Attached files
| file | filename |
|---|---|
| EX-32.2 - CERTIFICATION - Citius Pharmaceuticals, Inc. | f10k2017ex32-2_citiuspharma.htm |
| EX-32.1 - CERTIFICATION - Citius Pharmaceuticals, Inc. | f10k2017ex32-1_citiuspharma.htm |
| EX-31.2 - CERTIFICATION - Citius Pharmaceuticals, Inc. | f10k2017ex31-2_citiuspharma.htm |
| EX-31.1 - CERTIFICATION - Citius Pharmaceuticals, Inc. | f10k2017ex31-1_citiuspharma.htm |
| EX-21 - SUBSIDIARIES - Citius Pharmaceuticals, Inc. | f10k2017ex21_citiuspharma.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended September 30, 2017
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission File Number 333-170781
Citius Pharmaceuticals, Inc.
(Exact name of Registrant as specified in its Charter)
| Nevada | 27-3425913 | |
| (State
or other jurisdiction of incorporation or organization) |
(I.R.S.
Employer Identification No.) |
11 Commerce Drive, First Floor, Cranford, NJ 07016
(Address of principal executive offices) (Zip Code)
(908) 967-6677
(Registrant’s telephone number, including area code)
(Former name and address, if changed since last report)
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Common Stock, par value $0.001 per share | The NASDAQ Capital Market | |
Warrants to purchase Common Stock |
The NASDAQ Capital Market |
(Title or Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past ninety (90) days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ☒ Yes ☐ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendments to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ (Do not check if a smaller reporting company) | Smaller reporting company | ☒ |
| Emerging growth company | ☐ | ||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒ No
The aggregate market value of the voting and non-voting common equity held by non-affiliates* computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter (March 31, 2017) was $14,441,111.
* Affiliates for the purpose of this item refers to the issuer’s officers and directors and/or any persons or firms (excluding those brokerage firms and/or clearing houses and/or depository companies holding issuer’s securities as record holders only for their respective clienteles’ beneficial interest) owning 10% or more of the issuer’s Common Stock, both of record and beneficially.
APPLICABLE ONLY TO CORPORATE REGISTRANTS
Indicate the number of shares outstanding of each of the registrant’s classes of Common Stock, as of the latest practicable date:
8,423,391 shares as of December 1, 2017, all of one class of Common Stock, $0.001 par value.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Company’s Proxy Statement for the Annual Meeting of Shareholders to be held on February 7, 2018 are incorporated by reference in Part III of this Report.
Citius Pharmaceuticals, Inc.
FORM 10-K
September 30, 2017
TABLE OF CONTENTS
EXPLANATORY NOTE
In this annual report on Form 10-K, and unless the context otherwise requires the “Company,” “we,” “us” and “our” refer to Citius Pharmaceuticals, Inc. and its wholly-owned subsidiaries, Citius Pharmaceuticals, LLC and Leonard-Meron Biosciences, Inc., taken as a whole.
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements.” Forward-looking statements include, but are not limited to, statements that express our intentions, beliefs, expectations, strategies, predictions or any other statements relating to our future activities or other future events or conditions. These statements are based on current expectations, estimates and projections about our business based, in part, on assumptions made by management. These statements are not guarantees of future performance and involve risks, uncertainties and assumptions that are difficult to predict. Therefore, actual outcomes and results may, and are likely to, differ materially from what is expressed or forecasted in the forward-looking statements due to numerous factors discussed from time to time in this report, including the risks described under Item 1A - “Risk Factors,” and Item 7 - “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this report and in other documents which we file with the Securities and Exchange Commission. In addition, such statements could be affected by risks and uncertainties related to:
| ● | our ability to raise funds for general corporate purposes and operations, including our clinical trials; |
| ● | the commercial feasibility and success of our technology; |
| ● | our ability to recruit qualified management and technical personnel; | |
| ● | the success of our clinical trials; | |
| ● | our ability to obtain and maintain required regulatory approvals for our products; and | |
| ● | the other factors discussed in the “Risk Factors” section and elsewhere in this report. |
Any forward-looking statements speak only as of the date on which they are made, and except as may be required under applicable securities laws; we do not undertake any obligation to update any forward-looking statement to reflect events or circumstances after the filing date of this report.
| Item 1. | Business |
Overview
Citius Pharmaceuticals, Inc., headquartered in Cranford, New Jersey, is a specialty pharmaceutical company dedicated to the development and commercialization of critical care products targeting important medical needs with a focus on anti-infective products in adjunct cancer care and unique prescription products. Our goal is to achieve leading market positions by providing therapeutic products that address unmet medical needs yet have a lower development risk than is associated with new chemical entities. New formulations or combinations of previously approved drugs with substantial existing safety and efficacy data are a core focus as we seek to reduce development and clinical risks associated with drug development. Our strategy centers on products that have intellectual property and regulatory exclusivity protection, while providing competitive advantages over other existing therapeutic approaches.
The Company was founded as Citius Pharmaceuticals, LLC, a Massachusetts limited liability company, on January 23, 2007. On September 12, 2014, Citius Pharmaceuticals, LLC entered into a Share Exchange and Reorganization Agreement, with Citius Pharmaceuticals, Inc. (formerly Trail One, Inc.), a publicly traded company incorporated under the laws of the State of Nevada. Citius Pharmaceuticals, LLC became a wholly-owned subsidiary of Citius. On March 30, 2016, Citius acquired Leonard-Meron Biosciences, Inc. as a wholly-owned subsidiary. LMB was a pharmaceutical company focused on the development and commercialization of critical care products with a concentration on anti-infectives.
Since its inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, and raising capital. We are developing two proprietary products: Mino-Lok™, an antibiotic lock solution used to treat patients with catheter-related bloodstream infections by salvaging the infected catheter, and a hydrocortisone-lidocaine topical formulation that is intended to provide anti-inflammatory and anesthetic relief to individuals suffering from hemorrhoids. We believe the markets for our products are large, growing, and underserved by the current prescription products or procedures.
Citius is subject to a number of risks common to companies in the pharmaceutical industry including, but not limited to, risks related to the development by Citius or its competitors of research and development stage products, market acceptance of its products, competition from larger companies, dependence on key personnel, dependence on key suppliers and strategic partners, the Company’s ability to obtain additional financing and the Company’s compliance with governmental and other regulations.
Mino-LokTM
Overview
Mino-Lok is a patented solution containing minocycline, disodium ethylenediaminetetraacetic acid (edetate), and ethyl alcohol, all of which act synergistically to treat and salvage infected central venous catheters (“CVCs”) in patients with catheter related bloodstream infections (“CRBSIs”). Mino-Lok breaks down biofilm barriers formed by bacterial colonies, eradicates the bacteria, and provides anti-clotting properties to maintain patency in CVCs.
The administration of Mino-Lok consists of filling the lumen of the catheter with 0.8 ml to 2.0 ml of Mino-Lok solution. The catheter is then “locked”, meaning that the solution remains in the catheter without flowing into the vein. the lock is maintained for a dwell-time of two hours while the catheter is not in use. If the catheter has multiple lumens, all lumens may be locked with the Mino-Lok solution either simultaneously or sequentially. If patients are receiving continuous infusion therapy, the catheters alternate between being locked with the Mino-Lok solution and delivering therapy. The Mino-Lok therapy is two hours per day for at least five days, usually with two additional locks in the subsequent two weeks. After locking the catheter for two hours, the Mino-Lok solution is aspirated, and the catheter is flushed with normal saline. At that time, either the infusion will be continued, or will be locked with the standard-of-care lock solution until further use of the catheter is required. In a clinical study conducted by MD Anderson Cancer Center (“MDACC”), there were no serum levels of either minocycline or edetate detected in the sera of several patients who underwent daily catheter lock solution with minocycline and edetate (“M-EDTA”) at the concentration level proposed in Mino-Lok treatment. Thus, it has been demonstrated that the amount of either minocycline or edetate that leaks into the serum is very low or none at all.
Phase 2b Results
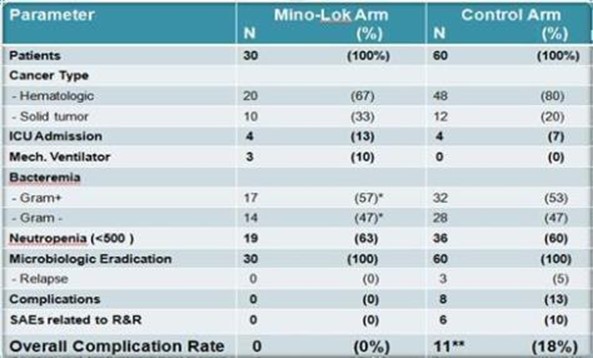
From April 2013 to July 2014, 30 patients with CVC-related bloodstream infection were enrolled at MDACC in a prospective Phase 2b study. Patients received Mino-Lok therapy for two hours once daily for a minimum of five days within the first week followed by two additional locks within the next two weeks. Patients were followed for one month post lock therapy. Demographic information, clinical characteristics, laboratory data, therapy, as well as adverse events and outcome were collected for each patient. Median age at diagnosis was 56 years (range: 21-73 years). In all patients, prior to the use of lock therapy, systemic treatment with a culture-directed, first-line intravenous antibiotic was started. Microbiological eradication was achieved at the end of therapy in all cases. None of the patients experienced any serious adverse event related to the lock therapy.
| 1 |
The active arm, which is the Mino-Lok treated group of patients, was then compared to 60 patients in a matched cohort that experienced removal and replacement of their CVCs within the same contemporaneous timeframe. The patients were matched for cancer type, infecting organism, and level of neutropenia. All patients were cancer patients and treated at the MDACC. The efficacy of Mino-Lok therapy was 100% in salvaging CVCs, demonstrating equal effectiveness to removing the infected CVC and replacing with a new catheter.
The main purpose of the study was to show that Mino-Lok therapy was at least as effective as the removal and replacement of CVCs when CRBSIs are present, and that the safety was better, that is, the complications of removing an infected catheter and replacing with a new one could be avoided. In addition to having a 100% efficacy rate with all CVCs being salvaged, Mino-Lok therapy had no significant adverse events (“SAEs”), compared to an 18% SAE rate in the matched cohort where patients had the infected CVCs removed and replaced (“R&R”) with a fresh catheter. There were no overall complication rates in the Mino-Lok arm group compared to 11 patients with events (18%) in the control group. These events included bacterial relapse (5%) at four (4) weeks post-intervention, and a number of complications associated with mechanical manipulation in the removal or replacement procedure for the catheter (10%) or development of deep seated infections such as septic thrombophlebitis and osteomyelitis (8%). As footnoted, six (6) patients had more than one (1) complication in the control arm group.
 |
Source: Dr. Issam Raad, Antimicrobial Agents and Chemotherapy, June 2016, Vol. 60 No. 6, Page 3429
Phase 3 Initiation
In November 2016, the Company initiated site recruitment for Phase 3 clinical trials. From initiation through first quarter 2017, the Company received input from several sites related to the control arm as being less than standard of care for some of the respective institutions. The Company worked closely with the FDA with respect to the design of the phase 3 trial, and received feedback on August 17, 2017. The FDA stated that they recognized that there is an unmet medical need in salvaging infected catheters and agreed that an open label, superiority design would address the Company’s concerns and would be acceptable to meet the requirements of a new drug application. The Company amended the phase 3 study design to remove the saline and heparin placebo control arm and to use an active control arm that conforms with today’s current standard of care. It is expected that patient enrollment will still commence in the fourth quarter 2017.
| 2 |
Fast Track Designation
In October 2017, the Company received official notice from FDA that the investigational program for Mino-Lok™ was granted “Fast Track” status. Fast Track is a designation that expedites FDA review to facilitate development of drugs which treat a serious or life-threatening condition and fill an unmet medical need. A drug that receives Fast Track designation is eligible for the following:
| ● | More frequent meetings with FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval; |
| ● | More frequent written correspondence from FDA about the design of the clinical trials; |
| ● | Priority review to shorten the FDA review process for a new drug from ten months to six months; and, |
| ● | Rolling Review, which means Citius can submit completed sections of its New Drug Application (NDA) for review by FDA, rather than waiting until every section of the application is completed before the entire application can be reviewed. |
Mino-Lok TM International Study
In October 2017, data from an international study on Mino-LokTM was presented at the Infectious Disease Conference, (“ID Week”), in San Diego, California. The 44 patient study was conducted in Brazil, Lebanon, and Japan and showed Mino-Lok™ therapy was an effective intervention to salvage long term, infected central venous catheters (CVCs) in catheter related bloodstream infections in patients who had cancer with limited vascular access. This study showed 95% effectiveness for Mino-Lok therapy in achieving microbiological eradication of the CVCs as compared to 83% for the control.
Market Opportunity
In spite of best clinical practice, catheters contribute to approximately 70% of blood stream infections that occur in the ICU, or are associated with hemodialysis or cancer patients (approximately 470,000 per year). Bacteria enter the catheter either from the skin or intraluminally through the catheter hub. Once in the catheter, bacteria tend to form a protective biofilm on the interior surface of the catheter that is resistant to most antimicrobial solutions. The most frequently used maintenance flush, heparin, actually stimulates biofilm formation. Heparin is widely used as a prophylactic lock solution, in spite of the evidence that it contributes to the promotion of biofilm formation. The formation of bacterial biofilm usually precedes CRBSIs.
The SOC in the management of CRBSI patients consists of removing the infected CVC and replacing it with a new catheter at a different vascular access site. However, in cancer and hemodialysis patients with long-term surgically implantable silicone catheters, removal of the CVC and reinsertion of a new one at a different site might be difficult, or even impossible, because of the unavailability of other accessible vascular sites and the need to maintain infusion therapy. Furthermore, critically ill patients with short-term catheters often have underlying coagulopathy, which makes reinsertion of a new CVC at a different site, in the setting of CRBSIs, risky in terms of mechanical complications, such as pneumothorax, misplacement, or arterial puncture. Studies have also revealed that CRBSI patients may be associated with serious complications, including septic thrombosis, endocarditis and disseminated infection, particularly if caused by Staphylococcus aureus or Candida species. Furthermore, catheter retention in patients with CRBSIs is associated with a higher risk of relapse and poor response to antimicrobial therapy.
According to Maki et al., published in the Mayo Clinic Proceedings in 2006, there are approximately 250,000 CRBSIs annually in the U.S. Subsequent to this study, our estimates have ranged upwards to over 450,000 CLABSIs annually (see analysis in the table below). CRBSIs are associated with a 12% to 35% mortality rate and an attributable cost of $35,000 to $56,000 per episode.
We estimate that the potential market for Mino-Lok in the U.S. to be approximately $500 million to $1 billion as shown in the table below based on a target price of up to $300 per dose of each salvage flush treatment.
| Short-Term CVC | Long-Term CVC | Total | ||||||||||
| No. of Catheters | 3 million | 4 million | 7 million | |||||||||
| Avg. Duration (Days) | 12 | 100 | N/A | |||||||||
| Catheter Days | 36 million | 400 million | 436 million | |||||||||
| Infection Rate | 2/1,000 days | 1/1,000 days | N/A | |||||||||
| Catheters Infected | 72,000 | 400,000 | 472,000 | |||||||||
| Flushes/Catheter | 5 | 7 | 6.7 | |||||||||
| Total Salvage Flushes | 360,000 | 2,800,000 | 3,160,000 | |||||||||
Sources: Ann Intern Med 2000; 132:391-402, Clev Clin J Med 2011; 78(1):10-17, JAVA 2007; 12(1):17-27, J Inf Nurs 2004;27(4):245-250, Joint Commission website Monograph, CLABSI and Internal Estimates.
Under various plausible pricing scenarios, we believe that Mino-Lok would be cost saving to the healthcare system given that the removal of an infected CVC and replacement of a new catheter in a different venous access site is estimated by the Company to cost between $8,000 and $10,000. Furthermore, there are potential additional medical benefits, a reduction in patient discomfort and avoidance of serious adverse events with the Mino-Lok approach since the catheter remains in place and is not subject to manipulation. We believe there will be an economic argument to enhance the adoption of Mino-Lok by infection control committees at acute care institutions.
| 3 |
In January of 2017, the Company commissioned a primary market research study with MEDACore, a subsidiary of Leerink, a healthcare focused network with more than 35,000 healthcare professionals, including key opinion leaders, experienced practitioners and other healthcare professionals throughout North America, Europe, Asia and other locations around the world. This network includes approximately 55 clinical specialties, 21 basic sciences and 20 business specialties a third party survey of 31 physicians to qualify the need for catheter salvage in patients with infected, indwelling central venous lines, especially when the catheter is a tunneled or an implanted port. There were 19 infectious disease experts and 12 intensivists surveyed who all agreed that salvage would be preferable to catheter exchange to avoid catheter misplacements, blood clots, or vessel punctures that can potentially occur during reinsertion. Most were also concerned that viable venous access may not be available in patients who were vitally dependent on a central line.
Hydro-Lido
Overview
Hydro-Lido is a topical formulation of hydrocortisone and lidocaine that is intended for the treatment of hemorrhoids. To our knowledge, there are currently no FDA-approved prescription drug products for the treatment of hemorrhoids. Some physicians are known to prescribe topical steroids for the treatment of hemorrhoids. In addition, there are various strengths of topical combination prescription products containing hydrocortisone along with lidocaine or pramoxine, each a topical anesthetic, that are prescribed by physicians for the treatment of hemorrhoids. These products contain drugs that were in use prior to the start of the Drug Efficacy Study Implementation (“DESI”) program and are commonly referred to as DESI drugs. However, none of these single-agent or combination prescription products have been clinically evaluated for safety and efficacy and approved by the FDA for the treatment of hemorrhoids. Further, many hemorrhoid patients use over the counter (“OTC”) products as their first line therapy. OTC products contain any one of several active ingredients including glycerin, phenylephrine, pramoxine, white petrolatum, shark liver oil and/or witch hazel, for symptomatic relief.
Development of Hemorrhoids Drugs
Hemorrhoids are a common gastrointestinal disorder, characterized by anal itching, pain, swelling, tenderness, bleeding and difficulty defecating. In the U.S., hemorrhoids affect nearly 5% of the population, with approximately 10 million persons annually admitting to having symptoms of hemorrhoidal disease. Of these persons, approximately one third visit a physician for evaluation and treatment of their hemorrhoids. The data also indicate that for both sexes a peak of prevalence occurs from age 45 to 65 years with a subsequent decrease after age 65 years. Caucasian populations are affected significantly more frequently than African Americans, and increased prevalence rates are associated with higher socioeconomic status in men but not women. Development of hemorrhoids before age 20 is unusual. In addition, between 50% and 90% of the general U.S., Canadian and European population will experience hemorrhoidal disease at least once in life. Although hemorrhoids and other anorectal diseases are not life-threatening, individual patients can suffer from agonizing symptoms which can limit social activities and have a negative impact on the quality of life.
Hemorrhoids are defined as internal or external according to their position relative to the dentate line. Classification is important for selecting the optimal treatment for an individual patient. Accordingly, physicians use the following grading system referred to as the Goligher’s classification of internal hemorrhoids:
| Grade I | Hemorrhoids not prolapsed but bleeding. |
| Grade II | Hemorrhoids prolapse and reduce spontaneously with or without bleeding. |
| Grade III | Prolapsed hemorrhoids that require reduction manually. |
| Grade IV | Prolapsed and cannot be reduced including both internal and external hemorrhoids that are confluent from skin tag to inner anal canal. |
Development Activities to Date
In the fall of 2015, we completed dosing patients in a double-blind dose ranging placebo controlled Phase 2 study where six different formulations containing hydrocortisone and lidocaine in various strengths were tested against the vehicle control. The objectives of this study were to: 1) demonstrate the safety and efficacy of the formulations when applied twice daily for two weeks in subjects with Grade I or II hemorrhoids and 2) assess the potential contribution of lidocaine hydrochloride and hydrocortisone acetate, alone or in combination for the treatment of symptoms of Goligher’s Classification Grade I or II hemorrhoids.
| 4 |
Symptom improvement was observed based on a global score of disease severity (“GSDS”), and based on some of the individual signs and symptoms of hemorrhoids, specifically itching and overall pain and discomfort. Within the first few days of treatment, the combination products (containing both hydrocortisone and lidocaine) were directionally favorable versus the placebo and their respective individual active treatment groups (e.g., hydrocortisone or lidocaine alone) in achieving ‘almost symptom free’ or ‘symptom free’ status according to the GSDS scale. These differences suggest the possibility of a benefit for the combination product formulation.
Overall, results from adverse event reporting support the safety profile of all test articles evaluated in this study and demonstrate similar safety profiles as compared to the vehicle. The safety findings were unremarkable. There was a low occurrence of adverse events and a similar rate of treatment related adverse events across all treatment groups. The majority of adverse events were mild and only one was severe. None of the adverse events were serious and the majority of adverse events were recovered/resolved at the end of the study. There were only two subjects who were discontinued from the study due to adverse events.
In addition to the safety and dose-ranging information, information was obtained relating to the use of the GSDS as an assessment tool for measuring the effectiveness of the test articles. Individual signs and symptoms were also assessed but can vary from patient to patient. Therefore, the goal of the GSDS was to provide an assessment tool that could be used for all patients regardless of which signs and symptoms they are experiencing. The GSDS proved to be a more effective tool for assessing the severity of the disease and the effectiveness of the drug when compared to the assessment of the individual signs and symptoms. Citius believes that we can continue to develop this assessment tool as well as other patient reported outcome endpoints for use in the next trials and in the pivotal trial.
Information was also obtained about the formulation of the drug and the vehicle. As a result of this study, we believe that the performance of the active arms of the study relative to the vehicle can be improved by re-formulating our topical preparation. Therefore, we have initiated work on vehicle formulation and evaluation of higher potency steroids.
In June and July 2016, the Company engaged the Dominion Group, a leading provider of healthcare and pharmaceutical marketing research services. The primary market research was conducted to understand the symptoms that are most bothersome to patients better in order to develop meaningful endpoints for the clinical trials. We also learned about the factors that drive patients to seek medical attention for hemorrhoids in an effort to understand the disease impact on quality of life. The results of this survey are able to help us develop patient reported outcome evaluation tools. These tools can be used in clinical trials to evaluate the patients’ conditions and to assess the performance of the test articles.
A Phase 2b study will begin once the new formulation is completed and the updated evaluation tools are developed. This study will be a 300 patient four arm study of individuals with Class II and III hemorrhoids. The cost is estimated at approximately $4.0 million and is expected to require approximately one year to complete.
Market Opportunity
The current market for OTC and topical DESI formulations of hydrocortisone and lidocaine is highly fragmented, and includes approximately 20 million units of OTC hemorrhoid products and over 4 million prescriptions for non-approved prescription treatments. Several topical combination prescription products for the treatment of hemorrhoids are available containing hydrocortisone in strengths ranging from 0.5% to 3.0%, combined with lidocaine in strengths ranging from 1.0% to 3.0%. The various topical formulations include creams, ointments, gels, lotions, enemas, pads, and suppositories. The most commonly prescribed topical combination gel is sold as a branded generic product and contains 2.5% hydrocortisone and 3.0% lidocaine.
We believe there are currently no FDA-approved prescription drug products for the treatment of hemorrhoids. Although there are numerous prescription and OTC products commonly used to treat hemorrhoids, none possess proven safety and efficacy data generated from rigorously conducted clinical trials. We believe that a novel topical formulation of hydrocortisone and lidocaine designed to provide anti-inflammatory and anesthetic relief and which has an FDA-approved label specifically claiming the treatment of hemorrhoids will become an important treatment option for physicians who want to provide their patients with a therapy that has demonstrated safety and efficacy in treating this uncomfortable and often recurring disease. We believe that our Hydro-Lido product represents an attractive, low-risk product opportunity with meaningful upside potential.
