Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - MEDICINOVA INC | mnov-ex322_10.htm |
| EX-32.1 - EX-32.1 - MEDICINOVA INC | mnov-ex321_8.htm |
| EX-31.2 - EX-31.2 - MEDICINOVA INC | mnov-ex312_11.htm |
| EX-31.1 - EX-31.1 - MEDICINOVA INC | mnov-ex311_6.htm |
| EX-23.1 - EX-23.1 - MEDICINOVA INC | mnov-ex231_7.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
Form 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from. to
Commission file number: 001-33185
MEDICINOVA, INC.
(Exact Name of Registrant as Specified in its Charter)
|
Delaware |
|
33-0927979 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
|
(I.R.S. Employer Identification No.) |
|
|
|
|
|
4275 Executive Square, Suite 300, La Jolla, CA |
|
92037 |
|
(Address of Principal Executive Offices) |
|
(Zip Code) |
(858) 373-1500
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Each Exchange on Which Registered |
|
Common Stock, par value $0.001 per share |
|
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
☐ |
|
Accelerated filer |
☒ |
|
|
Non-accelerated filer |
☐ |
|
Smaller reporting company |
☐ |
|
|
Emerging growth company |
☐ |
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes ☐ No ☒
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $172,754,300 based on the closing price of the registrant’s common stock on The NASDAQ Global Market of $5.26 per share on June 30, 2017. Shares of common stock held by each executive officer and director and each affiliated entity has been excluded from this calculation. This determination of affiliate status may not be conclusive for other purposes.
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, as of February 12, 2018 was 40,928,546.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A in connection with the registrant’s 2018 Annual Meeting of Stockholders, which will be filed subsequent to the date hereof, are incorporated by reference into Part III of this Form 10-K.
ii
FORM 10-K—ANNUAL REPORT
For the Fiscal Year Ended December 31, 2017
Table of Contents
|
|
|
Page |
|
PART I |
|
|
|
Item 1 |
4 |
|
|
Item 1A |
25 |
|
|
Item 1B |
46 |
|
|
Item 2 |
46 |
|
|
Item 3 |
46 |
|
|
tem 4 |
46 |
|
|
|
|
|
|
PART II |
|
|
|
Item 5 |
47 |
|
|
Item 6 |
49 |
|
|
Item 7 |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
50 |
|
Item 7A |
57 |
|
|
Item 8 |
57 |
|
|
Item 9 |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
77 |
|
Item 9A |
77 |
|
|
Item 9B |
81 |
|
|
|
|
|
|
PART III |
|
|
|
Item 10 |
82 |
|
|
Item 11 |
82 |
|
|
Item 12 |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
82 |
|
Item 13 |
Certain Relationships and Related Transactions, and Director Independence |
83 |
|
Item 14 |
83 |
|
|
|
|
|
|
PART IV |
|
|
|
Item 15 |
84 |
|
|
|
|
|
|
88 |
||
The MediciNova logo is a registered trademark of MediciNova, Inc. All other product and company names are registered trademarks or trademarks of their respective companies.
3
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K includes forward-looking statements that involve a number of risks and uncertainties, many of which are beyond our control. The forward-looking statements are contained principally in the sections titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” but are also contained elsewhere in this report. Forward-looking statements include all statements that are not historical facts and, in some cases, can be identified by terms such as "believe," "may," "will," "estimate," "continue," "anticipate," "design," "intend," "expect," "could," "plan," "potential," "predict," "seek," "should," "would" or the negative version of these words and similar expressions.
Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements, including those described in "Risk Factors" and elsewhere in this report. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our beliefs and assumptions only as of the date of this report. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. You should read this report completely and with the understanding that our actual future results may be materially different from what we expect.
The following factors are among those that may cause actual results to differ materially from our forward-looking statements:
4
|
• |
International trade or foreign exchange restrictions, increased tariffs, foreign currency exchange; |
|
|
|
• |
High quality material for our products may become difficult to obtain or expensive; |
|
|
• |
Strict government regulations on our business; |
|
|
• |
Regulations governing the production or marketing of our product candidates; |
|
|
• |
Loss of, or inability to attract, key personnel; and |
|
|
• |
Economic, political, foreign exchange and other risks associated with international operations. |
5
Overview
We are a biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of serious diseases with unmet medical needs and a commercial focus on the United States market. Our current strategy is to focus our development activities on MN-166 (ibudilast) for neurological disorders such as progressive multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), glioblastoma, and substance dependence and addiction (e.g., methamphetamine dependence, opioid dependence, and alcohol dependence), and MN-001 (tipelukast) for fibrotic diseases such as nonalcoholic steatohepatitis (NASH) and idiopathic pulmonary fibrosis (IPF). Our pipeline also includes MN-221 (bedoradrine) for the treatment of acute exacerbation of asthma and MN-029 (denibulin) for solid tumor cancers.
MN-166 (ibudilast) is currently in development for several different neurological diseases as described below.
|
•Progressive Multiple Sclerosis: We completed a Phase 2b clinical trial of MN-166 (ibudilast) for the treatment of relapsing multiple sclerosis (MS), in which positive safety and neuroprotective efficacy indicators were observed. The data from this trial indicated that MN-166 (ibudilast) may have potential in the treatment of progressive MS. |
We partnered with investigators on a Phase 2b clinical trial of MN-166 (ibudilast) in primary progressive and secondary progressive MS which was conducted by NeuroNEXT and funded by the National Institute of Health’s (NIH) National Institute of Neurological Diseases and Stroke (NINDS). The progressive MS trial completed randomization of 255 subjects in 2015, which exceeded the goal of 250 subjects that were planned for participation. In October 2017, we announced the presentation of positive top-line results from the SPRINT-MS Phase 2b clinical trial of MN-166 (ibudilast) in progressive MS. The trial achieved both primary endpoints of whole brain atrophy and safety and tolerability. MN-166 (ibudilast) demonstrated a statistically significant 48% reduction in the rate of progression of whole brain atrophy compared to placebo (p=0.04) as measured by MRI analysis using brain parenchymal fraction (BPF) and there was not an increased rate of serious adverse events in the MN-166 (ibudilast) group compared to the placebo group. In February 2018, we announced the presentation of positive clinical efficacy trends from this trial regarding the important secondary endpoint of confirmed disability progression. MN-166 (ibudilast) demonstrated a 26% reduction in the risk of confirmed disability progression compared to placebo (hazard ratio = 0.74), as measured by EDSS (Expanded Disability Status Scale).
The United States Food and Drug Administration (FDA) has granted Fast Track designation for the development of MN-166 (ibudilast) for the treatment of patients with progressive MS.
|
•Amyotrophic Lateral Sclerosis (ALS): We initiated a clinical trial of MN-166 (ibudilast) in amyotrophic lateral sclerosis (ALS) in the second half of 2014, and this trial was completed during the second half of 2017. In December 2017, we announced positive top-line results from this trial. The trial achieved the primary endpoint of safety and tolerability. In addition, there was a higher rate of responders on the ALSFRS-R total score in the MN-166 (ibudilast) group compared to the placebo group. The Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) total score measures the functional activity of an ALS subject. |
We have collaborated with Massachusetts General Hospital (MGH) to conduct a clinical trial to study the effects of MN-166 (ibudilast) on reducing brain microglial activation in ALS subjects which can be monitored by a biomarker. This ongoing clinical trial, which we refer to as the ALS / Biomarker study, will also evaluate several clinical outcomes.
The FDA has granted Fast Track designation to MN-166 (ibudilast) for the treatment of ALS as well as Orphan-Drug designation for the treatment of ALS, which will provide seven years of marketing exclusivity if it is approved for ALS. The European Commission also granted Orphan Medicinal Product Designation for MN-166 (ibudilast) for the treatment of ALS.
6
Investigators at Columbia University and the New York State Psychiatric Institute (NYSPI) previously completed a Phase 1b/2a clinical trial of MN-166 (ibudilast) in opioid withdrawal that was funded by NIDA. Investigators at Columbia University and the NYSPI also conducted a NIDA-funded, Phase 2a clinical trial to evaluate the efficacy of MN-166 (ibudilast) in the treatment of patients addicted to prescription opioids or heroin. In March 2016, we announced that positive findings from the results of this completed study in opioid dependence were presented at the Behavior, Biology and Chemistry: Translational Research in Addiction Meeting. Researchers at UCLA were granted approval and funding by the National Institute on Alcoholism and Alcohol Abuse (NIAAA) for a clinical trial to evaluate MN-166 (ibudilast) for the treatment of alcohol dependence. This clinical trial has been completed and results were presented at the American College of Neuropsychopharmacology (ACNP)’s 54th Annual Meeting in December 2015.
|
•Glioblastoma: We are planning to initiate clinical development to evaluate MN-166 (ibudilast) for the treatment of glioblastoma. In June 2017, we announced positive results from an animal model study that examined the potential clinical efficacy of MN-166 (ibudilast) for the treatment of glioblastoma. Results were presented at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting. |
MN-001 (tipelukast) is currently in development for fibrotic diseases including nonalcoholic steatohepatitis (NASH) and idiopathic pulmonary fibrosis (IPF), which are described below.
|
•Nonalcoholic Steatohepatitis (NASH) and Nonalcoholic Fatty Liver Disease (NAFLD): A clinical trial is currently ongoing to investigate MN-001 (tipelukast) for the treatment of hypertriglyceridemia in NASH and NAFLD patients. We announced positive results of MN-001 (tipelukast) in two different NASH mouse models in 2014 and we opened the IND (Investigational New Drug) application for MN-001 (tipelukast) for the treatment of NASH with the FDA in 2015. The FDA subsequently granted Fast Track designation to MN-001 (tipelukast) for the treatment of patients with NASH with fibrosis. |
We completed a Phase 2 clinical trial of MN-221 for the treatment of acute exacerbations of asthma treated in the emergency room and conducted an End-of-Phase 2 meeting with the United States Food and Drug Administration (FDA) in October 2012. In that meeting, the FDA identified the risk/benefit profile of MN-221 as a focal point for further development and advised that a clinical outcome, such as a reduction in hospitalizations, would need to be a pivotal trial primary endpoint. We believe the appropriate clinical development for MN-221 will involve conducting dose regimen and acute exacerbations of asthma trial design optimization studies prior to commencing pivotal trials. We are working to identify a partner for financial support before further clinical development is commenced.
We have acquired licenses to MN-166, MN-001, MN-221, and MN-029 for the development of these product candidates. We have pursued development of these product candidates in various indications including progressive MS, ALS, various addictions, NASH, IPF, acute exacerbations of asthma, and solid tumor cancers.
7
Our goal is to build a sustainable biopharmaceutical business through the successful development of differentiated products for the treatment of serious diseases with unmet medical needs in high-value therapeutic areas. Key elements of our strategy are as follows:
|
•Pursue the development of MN-166 (ibudilast) for multiple potential indications with the support of non-dilutive financings. |
We intend to advance our diverse MN-166 (ibudilast) program through a combination of investigator-sponsored clinical trials, trials funded through government grants or other grants, and trials funded by us. In addition to providing drug supply and regulatory support, we are funding portions of the consortium-sponsored trials. For example, we contributed financially to the Secondary and Primary Progressive Ibudilast NeuroNEXT Trial in Multiple Sclerosis (SPRINT-MS) Phase 2b clinical trial of MN-166 (ibudilast) for the treatment of progressive MS, which was primarily funded by the NIH. In addition, we contributed financially to the clinical trial of MN-166 (ibudilast) for the treatment of ALS as well as the ongoing ALS / Biomarker study. We intend to pursue additional strategic alliances to help support further clinical development of MN-166 (ibudilast).
|
•Pursue the development of MN-001 (tipelukast) for fibrotic diseases such as NASH and IPF. |
We intend to advance development of MN-001 (tipelukast) through a variety of means, which may include investigator-sponsored trials with or without grant funding as well as trials funded by us.
|
•Consider strategic partnerships with one or more leading pharmaceutical companies to complete late-stage product development and successfully commercialize our products. |
We develop and maintain relationships with pharmaceutical companies that are therapeutic category leaders. Upon completion of proof-of-concept Phase 2 clinical trials, we intend to discuss strategic alliances with leading pharmaceutical companies who seek late-stage product candidates, such as MN-166, MN-001, MN-221 and MN-029, which could support further clinical development and product commercialization.
Our Product Candidates and Programs
Our product development programs address diseases that we believe are not well served by currently available therapies and represent significant commercial opportunities. We believe that we have product candidates that offer innovative therapeutic approaches that may provide significant advantages relative to current therapies.
Our product acquisitions have focused primarily on product candidates with significant preclinical and early clinical testing data that have been developed by the licensors outside of the United States. We utilize the existing data in preparing Investigational New Drug (INDs) Applications or their foreign equivalents, and in designing and implementing additional preclinical or clinical trials to advance the development programs in the United States or abroad.
Following are the details of our product development programs:
MN-166 (ibudilast)
MN-166 (ibudilast) is a novel, first-in-class, oral, anti-inflammatory and neuroprotective agent. MN-166 (ibudilast) inhibits macrophage migration inhibitory factor (MIF) and certain phosphodiesterases (PDEs). MN-166 (ibudilast) also attenuates activated glia cells, which play a major role in certain neurological conditions. While it has been in use for more than 20 years in Japan and Korea for the treatment of asthma and post-stroke dizziness, we are developing MN-166 (ibudilast) for the treatment of primary progressive and secondary progressive MS, ALS, glioblastoma, and substance dependence. We licensed MN-166 (ibudilast) from Kyorin Pharmaceuticals (Kyorin) in 2004.
The FDA has granted Fast Track designations to MN-166 (ibudilast) for three separate indications: the treatment of progressive MS, the treatment of ALS, and the treatment of methamphetamine dependence. Fast track designation is a process designed to facilitate the development and expedite the review of drugs that are intended to treat serious diseases and have the potential to fill an unmet medical need. An important feature of the FDA’s Fast
8
Track program is that it emphasizes early and frequent communication between the FDA and the sponsor throughout the entire drug development and review process to improve the efficiency of product development. Accordingly, Fast Track status can potentially lead to a shortened timeline to ultimate drug approval.
The FDA has granted Orphan-Drug designation to MN-166 (ibudilast) for the treatment of ALS, which will provide seven years of marketing exclusivity if it is approved for ALS in the U.S. The European Commission also granted Orphan Medicinal Product Designation for MN-166 (ibudilast) for the treatment of ALS which offers potential benefits including 10 years of marketing exclusivity if it is approved for ALS in Europe.
We have filed patent applications for multiple uses of MN-166 (ibudilast) for the treatment of neurological conditions. Some of the patent estate has received allowance in the United States and foreign countries. For example, we have been granted separate U.S. patents that cover the use of MN-166 (ibudilast) for the treatment of progressive MS, for the treatment of ALS, and for the treatment of drug addiction or dependence.
Primary and Secondary Progressive Multiple Sclerosis: MS is a complex disease with predominantly unknown etiology and affects approximately 2.3 million people worldwide, according to the National Multiple Sclerosis Society, or NMSS. Also, according to NMSS, approximately 85 percent of people with MS are initially diagnosed with relapsing-remitting MS, or RRMS, and most people who are initially diagnosed with RRMS will eventually transition to secondary progressive MS, or SPMS. About 15 percent of people with MS are diagnosed with primary progressive MS, or PPMS. There is only one approved drug for PPMS and it is administered by intravenous infusion. There are no approved drugs generally considered safe and efficacious for SPMS in the absence of relapses. There is a significant medical need for a safe, effective, and conveniently administered therapy for patients with PPMS and SPMS. MN-166 (ibudilast) may meet these needs.
Based on promising results from a Phase 2 trial in relapsing MS completed in 2008, investigators from NeuroNEXT, a NIH-funded Phase 2 clinical trial network, evaluated MN-166 (ibudilast) in PPMS and SPMS patients in the United States. SPRINT-MS is the name of the Phase 2b, randomized, double-blind, placebo-controlled trial that evaluated the safety and tolerability of MN-166 (ibudilast) (up to 100 mg/day) in PPMS and SPMS patients. Recruitment and enrollment at 28 medical centers in the United States commenced in late 2013 and randomization of 255 subjects was completed in June 2015. In October 2017, we announced the presentation of positive top-line results from the SPRINT-MS Phase 2b clinical trial of MN-166 (ibudilast) in progressive MS. The trial achieved both primary endpoints of whole brain atrophy and safety and tolerability. MN-166 (ibudilast) demonstrated a statistically significant 48% reduction in the rate of progression of whole brain atrophy compared to placebo (p=0.04) as measured by MRI analysis using brain parenchymal fraction (BPF) and there was not an increased rate of serious adverse events in the MN-166 (ibudilast) group compared to the placebo group. In February 2018, we announced the presentation of positive clinical efficacy trends from this trial regarding the important secondary endpoint of confirmed disability progression. MN-166 (ibudilast) demonstrated a 26% reduction in the risk of confirmed disability progression compared to placebo (hazard ratio = 0.74), as measured by EDSS (Expanded Disability Status Scale. We were granted Fast Track designation from the FDA for MN-166 (ibudilast) for the treatment of progressive MS in 2016.
Amyotrophic Lateral Sclerosis (ALS): ALS, also known as Lou Gehrig’s disease, is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. The nerves lose the ability to trigger specific muscles, which causes the muscles to become weak. As a result, ALS affects voluntary movement and patients in the later stages of the disease may become totally paralyzed. Life expectancy of an ALS patient is usually two to five years. According to the ALS Association, there are approximately 20,000 ALS patients in the United States and approximately 6,000 people in the United States are diagnosed with ALS each year.
We have worked with Carolinas Neuromuscular/ALS-MDA Center at Carolinas HealthCare System Neurosciences Institute, which has conducted a clinical trial of MN-166 (ibudilast) in ALS. The trial was a randomized, double-blind, placebo-controlled study which included a six-month treatment period followed by a six-month open-label extension. The study evaluated the safety and tolerability of MN-166 (ibudilast) 60 mg/day versus placebo when administered in combination with riluzole in subjects with ALS, as well as several efficacy endpoints. Subject enrollment began in October 2014.
In December 2015, we announced that the FDA granted Fast Track designation to MN-166 (ibudilast) for the treatment of patients with ALS. In March 2016, we announced that we received a Notice of Allowance from the United States Patent and Trademark Office (PTO) for a new patent which covers MN-166 (ibudilast) for the
9
treatment of amyotrophic lateral sclerosis (ALS). In April 2016, we announced that interim efficacy data from a mid-study analysis of the clinical trial of MN-166 (ibudilast) in ALS was presented at the American Academy of Neurology (AAN) 68th Annual Meeting.
In December 2017, we announced positive top-line results from the ALS trial at Carolinas Neuromuscular/ALS-MDA Center. The trial achieved the primary endpoint of safety and tolerability. In addition, there was a higher rate of responders on the ALSFRS-R total score in the MN-166 (ibudilast) group compared to the placebo group. The Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) total score measures the functional activity of an ALS subject. There was also a higher rate of responders on the ALSAQ-5 score in the MN-166 (ibudilast) group compared to the placebo group. The Amyotrophic Lateral Sclerosis Assessment Questionnaire (ALSAQ-5) score measures the physical mobility, activities of daily living and independence, eating and drinking, communication, and emotional functioning of an ALS subject.
In October 2016, we announced that the FDA granted Orphan-Drug designation to MN-166 (ibudilast) for the treatment of ALS, which will provide seven years of marketing exclusivity if it is approved for ALS. In December 2016, we announced that the European Commission granted Orphan Medicinal Product Designation for MN-166 (ibudilast) for the treatment of ALS.
In February 2016, we entered into an agreement to collaborate with Massachusetts General Hospital (MGH) to study the effects of MN-166 (ibudilast) on reducing brain microglial activation in ALS subjects measured by a positron emission tomography (PET) biomarker. This ongoing clinical trial, which we refer to as the ALS / Biomarker study, will also evaluate safety and tolerability as well as several clinical outcomes including ALS functional rating scale (ALSFRS-R), slow vital capacity (SVC), and muscle strength measured by hand-held dynamometry (HHD).
Methamphetamine Addiction: Methamphetamine is a central nervous system stimulant drug that is similar in structure to amphetamine. It is a Schedule II drug, meaning that it has high abuse potential and low therapeutic potential. According to the Substance Abuse and Mental Health Services Administration’s (SAMHSA) 2016 National Survey on Drug Use and Health, there are approximately 684,000 people aged 12 or older with methamphetamine use disorder (includes those with dependence or abuse) in the United States. According to the Rand Corporation, the estimate of the economic burden in the United States of methamphetamine use, based on the most recent year for which data are available, is approximately $23.4 billion. Currently, there is no pharmaceutical treatment approved for methamphetamine dependence. Based on non-clinical results of the effects of MN-166 (ibudilast) in an animal model of methamphetamine relapse, investigators at UCLA conducted a Phase 1b clinical trial funded by NIDA to examine the safety and preliminary efficacy of MN-166 (ibudilast) in non-treatment-seeking, methamphetamine-dependent users in an inpatient trial that was completed in 2012. Subsequently, UCLA investigators received NIDA grant funding for a Phase 2 clinical trial to evaluate MN-166 (ibudilast) in methamphetamine-dependent users in an outpatient trial setting that commenced in 2013. Enrollment in this trial was completed in September 2017 and we expect results of this trial during the first quarter of 2018. In November 2017, we announced a collaboration with Oregon Health & Science University to initiate a biomarker study to evaluate MN-166 (ibudilast) in methamphetamine use disorder. We were granted Fast Track designation from the FDA for MN-166 (ibudilast) for the treatment of methamphetamine dependence in 2013.
Opioid Withdrawal and Dependency: According to the SAMHSA’s 2016 National Survey on Drug Use and Health, there are approximately 1.8 million people aged 12 or older with pain reliever use disorder (includes those with dependence or abuse) and approximately 626,000 people aged 12 or older with heroin use disorder (includes those with dependence or abuse) in the United States. Access to prescription opioids has recently become more difficult due to more stringent policies on prescribing opioids. An unintended consequence of this policy is increased use of heroin. Heroin is attractive to prescription opioid addicts because it is less expensive and more accessible than prescription opioids. Heroin poses serious health issues, such as risk of HIV and Hepatitis C infection, overdose and death (Knopf, 2012). The economic costs of nonmedical use of prescription opioids in the United States. in 2006 (Hansen et al., 2011), the most recent year for which data is available, was estimated to total more than $50 billion annually, with lost productivity and crime accounting for 94% of the total economic burden. There is an urgent, significant unmet medical need for a safe, effective non-addictive, non-opioid therapy for the treatment of prescription opioid and heroin addiction. Investigators at Columbia University and NYSPI previously completed a NIDA-funded, double-blind, randomized, placebo-controlled in-unit Phase 1b/2a clinical trial to evaluate the ability of MN-166 (ibudilast) to reduce opioid withdrawal symptoms in humans. Subsequently, investigators at Columbia University and NYSPI conducted a NIDA-funded Phase 2a clinical trial of MN-166 (ibudilast) for the treatment of prescription opioid or heroin dependency. In March 2016, we announced that positive
10
findings from the results of this completed study in opioid dependence were presented at the Behavior, Biology and Chemistry: Translational Research in Addiction Meeting.
Alcohol Addiction: According to SAMHSA’s 2016 National Survey on Drug Use and Health, there are approximately 15.1 million people aged 12 or older with alcohol use disorder (includes those with dependence or abuse) in the United States. The Centers for Disease Control and Prevention (CDC) reports that excessive alcohol use cost the United States $249 billion in 2010, the latest year for which complete data are available. Currently, medicines approved by the FDA to treat alcohol dependence include Antabuse®, Vivitrol®, Campral® and Revia®. However, the search for a safe and effective drug remains elusive due to limited success of these FDA-approved compounds (Witkiewitz et al., 2012). In a non-clinical trial (Bell et al., 2013), MN-166 (ibudilast) was examined in rats and mice and was found to reduce alcohol drinking in alcohol-preferring P rats and high-alcohol drinking (HAD1) rats by 50%, and in mice made dependent on alcohol at doses which had no effect on non-dependent mice. Investigators at UCLA received funding from the NIAAA to conduct a study to evaluate MN-166 (ibudilast) in a randomized, double-blind, placebo-controlled within-subject crossover design to determine the safety, tolerability and initial human laboratory efficacy of MN-166 (ibudilast) in a sample of 24 non-treatment seeking individuals with either alcohol abuse or dependence. The study was initiated in early 2014 and completed enrollment of 24 subjects in June 2015. Results of the alcohol dependence study were presented at the American College of Neuropsychopharmacology (ACNP)’s 54th Annual Meeting in December 2015. MN-166 (ibudilast), but not placebo, significantly decreased basal, daily alcohol craving over the course of the study (p<0.05). MN-166 (ibudilast) did not affect cue- and stress-induced alcohol craving. However, MN-166 (ibudilast) increased positive mood during both the cue reactivity and stress procedures. MN-166 (ibudilast) was safe and well-tolerated during the study.
Glioblastoma: Malignant primary brain tumors represent the most frequent cause of cancer death in children. According to the American Association of Neurological Surgeons, glioblastoma (GBM) is an aggressive, extremely lethal form of brain malignancy that develops from glial cells (astrocytes and oligodendrocytes) and rapidly grows and commonly spreads into nearby brain tissue. GBM is classified as Grade IV, the highest grade, in the World Health Organization (WHO) brain tumor grading system. The American Brain Tumor Association reports that GBM represents 15% of all primary brain tumors and 55% of all gliomas and has the highest number of cases of all malignant tumors, with an estimated 12,390 new cases predicted for 2017. Despite decades of advancements in neuroimaging, neurosurgery, chemotherapy, and radiation therapy, only modest improvements have been achieved and the prognosis has not improved for individuals diagnosed with GBM. Median survival of GBM patients is 14.6 months. In June 2017, we announced positive results from an animal model study that examined the potential clinical efficacy of MN-166 (ibudilast) for the treatment of GBM which were presented at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting. Results of the GBM mouse model study showed that median survival was higher in the group that received combination treatment with MN-166 (ibudilast) plus temozolomide (TMZ) compared to the group that received TMZ alone.
