Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Intersect ENT, Inc. | d470105dex321.htm |
| EX-31.2 - EX-31.2 - Intersect ENT, Inc. | d470105dex312.htm |
| EX-31.1 - EX-31.1 - Intersect ENT, Inc. | d470105dex311.htm |
| EX-23.1 - EX-23.1 - Intersect ENT, Inc. | d470105dex231.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| ☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission file number: 001-36545
INTERSECT ENT, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 20-0280837 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 1555 Adams Drive Menlo Park, CA |
94025 | |
| (Address of principal executive offices) | (zip code) | |
Registrant’s telephone number, including area code:
(650) 641-2100
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Exchange on Which Registered | |
| Common Stock, $0.001 par value | The NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☑
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☑
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer | ☑ |
Accelerated filer | ☐ | |||||
| Non-accelerated filer | ☐ |
(Do not check if a smaller reporting company) | Smaller reporting company | ☐ | ||||
| Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yes ☐ No ☑
As of June 30, 2017, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the voting common stock held by non-affiliates, was approximately $702,909,000. Shares of common stock held by each officer, director and our affiliates have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares of common stock outstanding as of January 31, 2018 was 29,804,937.
Documents Incorporated by Reference
Portions of the registrant’s definitive Proxy Statement for its 2018 Annual Stockholders’ Meeting are incorporated by reference into Part III of this Annual Report on Form 10-K, to be filed within 120 days of the registrant’s fiscal year ended December 31, 2017.
Table of Contents
INTERSECT ENT, INC.
Annual Report on Form 10-K
For the Fiscal Year Ended
December 31, 2017
i
Table of Contents
CAUTIONARY INFORMATION REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K for the year ended December 31, 2017, or “Form 10-K,” contains forward-looking statements concerning our business, operations, and financial performance and condition as well as our plans, objectives, and expectations for business operations and financial performance and condition. Any statements contained herein that are not of historical facts may be deemed to be forward-looking statements. You can identify these statements by words such as “anticipate,” “assume,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “should,” “will,” “would,” and other similar expressions that are predictions of or indicate future events and future trends. These forward-looking statements are based on current expectations, estimates, forecasts, and projections about our business and the industry in which we operate and management’s beliefs and assumptions and are not guarantees of future performance or development and involve known and unknown risks, uncertainties, and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this Form 10-K may turn out to be inaccurate. Factors that could materially affect our business operations and financial performance and condition include, but are not limited to, those risks and uncertainties described herein under “Item 1A — Risk Factors.” You are urged to consider these factors carefully in evaluating the forward-looking statements and are cautioned not to place undue reliance on the forward-looking statements. The forward-looking statements are based on information available to us as of the filing date of this Form 10-K. Unless required by law, we do not intend to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise. You should, however, review the factors and risks we describe in the reports we will file from time to time with the Securities and Exchange Commission, or SEC, after the date of this Form 10-K.
PART I
| Item 1. | Business |
Overview
We are a commercial drug delivery company committed to improving the quality of life for patients with ear, nose and throat conditions. Our approved products are steroid releasing implants designed to treat the spectrum of needs among patients who are managed by ENT physicians for chronic sinusitis, one of the most prevalent chronic diseases in the United States and one of the most costly conditions for U.S. employers. We are currently marketing our PROPEL® family of products, consisting of PROPEL®, PROPEL® Mini and PROPEL® Contour, which are used following sinus surgery to deliver steroid locally to treat inflammation and improve surgical outcomes. In addition, we received FDA approval in December 2017 for our SINUVA™ Sinus Implant, a new targeted approach to treating nasal polyp disease in patients who have had previous ethmoid sinus surgery.
Our PROPEL family of steroid releasing implants are clinically proven to improve outcomes for chronic sinusitis patients following sinus surgery. PROPEL implants mechanically prop open the sinuses and release mometasone furoate, an advanced corticosteroid with anti-inflammatory properties, directly into the sinus lining, and then dissolve. PROPEL’s safety and effectiveness is supported by Level 1-A clinical evidence from multiple clinical trials, which demonstrates that PROPEL implants reduce inflammation and scarring after surgery, thereby reducing the need for postoperative oral steroids and repeat surgical interventions. More than 200,000 patients have been treated with PROPEL products to-date. The PROPEL family of products are used today predominantly in hospitals and ambulatory surgical settings, although they may also be used in the physician office setting of care.
| • | PROPEL has been proven in a meta-analysis of prospective, multicenter, randomized, controlled, double-blind clinical studies to improve surgical outcomes, demonstrating a 35% relative reduction in the need for postoperative oral steroid and surgical intervention compared to surgery alone. A physician may treat a patient with PROPEL by inserting it into the ethmoid sinuses. PROPEL is a self-expanding implant designed to conform to and hold open the surgically enlarged sinus while gradually releasing an anti-inflammatory steroid over a period of approximately 30 days and is absorbed into the body in approximately six weeks. |
1
Table of Contents
| • | PROPEL Mini has also been shown by our clinical studies to reduce the need for postoperative interventions, including a 38% relative reduction in the need for postoperative interventions in the frontal sinus, compared to surgery alone with standard postoperative care. PROPEL Mini is a smaller version of PROPEL, and is approved for use both in the ethmoid and frontal sinuses. PROPEL Mini is preferentially used by physicians compared with PROPEL when treating smaller anatomies or following less extensive procedures. |
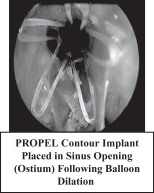
| • | PROPEL Contour is designed to facilitate treatment of the frontal and maxillary sinus ostia, or openings, of the dependent sinuses in procedures performed in both the operating room and in the office setting of care. PROPEL Contour’s lower profile, hourglass shape and malleable delivery system are designed for use in the narrow and difficult to access sinus ostia. In PROPEL Contour’s pivotal clinical study, the product demonstrated a 65% relative reduction in the need for postoperative interventions in the frontal sinus ostia compared to surgery alone with standard postoperative care. |
SINUVA, when placed during a routine physician office visit, expands into the sinus cavity and delivers an anti-inflammatory steroid directly to the site of polyp disease for 90 days. We have studied SINUVA in 4 clinical trials in over 400 patients to-date. Results from the pivotal RESOLVE II randomized clinical trial demonstrated a 74% relative reduction in bilateral polyp grade (a measurement of the extent of ethmoid polyp disease) and a 30% relative reduction in nasal obstruction and congestion for patients treated with SINUVA compared to a control group treated with a sham procedure, receiving no implant. Patients in both arms of the study were required to use intranasal steroid sprays daily. In addition, the study demonstrated a 61% reduction in the proportion of patients indicated for revision surgery at day 90. To complement clinical trials performed with SINUVA to-date, in which one course of SINUVA treatment was evaluated, we commenced the ENCORE study in November 2017. ENCORE is a 50-patient prospective, multicenter, open-label study focused on evaluation of the safety of repeat placement of the SINUVA implant in chronic sinusitis patients with nasal polyps. We completed enrollment of this study in January 2018.
According to the Centers for Disease Control and Prevention, or CDC, approximately 12% of the U.S. adult population, or 29 million people, are affected by chronic sinusitis, making it more prevalent than heart disease and asthma. Chronic sinusitis is an inflammatory condition in which the sinus lining becomes swollen and inflamed, leading to significant patient morbidity. Chronic sinusitis significantly impacts the quality of life of patients, including difficulty breathing, chronic headaches, recurrent infections, bodily pain and loss of sense of smell and taste. These persistent symptoms can severely impact a patient’s day-to-day well-being, resulting in frequent doctor visits and lost work productivity and can lead to chronic fatigue and depression. Chronic sinusitis is managed by a combination of medical management and surgical intervention. The first line of therapy is medical management involving antibiotics, anti-inflammatory steroids and decongestants. Sinusitis is the most common reason for adult outpatient antibiotic use in the United States, comprising 11% of all antibiotic prescriptions. Patients whose symptoms persist despite medical management are recommended to undergo functional endoscopic sinus surgery, or FESS. FESS is performed in the operating room to open the blocked sinus pathways by removing and/or displacing inflamed tissue and bone using surgical tools. Although sinus surgery can be effective, a majority of patients experience recurrent symptoms which commonly necessitate additional treatment with medications and surgery.
We estimate that there are more than 3.5 million people with chronic sinusitis who are managed by ENT physicians in the United States each year, many of whom we believe could benefit from products that incorporate our drug releasing bioabsorbable implant technology. The target market for our PROPEL family of products includes approximately 540,000 patients who undergo FESS for chronic sinusitis each year in the United States, with about 85% of those patients receiving treatment of the ethmoid sinus, approximately 30% receiving treatment of the frontal sinus and approximately 85% receiving treatment of the maxillary sinus, and with most patients receiving treatment for multiple sinuses. PROPEL Contour may also expand the availability of local steroid delivery in the office setting such as with sinus balloon dilation procedures, of which there are approximately 50,000 performed per year today. Long term, we believe that PROPEL Contour may provide a
2
Table of Contents
treatment option for the approximately 800,000 patients that are seen by an otolaryngologist each year for chronic sinusitis symptoms but choose not to undergo a surgical procedure. Our target market for SINUVA includes approximately 635,000 patients who have previously undergone FESS but continue to suffer with nasal polyps.
While our primary commercial focus is the U.S. market, both PROPEL and PROPEL Mini received CE Markings, permitting them to be marketed in Europe. We have initiated efforts intended to support future expansion into international geographies. Approximately 450,000 and 250,000 FESS procedures are performed annually in the Asia Pacific and European regions, respectively. Our commercialization strategy will consider several factors including regulatory requirements, reimbursement coverage for our products, and key opinion leader support. Our initial focus is on Germany, where we are working to build broad reimbursement, and Japan, where we are working through the regulatory process.
As of December 31, 2017, we estimate that approximately 2,700 accounts have stocked our PROPEL family of products for use by ENT physicians. Based on the number of units shipped as of December 31, 2017, we estimate that physicians have treated over 200,000 patients with our PROPEL family of products. For the years ended December 31, 2017, 2016 and 2015, we generated revenue of $96.3 million, $78.7 million and $61.6 million, respectively, and incurred a net loss of $16.4 million, $25.2 million and $26.6 million, for each respective year. As of December 31, 2017, we had an accumulated deficit of $164.8 million.
We have expanded our sales organization and we intend to continue to grow our sales force in order to expand our communication of the benefits of our steroid releasing implants to our physician customers. We seek to grow our revenue by increasing the frequency of use of our products among current physician customers and by adding new physician users.
Overview of Sinusitis
The sinuses are a system of connected air-filled cavities located within the bones around the nose and eyes that allow for natural ventilation and drainage of mucus. There are four sinus cavities: ethmoid, frontal, maxillary and sphenoid. One of each type of sinus lies on either side of the face. The sinuses are lined with soft, pink tissue called mucosa, which serves to constantly cleanse the sinuses of impurities such as dust, dirt, allergens, pollutants and bacteria. To clear these inhaled pathogens, the sinus lining secretes mucus which is then cleared away by small, hair-like structures called cilia, which act in coordination to sweep the mucus through the sinus pathways and out through the back of the throat.
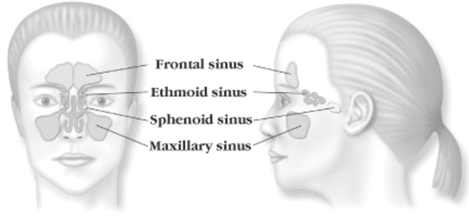
The ethmoid sinuses, which lie between the eyes, are a series of small cells with multiple, often interconnected openings in a honeycomb-like formation. These sinuses serve as the central aeration and drainage pathway for all other sinuses. The frontal, maxillary and sphenoid sinuses are known as dependent sinuses, as they each consist of one large cell that drains through an opening, or ostium, into the ethmoid sinus.
3
Table of Contents
Chronic sinusitis is an inflammatory condition in which the sinus lining becomes swollen and inflamed, leading to significant patient morbidity including difficulty breathing, chronic headaches, recurrent infections, bodily pain and loss of sense of smell and taste. These persistent symptoms can severely impact a patient’s day-to-day well-being, resulting in frequent doctor visits and can lead to chronic fatigue and depression. The condition significantly reduces work productivity from absenteeism and reduced on-the-job effectiveness, especially meaningful given the average chronic sinusitis patient age of approximately 37 years.
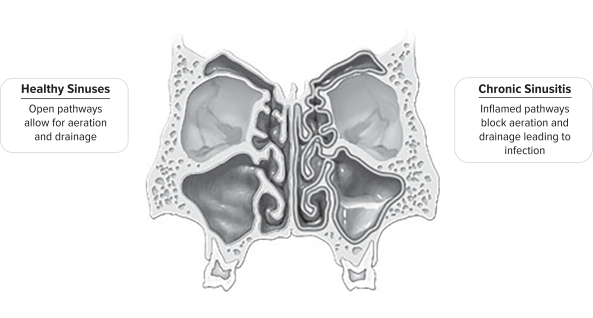
The debilitating patient symptoms and quality of life impairments attributed to chronic sinusitis create a significant healthcare burden to patients, insurers and employers.
Current Treatments for Chronic Sinusitis and Their Limitations
The treatment of chronic sinusitis often entails a combination of medical management and surgical intervention to treat the underlying inflammation of the sinus lining, while addressing the secondary symptoms caused by obstruction of the natural drainage pathways.
Medical Management
The first line of therapy for chronic sinusitis is medical management, which typically includes prescribed antibiotics, anti-inflammatory steroids and decongestants. Despite limited efficacy of use of antibiotics in this patient population and the consequence of increasing bacterial resistance, we believe there is pervasive overuse of these drugs, which could lead to patient resistance and has resulted in sinusitis being identified as a major target in national efforts to reduce unnecessary medical intervention. Sinusitis is the most common reason for adult outpatient antibiotic use in the United States, comprising 11% of all antibiotic prescriptions. In addition, physicians often prescribe decongestants and other drugs to target mucus accumulation.
Steroids are prescribed in two forms, oral steroid pills and nasal steroid sprays, both of which have serious limitations. Oral steroid therapy is effective at reaching the sinus lining, but it does so by means of systemic exposure and therefore carries the risk of serious side effects, including glaucoma, bone loss, weight gain, psychosis and difficulty in controlling blood glucose levels in patients with diabetes. Nasal steroid sprays, commonly indicated for rhinitis, or inflammation of the nasal passage, are routinely prescribed for chronic
4
Table of Contents
sinusitis patients. While nasal steroid sprays avoid systemic exposure and thus lack such serious side effects, an estimated 70% of the drug is swallowed and the remainder is directed to the nasal passages, instead of the sinuses, which limits efficacy. In a published study, the fraction of drug deposited in the sinuses from a nasal steroid spray was measured to be less than 2%. Poor patient compliance further limits the effectiveness of nasal steroid sprays. Although medical management can reduce symptoms, an estimated 20% or more of chronic sinusitis patients who receive medical therapy are unresponsive.
Of note, medical management is not only used as a first line of therapy for patients afflicted with primary chronic sinusitis, situations in which patients have not had sinus surgery, but also for patients who have recurrent symptoms despite having had sinus surgery. Patients in both stages of the condition are managed medically and hence are subject to the limitations described above.
Sinus Surgery
In cases where patients are symptomatic despite medical management, a physician may recommend FESS. In the FESS procedure, the physician enlarges the inflamed and obstructed sinus pathways by displacing and/or removing inflamed tissue and bone in order to facilitate normal sinus drainage and aeration. First introduced in the United States in the 1980s, FESS is considered the standard of care for surgical intervention to treat chronic sinusitis. During most procedures, the honeycomb-like cells of the ethmoid sinuses are removed, resulting in one large open cavity. ENTs may also enlarge the frontal and other sinuses by either surgically removing tissue or dilating the ostia, or opening, with a balloon.
FESS is typically performed under general anesthesia in an operating room. During the procedure, a physician inserts an endoscope into the nasal cavity to provide visualization of the patient’s anatomy. Surgical instruments, powered cutting tools and balloon dilation devices are used to remove or dilate obstructive tissue and bone. Following the surgical intervention, physicians often pack the newly opened ethmoid sinuses with gauze or other obstructive sinus packing materials to hold the sinus cavities open. A follow-up office visit may occur several days after the procedure, and an additional one or two follow-up visits typically occur over the first four to six weeks following surgery to monitor for and treat complications.
While FESS is the standard of care for treating medically-refractory chronic sinusitis, it has several limitations:
| • | Limited effectiveness. Inflammation and scarring in the postoperative period are common and can compromise the surgical result by negatively impacting the ability of the sinuses to heal. This increases the need for continued medical management and additional surgical procedures. Within the first year after surgery, approximately 64% of patients experience recurrent symptoms. |
| • | Limited ability to address postoperative inflammation. While oral steroids prescribed postoperatively can be effective at addressing inflammation and scarring, the required doses are significant and can result in serious systemic side effects, including glaucoma, bone loss, weight gain and psychosis. Further, use of oral steroids is restricted in patients with diabetes, glaucoma and certain psychological disorders. As a result, we believe only 20% of physicians prescribe them routinely after surgery. The absence of effective anti-inflammatory steroid therapy leaves the surgical wound susceptible to postoperative complications. |
| • | Pain and discomfort during postoperative period. During surgery, an ENT physician typically places sinus packing materials into the ethmoid sinuses to physically separate tissues in an attempt to prevent scarring and adhesions. Following surgery, physicians see patients two to three times in order to monitor for and, if necessary, to treat complications. |
| • | Potential for revision surgery. Within the first year after FESS, approximately 10% of patients will return to the operating room to undergo a revision procedure, while additional patients will return for a revision procedure after one year. We believe the risk of potential revision surgery is a significant deterrent to some patients that would otherwise undergo FESS for chronic sinusitis. |
5
Table of Contents
Trend for treatment in the physician’s office setting of care
Multiple technological advances, including balloon sinus dilation devices, have expanded the treatable chronic sinusitis patient population. Sinus dilation is now utilized by physicians in their offices to treat patients with mild chronic sinusitis who may not be willing to undergo or are not candidates for sinus surgery performed under general anesthesia in the operating room setting. The ability to treat patients in the office with sinus dilation has spurred interest in the ENT physician community for additional products that facilitate treatment of patients in the office setting of care.
We believe that the limitations of medical management and lack of disease resolution after FESS lead to undertreatment of many chronic sinusitis patients. We estimate that only a third of patients recommended for sinus surgery proceed with the potentially beneficial procedure, which we believe is due to its limitations and high risk for additional medical management and surgical revision. While balloon dilation has been introduced to open frontal, maxillary and sphenoid sinuses, or dependent sinuses, in a less invasive manner, balloon dilation procedures are not designed to treat disease in the most commonly involved sinuses, the ethmoids, and this procedure does not address the underlying inflammation associated with chronic sinusitis. We believe an opportunity exists to reach these undertreated patients by providing a more effective option to address inflammatory disease, while improving the overall outcomes of FESS.
Our Solution
Our PROPEL family of products offers ENT physicians a choice of three distinct drug releasing bioabsorbable implants to best meet the needs of surgical patients. We plan to launch SINUVA for use in the office setting of care to treat patients who have had prior ethmoid sinus surgery but have recurrent polyp disease requiring intervention and we continue to invest in ongoing research and development to develop additional new innovative solutions to further expand our ENT-focused business.
Our Technology Platform
Our drug releasing bioabsorbable implant technology consists of a polymer-based implant that is coated with a drug and polymer matrix. In fabricating the implant, we use polymers that are bioabsorbable and, over time, gradually and fully absorb into the body. The polymers chosen are materials with established safety profiles and have been used in medical devices for over 30 years.
Our innovative design process enables us to develop the mechanical and drug delivery features independently, lending to our customization capability. Our highly specialized bioabsorbable polymer engineering capability enables us to design each product with different physical characteristics such as size, radial strength and other attributes for a tailored approach to treating various sinuses. Our implants are designed to be self-expanding, which facilitates insertion when compressed, and expand to conform to the surrounding anatomy after insertion. The ability to control radial strength is important in enabling us to address different diseases at different states. For example, in some instances an implant may be used to maintain an already open passageway. In other situations, an implant with significantly greater strength may mechanically dilate a diseased passageway.
Our expertise in drug delivery allows us to effectively pair appropriate polymer delivery matrices with desired therapeutic agents. This allows selection of a therapeutic agent based on its clinical effectiveness and tailoring of the platform accordingly. In the case of PROPEL, we considered the wide range of off-patent corticosteroids, chose the one best suited for treatment of sinus inflammation, then customized the polymer coating to achieve the desired drug delivery. Once a drug is selected, our specialized capability in drug formulation enables us to control the rate at which the drug elutes, allowing us to design implants from which the drug is released over a matter of weeks to even longer durations. As a result, we have the flexibility to select the type of drug to be used on the implant and then engineer the implant to control the amount of drug delivered over time.
6
Table of Contents
Our Commercial Products
We currently market the PROPEL family of products, consisting of PROPEL, PROPEL Mini and PROPEL Contour. These products are indicated for use following sinus surgery, typically performed in the hospital out-patient or ambulatory surgery center setting of care. Our SINUVA product was approved by the FDA in December 2017 for the treatment of recurrent nasal polyp disease in patients 18 years or older who have had previous ethmoid sinus surgery. SINUVA provides a treatment for these patients designed to be administered in the physician’s office setting of care.

The PROPEL Family
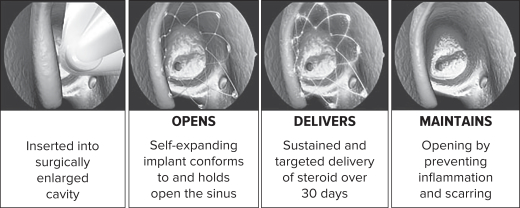
Our PROPEL family of steroid releasing implants are the first and only FDA-approved drug releasing sinus implants for chronic sinusitis sufferers 18 years or older. PROPEL is indicated for use following ethmoid sinus surgery, PROPEL Mini is indicated for use following ethmoid or frontal sinus surgery and PROPEL Contour is indicated for use following frontal or maxillary sinus surgery. Our PROPEL implants are designed to improve the outcomes of sinus surgery by holding open the sinus passageways, thereby reducing postoperative inflammation and scarring. These implants are inserted by a physician under endoscopic visualization following sinus surgery. Once inserted, the self-expanding implants conform to and hold open the surgically enlarged sinus, while gradually releasing an anti-inflammatory steroid, mometasone furoate, which is available generically, directly to the sinus lining over a period of approximately 30 days. The implants fully absorb into the body over a period of four to six weeks or are removed at the discretion of a physician during a routine office visit. Once absorbed or removed, the implant no longer provides structural support.
The graphic below illustrates the operation of PROPEL and PROPEL Mini in the ethmoid sinuses:
7
Table of Contents
We designed the steroid drug release of the PROPEL products to have a duration of approximately 30 days to match the postoperative healing cycle characterized in published medical literature. We selected mometasone furoate as the anti-inflammatory agent among numerous evaluated compounds based on three important characteristics: absorbability, binding affinity and low systemic bioavailability. The compound preferentially absorbs into the sinus lining instead of the surrounding mucous fluid. The drug has the highest glucocorticoid receptor binding affinity, making it highly potent in preventing inflammation once within tissue. Glucocorticoid receptors are the molecules in the surface membranes of cells throughout the body to which corticosteroids chemically bind. Additionally, the compound has low systemic bioavailability, meaning that it has negligible systemic safety side effects.
We believe the principal benefits of our PROPEL family of steroid releasing implants include:
| • | Improved surgical outcomes. Our implants have been clinically proven to improve FESS results by reducing postoperative inflammation and scarring. In a meta-analysis of prospective, multicenter, randomized, controlled, double-blind clinical studies, our PROPEL implants placed following ethmoid sinus surgery provided a 46% relative reduction in inflammation and a 70% relative reduction in scarring compared to the control implant. Postoperative complications, such as inflammation and polyps as well as scarring or adhesions, are common reasons for FESS failure. |
| • | Targeted steroid therapy to address postoperative inflammation. Our implants are the first and only FDA approved drug releasing bioabsorbable implants. They deliver an anti-inflammatory steroid postoperatively directly to the sinus lining in a controlled fashion over a period of approximately 30 days, which helps in the wound healing process. In a meta-analysis of prospective, multicenter, randomized, controlled, double-blind clinical studies, our PROPEL implants placed following ethmoid sinus surgery reduced the need for oral steroids by 40% compared to the control implant. |
| • | Improved healing without obstruction. Our implants improve postoperative care. Once inserted, the self-expanding implants are designed to conform to and hold open the surgically enlarged ethmoid sinuses until fully absorbed into the body, which improves wound healing without obstructing the sinuses and causing congestion. Our steroid releasing implants are designed to obviate the need for sinus packing materials, which can be a significant source of postoperative pain and discomfort. Our implants significantly reduce scarring and adhesions, which reduces the potential for pain in postoperative treatments. |
| • | Reduced need for postoperative surgical interventions. In clinical studies, our implants demonstrated a significant reduction in the need for postoperative surgical intervention. In a meta-analysis of prospective, multicenter, randomized, controlled, double-blind clinical studies of over 200 patients, PROPEL provided a 35% relative reduction in the need for postoperative oral steroids and surgical intervention compared to the control implant when placed in the ethmoid sinus. In addition, the PROGRESS study, an 80 patient prospective, randomized, blinded, multicenter trial of PROPEL Contour, demonstrated a statistically significant 65% relative reduction in the need for postoperative interventions compared to surgery alone when PROPEL Contour was placed in the frontal sinus. We believe that patients who have been deterred by the high revision rates associated with FESS may now consider surgical intervention to treat their chronic sinusitis condition. |
In February 2017, we received FDA approval for PROPEL Contour, a steroid releasing implant designed to facilitate treatment of the frontal and maxillary sinus ostia, or openings, of the dependent sinuses, which we believe represents an opportunity for adoption in a variety of settings for chronic sinusitis sufferers 18 years or older undergoing sinus surgery. In the operating room, PROPEL Contour has demonstrated the ability to expand adoption of steroid releasing implants overall by providing physicians with a range of products needed to customize treatment based on their patients’ disease and anatomy. In particular, we believe PROPEL Contour will be seen as the right fit for many of the 30% of sinus surgeries that involve the frontal sinuses today and the
8
Table of Contents
lower profile, malleable delivery system will increase usage particularly in those patients whose frontal sinuses are more challenging to access.
|
|
SINUVA
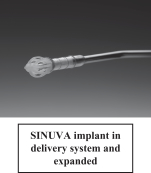
Following sinus surgery, the underlying chronic inflammation associated with chronic sinusitis can lead to recurrent obstruction of the sinus cavity over time, especially in patients afflicted with polyps, a sign of severe inflammation. Improving care of such chronic patients holds meaningful opportunity to significantly reduce healthcare costs by reducing the need for revision surgery. We have designed the SINUVA steroid releasing implant to be placed in the physician office setting following a routine visit as an alternative treatment option for patients who are candidates for revision surgery. The implant is based on the same drug releasing bioabsorbable implant technology as the PROPEL family of products, but is designed to have greater radial strength in order to dilate an obstructed, polyp-filled sinus cavity, and deliver drug for an extended period of time. SINUVA was subject to regulation as a drug product and we submitted a New Drug Application, or NDA, to seek approval from the FDA to commercialize SINUVA in the U.S. We believe SINUVA could be an appealing alternative to patients who have previously undergone FESS but continue to suffer from polyp recurrence.
|
|

Our family of drug releasing implants consists of polymers that control local drug release and provide structural support to adjacent tissues during the healing process. We believe the development, manufacturing and regulatory approval for products incorporating this technology requires capabilities in polymer science, drug delivery, analytical testing and combination products. These competencies allow our technical team to tailor drug formulation, polymer design, drug release duration, implant radial strength and degradation period to meet different clinical needs. We may apply these competencies to the development of new products over time. Such new products, or changes that we make in the therapeutic agent used in our products will require FDA approval prior to commercialization in the United States.
