Attached files
| file | filename |
|---|---|
| EX-99.1 - EX-99.1 - Ra Pharmaceuticals, Inc. | a18-5952_1ex99d1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of The Securities Exchange Act of 1934
Date of Report (Date of Earliest Event Reported): February 12, 2018
Ra Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
|
DELAWARE |
|
001-37926 |
|
26-2908274 |
|
(State or other jurisdiction |
|
(Commission |
|
(I.R.S. Employer |
|
87 Cambridge Park Drive |
|
02140 |
|
(Address of principal executive offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code (617) 401-4060
Not Applicable
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
o Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
o Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
o Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
o Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 or Rule 12b-2 of the Securities Exchange Act of 1934.
Emerging growth company x
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. x
Item 2.02. Results of Operations and Financial Condition.
On February 13, 2018, Ra Pharmaceuticals, Inc. (the “Company”) issued a press release announcing the commencement of an offering of shares of its common stock (the “Offering”). In connection with the Offering, the Company filed a preliminary prospectus supplement to the base prospectus included in the Company’s shelf registration statement on Form S-3 (No. 333-221266), filed with the SEC on November 1, 2017 and declared effective by the SEC on November 14, 2017. In the preliminary prospectus supplement, the Company announced that its cash and cash equivalents is expected to be approximately $70.4 million at December 31, 2017, as compared to $84.1 million at September 30, 2017. This financial data as of December 31, 2017 is preliminary and is based on information available to the Company’s management as of the date of this current report on Form 8-K and is subject to completion by management of the Company’s financial statements as of and for the year ended December 31, 2017. The Company’s independent registered public accountants have not audited, reviewed or performed any procedures with respect to such preliminary financial data and accordingly do not express an opinion or any other form of assurance with respect thereto. These results could change as a result of further review. Complete yearly results will be announced during the Company’s annual financial results earnings conference call and included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2017.
The information set forth in this Item 2.02 shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liabilities of that section, nor shall it be incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as expressly set forth by specific reference in such a filing.
Item 8.01. Other Events.
The preliminary prospectus supplement also described certain recent developments in the Company’s business, including those set forth below. On February 12, 2018, the Company announced completion of dosing in its Phase 2 clinical program of RA101495 SC in Paroxysmal Nocturnal Hemoglobinuria. The global, dose-finding Phase 2 clinical program enrolled a total of 29 patients in three cohorts, including patients naïve to eculizumab (n=10) and patients with prior eculizumab exposure, including eculizumab switch patients (n=16) and eculizumab inadequate responders (n=3). A total of 21 patients completed the initial 12-week dosing period and 16 of those patients (or 76%) are continuing treatment with RA101495 SC in the Company’s long-term extension study (8 naïve patients and 8 switch patients, including all 3 inadequate responders).
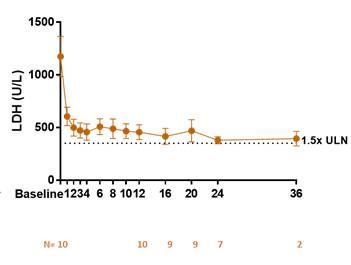
All 10 eculizumab naïve patients successfully completed 12 weeks of dosing. RA101495 SC met the primary endpoint, demonstrating a rapid, robust, and sustained reduction in lactate dehydrogenase (LDH) levels from baseline to the mean of Weeks 6-12 (p=0.002) and near-complete suppression of complement activity. Fifty percent of eculizumab naïve patients (3/6) who were transfusion-dependent prior to enrollment remained transfusion-free while on study. Meaningful improvements in standard measures of quality of life, as shown by the Functional Assessment of Chronic Illness Therapy (FACIT) fatigue score were observed, as well as a high level of patient satisfaction with subcutaneous self-administration based on patient surveys. Eight of the 10 patients in the naïve cohort continue in the long-term extension study, with the longest treated patients (n=2) dosed through 36 weeks.
