Attached files
| file | filename |
|---|---|
| EX-10.1 - FORM OF PURCHASE AGREEMENT, DATED AS OF SEPTEMBER 20, 2017 AMONG DELMAR PHARMACE - Kintara Therapeutics, Inc. | f8k0917ex10-1_delmarpharma.htm |
| EX-99.1 - PRESS RELEASE OF DELMAR PHARMACEUTICALS, INC. ISSUED SEPTEMBER 20, 2017 - Kintara Therapeutics, Inc. | f8k0917ex99-1_delmarpharma.htm |
| EX-10.2 - ENGAGEMENT LETTER, DATED SEPTEMBER 17, 2017 BETWEEN DELMAR PHARMACEUTICALS, INC. - Kintara Therapeutics, Inc. | f8k0917ex10-2_delmarpharma.htm |
| EX-5.1 - OPINION OF FENNEMORE CRAIG, P.C - Kintara Therapeutics, Inc. | f8k0917ex5-1_delmarpharma.htm |
| EX-4.1 - FORM OF WARRANT - Kintara Therapeutics, Inc. | f8k0917ex4-1_delmarpharma.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
PURSUANT TO SECTION 13 OR 15(d) OF
THE SECURITIES EXCHANGE ACT OF 1934
Date of Report (Date of earliest event reported): September 20, 2017
DELMAR PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Nevada | 000-54801 | 99-0360497 | ||
| (State
or other jurisdiction of incorporation) |
(Commission
File Number) |
(I.R.S.
Employer Identification Number) |
Suite 720-999 West Broadway
Vancouver, British Columbia
Canada V5Z 1K5
(Address of principal executive offices) (zip code)
(604) 629-5989
(Registrant's telephone number, including area code)
(Former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
☐ Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
☐ Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
☐ Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
☐ Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
| Item 1.01 | Entry into a Material Definitive Agreement. |
On September 20, 2017, DelMar Pharmaceuticals, Inc. (the “Company”) entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain institutional investors for the sale by the Company of shares of the Company’s common stock, par value $0.001 per share (the “Common Stock”) and warrants to purchase shares of Common Stock, in a registered direct offering. The investors in this offering have agreed to purchase, and the Company has agreed to sell, an aggregate of 8,000,000 shares of the Common Stock and warrants to purchase an aggregate of 8,000,000 shares of Common Stock (the “Warrants”), at a purchase price of $1.25 per share and related warrant. Subject to certain ownership limitations, the Warrants will be exercisable commencing on the issuance date at an exercise price equal to $1.25 per whole share of Common Stock, subject to adjustments as provided under the terms of the Warrants. The Warrants are exercisable for five years from the date of issuance. The aggregate gross proceeds for the sale of the shares of Common Stock and Warrants will be approximately $10,000,000. The closing of the sales of the shares of Common Stock and Warrants is expected to occur on or about September 22, 2017 and is subject to customary closing conditions.
H.C. Wainwright & Co, LLC (the “Placement Agent”), acted as the exclusive placement agent in connection with the offering.
The net proceeds to the Company from the transaction, after deducting the placement agent’s fees and expenses (not including the Placement Agent Warrants, as defined below), and the Company’s estimated offering expenses, and excluding the proceeds, if any, from the exercise of the Warrants, are expected to be approximately $9.0 million. The Company intends to use the net proceeds of this offering for its clinical trials and for general corporate purposes, which may include working capital, capital expenditures, research and development and other commercial expenditures. In addition, the Company may use the net proceeds from this offering for acquisitions or investments in businesses, products or technologies that are complementary to its business.
The securities sold in the offering were offered and sold by the Company pursuant to an effective shelf registration statement on Form S-3, which was filed with the Securities and Exchange Commission (the “SEC”) on September 13, 2016 and subsequently declared effective on September 27, 2016 (File No. 333-213601) (the “Registration Statement”), and the base prospectus dated as of September 27, 2016 contained therein. The Company will file a prospectus supplement with the SEC in connection with the sale of the securities.
The representations, warranties and covenants contained in the Purchase Agreement were made solely for the benefit of the parties to the Purchase Agreement. In addition, such representations, warranties and covenants (i) are intended as a way of allocating the risk between the parties to the Purchase Agreement and not as statements of fact, and (ii) may apply standards of materiality in a way that is different from what may be viewed as material by stockholders of, or other investors in, the Company. Accordingly, the Purchase Agreement is included with this filing only to provide investors with information regarding the terms of transaction, and not to provide investors with any other factual information regarding the Company. Stockholders should not rely on the representations, warranties and covenants or any descriptions thereof as characterizations of the actual state of facts or condition of the Company or any of its subsidiaries or affiliates. Moreover, information concerning the subject matter of the representations and warranties may change after the date of the Purchase Agreement, which subsequent information may or may not be fully reflected in public disclosures.
The Company also entered into an exclusive engagement letter (the “Engagement Letter”) with the Placement Agent. The engagement letter expires October 17, 2017. The Company has agreed to pay the Placement Agent an aggregate fee equal to 7% of the aggregate gross proceeds received by the Company from the sale of the shares of Common Stock and Warrants in the transactions plus a management fee equal to 1% of the gross proceeds from the offering. In addition, the Company will also pay the Placement Agent a $10,000 non-accountable expense allowance and reimburse the Placement Agent’s legal expenses up to $70,000. Pursuant to the Engagement Letter, the Company also agreed to issue to the Placement Agent, or its designees, warrants to purchase that number of shares of Common Stock equal to 5% of the aggregate number of shares of Common Stock placed in this offering (but not with respect to any shares of common stock issuable upon exercise of Warrants issued in this offering) at an exercise price of $1.25 per share (the “Placement Agent Warrants”). The Placement Agent Warrants are being issued in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”) and will be restricted from transfer for 180 days pursuant to FINRA Rule 5110(g), but are otherwise identical to the Warrants offered to investors. The Engagement Letter provides for a tail period through January 31, 2018 and right of first refusal period for up to five months, indemnity and other customary provisions for transactions of this nature.
2
The forms of the Purchase Agreement, the Warrant and the Engagement Letter are filed as Exhibits 10.1, 4.1 and 10.2, respectively, to this Current Report on Form 8-K. The foregoing summaries of the terms of these documents are subject to, and qualified in their entirety by, such documents, which are incorporated herein by reference.
A copy of the opinion of Fennemore Craig, P.C. relating to the legality of the securities offered by us is attached as Exhibit 5.1 hereto.
| Item 2.02 | Results of Operations and Financial Condition. |
At June 30, 2017, the Company had cash on hand of approximately $6.6 million (unaudited) and as of September 20, 2017, the Company had cash on hand of approximately $4.3 million (unaudited), not including the net proceeds from this offering.
On September 20, 2017, the Company issued a press release announcing the offering. A copy of the press release is attached hereto as Exhibit 99.1 and is incorporated herein by reference. The information in this Current Report on Form 8-K under Item 2.02, including the information contained in Exhibit 99.1, is being furnished to the Securities and Exchange Commission, and shall not be deemed to be “filed” for the purposes of Section 18 of the Securities Exchange Act of 1934 or otherwise subject to the liabilities of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except as shall be expressly set forth by a specific reference in such filing.
| Item 3.02 | Unregistered Sale of Equity Securities. |
Reference is made to the disclosure set forth in Item 1.01 above as to the Placement Agent Warrants. The Placement Agent Warrants and the shares issuable upon exercise of the Placement Agent Warrants will be issued in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and in reliance on similar exemptions under applicable state laws.
| Item 7.01 | Regulation FD Disclosure. |
On September 20, 2017, the Company issued a press release announcing the offering. A copy of the press release is attached hereto as Exhibit 99.1 and is incorporated herein by reference. The information in this Current Report on Form 8-K under Item 7.01, including the information contained in Exhibit 99.1, is being furnished to the Securities and Exchange Commission, and shall not be deemed to be “filed” for the purposes of Section 18 of the Securities Exchange Act of 1934 or otherwise subject to the liabilities of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except as shall be expressly set forth by a specific reference in such filing.
| Item 8.01 | Other Information. |
Business Update
Background
DelMar Pharmaceuticals, Inc. (the “Company”) is a clinical stage drug development company with a focus on the treatment of cancer. Our mission is to benefit patients and create shareholder value by developing and commercializing anti-cancer therapies for patients whose tumors exhibit features that make them resistant to, or unlikely to respond to, currently available therapies, particularly for orphan cancer indications where patients have failed, or are unlikely to respond to, modern therapy.
3
Our lead product candidate, VAL-083, is a first-in-class DNA-targeting chemotherapeutic that demonstrated activity against a range of tumor types on prior Phase 1 and Phase 2 clinical trials sponsored by the US National Cancer Institute (“NCI”). Our research suggests that VAL-083’s mechanism of action is different than other agents targeting DNA that are widely used in the treatment of cancer such as temozolomide, nitrosoureas, platinum-based drugs, topoisomerase inhibitors and PARP inhibitors. NCI clinical research and data from our own clinical trials suggest that VAL-083 may offer a superior safety profile to these other agents.
We have recently initiated a pivotal randomized Phase 3 clinical trial with VAL-083 for recurrent glioblastoma multiforme (“GBM”). The trial, entitled VAL-083 Phase 3 Study in Temozolomide-Avastin (bevacizumab) Recurrent GBM (“STAR-3”) is intended to enroll approximately 180 patients at approximately 25 centers in the United States. Patients in the trial will have GBM that has recurred following surgery, chemo-radiation with temozolomide, and bevacizumab (Avastin™). The trial will compare the overall survival of these patients following treatment with VAL-083 versus standard-of-care chemotherapy. If successful, the results of this trial will position us to file a new drug application (“NDA”) for the approval of VAL-083 in the United States for the treatment of recurrent GBM. Subject to the availability of capital, we anticipate that the trial will take approximately two years from first enrollment.
We have also initiated two open-label, bio-marker driven Phase 2 trials in MGMT-unmethylated GBM. MGMT is a DNA-repair enzyme that is associated with resistance to temozolomide, the current standard-of-care chemotherapy used in the treatment of GBM. Approximately two out of three GBM patients have MGMT-unmethylated tumors and exhibit a high expression of MGMT, which is correlated with temozolomide treatment failure and poor patient outcomes. Our research demonstrates that VAL-083’s anti-tumor activity is independent of MGMT expression. In these studies, we are using MGMT as a biomarker to identify patients for treatment with VAL-083. If successful, the result of these trials will position VAL-083 for advancement to pivotal clinical trials as a potential replacement for temozolomide in MGMT-unmethylated GBM. Funding for both of these trials is substantially supported through collaborations. We anticipate presenting interim data from these trials at peer reviewed scientific meetings during calendar 2018.
We have received notice of allowance from the FDA for a Phase 1/2, Open-Label, Multicenter, Study of VAL-083 in Patients with Recurrent Platinum Resistant Ovarian Cancer (“REPROVe”). Platinum-based chemotherapy is standard-of-care in the treatment of ovarian cancer. Nearly all ovarian cancer patients eventually become resistant to platinum (“Pt”) -based chemotherapy leading to treatment failure and poor patient outcomes. We have demonstrated that VAL-083 is active against Pt-resistant ovarian cancer in vitro. The Phase 1 portion of the REPROVe trial will enroll approximately 24 patients with Pt-resistant ovarian cancer to evaluate the overall response rate (“ORR”) following treatment with VAL-083. We plan to request a meeting with the FDA following completion of the Phase 1 portion of the REPROVe trial. If successful, data from this trial would lead to a confirmatory Phase 2 study of approximately 60 patients, which if successful, and subject to feedback from FDA may position us to potentially file an application for accelerated approval or to advance to a pivotal Phase 3 trial. Subject to availability of capital, we anticipate completing the Phase 1 portion of the VAL-083 REPROVe trial in approximately 18 months from the initiation of patient recruitment and we will present updates on the progress of the trial at peer reviewed scientific meetings.
In addition to our clinical development activities in the United States, pursuant to our collaboration with Guangxi Wuzhou Pharmaceutical Company, we have obtained certain exclusive commercial rights to VAL-083 in China where it is approved as a chemotherapy for the treatment of chronic myelogenous leukemia (“CML”) and lung cancer. We have entered into a collaboration agreement with the only manufacturer presently licensed by the China Food and Drug Administration (“CFDA”) to produce the product for the China market. This agreement potentially positions us to generate future royalty revenue through product sales or royalties for its approved indications in China while we seek global approval in new indications. To date, we have received no revenue from this collaboration.
We have filed a broad portfolio of patent applications to protect our intellectual property. Our patent applications claim compositions and methods of use of VAL-083 and related compounds, synthetic methods, and quality controls for the manufacturing process of VAL-083. We believe that our portfolio of intellectual property rights provides a defensible market position for the commercialization of VAL-083. In addition, VAL-083 has been granted protection under the Orphan Drug Act by the FDA and the European Medicines Agency (“EMA”) for the treatment of glioma, including GBM. In 2016, the FDA also granted Orphan Drug protection to VAL-083 for the treatment of medulloblastoma and ovarian cancer.
4
Our drug discovery research focuses on identifying well-validated preclinical, clinical and commercial-stage compounds and establishing a scientific rationale for development in cancer indications for patients whose tumors exhibit features that make them resistant to, or unlikely to respond to, currently available therapies. Through our relationship with Valent Technologies, LLC (“Valent”), a company owned by Dr. Dennis Brown, our Chief Scientific Officer, we are able to utilize Valent’s proprietary ChemEstate bioinformatics tools to screen and identify potential candidates. Promising candidates are further researched through our network of consultants, academic centers, and contract research organizations. This approach allows us to identify and advance potential drug candidates without significant investment in “wet lab” infrastructure. Based on this strategy, we acquired the initial VAL-083 intellectual property and prototype drug product from Valent and advanced into Phase 2 and 3 clinical trials and have also identified additional drug candidates that we may have the opportunity to license or acquire in the future.
Our corporate development strategy is to advance our lead candidate into a Phase 3 registration-directed clinical trial and then to consider licensing or acquiring additional product candidates in order to establish a product pipeline and position for long-term sustainability and growth of shareholder value. We believe the experience of our clinical development team will position us to efficiently develop drug candidates that we may acquire, or license in the future.
We plan to seek marketing partnerships to supplement our own commercialization efforts and potentially generate future royalty revenue.
Recent Highlights
| ● | In April 2017, we completed a public offering of common stock and warrants for gross proceeds of approximately $9.0 million. In addition, during the year ended June 30, 2017 we received $545,026 in proceeds from the exercise of warrants. We plan to use these funds to support the initiation of the STAR-3 pivotal clinical trial of VAL-083 in refractory GBM, and for general corporate and research purposes. |
| ● | In July 2017, we initiated patient recruitment for the STAR-3 pivotal Phase 3 clinical trial of VAL-083 in refractory GBM and hope to enroll our first patient in September or October 2017. |
| ● | In September 2017, we initiated patient recruitment for an open label Phase 1 - 2 clinical trial of VAL-083 in newly diagnosed patients MGMT-unmethylated GBM, which is being conducted with funding support through our collaboration with Guangxi Wuzhou Pharmaceutical (Group) Co. Ltd. This trial complements our ongoing open label Phase 2 clinical trial in patients with MGMT-unmethylated GBM whose tumors have recurred following treatment with temozolomide (bevacizumab naïve), which is being conducted in collaboration with the University of Texas MD Anderson Cancer Center. |
| ● | In September 2017, we received notice of allowance from the FDA for our Phase 1-2 VAL-083 REPROVe clinical trial in Pt-resistant ovarian cancer. |
| ● | We presented promising research results supporting the potential of VAL-083 in the treatment of a broad range of cancers for patients whose tumors exhibit features making them resistant or unlikely to currently available therapies. For example: |
| o | We presented data supporting the effectiveness of VAL-083 in the treatment of GBM at the Annual meetings of the American Society for Clinical Oncology (“ASCO”), the American Association of Cancer Research (“AACR”), the World Federation of NeuroOncology Societies (“WFNOS”), the European Association for NeuroOncology and the Society for NeuroOncology (“SNO”); |
5
| o | We presented data supporting the effectiveness of VAL-083 in the treatment of lung cancer at the AACR Annual Meeting, the 17th World Congress on Lung Cancer and the AACR New Horizons in Cancer Research Conference; |
| o | We presented data supporting the activity of VAL-083 in treatment-resistant medulloblastoma both as a single agent and in combination with topoisomerase inhibitors at the SNO Pediatric Oncology Symposium and at the AACR Advances in Pediatric Research: From Mechanisms and Models to Treatment and Survivorship Conference; and |
| o | We presented data supporting the effectiveness of VAL-083 against chemotherapy-resistant ovarian cancers at the 11th Biennial Ovarian Cancer Research Symposium. |
| ● | We continued to strengthen and expand our network of research collaborations with leading academic institutions including the announcement of a major sponsored research agreement with Duke University to evaluate VAL-083 as a front-line treatment for newly diagnosed patients with GBM. |
| ● | We continued to strengthen our intellectual property portfolio. DelMar now holds eight issued US patents and eight issued patents outside of the US. We have fourteen patent families in various stages of prosecution, and over 100 patent filings in total. |
| ● | We strengthened our Board of Directors and corporate governance with the addition of Saiid Zarrabian and the appointment of Dr. Erich Mohr as independent chairman. |
VAL-083
Our product candidate, VAL-083, is a “first-in-class” small molecule chemotherapeutic, which means that the molecular structure of VAL-083 is not an analogue or derivative of a product approved, or in development for the treatment of cancer. VAL-083 is a DNA-targeting agent that was originally discovered in the 1960’s. It was assessed in more than 40 NCI-sponsored Phase 1 and Phase 2 clinical trials as a treatment against various cancers including lung, brain, cervical, ovarian tumors and leukemia. Published preclinical and clinical data suggest that VAL-083 may be active against a range of tumor types. VAL-083 is approved as a cancer chemotherapeutic in China for the treatment of CML and lung cancer. VAL-083 has not been approved for any indications outside of China.
