Attached files
| file | filename |
|---|---|
| EX-32 - Sintx Technologies, Inc. | ex32.htm |
| EX-31.2 - Sintx Technologies, Inc. | ex31-2.htm |
| EX-31.1 - Sintx Technologies, Inc. | ex31-1.htm |
| EX-23.2 - Sintx Technologies, Inc. | ex23-2.htm |
| EX-23.1 - Sintx Technologies, Inc. | ex23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
| [X] | Annual report pursuant to section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 2016
or
| [ ] | Transition report pursuant to section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from _______ to _________
Commission File No. 001-33624
Amedica Corporation
(Exact name of registrant as specified in its charter)
| Delaware | 84-1375299 | |
(State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) |
1885 West 2100 South, Salt Lake City, UT 84119
(Address of principal executive offices and Zip Code)
(801) 839-3500
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.01 par value | The NASDAQ Capital Market |
Securities registered under Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [ ] No [X]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.[X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company.
| Large Accelerated Filer | [ ] | Accelerated Filer | [ ] |
| Non-Accelerated Filer | [ ] [Do not check if a smaller reporting company] | Smaller reporting company | [X] |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter was $46,002,726.
The number of shares outstanding of the registrant’s common stock, $0.01 par value per share, as of September 19, 2017 was 36,264,881.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the Registrant’s definitive Proxy Statement for its 2017 Annual Meeting of Stockholders are incorporated by reference into Part III of this Form 10-K.
TABLE OF CONTENTS
| 2 |
CAUTIONARY NOTE CONCERNING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). All statements other than statements of historical fact are forward-looking statements. We have tried to identify forward-looking statements by using words such as “believe,” “may,” “might,” “could,” “will,” “aim,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “plan” and similar words. These forward-looking statements are based on our current assumptions, expectations and estimates of future events and trends. Forward-looking statements are only predictions and are subject to many risks, uncertainties and other factors that may affect our businesses and operations and could cause actual results to differ materially from those predicted. These risks and uncertainties include, but are not limited to, factors affecting our quarterly and annual results, our ability to manage our growth, our ability to sustain our profitability, demand for our products, our ability to compete successfully (including without limitation our ability to convince surgeons to use our products and our ability to attract and retain sales and other personnel), our ability to rapidly develop and introduce new products, our ability to develop and execute on successful business strategies, our ability to comply with changes and applicable laws and regulations that are applicable to our businesses, our ability to safeguard our intellectual property, our success in defending legal proceedings brought against us, trends in the medical device industry, and general economic conditions, and other risks set forth throughout this Annual Report, including under “Item 1, Business,” “Item 1A, Risk Factors,” and “Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and those discussed in other documents we file with the Securities and Exchange Commission (the “SEC”). Moreover, we operate in an evolving environment. New risk factors and uncertainties emerge from time to time and it is not possible for us to predict all risk factors and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Given these risks and uncertainties, readers are cautioned not to place undue reliance on any forward-looking statements. Forward-looking statements contained in this Annual Report speak only as of the date of this Annual Report. We undertake no obligation to update any forward-looking statements as a result of new information, events or circumstances or other factors arising or coming to our attention after the date hereof.
WHERE YOU CAN FIND MORE INFORMATION
We are subject to the informational requirements of the Exchange Act. Accordingly, we file periodic reports and other information with the SEC. We will make our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports available through our Internet site, http://investors.amedica.com/sec.cfm as soon as reasonably practicable after electronically filing such materials with the SEC. They may also be obtained free of charge by writing to Amedica Corporation, Attn: Investor Relations, 1885 West 2100 South, Salt Lake City, UT 84119. In addition, copies of these reports may be obtained through the SEC’s website at www.sec.gov or by visiting the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549 or by calling the SEC at 800-SEC-0330. Our common stock trades on The NASDAQ Capital Market under the symbol “AMDA.”
Unless otherwise indicated, all information contained in this Annual Report reflects a 1-for-15 reverse split of our common stock which was effected on January 25, 2016.
| 3 |
Overview
We are a materials company focused on developing, manufacturing and selling silicon nitride ceramics that are used in medical implants and in a variety of industrial devices. At present, we commercialize silicon nitride in the spine implant market. We believe that our facile silicon nitride manufacturing expertise positions us favorably to introduce new and innovative devices in the medical and non-medical fields. We also believe that we are the first and only company to commercialize silicon nitride medical implants.
We have received 510(k) regulatory clearance in the United States, a CE mark in Europe, and ANVISA approval in Brazil for a number of our devices that are designed for spinal fusion surgery. To date, more than 28,000 of our silicon nitride devices have been implanted into patients, with an 8-year successful track record. In December 2016, we re-filed an FDA 510(k) submission for clearance in the United States of a modified novel composite spinal fusion device that combines porous and solid silicon nitride, and obviates the need for bone grafts that is comparable to our commercially-available Valeo®C cervical implants.
We believe that silicon nitride has a superb combination of properties that make it ideally suited for human implantation. Other biomaterials are based on bone grafts, metal alloys, and polymers; all of which have practical limitations. In contrast, silicon nitride has a legacy of success in the most demanding and extreme industrial environments. As a human implant material, silicon nitride offers bone ingrowth, resistance to bacterial infection, resistance to corrosion, superior strength and fracture resistance, and ease of diagnostic imaging, among other advantages.
We market and sell our Valeo brand of silicon nitride implants to surgeons and hospitals in the United States and to selected markets in Europe and South America through more than 50 independent sales distributors who are supported by an in-house sales and marketing management team. These implants are designed for use in cervical (neck) and thoracolumbar (lower back) spine surgery. In 2016 we entered into a 10-year exclusive distribution agreement with Shandong Weigao Orthopaedic Device Company Limited (“Weigao”) to sell Amedica-branded silicon nitride spinal fusion devices within the People’s Republic of China (“China”). Weigao, a large orthopedic company, has expertise in acquiring Chinese Food and Drug Administration (“CFDA”) approval of medical devices, and will assist us in obtaining regulatory approval. Weigao has committed to minimum purchase requirements totaling 225,000 implants in the first six years following CFDA clearance. We are also working with other partners in Japan to obtain regulatory approval for silicon nitride in that country. China and Japan are relevant because historically, ceramic implants are more familiar to, and more readily accepted by surgeons outside the United States, i.e., in Asia and Europe.
In addition to silicon nitride, we also sell metal-based products in the United States that provide surgeons and hospitals with a complete package for spinal surgery. These metal products are designed to address spinal deformity and degenerative conditions. Although these metal products have accounted for approximately 48% of our product revenues for each of the years ended December 31, 2016 and 2015, respectively, we remain focused on developing and promoting silicon nitride, and driving its adoption through a scientifically-intense, data-driven strategy.
In addition to direct sales, we have targeted original equipment manufacturer (“OEM”) and private label partnerships in order to accelerate adoption of silicon nitride, both in the spinal space, and also in future markets such as hip and knee replacements, dental, extremities, trauma, and sports medicine. Existing biomaterials, based on plastics, metals, and bone grafts have well-recognized limitations that we believe are addressed by silicon nitride, and we are uniquely positioned to convert existing, successful implant designs made by other companies into silicon nitride. We believe OEM and private label partnerships will allow us to work with a variety of partners, accelerate the adoption of silicon nitride, and realize incremental revenue at improved operating margins, when compared to the cost-intensive direct sales model.
We believe that silicon nitride addresses many of the biomaterial-related limitations in fields such as hip and knee replacements, dental implants, sports medicine, extremities, and trauma surgery. We further believe that the inherent material properties of silicon nitride, and the ability to formulate the material in a variety of compositions, combined with precise control of the surface properties of the material, opens up a number of commercial opportunities across orthopedic surgery, neurological surgery, maxillofacial surgery, and other medical disciplines.
We operate a 30,000 square foot manufacturing facility at our corporate headquarters in Salt Lake City, Utah, and we believe we are the only vertically integrated silicon nitride medical device manufacturer in the world.
| 4 |
Biomaterials
Biomaterials are natural or synthetic biocompatible materials that are used in virtually every medical specialty to improve or preserve body functionality. Various types of biomaterials are used as essential components in medical devices, drug delivery systems, replacement and tissue repair technologies, prostheses, and diagnostic technologies.
There are four general categories of biomaterials:
| ● | Ceramics. Ceramics are hard, non-metallic, non-corrosive, heat-resistant materials made by shaping and then applying high temperatures. Traditional ceramics commonly used as biomaterials include carbon, oxides of aluminum, zirconium and titanium, calcium phosphate and zirconia-toughened alumina. Examples of medical uses of ceramics include repair, augmentation or stabilization of fractured bones, bone and joint replacements, spinal fusion devices, dental implants and restorations, heart valves and surgical instruments. | |
| ● | Metals. Metals commonly used as biomaterials include titanium, stainless steel, cobalt, chrome, gold, silver and platinum, and alloys of these metals. Examples of medical uses of metals include the repair or stabilization of fractured bones, stents, surgical instruments, bone and joint replacements, spinal fusion devices, dental implants and restorations and heart valves. | |
| ● | Natural biomaterials. Natural biomaterials are derived from human donors, animal or plant sources and include human bone, collagen, gelatin, cellulose, chitin, alginate and hyaluronic acid. Examples of medical uses of natural biomaterials include the addition or substitution of hard and soft tissue, cornea protectors, vascular grafts, repair and replacement of tendons and ligaments, bone and joint replacements, spinal fusion devices, dental restorations and heart valves. | |
| ● | Polymers. Polymers are synthetic compounds consisting of similar molecules linked together that can be created to have specific properties. Polymers commonly used as biomaterials include nylon, silicon rubber, polyester, polyethylene, cross-linked polyethylene (a stronger version), polymethylmethacrylate, polyvinyl chloride and polyetheretherketone – which is commonly referred to as PEEK. Examples of medical uses of polymers include soft-tissue replacement, sutures, drug delivery systems, joint replacements, spinal fusion devices and dental restorations. |
Within orthopedics, biomaterials are extensively used in spinal fusion procedures, hip and knee replacements and the repair or stabilization of fractured bones. Currently, Amedica is the only FDA-cleared and ISO 13485 certified silicon nitride medical device manufacturing facility in the world. We believe we are the only provider of ceramics-based medical devices used for spinal fusion applications.
Market Opportunity
Overview
We believe our silicon nitride biomaterial technology platform provides us with numerous competitive advantages in the orthopedic biomaterials market. We market interbody spinal fusion devices and related products and are developing products for use as components in total hip and knee joint replacements, as well as dental applications. We believe we can also utilize our silicon nitride technology platform to develop future products in additional markets, such as the sports medicine, extremities, and trauma markets.
Of the interbody spinal fusion procedures conducted in the United States today, a significant majority utilized interbody devices comprised of PEEK and bone, with occasional use of metals and other materials including ceramics. The market for interbody spinal fusion devices has shifted over time as new biomaterials with superior characteristics have been incorporated into these devices and have launched into the market. We believe the market has reached another inflection point as surgeons and hospitals recognized the limitations of devices currently available. Similarly, we believe our silicon nitride interbody spinal fusion products address the key limitations of other biomaterials currently used in interbody spinal fusion devices and demonstrate superior characteristics needed to improve clinical outcomes.
We believe that the main drivers for growth within the orthopedic biomaterials market and, in particular, the spinal fusion and joint replacement markets, are the following:
| 5 |
| ● | Introduction of New Technologies. Better performing and longer-lasting biomaterials, improved diagnostics, and advances in surgical procedures allow for surgical intervention earlier in the continuum of care and better outcomes for patients. We believe surgical options using better performing and longer-lasting biomaterials will gain acceptance among surgeons and younger patients and drive accelerated growth and increase the size of the spinal fusion and joint replacement markets. | |
| ● | Favorable and Changing Demographics. With the growing number of elderly people, age-related ailments are expected to rise sharply, which we believe will increase the demand and need for biomaterials and devices with improved performance capabilities. Also, middle-aged and older patients increasingly expect to enjoy active lifestyles, and consequently demand effective treatments for painful spine and joint conditions, including better performing and longer-lasting interbody spinal fusion devices and joint replacements. | |
| ● | Market Expansion into New Geographic Areas. We anticipate that demand for biomaterials and the associated medical devices will increase as the applications in which biomaterials are used are introduced to and become more widely accepted in underserved countries, such as Brazil and China. |
The Interbody Spinal Fusion Market
The human spinal canal is made up of 33 interlocking bones, referred to as vertebrae, separated by 23 intervertebral discs comprised of a hard outer ring made of collagen with a soft inner core, that act as shock absorbers between vertebrae. Disorders of the spine can result from degenerative conditions, deformities and trauma or tumor-related damage. Spinal fusion is the standard of care used to treat most spinal disorders and typically involves the placement of an interbody device between vertebrae to reestablish spacing between vertebrae and alignment of the spine. Generally, the interbody device is stabilized by screws and, in some procedures, plates or rods. To enhance bone attachment, surgeons often pack the interbody device with a biomaterial that induces bone growth. Following successful treatment, new bone tissue grows in and around the interbody device over time, which helps fuse the vertebrae and create long-term stability of the interbody device, leading to the alleviation of pain and increase in mobility. We selected this market as the first application for our silicon nitride technology because of the limitations of currently available products, its size, and the key characteristics silicon nitride possesses, which are critical for a superior interbody spinal fusion outcomes.
| ● | Promotion of Bone Growth. The biomaterial should be both osteoconductive and create an osteoinductive environment to promote bone growth in and around the interbody device to further support fusion and stability. Osteoconduction occurs when material serves as a scaffold to support the growth of new bone in and around the material. Osteoinduction involves the stimulation of osteoprogenitor cells to develop, or differentiate, into osteoblasts, which are cells that are needed for bone growth. A material which stimulates bone growth and accelerates fusion rates is ideal in spinal fusion procedures. | |
| ● | Antibacterial. Spinal fusion devices can become colonized with bacteria, which may limit fusion to adjacent vertebrae or cause serious infection. Treating device-related infection is costly and generally requires repeat surgery, including surgery to replace the device, referred to as revision surgery, which may extend hospital stays, suffering and disability for patients. A biomaterial that has antibacterial properties can reduce the incidence of bacteria colonization in and around the interbody device that can lead to infection, revision surgery and associated increased costs. | |
| ● | Imaging Compatibility. The biomaterial should be visible through, and not inhibit the effective use of, common surgical and diagnostic imaging techniques, such as X-ray, CT and MRI. These imaging techniques are used by surgeons during and after spinal fusion procedures to assist in the proper placement of interbody devices and to assess the quality of post-operative bone fusion. | |
| ● | Strength and Resistance to Fracture. The biomaterial should be strong and resistant to fracture during implantation of the device and to successfully restore intervertebral disc space and spinal alignment during the fusion process. The biomaterial should have high flexural strength, which is the ability to resist breakage during bending, and high compressive strength, which is the ability to resist compression under pressure, to withstand the static and dynamic forces exerted on the spine during daily activities over the long term. |
Limitations of Biomaterials used in Interbody Spinal Fusion Devices
The three biomaterials most commonly used in interbody spinal fusion devices are PEEK, human cadaver bone, also referred to as allograft bone, and metals. We believe these materials do not possess the key characteristics required to form the optimal interbody spinal fusion device and are susceptible to potential fracture, implant-related infection, pain, limited fusion and instability, which have resulted in revision surgeries.
| 6 |
PEEK (polyetheretherketone)
We believe PEEK is the most frequently used biomaterial for interbody spinal devices and accounted for the majority of interbody spinal devices implanted in the United States in 2016. We believe PEEK has the following limitations:
| ● | Restricts Bone Growth. Due to PEEK’s hydrophobic nature, the human body may recognize PEEK as a foreign substance and, therefore, may encapsulate the device with fibrous tissue. Although it is still possible for bone to grow through the device, bone may not adhere to the surface of the device if this tissue develops. This fibrous layer could cause a non-fusion, allow bacterial colonization, and/or potentially lead to costly revision surgery. | |
| ● | Lacks Imaging Compatibility. PEEK is invisible on X-rays. As a result, manufacturers of PEEK devices add metal markers to their devices so surgeons can see the general location of the devices by X-ray. These markers, however, do not show the full outline of the device, which makes it difficult to assess the accuracy of the placement of the device. In addition, the metal markers cause artifacts on CT and MRI that can compromise the quality of the image. | |
| ● | Lacks Strength and Resistance to Fracture. PEEK lacks sufficient flexural strength, compressive strength and resistance to fracture necessary to reduce the risk of deformity or fracture during the fusion process. In addition, PEEK devices may fracture during implantation in certain interbody spinal fusion procedures. For example, in December 2012, Zimmer Spine recalled its PEEK Ardis® Interbody System Inserter, a surgical instrument used to implant a PEEK interbody spinal fusion device, because it resulted in the PEEK implants being susceptible to breakage when too much lateral force was applied to the inserter during implantation. Due to radiographic X-rays being the most common way for surgeons to assess fusion, and PEEK being invisible on X-rays, it is extremely difficult to clearly assess the extent to which fracture rates occur with PEEK interbody fusion devices. | |
| ● | Lacks Antibacterial Properties. PEEK does not have any inherent antibacterial properties. In fact, a biofilm may form around a PEEK device after implantation, which could allow for the colonization of bacteria, leading to infection and costly revision surgeries. |
Allograft Bone
We believe allograft bone was the second most frequently used biomaterial in interbody spinal fusion devices in the United States in 2016. We believe allograft bone has the following limitations:
| ● | Limited Promotion of Bone Growth. Allograft bone has limited osteoinductive characteristics and therefore may not effectively promote bone growth in and around the interbody device. | |
| ● | Inconsistent Quality. Generally, allograft bone is not as strong as live bone within the body or other materials used in interbody devices. Because the cadaveric bone can be harvested from a wide variety of sources, this often leads to inconsistent patient outcomes. Allograft bone is subject to inconsistent quality and size, which may require surgeons to make compromises on the fit of the device during surgery. In addition, techniques used to sterilize allograft bone, like gamma irradiation, can cause the allograft to become brittle and more susceptible to fracture. | |
| ● | Lacks Antibacterial Properties and Risk of Disease Transmission. In addition to not having inherent antibacterial properties, allograft bone exposes patients to a greater risk of disease transmission and an adverse auto-immune response. |
Metals
We believe metal interbody devices accounted for a fraction of the devices implanted in the United States in 2016. We believe metal-based interbody fusion devices have the following limitations:
| ● | Limited Promotion of Bone Growth. Metals have limited osteoinductive characteristics and therefore do not effectively promote bone growth in and around the interbody device. | |
● |
Lack Antibacterial Properties. Metals do not have inherent antibacterial properties and do not suppress the colonization of bacteria in and around the device, which can lead to infection and/or costly revision surgeries. | |
| ● | Lack Imaging Compatibility. Metals are opaque in X-rays and can cause significant imaging artifacts in CTs and MRIs. This can make it difficult for surgeons to detect the extent and quality of bone growth in and around the device in post-operative diagnostic imaging procedures. |
| 7 |
The Hip and Knee Joint Replacement Market
Total joint replacement involves removing the diseased or damaged joint and replacing it with an artificial implant consisting of components made from several different types of biomaterials. The key components of a total hip implant include an artificial femoral head, consisting of a ball mounted on an artificial stem attached to the femur, and a liner, which is placed inside a cup affixed into the pelvic bone. The femoral head and liner move against each other to replicate natural motion in what is known as an articulating implant. Total knee replacement implants also use articulating components and are comprised of the following four main components: a femoral condyle, which is a specially shaped bearing that is affixed to the lower end of the femur; a tibial tray that is affixed to the upper-end of the tibia; a tibial insert that is rigidly fixed to the tibial tray and serves as the surface against which the femoral condyle articulates; and a patella, or knee cap, which also articulates against the femoral condyle.
Implants for total hip and knee replacements are primarily differentiated by the biomaterials used in the components that articulate against one another. The combinations of biomaterials most commonly used in hip and knee replacement implants in the United States are metal-on-cross-linked polyethylene and traditional oxide ceramic-on-cross-linked polyethylene. The use of hip replacement implants incorporating metal-on-metal and traditional oxide ceramic-on-traditional ceramic biomaterials experienced a steep decline in the United States over the last several years due to their significant limitations. We believe that the most commonly used biomaterials in joint replacement implants also have limitations, and do not possess all of the following key characteristics required for optimal total joint replacement implants:
| ● | Resistance to Wear. The biomaterials should have sufficient hardness and toughness, as well as extremely smooth surfaces, to effectively resist wear. Because the articulating implants move against each other, they are subject to friction, which frequently leads to abrasive wear and the release of small wear particles. This may cause an inflammatory response which results in osteolysis, or bone loss. Surgeons have identified osteolysis as a leading cause of joint implant failure, resulting in the need for costly revision surgery to replace the failed implant. One of the most commonly used combinations of biomaterials, metal-on-cross-linked polyethylene, as well as metal-on-metal implants, tends to generate a large number of metal wear particles, which can cause osteolysis and a moderate to severe allergic reaction to the metal, referred to as metal sensitivity. While less common, metal implants may also cause a serious medical condition called metallosis, which involves the deposition and build-up of metal debris in the soft tissues of the body. Both metal sensitivity and metallosis can result in revision surgery. In addition, we believe traditional oxide ceramics currently used in total joint replacements accelerate wear of the cross-linked polyethylene liner as compared to our non-oxide ceramic composition found in our silicon nitride biomaterial platform. | |
● |
Non-Corrosive. The biomaterials should be non-corrosive and should not cause adverse patient reactions. Metal placed in the human body corrodes over time and also results in the formation of metal ions, which leads to metal sensitivity in approximately 10% to 15% of the population and, less commonly, metallosis. As a result, there are significant increased risks from using metal-on-cross-linked polyethylene and metal-on-metal implants. | |
● |
Hardness, Strength and Resistance to Fracture. The biomaterials should be hard, strong and resistant to fracture to adequately bear the significant loads placed on the hip and knee joints during daily activities. We believe there are strength limitations associated with traditional oxide ceramic-on-cross-linked polyethylene and traditional oxide ceramic-on-traditional oxide ceramic implants. | |
● |
Antibacterial. The biomaterials should have antibacterial properties to reduce the risk of bacteria colonization, infection, revision surgeries and associated increased costs. None of the most commonly used biomaterials in joint replacement implants have antibacterial properties. |
Our Silicon Nitride Technology Platform
We believe we are the only FDA-cleared and ISO 13485 certified silicon nitride medical device manufacturing facility in the world, and the only provider of ceramics-based medical devices used for spinal fusion applications. Silicon nitride is a chemical compound comprised of the elements silicon and nitrogen, with the chemical formula Si3N4. Silicon nitride, an advanced ceramic, is lightweight, resistant to fracture and strong, and is used in many demanding mechanical, thermal and wear applications, such as in space shuttle bearings, jet engine components and body armor.
We believe our silicon nitride is ideally suited for use in many medical applications and has the following characteristics that make it superior to other biomaterials, including PEEK, bone, metal and traditional oxide ceramics, which do not possess all of these characteristics:
| 8 |
| ● | Promotes Bone Growth. Our silicon nitride is osteoconductive through its inherent surface topography that provides scaffolding for new bone growth. We believe our silicon nitride promotes an ideal environment for osteoinduction. As a hydrophilic material, silicon nitride attracts protein cells and nutrients that stimulate osteoprogenitor cells to differentiate into osteoblasts, which are needed for optimal bone growth environments. Our silicon nitride has an inherent surface chemistry that is more similar to bone than PEEK and metals. These properties are highlighted in an in vivo study, where we measured the force required to separate devices from the spine after being implanted for three months, which indicates the level of osteointegration. In the absence of bacteria, the force required to separate our silicon nitride from its surrounding bone was approximately three times that of PEEK, and nearly two times that of titanium. In the presence of bacteria, the force required to separate our silicon nitride from its surrounding bone was over five times that of titanium, while there was effectively no separation force required for PEEK, indicating essentially no osteointegration. |
| ● | Hard, Strong and Resistant to Fracture. Our silicon nitride is hard, strong and possesses superior resistance to fracture over traditional ceramics and greater strength than polymers currently on the market. For example, our silicon nitride’s flexural strength is more than five times that of PEEK and our silicon nitride’s compressive strength is over twenty times that of PEEK. Unlike PEEK interbody spinal fusion devices, we believe our silicon nitride interbody spinal fusion devices can withstand the forces exerted during implantation and daily activities over the long term. |
| ● | Antibacterial. We have demonstrated in in vitro and in vivo studies that silicon nitride has inherent antibacterial properties, which reduce the risk of infection in and around a silicon nitride device. PEEK, traditional ceramics, metals and bone do not have inherent antibacterial characteristics. These properties were highlighted in an in vitro study (Acta Biomater. 2012 Dec;8(12):4447-54. doi: 10.1016/j.actbio.2012.07.038. Epub 2012 Jul 31.), where live bacteria counts were between 8 and 30 times lower on our silicon nitride than PEEK and up to 8 times lower on our silicon nitride than titanium. In addition to improving patient outcomes, we believe the antibacterial properties of our silicon nitride should make it an attractive biomaterial to hospitals and surgeons who are not reimbursed by third-party payors for the treatment of hospital-acquired infections. Additionally, silicon nitride is synthetic and, therefore, there is a lower risk of disease transmission through cross-contamination or of an adverse auto-immune response, sometimes associated with the use of allograft bone. |
| ● | Imaging Compatible. Our silicon nitride interbody spinal fusion devices are semi-radiolucent, clearly visible in X-rays, and produce no distortion under MRI and no scattering under CT. These characteristics enable an exact view of the device for precise intra-operative placement and post-operative bone fusion assessment in spinal fusion procedures. These qualities provide surgeons with greater certainty of outcomes with our silicon nitride devices than with other biomaterials, such as PEEK and metals. |
| ● | Resistant to Wear. We believe our silicon nitride joint implant product candidates could have higher resistance to wear than metal-on-cross-linked polyethylene and traditional oxide ceramic-on-cross-linked polyethylene joint implants, the two most commonly used total hip replacement implants. Wear debris associated with metal implants increases the risk of metal sensitivity and metallosis. It is a primary reason for early failures of metal and polymer articulating joint components. |
| ● | Non-Corrosive. Our silicon nitride does not have the drawbacks associated with the corrosive nature of metal within the body, including metal sensitivity and metallosis, nor does it result in the release of metal ions into the body. As a result, we believe our silicon nitride products will have lower revision rates and fewer complications than comparable metal and traditional oxide ceramic products. |
Our Forms of Silicon Nitride
The chemical composition of our in-house formulation of silicon nitride and our processing and manufacturing experience allow us to produce silicon nitride in four distinct forms. This capability provides us with the ability to utilize our silicon nitride biomaterial in a variety of ways depending on the intended application, which, together with our silicon nitride’s key characteristics, distinguishes us from manufacturers of products using other biomaterials.
| 9 |
We currently produce silicon nitride for use in our commercial products and product candidates in the following forms:
| ● | Solid Silicon Nitride. This form of silicon nitride is a fully dense, load-bearing solid used for devices that require high strength, toughness, fracture resistance and low wear, including interbody spinal fusion devices, hip and knee replacement implants, and dental implants. |
 | |
● |
Porous Silicon Nitride. While this form of silicon nitride has a chemical composition that is identical to that of our monolithic solid silicon nitride, this formulation has a porous structure, which is engineered to mimic cancellous bone, the spongy bone tissue that typically makes up the interior of human bones. Our porous silicon nitride has interconnected pores ranging in size between about 90 and 600 microns, which is similar to that of cancellous bone. This form of silicon nitride can be used for the promotion of bone in-growth and attachment. We believe our porous silicon nitride can act as a substitute for the orthobiologics currently used to fill interbody devices in an effort to stimulate fusion, as a bone void filler, and as a porous scaffold for medical devices. |
 | |
| ● | Composite Silicon Nitride. This form of silicon nitride is a combination, or composite, of our solid monolithic and porous formulations of silicon nitride. This composite may be used to manufacture devices and implants that mimic the structure of natural bone by incorporating both a fully dense, load-bearing solid component on the outside and a porous component intended to promote bone in-growth on the inside. This composite form of silicon nitride is used in interbody spinal fusion devices and can be used in components for total hip and knee replacement implants. |
 | |
● |
Silicon Nitride Coating. With a similar chemical composition as our other forms of silicon nitride, this form of silicon nitride can be applied as an adherent coating to metallic substrates, including cobalt-chromium, titanium and steel alloys. We believe applying an extremely thin layer of silicon nitride as a coating may provide a highly wear-resistant articulation surface, such as on femoral heads, which may reduce problems associated with metal or polymer wear debris. We also believe that the silicon nitride coating can be applied to devices that require firm fixation and functional connections between the device or implant and the surrounding tissue, such as hip stems and screws. The use of silicon nitride coating may also create an antibacterial barrier between the device and the adjacent bone or tissue. |
 |
| 10 |
Our Competitive Strengths
We believe we can use our silicon nitride technology platform to become a leading biomaterial company and have the following principal competitive strengths:
| ● | Sole Provider of Silicon Nitride Medical Devices. We believe we are the only company that designs, develops, manufactures and sells medical grade silicon nitride-based products. Due to its key characteristics, we believe our silicon nitride enables us to offer new and transformative products across multiple medical specialties. In addition, with the FDA clearance of our silicon nitride Valeo products, we are the only company to develop and manufacture a ceramic for use in FDA cleared spinal fusion medical devices in the United States. |
● |
In-House Manufacturing Capabilities. We operate a 30,000 square foot manufacturing facility located at our corporate headquarters in Salt Lake City, Utah. This operation complies with the FDA’s quality system regulation, or QSR, and is certified under the International Organization for Standardization’s, or ISO, standard 13485 for medical devices. This facility allows us to rapidly design and produce silicon nitride products, while controlling the entire manufacturing process from raw material to finished goods. We have also entered to a manufacturing, development and supply agreement with Kyocera Industrial Ceramics Corporation, or Kyocera, under which Kyocera has become a qualified secondary manufacturer of our silicon nitride-based spinal fusion products and product candidates. |
| ● | Established Commercial Infrastructure. We market and sell our products to surgeons and hospitals in the United States and select markets in Europe and South America through our established network of more than 50 independent sales distributors who are managed by our experienced in-house sales and marketing management team. Our control over the sales and marketing processes also allows us greater flexibility to selectively collaborate with distributors when we believe their experience or geographic reach can be beneficial to us. |
● |
Portfolio of Non-Silicon Nitride Products. In addition to designing, developing, manufacturing and commercializing silicon nitride interbody spinal fusion devices, we sell a complementary line of non-silicon nitride spinal fixation products. We offer a full suite of spinal fusion solutions, which increases our access to surgeons and hospitals, and allows us to more effectively market our silicon nitride spinal fusion products to our customers. Product revenue from the sale of these non-silicon nitride products also supports further development of our silicon nitride products and product candidates. |
| ● | Highly Experienced Management and Surgeon Advisory Team. Members of our management team have experience in product development, launching of new products into the orthopedics market and selling to hospitals through direct sales organizations, distributors, manufacturers and other orthopedic companies. We also collaborate with a network of leading surgeon advisors in the design, development and use of our silicon nitride products and product candidates. |
Our Strategy
Our goal is to become a leading biomaterial company focused on using our silicon nitride technology platform to develop, manufacture and commercialize a broad range of medical devices. Key elements of our strategy to achieve this goal are the following:
| ● | Drive Further Adoption of our Silicon Nitride Interbody Spinal Fusion Devices. We believe that increasing the awareness of our silicon nitride technology by educating surgeons about its key benefits, and the design improvements to our silicon nitride products and related instruments will accelerate the adoption of our products and ultimately help improve patient outcomes. To drive further awareness of our products and the associated benefits offered by our silicon nitride technology, we will continue to educate surgeons through multiple channels, including industry conferences and meetings, media outlets and through our sales and marketing efforts. |
| ● | Continue Establishing and Cultivating OEM and Private Label Partnerships. Because we believe silicon nitride is a superior platform and technology for application in the spine, total joint, dental, and extremities markets, we have established, and will seek to establish, additional partnerships with other medical device companies to replace their inferior materials and products with products manufactured from silicon nitride. For example, under an OEM arrangement, we would manufacture the company’s spinal fusion implant designs with silicon nitride and leverage their existing instrumentation, allowing the company to convert their existing line of spinal fusion devices with limited capital expenditures. Additionally, a private label arrangement would allow our partners to sell Amedica’s Valeo line of silicon nitride interbody spinal fusion devises under their own brand name. The private label agreements typically provide a quicker pathway to revenue as compared to the OEM arrangements. |
| 11 |
| ● | Enhance our Commercial Infrastructure. We expect to increase the productivity of our sales and marketing team by continuing to engage experienced independent sales distributors with strong orthopedic surgeon relationships. For example, we have entered into a European sales agent agreement with K2M, Inc. as well as a sales agent agreement with a Brazilian medical device distributor to distribute our Valeo line of silicon nitride interbody implants. We may also establish distribution collaborations in the United States and abroad when access to large or well-established sales and marketing organizations may help us gain access to new markets, increase sales in our existing markets, or accelerate market penetration for selected products. |
| ● | Develop Silicon Nitride for Total Joint Components. We are incorporating our silicon nitride technology into silicon nitride-coated metal components and solid silicon nitride components for use in total hip and knee replacement product candidates that we plan to develop in collaboration with a strategic partner. We are also working with the FDA to define the regulatory pathway required for development and commercialization of these components. |
| ● | Apply our Silicon Nitride Technology Platform to Other OEM Opportunities. Our silicon nitride technology platform is adaptable and we believe it may be used to develop products to address other significant opportunities, such as in the dental, extremities, sports medicine, cardiovascular and trauma markets. We have manufactured prototypes of dental implants, extremities, sports medicine and trauma products, and have developed a process to coat metals with our silicon nitride to enhance current medical devices and instruments. We plan to collaborate with other companies to develop and commercialize future products in these areas. |
Spinal Fusion Products and Product Candidates
Our Valeo Silicon Nitride Products and Product Candidates
Our first generation Valeo silicon nitride spinal fusion device received 510(k) regulatory clearance and a CE mark in 2008. Based on surgeon feedback for our first generation spinal fusion devices, we developed a second generation of Valeo products. In 2012, we received 510(k) clearance to market this second generation family of Valeo interbody spinal fusion devices which we launched with a select number of surgeons in 2013. Our second generation Valeo interbody spinal fusion devices offer distinct improvements over the first generation. The instrumentation of the second generation devices allow for better control of the device during implantation. The device allows for improved stability and potentially improved fusion after implantation and is offered in a broad selection of sizes. We completed the full launch of our second generation AL, PL, OL and TL Valeo interbody spinal fusion devices in the United States in 2014, our second generation LL Valeo interbody spinal fusion devices in August 2015 and our second generation C Valeo interbody spinal fusion devices in February 2016.