Market Exclusivity
We believe that we will be the first company to conduct rigorous clinical trials and receive FDA approval of a topical hydrocortisone-lidocaine combination product for the treatment of hemorrhoids. If we receive FDA approval, we will qualify for 3 years of market exclusivity for our dosage strength and formulation. In addition, we will also be the only product on the market specifically proven to be safe and effective for the treatment of hemorrhoids. Generally, if a company conducts clinical trials and receives FDA approval of a product for which there are similar, but non FDA-approved, prescription products on the market, the manufacturers of the unapproved but marketed products are required to withdraw them from the market. However, the FDA has significant latitude in determining how to enforce its regulatory powers in these circumstances. We have not had any communication with the FDA regarding this matter and cannot predict what action, if any, the FDA will take with respect to the unapproved products.
| 5 |
We believe that should our product receive an FDA approval and demonstrate, proven safety and efficacy data, and if our products obtain 3 years of market exclusivity based on our dosage strength and formulation, Citius is likely to have a meaningful advantage in its pursuit of achieving a significant position in the market for topical combination prescription products for the treatment of hemorrhoids.
Sales and Marketing
We are primarily focused on identifying opportunities within the critical care and cancer care market segments. In our product acquisition criteria, we concentrate on markets that are highly influenced by key opinion leaders (KOLs) and have products that are prescribed by a relatively small number of physicians, yet provide large opportunities for growth and market share. This strategy allows for a manageable commercialization effort for our Company in terms of resources and capital. We also seek to provide cost-effective therapies that would be endorsed by payers, patients, and providers. We believe that we will be able to commercialize products within the scope of these criteria ourselves, and that we can create marketing synergies by having a common narrow audience for our marketing efforts (“several products in the bag for the same customer”).
For products that we own that fall out of the narrow scope criteria, we have identified pharmaceutical companies with large sales forces, experienced sales and marketing management teams, direct-to-consumer (“DTC”) capabilities, significantly larger resources than ours, and non-competing product portfolios that we believe would make excellent sales and marketing partners for us. We intend to license our mass audience, non-specialty products to such companies for sales and marketing.
Intellectual Property
We rely on a combination of patent, trade secret, copyright, and trademark laws, as well as confidentiality, licensing and other agreements, to establish and protect our proprietary rights. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates both in the U.S. and abroad. However, patent protection may not provide us with complete protection against competitors who seek to circumvent our patents. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely and will in the future rely on trade secret protection and confidentiality agreements to protect our interests.
Mino-Lok Intellectual Property
Mino-Lok is covered by an issued U.S. patent (no. 7,601,731), “Antimicrobials in Combination with Chelators and Ethanol for the Rapid Eradication of Microorganisms Embedded in Biofilm,” which was issued on October 13, 2009. This patent is a composition of matter patent and provides intellectual property protection until June 7, 2024. There are corresponding applications pending in Europe and Canada (European Application No. EP 1644024; Canadian Patent Application No. 0252852). On April 15, 2014, a patent application was filed for an enhanced formulation that provides greater stability of the reconstituted Mino-Lok solution. In June 2017, the Company was notified that US Patent Application 15/344,113 has been published by the US Patent Office with a publication date of June 1, 2017. This patent is a step forward for Mino-Lok as it overcomes limitations in mixing antimicrobial solutions where components may precipitate because of physical and/or chemical factors, thus limiting the stability of the post-mix solutions.
On May 14, 2014, LMB entered into a patent and technology license agreement with Novel Anti-Infective Therapeutics, Inc., (“NAT”) to develop and commercialize Mino-Lok on an exclusive, worldwide (except for South America), sub licensable basis. LMB incurred a one-time license fee in May 2014. On March 20, 2017, LMB entered into an amendment to the license agreement that expanded the licensed territory to include South America, providing LMB with worldwide rights. Under the license agreement, the Company will pay (i) an annual maintenance fee until commercial sales of a product subject to the license, (ii) upon commercialization, we will pay annual royalties on net sales of licensed products, (iii) and certain regulatory and milestone payments. Unless earlier terminated by NAT based on the failure to achieve certain development or commercial milestones, the license agreement remains in effect until the date that all patents licensed under the agreement have expired and all patent applications within the licensed patent rights have been cancelled, withdrawn or expressly abandoned.
Mino-Lok has received a Qualified Infectious Disease Product (“QIDP”) designation. QIDP provides New Drug Applications an additional 5 years of market exclusivity with Hatch-Waxman for a combined total of 8 ½ years regardless of patent protection.
Hydro-Lido Intellectual Property
We are developing a new formulation of Hydro-Lido which will have a unique combination of excipients as well as unique concentrations of the active ingredients. The goal is to have a product that is optimized for stability and activity. Once the formulation development is completed and data is obtained, we will apply for a patent on this new topical formulation.
| 6 |
We seek to achieve approval for Hydro-Lido by utilizing the FDA’s 505(b)(2) pathway. This pathway will provide 3 years of market exclusivity.
Competition
We operate in a highly competitive and regulated industry which is subject to rapid and frequent changes. We face significant competition from organizations that are pursuing drugs that would compete with the drug candidates that we are developing and the same or similar products that target the same conditions we intend to treat. Due to our limited resources, we may not be able to compete successfully against these organizations, which include many large, well-financed and experienced pharmaceutical and biotechnology companies, as well as academic and research institutions and government agencies.
Mino-Lok Competition
Currently, the only alternative to Mino-Lok in the treatment of infected CVCs in CRBSI/CLABSI patients of which we are aware, is the SOC of removing the culprit CVC and replacing a new CVC at a different vascular site. Citius is not aware of any Investigational New Drug Applications (“INDs”) for a salvage antibiotic lock solution and does not expect any to be forthcoming due to the difficulty of meeting the necessary criteria to be effective and practical.
At this time, there are no pharmacologic agents approved in the U.S. for the prevention or treatment of CLABSIs in central venous catheters. Citius is aware that there are several agents in development for prevention but none for salvage. The most prominent of these appear to be Neutrolin from CorMedix and B-Lock from Great Lakes Pharmaceuticals, Inc. (“GLP”).
Neutrolin® (CorMedix Inc.)
Neutrolin is a formulation of Taurolidine 1.35%, Citrate 3.5%, and Heparin 1000 units/mL. Neutrolin is an anti-microbial catheter lock solution being developed by CorMedix to prevent CRBSIs and to prevent clotting. In January 2015, the U.S. Food and Drug Administration (the “FDA”) granted Fast Track and Qualified Infectious Disease Product (“QIDP”) designations for Neutrolin. In December 2015, CorMedix initiated its Phase 3 clinical trial in hemodialysis patients in the United States. The clinical trial named Catheter Lock Solution Investigational Trial, or LOCK-IT-100 is a prospective, multicenter, randomized, double-blind, placebo-controlled, active control trial designed to show efficacy and safety of Neutrolin in preventing CRBSIs in subjects receiving hemodialysis therapy. On April 20, 2017, CorMedix provided an update on the LOCK-IT-100 trial. CorMedix had enrolled 368 patients to date and completed a safety review by an independent Data and Safety Monitoring Board (“DSMB”) of the first 279 patients. The DSMB concluded that it was safe to continue the trial as designed; however, CorMedix initiated discussions with the FDA to make some protocol changes to include one or more interim efficacy analyses. According to CorMedix, the FDA accepted the CorMedix proposal. Recently, CorMedix stated that the LOCK-IT-100 is an event-driven study and that study completion would be dependent upon capturing 56 total CRBSI events. CorMedix now believes that an interim efficacy analysis will occur in the fourth quarter 2017, followed by enrollment completion in the second quarter 2018. The study is expected to conclude around year end 2018.
CorMedix is assessing the structure of its second planned Phase 3 study to seek possible efficiencies and improvements in design and execution.
B-Lock™ (Great Lakes Pharmaceuticals, Inc.)
B-Lock is a triple combination of trimethoprim, EDTA and ethanol from Great Lakes Pharmaceuticals, Inc. (“GLP”). On July 24, 2012, GLP announced the initiation of a clinical study of B-Lock. We are unaware as to the progress or results of these studies. In addition, we are not aware of any IND being filed in the US for B-Lock, nor are we aware of any clinical studies to support salvage of infected catheters in bacteremic patients.
Neither of these lock solutions have been shown to be effective in salvaging catheters in bacteremic patients as Mino-Lok is intended to do, and Citius does not expect that either would be pursued for this indication.
Hydro-Lido Competition
The primary competition in the hemorrhoid market is non-prescription over the counter products. When approved, Hydro-Lido will be the only prescription product for the treatment of hemorrhoids.
Supply and Manufacturing
We do not currently have and we do not intend to set up our own manufacturing facilities. We expect to use approved contract manufacturers for manufacturing our products in all stages of development after we file for FDA approval. Each of our domestic and foreign contract manufacturing establishments, including any contract manufacturers we may decide to use, must be listed in the New Drug Application (“NDA”) and must be registered with the FDA. Also, the FDA imposes substantial annual fees on manufacturers of branded products.
| 7 |
In general, our suppliers purchase raw materials and supplies on the open market. Substantially all such materials are obtainable from a number of sources so that the loss of any one source of supply would not have a material adverse effect on us.
If we elect to conduct product development and manufacturing, we will be subject to regulation under various federal and state laws, including the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, the Controlled Substances Act and other present and potential future federal, state or local regulations.
We have contracted with proven suppliers and manufacturers for active pharmaceutical ingredient, development and packaging. We are confident that all materials meet or will meet specifications discussed at the chemistry, manufacturing and controls meeting with the FDA.
Regulatory Strategy
United States Government Regulation
The research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing, among other things, of our products are extensively regulated by governmental authorities in the United States and other countries. Citius’ products may be classified by the FDA as a drug or a medical device depending upon the indications for use or claims. Because certain of our product candidates are considered as medical devices and others are considered as drugs for regulatory purposes, we intend to submit applications to regulatory agencies for approval or clearance of both medical devices and pharmaceutical product candidates.
In the United States, the FDA regulates drugs and medical devices under the Federal Food, Drug, and Cosmetic Act and the agency’s implementing regulations. If Citius fails to comply with the applicable United States requirements at any time during the product development process, clinical testing, and the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, adverse publicity, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on Citius.
Foreign Regulatory Requirements
Citius and any collaborative partners may be subject to widely varying foreign regulations, which may be different from those of the FDA, governing clinical trials, manufacture, product registration and approval and pharmaceutical sales. Whether or not FDA approval has been obtained, Citius or its collaboration partners must obtain a separate approval for a product by the comparable regulatory authorities of foreign countries prior to the commencement of product marketing in such countries. In certain countries, regulatory authorities also establish pricing and reimbursement criteria. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. In addition, under current United States law, there are restrictions on the export of products not approved by the FDA, depending on the country involved and the status of the product in that country.
International sales of medical devices manufactured in the U.S. that are not approved by the FDA for use in the U.S., or are banned or deviate from lawful performance standards, are subject to FDA export requirements. Exported devices are subject to the regulatory requirements of each country to which the device is exported. Some countries do not have medical device regulations, but in most foreign countries, medical devices are regulated. Frequently, regulatory approval may first be obtained in a foreign country prior to application in the U.S. to take advantage of differing regulatory requirements. Most countries outside of the U.S. require that product approvals be recertified on a regular basis, generally every 5 years. The recertification process requires that Citius evaluate any device changes and any new regulations or standards relevant to the device and conduct appropriate testing to document continued compliance. Where recertification applications are required, they must be approved in order to continue selling Citius’ products in those countries.
In the European Union, in order for a product to be marketed and sold, it is required to comply with the Medical Devices Directive and obtain CE Mark certification. The CE Mark certification encompasses an extensive review of the applicant’s quality management system which is inspected by a notified body’s auditor as part of a stage 1 and 2 International Organization for Standardization (“ISO”) 13485:2016 audit, in accordance with worldwide recognized ISO standards and applicable European Medical Devices Directives for quality management systems for medical device manufacturers. Once the quality management system and design dossier has been successfully audited by a notified body and reviewed and approved by a competent authority, a CE certificate for the medical device will be issued. Applicants are also required to comply with other foreign regulations such as the requirement to obtain Ministry of Health, Labor and Welfare approval before a new product can be launched in Japan. The time required to obtain these foreign approvals to market Citius’ products may vary from U.S. approvals, and requirements for these approvals may differ from those required by the FDA.
| 8 |
Medical device laws and regulations are in effect in many of the countries in which Citius may do business outside the United States. These laws and regulations range from comprehensive device approval requirements for Citius’ medical device product to requests for product data or certifications. The number and scope of these requirements are increasing. Citius may not be able to obtain regulatory approvals in such countries and may be required to incur significant costs in obtaining or maintaining its foreign regulatory approvals. In addition, the export of certain of Citius’ products which have not yet been cleared for domestic commercial distribution may be subject to FDA export restrictions. Any failure to obtain product approvals in a timely fashion or to comply with state or foreign medical device laws and regulations may have a serious adverse effect on Citius’ business, financial condition or results of operations.
Employees
As of September 30, 2017, the Company had 7 employees and various consultants providing support. Through our consulting and collaboration arrangements, and including our Scientific Advisory Board, we have access to more than 30 additional professionals, who possess significant expertise in business development, legal, accounting, regulatory affairs, clinical operations and manufacturing. We also rely upon a network of consultants to support our clinical studies and manufacturing efforts.
Executive Officers of Citius
Myron Holubiak, President, Chief Executive Officer and Director – Mr. Holubiak, 70, was appointed President, Chief Executive Officer and Director in March 2016. He previously served as a Director of Citius since October 2015 and was the founder and Chief Executive Officer and President of Leonard-Meron Biosciences, Inc., an acquired subsidiary of Citius, from March 2013 until March 2016.
Leonard Mazur, Executive Chairman and Secretary – Mr. Mazur, 72, has been a member of the Board since September 2014. Mr. Mazur previously served as Chief Executive Officer, President, and Chief Operating Officer from September 2014 until March 2016.
Jaime Bartushak, Chief Financial Officer and Principal Financial Officer – Mr. Bartushak, 50, was appointed as Chief Financial Officer in November 2017. Previously, he was one of the founders and Chief Financial Officer of Leonard-Meron Biosciences, Inc., an acquired subsidiary of Citius,
Other Information
While the Company was not previously subject to the filing requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”), it filed certain reports with the Securities and Exchange Commission (“SEC”) on a voluntarily basis. On October 22, 2015, the Company registered its Common Stock under the Exchange Act and the filing of the reports with the SEC became mandatory. You may read and copy these reports and other information at the SEC’s Public Reference Room at 100 F Street N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 or e-mail the SEC at publicinfo@sec.gov for more information on the operation of the public reference room. Our SEC filings are also available at the SEC’s website at http://www.sec.gov. Our internet address is http://www.citiuspharma.com.
Our business is subject to numerous risks. The following important factors, among others, could have a material adverse effect on our business, financial condition or results of operations.
| 9 |
Risks related to our Business and our Industry
Citius has a history of net losses and expects to incur losses for the foreseeable future. We may never generate revenues or, if we are able to generate revenues, achieve profitability.
Citius was formed as a limited liability company in 2007 and since its inception has incurred a net loss in each of its previous operating years. Our ability to become profitable depends upon our ability to generate revenues from sales of our product candidates. Citius has been focused on product development and has not generated any revenues to date. Citius has incurred losses in each period of our operations, and we expect to continue to incur losses for the foreseeable future. These losses are likely to continue to adversely affect our working capital, total assets and shareholders’ equity (deficit). The process of developing our products requires significant clinical, development and laboratory testing and clinical trials. In addition, commercialization of our product candidates will require that we obtain necessary regulatory approvals and establish sales, marketing and manufacturing capabilities, either through internal hiring or through contractual relationships with others. We expect to incur substantial losses for the foreseeable future as a result of anticipated increases in our research and development costs, including costs associated with conducting preclinical testing and clinical trials, and regulatory compliance activities. Citius incurred net losses of $10,384,953, $8,295,698 and $2,902,268 for the years ended September 30, 2017, 2016 and 2015, respectively. At September 30, 2017, Citius had stockholders’ equity of $21,947,388 and an accumulated deficit of $27,721,200. Citius’ net cash used for operating activities was $7,971,205, $5,900,421 and $2,385,416 for the years ended September 30, 2017, 2016 and 2015, respectively.
Our ability to generate revenues and achieve profitability will depend on numerous factors, including success in:
| ● | developing and testing product candidates; | |
| ● | receiving regulatory approvals; | |
| ● | commercializing our products; | |
| ● | manufacturing commercial quantities of our product candidates at acceptable cost levels; and | |
| ● | establishing a favorable competitive position. |
Many of these factors will depend on circumstances beyond our control. We cannot assure you that any of our products will be approved by the FDA, that we will successfully bring any product to market or, if so, that we will ever become profitable.
There is substantial doubt about our ability to continue as a going concern.
Our independent registered accountants report on our September 30, 2017 consolidated financial statements contains an emphasis of a matter regarding substantial doubt about our ability to continue as a going concern, that the consolidated financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets, or the amounts and classification of liabilities that may result if we do not continue as a going concern. Currently, we do not have sufficient capital to continue our operations after the next six months. You should not rely on our consolidated balance sheet as an indication of the amount of proceeds that would be available to satisfy claims of creditors, and potentially be available for distribution to shareholders, in the event of liquidation.
We need to secure additional financing.
We anticipate that we will incur operating losses for the foreseeable future. We have received gross proceeds of approximately $14.6 million from our public and private placement offerings through September 2017. Additionally, in connection with the acquisition of LMB our Executive Chairman, Leonard Mazur, made an equity investment of $3.0 million in March 2016. Mr. Mazur has also loaned the Company $4,710,000 pursuant to convertible promissory notes. On August 8, 2017, these notes and accrued interest of $76,240 were converted into 1,547,067 shares of common stock at a price of $3.09 per share as part of an underwritten public offering which closed on the same date.
The Company has engaged Paulson Investment Company, LLC to secure debt financing. We may need to seek additional financing, including from affiliates, to continue our clinical programs and manufacturing for clinical programs.
The amount and timing of our future funding requirements will depend on many factors, including, but not limited to:
| ● | the rate of progress and cost of our trials and other product development programs for our product candidates; | |
| ● | the costs and timing of obtaining licenses for additional product candidates or acquiring other complementary technologies; | |
| ● | the timing of any regulatory approvals of our product candidates; | |
| ● | the costs of establishing sales, marketing and distribution capabilities; and | |
| ● | the status, terms and timing of any collaborative, licensing, co-promotion or other arrangements. |
| 10 |
We will need to access the capital markets in the future for additional capital for research and development and for operations. Traditionally, pharmaceutical companies have funded their research and development expenditures through raising capital in the equity markets. Declines and uncertainties in these markets over the past several years have severely restricted raising new capital and have affected companies’ ability to continue to expand or fund existing research and development efforts. If these economic conditions continue or become worse, our future cost of equity or debt capital and access to the capital markets could be adversely affected. If we are not successful in securing additional financing, we may be required to delay significantly, reduce the scope of or eliminate one or more of our research or development programs, downsize our general and administrative infrastructure, or seek alternative measures to avoid insolvency, including arrangements with collaborative partners or others that may require us to relinquish rights to certain of our technologies or product candidates.
We are a late-stage development company with an unproven business strategy and may never achieve commercialization of our therapeutic products or profitability.
Our strategy of using collaborative partners to assist us in the development of our therapeutic products is unproven. Our success will depend upon our ability to enter into additional collaboration agreements on favorable terms and to select an appropriate commercialization strategy for each potential therapeutic product we and our collaborators choose to pursue. If we are not successful in implementing our strategy to commercialize our potential therapeutic products, we may never achieve, maintain or increase profitability. Our ability to successfully commercialize any of our products or product candidates will depend, among other things, on our ability to:
| ● | successfully complete our clinical trials; | |
| ● | produce, through a validated process, sufficiently large quantities of our drug compound(s) to permit successful commercialization; | |
| ● | receive marketing approvals from the FDA and similar foreign regulatory authorities; | |
| ● | establish commercial manufacturing arrangements with third-party manufacturers; | |
| ● | build and maintain strong sales, distribution and marketing capabilities sufficient to launch commercial sales of the drug(s) or establish collaborations with third parties for such commercialization; | |
| ● | secure acceptance of the drug(s) from physicians, health care payers, patients and the medical community; and | |
| ● | manage our spending as costs and expenses increase due to clinical trials, regulatory approvals and commercialization. |
There are no guarantees that we will be successful in completing these tasks. If we are unable to successfully complete these tasks, we may not be able to commercialize any of our product candidates in a timely manner, or at all, in which case we may be unable to generate sufficient revenues to sustain and grow our business. If we experience unanticipated delays or problems, our development costs could substantially increase and our business, financial condition and results of operations will be adversely affected.
We may fail to realize any of the anticipated benefits of the recent merger.
The success of our recent merger with Leonard-Meron Biosciences, Inc. will depend on, among other things, our ability to realize anticipated benefits and to combine the businesses of the Company and LMB in a manner that achieves synergy and a shared strategy but that does not materially disrupt the existing activities of the companies. If we are not able to successfully achieve these objectives, the anticipated benefits of the merger may not be realized fully, if at all, or may take longer to realize than expected.
We face significant risks in our product candidate development efforts.
Our business depends on the successful development and commercialization of our product candidates. We are not permitted to market any of our product candidates in the United States until we receive approval of an NDA from the FDA, or in any foreign jurisdiction until we receive the requisite approvals from such jurisdiction. The process of developing new drugs and/or therapeutic products is inherently complex, unpredictable, time-consuming, expensive and uncertain. We must make long-term investments and commit significant resources before knowing whether our development programs will result in drugs that will receive regulatory approval and achieve market acceptance. Product candidates that appear to be promising at all stages of development may not reach the market for a number of reasons that may not be predictable based on results and data of the clinical program. Product candidates may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than had been anticipated, may not be able to achieve the pre-defined clinical endpoints due to statistical anomalies even though clinical benefit may have been achieved, may fail to receive necessary regulatory approvals, may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality, or may fail to achieve market acceptance.
| 11 |
We cannot predict whether or when we will obtain regulatory approval to commercialize our product candidates that are under development and will be further developed using the proceeds of our private placements and we cannot, therefore, predict the timing of any future revenues from these product candidates, if any. The FDA has substantial discretion in the drug approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons. For example, the FDA:
| ● | could determine that we cannot rely on Section 505(b)(2) for any of our product candidates; | |
| ● | could determine that the information provided by us was inadequate, contained clinical deficiencies or otherwise failed to demonstrate the safety and effectiveness of any of our product candidates for any indication; | |
| ● | may not find the data from clinical trials sufficient to support the submission of an NDA or to obtain marketing approval in the United States, including any findings that the clinical and other benefits of our product candidates outweigh their safety risks; | |
| ● | may disagree with our trial design or our interpretation of data from preclinical studies or clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our trials; | |
| ● | may determine that we have identified the wrong reference listed drug or drugs or that approval of our Section 505(b)(2) application for any of our product candidates is blocked by patent or non-patent exclusivity of the reference listed drug or drugs; | |
| ● | may identify deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for the manufacturing of our product candidates; | |
| ● | may approve our product candidates for fewer or more limited indications than we request, or may grant approval contingent on the performance of costly post-approval clinical trials; | |
| ● | may change its approval policies or adopt new regulations; or | |
| ● | may not approve the labeling claims that we believe are necessary or desirable for the successful commercialization of our product candidates. |
Any failure to obtain regulatory approval of our product candidates would significantly limit our ability to generate revenues, and any failure to obtain such approval for all of the indications and labeling claims we deem desirable could reduce our potential revenues.
The results of pre-clinical studies and completed clinical trials are not necessarily predictive of future results, and our current product candidates may not have favorable results in later studies or trials.
Pre-clinical studies and Phase 1 and Phase 2 clinical trials are not primarily designed to test the efficacy of a product candidate in the general population, but rather to test initial safety, to study pharmacokinetics and pharmacodynamics, to study limited efficacy in a small number of study patients in a selected disease population, and to identify and attempt to understand the product candidate’s side effects at various doses and dosing schedules. Success in pre-clinical studies or completed clinical trials does not ensure that later studies or trials, including continuing pre-clinical studies and large-scale clinical trials, will be successful nor does it necessarily predict future results. Favorable results in early studies or trials may not be repeated in later studies or trials, and product candidates in later stage trials may fail to show acceptable safety and efficacy despite having progressed through earlier trials. In addition, the placebo rate in larger studies may be higher than expected.