Our research demonstrates that the mechanism of action of VAL-083 is distinct from other DNA-targeting agents used in the treatment of cancer. VAL-083 exhibits its anti-cancer activity by forming DNA-cross links leading to DNA double strand breaks, cell-cycle arrest and cancer cell death, DNA-targeting agents are among the most widely used treatments for cancer. They exhibit anti-cancer effects by binding to DNA and interfering with normal processes within the cancer cell which prevents the cell from making the proteins needed to grow and survive. We have presented research at peer-reviewed scientific meetings demonstrating that VAL-083 is active in patient-derived tumor cell lines and cancer stem cells that are resistant to other chemotherapies. These data, combined with clinical activity demonstrated against various cancers in prior NCI-sponsored clinical trials gives us confidence that VAL-083 may offer an opportunity as a new treatment option for patients whose tumors are resistant to currently available chemotherapies.
We are currently studying VAL-083 in clinical trials for the treatment of GBM, the most common and aggressive form of brain cancer. We have also recently received notice of allowance from the FDA for an IND to initiate clinical trials with VAL-083 in the treatment of ovarian cancer. Upon obtaining regulatory approval, we intend to commercialize VAL-083 for the treatment of orphan cancer indications such as refractory GBM. We also plan to seek collaborative development and commercialization partnerships to accelerate and expand the development of VAL-083 in newly diagnosed GBM and other non-orphan cancer indications.
The FDA Office of Orphan Products Development (“OOPD”) has granted orphan drug designations to VAL-083 for the treatment of glioma, ovarian cancer and medulloblastoma. VAL-083 has also been granted an orphan drug designation for in the treatment of glioma in Europe. Orphan diseases are defined in the United States under the Rare Disease Act of 2002 as “any disease or condition that affects fewer than 200,000 persons in the United States”. The Orphan Drug Act of 1983 is a federal law that provides financial and other incentives including a seven-year period of market exclusivity in the United States to encourage the development of new treatments for orphan diseases.
6
VAL-083 Mechanism of Action and the Opportunity in the Treatment of Cancer
Chemotherapy forms the basis of treatment in nearly all cancers. We believe that VAL-083 may be effective in treating cancer patients whose tumors exhibit features that cause resistance to currently available chemotherapy or that have failed, or become resistant to, other chemotherapies.
Our research suggests that VAL-083 attacks cancer cells via a unique mechanism of action which is distinct from other chemotherapies used in the treatment of cancer. Our data indicate that VAL-083 forms a crosslink at the N7 position of guanine on the DNA of cancer cells. Our data also indicate that this crosslink forms rapidly and is not easily repaired by the cancer cell resulting in cell-cycle arrest and lethal double-strand DNA breaks in cancer cells. VAL-083 readily crosses the blood brain barrier where it maintains a long half-life in comparison to the plasma. Published preclinical and clinical research demonstrate that VAL-083 is absorbed more readily in tumor cells versus normal cells.
Based on published research and our own data, the cytotoxic functional groups and the mechanism of action of VAL-083 are understood to be functionally different from alkylating agents commonly used in the treatment of cancer. VAL-083 has previously demonstrated activity in cell-lines that are resistant to other types of chemotherapy. No evidence of cross-resistance has been reported in published clinical studies.
Our data also demonstrate that VAL-083’s mechanism is distinct from current standard-of-care chemotherapy and is able to overcome drug resistance against a range of cancers in vitro. For example, VAL-083 is active against MGMT-unmethylated GBM cells which are resistant to treatment with temozolomide and nitrosoureas. VAL-083 also retains a high level of activity in p53 mutated non-small cell lung cancer (“NSCLC”), ovarian cancer and medulloblastoma cell lines that are resistant to platinum-based chemotherapy.
Importantly, clinical activity against each of the tumors mentioned above was established in prior NCI-sponsored Phase 2 clinical trials. We believe that these historical clinical data and our own research support the development of VAL-083 as a potential new treatment for multiple cancers.
The main dose-limiting toxicity (“DLT”) related to the administration of VAL-083 in previous NCI-sponsored clinical studies and our own clinical trials is myelosuppression. Myelosuppression is the decrease in cells responsible for providing immunity, carrying oxygen, and those responsible for normal blood clotting. Myelosuppression is a common side effect of chemotherapy. There is no evidence of lung, liver or kidney toxicity even with prolonged treatment by VAL-083. Commercial data from the Chinese market where the drug has been approved for more than 15 years supports the safety findings of the NCI studies.
Modern medicine allows for better management of myelosuppressive side effects. We believe this offers the potential opportunity to improve upon the drug’s already established efficacy profile by substantially increasing the dose of VAL-083 that can be safely administered to cancer patients.
Gliomas and Glioblastoma Multiforme (“GBM”)
Worldwide, there are an estimated 240,000 new cases of brain and central nervous system (“CNS”) tumors each year. Gliomas are a type of CNS tumor that arises from glial cells in the brain or spine. Glial cells are the cells surrounding nerves. Their primary function is to provide support and protection for neurons in the CNS.
GBM is the most common and the most lethal form of glioma. According to the World Health Organization, GBM occurs with an incidence of 3.17 per 100,000 person-years. Approximately 18,000 new cases of GBM are expected to be diagnosed in the United States and 26,000 in Europe during 2017.
GBM progresses quickly and patients’ conditions deteriorate rapidly progressing to death in less than two years for most patients. Common symptoms include headaches, seizures, nausea, weakness, paralysis and personality or cognitive changes such as loss of speech or difficulty in thinking clearly. The median survival in newly diagnosed patients with best available treatments is less than 15 months.
7
Standard treatment following diagnosis includes surgical resection to remove as much of the tumor as possible (“debulking”) followed by radiotherapy with concomitant and adjuvant chemotherapy with Temodar® (temozolomide “TMZ”). Nearly all patients diagnosed with GBM will relapse following first-line treatment, with a 1-year survival rate of approximately 25% following failure of front-line therapy, and with an average 5-year survival rate of less than 3%.
Avastin® (bevacizumab, an anti-VEGF antibody) is approved as a single agent for patients with recurrent GBM following prior therapy as an alternative to corticosteroids to relieve disease symptoms in the US, Canada, Australia and Japan. Avastin® carries a “black-box warning” related to severe, sometimes fatal, side effects such as gastrointestinal perforations, wound healing complications and hemorrhage. There are no data demonstrating an improvement in disease-related symptoms or increased survival in refractory GBM with Avastin®.
TMZ and the nitrosoureas, including carmustine, lomustine, and nimustine, are alkylating agents that readily cross the blood-brain-barrier and are used in the treatment of CNS cancers, including GBM. Alkylating agents are among the oldest type of cancer chemotherapies in use today. Alkylating agents bind to DNA to cause damage to cancer cells. Their anti-tumor mechanism is via alkylation of DNA resulting in base-pair mismatch or strand-mediated crosslinks between base pairs. The DNA damage caused by alkylating agents mimics naturally occurring errors, resulting in apoptosis and tumor cell death.
The primary anti-cancer mechanism of TMZ and the nitrosoureas is to attack the tumor’s DNA via alkylation of the O6-position of the DNA base residue, guanine. TMZ treatment causes DNA damage mainly by methylation at the O6-position of guanine resulting in guanine-thymine base pair mismatches during replication. Nitrosoureas mediate their cytotoxic effect by methylation at the O6-position of guanine which produces a cross-link to cytosine residues resulting in double-strand DNA breaks during mitosis.
A majority of GBM patients’ tumors are resistant to TMZ or nitrosourea therapy due to high expression of a naturally occurring enzyme called O6-DNA methylguanine methyl-transferase (“MGMT”) which repairs O6-guanine lesions. MGMT repair in turn inhibits the activity of TMZ and nitrosoureas and allows a patients’ GBM tumor to continue to grow in spite of treatment.
Consistent with the importance of its repair activity, high expression of MGMT is strongly correlated with poor patient outcomes. Several clinical studies have established that MGMT is an important prognostic biomarker of response to TMZ and patient survival.
8
Probability of GBM Patient Survival Correlated to Expression of MGMT Enzyme
(Unmethylated promoter = High MGMT Expression and Significantly Shorter Survival)

VAL-083 in GBM
VAL-083 is first-in-class DNA targeting agent which readily crosses the blood-brain-barrier. Data from prior NCI-sponsored clinical trials with VAL-083 demonstrate activity against GBM and other central nervous system tumors. In general, historical NCI-sponsored trials demonstrate tumor regression in brain cancer was achieved in 40% of patients treated and stabilization was achieved in an additional 20% to 30% of brain tumor patients following treatment with VAL-083.
VAL-083 demonstrated statistically significant improvement in the median survival of high grade glioma brain tumors, including GBM when combined with radiation versus radiation alone (p value = <0.05) with results similar, or superior to, other chemotherapies approved for the treatment of GBM.
A Summary of Published Data adapted from Separate Sources Comparing the Efficacy of VAL-083
and Other Therapies in the Treatment of GBM
| Comparative Therapy | ||||||
| Chemotherapy | Radiation (XRT) Alone | Radiation + Chemotherapy | Median Survival Benefit vs. XRT alone | |||
VAL-083 (Eagan 1979) | 8.4 months | 16.8 months | 8.4 months | |||
Temozolomide (Temodar®) (Stupp 2005) | 12.1 months | 14.6 months | 2.5 months | |||
| Lomustine (CCNU) (Walker 1976) | 11.8 months | 13 months | 1.2 months | |||
| Carmustine (BCNU) (Reagan 1976) | 10 months | 12.5 months | 2.5 months | |||
| Semustine (ACNU) (Takakura 1986) | 12 months | 14 months | 2.0 months | |||
Our research demonstrates that VAL-083’s unique cytotoxic mechanism forms DNA cross-links at the N7 position of guanine and retains cytotoxic activity independent of MGMT expression in vitro. This mechanism is distinct from that of temozolomide and nitrosoureas, which are DNA-targeting agents commonly used in the treatment of GBM. Of particular importance is in the treatment of GBM resistance to temozolomide, or nitrosoureas, due to activity of the repair enzyme MGMT, which results in chemoresistance in many GBM patients.
9
We have presented data demonstrating that VAL-083 is active independent of MGMT resistance in laboratory studies. VAL-083 has more potent activity against brain tumor cells in comparison to TMZ and overcomes resistance associated with MGMT suggesting the potential to surpass the current standard-of-care in the treatment of GBM.
A
Summary of Our Data Demonstrating that VAL-083’s Anti-Tumor Mechanism is Distinct from, and can
Overcome, MGMT-Related Chemoresistance in the Treatment of GBM

In addition, historical NCI clinical trial data and our own research support the activity of VAL-083 as a potentiator of radiotherapy. Radiotherapy in combination with temozolomide is the current standard of care in the treatment of GBM. Our research demonstrates that temozolomide and radiotherapy are ineffective against GBM cells exhibiting a high expression of MGMT, whereas VAL-083 potentiates the tumor-killing effect of radiation in these cells. Furthermore, the combination of VAL-083 and radiation has been demonstrated to be active against GBM cancer stem cells (CSCs) in vitro. CSCs are often resistant to chemotherapy and form the basis for tumor recurrence and metastasis. GBM CSCs display strong resistance to TMZ, even where MGMT expression is low. However, our data demonstrates that GBM CSCs are susceptible to VAL-083 independent of MGMT expression.

We believe that VAL-083’s more potent activity against brain tumor cells in comparison to TMZ, the ability to overcome MGMT-mediated resistance, and activity against GBM cancer stem cells suggests the potential of VAL-083 to surpass the current standard-of-care in the treatment of GBM.
10
Based on our research demonstrating a novel anti-tumor mechanism and the historical clinical data demonstrating activity against GBM, we have initiated clinical trials in refractory GBM and in MGMT-unmethylated GBM. Our clinical trials in the United States are being conducted under an investigational new drug (“IND”) application with the FDA. If successful, we believe data from these trials will support a potential paradigm shift in the treatment of GBM where VAL-083 could become the chemotherapy of choice in the treatment of the majority of GBM patients.
Clinical Trials of VAL-083 in Refractory GBM
Phase 3: VAL-083 STAR-3 GBM Trial
We recently initiated VAL-083 STAR-3 GBM trial is an adaptive, randomized, controlled pivotal Phase 3 clinical trial in patients with refractory GBM. The trial is designed to assess the efficacy and safety of VAL-083 versus salvage therapy in GBM patients whose disease has progressed following prior treatment with temozolomide and bevacizumab. There is currently no approved standard-of-care therapy for these patients.
A total of up to 180 eligible patients will be randomized at approximately 25 centers in the United States to receive either the investigational drug (VAL-083) or "investigator's choice salvage therapy" in a 2:1 fashion. Up to 120 eligible patients will be randomized to receive intravenous VAL-083 at 40 mg/m2 on days 1, 2, and 3 of a 21-day treatment cycle, for up to 12 21-day treatment cycles or until they fulfill one of the criteria for study discontinuation.
Up to 60 patients will be randomized to "investigator's choice" control, limited to temozolomide, lomustine, or carboplatin, until they fulfill one of the criteria for study discontinuation.
The primary endpoint of the STAR-3 trial is overall survival. The statistical design between the two arms of the study is 90% power, and includes an interim analysis at 50% of events for futility and superiority with O’Brien-Fleming boundary and non-binding, gamma (-5) futility boundary. We have based our assumptions for outcomes for the STAR-3 control arm on published literature. We are also undertaking a review of recent patient data to validate our control arm assumptions. In the event that this analysis suggests that a more conservative assumption is required, we may consider revising the trial design to maintain 90% power for the primary endpoint.
The study is estimated to complete in approximately two years from initiation. A detailed description of the STAR-3 trial can be found at clinicaltrials.gov, Identifier Number: NCT03149575.
Based on our current working capital, we do not have sufficient funding to complete the STAR-3 trial. Until additional funds are available, we plan to initiate the first 8-10 sites and only enroll a subset of patients for whom our cash resources will allow for completion of treatment and follow-up. We believe this strategy will best allow us to maintain timelines for trial completion, NDA submission and FDA approval while seeking further funding through the capital markets, grant funding or strategic partnerships.
Phase 1 – 2 Clinical Trial Overview and Summary of Results
Forty-eight GBM patients were enrolled in our Phase 1/2 clinical trial at five centers: the Mayo Clinic in Rochester, Minnesota; the Brain Tumor Center at University of California, San Francisco; the Sarah Cannon Cancer Research Center in Nashville, Tennessee, Denver, Colorado; and the SCRI affiliate site at the Florida Cancer Specialist Research Institute in Sarasota, Florida.
The Phase 1/2 trial was an open-label, single arm dose-escalation study designed to evaluate the safety, tolerability, pharmacokinetics and anti-cancer activity of VAL-083 in patients with refractory GBM. The trial enrolled GBM patients whose disease has progressed following prior treatment with temozolomide and bevacizumab, unless either or both were contra-indicated.
The overall goal of our Phase 1/2 clinical trial was to determine a modernized dosing regimen for advancement into a pivotal registration-directed Phase 3 clinical trial.
