Attached files
| file | filename |
|---|---|
| EX-21.1 - PROLUNG INC | ex21-1.htm |
UNITED
STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K/A
Amendment No. 2
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE FISCAL YEAR ENDED DECEMBER 31, 2016 |
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM _________ TO ___________ |
| 000-54600 | ||
| (Commission File No.) |
PROLUNG, INC. (FORMERLY FRESH MEDICAL LABORATORIES, INC.) |
||
| (Exact name of registrant as specified in its charter) |
| Delaware | 20-1922768 | |
(State or other jurisdiction of incorporation) |
(IRS Employer Identification No.) |
757 East South Temple, Suite 150 Salt Lake City, Utah 84012 |
||
| (Address of principal executive offices, including zip code) |
Registrant’s telephone number, including area code: (801) 736-0729
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: Common Stock, par value $.001 per share
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
YES [ ] NO [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
YES [ ] NO [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES [X] NO [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES [X] NO [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Report or any amendment to this Report. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “accelerated filer”, “large accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one):
| Large Accelerated Filer [ ] | Accelerated Filer [ ] |
| Non-accelerated Filer [ ] | Smaller reporting Company [X] Emerging Growth Company [X] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act . [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act): YES [ ] NO [X]
The aggregate market value of the shares of common stock held by non-affiliates of the Registrant on June 30, 2016 was approximately $13,367,160, based upon 22,620,078 shares held by non-affiliates and an assumed fair market value of $0.88 per share. The Registrant’s common stock does not trade on an established market; accordingly, fair market value is estimated based upon the last private purchase of the Company’s common stock prior to June 30, 2016. Shares of common stock held by each officer and director and by each other person who may be deemed to be an affiliate of the Registrant have been excluded.
As of April 17, 2017, the Registrant had 25,659,409 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE. None.
EXPLANATORY NOTE
ProLung, Inc. (the “Company”) is filing this Amendment No. 2 on Form 10-K/A (this “Amendment”, together with the portions of the Amendment No. 1 on Form 10-K/A not superseded hereby, this “Report”) in order to amend the disclosure set forth in the Amendment No. 1 on Form 10-K/A filed by the Company on April 19, 2017 (the “First Amendment”) with respect to the following Items.
| ● | Item 1 Business, in order to revise its description of the status and process related to FDA marketing approval; | |
| ● | Item 8 Financial Statements and Supplementary Data, in order to reflect an updated audit report and reflect in the notes the potential effects of ASU 2014-09 Revenue from Contracts with Customers. | |
| ● | Item 15 Exhibits, Financial Statement Schedules, in order to reflect updated certifications and the filing of certain Exhibits. |
The modifications are in response to comments by the SEC staff on the First Amendment.
In this Amendment, we set forth in their entirety Items that are being amended and have omitted Items that are not being amended. Any Items set forth in this Amendment supersede in its entirety the same Item included in the Initial Amendment. This Amendment should be read as an integrated Report together with the portions of the First Amendment that are not superseded hereby.
| 2 |
Item 1. Business
This Report contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve risks and uncertainties. Purchasers of any of the shares of common stock of ProLung, Inc. (formerly Fresh Medical Laboratories, Inc.) are cautioned that our actual results will differ (and may differ significantly) from the results discussed in the forward-looking statements. The reader is also encouraged to review other filings made by us with the Securities and Exchange Commission (the “SEC”) describing other factors that may affect future results.
In this filing, ProLung, Inc. (formerly Fresh Medical Laboratories, Inc.) and its consolidated subsidiary are referred to as “ProLung” in addition to as the “Company” versions of “we” or “us.” Current and all granted trademarks include ProLungdx®, Fresh Medical Laboratories®, ProLung®, EPN Scan®, Electro Pulmonary Nodule Scanner (EPN Scan)® and EPN Scanner®. Any other trademarks and service marks used in this Report are the property of their respective holders
Overview
We are a medical technology company specializing in predictive analytic, early stage lung cancer risk testing, which we refer to as the “ProLung Test.” Our noninvasive, painless and radiation-free ProLung Test was developed to immediately assess the risk of malignancy in lung nodules found in the chest by a CT scan, which is currently the primary method used for the early detection of lung cancer. As lung cancer is the leading cause of cancer death, early detection makes a substantial improvement in survival in a large population group. Timely identification of malignancy is essential for patients and their families. Currently, patients often wait from three months to three and one-half years to have the risk of malignancy assessed through periodic CT scan surveillance. Until malignancy is determined to be likely, invasive biopsy and treatment are significantly delayed. Current statistics reflect a 17% survival rate at five years for those diagnosed with lung cancer.
We believe the ProLung Test, in conjunction with the discovery of a nodule by CT scan, provides a more rapid assessment of the risk of malignancy, which must be determined prior to biopsy. Since a lung biopsy is invasive and may require life threatening thoracic surgery, physicians, patients, and insurance companies typically delay biopsy and therapy until the risk of malignancy outweighs the risk of further diagnostic procedures. For these patients, the delay reduces the treatment opportunity window and may cause sustained emotional trauma.
The ProLung Test enables the practitioner to promptly assess the risk of malignancy in patients with lung nodules. The ProLung Test utilizes mass averaging bioconductive technology which is similar to other bioconductive technologies utilized frequently in health care. Mass averaging bioconductive technology involves a scanning process that measures significant differences in electrical conductance between cancerous and benign tissue. We plan to introduce the ProLung Test to the market as a standard predictive analytic test, without the need for transmission of a physical sample or specimen to a lab for analysis.
The ProLung Test acquires bioconductance measurement data by means of a patented probe and disposable diaphoretic electrodes placed on the patient’s back and arms. The ProLung Test registers and evaluates measurement data derived from 62 pathways through the chest and is processed by a patented predictive analytic algorithm. The results are summarized in a report that can be used by the physician, in concert with other risk factors such as nodule size, family history, smoking history and gender, to evaluate patients with nodules. The ProLung Test requires minimal preparation and can be completed in fewer than 30 minutes. Most importantly, it guides the physician decision making without the time consuming, expensive and watchful waiting period. We believe the ProLung Test provides considerable cost savings when compared with periodic CT imaging studies, repeated follow-up and potentially unnecessary surgery.
ProLung licensed and developed the intellectual property and established the clinical research plan for the ProLung Test. Beginning in 2005, we embarked on clinical research which revealed the potential of our technology. In 2011, our research demonstrated the utility of the ProLung Test in lung cancer patients. To date, more than 545 patients have been tested using the ProLung Test in major cancer centers such as Stanford, UCLA, Loyola, MD Anderson and Huntsman, among others.
In the US, the push for early detection was greatly accelerated in 2013. Recognizing the dismal rate of lung cancer survival in the US, and the potential value of early detection, Federal guidelines were established for CT screening. The regulations provided for CT screening for lung cancer in asymptomatic adults aged 55 to 80 who have a 30 pack-year history of smoking and who currently smoke, or have quit smoking in the past 15 years. This demographic group addresses a substantial portion of individuals of high risk of lung cancer. The US health care industry has generally recognized the need for technologies that will provide for earlier detection of cancers at a lower cost. Genetic biomarkers, protein panels, and breath analysis, among others, are in various stages of development. To our knowledge, the ProLung Test is the first bioconductive technology that has been developed for the risk stratification of lung cancer. In February 2015, the US Center for Medicare and Medicaid Services announced its coverage of lung cancer screening by CT. This newly reimbursed screening procedure increased the number of individuals with suspicious lung nodules who may be candidates for the ProLung Test.
| 3 |
With the arrival of lung cancer screening recommendations, the large US market and government-backed reimbursement represent near term opportunities to accelerate diagnosis and treatment of lung cancer while reducing invasive biopsies and costs. We made US approval and recognition of the ProLung Test our major priority, targeting lung cancer risk stratification and reducing time to treatment. We intend to seek government-backed reimbursement after FDA approval. We believe the ProLung Test can be offered at a fraction of the cost of current standard of care which is repeat periodic imaging studies.
In May 2013, we achieved an important validation of our ProLung Test by receiving the “CE” mark in Europe. This certification verifies that the ProLung Test meets the regulatory requirements for the marketing and sale of the ProLung Test in the European Economic Area and European Free Trade Association Countries representing 510 million individuals and 31 member states. Our European clinical research includes testing more than 154 patients in Italy, Switzerland and Germany. We intend to seek European reimbursement approval and accelerate our marketing in Europe following receipt of US Food and Drug Administration, (“FDA”) market approval. We believe CT screening is likely to be implemented in Europe following the completion of several lung cancer screening trials already underway.
In early 2015, we submitted an application for marketing approval under Section 510(k) from the FDA. In February 2015, we received a “substantive review” from the FDA requesting additional information, regarding the risk classification of the test, the study design and study analysis. We held various meetings with the FDA and agreed to complete and include an additional clinical study which was already underway. Before the FDA can grant approval of our 510(k) or de novo application, we must resubmit the application with positive results of the requested study and resolve any remaining issues previously identified by the FDA as well as address possible issues that may be identified in the future. We are in the process of preparing the necessary information requested by the FDA.
We have developed the quality management system as well as supply chain and the ability to fully manufacture the entire ProLung System in our own Salt Lake City facility. We have received ISO 13485 and other approvals, and made certain refinements to the intellectual property that will further our capabilities, especially the development of the underlying predictive analytic algorithm and refinements to various software and physical components. Over the last five years, we have expanded our intellectual property portfolio, completed the development of the ProLung Test and manufacturing of the ProLung System and embarked upon clinical trials to provide validation to the medical community. The current clinical trial has 350 enrolled patients at 13 cancer and medical centers across the US. We are also enrolling up to 70 additional patients to replace those lost to follow-up or non-evaluable, as provided in the study protocol. When complete, data from our trial will be submitted to the FDA for the 510(K) or de novo market approval.
In addition, proceeds of the $8.22 million has allowed for a more definitive development of our marketing and sales initiatives described below. Our financial strength has been reinforced by the elimination of external and related party debt with the exception of two convertible notes remain owned by a single shareholder. All other convertible securities have been converted or repaid. Importantly, the Company has created a corporate infrastructure supported by a strong Board of Directors, an Audit Committee, a Nomination and Governance Committee, a Compensation Committee as well as a Science and Technology Committee to the Board that is in place to provide the necessary drive to commercialization, research oversight, regulatory direction, and financial reporting initiatives. Recently, the Company announced the appointment of Dr. Rex Yung, as Chief Science Officer, who was formerly director of Pulmonary Oncology at Johns Hopkins School of Medicine to our senior management together with our Chief Medical Officer, Dr. Jeff O’Driscoll. Dr. Yung will oversee various aspects of the development process and manage our extensive pulmonology network. Dr. Yung has published several studies and lectured widely on the application of bioconductive technology to the early detection of lung cancer.
Our Competitive Strengths
| ● | The only predictive analytic technology available for the lung utilizing bioconductive measurement technology. | |
| ● | More than 545 US patients tested with the ProLung Test in five well controlled clinical studies. | |
| ● | CE Mark approval in the European Economic Area. 154 patients tested with the ProLung Test in physician registries. |
| 4 |
| ● | ISO 13845 manufacturing capacity for the completion of the ProLung System and ProLung Test, including supply chain management, computerized drawing control, purchasing management and inventory control. | |
| ● | Patent portfolio that includes six US patents and 14 US and foreign applications. | |
| ● | A US and European network of key opinion leadership projecting influence throughout these markets. | |
| ● | Currently, conducting trials and on the path to FDA 510(k) or de novo near term application. |
Our Business Strategy
| ● | Complete the current multi-site US study of the ProLung Test. Resubmit the 510(k) or de novo application to the FDA including the results from the US study. Obtain FDA regulatory clearance to sell the ProLung Test in the US. | |
| ● | In conjunction with FDA approval pursue foreign market approvals and sales including the continuing development of key distributors and Key Network Leaders in these various markets. | |
| ● | Drive adoption through established KOLs: |
| ○ | ProLung has long established relationships with KOLs in the lung cancer field. KOLs influence large, sometimes national, networks and drive adoption of new technology. These networks consist of major cancer centers and veterans integrated system networks which have contract relationships with affiliate hospitals which adopt the protocols of the primary cancer center, creating a multiplier effect in terms of access and acceptance of the ProLung Test across the network. This strategy will be executed by ProLung’s sales representatives and distributors dedicated for each respective network. | |
| ○ | ProLung’s KOLs already have vital experience with the ProLung Test. The KOLs and their staff have installed ProLung Systems at their centers, completed training on the device and have used the ProLung Test on their patients in sponsored clinical studies. |
| ● | Transition existing hospital installations using the ProLung Test for investigational use to serve commercial paying customers. Leverage the multi-center study results, existing ProLung System installations and physician KOLs to acquire additional customer sites. | |
| ● | Continue to build our relationships with the medical community and patient advocacy groups in general. We are actively involved in scientific, medical and commercial organizations and communities such as the Medical Device Manufacturers Association, Society of Clinical Research Associates, the International Association for the Study of Lung Cancer and the Lung Cancer Alliance. We anticipate that we will be able to leverage our involvement in these organizations to increase awareness of the benefits of our ProLung Test. |
Add additional cancer risk stratification technologies to the Company’s product portfolio and build upon the existing platform utilizing other available data sources.
