Attached files
| file | filename |
|---|---|
| EX-99.1 - EX-99.1 - INSMED Inc | a17-21316_1ex99d1.htm |
| 8-K - 8-K - INSMED Inc | a17-21316_18k.htm |

Safe Harbor Statement This presentation contains forward looking statements. "Forward-looking statements," as that term is defined in the Private Securities Litigation Reform Act of 1995, are statements that are not historical facts and involve a number of risks and uncertainties. Words herein such as "may," "will," "should," "could," "would," "expects," "plans," "anticipates," "believes," "estimates," "projects," "predicts," "intends," "potential," "continues," and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) may identify forward-looking statements. The forward-looking statements in this press release are based upon the Company’s current expectations and beliefs, and involve known and unknown risks, uncertainties and other factors, which may cause the Company’s actual results, performance and achievements and the timing of certain events to differ materially from the results, performance, achievements or timing discussed, projected, anticipated or indicated in any forward-looking statements. Such factors include, among others: risks that the full six-month data from the CONVERT study or subsequent data from the remainder of the study’s treatment and off-treatment phases will not be consistent with the top-line six-month results of the study; uncertainties in the research and development of our existing product candidates, including due to delays in data readouts, such as the full data from the CONVERT study, patient enrollment and retention or failure of our preclinical studies or clinical trials to satisfy pre-established endpoints, including secondary endpoints in the CONVERT study and endpoints in the INS-312 study; lack of safety and efficacy of our product candidates; failure to develop, or to license for development, additional product candidates, including a failure to attract experienced third-party collaborators; failure to obtain, or delays in obtaining, regulatory approval from the United States Food and Drug Administration, Japan’s Ministry of Health, Labour and Welfare, the European Medicines Agency, and other regulatory authorities for our product candidates or their delivery devices, including due to insufficient clinical data or selection of endpoints that are not satisfactory to regulators, complexity in the review process for combination products or inadequate or delayed data from a human factors study required for US regulatory approval; lack of experience in conducting and managing preclinical development activities and clinical trials necessary for regulatory approval, including the regulatory filing and review process; failure of third parties on which we are dependent to conduct our clinical trials, to manufacture sufficient quantities of our product candidates for clinical or commercial needs, or to comply with our agreements or laws and regulations that impact our business; failure to comply with license agreements that are critical for our product development, including our license agreements with PARI Pharma GmbH and AstraZeneca AB; inaccuracies in our estimate of the size of the potential markets for our product candidates; failure to maintain regulatory approval for our product candidates, if received, due to a failure to satisfy post-approval regulatory requirements, such as the submission of sufficient data from confirmatory clinical trials; uncertainties in the rate and degree of market acceptance of product candidates, if approved; uncertainties in the timing, scope and rate of reimbursement for our product candidates; competitive developments affecting our product candidates; inaccurate estimates regarding our future capital requirements, including those necessary to fund our ongoing clinical development, regulatory and commercialization efforts as well as milestone payments or royalties owed to third parties; inability to repay our existing indebtedness or to obtain additional financing when needed; failure to obtain, protect and enforce our patents and other intellectual property; inability to create an effective direct sales and marketing infrastructure or to partner with third parties that offer such an infrastructure for distribution of our product candidates, if approved; the cost and potential reputational damage resulting from litigation to which we are a party, including, without limitation, the class action lawsuit pending against us; failure to comply with the laws and regulations that impact our business; loss of key personnel; and changes in laws and regulations applicable to our business, including those related to pricing and reimbursement of our product candidates. For additional information about the risks and uncertainties that may affect our business, please see the factors discussed in Item 1A, "Risk Factors," in the Company’s Annual Report on Form 10-K for the year ended December 31, 2016. The Company cautions readers not to place undue reliance on any such forward-looking statements, which speak only as of the date of this presentation. The Company disclaims any obligation, except as specifically required by law and the rules of the Securities and Exchange Commission, to publicly update or revise any such statements to reflect any change in expectations or in events, conditions or circumstances on which any such statements may be based, or that may affect the likelihood that actual results will differ from those set forth in the forward-looking statements. 2