Clinical Trial Findings and In-Process Studies
PROPEL and PROPEL Mini
PROPEL Ethmoid Sinus Studies. The safety and efficacy of PROPEL in the ethmoid sinuses has been studied in three prospective, multicenter clinical trials conducted in the United States enrolling a total of 205
9
Table of Contents
patients. The principal safety and efficacy information is derived from the ADVANCE II randomized clinical trial and is supported by the ADVANCE clinical trial and an initial pilot study. A meta-analysis that pooled data from the ADVANCE II study and the initial pilot study provides further evidence of efficacy. In all three studies, implants were placed following ethmoid sinus surgery, or ethmoidectomy, which entails removal of the honeycomb-like partitions between the ethmoid sinuses in order to create larger sinus cavities.
All three studies were designed to measure PROPEL’s ability to improve the outcomes of sinus surgery. This included measuring the need for postoperative interventions such as adhesion lysis, which is a procedure to separate scar tissue and adhesions within the sinus cavity, and the need to prescribe oral steroids to treat inflammation. The studies also measured the impact of PROPEL on other postoperative complications, such as occurrence of polyposis and middle turbinate lateralization. Prevention of these complications contributes to the long-term success of sinus surgery. Polyposis represents a higher level inflammatory disease. The middle turbinate is a bony structure in the middle of the sinus, responsible for filtration, heating, and humidification of nasal air flow. Middle turbinate lateralization is an undesired complication where this structure curves towards the outer, or lateral, wall of the nose resulting in blockage of the ethmoid sinus passage.
In a meta-analysis, the two prospective, multicenter, randomized, controlled, double-blind studies enrolled a total of 143 patients utilizing an intra-patient control design. The results of these two trials, the ADVANCE II study and the initial Pilot study, were then pooled which represents the first and only Level 1a evidence for any devices used in sinus surgery today. Level 1a evidence is the highest level of evidence according to the criteria of the Centre for Evidence-Based Medicine at the University of Oxford. For evidence Level 1a, a meta-analysis of multiple randomized controlled trials is required.
Compared to the control implant, the drug releasing implant provided a 35% relative reduction in postoperative interventions, a 51% relative reduction in adhesion lysis and a 40% relative reduction in oral steroid intervention. The relative reduction in frank polyposis was 46%. Additional efficacy endpoints of significant, or severe, adhesions and middle turbinate lateralization, determined by clinical investigators at the study centers, were reduced by 70% (p=0.0013) and 75% (p=0.0225), respectively.
| Principal Efficacy Results at Day 30 as graded by independent, blinded panel of physicians |
||||||||||||||||||||
| Outcome Measure |
Evaluable Patients* |
Treatment Sides (n=143) No. (%) |
Control Sides (n=143) No. (%) |
Relative Reduction (%) |
P Value | |||||||||||||||
| Postoperative intervention |
128 | 42 (32.8) | 65 (50.8) | -35 | 0.0008 | |||||||||||||||
| Adhesion lysis |
134 | 19 (14.2) | 39 (29.1) | -51 | 0.0016 | |||||||||||||||
| Oral steroid intervention |
113 | 25 (22.1) | 42 (37.2) | -40 | 0.0023 | |||||||||||||||
| Frank polyposis |
111 | 22 (19.8) | 41 (36.9) | -46 | <0.0001 | |||||||||||||||
| * | Evaluable subjects were those with gradable sinuses on both sides |
PROPEL Mini Frontal Sinus Study. We have completed a prospective, randomized blinded multicenter clinical trial to support an expanded indication for placement of PROPEL Mini in the frontal sinuses called PROGRESS. Approximately 30% of patients undergoing sinus surgery for chronic sinusitis suffers from frontal sinus disease. We enrolled 80 patients in the study using an intra-patient control design to assess both safety and efficacy of PROPEL Mini when placed following surgery of the frontal sinus, compared to surgery alone. The primary efficacy endpoint is the reduction in need for postoperative interventions such as the need for surgical intervention or oral steroids. In August 2015, we announced preliminary topline data from the PROGRESS trial, designed to evaluate the safety and efficacy of PROPEL Mini when placed in the frontal sinuses following surgery, showing that the study met its primary efficacy endpoint and demonstrating a statistically significant 38% relative reduction in the need for postoperative interventions compared to surgery alone. In March 2016, we received approval to expand the indication of PROPEL Mini to treat patients undergoing frontal sinus surgery.
10
Table of Contents
PROPEL Contour
In February 2017, we received FDA approval for PROPEL Contour, a steroid releasing implant designed to facilitate treatment of the frontal and maxillary sinus ostia, or openings, of the dependent sinuses, which we believe represents opportunity for adoption in a variety of settings. In the operating room, PROPEL Contour has the potential to lead to expanded adoption of steroid releasing implants overall by providing physicians with a range of products needed to customize treatment based on their patients’ disease and anatomy. In particular, we believe PROPEL Contour’s lower profile, malleable delivery system will increase usage particularly in those patients whose frontal sinuses are more challenging to access. Since sinus surgeries typically involve treatment of one or more of the ethmoid, maxillary or frontal sinuses, we believe the PROPEL Contour greatly increases the chance that a PROPEL product will be used. We announced results of the second cohort of patients in the PROGRESS study in May 2016. This phase of the PROGRESS study was an 80-patient prospective randomized blinded multicenter trial designed to assess the safety and efficacy of PROPEL Contour when placed in the frontal sinuses following sinus surgery. This study demonstrated a statistically significant 65% relative reduction in the need for post-operative interventions, such as the need for additional surgical procedures or need for oral steroid prescription, compared to surgery alone with standard post-operative care.
SINUVA
In December 2017, we received FDA approval for SINUVA, a steroid releasing implant for the treatment of nasal polyposis in adult patients who have had ethmoid surgery. The SINUVA implant is intended to be placed in the physician office setting of care. This product’s primary mode of action is as a drug, and for this reason we were required to obtain an NDA approval from the FDA, rather than a PMA approval. In order to support the NDA application with the FDA, we completed four studies of SINUVA: a pilot study, a pharmacokinetic study, RESOLVE and RESOLVE II. In July 2016, we completed enrollment of the RESOLVE II pivotal trial, which was a prospective, multicenter, randomized, controlled, blinded study of 300 patients. Both co-primary endpoints were met, including improvement in patient-reported nasal obstruction/congestion score (p=0.0074) and reduction in bilateral polyp grade as evaluated by a panel of three sinus surgeons (p=0.0073). In addition, several pre-specified secondary endpoints were met, including the reduction in the proportion of patients still indicated for repeat sinus surgery, reduction in ethmoid obstruction and improvement in sense of smell. The RESOLVE study (n=100) included ocular exams, and patients were followed for six months to assess longer-term outcomes. Compared to the control group, the treatment group demonstrated greater reduction from baseline to day 90 in nasal congestion/obstruction score and bilateral polyp grade (judged by an independent panel), but these primary endpoint results did not reach statistical significance (p=0.1365 and 0.0985, respectively). According to clinical investigator grading, the treatment group demonstrated statistically significant improvements in both bilateral polyp grade (p<0.02) and percent ethmoid sinus obstruction (p<0.0001) throughout the entire six month study period. In a post-hoc analysis of nasal congestion/obstruction scores in a subset of 67 patients with at least grade 2 polyposis on each side at baseline, this outcome trended towards statistical significance in favor of the treatment group (p=0.0505). Longer-term, the study showed that at six months, control patients were at 3.6x higher risk of remaining indicated for revision surgery than treated patients. The findings from the RESOLVE study were used to inform the pivotal RESOLVE II study design.
In November 2017, we commenced the ENCORE study, a 50 patient prospective, multicenter, open-label study focused on evaluation of the safety of repeat placement of the SINUVA implant in chronic sinusitis patients with nasal polyps, and completed enrollment in January 2018.
Seasonality
We expect revenue from our PROPEL family of products to fluctuate from quarter to quarter due to seasonal variations in the volume of sinus surgery procedures performed, which has been impacted historically by factors including the status of patient healthcare insurance plan deductibles and the seasonal nature of allergies which can impact sinus-related symptoms.
11
Table of Contents
Competition
Our industry is highly competitive, subject to change and significantly affected by new product introductions and other activities of industry participants. Many of the companies developing or marketing ENT products are publicly traded or are divisions of publicly-traded companies and may enjoy competitive advantages including:
| • | greater financial and human capital resources; |
| • | significantly greater name recognition; |
| • | established relationships with ENT physicians, referring physicians, customers and third-party payors; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer greater discounts or incentives to gain a competitive advantage; and |
| • | established sales, marketing and worldwide distribution networks. |
Because of the size of the market opportunity for the treatment of chronic sinusitis, potential competitors have historically dedicated and will continue to dedicate significant resources to aggressively promote their products or develop new products. New product developments that could compete with us more effectively are possible because of the prevalence of chronic sinusitis and the extensive research efforts and technological progress that exist within the market. Large medical device companies with ENT divisions, such as Medtronic, also have capability in drug releasing stents.
Our commercially available products are designed to be used following sinus surgery. If another company successfully develops a surgical technique, drug, drug delivery system (including intranasal steroid sprays) or device that is more efficacious than our steroid releasing implant solution, sales of our products would be significantly and adversely affected.
One alternative to delivering steroids to the sinuses postoperatively with our PROPEL family of products, and SINUVA, which was approved by the FDA in December 2017, is the prescription of oral steroids. While oral steroids prescribed postoperatively can be effective at addressing inflammation and scarring, the required doses are significant and can result in serious systemic side effects, including glaucoma, bone loss, weight gain and psychosis. Further, oral steroids have restricted use in diabetic individuals, patients with glaucoma and some psychological disorders. We believe, as a result, only 20% of physicians prescribe them routinely after surgery. Additionally, there are commercially available packing materials and spacers on the market that provide a spacing function and are less expensive than our products. During surgery, approximately 60% of ENT physicians place sinus packing materials, either absorbable or non-absorbable, into the ethmoid sinuses to physically separate tissues in an attempt to prevent scarring and adhesions. Non-absorbable spacers are sometimes placed in the frontal sinuses to maintain patency. Following surgery, the sinus packing materials or spacers are removed by pulling or suctioning them from the newly opened cavity, a painful and time-consuming process, often necessitating pain medication. Despite the use of packing materials, scarring and adhesions are common, sometimes necessitating painful removal of additional tissue during postoperative treatments. Some physicians choose to soak packing materials with steroid in liquid form in an effort to deliver steroid to the sinus. This practice is off-label and is not supported by clinical data. However, although we believe our products have significant advantages over sinus packing materials, spacers and other treatment options, they are expensive relative to packing materials and may not be reimbursed by third-party payors. As a result, ENT physicians may choose to use oral steroid delivery or packing/spacing materials or a combination of the two, which are less expensive, in lieu of our products.
We believe that our continued ability to compete favorably depends on:
| • | successfully expanding our commercial operations; |
| • | continuing to innovate and maintain scientifically-advanced technology; |
12
Table of Contents
| • | having reimbursement in place to support broad adoption of our products; |
| • | developing technologies for applications in the sinuses and other areas of ENT; |
| • | attracting and retaining skilled personnel; |
| • | obtaining patents or other intellectual property protection for our products; and |
| • | conducting clinical studies and obtaining and maintaining regulatory approvals. |
Intellectual Property
As of December 31, 2017, we owned 77 issued patents globally, of which 31 were issued U.S. patents, and we owned 25 pending patent applications globally, of which 6 were pending patent applications in the United States. Subject to payments of required maintenance fees, annuities and other charges, our issued patents have expiration dates between 2018 and 2032, of which one will expire by 2020, 18 will expire between 2021 and 2025, and the remaining 58 will expire after 2025.
As of December 31, 2017, our trademark portfolio contains 32 trademark registrations, six of which are U.S. trademark registrations, as well as 28 foreign and three U.S. pending trademark applications.
We also rely upon trade secrets, know-how, continuing technological innovation, and may rely upon licensing opportunities in the future, to develop and maintain our competitive position. We protect our proprietary rights through a variety of methods, including confidentiality agreements and proprietary information agreements with suppliers, employees, consultants and others who may have access to proprietary information.
Manufacturing and Supply
We manufacture our steroid releasing implants at our facility in Menlo Park, California with components supplied by external suppliers. We perform inspections of these components before use in our manufacturing operations. Using these components, we assemble, inspect, test and package our implants, and send them to a third-party sterilization vendor. After sterilization, we perform inspections of the finished implants internally and via third-party laboratories to determine compliance with our specifications, after which we place the implants into our inventory and ultimately ship the finished products to customers.
The active pharmaceutical ingredient, or API, and a number of our critical components used in our implants are supplied to us from single source suppliers. We rely on single source suppliers for some of our polymer materials, some extrusions and molded components and some off-the-shelf components. Our ability to commercially supply our products and to develop our product candidates depends, in part, on our ability to successfully obtain the API and polymer materials used in these products in accordance with regulatory requirements and in sufficient quantities for commercialization and clinical testing. We have entered into manufacturing, supply or quality agreements with a number of our single source suppliers pursuant to which they supply the API and components we need. We generally acquire our single source components pursuant to purchase orders placed in the ordinary course of business. However, we are not certain that our single source suppliers will be able to meet our demand for their products, whether because of the nature of our agreements with those suppliers, our limited experience with those suppliers or our relative importance as a customer to those suppliers. It may be difficult for us to assess their ability to timely meet our demand in the future. To date, we have not experienced any significant supply constraints or delays in procuring components and materials and while our suppliers have generally met our demand for their products on a timely basis in the past, they may subordinate our needs in the future to the needs of their other customers.
We continue to improve our manufacturing capabilities and increasing capacity as we increase the extent of our commercialization efforts. We believe our manufacturing operations are in compliance with regulations mandated by the FDA. We are an FDA-registered medical device and drug manufacturer and we were granted
13
Table of Contents
PMA approval for PROPEL in August 2011. Our manufacturing facilities and processes are subject to periodic inspections and audits by various federal, state and foreign regulatory agencies. For example, our facilities were inspected by the FDA in April and May 2011, December 2012, March 2013, June 2014 and April 2017. The FDA also performed a pre-approval inspection for SINUVA in November 2017. The State of California regulatory authority audited our manufacturing facility in connection with granting a California Device Manufacturing License to us in August 2009 and a pharmacological manufacturing license in December 2017. We have maintained ISO 13485 certification since January 2014 and have been audited by the European Notified Body in Ireland, National Standards Authority of Ireland, or NSAI on at least an annual basis since 2014. Our facility was last inspected in November 2017 by NSAI and no major nonconformance reports were issued as a result of that inspection.
We have manufacturing, supply or quality agreements with a number of our single source suppliers:
| • | In April 2014, we entered into an agreement with Hovione Inter Ltd., or Hovione, pursuant to which we are required to purchase 80% of our API from Hovione, in quantities to be specified in 12-month forecasts provided by us and updated on a quarterly basis. This agreement extends until April 2019. Either we or Hovione may terminate the agreement prior to that date for uncured material breach by or insolvency of the other party. We may also terminate the agreement in the event Hovione loses any required FDA approval rendering it unable to fulfill its contractual obligations, or if Hovione is engaged in felonious or fraudulent activities. Either we or Hovione may terminate the Agreement in the event regulatory agencies require changes to the product specifications that materially affect Hovione’s cost of production and we are unable to reach an agreement with Hovione regarding an equitable pricing adjustment. |
| • | In April 2014, we entered into an agreement with Polymer Solutions Incorporated, or PSI, pursuant to which PSI supplies us with analytical testing services for our polymer materials. The agreement has an indefinite term, but may be terminated by us at any time upon 30 days’ notice to PSI or immediately upon written notice in the event of an uncured material breach by PSI. In April 2016, this agreement was amended with testing services pricing. |
| • | In January 2014, we entered into an agreement with AIM Plastics, Inc., or AIM, pursuant to which AIM provides us with injection molded components in quantities to be specified in rolling quarterly forecasts provided by us. The agreement extends for an initial period of one year from its effective date, with automatic one-year renewal periods applying thereafter unless we or AIM provide written notice of non-renewal at least 30 days prior to the end of the then-current term. Either we or AIM may terminate the agreement for an uncured material breach by the other party. We may also terminate the agreement upon twelve months written notice in the event of a change in control of AIM. In February 2016, this agreement was amended with updated forecasts and component pricing. |
| • | In October 2015, we entered into an agreement with Exova Group Limited, or Exova, pursuant to which Exova supplies us with analytical testing services for our finished product. This agreement expired in December 2017, however, we are continuing to conduct business pursuant to the terms of our old agreement and are working on a new agreement. |
| • | In November 2013, we entered into an agreement with Stephen Gould Corporation, or SGC, pursuant to which SGC supplies us with packaging components in accordance with a rolling quarterly forecast provided by us. The agreement extends for an initial period of two years from the effective date, with automatic one-year renewal periods applying thereafter unless we or SGC provide written notice of intent not to renew at least 30 days prior to the end of the then current term. Either we or SGC may terminate the agreement for uncured material breach by the other party. We may also terminate the agreement at will upon 90 days prior written notice to SGC, or upon twelve months prior written notice in the event of a change in control of SGC. In October 2015, this agreement was amended with updated rolling forecasts and component pricing, and in August 2016, this agreement was amended with component pricing. |
14
Table of Contents
Each of these suppliers manufactures the components they produce for us or tests our components and devices to our specifications. We typically seek to negotiate new agreements with these vendors in advance of the expiration of the current agreements. We intend to maintain sufficient supplies of the API and components from these single source suppliers in the event that our agreements with one or more of these suppliers were to terminate to enable us to continue to manufacture our implants for a sufficient amount of time necessary to obtain another source of API or components.
Government Regulation
United States Regulation of Medical Devices and Drugs
Our products and any product candidates are drug releasing bioabsorbable implants that are regulated as combination products by the FDA. FDA’s Office of Combination Products designates a primary mode of action for such drug-device combination products, with the respective primary Center within the FDA leading the regulatory review for the product, in consultation with the secondary designated Center. FDA determined that the primary mode of action for our PROPEL family of products was that of a medical device, so these products have been approved and are regulated as medical devices. By comparison, the primary mode of action of SINUVA was designated to be its drug properties, so this product has been FDA approved and is regulated as a pharmaceutical.
FDA regulations require us to register as a medical device and drug product manufacturer with the FDA. Additionally, the California Department of Health Services, or CDHS, requires us to register as a medical device and drug manufacturer within the state. In order to maintain CE Markings, we must maintain compliance with ISO 13485. Because of this, the FDA and the NSAI inspect us on a routine basis for compliance with current good manufacturing practices. These regulations require that we manufacture our products and maintain related documentation in a prescribed manner with respect to manufacturing, testing and control activities, and product release for distribution. We have undergone and expect to continue to undergo regular current good manufacturing practice inspections in connection with the manufacture of our products at our facility.
Medical Devices
Our PROPEL family of products are regulated in the United States as Class III medical devices by the FDA under the Federal Food, Drug and Cosmetic Act, or FDCA. The FDA classifies medical devices into one of three classes based upon controls the FDA considers necessary to reasonably ensure their safety and effectiveness. Class I devices are subject to general controls such as labeling, adherence to good manufacturing practices and maintenance of product complaint records, but are usually exempt from premarket notification requirements. Class II devices are subject to the same general controls and also are subject to special controls such as performance standards, and FDA guidelines, and may also require clinical testing prior to approval. Class III devices are subject to the highest level of controls and rigorous clinical testing prior to their approval and generally require a PMA, or a PMA supplement approval prior to their sale.
Manufacturers must file an Investigational Device Exemption, or IDE, application if human clinical studies of a device are required and if the investigational use of the device represents a potential for significant risk to the patient. The IDE application must be supported by data, typically including the results of animal and engineering testing of the device. If the IDE application is approved by the FDA, human clinical studies may begin at a specific number of investigational sites with a maximum number of patients, as approved by the FDA. The clinical studies must be conducted under the review of an independent institutional review board to ensure the protection of the patients’ rights.
Generally, upon completion of these human clinical studies, a manufacturer seeks approval of a Class III medical device from the FDA by submitting a PMA application. A PMA application must be supported by extensive data, including the results of the clinical studies, as well as testing and literature to establish the safety and effectiveness of the device. PMA approval may be conditioned upon the conduct of certain post-approval studies, such as long-term follow-up studies.
15
Table of Contents
Drugs
The clinical testing, manufacturing, labeling, storage, distribution, record keeping, advertising, promotion, import, export and marketing, among other things, of our product SINUVA and any future drugs we may develop and seek to commercialize, are subject to the FDA’s drug authority and are governed by extensive regulation by governmental authorities in the United States and other countries. The FDA, under the FDCA, regulates pharmaceutical products in the United States. The steps required before a drug may be approved for marketing in the United States generally include:
| • | preclinical laboratory tests and animal tests conducted under Good Laboratory Practices, or GLP; |
| • | submission to the FDA of an Investigational New Drug, or IND, application for human clinical testing, which must become effective before human clinical trials commence; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product and conducted in accordance with Good Clinical Practices, or GCP; |
| • | the submission to the FDA of an NDA; |
| • | FDA acceptance, review and approval of the NDA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facilities at which the product is made to assess compliance with current Good Manufacturing Practices, or cGMPs: and |
| • | state pharmacological distribution licenses must be obtained. |
Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any products for which we have or may obtain regulatory approval. Sales of our PROPEL family of products, SINUVA, and any of our other product candidates, will depend, in part, on the extent to which the products will be covered and the costs of the products will be adequately reimbursed by third-party payors, including government healthcare programs such as Medicare and Medicaid, commercial health insurers and managed care organizations. The process for securing coverage for a product is separate from the process for establishing a reimbursement rate for the product if separately payable. Third-party payors may limit coverage to specific patient subpopulations and may limit coverage based on product inclusion on an approved list, or formulary, which might not include all FDA-approved products for a particular indication. A payor’s decision to provide coverage for a product does not ensure an adequate reimbursement rate.
Medical Devices
Our PROPEL family of products are used almost exclusively in the operating room of a hospital or ambulatory surgery center where they are commonly treated as adjunct surgical supplies utilized in sinus surgery and the cost is included in the reimbursement to the facility for the FESS procedure. In the event these procedures are performed in the physician office setting, our PROPEL family of products have been assigned a code under the Healthcare Common Procedure Coding System, S1090, which may be used to submit requests for product reimbursement to commercial payors. Several existing procedure codes may apply to describe the procedure associated with implant placement. However, payment is subject to payor coverage on the basis of either written medical policies related to the product or individual patient medical necessity. If, as a result of policies the payor has in place regarding these products, hospitals or other service providers are unable to receive adequate reimbursement to support the use of our products, this will negatively impact our revenues and our gross margins will decrease, which will adversely affect our ability to invest in and grow our business.
Drugs
Drugs that are administered incident to a physician service and are separately reportable, are generally billed by providers with HCPCS J codes and are paid based on quarterly price data submitted to CMS, pricing
16
Table of Contents
compendia, or provider invoice. For SINUVA, we have applied for a product specific HCPCS J code consistent with CMS timelines and requirements for physician-administered drugs used mostly in the office setting of care. Since J codes are assigned by CMS only once per calendar year, until such a product-specific code is assigned, we expect providers will submit claims for reimbursement of SINUVA using an unassigned J code such as J3490, along with the designated National Drug Code for SINUVA. This is the typical path for physician-administered drugs during the interim period between FDA approval and J code assignment.
For more information, see “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Research and Development
Our research and development expenses totaled $18.4 million, $18.9 million and $16.6 million in the years ended December 31, 2017, 2016 and 2015, respectively. For more information, see “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Employees
As of December 31, 2017, we had 327 employees, consisting of 64 in manufacturing, 71 in research and development and 192 in sales, general and administrative.
Information about Segment and Geographic Revenue
All of our revenues have been generated from the sale of our PROPEL family of products. Information about our assets and revenues, including segment and geographic revenue, is set forth in the Financial Statements, including Note 2, included in this Annual Report, which information is incorporated by reference here.
Corporate Information
We were incorporated in Delaware in October 2003 as Sinexus, Inc. We changed our name to Intersect ENT, Inc. in November 2009. Our offices are located at 1555 Adams Drive, Menlo Park, California 94025 and our telephone number is (650) 641-2100. Our website is www.intersectent.com. We completed our initial public offering in July 2014, and our common stock is listed on The NASDAQ Global Market under the symbol “XENT.”