Market Opportunity
According to the American Cancer Society (“ACS”), lung cancer is the leading cause of cancer death among both men and women; about one out of four cancer deaths are from lung cancer. ACS estimates that in 2017 more people in the United States will die of lung cancer than of colon, breast, and prostate cancers combined.
According to the World Health Organization (“WHO”), lung cancer is the most common cause of death from cancer worldwide and is estimated to be responsible for nearly one in five cancer related deaths. The overall ratio of mortality to incidence is 87%. Each year there are over 1.8 million new cases of lung cancer worldwide, as well as nearly 1.6 million deaths. The lifetime chance of developing lung cancer is 1:17 in women and 1:14 in men.
Until recently, asymptomatic lung cancer was detected only incidentally when looking for something else. Currently, a lung cancer screen now reimbursed by Medicare, is performed by low-dose computed tomography. This has led to a dramatic increase in number of individuals with lung nodules detected, which is intensifying the need for a risk stratification test such as the ProLung Test. The following is a summary of the principal markets for the Company’s ProLung Test.
| 5 |
Lung Cancer Incidence and Mortality
| New Cases | Deaths | |||||||
| United States | 222,500 | 155,870 | ||||||
| European Union | 313,000 | 268,000 | ||||||
| China | 653,000 | 597,000 | ||||||
| World | 1,825,000 | 1,590,000 | ||||||
Lung cancer patients face median five-year survival rates of only 16% (compared to 89% for breast cancer and 98% for prostate cancer). Survival rates of lung cancer lags behind that of other cancer sites due to a lack of early and effective detection, and a challenging biopsy. A significant amount of time is required to assess the risk under current guidelines. Should innovation reduce the time required for assessing the risk of malignancy, lung cancer mortality would approach that of other cancer sites. In those instances when lung cancer was detected in its earliest stage, five-year survival improves by 38%. Experts project that with accurate and early diagnosis, ten-year survival could approach 80%.
U.S. Market
Americans at high risk:
| Region | Population (in millions) | At high risk (in millions) | Market Channel | |||||||
| United States | 319 | 123 | Direct & Indirect | |||||||
Symptomatic:
Each year 225,500 are diagnosed with lung cancer. Approximately 87% of lung cancer patients are symptomatic at presentation.
Asymptomatic /Incidental:
In addition, an estimated 13.5 million chest CT scans are performed annually, primarily for other purposes, of which 18% reveal incidental non-calcified solitary pulmonary nodules resulting in an estimated 2.4 million patients without lung cancer symptoms whose indeterminate masses require follow-up.
Lung Cancer Screening:
Given the size of the US market and the progression of CT scan use in early detection, approval and acceptance of the ProLung Test in the US is the major priority. The CDC estimates that there are 123 million Americans at risk of lung cancer (which includes 94 million current and former smokers plus 27 million exposed to carcinogenic agents at home or in industry). In the National Lung Cancer Screening Trial of 53,454 patients, approximately 24% of the CT scans performed were positive revealing a lung nodule suspicious for lung cancer that required follow-up. CT screening was recommended by the US Preventive Services Task Force on December 31, 2013 and Medicare began to pay for lung cancer screening on February 5, 2016. Based on these estimates, if the approximately 123 million Americans at risk for lung cancer received a low dose CT screen approximately 24% (29 million) Americans may reveal lung nodules requiring follow up. We believe these patients would be eligible to receive the ProLung Test.
In the US, 14 hospital groups are currently using ProLung’s Test in lung cancer research, and we have plans to expand to an additional two hospitals and clinics for pre-and post-market related research. If our 510(k) or de novo FDA clearance is granted, of which there can be no assurance, we plan to transition hospitals involved in research to commercial placements of the ProLung Test System and consumable test kit.
European Market
ProLung plans to utilize its CE mark in conjunction with US approval in the European Union and European Free Trade Association Countries which represents 510 million individuals and 31-member states including the UK. Europe has some of the highest smoking prevalence of any region in the world which has led to a high incidence of lung cancer. In 2012, the World Health Organization estimated that 268,000 individuals died from lung cancer and that more than 313,000 cases were diagnosed in the European Union.
It is estimated that 28% of Europeans smoke and approximately 133 million individuals are at high-risk of lung cancer. Applying the US rates in the published National Lung Screening Trial (2011), over 30 million of these individuals are estimated to have an indeterminate lung nodule and require follow-up to determine the risk of malignancy. As the number of individuals with indeterminate lung nodules continues to increase in Europe, risk stratification tools such as the ProLung Test are needed to close the gap between discovery of a nodule and the determination of malignancy.
| 6 |
China Market
According to the World Health Organization, the number of smokers in China is steadily growing and increasing at higher rates than any other world region. One in three of the world’s cigarettes is smoked in China. The average Chinese smoker consumes 22 cigarettes per day. This is nearly a 50% increase from 1980. Overall, more cigarettes are smoked in China than in the next top 29 cigarette-consuming countries combined. Lung cancer is epidemic in China with 653,000 cases in 2012 and an estimated 597,000 deaths.
The government’s smoking cessation campaign and interventions are poorly funded and weakly enforced and certain provincial governments are somewhat dependent upon state-owned tobacco sales and taxation. However, China’s Government is collaborating with pulmonology and radiology leadership to study low-dose CT screening for earlier detection of lung cancer. The government has also sponsored economic studies to investigate the reimbursement of lung cancer screening in the health insurance system.
As the number of individuals with indeterminate lung nodules continues to increase in China, risk stratification tools such as the ProLung Test will be needed to close the gap between discovery of a nodule and the determination of malignancy. This clinical need for risk stratification will be multiplied if a lung cancer screening program is implemented in the Chinese healthcare system.
Latin American Market
Nearly 10% of the world’s smokers live in Latin America (i.e., more than 120 million). As yet, the lung cancer screening is not widespread. As the number of individuals with indeterminate lung nodules increases in Latin America, another growing market will be available to the ProLung Test.
Latin America has a population at-risk for lung cancer of at least 120 million. In accordance with rates from the National Lung Screening Trial (2010), roughly 25 million individuals will have an indeterminate pulmonary lesion if screened and require follow up to determine the risk of malignancy. As the number of individuals with indeterminate lung nodules increases in Latin America, risk stratification tools such as the ProLung Test are needed to close the gap between discovery of a nodule and the determination of malignancy.
Our Lead Product
ProLung Test
The ProLung Test has shown utility to evaluate the risk of lung cancer in patients with lung nodules in well-controlled clinical trials. See “Business – Research and Clinical Trial Results.” ProLung’s novel mass-averaging bioconductive technology simultaneously considers data from multiple measurement pathways and utilizes a patented predictive analytic algorithm to combine the individual measurements into a weighted average composite score that indicates an increased or decreased risk of malignancy in the individual in which the nodule has been detected. No images are generated by the ProLung Test and extensive training is not required to interpret the composite score.
The ProLung Test, will be introduced to the market as a standard predictive analytic test without the need for transmission of a physical sample or specimen. Instead, the ProLung Test acquires bioconductive measurement data by means of a patented probe and disposable diaphoretic electrodes placed on the back and arms. The data containing precision measurements is processed by a patented predictive analytic algorithm and a report is generated that may be used by the physician in addition to other risk factors such as nodule size, family history, smoking history, and gender to evaluate patients with suspicious masses or lesions identified by CT scan. The ProLung Test is rapid, non-invasive, and non-radiating. The ProLung Test can be completed in fewer than 30 minutes.
The ProLung Test is comprised of the following components:
| ● | ProLung System - Each system, which will be sold to the customer, consists of the probe, scanner, tower, monitor, and keyboard which are all medical grade components available for sale in English, French, German, Spanish, and Italian versions. The pricing of the ProLung System varies depending upon the volume of the ProLung Test Kits sold. | |
| ● | ProLung Test Kit – ProLung Test Kit sales should provide near term and continual cash flow. Each single-use, disposable, ProLung Test Kit is sold in a hygienic envelope that displays a unique identifier code that is required for access to a ProLung Test report, together with all the components necessary to assure precision test performance, patient comfort and hygiene. Each ProLung Test Kit includes six diaphoretic electrodes, one probe tip and one moistening sponge. Initially, ProLung plans to sell the ProLung Test Kit for $400 each, available in boxes of 10 and 40. Each ProLung Test Kit is encoded with a unique identifier number and bar code that releases a written test result to the ordering physician. |
| 7 |
The ProLung Test Procedure
| 1. | The ProLung Test System is connected to the probe, to the electrode cables, and to the power supply. Following a brief power-on sequence, the ProLung Test completes self-diagnostics. | |
| 2. | The patient is seated. | |
| 3. | ProLung Test kit is opened and removed from its tamper-proof packaging. | |
| 4. | Single-use diaphoretic electrodes are placed at sites on the patient’s back and arms. | |
| 5. | Session data is entered including technician name, physician name, report delivery method and patient data. | |
| 6. | Testing begins, as prompted by the device, by applying the probe to acquire measurement data from sites on the chest, shoulders and arms. | |
| 7. | Monitors the acquisition of real-time data. Should re-measurement be required, the device provides visual and audible notification that it has not received usable data. |
Research and Clinical Trial Results
Our ProLung Test has been evaluated in four clinical trials and is in the process of its fifth clinical trial. The ProLung Test is currently being evaluated in a US multicenter trial. We made modifications to the ProLung Test throughout the research process. A description of each clinical trial is below:
Proof of Principle - McHenry, IL (2005)
| ● | Description. A blinded single-site study of 36 subjects was designed to detect differences in bioelectrical impedance measurements between biopsy-confirmed lung cancer subjects and age- and gender-matched control subjects. The trial was configured as a sequential design consisting of three individual cohorts. Following the completion of each cohort, the data was evaluated for the presence of a predictive model which would discriminate between the lung cancer patients and control subjects. | |
| ● | Results. The First Cohort of 12 subjects could not be utilized for statistical analysis because of an incorrectly calibrated device. An algorithm or predictive model was derived in the Second Cohort of 14 patients which fully discriminated between lung cancer patients and healthy volunteers. | |
| Subsequent analysis of the Third Cohort offered potentially confounding results, but ProLung felt the hypothesis of feasibility of the device had been successfully demonstrated and that sufficient evidence of feasibility existed to proceed with further research. |
Reliability and Repeatability ― Salt Lake City, UT (2006)
| ● | Description. A single-site study to evaluate the variability of the ProLung Test in 22 healthy volunteers. | |
| ● | Results. Measurement variables evaluated were the maximum and minimum conductance. The maximum and minimum conductance values obtained from one operator making repeated measurements with the same device on volunteer subjects over two days of testing were comparable, with slightly lower standard deviations for maximum conductance readings and extremely high reliability indices for both measures. For both data sets, the same measurement points were found to have minimal variability (and maximal reliability) indices. The Electro Pulmonary Nodule Scan showed a reliability index of 0.99 and a correlation between device replicates of 0.98. |
ProLung conducted another internal research study relative to reliability and repeatability. The study was discontinued prior to completing the analysis due to issues with the study design and statistical analysis. No formal conclusions were reached.
| 8 |
Efficacy and Safety in the Target Indication ― Baltimore, MD (2012)
| ● | Description. This single arm, single site algorithm finding and internal validation trial was designed to assess efficacy and safety in the risk stratification of the presence of or absence of malignancy in patients symptomatic of lung cancer who have a suspicious mass as confirmed by CT scan. | |
| ● | Results. Final results included the identification of an algorithm capable of 90% sensitivity (correctly identifying 26 of 29 malignant masses), 92% specificity (correctly identifying 11 of 12 non-malignant masses), and Receiver Operating Characteristic (“ROC”) area (combined sensitivity and specificity) of 90% (correctly identifying 37 of 41 patients overall). Final results were presented in 2011 at the World Conference of the International Association for the Study of Lung Cancer and at the Annual Congress of the European Respiratory Society and were published in the April 2012 edition of the Journal of Thoracic Oncology. The ROC graph is presented below. |
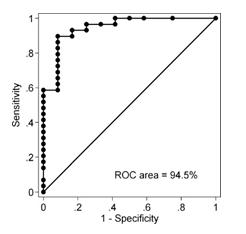
Though not part of the original study, a subsequent subset analysis was performed on Study subjects who had indeterminate results on FDG-PET scans (n=7). In this subset (3 benign, 4 malignant) the ProLung Test correctly predicted the risk of malignancy in the index nodule being assessed. These results were presented at the International Association for the Study of Lung Cancer World Congress in Denver, CO, in September 2015 and published in volume 10, number 9, Supplement 2, Journal of Thoracic Oncology, p. S305).
