Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Bioverativ Inc. | bivv-20171231ex321e3749c.htm |
| EX-31.2 - EX-31.2 - Bioverativ Inc. | bivv-20171231ex31236a75e.htm |
| EX-31.1 - EX-31.1 - Bioverativ Inc. | bivv-20171231ex311bd94bc.htm |
| EX-23.1 - EX-23.1 - Bioverativ Inc. | bivv-20171231ex2318c9749.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10‑K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2017 |
|
|
or |
|
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
|
Commission file number: 001‑37859

Bioverativ Inc.
(Exact name of Registrant as Specified in its Charter)
|
Delaware |
81‑3461310 |
|
|
|
|
225 Second Avenue, Waltham, Massachusetts |
02451 |
(781) 663‑4400
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Each Exchange on which registered |
|
Common Stock, par value $0.001 per share |
|
The Nasdaq Stock Market |
Securities to be registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S‑T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files): Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S‑K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10‑K or any amendment to this Form 10‑K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b‑2 of the Exchange Act. (Check one):
|
Large accelerated filer ☒ |
Accelerated filer ☐ |
Non‑accelerated filer ☐ |
Smaller reporting company ☐ |
Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in rule 12b‑2) of the Act. Yes ☐ No ☒
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) computed by reference to the price at which the common stock was last sold as of the last business day of the registrant’s most recently completed second fiscal quarter was $6,433.3 million.
The number of shares of the registrant’s common stock, $0.001 par value, outstanding as of February 9, 2018 was 108,223,644.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the registrant’s 2018 annual meeting of stockholders to be filed within 120 days of the end of its fiscal year ended December 31, 2017 are incorporated into Part III (Items 10, 11, 12, 13 and 14) of this Annual Report on Form 10-K where indicated.
BIOVERATIV INC.
ANNUAL REPORT ON FORM 10‑K
For the Year Ended December 31, 2017
2
NOTE REGARDING FORWARD‑LOOKING STATEMENTS
This report contains forward‑looking statements that are being made pursuant to the provisions of the Private Securities Litigation Reform Act of 1995 (the Act) with the intention of obtaining the benefits of the "Safe Harbor" provisions of the Act. Use by Bioverativ of the words “may,” “will,” “would,” “could,” “should,” “believes,” “estimates,” “projects,” “potential,” “expects,” “plans,” “seeks,” “intends,” “evaluates,” “pursues,” “anticipates,” “continues,” “designs,” “impacts,” “affects,” “forecasts,” “target,” “outlook,” “initiative,” “objective,” “designed,” “priorities,” or the negative of those words or other similar expressions is intended to identify forward‑looking statements.
These forward‑looking statements may include statements with respect to:
|
· |
the pending acquisition by Sanofi and our ability to satisfy all of the conditions under the merger agreement we entered into with Sanofi in the anticipated timeframe or at all; |
|
· |
accounting estimates, assumptions and policies, and the expected impact of the adoption of new accounting standards; |
|
· |
estimates of liabilities; |
|
· |
separation related adjustments and ongoing costs of certain services provided by Biogen post-separation; |
|
· |
expected timing of the completion of certain transition and other services provided by Biogen to us; |
|
· |
purchase price allocation determinations in connection with acquisitions, including accounting assumptions, judgments and estimates used in connection such determinations; |
|
· |
anticipated contingent payments, including milestone and royalty payment obligations, and the timing thereof; |
|
· |
our exposure to market volatility and foreign currency and interest rate risks, and the anticipated impact of actions taken to mitigate such risks; |
|
· |
revenue growth and associated costs, discounts or rebates in connection with our products; |
|
· |
tax rates; |
|
· |
future cash flows, working capital needs, capital expenditures, inventory purchases, strategic investments and access to capital markets; |
|
· |
future transactions in our securities and debt issuances; |
|
· |
dividends; |
|
· |
litigation related matters, including outcomes and expenses relating thereto; |
|
· |
the impact of healthcare reform, including any repeal, substantial modification or invalidation of the U.S. Patient Protection and Affordable Care Act; |
|
· |
business and strategic objectives; |
|
· |
business development activities; |
|
· |
our anticipated investments in infrastructure and personnel; |
|
· |
our expectation to invest in research and development activities; |
|
· |
expected clinical trials and timing thereof; |
|
· |
receipt of necessary regulatory authorization and approvals; |
|
· |
our anticipated geographic expansion; |
|
· |
our growth, including patient share growth; |
|
· |
our manufacturing, supply and distribution arrangements; |
|
· |
the sufficiency of our facilities; |
|
· |
our relationships with third parties, collaborators and our employees; and |
|
· |
our ability to operate as a standalone company. |
These forward‑looking statements involve risks and uncertainties, including those that are described in Item 1A. Risk Factors and elsewhere in this report, that could cause actual results to differ materially from those reflected in such statements. You should not place undue reliance on these statements. Forward‑looking statements speak only as of the date of this report. Except as required by law, we do not undertake any obligation to publicly update any forward‑looking statements, whether as a result of new information, future developments or otherwise.
3
NOTE REGARDING PRESENTATION OF INFORMATION
Unless the context otherwise requires, references in this report to the following terms shall have the following respective meanings:
|
· |
“Biogen” refers to Biogen Inc., a Delaware corporation, and its consolidated subsidiaries; |
|
· |
“distribution” refers to the distribution by Biogen to Biogen stockholders of all of the outstanding shares of Bioverativ, as further described in this report; |
|
· |
“hemophilia business” includes Biogen’s hemophilia business and certain additional assets and liabilities associated with Biogen’s pipeline programs related to hemophilia and other blood disorders; |
|
· |
“separation” refers to the separation of Biogen’s hemophilia business from Biogen’s other businesses and the creation, as a result of the distribution, of an independent, publicly traded company, Bioverativ Inc., that holds the hemophilia business, as further described in this report; |
|
· |
“separation date” is February 1, 2017; and |
|
· |
“Bioverativ,” “we,” “us,” “our,” “our company” and “the company” refer to Bioverativ Inc., a Delaware corporation, or Bioverativ Inc., together with its consolidated subsidiaries, as the context requires. |
See “Glossary of Scientific Terms” starting on page 62 of this report for definitions of certain additional terms as they are used in this report.
This report describes the business transferred to Bioverativ by Biogen in the separation as if the transferred business was Bioverativ’s business for all historical periods described. References in this report to Bioverativ’s historical assets, liabilities, products, businesses or activities of Bioverativ’s business are generally intended to refer to the historical assets, liabilities, products, businesses or activities of the transferred business as the business was conducted as part of Biogen prior to the separation. Since the separation date, Bioverativ has operated as a standalone company.
NOTE REGARDING TRADEMARKS, TRADE NAMES AND SERVICE MARKS
Bioverativ owns or has rights to use the trademarks, service marks and trade names that it uses in conjunction with the operation of its business. Some of the trademarks that Bioverativ owns or has rights to use that appear in this report include: ALPROLIX® and ELOCTATE®, which may be registered or trademarked in the United States and other jurisdictions. Bioverativ’s rights to some of its trademarks may be limited to select markets. Each trademark, trade name or service mark of any other company appearing in this report is, to Bioverativ’s knowledge, owned by such other company. References to ELOCTATE in this report shall also refer to ELOCTA, the approved trade name for ELOCTATE in the European Union, as the context may require.
4
Merger Agreement with Sanofi
On January 21, 2018, we entered into an Agreement and Plan of Merger (the “Merger Agreement”), with Sanofi (“Parent” or “Sanofi”), a French société anonyme, and Blink Acquisition Corp. (“Merger Sub”), a Delaware corporation and indirect wholly-owned subsidiary of Parent. See Note 21, Subsequent Events to our audited consolidated financial statements included in this Form 10-K.
Pursuant to the terms of Merger Agreement, upon the terms and subject to the conditions thereof, Merger Sub commenced a tender offer on February 7, 2018 (the “Offer”), to acquire all of the outstanding shares of common stock of the Company, par value $0.001 per share (the “Shares”), at a purchase price of $105.0 per Share in cash, net of applicable withholding taxes and without interest (the “Offer Price”). Unless extended by Merger Sub in accordance with the Merger Agreement, the offering period for the Offer will expire at 11:59 p.m. (New York City time), on March 7, 2018.
As soon as practicable following (but on the same day as) the consummation of the Offer and subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, Merger Sub will be merged with and into the Company (the “Merger”), with the Company surviving the Merger as an indirect wholly-owned subsidiary of Parent. The Merger will be governed by Section 251(h) of the Delaware General Corporation Law (“DGCL”) and effected without a vote of the Company’s stockholders.
The Merger Agreement includes representations and warranties, and covenants of the parties customary for a transaction of this nature.
We have prepared this Form 10-K and the forward-looking statements contained in this Form 10-K as if we were going to remain an independent company. If the Offer and Merger are consummated, many of the forward-looking statements contained in this Form 10-K will no longer be applicable.
Business Overview
Bioverativ Inc., a Delaware corporation, was formed to hold the hemophilia business of Biogen Inc. On February 1, 2017, we separated from Biogen and became an independent public company.
Bioverativ is a global biopharmaceutical company focused on the discovery, research, development and commercialization of innovative therapies for the treatment of rare blood diseases including hemophilia.
Our strategy is to lead in hemophilia, build a complement disease franchise, and transform the treatment of hemoglobinopathies. We aim to raise the standards of care and improve outcomes for patients as we pursue our vision to become the leading rare blood disease company.
Lead in Hemophilia. We have two marketed products, ELOCTATE [Antihemophilic Factor (Recombinant), Fc Fusion Protein] and ALPROLIX [Coagulation Factor IX (Recombinant), Fc Fusion Protein], extended half‑life clotting‑factor therapies for the treatment of hemophilia A and hemophilia B, respectively. These therapies, when originally introduced in 2014, were the first major advancements in the treatment of hemophilia A and B in nearly two decades. We continue to research and explore the potential benefits and science behind these therapies to address areas of serious need, including long-term joint health and immune tolerance induction in people with hemophilia who develop inhibitors. We currently market our products primarily in the United States, Japan, Canada and Australia, and our collaboration partner, Swedish Orphan Biovitrum AB (publ) (Sobi), markets the products primarily in the European Union. Geographic expansion in additional markets is a priority, with a primary focus on certain countries in Latin America.
5
Our new product development for the treatment of hemophilia includes discovery, preclinical and clinical programs studying next generation extended half-life hemophilia product candidates, gene therapies for both hemophilia A and B, and non-factor products to treat hemophilia leveraging mimetic bi-specific antibody technology and a novel bicyclic peptide product platform.
Build a Complement Disease Franchise. We plan to build a complement disease franchise with our initial focus on developing a treatment for cold agglutinin disease. Cold agglutinin disease is a global, chronic rare blood disease characterized by hemolytic anemia, with a significant portion of patients requiring transfusions and having crippling fatigue, poor quality of life and the potential for life-threatening complications such as thrombotic events, including pulmonary embolism and stroke. Our efforts in this therapeutic area are initially focused on advancing the development of BIVV009, a first-in-class monoclonal antibody for cold agglutinin disease in Phase 3 trials, which we obtained through the acquisition of True North Therapeutics in June 2017.
Transform Treatment of Hemoglobinopathies. We have early and research-stage programs for the treatment of hemoglobinopathies, including gene-edited cell therapies for the treatment of transfusion-dependent beta-thalassemia and sickle cell disease and several discovery programs seeking to target the root cause of sickle cell disease. We believe there is a significant unmet need for therapies to treat beta-thalassemia and sickle cell disease, which are life-long rare blood disorders linked to shorter life expectancies and with few treatment options.
2017 Highlights
In 2017, we focused on three key business priorities: maximizing our potential in hemophilia, advancing our pipeline, and executing on strategic business development.
Maximizing Our Potential In Hemophilia
|
· |
Revenue Growth – We delivered 31.7% revenue growth from 2016 to 2017. This growth was due primarily to strong commercial execution and differentiation of our Fc fusion technology and our extended half-life products. |
|
· |
Focus on Patient Outcomes – We aim to raise the standard of care and improve outcomes for patients with hemophilia through our extended half-life products. In particular, we are focused on better outcomes for patients based on improvement in joint health and prophylaxis usage of our products as evidenced by our growing body of data. |
|
· |
In October 2017, interim results from a longitudinal study of joint health in patients treated prophylactically with ELOCTATE were published in Haemophilia. These interim results show that participants enrolled in the ASPIRE extension study demonstrated continuous improvement in joint health over a nearly three-year period with prophylactic dosing of ELOCTATE, regardless of prior treatment regimen, severity of joint damage or target joints. Joint health improvements were most notable in hemophilia A patients with poor joint health. |
|
· |
In July 2017, we presented data at the International Society on Thrombosis and Haemostasis conference, which showed that long-term prophylactic use of ALPROLIX resolved target joints in 100% of adults and adolescents with severe hemophilia B in the extension study B-YOND. |
Advancing Our Pipeline
|
· |
Phase 3 Cold Agglutinin Disease Trials – In the fourth quarter of 2017, we initiated site start-up for two parallel Phase 3 trials, Cardinal and Cadenza, to investigate BIVV009 in primary cold agglutinin disease. |
|
· |
BIVV001 in Phase 1/2a Trial – In the fourth quarter of 2017, we enrolled the first patient in the Phase 1/2a trial of BIVV001 (also known as rFVIIIFc-VWF-XTEN), an investigational factor VIII therapy designed to |
6
potentially extend protection from bleeds with prophylactic dosing of once weekly or longer for people with hemophilia A. |
|
· |
IND for ST-400 – In October 2017, we and Sangamo Therapeutics, Inc. (Sangamo) announced that the U.S. Food and Drug Administration (FDA) accepted the Investigational New Drug (IND) application for ST-400, a gene-edited cell therapy candidate for people with transfusion-dependent beta-thalassemia. The IND enables Sangamo to initiate a Phase 1/2 clinical trial to assess the safety, tolerability and efficacy of ST-400 in adults with transfusion-dependent beta-thalassemia. |
Executing on Strategic Business Development
|
· |
True North Acquisition – In June 2017, we acquired True North, a clinical-stage biopharmaceutical company focused on the discovery, development and commercialization of first-in-class product candidates for complement-mediated diseases. With the acquisition, we added BIVV009 to our pipeline. BIVV009 has received breakthrough therapy designation from the FDA for the treatment of hemolysis in patients with primary cold agglutinin disease, and orphan drug designation from the FDA and the European Medicines Agency (EMA). |
|
· |
Bicycle Therapeutics Research Collaboration – In August 2017, we entered into a research collaboration with Bicycle Therapeutics (Bicycle), a biotechnology company that is pioneering a new class of medicines based on its novel bicyclic peptide (Bicycle®) product platform. The collaboration focuses on the discovery, development and commercialization of therapies for hemophilia and sickle cell disease. Under our agreement, Bicycle is responsible for leading initial discovery activities through lead optimization to candidate selection for two programs, and we will lead preclinical and clinical development, as well as subsequent marketing and commercialization efforts. |
Background on Hemophilia A and B
Hemophilia A and hemophilia B are rare, x‑linked genetic disorders that impair the ability of a person’s blood to clot due to reduced levels of a protein known as factor VIII or factor IX, respectively. This impairment can lead to recurrent and extended bleeding episodes that may cause pain, irreversible joint damage and life‑threatening hemorrhages. In its Annual Global Survey 2016, the World Federation of Hemophilia (WFH) estimated that nearly 150,000 people worldwide were identified as living with hemophilia A and nearly 30,000 people were diagnosed with hemophilia B.
Hemophilia is usually diagnosed at birth or at a very young age, and predominantly affects males. An individual’s hemophilia is classified as mild, moderate or severe and is based on the level of factor activity in the blood. Although hemophilia care varies widely across the globe, in the United States a majority of patients receive care from specialized hemophilia treatment centers.
Joint disease, which is caused by frequent bleeds into joints over time, is the leading cause of morbidity for people with hemophilia, often resulting in chronic pain and disability. The ability to minimize joint damage over the long term is a critical unmet need that could have a positive impact on patient outcomes.
Hemophilia is treated by injecting the missing clotting factor directly into the patient’s bloodstream. Therapies can be administered either on a schedule to help prevent or reduce bleeding episodes (prophylaxis) or to control bleeding when it occurs (on‑demand). Over time, regimens have shifted from on‑demand treatment to routine prophylaxis due to observed improvements in long‑term clinical outcomes, such as joint damage. In the United States, the February 2016 guidelines of the Medical and Scientific Advisory Council of the National Hemophilia Foundation recommend routine prophylaxis as optimal for the treatment of people with severe hemophilia.
Historically, hemophilia treatments were derived from factors taken from human blood plasma. In the early 1990s, recombinant factor products, developed in a lab through the use of recombinant DNA technology, became available. In 2016, use of recombinant factor product accounted for over 75% of sales globally. In 2014, ELOCTATE
7
and ALPROLIX became the first available extended half‑life recombinant factor therapies in the United States with the benefit of an effective treatment even with less frequent, more convenient dosing requirements.
Patients may experience complications with factor therapies. In some cases, patients may develop inhibitors that recognize the infused factor as a foreign protein and create antibodies to it. Inhibitor antibodies occur when a person with hemophilia has an immune response to treatment with clotting factor concentrates. According to the WFH, an inhibitor usually occurs within the first 75 exposures to factor concentrates and thus is most often seen in children with severe hemophilia. In 2014, the WFH estimated that approximately 25% to 30% of children with severe hemophilia A and approximately 1% to 6% of individuals with hemophilia B will develop inhibitors. A common treatment to eradicate inhibitors is immune tolerance induction. Immune tolerance induction involves exposure to frequent and higher doses of recombinant factor until the body can tolerate the factor. While this treatment can be effective, the treatment burden is high as it can take months or even years for the inhibitor antibodies to be removed.
Our Marketed Products
Our marketed products, ELOCTATE and ALPROLIX, leverage expertise in Fc fusion technology that was originally acquired by Biogen from Syntonix Pharmaceuticals (Syntonix, and now known as Bioverativ Therapeutics Inc., our wholly owned subsidiary) in 2007. Fc fusion is a proprietary technology used to link recombinant factor VIII and factor IX in the case of ELOCTATE and ALPROLIX, respectively, to a protein fragment in the body known as Fc. The fusion with Fc uses a naturally occurring pathway and is designed to extend the half‑life of the factor, thereby making the product last longer in a patient’s blood than traditional factor therapies. ELOCTATE consists of the Coagulation Factor VIII molecule (historically known as Antihemophilic Factor) linked to Fc and ALPROLIX consists of the Coagulation Factor IX molecule linked to Fc.
We collaborate with Sobi to develop and commercialize ELOCTATE and ALPROLIX globally. Under the collaboration, we have rights to commercialize ELOCTATE and ALPROLIX in the United States, Japan, Canada, Australia, Latin American countries and all other markets excluding Sobi’s commercialization territory. Sobi’s commercialization territory includes Europe, Russia and certain countries in Northern Africa and the Middle East. ELOCTATE and ALPROLIX were approved in the United States, Canada and Japan in 2014, and in the European Union in 2015 and 2016, respectively. For a further description of our development and collaboration agreement with Sobi, see “—Our Development and Commercialization Arrangements with Sobi” below.
|
Product |
|
General Description |
|
|
|
ELOCTATE is approved in the United States, Japan, Canada, Australia, Brazil, Colombia, the European Union and certain other countries for the treatment of adults and children with hemophilia A to control and prevent bleeding episodes. In the United States, it is indicated for use in adults and children with hemophilia A for on‑demand treatment and control of bleeding episodes, perioperative management of bleeding and routine prophylaxis to reduce the frequency of bleeding episodes. ELOCTATE has received orphan designation in the United States. |

8
|
|
|
ALPROLIX is approved in the United States, Japan, Canada, Australia, Brazil, Colombia, the European Union and certain other countries for the treatment of adults and children with hemophilia B to control and prevent bleeding episodes. In the United States, it is indicated for use in adults and children with hemophilia B for control and prevention of bleeding episodes, perioperative management and routine prophylaxis to reduce the frequency of bleeding episodes. ALPROLIX has received orphan designation in the United States and the European Union.
|

Product sales for ELOCTATE and ALPROLIX each accounted for approximately 62% and 31%, respectively, of our total revenue for the year ended December 31, 2017, 58% and 38%, respectively of our total revenue for the year ended December 31, 2016, and approximately 57% and 42%, respectively of our total revenue for the year ended December 31, 2015. For additional financial information about our product and other revenues, long lived assets and geographic areas in which we operate, please read Note 19, Segment Information to our audited consolidated financial statements, Item 6. Selected Financial Data and Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations included in this report. A discussion of the risks attendant to our global operations is set forth in this report under Item 1A. Risk Factors.
Our research activities relating to ELOCTATE and ALPROLIX include ongoing and planned post‑marketing studies exploring the potential impact of the Fc fusion technology on long‑term joint health, immunogenicity and immune tolerance induction in hemophilia patients who develop inhibitors.
Research and Development Activities
We are engaged in discovery, preclinical, and clinical programs focused on advancing new technologies for the treatment of hemophilia and other blood disorders, such as cold agglutinin disease, sickle cell disease, and beta‑thalassemia. Our scientific and medical leaders are focused on advancing large and small molecules and cell and gene therapy in the hopes of developing treatments for these diseases.
A brief description of certain of our research and development programs, together with certain related business relationships and collaborations, is described below.
BIVV009
We have initiated site start-up for two parallel BIVV009 Phase 3 clinical trials for primary cold agglutinin disease, CARDINAL and CADENZA. BIVV009 is designed to selectively target C1s, the proximal initiator of the classical complement cascade, while leaving other complement cascade pathways intact.
Cold agglutinin disease is a global, chronic rare blood disease characterized by hemolytic anemia, with a significant portion of patients requiring transfusions and having crippling fatigue, poor quality of life and the potential for life-threatening complications such as thrombotic events, including pulmonary embolism and stroke. People living with cold agglutinin disease currently have no approved treatment options, and risk chronic iron overload from frequent blood transfusions to normalize hemoglobin levels. In addition, natural history data we published at the 2017 Annual Meeting for the American Society of Hematology (ASH) show a statistically significant increase in risk of thromboembolic events, like stroke, for people living with cold agglutinin disease. According to Berensten et al 2006, the diagnosed prevalence is approximately 16 people per million globally, leading to an estimated 10,000 people in the United States and Europe.
Cold agglutinin disease is caused by auto-immune mediated destruction of red blood cells. It is triggered by IgM auto-antibodies that bind to the surface of red blood cells and activate the classical complement pathway, leading to
9
red blood cell destruction. The classical complement pathway is a normal component of the innate immune system. However, when dysregulated, it is associated with multiple disease processes, including cold agglutinin disease.
We also have an ongoing Phase 1b clinical trial of BIVV009 for refractory immune thrombocytopenia.
Hemophilia Programs
BIVV001 (rFVIIIFc‑VWF‑XTEN). A Phase 1/2a clinical program of the combination of recombinant factor VIII‑Fc fusion protein with part of Von Willebrand factor, another component of the clotting cascade, attached with proprietary XTEN technology. The product candidate is being developed with the objective of achieving once weekly or less frequent dosing by intravenous administration in patients with hemophilia A.
BIVV002 (rFIXFc‑XTEN). A preclinical program for a next generation recombinant factor IX replacement product using XTEN technology exploring the use of subcutaneous dosing for patients with hemophilia B with the objective of achieving once weekly or less frequent dosing, which we believe would simplify the administration process for patients with hemophilia B.
Gene Therapy Programs. We are collaborating with Fondazione Telethon and Ospedale San Raffaele S.r.l. in preclinical programs to develop gene therapies for hemophilia A and B. This collaboration centers on advanced lentiviral gene transfer technology of the San Raffaele Telethon Institute for Gene Therapy.
Bi‑specific Antibody Program. A preclinical program to develop a non‑factor bi‑specific antibody for the treatment of patients with hemophilia A with inhibitors and the general hemophilia A population.
Beta Thalassemia and Sickle Cell Disease
ST-400. A Phase 1/2 clinical trial is planned to assess the safety, tolerability and efficacy of ST-400, a gene-edited cell therapy candidate, in adults with transfusion-dependent beta-thalassemia. This program is being developed pursuant to an exclusive worldwide research, development and commercialization collaboration and license agreement with Sangamo under which both companies are working to develop and commercialize product candidates for the treatment of sickle cell disease and beta‑thalassemia. For more information on our collaboration with Sangamo, refer to Item 7. Management’s Discussion and Analysis and of Financial Condition and Results of Operations—Contractual Obligations—Funding Commitments—Sangamo Therapeutics, Inc.
We are also building a clinical-stage pipeline in sickle cell disease by developing programs that intersect at all points of the biology, and these include a gene-edited cell therapy program and multiple small molecule programs.
Our expenses for research and development activities were $224.6 million in 2017, $210.1 million in 2016, and $236.9 million in 2015. For the periods prior to separation, these expenses include costs associated with research and development activities performed while part of Biogen. These expenses include allocations from Biogen to us for depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. As a result, these expenses are not necessarily indicative of Bioverativ’s expenses for research and development activities as a standalone company.
Investment in research and development is critical to our future growth and our ability to remain competitive in the markets in which we participate. We intend to continue to make significant investment in research and development programs in addition to seeking to enhance future growth through internal efforts, acquisitions and collaborations with third parties.
Our Development and Commercialization Arrangements with Sobi
Bioverativ Therapeutics Inc., our wholly owned subsidiary, is a party to a development and commercialization agreement with Sobi. Under this agreement, the parties develop and commercialize in defined territories ELOCTATE, ALPROLIX and certain compound constructs that Sobi elects to designate as subject to the parties’ collaboration.
10
Generally, these compound constructs include fusion proteins containing both a recombinant factor and the Fc portion of an immunoglobulin, including certain constructs that may be developed using technology we obtained from Amunix.
The agreement generally defines Bioverativ’s commercialization territory as the United States, Japan, Canada, Australia, Latin American countries and all other markets excluding Sobi’s commercialization territory, and Sobi’s commercialization territory as Europe, Russia and certain countries in Northern Africa and the Middle East.
Under the agreement, prior to May 5, 2024, either Bioverativ or Sobi may present a compound construct as a potential product candidate that the parties may consider developing and commercializing under the collaboration. Upon Sobi’s election to treat a compound construct as a product, and in the case of a novel compound construct Sobi’s payment of an upfront fee to us, Sobi is granted the right to opt‑in to such compound construct and become responsible for final development and commercialization of that compound construct in Sobi’s commercialization territory. Generally, upon opt‑in, Sobi becomes obligated to make an advance payment and reimburse Bioverativ for certain development expenses incurred with respect to the compound construct. Until Sobi’s portion of the development expenses are fully paid, Sobi’s royalty rate payable to Bioverativ is increased, and the royalty payment payable by Bioverativ to Sobi for the sale of products in Bioverativ’s territory is decreased.
In 2016, Sobi assumed final development and commercialization activities and became the marketing authorization holder for ELOCTA and ALPROLIX in the European Union. Sobi has also elected to treat BIVV001 (rFVIIIFc-VWF-XTEN) and BIVV002 (rFIXFc‑XTEN), each compound constructs developed using the XTEN technology we obtained from Amunix, as subject to the collaboration.
The agreement provides for royalty payments between the parties for sales of collaboration products, including ELOCTATE and ALPROLIX, that vary based upon, among other things, the territory in which the sale was made and how the product is commercialized.
The agreement is terminable in its entirety or with respect to a product developed under the collaboration by either party upon six months’ written notice. The agreement is also terminable in its entirety under certain conditions and subject to certain dispute resolution procedures following a party’s uncured material breach of a material obligation of the agreement. Unless earlier terminated, the duration of the agreement continues with respect to each product, for so long as such product is being sold anywhere in the world.
Bioverativ and Sobi are also parties to ancillary agreements, including manufacturing and supply agreements for ELOCTATE and ALPROLIX pursuant to which Sobi forecasts, orders and purchases drug substance and drug product that is supplied to Sobi by Bioverativ.
For more information on our collaboration with Sobi, see Note 4, Collaborations, to the audited consolidated financial statements included elsewhere in this report.
Our International Operations
Outside of the United States, Japan remains a significant near term focus for growth of ELOCTATE and ALPROLIX. We have conducted research and development activities for hemophilia treatments in Japan since 2010. Through our dedicated Japanese sales force and marketing team, we have sold ELOCTATE and ALPROLIX in Japan since receipt of marketing approval in December 2014 and June 2014, respectively. For the years ended December 31, 2017 and 2016, we generated revenue of approximately $138.8 and $105.7 million, respectively, from our sales of ELOCTATE and ALPROLIX in Japan. In addition to Japan, we continue to make progress in other geographic areas, including Canada, Australia and countries in Latin America.
Intellectual Property
We rely on patents and other proprietary rights to develop, maintain and strengthen our competitive position. We own a number of patents and trademarks throughout the world and have entered into license arrangements relating to various third party patents and technologies.
11
Patents
Patents are important to obtaining and protecting exclusivity in our products and product candidates. We regularly seek patent protection in the U.S. and in selected countries outside of the U.S. for inventions originating from our research and development efforts. In addition, we license rights from others to various patents and patent applications.
U.S. patents, as well as most non‑U.S. patents, are generally effective for 20 years from the date the earliest application was filed; however, U.S. patents that issue on applications filed before June 8, 1995 may be effective until 17 years from the issue date, if that is later than the 20 year date. In some cases, the patent term may be extended to recapture a portion of the term lost during regulatory review of the claimed therapeutic, and in the case of the United States, also because of U.S. Patent and Trademark Office (USPTO) delays in prosecuting the application. Specifically, in the U.S., under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the Hatch‑Waxman Act, a patent that covers a U.S. Food and Drug Administration (FDA) approved drug may be eligible for patent term extension (for up to five years, but not beyond a total of 14 years from the date of product approval) as compensation for patent term lost during the FDA regulatory review process, but only one patent per approved drug product may be so extended. The duration and extension of the term of foreign patents varies, in accordance with local law.
Our patent portfolio includes issued patents and pending applications relating to our marketed products and our product pipeline. We hold patents for ELOCTATE and ALPROLIX that cover the composition of matter and methods of treatment of those therapies. Patents of primary importance to ELOCTATE and ALPROLIX have issued in the United States, Europe and Japan, and based on the applicable patent statutes and in the ordinary course, generally expire between 2024 and 2032. We also continue to pursue additional patents and patent term extensions in the United States and other territories covering various aspects of our products that may, if issued, extend exclusivity beyond the expiration of these patents.
The existence of patents does not guarantee our right to practice the patented technology or commercialize the patented product. Patents relating to biopharmaceutical and biotechnology products, compounds and processes, such as those that cover our existing compounds, products and processes and those that we will likely file in the future, do not always provide complete or adequate protection. Our patents may be invalidated earlier based on a competitor’s challenge in an applicable patent office or court proceeding.
Regulatory Exclusivity
In addition to patent protection, certain of our products are entitled to regulatory exclusivity which may consist of regulatory data protection and market protection. The expected expiration of this regulatory exclusivity in the United States and the European Union is set forth below:
|
|
|
|
|
Expected |
|
Product |
|
Territory |
|
Expiration |
|
ELOCTATE |
|
United States |
|
2026 |
|
ELOCTA(1) |
|
European Union |
|
2025 |
|
ALPROLIX |
|
United States |
|
2026 |
|
ALPROLIX(1) |
|
European Union |
|
2026(2) |
|
(1) |
Sobi has assumed responsibility for commercializing ELOCTA and ALPROLIX in Sobi’s commercialization territory pursuant to our development and commercialization agreement with Sobi. |
|
(2) |
This date has the potential to be extended by two years subject to EMA review and certification of activities conducted under our pediatric investigational plan. |
Regulatory data protection provides to the holder of a drug or biologic marketing authorization, for a set period of time, the exclusive use of the proprietary preclinical and clinical data that it created at significant cost and submitted
12
to the applicable regulatory authority to obtain approval of its product. After the applicable set period of time, third parties are then permitted to rely upon our data to file for approval of their abbreviated applications for, and to market (subject to any applicable market protection), their generic drugs and biosimilars referencing our data. Market protection provides to the holder of a drug or biologic marketing authorization the exclusive right to commercialize its product for a set period of time, thereby preventing the commercialization of another product containing the same active ingredient(s) during that period. Although the World Trade Organization’s agreement on trade‑related aspects of intellectual property rights requires signatory countries to provide regulatory exclusivity to innovative pharmaceutical products, implementation and enforcement varies widely from country to country. In the United States, biologics, such as ELOCTATE and ALPROLIX, are entitled to exclusivity under the Biologics Price Competition and Innovation Act, which was passed on March 23, 2010 as Title VII to the Patient Protection and Affordable Care Act (PPACA). PPACA provides a pathway for approval of biosimilars following the expiration of 12 years of exclusivity for the innovator biologic and a potential additional 180 day‑extension term for conducting pediatric studies. Under this framework, FDA cannot make a product approval effective for any biosimilar application until at least 12 years after the reference product’s date of first licensure. The PPACA also includes an extensive process for the innovator biologic and biosimilar manufacturer to litigate patent infringement, validity, and enforceability prior to the approval of the biosimilar. The PPACA does not, however, change the duration of patents granted on biologic products.
Japan also provides for market exclusivity through a re‑examination system, which prevents the entry of generics and biosimilars until the end of the re‑examination period (REP), which can be up to eight years from marketing approval. ELOCTATE and ALPROLIX are expected to have REPs ending in 2022.
Other Proprietary Rights
We also rely upon other forms of unpatented confidential information to remain competitive. We protect such information principally through confidentiality and non‑use agreements with our employees, consultants, outside scientific collaborators and scientists whose research we sponsor and other advisers. In the case of our employees, these agreements also provide, in compliance with relevant law, that inventions and other intellectual property conceived by such employees during their employment shall be our exclusive property.
Our trademarks are important to us and are generally covered by trademark applications or registrations in the USPTO and the patent or trademark offices of other countries. Trademark protection varies in accordance with local law, and continues in some countries as long as the trademark is used and in other countries as long as the trademark is registered. Trademark registrations generally are for fixed but renewable terms.
Litigation, interferences, oppositions, inter partes reviews or other proceedings are, have been and may in the future be necessary in some instances to determine the validity and scope of certain of our patents, regulatory exclusivities or other proprietary rights, and in other instances to determine the validity, scope or non‑infringement of certain patent rights claimed by others to be pertinent to the manufacture, use or sale of our products. We may also face challenges to our patents, regulatory exclusivities and other proprietary rights covering our products by manufacturers of generic drugs and biosimilars. A discussion of certain risks and uncertainties that may affect our patent position, regulatory exclusivities and other proprietary rights is set forth in Item 1A. Risk Factors.
Manufacturing and Facilities
ELOCTATE and ALPROLIX are currently manufactured at Biogen‑owned facilities located in North Carolina. The manufacturing process for bulk drug substance includes protein production, purification and viral clearance. Manufacture and supply of drug product, which includes fill finish, labeling and packaging, are provided primarily through third party contract manufacturing organizations.