Our periodic and current reports, registration statements, proxy and information statements and other information are available for inspection and copying at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549 or may be obtained by calling the SEC at 1-800-SEC-0330. The SEC also maintains a website containing such information available free of charge to the public at www.sec.gov. We make available free of charge on or through our Internet website, our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
17
Table of Contents
| Item 1A. | Risk Factors |
RISK FACTORS
Before deciding to invest in us or to maintain or increase your investment, you should carefully consider the risks described below, in addition to the other information contained in this Annual Report on Form 10-K and in our other filings with the SEC. If any of the risks discussed in this report actually occur, they may materially harm our business, financial condition, operating results, cash flows or growth prospects. As a result, the market price of our common stock could decline, and you could lose all or part of your investment. Additional risks and uncertainties that are not yet identified or that we think are currently immaterial may also materially harm our business, financial condition, operating results, cash flows or growth prospects and could result in a partial or complete loss of your investment.
Risks Related to Our Business
We have incurred significant operating losses since inception and may not be able to achieve profitability.
We have incurred net losses since our inception in 2003. We incurred a net loss of $16.4 million, $25.2 million and $26.6 million for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, we had an accumulated deficit of $164.8 million. To date, we have financed our operations primarily through sales of our capital stock, certain debt-related financing arrangements and from sales of our approved products. We have devoted substantially all of our resources to research and development of our products, including clinical and regulatory initiatives to obtain approvals for our products, and sales and marketing activities. Our ability to generate sufficient revenue from our existing products or from any of our product candidates in development, and to transition to profitability and generate consistent positive cash flows is uncertain. We expect that our operating expenses will continue to increase as we continue to build our commercial infrastructure, develop, enhance and commercialize new products and incur additional operational and reporting costs associated with being a public company. As a result, we expect to continue to incur operating losses for the foreseeable future and may never achieve profitability.
All of our revenue has been generated from our PROPEL® family of steroid releasing implants. Our revenue is completely dependent on the success of these products and of our newly approved product, SINUVA, and if these products fail to grow or to continue experiencing expanded adoption, our business will suffer.
We started selling PROPEL® in August 2011, PROPEL® Mini in November 2012 and PROPEL® Contour in February 2017, collectively referred to as our PROPEL family of products. We expect that sales of these products, together with SINUVATM, will account for all of our revenue for the foreseeable future. In addition, our ability to become profitable will depend upon the commercial success of these products. We market our products primarily to ear, nose and throat, or ENT, physicians who may be slow or fail to adopt our products or who may use our products in only a small percentage of their eligible patients for a variety of reasons, including, among others:
| • | lack of experience with our products; |
| • | lack of adequate reimbursement or cost to the patient; |
| • | lack of conviction regarding evidence supporting cost benefits or cost effectiveness of our products over existing alternatives; |
| • | lack of clinical data supporting longer-term patient benefits or, in the case of SINUVA, repeated use; and |
| • | liability risks generally associated with the use of new products and procedures. |
If we are unable to effectively demonstrate to ENT physicians and patients the benefits of our products or our products fail to achieve growing market acceptance, our future revenue will be adversely impacted.
18
Table of Contents
Because of the numerous risks and uncertainties associated with our commercialization efforts, we are unable to predict the extent to which we will continue to generate revenue from our products or the timing for when or the extent to which we will become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis.
Pricing pressure from our hospital and ambulatory surgery center customers due to cost sensitivities resulting from healthcare cost containment pressures and reimbursement changes could decrease demand for our PROPEL family of products, the prices that customers are willing to pay and the frequency of use of our products, which could have an adverse effect on our business.
Hospitals and ambulatory surgery centers that purchase our PROPEL family of products typically bill various third-party payors for a facility fee to cover the costs of supplies, including our PROPEL family of products, used in sinus surgery procedures. Because there is often no separate reimbursement for supplies used in surgical procedures, the additional cost associated with the use of our steroid releasing implants can impact the profit margin of the hospital or surgery center where the sinus surgery is performed. Some of our target customers may be unwilling to adopt or use broadly our steroid releasing implants in light of the additional associated cost. Further, any decline in the amount payors reimburse our customers for sinus surgery procedures could make it difficult for existing customers to continue using, or to adopt, our steroid releasing implants. This could create additional pricing pressure for us.
All third-party payors, whether governmental or commercial, whether inside the United States or outside, are developing increasingly sophisticated methods of controlling healthcare costs. These cost-control methods include prospective payment systems, bundled payment models, value-based payment models, capitated arrangements, group purchasing, benefit redesign, prior authorization processes and requirements for second opinions prior to major surgery. These cost-control methods also potentially limit the amount that healthcare providers may be willing to pay for medical devices.
Effective January 1, 2017, CMS assigned upper airway procedures, which includes sinus surgery, to a comprehensive Ambulatory Payment Classification, or APC, for procedures performed in the hospital outpatient department setting. With this assignment, the reimbursement per case was set at a fixed amount regardless of the number of procedures performed during that encounter. As a result, for Medicare patients, while payment increased for encounters involving one or two procedures, payment for encounters with three or more procedures, which are commonly associated with the use of our products, declined significantly below the prior average reimbursement amount. Some commercial payors may peg their rates directly to Medicare rates or use these rates as a reference for facility contract negotiations. If, as a result of this CMS ruling, hospitals are unable to receive adequate reimbursement to support the use of our products, or if we are forced to lower the price we charge for our products, this will negatively impact our revenues and our gross margins will decrease, which will adversely affect our ability to invest in and grow our business. We cannot predict how pending and future healthcare legislation and regulations will impact our business and any changes that further restricts coverage of our products or lowers reimbursement for procedures using our products could materially affect our business.
The existence of adequate coverage and reimbursement will be important for sales of our products in the office setting of care. Inadequate coverage and reimbursement policies for our products could affect the adoption of our products and our future revenue.
We will be commercializing SINUVA for use in the office setting of care. SINUVA is designated as a drug by the FDA and as such, providers or specialty pharmacies will seek reimbursement for the product using an unassigned J Code. We do not have experience with this reimbursement and do not know how effective this approach will be in securing reimbursement from payors to cover the cost of SINUVA or if the level of reimbursement will be sufficient to support usage. We have applied for a product specific code from CMS and we do not know if we will be successful in securing a product specific code for SINUVA. Furthermore, patients may have to pay a co-pay toward the product and the procedure, and we do not know if they will want to do that, which could impact the adoption of SINUVA.
19
Table of Contents
While our PROPEL family of products are used principally in the operating room setting in hospitals and ASCs, these products, particularly PROPEL Contour, could be used in the office setting of care following sinus opening. For example, following a balloon sinuplasty procedure. Successful sales of our PROPEL family of products in the physician’s office setting of care depends on the availability of adequate coverage and reimbursement from third-party payors for either the products specifically, the procedures associated with the use of the products, or both. Providers that purchase our products generally rely on third-party payors to reimburse all or part of the costs and fees associated with the procedures performed with these medical devices or the devices themselves. Adequate coverage and reimbursement from third-party payors, including governmental payors, such as Medicare and Medicaid, therefore, is important for obtaining product acceptance and widespread adoption in the marketplace.
To receive payment for procedural work associated with the office placement of our PROPEL family of products, physicians may use a variety of existing Current Procedure Terminology, or CPT, codes. CMS has also assigned a Healthcare Common Procedure Coding System, or HCPCS, code of S1090 to seek reimbursement for the PROPEL implant itself. This code applies to the entire PROPEL family of products.
The HCPCS code S1090 cannot be reported to all payors, including Medicare. Separate payment for this code is dependent upon payor coverage. Since PROPEL Contour has clinical applications that include placement in the office setting, the inability to report HCPCS code S1090 to all payors may be a limiting factor to our sales of PROPEL Contour in the office setting. While PROPEL Contour can be reported with an unlisted HCPCS code J3490 to such payors in the office setting, payment for an unlisted code varies by payor and is also dependent upon favorable coverage by the payor.
In the United States, coverage and reimbursement for medical devices vary among payors. In addition, payors review coverage policies on an ongoing basis and can, without notice, change or deny coverage for these new products and procedures. We estimate that commercial payors covering a significant number of U.S. covered lives currently have non-coverage policies relating to our PROPEL family of products, designating these products investigational or experimental. Some governmental and commercial payors do not currently cover or reimburse our products because they have determined insufficient evidence of favorable clinical outcomes is available. Although some consider the steroid releasing implants investigational or experimental at this time, these payors may in the future determine sufficient evidence has been developed to cover and reimburse our products and related procedures. We are actively working to reverse these non-coverage decisions but cannot provide assurance that we will be successful in these efforts. If we are not successful in reversing existing non-coverage policies, or if other third-party payors issue similar policies, this could have a material adverse effect on our business and operations. Further, third-party payors who currently cover and reimburse customers for procedures using our products may in the future choose to decrease current levels of reimbursement or eliminate reimbursement altogether, either of which will cause our business to suffer.
Our future growth depends on physician awareness and adoption of our steroid releasing implants.
We focus our sales, marketing and education efforts primarily on ENT physicians. We train physicians on the patient population that would benefit from our steroid releasing implants. This patient population is based on those included in our clinical studies and includes, for example, patients with or without polyps as well as patients undergoing either primary or revision surgery. Some physicians may choose to utilize our products on a subset of their patients such as patients with severe polyp disease that they deem at higher risk for postoperative complications. If we are not able to effectively demonstrate to those physicians that our products are beneficial in a broad range of patients on which they operate, their adoption of our products will be limited.
We train our physician customers on the proper techniques in using our devices to achieve the intended outcome. The successful use of our steroid releasing implants depends in large part on the physician’s adherence to the techniques that they are provided in training by our sales representatives. In the event that physicians do not adhere to these techniques or if they perceive that our products are too cumbersome for them to use, we may
20
Table of Contents
have difficulty facilitating adoption. Additionally, physicians may develop their own techniques for use of our products during insertion and during the period in which the drug is delivered and is absorbed. For example, we are aware some physicians are removing our steroid releasing implants before all of the drug has been released into the surrounding tissue. While physicians were allowed to remove the implant at any time at their discretion in our clinical studies, early removal could lead to suboptimal outcomes. In addition, if physicians utilize our products in a manner that is inconsistent with how they were studied clinically, their outcomes may not be consistent with the outcomes achieved in our clinical studies, which may impact their perception of patient benefit and limit their adoption of our products.
Our clinical studies were designed to demonstrate the safety and efficacy of our steroid releasing implants based on FDA requirements and may not be seen as compelling to physicians. Any subsequent clinical studies that are conducted and published may not be positive or consistent with our existing data, which would affect the rate of adoption of our products.
Our success depends on the medical community’s acceptance of our steroid releasing implants as tools that are useful to ENT physicians treating patients with chronic sinusitis. We have sponsored fourteen multicenter, prospective studies of over 900 patients to track outcomes of treatment with our steroid releasing implants across multiple sinuses and settings of care. These clinical data have resulted in the highest level of evidence generated for any medical device used to improve the outcomes of sinus surgery. While the results of these studies collectively indicate a favorable safety and efficacy profile, the study designs and results may not be viewed as compelling to our physician customers. If physicians do not find our data compelling, they may choose not to use our products or limit their use. Additionally, the long-term effects of sinus interventions in conjunction with our steroid releasing implants beyond six months are not known. Certain ENT physicians, hospitals and surgery centers may prefer to see longer term efficacy data than we have produced. We cannot assure that any data that we or others generate will be consistent with that observed in these studies or meet the endpoints, nor that the results will be maintained beyond the time points studied. We also cannot assure that any data that may be collected will be compelling to the medical community because the data may not be scientifically meaningful and may not demonstrate that sinus procedures using our steroid releasing implants are an attractive option when compared against data from alternative treatments.
Each ENT physician’s individual experience with our steroid releasing implants will vary, and we believe that physicians will compare actual long-term outcomes in their own practices using our steroid releasing implants against sinus surgery used in conjunction with traditional sinus packing techniques. A long-term, adequately-controlled clinical study comparing sinus surgery performed in conjunction with our steroid releasing implants against sinus surgery performed in conjunction with the variety of traditional sinus packing techniques incorporated by physicians would be expensive and time-consuming and we have not conducted, and are not currently planning to conduct, such a study. If the experience of physicians indicates that the use of our steroid releasing implants in FESS is not as safe or effective as other treatment options or does not provide a lasting solution to patients with chronic sinusitis, adoption of our products may suffer and our business would be harmed.
Following commercial launch, SINUVA will be available to a larger number of patients, and we do not know whether the results of SINUVA’s use in such larger number of patients will be consistent with the results from our clinical studies.
While the FDA granted approval of SINUVA based on the data included in its NDA, including data from our completed clinical trials, we do not know whether the results, when a large number of patients are exposed to SINUVA, including results related to safety and efficacy, will be consistent with the results from the clinical trials of SINUVA that served as the basis for the approval of SINUVA. During research and development, SINUVA’s use was limited principally to clinical trial patients under controlled conditions and under the care of expert physicians. New data relating to SINUVA, including from adverse event reports, may result in changes to the product label and may adversely affect sales, or result in withdrawal of SINUVA from the market. The FDA
21
Table of Contents
and regulatory authorities in other jurisdictions may also consider any new data in connection with further marketing approval applications. In addition, in patients who take multiple medications, drug interactions could occur that can be difficult to predict. If SINUVA or any additional approved products cause serious or unexpected side effects after receiving market approval, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw their approval of SINUVA or impose restrictions on its distribution; |
| • | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| • | we may be required to change the way SINUVA is promoted or administered, or conduct additional clinical studies; |
| • | we could be sued and held liable for harm caused to patients; or |
| • | our reputation may suffer. |
Any of these events could prevent us from maintaining market acceptance of the affected product and could substantially increase the costs of commercializing SINUVA or any additional products.
We utilize third-party, single source suppliers and service providers for many of the components, materials and services used in the production of our steroid releasing implants, and the loss of, or disruption by, any of these suppliers or service providers could harm our business.
The active pharmaceutical ingredient, or API, and a number of our critical components used in our steroid releasing implants are supplied to us from single source suppliers. We rely on single source suppliers for some of our polymer materials, some extrusions and molded components, and some off-the-shelf components. If a supplier delivers products of insufficient quality, it could lead to lot issues, failures or recalls. Our ability to supply our products commercially and to develop our product candidates depends, in part, on our ability to obtain these components in accordance with regulatory requirements and in sufficient quantities and quality for commercialization and clinical testing. We have entered into manufacturing, supply or quality agreements with a number of our single source suppliers pursuant to which they supply the components we need. We are not certain that our single source suppliers will be able to meet our demand for their products, either because of the nature of our agreements with those suppliers, our limited experience with those suppliers or our relative importance as a customer to those suppliers. It may be difficult for us to assess their ability to timely meet our demand in the future based on past performance. While our suppliers have generally met our demand for their products on a timely basis in the past, they may subordinate our needs in the future to their other customers.
Establishing additional or replacement suppliers for the API or any of the components or processes used in our products, if required, may not be accomplished quickly. If we are able to find a replacement supplier, the replacement supplier would need to be qualified and may require additional regulatory authority approval, which could result in further delay. For example, the FDA, could require additional supplemental data if we rely upon a new supplier for the API used in our PROPEL family of products and SINUVA. While we seek to maintain adequate inventory of the single source components and materials used in our products, any interruption or delay in the supply of components or materials, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders.
If our third-party suppliers fail to deliver the required commercial quantities of materials, on a timely basis and at commercially reasonable prices, and we are unable to find one or more replacement suppliers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and on a timely basis, the continued commercialization of our products and the development of our product candidates would be impeded, delayed, limited or prevented, which could harm our business, results of operations, financial condition and prospects.
22
Table of Contents
We plan to rely on specialty pharmacies and specialty distributors for distribution of SINUVA in the United States, and the failure of those specialty pharmacies and specialty distributors to distribute SINUVA effectively would adversely affect sales of SINUVA.
We have historically relied on our internal sales channel to sell our products. However, we plan to rely on specialty pharmacies and specialty distributors for the distribution of SINUVA in the United States. A specialty pharmacy is a pharmacy that specializes in the dispensing, and a specialty distributor that specializes in the distribution, of medications for complex or chronic conditions, which often require a high level of patient education, physician administration and ongoing management. The use of specialty pharmacies and specialty distributors involves certain risks, including, but not limited to, risks that these specialty entities will:
| • | not provide us accurate or timely information regarding their inventories, the number of patients who are using our products or complaints about our products; |
| • | reduce or discontinue their efforts to sell or support or otherwise not effectively sell or support our products; |
| • | not devote the resources necessary to sell our products in the volumes and within the time frames that we expect; |
| • | engage in unlawful or inappropriate business practices that result in legal or regulatory enforcement activity which could result in liability to the Company or damage its goodwill with customers; or |
| • | be unable to satisfy financial obligations to us or others. |
In the event that any of the specialty pharmacies or specialty distributors whom we work with do not fulfill their contractual obligations to us or refuses to or fails to adequately serve patients, or the agreements are terminated without adequate notice, shipments of SINUVA, and associated revenues, would be adversely affected.
It is difficult to forecast future performance, which may cause our financial results to fluctuate unpredictably.
It is difficult for us to predict future performance. As we gain additional commercial experience, a number of factors over which we have limited control may contribute to fluctuations in our financial results, such as seasonal variations in revenue. Demand for our products may be impacted adversely by weather and the annual resetting of patient healthcare insurance plan deductibles, both of which may cause patients to delay or decline elective procedures such as FESS and SINUVA implantation. Demand may also be impacted by the seasonal nature of allergies and cold and flu season and the resultant onset of sinus-related symptoms. Other factors that may impact our quarterly results include:
| • | ENT physician adoption of our steroid releasing implants; |
| • | fluctuations in revenue due to changes in or from estimated gross-to-net deductions, including distributor fees and prompt payment discounts, discounts related to commercial agreements or government mandated programs, returns and replacements and, should we elect to offer such support, patient or payor assistance programs, and other related deductions and adjustments; |
| • | unanticipated pricing pressure; |
| • | the hiring, retention and continued productivity of our sales representatives; |
| • | our ability to expand the geographic reach of our sales and marketing efforts; |
| • | our ability to obtain or maintain regulatory approval and reimbursement coverage for our products in development or for our current products outside the United States; |
| • | fluctuations in revenue due to changes in third party payor reimbursement for procedures associated with the use of our products; |
23
Table of Contents
| • | our ability to maintain intellectual property protection for our products and our competitors being granted patents for competing products; |
| • | results of clinical research and trials on our existing products and products in development; |
| • | delays in receipt of anticipated purchase orders; |
| • | timing of new product offerings, acquisitions, licenses or other significant events by us or our competitors; |
| • | delays in, failure of, or quality issues with, component and raw material deliveries by our suppliers or service providers; |
| • | manufacturing issues or lot failures; and |
| • | positive or negative coverage in the media or clinical publications of our steroid releasing implants or products of our competitors or our industry. |
In the event our actual revenue and operating results do not meet our forecasts for a particular period, the market price of our common stock may decline substantially.
Our long-term growth depends on our ability to develop and commercialize additional ENT products.
It is important to our business that we continue to build a more complete product offering within the ENT market. We are using our drug releasing bioabsorbable technology to develop new products for use in the physician office setting. Developing additional products is expensive and time-consuming and could divert management’s attention away from our current sinus surgery products and harm our business. Even if we are successful in developing additional products, the success of any new product offering or enhancement to an existing product will depend on several factors, including our ability to:
| • | properly identify and anticipate ENT physician and patient needs; |
| • | receive adequate reimbursement for such products; |
| • | develop and introduce new products or product enhancements in a timely manner; |
| • | avoid infringing upon the intellectual property rights of third parties; |
| • | demonstrate, if required, the safety and efficacy of new products with data from preclinical studies and clinical trials; |
| • | obtain the necessary regulatory clearances or approvals for new products or product enhancements; |
| • | be fully FDA-compliant with marketing and manufacturing of new devices or modified products; |
| • | provide adequate training to potential users of our products; and |
| • | develop an effective and FDA-compliant, dedicated sales and marketing team. |
If we are unsuccessful in developing and commercializing additional products in other areas of ENT, our ability to increase our revenue may be impaired.
Consolidation in the healthcare industry could lead to demands for price concessions, which may impact our ability to sell our products at prices necessary to support our current business strategies.
Healthcare costs have risen significantly over the past several decades, which has driven numerous cost reform initiatives by legislators, regulators and third-party payors. Cost reform has elicited a consolidation trend in the healthcare industry to aggregate purchasing power, which may create more requests for pricing concessions in the future. Additionally, group purchasing organizations, independent delivery networks and large
24
Table of Contents
single accounts may continue to use their market power to consolidate purchasing decisions for hospitals and ambulatory surgery centers. We expect that market demand, government regulation, third-party coverage and reimbursement policies and societal pressures will continue to change the healthcare industry worldwide, resulting in further business consolidations and alliances among our customers, which may exert further downward pressure on the prices of our products and may adversely impact our business, results of operations, financial condition and prospects.
We compete or may compete in the future against other companies, some of which have longer operating histories, more established products and greater resources, which may prevent us from achieving significant market penetration or improved operating results.
Our industry is highly competitive, subject to change and significantly affected by new product introductions and other activities of industry participants. Many of the companies developing or marketing ENT products are publicly traded companies, including Medtronic, Olympus, Johnson & Johnson, Stryker and Smith & Nephew. These companies could develop drug releasing products that could compete with our products and most of these companies enjoy several competitive advantages, including:
| • | greater financial and human capital resources; |
| • | significantly greater name recognition; |
| • | established relationships with ENT physicians, referring physicians, customers and third-party payors; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer greater discounts or incentives to gain a competitive advantage; and |
| • | established sales, marketing and worldwide distribution networks. |
In addition, there are and have been venture companies seeking to develop competitive products. Companies may also market alternatives to current modes of treatment, such as OptiNose. Finally, there are established pharmaceutical companies evaluating monoclonal antibodies for the treatment of chronic sinusitis.
If another company successfully develops an approach for the treatment of chronic sinusitis, including alternative device, drug delivery or pharmaceutical agent, our business could be significantly and adversely affected.
If physicians treat more patients in their offices instead of performing surgery in the operating room, our ability to sell our PROPEL family of products may be harmed.
The prevalence of sinus procedures being performed in the office has increased since sinus dilation products for use in the office setting received Category I CPT codes in 2011. As a result, the number of companies selling sinus dilation products has increased and well-known companies such as Medtronic, Entellus (which has agreed to be acquired by Stryker) and Johnson & Johnson have begun to sell sinus dilation products. This has led to increased marketing investments to sell these sinus dilation products in an attempt to not only grow the overall sinus procedure market but also to shift procedures from the operating room to the office. If more patients are treated for chronic sinusitis in a physician’s office with a sinus dilation product rather than through FESS procedures in the operating room, the volume of FESS procedures performed may not grow as anticipated and our ability to sell our products may be harmed.
We face the risk of product liability claims that could be expensive, divert management’s attention and harm our reputation and business. We may not be able to maintain adequate product liability insurance.
Our business exposes us to the risk of product liability claims that are inherent in the testing, manufacturing and marketing of medical devices and drug products. This risk exists even if a device or product is approved for
25
Table of Contents
commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA, such as the case with our PROPEL family of products and SINUVA, or an applicable foreign regulatory authority. Our products and product candidates are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our products or our product candidates could result in patient injury or death. The medical device industry has historically been subject to extensive litigation over product liability claims, and we cannot offer any assurance that we will not face product liability suits. We may be subject to product liability claims if our steroid releasing implants cause, or merely appear to have caused, patient injury or death. In addition, an injury that is caused by the activities of our suppliers, such as those who provide us with components and raw materials, may be the basis for a claim against us. Product liability claims may be brought against us by consumers, healthcare providers or others selling or otherwise coming into contact with our products or product candidates, among others. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| • | costs of litigation; |
| • | distraction of management’s attention from our primary business; |
| • | the inability to commercialize our products or, if approved, our product candidates; |
| • | decreased demand for our products or, if approved, product candidates; |
| • | impairment of our business reputation; |
| • | product recall or withdrawal from the market; |
| • | withdrawal of clinical trial participants; |
| • | substantial monetary awards to patients or other claimants; or |
| • | loss of revenue. |
While we may attempt to manage our product liability exposure by proactively recalling or withdrawing from the market any defective products, any recall or market withdrawal of our products may delay the supply of those products to our customers and may impact our reputation. We can provide no assurance that we will be successful in initiating appropriate market recall or market withdrawal efforts that may be required in the future or that these efforts will have the intended effect of preventing product malfunctions and the accompanying product liability that may result. Such recalls and withdrawals may also be used by our competitors to harm our reputation for safety or be perceived by patients as a safety risk when considering the use of our products, either of which could have an adverse effect on our business.
In addition, although we have product liability and clinical study liability insurance that we believe is appropriate, this insurance is subject to deductibles and coverage limitations. Our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, coverage may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost or on acceptable terms with adequate coverage or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may harm our business. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have a material adverse effect on our business, financial condition and results of operations.
The misuse or off-label use of our products may harm our image in the marketplace, result in injuries that lead to product liability suits or result in costly investigations and sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
The products we currently market have been approved by the FDA for specific treatments. We train our marketing and direct sales force to not promote our products for uses outside of the FDA-approved indications
26
Table of Contents
for use, known as “off-label uses.” We cannot, however, prevent a physician from using our products off-label, when in the physician’s independent professional medical judgment he or she deems it appropriate. There may be increased risk of injury to patients if physicians attempt to use our products off-label. Furthermore, the use of our products for indications other than those approved by the FDA or any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
Physicians may also misuse our products or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Product liability claims could divert management’s attention from our core business, be expensive to defend, and result in sizable damage awards against us that may not be covered by insurance. In addition, if the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, and the curtailment of our operations. Any of these events could significantly harm our business and results of operations and cause our stock price to decline.