Reliability and Repeatability —Salt Lake City, Utah, (2015)
| ● | Description. A single-site study to evaluate the variability in the ProLung Test in 60 healthy volunteers. Two measurements were taken on each subject in each of two measurement sessions on two different days, for a total of four measurements on each subject. Measurements were taken by the same operator using the same machine for all measurements. | |
| ● | Study objectives. (1) quantify scan variability when measured twice on the same subject by the same operator on the same day; (2) quantify day-to-day within-subject variability when the same operator uses the same scanner on the same subject on two different days within one week; (3) quantify effects of body mass index and gender on measurements; and (4) assess scan tolerability from subjects’ perspective. | |
| ● | Preliminary Results. While some portions of the data reproduced the results of the 2006 Reliability and Repeatability Study, other portions raised concerns about the reliability and repeatability of other measurement points and of the overall composite scores. Questions about the quality of the data arose from concerns about the measurement technique used by the operator in this study. Those issues, along with limited resources at the time, precluded final results or definitive conclusions. |
Multicenter Study of the ProLung Test – Multicenter (ongoing)
| ● | Description. ProLung is presently engaged in a multicenter study to demonstrate safety and efficacy of the ProLung Test in the lung cancer risk stratification of patients with pulmonary lesions identified by CT. This study commenced in 2012 and can be found on clinical trials.gov ID NCT01566682. Prior to commencing this study, we improved the usability and quality of the ProLung Test by replacing hand-held brass electrodes with adhesive diaphoretic electrodes. We researched the available adhesive electrodes and conducted equivalency testing to affect the improvement without compromising performance. There have been 350 patients have been enrolled. Currently, 70 replacement patients are being enrolled as provided for in the study protocol. The centers include: MD Anderson, Stanford, Huntsman Cancer Institute, Henry Ford Hospital, University of California Los Angeles Medical Center, Loyola, Greater Baltimore Medical Center, Intermountain Healthcare, University of California San Diego, Wake Forest, University of Minnesota Masonic Cancer Center and Providence Healthcare, Beth Israel Deaconess, and Medical University of South Carolina. |
| 9 |
There are three Specific Aims of this study:
| ○ | Optimize and confirm the stability of the ProLung Test risk-stratification algorithm in patients with a diagnosis. | |
| ○ | Externally validate the efficacy of the ProLung Test risk-stratification algorithm by comparing the test result to the conclusive patient diagnosis. | |
| ○ | Assess the safety and tolerability of the ProLung Test procedures. |
Status. We anticipate completion of enrollment and preliminary clinical results by the end of 2017. Our final clinical results are anticipated to be complete by the end of the first quarter of 2018. These results must present sufficient evidence of safety and effectiveness for the intended use. These results will not be known until the end of the clinical trial. If the results are favorable, they will then be included in our amended FDA application which we anticipate to submit during the second quarter of 2018.
Other Research
Mexico. In 2011, ProLung supported a study with a hospital located in Mexico City. The study was administered by ProLung’s partner who was pursuing a joint venture license for the Mexico territory. The partner eventually abandoned the study. After receiving preliminary test results, ProLung had reason to question the quality of the data being gathered and withdrew its support of the study.
China. ProLung has issued a nonexclusive license to an entity conducting research in China. This Chinese researcher has independently changed the classifier algorithm of the device. Results of research in China have been presented in the 2017 American Thoracic Society International Conference Poster Session. These results, however, were derived from a new device developed by the licensee and, therefore, may not be applicable to the ProLung Test.
Italy and Switzerland. Four centers in Italy and one center in Switzerland conducted research with the ProLung Test under the direction of local clinicians. At three of these sites, the research was part of a sales evaluation program for potential sale of the ProLung Test. Subject enrollment at these sites did not conform to research protocols utilized by ProLung. Consequently, the data generated by these clinics were not published by the Company.
At two other sites, Geneva and Florence, additional physician-sponsored research was conducted. It is not known whether these sites conducted research with the ProLung Test that was compliant with Good Clinical Practice or whether these patients conformed with the ProLung Test patient selection criteria. However, at the World Congress of Thoracic Imaging in 2017 the Geneva site posted results indicating Test sensitivity of 66% and a specificity of 66%. The positive predictive value was 94% and negative predictive value was 20%. Geneva researchers concluded the ProLung Test could lower the need for invasive biopsies, especially in high risk patients. The small number of patients (n=27) precludes definitive conclusions.
Similarly, at a center in Florence, Italy, a study looked at 22 subjects undergoing the ProLung Test and PET CT scans. They reported a sensitivity of 75% and a specificity of 50%, with a positive predictive value of 94% and a negative predictive value of 17%. Researchers concluded that the high positive predictive value of the ProLung Test suggested utility in the evaluation of solitary pulmonary nodules, adding that further research was warranted.
Competition
The development and commercialization of new products to improve the accuracy and efficiency of risk stratification of lung cancer is competitive and we expect considerable competition from major medical device companies, laboratory biomarker tests, and academic institutions that are conducting research in lung cancer. Extensive research and financial resources have been invested in the discovery and development of new lung cancer detection tests. Potential competing technologies include, but are not limited to, breath markers, sputum cytology and DNA related markers, blood markers, radiography and CT imaging.
The timing of market introduction of some of our potential products or of competitors’ products may be an important competitive factor. We believe the speed with which we can develop products, complete clinical trials and approval processes, and supply commercial quantities to market are important competitive factors. We expect that competition among products approved for sale will be based on various factors including product efficacy, safety, reliability, availability, price, reimbursement, and patent position. We believe that our ProLung Test is superior or equivalent to existing alternatives in all of these areas, other than availability (in the US due to lack of FDA approval) and reimbursement. We are in the process of seeking reimbursement approval in the European Union and expect to seek reimbursement approval in the US when we obtain marketing approval.
| 10 |
Intellectual Property
Protecting our intellectual property, exclusively licensed and owned, is essential to the creation of value in our business. We protect our intellectual property through confidentiality and trade secret agreements. We also have filed, and intend to continue to file, patent applications to protect key aspects of our technology.
Key Patents
Our patent protection is focused upon two key elements of the ProLung Test:
| 1. | The proprietary design of the ProLung Test probe and related computer algorithm used to prepare its report. | |
| 2. | The proprietary design of a group of algorithms or bioconductance profiles derived from our clinical research. |
We intend to actively pursue our patent opportunities in the US and abroad. We have 3 issued US patents and license 3 additional US patents. Product specific patents may be filed for all final products and issuance may correspond closely with regulatory agency approval to provide the longest proprietary protection. Existing patent applications of ours and BMC, from whom we have exclusive licenses, are set forth below:
| Title | Country | Type | Filed (6) | Application # | Patent # | |||||||
| Company Owned Patents | ||||||||||||
| Enhanced surface and tip for obtaining Bioelectrical signals | US | ORD (1) | 5/5/2014 | 14/269,248 | 9,526,432 | |||||||
| Method for diagnosing a disease | US | ORD (1) | 10/25/2007 | 11/978,045 | 7,603,171 | |||||||
| US | CON (2) | 10/13/2009 | 11/978,045 | 8,121,677 | ||||||||
| Licensed Patents | ||||||||||||
| Methods for obtaining quick, repeatable and non-invasive bioelectrical signals in living organisms | US | DIV (3) | 11/26/2007 | 11/944,696 | 7,536,220 | |||||||
| US | ORD (1) | 7/16/2003 | 10/621,178 | 7,542,796 | ||||||||
| Systems and methods of utilizing electrical readings in the determination of treatment | US | ORD (1) | 7/20/2004 | 10/895,149 | 7,937,139 | |||||||
| AU (5) | PCT(4) | 9/21/2004 | 2004322306 | |||||||||
| JP | PCT (5) | 1/15/2007 | JP2007-522475 | |||||||||
| Mexico | PCT (5) | 1/19/2007 | MX/a/2007/000798 | |||||||||
(1) Ordinary patent application - The first application for patent filed in the Patent Office without claiming priority from any application or without any reference to any other application under process in the Patent Office.
(2) Continuing patent application - A patent application which follows, and claims priority to, an earlier filed patent application.
(3) Divisional patent application - A patent application which has been divided from an existing application.
(4) International patent application - An international agreement for filing patent applications.
(5) Patent Cooperation Treaty Agreement under the laws of Australia.
(6) All patents expire 20 years from the date filed.
ProLung Patent Applications
| Country | Patent (Appln.) No. | Title | ||
| US | 13/970496 | Method for Diagnosing a Malignant Lung Tumor | ||
| EU | 13/789409.3 | Method for Diagnosing a Malignant Lung Tumor | ||
| Australia | 2013398354 | Method for Diagnosing a Malignant Lung Tumor | ||
| Canada | 2921690 | Method for Diagnosing a Malignant Lung Tumor | ||
| China | 201380079729.6 | Method for Diagnosing a Malignant Lung Tumor | ||
| EP | 2013789409 | Method for Diagnosing a Malignant Lung Tumor | ||
| India | 201617005691 | Method for Diagnosing a Malignant Lung Tumor | ||
| Japan | 2016-536073 | Method for Diagnosing a Malignant Lung Tumor | ||
| Korea | 10-2016-7006923 | Method for Diagnosing a Malignant Lung Tumor | ||
| Mexico | MX/a/2016/001948 | Method for Diagnosing a Malignant Lung Tumor | ||
| New Zealand | 716918 | Method for Diagnosing a Malignant Lung Tumor | ||
| US | 14/269,253 | Probe for Obtaining Bioelectric Signals |
| 11 |
Exclusive License Agreements
Effective November 2, 2006, we entered into an exclusive, worldwide, royalty-bearing License Agreement with BioMeridian Corporation (“BMC License”) to use certain patents. Under the agreement, we have the right to the exclusive use of certain patents, patents pending, and related technology in its medical devices and other products until such time that we are no longer utilizing any form, in whole or in part, of the licensed technology to develop, market or sell our products or generate revenues. In return, we agree to incur, and have incurred, a minimum of $4,750,000 in costs to develop and market our products worldwide and to make royalty payments based on a percentage of the aggregate worldwide net sales (as defined in the agreement) of our medical device and other products to the extent they utilize the licensed technology. Specifically, we have licensed from BMC certain design features of the ProLung Test including the probe and system, which are described in US patent numbers 7536220, 7542796, and 7937139. In addition, pursuant to the BMC License, we have licensed from BMC the rights to the technology that controls the functionality of the probe.
Governmental Regulations
Our business is subject to extensive federal, state, local and foreign laws and regulations, including those relating to the protection of the environment, health and safety. Some of the pertinent laws have not been definitively interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of subjective interpretations. In addition, these laws and their interpretations are subject to change, or new laws may be enacted.
Both federal and state governmental agencies continue to subject the healthcare industry to intense regulatory scrutiny, including heightened civil and criminal enforcement efforts. We believe that we have structured our business operations and relationships with our customers to comply with all applicable legal requirements. However, it is possible that governmental entities or other third parties could interpret these laws differently and assert otherwise. We discuss below the statutes and regulations most relevant to our business.
US Food and Drug Administration regulation of medical devices.
The Federal Food, Drug and Cosmetic Act (the “FDCA”) and FDA regulations establish a comprehensive system for the regulation of medical devices intended for human use. Our products include medical devices that are subject to these, as well as other federal, state, local and foreign, laws and regulations. The FDA is responsible for enforcing the laws and regulations governing medical devices in the United States.