We may be required to demonstrate through large, long-term outcome trials that our product candidates are safe and effective for use in a broad population prior to obtaining regulatory approval.
| 12 |
There is typically a high rate of attrition from the failure of product candidates proceeding through clinical trials. In addition, certain subjects in our clinical trials may respond positively to placebo treatment - these subjects are commonly known as “placebo responders” - making it more difficult to demonstrate efficacy of the test drug compared to placebo. This effect is likely to be observed in the treatment of hemorrhoids. If any of our product candidates fail to demonstrate sufficient safety and efficacy in any clinical trial, we will experience potentially significant delays in, or may decide to abandon development of that product candidate. If we abandon or are delayed in our development efforts related to any of our product candidates, we may not be able to generate any revenues, continue our operations and clinical studies, or become profitable. Our reputation in the industry and in the investment community would likely be significantly damaged. It may not be possible for us to raise funds in the public or private markets, and our stock price would likely decrease significantly.
If we are unable to file for approval under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act or if we are required to generate additional data related to safety and efficacy in order to obtain approval under Section 505(b)(2), we may be unable to meet our anticipated development and commercialization timelines.
Our current plans for filing additional NDAs for our product candidates include efforts to minimize the data we will be required to generate in order to obtain marketing approval for our additional product candidates and therefore possibly obtain a shortened review period for the applications. The timeline for filing and review of our NDAs is based upon our plan to submit those NDAs under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, wherein we will rely in part on data in the public domain or elsewhere. Depending on the data that may be required by the FDA for approval, some of the data may be related to products already approved by the FDA. If the data relied upon is related to products already approved by the FDA and covered by third-party patents we would be required to certify that we do not infringe the listed patents or that such patents are invalid or unenforceable. As a result of the certification, the third party would have 45 days from notification of our certification to initiate an action against us. In the event that an action is brought in response to such a certification, the approval of our NDA could be subject to a stay of up to 30 months or more while we defend against such a suit. Approval of our product candidates under Section 505(b)(2) may therefore be delayed until patent exclusivity expires or until we successfully challenge the applicability of those patents to our product candidates. Alternatively, we may elect to generate sufficient additional clinical data so that we no longer rely on data which triggers a potential stay of the approval of our product candidates. Even if no exclusivity periods apply to our applications under Section 505(b)(2), the FDA has broad discretion to require us to generate additional data on the safety and efficacy of our product candidates to supplement third-party data on which we may be permitted to rely. In either event, we could be required, before obtaining marketing approval for any of our product candidates, to conduct substantial new research and development activities beyond those we currently plan to engage in order to obtain approval of our product candidates. Such additional new research and development activities would be costly and time consuming.
We may not be able to obtain shortened review of our applications, and the FDA may not agree that our products qualify for marketing approval. If we are required to generate additional data to support approval, we may be unable to meet our anticipated development and commercialization timelines, may be unable to generate the additional data at a reasonable cost, or at all, and may be unable to obtain marketing approval of our product candidates. In addition, notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, some pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA’s interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) application that we submit.
Even if we receive regulatory approval to commercialize our product candidates, post-approval marketing and promotion of products is highly regulated by the FDA, and marketing campaigns which violate FDA standards may result in adverse consequences including regulatory enforcement action by the FDA as well as follow-on actions filed by consumers and other end-payers, which could result in substantial fines, sanctions and damage awards against us, any of which could harm our business.
Post-approval marketing and promotion of drugs, standards and regulations for direct-to-consumer advertising, dissemination of off-label product information, industry-sponsored scientific and educational activities and promotional activities via the Internet are heavily scrutinized and regulated by the FDA. Drugs may only be marketed for approved indications and in accordance with provisions of the FDA approved labels. Failure to comply with such requirements may result in adverse publicity, warning letters issued by the FDA, and civil or criminal penalties.
In the event the FDA discovers new violations, we could face penalties in the future including the FDA’s issuance of a cease and desist order, impounding of our products, and civil or criminal penalties. As a follow-on to such governmental enforcement activities, consumers and other end-payers of the product may initiate action against us claiming, among other things, fraudulent misrepresentation, civil RICO, unfair competition, violation of various state consumer protection statues and unjust enrichment. If the plaintiffs in such follow-on actions are successful, we could be subject to various damages, including compensatory damages, treble damages, punitive damages, restitution, disgorgement, prejudgment and post-judgment interest on any monetary award, and the reimbursement of the plaintiff’s legal fees and costs, any of which could have an adverse effect on our revenue, business and financial prospects.
| 13 |
Even if we receive regulatory approval to commercialize our product candidates, our ability to generate revenues from any resulting drugs will be subject to a variety of risks, many of which are out of our control.
Even if our product candidates obtain regulatory approval, those drugs may not gain market acceptance among physicians, patients, healthcare payers or the medical community. The indication may be limited to a subset of the population or we may implement a distribution system and patient access program that is limited. Coverage and reimbursement of our product candidates by third-party payers, including government payers, generally is also necessary for optimal commercial success. We believe that the degree of market acceptance and our ability to generate revenues from such drugs will depend on a number of factors, including:
| ● | prevalence and severity of any side effects; | |
| ● | results of any post-approval studies of the drug; | |
| ● | potential or perceived advantages or disadvantages over alternative treatments including generics; | |
| ● | the relative convenience and ease of administration and dosing schedule; | |
| ● | strength of sales, marketing and distribution support; | |
| ● | price of any future drugs, if approved, both in absolute terms and relative to alternative treatments; | |
| ● | the effectiveness of our or any future collaborators’ sales and marketing strategies; | |
| ● | the effect of current and future healthcare laws on our product candidates; | |
| ● | availability of coverage and reimbursement from government and other third-party payers; | |
| ● | patient access programs that require patients to provide certain information prior to receiving new and refill prescriptions; | |
| ● | requirements for prescribing physicians to complete certain educational programs for prescribing drugs; | |
| ● | the willingness of patients to pay out of pocket in the absence of government or third-party coverage; and | |
| ● | product labeling or product insert requirements of the FDA or other regulatory authorities. |
If approved, our product candidates may fail to achieve market acceptance or generate significant revenue to achieve or sustain profitability. In addition, our efforts to educate the medical community and third-party payers on the benefits of our product candidates may require significant resources and may never be successful.
Even if approved for marketing by applicable regulatory bodies, we will not be able to create a market for any of our products if we fail to establish marketing, sales and distribution capabilities, or fail to enter into arrangements with third parties.
Our strategy with our product candidates is to outsource to third parties, all or most aspects of the product development process, as well as marketing, sales and distribution activities. Currently, we do not have any sales, marketing or distribution capabilities. In order to generate sales of any product candidates that receive regulatory approval, we must either acquire or develop an internal marketing and sales force with technical expertise and with supporting distribution capabilities or make arrangements with third parties to perform these services for us. The acquisition or development of a sales and distribution infrastructure would require substantial resources, which may divert the attention of our management and key personnel and defer our product development efforts. To the extent that we enter into marketing and sales arrangements with other companies, our revenues will depend on the efforts of others. These efforts may not be successful. If we fail to develop sales, marketing and distribution channels, or enter into arrangements with third parties, we will experience delays in product sales and incur increased costs.
| 14 |
The markets in which we operate are highly competitive and we may be unable to compete successfully against new entrants or established companies.
Competition in the pharmaceutical and medical products industries is intense and is characterized by costly and extensive research efforts and rapid technological progress. We are aware of several pharmaceutical companies also actively engaged in the development of therapies for the same conditions we are targeting. Many of these companies have substantially greater research and development capabilities as well as substantially greater marketing, financial and human resources than we do. In addition, many of these companies have significantly greater experience than us in undertaking pre-clinical testing, human clinical trials and other regulatory approval procedures. Our competitors may develop technologies and products that are more effective than those we are currently marketing or researching and developing. Such developments could render our products, if approved, less competitive or possibly obsolete. We are also competing with respect to marketing capabilities and manufacturing efficiency, areas in which we have limited experience. Mergers, acquisitions, joint ventures and similar events may also significantly increase the competition. New developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical and medical technology industries at a rapid pace. These developments may render our products and product candidates obsolete or noncompetitive. Compared to us, many of our potential competitors have substantially greater:
| ● | research and development resources, including personnel and technology; | |
| ● | regulatory experience; | |
| ● | product candidate development and clinical trial experience; | |
| ● | experience and expertise in exploitation of intellectual property rights; and | |
| ● | access to strategic partners and capital resources. |
As a result of these factors, our competitors may obtain regulatory approval of their products more rapidly than we can or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs or surgical approaches that are more effective, more useful and less costly than ours and may also be more successful in manufacturing and marketing their products. In addition, our competitors may be more effective than us in commercializing their products and as a result, our business and prospects might be materially harmed.
Physicians and patients might not accept and use any of our products for which regulatory approval is obtained.
Even if the FDA approves one of our product candidates, physicians and patients might not accept and use it. Acceptance and use of our products will depend upon a number of factors, including:
| ● | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our products; | |
| ● | cost-effectiveness of our product relative to competing product or therapies; | |
| ● | availability of reimbursement for our product from government or other healthcare payers; and | |
| ● | effective marketing and distribution efforts by us and our licensees and distributors, if any. |
If our current product candidates are approved, we expect their sales to generate substantially all of our revenues for the foreseeable future, and as a result, the failure of these products to find market acceptance would harm our business and would require us to seek additional financing.
Our two product candidates, Mino-Lok and Hydro-Lido, are combination products consisting of components that have each been separately approved by the FDA for other indications and which are commercially available and marketed by other companies. Our approval under 505(b)(2) does not preclude physicians, pharmacists and patients from obtaining individual drug products and titrating the dosage of these drug products as close to our approved dose as possible.
Our Hydro-Lido product candidate for the treatment of hemorrhoids is a combination product consisting of two drugs, hydrocortisone and lidocaine, that have each been separately approved by the FDA for other indications and which are commercially available and marketed by other companies. Hydrocortisone creams are available from strengths ranging from 0.5% to 2.5% and lidocaine creams are also available in strengths up to 5%. From our market analysis and discussions with a limited number of physicians, we know that patients sometimes obtain two separate cream products and co-administer them as prescribed, giving them a combination treatment which could be very similar to what we intend to study and seek approval for. As a branded, FDA-approved product with safety and efficacy data, we intend to price our product substantially higher than the generically available individual creams. We will then have to convince third-party payers and pharmacy benefit managers of the advantages of our product and justify our premium pricing. We may encounter resistance from these entities and will then be dependent on patients’ willingness to pay the premium and not seek alternatives. In addition, pharmacists often suggest lower cost prescription treatment alternatives to both physicians and patients. Our 505(b)(2) approval and the market exclusivity we may receive will not guarantee that such alternatives will not exist, that substitution will not occur, or that there will be immediate acceptance to our pricing by payer formularies.
| 15 |
Our Mino-Lok solution contains minocycline, disodium ethylenediaminetetraacetic acid (edetate), and ethyl alcohol, all of which have been separately approved by the FDA for other indications, or are used as excipients in other parenteral products.
Our ability to generate product revenues will be diminished if our products sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our products, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
| ● | government and health administration authorities; | |
| ● | private health maintenance organizations and health insurers; and | |
| ● | other healthcare payers. |
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payers increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if our product candidates are approved by the FDA, insurance coverage might not be available, and reimbursement levels might be inadequate, to cover our products. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for our products, once approved, market acceptance of such products could be reduced. Proposals to modify the current health care system in the U.S. to improve access to health care and control its costs are continually being considered by the federal and state governments. In March 2010, the U.S. Congress passed landmark healthcare legislation. We cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically. Members of the U.S. Congress and some state legislatures are seeking to overturn at least portions of the legislation and we expect they will continue to review and assess this legislation and possibly alternative health care reform proposals. We cannot predict whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
Health administration authorities in countries other than the U.S. may not provide reimbursement for our products at rates sufficient for us to achieve profitability, or at all. Like the U.S., these countries have considered health care reform proposals and could materially alter their government-sponsored health care programs by reducing reimbursement rates. Any reduction in reimbursement rates under Medicare or foreign health care programs could negatively affect the pricing of our products. If we are not able to charge a sufficient amount for our products, then our margins and our profitability will be adversely affected.
We rely exclusively on third parties to formulate and manufacture our product candidates.
We do not have and do not intend to establish our own manufacturing facilities. Consequently, we lack the physical plant to formulate and manufacture our own product candidates, which are currently being manufactured entirely by a commercial third party. If any additional product candidate we might develop or acquire in the future receives FDA approval, we will rely on one or more third-party contractors to manufacture our products. If, for any reason, we become unable to rely on our current source or any future source to manufacture our product candidates, either for clinical trials or, for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers to manufacture compounds for preclinical, clinical and commercial purposes. We might not be successful in identifying additional or replacement third-party manufacturers, or in negotiating acceptable terms with any that we do identify. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance might be materially affected.
In addition, before any of our collaborators can begin to commercially manufacture our product candidates, each must obtain regulatory approval of the manufacturing facility and process. Manufacturing of drugs for clinical and commercial purposes must comply with the FDA’s Current Good Manufacturing Practices, or cGMP, and applicable non-U.S. regulatory requirements. The cGMP requirements govern quality control and documentation policies and procedures. Complying with cGMP and non-U.S. regulatory requirements will require that we expend time, money, and effort in production, recordkeeping, and quality control to assure that the product meets applicable specifications and other requirements. Our contracted manufacturing facilities must also pass a pre-approval inspection prior to FDA approval. Failure to pass a pre- approval inspection might significantly delay FDA approval of our products. If any of our collaborators fails to comply with these requirements, we would be subject to possible regulatory action which could limit the jurisdictions in which we are permitted to sell our products. As a result, our business, financial condition, and results of operations might be materially harmed.
| 16 |
Our reliance on a limited number of third-party manufacturers exposes us to the following risks:
| ● | We might be unable to identify manufacturers for commercial supply on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would generally require compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any; | |
| ● | Our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical and commercial needs, if any; | |
| ● | Our contract manufacturers might not perform as agreed or might not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products; | |
| ● | Currently, our contract manufacturer is foreign, which increases the risk of shipping delays and adds the risk of import restrictions; | |
| ● | Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have complete control over third-party manufacturers’ compliance with these regulations and standards; | |
| ● | If any third-party manufacturer makes improvements in the manufacturing process for our products, we might not own, or might have to share, the intellectual property rights to the innovation with our licensors; |
| ● | Operations of our third-party manufacturers or suppliers could be disrupted by conditions unrelated to our business or operations, including a bankruptcy of the manufacturer or supplier, and | |
| ● | We might compete with other companies for access to these manufacturers’ facilities and might be subject to manufacturing delays if the manufacturers give other clients higher priority than us. |
Each of these risks could delay our clinical trials or the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates and could result in higher costs or deprive us of potential product revenues. As a result, our business, financial condition, and results of operations might be materially harmed.
We will be dependent on third-party contract research organizations to conduct all of our future human studies.
We will be dependent on third-party research organizations to conduct all of our human studies with respect to pharmaceutical products that we may develop in the future. If we are unable to obtain any necessary testing services on acceptable terms, we may not complete our product development efforts in a timely manner. If we rely on third parties for human studies, we may lose some control over these activities and become too dependent upon these parties. These third parties may not complete testing activities on schedule or when we so request. We may not be able to secure and maintain suitable research organizations to conduct our human studies. We are responsible for confirming that each of our clinical trials is conducted in accordance with our general plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for our future product candidates.
| 17 |
Any termination or breach by or conflict with our strategic partners or licensees could harm our business.
If we or any of our collaborators or licensees fail to renew or terminate any of our collaboration or license agreements or if either party fails to satisfy its obligations under any of our collaboration or license agreements or complete them in a timely manner, we could lose significant sources of revenue, which could result in volatility in our future revenue. In addition, our agreements with our collaborators and licensees may have provisions that give rise to disputes regarding the rights and obligations of the parties. These and other possible disagreements could lead to termination of the agreement or delays in collaborative research, development, supply or commercialization of certain products, or could require or result in litigation or arbitration. Any such conflicts with our collaborators could reduce our ability to obtain future collaboration agreements and could have a negative impact on our relationship with existing collaborators, adversely affecting our business and revenues. Finally, any of our collaborations or license agreements may prove to be unsuccessful.
If we are unable to retain or hire additional qualified personnel, our ability to grow our business might be harmed.
We utilize the services of a clinical management team on part-time basis to assist us in managing our Phase 2 and Phase 3 trials. While we believe this will provide us with sufficient staffing for our current development efforts, we will need to hire or contract with additional qualified personnel with expertise in preclinical testing, clinical research and testing, government regulation, formulation and manufacturing and sales and marketing in connection with the continued development, regulatory approval and commercialization of our product candidates. We compete for qualified individuals with numerous pharmaceutical and biopharmaceutical companies, universities and other research institutions. Competition for these individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
In addition, we may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers. Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. The Company may have difficulty attracting and retaining directors with the requisite qualifications. If we are unable to attract and retain qualified officers and directors, the management of our business could be adversely affected.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
We will need to manage our anticipated growth and increased operational activity. Our personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our growth strategy will require that we:
| ● | manage our regulatory approval trials effectively; | |
| ● | manage our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors, collaborators and other third parties; | |
| ● | develop internal sales and marketing capabilities or establish collaborations with third parties with such capabilities; | |
| ● | commercialize our product candidates; | |
| ● | improve our operational, financial and management controls, reporting systems and procedures; and | |
| ● | attract and motivate sufficient numbers of talented employees. |
This future growth could place a strain on our administrative and operational infrastructure and may require our management to divert a disproportionate amount of its attention away from our day-to-day activities. We may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel, which may result in weaknesses in our infrastructure, and give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. We may not be able to make improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls. If our management is unable to effectively manage our expected growth, our expenses may increase more than expected, our ability to generate or increase our revenues could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to compete effectively will depend, in part, on our ability to effectively manage any future growth.
| 18 |
Risks Related to Our Regulatory and Legal Environment
We are subject to extensive and costly government regulation.
Product candidates and approved products such as ours are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the U.S. Department of Health and Human Services, the U.S. Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of pharmaceutical products. The FDA regulates small molecule chemical entities, whether administered orally, topically or by injection, as drugs, subject to an NDA, under the Federal Food, Drug, and Cosmetic Act. If product candidates and approved products such as ours are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval. Such foreign regulation might be equally or more demanding than corresponding U.S. regulation. Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our products. The regulatory review and approval process, which includes preclinical testing and clinical trials of each product candidate, is lengthy, expensive, and uncertain. Our collaborators or we must obtain and maintain regulatory authorization to conduct clinical trials and approval for each product we intend to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires submitting extensive preclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product’s safety and efficacy for each intended use. The development and approval process might take many years, requires substantial resources, and might never lead to the approval of a product. Even if we are able to obtain regulatory approval for a particular product, the approval might limit the indicated medical uses for the product, limit our ability to promote, sell, and distribute the product, require that we conduct costly post-marketing surveillance, and/or require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling, might require further regulatory review and approval. Once obtained, any approvals might be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators, or our contract manufacturers fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in, among other things, delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; warning letters; fines; import and export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
We might not obtain the necessary U.S. regulatory approvals to commercialize any product candidates.
We cannot assure you that we will receive the approvals necessary to commercialize for sale any product candidates we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. In order to obtain FDA approval of any product candidate, we must submit to the FDA an NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research, pre-clinical studies, and clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in additional drugs that the FDA considers safe for humans and effective for their indicated uses. The FDA has substantial discretion in the product approval process and might require us to conduct additional pre-clinical and clinical testing, perform post-marketing studies or otherwise limit or impose conditions on any additional approvals we obtain. The approval process might also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals might:
| ● | delay commercialization of, and our ability to derive product revenues from, our product candidates; | |
| ● | impose costly procedures on us; and | |
| ● | diminish any competitive advantages that we might otherwise enjoy. |
| 19 |
Even if we comply with all FDA requests, the FDA might ultimately reject one or more of our NDAs. We cannot be sure that we will ever obtain regulatory clearance for our product candidates. Failure to obtain FDA approval of our product candidates will severely undermine our business by leaving us without saleable products, and therefore without any potential sources of revenues, until another product candidate could be developed or obtained. There is no guarantee that we will ever be able to develop or acquire another product candidate
Following regulatory approval of any product candidates, we will be subject to ongoing regulatory obligations and restrictions, which may result in significant expense and limit our ability to commercialize our potential drugs.
If one of our product candidates is approved by the FDA or by another regulatory authority for a territory outside of the U.S., we will be required to comply with extensive regulations for product manufacturing, labeling, packaging, adverse event reporting, storage, distribution, advertising, promotion and record keeping. Regulatory approvals may also be subject to significant limitations on the indicated uses or marketing of the product candidates or to whom and how we may distribute our products. Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a drug’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. For example, the label ultimately approved for our products, if any, may include restrictions on use, including restrictions based on level of obesity and duration of treatment. If so, we may be subject to ongoing regulatory obligations and restrictions, which may result in significant expense and limit our ability to commercialize our products. The FDA could also require a registry to track the patients utilizing the drug or implement a Risk Evaluation and Mitigation Strategy, or REMS, that could restrict access to the drug, reduce our revenues and/or increase our costs. Potentially costly post-marketing clinical studies may be required as a condition of approval to further substantiate safety or efficacy, or to investigate specific issues of interest to the regulatory authority.
Manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, regulations, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our future approved drugs, if any, and these facilities are subject to ongoing regulatory inspections. In addition, regulatory agencies subject a drug, its manufacturer and the manufacturer’s facilities to continual review and inspections. The subsequent discovery of previously unknown problems with a drug, including adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured, may result in restrictions on the marketing of that drug, up to and including, withdrawal of the drug from the market. If the manufacturing facilities of our suppliers fail to comply with applicable regulatory requirements, it could result in regulatory action and additional costs to us. Failure to comply with applicable FDA and other regulatory requirements may, either before or after product approval, if any, subject our company to administrative or judicially imposed sanctions, including:
| ● | issuance of Form 483 notices, warning letters and adverse publicity by the FDA or other regulatory agencies; | |
| ● | imposition of fines and other civil penalties due to product liability or other issues; | |
| ● | criminal prosecutions; | |
| ● | injunctions, suspensions or revocations of regulatory approvals; | |
| ● | suspension of any ongoing clinical trials; | |
| ● | total or partial suspension of manufacturing; | |
| ● | delays in commercialization; | |
| ● | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our collaborators; | |
| ● | refusals to permit drugs to be imported into or exported from the U.S.; |
| ● | restrictions on operations, including costly new manufacturing requirements; and | |
| ● | product recalls or seizures. |
| 20 |
In addition, the law or regulatory policies governing pharmaceuticals may change. New statutory requirements may be enacted or additional regulations may be enacted that could prevent or delay regulatory approval of our product candidates. Contract Manufacturing Organizations, or CMOs, and their vendors or suppliers may also face changes in regulatory requirements from governmental agencies in the U.S. and other countries. We cannot predict the likelihood, nature, extent or effects of government regulation that may arise from future legislation or administrative action, either in the U.S. or elsewhere. If we are not able to maintain regulatory compliance, we might not be permitted to market any future approved products and our business could suffer.
We could be forced to pay substantial damage awards if product liability claims that may be brought against us are successful.
The use of any of our product candidates in clinical trials, and the sale of any approved products, may expose us to liability claims and financial losses resulting from the use or sale of our products. We have obtained limited product liability insurance coverage for our clinical trials of $2 million per occurrence and in the aggregate, subject to a deductible of $50,000 per occurrence. There can be no assurance that our existing insurance coverage will extend to our other products in the future. Any product liability insurance coverage may not be sufficient to satisfy all liabilities resulting from product liability claims. A successful claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable terms, if at all. Even if a claim is not successful, defending such a claim would be time consuming and expensive, may damage our reputation in the marketplace, and would likely divert management’s attention.