Our current products are:
| Valeo Interbody Fusion Devices | Generation | |
| AL: Anterior Lumbar | 1st and 2nd | |
| PL: Posterior Lumbar | 1st and 2nd | |
| OL: Oblique Lumbar | 1st and 2nd | |
| TL: Transforaminal Lumbar | 1st and 2nd | |
| LL: Lateral Lumbar | 2nd | |
| C: Cervical | 1st and 2nd | |
| CORP: Corpectomy | 1st |
In 2009, we received a CE Mark to commercialize the Valeo interbody spinal fusion devices made from our composite silicon nitride. The porous silicon nitride center of these devices is designed to facilitate bone growth into the device, which we believe will allow surgeons to reduce or eliminate the use of allograft bone and other osteoconductive biomaterials. We are currently marketing these devices in the Netherlands, Spain and Germany. Additionally, we conducted a prospective clinical trial in Europe, named CASCADE, comparing our Valeo composite silicon nitride interbody devices to PEEK interbody devices filled with autograft bone to obtain additional safety and efficacy data to support a 510(k) clearance in the United States. The CASCADE study enrolled 104 patients in a prospective clinical trial that independently scored fusion rates and clinical outcomes at 12 and 24 months follow-up. Neck Disability Index scores decreased similarly in both patient groups, consistent with clinical improvements reported in the literature. Importantly, the incidence of cervical spine fusion was statistically identical between study groups, and consistent with figures reported in other studies. The company previously submitted to the FDA a 510(k) premarket application to commercialize a composite silicon nitride implant. The device was not cleared for commercialization by the FDA, even though the device is marketed in Europe with successful outcomes. We have since re-filed a 510(k) premarket application with the FDA, for a modified CsC-based cervical implant that is comparable to our own commercially-available Valeo®C cervical implants.
| 12 |

Valeo Composite (Monolithic + Porous Silicon Nitride)
Our Non-Silicon Nitride Spinal Fixation Products
We sell a line of complementary non-silicon nitride spinal fixation products to provide surgeons and hospitals with a broader range of products. Product revenue from the sale of our non-silicon nitride spinal fixation products further supports development of our silicon nitride products and product candidates. In November 2016 the FDA notified the company that the new pedicle screw system, which we refer to as our Taurus™ Pedicle Screw System, was cleared for commercialization. The Taurus Pedicle Screw System is intended to immobilize and stabilize the spinal segments to supplement fusion of the lumbar and/or sacral spine. Taurus™ is a modular degenerative system, connecting strength with intra-operative flexibility. The Taurus™ modular screw can be attached in-situ facilitating screw-to-screw distraction, improving disc space visualization. The dual-lead screw design maintains a rapid insertion speed, while improving screw pullout strength. Additionally, the tension head-body holds its position at any angle, and the patented helical flange technology eliminates head splay and cross-threading.
Our Total Hip and Knee Joint Replacement Product Candidates
Our Total Hip Implant Product Candidates
We have developed a femoral head that is made from our solid silicon nitride, which could be used for total hip replacement product candidates. This femoral head is expected to articulate against a cross-linked polyethylene liner fixed into a metal acetabular cup. Together with a strategic partner, we have initiated biomechanical testing of our solid silicon nitride femoral heads. The results of this test will be released in 2017. If the tests indicate that silicon nitride femoral heads are superior in terms of wear performance, taper corrosion, strength and in vitro hydrothermal stability, we eventually intend to commercialize this product in cooperation with a strategic partner. However, clearance of these types of devices by the FDA will be required. Currently, the FDA has indicated that a limited one to two year clinical trial may be necessary to obtain clearance. If clearance is eventually obtained, we intend to commercially launch products for use in total hip replacement in 2018 or 2019.
Our Total Knee Implant Product Candidates
We have developed a femoral condyle design made from our solid silicon nitride. The femoral condyle component will attach to the lower end of the femur. The femoral condyle is expected to articulate against a cross-linked polyethylene tibial insert that will attach to the tibial tray at the upper end of the tibia, which we expect will be made from metal. We have successfully made prototypes of this design. Following the potential clearance of the femoral head components (discussed above), we intend to initiate biomechanical testing with a strategic partner for silicon nitride components for use in knee replacement procedures to support a 510(k) submission to the FDA. If this clearance is eventually obtained, we intend to commercialize our products for use in total knee replacement surgeries post-FDA clearance.
Other Product Opportunities
Our silicon nitride technology platform is adaptable and we believe it may be used to develop products to address other significant opportunities, such as in the dental, extremities, sports medicine and trauma markets.
We have entered into a joint development agreement with a dental implant design company and distributor of dental technologies for the development of a silicon nitride based dental implant system and devices.
We also believe our coating technology may be used to enhance our marketed metal products as well as other commercially available metal spinal fusion and joint replacement products. We have produced feasibility prototypes of dental implants, other components for use in total hip implants in addition to our total hip and knee implant product candidates discussed above, a suture anchor for sports medicine applications, an osteotomy wedge for extremities applications, and prototypes of silicon nitride-coated plates for potential trauma applications. We have also developed a process to apply our silicon nitride as a coating on other biomaterials.
| 13 |
The FDA has not evaluated any of these potential products and we are not currently advancing the development of any of these product candidates. We plan to collaborate with medical device companies to complete the development of and commercialize any product candidates we advance in these areas or develop any one of them ourselves if sufficient resources should become available.
Supporting Data
We and a number of independent third parties have conducted extensive biocompatibility, biomechanical, in vivo and in vitro testing on our silicon nitride composition to establish its safety and efficacy in support of regulatory clearance of our biomaterial, products and product candidates. We have also completed additional testing of our silicon nitride products and product candidates. The results of this testing have been published in peer reviewed publications. We believe our product development strategy is consistent with the manner in which other biomaterials have been successfully introduced into the market and adopted as the standard of care. Listed below is an overview of some of the key testing completed on our silicon nitride biomaterial, products and product candidates to date, as well as other information about our silicon nitride and other biomaterials.
Biocompatibility
Before our silicon nitride was cleared by the FDA in 2008, we conducted a series of biocompatibility tests following the guidelines of the FDA and ISO and submitted the results to the FDA as part of the regulatory clearance process. These tests confirmed that our silicon nitride products meet required biocompatibility standards for human use.
Promotion of Bone Growth
In 2012, we conducted two separate studies at Brown University, the results of which suggest that the chemistry and inherent surface topography of our solid silicon nitride provides an optimal environment for bone growth onto and around the device.
The first study was a series of in vitro analyses of protein adsorption, or presence on the surface of the biomaterial, onto silicon nitride, PEEK and titanium. The results of this study indicated that adsorption of two key proteins necessary for bone growth (fibronectin and vitronectin) were up to eight times greater on our silicon nitride than on PEEK, and up to four times greater than on titanium. A third important protein (laminin) had up to two times greater adsorption on our silicon nitride than on PEEK, and up to two-and-one-half times greater adsorption than on titanium.
The second study was an in vivo investigation of the osteointegration characteristics of these same three biomaterials after they had been surgically implanted into the skulls of laboratory rats. This study included an examination of the effect of Staphylococcus epidermidis bacteria on osteointegration. At time intervals of up to three months after implantation of the biomaterial, the amount of new bone growth within the surgical site and in direct contact with the implanted biomaterial was evaluated. In the absence of bacteria, new bone formation within the surgical site surrounding our silicon nitride was approximately 69%, compared with 36% and 24% for titanium and PEEK, respectively. Similarly, bone in direct contact, or apposition, with our silicon nitride, titanium and PEEK was 59%, 19% and 8%, respectively. As is common, in the presence of bacteria, new bone formation within the surgical site was suppressed, but still significantly greater for our silicon nitride than for the other two biomaterials. Observed new bone growth within the surgical site surrounding our silicon nitride was 41%, compared with 26% and 21% for titanium and PEEK, respectively. At the implant interface, the bone apposition for our silicon nitride, titanium and PEEK was 23%, 9% and 5%, respectively. To further characterize the extent of osteointegration, the force needed to separate each implant from its surrounding bone was measured. A larger force needed to separate the implant is an indication of improved osteointegration. At three months after implantation, in the absence of bacteria, the force required to separate our silicon nitride from its surrounding bone was approximately three times that of PEEK, and nearly two times that of titanium. In the presence of bacteria, there was effectively no separation force required for PEEK, indicating essentially no osteointegration. Our silicon nitride required over five times the force to separate it from its surrounding bone in the presence of bacteria in comparison to titanium.
In 2008, we conducted an animal study in which we evaluated the level of osteointegration of our porous silicon nitride with a knee-defect model in adult sheep. At three months after implantation, three out of five of the silicon nitride implants had extensive new bone formation at and into the implant surface, showing that the bone had grown into our porous silicon nitride to a depth of 3 millimeters, or mm. This animal study demonstrated the rapid osteointegration potential of our porous silicon nitride composition.
| 14 |
Hardness, Strength and Resistance to Fracture
Comparative Information
As shown in the table of comparative information publicly available about various biomaterials below:
| ● | the hardness, or a material’s resistance to deformity, of silicon nitride is comparable to traditional ceramics, but is substantially higher than either polymers or metals; | |
● |
the strength of silicon nitride is comparable or higher than metals and traditional ceramics, and is about sixteen to fifty-five times stronger than highly-cross-linked polyethylene, and four to eight times stronger than PEEK; and | |
| ● | silicon nitride has the highest fracture resistance of any medical ceramic material and is three to eleven times more resistant to fracture than PEEK or highly-cross-linked polyethylene. This is due to the interwoven microstructure of silicon nitride. Metals have the highest fracture resistance. |
Comparison of Mechanical Properties Among Orthopedic Biomaterials
| Material | Hardness (GPa)(1) | Strength (MPa)(1) | Fracture Resistance (MPam1/2)(1) | |||||||||
| Silicon Nitride | 13 – 16 | 800 – 1200 | 8 – 11 | |||||||||
| Aluminum Oxide Ceramic | 14 – 19 | 300 – 500 | 3 – 5 | |||||||||
| Zirconia-Toughened Alumina Ceramic | 12 – 19 | 700 – 1150 | 5 – 10 | |||||||||
| PEEK | 0.09 – 0.28 | 160 – 180 | 2 – 3 | |||||||||
| Highly-Cross-Linked Polyethylene Polymer | 0.03 – 0.07 | 22 – 48 | 1 – 2 | |||||||||
| Cobalt-Chromium Metal | 3 – 4 | 700 – 1000 | 50 – 100 | |||||||||
| Titanium Alloy Metal | 3 – 4 | 920 – 980 | 75 | |||||||||
| (1) | GPa is a giga-pascal. Pascals are a measure of pressure. MPam1/2 is mega-pascal times a square root meter and is a measure related to the energy required to initiate fracture of a material. |
We believe that the combination of high hardness, strength and fracture resistance positions our silicon nitride as an ideal biomaterial for many medical applications.
Burst Strength
In 2006, we conducted in-house comparative “burst strength” tests on femoral heads made from our silicon nitride produced by a contract manufacturer to our specifications and femoral heads made from one of the strongest commercially available ceramics, BIOLOX® delta (zirconia-toughened alumina). These tests were performed on three designs of 28 mm femoral heads using accepted testing protocols. The tests involved applying a load to each femoral head while mounted on a cobalt-chromium simulated hip implant stem, until the head burst. This enabled us to directly compare the strength of the femoral heads made of the two biomaterials. The results also provided an indication of each biomaterial’s resistance to fracture. The results of these tests are shown in the chart below.
| 15 |
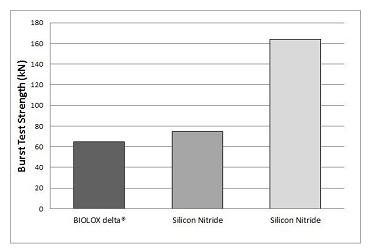
The average burst test strength for the silicon nitride femoral heads in these tests was 75 kilonewtons, or kNs, compared with 65 kN for BIOLOX® delta, or about a 15% improvement. The burst strengths observed in our tests for BIOLOX® delta femoral heads are comparable to those observed by an independent party testing the same design BIOLOX® delta femoral heads as we did. We also conducted burst strength tests of 36 mm femoral heads made from our silicon nitride which showed those femoral heads had burst strengths that averaged 164 kN.
Resistance to Wear
In 2011, we commissioned an independent laboratory to conduct a wear study using our silicon nitride femoral heads. We tested our 28 mm silicon nitride femoral heads articulated against cross-linked polyethylene acetabular liners and our 40 mm silicon nitride femoral heads articulated against cross-linked polyethylene acetabular liners using well-established protocols in a hip simulator for their wear performance over 5 million cycles. We then compared the results for our silicon nitride product candidates to the results for the cobalt chrome femoral head and publicly available data from other commonly paired products. The results and comparison showed that:
| ● | our silicon nitride-on-cross-linked polyethylene had approximately half the wear rate of that publicly reported for cobalt chrome-on-cross-linked polyethylene articulating hip components; and | |
● |
our silicon nitride-on-cross-linked polyethylene had comparable wear to that publicly reported for traditional oxide ceramic-on-cross-linked polyethylene articulating hip components. |
Antibacterial Properties
The results of the two studies at Brown University in 2012, demonstrate that our solid silicon nitride has antibacterial properties. The objective of the in vitro study was to determine how our silicon nitride, PEEK and titanium interact with bacteria, protein and bone cells without the use of antibiotics and compared the growth of five different types of bacteria on silicon nitride, PEEK and titanium over time. Live bacteria counts were between 8 to 30 times lower on silicon nitride than PEEK and up to 8 times lower on silicon nitride than titanium.
In the in vivo study, bacteria were applied to the biomaterials before implantation. Three months after implantation, no infection was observed with silicon nitride, whereas both PEEK and titanium showed infection. The data demonstrate that our silicon nitride inhibits biofilm formation and bacterial colonization around the biomaterial.
Imaging Compatibility
In 2007, we conducted a study to compare the imaging characteristics of test blanks made of PEEK, the metals titanium and tantalum, and silicon nitride using a cadaver human vertebral body. Images of the vertebral body and the blanks were obtained using X-ray, CT and MRI under identical conditions. We assessed the radiolucent characteristics of the blanks in X-ray images quantitatively, assessed the presence of scatter in CT qualitatively and assessed distortion in MRI quantitatively. In X-ray, the metal blanks did not permit visualization of the underlying bone of the vertebral body, while PEEK was transparent, rendering its location difficult to determine. The silicon nitride blank had an intermediate radiolucency that rendered it visible and allowed a visual assessment of the underlying bone of the vertebral body. CT and MRI of the metal blanks indicated the presence of distortion while silicon nitride and PEEK exhibited no scattering.
| 16 |
Sales and Marketing
We market and sell our products to surgeons and hospitals through our established network of more than 50 independent sales distributors who are managed by our experienced in-house sales and marketing management team. Our sales efforts to-date have been in the United States and select markets in Europe and South America. To supplement our independent sales distributors, in select international markets, such as Europe, China, Japan, Australia, Latin America and Canada, we may also seek to establish collaborations with leading orthopedic companies where we believe that a large, well-established partner may provide better access to those markets. For example, we have entered into a European sales agent agreement with K2M, Inc., a distribution agreement with a Brazilian medical device distributor for distribution of our Valeo line of products in Brazil, and we are working with strategic partners in China and Japan to gain approval of our products for commercialization in those countries. In addition, we may establish collaborations in the United States under circumstances where access to a larger sales and marketing organization may help to expand the market or accelerate penetration for selected products.
In addition to leveraging the strong existing surgeon relationships of our distribution network, we market our products through a combination of initiatives that are designed to establish and increase awareness of our silicon nitride products and their benefits over alternative products. We attend and make presentations at major industry events, including society meetings, to educate surgeons and distributors about our products and product candidates. We advertise in trade journals and publications, and offer unique pricing strategies, including product bundling and incentivizing our distribution network to create and maintain long-term relationships with surgeons and hospitals. We also use surgeon advisors to assist in product development and to help implement awareness campaigns aimed at educating surgeons about our products. As part of these campaigns, we provide educational materials for hospitals and surgeons. We also conduct regional training seminars where our product managers, trainers, engineers, sales and marketing staff members work together with our surgeon advisors to educate surgeons and our distribution network in the use of our products.
Original Equipment Manufacturing and Private Label
In addition to the markets into which we directly sell our products, we are utilizing our silicon nitride technology platform to expand our current penetration in the spinal fusion market through original equipment manufacturer (“OEM”) and private label partnerships. To that effect, we have entered into both a private label agreement and an OEM agreement with Spinal Kinetics, a privately-held medical device company focused on developing innovative and practical motion preservation systems for treating degenerative diseases of the spine. Pursuant to the private label agreement, Spinal Kinetics sells our Valeo line of products under their own label. Pursuant to the OEM agreement, we will be working together with Spinal Kinetics to develop their proprietary spinal implants to be manufactured with silicon nitride. We have also entered into private label agreements with BoTEC Medical, a Chinese orthopedic company, as well with another US regional medical device company providing for the distribution of our Valeo line of silicon nitride interbody fusion devises under their respective proprietary brand names. We also expect to do the same in other markets such as total hip and knee joint replacements, dental, extremities, and sports medicine. We believe our biomaterial expertise, strong intellectual property and formulaic manufacturing process will allow us to transition currently available medical device products made of inferior biomaterials and manufacture them using silicon nitride and our technology platform to improve their characteristics.
Manufacturing
Silicon Nitride Manufacturing
To control the quality, cost and availability of our silicon nitride products and product candidates, we operate our own manufacturing facility. Our 54,000 square foot corporate building includes a 30,000 square foot ISO 13485 certified medical device manufacturing space. It is equipped with state-of-the-art powder processing, spray drying, pressing and computerized machining equipment, sintering furnaces, and other testing equipment that enables us to control the entire manufacturing process for our silicon nitride products and product candidates. To our knowledge, we are the only vertically integrated silicon nitride orthopedic medical device manufacturer in the world. All operations with the exceptions of raw material production, cleaning, packaging and sterilization are performed in-house. We purchase raw materials, consisting of silicon nitride ceramic powder and dopant chemical compounds, from several vendors which are ISO registered and approved by us. These raw materials are characterized and tested in our facility in accordance with our specifications and then blended to formulate our silicon nitride. We believe that there are multiple vendors that can supply us these raw materials and we continually monitor the quality and pricing offered by our vendors to ensure high quality and cost-effective supply of these materials.
In June 2014, we also entered to a manufacturing, development and supply agreement with Kyocera Industrial Ceramics Corporation, or Kyocera, under which Kyocera has become a qualified secondary manufacturer of our silicon nitride-based spinal fusion products and product candidates.
| 17 |
Non-Silicon Nitride and Instruments Manufacturing
We obtain our non-silicon nitride spinal fixation products and instruments from third-party manufacturers. We also plan to rely on third-party manufacturers for the supply of the metal components of our silicon nitride hip and knee joint replacement product candidates. We only use manufacturers that operate under QSR and are ISO 13485 certified. Our in-house quality control group examines subcontracted components to ensure that they meet our required specifications. We believe that the use of third-party sources for non-silicon nitride spinal fixation products and instruments will reduce our capital investment requirements and allow us to strategically focus our resources on the manufacture of our silicon nitride products and product candidates.
Intellectual Property
We rely on a combination of patents, trademarks, trade secrets, nondisclosure agreements, proprietary information ownership agreements and other intellectual property measures to protect our intellectual property rights. We believe that in order to have a competitive advantage, we must continue to develop and maintain the proprietary aspects of our technologies.
As of September 19 , 2017, we had 62 issued U.S. patents, 8 pending U.S. patent applications, 6 granted foreign patents and 2 pending foreign patent applications. Our first issued patent expired in 2016, with the last of these patents expiring in 2032. The first core patents do not expire until 2022; these include US 6,881,229 and US 6,790,233.
We have fourteen U.S. patents and one pending U.S. patent application directed to articulating implants using our high-strength, high toughness doped silicon nitride solid ceramic. The issued patents, which include US 6,881,229; US 7,666,229; US 7,758,646; US 7,695,521; US 7,771,481; US 7,776,085; US 7,780,738; US 8,016,890; US 8,123,812; US 8,133,284, US 8,377,134; US 8,747,540; US 9,051,639; US 9,517,136; begin to expire in 2022. The pending U.S. patent application has been assigned serial no. 15/162,363 and is presently awaiting examination by the U.S. Patent & Trademark Office.
We also have six U.S. patents, and two foreign patents (Mexico and Europe) related to our CSC technology that are directed to implants that have both a dense load-bearing, or cortical, component and a porous, or cancellous, component, together with a surface coating. These issued patents, which include US 6,790,233; US 6,846,327; US 7,695,521; US 8,133,284; US 8,747,540; US 9,649,197; MX 193270; and EP 1389978, begin to expire in 2022. EP 1389978 was registered in Switzerland, Germany, France, Greece and the Netherlands.
We also have three U.S. patents that we acquired in July 2012 from Dytech Corporation Ltd., or Dytech, directed to manufacturing processes for the production of porous ceramics for use in our orthopedic implants. These patents include US 5,563,106 and US 5,705,448, which have now expired; these patents also include US 6,617,270, which expires in 2019. Under our acquisition agreement with Dytech, Dytech granted to us a perpetual, irrevocable and exclusive license, including the right to grant sublicenses, to certain improvements and know-how related to the acquired patents. In return, we are required to pay Dytech a low single-digit royalty on net sales of products sold by us, our affiliates, or our licensees that are covered by one or more valid claims of these patents, and a percentage of any non-royalty licensing income we may receive in the event we grant a license to others.
Our remaining issued patents and pending applications are directed to additional aspects of our products and technologies including, among other things:
| ● | designs for pedicle screws; | |
● |
designs for intervertebral fusion devices; | |
● |
designs for hip implants; and | |
● |
designs for knee implants. |
We also expect to rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our intellectual property position. However, trade secrets are difficult to protect. We seek to protect the trade secrets in our proprietary technology and processes, in part, by entering into confidentiality agreements with commercial partners, collaborators, employees, consultants, scientific advisors and other contractors and into invention assignment agreements with our employees and some of our commercial partners and consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of the technologies that are developed.
Competition
The main alternatives to our silicon nitride biomaterial include: PEEK, which is predominantly manufactured by Invibio; BIOLOX® delta, which is a traditional oxide ceramic manufactured by CeramTec; allograft bone; metals; and coated metals.
We believe our main competitors in the orthopedic implant market, which utilize a variety of competitive biomaterials, include: Medtronic, Inc.; DePuy Synthes Companies, a group of Johnson & Johnson companies; Stryker Corporation; Biomet, Inc.; Zimmer Holdings, Inc.; Smith & Nephew plc; and Aesculap Inc. Presently, these companies buy ceramic components on an OEM basis from manufacturers such as CeramTec, Kyocera and CoorTek, Inc., among others. We anticipate that these and other orthopedic companies and OEMs will seek to introduce new biomaterials and products that compete with ours.
| 18 |
Competition within the industry is primarily based on technology, innovation, product quality, and product awareness and acceptance by surgeons. Our principal competitors have substantially greater financial, technical and marketing resources, as well as significantly greater manufacturing capabilities than we do, and they may succeed in developing products that render our implants and product candidates non-competitive. Our ability to compete successfully will depend upon our ability to develop innovative products with advanced performance features based on our silicon nitride technologies.
Government Regulation of Medical Devices
Governmental authorities in the United States, at the federal, state and local levels, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing and export and import of products such as those we are commercializing and developing. Failure to obtain approval or clearance to market our products and products under development and to meet the ongoing requirements of these regulatory authorities could prevent us from continuing to market or develop our products and product candidates.
United States
Pre-Marketing Regulation
In the United States, medical devices are regulated by the FDA. Unless an exemption applies, a new medical device will require either prior 510(k) clearance or approval of a premarket approval application, or PMA, before it can be marketed in the United States. The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three classes on the basis of the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices, which are those that have the lowest level or risk associated with them, are subject to general controls, including labeling, premarket notification and adherence to the QSR. Class II devices are subject to general controls and special controls, including performance standards. Class III devices, which have the highest level of risk associated with them, are subject to most of the previously identified requirements as well as to premarket approval. Most Class I devices and some Class II devices are exempt from the 510(k) requirement, although manufacturers of these devices are still subject to registration, listing, labeling and QSR requirements.
A 510(k) premarket notification must demonstrate that the device in question is substantially equivalent to another legally marketed device, or predicate device, that did not require premarket approval. In evaluating the 510(k), the FDA will determine whether the device has the same intended use as the predicate device, and (a) has the same technological characteristics as the predicate device, or (b) has different technological characteristics, and (i) the data supporting the substantial equivalence contains information, including appropriate clinical or scientific data, if deemed necessary by the FDA, that demonstrates that the device is as safe and as effective as a legally marketed device, and (ii) does not raise different questions of safety and effectiveness than the predicate device. Most 510(k)s do not require clinical data for clearance, but the FDA may request such data. The FDA’s goal is to review and act on each 510(k) within 90 days of submission, but it may take longer based on requests for additional information. In addition, requests for additional data, including clinical data, will increase the time necessary to review the notice. If the FDA does not agree that the new device is substantially equivalent to the predicate device, the new device will be classified in Class III, and the manufacturer must submit a PMA. Since July 2012, however, with the enactment of the Food and Drug Administration Safety and Innovation Act, or FDASIA, a de novo pathway is directly available for certain low to moderate risk devices that do not qualify for the 510(k) pathway due to lack of a predicate device. Modifications to a 510(k)-cleared medical device may require the submission of another 510(k) or a PMA if the changes could significantly affect the safety or effectiveness or constitute a major change in the intended use of the device.
Modifications to a 510(k)-cleared device frequently require the submission of a traditional 510(k), but modifications meeting certain conditions may be candidates for FDA review under a Special 510(k). If a device modification requires the submission of a 510(k), but the modification does not affect the intended use of the device or alter the fundamental scientific technology of the device, then summary information that results from the design control process associated with the cleared device can serve as the basis for clearing the application. A Special 510(k) allows a manufacturer to declare conformance to design controls without providing new data. When the modification involves a change in material, the nature of the “new” material will determine whether a traditional or Special 510(k) is necessary. For example, in its Device Advice on How to Prepare a Special 510(k), the FDA uses the example of a change in a material in a finger joint prosthesis from a known metal alloy to a ceramic that has not been used in a legally marketed predicate device as a type of change that should not be submitted as a Special 510(k). However, if the “new” material is a type that has been used in other legally marketed devices within the same classification for the same intended use, a Special 510(k) is appropriate. The FDA gives as an example a manufacturer of a hip implant who changes from one alloy to another that has been used in another legally marketed predicate. Special 510(k)s are typically processed within 30 days of receipt.
| 19 |
The PMA process is more complex, costly and time consuming than the 510(k) clearance procedure. A PMA must be supported by extensive data including, but not limited to, technical, preclinical, clinical, manufacturing, control and labeling information to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use. After a PMA is submitted, the FDA has 45 days to determine whether it is sufficiently complete to permit a substantive review. If the PMA is complete, the FDA will file the PMA. The FDA is subject to performance goal review times for PMAs and may issue a decision letter as a first action on a PMA within 180 days of filing, but if it has questions, it will likely issue a first major deficiency letter within 150 days of filing. It may also refer the PMA to an FDA advisory panel for additional review, and will conduct a preapproval inspection of the manufacturing facility to ensure compliance with the QSR, either of which could extend the 180-day response target. While the FDA’s ability to meet its performance goals has generally improved during the past few years, it may not meet these goals in the future. A PMA can take several years to complete and there is no assurance that any submitted PMA will ever be approved. Even when approved, the FDA may limit the indication for which the medical device may be marketed or to whom it may be sold. In addition, the FDA may request additional information or request the performance of additional clinical trials before it will reconsider the approval of the PMA or as a condition of approval, in which case the trials must be completed after the PMA is approved. Changes to the device, including changes to its manufacturing process, may require the approval of a supplemental PMA.
If a medical device is determined to present a “significant risk,” the manufacturer may not begin a clinical trial until it submits an investigational device exemption, or IDE, to the FDA and obtains approval of the IDE from the FDA. The IDE must be supported by appropriate data, such as animal and laboratory testing results and include a proposed clinical protocol. These clinical trials are also subject to the review, approval and oversight of an institutional review board, or IRB, which is an independent and multi-disciplinary committee of volunteers who review and approve research proposals, and the reporting of adverse events and experiences, at each institution at which the clinical trial will be performed. The clinical trials must be conducted in accordance with applicable regulations, including but not limited to the FDA’s IDE regulations and current good clinical practices. A clinical trial may be suspended by the FDA, the IRB or the sponsor at any time for various reasons, including a belief that the risks to the study participants outweigh the benefits of participation in the trial. Even if a clinical trial is completed, the results may not demonstrate the safety and efficacy of a device, or may be equivocal or otherwise not be sufficient to obtain approval.
Post-Marketing Regulation
After a device is placed on the market, numerous regulatory requirements apply. These include:
| ● | compliance with the QSR, which require manufacturers to follow stringent design, testing, control, documentation, record maintenance, including maintenance of complaint and related investigation files, and other quality assurance controls during the manufacturing process; |
● |
labeling regulations, which prohibit the promotion of products for uncleared or unapproved or “off-label” uses and impose other restrictions on labeling; and |
| ● | medical device reporting obligations, which require that manufacturers investigate and report to the FDA adverse events, including deaths, or serious injuries that may have been or were caused by a medical device and malfunctions in the device that would likely cause or contribute to a death or serious injury if it were to recur. |
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
| ● | warning letters; |
| ● | fines, injunctions, and civil penalties; |
| ● | recall or seizure of our products; |
| ● | operating restrictions, partial suspension or total shutdown of production; |
| ● | refusal to grant 510(k) clearance or PMA approvals of new products; |
| ● | withdrawal of 510(k) clearance or PMA approvals; and |
| ● | criminal prosecution. |
| 20 |
To ensure compliance with regulatory requirements, medical device manufacturers are subject to market surveillance and periodic, pre-scheduled and unannounced inspections by the FDA, and these inspections may include the manufacturing facilities of our subcontractors.
International Regulation
International sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA approval, and the requirements may differ. For example, the primary regulatory authority with respect to medical devices in Europe is that of the European Union. The European Union consists of 28 countries and has a total population of over 500 million people. The unification of these countries into a common market has resulted in the unification of laws, standards and procedures across these countries, which may expedite the introduction of medical devices like those we are offering and developing. Norway, Iceland, Lichtenstein and Switzerland are not members of the European Union, but have transposed applicable European medical device laws into their national legislation. Thus, a device that is marketed in the European Union may also be recognized and accepted in those four non-member European countries as well.
The European Union has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices. Devices that comply with the requirements of relevant directives will be entitled to bear CE Conformity Marking, indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout the European Union. Actual implementation of these directives, however, may vary on a country-by-country basis. The CE Mark is a mandatory conformity mark on medical devices distributed and sold in the European Union and certifies that a medical device has met applicable requirements.
The method of assessing conformity varies, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a “Notified Body.” Notified Bodies are independent testing houses, laboratories, or product certifiers authorized by the European Union member states to perform the required conformity assessment tasks, such as quality system audits and device compliance testing. An assessment by a Notified Body based within the European Union is required in order for a manufacturer to distribute the product commercially throughout the European Union. Medium and higher risk devices require the intervention of a Notified Body which will be responsible for auditing the manufacturer’s quality system. The Notified Body will also determine whether or not the product conforms to the requirements of the applicable directives. Devices that meet the applicable requirements of E.U. law and have undergone the appropriate conformity assessment routes will be granted CE “certification.” The CE Mark is mandatory for medical devices sold not only within the countries of the European Union but more generally within most of Europe. As many of the European standards are converging with international standards, the CE Mark is often used on medical devices manufactured and sold outside of Europe (notably in Asia that exports many manufactured products to Europe). CE Marking gives companies easier access into not only the European market but also to Asian and Latin American markets, most of whom recognize the CE Mark on medical device as a mark of quality and adhering to international standards of consumer safety, health or environmental requirements.
Compliance with Healthcare Laws
We must comply with various U.S. federal and state laws, rules and regulations pertaining to healthcare fraud and abuse, including anti-kickback and false claims laws, rules, and regulations, as well as other healthcare laws in connection with the commercialization of our products. Fraud and abuse laws are interpreted broadly and enforced aggressively by various state and federal agencies, including the U.S. Department of Justice, the U.S. Office of Inspector General for the Department of Health and Human Services and various state agencies.
We have entered into agreements with certain surgeons for assistance with the design of our products, some of whom we anticipate may make referrals to us or order our products. A majority of these agreements contain provisions for the payments of royalties. In addition, some surgeons currently own shares of our stock. We have structured these transactions with the intention of complying with all applicable laws, including fraud and abuse, data privacy and security, and transparency laws. Despite this intention, there can be no assurance that a particular government agency or court would determine our practices to be in full compliance with such laws. We could be materially impacted if regulatory or enforcement agencies or courts interpret our financial arrangements with surgeons to be in violation of healthcare laws, including, without limitation, fraud and abuse, data privacy and security, or transparency laws.
| 21 |
The U.S. federal Anti-Kickback Statute prohibits persons, including a medical device manufacturer (or a party acting on its behalf), from knowingly or willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for a service or product or the purchasing, ordering, arranging for, or recommending the ordering of, any service or product for which payment may be made by Medicare, Medicaid or any other federal healthcare program. This statute has been interpreted to apply to arrangements between medical device manufacturers on one hand and healthcare providers on the other. The term “remuneration” is not defined in the federal Anti-Kickback Statute and has been broadly interpreted to include anything of value, such as cash payments, gifts or gift certificates, discounts, waiver of payments, credit arrangements, ownership interests, the furnishing of services, supplies or equipment, and the provision of anything at less than its fair market value. Courts have broadly interpreted the scope of the law, holding that it may be violated if merely “one purpose” of an arrangement is to induce referrals, irrespective of the existence of other legitimate purposes. The Anti-Kickback Statute prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain business arrangements from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from federal Anti-Kickback Statute liability. The reach of the Anti-Kickback Statute was broadened by the recently enacted Patient Protection and Affordable Care Act of 2010 and the Health Care and Education Affordability Reconciliation Act of 2010, collectively, the Affordable Care Act or ACA, which, among other things, amends the intent requirement of the federal Anti-Kickback Statute such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act (discussed below) or the civil monetary penalties statute, which imposes fines against any person who is determined to have presented or caused to be presented claims to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. In addition to the federal Anti-Kickback Statute, many states have their own anti-kickback laws. Often, these laws closely follow the language of the federal law, although they do not always have the same scope, exceptions, safe harbors or sanctions. In some states, these anti-kickback laws apply not only to payments made by government healthcare programs but also to payments made by other third-party payors, including commercial insurance companies.