The FDA classifies medical devices into one of three classes - Class I, Class II, or Class III depending on their level of risk and the types of controls that are necessary to ensure device safety and effectiveness. The class assignment is a factor in determining the type of premarketing submission or application, if any, that will be required before marketing in the United States. We currently anticipate that the ProLung System will be classified as a Class II medical device.
| ● | Class I devices present a low risk and are not life-sustaining or life-supporting. The majority of Class I devices are subject only to “general controls” -e.g., prohibition against adulteration and misbranding, registration and listing, good manufacturing practices, labeling, and adverse event reporting. General controls are baseline requirements that apply to all classes of medical devices. | |
| ● | Class II devices present a moderate risk and are devices for which general controls alone are not sufficient to provide a reasonable assurance of safety and effectiveness. Devices in Class II are subject to both general controls and “special controls” -e.g., special labeling, compliance with industry standards, and post market surveillance. Unless exempted, Class II devices typically require FDA clearance before marketing, through the premarket notification (510(k) process.) | |
| ● | De novo application process provides a pathway to Class II classification for medical devices for general and special controls provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. | |
| ● | Class III devices present the highest risk. These devices generally are life-sustaining, life-supporting, or for a use that is of substantial importance in preventing impairment of human health, or present a potential unreasonable risk of illness or injury. Class III devices are devices for which general controls, by themselves, are insufficient and for which there is insufficient information to establish special controls to provide a reasonable assurance of safety and effectiveness. Class III devices are subject to general controls and typically require FDA approval of a premarket approval (“PMA”) application before marketing. |
| 12 |
Unless it is exempt from premarket review requirements, a medical device must receive marketing authorization from the FDA prior to being commercially marketed, distributed or sold in the United States. The most common pathways for obtaining marketing authorization are 510(k) clearance and PMA.
510(k) pathway
The 510(k)-review process compares a new device to a legally marketed device. Through the 510(k) process, the FDA determines whether a new medical device is “substantially equivalent” to a legally marketed device (i.e., predicate device) that is not subject to PMA requirements. “Substantial equivalence” means that the proposed device has the same intended use as the predicate device, and the same or similar technological characteristics, or if there are differences in technological characteristics, the differences do not raise different questions of safety and effectiveness as compared to the predicate, and the information submitted in the 510(k) demonstrates that the proposed device is as safe and effective as the predicate device.
To obtain 510(k) clearance, a company must submit a 510(k)-application containing sufficient information and data to demonstrate that its proposed device is substantially equivalent to a legally marketed predicate device. These data generally include non-clinical performance testing (e.g., software validation, animal testing electrical safety testing), but may also include clinical data. Typically, it takes three to twelve months for the FDA to complete its review of a 510(k) submission; however, it can take significantly longer and clearance is never assured. During its review of a 510(k), the FDA may request additional information, including clinical data, which may significantly prolong the review process. After completing its review of a 510(k), the FDA may issue an order, in the form of a letter, that finds the device to be either (1) substantially equivalent and states that the device can be marketed in the United States, or (2) not substantially equivalent and states that device cannot be marketed in the United States. Depending upon the reasons for the not substantially equivalent finding, the device may need to be approved through the PMA pathway (discussed below) prior to commercialization.
After a device receives 510(k) clearance, any modification that could significantly affect the safety or effectiveness of the device, or that would constitute a major change in its intended use, including significant modifications to any of our products or procedures, requires submission and clearance of a new 510(k). The FDA relies on each manufacturer to make and document this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. We may make minor product enhancements that we believe do not require new 510(k) clearance. If the FDA disagrees with our determination regarding whether a new 510(k) clearance was required for these modifications, we may need to cease marketing and/or recall the modified device. The FDA may also subject us to other enforcement actions, including, but not limited to, issuing a warning letter or untitled letter to us, seizing our products, imposing civil penalties, or initiating criminal prosecution.
Premarket approval pathway
Unlike the comparative standard of the 510(k) pathway, the PMA approval process requires an independent demonstration of the safety and effectiveness of a device. PMA is the most stringent type of device marketing application required by the FDA. PMA approval is based on a determination by the FDA that the PMA contains sufficient valid scientific evidence to ensure that the device is safe and effective for its intended use(s). A PMA application generally includes extensive information about the device including the results of clinical testing conducted on the device and a detailed description of the manufacturing process.
After a PMA application is accepted for review, the FDA begins an in-depth review of the submitted information. FDA regulations provide 180 days to review the PMA and make a determination; however, the review time is normally longer (e.g., 1-3 years). During this review period, the FDA may request additional information or clarification of information already provided. Also, during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the data supporting the application and provide recommendations to the FDA as to whether the data provide a reasonable assurance that the device is safe and effective for its intended use. In addition, the FDA generally will conduct a preapproval inspection of the manufacturing facility to ensure compliance with QSR, which imposes comprehensive development, testing, control, documentation and other quality assurance requirements for the design and manufacturing of a medical device.
Based on its review, the FDA may (1) issue an order approving the PMA, (2) issue a letter stating the PMA is “approvable” (e.g., minor additional information is needed), (3) issue a letter stating the PMA is “not approvable,” or (4) issue an order denying PMA. A company may not market a device subject to PMA review until the FDA issues an order approving the PMA. As part of a PMA approval, the FDA may impose post-approval conditions intended to ensure the continued safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution, and requiring the collection of additional clinical data. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including withdrawal of the approval.
| 13 |
Most modifications to a PMA approved device, including changes to the design, labeling, or manufacturing process, require prior approval before being implemented. Prior approval is obtained through submission of a PMA supplement. The type of information required to support a PMA supplement and the FDA’s time for review of a PMA supplement vary depending on the nature of the modification.
Clinical trials
Clinical trials of medical devices in the United States are governed by the FDA’s Investigational Device Exemption (“IDE”) regulation. This regulation places significant responsibility on the sponsor of the clinical study including, but not limited to, choosing qualified investigators, monitoring the trial, submitting required reports, maintaining required records, and assuring investigators obtain informed consent, comply with the study protocol, control the disposition of the investigational device, submit required reports, etc.
Clinical trials of significant risk devices (e.g., implants, devices used in supporting or sustaining human life, devices of substantial importance in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health) require FDA and Institutional Review Board (“IRB”), approval prior to starting the trial. FDA approval is obtained through submission of an IDE application. Clinical trials of non-significant risk (“NSR”), devices (i.e. devices that do not meet the regulatory definition of a significant risk device) only require IRB approval before starting. The clinical trial sponsor is responsible for making the initial determination of whether a clinical study is significant risk or NSR; however, a reviewing IRB and/or FDA may review this decision and disagree with the determination.
An IDE application must be supported by appropriate data, such as performance data, animal and laboratory testing results, showing that it is safe to evaluate the device in humans and that the clinical study protocol is scientifically sound. There is no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to an unacceptable health risk.
As noted above, the FDA may require a company to collect clinical data on a device in the post market setting.
The collection of such data may be required as a condition of PMA approval. The FDA also has the authority to order, via a letter, a post market surveillance study for certain devices at any time after they have been cleared or approved.
Pervasive and continuing FDA regulation
After a device is placed on the market, regardless of its classification or premarket pathway, numerous additional FDA requirements generally apply. These include, but are not limited to:
| ● | Establishment registration and device listing requirements; | |
| ● | Quality System Regulation (“QSR”), which governs the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation, and servicing of finished devices; | |
| ● | Labeling requirements, which mandate the inclusion of certain content in device labels and labeling, and generally require the label and package of medical devices to include a unique device identifier (“UDI”), and which also prohibit the promotion of products for uncleared or unapproved, i.e., “off-label,” uses; | |
| ● | Medical Device Reporting (“MDR”), regulation, which requires that manufacturers and importers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and | |
| ● | Reports of Corrections and Removals regulation, which requires that manufacturers and importers report to the FDA recalls (i.e., corrections or removals) if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; manufacturers and importers must keep records of recalls that they determine to be not reportable. |
The FDA enforces these requirements by inspection and market surveillance. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include, but is not limited to, the following sanctions:
| ● | Untitled letters or warning letters; | |
| ● | Fines, injunctions and civil penalties; | |
| ● | Recall or seizure of our products; | |
| ● | Operating restrictions, partial suspension or total shutdown of production; | |
| ● | Refusing our request for 510(k) clearance or premarket approval of new products; | |
| ● | Withdrawing 510(k) clearance or premarket approvals that are already granted; and | |
| ● | Criminal prosecution. |
| 14 |
We are subject to unannounced device inspections by the FDA, as well as other regulatory agencies overseeing the implementation of and compliance with applicable state public health regulations. These inspections may include our suppliers’ facilities.
Marketing Approvals Outside the United States
Sales of medical devices outside the United States are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ.
Europe
Under the European Union Medical Device Directive, or EU MDD, medical devices must meet the EU MDD requirements and receive a CE marking certification prior to marketing in the European Union, or EU, which we received for the ProLung Test in May 2013. CE marking is the uniform labeling system of products designed to facilitate the supervision and control of the EU concerning manufacturers’ compliance with the various regulations and directives of the EU and to clarify the obligations imposed in the various legislative provisions in the EU. Use of a uniform product labeling indicates compliance with all the directives and regulations required for the application of such labeling, and it is effective as a manufacturer’s declaration that the product meets the required criteria and technical specifications of the relevant authorities such as health, safety, and environmental protection. CE marking ensures free trade between the EU and European Free Trade Association countries (Switzerland, Iceland, Liechtenstein, and Norway) and permits the enforcement and customs authorities in European countries not to allow the marketing of similar products that do not bear the CE marking sign. Such certification allows, among other things, marking the products (according to various categories) with the CE marking and their sale and marketing in the EU.
CE marking certification requires a comprehensive quality system program, comprehensive technical documentation and data on the product, which are then reviewed by a Notified Body, or NB. An NB is an organization designated by the national governments of the EU member states to make independent judgments about whether a product complies with the EU MDD requirements and to grant the CE marking if we, and our product, comply with specified terms. After receiving the CE marking, we must pass a review carried out by the competent NB annually, under which it audits our facilities to verify our compliance with the ISO 13485 quality system standard.
Compliance with the ISO 13485 standard, for medical device quality management systems, is required for regulatory purposes. ISO standards are recognized international quality standards that are designed to ensure that we develop and manufacture quality medical devices. Other countries are also instituting regulations regarding medical devices. Compliance with these regulations requires extensive documentation and clinical reports for all our product candidates, revisions to labeling, and other requirements such as facility inspections to comply with the registration requirements.
China
China’s medical device market, currently in a rapid state of expansion, is overseen by the China Food and Drug Administration, or CFDA (formerly the State Food and Drug Administration). The CFDA issues registration certificates required for all medical devices sold in China. The CFDA uses a risk-based system, and its approval process requires mandatory testing for Class II and III devices. Class II devices are moderate-risk devices and Class III devices are high-risk medical devices. Third-party review of devices is currently not allowed in China; only the CFDA is authorized to approve devices. The registration process requires the submission of a registration standard along with device samples for testing. Manufacturers of Class II and Class III medical devices are also required to demonstrate that the device has been approved by the country of origin with documents like a CE certificate, 510(k) letter and PMA approval and compliance with ISO 13485, and they may also be required to submit clinical data in support of their application. In addition to these requirements, all medical device manufacturers must also include product information in Chinese on all packaging and labeling. Manufacturers exporting medical devices to China must appoint several China-based agents to act on their behalf. These include a registration agent to coordinate the CFDA registration process, a legal agent to handle any adverse events reported with a registered device, including a product recall, and an after-sales agent to provide technical service and maintenance support.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the CMS, other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments. These regulations include:
| 15 |
| ● | the federal healthcare program anti-kickback law which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; | |
| ● | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government reimbursement programs that are false or fraudulent. The government may assert that a claim including items or services resulting from a violation of the federal healthcare program anti-kickback law or related to off-label promotion constitutes a false or fraudulent claim for purposes of the federal false claims laws; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996 which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; | |
| ● | the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; | |
| ● | the Federal Physician Payments Sunshine Act within the Patient Protection and Affordable Care Act, and its implementing regulations, require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members; and | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The Health Insurance Portability and Accountability Act, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Post-Marketing Regulations
Following approval of a new product, a company and the approved product are subject to continuing regulation by the FDA and other federal and state regulatory authorities, including, among other things, monitoring and recordkeeping activities, reporting to applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting for uses or in patient populations not described in the product’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available products for off-label uses, manufacturers may not market or promote such off label uses. Modifications or enhancements to the products or labeling or changes of site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Centers for Medicare & Medicaid Services (“CMS”), other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency, and state and local governments. Sales, marketing and scientific/educational programs must also comply with federal and state fraud and abuse laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the US Omnibus Budget Reconciliation Act of 1990. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair completion laws.
| 16 |
The distribution of medical device products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of medical device products.
Our Marketing Approvals
We must receive separate regulatory approvals from the FDA and equivalent regulatory bodies in other countries for each of the devices before we can sell them commercially in the US or internationally. We cannot make the claims necessary to market any of our product candidates until we have completed the requirements for regulatory approval. We do not know whether regulatory authorities will grant approval for any of the products that we, our marketing partners, or distribution partners will develop.