Risks Related to our Intellectual Property
Our business depends on protecting our intellectual property.
If we do not obtain protection for our intellectual property rights, our competitors might be able to take advantage of our research and development efforts to develop competing drugs. Our success, competitive position and future revenues, if any, depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We anticipate filing additional patent applications both in the U.S. and in other countries, as appropriate. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following:
| ● | Our patent rights might be challenged, invalidated, or circumvented, or otherwise might not provide any competitive advantage; | |
| ● | Our competitors, many of which have substantially greater resources than we do and many of which might make significant investments in competing technologies, might seek, or might already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the U.S. or in international markets; | |
| ● | As a matter of public policy regarding worldwide health concerns, there might be significant pressure on the U.S. government and other international governmental bodies to limit the scope of patent protection both inside and outside the U.S. for disease treatments that prove successful; and | |
| ● | Countries other than the U.S. might have less restrictive patent laws than those upheld by U.S. courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products. | |
In addition, the U.S. Patent and Trademark Office and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents might be substantially narrower than anticipated.
Because the time period from filing a patent application to the issuance, if ever, of the patent is often more than three years and because any regulatory approval and marketing for a drug often occurs several years after the related patent application is filed, the resulting market exclusivity afforded by any patent on our drug candidates and technologies will likely be substantially less than 20 years. In the United States, the European Union and some other jurisdictions, patent term extensions are available for certain delays in either patent office proceedings or marketing and regulatory approval processes. However, due to the specific requirements for obtaining these extensions, there is no assurance that our patents will be granted extensions even if we encounter significant delays in patent office proceedings or marketing and regulatory approval.
| 21 |
In addition to patents, we also rely on trade secrets and proprietary know-how. Although we take measures to protect this information by entering into confidentiality and inventions agreements with our employees, scientific advisors, consultants, and collaborators, we cannot provide any assurances that these agreements will not be breached, that we will be able to protect ourselves from the harmful effects of disclosure if they are breached, or that our trade secrets will not otherwise become known or be independently discovered by competitors. If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of this information may be greatly reduced.
Patent and other intellectual property protection is crucial to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate. Our business and prospects will be harmed if these protections prove insufficient.
We rely on trade secret protections through confidentiality agreements with our employees, customers and other parties, and the breach of these agreements could adversely affect our business and prospects.
We rely on trade secrets, which we seek to protect, in part, through confidentiality and non-disclosure agreements with our employees, collaborators, suppliers, and other parties. There can be no assurance that these agreements will not be breached, that we would have adequate remedies for any such breach or that our trade secrets will not otherwise become known to or independently developed by our competitors. We might be involved from time to time in litigation to determine the enforceability, scope and validity of our proprietary rights. Any such litigation could result in substantial cost and divert management’s attention from our operations.
If we infringe the rights of third parties we might have to forgo selling our future products, pay damages, or defend against litigation.
If our product candidates, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we might have to:
| ● | obtain licenses, which might not be available on commercially reasonable terms, if at all; | |
| ● | abandon an infringing product candidate; | |
| ● | redesign our products or processes to avoid infringement; | |
| ● | stop using the subject matter claimed in the patents held by others; | |
| ● | pay damages, and/or | |
| ● | defend litigation or administrative proceedings which might be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
Any of these events could substantially harm our earnings, financial condition and operations.
Risks Related to Our Securities and Liquidity Risks
Nasdaq may delist our common stock and warrants from quotation on its exchange. Failure to maintain NASDAQ listing could limit investors’ ability to make transactions in our common stock and warrants and subject us to additional trading restrictions.
Our common stock and warrants are currently listed on Nasdaq. We may not be able to meet the continued listing requirements for our common stock and warrants in the future. Failure to meet the continued listing requirements could result in Nasdaq delisting our ordinary shares from trading on its exchange. If this should happen, we could face significant material adverse consequences, including:
| ● | a limited availability of market quotations for our securities; |
| 22 |
| ● | a limited amount of news and analyst coverage for us; and | |
| ● | a decreased ability to issue additional securities or obtain additional financing in the future. |
If our common stock were delisted and determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock in the secondary market.
If our common stock were removed from listing with the Nasdaq Capital Market, it may be subject to the so-called “penny stock” rules. The SEC has adopted regulations that define a “penny stock” to be any equity security that has a market price per share of less than $5.00, subject to certain exceptions, such as any securities listed on a national securities exchange. For any transaction involving a “penny stock,” unless exempt, the rules impose additional sales practice requirements on broker-dealers, subject to certain exceptions. If our common stock were delisted and determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock on the secondary market. Investors in penny stocks should be prepared for the possibility that they may lose their whole investment.
Compliance with the reporting requirements of federal securities laws can be expensive.
While the Company was not previously subject to the filing requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, it filed certain reports with the Securities and Exchange Commission on a voluntary basis. On October 22, 2015, the Company registered its Common Stock under the Exchange Act and the filing of the reports with the SEC became mandatory. The quotation of the Company’s Common Stock on Nasdaq is contingent upon the Company staying current on such Exchange Act filings. The costs of preparing and filing annual and quarterly reports and other information with the SEC and furnishing audited reports to stockholders will cause our expenses to be higher than they would be if we remained privately-held.
If the Company fails to maintain an effective system of internal controls, it may not be able to accurately report its financial results or detect fraud. Consequently, shareholders could lose confidence in the Company’s financial reporting and this may decrease the trading price of its stock.
The Company must maintain effective internal controls to provide reliable financial reports and to be able to detect fraud. The Company has been assessing its internal controls to identify areas that need improvement and as of September 30, 2017, management identified material weaknesses in its internal controls over financial reporting. While the Company is in the process of implementing changes to internal controls, it has not yet completed implementing these changes and there is no assurance that the changes will remediate the material weakness or that the controls will prevent or defect future material weakness. Failure to implement these changes to the Company’s internal controls or any others that it identifies as necessary to maintain an effective system of internal controls could harm its operating results and cause shareholders to lose confidence in the Company’s reported financial information. Any such loss of confidence would have a negative effect on the trading price of the Company’s stock.
The price of our securities may become volatile, which could lead to losses by shareholders and costly securities litigation.
The trading price of our securities is likely to be highly volatile and could fluctuate in response to factors such as:
| ● | actual or anticipated variations in the Company’s operating results; | |
| ● | announcements of developments by the Company or its competitors; | |
| ● | the completion and/or results of the Company’s clinical trials; | |
| ● | regulatory actions regarding the Company’s products; | |
| ● | announcements by the Company or its competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| ● | adoption of new accounting standards affecting the Company’s industry; |
| 23 |
| ● | additions or departures of key personnel; | |
| ● | introduction of new products by the Company or its competitors; | |
| ● | sales of the Company’s Common Stock or other securities in the open market; and | |
| ● | other events or factors, many of which are beyond the Company’s control. |
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been initiated against such a company. Litigation initiated against the Company, whether or not successful, could result in substantial costs and diversion of its management’s attention and resources, which could harm the Company’s business and financial condition.
We completed a Reverse Stock Split of our shares of common stock, which may reduce and may limit the market trading liquidity of the shares due to the reduced number of shares outstanding, and may potentially have an anti-takeover effect.
We completed the Reverse Stock Split of our Common Stock by a ratio of 1-for-15 effective June 9, 2017. The liquidity of our Common Stock may be adversely affected by the Reverse Stock Split as a result of the reduced number of shares outstanding following the Reverse Stock Split. In addition, the Reverse Stock Split may increase the number of stockholders who own odd lots of our Common Stock, creating the potential for such stockholders to experience an increase in the cost of selling their shares and greater difficulty affecting such sales. Reducing the number of outstanding shares of our Common Stock through the Reverse Stock Split is intended, absent other factors, to increase the per share market price of our Common Stock. However, other factors, such as our financial results, market conditions and the market perception of our business may adversely affect the market price of our Common Stock. As a result, there can be no assurance that the Reverse Stock Split will result in the intended benefits, that the market price of our Common Stock will remain higher following the Reverse Stock Split or that the market price of our Common Stock will not decrease in the future. Further, since the Reverse Stock Split was not accompanied by a corresponding decrease in the number of shares authorized for issuance under our Amended and Restated Articles of Incorporation, the relative increase in the number of shares authorized for issuance could, under certain circumstances, have an anti-takeover effect by enabling the Board of Directors to issue additional shares of Common Stock in a transaction making it more difficult for a party to obtain control of us by tender offer or other means.
You may experience dilution of your ownership interests because of the future issuance of additional shares of the Common Stock.
In the future, the Company may issue additional authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of its present stockholders. The Company is currently authorized to issue an aggregate of 200,000,000 shares of Common Stock and 10,000,000 shares of preferred stock. As of September 30, 2017, there are 8,345,844 shares of Common Stock outstanding, 3,346,920 shares underlying warrants with a weighted average exercise price of $5.77 per share, and 861,039 shares underlying options with a weighted average exercise price of $6.69 per share. The Company may also issue additional shares of its Common Stock or other securities that are convertible into or exercisable for Common Stock in connection with hiring or retaining employees, future acquisitions, future sales of its securities for capital raising purposes, or for other business purposes. The future issuance of any such additional shares of Common Stock may create downward pressure on the trading price of the Common Stock.
The Common Stock is controlled by insiders.
As of September 30, 2017, the former managing members of Citius Pharmaceuticals, LLC beneficially own approximately 14.4% of our outstanding shares of Common Stock and the Company’s current officers and directors beneficially own approximately 51.7% of our outstanding shares of Common Stock. Such concentrated control of the Company may adversely affect the price of the Common Stock. If you acquire Common Stock, you may have no effective voice in the management of the Company. Sales by insiders or affiliates of the Company, along with any other market transactions, could affect the market price of the Common Stock.
We do not intend to pay dividends for the foreseeable future.
We have paid no dividends on our Common Stock to date and it is not anticipated that any dividends will be paid to holders of our Common Stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. The lack of a dividend can further affect the market value of our stock, and could significantly affect the value of any investment in our Company.
| 24 |
Our Certificate of Incorporation allows for the board of directors to create new series of preferred stock without further approval by stockholders, which could adversely affect the rights of the holders of the Common Stock.
The Company’s Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. The Company’s Board of Directors has the authority to issue up to 10,000,000 shares of preferred stock without further stockholder approval. As a result, the Company’s Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of Common Stock and the right to the redemption of the shares, together with a premium, prior to the redemption of the Common Stock. In addition, the Company’s Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than the Common Stock or that is convertible into our Common Stock, which could decrease the relative voting power of the Common Stock or result in dilution to our existing stockholders.
There are a significant number of shares of Common Stock eligible for sale, which could depress the market price of such shares.
A large number of shares of Common Stock will be available for sale in the public market, which could harm the market price of the stock. Further, shares may be offered from time to time in the open market pursuant to Rule 144, and these sales may have a depressive effect as well.
Risks Related to Ownership of our Securities
There is not an active liquid trading market for the Company’s Common Stock.
The Company files reports under the Exchange Act and is listed on Nasdaq. However, there has not been a regular active trading market in the Company’s Common Stock, and we cannot give any assurance that an active trading market will develop. If an active market for the Company’s Common Stock develops, there is a significant risk that the Company’s stock price may fluctuate dramatically in the future in response to any of the following factors, some of which are beyond our control:
| ● | variations in our quarterly operating results; | |
| ● | announcements that our revenue or income are below analysts’ expectations; | |
| ● | general economic slowdowns; | |
| ● | sales of large blocks of the Company’s Common Stock; and | |
| ● | announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments. |
Because we became a public company by means of a reverse acquisition, we may not be able to attract the attention of brokerage firms.
Because we became public through a “reverse acquisition”, securities analysts of brokerage firms may not provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our Common Stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on behalf of the Company in the future.
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for the Company to retain or attract qualified officers and directors, which could adversely affect the management of its business and its ability to obtain or retain listing of its Common Stock and warrants.
The Company may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by the stock exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
| 25 |
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. The Company may have difficulty attracting and retaining directors with the requisite qualifications. If the Company is unable to attract and retain qualified officers and directors, the management of its business and its ability to obtain or retain listing of our shares of Common Stock on any stock exchange (assuming the Company is successful in obtaining such listing) could be adversely affected.
Sales of a substantial number of shares of our common stock in the public market, or the perception such sales may occur, could cause the market price of shares of our common stock to fall.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market of such sales or that the holders of a large number of shares intend to sell shares, could reduce the market price of our shares of our common stock. As of September 30, 2017, we have 8,345,844 shares of common stock outstanding. This includes registered shares of common stock as well as 3,750,998 shares of our common stock which are available for resale under Rule 144 of the Securities Act of 1933, as amended, or the “Securities Act”. On August 8, 2017, our executive officers and directors entered into lock-up agreements pursuant to which they agreed not to sell any of our shares for a period of 90 days from the effective date of our recent public offering. As representative of the underwriters, Aegis Capital Corp. may, in its sole discretion, allow early releases under the referenced lock-up restrictions.
Our failure to meet the continued listing requirements of the Nasdaq Capital Market could result in a delisting of our common stock and warrants.
If we fail to satisfy the continued listing requirements of the Nasdaq Capital Market, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock and warrants. Such a delisting would likely have a negative effect on the price of our common stock and warrants and would impair your ability to sell or purchase our common stock and warrants when you wish to do so. In the event of a delisting, we would take actions to restore our compliance with Nasdaq’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s listing requirements.
Risks Related to Our Reverse Stock Split
We completed the Reverse Stock Split in order to meet the initial listing requirements of Nasdaq. However, the Reverse Stock Split may not result in our stock price remaining compliant with the minimum price requirements of Nasdaq.
We completed the Reverse Stock Split in order to achieve the requisite increase in the market price of our common stock to be in compliance with the minimum price requirements of Nasdaq. We cannot assure you that the market price of our common stock following the Reverse Stock Split will remain at the level required for the period of time required for listing or for continuing compliance with that requirement. It is not uncommon for the market price of a Company’s common stock to decline in the period following a Reverse Stock Split. If the market price of our common stock declines following the Reverse Stock Split, the percentage decline may be greater than would occur in the absence of a reverse stock split. In any event, other factors unrelated to the number of shares of our common stock outstanding, such as negative financial or operational results, could adversely affect the market price of our common stock and jeopardize our ability to maintain Nasdaq’s minimum price requirements. In addition to specific listing and maintenance standards, Nasdaq has broad discretionary authority over the continued listing of securities, which it could exercise with respect to the listing of our common stock.
Even if the Reverse Stock Split increases the market price of our common stock, there can be no assurance that we will be able to comply with other continued listing standards of Nasdaq.
We cannot assure you that we will be able to comply with the other standards that we are required to meet in order to maintain a listing of our common stock and warrants on Nasdaq. Our failure to meet these requirements may result in our common stock and warrants being delisted from Nasdaq, irrespective of our compliance with the minimum bid price requirement.
The Reverse Stock Split may decrease the liquidity of the shares of our common stock.
The liquidity of the shares of our common stock may be affected adversely by the Reverse Stock Split given the reduced number of shares that will be outstanding following the Reverse Stock Split, especially if the market price of our common stock does not increase as a result of the Reverse Stock Split. In addition, the Reverse Stock Split may increase the number of stockholders who own odd lots (less than 100 shares) of our common stock, creating the potential for such stockholders to experience an increase in the cost of selling their shares and greater difficulty affecting such sales.
| 26 |
Following the Reverse Stock Split, the resulting market price of our common stock may not attract new investors, including institutional investors, and may not satisfy the investing requirements of those investors. Consequently, the trading liquidity of our common stock may not improve.
Although we believe that a higher market price of our common stock may help generate greater or broader investor interest, there can be no assurance that the Reverse Stock Split will result in a share price that will attract new investors, including institutional investors. In addition, there can be no assurance that the market price of our common stock will satisfy the investing requirements of those investors. As a result, the trading liquidity of our common stock may not necessarily improve.
Item 1 B. Unresolved Staff Comments
Not Applicable
We maintain our offices at 11 Commerce Drive, Cranford, NJ 07016. We do not intend to expand our operations for the foreseeable future and do not intend to lease additional space.
The Company is not involved in any litigation that we believe could have a material adverse effect on our financial position or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of our executive officers, threatened against or affecting our company or our officers or directors in their capacities as such.
Item 4. Mine Safety Disclosures
Not applicable.
| 27 |
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our Common Stock was not traded during the nine months ended September 30, 2014 and traded on a limited basis during the year ended September 30, 2015 and through the six months ended March 31, 2016. Since the acquisition of Leonard-Meron Biosciences, Inc. on March 30, 2016, the trading volume of our Common Stock has started to increase. We were quoted under the ticker symbol TRLO.QB through October 9, 2014 and on October 10, 2014, our ticker symbol changed to CTXR.QB. On August 3, 2017 our Common Stock began trading on the Nasdaq Capital Market (“Nasdaq”) under the symbol CTXR.
The following table sets forth the range of the high and low bid quotations of our Common Stock for the last eight fiscal quarters, as reported by the OTCQB or Nasdaq, as applicable after giving retroactive effect to the Reverse Stock Split. The over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
| High | Low | |||||||
| Quarter ended December 31, 2015 | $ | 27.75 | $ | 15.00 | ||||
| Quarter ended March 31, 2016 | $ | 37.50 | $ | 23.25 | ||||
| Quarter ended June 30, 2016 | $ | 37.50 | $ | 11.70 | ||||
| Quarter ended September 30, 2016 | $ | 18.00 | $ | 8.70 | ||||
| Quarter ended December 31, 2016 | $ | 14.85 | $ | 2.55 | ||||
| Quarter ended March 31, 2017 | $ | 14.63 | $ | 5.40 | ||||
| Quarter ended June 30, 2017 | $ | 11.40 | $ | 4.75 | ||||
| Quarter ended September 30, 2017 | $ | 6.37 | $ | 2.60 | ||||
On December 1, 2017, the closing bid price of our Common Stock as reported by the Nasdaq was $ 5.03 per share.
Holders of Common Stock
We are authorized to issue 200,000,000 shares of Common Stock, $0.001 par value per share. As of December 1, 2017, we have 8,423,391 shares of Common Stock issued and outstanding and there are approximately 2,400 shareholders of the Company’s Common Stock.
Each share of Common Stock shall have one (1) vote per share for all purposes. The holders of a majority of the shares entitled to vote, present in person or represented by proxy shall constitute a quorum at all meetings of our shareholders. Our Common Stock does not provide preemptive, subscription or conversion rights and there are no redemption or sinking fund provisions or rights. Our Common Stock holders are not entitled to cumulative voting for election of the board of directors.
Holders of Common Stock are entitled to receive ratably such dividends as may be declared by the board of directors out of funds legally available therefore as well as any distributions to the security holder. We have never paid cash dividends on our Common Stock, and do not expect to pay such dividends in the foreseeable future.
In the event of a liquidation, dissolution or winding up of our company, holders of Common Stock are entitled to share ratably in all of our assets remaining after payment of liabilities. Holders of Common Stock have no preemptive or other subscription or conversion rights. There are no redemption or sinking fund provisions applicable to the Common Stock.
Dividends
We have never paid dividends on our Common Stock. We intend to follow a policy of retaining earnings, if any, to finance the growth of our business and do not anticipate paying any cash dividends in the foreseeable future. The declaration and payment of future dividends on the Common Stock will be at sole discretion of the Board of Directors and will depend on the our profitability and financial condition, capital requirements, statutory and contractual restrictions, future prospects and other factors deemed relevant.
Securities Authorized for Issuance under Equity Compensation Plans
On September 12, 2014, we adopted the 2014 Stock Incentive Plan (the “2014 Plan”). Under the 2014 Plan we are authorized to issue up to 866,667 shares of our Common Stock to employees, directors, consultants and advisors in exchange for consideration in the form of services (See Item 11 – “Executive Compensation”). As of September 30, 2017, we have issued 861,039 options pursuant to the 2014 Plan.
| 28 |
Recent Sales of Unregistered Securities
On September 12, 2014, we sold 226,671 Units for a purchase price of $9.00 per Unit, each Unit consisting of one share of Common Stock and one five-year warrant (the “Investor Warrants”) to purchase one share of Common Stock at an exercise price of $9.00, (the “Private Offering”). As of September 12, 2014, we raised gross proceeds of $2,040,040. The exercise price of the Investor Warrants is subject to adjustment, for up to one year, in the event that we sell Common Stock at a price lower than the exercise price, subject to certain exceptions. The Investor Warrants are redeemable by us at a price of $0.015 per Investor Warrant at any time subject to the conditions that (i) our Common Stock has traded for twenty (20) consecutive trading days with a closing price of at least $22.50 per share with an average trading volume of 3,333 shares per day and (ii) we provide 20 trading days prior notice of the redemption and the closing price of our Common Stock is not less than $17.55 for more than any 3 days during such notice period and (iii) the underlying shares of Common Stock are registered.
On September 12, 2014, the Company issued its President and CEO options to purchase 220,000 shares of Common Stock at $6.75 per share pursuant to the 2014 Plan.
On December 31, 2014, note holders requested conversion of $600,000 in Promissory Notes and accrued interest of $33,333 into 70,371 shares of Common Stock at a conversion price of $9.00 per share.
During the year ended September 30, 2015, we sold an aggregate of 189,136 Units at $8.10 per Unit and an aggregate of 13,333 Units at a price of $9.00 per Unit.
During the year ended September 30, 2016, we sold an additional 290,000 Units for a purchase price of $8.10 per Unit and 17,778 Units for a purchase price of $9.00 per Unit.
On March 22, 2016, the Company sold 333,333 shares of Common Stock at $9.00 per share to its Chairman of the Board, Leonard Mazur.
In February 2017, the Company completed a private placement offering (the “2016 Offering”) and sold 128,017 units at $6.00 per unit for gross proceeds of $768,100. Each unit consisted of (i) one share of common stock and (ii) a five year warrant to purchase one share of common stock at an exercise price of $8.25 per share.
On June 7, 2017, the Company entered into a release agreement with the placement agent for the 2016 Offering. The placement agent consented to future financings and waived certain covenants contained in the 2016 Offering agreements. As consideration for the release, the Company issued 6,668 shares of common stock to the placement agent.
On June 8, 2017, the Company entered into release agreements with the investors in the 2016 Offering where each investor released the Company from the restrictions included in the unit purchase agreements. In exchange, the Company agreed that (i) in the event that a financing is conducted at a price per share or price per unit lower than $6.00, then the Company will issue additional shares to each investor sufficient to effectively reprice the sale of the 2016 Offering units to the lower price and in the event that the financing is conducted at a price per share or price per unit less than the $8.25 exercise price of the warrants issued in the 2016 Offering then the exercise price of the warrants shall be reduced to the lower price. On August 8, 2017, the Company completed an underwritten public offering (the “2017 offering) and issued 58,191 shares of common stock to the investors in the 2016 Offering to reprice the sale of the 2016 Offering units to $4.125 per unit and repriced the 2016 Offering Warrants to an exercise price of $4.125 per share.
Mr. Mazur has also loaned the Company $4,710,000 pursuant to convertible promissory notes. On August 8, 2017, these notes and accrued interest of $76,240 were converted into 1,547,067 shares of common stock at a price of $3.09 per share as part of the 2017 public offering.
The transactions described above were exempt from registration under Section 4(a)(2) of the Securities Act.
Issuer Purchases of Equity Securities
We did not make any purchases of our Common Stock during the three months ended September 30, 2017, which is the fourth quarter of our fiscal year.
Item 6. Selected Financial Data
Not
required.
| 29 |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read together with our financial statements and related notes included elsewhere in this annual report on Form 10-K. Management’s discussion and analysis contains forward-looking statements, such as statements of our plans, objectives, expectations and intentions. Any statements that are not statements of historical fact are forward-looking statements. When used, the words “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect” and the like, and/or future tense or conditional constructions (“will,” “may,” “could,” “should,” etc.), or similar expressions, identify certain of these forward-looking statements. These forward-looking statements are subject to risks and uncertainties including those under “Risk Factors” in Item 1A in this Form 10-K that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements. Our actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of several factors. The Company does not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the filing date of this report.