Sales, marketing, consulting, and advisory arrangements between medical device manufacturers and sales agents and physicians are subject to the Anti-Kickback Statute and other fraud and abuse laws. Government officials have focused recent enforcement efforts on, among other things, the sales and marketing activities of healthcare companies, including medical device manufacturers, and have brought cases against individuals or entities whose personnel allegedly offered unlawful inducements to potential or existing customers in an attempt to procure their business. We expect these activities to continue to be a focus of government enforcement efforts. Settlements of these cases by healthcare companies have involved significant fines and penalties and in some instances criminal plea agreements. We are also aware of governmental investigations of some of the largest orthopedic device companies reportedly focusing on consulting and service agreements between these companies and orthopedic surgeons. These developments are ongoing and we cannot predict the effects they will have on our business.
The federal False Claims Act imposes liability on any person that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam provisions of the False Claims Act allow a private individual to bring civil actions on behalf of the federal government alleging that the defendant has submitted a false claim, or has caused such a claim to be submitted, to the federal government, and to share in any monetary recovery. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when a person knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The False Claims Act has been used to assert liability on the basis of inadequate care, kickbacks, and other improper referrals, and allegations as to misrepresentations with respect to the services rendered. Qui tam actions have increased significantly in recent years, causing greater numbers of healthcare companies, including medical device manufacturers, to defend false claim actions, pay damages and penalties, or be excluded from participation in Medicare, Medicaid or other federal or state healthcare programs as a result of investigations arising out of such actions. In addition, various states have enacted similar laws analogous to the False Claims Act. Many of these state laws apply where a claim is submitted to any third-party payor and not merely a federal healthcare program. We are unable to predict whether we would be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the cost of defending such claims, as well as any sanctions imposed, could adversely affect our financial performance. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created several new federal crimes, including healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third party payors. The false statements statute prohibits knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious, or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items, or services.
| 22 |
In addition, we may be subject to, or our marketing or research activities may be limited by, data privacy and security regulation by both the federal government and the states in which we conduct our business. For example, HIPAA and its implementing regulations established uniform federal standards for certain “covered entities” (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information. The American Recovery and Reinvestment Act of 2009, commonly referred to as the economic stimulus package, included expansion of HIPAA’s privacy and security standards called the Health Information Technology for Economic and Clinical Health Act, or HITECH, which became effective on February 17, 2010. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”—independent contractors or agents of covered entities that create, receive, maintain, or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. These laws also require the reporting of breaches of protected health information to affected individuals, regulators and in some cases, local or national media. HIPAA and HITECH impose strict limits on our physician collaborators’ ability to use and disclose patient information on our behalf.
There are also an increasing number of state “sunshine” laws that require manufacturers to provide reports to state governments on pricing and marketing information. Several states have enacted legislation requiring medical device companies to, among other things, establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales and marketing activities, and to prohibit or limit certain other sales and marketing practices. In addition, a federal law known as the Physician Payments Sunshine Act, now requires medical device manufacturers to track and report to the federal government certain payments and other transfers of value made to physicians and teaching hospitals and ownership or investment interests held by physicians and their immediate family members. The first reporting period covered only payments or transfers of value made and ownership or investment interests held by physicians and their immediate family members from August 1, 2013 to December 31, 2013. The federal government disclosed the reported information on a publicly available website beginning in September 2014. For calendar year 2014, the Physician Payments Sunshine Act will require medical device manufacturers to report payments and transfers of values made and ownership or investment interests held by physicians and their immediate family members for the full calendar year. These laws may adversely affect our sales, marketing, and other activities by imposing administrative and compliance burdens on us. If we fail to track and report as required by these laws or to otherwise comply with these laws, we could be subject to the penalty provisions of the pertinent state and federal authorities.
Clinical research is heavily regulated by FDA regulations for the protection of human subjects (21 C.F.R. 50 and 56) and also the regulations of the U.S Department of Health and Human Services, or the Common Rule (45 C.F.R 46). Both FDA human subject regulations and the Common Rule impose restrictions on the involvement of human subjects in clinical research and require, among other things, the balancing of the risks and benefits of research, the documented informed consent of research participants, initial and ongoing review of research by an IRB. Similar regulations govern research conducted in foreign countries. Compliance with human subject protection regulations is costly and time consuming. Failure to comply could substantially and adversely impact our research program and the development of our products.
Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government healthcare programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product clearances and approvals, private “qui tam” actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations. Public disclosure of privacy and data security violations could cause significant reputational harm. Any of these events could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, implementation of corporate compliance programs, as well as laws and regulations requiring transparency of pricing and marketing information and governing the privacy and security of health information, such as the E.U.’s Directive 95/46 on the Protection of Individuals with regard to the Processing of Personal Data, or the Data Directive, and the wide variety of national laws implementing the Data Directive.
Healthcare Reform
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs.
In March 2010, President Obama signed into law the ACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers and impose additional health policy reforms. Among other things, the ACA imposes a 2.3% medical device excise tax on sales of many medical devices in the United States which became effective on January 1, 2013. Substantial new provisions affecting compliance have also been enacted, which may affect our business practices with healthcare practitioners and a significant number of provisions are not yet, or have only recently become, effective. Although it is too early to determine the full effect of the ACA, the new law appears likely to place downward pressure on pricing of medical devices, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
Modifications to or repeal of all or certain provisions of the Health Care Reform Act are expected as a result of the outcome of the recent presidential election and Republicans maintaining control of Congress, consistent with statements made by Donald Trump and members of Congress during the presidential campaign and following the election. We cannot predict the ultimate content, timing or effect of any changes to the Health Care Reform Act or other federal and state reform efforts. There is no assurance that federal or state healthcare reform will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative, judicial or administrative changes relating to healthcare reform will affect our business.
| 23 |
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, on August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or ATRA, which, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. On March 1, 2013, the President signed an executive order implementing the Budget Control Act’s 2% Medicare payment reductions, and on April 1, 2013, these reductions went into effect. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our financial operations.
We expect that the ACA, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for our products. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may affect our ability to generate revenue and profits or commercialize our product candidates.
Third-Party Reimbursement
Because we typically receive payment directly from hospitals and surgical centers, we do not anticipate relying directly on payment for any of our products from third-party payors, such as Medicare, Medicaid, private insurers, and managed care companies. However, our business will be affected by policies administered by federal and state healthcare programs, such as Medicare and Medicaid, as well as private third-party payors, which often follow the policies of the state and federal healthcare programs. For example, our business will be indirectly impacted by the ability of a hospital or medical facility to obtain coverage and third-party reimbursement for procedures performed using our products. Many hospitals and clinics in the United States belong to group purchasing organizations (that typically incentivize their hospital members to make a relatively large proportion of purchases from a limited number of vendors of similar products that have contracted to offer discounted prices). Such contracts often include exceptions for purchasing certain innovative new technologies, however. Accordingly, the commercial success of our products may also depend to some extent on our ability to either negotiate favorable purchase contracts with key group purchasing organizations or persuade hospitals and clinics to purchase our product “off contract.” These third-party payors may deny reimbursement if they determine that a device used in a procedure was not medically necessary; was not used in accordance with cost-effective treatment methods, as determined by the third-party payor; or was used for an unapproved use. A national or local coverage decision denying Medicare coverage for one or more of our products could result in private insurers and other third party payors also denying coverage. Even if favorable coverage and reimbursement status is attained for our products, less favorable coverage policies and reimbursement rates may be implemented in the future. The cost containment measures that third-party payors and providers are instituting, both within the United States and abroad, could significantly reduce our potential revenues from the sale of our products and any product candidates. We cannot provide any assurances that we will be able to obtain and maintain third party coverage or adequate reimbursement for our products and product candidates in whole or in part.
For inpatient and outpatient procedures, including those that will involve use of our products, Medicare and many other third-party payors in the United States reimburse hospitals at a prospectively determined amount. This amount is generally based on one or more diagnosis related groups, or DRGs, associated with the patient’s condition for inpatient treatment and generally based on ambulatory payment classifications, or APCs, associated with the procedures performed as an outpatient at an ambulation surgicenter. Each DRG or APC is associated with a level of payment and may be adjusted from time to time, usually annually. Prospective payments are intended to cover most of the non-physician hospital costs incurred in connection with the applicable diagnosis and related procedures. Implant products, such as those we plan to sell, represent part of the total procedure costs while labor, hospital room and board, and other supplies and services represent the balance of those costs. However, the prospective payment amounts are typically set independently of a particular hospital’s actual costs associated with treating a particular patient and implanting a device. Therefore, the payment that a hospital would receive for a particular hospital visit would not typically take into account the cost of our products.
Medicare has established a number of DRGs for inpatient procedures that involve the use of products similar to ours. Although Medicare has authority to create special DRGs for hospital services that more properly reflect the actual costs of expensive or new-technology devices implanted as part of a procedure, it has declined to do so in the past, and we do not expect that it will do so with respect to our current products and product candidates. Medicare’s DRG and APC classifications may have implications outside of Medicare, as many other U.S. third-party payors often use Medicare DRGs and APCs for purposes of determining reimbursement.
| 24 |
We believe that orthopedic implants generally have been well received by third-party payors because of the ability of these implants to greatly reduce long-term healthcare costs for patients with degenerative joint disease. However, coverage and reimbursement policies vary from payor to payor and are subject to change. As discussed above, hospitals that purchase medical devices for treatment of their patients generally rely on third-party payors to reimburse all or part of the costs and fees associated with the procedures performed with these devices. Both government and private third-party coverage and reimbursement levels are critical to new product acceptance. Neither hospitals nor surgeons are likely to use our products if they do not receive reimbursement for the procedures adequate to cover the cost of our products.
While it is expected that hospitals will be able to obtain coverage for procedures using our products, the level of payment available to them for such procedures may change over time. State and federal healthcare programs, such as Medicare and Medicaid, closely regulate provider payment levels and have sought to contain, and sometimes reduce, payment levels. Commercial insurers and managed care plans frequently follow government payment policies, and are likewise interested in controlling increases in the cost of medical care. These third-party payors may deny payment if they determine that a procedure was not medically necessary, a device used in a procedure was not used in accordance with cost-effective treatment methods, as determined by the third-party payor, or was used for an unapproved use.
In addition, some payors are adopting pay-for-performance programs that differentiate payments to healthcare providers based on the achievement of documented quality-of-care metrics, cost efficiencies, or patient outcomes. These programs are intended to provide incentives to providers to find ways to deliver the same or better results while consuming fewer resources. As a result of these programs, and related payor efforts to reduce payment levels, hospitals and other providers are seeking ways to reduce their costs, including the amounts they pay to medical device suppliers. Adverse changes in payment rates by payors to hospitals could adversely impact our ability to market and sell our products and negatively affect our financial performance.
In international markets, healthcare payment systems vary significantly by country and many countries have instituted price ceilings on specific product lines. There can be no assurance that our products will be considered cost-effective by third-party payors, that reimbursement will be available or, if available, that the third-party payors’ reimbursement policies will not adversely affect our ability to sell our products profitably.
Member countries of the European Union offer various combinations of centrally financed healthcare systems and private health insurance systems. The relative importance of government and private systems varies from country to country. Governments may influence the price of medical devices through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may be marketed only once a reimbursement price has been agreed upon. Some of these countries may require, as condition of obtaining reimbursement or pricing approval, the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Some E.U. member states allow companies to fix their own prices for devices, but monitor and control company profits. The choice of devices is subject to constraints imposed by the availability of funds within the purchasing institution. Medical devices are most commonly sold to hospitals or healthcare facilities at a price set by negotiation between the buyer and the seller. A contract to purchase products may result from an individual initiative or as a result of a competitive bidding process. In either case, the purchaser pays the supplier, and payment terms vary widely throughout the European Union. Failure to obtain favorable negotiated prices with hospitals or healthcare facilities could adversely affect sales of our products.
Employees
As of September 19, 2017, we had 35 employees. We believe that our success will depend, in part, on our ability to attract and retain qualified personnel. We have never experienced a work stoppage due to labor difficulties and believe that our relations with our employees are good. None of our employees are represented by labor unions.
| 25 |
Executive Officers
Our current executive officers and their respective ages and positions are as follows:
| Name | Age | Position | ||
| B. Sonny Bal, M.D. | 54 | Chairman of the Board of Directors, President and Chief Executive Officer, Principal Financial Officer | ||
| Bryan J. McEntire | 64 | Chief Technology Officer |
The following is a brief summary of the background of each of our current directors and executive officers.
B. Sonny Bal, M.D. has served on our board of directors since February 2012, as Chairman of our board of directors since August 2014 and as our President and Chief Executive Officer since October 2014. Dr. Bal is Professor & Chief of Adult Reconstruction at the University of Missouri, Columbia, and Adjunct Professor of Material Sciences at the University of Missouri at Rolla. Dr. Bal is a member of the American Academy of Orthopaedic Surgeons, the American Association of Hip and Knee Surgeons and the International Society of Technology in Arthroplasty. Dr. Bal received his M.D. degree from Cornell University and an M.B.A. from Northwestern University, and a J.D. from the University of Missouri. Dr. Bal is a licensed attorney and co-founder of the Bal Brenner law firm in North Carolina.
Bryan J. McEntire has served as our Chief Technology Officer since May 2012. From June 2004 to May 2012 he served as our Vice President of Manufacturing and as our Vice President of Research from December 2006 to May 2012. Mr. McEntire has worked in various advanced ceramic product development, quality engineering and manufacturing roles at Applied Materials, Inc., Norton Advanced Ceramics, a division of Saint-Gobain Industrial Ceramics Corporation, Norton/TRW Ceramics and Ceramatec, Inc., a small producer of ionic-conducting and structural ceramic components located in Salt Lake City, Utah. Mr. McEntire holds a B.S. degree in Materials Science and Engineering and an M.B.A. from the University of Utah.
In addition to the other information contained in this Annual Report, the following risk factors should be considered carefully in evaluating our company. Our business, financial condition, liquidity or results of operations could be materially adversely affected by any of these risks.
Risks Related to Our Business and Strategy
We have incurred net losses since our inception and anticipate that we will continue to incur substantial net losses for the foreseeable future. We may never achieve or sustain profitability.
We have incurred substantial net losses since our inception. For the years ended December 31, 2016 and 2015 we incurred a net loss of $16.6 million and $23.9 million, respectively, and used cash in operations of $7.2 million and $9.1 million, respectively. We have an accumulated deficit of $213.1 million at December 31, 2016. Our losses have resulted principally from costs incurred in connection with our sales and marketing activities, research and development activities, manufacturing activities, general and administrative expenses associated with our operations, impairments on intangible assets and property and equipment, interest expense, loss on extinguishment of debt and offering costs. Even if we are successful in launching additional products into the market, we expect to continue to incur substantial losses for the foreseeable future as we continue to sell and market our current products and research and develop, and seek regulatory approvals for, our product candidates.
If sales revenue from any of our current products or product candidates that receive marketing clearance from the FDA or other regulatory body is insufficient, if we are unable to develop and commercialize any of our product candidates, or if our product development is delayed, we may never become profitable. Even if we do become profitable, we may be unable to sustain or increase our profitability on a quarterly or annual basis.
Our success depends on our ability to successfully commercialize silicon nitride-based medical devices, which to date have experienced only limited market acceptance.
We believe we are the first and only company to use silicon nitride in medical applications. To date, however, we have had limited acceptance of our silicon nitride-based products and our product revenue has been derived substantially from our non-silicon nitride products. In order to succeed in our goal of becoming a leading biomaterial technology company utilizing silicon nitride, we must increase market awareness of our silicon nitride interbody spinal fusion products, continue to implement our sales and marketing strategy, enhance our commercial infrastructure and commercialize our silicon nitride joint replacement components and other products. If we fail in any of these endeavors or experience delays in pursuing them, we will not generate revenues as planned and will need to curtail operations or seek additional financing earlier than otherwise anticipated.
| 26 |
Our current products and our future products may not be accepted by hospitals and surgeons and may not become commercially successful.
Although we received 510(k) regulatory clearance from the FDA for our first silicon nitride spinal fusion products in 2008, we have not been able to obtain significant market share of the interbody spinal fusion market to date, and may not obtain such market share in the future. Even if we receive regulatory clearances or approvals for our product candidates in development, these product candidates may not gain market acceptance among orthopedic surgeons and the medical community. Orthopedic surgeons may elect not to use our products for a variety of reasons, including:
| ● | lack or perceived lack of evidence supporting the beneficial characteristics of our silicon nitride technology; | |
| ● | limited long-term data on the use of silicon nitride in medical devices; | |
| ● | lower than expected clinical benefits in comparison with other products; | |
| ● | the perception by surgeons that there are insufficient advantages of our products relative to currently available products; | |
| ● | hospitals may choose not to purchase our products; | |
| ● | group purchasing organizations may choose not to contract for our products, thus limiting availability of our products to hospital purchasers; | |
| ● | the price of our products, which may be higher than products made of the other commonly used biomaterials in the interbody spinal fusion market and total joint market; | |
| ● | lack of coverage or adequate payment from managed care plans and other third-party payers for the procedures that use our products; | |
| ● | Medicare, Medicaid or other third-party payers may limit or not permit reimbursement for procedures using our products; | |
| ● | ineffective marketing and distribution support; | |
| ● | the time and resources that may be required for training, or the inadequate training, of surgeons in the proper use of our products; | |
| ● | the development of alternative biomaterials and products that render our products less competitive or obsolete; and | |
| ● | the development of or improvement of competitive products. |
If surgeons do not perceive our silicon nitride products and product candidates as superior alternatives to competing products, we will not be able to generate significant revenues, if any.
Even if surgeons are convinced of the superior characteristics of our silicon nitride products and our product candidates that we successfully introduce compared to the limitations of the current commonly used biomaterials, surgeons may find other methods or turn to other biomaterials besides silicon nitride to overcome such limitations. For instance, with respect to interbody spinal fusion products, surgeons or device manufacturers may use more effective markers for enhancing the imaging compatibility of PEEK devices, more effective antibiotics to prevent or treat implant-related infections, and more effective osteoconductive and osteoinductive materials when implanting an interbody spinal fusion device. Device manufacturers may also coat metal with existing traditional ceramics to reduce the risk of metal wear particles and corrosion in total joint replacement implants. Additionally, surgeons may increase their use of metal interbody spinal fusion devices if there is an increasing perception that PEEK devices are limited by their strength and resistance to fracture.
| 27 |
If we are unable to increase the productivity of our sales and marketing infrastructure we will not be able to penetrate the spinal fusion market.
We market and sell our products to surgeons and hospitals in the United States and select markets in Europe and South America using a network of independent third-party distributors who have existing surgeon relationships. We manage this distribution network through our in-house sales and marketing management team. We may also establish distribution collaborations in the United States and abroad in instances where access to a large or well-established sales and marketing organization may help to expand the market or accelerate penetration for selected products.
We cannot assure you that we will succeed in entering into and maintaining productive arrangements with an adequate number of distributors that are sufficiently committed to selling our products. The establishment of a distribution network is expensive and time consuming. As we launch new products and increase our marketing effort with respect to existing products, we will need to continue to hire, train, retain and motivate skilled independent distributors with significant technical knowledge in various areas, such as spinal fusion and total hip and knee joint replacement. In addition, the commissions we pay our distributors have increased over time, which has resulted in higher sales and marketing expenses, and those commissions and expenses may increase in the future. Furthermore, current and potential distributors may market and sell the products of our competitors. Even if the distributors market and sell our products, our competitors may be able, by offering higher commission payments or other incentives, to persuade these distributors to reduce or terminate their sales and marketing efforts related to our products. The distributors may also help competitors solicit business from our existing customers. Some of our independent distributors account for a significant portion of our sales volume, and, if we were to lose them, our sales could be adversely affected.
Even if we engage and maintain suitable relationships with an adequate number of distributors, they may not generate revenue as quickly as we expect them to, commit the necessary resources to effectively market and sell our products, or ultimately succeed in selling our products. We have been unable to obtain meaningful market share in the interbody spinal fusion device market with our current silicon nitride products to date and we may not be successful in increasing the productivity of our sales and marketing team and distribution network to gain meaningful market share for our silicon nitride products, which could adversely affect our business and financial condition.
The orthopedic market is highly competitive and we may not be able to compete effectively against the larger, well-established companies that dominate this market or emerging and small innovative companies that may seek to obtain or increase their share of the market.
The markets for spinal fusions and total hip and knee implant products are intensely competitive, and many of our competitors are much larger and have substantially more financial and human resources than we do. Many have long histories and strong reputations within the industry, and a relatively small number of companies dominate these markets. Medtronic, Inc.; DePuy Synthes Companies, a group of Johnson & Johnson companies; Stryker Corporation; Biomet, Inc.; Zimmer Holdings, Inc.; and Smith & Nephew plc, account for a significant amount of orthopedic sales worldwide.
These companies enjoy significant competitive advantages over us, including:
| ● | broad product offerings, which address the needs of orthopedic surgeons and hospitals in a wide range of procedures; | |
| ● | products that are supported by long-term clinical data; | |
| ● | greater experience in, and resources for, launching, marketing, distributing and selling products, including strong sales forces and established distribution networks; | |
| ● | existing relationships with spine and joint reconstruction surgeons; | |
| ● | extensive intellectual property portfolios and greater resources for patent protection; | |
| ● | greater financial and other resources for product research and development; | |
| ● | greater experience in obtaining and maintaining FDA and other regulatory clearances and approvals for products and product enhancements; | |
| ● | established manufacturing operations and contract manufacturing relationships; | |
| ● | significantly greater name recognition and widely recognized trademarks; and | |
| ● | established relationships with healthcare providers and payers. |
| 28 |
Our products and any product candidates that we may introduce into the market may not enable us to overcome the competitive advantages of these large and dominant orthopedic companies. In addition, even if we successfully introduce additional product candidates incorporating our silicon nitride biomaterial into the market, emerging and small innovative companies may seek to increase their market share and they may eventually possess competitive advantages, which could adversely impact our business. Our competitors may also employ pricing strategies that could adversely affect the pricing of our products and pricing in the spinal fusion and total joint replacement market generally.
Moreover, many other companies are seeking to develop new biomaterials and products which may compete effectively against our products in terms of performance and price. For example, Smith & Nephew has developed a ceramic-coated metal, known as Oxinium, which may overcome certain of the limitations of metal joint replacement products and could directly compete with our silicon nitride and silicon nitride-coated product candidates.
We have significant customer concentration, so that economic difficulties or changes in the purchasing policies or patterns of our key customers could have a significant impact on our business and operating results.
A small number of customers account for a substantial portion of our product revenues. Our customers are primarily hospitals and surgical centers. At December 31, 2016 and 2015, our largest customer, Bon Secours St. Mary’s Hospital, or St. Mary’s, had a receivable balance of approximately 16% and 7%, respectively, of our total trade accounts receivable. In addition, St. Mary’s accounted for 18% and 12% of our product revenues for the years ended December 31, 2016 and 2015, respectively. Sales of our products to our customers, including St. Mary’s, are not based on long-term, committed-volume purchase contracts, and we may not continue to receive significant revenues from St. Mary’s or any customer. Because of our significant customer concentration, our revenue could fluctuate significantly due to changes in economic conditions, the use of competitive products, or the loss of, reduction of business with, or less favorable terms with St. Mary’s or any of our other significant customers. A significant portion of St. Mary’s’ purchases have been of our non-silicon nitride products, so it may be able to purchase competitive similar products from others. A reduction or delay in orders from St. Mary’s or any of our other significant customers, or a delay or default in payment by any significant customer, could materially harm our business and results of operations.
The manufacturing process for our silicon nitride products is complex and requires sophisticated state-of-the-art equipment, experienced manufacturing personnel and highly specialized knowledge. If we are unable to manufacture our silicon nitride products on a timely basis consistent with our quality standards, our results of operation will be adversely impacted.
In order to control the quality, cost and availability of our silicon nitride products, we developed our own manufacturing capabilities. We operate a 30,000 square foot manufacturing facility which is certified under the ISO 13485 medical device manufacturing standard for medical devices and operates under the FDA’s quality systems regulations, or QSRs. All operations with the exceptions of raw material production, cleaning, packaging and sterilization are performed at this facility.
In order to mitigate the risk associated with us being the sole manufacturer of our silicon nitride medical device products, in June 2014, we entered into a manufacturing development and supply agreement with Kyocera Industrial Ceramics Corporation, or Kyocera. We updated our material master file and submitted a 510(k) with the FDA in the third quarter of 2014 to qualify Kyocera as a second source supplier of our silicon nitride products. Kyocera has been qualified as a second source supplier of our silicon nitride products. Although we expect this arrangement with Kyocera to continue, if Kyocera ceases to continue as a qualified manufacturer of these products and product candidates, we will be the sole manufacturer of these products and will need to seek other potential secondary manufacturers. Our reliance solely on our internal resources to manufacture our silicon nitride products entails risks to which we would not be subject if we had secondary suppliers for their manufacture, including:
| ● | the inability to meet our product specifications and quality requirements consistently; | |
| ● | a delay or inability to procure or expand sufficient manufacturing capacity to meet additional demand for our products; | |
| ● | manufacturing and product quality issues related to the scale-up of manufacturing; | |
| ● | the inability to produce a sufficient supply of our products to meet product demands; | |
| ● | the disruption of our manufacturing facility due to equipment failure, natural disaster or failure to retain key personnel; and | |
| ● | our inability to ensure our compliance with regulations and standards of the FDA, including QSRs, and corresponding state and international regulatory authorities, including the CFDA. |
| 29 |
Any of these events could lead to a reduction in our product sales, product launch delays, failure to obtain regulatory clearance or approval or impact our ability to successfully sell our products and commercialize our products candidates.
We depend on a limited number of third-party suppliers for key raw materials used in the manufacturing of our silicon nitride products, and the loss of these third-party suppliers or their inability to supply us with adequate raw materials could harm our business.
We rely on a limited number of third-party suppliers for the raw materials required for the production of our silicon nitride products and product candidates. Our dependence on a limited number of third-party suppliers involves several risks, including limited control over pricing, availability, quality, and delivery schedules for raw materials. We have no supply agreements in place with any of our suppliers and cannot be certain that our current suppliers will continue to provide us with the quantities of raw materials that we require or that satisfy our anticipated specifications and quality requirements. Any supply interruption in limited or single sourced raw materials could materially harm our ability to manufacture our products until a new source of supply, if any, could be identified and qualified. We may be unable to find a sufficient alternative supply channel within a reasonable time or on commercially reasonable terms. Any performance failure on the part of our suppliers could delay the production of our silicon nitride products and product candidates and delay the development and commercialization of our product candidates, including limiting supplies necessary for commercial sale, clinical trials and regulatory approvals, which could have a material adverse effect on our business.
Use of third-party manufacturers increases the risk that we will not have adequate supplies of our non-silicon nitride products or instrumentation sets.
The majority of our product revenue is currently generated by sales of non-silicon nitride products. Our reliance on a limited number of third-party manufacturers to supply us with our non-silicon nitride products and instruments exposes us to risks that could delay our sales, or result in higher costs or lost product revenues. In particular, our manufacturers could:
| ● | encounter difficulties in achieving volume production, quality control and quality assurance or suffer shortages of qualified personnel, which could result in their inability to manufacture sufficient quantities of our commercially available non-silicon nitride products to meet market demand for those products, or they could experience similar problems that result in the manufacture of insufficient quantities of our non-silicon nitride product candidates; and | |
| ● | fail to follow and remain in compliance with the FDA-mandated QSRs, compliance which is required for all medical devices, or fail to document their compliance to QSRs, either of which could lead to significant delays in the availability of materials for our non-silicon nitride products or instrumentation sets. |
If we are unable to obtain adequate supplies of our non-silicon nitride products and related instrumentation sets that meet our specifications and quality standards, it will be difficult for us to compete effectively. We have no supply agreements in place with our manufacturers and they may change the terms of our future orders or choose not to supply us with products or instrumentation sets in the future. Furthermore, if a third-party manufacturer from whom we purchase fails to perform its obligations, we may be forced to purchase products or related instrumentation from other third-party manufacturers, which we may not be able to do on reasonable terms, if at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer or the re-verification of an existing manufacturer could negatively affect our ability to produce and distribute our non-silicon nitride products or instruments in a timely manner.
In order to be successful, we must expand our available product lines of silicon nitride-based medical devices by commercializing new product candidates, but we may not be able to do so in a timely fashion and at expected costs, or at all.
Although we are currently marketing our silicon nitride interbody spinal fusion implants, in order to be successful, we will need to expand our product lines to include other silicon nitride devices. Therefore, we are developing silicon nitride product candidates for total hip and knee replacement procedures and are exploring the application of our silicon nitride technology for other potential applications. However, we have yet to commercialize any silicon nitride products beyond our spinal fusion products. To succeed in our commercialization efforts, we must effectively continue product development and testing, obtain regulatory clearances and approvals, and enhance our sales and marketing capabilities. We may also have to write down significant inventory if existing products are replaced by new products. Because of these uncertainties, there is no assurance that we will succeed in bringing any of our current or future product candidates to market. If we fail in bringing our product candidates to market, or experience delays in doing so, we will not generate revenues as planned and will need to curtail operations or seek additional financing earlier than otherwise anticipated.
| 30 |
We will depend on one or more strategic partners to develop and commercialize our total joint replacement product candidates, and if our strategic partners are unable to execute effectively on our agreements with them, we may never become profitable.
We are seeking a strategic partner to develop and commercialize our total joint replacement product candidates. We will be reliant on our strategic partners to develop and commercialize a total hip or knee joint replacement product candidate that utilizes silicon nitride-coated components, although we have not yet entered into an agreement with any strategic partner to develop products with these silicon nitride-coated components and may be unable to do so on agreeable terms. In order to succeed in our joint commercialization efforts, we and any future partners must execute effectively on all elements of a combined business plan, including continuing to establish sales and marketing capabilities, manage certified, validated and effective commercial-scale manufacturing operations, conduct product development and testing, and obtain regulatory clearances and approvals for our product candidate. If we or any of our strategic partners fail in any of these endeavors, or experience delays in pursuing them, we will not generate revenues as planned and will need to curtail operations or seek additional financing earlier than otherwise anticipated.
Part of our strategy is to establish and develop OEM partnerships and arrangements, which subjects us to various risks.
Because we believe silicon nitride is a superior platform and technology for application in the spine, total joint and other markets, we are establishing OEM partnerships with other companies to replace their materials and products with silicon nitride. Sales of products to OEM customers will expose our business to a number of risks. Sales through OEM partners could be less profitable than direct sales. Sales of our products through multiple channels could also confuse customers and cause the sale of our products to decline. In addition, OEM customers will require that products meet strict standards. Our compliance with these requirements could result in increased development, manufacturing, warranty and administrative costs. A significant increase in these costs could adversely affect our operating results. If we fail to meet OEM specifications on a timely basis, our relationships with our OEM partners may be harmed. Furthermore, we would not control our OEM partners, and they could sell competing products, may not incorporate our technology into their products in a timely manner and may devote insufficient sales efforts to the OEM products.
If hospitals and other healthcare providers are unable to obtain coverage or adequate reimbursement for procedures performed with our products, it is unlikely our products will be widely used.
In the United States, the commercial success of our existing products and any future products will depend, in part, on the extent to which governmental payers at the federal and state levels, including Medicare and Medicaid, private health insurers and other third-party payers provide coverage for and establish adequate reimbursement levels for procedures utilizing our products. Because we typically receive payment directly from hospitals and surgical centers, we do not anticipate relying directly on payment from third-party payers for our products. However, hospitals and other healthcare providers that purchase our orthopedic products for treatment of their patients generally rely on third-party payers to pay for all or part of the costs and fees associated with our products as part of a “bundled” rate for the associated procedures. The existence of coverage and adequate reimbursement for our products and the procedures performed with them by government and private payers is critical to market acceptance of our existing and future products. Neither hospitals nor surgeons are likely to use our products if they do not receive adequate reimbursement for the procedures utilizing our products.
Many private payers currently base their reimbursement policies on the coverage decisions and payment amounts determined by the Centers for Medicare and Medicaid Services, or CMS, which administers the Medicare program. Others may adopt different coverage or reimbursement policies for procedures performed with our products, while some governmental programs, such as Medicaid, have reimbursement policies that vary from state to state, some of which may not pay for the procedures performed with our products in an adequate amount, if at all. A Medicare national or local coverage decision denying coverage for one or more of our products could result in private and other third-party payers also denying coverage for our products. Third-party payers also may deny reimbursement for our products if they determine that a product used in a procedure was not medically necessary, was not used in accordance with cost-effective treatment methods, as determined by the third-party payer, or was used for an unapproved use. Unfavorable coverage or reimbursement decisions by government programs or private payers underscore the uncertainty that our products face in the market and could have a material adverse effect on our business.
Many hospitals and clinics in the United States belong to group purchasing organizations, which typically incentivize their hospital members to make a relatively large proportion of purchases from a limited number of vendors of similar products that have contracted to offer discounted prices. Such contracts often include exceptions for purchasing certain innovative new technologies, however. Accordingly, the commercial success of our products may also depend to some extent on our ability to either negotiate favorable purchase contracts with key group purchasing organizations and/or persuade hospitals and clinics to purchase our product “off contract.”
| 31 |
The healthcare industry in the United States has experienced a trend toward cost containment as government and private payers seek to control healthcare costs by paying service providers lower rates. While it is expected that hospitals will be able to obtain coverage for procedures using our products, the level of payment available to them for such procedures may change over time. State and federal healthcare programs, such as Medicare and Medicaid, closely regulate provider payment levels and have sought to contain, and sometimes reduce, payment levels. Private payers frequently follow government payment policies and are likewise interested in controlling increases in the cost of medical care. In addition, some payers are adopting pay-for-performance programs that differentiate payments to healthcare providers based on the achievement of documented quality-of-care metrics, cost efficiencies, or patient outcomes. These programs are intended to provide incentives to providers to deliver the same or better results while consuming fewer resources. As a result of these programs, and related payer efforts to reduce payment levels, hospitals and other providers are seeking ways to reduce their costs, including the amounts they pay to medical device manufacturers. We may not be able to sell our implants profitably if third-party payers deny or discontinue coverage or reduce their levels of payment below that which we project, or if our production costs increase at a greater rate than payment levels. Adverse changes in payment rates by payers to hospitals could adversely impact our ability to market and sell our products and negatively affect our financial performance.