Our ability to maintain our competitive position depends on our ability to attract and retain highly qualified personnel.
We believe that our continued success depends, to a significant extent, upon the efforts and abilities of our key employees. All of our executive officers and other employees are at-will employees, and therefore may terminate employment with us at any time with no advance notice. The replacement of any of our key personnel or the turnover of a meaningful number of our employees within a particular function or throughout the company within a given period of time, likely would involve significant time and costs and may significantly delay or prevent the achievement of our business objectives and would harm our business.
Our future success also depends on our ability to continue to attract and retain our executive officers and other key employees. Many of our employees have become or will soon become vested in a substantial amount of stock or number of stock options. Our employees may be more likely to leave us if the shares they own or the shares underlying their vested options have significantly appreciated in value relative to the original purchase prices of the shares or the exercise prices of the options, or if the exercise prices of the options that they hold are significantly below the market price of our common stock. Further, our employees’ ability to exercise those options and sell their stock in a public market may result in a higher than normal turnover rate. We do not carry any “key person” insurance policies.
If our facilities or the facility of a supplier or customer become inoperable, we will be unable to continue to research, develop, manufacture, commercialize and sell our products and, as a result, our business will be harmed until we are able to secure a new facility.
We do not have redundant facilities. We perform substantially all of our research and development, manufacturing and commercialization activity and maintain all our raw material and a significant portion of our finished goods inventory in a single location in Menlo Park, California. Menlo Park is situated on or near earthquake fault lines. Our facility and equipment would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including, but not limited to, earthquakes, flooding, fire, water shortages and power outages, which may render it difficult or impossible for us to perform our research, development, manufacturing and commercialization activities for some period of time. The inability to perform those activities, combined with our limited inventory
27
Table of Contents
of raw materials and finished product reserve, may result in the inability to continue manufacturing our products during such periods and the loss of customers or harm to our reputation. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and this insurance may not continue to be available to us on acceptable terms, or at all. In addition, while we have a limited amount of inventory at a third party storage and fulfillment center, that inventory may not be sufficient to continue our operations if our primary facility is damaged. The occurrence of natural disasters or acts of terrorism could also cause delays in our customers’ supply chain, causing them to delay their requirements for our products until they resolve shortages from their other suppliers. Any such occurrences of natural disasters or acts of terrorism could have a material adverse effect on our business, our results of operations and our financial condition.
If we experience significant disruptions in our information technology systems, our business may be adversely affected.
We depend on our information technology systems for the efficient functioning of our business, including accounting, data storage, compliance, purchasing and inventory management. Our current systems provide physical and virtual redundancy while being operated from our physical location in Menlo Park. While we will attempt to mitigate interruptions in our information technology systems, we may experience events or circumstances which could disrupt our operations, including our ability to timely ship and track product orders, project inventory requirements, manage our supply chain and otherwise adequately service our customers. In the event we experience significant disruptions, such as natural disasters or security breaches, as a result of the current implementation of our information technology systems, we may not be able to repair our systems in an efficient and timely manner. Accordingly, such events may disrupt or reduce the efficiency of our entire operation and have a material adverse effect on our results of operations and cash flows.
We are increasingly dependent on sophisticated information technology for our infrastructure. Our information systems require an ongoing commitment of significant resources to maintain, protect and enhance existing systems. Failure to maintain or protect our information systems and data integrity effectively could have a materially adverse effect on our business. For example, third parties may attempt to hack into our information systems and may obtain our proprietary information.
We are expanding the scale and complexity of our operations by adding commercialization of a drug to our underlying device business. We may encounter difficulties in managing this expansion, which could disrupt our business.
SINUVA will be our first commercial product that is regulated as a drug. To sell this product, we are expanding the scope of our operations to comply with manufacturing and regulatory requirements of a drug. We are also adding a network of specialty pharmacies and specialty distributors to support product access, and adding internal or external capabilities to handle new operational requirements. We are relying on one integrated sales force to sell all our products. We will remain subject to ongoing inspection by regulatory agencies and must maintain compliance with both device and drug regulatory requirements for Quality Systems Regulation and Good Manufacturing Practice compliance, respectively.
To manage our anticipated future growth for SINUVA, our PROPEL family of products and our pipeline, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. We may not be able to effectively manage the expected expansion of our operations or recruit and train additional qualified personnel. Moreover, the expected expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
28
Table of Contents
If clinical studies of our future products or product indications do not produce results necessary to support regulatory clearance or approval in the United States or, with respect to our current or future products, elsewhere, we will be unable to commercialize these products.
We will likely need to conduct additional clinical studies in the future to support new product or product indication approvals, or for the approval of the use of our products in some foreign countries. Clinical testing takes many years, is expensive and carries uncertain outcomes. The initiation and completion of any of these studies may be prevented, delayed, or halted for numerous reasons, including, but not limited to, the following:
| • | the FDA, institutional review boards or other regulatory authorities do not approve a clinical study protocol, force us to modify a previously approved protocol, or place a clinical study on hold; |
| • | patients do not enroll in, or enroll at a lower rate than we expect, or do not complete a clinical study; |
| • | patients or investigators do not comply with study protocols; |
| • | patients do not return for post-treatment follow-up at the expected rate; |
| • | patients experience unexpected adverse event or side effects for a variety of reasons that may or may not be related to our products; |
| • | sites participating in an ongoing clinical study withdraw, requiring us to engage new sites; |
| • | difficulties or delays associated with establishing additional clinical sites; |
| • | third-party clinical investigators decline to participate in our clinical studies, do not perform the clinical studies on the anticipated schedule, or are inconsistent with the investigator agreement, clinical study protocol, good clinical practices or other agency requirements; |
| • | third-party organizations do not perform data collection and analysis in a timely or accurate manner; |
| • | regulatory inspections of our clinical studies or manufacturing facilities require us to undertake corrective action or suspend or terminate our clinical studies; |
| • | changes in federal, state, or foreign governmental statutes, regulations or policies; |
| • | interim results are inconclusive or unfavorable as to immediate and long-term safety or efficacy; |
| • | the study design is inadequate to demonstrate safety and efficacy; or |
| • | the study does not meet the primary endpoints. |
Clinical failure can occur at any stage of the testing. Our clinical studies may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and non-clinical testing in addition to those we have planned. Our failure to adequately demonstrate the safety and efficacy of any of our devices would prevent receipt of regulatory clearance or approval and, ultimately, the commercialization of that device or indication for use. Even if our future products are approved in the United States, commercialization of our products in foreign countries would require approval by regulatory authorities in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials. Any of these occurrences may harm our business, results of operations, financial condition and prospects.
Reimbursement in international markets may require us to undertake country-specific reimbursement activities, including additional clinical studies, which could be time-consuming and expensive and may not yield acceptable reimbursement rates.
In international markets, market acceptance of our products will likely depend in large part on the availability of reimbursement within prevailing healthcare payment systems. Reimbursement and healthcare
29
Table of Contents
payment systems in international markets vary significantly by country, and by region in some countries, and include both government-sponsored healthcare and private insurance. Securing separate payment for our products may require additional investment in clinical data to satisfy the requirements of health technology assessment organizations in these countries. We may not obtain international reimbursement approvals in a timely manner, if at all. In addition, even if we do obtain international reimbursement approvals, the level of reimbursement may not be enough to commercially justify expansion of our business into the approving jurisdiction. To the extent we or our customers are unable to obtain reimbursement for our steroid releasing implants in major international markets in which we seek to market and sell our products, our international revenue growth would be harmed, and our business and results of operations would be adversely affected.
Pricing for pharmaceutical products has come under increasing scrutiny by governments, legislative bodies and enforcement agencies. These activities may result in actions that have the effect of reducing our revenue or harming our business or reputation.
Many companies in our industry have received a governmental request for documents and information relating to drug pricing and patient support programs. We could receive a similar request, which would require us to incur significant expense and result in distraction for our management team. Additionally, to the extent there are findings, or even allegations, of improper conduct on the part of the company, such findings could further harm our business, reputation and/or prospects. It is possible that such inquiries could result in: negative publicity or other negative actions that could harm our reputation; changes in our product pricing and distribution strategies; reduced demand for our approved products; and/or reduced reimbursement of approved products, including by federal health care programs such as Medicare and Medicaid and state health care programs.
In addition, the current administration has indicated interest in taking regulatory and other policy actions pertaining to drug pricing, including potential proposals relating to Medicare price negotiations, importation of drugs from other countries and facilitating value-based arrangements between manufacturers and payers. At this time, it is unclear whether any of these proposals will be pursued and how they would impact our products or our future product candidates.
State and local governments continue to consider prescription drug pricing transparency proposals. In October 2017, California Governor Jerry Brown signed legislation requiring pharmaceutical manufacturers to disclose and provide justification for certain price increases; however, the regulations under which we will have to operate have not yet been promulgated. While we have taken and will continue to take appropriate actions to ensure compliance with this new law, without knowing the final regulations applicable to us, we cannot comprehensively assess the potential impact on our business. Additional legislation or ballot initiatives may be proposed by various states and municipalities in the future, and we cannot predict the outcome of any future proposals, the market’s perception of them or their potential impact on us.
Risks Relating to Regulatory Matters
Our products are subject to extensive regulation by the FDA, and other agencies, including the requirement to obtain approval prior to commercializing our products and the requirement to report adverse events and other ongoing reporting requirements. If we fail to obtain necessary FDA device or drug approvals for our products, or are subject to regulatory enforcement action as a result of our failure to properly report adverse events or otherwise comply with regulatory requirements, our commercial operations would be harmed.
Our steroid releasing implants are subject to extensive regulation by the FDA and various other federal, state and foreign governmental authorities. The Premarket Approval, or PMA, and New Drug Application, or NDA, approval processes can be expensive and lengthy. Despite the time, effort and cost required to obtain approval, there can be no assurance that any product that we intend to commercialize in the future will be approved by the FDA in a timely fashion, if at all.
30
Table of Contents
Our currently marketed products are subject to Medical Device Reporting, or MDR, and drug postmarketing safety reporting obligations, which require that we timely report any incidents to the FDA. In the European Union, our CE Marked products are subject to vigilance reporting.
The FDA and state authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA or state agencies, which may include any of the following sanctions:
| • | adverse publicity, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | repair, replacement, recall or seizure of our products; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | delaying or refusing our requests for approval of new products, new intended uses or modifications to our existing products; |
| • | refusal to grant export approval for our products; |
| • | withdrawing product approvals that have already been granted; and |
| • | criminal prosecution. |
If any of these enforcement actions were to be taken by the government, our business could be harmed.
We cannot predict whether or when we will obtain regulatory approval to commercialize product candidates and we cannot, therefore, predict the timing of any future revenue from product candidates. Regulatory approval of a product candidate is not guaranteed, and the approval process is expensive, uncertain and lengthy.
We cannot commercialize our product candidates until the appropriate regulatory authorities, such as the FDA, have reviewed and approved the product candidate. Regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval for product candidates. Additional delays may result if product candidates are brought before an FDA advisory committee, which could recommend restrictions on approval or recommend non-approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical studies and the review process. As a result, we cannot predict when, if at all, we will receive any future revenue from commercialization of product candidates. The FDA has substantial discretion in the drug approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons, including the following:
| • | we may be unable to demonstrate to the satisfaction of regulatory authorities that a product candidate is safe and effective for any indication; |
| • | regulatory authorities may not find the data from clinical studies sufficient or may differ in the interpretation of the data; |
| • | regulatory authorities may require additional clinical studies; |
| • | the FDA or foreign regulatory authority might not approve our manufacturing processes or facilities for clinical or commercial production; |
| • | the FDA or foreign regulatory authority may change its approval policies or adopt new regulations; |
| • | the FDA or foreign regulatory authorities may disagree with the design or implementation of our clinical studies; |
| • | the FDA or foreign regulatory authority may not accept clinical data from studies that are conducted in countries where the standard of care is potentially different from that in the U.S.; |
31
Table of Contents
| • | the results of clinical studies may not meet the level of statistical significance required by the FDA or foreign regulatory authorities for approval; |
| • | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; and |
| • | the data collection from clinical studies of our product candidates may not be sufficient to support the submission of a NDA or other submission or to obtain regulatory approval in the U.S. or elsewhere. |
In addition, events raising questions about the safety of certain marketed products may result in increased caution by the FDA and other regulatory authorities in reviewing new products based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals.
Even though SINUVA was approved by the FDA, this approval was limited to certain indications, and additional clinical studies and regulatory applications are required to expand SINUVA indications. We can provide no assurances that such additional clinical studies or regulatory applications will be successful.
We have developed SINUVA for use in the physician’s office for treatment of patients who have had sinus surgery but continue to suffer from symptoms of chronic sinusitis. In November 2017, we commenced the ENCORE study, a 50 patient prospective, multicenter, open-label study focused on evaluation of the safety of repeat placement of the SINUVA implant in chronic sinusitis patients with nasal polyps, and completed enrollment in January 2018. Additional clinical studies may be required to support any targeted indications, which will require additional time and expense and may not prove successful. Limitations in our label for SINUVA will reduce the number of patients for whom SINUVA is indicated and could reduce the size of the market and our financial prospects. Further, there is no guarantee that any efforts that we decide to undertake will meet the FDA’s requirements, and we may not receive approval the additional indications for SINUVA despite such efforts.
If we are able to successfully commercialize SINUVA, and if we participate in but fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program, or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition and results of operations.
If we participate in the Medicaid Drug Rebate Program, and other governmental pricing programs, we will be obligated to pay certain specified rebates and report pricing information with respect to SINUVA. Pricing and rebate calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. We cannot assure you that our submissions will not be found by the CMS to be incomplete or incorrect. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current average manufacturer price, or AMP, and best price, or BP, for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due, and CMS may request or require restatements for earlier periods as well. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities, such as safety-net providers, under the Public Health Service’s 340B drug pricing program, or 340B, and under other similar government pricing programs
We will also be liable for errors associated with our submission of pricing data. In addition to retroactive rebates and the potential for 340B refunds, if we are found to have knowingly submitted false AMP or BP information to the government, we may be liable for civil monetary penalties. If we are found to have made a
32
Table of Contents
misrepresentation in the reporting of our AMP, we may be liable for civil monetary penalties as well. Our failure to submit monthly or quarterly AMP and BP data on a timely basis could result in a civil monetary penalty for each day the information is late beyond the due date. Such failure also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid for SINUVA. A final regulation imposes a civil monetary penalty for each instance of knowingly and intentionally charging a 340B covered entity more than the 340B ceiling price.
Federal law requires that a company must participate in the U.S. Department of Veterans Affairs, or VA, Federal Supply Schedule, or FSS, pricing program to be eligible to have its products paid for with federal funds. As part of this program, we would be obligated to make SINUVA available for procurement on an FSS contract under which we must comply with standard government terms and conditions and charge a price that is no higher than the statutory Federal Ceiling Price, or FCP, to several federal agencies including the VA, the U.S. Department of Defense, the Public Health Service and the U.S. Coast Guard and others. The FCP is based on the Non-Federal Average Manufacturer Price, or Non-FAMP, which we calculate and report to the VA on a quarterly and annual basis. If we overcharge the government in connection with our FSS contract or Section 703 Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the U.S. civil False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time consuming, and could have a material adverse effect on our business, financial condition and results of operations.
If we materially modify our approved products, we may need to seek and obtain new approvals, which, if not granted, would prevent us from selling our modified products.
A component of our strategy is to continue to modify and upgrade our steroid releasing implants. Medical devices can be marketed only for the indications for which they are approved. We have received a number of PMA supplement approvals since the original approval of PROPEL. We may not be able to obtain additional regulatory approvals for new products or for modifications to, or additional indications for, our existing products in a timely fashion, or at all. Delays in obtaining future approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our revenue and potential future profitability.
We may fail to obtain foreign regulatory approvals to market our products in other countries.
We have only had limited sales outside the United States. Sales of our steroid releasing implants outside the United States are subject to foreign regulatory requirements that vary widely from country to country. In addition, the FDA regulates exports of medical devices from the United States. Complying with international regulatory requirements can be an expensive and time-consuming process and approval is not certain. The time required to obtain approvals, if required by other countries, may be longer than that required for FDA approvals, and requirements for such approvals may significantly differ from FDA requirements. In certain countries we may rely upon a third-party or third-party distributors to obtain all required regulatory approvals, and these distributors may be unable to obtain or maintain such approvals. Our distributors in these countries may also incur significant costs in attempting to obtain and in maintaining foreign regulatory approvals or qualifications, which could increase the difficulty of attracting and retaining qualified distributors. If these distributors experience delays in receiving necessary qualifications, clearances or approvals to market our products outside the United States, or if they fail to receive those qualifications, clearances or approvals, we may be unable to market our products or enhancements in certain international markets effectively, or at all.
International jurisdictions require separate regulatory approvals and compliance with numerous and varying regulatory requirements. The approval procedures vary among countries and may involve requirements for
33
Table of Contents
additional testing, and the time required to obtain approval may differ from country to country and from that required to obtain clearance or approval in the United States.
Approval in the United States does not ensure approval or certification by regulatory authorities in other countries or jurisdictions, and approval or certification by one foreign regulatory authority does not ensure approval or certification by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval or certification process may include all of the risks associated with obtaining FDA approval. In addition, some countries only approve or certify a product for a certain period of time, and we are required to re-approve or re-certify our products in a timely manner prior to the expiration of our prior approval or certification. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals or certifications and may not receive necessary approvals to commercialize our products in any market. If we fail to receive necessary approvals or certifications to commercialize our products in foreign jurisdictions on a timely basis, or at all, or if we fail to have our products re-approved or re-certified, our business, results of operations and financial condition could be adversely affected.
These and other factors may have a material adverse effect on our international operations or on our business, results of operations and financial condition generally.
If we, our suppliers or service providers fail to comply with ongoing FDA or foreign regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain approval, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory bodies. In particular, we and our third-party suppliers are required to comply with the FDA’s current good manufacturing practices and Quality Systems regulation. These FDA regulations cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. If we, or our suppliers, fail to adhere to current good manufacturing practice requirements in the United States, this could delay production of our products and lead to fines, difficulties in obtaining regulatory approvals, recalls, enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse effect on our financial condition or results of operations.
In addition, the FDA audits compliance through periodic announced and unannounced inspections of manufacturing and other facilities. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in any of the following enforcement actions:
| • | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | unanticipated expenditures to address or defend such actions; |
| • | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | refusing or delaying our requests for regulatory approvals of new products or modified products; |
| • | withdrawing PMA or NDA approvals that have already been granted; |
| • | refusal to grant export approval for our products; or |
| • | criminal prosecution. |
34
Table of Contents
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
If we expand our operations outside the United States, our products and operations will be required to comply with standards set by foreign regulatory bodies, and those standards, types of evaluation and scope of review differ among foreign regulatory bodies. We intend to comply with the standards enforced by such foreign regulatory bodies as needed to commercialize our products. If we fail to comply with any of these standards adequately, a foreign regulatory body may take adverse actions similar to those within the power of the FDA. For example, in Europe, we are subject to a conformity assessment procedure under which a so-called Notified Body, an organization accredited by a member state of the European Economic Area, or EEA, which will audit and examine our quality system for the manufacture, design, and release of our products and confirm adherence with applicable regulatory requirements. If we fail to maintain CE Markings in accordance with these requirements, we would be precluded from selling our products in the EEA. Any such action or circumstance may harm our reputation and business, and could have an adverse effect on our business, results of operations and financial condition.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our products in their respective jurisdictions in the event of material deficiencies or defects in the design or manufacture of our products. We may, under our own initiative, recall a product if any material deficiency in our steroid releasing implants is found. The FDA requires that recalls be reported to the FDA within 10 working days after the recall is initiated. A government-mandated or voluntary recall by us or one of our international distributors could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to generate profits. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action for failing to report the recalls when they were conducted.
If the third parties on which we rely to conduct our clinical trials do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or commercialize such product candidates.
We often must rely on third parties, such as medical institutions, clinical investigators and contract laboratories to conduct our clinical trials and provide data or prepare deliverables for our PMA or NDA submissions, including supplements thereto. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed, suspended or
35
Table of Contents
terminated, and/or we may not be able to obtain regulatory approval for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
We may be subject to enforcement action if we engage in improper marketing or promotion of our products.
Our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of unapproved, or off-label, use. Physicians may use our products off-label, as the FDA does not restrict or regulate a physician’s choice of treatment within the practice of medicine. However, if the FDA determines that our promotional materials or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products could be impaired. Although our policy is to refrain from statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion. In addition, the off-label use of our products may increase the risk of product liability claims. Product liability claims are expensive to defend and could divert our management’s attention, result in substantial damage awards against us, and harm our reputation.
If we fail to comply with U.S. federal and state healthcare regulatory laws, we could be subject to penalties, including, but not limited to, administrative, civil and criminal penalties, damages, fines, disgorgement, exclusion from participation in governmental healthcare programs, and the curtailment of our operations, any of which could adversely impact our reputation and business operations.
There are numerous U.S. federal and state healthcare regulatory laws, including, but not limited to, anti-kickback laws, false claims laws, privacy laws, and transparency laws. Our relationships with healthcare providers and entities, including but not limited to, physicians, hospitals, ambulatory surgery centers, group purchasing organizations and our international distributors are subject to scrutiny under these laws. Violations of these laws can subject us to penalties, including, but not limited to, administrative, civil and criminal penalties, damages, fines, disgorgement, imprisonment, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, exclusion from participation in federal and state healthcare programs, including the Medicare, Medicaid and Veterans Administration health programs, and the curtailment of our operations. Healthcare fraud and abuse regulations are complex, and even minor irregularities can potentially give rise to claims that a statute or prohibition has been violated. The laws that may affect our ability to operate include, but are not limited to:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in cash or in kind, in exchange for or to induce either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid; |
| • | the federal civil False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from federal health care programs, such as Medicare and Medicaid that are false or fraudulent; knowingly making, using, or causing to be made or used, a false record or statement to get a false or fraudulent claim paid or approved by the government; or knowingly making, using, or causing to be made or used, a false record or statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
36
Table of Contents
| • | the federal criminal False Claims Act, which imposes criminal fines or imprisonment against individuals or entities who make or present a claim to the government knowing such claim to be false, fictitious or fraudulent; |
| • | the civil monetary penalties statute, which imposes penalties against any person or entity who, among other things, is determined to have presented or caused to be presented, a claim to a federal healthcare program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended, which created federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans and healthcare clearinghouses as well as their business associates that perform services for them that involve individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization, including mandatory contractual terms as well as directly applicable privacy and security standards and requirements; |
| • | the Federal Trade Commission Act and similar laws regulating advertisement and consumer protections; |
| • | the federal Foreign Corrupt Practices Act of 1997, which prohibits corrupt payments, gifts or transfers of value to foreign officials; and |
| • | foreign or U.S. state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
Further, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the Affordable Care Act, among other things, amends the intent requirements of the federal Anti-Kickback Statute and certain criminal statutes governing healthcare fraud. A person or entity can now be found guilty of violating the statute without actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. Moreover, while we do not submit claims and our customers make the ultimate decision on how to submit claims, from time-to-time, we may provide reimbursement guidance to our customers. If a government authority were to conclude that we provided improper advice to our customers or encouraged the submission of false claims for reimbursement, we could face action against us by government authorities. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our reputation, business, results of operations and financial condition.
We have entered into consulting agreements with physicians, including some who influence the ordering of and use our products in procedures they perform. While we believe these transactions were structured to comply with all applicable laws, including state and federal anti-kickback laws, to the extent applicable, regulatory agencies may view these transactions as prohibited arrangements that must be restructured, or discontinued, or for which we could be subject to other significant penalties. We could be adversely affected if regulatory agencies interpret our financial relationships with ENT physicians who influence the ordering of and use our products to be in violation of applicable laws. This could subject us to the penalties described above.
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available under such laws, it is possible that some of our business activities, including our relationships
37
Table of Contents
with healthcare providers and entities, including, but not limited to, physicians, hospitals, ambulatory surgery centers, group purchasing organizations and our independent distributors and certain sales and marketing practices, including the provision of certain items and services to our customers, could be subject to challenge under one or more of such laws.
To enforce compliance with the healthcare regulatory laws, federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time and resource consuming and can divert management’s attention from the business. Additionally, as a result of these investigations, healthcare providers and entities may have to agree to additional onerous compliance and reporting requirements as part of a consent decree or corporate integrity agreement. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
In certain cases, federal and state authorities pursue actions for false claims on the basis that manufacturers and distributors are promoting off-label uses of their products. Pursuant to FDA regulations, we can only market our products for cleared or approved uses. Although physicians are permitted to use medical devices for indications other than those cleared or approved by the FDA in their professional medical judgment, we are prohibited from promoting products for off-label uses. We market our products and provide promotional materials and training programs to physicians regarding the use of our products. If it is determined that our business activities, including our marketing, promotional materials or training programs constitute promotion of unapproved uses, we could be subject to significant fines in addition to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure and criminal penalty.
In addition, there has been a recent trend of increased federal and state regulation of payments and transfers of value provided to healthcare professionals or entities. The Physician Payments Sunshine Act that imposes new annual reporting requirements on device manufacturers for payments and other transfers of value provided by them, directly or indirectly, to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their family members. A manufacturer’s failure to submit timely, accurately and completely the required information for all payments, transfers of value or ownership or investment interests may result in civil monetary penalties of up to an aggregate of $165,786 per year, and up to an aggregate of $1,105,241 per year for “knowing failures.” Manufacturers are required to report to CMS the detailed payment and transfers of value data and submit legal attestation to the accuracy of such data by the 90th day of each calendar year. Due to the difficulty in complying with the Physician Payments Sunshine Act, we cannot assure you that we will successfully report all payments and transfers of value provided by us, and any failure to comply could result in significant fines and penalties. Some states, such as California and Connecticut, also mandate implementation of commercial compliance programs, and other states, such as Massachusetts and Vermont, impose restrictions on device manufacturer marketing practices and tracking and reporting of gifts, compensation and other remuneration to healthcare professionals and entities. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may fail to comply fully with one or more of these requirements.
Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Most of these laws apply to not only the actions taken by us, but also to actions taken by our distributors. We have limited knowledge and control over the business practices of our distributors, and we may face regulatory action against us as a result of their actions which could have a material adverse effect on our reputation, business, results of operations and financial condition.
38
Table of Contents
In addition, the scope and enforcement of these laws are uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal or state regulatory authorities might challenge our current or future activities under these laws. Any such challenge could have a material adverse effect on our reputation, business, results of operations and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming. Additionally, we cannot predict the impact of any changes in these laws, whether or not retroactive.
Legislative or regulatory healthcare reforms may make it more difficult and costly for us to obtain regulatory approval of new products and to produce, market and distribute our products after approval is obtained.
FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of, or failure to receive, regulatory approvals for our new products would have a material adverse effect on our business, results of operations and financial condition.
Federal and state governments in the United States have recently enacted legislation to overhaul the nation’s healthcare system. While the goal of healthcare reform is to expand coverage to more individuals, it also involves increased government price controls, additional regulatory mandates and other measures designed to constrain medical costs. The Affordable Care Act significantly impacts the medical device and pharmaceutical industries, we will have to comply with its drug-specific provisions now that SINUVA has been approved. Among other things, the Affordable Care Act:
| • | imposes an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States beginning in 2013; |
| • | establishes a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; |
| • | implements payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models; and |
| • | creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
The medical device excise tax was recently suspended by the Consolidated Appropriations Act of 2016, or CAA, for calendar years 2016 and 2017. In January 2018, the medical device excise tax suspension was extended for calendar years 2018 and 2019. Absent further congressional action the excise tax will be reinstated for medical device sales beginning January 1, 2020. The CAA also temporarily delays implementation of other taxes intended to help fund Affordable Care Act programs.
Further, there have been judicial and congressional challenges to other aspects of the Affordable Care Act. For example, since January 2017, our current President of the United States has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the Affordable Care Act. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the Affordable Care Act have been signed into law. The recent resolution on appropriations for fiscal year 2018 that extended the suspension of the medial device excise tax also delayed the implementation of the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, as well as the annual fee imposed on certain health insurance providers based on market share. Additionally, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”.
39
Table of Contents
Further, the Bipartisan Budget Act of 2018, or the BBA, among other things, amends the Affordable Care Act, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. We expect there will be additional challenges and amendments to the Affordable Care Act in the future.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes reductions to Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013 and, following passage of subsequent legislative amendments to the statute, including the BBA, will stay in effect through 2027, unless additional congressional action is taken. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012 which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Given the current political environment, we expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
Our operations involve the use of hazardous and toxic materials, and we must comply with environmental laws and regulations, which can be expensive, and may affect our business and operating results.
We are subject to a variety of federal, state and local regulations relating to the use, handling, storage, disposal and human exposure to hazardous materials. Liability under environmental laws can be joint and several, and without regard to comparative fault, and environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations, which could harm our business. Although we believe that our activities conform in all material respects with environmental laws, there can be no assurance that violations of environmental and health and safety laws will not occur in the future as a result of human error, accident, equipment failure or other causes. The failure to comply with past, present or future laws could result in the imposition of fines, third-party property damage and personal injury claims, investigation and remediation costs, the suspension of production, or a cessation of operations. We also expect that our operations will be affected by other new environmental and health and safety laws on an ongoing basis. Although we cannot predict the ultimate impact of any such new laws, they will likely result in additional costs, and may require us to change how we manufacture our products, which could have a material adverse effect on our business.
Failure to comply with the United States Foreign Corrupt Practices Act, or FCPA, and similar laws associated with any activities outside the United States could subject us to penalties and other adverse consequences.
We are subject to the FCPA and other anti-bribery legislation around the world. The FCPA prohibits covered entities and their intermediaries from engaging in bribery or making other prohibited payments, offers or promises to foreign officials for the purpose of obtaining or retaining business or other advantages. In addition, the FCPA imposes recordkeeping and internal controls requirements on publicly traded corporations and their foreign affiliates, which are intended to, among other things, prevent the diversion of corporate funds to the payment of bribes and other improper payments, and to prevent the establishment of “off books” slush funds from which such improper payments can be made. Although we currently have very little commercial activity outside the United States, in the future we may face significant risks if we fail to comply with the FCPA and other laws that prohibit improper payments, offers or promises of payment to foreign governments and their
40
Table of Contents
officials and political parties by us and other business entities for the purpose of obtaining or retaining business or other advantages. In many foreign countries, particularly in countries with developing economies, some of which may represent attractive markets for us, it may be a local custom that businesses operating in such countries engage in business practices that are prohibited by the FCPA or other laws and regulations. Although we have implemented a company policy requiring our employees and consultants to comply with the FCPA and similar laws, such policy may not be effective at preventing all potential FCPA or other violations. There can be no assurance that none of our employees and agents, or those companies to which we outsource certain portions of our business operations, will not take actions that violate our policies or applicable laws, for which we may be ultimately held responsible. As a result of our focus on managing our growth, our development of infrastructure designed to identify FCPA matters and monitor compliance is at an early stage. Any violation of the FCPA and related policies could result in severe criminal or civil sanctions, which could have a material and adverse effect on our reputation, business, operating results and financial condition.
Risks Relating to Intellectual Property Matters
Intellectual property rights may not provide adequate protection, which may permit third parties to compete against us more effectively.
Our success depends significantly on our ability to protect our proprietary rights to the technologies and inventions used in, or embodied by, our products. To protect our proprietary technology, we rely on patent protection, as well as a combination of copyright, trade secret and trademark laws, as well as nondisclosure, confidentiality and other contractual restrictions in our consulting and employment agreements. However, these legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage.
Patents
The process of applying for patent protection itself is time consuming and expensive and we cannot assure you that all of our patent applications will issue as patents or that, if issued, they will issue in a form that will be advantageous to us. The rights granted to us under our patents, including prospective rights sought in our pending patent applications, may not be meaningful or provide us with any commercial advantage and they could be opposed, contested or circumvented by our competitors or be declared invalid or unenforceable in judicial or administrative proceedings.
We own numerous issued patents and pending patent applications that relate to the sinus delivery of sustained release therapeutics, sinus delivery of implants, implant designs, as well as individual components of our steroid releasing systems. The API contained in our steroid releasing implants is generic and is not the subject of independent patent protection. If any of our patents are challenged, invalidated or legally circumvented by third parties, and if we do not own other enforceable patents protecting our products, competitors could market products and use processes that are substantially similar to, or superior to, ours, and our business may suffer. In addition, the patents we own may not be of sufficient scope or strength to provide us with any meaningful protection or commercial advantage, and competitors may be able to design around our patents or develop products that provide outcomes comparable to ours without infringing on our intellectual property rights.
Recent patent reform legislation may increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a “first-to-invent” system to a “first-to-file” system. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The U.S. Patent
41
Table of Contents
and Trademark Office, or USPTO, recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation may increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which may have a material adverse effect on our business and financial condition. In addition, patent reform legislation may pass in the future that may lead to additional uncertainties and increased costs surrounding the prosecution, enforcement, and defense of our patents and applications.
We may be subject to a third-party pre-issuance submission of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review, or other patent office proceedings or litigation, in the United States or elsewhere, challenging our patent rights. An adverse determination in any such submission, proceeding or litigation may reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
Moreover, the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees on issued patents often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent. While an unintentional lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products or procedures, we may not be able to stop a competitor from marketing products that are the same as or similar to our products, which may have a material adverse effect on our business.
Competing products may also be sold in other countries in which our patent coverage might not exist or be as strong. We do not have patent rights in certain foreign countries in which a market may exist in the future, and the laws of many foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. Thus, we may not be able to stop a competitor from marketing and selling in foreign countries products that are the same as or similar to our products.
Trademarks
We rely on our trademarks as one means to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks. Our trademark applications may not be approved, however. Third parties may oppose our trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we may be forced to rebrand our products, which may result in loss of brand recognition and may require us to devote resources to advertising and marketing new brands. Our competitors may infringe our trademarks and we may not have adequate resources to enforce our trademarks.
Trade Secrets and Know-How
We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by consultants, vendors, former employees or current employees, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized uses and disclosures of our intellectual property is difficult, and we do not know whether the steps we have taken to protect our intellectual property will be effective.
42
Table of Contents
Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Competitors could purchase our steroid releasing implants and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If our intellectual property is not adequately protected so as to protect our market against competitors’ products and methods, our competitive position may be adversely affected, as may our business.
We may in the future be a party to patent and other intellectual property litigation and administrative proceedings that may be costly and may interfere with our ability to sell our steroid releasing implants.
The industries in which we operate in have been characterized by frequent and extensive intellectual property litigation. Additionally, the ENT market is extremely competitive. Our competitors, such as Medtronic, Olympus, Johnson & Johnson, Stryker, and Smith & Nephew, or other patent holders may assert that our steroid releasing implants and the methods employed in our steroid releasing implants are covered by their patents. If our steroid releasing implants or methods are found to infringe, we may be prevented from manufacturing or marketing our steroid releasing implants. In the event that we become involved in such a dispute, we may incur significant costs and expenses, may be prevented from marketing our products and may need to devote resources to resolving any claims, which would reduce the cash we have available for operations and may be distracting to management. If we lose a patent lawsuit, alleging our infringement of a competitor’s patents, we may be prevented from marketing our steroid releasing implants in one or more countries. We may also initiate litigation against third parties to protect our own intellectual property. Our intellectual property has not been tested in litigation. If we initiate litigation to protect our rights, we run the risk of having our patents invalidated, which may undermine our competitive position.
Litigation related to infringement and other intellectual property claims, with or without merit, is unpredictable, may be expensive and time-consuming and may divert management’s attention from our core business. If we lose this kind of litigation, a court may require us to pay substantial damages, treble damages and attorneys’ fees, and prohibit us from using technologies essential to our steroid releasing implants, any of which may have a material adverse effect on our business, results of operations and financial condition. If relevant patents are upheld as valid and enforceable and we are found to infringe, we may be prevented from selling our steroid releasing implants unless we can obtain licenses to use technology covered by such patents. We do not know whether any necessary licenses would be available to us on satisfactory terms, if at all. If we cannot obtain these licenses, we may be forced to design around those patents at additional cost or abandon our products altogether. As a result, our ability to grow our business and compete in the market may be harmed.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
Many of our employees were previously employed at other medical device companies, including our competitors or potential competitors, in some cases until recently. We may in the future be subject to claims that we or our employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of these former employers or competitors. In addition, we have been and may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation may result in substantial costs and may be a distraction to management. If our defense to those claims fails, in addition to paying monetary damages, a court may prohibit us from using technologies or features that are essential to our products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. An inability to incorporate technologies or features that are important or essential to our products may have a material adverse effect on our business, and may prevent us from selling our products. In addition, we may lose
43
Table of Contents
valuable intellectual property rights or personnel. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product may hamper or prevent our ability to commercialize our products, which may have an adverse effect on our business, results of operations and financial condition.
Risks Relating to Our Capital Requirements and Finances
We may need substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce, eliminate or abandon our commercialization efforts or product development programs.
Our ability to continue as a going concern may require us to obtain additional financing to fund our operations. We may need to raise substantial additional capital to:
| • | expand the commercialization of our products; |
| • | fund our operations and clinical studies; |
| • | continue our research and development activities; |
| • | defend, in litigation or otherwise, any claims that we infringe third-party patents or other intellectual property rights; |
| • | enforce our patent and other intellectual property rights; |
| • | address legal or enforcement actions by the FDA or other governmental agencies and remediate underlying problems; |
| • | commercialize our new products in development, if any such products receive regulatory clearance or approval for commercial sale; and |
| • | acquire companies and in-license products or intellectual property. |
We believe that our existing cash, cash equivalents and short-term investments, revenue and available debt financing arrangements will be sufficient to meet our capital requirements and fund our operations through at least twelve months after the date the financial statements are issued. However, we have based these estimates on assumptions that may prove to be wrong, and we could spend our available financial resources much faster than we currently expect. Any future funding requirements will depend on many factors, including:
| • | market acceptance of our products, including access to adequate reimbursement; |
| • | the cost of our research and development activities, including clinical studies; |
| • | the cost of filing and prosecuting patent applications and defending and enforcing our patent or other intellectual property rights; |
| • | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patents or other intellectual property rights; |
| • | the cost and timing of additional regulatory clearances or approvals; |
| • | the cost and timing of growing sales, marketing and distribution capabilities; |
| • | costs associated with any product recall that may occur; |
| • | the effect of competing technological and market developments; |
| • | the extent to which we acquire or invest in products, technologies and businesses, although we currently have no commitments or agreements relating to any of these types of transactions; and |
| • | the costs of operating as a public company. |
44
Table of Contents
If we raise additional funds by issuing equity securities, our stockholders may experience dilution. Any future debt financing into which we enter may impose upon us covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our stock, make certain investments and engage in certain merger, consolidation or asset sale transactions. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish some rights to our technologies or our products, or grant licenses on terms that are not favorable to us. If we are unable to raise adequate funds, we may have to liquidate some or all of our assets, or delay, reduce the scope of or eliminate some or all of our development programs.
We cannot be certain that additional funding will be available on acceptable terms, if at all. If we do not have, or are not able to obtain, sufficient funds, we may have to delay development or commercialization of our products or license to third parties the rights to commercialize products or technologies that we would otherwise seek to commercialize. We also may have to reduce marketing, customer support or other resources devoted to our products or cease operations. Any of these factors could harm our operating results.
Our ability to use our net operating losses and research and development credit carryforwards to offset future taxable income may be subject to certain limitations.
In general, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an “ownership change,” generally defined as a greater than 50% change by value in its equity ownership over a three-year period, is subject to limitations on its ability to utilize its pre-change net operating losses, or NOLs, and its research and development credit carryforwards to offset future taxable income. Our existing NOLs and research and development credit carryforwards may be subject to limitations arising from previous ownership changes, and if we undergo an ownership change, our ability to utilize NOLs and research and development credit carryforwards could be further limited by Sections 382 and 383 of the Code. Future changes in our stock ownership, some of which might be beyond our control, could result in an ownership change under Section 382 of the Code. For these reasons, in the event we experience a change of control, we may not be able to utilize a material portion of the NOLs and research and development credit carryforwards, even if we attain profitability.
Changes in generally accepted accounting principles may materially adversely affect our reported results of operation or financial condition.
From time to time, the Financial Accounting Standards Board, or FASB, issues new accounting principles. For example, in May 2014, the FASB issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers, with amendments in 2015 and 2016, which created a new Accounting Standards Codification Topic 606, or Topic 606, that will replace most existing revenue recognition guidance in U.S. generally accepted accounting principles, or GAAP, when it becomes effective for us on January 1, 2018. We have completed our assessment of the potential effects of the new standard and we do not believe this standard will have an effect on our financial statements, revenue recognition policies and disclosures. Under Topic 606, more judgment and estimates will be required within the revenue recognition process than were previously required under GAAP. Refer to Note 2, “Recent Accounting Pronouncements,” in the Notes to the Financial Statements for additional information about this and other recent accounting pronouncements. Changes to existing rules, or changes to the interpretations of existing rules, including, but not limited to, the changes within Topic 606, could lead to changes in our accounting policies and systems. Such changes could materially adversely affect our reported financial results and stock price.
Risks Related to Our Common Stock
We expect that the price of our common stock will fluctuate substantially.
The market price of our common stock has been, and is likely to continue to be, highly volatile. The stock market in general and the market for medical device companies in particular have experienced extreme volatility
45
Table of Contents
that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may experience losses on their investment in our common stock. The market price of our common stock may be influenced by many factors, including:
| • | volume and timing of sales of our steroid releasing implants; |
| • | changes in reimbursement or in coverage by commercial payors related to our products; |
| • | changes in governmental regulations or in the status of our regulatory approvals or applications; |
| • | the introduction of new products or product enhancements by us or others in our industry; |
| • | disputes or other developments with respect to our or others’ intellectual property rights; |
| • | our ability to develop, obtain regulatory clearance or approval for, and market new and enhanced products on a timely basis; |
| • | product liability claims or other litigation; |
| • | quarterly variations in our results of operations or those of others in our industry; |
| • | sales of large blocks of our common stock, including sales by our executive officers and directors; |
| • | media exposure of our steroid releasing implants or products of others in our industry; |
| • | changes in earnings estimates or recommendations by securities analysts; and |
| • | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors. |
In recent years, the stock markets generally have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may significantly affect the market price of our common stock, regardless of our actual operating performance. These fluctuations may be even more pronounced in the trading market for our common stock.
In addition, in the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Securities litigation brought against us following volatility in our stock price, regardless of the merit or ultimate results of such litigation, could result in substantial costs, which would hurt our financial condition and operating results and divert management’s attention and resources from our business.
These and other factors may make the price of our stock volatile and subject to unexpected fluctuation.
Securities analysts may not publish favorable research or reports about our business or may publish no information at all, which could cause our stock price or trading volume to decline.
The trading market for our common stock will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. As a newly public company, we may be slow to attract research coverage and the analysts who publish information about our common stock will have had relatively little experience with our company or industry, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of our company or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline.
46
Table of Contents
If we experience material weaknesses or otherwise fail to maintain an effective system of internal controls, we may not be able to accurately report our financial condition or results of operations which may adversely affect investor confidence in us and, as a result, the value of our common stock.
We are required, under Section 404 of the Sarbanes-Oxley Act to furnish a report by management on the effectiveness of our internal control over financial reporting, and our auditors are required to express an opinion on the effectiveness of our internal controls. This resulted in increased compliance fees. Our management assessment needs to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a company’s annual and interim financial statements will not be detected or prevented on a timely basis.
Though we have enhanced our internal controls, processes and related documentation necessary to perform the evaluation needed to comply with Section 404, future evaluations and tests may reveal material weaknesses. If during the evaluation and testing process, we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are effective. The effectiveness of our controls and procedures may be limited by a variety of factors, including:
| • | faulty human judgment and simple errors, omissions or mistakes; |
| • | fraudulent action of an individual or collusion of two or more people; |
| • | inappropriate management override of procedures; and |
| • | the possibility that any enhancements to controls and procedures may still not be adequate to assure timely and accurate financial control. |
If we are unable to confirm that our internal control over financial reporting is effective, or if our auditors are unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause the price of our common stock to decline.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act. We designed our disclosure controls and procedures to provide reasonable assurance that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their
47
Table of Contents
shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. Among others, these provisions include that:
| • | our board of directors has the right to expand the size of our board of directors and to elect directors to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
| • | our stockholders may not act by written consent or call special stockholders’ meetings; as a result, a holder, or holders, controlling a majority of our capital stock would not be able to take certain actions other than at annual stockholders’ meetings or special stockholders’ meetings called by the board of directors, the chairman of the board, the chief executive officer or the president; |
| • | our certificate of incorporation prohibits cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
| • | the affirmative vote of holders of at least 66-2/3% of the voting power of all of the then outstanding shares of voting stock, voting as a single class, will be required (a) to amend certain provisions of our certificate of incorporation, including provisions relating to the size of the board, removal of directors, special meetings, actions by written consent and cumulative voting and (b) to amend or repeal our bylaws, although our bylaws may be amended by a simple majority vote of our board of directors; |
| • | stockholders must provide advance notice and additional disclosures in order to nominate individuals for election to the board of directors or to propose matters that can be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquiror from conducting a solicitation of proxies to elect the acquiror’s own slate of directors or otherwise attempting to obtain control of our company; and |
| • | our board of directors may issue, without stockholder approval, shares of undesignated preferred stock; the ability to issue undesignated preferred stock makes it possible for our board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to acquire us. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We occupy approximately 50,400 square feet of leased office and laboratory space located in Menlo Park, California which expires on May 31, 2020. We believe that our facilities are sufficient to meet our current needs.
| Item 3. | Legal Proceedings |
We are not currently a party to any material legal proceedings.
48
Table of Contents
| Item 4. | Mine Safety Disclosures |
Not applicable.
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Shares of our common stock are traded on The NASDAQ Global Market, or NASDAQ, under the symbol XENT. The following table sets forth the high and low intraday prices per share of our common stock as reported by NASDAQ.
| High | Low | |||||||
| Fiscal Year Ended December 31, 2017 |
||||||||
| First quarter |
$ | 17.70 | $ | 11.55 | ||||
| Second quarter |
28.23 | 15.90 | ||||||
| Third quarter |
33.25 | 26.90 | ||||||
| Fourth quarter |
34.40 | 26.95 | ||||||
| Fiscal Year Ended December 31, 2016 |
||||||||
| First quarter |
$ | 22.01 | $ | 15.60 | ||||
| Second quarter |
20.64 | 11.88 | ||||||
| Third quarter |
17.00 | 12.50 | ||||||
| Fourth quarter |
18.00 | 7.65 | ||||||
As of January 31, 2018, the closing price of our common stock was $37.35 and there were approximately 20 stockholders of record. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock, and we do not currently intend to pay any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to support operations and to finance the growth and development of our business. Any future determination to pay dividends will be made at the discretion of our board of directors subject to applicable laws, and will depend upon, among other factors, our results of operations, financial condition, contractual restrictions and capital requirements. Our future ability to pay cash dividends on our capital stock may also be limited by the terms of any future debt or preferred securities or future credit facility.
Recent Sales of Unregistered Securities
There were no sales of equity securities by us that were not registered under the Securities Act of 1933, as amended, or the Securities Act, during fiscal year ended December 31, 2017, that have not been previously reported in a Quarterly Report on Form 10-Q or in a Current Report on Form 8-K.
Issuer Purchases of Equity Securities
None.
Performance Graph
The graph below compares the cumulative total return to security holders of our common stock with the comparable cumulative returns of the NASDAQ Composite and Biotechnology Indexes. The graph assumes the investment of $100 on July 24, 2014, the date on which our common stock began trading on The NASDAQ Global Market, through December 31, 2017. Points on the graph represent the performance at year-end.
49
Table of Contents
The information under the heading “Performance Graph” shall not be deemed filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended.
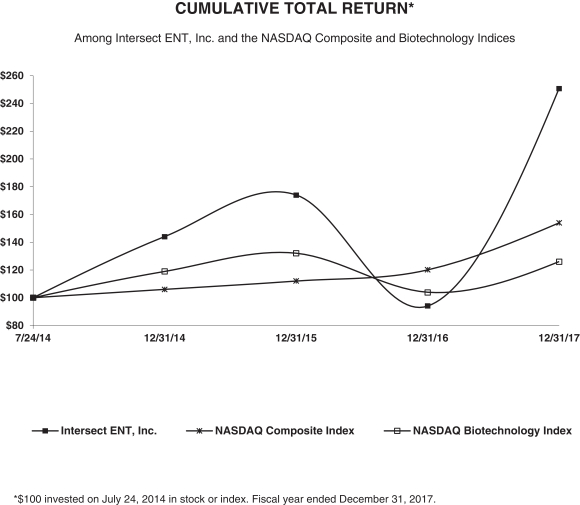
| Cumulative Total Return as of | ||||||||||||||||||||
| 7/24/14 | 12/31/14 | 12/31/15 | 12/31/16 | 12/31/17 | ||||||||||||||||
| Intersect ENT, Inc. |
$ | 100 | $ | 144 | $ | 174 | $ | 94 | $ | 251 | ||||||||||
| NASDAQ Composite Index |
100 | 106 | 112 | 120 | 154 | |||||||||||||||
| NASDAQ Biotechnology Index |
100 | 119 | 132 | 104 | 126 | |||||||||||||||
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
Since shares of our common stock have only been publicly traded since July 24, 2014, information surrounding stockholder returns in comparison to the NASDAQ Composite and Biotechnology Indices may not be meaningful to investors.
The material in this section is not “soliciting material” and is not deemed “filed” with the SEC and is not to be incorporated by reference into any filing of Intersect ENT, Inc. made under the Securities Act or the Exchange
50
Table of Contents
Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing except to the extent we specifically incorporate this section by reference.
| Item 6. | Selected Financial Data |
The following selected financial information has been derived from our audited financial statements. The financial information as of December 31, 2017 and 2016, and for each of the three years in the period ended December 31, 2017, is derived from audited financial statements included elsewhere in this Annual Report on Form 10-K. The financial information as of December 31, 2015, 2014 and 2013, and for each of the two years in the period ended December 31, 2014, is derived from audited financial statements not included in this Annual Report on Form 10-K. The information set forth below is not necessarily indicative of results of future operations and should not be relied upon as an indicator of our future performance. You should read the selected financial data set forth in the table below, together with the Financial Statements and related Notes to Financial Statements included in this Annual Report, as well as Part II, Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations, of this Annual Report.
| Fiscal Years Ended December 31, |
||||||||||||||||||||
| (in thousands, except per share data) |
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
| Statements of operations data: |
||||||||||||||||||||
| Revenue |
$ | 96,301 | $ | 78,708 | $ | 61,593 | $ | 38,587 | $ | 17,931 | ||||||||||
| Cost of sales |
15,499 | 13,003 | 12,288 | 10,223 | 8,150 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Gross profit |
80,802 | 65,705 | 49,305 | 28,364 | 9,781 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Selling, general and administrative |
80,045 | 72,926 | 59,637 | 36,111 | 18,229 | |||||||||||||||
| Research and development |
18,360 | 18,890 | 16,608 | 10,331 | 9,518 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
98,405 | 91,816 | 76,245 | 46,442 | 27,747 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(17,603 | ) | (26,111 | ) | (26,940 | ) | (18,078 | ) | (17,966 | ) | ||||||||||
| Interest and other income (expense), net |
1,240 | 889 | 306 | (284 | ) | (403 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (16,363 | ) | $ | (25,222 | ) | $ | (26,634 | ) | $ | (18,362 | ) | $ | (18,369 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss per share, basic and diluted |
$ | (0.56 | ) | $ | (0.89 | ) | $ | (1.02 | ) | $ | (1.61 | ) | $ | (12.57 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average common shares used to compute net loss per share, basic and diluted |
29,119 | 28,420 | 26,159 | 11,384 | 1,461 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| December 31, | ||||||||||||||||||||
| (in thousands) |
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
| Balance sheet data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 102,320 | $ | 103,945 | $ | 124,300 | $ | 48,443 | $ | 12,294 | ||||||||||
| Working capital |
112,614 | 110,928 | 128,142 | 52,167 | 13,118 | |||||||||||||||
| Total assets |
135,575 | 129,777 | 144,635 | 62,953 | 21,035 | |||||||||||||||
| Total liabilities |
18,356 | 15,380 | 14,404 | 9,141 | 6,892 | |||||||||||||||
| Convertible preferred stock |
— | — | — | — | 90,760 | |||||||||||||||
| Accumulated deficit |
(164,840 | ) | (148,477 | ) | (123,255 | ) | (96,621 | ) | (78,259 | ) | ||||||||||
| Total stockholders’ equity (deficit) |
117,219 | 114,397 | 130,231 | 53,812 | (76,617 | ) | ||||||||||||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis of our financial condition and results of operations together with the section entitled “Selected Financial Data,” should be read in conjunction with our Financial Statements and the related notes to those statements included elsewhere in this Annual Report. In addition to historical financial information, the following discussion and analysis contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. These forward-looking statements relate to future events or our future financial performance that involve risks, uncertainties and
51
Table of Contents
assumptions. Our actual results and timing of events may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those discussed under “Risk Factors” and elsewhere in this prospectus. Please see “Cautionary Information Regarding Forward-Looking Statements” at the beginning of this Form 10-K for additional information you should consider regarding forward-looking statements. We undertake no obligation to revise or update any forward-looking statements to reflect any event or circumstance that arises after the date of this report, or to conform such statements to actual results or changes in our expectations.
