Attached files
| file | filename |
|---|---|
| EX-10.10 - SEVERANCE AGREEMENT - Oncolix, Inc. | aepp_ex1010.htm |
| EX-23.1 - CONSENT OF INDEPENDENT - Oncolix, Inc. | aepp_ex231.htm |
| EX-21.1 - LIST OF SUBSIDIARIES - Oncolix, Inc. | aepp_ex211.htm |
| EX-10.12 - FORM OF OPTION AGREEMENT - Oncolix, Inc. | aepp_ex1012.htm |
| EX-10.11 - SEVERANCE AGREEMENT - Oncolix, Inc. | aepp_ex1011.htm |
| EX-5.1 - LEGAL OPINION - Oncolix, Inc. | aepp_ex51.htm |
| EX-4.1 - COMMON STOCK SPECIMEN - Oncolix, Inc. | aepp_ex41.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
| ADVANCED ENVIRONMENTAL PETROLEUM PRODUCERS INC. |
| (Exact name of registrant as specified in its charter) |
| Florida |
| 2834 |
| 46-3046340 |
| (State or other jurisdiction of |
| (Primary Standard Industrial |
| (I. R. S. Employer |
| incorporation or organization) |
| Classification Code Number) |
| Identification Number) |
14405 Walters Road, Suite 780
Houston, Texas 77014
(281) 402-3167
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Michael T. Redman
Chief Executive Officer
14405 Walters Road, Suite 780
Houston, Texas 77014
(281) 402-3167
(Name, address, including zip code, and telephone number, including area code, of agent for service)
With copy to:
Thomas C. Pritchard, Esq.
Thomas C. Pritchard, P.C.
800 Bering Dr., Suite 201
Houston, Texas 77057
Tel: (713) 209-2911
Fax: (832) 538-1265
As soon as practicable after this Registration Statement becomes effective
(Approximate date of commencement of proposed sale to the public)
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filed, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filed,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ |
| Non-accelerated filer | ¨ | Smaller reporting company | x |
| (Do not check if a smaller reporting company) | Emerging growth company | ¨ | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act.
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to Be Registered |
| Amount Being Registered (1) |
|
| Proposed Maximum Offering Price Per Share(2) |
|
| Proposed Maximum Aggregate Offering Price |
|
| Amount of Registration Fee |
| ||||
| Shares of Common Stock underlying the Principal Amount of Convertible Notes(3) |
|
| 55,872,837 |
|
| $ | 0.105 |
|
| $ | 5,866,647.89 |
|
| $ | 679.94 |
|
| Common Stock Underlying the Interest of the Convertible Notes(4) |
|
| 5,633,847 |
|
| $ | 0.105 |
|
| $ | 591,553.94 |
|
| $ | 68.56 |
|
| Shares underlying the Principal Amount of Interest of the Convertible Notes in the event of a price decline and concomitant reduction in conversion price(5) |
|
| 6,150,670 |
|
| $ | 0.105 |
|
| $ | 645,820.35 |
|
| $ | 74.85 |
|
| Shares of Common Stock underlying the Warrants(6) |
|
| 59,072,837 |
|
| $ | 0.105 |
|
| $ | 6,202,647.89 |
|
| $ | 718.89 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| TOTAL |
|
| 126,730,191 |
|
|
| -- |
|
| $ | 13,306,670.07 |
|
| $ | 1,542.24 |
|
Shares to Register:
|
| (1) | Pursuant to Rule 416 under the Securities Act of 1933, the Registrant is also registering such additional indeterminate number of shares as may become necessary to adjust the number of shares as a result of a stock split, stock dividend or similar adjustment of its outstanding Common Stock. Registrant is also registering additional shares of Common Stock to be available for issuance based on market fluctuation. |
|
|
|
|
|
| (2) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(c) under the Securities Act of 1933, as amended, based on the average high and low prices of the Common Stock as traded on the OTC Pink on August 24, 2017. |
|
|
|
|
|
| (3) | Represents shares of Common Stock issuable by the registrant upon the conversion of the convertible notes in the aggregate original principal amount of $4,190,463 issued to the selling security holders (the “Convertible Notes”) pursuant to the terms of the Convertible Notes dated August 3, 2017. |
|
|
|
|
|
| (4) | Represents shares of Common Stock issuable by the registrant upon the conversion of interest to be accrued pursuant to the terms of the Convertible Notes at a conversion price of $0.075 per share. |
|
|
|
|
|
| (5) | Represents shares of Common Stock as a buffer in the event the monthly redemption payment or interest payment calculations are based on the lower of $0.075 per share and 75% of the average of the five lowest closing bid prices for the twenty consecutive trading days prior to such determination date. |
|
|
|
|
|
| (6) | Represents shares of Common Stock issuable by the registrant upon exercise of Warrants to purchase shares of Common Stock to be issued to the selling security holders (the “Warrants”) upon the terms and conditions set forth in the warrant agreement dated August 3, 2017. |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
| 2 |
The information in this prospectus is not complete and may be amended. The selling security holders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION
Prospectus dated August 29, 2017
ADVANCED ENVIRONMENTAL PETROLEUM PRODUCERS INC.
126,730,191 Shares of Common Stock
This prospectus relates to the resale of up to 126,730,191 shares of common stock, par value $0.0001 (“Common Stock”), by certain shareholders (the “selling security holders”). The shares of Common Stock subject to this prospectus include:
|
| (1) | 67,657,354 shares of Common Stock issuable upon conversion of the Convertible Notes with the aggregate principal amounts of $4,190,463 issued to selling security holders (the “Convertible Notes”) pursuant to the Convertible Notes issued August 3, 2017, by and between the Company and selling security holders, and interest to be accrued until maturity. Based upon the current market price of the Company’s Common Stock, the amount of the Company’s Common Stock being registered may be in excess of the number of shares into which the Convertible Notes may currently be converted, however the parties have agreed upon the aggregate number of shares to be registered to account for market fluctuations. |
|
|
|
|
|
| (2) | 59,072,837 shares of Common Stock issuable upon exercise of warrants (“Warrants”) upon the terms and conditions set forth in the Warrant Agreement. |
We will not receive any proceeds from the resale of any of the shares offered hereby,
Our Common Stock is listed for quotation on the OTC Pink quotation systems under the symbol “AEPP.” The market for the Common Stock is limited, sporadic and volatile. The closing price of our Common Stock on August 24, 2017 was $0.11 per share.
We are not selling any of the Common Stock that we are registering. The Common Stock will be sold by the selling security holders as detailed in this prospectus. Such selling security holders may sell the Common Stock at the market price as of the date of sale or a price negotiated in a private sale. Our Common Stock is listed for quotation on the OTC Pink quotation system under the symbol “AEPP.”
This investment involves a high degree of risk. You should purchase shares only if you can afford a complete loss of your investment. You should read this prospectus in its entirety and carefully consider the risk factors beginning on page 9 of this prospectus and the financial data and related notes incorporated by reference before deciding to invest in the shares.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is _______, 2017
| 3 |
|
| 5 |
| |
|
| 5 |
| |
|
| 10 |
| |
|
| 23 |
| |
|
| 24 |
| |
|
| 24 |
| |
|
| 28 |
| |
|
| 30 |
| |
|
| 34 |
| |
| MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
| 49 |
|
|
| 56 |
| |
|
| 56 |
| |
|
| 58 |
| |
|
| 61 |
| |
| DISCLOSURE OF COMMISSION POSITION OF INDEMNIFICATION FOR SECURITIES ACT LIABILITIES |
| 63 |
|
|
| 63 |
| |
|
| 64 |
| |
|
| 64 |
| |
|
| F-1 |
|
You should rely only on the information contained in this prospectus. We have not authorized anyone to provide you with information that is different. This prospectus is not an offer to sell, nor is it seeking an offer to buy, these securities in any jurisdiction where the offer or sale of these securities is not permitted. You should assume that the information contained in this prospectus is accurate as of the date on the front of this prospectus only. Our business, financial condition, results of operations and prospects may have changed since that date. This prospectus will be updated as required by law.
| 4 |
| Table of Contents |
PROSPECTUS SUMMARY
ADVANCED ENVIRONMENTAL PETROLEUM PRODUCERS INC.
This summary highlights selected information about the Company and this offering. This summary is not complete and does not contain all of the information that may be important to you. You should read carefully the entire prospectus, including “Risk Factors” and the other information contained or incorporated by reference in this prospectus before making an investment decision. Unless otherwise indicated or the context otherwise requires: (i) “we,” “us,” “our,” or “Company” refer to Advanced Environmental Petroleum Producers Inc. and its wholly-owned subsidiary, Oncolix, Inc. (“Oncolix”) and (ii) in certain instances we refer to AEPP to describe AEPP’s operations or financial statements prior to the Merger (as described below) and Oncolix to describe Oncolix’s operations or financial statements prior to the Merger.
This prospectus contains certain “forward-looking statements” within the meaning of the federal securities laws, including the Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which statements involve substantial risks and uncertainties. In some cases, it is possible to identify forward-looking statements because they contain words such as “anticipates,” believes,” “contemplates,” “continue,” “could,” “estimates,” “expects,” “future,” “intends,” “likely,” “may,” “plans,” “potential,” “predicts,” “projects,” “seek,” “should,” “target” or “will,” or the negative of these words or other similar terms or expressions that concern our expectations, strategy, plans or intentions. Many factors could cause our actual operations or results to differ materially from the operations and results anticipated in forward-looking statements. These factors include, but are not limited to, those set forth under “Risk Factors” commencing on page 10.
· the early stage of development of our product candidate Prolanta™; · the timing, cost or results of our clinical trials; · our need for substantial additional funds and uncertainties regarding our ability to raise such funds on acceptable terms, if at all; · our ability to make arrangements to repay indebtedness, and the possible loss of assets or our ability to operate if we are unable to do so on acceptable terms; · uncertainties relating to patent and intellectual property matters; and · our dependence on third parties, including our contract manufacturer, clinical research organizations, and consultants.
Company
We are a clinical-stage biopharmaceutical company developing medical therapies for the treatment of cancer. We currently own issued or pending patent applications related to the use of Prolanta™, a recombinant protein analogue of human prolactin that is designed to bind to the prolactin receptor on human cells. There is substantial scientific evidence that prolactin is associated with various cancers, including ovarian, breast, uterine and prostate. We and our collaborators have conducted animal studies that indicate that our drug Prolanta™ may be effective in treating breast and ovarian cancers, either as a stand-alone therapy or in combination with other drugs, by blocking the effect of human prolactin. ProlantaTM may also affect the cellular mechanisms that assist cancer cells to develop resistance to chemotherapy.
Our initial focus is the development, clinical testing and commercialization of Prolanta™ for the treatment of ovarian cancer. The US Food and Drug Administration (“FDA”) has provided us with clearance to commence a Phase 1clinical trial of Prolanta™ in the treatment of ovarian cancer, and we have dosed three subjects in the first phase of this clinical trial. We chose ovarian cancer as the first human application of Prolanta™ because of the significant unmet medical need as well as favorable regulatory status. Because of the size of the US market, the FDA has granted Prolanta™ status as an Orphan Drug, providing potential tax benefits and market exclusivity. We intend to apply for Orphan designation in the European Union and in Japan.
If Prolanta™ is found to be safe and well tolerated in the Phase 1 study, we will seek regulatory clearance to conduct additional clinical trials in larger numbers of ovarian cancer patients to establish efficacy. We expect that at least two clinical studies will be required, and possibly more, to establish the efficacy and safety of Prolanta™ in sufficient numbers of ovarian cancer patients in order to seek approval to market Prolanta™ for this cancer. The design of these studies will be finalized after the completion of the Phase 1 study and after consultation with the FDA, but we currently believe we will first evaluate the use of Prolanta™ in combination with other chemotherapy drugs such as platinum-based drugs and taxanes. There is preclinical and laboratory evidence that Prolanta™ may be synergistic with these other therapies, allowing a broader market opportunity. We intend to seek a distribution partner to market Prolanta™, but may develop our own sales force in the event we cannot make suitable arrangements.
| 5 |
| Table of Contents |
Depending on the availability of resources, we also intend to evaluate Prolanta™ for the treatment of other cancers. These studies may be performed in collaboration with larger pharmaceutical companies, either through licenses or joint ventures. Our scientific collaborators have demonstrated in animal models the efficacy of Prolanta™ in treating breast cancer, and we intend to engage academic collaborators to develop data for the treatment of other cancer types.
We may also consider in-licensing or acquiring other drug candidates, preferably in oncology. We operate with a small staff of employees and key consultants and have utilized contractors for certain operations. We do not intend to develop the infrastructure or acquire the facilities necessary to manufacture Prolanta™, and instead we plan to utilize contract manufacturers. We also plan to utilize contract research organizations to assist us in conducting clinical trials. We intend to employ additional administrative and clinical personnel as necessary to conduct our clinical trials and commercialize Prolanta™.
Recent Developments
Acquisition of Oncolix
On August 3, 2017, the Company closed its agreement and plan of merger whereby AEPP Merger Sub, Inc., a wholly-owned subsidiary of the Company (“Merger Sub”) merged with and into Oncolix, and Oncolix became a wholly-owned subsidiary of the Company (“Merger”). Pursuant to the terms of the Merger, a total of 71,770,200 shares of Company Common Stock were issued to the former common stock holders of Oncolix and 62,138,680 shares of Company Series A Preferred Stock were issued to the former series A preferred stock holders of Oncolix. Each share of Series A Preferred Stock issued by the Company is convertible into one share of Company Common Stock, has voting rights, a liquidation preference, and other customary rights and preferences. As a result of the Merger, the shares of Company Common Stock owned by Oncolix were cancelled. In addition, pursuant to the Merger, all outstanding options and warrants to acquire Oncolix common stock and Series A Preferred Stock were converted into the right to acquire or convert into Company Common Stock or Company Series A Preferred Stock.
Post-Merger a warrant to purchase Series A Preferred Stock was exercised resulting in an additional amount of 899,604 shares of Series A Preferred Stock being issued. As of August 28, 2017, there are 103,536,703 shares of Company Common Stock issued and outstanding and 63,038,284 shares of Series A Preferred Stock issued and outstanding that are convertible into 63,038,284 shares of Company Common Stock.
The Merger was treated as a reverse acquisition of AEPP, a public shell company, for financial accounting and reporting purposes. As such, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes. Further, as a result, the financial statements for periods prior to the Merger that will be reflected in this filing and the Company’s future financial statements filed with the United States Securities and Exchange Commission (“SEC”) will be those of Oncolix, and the Company’s assets, liabilities and results of operations for periods after the Merger, including this filing, will be the consolidated assets, liabilities and results of operations of AEPP and Oncolix.
Debt and Warrant Financing
On August 3, 2017, AEPP and Oncolix entered into agreements with investors in connection with a $4,190,463 convertible debt and warrant financing (the “Units”). Such investment was effected through the issuance and sale of Units of Convertible Notes and Warrants for (i) $2,000,000 in cash for Convertible Notes in the principal amount of $2,352,941 and Warrants to acquire 31,372,547 shares of AEPP Common Stock (the “Cash Offering”) and (ii) (A) the exchange of outstanding secured indebtedness in the amount of $1,561,894 and warrants to acquire 17,608,280 shares of AEPP Common Stock (formerly Oncolix warrants prior to being assumed by AEPP in the Merger) issued by Oncolix to existing noteholders (“Existing Investors”), for (B) the issuance and sale of Units of Convertible Notes issued by the Company in the aggregate principal amount of $1,837,522 and Warrants to acquire 24,500,290 shares of Company Common Stock (the “Exchange Offering” and collectively with the Cash Offering, referred to as the “Unit Offering). The investors in the Unit Offering are collectively referred to as “Purchasers”. In addition, Warrants to acquire an additional 3,200,000 shares of Company Common Stock are held by a Purchaser and included in Warrants herein.