In international markets, medical device regulatory requirements and healthcare payment systems vary significantly from country to country, and many countries have instituted price ceilings on specific product lines. We cannot assure you that our products will be considered cost-effective by international third-party payers, that reimbursement will be available or, if available, that the third-party payers’ reimbursement policies will not adversely affect our ability to sell our products profitably. Any failure to receive regulatory or reimbursement approvals would negatively impact market acceptance of our products in any international markets in which those approvals are sought.
Modifications to or repeal of all or certain provisions of the Health Care Reform Act are expected as a result of the outcome of the recent presidential election and Republicans maintaining control of Congress, consistent with statements made by Donald Trump and members of Congress during the presidential campaign and following the election. We cannot predict the ultimate content, timing or effect of any changes to the Health Care Reform Act or other federal and state reform efforts. There is no assurance that federal or state healthcare reform will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative, judicial or administrative changes relating to healthcare reform will affect our business.
Prolonged negative economic conditions in domestic and international markets may adversely affect us, our suppliers, partners and consumers, and the global orthopedic market which could harm our financial position.
There is a risk that one or more of our current suppliers may not continue to operate. Any lender that is obligated to provide funding to us under any future credit agreement with us may not be able to provide funding in a timely manner, or at all, when we require it. The cost of, or lack of, available credit or equity financing could impact our ability to develop sufficient liquidity to maintain or grow our company. These negative changes in domestic and international economic conditions or additional disruptions of either or both of the financial and credit markets may also affect third-party payers and may have a material adverse effect on our business, results of operations, financial condition and liquidity.
In addition, we believe that various demographics and industry-specific trends will help drive growth in the orthopedics markets, but these demographics and trends are uncertain. Actual demand for orthopedic products generally, and our products in particular, could be significantly less than expected if our assumptions regarding these factors prove to be incorrect or do not materialize, or if alternative treatments gain widespread acceptance.
We are dependent on our senior management team, engineering team, sales and marketing team and surgeon advisors, and the loss of any of them could harm our business. We may not have sufficient personnel to effectuate our business strategy due to our recent reduction in force.
The members of our current senior management team have worked together in their new positions with us for a limited time and may not be able to successfully implement our strategy. In addition, we have not entered into employment agreements, other than change-in-control severance agreements, with any of the members of our senior management team. There are no assurances that the services of any of these individuals will be available to us for any specified period of time. The successful integration of our senior management team, the loss of members of our senior management team, sales and marketing team, engineering team and key surgeon advisors, or our inability to attract or retain other qualified personnel or advisors could have a material adverse effect on our business, financial condition and results of operations.
In October 2016, we implemented a substantial reduction in our workforce by approximately 38% to lower operating expenses. This most recent restructuring plan and other such efforts could result in disruptions to our operations. We may not have sufficient number of qualified personnel to effectuate our business strategy which could have a material adverse effect on our business, financial condition and results of operations.
| 32 |
If we experience significant disruptions in our information technology systems, our business, results of operations and financial condition could be adversely affected.
The efficient operation of our business depends on our information technology systems. We rely on our information technology systems to effectively manage our sales and marketing, accounting and financial functions; manufacturing processes; inventory; engineering and product development functions; and our research and development functions. As such, our information technology systems are vulnerable to damage or interruption including from earthquakes, fires, floods and other natural disasters; terrorist attacks and attacks by computer viruses or hackers; power losses; and computer systems, or Internet, telecommunications or data network failures. The failure of our information technology systems to perform as we anticipate or our failure to effectively implement new systems could disrupt our entire operation and could result in decreased sales, increased overhead costs, excess inventory and product shortages, all of which could have a material adverse effect on our reputation, business, results of operations and financial condition.
Risks Related to Our Capital Resources and Impairments
We will require additional financing and our failure to obtain additional funding would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We currently have limited committed sources of capital and we have limited liquidity. Our cash and cash equivalents as of December 31, 2016 was $6.9 million. We require substantial future capital in order to continue to conduct the research and development and regulatory clearance and approval activities necessary to bring our products to market, to establish effective marketing and sales capabilities. Our existing capital resources are not sufficient to enable us to fund the completion of the development and commercialization of all of our product candidates. We cannot determine with certainty the duration and completion costs of the current or future development and commercialization of our product candidates for spinal fusion procedures, joint replacement and coated metals or if, when, or to what extent we will generate revenues from the commercialization and sale of any of these product candidates for which we obtain regulatory approval. We may never succeed in achieving regulatory approval for certain or all of these product candidates. The duration, costs and timing of clinical trials and development of our spinal fusion, joint replacement and coated metal product candidates will depend on a variety of factors, including:
| ● | the scope, rate of progress, and expense of our ongoing, as well as any additional, clinical trials and other research and development activities; | |
| ● | future clinical trial results we may must or choose to conduct; | |
| ● | potential changes in government regulation; and | |
| ● | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of spinal fusion, joint replacement or coated metal product candidates could mean a significant change in the costs and timing associated with the development of these product candidates.
In addition, the repayment of the loan under the Loan and Security Agreement and the associated liquidity covenant limit (as described below) our ability to use our cash and cash equivalents to fund our operations and may restrict our ability to continue development of our product candidates. Additionally, the Loan and Security Agreement restricts our ability to incur additional pari passu indebtedness, which may reduce our ability to seek additional financing. If adequate funds are not available on a timely basis, we may terminate or delay the development of one or more of our product candidates, or delay activities necessary to commercialize our product candidates. Additional funding may not be available to us on acceptable terms, or at all. Any additional equity financing, if available, may not be available on favorable terms and will most likely be dilutive to our current stockholders, and debt financing, if available, may involve more restrictive covenants. Our ability to access capital when needed is not assured and, if not achieved on a timely basis, will materially harm our business, financial condition and results of operations or could cause us to cease operations.
As a result of our debt obligations, we will need additional funds to meet our operational needs and capital requirements for product development, clinical trials and commercialization. The timing and amount of our future capital requirements will depend on many factors, including:
| 33 |
| ● | our ability to satisfy our obligation to pay principal and interest on the Loan and Security Agreement; | |
| ● | our ability to comply with the minimum liquidity covenant related to the Loan and Security Agreement; | |
| ● | the level of sales of our current products and the cost of revenue and sales and marketing; | |
| ● | the extent of any clinical trials that we will be required to conduct in support of the regulatory clearance of our total hip and knee replacement product candidates; | |
| ● | the scope, progress, results and cost of our product development efforts; | |
| ● | the costs, timing and outcomes of regulatory reviews of our product candidates; | |
| ● | the number and types of products we develop and commercialize; | |
| ● | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; and | |
| ● | the extent and scope of our general and administrative expenses. |
We experienced additional risks and costs as a result of the delayed filing of this Form 10-K.
As a result of the circumstances giving rise to this delayed filing of this Form 10-K we experienced additional risks and costs. The audit was time-consuming, required us to incur significant incremental expenses and affected management’s attention and resources. Further, the measures to strengthen internal controls being implemented continued to require and will likely require in the future greater management time and company resources to implement and monitor. As a result of our delayed filing of this Form 10-K, we will be ineligible to register our securities on Form S-3 for sale by us or resale by others until we have timely filed all periodic reports under the Securities Exchange Act of 1934 for one year. The inability to use Form S-3 could adversely affect our ability to raise capital or complete acquisitions of other companies during this period. In addition, although we have now filed this Form 10-K, our failure to file it in a timely manner is a violation of the Securities Exchange Act of 1934 and may lead to further investigation and scrutiny by the SEC, which such investigation, and any results thereof, would require management time and company resources and could adversely affect our business and the trading price of our common stock.
If we do not adhere to the financial covenants set forth in the Loan and Security Agreement with Hercules Technology, we will be in default of the Loan and Security Agreement.
In June 2014 we entered into a Loan and Security Agreement with Hercules Technology Growth Capital, Inc., or Hercules Technology, as administrative and collateral agent for the lenders thereunder and as lender, and Hercules Technology III, LP, as lender. The Loan and Security Agreement provides us with a $20 million term loan with a maturity date of January 1, 2018 and is secured by substantially all of our assets and is described in more detail in the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of this Annual Report on Form 10-K.
The Loan and Security Agreement contains a minimum liquidity covenant that requires us to maintain cash and cash equivalents and availability under the Loan and Security Agreement of not less than an amount that varies based on the loan amount and reduces as the loan amount is reduced with a maximum cash requirement of $9.0 million if the loan amount exceeds $19.0 million and a potential minimum cash requirement of $2.5 million if the loan amount is $7.0 million or less. As of September 19, 2017, the minimum liquidity covenant was $2.5 million. We anticipate we will need to refinance the Loan and Security Agreement or obtain additional funding during the fourth quarter of 2017 to maintain compliance with the minimum liquidity covenant through the next twelve months. Furthermore, if we are unable to access additional funds prior to becoming non-compliant with the liquidity covenant, the entire remaining balance of the Loan and Security Agreement could become immediately due and payable at the option of Hercules Technology.
Hercules Technology could declare a default under the Loan and Security Agreement upon the occurrence of a material adverse effect, as defined under the credit facility, thereby requiring us to either repay the outstanding indebtedness immediately or attempt to reverse the declaration of default through negotiation or litigation. Any declaration of an event of default would significantly harm our business and prospectus and could cause the price of our common stock to decline.
Raising additional capital by issuing securities or through debt financings or licensing arrangements may cause dilution to existing stockholders, restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. Further, our current debt obligations to Hercules Technology could also impair our ability to raise future capital through equity or debt transactions. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or products or grant licenses on terms that are not favorable to us. Any of these events could adversely affect our ability to achieve our product development and commercialization goals and have a material adverse effect on our business, financial condition and results of operations.
Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements. We may be unable to continue to operate without the threat of liquidation for the foreseeable future
Our report from our independent registered public accounting firm for the year ended December 31, 2016 includes an explanatory paragraph stating that our recurring losses from operations and our need to obtain additional financing in order to satisfy our debt obligations and to be compliant with covenants under our debt obligations through 2017 raise substantial doubt about our ability to continue as a going concern. If we are unable to obtain sufficient additional funding, our business, prospects, financial condition and results of operations will be materially and adversely affected and we may be unable to continue as a going concern. If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our consolidated financial statements, and it is likely that investors will lose all or a part of their investment. Future reports from our independent registered public accounting firm may also contain statements expressing doubt about our ability to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all.
| 34 |
We have identified material weaknesses in our internal control over financial reporting and such weaknesses have led to a conclusion that our disclosure controls and procedures were not effective as of December 31, 2016. Our ability to remediate these material weaknesses, our discovery of additional weaknesses, and our inability to achieve and maintain effective disclosure controls and procedures and internal control over financial reporting, have and could continue to adversely affect our results of operations, our stock price and investor confidence in our company.
Section 404 of the Sarbanes-Oxley Act of 2002 requires that companies evaluate and report on their systems of internal control over financial reporting. As disclosed in more detail under "Controls and Procedures" in Part II, Item 9A of this Report, management has concluded that, as of December 31, 2016, our internal control over financial reporting was not effective due to the material weaknesses described below.
The design and operating effectiveness of our controls were inadequate to ensure that complex accounting matters are properly accounted for and reviewed in a timely manner. As a result, we failed to accurately record a complex equity transaction which caused the restatement of our third quarter filing. In addition we failed to properly evaluate and test certain long-lived assets for impairment, which ultimately resulted in recognition of an impairment charge. These errors are a result the following control deficiencies:
Control Environment and Risk Assessment – The Company did not have an effective control environment with the structure necessary for effective internal controls over financial reporting. Further, the Company did not have an effective risk assessment to identify and assess risks associated with changes to the Company’s structure and the impact on internal controls. With the dismissal of the Company’s CFO, the Company did not have appropriately qualified personnel to meet the Company’s control objectives. The Company does not have personnel with an appropriate level of GAAP knowledge and experience to properly review and evaluate the work performed by other Company personnel and experts related to complex accounting matters.
Control Activities – The Company did not have control activities that were designed and operating effectively including management review controls, controls related to monitoring and assessing the work of consultants, and controls to verify the completeness and adequacy of information. Specifically, the Company did not have procedures for competent personnel to review work performed by experts in relation to complex debt and equity transactions and impairment evaluations.
Monitoring Activities – The Company did not maintain effective monitoring controls related to the financial reporting process. The Company did not effectively monitor the changes in internal control related to changes in the roles and responsibilities associated with the changes in personnel and organizational structure. The failure to properly monitor impacted the timing, accuracy, and completion of the work related to significant accounting matters.
Our Chief Executive Officer and Principal Financial Officer is in the preliminary stage of a review of our controls relating to complex accounting matters. Although our analysis is not complete, we will be adding additional resources with expertise in accounting for complex accounting matters including timely review and evaluation of assets for potential impairment. We are also considering redesigning controls to add additional layers of review and approval whenever entering into or subsequently converting, exercising, amending, repricing, exiting or otherwise experiencing changes in or to complex financial instruments.
If we fail to remediate these material weaknesses and maintain an effective system of disclosure controls or internal control over financial reporting, our business and results of operations could be harmed, investors could lose confidence in our reported financial information and our ability to obtain additional financing, or additional financing on favorable terms, could be adversely affected. Any of these could result in delisting actions by the NASDAQ Stock Market, investigation and sanctions by regulatory authorities, and adversely affect our business and the trading price of our common stock. In addition, failure to maintain effective internal control over financial reporting could result in investigations or sanctions by regulatory authorities.
An impairment charge could have a material adverse effect on our financial condition and results of operations
We are required to test acquired goodwill for impairment on an annual basis. Goodwill represents the excess of the amount paid over the fair value of the net assets at the date of the acquisition. We have chosen to complete our annual impairment reviews of goodwill at the end of each calendar year. We also are required to test goodwill for impairment between annual tests if events occur or circumstances change that would more likely than not reduce our enterprise fair value below its book value. In addition, we are required to test our finite-lived intangible assets for impairment if events occur or circumstances change that would indicate the remaining net book value of the finite-lived intangible assets might not be recoverable. These events or circumstances could include a significant change in the business climate, including a significant sustained decline in our market value, legal factors, operating performance indicators, competition, sale or disposition of a significant portion of our business and other factors.
If the fair market value of our reporting unit is less than its book value, we could be required to record an impairment charge. The valuation of a reporting unit requires judgment in estimating future cash flows, discount rates and other factors. In making these judgments, we evaluate the financial health of our business, including such factors as industry performance, changes in technology and operating cash flows. Changes in our forecasts or decreases in the value of our common stock could cause book values of our reporting unit to exceed its fair value, which may result in goodwill impairment charges. The amount of any impairment could be significant and could have a material adverse effect on our reported financial results for the period in which the charge is taken.
Risks Related to Regulatory Approval of Our Products and Other Government Regulations
Our long-term success depends substantially on our ability to obtain regulatory clearance or approval and thereafter commercialize our product candidates; we cannot be certain that we will be able to do so in a timely manner or at all.
The process of obtaining regulatory clearances or approvals to market a medical device from the FDA or similar regulatory authorities outside of the United States can be costly and time consuming, and there can be no assurance that such clearances or approvals will be granted on a timely basis, or at all. The FDA’s 510(k) clearance process generally takes one to six months from the date of submission, depending on whether a special or traditional 510(k) premarket notification has been submitted, but can take significantly longer. An application for premarket approval, or PMA, must be submitted to the FDA if the device cannot be cleared through the 510(k) clearance process or is not exempt from premarket review by the FDA. The PMA process almost always requires one or more clinical trials and can take two to three years from the date of filing, or even longer. In some cases, including in the case of our interbody spinal fusion devices which incorporate our CSC technology and our solid silicon nitride femoral head component, the FDA requires clinical data as part of the 510(k) clearance process.
| 35 |
It is possible that the FDA could raise questions about our spinal fusion products, our spinal fusion product candidates and our total hip and knee joint replacement product candidates and could require us to perform additional studies on our products and product candidates. Even if the FDA permits us to use the 510(k) clearance process, we cannot assure you that the FDA will not require either supporting data from laboratory tests or studies that we have not conducted, or substantial supporting clinical data. If we are unable to use the 510(k) clearance process for any of our product candidates, are required to provide clinical data or laboratory data that we do not possess to support our 510(k) premarket notifications for any of these product candidates, or otherwise experience delays in obtaining or fail to obtain regulatory clearances, the commercialization of our product candidates in the United States will be delayed or prevented, which will adversely affect our ability to generate additional revenues. It also may result in the loss of potential competitive advantages that we might otherwise attain by bringing our products to market earlier than our competitors. Additionally, although the FDA allows modifications to be made to devices that have received 510(k) clearance with supporting documentation, the FDA may disagree with our decision to modify our cleared devices without submission of a new 510(k) premarket notification, subjecting us to potential product recall, field alerts and corrective actions. Any of these contingencies could adversely affect our business.
Similar to our compliance with U.S. regulatory requirements, we must obtain and comply with international requirements, including those of the CFDA, in order to market and sell our products outside of the United States and we may only promote and market our products, if approved, as permitted by applicable regulatory authorities. There is no guarantee that we will receive the necessary regulatory approvals for our product candidates either inside the United States or internationally, including approvals from the CFDA. If our product candidates do not receive necessary regulatory approvals, our business could be materially and adversely affected.
The safety of our products is not yet supported by long-term clinical data, and they may prove to be less safe and effective than our laboratory data indicate.
We obtained FDA clearance for each of our products that we currently market, and we have sought and intend to seek FDA clearance or approval through the FDA’s 510(k) or PMA process and, where applicable, CE marking for our product candidates. The 510(k) clearance process is based on the FDA’s agreement that a new product candidate is substantially equivalent to an already marketed product for which a PMA was not required. While most 510(k) premarket notifications do not require clinical data for clearance, the FDA may request that such data be provided. Long-term clinical data or marketing experience obtained after clearance may indicate that our products cause unexpected complications or other unforeseen negative effects. If this happens, we could be subject to the withdrawal of our marketing clearance and other enforcement sanctions by the FDA or other regulatory authority, product recalls, significant legal liability, significant negative publicity, damage to our reputation and a dramatic reduction in our ability to sell our products, any one of which would have a material adverse effect on our business, financial condition and results of operations.
We expect to be required to conduct clinical trials to support regulatory approval of some of our product candidates. We have little experience conducting clinical trials, they may proceed more slowly than anticipated, and we cannot be certain that our product candidates will be shown to be safe and effective for human use.
In order to commercialize our product candidates in the United States, we must submit a PMA for some of these product candidates, which will require us to conduct clinical trials. We also plan to provide the FDA with clinical trial data to support some of our 510(k) premarket notifications. We will receive approval or clearance from the FDA to commercialize products requiring a clinical trial only if we can demonstrate to the satisfaction of the FDA, through well-designed and properly conducted clinical trials, that our product candidates are safe and effective and otherwise meet the appropriate standards required for approval or clearance for specified indications.
Clinical trials are complex, expensive, time consuming, uncertain and subject to substantial and unanticipated delays. Before we may begin clinical trials, we must submit and obtain approval for an investigational device exemption, or IDE, that describes, among other things, the manufacture of, and controls for, the device and a complete investigational plan. Clinical trials generally involve a substantial number of patients in a multi-year study. Because we do not have the experience or the infrastructure necessary to conduct clinical trials, we will have to hire one or more contract research organizations, or CROs, to conduct trials on our behalf. CRO contract negotiations may be costly and time consuming and we will rely heavily on the CRO to ensure that our trials are conducted in accordance with regulatory and industry standards. We may encounter problems with our clinical trials and any of those problems could cause us or the FDA to suspend those trials, or delay the analysis of the data derived from them.
A number of events or factors, including any of the following, could delay the completion of our clinical trials in the future and negatively impact our ability to obtain FDA approval for, and to introduce our product candidates:
| ● | failure to obtain financing necessary to bear the cost of designing and conducting clinical trials; | |
| ● | failure to obtain approval from the FDA or foreign regulatory authorities to commence investigational studies; | |
| ● | conditions imposed on us by the FDA or foreign regulatory authorities regarding the scope or design of our clinical trials; | |
| ● | failure to find a qualified CRO to conduct our clinical trials or to negotiate a CRO services agreement on favorable terms; | |
| ● | delays in obtaining or in our maintaining required approvals from institutional review boards or other reviewing entities at clinical sites selected for participation in our clinical trials; | |
| ● | insufficient supply of our product candidates or other materials necessary to conduct our clinical trials; |
| 36 |
| ● | difficulties in enrolling patients in our clinical trials; | |
| ● | negative or inconclusive results from clinical trials, or results that are inconsistent with earlier results, that necessitate additional clinical studies; | |
| ● | failure on the part of the CRO to conduct the clinical trial in accordance with regulatory requirements; | |
| ● | our failure to maintain a successful relationship with the CRO or termination of our contractual relationship with the CRO before completion of the clinical trials; | |
| ● | serious or unexpected side effects experienced by patients in whom our product candidates are implanted; or | |
| ● | failure by any of our third-party contractors or investigators to comply with regulatory requirements or meet other contractual obligations in a timely manner. |
Our clinical trials may need to be redesigned or may not be completed on schedule, if at all. Delays in our clinical trials may result in increased development costs for our product candidates, which could cause our stock price to decline and limit our ability to obtain additional financing. In addition, if one or more of our clinical trials are delayed, competitors may be able to bring products to market before we do, and the commercial viability of our product candidates could be significantly reduced.
Our current and future relationships with third-party payers and current and potential customers in the United States and elsewhere may be subject, directly or indirectly, to applicable anti-kickback, fraud and abuse, false claims, transparency, health information privacy and security and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm administrative burdens and diminished profits and future earnings.
Our current and future arrangements with third-party payers and current and potential customers, including providers and physicians, as well as physician owned distributorships or PODs, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations, including, without limitation, the federal Anti-Kickback Statute and the federal False Claims Act, which may constrain the business or financial arrangements and relationships through which we sell, market and distribute our products. In addition, we may be subject to transparency laws and patient privacy regulations by U.S. federal and state governments and by governments in foreign jurisdictions in which we conduct our business. The applicable federal, state and foreign healthcare laws and regulations that may affect our ability to operate include:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal healthcare programs, such as Medicare and Medicaid; | |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, including the federal False Claims Act, which impose criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose obligations on covered healthcare providers, health plans, and healthcare clearinghouses, as well as their business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| 37 |
| ● | the Physician Payments Sunshine Act, which requires (i) manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to CMS information related to certain “payments or other transfers of value” made to physicians, which is defined to include doctors, dentists, optometrists, podiatrists and chiropractors, and teaching hospitals, with data collection beginning on August 1, 2013, (ii) applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held in such entities by physicians and their immediate family members, with data collection beginning on August 1, 2013, (iii) manufacturers to submit reports to CMS by March 31, 2014 and the 90th day of each subsequent calendar year, and (iv) disclosure of such information by CMS on a publicly available website beginning in September 2014; and | |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers; state and foreign laws that require medical device companies to comply with the medical device industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state and foreign laws that require medical device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, which could have a material adverse effect on our business. If any of the physicians or other healthcare providers or entities with whom we expect to do business, including our collaborators, are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from participation in government healthcare programs, which could also materially affect our business. |
Recently enacted and future legislation may increase the difficulty and cost for us to obtain and monitor regulatory approval or clearance of our product candidates and affect the prices we may obtain for our products.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay clearance and/or approval of our product candidates, restrict or regulate post-clearance and post-approval activities and affect our ability to profitably sell our products and any product candidates for which we obtain marketing approval or clearance.
In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of or failure to receive regulatory clearances or approvals for our new products would have a material adverse effect on our business, results of operations and financial condition. In addition, the FDA is currently evaluating the 510(k) process and may make substantial changes to industry requirements, including which devices are eligible for 510(k) clearance, the ability to rescind previously granted 510(k) clearances and additional requirements that may significantly impact the process.
Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access. In the United States, the medical device industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the ACA, a sweeping law intended, among other things, to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Among the provisions of the ACA of importance to our products and product candidates are:
| ● | a 2.3% medical device excise tax on the U.S. sales of most medical devices, for which a moratorium on the payment of the excise tax for 2016 and 2017 was enacted in December 2015; | |
| ● | expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, and new government investigative powers and enhanced penalties for non-compliance; |
| 38 |
| ● | new requirements under the federal Open Payments program and its implementing regulations; | |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and | |
| ● | creation of an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, on August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or ATRA, which, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. On March 1, 2013, the President signed an executive order implementing the Budget Control Act’s 2% Medicare payment reductions, and on April 1, 2013, these reductions went into effect. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our financial operations.
We expect that the ACA, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for our products. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may affect our ability to generate revenue and profits or commercialize our product candidates.
In the European Union and some other international markets, the government provides health care at a low cost to consumers and regulates prices of healthcare products, patient eligibility or reimbursement levels to control costs for the government-sponsored health care system. Many countries are reducing their public expenditures and we expect to see strong efforts to reduce healthcare costs in international markets, including patient access restrictions, suspensions on price increases, prospective and possibly retroactive price reductions and other recoupments and increased mandatory discounts or rebates and recoveries of past price increases. These cost control measures could reduce our revenues. In addition, certain countries set prices by reference to the prices in other countries where our products are marketed. Thus, our inability to secure adequate prices in a particular country may not only limit the marketing of our products within that country, but may also adversely affect our ability to obtain acceptable prices in other markets. This may create the opportunity for third-party cross border trade or influence our decision to sell or not to sell a product, thus adversely affecting our geographic expansion plans and revenues.
Risks Related to Our Intellectual Property and Litigation
If the combination of patents, trade secrets and contractual provisions that we rely on to protect our intellectual property is inadequate, our ability to commercialize our orthopedic products successfully will be harmed, and we may not be able to operate our business profitably.
Our success depends significantly on our ability to protect our proprietary rights to the technologies incorporated in our products. We rely on a combination of patent protection, trade secret laws and nondisclosure, confidentiality and other contractual restrictions to protect our proprietary technology. However, these may not adequately protect our rights or permit us to gain or keep any competitive advantage.
The issuance of a patent is not conclusive as to its scope, validity or enforceability. The scope, validity or enforceability of our issued patents can be challenged in litigation or proceedings before the U.S. Patent and Trademark Office, or the USPTO, or foreign patent offices. In addition, our pending patent applications include claims to numerous important aspects of our products under development that are not currently protected by any of our issued patents. We cannot assure you that any of our pending patent applications will result in the issuance of patents to us. The USPTO or foreign patent offices may deny or require significant narrowing of claims in our pending patent applications. Patents issued as a result of the pending patent applications, if any, may not provide us with significant commercial protection or be issued in a form that is advantageous to us. Proceedings before the USPTO or foreign patent offices could result in adverse decisions as to the priority of our inventions and the narrowing or invalidation of claims in issued patents. The laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States, if at all.
| 39 |
Our competitors may successfully challenge and invalidate or render unenforceable our issued patents, including any patents that may issue in the future, which could prevent or limit our ability to market our products and could limit our ability to stop competitors from marketing products that are substantially equivalent to ours. In addition, competitors may be able to design around our patents or develop products that provide outcomes that are comparable to our products but that are not covered by our patents.
We have also entered into confidentiality and assignment of intellectual property agreements with all of our employees, consultants and advisors as one of the ways we seek to protect our intellectual property and other proprietary technology. However, these agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements.
In the event a competitor infringes upon any of our patents or other intellectual property rights, enforcing our rights may be difficult, time consuming and expensive, and would divert management’s attention from managing our business. There can be no assurance that we will be successful on the merits in any enforcement effort. In addition, we may not have sufficient resources to litigate, enforce or defend our intellectual property rights.
We have no patent protection covering the composition of matter for our solid silicon nitride or the process we use for manufacturing our solid silicon nitride, and competitors may create silicon nitride formulations substantially similar to ours.
Although we have a number of U.S. and foreign patents and pending applications relating to our solid silicon nitride products or product candidates, we have no patent protection either for the composition of matter for our silicon nitride or for the processes of manufacturing solid silicon nitride. As a result, competitors may create silicon nitride formulations substantially similar to ours, and use their formulations in products that may compete with our silicon nitride products, provided they do not violate our issued product patents. Although we have, and will continue to develop, significant know-how related to these processes, there can be no assurance that we will be able to maintain this know-how as trade secrets, and competitors may develop or acquire equally valuable or more valuable know-how related to the manufacture of silicon nitride.
We could become subject to intellectual property litigation that could be costly, result in the diversion of management’s time and efforts, require us to pay damages, prevent us from marketing our commercially available products or product candidates and/or reduce the margins we may realize from our products that we may commercialize.
The medical devices industry is characterized by extensive litigation and administrative proceedings over patent and other intellectual property rights. Whether a product infringes a patent involves complex legal and factual issues, and the determination is often uncertain. There may be existing patents of which we are unaware that our products under development may inadvertently infringe. The likelihood that patent infringement claims may be brought against us increases as the number of participants in the orthopedic market increases and as we achieve more visibility in the market place and introduce products to market.
Any infringement claim against us, even if without merit, may cause us to incur substantial costs, and would place a significant strain on our financial resources, divert the attention of management from our core business, and harm our reputation. In some cases, litigation may be threatened or brought by a patent holding company or other adverse patent owner who has no relevant product revenues and against whom our patents may provide little or no deterrence. If we were found to infringe any patents, we could be required to pay substantial damages, including triple damages if an infringement is found to be willful, and royalties and could be prevented from selling our products unless we obtain a license or are able to redesign our products to avoid infringement. We may not be able to obtain a license enabling us to sell our products on reasonable terms, or at all, and there can be no assurance that we would be able to redesign our products in a way that would not infringe those patents. If we fail to obtain any required licenses or make any necessary changes to our technologies or the products that incorporate them, we may be unable to commercialize one or more of our products or may have to withdraw products from the market, all of which would have a material adverse effect on our business, financial condition and results of operations.
In addition, in order to further our product development efforts, we have entered into agreements with orthopedic surgeons to help us design and develop new products, and we expect to enter into similar agreements in the future. In certain instances, we have agreed to pay such surgeons royalties on sales of products which incorporate their product development contributions. There can be no assurance that surgeons with whom we have entered into such arrangements will not claim to be entitled to a royalty even if we do not believe that such products were developed by cooperative involvement between us and such surgeons. In addition, some of our surgeon advisors are employed by academic or medical institutions or have agreements with other orthopedic companies pursuant to which they have agreed to assign or are under an obligation to assign to those other companies or institutions their rights in inventions which they conceive or develop, or help conceive or develop.
| 40 |
There can be no assurance that one or more of these orthopedic companies or institutions will not claim ownership rights to an invention we develop in collaboration with our surgeon advisors or consultants on the basis that an agreement with such orthopedic company or institution gives it ownership rights in the invention or that our surgeon advisors on consultants otherwise have an obligation to assign such inventions to such company or institution. Any such claim against us, even without merit, may cause us to incur substantial costs, and would place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation.
We may be subject to damages resulting from claims that we, our employees, or our independent sales agencies have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition agreements with our competitors or non-solicitation agreements.
Many of our employees were previously employed at other orthopedic companies, including our competitors and potential competitors. Many of our distributors and potential distributors sell, or in the past have sold, products of our competitors. We may be subject to claims that either we, or these employees or distributors, have inadvertently or otherwise used or disclosed the trade secrets or other proprietary information of our competitors. In addition, we have been and may in the future be subject to claims that we caused an employee or sales agent to break the terms of his or her non-competition agreement or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money damages, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize products, which could have an adverse effect on our business, financial condition and results of operations.
If our silicon nitride products or our product candidates conflict with the rights of others, we may not be able to manufacture or market our products or product candidates, which could have a material and adverse effect on us.
Our commercial success will depend in part on not infringing the patents or violating the other proprietary rights of third parties. Issued patents held by others may limit our ability to develop commercial products. All issued patents are entitled to a presumption of validity under the laws of the United States. If we need suitable licenses to such patents to permit us to develop or market our product candidates, we may be required to pay significant fees or royalties and we cannot be certain that we would even be able to obtain such licenses. Competitors or third parties may obtain patents that may cover subject matter we use in developing the technology required to bring our products to market, that we use in producing our products, or that we use in treating patients with our products. We know that others have filed patent applications in various jurisdictions that relate to several areas in which we are developing products. Some of these patent applications have already resulted in patents and some are still pending. If we were found to infringe any of these issued patents or any of the pending patent applications, when and if issued, we may be required to alter our processes or product candidates, pay licensing fees or cease activities. If use of technology incorporated into or used to produce our product candidates is challenged, or if our processes or product candidates conflict with patent rights of others, third parties could bring legal actions against us, in Europe, the United States and elsewhere, claiming damages and seeking to enjoin manufacturing and marketing of the affected products. Additionally, it is not possible to predict with certainty what patent claims may issue from pending applications. In the United States, for example, patent prosecution can proceed in secret prior to issuance of a patent, provided such application is not filed in foreign jurisdiction. For U.S. patent applications that are also filed in foreign jurisdictions, such patent applications will not publish until 18 months from the filing date of the application. As a result, third parties may be able to obtain patents with claims relating to our product candidates which they could attempt to assert against us. Further, as we develop our products, third parties may assert that we infringe the patents currently held or licensed by them, and we cannot predict the outcome of any such action.
There has been extensive litigation in the medical devices industry over patents and other proprietary rights. If we become involved in any litigation, it could consume a substantial portion of our resources, regardless of the outcome of the litigation. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license, grant cross-licenses and pay substantial royalties in order to continue to manufacture or market the affected products.
We cannot assure you that we would prevail in any legal action or that any license required under a third party patent would be made available on acceptable terms, or at all. Ultimately, we could be prevented from commercializing a product, or forced to cease some aspect of our business operations, as a result of claims of patent infringement or violation of other intellectual property rights, which could have a material and adverse effect on our business, financial condition and results of operations.
| 41 |
Risks Related to Potential Litigation from Operating Our Business
We may become subject to potential product liability claims, and we may be required to pay damages that exceed our insurance coverage.
Our business exposes us to potential product liability claims that are inherent in the design, testing, manufacture, sale and distribution of our currently marketed products and each of our product candidates that we are seeking to introduce to the market. The use of orthopedic medical devices can involve significant risks of serious complications, including bleeding, nerve injury, paralysis, infection, and even death. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates or in our inability to secure coverage in the future on commercially reasonable terms, if at all. In addition, if our product liability insurance proves to be inadequate to pay a damage award, we may have to pay the excess of this award out of our cash reserves, which could significantly harm our financial condition. If longer-term patient results and experience indicate that our products or any component of a product causes tissue damage, motor impairment or other adverse effects, we could be subject to significant liability. A product liability claim, even one without merit, could harm our reputation in the industry, lead to significant legal fees, and result in the diversion of management’s attention from managing our business.