A summary of the status of our marketing approvals in the key initial markets we have identified is set forth below:
| ● | United States. In early 2015, we applied for marketing approval under Section 510(k) from the FDA. In February 2015, we received a Substantive Review from the FDA requesting clarification of research to date, updated and additional safety testing, clarification of the Indications For Use (IFU) statement, and results of the ongoing multisite trial. We communicated with the FDA by conference call, in writing, and in a July 16, 2015, face-to-face Submission Issue Meeting. We reached concurrence with the FDA on a revised IFU statement. We also clarified and updated the requested safety testing. Statutory requirements for an active FDA application mandated ProLung’s withdraw their application while awaiting results of the multisite trial . ProLung has completed the enrollment of the initial 350 subjects across the U.S. for this study. As is common in a study of this type and size, ProLung is now enrolling 70 replacement subjects as provided for in the study protocol. Before the FDA can grant approval of our 510(k) de novo application, we must resubmit the application with the results of the multisite trial and resolve or negotiate any new issues identified by the FDA. While ProLung is optimistic about the resolution of these issues, based on the face-to-face July 16, 2015 meeting with the FDA, submission of a new 510(k) de novo application and possible changes in the FDA review team make it impossible to predict approval with certainty. | |
| ● | European Union. CE marking was granted as of May 10, 2013 for the ProLung Test which permits the product to be sold throughout the European Economic Area (European Union member states plus Iceland, Liechtenstein and Norway), Switzerland, and Turkey. CE marking requires manufacturers to maintain an ISO 13485 Quality System. | |
| ● | Latin America. ProLung has planned sponsorship and speaking opportunities at pulmonary and lung cancer specific symposia in Latin America and has developed relationships with key regional opinion leaders in lung cancer management. ProLung is in discussion with distributors in the major Latin American markets for distribution and commercialization deals. Based on primary physician feedback and response, ProLung expects a viable and strong market for a predictive analytic device such as the ProLung Test. | |
| ● | China. State Food and Drug Administration (“SFDA”) roughly follows the FDA model and approval from the SFDA permits the marketing and sale of the device in China. To be sold in China, medical devices must be registered with Chinese health authorities. In February 2014, the Company’s licensor in China received approval to manufacture the device from the Beijing government. Additional approvals are required to market and sell the device in this market. |
After each respective regulatory approval is obtained, the next step in each of these markets is for insurance companies or government agencies, as applicable, to agree to reimburse for the ProLung Test. We have not commenced this process in the US or China, as we do not have marketing approval.
Manufacturing Requirements
As a manufacturer of medical devices, we must comply with the 21 CFR Part 820 Good Manufacturing Practice regulations established by the FDA. These requirements are meant to ensure that medical devices are safe and effective. We maintain a quality management system that includes standard operating procedures for key processes such as manufacturing, record keeping, post market surveillance, complaint handling and corrective and preventative action. Our quality management system is currently ISO 1348542 certified and complies with the 21 CFR Part 820 Good Manufacturing Practice regulations. We will also be subject to similar requirements imposed by other countries.
| 17 |
Manufacturing
We currently manufacture the ProLung Test and the ProLung Test Kit. When volume requirements exceed current manufacturing capacity, we intend to utilize contract manufacturers for the physical manufacturing of our products. This may afford us numerous benefits, including:
| ● | the ability to ramp up production quickly; | |
| ● | access to leading technologies, supply chain networks and best-in-class manufacturing processes for its products; | |
| ● | flexibility to use one or many manufacturers in many regions of the world to optimize costs, production volumes, material availability, lead times, and to meet various regional regulations. |
We have interviewed, performed site visits, and qualified multiple, redundant contract manufacturers which may be required to produce our products. As of June 30, 2017, we have no contractual obligations with such contract manufacturers for the manufacturing of our products.
Our prospective contract manufacturers will source our product components from multiple specialized vendors that supply plastics, sheet metal, machining, cables, wire harnesses, and other computer hardware components. We maintain our own design control and ISO 13485 quality system.
Research and Development
We spent $1,219,189 and $1,250,723 on company-sponsored research and development during fiscal years ending December 31, 2016 and 2015, respectively.
Employees
As of September 14, 2017, we had 15 employees.
Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”), and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
In addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are choosing to take advantage of the extended transition period for complying with new or revised accounting standards. As a result, our financial statements may not be comparable to those of companies that comply with public company effective dates.
We believe we will remain an “emerging growth company” through at least December 31, 2017.
Because of the exemptions from various reporting requirements provided to us as an “emerging growth company” and because we will have an extended transition period for complying with new or revised financial accounting standards, we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
| 18 |
Item 8. Financial Statements and Supplementary Data
Financial Statements
Reference is made to the consolidated financial statements and accompanying notes included in this report, which begin on page F-1.
Supplemental Financial Data
This item is not applicable to the Company because the Company is a smaller reporting company.
Item 15. Exhibits, Financial Statement Schedules
1. Financial Statements. The following Consolidated Financial Statements of the company and Auditors’ reports are filed as part of this Annual Report on Form 10-K:
| ● | Reports of Independent Registered Public Accounting Firms | |
| ● | Consolidated Balance Sheets as of December 31, 2016 and 2015 | |
| ● | Consolidated Statements of Operations for the years ended December 31, 2016 and 2015 | |
| ● | Consolidated Statements of Stockholders’ Deficit for the years ended December 31, 2016 and 2015 | |
| ● | Consolidated Statements of Cash Flows for the years ended December 31, 2016 and 2015 | |
| ● | Notes to the Consolidated Financial Statements |
2. Financial Statements Schedule. Not applicable.
3. Exhibits. The information required by this item is set forth on the exhibit index that follows the signature page of this report.
| 19 |
PROLUNG, INC. AND SUBSIDIARY
(FORMERLY FRESH MEDICAL LABORATORIES, INC.)
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
ProLung, Inc.
Salt Lake City, Utah
We have audited the accompanying consolidated balance sheets of ProLung, Inc. (formerly Fresh Medical Laboratories, Inc.) and its subsidiary (collectively, the “Company”) as of December 31, 2016 and 2015, and the related consolidated statements of operations, stockholders’ deficit, and cash flows for the years then ended. These consolidated financial statements are the responsibility of the entity’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform an audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of ProLung, Inc. and its subsidiary as of December 31, 2016 and 2015, and the consolidated results of their operations and their cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
/s/ MaloneBailey, LLP
www.malonebailey.com
Houston, Texas
April 17, 2017
| F-2 |
ProLung, Inc. and Subsidiary
(formerly Fresh Medical Laboratories, Inc.)
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash | $ | 28,922 | $ | 451,526 | ||||
| Accounts receivable, net of allowance for doubtful accounts of $0 and $194,467, respectively | - | - | ||||||
| Inventory | - | 35,174 | ||||||
| Prepaid expenses | 8,831 | 30,520 | ||||||
| Total Current Assets | 37,753 | 517,220 | ||||||
| Inventory, noncurrent | 291,559 | 206,722 | ||||||
| Property and equipment, net of accumulated depreciation | 82,917 | 106,541 | ||||||
| Intangible assets, net of accumulated amortization | 165,738 | 175,300 | ||||||
| Total Assets | $ | 577,967 | $ | 1,005,783 | ||||
| Liabilities and Stockholders’ Deficit | ||||||||
| Current Liabilities | ||||||||
| Accounts payable | $ | 358,477 | $ | 97,849 | ||||
| Accrued liabilities | 264,698 | 138,683 | ||||||
| Related-party notes payable | 105,000 | 25,000 | ||||||
| Current portion of long-term debt | 32,000 | 189,389 | ||||||
| Total Current Liabilities | 760,175 | 450,921 | ||||||
| Long-Term Liabilities | ||||||||
| Long-term debt, net of current portion | 2,653,370 | 3,206,931 | ||||||
| Total Long-Term Liabilities | 2,653,370 | 3,206,931 | ||||||
| Total Liabilities | 3,413,545 | 3,657,852 | ||||||
| Stockholders’ Deficit: | ||||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized; none issued and outstanding | - | - | ||||||
| Common stock, $0.001 par value; 40,000,000 shares authorized; 24,006,515 shares and 21,525,126 shares issued and outstanding, respectively | 24,007 | 21,525 | ||||||
| Additional paid-in capital | 13,226,048 | 10,636,583 | ||||||
| Accumulated deficit | (16,085,633 | ) | (13,310,177 | ) | ||||
| Total Stockholders’ Deficit | (2,835,578 | ) | (2,652,069 | ) | ||||
| Total Liabilities and Stockholder’ Deficit | $ | 577,967 | $ | 1,005,783 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
ProLung, Inc. and Subsidiary
(formerly Fresh Medical Laboratories, Inc.)
Consolidated Statements of Operations
| For the Years Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Revenues: | ||||||||
| Revenue | $ | 8,800 | $ | 19,450 | ||||
| Total revenue | 8,800 | 19,450 | ||||||
| Cost of revenue | 10,193 | 15,563 | ||||||
| Gross margin | (1,393 | ) | 3,887 | |||||
| Operating expenses: | ||||||||
| Research and development expense | 1,219,189 | 1,250,723 | ||||||
| Selling, general and administrative expense | 1,288,960 | 1,257,557 | ||||||
| Total operating expenses | 2,508,149 | 2,508,280 | ||||||
| Loss from operations | (2,509,542 | ) | (2,504,393 | ) | ||||
| Other expense: | ||||||||
| Interest expense | (265,914 | ) | (271,984 | ) | ||||
| Foreign currency exchange loss, net | - | (24,093 | ) | |||||
| Total other expense | (265,914 | ) | (296,077 | ) | ||||
| Net loss | $ | (2,775,456 | ) | $ | (2,800,470 | ) | ||
| Basic and diluted loss per share | $ | (0.12 | ) | $ | (0.14 | ) | ||
| Weighted-average common shares outstanding, basic and diluted | 22,739,569 | 20,344,262 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
ProLung, Inc. and Subsidiary
(formerly Fresh Medical Laboratories, Inc.)
Consolidated Statements of Stockholders’ Deficit
For the years ended December 31, 2015 and 2016
| Common Stock | Additional Paid-in | Accumulated | Total Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balance, December 31, 2014 | 19,730,052 | $ | 19,730 | $ | 9,075,590 | $ | (10,509,707 | ) | $ | (1,414,387 | ) | |||||||||
| Stock-based compensation | - | - | 255,915 | - | 255,915 | |||||||||||||||
| Common stock issued for cash | 294,000 | 294 | 146,706 | - | 147,000 | |||||||||||||||
| Common stock issued for cash | 1,235,278 | 1,235 | 925,225 | - | 926,460 | |||||||||||||||
| Common stock issued pursuant to bill of sale and patent assignment agreements | 150,000 | 150 | 112,350 | - | 112,500 | |||||||||||||||
| Common stock issued for conversion of note and accrued interest | 95,283 | 95 | 61,839 | - | 61,934 | |||||||||||||||
| Issuance of warrants under consulting agreement | - | - | 43,594 | - | 43,594 | |||||||||||||||
| Common stock issued for services | 20,513 | 21 | 15,364 | - | 15,385 | |||||||||||||||
| Net loss | - | - | - | (2,800,470 | ) | (2,800,470 | ) | |||||||||||||
| Balance, December 31, 2015 | 21,525,126 | 21,525 | 10,636,583 | (13,310,177 | ) | (2,652,069 | ) | |||||||||||||
| Stock-based compensation | - | - | 262,474 | - | 262,474 | |||||||||||||||
| Common stock issued for cash and warrants, net of offering costs | 1,106,952 | 1,107 | 1,497,624 | - | 1,498,731 | |||||||||||||||
| Common stock issued upon conversion of debt and accrued interest | 1,251,504 | 1,252 | 812,225 | - | 813,477 | |||||||||||||||
| Common stock issued to placement agent | 103,166 | 103 | (103 | ) | - | - | ||||||||||||||
| Common stock issued for service | 19,767 | 20 | 17,245 | - | 17,265 | |||||||||||||||
| Net loss | - | - | - | (2,775,456 | ) | (2,775,456 | ) | |||||||||||||
| Balance, December 31, 2016 | 24,006,515 | $ | 24,007 | $ | 13,226,048 | $ | (16,085,633 | ) | $ | (2,835,578 | ) | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
ProLung, Inc. and Subsidiary
(formerly Fresh Medical Laboratories, Inc.)