Historical Background
Citius Pharmaceuticals, Inc. (“Citius” or the “Company”) is a specialty pharmaceutical company dedicated to the development and commercialization of critical care products targeting unmet needs with a focus on anti-infectives, cancer care and unique prescription products. On September 12, 2014, we acquired Citius Pharmaceuticals, LLC as a wholly-owned subsidiary.
Citius Pharmaceuticals, LLC was founded in Massachusetts in January 2007. Activities since Citius Pharmaceuticals, LLC’s inception through September 30, 2017, were devoted primarily to the development and commercialization of therapeutic products for large and growing markets using innovative patented or proprietary formulations and novel drug delivery technology.
On March 30, 2016, the Company acquired all of the outstanding stock of Leonard-Meron Biosciences, Inc. (“LMB”) by issuing 1,942,456 shares of its common stock. As of March 30, 2016, the stockholders of LMB received approximately 41% of the issued and outstanding common stock of the Company. In addition, the Company converted the outstanding common stock warrants of LMB into 243,020 common stock warrants of the Company and converted the outstanding common stock options of LMB into 77,252 common stock options of the Company. Management estimated the fair value of the purchase consideration to be $19,015,073.
In connection with the acquisition, the Company acquired net assets of $17,428,277, including identifiable intangible assets of $19,400,000 related to in-process research and development and other assets and liabilities. The Company recorded goodwill of $1,586,796 for the excess of the purchase price over the net assets acquired.
In-process research and development represents the value of LMB’s leading drug candidate, which is an antibiotic solution used to treat catheter-related bloodstream infections. Goodwill represents the value of LMB’s industry relationships and its assembled workforce. In-process research and development is expected to be amortized on a straight-line basis over a period of eight years commencing upon revenue generation. Goodwill will not be amortized, but will be tested at least annually for impairment.
Through September 30, 2017, the Company has devoted substantially all of its efforts to product development, raising capital, building infrastructure through strategic alliances and coordinating activities relating to its proprietary products. On July 1, 2016, the Company announced that it was discontinuing Suprenza and was focusing on the Phase 3 development of Mino-Lok™, an antibiotic lock solution used to treat patients with catheter-related bloodstream infections, and the Phase 2b development of Hydro-Lido for hemorrhoids. The Company has not yet realized any revenues from its operations.
Patent and Technology License Agreement
LMB has a patent and technology license agreement with Novel Anti-Infective Therapeutics, Inc., (“NAT”) to develop and commercialize Mino-Lok™ on an exclusive worldwide sub licensable basis, as amended. Since May 2014, LMB has paid an annual maintenance fee of $30,000 that increases over five years to $90,000, until commercial sales of a product subject to the license. LMB will also pay annual royalties on net sales of licensed products, with royalties ranging from the mid-single digits to the low double digits. In limited circumstances in which the licensed product is not subject to a valid patent claim and a competitor is selling a competing product, the royalty rate is in the low-single digits. After a commercial sale is obtained, LMB must pay minimum aggregate annual royalties that increase in subsequent years. LMB must also pay NAT up to $1,390,000 upon achieving specified regulatory and sales milestones. Finally, LMB must pay NAT a specified percentage of payments received from any sub licensees.
| 30 |
Results of Operations for Year Ended September 30, 2017 compared to Year Ended September 30, 2016
| Year Ended September 30, 2017 | Year Ended September 30, 2016 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 2,936,252 | 2,933,199 | ||||||
| General and administrative | 6,063,439 | 3,783,941 | ||||||
| Stock-based compensation – general and administrative | 986,620 | 732,151 | ||||||
| Total operating expenses | 9,986,311 | 7,449,291 | ||||||
| Operating loss | (9,986,311 | ) | (7,449,291 | ) | ||||
| Interest income | - | 806 | ||||||
| Gain (loss) on revaluation of derivative warrant liability | 452,147 | (838,219 | ) | |||||
| Interest expense | (850,789 | ) | (8,994 | ) | ||||
| Net loss | $ | (10,384,953 | ) | $ | (8,295,698 | ) | ||
Revenues
We did not generate any revenues for the years ended September 30, 2017 and 2016.
Research and Development Expenses
For the year ended September 30, 2017, research and development expenses were $2,936,252 as compared to $2,933,199 during the year ended September 30, 2016. The $3,053 increase in 2017 was primarily due to an increase of $776,192 in costs incurred in the development of Mino-Lok™ offset by a decrease of $773,139 in costs incurred in the development of our product for the treatment of hemorrhoids and costs related to Suprenza, including $292,575 received in 2016 from Alpex as reimbursement for regulatory filing fees. We are actively seeking to raise additional capital in order to fund our research and development efforts.
General and Administrative Expenses
For the year ended September 30, 2017, general and administrative expenses were $6,063,439 as compared to $3,783,941 during the year ended September 30, 2016. The $2,279,498 increase in 2017 was primarily due to the acquisition of LMB on March 30, 2016, which resulted in increased compensation costs, increased consulting fees incurred for financing activities and corporate development services, and increased investor relations fees. In addition, the year ended September 30, 2016 only includes six months of expenses for LMB as the acquisition was completed on March 30, 2016.
Stock-based Compensation Expense
For the year ended September 30, 2017, stock-based compensation expense was $986,620 as compared to $732,151 for the year ended September 30, 2016. The $254,469 increase in expense includes the expense for unvested options assumed in the acquisition of LMB, as well as new grants to directors, employees and consultants.
Other Income (Expense)
There was no interest income earned on our cash balances for the year ended September 30, 2017 and only $806 in interest income earned for the year ended September 30, 2016.
Gain (loss) on revaluation of derivative warrant liability for the year ended September 30, 2017 was $452,147 compared to $(838,219) for the year ended September 30, 2016. The fair value of the derivative warrant liability fluctuates with changes in our stock price, volatility, remaining lives of the warrants, and interest rates. The gain for the year ended September 30, 2017 was primarily due to a decrease in the fair value of our stock from $9.45 per share at September 30, 2016 to $4.125 per share at August 8, 2017 when the final derivative warrants were reclassified to equity. The loss for the year ended September 30, 2016 was primarily due to an increase in the fair value of our common stock from $8.10 at September 30, 2015 to $9.45 at September 30, 2016. At September 30, 2017, the Company has no outstanding warrants that are considered to be derivative instruments.
Interest expense on the notes payable acquired in the acquisition of LMB and recent borrowings from our Chairman was $850,789 for the year ended September 30, 2017, and includes net non-cash interest expense of $762,078 due to the beneficial conversion feature on the conversion price of $1,595,411 and the amortization of the previously recorded modification premium of $833, 333. After the August 8, 2017 conversions of debt to common stock, the Company has $172,970 in outstanding notes payable at September 30, 2017. Interest expense on the notes payable acquired in the acquisition of LMB was $8,994 for the year ended September 30, 2016.
| 31 |
Net Loss
For the year ended September 30, 2017, we incurred a net loss of $10,384,953 compared to a net loss for the year ended September 30, 2016 of $8,295,698. The $2,089,255 increase in the net loss was primarily due to the $2,279,498 increase in general and administrative expenses and the 841,795 increase in interest expense offset by the $1,290,366 change in the (gain) loss on revaluation of the derivative warrant liability.
Results of Operations for Year Ended September 30, 2016 compared to Year Ended September 30, 2015
| Year Ended September 30, 2016 | Year Ended September 30, 2015 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 2,933,199 | 1,797,045 | ||||||
| General and administrative | 3,783,941 | 946,613 | ||||||
| Stock-based compensation – general and administrative | 732,151 | 486,271 | ||||||
| Total operating expenses | 7,449,291 | 3,229,929 | ||||||
| Operating loss | (7,449,291 | ) | (3,229,929 | ) | ||||
| Interest income | 806 | 3,066 | ||||||
| Gain (loss) on revaluation of derivative warrant liability | (838,219 | ) | 332,095 | |||||
| Interest expense | (8,994 | ) | (7,500 | ) | ||||
| Net loss | $ | (8,295,698 | ) | $ | (2,902,268 | ) | ||
Revenues
We did not generate any revenues for the years ended September 30, 2016 and 2015.
Research and Development Expenses
For the year ended September 30, 2016, research and development expenses were $2,933,199 as compared to $1,797,045 for the year ended September 30, 2015. The $1,136,154 increase in 2016 was primarily due to the $1,912,745 in costs incurred in the development of Mino-Lok™ offset by a decrease in the costs on our product for the treatment of hemorrhoids and the reimbursement of $292,575 from Alpex for regulatory filing fees. We are actively seeking additional capital in order to fund our research and development efforts.
General and Administrative Expenses
For the year ended September 30, 2016, general and administrative expenses were $3,783,941 as compared to $946,613 for the year ended September 30, 2015. The increase of $2,837,328 in 2016 was primarily due to the acquisition of LMB which resulted in increased compensation costs, increased consulting fees incurred for financing activities and corporate development services, and increased investor relations fees.
Stock-based Compensation Expense
For the year ended September 30, 2016, stock-based compensation expense was $732,151 as compared to $486,271 for the year ended September 30, 2015, an increase of $245,880. The $732,151 expense for the year ended September 30, 2016 includes the expenses for our Chairman’s options, an option granted to a consultant, options granted to six directors (including our current Chief Executive Officer), options granted to three employees, and options granted in connection with the acquisition of LMB. The $486,271 expense for the year ended September 30, 2015 was due to the stock options granted to our Chairman in connection with his employment agreement and options granted to two consultants.
Other Income (Expense)
Interest income earned was $806 for the year ended September 30, 2016 compared to $3,066 for the year September 30, 2015. The interest income was earned on the proceeds of our private offerings that were invested in money market accounts.
Loss on revaluation of derivative warrant liability for the year ended September 30, 2016 was $838,219 compared to a gain of $332,095 for the year ended September 30, 2015. The $838,219 loss for the year ended September 30, 2016 was primarily due to the increase in the fair value of our Common Stock from $8.10 per share at September 30, 2015 to $9.45 per share at September 30, 2016 and an increase in volatility from 57% at September 30, 2015 to 73% at September 30, 2016. The $332,095 gain for the year ended September 30, 2015 was primarily due to the decrease in our stock price used to calculate the fair value of the derivative liability from $9.00 at September 30, 2014 to $8.10 at September 30, 2015.
| 32 |
For the year ended September 30, 2016, interest expense increased by $1,494 in comparison to the year ended September 30, 2015. Interest expense for the year ended September 30, 2016 related to the demand notes payable assumed in the acquisition of LMB and the new $500,000 demand note payable issued in September 2016. For the year ended September 30, 2015, interest expense related to promissory notes issued to two existing investors. On December 31, 2014, the outstanding $600,000 promissory notes and accrued interest of $33,333 were converted into 1,055,554 shares of Common Stock at a conversion price of $0.60 per share. From December 31, 2014 to March 30, 2016, the Company had no outstanding interest bearing debt.
Net Loss
For the year ended September 30, 2016, we incurred a net loss of $8,295,698 compared to a net loss of $2,902,268 for the year ended September 30, 2015. The $5,393,430 increase in the net loss was primarily due to the $2,837,328 increase in general and administrative expenses, the $1,136,154 increase in research and development expenses and the $1,170,314 change in the gain (loss) on revaluation of derivative warrant liability.
LIQUIDITY AND CAPITAL RESOURCES
Going Concern Uncertainty and Working Capital
Citius has incurred losses of $10,384,953, $8,295,698 and $2,902,268 for the years ended September 30, 2017, 2016 and 2015, respectively. At September 30, 2017, Citius had an accumulated deficit of $27,721,200. Citius’ net cash used in operations during the years ended September 30, 2017, 2016 and 2015, was $7,971,205, $5,900,421 and $2,385,416, respectively.
Our independent registered accountants report on our September 30, 2017 consolidated financial statements contains an emphasis of a matter regarding substantial doubt about our ability to continue as a going concern and that the consolidated financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets, or the amounts and classification of liabilities that may result if we do not continue as a going concern.
As of September 30, 2017, Citius had working capital of $955,189. Our limited working capital was attributable to the operating losses incurred by the Company since inception offset by our capital raising activities. At September 30, 2017, Citius had cash and cash equivalents of $3,204,108 available to fund its operations. The Company’s only source of cash flow since inception has been from financing activities. During the years ended September 30, 2017, 2016 and 2015, the Company received net proceeds of $6,673,088, $5,427,688 and $1,509,493, respectively from the issuance of equity. We also received $4,210,000 from the issuance of notes payable to our Chairman of the Board, Mr. Leonard Mazur, during the year ended September 30, 2017. Mr. Mazur converted the notes payable to common stock on August 8, 2017. Our primary uses of operating cash were for product development and commercialization activities, regulatory expenses, employee compensation, consulting fees, legal and accounting fees, and insurance and travel expenses.
On September 12, 2014, the Company sold 226,671 units (“Units”) for a purchase price of $9.00 per Unit for gross proceeds of $2,040,040. Each Unit consists of one share of Common Stock and one five-year warrant (the “Investor Warrants”) to purchase one share of Common Stock at an exercise price of $9.00 (the “Private Offering”).
On December 31, 2014, the note holders requested conversion of $600,000 in Promissory Notes and accrued interest of $33,333 into 70,371 shares of Common Stock at a conversion price of $9.00 per share.
Between March 19, 2015 and September 14, 2015, the Company sold an additional 189,136 Units for a purchase price of $8.10 per Unit and 13,333 Units for a purchase price of $9.00 per Unit for gross proceeds of $1,652,000.
During the year ended September 30, 2016, the Company sold an additional 290,000 Units for a purchase price of $8.10 per Unit and 17,778 Units for a purchase price of $9.00 per Unit for gross proceeds of $2,509,000.
On March 22, 2016, the Company sold 333,333 shares of Common Stock at $9.00 per share to its Chairman of the Board, Leonard Mazur, for gross proceeds of $3,000,000.
The Board of Directors authorized revolving demand promissory notes with Leonard Mazur in an aggregate principal amount of up to $2,500,000 that accrue interest at the prime rate plus 1%. On September 7, 2016, the Company issued a $500,000 note. The Company issued $2,000,000 of additional notes through the period ended May 10, 2017. On May 10, 2017, the notes were converted into a $2,500,000 convertible promissory note that matures on June 30, 2018 and is convertible into shares of common stock, at the sole discretion of Mr. Mazur, at a conversion price equal to 75% of the price per share paid by investors in the Company’s 2017 registered public offering. In connection with the modification of the note, the Company recorded a charge of $833,333 to additional paid-in capital and increased the carrying value of the notes to $3,333,333 which is the fair value of the common stock issuable on conversion. On August 8, 2017, Leonard Mazur converted the $2,500,000 principal balance and accrued interest of $63,174 into 828,500 shares of common stock.
| 33 |
On May 10, 2017 and June 23, 2017, the Company executed a $1,500,000 future advance convertible promissory note and a $1,000,000 future advance convertible promissory note, respectively, with Leonard Mazur that both mature on December 31, 2017 and accrue interest at the prime rate plus 1%. The notes are convertible into shares of common stock, at the sole discretion of Mr. Mazur, at a conversion price equal to 75% of the price per share paid by investors in the Company’s 2017 registered public offering. On August 8, 2017, Leonard Mazur converted the outstanding $2,210,000 principal balances and accrued interest of $13,066 into 718,567 shares of common stock.
In February 2017, the Company completed an offering (the “2016 Offering”) and sold 128,017 units at $6.00 per unit for gross proceeds of $768,100. Each unit consisted of (i) one share of common stock and (ii) a five year warrant to purchase one share of common stock at an exercise price of $8.25 per share (the “2016 Offering Warrants”). The placement agent received a 10% cash commission on the gross proceeds, an expense allowance equal to 3% of the proceeds, and warrants to purchase 12,802 shares of common stock at an exercise price of $8.25 per share. The placement agent commissions and expense allowance was $99,853. Other costs of the placement were $176,896. On June 8, 2017, the Company entered into release agreements with the investors in the 2016 Offering where each investor released the Company from the restrictions included in the unit purchase agreements. In exchange, the Company agreed to reprice the sale of the 2016 Offering units to $4.125 per unit and reprice the 2016 Offering Warrants to an exercise price of $4.125 per share. During the year ended September 30, 2017, the Company issued an additional 58,191 shares of common stock to the investors.
On August 8, 2017, the Company closed an underwritten public offering of 1,648,484 shares of common stock and warrants to purchase 1,646,484 shares of common stock at an offering price of $4.125 per share and $0.01 per warrant. The warrants have a per share exercise price of $4.125, are exercisable immediately and will expire five years from the date of issuance. The gross proceeds to Citius from this offering were $6,802,469, before deducting underwriting discounts and commissions and other offering expenses of $685,573. The Company granted the underwriters a 45-day option to purchase up to an additional 247,272 shares of common stock and warrants to purchase 247,272 shares of common stock to cover over-allotments, if any. On August 8, 2017, the underwriters partially exercised the over-allotment to purchase an additional 247,272 warrants.
We expect that we will have sufficient capital to continue our operations for the next six months from September 30, 2017. We plan to raise additional capital in the future to support our operations. There is no assurance, however, that we will be successful in raising the needed capital or that the proceeds will be received in a timely manner to fully support our operations.
Inflation
Our management believes that inflation has not had a material effect on our results of operations.
Off Balance Sheet Arrangements
We do not have any off balance sheet arrangements.
CRITICAL ACCOUNTING POLICIES
Our discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates. We believe the judgments and estimates required by the following accounting policies to be critical in the preparation of our financial statements.
Research and Development
Research and development costs, including upfront fees and milestones paid to collaborators who are performing research and development activities under contractual agreement with us, are expensed as incurred. We defer and capitalize our nonrefundable advance payments that are for research and development activities until the related goods are delivered or the related services are performed. When we are reimbursed by a collaboration partner for work we perform, we record the costs incurred as research and development expenses and the related reimbursement as a reduction to research and development expenses in our statement of operations. Research and development expenses primarily consist of clinical and non-clinical studies, materials and supplies, third-party costs for contracted services, and payments related to external collaborations and other research and development related costs.
| 34 |
In-process Research and Development and Goodwill
In process research and development represents the value of LMB’s leading drug candidate, Mino-Lok TM, an antibiotic lock solution in phase 3 clinical development, which if approved, would be used to assist in the treatment of catheter related bloodstream infections and is expected to be amortized on a straight line basis over 8 years upon revenue generation. Goodwill represents the value of LMB’s industry relationships and its assembled workforce. Goodwill will not be amortized and will be tested at least annually for impairment.
The Company reviews intangible assets annually to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life of any intangible asset. If the carry value of an asset exceeds its undiscounted cash flows, the Company writes down the carrying value of the intangible asset to its fair value for the period identified. No triggering events occurred since the acquisition of LMB that would suggest a potential impairment may have occurred through September 30, 2017.
The Company evaluates the recoverability of goodwill annually or more frequently if events or changes in circumstances indicate that the carrying value of an asset may be impaired. Goodwill is first qualitatively assessed to determine whether further impairment testing is necessary. Factors that management considers in the assessment include macroeconomic conditions, industry and market conditions, overall financial performance, (both current and projected), changes in management and strategy as well as changes in the composition of the carrying amount of net assets. If this qualitative assessment indicates that it is more likely that not that the fair value of a reporting unit is less than its carrying amount, a two-step process is then performed.
The Company performed a qualitative assessment for its 2017 analysis of goodwill. Based on this assessment, management does not believe that it is more likely than not, that the carrying value of the reporting unit exceeds its fair value. Accordingly, no further testing was performed as management believes that there are no impairment issues with respect to goodwill as of September 30, 2017.
Derivative Warrant Liability
The FASB ASC 815-40: Derivatives and Hedging-Contracts in Entity’s Own Equity requires freestanding contracts that are settled in a company’s own stock, including common stock warrants, to be designated as an equity instrument, asset or a liability. Under the provisions of ASC 815-40, a contract designated as an asset or a liability must be carried at fair value on a company’s balance sheet, with any changes in fair value recorded in the company’s results of operations. A contract designated as an equity instrument must be included within equity, and no fair value adjustments are required from period to period. The issuance of certain warrants were classified as liabilities at issuance because the exercise price of the warrants was subject to adjustment in the event that the Company issued common stock for less than the original issuance price per share within one-year of the issuance of the warrants. Subsequent private placements did not result in an adjustment of the exercise price of these warrants.
The Company performed valuations of the warrants classified as derivative warrants using a probability weighted Black-Scholes Pricing Model which value was compared to a Binomial Option Pricing Model for reasonableness. The model uses market-sourced inputs such as underlying stock prices, risk-free interest rates, volatility, expected life and dividend rates and has also considered the likelihood of “down-round” financings. Selection of these inputs involves management’s judgment and may impact net income (loss). Due to our limited operating history and limited number of sales of our common stock, we estimate our volatility based on a number of factors including the volatility of comparable publicly traded pharmaceutical companies. The volatility factor used in the Black-Scholes Pricing Model has a significant effect on the resulting valuation of the derivative liabilities on our balance sheet. The volatility calculated at September 30, 2016 was 73%. We used a risk-free interest rate of 1.14% and estimated lives of 4.10 to 4.57 years, which are the remaining contractual lives of the warrants.
As of September 30, 2017, there were no outstanding warrants classified as a derivative warrant liability.
Income Taxes
We follow accounting guidance regarding the recognition, measurement, presentation and disclosure of uncertain tax positions in the financial statements. Tax positions taken or expected to be taken in the course of preparing our tax returns are required to be evaluated to determine whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authorities. Tax positions not deemed to meet a more-likely-than-not threshold would be recorded in the financial statements. There are no uncertain tax positions that require accrual or disclosure as of September 30, 2017.
Any interest or penalties are charged to expense. None have been recognized in these financial statements. Generally, we are subject to federal and state tax examinations by tax authorities for all years subsequent to December 31, 2013.
We recognize deferred tax assets and liabilities based on differences between the financial reporting and tax basis of assets and liabilities using the enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. We provide a valuation allowance for deferred tax assets for which we do not consider realization of such assets to be more likely than not.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not required.
| 35 |
Item 8. Financial Statements and Supplementary Data
CITIUS PHARMACEUTICALS, INC.
CONSOLIDATED FINANCIAL STATEMENTS
INDEX
| 36 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Citius Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Citius Pharmaceuticals, Inc. as of September 30, 2017 and 2016, and the related consolidated statements of operations, changes in stockholders' equity (deficit) and cash flows for each of the years in the three-year period ended September 30, 2017. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Citius Pharmaceuticals, Inc. as of September 30, 2017 and 2016, and the results of its operations and its cash flows for each of the years in the three-year period ended September 30, 2017, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has suffered recurring losses from operations, has negative cash flows from operations, a working capital deficit and a significant accumulated deficit. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Wolf & Company, P.C.
Boston, Massachusetts
December 13, 2017
| 37 |
CONSOLIDATED BALANCE SHEETS
SEPTEMBER 30, 2017 AND 2016
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 3,204,108 | $ | 294,351 | ||||
| Prepaid expenses | 220,246 | 598,484 | ||||||
| Total Current Assets | 3,424,354 | 892,835 | ||||||
| Property and equipment, net | 3,236 | 3,742 | ||||||
| Other Assets: | ||||||||
| Deposits | 2,167 | 2,167 | ||||||
| Deferred offering costs | — | 64,801 | ||||||
| In-process research and development | 19,400,000 | 19,400,000 | ||||||
| Goodwill | 1,586,796 | 1,586,796 | ||||||
| Total Other Assets | 20,988,963 | 21,053,764 | ||||||
| Total Assets | $ | 24,416,553 | $ | 21,950,341 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 602,431 | $ | 909,156 | ||||
| Accrued expenses | 560,918 | 958,101 | ||||||
| Accrued compensation | 1,063,000 | 903,250 | ||||||
| Accrued interest | 42,209 | 30,871 | ||||||
| Notes payable – related parties | 172,970 | 672,970 | ||||||
| Derivative warrant liability | — | 1,681,973 | ||||||
| Due to related party | 27,637 | 27,637 | ||||||
| Total Current Liabilities | 2,469,165 | 5,183,958 | ||||||
| Commitments and Contingencies | ||||||||
| Stockholders’ Equity: | ||||||||
| Preferred stock - $0.001 par value; 10,000,000 shares authorized; no shares issued and outstanding | — | — | ||||||
| Common stock - $0.001 par value; 200,000,000 shares authorized; 8,345,844 and 4,875,871 shares issued and outstanding at September 30, 2017 and 2016, respectively | 8,346 | 4,876 | ||||||
| Additional paid-in capital | 49,660,242 | 34,097,754 | ||||||
| Accumulated deficit | (27,721,200 | ) | (17,336,247 | ) | ||||
| Total Stockholders’ Equity | 21,947,388 | 16,766,383 | ||||||
| Total Liabilities and Stockholders’ Equity | $ | 24,416,553 | $ | 21,950,341 | ||||
See accompanying report of independent registered public accounting firm and notes to the consolidated financial statements.