Any claims relating to our improper handling, storage or disposal of biological or hazardous materials could be time consuming and costly.
Although we do not believe that the manufacture of our silicon nitride or non-silicon nitride products will involve the use of hazardous materials, it is possible that regulatory authorities may disagree or that changes to our manufacturing processes may result in such use. Our business and facilities and those of our suppliers and future suppliers may therefore be subject to foreign, federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We may incur significant expenses in the future relating to any failure to comply with environmental laws. Any such future expenses or liability could have a significant negative impact on our business, financial condition and results of operations.
Risks Related to Our Common Stock
The price of our common stock is volatile and is likely to continue to fluctuate due to reasons beyond our control.
The volatility of orthopedic company stocks, including shares of our common stock, often do not correlate to the operating performance of the companies represented by such stocks or our operating performance. Some of the factors that may cause the market price of our common stock to fluctuate include:
| ● | our ability to sell our current products and the cost of revenue; | |
| ● | our ability to develop, obtain regulatory clearances or approvals for, and market new and enhanced product candidates on a timely basis; | |
| ● | our ability to enter into OEM and private label partnership agreements and the terms of those agreements; | |
| ● | changes in governmental regulations or in the status of our regulatory approvals, clearances or future applications; | |
| ● | our announcements or our competitors’ announcements regarding new products, product enhancements, significant contracts, number and productivity of distributors, number of hospitals and surgeons using products, acquisitions or strategic investments; | |
| ● | announcements of technological or medical innovations for the treatment of orthopedic pathology; | |
| ● | delays or other problems with the manufacturing of our products, product candidates and related instrumentation; | |
| ● | volume and timing of orders for our products and our product candidates, if and when commercialized; | |
| ● | changes in the availability of third-party reimbursement in the United States and other countries; | |
| ● | quarterly variations in our or our competitors’ results of operations; |
| 42 |
| ● | changes in earnings estimates or recommendations by securities analysts, if any, who cover our common stock; | |
| ● | failure to meet estimates or recommendations by securities analysts, if any, who cover our stock; | |
| ● | changes in the fair value of our derivative liabilities resulting from changes in the market price of our common stock, which may result in significant fluctuations in our quarterly and annual operating results; | |
| ● | changes in healthcare policy in the United States and internationally; | |
| ● | product liability claims or other litigation involving us; | |
| ● | sales of a substantial aggregate number of shares of our common stock; | |
| ● | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders; | |
| ● | disputes or other developments with respect to intellectual property rights; | |
| ● | changes in accounting principles; | |
| ● | changes to tax policy; and | |
| ● | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors. |
These and other external factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent our stockholders from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the company that issued the stock. If our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit regardless of the merits of the case or the eventual outcome. Such a lawsuit also would divert the time and attention of our management from running our company.
Securities analysts may not continue to provide coverage of our common stock or may issue negative reports, which may have a negative impact on the market price of our common stock.
Since completing our initial public offering of shares of our common stock in February 2014, a limited number of securities analysts have begun providing research coverage of our common stock. If securities analysts do not continue to cover our common stock, the lack of research coverage may cause the market price of our common stock to decline. The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about our business. If one or more of the analysts who elect to cover us downgrade our stock, our stock price would likely decline rapidly. If one or more of these analysts cease coverage of us, we could lose visibility in the market, which in turn could cause our stock price to decline. In addition, under the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and a global settlement among the Securities and Exchange Commission, or the SEC, other regulatory agencies and a number of investment banks, which was reached in 2003, many investment banking firms are required to contract with independent financial analysts for their stock research. It may be difficult for a company such as ours, with a smaller market capitalization, to attract independent financial analysts that will cover our common stock. This could have a negative effect on the market price of our stock.
Anti-takeover provisions in our organizational documents and Delaware law may discourage or prevent a change in control, even if an acquisition would be beneficial to our stockholders, which could affect our stock price adversely and prevent attempts by our stockholders to replace or remove our current management.
Our restated certificate of incorporation and restated bylaws contain provisions that could discourage, delay or prevent a merger, acquisition or other change in control of our company or changes in our board of directors that our stockholders might consider favorable, including transactions in which you might receive a premium for your shares. These provisions also could limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove management. These provisions:
| 43 |
| ● | allow the authorized number of directors to be changed only by resolution of our board of directors; | |
| ● | provide for a classified board of directors, such that not all members of our board will be elected at one time; | |
| ● | prohibit our stockholders from filling board vacancies, limit who may call stockholder meetings, and prohibit the taking of stockholder action by written consent; | |
| ● | prohibit our stockholders from making certain changes to our restated certificate of incorporation or restated bylaws except with the approval of holders of 75% of the outstanding shares of our capital stock entitled to vote; | |
| ● | require advance written notice of stockholder proposals that can be acted upon at stockholders meetings and of director nominations to our board of directors; and | |
| ● | authorize our board of directors to create and issue, without prior stockholder approval, preferred stock that may have rights senior to those of our common stock and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our board of directors. |
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. Any delay or prevention of a change in control transaction or changes in our board of directors could cause the market price of our common stock to decline.
We do not intend to pay cash dividends.
We have never declared or paid cash dividends on our capital stock and we do not anticipate paying any cash dividends in the foreseeable future. We currently intend to retain all available funds and any future earnings for debt service and use in the operation and expansion of our business. The Loan and Security Agreement contains a negative covenant which prohibits us from paying dividends to our stockholders without the prior written consent of Hercules Technology. In addition, the terms of any future debt or credit facility may preclude us from paying any dividends.
Risks Related to Public Companies
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 and a “smaller reporting company” and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including (1) not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, (2) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (3) exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. Additionally, under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We are electing to delay such adoption of new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. As a result of this election, our financial statements may not be comparable to the financial statements of other public companies.
We may take advantage of these exemptions until we are no longer an emerging growth company. Under the JOBS Act, we may be able to maintain emerging growth company status for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700 million as of any June 30 before the end of such five-year period or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31. Additionally, if we issue more than $1.0 billion in non-convertible debt during any three-year period before that time, we would cease to be an emerging growth company immediately.
We are also currently a “smaller reporting company” as defined in the Securities Exchange Act of 1934, and in the event that we are still considered a smaller reporting company at such time as we cease being an emerging growth company, we will be required to provide additional disclosure in our SEC filings. However, similar to emerging growth companies, smaller reporting companies are able to provide simplified executive compensation disclosures in their filings, are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting, and have certain other decreased disclosure obligations in their SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. We cannot predict whether investors will find our common stock less attractive because of our reliance on any of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
| 44 |
Our common stock may be subject to delisting from The NASDAQ Stock Market because the bid price of our common stock price dropped below $1.00 for a period of 30 consecutive business days or because we have not timely filed our Quarterly Reports 10-Q for quarters ended April 30, 2017 and June 30, 2017.
On August 17, 2016, the Company received a letter from the Listing Qualifications Staff (the “Staff”) of The NASDAQ Stock Market LLC (“Nasdaq”) indicating that the bid price for the Company’s common stock had closed below the minimum $1.00 per share threshold for continued listing on The Nasdaq Capital, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Bid Price Rule”), for the prior 30 consecutive trading days. In accordance with Nasdaq Listing Rule 5810(c)(3)(A), the Company was ultimately provided with two consecutive 180 calendar day periods to demonstrate compliance with the Bid Price Rule, which expired on August 14, 2017. The Company did not regain compliance with the Bid Price Rule by August 14, 2017; accordingly, on August 22, 2017, the Company received notice from the Staff that based upon the Company’s continued non-compliance with the Rule, the Company’s securities would be subject to delisting from Nasdaq unless the Company timely requested a hearing before the Nasdaq Hearings Panel (the “Panel”). The Staff’s August 22, 2017 notice also indicated that because the Company is delinquent in filing its Form 10-K for the period ended December 31, 2016, and Forms 10-Q for the periods ended March 31, 2017 and June 30, 2017, it is not in compliance with Nasdaq Listing Rule 5250(c)(1) (the “Filing Rule”), which could also serve as a separate basis for delisting. The Company timely requested a hearing and has now received notice from Nasdaq indicating that a Nasdaq Hearings Panel (the "Panel") granted the Company's request to extend the stay of the suspension of trading in the Company's common stock pending the Company's scheduled hearing on October 12, 2017 before the Panel and a final determination regarding the Company's listing status. At the October 12, 2017 hearing, the Company will present its plan to evidence compliance with Nasdaq's listing requirements. The Company is diligently working to evidence compliance with Nasdaq 's listing requirements as soon as possible; however, there can be no assurance that the Panel will grant the Company's request for continued listing and a further stay of suspension. If our common stock is delisted, it would adversely impact liquidity of our common stock and potentially result in lower bid prices for our common stock.
We incur substantial costs as a result of being a public company and our management expects to devote substantial time to public company compliance programs.
As a public company, we incur significant legal, insurance, accounting and other expenses, including costs associated with public company reporting. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment will result in increased general and administrative expenses and may divert management’s time and attention from product development and commercialization activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us, and our business may be harmed. These laws and regulations could make it more difficult and costly for us to obtain director and officer liability insurance for our directors and officers, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified executive officers and qualified members of our board of directors, particularly to serve on our audit and compensation committees. In addition, if we are unable to continue to meet the legal, regulatory and other requirements related to being a public company, we may not be able to maintain the listing of our common stock on The NASDAQ Capital Market, which would likely have a material adverse effect on the trading price of our common stock.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
| ITEM 2. | PROPERTIES |
Our 54,000 square foot corporate office and manufacturing facilities are located in Salt Lake City, Utah. We occupy these facilities pursuant to a lease that expires in December 2019. Pursuant to the terms of the lease agreement, we may extend the lease for two additional periods of five years each. We believe that our existing facilities are adequate for our current and projected needs for the foreseeable future. The company is subleasing approximately 6,500 square feet to a third party through April of 2018.
| ITEM 3. | LEGAL PROCEEDINGS |
We are currently not a party to any material legal proceedings. However, our industry is characterized by frequent claims and litigation, including claims regarding intellectual property and product liability. As a result, we may be subject to various legal proceedings in the future.
| 45 |
| ITEM 4. | MINE SAFETY DISCLOSURES |
This item does not apply to our business.
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our shares of common stock are currently quoted on The NASDAQ Capital Market under the symbol “AMDA”.
The following table sets forth the high and low sale prices of our common stock, as reported by NASDAQ Capital Markets for the periods indicated:
| 2016 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 3.62 | $ | 1.16 | * | |||
| Second Quarter | $ | 2.80 | $ | 1.22 | ||||
| Third Quarter | $ | 1.40 | $ | 0.60 | ||||
| Fourth Quarter | $ | 1.09 | $ | 0.60 | ||||
| 2015 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 16.80 | $ | 4.86 | ||||
| Second Quarter | $ | 12.15 | $ | 3.06 | ||||
| Third Quarter | $ | 12.59 | $ | 4.52 | ||||
| Fourth Quarter | $ | 7.02 | $ | 1.35 | ||||
* Prices listed are adjusted to reflect the 1/25/16 reverse stock split
Holders of Record
As of September 19, 2017, we had approximately 388 holders of record of our common stock. Because many of our shares of common stock are held by brokers and other institutions on behalf of stockholders, this number is not indicative of the total number of stockholders represented by these stockholders of record.
Dividends
We have not declared or paid dividends to stockholders since inception and do not plan to pay cash dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance our growth.
Issuer Purchases of Equity Securities
None
| ITEM 6. | SELECTED FINANCIAL DATA |
Not applicable.
| 46 |
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes appearing elsewhere in this Annual Report. This discussion and analysis contains forward-looking statements based upon current beliefs, plans, expectations, intentions and projections that involve risks, uncertainties and assumptions, such as statements regarding our plans, objectives, expectations, intentions and projections. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under “Risk Factors” and elsewhere in this Annual Report.
Overview
We are a materials company focused on developing, manufacturing and selling silicon nitride ceramics that are used in medical implants and in a variety of industrial devices. At present, we commercialize silicon nitride in the spine implant market. We believe that our facile silicon nitride manufacturing expertise positions us favorably to introduce new and innovative devices in the medical and non-medical fields. We also believe that we are the first and only company to commercialize silicon nitride medical implants.
We have received 510(k) regulatory clearance in the United States, a CE mark in Europe, and ANVISA approval in Brazil for a number of our devices that are designed for spinal fusion surgery. To date, more than 28,000 of our silicon nitride devices have been implanted into patients, with an 8-year successful track record. In December 2016, we re-filed an FDA 510(k) submission for clearance in the United States of a modified novel composite spinal fusion device that combines porous and solid silicon nitride, and obviates the need for bone grafts that is comparable to our commercially-available Valeo®C cervical implants.
We believe that silicon nitride has a superb combination of properties that make it ideally suited for human implantation. Other biomaterials are based on bone grafts, metal alloys, and polymers; all of which have practical limitations. In contrast, silicon nitride has a legacy of success in the most demanding and extreme industrial environments. As a human implant material, silicon nitride offers bone ingrowth, resistance to bacterial infection, resistance to corrosion, superior strength and fracture resistance, and ease of diagnostic imaging, among other advantages.
We market and sell our Valeo brand of silicon nitride implants to surgeons and hospitals in the United States and to selected markets in Europe and South America through more than 50 independent sales distributors who are supported by an in-house sales and marketing management team. These implants are designed for use in cervical (neck) and thoracolumbar (lower back) spine surgery. In 2016 we entered into a 10-year exclusive distribution agreement with Shandong Weigao Orthopaedic Device Company Limited (“Weigao”) to sell Amedica-branded silicon nitride spinal fusion devices within the People’s Republic of China (“China”). Weigao, a large orthopedic company, has expertise in acquiring Chinese Food and Drug Administration (“CFDA”) approval of medical devices, and will assist us in obtaining regulatory approval. Weigao has committed to minimum purchase requirements totaling 225,000 implants in the first six years following CFDA clearance. We are also working with other partners in Japan to obtain regulatory approval for silicon nitride in that country. China and Japan are relevant because historically, ceramic implants are more familiar to, and more readily accepted by surgeons outside the United States, i.e., in Asia and Europe.
In addition to silicon nitride, we also sell metal-based products in the United States that provide surgeons and hospitals with a complete package for spinal surgery. These metal products are designed to address spinal deformity and degenerative conditions. Although these metal products have accounted for approximately 48% of our product revenues for each of the years ended December 31, 2016 and 2015, respectively, we remain focused on developing and promoting silicon nitride, and driving its adoption through a scientifically-intense, data-driven strategy.
In addition to direct sales, we have targeted original equipment manufacturer (“OEM”) and private label partnerships in order to accelerate adoption of silicon nitride, both in the spinal space, and also in future markets such as hip and knee replacements, dental, extremities, trauma, and sports medicine. Existing biomaterials, based on plastics, metals, and bone grafts have well-recognized limitations that we believe are addressed by silicon nitride, and we are uniquely positioned to convert existing, successful implant designs made by other companies into silicon nitride. We believe OEM and private label partnerships will allow us to work with a variety of partners, accelerate the adoption of silicon nitride, and realize incremental revenue at improved operating margins, when compared to the cost-intensive direct sales model.
| 47 |
We believe that silicon nitride addresses many of the biomaterial-related limitations in fields such as hip and knee replacements, dental implants, sports medicine, extremities, and trauma surgery. We further believe that the inherent material properties of silicon nitride, and the ability to formulate the material in a variety of compositions, combined with precise control of the surface properties of the material, opens up a number of commercial opportunities across orthopedic surgery, neurological surgery, maxillofacial surgery, and other medical disciplines.
We operate a 30,000 square foot manufacturing facility at our corporate headquarters in Salt Lake City, Utah, and we believe we are the only vertically integrated silicon nitride medical device manufacturer in the world.
Components of our Results of Operations
We manage our business within one reportable segment, which is consistent with how our management reviews our business, makes investment and resource allocation decisions and assesses operating performance.
Product Revenue
We derive our product revenue primarily from the sale of spinal fusion devices and related products used in the treatment of spine disorders. Our product revenue is generated from sales to three types of customers: (1) surgeons and hospitals; (2) stocking distributors; and (3) private label customers. Most of our products are sold on a consignment basis through a network of independent sales distributors; however, we also sell our products to independent stocking distributors and private label customers. Product revenue is recognized when all four of the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products has occurred; (3) the selling price of the product is fixed or determinable; and (4) collectability is reasonably assured. We generate the majority of our revenue from the sale of inventory that is consigned to independent sales distributors that sell our products to surgeons and hospitals. For these products, we recognize revenue at the time we are notified the product has been used or implanted and all other revenue recognition criteria have been met. For all other transactions, we recognize revenue when title and risk of loss transfer to the stocking distributor or private label customers, and all other revenue recognition criteria have been met. We generally recognize revenue from sales to stocking distributors and private label customers at the time the product is shipped to the distributor. Stocking distributors, who sell the products to their customers, take title to the products and assume all risks of ownership at time of shipment. Our stocking distributors are obligated to pay within specified terms regardless of when, if ever, they sell the products. In general, our customers do not have any rights of return or exchange.
We believe our product revenue from the sale of our silicon nitride based products will increase due to our sales and marketing efforts and as we introduce new silicon nitride based products into the market and product revenue from the sale of our non-silicon nitride products to remain flat. We expect that our product revenue will continue to be primarily attributable to sales of our products in the United States.
Costs of Revenue
The expenses that are included in costs of revenue include all direct product costs if we obtained the product from third-party manufacturers and our in-house manufacturing costs for the products we manufacture. We obtain our non-silicon nitride products, including our metal and orthobiologic products, from third-party manufacturers, while we currently manufacture our silicon-nitride products in-house.
Specific provisions for excess or obsolete inventory and the excise tax on the sale of medical devices in the United States, are also included in cost of revenue. In addition, we pay royalties attributable to the sale of specific products to some of our surgeon advisors that assisted us in the design, regulatory clearance or commercialization of a particular product, and these payments are recorded as cost of revenue. In addition the Company has incurred impairment charges relating to the ceramic equipment used to manufacture our implants.
Gross Profit
Our gross profit measures our product revenue relative to our costs of revenue. We expect our gross profit to decrease as we expand the penetration of our silicon nitride technology platform through OEM and private label partnerships.
Research and Development Expenses
Our net research and development costs are expensed as incurred. Research and development costs consist of engineering, product development, clinical trials, test-part manufacturing, testing, developing and validating the manufacturing process, manufacturing, facility and regulatory-related costs. Research and development expenses also include employee compensation, employee and non-employee stock-based compensation, supplies and materials, consultant services, and travel and facilities expenses related to research activities. To the extent that certain research and development expenses are directly related to our manufactured products, such expenses and related overhead costs are allocated to inventory.
| 48 |
We expect to incur additional research and development costs as we continue to develop new spinal fusion products, our product candidates for total joint replacements, such as our total hip replacement product candidate, and our silicon nitride-coated metals which may increase our total research and development expenses.
Sales and Marketing Expenses
Sales and marketing expenses consist of salaries, benefits and other related costs, including stock-based compensation, for personnel employed in sales, marketing, medical education and training. In addition, our sales and marketing expenses include commissions and bonuses, generally based on a percentage of sales, to our sales managers and independent sales distributors. We provide our products in kits or banks that consist of a range of device sizes and separate instruments necessary to complete the surgical procedure. We generally consign our instruments to our distributors or our hospital customers that purchase the device used in spinal fusion surgery. Our sales and marketing expenses include depreciation and impairment charges of the surgical instruments.
We expect our sales and marketing expenses will remain flat or slightly decline due to the recently implemented cost saving measures. Additionally, we expect our commissions to continue to increase in absolute terms over time but remain approximately the same or decrease as a percentage of product revenue.
General and Administrative Expenses
General and administrative expenses primarily consist of salaries, benefits and other related costs, including stock-based compensation, for certain members of our executive team and other personnel employed in finance, legal, compliance, administrative, information technology, customer service, executive and human resource departments. General and administrative expenses include allocated facility expenses, related travel expenses and professional fees for accounting, legal services, and impairment charges of leasehold improvements.
Results of Operations
Year Ended December 31, 2016 Compared to the Year Ended December 31, 2015
The following table sets forth, for the periods indicated, our results of operations for the years ended December 31, 2016 and 2015 (in thousands):
| Year Ended December 31, | ||||||||||||||||
| 2016 | 2015 | $ Change | % Change | |||||||||||||
| Product revenue | $ | 15,226 | $ | 19,453 | $ | (4,227 | ) | -21.73 | % | |||||||
| Costs of revenue | 3,777 | 6,250 | (2,473 | ) | -39.57 | % | ||||||||||
| Gross profit | 11,449 | 13,203 | (1,754 | ) | -13.28 | % | ||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 6,345 | 6,387 | (42) | -0.66 | % | |||||||||||
| General and administrative | 6,292 | 6,436 | (144 | ) | -2.24 | % | ||||||||||
| Sales and marketing | 10,347 | 12,421 | (2,074 | ) | -16.70 | % | ||||||||||
| Total operating expenses | 22,984 | 25,244 | (2,260 | ) | -8.95 | % | ||||||||||
| Loss from operations | (11,535 | ) | (12,041 | ) | 506 | 4.20 | % | |||||||||
| Other income (expense) | (5,063 | ) | (11,871 | ) | 6,808 | 57.35 | % | |||||||||
| Net loss before income taxes | (16,598 | ) | (23,912 | ) | 7,314 | 30.59 | % | |||||||||
| Provision for income taxes | - | - | - | N/A | ||||||||||||
| Net loss | $ | (16,598 | ) | $ | (23,912 | ) | $ | 7,314 | 30.59 | % | ||||||
| Deemed dividend related to beneficial conversion feature and accretion of discount on convertible series A preferred stock | (6,278 | ) | - | (6,278 | ) | N/A | ||||||||||
| Net loss attributable to common stockholders | $ | (22,876 | ) | $ | (23,912 | ) | $ | 1,036 | 4.33 | % | ||||||
| 49 |
Product Revenue
The following table sets forth our product revenue from sales of the indicated product category for the years ended December 31, 2016 and 2015 (in thousands):
| Year Ended December 31, | ||||||||||||||||
| 2016 | 2015 | $ Change | % Change | |||||||||||||
| Silicon Nitride | $ | 7,896 | $ | 10,121 | $ | (2,225 | ) | -22 | % | |||||||
| Non-Silicon Nitride | $ | 7,330 | $ | 9,332 | $ | (2,002 | ) | -21 | % | |||||||
| Total product revenue | $ | 15,226 | $ | 19,453 | $ | (4,227 | ) | -22 | % | |||||||
Total product revenue was $15.2 million in 2016 as compared to $19.4 million in 2015, a decrease of $4.2 million or 22%. This decline was due to silicon nitride sales decreasing by $2.2 million, or 22%, and non-silicon nitride sales decreasing by $2.0 million, or 21%, as compared to the same period in 2015. This decline was primarily attributable to the loss of a few surgeons during the year and consequences from our restructuring. The remaining decrease was due to reduced private label sales.
The following table sets forth, for the periods indicated, our product revenue by geographic area (in thousands):
| Year Ended December 31, | ||||||||||||||||
| 2016 | 2015 | $ Change | % Change | |||||||||||||
| Domestic | $ | 14,919 | $ | 19,293 | $ | (4,374 | ) | -23 | % | |||||||
| International | $ | 307 | $ | 160 | $ | 147 | 92 | % | ||||||||
| Total product revenue | $ | 15,226 | $ | 19,453 | $ | (4,227 | ) | -22 | % | |||||||
International revenue increased in 2016 as compared to 2015 primarily as a result of having received regulatory approval to begin selling our silicon nitride products in Brazil.
Costs of Revenue and Gross Profit
Our cost of revenue decreased $2.5 million, or 40%, as compared to the same period in 2015. The decrease was primarily due to the decline in sales and the moratorium on the medical device excise tax. This decrease was offset by an increase of $0.2 of impairment charges relating to our manufacturing assets.
Research and Development Expenses
Research and development expenses decreased $0.04 million, or 1 %, as compared to the same period in 2015. This decrease was primarily due to reductions in personnel related expenses.
General and Administrative Expenses
General and administrative expenses decreased $0.1 million, or 2%, as compared to the same period in 2015. This decrease was primarily due to a $0.6 million decrease in personnel related expenses, 40% of these reductions were in the fourth quarter, a $0.1 million decrease in franchise taxes, a $0.1 million decrease in board of director expenses, a $0.1 million decrease in investor relations expenses, $0.1 million decrease in insurance expenses, along with another $0.2 million decrease in various other expenses. These decreases were offset by a $0.9 million increase in legal fees and impairment charges for 2016 in the amount of $0.2 million from the impairment of leasehold improvements. There were no impairments during 2015.
Sales and Marketing Expenses
Sales and marketing expenses decreased $2.1 million, or 17%, as compared to the same period in 2015. This decrease was primarily due to a $1.8 million decrease in commissions due to lower sales, and the remaining decrease is due to $0.8 million reductions in personnel related costs, travel, consulting and other operating expenses as we reduced operating costs in 2016. These decreases were offset by a $0.5 million in impairment charges of instrument sets. There were no impairments during 2015.
Deemed Dividend
Deemed dividend in 2016 related to a beneficial conversion feature and accretion of discount on convertible preferred stock valued at $6.3 million, compared to none for 2015. A beneficial conversion amount was calculated in association with the 2016 issuance of certain convertible preferred stock and warrants that could convert to common stock at a discount below the trading price on the date of issuance. The preferred stock was actually converted to common stock during 2016. No such stock was issued or converted during 2015.
| 50 |
Other Income (Expense), Net
Other income (expenses), net decreased $6.8 million, or 57%. The decrease was primarily due to a $7.7 million decrease in the change in the fair value of derivative liabilities, a $1.3 million decrease in loss on extinguishment of derivative liabilities, and a $0.8 million decrease in offering costs. These improvements were offset by a $2.8 million dollar increase in loss on extinguishment of debt, and a $0.2 million increase in interest expense.
Liquidity and Capital Resources
The consolidated financial statements have been prepared assuming we will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and does not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to our ability to continue as a going concern within one year from the date of issuance of our consolidated financial statements.
For the years ended December 31, 2016 and 2015, we incurred a net loss of $16.6 million and $23.9 million, respectively, and used cash in operations of $7.2 million and $9.1 million, respectively. We had an accumulated deficit of $213.1 million and $196.5 million at December 31, 2016 and 2015, respectively. To date, our operations have been principally financed from proceeds from the issuance of preferred and common stock, convertible debt and bank debt and, to a lesser extent, cash generated from product sales. It is anticipated that we will continue to generate operating losses and use cash in operations for the foreseeable future. Our continuation as a going concern is dependent upon our ability to increase sales, implement cost saving measures, maintain compliance with debt covenants and/or raise additional funds through the capital markets. Whether and when we can attain profitability and positive cash flows from operations or obtain additional financing is uncertain.
In 2016 we implemented certain cost saving measures, including workforce and office space reductions, and will continue to evaluate additional cost savings alternatives during 2017. These additional cost savings measures may include additional workforce and research and development reductions, as well as reductions to certain other operating expenses including legal, patent, and travel expenses. In addition to these cost saving measures an experienced and highly successful leader for the Sales and Marketing team was recruited and hired. This individual has subsequently hired additional experienced personnel in Sales and Market Development. We are actively generating additional scientific and clinical data to have it published in leading industry publications. The unique features of our silicon nitride material are not well known, and the publication of such data would help sales efforts as we approach new prospects. We are also making additional changes to the sales strategy, including a focus on revenue growth of silicon nitride lateral lumbar implants and the newly developed pedicle screw system (known as Taurus).
As discussed further in Note 7 to the consolidated financial statements, in June 2014, we entered into a term loan with Hercules Technology Growth Capital, Inc. (“Hercules Technology”), as administrative and collateral agent for the lenders thereunder and as lender, and Hercules Technology III, LP, (“HT III” and, together with Hercules Technology, “Hercules”) as lender (the “Hercules Term Loan”). The Hercules Term Loan has a liquidity covenant that requires us to maintain a cash balance of not less than $3.0 million at December 31, 2016. At December 31, 2016, our cash balance was approximately $6.9 million. We believe we will be in position to maintain compliance with the liquidity covenant related to the Hercules Term Loan into the fourth quarter of 2017. To maintain compliance beyond that date, we would need to refinance the note or obtain additional funding in or prior to the fourth quarter of 2017. We have common stock that is publicly traded and have been able to successfully raise capital when needed since the time of our initial public offering. We are engaged in discussions with an investment banking firm to examine financing alternatives, including options to encourage the exercise of outstanding warrants. The Company is also seeking to refinance the Hercules Term Loan and is in discussions with banking firms to look at lending alternatives.
If we are unable to refinance the Hercules Term Loan or access additional funds prior to becoming non-compliant with the financial and liquidity covenants related to the Hercules Term Loan, the entire remaining balance of the debt under the Hercules Term Loan could become immediately due and payable at the option of the lender. Although we are seeking to refinance the note or obtain additional debt financing, such funding is not assured and may not be available to us on favorable or acceptable terms, and may involve restrictive covenants. Any additional equity financing is also not assured and, if available to us, will most likely be dilutive to its current stockholders. If we are not able to obtain additional debt or equity financing on a timely basis, the impact on us will be material and adverse.
These uncertainties create substantial doubt about our ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of these uncertainties.
Cash Flows
The following table summarizes, for the periods indicated, cash flows from operating, investing and financing activities (in thousands):
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Net cash used in operating activities | $ | (7,170 | ) | $ | (9,063 | ) | ||
| Net cash used by investing activities | (617 | ) | (658 | ) | ||||
| Net cash provided by financing activities | 3,217 | 2,959 | ||||||
| Net cash used | $ | (4,570 | ) | $ | (6,762 | ) | ||
Net Cash Used in Operating Activities
Net cash used in operating activities was $7.2 million in 2016, compared to $9.1 million used in 2015, a decrease of $1.9 million, or 21%. The decrease in cash used in operating activities during 2016 was primarily due to a decrease in the net loss of $8.2 million, which equated to a decrease in the net loss after non-cash add backs of $1.7 million. The net change in operating assets and liabilities had the effect of decreasing the net cash used in operating activities by another $200,000, which when added to the $1.7 million accounts for the $1.9 million overall decrease in net cash used in operating activities.
| 51 |
Net Cash Provided by Investing Activities
Net cash used in investing activities was $0.6 million during 2016, compared to $0.7 million used in investing activities during the same period in 2015, a decrease of $0.1 million. The decrease in net cash used in investing activities during 2016 was primarily attributable to decreased purchases of property and equipment and an increase in the proceeds from sale of property and equipment.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $3.2 million during 2016, compared to $3.0 million provided during the same period in 2015, an increase of $0.2 million. The increase in cash provided by financing activities in 2016 was primarily attributable to the July 2016 stock offering, which was offset by the principal debt payments to Hercules after the July stock offering.
Indebtedness
Magna Note
In August 2014, the Company entered into a Securities Purchase Agreement with Magna pursuant to which the Company sold to Magna an unsecured promissory note with an aggregate principal amount of $3.5 million (the “Magna Note”). In July 2016, the Company paid Magna $888,000 to redeem in full the remaining principal balance and interest related to the Magna Note. The outstanding principal amount of the Magna Note at extinguishment was $763,000. The Magna Note would have matured on August 11, 2016, and accrued interest at an annual rate of 6.0%.
Hercules and Riverside Debt Exchange
On April 4, 2016, the Company entered into an Assignment and Second Amendment to Loan and Security Agreement (the “Assignment Agreement”) with Riverside Merchant Partners, LLC (“Riverside”), and Hercules, pursuant to which Hercules sold $1.0 million of the principal amount outstanding under the Hercules Term Loan to Riverside. In addition, pursuant to the terms of the Assignment Agreement, Riverside acquired an option to purchase an additional $2.0 million of the principal amount outstanding under the Hercules Term Loan from Hercules. On April 18, 2016, Riverside exercised and purchased an additional $1.0 million of the principal amount of the Hercules Term Loan and on April 27, 2016, Riverside exercised the remainder of its option and purchased an additional $1.0 million of the principal amount of the Hercules Term Loan from Hercules.
Riverside Debt
On April 4, 2016, the Company entered into an exchange agreement (the “Exchange Agreement”) with Riverside, pursuant to which the Company agreed to exchange $1.0 million of the principal amount outstanding under the Hercules Term Loan held by Riverside for a subordinated convertible promissory note in the principal amount of $1.0 million (the “First Exchange Note”) and a warrant to purchase 100,000 shares of common stock of the Company at a fixed exercise price of $1.62 per share (the “First Exchange Warrant”) (the “Exchange”). All principal accrued under the Exchange Notes was convertible into shares of common stock at the election of the Holder at any time at a fixed conversion price of $1.43 per share (the “Conversion Price”). The closing stock price on April 4, 2016, was $1.63 and a beneficial conversion feature of $246,000 was recorded to equity and as a debt discount. The warrant value of $106,000 was recorded to equity and as a debt discount.
In addition, pursuant to the terms and conditions of the Exchange Agreement, the Company and Riverside had the option to exchange an additional $2.0 million of the principal amount of the Hercules Term Loan for an additional subordinated convertible promissory note in the principal amount of up to $2.0 million and an additional warrant to purchase 100,000 shares of common stock (the “Second Exchange Warrant”). The Exchange Agreement also provided that if the volume-weighted average price of the Company’s common stock was less than the Conversion Price, the Company would issue up to an additional 150,000 shares of common stock (the “True-Up Shares”) to Riverside, which was subsequently reduced to 140,000 shares of common stock.
| 52 |
On April 18, 2016, the Company and Riverside exercised their option to exchange an additional $1.0 million of the principal amount of the Hercules Term Loan for an additional subordinated convertible promissory note in the principal amount of $1.0 million (the “Second Exchange Note”). The closing stock price on April 18, 2016, was $2.02 and a beneficial conversion feature of $413,000 was recorded to equity and as a debt discount. Additionally, on April 27, 2016, the Company and Riverside exercised their option to exchange an additional $1.0 million of the principal amount of the Term Loan for an additional subordinated convertible promissory note in the principal amount of $1.0 million (the “Third Exchange Note”) and an additional warrant to purchase 100,000 shares of the Company’s common stock at a fixed exercise price of $1.66 per share. The warrant value of $107,000 was recorded to equity and as a debt discount. The closing stock price on April 27, 2016, was $1.66 and a beneficial conversion feature of $268,000 was recorded to equity and as a debt discount. Financing costs were $267,000 and were recorded to interest expense. The unamortized deferred financing costs and debt discount of the Hercules Term Loan exchanged were $244,000 at the time of the exchange and were recorded as a loss on extinguishment of debt related to the debt exchange. The First Exchange Note, the Second Exchange Note and the Third Exchange Note are collectively referred to herein as the “Exchange Notes.”