Overview
We are a commercial drug delivery company committed to improving the quality of life for patients with ear, nose and throat conditions. Our approved products are steroid releasing implants designed to treat the spectrum of needs among patients who are managed by ENT physicians for chronic sinusitis, one of the most prevalent chronic diseases in the United States and one of the most costly conditions for U.S. employers. We are currently marketing our PROPEL® family of products, consisting of PROPEL®, PROPEL® Mini and PROPEL® Contour, which are used following sinus surgery to deliver steroid locally to treat inflammation and improve surgical outcomes. In addition, we received FDA approval in December 2017 for our SINUVA™ Sinus Implant, a new targeted approach to treating nasal polyp disease in patients who have had previous ethmoid sinus surgery.
Our PROPEL family of steroid releasing implants are clinically proven to improve outcomes for chronic sinusitis patients following sinus surgery. PROPEL implants mechanically prop open the sinuses and release mometasone furoate, an advanced corticosteroid with anti-inflammatory properties, directly into the sinus lining, and then dissolve. PROPEL’s safety and effectiveness is supported by Level 1-A clinical evidence from multiple clinical trials, which demonstrates that PROPEL implants reduce inflammation and scarring after surgery, thereby reducing the need for postoperative oral steroids and repeat surgical interventions. More than 200,000 patients have been treated with PROPEL products to-date. The PROPEL family of products are used today predominantly in hospitals and ambulatory surgical settings, although they may also be used in the physician office setting of care.
| • | PROPEL has been proven in a meta-analysis of prospective, multicenter, randomized, controlled, double-blind clinical studies to improve surgical outcomes, demonstrating a 35% relative reduction in the need for postoperative oral steroid and surgical intervention compared to surgery alone. A physician may treat a patient with PROPEL by inserting it into the ethmoid sinuses. PROPEL is a self-expanding implant designed to conform to and hold open the surgically enlarged sinus while gradually releasing an anti-inflammatory steroid over a period of approximately 30 days and is absorbed into the body in approximately six weeks. |
| • | PROPEL Mini has also been shown by our clinical studies to reduce the need for postoperative interventions, including a 38% relative reduction in the need for postoperative interventions in the frontal sinus, compared to surgery alone with standard postoperative care. PROPEL Mini is a smaller version of PROPEL, and is approved for use both in the ethmoid and frontal sinuses. PROPEL Mini is preferentially used by physicians compared with PROPEL when treating smaller anatomies or following less extensive procedures. |
| • | PROPEL Contour is designed to facilitate treatment of the frontal and maxillary sinus ostia, or openings, of the dependent sinuses in procedures performed in both the operating room and in the office setting of care. PROPEL Contour’s lower profile, hourglass shape and malleable delivery system are designed for use in the narrow and difficult to access sinus ostia. In PROPEL Contour’s pivotal clinical study, the product demonstrated a 65% relative reduction in the need for postoperative interventions in the frontal sinus ostia compared to surgery alone with standard postoperative care. |
SINUVA, when placed during a routine physician office visit, expands into the sinus cavity and delivers an anti-inflammatory steroid directly to the site of polyp disease for 90 days. We have studied SINUVA in 4 clinical
52
Table of Contents
trials in over 400 patients to-date. Results from the pivotal RESOLVE II randomized clinical trial demonstrated a 74% relative reduction in bilateral polyp grade (a measurement of the extent of ethmoid polyp disease) and a 30% relative reduction in nasal obstruction and congestion for patients treated with SINUVA compared to a control group treated with a sham procedure, receiving no implant. Patients in both arms of the study were required to use intranasal steroid sprays daily. In addition, the study demonstrated a 61% reduction in the proportion of patients indicated for revision surgery at day 90. To complement clinical trials performed with SINUVA to-date, in which one course of SINUVA treatment was evaluated, we commenced the ENCORE study in November 2017. ENCORE is a 50-patient prospective, multicenter, open-label study focused on evaluation of the safety of repeat placement of the SINUVA implant in chronic sinusitis patients with nasal polyps. We completed enrollment of this study in January 2018.
We have expanded our sales organization and we intend to continue to grow our sales force in order to expand our communication of the benefits of our steroid releasing implants to our physician customers. We seek to grow our revenue by increasing the frequency of use of our products among current physician customers and by adding new physician users.
Components of Our Results of Operations
Revenue
Our revenue has been derived solely from the sales of our PROPEL family of products. We expect our revenue to increase as we continue to expand our sales, marketing and reimbursement efforts and increase awareness of our products. We also expect revenue from our PROPEL family of products to fluctuate from quarter to quarter due to seasonal variations in the volume of sinus surgery procedures performed, which has been impacted historically by factors including the status of patient healthcare insurance plan deductibles and the seasonal nature of allergies which can impact sinus-related symptoms.
Our PROPEL family of products are used almost exclusively in the operating room of a hospital or ambulatory surgery center. These providers receive a facility fee for the sinus surgery procedure which is intended to pay for supplies used in this procedure, including the PROPEL family of products. In the event these procedures are performed in the physician office setting of care, our PROPEL family of products have been assigned a code under the Healthcare Common Procedure Coding System, S1090, which may be used to submit requests for product cost reimbursement to commercial payors. In addition, several existing procedure codes may apply to describe the procedure associated with implant placement. However, the decision to reimburse by the payor is usually dependent on policies the payor has in place regarding these products.
Our revenue is almost entirely derived from within the U.S. and no single customer accounted for more than 10% of our revenue during the years ended December 31, 2017, 2016 and 2015.
Cost of Sales and Gross Profit
We manufacture our PROPEL family of products and SINUVA in our facility in Menlo Park, California. Cost of sales consists primarily of manufacturing overhead costs, material costs, direct labor and other direct costs such as shipping costs. A significant portion of our cost of sales currently consists of manufacturing overhead costs. These overhead costs include the cost of quality assurance, material procurement, inventory control, facilities, information technology, equipment and operations supervision and management. We expect overhead costs as a percentage of revenue to become less significant as our production volume increases. We expect cost of sales to increase in absolute dollars primarily as, and to the extent, our revenue grows.
We calculate gross margin as gross profit divided by revenue. Our gross margin has been and will continue to be affected by a variety of factors, including production volumes, average selling prices, manufacturing costs and product yields and, to a lesser extent, the implementation of cost-reduction strategies. We expect our gross
53
Table of Contents
margin to fluctuate based on changes in the average selling price and the manufacturing costs of our products. Manufacturing cost will change as our production volume changes. The per unit allocation of our manufacturing overhead costs may decrease as production volume increases until we increase our manufacturing capacity or introduce additional products, at which point the per unit allocation of our manufacturing overhead costs may increase due to the additional costs of our expanded manufacturing operations.
Selling, General and Administrative Expenses
Selling, general and administrative, or SG&A, expenses consist primarily of compensation for personnel, including stock-based compensation, related to selling, marketing, finance, reimbursement, business development, legal and human resource functions as well as costs related to any post-market studies. Additional SG&A expenses include commissions, training, travel expenses, promotional activities, conferences, trade shows, professional services fees, audit and Sarbanes-Oxley Act of 2002 compliance expenses, insurance costs and general corporate expenses including allocated facilities and information technology expenses. We expect SG&A expenses to continue to increase in absolute dollars for the foreseeable future as we expand our commercial and administrative infrastructure to drive and support the anticipated growth in revenue and incur additional legal, accounting, insurance and other professional services fees.
Research and Development Expenses
Research and development, or R&D, expenses consist primarily of compensation for personnel, including stock-based compensation, related to product development, clinical and medical affairs and regulatory affairs as well, and allocated facilities and information technology expenses. R&D expenses also may include expenses for clinical studies related to clinical trial design, site reimbursement, data management, travel expenses and the cost of manufacturing products for clinical trials. Finally, R&D expenses also include expenses related to the development of products and technologies such as consulting services and supplies. We expect R&D expenses to remain at a consistent level in absolute dollars for the foreseeable future as we continue to seek to develop and commercialize new products and enhance our current products.
Results of Operations
| Fiscal Years Ended December 31, |
||||||||||||
| (in thousands, except percentages) |
2017 | 2016 | 2015 | |||||||||
| Revenue |
$ | 96,301 | $ | 78,708 | $ | 61,593 | ||||||
| Cost of sales |
15,499 | 13,003 | 12,288 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
80,802 | 65,705 | 49,305 | |||||||||
| Gross margin |
84 | % | 83 | % | 80 | % | ||||||
| Operating expenses: |
||||||||||||
| Selling, general and administrative |
80,045 | 72,926 | 59,637 | |||||||||
| Research and development |
18,360 | 18,890 | 16,608 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
98,405 | 91,816 | 76,245 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(17,603 | ) | (26,111 | ) | (26,940 | ) | ||||||
| Interest income and other, net |
1,240 | 889 | 306 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (16,363 | ) | $ | (25,222 | ) | $ | (26,634 | ) | |||
|
|
|
|
|
|
|
|||||||
Comparison of Years Ended December 31, 2017 and 2016
Revenue
Revenue increased $17.6 million, or 22%, to $96.3 million during the year ended December 31, 2017, compared to $78.7 million during the year ended December 31, 2016. The growth in revenue was attributable to
54
Table of Contents
an increase in unit sales of our PROPEL family of products, driven by sales of PROPEL Contour which was approved by the FDA in February 2017, and to a lesser degree, an increase in the average selling price of our PROPEL family of products.
Cost of Sales and Gross Margin
Cost of sales increased $2.5 million, or 19%, to $15.5 million during the year ended December 31, 2017, compared to $13.0 million during the year ended December 31, 2016. The increase in cost of sales was primarily attributable to the growth in the number of units sold.
Gross margin for the year ended December 31, 2017, increased to 84%, compared to 83% for the year December 31, 2016. The increase in gross margin was primarily due to an increase in our average selling price of our PROPEL family of products.
Selling, General and Administrative Expenses
SG&A expenses increased $7.1 million, or 10%, to $80.0 million during the year ended December 31, 2017, compared to $72.9 million during the year ended December 31, 2016. The increase in SG&A expenses was primarily due to an increase in headcount to support the ongoing commercialization of our PROPEL family of products and to prepare for the commercial launch of SINUVA.
Research and Development Expenses
R&D expenses decreased $0.5 million, or 3%, to $18.4 million during the year ended December 31, 2017, compared to $18.9 million during the year ended December 31, 2016. The decrease in R&D expenses was due to a decrease in clinical trial costs, partially offset by an increase in personnel costs.
Interest Income and Other, Net
Interest income and other, net, increased $0.3 million to $1.2 million of income during the year ended December 31, 2017, compared to $0.9 million of income during the year ended December 31, 2016. The increase in interest income and other, net, was primarily attributable to higher interest rates earned on our investments.
Comparison of Years Ended December 31, 2016 and 2015
Revenue
Revenue increased $17.1 million, or 28%, to $78.7 million during the year ended December 31, 2016, compared to $61.6 million during the year ended December 31, 2015. The growth in revenue was attributable to an increase in unit sales of PROPEL and PROPEL Mini. The increase in units was driven by an expansion of our sales, marketing and reimbursement organizations as well as by the introduction of an expanded indication for PROPEL Mini following approval from the FDA to treat patients undergoing frontal sinus surgery. In addition, our average selling price increased during the year ended December 31, 2016.
Cost of Sales and Gross Margin
Cost of sales increased $0.7 million, or 6%, to $13.0 million during the year ended December 31, 2016, compared to $12.3 million during the year ended December 31, 2015. The increase in cost of sales was attributable to the growth in the number of PROPEL and PROPEL Mini units sold, partially offset by the reduced per unit overhead costs and increases in manufacturing efficiency.
Gross margin for the year ended December 31, 2016, increased to 83%, compared to 80% for the year December 31, 2015. The increase in gross margin was due to several factors including an increase in our average selling price, growth in unit volume during the year ended December 31, 2016, which allowed us to spread our manufacturing overhead costs over more production units, and ongoing increases in manufacturing efficiency.
55
Table of Contents
Selling, General and Administrative Expenses
SG&A expenses increased $13.3 million, or 22%, to $72.9 million during the year ended December 31, 2016, compared to $59.6 million during the year ended December 31, 2015. The increase in SG&A expenses was primarily due to the build out of our infrastructure to support the ongoing commercialization of PROPEL and PROPEL Mini.
The primary component of this increase was employee-related expenses of our sales, marketing and reimbursement organizations, which increased $10.1 million for the year ended December 31, 2016, compared to the year ended December 31, 2015, as we increased headcount to 145 as of December 31, 2016, compared to 129 as of December 31, 2015. In addition, other SG&A expenses increased $3.2 million for the year ended December 31, 2016, compared to the year ended December 31, 2015, primarily due to an increase in headcount.
Research and Development Expenses
R&D expenses increased $2.3 million, or 14%, to $18.9 million during the year ended December 31, 2016, compared to $16.6 million during the year ended December 31, 2015. The increase in R&D expenses was primarily due to increases in personnel costs as we increased headcount.
Interest Income and Other, Net
Interest income and other, net, increased $0.6 million to $0.9 million during the year ended December 31, 2016, compared to $0.3 million during the year ended December 31, 2015. The increase in interest income and other, net, was primarily attributable to higher interest rates and higher average cash, cash equivalents and short-term investments available-for-sale balances during the year ended December 31, 2016 from our follow-on offering.
Liquidity and Capital Resources
Overview
As of December 31, 2017, we had cash, cash equivalents and short-term investments of $102.3 million, compared to cash, cash equivalents and short-term investments of $103.9 million as of December 31, 2016.
Cash Flows
| Fiscal Years Ended December 31, |
||||||||||||
| (in thousands) |
2017 | 2016 | 2015 | |||||||||
| Net cash (used in) provided by: |
||||||||||||
| Operating activities |
$ | (8,041 | ) | $ | (20,059 | ) | $ | (20,087 | ) | |||
| Investing activities |
9,248 | (6,857 | ) | (56,669 | ) | |||||||
| Financing activities |
8,771 | 1,966 | 98,162 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
$ | 9,978 | $ | (24,950 | ) | $ | 21,406 | |||||
|
|
|
|
|
|
|
|||||||
Net Cash Used in Operating Activities
During the year ended December 31, 2017, net cash used in operating activities was $8.0 million, consisting primarily of a net loss of $16.4 million and an increase in net operating assets of $2.9 million, partially offset by non-cash charges of $11.3 million. The cash used in operations was due primarily to an increase in headcount to support the ongoing commercialization of our PROPEL family of products and to prepare for the launch of SINUVA. The non-cash charges consisted primarily of stock-based compensation expense. The increase in net operating assets is due primarily to an increase in inventory, accounts receivable and other assets, partially offset by an increase in accrued year-end bonuses and sales commissions.
56
Table of Contents
During the year ended December 31, 2016, net cash used in operating activities was $20.1 million, consisting primarily of a net loss of $25.2 million and an increase in net operating assets of $3.6 million, partially offset by non-cash charges of $8.7 million. The cash used in operations was due primarily to the ongoing commercialization of PROPEL and PROPEL Mini. To support the ongoing commercialization of these products, we continued to expand our sales, marketing and reimbursement organizations. The increase in net operating assets was due primarily to an increase in accounts receivable and inventory, partially offset by an increase in accounts payable. The non-cash charges consisted primarily of stock-based compensation expense.
During the year ended December 31, 2015, net cash used in operating activities was $20.1 million, consisting primarily of a net loss of $26.6 million, partially offset by non-cash charges of $6.4 million and a decrease in net operating assets of $0.1 million. The cash used in operations was due primarily to the ongoing commercialization of PROPEL and PROPEL Mini. To support the ongoing commercialization of these products, we continued to expand our sales, marketing and reimbursement organizations. The non-cash charges consisted primarily of stock-based compensation expense. The decrease in net operating assets was due primarily to an increase in accrued compensation, partially offset by an increase in accounts receivable and inventory.
Net Cash Provided by (Used in) Investing Activities
During the year ended December 31, 2017, net cash provided by investing activities was $9.2 million, consisting primarily of net maturities of short-term investments of $11.5 million, partially offset by purchases of property and equipment of $2.3 million.
During the year ended December 31, 2016, net cash used in investing activities was $6.9 million, consisting of net purchases of short-term investments and purchases of property and equipment.
During the year ended December 31, 2015, net cash used in investing activities was $56.7 million, consisting primarily of net purchases of short-term investments of $55.1 million.
Net Cash Provided by Financing Activities
During the year ended December 31, 2017, net cash provided by financing activities was $8.8 million, consisting of net proceeds from the issuance of common stock upon exercises of employee stock options and purchases under our employee stock purchase plan.
During the year ended December 31, 2016, net cash provided by financing activities was $2.0 million, consisting of net proceeds from the issuance of common stock upon the exercises of employee stock options and purchases under our employee stock purchase plan.
During the year ended December 31, 2015, net cash provided by financing activities was $98.2 million, consisting primarily of net proceeds from our follow-on offering of $96.4 million.
Liquidity
We currently believe that our existing cash, cash equivalents and short-term investments as of December 31, 2017, will be sufficient to meet our capital requirements and fund our operations through at least twelve months after the date the financial statements are issued. Beyond that, if these sources are insufficient to satisfy our liquidity requirements, we may seek to sell additional equity or debt securities or obtain credit facilities. If we raise additional funds by issuing equity securities, our stockholders would experience dilution. Debt financing, if available, may involve covenants restricting our operations or our ability to incur additional debt. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. Additional financing may not be available at all, or in amounts or on terms unacceptable to us. If we are unable to obtain additional financing, we may be required to delay the development, commercialization and marketing of our products.
57
Table of Contents
Off-Balance Sheet Arrangements
As of December 31, 2017 and 2016, we were not a party to any off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our financial condition, changes in financial condition, revenue or expenses, results of operations, liquidity, capital expenditures or capital resources.
Contractual Obligations
The following table sets out our contractual obligations due by period as of December 31, 2017.
| Due by Period | ||||||||||||||||||||
| Less Than 1 Year |
1-3 Years |
3-5 Years |
More Than 5 Years |
Total | ||||||||||||||||
| Operating lease obligations |
$ | 1,580 | $ | 2,310 | $ | — | $ | — | $ | 3,890 | ||||||||||
| Purchase commitments |
4,773 | 628 | — | — | 5,401 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| $ | 6,353 | $ | 2,938 | $ | — | $ | — | $ | 9,291 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
Related Parties
None.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note 2 of our Financial Statements included in this Annual Report, we believe the following discussion addresses our most critical accounting policies, which are those that are most important to the portrayal of our financial condition and results of operations and require our most difficult, subjective and complex judgments.
Revenue Recognition
We derive revenue from the sale of PROPEL family of products to hospitals and ambulatory surgery centers almost entirely in the United States. We recognize revenue when persuasive evidence of an arrangement exists, product delivery has occurred or there is no further obligation, pricing is fixed or determinable and collection is reasonably assured. We must make assumptions regarding the future collectability of amounts receivable from customers to determine whether revenue recognition criteria have been met. If collectability is not reasonably assured at the time of shipment, we defer revenue until such criteria have been met. In general, our standard terms and conditions of sale do not allow for product returns. We expense shipping and handling costs as incurred and include them in the cost of sales. In those cases where we bill shipping and handling costs to customers, we classify the amounts billed as a component of revenue.
Stock-based Compensation
We maintain an equity incentive plan to provide long-term incentive for employees, consultants and members of the board of directors. The plan allows for the issuance of non-statutory and incentive stock options to employees and non-statutory stock options to consultants and non-employee directors.
58
Table of Contents
We are required to determine the fair value of equity incentive awards and recognize compensation expense for all equity incentive awards made to employees, consultants and directors. Stock-based compensation expense is recognized over the requisite service period in the statements of operations and comprehensive loss. We use the straight-line method for expense attribution. Effective January 1, 2017, we adopted Accounting Standards Update (“ASU”) No. 2016-9, Improvements to Employee Share-Based Payment Accounting (“ASU 2016-9”), and elected to account for forfeitures when they occurred. Prior to the adoption of ASU 2016-9, we estimated the awards ultimately expected to vest based on our historical forfeiture experience and an analysis of similar companies, therefore reducing the amount of stock-based compensation expense for estimated forfeitures.
The valuation model we use for calculating the fair value of awards for stock-based compensation expense is the Black-Scholes option-pricing model, or the Black-Scholes model. The Black-Scholes model requires us to make assumptions and judgments about the variables used in the calculation, including the expected term weighted average period of time that the options granted are expected to be outstanding, the volatility of common stock and an assumed risk-free interest rate. The fair market value of our common stock is determined based on the closing price of our common stock on The NASDAQ Global Market.
Recent Accounting Pronouncements
Please see Note 2 to the Financial Statements included in this Annual Report.
| Item 7A. | Quantitative and Qualitative Disclosure About Market Risk |
Interest Rate Risk
The risk associated with fluctuating interest rates is primarily limited to our cash equivalents and short-term investments which are carried at fair market value. We do not currently use or plan to use financial derivatives in our investment portfolio.
As of December 31, 2017 and 2016, we had cash, cash equivalents and short-term investments of $102.3 million and $103.9 million, respectively. Cash equivalents and short-term investments are composed of money market funds, corporate debt securities and commercial paper. Our investment policy requires investments to be of high credit quality and generally limits the amount of credit exposure to any single issuer or group of issuers. Our objective is the preservation of capital and to maintain proper liquidity to meet our operating requirements while at the same time maximizing the income we receive from our financial instruments without significantly increasing risk. Because our short-term investments have a weighted average maturity of not more than one year, we believe the impact of a hypothetical 10% change in market interest rates at December 31, 2017 and 2016 would not have a material effect on our financial position, results of operations or cash flows.
Credit Risk
As of December 31, 2017 and 2016, our cash, cash equivalents and short-term investments were maintained with two financial institutions in the United States, and our current deposits are likely in excess of insured limits. We have reviewed the financial statements of these institutions and believe they have sufficient assets and liquidity to conduct their operations in the ordinary course of business with little or no credit risk to us.
Our accounts receivable relate to revenue from the sale of our PROPEL family of products to hospitals and ambulatory surgery centers almost entirely in the United States. No single customer represented more than 10% of our accounts receivable as of December 31, 2017 and 2016.
Foreign Currency Risk
Our business is almost entirely conducted in U.S. dollars. Transactions conducted in foreign currencies have not had, and are not expected to have, a material effect on our results of operations, financial position or cash flows.
59
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
Please see the Financial Statements included in this Annual Report on Form 10-K, beginning on Page F-l following the signature page to this Form 10-K, which are incorporated by reference here.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Management, with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the consolidated financial statements.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in “Internal Control — Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO. Our management concluded that our internal control over financial reporting was effective as of December 31, 2017.
Our independent registered public accounting firm, Ernst & Young LLP, has audited the effectiveness of our internal control over financial reporting as of December 31, 2017 as stated in their report which is included herein.
60
Table of Contents
Limitations on Effectiveness of Controls and Procedures and Internal Control over Financial Reporting
In designing and evaluating the disclosure controls and procedures and internal control over financial reporting, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures and internal control over financial reporting must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the three months ended December 31, 2017, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
61
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
Intersect ENT, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Intersect ENT, Inc.’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Intersect ENT, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the balance sheets of the Company as of December 31, 2017 and 2016, the related statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and financial statement schedule listed in the Index at Item 15(a), and our report dated February 28, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that
62
Table of Contents
Table of Contents
PART III
Certain information required by Part III is omitted from this Annual Report and is incorporated herein by reference from our Definitive Proxy Statement, relating to our 2018 Annual Meeting of Stockholders to be held on June 5, 2018, pursuant to Regulation 14A of the Exchange Act, or Proxy Statement, which will be filed with the SEC within 120 days of December 31, 2017.
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by this Item concerning our directors and executive officers is incorporated by reference to the sections of our Proxy Statement under the headings “Proposal 1 — Election of Directors,” “Board Committees and Meetings,” “Stockholder Communications with the Board of Directors,” “Management” and “Section 16(a) Beneficial Ownership Reporting Compliance.”
Our written Code of Ethics applies to all of our directors and employees, including our executive officers, including without limitation our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions. The Code of Ethics is available on our website at www.intersectent.com in the Investors section under “Corporate Governance.” Changes to or waivers of the Code of Ethics will be disclosed on the same website. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding any amendment to, or waiver of, any provision of the Code of Ethics by disclosing such information on the same website.
| Item 11. | Executive Compensation |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Compensation Discussion and Analysis,” “Executive Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Compensation of Non-Employee Board Members.”