MN-221 (bedoradrine)
MN-221 (bedoradrine) is a novel, highly selective ß2-adrenergic receptor agonist which has been developed for the treatment of acute exacerbations of asthma. We licensed MN-221 from Kissei Pharmaceutical Co., Ltd. (Kissei) in February 2004. Current inhaled beta-agonist treatments for asthma exacerbations are limited by bronchoconstriction or insufficient airflow due to inflammation and airway constriction, which reduces the amount of inhaled drug that can get into the lungs. In addition, the amount of inhaled treatments a patient can tolerate is limited due to the potential for cardiovascular side effects (e.g. increased heart rate).
MN-221 is designed to treat acute exacerbations of asthma via intravenous (i.v.) infusion, bypassing constricted airways to deliver the drug to the lungs. Preclinical studies showed MN-221 to have a high affinity for the ß2-adrenergic receptor, found primarily in the lungs, and a much lower affinity for the ß1-adrenergic receptor found primarily in cardiac tissue. MN-221’s improved delivery to the lungs and its cardiac safety profile may help fill an unmet need for patients with acute exacerbations of asthma, helping them to breathe easier and avoid a costly hospital stay.
Acute Exacerbation of Asthma: According to the most recent data available from the United States National Center for Health Statistics, there were 1.75 million emergency department visits, 439,000 hospitalizations, and
11
3,404 deaths due to asthma in 2010. According to the United States National Heart, Lung and Blood Institute, the direct costs associated with hospital care due to asthma were estimated at $5.5 billion in the United States in 2010.
We completed a Phase 2b randomized, double-blind, placebo-controlled clinical trial (N=175) evaluating MN-221 in patients with acute exacerbations of asthma in the emergency department setting. MN-221 did not statistically meet the primary endpoint, improvement in FEV1 (Forced Expiratory Volume in One Second) compared to placebo. However, MN-221 treatment demonstrated statistically significant improvements in endpoints associated with Dyspnea Index scores. MN-221 treatment significantly increased (improved) the change from baseline in Dyspnea Index scale score over Hours 0-3 compared to placebo (based on AUC [0-3 hr], p = 0.0405), significantly increased the change from baseline in Dyspnea Index scale scores at Hour 2 compared to placebo (based on mean score, p = 0.0042), and significantly increased the percentage of subjects who had improvement in the Dyspnea Index score ≥ 1 point at Hour 2 compared to placebo (p = 0.0323). A post-hoc analysis was performed to evaluate the Treatment Failure rate defined as the number of subjects who were either hospitalized or who returned to the emergency department during the course of the study. In subjects who received corticosteroids greater than 3 hours prior to study drug infusion, the number of treatment failures was significantly greater in the placebo group (74%) versus the MN-221 group (43%), p=0.0489. No safety/tolerability issues of clinical significance were observed.
In October 2012, we met with the FDA to review future development of this product candidate. The FDA identified the risk/benefit profile of MN-221 as a focal point for further development and advised that a clinical outcome, such as a reduction in hospitalizations, would need to be a pivotal trial primary endpoint. We have decided that any future MN-221 development will be designed based on the feedback received from the FDA and that any future MN-221 clinical trial development for asthma will be partner-dependent from a funding perspective.
MN-001 (tipelukast)
MN-001 (tipelukast) is a novel, orally bioavailable small molecule compound which exerts its effects through several mechanisms to produce its anti-fibrotic and anti-inflammatory activity in preclinical models, including leukotriene (LT) receptor antagonism, inhibition of PDEs (mainly 3 and 4), and inhibition of 5-lipoxygenase (5-LO). The 5-LO/LT pathway has been postulated as a pathogenic factor in fibrosis development and MN-001 (tipelukast)’s inhibitory effect on 5-LO and the 5-LO/LT pathway is considered to be a novel approach to treat fibrosis. MN-001 (tipelukast) has been shown to down-regulate expression of genes that promote fibrosis including LOXL2, Collagen Type 1 and TIMP-1. MN-001 (tipelukast) has also been shown to down-regulate expression of genes that promote inflammation including CCR2 and MCP-1. In addition, histopathological data shows that MN-001 (tipelukast) reduces fibrosis in multiple animal models. We licensed MN-001 (tipelukast) from Kyorin in 2002. In addition to granting MN-001 (tipelukast) Fast Track designation for the treatment of NASH with fibrosis, the FDA has also granted MN-001 (tipelukast) Orphan-Drug designation and Fast Track designation for treatment of idiopathic pulmonary fibrosis (IPF).
Previously, we evaluated MN-001 (tipelukast) for its potential clinical efficacy in asthma and completed a Phase 2 study in asthma with positive results. MN-001 (tipelukast) has been exposed to more than 600 subjects and is considered generally safe and well-tolerated.
Nonalcoholic Steatohepatitis (NASH) and Nonalcoholic Fatty Liver Disease (NAFLD): Nonalcoholic steatohepatitis (NASH) is a condition in which there is fat in the liver along with inflammation and damage to liver cells. NASH is a common liver disease that resembles alcoholic liver disease but occurs in people who drink little or no alcohol. According to the United States National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NASH prevalence in adults in the United States is 3-12%, and an additional 30-40% of adult Americans have nonalcoholic fatty liver disease (NAFLD). The underlying cause of NASH is unclear, but it most often occurs in persons who are middle-aged and overweight or obese. Many patients with NASH have elevated serum lipids, diabetes or pre-diabetes. Progression of NASH can lead to liver cirrhosis. Liver transplantation is the only treatment for advanced cirrhosis with liver failure. At this time, there is no treatment for NASH.
We completed a pre-clinical study evaluating MN-001 (tipelukast)’s potential clinical efficacy for the treatment of NASH. MN-001 (tipelukast) administered orally once daily (10, 30, and 100 mg/kg for three weeks) was evaluated in the STAM™ (NASH-HCC) mouse model of NASH, as measured by liver biochemistry and histopathology, NAFLD activity score, and percent of fibrosis and gene expression. MN-001 (tipelukast), in a dose-
12
dependent manner, significantly reduced fibrosis area compared with placebo (p<0.01) as demonstrated by a reduction in liver hydroxyproline content, supporting MN-001 (tipelukast)’s anti-fibrotic properties. MN-001 (tipelukast) significantly improved NAS (p<0.01). MN-001 (tipelukast), in this animal model, improved NASH pathology by inhibiting hepatocyte damage (p<0.01) and ballooning (p<0.01). At the same time, MN-001 (tipelukast) was also shown to reduce certain gene expression levels in the liver, thus implying that MN-001 (tipelukast) reduces the formation of fibrosis in the NASH model. We completed a second preclinical study that examined the potential clinical efficacy of MN-001 (tipelukast) for the treatment of advanced NASH. This study used mice in more advanced stages of NASH as compared to the first study of MN-001 (tipelukast) in a NASH mouse model. MN-001 (tipelukast) showed anti-NASH and anti-fibrotic effects in the advanced NASH mouse model. NAFLD activity score (NAS) was significantly reduced in the MN-001 (tipelukast)-treated group compared to the non-treated group (p<0.001). The reduction was observed consistently in all NAS components including hepatocyte ballooning score (p<0.001), lobular inflammation score (p<0.01), and steatosis score (p<0.05). Percent fibrosis area was also reduced in the MN-001 (tipelukast) treated group (p<0.01). In addition, alpha-SMA-positive staining area was significantly reduced in the MN-001 (tipelukast)-treated group (p<0.001). Collectively, these results provide compelling evidence that MN-001 (tipelukast) warrants further evaluation for the treatment of NASH in humans. We have an open IND and the FDA has approved two different Phase 2 clinical trial protocols for MN-001 (tipelukast) for the treatment of NASH in the United States. A Phase 2 clinical trial is currently ongoing to investigate MN-001 (tipelukast) for the treatment of hypertriglyceridemia in NASH patients and NAFLD patients. The FDA has granted Fast Track designation to MN-001 (tipelukast) for the treatment of patients with NASH with fibrosis.
Idiopathic Pulmonary Fibrosis (IPF): Pulmonary fibrosis (PF) is a progressive disease characterized by scarring of the lungs that thickens the lining, causing an irreversible loss of the tissue's ability to transport oxygen. The causes of PF vary and can be due to anti-cancer drug therapy or exposure to chemicals. Idiopathic pulmonary fibrosis (IPF) is one type of PF without a clear cause. According to the Pulmonary Fibrosis Foundation, IPF affects between 132,000 – 200,000 people in the United States, and an estimated 50,000 new cases are diagnosed annually. The prognosis for IPF is poor with a median survival of only two to three years following diagnosis and more than two-thirds of IPF patients die within five years.
We completed a pre-clinical study evaluating MN-001 (tipelukast)’s potential clinical efficacy for the treatment of pulmonary fibrosis. MN-001 (tipelukast), which was administered orally once daily (30, 100 and 300 mg/kg) for two weeks, was evaluated in a mouse model of bleomycin-induced pulmonary fibrosis (PF) as measured by CT evaluation of lung density, degree of pulmonary fibrosis using the Ashcroft score based on histopathological staining, and hydroxyproline content, which is an indicator of fibrosis or storage of collagen in tissue. MN-001 (tipelukast) significantly decreased the Ashcroft score compared to Vehicle group (p<0.05) after two weeks of treatment and MN-001 (tipelukast) reduced lung density when compared to the Vehicle-treated group. Moreover, lung hydroxyproline content was significantly reduced compared to the Vehicle group (p<0.01). These results show that treatment with MN-001 (tipelukast) has significant anti-fibrogenic effects in bleomycin-induced pulmonary fibrosis in mice. We have an open IND and the FDA approved a Phase 2 clinical trial protocol for MN-001 (tipelukast) for the treatment of moderate to severe IPF in the United States. A Phase 2 clinical trial of MN-001 (tipelukast) to treat moderate to severe IPF is currently ongoing at Penn State. The FDA has granted Orphan-Drug designation to MN-001 (tipelukast) for treatment of IPF. Orphan-Drug designation will provide seven years of marketing exclusivity for MN-001 (tipelukast) for the treatment of IPF if it is approved for this indication. The FDA has also granted Fast Track designation to MN-001 (tipelukast) for the treatment of patients with IPF.
MN-029 (denibulin)
MN-029 (denibulin) is a novel tubulin binding agent (TBA) under development for the treatment of solid tumors. It exerts its activity through reversible inhibition of tubulin polymerization resulting in disruption of the cell cytoskeleton, which causes the cancer cells to deform in shape and ultimately leads to extensive central necrosis of the solid tumor. We licensed MN-029 from Angiogene Pharmaceuticals, Ltd. (Angiogene) in 2002.
Several preclinical pharmacology studies have assessed the mechanism of action and anti-tumor activity of MN-029 in vivo in rodent models of breast adenocarcinoma, colon carcinoma, lung carcinoma and KHT sarcoma. In these studies, MN-029 damaged poorly formed tumor blood vessels by weakening tumor blood vessel walls and causing leakage, clotting and eventual vascular shutdown within the tumor, in addition to the direct effect over
13
tumor cells. These studies suggest that MN-029 acts quickly and is rapidly cleared from the body, which may reduce the potential for some adverse effects commonly associated with chemotherapy. Shutdown of tumor blood flow in tumor models was confirmed through the use of dynamic contrast-enhanced magnetic resonance imaging. In two Phase I clinical studies we conducted, MN-029 was well-tolerated at doses that reduced tumor blood flow.
The first Phase 1 trial determined the safety, tolerability, and maximum tolerated dose (MTD) level of single doses of MN-029 given every three weeks in 34 subjects with refractory cancer. The MTD was determined to be 180 mg/m2 and appeared to be safe as a single i.v. dose administered every three weeks for as many as 25 cycles. There were no clinically significant changes in routine laboratory assessments, vital signs, or ECG monitoring. The most commonly reported adverse events (AEs) were similar to other chemotherapies—vomiting, nausea, diarrhea, and fatigue. There were a total of nine serious adverse events (SAEs) and study discontinuations due to AEs. In a preliminary evaluation of anti-tumor activity, no patient had a complete response or partial response; however stable disease was seen in 12 patients. MN-029 had a desired vascular effect in seven of 11 patients that were administered drug at dose levels of ≥120 mg/m2. Nine patients continued into extended cycles of treatment.
The second Phase 1 study was conducted to determine the safety, tolerability and MTD of single doses of MN-029 given every seven days for a total of three doses (Days 1, 8 and 15), followed by 13-day recovery (Days 16-28) in subjects with advanced/metastatic solid tumor cancer. Subjects who tolerated treatment with MN-029 could receive additional cycles. All 20 subjects reported at least one AE related to study drug. The most common AEs considered related to study drug were vomiting, nausea, arthralgia and headache. There were no clinically significant changes in routine laboratory assessments, vital signs or ECG monitoring. There was one SAE considered unrelated to study drug. Consistent with the previous Phase 1 trial, MN-029 up to dose levels of 180 mg/m2 appeared to be safe and well tolerated. One subject had a partial response which lasted for 74 days. Stable disease was observed in seven subjects. The results suggested an effect of MN-029 on vascular perfusion; however, a larger sample size is warranted.
In January 2014, we were granted a new patent from the United States Patent and Trademark Office which covers MN-029 (denibulin) di-hydrochloride. The patent, which will expire no earlier than July 2032, has claims that cover a compound, pharmaceutical composition and method of treating certain cell proliferation diseases, including solid tumors, based on denibulin di-hydrochloride. We have filed patent applications based on this U.S. patent in certain foreign countries, and most of them have been granted. We plan to pursue further development of MN-029 for the treatment of solid tumors.
Table 1 Product Candidates and Programs—MN-166 (ibudilast)
|
Indication |
|
Clinical Study |
|
Principal Investigator /Institution /Funding Agency(s) |
|
Status |
|
|
|
|
|
|
|
|
|
Primary Progressive and Secondary Progressive Multiple Sclerosis |
|
A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability and Activity of Ibudilast (MN-166) in Subjects with Progressive Multiple Sclerosis |
|
Robert J. Fox, M.D., M.S., FAAN Cleveland Clinic National Institute on Neurological Diseases and Stroke MediciNova, Inc. |
|
Completed |
|
|
|
|
|
|
|
|
14
We currently have no marketing and sales capabilities and we expect to rely on strategic partners to commercialize our products.
Manufacturing
We rely on third parties to manufacture bulk active pharmaceutical ingredients (API) and finished investigational products for research, development, preclinical and clinical trials. We expect to continue to rely on third-party manufacturers for the manufacture of the API and finished products for our clinical and any future commercial production requirements. We believe that there are several manufacturing sources available at commercially reasonable terms to meet our clinical requirements and any future commercial production requirements for the API of our products and the finished drug products.
For the MN-166 (ibudilast) development program, we have sourced and imported delayed-release ibudilast capsules, marketed in Japan as Pinatos®, from Taisho Pharmaceutical Co., Ltd. (Taisho).
Pursuant to the terms of our license agreement with Kissei for MN-221, Kissei has the exclusive right to manufacture the commercial supply of the API for MN-221. If we enter into a supply agreement with Kissei, we will purchase from Kissei all API that we require for the commercial supply of MN-221, if this product candidate is approved for commercial sale by the FDA or other regulatory authorities.
Intellectual Property and License Agreements
Since our inception in September 2000, we have entered into license agreements with pharmaceutical companies which cover our current product candidates. We have also entered into license agreements with universities which cover additional intellectual property related to our product candidates. In general, we seek to procure patent protection for our anticipated products, or obtain such protection from the relevant patents owned by our licensors. We hold licensed rights to one issued U.S. patent and 19 issued foreign patents. In addition to these licensed rights, we hold 24 issued U.S. patents and have filed 6 additional U.S. patent applications. We also hold 36
15
issued foreign patents and 44 pending foreign patent applications corresponding to these U.S. patents and patent applications. We are not aware of any third-party infringement of the patents owned or licensed by us and are not party to any material claims by third parties of infringement by us of such third parties’ intellectual property rights. The following is a description of our existing license agreements and intellectual property rights for each of our product candidates.
MN-166 (ibudilast)
On October 22, 2004, we entered into an exclusive license agreement with Kyorin for the development and commercialization of MN-166 (ibudilast). Kyorin is a fully integrated Japanese pharmaceutical company and is listed on the First Section of the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan, China, South Korea and Taiwan), sub-licensable license to the patent rights and know-how related to MN-166 (ibudilast) for the treatment of MS, except for ophthalmic solution formulations. MN-166 (ibudilast) is not covered by a composition of matter patent. The United States method of use patent for MN-166 (ibudilast) in MS underlying the license is set to expire on August 10, 2018. Corresponding method of use patents in certain foreign countries are set to expire on August 10, 2018. Under the terms of the agreement, we granted to Kyorin an exclusive, royalty-free, sub-licensable license to use the preclinical, clinical and regulatory databases to develop ophthalmic products incorporating the MN-166 (ibudilast) compound anywhere in the world and non-ophthalmic products incorporating the MN-166 (ibudilast) compound outside of our territory.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party. We may terminate the agreement for any reason with 90 days’ written notice to Kyorin or, in the event that a third party claims that MN-166 (ibudilast) infringes upon such third party’s intellectual property rights, with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of the expiration of the obligation to make payments under the agreement or the last date on which the manufacture, use or sale of the product would infringe a valid patent claim held by Kyorin but for the license granted by the agreement or the last date of the applicable market exclusivity period. In the absence of a valid patent claim and generic competition in a particular country, the agreement will expire on the earlier of five years from the date of the first commercial sale of the product by us or the end of the second consecutive calendar quarter in which generic competition exists in such country.
Under the license agreement, we have paid Kyorin $700,000 to date, and we are obligated to make payments of up to $5.0 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
We own, co-own or hold licenses to seven issued U.S. patents and five pending U.S. patent applications as well as 22 issued foreign patents and seven pending foreign patent applications covering MN-166 (ibudilast) and its analogs. These patents and patent applications are related to our development portfolio and are primarily directed to methods of treating various indications using MN-166 (ibudilast) and its analogs.
We have been granted a U.S. patent which covers the use of MN-166 (ibudilast) for the treatment of progressive forms of MS. This patent will expire no earlier than November 2029, not including a potential extension under patent term restoration rules, and covers a method of treating PPMS or SPMS by administering MN-166 (ibudilast). Counterparts of this patent application have been granted in certain foreign jurisdictions. We have been granted a U.S. patent which covers the use of MN-166 (ibudilast) for the treatment of amyotrophic lateral sclerosis (ALS) and it expires no earlier than January 2029. We have been granted a patent which covers the use of MN-166 (ibudilast) for the treatment of drug addiction or drug dependence or withdrawal syndrome in the United States and it expires no earlier than January 2030. Counterparts of this patent application have been granted in certain foreign jurisdictions. We have been granted a patent which covers the use of MN-166 (ibudilast) for the treatment of neuropathic pain in the United States and it expires no earlier than December 2025.
MN-221 (bedoradrine)
16
On February 25, 2004, we entered into an exclusive license agreement with Kissei for the development and commercialization of MN-221. Kissei is a fully integrated Japanese pharmaceutical company and is listed on the Tokyo Stock Exchange. We obtained an exclusive, worldwide (excluding Japan), sub-licensable license to various patent rights and know-how related to MN-221 and other compounds disclosed or included in, or covered by, these patent rights, for all indications. This license includes an exclusive license under one U.S. patent and certain corresponding patents in foreign countries and is sub-licensable upon receipt of the written consent of Kissei. The United States composition of matter patent underlying the license issued on October 17, 2000 and it expired on February 18, 2017. Most of the corresponding composition of matter patents in various other countries also expired on February 18, 2017.
In addition to the licensed patents, we have filed patent applications in the United States and certain foreign countries regarding additional uses and formulations of MN-221. We have been granted a U.S. patent which covers the use of MN-221 for the treatment of acute exacerbations of asthma and it expires no earlier than November 2030. This patent includes claims covering the use of MN-221 (bedoradrine) in combination with a standard of care treatment regimen and covers different routes of administration, including intravenous, oral and inhalation. We have been granted a U.S. patent that covers the use of MN-221 for the treatment of irritable bowel syndrome and it expires no earlier than April 2031.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party, and we may terminate the agreement for scientific or commercial reasons upon 100 days’ prior written notice to Kissei during the development phase and 180 days’ prior written notice to Kissei during the commercialization phase.
The term of the agreement is determined on a country-by-country basis and extends until the expiration of the last Kissei patent (or equivalent) under license to expire or in the event that a valid claim does not exist or, if a valid claim expired more than ten years from the date of first commercial sale, ten years from the date of first commercial sale. In either case, the term of the agreement would not extend for any particular country past the date on which generic competition exists in such country.
Under the license agreement, we have paid Kissei $1.0 million to date, and we are obligated to make payments of up to $17.0 million based on the achievement of certain clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products. Under the terms of a letter agreement we entered into with Kissei in September 2011, we agreed to renegotiate in good faith with Kissei the existing levels of the milestone payment amounts and royalty rates.
MN-001 (tipelukast)
On March 14, 2002, we entered into an exclusive license agreement with Kyorin for the development and commercialization of MN-001 (tipelukast). We obtained an exclusive, worldwide (excluding Japan, China, South Korea and Taiwan) sub-licensable license to the patent rights and know-how related to MN-001 (tipelukast) and its active metabolite, MN-002, disclosed and included in, or covered by, these patents, in all indications, except for ophthalmic solution formulations. This license includes an exclusive, sub-licensable license under two U.S. patents and certain corresponding patents in foreign countries. The United States composition of matter patent for MN-001 (tipelukast) underlying the license expired on February 23, 2009, and the United States composition of matter patent for MN-002 underlying the license expired on December 30, 2011. Foreign composition of matter patents for MN-001 (tipelukast) and MN-002 have also expired. We have been granted 14 U.S. patents covering certain compositions, uses and manufacturing processes associated with MN-001 (tipelukast) and MN-002. Uses covered by these patents include nonalcoholic steatohepatitis (NASH), advanced NASH with fibrosis, nonalcoholic fatty liver disease (NAFLD), steatosis, hypertriglyceridemia, hypercholesterolemia, hyperlipoproteinemia, fibrosis, ulcerative colitis, interstitial cystitis, and irritable bowel syndrome. Patent applications corresponding to these U.S. patents have been filed in certain foreign countries and some of the foreign patents have issued.
Under the terms of the agreement, we granted to Kyorin an exclusive, royalty-free, sub-licensable license to use the preclinical, clinical and regulatory databases to develop ophthalmic products incorporating MN-001 (tipelukast) anywhere in the world and non-ophthalmic products incorporating MN-001 (tipelukast) outside of our territory. The license agreement may be terminated by either party following an uncured breach of any material
17
provision in the agreement by the other party, and we may terminate the agreement for any reason with 90 days’ written notice to Kyorin or, in the event that a third party claims that the licensed patent rights or know-how infringe upon such third party’s intellectual property rights, with 30 days’ written notice.
The term of this agreement is determined on a country-by-country basis and extends until the later of the expiration of the obligation to make payments under the agreement or the last date on which the manufacture, use or sale of the product would infringe a valid patent claim held by Kyorin but for the license granted by the agreement or the last date of the applicable market exclusivity period. In the absence of a valid patent claim and generic competition in a particular country, the agreement will expire on the earlier of five years from the date of the first commercial sale of the product by us or the end of the second consecutive calendar quarter in which generic competition exists in such country.
Under the license agreement, we have paid Kyorin $4.0 million to date, and we are obligated to make payments of up to $5.0 million based on the achievement of clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
On June 19, 2002, we entered into an exclusive license agreement with Angiogene for the development and commercialization of the ANG-600 series of compounds. Angiogene is a privately held, British drug discovery company. We obtained an exclusive, worldwide, sub-licensable license to the patent rights and know-how related to the ANG-600 series of compounds disclosed in and included or covered by these patents for all indications. MN-029 is one of the ANG-600 series compounds covered by this license. We have been granted a U.S. patent which covers MN-029 (denibulin) di-hydrochloride and expires no earlier than July 2032. The allowed claims cover a compound, pharmaceutical composition and method of treating certain cell proliferation diseases, including solid tumors, based on denibulin di-hydrochloride. Patent applications corresponding to this U.S. patent were filed in certain foreign countries and patents have been granted or allowed in some of those countries.
The license agreement may be terminated by either party following an uncured breach of any material provision in the agreement by the other party, and we may terminate the agreement at any time by giving 30 days’ advance written notice to Angiogene.
The term of this agreement is determined on a country-by-country basis and extends until the earlier of the expiration of the last Angiogene patent (or equivalent) under license which has a valid claim to expire or 15 years from the date of first commercial sale.
Under the license agreement, we have paid Angiogene $1.4 million to date and are obligated to make payments of up to $16.5 million based on the achievement of clinical and regulatory milestones. We are also obligated to pay a royalty on net sales of the licensed products.
General
Our proposed commercial activities may conflict with patents which have been or may be granted to competitors, universities and/or others. Third parties could bring legal action against us, our licensors or our sub-licensees claiming patent infringement and could seek damages or enjoin manufacturing and marketing of the affected product or its use or the use of a process for the manufacturing of such products. If any such actions were to be successful, in addition to any potential liability for indemnification, damages and attorneys’ fees in certain cases, we could be required to obtain a license, which may not be available on commercially reasonable terms or at all, in order to continue to manufacture, use or market the affected product. We also rely upon unpatented proprietary technology because, in some cases, our interests would be better served by reliance on trade secrets or confidentiality agreements than by patents. However, others may independently develop substantially equivalent proprietary information and techniques or gain access to or disclose such proprietary technology. We may not be able to meaningfully protect our rights in such unpatented proprietary technology. We may also conduct research on other pharmaceutical compounds or technologies, the rights to which may be held by, or be subject to patent rights of, third parties. Accordingly, if products based on such research are commercialized, such commercial activities may infringe patents or other rights, which may require us to obtain a license to such patents or other rights. We are
18
not aware of any third-party infringements of patents we hold or have licensed and have not received any material claims by third parties of infringement by us of such parties’ intellectual property rights.
There can be no assurance that patent applications filed by us or others, in which we have an interest as assignee, licensee or prospective licensee, will result in patents being issued or that, if issued, any of such patents will afford protection against competitors with similar technology or products or could not be circumvented or challenged. For example, we have U.S. patents covering the method of treating progressive MS with MN-166 (ibudilast), the method of treating ALS with MN-166 (ibudilast), the method of treating drug addiction or drug dependence with MN-166 (ibudilast), and the method of treating neuropathic pain with MN-166 (ibudilast), but we do not have any composition of matter patent claims for MN-166 (ibudilast) because that patent has expired. As a result, unrelated third parties may develop products with the same API as MN-166 (ibudilast) so long as such parties do not infringe our method of use patents, other patents we have exclusive rights to through our licensors or any patents we may obtain for MN-166 (ibudilast).