In connection with our separation from Biogen, we entered into a manufacturing and supply agreement with Biogen for hemophilia products, pursuant to which Biogen manufactures and supplies, exclusively for us, drug substance, drug product and finished goods with respect to ELOCTATE and ALPROLIX, as well as for certain of our pipeline product candidates. Fill finish, label and packaging, distribution and logistics services for ELOCTATE and ALPROLIX drug product are being provided by Biogen directly or through third party contract manufacturing
13
organizations. Additionally, we have increased and continue to increase our level of direct contractual responsibility with other third party contract manufacturing organizations, logistics providers and distributors as we scale up our internal supply management capabilities.
Our properties include facilities which, in our opinion, are suitable and adequate for development and distribution of our products. For additional information regarding our properties, see Item 2. Properties.
Raw Materials
We rely on Biogen for all supplies and raw materials used in the production of ELOCTATE and ALPROLIX drug substance. We also rely on third party contract manufacturers and suppliers for the materials we require for our clinical trials. A discussion of certain risks and uncertainties that may affect the availability of sufficient quantities of supplies and raw materials is set forth in Item 1A. Risk Factors.
Sales, Marketing and Distribution
We have our own direct sales force. Our products are distributed to and through specialty pharmacies, hemophilia treatment centers, public and private hospitals and independent distributors. For the year ended December 31, 2017, ASD Specialty Healthcare, a specialty distributor, and CVS Health Corporation, a specialty pharmacy, individually represent 16% and 13%, respectively, of total revenues. Our sales, particularly to specialty pharmacies and hemophilia treatment centers, are subject to discounted pricing. See “—Regulatory Matters—Pricing and Reimbursement” below. We review our sales channels from time to time, and from time to time will make changes in our sales and distribution model as we believe necessary to best implement our business plan and strategies.
In the United States, third parties warehouse and ship our products through their distribution centers. These centers are generally stocked with adequate inventories to facilitate prompt customer service. Sales and distribution methods include frequent contact by sales and customer service representatives, automated communications via various electronic purchasing systems, circulation of catalogs and merchandising bulletins, direct‑mail campaigns, trade publication presence and advertising. Our Japanese sales and product distributions are made on a direct basis.
We use and expect to continue to use a variety of collaboration, distribution and other marketing arrangements with one or more third parties to commercialize our products in certain other markets outside of the United States. Under our development and commercialization arrangement with Sobi, for example, Sobi has assumed responsibility for commercializing ELOCTATE and ALPROLIX in its territory. See “—Our Development and Commercialization Arrangements with Sobi” above.
Competition
We face substantial competition from biotechnology, biopharmaceutical and other companies of all sizes, in the United States and other countries, as such competitors continue to expand their manufacturing capacity and sales and marketing channels related to our hemophilia products. Many of our competitors are working to develop products similar to those we are developing or those that we already market. Competition is primarily focused on cost‑effectiveness, price, service, product effectiveness and quality, patient convenience and technological innovation. The introduction of new products by competitors and changes in medical practices and procedures can impact our products. The principal sources of competition for Bioverativ’s marketed products globally are as follows:
|
· |
ELOCTATE: ELOCTATE competes with recombinant Factor VIII products including: |
|
· |
ADVATE® (Antihemophilic Factor (Recombinant))—Shire |
|
· |
ADYNOVATE (Antihemophilic Factor (Recombinant), PEGylated)—Shire |
|
· |
AFSTYLA ® [Antihemophilic Factor (Recombinant) Single Chain]—CSL Behring |
|
· |
HELIXATE® FS (Antihemophilic Factor (Recombinant))—CSL Behring |
|
· |
HEMLIBRA® (emicizumab-kxwh) (for inhibitor patients)—Genentech |
14
|
· |
KOGENATE® FS (Antihemophilic Factor (Recombinant))—Bayer |
|
· |
KOVALTRY® Antihemophilic Factor (Recombinant)—Bayer |
|
· |
NovoEight® (Antihemophilic Factor (Recombinant))—Novo Nordisk |
|
· |
Nuwiq® Recombinant Factor VIII—Octapharma |
|
· |
RECOMBINATE™ (Antihemophilic Factor (Recombinant))—Shire |
|
· |
XYNTHA®/ReFacto AF® (Antihemophilic Factor (Recombinant), Plasma/Albumin‑Free)—Pfizer and Sobi |
|
· |
ALPROLIX: competes with recombinant Factor IX products including: |
|
· |
BENEFIX® (Coagulation Factor IX (Recombinant))—Pfizer |
|
· |
IDELVION® (Coagulation Factor IX (Recombinant), Albumin Fusion Protein)—CSL Behring |
|
· |
IXINITY® (Coagulation Factor IX (Recombinant))—Aptevo |
|
· |
REBINYN® (Coagulation Factor IX (Recombinant), GlycoPEGylated)—Novo Nordisk |
|
· |
RIXUBIS® (Coagulation Factor IX (Recombinant))—Shire |
Our products also compete with a number of plasma derived Factor VIII and IX products. We are also aware of other longer‑acting products and non-factor technologies, such as bi-specific antibodies and gene therapies that are in development and, if successfully developed and approved, would compete with our hemophilia products. New therapies and technologies have the potential to transform the standard of care for hemophilia patients, and our products may be unable to compete successfully with such new therapies and technologies that may be developed and marketed by other companies.
There are additional competitive products or alternative therapy regimens available on a more limited geographic basis throughout the world. For additional information regarding competition, see the discussion of such matters in Item 1A. Risk Factors, including the following: “Risk Factors—Risks Related to Our Business—If our hemophilia products fail to compete effectively, our business and market position would suffer.”
Regulatory Matters
Our operations and products are subject to extensive regulation by numerous government agencies, both within and outside of the United States. The FDA, the EMA, the Ministry of Health, Labour and Welfare in Japan (the MHLW) and other government agencies both inside and outside of the United States, regulate the testing, safety, effectiveness, manufacturing, labeling, promotion and advertising, distribution and post‑market surveillance of our products. We must obtain specific approval from the FDA and non‑U.S. regulatory authorities before we can market and sell our products in a particular country. The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country.
Clinical Trial and Approval Process
The FDA, the EMA and other regulatory agencies promulgate regulations and standards for designing, conducting, monitoring, auditing and reporting the results of clinical trials to ensure that the data and results are accurate and that the rights and welfare of trial participants are adequately protected commonly referred to as current good clinical practices (cGCPs). Regulatory agencies enforce cGCPs through inspections of trial sponsors, principal investigators and trial sites, contract research organizations (CROs), and institutional review boards. If studies fail to comply with applicable cGCPs, the clinical data generated may be deemed unreliable and relevant regulatory agencies may conduct additional audits or require additional clinical trials before approving a marketing application. Noncompliance can also result in civil or criminal sanctions. We rely on third parties, including CROs, to carry out many of our clinical trial‑related activities. Failure of such third parties to comply with cGCPs can likewise result in rejection of our clinical trial data or other sanctions.
Before new products may be sold in the United States, preclinical studies and clinical trials of the products must be conducted and the results submitted to the FDA for approval. With limited exceptions, the FDA requires companies
15
to register both pre‑approval and post‑approval clinical trials and disclose clinical trial results in public databases. Failure to register a trial or disclose study results within the required time periods could result in penalties, including civil monetary penalties. To support marketing approval, clinical trial programs must establish a product candidate’s efficacy, determine an appropriate dose and dosing regimen and define the conditions for safe use. This is a high‑risk process that requires stepwise clinical studies, usually conducted in three phases, in which the candidate product must successfully meet predetermined endpoints. The results of the preclinical and clinical testing of a product are then submitted to the FDA in the form of a marketing application. In response to a marketing application, the FDA may grant marketing approval, request additional information or deny the application if it determines the application does not provide an adequate basis for approval.
Product development and receipt of regulatory approval takes a number of years, involves the expenditure of substantial resources and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments, potential safety signals observed in preclinical or clinical tests and the risks and benefits of the product as demonstrated in clinical trials. Many research and development programs do not result in the commercialization of a product. The FDA has substantial discretion in the product approval process, and it is impossible to predict with any certainty whether and when the FDA will grant marketing approval, or whether an approval, if granted, will be subject to limitations based on the FDA’s interpretation of the relevant pre‑clinical or clinical data. The agency also may require the sponsor of a marketing application to conduct additional clinical studies or to provide other scientific or technical information about the product, and these additional requirements may lead to unanticipated delay or expense.
Most non‑U.S. jurisdictions have product approval and post‑approval regulatory processes that are similar in principle to those in the United States. In Europe, for example, there are several tracks for marketing approval, depending on the type of product for which approval is sought. Under the centralized procedure in Europe, a company submits a single application to the EMA that is similar to the marketing application in the United States. A marketing application approved by the EC is valid in all member states. In addition to the centralized procedure, Europe also has various other methods for submitting applications and receiving approvals. Regardless of the approval process employed, various parties share responsibilities for the monitoring, detection and evaluation of adverse events post‑approval, including national authorities, the EMA, the EC and the marketing authorization holder. In some regions, it is possible to receive an “accelerated” review whereby the national regulatory authority will commit to truncated review timelines for products that meet specific medical needs.
Under the U.S. Orphan Drug Act, the FDA may grant orphan drug designation to biologics intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than 200,000 individuals in the United States. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity. This means that the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years following marketing approval, except in certain very limited circumstances, such as if the later product is shown to be clinically superior to the orphan product. Legislation similar to the U.S. Orphan Drug Act has been enacted in other countries to encourage the research, development and marketing of medicines to treat, prevent or diagnose rare diseases. In the European Union, medicinal products intended for diagnosis, prevention or treatment of life‑threatening or very serious diseases affecting less than five in 10,000 people receive 10‑year market exclusivity, protocol assistance, and access to the centralized procedure for marketing authorization. ELOCTATE and ALPROLIX have each received an orphan drug designation in the United States and have received orphan exclusivity through June 2021 and March 2021, respectively. ALPROLIX has also received an orphan drug designation in the European Union.
Biologic products may be subject to increased competition from biosimilar formulations of reference biologic products in the future. The complex nature of biologic products has warranted the creation of biosimilar regulatory approval pathways with strict, science‑based approval standards that take into account patient safety considerations. These biosimilar approval pathways are considered to be more abbreviated than for new biologics, although they are significantly different from the abbreviated approval pathways available for “generic drugs” (small‑molecule drugs that are the same as, and bioequivalent to, an already‑approved small molecule drug). The European Union has created a pathway for the approval of biosimilars, and has published guidance for approval of certain biosimilar products. More recently, in 2010, the PPACA authorized the FDA to approve biosimilars, but only a small number of biosimilars have
16
been approved by the FDA to date and the U.S. approval pathway for biosimilars remains subject to ongoing guidance from the FDA. While mature pathways for regulatory approval of generic drugs and healthcare systems exist around the globe that support and promote the substitutability of generic drugs, the approval pathways for biosimilar products remain in various stages of development, as do private and public initiatives or actions supporting the substitutability of biosimilar products. Thus, the extent to which biosimilars will be viewed as readily substitutable, and in practice readily substituted, for the reference biologic product is largely yet to be determined.
Post‑Approval Requirements
The FDA may require a sponsor to conduct additional post‑marketing studies as a condition of approval to provide data on safety and effectiveness. If a sponsor fails to conduct the required studies, the agency may withdraw its approval. In addition, if the FDA concludes that a product that has been shown to be effective can be safely used only if distribution or use is restricted, it can mandate post‑marketing restrictions as necessary to assure safe use. These may include requiring the sponsor to establish rigorous systems, such as risk evaluation and mitigation strategies (REMS), to assure use of the product under safe conditions. The FDA can impose financial penalties for failing to comply with certain post‑marketing commitments, including REMS. In addition, any changes to approved REMS must be reviewed and approved by the FDA prior to implementation.
Changes to approved products, such as adding an indication, making certain manufacturing changes, or changing manufacturers or suppliers of certain ingredients or components, may be subject to rigorous review, including multiple regulatory submissions, and approvals are not certain. For example, to obtain a new indication, a company must demonstrate with additional clinical data that the product is safe and effective for the new use. FDA regulatory review may result in denial or modification of the planned changes, or requirements to conduct additional tests or evaluations that can substantially delay or increase the cost of the planned changes.
Even after a company obtains regulatory approval to market a product, the product and the company’s manufacturing processes and quality systems are subject to continued review by the FDA and other regulatory authorities globally. We and our contract manufacturers also must adhere to current good manufacturing practices (cGMPs) and product‑specific regulations enforced by regulatory agencies both before and after product approval. Regulatory agencies regulate and inspect equipment, facilities and processes used in the manufacturing and testing of products prior to approving a product, as well as periodically following the initial approval of a product. If, as a result of these inspections, it is determined that our equipment, facilities or processes or that of our manufacturers do not comply with applicable regulations and conditions of product approval, we may face civil, criminal or administrative sanctions or remedies, including significant financial penalties and the suspension of our manufacturing operations.
Manufacturers are also required to monitor information on side effects and adverse events reported during clinical studies and after marketing approval and report such information and events to regulatory agencies. Non‑compliance with the FDA’s safety reporting requirements may result in civil or criminal penalties. Side effects or adverse events that are reported during clinical trials can delay, impede or prevent marketing approval. Based on new safety information that emerges after approval, the FDA can mandate product labeling changes, impose a new REMS or the addition of elements to an existing REMS, require new post‑marketing studies (including additional clinical trials) or suspend or withdraw approval of the product. These requirements may affect a company’s ability to maintain marketing approval of its products or require a company to make significant expenditures to obtain or maintain such approvals.
Pricing and Reimbursement
In both U.S. and non‑U.S. markets, sales of our products depend, in part, on the availability and amount of reimbursement by third party payors, including governments, private health plans and other organizations. Substantial uncertainty exists regarding the coverage, pricing and reimbursement of our products. Governments may regulate coverage, reimbursement and pricing of our products to control healthcare cost or affect utilization of the products. The U.S. and non‑U.S. governments have enacted and regularly consider additional reform measures that affect health care and drug coverage and costs. Private health plans may also seek to manage cost and utilization by implementing coverage and reimbursement limitations. Other payors, including managed care organizations, health insurers, pharmacy benefit managers, government health administration authorities and private health insurers, seek price discounts or
17
rebates in connection with the placement of our products on their formularies and, in some cases, the imposition of restrictions on access or coverage of particular drugs or pricing determined based on perceived value.
Within the United States
|
· |
Medicaid: Medicaid is a joint federal and state program that is administered by the states for low‑income and disabled beneficiaries. Under the Medicaid Drug Rebate Program, we are required to pay a rebate for each unit of product reimbursed by the state Medicaid programs. For most brand name drugs, the amount of the basic rebate for each product is set by law as 17.1% for clotting factors and certain other products of the average manufacturer price (AMP) or the difference between AMP and the best price available from us to any customer (with limited exceptions). The rebate amount must be adjusted upward if AMP increases more than inflation (measured by the Consumer Price Index—Urban) from launch of the product. This adjustment can cause the total rebate amount to exceed the minimum 17.1% basic rebate amount. The rebate amount is calculated each quarter based on our report of current AMP and best price for each of our products to the Centers for Medicare & Medicaid Services (CMS). The requirements for calculating AMP and best price are complex. We are required to report any revisions to AMP or best price previously reported within a certain period, which revisions could affect our rebate liability for prior quarters. In addition, if we fail to provide information timely or are found to have knowingly submitted false information to the government, the statute governing the Medicaid Drug Rebate Program provides for civil monetary penalties. |
|
· |
Medicare: Medicare is a federal program that is administered by the federal government and covers individuals age 65 and over, as well as those with certain disabilities. Medicare Part B generally covers drugs that must be administered by physicians or other health care practitioners; are provided in connection with certain durable medical equipment; or certain oral anti‑cancer drugs and certain oral immunosuppressive drugs. Clotting factors for hemophilia are typically paid under Medicare Part B. Medicare Part B pays for such drugs under a payment methodology based on the average sales price (ASP) of the drugs. Manufacturers, including us, are required to provide ASP information to the CMS on a quarterly basis. The manufacturer‑submitted information is used to calculate Medicare payment rates. For drugs administered outside the hospital outpatient setting, the current payment rate for Medicare Part B drugs is ASP plus six percent (4.3% after sequestration). The payment rates for drugs in the hospital outpatient setting are subject to periodic adjustment. If a manufacturer is found to have made a misrepresentation in the reporting of ASP, the governing statute provides for civil monetary penalties. |
Medicare Part D provides coverage to enrolled Medicare patients for self‑administered drugs (i.e., drugs that are not administered by a physician). Medicare Part D is administered by private prescription drug plans approved by the U.S. government and each drug plan establishes its own Medicare Part D formulary for prescription drug coverage and pricing, which the drug plan may modify from time‑to‑time. The prescription drug plans negotiate pricing with manufacturers and may condition formulary placement on the availability of manufacturer discounts. In addition, manufacturers, including us, are required to provide to CMS a 50% discount on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries reach the coverage gap in their drug benefits.
|
· |
Federal Agency Discounted Pricing: Our products are subject to discounted pricing when purchased by federal agencies via the Federal Supply Schedule (FSS). FSS participation is required for our products to be covered and reimbursed by the Veterans Administration (VA), Department of Defense, Coast Guard and the U.S. Public Health Service (PHS). Coverage under Medicaid, Medicare and the PHS pharmaceutical pricing program is also conditioned upon FSS participation. FSS pricing is intended not to exceed the price that we charge our most‑favored non‑federal customer for a product. In addition, prices for drugs purchased by the VA, Department of Defense (including drugs purchased by military personnel and dependents through the TriCare retail pharmacy program), Coast Guard and PHS are subject to a cap on pricing equal to 76% of the non‑federal average manufacturer price (non‑FAMP). An additional discount applies if non‑FAMP increases more than inflation (measured by the Consumer Price Index—Urban). In addition, if we fail to provide information timely or are found to have knowingly submitted false information to the government, the governing statute provides for civil monetary penalties. |
18
|
· |
340B Discounted Pricing: To maintain coverage of our products under the Medicaid Drug Rebate Program and Medicare Part B, we are required to extend significant discounts to certain covered entities that purchase products under Section 340B of the PHS pharmaceutical pricing program. Purchasers eligible for discounts include hospitals that serve a disproportionate share of financially needy patients, community health clinics, hemophilia treatment centers and other entities that receive certain types of grants under the Public Health Service Act. For all of our products, we must agree to charge a price that will not exceed the amount determined under statute (the “ceiling price”) when we sell outpatient drugs to these covered entities. In addition, we may, but are not required to, offer these covered entities a price lower than the 340B ceiling price. The 340B discount formula is based on AMP and is generally similar to the level of rebates calculated under the Medicaid Drug Rebate Program. |
Outside of the United States
Outside of the United States, products are paid for by a variety of payors, with governments being the primary source of payment. Governments may determine or influence reimbursement of products. Governments may also set prices or otherwise regulate pricing. Negotiating prices with governmental authorities can delay commercialization of our products. Governments may use a variety of cost‑containment measures to control the cost of products, including price cuts, mandatory rebates, “value‑based pricing” and reference pricing (i.e., referencing prices in other countries and using those reference prices to set a price). Budgetary pressures in many countries, including in the European Union, are continuing to cause governments to consider or implement various cost‑containment measures, such as price freezes, increased price cuts and rebates and expanded generic substitution and patient cost‑sharing.
Japanese Regulatory Matters
In Japan, the MHLW is responsible for regulating biological and pharmaceutical products under the Pharmaceuticals and Medical Devices Law, which provides a regulatory framework similar to that of the United States. Specifically, with regard to the clinical trial and approval process, before a new biological product may be sold in Japan, clinical trials must be conducted for the product of which the MHLW and the Pharmaceuticals and Medical Devices Agency (PMDA), a governmental organization authorized by the MHLW, must be notified. For the product to be approved, the results of such clinical trials must then be submitted to the PMDA. Approved products are subject to regulatory requirements similar to those of the United States, including (i) the possibility of the MHLW requiring post‑marketing studies to gather data on a product’s safety and efficacy as a condition for approval, (ii) re‑examination of the approved product within a specific time period following approval (e.g., 8 years for a new product) to verify its safety and efficacy and (iii) the reporting of any adverse event to the PMDA. With regard to pricing and reimbursement, Japan has a single health insurance system (National Health Insurance), under which drugs are provided to patients at a price designated by the MHLW in its discretion after negotiations between the applicable biopharmaceutical company and the MHLW under the National Health Insurance Act.
Other Laws
We and our products are also subject to various other regulatory regimes both inside and outside of the United States. Various laws, regulations and recommendations relating to data privacy and protection, safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import, export and use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research work are or may be applicable to our activities. In the United States alone, we are subject to the oversight of the FDA, the Office of the Inspector General within the Department of Health and Human Services, the CMS, the Department of Justice, the Environmental Protection Agency, the Department of Defense and Customs and Border Protection, in addition to others. In jurisdictions outside of the United States, our activities are subject to regulation by government agencies including the EMA in Europe, and other agencies in other jurisdictions. Many of the agencies enforcing these laws have increased their enforcement activities with respect to healthcare companies in recent years. These actions appear to be part of a general trend toward increased enforcement activity globally. In addition, certain agreements entered into by us involving exclusive license rights may be subject to national or international antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
19
Patient Engagement, Patient Access and Humanitarian Aid
We interact with patients, advocacy organizations and healthcare societies in order to gain insights into unmet needs, particularly in the hemophilia treatment community. The insights gained from these engagements help develop services, programs and applications that are designed to help patients lead better lives.
We are dedicated to helping patients obtain access to our therapies. For example, we provide charitable contributions that may assist eligible patients to receive our products. We have also continued our commitment, together with Sobi, to donate up to one billion international units (IUs) of clotting factor therapy for humanitarian use over a ten-year period, of which up to 500 million IUs will be donated to the WFH over a period of five years. We are responsible for half of the committed donation. Through the end of 2017, we have donated over 262 million of IUs, treating over 15,000 people in the developing world.
Employees
We employ approximately 460 persons as of December 31, 2017. We believe that we have good relations with our employees.
Environmental Matters
Our environmental policies require compliance with all applicable environmental regulations and contemplate, among other things, appropriate capital expenditures for environmental protection. While it is impossible to predict accurately the future costs associated with environmental compliance and potential remediation activities, compliance with environmental laws is not expected to require significant capital expenditures and has not had, and is not expected to have, a material adverse effect on our operations or competitive position.
Available Information
Bioverativ’s principal executive offices are 225 Second Avenue, Waltham, MA 02451, and our telephone number is 781‑663‑4400. Our website address is www.bioverativ.com. We make available free of charge through the Investors section of our website our Annual Reports on Form 10‑K, Quarterly Reports on Form 10‑Q, Current Reports on Form 8‑K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. We include our website address in this report only as an inactive textual reference and do not intend it to be an active link to our website. The contents of our website are not incorporated into this report.
You should carefully consider the following risks and other information in this report in evaluating Bioverativ and Bioverativ’s common stock. Any of the following risks could materially and adversely affect our results of operations or financial condition and could adversely impact, or result in volatility to, our stock price.
Risks Related to the Merger
The conditions under the Merger Agreement to Sanofi’s consummation of the Offer and the subsequent Merger may not be satisfied at all or in the anticipated timeframe.
On January 21, 2018, Bioverativ entered into the Merger Agreement with Sanofi. The obligation of Merger Sub to purchase Shares tendered in the Offer is subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, including (i) that the number of Shares validly tendered and not withdrawn as of the expiration of the Offer, together with any Shares (if any) owned by Parent and its affiliates and excluding any Shares tendered pursuant to guaranteed delivery procedures that have not yet been “received” (as such term is defined in Section 251(h)(6)(f) of the DGCL), represents at least a majority of the then outstanding Shares (the “Minimum Tender Condition”); (ii) (x) the expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended and (y) that all consents of, and/or filings with, any governmental authority of
20
competent jurisdiction or pursuant to any antitrust laws have been obtained and are in full force and effect and any applicable waiting period with respect thereto has expired; (iii) the absence of any law or order prohibiting the consummation of the Offer or the Merger; (iv) the accuracy of the Company’s representations and warranties under the Merger Agreement (subject to customary materiality qualifiers); (v) the Company’s performance in all material respects of its obligations under the Merger Agreement; (vi) the absence, since the date of the Merger Agreement, of any fact, circumstance, condition, event, change, development, occurrence, result or event which, individually or in the aggregate, has had, or would reasonably be expected to have, a Company Material Adverse Effect (as defined in the Merger Agreement); (vii) the delivery to Parent and Merger Sub of a certificate, dated as of the expiration time of the Offer, certifying that the conditions in clauses (iv), (v) and (vi) have been satisfied; (viii) the Merger Agreement has not been terminated; and (ix) that the Company has received and delivered to Biogen and Parent a copy of the tax opinion required by a letter agreement dated January 21, 2018 between Biogen, Parent and the Company (the “Letter Agreement”) and the requirement that the Company deliver such tax opinion pursuant to the Tax Matters Agreement between Biogen and the Company dated January 31, 2017 has been satisfied pursuant to the Letter Agreement. These conditions are more specifically described in the Merger Agreement, which we filed as Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the SEC on January 22, 2018.
We intend to pursue all required approvals in accordance with the Merger Agreement. However, no assurance can be given that the required approvals will be obtained and, even if all such approvals are obtained, no assurance can be given as to the terms, conditions and timing of the approvals or that they will satisfy the terms of the Merger Agreement. In addition, there can be no assurances that our stockholders will tender their shares, or that any or all of the other conditions will be satisfied.
The announcement of the Offer and the Merger could negatively impact our business, financial condition, results of operations or our stock price.
Our announcement of having entered into the Merger Agreement and Sanofi and Merger Sub’s commencement of the Offer could cause a material disruption to our business and there can be no assurance that the conditions to the completion of the Offer and the Merger will be satisfied. The Merger Agreement may also be terminated by us or Sanofi in certain specified circumstances, including, subject to compliance with the terms of the Merger Agreement, by us in order to accept a third-party acquisition proposal that our board of directors determines constitutes a superior proposal upon payment of a termination fee to Sanofi of $326.0 million. We are subject to several risks as a result of the announcement of the Merger Agreement and the Offer, including, but not limited to, the following:
|
· |
Pursuant to the Merger Agreement, we are subject to certain restrictions on the conduct of our business prior to the consummation of the Merger, which restrictions could adversely affect our ability to realize certain of our business strategies or take advantage of certain business opportunities; |
|
· |
Third parties may determine to delay or defer purchase decisions with regard to our products or terminate and/or attempt to renegotiate their relationships with us as a result of the Offer and Merger, whether pursuant to the terms of their existing agreements with us or otherwise; |
|
· |
The attention of our management may be directed toward the consummation of the Offer, the Merger and related matters, and their focus may be diverted from the day-to-day business operations of our company, including from other opportunities that might otherwise be beneficial to us; |
|
· |
Current and prospective employees may experience uncertainty regarding their future roles with us (and, if the Offer is completed, Sanofi), which might adversely affect our ability to retain, recruit and motivate key personnel and may adversely affect the focus of our employees on development and sales of our products; and |
|
· |
Our inability to hire capable employees, given the uncertainty regarding the future of the company, in order to execute on our continuing business operations. |
|
· |
Any of the above matters could adversely affect our stock price or harm our financial condition, results of operations or business prospects. |
Any of the above matters could adversely affect our stock price or harm our financial condition, results of operations or business prospects.
21
If the proposed transactions are not consummated, our business and stock price could be adversely affected.
The consummation of the transactions contemplated by the Merger Agreement (the “Transactions”) is subject to the satisfaction or waiver of certain conditions set forth in the Merger Agreement. If the Offer and/or the Merger are delayed or otherwise not consummated, we could suffer a number of consequences that may adversely affect our business, results of operations and stock price, including the following:
|
· |
if the Offer and the Merger are not completed, the trading price of the Company Shares may change to the extent that the current trading price of the Company Shares reflects an assumption that the Offer and the Merger will be completed; |
|
· |
we have incurred and expect to continue to incur significant expenses related to the proposed Transactions. These transaction -related expenses include certain investment banking fees, legal, accounting and other professional fees. These fees must be paid even if the Offer is not consummated; |
|
· |
we could be obligated to pay (or cause to be paid) to Sanofi a $326.0 million termination fee in connection with the termination of the Merger Agreement in certain circumstances; |
|
· |
failure of the Offer and the Merger may result in negative publicity and/or a negative impression of us in our customers, prospective customers, the investment community or business community generally. The failure to consummate the Transactions may be viewed as a poor reflection on our business or prospects; |
|
· |
certain of our suppliers, customers, distributors, and other business, collaboration and strategic partners may seek to change or terminate their relationships with us as a result of the proposed Transactions; and |
|
· |
the market price of our common stock may decline, particularly to the extent that the current market price reflects a market assumption that the proposed Offer and Merger will be completed. |
Our executive officers and directors may have interests that are different from, or in addition to, those of our stockholders generally.
Our executive officers and directors may have interests in the Offer and Merger that are different from, or are in addition to, those of our stockholders generally. These interests include direct or indirect ownership of our common stock, stock options, and restricted stock units, and the receipt or potential receipt of change in control or other retention or severance payments in connection with the proposed transactions.
The Merger Agreement contains provisions that could make it difficult for a third party to acquire us prior to the completion of the Offer and the Merger.
The Merger Agreement contains restrictions on our ability to obtain a third-party proposal for an acquisition of our company. These provisions include our agreement not to initiate, solicit or knowingly encourage or knowingly facilitate any discussions with third parties regarding other proposals to acquire us, as well as restrictions on our ability to respond to such proposals, subject to fulfillment of certain fiduciary requirements of our board of directors. The Merger Agreement also contains certain termination rights, including, under certain circumstances, a requirement for us to pay (or cause to be paid) to Sanofi a termination fee of $326.0 million.
These provisions might discourage an otherwise-interested third party from considering or proposing an acquisition of our company, even one that may be deemed of greater value to our stockholders than the Offer. Furthermore, even if a third party elects to propose an acquisition, the concept of a termination fee may result in that third-party’s offering a lower value to our stockholders than such third party might otherwise have offered.
Risks Related to Our Business
We are dependent on revenues from our products, ELOCTATE and ALPROLIX. If we or Sobi are unable to successfully commercialize ELOCTATE or ALPROLIX, our results of operations would be materially harmed.
Net sales of ELOCTATE and ALPROLIX represent substantially all of our revenues, and this concentration makes us dependent on these two products. If we or our collaboration partner Sobi were to experience difficulty with the
22
commercialization of ELOCTATE or ALPROLIX, we could experience a significant reduction in revenue and may not be profitable.
The commercial success of ELOCTATE and ALPROLIX and our ability to generate revenues will depend on many factors, including the following:
|
· |
the effectiveness of our global commercial strategy for the marketing and sale of our products; |
|
· |
our ability to maintain our development and commercialization arrangements with Sobi; |
|
· |
our ability to demonstrate and differentiate the efficacy, safety and value of ELOCTATE and ALPROLIX; |
|
· |
our ability to maintain a positive reputation among hemophilia patients and in the hemophilia community; |
|
· |
the introduction and success of competing products and technologies; |
|
· |
our ability to obtain and maintain adequate pricing, coverage or reimbursement for our products; |
|
· |
our ability to manufacture and supply product without interruption; |
|
· |
the absence of adverse legal, administrative, regulatory or legislative developments; and |
|
· |
other factors described below in this “Risk Factors” section. |
Many of these factors are beyond our control, and success in any one of these factors will not guarantee success in any of the others. Accordingly, we cannot assure you that we will be able to continue to generate revenue through the sale of ELOCTATE or ALPROLIX.
If our hemophilia products fail to compete effectively, our business and market position would suffer.
Due to our dependence on sales of our hemophilia products, our business may be harmed if our products are unable to successfully compete in the hemophilia treatment market. The hemophilia treatment market is highly competitive. We compete in the marketing and sale of our products, and in the development of, and acquisition of rights to, new products and technologies.
We compete with biotechnology and biopharmaceutical companies that have greater financial, technological and other resources than we do. One or more of our competitors may benefit from significantly greater sales and marketing capabilities, may develop products that are accepted more widely than ours or may receive patent protection that dominates, blocks or adversely affects our product development or business.
Our ability to successfully compete with other hemophilia treatments may be adversely affected if our therapies are not regarded by patients, healthcare providers or payors as offering significant benefits and value as compared to other current treatments. If we fail to successfully educate the medical and scientific community about our products or if we fail to demonstrate that our products and technologies are differentiated from competing drugs, our business may be harmed.
A number of companies, including some mid-to-large biopharmaceutical companies, such as Alnylam Pharmaceuticals, Bayer AG, Novo Nordisk, CSL Ltd., Pfizer Inc., Roche Holding AG, Sanofi Genzyme and Shire Plc., currently market or are pursuing the development of products for the treatment of hemophilia. We are also aware of other extended half‑life factor products as well as other new technologies, such as gene therapies, bi‑specific antibodies and RNA interference technologies, that are in development and, if successfully developed and approved, would compete with ELOCTATE or ALPROLIX. New therapies and technologies have the potential to transform the standard of care for hemophilia patients, and our products may be unable to compete successfully with such new therapies and technologies that may be developed and marketed by other companies.
23
Our reliance on third parties for our manufacturing and distribution processes increases the risk that we will not have available sufficient quantities of ELOCTATE and ALPROLIX, or that such quantities may not be available at an acceptable cost, which could delay, prevent or impair our commercialization efforts and materially harm our business, results of operations and financial condition.
We rely, and expect to continue to rely, on third parties for the commercial manufacture and distribution of ELOCTATE and ALPROLIX. Biogen is currently our sole manufacturer and supplier of ELOCTATE and ALPROLIX. Biogen also relies on third parties with respect to certain aspects of its manufacturing process, including certain sole sources of raw materials. We also rely, and expect to continue to rely, on third parties to distribute our products, including global, regional, and specialty distribution and logistics providers.
Biogen and other third party providers are independent entities subject to their own unique operational and financial risks that are outside of our control. Any of these third parties may not perform their obligations in a timely and cost‑effective manner or in compliance with applicable regulations. A third party, for instance, may be unable or unwilling to increase production capacity commensurate with demand for our products. Finding alternative providers could take a significant amount of time and involve significant expense due to the specialized nature of these services.
We have entered into, and plan to enter into other agreements, with additional manufacturing partners. Establishing relationships with additional manufacturing partners could take a significant amount of time and involve significant expense. In the event we change manufacturing partners or the third parties providing raw materials, drug substance, drug product, fill finish, packaging, labeling and/or storage of our products, we may need to obtain approval from applicable regulatory authorities. Manufacturers are generally required to maintain compliance with current good manufacturing practices, or cGMPs, and other stringent requirements and are subject to inspections by the FDA and comparable agencies in other jurisdictions to confirm such compliance. These cGMP requirements and regulations are not prescriptive instructions on how to manufacture products, but rather a series of principles that must be observed during manufacturing; as a result, their implementation may not be clearly delineated and may present a challenging task as these regulatory requirements are complex, time‑consuming and expensive. Moreover, as our current products are biologics, they require processing steps that are more difficult than those required for most chemical pharmaceuticals. Any delay, interruption or other issues that arise in the manufacture, fill‑finish, packaging or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection could result in administrative sanctions by the FDA or other U.S. or non‑U.S. regulatory agencies. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions. We cannot be certain that we could reach agreement with alternative providers or that the FDA or other regulatory authorities would approve our use of alternative manufacturers or providers on a timely basis.