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Security Ownership of Certain Beneficial Owners and Management” and “Securities Authorized for Issuance under Equity Compensation Plans.”
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Proposal 1 — Election of Directors” and “Certain Relationships and Related Party Transactions.”
| Item 14. | Principal Accountant Fees and Services |
The information required by this Item is incorporated by reference to the section of the Proxy Statement under the heading “Principal Accountant Fees and Services.”
With the exception of the information specifically incorporated by reference in Part III to this Annual Report from our Proxy Statement, our Proxy Statement shall not be deemed to be filed as part of this report.
64
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a) The following documents are filed as part of this Annual Report on Form 10-K:
(1) Financial Statements
The Financial Statements of the Company are included herein as required under Part II, Item 8, Financial Statements and Supplementary Data, of this Annual Report. See Index to Financial Statements on page F-l.
(2) Financial Statement Schedule
For the three fiscal years ended December 31, 2017 — Schedule II Valuation and Qualifying Accounts
Schedules not listed above have been omitted because information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
(3) Exhibits (numbered in accordance with Item 601 of Regulation S-K)
See Part IV, Item 15(b) below.
(b) The following exhibits are filed or incorporated by reference into this Annual Report:
65
Table of Contents
66
Table of Contents
67
Table of Contents
| Incorporation By Reference |
||||||||||||||||||
|
Exhibit |
Description |
Form |
SEC File No. |
Exhibit |
Filing Date |
|||||||||||||
| 32.1* | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |||||||||||||||||
| 101.INS | XBRL Instance Document. | |||||||||||||||||
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |||||||||||||||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | |||||||||||||||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. | |||||||||||||||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document. | |||||||||||||||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | |||||||||||||||||
| ** | Management compensatory contract or arrangement. |
| # | Confidential Treatment Granted. |
| * | Exhibit 32.1 is being furnished and shall not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liability of that section, nor shall such exhibit be deemed to be incorporated by reference in any registration statement or other document filed under the Securities Act of 1933, as amended, or the Exchange Act, except as otherwise specifically stated in such filing. |
| Item 16. | Form 10-K Summary |
None.
68
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Intersect ENT, Inc. | ||||||
| Date: February 28, 2018 | By: | /S/ LISA D. EARNHARDT | ||||
| Lisa D. Earnhardt President and Chief Executive Officer | ||||||
| Date: February 28, 2018 | By: | /S/ JERYL L. HILLEMAN | ||||
| Jeryl L. Hilleman Chief Financial Officer | ||||||
69
Table of Contents
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Lisa D. Earnhardt and Jeryl L. Hilleman, jointly and severally, his or her attorneys-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name |
Title |
Date | ||
| /S/ LISA D. EARNHARDT Lisa D. Earnhardt |
President and Chief Executive Officer (Principal Executive Officer) and Director |
February 28, 2018 | ||
| /S/ JERYL L. HILLEMAN Jeryl L. Hilleman |
Chief Financial Officer (Principal Financial and Accounting Officer) |
February 28, 2018 | ||
| /S/ KIERAN T. GALLAHUE Kieran T. Gallahue |
Lead Director | February 28, 2018 | ||
| /S/ TERESA L. KLINE Teresa L. Kline |
Director | February 28, 2018 | ||
| /S/ CYNTHIA L. LUCCHESE Cynthia L. Lucchese |
Director | February 28, 2018 | ||
| /S/ DANA G. MEAD, JR. Dana G. Mead, Jr. |
Director | February 28, 2018 | ||
| /S/ FREDERIC H. MOLL Frederic H. Moll |
Director | February 28, 2018 | ||
| /S/ W. ANTHONY VERNON W. Anthony Vernon |
Director | February 28, 2018 | ||
70
Table of Contents
INTERSECT ENT, INC.
Index to Financial Statements
For the Three Fiscal Years Ended
December 31, 2017
| Page | ||||
| F-2 | ||||
| Financial Statements: |
||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
Intersect ENT, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Intersect ENT, Inc. (the Company) as of December 31, 2017 and 2016, the related statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and financial statement schedule listed in the Index at Item 15(a) (collectively referred to as the “financial statements”). In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 28, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatements of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2008.
Redwood City, California
February 28, 2018
F-2
Table of Contents
INTERSECT ENT, INC.
(in thousands, except per share data)
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Assets | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 19,837 | $ | 9,859 | ||||
| Short-term investments, available-for-sale |
82,483 | 94,086 | ||||||
| Accounts receivable, net |
16,589 | 14,421 | ||||||
| Inventory |
8,474 | 5,613 | ||||||
| Prepaid expenses and other current assets |
2,908 | 1,313 | ||||||
|
|
|
|
|
|||||
| Total current assets |
130,291 | 125,292 | ||||||
| Property and equipment, net |
4,848 | 4,127 | ||||||
| Other non-current assets |
436 | 358 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 135,575 | $ | 129,777 | ||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 3,400 | $ | 3,267 | ||||
| Accrued compensation |
13,152 | 10,152 | ||||||
| Other current liabilities |
1,125 | 945 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
17,677 | 14,364 | ||||||
| Deferred rent and other non-current liabilities |
679 | 1,016 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
18,356 | 15,380 | ||||||
| Commitments and contingencies (note 7) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; |
||||||||
| Authorized shares: 10,000 at December 31, 2017 and 2016; |
||||||||
| Issued and outstanding shares: none |
— | — | ||||||
| Common stock, $0.001 par value; |
||||||||
| Authorized shares: 150,000 at December 31, 2017 and 2016; |
||||||||
| Issued and outstanding shares: 29,678 and 28,673 at December 31, 2017 and 2016, respectively |
30 | 29 | ||||||
| Additional paid-in capital |
282,121 | 262,882 | ||||||
| Accumulated other comprehensive loss |
(92 | ) | (37 | ) | ||||
| Accumulated deficit |
(164,840 | ) | (148,477 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
117,219 | 114,397 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 135,575 | $ | 129,777 | ||||
|
|
|
|
|
|||||
See Accompanying Notes to Financial Statements
F-3
Table of Contents
INTERSECT ENT, INC.
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except per share data)
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Revenue |
$ | 96,301 | $ | 78,708 | $ | 61,593 | ||||||
| Cost of sales |
15,499 | 13,003 | 12,288 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
80,802 | 65,705 | 49,305 | |||||||||
| Operating expenses: |
||||||||||||
| Selling, general and administrative |
80,045 | 72,926 | 59,637 | |||||||||
| Research and development |
18,360 | 18,890 | 16,608 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
98,405 | 91,816 | 76,245 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(17,603 | ) | (26,111 | ) | (26,940 | ) | ||||||
| Interest income and other, net |
1,240 | 889 | 306 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
(16,363 | ) | (25,222 | ) | (26,634 | ) | ||||||
| Other comprehensive (loss) income: |
||||||||||||
| Unrealized (loss) gain on short-term investments |
(55 | ) | (45 | ) | 8 | |||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (16,418 | ) | $ | (25,267 | ) | $ | (26,626 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share, basic and diluted |
$ | (0.56 | ) | $ | (0.89 | ) | $ | (1.02 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average common shares used to compute net loss per share, basic and diluted |
29,119 | 28,420 | 26,159 | |||||||||
|
|
|
|
|
|
|
|||||||
See Accompanying Notes to Financial Statements
F-4
Table of Contents
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balance at December 31, 2014 |
23,379 | $ | 23 | $ | 150,410 | $ | — | $ | (96,621 | ) | $ | 53,812 | ||||||||||||
| Issuance of common stock upon exercise of stock options |
560 | 1 | 908 | — | — | 909 | ||||||||||||||||||
| Issuance of common stock through employee stock purchase plan |
53 | — | 811 | — | — | 811 | ||||||||||||||||||
| Issuance of common stock upon exercise of warrants |
48 | — | 69 | — | — | 69 | ||||||||||||||||||
| Issuance of common stock for follow-on offering, net of issuance costs |
4,119 | 4 | 96,388 | — | — | 96,392 | ||||||||||||||||||
| Stock-based compensation expense |
— | — | 4,864 | — | — | 4,864 | ||||||||||||||||||
| Unrealized gain on short-term investments |
— | — | — | 8 | — | 8 | ||||||||||||||||||
| Net loss |
— | — | — | — | (26,634 | ) | (26,634 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2015 |
28,159 | 28 | 253,450 | 8 | (123,255 | ) | 130,231 | |||||||||||||||||
| Issuance of common stock upon exercise of stock options |
379 | 1 | 551 | — | — | 552 | ||||||||||||||||||
| Issuance of common stock through employee stock purchase plan |
135 | — | 1,414 | — | — | 1,414 | ||||||||||||||||||
| Stock-based compensation expense |
— | — | 7,467 | — | — | 7,467 | ||||||||||||||||||
| Unrealized loss on short-term investments |
— | — | — | (45 | ) | — | (45 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (25,222 | ) | (25,222 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2016 |
28,673 | 29 | 262,882 | (37 | ) | (148,477 | ) | 114,397 | ||||||||||||||||
| Issuance of common stock upon exercise of stock options |
849 | 1 | 7,689 | — | — | 7,690 | ||||||||||||||||||
| Issuance of common stock through employee stock purchase plan |
156 | — | 1,730 | — | — | 1,730 | ||||||||||||||||||
| Stock-based compensation expense |
— | — | 9,820 | — | — | 9,820 | ||||||||||||||||||
| Unrealized loss on short-term investments |
— | — | — | (55 | ) | — | (55 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (16,363 | ) | (16,363 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2017 |
29,678 | $ | 30 | $ | 282,121 | $ | (92 | ) | $ | (164,840 | ) | $ | 117,219 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
See Accompanying Notes to Financial Statements
F-5
Table of Contents
STATEMENTS OF CASH FLOWS
(in thousands)
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Operating activities: |
||||||||||||
| Net loss |
$ | (16,363 | ) | $ | (25,222 | ) | $ | (26,634 | ) | |||
| Adjustments to reconcile net loss to cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
1,464 | 1,166 | 826 | |||||||||
| Stock-based compensation expense |
9,820 | 7,467 | 4,864 | |||||||||
| Amortization of net investment premium |
19 | 148 | 682 | |||||||||
| Loss on sales of short-term investments |
— | — | 20 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable, net |
(2,168 | ) | (2,953 | ) | (3,131 | ) | ||||||
| Inventory |
(2,861 | ) | (1,664 | ) | (1,402 | ) | ||||||
| Prepaid expenses and other assets |
(914 | ) | 202 | (544 | ) | |||||||
| Accounts payable |
203 | 1,180 | (230 | ) | ||||||||
| Accrued compensation |
3,000 | 564 | 4,503 | |||||||||
| Deferred rent and other liabilities |
(241 | ) | (947 | ) | 959 | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(8,041 | ) | (20,059 | ) | (20,087 | ) | ||||||
| Investing activities: |
||||||||||||
| Purchases of short-term investments |
(116,622 | ) | (153,919 | ) | (184,672 | ) | ||||||
| Sales of short-term investments |
— | — | 6,516 | |||||||||
| Maturities of short-term investments |
128,151 | 149,131 | 123,011 | |||||||||
| Purchases of property and equipment |
(2,281 | ) | (2,069 | ) | (1,524 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
9,248 | (6,857 | ) | (56,669 | ) | |||||||
| Financing activities: |
||||||||||||
| Proceeds from issuance of common stock |
8,771 | 1,966 | 1,770 | |||||||||
| Proceeds from public offering, net of issuance costs |
— | — | 96,392 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
8,771 | 1,966 | 98,162 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
9,978 | (24,950 | ) | 21,406 | ||||||||
| Cash and cash equivalents: |
||||||||||||
| Beginning of the period |
9,859 | 34,809 | 13,403 | |||||||||
|
|
|
|
|
|
|
|||||||
| End of the period |
$ | 19,837 | $ | 9,859 | $ | 34,809 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing activities: |
||||||||||||
| Property and equipment included in accounts payable |
$ | 146 | $ | 215 | $ | 43 | ||||||
See Accompanying Notes to Financial Statements
F-6
Table of Contents
NOTES TO FINANCIAL STATEMENTS
| 1. | Organization |
Description of Business
Intersect ENT, Inc. (the “Company”) is incorporated in the state of Delaware and its facilities are located in Menlo Park, California. The Company is a commercial drug delivery company committed to improving the quality of life for patients with ear, nose and throat conditions. The Company’s approved and in-development products are steroid releasing implants designed to treat the spectrum of needs among patients who are managed by ear, nose and throat (“ENT”) physicians for chronic sinusitis, one of the most prevalent chronic diseases in the United States. The Company’s current commercial products comprise the PROPEL® family of products, which are PROPEL®, PROPEL® Mini and PROPEL® Contour. The PROPEL family of products are used predominantly in hospitals and ambulatory surgical settings, although they may also be used in the physician office setting of care. In addition to these commercial products, the Company recently received approval from the U.S. Food and Drug Administration (“FDA”) to market another steroid releasing implant, SINUVATM, previously known as the RESOLVE product, in December 2017. SINUVA is designed for use in the physician’s office for treatment of patients who have had ethmoid sinus surgery yet suffer from recurrent sinus obstruction due to polyps.
Liquidity and Business Risks
As of December 31, 2017, the Company had cash, cash equivalents and short-term investments of $102.3 million, and an accumulated deficit of $164.8 million. The Company expects its cash, cash equivalents and short-term investments will be sufficient to fund its operations through at least twelve months after the date the financial statements are issued.
| 2. | Summary of Significant Accounting Policies |
Basis of Preparation
The financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) and pursuant to the rules and regulations of the United States Securities and Exchange Commission (“SEC”).
Follow-on Offering
In June 2015, the Company completed its follow-on offering by issuing 4,119,300 shares of common stock, including 537,300 shares pursuant to the full exercise by the underwriters of their option to purchase additional shares, at an offering price of $25.00 per share, for net proceeds of approximately $96.4 million, after deducting underwriting discounts and commissions of $6.2 million and offering expenses of $0.4 million.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts and disclosures reported in the financial statements. Management uses significant judgment when making estimates related to its stock-based compensation, as well as certain accrued liabilities. Management bases its estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from those estimates.
F-7
Table of Contents
Cash and Cash Equivalents
The Company considers all highly liquid securities, readily convertible to cash, that mature within 90 days or less from the date of purchase to be cash equivalents.
Short-term Investments, Available-for-Sale
Short-term investments, which are available-for-sale, represent highly liquid debt instruments with maturities greater than 90 days at date of purchase. Such investments are recorded at fair value and unrealized holding gains and losses are reported as a separate component of accumulated comprehensive income (loss) in stockholders’ equity until realized. The Company reviews its investment portfolio periodically to assess for other-than-temporary impairment. Should the Company determine that any unrealized losses on the investments are other-than-temporary, the amount of that impairment to be recognized in earnings will depend on whether the Company intends to sell the security or more likely than not will be required to sell the security before recovery of its amortized cost basis less any current period credit loss. The specific identification method is used to determine the cost of securities disposed of, with realized gains and losses reflected in interest and other income or expense, as appropriate, in the statement of operations.
Fair Value of Financial Instruments
The Company measures certain financial assets and liabilities at fair value on a recurring basis, including cash equivalents and short-term investments, available-for-sale. Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or a liability. A three-tier fair value hierarchy is established as a basis for considering such assumptions and for inputs used in the valuation methodologies in measuring fair value:
| Level 1 | — | Observable inputs such as quoted prices (unadjusted) for identical assets or liabilities in active markets. | ||
| Level 2 | — | Other inputs that are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant inputs are observable in the market or can be derived from observable market data. | ||
| Level 3 | — | Unobservable inputs that are supported by little or no market activities, which would require the Company to develop its own assumptions. | ||
The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
F-8
Table of Contents
The following is a summary of cash, cash equivalents and short-term investments, available-for-sale, by type of instrument measured at fair value on a recurring basis (in thousands):
| December 31, | ||||||||||||||||||||||||||||||||
| 2017 | 2016 | |||||||||||||||||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | Total | |||||||||||||||||||||||||
| Cash |
$ | 7,646 | $ | — | $ | — | $ | 7,646 | $ | 5,222 | $ | — | $ | — | $ | 5,222 | ||||||||||||||||
| Money market funds |
12,191 | — | — | 12,191 | 4,637 | — | — | 4,637 | ||||||||||||||||||||||||
| Corporate debt securities |
— | 61,627 | — | 61,627 | — | 51,738 | — | 51,738 | ||||||||||||||||||||||||
| Commercial paper |
— | 20,856 | — | 20,856 | — | 42,348 | — | 42,348 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| $ | 19,837 | $ | 82,483 | $ | — | $ | 102,320 | $ | 9,859 | $ | 94,086 | $ | — | $ | 103,945 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Reported as: |
||||||||||||||||||||||||||||||||
| Cash and cash equivalents |
$ | 19,837 | $ | 9,859 | ||||||||||||||||||||||||||||
| Short-term investments, available-for-sale |
82,483 | 94,086 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|||||||||||||||||||||||||||||
| $ | 102,320 | $ | 103,945 | |||||||||||||||||||||||||||||
|
|
|
|
|
|||||||||||||||||||||||||||||
There were no transfers in and out of Level 1 and Level 2 during the years ended December 31, 2017 and 2016.
Concentration of Credit Risk
Financial instruments, which potentially subject the Company to concentrations of credit risk, consist principally of cash equivalents, short-term investments and accounts receivable. The Company believes that the credit risk in its accounts receivable is mitigated by its credit evaluation process, relatively short collection terms and diversity of its customer base. The Company generally does not require collateral and losses on accounts receivable have historically been within management’s expectations.
The Company’s investment policy limits investments to certain types of debt securities issued by the U.S. government, its agencies, and institutions with investment-grade credit ratings, as well as corporate debt or commercial paper issued by the highest quality financial and non-financial companies, and places restrictions on maturities and concentration by type and issuer. The Company is exposed to credit risk in the event of a default by the financial institutions holding its cash and cash equivalents and issuers of investments to the extent recorded on the balance sheets. The Company has limited its credit risk associated with cash, cash equivalents and short-term investments by placing its investments with banks it believes are highly creditworthy and with highly rated investments.
Allowance for Doubtful Accounts
The Company provides for uncollectible accounts receivable by recording an allowance for doubtful accounts for balances deemed uncollectible. The Company evaluates the collectability of its accounts receivable based on known collection risks and historical experience. In circumstances where the Company is aware of a specific customer’s inability to meet its financial obligations to the Company (e.g., bankruptcy filings, substantial downgrading of credit ratings), the Company records a specific allowance for bad debts against amounts due to reduce the carrying amount of accounts receivable to the amount it reasonably believes will be collected. Based on the high creditworthiness of the customers that the Company sells to, the Company has not experienced any significant collection issues.
Inventory
Inventory is valued at the lower of cost or market, computed on a first-in, first-out basis, or net realizable value. This requires the Company to make estimates regarding the recoverability of its inventory, including an
F-9
Table of Contents
assessment of excess or obsolete inventory. The Company determines excess and obsolete inventory based on future demand for products within a specified time horizon, generally twelve months, product life cycles and any expiration of inventory prior to sale. Any inventory written down creates a new cost basis for the inventory value.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Depreciation is determined using the straight-line method over the estimated useful lives of the respective assets, generally three to five years. Leasehold improvements are amortized on a straight-line basis over the shorter of their estimated useful lives or the term of the lease. Any amortization of assets under capital leases is included in depreciation and amortization expense. Maintenance and repairs are charged to operations as incurred.
Impairment of Long-lived Assets
Long-lived assets consists primarily of property and equipment and are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If circumstances require that a long-lived asset be tested for possible impairment, the Company compares the undiscounted cash flows expected to be generated by the asset group to the carrying amount of the asset group. If the carrying amount of the long-lived asset is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying amount exceeds its fair value. The Company determines fair value using the income approach based on the present value of expected future cash flows or other appropriate measures of estimated fair value. The Company’s cash flow assumptions consider historical and forecasted revenue and operating costs and other relevant factors. Since inception, the Company has not recorded impairment charges on its long-lived assets.
Revenue Recognition
Revenue is derived from the sale of the PROPEL family of products to hospitals and ambulatory surgery centers almost entirely in the United States. The Company recognizes revenue when persuasive evidence of an arrangement exists, product delivery has occurred or there is no further obligation, pricing is fixed or determinable and collection is reasonably assured. The Company must make assumptions regarding the future collectability of amounts receivable from customers to determine whether revenue recognition criteria have been met. If collectability is not reasonably assured at the time of shipment, the Company defers revenue until such criteria have been met. In general, the Company’s standard terms and conditions of sale do not allow for product returns. The Company expenses shipping and handling costs as incurred and includes them in the cost of sales. In those cases where shipping and handling costs are billed to customers, the Company classifies the amounts billed as a component of revenue.
Cost of Sales
Cost of sales consists primarily of manufacturing overhead costs, material costs and direct labor. A significant portion of the Company’s cost of sales currently consists of manufacturing overhead costs. These overhead costs include the cost of quality assurance, material procurement, inventory control, facilities, information technology, equipment and operations supervision and management. Cost of sales also includes depreciation expense for production equipment and certain direct costs such as shipping costs.
Research and Development
Research and development expenses consist primarily of product development, clinical and regulatory affairs, consulting services and other costs associated with products and technologies in development. These expenses include employee compensation, stock-based compensation, supplies, quality assurance and related travel and allocated facilities and information technology expenses. Clinical expenses include clinical trial design, clinical site reimbursement, data management and travel expenses, and the cost of manufacturing products for clinical trials.
F-10
Table of Contents
Stock-based Compensation
The Company maintains equity incentive plans to provide long-term incentives for employees, consultants and members of the board of directors. The plans allow for the issuance of non-statutory and incentive stock options to employees and non-statutory stock options to consultants and non-employee directors.
The Company is required to determine the fair value of equity incentive awards and recognize compensation expense for all equity incentive awards made to employees and directors, including employee stock options. Stock-based compensation expense is recognized over the requisite service period in the statements of operations and comprehensive loss. The Company uses the straight-line method for expense attribution. Effective January 1, 2017, the Company adopted Accounting Standards Update (“ASU”) No. 2016-9, Improvements to Employee Share-Based Payment Accounting (“ASU 2016-9”), and elected to account for forfeitures when they occur. Prior to the adoption of ASU 2016-9, the Company estimated the awards ultimately expected to vest based on the Company’s historical forfeiture experience and an analysis of similar companies, therefore reducing the amount of stock-based compensation expense for estimated forfeitures. To the extent actual forfeitures differed from the estimates, the Company recorded the difference as a cumulative adjustment in the period that any estimate was revised.
The valuation model used for calculating the fair value of awards for stock-based compensation expense is the Black-Scholes option-pricing model (the “Black-Scholes model”). The Black-Scholes model requires the Company to make assumptions and judgments about the variables used in the calculation, including the expected term (weighted average period of time that the options granted are expected to be outstanding), the volatility of common stock and an assumed risk-free interest rate. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected term of the option.
Deferred Rent
Rent expense is recognized on a straight-line basis over the non-cancelable term of the Company’s operating lease and, accordingly, the Company records the difference between cash rent payments and the recognition of rent expense as a deferred rent liability. The Company also records lessor-funded lease incentives, such as leasehold improvements, as a deferred rent liability, which is amortized as a reduction of rent expense over the non-cancelable term of the respective operating lease.
Advertising Expenses
The Company expenses the costs of advertising, including promotional expenses, as incurred. Advertising expenses were $0.6 million, $0.6 million and $0.3 million during the years ended December 31, 2017, 2016 and 2015, respectively.
Income Taxes
The Company accounts for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates that will be in effect when the differences are expected to reverse. Valuation allowances against deferred tax assets are established when necessary to reduce deferred tax assets to the amounts expected to be realized. Currently, the Company has recorded a full valuation allowance against its deferred tax assets and there is no provision for income taxes, as the Company has incurred operating losses to-date. The Company’s policy is to record interest and penalties expense related to uncertain tax positions as “other expense” within interest income and other, net in the statement of operations.
Net Loss per Share
Basic net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock outstanding during the period. Diluted net loss per share is computed by dividing the net loss by
the weighted average number of shares of common stock and dilutive potential shares of common stock
F-11
Table of Contents
outstanding during the period. Because the Company has reported a net loss for all periods presented, diluted net loss per share is the same as basic net loss per share for those periods as all potentially dilutive securities were antidilutive in those periods.
The following potentially dilutive securities outstanding have been excluded from the computations of weighted average shares outstanding because such securities have an antidilutive impact due to losses reported (in common stock equivalent shares, in thousands):
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Common stock options |
3,788 | 3,585 | 2,946 | |||||||||
| RSUs |
275 | — | — | |||||||||
| ESPP shares |
190 | 221 | 252 | |||||||||
|
|
|
|
|
|
|
|||||||
| 4,253 | 3,806 | 3,198 | ||||||||||
|
|
|
|
|
|
|
|||||||
Comprehensive Loss
Comprehensive loss consists of net loss and changes in unrealized gains and losses on short-term investments, available-for-sale.
Segment, Geographical and Customer Concentration
The Company has one operating segment. The Company’s assets and revenue are almost entirely based in the U.S. No single customer accounted for more than 10% of revenue during the years ended December 31, 2017, 2016 and 2015, and no single customer accounted for more than 10% of accounts receivable at December 31, 2017 and 2016.
Recent Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-13, Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”). ASU 2016-13 requires that credit losses be presented as an allowance rather than as a write-down for available-for-sale debt securities and allows for the reversal of estimated credit losses in the current period, aligning the income statement recognition of credit losses with the reporting period in which changes occur. ASU 2016-13 also broadens the information an entity must consider in developing its expected credit loss estimate for assets measured at amortized costs. ASU 2016-13 is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption is permitted for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The Company is evaluating the effect that ASU 2016-13 will have on its financial statements and related disclosures.