The Unit Offering is documented with the Purchasers pursuant to the following: (i) a Securities Purchase Agreement, dated August 3, 2017, by and among AEPP, Purchasers and Oncolix (the “Purchase Agreement”); (ii) 10% Senior Secured Convertible Notes of AEPP issued in favor of the Purchasers (the “Convertible Notes”); (iii) a Registration Rights Agreement, dated August 3, 2017, by and between AEPP and the Purchasers (the “Registration Rights Agreement”); (iv) a Security Agreement, dated August 3, 2017, by and among AEPP, Oncolix and Purchasers (the “Security Agreement”); (v) an IP Security Agreement, dated August 3, 2017, by and among AEPP, Oncolix and the collateral agent of the Unit Offering (the “IP Security Agreement”); (vi) five-year warrants to purchase AEPP Common Stock dated August 3, 2017, issued by AEPP to the Purchasers (“Warrants”); (vii) a Lock-Up Agreement, dated August 3, 2017, by and among AEPP, Oncolix and certain existing equity holders (the “Lock-Up Agreement”); and (viii) a Subsidiary Guarantee, dated August 3, 2017, by and between Oncolix and Purchasers (the “Guarantee” and collectively with the Purchase Agreement, Convertible Notes, Registration Rights Agreement, Security Agreement, IP Security Agreement, Warrants and Lock-Up Agreement, the “Transaction Documents”), pursuant to which AEPP issued and sold the Convertible Notes and the Warrants to the Purchasers pursuant to the Transaction Documents.
| 6 |
| Table of Contents |
Convertible Notes
Pursuant to the Purchase Agreement, the Company issued Convertible Notes to the Purchasers in an aggregate principal amount of $4,190,463 for a purchase price of $3,561,894, consisting of gross cash proceeds of $2,000,000 and the exchange of existing convertible debt and related accrued interest of Oncolix totaling $1,561,894. The Convertible Notes were issued at a 15% discount.
The Convertible Notes bear interest at a rate of 10% per annum, payable quarterly on November 1, 2017, February 1, 2018, and May 1, 2018 and thereafter, on a monthly basis until maturity. The interest is payable in cash or, at the option of the Company, subject to compliance with Equity Conditions (as defined below), in fully tradable Company Common Stock at the lesser of the then conversion price or a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the interest payment date. The final maturity of the Convertible Notes is November 1, 2018, but commencing on May 1, 2018 and on the first day of each month thereafter until maturity, we are required to redeem an amount of the Convertible Notes equal to 1/7th of the original principal amount, plus 10% of such monthly redemption amount as a bonus. The principal payments shall be made in cash or, subject to the satisfaction of Equity Conditions, in our Common Stock valued at the lesser of the then conversion price or a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the amortization date.
The Convertible Notes are convertible at the option of the Purchasers at a conversion price equal to the lesser of (i) $0.075 per share of our Common Stock, (ii) 75% of the 10-day average closing bid price of our Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of our Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share). The Convertible Notes contain customary anti-dilution protection, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both conversion price and share amounts to be issued upon conversion).
In the event of a closing of an $8 million financing in connection with a change in listing of our Common Stock to Nasdaq or the NYSE, the Convertible Notes shall be subject to mandatory conversion into our Common Stock at a 30% discount to the offering price.
We shall be required to offer to prepay in cash the aggregate principal amount of the Convertible Notes at 120% of the principal amount thereof plus any unpaid accrued interest to the date of repayment, at maturity, on the sale of substantially all of our assets, upon a change of control, upon a qualified offering, or upon a “fundamental transaction” (tender offer, reclassification, sale of substantially all assets and merger); in such an event, the Purchasers shall have the right to convert the Convertible Notes prior to the date of any such prepayment.
The Convertible Notes contain standard negative covenants customary for transactions of this type. The events of default are also customary for transactions of this type, including default in timely payment of principal or interest, failure to observe or perform any covenant or agreement contained in the Convertible Note and Transaction Documents (including the Registration Rights Agreement), the commencement of bankruptcy or insolvency proceedings, failure to timely deliver conversion shares underlying the Convertible Notes, failure to timely file Exchange Act filings, and failure to satisfy certain Equity Conditions.
Equity Conditions mean, during the period in question, (a) the Company shall have duly honored all conversions and redemptions of the Convertible Notes, (b) we shall have complied with all conversion and other provisions of the Convertible Notes and shall have paid all liquidated damages and other amounts owing in respect of this Convertible Note, (c)(A) there is an effective registration statement pursuant to which the holder is permitted to utilize the prospectus thereunder to resell at least the number of shares of Common Stock issuable pursuant to the Transaction Documents at such time or (B) all of the underlying shares may be resold pursuant to Rule 144 without volume or manner-of-sale restrictions or current public information requirements, (d) our Common Stock is trading on a trading market, (e) there is a sufficient number of authorized but unissued and otherwise unreserved shares of our Common Stock for the issuance of five times all of the shares issuable pursuant to the Convertible Notes and Warrants, (f) there is no existing event of default or no existing event which, with the passage of time or the giving of notice, would constitute an event of default, (g) the issuance of the shares in question would not violate the beneficial ownership limitations in the Transaction Documents, (h) there has been no public announcement of a pending or proposed fundamental transaction or change of control transaction that has not been consummated, (i) the holder is not in possession of any information provided by the Company that constitutes, or may constitute, material non-public information, (j) our Common Stock is DWAC eligible and not subject to a DTC “chill;” and (k) for each trading day in a period of 10 consecutive trading days prior to the applicable date in question (A) the daily dollar trading volume for our Common Stock on the principal trading market exceeds $75,000 per trading day and (B) the weighted average stock price for our Common Stock on the principal trading market is above 50% of the closing stock price on August 3, 2017 (as adjusted for all stock splits stock dividends, stock combinations, reclassifications and other transactions).
We have the right at any time to redeem all, but not less than all, of the total outstanding amount of principal amount then remaining under the Convertible Notes at a price equal to 120% of such principal amount.
| 7 |
| Table of Contents |
Warrants
In connection with the Purchase Agreement, the Company issued the Purchasers five-year Warrants to purchase an aggregate of 55,872,837 shares of our Common Stock at a purchase price of $0.09 per share, exercisable for cash or on a cashless basis. In addition, in connection with the debt converted in the Exchange Offering, one Purchaser was issued five-year Warrants to acquire 3,200,000 shares of our Common Stock at a purchase price of $0.0825 per share, exercisable for cash or on a cashless basis. The Warrants contain customary anti-dilution provisions, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both exercise price and share amounts to be issued upon exercise).
Security Agreements and Guarantee
In connection with the issuance of the Convertible Notes, the Company and its wholly-owned subsidiary Oncolix, pledged substantially all of their assets as security and collateral for the Convertible Notes, including all intellectual property of Oncolix and the shares of capital stock of Oncolix owned by the Company. Additionally, Oncolix guaranteed the obligations of the Convertible Notes.
Registration Rights Agreement
In connection with the execution of the Purchase Agreement, the Company and the Purchasers also entered into the Registration Rights Agreement. Pursuant to the Registration Rights Agreement, the Company has agreed to file an initial registration statement (“Initial Registration Statement”) with the SEC to register the resale of all shares of Common Stock issuable under conversion of the Convertible Notes and upon exercise of the Warrants and any securities issued or then issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect to the foregoing (the “Registrable Shares”) on or prior to September 2, 2017, and have it declared effective at the earlier of (i) the 90th calendar day after August 3, 2017 (120th calendar day after August 3, 2017 in the event that the registration statement is subject to review by the SEC) and (ii) the 5th business day after the date the Company is notified by the SEC that the registration statement will not be reviewed or will not be subject to further review.
The Company also agreed to file additional registration statements to cover all of the Registrable Securities that are not covered by the Initial Registration Statement on or prior to 30 days following the date the Company first knows that such additional registration statement is required to be filed (the “Additional Registration Statements”) and have it declared effective no later than the earlier of (i) the 60th calendar day following the date the Company first knows that it is required to file such Additional Registration Statement and (ii) the 5th business day after the date the Company is notified by the SEC that the registration statement will not be reviewed or will not be subject to further review. If (i) the Initial Registration Statement is not declared effective on or before the 90th calendar day following the date of the Purchase Agreement (120th day in the event of a SEC review), (ii) any Additional Registration Statement is not declared effective on or before its effectiveness deadline, or (iii) any registration statement is declared effective by the SEC but shall thereafter cease to be effective for a period of time of 10 consecutive calendar days but not more than an aggregate of 15 calendar days in any one 12-month period (each such event, a “Non-Registration Event”), then the Company shall deliver to the Purchasers, as liquidated damages, an amount equal to ten percent of the outstanding principal amount of the Convertible Notes then held by the Purchasers for the first 30 days (or part thereof) and after such 30 days, an additional 10% of the outstanding principal amount of the Convertible Notes for a subsequent 30-day period (or part thereof); provided that the maximum amount of liquidated damages shall not exceed 18% of the aggregate outstanding principal amount of the Convertible Notes. The Company may pay the liquidated damages in cash or through the issuance of shares of Common Stock (valued at 75% of the average of the five lowest closing bid prices for the twenty consecutive trading days prior to the Non-Registration Event) provided the Equity Conditions are met. In addition, if there is not an effective registration statement covering all the Registrable Securities and the Company elects to register the issuance or resale of other Company securities, the Company is required to include such Registrable Securities in such registration statement filed with the SEC.
The Company also agreed, among other things, to indemnify the Purchasers from certain liabilities and fees and expenses of the Purchasers incident to the Company’s obligations under the Registration Rights Agreement, including certain liabilities under the Securities Act. The Purchasers have agreed to indemnify and hold harmless the Company and each of its directors, officers and persons who control the Company against certain liabilities that may be based upon written information furnished by the Purchasers to the Company for inclusion in a registration statement pursuant to the Registration Rights Agreement, including certain liabilities under the Securities Act.
| 8 |
| Table of Contents |
Other Terms
The Purchase Agreement contains customary representations, warranties and covenants by, among and for the benefit of the parties. Additionally, the Purchase Agreement provides for (i) liquidated damages in the event of the failure to timely (A) deliver our Common Stock upon conversion of the Notes or issuance of shares upon exercise of Warrants and (B) remove restrictive legends, (ii) a right to participate in future financings, (iii) a most-favored nation’s right with respect to future financings, and (iv) certain “down-round” protection. The Purchase Agreement also provides for indemnification of the Purchasers and its affiliates in the event that the Purchasers incur losses, liabilities, obligations, claims, contingencies, damages, costs and expenses related to the Company’s breach of any of its representations, warranties or covenants under the Purchase Agreement.
The officers and directors of the Company, Michael T. Redman and J. Donald Payne, as well as GHC Research Development Corporation (“GHC”), agreed for a period of fifteen months from August 3, 2017, not to sell, assign or otherwise transfer any of their shares of our Common Stock or our Series A Preferred Stock.
At no time will the Purchasers be entitled to convert any portion of the Convertible Notes or exercise any portion of the Warrants, to the extent that after such conversion and/or exercise, the Purchasers (together with their affiliates) would beneficially own more than 4.99% of the outstanding shares of our Common Stock as of such date, which limit may be increased to 9.99% at the election of the Purchasers but no greater than 9.99%.
Risk Factors
Investing in our Common Stock involves risks that include the speculative nature of the biopharmaceutical business, competition, need for additional capital and other material factors. You should read carefully the section of this prospectus entitled “Risk Factors” beginning on page 10 for an explanation of these risks before investing in our Common Stock. In particular, the following considerations may offset our competitive strengths or have a negative effect on our strategy or operating activities, which could cause a decrease in the price of our Common Stock and a loss of all or part of your investment:
· Our business is difficult to evaluate because of our limited operating history; · We have incurred net losses from inception and expect to continue to incur net losses; · We have no product revenue and don’t expect product revenue in the near future; · We need to raise capital to continue to fund working capital obligations; · The terms of our Convertible Notes may adversely affect our ability to raise additional capital; · The biopharmaceutical industry is intensely competitive; · Regulations that govern the biopharmaceutical industry are significant; · Our intellectual property rights may be challenged; · The selling security holders’ ability to sell a significant percentage of our Common Stock from time to time under this prospectus may have an adverse effect on the public market of our Common Stock; and · We may be unable to repay or redeem the non-converted portion of the Convertible Notes when due.
About This Offering
| Common Stock offered by selling security holder |
| 126,730,191 shares(1) beneficially held by the selling security holders. The selling security holders may from time to time sell some, all or none of the shares of Common Stock pursuant to which this prospectus is a part. |
|
|
|
|
| Shares outstanding prior to the offering |
| 103,536,703 shares of Company Common Stock and 63,038,284 shares of Series A Preferred Stock convertible into 63,038,284 shares of Company Common Stock, aggregating 166,574,987 voting capital stock issued and outstanding as of August 28, 2017. In addition, the selling security holders must convert indebtedness or exercise Warrants for the 126,730,191 shares beneficially held in order to sell the shares of Common Stock offered hereby. |
|
|
|
|
| Shares to be outstanding after the offering |
| 230,266,894 shares of Company Common Stock and 63,038,284 shares of Series A Preferred Stock convertible into 63,038,284 shares of Company Common Stock, aggregating 293,305,178 voting capital stock issued and outstanding as of August 28, 2017(2) |
|
|
|
|
| Use of proceeds |
| The selling security holder will receive all of the proceeds from the sale of shares of our Common Stock. We will not receive any proceeds from the sale of the Common Stock. |
|
|
|
|
| Risk Factors |
| The securities offered hereby involve a high degree of risk. See “Risk Factors.” |
|
|
| |
| Stock symbol |
| AEPP |
| 9 |
| Table of Contents |
__________
(1) The shares of Common Stock are comprised of the following: (i) with respect to the shares underlying the Convertible Notes, (A) 55,872,837 shares of Common Stock issuable upon conversion of the principal amount of $4,190,463 based on a conversion price of $0.075 per share pursuant to the monthly redemption payment schedule assuming timely payment and no default either in principal or interest payment obligations, (B) 5,633,847 shares of Common Stock issuable as payment of interest in lieu of cash, assuming timely payment of all interest obligations and no default either in principal or interest payment obligations, based on an interest conversion rate of $0.75 per share, and (C) 6,150,670 shares of Common Stock as a buffer in the event the monthly redemption payment or interest payment calculations are based on the lower of $0.075 per share and 75% of the average of the five lowest closing bid prices for the twenty consecutive trading days prior to such determination date; and (ii) 59,072,837 shares of Common Stock underlying the Warrants. (2) Assuming all shares offered hereby are issued upon conversion of the Convertible Notes and exercise of the Warrants as of the date of the prospectus.
Corporate Address
Our executive offices are located at 14405 Walters Road, Suite 780, Houston, Texas 77014, and our telephone number is (281) 402-3167.
This investment has a high degree of risk. Before you invest you should carefully consider the risks and uncertainties described below and the other information in this prospectus. If any of the following risks actually occur, our business, operating results and financial condition could be harmed and the value of our stock could go down. This means you could lose all or a part of your investment.
Risks Related to Our Financial Position and Capital Needs
We have incurred net losses since our inception and anticipate that we will continue to incur net losses for the foreseeable future.
Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval or be commercially viable. We are a clinical stage biopharmaceutical company with limited operating history. We have not completed our Phase I clinical trials and no assurances can be given that when, if ever, we will be able to. As a result we have not yet demonstrated an ability to complete clinical trials, obtain regulatory approvals, manufacture a commercial-scale drug or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. We may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown difficulties in achieving our business objectives. We have no product approved for commercial sale and have not generated any product revenues to date, and we continue to incur significant research and development and other expenses related to our ongoing operations and clinical development of Prolanta™. As a result, we are not and have never been profitable and have incurred losses in each period since our commencement of operations in 2007. We will need to raise substantial additional capital to fund operations and the failure to obtain capital may cause us to curtail or cease operations.
For the year ended December 31, 2016, we reported a net loss of $1.4 million; and as of December 31, 2016, we had an accumulated deficit of $19.0 million. For the six months ended June 30, 2017, we reported a net loss of $1.3 million; and as of June 30, 2017, we had an accumulated deficit of $20.3 million. We expect to continue to incur significant losses for the foreseeable future, and we expect these losses to increase as we continue our research and development of, and seek regulatory approvals for, Prolanta™. We may also encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues, if any. Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity and working capital.
We currently have no source of product revenue and may never achieve or maintain profitability.
Our ability to generate product revenue and become profitable depends upon our ability to successfully commercialize our sole product, Prolanta™. We do not anticipate generating revenue from the sale of Prolanta™ for the foreseeable future. Our ability to generate future product revenue also depends on a number of additional factors, including, but not limited to, our ability to:
· successfully complete the research and clinical development of, and receive regulatory approval for, Prolanta™;
| 10 |
| Table of Contents |
· launch, commercialize and achieve market acceptance of Prolanta™, and if launched independently, successfully establish a sales, marketing and distribution infrastructure; · continue to build a portfolio of product candidates through the acquisition or in-license of products, product candidates or technologies; · initiate preclinical and clinical trials for any additional product candidates that we may pursue in the future; · establish and maintain supplier and manufacturing relationships with third parties, and ensure adequate and legally compliant manufacturing of bulk drug substances and drug products to maintain that supply; · obtain coverage and adequate product reimbursement from third-party payors, including government payors; · establish, maintain, expand and protect our intellectual property rights; and · attract, hire and retain additional qualified personnel.
In addition, because of the numerous risks and uncertainties associated with drug development, we are unable to predict the timing or amount of increased expenses, and if or when we will achieve or maintain profitability. In addition, our expenses could increase beyond expectations if we decide to or are required by the FDA or foreign regulatory authorities to perform studies or trials in addition to those that we currently anticipate. Even if we complete the development and regulatory processes described above, we anticipate incurring significant costs associated with launching and commercializing Prolanta™. We will need to raise substantial additional capital to fund operations and the failure to obtain capital may cause us to curtail or cease operations.
Even if we generate revenues from the sale of Prolanta™, we may not become profitable and may need to obtain additional funding to continue operations or acquire additional products that will require additional funding to develop them. If we fail to become profitable or do not sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations or curtail operations.
We have limited cash on hand, and we will need substantial additional funding and if we are unable to raise additional capital when needed, we would be forced to delay, reduce or eliminate our clinical trials and curtail operations.
The Company believes that it has sufficient cash on hand to fund its operations through calendar 2017, and into early 2018. The exact duration that our liquidity will be sufficient to fund operations depends upon many factors, some of which are outside the control of the Company, and is difficult to predict. We expect our research and development expenses to increase in connection with our ongoing activities relating to Prolanta™, particularly as we focus on and proceed with our clinical trials for our Prolanta™ product. In addition, our expenses could increase beyond current expectations if applicable regulatory authorities, including the FDA, require that we perform studies in addition to those that we currently anticipate, assuming we successfully complete our Phase I clinical trials, commence and complete our proposed Phase II clinical trials. We currently estimate that we require approximately $2 million to complete the Phase I clinical trial, depending on the cost and availability of drug supplies. We will still need substantial additional capital in the future in order to complete the clinical trials and obtain regulatory approval of Prolanta™. Accordingly, we will need to raise additional capital to fund future operations and we expect to finance future cash needs through best-efforts public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Such funding may not be available on favorable terms, if at all. Failure to raise additional capital could cause us to curtail or cease operations and result in the loss of your investment in our Common Stock.
If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or to grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
Our forecast of the period of time through which our current capital will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
The terms of the Convertible Notes will restrict our operating flexibility.
Pursuant to the Purchase Agreement, the Company issued Convertible Notes to the Purchasers in an aggregate principal amount of $4,190,463 at a 15% discount. The Convertible Notes bear interest at a rate of 10% per annum, payable quarterly on November 1, 2017, February 1, 2018, and May 1, 2018 and thereafter, on a monthly basis until maturity. The interest is payable in cash or, at our option, subject to compliance with the Equity Conditions, in fully tradable Common Stock at the lesser of the then conversion price or a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the interest payment date. The final maturity of the Convertible Notes is November 1, 2018, but commencing on May 1, 2018 and on the first day of each month thereafter until maturity, we are required to redeem an amount of the Convertible Notes equal to 1/7th of the original principal amount, plus 10% of such monthly redemption amount. The principal payments shall be made in cash or, subject to the satisfaction of Equity Conditions, in our Common Stock valued at the lesser of the then conversion price or a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the amortization date. Accordingly, in the event that the Company issues shares of its Common Stock to pay principal and/or interest on the Convertible Notes, such payment through the issuance of shares of Common Stock may be dilutive to the Company. Moreover, satisfaction of the Equity Conditions may be difficult; therefore investors should assume that the Company will be required to utilize cash for its payment obligations pursuant to the Convertible Notes. The Company will need to raise additional capital to fund principal and interest obligations of the Convertible Notes and there is no assurance that it will be successful in this endeavor. Failure to raise additional capital could cause a default in the Convertible Notes and result in a foreclosure by the Convertible Note holders on substantially all of our assets.
| 11 |
| Table of Contents |
The Convertible Notes are convertible at the option of the Purchasers at a conversion price equal to the lesser of (i) $0.075 per share of Common Stock, (ii) 75% of the 10-day average closing bid price of our Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of our Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share). The Convertible Notes contain customary anti-dilution protection, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both conversion price and share amounts to be issued upon conversion). Accordingly, the conversion price of the Convertible Notes is subject to future market prices of the Company Common Stock (of which the Company has no control) as well as the price of future Common Stock issuances (either in connection with any potential financings or in lieu of cash for services to be rendered) which could result in the conversion price being further adjusted to a price lower than the floor of $0.0375 per share with additional shares being issued upon conversion per the anti-dilution provisions. These provisions may make it more difficult and more expensive for us to raise capital in the future and may result in further dilution to shareholders.
In the event of a closing of an $8 million financing in connection with a change in listing of our Common Stock to Nasdaq or the NYSE, the Convertible Notes shall be subject to mandatory conversion into our Common Stock at a 30% discount to the offering price. There can be no assurance that the Company will be able to satisfy initial listing requirements of the Nasdaq or NYSE. We shall be required to offer to prepay in cash the aggregate principal amount of the Convertible Notes at 120% of the principal amount thereof plus any unpaid accrued interest to the date of repayment, at maturity, on the sale of substantially all of our assets, upon a change of control, upon a qualified offering, or upon a “fundamental transaction”; in such an event, the Purchasers shall have the right to convert the Convertible Notes prior to the date of any such prepayment.
The Convertible Notes contain standard negative covenants customary for transactions of this type. These negative covenants may preclude or restrict the ability of the Company to effect future debt and convertible debt financings without the prior approval of holders of the majority principal amount of the Convertible Notes. The events of default are also customary for transactions of this type, including default in timely payment of principal or interest, failure to observe or perform any covenant or agreement contained in the Convertible Note and Transaction Documents, the commencement of bankruptcy or insolvency proceedings, failure to timely deliver conversion shares underlying the Convertible Notes, failure to timely file Exchange Act filings, and failure to satisfy certain Equity Conditions. In the event that the Company triggers one of these event of default provisions, the holders of the Convertible Notes have the ability to foreclose on substantially all of the assets of the Company which would result in the cessation of our operations.
Equity Conditions mean, during the period in question, (a) we shall have duly honored all conversions and redemptions of the Convertible Notes, (b) we shall have complied with all conversion and other provisions of the Convertible Notes and shall have paid all liquidated damages and other amounts owing in respect of this Convertible Note, (c)(A) there is an effective registration statement pursuant to which the holder is permitted to utilize the prospectus thereunder to resell at least the number of shares of Common Stock issuable pursuant to the Transaction Documents at such time or (B) all of the underlying shares may be resold pursuant to Rule 144 without volume or manner-of-sale restrictions or current public information requirements, (d) our Common Stock is trading on a trading market, (e) there is a sufficient number of authorized but unissued and otherwise unreserved shares of AEPP Common Stock for the issuance of five times all of the shares issuable pursuant to the Convertible Notes and Warrants, (f) there is no existing event of default or no existing event which, with the passage of time or the giving of notice, would constitute an event of default, (g) the issuance of the shares in question would not violate the beneficial ownership limitations in the Transaction Documents, (h) there has been no public announcement of a pending or proposed fundamental transaction or change of control transaction that has not been consummated, (i) the holder is not in possession of any information provided by the Company that constitutes, or may constitute, material non-public information, (j) our Common Stock is DWAC eligible and not subject to a DTC “chill;” and (k) for each trading day in a period of 10 consecutive trading days prior to the applicable date in question (A) the daily dollar trading volume for our Common Stock on the principal trading market exceeds $75,000 per trading day and (B) the weighted average stock price for our Common Stock on the principal trading market is above 50% of the closing stock price on August 3, 2017 (as adjusted for all stock splits stock dividends, stock combinations, reclassifications and other transactions). The satisfaction of the Equity Conditions may be difficult to meet as many of these conditions involve events outside of the Company’s control. The failure to meet these Equity Conditions precludes the Company’s ability to utilize its shares of Common Stock to satisfy payment obligations pursuant to the Convertible Notes.
We have the right at any time to redeem all, but not less than all, of the total outstanding amount of principal amount then remaining under the Convertible Notes at a price equal to 120% of such principal amount.
| 12 |
| Table of Contents |
Terms of our existing $4,190,463 senior debt and warrants may impact our ability to obtain additional financing.
The Convertible Notes are senior secured obligations of the Company. The terms of the Convertible Notes preclude the Company from obtaining any additional debt senior to or pari passu with the Convertible Notes. Additionally, the Convertible Notes are secured by substantially all of our assets and guaranteed by Oncolix. In the event of our default with respect to the Convertible Notes, the holders have the right to foreclose on substantially all of our assets which likely would result in us ceasing operations. The terms of the Convertible Notes may adversely affect the Company’s ability to obtain additional debt or equity financing to fund additional working capital needs, including the payment obligations of the Convertible Notes.
Registration Rights Agreement will affect future capital raises.
The Registration Rights Agreement entered into in connection with the issuance of the Convertible Notes requires the Company to register the resale of all the underlying shares of our Common Stock issuable upon conversion of the Convertible Notes and exercise of the Warrants. The Company has filed this registration statement, and intends to use its best efforts to file any additional registration statements, to register the resale of these underlying shares under the Securities Act as soon as practical. The availability of these shares for sale in the public markets will likely have a depressive effect on any market price of our Common Stock. The Registration Rights Agreement contains liquidated damage provisions in the event that the Company fails to timely comply with the registration requirements therein. Additionally, the registration obligations may adversely impact our ability to issue additional equity or convertible debt securities to fund additional working capital needs, including the payment obligations of the Convertible Notes.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
We have incurred substantial losses during our history. We do not expect to become profitable in the near future, and we may never achieve profitability. Unused losses generally are available to be carried forward to offset future taxable income, if any, until such unused losses expire. Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research tax credits) to offset its post-change taxable income or taxes may be limited. Our largest stockholder, GHC, is considering a reorganization that would limit our ability to utilize certain tax attributes. As a result, our ability to use our pre-change NOLs to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us.
The current economic climate and legislative and regulatory changes in the U.S. and internationally creates uncertainties for the Company.
The uncertain economic climate in the U.S. and internationally may, therefore, have a material adverse effect on the healthcare industry in general and the Company in particular. In addition, proposed healthcare legislation in the United States, and the uncertainty of its future regulatory framework, may have a substantial impact on the Company and its operations. The Company cannot predict at this time what the potential impact may be or whether the effects on the Company will be positive or negative. If the Company suffers adverse effects it could cause the Company to fail to meet, or face delays in meeting, its marketing and sales objectives. Any such failures or delays will create additional financial uncertainties and difficulties for the Company.
We face significant competition from other pharmaceutical and biotechnology companies and they or others may also discover, develop or commercialize products before or more successfully than we do.
The development and commercialization of a drug candidate is highly competitive. We face competition with respect to our current product candidate, and will face competition with respect to any product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. We have competitors many of which have substantially greater name recognition, commercial infrastructure and financial, technical and personnel resources than we have. Established competitors may invest heavily to quickly discover and develop products that could make our products obsolete or uneconomical. Any new product that competes with an approved product may need to demonstrate compelling advantages in efficacy, cost, convenience, tolerability and safety to be commercially successful. Other competitive factors, including generic competition, could force us to lower prices or could result in reduced sales. In addition, new products developed by others could emerge as competitors to our products. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
| 13 |
| Table of Contents |
There can be no assurance that Prolanta™ will successfully complete its Phase 1 and later stage clinical trials.
There can be no assurance that we will be able to successfully complete such Phase 1 clinical trials or if so, that we will obtain regulatory approval for our later stage clinical trials and/or successfully complete such later stage clinical trials and/or obtain all required regulatory approvals. Moreover, no assurances can be given we will be able to commercialize Prolanta™ or that we will ever generate sustained revenue or achieve profitability. Our ability to operate profitably in the future will depend upon, among other items, our ability to (i) successfully complete all required clinical trials for Prolanta™, (ii) obtain regulatory approval from the FDA or other regulatory agencies for Prolanta™, (iii) scale up our business and operational structure, (iv) commercialize and market Prolanta™, and (v) successfully gain market acceptance of Prolanta™. There can be no assurance that we will successfully achieve any of the foregoing. Our prospects must be considered in light of the numerous risks, expenses, delays and difficulties frequently encountered in the intensely competitive and high risk markets in which Prolanta™ is intended to compete. If we are unable to develop and commercialize Prolanta™, we will not achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability.
We will need substantial additional funding and if we are unable to raise capital when needed, we would be forced to delay, reduce or eliminate our product development and commercialization plans.
We expect our research and development expenses to increase in connection with our ongoing activities. In addition, our expenses could increase beyond expectations if applicable regulatory authorities, including the FDA, require that we perform studies in addition to those that we currently anticipate, in which case the timing of any potential product approval may be delayed. We will still need substantial additional capital in the future in order to complete the development and commercialization of Prolanta™. Until we can generate a sufficient amount of product revenue, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Such funding, if needed, may not be available on favorable terms, if at all. Failure to obtain additional capital could cause us to curtail or cease operations.
If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or to grant licenses on terms that may not be favorable to us.
We contract with a third party for the manufacture of Prolanta™ for clinical trials and we expect to continue to do so in connection with the commercialization of Prolanta™ and for clinical trials and commercialization of any other product candidates that we develop. Any failure by the Manufacturer to produce acceptable supplies for us or to comply with FDA or comparable foreign regulations may delay or impair our ability to initiate or complete our clinical trials, obtain regulatory approvals to market, or sell any resulting product.
We do not currently have nor do we plan to build the internal infrastructure or capability to manufacture Prolanta™ or any future product candidates for use in the conduct of our clinical trials or for commercial supply. We currently rely on a third-party contract manufacturer to manufacture clinical supplies of Prolanta™. We expect to rely upon this manufacturer or another contract manufacturer to manufacture clinical trial supplies and commercial quantities of Prolanta™ if and when we receive approval for marketing by applicable regulatory authorities. Our contract manufacturer subcontracts to other third-party manufacturers certain aspects of the manufacturing of Prolanta™, including the filling of vials and the lyophilization (freeze-drying) of the final drug product.