Reflects a 1-for-15 reverse stock split effective June 9, 2017
| 38 |
CONSOLIDATED STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED SEPTEMBER 30, 2017, 2016 AND 2015
| 2017 | 2016 | 2015 | ||||||||||
| Revenues | $ | — | $ | — | $ | — | ||||||
| Operating Expenses: | ||||||||||||
| Research and development | 2,936,252 | 2,933,199 | 1,797,045 | |||||||||
| General and administrative | 6,063,439 | 3,783,941 | 946,613 | |||||||||
| Stock-based compensation – general and administrative | 986,620 | 732,151 | 486,271 | |||||||||
| Total Operating Expenses | 9,986,311 | 7,449,291 | 3,229,929 | |||||||||
| Operating Loss | (9,986,311 | ) | (7,449,291 | ) | (3,229,929 | ) | ||||||
| Other Income (Expense), Net: | ||||||||||||
| Interest income | — | 806 | 3,066 | |||||||||
| Gain (loss) on revaluation of derivative warrant liability | 452,147 | (838,219 | ) | 332,095 | ||||||||
| Interest expense | (850,789 | ) | (8,994 | ) | (7,500 | ) | ||||||
| Total Other Income (Expense), Net | (398,642 | ) | (846,407 | ) | 327,661 | |||||||
| Loss before Income Taxes | (10,384,953 | ) | (8,295,698 | ) | (2,902,268 | ) | ||||||
| Income tax benefit | — | — | — | |||||||||
| Net Loss | $ | (10,384,953 | ) | $ | (8,295,698 | ) | $ | (2,902,268 | ) | |||
| Net Loss Per Share - Basic and Diluted | $ | (1.89 | ) | $ | (2.29 | ) | $ | (1.37 | ) | |||
| Weighted Average Common Shares Outstanding | ||||||||||||
| Basic and diluted | 5,482,494 | 3,623,208 | 2,122,363 | |||||||||
See accompanying report of independent registered public accounting firm and notes to the consolidated financial statements.
Reflects a 1-for-15 reverse stock split effective June 9, 2017
| 39 |
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY (DEFICIT)
FOR THE YEARS ENDED SEPTEMBER 30, 2017, 2016 AND 2015
| Total | ||||||||||||||||||||||||
| Additional | Stockholders' | |||||||||||||||||||||||
| Preferred | Common Stock | Paid-In | Accumulated | Equity | ||||||||||||||||||||
| Stock | Shares | Amount | Capital | Deficit | (Deficit) | |||||||||||||||||||
| Balance, September 30, 2014 | $ | — | 2,001,686 | $ | 2,002 | $ | 5,394,344 | $ | (6,138,281 | ) | $ | (741,935 | ) | |||||||||||
| Conversion of promissory notes and accrued interest | — | 70,371 | 70 | 633,263 | — | 633,333 | ||||||||||||||||||
| Issuance of common stock in private placement, net of costs | — | 202,469 | 203 | 740,855 | — | 741,058 | ||||||||||||||||||
| Reclassification of derivative warrant liability to additional paid-in capital | — | — | — | 1,148,328 | — | 1,148,328 | ||||||||||||||||||
| Stock-based compensation | — | — | — | 486,271 | — | 486,271 | ||||||||||||||||||
| Net loss | — | — | — | — | (2,902,268 | ) | (2,902,268 | ) | ||||||||||||||||
| Balance, September 30, 2015 | — | 2,274,526 | 2,275 | 8,403,061 | (9,040,549 | ) | (635,213 | ) | ||||||||||||||||
| Issuance of common stock in private placement, net of costs | — | 641,111 | 641 | 4,228,483 | — | 4,229,124 | ||||||||||||||||||
| Issuance of common stock for services | — | 17,778 | 18 | 149,982 | — | 150,000 | ||||||||||||||||||
| Issuance of common stock, warrants and stock options for acquisition | — | 1,942,456 | 1,942 | 19,013,131 | — | 19,015,073 | ||||||||||||||||||
| Issuance of warrants for services | — | — | — | 477,181 | — | 477,181 | ||||||||||||||||||
| Reclassification of derivative warrant liability to additional paid-in capital | — | — | — | 1,093,765 | — | 1,093,765 | ||||||||||||||||||
| Stock-based compensation | — | — | — | 732,151 | — | 732,151 | ||||||||||||||||||
| Net loss | — | — | — | — | (8,295,698 | ) | (8,295,698 | ) | ||||||||||||||||
| Balance, September 30, 2016 | — | 4,875,871 | 4,876 | 34,097,754 | (17,336,247 | ) | 16,766,383 | |||||||||||||||||
| Issuance of common stock in private placement, net of costs | — | 128,016 | 128 | 491,223 | — | 491,351 | ||||||||||||||||||
| Issuance of common stock in public offering, net of costs | — | 1,648,484 | 1,648 | 6,115,248 | 6,116,896 | |||||||||||||||||||
| Issuance of common stock for services and release agreements | — | 140,843 | 141 | 703,878 | — | 704,019 | ||||||||||||||||||
| Issuance of fractional shares for 1-for-15 reverse stock split | — | 734 | 1 | (1 | ) | — | — | |||||||||||||||||
| Stock options exercised | — | 4,829 | 5 | 35 | — | 40 | ||||||||||||||||||
| Conversion of convertible promissory notes – related party to common stock | 1,547,067 | 1,547 | 4,784,693 | — | 4,786,240 | |||||||||||||||||||
| Beneficial conversion feature on convertible promissory notes – related party | — | — | — | 1,595,411 | — | 1,595,411 | ||||||||||||||||||
| Premium on convertible promissory notes – related party | — | — | — | (833,333 | ) | — | (833,333 | ) | ||||||||||||||||
| Issuance of unit purchase options | — | — | — | 297,998 | — | 297,998 | ||||||||||||||||||
| Issuance of warrants in settlement of liabilities | — | — | — | 190,890 | — | 190,890 | ||||||||||||||||||
| Reclassification of derivative warrant liability to additional paid-in capital, net | — | — | — | 1,229,826 | — | 1,229,826 | ||||||||||||||||||
| Stock-based compensation | — | — | — | 986,620 | — | 986,620 | ||||||||||||||||||
| Net loss | — | — | — | — | (10,384,953 | ) | (10,384,953 | ) | ||||||||||||||||
| Balance, September 30, 2017 | — | 8,345,844 | $ | 8,346 | $ | 49,660,242 | $ | (27,721,200 | ) | $ | 21,947,388 | |||||||||||||
See accompanying report of independent registered public accounting firm and notes to the consolidated financial statements.
Reflects a 1-for-15 reverse stock split effective June 9, 2017
| 40 |
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED SEPTEMBER 30, 2017, 2016 AND 2015
| 2017 | 2016 | 2015 | ||||||||||
| Cash Flows From Operating Activities: | ||||||||||||
| Net loss | $ | (10,384,953 | ) | $ | (8,295,698 | ) | $ | (2,902,268 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Stock-based compensation | 986,620 | 732,151 | 486,271 | |||||||||
| (Gain) loss on revaluation of derivative warrant liability | (452,147 | ) | 838,219 | (332,095 | ) | |||||||
| Fair value of stock issued for services and release agreements | 704,019 | 150,000 | — | |||||||||
| Fair value of options issued to purchase units of common stock | 104,138 | — | — | |||||||||
| Warrants issued and repriced in settlement agreements | 190,890 | — | — | |||||||||
| Non-cash interest expense | 762,078 | — | — | |||||||||
| Depreciation | 2,632 | 1,343 | — | |||||||||
| Write-off of abandoned trademarks | — | 5,401 | — | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Prepaid expenses | 572,098 | (40,759 | ) | (60,000 | ) | |||||||
| Accounts payable | (306,725 | ) | 105,230 | 452,981 | ||||||||
| Accrued expenses | (397,183 | ) | 351,182 | (52,057 | ) | |||||||
| Accrued compensation | 159,750 | 288,250 | — | |||||||||
| Accrued interest | 87,578 | 7,009 | 7,500 | |||||||||
| Due to related party | — | (42,749 | ) | 14,252 | ||||||||
| Net Cash Used In Operating Activities | (7,971,205 | ) | (5,900,421 | ) | (2,385,416 | ) | ||||||
| Cash Flows From Investing Activities: | ||||||||||||
| Cash acquired in acquisition | — | 255,748 | — | |||||||||
| Purchase of property and equipment | (2,126 | ) | — | — | ||||||||
| Net Cash Provided By (Used In) Investing Activities | (2,126 | ) | 255,748 | — | ||||||||
| Cash Flows From Financing Activities: | ||||||||||||
| Proceeds from notes payable – related parties | 4,210,000 | 500,000 | — | |||||||||
| Repayment of notes payable – related parties | — | (600,000 | ) | — | ||||||||
| Proceeds from stock option exercise | 40 | — | — | |||||||||
| Net proceeds from private placement | 556,152 | 5,427,688 | 1,509,493 | |||||||||
| Net proceeds from public offering | 6,116,896 | — | — | |||||||||
| Deferred offering costs | — | (64,801 | ) | — | ||||||||
| Net Cash Provided By Financing Activities | 10,883,088 | 5,262,887 | 1,509,493 | |||||||||
| Increase (Decrease) in Cash and Cash Equivalents | 2,909,757 | (381,786 | ) | (875,923 | ) | |||||||
| Cash and Cash Equivalents – Beginning of Year | 294,351 | 676,137 | 1,552,060 | |||||||||
| Cash and Cash Equivalents – End of Year | $ | 3,204,108 | $ | 294,351 | $ | 676,137 | ||||||
| Supplemental Disclosures of Cash Flow Information and Non-cash Transactions: | ||||||||||||
| Interest paid | $ | 1,133 | $ | 1,985 | $ | — | ||||||
| Premium on convertible promissory notes – related party | $ | 833,333 | $ | — | $ | — | ||||||
| Fair value of unit purchase option issued for future services | $ | 193,860 | $ | — | $ | — | ||||||
| Fair value of warrants recorded as derivative warrant liability | $ | 641,385 | $ | 1,198,564 | $ | 768,435 | ||||||
| Fair value of warrants issued for future services | $ | — | 477,181 | — | ||||||||
| Reclassification of derivative warrant liability to additional paid-in capital, net | $ | 1,229,826 | $ | 1,093,765 | $ | 1,148,328 | ||||||
| Beneficial conversion feature on convertible promissory notes – related party | $ | 1,595,411 | $ | — | $ | — | ||||||
| Conversion of on convertible promissory notes – related party and related accrued interest into common stock | $ | 4,786,240 | $ | — | $ | 633,333 | ||||||
See Note 1 for supplemental cash flow information related to the acquisition of Leonard-Meron Biosciences, Inc.
See accompanying report of independent registered public accounting firm and notes to the consolidated financial statements.
Reflects a 1-for-15 reverse stock split effective June 9, 2017
| 41 |
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED SEPTEMBER 30, 2017, 2016 AND 2015
1. NATURE OF OPERATIONS AND BASIS OF PRESENTATION
Business
Citius Pharmaceuticals, Inc. (“Citius” or the “Company”) is a specialty pharmaceutical company dedicated to the development and commercialization of critical care products targeting unmet needs with a focus on anti-infectives, cancer care and unique prescription products. The Company was founded as Citius Pharmaceuticals, LLC, a Massachusetts limited liability company, on January 23, 2007. On September 12, 2014, Citius Pharmaceuticals, LLC entered into a Share Exchange and Reorganization Agreement (the “Exchange Agreement”), with Citius Pharmaceuticals, Inc. (formerly Trail One, Inc.), a publicly traded company incorporated under the laws of the State of Nevada. Citius Pharmaceuticals, LLC became a wholly-owned subsidiary of Citius.
On March 30, 2016, Citius acquired Leonard-Meron Biosciences, Inc. (“LMB”) as a wholly-owned subsidiary (see “Acquisition of Leonard-Meron Biosciences, Inc.” below).
The Company had one approved and marketed product, Suprenza (phentermine hydrochloride), which it licensed out for promotion in the United States, Canada and Mexico. On July 1, 2016, the Company announced that it was discontinuing Suprenza. Since its inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, and raising capital.
Citius is subject to a number of risks common to companies in the pharmaceutical industry including, but not limited to, risks related to the development by Citius or its competitors of research and development stage products, market acceptance of its products, competition from larger companies, dependence on key personnel, dependence on key suppliers and strategic partners, the Company’s ability to obtain additional financing and the Company’s compliance with governmental and other regulations.
Reverse Stock Split
On June 9, 2017, the Company affected a 1-for-15 reverse stock split of its issued and outstanding shares of common stock, $0.001 par value. Under the terms of the reverse stock split, fractional shares issuable to stockholders were rounded up to the nearest whole share, resulting in a reverse split slightly less than 1-for-15 in the aggregate. All per share amounts and number of shares (other than authorized shares) in these consolidated financial statements and related notes have been retroactively restated to reflect the reverse stock split.
Acquisition of Leonard-Meron Biosciences, Inc.
On March 30, 2016, the Company acquired all of the outstanding stock of Leonard-Meron Biosciences, Inc. (“LMB”) by issuing 1,942,456 shares of its common stock. As of March 30, 2016, the stockholders of LMB received approximately 41% of the issued and outstanding common stock of the Company. In addition, the Company converted the outstanding common stock warrants of LMB into 243,020 common stock warrants of the Company and converted the outstanding common stock options of LMB into 77,252 common stock options of the Company.
The Company acquired tangible assets consisting of cash of $255,748, prepaid expenses of $20,544, property and equipment of $5,085, deposits of $2,167, and identifiable intangible assets of $19,400,000 related to in-process research and development. The Company assumed accounts payable of $244,776, accrued expenses of $598,659, accrued compensation of $615,000, accrued interest of $23,862, and notes payable of $772,970. Accordingly, the net assets acquired amounted to $17,428,277.
The fair value of LMB’s net assets acquired on the date of the acquisition, based on management’s analysis of the fair value of the 1,942,456 shares of the Company’s common stock issued for LMB’s outstanding stock, the 243,020 Company common stock warrants issued for LMB’s outstanding common stock warrants, and the vested portion of the 77,252 Company common stock options issued for LMB’s outstanding common stock options was $19,015,073. The fair value of the common stock issued was estimated at $17,482,093, the fair value of the warrants issued was estimated at $1,071,172 and the fair value of the vested options was estimated at $461,808.
The Company recorded goodwill of $1,586,796 for the excess of the purchase price of $19,015,073 over the net assets acquired of $17,428,277.
In-process research and development represents the value of LMB’s leading drug candidate which is an antibiotic solution used to treat catheter-related bloodstream infections (Mino-Lok™) and is expected to be amortized on a straight-line basis over a period of eight years commencing upon revenue generation. Goodwill represents the value of LMB’s industry relationships and its assembled workforce. Goodwill will not be amortized but will be tested at least annually for impairment.
See report of independent accounting firm
| 42 |
Unaudited pro forma operating results, assuming the acquisition of LMB had been made as of October 1, 2015, are as follows:
| Year Ended September 30, | ||||||||
| 2016 | 2015 | |||||||
| Revenues | $ | — | $ | — | ||||
| Net loss | $ | (11,548,647 | ) | $ | (6,640,600 | ) | ||
| Net loss per share – basic and diluted | $ | (2.52 | ) | $ | (1.64 | ) | ||
Basis of Presentation
The accompanying consolidated financial statements include the operations of Citius Pharmaceuticals, Inc., and its wholly-owned subsidiaries, Citius Pharmaceuticals, LLC and Leonard-Meron Biosciences, Inc. (“LMB”) since the March 30, 2016 acquisition. All significant inter-company balances and transactions have been eliminated in consolidation.
2. GOING CONCERN UNCERTAINTY AND MANAGEMENT’S PLAN
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company experienced negative cash flows from operations of $7,971,205, $5,900,421 and $2,385,416, for the years ended September 30, 2017, 2016 and 2015, respectively. The Company has no revenue and has relied on proceeds from equity transactions and debt to finance its operations. At September 30, 2017, the Company had limited capital to fund its operations The Company plans to raise capital through equity financings from outside investors as well as raise additional funds from existing investors. There is no assurance, however, that that the Company will be successful in raising the needed capital and, if funding is available, that it will be available on terms acceptable to the Company. There is substantial doubt about the Company’s ability to continue as a going concern within a year after the date that the consolidated financial statements are available to be issued and these financial statements do not include any adjustments that might result from the outcome of this uncertainty.
3. BUSINESS AGREEMENTS
Alpex Pharma S.A.
On June 12, 2008, the Company entered into a collaboration and license agreement with Alpex Pharma S.A. (“Alpex”), in which Alpex granted the Company an exclusive right and license to use certain Alpex intellectual property in order to develop and commercialize orally disintegrating tablet formulations of pharmaceutical products in United States, Canada and Mexico. In addition, Alpex manufactured Suprenza, the Company’s commercialized pharmaceutical product, on a contract basis. The agreement was amended on November 15, 2011 (see the “Three-Party Agreement” below).
No milestone, royalty or other payments were earned or received by the Company except for the reimbursement of regulatory fees under the Three-Party Agreement. On July 1, 2016, the Company announced that it notified the Food and Drug Administration and Alpex that it was discontinuing Suprenza.
Prenzamax, LLC
On November 15, 2011, the Company entered into an exclusive license agreement (the “Sublicense Agreement”) with Prenzamax, LLC (“Prenzamax”), in which the Company granted Prenzamax and its affiliates the exclusive right to commercialize Suprenza in the United States. Prenzamax is an affiliate of Akrimax, a related party (see Note 8) and was formed for the specific purpose of managing the Sublicense Agreement. The Company was not reimbursed for any development costs and it did not earn any revenue under the agreement. On July 1, 2016, the Company announced that it notified Prenzamax that it was discontinuing Suprenza.
Three-Party Agreement
On November 15, 2011, the Company, Alpex and Prenzamax entered into an agreement wherein the terms of the Alpex agreement were modified, and Prenzamax and the Company agreed to each pay a portion of certain regulatory filing fees for as long as Prenzamax was purchasing Suprenza from Alpex pursuant to the agreement. During the three months ended March 31, 2016, the Company received $292,575 from Alpex as reimbursement for regulatory filing fees. The reimbursement was recorded as a reduction of research and development expenses. On July 1, 2016, the Company announced that it notified Alpex and Prenzamax that it was discontinuing Suprenza.
See report of independent accounting firm
| 43 |
Patent and Technology License Agreement
LMB has a patent and technology license agreement with Novel Anti-Infective Therapeutics, Inc., (“NAT”) to develop and commercialize Mino-Lok™ on an exclusive, worldwide sub licensable basis, as amended. Since May 2014, LMB has paid an annual maintenance fee of $30,000 that increases over five years to $90,000, until commercial sales of a product subject to the license. Since the acquisition of LMB, the Company recorded maintenance fee expense of $50,000 and $45,000 in 2017 and 2016, respectively under the terms of this agreement.
LMB will also pay annual royalties on net sales of licensed products, with royalties ranging from the mid-single digits to the low double digits. In limited circumstances in which the licensed product is not subject to a valid patent claim and a competitor is selling a competing product, the royalty rate is in the low-single digits. After a commercial sale is obtained, LMB must pay minimum aggregate annual royalties of $100,000 in the first commercial year which is prorated for a less than 12-month period, increasing $25,000 per year to a maximum of $150,000 annually. LMB must also pay NAT up to $1,390,000 upon achieving specified regulatory and sales milestones. Finally, LMB must pay NAT a specified percentage of payments received from any sub licensees.
Unless earlier terminated by NAT, based on the failure to achieve certain development and commercial milestones, the license agreement remains in effect until the date that all patents licensed under the agreement have expired and all patent applications within the licensed patent rights have been cancelled, withdrawn or expressly abandoned.
4. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies followed by the Company in the preparation of the consolidated financial statements is as follows:
Use of Estimates
The process of preparing financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of financial statements and the reported amounts of revenues and expenses during the reporting period. Estimates having relatively higher significance include the accounting for acquisitions, stock-based compensation, valuation of warrants, and income taxes. Actual results could differ from those estimates and changes in estimates may occur.
Cash and Cash Equivalents
The Company considers all highly liquid instruments with maturities of less than three months at the time of purchase to be cash equivalents. From time to time, the Company may have cash balances in financial institutions in excess of insurance limits. The Company has never experienced any losses related to these balances.
Property and Equipment
Property and equipment are valued at cost and are being depreciated over their useful lives using the straight-line method for financial reporting purposes. Routine maintenance and repairs are charged to expense as incurred. Expenditures which materially increase the value or extend useful lives are capitalized. Property and equipment are depreciated over estimated useful lives of three to five years.
Property and equipment consisted of the following at September 30, 2017 and 2016:
| 2017 | 2016 | |||||||
| Computer equipment | $ | 10,648 | $ | 8,522 | ||||
| Less accumulated depreciation | (7,412 | ) | (4,780 | ) | ||||
| $ | 3,236 | $ | 3,742 | |||||
Depreciation and amortization expense for the years ended September 30, 2017, 2016 and 2015 was $2,632, $1,343 and $0, respectively.
See report of independent accounting firm
| 44 |
Research and Development
Research and development costs, including upfront fees and milestones paid to collaborators who are performing research and development activities under contractual agreement with the Company, are expensed as incurred. The Company defers and capitalizes its nonrefundable advance payments that are for research and development activities until the related goods are delivered or the related services are performed. When the Company is reimbursed by a collaboration partner for work the Company performs, it records the costs incurred as research and development expenses and the related reimbursement as a reduction to research and development expenses in its consolidated statement of operations. Research and development expenses primarily consist of clinical and non-clinical studies, materials and supplies, third-party costs for contracted services, and payments related to external collaborations and other research and development related costs.
In-process Research and Development and Goodwill
In-process research and development represents the value of LMB’s leading drug candidate which is an antibiotic solution used to treat catheter-related bloodstream infections (Mino-Lok™) and is expected to be amortized on a straight-line basis over a period of eight years commencing upon revenue generation. Goodwill represents the value of LMB’s industry relationships and its assembled workforce. Goodwill will not be amortized but will be tested at least annually for impairment.
The Company reviews intangible assets annually to determine if any adverse conditions exist or a change in circumstances has occurred that would indicate impairment or a change in the remaining useful life of any intangible asset. If the carrying value of an asset exceeds its undiscounted cash flows, the Company writes down the carrying value of the intangible asset to its fair value in the period identified. No triggering events occurred since the acquisition of LMB that would suggest that a potential impairment may have occurred through September 30, 2017.
The Company evaluates the recoverability of goodwill annually or more frequently if events or changes in circumstances indicate that the carrying value of an asset might be impaired. Goodwill is first qualitatively assessed to determine whether further impairment testing is necessary. Factors that management considers in this assessment include macroeconomic conditions, industry and market considerations, overall financial performance (both current and projected), changes in management and strategy and changes in the composition or carrying amount of net assets. If this qualitative assessment indicates that it is more likely than not that the fair value of a reporting unit is less than its carrying amount, a two-step process is then performed.