Patients received VAL-083 on days 1,2 and 3 on a 21-day treatment cycle. The Phase 1 portion of the study involved dose escalation cohorts until a maximum tolerated dose (“MTD”) was established at 40mg/m2. A further 14-patient, Phase 2 expansion was then enrolled at the MTD to gather further safety data at our chosen therapeutic dose and to further explore the outcomes in this patient population.
11
In May 2016, we held an end of Phase 2 meeting with the FDA where design of a Phase 3, registration-directed clinical program for VAL-083 in refractory GBM was discussed. Based on the input we received from the FDA, the agency confirmed that it would consider the totality of data available, including data obtained from DelMar's other planned clinical trials in related GBM populations, when assessing the New Drug Application (“NDA”). The FDA also noted that DelMar can rely on prior NCI studies and historical literature to support nonclinical data required for an NDA filing and that DelMar will have the option to file under a 505(b)(2) strategy which allows a sponsor to rely on already established safety and efficacy data in support of an NDA.
We reported updated results of our Phase 1/2 clinical trial at the 2016 ASCO annual meeting. In summary, these data are as follows:
Tumor Response and Outcomes
GBM patients in our Phase 1/2 clinical trial were not re-resected prior to treatment with VAL-083 and therefore had a growing recurrent GBM tumor at the time of enrollment. Patients were monitored for tumor response by MRI.
Consistent with un-resected refractory GBM, median progression free survival (“PFS”) was short at 1.2 months (range: 0.2 – 20.1 months). Five GBM patients treated with VAL-083 were reported to have stable disease as their best response following treatment; the remainder reported progressive disease.
Disease progression is typical in a refractory GBM population with non-resected tumors. However, we believe that slowed progression may provide meaningful clinical benefit in this patient population through prolonged overall survival and improved quality of life. According to published literature, GBM patients failing bevacizumab have a poor prognosis with expected survival under five months.
Ad-hoc subgroup analysis of the Phase 1 dose-escalation data indicated a dose response trend. Increased survival was observed following initiation of treatment in a high dose (30 and 40mg/m2, n=9) sub-group vs. a low dose (≤5mg/m2, n=6) sub-group with median survival of >9 months vs. 4.4 months for the high and low dose groups, respectively.
Observed Survival Based on Phase 1 Sub-Group Analysis

An additional 14 patients were enrolled in an expansion cohort at the MTD (40mg/m2). Analysis of patients receiving an assumed therapeutic dose of VAL-083 (≥20mg/m2) demonstrated median survival of 8.35 months following bevacizumab failure. At the time of the analysis, more than half of patients receiving an assumed therapeutic dose survived more than six months following bevacizumab failure; more than 40% survived for nine months or are currently alive and more than 20% have survived for twelve months or more.
12
ASCO 2016: VAL-083 compared to published literature
| Reference | Post Avastin Salvage Therapy | Median Survival following Bevacizumab Failure | ||
| Shih (2016) | VAL-083 | 8.35 months | ||
| Rahman (2014) | nitrosourea | 4.3 months | ||
| Mikkelson (2011) | TMZ + irinotecan | 4.5 months | ||
| Lu (2011) | dasatinib | 2.6 months | ||
| Reardon (2011) | etoposide | 4.7 months | ||
| Reardon (2011) | TMZ | 2.9 months | ||
| Iwomoto (2009) | various | 5.1 months |
While recognizing these data are representative of a relatively small, non-controlled Phase 1/2 clinical trial, we believe these outcomes support the potential of VAL-083 to offer meaningful clinical benefit to GBM patients who have failed bevacizumab, compared to currently available therapy.
Safety and Tolerability
In the Phase 1 dose escalation regimen, no serious adverse events (“SAE”) related to VAL-083 were encountered at doses up to 40 mg/m2/day.
Increasing frequency of, and higher grade, hematologic toxicities were observed at doses above 40 mg/m2/day. Consistent with the published literature, the observed dose limiting toxicity for VAL-083 is primarily thrombocytopenia (low platelets). Observed platelet nadir occurred at approximately day 18, and recovery was rapid and spontaneous following treatment.
Based on Phase 1 observations, fourteen additional patients were enrolled in a Phase 2 expansion cohort at 40mg/m2, which was established at the MTD. Consistent with Phase 1, the dose of VAL-083 of 40 mg/m2 on days 1, 2 and 3 of a 21-day cycle was generally well tolerated in Phase 2. At this dose, one subject previously treated with CCNU, a nitrosourea agent, reported severe (Grade 4) thrombocytopenia. As a result of this observation, the protocol inclusion criterion for platelet count was increased from 100,000/μL to 150,000/μL for patients receiving prior nitrosoureas within 12 weeks preceding enrollment. No other dose limiting toxicities were observed in Phase 2.
VAL-083 Safety Observations From Phase 1/2 Clinical Trial
| Hematologic parameter and CTCAE grade | dose | ≤30 mg/m2 | 40 mg/m2 | 45 mg/m2 | 50 mg/m2 | |||||||||||||||||||||||||||||||
| n = | 20 | 17 | 4 | 7 |
| |||||||||||||||||||||||||||||||
| Anemia | ≤G2 | 11 | 55 | % | 2 | 12 | % | 2 | 50 | % | 6 | 86 | % | |||||||||||||||||||||||
| G3 | 2 | 10 | % | - | 0 | % | - | 0 | % | - | 0 | % | ||||||||||||||||||||||||
| G4 | - | 0 | % | - | 0 | % | - | 0 | % | - | 0 | % | ||||||||||||||||||||||||
| Leukopenia | ≤G2 | 5 | 25 | % | 2 | 12 | % | - | 0 | % | 5 | 71 | % | |||||||||||||||||||||||
| G3 | 1 | 5 | % | - | 0 | % | - | 0 | % | 3 | 43 | % | ||||||||||||||||||||||||
| G4 | - | 0 | % | - | 0 | % | 2 | 50 | % | - | 0 | % | ||||||||||||||||||||||||
| Neutropenia | ≤G2 | 4 | 20 | % | - | 0 | % | - | 0 | % | - | 0 | % | |||||||||||||||||||||||
| G3 | - | 0 | % | - | 0 | % | - | 0 | % | 3 | 43 | % | ||||||||||||||||||||||||
| G4 | - | 0 | % | - | 0 | % | 2 | 50 | % | 1 | 14 | % | ||||||||||||||||||||||||
| Thrombocytopenia | ≤G2 | 9 | 45 | % | 3 | 18 | % | - | 0 | % | 3 | 43 | % | |||||||||||||||||||||||
| G3 | - | 0 | % | - | 0 | % | 1 | 25 | % | 3 | 43 | % | ||||||||||||||||||||||||
| G4 | - | 0 | % | 1 | 6 | % | 2 | 50 | % | 1 | 14 | % | ||||||||||||||||||||||||
| DLT Observed | nil | 1 | 2 | 2 | ||||||||||||||||||||||||||||||||
13
Doses Achieved
We confirmed that we achieved doses of VAL-083 that are substantially higher than were utilized in the original published NCI-sponsored clinical trials. A summary in comparison to the NCI’s historical regimen is as follows:
Dosing Regimen & Study | Single Dose | Acute Regimen (single cycle) | Comparative Cumulative Dose (@ 35 days) | Dose Intensity (dose per week) | ||||||||
NCI GBM historical regimen (Eagan etal) daily x 5 q 5wks (cycle = 35 days) | 25 mg/m2 | x5 days = | 125 mg/m2 | 125 mg/m2 | 25mg/m2/wk | |||||||
| DelMar VAL-083 optimized regimen daily x 3 q 3wks (cycle = 21 days) | 40 mg/m2 | x3 days = | 120 mg/m2 | 240 mg/m 2 | 40mg/m2/wk | |||||||
Daily x 5 q 5wks refers to a dosing regimen of once per day for five consecutive days every five weeks (35-day cycle); while daily x 3 q 3wks refers to a dosing regimen of once per day for three consecutive days every three weeks (21-day cycle).
Our optimized dosing regimen increases the amount of VAL-083 delivered to the CNS by 60% over historical regimens without increased toxicity. Thus, the DelMar regimen achieves both a higher maximum concentration and higher overall exposure, which we believe may increase the likelihood of successful treatment outcomes in glioblastoma and other brain tumors.
Pharmacokinetics
Pharmacokinetic (“PK”) analyses showed dose-dependent linear systemic exposure with a short (1-2h) plasma terminal half-life; average Cmax at 40 mg/m2/day was 781 ng/mL (5.3µM). The observed PK profile is comparable to published literature. Prior NCI-sponsored studies demonstrated that VAL-083 readily crosses the blood brain barrier and has a long (>20 hour) half-life in the central nervous system (“CNS”).
We believe that this PK profile is optimal for the treatment of brain tumors: A long CNS half-life is expected to maximize exposure of the drug in the brain increasing the likelihood of successful treatment outcomes, while a short plasma half-life is desirable to minimize systemic side effects.
14
Observed pharmacokinetics from VAL-083 Phase 1 clinical trial dose vs. AUC
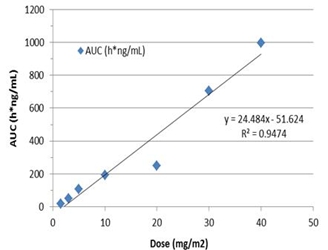
Based on observed and previously published pharmacokinetics, DelMar believes that therapeutic doses equal to, or above, 20 mg/m2 daily on days 1, 2 and 3 of a 21-day cycle should deliver sufficient levels of VAL-083 to brain tumors to achieve a therapeutic benefit.
MGMT & IDH1
High expression of MGMT and wild-type form of the enzyme isocitrate dehydrogenase (“IDH1”) have been previously shown to be diagnostic markers that correlate with resistance to currently available chemotherapies (e.g. temozolomide or nitrosourea) in the treatment of GBM and poor patient outcomes. Measurement of these biomarkers has become routine in clinical practice.
Notably, we have previously demonstrated that VAL-083’s anti-tumor mechanism is active independent from the MGMT status in vitro. While the science behind their importance in the disease pathway and their ultimate predictive value are still being explored, we believe we will ultimately be able to use such biomarkers in a prognostic fashion to select the patients most likely to respond to treatment as we expand the clinical development of VAL-083.
MGMT expression was characterized by PCR and/or ELISA for nineteen GBM patients enrolled in our Phase 1/2 study. IDH1 status was reported in eleven patients; both MGMT and IDH1 status were reported in four patients.
| Biomarker | Observation in Phase 1 /2 clinical trial | ||
| High MGMT (n=19) | 84% | ||
| IDH-WT (n=11) | 90% | ||
Notably, all patients whose samples were tested for both markers were MGMT-unmethylated by PCR and wild-type IDH1, a phenotype that is correlated with particularly poor prognosis.
Clinical Trials of VAL-083 in MGMT-unmethylated GBM
MGMT methylation status has been previously shown to be a diagnostic marker that correlates with patient outcomes and survival in GBM. GBM patients whose tumors are characterized as MGMT-unmethylated exhibit high expression of the DNA-repair enzyme MGMT. High MGMT levels have correlated resistance to currently available chemotherapies (e.g. temozolomide or nitrosourea) and significantly reduced survival. The development of new therapies for MGMT-unmethylated GBM is a significant unmet medical need.
15
Approximately two-thirds of newly diagnosed GBM patients have tumors assessed as MGMT-unmethylated. This represents a potential treatment population of approximately 12,000 patients in the United States and 18,000 patients in Europe annually.
Notably, we have previously demonstrated that VAL-083’s anti-tumor mechanism is active independent from the MGMT status in vitro. This suggests the potential of VAL-083 as a replacement for currently available chemotherapies in MGMT-unmethylated GBM.
Measurement of MGMT methylation status has become routine in clinical practice. We can therefore utilize MGMT-methylation status to identify newly diagnosed GBM patients who are least likely to respond to temozolomide and instead treat them with VAL-083.
We have initiated two Phase 2 clinical trials to explore the potential of VAL-083 in the treatment of MGMT-unmethylated GBM. Expenditures related to our ongoing clinical trials in MGMT-unmethylated GBM are substantially supported through collaborations, which allows us to implement these protocols with minimal impact to our own working capital balance.
Phase 2 Trial in Newly Diagnosed MGMT-unmethylated GBM
In September 2017, we initiated a single arm, biomarker driven open-label Phase 2 study in newly diagnosed MGMT-unmethylated GBM patients at Sun Yat-sen University Cancer Center in Guangzhou, China. The trial is being conducted in the context of our 2012 collaboration agreement with Guangxi Wuzhou Pharmaceutical (Group) Co. Ltd. Under the terms of this agreement, Guangxi Wuzhou Pharmaceutical (Group) Co. Ltd. is responsible for funding VAL-083 clinical trials that we conduct in China.
In this study, VAL-083 will be combined with radiotherapy as a potential replacement for temozolomide in patients with high expression of MGMT. The main goal of the trial will be to confirm the safety of DelMar’s optimized dosing regimen in combination with radiotherapy and to investigate outcomes of the combination of VAL-083 and radiotherapy in MGMT-unmethylated GBM patients.
Up to 30 newly diagnosed MGMT-unmethylated GBM patients will be enrolled in this trial. The primary efficacy endpoint is the determination of tumor response in patients measured by progression free survival. Assessments of safety and tolerability will be used to support further clinical development of VAL-083 in combination with radiotherapy. Pharmacokinetic assessments of VAL-083 in plasma and cerebral spinal fluid (“CSF”) will be used to correlate drug exposure in the central nervous system with patient outcomes.
Outcomes following treatment with VAL-083 will be compared to MGMT-unmethylated patients in the RTOG0525 trial. We anticipate obtaining safety data from the trial within nine months and top-line outcomes data within 18 months.
Data from the trial will be used to establish a dosing regimen and trial design for advanced registration-directed clinical trials with VAL-083 in newly diagnosed MGMT-unmethylated GBM. If successful, data from the trial will strongly position VAL-083 as a potential replacement for current standard-of-care chemotherapy in the treatment of GBM.
Phase 2 Study in Recurrent MGMT-unmethylated GBM in Collaboration with University of Texas MD Anderson Cancer Center
In January 2017, we initiated a biomarker driven, open-label single-arm Phase 2 study in collaboration with the University of Texas with MD Anderson Cancer Center. This trial will enroll up to 48 MGMT-unmethylated GBM patients whose tumors have recurred following treatment with temozolomide. These patients will not have been treated with prior bevacizumab.
The primary endpoint of the trial is overall survival. Outcomes following treatment with VAL-083 will be compared to the outcome of MGMT-unmethylated patients who had been treated with lomustine (CCNU) following temozolomide failure in the recently published EORTC20601 trial.
16
Safety data from this trial will become part of the overall safety dossier to support future filings with the FDA and other regulatory agencies. A positive outcome will establish a strong position for VAL-083 in the treatment of MGMT-unmethylated GBM.
We anticipate presenting interim data from this trial at peer reviewed meetings during calendar 2018.
Ovarian Cancer
Ovarian cancer is the fifth most common cancer in women and is the leading cause of death among women diagnosed with gynecological malignancies. In 2016, approximately 22,300 women in the US were diagnosed with ovarian cancer and 14,300 died from their disease. If detected early, ovarian cancer can often be cured with surgery. When detected early, up to 90% of patients are likely to survive for >5 years.
Unfortunately, the initial symptoms of ovarian cancer such as abdominal bloating, indigestion, pelvic pain or nausea are often attributed to symptoms caused by less a serious situation. Therefore, in most cases, ovarian cancer isn’t diagnosed until it has progressed to an advanced stage when it is no longer possible to surgically remove all tumor tissue.
Without treatment, ovarian cancer spreads within the pelvic region and metastasizes to distant sites such as the lungs, liver, spleen and, rarely, the brain. When diagnosed at an advanced stage the 5-year survival rate is less than 40%. Women with ovarian cancer receive chemotherapy following surgery to treat residual disease. Pt-based chemotherapy is the standard-of-care in the treatment of advanced ovarian cancer.
Ovarian cancer patients whose tumors are sensitive to Pt-based chemotherapy have the most favorable outcome. Recently, the introduction of PARP inhibitors in the treatment of ovarian cancer patients with Pt-sensitive disease demonstrated significant improvements in overall survival.
Unfortunately, the development of resistance to Pt-based agents is nearly inevitable, leading to disease recurrence and increased mortality. Ultimately, most women with advanced ovarian cancer develop recurrent disease with progressively shorter disease-free intervals. Those whose tumors recur within 6 months of Pt-based therapy are considered Pt-resistant/refractory and have a very poor prognosis.