Reliance on a contract manufacturer to manufacture Prolanta™ entails risks, including:
· manufacturing delays if the contract manufacturer gives greater priority to the supply of other products over Prolanta™; · manufacturing delays or increased costs if the contract manufacturer does not satisfactorily perform according to the terms of any agreement between us; · the failure of the contract manufacturer to comply with applicable regulatory requirements; · the possible misappropriation of our proprietary information, including our trade secrets and know-how.
| 14 |
| Table of Contents |
We are currently conducting a Phase I clinical trial of Prolanta™ for the treatment of ovarian cancer. The first of three dosing groups has been completed, and we must obtain additional supplies of Prolanta™ to complete this and future clinical trials. We will seek these drug supplies from our contract manufacturer. We also intend to pursue alternative sources of manufacturing to evaluate the feasibility and cost of other suppliers.
In the event we develop or acquire additional product candidates, we expect to make similar arrangements with a other third-party contract manufacturers for such product candidates, and these arrangements will entail similar risks.
If we are unable to make satisfactory arrangements with contract manufacturers, we may be required to alter our plans and we may have to establish our own manufacturing capabilities. This would require substantial additional funds, personnel and other resources to build, lease or operate any manufacturing facility.
Third-party manufacturers are required to comply with Current Good Manufacturing Practices (“cGMPs”) and similar regulatory requirements outside the United States. Facilities used by our contract manufacturer must be approved by the FDA before potential approval of Prolanta™. Similar regulations apply for use or sale in foreign countries. We do not control the manufacturing process and are completely dependent on contract manufacturers for compliance with the applicable regulatory requirements for the manufacture of Prolanta™. If a contract manufacturer cannot successfully manufacture material that conforms to the strict regulatory requirements of the FDA and any applicable foreign regulatory authority, they will not be able to secure the applicable approval for their manufacturing facilities. If these facilities are not approved for commercial manufacture, we may need to find alternative manufacturing facilities, which could result in delays in obtaining approval for the applicable product candidate as well as substantial additional expense.
In addition, contract manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. Failure by contract manufacturers to comply with applicable cGMPs or other regulatory requirements could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply and criminal prosecutions, any of which could significantly and adversely affect supplies of our Prolanta™ and have a material adverse impact on our business, financial condition and results of operations.
Our current and anticipated future dependence upon others for the manufacture of Prolanta™ and any other product candidate that we develop may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
If clinical trials of Prolanta™ or of any future product candidate that we advance to clinical trials fail to demonstrate safety and efficacy to the satisfaction of the FDA or comparable foreign regulatory authorities or do not otherwise produce favorable results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of Prolanta™ or any other product candidate.
We are not permitted to commercialize, market, promote, or sell Prolanta™ in the United States without obtaining marketing approval from the FDA or in other countries without obtaining approvals from comparable foreign regulatory authorities, such as the EMA, and we may never receive such approvals. We must complete extensive clinical trials to demonstrate the safety and efficacy of Prolanta™ in humans before we will be able to obtain these approvals. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We have not previously submitted an NDA to the FDA or similar drug approval filings to comparable foreign regulatory authorities for Prolanta™.
The clinical development of Prolanta™ and any other product candidates is susceptible to the risk of failure inherent at any stage of drug development, including failure to achieve efficacy in a trial or across a broad population of patients, the occurrence of severe adverse events, failure to comply with protocols or applicable regulatory requirements, and determination by the FDA or any comparable foreign regulatory authority that a drug product is not approvable. The outcome of preclinical studies and any early clinical trial may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. For example, although Prolanta™ demonstrated an acceptable safety profile and efficacy in preclinical studies, we may nonetheless fail to demonstrate the safety or tolerability of Prolanta™ in our Phase 1 clinical trial, and may fail to demonstrate efficacy in any of our clinical trials.
In addition, preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for the product candidates. Even if we believe that the results of our clinical trials warrant marketing approval, the FDA or comparable foreign regulatory authorities may disagree and may not grant marketing approval of our product candidates.
| 15 |
| Table of Contents |
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We cannot assure you that any of the clinical trials that we may conduct will demonstrate consistent or adequate efficacy and safety to obtain regulatory approval to market our product candidates.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent us from obtaining regulatory approval for Prolanta™, including:
· clinical trials of our product candidates may produce unfavorable or inconclusive results; · we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs; · the number of patients required for clinical trials may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; · our third-party contractors, including the manufacturer for Prolanta™ or the CROs conducting clinical trials on our behalf, may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; · regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; · we may have delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; · we may have to suspend or terminate clinical trials of Prolanta™ for various reasons, including lack of capital, a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of our product candidate; · regulators or institutional review boards may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks, undesirable side effects or other unexpected characteristics of the product candidate; · the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of our third-party manufacturer; · the supply or quality of Prolanta™ or other materials necessary to conduct clinical trials may be insufficient or inadequate; and · the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval
If we are required to conduct additional clinical trials or other testing of Prolanta™ beyond the trials and testing that we contemplate, if we are unable to successfully complete clinical trials of Prolanta™ or other testing, if the results of these trials or tests are unfavorable or are only modestly favorable or if there are safety concerns associated with Prolanta™, we may:
· be delayed in obtaining marketing approval for Prolanta™; · not obtain marketing approval at all; · obtain approval for indications or patient populations that are not as broad as intended or desired; obtain approval with labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings; · be subject to additional post-marketing testing or other requirements; · remove the product from the market after obtaining marketing approval; or · curtail operations.
Our product development costs will also increase if we experience delays in testing or marketing approvals and we may be required to obtain additional funds to complete clinical trials. We cannot assure you that our clinical trials will begin as planned or be completed on schedule, if at all, or that we will not need to restructure our trials after they have begun. Significant clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize Prolanta™ or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates, which may harm our business and results of operations. In addition, many of the factors that cause, or lead to, clinical trial delays may ultimately lead to the denial of regulatory approval of Prolanta™.
If we experience delays or difficulties in the enrollment of patients in clinical trials, if enrolled patients do not comply with the clinical trial protocol, or if clinicians participating in our clinical trials do not follow acceptable procedures, our development program will be delayed or halted and the receipt of necessary regulatory approvals could be delayed or prevented and we could have insufficient resources to complete our commercialization activities.
| 16 |
| Table of Contents |
We may not be able to initiate or continue clinical trials for Prolanta™ if we are unable to locate and enroll a sufficient number of eligible patients to participate in clinical trials as required by the FDA or comparable foreign regulatory authorities, such as the EMA. Patient enrollment is a significant factor in the timing of clinical trials, and is affected by many factors, including:
· the size and nature of the patient population; · the severity of the disease under investigation; · the proximity of patients to clinical sites; · the eligibility criteria for the trial; · the design of the clinical trial; and · competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages and risks of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating.
There is substantial competition among drug companies for the enrollment of ovarian cancer patients. The designation of Prolanta™ as a Orphan Drug relates to the limited number of such patients, which makes enrollment in clinical trials highly competitive. Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays, increasing our costs, and would have an adverse effect on our Company.
Once enrolled in a clinical trial, each patient is required to comply with the clinical protocol, including the proper dosing of Prolanta™, scheduled physician visits, and required medical and laboratory tests. The clinicians who participate in the study must also follow the procedures in the clinical protocol, including maintaining proper records. The failure of any enrolled patient or the participating clinician to comply with the clinical protocol may delay the completion of the clinical trial, require an increase in enrolled patients, increase our development costs, affect the results of the study, and potentially adversely affect our commercialization timelines and plans.
Serious adverse events or undesirable side effects or other unexpected properties of Prolanta™ or any other product candidate may be identified during development or after approval, if obtained, that could delay, prevent or cause the withdrawal of the product candidates’ regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if obtained.
Serious adverse events or undesirable side effects caused by, or other unexpected properties of, of Prolanta™ or any other product candidate could cause us, an institutional review board, or regulatory authorities to interrupt, delay or halt our clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or comparable foreign regulatory authorities. If Prolanta™ or any of our other product candidates are associated with serious adverse events or undesirable side effects or have properties that are unexpected, we may need to abandon their development or limit development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Many drugs that initially showed promise in clinical or earlier stage testing have later been found to cause undesirable or unexpected side effects that prevented further development of the drug
If an event occurs after Prolanta™ or any other product candidates are approved, a number of potentially significant negative consequences may result, including:
· regulatory authorities may withdraw the approval of such product; · regulatory authorities may require additional warnings on the label; · we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; · we could be sued and held liable for harm caused to patients; and · our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance, if approved, or could substantially increase commercialization costs and expenses, which could delay or prevent us from generating revenues from the sale of our products and harm our business and results of operations.
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time-consuming and inherently unpredictable, and if we or our collaborators are ultimately unable to obtain timely regulatory approval for our product candidates, our business will be substantially harmed.
The regulatory approval process is expensive and, while the time required to gain FDA and foreign regulatory approval is uncertain, it may take years. Regulatory approval is unpredictable and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of preclinical and clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We may be required to undertake and complete certain additional preclinical studies to generate toxicity and other data required to support the submission of a New Drug Application, or NDA, to the FDA or other regulatory authority. We have not obtained regulatory approval for Prolanta™. and it is possible that Prolanta™ or any future product candidate will ever obtain regulatory approval. Furthermore, approval in the United States by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA.
| 17 |
| Table of Contents |
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
· the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; · we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that Prolanta™ is safe and effective for its proposed indication; · the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; · we may be unable to demonstrate that Prolanta™’s clinical and other benefits outweigh its safety risks; · the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; · the data collected from clinical trials may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; · the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of our third-party manufacturer; and · the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval
We cannot be assured that after spending substantial time and resources, we will obtain regulatory approval. Even if we were to obtain approval, regulatory authorities may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve Prolanta™ with a label that does not include the labeling claims necessary or desirable for the successful commercialization.
The designation of Prolanta™ as an Orphan Drug for the treatment of ovarian cancer may not be affirmed by FDA if and when we commence any marketing of the product.
The FDA has granted Orphan Drug designation to Prolanta™ for the treatment of ovarian cancer. Under the Orphan Drug Act in the United States, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is defined by the FDA as a disease or condition that affects fewer than 200,000 individuals in the United States. In the EU, orphan drug designation is provided for a drug that is intended to diagnose, prevent or treat a life-threatening or chronically debilitating condition which affects no more than 5 in 10,000 individuals in the EU (approximately 245,000 individuals) and for which no satisfactory method of diagnosis, prevention or treatment of the condition already exists, or if such method does exist, that the orphan product must be of significant benefit to the patient population over existing products. The company that obtains the first FDA approval for a designated orphan drug indication receives marketing exclusivity for use of that drug for that indication for a period of seven years in the US and 10 years for the 27 member states in the EU. The orphan drug designation also allows a reduction in select regulatory fees. Orphan drug exclusive marketing rights may be lost if the FDA later determines that the request for designation was materially defective, or if the manufacturer is unable to assure sufficient quantity of the drug. Orphan drug designation does not shorten the development or regulatory review time of a drug.
Orphan drug exclusivity may not prevent other market entrants. A different drug, or, under limited circumstances, the same drug may be approved by the FDA for the same orphan indication. The limited circumstances include an inability to supply the drug in sufficient quantities or where a new formulation of the drug has shown superior safety or efficacy. As a result, if Prolanta™ were approved for the treatment of ovarian cancer, other drugs could still be approved for use in treating the same indication covered by our product, which could create a more competitive market for us.
Moreover, due to the uncertainties associated with developing pharmaceutical products, we may not be the first to obtain marketing approval for any orphan drug indication, and as a result we may not be able to obtain marketing exclusivity. In such case, our competitive position would be limited to claims we can assert, if any, under our existing or pending patents. If a competitor obtains regulatory approval for a prolactin receptor antagonist equivalent to Prolanta™ for the same indication we are targeting before we do, and we are unsuccessful in challenging such product using our patent rights, we would be blocked from obtaining approval for that indication for seven years, unless our product is a new formulation of the drug that has shown superior safety or efficacy, or the competitor is unable to supply sufficient quantities.
There can be no assurances that products for the treatment of ovarian cancer will continue to be eligible for Orphan Drug designation, that the rules governing orphan drugs will not be amended or revoked, or that Prolanta™ will continue to qualify as an orphan drug. Accordingly, we may not receive any future benefits from the FDA designation of Prolanta™ as an orphan drug. Such as tax credits, reduced filing fees or market exclusivity. Additionally, other regulatory authorities, such as the EMA, may not grant Prolanta™ a similar designation.
| 18 |
| Table of Contents |
We are dependent on our executive officers to manage our business effectively and if we are not able to retain such persons, our business will be materially adversely affected.
Our operations are wholly dependent on the skills, efforts and continued services of our two executive officers. These persons also are our only employees. While we have entered into employment agreements with such persons, any of them may terminate their employment with us at any time on short notice. Accordingly, there can be no assurance that such persons will remain associated with us. The loss of one or both of such persons would have a material adverse effect on our business.
Risks Related to Regulatory Approval and Other Governmental Regulations
We are subject to government regulation, and we may not receive approval for our proposed products in the United States or other major markets on a timely basis, if at all.
All aspects of our development, manufacturing and clinical trials of Prolanta™ are, and will continue to be, subject to extensive and rigorous scrutiny and regulation by the FDA and by comparable agencies in foreign countries. In the United States, the FDA regulates the introduction of biopharmaceutical products, as well as manufacturing, labeling and record keeping procedures for such products. The process of obtaining marketing clearance or approval for new medical products from the FDA is costly and time consuming, and there can be no assurance that such approval will be granted for our proposed products on a timely basis, if at all, or that the FDA review will not involve delays that would adversely affect our ability to commercialize our proposed products. Even if regulatory clearance or approval to market a product is obtained from the FDA, this clearance or approval may entail limitations on the indicated uses of the product. Marketing clearance or approval can also be withdrawn by the FDA due to failure to comply with regulatory standards or the occurrence of unforeseen problems following initial clearance.
Even if our proposed products are approved by regulatory authorities, if we fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our proposed products, it could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continual review and periodic inspections by the FDA and other regulatory bodies. Even if regulatory approval of our proposed products is granted in the United States, the approval may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for costly post-marketing testing and surveillance to monitor the safety or effectiveness of the product. Later discovery of previously unknown problems with our proposed products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturer or manufacturing processes, or failure to comply with regulatory requirements, may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recall, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
Risks Related to Our Intellectual Property
Our competitive position is contingent upon the protection of our intellectual property.
Our current patents, and any patents we obtain in the future might be invalidated or circumvented by third parties. If any challenges are successful, competitors might be able to market products and use manufacturing processes that are substantially similar to ours. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by consultants, vendors or former or current employees, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized use and disclosure of our intellectual property is difficult, and we do not know whether the steps we have taken to protect our intellectual property will be adequate. In addition, the laws of many foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. If our intellectual property is not adequately protected against competitors' products and methods, our competitive position could be adversely affected, as could our business.
Our commercial success, if any, will be significantly harmed if we infringe the patent rights of third parties or if we breach any agreements that we have entered into with regard to our technology or business.