Any adverse developments affecting our supply chain may result in development delays, shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the commercial supply of our products and/or the timing of our clinical trials. In addition, loss or damage to a manufacturing facility or storage site due to a natural disaster or otherwise could adversely affect our ability to manufacture sufficient quantities of our products or to deliver products to meet customer demand or contractual requirements, any of which may result in a loss of revenue and other adverse business consequences. We may also have to take inventory write‑offs and incur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Such developments could increase our manufacturing or development costs, cause us to lose revenue or market share as patients and physicians turn to competing therapeutics, diminish our profitability or damage our reputation. Moreover, any failure of Biogen or other third party to supply ELOCTATE and ALPROLIX could cause us to breach our supply agreements to Sobi for these products, which may subject us to liability under those agreements and impair our relationship with Sobi.
Issues with product quality or safety, including the perception of such issues, could negatively affect our business, subject us to regulatory or other actions and cause a loss of confidence in us or our products.
Our success depends upon the quality and safety of our products. Even after a product is approved for marketing, new safety data may emerge from adverse event reports or post‑marketing studies. Previously unknown risks and adverse effects of our products may also be discovered in connection with unapproved or off‑label uses of our
24
products. Additionally, global regulatory authorities, at their discretion, may review and raise new or additional questions related to approved product information. This could result in changes to the approved product prescribing information or to the product approval itself.
A quality or safety issue, including the perception of such issues, may result in one or more adverse events such as:
|
· |
investigations by regulatory authorities; |
|
· |
refusal of a regulatory authority to grant approvals and licenses; |
|
· |
fines, suspension or withdrawal of existing regulatory approvals and licenses; |
|
· |
restrictions on operations, including injunctions to halt manufacture and distribution of products; |
|
· |
requirements for additional labeling or safety monitoring; |
|
· |
product liability; |
|
· |
adverse inspection reports, warning letters, product recalls or seizures; |
|
· |
monetary, civil or criminal fines, sanctions; and |
|
· |
costly litigation. |
An inability to address any of these issues in an effective and timely manner may cause negative publicity, loss of physician and patient confidence in the company or its current or future products and may negatively impact physicians’ decisions to prescribe our products. These issues could also result in liabilities, loss of revenue, material write‑offs of inventory, withdrawal or voluntary recall of our products from the marketplace, delays or limitations in regulatory approvals, material impairments of intangible assets, goodwill and fixed assets, material restructuring charges, difficulty in successfully launching new products and other adverse impacts on our results of operations.
Our inability to maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business and results of operations.
Sales of ELOCTATE and ALPROLIX are dependent, in large part, on the availability and extent of coverage, pricing and reimbursement from government health administration authorities, private health insurers and other organizations. When a new biopharmaceutical product is approved, the availability of government and private insurance coverage for that product may be uncertain, as may be the pricing of the product and extent to which the product will be reimbursed. Failure to maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business and results of operations.
Pricing and reimbursement for our products may be adversely affected by a number of factors, including:
|
· |
changes in federal, state or foreign government regulations or private third party payors’ reimbursement policies; |
|
· |
pressure by employers on private health insurance plans to reduce costs; |
|
· |
consolidation and increasing assertiveness of payors, including managed care organizations, health insurers, pharmacy benefit managers, government health administration authorities, private health insurers and other organizations seeking price discounts or rebates in connection with the placement of our products on their formularies and, in some cases, imposing restrictions on access to, coverage of or pricing for particular drugs based on perceived value; and |
|
· |
the influence of third party organizations advocating for discounted pricing with respect to our products. |
Our ability to set the price for our products can vary significantly from country to country and, as a result, so can the price of our products. For example, in some countries, pricing, coverage and level of reimbursement or funding of prescription drugs, are subject to governmental control, and we may be unable to timely or successfully negotiate coverage, pricing, and reimbursement on terms that are favorable to us. Further, we are subject to risks due to the tendering process required in certain countries, as well as the comparison of dose pricing of our products against other available treatments. If we are unable to demonstrate to healthcare providers and payors the value of our products, our products may not be accepted or we may not secure adequate prices in a particular country. Our inability to secure
25
adequate prices in a particular country may limit the marketing of our products within that country, and may also adversely affect our ability to obtain acceptable prices in other markets. This may create the opportunity for third party cross‑border trade or influence our decision to sell or not to sell a product in a particular country, thus adversely affecting our geographic expansion plans and revenues.
Pricing for therapies and other health care costs are under significant scrutiny in the markets in which our products are prescribed and continue to be subject to intense political and societal pressures, which we anticipate will continue and escalate. For example, there is significant discussion and proposed legislation in the U.S. designed to bring more transparency to drug pricing, review of relationships between pricing and manufacturer patient programs, and reform around government program reimbursement methodologies. We cannot predict the extent to which our business may be affected by these or other potential future legislative or regulatory developments. However, future price controls or other changes in pricing regulation or negative publicity related to our product pricing or the pricing of pharmaceutical drugs generally could restrict the amount that we are able to charge, which would adversely affect our revenue and results of operations.
If we are unable to obtain and maintain adequate protection for our intellectual property and other proprietary rights, or if we are unable to avoid violation of the intellectual property or proprietary rights of others, we may be subject to liability, the operation of our business may be interrupted or our business or prospects may be otherwise harmed.
Our commercial success depends in part on our ability to obtain and defend patent and other intellectual property rights that are important to the development, manufacture and commercialization of our products and product candidates. The degree of patent protection that will be afforded to our products and processes in the United States and in other important non‑U.S. markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts and lawmakers in those countries. We can provide no assurance that we will successfully obtain or preserve patent protection for the technologies incorporated into our products and processes, or that the scope of patent protection obtained will be sufficient to protect our commercial interests in all countries where we conduct business. If we cannot prevent others from exploiting our inventions, we will not derive the benefit from them that we currently expect.
We rely on third party licenses for certain aspects of our products and product candidates, including for the treatment of hemophilia and cold agglutinin disease. If an agreement with a third party was to be terminated or if we otherwise lost our rights to such technology, our ability to develop, manufacture and commercialize products and product candidates could be adversely affected, and could materially harm our business prospects.
We also rely on regulatory exclusivity for protection of our products. Implementation and enforcement of regulatory exclusivity varies widely from country to country. Failure to qualify for regulatory exclusivity, or failure to obtain or maintain the extent or duration of such protections that we expect for our products in each of these markets due to challenges, changes of interpretations in the law or otherwise, could affect the revenue for our products, our decision on whether to market our products in a particular country or countries or could otherwise have an adverse impact on our results of operations.
Additionally, we rely in part on confidentiality and non‑use agreements with our employees, consultants, collaborators and other business partners to protect our proprietary technology and processes. If any of these individuals or entities breaches their confidentiality, non‑use or similar agreements with us, we may not have adequate remedies for that breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors and even patented by them. If that happens, the potential competitive advantages provided by our intellectual property may be adversely affected. We may then need to license such competing technologies, and we may not be able to obtain licenses on reasonable terms, if at all, which could cause material harm to our business. Moreover, to the extent that our employees, consultants, parties to collaboration agreements and other business partners use intellectual property owned by others in their work for the company, disputes may arise as to the rights in related or resulting know‑how and inventions.
Our success also depends in part on our, and on the people with whom we collaborate and do business, not infringing patents and proprietary rights of others, and not breaching any licenses or other agreements that we or they
26
have entered into with regard to our technologies, products and business. We cannot be certain that patents have not or will not be issued to others that would block our ability to obtain patents or to operate our business as we would like or at all. There may be patents in some countries that, if valid and if we are unsuccessful in circumventing or acquiring rights to them, could block our ability to commercialize products in those countries. There also may be claims in patent applications filed in some countries that, if granted and valid, and if we are unable to circumvent or license them, could also block our ability to commercialize products or processes in those countries.
Litigation and other disputes over intellectual property and other proprietary rights in the pharmaceutical and biotechnology industry is inherently costly and unpredictable.
Litigation, interferences, oppositions, inter partes reviews or other proceedings are, and may in the future be, necessary in some instances to determine the validity and scope of certain of our proprietary rights, and in other instances to determine the validity, scope or non‑infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. Patent‑related claims could include challenges to the scope and validity of our patents on products or processes as well as allegations that our products infringe patents held by competitors or others. We may also face challenges to regulatory or patent protections covering our products by manufacturers of biosimilars that may choose to launch or attempt to launch their products before the expiration of our regulatory or patent exclusivity.
In July 2017, we filed complaints in the U.S. District Court for the District of Delaware and the U.S. International Trade Commission (“ITC”) against CSL Behring LLC, CSL Behring GmbH, and CSL Behring Recombinant Facility AG (“CSL Behring”), alleging that the use of Idelvion®, CSL Behring’s [Coagulation Factor IX (Recombinant), Albumin Fusion Protein], infringes three of our U.S. patents. The District Court complaint seeks injunctive relief and damages. The District Court action was stayed pending final determination in the ITC. Our ITC complaint requested that the ITC issue a limited exclusion order and cease and desist orders covering CSL Behring’s alleged infringing articles, and impose a bond for imports of such articles made during the applicable review period. On February 6, 2018, we filed a motion to terminate the ITC investigation in order to proceed without further delay in the District Court. Patent enforcement proceedings can be expensive and time consuming, with no assurance of success.
The disposition of claims or proceedings is unpredictable and, regardless of the merits or the outcome, may be protracted, expensive and distracting to management. Moreover, the disposition and outcome of any such claims or proceedings could, among other things:
|
· |
adversely affect the validity and scope of our patent or other proprietary rights; |
|
· |
hinder our ability to manufacture, market and sell our products; |
|
· |
lead to attempts on the part of other parties to pursue similar claims; |
|
· |
force us to redesign those products or processes that use any allegedly infringing or misappropriated technology, which may result in significant cost or delay to us, or which the redesign of could be technically infeasible; |
|
· |
require us to seek a license for the impacted product or technology and pay royalties; or |
|
· |
result in the assessment of significant monetary damages against us that may exceed amounts, if any, accrued in our financial statements, including the possibility of treble damages in a patent case if a court finds us to have willfully infringed certain intellectual property rights. |
In addition, payments under any licenses that we are able to obtain would reduce our profits derived from the covered products and services. Furthermore, many of our collaboration agreements, including with Sobi, require us to indemnify the collaboration parties for third party intellectual property infringement claims, which would increase the cost to us of any such claim. Any of these adverse effects may be material and, consequently, may adversely impact our cash flow, financial position and results of operations.
27
Our presence in international markets subjects us to certain risks of doing business in such markets, which could adversely impact our business, results of operations and financial condition.
Our presence in international markets subjects us to many risks that could adversely affect our business and revenues, such as:
|
· |
the inability to obtain necessary regulatory or pricing approvals of products in a timely manner; |
|
· |
timing and complexity of local tendering processes; |
|
· |
differing local product preferences and product requirements; |
|
· |
difficulties or delays in establishing commercial operations in countries where we have not previously operated; |
|
· |
changes in medical reimbursement policies and programs; |
|
· |
fluctuations in foreign currency exchange rates; |
|
· |
difficulties in staffing and managing non‑U.S. operations; |
|
· |
unexpected difficulties in establishing effective financial and other controls in non-US jurisdictions; |
|
· |
uncertainties regarding the collectability of accounts receivable; |
|
· |
differing and increased labor regulations, including non‑U.S. work councils; |
|
· |
the imposition of governmental controls; |
|
· |
less favorable intellectual property or other applicable laws; |
|
· |
increasingly complex standards for complying with non‑U.S. laws and regulations that may differ substantially from country to country and may conflict with corresponding U.S. laws and regulations; |
|
· |
government involvement in funding health care in major overseas markets; |
|
· |
the far‑reaching anti‑bribery and anti‑corruption legislation in the United Kingdom and elsewhere, including the U.K. Bribery Act 2010, and the escalation of investigations and prosecutions pursuant to such laws; |
|
· |
compliance with complex import and export control laws; |
|
· |
restrictions on direct investments by non‑U.S. entities and trade restrictions; |
|
· |
compliance with complex local tax and regulatory requirements; |
|
· |
greater political or economic instability; and |
|
· |
changes in tax laws and tariffs. |
We cannot guarantee that our efforts to initiate or expand sales in these markets will succeed. Some non‑U.S. markets may be especially vulnerable to periods of financial instability or may have very limited resources to spend on health care. To successfully implement our strategy in non‑U.S. markets, we must attract and retain qualified personnel or may be required to increase our reliance on third party distributors within those markets. In addition, many of the countries in emerging markets have currencies that fluctuate substantially. If such currencies devalue and we cannot offset the devaluations, our financial performance within those countries could be adversely affected. In addition, price and currency exchange controls, limitations on participation in local enterprises, expropriation, nationalization and other governmental actions could affect our business and results of operations in these markets.
In addition, our non‑U.S. operations are subject to regulation under U.S. law. For example, the U.S. Foreign Corrupt Practices Act (FCPA) prohibits U.S. companies and their representatives from offering, promising, authorizing or making payments to foreign officials for the purpose of obtaining or retaining business abroad. In many countries, the health care professionals we regularly interact with may meet the definition of a foreign government official for purposes of the FCPA. Failure to comply with U.S. or non‑U.S. laws could result in various adverse consequences, including: possible delay in approval or refusal to approve a product; recalls, seizures or withdrawal of an approved product from the market; disruption in the supply or availability of our products or suspension of export or import privileges; the imposition of civil or criminal sanctions; the prosecution of executives overseeing our international operations; and damage to our reputation. Any significant impairment of our ability to sell products outside of the U.S. could adversely impact our business and financial results.
28
Development of our product candidates is expensive and uncertain. If we are unable to successfully develop and test our product candidates, our business, financial condition, results of operations and prospects will be harmed.
A part of our business strategy is the continued development and support of our marketed products and our product pipeline programs. Clinical trials and the research and development of drug treatments are subject to numerous risks and uncertainties and requires significant capital expenditures and management resources. Only a small percentage of product candidates that enter the development process ever receive regulatory approval. The process of conducting the preclinical and clinical testing required to establish safety and efficacy and obtain regulatory approval is expensive and uncertain and takes many years. The FDA and non‑U.S. regulatory agencies generally require pre‑clinical (animal) testing as well as multiple stages of clinical (human) testing before a product gains regulatory approval, and failure may occur at any stage of testing. It is possible that positive results in a trial may not be replicated in subsequent or confirmatory trials, and success in preclinical work or early stage clinical trials does not ensure that later stage or larger scale clinical trials will be successful or that regulatory approval will be obtained.
Our ability to commence and complete clinical trials may be delayed, and our existing trials may be stopped, due to various factors, including:
|
· |
ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials and the number of clinical trials we must conduct; |
|
· |
delays in enrolling volunteers or patients into clinical trials, including as a result of low numbers and types of patients that meet eligibility criteria for each study; |
|
· |
lower than anticipated retention rate of volunteers or patients in clinical trials; |
|
· |
difficulty in maintaining contact with patients after treatment, resulting in incomplete data, unforeseen safety issues or side effects; |
|
· |
the need to repeat clinical trials as a result of inconclusive results, unforeseen complications in testing or clinical investigator errors; |
|
· |
inadequate supply or deficient quality of drug candidate materials or other materials necessary for the conduct of our clinical trials; |
|
· |
unfavorable FDA or foreign regulatory authority inspection and review of a manufacturing facility that supplied clinical trial materials or its relevant manufacturing records or a clinical trial site or records of any clinical or preclinical investigation; |
|
· |
varying interpretations of clinical trial data; |
|
· |
poor or unanticipated effectiveness of product candidates, or adverse or unexpected safety events or side effects experienced by participants in our clinical trials; |
|
· |
favorable results in testing of our competitors’ product candidates, or FDA or foreign regulatory authority approval of our competitors’ product candidates; or |
|
· |
other government or regulatory delays. |
The occurrence of any of these events could result in significant costs and expenses and lost market opportunities, and could result in failure of a regulatory agency to grant approval of a product candidate in a timely manner, or at all.
Our product research and development efforts may be harmed if the third parties on whom we rely fail to perform satisfactorily.
Our reliance on third parties for aspects of the research and development process subjects our product research and development efforts to risk of failure or delay if such third parties do not perform satisfactorily. For example, we rely, and expect to continue to rely, on third parties to store and distribute drug supplies for our clinical trials as well as contract research organizations (CROs), clinical data management organizations, medical institutions and clinical investigators to conduct and manage our preclinical and clinical trials and to accurately report their results. Reduced control over these activities may impact our ability to control the timing, conduct, expense, reliability and quality of our clinical trials, but does not relieve us of our regulatory responsibility for trials that we sponsor. For example, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan
29
and protocols for the trial as well as regulatory standards such as current cGCPs. Failure to fully comply with the study protocol or applicable regulations or regulatory standards could result in the clinical data generated in those studies being deemed unreliable. This failure may also result in the rejection of our product candidates by the FDA or a non‑U.S. regulatory agency, or may result in our having to conduct additional audits or require additional clinical studies, which would delay our development programs, require us to incur additional costs and could substantially harm our business and financial condition. If the third parties we rely on for research and development activities do not successfully carry out their contractual duties, do not meet expected deadlines, experience work stoppages, terminate their agreements with us, need to be replaced or do not conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we may need to enter into new arrangements with alternative third parties, which could be difficult, costly or impossible. As a result, our clinical trials may be extended, delayed, terminated or may need to be repeated.
We also rely on third party contract manufacturers and suppliers for the materials we require for our clinical trials. Reliance on third parties for manufacturing and supply of materials for our clinical trials subject us to risks similar to those we face due to our reliance on third parties for manufacturing of our commercial products. If we suffer any manufacturing or supply disruptions, due to among other things, shortages in product raw materials, labor or technical difficulties, regulatory inspections or restrictions, shipping or customs delays or any other performance failure by any third party manufacturer on which we rely, our clinical trials may be adversely affected, including the timing and completion of the trials.
The occurrence of any of these events could result in significant costs and expenses and lost market opportunities, and could result in failure of a regulatory agency to grant approval of a product candidate in a timely manner, or at all.
If our business development activities are unsuccessful, our business could suffer and our financial performance could be adversely affected.
We have engaged and intend to continue to engage in business development activities, including evaluating potential acquisitions, strategic alliances, collaborations, technology licensing arrangements and other opportunities. An example of these activities includes our recent acquisition of True North Therapeutics (now known as Bioverativ USA Inc.), through which we acquired rights to BIVV009, a monoclonal antibody in development for cold agglutinin disease, a rare and chronic autoimmune hemolytic condition that often leads to severe anemia.
Our business development activities may require a substantial investment of our resources, with no certainty of success. Further, our success in developing products or expanding our product portfolio from such business development activities will depend on a number of factors, including:
|
· |
our ability to find suitable opportunities for acquisition, investment or alliance; |
|
· |
our ability to successfully complete due diligence processes and identify material contingencies, challenges or liabilities of a target company, compound, product or technology; |
|
· |
whether we are able to complete an acquisition, investment or alliance on terms that are satisfactory to us; |
|
· |
the strength of the technology and products being licensed or acquired; |
|
· |
any intellectual property and litigation related to these products or technology; |
|
· |
our ability to successfully integrate the acquired company, personnel, business, product, technology or research into our existing operations, including the ability to adequately fund, develop and commercialize acquired in‑process research and development projects and to maintain adequate controls over the consolidated operations; |
|
· |
our ability to enter into markets or disease areas in which we have no or limited prior experience or where competitors in such markets have stronger market positions; and |
|
· |
our ability to access capital markets or incur indebtedness at terms that are satisfactory to us. |
If we are unsuccessful in our business development activities, we may be unable to grow or meet our financial targets and our business and financial performance could be adversely affected.
30
Further, even if we are able to successfully identify and complete acquisitions and other strategic alliances, licenses and collaborations, we may face unanticipated costs or liabilities in connection with the transaction or we may not be able to integrate them or take full advantage of them or otherwise realize the benefits that we expect.
We depend on relationships with collaborators and other third parties for revenue, and for the development, regulatory approval, commercialization and marketing of certain products, which are outside our full control. If our collaborative efforts are unsuccessful, our commercialization strategies or product development may be delayed, which could have an adverse impact on our business, prospects and results of operations.
We rely, and expect to continue to rely, on a number of significant collaborative and other third party relationships for revenue, and for the development, regulatory approval, commercialization and marketing of our products and product candidates. These third parties may include other biotechnology and biopharmaceutical companies, academic and research institutions, governments and government agencies and other public and private research organizations. For example, in addition to our collaboration with Sobi, we are pursuing programs with other third parties in hemophilia using different technologies, including XTEN technology, gene therapy, gene editing, and non‑factor modalities. We also work with third parties to develop products and product candidates in other rare blood disorders.
Reliance on collaborative and other third party relationships subjects us to a number of risks, including:
|
· |
we may be unable to control the resources our collaborator devotes to our programs or product candidates; |
|
· |
disputes may arise with respect to ownership of rights to technology developed with our collaborator, and the underlying contract with our collaborator may fail to provide us with significant protection or may fail to be effectively enforced if the collaborator fails to perform; |
|
· |
our collaborators’ interests may not always be aligned with our interests and a collaborator may not pursue regulatory approvals or market a product in the same manner or to the same extent that we would, which could adversely affect our revenues; |
|
· |
the failure to effectively cooperate with our collaborators could adversely affect product sales or the clinical development or regulatory approvals of product candidates under joint control and could also result in termination of the research, development or commercialization of product candidates, litigation or arbitration; and |
|
· |
any failure on the part of our collaborator to comply with applicable laws and regulatory requirements in the marketing, sale and maintenance of the market authorization of our products or to fulfill any responsibilities our collaborator may have to protect and enforce any intellectual property rights underlying our products or product candidates could have an adverse effect on our revenues and involve us in legal proceedings. |
Given these risks, there is considerable uncertainty regarding the success of our current and future collaborative efforts. If these efforts fail, our product development or commercialization of new products could be delayed or revenues from products could decline.
We may not be able to access the capital and credit markets on terms that are favorable to us.
We may need to raise additional capital to supplement our existing funds and cash generated from operations for working capital, capital expenditures, and strategic investments. Funding needs may shift and the amount of capital we may need depends on many factors, such as the cost of any new acquisition or collaborative, licensing or other commercial relationships that we may establish, and the progress, timing and scope of our research and development. The capital and credit markets can be volatile and, as a result, we may not receive additional funding when we need it or funding may only be available on unfavorable terms.
If we or third parties with whom we do business fail to comply with the extensive legal and regulatory requirements affecting the health care industry, we could face increased costs, penalties and harm to our business.
Our activities, and the activities of our collaborators, distributors and other third party providers, are subject to extensive government regulation and oversight both in the U.S. and in non‑U.S. jurisdictions.
31
To be approved for marketing, a potential product must undergo lengthy and rigorous testing and other extensive, costly and time‑consuming procedures mandated by the FDA and non‑U.S. regulatory authorities. Satisfaction of these regulatory requirements typically takes many years. Moreover, regulatory oversight continues to apply after product marketing approval and covers, among other things, testing, manufacturing, distribution, quality control, labeling, advertising, promotion, risk mitigation and adverse event reporting requirements. Our facilities, or those of third parties on which we rely, must be licensed prior to production and remain subject to inspection from time to time thereafter. Separately, if previously unknown problems occur with regard to our marketed products, any of our products may have to be withdrawn from the market or subject to restrictions. Regulatory agencies may also require additional clinical trials or testing for our products, and our products may be recalled or may be subject to reformulation, changes in labeling, warnings to the public and negative publicity.
Further, even if we are successful in gaining regulatory approval of any of our product candidates, the extent to which we are able to commercialize the product may be less than we anticipate. Regulatory authorities may grant marketing approval that is more restricted than anticipated. These restrictions may include limiting indications to narrow patient populations and imposing safety monitoring, educational requirements and risk evaluation and mitigation strategies (REMS). In addition, if we seek to expand or change the use of any of our marketed products, those changes may be subject to vigorous review and include multiple regulatory submissions, and approvals are not certain.
In addition to FDA and related regulatory requirements, we are subject to health care “fraud and abuse” laws governing our interactions in the U.S. and non‑U.S. jurisdictions with physicians or other health care providers that prescribe or purchase our products. In the United States, these laws include the federal False Claims Act, the anti‑kickback provisions of the federal Social Security Act, the Physician Payment Sunshine provisions, and other state and federal laws and regulations. In both the United States and other jurisdictions, health care companies such as ours are facing heightened scrutiny of their relationships with health care providers from anti‑corruption enforcement officials and private individuals. Many biotechnology and biopharmaceutical companies have been the target of lawsuits and investigations alleging violations of government regulation, including claims asserting submission of incorrect pricing information, impermissible off‑label promotion of biotechnology and biopharmaceutical products, payments intended to influence the referral of health care business, submission of false claims for government reimbursement, antitrust violations or violations related to environmental matters. There also recently has been enhanced scrutiny of company‑sponsored patient assistance programs, including insurance premium and co‑pay assistance programs and donations to third party charities that provide such assistance. If we, or our vendors or donation recipients, fail to comply with relevant laws, regulations or government guidance in the operation of these programs, we could be subject to significant fines or penalties. Our risks under health care fraud and abuse laws may be heightened as we continue to expand our global operations and if we enter new therapeutic areas with different patient populations, which may have product distribution methods distinct from those we currently utilize.
Violations of governmental regulation, such as a failed inspection or a failure in our adverse event reporting system, or any health care fraud and abuse law may be punishable by criminal, civil and administrative sanctions against us as well as against executives overseeing our business. These may include adverse inspection reports; refusal to grant approvals or licenses; warning letters; fines and civil monetary penalties; withdrawal of regulatory approval or licenses; interruption of production; operating restrictions; product recall or seizure; injunctions; criminal prosecution and exclusion from participation in government programs, including Medicare and Medicaid, as well as against executives overseeing our business. In addition to penalties for violation of laws and regulations, we could be required to repay amounts we received from government payors, or pay additional rebates and interest if we are found to have miscalculated the pricing information we have submitted to the government. We cannot ensure that our compliance controls, policies and procedures will, in every instance, protect us from acts committed by our employees, collaborators, partners or third party providers that would violate the laws or regulations of the jurisdictions in which we operate. Whether or not we have complied with the law, an investigation into alleged unlawful conduct could increase our expenses, damage our reputation, divert management time and attention and adversely affect our business. Any of these actions could cause a loss of confidence in us and our products, which could adversely affect our sales. Even if it is later determined that we are not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our position and have to divert significant management resources from other matters.
32
Our business and results of operations may be adversely affected by current and potential future health care reforms.
In the United States, federal and state legislatures, health agencies and third party payors continue to focus on containing the cost of health care. Legislative and regulatory proposals and enactments to reform health care insurance programs could significantly influence the manner in which our products are prescribed and purchased. We continue to face uncertainties as a result of federal and administrative efforts to repeal, substantially modify or invalidate or replace some or all of the provisions of the Patient Protection and Affordable Care Act (PPACA). There is no assurance that the PPACA, as currently enacted or as amended or replaced in the future, will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform will affect our business.
There is also significant economic pressure on U.S. state budgets that may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for our drugs. Some states have considered legislation and ballot initiatives that would control the prices of drugs, including laws to allow importation of biotechnology and biopharmaceutical products from lower cost jurisdictions outside of the United States and laws intended to impose price controls on state drug purchases. State Medicaid programs are increasingly requesting manufacturers pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not paid. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
In the European Union and some other non‑U.S. markets, the government provides health care at low cost to consumers and regulates biotechnology and biopharmaceutical prices, patient eligibility or reimbursement levels to control costs for the government‑sponsored health care system. Many countries have announced or implemented measures to reduce health care costs to constrain their overall level of government expenditures. These measures vary by country and may include, among other things, patient access restrictions, suspensions on price increases, prospective and possibly retroactive price reductions and other recoupments and increased mandatory discounts or rebates, recoveries of past price increases and greater importation of drugs from lower‑cost countries. These measures have negatively impacted our revenues and may continue to adversely affect our revenues and results of operations in the future.
If we fail to attract and retain key personnel, our business may suffer.
Our success depends in large part upon the leadership and performance of our management team and other key employees. Operating as an independent company demands a significant amount of time and effort from our management and other employees and may give rise to increased employee turnover. If we lose the services of members of our management team or other key employees, we may not be able to successfully manage our business or achieve our business objectives.
Our ability to attract, recruit and retain such talent will depend on a number of factors, including the hiring practices of our competitors, the performance of our commercial products and pipeline programs, our compensation and benefits, work location and work environment and economic conditions affecting our industry generally, and, as described above, uncertainty as a result of the potential merger with Sanofi. If we cannot effectively hire and retain qualified employees, our business, results of operations and prospects could suffer.
A breakdown or breach of our or third parties’ technology systems could subject us to liability or interrupt the operation of our business.
We are increasingly dependent upon technology systems and data, many of which are new or unfamiliar systems. Our intellectual property, computer systems, other proprietary technology and other sensitive company data is potentially vulnerable to loss, damage or misappropriation from system malfunction, computer viruses, unauthorized access to data or misappropriation or misuse thereof by those with permitted access and other events. Likewise, data privacy or security breaches by individuals authorized to access our technology systems or others may pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to us, our patients,
33
customers or other business partners, may be exposed to unauthorized persons or to the public. The increasing use and evolution of technology, including cloud‑based computing, creates additional opportunities for the unintentional dissemination of information and the intentional destruction of confidential information stored in the company’s systems or in non‑encrypted portable media or storage devices. Cyber attacks continue to increase in their frequency, sophistication and intensity. While we believe we take appropriate security measures to reduce these risks to our intellectual property, data and information technology systems, there can be no assurance that our efforts will prevent breakdowns, breaches, cyber incidents or other events. Such events could have a negative effect on our reputation, business, financial condition or results of operations. Further, the misappropriation or other loss of our intellectual property from any of the foregoing could have an adverse effect on our competitive position and may cause us to incur substantial litigation costs.
Our business involves environmental risks, which include the cost of compliance and the risk of contamination or injury, which could harm our business.
Our business and the business of several of our third party contractors involve the controlled use of hazardous materials, chemicals, biologics and radioactive compounds. Although we believe that our and their safety procedures for the handling and disposing of such materials comply with state, federal and non‑U.S. laws and standards, there will always be the risk of accidental contamination or injury. If we were to become liable for an accident, or if a facility in which our products or product candidates were manufactured suffered an extended shutdown, we could incur significant costs, damages or penalties that could harm our business. Manufacturing, distribution and disposal of our products and product candidates also requires compliance with environmental laws and may require permits from government agencies, including governmental authorizations or permits for water supply, wastewater discharge and waste disposal. If we or our contract parties do not obtain or comply with appropriate permits and other requirements of environmental laws, we or they could incur significant penalties and other costs and limits on manufacturing volumes that could harm our business.
Significant legal proceedings may adversely affect our results of operations or financial condition.
We are subject to the risk of litigation, derivative claims, securities class actions, regulatory and governmental investigations and other proceedings, including proceedings arising from investor dissatisfaction with us or our performance. If any claims were brought against us and resulted in a finding of substantial legal liability, the finding could materially adversely affect our business, financial condition or results of operations or cause significant reputational harm to us, which could seriously adversely impact our business. Allegations of improper conduct by private litigants or regulators, regardless of veracity, may harm our reputation and adversely impact our ability to grow our business.
Risks Related to the Separation
We face a number of risks as a standalone, independent public company following our separation from Biogen.