Pursuant to the terms of the Exchange Notes, since the volume-weighted average price of the Company’s common stock was less than the Conversion Price on May 6, 2016, the Company issued an additional 140,000 shares of common stock to Riverside and recorded the value of the True-Up Shares of $199,000 to interest expense and equity.
All principal outstanding under each of the Exchange Notes was to be due on April 3, 2018 (the “Maturity Date”). Each of the Exchange Notes bore interest at a rate of 6% per annum, with the interest that would accrue on the initial principal amount of the Exchange Notes during the first 12 months being guaranteed and deemed earned as of the date of issuance. Prior to the Maturity Date, all interest accrued under the Exchange Notes was payable in cash or, if certain conditions were met, payable in shares of common stock at the Company’s option, at a conversion price of $1.34 per share. The entire principal amount of the First and Second Exchange Notes, $300,000 of the Third Exchange Note, and the interest related to the First, Second, and Third Exchange Notes has been converted into 1,742,718 shares of common stock. In July 2016, the Company paid Riverside $840,000 to redeem in full the remaining principal balance of the Third Exchange Note. The debt discounts associated with the converted debt was recorded to interest expense.
Contractual Obligations and Commitments
The following table summarizes our outstanding contractual obligations as of December 31, 2016 (in thousands):
| Total | Less Than 1 Year | 1 Year | 2-3 Years | 4-5 Years | After 5 Years | |||||||||||||||||||
| Long-term debt, gross | $ | 7,421 | $ | 6,779 | $ | 642 | $ | - | $ | - | ||||||||||||||
| Operating leases, net | 2,857 | 925 | 952 | 980 | ||||||||||||||||||||
| Total contractual obligations | $ | 10,278 | $ | 7,704 | $ | 1,594 | $ | 980 | $ | - | ||||||||||||||
The information above reflects only payment obligations that are fixed and determinable. The long-term debt disclosed above is gross of debt issuance costs, and outlines the contractual maturities before considering maturity acceleration due to non-compliance with loan covenants. Our commitments for long-term debt relate to our term loan with Hercules, our commitments for operating leases relate to our operating lease for our corporate headquarters and manufacturing facility in Salt Lake City, Utah and other equipment leases. The Hercules loan requires a liquidity covenant of $3.0 million. The Company believes that it is in a position to maintain compliance with the liquidity covenant into the fourth quarter of 2017, and has therefore classified the entire obligation as a current liability. The above table does not include any of the contractual obligations with respect to royalties payable upon sales of certain of our products as none of our arrangements contain minimum royalty payments. We also do not have contractually minimum purchase commitments for the supply of any of our raw materials, products or instruments.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, as defined in Item 303(a)(4) of Regulation S-K.
Related-Party Transactions
For a description of our related-party transactions, see “Certain Relationships and Related Party Transactions.”
Seasonality and Backlog
Our business is generally not seasonal in nature. Our sales generally consist of products that are in stock with us or maintained at hospitals or with our sales distributors. Accordingly, we do not have a backlog of sales orders.
Critical Accounting Policies and Estimates
The preparation of the consolidated financial statements requires us to make assumptions, estimates and judgments that affect the reported amounts of assets and liabilities, the disclosures of contingent assets and liabilities as of the date of the consolidated financial statements, and the reported amounts of product revenues and expenses during the reporting periods. Certain of our more critical accounting policies require the application of significant judgment by management in selecting the appropriate assumptions for calculating financial estimates. By their nature, these judgments are subject to an inherent degree of uncertainty. On an ongoing basis, we evaluate our judgments, including those related to inventories, recoverability of long-lived assets the fair value of our common stock, and goodwill. We use historical experience and other assumptions as the basis for our judgments and making these estimates. Because future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Any changes in those estimates will be reflected in our consolidated financial statements as they occur. As an “emerging growth company,” we have elected to delay the adoption of new or revised accounting standards until those standards would otherwise apply to private companies. As a result, our financial statements may not be comparable to those of other public companies. While our significant accounting policies are more fully described in the footnotes to our consolidated financial statements included elsewhere in this Annual Report, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results. The critical accounting policies addressed below reflect our most significant judgments and estimates used in the preparation of our consolidated financial statements.
| 53 |
Revenue Recognition
We derive our product revenue primarily from the sale of spinal fusion devices and related products used in the treatment of spine disorders. Our product revenue is generated from sales to three types of customers: (1) surgeons and hospitals; (2) stocking distributors; and (3) private label customers. Most of our products are sold on a consignment basis through a network of independent sales distributors; however, we also sell our products to independent stocking distributors. Product revenue is recognized when all four of the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products has occurred; (3) the selling price of the product is fixed or determinable; and (4) collectability is reasonably assured. We generate the majority of our revenue from the sale of inventory that is consigned to independent sales distributors that sell our products to surgeons and hospitals. For these products, we recognize revenue at the time we are notified the product has been used or implanted and all other revenue recognition criteria have been met. For all other transactions, we recognize revenue when title and risk of loss transfer to the stocking distributor, and all other revenue recognition criteria have been met. We generally recognize revenue from sales to stocking distributors at the time the product is shipped to the distributor. Stocking distributors, who sell the products to their customers, take title to the products and assume all risks of ownership at time of shipment. Our stocking distributors are obligated to pay within specified terms regardless of when, if ever, they sell the products. In general, our customers do not have any rights of return or exchange.
Accounts Receivable and Allowance for Doubtful Accounts
The majority of our accounts receivable is composed of amounts due from hospitals or surgical centers. Accounts receivable are carried at cost less an allowance for doubtful accounts. On a regular basis, we evaluate accounts receivable and estimate an allowance for doubtful accounts, as needed, based on various factors such as customers’ current credit conditions, length of time past due, and the general economy as a whole. Receivables are written off against the allowance when they are deemed uncollectible.
Inventories
Inventories are stated at the lower of cost or market, with cost for manufactured inventory determined under the standard cost method which approximates the first-in first-out method. Manufactured inventory consists of raw material, direct labor and manufacturing overhead cost components. Inventories purchased from third-party manufacturers are stated at the lower of cost or market using the first-in, first out method. We review the carrying value of inventory on a periodic basis for excess or obsolete items and record an expense for the identified items as necessary. We have made adjustments to, and it is reasonably possible that we may be required to make further adjustments to, the carrying value of inventory in future periods. We hold some consigned inventory at distributors and other customer locations where revenue recognition criteria have not yet been met.
Long-Lived Assets and Goodwill
The Company periodically evaluates the carrying value of definitely-lived intangibles when events or changes in circumstances indicate that the carrying value may not be recoverable. Factors the Company considers important which could trigger an impairment review include, but are not limited to, significant under-performance relative to historical or projected future operating results, significant changes in the manner of its use of acquired assets or its overall business strategy, and significant industry or economic trends. The Company amortizes finite-lived intangible assets on a straight-line basis over their useful lives. The Company recorded no impairment loss for definite-lived intangible assets during the years ended December 31, 2016 and 2015.
When the Company determines that the carrying value of a long-lived asset may not be recoverable based upon the existence of one or more of the above indicators, the Company determines the recoverability by comparing the carrying amount of the asset to net future undiscounted cash flows that the asset is expected to generate and recognizes an impairment charge equal to the amount by which the carrying amount exceeds the fair market value of the asset.
If our revenues or other estimated operating results are not achieved at or above our forecasted level, and we are unable to recover such costs through price increases, the carrying value of certain of our assets may prove to be unrecoverable and we may incur impairment charges of definitive-live intangible assets.
In accordance with ASC 350, Goodwill and Other Intangible Assets, goodwill is not amortized but is required to be reviewed for impairment at least annually or when events or circumstances indicate that carrying value may exceed fair value. The Company is permitted the option to first assess qualitative factors to determine whether the existence of events and circumstances indicates that it is more likely than not that the fair value of any reporting unit is less than its corresponding carrying value. If, after assessing the totality of events and circumstances, the Company concludes that it is not more likely than not that the fair value of any reporting unit is less than its corresponding carrying value then the Company is not required to take further action. However, if the Company concludes otherwise, then it is required to perform a quantitative impairment test, including computing the fair value of the reporting unit and comparing that value to its carrying value. The Company considers valuation factors and an estimated control premium. The estimated fair value of the reporting unit exceeded the carrying value by approximately 20%. The declining price of the Company’s stock is an early indicator that goodwill impairment may be a factor during 2017. We will continue to monitor our market capitalization and impairment indicators.
If the fair value is less than its carrying value, a second step of the test is required to determine if recorded goodwill is impaired. In the event that goodwill is impaired, an impairment charge to earnings would become necessary.
| 54 |
Property and Equipment
Property and equipment, including surgical instruments and leasehold improvements, are stated at cost, less accumulated depreciation and amortization. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets, which range from three to five years. Leasehold improvements are amortized over the shorter of their estimated useful lives or the related lease term, generally five years.
Periodically we review the carrying value of our property and equipment that are held and used in our operations for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of these assets is determined based upon expected undiscounted future net cash flows from the operations to which the assets relate, utilizing management’s best estimate, assumptions, and projections at the time. If the carrying value is determined to be unrecoverable from future operating cash flows, the asset is deemed impaired and an impairment charge would be recognized to the extent the carrying value exceeded the estimated fair value of the asset. We estimate the fair value of assets based on the estimated future discounted cash flows of the asset. Management has evaluated its property and equipment and has identified asset impairment during the year ended December 31, 2016.
Income Taxes
We recognize deferred tax assets and liabilities for the future tax consequences attributable to the differences between the financial statement carrying value of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates in effect for the fiscal year in which those temporary differences are expected to be recovered or settled. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
We operate in various tax jurisdictions and are subject to audit by various tax authorities. We provide for tax contingencies whenever it is deemed probable that a tax asset has been impaired or a tax liability has been incurred for events such as tax claims or changes in tax laws. Tax contingencies are based upon their technical merits relative tax law and the specific facts and circumstances as of each reporting period. Changes in facts and circumstances could result in material changes to the amounts recorded for such tax contingencies.
We recognize uncertain income tax positions taken on income tax returns at the largest amount that is more-likely than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
Our policy for recording interest and penalties associated with uncertain tax positions is to record such items as a component of our income tax provision. For the years ended December 31, 2016 and 2015, we did not record any material interest income, interest expense or penalties related to uncertain tax positions or the settlement of audits for prior periods.
Stock-Based Compensation
We apply the fair value recognition provisions of Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 718, Compensation-Stock Compensation, or ASC 718. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of the fair value of stock options and other equity awards as of their grant date. Stock-based compensation expense is recognized ratably over the requisite service period, which in most cases is the vesting period of the award. Calculating the fair value of stock-based awards requires that we make highly subjective assumptions. Use of this valuation methodology requires that we make assumptions as to the volatility of our common stock, the expected term of our stock options, the risk-free rate of return for a period that approximates the expected term of our stock options and our expected dividend yield. Because we were a privately-held company with no trading history prior to February 2014 and have limited stock history since February 2014, we utilize the historical stock price volatility from a representative group of public companies to estimate expected stock price volatility and our historical stock price. We selected companies from the medical device industry, specifically those who are focused on the design, development and commercialization of products for the treatment of spine disorders, and who have similar characteristics to us, such as stage of life cycle and size. We intend to continue to utilize the historical volatility of the same or similar public companies to estimate expected volatility until a sufficient amount of historical information regarding the price of our publicly traded stock becomes available. We use the simplified method as prescribed by the Securities and Exchange Commission Staff Accounting Bulletin No. 107, Share-based Payment, to calculate the expected term of stock option grants to employees as we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term of stock options granted to employees. We utilize a dividend yield of zero because we have never paid cash dividends and have no current intention to pay cash dividends. The risk-free rate of return used for each grant is based on the U.S. Treasury yield curve in effect at the time of grant for instruments with a similar expected life.
| 55 |
The estimated fair value of stock-based awards for employee and non-employee director services is expensed over the requisite service period. Option awards issued to non-employees, excluding non-employee directors, are recorded at their fair value as determined in accordance with authoritative guidance, are periodically revalued as the options vest and are recognized as expense over the related service period. As a result, the charge to operations for non-employee awards with vesting conditions is affected each reporting period by changes in the fair value of our common stock.
We are required to estimate the level of forfeitures expected to occur and record stock-based compensation expense only for those awards that we ultimately expect will vest. We estimate our forfeiture rate based on the type of award, employee class and historical experience.
Derivative Liabilities
Derivative liabilities includes the fair value of instruments such as common stock warrants, preferred stock warrants and convertible features of notes, that are initially recorded at fair value and are required to be re-measured to fair value at each reporting period under provisions of ASC 480, Distinguishing Liabilities from Equity, or ASC 815, Derivatives and Hedging. The change in fair value of the instruments is recognized as a component of other income (expense), net in our consolidated statements of operations until the instruments settle or expire. We estimate the fair value of these instruments using the Black-Scholes-Merton or Monte-Carlo valuation models depending on the complexity of the underlying instruments. The significant assumptions used in estimating the fair value include the exercise price, volatility of the stock underlying the instrument, risk-free interest rate, estimated fair value of the stock underlying the instrument and the estimated life of the instrument.
Recently Issued Accounting Pronouncements
In July 2015, the FASB issued ASU 2015-11, “Inventory (Topic 330) Simplifying the Measurement of Inventory”. The amendments clarify that an entity should measure inventory within the scope of this update at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Substantial and unusual losses that result from subsequent measurement of inventory should be disclosed in the financial statements. This guidance is effective for fiscal years beginning after December 15, 2016, including interim periods within those annual periods. The amendments are to be applied prospectively with earlier application permitted as of the beginning of an interim or annual reporting period. This guidance is not expected to have a material impact on the consolidated financial statements.
In August 2016, the Financial Accounting Standards Board (“FASB”) updated accounting guidance on the following eight specific cash flow classification issues: (1) debt prepayment or debt extinguishment costs; (2) settlement of zero-coupon debt instruments or other debt instruments with coupon interest rates that are insignificant in relation to the effective interest rate of the borrowing; (3) contingent consideration payments made after a business combination; (4) proceeds from the settlement of insurance claims; (5) proceeds from the settlement of corporate-owned life insurance policies, including bank-owned life insurance policies; (6) distributions received from equity method investees; (7) beneficial interests in securitization transactions; and (8) separately identifiable cash flows and application of the predominance principle. Current GAAP does not include specific guidance on these eight cash flow classification issues. These updates are effective for the Company for its annual period beginning January 1, 2019, and interim periods therein, with early adoption permitted. The guidance in this standard is not expected to have a material impact on the consolidated financial statements.
In March 2016 the FASB updated the accounting guidance related to stock compensation. This update simplifies the accounting for employee share-based payment transactions, including the accounting for income taxes, forfeitures, and statutory tax withholding requirements, as the well as classification in the statement of cash flows. The standard is effective for the Company for its annual period beginning January 1, 2018. The guidance in this standard is not expected to have a material impact on the consolidated financial statements of the Company.
| 56 |
In February 2016, the FASB updated the accounting guidance related to leases as part of a joint project with the International Accounting Standards Board (“IASB”) to increase transparency and comparability among organizations by recognizing lease assets and lease liabilities on the balance sheet and disclosing key information about leasing arrangements. Under the new guidance, a lessee will be required to recognize assets and liabilities for capital and operating leases with lease terms of more than 12 months. Additionally, this update will require disclosures to help investors and other financial statement users better understand the amount, timing, and uncertainty of cash flows arising from leases, including qualitative and quantitative requirements. The standard is effective for the Company for its annual period beginning January 1, 2020, and interim periods therein, with early adoption permitted. The Company is currently evaluating the potential impact this new standard may have on its consolidated financial statements, but believes the most significant change will relate to building leases.
In May 2014, in addition to several amendments issued during 2016, the FASB updated the accounting guidance related to revenue from contracts with customers, which supersedes nearly all existing revenue recognition guidance under U.S. GAAP. The core principle is that a company should recognize revenue when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. The standard defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP. The standard is effective for the Company for its annual period beginning January 1, 2019, and interim periods therein, and shall be applied either retrospectively to each period presented or as a cumulative-effect adjustment as of the date of adoption. The Company has yet to begin evaluation of the new accounting standard and therefore has yet to determine the impact, if any, that the new standard will have on its consolidated financial statements.
In January of 2017, the FASB issued ASU 2017-04—Intangibles—Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment. The amendments in this guidance to eliminate the requirement to calculate the implied fair value of goodwill to measure goodwill impairment charge (Step 2). As a result, an impairment charge will equal the amount by which a reporting unit’s carrying amount exceeds its fair value, not to exceed the amount of goodwill allocated to the reporting unit. An entity still has the option to perform the qualitative assessment for a reporting unit to determine if the quantitative impairment test is necessary. The amendment should be applied on a prospective basis. The guidance is effective for goodwill impairment tests in fiscal years beginning after December 15, 2021. Early adoption is permitted for goodwill impairment tests performed after January 1, 2017. The impact of this guidance for the Company will depend on the outcomes of future goodwill impairment tests.
The Company has reviewed all other recently issued, but not yet adopted, accounting standards, in order to determine their effects, if any, on its results of operations, financial position or cash flows. Based on that review, the Company believes that none of these pronouncements will have a significant effect on its consolidated financial statements.
Jumpstart Our Business Startups Act of 2012
On April 5, 2012, the Jumpstart Our Business Startups Act of 2012, or JOBS Act, was enacted. Section 107 of the JOBS Act, provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are electing to delay such adoption of new or revised accounting standards, and as a result, we may not comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. As a result of this election, our financial statements may not be comparable to the financial statements of other public companies. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including without limitation, (1) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (2) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the consolidated financial statements, known as the auditor discussion and analysis. We may be able to remain an “emerging growth company” until the earliest of (a) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more, (b) the last day of our fiscal year following the fifth anniversary of the date of our IPO, (c) the date on which we have issued more than $1 billion in non-convertible debt during the previous three years or (d) the date on which we are deemed to be a large accelerated filer under the rules of the SEC. Additionally, we are also currently a “smaller reporting company” as defined in the Securities Exchange Act of 1934, and in the event that we are still considered a smaller reporting company at such time as we cease being an emerging growth company, we will be exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting.
| 57 |
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Not applicable.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Financial Statements
The consolidated financial statements of Amedica appear at the end of this Annual Report beginning with the index to Financial Statements on page F-1 (see Part IV, Item 15 “Financial Statements”), and are incorporated herein.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
As previously reported in our Current Report on Form 8-K filed on July 22, 2016, on July 1, 2016, Mantyla McReynolds, LLC (“Mantyla”) merged with BDO USA, LLP (“BDO”). As a result of this transaction, Mantyla resigned, effective as of July 19, 2016 (the “Resignation Date”), as the Company’s independent registered public accounting firm.
Mantyla’s reports on the Company’s consolidated financial statements as of and for the fiscal years ended December 31, 2015 and 2014 contained an emphasis paragraph that raised substantial doubt about its ability to continue as a going concern. Other than the going concern matter, the reports of Mantyla on the consolidated financial statements of the Company for the fiscal years ended December 31, 2015 and 2014 did not contain any other adverse opinion or disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
During the Company’s two most recent fiscal years and the subsequent interim period through the Resignation Date, the Company and Mantyla did not have any disagreements on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure which, if not resolved to the satisfaction of Mantyla, would have caused Mantyla to make reference to the matter in its reports on the Company’s consolidated financial statements during such periods; and there were no “reportable events” as the term is described in Item 304(a)(1)(v) of Regulation S-K.
The Company requested Mantyla furnish a letter addressed to the Securities and Exchange Commission, pursuant to Item 304(a)(3) of Regulation S-K, stating whether or not Mantyla agrees with the above statements, which letter we filed as Exhibit 16.1 to our Current Report on Form 8-K filed on July 22, 2016.
The Audit Committee recommended and approved the engagement of BDO as the successor independent registered public accounting firm, effective upon July 22, 2016. At no time during the Company’s fiscal years ended December 31, 2015 and 2014 and during any subsequent interim period through the Resignation Date, did the Company consult with BDO regarding (i) the application of accounting principles to a specific completed or contemplated transaction, or the type of audit opinion that might be rendered on the Company’s consolidated financial statements, and no written report or oral advice was provided to the Company that BDO concluded was an important factor considered by the Company in reaching a decision as to any accounting, auditing or financial reporting issue or (ii) any matter that was the subject of a disagreement as defined in Item 304(a)(1)(iv) and related instructions of Regulation S-K or a “reportable event” as described in Item 304(a)(1)(v) of Regulation S-K.
| ITEM 9A. | CONTROLS AND PROCEDURES |
(a) Disclosure Controls and Procedures
We maintain disclosure controls and procedures, as such term is defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934 (the “Exchange Act”), that are designed to ensure that information required to be disclosed in the reports filed or submitted under the Exchange Act, is recorded, processed, summarized, and reported within the time periods specified by the Commission’s rules and forms. Disclosure controls and procedures include controls and procedures designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act are properly recorded, processed, summarized and reported within the time periods required by the Commission’s rules and forms.
We carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer (principal executive officer and principal financial officer), of the effectiveness of the design and operation of these disclosure controls and procedures, as such term is defined in Exchange Act Rule 13a-15(e), as of December 31, 2016. Based on this evaluation, the Chief Executive Officer concluded that our disclosure controls and procedures were not effective as of December 31, 2016, the end of the period covered by this Annual Report on Form 10-K due to the material weaknesses described below.
(b) Management’s Report on Internal Control over Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act.
Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness of internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our internal control over financial reporting is designed to provide reasonable assurance of achieving its objectives as specified above. Management does not expect, however, that our internal control over financial reporting will prevent or detect all error and fraud. Any control system, no matter how well designed and operated, is based upon certain assumptions and can provide only reasonable, not absolute, assurance that its objectives will be met. Further, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within the Company have been detected.
Management, including our Chief Executive Officer, has assessed the effectiveness of our internal control over financial reporting as of December 31, 2016. In making our assessment of the effectiveness of internal control over financial reporting, management used the criteria set forth in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, management has concluded that, as of December 31, 2016, our internal control over financial reporting was not effective due to the material weaknesses described below.
(c) Material Weaknesses
As defined in SEC Regulation S-X, a material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. Based on this assessment, management determined that, as of December 31, 2016, the Company's internal control over financial reporting was not effective because of the material weaknesses described below.
The design and operating effectiveness of our controls were inadequate to ensure that complex accounting matters are properly accounted for and reviewed in a timely manner. As a result, we failed to accurately record a complex equity transaction which caused the restatement of our third quarter filing. In addition we failed to properly evaluate and test certain long-lived assets for impairment, which ultimately resulted in recognition of an impairment charge. These errors are a result the following control deficiencies:
Control Environment and Risk Assessment – The Company did not have an effective control environment with the structure necessary for effective internal controls over financial reporting. Further, the Company did not have an effective risk assessment to identify and assess risks associated with changes to the Company’s structure and the impact on internal controls. With the dismissal of the Company’s CFO, the Company did not have appropriately qualified personnel to meet the Company’s control objectives. The Company does not have personnel with an appropriate level of GAAP knowledge and experience to properly review and evaluate the work performed by other Company personnel and experts related to complex accounting matters.
Control Activities – The Company did not have control activities that were designed and operating effectively including management review controls, controls related to monitoring and assessing the work of consultants, and controls to verify the completeness and adequacy of information. Specifically, the Company did not have procedures for competent personnel to review work performed by experts in relation to complex debt and equity transactions and impairment evaluations.
Monitoring Activities – The Company did not maintain effective monitoring controls related to the financial reporting process. The Company did not effectively monitor the changes in internal control related to changes in the roles and responsibilities associated with the changes in personnel and organizational structure. The failure to properly monitor impacted the timing, accuracy, and completion of the work related to significant accounting matters.
Our Chief Executive Officer is in the preliminary stage of a review of our controls relating to complex accounting matters. Although our analysis is not complete, we will be adding additional resources with expertise in accounting for complex accounting matters including timely review and evaluation of assets for potential impairment. We are also considering redesigning controls to add additional layers of review and approval whenever entering into or subsequently converting, exercising, amending, repricing, exiting or otherwise experiencing changes in or to complex financial instruments.
Notwithstanding the identified material weaknesses, the Company believes the consolidated financial statements included in this Annual Report on Form 10-K fairly represent in all material respects our financial condition, results of operations and cash flows at and for the periods presented in accordance with accounting principles generally accepted in the United States of America.
| 58 |
(d) Changes in Internal Control Over Financial Reporting
Other than described above in the Item 9A. Controls and Procedures, there were no changes in our internal control over financial reporting that occurred during the fourth quarter of 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Information required by this Item 10 will be presented in our Proxy Statement for the 2016 annual meeting of Shareholders and is incorporated herein by reference. The information required pursuant to General Instructions G(3) of Form 10-K on our executive officers is presented under Item 1 of this report on Form 10-K.
| ITEM 11. | EXECUTIVE COMPENSATION |
Information required by this Item 11 will be presented in our Proxy Statement for the 2017 annual meeting of Shareholders and is incorporated herein by reference.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Information required by this Item 12 will be presented in our Proxy Statement for the 2017 annual meeting of Shareholders and is incorporated herein by reference.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Information required by this Item 13 will be presented in our Proxy Statement for the 2017 annual meeting of Shareholders and is incorporated herein by reference.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
Information required by this Item 14 will be presented in our Proxy Statement for the 2017 annual meeting of Shareholders and is incorporated herein by reference.
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
Reference is made to the Index to Consolidated Financial Statements beginning on Page F-1 hereof.
| (1) | Financial Statements. The following consolidated financial statements and the notes thereto, and the Report of Independent Registered Public Accounting Firms are incorporated by reference as provided in Item 8 of this report: |
| 59 |
(2) Consolidated Financial Statement Schedules
Consolidated Financial Statement Schedules have been omitted because they are either not required or not applicable, or because the information required to be presented is included in the consolidated financial statements or the notes thereto included in this Annual Report.
(3) Exhibits
The exhibits listed on the accompanying Exhibit Index are filed or incorporated by reference as part of this Annual Report and such Exhibit Index is incorporated by reference.
| 60 |
| 61 |
| 62 |
| 63 |
| 10.26 | Warrant Agency Agreement dated January 24, 2017, by and between Amedica Corporation and American Stock Transfer & Trust Company, LLC | Form 8-K (Exhibit 8-K) |
1/24/17 | 001-33624 | ||||||
| 21.1 | List of Subsidiaries of the Registrant | Form S-1 (Exhibit 21.1) |
11/8/13 | 333-192232 | ||||||
| 23.1 | Consent of Independent Registered Public Accounting Firm, Mantyla McReynolds, LLC |
X | ||||||||
| 23.2 | Consent of Independent Registered Public Accounting Firm, BDO USA, LLP |
X | ||||||||
| 31.1 | Certification of Chief Executive Officer | X | ||||||||
| 31.2 | Certification of Principal Financial Officer | X | ||||||||
| 32 | Certification pursuant to Section 906 of the Sarbanes Oxley Act of 2002 | X | ||||||||
| * | Management contract or compensatory plan or arrangement. |
| ITEM 16. | |
| None. |
| 64 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AMEDICA CORPORATION | |||
| Date: September 19, 2017 | /s/ B. Sonny Bal | ||
| B. Sonny Bal | |||
| Chief Executive Officer | |||
| (Principal Executive Officer and Principal Financial Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Date: September 19, 2017 | /s/ B. Sonny Bal | ||
| B. Sonny Bal, M.D., Director | |||
| Date: September 19, 2017 | /s/ David W. Truetzel | ||
| David W. Truetzel, Director | |||
| Date: September 19, 2017 | /s/ Jeffrey S. White | ||
| Jeffrey S. White, Director | |||
| Date: September 19, 2017 | /s/ Eric A. Stookey | ||
| Eric A. Stookey, Director |
| 65 |
AMEDICA CORPORATION
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Years ended December 31, 2016 and 2015 | ||
| Reports of Independent Registered Public Accounting Firms | F-2, F-3 | |
| Consolidated Balance Sheets | F-4 | |
| Consolidated Statements of Operations | F-5 | |
| Consolidated Statements of Stockholders’ Equity | F-6 | |
| Consolidated Statements of Cash Flows | F-7 | |
| Notes to Consolidated Financial Statements | F-8 |
| F-1 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Amedica Corporation
Salt Lake City, Utah
We have audited the accompanying consolidated balance sheet of Amedica Corporation (the “Company”) as of December 31, 2016 and the related consolidated statements of operations, stockholders’ equity, and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Amedica Corporation at December 31, 2016, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has recurring losses from operations and negative operating cash flows and needs to obtain additional financing to be compliant with debt covenants and to finance its operations. These issues raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ BDO USA, LLP
Salt Lake City, Utah
September 19, 2017
| F-2 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Amedica Corporation
Salt Lake City, Utah
We have audited the accompanying consolidated balance sheet of Amedica Corporation (the “Company”) as of December 31, 2015 and the related consolidated statements of operations, stockholders’ equity, and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company has determined that it is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Amedica Corporation at December 31, 2015, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has recurring losses from operations and negative operating cash flows and needs to obtain additional financing to be compliant with debt covenants through 2016. These issues raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Mantyla McReynolds, LLC
Salt Lake City, Utah
March 23, 2016
| F-3 |
Consolidated Balance Sheets
(in thousands, except share and per share data)
| As of December 31, | ||||||||
| 2016 | 2015 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 6,915 | $ | 11,485 | ||||
| Trade accounts receivable, net of allowance of $22 and $49, respectively | 1,620 | 2,660 | ||||||
| Prepaid expenses and other current assets | 239 | 229 | ||||||
| Inventories | 7,213 | 9,131 | ||||||
| Total current assets | 15,987 | 23,505 | ||||||
| Property and equipment, net | 889 | 2,472 | ||||||
| Intangible assets, net | 3,187 | 3,687 | ||||||
| Goodwill | 6,163 | 6,163 | ||||||
| Other long-term assets | 35 | 35 | ||||||
| Total assets | $ | 26,261 | $ | 35,862 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 658 | $ | 643 | ||||
| Accrued liabilities | 3,183 | 3,421 | ||||||
| Current portion of long-term debt | 7,012 | 16,365 | ||||||
| Total current liabilities | 10,853 | 20,429 | ||||||
| Deferred rent | 319 | 432 | ||||||
| Other long-term liabilities | 188 | 171 | ||||||
| Derivative liabilities | 528 | 598 | ||||||
| Total liabilities | 11,888 | 21,630 | ||||||
| Commitments and contingencies (Note 11) | ||||||||
| Stockholders’ equity: | ||||||||
| Convertible preferred stock, $0.01 par value, 130,000,000 shares authorized; no shares issued and outstanding at December 31, 2016 and 2015 | - | - | ||||||
| Common stock, $0.01 par value, 250,000,000 shares authorized, 27,364,881 and 10,886,248 shares issued and outstanding at December 31, 2016 and 2015, respectively | 274 | 109 | ||||||
| Additional paid-in capital | 227,234 | 210,660 | ||||||
| Accumulated deficit | (213,135 | ) | (196,537 | ) | ||||
| Total stockholders’ equity | 14,373 | 14,232 | ||||||
| Total liabilities and stockholders’ equity | $ | 26,261 | $ | 35,862 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
Consolidated Statements of Operations
(in thousands, except share and per share data)
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Product revenue | $ | 15,226 | $ | 19,453 | ||||
| Costs of revenue | 3,777 | 6,250 | ||||||
| Gross profit | 11,449 | 13,203 | ||||||
| Operating expenses: | ||||||||
| Research and development | 6,345 | 6,387 | ||||||
| General and administrative | 6,292 | 6,436 | ||||||
| Sales and marketing | 10,347 | 12,421 | ||||||
| Total operating expenses | 22,984 | 25,244 | ||||||
| Loss from operations | (11,535 | ) | (12,041 | ) | ||||
| Other income (expenses): | ||||||||
| Interest expense | (4,511 | ) | (4,339 | ) | ||||
| Gain (loss) on extinguishment of debt | (661 | ) | 2,171 | |||||
| Change in fair value of derivative liabilities | 70 | (7,605 | ) | |||||
| Loss on extinguishment of derivative liabilities | - | (1,263 | ) | |||||
| Offering costs | - | (821 | ) | |||||
| Other income (expense) | 39 | (14 | ) | |||||
| Total other expense, net | (5,063 | ) | (11,871 | ) | ||||
| Net loss before income taxes | (16,598 | ) | (23,912 | ) | ||||
| Provision for income taxes | - | - | ||||||
| Net loss | (16,598 | ) | (23,912 | ) | ||||
| Deemed dividend related to beneficial conversion feature and accretion of discount on convertible series A preferred stock | (6,278 | ) | - | |||||
| Net loss attributable to common stockholders | $ | (22,876 | ) | $ | (23,912 | ) | ||
| Net loss per share attributable to common stockholders: | ||||||||
| Basic and diluted | $ | (1.23 | ) | $ | (5.50 | ) | ||
| Weighted average common shares outstanding: | ||||||||
| Basic and diluted | 18,524,815 | 4,344,253 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
Consolidated Statements of Stockholders’ Equity
(in thousands, except share data)
| Total | ||||||||||||||||||||||||||||
| Convertible | Additional | Stockholders | ||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In | Accumulate | Equity | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||||||||
| Balance at December 31, 2014 | - | $ | - | 1,756,911 | $ | 16 | $ | 179,396 | $ | (172,505 | ) | $ | 6,907 | |||||||||||||||
| Issuance of common stock upon cashless exercise of warrants | - | - | 2,546,856 | 26 | 11,563 | - | 11,589 | |||||||||||||||||||||
| Issuance of common stock with offering | - | - | 874,891 | 9 | 122 | - | 131 | |||||||||||||||||||||
| Issuance of common stock upon exercise of warrants, net of issuance costs | - | - | 5,607,839 | 56 | 15,816 | - | 15,872 | |||||||||||||||||||||
| Issuance of common stock upon conversion of notes payable | - | - | 24,860 | 1 | 376 | - | 377 | |||||||||||||||||||||
| Stock-based compensation | - | - | 74,891 | 1 | 984 | (120 | ) | 865 | ||||||||||||||||||||
| Reclassification of warrant derivative liability to equity | - | - | - | - | 2,403 | - | 2,403 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (23,912 | ) | (23,912 | ) | |||||||||||||||||||
| Balance at December 31, 2015 | - | - | 10,886,248 | 109 | 210,660 | (196,537 | ) | 14,232 | ||||||||||||||||||||
| Issuance of common stock upon cashless exercise of warrants | - | - | 536,388 | 5 | (5 | ) | - | - | ||||||||||||||||||||
| Issuance of common stock and warrants with offering, net of issuance costs | - | - | 5,258,000 | 53 | 4,649 | - | 4,702 | |||||||||||||||||||||
| Issuance of convertible preferred stock and warrants with offering, net of issuance costs | 7,392 | - | - | - | 6,706 | 6,706 | ||||||||||||||||||||||
| Accretion of convertible preferred stock discount | 6,278 | 6,278 | ||||||||||||||||||||||||||
| Deemed dividend on convertible preferred stock | (6,278 | ) | (6,278 | ) | ||||||||||||||||||||||||
| Preferred stock converted to common stock | (7,392) | - | 7,392,000 | 74 | (74) | - | - | |||||||||||||||||||||
| Issuance of common stock upon exercise of warrants, net of issuance costs | 447,147 | 4 | 443 | - | 447 | |||||||||||||||||||||||
| Beneficial conversion feature associated with issuance of convertible notes | 1,138 | 1,138 | ||||||||||||||||||||||||||
| Issuance of common stock upon conversion of notes payable | - | - | 1,882,718 | 19 | 2,660 | - | 2,679 | |||||||||||||||||||||
| Issuance of common stock in connection with settlement of litigation | - | - | 962,380 | 10 | 784 | - | 794 | |||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 273 | - | 273 | |||||||||||||||||||||
| Net loss | (16,598 | ) | (16,598 | ) | ||||||||||||||||||||||||
| Balance at December 31, 2016 | - | $ | - | 27,364,881 | $ | 274 | $ | 227,234 | $ | (213,135 | ) | $ | 14,373 | |||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
Consolidated Statements of Cash Flow
(in thousands)
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Cash flow from operating activities | ||||||||
| Net loss | $ | (16,598 | ) | $ | (23,912 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation expense | 1,435 | 1,652 | ||||||
| Amortization of intangible assets | 501 | 501 | ||||||
| Amortization of lease incentive for tenant improvements | 20 | 20 | ||||||
| Impairment of property and equipment | 852 | - | ||||||
| Non cash interest expense | 2,904 | 2,194 | ||||||
| Non cash issuance of stock to settle litigation | 794 | - | ||||||
| (Gain) loss on extinguishment of debt | 661 | (2,171 | ) | |||||
| Stock based compensation | 273 | 911 | ||||||
| Change in fair value of derivative liabilities | (65 | ) | 7,605 | |||||
| Loss on extinguishment of derivative liabilities | (6 | ) | 1,263 | |||||
| (Gain) loss on disposal of equipment | (27 | ) | (21 | ) | ||||
| Provision for inventory reserve | 1,161 | 1,333 | ||||||
| Bad debt expense | - | (27 | ) | |||||
| Offering costs | - | 821 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Trade accounts receivable | 1,040 | (120 | ) | |||||
| Prepaid expenses and other current assets | (3 | ) | (74 | ) | ||||
| Inventories | 749 | 1,357 | ||||||
| Accounts payable and accrued liabilities | (861 | ) | (395 | ) | ||||
| Net cash used in operating activities | (7,170 | ) | (9,063 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of property and equipment | (671 | ) | (695 | ) | ||||
| Proceeds from sale of property and equipment | 54 | 37 | ||||||
| Net cash used in investing activities | (617 | ) | (658 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from issuance of common stock, net of issuance costs ( $601) | 4,702 | 4,337 | ||||||
| Proceeds from issuance of preferred stock, net of issuance costs ( $641) | 6,706 | - | ||||||
| Proceeds from issuance of stock in connection with exercise of warrants, net of issuance costs | 447 | 5,863 | ||||||
| Payments on long-term debt | (6,630 | ) | (2,949 | ) | ||||
| Debt extinguishment payments | (1,728 | ) | (4,112 | ) | ||||
| Payments for capital lease | (13 | ) | - | |||||
| Deferred costs paid for debt | (267 | ) | (60 | ) | ||||
| Purchase of treasury stock | - | (120 | ) | |||||
| Net cash provided by financing activities | 3,217 | 2,959 | ||||||
| Net increase (decrease) in cash and cash equivalents | (4,570 | ) | (6,762 | ) | ||||
| Cash and cash equivalents at beginning of period | 11,485 | 18,247 | ||||||
| Cash and cash equivalents at end of period | 6,915 | 11,485 | ||||||
| Noncash investing and financing activities | ||||||||
| Preferred stock converted to common stock | $ | 74 | $ | - | ||||
| Reclassification of derivative liability | - | 4,229 | ||||||
| Capital lease for property and equipment | 60 | |||||||
| Notes payable and accrued interest converted to common stock | 2,679 | 202 | ||||||
| Debt exchange - Riverside | 3,000 | - | ||||||
| Debt discount for Riverside not - beneficial conversion feature (BCF) and | ||||||||
| warrants | 1,138 | - | ||||||
| Common stock issued for cashless exercise of warrant derivative liabilities | 19,772 | |||||||
| Issuance of treasury stock upon conversion of RSUs to common stock | 120 | |||||||
| Derivative liabilities recorded as a debt discount | 382 | |||||||
| Deemed dividend on convertible preferred stock | $ | 6,278 | $ | - | ||||
| Supplemental cash flow information | ||||||||
| Cash paid for interest | 1,606 | 2,379 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-7 |
1. Organization and Summary of Significant Accounting Policies
Amedica Corporation (“Amedica” or “the Company”) was incorporated in the state of Delaware on December 10, 1996. Amedica is a commercial-stage biomaterial company focused on using its silicon nitride technology platform to develop, manufacture, and commercialize a broad range of medical devices. The Company believes it is the first and only manufacturer to use silicon nitride in medical applications. The Company acquired US Spine, Inc. (“US Spine”), a Delaware spinal products corporation with operations in Florida, on September 20, 2010. The Company’s products are primarily sold in the U.S.