The biopharmaceutical industry is characterized by frequent and extensive litigation regarding patents and other intellectual property rights. Many biopharmaceutical companies with substantially greater resources than us have employed intellectual property litigation as a way to gain a competitive advantage. We may become involved in litigation, interference proceedings, oppositions, reexamination, protest or other potentially adverse intellectual property proceedings as a result of alleged infringement by us of the rights of others or as a result of priority of invention disputes with third parties. Third parties may also challenge the validity of any of our issued patents. Similarly, we may initiate proceedings to enforce our patent rights and prevent others from infringing our intellectual property rights. In any of these circumstances, we might have to spend significant amounts of money, time and effort defending our position and we may not be successful. In addition, any claims relating to the infringement of third-party proprietary rights or proprietary determinations, even if not meritorious, could result in costly litigation, lengthy governmental proceedings, divert management's attention and resources, or require us to enter into royalty or license agreements that are not advantageous to us. We may not have sufficient resources to enforce our intellectual property rights or to defend our patents against a challenge.
| 19 |
| Table of Contents |
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We require our employees to execute appropriate confidentiality and assignment-of-inventions agreements with us. These agreements typically provide that all materials and confidential information developed or made known to the individual during the course of the individual's relationship with us be kept confidential and not disclosed to third parties except in specific circumstances and that all inventions arising out of the individual's relationship with us will be our exclusive property. Additionally, we seek to have our consultants and advisors execute similar confidentiality and assignment-of-inventions agreements with us, but in some instances these agreements have not included assignment-of-invention provisions. These agreements may be breached, and in some instances, we may not have an appropriate remedy available for breach of the agreements. Furthermore, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology.
Claims of infringement or misappropriation of the intellectual property rights of others could prohibit us from protecting our rights to, or use and commercialization of, our proposed products, which could harm our business.
The biopharmaceutical industry has been characterized by extensive litigation regarding patents and other intellectual property rights, and companies in the industry have used intellectual property litigation to gain a competitive advantage. We may become a party to patent infringement claims and litigation or interference proceedings declared by the U.S. Patent and Trademark Office to determine the priority of inventions. The defense and prosecution of these matters are both costly and time consuming. Additionally, we may need to commence proceedings against others to enforce our patents, to protect our trade secrets or know-how or to determine the enforceability, scope and validity of the proprietary rights of others. These proceedings would result in substantial expense to us and significant diversion of effort by our technical and management personnel.
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our product candidates and the methods used to manufacture them, as well as successfully defending these patents against third-party challenges, should any be brought. Our ability to stop third parties from making, using, selling, offering to sell or importing our products is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We have filed and are actively pursuing patent applications for our product candidates and manufacturing processes. The patent positions of pharmaceutical and biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biopharmaceutical patents has emerged to date in the United States. The biopharmaceutical patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents we own or to which we have a license from a third-party. Further, if any of our patents are deemed invalid and unenforceable, it could impact our ability to license our technology and, as noted previously, fend off competitive challenges.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
· others may be able to make compositions or formulations that are similar to our product candidates but that are not covered by the claims of our patents; · we might not have been the first to make the inventions covered by our issued patents or pending patent applications; · we might not have been the first to file patent applications for these inventions; · others may independently develop similar or alternative technologies or duplicate any of our technologies; · it is possible that our pending patent applications will not result in issued patents; · our issued patents may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties; · we may not develop additional proprietary technologies that are patentable; or · the patents of others may have an adverse effect on our business.
| 20 |
| Table of Contents |
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
We may infringe or be alleged to infringe intellectual property rights of third parties.
Our products or product candidates may infringe on, or be accused of infringing on, one or more claims of an issued patent or may fall within the scope of one or more claims in a published patent application that may be subsequently issued and to which we do not hold a license or other rights. Third parties may own or control these patents or patent applications in the United States and abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we are found to infringe the patent rights of a third party, or in order to avoid potential claims, we or our collaborators may choose or be required to seek a license from a third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms.
Risks Associated with Our Common Stock
There is not now, and there may never be, an active market for our Common Stock.
Shares of our Common Stock have historically been thinly traded. Currently there is a limited, sporadic and highly volatile market for our Common Stock, and no active market for our Common Stock may develop in the future. As a result, our stock price as quoted by the OTC Pink may not reflect an actual or perceived value. Moreover, several days may pass before any shares are traded; meaning that the number of persons interested in purchasing our common shares at or near ask prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including, but not limited to:
· we are a small company that is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume; and · stock analysts, stock brokers and institutional investors may be risk-averse and reluctant to follow a company such as ours that faces substantial doubt about its ability to continue as a going concern or to purchase or recommend the purchase of our shares until such time as we become more viable.
As a result, an investor may find it difficult to dispose of, or to obtain accurate quotations of the price of, our Common Stock. Accordingly, investors must assume they may have to bear the economic risk of an investment in our Common Stock for an indefinite period of time, and may lose their entire investment. There can be no assurance that a more active market for our Common Stock will develop, or if one should develop, there is no assurance that it will be sustained. This severely limits the liquidity of our Common Stock and would likely have a material adverse effect on the market price of our Common Stock and on our ability to raise additional capital.
We cannot assure that our Common Stock will become liquid or that it will be listed on a national securities exchange.
Until our Common Stock is listed on a national securities exchange such as Nasdaq or the NYSE, we expect our Common Stock to remain eligible for quotation on the OTC Pink. If we fail to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our Common Stock, which may further affect the liquidity of our Common Stock. This would also make it more difficult for us to raise capital.
We may issue additional shares of preferred stock.
Our Articles of Incorporation authorizes the issuance of blank check preferred stock with designations, rights and preferences determined from time to time by the board of directors. There are currently 62,138,680 shares of Series A Preferred Stock issued and outstanding and the Company has reserved the balance of the preferred shares for issuance upon exercise of outstanding warrants to purchase shares of Series A Preferred Stock. The board of directors expects to increase the amount of blank-check preferred stock (this will require shareholder approval) to reserve additional shares of Series A Preferred Stock as well as additional shares of “blank-check” preferred stock for future issuance. Accordingly, our board of directors is expected to be empowered, without stockholder approval, to issue additional preferred stock with dividend, liquidation, conversion, voting, or other rights which could adversely affect the voting power or other rights of the holders of the Common Stock. In the event of issuance, the preferred stock could be utilized, under certain circumstances, as a method of discouraging, delaying or preventing a change in control of the Company.
| 21 |
| Table of Contents |
Future sales of our Common Stock could lower our stock price.
We expect to sell additional shares of Common Stock to fund future working capital obligations. We cannot predict the size of future issuances of our Common Stock or the effect, if any, that future issuances and sales of shares of our Common Stock will have on the market price of our Common Stock. Sales of substantial amounts of our Common Stock, or the perception that such sales could occur, may adversely affect prevailing market prices for our Common Stock. Moreover, sales of our Common Stock by existing shareholders could also depress the price of our Common Stock.
Rule 144 will not be available until August 2018 and this may affect trading in our stock before and after this date.
As a result of the Merger, AEPP ceased being a “shell” company under the SEC’s rules and Rule 144 will be available commencing in August 2018. The unavailability of Rule 144 until August 2018 may impact the trading market for the Company Common Stock prior to and after August 2018.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the near future, which could cause the market price of our Common Stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our Common Stock in the public market could occur at any time, either from holders of shares that are in the public float or as a result of registering the issuance of shares to be issued upon conversion of the Convertible Notes and exercise of the Warrants. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our Common Stock. As of this date, there are approximately 103,536,703 shares of Common Stock outstanding. Of such number, approximately 31,765,903 shares may be sold in the public market immediately without restriction and the balance are currently restricted as a result of securities laws or lock-up agreements until at least August 2018. Additionally, there are 63,038,284 shares of Series A Preferred Stock that are currently convertible into 63,038,284 shares of Common Stock eligible to be resold pursuant to Rule 144 commencing in August 2018. In addition, upon the exercise of warrants to acquire Series A Preferred Stock or the exercise of other warrants and options to acquire Common Stock, 62,064,664 shares of Common Stock could be issued and become eligible to be resold pursuant to Rule 144 commencing in August 2018.
Our Common Stock is subject to the “penny stock” rules of the SEC, which makes transactions in our Common Stock cumbersome and may reduce the value of an investment in the stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require:
· that a broker or dealer approve a person’s account for transactions in penny stocks; and · the broker or dealer receives from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased.
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
· obtain financial information and investment experience objectives of the person; and · make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth:
· the basis on which the broker or dealer made the suitability determination; and · that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
22 Table of Contents
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of Common Stock and cause a decline in the market value of stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
The price of our Common Stock will remain volatile, which could lead to losses by investors and costly securities litigation.
The trading price of our Common Stock is likely to be highly volatile and could fluctuate in response to factors such as:
· actual or anticipated variations in our operating results; · announcements of developments by us, our strategic partners or our competitors; · announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; · adoption of new accounting standards affecting our Company’s industry; · additions or departures of key personnel; · sales of our Common Stock or other securities in the open market; · our ability to acquire seismic data and other intellectual property on commercially reasonable terms and to defend such intellectual property from third party claims; · litigation; and · other events or factors, many of which are beyond our control.
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of companies’ securities, securities class action litigation has often been initiated against those companies. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management’s attention and resources, which could harm our business and financial condition.
We do not anticipate dividends to be paid on our Common Stock.
Cash dividends have never been declared or paid on our Common Stock, and we do not anticipate such a declaration or payment for the foreseeable future.
The selling security holders will have the potential of selling a significant percentage of our Common Stock from time to time under this prospectus, which may have an adverse impact on our trading and the market price of our shares.
The potential number of shares of Common Stock that may be sold by the selling security holders may have an adverse effect on the public market of our stock. The holders, however, have entered into an agreement with the Company that they will not at any time hold more than 4.99% of our Common Stock, which amount may be increased to 9.99% at the election of the holders, but not in excess of 9.99%, which is referred to as the blocker provision. Because of the large number of shares that may be issued from time to time on conversion of the Convertible Notes and/or exercise of the Warrants, there may be an adverse effect on the market because of the quantity and regularity of conversion and/or exercise and sale of those shares, or even the potential of those shares being sold. Although there are the blocker provisions, investors may disregard these limits when evaluating our Common Stock available to be sold in the public market. Therefore, there may be limited demand and excessive price and volume volatility.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus includes forward-looking statements. These statements relate to future events or future financial performance of the Company including among others (i) completion of our Phase I and future clinical trials, (ii) anticipated trends in the Company’s financial condition and results of operations, (iii) the financial projections contained herein, (iv) the possible and actual results of the Company’s efforts to obtain and maintain various approvals from the FDA relating to its products, and (v) the Company’s business strategy for, among other things, testing, developing, producing, distributing and in the future possibly selling and/or leasing its product candidates and its other proposed products. These forward-looking statements are based on the Company’s expectations as of the date of this prospectus and are subject to a number of known and unknown risks and uncertainties. Actual results could differ significantly from these forward-looking statements. In some cases, you can identify forward-looking statements by words including, but not limited to, “estimate,” “project,” “intend,” “forecast,” “anticipate,” “plan,” “planning, “expect,” “believe,” “will,” “will likely,” “should,” “could,” “would,” “may” or the negative of such terms and other comparable terminology. All such forward-looking statements involve risks and uncertainties and are not guaranties of future performance. These factors may include risks unknown to us currently and/or that may arise in the future as well as certain known risks including, but not limited to, those risks described in detail in the section herein captioned “Risk Factors,” and other information set forth herein. In addition to the items described in this prospectus under the heading “Risk Factors,” many important factors affect our ability to achieve our stated objectives, including, among other things:
· failure to complete our Phase I clinical trials, or any other clinical trials; · failure to obtain FDA approval;
| 23 |
| Table of Contents |
· failure to commercially sell the Company’s product at all or in quantities or at prices necessary to achieve profitability; · the early stage of development of our product candidate Prolanta™; · the timing, cost or results of our clinical trials; · our need for substantial additional funds and uncertainties regarding our ability to raise such funds on acceptable terms, if at all; · our ability to make arrangements to repay indebtedness, and the possible loss of assets or our ability to operate if we are unable to do so on acceptable terms; · uncertainties relating to patent and intellectual property matters; · our dependence on third parties, including our contract manufacturer, clinical research organizations, and consultants; · changes in, and uncertainty in decision-making involving, governmental authorities including the FDA and its regulations and requirements; · changes in the Company’s business strategy or an inability to execute its strategy due to lack of capital or unanticipated changes in the industries in which it proposes to operate; and · various competitive factors that may prevent the Company from competing successfully in the marketplace.
In light of these risks and uncertainties, many of which are described in greater detail elsewhere in this prospectus, there can be no assurance that the events predicted in forward-looking statements contained in the prospectus will in fact transpire.
Should any of these risks or uncertainties materialize (in whole or in part), or should any of assumptions prove incorrect, actual results may differ materially from those included within these forward-looking statements. Except as may be required under applicable securities laws, the Company undertakes no obligation to update or publicly release the result of any revision to these forward-looking statements to reflect events or circumstances occurring after the date they are made or to reflect the occurrence of unanticipated events.
An investment in our capital stock involves significant risks, including the risk of a loss of your entire investment. You should carefully consider the risks and uncertainties described below before purchasing our capital stock. The risks set forth below are not the only ones facing our Company. Additional risks and uncertainties may exist and others could arise that could also adversely affect our business, financial condition, operations and prospects. If any of the following risks actually materialize, our business, financial condition, prospects and operations would suffer. In such event, the value of our capital stock would decline, and you could lose all or a substantial portion of your investment.
The selling security holders are selling shares of Common Stock underlying the Convertible Notes and Warrants covered by this prospectus for their own accounts. We will not receive any of the proceeds from the resale of these shares.
The following table details the name of the selling security holders, the number of shares beneficially owned by the selling security holders, and the number of shares that may be offered by the selling security holders for resale under this prospectus. The selling security holders may sell up to 126,730,191 shares of our Common Stock from time to time in one or more offerings under this prospectus. Because the selling security holders may offer all, some or none of the shares they hold, and because, based upon information provided to us, there are currently no agreements, arrangements, or understandings with respect to the sale of any of the shares, no definitive estimate as to the number of shares that will be held by the selling security holders after the offering can be provided. The selling security holders have informed us that they are not registered broker-dealers and do not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities. Furthermore, none of the selling security holders are an affiliate of a broker-dealer. The following table has been prepared on the assumption that all shares offered under this prospectus will be sold to parties unaffiliated with the selling security holders.
This prospectus covers the resale of 126,730,191 of the number of shares of Common Stock comprised of the following: (i) with respect to the shares underlying the Convertible Notes, (A) 55,872,837 shares of Common Stock issuable upon conversion of the principal amount of $4,190,463 based on a conversion price of $0.075 per share pursuant to the monthly redemption payment schedule assuming timely payment and no default either in principal or interest payment obligations, (B) 5,633,847 shares of Common Stock issuable as payment of interest in lieu of cash, assuming timely payment of all interest obligations and no default either in principal or interest payment obligations, based on an interest conversion rate of $0.75 per share, and (C) 6,150,670 shares of Common Stock as a buffer in the event the monthly redemption payment or interest payment calculations are based on the lower of $0.075 per share and 75% of the average of the five lowest closing bid prices for the twenty consecutive trading days prior to such determination date; and (ii) 59,072,837 shares of Common Stock underlying the Warrants.
| 24 |
| Table of Contents |
Under the terms of the Convertible Notes, the selling security holders may not convert the Convertible Notes or exercise the Warrants to the extent such conversion would cause a selling security holder, together with its affiliates, to beneficially own a number of shares of Common Stock which would exceed 4.99% of our then outstanding shares of Common Stock following the conversion or exercise, which limit may be increased to 9.99% at the election of the selling security holder. The number of shares in the second column does not reflect this limitation (the “Maximum Percentage”). The selling security holders may sell all, some or none of their shares in this offering. See “Plan of Distribution.”