Prior to our separation from Biogen in February 2017, we were fully part of Biogen’s organizational structure, including from an operational and financial perspective. Following the separation, we have worked to develop our own systems, processes and operations, though we continue to rely on Biogen for certain activities. As a result of the separation, we remain subject to risks as a standalone, independent public company. These risks include the following:
|
· |
We may not achieve all the benefits we expect as a standalone company and we may be more susceptible to market fluctuations and other adverse events than we would have if we were still part of Biogen’s organizational structure, particularly because we may lack Biogen’s operating diversity, purchasing power, integrated strategies with Biogen’s other businesses, access to capital markets, market reputation, performance and brand identity. If we fail to achieve some or all of the benefits that we expect to achieve as an independent company, or do not achieve them in the time we expect, our business, operating results, financial condition or prospects may suffer. |
34
|
· |
We may be unable to make, on a timely or cost‑effective basis, the changes necessary to operate as a standalone company, and we remain reliant on Biogen for the provision of certain transition, manufacturing and supply services. If Biogen is unable or unwilling to satisfy its obligations to provide us with certain services under our agreements with them, we could incur operational difficulties or losses that could have a material and adverse effect on our business, operating results and financial condition. Furthermore, Biogen is only obligated to provide certain services for a limited period of time from the separation. If we do not have in place our own systems and services, or if we do not have agreements with other providers to provide these services in a timely manner or on terms and conditions as favorable as those we receive from Biogen, we may not be able to operate our business effectively and our profitability may decline. Additionally, if we fail to obtain the quality of services necessary to operate effectively or incur greater costs in obtaining these services, our profitability, operating results and financial condition may be materially and adversely affected. |
|
· |
Our historical financial information for periods prior to the separation is not necessarily representative of the results that we would have achieved as a separate, publicly traded company and should not be relied upon as an indicator of our future results. Periods prior to the separation reflect shared economies of scope and scale in costs, employees, vendor relationships and customer relationships with Biogen. Although we entered into certain agreements with Biogen in connection with the separation, these arrangements were entered into in the context of the separation while we were still controlled by Biogen and may not reflect terms that would have resulted from negotiations between unaffiliated third parties. These arrangements may also not fully capture the benefits that we have enjoyed as a result of being integrated with Biogen and may result in us incurring higher costs than in the past, and we may lose certain synergies and benefits we enjoyed as a result of being a part of Biogen, such as access to sufficient working capital for general corporate purposes as well as acquisitions, investments and capital expenditures. In addition, being a part of Biogen enabled us to leverage Biogen’s technological capabilities, data and commerce platforms. After the separation, our cost of capital may be higher than Biogen’s cost of capital prior to the separation. We may also face other significant changes in our cost structure, management, financing and business operations as a result of operating as a company separate from Biogen. |
|
· |
The separation may result in disruptions to, and negatively impact our relationships with, our customers and other business partners. Specifically, uncertainty related to the separation may lead customers and other parties with which we currently do business or may do business in the future to terminate or attempt to negotiate changes in our existing business relationships, or cause them to delay entering into business relationships with us or consider entering into business relationships with parties other than us. The effect of such disruptions could be exacerbated by any delays in the transition following the separation. |
|
· |
We have implemented numerous financial and management controls, reporting systems and procedures, and added accounting, finance and information technology staff to assist us in meeting our financial reporting and other requirements to which we are now subject. These requirements include, among other things, our annual management assessment of the effectiveness of our internal control over financial reporting and a report by our independent registered public accounting firm addressing these assessments. These and other obligations have placed significant demands on our management, administrative and operational resources, including accounting and information technology resources. If we fail to successfully implement our systems and controls, our ability to comply with our financial reporting requirements and other rules that apply to reporting companies could be impaired and our business could be harmed. |
|
· |
Upon completion of the separation, Biogen received an opinion from its tax counsel that the distribution qualified as a tax-free distribution for U.S. federal income tax purposes. The opinion of tax counsel is not binding on the Internal Revenue Service or any court, and the Internal Revenue Service could determine on audit that the distribution is taxable for U.S. federal income tax purposes. If the distribution does not qualify as a transaction that is tax‑free for U.S. federal income tax purposes, Biogen and its stockholders could be subject to significant tax liabilities. Under our agreements with Biogen, we could be required to indemnify Biogen for material taxes pursuant to indemnification obligations under the tax matters agreement. If we are required to indemnify Biogen under the circumstances set forth in the tax matters agreement, we may be subject to substantial liabilities, which could materially and adversely affect our financial condition. |
35
|
· |
Following the separation from Biogen, we remain subject to numerous restrictions to preserve the tax‑free treatment of the separation transactions in the U.S., which may reduce our strategic and operating flexibility. More specifically, even if the distribution otherwise qualifies for tax‑free treatment, the distribution may result in corporate‑level taxable gain to Biogen under Section 355(e) of the Code if 50% or more, by vote or value, of shares of our stock or Biogen’s stock are acquired or issued as part of a plan or series of related transactions that includes the distribution. The process for determining whether an acquisition or issuance triggering these provisions has occurred is complex, inherently factual and subject to interpretation of the facts and circumstances of a particular case. Any acquisitions or issuances of our stock or Biogen’s stock within a two‑year period after the distribution generally are presumed to be part of such a plan, although we or Biogen, as applicable, may be able to rebut that presumption. Accordingly, under the tax matters agreement we entered into with Biogen, for the two‑year period following the distribution, we are prohibited, except in certain circumstances, from: |
|
· |
entering into any transactions resulting in the acquisition of 40% or more of our stock or substantially all of our assets, whether by merger or otherwise; |
|
· |
merging, consolidating or liquidating; |
|
· |
issuing equity securities beyond certain thresholds; |
|
· |
repurchasing our capital stock; or |
|
· |
ceasing to actively conduct our business. |
These restrictions may limit our ability to pursue certain strategic transactions or other transactions that we may believe to otherwise be in the best interests of our stockholders or that might increase the value of our business. In addition, under the tax matters agreement, we will be required to indemnify Biogen against any such tax liabilities as a result of the acquisition of our stock or assets, even if we do not participate in or otherwise facilitate the acquisition. In connection with the transactions contemplated by the Merger, we entered into a letter agreement with Biogen and Sanofi to waive provisions under the Tax Matters Agreement that would have otherwise prohibited us from entering into the Merger Agreement. Further, so long as we receive the tax opinion referenced in the letter agreement, Biogen has agreed to waive the restrictions as they apply to the consummation of the Merger. See also the risks described under the heading “—Risks Related to the Merger”.
|
· |
After the distribution, there are several significant areas where the liabilities of Biogen may become our obligations, including contingent tax related liabilities of Biogen. For example, under the tax matters agreement we entered into with Biogen, if Biogen were unable to pay any prior period taxes for which it is responsible, under applicable law we could be required to pay the entire amount of such taxes, and such amounts could be significant. Other provisions of federal, state, local or foreign law may establish similar liability for other matters, including laws governing tax‑qualified pension plans, as well as other contingent liabilities. |
|
· |
Pursuant to the separation agreement and certain other agreements we entered into with Biogen, we assumed and agreed to indemnify Biogen for certain liabilities for uncapped amounts, which may include, among other items, associated defense costs, settlement amounts and judgments. Payments pursuant to these indemnities may be significant and could negatively impact our business, particularly indemnities relating to our actions that could impact the tax‑free nature of the distribution and certain related transactions. |
Risks Related to Our Common Stock
The market price for our common stock may fluctuate widely.
The market price of our shares of common stock may fluctuate widely, depending upon many factors, some of which are beyond our control, including the following:
|
· |
completion of the Merger with Sanofi, as described above under the heading “—Risks Related to the Merger”; |
|
· |
our quarterly or annual earnings, or those of other comparable companies; |
36
|
· |
actual or anticipated fluctuations in our operating results; |
|
· |
changes in accounting standards, policies, guidance, interpretations or principles; |
|
· |
developments in our pipeline or the pipeline of competitors; |
|
· |
announcements by us or our competitors of significant clinical trial results or regulatory approvals, investments, acquisitions or dispositions; |
|
· |
the failure of securities analysts to cover our shares of common stock; |
|
· |
changes in ratings or earnings estimates by securities analysts or our ability to meet those estimates; |
|
· |
the operating and stock price performance of other comparable companies; |
|
· |
overall market fluctuations; and |
|
· |
general economic conditions. |
Stock markets in general often experience volatility that is unrelated to the operating performance of a particular company. These broad market fluctuations may adversely affect the trading price of our shares of common stock. You may not be able to resell your shares of common stock following periods of volatility because of the market’s adverse reaction to volatility. When the market price of a company’s common stock drops significantly, stockholders often institute securities class action lawsuits against the company. This type of lawsuit against us could cause us to incur substantial costs and could divert the time and attention of management and other resources.
Your percentage ownership in the company may be diluted in the future.
In the future, your percentage ownership in the company may be diluted because of equity issuances for acquisitions, capital market transactions or otherwise, including equity awards that we grant to our directors, officers and employees. Such awards will have a dilutive effect on our earnings per share, which could adversely affect the market price of our common stock. From time to time, we issue stock options or other share‑based awards to employees under our employee benefits plans.
In addition, our amended and restated certificate of incorporation authorizes us to issue, without the approval of stockholders, one or more classes or series of preferred stock having such designation, powers, preferences and relative, participating, optional and other special rights, including preferences over the company’s common stock respecting dividends and distributions, as the board of directors generally may determine. The terms of one or more classes or series of preferred stock could dilute the voting power or reduce the value of our common stock. For example, we could grant the holders of preferred stock the right to elect some number of directors in all events or on the happening of specified events or the right to veto specified transactions. Similarly, the repurchase or redemption rights or liquidation preferences we could assign to holders of preferred stock could affect the residual value of the common stock.
We do not expect to declare any dividends in the foreseeable future.
We do not anticipate declaring any cash dividends to holders of our common stock in the foreseeable future. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on the value of their shares of our common stock.
Provisions contained in our amended and restated certificate of incorporation and amended and restated bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our amended and restated certificate of incorporation and amended and restated bylaws contain certain provisions that could have the effect of rendering more difficult or discouraging an acquisition deemed undesirable by our board of directors. For example, our corporate governance documents include provisions:
|
· |
authorizing blank check preferred stock, which could be issued with voting, liquidation, dividend and other rights superior to our common stock; |
|
· |
limiting the liability of, and providing indemnification to, our directors and officers; |
|
· |
limiting the ability of our stockholders to call and bring business before special meetings; |
|
· |
eliminating the ability of our stockholders to act by written consent in lieu of a meeting; |
37
|
· |
requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our board of directors; and |
|
· |
limiting the determination of the number of directors on our board of directors and the filling of newly created seats on the board to our board of directors then in office. |
These provisions, alone or together, could delay hostile takeovers and changes in control of our company or changes in our management.
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation Law (DGCL), which prevents some stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of substantially all of our outstanding common stock. Any provision of our amended and restated certificate of incorporation or amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
In addition, an acquisition or further issuance of our stock could trigger the application of Section 355(e) of the Code. Under the tax matters agreement we entered into with Biogen in connection with the separation, we would be required to indemnify Biogen for any resulting taxes, and this indemnity obligation might discourage, delay or prevent a change of control that our stockholders may consider favorable. In connection with the transactions contemplated by the Merger, we entered into a letter agreement with Biogen and Sanofi to waive provisions under the Tax Matters Agreement that would have otherwise prohibited us from entering into the Merger Agreement. Further, so long as we receive the tax opinion referenced in the letter agreement, Biogen has agreed to waive the restrictions as they apply to the consummation of the Merger. See also the risks described under the heading “—Risks Related to the Merger”.
Our amended and restated certificate of incorporation designates the state courts of the State of Delaware, or, if no state court located in the State of Delaware has jurisdiction, the federal court for the District of Delaware, as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could discourage lawsuits against us and our directors and officers.
Our amended and restated certificate of incorporation provides that unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware, to the fullest extent permitted by law, will be the sole and exclusive forum for:
|
· |
any derivative action or proceeding brought on behalf of us; |
|
· |
any action asserting a claim for breach of a fiduciary duty owed by any of our directors, officers or other employees of the company to us or our stockholders; |
|
· |
any action asserting a claim arising pursuant to any provision of the DGCL; or |
|
· |
any action asserting a claim governed by the internal affairs doctrine under Delaware state corporate law. |
This exclusive forum provision may limit the ability of our stockholders to bring a claim in a judicial forum that such stockholders find favorable for disputes with us or our directors or officers, which may discourage such lawsuits against the company and our directors and officers. Alternatively, if a court outside of Delaware were to find this exclusive forum provision inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings described above, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, operating results or financial condition.
Item 1B. Unresolved Staff Comments
None.
38
Below is a summary of our leased properties as of December 31, 2017.
United States
In Waltham, Massachusetts, we lease approximately 125,000 square feet of real estate space, consisting of a building that houses a research laboratory and general office space, pursuant to a ten-year lease. We also lease space in South San Francisco, California, for general office, research and development, laboratory uses.
International
We sublease office space from Biogen in Canada and Japan. In addition, we lease office space in Australia, Brazil and Colombia. Our international lease agreements expire at various dates through the year 2020.
For a discussion of legal matters as of December 31, 2017, please read Note 16, Litigation to our audited consolidated financial statements included in this report, which is incorporated into this item by reference.
Item 4. Mine Safety Disclosures
Not applicable.
39
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market and Stockholder Information
Our common stock trades on the NASDAQ Global Select Market under the symbol “BIVV.” A “when issued” trading market for our common stock began on the NASDAQ Global Select Market on January 12, 2017, and “regular way” trading of our common stock began on February 2, 2017. The following table shows the high and low sales price for our common stock as reported by The NASDAQ Global Select Market for each quarter in the years ended December 31, 2017:
|
|
|
Common Stock Price |
||||
|
|
|
2017 |
||||
|
|
|
High |
|
Low |
||
|
First Quarter |
|
$ |
55.16 |
|
$ |
41.88 |
|
Second Quarter |
|
$ |
63.23 |
|
$ |
51.06 |
|
Third Quarter |
|
$ |
64.41 |
|
$ |
54.12 |
|
Fourth Quarter |
|
$ |
60.74 |
|
$ |
48.28 |
As of February 9, 2018, there were approximately 663 stockholders of record of our common stock.
Dividends
We do not expect to pay a regular cash dividend. The payment of any dividends in the future, and the timing and amount thereof, is within the discretion of our board of directors. Our board of directors’ decisions regarding the payment of dividends will depend on many factors, such as our financial condition, earnings, capital requirements, industry practice, legal requirements, regulatory constraints and other factors that our board of directors deems relevant. Our ability to pay dividends will depend on our ongoing ability to generate cash from operations and on our access to the capital markets. We cannot guarantee that we will pay a dividend in the future or continue to pay any dividends if and when we commence paying dividends.
Item 6. Selected Financial Data
The following table sets forth Bioverativ’s selected financial information derived from its (i) audited consolidated financial statements as of and for the year ended December 31, 2017; and (ii) audited consolidated financial statements for the hemophilia business of Biogen for the years ended December 31, 2016, 2015, 2014 and 2013. For the periods prior to the separation, the historical consolidated financial statements have been prepared on a carve‑out basis for the purpose of presenting what our historical financial position, results of operations and cash flows would have been for the periods presented had Bioverativ operated the hemophilia business as a standalone entity. Bioverativ did not operate as a standalone entity in the past and accordingly the selected historical financial data presented herein is not necessarily indicative of the company’s future performance and does not reflect what the company’s performance would have been had Bioverativ operated as an independent, publicly traded company during the periods presented, and accordingly should not be relied upon as an indicator of our future results.
The selected financial information should be read in conjunction with Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations, and the audited consolidated financial statements and the corresponding notes included elsewhere in this report.
40
|
|
|
For the Years Ended |
|||||||||||||
|
|
|
December 31, |
|||||||||||||
|
(In millions, except per share amounts) |
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|||||
|
Consolidated Statement of Income (Loss) Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues |
|
$ |
1,168.5 |
|
$ |
887.4 |
|
$ |
560.3 |
|
$ |
134.4 |
|
$ |
— |
|
Net income (loss) |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
$ |
(360.3) |
|
$ |
(344.6) |
|
Diluted earnings per share |
|
$ |
3.28 |
|
$ |
4.07 |
|
$ |
1.01 |
|
$ |
(3.34) |
|
$ |
(3.19) |
|
Shares used in calculating diluted earnings per share |
|
|
108.5 |
|
|
108.0 |
|
|
108.0 |
|
|
108.0 |
|
|
108.0 |
|
|
|
As of |
|||||||||||||
|
|
|
December 31, |
|||||||||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|||||
|
Consolidated Balance Sheet Data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets |
|
$ |
1,618.3 |
|
$ |
731.9 |
|
$ |
475.6 |
|
$ |
376.4 |
|
$ |
180.7 |
|
Total long term liabilities |
|
$ |
330.6 |
|
$ |
63.7 |
|
$ |
30.7 |
|
$ |
17.1 |
|
$ |
9.2 |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion should be read in conjunction with the audited consolidated financial statements and the corresponding notes included elsewhere in this report. This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains forward‑looking statements. The matters discussed in these forward‑looking statements are subject to risk, uncertainties and other factors that could cause actual results to differ materially from those made, projected or implied in the forward‑looking statements. Please see Item 1A. Risk Factors and “Note Regarding Forward‑Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements.
On May 3, 2016, Biogen announced its plans to separate into two independent, publicly traded companies. For purposes of the following discussion, Bioverativ refers to the hemophilia business of Biogen prior to the separation. To accomplish this separation, Biogen created a new company, Bioverativ Inc., to be the parent company for the hemophilia business. Bioverativ Inc. was incorporated in the State of Delaware on August 4, 2016. To effect the separation, Biogen made a 2‑1 pro rata distribution of Bioverativ Inc.’s common stock to Biogen’s stockholders. The distribution occurred on February 1, 2017 and since the distribution, Bioverativ Inc. has operated as an independent, publicly traded company.
Overview
We are a global biotechnology company focused on the discovery, research, development and commercialization of innovative therapies for the treatment of hemophilia and other blood disorders. We have two marketed products, ELOCTATE [Antihemophilic Factor (Recombinant), Fc Fusion Protein] and ALPROLIX [Coagulation Factor IX (Recombinant), Fc Fusion Protein], and an innovative product pipeline.
Our business strategy is aimed at improving treatment and standards of care for hemophilia and other blood disorder patients by further increasing sales and market share of our marketed products, advancing treatment attributes for our marketed products, leveraging our internal expertise to develop new products that meaningfully advance treatment and opportunistically pursuing strategic alliances and tactical acquisitions.
For the periods prior to the separation, the audited consolidated financial statements have been prepared on a carve‑out basis for the purpose of presenting our historical financial position, results of operations and cash flows. We did not operate on a standalone basis prior to the separation.
Our revenues are primarily derived from sales of ELOCTATE and ALPROLIX in the United States, Japan and Canada. We also earn revenue from the supply of ELOCTATE and ALPROLIX to Sobi and royalties on sales of ELOCTATE and ALPROLIX by Sobi in its commercialization territory, which is Europe, Russia and certain countries
41
in Northern Africa and the Middle East. See Item 1. Business—Our Development and Commercialization Arrangements with Sobi.
Merger Agreement with Sanofi
On January 21, 2018, we entered into an Agreement and Plan of Merger (the “Merger Agreement”), with Sanofi (“Parent” or “Sanofi”), a French société anonyme, and Blink Acquisition Corp. (“Merger Sub”), a Delaware corporation and indirect wholly-owned subsidiary of Parent. See Note 21, Subsequent Events to our audited consolidated financial statements included in this Form 10-K.
Pursuant to the terms of Merger Agreement, upon the terms and subject to the conditions thereof, Merger Sub commenced a tender offer on February 7, 2018 (the “Offer”), to acquire all of the outstanding shares of common stock of the Company, par value $0.001 per share (the “Shares”), at a purchase price of $105.0 per Share in cash, net of applicable withholding taxes and without interest (the “Offer Price”). Unless extended by Merger Sub in accordance with the Merger Agreement, the offering period for the Offer will expire at 11:59 p.m. (New York City time), on March 7, 2018.
As soon as practicable following (but on the same day as) the consummation of the Offer and subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, Merger Sub will be merged with and into the Company (the “Merger”), with the Company surviving the Merger as an indirect wholly-owned subsidiary of Parent. The Merger will be governed by Section 251(h) of the Delaware General Corporation Law (“DGCL”) and effected without a vote of the Company’s stockholders.
Financial Results Overview—Years Ended December 31, 2017, 2016 and 2015
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
Total revenues |
|
$ |
1,168.5 |
|
$ |
887.4 |
|
$ |
560.3 |
|
31.7 |
% |
|
58.4 |
% |
|
|
Net income |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
(19.1) |
% |
|
304.8 |
% |
|
Refer to “—Results of Operations—Years Ended December 31, 2017, 2016 and 2015” section below for further discussion of our results.
Commercial Highlights
The United States, Japan and Canada are currently the principal markets outside of Sobi’s commercialization territory for our marketed products. We expect to continue to drive revenue growth and increased patient share of ELOCTATE and ALPROLIX by expanding into new geographies and continuing to penetrate our existing geographies. In addition, in 2016 we began earning royalties from Sobi on sales of ELOCTA and ALPROLIX following Sobi’s commercial launch of ELOCTA and ALPROLIX in the European Union.
Business Development Highlights
True North Acquisition - On June 28, 2017, we acquired all of the outstanding equity of True North Therapeutics, Inc. (True North), a clinical stage biopharmaceutical company focused on the discovery, development and commercialization of first-in-class product candidates for complement-mediated diseases. At closing, we paid True North equityholders upfront consideration of $395.7 million plus assumed cash. True North equityholders are also eligible to receive additional payments of up to $425.0 million contingent on the achievement of future development, regulatory and sales milestones. With the acquisition, we added BIVV009 (formerly TNT009), a first-in-class monoclonal antibody for cold agglutinin disease (CAgD), to our pipeline. BIVV009 has received breakthrough therapy designation from the FDA for the treatment of hemolysis in patients with primary CAgD, and orphan drug designation from the FDA and the European Medicines Agency. Late-stage clinical development planning for BIVV009, including a
42
phase 3 registrational program, is underway. Refer to Note 3, Acquisition of True North Therapeutics, Inc., for further discussion.
Bicycle Therapeutics Research Collaboration - In August 2017, we entered into a research collaboration agreement with Bicycle Therapeutics (Bicycle), a biotechnology company that is pioneering a new class of medicines based on its novel bicyclic peptide (Bicycle®) product platform. The collaboration will focus on the discovery, development and commercialization of therapies for hemophilia and sickle cell disease. Under this agreement, Bicycle will be responsible for leading initial discovery activities through lead optimization to candidate selection for three programs. Bioverativ will lead preclinical and clinical development, as well as subsequent marketing and commercialization efforts. Under the agreement, we paid Bicycle a $10.0 million upfront payment. Bicycle is eligible to receive up to $410.0 million related to development, regulatory and commercialization milestones as well as tiered single to low double-digit royalties related to product sales. Bioverativ will also reimburse Bicycle for internal and external program-related costs.
Research and Development Highlights
IND for BIVV001 - On June 12, 2017, we announced that the U.S. Food and Drug Administration (FDA) accepted our Investigational New Drug (IND) application for BIVV001 (also known as rFVIIIFc-VWF-XTEN), an investigational factor VIII therapy designed to potentially extend protection from bleeds with prophylactic dosing of once weekly or longer for people with hemophilia A.
IND for ST-400 - In October 2017, we and Sangamo announced that the FDA accepted the IND application for ST-400, a gene-edited cell therapy candidate for people with transfusion-dependent beta-thalassemia. The IND enables Sangamo to initiate a Phase 1/2 clinical trial to assess the safety, tolerability and efficacy of ST-400 in adults with transfusion-dependent beta-thalassemia.
We expect to make substantial investments in research and development in support of our ongoing proprietary research programs and through collaborations with third parties for the development of new products and therapies. Research and development expenses were $224.6 million, $210.1 million and $236.9 million, or 19.2%, 23.7% and 42.3% of total revenue, respectively, during the years ended December 31, 2017, 2016 and 2015.
Our overall research and development strategy includes the continued pursuit of collaborations and strategic relationships with third parties that are developing new products and therapies. These collaborations and relationships generally involve the granting or obtaining development and commercialization rights to or from third parties in exchange for an upfront payment upon execution of the agreement and potential future payments related to the achievement of development, regulatory approval or commercial milestones, as well as royalties. Our most significant collaboration is our relationship with Sobi for the development and commercialization of ELOCTATE and ALPROLIX. Please refer to Note 4, Collaborations, to the audited consolidated financial statements included elsewhere in this report for additional details on our collaboration with Sobi.
Key Factors Affecting Results of Operations
Separation from Biogen on February 1, 2017
Bioverativ separated from Biogen on February 1, 2017 as a result of a special dividend distribution of all the outstanding shares of common stock of Bioverativ to Biogen stockholders. The distribution was made to Biogen stockholders of record as of the close of business on January 17, 2017, who received one share of Bioverativ common stock for every two shares of Biogen common stock held as of such date. As a result of the distribution, Bioverativ is now an independent public company.
Separation from Biogen
We did not operate as an independent, standalone company before the completion of the separation, but rather operated as a part of Biogen’s overall business. Accordingly, financial information for periods prior to the separation are
43
shown on a carve-out basis for the hemophilia business as part of Biogen. There are limitations inherent in the preparation of all carve‑out financial statements due to the fact that the business was previously part of a larger organization. The basis of preparation included in the notes to our audited consolidated financial statements included in this Form 10-K provides a detailed description of the treatment of historical transactions prior to the separation. Our historical net income for periods prior to the separation may not be indicative of future results. Our historical net income for periods prior to the separation has been most notably impacted by the following consequences of carve‑out accounting and the separation:
|
· |
Biogen utilized a centralized treasury management system and cash or debt was not allocated to Bioverativ in the carve‑out financial statements. In connection with the separation, the capital structures of both companies were re‑aligned, resulting in Bioverativ having adequate cash to fund its operations. |
|
· |
The consolidated statements of income include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. The research and development costs include depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. The selling, general and administrative costs include certain services provided by Biogen, which include executive oversight, treasury, finance, legal, human resources, tax planning, internal audit, financial reporting, information technology, investor relations, shared services, insurance, employee benefits and incentives and share‑based compensation. The amounts of these allocations may not necessarily be indicative of the similar costs we are incurring subsequent to the separation. The total amount of research and development, selling, general and administrative costs and other income (expense) allocated to us from Biogen for the years ended December 31, 2017, 2016 and 2015 were $11.8 million, $150.0 million and $148.6 million, respectively. |
|
· |
We incurred certain one‑time separation costs, which are primarily associated with the design and establishment of us as a standalone public company. |
|
· |
Income tax expense (benefit) is computed on a “separate return basis”, as if operated as a standalone entity or a separate entity or a separate consolidated group in each material jurisdiction in which we operate. Income tax expense (benefit) prior to the separation included in the condensed consolidated financial statements may not be indicative of our future expected effective income tax rate. |
|
· |
Concurrent with the separation, we entered into a manufacturing and supply agreement with Biogen whereby Biogen continues to produce ELOCTATE and ALPROLIX for us on terms on cost plus basis. As products were historically transferred at cost between Biogen and Bioverativ, these manufacturing and supply arrangements will result in changes to cost of goods sold in future periods. |
Results of Operations—Years Ended December 31, 2017, 2016 and 2015
Revenue
Total Revenues
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
Product revenues |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
905.7 |
|
$ |
713.2 |
|
$ |
517.1 |
|
27.0 |
% |
|
37.9 |
% |
|
|
All Other Markets |
|
|
183.4 |
|
|
133.4 |
|
|
37.0 |
|
37.5 |
% |
|
260.5 |
% |
|
|
Total product revenues |
|
|
1,089.1 |
|
|
846.6 |
|
|
554.1 |
|
28.6 |
% |
|
52.8 |
% |
|
|
Collaboration revenue |
|
|
79.4 |
|
|
40.8 |
|
|
6.2 |
|
94.6 |
% |
|
558.1 |
% |
|
|
Total revenues |
|
$ |
1,168.5 |
|
$ |
887.4 |
|
$ |
560.3 |
|
31.7 |
% |
|
58.4 |
% |
|
44
Revenues totaled $1,168.5 million in 2017, an increase of 31.7% over 2016. In 2017, product revenues in the United States totaled $905.7 million, an increase of 27.0% over 2016 and sales outside of the United States totaled $183.4 million, an increase of 37.5% over 2016. Collaboration revenue totaled $79.4 million in 2017, an increase of 94.6% over 2016, due primarily to Sobi’s continued launch of ELOCTATE and ALPROLIX in additional markets in 2017.
Revenues totaled $887.4 million in 2016, an increase of 58.4% over 2015. In 2016, product revenues in the United States totaled $713.2 million, an increase of 37.9% over 2015 and sales outside of the United States totaled $133.4 million, an increase of 260.5% over 2015. In the first and second quarters of 2016, Sobi had its first commercial sales of ELOCTATE and ALPROLIX, respectively. As a result, collaboration revenue, consisting of manufacturing and royalty revenues totaled $40.8 million, an increase of 558.1% over 2015.
Product Revenues
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
ELOCTATE |
|
$ |
724.5 |
|
$ |
513.0 |
|
$ |
319.7 |
|
41.2 |
% |
|
60.5 |
% |
|
|
ALPROLIX |
|
|
364.6 |
|
|
333.6 |
|
|
234.4 |
|
9.3 |
% |
|
42.3 |
% |
|
|
Total product revenues |
|
$ |
1,089.1 |
|
$ |
846.6 |
|
$ |
554.1 |
|
28.6 |
% |
|
52.8 |
% |
|
45
|
ELOCTATE |
|
ALPROLIX |
|
|
|
|
|
|
|
|
|
Sales of ELOCTATE for the year ended December 31, 2017 increased $211.5 million or 41.2%, compared to the same period in 2016. The increase in ELOCTATE revenues in the U.S. was $170.3 million, due to increases in unit sales volume of 40.5%. The increase in ELOCTATE revenues in all other markets revenues was $41.2 million, due to an increase in unit sales volume in Japan of 57.7% compared to the same period in 2016.
Sales of ELOCTATE for the year ended December 31, 2016 increased $193.3 million or 60.5%, compared to the same period in 2015. The increase in ELOCTATE revenues in the U.S. was $137.0 million, due to increases in unit sales volume of approximately 45%. The increase in ELOCTATE revenues in all other markets revenues was $56.3 million, due to an increase in unit sales volume in Japan of approximately 313%, due to its uptake following launch in the second quarter of 2015, as well as uptake following ELOCTATE’s launch in Canada in the first quarter of 2016. |
|
Sales of ALPROLIX for the year ended December 31, 2017 increased $31.0 million or 9.3%, compared to the same period in 2016. The increase in ALPROLIX revenues in the U.S. was $22.2 million, primarily due to increases in unit sales volume of 6.5%. The increase in ALPROLIX revenues in all other markets was $8.8 million, due to an increase in unit sales volume in Japan of 15.1% compared to the same period in 2016. We expect that ALPROLIX will continue to face a competitive environment.
Sales of ALPROLIX for the year ended December 31, 2016 increased $99.2 million or 42.3%, compared to the same period in 2015. The increase in ALPROLIX revenues in the U.S. was $59.1 million, primarily due to increases in unit sales volume of approximately 28%. The increase in ALPROLIX revenues in all other markets was $40.1 million, due to an increase in unit sales volume in Japan of approximately 68%, as well as uptake following ALPROLIX’s launch in Canada in the first quarter of 2016. |
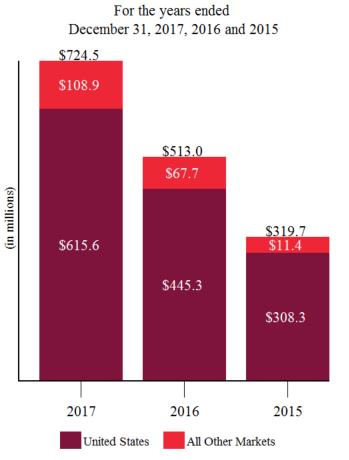
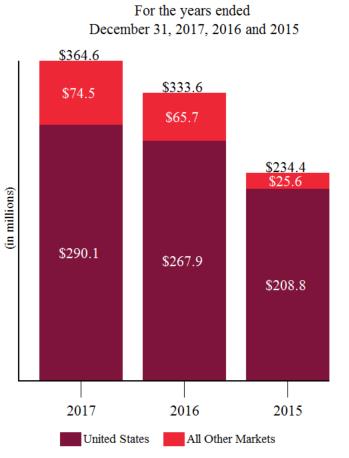
Discounts and allowances
Discounts and allowances for the year ended December 31, 2017 was approximately 31% of gross sales, and approximately 28% of gross sales for the years ended December 31, 2016 and 2015.
46
Revenue from collaborative partners
For the year ended December 31, 2017, compared to the same period in 2016, the increase in revenue from collaborative partners is attributable to a $12.8 million increase in contract manufacturing revenue and $25.8 million of royalty revenue from Sobi as Sobi continued to expand product sales in additional markets in their territory.
For the year ended December 31, 2016, compared to the same period in 2015, the increase in revenue from collaborative partners is attributable to a $29.5 million increase in contract manufacturing revenue and $5.1 million of royalty revenue from Sobi. Revenue from collaborative partners for the year ended December 31, 2015 was solely from contract manufacturing revenue from Sobi.
See Note 4, Collaborations, to the audited consolidated financial statements included elsewhere in this report for additional information on our collaboration with Sobi.
Costs and Expenses
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of sales |
|
$ |
279.6 |
|
$ |
237.9 |
|
$ |
52.9 |
|
17.5 |
% |
|
349.7 |
% |
|
|
Research and development |
|
|
224.6 |
|
|
210.1 |
|
|
236.9 |
|
6.9 |
% |
|
(11.3) |
% |
|
|
Selling, general and administrative |
|
|
217.1 |
|
|
147.0 |
|
|
172.5 |
|
47.7 |
% |
|
(14.8) |
% |
|
|
Total costs and expenses |
|
$ |
721.3 |
|
$ |
595.0 |
|
$ |
462.3 |
|
21.2 |
% |
|
28.7 |
% |
|
Cost of Sales
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
Product |
|
$ |
146.6 |
|
$ |
145.5 |
|
$ |
34.7 |
|
0.8 |
% |
|
319.3 |
% |
|
|
Royalty |
|
|
127.3 |
|
|
87.6 |
|
|
15.2 |
|
45.3 |
% |
|
476.3 |
% |
|
|
Amortization of developed technology |
|
|
5.7 |
|
|
4.8 |
|
|
3.0 |
|
18.8 |
% |
|
60.0 |
% |
|
|
Total cost of sales |
|
$ |
279.6 |
|
$ |
237.9 |
|
$ |
52.9 |
|
17.5 |
% |
|
349.7 |
% |
|
For the year ended December 31, 2017 cost of sales increased $41.7 million compared to the same period in 2016 as a result of increased unit sales of both ELOCTATE and ALPROLIX in our territory and manufacturing revenue in Sobi’s territory partially offset by impairment charges associated with the shutdown of a Biogen Cambridge manufacturing facility in 2016 and lower inventory write-offs.
For the year ended December 31, 2016 cost of sales increased $185.0 million compared to the same period in 2015 as a result of increased unit sales of both ELOCTATE and ALPROLIX, an increase in royalty rate as a result of Sobi’s first commercial sales of ELOCTATE and ALPROLIX, and higher inventory write-offs in 2016 compared to 2015 attributable to excess and obsolete inventory. Also included in product cost of sales in the year ended December 31, 2016 is approximately $41 million related to the shutdown of Biogen’s Cambridge manufacturing facility.
Royalty cost of sales consists mainly of our royalty to Sobi. Upon Sobi’s first commercial sale in 2016, and during the Reimbursement Period, the royalty rate the company will pay Sobi on sales of ELOCTATE and ALPROLIX in the Bioverativ North American Territory is 7% and Bioverativ Direct Territory is 12%. After the Reimbursement Period concludes, the royalty rate we pay to Sobi increases by 5%. For the year ended December 31, 2017, we are recording cost of sales at the effective royalty rate of approximately 11%. For the year ended December 31, 2016, the
47
effective royalty rate was approximately 10%. Please refer to Note 4, Collaborations, to the audited consolidated financial statements included elsewhere in this report for further information regarding our royalty structure with Sobi.
Inventory and inventoriable costs written down as a result of excess, obsolescence, unmarketability or other reasons are charged to cost of sales. There were $8.8 million, $13.5 million and $1.3 million of write-offs in the years ended December 31, 2017, 2016 and 2015, respectively.
Research and Development Expenses
|
|
|
For the Years Ended |
|
|||||||
|
|
|
December 31, |
|
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Upfront and milestone payments |
|
$ |
16.9 |
|
$ |
1.3 |
|
$ |
— |
|
|
Research and discovery |
|
|
63.7 |
|
|
49.5 |
|
|
30.8 |
|
|
Early stage programs |
|
|
26.4 |
|
|
— |
|
|
— |
|
|
Late stage programs |
|
|
17.6 |
|
|
— |
|
|
— |
|
|
Marketed programs |
|
|
76.5 |
|
|
70.6 |
|
|
121.6 |
|
|
Changes in contingent consideration |
|
|
9.9 |
|
|
— |
|
|
— |
|
|
Other research and development expenses |
|
|
13.6 |
|
|
88.7 |
|
|
84.5 |
|
|
Total research and development |
|
$ |
224.6 |
|
$ |
210.1 |
|
$ |
236.9 |
|
Research and discovery includes costs incurred to support our discovery research and translational science efforts up to the initiation of Phase 1 development. Early stage programs are programs in Phase 1 or Phase 2 development activities. Late stage programs are programs in Phase 3 development or in registration stage. Marketed programs are programs in support of our marketed products, including costs associated with product lifecycle management activities and, if applicable, costs associated with the development of new indications for existing products. Other research and development expenses consist mainly of allocations from Biogen prior to the separation and include costs not directly attributable to individual projects and depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. Costs are reflected in the development stage based upon the program status when incurred. Therefore, the same program could be reflected in different development stages in the same year.
For 2017 compared to 2016, the decrease in research and development was mainly due to a decrease in allocations from Biogen offset by an increase due to incremental investments in research collaborations and incremental investments to advance the pipeline.