Basis of Presentation and Principles of Consolidation
These consolidated financial statements have been prepared by management in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”), and include all assets and liabilities of the Company and its wholly-owned subsidiary, US Spine. All material intercompany transactions and balances have been eliminated in consolidation.
Liquidity and Capital Resources
The consolidated financial statements have been prepared assuming the Company will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and does not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to its ability to continue as a going concern within one year from the date of issuance of these consolidated financial statements.
For the years ended December 31, 2016 and 2015, the Company incurred a net loss of $16.6 million and $23.9 million, respectively, and used cash in operations of $7.2 million and $9.1 million, respectively. The Company had an accumulated deficit of $213.1 million and $196.5 million at December 31, 2016 and 2015, respectively. To date, the Company’s operations have been principally financed from proceeds from the issuance of preferred and common stock, convertible debt and bank debt and, to a lesser extent, cash generated from product sales. It is anticipated that the Company will continue to generate operating losses and use cash in operations. The Company’s continuation as a going concern is dependent upon its ability to increase sales, implement cost saving measures, maintain compliance with debt covenants and/or raise additional funds through the capital markets. Whether and when the Company can attain profitability and positive cash flows from operations or obtain additional financing is uncertain.
In 2016 the Company implemented certain cost saving measures, including workforce and office space reductions, and will continue to evaluate additional cost savings alternatives during 2017. These additional cost savings measures may include additional workforce and research and development reductions, as well as cuts to certain other operating expenses. In addition to these cost saving measures an experienced and highly successful leader for the Sales and Marketing team was recruited and hired. This individual has subsequently hired additional experienced personnel in Sales and Market Development. The Company is actively generating additional scientific and clinical data to have it published in leading industry publications. The unique features of our silicon nitride material are not well known, and such the publication of such data would help sales efforts as the Company approaches new prospects. The Company is also making additional changes to the sales strategy, including a focus on revenue growth of silicon nitride lateral lumbar implants and the newly developed pedicle screw system (known as Taurus).
As discussed further in Note 7, in June 2014, the Company entered into a term loan with Hercules Technology Growth Capital, Inc. (“Hercules Technology”), as administrative and collateral agent for the lenders thereunder and as lender, and Hercules Technology III, LP, (“HT III” and, together with Hercules Technology, “Hercules”) as lender (the “Hercules Term Loan”). The Hercules Term Loan has a liquidity covenant that requires the Company to maintain a cash balance of not less than $3.0 million at December 31, 2016. At December 31, 2016, the Company’s cash balance was approximately $6.9 million. The Company believes it will be in position to maintain compliance with the liquidity covenant related to the Hercules Term Loan into the fourth quarter of 2017. To maintain compliance beyond that date, the Company would need to refinance the note or obtain additional funding in or prior to the fourth quarter of 2017. The Company has common stock that is publicly traded and has been able to successfully raise capital when needed since the time of its initial public offering. The Company is engaged in discussions with an investment banking firm to examine financing alternatives, including options to encourage the exercise of outstanding warrants. The Company is also seeking to refinance the Hercules Term Loan and is in discussions with banking firms to look at lending alternatives.
If the Company is unable to refinance the Hercules Term Loan or access additional funds prior to becoming non-compliant with the financial and liquidity covenants related to the Hercules Term Loan, the entire remaining balance of the debt under the Hercules Term Loan could become immediately due and payable at the option of the lender. Although the Company is seeking to refinance the note or obtain additional debt financing, such funding is not assured and may not be available to the Company on favorable or acceptable terms, and may involve restrictive covenants. Any additional equity financing is also not assured and, if available to the Company, will most likely be dilutive to its current stockholders. If the Company is not able to obtain additional debt or equity financing on a timely basis, the impact on the Company will be material and adverse.
These uncertainties create substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of these uncertainties.
Reverse Stock Split
On January 25, 2016, the Company effected a 1 for 15 reverse stock split of the Company’s common stock. The par value and the authorized shares of the common and convertible preferred stock were not adjusted as a result of the reverse stock split. All common stock share and per-share amounts for all periods presented in these consolidated financial statements have been adjusted to reflect the reverse stock split.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the period. Actual results could differ from those estimates. Some of the more significant estimates relate to inventory, stock-based compensation, long-lived and intangible assets, goodwill, and derivative liabilities.
| F-8 |
Concentrations of Credit Risk and Significant Customers
Financial instruments which potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents, marketable securities, accounts receivable and restricted cash. The Company limits its exposure to credit loss by placing its cash and cash equivalents with high credit-quality financial institutions in bank deposits, money market funds, U.S. government securities and other investment grade debt securities that have strong credit ratings. The Company has established guidelines relative to diversification of its cash and marketable securities and their maturities that are intended to secure safety and liquidity. These guidelines are periodically reviewed and modified to take advantage of trends in yields and interest rates and changes in the Company’s operations and financial position. Although the Company may deposit its cash and cash equivalents with multiple financial institutions, its deposits, at times, may exceed federally insured limits.
At December 31, 2016, one customer receivable balance was 16% of the Company’s total trade accounts receivable. At December 31, 2015, one customer receivable balance was 11% of the Company’s total trade accounts receivable. There was one customer that accounted for 10% or more of the Company’s revenue representing 17% of revenue for the year ended December 31, 2016. There was one customer that accounted for 10% or more of the Company’s revenue representing 12% of revenue for the year ended December 31, 2015.
Revenue Recognition
The Company derives its product revenue primarily from the sale of spinal fusion devices and related products used in the treatment of spine disorders. The Company’s product revenue is generated from sales to three types of customers: (1) surgeons and hospitals; (2) stocking distributors; and (3) private label customers. Most of our products are sold on a consignment basis through a network of independent sales distributors; however, the Company also sells its products to independent stocking distributors and private label customers. Product revenue is recognized when all four of the following criteria are met: (1) persuasive evidence that an arrangement exists; (2) delivery of the products has occurred; (3) the selling price of the product is fixed or determinable; and (4) collectability is reasonably assured. The Company generates the majority of its revenue from the sale of inventory that is consigned to independent sales distributors that facilitate sells of the Company’s products to surgeons and hospitals. For these products, we recognize revenue at the time we are notified the product has been used or implanted and all other revenue recognition criteria have been met. For all other transactions, the Company recognizes revenue when title and risk of loss transfer to the stocking distributor or private label customer, and all other revenue recognition criteria have been met. The Company recognizes revenue from sales to stocking distributors and private label customers at the time the product is shipped. Stocking distributors, who sell the products to their customers, take title to the products and assume all risks of ownership at time of shipment. The Company’s stocking distributors are obligated to pay within specified terms regardless of when, if ever, they sell the products. The Company’s policy is to classify shipping and handling costs billed to customers as an offset to total shipping expense in the consolidated statements of operations, primarily within sales and marketing. In general, the Company’s customers do not have any rights of return or exchange.
Costs of Revenue
The expenses that are included in costs of revenue include all direct product costs if we obtained the product from third-party manufacturers and our in-house manufacturing costs for the products we manufacture. We obtain our non-silicon nitride products, including our metal and orthobiologic products, from third-party manufacturers, while we currently manufacture the majority of our silicon-nitride products in-house.
Specific provisions for excess or obsolete inventory and, beginning in 2013, the excise tax on the sale of medical devices in the United States, are also included in costs of revenue. In addition, we pay royalties attributable to the sale of specific products to some of our surgeon advisors that assisted us in the design, regulatory clearance or commercialization of a particular product, and these payments are recorded as costs of revenue.
Cash and Cash Equivalents
The Company considers all cash on deposit, money market accounts and highly-liquid debt instruments purchased with original maturities of three months or less to be cash and cash equivalents.
Inventories
Inventories are stated at the lower of cost or market, with cost for manufactured inventory determined under the standard costs, which approximate actual costs, determined on the first-in first-out (“FIFO”) method. Manufactured inventory consists of raw material, direct labor and manufacturing overhead cost components. Inventories purchased from third-party manufacturers are stated at the lower of cost or market using the first-in, first-out method. The Company reviews the carrying value of inventory on a periodic basis for excess or obsolete items, and records any write-down as a cost of revenue, as necessary. It is reasonably possible that the Company may be required to make adjustments to the carrying value of inventory in future periods. Inventory write-downs for excess or obsolete inventory are recorded as a cost of revenue. The Company holds consigned inventory at distributor and other customer locations where revenue recognition criteria have not yet been achieved.
| F-9 |
Property and Equipment
Property and equipment, including surgical instruments and leasehold improvements, are stated at cost, less accumulated depreciation and amortization. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets, which range from three to five years. Leasehold improvements are amortized over the shorter of their estimated useful lives or the related lease term, generally five years.
Periodically we review the carrying value of our property and equipment that are held and used in our operations for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of these assets is determined based upon expected undiscounted future net cash flows from the operations to which the assets relate, utilizing management’s best estimate, assumptions, and projections at the time. If the carrying value is determined to be unrecoverable from future operating cash flows, the asset is deemed impaired and an impairment charge would be recognized to the extent the carrying value exceeded the estimated fair value of the asset. We estimate the fair value of assets based on the estimated future discounted cash flows of the asset. Management has evaluated its property and equipment and has identified asset impairment during the year ended December 31, 2016.
Accounts Receivable and Allowance for Doubtful Accounts
The majority of our accounts receivable is composed of amounts due from hospitals or surgical centers. Accounts receivable are carried at invoiced amount less an allowance for doubtful accounts. On a regular basis, we evaluate accounts receivable and estimate an allowance for doubtful accounts, as needed, based on various factors such as customers’ current credit conditions, length of time past due, and the general economy as a whole. Receivables are written off against the allowance when they are deemed uncollectible.
Long Lived Intangible Assets and Goodwill
The Company periodically evaluates the carrying value of definitely-lived intangibles when events or changes in circumstances indicate that the carrying value may not be recoverable. Factors the Company considers important which could trigger an impairment review include, but are not limited to, significant under-performance relative to historical or projected future operating results, significant changes in the manner of its use of acquired assets or its overall business strategy, and significant industry or economic trends. The Company amortizes finite-lived intangible assets on a straight-line basis over their useful lives. The Company recorded no impairment loss for definite-lived intangible assets during the years ended December 31, 2016 and 2015.
When the Company determines that the carrying value of a long-lived asset may not be recoverable based upon the existence of one or more of the above indicators, the Company determines the recoverability by comparing the carrying amount of the asset to net future undiscounted cash flows that the asset is expected to generate and recognizes an impairment charge equal to the amount by which the carrying amount exceeds the fair market value of the asset.
If our revenues or other estimated operating results are not achieved at or above our forecasted level, and we are unable to recover such costs through price increases, the carrying value of certain of our assets may prove to be unrecoverable and we may incur impairment charges of definitive-live intangible assets.
In accordance with ASC 350, Goodwill and Other Intangible Assets, goodwill is not amortized but is required to be reviewed for impairment at least annually or when events or circumstances indicate that carrying value may exceed fair value. The Company is permitted the option to first assess qualitative factors to determine whether the existence of events and circumstances indicates that it is more likely than not that the fair value of any reporting unit is less than its corresponding carrying value. If, after assessing the totality of events and circumstances, the Company concludes that it is not more likely than not that the fair value of any reporting unit is less than its corresponding carrying value then the Company is not required to take further action. However, if the Company concludes otherwise, then it is required to perform a quantitative impairment test, including computing the fair value of the reporting unit and comparing that value to its carrying value. The Company considers valuation factors and an estimated control premium. The estimated fair value of the reporting unit exceeded the carrying value by approximately 20%. The declining price of the Company’s stock is an early indicator that goodwill impairment may be a factor during 2017. We will continue to monitor our market capitalization and impairment indicators.
If the fair value is less than its carrying value, a second step of the test is required to determine if recorded goodwill is impaired. In the event that goodwill is impaired, an impairment charge to earnings would become necessary.
Derivative Liabilities
Derivative liabilities includes the fair value of instruments such as common stock warrants, preferred stock warrants and convertible features of notes, that are initially recorded at fair value and are required to be re-measured to fair value at each reporting period under provisions of ASC 480, Distinguishing Liabilities from Equity, or ASC 815, Derivatives and Hedging. The change in fair value of the instruments is recognized as a component of other income (expense) in the Company’s consolidated statements of operations until the instruments settle, expire or are no longer classified as derivative liabilities. The Company estimates the fair value of these instruments using the Black-Scholes-Merton or Monte-Carlo valuation models depending on the complexity of the underlying instrument. The significant assumptions used in estimating the fair value include the exercise price, volatility of the stock underlying the instrument, risk-free interest rate, estimated fair value of the stock underlying the instrument and the estimated life of the instrument.
| F-10 |
Research and Development
All research and development costs, including those funded by third parties, are expensed as incurred. Research and development costs consist of engineering, product development, test-part manufacturing, testing, developing and validating the manufacturing process, and regulatory related costs. Research and development expenses also include employee compensation, employee and nonemployee stock-based compensation, supplies and materials, consultant services, and travel and facilities expenses related to research activities.
Advertising Costs
Advertising costs are expensed as incurred. The primary component of the Company’s advertising expenses is advertising in trade periodicals. Advertising costs were approximately $84,000 and $31,000 for the years ended December 31, 2016 and 2015, respectively.
Income Taxes
The Company recognizes a liability or asset for the deferred tax consequences of all temporary differences between the tax basis of assets and liabilities and their reported amounts in the consolidated financial statements that will result in taxable or deductible amounts in future years when the reported amounts of the assets and liabilities are recovered or settled. The Company recognizes interest and penalties as a component of the provision for income taxes. No interest or penalties were recognized in the years ended December 31, 2016 and 2015.
The Company operate in various tax jurisdictions and are subject to audit by various tax authorities. The Company provides for tax contingencies whenever it is deemed probable that a tax asset has been impaired or a tax liability has been incurred for events such as tax claims or changes in tax laws. Tax contingencies are based upon their technical merits relative tax law and the specific facts and circumstances as of each reporting period. Changes in facts and circumstances could result in material changes to the amounts recorded for such tax contingencies.
The Company recognizes uncertain income tax positions taken on income tax returns at the largest amount that is more-likely than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
The Company’s policy for recording interest and penalties associated with uncertain tax positions is to record such items as a component of our income tax provision. For the years ended December 31, 2016 and 2015, the Company did not record any interest expense or penalties related to uncertain tax positions or the settlement of audits for prior periods.
Stock-Based Compensation
The Company measures stock-based compensation expense related to employee stock-based awards based on the estimated fair value of the awards as determined on the date of grant and is recognized as expense over the remaining requisite service period. The Company utilizes the Black-Scholes-Merton option pricing model to estimate the fair value of employee stock options. The Black-Scholes-Merton model requires the input of highly subjective and complex assumptions, including the estimated fair value of the Company’s common stock on the date of grant, the expected term of the stock option, and the expected volatility of the Company’s common stock over the period equal to the expected term of the grant. The Company estimates forfeitures at the date of grant and revises the estimates, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company accounts for stock options to purchase shares of stock that are issued to non-employees based on the estimated fair value of such instruments using the Black-Scholes-Merton option pricing model. The measurement of stock-based compensation expense for these instruments is variable and subject to periodic adjustments to the estimated fair value until the awards vest. Any resulting change in the estimated fair value is recognized in the Company’s consolidated statements of operations during the period in which the related services are rendered.
| F-11 |
Because the Company was a privately-held company with no trading history prior to February 2014 and has limited stock history since February 2014, the Company utilizes the historical stock price volatility from a representative group of public companies to estimate expected stock price volatility and our historical stock price. The Company selected companies from the medical device industry, specifically those who are focused on the design, development and commercialization of products for the treatment of spine disorders, and who have similar characteristics to us, such as stage of life cycle and size. The Company intends to continue to utilize the historical volatility of the same or similar public companies to estimate expected volatility until a sufficient amount of historical information regarding the price of our publicly traded stock becomes available. The Company uses the simplified method as prescribed by the Securities and Exchange Commission Staff Accounting Bulletin No. 107, Share-based Payment, to calculate the expected term of stock option grants to employees as the Company does not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term of stock options granted to employees. The Company utilizes a dividend yield of zero because the Company has never paid cash dividends and has no current intention to pay cash dividends. The risk-free rate of return used for each grant is based on the U.S. Treasury yield curve in effect at the time of grant for instruments with a similar expected life.
The Company accounts for stock options to purchase shares of stock that are issued to non-employees based on the estimated fair value of such instruments using the Black-Scholes-Merton option pricing model. The measurement of stock-based compensation expense for these instruments is variable and subject to periodic adjustments to the estimated fair value until the awards vest. Any resulting change in the estimated fair value is recognized in the Company’s consolidated statements of operations during the period in which the related services are rendered.
Offering Costs
Offering costs consist of legal, accounting, and other advisory costs related to the Company’s efforts to raise debt and equity capital.
Offering costs paid in cash or by issuing warrants associated with the Company’s equity fundraising activities are either recorded to additional paid in capital as a reduction of the proceeds or immediately expensed depending on the amount of the offering costs compared to the gross proceeds.
Offering costs paid in cash or by issuing warrants associated with the Company’s debt fundraising activities are recorded as a debt discount and amortized as interest expense over the lie of the debt or immediately expensed depending on the amount of offering costs compared to debt, with the offset to additional paid in capital.
New Accounting Pronouncement, Not Yet Adopted
In July 2015, the FASB issued ASU 2015-11, “Inventory (Topic 330) Simplifying the Measurement of Inventory”. The amendments clarify that an entity should measure inventory within the scope of this update at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Substantial and unusual losses that result from subsequent measurement of inventory should be disclosed in the consolidated financial statements. This guidance is effective for fiscal years beginning after December 15, 2016, including interim periods within those annual periods. The amendments are to be applied prospectively with earlier application permitted as of the beginning of an interim or annual reporting period. This guidance is not expected to have a material impact on the consolidated financial statements.
In August 2016, the Financial Accounting Standards Board (“FASB”) updated accounting guidance on the following eight specific cash flow classification issues: (1) debt prepayment or debt extinguishment costs; (2) settlement of zero-coupon debt instruments or other debt instruments with coupon interest rates that are insignificant in relation to the effective interest rate of the borrowing; (3) contingent consideration payments made after a business combination; (4) proceeds from the settlement of insurance claims; (5) proceeds from the settlement of corporate-owned life insurance policies, including bank-owned life insurance policies; (6) distributions received from equity method investees; (7) beneficial interests in securitization transactions; and (8) separately identifiable cash flows and application of the predominance principle. Current GAAP does not include specific guidance on these eight cash flow classification issues. These updates are effective for the Company for its annual period beginning January 1, 2019, and interim periods therein, with early adoption permitted. The guidance in this standard is not expected to have a material impact on the consolidated financial statements.
In March 2016 the FASB updated the accounting guidance related to stock compensation. This update simplifies the accounting for employee share-based payment transactions, including the accounting for income taxes, forfeitures, and statutory tax withholding requirements, as the well as classification in the statement of cash flows. The standard is effective for the Company for its annual period beginning January 1, 2018. The guidance in this standard is not expected to have a material impact on the consolidated financial statements of the Company.
In February 2016, the FASB updated the accounting guidance related to leases as part of a joint project with the International Accounting Standards Board (“IASB”) to increase transparency and comparability among organizations by recognizing lease assets and lease liabilities on the balance sheet and disclosing key information about leasing arrangements. Under the new guidance, a lessee will be required to recognize assets and liabilities for capital and operating leases with lease terms of more than 12 months. Additionally, this update will require disclosures to help investors and other financial statement users better understand the amount, timing, and uncertainty of cash flows arising from leases, including qualitative and quantitative requirements. The standard is effective for the Company for its annual period beginning January 1, 2020, and interim periods therein, with early adoption permitted. The Company is currently evaluating the potential impact this new standard may have on its consolidated financial statements, but believes the most significant change will relate to building leases.
| F-12 |
In May 2014, in addition to several amendments issued during 2016, the FASB updated the accounting guidance related to revenue from contracts with customers, which supersedes nearly all existing revenue recognition guidance under U.S. GAAP. The core principle is that a company should recognize revenue when promised goods or services are transferred to customers in an amount that reflects the consideration to which an entity expects to be entitled for those goods or services. The standard defines a five step process to achieve this core principle and, in doing so, more judgment and estimates may be required within the revenue recognition process than are required under existing U.S. GAAP. The standard is effective for the Company for its annual period beginning January 1, 2019, and interim periods therein, and shall be applied either retrospectively to each period presented or as a cumulative-effect adjustment as of the date of adoption. The Company has yet to begin evaluation of the new accounting standard and therefore has yet to determine the impact, if any, that the new standard will have on its consolidated financial statements.
In January of 2017, the FASB issued ASU 2017-04—Intangibles—Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment. The amendments in this guidance to eliminate the requirement to calculate the implied fair value of goodwill to measure goodwill impairment charge (Step 2). As a result, an impairment charge will equal the amount by which a reporting unit’s carrying amount exceeds its fair value, not to exceed the amount of goodwill allocated to the reporting unit. An entity still has the option to perform the qualitative assessment for a reporting unit to determine if the quantitative impairment test is necessary. The amendment should be applied on a prospective basis. The guidance is effective for goodwill impairment tests in fiscal years beginning after December 15, 2021. Early adoption is permitted for goodwill impairment tests performed after January 1, 2017. The impact of this guidance for the Company will depend on the outcomes of future goodwill impairment tests.
The Company has reviewed all other recently issued, but not yet adopted, accounting standards, in order to determine their effects, if any, on its results of operations, financial position or cash flows. Based on that review, the Company believes that none of these pronouncements will have a significant effect on its consolidated financial statements.
New Accounting Pronouncement, Adopted in 2016
In April 2015, the FASB issued ASU 2015-03, Interest - Imputation of Interest, which amends the current guidance to change the manner in which debt issuance costs are presented on an entity’s balance sheet. This new guidance requires the Company to present debt issuance costs related to recognized debt liabilities on the balance sheet as a direct deduction from the debt liability, as opposed to the previous guidance that provides for presentation of the cost of issuing debt as a separate asset. ASU 2015-03 required retrospective application to all prior periods presented in the consolidated financial statements. The Company adopted this new guidance effective first quarter of 2016. As a result of adopting this standard on January 1, 2016, deferred financing costs of $592,000 as of December 31, 2015 previously reported within current assets, were reclassified to current portion of long-term debt in the consolidated balance sheets. The impact of this adoption was not material to the consolidated financial statements.
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by dividing the net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method. Dilutive common stock equivalents are comprised of convertible preferred stock, warrants for the purchase of convertible preferred stock and common stock, convertible notes, and stock options and restricted stock units outstanding under the Company’s equity incentive plans. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding due to the Company’s net loss position.
| F-13 |
Potentially dilutive securities not included in the calculation of diluted net loss per share because to do so would be anti-dilutive are as follows (in common stock equivalent shares):
| As of December 31, | ||||||||
| 2016 | 2015 | |||||||
| Common stock warrants | 13,427,259 | 1,510,953 | ||||||
| Common stock options | 137,347 | 112,639 | ||||||
| 13,564,606 | 1,623,592 | |||||||
2. Inventories
The components of inventory were as follows (in thousands):
| As of December 31, | ||||||||
| 2016 | 2015 | |||||||
| Raw materials | $ | 761 | $ | 819 | ||||
| WIP | 75 | 235 | ||||||
| Finished goods | 6,377 | 8,077 | ||||||
| $ | 7,213 | $ | 9,131 | |||||
Finished goods include consigned inventory of approximately $5.6 million and $3.8 million as of December 31, 2016 and 2015, respectively.
3. Property and Equipment
The following is a summary of the components of property and equipment (in thousands):
| Year Ended December 31 , | ||||||||
| 2016 | 2015 | |||||||
| Manufacturing and lab equipment | $ | 223 | $ | 7,463 | ||||
| Surgical instruments | 5,269 | 8,672 | ||||||
| Leasehold improvements | 863 | 1,439 | ||||||
| Software and computer equipment | 816 | 845 | ||||||
| Furniture and equipment | 629 | 629 | ||||||
| 7,800 | 19,048 | |||||||
| Less: accumulated depreciation | (6,911 | ) | (16,576 | ) | ||||
| $ | 889 | $ | 2,472 | |||||
Depreciation expense for 2016 and 2015 was approximately $1.4 million and $1.7 million, respectively.
Management analyzed the undiscounted cash flows expected to be generated by the ceramic product line asset group and concluded that due to continuing declining revenues that the carrying value of ceramic related assets exceeded the total of the undiscounted cash flows. As a result, the Company recognized impairment charges of approximately $0.22 million related to its ceramics equipment, approximately $0.17 million related to leasehold improvements, and approximately $0.46 related to its ceramics surgical instrument sets, totaling $0.85 million of impairment charges for the year ended December 31, 2016 to bring the carrying value to the estimated fair value. No impairment charge was recorded during the year ended December 31, 2015.
4. Intangible Assets
Intangible assets consisted of the following (in thousands):
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Customer relationships | $ | 3,990 | $ | 3,990 | ||||
| Developed technology | 4,685 | 4,685 | ||||||
| Other patents and patent applications | 562 | 562 | ||||||
| Trademarks | 350 | 350 | ||||||
| 9,587 | 9,587 | |||||||
| Less: accumulated amortization | (6,400 | ) | (5,900 | ) | ||||
| $ | 3,187 | $ | 3,687 | |||||
Based on the recorded intangibles at December 31, 2016, the estimated amortization expense is expected to be approximately $501,000 per year through 2021 and $332,000 through 2022.
| F-14 |
5. Fair Value Measurements
Financial Instruments Measured and Recorded at Fair Value on a Recurring Basis
The Company measures and records certain financial instruments at fair value on a recurring basis. Fair value is based on the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, under a three-tier fair value hierarchy which prioritizes the inputs used in measuring fair value as follows:
| Level 1 | - | quoted market prices for identical assets or liabilities in active markets. | |
Level 2 |
- |
observable prices that are based on inputs not quoted on active markets, but corroborated by market data. | |
Level 3 |
- |
unobservable inputs reflecting management’s assumptions, consistent with reasonably available assumptions made by other market participants. These valuations require significant judgment. |
The Company classifies assets and liabilities measured at fair value in their entirety based on the lowest level of input that is significant to their fair value measurement. No financial assets were measured on a recurring basis at December 31, 2016 and 2015. The following tables set forth the financial liabilities measured at fair value on a recurring basis by level within the fair value hierarchy at December 31, 2016 and 2015.
| Fair
Value Measurements at December 31, 2016 (in thousands) | ||||||||||||||||
| Description | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Derivative liability | ||||||||||||||||
| Common stock warrants | $ | - | $ | - | $ | 528 | $ | 528 | ||||||||
| Fair
Value Measurements at December 31, 2015 (in thousands) | ||||||||||||||||
| Description | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Derivative liability | ||||||||||||||||
| Common stock warrants | $ | - | $ | - | $ | 598 | $ | 598 | ||||||||
The Company did not have any transfers of assets and liabilities between Level 1 and Level 2 of the fair value measurement hierarchy during the years ended December 31, 2016 and 2015. The following table presents a reconciliation of the derivative liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) during the years ended December 31, 2016 and 2015 (in thousands):
| Preferred | Conversion | Total | ||||||||||||||
| Common Stock | Stock | Feature of | Derivative | |||||||||||||
| Warrants | Warrants | Notes | Liability | |||||||||||||
| Balance at December 31, 2014 | $ | (11,358 | ) | $ | - | $ | (2,612 | ) | $ | (13,970 | ) | |||||
| Issuances of derivatives | (14,556 | ) | - | -- | (14,556 | ) | ||||||||||
| Modification of terms | (382 | ) | - | - | $ | (382 | ) | |||||||||
| Decrease in liability due to debt conversions | - | - | 179 | 179 | ||||||||||||
| Decrease in liability due to warrants being exercised | 20,335 | - | - | 20,335 | ||||||||||||
| Reclassification from liability to equity | 2,403 | - | - | 2,403 | ||||||||||||
| Extinguishment of derivative liabilities | - | - | 3,468 | 3,468 | ||||||||||||
| Change in fair value | 2,960 | - | (1,035 | ) | 1,925 | |||||||||||
| Balance at December 31, 2015 | $ | (598 | ) | $ | - | $ | - | $ | (598 | ) | ||||||
| Balance at December 31, 2015 | (598 | ) | - | - | (598 | ) | ||||||||||
| Change in fair value | 70 | - | - | 70 | ||||||||||||
| Balance at December 31, 2016 | $ | (528 | ) | $ | - | $ | - | $ | (528 | ) | ||||||
There were no warrant derivatives issued in 2016. In 2015 there were $9.5 million of warrant derivatives issued for the September 2015 offering were recorded as a loss and included in the change in fair value of derivative liabilities per the consolidated statements of operations since the value of the derivative liabilities issued exceeded the proceeds received from the issuance of common stock and warrants. See Note 8 for additional information.
| F-15 |
Common Stock Warrants
The Company has issued certain warrants to purchase shares of common stock, which are considered mark-to-market liabilities and are re-measured to fair value at each reporting period in accordance with accounting guidance.