In March 2016, the FASB issued ASU No. 2016-9, Improvements to Employee Share-Based Payment Accounting (“ASU 2016-9”). ASU 2016-9 simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, classification on the statement of cash flows and an entity can now make an entity-wide election to either estimate the number of awards expected to vest or account for forfeitures when they occur. ASU 2016-9 was effective for annual periods beginning after December 15, 2016 and interim periods within those annual periods. The Company adopted ASU 2016-9 effective January 1, 2017, which did not have a material impact on the Company’s financial condition or results of operations.
In February 2016, the FASB issued ASU No. 2016-2, Leases: Amendments to the FASB Accounting Standards Codification (“ASU 2016-2”). Under ASU 2016-02, a lessee will be required to recognize assets and
F-12
Table of Contents
liabilities for leases with lease terms of more than twelve months. Recognition, measurement, and presentation of expenses and cash flows arising from a lease by a lessee primarily will depend on its classification as a finance or operating lease. ASU 2016-02 will require both types of leases to be recognized on the balance sheet. ASU 2016-02 also will require disclosures to help investors and other financial statement users to better understand the amount, timing and uncertainty of cash flows arising from the leases. These disclosures include qualitative and quantitative requirements, providing additional information about the amounts recorded in the financial statements. ASU 2016-2 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early adoption is permitted. The Company is evaluating the effect that ASU 2016-2 will have on its financial statements and related disclosures.
In August 2015, the FASB issued ASU No. 2015-14, Revenue from Contracts with Customers: Deferral of the Effective Date (“ASU 2015-14”), which defers the effective date of ASU No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”) by one year, and is now effective for all entities for annual reporting periods beginning after December 15, 2017. ASU 2014-09, requires an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers and will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective. In doing so, companies may need to use more judgment and make more estimates than under current guidance. These include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. The Company plans to adopt ASU 2014-09 on January 1, 2018 using the cumulative effect transition method and has completed its evaluation of the potential impact. Based on the Company’s work to-date, the Company does not believe the adoption of ASU 2014-09 will have an effect on the Company’s financial condition or results of operations. The Company will continue to monitor industry activities and any additional guidance provided by regulators, standards setters or the accounting profession and may adjust the Company’s assessment and implementation plans accordingly.
| 3. | Composition of Certain Financial Statement Items |
Accounts Receivable (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Accounts receivable |
$ | 16,739 | $ | 14,583 | ||||
| Allowance for doubtful accounts |
(150 | ) | (162 | ) | ||||
|
|
|
|
|
|||||
| $ | 16,589 | $ | 14,421 | |||||
|
|
|
|
|
|||||
Inventory (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Raw materials |
$ | 1,221 | $ | 778 | ||||
|
Work-in-process |
302 | 247 | ||||||
| Finished goods |
6,951 | 4,588 | ||||||
|
|
|
|
|
|||||
| $ | 8,474 | $ | 5,613 | |||||
|
|
|
|
|
|||||
F-13
Table of Contents
Property and Equipment (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Computer equipment and software |
$ | 1,330 | $ | 1,137 | ||||
| Furniture and office equipment |
548 | 423 | ||||||
| Laboratory equipment |
5,892 | 4,314 | ||||||
| Leasehold improvements |
1,739 | 1,618 | ||||||
|
|
|
|
|
|||||
| 9,509 | 7,492 | |||||||
| Less: accumulated depreciation and amortization |
(4,661 | ) | (3,365 | ) | ||||
|
|
|
|
|
|||||
| $ | 4,848 | $ | 4,127 | |||||
|
|
|
|
|
|||||
| 4. | Cash, Cash Equivalents and Short-term Investments |
The following is a summary of cash, cash equivalents and short-term investments, available-for-sale, by type of instrument (in thousands):
| December 31, | ||||||||||||||||||||||||||||||||
| 2017 | 2016 | |||||||||||||||||||||||||||||||
| Amortized Cost |
Gross Unrealized | Estimated Fair Value |
Amortized Cost |
Gross Unrealized | Estimated Fair Value |
|||||||||||||||||||||||||||
| Gains | Losses | Gains | Losses | |||||||||||||||||||||||||||||
| Cash |
$ | 7,646 | $ | — | $ | — | $ | 7,646 | $ | 5,222 | $ | — | $ | — | $ | 5,222 | ||||||||||||||||
| Money market funds |
12,191 | — | — | 12,191 | 4,637 | — | — | 4,637 | ||||||||||||||||||||||||
| Corporate debt securities |
61,695 | 1 | (69 | ) | 61,627 | 51,761 | 3 | (26 | ) | 51,738 | ||||||||||||||||||||||
| Commercial paper |
20,880 | — | (24 | ) | 20,856 | 42,362 | 6 | (20 | ) | 42,348 | ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| $ | 102,412 | $ | 1 | $ | (93 | ) | $ | 102,320 | $ | 103,982 | $ | 9 | $ | (46 | ) | $ | 103,945 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Reported as: |
||||||||||||||||||||||||||||||||
| Cash and cash equivalents |
$ | 19,837 | $ | 9,859 | ||||||||||||||||||||||||||||
| Short-term investments, available-for-sale |
82,483 | 94,086 | ||||||||||||||||||||||||||||||
|
|
|
|
|
|||||||||||||||||||||||||||||
| $ | 102,320 | $ | 103,945 | |||||||||||||||||||||||||||||
|
|
|
|
|
|||||||||||||||||||||||||||||
As of December 31, 2017 and 2016, the Company had no investments with a contractual maturity of greater than one year.
Based on an evaluation of securities that have been in a loss position, the Company did not recognize any other-than-temporary impairment charges during the years ended December 31, 2017, 2016 and 2015. The Company considered various factors which included a credit and liquidity assessment of the underlying securities and the Company’s intent and ability to hold the underlying securities until the estimated date of recovery of its amortized cost.
| 5. | Stockholders’ Equity |
2014 Equity Incentive Plan
In July 2014, the Company’s board of directors approved the 2014 Equity Incentive Plan (the “2014 Plan”). Under the 2014 Plan, the Company may grant stock options, stock appreciation rights, restricted stock, restricted stock units and certain other awards to individuals who are employees, officers, directors or consultants of the Company. A total of 4,750,000 shares of common stock were initially reserved for issuance under the 2014 Plan. The number of shares of common stock reserved for issuance under the 2014 Plan will automatically increase on January 1 of each year, beginning on January 1, 2015, and continuing through and including January 1, 2024, by
F-14
Table of Contents
3% of the total number of shares of the Company’s capital stock outstanding on December 31 of the preceding calendar year, or a lesser number of shares determined by the Company’s board of directors. The maximum number of shares that may be issued upon the exercise of ISOs under the 2014 Plan is 10.0 million. Incentive stock options (“ISOs”) and non-statutory stock options (“NSOs”) may be granted with exercise prices at no less than 100% of the fair value of the common stock on the date of grant. ISOs granted under the 2014 Plan generally vest 25% after the completion of twelve months of service and the balance vests in equal monthly installments over the next 36 months of service and expire 10 years from the grant date. NSOs vest per the specific agreement and expire 10 years from the date of grant. New shares are issued upon exercise of options under the stock plan. On January 1, 2017, the total number of shares of common stock reserved for issuance increased by 860,019 shares to 7,156,121 shares. In January 2017, the Company began issuing restricted stock units (“RSUs”) under the 2014 Plan. The RSUs generally vest annually over three years.
A summary of the Company’s stock option activity and related information is as follows (in thousands, except price data):
| Fiscal Year Ended December 31, 2017 |
||||||||
| Options | Weighted Average Exercise Price |
|||||||
| Outstanding, beginning of period |
3,585 | $ | 14.81 | |||||
| Granted |
1,286 | 16.04 | ||||||
| Exercised |
(849 | ) | 9.06 | |||||
| Forfeited |
(230 | ) | 18.84 | |||||
| Expired |
(4 | ) | 2.62 | |||||
|
|
|
|||||||
| Outstanding, end of period |
3,788 | 16.28 | ||||||
|
|
|
|||||||
| Exercisable |
1,881 | 14.77 | ||||||
|
|
|
|||||||
As of December 31, 2017, the aggregate pre-tax intrinsic value of options outstanding was $61.0 million and options outstanding and exercisable was $33.2 million, the weighted-average remaining contractual term of options outstanding was 7.9 years and options outstanding and exercisable was 7.2 years. The aggregate pre-tax intrinsic value of options exercised was $14.6 million, $5.5 million and $12.7 million during the years ended December 31, 2017, 2016 and 2015, respectively.
A summary of the Company’s RSU activity and related information (RSUs in thousands):
| Fiscal Year Ended December 31, 2017 |
||||||||
| RSUs | Weighted Average Fair Value |
|||||||
| Outstanding, beginning of period |
— | $ | — | |||||
| Awarded |
288 | 13.74 | ||||||
| Forfeited |
(13 | ) | 15.57 | |||||
|
|
|
|||||||
| Outstanding, end of period |
275 | 13.65 | ||||||
|
|
|
|||||||
As of December 31, 2017, the aggregate pre-tax intrinsic value of RSUs outstanding was $8.9 million, calculated based on the closing price of the Company’s common stock at the end of the period, and the weighted-average remaining contractual term of RSUs outstanding was 2.0 years.
2014 Employee Stock Purchase Plan
In July 2014, the Company’s board of directors approved the 2014 Employee Stock Purchase Plan (“2014 ESPP”). The 2014 ESPP became effective on the effective date of the IPO. A total of 496,092 shares were
F-15
Table of Contents
initially reserved for issuance under the 2014 ESPP. Shares issued during each of the years ended December 31, 2017, 2016 and 2015 were 0.2 million, 0.1 million and 0.1 million, respectively.
| 6. | Stock-Based Compensation Expense |
Total stock-based compensation expense recognized is as follows (in thousands):
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Cost of sales |
$ | 892 | $ | 708 | $ | 417 | ||||||
| Selling, general and administrative |
7,292 | 5,606 | 3,812 | |||||||||
| Research and development |
1,636 | 1,153 | 635 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 9,820 | $ | 7,467 | $ | 4,864 | |||||||
|
|
|
|
|
|
|
|||||||
As of December 31, 2017, the amount of unearned stock-based compensation currently estimated to be expensed through the year 2021 related to unvested employee stock-based awards was $18.1 million and the weighted average period over which the unearned stock-based compensation is expected to be recognized was 2.4 years. If there are any modifications or cancellations of the underlying unvested securities, the Company may be required to accelerate, increase or cancel any remaining unearned stock-based compensation expense. Future stock-based compensation expense and unearned stock-based compensation will increase to the extent that the Company grants additional share-based payments.
The Company estimates the fair value of stock-based compensation on the date of grant using the Black-Scholes option-pricing model. The Black-Scholes model determines the fair value of stock-based payment awards based on the fair market value of the Company’s common stock on the date of grant and is affected by assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, the fair market value of the Company’s common stock, volatility over the expected term of the awards and actual and projected employee stock option exercise behaviors. The Company has opted to use the “simplified method” for estimating the expected term of options, whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the option. Due to the Company’s limited operating history and a lack of company specific historical and implied volatility data, the Company has based its estimate of expected volatility on the historical volatility of a group of similar companies that are publicly traded. When selecting these public companies on which it has based its expected stock price volatility, the Company generally selected companies with comparable characteristics to it, including enterprise value, stages of clinical development, risk profiles, position within the industry and with historical share price information sufficient to meet the expected life of the stock-based awards. The historical volatility data was computed using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of the share-based payments. The Company will continue to analyze the historical stock price volatility and expected term assumptions as more historical data for the Company’s common stock becomes available. The risk-free rate assumption is based on the U.S. Treasury instruments with maturities similar to the expected term of the Company’s stock options. The expected dividend assumption is based on the Company’s history of not paying dividends and its expectation that it will not declare dividends for the foreseeable future.
Effective January 1, 2017, the Company adopted ASU 2016-9 and elected to account for forfeitures when they occurred. Prior to the adoption of ASU 2016-9, the Company estimated the awards ultimately expected to vest based on the Company’s historical forfeiture experience and an analysis of similar companies, therefore reducing the amount of stock-based compensation expense for estimated forfeitures. To the extent actual forfeitures differed from the estimates, the Company recorded the difference as a cumulative adjustment in the period that any estimate was revised.
F-16
Table of Contents
The fair value of options granted to employees or directors during the periods presented below were estimated as of the grant date using the Black-Scholes model assuming the weighted average assumptions listed in the following table:
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Expected term (years) |
6.0 | 6.0 | 6.0 | |||||||||
| Expected volatility |
45 | % | 44 | % | 48 | % | ||||||
| Risk-free interest rate |
2.1 | % | 1.5 | % | 1.6 | % | ||||||
| Dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Fair value |
$ | 7.24 | $ | 7.83 | $ | 10.66 | ||||||
The fair value of options granted under the 2014 ESPP to employees was estimated as of the grant date using the Black-Scholes model assuming the weighted average assumptions listed in the following table:
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Expected term (years) |
1.3 | 1.3 | 1.3 | |||||||||
| Expected volatility |
44 | % | 48 | % | 38 | % | ||||||
| Risk-free interest rate |
1.3 | % | 0.7 | % | 0.4 | % | ||||||
| Dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Fair value |
$ | 8.71 | $ | 4.46 | $ | 7.29 | ||||||
| 7. | Commitments and Contingencies |
Contingencies
In the normal course of business, the Company enters into contracts and agreements that contain a variety of representations and warranties and provide for general indemnifications. The Company’s exposure under these agreements is unknown because it involves claims that may be made against the Company in the future, but have not yet been made. The Company accrues a liability for such matters when it is probable that future expenditures will be made and such amounts can be reasonably estimated.
Indemnification
The Company’s amended and restated certificate of incorporation contains provisions limiting the liability of directors, and its amended and restated bylaws provide that the Company will indemnify each of its directors to the fullest extent permitted under Delaware law. The Company’s amended and restated certificate of incorporation and amended and restated bylaws also provide its board of directors with discretion to indemnify its officers and employees when determined appropriate by the board. In addition, the Company has entered and expects to continue to enter into agreements to indemnify its directors and executive officers.
Litigation
The Company is not currently a party to any material legal proceedings. The Company may at times be involved in litigation and other legal claims in the ordinary course of business. When appropriate in the Company’s estimation, it may record reserves in its financial statements for pending litigation and other claims.
Building Lease
As of December 31, 2017, the Company has one leased facility under an operating lease agreement entered into in March 2012. In December 2014, the operating lease agreement was amended for an additional 17,900
F-17
Table of Contents
square feet for a total of 50,400 square feet and the expiration was extended to May 31, 2020. Rental payments are charged to expense on a straight-line basis over the period of the lease. The lease agreement requires the Company to pay executory costs such as real estate taxes, insurance and repairs, and includes a renewal provision allowing the Company to extend this lease for an additional three years at 95% of the then-current fair market rental rate. Because of the terms of the amended lease agreement, the Company is the deemed owner, for accounting purposes only, of the building improvements. Accordingly, the Company recorded an asset of $1.0 million, representing the total costs of the building improvements payable by the lessor, the legal owner of the building, with a corresponding offset recorded as deferred rent. The building improvements are being amortized over the life of the lease.
Future minimum annual operating lease payments are as follows (in thousands):
| Fiscal Years Ending December 31, |
December 31, 2017 |
|||
| 2018 |
$ | 1,580 | ||
| 2019 |
1,625 | |||
| 2020 |
685 | |||
| Thereafter |
— | |||
|
|
|
|||
| $ | 3,890 | |||
|
|
|
|||
Rent expense was $1.9 million, $2.0 million and $1.9 million during the years ended December 31, 2017, 2016 and 2015, respectively.
Purchase Commitments
As of December 31, 2017, the Company had commitments to suppliers for purchases totaling $5.4 million.
| 8. | Employee Retirement Plan |
In January 2007, the Company established a qualified retirement plan under section 401(k) of the Internal Revenue Code (“IRC”) under which participants may contribute up to 100% of their eligible compensation, subject to maximum deferral limits specified by the IRC. The Company may make a discretionary profit sharing contribution to each eligible employee, subject to limits specified by the IRC, on an annual basis, provided the employee is employed with the Company on the last day of the plan year which is December 31. In addition, the Company may also make matching contributions of up to 3% of an employee’s eligible compensation. The Company’s contributions will vest 25% per year over four years. Total matching contributions were $0.3 million, $0.3 million and $0.2 million during the years ended December 31, 2017, 2016 and 2015, respectively.
| 9. | Income Taxes |
The Company has a history of losses and therefore has made no provision for income taxes.
F-18
Table of Contents
The amount computed by applying the federal statutory rate to loss before income taxes reconciles to the provision for income taxes is as follows (in thousands):
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Tax at federal statutory rate |
$ | (5,563 | ) | $ | (8,575 | ) | $ | (9,056 | ) | |||
| State tax, net of federal benefit |
(575 | ) | (1,210 | ) | (589 | ) | ||||||
| Permanent items |
460 | 549 | 463 | |||||||||
| Stock-based compensation |
(2,919 | ) | 295 | 212 | ||||||||
| R&D tax credit |
(627 | ) | (536 | ) | (559 | ) | ||||||
| Change in U.S. federal tax rate |
20,974 | — | — | |||||||||
| Change in valuation allowance |
(11,750 | ) | 9,477 | 9,529 | ||||||||
|
|
|
|
|
|
|
|||||||
| $ | — | $ | — | $ | — | |||||||
|
|
|
|
|
|
|
|||||||
Significant components of net deferred tax assets are as follows (in thousands):
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: |
||||||||
| Net operating losses |
$ | 38,531 | $ | 46,478 | ||||
| R&D tax credit |
5,983 | 4,612 | ||||||
| Accruals and other |
6,730 | 7,456 | ||||||
|
|
|
|
|
|||||
| 51,244 | 58,546 | |||||||
| Deferred tax liabilities: |
||||||||
| Depreciation and amortization |
(212 | ) | (337 | ) | ||||
|
|
|
|
|
|||||
| (212 | ) | (337 | ) | |||||
|
|
|
|
|
|||||
| Net deferred tax assets: |
51,032 | 58,209 | ||||||
| Valuation allowance |
(51,032 | ) | (58,209 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
In December 2017, the U.S. government enacted comprehensive tax legislation, commonly referred to as the Tax Cuts and Jobs Act (“Tax Act”). The Tax Act makes broad and complex changes to the U.S. tax code, including, but not limited to, (1) reducing the U.S. federal corporate tax rate from 35 percent to 21 percent; (2) requiring companies to pay a one-time transition tax on certain unrepatriated earnings of foreign subsidiaries; (3) generally eliminating U.S. federal income taxes on dividends from foreign subsidiaries; (4) requiring a current inclusion in U.S. federal taxable income of certain earnings of controlled foreign corporations; (5) eliminating the corporate alternative minimum tax (“AMT”) and changing how existing AMT credits can be realized; (6) creating the base erosion anti-abuse tax (“BEAT”), a new minimum tax; (7) creating a new limitation on deductible interest expense; and (8) changing the rules related to uses and limitations of net operating loss (“NOL”) carryforwards created in tax years beginning after December 31, 2017.
The Company has not historically had significant non-U.S. operations and, as such, the only significant impact of the Tax Act for the Company will be the reduction in the U.S. corporate tax rate. The Act reduces the corporate tax rate to 21 percent, effective January 1, 2018. Consequently, we have recorded a decrease in our net deferred tax asset balance of $21.0 million, with a corresponding and fully offsetting adjustment to our valuation allowance for the year ended December 31, 2017.
In December 2017, Staff Accounting Bulletin No. 118 (“SAB 118”) was issued to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared, or
F-19
Table of Contents
analyzed in reasonable detail to complete the accounting for certain income tax effects of the Tax Act. As of December 31, 2017, due to the complexities of the new law, we have not completed our accounting for all the tax effects of the Tax Act, however, as noted above we have made a reasonable estimate of the effects on our existing deferred tax balances. We are still analyzing certain aspects of the Tax Act and refining our calculations, which could potentially affect the measurement of these balances or potentially give rise to new deferred tax amounts. In all cases, we will continue to make and refine our calculations as additional analysis is completed. In addition, our provisional estimates may also be adjusted as we gain a more thorough understanding of the tax law.
Deferred income taxes reflect the tax effects of NOLs and tax credit carryforwards and the net temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
Realization of the deferred tax assets is dependent upon the generation of future taxable income, if any, the amount and timing of which are uncertain. Based on available objective evidence, management believes it is more likely than not that the deferred tax assets are not recognizable and will not be recognizable until the Company has sufficient taxable income. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by $7.2 million and increased by $9.5 million during the years ended December 31, 2017 and 2016, respectively.
On January 1, 2017, the Company adopted ASU 2016-09, which simplified several aspects of accounting for employee share-based payment transactions, including the accounting for income taxes, forfeitures, statutory tax withholding requirements and classification in the statement of cash flows. The adoption of ASU 2016-09 did not have an impact on our balance sheet, results of operations, cash flows or statement of stockholders’ equity because we have a full valuation allowance on our deferred tax assets. Upon adoption, the Company recognized the previously unrecognized excess tax benefits using the modified retrospective transition method. The previously unrecognized excess tax effects were recorded as a deferred tax asset, which was fully offset by a valuation allowance. Without the valuation allowance, the Company’s deferred tax assets would have increased by $4.6 million.
As of December 31, 2017, the Company’s federal NOL carryforwards of $149.1 million will expire at various dates beginning in 2026, if not utilized, and federal research and development tax credits of $4.4 million will begin to expire in 2026. In addition, NOL carryforwards for state income tax purposes of $53.0 million will begin to expire in 2028 and state research and development tax credits of $3.9 million do not expire.
Utilization of the NOL carryforwards may be subject to an annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986 and similar state provisions. The annual limitation may result in the expiration of the NOL before utilization.
Due to the Company’s full valuation allowance against all net deferred tax assets, the Company’s unrecognized tax benefits, if recognized, would not affect the effective tax rate.
A reconciliation of the change in the unrecognized tax benefit during the year is as follows (in thousands):
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Beginning of year |
$ | 1,374 | $ | 1,094 | $ | 861 | ||||||
| Additions for tax positions related to: |
||||||||||||
| Current year |
286 | 294 | 227 | |||||||||
| Prior years |
— | (14 | ) | 6 | ||||||||
|
|
|
|
|
|
|
|||||||
| End of year |
$ | 1,660 | $ | 1,374 | $ | 1,094 | ||||||
|
|
|
|
|
|
|
|||||||
F-20
Table of Contents
The Company does not expect a significant change to its unrecognized tax benefits over the next twelve months.
The Company files income tax returns in the U.S. federal and various state jurisdictions. Tax years beginning in 2004 through 2017 remain open to examination by the major taxing authorities to which the Company is subject to. The Company’s policy is to record interest related to uncertain tax positions as interest expense and any penalties as other expense in its statements of operations and comprehensive loss. The Company has not recorded any interest expense or penalties associated with unrecognized tax benefits.
Supplemental Quarterly Financial Information (unaudited)
The following table sets forth unaudited statements of operations data for each of the Company’s last eight quarters. This quarterly information is unaudited and has been prepared on the same basis as the annual financial statements. In the Company’s opinion, this quarterly information reflects all adjustments necessary for a fair presentation of the periods presented. The operating results for any quarter are not necessarily indicative of results for any future period.
| Fiscal Years Ended December 31, | ||||||||||||||||||||||||||||||||
| 2017 | 2016 | |||||||||||||||||||||||||||||||
| Q1 | Q2 | Q3 | Q4 | Q1 | Q2 | Q3 | Q4 | |||||||||||||||||||||||||
| Revenue |
$ | 20,474 | $ | 23,985 | $ | 22,313 | $ | 29,529 | $ | 16,692 | $ | 19,317 | $ | 18,468 | $ | 24,231 | ||||||||||||||||
| Cost of sales |
2,884 | 3,684 | 3,808 | 5,123 | 3,210 | 3,117 | 2,788 | 3,888 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Gross profit |
17,590 | 20,301 | 18,505 | 24,406 | 13,482 | 16,200 | 15,680 | 20,343 | ||||||||||||||||||||||||
| Gross margin |
86 | % | 85 | % | 83 | % | 83 | % | 81 | % | 84 | % | 85 | % | 84 | % | ||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||||||||||
| Sales, general and administrative |
20,319 | 18,682 | 18,746 | 22,298 | 17,393 | 17,795 | 17,905 | 19,833 | ||||||||||||||||||||||||
| Research and development |
4,220 | 4,176 | 4,346 | 5,618 | 4,495 | 4,588 | 4,237 | 5,570 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Total operating expenses |
24,539 | 22,858 | 23,092 | 27,916 | 21,888 | 22,383 | 22,142 | 25,403 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Loss from operations |
(6,949 | ) | (2,557 | ) | (4,587 | ) | (3,510 | ) | (8,406 | ) | (6,183 | ) | (6,462 | ) | (5,060 | ) | ||||||||||||||||
| Interest and other income, net |
268 | 288 | 326 | 358 | 185 | 224 | 236 | 244 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net loss |
$ | (6,681 | ) | $ | (2,269 | ) | $ | (4,261 | ) | $ | (3,152 | ) | $ | (8,221 | ) | $ | (5,959 | ) | $ | (6,226 | ) | $ | (4,816 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Basic and diluted net loss per share |
$ | (0.23 | ) | $ | (0.08 | ) | $ | (0.15 | ) | $ | (0.11 | ) | $ | (0.29 | ) | $ | (0.21 | ) | $ | (0.22 | ) | $ | (0.17 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Shares used to compute basic and diluted net loss per share |
28,708 | 28,950 | 29,269 | 29,538 | 28,207 | 28,379 | 28,479 | 28,610 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
F-21
Table of Contents
INTERSECT ENT, INC.
SCHEDULE II — VALUATION AND QUALIFYING ACCOUNTS
(in thousands)
| Fiscal Years Ended December 31, |
||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Allowance for doubtful accounts: |
||||||||||||
| Beginning |
$ | 162 | $ | 86 | $ | — | ||||||
| Charges |
62 | 155 | 92 | |||||||||
| Write-offs |
(74 | ) | (79 | ) | (6 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Ending |
$ | 150 | $ | 162 | $ | 86 | ||||||
|
|
|
|
|
|
|
|||||||