In addition, if we develop certain products that are not covered by any patents, we will be dependent on obtaining market exclusivity under the new chemical entity exclusivity provisions of Hatch-Waxman Act for such products in the United States and/or data exclusivity provisions in Europe. If we are unable to obtain strong proprietary protection for our products after obtaining regulatory approval, competitors may be able to market competing generic products by taking advantage of an abbreviated procedure for obtaining regulatory clearance, including the ability to demonstrate bioequivalency to our product(s) without being required to conduct lengthy clinical trials. Certain of our license agreements provide for reduced or foregone royalties in the event of generic competition.
Competition
The development and commercialization of new drugs is extremely competitive and characterized by extensive research efforts and rapid technological progress. Competition in our industry occurs on a variety of fronts, including developing and bringing new products to market before others, developing new products to provide the same benefits as existing products at lower cost and developing new products to provide benefits superior to those of existing products. We face competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies in the United States and abroad. Some of these competitors have products or are pursuing the development of drugs that target the same diseases and conditions that are the focus of our product development programs. Many of our competitors have products that have been approved or are in advanced development and may succeed in developing drugs that are more effective, safer and more affordable or more easily administered than ours or that achieve patent protection or commercialization sooner than our products. Our competitors may also develop alternative therapies that could further limit the market for any products that we are able to obtain approval for, if at all.
In many of our target disease areas, potential competitors are working to develop new compounds with different mechanisms of action and attractive efficacy and safety profiles. Many of our competitors have substantially greater financial, research and development resources (including personnel and technology), clinical trial experience, manufacturing, sales and marketing capabilities and production facilities than we do. Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaboration arrangements with large pharmaceutical and established biotechnology companies.
MN-166 (ibudilast) for Progressive Multiple Sclerosis (Progressive MS)
Our MN-166 (ibudilast) product candidate is in development for the treatment of progressive MS. Only one drug, mitoxantrone, is approved for the treatment of secondary progressive MS. However, mitoxantrone cannot be used on a long-term basis because of the potential for cardiac toxicity. Only one drug, Ocrevus (ocrelizumab) is approved for the treatment of primary progressive MS. Other programs in clinical development for progressive MS include Novartis’s BAF312 (siponimod), Teva’s Nerventra (laquinimod), and AB Science’s masitinib.
MN-166 (ibudilast) for Amyotrophic Lateral Sclerosis (ALS)
19
Our MN-166 (ibudilast) product candidate is also in development for the treatment of ALS. Riluzole and Radicava (edaravone) are approved for the treatment of ALS. We are aware of additional compounds in clinical development for the treatment of ALS at other companies including Cytokinetics, BrainStorm Cell Therapeutics Inc., AB Science, Mallinckrodt, Biogen, Neuraltus Pharmaceuticals, and Amylyx Pharmaceuticals.
MN-166 (ibudilast) for Substance Dependence and Addiction
Our MN-166 (ibudilast) product candidate is also in development for treatment of opioid dependence, methamphetamine addiction, and alcohol dependence. Current treatments for opioid withdrawal symptoms include narcotics such as generic methadone and Indivior, Inc.’s Suboxone® Film (buprenorphine + the opioid antagonist naloxone). Other products approved for opioid dependence include Alkermes’ Vivitrol® (naltrexone monthly injection), Orexo’s Zubsolv® (buprenorphine and naloxone), BioDelivery Sciences’s Bunavail® (buprenorphine and naloxone), Braeburn Pharmaceuticals Inc.’s Probuphine (buprenorphine) implant, and Indivior’s Sublocade (buprenorphine extended-release injection). Braeburn Pharmaceuticals is developing an injectable buprenorphine product for the treatment of opioid dependence. Limited non-narcotic drug candidates for opioid withdrawal symptoms exist. Britannia Pharmaceuticals Limited’s BritLofex® (Lofexidine), an a2 adrenergic receptor agonist like clonidine which may have somewhat less orthostatic hypotension limitations, was licensed to US WorldMeds LLC for development in the United States for opioid withdrawal symptoms. There are no pharmaceuticals currently approved for the treatment of methamphetamine addiction. Current treatments for alcohol dependence include Antabuse® (disulfiram), Vivitrol® (naltrexone), and generic acamprosate. We are aware of additional compounds in development for the treatment of alcohol dependence at other companies including Indivior.
MN-221 (bedoradrine) for Acute Exacerbations of Asthma
Our MN-221 product candidate has been developed for the treatment of acute exacerbations of asthma in the emergency room setting. The current standard of care for acute exacerbations of asthma is inhaled albuterol (a ß 2 -adrenergic receptor agonist), inhaled ipratropium (an anticholinergic) and oral or injected corticosteroids. In addition, subcutaneously administered terbutaline (a ß 2 -adrenergic receptor agonist) is sometimes used to treat this condition, particularly in pediatric patients.
MN-001 (tipelukast) for Nonalcoholic Steatohepatitis (NASH)
Our MN-001 (tipelukast) product candidate is being developed for the treatment of NASH. There are currently no therapeutic products approved for the treatment of NASH. We are aware of compounds in clinical development for the treatment of NASH at other companies including Intercept Pharmaceuticals, Genfit, Galectin Therapeutics, Gilead Sciences, Allergan (which acquired Tobira Therapeutics), Galmed Pharmaceuticals, Bristol-Myers Squibb, Shire and Conatus Pharmaceuticals.
MN-001 (tipelukast) for Idiopathic Pulmonary Fibrosis (IPF)
Our MN-001 (tipelukast) product candidate is also being developed for the treatment of IPF. Products approved in the United States for treatment of IPF include Roche’s (formerly InterMune) Esbriet® (pirfenidone) and Boehringer Ingelheim’s OFEV® (nintedanib). Companies working on clinical development programs for treatment of IPF include Roche, Biogen and FibroGen.
MN-029 (denibulin) for Solid Tumor Cancer
Our MN-029 product candidate is being developed for the treatment of solid tumor cancers. Roche’s Kadcyla®, a HER2-targeted antibody and microtubule inhibitor conjugate, is approved for treatment of patients with HER2-positive metastatic breast cancer who previously were treated with trastuzumab and/or a taxane. Bayer’s Stivarga®, a kinase inhibitor approved for metastatic colorectal cancer, was also approved for patients with advanced, unresectable (not subject to surgical removal) or metastatic gastrointestinal stromal tumor. Other drugs approved for solid tumor cancers include Roche’s Avastin and Xeloda, Amgen’s Xgeva, Pfizer’s Sutent, and Novartis’s Afinitor. We are aware of additional compounds in development for the treatment of solid tumor cancers at companies including Eli Lilly, Roche, Novartis, Pfizer, Amgen and Celgene.
20
Government authorities in the United States and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing and import and export of pharmaceutical products and biologics such as those we are developing. In the United States, the FDA, under the Federal Food, Drug and Cosmetic Act, as amended, and other federal statutes and regulations, subjects pharmaceutical products to extensive and rigorous review. Any failure to comply with applicable requirements, both before and after approval, may subject us, our third-party manufacturers, contractors, suppliers and partners to administrative and judicial sanctions, such as a delay in approving or refusal to approve pending applications, fines, warning letters, product recalls, product seizures, total or partial suspension of manufacturing or marketing, injunctions and/or criminal prosecution.
United States Regulatory Approval
Overview. In the United States, drugs and drug testing are regulated by the FDA under the Federal Food, Drug and Cosmetic Act, or FDCA, as well as state and local government authorities. All of our product candidates in development will require regulatory approval by government agencies prior to commercialization. To obtain approval of a new product from the FDA, we must, among other requirements, submit data supporting safety and efficacy, as well as detailed information on the manufacture and composition of the product and proposed labeling. Our product candidates are in the early stages of testing and none has been approved. The steps required before a drug can be approved generally involve the following:
|
|
• |
completion of nonclinical laboratory, animal studies, and formulation studies; |
|
|
• |
submission of an IND which must become effective before human clinical trials may begin in the United States; |
|
|
• |
completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each indication for which approval is sought; |
|
|
• |
submission to the FDA of a New Drug Application (NDA) accompanied by a substantial user fee; |
|
|
• |
development of manufacturing processes which conform to FDA-mandated commercial good manufacturing practices (cGMPs) and satisfactory completion of FDA inspections to assess cGMP compliance and clinical investigator compliance with good clinical practices; and |
|
|
• |
FDA review and approval of an NDA, which process may involve input from advisory committees to the FDA and may include post-approval commitments for further clinical studies and distribution restrictions intended to mitigate drug risks. |
The testing, collection of data, preparation of necessary applications and approval process requires substantial time, effort and financial resources. Additionally, statutes, rules, regulations and policies may change and new regulations may be issued that could delay approvals of our drugs. The FDA may not act quickly or favorably in reviewing our applications, and we may encounter significant difficulties and costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing our product candidates.
Preclinical Tests. Preclinical tests include laboratory evaluation of the product candidate, its chemistry, toxicity, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the product candidate. The results of the preclinical tests, together with manufacturing information, analytical data and other available information about the product candidate, are submitted to the FDA as part of an IND. Preclinical tests and studies can take several years to complete and, despite completion of those tests and studies, the FDA may not permit clinical testing to begin.
The IND Process. An IND must be effective to administer an investigational drug to humans. The IND will automatically become effective 30 days after its receipt by the FDA unless the FDA, before that time, places the IND on clinical hold. At any time thereafter, the FDA may raise concerns or questions about the conduct of the trials
21
as outlined in the IND and impose a clinical hold if the FDA deems it appropriate. In such case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin or continue. The IND application process may become extremely costly and substantially delay development of our product candidates. Moreover, positive results in preclinical tests or prior human studies do not necessarily predict positive results in subsequent clinical trials.
Annual progress reports detailing the results of the clinical trials must be submitted to the FDA and written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events or any findings from tests in laboratory animals that suggest a significant risk for human subjects.
Clinical Trials. Human clinical trials are typically conducted in three sequential phases that may overlap:
|
|
• |
Phase 1: The drug candidate is initially introduced into a small number of healthy human subjects or patients and tested for safety, dosage tolerance, absorption, distribution, excretion and metabolism. If the investigational product is considered inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in the target population. |
|
|
• |
Phase 2: The drug candidate is introduced into a limited patient population to assess the efficacy of the drug in specific, targeted indications, assess dosage tolerance and optimal dosage, and to identify possible adverse effects and safety risks. |
|
|
• |
Phase 3: The drug candidate is introduced into an expanded patient population at geographically dispersed clinical trial sites to further evaluate clinical efficacy and safety. The purpose of the Phase 3 trial is to conduct a risk/benefit analysis of the potential drug and provide an adequate basis for product labeling. It is common to have two adequate and well-controlled Phase 3 trials for the FDA to approve an NDA. |
Prior to initiation of each clinical trial, an independent Institutional Review Board (IRB) for each medical site proposing to conduct the clinical trials must review and approve the study protocol and study subjects must provide informed consent for participation in the study.
We cannot be certain that we will successfully complete Phase 1, 2 or 3 testing of our drug candidates within any specific time period, if at all. Clinical trials must be conducted in accordance with the FDA’s good clinical practices (GCP) requirements. The FDA may order the partial, temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The IRB may also require the clinical trial at that site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions. In addition, we may suspend or discontinue a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk.
During the development of a new drug, we may request to meet with the FDA at times such as prior to submitting an IND, at the End-of-Phase 2 meeting, and before an NDA is submitted, and meetings are not limited to these certain times. The purpose of the End-of-Phase 2 meeting is to discuss the Phase 2 clinical trial results and present plans for a pivotal Phase 3 trial that, in our opinion, will support the approval of the new drug. Additional animal safety studies, formulation studies and pharmacology studies are concurrently conducted with the ongoing clinical trials. Also, in compliance with cGMP requirements, the process for manufacturing commercial quantities of the new drug is finalized, with the expectation that the quality, purity, and potency of the drug will meet standards. A sponsor may also request a Special Protocol Assessment (SPA), the purpose of which is to reach agreement with the FDA on the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
Fast track designation: The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation
22
applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
Any product submitted to the FDA for marketing, including a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an NDA designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
United States patent term restoration and marketing exclusivity: Depending upon the timing, duration and specifics of the FDA approval of a drug candidate, some U.S. patents covering the product candidates may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent terms for one or more of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (ANDA) or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. Pediatric exclusivity is another type of regulatory market exclusivity in the United States
23
Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Regulation outside the United States: In addition to regulations in the United States, we and our strategic alliance partners will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a clinical trial application (CTA) must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug under European Union regulatory systems, we or our strategic alliance partners must submit a marketing authorization application. The application used to file the NDA in the United States is similar to that required in the European Union, with the exception of, among other things, country-specific document requirements.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we or our strategic alliance partners fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Employees
We have assembled an experienced and cohesive management and support team, with core competencies in general management, clinical development, regulatory affairs and corporate development. We have 9 full-time employees as of the date of this report. We believe that our relations with our employees are good, and we have no history of work stoppages.
Company Information
We were originally incorporated in the State of Delaware in September 2000. Our principal executive offices are located at 4275 Executive Square, Suite 300, La Jolla, CA 92037. Our telephone number is 858-373-1500. Our website is www.medicinova.com, which includes links to reports we have filed with the Securities and Exchange Commission, or SEC. The information contained in, or that can be accessed through, our website is not part of, and is not incorporated into, this Annual Report on Form 10-K.
24
We operate in a dynamic and rapidly changing environment that involves numerous risks and uncertainties. Certain factors may have a material adverse effect on our business, financial condition and results of operations, and you should carefully consider them. Accordingly, in evaluating our business, we encourage you to consider the following discussion of risk factors, in its entirety, in addition to other information contained in this Annual Report on Form 10-K and our other public filings with the SEC. Other events that we do not currently anticipate or that we currently deem immaterial may also affect our results of operations and financial condition.
Risks Related to Our Business and Industry
We have incurred significant operating losses since our inception and expect that we will incur continued losses for the foreseeable future.
We have incurred significant net losses since our inception. For the year ended December 31, 2017, we had a net loss of $11.2 million. At December 31, 2017, from inception, our accumulated deficit was $341.5 million. We expect to incur substantial net losses for the next several years as we continue to develop certain of our existing product development candidates, and over the long-term if we expand our research and development programs and acquire or in-license products, technologies or businesses that are complementary to our own. As of December 31, 2017, we had available cash and cash equivalents of $28.0 million and working capital of $25.4 million. There can be no assurances that there will be adequate financing available to us in the future on acceptable terms, or at all. If we are unable to obtain additional financing, we may have to out-license or sell one or more of our programs or cease operations.
Our future cash requirements will also depend on many factors, including:
|
|
• |
progress in, and the costs of future planned clinical trials and other research and development activities; |
|
|
• |
the scope, prioritization and number of our product development programs; |
|
|
• |
our obligations under our license agreements, pursuant to which we may be required to make future milestone payments upon the achievement of various milestones related to clinical, regulatory or commercial events; |
|
|
• |
our ability to establish and maintain strategic collaborations, including licensing agreements and other arrangements; |
|
|
• |
the time and costs involved in obtaining regulatory approvals; |
|
|
• |
the costs of securing manufacturing arrangements for clinical or commercial production of our product candidates; |
|
|
• |
the costs associated with any expansion of our management, personnel, systems and facilities; |
|
|
• |
the costs associated with any litigation; |
|
|
• |
the costs associated with the operations or wind-down of any business we may acquire; |
|
|
• |
the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; and |
|
|
• |
the costs of establishing or contracting for sales and marketing capabilities and commercialization activities if we obtain regulatory approval to market our product candidates. |
We expect our research and development expenses to increase in 2018 relative to 2017 as we continue our focus on the development of MN-166 (ibudilast) and MN-001 (tipelukast) in 2018. Our estimate of cash
25
requirements for future operating expenses assumes that we do not incur significant additional new clinical development expenditures unless we raise additional capital and/or enter into one or more strategic alliances. We do expect to continue to incur significant operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing drug products, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
If we have taxable income in the future, utilization of the net operating losses, or NOL, and tax credit carry-forwards will be subject to a substantial annual limitation under Sections 382 and 383 of the Internal Revenue Code of 1986, and similar state provisions due to ownership change limitations that have occurred. These ownership changes will limit the amount of NOL and tax credit carry-forwards that can be utilized to offset future taxable income and tax, respectively.
If we fail to obtain the capital necessary to fund our operations, we will be unable to develop and commercialize our product candidates.
We have consumed substantial amounts of capital since our inception.
Our business will continue to require us to incur substantial research and development expenses. We believe that without raising additional capital from accessible sources of financing, we will not otherwise have adequate funding to continue our operations and to complete the development of our existing product candidates or the commercialization of any products we successfully develop. There is no guarantee that adequate funds will be available when needed from debt or equity financings, arrangements with partners, or from other sources, on terms attractive to us. The inability to obtain sufficient additional funds when needed to fund our operations would require us to significantly delay, scale back, or eliminate some or all of our clinical or regulatory activities and reduce general and administrative expenses.
We do not have any products that are approved for commercial sale and therefore do not expect to generate any revenues from product sales in the foreseeable future, if ever.
To date, we have funded our operations primarily from sales of our securities and, to a lesser extent, debt financing. We do not expect to receive any revenues from the commercialization of our product candidates for at least the next several years, if at all. We anticipate that, prior to our commercialization of a product candidate, out-licensing upfront and milestone payments will be our primary source of revenue if we can enter into collaborations, strategic alliances or other agreements that would provide us with such revenues. To obtain revenues from sales of our product candidates, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing drugs with commercial potential. We may never succeed in these activities, and we may not generate sufficient revenues to continue our business operations or achieve and maintain profitability.
We are largely dependent on the success of our MN-166 (ibudilast) and MN-001 (tipelukast) product candidates and we cannot be certain that these product candidates will receive regulatory approval or be successfully commercialized.
We currently have no products for sale, and we cannot guarantee that we will ever have any drug products approved for sale. The research, testing, manufacturing, labeling, approval, sales, marketing and distribution of drug products are subject to extensive regulation by the FDA and comparable regulatory authorities in other countries. We are not permitted to market any of our product candidates in the United States until we submit and receive approval of a New Drug Application, or NDA, for a product candidate from the FDA or its foreign equivalent from a foreign regulatory authority. Obtaining FDA approval is a lengthy, expensive and uncertain process. The success of our business currently depends primarily on the successful development and commercialization of our MN-166 (ibudilast) and MN-001 (tipelukast) product candidates. These product candidates have not completed the clinical development process, and therefore we have not submitted an NDA or foreign equivalent or received marketing approval.
26
The clinical development program for our product candidates may not lead to commercial products for a number of reasons, including our clinical trials’ failure to demonstrate to the FDA’s satisfaction that this product candidate is safe and effective, or our failure to obtain necessary approvals from the FDA or similar foreign regulatory authorities for any reason. We may also fail to obtain the necessary approvals if we have inadequate financial or other resources to advance our product candidates through the clinical trial process or are unable to secure a strategic collaboration or partnership with a third party. Any failure or delay in completing clinical trials or obtaining regulatory approval for our product candidates in a timely manner would have a material and adverse impact on our business and our stock price.
Because the results of early clinical trials are not necessarily predictive of future results, our product candidates we advance into clinical trials in any indication may not have favorable results in later clinical trials, if any, or receive regulatory approval.
Our product candidates are subject to the risks of failure inherent in drug development. We will be required to demonstrate through well-controlled clinical trials that our product candidates are safe and effective for use in a diverse population for its target indications before we can seek regulatory approvals for their commercial sale. Success in early clinical trials does not mean that later clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety or efficacy despite having progressed through initial clinical testing, even at statistically significant levels.
Companies frequently suffer significant setbacks in advanced clinical trials, even after earlier clinical trials have shown promising results. Any of our planned clinical trials for our product candidates may not be successful for a variety of reasons, including the clinical trial designs, the failure to enroll a sufficient number of patients, undesirable side effects and other safety concerns and the inability to demonstrate sufficient efficacy. If a product candidate fails to demonstrate sufficient safety or efficacy, we would experience potentially significant delays in, or be required to abandon, development of such product candidate.
Our attempts to develop MN-001 (tipelukast) in NASH and IPF may detract from our efforts to develop other product candidates and may limit the effectiveness of our product development efforts as a whole.
We have decided to pursue development of MN-001 (tipelukast) in NASH and IPF. These activities will divert financial and management resources from our other product development activities and may limit our ability to complete or continue those other programs.
In order to commercialize a therapeutic drug successfully, a product candidate must receive regulatory approval after the successful completion of clinical trials, which are long, complex and costly, have a high risk of failure and can be delayed or terminated at any time.
Our product candidates are subject to extensive government regulations related to development, clinical trials, manufacturing and commercialization. The process of obtaining FDA and other regulatory approvals is costly, time-consuming, uncertain and subject to unanticipated delays. To receive regulatory approval for the commercial sale of any of our product candidates, we must conduct, at our own expense, adequate and well-controlled clinical trials in human patients to demonstrate the efficacy and safety of the product candidate. Clinical testing is expensive, takes many years and has an uncertain outcome. To date, we have obtained regulatory authorization to conduct clinical trials for our product development programs. INDs were approved by the FDA and are active for our product candidates.
It may take years to complete the clinical development necessary to commercialize a drug, and delays or failure can occur at any stage, which may result in our inability to market and sell any of our product candidates that are ultimately approved by the FDA or foreign regulatory authorities. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or non-clinical testing. Interim results of clinical trials do not necessarily predict final results, and success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials even after obtaining promising results in earlier clinical trials. In addition, any delays in completing clinical trials or the rejection of data
27
from a clinical trial by a regulatory authority will result in increased development costs and could have a material adverse effect on the development of the impacted product candidate.
In connection with the conduct of clinical trials for each of our product candidates, we face many risks, including the risks that:
|
|
• |
the product candidate may not prove to be effective in treating the targeted indication; |
|
|
• |
clinical trial participants and/or patients may experience serious adverse events or other undesirable drug-related side effects; |
|
|
• |
the results may not confirm the positive results of earlier trials; |
|
|
• |
the FDA or other regulatory authorities may not agree with our proposed development plans or accept the results of completed clinical trials; and |
|
|
• |
our planned clinical trials and the data collected from such clinical trials may be deemed by the FDA or other regulatory authorities not to be sufficient, which would require additional development for the product candidate before it can be evaluated in late stage clinical trials or before the FDA will consider an application for marketing approval. |
If we do not complete clinical development of our product candidates successfully, we will be unable to obtain regulatory approval to market products and generate revenues from such product candidates. We may also fail to obtain the necessary regulatory approvals if we have inadequate financial or other resources to advance our product candidates through the clinical trial process. In addition, even if we believe that the preclinical and clinical data are sufficient to support regulatory approval for a product candidate, the FDA and foreign regulatory authorities may not ultimately approve such product candidate for commercial sale in any jurisdiction, which would limit our ability to generate revenues and adversely affect our business. In addition, even if our product candidates receive regulatory approval, they remain subject to ongoing FDA regulations, including obligations to conduct additional clinical trials, changes to the product label, new or revised regulatory requirements for manufacturing practices, written advisements to physicians, and/or a product recall or withdrawal.
We are subject to stringent regulation of our product candidates, which could delay the development and commercialization of our product candidates.
We, our third-party manufacturers, service providers, suppliers and partners, if any, and our product candidates are subject to stringent regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. None of our product candidates can be marketed in the United States until it has been approved by the FDA. None of our product candidates has been approved by the FDA to date, and we may never receive FDA approval for any of our product candidates. Obtaining FDA approval for a product takes many years of clinical development and requires substantial resources. Additionally, changes in regulatory requirements and guidance may occur or new information regarding the product candidate or the target indication may emerge, and we may need to perform additional, unanticipated non-clinical or clinical testing of our product candidates or amend clinical trial protocols to reflect these changes. Any additional unanticipated testing would add costs and could delay or result in the denial of regulatory approval for a product candidate. These regulatory requirements may limit the size of the market for the product candidate or result in the incurrence of additional costs. Any delay or failure in obtaining required approvals could substantially reduce or negate our ability to generate revenues from the particular product candidate.
In addition, both before and after regulatory approval, we, our partners and our product candidates are subject to numerous FDA requirements, including requirements related to testing, manufacturing, quality control, labeling, advertising, promotion, distribution and export. The FDA’s requirements may change and additional government regulations may be promulgated that could affect us, our partners and our product candidates. Given the number of recent high profile adverse safety events with certain drug products, the FDA may require, as a condition of approval, costly risk management programs, which may include safety surveillance, restricted distribution and
28
use, patient education, enhanced labeling, special packaging or labeling, expedited reporting of certain adverse events, preapproval of promotional materials and restrictions on direct-to-consumer advertising. Furthermore, we cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad.
In order to market any of our products outside of the United States, we and our strategic partners and licensees must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods beyond the requirements of the FDA and the time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States Regulatory approval in one country, including FDA approval in the United States, does not ensure regulatory approval in another. In addition, a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. A product candidate may not be approved for all indications that we request, which would limit the uses of our product and adversely impact our potential royalties and product sales, and any approval that we receive may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
If we fail to comply with applicable regulatory requirements in the United States or other countries, we may be subject to regulatory and other consequences, including fines and other civil penalties, delays in approving or failure to approve a product, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, interruption of manufacturing or clinical trials, injunctions and criminal prosecution, any of which would harm our business.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies, including additional research and development and clinical trials. Any of these restrictions or requirements could adversely affect our potential product revenues. For example, the label ultimately approved for any of our other product candidates or any other product candidates that we may in-license or acquire, if any, may include a restriction on the terms of its use, or it may not include one or more of our intended indications.