For 2016 compared to 2015, the decrease in research and development is primarily related to a decrease in workforce and clinical costs associated with post-marketing studies partially offset by an increase in research and discovery driven by an increase in work force associated with a clinical product manufacturing campaign and increased allocations from Biogen.
Selling, General and Administrative Expenses
|
|
|
For the Years Ended |
|
Percent Change |
|
|||||||||||
|
|
|
December 31, |
|
2017 compared |
|
2016 compared |
|
|||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
2015 |
|
to 2016 |
|
to 2015 |
|
|||||
|
Selling, general and administrative |
|
$ |
217.1 |
|
$ |
147.0 |
|
$ |
172.5 |
|
47.7 |
% |
|
(14.8) |
% |
|
For 2017 compared to 2016, the increase in selling, general and administrative is due to an increase in workforce and higher fees and services partially offset by a decrease in allocations from Biogen.
For 2016 compared to 2015, the decrease in selling, general and administrative expenses was mainly due to decreases in workforce and third party service providers as well as decreases in allocations from Biogen. The decrease in allocations from Biogen is due to lower Biogen expenses partially offset by higher Bioverativ allocation rates.
48
Income Taxes
We recorded income tax expense (benefit) of $96.3 million, $(147.7) million and $(10.0) million for the years ended December 31, 2017, 2016 and 2015, respectively. Our effective income tax rate was 21.3%, (50.6)% and (10.2)% of income before income taxes for the years ended December 31, 2017, 2016 and 2015, respectively. See Note 11, Income Taxes, in our audited consolidated financial statements included elsewhere in this report for further information regarding our income taxes.
The factors that most significantly impact our effective tax rate include the levels of certain deductions and credits, and changes in tax laws.
The Tax Cuts and Jobs Act of 2017 (the 2017 Tax Act), which was signed into law on December 22, 2017, has resulted in significant changes to the U.S. corporate income tax system. These changes include a federal statutory rate reduction from 35% to 21%, the elimination or reduction of certain domestic deductions and credits and limitations on the deductibility of interest expense and executive compensation. The 2017 Tax Act also transitions international taxation from a worldwide system to a modified territorial system and includes base erosion prevention measures on non-U.S. earnings. These changes are effective beginning in 2018. The 2017 Tax Act also includes a one-time mandatory deemed repatriation tax on accumulated foreign subsidiaries' previously untaxed foreign earnings (the Transition Toll Tax). Changes in tax rates and tax laws are accounted for in the period of enactment. During the year ended December 31, 2017, we recorded a benefit totaling $58.8 million related to our preliminary estimate of the provisions of the 2017 Tax Act. The preliminary estimate of Transition Toll Tax was immaterial.
Our preliminary estimate of the Transition Toll Tax and the remeasurement of our deferred tax assets and liabilities is subject to the finalization of management's analysis related to certain matters, such as developing interpretations of the provisions of the 2017 Tax Act, changes to certain estimates and amounts related to the earnings and profits of certain subsidiaries and the filing of our tax returns. U.S. Treasury regulations, administrative interpretations or court decisions interpreting the 2017 Tax Act may require further adjustments and changes in our estimates, which could have a material adverse effect on our business, results of operations or financial conditions. The final determination of the Transition Toll Tax and the remeasurement of our deferred tax assets and liabilities will be completed as additional information becomes available, but no later than one year from the enactment of the 2017 Tax Act.
Our effective tax rate for 2017 compared to 2016 fluctuated primarily due to a $58.8 million one-time deferred tax revaluation based on recent tax law changes in 2017 and the release of the $247.3 million valuation allowance on our domestic deferred tax assets in 2016. Our effective tax rate for 2016 compared to 2015 benefitted primarily due to the release of a $247.3 million valuation allowance release in 2016.
Liquidity and Capital Resources
Prior to the separation from Biogen, we participated in Biogen’s centralized treasury management, including centralized cash pooling and overall financing arrangements. Since the third quarter of 2015, we have generated and expect to continue to generate positive cash flow from operations on an annual basis. Net cash used for financing activities in the historical periods prior to the separation primarily reflects changes in Biogen’s investment in us. We had no reported cash or cash equivalents on our balance sheet as of December 31, 2016 due to our participation in Biogen’s centralized treasury management.
Subsequent to the separation from Biogen, we are no longer participating in cash management and funding arrangements with Biogen. Our ability to fund our operations and capital needs depends on our ongoing ability to generate cash from operations and access to capital markets, as further described under the “Debt and Capital” caption directly below. We anticipate that our principal uses of cash in the future will be primarily to fund our operations, working capital needs, capital expenditures and strategic investments.
49
Debt and Capital
On June 28, 2017, we entered into a credit agreement with Bank of America, which provides for a $175.0 million unsecured, revolving credit facility. See Note 5, Debt and Credit Agreements. As of December 31, 2017, there were no amounts outstanding under the Credit Facility.
Historical Cash Flow Trends
|
|
|
For the Years Ended |
|
|||||||
|
|
|
December 31, |
|
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Net cash flows provided by operating activities |
|
$ |
590.7 |
|
$ |
301.6 |
|
$ |
41.4 |
|
|
Net cash flows used in investing activities |
|
$ |
(408.8) |
|
$ |
(35.2) |
|
$ |
(10.6) |
|
|
Net cash flows provided by (used in) financing activities |
|
$ |
253.6 |
|
$ |
(266.4) |
|
$ |
(30.8) |
|
Net cash provided by operations increased $289.1 million during the year ended December 31, 2017 as compared to the prior period driven primarily by changes in working capital of $294.9 million.
Net cash used for investing activities increased $373.6 million during the year ended December 31, 2017 as compared to the prior period mainly due to the acquisition of True North and purchases of property, plant and equipment.
The $520.0 million increase in net cash provided (used) for financing activities for the year ended December 31, 2017 as compared to the same period in 2016 is mainly due to the initial capitalization from Biogen of $325.0 million and decrease of $221.1 million in pre-separation transfers to Biogen partially offset by working capital payments between us and Biogen of $23.5 million.
Net cash provided by operations increased $260.2 million during the year ended December 31, 2016 as compared to the prior period driven primarily by increases in net income of $331.0 million, increases in depreciation and amortization expense of $37.8 million and increases in working capital of $24.1 million partially offset by a $154.2 million decrease in deferred taxes.
Net cash used for investing activities increased $24.6 million during the year ended December 31, 2016 as compared to the prior period mainly due to the acquisition of intangible assets of $26.5 million in 2016.
Net cash used for financing activities represents amounts due to Biogen resulting from our operations. The $235.6 million increase during the year ended December 31, 2016 as compared to the prior period is mainly due to an increase in cash provided by operations.
We expect that our future cash from operations and access to capital markets will provide adequate resources to fund our ongoing cash flow obligations for the foreseeable future.
50
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2017, excluding funding commitments and contingent regulatory milestone payments, as described below.
|
|
|
Payments due by period |
|||||||||||||
|
(In millions) |
|
Total |
|
Less than 1 year |
|
1 to 3 years |
|
3 to 5 years |
|
After 5 years |
|||||
|
Non-cancellable operating leases(1) |
|
$ |
56.4 |
|
$ |
6.6 |
|
$ |
12.8 |
|
$ |
12.1 |
|
$ |
24.9 |
|
Purchase and other obligations(2) |
|
|
426.3 |
|
|
426.3 |
|
|
— |
|
|
— |
|
|
— |
|
Net minimum payments |
|
$ |
482.7 |
|
$ |
432.9 |
|
$ |
12.8 |
|
$ |
12.1 |
|
$ |
24.9 |
|
(1) |
We lease property and equipment for use in our operations. Amounts reflected in the table above detail future minimum rental commitments under non‑cancelable operating leases as of December 31, 2017 for each period presented. In addition to the minimum rental commitments, these leases may require us to pay additional amounts for taxes, insurance, maintenance and other operating expenses. |
|
(2) |
Purchase and other obligations primarily include our obligations to purchase materials or services, which include approximately $410 million of future inventory purchases from Biogen. |
Funding Commitments
True North Therapeutics
In connection with our acquisition of True North, the True North equityholders are eligible to receive additional payments of up to $425.0 million contingent on the achievement of future development, regulatory and sales milestones.
Contingent Development, Regulatory and Commercial Milestone Payments
Based on our development plans primarily in gene editing and gene therapy for hemophilia and other blood disorders as of December 31, 2017, we could make potential future milestone payments to third party collaborators of up to approximately $755 million. Payments under these agreements generally become due and payable upon achievement of certain development, regulatory or commercial milestones. Amounts related to contingent milestone payments are not included in the contractual obligations table as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones. The milestones are comprised of the following:
|
|
|
As of |
|
|
|
|
|
December 31, |
|
(In millions) |
|
2017 |
|
|
Development |
|
$ |
215.0 |
|
Regulatory |
|
|
238.0 |
|
Commercial |
|
|
302.0 |
|
Total |
|
$ |
755.0 |
Sangamo Therapeutics, Inc.
In January, 2014, Biogen MA Inc., a subsidiary of Biogen, entered into an exclusive worldwide research, development and commercialization collaboration and license agreement with Sangamo under which both companies agreed to develop and commercialize product candidates for the treatment of two inherited blood disorders, sickle cell disease and beta-thalassemia. In connection with the separation, we succeeded to Biogen's rights and obligations under this agreement.
Under the collaboration, Sangamo is responsible for identifying a product candidate for the treatment of beta-thalassemia and advancing that candidate through a completed Phase 1 human clinical trial, at which point we would assume responsibility for development. The collaboration contemplates our joint development of a sickle cell disease
51
candidate through the potential filing of an investigational new drug application, after which we would assume clinical responsibilities. We have the right to lead the global development and commercialization efforts and Sangamo has the option to assume co-promotion responsibilities in the United States. The collaboration is currently in the pre-clinical stage of development.
Under the terms of the agreement, Sangamo is eligible to receive up to $215.0 million related to development, regulatory and commercialization milestones as well as tiered low to mid double-digit royalties related to product sales.
Bicycle Therapeutics Research Collaboration
In August 2017, we entered into a research collaboration agreement with Bicycle Therapeutics (Bicycle), a biotechnology company that is pioneering a new class of medicines based on its novel bicyclic peptide (Bicycle®) product platform. The collaboration will focus on the discovery, development and commercialization of therapies for hemophilia and sickle cell disease. Under this agreement, Bicycle will be responsible for leading initial discovery activities through lead optimization to candidate selection for three programs. Bioverativ will lead preclinical and clinical development, as well as subsequent marketing and commercialization efforts. Under the agreement, we paid Bicycle a $10.0 million upfront payment. Bicycle is eligible to receive up to $410.0 million related to development, regulatory and commercialization milestones as well as tiered single to low double-digit royalties related to product sales. Bioverativ will also reimburse Bicycle for internal and external program-related costs.
Payments under these agreements generally become due and payable upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones had not occurred as of December 31, 2017, such contingencies have not been recorded in our financial statements. Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones.
Critical Accounting Policies and Estimates
The preparation of our consolidated financial statements, which have been prepared in accordance with U.S. GAAP requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. A summary of our significant accounting policies is included in Note 2, Summary of Significant Accounting Policies, to the audited consolidated financial statements included elsewhere in this report. Certain of our accounting policies are considered critical because these policies are the most important to the depiction of our financial statements and require significant, difficult or complex judgments by us, often requiring the use of estimates about the effects of matters that are inherently uncertain. On an ongoing basis we evaluate our estimates, judgments and methodologies. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable, the results of which form the basis for making judgments about the carrying value of assets, liabilities and equity and the amount of revenues and expenses. Actual results that differ from our estimates could have an unfavorable effect on our results of operations and financial position. We apply estimation methodologies consistently from year to year. The following is a summary of accounting policies that we consider critical to the consolidated financial statements.
Revenue Recognition
The company recognizes revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the price to the customer is fixed or determinable; and collectability is reasonably assured.
Product Revenues
We sell mainly to specialty pharmacies, hemophilia treatment centers, public and private hospitals and independent specialty distributors. Any discounts offered to these customers are reflected as on‑invoice discounts. We also sell to specialty distributors who receive both on‑invoice discounts as well as chargebacks for sales to various U.S. government agencies such as U.S. Public Health Service (PHS). Provisions for rebates, chargebacks to distributors, and discounts are provided for at the time the related sales are recorded, and are reflected as a reduction of sales. Reserves
52
established for these discounts and allowances are classified as reductions of accounts receivable (if the amount is payable to our customer) or a liability (if the amount is payable to a party other than our customer). Our estimates take into consideration our historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Actual amounts may ultimately differ from our estimates. If actual results vary, we adjust these estimates, which could have an effect on earnings in the period of adjustment. To date historical adjustments have not been material.
Product revenue reserves are categorized as follows: discounts and contractual adjustments. Discounts include trade term discounts and volume discounts. Trade term discounts relate to estimated obligations for credits to be granted to customers for remitting payment on their purchases within established incentive periods. Volume discounts are earned as customers reach certain tier levels based upon their purchases. Contractual adjustments primarily relate to Medicaid and PHS discounts. Product returns are insignificant and mainly resulting from product damaged in transit or incorrectly shipped.
Accounts Receivable
The majority of accounts receivable arise from product sales and primarily represent amounts due from specialty pharmacies, hemophilia treatment centers, public and private hospitals and independent specialty distributors as well as contract manufacturing revenue from Sobi. The company monitors the financial performance and creditworthiness of its customers so that the company can properly assess and respond to changes in their credit profile. The company provides reserves against trade receivables for estimated losses that may result from a customer’s inability to pay. Amounts determined to be uncollectible are charged or written‑off against the reserve.
Concentration of Credit Risk
CVS Health Corporation (CVS), a specialty pharmacy and ASD Specialty Healthcare (ASD), a specialty distributor, individually represent 13% and 12%, respectively, of outstanding accounts receivable for the year ended December 31, 2017; CVS, ASD and Accredo Health Incorporated, a specialty pharmacy, individually represent 24%, 14% and 11%, respectively, of outstanding accounts receivable for the year ended December 31, 2016. Other than these significant customers, concentration of credit risk with respect to receivables, which are typically unsecured, is largely mitigated due to the wide variety of customers. The majority of accounts receivable currently arise from product sales in the United States, Japan and Canada and have standard payment terms which generally require payment within 30 to 90 days. We monitor the financial performance and creditworthiness of our customers so that we can properly assess and respond to changes in their credit profile. We continue to monitor these conditions and assess their possible impact on our business.
Inventory
Inventories are stated at the lower of cost or market with cost based on the first‑in, first‑out (FIFO) method. Inventory that can be used in either the production of clinical or commercial products is expensed as research and development costs when selected for use in a clinical manufacturing campaign.
Capitalization of Inventory Costs
The company capitalizes inventory costs associated with its products prior to regulatory approval, when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. In determining whether or not to capitalize such costs, the company evaluates, among other factors, information regarding the drug candidate’s safety and efficacy, the status of regulatory submissions and communications with regulatory authorities and the outlook for commercial sales, including the existence of current or anticipated competitive drugs and the availability of reimbursement. In addition, the company evaluates risks associated with manufacturing the drug candidate and the remaining shelf‑life of the inventories.
53
Obsolescence and Unmarketable Inventory
The company periodically reviews its inventories for excess or obsolescence and write‑down obsolete or otherwise unmarketable inventory to its estimated net realizable value.
Acquired Intangibles
Acquired intangibles include product rights, patents and in-process research and development (IPR&D) acquired in a business combination. IPR&D represents the fair value of research and development assets that have not reached technological feasibility. The value assigned to acquired IPR&D is determined by estimating the costs to develop the acquired technology into a commercial product, estimating revenue and costs associated with the product, adjusting the net cash flows for probability of success and discounting to present value. The estimated net cash flows consider the relevant market sizes and growth factors, expected trends in technology and the nature and expected timing of new product introductions by us and our competitors. The rates utilized to discount the net cash flows to their present value are commensurate with the stage of development of the projects and uncertainties in the economic estimates used in the projections. In estimating the probability of success, we utilize data regarding similar assets events from several sources, including industry studies and our own experience.
Upon the acquisition of intangible assets, we complete an assessment of whether our acquisition constitutes the purchase of a single asset or a group of assets. We consider multiple factors in this assessment, including the nature of the technology acquired, the presence or absence of separate cash flows, the development process and stage of completion, quantitative significance and our rationale for entering into the transaction. If we acquire a business as defined under applicable accounting standards, then the acquired IPR&D is capitalized as an intangible asset. If we acquire an asset or group of assets that do not meet the definition of a business, then the acquired IPR&D is expensed on its acquisition date if it has no future alternative use. Future costs to develop these assets are recorded to research and development as they are incurred. When performing our impairment assessment, we calculate the fair value using the same methodology as described above. If the carrying value of our acquired IPR&D exceeds its fair value, then the intangible asset is written-down to its fair value.
Acquired product rights and patents are recorded at fair value, assigned an estimated useful life, and are amortized over their estimated useful lives of approximately twelve years. See Note 9, Intangible Assets. Amortization is included in cost of sales in the consolidated statements of income.
Goodwill
Goodwill represents the difference between the purchase price and the fair value of the identifiable tangible and intangible net assets when accounted for using the purchase method of accounting. Goodwill is not amortized, but reviewed for impairment. Goodwill is reviewed annually and whenever events or changes in circumstances indicate that the carrying value of the goodwill might not be recoverable.
We compare the fair value of our reporting unit to its carrying value. If the carrying value of the net assets assigned to the reporting unit exceeds the fair value of our reporting unit, then we would need to determine the implied fair value of our reporting unit’s goodwill. If the carrying value of our reporting unit’s goodwill exceeds its implied fair value, then we would record an impairment loss equal to the difference. As described above in Note 1, Nature of Business and Basis of Preparation, we operate in one operating segment which we consider our only reporting unit.
Impairment of Long‑Lived Assets
Long‑lived assets to be held and used, including property, plant and equipment and definite‑lived intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets or asset group may not be recoverable.
Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover
54
the carrying amount of the assets, the assets are written‑down to their fair values. Long‑lived assets to be disposed of are carried at fair value less costs to sell.
Contingent Consideration
The consideration for our acquisition includes future payments that are contingent upon the occurrence of various events. We record a liability for such contingent payments at fair value on the acquisition date. We estimate the fair value of contingent consideration through valuation models that incorporate probability-adjusted assumptions related to the likelihood of achieving the milestones and making related payments. Our contingent consideration is remeasured each reporting period. Changes in the fair value of our contingent consideration are recognized in our condensed consolidated statements of income. Changes in the fair value of the contingent consideration can result from changes to one or multiple inputs, including changes to the discount rates, changes in the probability of certain clinical events, regulatory approval, and the amount or timing of development and sales based milestones. Discount rates in our valuation models represent a measure of the weighted average cost of capital of the target associated with settling the liability. The period over which we discount our contingent consideration is based on the current development stage of the product candidates and our specific development plan for that product candidate. In estimating the probability of success, we utilize data regarding similar milestone events from several sources, including industry studies and our own experience. These fair value measurements are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we record in any given period.
Income Taxes
Deferred taxes are recognized for the future tax effects of temporary differences between financial and income tax reporting based on enacted tax laws and rates. With respect to uncertain tax positions, the company determines whether the position is more likely than not to be sustained upon examination, based on the technical merits of the position. Any tax position that meets the more‑likely‑than‑not recognition threshold is measured and recognized in the consolidated financial statements at the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement. The liability relating to uncertain tax positions is classified as current in the consolidated balance sheets to the extent the company anticipates making a payment within one year. Interest and penalties associated with income taxes are classified in the income tax expense line in the consolidated statements of income.
Prior to the separation, the company’s income tax expense and deferred tax balances were calculated on a separate tax return basis although the company’s operations had historically been included in the tax returns filed by the respective Biogen entities, of which the company’s business was a part. For periods subsequent to the separation, Bioverativ files tax returns on its own behalf and its income tax expense and deferred income tax balances are recorded in accordance with the company’s standalone income tax positions. Biogen and Bioverativ entered into a tax matters agreement effective as of the date of separation.
Accounting for Share‑Based Compensation
In connection with the separation, we adopted our own share‑based compensation plans. Outstanding Biogen Cash-Settled Performance Units (CSPUs), Market Stock Units (MSUs) and Performance-Vested Restricted Stock Units (PUs) were converted to Bioverativ Restricted Stock Units (RSUs), and outstanding Biogen stock options were converted to Bioverativ stock options. See Note 13, Share‑Based Compensation.
We charge the estimated fair value of awards against income over the requisite service period, which is generally the vesting period. Where awards are made with non‑substantive vesting periods (for instance, where a portion of the award vests upon retirement eligibility), we estimate and recognize expense based on the period from the grant date to the date on which the employee is retirement eligible. We recognize forfeitures as they occur.
The fair values of our stock option grants are estimated as of the date of grant using a Black‑Scholes option valuation model. The estimated fair values of the stock options are then expensed over the options’ vesting periods. The
55
fair values of RSUs are based on the market value of our stock on the date of grant. Compensation expense for RSUs is recognized on a straight‑line basis over the applicable service period.
Fair Value Measurements
We have certain financial assets and liabilities recorded at fair value which have been classified as Level 1, 2 or 3 within the fair value hierarchy as described in the accounting standards for fair value measurements.
|
· |
Level 1 – Fair values are determined by utilizing quoted prices (unadjusted) in active markets for identical assets or liabilities that we have the ability to access; |
|
· |
Level 2 – Fair values are determined by utilizing quoted prices for similar assets and liabilities in active markets or other market observable inputs such as interest rates, yield curves and foreign currency spot rates; and |
|
· |
Level 3 – Prices or valuations that require inputs that are both significant to the fair value measurement and unobservable. |
As noted in Note 6, Financial instruments and fair value measurements to our consolidated financial statements, the majority of our financial assets have been classified as Level 2. Our financial assets (which include our cash equivalents, plan assets for deferred compensation and derivative contracts) have been initially valued at the transaction price and subsequently valued, at the end of each reporting period, utilizing market observable data. These observable market inputs include reportable trades, benchmark yields, credit spreads, broker/dealer quotes, bids, offers, current spot rates and other industry and economic events.
For contingent consideration obligations, the fair value measurements are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the applicable acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we record in any given period. As of December 31, 2017, we have the contingent consideration of $198.4 million, which represents substantially all liabilities measured at fair value.
New Accounting Standards
For a discussion of new accounting standards please read Note 2, Summary of Significant Accounting Policies, to the audited consolidated financial statements included elsewhere in this report.
Off‑Balance Sheet Arrangements
We do not have any relationships with entities often referred to as structured finance or special purpose entities that were established for the purpose of facilitating off‑balance sheet arrangements. As such, we are not exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Market Risk
We are subject to certain risks which may affect our results of operations, cash flows and fair values of assets and liabilities, including volatility in foreign currency exchange rates, interest rate movements, pricing pressures worldwide and weak economic conditions in the foreign markets in which we operate.
56
Foreign Currency Exchange Risk
Our results of operations are subject to foreign currency exchange rate fluctuations due to the global nature of our operations. We currently have operations in the United States, Japan, Canada, Australia, Brazil and Colombia. As a result, our financial position, results of operations and cash flows can be affected by market fluctuations in foreign exchange rates. While the financial results of our global activities are reported in U.S. dollars, the functional currency for our foreign subsidiaries is their respective local currency. Fluctuations in the foreign currency exchange rates of the countries in which we do business will affect our operating results, often in ways that are difficult to predict. In particular, as the U.S. dollar strengthens versus other currencies, the value of the non‑U.S. revenue will decline when reported in U.S. dollars. The impact to net income as a result of a strengthening U.S. dollar will be partially mitigated by the value of non‑U.S. expense which will also decline when reported in U.S. dollars. As the U.S. dollar weakens versus other currencies, the value of the non‑U.S. revenue and expenses will increase when reported in U.S. dollars.
Currency forward contracts are used as a part of our strategy to manage exposure related to foreign currency denominated monetary assets and liabilities. These currency forward contracts are not designated as cash flow, fair value or net investment hedges. The currency forward contracts are marked-to-market with changes in fair value recorded to other income (expense) and are entered into for periods consistent with currency transaction exposures, generally less than one year.
Pricing Pressure
In the United States, federal and state legislatures, health agencies and third party payors continue to focus on containing the cost of health care. Legislative and regulatory proposals, enactments to reform health care insurance programs and increasing pressure from political and social sources could significantly influence the manner in which our products are prescribed and purchased. It is possible that additional federal health care reform measures will be adopted in the future, which could result in increased pricing pressure and reduced reimbursement for our products and otherwise have an adverse impact on our financial position or results of operations.
There is also significant economic pressure on state budgets that may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for our drugs.
Governments in some international markets in which we operate have also implemented measures aimed at reducing healthcare costs to constrain the overall level of government expenditures. These implemented measures vary by country and include, among other things, mandatory rebates and discounts, prospective and possible retroactive price reductions and suspensions on price increases of pharmaceuticals.
Interest Rate Risk
In June 2017, we entered into a $175.0 million revolving credit facility. Borrowings under the Credit Facility bear interest, at our option, at either a base rate or a Eurocurrency rate, in each case plus an applicable margin. Under the Credit Facility, the applicable margin on base rate loans ranges from 0.50% to 1.00% and the applicable margin on Eurocurrency loans ranges from 1.50% to 2.00%, in each case, based on our consolidated leverage ratio (the ratio of our total consolidated funded indebtedness to our consolidated EBITDA for the most recently completed four fiscal quarter period).
Credit Risk
We are subject to credit risk from our accounts receivable related to our product sales and contract manufacturing and royalty revenue from Sobi. The majority of our accounts receivable arise from product sales in the U.S. and Japan as well as contract manufacturing and royalty revenue from Sobi with concentration of credit risk limited due to the wide variety of customers using our products. Our accounts receivable are primarily due from wholesale distributors, public hospitals and other government entities. We monitor the financial performance and creditworthiness of our customers so that we can properly assess and respond to changes in their credit profile. To date, we have not experienced any significant losses with respect to the collection of our accounts receivable.
57
Item 8. Financial Statements and Supplementary Data
The information required by this Item 8 is contained on pages F‑1 through F‑36 of this report and is incorporated herein by reference.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Bioverativ has carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer (principal executive officer) and Chief Financial Officer (principal financial officer), of the effectiveness of the design and operation of Bioverativ's disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the Exchange Act)) as of December 31, 2017. Based on that evaluation the Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2017, the company's disclosure controls and procedures were effective to provide reasonable assurance that (a) the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and (b) such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, a company’s principal executive and principal financial officers and effected by a company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
|
· |
pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets |
|
· |
provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
|
· |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
58
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in its 2013 Internal Control — Integrated Framework.
Based on our assessment, our management has concluded that, as of December 31, 2017, our internal control over financial reporting was effective based on those criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
None.
59
Item 10. Directors, Executive Officers and Corporate Governance
Executive Officers
The following sets forth information regarding our executive officers, including their positions and age as of February 9, 2018.
|
Name |
|
Age |
|
Title |
|
John G. Cox |
|
55 |
|
Director and Chief Executive Officer |
|
John T. Greene |
|
52 |
|
Executive Vice President, Chief Financial Officer |
|
Rogério Vivaldi Coelho, M.D. |
|
54 |
|
Executive Vice President and Chief Global Therapeutic Operations Officer |
|
Richard Brudnick |
|
61 |
|
Executive Vice President of Business Development |
|
Lucia Celona |
|
51 |
|
Executive Vice President, Chief Human Resources and Corporate Communications Officer |
|
Andrea DiFabio |
|
49 |
|
Executive Vice President and Chief Legal Officer |
|
Timothy Harris |
|
67 |
|
Executive Vice President of Research and Development |
John G. Cox became a director and the Chief Executive Officer of Bioverativ in August 2016. Previously, from June 2010 through June 2016, Mr. Cox served on Biogen’s executive committee as Executive Vice President, Pharmaceutical Operations & Technology, where he oversaw Biogen’s global manufacturing facilities, supply chain operations, technical development, quality and engineering. Mr. Cox also had responsibility for the development and commercialization of Biogen’s biosimilar business. From October 2015 to May 2016, Mr. Cox also served as interim Executive Vice President, Global Therapeutic Operations, responsible for Biogen’s therapeutic groups of Specialty Medicines and Rare Diseases, which included responsibility for the global commercial performance of Biogen’s marketed products. Mr. Cox joined Biogen in 2003, holding several senior executive positions including Senior Vice President of Technical Operations, Senior Vice President of Global Manufacturing and Vice President of Manufacturing and General Manager of Biogen’s operations in Research Triangle Park, North Carolina. He reported to Biogen’s Chief Executive Officer as a member of Biogen’s executive committee, since 2010. Mr. Cox’s qualifications for service on our board of directors include his substantial executive, operational, business and technical experience and acumen in the biotechnology industry. Mr. Cox serves on the board of directors of Repligen Corporation, a life sciences company.
John T. Greene became Executive Vice President, Chief Financial Officer of Bioverativ in November 2016. Prior to joining Bioverativ, Mr. Greene served as the Chief Financial Officer of Willis Group Holdings (Willis), a multinational risk advisory, insurance and reinsurance brokerage firm, from June 2014 until January 2016. Following the merger of Willis with Towers Watson in January 2016, Mr. Greene served as a transition advisor to the new management team until May 2016. Before joining Willis, he served for over eight years in senior executive roles at HSBC, a global financial services company, including as Chief Financial Officer of HSBC Retail Bank and Wealth Management from May 2011 to May 2014, Chief Financial Officer of HSBC Insurance from August 2009 until May 2011 and Chief Financial Officer of HSBC’s Consumer and Mortgage Lending business prior to August 2009. Before joining HSBC, Mr. Greene served for 12 years in various roles at General Electric Company, including as Chief Financial Officer for GE Global Business Finance.
Rogério Vivaldi Coelho, M.D. became Executive Vice President, Chief Global Therapeutic Operations Officer of Bioverativ in October 2016. Prior to joining Bioverativ, Dr. Vivaldi Coelho served as Chief Commercial Officer of Spark Therapeutics, Inc. (Spark) from December 2014 to October 2016. Prior to joining Spark, Dr. Vivaldi Coelho was Chief Executive Officer and President of Minerva Neurosciences, Inc. from November 2013 to December 2014. Prior to joining Minerva, Dr. Vivaldi Coelho served as Senior Vice President—Head of the Rare Diseases business unit at Genzyme Corporation (Genzyme), from October 2011 to October 2013. From July 2010 to September 2011, he was the Senior Vice President—Head of the Renal and Endocrinology business unit at Genzyme and from January 2004 to June 2010 he was the Senior Vice President—Head of Genzyme Latin America. Previously, Dr. Vivaldi Coelho founded Genzyme Brazil in 1997. Dr. Vivaldi Coelho served on Spark’s board of directors from April 2014 to December 2014 and served on the board of directors of Minerva Neurosciences from November 2013 to December 2014. Dr. Vivaldi Coelho holds a medical degree from the University of Rio de Janeiro and his M.B.A. from Federal University of Rio de
60
Janeiro. Dr. Vivaldi Coelho completed his residency in metabolism and endocrinology at Rio de Janeiro State University and his fellowship at Mount Sinai Hospital Center in New York, department of genetics, with an emphasis on Gaucher’s disease.
Richard Brudnick became the Executive Vice President, Business Development of Bioverativ in January 2017. Prior to that, Mr. Brudnick served as Biogen’s Senior Vice President of Corporate Development since 2014. Mr. Brudnick joined Biogen in 2001 and held senior positions in the areas of Portfolio Strategy & Business Development and Corporate Development. Before joining Biogen, Mr. Brudnick was the Chief Executive Officer of a regional pharmaceutical distribution business, a co‑founder of two companies and a strategy consultant at Bain & Company.
Lucia Celona became Executive Vice President, Chief Human Resources and Corporate Communications Officer of Bioverativ in January 2017. Prior to that, Ms. Celona served as the Vice President of Human Resources for the Pharmaceutical Operations and Technology function of Biogen from 2013 through June 2016. Ms. Celona joined Biogen in 2006 and held senior level positions including Vice President, Talent Acquisition & Global Mobility, interim Vice President of Compensation & Benefits, Chief of Staff to the Executive Vice President of Human Resources and Executive Vice President of Global Therapeutic Operations. Before joining Biogen, Ms. Celona was head of human resources for Sentillion Inc., a privately held company that focused on single‑sign on solutions for healthcare providers, and the human resources director for two manufacturing sites at Philips Medical Systems in Andover, Massachusetts.
Andrea DiFabio became Executive Vice President and Chief Legal Officer of Bioverativ in January 2017. Prior to that, Ms. DiFabio served as Biogen’s Senior Vice President, Chief Research and Business Development Counsel since 2015. Ms. DiFabio joined Biogen in 2006 and held various positions including serving as Chief U.S. Counsel, Interim Chief Compliance Officer and Chief Research and Development Counsel. Prior to Biogen, she was Corporate Vice President and Deputy General Counsel at Parexel International, a global contract research organization. Prior to her work at Parexel International, Ms. DiFabio was a trial lawyer for two Boston law firms, Brown Rudnick and Robins, Kaplan Miller & Ceresi.
Timothy J.R. Harris, Ph.D., D.Sc. joined Bioverativ in February 2017 as interim Head of Research & Development, and was appointed Executive Vice President, Research and Development in April 2017. Prior to joining Bioverativ, Dr. Harris was a venture partner at SV Health Investors, since March 2016. Prior to that, Dr. Harris was Senior Vice President for Precision Medicine at Biogen Inc. from March 2015 until February 2016. Prior to such position, he was Senior Vice President for Translational Medicine and Technology at Biogen from June 2011 to February 2015. Before joining Biogen, he was the Chief Technical Officer and Director of the Advanced Technology Program at SAIC-Frederick in Maryland from January 2007 to June 2011. His professional experience includes senior executive positions at a number of companies, including Novasite Pharmaceuticals, where he served as President and Chief Executive Officer from 2005 to September 2006. Dr. Harris founded SGX Pharmaceuticals, Inc. (formerly Structural GenomiX Inc.) (SGX) in 1999, and was Chief Executive Officer of that company until 2005. Before founding SGX, Dr. Harris was Senior Vice President, Research and Development at Axys Pharmaceuticals Inc. (formerly Sequana Therapeutics Inc.). Dr. Harris serves on the Board of Directors of Opgen Inc. and during the past five years, previously served on the board of directors of BG Medicine, Inc. He is a visiting Professor at Columbia University. Dr. Harris is a molecular biologist and biochemist and an executive with nearly 40 years of scientific and leadership expertise in the biotechnology industry. He holds a Ph. D in molecular virology and a D.Sc., both from the University of Birmingham, UK, and is credited with over 100 publications and five issued patents.
Code of Conduct
We have adopted a code of conduct setting forth standards applicable to all of our companies and our directors, officers and employees. The code of conduct is available on Bioverativ’s website at www.bioverativ.com. Any amendment to the code, or any waiver of its requirements, that applies to our Chief Executive Officer, Chief Financial Officer, Controller or persons performing similar functions will be disclosed on our website.