As shown in Figure 1, the clinically meaningful reduction in mean LDH observed during the 12 week Phase 2 dosing period has been sustained in the long-term extension study.
Figure 1.
LDH in Eculizumab Naïve Cohort
In eculizumab switch patients, transfusion-independent patients (n=5) switching to RA101495 SC maintained an overall stable mean LDH level, with one patient withdrawing early due to breakthrough hemolysis and reverting to eculizumab without complications. Among switch patients who were transfusion-dependent at baseline (n=11), breakthrough hemolysis occurred after switching in seven patients (7/11), who all reverted to eculizumab treatment without complications. Persistent transfusion-dependence, which occurs in up to 20 percent of the eculizumab-treated population, is most commonly attributable to extravascular hemolysis driven by intense C3 deposition on erythrocytes. This condition, a unique complication of long-term eculizumab therapy in certain PNH patients, may not be adequately addressed by inhibition at the level of C5, and published data suggest that targeting complement upstream of C5 (e.g. Factor D or C3) may have greater utility in this small subset of PNH patients.
In the U.S.-based cohort of patients who were inadequate responders to eculizumab and have a history of elevated LDH levels, all three patients (two transfusion-independent, one transfusion-dependent) have completed 12 weeks of dosing and maintained stable mean LDH levels.
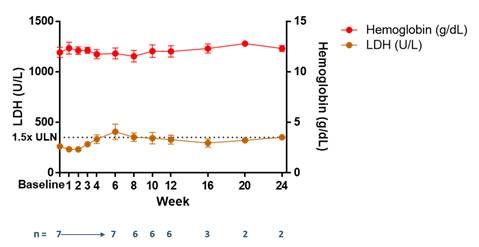
As shown in Figure 2, in transfusion-independent patients switching from eculizumab to RA101495 SC, pooled from both the switch cohort and the U.S.-based inadequate responder cohort (collectively, n=7), mean LDH and hemoglobin levels have remained stable in patients enrolled in the long-term extension study through the data cut-off date of February 7, 2018.
Figure 2.
Stabilization of LDH and Hemoglobin in Transfusion-Independent Patients
Eculizumab Switch & Inadequate Responder Cohorts — Pooled Data
No major safety or tolerability concerns have been reported after more than 500 patient weeks of cumulative exposure across all cohorts. No meningococcal infections or thromboembolic events have been observed. Out of more than 3,500 doses administered to date, only nine mild (grade 1) injection site reactions have occurred in a total of five patients. As of February 7, 2018, full compliance with once daily subcutaneous self-administration of RA101495 SC has been observed.
The Phase 2 study provided evidence supporting the dose required for near-complete complement inhibition. Whereas the 0.1 mg/kg dose of RA101495 SC administered in the Phase 2 study in PNH resulted in sub-maximal inhibition of hemolysis (<95% at trough), the 0.3 mg/kg dose of RA101495 SC resulted in near-complete inhibition of hemolysis (> 95% at trough at all timepoints). These data support the selection of 0.3 mg/kg RA101495 SC daily as the recommended dose in the plannaed Phase 3 studies in PNH.
Based on the results of the Phase 2 studies, the Company is planning to conduct a Phase 3 program to support the approval of RA101495 SC in PNH. Both treatment naïve and switch patients who were transfusion-independent on eculizumab are planned for inclusion in Company’s Phase 3 program.
A copy of the press release that the Company issued in connection with the announcement of the Offering is attached hereto as Exhibit 99.1 and is incorporated herein by reference.
Item 9.01. Financial Statements and Exhibits.
(d) Exhibits
|
Exhibit |
|
Description |
|
|
|
|
|
99.1 |
|
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
|
Date: February 13, 2018 |
RA PHARMACEUTICALS, INC. | |
|
|
|
|
|
|
By: |
/s/ David C. Lubner |
|
|
|
David C. Lubner |
|
|
|
Executive Vice President and Chief Financial Officer |