Currently, there are no high-efficacy therapeutic options for Pt-resistant ovarian tumors, leaving these cancer patients with a very poor prognosis. The response rate to second line therapy for Pt-resistant ovarian cancer patients is in the 10-15% range and overall survival is approximately 12-months. The development of new chemotherapies and targeted agents to overcome Pt resistance in ovarian cancer is a significant unmet medical need.
Treatment Resistance to Pt-based Chemotherapy in Ovarian Cancer
Pt-based chemotherapy is employed in the treatment of nearly 50% of all cancer patients and forms the mainstay as part of the front-line treatment regimen against a range of solid tumors including testicular, ovarian, cervical, bladder, colorectal, head-and-neck, and lung cancer. Pt-based chemotherapy is used to treat nearly all advanced-stage ovarian cancer patients.
Pt-based chemotherapies function by causing extensive damage to a cancer cell’s DNA. When a cell is ready to divide, cellular mechanisms assess potential DNA damage, and if severe damage is identified, the cell will halt the division process and may even be directed to self-destruct. Thus, chemotherapies that target DNA are intended to be lethal to cancer cells, or at least prevent them from dividing to inhibit a tumor’s growth.
Unfortunately, cancer cells are adept at overcoming DNA damage or employing mechanisms to repair damaged DNA. These factors limit the damage that DNA-damaging drugs can do or allow cancer cells to become resistant to chemotherapy. One of the most common obstacles to DNA-damaging chemotherapy is mutations to a gene called p53. Cellular processes governed by the p53 gene are critical in assessing DNA damage and determining if a cell should cease from dividing or self-destruct. When p53 does not function properly, cancer cells continue to divide despite the treatment with DNA-damaging chemotherapy, making these drugs ineffective and leading to treatment resistance. This occurs in nearly all cases of the most difficult ovarian cancer to treat – high grade serous ovarian cancer (HGSOC) – which accounts for up to 70% of ovarian cancer cases and approximately 90% of ovarian cancer deaths. P53 mutations are associated with resistance to Pt-based chemotherapy, which leads to treatment failure and increased mortality. Solving this problem is a major goal in the development of new treatments for ovarian cancer.
17
VAL-083 in Ovarian Cancer
VAL-083 is a first-in-class, DNA-targeting agent that demonstrated activity in prior NCI-sponsored clinical trials. Activity against ovarian epithelial adenocarcinoma (OEA) and squamous cell carcinoma of the cervix (SCC) was reported in multiple studies. Importantly, NCI-researchers recommended VAL-083 for further advanced studies in the treatment of ovarian cancer.
We have presented data demonstrating that VAL-083’s distinct mechanism of action allows activity in tumors that are resistant to other therapies. We have shown that cytotoxicity of VAL-083 against ovarian cancer is independent of sensitivity to cisplatin or p53 status in vitro. We have demonstrated that VAL-083 is active in Pt-resistant ovarian cells harboring a range of p53-mutations. Similar results were observed comparing activity of VAL-083, cisplatin and oxaliplatin in Pt-sensitive and -resistant non-small cell lung cancer (NSCLC) cell lines.

Our research has demonstrated that VAL-083 not only overcomes Pt resistance, but the combination of VAL-083 with Pt-based chemotherapy displays synergy in multiple models in vitro and in vivo. This further suggests a distinct mechanism of action and potential use as part of a VAL-083/Pt-combination therapy.

The combination of VAL-083 with either cisplatin (A) or oxaliplatin (B) in the human H460 (WT p53) NSCLC model demonstrated significant superadditivity (p≤0.05) and/or synergism (CI<1) for both combinations. This cytotoxic effect of VAL-083 in combination with either platinum drug was observed also in A549 (WT p53) and H1975 (mutant p53) NSCLC cells, independently of p53 status (not shown). Data, where applicable, are shown as mean ± SE; N=7.
18
While Pt-based chemotherapy is the standard treatment for ovarian cancer, PARP inhibitors have recently provided a new treatment option for a subset of patients with platinum-sensitive recurrent ovarian cancer. VAL-083 also demonstrates synergistic activity with the PARP inhibitor olaparib in vitro, suggesting VAL-083 may have utility in the treatment of ovarian cancer in combination with PARP inhibitors.
We believe that these data demonstrate the potential of VAL-083 to treat platinum-resistant ovarian cancers as a single-agent against platinum-resistant tumors, combination with platinum-based chemotherapeutic regimens or in combination with PARP inhibitors.
In April 2016, the FDA granted orphan drug designation for the use of VAL-083 in the treatment of ovarian cancer.
We plan to work with our advisors to develop a strategy to advance VAL-083 into clinical trials for the treatment of ovarian cancer, either as a single-agent or in combination with other approved agents.
VAL-083 REPROVe Ovarian Cancer Trial
We have also recently received notice of allowance from the FDA of our IND for a Phase 1/2, Open-Label, Multicenter, Study of VAL-083 in Patients with Recurrent Platinum Resistant Ovarian Cancer (REPROVe).
The Phase 1 portion of the trial will enroll approximately 24 patients with Pt-resistant ovarian cancer to evaluate the response to treatment with VAL-083.
Ovarian cancer patients enrolled in the trial will have been previously treated with at least two lines of Pt-based chemotherapy and up to two other cytotoxic regimens, whose cancer has recurred within 6 months of prior Pt-based chemotherapy.
The primary efficacy of the trial will be overall response rate (“ORR”) based on Response Evaluation Criteria In Solid Tumors (RECIST) criteria. RECIST is a set of published rules that define when tumors in cancer patients improve ("respond"), stay the same ("stabilize"), or worsen ("progress") during treatment.
We plan to request a meeting with FDA following completion of the Phase 1 portion of the REPROVe trial. If successful, data from this trial would lead to a confirmatory Phase 2 study of approximately 60 patients, which if successful, and subject to feedback from the FDA may position us to potentially file an application for accelerated approval or to advance to a pivotal Phase 3 trial.
We anticipate completing the Phase 1 portion of the trial in approximately 18 months from the initiation of patient recruitment and presenting updates on the progress of this trial at peer reviewed meetings.
Based on our current working capital, we do not have sufficient funding to initiate patient enrollment in the REPROVe trial. We have identified sites and will complete certain contracting and site-initiation activities, but will not initiate patient enrollment until we can appropriately fund the treatment and follow-up of patients enrolled in the trial. We believe this strategy will best allow us to maintain timelines for trial completion, NDA submission and FDA approval while seeking further funding through the capital markets, grant funding or strategic partnerships.
Other Indications for VAL-083
VAL-083 in Lung Cancer
Lung cancer is a leading cause of cancer death around the world and effective treatment for lung cancer remains a significant global unmet need despite advances in therapy. In general, prognosis for lung cancer patients remains poor, with a 5-year survival rate of less than 14% among males and less than 18% among females in most countries.
Incidence of lung cancer in the United States is approximately 59 per 100,000 with the majority (52:100,000) being NSCLC. World Health Organization projects that the incidence of lung cancer in China is expected to exceed one million (1,000,000) new cases per year by 2025. Globally, the market for lung cancer treatment may exceed $24 billion by 2033 according to a report published by Evaluate Pharma.
19
The activity of VAL-083 against solid tumors, including lung cancer, has been established in both preclinical and human clinical trials conducted by the NCI. VAL-083 is approved for the treatment of lung cancer in China; however, sales of VAL-083 in China have been limited by a lack of modern data, poor distribution, and preference for targeted therapies such as tyrosine kinase inhibitors (“TKIs”) in the modern era.
Non-small cell lung cancer (“NSCLC”) is the most common type of lung cancer. There are three common forms of NSCLC: adenocarcinomas are often found in an outer area of the lung; squamous cell carcinomas are usually found in the center of the lung next to an air tube (bronchus); and large cell carcinomas, which can occur in any part of the lung and tend to grow and spread faster than adenocarcinoma. NSCLC accounts for 85% of all lung cancer cases in the United States and approximately 90% of lung cancer cases diagnosed in China.
Recently approved immunotherapy drugs such as nivolumab (Opdivo®) and pembrolizumab (Keytruda®) have shown benefit in a subset of patients with recurrent NSCLC whose tumors exhibit immunogenic targets such as PD-L1. Many NSCLC patients’ tumors do not express immunotherapy targets at sufficient levels to trigger an immunotherapy treatment response and the development of resistance to immunotherapy has begun to emerge.
DelMar has developed new nonclinical data to support the utility of VAL-083 in the modern treatment of lung cancer. We have announced results of preclinical studies designed to evaluate the activity of VAL-083 in models of drug-resistant NSCLC in comparison to cisplatin. In an established murine xenograft model of NSCLC, the activity of VAL-083 was compared to standard platinum-based therapy with cisplatin against human NSCLC cell lines A549 (TKI-sensitive) and H1975 (TKI-resistant). In the study, VAL-083 demonstrated superior efficacy and safety in the treatment of TKI-susceptible (A549) tumors and in TKI-resistant (H1975) tumors.
Based on these data, we believe VAL-083’s unique mechanism of action could make it a valuable drug of choice in NSCLC patients who are or become resistant to platinum-based chemotherapy and TKI therapy. In addition, VAL-083 readily crosses the blood brain barrier suggesting that it may be possible for VAL-083 to treat patients whose lung cancer has spread to the brain.
We have developed a clinical trial protocol to explore the activity of VAL-083 in recurrent lung cancer. If successful, we believe data from this trial would support the potential to establish global partnerships and collaborations with larger pharmaceutical companies who have the resources and commercial infrastructure to effectively develop and commercialize VAL-083 as a treatment for NSCLC on a worldwide basis.
It is our current intention to conduct this trial with leading investigators in China under the terms of our collaboration with Guangxi Wuzhou Pharmaceutical Group Co. Ltd. (“Guangxi Wuzhou Pharmaceuticals”), which would allow us to enhance the potential value of VAL-083 without significantly increasing our own planned cash expenditures.
We have determined, in consultation with Guangxi Wuzhou Pharmaceuticals, that initiation of a lung cancer trial should be delayed until our planned China-based MGMT-unmethylated GBM trial had received regulatory approval for initiation. In July 2017, the Human Genetic Resources Administration of China (“HGRAC”) approved the GBM trial, so it is now our intention to work with Guangxi Wuzhou Pharmaceuticals to determine the appropriate strategy and timing for initiation of VAL-083 in clinical trials in lung cancer.
Central Nervous System Metastases of Solid Tumors
In June 2013, we split our Phase 1/2 clinical trial protocol into two separate studies: one focusing solely on refractory GBM and the other focusing on secondary brain cancers caused by other tumors that have spread to the brain. The successful management of systemic tumors by modern targeted therapies has led to increased incidence of mortality due to CNS metastases of lung cancer and other solid tumors.
Based on historical clinical activity and our own research, we believe that VAL-083 may be suitable for the treatment of patients with CNS metastases who currently have limited treatment options. Subject to the availability of financial and operating resources, we plan to develop a separate protocol for the continued exploration of VAL-083 in patients with secondary brain cancer caused by a solid tumor spreading to the brain.
20
Pediatric Brain Tumors
Tumors of the brain and spine make up approximately 20 percent of all childhood cancers and they are the second most common form of childhood cancer after leukemia.
The activity of VAL-083 against childhood and adolescent brain tumors has been established in both preclinical and human clinical trials conducted by the NCI. We have presented data indicating that VAL-083 offers potential therapeutic alternatives for the treatment of pediatric brain tumors including SHH-p53 mutated medulloblastoma. In March 2016, the FDA granted orphan drug designation for the use of VAL-083 in the treatment of medulloblastoma. Subject to the availability of resources, we intend to collaborate with leading academic researchers for the continued exploration of VAL-083 as a potential treatment of childhood brain tumors.
Additional Indications for VAL-083
In historical studies sponsored by the NCI in the United States, VAL-083 exhibited clinical activity against a range of tumor types including central nervous system tumors, solid tumors and hematologic malignancies. We have established new nonclinical data supporting the activity of VAL-083 in different types of cancer that are resistant to modern targeted therapies and we believe that the unique cytotoxic mechanism of VAL-083 may provide benefit to patients in a range of indications. We intend to continue to research these opportunities, and if appropriate, expand our clinical development efforts to include additional indications.
Other Product Opportunities
Through our relationship with Valent Technologies, LLC (“Valent”), a company owned by Dr. Dennis Brown, our Chief Scientific Officer, we have identified additional drug candidates that we may have the opportunity to license or acquire in the future.
VAL-083 Target Markets
DNA-targeting agents such as alkylating agents or platinum-based chemotherapy form the mainstay of chemotherapy treatments used in the treatment of cancers. Global sales of platinum-based chemotherapies reached nearly $2.5 billion in 2011 and declined to $600 million following the expiry of key patents. Alkylating agents such as temozolomide, bendamustine, nitrosoureas, and cyclophosphamide generated more than $1.3 billion in sales in 2016 after reaching a peak of $1.7 billion in 2014 (evaluate pharma).

Fig X: Peak sales of selected DNA-targeting Agents
Our lead product candidate, VAL-083, is a first-in-class DNA targeting agent with a novel mechanism of action. VAL-083’s anti-cancer activity was established in a range of tumor types in prior NCI-sponsored clinical trials. Based on this novel mechanism, we have demonstrated that the anti-cancer activity is maintained against tumor cells that are resistant to other DNA-targeting agents. We believe this positions VAL-083 as a potential chemotherapy-of-choice for patients whose tumors are resistant to current standard-of-care chemotherapy in orphan and major cancer indications.
21
Our ongoing research and development activities are focused on indications where VAL-083 demonstrated promising activity in prior NCI-sponsored trials and where our research suggests an opportunity to address significant unmet medical needs due to the failure of existing treatments.
| VAL-083 target markets | 2022 Estimated Global Sales | |
| Glioblastoma multiforme (GBM) | $1.5B | |
| Ovarian Cancer | $4.6 B | |
| Non-small cell lung cancer (NSCLC) | $24.8 B | |
| Source: Evaluate Pharma |
Glioblastoma Multiforme
GBM is the most common and the most lethal form of glioma. According to the World Health Organization, GBM occurs with an incidence of 3.17 per 100,000 person-years. Approximately 18,000 new cases of GBM are expected to be diagnosed in the United States and 26,000 in Europe during 2017.
Newly diagnosed patients suffering from GBM are initially treated through invasive brain surgery, although disease progression following surgical resection is nearly 100%. Temozolomide (Temodar®) in combination with radiation is the front-line therapy for GBM following surgery. Global revenues of branded Temodar reached $1.1 billion in 2009. Following patent expiry in 2013, global revenue for generic temozolomide exceeded $400 million in 2014 even though most patients fail to gain long-term therapeutic benefits. Approximately 60% of GBM patients treated with Temodar® experience tumor progression within one year.
Bevacizumab (Avastin®) has been approved for the treatment of GBM in patients failing Temodar®. In clinical studies, only about 20% of patients failing Temodar® respond to Avastin® therapy and no improvement in median survival was reported. In spite of these low efficacy results, Avastin revenues exceeded $600 million in 2014.
The market for refractory (Avastin-failed) GBM is limited to those jurisdictions where Avastin is approved for the treatment of GBM. The United States, Canada, Australia, Japan and Switzerland represent the major markets where Avastin is used in the treatment of GBM. Based on our estimates, we believe that VAL-083 could generate sales for the treatment of refractory GBM in the $100’s of millions annually.
The market for MGMT-unmethylated GBM represents approximately two-thirds of all GBM patients worldwide. Based on our estimates, we believe that sales of VAL-083 for the treatment of MGMT-unmethylated GBM could exceed $1 billion annually.
Ovarian Cancer
According to Evaluate Pharma, the annual market for ovarian cancer therapies is projected to exceed $4.6 billion in 2022. The American Cancer Society estimates that approximately 22,000 women will receive a new diagnosis of ovarian cancer and approximately 14,000 women will die from ovarian cancer in the United States each year. Ovarian cancer ranks fifth in cancer deaths among women, accounting for more deaths than any other cancer of the female reproductive system.
The potential of VAL-083 in the treatment of ovarian cancer has been established in prior NCI-sponsored clinical trials and by our recent research. The FDA has granted orphan drug status to VAL-083 as a potential treatment for ovarian cancer and we have recently received notice of allowance for our IND to initiate a Phase 1-2 clinical trial to investigate the safety and effectiveness of VAL-083 in patients with recurrent platinum resistant ovarian cancer (VAL-083 REPROVe trial).
Ovarian cancers are commonly treated with a platinum-based chemotherapy regimen. Initial tumor response rates are relatively high. However, the development of resistance to Pt-based chemotherapy in ovarian cancer patients is nearly inevitable. Our research suggests that VAL-083 may offer a potential treatment option for ovarian cancer patients who are resistant to platinum-based chemotherapy and as a potential combination therapy with other agents. We believe the profile of VAL-083 offers the potential to capture meaningful market share in the multi-billion ovarian cancer market.