The response to the remainder of this item is incorporated by reference from the discussion responsive thereto in the sections entitled “Proposal 1 - Election of Directors,” “Corporate Governance at Bioverativ,” “Stock Ownership -
61
Section 16(a) Beneficial Ownership Reporting Compliance” and “Miscellaneous - Stockholder Proposals” contained in an amendment to this Form 10-K or in the proxy statement for our 2018 annual meeting of stockholders.
Item 11. Executive Compensation
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled “Executive Compensation Matters” and “Corporate Governance at Bioverativ” contained in an amendment to this Form 10-K or in the proxy statement for our 2018 annual meeting of stockholders.
Item 12. Security Ownership By Certain Beneficial Owners and Management and Related Stockholder Matters
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled “Stock Ownership” and “Equity Compensation Plan Information” contained in an amendment to this Form 10-K or in the proxy statement for our 2018 annual meeting of stockholders.
Item 13. Certain Relationships, Related Person Transactions, and Director Independence
The response to this item is incorporated by reference from the discussion responsive thereto in the sections entitled “Certain Relationships and Related Person Transactions” and “Corporate Governance at Bioverativ” contained in an amendment to this Form 10-K or in the proxy statement for our 2018 annual meeting of stockholders.
Item 14. Principal Accountant Fees and Services
The response to this item is incorporated by reference from the discussion responsive thereto in the section entitled “Proposal 2 — Ratification of the Selection of our Independent Registered Public Accounting Firm” contained in an amendment to this Form 10-K or in the proxy statement for our 2018 annual meeting of stockholders.
GLOSSARY OF SCIENTIFIC TERMS
Below is a list of additional scientific terms and their respective meanings which are used throughout this report.
ALPROLIX: [Coagulation Factor IX (Recombinant), Fc Fusion Protein], our recombinant factor IX therapy for the treatment of hemophilia B.
Beta-Thalassemia: An inherited blood disorder caused by mutations in the gene for beta-globin, a sub-unit of the oxygen-carrying protein of red blood cells, that results in excessive destruction of red blood cells and their precursors that could lead to life-threatening anemia, enlarged spleen, liver and heart, and bone abnormalities.
Biologics: Medical products made from a variety of natural sources (human, animal or microorganism) intended to treat diseases and medical conditions or used to prevent or diagnose diseases; products include vaccines, blood and blood products, allergenic extracts, human cells and tissues, gene therapies and cellular therapies.
Biosimilars: A biological product that is highly similar to a U.S.-licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product.
Bi-specific antibodies: A class of antibodies that combine two antigen-recognizing elements into a single construct to bind two targets at the same time.
Cold agglutinin disease: A global, chronic rare blood disease characterized by hemolytic anemia, with a significant portion of patients requiring transfusions and having crippling fatigue, poor quality of life and the potential for life-threatening complications such as thrombotic events, including pulmonary embolism and stroke.
62
ELOCTATE: [Antihemophilic Factor (Recombinant), Fc Fusion Protein], our recombinant factor VIII therapy for the treatment of hemophilia A. ELOCTA is the approved trade name for ELOCTATE in the European Union.
Extended Half-Life: Prolonged circulation of the replacement clotting factor therapy in the body.
Generic Drug: A small-molecule drug that is the same as, and bioequivalent to, an already-approved small molecule drug.
Hemophilia A: A medical condition that occurs when clotting factor VIII, a naturally occurring protein in blood that controls bleeding, is not present in sufficient amounts or is absent.
Hemophilia B: A medical condition that occurs when clotting factor IX, a naturally occurring protein in blood that controls bleeding, is not present in sufficient amounts or is absent.
Sickle Cell Disease: A group of genetic disorders that occurs due to inheritance of mutations in hemoglobin, the oxygen carrying molecule inside red blood cells, resulting in chronic anemia and vascular obstructive complications.
von Willebrand Factor: A naturally occurring protein in blood and the lining of blood vessels involved in bleeding and clotting that also binds to and stabilizes clotting factor VIII.
63
Item 15. Exhibits and Financial Statement Schedules
The following financial statements are filed as part of this report:
|
|
|
|
F-2 |
|
|
F-3 |
|
|
F-4 |
|
|
F-5 |
|
|
F-6 |
|
|
F-35 |
|
· |
Financial Statement Schedules |
Schedules are omitted because they are not applicable, or are not required, or because the information is included in the consolidated financial statements and notes thereto.
|
· |
Exhibits |
The exhibits listed on the Exhibit Index beginning on page 65, which is incorporated herein by reference, are filed or furnished as part of this report or are incorporated into this report by reference.
None.
64
|
Exhibit |
|
Exhibit Description |
|
2.1** |
|
|
|
2.2** |
|
|
|
2.3** |
|
|
|
2.4** |
|
|
|
2.5** |
|
|
|
2.6** |
|
|
|
2.7** |
|
|
|
2.8** |
|
|
| 3.1 |
|
|
| 3.2 |
|
|
|
10.1+ |
|
|
|
10.2+ |
|
|
|
10.3+ |
|
|
|
10.4+ |
|
|
|
10.5+ |
|
|
|
10.6+ |
|
|
|
10.7+ |
|
|
|
10.8+ |
|
|
|
10.9+ |
|
|
|
10.10+ |
|
|
|
10.11+ |
|
65
|
Exhibit |
|
Exhibit Description |
|
10.12+ |
|
|
|
10.13+ |
|
|
|
10.14+ |
|
|
|
10.15+ |
|
|
|
10.16# |
|
|
| 10.17 |
|
|
| 10.18 |
|
|
| 10.19 |
|
|
|
10.20+ |
|
|
| 21.1 |
|
|
|
23.1* |
|
Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm |
|
31.1* |
|
|
|
31.2* |
|
|
|
32.1*++ |
|
|
| 99.1 |
|
|
| 99.2 |
|
* Filed herewith
** Schedules and exhibits have been omitted pursuant to Item 601(b)(2) of Regulation S‑K. The registrant hereby undertakes to furnish copies of any of the omitted schedules and exhibits upon request by the U.S. Securities and Exchange Commission.
# Confidential treatment has been granted with respect to certain portions of this exhibit
+ Management contract or compensatory plan or arrangement
++ Furnished herewith
66
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
BIOVERATIV INC. |
|
|
|
|
|
|
|
|
|
|
|
By: |
/s/ John G. Cox |
|
|
|
John G. Cox |
|
|
|
Chief Executive Officer |
Date: February 14, 2018
Pursuant to the requirements the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Name |
|
Capacity |
|
Date |
|
|
|
|
|
|
|
/s/ John G. Cox |
|
Director and Chief Executive Officer (principal executive officer) |
|
February 14, 2018 |
|
John G. Cox |
|
|
||
|
|
|
|
|
|
|
/s/ John T. Greene |
|
Executive Vice President, Chief Financial Officer (principal financial officer) |
|
February 14, 2018 |
|
John T. Greene |
|
|
||
|
|
|
|
|
|
|
/s/ Diantha Duvall |
|
Vice President, Chief Accounting Officer (principal accounting officer) |
|
February 14, 2018 |
|
Diantha Duvall |
|
|
||
|
|
|
|
|
|
|
/s/ Brian S. Posner |
|
Director and Chairman of Board of Directors |
|
February 14, 2018 |
|
Brian S. Posner |
|
|
||
|
|
|
|
|
|
|
/s/ Alexander J. Denner |
|
Director |
|
February 14, 2018 |
|
Alexander J. Denner |
|
|
||
|
|
|
|
|
|
|
/s/ Louis J. Paglia |
|
Director |
|
February 14, 2018 |
|
Louis J. Paglia |
|
|
||
|
|
|
|
|
|
|
/s/ Geno Germano |
|
Director |
|
February 14, 2018 |
|
Geno Germano |
|
|
||
|
|
|
|
|
|
|
/s/ Anna Protopapas |
|
Director |
|
February 14, 2018 |
|
Anna Protopapas |
|
|
67
INDEX TO FINANCIAL STATEMENTS
BIOVERATIV INC. AND SUBSIDIARIES
Consolidated Financial Statements
|
F-2 |
|
|
F-3 |
|
|
F-4 |
|
|
F-5 |
|
|
F-6 |
|
|
F-35 |
F-1
BIOVERATIV INC. AND SUBSIDIARIES
Consolidated Statements of Income and Comprehensive Income
(In millions, except per share amounts)
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|||
|
Revenues: |
|
|
|
|
|
|
|
|
|
|
Product, net |
|
$ |
1,089.1 |
|
$ |
846.6 |
|
$ |
554.1 |
|
Collaboration |
|
|
79.4 |
|
|
40.8 |
|
|
6.2 |
|
Total revenues |
|
|
1,168.5 |
|
|
887.4 |
|
|
560.3 |
|
Cost and expenses: |
|
|
|
|
|
|
|
|
|
|
Cost of sales |
|
|
279.6 |
|
|
237.9 |
|
|
52.9 |
|
Research and development |
|
|
224.6 |
|
|
210.1 |
|
|
236.9 |
|
Selling, general and administrative |
|
|
217.1 |
|
|
147.0 |
|
|
172.5 |
|
Total cost and expenses |
|
|
721.3 |
|
|
595.0 |
|
|
462.3 |
|
Income from operations |
|
|
447.2 |
|
|
292.4 |
|
|
98.0 |
|
Other income (expense), net |
|
|
4.7 |
|
|
(0.5) |
|
|
0.6 |
|
Income before income tax expense (benefit) |
|
|
451.9 |
|
|
291.9 |
|
|
98.6 |
|
Income tax expense (benefit) |
|
|
96.3 |
|
|
(147.7) |
|
|
(10.0) |
|
Net income |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
Net income per share: |
|
|
|
|
|
|
|
|
|
|
Basic earnings per share |
|
$ |
3.29 |
|
$ |
4.07 |
|
$ |
1.01 |
|
Diluted earnings per share |
|
$ |
3.28 |
|
$ |
4.07 |
|
$ |
1.01 |
|
Weighted average shares used in calculating: |
|
|
|
|
|
|
|
|
|
|
Basic earnings per share |
|
|
108.1 |
|
|
108.0 |
|
|
108.0 |
|
Diluted earnings per share |
|
|
108.5 |
|
|
108.0 |
|
|
108.0 |
|
Other comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
Currency translation adjustment |
|
|
(1.8) |
|
|
1.5 |
|
|
(0.2) |
|
Total other comprehensive income (loss) |
|
|
(1.8) |
|
|
1.5 |
|
|
(0.2) |
|
Comprehensive income |
|
$ |
353.8 |
|
$ |
441.1 |
|
$ |
108.4 |
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-2
BIOVERATIV INC. AND SUBSIDIARIES
(In millions)
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
|
|
2017 |
|
2016 |
||
|
ASSETS |
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
436.5 |
|
$ |
— |
|
Accounts receivable, net |
|
|
189.4 |
|
|
149.4 |
|
Inventory |
|
|
40.6 |
|
|
302.0 |
|
Restricted cash |
|
|
40.0 |
|
|
— |
|
Other current assets |
|
|
71.8 |
|
|
24.2 |
|
Total current assets |
|
|
778.3 |
|
|
475.6 |
|
Property, plant and equipment, net |
|
|
24.0 |
|
|
28.4 |
|
Intangible assets, net |
|
|
635.9 |
|
|
51.7 |
|
Goodwill |
|
|
170.7 |
|
|
— |
|
Deferred tax assets, net |
|
|
1.0 |
|
|
154.2 |
|
Other long-term assets |
|
|
8.4 |
|
|
22.0 |
|
Total assets |
|
$ |
1,618.3 |
|
$ |
731.9 |
|
LIABILITIES AND EQUITY |
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
Accounts payable |
|
$ |
26.1 |
|
$ |
12.7 |
|
Accrued expenses and other current liabilities |
|
|
233.5 |
|
|
89.3 |
|
Due to True North Therapeutics equityholders |
|
|
40.0 |
|
|
— |
|
Due to Biogen |
|
|
90.5 |
|
|
— |
|
Total current liabilities |
|
|
390.1 |
|
|
102.0 |
|
Other long-term liabilities |
|
|
92.5 |
|
|
63.7 |
|
Deferred tax liabilities, net |
|
|
89.0 |
|
|
— |
|
Contingent consideration |
|
|
149.1 |
|
|
— |
|
Total liabilities |
|
$ |
720.7 |
|
$ |
165.7 |
|
Commitments and contingencies |
|
|
|
|
|
|
|
Equity: |
|
|
|
|
|
|
|
Preferred stock, $0.001 par value (shares authorized of 50,000,000 at December 31, 2017 and 0 at December 31, 2016; no shares issued and outstanding at December 31, 2017 or at December 31, 2016) |
|
|
— |
|
|
— |
|
Common stock, $0.001 par value (shares authorized of 800,000,000 at December 31, 2017 and 1,000 at December 31, 2016; shares issued and outstanding of 108,203,439 at December 31, 2017 and 1,000 at December 31, 2016) |
|
|
0.1 |
|
|
— |
|
Additional paid-in capital |
|
|
565.2 |
|
|
— |
|
Retained earnings |
|
|
332.3 |
|
|
— |
|
Net parent company investment |
|
|
— |
|
|
564.4 |
|
Accumulated other comprehensive loss |
|
|
— |
|
|
1.8 |
|
Total equity |
|
$ |
897.6 |
|
$ |
566.2 |
|
Total liabilities and equity |
|
$ |
1,618.3 |
|
$ |
731.9 |
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-3
BIOVERATIV INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
(In millions)
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
Adjustments to reconcile net income to net cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
14.5 |
|
|
53.4 |
|
|
15.6 |
|
Stock-based compensation |
|
|
32.0 |
|
|
15.3 |
|
|
13.2 |
|
Deferred taxes |
|
|
(59.5) |
|
|
(154.2) |
|
|
— |
|
Remeasurement of contingent consideration |
|
|
9.9 |
|
|
— |
|
|
— |
|
Changes in operating assets and liabilities, net: |
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
(39.9) |
|
|
(55.0) |
|
|
(27.0) |
|
Inventory |
|
|
82.4 |
|
|
(49.9) |
|
|
(72.9) |
|
Due from Biogen, net |
|
|
90.5 |
|
|
— |
|
|
— |
|
Other assets |
|
|
(45.6) |
|
|
(22.6) |
|
|
(5.4) |
|
Accounts payable |
|
|
11.1 |
|
|
2.0 |
|
|
(2.3) |
|
Accrued expenses and other current liabilities |
|
|
110.9 |
|
|
40.0 |
|
|
(2.0) |
|
Other liabilities |
|
|
28.8 |
|
|
33.0 |
|
|
13.6 |
|
Net cash flows provided by operating activities |
|
|
590.7 |
|
|
301.6 |
|
|
41.4 |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
Purchases of property, plant and equipment |
|
|
(13.1) |
|
|
(8.7) |
|
|
(10.6) |
|
Acquisition of True North Therapeutics, net of cash |
|
|
(395.7) |
|
|
— |
|
|
— |
|
Acquisition of intangible assets |
|
|
— |
|
|
(26.5) |
|
|
— |
|
Net cash flows used in investing activities |
|
|
(408.8) |
|
|
(35.2) |
|
|
(10.6) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
Transfers to Biogen |
|
|
(45.3) |
|
|
(266.4) |
|
|
(30.8) |
|
Cash from Biogen upon separation |
|
|
325.0 |
|
|
— |
|
|
— |
|
Working capital adjustments paid to Biogen, net |
|
|
(23.5) |
|
|
— |
|
|
— |
|
Shares issued under employee benefit plans |
|
|
(2.6) |
|
|
— |
|
|
— |
|
Net cash flows provided by (used in) financing activities |
|
|
253.6 |
|
|
(266.4) |
|
|
(30.8) |
|
Effect of foreign exchange rate changes on cash and equivalents |
|
|
1.0 |
|
|
— |
|
|
— |
|
Net increase in cash and cash equivalents |
|
|
436.5 |
|
|
— |
|
|
— |
|
Cash and cash equivalents, beginning of the year |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Cash and cash equivalents, end of the year |
|
$ |
436.5 |
|
$ |
— |
|
$ |
— |
|
Supplemental cash flow disclosure: |
|
|
|
|
|
|
|
|
|
|
Cash paid for income taxes |
|
$ |
144.5 |
|
$ |
— |
|
$ |
— |
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-4
BIOVERATIV INC. AND SUBSIDIARIES
Consolidated Statements of Equity
(In millions)
|
|
|
|
|
|
|
|
Additional |
|
|
|
Net Parent |
|
Accumulated Other |
|
|
|||||
|
|
|
Common Stock |
|
Paid-In |
|
Retained |
|
Company |
|
Comprehensive |
|
|
||||||||
|
|
|
Shares |
|
|
Amount |
|
Capital |
|
Earnings |
|
Investment |
|
Income |
|
Total equity |
|||||
|
Balance, December 31, 2015 |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
384.4 |
|
$ |
0.3 |
|
$ |
384.7 |
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
439.6 |
|
|
— |
|
|
439.6 |
|
Transfers to Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(259.6) |
|
|
— |
|
|
(259.6) |
|
Foreign currency translation adjustments |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
1.5 |
|
|
1.5 |
|
Balance, December 31, 2016 |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
564.4 |
|
$ |
1.8 |
|
$ |
566.2 |
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
332.3 |
|
|
23.3 |
|
|
— |
|
|
355.6 |
|
Separation-related adjustments |
|
— |
|
|
— |
|
|
2.1 |
|
|
— |
|
|
(312.4) |
|
|
— |
|
|
(310.3) |
|
Transfers to Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(43.0) |
|
|
— |
|
|
(43.0) |
|
Cash from Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
325.0 |
|
|
— |
|
|
325.0 |
|
Working capital adjustments paid to Biogen, net |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(23.5) |
|
|
— |
|
|
(23.5) |
|
Reclassification of net parent company investment to additional paid-in capital |
|
— |
|
|
— |
|
|
533.8 |
|
|
— |
|
|
(533.8) |
|
|
— |
|
|
— |
|
Issuance of common stock upon separation |
|
108.0 |
|
|
0.1 |
|
|
(0.1) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Share-based compensation expense |
|
— |
|
|
— |
|
|
32.0 |
|
|
— |
|
|
— |
|
|
— |
|
|
32.0 |
|
Shares issued under employee benefit plans |
|
0.2 |
|
|
— |
|
|
(2.6) |
|
|
— |
|
|
— |
|
|
— |
|
|
(2.6) |
|
Foreign currency translation adjustments |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(1.8) |
|
|
(1.8) |
|
Balance, December 31, 2017 |
|
108.2 |
|
$ |
0.1 |
|
$ |
565.2 |
|
$ |
332.3 |
|
$ |
— |
|
$ |
— |
|
$ |
897.6 |
The accompanying notes are an integral part of these Consolidated Financial Statements
F-5
BIOVERATIV INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
1. Nature of Business and Basis of Preparation
Nature of Business
Bioverativ Inc. (Bioverativ) separated from Biogen Inc. (Biogen) on February 1, 2017 as a result of a special dividend distribution of all the outstanding shares of common stock of Bioverativ to Biogen stockholders. The distribution was made to Biogen stockholders of record as of the close of business on January 17, 2017, who received one share of Bioverativ common stock for every two shares of Biogen common stock held as of such date. As a result of the distribution, Bioverativ is now an independent public company.
Bioverativ holds the assets and liabilities of Biogen’s former hemophilia business. Bioverativ is focused on the discovery, research, development and commercialization of innovative therapies for the treatment of hemophilia and other blood disorders.
Bioverativ’s marketed products include ELOCTATE and ALPROLIX, extended half‑life factors for the treatment of hemophilia A and hemophilia B, respectively. Pursuant to a development and commercialization agreement, Bioverativ collaborates with Swedish Orphan Biovitrum AB (publ) (Sobi) to jointly develop and commercialize ELOCTATE and ALPROLIX globally. Sobi has responsibility for commercialization of ELOCTATE and ALPROLIX in Europe, Russia and certain countries in Northern Africa and the Middle East, while Bioverativ retains rights to commercialize those therapies in the United States, Japan, Canada, Australia, Latin American countries and all other markets excluding Sobi’s commercialization territory. See Note 4, Collaborations, for further information on Bioverativ’s collaboration with Sobi.
On January 21, 2018, we entered into an Agreement and Plan of Merger (the “Merger Agreement”), with Sanofi (“Parent” or “Sanofi”), a French société anonyme, and Blink Acquisition Corp. (“Merger Sub”), a Delaware corporation and indirect wholly-owned subsidiary of Parent. See Note 21, Subsequent Events to our audited consolidated financial statements included in this Form 10-K.
Basis of Preparation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (GAAP). The accompanying consolidated financial statements include those of the company and its subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation.
We operate as one operating segment, which is discovering, researching, developing and commercializing innovative therapies for the treatment of hemophilia and other rare blood disorders.
The accompanying consolidated financial statements reflect the consolidated financial position and consolidated results of operations of the company as an independent, publicly-traded company for the period after the February 1, 2017 separation. The consolidated financial statements also reflect the consolidated financial position and consolidated results of operations of the company as a consolidated reporting entity of Biogen for periods prior to the separation.
In 2017, the company recorded certain separation related adjustments in its consolidated statement of equity. The separation related adjustments primarily related to differences between assets and liabilities transferred to Bioverativ as a result of the separation and assets and liabilities reported in the company’s consolidated balance sheet as of January 31, 2017. Separation related adjustments consists primarily of inventory and deferred tax assets retained by Biogen upon separation. Additional separation related adjustments could be recorded in future periods.
F-6
Prior to the separation, the consolidated financial statements were prepared on a standalone basis and were derived from Biogen’s consolidated financial statements and accounting records as if the former hemophilia business of Biogen had been standalone business. Accordingly, financial information for periods prior to the separation is shown on a carve-out basis for the hemophilia business as part of Biogen. The consolidated financial statements reflected the company’s financial position, results of operations and cash flows as the business was operated as part of Biogen prior to the distribution.
Prior to the separation, the consolidated financial statements included the attribution of certain assets and liabilities that were historically held at the Biogen corporate level but which were specifically identifiable or attributable to the company. All intercompany transactions and accounts within the company were eliminated. All transactions between the company and Biogen were considered to be effectively settled in the consolidated financial statements at the time the transaction was recorded. The total net effect of the settlement of the transactions with Biogen is reflected in the consolidated statements of cash flows in periods prior to the separation as a financing activity and in the consolidated balance sheet as net parent company investment.
Prior to the separation, these consolidated financial statements include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. The research and development costs include depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. The selling, general and administrative costs include certain services provided by Biogen, which include, but are not limited to, executive oversight, treasury, finance, legal, human resources, tax planning, internal audit, financial reporting, information technology, investor relations, shared services, insurance, employee benefits and incentives and share‑based compensation. Allocated amounts have been included in research and development, selling, general and administrative and other income and expense. These expenses have been allocated to the company based on direct usage or benefit where specifically identifiable, with the remainder allocated primarily based on hours or direct costs. The company considers the expense methodology and results to be reasonable for all periods presented. However, the allocations may not be indicative of the actual expense that would have been incurred had the company operated as an independent, publicly traded company for the periods presented.
In periods prior to the separation, Bioverativ’s employees participated in various benefit and share-based compensation plans maintained by Biogen. A portion of the cost of those plans was included in the company’s financial statements. However, the consolidated balance sheets in periods prior to the separation did not include any equity related to share-based compensation plans.
Prior to the separation, the company’s equity balance represented the excess of total assets over total liabilities, including the due to/from balances between the company and Biogen (net parent company investment) and cumulative translation adjustment. In connection with the separation, the company’s net parent company investment balance was reclassified to additional paid-in capital.
2. Summary of Significant Accounting Policies
The accompanying consolidated financial statements reflect the application of certain significant accounting policies as described below and elsewhere in these notes to the consolidated financial statements.
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current period presentation. These reclassifications had no effect on the reported results of operations. In first quarter of 2017, the company concluded that it was appropriate to classify costs associated with medical affairs as research and development to better align with Bioverativ’s organizational structure. Previously, such costs had been classified as selling, general and administrative.
F-7
As a result, the amounts that were previously presented in selling, general and administrative were reclassified in research and development for the years ended December 31, 2016 and 2015.
|
|
|
For the Years Ended |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2016 |
|
2015 |
||
|
Medical reclassification |
|
$ |
31.2 |
|
$ |
50.8 |
Use of Estimates
The preparation of our condensed consolidated financial statements requires us to make estimates, judgments, and assumptions that may affect the reported amounts of assets, liabilities, equity, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis we evaluate our estimates, judgments and methodologies. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenues and expenses. Actual results may differ from these estimates under different assumptions or conditions.
Cash and Cash Equivalents
We consider only those investments which are highly liquid, readily convertible to cash and that mature within three months from date of purchase to be cash equivalents. The carrying values of money market funds approximate fair value due to their short-term maturities.
Accounts Receivable
The majority of accounts receivable arise from product sales and primarily represent amounts due from specialty pharmacies, hemophilia treatment centers, public and private hospitals and independent specialty distributors as well as contract manufacturing and royalty revenue from Sobi. The company monitors the financial performance and creditworthiness of its customers so that the company can properly assess and respond to changes in their credit profile. The company provides reserves against trade receivables for estimated losses that may result from a customer’s inability to pay. Amounts determined to be uncollectible are charged or written‑off against the reserve.
Concentration of Credit Risk
CVS Health Corporation (CVS), a specialty pharmacy and ASD Specialty Healthcare (ASD), a specialty distributor, individually represent 13% and 12%, respectively, of outstanding accounts receivable for the year ended December 31, 2017; CVS, ASD and Accredo Health Incorporated, a specialty pharmacy, individually represent 24%, 14% and 11%, respectively, of outstanding accounts receivable for the year ended December 31, 2016. Other than these significant customers, concentration of credit risk with respect to receivables, which are typically unsecured, are largely mitigated due to the wide variety of customers. The majority of accounts receivable arise from product sales in the United States and Japan and have standard payment terms which generally require payment within 30 to 90 days. The company monitors the financial performance and creditworthiness of its customers so that the company can properly assess and respond to changes in their credit profile. The company continues to monitor these conditions and assess their possible impact on its business.
Inventory
Inventories are stated at the lower of cost or market with cost based on the first‑in, first‑out (FIFO) method. Inventory that can be used in either the production of clinical or commercial products is expensed as research and development costs when selected for use in a clinical manufacturing campaign.
F-8
Capitalization of Inventory Costs
The company capitalizes inventory costs associated with its products prior to regulatory approval, when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized. In determining whether or not to capitalize such costs, the company evaluates, among other factors, information regarding the drug candidate’s safety and efficacy, the status of regulatory submissions and communications with regulatory authorities and the outlook for commercial sales, including the existence of current or anticipated competitive drugs and the availability of reimbursement. In addition, the company evaluates risks associated with manufacturing the drug candidate and the remaining shelf‑life of the inventories.
Obsolescence and Unmarketable Inventory
The company periodically reviews its inventories for excess or obsolescence and write‑down obsolete or otherwise unmarketable inventory to its estimated net realizable value.
Property, Plant and Equipment
Property, plant and equipment are carried at cost, subject to review for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. The cost of normal, recurring, or periodic repairs and maintenance activities related to property, plant and equipment are expensed as incurred. The cost for planned major maintenance activities, including the related acquisition or construction of assets, is capitalized if the repair will result in future economic benefits.
Prior to the separation, Property, plant and equipment comprised of assets whereby a majority of its use was dedicated to the hemophilia business.
The company generally depreciates or amortizes the cost of its property, plant and equipment using the straight‑line method over the estimated useful lives of the respective assets, which are summarized as follows:
|
Asset Category |
|
Useful Lives |
|
Land |
|
Not depreciated |
|
Buildings |
|
15 to 40 years |
|
Leasehold Improvements |
|
Lesser of the useful life or the term of the respective lease |
|
Furniture and Fixtures |
|
5 to 7 years |
|
Machinery and Equipment |
|
5 to 20 years |
|
Computer Software and Hardware |
|
3 to 5 years |
Acquired Intangibles
Acquired intangibles include product rights, patents and in-process research and development (IPR&D) acquired in a business combination. IPR&D represents the fair value of research and development assets that have not reached technological feasibility. The value assigned to acquired IPR&D is determined by estimating the costs to develop the acquired technology into a commercial product, estimating revenue and costs associated with the product, adjusting the net cash flows for probability of success and discounting to present value. The estimated net cash flows consider the relevant market sizes and growth factors, expected trends in technology and the nature and expected timing of new product introductions by us and our competitors. The rates utilized to discount the net cash flows to their present value are commensurate with the stage of development of the projects and uncertainties in the economic estimates used in the projections. In estimating the probability of success, we utilize data regarding similar assets events from several sources, including industry studies and our own experience.
Upon the acquisition of intangible assets, we complete an assessment of whether our acquisition constitutes the purchase of a single asset or a group of assets. We consider multiple factors in this assessment, including the nature of the technology acquired, the presence or absence of separate cash flows, the development process and stage of completion, quantitative significance and our rationale for entering into the transaction. If we acquire a business as
F-9
defined under applicable accounting standards, then the acquired IPR&D is capitalized as an intangible asset. If we acquire an asset or group of assets that do not meet the definition of a business, then the acquired IPR&D is expensed on its acquisition date if it has no future alternative use. Future costs to develop these assets are recorded to research and development as they are incurred. We perform a qualitative assessment to determine whether any indicators for impairment exist. As a result of the assessment, the company concluded the carrying value of our acquired IPR&D exceeds its fair value.
Acquired product rights and patents are recorded at fair value, assigned an estimated useful life, and are amortized over their estimated useful lives of approximately twelve years. See Note 9, Intangible Assets. Amortization is included in cost of sales in the consolidated statements of income.
Goodwill
Goodwill represents the difference between the purchase price and the fair value of the identifiable tangible and intangible net assets when accounted for as a business combination using the purchase method of accounting. Goodwill is not amortized, but reviewed for impairment. Goodwill is reviewed annually and whenever events or changes in circumstances indicate that the carrying value of the goodwill might not be recoverable.
We compare the fair value of our reporting unit to its carrying value. If the carrying value of the net assets assigned to the reporting unit exceeds the fair value of our reporting unit, then we would need to determine the implied fair value of our reporting unit’s goodwill. If the carrying value of our reporting unit’s goodwill exceeds its implied fair value, then we would record an impairment loss equal to the difference. As described above in Note 1, Nature of Business and Basis of Preparation, we operate in one operating segment which we consider our only reporting unit.
Impairment of Long‑Lived Assets
Long‑lived assets to be held and used, including property, plant and equipment and definite‑lived intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets or asset group may not be recoverable.
Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written‑down to their fair values. Long‑lived assets to be disposed of are carried at fair value less costs to sell.
Contingent Consideration
The consideration for our acquisition includes future payments that are contingent upon the occurrence of various events. We recorded a liability for such contingent payments at fair value on the acquisition date. We estimated the fair value of contingent consideration through valuation models that incorporate probability-adjusted assumptions related to the likelihood of achieving the milestones and making related payments. Our contingent consideration is remeasured each reporting period. Changes in the fair value of our contingent consideration are recognized in our condensed consolidated statements of income. Changes in the fair value of the contingent consideration can result from changes to one or multiple inputs, including changes to the discount rates, changes in the probability of certain clinical events, regulatory approval, and the amount or timing of development and sales based milestones. Discount rates in our valuation models represent a measure of the weighted average cost of capital of the target associated with settling the liability. The period over which we discount our contingent consideration is based on the current development stage of the product candidates and our specific development plan for that product candidate. In estimating the probability of success, we utilize data regarding similar milestone events from several sources, including industry studies and our own experience. These fair value measurements are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we record in any given period.
F-10
Non-designated Foreign Currency Contracts
Currency forward contracts are used as a part of our strategy to manage exposure related to foreign currency denominated monetary assets and liabilities. These currency forward contracts are not designated as cash flow, fair value or net investment hedges. The currency forward contracts are marked-to-market with changes in fair value recorded to other income (expense) and are entered into for periods consistent with currency transaction exposures, generally less than one year.
Translation of Foreign Currencies
The functional currency for the company’s non‑U.S. subsidiaries is their local currency. For non‑U.S. subsidiaries that transact in a functional currency other than the U.S. dollar, assets and liabilities are translated at current rates of exchange at the balance sheet date. Income and expense items are translated at the average foreign exchange rates for the period. Adjustments resulting from the translation of the financial statements of non‑U.S. operations into U.S. dollars are excluded from the determination of net income and are recorded in accumulated other comprehensive income, a separate component of equity.
Contingencies
We are currently involved in various claims and legal proceedings. Loss contingency provisions are recorded if the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated or a range of loss can be determined. These accruals represent management’s best estimate of probable loss. Disclosure also is provided when it is reasonably possible that a loss will be incurred or when it is reasonably possible that the amount of a loss will exceed the recorded provision. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. Significant judgment is required in both the determination of probability and the determination as to whether an exposure is reasonably estimable. Because of uncertainties related to these matters, accruals are based only on the best information available at the time. As additional information becomes available, we reassess the potential liability related to pending claims and litigation and may change our estimates. These changes in the estimates of the potential liabilities could have a material impact on our consolidated results of operations and financial position.
Revenue Recognition
The company recognizes revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the price to the customer is fixed or determinable; and collectability is reasonably assured.
Product Revenues
We sell mainly to specialty pharmacies, hemophilia treatment centers, public and private hospitals and independent specialty distributors. Any discounts offered to these customers are reflected as on‑invoice discounts. We also sell to specialty distributors who receive both on‑invoice discounts as well as chargebacks for sales to various U.S. government agencies such as U.S. Public Health Service (PHS). Provisions for rebates, chargebacks to distributors, and discounts are provided for at the time the related sales are recorded, and are reflected as a reduction of sales. Reserves established for these discounts and allowances are classified as reductions of accounts receivable (if the amount is payable to our customer) or a liability (if the amount is payable to a party other than our customer). Our estimates take into consideration our historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Actual amounts may ultimately differ from our estimates. If actual results vary, we adjust these estimates, which could have an effect on earnings in the period of adjustment. To date historical adjustments have not been material.
Product revenue reserves are categorized as follows: discounts and contractual adjustments. Discounts include trade term discounts and volume discounts. Trade term discounts relate to estimated obligations for credits to be granted to customers for remitting payment on their purchases within established incentive periods. Volume discounts are earned
F-11
as customers reach certain tier levels based upon their purchases. Contractual adjustments primarily relate to Medicaid and PHS discounts. Product returns are insignificant and mainly resulting from product damaged in transit or incorrectly shipped.
Collaborations
Our development and commercialization arrangement with Sobi represents a collaborative arrangement because both Sobi and us are active participants and exposed to significant risks and rewards of the arrangement. Where we are the principal on sales transactions with third parties, we recognize revenue, cost of sales, including royalty cost of sales, and operating expenses on a gross basis in our income statement. We recognize payments between Sobi and us based upon their nature. These payments consist of royalty cost of sales, royalty revenue and contract manufacturing revenue. Royalty revenue and contract manufacturing revenue represent collaboration revenue in our income statement. See Note 4, Collaborations.
Research and Development Expenses
Research and development costs are expensed as incurred. Milestone payments prior to regulatory approval are expensed when the milestone is achieved. Payments made to counterparties on or after regulatory approval are capitalized and amortized over the remaining useful life of the related product. Amounts capitalized for such payments are included in acquired intangible assets, net of accumulated amortization. Also included in research and development expenses are allocations from Biogen. See Note 14, Related Parties.