The Company performed a qualitative assessment for our 2017 analysis of goodwill. Based on this assessment, management does not believe that it is more likely than not that the carrying value of the reporting unit exceeds its fair value. Accordingly, no further testing was performed as management believes that there are no impairment issues in regards to goodwill as of September 30, 2017.
Patents and Trademarks
Certain costs of outside legal counsel related to obtaining trademarks for the Company are capitalized. Patent costs are amortized over the legal life of the patents, generally twenty years, starting at the patent issuance date. There are no capitalized patents and trademarks as of September 30, 2017.
The costs of unsuccessful and abandoned applications are expensed when abandoned. The cost of maintaining existing patents are expensed as incurred.
Stock-Based Compensation
The Company recognizes compensation costs resulting from the issuance of stock-based awards to employees and directors, net of expected forfeitures, as an expense in the consolidated statement of operations over the requisite service period based on the fair value for each stock award on the grant date. The fair value of each option grant is estimated as of the date of grant using the Black-Scholes option pricing model. Due to its limited operating history, limited number of sales of its common stock and limited history of its shares being publicly traded, the Company estimates its volatility in consideration of a number of factors including the volatility of comparable public companies. The estimated forfeiture rate is based on historical forfeiture information as well as subsequent events occurring prior to the issuance of the financial statements. Because our stock options have characteristics significantly different from those of traded options, and because changes in the input assumptions can materially affect the fair value estimate, the existing model may not necessarily provide a reliable single measure of fair value of our stock options
See report of independent accounting firm
| 45 |
The Company recognizes compensation costs resulting from the issuance of stock-based awards to non-employees as an expense in the consolidated statement of operations over the service period based on the measurement of fair value for each stock award.
Derivative Instruments
The Company generally does not use derivative instruments to hedge exposures to cash-flow or market risks; however, certain warrants to purchase common stock that do not meet the requirements for classification as equity are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. Such financial instruments are initially recorded at fair value with subsequent changes in fair value charged (credited) to operations in each reporting period. If these instruments subsequently meet the requirements for classification as equity, the Company reclassifies the fair value to equity.
Income Taxes
The Company follows accounting guidance regarding the recognition, measurement, presentation and disclosure of uncertain tax positions in the consolidated financial statements. Tax positions taken or expected to be taken in the course of preparing our tax returns are required to be evaluated to determine whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authorities. Tax positions not deemed to meet a more-likely-than-not threshold would be recorded in the consolidated financial statements. There are no uncertain tax positions that require accrual or disclosure as of September 30, 2017.
Any interest or penalties are charged to expense. During the years ended September 30, 2017, 2016 and 2015, the Company did not recognize any interest and penalties. Tax years subsequent to December 31, 2013 are subject to examination by federal and state authorities.
We recognize deferred tax assets and liabilities based on differences between the financial reporting and tax basis of assets and liabilities, and operating loss and tax credit carry forwards. Deferred tax assets and liabilities are measured using the enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. We provide a valuation allowance, if necessary, for deferred tax assets for which we do not consider realization of such assets to be “more-likely-than-not”. The deferred tax benefit or expense for the period represents the change in the deferred tax asset or liability from the beginning to the end of the period.
Basic and Diluted Net Loss per Common Share
Basic and diluted net loss per common share is computed by dividing net loss in each period by the weighted average number of shares of common stock outstanding during such period. For the periods presented, common stock equivalents, consisting of options, warrants and convertible securities were not included in the calculation of the diluted loss per share because they were anti-dilutive.
Fair Value of Financial Instruments
The financial statements include various estimated fair value information. Financial instruments are initially recorded at historical cost. If subsequent circumstances indicate that a decline in the fair value of a financial asset is other than temporary, the financial asset is written down to its fair value.
Unless otherwise indicated, the fair values of financial instruments approximate their carrying amounts. By their nature, all financial instruments involve risk, including credit risk for non-performance by counterparties. The fair values of cash and cash equivalents, accounts payable, accrued interest, accrued expenses, notes payable and due to related party approximate their recorded amounts because of their relatively short settlement terms.
The Company groups its financial assets and financial liabilities generally measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
| Level 1: | Valuation is based on quoted prices in active markets for identical assets or liabilities. Level 1 assets and liabilities generally include debt and equity securities that are traded in an active exchange market. Valuations are obtained from readily available pricing sources for market transactions involving identical assets or liabilities. |
| Level 2: | Valuation is based on observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. For example, Level 2 assets and liabilities may include debt securities with quoted prices that are traded less frequently than exchange-traded instruments. |
| Level 3: | Valuation is based on unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation. This category generally includes certain private equity investments and long-term derivative contracts. |
See report of independent accounting firm
| 46 |
The Company's financial liabilities measured at fair value consist solely of the derivative warrant liability which is classified as Level 3 in fair value hierarchy (see Note 6). The Company uses a valuation method, the Black-Scholes option pricing model, and the requisite assumptions in estimating the fair value for the warrants considered to be derivative instruments. The Company has no financial assets measured at fair value.
The Company may also be required, from time to time, to measure certain other financial assets at fair value on a nonrecurring basis. These adjustments to fair value usually result from application of lower-of-cost-or-market accounting or write-downs of individual assets. There were no such adjustments in the years ended September 30, 2017, 2016 and 2015.
Segment Reporting
The Company currently operates as a single segment.
Concentrations of Credit Risk
The Company has no significant off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other hedging arrangements.
Recently Issued Accounting Standards
In January 2017, the FASB issued ASU No. 2017-04, Intangibles – Goodwill and Other (Topic 350). This ASU eliminates step 2 from the goodwill impairment test by comparing the fair value of a reporting unit with the carrying amount of the reporting unit. If the carrying amount exceeds the fair value, an impairment charge for the excess is recorded. The amendments of this ASU are effective for annual or any interim goodwill impairment tests in fiscal years beginning after December 15, 2019. Early adoption is permitted for interim or annual goodwill impairment tests performed on testing dates after January 1, 2017. The Company is evaluating the impact of the adoption of this guidance on its financial statements but does not expect it to have a material impact.
In May 2017, the FASB issued ASU No. 2017-09, Compensation - Stock Compensation (Topic 718): Scope of Modification Accounting, which clarifies when changes to the terms or conditions of a share-based payment award must be accounted for as modifications. The new guidance will reduce diversity in practice and result in fewer changes to the terms of an award being accounted for as modifications. Under ASU 2017-09, an entity will not apply modification accounting to a share-based payment award if the award’s fair value, vesting conditions and classification as an equity or liability instrument are the same immediately before and after the change. ASU 2017-09 will be applied prospectively to awards modified on or after the adoption date. The guidance is effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption is permitted. The Company is evaluating the impact of the adoption of this guidance on its financial statements but does not expect it to have a material impact.
In July 2017, FASB issued ASU No. 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Non-controlling Interests with a Scope Exception. Part I of this Update addresses the complexity of accounting for certain financial instruments with down round features by simplifying the accounting for these instruments. This Update requires companies to disregard the down round feature when assessing whether an instrument, such as a warrant, is indexed to its own stock, for purposes of determining liability or equity classification. This will change the classification of certain warrants with down round features from a liability to equity. Also, entities must adjust their basic EPS calculation for the effect of the down round provision when triggered (that is, when the exercise price of the related equity-linked financial instrument is adjusted downward because of the down round feature). That effect is treated as a dividend and as a reduction of income available to common shareholders in basic EPS. An entity will also recognize the effect of the trigger within equity. The guidance is effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption is permitted. The Company is evaluating the impact of the adoption of this guidance on its financial statements but does not expect it to have a material impact. Part II of this Update addresses the difficulty of navigating Topic 480, Distinguishing Liabilities from Equity, because of the existence of extensive pending content in the FASB Accounting Standards Codification. The amendments in Part II of this Update re-characterize the indefinite deferral of certain provisions of Topic 480, Distinguishing Liabilities from Equity that previously were presented as pending content in the Codification, to a scope exception, and do not have any accounting effect.
See report of independent accounting firm
| 47 |
5. NOTES PAYABLE
A summary of notes payable outstanding as of September 30, 2017 and 2016 is as follows:
| 2017 | 2016 | |||||||
| Demand notes payable – Leonard Mazur | $ | 160,470 | $ | 160,470 | ||||
| Demand notes payable – Myron Holubiak | 12,500 | 12,500 | ||||||
| Revolving demand promissory notes – Leonard Mazur | — | 500,000 | ||||||
| Notes payable | $ | 172,970 | $ | 672,970 | ||||
Promissory Notes
In November 2013, the Company issued two 5% promissory notes (the “Promissory Notes”) to two existing investors in aggregate total principal amount of $600,000. On December 31, 2014, the note holders requested conversion of the outstanding $600,000 Promissory Notes and accrued interest of $33,333 into 70,371 shares of common stock at a conversion price of $9.00 per share.
Notes Payable - Related Parties
On March 30, 2016, the Company assumed $772,970 of demand notes payable in the acquisition of LMB, including $760,470 to our Chairman, Leonard Mazur, and $12,500 to our Chief Executive Officer, Myron Holubiak. Notes with a principal balance of $704,000 accrue interest at the “Prime Rate”, as published in the Wall Street Journal on the last day of each month plus 1% and notes with a principal balance of $68,970 accrue interest at 12% per annum. In April 2016, $600,000 of the “Prime Rate” plus 1% demand notes payable and accrued interest of $1,985 was repaid to Leonard Mazur.
The Board of Directors authorized revolving demand promissory notes with Leonard Mazur in an aggregate principal amount of up to $2,500,000 that accrue interest at the prime rate plus 1%. On September 7, 2016, the Company issued a $500,000 note. The Company issued $2,000,000 of additional notes through the period ended May 10, 2017. On May 10, 2017, the notes were converted into a $2,500,000 convertible promissory note that matures on June 30, 2018 and is convertible into shares of common stock, at the sole discretion of Mr. Mazur, at a conversion price equal to 75% of the price per share paid by investors in the Company’s 2017 registered public offering. In connection with the modification of the note, the Company recorded a charge of $833,333 to additional paid-in capital and increased the carrying value of the notes to $3,333,333 which is the fair value of the common stock issuable on conversion. On August 8, 2017, Leonard Mazur converted the $2,500,000 principal balance and accrued interest of $63,174 into 828,500 shares of common stock.
On May 10, 2017 and June 23, 2017, the Company executed a $1,500,000 future advance convertible promissory note and a $1,000,000 future advance convertible promissory note, respectively, with Leonard Mazur that both mature on December 31, 2017 and accrue interest at the prime rate plus 1%. The notes are convertible into shares of common stock, at the sole discretion of Mr. Mazur, at a conversion price equal to 75% of the price per share paid by investors in the Company’s 2017 registered public offering. On August 8, 2017, Leonard Mazur converted the outstanding $2,210,000 principal balances and accrued interest of $13,066 into 718,567 shares of common stock.
In connection with the conversions, the Company recorded net non-cash interest expense of $762,078 due to the beneficial conversion feature on the conversion price of $1,595,411 and the amortization of the previously recorded modification premium of $833,333.
The Company evaluated all terms of the future advance convertible promissory notes, including the Change in Control provision, to identify any embedded features that required bifurcation and recording as derivative instruments. The Company determined that there were no such features requiring separate accounting.
Interest Expense
Interest expense on notes payable for the years ended September 30, 2017, 2016 and 2015 was $850,789, $8,994, and $7,500, respectively.
6. DERIVATIVE WARRANT LIABILITY
Derivative financial instruments are recognized as a liability on the consolidated balance sheet and measured at fair value. At September 30, 2016, the Company had outstanding warrants to purchase 307,778 shares, of its common stock that were considered to be derivative instruments since the agreements contained “down round” provisions whereby the exercise price of the warrants was subject to adjustment in the event that the Company issues common stock for a lower price per share than the investors paid within a specified time period after the original issuance of the warrants (see Note 7).
See report of independent accounting firm
| 48 |
On September 12, 2015, anti-dilution rights related to warrants to purchase 338,672 shares of common stock expired which resulted in a reclassification from derivative warrant liability to additional paid-in capital of $1,148,328. During the year ended September 30, 2016, anti-dilution rights related to warrants to purchase 202,469 shares of common stock expired which resulted in a reclassification from derivative warrant liability to additional paid-in capital of $1,093,765. During the year ended September 30, 2017, anti-dilution rights related to warrants to purchase 307,778 shares of common stock expired which resulted in a reclassification from derivative warrant liability to additional paid-in capital of $1,433,316.
On June 8, 2017, the Company granted anti-dilution rights to the investors and the placement agent for the 2016 Offering (see Note 7) in connection with a release agreement. The investors and placement agent hold 140,819 warrants to purchase common stock at $8.25 per share. The exercise price of the warrants was subject to adjustment in the event that the price per share paid by investors in the Company’s 2017 public offering was lower than the $8.25 exercise price of the warrants. On June 8, 2017, the Company reclassified the $641,385 fair value of the warrants to derivative warrant liability. The Company recorded a gain of $203,490 based on the change in the estimated fair value of the warrants between June 8, 2017 and August 8, 2017. On August 8, 2017, the Company adjusted the exercise price of 2016 Offering warrants to $4.125 per share and reclassified the $437,895 derivative warrant liability to additional paid-in capital.
The Company performs valuations of the warrants using a probability weighted Black-Scholes option pricing model which value was also compared to a Binomial Option Pricing Model for reasonableness. This model requires input of assumptions including the risk-free interest rates, volatility, expected life and dividend rates, and has also considered the likelihood of “down-round” financings. Selection of these inputs involves management’s judgment and may impact net income. Due to our limited operating history and limited number of sales of our common stock, we estimate our volatility based on a number of factors including the volatility of comparable publicly traded pharmaceutical companies. The volatility factor used in the Black-Scholes option pricing model has a significant effect on the resulting valuation of the derivative liabilities on our balance sheet. The volatility calculated at September 30, 2016 was 73% and we used a risk-free interest rate of 1.14%, estimated lives of 4.10 to 4.57 years, which were the remaining contractual lives of the warrants subject to “down-round” provisions, and no dividends to our common stock. No warrants are classified as derivative warrant liabilities as of September 30, 2017.
The table below presents the changes in the derivative warrant liability for the years ended September 30, 2017, 2016 and 2015, which were measured at fair value on a recurring basis and classified as Level 3 in the fair value hierarchy (see Note 4):
| 2017 | 2016 | 2015 | ||||||||||
| Derivative warrant liability, beginning of year | $ | 1,681,973 | $ | 738,955 | $ | 1,450,943 | ||||||
| Fair value of warrants issued | 641,385 | 1,198,564 | 768,435 | |||||||||
| Total realized/unrealized losses (gains) included in net loss | (452,147 | ) | 838,219 | (332,095 | ) | |||||||
| Reclassification of liability to additional paid-in capital | (1,871,211 | ) | (1,093,765 | ) | (1,148,328 | ) | ||||||
| Derivative warrant liability, end of year | $ | - | $ | 1,681,973 | $ | 738,955 | ||||||
7. COMMON STOCK, STOCK OPTIONS AND WARRANTS
Common Stock
On September 15, 2016, the stockholders approved an increase in the number of shares of authorized common stock from 90,000,000 shares to 200,000,000 shares. On June 9, 2017, the Company affected a 1-for-15 reverse stock split of its issued and outstanding shares of common stock, $0.001 par value. Under the terms of the reverse stock split, fractional shares issuable to stockholders were rounded up to the nearest whole share, resulting in a reverse split slightly less than 1-for-15 in the aggregate. All per share amounts and number of shares (other than authorized shares) in these consolidated financial statements and related notes have been retroactively restated to reflect the reverse stock split.
See report of independent accounting firm
| 49 |
Private Offerings
On September 12, 2014, the Company sold 226,671 Units for a purchase price of $9.00 per Unit for gross proceeds of $2,040,040. Each Unit consists of one share of common stock and one five-year warrant (the “Investor Warrants”) to purchase one share of common stock at an exercise price of $9.00, (the “Private Offering”). The Investor Warrants will be redeemable by the Company at a price of $0.015 per Investor Warrant at any time subject to the conditions that (i) the common stock has traded for twenty (20) consecutive trading days with a closing price of at least $22.50 per share with an average trading volume of 3,333 shares per day and (ii) the Company provides 20 trading days prior notice of the redemption and the closing price of the common stock is not less than $17.55 for more than any 3 days during such notice period and (iii) the underlying shares of common stock are registered.
The Company issued the Placement Agent and their designees five-year warrants (the “Placement Agent Unit Warrants”) to purchase 45,334 Units at an exercise price of $9.00 per Unit on a cash or cashless basis with respect to purchase of the Units, and exercisable only for cash with respect to warrants received as part of the Units.
In addition, the Placement Agent was issued warrants to purchase 66,667 shares of common stock exercisable for cash at $9.00 per share for investment banking services provided in connection with the transaction (the “Placement Agent Share Warrants”).
In connection with the Private Offering, the Company entered into an agreement pursuant to which the Company filed a registration statement, registering for resale all shares of common stock (i) included in the Units; and (ii) issuable upon exercise of the Investor Warrants. The registration statement was declared effective on January 21, 2016.
During the year ended September 30, 2015, the Company sold 189,136 Units for a purchase price of $8.10 per Unit and 13,333 Units for a purchase price of $9.00 per Unit for gross proceeds of $1,652,000. Each Unit consists of one share of common stock and one Investor Warrant (see description above). There was no placement agent for the 2015 private placements and other cash expenses related to the placements were $142,507. In connection with these placements, the Company credited $741,058 to stockholders’ equity (deficit) and $768,435 to derivative warrant liability.
During the year ended September 30, 2016, the Company sold 290,000 Units for a purchase price of $8.10 per Unit and 17,778 Units for a purchase price of $9.00 per Unit for gross proceeds of $2,509,000. Each Unit consists of one share of common stock and one Investor Warrant (see description above). There was no placement agent for these private placements and other cash expenses related to the placements were $81,312. In connection with these placements, the Company credited $1,229,124 to stockholders’ equity (deficit) and $1,198,564 to derivative warrant liability.
On March 22, 2016, the Company sold 333,333 shares of common stock at $9.00 per share to its Chairman of the Board, Leonard Mazur, for gross proceeds of $3,000,000. There were no expenses related to this placement.
In February 2017, the Company completed an offering (the “2016 Offering”) and sold 128,017 units at $6.00 per unit for gross proceeds of $768,100. Each unit consisted of (i) one share of common stock and (ii) a five year warrant to purchase one share of common stock at an exercise price of $8.25 per share (the “2016 Offering Warrants”). The placement agent received a 10% cash commission on the gross proceeds, an expense allowance equal to 3% of the proceeds, and warrants to purchase 12,802 shares of common stock at an exercise price of $8.25 per share. The estimated fair value of the 128,017 warrants issued to the investors was $587,592 and the estimated fair value of the 12,802 warrants issued to the placement agent was $58,759. The placement agent commissions and expense allowance was $99,853. Other costs of the placement were $176,896.
During January 2017, the Company issued 29,729 shares of its common stock for investor relations services. The $298,774 fair value of the common stock was expensed during the year ended September 30, 2017.
On May 5, 2017, the Company issued 11,400 shares of common stock valued at $77,748 in connection with a settlement agreement and release with a consultant that had an agreement with Leonard-Meron Biosciences. The Company expensed the $77,748 as a settlement expense during the year ended September 30, 2017.
On June 7, 2017, the Company entered into a release agreement with the placement agent for the 2016 Offering. The placement agent consented to future financings and waived certain covenants contained in the 2016 Offering agreements. As consideration for the release, the Company issued 6,668 shares of common stock valued at $45,476 to the placement agent. The Company expensed the $45,476 as a settlement expense during the year ended September 30, 2017.
See report of independent accounting firm
| 50 |
On June 8, 2017, the Company entered into release agreements with the investors in the 2016 Offering where each investor released the Company from the restrictions included in the unit purchase agreements. In exchange, the Company agreed that (i) in the event that a financing is conducted at a price per share or price per unit lower than $6.00, then the Company will issue additional shares to each investor sufficient to effectively reprice the sale of the 2016 Offering units to the lower price; (ii) in the event that the financing is conducted at a price per share or price per unit less than the $8.25 exercise price of the warrants issued in the 2016 Offering then the exercise price of the warrants shall be reduced to the lower price; and (iii) the Company will give each investor no less than 6 hours of notice before the closing of any subsequent financing, through and including the Company’s 2017 registered public offering, and each investor shall have a 6-hour option to purchase up to 20% of the securities sold in such offering. In connection with these agreements the Company reclassified the $641,385 fair value of the 140,819 warrants issued in the 2016 Offering to derivative warrant liability on June 8, 2017 (see Note 6). On August 8, 2017, the Company completed the 2017 public offering and issued 58,191 shares of common stock to the investors in the 2016 Offering to reprice the sale of the 2016 Offering units to $4.125 per unit and repriced the 2016 Offering Warrants to an exercise price of $4.125 per share. During the year ended September 30, 2017, the Company recorded a settlement expense of $161,771 in connection with the issuance of the additional 58,191 shares of common stock and reclassified the current fair value of the warrants to additional paid-in capital.
2017 Public Offering
On August 8, 2017, the Company closed an underwritten public offering of 1,648,484 shares of common stock and warrants to purchase 1,648,484 shares of common stock at an offering price of $4.125 per share and $0.01 per warrant. The warrants have a per share exercise price of $4.125, are exercisable immediately and will expire five years from the date of issuance. The gross proceeds to Citius from this offering were $6,802,469, before deducting underwriting discounts and commissions and other estimated offering expenses of $685,573. The Company granted the underwriters a 45-day option to purchase up to an additional 247,272 shares of common stock and warrants to purchase 247,272 shares of common stock to cover over-allotments, if any. On August 8, 2017, the underwriters partially exercised the over-allotment to purchase an additional 247,272 warrants. The estimated fair value of the 1,895,756 warrants issued to the investors was $4,160,195 and the estimated fair value of the 65,940 warrants issued to the underwriters was $142,419.
Unit Purchase Options
On April 7, 2017, the Company issued a three year Unit Purchase Option Agreement to a consultant for the purchase of 38,000 units at a purchase price of $9.00 per unit. Each unit consists of one share of common stock and a warrant to purchase one share of common stock at an exercise price of $9.00 per share which expires on the earlier of three years after exercise of the Unit Purchase Option Agreement or April 7, 2023. The consultant provided the Company with business development and financing assistance for the three months ended June 30, 2017. The Company estimated the fair value of the unit purchase option agreement at $104,138 and expensed it during the year ended September 30, 2017.
On June 29, 2017, the Company issued a three year Unit Purchase Option Agreement to a consultant for the purchase of 62,667 units at a purchase price of $9.00 per unit. Each unit consists of one share of common stock and a warrant to purchase one share of common stock at an exercise price of $9.00 per share which expires on the earlier of three years after exercise of the Unit Purchase Option Agreement or June 29, 2022. The consultant will provide the Company with business development and financing assistance through December 31, 2017. The Company estimated the fair value of the unit purchase option agreement at $193,860 and recorded it as a prepaid expense at June 30, 2017. The Company recorded an expense of $96,930 for this agreement during the year ended September 30, 2017.
Stock Options
On September 12, 2014, the Board of Directors adopted the 2014 Stock Incentive Plan (the “2014 Plan”) and reserved 866,667 shares of common stock for issuance to employees, directors and consultants. On September 12, 2014, the stockholders approved the plan. Pursuant to the 2014 Plan, the Board of Directors (or committees and/or executive officers delegated by the Board of Directors) may grant stock options, stock appreciation rights, restricted stock, restricted stock units, other stock-based awards and cash-based awards. As of September 30, 2017, there were options to purchase an aggregate of 861,039 shares of common stock outstanding under the 2014 Plan, options to purchase 4,829 were exercised and 799 shares available for future grants.
The fair value of each stock option award is estimated on the date of grant using the Black-Scholes option pricing model. Due to its limited operating history and limited number of sales of its common stock, the Company estimated its volatility in consideration of a number of factors including the volatility of comparable public companies. The Company uses historical data, as well as subsequent events occurring prior to the issuance of the consolidated financial statements, to estimate option exercises and employee terminations within the valuation model. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected term assumption. The expected term of stock options granted to employees and directors, all of which qualify as “plain vanilla,” is based on the average of the contractual term (generally 10 years) and the vesting period. For non-employee options, the expected term is the contractual term.