Our product candidates, if approved, will also be subject to ongoing FDA requirements for the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information on the drug. In addition, approved products, manufacturers and manufacturers’ facilities are subject to continual review and periodic inspections. If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our product candidates fail to comply with applicable regulatory requirements, such as cGMPs, a regulatory agency may:
|
|
• |
issue warning letters or untitled letters; |
|
|
• |
require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
|
|
• |
impose other civil or criminal penalties; |
|
|
• |
suspend regulatory approval; |
|
|
• |
|
|
|
• |
refuse to approve pending applications or supplements to approved applications filed by us; |
29
|
• |
impose restrictions on operations, including costly new manufacturing requirements; or |
|
|
|
• |
seize or detain products or require a product recall. |
Any of our product candidates that we advance into clinical trials may cause undesirable side effects or have other properties that could delay or prevent regulatory approval or commercialization or limit its commercial potential.
Undesirable side effects caused by any of our product candidates that we advance into clinical trials could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, or cause us to evaluate the future of our development programs. This, in turn, could prevent us from commercializing the affected product candidate and generating revenues from its sale.
In addition, if any product candidates we may develop receives marketing approval and we or others later identify undesirable side effects caused by the product, a number of significant negative consequences could result, including:
|
|
• |
regulatory authorities may withdraw their approval of the product or place restrictions on the way it is prescribed; |
|
|
• |
regulatory authorities may require a larger clinical benefit for approval to offset the risk; |
|
|
• |
regulatory authorities may require the addition of labeling statements that could diminish the usage of the product or otherwise limit the commercial success of the product; |
|
|
• |
we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product or implement a risk evaluation and mitigation strategy; |
|
|
• |
we may choose to discontinue sale of the product; |
|
|
• |
we could be sued and held liable for harm caused to patients; |
|
|
• |
we may not be able to enter into collaboration agreements on acceptable terms and execute our business model; and |
|
|
• |
our reputation may suffer. |
Delays in the commencement or completion of clinical trials, or suspension or termination of our clinical trials, could result in increased costs to us and delay or limit our ability to obtain regulatory approval for our product candidates.
If we experience delays in the commencement or completion of our clinical trials, we could incur significantly higher product development costs and our ability to obtain regulatory approvals for our product candidates could be delayed or limited. The commencement and completion of clinical trials requires us to identify and maintain a sufficient number of study sites and enroll a sufficient number of patients at such sites. We do not know whether enrollment in our future clinical trials for our product candidates will be completed on time, or whether our additional planned and ongoing clinical trials for our product candidates will be completed on schedule, if at all.
The commencement and completion of clinical trials can be delayed for a variety of other reasons, including delays in:
|
|
• |
obtaining regulatory approval to commence or amend a clinical trial; |
30
In addition, a clinical trial may be delayed, suspended or terminated by us, the FDA or other regulatory authorities due to a number of factors, including:
|
|
• |
ongoing discussions with regulatory authorities regarding the scope or design of our clinical trials or requests by them for supplemental information with respect to our clinical trial results, which may result in the imposition of a clinical hold on the IND for any clinical trial, as well as the inability to resolve any outstanding concerns with the FDA so that a clinical hold already placed on the IND may be lifted and the clinical trial may begin; |
|
|
• |
inspections of our own clinical trial operations, the operations of our CROs or our clinical trial sites by the FDA or other regulatory authorities, which may result in the imposition of a clinical hold or potentially prevent us from using some of the data generated from our clinical trials to support requests for regulatory approval of our product candidates; |
|
|
• |
our failure or inability, or the failure or inability of our CROs, clinical trial site staff or other third party service providers involved in the clinical trial, to conduct clinical trials in accordance with regulatory requirements or our clinical protocols; |
|
|
• |
lower than anticipated enrollment or retention rates of patients in clinical trials; |
|
|
• |
new information suggesting unacceptable risk to subjects or unforeseen safety issues or any determination that a clinical trial presents unacceptable health risks; |
|
|
• |
insufficient supply or deficient quality of product candidates or other materials necessary for the conduct of our clinical trials; |
|
|
• |
lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and increased expenses associated with the services of our CROs and other third parties; and |
|
|
• |
the formulation or dosing regimen of a product candidate may result, unintentionally, in patient non-compliance, leading to low patient retention rates, incomplete data to conduct an adequate analysis, and failure to complete the trial. |
If we experience delays in the completion of our clinical trials for a product candidate, the commercial prospects for such product candidate may be harmed, we may incur increased costs for development of such product candidate and our ability to obtain regulatory approval for such product candidate could be delayed or limited. Many of the factors that cause or lead to delays in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval for a product candidate. In addition, any amendment to a clinical trial
31
protocol may require us to resubmit our clinical trial protocols to IRBs or their foreign counterparts for reexamination, which may delay or otherwise impact the costs, timing or successful completion of a clinical trial.
The loss of any rights to develop and market any of our product candidates could significantly harm our business.
We license the rights to certain compounds to develop and market our product candidates.
We are obligated to develop and commercialize certain product candidates in accordance with mutually agreed upon terms and conditions. Our ability to satisfy some or all of the terms and conditions of our license agreements is dependent on numerous factors, including some factors that are outside of our control. Any of our license agreements may be terminated if we breach our obligations under the agreement materially and fail to cure any such breach within a specified period of time.
If any of our license agreements is terminated, we would have no further rights to develop and commercialize the product candidate that is the subject of the license. The termination of any of our license agreements could materially and adversely affect our business.
If our competitors develop and market products that are more effective than our product candidates, they may reduce or eliminate our commercial opportunities.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies, in the United States and abroad. Some of these competitors have products or are pursuing the development of drugs that target the same diseases and conditions that are the focus of our product development programs. We cannot assure you that developments by others will not render our product candidates obsolete or noncompetitive. Many of our competitors have products that have been approved or are in advanced development and may succeed in developing drugs that are more effective, safer, more affordable or more easily administered than ours, or that achieve patent protection or commercialization sooner than our products. Our competitors may also develop alternative therapies that could further limit the market for any product candidates that we are able to obtain approval for, if at all. In addition, new developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical industry at a rapid pace. These developments may render our product candidates obsolete or noncompetitive.
In many of our target disease areas, potential competitors are working to develop new compounds with different mechanisms of action and attractive efficacy and safety profiles. Many of our competitors have substantially greater financial, research and development resources, including personnel and technology, clinical trial experience, manufacturing, sales and marketing capabilities and production facilities than we do. Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaboration arrangements with large pharmaceutical and established biotechnology companies.
Our competitors may obtain regulatory approval of their products more rapidly than we are able to or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are more effective and less costly than ours and may also be more successful than us in manufacturing and marketing their products. We also expect to face similar competition in our efforts to identify appropriate collaborators or partners to help develop or commercialize our product candidates.
We will depend on strategic collaborations with third parties to develop and commercialize selected product candidates and will not have control over a number of key elements relating to the development and commercialization of these product candidates if we are able to achieve such third-party arrangements.
A key aspect of our strategy is to seek collaborations with partners, such as large pharmaceutical companies, that are willing to conduct later-stage clinical trials and further develop and commercialize selected product
32
candidates. To date, we have not entered into any such collaborative arrangements, and we may not be able to enter into any collaborations or otherwise monetize these product candidates on acceptable terms, if at all.
By entering into a strategic collaboration with a partner, we may rely on the partner for financial resources and for development, regulatory and commercialization expertise. Even if we are successful in entering into a strategic collaboration for one of our product candidates, our partner may fail to develop or effectively commercialize the product candidate because such partner:
|
|
• |
does not have sufficient resources or decides not to devote the necessary resources due to internal constraints such as limited cash or human resources; |
|
|
• |
decides to pursue a competitive potential product developed outside of the collaboration; |
|
|
• |
cannot obtain the necessary regulatory approvals; |
|
|
• |
determines that the market opportunity is not attractive; or |
|
|
• |
cannot manufacture the necessary materials in sufficient quantities from multiple sources or at a reasonable cost. |
We also face competition in our search for partners from other biotechnology and pharmaceutical companies worldwide, many of whom are larger and able to offer more attractive deals in terms of financial commitments, contribution of human resources, or development, manufacturing, regulatory or commercial expertise and support.
If we are not successful in attracting partners and entering into collaborations on acceptable terms for these product candidates or otherwise monetizing these product candidates, we may not be able to complete development of or obtain regulatory approval for such product candidates. In such event, our ability to generate revenues from such products and achieve or sustain profitability would be significantly hindered.
The terms under which we enter collaborations or raise additional equity or debt financing may harm our business and may significantly dilute stockholders’ ownership interests.
If we raise additional funds through collaborations or licensing arrangements with third parties, we may need to relinquish some rights to our product candidates, including commercialization rights, which may hinder our ability to generate revenues and achieve or sustain profitability. If we raise additional funds by issuing equity securities, including as part of a debt financing, stockholders may experience substantial dilution. Debt financing, if available, may involve significant cash payment obligations and restrictive covenants and other financial terms that may impede our ability to operate our business. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders.
We rely on third parties to conduct our clinical trials, and we may incur additional development costs, experience delays in the commencement and completion of clinical trials, and be unable to obtain regulatory approval for or commercialize our product candidates on our anticipated timeline if these third parties do not successfully carry out their contractual duties or meet expected deadlines.
We rely extensively on CROs, medical institutions, clinical investigators, contract laboratories and other service providers to perform important functions related to the conduct of our clinical trials, the collection and analysis of data and the preparation of regulatory submissions. Although we design/or and manage our current clinical trials to ensure that each clinical trial is conducted in accordance with its investigational plan and protocol, we do not have the ability to conduct all aspects of our clinical trials directly for our product candidates.
The FDA requires us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on CROs does not relieve us of these
33
responsibilities and requirements. The CROs, medical institutions, clinical investigators, contract laboratories and other service providers that we employ in the conduct of our clinical trials are not our employees, and we cannot control the amount or timing of resources that they devote to our product development programs. If any of these third parties fails to devote sufficient care, time and resources to our product development programs, if its performance is substandard, or if any third party is inspected by the FDA and found not to be in compliance with GCPs, it will delay the completion of the clinical trial in which they are involved and the progress of the affected development program. The CROs with which we contract for execution of our clinical trials play a significant role in the conduct of the clinical trials and the subsequent collection and analysis of data. Any failure of the CROs to meet their obligations could adversely affect clinical development of our product candidates. Moreover, the CROs, clinical investigators and other service providers may have relationships with other commercial entities, some of which may have competitive products under development or currently marketed, and our competitive position could be harmed if they assist our competitors. If any of these third parties does not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality or accuracy of the clinical data is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for our product candidates. In addition, while we believe that there are numerous alternative sources to provide these services, we might not be able to enter into replacement arrangements without delays or additional expenditures if we were to seek such alternative sources.
We rely on third-party manufacturers to produce our product candidates, which may result in delays in our clinical trials and the commercialization of products, as well as increased costs.
We have no manufacturing facilities, and we do not intend to develop facilities for the manufacture of our product candidates for clinical trials or commercial purposes in the foreseeable future. We contract with third-party manufacturers to produce, in collaboration with us, sufficient quantities of our product candidates for clinical trials, and we plan to contract with third-party manufacturers to produce sufficient quantities of any product candidates that may be approved by the FDA or other regulatory authorities for commercial sale. While we believe that there are competitive sources available to manufacture our product candidates, we may not be able to enter into arrangements without delays or additional expenditures. We cannot estimate these delays or costs with certainty.
Reliance on third-party manufacturers limits our ability to control certain aspects of the manufacturing process and therefore exposes us to a variety of significant risks, including risks related to our ability to commercialize any products approved by regulatory authorities or conduct clinical trials, reliance on such third parties for regulatory compliance and quality assurance, and the refusal or inability of a third-party manufacturer to supply our requirements on a long-term basis. In addition, manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, shortages of qualified personnel and compliance with federal, state and foreign regulations. Also, our manufacturers may not perform as agreed. If our manufacturers were to encounter any of these difficulties, our ability to timely produce our product candidates for clinical trials and commercial sale may be interrupted, which could result in delayed clinical trials or delayed regulatory approval and lost or delayed revenues.
We may not be able to establish or maintain any commercial manufacturing and supply arrangements on commercially reasonable terms that we require for purposes of commercializing a product. Any failure by us to secure or maintain any such required commercial supply agreements could result in interruption of supply and lost or delayed revenues, which would adversely affect our business. Any problems or delays we experience in preparing for commercial-scale manufacturing of a product candidate may result in a delay in FDA or other regulatory approval of the product candidate or may impair our ability to manufacture commercial quantities, which would adversely affect our business. For example, our manufacturers will need to produce specific batches of a product candidate to demonstrate acceptable stability under various conditions and for commercially viable lengths of time. We and our third-party manufacturers will need to demonstrate to the FDA and other regulatory authorities this acceptable stability data for the product candidate, as well as validate methods and manufacturing processes, in order to receive regulatory approval to commercialize such product candidate.
Our manufacturers are obligated to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs and, in some cases, International Convention on Harmonization, or ICH, standards. A failure of any of our third-party manufacturers to establish and follow cGMPs and/or ICH standards and to document their
34
adherence to such practices may lead to significant delays in our ability to timely conduct and complete clinical trials, obtain regulatory approval of product candidates or launch of our products into the market. In addition, changing third-party manufacturers is difficult. For example, a change in third-party manufacturer for a particular product candidate requires re-validation of the manufacturing processes and procedures in accordance with cGMPs, which may be costly and time-consuming and, in some cases, our manufacturers may not provide us with adequate assistance to transfer the manufacturing processes and procedures for our product candidates to new manufacturers or may possess intellectual property rights covering parts of these processes or procedures for which we may need to obtain a license. Failure by our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of regulatory approvals, seizures or recalls of products, operating restrictions and criminal prosecutions.
We may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities for preclinical studies and clinical trials. If any of our product candidates is approved by the FDA or comparable regulatory authorities in other countries for commercial sale, we will need to manufacture such product candidate in larger quantities. We may not be able to increase successfully the manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to increase successfully the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in supply. Our product candidates require precise, high quality manufacturing. Our failure to achieve and maintain these high manufacturing standards in collaboration with our third-party manufacturers, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could harm our business, financial condition and results of operations.
Materials necessary to manufacture our product candidates may not be available on commercially reasonable terms, or at all, which may delay the development and commercialization of our product candidates.
We rely on the third-party manufacturers of our product candidates to purchase from third-party suppliers the materials necessary to produce the API and product candidates for our clinical trials, and we will rely on such manufacturers to purchase such materials to produce the API and finished product for any commercial distribution of our products if we obtain marketing approval. Suppliers may not sell these materials to our manufacturers at the time they need them in order to meet our required delivery schedule or on commercially reasonable terms, if at all. We do not have any control over the process or timing of the acquisition of these materials by our manufacturers. Moreover, we currently do not have any agreements for the production of these materials. If our manufacturers are unable to obtain these materials for our clinical trials, testing of the affected product candidate would be delayed, which may significantly impact our ability to develop the product candidate. If we or our manufacturers are unable to purchase these materials after regulatory approval has been obtained for one of our products, the commercial launch of such product would be delayed or there would be a shortage in supply of such product, which would harm our ability to generate revenues from such product and achieve or sustain profitability.
Our product candidates, if approved for sale, may not gain acceptance among physicians, patients and the medical community, thereby limiting our potential to generate revenues.
If one of our product candidates is approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product by physicians, healthcare professionals and third-party payers and our profitability and growth will depend on a number of factors, including:
|
|
• |
demonstration of efficacy; |
|
|
• |
changes in the standard of care for the targeted indication; |
|
|
• |
relative convenience and ease of administration; |
35
If any product candidate that we develop does not provide a treatment regimen that is as beneficial as, or is perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that product candidate, if approved for commercial sale by the FDA or other regulatory authorities, likely will not achieve market acceptance. Our ability to effectively promote and sell any approved products will also depend on pricing and cost-effectiveness, including our ability to produce a product at a competitive price and our ability to obtain sufficient third-party coverage or reimbursement. If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, patients and third-party payers, our ability to generate revenues from that product would be substantially reduced. In addition, our efforts to educate the medical community and third-party payers on the benefits of our product candidates may require significant resources and may never be successful.
If our products are not accepted by the market or if users of our products are unable to obtain adequate coverage of and reimbursement for our products from government and other third-party payers, our revenues and profitability will suffer.
Our ability to commercialize our products successfully will depend in significant part on pricing and cost effectiveness, including our ability to produce a product at a competitive price and our ability to obtain appropriate coverage of and reimbursement for our products and related treatments from governmental authorities, private health insurers and other organizations, such as health maintenance organizations, or HMOs. Third-party payers are increasingly challenging the prices charged for medical products and services. We cannot provide any assurances that third-party payers will consider our products cost-effective or provide coverage of and reimbursement for our products, in whole or in part.
Uncertainty exists as to the coverage and reimbursement status of newly approved medical products and services and newly approved indications for existing products. Third-party payers may conclude that our products are less safe, less clinically effective or less cost-effective than existing products, and third-party payers may not approve our products for coverage and reimbursement. If we are unable to obtain adequate coverage of and reimbursement for our products from third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them. Such reduction or limitation in the use of our products could cause our sales to suffer. Even if third-party payers make reimbursement available, payment levels may not be sufficient to make the sale of our products profitable.
Market acceptance and sales of our current or future product candidates will depend in large part on global reimbursement policies and may be affected by future healthcare reform measures, both in the United States and other key international markets. For example, continuing health care reform in the United States will control or significantly influence the purchase of medical services and products, and may result in inadequate coverage of and reimbursement for our products. Many third-party payers are pursuing various ways to reduce pharmaceutical costs, including the use of formularies. The market for our products depends on access to such formularies, which are lists of medications for which third-party payers provide reimbursement. These formularies are increasingly restricted, and pharmaceutical companies face significant competition in their efforts to place their products on formularies.
36
This increased competition has led to a downward pricing pressure in the industry. The cost containment measures that third-party payers, including government payers, are instituting could have a material adverse effect on our ability to operate profitably.
We are dependent on our management team, particularly our President and Chief Executive Officer, and our and experienced scientific staff, and if we are unable to retain, motivate and attract key personnel, our product development programs may be delayed and we may be unable to develop successfully or commercialize our product candidates.
We are dependent upon the continued services of our executive officers and other key personnel, particularly Yuichi Iwaki, M.D., Ph.D., a founder and our President and Chief Executive Officer, who has been instrumental in our ability to in-license product candidates from Japanese pharmaceutical companies and secure financing from Japanese institutions. The relationships that certain of our key managers have cultivated with pharmaceutical companies from whom we license product candidates and to whom we expect to out-license product candidates make us particularly dependent upon their continued services with us, whether through employment, service on our board of directors or a consulting agreement. We are also substantially dependent on the continued services of clinical development personnel because of the highly technical nature of our product development programs. We are not presently aware of any plans of our executive officers or key personnel to retire or leave employment. Following termination of employment, these individuals may engage in other businesses that may compete with us.
If we acquire or license new product candidates, our success may depend on our ability to attract, retain and motivate highly qualified management and scientific personnel to manage the development of these new product candidates. In particular, our product development programs depend on our ability to attract and retain highly experienced clinical development personnel. However, we face competition for experienced professional personnel from numerous companies and academic and other research institutions. Competition for qualified personnel is particularly intense in the San Diego, California area, where our corporate headquarters is located. Our short operating history and the uncertainties could impair our ability to attract and retain personnel and impede the achievement of our development and commercialization objectives. In addition, we have scientific and clinical advisors who assist us in our product development and clinical strategies. These third parties are not our employees and may have commitments to, or contracts with, other entities that may limit their availability to us, or may have arrangements with other companies to assist in the development of products that may compete with our product candidates.
Although we have employment agreements with key members of management, each of our employees, subject to applicable notice requirements, may terminate his or her employment at any time. We do not carry “key person” insurance covering members of senior management. If we lose any of our key management personnel, we may not be able to find suitable replacements, which would adversely affect our business.
If we are unable to establish sales, marketing and distribution capabilities, whether independently or with third parties, we will be unable to commercialize our product candidates successfully.
To date, we have not sold, marketed or distributed any pharmaceutical products. If we are successful in obtaining regulatory approvals for any of our product candidates or acquiring other approved products, we will need to establish sales, marketing and distribution capabilities on our own or with partners in order to commercialize an approved product. The acquisition or development of an effective sales and marketing infrastructure will require a significant amount of our financial resources and time and could negatively impact our commercialization efforts, including delay of a product launch. We may be unable to establish and manage a sufficient or effective sales force in a timely or cost-effective manner, if at all, and any sales force we do establish may not be capable of generating demand for our products, therefore hindering our ability to generate revenues and achieve or sustain profitability. In addition, if we are unable to develop internal sales capabilities, we will need to contract with third parties or establish a partnership to market and sell the product. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate any product revenues, may generate increased expenses and may never become profitable. In addition, although we intend to establish strategic collaborations to market any products approved for sale by regulatory authorities outside of the United States, we may be required to market our product candidates outside of the United States directly if we
37
are unable to establish such collaborations. In that event, we may need to build a corresponding international sales and marketing capability with technical expertise and with supporting distribution capabilities.
Health care reform measures could adversely affect our business.
The business and financial condition of pharmaceutical and biotechnology companies are affected by the efforts of governmental and third-party payers to contain or reduce the costs of health care. In the United States and in foreign jurisdictions, there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the health care system. For example, in some countries, pricing of prescription drugs is subject to government control, and we expect to continue to see proposals to implement similar controls in the United States to continue. Another example of proposed reform that could affect our business is drug reimportation into the United States. Moreover, the pendency or approval of such proposals could result in a decrease in our stock price or our ability to raise capital or to obtain strategic partnerships or licenses. More recently, the Patient Protection and Affordable Care Act imposed numerous reforms that may impact the costs, legal requirements and potential success of our operations.
We may be sued for product liability, which could result in substantial liabilities that exceed our available resources and damage our reputation.
The development and commercialization of drug products entails significant product liability risks. Product liability claims may arise from use of any of our product candidates in clinical trials and the commercial sale of any approved products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
|
|
• |
withdrawal of clinical trial participants; |
|
|
• |
termination of clinical trial sites or entire clinical trial programs; |
|
|
• |
decreased demand for our product candidates; |
|
|
• |
impairment of our business reputation; |
|
|
• |
costs of related litigation; |
|
|
• |
substantial monetary awards to patients or other claimants; |
|
|
• |
loss of revenues; and |
|
|
• |
the inability to commercialize our product candidates. |
We currently have insurance that covers our clinical trials. We believe our current insurance coverage is reasonably adequate at this time; however, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for all expenses or losses we may suffer. In addition, we will need to increase and expand this coverage as we commence additional clinical trials, as well as larger scale clinical trials, and in the event that any of our product candidates is approved for commercial sale. This insurance may be prohibitively expensive or may not fully cover our potential liabilities. In addition, our inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the regulatory approval or commercialization of products that we or one of our collaborators develop. Successful product liability claims could have a material adverse effect on our business and results of operations. Liability from such claims could exceed our total assets if we do not prevail in any lawsuit brought by a third party alleging that an injury was caused by one of our product candidates.
We expect that our results of operations will fluctuate, which may make it difficult to predict our future performance from period to period.
38
Our quarterly operating results have fluctuated in the past and are likely to continue to do so in the future. Some of the factors that could cause our operating results to fluctuate from period to period include:
|
|
• |
the status of development of our product candidates and, in particular, the advancement or termination of activities related to our product development programs and the timing of any milestone payments payable under our licensing agreements; |
|
|
• |
the execution of other collaboration, licensing and similar arrangements and the timing of payments we may make or receive under these arrangements; |
|
|
• |
variations in the level of expenses related to our product development programs; |
|
|
• |
the unpredictable effects of collaborations during these periods; |
|
|
• |
the timing of our satisfaction of applicable regulatory requirements, if at all; |
|
|
• |
the rate of expansion of our clinical development and other internal research and development efforts; |
|
|
• |
the costs of any litigation; |
|
|
• |
the effect of competing technologies and products and market developments; and |
|
|
• |
general and industry-specific economic conditions. |
We believe that quarterly or yearly comparisons of our financial results are not necessarily meaningful and should not be relied upon as indications of our future performance.
We will continue to incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we are required to comply with the Sarbanes-Oxley Act of 2002, as well as rules and regulations implemented by the SEC, The NASDAQ Stock Market, or NASDAQ, and Japanese securities laws, and incur significant legal, accounting and other expenses as a result. These rules impose various requirements on public companies, including requiring the establishment and maintenance of effective disclosure and financial controls and appropriate corporate governance practices. Our management and other personnel have devoted and will continue to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and may make it more difficult and expensive for us to renew our director and officer liability insurance, and may result in imposition of reduced policy limits and coverage.
The Sarbanes-Oxley Act requires that we (i) maintain effective internal controls for financial reporting and disclosure controls and procedures and (ii) perform an evaluation of our internal control over financial reporting to allow management to report on the effectiveness of those controls, as required by Section 404. Our listing obligations under the JASDAQ Market of the Tokyo Stock Exchange, or TSE, also require that we comply either with Section 404 of the Sarbanes-Oxley Act or equivalent regulations in Japan and we elected to comply with Section 404. Additionally, we are subject to attestation by our independent registered public accounting firm regarding our internal controls over financial reporting as of December 31, 2017 under Japanese securities laws. Our efforts to comply with Section 404 and related regulations have required, and continue to require, the commitment of significant financial and managerial resources. We cannot be certain that a material weakness will not be identified when we test the effectiveness of our controls in the future. If a material weakness is identified, we could be subject to sanctions or investigations by NASDAQ, the SEC, the TSE or other regulatory authorities, which would require additional financial and management resources, costly litigation or a loss of public confidence in our internal controls, which could have an adverse effect on the market price of our stock.
Additionally, in July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation related provisions
39
in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas. To maintain high standards of corporate governance and public disclosure, we intend to invest all reasonably necessary resources to comply with such compliance programs and rules and all other evolving standards. These investments may result in increased general and administrative costs and a diversion of our management’s time and attention from strategic revenue generating and cost management activities.
Our business and operations would suffer in the event of system failures and natural disasters.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our drug development programs, including delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability and the further development of our product candidates may be delayed.
A variety of risks associated with operating our business and marketing our products internationally could materially adversely affect our business.