The number and percentage of shares beneficially owned is determined in accordance with Rule 13d-3 of the Exchange Act, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rule, beneficial ownership includes any shares as to which the selling security holder has sole or shared voting power or investment power and also any shares, which the selling security holder has the right to acquire within 60 days.
| Name of Selling Security Holder |
| Number of Shares of Common Stock Beneficially Owned Prior to the Offering(1) |
|
| Shares of Common Stock Included in Prospectus |
|
| Number of Shares of Common Stock Beneficially Owned After the Offering(2) |
|
| Percentage of Ownership Beneficially After Completion of Offering(2)(3) |
| ||||
| James Besser |
|
| 21,900 |
|
|
| 1,750,429 |
|
|
| 21,900 |
|
| * |
| |
| Joseph R. Borda & Marlene Borda, co-Trustees of the Borda Family Trust, 9/7/2016(4) |
|
| -- |
|
|
| 3,468,104 |
|
|
| -- |
|
|
| -- |
|
| Cavalry Fund I LP(5) |
|
| -- |
|
|
| 10,404,313 |
|
|
| -- |
|
|
| -- |
|
| Concordia Capital Partners LLC(6) |
|
| -- |
|
|
| 2,947,889 |
|
|
| -- |
|
|
| -- |
|
| Sharon T. Ewton |
|
| 123,280 |
|
|
| 8,752,145 |
|
|
| 123,280 |
|
| * |
| |
| Nicole R. Hoagland |
|
| 13,120 |
|
|
| 1,050,256 |
|
|
| 13,120 |
|
| * |
| |
| Jeb Partners, L.P. (7) |
|
| 235,600 |
|
|
| 15,753,865 |
|
|
| 235,600 |
|
| * |
| |
| L1 Capital Global Opportunities Master Fund(8) |
|
| -- |
|
|
| 20,808,629 |
|
|
| -- |
|
|
| -- |
|
| Robin and Leonidas Lemonidas JTWROS |
|
| 10,280 |
|
|
| 1,050,256 |
|
|
| 10,280 |
|
| * |
| |
| Caroline J. Lester |
|
| 8,560 |
|
|
| 875,214 |
|
|
| 8,560 |
|
| * |
| |
| Gregory G. Mario |
|
| -- |
|
|
| 3,468,104 |
|
|
| -- |
|
|
| -- |
|
| Mario Family Partners LP(9) |
|
| 104,460 |
|
|
| 11,716,867 |
|
|
| 104,460 |
|
| * |
| |
| Steven E. Nathanson |
|
| 17,160 |
|
|
| 1,750,429 |
|
|
| 17,160 |
|
| * |
| |
| Northbridge Financial Inc. (10) |
|
| -- |
|
|
| 9,537,287 |
|
|
| -- |
|
|
| -- |
|
| OEP Opportunities, LP(11) |
|
| -- |
|
|
| 1,734,053 |
|
|
| -- |
|
|
| -- |
|
| Pacific Capital Management LLC(12) |
|
| 82,540 |
|
|
| 7,001,719 |
|
|
| 82,540 |
|
| * |
| |
| Puritan Partners LLC(13) |
|
| -- |
|
|
| 23,273,390 |
|
|
| -- |
|
|
| -- |
|
| Laurence Stewart |
|
| -- |
|
|
| 1,387,242 |
|
|
| -- |
|
|
| -- |
|
_____
* Less than one percent
| (1) | Does not give any effect to any beneficial ownership of certain selling security holders in Company Common Stock that existed prior to the Exchange Offering, where such securities were exchanged for Convertible Notes and Warrants in the Unit Offering; disclosure of such ownership is reflected in the table below set forth in “-Other Information.” |
|
|
|
| (2) | Assumes the sale of all shares of Common Stock registered pursuant to this prospectus without regard to any conversion or exercise limitations contained therein, although the selling security holders are under no obligations known to us to sell any shares of Common Stock at this time. |
|
|
|
| (3) | This percentage is based upon 166,574,987 voting shares of capital stock issued and outstanding as of August _, 2017, plus the additional shares that a selling security holder is deemed to beneficially own. |
|
|
|
| (4) | The shares are directly owned by the Borda Family Trust, 9/7/2016, and each of Joseph R. Borda & Marlene Borda are co-Trustees and have voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (5) | The shares are directly owned by Cavalry Fund I LP, and Thomas Walsh is the general partner of Cavalry Fund I LP and has voting and investment control over the shares of Common Stock held by the selling security holder. |
| 25 |
| Table of Contents |
| (6) | The shares are directly owned by Concordia Capital Partners LLC, and Michael S. Liss is the managing member of Concordia Capital Partners LLC and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (7) | The shares are directly owned by Jeb Partners, L.P., and James Besser is the general partner of Jeb Partners, L.P. and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (8) | The shares are directly owned by L1 Capital Global Opportunities Master Fund, and David Feldman is the director of L1 Capital Global Opportunities Master Fund and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (9) | The shares are directly owned by Mario Family Partners LP, and Christopher Mario is manager of Melmotte LLC, the general partner of Mario Family Partners LP, and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (10) | The shares are directly owned by Northbridge Financial Inc., and Bryan Collins is president of Northbridge Financial Inc. and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (11) | The shares are directly owned by OEP Opportunities, LP, and James Cacioppo is the managing partner of OEP Opportunities, LP and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (12) | The shares are directly owned by Pacific Capital Management LLC, and Jonathan Glaser is the managing member of Pacific Capital Management LLC and has voting and investment control over the shares of Common Stock held by the selling security holder. |
|
|
|
| (13) | The shares are directly owned by Puritan Partners LLC, and Richard Smithline is the managing member of Puritan Partners LLC and has voting and investment control over the shares of Common Stock held by the selling security holder. |
Dollar Value of Underlying Securities and Potential Profits on Conversion
The following tables set forth the potential profit to be realized upon conversion by the selling security holders of the principal (excluding interest) of the Convertible Notes based on the applicable conversion price on August 3, 2017.
Potential Profit from Conversion of the Convertible Notes at the Option of the Selling Security Holders
| Per share market price on August 3, 2017 |
| $ | 0.11 |
|
| Per share conversion price calculated August 3, 2017 |
| $ | 0.075 |
|
| Total shares underlying Convertible Notes based on conversion price on August 3, 2017 |
|
| 55,872,837 |
|
| Aggregate market value of underlying shares based on market price on August 3, 2017 |
| $ | 6,146,012 |
|
| Aggregate purchase price for the Convertible Notes paid by the selling security holders(1) |
| $ | 3,561,894 |
|
| Selling security holders’ potential gross profits |
| $ | 2,584,118 |
|
| Percent of potential profit for selling security holders on conversion of the Convertible Notes |
|
| 73 | % |
______________
(1) The aggregate of the cash paid and existing indebtedness exchanged being deemed the purchase price.
Potential Payments to Selling Security Holders in Addition to Repayment of the Purchase Price
The selling security holders paid an aggregate of $3,586,621 in cash or through the exchange of existing indebtedness for a principal amount of $4,190,463 of Convertible Notes. In connection with the Convertible Notes, we are or may be required to make the following payments to the selling security holders:
| Payee |
| Interest Payments(1) |
|
| Mandatory Repayment(2) |
|
| Monthly Mandatory Principal Redemption Payments(3) |
|
| Optional Redemption Payments(4) |
|
| Maximum Registration Penalties Payments(5) |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Selling Security Holders |
| $ | 422,539 |
|
| $ | 5,028,556 |
|
| $ | 4,609,509 |
|
| $ | 5,028,556 |
|
| $ | 754,283 |
|
____________
| (1) | Represents the maximum amount of interest payable by the Company to the selling security holders under the Convertible Notes at 10% annually, assuming (a) that all payments thereunder are timely made and that no payments are accelerated or deferred, (b) that no payments of interest will be made in advance, (c) that the Convertible Notes are not otherwise converted prior to the monthly redemption dates and maturity date, (d) that interest is paid in cash, and (e) that no event of default thereunder occurs. |
| Table of Contents |
| (2) | Represents the cash amount that would be payable by the Company if it were required to redeem the Convertible Notes as a result of an event of default, change of control or fundamental transaction, a qualified offering ($8 million debt or equity financing), sale of substantially all of the assets or at maturity, assuming (a) that the applicable premium is 120%, (b) that the triggering event occurs on August 3, 2017, and (c) no interest had accrued on the Convertible Notes. The default interest rate is 24% per annum upon the occurrence and continuance of an event of default. |
|
|
|
| (3) | Represents the cash amount payable by the Company per the mandatory monthly principal redemption payments of 1/7th of the original principal amount on each of the 1st calendar day of the seven consecutive months, commencing on May 1, 2018, assuming that the applicable payment is 110% of the monthly redemption amount. |
|
|
|
| (4) | Represents the cash amount payable by the Company if it were to redeem, at its option, the Convertible Notes, assuming (a) that the applicable premium is 120%, (b) that the triggering event occurs on August 3, 2017, and (c) no interest had accrued on the Convertible Notes. |
|
|
|
| (5) | Represents 18% of the aggregate outstanding principal amount of the Convertible Notes assuming that the Company fails to satisfy its obligations under the Registration Rights Agreement. The Company is obligated to make a penalty payment, if the Initial Registration Statement which contains this prospectus is not declared effective by the SEC by the 90th calendar day after August 3, 2017 (120th day in the event of a SEC review), if an Additional Registration Statement is not timely filed with, or declared effective by, the SEC, or if, after a registration statement is declared effective it ceases to be effective for a period of time which shall exceed 10 consecutive calendar days or more than an aggregate of 15 calendar days in any one 12-month period, then the Company has agreed to pay to the selling security holders, an amount equal to 10% of the outstanding principal amount of the Convertible Notes for the first 30-day period (or part thereof) and after such 30 days, an additional 10% of outstanding principal amount of the Convertible Notes for a subsequent 30-day period (or part thereof), not to exceed 18% of the outstanding principal amount of the Convertible Notes. |
The Company’s Estimated Net Proceeds from the Convertible Notes
The following table sets forth the gross proceeds of $3,586,894 received by the Company ($2,000,000 in cash and $1,561,894 of existing indebtedness exchanged) as a result of the private placement of the Convertible Notes (the Convertible Notes were issued at a 15% discount), and then calculates the estimated net proceeds thereof after deduction of the interest (assuming it is paid in cash and not converted into shares) accruing on the Convertible Notes and monthly redemption payment premiums (assuming they are paid in cash and not converted into shares) pursuant to the terms of the Convertible Notes. The estimated net proceeds do not include the payment of any contingent payments, such as liquidated damages or redemption premiums in the case of default, change of control, uplisting, fundamental transaction, or optional redemption. The estimated net proceeds assumes that all principal will be paid in cash without any prepayment premium. The interest amount reflected below assumes that it is paid during the term of the Convertible Note, when due without any event of default, and the table assumes that none of the Convertible Notes are converted prior to maturity. Based on the foregoing assumptions, the estimated net proceeds from the Convertible Notes represent approximately 74% of the gross proceeds.
| Gross Proceeds Received |
| $ | 3,561,894 |
|
| Approximate Aggregate Interest Payments |
| $ | (422,539 | ) |
| Approximate Aggregate Mandatory Monthly Redemption Payments |
| $ | (419,046 | ) |
| Approximate Transaction Costs |
| $ | (100,000 | ) |
| Estimated Net Proceeds |
| $ | 2,620,309 |
|
Comparison of Registered Shares to Outstanding Shares
The following table compares the number of shares held by persons other than the selling security holders (including affiliates) and Company affiliates with the number of shares registered for resale.
| Voting capital stock outstanding, comprised of 103,536,703 shares of Company Common Stock and 63,038,284 shares of Series A Preferred Stock, convertible into 63,038,284 shares of Common Stock, issued and outstanding as of August 28, 2017 |
|
| 166,574,987 |
|
| Shares outstanding held by persons other than the (a) selling security holders and (b) affiliates of the Company |
|
| 56,736,703 |
|
| Shares registered for resale on behalf of the selling security holders |
|
| 126,730,191 |
|
| 27 |
| Table of Contents |
Other Information
As of the date of this prospectus, the Company does not believe that it will have the financial ability to make the payments under the terms of the Convertible Notes in cash when due. Accordingly, the Company expects that the selling security holders will convert the Convertible Notes into shares of Company Common Stock to the greatest extent possible.
The selling security holders have advised the Company that they may enter into short sales in connection with the Common Stock to be received on conversion of the Convertible Notes or exercise of the Warrants.
The Company has not had any material relationships or arrangements with any selling security holder, their affiliates, or any person with whom any selling security holder has a contractual relationship prior to the August 3, 2017 private placement (or any predecessors of those persons), other than the indebtedness (principal and accrued interest) incurred by Oncolix in 2016 and 2017 pursuant to negotiated private placements in the aggregate amount of $1,561,894 that was exchanged for Convertible Notes and Warrants in the Exchange Offering, a condition precedent to the funding of the Cash Offering. The prior debt investment in, and warrants issued by, Oncolix prior to the Merger that were exchanged for Convertible Notes and Warrants in the Exchange Offering is reflected in the table below:
| Name |
| Existing Indebtedness Exchanged |
|
| Principal Amount of Convertible Notes Issued |
|
| Shares Underlying Warrants Issued |
| |||
| Jeb Partners, L.P. |
| $ | 454,250 |
|
| $ | 534,412 |
|
|
| 7,125,490 |
|
| James Besser |
| $ | 50,472 |
|
| $ | 59,379 |
|
|
| 791,721 |
|
| Mario Family Partners LP |
| $ | 237,846 |
|
| $ | 279,819 |
|
|
| 3,730,924 |
|
| Sharon T. Ewton |
| $ | 252,361 |
|
| $ | 296,895 |
|
|
| 3,958,605 |
|
| Nicole R. Hoagland |
| $ | 30,283 |
|
| $ | 35,627 |
|
|
| 475,032 |
|
| Pacific Capital Management LLC |
| $ | 201,889 |
|
| $ | 237,516 |
|
|
| 3,166,885 |
|
| Robin and Leonidas Lemonidis JTWROS |
| $ | 30,283 |
|
| $ | 35,627 |
|
|
| 475,032 |
|
| Steven E. Nathanson |
| $ | 50,472 |
|
| $ | 59,379 |
|
|
| 791,721 |
|
| Caroline J. Lester |
| $ | 25,236 |
|
| $ | 29,690 |
|
|
| 395,860 |
|
| Puritan Partners LLC |
| $ | 228,800 |
|
| $ | 269,176 |
|
|
| 6,789,020 |
|
Each selling security holder and any of their pledgees, donees, transferees, assignees and successors-in-interest may, from time to time, sell any or all of their shares of Common Stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These sales may be at fixed or negotiated prices. A selling security holder may use any one or more of the following methods when selling shares:
· ordinary brokerage transactions and transactions in which the broker-dealer solicits investors; · block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction; · purchases by a broker-dealer as principal and resale by the broker-dealer for its account; · an exchange distribution in accordance with the rules of the applicable exchange; · privately negotiated transactions; · to cover short sales made after the date that this registration statement is declared effective by the SEC; · broker-dealers may agree with the selling security holder to sell a specified number of such shares at a stipulated price per share; · a combination of any such methods of sale; and · any other method permitted pursuant to applicable law.
| 28 |
| Table of Contents |
The selling security holders may also sell shares under Rule 144 under the Securities Act, if available, rather than under this prospectus.
Broker-dealers engaged by a selling security holder may arrange for other broker-dealers to participate in sales. Broker-dealers may receive commissions or discounts from a selling security holder (or, if any broker-dealer acts as agent for the purchaser of shares, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this Prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2440; and in the case of a principal transaction a markup or markdown in compliance with FINRA IM-2440.
