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2017 filed on August 9, 2017).* | Incorporated by Reference | ||
Form of Restricted Stock (Stock Bonus) Award Agreement under the Impax Laboratories, Inc. Fourth Amended and Restated 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2017 filed on August 9, 2017).* | Incorporated by Reference | ||
Impax Laboratories, Inc. Executive Non-Qualified Deferred Compensation Plan, amended and restated effective January 1, 2008 (incorporated by reference to Exhibit 10.1.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 filed on May 6, 2010).* | Incorporated by Reference | ||
Amendment to Impax Laboratories, Inc. Executive Non-Qualified Deferred Compensation Plan, effective as of January 1, 2009 (incorporated by reference to Exhibit 10.1.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 filed on May 6, 2010).* | Incorporated by Reference | ||
Employment Agreement, dated as of March 24, 2017, between the Company and Paul M. Bisaro (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017).* | Incorporated by Reference | ||
Stock Option Agreement with Paul M. Bisaro (incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017 filed on May 10, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of December 16, 2017, by and among Amneal Pharmaceuticals LLC, Atlas Holdings, Inc., a wholly owned subsidiary of the Company, and Robert A. Stewart (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 20, 2017).* | Incorporated by Reference | ||
Memorandum of Understanding, dated as of December 16, 2017, by and among Amneal Pharmaceuticals LLC, Paul M. Bisaro, the Company and Atlas Holdings, Inc., a wholly owned subsidiary of the Company (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on December 20, 2017).* | Incorporated by Reference | ||
Letter Agreement, dated as of December 19, 2016, between the Company and J. Kevin Buchi (incorporated by reference to Exhibit 10.19 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 filed on March 1, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of April 21, 2014, by and between the Company and G. Frederick Wilkinson (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 24, 2014).* | Incorporated by Reference | ||
General Release and Waiver, dated as of December 19, 2016, by and between the Company and G. Frederick Wilkinson (incorporated by reference to Exhibit 10.20.2 of the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 filed on March 1, 2017).* | Incorporated by Reference | ||
Employment Agreement, dated as of January 1, 2010, between the Company and Michael J. Nestor (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on January 14, 2010).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of January 1, 2014, between the Company and Michael Nestor (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Separation Agreement, dated as of January 8, 2018, between the Company and Michael J. Nestor (incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K filed on January 9, 2018).* | Incorporated by Reference | ||
Employment Agreement, dated as of July 14, 2016, between the Company and Douglas S. Boothe (incorporated by reference to Exhibit 10.6 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2016 filed on August 9, 2016).* | Incorporated by Reference | ||
Offer of Employment Letter, dated as of March 17, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.13.1 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2011 filed on February 28, 2012).* | Incorporated by Reference | ||
Employment Agreement, dated as of May 2, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.13.2 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2011 filed on February 28, 2012).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of May 2, 2011, between the Company and Mark A. Schlossberg (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Employment Agreement, dated as of December 12, 2012, between the Company and Bryan M. Reasons (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 13, 2012).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of December 12, 2012 between the Company and Bryan M. Reasons (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 2, 2014).* | Incorporated by Reference | ||
Employment Agreement, dated as of November 28, 2011, by and between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
Amendment, dated as of April 1, 2014, to the Employment Agreement, dated as of November 28, 2011, by and between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
Letter Agreement, dated as of April 1, 2014, between the Company and Jeffrey Nornhold (incorporated by reference to Exhibit 10.6.3 of the Company’s Quarterly Report on Form 10-Q to the quarter ended June 30, 2014 filed on August 6, 2014).* | Incorporated by Reference | ||
11.1 | Statement re computation of per share earnings (incorporated by reference to Note 12 to the Notes to Consolidated Financial Statements in this Annual Report on Form 10-K). | ||
Subsidiaries of the registrant. | Filed Herewith | ||
Consent of Independent Registered Public Accounting Firm. | Filed Herewith | ||
24.l | Powers of Attorney (included on the signature page of this Annual Report on Form 10-K) | ||
Certification of the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
Certification of the Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | Filed Herewith | ||
101 | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2017, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets as of December 31, 2017 and 2016, (ii) Consolidated Statements of Operations for each of the three years in the period ended December 31, 2017, (iii) Consolidated Statements of Comprehensive (Loss) Income for each of the three years in the period ended December 31, 2017, (iv) Consolidated Statements of Changes in Stockholders’ Equity for each of the three years in the period ended December 31, 2017, (v) Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2017 and (vi) Notes to Consolidated Financial Statements for each of the three years in the period ended December 31, 2017. | ||
* | Management contract, compensatory plan or arrangement. | ||
** | Confidential treatment granted for certain portions of this exhibit pursuant to Rule 24b-2 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which portions are omitted and filed separately with the SEC. | ||
*** | Confidential treatment requested for certain portions of the exhibit pursuant to Rule 24b-2 under the Exchange Act, which portions are omitted and filed separately with the SEC. | ||
† | Schedules omitted pursuant to Item 601(b)(2) of Regulation S-K. The Company agrees to furnish a supplemental copy of any omitted schedule to the SEC upon request. | ||