A significant amount of our business activity is outside of the United States. We face risks associated with our international operations, including possible unfavorable regulatory, pricing and reimbursement, political, tax and labor conditions, which could harm our business. We are subject to numerous risks associated with international business activities, including, but not limited to:
|
|
• |
compliance with differing or unexpected regulatory requirements for our products; |
|
|
• |
difficulties in staffing and managing foreign operations; |
|
|
• |
in certain circumstances, including with respect to the commercialization of our product candidates in Europe, increased dependence on the commercialization efforts of our distributors or strategic partners; |
|
|
• |
foreign government taxes, regulations and permit requirements; |
|
|
• |
United States and foreign government tariffs, trade restrictions, price and exchange controls and other regulatory requirements; |
|
|
• |
economic weakness, including inflation, natural disasters, war, events of terrorism or political instability in particular foreign countries; |
|
|
• |
fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenues, and other obligations related to doing business in another country; |
|
|
• |
compliance with tax, employment, immigration and labor laws, regulations and restrictions for employees living or traveling abroad; |
|
|
• |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
|
|
• |
changes in diplomatic and trade relationships; and |
|
|
• |
challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States |
These and other risks associated with our international operations may materially adversely affect our business, financial condition and results of operations.
40
Risks Related to Our Intellectual Property
Our ability to compete may decline if we do not adequately protect our proprietary rights.
There is the risk that our patents (both those owned by us and those in-licensed) may not provide a competitive advantage, including the risk that our patents expire before we obtain regulatory and marketing approval for one or more of our product candidates, particularly our in-licensed patents. Also, our competitors may develop products similar to ours using methods and technologies that are beyond the scope of our intellectual property rights. Composition of matter patents on APIs may provide protection for pharmaceutical products without regard to formulation, method of use, or other type of limitation. We do not have compound patent protection for the API in our MN-166 (ibudilast), MN-001 (tipelukast), and MN-221 product candidates, although we do have patent protection for a particular crystalline polymorph of MN-001 (tipelukast) and we have composition of matter protection on an analog of MN-166 (ibudilast). As a result, competitors that obtain the requisite regulatory approval will be able to offer products with the same API as found in our MN-166 (ibudilast), MN-001 (tipelukast), and MN-221 product candidates so long as such competitors do not infringe any methods of use, methods of manufacture, formulation or, in the case of MN-001 (tipelukast), specific polymorph patents that we hold or have exclusive rights to through our licensors. For example, we currently rely on method of use patents for MN-166 (ibudilast), MN-001 (tipelukast), and MN-221 although we have a compound patent for MN-029.
It is our policy to consult with our licensors in the maintenance of granted patents we have licensed and in their pursuit of patent applications that we have licensed, but each of our licensors generally remains primarily responsible for or in control of the maintenance of the granted patents. We have limited control, if any, over the amount or timing of resources that each licensor devotes on our behalf. As a result of this lack of control, we cannot be sure that our licensed patents will be maintained and that any additional patents will ever mature from our licensed applications. Issued U.S. patents require the payment of maintenance fees to continue to be in force. We typically rely on our licensors to do this and their failure to do so could result in the forfeiture of patents not timely maintained. Many foreign patent offices also require the payment of periodic annuities to keep patents and patent applications in good standing. As we generally do not maintain control over the payment of annuities, we cannot be certain that our licensors will timely pay such annuities and that the granted patents will not become abandoned. For example, certain annuities were not paid in a timely manner with respect to foreign patents licensed under MN-002 (the active metabolite of MN-001) and, as a result, our patent rights may be impaired in those territories. In addition, our licensors may have selected a limited amount of foreign patent protection, and therefore applications have not been filed in, and foreign patents may not have been perfected in, all commercially significant countries.
The patent protection of our product candidates and technology involves complex legal and factual questions. Most of our license agreements give us a right, but not an obligation, to enforce our patent rights. To the extent it is necessary or advantageous for any of our licensors’ cooperation in the enforcement of our patent rights, we cannot control the amount or timing of resources our licensors devote on our behalf or the priority they place on enforcing our patent rights. We may not be able to protect our intellectual property rights against third party infringement, which may be difficult to detect, especially for infringement of patent claims for methods of manufacturing. Additionally, challenges may be made to the ownership of our intellectual property rights, our ability to enforce them or our underlying licenses, which in some cases have been made under foreign laws and may provide different protections than that of U.S. law.
We cannot be certain that any of the patents or patent applications owned by us or our licensors related to our product candidates and technology will provide adequate protection from competing products. Our success will depend, in part, on whether we or our licensors can:
|
|
• |
obtain and maintain patents to protect our product candidates; |
|
|
• |
obtain and maintain any required or desirable licenses to use certain technologies of third parties, which may be protected by patents; |
|
|
• |
protect our trade secrets and know-how; |
|
|
• |
operate without infringing the intellectual property and proprietary rights of others; |
41
|
• |
||
|
|
• |
develop additional proprietary technologies that are patentable. |
The degree of future protection for our proprietary rights is uncertain. For example:
|
|
• |
we or our licensor might not have been the first to make the inventions covered by each of our pending patent applications or issued patents; |
|
|
• |
we or our licensor might not have been the first to file patent applications for these inventions; |
|
|
• |
others may independently develop similar or alternative technologies or duplicate any of our technologies; |
|
|
• |
it is possible that none of our pending patent applications will result in issued patents; |
|
|
• |
any patents under which we hold rights may not provide us with a basis for maintaining market exclusivity for commercially viable products, may not provide us with any competitive advantages or may be challenged by third parties as invalid, not infringed or unenforceable under U.S. or foreign laws; or |
|
|
• |
any of the issued patents under which we hold rights may not be valid or enforceable or may be circumvented successfully in light of the continuing evolution of domestic and foreign patent laws. |
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
Because we operate in the highly technical field of research and development of small molecule drugs, we rely in part on trade secret protection in order to protect our proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and we cannot be certain that others will not develop the same or similar technologies on their own. We have taken steps, including entering into confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors, to protect our trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. We also typically obtain agreements from these parties which provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Further, we have limited control, if any, over the protection of trade secrets developed by our licensors. Enforcing a claim that a party illegally obtained and is using our trade secrets or know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in our industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our product candidates, their methods of use, manufacturing or other technologies or activities infringe the intellectual property rights of such third parties. There are many patents relating to chemical compounds and methods of use. If our compounds or their methods of use or manufacture are found to infringe any such patents, we may have to pay significant damages or seek licenses under such patents. We have not conducted comprehensive searches for unexpired patents issued to third parties relating to our product candidates. Consequently, no assurance can be given that unexpired, third-party
42
patents containing claims covering our product candidates, their methods of use or manufacture do not exist. Moreover, because some patent applications in the United States may be maintained in secrecy until the patents are issued, and because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing, we cannot be certain that others have not filed patent applications that will mature into issued patents that relate to our current or future product candidates and which could have a material effect in developing and commercializing one or more of our product candidates. The owner of a patent that is arguably infringed can bring a civil action seeking to enjoin an accused infringer from importing, making, marketing, distributing, using or selling an infringing product. We may need to resort to litigation to enforce our intellectual property rights or to seek a declaratory judgment concerning the scope, validity or enforceability of third-party proprietary rights. Similarly, we may be subject to claims that we have inappropriately used or disclosed trade secrets or other proprietary information of third parties. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. Some of our competitors may be able to sustain the costs of complex intellectual property litigation more effectively than we can because they have substantially greater resources. We may not be able to afford the costs of litigation. Any legal action against us or our collaborators could lead to:
|
|
• |
payment of actual damages, royalties, lost profits, potential enhanced damages and attorneys’ fees, if any infringement for which we are found liable is deemed willful, or a case against us is determined by a judge to be exceptional; |
|
|
• |
injunctive or other equitable relief that may effectively block our ability to further develop, commercialize and sell our products; |
|
|
• |
having to enter into license arrangements that may not be available on reasonable or commercially acceptable terms; or |
|
|
• |
significant cost and expense, as well as distraction of our management from our business. |
As a result, we could lose our ability to develop and commercialize current or future product candidates.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. From time to time, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to the Securities Markets and Investment in Our Common Stock
Our stock price may be volatile, and you may not be able to resell our shares at a profit or at all.
Despite the listing of our common stock on The NASDAQ Global Market and the JASDAQ Market of the Tokyo Stock Exchange in Japan, trading volume in our securities has been light and an active trading market may not develop for our common stock. In 2017, our average trading volume was approximately 70,398 shares per day on The NASDAQ Global Market and approximately 136,536 shares per day on the JASDAQ Market.
The market prices for securities of biopharmaceutical and biotechnology companies, and early-stage drug discovery and development companies like us in particular, have historically been highly volatile and may continue to be highly volatile in the future. For example, since the date of our initial public offering in Japan on February 8, 2005 through December 31, 2017, our common stock has traded as high as approximately $42.00 and as low as approximately $1.30. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
43
|
• |
the development status of our product candidates, including clinical trial results and determinations by regulatory authorities with respect to our product candidates; |
|
|
|
• |
the initiation, termination, or reduction in the scope of any collaboration arrangements or any disputes or developments regarding such collaborations; |
|
|
• |
FDA or foreign regulatory actions, including failure to receive regulatory approval for any of our product candidates; |
|
|
• |
announcements of technological innovations, new commercial products or other material events by us or our competitors; |
|
|
• |
disputes or other developments concerning our intellectual property rights; |
|
|
• |
market conditions in the pharmaceutical and biotechnology sectors; |
|
|
• |
actual and anticipated fluctuations in our quarterly or annual operating results; |
|
|
• |
price and volume fluctuations in the overall stock markets; |
|
|
• |
changes in, or failure to meet, securities analysts’ or investors’ expectations of our financial performance; |
|
|
• |
additions or departures of key personnel; |
|
|
• |
discussions of our business, management, products, financial performance, prospects or stock price by the financial and scientific press and online investor communities; |
|
|
• |
litigation or public concern about the safety of our potential products; |
|
|
• |
public concern as to, and legislative action with respect to, the pricing and availability of prescription drugs or the safety of drugs and drug delivery techniques; or |
|
|
• |
regulatory developments in the United States and in foreign countries. |
Broad market and industry factors, as well as economic and political factors, also may materially adversely affect the market price of our common stock.
Our common stock may be delisted on The NASDAQ Global Market or the JASDAQ Market of the Tokyo Stock Exchange.
In addition to the risks identified immediately above, the market price of our common stock, and your ability to sell your shares at a profit, or at all, may be affected by the delisting of our shares for failure to meet applicable listing standards. For example, price per share minimums are maintained by The NASDAQ Global Market, and our share price has, in the past, fallen below the required minimum. In addition, JASDAQ Market listing requirements currently mandate that listed companies achieve a profit or positive cash flow from operations within a five-year period. Failure to meet these or other listing requirements for either of the stock exchanges on which our common stock is listed could adversely affect the market price for our common stock and your ability to sell your shares at a profit, or at all.
The sale of additional common stock under our existing at-the-market issuance sales agreement may cause substantial dilution to our existing stockholders and/or the price of our common stock to decline.
44
On May 22, 2015, we entered into an at-the-market issuance sales agreement, or the ATM Agreement, with MLV & Co. LLC, or MLV, pursuant to which we may sell common stock through MLV from time to time up to an aggregate offering price of $30.0 million. On September 16, 2016, we entered into an amendment to the ATM Agreement to also include FBR Capital Markets & Co as a sales agent thereunder. From time to time, we may sell additional shares of our common stock under the ATM Agreement. Depending upon market liquidity at the time, sales of shares of our common stock under the ATM Agreement may cause the trading price of our common stock to decline and may result in substantial dilution to the interests of other holders of our common stock. The sale of a substantial number of shares of our common stock under the ATM Agreement, or anticipation of such sales, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of biotechnology and biopharmaceutical companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have in the past experienced significant stock price volatility. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could adversely affect our business.
Future sales of our common stock may cause our stock price to decline and may make it difficult for us to raise additional capital or for you to sell your shares.
Sales of substantial amounts of our common stock, or the availability of such common stock for sale, could adversely affect the prevailing market prices for our common stock. If this occurs and continues, it could impair our ability to raise additional capital through the sale of securities if we should desire to do so. In addition, it may be difficult, or even impossible, to find a buyer for shares of our common stock.
We have also registered all common stock that we may issue under our current employee benefits plans and upon exercise of warrants. As a result, these shares can be freely sold in the public market upon issuance, subject to the terms of the underlying agreements governing the grants and the restrictions of the securities laws. In addition, our directors and officers may in the future establish programmed selling plans under Rule 10b5-1 of the Exchange Act, for the purpose of effecting sales of our common stock. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us more complicated and the removal and replacement of our directors and management more difficult.
Our restated certificate of incorporation and amended and restated bylaws contain provisions that may delay or prevent a change in control, discourage bids at a premium over the market price of our common stock or adversely affect the market price of our common stock and the voting and other rights of the holders of our common stock. These provisions may also make it difficult for stockholders to remove and replace our board of directors and management. These provisions:
|
|
• |
establish that members of the board of directors may be removed only for cause upon the affirmative vote of stockholders owning at least a majority of our capital stock; |
|
|
• |
authorize the issuance of “blank check” preferred stock that could be issued by our board of directors in a discriminatory fashion designed to increase the number of outstanding shares and prevent or delay a takeover attempt; |
|
|
• |
limit who may call a special meeting of stockholders; |
45
|
• |
establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; |
|
|
|
• |
prohibit our stockholders from making certain changes to our restated certificate of incorporation or amended and restated bylaws except with 66-2/3% stockholder approval; and |
|
|
• |
provide for a classified board of directors with staggered terms. |
We also may be subject to provisions of the Delaware corporation law that, in general, prohibit any business combination with a beneficial owner of 15% or more of our common stock for three years unless the holder’s acquisition of our stock was approved in advance by our board of directors. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In any event, these provisions may delay or prevent a third party from acquiring us. Any such delay or prevention could cause the market price of our common stock to decline.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
Item 1B. Unresolved Staff Comments
Not applicable.
We executed a sublease agreement for our headquarters effective December 1, 2017 (the “Sublease”) with Cardinal Health 127 Inc., the sublessor, to which Irvine Company, the master lessor, has provided its consent. The Sublease is for approximately 4,400 square feet, and has a term of four years and one month. In June 2005, we leased office space in Tokyo, Japan under a non-cancelable operating lease with an original expiration date of May 2013 and an auto-renewal two-year extension, which we have extended through May 2019, with the acceptance of the extensions for those given periods. We have no laboratory, research or manufacturing facilities, and we currently do not plan to purchase or lease any such facilities, as such services are provided to us by third-party service providers. We believe that our current facilities are adequate for our needs for the immediate future and that, should it be needed, suitable additional space will be available to accommodate expansion of our operations on commercially reasonable terms.
We are not involved in any material legal proceedings as of the date of this report. We may become involved in various disputes and legal proceedings which arise in the ordinary course of business. Our assessment of the likely impact of our pending litigation may change over time. An adverse result in any of these matters may occur which could harm our business and result in a material liability.
Item 4. Mine Safety Disclosures
Not applicable.
46
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed on the JASDAQ Market of the Tokyo Stock Exchange and trades under the code “4875,” and is listed on The NASDAQ Global Market and trades under the symbol “MNOV.” Our stock had been traded on the Hercules Market since February 8, 2005 (through the Hercules Market’s closure in 2010) and now is currently traded on the JASDAQ Market and on The NASDAQ Global Market since December 7, 2006.
The following table sets forth the high and low sale prices per share of our common stock as reported on The NASDAQ Global Market.
|
|
|
Common Stock Price |
|
|||||
|
|
|
High |
|
|
Low |
|
||
|
Fiscal year ended December 31, 2016 |
|
|
|
|
|
|
|
|
|
First quarter |
|
$ |
8.34 |
|
|
$ |
3.50 |
|
|
Second quarter |
|
$ |
10.16 |
|
|
$ |
5.66 |
|
|
Third quarter |
|
$ |
8.00 |
|
|
$ |
5.65 |
|
|
Fourth quarter |
|
$ |
7.78 |
|
|
$ |
6.00 |
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal year ended December 31, 2017 |
|
|
|
|
|
|
|
|
|
First quarter |
|
$ |
6.60 |
|
|
$ |
5.49 |
|
|
Second quarter |
|
$ |
6.26 |
|
|
$ |
4.85 |
|
|
Third quarter |
|
$ |
6.60 |
|
|
$ |
4.40 |
|
|
Fourth quarter |
|
$ |
7.85 |
|
|
$ |
5.75 |
|
Holders of Common Stock
As of December 31, 2017, there were approximately 13,200 holders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future. We expect to retain our future earnings, if any, to fund the growth and development of our business.
Securities Authorized for Issuance Under Equity Compensation Plans
Information regarding the securities authorized for issuance under our equity compensation plans can be found under Item 12 of this Annual Report on Form 10-K.
47
Performance Measurement Comparison
The following graph illustrates the total stockholder return of an investment of $100 in cash made on December 31, 2012 in each of our common stock and two indices: the Nasdaq Composite Index and the Nasdaq Biotechnology Index.
The performance graph assumes an initial investment of $100 on December 31, 2012 and that all dividends were reinvested. No dividends have been declared nor paid on our common stock. The comparisons in the graph below are required by the SEC and are not intended to forecast or be indicative of possible future performance of our common stock.
This performance graph is furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or incorporated by reference into any of our other filings under the Exchange Act or the Securities Act except to the extent we specifically incorporate it by reference into such filing.
|
Comparison of Cumulative Total Return on Investment |
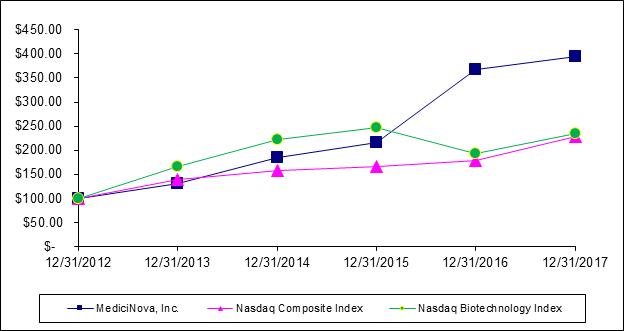
|
|
12/31/2012 |
|
12/31/2013 |
|
12/31/2014 |
|
12/31/2015 |
|
12/31/2016 |
|
12/31/2017 |
|
||||||
|
MediciNova, Inc. |
$ |
100.00 |
|
$ |
130.49 |
|
$ |
185.37 |
|
$ |
216.46 |
|
$ |
367.68 |
|
$ |
394.51 |
|
|
Nasdaq Composite Index |
$ |
100.00 |
|
$ |
138.32 |
|
$ |
156.85 |
|
$ |
165.84 |
|
$ |
178.28 |
|
$ |
228.63 |
|
|
Nasdaq Biotechnology Index |
$ |
100.00 |
|
$ |
165.61 |
|
$ |
222.08 |
|
$ |
247.44 |
|
$ |
193.79 |
|
$ |
234.60 |
|
48
Item 6. Selected Financial Data
The selected financial data set forth below is derived from our audited consolidated financial statements and may not be indicative of future operating results. The following selected financial data should be read in conjunction with the Consolidated Financial Statements and notes thereto and Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K. Amounts are in thousands, except per share amounts.
|
|
|
Years ended December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
6,003 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and patent |
|
|
4,224 |
|
|
|
3,519 |
|
|
|
3,017 |
|
|
|
3,260 |
|
|
|
3,366 |
|
|
General and administrative |
|
|
8,803 |
|
|
|
7,363 |
|
|
|
5,805 |
|
|
|
5,963 |
|
|
|
6,658 |
|
|
Total operating expenses |
|
|
13,027 |
|
|
|
10,882 |
|
|
|
8,822 |
|
|
|
9,223 |
|
|
|
10,024 |
|
|
Operating loss |
|
|
(13,027 |
) |
|
|
(10,882 |
) |
|
|
(8,822 |
) |
|
|
(9,223 |
) |
|
|
(4,021 |
) |
|
Other expense |
|
|
(25 |
) |
|
|
(47 |
) |
|
|
(54 |
) |
|
|
(12 |
) |
|
|
(25 |
) |
|
Interest expense |
|
|
(1 |
) |
|
|
0 |
|
|
|
(1 |
) |
|
|
(1 |
) |
|
|
— |
|
|
Other income |
|
|
146 |
|
|
|
67 |
|
|
|
39 |
|
|
|
37 |
|
|
|
21 |
|
|
Loss before income taxes |
|
|
(12,907 |
) |
|
|
(10,862 |
) |
|
|
(8,838 |
) |
|
|
(9,199 |
) |
|
|
(4,025 |
) |
|
Income tax benefit (expense) |
|
|
1,744 |
|
|
|
(4 |
) |
|
|
(7 |
) |
|
|
4 |
|
|
|
(4 |
) |
|
Net loss |
|
$ |
(11,163 |
) |
|
$ |
(10,866 |
) |
|
$ |
(8,845 |
) |
|
$ |
(9,195 |
) |
|
$ |
(4,029 |
) |
|
Net loss applicable to common stockholders |
|
$ |
(11,163 |
) |
|
$ |
(10,866 |
) |
|
$ |
(8,845 |
) |
|
$ |
(9,195 |
) |
|
$ |
(4,029 |
) |
|
Basic and diluted net loss per share |
|
$ |
(0.32 |
) |
|
$ |
(0.33 |
) |
|
$ |
(0.33 |
) |
|
$ |
(0.38 |
) |
|
$ |
(0.19 |
) |
|
Shares used to compute basic and diluted net loss per share |
|
|
35,137,028 |
|
|
|
32,986,740 |
|
|
|
26,578,770 |
|
|
|
24,067,781 |
|
|
|
20,697,440 |
|
|
|
|
As of December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
27,992 |
|
|
$ |
24,118 |
|
|
$ |
22,077 |
|
|
$ |
11,669 |
|
|
$ |
6,700 |
|
|
Working capital |
|
|
25,447 |
|
|
|
23,074 |
|
|
|
21,236 |
|
|
|
10,539 |
|
|
|
13,922 |
|
|
Total assets |
|
|
43,419 |
|
|
|
39,813 |
|
|
|
37,906 |
|
|
|
27,273 |
|
|
|
29,546 |
|
|
Accumulated deficit |
|
|
(341,456 |
) |
|
|
(330,293 |
) |
|
|
(319,427 |
) |
|
|
(310,582 |
) |
|
|
(301,387 |
) |
|
Total stockholders’ equity |
|
|
38,642 |
|
|
|
34,532 |
|
|
|
32,753 |
|
|
|
22,011 |
|
|
|
25,426 |
|
49
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis together with “Item 6. Selected Financial Data” and the consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K. The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those expressed or implied in any forward-looking statements as a result of various factors, including those set forth under the caption “Item 1A. Risk Factors.”
Overview
Background
We are a biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of serious diseases with unmet medical needs and a commercial focus on the United States market. We were incorporated in Delaware in September 2000.
We have incurred significant net losses since our inception. For the year ended December 31, 2017, we had a net loss of $11.2 million. At December 31, 2017, from inception, our accumulated deficit was $341.5 million. We expect to incur substantial net losses for the next several years as we continue to develop certain of our existing product development programs, and over the long-term if we expand our research and development programs and acquire or in-license products, technologies or businesses that are complementary to our own.
Our current strategy is to focus our development activities on MN-166 (ibudilast) for neurological disorders such as progressive multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), glioblastoma, and substance dependence and addiction (e.g., methamphetamine dependence, opioid dependence, and alcohol dependence), and MN-001 (tipelukast) for fibrotic diseases such as nonalcoholic steatohepatitis (NASH) and idiopathic pulmonary fibrosis (IPF). Our pipeline also includes MN-221 (bedoradrine) for the treatment of acute exacerbation of asthma and MN-029 (denibulin) for solid tumor cancers.
Upon completion of proof-of-concept Phase 2 clinical trials, we intend to enter into discussions regarding strategic alliances with leading pharmaceutical or biotechnology companies who seek late stage product candidates to support further clinical development and product commercialization. Depending on decisions we may make as to further clinical development, we may seek to raise additional capital. We may also pursue potential partnerships and potential acquirers of license rights to our programs in markets outside the United States.
We entered into an agreement to form a joint venture company with Zhejiang Medicine Co., Ltd. and Beijing Medfron Medical Technologies Co. Ltd., (formerly Beijing Make-Friend Medicine Technology Co., Ltd.) effective September 27, 2011. The joint venture agreement provides for the joint venture company, Zhejiang Sunmy Bio-Medical Co., Ltd. (Zhejiang Sunmy), to develop and commercialize MN-221 in China and search for additional compounds to develop. On July 24, 2017, the Company and Beijing Medfron Medical Technologies Co., Ltd. agreed to dissolve Zhejiang Sunmy, subject to approval by applicable Chinese regulatory authorities which was granted on December 11, 2017. At December 31, 2017, we reflect a long-term asset on our consolidated balance sheet which represents our investment in Zhejiang Sunmy, net of our portion of any generated loss or income
Revenues and Cost of Revenues
We did not recognize any revenue for the years ended December 31, 2017, 2016 or 2015.
In October 2011, we entered into an agreement with Kissei to perform research and development services relating to MN-221 in exchange for a non-refundable upfront payment of $2.5 million. Under the terms of the agreement, we are responsible for all costs incurred and to be incurred in the performance of these services. Certain of the development services were completed in 2013 and 2012, and the remaining services are expected to be delivered and completed at a future date. We assessed the deliverables in accordance with the authoritative guidance and concluded the existence of one deliverable, which was research and development services. The $2.5 million was initially recorded as deferred revenue of which $0.8 million was recognized through 2013. No revenue was recorded in 2017, 2016 and 2015 associated with the Kissei agreement.
50
Research, Development and Patent Expenses
Our research, development and patent expenses consist primarily of the license fees related to our product candidates, salaries and related employee benefits, costs associated with the preclinical and clinical development of our product development programs, costs associated with non-clinical activities, such as regulatory expenses, and pre-commercialization manufacturing development activities. We use external service providers to manufacture our compounds to be used in clinical trials and for the majority of the services performed in connection with the preclinical and clinical development of our product candidates. Research, development and patent expenses include fees paid to consultants, contract research organizations, contract manufacturers and other external service providers, including professional fees and costs associated with legal services, patents and patent applications for our intellectual property. Internal research and development expenses include costs of compensation and other expenses for research and development personnel, supplies, facility costs and depreciation. Research, development and patent costs are expensed as incurred, and we expect to increase such costs in 2018 as our development programs progress.