3 To transform the lives of patients battling serious rare diseases OUR MISSION Top-line data indicates Phase 3 CONVERT trial met primary endpoint of culture conversion Company plans to pursue accelerated approval and request priority review

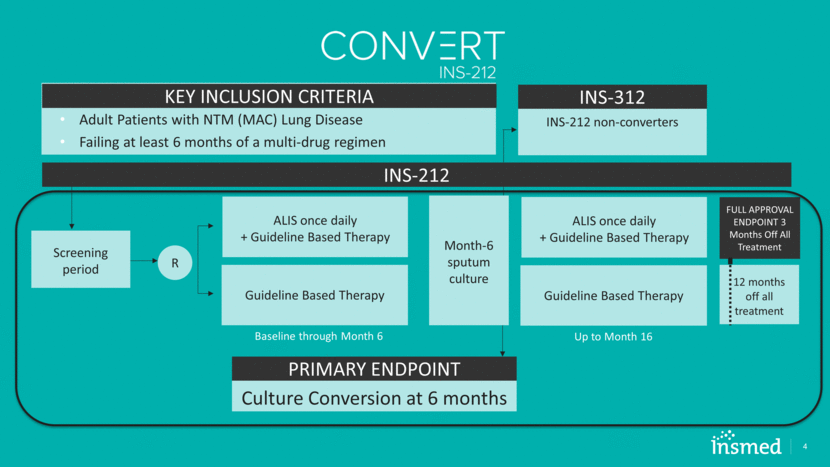
ALIS once daily + Guideline Based Therapy 4 Adult Patients with NTM (MAC) Lung Disease Failing at least 6 months of a multi-drug regimen KEY INCLUSION CRITERIA Guideline Based Therapy R Screening period Month-6 sputum culture PRIMARY ENDPOINT Culture Conversion at 6 months INS-212 non-converters INS-312 ALIS once daily + Guideline Based Therapy Guideline Based Therapy Baseline through Month 6 Up to Month 16 12 months off all treatment FULL APPROVAL ENDPOINT 3 Months Off All Treatment INS-212
Top-line Data Indicates CONVERT Study Met Primary Endpoint Addition of ALIS to GBT* eliminated evidence in sputum of NTM lung disease caused by MAC by Month 6 in 29% of patients, compared to 9% of patients on GBT alone (p <0.0001) 20% absolute difference in treatment groups in favor of ALIS arm 5 p <0.0001 Percentage of Patients with Negative NTM Sputum Culture BY Month 6 (3 Consecutive Negative Sputum Cultures) *Guideline Based Therapy 29% 9% ALIS + GBT GBT

Top-line CONVERT Study Results – Secondary Endpoints at Month 6 6 Time to Conversion Patients in the GBT-only arm took approximately 30% longer to convert when compared to patients on ALIS plus GBT (p<0.0001) 6-Minute Walk Distance (6MWD) No statistically significant difference between patients in the two arms Analysis per a pre-specified endpoint shows that patients who achieved culture conversion across both arms demonstrated an improvement in 6-minute walk distance vs. patients who did not culture convert (p=0.0108)