In connection with the sale of the securities or interests therein, the selling security holders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The selling security holders may also sell securities short and deliver these securities to close out their short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The selling security holders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The selling security holders and any broker-dealers or agents that are involved in selling the securities may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each selling security holder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities. In no event shall any broker-dealer receive fees, commissions and markups which, in the aggregate, would exceed eight percent (8%).
Because selling security holders may be deemed to be “underwriters” within the meaning of the Securities Act, they will be subject to the prospectus delivery requirements of the Securities Act including Rule 172 thereunder. In addition, any securities covered by this prospectus which qualify for sale pursuant to Rule 144 under the Securities Act may be sold under Rule 144 rather than under this prospectus. The selling security holders have advised us that there is no underwriter or coordinating broker acting in connection with the proposed sale of the resale securities by the selling security holders.
We agreed to keep this prospectus effective until the earliest of (i) one (1) year from the date the registration statement is declared effective by the SEC, (ii) the date on which the securities may be resold by the selling security holders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (iii) the date on which all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously engage in market making activities with respect to the Common Stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the selling security holders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of securities of the Common Stock by the selling security holders or any other person. We will make copies of this prospectus available to the selling security holders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
The Company is required to pay all fees and expenses incident to the registration of the shares, but the Company will not receive any proceeds from the sale of the Common Stock. The Company has agreed to indemnify the selling security holders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
| 29 |
| Table of Contents |
DESCRIPTION OF OUR CAPITAL STOCK
We are authorized to issue 500,000,000 shares of Common Stock, par value $0.0001, of which 103,536,703 shares of Common Stock are issued and outstanding as of August 28, 2017, and 100,000,000 shares of preferred stock, par value $0.001, all of which are designated Series A Preferred Stock, of which 63,038,204 shares of Series A Preferred Stock are issued and outstanding as of August 28, 2017. In addition, an aggregate of 240,048,622 shares of Company Common Stock are reserved for issuance upon (i) the conversion of the 63,038,204 shares of Series A Preferred Stock issued and outstanding, (ii) the exercise of warrants to acquire 49,477,380 shares of Series A Preferred Stock and the conversion of such shares into 49,477,380 shares of Common Stock, (iii) the exercise of other outstanding options and warrants to acquire 12,587,284 shares of Common Stock, and (iv) the conversion of the Convertible Notes and exercise of the Warrants held by the selling shareholders.
Common Stock
The holders of Common Stock are entitled to one vote per share with respect to all matters required by law to be submitted to stockholders. The holders of Common Stock have the sole right to vote, except as otherwise provided by law or by our Articles of Incorporation. The Common Stock does not have any cumulative voting, preemptive, subscription or conversion rights. Election of directors requires the affirmative vote of a plurality of shares represented at a meeting, and other general stockholder action requires the affirmative vote of a majority of shares represented at a meeting in which a quorum is represented. The outstanding shares of Common Stock are validly issued, fully paid and non-assessable.
The holders of Common Stock are entitled to receive dividends, if declared by our Board of Directors, out of funds legally available, subject to the dividend preference of the Series A Preferred Stock as discussed below. In the event of liquidation, dissolution or winding up of the affairs of the Company, the holders of Common Stock are entitled to share ratably in all assets remaining available for distribution to them after payment or provision for all liabilities, including the Series A Preferred Stock liquidation preference discussed below.
The authorized but unissued shares of our Common Stock are available for future issuance without stockholder approval. These additional shares may be used for a variety of corporate purposes, including future offerings to raise additional capital, corporate acquisitions and employee benefit plans. The existence of authorized but unissued shares of Common Stock may enable our Board to issue shares of stock to persons friendly to existing management, which may deter or frustrate a takeover of the Company.
Preferred Stock
The terms of the Series A Preferred Stock are set forth below.
Seniority: The shares of Series A Preferred Stock shall rank senior to the Common Stock and any other preference shares.
Dividends: The Company shall not declare, pay or set aside any dividends on shares of any other class or series of capital stock (whether payable in cash or common stock) unless the holders of the Series A Preferred Stock then outstanding shall first receive, or simultaneously receive, a dividend on each outstanding share of Series A Preferred Stock in an amount at least equal to (i) in the case of a dividend on Common Stock or any class or series that is convertible into Common Stock, that dividend per share of Series A Preferred Stock as would equal the product of (A) the dividend payable on each share of such class or series determined, if applicable, as if all shares of such class or series had been converted into Common Stock and (B) the number of shares of Common Stock issuable upon conversion of a share of Series A Preferred Stock, in each case calculated on the record date for determination of holders entitled to receive such dividend or (ii) in the case of a dividend on any class or series that is not convertible into Common Stock, at a rate per share of Series A Preferred Stock determined by (A) dividing the amount of the dividend payable on each share of such class or series of capital stock by the original issuance price of such class or series of capital stock (subject to appropriate adjustment in the event of any stock dividend, stock split, combination or other similar recapitalization with respect to such class or series) and (B) multiplying such fraction by an amount equal to $0.075.
| 30 |
| Table of Contents |
Liquidation Preference: A liquidation event (“Liquidation”) shall occur in the event of (i) the closing of the sale, transfer, exclusive license or other disposition of all or substantially all of the Company’s assets, (ii) the consummation of the merger or consolidation of the Company with or into another entity (except a merger or consolidation in which the holders of capital stock of the Company immediately prior to such merger or consolidation continue to hold at least 50% of the voting power of the capital stock of the Company or the surviving or acquiring entity), (iii) the closing of the transfer (whether by merger, consolidation or otherwise), in one transaction or a series of related transactions, to a person or group of affiliated persons (other than an underwriter of the Company’s securities), of the Company’s issued and outstanding securities if, after such closing, such person or group of affiliated persons would hold 50% or more of the outstanding voting stock of the Company and, as a result of such transaction(s), the persons who were directors of the Company before such transaction(s) shall cease to constitute a majority of the Board of Directors or (iv) a liquidation, dissolution or winding up of the Company. In any Liquidation, each holder of Series A Preferred Stock shall be entitled to receive, for each share of Series A Preferred Stock then held, out of the proceeds of such Liquidation Event, the greater of (A) the sum of (i) an amount per share equal to $0.075, plus any then accrued but unpaid dividend on such share of Series A Preferred Stock and (ii) an amount per share based upon the distribution of the remaining assets to the holders of the Series A Preferred Stock and Common Stock, pro rata based on the number of shares of Common Stock held by each (assuming conversion of all Series A Preferred Stock), or (B) the amount of cash, securities or other property to which such holder would be entitled to receive in a Liquidation with respect to such shares if such shares had been converted to Common Stock immediately prior to such Liquidation.
Voting. Each holder of outstanding shares of Preferred Stock shall be entitled to cast the number of votes equal to the number of whole shares of Common Stock into which the shares of Preferred Stock held by such holder are then convertible.
Optional Conversion. Each share of Preferred Stock shall be convertible, at the option of the holder thereof, at any time, into one share of Common Stock, subject to customary conversion price adjustments, including weighted average adjustments for below conversion price issuances with standard carve-outs.
Mandatory Conversion. All outstanding shares of Preferred Stock shall automatically be converted into shares of Common Stock, at the then effective conversion rate, in the event of a closing of an underwritten public offering resulting in gross proceeds of at least $10 million.
Election of a Director. The Series A Preferred Stock holders have the right to appoint one director to the Company. To date, the holders of Series A Preferred Stock have not appointed a director.
Convertible Notes
Pursuant to the Purchase Agreement, the Company issued Convertible Notes to the Purchasers in an aggregate principal amount of $4,190,463 for a purchase price of $3,561,894, consisting of gross cash proceeds of $2,000,000 and the exchange of existing convertible debt and related accrued interest of Oncolix totaling $1,561,894. The Convertible Notes were issued at a 15% discount.
The Convertible Notes bear interest at a rate of 10% per annum, payable quarterly on November 1, 2017, February 1, 2018, and May 1, 2018 and thereafter, on a monthly basis until maturity. The interest is payable in cash or, at the option of the Company, subject to compliance with Equity Conditions (as defined below), in fully tradable Company Common Stock at the lesser of the then conversion price and a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the interest payment date. The final maturity of the Convertible Notes is November 1, 2018, but commencing on May 1, 2018 and on the first day of each month thereafter until maturity, we are required to redeem an amount of the Convertible Notes equal to 1/7th of the original principal amount, plus 10% of such monthly redemption amount as a bonus. The principal payments shall be made in cash or, subject to the satisfaction of Equity Conditions, in our Common Stock valued at the lesser of the then conversion price and a 25% discount to the average of the five lowest closing bid prices for the twenty trading days prior to the amortization date.
| 31 |
| Table of Contents |
The Convertible Notes are convertible at the option of the Purchasers at a conversion price equal to the lesser of (i) $0.075 per share of our Common Stock, (ii) 75% of the 10-day average closing bid price of our Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of our Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share). The Convertible Notes contain customary anti-dilution protection, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both conversion price and share amounts to be issued upon conversion).
In the event of a closing of an $8 million financing in connection with a change in listing of our Common Stock to Nasdaq or the NYSE, the Convertible Notes shall be subject to mandatory conversion into our Common Stock at a 30% discount to the offering price.
We shall be required to offer to prepay in cash the aggregate principal amount of the Convertible Notes at 120% of the principal amount thereof plus any unpaid accrued interest to the date of repayment, at maturity, on the sale of substantially all of our assets, upon a change of control, upon a qualified offering, or upon a “fundamental transaction” (tender offer, reclassification, sale of substantially all assets and merger); in such an event, the Purchasers shall have the right to convert the Convertible Notes prior to the date of any such prepayment.
The Convertible Notes contain standard negative covenants customary for transactions of this type. The events of default are also customary for transactions of this type, including default in timely payment of principal or interest, failure to observe or perform any covenant or agreement contained in the Convertible Note and Transaction Documents (including the Registration Rights Agreement), the commencement of bankruptcy or insolvency proceedings, failure to timely deliver conversion shares underlying the Convertible Notes, failure to timely file Exchange Act filings, and failure to satisfy certain Equity Conditions.
Equity Conditions mean, during the period in question, (a) the Company shall have duly honored all conversions and redemptions of the Convertible Notes, (b) we shall have complied with all conversion and other provisions of the Convertible Notes and shall have paid all liquidated damages and other amounts owing in respect of this Convertible Note, (c)(A) there is an effective registration statement pursuant to which the holder is permitted to utilize the prospectus thereunder to resell at least the number of shares of Common Stock issuable pursuant to the Transaction Documents at such time or (B) all of the underlying shares may be resold pursuant to Rule 144 without volume or manner-of-sale restrictions or current public information requirements, (d) our Common Stock is trading on a trading market, (e) there is a sufficient number of authorized but unissued and otherwise unreserved shares of our Common Stock for the issuance of five times all of the shares issuable pursuant to the Convertible Notes and Warrants, (f) there is no existing event of default or no existing event which, with the passage of time or the giving of notice, would constitute an event of default, (g) the issuance of the shares in question would not violate the beneficial ownership limitations in the Transaction Documents, (h) there has been no public announcement of a pending or proposed fundamental transaction or change of control transaction that has not been consummated, (i) the holder is not in possession of any information provided by the Company that constitutes, or may constitute, material non-public information, (j) our Common Stock is DWAC eligible and not subject to a DTC “chill;” and (k) for each trading day in a period of 10 consecutive trading days prior to the applicable date in question (A) the daily dollar trading volume for our Common Stock on the principal trading market exceeds $75,000 per trading day and (B) the weighted average stock price for our Common Stock on the principal trading market is above 50% of the closing stock price on August 3, 2017 (as adjusted for all stock splits stock dividends, stock combinations, reclassifications and other transactions).
We have the right at any time to redeem all, but not less than all, of the total outstanding amount of principal amount then remaining under the Convertible Notes at a price equal to 120% of such principal amount.
| 32 |
| Table of Contents |
Warrants
In connection with the Purchase Agreement, the Company issued the Purchasers five-year Warrants, exercisable for cash at $0.09 per share or on a cashless basis. In addition, in connection with the debt converted in the Exchange Offering, one Purchaser was issued five-year Warrants to acquire 3,200,000 shares of our Common Stock at a purchase price of $0.0825 per share, exercisable for cash or on a cashless basis. The Warrants contain customary anti-dilution provisions, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both exercise price and share amounts to be issued upon exercise).
Series A Preferred Stock Warrants
In connection with the issuance of the Series A Preferred Stock by Oncolix, there are outstanding warrants to purchase up to 49,477,380 shares of Series A Preferred Stock at an exercise price of $0.0825 per share for cash or on a cashless basis that expire in January 2020.
Other Warrants and Options
The Company has other warrants and options issued and outstanding to purchase an aggregate of 12,587,284 shares of Company Common Stock at prices ranging from $0.005 to $0.09, expiring on various dates through August 2022.
Registration Rights Agreement
In connection with the execution of the Purchase Agreement, the Company and the Purchasers also entered into the Registration Rights Agreement. Pursuant to the Registration Rights Agreement, the Company has agreed to file the Initial Registration Statement with the SEC to register the resale of the Registrable Securities, being all shares of Common Stock issuable under conversion of the Notes and upon exercise of the Warrants and any securities issued or then issuable upon any stock split, dividend or other distribution, recapitalization or similar event with respect to the foregoing, on or prior to September 2, 2017, and have it declared effective at the earlier of (i) the 90th calendar day after August 3, 2017 (120th calendar day after August 3, 2017 in the event that the registration statement is subject to review by the SEC) and (ii) the 5th business day after the date the Company is notified by the SEC that the registration statement will not be reviewed or will not be subject to further review.
The Company also agreed to file Additional Registration Statements to cover all of the Registrable Securities that are not covered by the Initial Registration Statement on or prior to 30 days following the date the Company first knows that such additional registration statement is required to be filed and have it declared effective no later than the earlier of (i) the 60th calendar day following the date the Company first knows that it is required to file such Additional Registration Statement and (ii) the 5th business day after the date the Company is notified by the SEC that the registration statement will not be reviewed or will not be subject to further review. If (i) the Initial Registration Statement is not declared effective on or before the 90th calendar day following the date of the Purchase Agreement (120th day in the event of a SEC review), (ii) any Additional Registration Statement is not declared effective on or before its effectiveness deadline, or (iii) any registration statement is declared effective by the SEC but shall thereafter cease to be effective for a period of time of 10 consecutive calendar days but not more than an aggregate of 15 calendar days in any one 12-month period, each such event being a Non-Registration Event, then the Company shall deliver to the Purchasers, as liquidated damages, an amount equal to ten percent of the outstanding principal amount of the Convertible Notes then held by the Purchasers for the first 30 days (or part thereof) and after such 30 days, an additional 10% of the outstanding principal amount of the Convertible Notes for a subsequent 30-day period (or part thereof); provided that the maximum amount of liquidated damages shall not exceed 18% of the aggregate outstanding principal amount of the Convertible Notes. The Company may pay the liquidated damages in cash or through the issuance of shares of Common Stock (valued at 75% of the average of the 5 lowest closing bid prices for the twenty consecutive trading days prior to the Non-Registration Event) provided the Equity Conditions are met. In addition, if there is not an effective registration statement covering all the Registrable Securities and the Company elects to register the issuance or resale of other Company securities, the Company is required to include such Registrable Securities in such registration statement filed with the SEC.
| 33 |
| Table of Contents |
The Company also agreed, among other things, to indemnify the Purchasers from certain liabilities and fees and expenses of the Purchasers incident to the Company’s obligations under the Registration Rights Agreement, including certain liabilities under the Securities Act. The Purchasers have agreed to indemnify and hold harmless the Company and each of its directors, officers and persons who control the Company against certain liabilities that may be based upon written information furnished by the Purchasers to the Company for inclusion in a registration statement pursuant to the Registration Rights Agreement, including certain liabilities under the Securities Act.