The following table summarizes our research, development and patent expenses for the periods indicated for each of our product development programs. To the extent that costs, including personnel costs, are not tracked to a specific product development program, such costs are included in the “Other R&D expense” category (in thousands):
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
External development expense: |
|
|
|
|
|
|
|
|
|
|
|
|
|
MN-221 |
|
$ |
12 |
|
|
$ |
6 |
|
|
$ |
9 |
|
|
MN-166 |
|
|
1,055 |
|
|
|
707 |
|
|
|
802 |
|
|
MN-001 |
|
|
420 |
|
|
|
364 |
|
|
|
191 |
|
|
MN-029 |
|
|
3 |
|
|
|
3 |
|
|
|
14 |
|
|
Total external development expense |
|
|
1,490 |
|
|
|
1,080 |
|
|
|
1,016 |
|
|
R&D personnel expense |
|
|
2,264 |
|
|
|
1,951 |
|
|
|
1,404 |
|
|
R&D facility expense |
|
|
57 |
|
|
|
57 |
|
|
|
55 |
|
|
Patent expense |
|
|
311 |
|
|
|
369 |
|
|
|
356 |
|
|
Other R&D expense |
|
|
102 |
|
|
|
62 |
|
|
|
186 |
|
|
Total research, development and patent expense |
|
$ |
4,224 |
|
|
$ |
3,519 |
|
|
$ |
3,017 |
|
Our goal is to build a sustainable biopharmaceutical business through the successful development of differentiated products for the treatment of serious diseases with unmet medical needs in high-value therapeutic areas. Key elements of our strategy are as follows:
|
•Pursue the development of MN-166 (ibudilast) for multiple potential indications with the support of non-dilutive financings. |
We intend to advance our diverse MN-166 (ibudilast) program through a combination of investigator-sponsored clinical trials, trials funded through government grants or other grants, and trials funded by us. In addition to providing drug supply and regulatory support, we are funding portions of the consortium-sponsored trials. For example, we contributed financially to the Secondary and Primary Progressive Ibudilast NeuroNEXT Trial in Multiple Sclerosis (SPRINT-MS) Phase 2b clinical trial of MN-166 (ibudilast) for the treatment of progressive MS, which was primarily funded by the NIH. In addition, we contributed financially to the clinical trial of MN-166 (ibudilast) for the treatment of ALS as well as the ongoing ALS / Biomarker study. We intend to pursue additional strategic alliances to help support further clinical development of MN-166 (ibudilast).
|
•Pursue the development of MN-001 (tipelukast) for fibrotic diseases such as NASH and IPF. |
We intend to advance development of MN-001 (tipelukast) through a variety of means, which may include investigator-sponsored trials with or without grant funding as well as trials funded by us.
51
|
•Consider strategic partnerships with one or more leading pharmaceutical companies to complete late-stage product development and successfully commercialize our products. |
We develop and maintain relationships with pharmaceutical companies that are therapeutic category leaders. Upon completion of proof-of-concept Phase 2 clinical trials, we intend to discuss strategic alliances with leading pharmaceutical companies who seek late-stage product candidates, such as MN-166, MN-001, MN-221 and MN-029, which could support further clinical development and product commercialization.
General and Administrative
Our general and administrative costs primarily consist of salaries, benefits and consulting and professional fees related to our administrative, finance, human resources, business development, legal, information systems support functions, facilities and insurance costs. General and administrative costs are expensed as incurred.
Our general and administrative expenses may increase in future periods if we are required to expand our infrastructure based on the success of our product development programs and in raising capital to support our product development programs or otherwise in connection with increased business development activities related to partnering, out-licensing or product disposition.
Other Income and Expense
Other income primarily consists of interest earned on our cash and cash equivalents. In 2017, 2016 and 2015, other expense primarily consists of losses from the joint venture and net foreign exchange losses related to vendor invoices denominated in foreign currencies.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of the consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses and the related disclosure of contingent liabilities. We review our estimates on an ongoing basis, including those related to our significant accruals. We base our estimates on historical experience and on various other assumptions that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates.
Our significant accounting policies are more fully described in Note 1 to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K. Our most critical accounting estimates include research, development and patent expenses which impacts operating expenses and accrued liabilities, stock-based compensation which impacts operating expenses and goodwill and purchased intangibles. We review our estimates and assumptions periodically and reflect the effects of revisions in the period in which they are deemed to be necessary. We believe that the following accounting policies are critical to the judgments and estimates used in preparation of our consolidated financial statements.
Research, Development and Patent Expenses
Research, development and patent costs are expensed as incurred based on certain contractual factors such as estimates of work performed, milestones achieved, patient enrollment and experience with similar contracts. As actual costs become known, accruals are adjusted. To date, our accrued research, development and patent expenses have not differed significantly from the actual expenses incurred.
Stock-based Compensation
We grant options to purchase our common stock to our employees and directors under our 2013 Stock Incentive Plan. Additionally, we have outstanding stock options that were granted under our Amended and Restated 2004 Stock Incentive Plan. Under our 2007 Employee Stock Purchase Plan, full-time employees are permitted to
52
purchase common stock through payroll deduction at the lower of 85% of fair market value at the beginning of the offering period or the end of each six-month offering period. The benefits provided under all of these plans requires stock-based compensation for an award of equity instruments, including stock options and employee stock purchase rights issued to employees to be recognized as a cost in the consolidated financial statements. The cost of these awards is measured according to the grant date fair value of the stock award and is recognized on a straight-line basis over the period during which an employee is required to provide service in exchange for the award, which is usually the vesting period. We occasionally issue employee performance-based stock options, the vesting of which is subsequently based on a determination made by our board of directors as to the achievement of certain corporate objectives. The grant date of such awards is the date on which our board of directors makes its determination. For periods preceding the grant date, the cost of these awards is measured according to their fair value at each reporting date.
Valuation of our stock option grants requires us to estimate certain variables, such as estimated volatility and expected life. If any of our estimations change, such changes could have a significant impact on the stock-based compensation amount we recognize.
Goodwill and Purchased Intangibles
Goodwill is recorded when the consideration paid for an acquisition exceeds the fair value of the identified net tangible and intangible assets of acquired businesses. The allocation of purchase price for acquisitions require extensive use of accounting estimates and judgments to allocate the purchase price to the identifiable tangible and intangible assets acquired and liabilities assumed based on their respective fair values. Additionally, we must determine whether an acquired entity is considered to be a business or a set of net assets as a portion of the purchase price can only be allocated to goodwill in a business combination. Goodwill and intangible assets deemed to have indefinite lives are not amortized, but are subject to annual impairment tests. The amounts and useful lives assigned to intangible assets that have finite useful lives require the use of estimates and the exercise of judgment. These judgments can significantly affect our net operating results. Goodwill and In-Process Research and Development, or IPR&D are considered to have indefinite lives and are carried at cost. As of December 31, 2017 and 2016, we have goodwill and IPR&D, of $9.6 million and $4.8 million, respectively.
At least annually in the fourth quarter, or more frequently if indicators of impairment exist, we complete an impairment test for goodwill and purchased indefinite life intangibles. The impairment evaluation is performed assuming that the Company operates in a single operating segment and reporting unit. When impaired, the carrying value of goodwill is written down to fair value. The goodwill impairment test involves consideration of qualitative information to determine if it is more likely than not, that the fair value of a reporting unit is less than its carrying value. If such a determination is made, then the traditional two-step goodwill impairment test is applied. The first step, identifying a potential impairment, compares the fair value of a reporting unit with its carrying amount, including goodwill. If the carrying value of the reporting unit exceeds its fair value, the second step would need to be conducted; otherwise, no further steps are necessary as no potential impairment exists. The second step, measuring the impairment loss, compares the implied fair value of the reporting unit goodwill with the carrying amount of that goodwill. Any excess of the reporting unit goodwill carrying value over the respective implied fair value is recognized as an impairment loss. There was no impairment of goodwill for all periods presented.
We periodically re-evaluate the original assumptions and rationale utilized in the establishment of the carrying value and estimated lives of our long-lived assets. The criteria used for these evaluations include management’s estimate of the asset’s continuing ability to generate income from operations and positive cash flows in future periods as well as the strategic significance of any intangible assets in our business objectives. If assets are considered to be impaired, the impairment recognized is the amount by which the carrying value of the assets exceeds the fair value of the assets.
Recent Accounting Pronouncements
The impact of recent accounting pronouncements is more fully described in Note 1 of our consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
53
Comparison of the Years ended December 31, 2017 and 2016
Revenues
We did not recognize any revenue for the years ended December 31, 2017 and 2016.
Research, Development and Patent Expenses
Research, development and patent expenses for the year ended December 31, 2017 increased by $0.7 million as compared to the same period in 2016, primarily due to an increase in clinical trial activities related to the MN-001 and MN-166 trials as well as higher stock compensation expense for performance-based stock options.
General and Administrative
General and administrative expenses for the year ended December 31, 2017 increased by $1.4 million compared to the same period in 2016, primarily driven by higher stock compensation expense for performance-based stock options as well as an increase in legal fees related to the Company’s SEC filings and other legal matters.
Other Expense
Other expense for the years ended 2017 and 2016 was approximately $25,000 and $47,000, respectively. Other expense consisted of losses from the joint venture accounted for under the equity method according to our percentage ownership, and net transaction losses related to vendor invoices denominated in foreign currencies.
Other Income
Other income for the year ended December 31, 2017 was approximately $146,000, as compared to approximately $67,000 for the same period in 2016. Other income consisted of interest income on our cash and cash equivalents in 2017 and 2016.
Comparison of the Years ended December 31, 2016 and 2015
Revenues
We did not recognize any revenue for the years ended December 31, 2016 and 2015.
Research, Development and Patent Expenses
Research, development and patent expenses for the year ended December 31, 2016 increased by $0.5 million compared to the same period in 2015, primarily due to higher stock compensation expense for performance-based stock options due in part to an increase in our stock price.
General and Administrative
General and administrative expenses for the year ended December 31, 2016 increased by $1.6 million compared to the same period in 2015 primarily due to higher stock compensation expense for performance-based stock options due to an increase in our stock price as well as an increase in legal fees related to corporate matters.
Other Expense
Other expense for the years ended 2016 and 2015 was approximately $47,000 and $54,000, respectively. Other expense consisted of losses from the joint venture accounted for under the equity method according to our percentage ownership, and net transaction losses related to vendor invoices denominated in foreign currencies.
Other Income
54
Other income for the year ended December 31, 2016 was approximately $67,000, as compared to approximately $39,000 for the same period in 2015. Other income consisted of interest income on our cash and cash equivalents in 2016 and 2015.
Liquidity and Capital Resources
We incurred losses of $11.2 million, $10.9 million, and $8.8 million for the years ended December 31, 2017, 2016, and 2015, respectively. At December 31, 2017, our accumulated deficit was $341.5 million. Our operating losses to date have been funded primarily through the private placement of our equity securities, the public sale of our common stock, long-term debt, development agreements with partners and the exercise of founder’s warrants, net of treasury stock repurchases.
The following table shows a summary of our cash flows for the years ended December 31:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net cash (used in) provided by: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
|
(6,924 |
) |
|
|
(6,546 |
) |
|
|
(7,152 |
) |
|
Investing activities |
|
|
— |
|
|
|
(84 |
) |
|
|
(2 |
) |
|
Financing activities |
|
|
10,797 |
|
|
|
8,666 |
|
|
|
17,564 |
|
|
Total |
|
$ |
3,873 |
|
|
$ |
2,036 |
|
|
$ |
10,410 |
|
Equity Financing
On May 22, 2015, we entered into an at-the-market issuance sales agreement (the “ATM Agreement”) with MLV & Co. LLC, or MLV, pursuant to which we may sell common stock through MLV from time to time up to an aggregate offering price of $30.0 million. Sales of our common stock through MLV, if any, will be made by any method that is deemed to be an “at-the-market” equity offering as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including sales made directly on NASDAQ, on any other existing trading market for the common stock or to or through a market maker. MLV may also sell the common stock in privately negotiated transactions, subject to our prior approval. We agreed to pay MLV an aggregate commission rate of up to 4.0% of the gross proceeds of any common stock sold under this agreement. Proceeds from sales of common stock will depend on the number of shares of common stock sold to MLV and the per share purchase price of each transaction. We are not obligated to make any sales of common stock under the sales agreement and may terminate the sales agreement at any time upon written notice.
On August 24, 2015, we completed a firm-commitment underwritten public offering of 5,000,000 shares of common stock at a purchase price of $3.50 per share for gross proceeds of $17.5 million, and received net proceeds of approximately $16.0 million, net of underwriting discounts and commissions and offering expenses.
On September 16, 2016, we entered into an amendment No. 1 to the ATM Agreement with MLV to also include FBR Capital Markets & Co (“FBR”) as a sales agent.
For the year ended December 31, 2017, we generated gross and net proceeds of $10.3 million and $9.9 million, respectively, on sales of 1,689,436 shares of our common stock at prices ranging from $5.30 to $6.98 per share. For the year ended December 31, 2016, we generated gross and net proceeds of $264,000 and $159,529, respectively, on sales of 36,248 shares of our common stock at prices ranging from $6.90 to $7.54 per share.
Warrants
During the years ended December 31, 2017 and 2016, warrants to purchase 119,047 and 2,131,700 shares were exercised for gross proceeds of $0.4 million and $7.6 million respectively. On May 10, 2017, warrants to purchase 198,020 shares expired unexercised.
As of December 31, 2017, we have 750,000 common stock warrants outstanding at an exercise price of $3.15 per share, which expire on May 9, 2018.
55
Factors That May Affect Future Financial Condition and Liquidity
As of December 31, 2017, we had available cash and cash equivalents of $28.0 million and working capital of $25.4 million. As of the date of this report, we believe we have working capital sufficient to fund operations through the end of 2019.
Our future funding requirements will depend on many factors, including, but not limited to:
|
|
• |
progress in, and the costs of, future planned clinical trials and other research and development activities; |
|
|
• |
the scope, prioritization and number of our product development programs; |
|
|
• |
our obligations under our license agreements, pursuant to which we may be required to make future milestone payments upon the achievement of various milestones related to clinical, regulatory or commercial events; |
|
|
• |
our ability to establish and maintain strategic collaborations, including licensing and other arrangements, and to complete acquisitions of additional product candidates; |
|
|
• |
the time and costs involved in obtaining regulatory approvals; |
|
|
• |
the costs of securing manufacturing arrangements for clinical or commercial production of our product candidates; |
|
|
• |
the costs associated with any expansion of our management, personnel, systems and facilities; |
|
|
• |
the costs associated with any litigation; |
|
|
• |
the costs associated with the operations or wind-down of any business we may acquire; |
|
|
• |
the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; and |
|
|
• |
the costs of establishing or contracting for sales and marketing capabilities and commercialization activities if we obtain regulatory approval to market our product candidates. |
Other Significant Contractual Obligations
The following summarizes our scheduled long-term contractual obligations that may affect our future liquidity as of December 31, 2017 (in thousands):
|
Contractual Obligations |
|
Total |
|
|
Less than 1 Year |
|
|
1-3 Years |
|
|
3-5 Years |
|
|
More than 5 Years |
|
|||||
|
Operating leases |
|
$ |
690 |
|
|
$ |
211 |
|
|
$ |
479 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Research and development services(1) |
|
|
2,351 |
|
|
|
— |
|
|
|
2,351 |
|
|
|
— |
|
|
|
— |
|
|
Total(2) |
|
$ |
3,041 |
|
|
$ |
211 |
|
|
$ |
2,830 |
|
|
$ |
— |
|
|
$ |
— |
|
|
(1) |
In October 2011, we entered into an agreement with Kissei to perform research and development services relating to MN-221 in exchange for a non-refundable upfront payment of $2.5 million. We are responsible for all costs to be incurred in the performance of these services. The estimated remaining costs to be incurred in the performance of all such remaining services are included above. |
|
(2) |
We also enter into agreements with third parties to conduct our clinical trials, manufacture our product candidates, and perform data collection, analysis and other services in connection with our product development programs. As our payment obligations under these agreements depend upon the progress of our product development programs, we are unable at this time to estimate the future costs we might incur under these agreements. |
56
Off-Balance Sheet Arrangements
At December 31, 2017, we did not have any relationship with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance variable interest, or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we did not engage in trading activities involving non-exchange traded contracts. As a result, we are not exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships. We do not have relationships and transactions with persons and entities that derive benefits from their non-independent relationship with us or our related parties except as disclosed herein.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable.
Item 8. Financial Statements and Supplementary Data
57
Report of Independent Registered Public Accounting Firm
Shareholders and Board of Directors
MediciNova, Inc.
La Jolla, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of MediciNova, Inc. (the “Company”) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and our report dated February 13, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ BDO USA, LLP
We have served as the Company's auditor since 2015.
San Diego, California
February 13, 2018
58
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Assets: |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
27,991,743 |
|
|
$ |
24,118,037 |
|
|
Prepaid expense and other current assets |
|
|
336,580 |
|
|
|
585,810 |
|
|
Total current assets |
|
|
28,328,323 |
|
|
|
24,703,847 |
|
|
Goodwill |
|
|
9,600,240 |
|
|
|
9,600,240 |
|
|
In-process research and development |
|
|
4,800,000 |
|
|
|
4,800,000 |
|
|
Investment in joint venture |
|
|
616,657 |
|
|
|
618,330 |
|
|
Property and equipment, net |
|
|
62,886 |
|
|
|
90,717 |
|
|
Other long-term assets |
|
|
10,958 |
|
|
|
— |
|
|
Total assets |
|
$ |
43,419,064 |
|
|
$ |
39,813,134 |
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
1,520,225 |
|
|
$ |
367,275 |
|
|
Accrued liabilities |
|
|
1,360,744 |
|
|
|
1,262,800 |
|
|
Total current liabilities |
|
|
2,880,969 |
|
|
|
1,630,075 |
|
|
Long-term deferred rent and lease liability |
|
|
— |
|
|
|
967 |
|
|
Deferred tax liability |
|
|
201,792 |
|
|
|
1,956,000 |
|
|
Long-term deferred revenue |
|
|
1,694,163 |
|
|
|
1,694,163 |
|
|
Total liabilities |
|
|
4,776,924 |
|
|
|
5,281,205 |
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Common stock, $0.001 par value; 100,000,000 shares authorized at December 31, 2017 and December 31, 2016; 36,452,893 and 34,523,678 shares issued and outstanding at December 31, 2017 and December 31, 2016, respectively |
|
|
36,453 |
|
|
|
34,525 |
|
|
Additional paid-in capital |
|
|
380,156,510 |
|
|
|
364,886,468 |
|
|
Accumulated other comprehensive loss |
|
|
(94,623 |
) |
|
|
(96,000 |
) |
|
Accumulated deficit |
|
|
(341,456,200 |
) |
|
|
(330,293,064 |
) |
|
Total stockholders’ equity |
|
|
38,642,140 |
|
|
|
34,531,929 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
43,419,064 |
|
|
$ |
39,813,134 |
|
See accompanying notes to consolidated financial statements.
59
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
|
|
|
Years ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and patent |
|
$ |
4,223,746 |
|
|
$ |
3,519,172 |
|
|
$ |
3,017,169 |
|
|
General and administrative |
|
|
8,803,347 |
|
|
|
7,362,662 |
|
|
|
5,805,217 |
|
|
Total operating expenses |
|
|
13,027,093 |
|
|
|
10,881,834 |
|
|
|
8,822,386 |
|
|
Operating loss |
|
|
(13,027,093 |
) |
|
|
(10,881,834 |
) |
|
|
(8,822,386 |
) |
|
Other expense |
|
|
(25,303 |
) |
|
|
(46,584 |
) |
|
|
(54,206 |
) |
|
Interest expense |
|
|
(298 |
) |
|
|
(454 |
) |
|
|
(514 |
) |
|
Other income |
|
|
145,508 |
|
|
|
66,647 |
|
|
|
39,386 |
|
|
Loss before income taxes |
|
|
(12,907,186 |
) |
|
|
(10,862,225 |
) |
|
|
(8,837,720 |
) |
|
Income tax benefit (expense) |
|
|
1,744,050 |
|
|
|
(3,754 |
) |
|
|
(7,359 |
) |
|
Net loss applicable to common stockholders |
|
$ |
(11,163,136 |
) |
|
$ |
(10,865,979 |
) |
|
$ |
(8,845,079 |
) |
|
Basic and diluted net loss per common share |
|
$ |
(0.32 |
) |
|
$ |
(0.33 |
) |
|
$ |
(0.33 |
) |
|
Shares used to compute basic and diluted net loss per share |
|
|
35,137,028 |
|
|
|
32,986,740 |
|
|
|
26,578,770 |
|
|
Net loss applicable to common stockholders |
|
$ |
(11,163,136 |
) |
|
$ |
(10,865,979 |
) |
|
$ |
(8,845,079 |
) |
|
Other comprehensive income (loss), net of tax: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation adjustments |
|
|
1,377 |
|
|
|
6,765 |
|
|
|
(1,788 |
) |
|
Comprehensive loss |
|
$ |
(11,161,759 |
) |
|
$ |
(10,859,214 |
) |
|
$ |
(8,846,867 |
) |
See accompanying notes to consolidated financial statements.
60
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
|
|
|
Preferred stock |
|
|
Common stock |
|
|
Additional paid-in |
|
|
Accumulated other comprehensive |
|
|
Accumulated |
|
|
Total stockholders’ |
|
||||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
capital |
|
|
income (loss) |
|
|
deficit |
|
|
equity |
|
||||||||
|
Balance at December 31, 2014 |
|
|
220,000 |
|
|
$ |
2,200 |
|
|
|
24,436,317 |
|
|
$ |
24,437 |
|
|
$ |
332,666,935 |
|
|
$ |
(100,977 |
) |
|
$ |
(310,582,006 |
) |
|
$ |
22,010,589 |
|
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,025,500 |
|
|
|
— |
|
|
|
— |
|
|
|
2,025,500 |
|
|
Issuance of shares under an employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
35,178 |
|
|
|
35 |
|
|
|
89,874 |
|
|
|
— |
|
|
|
— |
|
|
|
89,909 |
|
|
Issuance of common stock under at-the-market equity distribution and sales agreements |
|
|
— |
|
|
|
— |
|
|
|
232,800 |
|
|
|
233 |
|
|
|
607,295 |
|
|
|
— |
|
|
|
— |
|
|
|
607,528 |
|
|
Issuance of common stock, net of offering costs |
|
|
— |
|
|
|
— |
|
|
|
5,000,000 |
|
|
|
5,000 |
|
|
|
15,988,683 |
|
|
|
— |
|
|
|
— |
|
|
|
15,993,683 |
|
|
Exercise of warrants |
|
|
— |
|
|
|
— |
|
|
|
252,200 |
|
|
|
252 |
|
|
|
872,380 |
|
|
|
— |
|
|
|
— |
|
|
|
872,632 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(8,845,079 |
) |
|
|
(8,845,079 |
) |
|
Foreign currency translation adjustments |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(1,788 |
) |
|
|
|
|
|
|
(1,788 |
) |
|
Balance at December 31, 2015 |
|
|
220,000 |
|
|
|
2,200 |
|
|
|
29,956,495 |
|
|
|
29,957 |
|
|
|
352,250,667 |
|
|
|
(102,765 |
) |
|
|
(319,427,085 |
) |
|
|
32,752,974 |
|
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,972,533 |
|
|
|
— |
|
|
|
— |
|
|
|
3,972,533 |
|
|
Issuance of shares under an employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
26,650 |
|
|
|
27 |
|
|
|
87,702 |
|
|
|
— |
|
|
|
— |
|
|
|
87,729 |
|
|
Issuance of common stock under at-the-market equity distribution and sales agreements, net of offering costs |
|
|
— |
|
|
|
— |
|
|
|
36,248 |
|
|
|
36 |
|
|
|
159,493 |
|
|
|
— |
|
|
|
— |
|
|
|
159,529 |
|
|
Conversion of preferred stock to common stock |
|
|
(220,000 |
) |
|
|
(2,200 |
) |
|
|
2,200,000 |
|
|
|
2,200 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Issuance of common stock for option exercises |
|
|
|
|
|
|
|
|
|
|
172,585 |
|
|
|
173 |
|
|
|
829,353 |
|
|
|
— |
|
|
|
— |
|
|
|
829,526 |
|
|
Exercise of warrants |
|
|
— |
|
|
|
— |
|
|
|
2,131,700 |
|
|
|
2,132 |
|
|
|
7,586,720 |
|
|
|
— |
|
|
|
— |
|
|
|
7,588,852 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(10,865,979 |
) |
|
|
(10,865,979 |
) |
|
Foreign currency translation adjustments |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
6,765 |
|
|
|
— |
|
|
|
6,765 |
|
|
Balance at December 31, 2016 |
|
|
— |
|
|
|
— |
|
|
|
34,523,678 |
|
|
|
34,525 |
|
|
|
364,886,468 |
|
|
|
(96,000 |
) |
|
|
(330,293,064 |
) |
|
|
34,531,929 |
|
|
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
4,474,945 |
|
|
|
— |
|
|
|
— |
|
|
|
4,474,945 |
|
|
Issuance of shares under an employee stock purchase plan |
|
|
— |
|
|
|
— |
|
|
|
15,153 |
|
|
|
15 |
|
|
|
77,304 |
|
|
|
— |
|
|
|
— |
|
|
|
77,319 |
|
|
Issuance of common stock under at-the-market equity distribution and sales agreements, net of offering costs |
|
|
— |
|
|
|
— |
|
|
|
1,689,436 |
|
|
|
1,689 |
|
|
|
9,889,626 |
|
|
|
— |
|
|
|
— |
|
|
|
9,891,315 |
|
|
Issuance of common stock for option exercises |
|
|
|
|
|
|
|
|
|
|
105,579 |
|
|
|
105 |
|
|
|
425,908 |
|
|
|
— |
|
|
|
— |
|
|
|
426,013 |
|
|
Exercise of warrants |
|
|
— |
|
|
|
— |
|
|
|
119,047 |
|
|
|
119 |
|
|
|
402,259 |
|
|
|
— |
|
|
|
— |
|
|
|
402,378 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(11,163,136 |
) |
|
|
(11,163,136 |
) |
|
Foreign currency translation adjustments |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,377 |
|
|
|
|
|
|
|
1,377 |
|
|
Balance at December 31, 2017 |
|
|
— |
|
|
$ |
— |
|
|
|
36,452,893 |
|
|
$ |
36,453 |
|
|
$ |
380,156,510 |
|
|
$ |
(94,623 |
) |
|
$ |
(341,456,200 |
) |
|
$ |
38,642,140 |
|
See accompanying notes to consolidated financial statements.