The following assumptions were used in determining the fair value of stock option grants for the years ended September 30, 2017, 2016 and 2015:
| 2017 | 2016 | 2015 | ||||||||||
| Risk-free interest rate | 1.79 – 1.90 | % | 0.95 – 1.40 | % | 1.37 – 1.52 | % | ||||||
| Expected dividend yield | 0 | % | 0 | % | 0 | % | ||||||
| Expected term | 6.50 – 10 years | 4.75 – 9 years | 2.5 – 6 years | |||||||||
| Forfeiture rate | 0 | % | 0 | % | 0 | % | ||||||
| Expected volatility | 85 - 108 | % | 57 – 74 | % | 53 – 58 | % | ||||||
See report of independent accounting firm
| 51 |
A summary of option activity under the 2014 Plan is presented below:
| Options | Shares | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Term | Aggregate Intrinsic Value | ||||||||||
| Outstanding at September 30, 2014 | 220,000 | $ | 6.75 | 9.96 years | $ | 495,000 | ||||||||
| Granted | 40,000 | 9.00 | ||||||||||||
| Exercised | — | — | ||||||||||||
| Forfeited or expired | — | — | ||||||||||||
| Outstanding at September 30, 2015 | 260,000 | 7.10 | 8.94 years | $ | 297,000 | |||||||||
| Granted | 244,933 | 11.41 | ||||||||||||
| Assumed in acquisition | 77,252 | 1.08 | ||||||||||||
| Exercised | — | — | ||||||||||||
| Forfeited or expired | — | — | ||||||||||||
| Outstanding at September 30, 2016 | 582,185 | 8.11 | 8.59 years | $ | 1,355,924 | |||||||||
| Granted | 283,669 | 3.65 | ||||||||||||
| Fractional share adjustment for 1-for-15 reverse stock split | 14 | — | ||||||||||||
| Exercised | (4,829 | ) | 0.01 | |||||||||||
| Forfeited or expired | — | — | ||||||||||||
| Outstanding at September 30, 2017 | 861,039 | $ | 6.69 | 8.37 years | $ | 208,151 | ||||||||
| Exercisable at September 30, 2017 | 513,997 | $ | 7.71 | 7.47 years | $ | 208,151 | ||||||||
On April 1, 2015, the Board of Directors granted stock options to purchase 6,667 shares of common stock at an exercise price of $9.00 per share. The weighted average grant-date fair value of the options granted was estimated at $2.44 per share. These options vested immediately and have a term of 5 years. On June 1, 2015, the Board of Directors granted stock options to purchase 33,333 shares of common stock at an exercise price of $9.00 per share. The weighted average grant-date fair value of the options granted was estimated at $4.10 per share. These options vest over three years and have a term of 10 years.
In October 2015, the Company appointed two new directors. Each director received an option to purchase 26,667 shares of common stock at an exercise price of $8.10 per share in consideration for their services as members of the Board of Directors. The weighted average grant-date fair value of the options was estimated at $4.17 per share. These options vest over 14 months and have a term of 10 years.
On March 30, 2016, the Company assumed stock options to purchase 77,252 shares of common stock in connection with the acquisition of LMB. The LMB option holders received stock options to purchase 71,217 shares at an exercise price of $0.01 per share and 6,035 shares at an exercise price of $13.65 per share. Pursuant to the original grants, options to purchase 4,829 shares were immediately vested and options to purchase 72,423 shares vest over three years. The March 30, 2016 estimated fair value of the stock options was $670,242. The fair value of the vested options was estimated at $461,808 and has been included in the purchase price of LMB. The March 30, 2016 fair value of the unvested options was estimated at $208,434 per share and will be expensed over the remaining vesting period of the options. These options all had original terms of 10 years.
On June 23, 2016, the Board of Directors granted stock options to four directors. Each director received an option to purchase 13,333 shares of common stock at an exercise price of $12.00 per share in consideration for their services as members of the Board of Directors. The weighted average grant-date fair value of the options was estimated at $6.58 per share. These options vest in full on June 23, 2017 and have a term of 10 years.
In July 2016, the Board of Directors granted stock options to purchase a total of 138,267 shares to three employees at prices ranging from $10.50 to $13.50 per share. The weighted average grant date fair value of the options was estimated at $7.70 per share. These options vest over terms of 19 to 36 months and have a term of 10 years.
See report of independent accounting firm
| 52 |
On January 1, 2017, the Board of Directors granted stock options to purchase a total of 8,669 shares to four consultants at $10.05 per share. The weighted average grant date fair value of the options was estimated at $8.41 per share. These options vest over terms of 12 to 36 months and have a term of 10 years.
In September 2017, the Board of Directors granted stock options to purchase a total of 225,000 shares to 12 employees and 50,000 options to two consultants at $3.45 per share. The weighted average grant date fair value of the options was estimated at $2.95 per share. These options vest over terms of 12 to 36 months and have a term of 10 years.
Stock-based compensation expense for the years ended September 30, 2017, 2016 and 2015 was $986,620, $732,151 and $486,271, respectively.
At September 30, 2017, unrecognized total compensation cost related to unvested awards of $1,183,113 is expected to be recognized over a weighted average period of 1.79 years.
Warrants
The Company has reserved 3,346,891 shares of common stock for the exercise of outstanding warrants. The following table summarizes the warrants outstanding at September 30, 2017:
| Exercise price | Number | Expiration Dates | ||||||||
| Investor Warrants | $ | 9.00 | 226,671 | September 12, 2019 | ||||||
| Placement Agent Unit Warrants | 9.00 | 45,334 | September 12, 2019 | |||||||
| Warrants underlying Placement Agent Unit Warrants | 9.00 | 45,334 | September 12, 2019 | |||||||
| Placement Agent Share Warrants | 9.00 | 66,667 | September 12, 2019 | |||||||
| Investor Warrants | 9.00 | 202,469 | March 19, 2020 – September 14, 2020 | |||||||
| Investor Warrants | 9.00 | 307,778 | November 5, 2020 – April 25, 2021 | |||||||
| LMB Warrants | 6.15 | 90,151 | June 12, 2019 – March 2, 2021 | |||||||
| LMB Warrants | 9.90 | 8,155 | September 30, 2019 – January 8, 2020 | |||||||
| LMB Warrants | 20.70 | 17,721 | November 3, 2019 – March 6, 2020 | |||||||
| LMB Warrants | 7.50 | 73,883 | August 18, 2020 – March 14, 2021 | |||||||
| LMB Warrants | 13.65 | 53,110 | March 24, 2022 – April 29, 2022 | |||||||
| Financial Advisor Warrants | 3.00 | 66,667 | August 15, 2021 | |||||||
| 2016 Offering Warrants | 4.125 | 128,017 | November 23, 2021 – February 27, 2022 | |||||||
| 2016 Offering Placement Agent Warrants | 4.125 | 12,802 | November 23, 2021 – February 27, 2022 | |||||||
| Convertible Note Warrants | 9.75 | 40,436 | September 12, 2019 | |||||||
| 2017 Public Offering Warrants | 4.125 | 1,895,756 | August 2, 2022 | |||||||
| 2017 Public Offering Underwriter Warrants | 4.5375 | 65,940 | February 2, 2023 | |||||||
| 3,346,891 | ||||||||||
On March 30, 2016, the Company granted warrants to purchase 243,020 shares of common stock in connection with the acquisition of LMB. The warrants have exercise prices between $6.15 and $20.70 per share. All warrants were vested at March 30, 2016. The fair value of the warrants was estimated at $1,071,172 and has been included in the purchase price of LMB.
On August 16, 2016, the Company granted warrants to purchase 66,667 shares of common stock in connection with a one year financial advisory agreement. The warrants were vested on issuance, have an exercise price of $3.00 per share and are exercisable on a cash or cashless basis. The fair value of the warrants was estimated at $477,181 and recorded as a prepaid expense on the issuance date. During the years ended September 30, 2017 and 2016, the Company expensed $417,181 and $60,000, respectively, in connection with the agreement.
During the year ended September 30, 2017, the Company sold 128,017 2016 Offering Units, at a price of $6.00 per Unit, consisting of (i) one share of common stock and (ii) a warrant to purchase one share of common stock. Each 2016 Offering Warrant has an exercise price of $8.25 and is exercisable for five years from the date of issuance. Additionally, warrants to purchase 12,802 shares of common stock were granted to the Placement Agent pursuant to the above pricing terms.
On June 7, 2017, the Company issued a warrant to purchase 40,436 shares of common stock at $9.75 per share in settlement of issues related to the July 31, 2014 conversion of a subordinated convertible promissory note. The Company charged the $119,402 estimated fair value of the warrant to settlement expenses during the year ended September 30, 2017.
See report of independent accounting firm
| 53 |
On June 8, 2017, the Company entered into release agreements with the investors in the 2016 Offering where each investor released the Company from the restrictions included in the unit purchase agreements. In exchange, the Company agreed that (i) in the event that a financing is conducted at a price per share or price per unit lower than $6.00, then the Company will issue additional shares to each investor sufficient to effectively reprice the sale of the 2016 Offering units to the lower price; (ii) in the event that the financing is conducted at a price per share or price per unit less than the $8.25 exercise price of the warrants issued in the 2016 Offering then the exercise price of the warrants shall be reduced to the lower price; and (iii) the Company will give each investor no less than 6 hours of notice before the closing of any subsequent financing, through and including the Company’s 2017 registered public offering, and each investor shall have a 6-hour option to purchase up to 20% of the securities sold in such offering. In connection with these agreements the Company reclassified the fair value of the 140,819 warrants issued in the 2016 Offering to derivative warrant liability on June 8, 2017 (see Note 5). On August 8, 2017, the Company repriced the 2016 Offering Warrants to an exercise price of $4.125 per share.
Effective June 16, 2017, the Company amended warrants associated with the Leonard-Meron Biosciences, Inc. 2015 private placement offering. The warrant amendments removed the exercise price reset provisions, adjusted the exercise price of the warrants to $7.50 per share and extended the term of the warrants by three years. The estimated fair value of the warrants on June 16, 2017 after the amendments was $250,733. As a result of the amendment, the Company recorded an incremental cost of $71,488 as a settlement expense during the year ended September 30, 2017.
See Note 7 (2017 Public Offering) for a description of the 2017 public offering warrants and underwriter warrants.
At September 30, 2017, the weighted average remaining life of all of the outstanding warrants is 4.08 years, all warrants are exercisable, and the aggregate intrinsic value for the warrants outstanding was $10,000.
Common Stock Reserved
A summary of common stock reserved for future issuances as of September 30, 2017 and 2016 is as follows:
| 2017 | 2016 | |||||||
| 2014 Stock Incentive Plan options outstanding | 861,039 | 582,185 | ||||||
| 2014 Stock Incentive Plan available for future grants | 799 | 284,482 | ||||||
| Warrants | 3,346,891 | 1,203,940 | ||||||
| Unit purchase options | 201,334 | — | ||||||
| Total | 4,410,063 | 2,070,607 | ||||||
8. RELATED PARTY TRANSACTIONS
The Company’s headquarters were previously located in Maynard, MA in the office space of a company affiliated through common ownership. In connection with the March 30, 2016 acquisition of LMB, the Company moved its principal executive offices to Cranford, NJ. The Company did not record any revenue or expense related to the use of the Maynard, MA office space as management has determined the usage to be immaterial and the affiliate has not charged for the usage.
As of September 30, 2017 and 2016, the Company owed $27,637, respectively, to a company affiliated through common ownership for the expenses the related party paid on the Company’s behalf and services performed by the related party.
Our Chairman of the Board, Leonard Mazur, is the cofounder and Vice Chairman of Akrimax Pharmaceuticals, LLC (“Akrimax”), a privately held pharmaceutical company specializing in producing cardiovascular and general pharmaceutical products (see Note 3). The Company leases office space from Akrimax (see Note 9).
Our Chairman of the Board, Leonard Mazur, and our Chief Executive Officer, Myron Holubiak, are co-founders and were significant shareholders in LMB. In connection with the acquisition of LMB, our Chairman purchased an additional 333,333 shares of the Company. See Note 5 for a description of related party debt transactions.
In connection with the 2017 Public Offering, Mr. Mazur purchased 421,400 units consisting of 421,400 shares of common stock at $4.125 per share and 421,400 warrants at $0.01 per warrant and converted certain notes payable to common stock (See Note 5).
See report of independent accounting firm
| 54 |
9. EMPLOYMENT AND CONSULTING AGREEMENTS
Employment Agreements
The Company entered into a three year employment agreement with its Chief Executive Officer, Leonard Mazur, effective September 12, 2014. Upon expiration, the agreement automatically renews for successive periods of one-year. The agreement requires the Company to pay base compensation plus incentives over the employment term plus severance benefits upon the occurrence of certain events as described in the agreement. Under the agreement, Leonard Mazur was granted options to purchase 220,000 shares of common stock. On March 30, 2016, in connection with the acquisition of LMB, Leonard Mazur resigned as Chief Executive Officer but will continue to serve as Chairman of the Board under the current employment agreement. On October 19, 2017, the Company and Mr. Mazur, entered into an amended employment agreement with a three year term. Under the terms of the amended agreement, the Company is required to pay base compensation plus incentives over the employment term plus severance benefits upon the occurrence of certain events as described in the agreement.
On March 30, 2016, in connection with the acquisition of LMB, the Company entered into a three year employment agreement with Myron Holubiak to serve as Chief Executive Officer. Upon expiration, the agreement automatically renews for successive periods of one-year. The agreement requires the Company to pay base compensation plus incentives over the employment term plus severance benefits upon the occurrence of certain events as described in the agreement.
The Company has employment agreements with certain other employees that require the Company to pay base compensation plus incentives over the employment term plus severance benefits upon the occurrence of certain events as described in the agreement.
Consulting Agreements
Effective September 1, 2014, the Company entered into three consulting agreements. Two of the agreements are for financial consulting services including accounting, preparation of financial statements and filings with the SEC. The third agreement is for financing activities, product development strategies and corporate development. The agreements may be terminated by the Company or the consultant with 90 days written notice.
Consulting expense under the agreements for the years ended September 30, 2017, 2016 and 2015 was $372,000, $460,000 and $348,000, respectively. Consulting expense for the years ended September 30, 2017, 2016 and 2015 includes $48,000, $48,000 and $48,000, respectively, paid to a financial consultant who is a stockholder of the Company. In addition, one financial consulting services agreement provides for the grant of options to purchase 33,333 shares of common stock contingent upon approval by the Board of Directors. The options were granted on June 1, 2015.
10. COMMITMENTS AND CONTINGENCIES
Operating Lease
The Company leases office space from Akrimax, a related party (see Note 8), in Cranford, New Jersey at a monthly rental rate of $2,167 pursuant to an agreement which currently expires on October 31, 2018. Rent expense for the years ended September 30, 2017 and 2016 was $26,000 and $13,002. There was no rent expense for the year ended September 30, 2015. Future minimum rentals for the years ending September 30, 2018 and 2019 are $26,000 and $2,167, respectively.
Legal Proceedings
The Company is not involved in any litigation that we believe could have a material adverse effect on our financial position or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of our executive officers, threatened against or affecting our company or our officers or directors in their capacities as such.
11. INCOME TAXES
There was no provision for federal or state income taxes for the years ended September 30, 2017, 2016 and 2015 due to the Company’s operating losses and a full valuation reserve on deferred tax assets.
See report of independent accounting firm
| 55 |
The income tax benefit differs from the amount of income tax determined by applying the U.S. federal income tax rate to pretax income for the years ended September 30, 2017, 2016 and 2015 due to the following:
| 2017 | 2016 | 2015 | ||||||||||
| Computed “expected” tax benefit | (35.0 | %) | (35.0 | %) | (35.0 | %) | ||||||
| Increase (decrease) in income taxes resulting from: | ||||||||||||
| State taxes, net of federal benefit | (5.2 | %) | (5.2 | %) | (5.2 | %) | ||||||
| Permanent differences | 1.3 | % | 4.2 | % | (4.6 | %) | ||||||
| Increase in the valuation reserve | 38.9 | % | 36.0 | % | 44.8 | % | ||||||
| 0.0 | % | 0.0 | % | 0.0 | % | |||||||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company's deferred tax assets and liabilities are as follows:
| September 30, 2017 | September 30, 2016 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforward | $ | 7,123,000 | $ | 3,801,000 | ||||
| Stock-based compensation | 1,425,000 | 703,000 | ||||||
| Valuation allowance | (8,548,000 | ) | (4,504,000 | ) | ||||
| Deferred tax assets | $ | — | $ | — | ||||
The Company has recorded a valuation allowance against deferred tax assets as the utilization of the net operating loss carryforward and other deferred tax assets is uncertain. During the years ended September 30, 2017, 2016 and 2015, the valuation allowance increased by $4,044,000, $2,989,000 and $1,299,000, respectively. The increase in the valuation allowance during the years ended September 30, 2017, 2016 and 2015 was due to the Company’s net operating loss. At September 30, 2017, the Company has a net operating loss carryforward of approximately $17,719,000 which begins expiring in 2034.
12. SUBSEQUENT EVENTS
Release and Termination of Underwriting Agreement
On November 7, 2017, the Company entered into a release agreement (the “Release”) with Aegis Capital Corp. (“Aegis”). Pursuant to the previously disclosed underwriting agreement dated August 3, 2017 between the Company and Aegis (the “Underwriting Agreement”), the Company granted Aegis a right of first refusal to underwrite all public and private equity and debt offerings for a period of twelve months following completion of the public offering (the “Right of First Refusal”). Under the Release, the Company agreed to pay Aegis $100,000 in cash and to issue an aggregate of 60,000 shares of restricted Company common stock to certain designees of Aegis in exchange for a full release of the Company from any and all obligations related to the Right of First Refusal.
See report of independent accounting firm
| 56 |
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
We maintain disclosure controls and procedures designed to provide reasonable assurance that information required to be disclosed in reports filed under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the specified time periods and accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding disclosure.
Our Chief Executive Officer and Principal Financial Officer (“CEO”), evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) promulgated under the Exchange Act) as of September 30, 2017, the end of our fiscal year. In designing and evaluating disclosure controls and procedures, we recognize that any disclosure controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objective. As of September 30, 2017, based on the evaluation of these disclosure controls and procedures, and in light of the material weaknesses found in our internal controls, the CEO concluded that our disclosure controls and procedures were not effective.
In light of the conclusion that our internal controls over financial reporting were ineffective as of September 30, 2017, we have applied procedures and processes as necessary to ensure the reliability of our financial reporting in regards to this annual report. Accordingly, the Company believes, based on its knowledge, that: (i) this annual report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which they were made, not misleading with respect to the period covered by this report; and (ii) the financial statements, and other financial information included in this annual report, fairly present in all material respects our financial condition, results of operations and cash flows as of and for the periods presented in this annual report.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining effective internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Because of its inherent limitations, internal control over financial reporting is not intended to provide absolute assurance that a misstatement of our financial statements would be prevented or detected. Under the supervision of our CEO, the Company conducted an evaluation of the effectiveness of our internal control over financial reporting as of September 30, 2017 using the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) (2013 Framework).
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. In our assessment of the effectiveness of internal control over financial reporting as of September 30, 2017, we determined that control deficiencies existed that constituted material weaknesses, as described below:
| 1) | lack of documented policies and procedures; |
| 2) | the financial reporting function is carried out by consultants; and |
| 3) | ineffective separation of duties due to limited staff. |
| 57 |
Subject to our ability to obtain additional financing and hire additional employees, the Company expects to be able to design and implement effective internal controls in the future that address these material weaknesses.
Accordingly, we concluded that these material weaknesses resulted in a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis by the Company's internal controls.
As a result of the material weaknesses described above, our CEO concluded that the Company did not maintain effective internal control over financial reporting as of September 30, 2017 based on criteria established in Internal Control—Integrated Framework issued by COSO (2013 Framework).
Changes in Internal Controls over Financial Reporting
There were no changes in our internal controls over financial reporting during the fourth quarter of fiscal 2017 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the Effectiveness of Controls
Our CEO does not expect that our disclosure controls or our internal control over financial reporting will prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. The design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Further, because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Projections of any evaluation of controls effectiveness to future periods are subject to risks. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
None.
| 58 |
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by Item 401 of Regulation S-K regarding directors is included under “Election of Directors” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference. The information required by Item 401 of Regulation S-K regarding executive officers is included under “Executive Officers of Citius” in Item 1 of this Report. The information required by Item 405 of Regulation S-K is included under “Election of Directors — Section 16(a) Beneficial Ownership Reporting Compliance” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference. The information required by Item 406 of Regulation S-K is included under “Corporate Governance — Code of Business Conduct and Ethics” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference. The information required by Items 407(d)(4) and (d)(5) of Regulation S-K regarding the Audit Committee of the Board of Directors is included under “Corporate Governance — Board Committees” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by Items 402, 407(e)(4) and 407(e)(5) of Regulation S-K regarding executive compensation is included under “Election of Directors — Director Compensation,” “Compensation Discussion and Analysis,” “Executive Compensation,” “Election of Directors — Compensation Committee Interlocks and Insider Participation,” and “Compensation Committee Report” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 201(d) of Regulation S-K is included under “Executive Compensation — Equity Compensation Plans” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference. The information required by Item 403 of Regulation S-K is included under “Election of Directors — Stock Holdings of Certain Owners and Management” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Items 404 and 407(a) of Regulation S-K is included under “Election of Directors — Transactions with Related Persons” and “Corporate Governance — Director Independence” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by Item 9(e) of Schedule 14A is included under “Ratification of Independent Registered Public Accounting Firm” in the definitive Proxy Statement for our 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
| 59 |
Item 15. Exhibits, Financial Statement Schedules
All references to registrant’s Forms 8-K, 10-K and 10-Q include reference to File No. 333-170781
| 60 |
| (1) | Incorporated by Reference to the Current Report on form 8-K filed by the Company on September 18, 2014. |
| (2) | Incorporated by Reference to the Company’s Annual Report on Form 10-K filed by the Company on December 29, 2014. |
| (3) | Incorporated by Reference to the Company’s Registration Statement on Form S-1 (Reg. No. 333-206903). |
| (4) | Incorporated by Reference to the Company’s Current Report on Form 8-K filed by the Company on April 5, 2016. |
| (5) | Incorporated by Reference to the Company’s Quarterly Report on Form 10-Q filed by the Company on May 15, 2017. |
| (6) | Incorporated by Reference to the Company’s Current Report on Form 8-K filed by the Company on August 4, 2017. |
| (7) | Incorporated by Reference to the Company’s Current Report on Form 8-K filed by the Company on August 4, 2017. |
| (8) |
* Filed herewith.
| 61 |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| CITIUS PHARMACEUTICALS, INC. | ||
| Date: December 13, 2017 | By: | /s/ Myron Holubiak |
| Myron Holubiak | ||
President and Chief Executive Officer (Principal Executive Officer) | ||
In accordance with the Exchange Act, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Leonard Mazur | Executive Chairman of the Board of Directors | December 13, 2017 | ||
| Leonard Mazur | ||||
| /s/ Myron Holubiak | President and Chief Executive Officer and Director | December 13, 2017 | ||
| Myron Holubiak | ||||
| /s/ Jaime Bartushak | Chief Financial Officer and Chief Accounting | December 13, 2017 | ||
| Jaime Bartushak | Officer (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Suren Dutia | Director | December 13, 2017 | ||
| Suren Dutia | ||||
| /s/ Carol Webb | Director | December 13, 2017 | ||
| Carol Webb | ||||
| /s/ William Kane | Director | December 13, 2017 | ||
| William Kane | ||||
| Director | December __, 2017 | |||
| Howard Safir | ||||
| /s/ Eugene Holuka | Director | December 13, 2017 | ||
| Eugene Holuka |
| 62 |