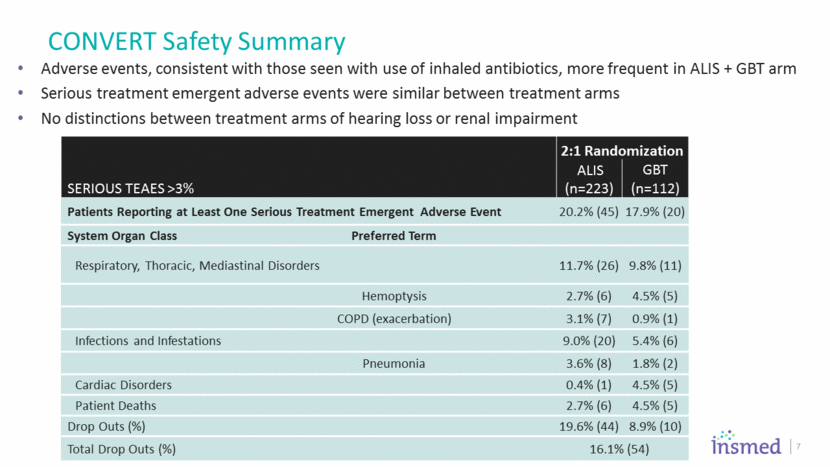
CONVERT Safety Summary Adverse events, consistent with those seen with use of inhaled antibiotics, more frequent in ALIS + GBT arm Serious treatment emergent adverse events were similar between treatment arms No distinctions between treatment arms of hearing loss or renal impairment 7 2:1 Randomization Serious TEAEs >3% ALIS (n=223) GBT (n=112) Patients Reporting at Least One Serious Treatment Emergent Adverse Event 20.2% (45) 17.9% (20) System Organ Class Preferred Term Respiratory, Thoracic, Mediastinal Disorders 11.7% (26) 9.8% (11) Hemoptysis 2.7% (6) 4.5% (5) COPD (exacerbation) 3.1% (7) 0.9% (1) Infections and Infestations 9.0% (20) 5.4% (6) Pneumonia 3.6% (8) 1.8% (2) Cardiac Disorders 0.4% (1) 4.5% (5) Patient Deaths 2.7% (6) 4.5% (5) Drop Outs (%) 19.6% (44) 8.9% (10) Total Drop Outs (%) 16.1% (54)
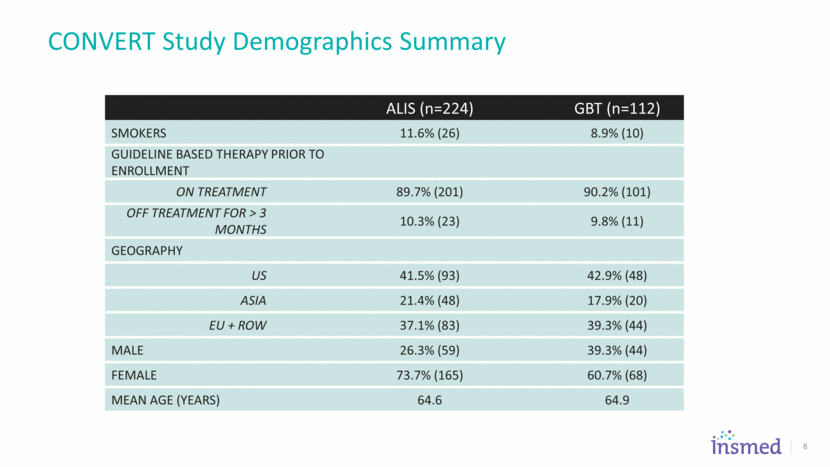
CONVERT Study Demographics Summary ALIS (n=224) GBT (n=112) Smokers 11.6% (26) 8.9% (10) Guideline Based Therapy Prior to Enrollment On Treatment 89.7% (201) 90.2% (101) Off Treatment for > 3 months 10.3% (23) 9.8% (11) Geography US 41.5% (93) 42.9% (48) Asia 21.4% (48) 17.9% (20) EU + ROW 37.1% (83) 39.3% (44) Male 26.3% (59) 39.3% (44) Female 73.7% (165) 60.7% (68) Mean Age (years) 64.6 64.9 8
NDA Submission planned based on primary endpoint under Subpart H Pathway to US Submission – Division of Anti-Infective Products 9 Breakthrough Designation Orphan and QIDP Accelerated Approval Full Approval Requirement 12 years of exclusivity (7 orphan and 5 QIDP) and likely priority review Guidance on efficient drug development; allows for rolling submission Confirmatory endpoint contained within study design

Insmed: A Global Biopharmaceutical Focused on Rare Disease 10 POSITIVE TOP-LINE PHASE 3 RESULTS FOR ALIS GLOBAL COMMERCIAL OPPORTUNITY ALIS LIFE CYCLE MANAGEMENT
[LOGO]