61
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
|
Years ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(11,163,136 |
) |
|
$ |
(10,865,979 |
) |
|
$ |
(8,845,079 |
) |
|
Adjustments to reconcile net loss to net cash (used in) provided by operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash share-based compensation |
|
|
4,474,945 |
|
|
|
3,972,533 |
|
|
|
2,025,500 |
|
|
Depreciation and amortization |
|
|
28,098 |
|
|
|
14,127 |
|
|
|
26,704 |
|
|
Tax benefit from fluctuations in other comprehensive income |
|
|
— |
|
|
|
(1,901 |
) |
|
|
— |
|
|
Change in equity method investment |
|
|
1,673 |
|
|
|
32,139 |
|
|
|
34,319 |
|
|
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Receivables, prepaid expenses and other assets |
|
|
242,064 |
|
|
|
175,495 |
|
|
|
(284,538 |
) |
|
Accounts payable, accrued liabilities and other current liabilities |
|
|
1,246,630 |
|
|
|
127,373 |
|
|
|
(109,276 |
) |
|
Deferred tax liability, deferred revenue and other long term liabilities |
|
|
(1,754,208 |
) |
|
|
— |
|
|
|
— |
|
|
Net cash used in operating activities |
|
|
(6,923,934 |
) |
|
|
(6,546,213 |
) |
|
|
(7,152,370 |
) |
|
Investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Acquisitions of property and equipment |
|
|
— |
|
|
|
(84,483 |
) |
|
|
(2,320 |
) |
|
Net cash provided by (used) in investing activities |
|
|
— |
|
|
|
(84,483 |
) |
|
|
(2,320 |
) |
|
Financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock, exercise of common stock options and warrants, net of issuance costs |
|
|
10,719,707 |
|
|
|
8,577,907 |
|
|
|
17,473,843 |
|
|
Proceeds from issuance of equity under ESPP |
|
|
77,319 |
|
|
|
87,729 |
|
|
|
89,909 |
|
|
Net cash provided by financing activities |
|
|
10,797,026 |
|
|
|
8,665,636 |
|
|
|
17,563,752 |
|
|
Effects of foreign exchange rates on cash |
|
|
614 |
|
|
|
6,348 |
|
|
|
(1,748 |
) |
|
Net increase in cash and cash equivalents |
|
|
3,873,706 |
|
|
|
2,041,288 |
|
|
|
10,407,314 |
|
|
Cash and cash equivalents, beginning of period |
|
|
24,118,037 |
|
|
|
22,076,749 |
|
|
|
11,669,435 |
|
|
Cash and cash equivalents, end of period |
|
$ |
27,991,743 |
|
|
$ |
24,118,037 |
|
|
$ |
22,076,749 |
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Income taxes paid |
|
$ |
9,203 |
|
|
$ |
6,035 |
|
|
$ |
7,443 |
|
See accompanying notes to consolidated financial statements.
62
Notes to Consolidated Financial Statements
1. Organization and Summary of Significant Accounting Policies
Organization and Business
The Company was incorporated in the state of Delaware in September 2000 and is a public company. The Company’s common stock is listed in both the United States and Japan and trades on The NASDAQ Global Market and the JASDAQ Market of the Tokyo Stock Exchange. The Company is a biopharmaceutical company focused on acquiring and developing novel, small molecule therapeutics for the treatment of serious diseases with unmet medical needs with a commercial focus on the United States market. The Company’s current strategy is to focus its development activities on MN-166 (ibudilast) for neurological disorders such as progressive multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and substance dependence and addiction (e.g., methamphetamine dependence, opioid dependence, and alcohol dependence), and MN-001 (tipelukast) for fibrotic diseases such as nonalcoholic steatohepatitis (NASH) and idiopathic pulmonary fibrosis (IPF). The Company’s pipeline also includes MN-221 (bedoradrine) for the treatment of acute exacerbation of asthma and MN-029 (denibulin) for solid tumor cancers.
As of December 31, 2017, the Company had available cash and cash equivalents of $28.0 million and working capital of $25.4 million.
Principles of Consolidation
The consolidated financial statements include the accounts of MediciNova, Inc. and its wholly-owned subsidiaries MediciNova (Europe) Limited, MediciNova Japan, Inc. and Avigen, Inc. All intercompany transactions and balances are eliminated in consolidation. MediciNova (Europe) Limited was incorporated under the laws of England in 2006. As of December 31, 2017, there have been no significant transactions related to MediciNova (Europe) Limited. MediciNova Japan, Inc. was incorporated in Japan in 2007.
Segment Reporting
The Company operates in a single operating segment – the acquisition and development of small molecule therapeutics for the treatment of serious diseases with unmet medical needs.
Use of Estimates
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”). The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and other highly liquid investments with original maturities of three months or less from the date of purchase. Cash equivalents at December 31, 2017 consisted of money market funds.
Concentrations and Credit Risk
The Company maintains cash balances at various financial institutions and such balances commonly exceed the $250,000 amount insured by the Federal Deposit Insurance Corporation. The Company also maintains money market funds at various financial institutions which are not federally insured although are invested primarily in U.S.
63
government securities. The Company has not experienced any losses in such accounts and management believes that the Company does not have significant credit risk with respect to such cash and cash equivalents.
Fair Value of Financial Instruments
Financial instruments, including cash and cash equivalents, prepaid expenses and other current assets, accounts payable and accrued liabilities, are carried at cost, which management believes approximates fair value because of the short-term maturity of these instruments.
Goodwill and Purchased Intangibles
The Company records goodwill and other intangible assets based on the fair value of the assets acquired. In determining the fair value of the assets acquired, the Company utilizes extensive accounting estimates and judgments to allocate the purchase price to the fair value of the net tangible and intangible assets acquired. The Company uses the discounted cash flow method to estimate the value of intangible assets acquired.
The Company assesses goodwill and indefinite lived intangible assets for impairment using fair value measurement techniques on an annual basis during the fourth quarter of the year, or more frequently if indicators of impairment exist. The impairment evaluation is performed assuming that the Company operates in a single operating segment and reporting unit. When impaired, the carrying value of goodwill is written down to fair value. The goodwill impairment test involves consideration of qualitative information to determine if it is more likely than not that the fair value of a reporting unit is less than its carrying value. If such a determination is made, then the traditional two-step goodwill impairment test is applied. The first step, identifying a potential impairment, compares the fair value of a reporting unit with its carrying amount, including goodwill. If the carrying value of the reporting unit exceeds its fair value, the second step would need to be conducted; otherwise, no further steps are necessary as no potential impairment exists. The second step, measuring the impairment loss, compares the implied fair value of the reporting unit goodwill with the carrying amount of that goodwill. Any excess of the reporting unit goodwill carrying value over the respective implied fair value is recognized as an impairment loss. There was no impairment of goodwill for all periods presented.
The Company periodically re-evaluates the original assumptions and rationale utilized in the establishment of the carrying value and estimated lives of its long-lived assets. The criteria used for these evaluations include management’s estimate of the asset’s continuing ability to generate income from operations and positive cash flows in future periods as well as the strategic significance of any intangible assets in the Company’s business objectives. If assets are considered to be impaired, the impairment recognized is the amount by which the carrying value of the assets exceeds the fair value of the assets. There was no impairment of long-lived assets for the periods presented.
Research, Development and Patents
Research and development costs are expensed in the period incurred. Research and development costs primarily consist of salaries and related expenses for personnel, facilities and depreciation, research and development supplies, licenses and outside services. Such research and development costs totaled $3.9 million, $3.1 million and $2.7 million for the years ended December 31, 2017, 2016 and 2015, respectively.
Costs related to filing and pursuing patent applications are expensed as incurred, as recoverability of such expenditures is uncertain. The Company includes all external costs related to the filing of patents on developments in Research, Development and Patents expenses. Such patent-related expenses totaled $0.3 million, $0.4 million and $0.3 million for the years ended December 31, 2017, 2016, and 2015, respectively.
Share-Based Compensation
The Company estimates the fair value of stock options using the Black-Scholes option pricing model on the date of grant. The fair value of equity instruments expected to vest are recognized and amortized on a straight-line
64
basis over the requisite service period of the award, which is generally three to four years; however, the Company’s equity compensation plans provide for any vesting schedule as the board may deem appropriate.
Net Loss Per Share
The Company computes basic net loss per share using the weighted average number of common shares outstanding during the period. Diluted net income per share is based upon the weighted average number of common shares and potentially dilutive securities (common share equivalents) outstanding during the period. Common share equivalents outstanding, determined using the treasury stock method, are comprised of shares that may be issued under the Company’s stock option agreements and warrants. Common share equivalents were excluded from the diluted net loss per share calculation because of their anti-dilutive effect for all periods presented.
Potentially dilutive outstanding securities excluded from diluted net loss per common share because of their anti-dilutive effect for the periods presented are as follows:
|
|
|
December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Convertible preferred stock, as converted |
|
|
— |
|
|
|
— |
|
|
|
2,200,000 |
|
|
Stock options |
|
|
5,514,038 |
|
|
|
4,432,017 |
|
|
|
4,133,969 |
|
|
Warrants |
|
|
750,000 |
|
|
|
1,067,067 |
|
|
|
3,406,367 |
|
|
Total |
|
|
6,264,038 |
|
|
|
5,499,084 |
|
|
|
9,740,336 |
|
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) amended the existing accounting standards for revenue recognition. The amendments are based on the principle that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for said goods or services. The Company is required to adopt the amendments beginning January 1, 2018. The amendments may be applied retrospectively to each prior period presented or retrospectively with the cumulative effect recognized as of the date of initial application (“modified retrospective method”). We have adopted the standard as of January 1, 2018 and determined that there will be no impact on our financial statements as we currently do not have any revenue contracts within the scope of the new guidance. We will continue to account for our agreement to perform research and development services for Kissei Pharmaceutical Co., Ltd as a collaborative arrangement with no change to the method of recognizing revenues for this contract.
In February 2016, the FASB issued Accounting Standards Update (“ASU”) 2016-02, Leases, which introduces the recognition of lease assets and lease liabilities by lessees for those leases classified as operating leases under previous guidance. The new standard establishes a right-of-use ("ROU") model that requires a lessee to record an ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. The new standard is effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years with early adoption permitted. We are evaluating the impact that the adoption of this standard will have on our financial statements.
In March 2016, the FASB issued Accounting Standard Update No. 2016-09, "Improvements to Employee Share-Based Payment Accounting."Current GAAP requires an entity to recognize excess tax benefit or deficiency as additional paid-in capital. To simplify the presentation of stock compensation, the amendments in this Update require that the excess tax benefit or deficiency is recognized as expense. For public business entities, the amendments in this update are effective for financial statements issued for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The FASB further provides that this Update may be applied on a modified retrospective basis to all periods presented. The Company chose to adopt the update prospectively beginning in the year ended December 31, 2017. Prior periods have not been retrospectively adjusted, and given the Company's full valuation position there is no quantitative impact to the financial statements.
65
In November 2015, the FASB issued Accounting Standard Update No. 2015-17, "Balance Sheet Classification of Deferred Taxes", an update to ASC 740, Income Taxes. Current GAAP requires an entity to separate deferred income tax liabilities and assets into current and noncurrent amounts in a classified statement of financial position. To simplify the presentation of deferred income taxes, the amendments in this Update require that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. The current requirement that deferred tax liabilities and assets of a tax-paying component of an entity be offset and presented as a single amount is not affected by the amendments in this Update. For public business entities, the amendments in this update are effective for financial statements issued for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The FASB further provides that this update may be applied to all deferred tax liabilities and assets prospectively to all periods presented. The Company has chosen to adopt the update prospectively for the year ended December 31, 2017 and thereafter.
2. Revenue Recognition
Revenue Recognition Policy
Revenues consist of milestone payments and research and development services. Milestone payments are recognized as revenue upon achievement of pre-defined scientific events, which require substantive effort, and for which achievement of the milestone was not readily assured at the inception of the agreement. Milestones that do not meet the criteria for accounting under the milestone method because the payments are solely contingent upon the performance of a third party are accounted for as contingent revenue. Research and development services are recognized as research costs are incurred over the period the services are performed. For all other revenue the Company recognizes revenues when all four of the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products and/or services has occurred; (3) the selling price is fixed or determinable; and (4) collectability is reasonably assured.
Kissei Pharmaceutical Co., Ltd
In October 2011, the Company entered into a collaboration agreement with Kissei Pharmaceutical Co., Ltd., or Kissei, to perform research and development services relating to MN-221 in exchange for a non-refundable upfront payment of $2.5 million. Under the terms of the agreement the Company is responsible for all costs to be incurred in the performance of these services. Certain of these research and development services were completed in 2013 and 2012, and the remaining services are expected to be delivered and completed after 2018. The Company assessed the deliverables in accordance with the authoritative guidance and concluded the existence of one deliverable, research and development services. As such, revenue is being recognized as the research and development services are performed. The amount received from Kissei, net of the amount recorded as revenue, is included on the balance sheet as long-term deferred revenue and will be recognized as revenue as the remaining services are performed. No revenue was recorded in 2017, 2016 or 2015 in connection with the collaboration agreement with Kissei.
3. Fair Value Measurements
Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability. As a basis for considering such assumptions, a three-tier fair value hierarchy has been established, which prioritizes the inputs used in measuring fair value as follows:
|
Level 1: |
|
Observable inputs such as quoted prices in active markets; |
|
|
|
|
|
Level 2: |
|
Inputs are quoted prices for similar items in active markets or inputs are quoted prices for identical or similar items in markets that are not active near the measurement date; and |
|
|
|
|
|
Level 3: |
|
Unobservable inputs due to little or no market data, which require the reporting entity to develop its own assumptions |
66
Cash equivalents including money market accounts of $666,265 and $661,287 measured at fair value as of December 31, 2017 and 2016, respectively, are classified within Level 1.
4. Balance Sheet Details
Property and Equipment
Property and equipment, net, consist of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Leasehold improvements |
|
$ |
15,742 |
|
|
$ |
20,157 |
|
|
Furniture and equipment |
|
|
233,441 |
|
|
|
284,585 |
|
|
Software |
|
|
285,418 |
|
|
|
285,375 |
|
|
|
|
|
534,601 |
|
|
|
590,117 |
|
|
Less accumulated depreciation and amortization |
|
|
(471,715 |
) |
|
|
(499,400 |
) |
|
Property and equipment, net |
|
$ |
62,886 |
|
|
$ |
90,717 |
|
The Company uses the straight-line method to record depreciation expense with useful lives of three to five years. Depreciation and amortization of property and equipment of $28,098, $14,127, $26,704, was recorded for the years ended December 31, 2017, 2016 and 2015, respectively.
Accrued Liabilities
Accrued liabilities consist of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Accrued compensation |
|
$ |
952,470 |
|
|
$ |
882,090 |
|
|
Research and development costs |
|
|
122,665 |
|
|
|
191,343 |
|
|
Professional services fees |
|
|
144,105 |
|
|
|
43,767 |
|
|
Other |
|
|
141,504 |
|
|
|
145,600 |
|
|
|
|
$ |
1,360,744 |
|
|
$ |
1,262,800 |
|
5. Related Party Transactions
On October 13, 2011, the Company entered into a services agreement with Kissei to perform two separate studies relating to MN-221 in exchange for $2.5 million paid to the Company in October 2011. The Company is responsible for all costs to be incurred in the performance of these studies. The amount received from Kissei, net of the amount recorded as revenue through December 31, 2017, is included on the balance sheet at December 31, 2017 and 2016 as deferred revenue and will be recognized as revenue in future periods as the Company performs the remaining services. On September 26, 2011, the Company issued and sold to Kissei 220,000 shares of Series B Convertible Preferred Stock. Each share of the Series B Preferred stock was convertible into shares of common stock at a conversion rate of 1:10 at the option of the holder. On June 15, 2016, Kissei elected to convert all 220,000 preferred shares into 2,200,000 shares of common stock. As of December 31, 2016, Kissei is no longer considered a related party.
6. Commitments and Contingencies
Lease Commitments
The Company subleases its office space under an operating lease with an initial term of four years and one month, expiring in December 2021. Rent expense for the years ended December 31, 2017, 2016 and 2015 was $254,593, $256,314 and $240,419, respectively. The difference between the minimum lease payments and the
67
straight-line amount of total rent expense is recorded as deferred rent. Deferred rent at December 31, 2017 and 2016 was $253 and $11,042, respectively.
As of December 31, 2017, the total estimated future annual minimum lease payments under the Company’s non-cancelable building and copier leases for the years ending after December 31, 2017 are as follows:
|
Years ending December 31: |
|
|
|
|
|
2018 |
|
$ |
211,663 |
|
|
2019 |
|
|
187,574 |
|
|
2020 |
|
|
142,703 |
|
|
2021 |
|
|
148,419 |
|
|
Total minimum payments |
|
$ |
690,359 |
|
Product Liability
The Company’s business exposes it to liability risks from its potential drug products. A successful product liability claim or series of claims brought against the Company could result in the payment of significant amounts of money and divert management’s attention from running the business. The Company may not be able to maintain insurance on acceptable terms, or the insurance may not provide adequate protection in the case of a product liability claim. To the extent that product liability insurance, if available, does not cover potential claims, the Company would be required to self-insure the risks associated with such claims. The Company believes it carries reasonably adequate insurance for product liability.
License and Research Agreements
The Company has entered into in-licensing agreements with various pharmaceutical companies. Under the terms of these agreements, the Company has received licenses to research, know-how and technology claimed, in certain patents or patent applications. Under these license agreements, the Company is generally required to make upfront payments and additional payments upon the achievement of milestones and/or royalties on future sales of products until the later of the expiration of the applicable patent or the applicable last date of market exclusivity after the first commercial sale, on a country-by-country basis.
No amounts have been expended under these agreements during the years ended December 31, 2017, 2016 or 2015. For products currently in development, future potential milestone payments based on product development are $10.0 million as of December 31, 2017. For all other products, future potential milestone payments related to development milestones and commercialization milestones totaled $33.5 million as of December 31, 2017. There are no minimum royalties required under any of the license agreements. The Company is unable to estimate with certainty the timing on when these milestone payments will occur as these payments are dependent upon the progress of the Company’s product development programs.
Legal Proceedings
From time to time, the Company may be subject to legal proceedings and claims in the ordinary course of business. The Company is not aware of any such proceedings or claims that it believes will have, individually or in aggregate, a material adverse effect on its business, financial condition or results of operations.
7. Joint Venture
The Company entered into an agreement to form a joint venture company with Zhejiang Medicine Co., Ltd. and Beijing Medfron Medical Technologies Co., Ltd. (formerly Beijing Make-Friend Medicine Technology Co., Ltd.) effective September 27, 2011. The joint venture agreement provides for the joint venture company, Zhejiang Sunmy Bio-Medical Co., Ltd. (Zhejiang Sunmy), to develop and commercialize MN-221 in China and pursue additional compounds to develop, each with a 50% interest in Zhejiang Sunmy.
68
Zhejiang Sunmy was a variable interest entity for which the Company is not the primary beneficiary as the Company does not have a majority of the board seats and does not have power to direct or significantly influence the actions of the entity. The activities of Zhejiang Sunmy are accounted for under the equity method whereby the Company absorbs any loss or income generated by Zhejiang Sunmy according to the Company’s percentage ownership.
. At December 31, 2016, the investment is reflected as a long-term asset on the Company’s consolidated balance sheets which represents the investment and maximum loss exposure in Zhejiang Sunmy, net of the Company’s portion of any generated loss or income.
On July 24, 2017, the Company and Beijing Medfron Medical Technologies Co., Ltd. agreed to dissolve Zhejiang Sunmy, subject to approval by applicable Chinese regulatory authorities which was granted on December 11, 2017. At December 31, 2017, we reflect a long-term asset on our consolidated balance sheet which represents our investment in Zhejiang Sunmy, net of our portion of any generated loss or income.
8. Share-based Compensation
Stock Incentive Plans
In June 2013, the Company adopted the 2013 Equity Incentive Plan, or 2013 Plan, under which the Company may grant stock options, stock appreciation rights, restricted stock, restricted stock units and other awards to individuals who are then employees, officers, non-employee directors or consultants of the Company or its subsidiaries. The 2013 Plan is the successor to the Company’s Amended and Restated 2004 Stock Incentive Plan, or 2004 Plan. A total of 2,500,000 shares of common stock were initially reserved for issuance under the 2013 Plan. At the annual meeting of stockholders held in June 2017, the Company’s stockholders approved an amendment to the 2013 Plan to increase the number of shares of common stock reserved for issuance under the plan by 1,200,000 shares. In addition, “returning shares” that may become available from time to time are added back to the plan. “Returning shares” are shares that are subject to outstanding awards granted under the 2004 Plan that expire or terminate prior to exercise or settlement, are forfeited because of the failure to vest, are repurchased, or are withheld to satisfy tax withholding or purchase price obligations in connection with such awards. Although the Company no longer grants equity awards under the 2004 Plan, all outstanding stock awards granted under the 2004 Plan will continue to be subject to the terms and conditions as set forth in the agreements evidencing such stock awards and the terms of the 2004 Plan. As of December 31, 2017, 1,215,592 shares remain available for future grant under the 2013 Plan.
We occasionally issue employee performance-based stock options, the vesting of which is based on a determination made by our board of directors as to the achievement of certain corporate objectives at the end of the performance period. The grant date of such awards is the date on which our board of directors makes its determination. For periods preceding the grant date, the expense related to these awards is measured based on their fair value at each reporting date.
Stock Options
Options granted under the 2013 Plan and 2004 Plan have terms of ten years from the date of grant unless earlier terminated and generally vest over a three or four-year period.
The exercise price of all options granted during the years ended December 31, 2017, 2016 and 2015 was equal to the market value of the Company’s common stock on the date of grant.
69
A summary of stock option activity and related information for the years ended December 31, 2017, 2016 and 2015 is as follows:
|
|
|
Number of Option Shares |
|
|
Weighted Average Exercise Price |
|
||
|
Outstanding at December 31, 2014 |
|
|
3,447,969 |
|
|
$ |
5.00 |
|
|
Granted |
|
|
689,000 |
|
|
$ |
3.14 |
|
|
Exercised |
|
|
— |
|
|
$ |
- |
|
|
Cancelled |
|
|
(3,000 |
) |
|
$ |
9.27 |
|
|
Outstanding at December 31, 2015 |
|
|
4,133,969 |
|
|
$ |
4.69 |
|
|
Granted |
|
|
1,158,000 |
|
|
$ |
4.00 |
|
|
Exercised |
|
|
(172,585 |
) |
|
$ |
4.81 |
|
|
Cancelled |
|
|
(687,367 |
) |
|
$ |
11.27 |
|
|
Outstanding at December 31, 2016 |
|
|
4,432,017 |
|
|
$ |
3.47 |
|
|
Granted |
|
|
1,195,000 |
|
|
$ |
6.10 |
|
|
Exercised |
|
|
(105,579 |
) |
|
$ |
4.04 |
|
|
Cancelled |
|
|
(7,400 |
) |
|
$ |
7.76 |
|
|
Outstanding at December 31, 2017 |
|
|
5,514,038 |
|
|
$ |
4.03 |
|
|
Exercisable at December 31, 2017 |
|
|
4,252,201 |
|
|
$ |
3.47 |
|
|
Vested and expected to vest at December 31, 2017 |
|
|
5,514,038 |
|
|
$ |
4.03 |
|
Cash received from stock option exercises for the years ended December 31, 2017, 2016 and 2015 was $426,013, $829,526 and $0, respectively. The aggregate intrinsic value of options exercised was $198,164, $414,572 and $0 for the years ended December 31, 2017, 2016 and 2015, respectively. Options outstanding and exercisable at December 31, 2017 had a weighted average contractual life of 6.58 and 6.53 years, respectively.
As of December 31, 2017 and 2016, the total intrinsic value of options outstanding was $13.6 million and $11.5 million, respectively. Total intrinsic value of options exercisable was $12.9 million and $9.2 million as of December 31, 2017 and 2016, respectively.
Employee Stock Purchase Plan
Under the Company’s 2007 Employee Stock Purchase Plan, or ESPP, 300,000 shares of common stock were originally reserved for issuance. In addition, the shares reserved automatically increase each year by a number equal to the lesser of: (i) 15,000 shares; (ii) 1% of the outstanding shares of common stock on the last day of the immediately preceding fiscal year; or (iii) such lesser amount as determined by the Board. The ESPP permits full-time employees to purchase common stock through payroll deductions (which cannot exceed 15% of each employee’s compensation) at the lower of 85% of fair market value at the beginning of the offering period or the end of each six-month offering period. The ESPP is considered a compensatory plan and the Company records compensation expense.
For the year ended December 31, 2017, an aggregate of 15,153 shares were issued under the ESPP, leaving 183,972 shares available for future issuance.
70
The estimated fair value of each stock option award was determined on the date of grant using the Black-Scholes option valuation model with the following weighted-average assumptions for stock option grants:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Stock Options |
|
|
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
2.06 |
% |
|
|
1.60 |
% |
|
|
1.48 |
% |
|
Expected volatility of common stock |
|
|
72.55 |
% |
|
|
78.30 |
% |
|
|
79.28 |
% |
|
Dividend yield |
|
|
0.00 |
% |
|
|
0.00 |
% |
|
|
0.00 |
% |
|
Expected option term (in years) |
|
|
5.67 |
|
|
|
5.57 |
|
|
|
5.49 |
|
The estimated fair value of employee stock purchase rights under the Company’s ESPP was determined on the date of grant using the Black-Scholes option valuation model with the following weighted-average assumptions for stock option grants:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Employee Stock Purchase Plan |
|
|
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
|
1.09 |
% |
|
|
0.44 |
% |
|
|
0.10 |
% |
|
Expected volatility of common stock |
|
|
38.34 |
% |
|
|
51.65 |
% |
|
|
76.52 |
% |
|
Dividend yield |
|
|
0.00 |
% |
|
|
0.00 |
% |
|
|
0.00 |
% |
|
Expected option term (in years) |
|
|
0.5 |
|
|
|
0.5 |
|
|
|
0.5 |
|
The risk-free interest rate assumption is based upon observed interest rates appropriate for the expected term of employee stock options. The expected volatility is based on the historical volatility of the Company’s common stock. The Company has not paid nor does the Company anticipate paying dividends on its common stock in the foreseeable future. The expected term of employee stock options is based on the simplified method as provided by the authoritative guidance on stock compensation, as the historical stock option exercise experience does not provide a reasonable basis to estimate the expected term.