The assumptions used in estimating the common stock warrant liability at December 31, 2016 and 2015 were as follows:
| December 31, 2016 | December 31, 2015 | |||||||
| Weighted-average risk free interest rate | 0.92 | % | 1.71 | % | ||||
| Weighted-average expected life (in years) | 2.5 | 3.7 | ||||||
| Expected dividend yield | 0 | % | 0 | % | ||||
| Weighted-average expected volatility | 136 | % | 119 | % | ||||
Other Financial Instruments
The Company’s recorded values of cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities approximate their fair values based on their short-term nature. The recorded value of notes payable approximates the fair value as the interest rate approximates market interest rates.
6. Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Commissions | $ | 466 | $ | 867 | ||||
| Payroll and related expenses | 461 | 683 | ||||||
| Royalties | 416 | 515 | ||||||
| Interest payable | 76 | 222 | ||||||
| Final loan payment fees | 1,333 | 783 | ||||||
| Other | 431 | 351 | ||||||
| $ | 3,183 | $ | 3,421 | |||||
7. Debt
Hercules Term Loan
On June 30, 2014, the Company entered into a Loan and Security Agreement with Hercules which provided the Company with a $20 million term loan. The Hercules Term Loan matures on January 1, 2018. The Hercules Term Loan included a $200,000 closing fee, which was paid to Hercules on the closing date of the loan. The closing fee was recorded as a debt discount and is being amortized to interest expense over the life of the loan. The Hercules Term Loan also includes a non-refundable final payment fee of $1.7 million. The final payment fee is being accrued and recorded to interest expense over the life of the loan. The Hercules Term Loan bears interest at the rate of the greater of either (i) the prime rate plus 9.2%, and (ii) 12.5%, and was 12.7% at December 31, 2016. Interest accrues from the closing date of the loan and interest payments are due monthly. Principal payments commenced August 1, 2015 and are currently being made in equal monthly installments of approximately $500,000, with the remainder due at maturity. The Company’s obligations to Hercules are secured by a first priority security interest in substantially all of its assets, including intellectual property. The Hercules Term Loan contains certain covenants related to restrictions on payments to certain Company affiliates and financial reporting requirements.
On September 8, 2015, the Company entered into a Consent and First Amendment to Loan and Security Agreement (the “Amendment”) with Hercules. The Amendment modified the liquidity covenant to reduce the minimum cash balance required by $500,000 for every $1.0 million paid in principal to a minimum of $2.5 million. The minimum cash and cash equivalents balance required to maintain compliance with the minimum liquidity covenant at December 31, 2016, was $3.0 million. The Company believes it is in position to maintain compliance with the liquidity covenant related to the Hercules Term Loan into the fourth quarter of 2017. To maintain compliance beyond that date, the Company would need to refinance the note or obtain additional funding in or prior to the fourth quarter of 2017, and has therefore classified the entire obligation as a current liability.
| F-16 |
See discussion below with respect to the assignment of $3.0 million of the principal balance of the Hercules Term Loan to Riverside Merchant Partners, LLC (“Riverside”) and the subsequent agreement between the Company and Riverside to exchange the $3.0 million of the Hercules Term Loan held by Riverside for subordinated convertible promissory notes in the aggregate principal amount of $3.0 million.
Magna Note
On April 2, 2015, we entered into an Amendment and Exchange Agreement (the “Amendment Agreement”) with Magna. The Amendment Agreement provides for the issuance by the Company to Magna of two new senior convertible notes, one with a maturity date in June 2016 and one with a maturity date in August 2016 (the “June Note”, the “August Note,” and collectively the “Exchange Convertible Notes”) in exchange for the Initial Convertible Note, the Additional Convertible Note and a warrant issued to Magna (“Magna Warrant”) to purchase 37,926 shares of the Company’s common stock at an exercise price of $69.75. The exchange resulted in the cancellation of the Initial Convertible Note, Additional Convertible Note and Magna Warrant.
On June 19, 2015, the Company received written notice from Magna that an event of default had occurred with respect to the Exchange Convertible Notes and underlying agreements. On September 8, 2015, the Company entered into a Settlement and Waiver Agreement (“Settlement Agreement”) with Magna. Pursuant to the Settlement Agreement, the Company paid Magna $2.5 million from the September 2015 Offering discussed in Footnote 8 to redeem the entire $797,000 of outstanding principal amount and accrued interest of the June Note and to partially redeem $1.4 million principal amount of the August Note and any accrued interest. On November 18, 2015, the Company paid Magna $1.3 million of the funds raised from exercise of the Series B warrants discussed in Footnote 8 to redeem $1.1 million of outstanding principal and the associated accrued interest. On December 31, 2015, the Company paid Magna $368,000 of the funds raised from exercise of the Series C warrants discussed in Footnote 8 to redeem $311,000 of outstanding principal and the associated accrued interest. As part of the Settlement Agreement, Magna waived its event of default notice delivered to the Company on June 19, 2015 and its right to convert the August Note into shares of common stock.
The Settlement Agreement was accounted for as a debt extinguishment and the Company recorded a gain on extinguishment of debt of $2.2 million during the year ended December 31, 2015. Since the conversion features of the Exchange Convertible Notes were eliminated, the estimated fair value of the conversion features of $3.5 million was extinguished and included in the calculation of the gain on extinguishment of debt.
During the year ended December 31, 2015, Magna converted a total of $202,000 of the principal amount of the Initial Convertible Note into 24,867 shares of common stock. The Company recorded a loss upon extinguishment of $79,000 during the year ended December 31, 2015 related to the conversion into shares of common stock.
The outstanding principal amount of the remaining Magna August Note was $763,000 at December 31, 2015. The Magna August Note matures on August 11, 2016.
In July 2016, the Company paid Magna $888,000 to redeem in full the remaining principal balance and interest related to the Magna Note. The outstanding principal amount of the Magna Note at extinguishment was $763,000. The Magna Note would have matured on August 11, 2016, and accrued interest at an annual rate of 6.0%.
Hercules and Riverside Debt Exchange
On April 4, 2016, the Company entered into an Assignment and Second Amendment to Loan and Security Agreement (the “Assignment Agreement”) with Riverside Merchant Partners, LLC (“Riverside”), and Hercules, pursuant to which Hercules sold $1.0 million of the principal amount outstanding under the Hercules Term Loan to Riverside. In addition, pursuant to the terms of the Assignment Agreement, Riverside acquired an option to purchase an additional $2.0 million of the principal amount outstanding under the Hercules Term Loan from Hercules. On April 18, 2016, Riverside exercised and purchased an additional $1.0 million of the principal amount of the Hercules Term Loan and on April 27, 2016, Riverside exercised the remainder of its option and purchased an additional $1.0 million of the principal amount of the Hercules Term Loan from Hercules.
Riverside Debt
On April 4, 2016, the Company entered into an exchange agreement (the “Exchange Agreement”) with Riverside, pursuant to which the Company agreed to exchange $1.0 million of the principal amount outstanding under the Hercules Term Loan held by Riverside for a subordinated convertible promissory note in the principal amount of $1.0 million (the “First Exchange Note”) and a warrant to purchase 100,000 shares of common stock of the Company at a fixed exercise price of $1.62 per share (the “First Exchange Warrant”) (the “Exchange”). All principal accrued under the Exchange Notes was convertible into shares of common stock at the election of the Holder at any time at a fixed conversion price of $1.43 per share (the “Conversion Price”). The closing stock price on April 4, 2016, was $1.63 and a beneficial conversion feature of $245,000 was recorded to equity and as a debt discount. The warrant value of $106,000 was recorded to equity and as a debt discount.
In addition, pursuant to the terms and conditions of the Exchange Agreement, the Company and Riverside had the option to exchange an additional $2.0 million of the principal amount of the Hercules Term Loan for an additional subordinated convertible promissory note in the principal amount of up to $2.0 million and an additional warrant to purchase 100,000 shares of common stock (the “Second Exchange Warrant”). The Exchange Agreement also provided that if the volume-weighted average price of the Company’s common stock was less than the Conversion Price, the Company would issue up to an additional 150,000 shares of common stock (the “True-Up Shares”) to Riverside, which was subsequently reduced to 140,000 shares of common stock.
On April 18, 2016, the Company and Riverside exercised their option to exchange an additional $1.0 million of the principal amount of the Hercules Term Loan for an additional subordinated convertible promissory note in the principal amount of $1.0 million (the “Second Exchange Note”). The closing stock price on April 18, 2016, was $2.02 and a beneficial conversion feature of $413,000 was recorded to equity and as a debt discount. Additionally, on April 27, 2016, the Company and Riverside exercised their option to exchange an additional $1.0 million of the principal amount of the Term Loan for an additional subordinated convertible promissory note in the principal amount of $1.0 million (the “Third Exchange Note”) and an additional warrant to purchase 100,000 shares of the Company’s common stock at a fixed exercise price of $1.66 per share. The warrant value of $107,000 was recorded to equity and as a debt discount. The closing stock price on April 27, 2016, was $1.66 and a beneficial conversion feature of $268,000 was recorded to equity and as a debt discount. Financing costs were $267,000 and were recorded to interest expense. The unamortized deferred financing costs and debt discount of the Hercules Term Loan exchanged were $244,000 at the time of the exchange and were recorded as a loss on extinguishment of debt related to the debt exchange. The First Exchange Note, the Second Exchange Note and the Third Exchange Note are collectively referred to herein as the “Exchange Notes.”
| F-17 |
Pursuant to the terms of the Exchange Notes, since the volume-weighted average price of the Company’s common stock was less than the Conversion Price on May 6, 2016, the Company issued an additional 140,000 shares of common stock to Riverside and recorded the value of the True-Up Shares of $199,000 to interest expense and equity.
All principal outstanding under each of the Exchange Notes was to be due on April 3, 2018 (the “Maturity Date”). Each of the Exchange Notes bore interest at a rate of 6% per annum, with the interest that would accrue on the initial principal amount of the Exchange Notes during the first 12 months being guaranteed and deemed earned as of the date of issuance. Prior to the Maturity Date, all interest accrued under the Exchange Notes was payable in cash or, if certain conditions were met, payable in shares of common stock at the Company’s option, at a conversion price of $1.34 per share. The entire principal amount of the First and Second Exchange Notes, $300,000 of the Third Exchange Note, and the interest related to the First, Second, and Third Exchange Notes had been converted into 1,742,718 shares of common stock. In July 2016, the Company paid Riverside $840,000 to redeem in full the remaining principal balance of the Third Exchange Note. The debt discounts associated with the converted debt was recorded to interest expense. As discussed above, the Company classifies all future debt obligations as current due to uncertainties in their ability to comply with debt covenant requirements.
| F-18 |
Outstanding long-term debt consisted of the following (in thousands):
| December 31, 2016 | December 31, 2015 | |||||||||||||||||||||||
| Unamortized | Unamortized | |||||||||||||||||||||||
| Outstanding | Discount and Debt | Net Carrying | Outstanding | Discount and Debt | Net Carrying | |||||||||||||||||||
| Principle | Issuance Costs | Amount | Principle | Issuance Costs | Amount | |||||||||||||||||||
| Hercules Term Loan | $ | 7,421 | $ | (409 | ) | $ | 7,012 | $ | 17,051 | $ | (1,420 | ) | $ | 15,631 | ||||||||||
| Convertible Note | - | - | - | - | - | - | ||||||||||||||||||
| Magna Note | - | - | - | 763 | (29 | ) | 734 | |||||||||||||||||
| Total debt | 7,421 | (409 | ) | 7,012 | 17,814 | (1,449 | ) | 16,365 | ||||||||||||||||
| Less: Current portion | (7,421 | ) | 409 | (7,012 | ) | (17,814 | ) | 1,449 | (16,365 | ) | ||||||||||||||
| Long-term debt | $ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
The following summarizes, by year, the future contractual principle payment obligations on the Hercules term loan as of December 31, 2016, before considering acceleration of maturity payments due to non-compliance with loan covenants (in thousands):
| Hercules Term | ||||
| Years Ending December 31, | Loan | |||
| 2017 | $ | 6,779 | ||
| 2018 | 642 | |||
| Total future principle payments | $ | 7,421 | ||
8. Equity
July 2016 Offering
In July 2016, the Company completed a secondary offering in which the Company sold 5,258,000 Class A Units, including 1,650,000 units sold pursuant to the exercise by the underwriters of their over-allotment option, priced at $1.00 per unit, and 7,392 Class B Units, priced at $1,000 per unit. Each Class A Unit consisted of one share of common stock and one warrant to purchase one share of common stock. Each Class B Unit consisted of one share of preferred stock convertible into 1,000 shares of common stock and warrants to purchase 1,000 shares of common stock. The securities comprising the units were immediately separable and were issued separately. In total, the Company issued 5,258,000 shares of common stock, 7,392 shares of preferred stock convertible into 7,392,000 shares of common stock, and warrants to purchase 12,650,000 shares of common stock at a fixed exercise price of $1.00 per share. The Company received proceeds of approximately $11.4 million, net of underwriting and other offering costs.
The Company raised $4.7 million, net of issuance costs of $0.6 million, associated with the Class A Units which were recorded as common stock and additional paid in capital, with $2.5 million allocated to the common stock and $2.2 million allocated to the warrants. The Company also raised $6.7 million, net of issuance costs of $0.7 million, associated with the Class B Units of which it allocated and recorded $3.6 million to preferred stock and allocated $3.1 million to the warrants which were recorded to additional paid in capital. The 7,392 preferred shares were convertible into 7,392,000 shares of common stock and had an effective conversion rate of $0.50 per share based on the proceeds that were allocated to them. The stock price on July 8, 2016, was $0.88 per share which resulted in a fair value in excess of carrying value of $0.38 per share or $2.5 million in total. The fair value in excess of carrying value, or beneficial conversion feature, was recorded as an adjustments within equity (e.g., deemed dividend). The Company recorded a non-cash, deemed dividend of $6.3 million related to a beneficial conversion feature and accretion of a discount on convertible preferred stock.
Subsequent to the secondary offering, all 7,392 shares of convertible preferred stock have been converted into 7,392,000 shares of common stock. Furthermore, the Company received $447,000 and issued 446,500 shares of common stock upon the exercise of certain warrants issued in the secondary offering.
September 2015 Offering
In September 2015, the Company entered into a Securities Purchase Agreement whereby it issued to certain investors 874,891 shares of common stock at a price of $5.72 per share for gross proceeds of $5.0 million before deducting placement agent fees and related offering expenses of $663,000. Pursuant to the terms of the Securities Purchase Agreement the company also issued to the investors 874,891 each of Series A warrants, Series B warrants and Series C warrants.
Shareholder approval was required for the issuance of the common shares underlying the Series B and Series C warrants. On November 3, 2015, the stockholders approved the proposal to allow the Company to issue the underlying shares upon exercise of the Series B and Series C warrants. In November 2015, the automatic exercise provision of the Series B warrants triggered and the Company received gross proceeds of $5.0 million and issued 3,324,192 shares of common stock in exchange for all 874,891 of the Series B warrants. Furthermore, pursuant to the terms of the warrant agreement, the number of Series A warrants increased by 3,324,192 to 4,199,082 and the exercise price of the Series A warrant was adjusted from $7.05 to $1.50. In December 2015, the Company amended the Series A and Series C warrants, whereby the exercise prices of the Series A and Series C warrants were fixed at $1.50 and the number of Series C warrants was fixed at 1,093,613. The Company received gross proceeds of $1.4 million and issued 962,969 shares of common stock upon exercise of 962,969 Series C warrants. The remaining 130,644 Series C warrants expired on December 30, 2015. Furthermore, pursuant to the terms of the warrant agreement, the number of Series A warrants increased by 962,969. During the year ended December 31, 2015, the Company issued 1,315,781 shares of common stock upon the cashless exercise of 3,924,687 Series A warrants. There were 1,237,365 outstanding Series A warrants at December 31, 2015 that terminate on December 11, 2020. The Company paid $585,000 in offering costs in connection with the proceeds received from the exercise of the Series B and C warrants.
| F-19 |
The Series A warrants, Series B warrants and Series C warrants were initially considered to be liabilities and were marked to market at each reporting period until they were exercised, terminated or were no longer classified as liabilities. At December 31, 2015, the remaining Series A warrants were no longer considered liabilities. The Company estimated the fair value of these warrants to be $14.4 million at issuance. The Company recorded $4.9 million of the $5.0 million gross proceeds from the offering to derivative liabilities and $131,000 to equity and recorded a loss of $9.5 million, which was included in the change in fair value of derivative liabilities per the Consolidated Statements of Operations. Furthermore, all of the $821,000 of the September 2015 offering costs were expensed since the value of the warrants exceeded the gross proceeds.
The Company entered into a placement agent agreement in connection with the September 2015 Offering. As part of the placement agent agreement, the Company issued a warrant to the placement agent to purchase 43,745 shares of common stock at an exercise price of $7.05. The warrant was determined to be a liability at issuance and the estimated fair value of $157,000 was included in offering costs.
During the year ended December 31, 2016 536,388 shares of common stock were issued upon the cashless exercise of 1,137,365 Series A warrants issued in September 2015 and 647 shares of common stock were issued upon warrants exercised for cash.
Other Issuances
During the year ended December 31, 2015, the Company issued 18,000 shares of common stock to a service provider as consideration for services to be rendered under a consulting agreement. Furthermore, 56,891 shares of common stock were issued upon the conversion of restricted stock units into common stock, of which 13,789 shares of common stock were withheld to satisfy the employees’ tax withholding obligations associated with the conversion of the restricted stock units into common stock. The withheld shares were included in treasury stock at a total value of $120,000, which was based on the market price of the common stock on the date the shares were issued. During June 2015, these treasury shares were issued upon the conversion of restricted stock units into common stock. Additionally, during the year ended December 31, 2015, 17 shares of common stock were issued upon the exercise of other warrants.
On April 1, 2016, Hampshire MedTech Partners II, GP (“Hampshire GP”) filed suit against the Company in the Travis County, Texas 200th Judicial District Court relating to a Warrant to Purchase Shares of Common Stock issued to Hampshire MedTech Partners II, LP (“Hampshire LP”) on November 6, 2014 (the “Warrant”). Hampshire GP alleged that as a result of a subsequent financing the Company breached the anti-dilution provision of the Warrant by failing to increase the number of shares subject to the Warrant as well as failing to reduce the exercise price of the Warrant. In November 2016, the Company and Hampshire GP settled the lawsuit through the execution of a Settlement Agreement and Release. Pursuant to the terms of settlement, the Company issued 962,380 shares with a fair value of $794,000 and the pre-existing warrants held by Hampshire GP were cancelled. The value of the shares issued were recognized as loss on settlement in the consolidated statements of operations.
1,882,718 shares of common stock were issued related to the Riverside Debt discussed in Note 7.
9. Stock-Based Compensation
Option and Equity Plans
In May 2016, the stockholders of the Company approved common shares issuable under the 2012 Employee, Director and Consultant Equity Incentive Plan (the “2012 Plan”) of 1,142,425 shares. The total number of shares available for grant under the 2012 Plan at December 31, 2016 was 907,200.
Stock Options
A summary of the Company’s stock option activity for the years ended December 31, 2016 and 2015 is as follows:
| F-20 |
| December 31, 2016 | ||||||||||||||||
| Weighted-Average | Weighted-Average Remaining Contractual Life | |||||||||||||||
| Options | Exercise Price | (Years) | Intrinsic Value | |||||||||||||
| Outstanding at December 31, 2015 | 112,373 | $ | 41.55 | |||||||||||||
| Granted | 39,354 | $ | 1.37 | |||||||||||||
| Exercised | ||||||||||||||||
| Forfeited | ||||||||||||||||
| Expired | (14,380 | ) | $ | 41.33 | ||||||||||||
| Outstanding at December 31, 2016 | 137,347 | $ | 30.59 | 8.2 | $ | - | ||||||||||
| Exercisable at December 31, 2016 | 107,954 | $ | 42.31 | 7.9 | $ | - | ||||||||||
| Vested and expected to vest at December 31, 2016 | 137,347 | $ | 30.59 | 8.7 | $ | - | ||||||||||
| December 31, 2015 | ||||||||||||||||
| Weighted-Average | Weighted-Average Remaining Contractual Life | |||||||||||||||
| Options | Exercise Price | (Years) | Intrinsic Value | |||||||||||||
| Outstanding at December 31, 2014 | 92,289 | $ | 76.95 | |||||||||||||
| Granted | 115,690 | $ | 13.20 | |||||||||||||
| Exercised | - | $ | - | |||||||||||||
| Forfeited | (70,447 | ) | $ | 41.10 | ||||||||||||
| Expired | (25,159 | ) | $ | 63.65 | ||||||||||||
| Outstanding at December 31, 2015 | 112,373 | $ | 41.55 | 7.7 | $ | - | ||||||||||
| Exercisable at December 31, 2015 | 44,879 | $ | 83.10 | 5.5 | $ | - | ||||||||||
| Vested and expected to vest at December 31, 2015 | 109,996 | $ | 42.15 | 7.7 | $ | - | ||||||||||
The aggregate intrinsic value in the table above is calculated as the difference between the estimated fair value of the Company’s stock at December 31, 2016 and the exercise price of each option.
The weighted average grant date fair value of options granted during the years ended December 31, 2016 and 2015 was $0.84 and $5.32, respectively.
The Company estimates the fair value of each stock option on the grant date using the Black-Scholes valuation model, which requires several estimates including an estimate of the fair value of the underlying common stock on grant date. The expected volatility was based on an average of the historical volatility of a peer group of similar companies. The expected term was calculated utilizing the simplified method. The risk-free interest rate was based on the U.S. Treasury yield curve in effect at the time of grant for the expected term of the option. The following weighted average assumptions were used in the calculation to estimate the fair value of options granted to employees for the years ended December 31, 2016 and 2015:
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Weighted-average risk-free interest rate | 1.63 | % | 1.64 | % | ||||
| Weighted-average expected life (in years) | 6.30 | 6.30 | ||||||
| Expected dividend yield | 0 | % | 0 | % | ||||
| Weighted-average expected volatility | 66 | % | 48 | % | ||||
Restricted Stock Award
During the year ended December 31, 2015, the Company issued 18,000 Restricted stock awards (“RSA”) with a weighted average grant date fair value of $4.80 per share. Such shares vested in 2015 and the Company recorded $87,000 of stock-based compensation for RSAs during the year ended December 31, 2015.
There was not RSA activity during the year ended December 31, 2016.
| F-21 |
Stock-Based Awards Granted to Nonemployees
The Company from time to time grants options to purchase common stock or restricted stock to non-employees for services rendered and records expense ratably over the vesting period of each award. The Company estimates the fair value of the stock options using the Black-Scholes valuation model at each reporting date. No stock options were granted to non-employees during the year ended December 31, 2015. The Company granted 18,000 RSAs to non-employees and recorded stock-based compensation expense of $87,000 during the year ended December 31, 2015. The Company did not grant any RSA’s or stock options to non-employees in 2016. Total non-employee options were 10,766 number of shares with a total expense of $6,795 in 2016. These amounts are included in the tables above.
Summary of Stock-Based Compensation Expense
Total stock-based compensation expense included in the consolidated statements of operations was allocated as follows (in thousands):
| As of December 31, | ||||||||
| 2016 | 2015 | |||||||
| Cost of revenue | $ | 17 | $ | 50 | ||||
| Research and development | 104 | 177 | ||||||
| General and administrative | 132 | 514 | ||||||
| Selling and marketing | 17 | 170 | ||||||
| Capitalized into inventory | 3 | 75 | ||||||
| $ | 273 | $ | 986 | |||||
Unrecognized stock-based compensation at December 31, 2016 and 2015 were as follows (in thousands):
| As of December 31, 2016 | ||||||||
| Weighted | ||||||||
| Average | ||||||||
| Unrecognized | Remaining Period | |||||||
| Stock-Based | of Recognition | |||||||
| Compensation | (in years) | |||||||
| Stock options | $ | 256 | 1.13 | |||||
| As of December 31, 2015 | ||||||||
| Weighted | ||||||||
| Average | ||||||||
| Unrecognized | Remaining Period | |||||||
| Stock-Based | of Recognition | |||||||
| Compensation | (in years) | |||||||
| Stock options | $ | 484 | 1.9 | |||||
10. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
| F-22 |
The following is a reconciliation of the expected statutory federal income tax provision to the actual income tax expense:
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Federal statutory rate | (35.0 | )% | (35.0 | )% | ||||
| State taxes, net of federal benefits | (3.0 | )% | (2.3 | )% | ||||
| Research and development credits | (0.0 | )% | 1.5 | % | ||||
| Equity related expenses | 2.3 | % | 10.7 | % | ||||
| Change in valuation allowance | 35.8 | % | 25.10 | % | ||||
| Other | -0.10 | % | 0.00 | % | ||||
| Total income tax expense | 0.00 | % | 0.00 | % | ||||
Significant components of the Company's deferred tax assets and liabilities were as follows (in thousands):
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 61,279 | $ | 56,679 | ||||
| Depreciation | 474 | 37 | ||||||
| Federal R&D Credit | 2,222 | 2,222 | ||||||
| Other | 7,107 | 6,308 | ||||||
| Total deferred tax assets | 71,082 | 65,246 | ||||||
| Deferred tax liabilities: | ||||||||
| Amortization of intangibles | (697 | ) | (807 | ) | ||||
| Total deferred tax liabilities | (697 | ) | (807 | ) | ||||
| Net deferred tax asset | 70,385 | 64,439 | ||||||
| Less valuation allowance | (70,519 | ) | (64,573 | ) | ||||
| Net deferred tax liabilities | $ | (134 | ) | $ | (134 | ) | ||
At December 31, 2016 and 2015, the Company had net operating loss carryforwards for federal and state income tax purposes of approximately $160.2 million and $148.2 million, respectively. The federal and state net operating loss carryforwards will expire from 2023 to 2036, unless previously utilized. Additionally, the Company believes an ownership change has occurred that would trigger the limitation on usage of net operating losses imposed by Internal Revenue Code section 382. Because of this limitation, a significant portion of the net operating losses would more likely than not expire unused.
During the years ended December 31, 2016 and 2015, the Company recognized no amounts related to tax interest or penalties related to uncertain tax positions. The Company is subject to taxation in the United States and various state jurisdictions. The Company currently has no years under examination by any jurisdiction.
A valuation allowance has been established as realization of such deferred tax assets has not met the more likely-than-not threshold requirement. If the Company’s judgment changes and it is determined that the Company will be able to realize these deferred tax assets, the tax benefits relating to any reversal of the valuation allowance on deferred tax assets will be accounted for as a reduction to income tax expense. The tax valuation allowance increased by approximately $5.6 million and $6.0 million for the years ended December 31, 2016 and 2015, respectively.
11. Commitment and Contingencies
The Company currently leases laboratory, manufacturing and office space and equipment under noncancelable operating leases which provide for rent holidays and escalating payments; this lease ends 2019. Lease incentives, including rent holidays, allowances for tenant improvements and rent escalation provisions, are recorded as deferred rent. Rent under operating leases is recognized on a straight-line basis beginning with lease commencement through the end of the lease term. Sublease income is recorded as a reduction of rent expense. For each of the years ended December 31, 2016 and 2015, rental expense was $661,000 and $734,000, respectively. Sublease income was $133,000 and $84,000 during the year ended December 31, 2016 and 2015, respectively.
| F-23 |
The following table summarizes future minimum rental payments required under operating leases that have initial or remaining non-cancelable lease terms in excess of one year as of December 31, 2016 (in thousands):
| Year ending December 31: | Operating Leases | Sublease Income | Total | ||||||||||
| 2017 | $ | 925 | $ | (129 | ) | $ | 796 | ||||||
| 2018 | 952 | (43 | ) | 909 | |||||||||
| 2019 | 980 | - | 980 | ||||||||||
| Total minimum lease payments | $ | 2,857 | $ | (172 | ) | $ | 2,685 | ||||||
The Company has entered into consulting and development agreements with some of its advisors, including some surgeon advisors. The Company has agreed to pay some of the surgeon advisors a portion of the net profits attributable to the sale of specific spine products for which the surgeon advisors provided the Company with consulting and related services related to the conceptualization, development, testing, clearance, approval and/or related matters involving implant products. The Company is obligated to pay royalties to different surgeon advisors in connection with the sale of certain of its implant products. These agreements generally continue until the later of (a) ten years from the date of the agreements, and (b) the expiration of the patent rights relating to the devices covered by the agreements, when rights have been assigned by the individuals to the Company. The Company incurred royalties of $607,000 and $775,000 related to these agreements for the years ended December 31, 2016 and 2015, respectively. None of the royalty arrangements contain minimum royalty payments.
On May 13, 2015, the Company entered into a joint agreement for research and development of silicon nitride based devices. This agreement is effective for a period of five years from the date of commencement. The Company incurred payments of $270,000 and $270,000 related to this agreement for the years ended December 31, 2016 and 2015, respectively.
The Company has executed agreements with certain executive officers of the Company which, upon the occurrence of certain events related to a change in control, call for payments to the executives up to three times their annual salary and accelerated vesting of previously granted stock options.
From time to time, the Company is subject to various claims and legal proceedings covering matters that arise in the ordinary course of its business activities. Management believes any liability that may ultimately result from the resolution of these matters will not have a material adverse effect on the Company’s consolidated financial position, operating results or cash flows.
12. 401(k) Plan
Effective June 1, 2004, the Company adopted a defined contribution retirement plan under Section 401(k) of the Internal Revenue Code. The plan covers substantially all employees. Eligible employees may contribute amounts to the plan, via payroll withholdings, subject to certain limitations. The plan permits, but does not require, additional matching contributions to the plan by the Company on behalf of the participants in the plan. The Company incurred approximately $145,000 and $175,000 relating to retirement contributions for the years ended December 31, 2016 and 2015, respectively.
13. Restatement to previously issued condensed consolidated financial statements
Subsequent to the issuance of the condensed consolidated financial statements as of September 30, 2016, and for the three and nine month periods then ended, the Company identified errors related to a failure to record a one-time, non-cash $3.8 million charge attributable to the deemed dividend related to the accretion of a discount on Series A convertible preferred stock upon conversion into the Company’s common stock, which occurred in July 2016. To correctly present the deemed dividend on Series A Preferred Stock, net loss attributable to common stockholders and per share net loss attributable to common stockholders, the Company’s previously issued condensed consolidated financial statements as of and for the three and nine month periods ended September 30, 2016 were restated in an amended Quarterly Report on Form 10-Q/A.
The impact of this change as of and for the three and nine month periods ended September 30, 2016, is as follows (in thousands, except share and per share data):
| Three
months ended September 30, 2016 | ||||||||
| As
Previously Reported | As Restated | |||||||
| Total comprehensive loss | $ | (4,338 | ) | $ | (4,338 | ) | ||
| Deemed dividend related to Series A convertible preferred stock | (2,499 | ) | (6,278 | ) | ||||
| Net loss attributable to common stockholders | $ | (6,837 | ) | $ | (10,616 | ) | ||
| Basic and diluted net loss per common share | $ | (0.30 | ) | $ | (0.46 | ) | ||
| Shares used to compute basic and diluted net loss per common share | 23,048,941 | 23,048,941 | ||||||
| Nine
months ended September 30, 2016 | ||||||||
| As
Previously Reported | As Restated | |||||||
| Total comprehensive loss | $ | (12,790 | ) | $ | (12,790 | ) | ||
| Deemed dividend related to Series A convertible preferred stock | (2,499 | ) | (6,278 | ) | ||||
| Net loss attributable to common stockholders | $ | (15,289 | ) | $ | (19,068 | ) | ||
| Basic and diluted net loss per common share | $ | (0.97 | ) | $ | (1.21 | ) | ||
| Shares used to compute basic and diluted net loss per common share | 15,711,429 | 15,711,429 | ||||||
The above-mentioned corrections do not have an effect on consolidated total comprehensive loss, and the consolidated balance sheets or consolidated statements of cash flows, except for the non-cash financing activities presented on the consolidated statements of cash flows.
14. Subsequent Events
In January 2017, the Company completed a secondary offering in which the Company sold 8,900,000 shares of common stock and warrants to purchase 4,005,000 shares of common stock for $0.51 per unit (each unit consisting of one share and 0.45 warrants). The Company received approximately $4.1 million in net proceeds from the offering after deducting underwriting expenses, commission and other offering expenses. The warrants became exercisable on the closing date and expire on the five-year anniversary of the closing date and have an initial exercise price per share equal to $0.55 per share, subject to appropriate adjustment in the event of recapitalization events, stock dividends, stock splits, stock combinations, reclassifications, reorganizations or similar events affecting the Company’s Common Stock.
In connection with the January 2017 secondary offering of shares of common stock and warrants, on February 24, 2017, the underwriter in the offering exercised its option to purchase additional warrants to purchase 360,000 shares of common stock.
On May 12, 2016, the Company entered into an engagement letter (“the “Engagement Letter”) with Ladenburg Thalmann to serve as underwriter in the Company’s July 2016 secondary offering of Class A Units and Class B Units. The Engagement Letter required the Company to pay to Ladenburg Thalmann an additional fee, if, during the term of the Engagement Letter or within six months after the date of termination or expiration of the Engagement Letter, the Company sells securities to investors contacted by Ladenburg Thalmann during the term of the Engagement Letter. In connection with the closing of the January 2017 secondary offering, the Company incurred an obligation under the Engagement Letter to pay to Ladenburg Thalmann, a cash fee in the amount of $314,160.
On July 28, 2017, the Company closed on a $2.5 million term loan (the Loan”) with North Stadium Investments, LLC, a company owned and controlled by the Company’s Chief Executive Officer, Chairman of the Board, and Principal Financial Officer. The Loan bears interest at the rate of 10% per annum, requires the Company to make monthly interest payments for a period of 12 months, starting September 5, 2017, with principal and any unpaid interest being due and payable 12 months from the effective date of July 28, 2017, and is secured by substantially all of the assets of the Company, junior to the already existing security interest in such assets held by Hercules Capital, Inc. In connection with the Loan the Company also issued to North Stadium Investments a warrant to acquire up to 660,000 common shares with a purchase price set at $0.42 per share and a 5 year term.
| F-24 |