22
Lung Cancer
According to Evaluate Pharma, the annual market for lung cancer therapies is projected to reach nearly $25 billion in 2022. Lung cancer is the most common cancer in the world with 1.8 million cases in 2012, representing 13% of all cancers according to a report published by the World Cancer Research Fund International. Lung cancer has a higher mortality rate than the next top three cancers combined and it is responsible for 1.6 million deaths annually, representing 19% of all cancer deaths. NSCLC represents approximately 90% of newly diagnosed lung cancers.
The potential of VAL-083 in the treatment of NSLSC has been established in both human clinical trials conducted by the NCI and by the drug’s commercial approval in China. We believe the profile of VAL-083 offers the potential to capture meaningful market share in the multi-billion NSCLC market.
VAL-083 Manufacturing
VAL-083 is a small-molecule chemotherapeutic. Chemical synthesis of the active pharmaceutical ingredient (“API”) was initially established by the NCI. We have made improvements to this process and have obtained patents on these improvements. The current manufacturing process involves fewer than five synthetic steps.
VAL-083 drug product is a lyophilized (freeze-dried) formulation that is reconstituted for intravenous injection. We anticipate that overall cost of goods for an eventual commercial product will be similar to other injectable, small-molecule pharmaceuticals.
For our Phase 3 clinical trial, we have engaged third-party contract manufacturers with the capabilities to establish the processes, procedures and quality systems necessary to meet U.S., Canadian, E.U. and other international manufacturing requirements in accordance with Good Manufacturing Practice (“cGMP”) regulations.
Supply of VAL-083 for our clinical trials to date has been provided through a collaboration with Guangxi Wuzhou Pharmaceutical (Group) Co. Ltd. Guangxi Wuzhou Pharma as a manufacturer has established a commercial-scale manufacturing process based on the North American process originally developed for the NCI that has been licensed by the Chinese FDA (“CFDA”) for commercial supply of VAL-083 in China. DelMar has developed and patented certain intellectual property related to quality controls that are used in the release of VAL-083 for our clinical trials in the United States. This intellectual property is also required for product release under CFDA guidelines and we have granted access to our intellectual property for this purpose.
Research & Development Collaborations
Guangxi Wuzhou Pharmaceutical Company
Pursuant to a memorandum of understanding and collaboration agreement, dated October 25, 2012, we have established a strategic collaboration with Guangxi Wuzhou Pharmaceutical Company (“Guangxi Wuzhou Pharmaceuticals”), a subsidiary of publicly traded Guangxi Wuzhou Zhongheng Group Co., Ltd. (SHG: 600252) (the “Guangxi Agreement”). VAL-083 is approved for the treatment of chronic myelogenous leukemia (“CML”) and lung cancer in China and Guangxi Wuzhou Pharmaceuticals is the only manufacturer licensed by the CFDA to produce the product for the China market. Through the Guangxi Agreement, we have been provided with exclusive access to drug product at the production price for our VAL-083 clinical trials in the United States and we have also secured certain commercial rights in China.
Pursuant to the Guangxi Agreement, we granted to Guangxi Wuzhou Pharmaceuticals a royalty-free license to certain of our intellectual property, as it relates to quality control and drug production methods for VAL-083, and we agreed that Guangxi Wuzhou Pharmaceuticals will be our exclusive supplier of VAL-083 for clinical trials and commercial sales, subject to Guangxi Wuzhou Pharmaceuticals obtaining and maintaining cGMP certification by the FDA, EMA or other applicable regulatory agencies, and Guangxi Wuzhou Pharmaceuticals being able to meet volumes ordered by us. In accordance with this agreement, we have contracted with established third-party suppliers for our Phase 3 clinical trials. We will continue to work with Guangxi Wuzhou Pharmaceuticals to achieve FDA compliance in order to potentially have them as our future supplier for global sales of VAL-083.
23
This Guangxi Agreement also provides us with certain exclusive commercial rights related to drug supply. Specifically, the Guangxi Agreement establishes an exclusive supply relationship between us and Guangxi Wuzhou Pharmaceuticals for the Chinese market and all markets outside China. Guangxi Wuzhou Pharmaceuticals agreed that it may not sell VAL-083 for markets outside of China to any other purchaser other than us, provided that, during the first three years following regulatory clearance for marketing of VAL-083 in a particular country or region, we meet proposed sales volumes set by Guangxi Wuzhou Pharmaceuticals for the country or region. In addition, Guangxi Wuzhou Pharmaceuticals granted us a pre-emptive right in China (subject to our acceptance of proposed sales volume and prices) to purchase VAL-083 produced by Guangxi Wuzhou Pharmaceuticals.
Our collaboration with Guangxi Wuzhou Pharmaceuticals positions us with the potential to generate future revenue through product sales or royalties for its approved indications in China while we seek global approval in new indications.
Under the terms of the Guangxi Agreement, Guangxi Wuzhou Pharmaceuticals will provide funding support for clinical trials conducted in China and we are responsible for development and commercialization. We anticipate establishing sales channels in China through a third-party marketing partner in collaboration with Guangxi Wuzhou Pharmaceuticals in order to obtain sales or royalty revenue from China.
The term of the Guangxi Agreement (except as it relates to the exclusive rights in the China market) is indefinite, subject to termination upon written agreement of all parties, or if either party breaches any material term and fails to remedy such breach within 30 days of receipt of notice of the breach, or if any action to be taken thereunder is not agreed to by both parties, provided that such matter is referred to the chief executive officer of both parties, and they are unable to resolve such matter within 90 days. No payments have been made to date under the Guangxi Agreement.
Accurexa Collaboration
We have entered into a collaboration agreement with Accurexa, Inc. (“Accurexa”). Accurexa is a biotechnology company focused on developing novel neurological therapies to be directly delivered to specific regions of the brain. Under the terms of the agreement, we and Accurexa will undertake collaborative research activities for the purpose of evaluating formulations of VAL-083 and one or more of temozolomide and BCNU for local delivery. Under the terms of the agreement, we will supply VAL-083 and Accurexa will conduct experiments related to the development and validation of a novel formulation for the combined local delivery of VAL-083 and temozolomide. We have been granted an exclusive right to license or acquire any product candidates and related intellectual property that results from research conducted under the agreement for further development and commercialization on an exclusive worldwide basis, or other terms that may be agreed upon between the parties. The initial financial commitment by us is not significant.
Duke University Collaboration
In April 2017, we entered into a three-year collaboration with Duke University to evaluate VAL-083 as a front-line treatment for newly diagnosed patients with GBM. Under the terms of the collaboration, we will fund a series of preclinical studies to be conducted by Duke University’s Glioblastoma Drug Discovery Group to identify molecular characteristics of GBM tumors that are more likely to respond to VAL-083, and not the standard of care, temozolomide, as a front-line treatment or through combination therapies.
Patents and Proprietary Rights
Our success will depend in part on our ability to protect our existing product candidate and the products we acquire or license by obtaining and maintaining a strong proprietary position. To develop and maintain our position, we intend to continue relying upon patent protection, orphan drug status, Hatch-Waxman exclusivity, trade secrets, know-how, continuing technological innovations and licensing opportunities.
24
We have filed patent applications claiming the use of, and improvements related to VAL-083. Our patent filings also include proposed treatment regimens, improvements to the manufacturing process, formulation and composition of the active pharmaceutical ingredient, and finished dosage forms of VAL-083. We are prosecuting our patent applications in the United States and other jurisdictions which we deem important for the potential commercial success of VAL-083.
Our patents and patent applications can be summarized in fourteen series as follows:
| ● | Series
I is generally directed to synthesis of VAL-083. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent No. 8,563,758 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||||
| United States Patent No. 8,921,585 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||||
| United States Patent No. 9,085,544 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||||
| United States Patent No. 9,630,938 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol | 2031 | ||||
| PCT Patent Application Serial No. PCT/US2011/048032 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol. National phase applications pending in various countries. | 2031 | ||||
| PCT Patent Application Serial No. PCT/US2011/048032 | Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol: Patents granted in the following countries: Australia, China, Israel, Japan, Mexico, Singapore. | 2031 |
| ● | Series II is generally directed to use of VAL-083 to treat a range of diseases and conditions, including but not limited to malignancies. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent No. 9,066,918 | Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol | 2031 | ||||
| United States Patent Application Serial No. 14/753,911 | Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol | |||||
| PCT Patent Application Serial No. PCT/US2011/048031 | Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol. National phase applications pending in various countries. | 2031 |
25
| ● | Series III is generally directed to analytical methods for VAL-083. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent No. 9,759,698 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol | |||||
| United States Patent Application Serial No. 14/380,924 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol | |||||
| United States Patent No. 9,029,164 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol | 2033 | ||||
| PCT Patent Application Serial No. PCT/IB2013/000793 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol. National phase applications pending in various countries. | 2033 | ||||
| PCT Patent Application Serial No. PCT/IB2013/000793 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol granted in Australia | 2033 | ||||
| Patent or Patent Application No. | Title | Expiry | ||||
| PCT Patent Application Serial No. PCT/US2014/066087 | Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol. | 2034 |
| ● | Series IV is generally directed to the use of VAL-083 to treat GBM or medulloblastoma. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 14/373,552 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma | |||||
| United States Patent No. 9,687,466 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma | 2033 | ||||
| United States Patent Application Serial No. 15/617,756 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma | |||||
| PCT Patent Application Serial No. PCT/US2013/022505 | Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma. National phase applications pending in various countries. | 2033 |
26
| ● | Series V is generally directed to the veterinary use of VAL-083. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 14/400,271 | Veterinary Use Of Dianhydrogalactitol, Diacetyldianhydrogalactitol, And Dibromodulcitol To Treat Malignancies | |||||
| ● | Series VI is generally directed to the use of VAL-083 to treat tyrosine-kinase-inhibitor-resistant malignancies. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 14/409,909 | Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof | |||||
| PCT Patent Application Serial No. PCT/US2013/047320 | Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof. National phase applications pending in various countries. | 2033 |
| ● | Series VII is generally directed to the use of VAL-083 to treat recurrent malignant glioma and progressive secondary brain tumor. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 14/682,226 | Use Of Dianhydrogalactitol And Analogs And Derivatives Thereof To Treat Recurrent Malignant Glioma Or Progressive Secondary Brain Tumor | |||||
| PCT Application Serial No. PCT/US2014/040461 | Use Of Dianhydrogalactitol And Analogs And Derivatives Thereof To Treat Recurrent Malignant Glioma Or Progressive Secondary Brain Tumor. National phase applications pending in various countries. | 2034 |
27
| ● | Series VIII is generally directed to the use of VAL-083 to treat non-small-cell lung cancer. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 14/710,240 | Use of Dianhydrogalactitol and Analogs or Derivatives Thereof in Combination With Platinum-Containing Antineoplastic Agents to Treat Non Small-Cell Carcinoma of the Lung and Brain Metastases | |||||
| PCT Patent Application Serial No. PCT/US2015/024462 | Use of Dianhydrogalactitol and Analogs or Derivatives Thereof to Treat Non-Small Cell Carcinoma of the Lung and Ovarian Cancer. National phase applications pending in various countries. | 2035 | ||||
| PCT Patent Application Serial No. PCT/US2016/032120 | Combination of Analogs or Derivatives of Dianhydrogalactitol with Platinum-Containing Antineoplastic Agents to Treat Cancer. | 2035 |
| ● | Series IX is generally directed to the use of VAL-083 and radiation to treat NSCLC and GBM. |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 15/525,933 | Dianhydrogalactitol Together with Radiation to Treat Non-Small-Cell Carcinoma of the Lung and Glioblastoma Multiforme. | |||||
| PCT Patent Application Serial No. PCT/US2015/059814 | Dianhydrogalactitol Together with Radiation to Treat Non-Small-Cell Carcinoma of the Lung and Glioblastoma Multiforme | 2035 |
| ● | Series X is generally directed to the use of VAL-083 in NSCLC and ovarian carcinoma by induction of DNA damage and stalling of cell cycle: |
| Patent or Patent Application No. | Title | Expiry | ||||
| PCT Patent Application Serial No. PCT/IB2016/001436 | Use of Dianhydrogalactitol or Derivatives and Analogs Thereof for Treatment of Non-Small-Cell Lung Carcinoma, Glioblastoma, and Ovarian Carcinoma by Induction of DNA Damage and Stalling of Cell Cycle. | 2036 |
| ● | Series XI is generally directed to the use of VAL-083 in the treatment of pediatric CNS malignancies: |
| Patent or Patent Application No. | Title | Expiry | ||||
| United States Patent Application Serial No. 15/624,200 | Use of Dianhydrogalactitol or Derivatives and Analogs Thereof for Treatment of Pediatric Central Nervous System Malignancies. | |||||
| PCT Patent Application Serial No. PCT/US2016/058661 | Use of Dianhydrogalactitol or Derivatives and Analogs Thereof for Treatment of Pediatric Central Nervous System Malignancies. | 2036 |
| ● | Series XII is generally directed to the analysis and resolution of VAL-083 preparations: |
| Patent or Patent Application No. | Title | Expiry | ||||
| PCT Patent Application Serial No. PCT/US2016/063362 | Methods for analysis and Resolution of Preparations of Dianhydrogalactitol and Derivatives and Analogs Thereof. | 2036 |
| ● | Series XIII-XIV –Provisional U.S. patent applications |
| Patent or Patent Application No. | Title | Expiry | ||||
| Two provisional U.S. patent applications have been filed | ||||||
28
One of the inventors listed in our Series IX applications is an employee of the University of California, San Francisco. If a patent issues from a patent application in this series with a claim that the University of California employee conceived of, in whole or in part, then the Regents of the University of California will share ownership of any such patent with us. Our research agreements with the University of California address this issue by providing the Company with an exclusive option, for a limited period of time, to negotiate a royalty-bearing exclusive license for commercialization of the invention covered by that patent.
In addition to patent protection, we may also seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and Canada, and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for a different clinical indication.
In February 2012, the FDA granted orphan drug status to VAL-083 for the treatment of glioma. In January 2013, the EMA also granted orphan drug protection to VAL-083 for the treatment of glioma. In the spring of 2016, the FDA Office of Orphan Products Development granted orphan drug designations to VAL-083 for the treatment of ovarian cancer and medulloblastoma.
In addition to our patents and orphan drug protection, we intend to rely on the Hatch-Waxman Amendments for five years of data exclusivity for VAL-083.Under the Hatch-Waxman Amendments, newly approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. These amendments provide five-year data exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active ingredient. The Hatch-Waxman Amendments prohibit the submission of an abbreviated new drug application, also known as an ANDA or generic drug application, during the five-year exclusive period if no patent is listed. If there is a patent listed and the ANDA applicant certifies that the NDA holder’s listed patent for the product is invalid or will not be infringed, the ANDA can be submitted four years after NDA approval. Protection under the Hatch-Waxman Amendments will not prevent the filing or approval of another full NDA; however, the applicant would be required to conduct its own pre-clinical studies and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Amendments also provide three years of data exclusivity for the approval of NDAs with new clinical trials for previously approved drugs and supplemental NDAs, for example, for new indications, dosages or strengths of an existing drug, if new clinical investigations were conducted by or on behalf of the sponsor and were essential to the approval of the application. This three-year exclusivity covers only the new changes associated with the supplemental NDA and does not prohibit the FDA from approving ANDAs for drugs containing the original active ingredient.
We also rely on trade secret protection for our confidential and proprietary information. We believe that the substantial costs and resources required to develop technological innovations, such as the manufacturing processes associated with VAL-083, will help us to protect the competitive advantage of our product candidate.
The protection of intellectual property rights in China (where our clinical product candidate, VAL-083, is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the CFDA to produce the product for the China market, and where VAL-03 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate revenue from VAL-083 in China.
29
Our policy is to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and consultants, the agreements provide that all inventions conceived by the individual shall be our exclusive property.
Government Regulation and Product Approval
Regulation by governmental authorities in the U.S. and other countries is a significant factor, affecting the cost and time of our research and product development activities, and will be a significant factor in the manufacture and marketing of any approved products. Our product candidates will require regulatory approval by governmental agencies prior to commercialization. In particular, our products are subject to rigorous preclinical and clinical testing and other approval requirements by the FDA and similar regulatory authorities in other countries. Various statutes and regulations also govern or influence the manufacturing, safety, reporting, labeling, transport and storage, record keeping and marketing of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, the necessary regulatory approvals could harm our business.