The weighted-average fair value of each stock option granted during the years ended December 31, 2017, 2016 and 2015, estimated as of the grant date using the Black-Scholes option valuation model, was $3.78 per option, $2.64 per option and $2.08 per option, respectively.
Share-based compensation expense for stock option awards and ESPP shares are reflected in total operating expense for each respective year. For the years ended December 31, 2017, 2016 and 2015, share-based compensation expense related to stock options and the ESPP was $4.5 million, $4.0 million and $2.0 million, respectively, of which $3.1 million, $2.9 million and $1.5 million was recorded as a component of general and administrative expense, respectively, and $1.4 million, $1.1 million and $0.5 million was recorded as a component of research, development and patents expense, respectively.
As of December 31, 2017, there was $0.4 million of unamortized compensation cost related to unvested stock option awards which is expected to be recognized over a remaining weighted-average vesting period of 0.91 years, on a straight-line basis.
9. Stockholders’ Equity
Equity Offerings
On October 16, 2013, the Company entered into an at-the-market equity distribution agreement with Macquarie Capital (USA) Inc., or MCUSA, pursuant to which the Company could sell common stock through
71
MCUSA from time to time up to an aggregate offering price of $10.0 million. The at-the-market equity distribution agreement with MCUSA was terminated on May 22, 2015, and as of such date, the Company had completed sales to MCUSA totaling 2,127,500 shares of common stock at prices ranging from $2.01 to $4.45 per share, generating gross and net proceeds of $5.3 million and $4.5 million, respectively. For the year ended December 31, 2015, the Company generated gross and net proceeds of $0.9 million and $0.7 million, respectively, on the sale of 225,000 shares of common stock under this agreement.
On May 22, 2015, the Company entered into an at-the-market issuance sales agreement (the “ATM Agreement”) with MLV & Co. LLC, or MLV, pursuant to which the Company may sell common stock through MLV from time to time up to an aggregate offering price of $30.0 million. Sales of the Company’s common stock through MLV, if any, can be made by any method that is deemed to be an “at-the-market” equity offering as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including sales made directly on NASDAQ, on any other existing trading market for the common stock or to or through a market maker. MLV may also sell the common stock in privately negotiated transactions, subject to the Company’s prior approval. The Company agreed to pay MLV an aggregate commission rate of up to 4.0% of the gross proceeds of any common stock sold under this agreement. Proceeds from sales of common stock will depend on the number of shares of common stock sold to MLV and the per share purchase price of each transaction. The Company is not obligated to make any sales of common stock under the sales agreement and may terminate the sales agreement at any time upon written notice. On September 16, 2016, the Company entered into an amendment No. 1 to the ATM Agreement to also include FBR Capital Markets & Co as a sales agent.
The following table summarizes the activity under the ATM agreements for the following periods (in thousands except share price and shares sold):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Gross proceeds |
$ |
10,303.0 |
|
|
$ |
264.0 |
|
|
$ |
884.0 |
|
|
Net proceeds |
$ |
9,891.3 |
|
|
$ |
159.5 |
|
|
$ |
607.5 |
|
|
Shares sold |
|
1,689,436 |
|
|
|
36,248 |
|
|
|
232,800 |
|
|
Price range |
$5.30 - 6.98 |
|
|
$6.90 - 7.54 |
|
|
$4.16 - 4.23 |
|
|||
Warrants
During the year ended December 31, 2017, 119,047 shares of the Company’s common stock were issued upon exercise of warrants for gross proceeds of $0.4 million with warrants exercisable for 198,020 shares expiring unexercised on May 10, 2017. During the years ended December 31, 2016 and 2015, 2,131,700 and 252,200 shares of the Company’s common stock were issued upon exercise of warrants for gross proceeds of $7.6 million and $0.9 million, respectively, with warrants exercisable for 207,600 shares expiring unexercised in 2016.
As of December 31, 2017, the Company has warrants exercisable for 750,000 shares outstanding at an exercise price of $3.15 per share, which expire on May 9, 2018.
These warrants were accounted for as equity at issuance.
72
Common Stock Reserved for Future Issuance
The following table summarizes common stock reserved for future issuance at December 31, 2017:
|
Common Stock reserved for issuance under the ESPP |
|
|
183,972 |
|
|
Common stock reserved for issuance upon exercise of warrants outstanding |
|
|
750,000 |
|
|
Common stock reserved for issuance upon exercise of options outstanding (under the 2004 Plan and 2013 Plan) |
|
|
5,514,038 |
|
|
Common stock reserved for future equity awards (under the 2013 Plan) |
|
|
1,215,592 |
|
|
|
|
|
7,663,602 |
|
Public Offering
On August 24, 2015, the Company completed a firm-commitment underwritten public offering of 5,000,000 shares of common stock at a purchase price of $3.50 per share for gross proceeds of $17.5 million, and received net proceeds of approximately $16.0 million, net of underwriting discounts and commissions and offering expenses.
10. Income Taxes
A reconciliation of loss before income taxes for domestic and foreign locations for the years ended December 31, 2017, 2016 and 2015 is as follows:
|
|
|
|
|
|
|
|
|
|||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
(12,940,362 |
) |
|
$ |
(10,892,275 |
) |
|
$ |
(8,866,201 |
) |
|
Foreign |
|
|
33,176 |
|
|
|
30,050 |
|
|
|
28,481 |
|
|
Loss before income taxes |
|
$ |
(12,907,186 |
) |
|
$ |
(10,862,225 |
) |
|
$ |
(8,837,720 |
) |
A reconciliation of income tax benefit (expense) for the years ended December 31, 2017, 2016 and 2015 is as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Current: |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Federal |
|
$ |
— |
|
|
$ |
1,489 |
|
|
$ |
— |
|
|
State |
|
|
— |
|
|
|
412 |
|
|
|
— |
|
|
Foreign |
|
|
(10,158 |
) |
|
|
(5,655 |
) |
|
|
(7,359 |
) |
|
Total current income tax benefit (expense) |
|
|
(10,158 |
) |
|
|
(3,754 |
) |
|
|
(7,359 |
) |
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
1,329,888 |
|
|
|
— |
|
|
|
— |
|
|
State |
|
|
424,320 |
|
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total deferred income tax benefit (expense) |
|
|
1,754,208 |
|
|
|
— |
|
|
|
— |
|
|
Total income tax benefit (expense) |
|
$ |
1,744,050 |
|
|
$ |
(3,754 |
) |
|
$ |
(7,359 |
) |
73
The significant components of deferred income taxes at December 31, 2017, 2016 and 2015 are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred tax assets: |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net operating loss carryforwards |
|
$ |
59,480,000 |
|
|
$ |
90,211,000 |
|
|
$ |
88,900,000 |
|
|
Capitalized licenses |
|
|
429,000 |
|
|
|
838,000 |
|
|
|
1,084,000 |
|
|
Research tax credits |
|
|
8,160,000 |
|
|
|
7,776,000 |
|
|
|
7,677,000 |
|
|
Stock options |
|
|
2,585,000 |
|
|
|
2,384,000 |
|
|
|
2,624,000 |
|
|
Other, net |
|
|
572,000 |
|
|
|
777,000 |
|
|
|
763,000 |
|
|
Total deferred tax assets |
|
|
71,226,000 |
|
|
|
101,986,000 |
|
|
|
101,048,000 |
|
|
Deferred tax liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
In process R&D |
|
|
(1,343,000 |
) |
|
|
(1,956,000 |
) |
|
|
(1,956,000 |
) |
|
Total deferred tax liabilities |
|
|
(1,343,000 |
) |
|
|
(1,956,000 |
) |
|
|
(1,956,000 |
) |
|
Net deferred tax assets |
|
|
69,883,000 |
|
|
|
100,030,000 |
|
|
|
99,092,000 |
|
|
Valuation allowance |
|
|
(70,086,000 |
) |
|
|
(101,986,000 |
) |
|
|
(101,048,000 |
) |
|
Net deferred tax liability |
|
$ |
(203,000 |
) |
|
$ |
(1,956,000 |
) |
|
$ |
(1,956,000 |
) |
The Company has established a valuation allowance against net deferred tax assets due to the uncertainty that such assets will be realized. The Company periodically evaluates the recoverability of the deferred tax assets. At such time as it is determined that it is more likely than not that deferred tax assets will be realizable, the valuation allowance will be reduced.
At December 31, 2017, the Company has federal and California net operating losses, or NOL, carryforwards of approximately $239.9 million and $130.5 million, respectively. The federal NOL carryforwards begin to expire in 2020, and the California NOL carryforwards begin to expire in 2028. At December 31, 2017, the Company also had federal and California research tax credit carryforwards of approximately $6.8 million and $1.8 million, respectively. The federal research tax credit carryforwards begin to expire in 2024, and the California research tax credit carryforward does not expire and can be carried forward indefinitely until utilized.
The above NOL carryforward and the research tax credit carryforwards are subject to an annual limitation under Section 382 and 383 of the Internal Revenue Code of 1986, and similar state provisions due to ownership change limitations that have occurred which will limit the amount of NOL and tax credit carryforwards that can be utilized to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382 and 383, results from transactions increasing ownership of certain stockholders or public groups in the stock of the corporation by more than 50 percentage points over a three-year period. The Company has not completed an IRC Section 382/383 analysis since 2011 regarding the limitation of net operating loss and research and development credit carryforwards. There is a risk that additional changes in ownership have occurred since the completion of the Company’s analysis, which was through December 2011. If a change in ownership were to have occurred, additional NOL and tax credit carryforwards could be eliminated or restricted. If eliminated, the related asset would be removed from the deferred tax asset schedule with a corresponding reduction in the valuation allowance. Due to the existence of the valuation allowance, limitations created by future ownership changes, if any, related to the Company’s operations in the United States will not impact the Company’s effective tax rate.
74
A reconciliation of the federal statutory income tax rate to the Company’s effective income tax rate is as follows:
|
|
|
|
|
|
|
|
|
|||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Federal statutory income tax rate |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
State income taxes, net of federal benefit |
|
|
5.2 |
|
|
|
3.5 |
|
|
|
5.2 |
|
|
Tax credits |
|
|
1.1 |
|
|
|
0.9 |
|
|
|
1.4 |
|
|
Change in valuation allowance |
|
|
247.4 |
|
|
|
(8.4 |
) |
|
|
(13.5 |
) |
|
Permanent differences |
|
|
(0.3 |
) |
|
|
(0.1 |
) |
|
|
(0.1 |
) |
|
Expiration of attributes |
|
|
(18.7 |
) |
|
|
(17.2 |
) |
|
|
(24.6 |
) |
|
Tax Cuts and Jobs Act |
|
|
(253.7 |
) |
|
|
— |
|
|
|
— |
|
|
Stock compensation |
|
|
(2.9 |
) |
|
|
(13.6 |
) |
|
|
(3.4 |
) |
|
Other |
|
|
0.4 |
|
|
|
(0.1 |
) |
|
|
(0.1 |
) |
|
Provision for income taxes |
|
|
13.5 |
% |
|
|
0.0 |
% |
|
|
(0.1 |
)% |
On December 22, 2017, the Tax Cuts and Jobs Act of 2017 (the “Act”) was signed into law making significant changes to the Internal Revenue Code. Changes include, but are not limited to, a corporate tax rate decrease from 35% to 21% effective for tax years beginning after December 31, 2017, the transition of U.S. international taxation from a worldwide tax system to a territorial system, and a one-time transition tax on the mandatory deemed repatriation of cumulative foreign earnings as of December 31, 2017. Under the Act, NOL deductions are limited to 80% of taxable income in future periods and NOLs can be carried forward indefinitely. The Company has calculated its best estimate of the impact of the Act in its year end income tax provision in accordance with its understanding of the Act and guidance available as of the date of this filing and as a result of the rate reduction, the company has reduced the deferred tax asset balance as of December 31, 2017 by $32.7 million and the valuation allowance by $33.4 million.
Due to uncertainties which currently exist in the interpretation of the provisions of the Tax Cuts and Jobs Act of 2017 regarding Internal Revenue Code Section 162(m), the Company has not evaluated the potential impacts of IRC Section 162(m) as amended by the Tax Cuts and Jobs Act of 2017 on its financial statements. The provisional amount related to the one-time transition tax on the mandatory deemed repatriation of foreign earnings was $0.1 million additional taxable income based on cumulative foreign earnings of $0.2 million.
On December 22, 2017, Staff Accounting Bulletin No. 118 ("SAB 118") was issued to address the application of US GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Act. In accordance with SAB 118, the Company has determined that the $0.6 million of the deferred tax benefit recorded in connection with the remeasurement of certain deferred tax assets and liabilities and the $0.1 million of additional taxable income recorded in connection with the transaction tax on the mandatory deemed repatriation of foreign earnings was a provisional amount and a reasonable estimate at December 31, 2017. Additional work is necessary to do a more detailed analysis of historical foreign earnings as well as potential correlative adjustments. Any subsequent adjustments to these amounts will be recorded in the third quarter of 2018 when the analysis is complete.
The Company recorded a deferred tax benefit of $1.1 million of income tax benefit as management has reassessed the ability to utilize deferred tax liabilities associated with indefinite-lived intangibles as a source of income against tax attributes that can be carried forward indefinitely.
The Company files income tax returns in the United States, California and foreign jurisdictions. Due to the Company’s losses incurred, the Company is essentially subject to income tax examination by tax authorities from inception to date. The Company’s policy is to recognize interest expense and penalties related to income tax matters as tax expense. At December 31, 2017, there are no unrecognized tax benefits, and there are not significant accruals for interest related to unrecognized tax benefits or tax penalties.
75
11. Employee Savings Plan
The Company has an employee savings plan available to substantially all employees. Under the plan, an employee may elect salary reductions which are contributed to the plan. The plan provides for discretionary contributions by the Company, which totaled $65,995, $66,289 and $64,749 for the years ended December 31, 2017, 2016 and 2015, respectively.
12. Quarterly Financial Data (Unaudited)
The following table presents certain quarterly financial data for eight consecutive quarters ended December 31, 2017.and 2016. The unaudited quarterly information has been prepared on the same basis as the audited consolidated financial statements and, in the opinion of management, includes all adjustments, necessary for a fair presentation of this data (in thousands, except per share data).
|
|
|
Year Ended December 31, 2017 |
|
|||||||||||||
|
|
|
1st Quarter |
|
|
2nd Quarter |
|
|
3rd Quarter |
|
|
4th Quarter |
|
||||
|
Selected quarterly financial data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses |
|
$ |
3,024 |
|
|
$ |
2,805 |
|
|
$ |
3,792 |
|
|
$ |
3,406 |
|
|
Net loss |
|
|
(3,017 |
) |
|
|
(2,789 |
) |
|
|
(3,755 |
) |
|
|
(1,602 |
) |
|
Net loss applicable to common stockholders |
|
|
(3,017 |
) |
|
|
(2,789 |
) |
|
|
(3,755 |
) |
|
|
(1,602 |
) |
|
Basic and diluted net loss per common share(1) |
|
|
(0.09 |
) |
|
|
(0.08 |
) |
|
|
(0.11 |
) |
|
|
(0.02 |
) |
|
|
|
Year Ended December 31, 2016 |
|
|||||||||||||
|
|
|
1st Quarter |
|
|
2nd Quarter |
|
|
3rd Quarter |
|
|
4th Quarter |
|
||||
|
Selected quarterly financial data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses |
|
$ |
3,392 |
|
|
$ |
3,205 |
|
|
$ |
2,845 |
|
|
$ |
1,440 |
|
|
Net loss |
|
|
(3,382 |
) |
|
|
(3,199 |
) |
|
|
(2,836 |
) |
|
|
(1,449 |
) |
|
Net loss applicable to common stockholders |
|
|
(3,382 |
) |
|
|
(3,199 |
) |
|
|
(2,836 |
) |
|
|
(1,449 |
) |
|
Basic and diluted net loss per common share(1) |
|
|
(0.11 |
) |
|
|
(0.10 |
) |
|
|
(0.08 |
) |
|
|
(0.04 |
) |
|
(1) |
Net loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly net income and loss per share will not necessarily equal the Annual Per Share Calculation. |
13. Subsequent Events
Public Offering
On February 12, 2018, the Company completed a firm-commitment underwritten public offering of 4,419,890 shares of common stock at a purchase price of $9.05 per share for aggregate gross proceeds of $40.0 million, and received aggregate net proceeds of approximately $37.4 million, net of underwriting discounts and commissions and offering expenses. Additionally, the Company granted the underwriters a 30-day option to purchase up to an additional 662,983 shares of commons stock at the public offering price.
76
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
An evaluation was performed by our Chief Executive Officer and Chief Financial Officer of the effectiveness of the design and operation of our disclosure controls and procedures as defined in the Rules 13(a)-15(e) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Disclosure controls and procedures are those controls and procedures designed to provide reasonable assurance that the information required to be disclosed in our Exchange Act filings is (1) recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission’s rules and forms, and (2) accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2017, our disclosure controls and procedures were effective.
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our procedures or our internal controls will prevent or detect all errors and all fraud. An internal control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of our controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Changes in Internal Control over Financial Reporting
There was no change in internal control over financial reporting (as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) during our fourth fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
(1) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(2) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
(3) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not
77
eliminate, this risk. Management is responsible for establishing and maintaining adequate internal control over financial reporting for the company.
Management has used the framework set forth in the report entitled Internal Control-Integrated Framework (2013 framework) published by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), known as COSO, to evaluate the effectiveness of our internal control over financial reporting. Based on this assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2017.
BDO USA, LLP, an independent registered public accounting firm, has audited our consolidated financial statements included in this Annual Report on Form 10-K and has issued an attestation report, included herein, on the effectiveness of our internal control over financial reporting as of December 31, 2017.
78
Report of Independent Registered Public Accounting Firm
Shareholders and Board of Directors
MediciNova, Inc.
La Jolla, California
Opinion on Internal Control over Financial Reporting
We have audited MediciNova, Inc.’s (the “Company’s”) internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated balance sheets of the Company and subsidiaries as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and our report dated February 13, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Item 9A, “Management’s Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
79
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ BDO USA, LLP
San Diego, California
February 13, 2018
80
None.
81
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item and not set forth below will be contained in the sections titled “Election of Directors,” “Section 16(a) Beneficial Ownership Reporting Compliance, “Corporate Governance,” “Meetings and Committees of the Board, and “Executive Officers” in our definitive proxy statement for our 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the conclusion of our fiscal year ended December 31, 2017 and is incorporated in this Annual Report on Form 10-K by reference.
We have adopted a Code of Ethics for Senior Officers, or Code of Ethics, that applies to our Chief Executive Officer, President, Chief Financial Officer and key management employees (including other senior financial officers) who have been identified by our Board of Directors. We have also adopted a Code of Business Conduct that applies to all of our officers, directors, employees, consultants and representatives. Each of the Code of Ethics and Code of Business Conduct are available on our website at www.medicinova.com under the Corporate Governance section of our Investor Relations page. We will promptly post on our website (i) any waiver, if and when granted, to any provision of the Code of Ethics or Code of Business Conduct (for executive officers or directors) and (ii) any amendment to the Code of Ethics or Code of Business Conduct.
Item 11. Executive Compensation
The information required by this item will be contained in the section titled “Executive Compensation” in our Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Certain of the information required by this item will be contained in section titled “Security Ownership of Certain Beneficial Owners and Management” the Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
The following table provides information as of December 31, 2017 with respect to the shares of our common stock that may be issued under our existing equity compensation plans.
Equity Compensation Plan Information
|
Plan Category |
|
Number of Securities to be Issued Upon Exercise of Outstanding Options and Rights |
|
|
Weighted Average Exercise Price of Outstanding Options and Rights |
|
|
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans |
|
|||
|
Equity Compensation Plans Approved by Stockholders(1) |
|
|
5,514,038 |
|
|
$ |
4.03 |
|
|
|
1,399,564 |
|
|
Equity Compensation Plans Not Approved by Stockholders |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
|
5,514,038 |
|
|
$ |
4.03 |
|
|
|
1,399,564 |
|
|
(1) |
Consists of the Amended and Restated 2004 Stock Incentive Plan, the 2013 Equity Incentive Plan and the 2007 Employee Stock Purchase Plan, or ESPP. Under the ESPP, the shares reserved automatically increase by a number equal to the lesser of: (i) 15,000 shares; (ii) 1% of the outstanding shares of our common stock on the last day of the immediately preceding fiscal year; or (iii) such lesser amount as determined by the Board. |
82
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be contained in the sections titled “Certain Relationships and Related Transactions” and “Corporate Governance” in our Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
Item 14. Principal Accountant Fees and Services
The information required by this item will be contained in the section titled “Ratification of Appointment of Independent Registered Public Accounting Firm” in our Proxy Statement and is incorporated in this Annual Report on Form 10-K by reference.
83
Item 15. Exhibits and Financial Statement Schedules
(a) Documents filed as part of this report.
1. Financial Statements. The following financial statements of MediciNova, Inc. and Reports of BDO USA, LLP, an independent registered public accounting firm, are included in this Annual Report on Form 10-K:
|
|
|
Page |
|
|
58 |
|
|
|
59 |
|
|
Consolidated Statements of Operations and Comprehensive Loss |
|
60 |
|
|
61 |
|
|
|
||
|
|
63 |
2. Financial Statement Schedules. None.
3. Exhibits.
|
Exhibit Number |
|
Description |
|
|
|
|
|
|
|
3.1 |
|
||
|
|
|
|
|
|
3.2 |
|
||
|
|
|
|
|
|
4.1 |
|
||
|
|
|
|
|
|
4.2 |
|
||
|
|
|
|
|
|
4.3 |
|
||
|
|
|
|
|
|
4.4 |
|
||
|
|
|
|
|
|
10.1* |
|
||
|
|
|
|
|
|
10.2* |
|
||
|
|
|
|
|
|
10.3† |
|
||
|
|
|
|
|
|
10.4† |
|
||
|
|
|
|
|
84
|
Exhibit Number |
|
Description |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
10.6† |
|
||
|
|
|
|
|
|
10.7† |
|
||
|
|
|
|
|
|
10.8† |
|
||
|
|
|
|
|
|
10.9* |
|
||
|
|
|
|
|
|
10.10† |
|
||
|
|
|
|
|
|
10.11† |
|
||
|
|
|
|
|
|
10.12* |
|
||
|
|
|
|
|
|
10.13* |
|
||
|
|
|
|
|
|
10.14† |
|
||
|
|
|
|
|
|
10.15† |
|
||
|
|
|
|
|
|
10.16† |
|
||
|
|
|
|
|
|
10.17* |
|
||
|
|
|
|
|
|
10.18† |
|
||
|
|
|
|
|
|
10.19 |
|
||
|
|
|
|
|
85
|
Exhibit Number |
|
Description |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
10.21* |
|
||
|
|
|
|
|
|
10.22* |
|
||
|
|
|
|
|
|
10.23* |
|
||
|
|
|
|
|
|
10.24* |
|
||
|
|
|
|
|
|
10.25* |
|
||
|
|
|
|
|
|
10.26* |
|
||
|
|
|
|
|
|
10.27* |
|
||
|
|
|
|
|
|
10.28 |
|
|
|
|
10.29 |
|
||
|
|
|
|
|
|
10.30 |
|
||
|
|
|
|
|
|
23.1 |
|
||
|
|
|
|
|
|
24.1 |
|
||
|
|
|
|
|
|
31.1 |
|
||
|
|
|
|
|
|
31.2 |
|
||
|
|
|
|
|
|
32.1 |
|
||
|
|
|
|
|
|
32.2 |
|
||
|
|
|
|
|
86
|
Exhibit Number |
|
Description |
|
|
|
|
|
|
|
|
The following financial statements from MediciNova, Inc. on Form 10-K as of and for the year ended December 31, 2017 formatted in Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets; (ii) Consolidated Statements of Operations; (iii) Consolidated Statements of Stockholders’ Equity and Comprehensive Loss; (iv) Consolidated Statements of Cash Flows; and (v) the notes to the consolidated financial statements. |
||
|
† |
Portions of this Exhibit have been omitted pursuant to a grant of confidential treatment by the SEC. |
|
* |
Indicates management contract or compensatory plan. |
87
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
MEDICINOVA, INC. |
||
|
|
|
A Delaware Corporation |
||
|
|
|
|
|
|
|
Date: February 13, 2018 |
|
By: |
|
/s/ Yuichi Iwaki |
|
|
|
|
|
Yuichi Iwaki, M.D., Ph.D. |
|
|
|
|
|
President & Chief Executive Officer |
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Yuichi Iwaki his true and lawful attorney-in-fact, with full power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact or his substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Yuichi Iwaki Yuichi Iwaki, M.D., Ph.D. |
|
Director, President and Chief Executive Officer (Principal executive officer) |
|
February 13, 2018 |
|
|
|
|
|
|
|
/s/ Ryan Selhorn Ryan Selhorn |
|
Chief Financial Officer (Principal financial and accounting officer) |
|
February 13, 2018 |
|
|
|
|
|
|
|
/s/ Jeff Himawan Jeff Himawan, Ph.D. |
|
Chairman of the Board of Directors |
|
February 13, 2018 |
|
|
|
|
|
|
|
/s/ Yoshio Ishizaka Yoshio Ishizaka |
|
Director |
|
February 13, 2018 |
|
|
|
|
|
|
|
/s/ Yutaka Kobayashi Yutaka Kobayashi |
|
Director |
|
February 13, 2018 |
|
|
|
|
|
|
|
/s/ Hideki Nagao |
|
Director |
|
February 13, 2018 |
|
Hideki Nagao |
|
|
88