Income Taxes
Deferred taxes are recognized for the future tax effects of temporary differences between financial and income tax reporting based on enacted tax laws and rates. With respect to uncertain tax positions, the company determines whether the position is more likely than not to be sustained upon examination, based on the technical merits of the position. Any tax position that meets the more‑likely‑than‑not recognition threshold is measured and recognized in the consolidated financial statements at the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement. The liability relating to uncertain tax positions is classified as current in the consolidated balance sheets to the extent the company anticipates making a payment within one year. Interest and penalties associated with income taxes are classified in the income tax expense line in the consolidated statements of income.
Prior to the separation, the company’s income tax expense and deferred tax balances were calculated on a separate tax return basis although the company’s operations had historically been included in the tax returns filed by the respective Biogen entities, of which the company’s business was a part. For periods subsequent to the separation, Bioverativ files tax returns on its own behalf and its income tax expense and deferred income tax balances are recorded in accordance with the company’s standalone income tax positions. Biogen and Bioverativ entered into a tax matters agreement effective as of the date of separation.
Accounting for Share‑Based Compensation
In connection with the separation, we adopted our own share‑based compensation plans. Outstanding Biogen CSPUs, MSUs and PUs were converted to Bioverativ RSUs and outstanding Biogen stock options were converted to Bioverativ stock options. See Note 13, Share‑Based Compensation.
We charge the estimated fair value of awards against income over the requisite service period, which is generally the vesting period. Where awards are made with non‑substantive vesting periods (for instance, where a portion of the award vests upon retirement eligibility), we estimate and recognize expense based on the period from the grant date to the date on which the employee is retirement eligible. We recognize forfeitures as they occur.
The fair values of our stock option grants are estimated as of the date of grant using a Black‑Scholes option valuation model. The estimated fair values of the stock options are then expensed over the options’ vesting periods. The
F-12
fair values of RSUs are based on the market value of our stock on the date of grant. Compensation expense for RSUs is recognized on a straight‑line basis over the applicable service period.
New Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) or other standard setting bodies that we adopt as of the specified effective date.
The following new standards issued by FASB were adopted by the company on January 1, 2017:
|
· |
Accounting Standards Update (ASU) No. 2015‑11, Inventory (Topic 330): Simplifying the Measurement of Inventory. The new standard applies only to inventory for which cost is determined by methods other than last-in, first-out and the retail inventory method, which includes inventory that is measured using first in, first out (FIFO) or average cost. Inventory within the scope of this standard is required to be measured at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal and transportation. The adoption of this standard did not have a material impact on our financial position, results of operations or statements of cash flows upon adoption. |
|
· |
ASU No. 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes. The new standard requires that deferred tax assets and liabilities be classified as noncurrent in a classified statement of financial position. The adoption of this standard did not have a material impact on our financial position, results of operations or statements of cash flows upon adoption. |
|
· |
ASU No. 2016‑09, Compensation‑Stock Compensation (Topic 718): Improvements to Employee Share‑Based Payment Accounting. The new standard requires recognition of the income tax effects of vested or settled awards in the income statement and involves several other aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. The adoption of this standard did not have a material impact on our financial position, results of operations or statements of cash flows upon adoption. |
In the fourth quarter of 2017 we adopted the following standard:
|
· |
In January 2017 the FASB issued ASU No. 2017-04, Intangibles - Goodwill and Other (Topic 350): Simplifying the Test of Goodwill Impairment. This new standard simplifies how an entity is required to test goodwill for impairment by eliminating Step 2 from the goodwill impairment test. Step 2 measures a goodwill impairment loss by comparing the implied fair value of a reporting unit’s goodwill with the carrying amount of that goodwill. The adoption of this standard did not have a material impact on our financial position or results of operations. |
The following new standards have been issued by FASB but are not yet effective as of December 31, 2017:
|
· |
In May 2014 the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606), which supersedes all existing revenue recognition requirements, including most industry-specific guidance. The new standard requires a company to recognize revenue when it transfers goods or services to customers in an amount that reflects the consideration that the company expects to receive for those goods or services. The FASB has subsequently issued amendments to ASU No. 2014-09 that have the same effective date and transition date of January 1, 2018. We expect to adopt these standards using the full retrospective method. We will apply all of the requirements of the new standards to each comparative period presented in accordance with the requirements on accounting changes. |
We are finalizing our assessment of the effect of adoption. Based on this assessment, we expect changes in our revenue recognition policy relating to the opt-in consideration payable by Sobi to us under our collaboration
F-13
agreement with Sobi (See Note 4, Collaborations). Currently the payment by Sobi of the opt-in consideration is recognized over the term of the commercialization period using a cross-royalty payment structure. Upon adoption of these standards, for the year ended December 31, 2016, the net present value of the remaining opt-in consideration will be recognized as revenue and note receivable. Commencing in 2017, royalty revenue will be recognized at the contractual 12% rate rather than the effective rate of 13-15%. Royalty cost of sales will be recognized at the contractual rate of 12% in North America and 17% outside North America instead of the effective rate of 10-11% and 15-16%, respectively. Upon adoption in Q1 2018, our balance sheet at December 31, 2017 is expected to increase by approximately $190 million due to the note receivable from Sobi.
|
· |
In January 2016 the FASB issued ASU No. 2016-01, Financial Instruments - Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities. The new standard amends certain aspects of accounting and disclosure requirements of financial instruments, including the requirement that equity investments with readily determinable fair values be measured at fair value with changes in fair value recognized in a company's results of operations. The new standard will be effective for us on January 1, 2018. The adoption of this standard is not expected to have a material impact on our financial position or results of operations. |
|
· |
In February 2016 the FASB issued ASU No. 2016-02, Leases (Topic 842). The new standard requires that all lessees recognize the assets and liabilities that arise from leases on their balance sheet and disclose qualitative and quantitative information about their leasing arrangements. The new standard will be effective for us on January 1, 2019. The adoption of this standard is not expected to have a material impact on our results of operations but may impact our financial position. |
|
· |
In October 2016 the FASB issued ASU No. 2016-16, Income Taxes (Topic 740): Intra-Entity Transfer of Assets Other Than Inventory. This new standard eliminates the deferral of the tax effects of intra-entity transfers of an asset other than inventory. Under the new standard, entities should recognize the income tax consequences on an intra-entity transfer of an asset other than inventory when the transfer occurs. This new standard will be effective for us on January 1, 2018 and will be applied on a modified retrospective basis through a cumulative-effect adjustment directly to retained earnings as of the beginning of the period of adoption. The adoption of this standard is not expected to have a material impact on our financial position. |
|
· |
In November 2016 the FASB issued ASU No. 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash. The amendments in this update require that a statement of cash flows explain the change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. Therefore, amounts generally described as restricted cash and restricted cash equivalents should be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. This new standard will be effective for us on January 1, 2018. The adoption of this standard is not expected to have a material impact on our financial position. |
|
· |
In January 2017 the FASB issued ASU No. 2017-01, Business Combinations (Topic 805): Clarifying the Definition of a Business. This new standard clarifies the definition of a business and provides a screen to determine when an integrated set of assets and activities is not a business. The screen requires that when substantially all of the fair value of the gross assets acquired (or disposed of) is concentrated in a single identifiable asset or a group of similar identifiable assets, the set of assets and activities is not a business. This new standard will be effective for us on January 1, 2018, however early adoption is permitted. The adoption of this standard is not expected to have a material impact on our financial position. |
|
· |
In May 2017 the FASB issued ASU No. 2017-09, Compensation-Stock Compensation (Topic 718): Scope of Modification Accounting. This new standard provides clarity and reduces both (1) diversity in practice and (2) cost and complexity when applying the guidance in Topic 718, Compensation-Stock Compensation, to a change to the terms or conditions of a share-based payment award. This new standard will be effective for us on January 1, 2018. The adoption of this standard is not expected to have a material impact on our financial position or results of operations. |
F-14
3. Acquisition of True North Therapeutics, Inc.
On June 28, 2017, Bioverativ acquired all of the outstanding equity of True North Therapeutics, Inc. (True North), a clinical stage biopharmaceutical company focused on the discovery, development and commercialization of first-in-class product candidates for complement-mediated diseases. At closing, we paid True North equityholders as of the acquisition date (True North equityholders) upfront consideration of $395.7 million plus assumed cash. True North equityholders are also eligible to receive additional payments of up to $425.0 million contingent on the achievement of future development, regulatory and sales milestones. The upfront consideration was paid with cash.
The acquisition of True North was accounted for as a business combination using the acquisition method of accounting. The assets acquired and the liabilities assumed from True North have been recorded at their preliminary fair value as of June 28, 2017, the date of acquisition. The company’s audited consolidated financial statements included the results of True North from the date of acquisition.
The company completed its evaluation of information, assumptions and valuation methodologies it used in its fair value of the purchase price consideration.
The fair value of the purchase price consideration consisted of the following:
|
(In millions) |
|
Estimated Fair Value |
|
|
Cash paid, net of cash acquired |
|
$ |
395.7 |
|
Contingent consideration |
|
|
188.5 |
|
Total purchase price consideration |
|
$ |
584.2 |
The company’s allocation of the purchase price to the assets acquired and liabilities assumed as of December 31, 2017 are outlined below.
|
|
|
As of |
|
|
|
|
December 31, |
|
|
(In millions) |
|
2017 |
|
|
Current assets |
|
$ |
0.6 |
|
Property, plant and equipment, net |
|
|
0.6 |
|
In-process research and development (IPR&D) |
|
|
590.0 |
|
Goodwill |
|
|
170.7 |
|
Accounts payable and accrued expenses |
|
|
(4.0) |
|
Deferred tax liabilities |
|
|
(173.7) |
|
Final fair value of identifiable assets acquired and liabilities assumed |
|
$ |
584.2 |
Intangible Assets
IPR&D intangible assets totaling $590.0 million represent the value assigned to research and development (R&D) projects acquired from True North. These IPR&D intangible assets are capitalized and accounted for as indefinite-lived intangible assets and will be subject to impairment testing until completion or abandonment of the projects. Upon successful completion of each project, the company will make a separate determination of the estimated useful life of the IPR&D intangible assets and the related amortization will be recorded as an expense over the estimated useful life.
Some of the more significant assumptions inherent in the development of those asset valuations include the estimated net cash flows for each year for each asset (including net revenues, cost of sales, R&D costs, selling and marketing costs, working capital/asset contributory asset charges and other cash flow assumptions), probability of success, the appropriate discount rate to select in order to measure the risk inherent in each future cash flow stream, the assessment of each asset’s life cycle, the potential regulatory and commercial success risks, competitive trends impacting the asset and each cash flow stream as well as other factors.
F-15
The discount rate used to arrive at the present value at the acquisition date of the IPR&D intangible assets was 18.0% to reflect the internal rate of return and uncertainty in the cash flow projections. No assurances can be given that the underlying assumptions used to prepare the discounted cash flow analysis will not change. For these and other reasons, actual results may vary significantly from estimated results.
Goodwill
Goodwill, of $170.7 million as of December 31, 2017, which is not deductible for tax purposes, includes the value of net deferred tax liabilities associated with IPR&D.
The table below presents a rollforward of goodwill:
|
(In millions) |
|
|
|
|
Balance at December 31, 2016 |
|
$ |
— |
|
Additions |
|
|
170.7 |
|
Balance at December 31, 2017 |
|
$ |
170.7 |
Deferred tax liabilities
Deferred tax liabilities includes the impact of IPR&D intangible assets which have no tax basis, partially offset by deferred tax assets associated with acquired net operating losses and research and development credits.
Contingent Consideration
True North equityholders are also eligible to receive additional payments of up to $425.0 million contingent on the achievement of future development, regulatory and sales milestones. The company estimated the fair value of the contingent consideration to be $188.5 million using a probability adjusted approach that considered the possible outcomes based on assumptions related to the timing and probability of the product launch, discount rates matched to the timing of first payment, and probability of success rates and discount adjustments on the related cash flows. Changes to assumptions used in the estimated fair value of contingent consideration were refinements based on current information as of December 31, 2017. The impact of these refinements was an increase of $9.9 million in the fair value of contingent consideration, which has been recorded as an increase in the contingent consideration liability and a research and development expense.
Due to True North Equityholders
In connection with the acquisition, we assumed $40.0 million of True North cash which is being held in escrow to satisfy specified indemnification obligations that arise within 18 months post closing. Any amounts remaining in escrow are payable to True North equityholders. The amounts in escrow are classified as short term restricted cash and amounts due to True North equityholders are classified as a short term liability as of December 31, 2017.
Integration and Acquisition Costs
During the year ended December 31, 2017, the company expensed $9.0 million relating to the acquisition and integration of True North, which has been recorded within selling, general and administrative in the company’s consolidated statements of income.
Supplemental Disclosure of Pro Forma Information
The following unaudited pro forma financial information presents the combined results of the operations of Bioverativ and True North as if the acquisition of True North had occurred as of January 1, 2016. The unaudited pro forma financial information is not necessarily indicative of what the consolidated results of operations actually would
F-16
have been had the acquisitions been completed on January 1, 2016. In addition, the unaudited pro forma financial information does not purport to project the future results of operations of the company.
|
|
|
For the Years Ended |
||||
|
|
|
December 31, |
||||
|
(In millions, except per share amounts) |
|
2017 |
|
2016 |
||
|
Revenues |
|
$ |
1,168.6 |
|
$ |
887.4 |
|
Net income |
|
|
341.0 |
|
|
407.7 |
|
Per share amounts: |
|
|
|
|
|
|
|
Net income per share - basic |
|
|
3.15 |
|
|
3.78 |
|
Net income per share - diluted |
|
|
3.14 |
|
|
3.78 |
The unaudited pro forma financial information above reflects a pro forma adjustment for the year ended December 31, 2017 of $11.6 million, related to integration and acquisition costs, net of tax incurred by Bioverativ and True North, including costs incurred by True North associated with its preparation for an initial public offering.
4. Collaborations
In connection with the company’s business strategy, the company has entered into various collaboration agreements which provide the company with rights to develop, produce and market products using certain know‑how, technology and patent rights maintained by the company’s collaborative partners. Terms of the various collaboration agreements may require the company to make milestone payments upon the achievement of certain product research and development objectives and pay royalties on future sales, if any, of commercial products resulting from the collaboration.
Swedish Orphan Biovitrum AB (publ)
In January 2007, Biogen acquired 100% of the stock of Syntonix Pharmaceuticals (Syntonix). Syntonix, now known as Bioverativ Therapeutics Inc. (formerly Biogen Hemophilia Inc.), had previously entered into a development and commercialization agreement with Sobi to jointly develop and commercialize Factor VIII and Factor IX hemophilia products, including ELOCTATE and ALPROLIX. Under the development and commercialization agreement, as has been amended and restated, Bioverativ has commercial rights for North America (the Bioverativ North America Territory) and for all other markets outside of the Sobi territory (the Bioverativ Direct Territory), which consists of Europe, Russia and certain countries in Northern Africa and the Middle East (the Sobi Territory).
Under the development and commercialization agreement, prior to May 5, 2024, either Bioverativ or Sobi may present a compound construct as a potential product candidate that the parties may consider developing and commercializing under the collaboration. Upon Sobi's election to treat a compound construct as a product, and in the case of a novel compound construct upon Sobi's payment of an upfront fee, Sobi is granted the right to opt-in to such compound construct and become responsible for final development and commercialization of that compound construct in Sobi's commercialization territory. Generally, upon opt-in, Sobi becomes obligated to make an advance payment and reimburse Bioverativ for certain development expenses incurred with respect to the compound construct. Until Sobi's portion of the development expenses are fully paid, Sobi's royalty rate payable to Bioverativ is increased, and the royalty payment payable by Bioverativ to Sobi for the sale of products in Bioverativ's territory is decreased.
In November 2014, Sobi exercised its option under the agreement to assume final development and commercialization activities in the Sobi Territory for ELOCTA (the trade name for ELOCTATE in the European Union). In July 2015, Sobi exercised its option under the agreement to assume final development and commercialization of ALPROLIX within the Sobi Territory. Upon each exercise of opt‑in right under the terms of the development and commercialization agreement, Sobi made a $10.0 million payment.
Upon European Medicines Agency (EMA) regulatory approval of each of ELOCTA and ALPROLIX, Sobi was obligated to reimburse Bioverativ 50% of all shared manufacturing and development expenses incurred by Bioverativ from October 1, 2009 through the earlier of the date on which Sobi is registered as the marketing authorization holder for
F-17
the applicable product or 90 days post‑regulatory approval, as well as 100% of certain development expenses incurred exclusively for the benefit of the Sobi Territory (the Opt‑In Consideration).
ELOCTA was approved by the European Commission (EC) in November 2015. The Opt‑In Consideration and aggregate amount reimbursable by Sobi for ELOCTA was $211.0 million. As of December 31, 2017, approximately $90.5 million remained reimbursable by Sobi.
ALPROLIX was approved by the EC in May 2016. The Opt‑In Consideration and aggregate amount reimbursable by Sobi for ALPROLIX was $184.7 million. As of December 31, 2017, approximately $97.4 million remained reimbursable by Sobi.
The Opt‑In Consideration for each product is being paid by Sobi using a cross‑royalty cash payment structure for sales in each company’s respective territories. If the reimbursement of the Opt‑in Consideration for a product has not been achieved within six years of the first commercial sale of such product (the Reimbursement Period), the company maintains the right to require Sobi to pay any remaining balances due to us within 90 days of the six-year anniversary date of the first commercial sale. After Sobi’s Opt‑In Consideration has been repaid, the royalty paid and received by the company resets to the contractual royalty rate of 12%.
Upon Sobi’s first commercial sale in 2016, and during the Reimbursement Period, the royalty rate the company will pay Sobi on sales of ELOCTATE and ALPROLIX in the Bioverativ North American Territory is 7% and Bioverativ Direct Territory is 12%. After the Reimbursement Period concludes, the royalty rate we pay to Sobi increases by 5%. For the year ended December 31, 2017, we are recording cost of sales at the effective royalty rate of approximately 11%. For the year ended December 31, 2016, the effective royalty rate was approximately 10%.
The company is accounting for the development and commercialization agreement under a right to use model and is recognizing revenue over the commercialization period.
The royalty rate received by the company, during the Reimbursement Period on sales of ELOCTATE and ALPROLIX in Sobi’s territory is 17%. After the Reimbursement Period concludes, the royalty we receive decreases to 12%. We are recording revenue at an effective royalty rate of approximately 14%.
In September 2014, Sobi elected to treat BIVV001 (rFVIIIFc‑VWF‑XTEN), a compound construct developed using the XTEN technology, as subject to the collaboration.
In February 2017, Sobi elected to treat BIVV002 (rFIXFc-XTEN), a preclinical compound construct developed using the XTEN technology, as subject to the collaboration. In consideration for its election, Sobi paid $6.2 million, which has been included as a component of research and development expense.
If Sobi exercises its opt-in right for BIVV001 or BIVV002, as the case may be, it will become responsible for final development and commercialization of the applicable product in the Sobi Territory. Further, in general upon opt-in, Sobi will be obligated to reimburse Bioverativ 50% of all shared manufacturing and development expenses incurred by Bioverativ from the period of opt-in through the earlier of the date on which Sobi is registered as the marketing authorization holder for the applicable product or 90 days post‑regulatory approval, as well as 100% of certain development expenses incurred exclusively for the benefit of the Sobi Territory.
Sangamo Therapeutics, Inc.
In January, 2014, Biogen MA Inc., a subsidiary of Biogen, entered into an exclusive worldwide research, development and commercialization collaboration and license agreement with Sangamo under which both companies agreed to develop and commercialize product candidates for the treatment of two inherited blood disorders, sickle cell disease and beta-thalassemia. In connection with the separation, we succeeded to Biogen's rights and obligations under this agreement.
F-18
Under the collaboration, Sangamo is responsible for identifying a product candidate for the treatment of beta-thalassemia and advancing that candidate through a completed Phase 1 human clinical trial, at which point we would assume responsibility for development. The collaboration contemplates our joint development of a sickle cell disease candidate through the potential filing of an investigational new drug application, after which we would assume clinical responsibilities. We have the right to lead the global development and commercialization efforts and Sangamo has the option to assume co-promotion responsibilities in the United States. The collaboration is currently in the pre-clinical stage of development.
Under the terms of the agreement, Sangamo is eligible to receive up to $215.0 million related to development, regulatory and commercialization milestones as well as tiered low to mid double-digit royalties related to product sales.
Bicycle Therapeutics Research Collaboration
In August 2017, we entered into a research collaboration agreement with Bicycle Therapeutics (Bicycle), a biotechnology company that is pioneering a new class of medicines based on its novel bicyclic peptide (Bicycle®) product platform. The collaboration will focus on the discovery, development and commercialization of therapies for hemophilia and sickle cell disease. Under this agreement, Bicycle will be responsible for leading initial discovery activities through lead optimization to candidate selection for three programs. Bioverativ will lead preclinical and clinical development, as well as subsequent marketing and commercialization efforts. Under the agreement, we paid Bicycle a $10.0 million upfront payment. Bicycle is eligible to receive up to $410.0 million related to development, regulatory and commercialization milestones as well as tiered single to low double-digit royalties related to product sales. Bioverativ will also reimburse Bicycle for internal and external program-related costs.
5. Debt and Credit Agreements
On June 28, 2017, we entered into a $175.0 million revolving credit facility (Credit Facility), by and among the company and certain subsidiaries as borrowers, and Bank of America, N.A. and other lenders party thereto (the Lenders). The Credit Facility matures on June 28, 2020.
Borrowings under the Credit Facility bear interest, at our option, at either a base rate or a Eurocurrency rate, in each case plus an applicable margin. Under the Credit Facility, the applicable margin on base rate loans ranges from 0.50% to 1.00% and the applicable margin on Eurocurrency loans ranges from 1.50% to 2.00%, in each case, based on our consolidated leverage ratio (the ratio of our total consolidated funded indebtedness to our consolidated EBITDA for the most recently completed four fiscal quarter period). Borrowings under the Credit Facility are guaranteed by certain of our domestic subsidiaries.
The Credit Facility contains customary representations and warranties and affirmative and negative covenants, including financial covenants to maintain (x) a maximum consolidated leverage ratio of 3.00 to 1.00 (which ratio may, at the election of the Company, be increased to 4.00 to 1.00 upon consummation of a material acquisition, and which shall step down by 0.25x after each full fiscal quarter following such acquisition and return to 3.00 to 1.00 after four full fiscal quarters) and (y) a minimum consolidated interest coverage ratio (the ratio of our EBITDA to consolidated interest charges) of 3.00 to 1.00. As of December 31, 2017, the company was in compliance with the financial covenants under the Credit Facility.
As of December 31, 2017, there were no amounts outstanding under the Credit Facility.
6. Financial instruments and fair value measurements
We have certain financial assets and liabilities recorded at fair value which have been classified as Level 1, 2 or 3 within the fair value hierarchy as described in the accounting standards for fair value measurements.
|
· |
Level 1 – Fair values are determined by utilizing quoted prices (unadjusted) in active markets for identical assets or liabilities that we have the ability to access; |
F-19
|
· |
Level 2 – Fair values are determined by utilizing quoted prices for similar assets and liabilities in active markets or other market observable inputs such as interest rates, yield curves and foreign currency spot rates; and |
|
· |
Level 3 – Prices or valuations that require inputs that are both significant to the fair value measurement and unobservable. |
The majority of our financial assets have been classified as Level 2. Our financial assets (which include our cash equivalents, plan assets for deferred compensation and derivative contracts) have been initially valued at the transaction price and subsequently valued, at the end of each reporting period, utilizing market observable data. These observable market inputs include reportable trades, benchmark yields, credit spreads, broker/dealer quotes, bids, offers, current spot rates and other industry and economic events.
For contingent consideration obligations, the fair value measurements are based on significant inputs not observable in the market. Significant judgment is employed in determining the appropriateness of these assumptions as of the applicable acquisition date and for each subsequent period. Accordingly, changes in assumptions could have a material impact on the amount of contingent consideration expense we record in any given period.
The table below presents information about our assets that are regularly measured and carried at fair value and indicate the level within the fair value hierarchy of the valuation techniques we utilized to determine such fair value:
|
As of December 31, 2017 (In millions) |
|
Total |
|
Quoted Prices |
|
Significant Other |
|
Significant |
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents |
|
$ |
411.1 |
|
$ |
— |
|
$ |
411.1 |
|
$ |
— |
|
Plan assets for deferred compensation |
|
|
18.7 |
|
|
— |
|
|
18.7 |
|
|
— |
|
Derivative contracts |
|
|
1.1 |
|
|
— |
|
|
1.1 |
|
|
— |
|
Total |
|
$ |
430.9 |
|
$ |
— |
|
$ |
430.9 |
|
$ |
— |
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Derivative contracts |
|
|
0.7 |
|
|
— |
|
|
0.7 |
|
|
— |
|
Contingent consideration obligations |
|
$ |
198.4 |
|
$ |
— |
|
$ |
— |
|
$ |
198.4 |
|
Total |
|
$ |
199.1 |
|
$ |
— |
|
$ |
0.7 |
|
$ |
198.4 |
As of December 31, 2017, cash equivalents were comprised of money market funds with maturities less than 90 days from the date of purchase.
As of December 31, 2017, the notional amount of outstanding undesignated foreign currency derivative instruments was $123.1 million. The fair value of derivative contracts assets of $1.1 million was a component of other current assets and the fair value of derivative contracts liabilities of $0.7 million was a component of other current liabilities. For the year ended December 31, 2017, foreign currency derivative gain was $2.2 million, and was a component of other income (expense). There were no hedge gains or losses in any other period presented.
There have been no material impairments of our assets measured and carried at fair value during the year ended December 31, 2017. In addition, there were no changes in valuation techniques or inputs utilized or transfers between fair value measurement levels during the year ended December 31, 2017.
The table below presents a rollforward of contingent consideration obligations:
|
(In millions) |
|
|
|
|
Balance at December 31, 2016 |
|
$ |
— |
|
Additions |
|
|
188.5 |
|
Research and development expenses recognized due to change in fair value |
|
|
9.9 |
|
Balance at December 31, 2017 |
|
$ |
198.4 |
F-20
As of December 31, 2016, we had no financial assets and liabilities that were recorded at fair value.
7. Reserves for Discounts and Allowances
Following the company’s product launches, the company began recognizing reserves for discounts and allowances related to products revenue.
An analysis of the change in reserves is summarized as follows:
|
|
|
|
|
|
Contractual |
|
|
|
|
|
|
|
|
(In millions) |
|
Discounts |
|
Adjustments |
|
Returns |
|
Total |
||||
|
Balance at December 31, 2015 |
|
$ |
5.8 |
|
$ |
16.0 |
|
|
— |
|
$ |
21.8 |
|
Provision related to current period sales |
|
|
160.9 |
|
|
165.2 |
|
|
1.3 |
|
|
327.4 |
|
Adjustment related to prior period sales |
|
|
(2.0) |
|
|
(0.7) |
|
|
— |
|
|
(2.7) |
|
Credits/payments made |
|
|
(158.7) |
|
|
(154.0) |
|
|
(1.0) |
|
|
(313.7) |
|
Balance at December 31, 2016 |
|
$ |
6.0 |
|
$ |
26.5 |
|
|
0.3 |
|
$ |
32.8 |
|
Provision related to current period sales |
|
|
178.0 |
|
|
304.1 |
|
|
0.7 |
|
|
482.8 |
|
Adjustment related to prior period sales |
|
|
(1.3) |
|
|
3.2 |
|
|
0.3 |
|
|
2.2 |
|
Credits/payments made |
|
|
(176.9) |
|
|
(282.2) |
|
|
(0.3) |
|
|
(459.4) |
|
Balance at December 31, 2017 |
|
$ |
5.8 |
|
$ |
51.6 |
|
$ |
1.0 |
|
$ |
58.4 |
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Reduction of accounts receivable |
|
$ |
10.5 |
|
$ |
11.1 |
|
Current liability |
|
|
47.9 |
|
|
21.7 |
|
Total reserves |
|
$ |
58.4 |
|
$ |
32.8 |
8. Inventory
The components of inventory are summarized as follows:
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Raw materials |
|
$ |
0.4 |
|
$ |
42.9 |
|
Work in process |
|
|
11.4 |
|
|
227.2 |
|
Finished goods |
|
|
28.8 |
|
|
31.9 |
|
Total inventory |
|
$ |
40.6 |
|
$ |
302.0 |
Inventory and inventoriable costs written down as a result of excess, obsolescence, unmarketability or other reasons are charged to cost of sales. There were $8.8 million, $13.5 million and $1.3 million of write-offs in the years ended December 31, 2017, 2016 and 2015, respectively.
Inventory consists of amounts contributed as part of the separation. Since the separation, we have drawn down contributed inventory. In connection with the separation, certain raw material and work in process inventory totaling $179.0 million was retained by Biogen. This inventory is subject to the terms and conditions of our manufacturing and supply agreement with Biogen. In the fourth quarter of 2017, we commenced purchasing inventory from Biogen pursuant to a manufacturing and supply agreement.
F-21
9. Intangible Assets
Our intangible assets consist of developed technology milestones and IPR&D. Developed technology primarily relates to approval milestones for ALPROLIX paid to the former Syntonix shareholders in 2014 and 2016. IPR&D consists of assets acquired as a result of our acquisition of True North and primarily relates to BIVV009 (formerly TNT009), a first-in-class monoclonal antibody in development for the treatment of cold agglutinin disease (CAgD).
Intangible assets, net of accumulated amortization are summarized as follows:
|
|
|
|
|
As of |
|
As of |
||||||||||||||
|
|
|
|
|
December 31, 2017 |
|
December 31, 2016 |
||||||||||||||
|
|
|
Estimated |
|
|
|
Accumulated |
|
|
|
|
|
Accumulated |
|
|
||||||
|
(In millions except for estimated life) |
|
Life |
|
Cost |
|
Amortization |
|
Net |
|
Cost |
|
Amortization |
|
Net |
||||||
|
Developed technology |
|
11 years |
|
$ |
61.7 |
|
$ |
(15.8) |
|
$ |
45.9 |
|
$ |
61.7 |
|
$ |
(10.0) |
|
$ |
51.7 |
|
IPR&D |
|
Indefinite |
|
|
590.0 |
|
|
— |
|
|
590.0 |
|
|
— |
|
|
— |
|
|
— |
|
Total |
|
|
|
$ |
651.7 |
|
$ |
(15.8) |
|
$ |
635.9 |
|
$ |
61.7 |
|
$ |
(10.0) |
|
$ |
51.7 |
During the years ended December 31, 2017, 2016 and 2015, amortization expense associated with developed technology assets was $5.8 million, $4.8 million, and $3.0 million, respectively.
10. Property, Plant and Equipment
Property, plant and equipment are recorded at historical cost, net of accumulated depreciation. Components of property, plant and equipment, net are summarized as follows:
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Buildings |
|
$ |
— |
|
$ |
8.0 |
|
Leasehold improvements |
|
|
8.7 |
|
|
— |
|
Machinery and equipment |
|
|
30.8 |
|
|
21.0 |
|
Computer software and hardware |
|
|
6.6 |
|
|
5.9 |
|
Furniture and fixtures |
|
|
0.9 |
|
|
0.5 |
|
Construction in progress |
|
|
1.4 |
|
|
6.9 |
|
Total cost |
|
|
48.4 |
|
|
42.3 |
|
Less: accumulated depreciation |
|
|
24.4 |
|
|
13.9 |
|
Total property, plant and equipment, net |
|
$ |
24.0 |
|
$ |
28.4 |
In connection with the separation, certain property, plant and equipment, mainly buildings and associated accumulated depreciation totaling $9.8 million was retained by Biogen. During the years ended December 31, 2017, 2016 and 2015, depreciation expense associated with property, plant and equipment was $8.7 million, $48.6 million, and $12.6 million, respectively.
11. Income Taxes
The company’s effective tax rate was 21.3% during the year ended December 31, 2017 as compared to (50.6)% in the prior year comparative period. The company’s effective income tax rate for year ended December 31, 2017 differs from the U.S. federal statutory rate mainly due to state and local taxes partially offset by credits, permanent items, and the impact of a one-time deferred tax revaluation under the 2017 Tax Act. The company’s effective income tax rate for year ended December 31, 2016 differ from the U.S. federal statutory rates mainly due to the release of a previously established valuation allowance.
Prior to the separation, the company’s income tax expense and deferred tax balances were calculated on a separate tax return basis although the company’s operations had historically been included in the tax returns filed by the
F-22
respective Biogen entities, of which the company’s business was a part. For periods subsequent to the separation, Bioverativ files tax returns on its own behalf and its income tax expense and deferred income tax balances are recorded in accordance with the company’s standalone income tax positions. Biogen and Bioverativ entered into a tax matters agreement effective as of the date of separation. Under the terms of the tax matters agreement Bioverativ may be required to indemnify Biogen for certain tax matters. To date, no amounts have been identified.
The 2017 Tax Act, which was signed into law on December 22, 2017, has resulted in significant changes to the U.S. corporate income tax system. These changes include a federal statutory rate reduction from 35% to 21%, the elimination or reduction of certain domestic deductions and credits and limitations on the deductibility of interest expense and executive compensation. The 2017 Tax Act also transitions international taxation from a worldwide system to a modified territorial system and includes base erosion prevention measures on non-U.S. earnings. These changes are effective beginning in 2018. The 2017 Tax Act also includes a one-time mandatory deemed repatriation tax on accumulated foreign subsidiaries' previously untaxed foreign earnings (the Transition Toll Tax). Changes in tax rates and tax laws are accounted for in the period of enactment. During the year ended December 31, 2017, we recorded a benefit totaling $58.8 million related to our current estimate of the provisions of the 2017 Tax Act. The preliminary estimate of Transition Toll Tax was immaterial.
Our preliminary estimate of the Transition Toll Tax and the remeasurement of our deferred tax assets and liabilities is subject to the finalization of management's analysis related to certain matters, such as developing interpretations of the provisions of the 2017 Tax Act, changes to certain estimates and amounts related to the earnings and profits of certain subsidiaries and the filing of our tax returns. U.S. Treasury regulations, administrative interpretations or court decisions interpreting the 2017 Tax Act may require further adjustments and changes in our estimates, which could have a material adverse effect on our business, results of operations or financial conditions. The final determination of the Transition Toll Tax and the remeasurement of our deferred tax assets and liabilities will be completed as additional information becomes available, but no later than one year from the enactment of the 2017 Tax Act.