The regulatory requirements relating to the testing, manufacturing and marketing of our products may change from time to time and this may impact our ability to conduct clinical trials and the ability of independent investigators to conduct their own research with support from us.
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, in Canada, Health Canada, and, in the E.U., the EMA. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act in the U.S. and numerous directives, regulations, local laws and guidelines in Canada and the E.U. govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years and involves the expenditure of substantial resources.
Regulatory approval will be required in all the major markets in which we seek to develop our products. At a minimum, approval requires the generation and evaluation of data relating to the quality, safety, and efficacy of an investigational product for its proposed use. The specific types of data required and the regulations relating to this data will differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In general, new chemical entities are tested in animals until adequate evidence of safety is established to support the proposed clinical study protocol designs. Clinical trials for new products are typically conducted in three sequential phases that may overlap. In Phase 1, the initial introduction of the pharmaceutical into either healthy human volunteers or patients with the disease (20 to 50 subjects), the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase 2 involves studies in a limited patient population (50 to 200 patients) to determine the initial efficacy of the pharmaceutical for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a compound shows preliminary evidence of some effectiveness and is found to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to more fully evaluate clinical outcomes in a larger patient population in adequate and well-controlled studies designed to yield statistically sufficient clinical data to demonstrate efficacy and safety.
30
In the U.S., specific preclinical data, manufacturing and chemical data, as described above, need to be submitted to the FDA as part of an IND application, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase 1 studies in human volunteers may commence only after the application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the E.U. Currently, in each member state of the E.U., following successful completion of Phase 1 studies, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase 2 studies. The regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed study, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase 1 studies, further submissions to regulatory authorities are necessary in relation to Phase 2 and III studies to update the existing IND. Authorities may require additional data before allowing the studies to commence and could demand that the studies be discontinued at any time if there are significant safety issues. In addition to the regulatory review, studies involving human subjects must be approved by an independent body. The exact composition and responsibilities of this body will differ from country to country. In the U.S., for example, each study will be conducted under the auspices of an independent institutional review board (IRB) at each institution at which the study is conducted. The IRB considers among other things, the design of the study, ethical factors, the privacy of protected health information as defined under the Health Insurance Portability and Accountability Act, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules to protect subjects’ rights and welfare apply in each member state of the E.U. where one or more independent ethics committees, which typically operate similarly to an IRB, will review the ethics of conducting the proposed research. Other regulatory authorities around the rest of the world have slightly differing requirements involving both the execution of clinical trials and the import/export of pharmaceutical products. It is our responsibility to ensure we conduct our business in accordance with the regulations of each relevant territory.
In order to gain marketing approval, we must submit a dossier to the relevant authority for review, which is known in the U.S. as a new drug application (NDA) and in the E.U. as a marketing authorization application (MAA). The format is usually specific and laid out by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as the nonclinical and clinical data. Once the submitted NDA is accepted for filing by the FDA, it undertakes the review process that takes 10 months, unless an expedited priority review is granted which takes six months to complete. Approval can take several months to several years, if multiple 10-month review cycles are needed before final approval is obtained, if at all.
The approval process can be affected by a number of factors. The NDA may be approvable requiring additional preclinical, manufacturing data or clinical trials which may be requested at the end of the 10-month NDA review cycle, thereby delaying approval until additional data are submitted and may involve substantial unbudgeted costs.
In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. The regulatory authorities usually will conduct an inspection of relevant manufacturing facilities, and review manufacturing procedures, operating systems and personnel qualifications. Further inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies may be necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs in the indications on which we are focusing our efforts. These include accelerated approval under Subpart H of the agency’s NDA approval regulations, fast track drug development procedures and priority review. At this time, we have not determined whether any of these approval procedures will apply to our current drug candidate.
By leveraging existing preclinical and clinical safety and efficacy data, we seek to build upon an existing knowledge and data to accelerate our research. In addition, through our focus on end-stage population which has no current treatment options, regulatory approval for commercialization may sometimes be achieved in an accelerated manner. Accelerated approval by the FDA in this category may be granted on objective response rates and duration of responses rather than demonstration of survival benefit. As a result, trials of drugs to treat end-stage refractory cancer indications have historically involved fewer patients and generally have been faster to complete than trials of drugs for other indications. We are aware that the FDA and other similar agencies are regularly reviewing the use of objective endpoints for commercial approval and that policy changes may impact the size of trials required for approval, timelines and expenditures significantly.
31
The U.S., E.U. and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the U.S., is generally a disease or condition that affects no more than 200,000 individuals. In the E.U., orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than 50 in 100,000 persons in the E.U.; without incentive, it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. Orphan drug designation must be requested before submitting an NDA or MAA. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process. However, this designation provides an exemption from marketing and authorization fees charged to NDA sponsors under the Prescription Drug User Fee Act (PDUFA Fees).
We are also subject to numerous environmental and safety laws and regulations, including those governing the use and disposal of hazardous materials. The cost of compliance with and any violation of these regulations could have a material adverse effect on our business and results of operations. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. Compliance with laws and regulations relating to the protection of the environment has not had a material effect on our capital expenditures or our competitive position. However, we are not able to predict the extent of government regulation, and the cost and effect thereof on our competitive position, which might result from any legislative or administrative action pertaining to environmental or safety matters.
Competition
The development and commercialization of new drugs is highly competitive and we may face competition from established pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies and private and public research institutions worldwide.
Various products currently are marketed for the treatment of the cancers that we may target with VAL-083 or future product candidates and a number of companies are developing new treatments. Companies also developing products for GBM include but are not limited to Celgene Corp., Celldex Therapeutics, Northwest Biotherapeutics, Inc., Immunocellular Therapeutics Ltd., and many major pharmaceutical companies. Our success will be based in part on our ability to build and actively manage a portfolio of drugs that addresses unmet medical needs and create value in patient therapy.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than products that we may develop. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
32
We expect that our ability to compete effectively will depend upon our ability to:
| ● | successfully and rapidly complete adequate and well-controlled clinical trials that demonstrate statistically significant safety and efficacy and to obtain all requisite regulatory approvals in a cost-effective manner; |
| ● | maintain a proprietary position for our manufacturing processes and other technology; |
| ● | produce our products in accordance with United States FDA and international regulatory guidelines; |
| ● | attract and retain key personnel; and |
| ● | build or access an adequate sales and marketing infrastructure for any approved products. |
Failure to do one or more of these activities could have an adverse effect on our business, financial condition or results of operations.
Corporate History
The Company is a Nevada corporation formed on June 24, 2009 under the name Berry Only Inc. On January 25, 2013, the Company entered into and closed an exchange agreement (the “Exchange Agreement”), with Del Mar Pharmaceuticals (BC) Ltd. (“DelMar (BC)”), 0959454 B.C. Ltd. (“Callco”), and 0959456 B.C. Ltd. (“Exchangeco”) and the security holders of DelMar (BC). Upon completion of the Exchange Agreement, DelMar (BC) became a wholly-owned subsidiary of the Company.
DelMar Pharmaceuticals, Inc. is the parent company of DelMar (BC), a British Columbia, Canada corporation incorporated on April 6, 2010, which is a clinical stage company with a focus on the development of drugs for the treatment of cancer. The Company is also the parent company to Callco and Exchangeco which are British Columbia, Canada corporations. Callco and Exchangeco were formed to facilitate the Reverse Acquisition.
On May 20, 2016, the Company effected a 1-for-4 reverse split of its common stock. All share amounts in this report give effect to the reverse split unless otherwise indicated.
Research and Development
During the year ended June 30, 2017 and 2016, we recognized $5,003,640 (unaudited) and $3,360,878, respectively in research and development expenses.
Employees
We have four full-time employees and retain the services of approximately 15 persons on an independent contractor/consultant and contract-employment basis. As such, we currently operate in a “virtual” corporate structure in order to minimize fixed personnel costs. Over time, we plan to establish a base of full time employees and corporate infrastructure.
Risk Factors
Risks Related to Our Business
We expect our independently audited June 30, 2017 consolidated financial statements contain going concern disclosure.
We expect our audited financial statements for the fiscal year ended June 30, 2017 will include an explanatory paragraph regarding our going concern risk. The consolidated financial statements have been prepared on a going concern basis which assumes that the Company will continue its operations for the foreseeable future and contemplates the realization of assets and the settlement of liabilities in the normal course of business. We expect to report a net loss for the year ended June 30, 2017.
The Company is in the development stage and has not generated any revenues to date. The Company does not have the prospect of achieving revenues until such time that its product candidate is commercialized, or partnered, which may not ever occur. In the near future, the Company will require additional funding to maintain its research and development projects and for general operations. These circumstances indicate the existence of a material uncertainty that may cast substantial doubt as to the ability of the Company to meet its obligations as they come due.
33
Consequently, management is pursuing various financing alternatives to fund the Company’s operations so it can continue as a going concern. Management plans to secure the necessary financing through the issue of new equity and/or the entering into of strategic partnership arrangements. Nevertheless, there is no assurance that these initiatives will be successful.
The financial statements do not give effect to any adjustments to the amounts and classification of assets and liabilities that may be necessary should the Company be unable to continue as a going concern. Such adjustments could be material.
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
For the three and nine months ended March 31, 2017, the Company reported net losses of $1,868,460 and $5,480,772 respectively, and negative cash flow from operations of $4,056,858. We had an accumulated deficit of $38,401,763 as of March 31, 2017. As of March 31, 2017, the Company had cash on hand of $2,100,406 and a working capital balance of $1,297,239. We are an early stage company and there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We expect to incur substantial additional net expenses over the next several years as our research, development and commercial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidate; obtaining necessary regulatory approvals from the FDA and international regulatory agencies; successful manufacturing, sales and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects and results of operations may be materially adversely affected.
We will need to raise additional capital, which may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings and/or license and development agreements with collaboration partners. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, then-existing stockholders’ interests may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely their rights as common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring dividends. In addition, debt financing would result in fixed payment obligations.
If we raise funds through collaborations, strategic partnerships or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Our inability to obtain additional financing could adversely affect our ability to meet our obligations under our planned clinical trials and could negatively impact the timing of our clinical results.
Our ability to meet our obligations and continue the research and development of our product candidate is dependent on our ability to continue to raise adequate financing. We may not be successful in obtaining such additional financing in the amount required at any time, or for any period, or, if available, that it can be obtained on terms satisfactory to us. In the event that we are unable to obtain such additional financing, we may be unable to meet our obligations under our planned clinical trials and we may have to tailor our drug candidate development programs based on the amount of funding we raise which could negatively impact the timing of our clinical results.
34
If we are unable to effectively implement or maintain a system of internal control over financial reporting, we may not be able to accurately or timely report our financial results and our stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 and related regulations require us to evaluate the effectiveness of our internal control over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal control over financial reporting in our Annual Report on Form 10-K for that fiscal year. Management determined that as of June 30, 2017, our disclosure controls and procedures and internal control over financial reporting were not effective due to material weaknesses in our internal control over financial reporting related to our limited number of employees in our accounting department and inadequate segregation of duties over authorization, review and recording of transactions, as well as the financial reporting of such transactions. Any failure to implement new or improved controls necessary to remedy the material weaknesses described above, or difficulties encountered in the implementation or operation of these controls, could harm our operations, decrease the reliability of our financial reporting, and cause us to fail to meet our financial reporting obligations, which could adversely affect our business and reduce our stock price.
We are an early-stage company and may never achieve commercialization of our candidate products or profitability.
We are at an early stage of development and commercialization of our technologies and product candidate. We have not yet begun to market any products and, accordingly, have not begun or generate revenues from the commercialization of our product. Our product will require significant additional clinical testing and investment prior to commercialization. A commitment of substantial resources by ourselves and, potentially, our partners to conduct time-consuming research and clinical trials will be required if we are to complete the development of our product candidate. There can be no assurance that our product candidate will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs or be successfully marketed. Our product candidate is not expected to be commercially available for several years, if at all.
We are currently focused on the development of a single product candidate.
Our product development efforts are currently focused on a single product, VAL-083, for which we are researching multiple indications. If VAL-083 fails to achieve clinical endpoints or exhibits unanticipated toxicity or if a superior product is developed by a competitor, our prospects for obtaining regulatory approval and commercialization may be negatively impacted. In the long-term, we hope to establish a pipeline of product candidates, and we have identified additional product candidates that we may be able to acquire or license in the future. However, at this time we do not have any formal agreements granting us any rights to such additional product candidates.
Even if we are able to commercialize any product candidate that we develop, the product may become subject to unfavorable pricing regulations, third-party payor reimbursement practices or healthcare reform initiatives that could harm our business.
The commercial success of our current or future product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidate will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities (such as Medicare and Medicaid), private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our products. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish and maintain pricing sufficient to realize a meaningful return on our investment.
35
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved drugs. Marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some non-U.S. markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay commercial launch of the product, possibly for lengthy time periods, which may negatively impact the revenues we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Our ability to commercialize VAL-083 or any other product candidates will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will cover and establish reimbursement levels. The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications, which could affect our ability to sell our product candidate profitably. These payors may not view our products, if any, as cost-effective, and coverage and reimbursement may not be available to our customers, or may not be sufficient to allow our products, if any, to be marketed on a competitive basis. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated product revenues. If the prices for our products, if any, decrease or if governmental and other third-party payors do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
There may also be delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA or comparable non-U.S. regulatory authorities. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Reimbursement rates may vary, by way of example, according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based on reimbursement levels already set for lower cost drugs or may be incorporated into existing payments for other services.
In addition, increasingly, third-party payors are requiring higher levels of evidence of the benefits and clinical outcomes of new technologies and are challenging the prices charged. We cannot be sure that coverage will be available for any product candidate that we, or they, commercialize and, if available, that the reimbursement rates will be adequate. Further, the net reimbursement for drug products may be subject to additional reductions if there are changes to laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for any our product candidates for which we obtain marketing approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
We are dependent on obtaining certain patents and protecting our proprietary rights.
Our success will depend, in part, on our ability to obtain patents, maintain trade secret protection and operate without infringing on the proprietary rights of third parties or having third parties circumvent our rights. We have filed and are actively pursuing patent applications for our products. The patent positions of biotechnology, biopharmaceutical and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. Thus, there can be no assurance that any of our patent applications will result in the issuance of patents, that we will develop additional proprietary products that are patentable, that any patents issued to us or those that already have been issued will provide us with any competitive advantages or will not be challenged by any third parties, that the patents of others will not impede our ability to do business or that third parties will not be able to circumvent our patents. Furthermore, there can be no assurance that others will not independently develop similar products, duplicate any of our products not under patent protection, or, if patents are issued to us, design around the patented products we developed or will develop.
36
We may be required to obtain licenses from third parties to avoid infringing patents or other proprietary rights. No assurance can be given that any licenses required under any such patents or proprietary rights would be made available, if at all, on terms we find acceptable. If we do not obtain such licenses, we could encounter delays in the introduction of products or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
A number of pharmaceutical, biopharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to or affect our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. Such conflict could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our patent applications. In addition, if patents that cover our activities are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited. In addition, we could incur substantial costs in defending ourselves in suits brought against us on patents it might infringe or in filing suits against others to have such patents declared invalid.
Patent applications in the U.S. are maintained in secrecy and not published if either: i) the application is a provisional application or, ii) the application is filed and we request no publication, and certify that the invention disclosed “has not and will not” be the subject of a published foreign application. Otherwise, U.S. applications or foreign counterparts, if any, publish 18 months after the priority application has been filed. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor was the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost to us, even if the eventual outcome were favorable to us. There can be no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Moreover, we may be subject to third-party preissuance submissions of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us, or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate, or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, the protection of intellectual property rights in China (where our clinical product candidate, VAL-083, is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the China Food and Drug Administration to produce the product for the China market, and where VAL-083 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate royalty revenue from sales of VAL-083 in China.
37
Much of our know-how and technology may not be patentable. To protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, our business may be adversely affected by competitors who independently develop competing technologies, especially if we obtain no, or only narrow, patent protection.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to United States patent law. The Leahy-Smith Act includes provisions that affect the way patent applications are prosecuted and affect patent litigation. The United States Patent and Trademark Office, or PTO, recently developed new regulations and procedures to govern administration of the Leahy-Smith Act. However, many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition.
We may be unable to protect our patents and proprietary rights.