Consolidated Statements of Cash Flows
| For the Years Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (2,775,456 | ) | $ | (2,800,470 | ) | ||
| Adjustments to reconcile net loss to net cash flows from operating activities: | ||||||||
| Depreciation and amortization | 33,186 | 10,923 | ||||||
| Stock-based compensation | 279,739 | 343,488 | ||||||
| Obsolete inventory | 10,193 | - | ||||||
| Impairment loss | - | 50,000 | ||||||
| Provision for doubtful accounts | - | 102,282 | ||||||
| Change in assets and liabilities: | ||||||||
| Accounts receivable | - | 52,517 | ||||||
| Inventory | (59,856 | ) | (31,422 | ) | ||||
| Prepaid expenses | 21,689 | (20,474 | ) | |||||
| Accounts payable | 260,628 | (7,467 | ) | |||||
| Accrued liabilities | 196,542 | (255,719 | ) | |||||
| Net cash flows from operating activities | (2,033,335 | ) | (2,556,342 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Payments for property and equipment | - | (164,489 | ) | |||||
| Net cash flows from investing activities | - | (164,489 | ) | |||||
| Cash flows from financing activities: | ||||||||
| Issuance of common stock and warrants for cash, net of offering costs | 1,498,731 | 1,073,460 | ||||||
| Proceeds from issuance of convertible debentures | - | 2,000,000 | ||||||
| Proceeds from issuance of convertible notes payable | - | 1,206,931 | ||||||
| Payments on convertible notes payable | - | (40,000 | ) | |||||
| Payments on notes payable | - | (1,097,078 | ) | |||||
| Proceeds from notes payable | 32,000 | - | ||||||
| Proceeds from related party debt | 210,000 | 50,000 | ||||||
| Payments on related party debt | (130,000 | ) | (25,000 | ) | ||||
| Net cash flows from financing activities | 1,610,731 | 3,168,313 | ||||||
| Net increase (decrease) in cash | (422,604 | ) | 447,482 | |||||
| Cash at beginning of period | 451,526 | 4,044 | ||||||
| Cash at end of period | $ | 28,922 | $ | 451,526 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for interest | $ | 76,170 | $ | 524,544 | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Conversion of convertible debt and interest | $ | 813,477 | $ | 61,934 | ||||
| Stock issued to placement agent | $ | 103 | $ | - | ||||
| Common stock issued to acquire property and equipment, and intangible assets | $ | - | $ | 112,500 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Note 1 – Organization and Summary of Significant Accounting Policies
Organization – ProLung, Inc. (formerly Fresh Medical Laboratories, Inc.) (the “Company”) is a Delaware corporation that was incorporated on November 22, 2004 and is doing business as “ProLungdx.” The Company’s headquarters are located in Salt Lake City, Utah. The Company’s business is the marketing and sales of precision predictive analytical medical devices specializing in the lung cancer. The Company’s principal activities are primarily developing markets for its products, securing strategic alliances and obtaining financing.
Principles of Consolidation – During the year ended December 31, 2012, the Company formed a wholly-owned subsidiary, Hilltop Acquisition Corporation, Inc., which has had no activity since its inception and is included in the accompanying financial statements from the date of its formation.
Basis of Presentation – The Company has incurred losses for the past several years while pursuing the development of its primary predictive analytical medical device, and approval from the U.S. Food and Drug Administration (FDA) to market the device, while also developing markets outside the United States. The Company incurred net losses of $2.8 million in 2016 and 2015. Cash used in operating activities was $2.0 million and $2.6 million in 2016 and 2015, respectively. Historically, operations have been funded primarily through the sale of equity or debt securities. Should management continue to fund operations at similar levels, additional equity or debt securities would need to be sold, or other financing arrangements made.
The Company has the ability to maintain current levels of spending or reduce expenditures significantly if funding is not available. Additionally, should FDA approval be obtained, the Company could execute on an aggressive marketing plan that would require significant additional funding; however, this plan would not begin until funding is in place.
As discussed in Note 13, subsequent to December 31, 2016, the Company sold 2,556,634 units from its on-going Private Placement Memorandum for approximately $3.4 million. Additionally, the Company converted outstanding debt of approximately $1.3 million to equity, and paid debt and accrued interest of approximately $0.5 million. Therefore, approximately $1.8 million of liabilities on the December 31, 2016 balance sheet were converted to equity or repaid subsequent to December 31, 2016.
The Company’s financial statements for the prior year ended December 31, 2015 disclosed substantial doubt about the Company’s ability to continue as a going concern. Based on management’s plans and the significant capital raised during subsequent to the year ended December 31, 2016, that substantial doubt has been alleviated.
Use of Estimates – The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect certain reported amounts and disclosures. Accordingly, actual results could differ from those estimates.
Fair Value of Financial Instruments – Certain notes payable bear interest rates that are not market interest rates given the risks associated with a company in the early stage of its development. However, for notes payable which are classified among current liabilities due to their relatively short terms remaining to the notes’ maturity dates as of December 31, 2016, the carrying value of those notes payable approximates their fair value. For the notes payable and convertible debentures classified as long-term liabilities, the estimated fair value is approximately equal to the carrying value based on the interest rates and other terms of debt.
Research and Development – The Company expenses research and development costs as incurred. Research and development costs primarily consist of clinical study costs, consulting fees, compensation of employees related to activities to obtain regulatory approval for the Company’s devices, and materials and supplies.
Cash and Cash Equivalents – The Company considers all unrestricted highly liquid investments purchased with a maturity of three months or less to be cash equivalents. The Company had no cash equivalents as of December 31, 2016 or 2015.
Inventory – Inventory is valued at the lower of cost or market value, with cost determined based on the first-in-first-out method. The estimated cost of inventory not expected to be converted to cash within one year is reflected as “Inventory, noncurrent” in the consolidated balance sheets although all inventory is ready and available for sale at any moment. During 2016 and 2015, the Company critically reviewed all inventory for impairment.
Property and Equipment – Property and Equipment is stated at cost and depreciated using the straight-line method over useful lives of 3 to 5 years.
| F-7 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Intangible Assets – As further discussed in Note 9 to these consolidated financial statements, intangible assets consist of rights to certain patent applications acquired in December 2015 under a Patent Assignment Agreement. These intangible assets will be amortized over an estimated useful life of eighteen years, with periodic evaluation for impairment.
Revenue Recognition – The Company commenced selling the EPN Scan during the year ended December 31, 2014. The Company recognizes revenue from the sale of the EPN Scan when it is realized or realizable and earned. The Company considers revenue realized or realizable and earned when (1) it has persuasive evidence of an arrangement, (2) delivery has occurred, (3) the sales price is fixed or determinable, and (4) collectability is reasonably assured. The Company recognizes revenue from licensing arrangements on a straight-line basis over the contractual term of the arrangement or the expected period during which the specified services will be performed, whichever is longer. However, for licensing arrangements where there are no future service obligations, the licensing income is recognized upon receipt of the consideration under the arrangement.
Trade Receivables and Credit Policies – Accounts receivable are recorded at the invoiced amount, with foreign currencies reflected in U.S. dollars (based on the exchange rate on the date of sale and adjusted to current exchange rates at the end of each reporting period), and do not bear interest. The Company uses an allowance for doubtful accounts to reflect the Company’s best estimate of the amount of probable credit losses in accounts receivable. Account balances will be charged off against the allowance when the account receivable is considered uncollectible. The allowance for doubtful accounts is an estimate that is particularly susceptible to change in the near term. During the years ended December 31, 2016 and 2015, the Company recorded a provision for doubtful accounts in the amount of $0 and $102,282, respectively, for accounts receivable that had not been collected and were overdue at that date. At December 31, 2016 and 2015, the allowance for doubtful accounts is $0 and $194,467, respectively.
Employee Stock-based Compensation – The Company accounts for employee stock-based compensation in accordance with ASC 718, “Compensation-Stock Compensation.” ASC 718 requires companies to measure the cost of employee services received in exchange for an award of equity instruments based on the grant-date fair value of the award and to recognize it as compensation expense over the period the employee is required to provide service in exchange for the award, usually the vesting period.
Non-Employee Stock-based Compensation – The Company accounts for non-employee stock-based compensation in accordance with the provision of ASC 505, “Equity Based Payments to Non-Employees,” which requires that such equity instruments are recorded at their fair value on the measurement date. The measurement of stock-based compensation is subject to periodic adjustment as the underlying equity instruments vest.
Income Taxes – The Company accounts for income taxes under the asset and liability method. Deferred income tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, and for operating loss and tax credit carry-forwards. Deferred income tax assets and liabilities are measured using the enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred income tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. The Company has established a valuation allowance to reduce deferred income tax assets to their realizable values based on whether it is more likely than not that such deferred income tax assets will be realized. At December 31, 2016 and 2015, the Company has recorded a full valuation allowance against the net deferred tax assets related to temporary differences and operating losses because there is significant uncertainty as to the realizability of the deferred tax assets. The Company recognizes the tax benefit from an uncertain tax position only if it is more likely than not the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such positions are then measured based on the largest benefit that has a greater than 50% likelihood of being realized upon settlement.
| F-8 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Basic and Diluted Loss Per Share – The Company computes basic loss per share by dividing net loss by the weighted-average number of common shares outstanding during the period. The Company computes diluted loss per share by dividing net loss by the sum of the weighted-average number of common shares outstanding and the weighted-average dilutive common share equivalents outstanding. The computation of diluted loss per share does not assume exercise or conversion of securities that would have an anti-dilutive effect. As of December 31, 2016 and 2015, the following items were excluded from the computation of diluted net loss per common share as their effect is anti-dilutive:
| For the Years Ended | ||||||||
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Warrants to purchase shares | 3,447,386 | 1,423,211 | ||||||
| Restricted common stock grants | 872 | 253,670 | ||||||
| Convertible debentures | 2,198,850 | 3,253,279 | ||||||
| Convertible notes | 1,641,692 | 1,609,242 | ||||||
Foreign Currency Policy – Transactions in foreign currencies are initially recorded at the rates of exchange prevailing on the dates of the transactions. Monetary assets and liabilities denominated in foreign currencies are retranslated into the Company’s functional currency at the rates prevailing on the balance sheet date. Exchange differences arising on the settlement of monetary items, and on the retranslation of monetary items, are reported as other income (expense) and included in Net loss for the period. The Company recorded a foreign currency exchange loss of $24,093 for the year ended December 31, 2015.
Related Parties – The Company discloses related party transactions in accordance with ASC 850, “Related Party Disclosures.” All transactions with related parties are in the normal course of operations and are measured at the exchange amount.
Recent Accounting Pronouncements – In May 2014, the FASB issued Accounting Standards Update (ASU) No. 2014-09, Revenue from Contracts with Customers (Topic 606), which was amended with ASU No. 2015-14, ASU No. 2016-08, ASU No. 2016-10, ASU No. 2016-12 and ASU No. 2016-20. These new standards supersede all existing revenue recognition requirements, including most industry specific guidance. The new standard requires a company to recognize revenue when it transfers goods or services to customers in an amount that reflects the consideration that the company expects to receive for those goods or services. The Company is evaluating the guidance but does not at this time expect to have a material impact on its revenue recognition; however, the Company does expect to have significant changes to the footnote disclosures related to revenue recognition as a result of implementing these new standards. As the Company has elected to be treated as an emerging growth company, this standard will be implemented effective January 1, 2019.
In March 2016, the FASB issued ASU 2016-09, Stock Compensation (“ASU 2016-09”), which simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. ASU 2016-09 will be effective for annual periods beginning after December 15, 2016 and interim periods within those annual periods. It is not anticipated that this update will have a material effect on the Company’s consolidated financial statements.
Note 2 – Inventory
Inventory principally consists of the cost of materials purchased and assembled during the years ended December 31, 2016 and 2015. The cost of inventory also includes the costs of direct labor for the assembly and certain indirect costs incurred in connection with purchasing of parts and the assembly of products. Inventory consists of the following:
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Raw materials | $ | 69,264 | $ | 76,925 | ||||
| Work in progress | 31,185 | 58,376 | ||||||
| Finished goods | 191,110 | 106,595 | ||||||
| Total inventory | 291,559 | 241,896 | ||||||
| Less carrying value of inventory not deemed to be a current asset | 291,559 | 206,722 | ||||||
| Inventory, included in current assets | $ | - | $ | 35,174 | ||||
| F-9 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
In an effort to create a unified marketing image, inventory recorded at $10,193, which consisted of older packaging materials was written off during the year ended December 31, 2016 and is reported as cost of revenues in the accompanying statement of operations.
Note 3 – Property and Equipment
Property and equipment consists of the following at December 31, 2016 and 2015:
| December 31, | ||||||||||
| Life | 2016 | 2015 | ||||||||
| Computer equipment | 3 years | $ | 19,787 | $ | 19,787 | |||||
| Office equipment | 3 to 5 years | 13,852 | 13,852 | |||||||
| Tooling | 5 years | 92,228 | 92,228 | |||||||
| 125,867 | 125,867 | |||||||||
| Less accumulated depreciation | (42,950 | ) | (19,326 | ) | ||||||
| Property and equipment, net | $ | 82,917 | $ | 106,541 | ||||||
Depreciation expense for the years ended December 31, 2016 and 2015 was $23,624 and $10,923, respectively.