Income before Income Tax Provision and Income Tax Expense
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|||
|
Income (loss) before income tax expense (benefit): |
|
|
|
|
|
|
|
|
|
|
Domestic |
|
$ |
444.0 |
|
$ |
284.1 |
|
$ |
94.2 |
|
Foreign |
|
|
7.9 |
|
|
7.8 |
|
|
4.4 |
|
Total |
|
$ |
451.9 |
|
$ |
291.9 |
|
$ |
98.6 |
|
Income tax expense (benefit): |
|
|
|
|
|
|
|
|
|
|
Current |
|
|
|
|
|
|
|
|
|
|
Domestic |
|
$ |
140.8 |
|
$ |
8.4 |
|
$ |
— |
|
Foreign |
|
|
3.1 |
|
|
2.4 |
|
|
1.4 |
|
Deferred |
|
|
|
|
|
|
|
|
|
|
Domestic |
|
|
(47.3) |
|
|
(158.5) |
|
|
(11.4) |
|
Foreign |
|
|
(0.3) |
|
|
— |
|
|
— |
|
Total income tax expense (benefit) |
|
$ |
96.3 |
|
$ |
(147.7) |
|
$ |
(10.0) |
F-23
Deferred Tax Assets and Liabilities
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Deferred tax assets |
|
|
|
|
|
|
|
Net operating loss |
|
$ |
21.6 |
|
$ |
85.5 |
|
Tax credits |
|
|
3.6 |
|
|
57.4 |
|
Inventory, other reserves, and accruals |
|
|
12.8 |
|
|
14.5 |
|
Share-based compensation |
|
|
6.7 |
|
|
3.4 |
|
Intangibles, net |
|
|
— |
|
|
9.5 |
|
Deferred revenue |
|
|
9.2 |
|
|
3.6 |
|
Other |
|
|
1.2 |
|
|
— |
|
Valuation allowance |
|
|
— |
|
|
— |
|
Total deferred tax assets |
|
$ |
55.1 |
|
$ |
173.9 |
|
Deferred tax liabilities |
|
|
|
|
|
|
|
Purchased intangible assets |
|
$ |
(140.4) |
|
$ |
(16.3) |
|
Depreciation and amortization, net |
|
|
(2.7) |
|
|
(3.4) |
|
Total deferred tax liabilities |
|
$ |
(143.1) |
|
$ |
(19.7) |
In addition to deferred tax assets and liabilities, we have recorded prepaid tax and deferred charges related to intercompany transactions. As of December 31, 2017 the total deferred charges and prepaid taxes were $26.0 million.
Income Tax Expense Reconciliation
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|||
|
Statutory rate |
|
35.0 |
% |
|
35.0 |
% |
|
35.0 |
% |
|
State taxes |
|
2.1 |
|
|
0.8 |
|
|
1.0 |
|
|
Taxes on foreign earnings |
|
— |
|
|
(0.1) |
|
|
(0.3) |
|
|
Tax rate change |
|
(13.0) |
|
|
— |
|
|
— |
|
|
Credits and NOLs |
|
(5.5) |
|
|
(0.7) |
|
|
(3.0) |
|
|
Changes in valuation allowance |
|
— |
|
|
(84.8) |
|
|
(42.7) |
|
|
Other permanent items |
|
2.2 |
|
|
(1.3) |
|
|
(0.2) |
|
|
Other |
|
0.5 |
|
|
0.5 |
|
|
— |
|
|
Effective income tax rate |
|
21.3 |
% |
|
(50.6) |
% |
|
(10.2) |
% |
Our effective tax rate for 2017 compared to 2016 fluctuated primarily due to a $58.8 million one-time deferred tax revaluation based on recent tax law changes in 2017 and the release of the $247.3 million valuation allowance on our domestic deferred tax assets in 2016. Our effective tax rate for 2016 compared to 2015 benefitted primarily due to the release of a $247.3 million valuation allowance release in 2016.
As of December 31, 2017, we had acquired net operating losses and general business credit carry forwards for federal income tax purposes of approximately $68.8 million and $4.0 million, respectively, which begin to expire in 2025 and 2020, respectively. Additionally, for state income tax purposes, we had acquired net operating loss carry forwards of approximately $102.3 million, which begin to expire in 2033. Internal Revenue Code Section 382 limits the use of net operating loss and tax credit carryforwards in the case of an “ownership change” of a corporation. Any ownership changes, as defined, may restrict utilization of carryforwards.
In assessing the realizability of our deferred tax assets, we have considered whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. In making this determination, under the applicable financial reporting standards, we are allowed to consider
F-24
the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies. Our estimates of future taxable income take into consideration, among other items, our estimates of future income tax deductions related to the exercise of stock options. Based upon the reversal of taxable temporary differences, its level of historical taxable income and income tax liability and projections for future taxable income over the periods in which the deferred tax assets are utilizable, we believe it is more likely than not that we will realize the net benefits of the deferred tax assets of our wholly owned subsidiaries. In the event that actual results differ from our estimates or we adjust our estimates in future periods, we may need to establish a valuation allowance, which could materially impact our financial position and results of operations.
As of December 31, 2017, we are subject to taxation in the U.S. federal jurisdiction, various U.S. states, and foreign jurisdictions. The Company is subject to examination for tax years beginning in 2017.
12. Earnings per Share
The denominator for basic earnings per common share (EPS) is the weighted-average number of common shares outstanding during the period. The dilutive effect of outstanding stock options and time-vested restricted stock units is reflected in the denominator for diluted EPS using the treasury stock method. The numerator for both basic and diluted EPS is net income.
On February 1, 2017, Biogen distributed approximately 108 million shares of Bioverativ common stock to its shareholders. The computation of basic and dilutive EPS for periods prior to the separation was calculated using the shares distributed.
Basic and diluted earnings per share are calculated as follows:
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|||
|
Numerator: |
|
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
355.6 |
|
$ |
439.6 |
|
$ |
108.6 |
|
Denominator: |
|
|
|
|
|
|
|
|
|
|
Weighted average number of common shares outstanding |
|
|
108.1 |
|
|
108.0 |
|
|
108.0 |
|
Effect of dilutive securities: |
|
|
|
|
|
|
|
|
|
|
Stock options |
|
|
— |
|
|
— |
|
|
— |
|
Time-vested restricted stock units |
|
|
0.4 |
|
|
— |
|
|
— |
|
Dilutive potential common shares |
|
|
0.4 |
|
|
— |
|
|
— |
|
Shares used in calculating diluted earnings per share |
|
|
108.5 |
|
|
108.0 |
|
|
108.0 |
The computation of diluted earnings per share excludes 2.2 million of outstanding common share equivalents for stock options for the year ended December 31, 2017, as compared to 2.5 million of outstanding common share equivalents for stock options and time-vested restricted stock units for the years ended December 31, 2016 and 2015 as the effect is anti-dilutive.
Bioverativ Share‑Based Compensation Plans
In connection with the separation, we adopted our own share‑based compensation plans. Specifically, we adopted the Bioverativ Inc. 2017 Omnibus Equity Plan (the Omnibus Plan); the Bioverativ Inc. 2017 Non‑Employee Directors Equity Plan (the Directors Plan); and the Bioverativ Inc. 2017 Employee Stock Purchase Plan (ESPP). Cash-settled performance units (CSPUs), market stock units (MSUs), performance‑vested restricted stock units (PUs) and restricted stock units (RSUs) granted under Biogen 2008 Omnibus Equity Plan to Biogen employees who became employees of Bioverativ were converted into Bioverativ RSUs, and stock options held by Biogen employees who became employees of Bioverativ were converted into Bioverativ stock options, each according to the terms of an
F-25
employee matters agreement entered into between Biogen and Bioverativ in connection with the separation (converted awards). Converted awards, as well as future awards issued to Bioverativ officers, directors and employees, will be awarded under, and subject to the terms of, the Omnibus Plan, the Directors Plan and the ESPP, as the case may be. Unless extended or earlier terminated, the term of the awards under these plans shall be 10 years from the effective date. The maximum number of shares of common stock subject to options granted to any participant, and that may be granted as RSUs, shall not exceed an aggregate of 2,250,000 in any calendar year. The maximum aggregate number of shares of common stock available for purchase under the ESPP by eligible employees will be 1,625,000 shares.
The following table provides share-based compensation expense by statement of income line item for the years ended December 31, 2017, 2016 and 2015:
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|||
|
Cost of sales |
|
$ |
— |
|
$ |
7.3 |
|
$ |
2.1 |
|
Research and development |
|
|
5.0 |
|
|
3.7 |
|
|
4.8 |
|
Selling, general and administrative |
|
|
27.0 |
|
|
4.3 |
|
|
6.3 |
|
Total share-based compensation expense |
|
$ |
32.0 |
|
$ |
15.3 |
|
$ |
13.2 |
During the year ended December 31, 2017 and subsequent to the separation, Bioverativ awarded share-based compensation grants consisting of 2.3 million stock options and 0.4 million RSUs, with respect to the company’s employees.
Post-Separation
Stock Options
Stock options awarded to employees generally vest one-third per year over three years on the anniversary of the date of grant, or upon the third anniversary of the date of the grant, provided the employee remains continuously employed with us, except as otherwise provided in the plan. Options granted are exercisable at a price per share not less than the fair market value of the underlying common stock on the date of grant. The estimated fair value of stock option, including the effect of forfeitures, is recognized over the options’ vesting periods. The fair value of the stock options granted in 2017 was estimated as of the date of grant using a Black-Scholes option valuation model.
The weighted-average Black-Scholes assumptions used in estimating the fair value of stock options granted by Bioverativ following the separation during the year ended December 31, 2017, along with the weighted-average grant-date fair values, were as follows:
|
|
|
For the Years Ended |
|
|
|
December 31, |
|
|
|
2017 |
|
Expected volatility |
|
40.0% |
|
Range of expected life (in years) |
|
4 ̶ 5 |
|
Range of risk-free interest rate |
|
1.72% ̶ 2.13% |
|
Dividend yield |
|
̶ |
|
Range of fair value per stock option |
|
$16.78 ̶ $23.38 |
Expected volatility was determined using an average of peer companies’ expected volatility. As of December 31, 2017, the unrecognized compensation cost related to all unvested stock options held by Bioverativ’s employees of $19.8 million is expected to be recognized as expense over a weighted-average period of 2.2 years. As of December 31, 2017, no stock options were exercisable.
F-26
The following table summarizes our stock option activity:
|
|
|
|
|
Weighted Average |
|
|
|
|
|
|
Per Share Grant |
|
|
(In thousands, except per share amounts) |
|
Shares |
|
Date Fair Value |
|
|
Outstanding at December 31, 2016 |
|
— |
|
$ |
— |
|
Granted |
|
2,254.5 |
|
|
16.98 |
|
Exercised |
|
— |
|
|
— |
|
Cancelled |
|
(91.2) |
|
|
17.61 |
|
Outstanding at December 31, 2017 |
|
2,163.3 |
|
$ |
16.97 |
In connection with the merger with Sanofi, the stock options outstanding as of December 31, 2017 will accelerate and become fully vested and will be canceled and converted into the right to receive the $105.0 per share less the applicable exercise price in cash. See Note 21, Subsequent Events to our audited consolidated financial statements included in this Form 10-K.
Restricted Stock Units
RSUs awarded to employees generally vest one-third per year over three years on the anniversary of the date of grant, or upon the third anniversary of the date of the grant. Shares of our common stock will be delivered to the employee upon vesting, subject to payment of applicable withholding taxes. The vesting of these awards is subject to the respective employee's continued employment. The fair value of all RSUs is based on the market value of our stock on the date of grant. Compensation expense, including the effect of forfeitures, is recognized over the applicable service period.
As of December 31, 2017, the unrecognized compensation cost related to all unvested RSUs held by Bioverativ employees of $13.8 million is expected to be recognized as expense over a weighted-average period of 1.4 years.
The following table summarizes our RSU activity:
|
|
|
|
|
Weighted Average |
|
|
|
|
|
|
Per Share Grant |
|
|
(In thousands, except per share amounts) |
|
Shares |
|
Date Fair Value |
|
|
Unvested at December 31, 2016 |
|
348.6 |
|
$ |
46.86 |
|
Granted |
|
401.4 |
|
|
46.79 |
|
Conversions |
|
337.4 |
|
|
44.98 |
|
Vested |
|
(227.5) |
|
|
47.69 |
|
Forfeited |
|
(55.2) |
|
|
46.30 |
|
Unvested at December 31, 2017 |
|
804.7 |
|
$ |
45.84 |
Conversions relate to Biogen MSUs, Biogen CSPUs and Biogen PUs that were converted to RSUs upon separation. The components of conversions are as follows:
|
|
|
|
|
|
(In thousands) |
|
Shares |
|
|
RSUs converted from MSUs |
|
155.0 |
|
|
RSUs converted from CSPUs |
|
43.6 |
|
|
RSUs converted from PUs |
|
138.8 |
|
|
Total |
|
337.4 |
|
F-27
In connection with the merger with Sanofi, the RSUs outstanding as of December 31, 2017 will accelerate and become fully vested and will be canceled and converted into the right to receive the $105.0 per share in cash. See Note 21, Subsequent Events to our audited consolidated financial statements included in this Form 10-K.
Pre-separation
Market Stock Units
MSUs awarded by Biogen to Bioverativ employees vest in three equal annual increments beginning on the first anniversary of the grant date. The vesting of these awards is subject to the respective employee's continued employment. Compensation expense, including the effect of forfeitures, is recognized over the applicable service period.
MSUs were valued using a lattice model with a Monte Carlo simulation. This valuation methodology utilized several key assumptions, the 30 calendar day average Biogen closing stock price on the date of grant, expected volatility of Biogen’s stock price, risk-free rates of return and expected dividend yield. The assumptions used in Biogen’s valuation are summarized as follows:
|
|
|
For the Years Ended |
||
|
|
|
December 31, |
||
|
|
|
2016 |
|
2015 |
|
Expected dividend yield |
|
̶ |
|
̶ |
|
Range of expected stock price volatility |
|
38.2% - 40.7% |
|
31.0% - 33.2% |
|
Range of risk-free interest rates |
|
0.6% - 0.9% |
|
0.2% - 1.0% |
|
30 calendar day average stock price on grant date |
|
$260.67 - $304.86 |
|
$277.35 - $426.27 |
|
Weighted-average per share grant date fair value |
|
$326.65 |
|
$499.25 |
Cash-Settled Performance Units
CSPUs awarded by Biogen to Bioverativ employees vest in three equal annual increments beginning on the first anniversary of the grant date. The vesting of these awards is subject to the respective employee's continued employment with such awards settled in cash. Compensation expense, including the effect of forfeitures, is recognized over the applicable service period.
Performance-Vested Restricted Stock Units
PUs awarded by Biogen to certain Bioverativ employees in the form of restricted stock units that may be settled in cash or shares of Biogen common stock at the sole discretion of the Compensation and Management Development Committee of Biogen's board of directors. These awards were structured and accounted for the same way as the cash settled performance units, and vest in three equal annual increments beginning on the first anniversary of the grant date. Compensation expense, including the effect of forfeitures, is recognized over the applicable service period. The vesting of these awards is subject to the respective employee's continued employment.
14. Related Parties
Under the terms of the transition services agreement entered into with Biogen in connection with the separation, Biogen provides various services on an interim, transitional basis. The services provided by Biogen to Bioverativ consist of certain information technology, quality, regulatory, research and supply chain. These services are generally provided on a cost-plus basis. The services are generally expected to extend for up to 2 years following the separation.
For the company’s operations in the United States and Canada transfer of marketing authorizations and other regulatory requirements did not occur by the separation date of February 1, 2017. As a result, the company entered into a commercial operations agreement with Biogen for the United States and Canada. Under the terms of the commercial operations agreements, until the Company has obtained the various required authorizations, Biogen agreed to perform certain services related to the distribution of finished goods to the company’s customers. The company is responsible for
F-28
the business activities conducted by Biogen on its behalf, and is subject to the risks and entitled to the benefits generated by these operations and assets. As a result, the related liabilities and results of operations have been reported in the company’s consolidated financial statements as of and for the year ended December 31, 2017. Net sales related to these operations totaled $668.8 million during the year ended December 31, 2017. In May 2017, the Canadian marketing authorizations transferred to Bioverativ and in September 2017, the U.S. marketing authorizations transferred to Bioverativ. In July 2017 and October 2017, the commercial operations agreements in Canada and the United States, respectively, were completed.
The company and Biogen also entered into a manufacturing and supply agreement whereby Biogen continues to produce ELOCTATE and ALPROLIX for us on a cost‑plus basis. As products were historically transferred at cost between Biogen and Bioverativ, these manufacturing and supply arrangements will result in changes to cost of goods sold in future periods.
The company and Biogen also entered into a separation and distribution agreement, tax matters agreement, an employee matters agreement.
As of December 31, 2017, total amount due to Biogen associated with transition service and manufacturing and supply agreements was $90.5 million.
Corporate Overhead and Other Allocations from Biogen
Prior to the separation, the company did not historically operate as a standalone business and had various relationships with Biogen whereby Biogen provided services to the company. These consolidated financial statements include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. For more information, see Note 1, Nature of Business and Basis of Preparation. For the years ended December 31, 2017, the allocations are for the period from January 1, 2017 through the separation date. These allocations were reflected as follows in the consolidated financial statements:
|
|
|
For the Years Ended |
|
|||||||
|
|
|
December 31, |
|
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Research and development allocations |
|
$ |
6.5 |
|
$ |
88.7 |
|
$ |
84.5 |
|
|
Selling, general and administrative allocations |
|
|
5.1 |
|
|
60.7 |
|
|
64.0 |
|
|
Other (income) expense, net allocations |
|
|
0.2 |
|
|
0.6 |
|
|
0.1 |
|
|
Total corporate overhead and other allocations from Biogen |
|
$ |
11.8 |
|
$ |
150.0 |
|
$ |
148.6 |
|
F-29
15. Other Consolidated Financial Statement Detail
Accrued Expenses and Other Current Liabilities
Accrued expenses and other consists of the following:
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Revenue-related rebates |
|
$ |
47.9 |
|
$ |
21.7 |
|
Employee compensation and benefits |
|
|
52.4 |
|
|
15.9 |
|
Research and development |
|
|
9.9 |
|
|
5.5 |
|
Royalty and collaboration |
|
|
38.8 |
|
|
26.6 |
|
Short-term portion of contingent consideration |
|
|
49.2 |
|
|
— |
|
Taxes payable |
|
|
9.4 |
|
|
10.8 |
|
Other |
|
|
25.9 |
|
|
8.8 |
|
Total accrued expenses and other current liabilities |
|
$ |
233.5 |
|
$ |
89.3 |
Long‑Term Liabilities
Long‑term liabilities consist of the following:
|
|
|
As of |
||||
|
|
|
December 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Employee compensation and benefits |
|
$ |
— |
|
$ |
18.7 |
|
Sobi payments |
|
|
87.8 |
|
|
45.0 |
|
Other |
|
|
4.7 |
|
|
— |
|
Total long term liabilities |
|
$ |
92.5 |
|
$ |
63.7 |
16. Litigation
From time to time, we may be involved in various claims and legal proceedings.
With respect to some loss contingencies, an estimate of the possible loss or range of loss cannot be made until management has further information, including, for example, (i) which claims, if any, will survive dispositive motion practice; (ii) information to be obtained through discovery; (iii) information as to the parties’ damages claims and supporting evidence; (iv) the parties’ legal theories; and (v) the parties’ settlement positions.
Subject to the terms of the separation agreement we entered into with Biogen, the company may be responsible for certain liabilities relating to the following legal matters involving Biogen prior to the separation, solely to the extent such liabilities relate to, arise out of or result from hemophilia business. The imposition of any such liability would be carefully evaluated by the company under the terms of the separation agreement.
Government Matters
On March 4, 2016, Biogen received a subpoena from the federal government for documents relating to Biogen’s relationship with non‑profit organizations that provide assistance to patients taking drugs sold by Biogen.
On July 1, 2016, Biogen received civil investigative demands from the federal government for documents and information relating to Biogen’s treatment of certain service agreements with wholesalers when calculating and reporting Average Manufacturer Prices in connection with the Medicaid Drug Rebate Program.
F-30
17. Commitments and Contingencies
True North Therapeutics
In connection with our acquisition of True North, the True North equityholders are eligible to receive additional payments of up to $425.0 million contingent on the achievement of future development, regulatory and sales milestones. See Note 3, Acquisition of True North Therapeutics, Inc.
Contingent Development, Regulatory and Commercial Milestone Payments
Based on our development plans primarily in gene editing and gene therapy for hemophilia and other blood disorders as of December 31, 2017, we could make potential future milestone payments to third party collaborators of up to approximately $755 million. Payments under these agreements generally become due and payable upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones had not occurred as of December 31, 2017, such contingencies have not been recorded in our financial statements. Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones.
Leases
We rent laboratory and office space under non-cancelable operating leases. These lease agreements contain various clauses for renewal at our option and, in certain cases, escalation clauses typically linked to rates of inflation. Rental expense under these leases amounted to $6.8 million in 2017. In addition to rent, the leases may require us to pay additional amounts for taxes, insurance, maintenance and other operating expenses.
As of December 31, 2017, minimum rental commitments under non-cancelable leases for each of the next five years and total thereafter were as follows:
|
(in millions) |
|
2018 |
|
2019 |
|
2020 |
|
2021 |
|
2022 |
|
Thereafter |
|
Total |
|||||||
|
Minimum lease payments |
|
$ |
6.6 |
|
$ |
6.7 |
|
$ |
6.1 |
|
$ |
6.0 |
|
$ |
6.1 |
|
$ |
24.9 |
|
$ |
56.4 |
18. Guarantees
As of December 31, 2017 and 2016, the company did not have significant liabilities recorded for guarantees.
The company enters into indemnification provisions under agreements with other companies in the ordinary course of business, typically with business partners, contractors, clinical sites and customers. Under these provisions, the company generally indemnifies and holds harmless the indemnified party for losses suffered or incurred by the indemnified party as a result of our activities. These indemnification provisions generally survive termination of the underlying agreement. The maximum potential amount of future payments we could be required to make under these indemnification provisions is unlimited. However, to date the company has not incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. As a result, the estimated fair value of these agreements is minimal. Accordingly, the company has no liabilities recorded for these agreements as of December 31, 2017 and 2016.
19. Segment Information
We operate as one operating segment, which is discovering, researching, developing and commercializing innovative therapies for the treatment of rare blood disorders, and, therefore, our chief operating decision‑maker manages the operations of our company as a single operating segment. Enterprise‑wide disclosures about product revenues, other revenues and long‑lived assets by geographic area and information relating to major customers are presented below. Revenues are primarily attributed to individual countries based on location of the customer or licensee.
F-31
Revenue by product is summarized as follows:
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|||
|
United States |
|
|
|
|
|
|
|
|
|
|
ELOCTATE |
|
$ |
615.6 |
|
$ |
445.3 |
|
$ |
308.3 |
|
ALPROLIX |
|
|
290.1 |
|
|
267.9 |
|
|
208.8 |
|
Total product revenues |
|
$ |
905.7 |
|
$ |
713.2 |
|
$ |
517.1 |
|
All Other Markets |
|
|
|
|
|
|
|
|
|
|
ELOCTATE |
|
$ |
108.9 |
|
$ |
67.7 |
|
$ |
11.4 |
|
ALPROLIX |
|
|
74.5 |
|
|
65.7 |
|
|
25.6 |
|
Total product revenues |
|
$ |
183.4 |
|
$ |
133.4 |
|
$ |
37.0 |
|
Total |
|
|
|
|
|
|
|
|
|
|
ELOCTATE |
|
$ |
724.5 |
|
$ |
513.0 |
|
$ |
319.7 |
|
ALPROLIX |
|
|
364.6 |
|
|
333.6 |
|
|
234.4 |
|
Total product revenues |
|
$ |
1,089.1 |
|
$ |
846.6 |
|
$ |
554.1 |
Geographic Inforrmation
|
|
|
For the Years Ended |
|||||||
|
|
|
December 31, |
|||||||
|
(In millions) |
|
2017 |
|
2016 |
|
2015 |
|||
|
U.S. |
|
|
|
|
|
|
|
|
|
|
Product revenues from external customers |
|
$ |
905.7 |
|
$ |
713.2 |
|
$ |
517.1 |
|
Revenue from collaborative partners |
|
$ |
79.4 |
|
$ |
40.8 |
|
$ |
6.2 |
|
Long-lived assets |
|
$ |
22.9 |
|
$ |
27.7 |
|
$ |
75.0 |
|
Japan |
|
|
|
|
|
|
|
|
|
|
Product revenues from external customers |
|
$ |
138.8 |
|
$ |
105.7 |
|
$ |
37.0 |
|
Long-lived assets |
|
$ |
0.8 |
|
$ |
0.7 |
|
$ |
0.5 |
|
Other |
|
|
|
|
|
|
|
|
|
|
Product revenues from external customers |
|
$ |
44.6 |
|
$ |
27.7 |
|
$ |
— |
|
Long-lived assets |
|
$ |
0.3 |
|
$ |
— |
|
$ |
— |
|
Total |
|
|
|
|
|
|
|
|
|
|
Product revenues from external customers |
|
$ |
1,089.1 |
|
$ |
846.6 |
|
$ |
554.1 |
|
Revenue from collaborative partners |
|
$ |
79.4 |
|
$ |
40.8 |
|
$ |
6.2 |
|
Long-lived assets |
|
$ |
24.0 |
|
$ |
28.4 |
|
$ |
75.5 |
20. Quarterly Financial Data (Unaudited)
|
|
|
First |
|
Second |
|
Third |
|
Fourth |
|
Total |
|||||
|
(in millions) |
|
Quarter |
|
Quarter |
|
Quarter |
|
Quarter |
|
Year |
|||||
|
2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenue, net |
|
$ |
242.0 |
|
$ |
263.9 |
|
$ |
274.8 |
|
$ |
308.4 |
|
$ |
1,089.1 |
|
Collaboration revenue |
|
|
17.1 |
|
|
25.2 |
|
|
16.8 |
|
|
20.3 |
|
|
79.4 |
|
Total revenues |
|
|
259.1 |
|
|
289.1 |
|
|
291.6 |
|
|
328.7 |
|
|
1,168.5 |
|
Gross profit(1) |
|
|
195.8 |
|
|
208.6 |
|
|
226.1 |
|
|
258.4 |
|
|
888.9 |
|
Net income |
|
$ |
69.3 |
|
$ |
77.1 |
|
$ |
67.9 |
|
$ |
141.3 |
|
$ |
355.6 |
F-32
|
|
|
First |
|
Second |
|
Third |
|
Fourth |
|
Total |
|||||
|
(in millions) |
|
Quarter |
|
Quarter |
|
Quarter |
|
Quarter |
|
Year |
|||||
|
2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenue, net |
|
$ |
182.8 |
|
$ |
204.9 |
|
$ |
217.1 |
|
$ |
241.8 |
|
$ |
846.6 |
|
Collaboration revenue |
|
|
8.9 |
|
|
5.4 |
|
|
12.1 |
|
|
14.4 |
|
|
40.8 |
|
Total revenues |
|
|
191.7 |
|
|
210.3 |
|
|
229.2 |
|
|
256.2 |
|
|
887.4 |
|
Gross profit(1) |
|
|
159.2 |
|
|
152.1 |
|
|
157.7 |
|
|
180.5 |
|
|
649.5 |
|
Net income (loss) |
|
$ |
66.7 |
|
$ |
63.5 |
|
$ |
80.5 |
|
$ |
228.9 |
|
$ |
439.6 |
|
(1) |
Gross profit is calculated as total revenues less cost of sales. |
21. Subsequent Events
Agreement and Plan of Merger
On January 21, 2018, Bioverativ entered into an Agreement and Plan of Merger (the “Merger Agreement”) with Sanofi (“Parent” or “Sanofi”), a French société anonyme, and Blink Acquisition Corp. (“Merger Sub”), a Delaware corporation and indirect wholly-owned subsidiary of Parent. The Board of Directors of the Company unanimously approved the Merger Agreement.
Pursuant to the Merger Agreement, upon the terms and subject to the conditions thereof, Merger Sub commenced a tender offer on February 7, 2018 (the “Offer”), to acquire all of the outstanding shares of common stock of the Company, par value $0.001 per share (the “Shares”), at a purchase price of $105.0 per Share in cash, net of applicable withholding taxes and without interest (the “Offer Price”).
The obligation of Merger Sub to purchase Shares tendered in the Offer is subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, including (i) that the number of Shares validly tendered and not withdrawn as of the expiration of the Offer, together with any Shares (if any) owned by Parent and its affiliates and excluding any Shares tendered pursuant to guaranteed delivery procedures that have not yet been “received” (as such term is defined in Section 251(h)(6)(f) of the Delaware General Corporation Law, as amended (“DGCL”)), represents at least a majority of the then outstanding Shares (the “Minimum Tender Condition”); (ii) (x) the expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended and (y) that all consents of, and/or filings with, any governmental authority of competent jurisdiction or pursuant to any antitrust laws have been obtained and are in full force and effect and any applicable waiting period with respect thereto has expired; (iii) the absence of any law or order prohibiting the consummation of the Offer or the Merger; (iv) the accuracy of the Company’s representations and warranties (subject to customary materiality qualifiers); (v) the Company’s performance in all material respects of its obligations under the Merger Agreement; (vi) the absence, since the date of the Merger Agreement, of any fact, circumstance, condition, event, change, development, occurrence, result or event which, individually or in the aggregate, has had, or would reasonably be expected to have, a Company Material Adverse Effect (as defined in the Merger Agreement); (vii) the delivery to Parent and Merger Sub of a certificate, dated as of the expiration time of the Offer, certifying that the conditions in clauses (iv), (v) and (vi) have been satisfied; (viii) the Merger Agreement has not been terminated; and (ix) that the Company has received and delivered to Biogen and Parent a copy of the tax opinion required by a letter agreement dated January 21, 2018 between Biogen, Parent and the Company (the “Letter Agreement”) and the requirement that the Company deliver such tax opinion pursuant to the Tax Matters Agreement between Biogen and the Company dated January 31, 2017 has been satisfied pursuant to the Letter Agreement. These conditions are more specifically described in the Merger Agreement, which we filed as Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the SEC on January 22, 2018.
As soon as practicable following (but on the same day as) the consummation of the Offer and subject to the satisfaction or waiver of certain conditions set forth in the Merger Agreement, Merger Sub will be merged with and into the Company (the “Merger”), with the Company surviving the Merger as an indirect wholly-owned subsidiary of Parent. The Merger will be governed by Section 251(h) of the DGCL and effected without a vote of the Company stockholders.
As a result of the Merger, each Share (other than any Shares (i) owned by Parent, Merger Sub or any other direct or indirect wholly-owned subsidiary of Parent immediately prior to the Effective Time (as defined below) (other
F-33
than Shares tendered and accepted for payment by Merger Sub in connection with the Offer), (ii) owned by the Company or held in the Company’s treasury immediately prior to the Effective Time, (iii) owned by any direct or indirect wholly-owned Subsidiary of the Company immediately prior to the Effective Time or (iv) issued and outstanding immediately prior to the Effective Time and held by a holder who is entitled to demand appraisal and who has properly exercised and perfected a demand for appraisal of such Shares in accordance with Section 262 of the DGCL and, as of the Effective Time, has neither effectively withdrawn nor lost such holder’s right to appraisal and payment under the DGCL with respect to such Shares) will (x) be converted automatically into the right to receive the Offer Price in cash, net of applicable withholding taxes and without interest and (y) cease to be outstanding and will automatically be cancelled and cease to exist and each holder of a certificate or a book-entry account statement representing any such Shares will have only the right to receive the Offer Price in accordance with the Merger Agreement.
The Merger Agreement also provides that each Company stock option and Company restricted stock unit (collectively, the “Company Equity Awards”) that is outstanding as of immediately prior to the consummation of the Offer will accelerate and become fully vested and will be canceled and converted into the right to receive the Offer Price (less the applicable exercise price in the case of Company stock options) in cash (net of applicable withholding taxes and without interest) payable in respect of each Share subject to such Company Equity Award, except that the cash payment with respect to any Company restricted stock unit granted after the date of the Merger Agreement in accordance with the terms thereof to employees who are not executive officers will not be vested, and instead will remain outstanding and become vested and payable on December 31, 2018, subject to the employee’s continued employment with the Company or its affiliates, or upon any earlier termination without cause or constructive termination under the applicable severance plan. At or prior to the consummation of the Offer, the Company, its Board of Directors and the Compensation Committee of the Board of Directors, as applicable, are required to adopt resolutions and take any actions that are necessary to effectuate the provisions of the Merger Agreement with respect to the Company Equity Awards. In addition, pursuant to the Merger Agreement, the Company is required, among other things, to take all reasonable actions to terminate its 2017 Employee Stock Purchase Plan prior to the date that the Offer, the Merger and the other transactions contemplated by the Merger Agreement (collectively, the “Transactions”) close and to ensure no new offering period is commenced.
The Merger Agreement includes representations and warranties, and covenants of the parties customary for a transaction of this nature. Until the earlier of the termination of the Merger Agreement and the date on which the Merger becomes effective (the “Effective Time”), the Company has agreed to operate its business and the business of its subsidiaries in the ordinary course, consistent with past practice, and has agreed to certain other restrictive operating covenants, as set forth more fully in the Merger Agreement. Subject to the terms and conditions of the Merger Agreement, the Company has also agreed not to solicit or engage in discussions with any third party regarding acquisition proposals.
The Merger Agreement includes a remedy of specific performance for the Company, Parent and Merger Sub in the event one of the other parties breaches or fails to perform its obligations thereunder. The Merger Agreement also includes customary termination provisions for both the Company and Parent and provides that, in connection with the termination of the Merger Agreement under specified circumstances, including termination by the Company to accept and enter into a definitive agreement with respect to a “superior proposal” or by Parent in the event our Board of Directors changes its recommendation, the Company will be required to pay (or cause to be paid) a termination fee of $326.0 million (the “Termination Fee”). Any termination of the Merger Agreement by the Company in connection with a superior proposal is subject to certain conditions, including the Company’s compliance with certain procedures set forth in the Merger Agreement and a determination by our Board of Directors that the failure to take such action would reasonably be expected to be inconsistent with its fiduciary duties, payment of the Termination Fee by (or on behalf of) the Company and the concurrent execution of a definitive agreement by the Company with such third party. A “superior proposal” is defined in the Merger Agreement as a bona fide written “acquisition proposal”, which generally means a proposal made by a third party to acquire (x) 50% of the outstanding equity interests of the Company or (y) assets of the Company and/or its subsidiaries representing at least 50% of the fair market value of the assets of the Company and its subsidiaries, taken as a whole, that did not result from the Company’s breach of the non-solicitation provisions set forth in the Merger Agreement and that the Board of Directors determines in its good faith judgment (after consultation with its financial advisors and outside legal counsel) taking into account certain factors, is reasonably likely to be consummated in accordance with its terms and would, if consummated, result in a transaction that is more favorable to the Company’s stockholders from a financial point of view than the Transactions.
F-34
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Bioverativ Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of Bioverativ Inc. and its subsidiaries as of December 31, 2017 and 2016, and the related consolidated statements of income and comprehensive income, of equity and of cash flows for each of the three years in the period ended December 31, 2017, including the related notes (collectively referred to as the “consolidated financial statements”). We also have audited the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2017 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Annual Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company's internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the
F-35
transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
Februay 14, 2018
We have served as the Company’s auditor since 2016.
F-36