Our future success will depend to a significant extent on our ability to:
| ● | obtain and keep patent protection for our products and technologies on an international basis; |
| ● | enforce our patents to prevent others from using our inventions; |
| ● | maintain and prevent others from using our trade secrets; and |
| ● | operate and commercialize products without infringing on the patents or proprietary rights of others. |
We can provide no assurance that our patent rights will afford any competitive advantages and these rights may be challenged or circumvented by third parties. Further, patents may not be issued on any of our pending patent applications in the U.S. or abroad. Because of the extensive time required for development, testing and regulatory review of a product candidate, it is possible that before a product candidate can be commercialized, any related patent may expire, or remain in existence for only a short period following commercialization, reducing or eliminating any advantage of the patent.
If we sue others for infringing our patents, a court may determine that such patents are invalid or unenforceable. Even if the validity of our patent rights is upheld by a court, a court may not prevent the alleged infringement of our patent rights on the grounds that such activity is not covered by our patent claims.
In addition, third parties may sue us for infringing their patents. In the event of a successful claim of infringement against us, we may be required to:
| ● | defend litigation or administrative proceedings; |
| ● | pay substantial damages; |
| ● | stop using our technologies and methods; |
| ● | stop certain research and development efforts; |
| ● | develop non-infringing products or methods; and |
| ● | obtain one or more licenses from third parties. |
38
If required, we can provide no assurance that we will be able to obtain such licenses on acceptable terms, or at all. If we are sued for infringement, we could encounter substantial delays in development, manufacture and commercialization of our product candidates. Any litigation, whether to enforce our patent rights or to defend against allegations that we infringe third-party rights, will be costly, time consuming, and may distract management from other important tasks.
As is commonplace in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. To the extent our employees are involved in research areas which are similar to those areas in which they were involved at their former employers, we may be subject to claims that such employees and/or we have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of the former employers. Litigation may be necessary to defend against such claims, which could result in substantial costs and be a distraction to management and which may have a material adverse effect on us, even if we are successful in defending such claims.
We are subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S., Canada and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current cGMP during production and storage, and control of marketing activities, including advertising and labeling.
VAL-083 and any other products may develop will require significant development, preclinical and clinical testing and investment of substantial funds prior to its commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that we will successfully develop any future products that will prove to be safe and effective in clinical trials or receive applicable regulatory approvals. Markets other than the U.S. and Canada have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
We may request priority review for our product candidate in the future. The FDA may not grant priority review for our product candidate. Moreover, even if the FDA designated such product for priority review, that designation may not lead to a faster regulatory review or approval process and, in any event, would not assure FDA approval.
We may be eligible for priority review designation for our product candidate if the FDA determines such product candidate offers major advances in treatment or provides a treatment where no adequate therapy exists. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months. The FDA has broad discretion with respect to whether or not to grant priority review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status, the FDA may decide not to grant it. Thus, while the FDA has granted priority review to other oncology disease products, our product candidate, should we determine to seek priority review, may not receive similar designation. Moreover, even if our product candidate is designated for priority review, such a designation does not necessarily mean a faster regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval within an accelerated timeline or thereafter.
39
We believe we may in some instances be able to secure approval from the FDA or comparable non-U.S. regulatory authorities to use accelerated development pathways. If we are unable to obtain such approval, we may be required to conduct additional preclinical studies or clinical trials beyond those that we contemplate, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals.
We anticipate that we may seek an accelerated approval pathway for our product candidate. Under the accelerated approval provisions in the Federal Food, Drug, and Cosmetic Act, or FDCA, and the FDA’s implementing regulations, the FDA may grant accelerated approval to a product designed to treat a serious or life-threatening condition that provides meaningful therapeutic benefit over available therapies upon a determination that the product has an effect on a surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit. The FDA considers a clinical benefit to be a positive therapeutic effect that is clinically meaningful in the context of a given disease, such as irreversible morbidity or mortality. For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. The accelerated approval pathway may be used in cases in which the advantage of a new drug over available therapy may not be a direct therapeutic advantage, but is a clinically important improvement from a patient and public health perspective. If granted, accelerated approval is usually contingent on the sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. If such post-approval studies fail to confirm the drug’s clinical benefit, the FDA may withdraw its approval of the drug.
Prior to seeking such accelerated approval, we will seek feedback from the FDA and will otherwise evaluate our ability to seek and receive such accelerated approval. There can also be no assurance that after our evaluation of the feedback and other factors we will decide to pursue or submit a New Drug Application (“NDA”), for accelerated approval or any other form of expedited development, review or approval. Similarly, there can be no assurance that after subsequent FDA feedback that we will continue to pursue or apply for accelerated approval or any other form of expedited development, review or approval, even if we initially decide to do so. Furthermore, if we decide to submit an application for accelerated approval or under another expedited regulatory designation (e.g., breakthrough therapy designation), there can be no assurance that such submission or application will be accepted or that any expedited development, review or approval will be granted on a timely basis, or at all. The FDA or other non-U.S. authorities could also require us to conduct further studies prior to considering our application or granting approval of any type. A failure to obtain accelerated approval or any other form of expedited development, review or approval for any of our product candidates that we determine to seek accelerated approval for would result in a longer time period to commercialization of such product candidate, could increase the cost of development of such product candidate and could harm our competitive position in the marketplace.
We have conducted, and may in the future conduct, clinical trials for certain of our product candidate at sites outside the United States, and the FDA may not accept data from trials conducted in such locations.
We have conducted and may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical trials conducted outside of the United States must be representative of the population for whom we intend to seek approval in the United States. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials also complied with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept the data from any of our clinical trials that we determine to conduct outside the United States, it would likely result in the need for additional trials, which would be costly and time-consuming and delay or permanently halt our development of the product candidate.
40
In addition, the conduct of clinical trials outside the United States could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| ● | foreign regulatory requirements that could restrict or limit our ability to conduct our clinical trials; |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| ● | foreign exchange fluctuations; and |
| ● | diminished protection of intellectual property in some countries. |
If clinical our trials fail to demonstrate safety and efficacy to the satisfaction of the FDA and comparable non-U.S. regulators, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidate.
We are not permitted to commercialize, market, promote or sell any product candidate in the United States without obtaining marketing approval from the FDA. Comparable non-U.S. regulatory authorities, such as the EMA, impose similar restrictions. We may never receive such approvals. We must complete extensive preclinical development and clinical trials to demonstrate the safety and efficacy of our product candidate in humans before we will be able to obtain these approvals.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We have not previously submitted an NDA to the FDA or similar drug approval filings to comparable non-U.S. regulatory authorities for any product candidate.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us and impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if (1) we are required to conduct additional clinical trials or other testing of our product candidate beyond the trials and testing that we contemplate, (2) we are unable to successfully complete clinical trials of our product candidate or other testing, (3) the results of these trials or tests are unfavorable, uncertain or are only modestly favorable, or (4) there are unacceptable safety concerns associated with our product candidate, we, in addition to incurring additional costs, may:
| ● | be delayed in obtaining marketing approval for our product candidates; |
| ● | not obtain marketing approval at all; |
| ● | obtain approval for indications or patient populations that are not as broad as we intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings; |
| ● | be subject to additional post-marketing testing or other requirements; or |
| ● | be required to remove the product from the market after obtaining marketing approval. |
If we experience any of a number of possible unforeseen events in connection with clinical trials of our product candidate, potential marketing approval or commercialization of our product candidate could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent marketing approval of our product candidate, including:
| ● | clinical trials of our product candidate may produce unfavorable or inconclusive results; |
| ● | we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; |
| ● | the number of patients required for clinical trials of our product candidate may be larger than we anticipate, patient enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
41
| ● | data safety monitoring committees may recommend suspension, termination or a clinical hold for various reasons, including concerns about patient safety; |
| ● | regulators or IRBs may suspend or terminate the trial or impose a clinical hold for various reasons, including noncompliance with regulatory requirements or concerns about patient safety; |
| ● | patients with serious, life-threatening diseases included in our clinical trials may die or suffer other adverse medical events for reasons that may not be related to our product candidate; |
| ● | participating patients may be subject to unacceptable health risks; |
| ● | patients may not complete clinical trials due to safety issues, side effects, or other reasons; |
| ● | changes in regulatory requirements and guidance may occur, which require us to amend clinical trial protocols to reflect these changes; |
| ● | our third-party contractors, including those manufacturing our product candidate or components or ingredients thereof or conducting clinical trials on our behalf, may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner or at all; |
| ● | regulators or institutional review boards, or IRBs may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| ● | we may experience delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; |
| ● | patients who enroll in a clinical trial may misrepresent their eligibility to do so or may otherwise not comply with the clinical trial protocol, resulting in the need to drop the patients from the clinical trial, increase the needed enrollment size for the clinical trial or extend the clinical trial’s duration; |
| ● | we may have to suspend or terminate clinical trials of our product candidate for various reasons, including a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of a product candidate; |
| ● | regulators or IRBs may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or their respective standards of conduct, a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of the product candidate or findings of undesirable effects caused by a chemically or mechanistically similar drug or drug candidate; |
| ● | the FDA or comparable non-U.S. regulatory authorities may disagree with our clinical trial design or our interpretation of data from preclinical studies and clinical trials; |
| ● | the FDA or comparable non-U.S. regulatory authorities may fail to approve or subsequently find fault with the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for clinical and commercial supplies; |
| ● | the supply or quality of raw materials or manufactured product candidate or other materials necessary to conduct clinical trials of our product candidate may be insufficient, inadequate, delayed, or not available at an acceptable cost, or we may experience interruptions in supply; and |
| ● | the approval policies or regulations of the FDA or comparable non-U.S. regulatory authorities may significantly change in a manner rendering our clinical data insufficient to obtain marketing approval. |
42
Product development costs for us will increase if we experience delays in testing or pursuing marketing approvals and we may be required to obtain additional funds to complete clinical trials and prepare for possible commercialization of our product candidate. We do not know whether any preclinical tests or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidate or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidate and may harm our business and results of operations. In addition, many of the factors that cause, or lead to, clinical trial delays may ultimately lead to the denial of marketing approval of our product candidate.
If we experience delays or difficulties in the enrollment of patients in clinical trials, we may not achieve our clinical development on our anticipated timeline, or at all, and our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for VAL-083 or any other product candidate if we are unable to locate and enroll a sufficient number of eligible patients to participate in clinical trials. Patient enrollment is a significant factor in the timing of clinical trials, and is affected by many factors, including:
| ● | the size and nature of the patient population; |
| ● | the severity of the disease under investigation; |
| ● | the proximity of patients to clinical sites; |
| ● | the eligibility criteria for the trial; |
| ● | the design of the clinical trial; |
| ● | efforts to facilitate timely enrollment; |
| ● | competing clinical trials; and |
| ● | clinicians’ and patients’ perceptions as to the potential advantages and risks of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. |
Our inability to enroll a sufficient number of patients for our clinical trials could result in significant delays or may require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidate, delay or halt the development of and approval processes for our product candidate and jeopardize our ability to achieve our clinical development timeline and goals, including the dates by which we will commence, complete and receive results from clinical trials. Enrollment delays may also delay or jeopardize our ability to commence sales and generate revenues from our product candidate. Any of the foregoing could cause the value of the Company to decline and limit our ability to obtain additional financing, if needed.
Positive results in previous clinical trials of VAL-083 may not be replicated in future clinical trials, which could result in development delays or a failure to obtain marketing approval.
Positive results in previous clinical studies of VAL-083 may not be predictive of similar results in future clinical trials. Also, interim results during a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical trials for VAL-083 may not be predictive of the results we may obtain in later stage trials. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain FDA or EMA, or other regulatory agency, approval for their products.
43
FDA approval of VAL-083 or future product candidates may be denied.
There can be no assurance that the FDA will ultimately approve our NDA. The FDA may deny approval of VAL-083 for many reasons, including:
| ● | we may be unable to demonstrate to the satisfaction of the FDA that our products are safe and effective for its intended uses; |
| ● | the FDA may disagree with our interpretation of data from the clinical trials; |
| ● | we may be unable to demonstrate that any clinical or other benefits our products outweigh any safety or other perceived risks; or |
| ● | we may not be able to successfully address any other issues raised by the FDA. |
| If VAL-083 fails to receive FDA approval, our business and prospects will be materially adversely impacted. |
We expect to rely on orphan drug status to develop and commercialize our product candidate, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits as anticipated.
Market exclusivity afforded by orphan drug designation is generally offered as an incentive to drug developers to invest in developing and commercializing products for unique diseases that impact a limited number of patients. The FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. Qualification to maintain orphan drug status is generally monitored by the regulatory authorities during the orphan drug exclusivity period, currently seven years from the date of approval in the United States.
We have been granted orphan drug designation in the United States for GBM, ovarian cancer, and medulloblastoma, and in Europe for GBM. We expect to rely on orphan drug exclusivity for our product candidate. It is possible that the incidence and prevalence numbers for GBM could change. Should the incidence and prevalence of GBM patients materially increase, it is possible that the orphan drug designation, and related market exclusivity, in the United States could be lost. Further, while we have been granted this orphan designation, the FDA can still approve different drugs for use in treating the same indication or disease, which would create a more competitive market for us and our revenues will be diminished.
Further, it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company’s period of exclusivity expires. Even if we are the first to obtain marketing authorization for an orphan drug indication, there are circumstances under which a competing product may be approved for the same indication during the seven-year period of marketing exclusivity, such as if the later product is shown to be clinically superior to the orphan product, or if the later product is deemed a different product than ours. Further, the seven-year marketing exclusivity would not prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation, or for the use of other types of products in the same indications as our orphan product.
44
If the market opportunities for our product candidate are smaller than we believe they are, our revenues may be adversely affected and our business may suffer. Because the target patient populations of our product candidate are small, we must be able to successfully identify patients and capture a significant market share to achieve and maintain profitability.
We focus our research and product development on treatments for orphan cancer indications. Our projections of both the number of people who have failed other therapies or have limited medical options, are based on estimates. These estimates may prove to be incorrect and new studies may change the estimated incidence or prevalence. The number of patients in the United States, Europe and elsewhere may turn out to be lower than expected or may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. Additionally, because our target patient populations are small, we will be required to capture a significant market share to achieve and maintain profitability.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our products.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects or even death as a result of participating in our clinical trials.
We may not receive regulatory approvals for our product candidate or there may be a delay in obtaining such approvals.
Our product and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the EMA, will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval of VAL-083 or any future product candidates.
The time required to obtain regulatory approval varies between countries. In the U.S., for products without “Fast Track” status, it can take up to eighteen (18) months after submission of an application for product approval to receive the FDA's decision. Even with Fast Track status, FDA review and decision can take up to twelve (12) months. At present, we do not have Fast Track status for VAL-083.
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
45
Even if our product candidate receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success and the market opportunity for the product candidate may be smaller than we estimate.
We have never commercialized a product. Even if VAL-083 or any other product candidate is approved by the appropriate regulatory authorities for marketing and sale, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. For example, physicians are often reluctant to switch their patients from existing therapies even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of reimbursement for existing therapies.
Efforts to educate the medical community and third-party payors on the benefits of our product candidate may require significant resources and may not be successful. If our product candidate is approved but does not achieve an adequate level of market acceptance, we may not generate significant revenues and we may not become profitable. The degree of market acceptance of VAL-083 or any other product candidate, if approved for commercial sale, will depend on a number of factors, including:
| ● | the efficacy and safety of the product; |
| ● | the potential advantages of the product compared to alternative treatments; |
| ● | the prevalence and severity of any side effects; |
| ● | the clinical indications for which the product is approved; |
| ● | whether the product is designated under physician treatment guidelines as a first-line therapy or as a second- or third-line therapy; |
| ● | limitations or warnings, including distribution or use restrictions, contained in the product’s approved labeling; |
| ● | our ability to offer the product for sale at competitive prices; |
| ● | our ability to establish and maintain pricing sufficient to realize a meaningful return on our investment; |
| ● | the product’s convenience and ease of administration compared to alternative treatments; |
| ● | the willingness of the target patient population to try, and of physicians to prescribe, the product; |
| ● | the strength of sales, marketing and distribution support; |
| ● | the approval of other new products for the same indications; |
| ● | changes in the standard of care for the targeted indications for the product; |
| ● | the timing of market introduction of our approved products as well as competitive products and other therapies; |
| ● | availability and amount of reimbursement from government payors, managed care plans and other third-party payors; |
| ● | adverse publicity about the product or favorable publicity about competitive products; and |
| ● | potential product liability claims. |
46
The potential market opportunities for our product candidate are difficult to estimate precisely. Our estimates of the potential market opportunities are predicated on many assumptions, including industry knowledge and publications, third-party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve the exercise of significant judgment on the part of our management, are inherently uncertain and the reasonableness of these assumptions has not been assessed by an independent source. If any of the assumptions proves to be inaccurate, the actual markets for our product candidate could be smaller than our estimates of the potential market opportunities.