Effective January 2014, the Company entered into a Master Services Agreement (the “Agreement”) with an entity that provides consulting and professional services to develop an internet-based customer service portal. The entity is owned and managed by a former director of the Company. By December 31, 2015, the Company had paid a total of $50,000 under the Agreement in full satisfaction of amounts owed for services provided under the Agreement. With this payment, the Agreement was terminated. With the termination of the Agreement, management evaluated the status of this project in light of its plan for the future development and completion of the project and concluded that the $50,000 of costs paid and recorded will not have a significant future benefit. Accordingly, an impairment loss of $50,000 was recorded at December 31, 2015.
Note 4 – Accrued Liabilities
Accrued liabilities consisted of the following at December 31, 2016 and 2015:
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Accrued interest | $ | 234,405 | $ | 115,627 | ||||
| Accrued royalties | 17,873 | 5,183 | ||||||
| Accrued payroll and payroll taxes | 12,420 | 17,873 | ||||||
| Total accrued liabilities | $ | 264,698 | $ | 138,683 | ||||
Related party accrued interest was 35,519 and 1,012 at December 31, 2016 and 2015 respectively.
| F-10 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Note 5 –Short and Long-term Debt
Short and Long-term debt is summarized as follows:
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Convertible debentures; unsecured; interest at 8.00% per annum; due May 1, 2018; $742,950 was converted to common stock during the year ended December 31, 2016 | $ | 1,257,050 | $ | 2,000,000 | ||||
| Convertible notes payable; unsecured; interest at 8.00% per annum; due November 6, 2020 | 1,206,931 | 1,206,931 | ||||||
| Note payable secured by all the assets of the Company; interest at 15.00% per annum; due June 30, 2018 | 189,389 | 189,389 | ||||||
| Unsecured Note payable; interest at 10.00% per annum; due on demand | 32,000 | - | ||||||
| Total long-term debt | 2,685,370 | 3,396,320 | ||||||
| Less: current portion | 32,000 | 189,389 | ||||||
| Long-term debt, net of current portion | $ | 2,653,370 | $ | 3,206,931 | ||||
During the year ended December 31, 2016, notes totaling $32,000 became due. These notes are now considered due on demand and are recorded as current notes payable.
Maturities on long-term debt are as follows:
| Year ending December 31, | ||||
| 2017 | $ | 32,000 | ||
| 2018 | 1,446,439 | |||
| 2019 | - | |||
| 2020 | 1,206,931 | |||
| 2021 | - | |||
Note Payable Secured by the Assets of the Company
During the year ended December 31, 2015, the Company paid off the remaining principal of a master note to a shareholder of $929,536 and accrued interest of $310,770. Total interest expense related to this note for the year ended December 31, 2015 was $87,028.
Other Convertible Notes
During the year ended December 31, 2015, one note payable in the amount of $40,000 and related accrued interest of $9,837 were paid off for cash. During the year ended December 31, 2015, a note payable in the amount of $50,000 and related accrued interest of $11,934 was converted into 95,283 shares of the Company’s common stock, at $0.65 per share.
| F-11 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Note Payable to a Relative of an Executive Officer
At December 31, 2016 and 2015, the Company was obligated under the terms of a master note to an individual related to an executive officer of the Company in the amount of $189,389. During the year ended December 31, 2015, the Company paid $356,931 to the note holder, which paid all accrued interest in the amount of $189,389 as of the date of the payment and the remainder of the payment was applied to reduce the principal of the note by $167,542, leaving a balance of $189,389. The note is secured by all the assets of the Company, bears interest at 15 percent per annum, and requires the board of directors to retain the current management as long as the note is outstanding. The note was extended on June 30, 2016 and is now due September 30, 2018. The balance of accrued interest at December 31, 2016 and 2015 was $29,498 and $1,012, respectively. As part of the extension of the due date, the Company analyzed the note and determined that the change in due date did not qualify as a debt modification under generally accepted accounting principles and accordingly, classified the note as long-term. As described in Note 13, the remainder of the principal and interest was either converted or repaid subsequent to year end.
Convertible Debentures
In 2015, the Company issued $2,000,000 in Convertible Debentures. The Convertible Debentures are unsecured and bear interest at the rate of 8% per annum. Principal and accrued interest are due on the maturity date, which is May 1, 2018. The holder of the Convertible Debenture is entitled, at its option, to convert all or any portion of the outstanding principal of the Convertible Debenture into shares of the Company’s common stock at a conversion price of $0.65 per share. Interest accruing from the date of issuance to the conversion date shall be paid on the maturity date. The Company evaluated the Convertible Debentures for consideration of any beneficial conversion features as required under generally accepted accounting principles. The Company determined that there was no beneficial conversion feature.
As further described in Note 6 to these consolidated financial statements, the Company entered into a Placement Agent Agreement, effective December 28, 2015, that provides for compensation to a Placement Agent in connection with an offering of common stock. Additionally, the Placement Agent Agreement provides for potential compensation to the Placement Agent in connection with the future conversion of the Convertible Debentures into shares of common stock of the Company. Upon the conversion of the Convertible Debentures, the Company shall issue the Placement Agent warrants to acquire shares of the Company’s common stock at an exercise price of $0.65 per share. On a quarterly basis, the Placement Agent will be issued a warrant to purchase one share of common stock for each $0.81 of the principal amount of the Convertible Debentures converted into common stock during the quarter, with the maximum number of warrants issuable under the Placement Agreement limited to 2,463,460 shares of the Company’s common stock. The term of the warrants shall be for a period of 36 months from the date of issuance.
As of December 31, 2016, $742,950 of principal and accrued interest of $70,525 were converted into 1,251,504 shares of common stock. As described in Note 13, the remainder of the principal and interest was either converted or repaid subsequent to year end.
Convertible Notes Payable
On November 6, 2015, the Company issued two convertible promissory notes (the “Convertible Notes”) in the aggregate principal amount of $1,206,931 to two investment entities controlled by a single family. In the same transaction, the investment entities purchased an aggregate of 66,666 shares of common stock for a purchase price of $50,000, or $0.75 per share. The Convertible Notes are unsecured and accrue interest at the rate of 8% per annum, with interest payable on the last day of each calendar quarter. The principal amount under the Convertible Notes is due on the five-year anniversary of the issue date. The Convertible Notes are convertible at any time prior to maturity at the option of the holders at a conversion rate of $0.75 per share. If the Company’s common stock commences trading and closes at a price of $3.50 per share for five consecutive trading days, the principal amount under the Convertible Notes automatically converts into common stock at the rate of $0.75 per share. Proceeds from the Convertible Notes were to be used for the purpose of retirement of long-term debt. The Company evaluated the Convertible Notes for consideration of any beneficial conversion features as required under generally accepted accounting principles. The Company determined that there was no beneficial conversion feature.
Other Notes Payable
On August 16, 2016, the Company issued an unsecured bridge note to an individual for $32,000 with an interest rate of 8%. This note was originally due on September 30, 2016, and is now due on demand. As of December 31, 2016, there is a balance of $1,461 in accrued interest related to this note. As described in Note 13, this principal and interest was repaid subsequent to year end.
| F-12 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Related-Party Notes Payable
During the year ended December 31, 2016 the Company issued notes to related parties for $210,000. Also during the year ended December 31, 2016, $105,000 of those notes were paid back along with interest and fees of $3,089.
On December 18, 2015, the Company entered into a Patent Assignment Agreement for the acquisition of certain patent application rights. Prior to the execution of the Patent Assignment Agreement, a member of the Company’s board of directors advanced $50,000 on behalf of the Company to the seller under the Patent Assignment Agreement. The advance did not bear interest, was unsecured, and did not offer conversion terms at any time. In December 2015, the Company repaid $25,000, and as described in Note 13, the remainder of the principal and interest was repaid subsequent to year end.
Note 6 – Preferred Stock
The stockholders of the Company have authorized 10,000,000 shares of preferred stock, par value $0.001 per share. The preferred stock may be issued in one or more series. The board of directors has the right to fix the number of shares of each series (within the total number of authorized shares of the preferred stock available for designation as a part of such series), and designate, in whole or part, the preferences, limitations and relative rights of each series of preferred stock. As of December 31, 2016 and 2015, the board of directors has not designated any series of preferred stock and there are no shares of preferred stock issued or outstanding.
Note 7 – Common Stock
Common Stock Issued for Cash
The Company signed a Private Placement Memorandum dated December 28, 2015 to offer a maximum of 3,500,000 shares of its common stock at a price of $1.50 per share. On July 7, 2016, the board of directors authorized changing the offering to be units of one share of common stock and one warrant, sold for a price of $1.50 per unit. This change was applied retroactively to all purchasers under the Private Placement Memorandum. The units are being offered on a “best efforts” basis. During the year ended December 31, 2016, 1,106,952 units were subscribed, conditions for the minimum offering were met, and the Company received net proceeds of $1,498,731 from the offering.
Concurrently with the Private Placement Memorandum, the Company entered into a Placement Agent Agreement, effective December 28, 2015, that provides for compensation to a Placement Agent in connection with the offering of common stock. Pursuant to the Placement Agent Agreement, the Company will pay the Placement Agent a cash commission of ten percent of the issuance price of the common stock sold in the offering, and one share of common stock of the Company for each ten shares of the Company’s common stock sold in the offering. Pursuant to these provisions, with the release of shares described in the previous paragraph, the Company incurred commission fees to the Placement Agent of $166,043 and has issued the Placement Agent 103,166 shares of common stock. The Placement Agent will also receive an expense allowance of up to $10,000 to reimburse it for direct out-of-pocket costs related to the offering and the Escrow Agent was paid $1,000 for services in connection with the offering. Legal fees of $6,949 were also paid in connection with the offering.
During the three months ended March 31, 2015, the Company issued 294,000 shares of common stock for cash. Proceeds from these issuances total $147,000, or $0.50 per share.
During the nine months ended December 31, 2015, the Company issued 1,235,278 shares of common stock for cash. Proceeds from these issuances total $926,460, or $0.75 per share. Certain of these issuances were the result of the Company receiving proceeds in excess of the number of Convertible Debentures authorized by the Company’s board of directors. These investors opted to purchase shares of common stock in the Company at $0.75 per share in accordance with the provisions of the convertible debentures.
Common Stock Issued for Conversion of Debt
During the year ended December 31, 2016, certain convertible debenture holders exercised their right and converted $742,950 of principal and $70,527 of accrued interest into common stock. The Company issued 1,251,504 shares of common stock at $0.65 per share in accordance with the provisions of the convertible debentures.
During the year ended December 31, 2015, a convertible note payable in the amount of $50,000 and related accrued interest of $11,934 was converted into 95,283 shares of the Company’s common stock, at $0.65 per share.
| F-13 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Common Stock Issued Pursuant to Bill of Sale and Patent Assignment Agreements
On December 18, 2015, the Company entered into a Bill of Sale Agreement and a Patent Assignment Agreement with an individual. Pursuant to the two agreements, the Company acquired a) inventory with an estimated value of $2,200; b) molds with an estimated value of $35,000; and c) certain patent application rights with an estimated value of $175,300. Total consideration given for these assets was cash in the amount of $100,000 and 150,000 shares of the Company’s common stock, valued at $0.75 per share, or $112,500. The value assigned to the common stock was based on the price per share that common stock was most-recently issued to third parties for cash.
Common Stock Issued for Services
Periodically, the Company issues restricted common stock grants to directors, officers and consultants as compensation for future services. During the year ended December 31, 2016, the Company recognized $126,400 in stock compensation expense related to the amortization of this deferred compensation. During the year ended December 31, 2015 the Company issued 20,513 shares to employees, directors, and consultants as compensation for current services. The Company recognized stock-based compensation of $15,385 ($0.75 per share) for the year ended December 31, 2015.
In addition, during the year ended December 31, 2016, the Company issued 19,767 shares of common stock with a total value of $17,265 to two consultants for services rendered.
The Company recognized stock-based compensation related to the shares issued to directors, officers and consultants for the year ended December 31, 2015 of $255,915.