If our product candidate receives marketing approval and we, or others, later discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, our ability to market the drug could be compromised.
Clinical trials of our product candidate are conducted in carefully defined subsets of patients who have agreed to enter into clinical trials. Consequently, it is possible that our clinical trials may indicate an apparent positive effect of a product candidate that is greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of our product candidate, we, or others, discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, any of the following adverse events could occur:
| ● | regulatory authorities may withdraw their approval of the drug or seize the drug; |
| ● | we may be required to recall the drug or change the way the drug is administered; |
| ● | additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the particular drug; |
| ● | we may be subject to fines, injunctions or the imposition of civil or criminal penalties; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| ● | we may be required to create a Medication Guide outlining the risks of the previously unidentified side effects for distribution to patients; |
| ● | we could be sued and held liable for harm caused to patients; |
| ● | the drug may become less competitive; and |
| ● | our reputation may suffer. |
Any of these events could have a material and adverse effect on our operations and business and could adversely impact our stock price.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, qualification testing, post-approval clinical data, labeling and promotional activities for such product, will be subject to continual and additional requirements of the FDA and other regulatory authorities.
These requirements include submissions of safety and other post-marketing information, reports, registration and listing requirements, good manufacturing practices, or GMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and recordkeeping. Even if marketing approval of our product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates the post-approval marketing and promotion of pharmaceutical products to ensure such products are marketed only for the approved indications and in accordance with the provisions of the approved labeling.
47
In addition, later discovery of previously unknown problems with our products, manufacturing processes, or failure to comply with regulatory requirements, may lead to various adverse results, including:
| ● | restrictions on such products, manufacturers or manufacturing processes; |
| ● | restrictions on the labeling or marketing of a product; |
| ● | restrictions on product distribution or use; |
| ● | requirements to conduct post-marketing clinical trials; |
| ● | requirements to institute a risk evaluation mitigation strategy, or REMS, to monitor safety of the product post-approval; |
| ● | warning letters issued by the FDA or other regulatory authorities; |
| ● | withdrawal of the products from the market; |
| ● | refusal to approve pending applications or supplements to approved applications that we submit; |
| ● | recall of products, fines, restitution or disgorgement of profits or revenue; |
| ● | suspension, revocation or withdrawal of marketing approvals; |
| ● | refusal to permit the import or export of our products; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
If we are unable to establish sales, marketing and distribution capabilities or enter into acceptable sales, marketing and distribution arrangements with third parties, we may not be successful in commercializing any product candidates that we develop, if and when those product candidates are approved.
We do not have a sales, marketing or distribution infrastructure and have limited experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop a sales and marketing organization, outsource these functions to third parties, or license our product candidates to others. If approved, we may seek to license VAL-083 to a large pharmaceutical company with greater resources and experience than us. We may not be able license the VAL-083 on reasonable terms, if at all. The development of sales, marketing and distribution capabilities will require substantial resources, will be time-consuming and could delay any product launch. We expect that we will commence the development of these capabilities prior to receiving approval of our product candidate. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and distribution capabilities is delayed or does not occur for any reason, we could have prematurely or unnecessarily incurred these commercialization costs. Such a delay may be costly, and our investment could be lost if we cannot retain or reposition our sales and marketing personnel. In addition, we may not be able to hire or retain a sales force in the United States that is sufficient in size or has adequate expertise in the medical markets that we plan to target. If we are unable to establish or retain a sales force and marketing and distribution capabilities, our operating results may be adversely affected. If a potential partner has development or commercialization expertise that we believe is particularly relevant to our product candidate, then we may seek to collaborate with that potential partner even if we believe we could otherwise develop and commercialize the product independently.
We expect to seek one or more strategic partners for commercialization of our product candidate outside the United States. As a result of entering into arrangements with third parties to perform sales, marketing and distribution services, our product revenues or the profitability of these product revenues may be lower, perhaps substantially lower, than if we were to directly market and sell products in those markets. Furthermore, we may be unsuccessful in entering into the necessary arrangements with third parties or may be unable to do so on terms that are favorable to us. In addition, we may have little or no control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively.
48
If we do not establish sales and marketing capabilities, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidate.
We face substantial competition from other pharmaceutical and biotechnology companies and our operating results may suffer if we fail to compete effectively.
The development and commercialization of new drug products is highly competitive. We expect that we will face significant competition from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide with respect to VAL-083 and any other product candidates that we may seek to develop or commercialize in the future. Specifically, due to the large unmet medical need, global demographics and relatively attractive reimbursement dynamics, the oncology market is fiercely competitive and there are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates for the treatment of cancer. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective, have fewer or more tolerable side effects or are less costly than any product candidates that we are currently developing or that we may develop, which could render our product candidates obsolete and noncompetitive.
All of the top ten global pharmaceutical companies and most of the mid-size pharmaceutical companies have a strong research and development and commercial presence in oncology. Smaller companies also focus on oncology, including companies such as ARIAD Pharmaceuticals, Inc., Agios Pharmaceuticals, Inc., BIND Therapeutics, Inc., Clovis Oncology, Inc., Endocyte, Inc., Epizyme, Inc., ImmunoGen, Inc., Incyte Corporation, Infinity Pharmaceuticals, Inc., MacroGenics, Inc., Merrimack Pharmaceuticals, Inc., OncoMed Pharmaceuticals, Inc., Onconova Therapeutics, Inc., Pharmacyclics, Inc., Puma Biotechnology, Inc., Seattle Genetics, Inc. and TESARO, Inc.
Several companies are marketing and developing oncology immunotherapy products. Companies with approved marketed oncology products for GBM are Merck (Temodar®) and Genentech (Avastin®). Companies with oncology immunotherapy product candidates in clinical development include Northwest Biotherapeutics (DCVax-L), Celldex Therapeutics (Rindopepimut (CDX-110)) and ImmunoCellular Therapeutics (ICT-107).
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other marketing approval for their products before we are able to obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of our existing and potential future competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining marketing approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we are unable to or delayed in obtaining state regulatory licenses for the distribution of our product, we would not be able to sell our product candidate.
The majority of states require manufacturer and/or wholesaler licenses for the sale and distribution of drugs into that state. The application process is complicated, time consuming and requires dedicated personnel or a third-party to oversee and manage. If we are delayed in obtaining these state licenses, or denied the licenses, even with FDA approval, we would not be able to sell or ship product into that state which would adversely affect our sales and revenues.
49
We rely on key personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to grow effectively.
We are dependent on certain members of our management, scientific and drug development staff and consultants, the loss of services of one or more of whom could materially adversely affect us.
We currently have four full-time employees, and retain the services of approximately 15 persons on an independent contractor/consultant and contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel.
Our success depends in large part upon our ability to attract and retain highly qualified personnel. We compete in our hiring efforts with other pharmaceutical and biotechnology companies, as well as universities and nonprofit research organizations, and we may have to pay higher salaries to attract and retain personnel, which would be very costly.
We may be subject to foreign exchange fluctuation.
Our functional and reporting currency is the United States dollar. We maintain bank accounts in United States and Canadian dollars. A portion of our expenditures are in foreign currencies, most notably in Canadian dollars, and therefore we are subject to foreign currency fluctuations, which may, from time to time, impact our financial position and results. We may enter into hedging arrangements under specific circumstances, typically through the use of forward or futures currency contracts, to minimize the impact of increases in the value of the Canadian dollar. In order to minimize our exposure to foreign exchange fluctuations we may hold sufficient Canadian dollars to cover our expected Canadian dollar expenditures.
Product liability lawsuits against us could divert our resources, cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
We face an inherent risk of product liability claims as a result of the clinical testing of our product candidate despite obtaining appropriate informed consents from our clinical trial participants. We will face an even greater risk if we commercially sell any product that we may develop. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidate. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our product candidate or products that we may develop; |
| ● | injury to our reputation and significant negative media attention; |
| ● | withdrawal of clinical trial participants; |
| ● | significant costs to defend resulting litigation; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | loss of revenue; |
| ● | reduced resources of our management to pursue our business strategy; and |
| ● | the inability to commercialize any products that we may develop. |
50
Although we maintain general liability insurance, this insurance may not fully cover potential liabilities that we may incur. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. We will need to increase our insurance coverage if and when we begin selling any product candidate that receives marketing approval. In addition, insurance coverage is becoming increasingly expensive. If we are unable to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims, it could prevent or inhibit the development and commercial production and sale of our product candidate, which could adversely affect our business, financial condition, results of operations and prospects.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct clinical trials for our product candidate. Any failure by a third-party to meet its obligations with respect to the clinical development of our product candidate may delay or impair our ability to obtain regulatory approval for our product candidate.
We rely on academic institutions and private oncology centers to conduct our clinical trial. Our reliance on third parties to conduct clinical trials could, depending on the actions of such third parties, jeopardize the validity of the clinical data generated and adversely affect our ability to obtain marketing approval from the FDA or other applicable regulatory authorities.
Such clinical trial arrangements provide us with information rights with respect to the clinical data, including access to and the ability to use and reference the data, including for our own regulatory filings, resulting from the clinical trials. If investigators or institutions breach their obligations with respect to the clinical trials of our product candidate, or if the data proves to be inadequate, then our ability to design and conduct any future clinical trials may be adversely affected.
We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
We currently rely on third-party clinical research organizations, or CROs, to conduct our clinical trials. We expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct our clinical trials. Our agreements with these third parties generally allow the third-party to terminate the agreement at any time. If we are required to enter into alternative arrangements because of any such termination the introduction of our product candidates to market could be delayed.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we design our clinical trials and will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as good clinical practices, or GCPs, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidate or commercialization of our products, producing additional losses and depriving us of potential product revenue.
51
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidate. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our product candidate.
We may seek third-party collaborators for development and commercialization of our product candidate. Our likely collaborators for any marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. We are currently party to a limited number of such arrangements and have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidate. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidate currently pose, and will continue to pose, the following risks to us:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| ● | collaborators may not pursue development and commercialization of our product candidate or may elect not to continue or renew development or commercialization programs based on preclinical or clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; | |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidate if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| ● | collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
| ● | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| ● | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidate or that result in costly litigation or arbitration that diverts management attention and resources; and |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of our product candidate in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
52
If we are not able to establish collaborations, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidate will require substantial additional cash to fund expenses. We may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of our product candidate.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of preclinical studies or clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of our product candidate, reduce or delay its development program, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidate or bring it to market and generate product revenue.
We currently manufacture our clinical supplies at a single location. Any disruption at this facility could adversely affect our business and results of operations.
We have engaged a single manufacturer to produce active pharmaceutical ingredient and drug product for our STAR-3 Phase 3 clinical trial. In addition, we rely on our manufacturing partner, Guangxi Wuzhou Pharmaceuticals (Group) Co. Ltd., for the manufacture of clinical supply of VAL-083 for our preclinical and Phase 2 clinical studies. If our manufacturer’s facility were damaged or destroyed, or otherwise subject to disruption, it would require substantial lead-time to replace our clinical supply. In such event, we would be forced to rely entirely on other third-party contract manufacturers for an indefinite period of time. We have established a relationship with a back-up manufacturer, which has produced quantities of the active pharmaceutical ingredient contained in VAL-083. However, at this time no drug product has been manufactured by a third-party back-up manufacturer. Any disruptions or delays by our third-party manufacturers or Guangxi Wuzhou Pharmaceuticals or their failure to meet regulatory compliance could impair our ability to develop VAL-083, which would adversely affect our business and results of operations.
We rely on these third-party manufacturers to provide drug product supply for our Phase 3 clinical trial. There is no assurance that such a supplier will be able to meet our needs from a technical, timing, or cost-effective manner. Our failure to enter into appropriate agreements with such a third-party manufacturer would delay the initiation of our pivotal Phase 3 clinical trial.
53
We may become subject to liabilities related to risks inherent in working with hazardous materials.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. We are not specifically insured with respect to this liability. Although we believe that we are in compliance in all material respects with applicable environmental laws and regulations and currently do not expect to make material capital expenditures for environmental control facilities in the near-term, there can be no assurance that we will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that our operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Risks Related to Our Common Stock
The market price of our common stock is, and is likely to continue to be, highly volatile and subject to wide fluctuations.
The market price of our common stock is highly volatile and could be subject to wide fluctuations in response to a number of factors that are beyond our control, including:
| ● | variations in our quarterly operating results; |
| ● | announcements that our revenue or income are below analysts’ expectations; |
| ● | general economic slowdowns; |
| ● | sales of large blocks of our common stock; and |
| ● | announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments. |
Because we became public by means of a reverse acquisition, we may not be able to attract, or maintain, the attention of brokerage firms.
Because we became public through a “reverse acquisition”, securities analysts of brokerage firms may not provide or continue to provide coverage of us since there is little incentive to brokerage firms to recommend the purchase of our common stock. No assurance can be given that brokerage firms will want to conduct any follow-on offerings on behalf of the Company in the future.
Voting power of our shareholders is highly concentrated by insiders.
Our officers, directors, and 5% shareholders control, either directly or indirectly, a substantial portion of our voting securities. Therefore, our management may significantly affect the outcome of all corporate actions and decisions for an indefinite period of time including election of directors, amendment of charter documents and approval of mergers and other significant corporate transactions.
We do not intend to pay dividends on our common stock for the foreseeable future.
We have paid no dividends on our common stock to date and we do not anticipate paying any dividends to holders of our common stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, we currently anticipate that any earnings will be retained to finance our future expansion and for the implementation of our business plan. Investors should take note of the fact that a lack of a dividend can further affect the market value of our common stock, and could significantly affect the value of any investment in the Company.
54
Our articles of incorporation allow for our board to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board of Directors has the authority to issue up to 5,000,000 shares of our preferred stock (of which 1 share has been designated Special Voting Preferred Stock and is issued and outstanding, 278,530 shares have been designated Series A Preferred Stock and are issued and outstanding, and 1,000,000 shares have been designated as Series B Preferred Stock, of which 881,113 shares are issued and outstanding) without further stockholder approval. As a result, our Board of Directors could authorize the issuance of additional series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock. In addition, our Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders. Although we have no present intention to issue any additional shares of preferred stock or to create any additional series of preferred stock, we may issue such shares in the future.
Our issuance of common stock upon exercise of warrants or options, exchange of Exchangeable Shares, or conversion of Series B Preferred Stock may depress the price of our common stock.
As of September 19, 2017, the Company has 13,551,872 shares of common stock issued and outstanding, 957,761 shares of common stock issuable upon exchange of the Exchangeable Shares of Exchangeco (which entitle the holder to require Exchangeco to redeem (or, at the option of the Company or Callco, to have the Company or Callco purchase) the Exchangeable Shares, and upon such redemption or purchase to receive an equal number of shares of common stock of the Company) (the Exchangeable Shares are recognized on an as-exchanged for common stock basis for financial statement purposes), outstanding warrants to purchase 6,628,906 shares of common stock, outstanding Series B convertible preferred shares that are convertible into 2,202,792 shares of common stock, and outstanding options to purchase 1,300,850 shares of common stock. All Exchangeable Shares, warrants, and options are convertible or exercisable into one share of common stock. Each share of Series B preferred stock is convertible into 2.5 shares of common stock. The issuance of shares of common stock upon exercise of outstanding warrants or options or exchange of Exchangeable Shares could result in substantial dilution to our stockholders, which may have a negative effect on the price of our common stock.
Forward-Looking Statements
Certain of the foregoing statements are forward-looking statements that involve a number of risks and uncertainties, including statements relating to expectations regarding the completion of the Offering. Such forward-looking statements are within the meaning of that term in Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. Reference is made to the Company’s filings under the Securities Exchange Act for additional factors that could cause actual results to differ materially, including the Risk Factors described in the Form 10-K for the fiscal year ended June 30, 2016. The Company undertakes no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events, or otherwise. Readers are cautioned that any such forward-looking statements are not guarantees of future performance and involve risks and uncertainties, and that actual results may differ materially from those indicated in the forward-looking statements as a result of various factors. Readers are cautioned not to place undue reliance on these forward-looking statements.
| Item 9.01 | Financial Statements and Exhibits. |
(d) Exhibits
55
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| DELMAR PHARMACEUTICALS, INC. | ||
| Dated: September 21, 2017 | By: | /s/ Jeffrey Bacha |
| Name: Jeffrey Bacha | ||
| Title: Chief Executive Officer | ||
56
Exhibit Index
58