A summary of the status of the Company’s restricted common stock grants as of December 31, 2016 and changes during the year then ended, is presented below:
| Weighted | ||||||||
| Restricted | Average | |||||||
| Common | Common | |||||||
| Stock | Stock | |||||||
| Grants | Price | |||||||
| Balance at December 31, 2014 | 765,500 | $ | 0.50 | |||||
| Awarded | - | - | ||||||
| Vested | (511,830 | ) | 0.50 | |||||
| Balance at December 31, 2015 | 253,670 | 0.50 | ||||||
| Awarded | - | - | ||||||
| Vested | (252,798 | ) | 0.50 | |||||
| Balance at December 31, 2016 | 872 | $ | 0.50 | |||||
As of December 31, 2016, there was $436 of total unrecognized compensation cost related to the restricted common stock grants and the stock-based compensation arrangements awarded to directors, officers, and consultants. That cost is expected to be recognized over a weighted-average period of 0.02 years.
| F-14 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Total stock-based compensation expense from all sources for the year ended December 31, 2016 and 2015, including stock-based compensation for the warrants and related amortization discussed below in Note 8, has been included in the consolidated statements of operations as follows:
| For the Years Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Research and development expense | $ | 166,626 | $ | 165,342 | ||||
| Selling, general and administrative expense | 113,114 | 178,146 | ||||||
| Total share-based compensation | $ | 279,739 | $ | 343,488 | ||||
Note 8 – Common Stock Warrants
The Company has issued warrants to purchase its common stock for payment of consulting services, in connection with the extension of a note payable, as incentives to investors, and for cash. The fair value of warrants issued for consulting services is recognized as consulting expense at the date the warrants become exercisable. The Company values warrants based on the fair value of the stock on the date of issuance and records compensation over the requisite service period which is usually the vesting period. The non-vested shares are included in the total outstanding shares recorded in the consolidated financial statements. The fair value of warrants was estimated using the Black-Scholes option pricing model with volatility based on peer group companies. The fair value of the warrants that vested during the year ended December 31, 2016 was $0.76 per share. The fair value of the warrants that vested during the year ended December 31, 2015 was $0.402 per share. Management used the following inputs to value the warrants for the year ended December 31, 2016:
For the Years Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Expected life | 4.5 years | 5.2 years | ||||||
| Exercise price | $ | 0.50 | $ | 0.50 | ||||
| Expected volatility | 124 | % | 71 | % | ||||
| Expected dividends | None | None | ||||||
| Risk-free interest rate | 1.33 | % | 1.70 | % | ||||
The Company recognized $136,074 of share-based compensation and additional paid in capital during the year ended December 31, 2016 and $43,594 of share-based compensation and additional paid-in capital in addition to $28,594 in amortization related to the vesting of warrants for the year ended December 31, 2015.
Pursuant to the Private Placement Memorandum discussed in Note 7, the Company issued, to the investors, one warrant to purchase a share of common stock at a price of $1.50 for each share purchased. The Company issued 1,106,952 warrants under these terms. The fair value of warrants was estimated using the Black-Scholes option pricing model with the following weighted-average assumptions: risk-free interest rate of 1.02%, expected volatility 141%, expected life 2.12 years, expected dividend yield of zero. The proceeds of the private placement were allocated to the stock and warrants based on their relative fair values with $688,644 being allocated to the warrants.
In addition, as noted in Note 7 above, the Private Placement Memorandum requires the Company to issue, to the Placement Agent, a warrant to purchase one share of common stock at a price of $0.65 for each $0.81 of the principal amount of the outstanding 8% Convertible Debentures that is converted into Common Stock of the Company. During the year ended December 31, 2016, certain convertible debenture holders exercised their right and converted $742,950 of principal which resulted in 917,223 warrants issued to the Placement Agent.
| F-15 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
A summary of warrant activity for the years ended December 31, 2016 and 2015 is presented below:
| Weighted | Aggregate | |||||||||||||||
| Weighted | Average | Intrinsic | ||||||||||||||
| Shares | Average | Remaining | Value of | |||||||||||||
| Under | Exercise | Contractual | Vested | |||||||||||||
| Warrants | Price | Life | Warrants | |||||||||||||
| Outstanding at December 31, 2014 | 1,423,211 | $ | 0.54 | 8.3 years | $ | 17,640 | ||||||||||
| Issued | - | - | ||||||||||||||
| Exercised | - | - | ||||||||||||||
| Expired | - | - | ||||||||||||||
| Outstanding at December 31, 2015 | 1,423,211 | $ | 0.54 | 7.3 years | $ | 213,364 | ||||||||||
| Issued | 2,024,175 | 1.26 | ||||||||||||||
| Exercised | - | - | ||||||||||||||
| Expired | - | - | ||||||||||||||
| Outstanding at December 31, 2016 | 3,447,386 | $ | 0.88 | 4.2 years | $ | 546,333 | ||||||||||
The intrinsic value at December 31, 2016 is calculated at $0.85 per share less the exercise price, based on management’s latest estimate of the fair value of the shares of common stock, which is the latest price the Company issued shares of common stock for cash.
Note 9 – Intangible Assets
In December 2015, the Company purchased patents for a probe as well as enhanced surface and tips for obtaining bioelectrical signals for $175,300 comprised of $62,800 in cash and 150,000 shares of common stock. These patents will be amortized at a rate of $797 per month, or $9,562 per year, over the 220-month remaining life of the patents. During the years ended December 31, 2016 and 2015 the Company recognized amortization expense of $9,562 and $0, respectively.
Note 10 – Commitments and Contingencies
Consulting Representation Agreement
On January 1, 2016, the Company entered into a Consulting Representation Agreement with two consultants located in the European Union. Pursuant to the Consulting Representation Agreement, the consultants agreed to complete certain marketing milestones related to relationship development with key government and regulatory officials in the European Union and the introduction and marketing of the Company’s products to potential medical, clinical and hospital customers of the member states of the European Union. This Consulting Representation Agreement was terminated during the year ended December 31, 2016 due to failure of the consultants to perform. During the year ended December 31, 2016, the Company has issued 10,000 shares of common stock in accordance with this agreement.
Lease Agreement
The Company leases office space under an agreement that expires in 2017, with an option to renew with a 3% annual rent escalation. Monthly rental payments as of December 31, 2016 are $3,940 per month.
Lease expense charged to operations for the years ended December 31, 2016 and 2015 was $49,469 and $48,649, respectively.
| F-16 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
License Agreement
The Company has a license agreement with a party related through a shareholder and former member of the board of directors. Under the agreement, the Company has the right to the exclusive use of certain patents pending and related technology (the “technology”) in its medical devices and other products for an indefinite term. In return, the Company agreed to incur a minimum of $4,750,000 in development costs by the year 2014 to develop and market its products worldwide based on a graduated schedule and to make royalty payments based on a percentage of the aggregate worldwide net sales (as defined in the agreement) of its medical device and other products that utilize the technology. The minimum expenditure of $4,750,000 was achieved. At December 31, 2016 and 2015, accrued royalties under this license agreement total $17,873, respectively.
Note 11 – Income Taxes
The Company provides for income taxes using an asset and liability based approach. Deferred income tax assets and liabilities are recorded to reflect the future tax consequences of temporary differences between the financial statement and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
The significant components of net deferred tax assets (liabilities) were as follows at December 31, 2016 and 2015:
| 2016 | 2015 | |||||||
| Net operating losses | $ | 4,841,700 | $ | 3,776,478 | ||||
| Research and development credit carryforward | 129,500 | 75,004 | ||||||
| Related-party accruals | 2,300 | - | ||||||
| Allowance for doubtful accounts | - | 75,842 | ||||||
| Stock based compensation | - | 57,253 | ||||||
| Depreciation and amortization | 15,500 | (4,477 | ) | |||||
| Change in valuation allowance | (4,989,000 | ) | (3,980,100 | ) | ||||
| Net Deferred Tax Asset | $ | - | $ | - | ||||
As of December 31, 2016, the Company had no unrecognized tax benefits that, if recognized, would affect the Company’s effective income tax rate over the next 12 months. A reconciliation of the expected income tax benefit at the U.S. Federal income tax rate to the income tax benefit actually recognized for the years ended December 31, 2016 and 2015 is set forth below:
| 2016 | 2015 | |||||||
| Net loss | $ | (1,082,400 | ) | $ | (952,160 | ) | ||
| Non-deductible expenses | 110,300 | (8,824 | ) | |||||
| Valuation allowance | 972,100 | 960,984 | ||||||
| Benefit from Income Taxes | $ | - | $ | - | ||||
As of December 31, 2016, the Company has a net operating loss carry-forward for U.S. federal income tax purposes of approximately $12.2 million. This carry-forward is available to offset future taxable income, if any, and will expire, if not used, from 2017 through 2036. The utilization of the net operating loss carry-forward is dependent upon the tax laws in effect at the time the net operating loss carry-forward can be utilized and may be limited by changes in ownership control of the Company. The Company’s U.S. federal and Utah income tax returns, constituting the returns of the major taxing jurisdictions, are subject to examination by the taxing authorities for all open years as prescribed by applicable statute. No income tax waivers have been executed that would extend the period subject to examination beyond the period prescribed by statute. The Company is no longer subject to U.S. federal tax examinations for tax years before and including December 31, 2012. The Company is no longer subject to Utah state tax examinations for tax years before and including December 31, 2010. During the years ended December 31, 2016 and 2015, the Company did not incur interest and penalties.
| F-17 |
ProLung, Inc. and Subsidiary
Notes to Consolidated Financial Statements
Note 12 – Other Related Party Transactions
During the year ended December 31, 2016, the Company has consulting agreements in place with two of the members of its board of directors. These directors provide marketing and medical advisory services. One of the agreements was terminated during the year ended December 31, 2016. The remaining consulting agreement may be terminated by either the Company or by the consultant at any time and for any reason. During the year ended December 31, 2016, these directors were paid a total of $161,000 under these agreements.
Note 13 – Subsequent Events
The Company evaluated all subsequent events that occurred after the balance sheet date through April 17, 2017, the date its financial statements were available to be issued, and concluded there were additional events and transactions occurring during this period that required recognition or disclosure in the financial statements.
Subsequent to December 31, 2016, the Company sold 2,256,634 shares under the Private Placement Memorandum discussed in Note 7 for cash received of $3,384,952.
Subsequent to December 31, 2016, the remaining balance of the note payable to a relative of an executive officer was converted or repaid as follows: 1) the Company issued 66,667 shares of common stock as well as 66,667 warrants to purchase stock at a price of $1.50 for conversion of debt principal of $100,000, and 2) the Company repaid the remaining principal of $89,389 and accrued interest payable of $39,071. Any and all security interest held by the noteholder was released to the Company.
Subsequent to December 31, 2016, the remaining balance of convertible debentures was converted or repaid as follows: 1) the Company issued 1,752,274 shares of common stock for conversion of debenture principal of $991,550 and accrued interest payable of $147,428, and 2) the Company repaid the remaining principal of $265,500 and accrued interest payable of $41,607.
Subsequent to December 31, 2016, the Company repaid the other note payable of $32,000.
Subsequent to December 31, 2016, the remaining balance of the related-party notes payable were converted or repaid as follows: 1) the Company issued 40,000 shares of common stock as well as 40,000 warrants to purchase stock at a price of $1.50 for conversion of debt principal of $60,000, and 2) the Company repaid the remaining principal, interest and fees of $52,300.
| F-18 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this Amendment No. 2 to Annual Report on Form 10-K/A to be signed on its behalf by the undersigned thereunto duly authorized.
| PROLUNG, INC. (FORMERLY FRESH MEDICAL LABORATORIES, INC.) | |||
| September 14, 2017 | By: | /s/ Steven C. Eror | |
| Date | Steven C. Eror, | ||
Chief Executive Officer and President (Principal Executive Officer) | |||
Exhibit Index
| * | Filed herewith |
| # | Management compensation agreement. |
| (1) | Incorporated by reference with Form 10 filed February 10, 2012, File No. 12750426. |
| (2) | Incorporated by reference with Form 10/A filed April 10, 2012, File No. 12594347. |
| (3) | Incorporated by reference from an exhibit to our Annual Report on Form 10-K filed on April 3, 2014. |
| (4) | Incorporated by reference from an exhibit to our Quarterly Report on Form 10-Q filed on May 14, 2014. |
| (5) | Incorporated by reference from an exhibit to our Current Report on Form 8-K filed on July 8, 2014. |
| (6) | Incorporated by reference from an exhibit to our Quarterly Report on Form 10-Q filed on November 14, 2014. |
| (7) | Incorporated by reference from an exhibit to our Current Report on Form 8-K filed on December 9, 2014. |
| (8) | Incorporated by reference from an exhibit to our Annual Report on Form 10-K filed on March 31, 2015. |
| (9) | Incorporated by reference from an exhibit to our Current Report on Form 8-K filed on May 5, 2015. |
| (10) | Incorporated by reference from an exhibit to our Quarterly Report on Form 10-Q filed on November 16, 2015. |
| (11) | Incorporated by reference from an exhibit to our Annual Report on Form 10-K filed on April 14, 2016. |