Control Share Acquisition Provisions of the Florida Business Corporation Act
The “control share” provisions of Section 607.0902 of the Florida Business Corporation Act prohibits an acquirer, under certain circumstances, from voting its shares of Common Stock after crossing certain ownership threshold percentages, unless the acquirer obtains approval from the disinterested shareholders. The Company’s articles of incorporation opts out of this provision, as provided for in the Florida Business Corporation Act, and accordingly, the control share statute is not applicable to us.
Transfer Agent
The transfer agent and registrar for our Common Stock is VStock Transfer, LLC whose address is 18 Lafayette Place, Woodmere, NY 11598.
Overview
We are a clinical-stage biopharmaceutical company developing medical therapies for the treatment of cancer. Since 2008, our activities have been focused on the development of Prolanta™, a biological drug for the treatment of cancer. Our initial focus is ovarian cancer, and we have commenced a Phase I clinical trial of Prolanta™ for the treatment of ovarian cancer after we received clearance from the FDA. We also believe that Prolanta™ may be effective in the treatment of breast, uterine, prostate and other cancers.
Our drug candidate Prolanta™ is a genetically modified form of human prolactin, a hormone associated with various types of cancers. Prolanta™ has been designed to bind to the prolactin receptor on cells to block the effects of human prolactin. Human prolactin has been documented to cause cancer cell proliferation by interacting with cancer cells. We and our collaborators have conducted animal studies to determine the efficacy of Prolanta™ in treating breast and ovarian cancers, and we expect to also investigate animal models of other types of cancer. We have also conducted animal studies to evaluate the toxicity and tolerability of Prolanta™, which was included in our Investigation New Drug Application (“IND”) with the FDA, which has granted us clearance to commence an initial human trial of Prolanta™ in ovarian cancer patients. There is no evidence of dose-limiting toxicities or serious adverse events to date in this clinical trial.
Ovarian cancer is the fourth leading cause of cancer-related death in women in the U.S. and the leading cause of gynecologic death. According to the National Cancer Institute, in 2017 approximately 22,000 women will be diagnosed with ovarian cancer in the U.S., and 14,000 are expected to die from this disease. We chose ovarian cancer as the first human application of Prolanta™ because of the significant unmet medical need as well as favorable regulatory status. Because of the size of the US market, the FDA has granted Prolanta™ favorable regulatory status as an Orphan Drug. As such, Prolanta™ may qualify for federal income tax credits, reduced filing fees and an extended period of marketing exclusivity.
| 34 |
| Table of Contents |
Company History
The Company was incorporated in the State of Florida in June 2013 and was a shell company immediately prior to August 13, 2017. We had no assets or operations prior to August 2017. Our wholly owned subsidiary, Oncolix, was incorporated in the State of Delaware in December 2006, and commenced operations in 2007. At formation, our intellectual property was transferred to Oncolix by GHC.
Summary of Our Business Strategy
We are a clinical-stage biopharmaceutical company developing medical therapies for the treatment of cancer. We currently own issued or pending patent applications related to the use of Prolanta™, a recombinant protein analogue of human prolactin that is designed to bind to the prolactin receptor on human cells. There is substantial scientific evidence that prolactin is associated with various cancers, including ovarian, breast, uterine and prostate. We and our collaborators have conducted animal studies that indicate that our drug Prolanta™ may be effective in treating breast and ovarian cancers, either as a stand-alone therapy or in combination with other drugs, by blocking the effect of human prolactin. Prolanta™ may also affect the cellular mechanisms that assist cancer cells to develop resistance to chemotherapy.
Our initial focus is the development, clinical testing and commercialization of Prolanta™ for the treatment of ovarian cancer. The FDA has provided us with clearance to commence a Phase I clinical trial of Prolanta™ in the treatment of ovarian cancer, and we have dosed three subjects in the first phase of this clinical trial. We chose ovarian cancer as the first human application of Prolanta™ because of the significant unmet medical need as well as favorable regulatory status. Because of the size of the US market, the FDA has granted Prolanta™ status as an Orphan Drug. We intend to apply for Orphan designation in the European Union and in Japan.
If Prolanta™ is found to be safe and well tolerated in the Phase I study, we will seek regulatory clearance to conduct additional clinical trials in larger numbers of ovarian cancer patients to establish efficacy. We expect that at least two clinical studies will be required, and possibly more, to establish the efficacy and safety of Prolanta™ in sufficient numbers of ovarian cancer patients in order to seek approval to market Prolanta™ for this cancer. The design of these studies will be finalized after the completion of the Phase I study and after consultation with the FDA, but we currently believe we will first evaluate the use of Prolanta™ in combination with other chemotherapy drugs such as platinum-based drugs and taxanes. There is preclinical and laboratory evidence that Prolanta™ may be synergistic with these other therapies, allowing a broader market opportunity. We intend to seek a distribution partner to market Prolanta™, but may develop our own sales force in the event we cannot make suitable arrangements.
Depending on the availability of resources, we also intend to evaluate Prolanta™ for the treatment of other cancers. These studies may be performed in collaboration with larger pharmaceutical companies, either through licenses or joint ventures. Our scientific collaborators have demonstrated in animal models the efficacy of Prolanta™ in treating breast cancer, and we intend to engage academic collaborators to develop data for the treatment of other cancer types.
We may also consider in-licensing or acquiring other drug candidates, preferably in oncology.
We operate with a small staff of employees and key consultants and have utilized contractors for certain operations. We do not intend to develop the infrastructure or acquire the facilities necessary to manufacture Prolanta™, and instead we plan to utilize contract manufacturers. We also plan to utilize contract research organizations to assist us in conducting clinical trials. We intend to employ additional administrative and clinical personnel as necessary to conduct our clinical trials and commercialize Prolanta™.
Scientific Background on Human Prolactin
Scientific studies by other researchers have indicated that human prolactin is associated with the growth of ovarian, breast and other cancers. Additionally, other scientific studies have noted that human prolactin is associated with increased resistance of some cancer cells to certain therapies that are first-line treatments for cancer. Accordingly, we believe that blocking the effects of human prolactin may be an effective strategy for treating ovarian and other cancers and may improve the effectiveness of existing drugs.
Human prolactin is a single-chain polypeptide of 199 amino acids with a molecular weight of approximately 23,000 Daltons. Prolactin is mainly synthesized and secreted by cells in the anterior pituitary gland, but is also produced at sites outside the pituitary gland, such as mammary gland, ovary, uterus, prostate, lymphocytes, and several types of tumor cells. The most well-known function of prolactin is the control of lactation during pregnancy, but research by others also indicates that this protein may be involved in angiogenesis and regulation of the immune system.
| 35 |
| Table of Contents |
Human prolactin activates certain intracellular activities by binding with two receptors on the surface of a cell (also referred to as “dimerization”). When the two receptors are bound by prolactin, the receptors come closer together triggering a complex web of intracellular activity, including Janus kinase 2 (JAK2), Src kinase, phosphatidylinositol 3’-kinase (PI3K), protein kinase C (PKC) and mitogen-activated protein kinase (MAPK). The best-characterized signaling pathway is the JAK2/STAT5 pathway, which has been shown to play a central role in cell proliferation, differentiation and cell death. In addition, the ras/raf/MAPK pathway is also activated by prolactin and may be involved in the effects of the protein on cell proliferation.
Scientific research has shown that serum levels of human prolactin are significantly elevated in women with a strong family history of ovarian and other gynecological cancers. Increased expression of the prolactin receptor has been observed in ovarian and endometrial tumors, signifying the importance of prolactin signaling in malignant conditions. Additionally, prolactin mRNA has been detected in ovarian and endometrial tumors, indicating the presence of an autocrine loop. Prolactin inhibited cell death (apoptosis) in several ovarian and endometrial cancer cell lines. Prolactin also activated the ras oncogenic signaling in normal ovarian epithelial cells such that the malignantly transformed cells were able to form tumors in immunodeficient mice.
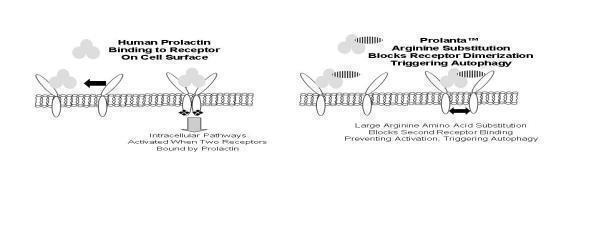
Consistent with this prior evidence, laboratory and preclinical studies have demonstrated that blocking the effects of human prolactin was effective in reducing the size and number of human ovarian cancer tumors in orthotopic mouse models.
In addition to the association of human prolactin with tumor cell growth, scientific studies have also demonstrated that prolactin may protects cells from some chemotherapy drugs by activating a detoxification enzyme. One study demonstrated that human prolactin can reduce the effectiveness of the platinum-based drug cisplatin on ovarian cancer cells, and another study demonstrated that human prolactin can reduce the effectiveness of cisplatin, taxol, vinblastine and doxorubicin on breast cancer cells.
Accordingly, we believe that blocking the effect of prolactin at the cancer cell level may provide an effective therapy for ovarian and other cancers and may improve the effectiveness of existing drugs.
Description of Prolanta™
Our drug Prolanta™ is a recombinant human prolactin analogue with an amino acid substitution of arginine for glycine at position 129. As such, Prolanta™ has also been referred to as G129R in the scientific literature. As a result of this amino acid substitution, Prolanta™ acts as an antagonist to the human prolactin receptor. Prolanta™ will bind to one prolactin receptor on a cancer cell, but the arginine amino acid replacement is designed to prevent binding to a second receptor. As a result, Prolanta™ will compete with normal human prolactin and may prevent the dimerization (the binding to two prolactin receptors), preventing the intracellular activity normally triggered by prolactin binding.
We funded a study by scientists at The University of Texas MD Anderson Cancer Center (“MD Anderson”) to evaluate the effects of Prolanta™ on ovarian cancer cells. This study concluded that Prolanta™ induces ovarian cancer cell death through a process known as sustained autophagy, or autophagocytosis. Autophagy is a lysosome-dependent, intracellular pathway for protein and organelle degradation. Sustained autophagy, or Type II programmed cell death, also has been proposed by others as a strategy for cancer therapy. We are not aware of any products currently marketed that utilize autophagy as a mechanism of action.
| 36 |
| Table of Contents |
Scientists at MD Anderson also conducted animal studies (xenografts) using Prolanta™ alone or in combination with taxane drugs, which are commonly used to treat ovarian cancer in humans. Prolanta™ alone reduced the numbers and sizes of ovarian tumors in these studies, and also was synergistic with taxanes.
Our scientific founder at Clemson has conducted various studies of the use of Prolanta™ to treat breast cancer. In these studies, Prolanta™ alone was effective in the treatment of breast cancer, and was synergistic with a platinum-based drug as well as Herceptin®, common therapies for breast cancer.
The Cancer Market
The American Cancer Society estimates that over 1.6 million people in the U.S. are expected to be diagnosed with cancer in 2017, excluding basal and squamous cell skin cancers and in situ carcinomas (other than urinary bladder carcinomas).
Despite continuous advances every year in the field of cancer research, there remains a significant unmet medical need in the treatment of cancer, as the overall five-year survival rate for a cancer patient is approximately 68% according to the American Cancer Society. According to that same source, cancer is the second leading cause of mortality in the U.S. behind heart disease. Overall, the American Cancer Society estimates that approximately one in four deaths in the U.S. is due to cancer.
Treatments for advanced cancer may include, among other approaches: chemotherapy (toxic drugs generally selected to kill rapidly growing cells); biological therapies (including antibodies, proteins or other molecules designed to alter cancer cell processes, recruit the patient’s body to assist in fighting the disease, or act as a carrier for other toxic molecules); surgical intervention to remove the cancerous areas; and radiation therapies to damage rapidly growing cells like cancer. The treatment varies widely by cancer type, and the effectiveness of different drugs or therapies will vary both by type of cancer as well as by patient.
Ovarian Cancer
Our initial focus is to develop Prolanta™ for the treatment of ovarian cancer, which is the fourth leading cause of cancer-related death in women in the US and the leading cause of gynecologic death. According to the National Cancer Institute, in 2017 approximately 22,000 women will be diagnosed with ovarian cancer in the U.S., and 14,000 are expected to die from this disease in the U.S. The incidence of ovarian cancer in Europe is estimated to be twice as high as the US.
Because of the difficulties with the early detection of ovarian cancer, approximately 80 percent of patients in the U.S. are diagnosed with advanced stage disease. Of these patients, 80 to 90 percent will initially respond to chemotherapy, but less than 10 to 15 percent will remain in permanent remission. Although advances in treatment have led to an improved five-year survival rate approaching 47%, overall survival has not improved. The five-year survival rate for patients with distant disease is only 29 percent.
The current standard of care for ovarian cancer is surgical debulking, with a total hysterectomy and oophorectomy typically performed at the same time. The majority of patients require postoperative chemotherapy to eradicate residual disease. Six cycles of a taxane, such as paclitaxel, combined with a platinum-based therapy, such as carboplatin or cisplatin, is now standard treatment. Even in patients that respond to therapy by going into remission, the majority of those patients with advanced disease will have a relapse. Only 10% to 30% of such patients have long-term survival. Approximately 20% to 30% of patients never have a clinical remission despite receiving first-line chemotherapy or platinum-based therapy. This residual or progressive disease is generally not curable with current treatments.
Both taxanes and the platinum-based regimens cause severe side effects, including progressive neuropathy, cumulative thrombocytopenia, anemia, neutropenia, platinum allergy, cardiovascular effects, nausea and renal toxicity. Severe or cumulative side effects can necessitate a switch to alternative therapies, such as liposomal doxorubicin, topotecan, gemcitabine, etoposide and vinorelbine; however, the response rate for each of these drugs is only 10% to 20% in patients with platinum-resistant disease.
The inability to cure most patients with current therapies, including second-line therapies, indicates the need for more effective therapeutic options.
As noted above, various scientific studies have indicated a relationship between human prolactin and the growth of ovarian cancer cells, as well as the development by ovarian cancer cells of resistance to common drug therapies. Our scientific collaborators have demonstrated that our prolactin antagonist, Prolanta™, was effective in inhibiting the growth of ovarian cancer cells, either as a stand-alone therapy or in combination with taxanes. As a result, we believe Prolanta™ may be an effective new therapy for ovarian cancer.
| 37 |
| Table of Contents |
Drugs to treat ovarian cancer may qualify for Orphan Drug designation in the US and the EU, and this designation offers significant benefits through reduced regulatory fees, substantial tax credits and potential marketing exclusivity. The FDA granted Prolanta™ Orphan Drug status for the treatment of ovarian cancer, and we intend to seek EU designation.
Plan of Operations for the Next Twelve Months
Our initial commercial focus is the development of Prolanta™ for the treatment of ovarian cancer. We have commenced a Phase 1 clinical trial after receiving clearance from the FDA. We completed the first dosing group of 3 subjects in the Phase I clinical trial, with no evidence of serious adverse events or dose-limiting toxicities. In order to proceed with the next doing group, we must have a contract manufacturer make a supply of new Prolanta™. We are working with contract manufacturers at this time to determine the cost and timing of new drug supplies.
This Phase I trial is an open-label safety and pharmacokinetic study in up to 18 women with recurrent or persistent epithelial ovarian cancer, primary peritoneal cancer, or fallopian tube cancer. The primary objectives of this study will be to evaluate the safety and tolerability, to determine the pharmacokinetic profile, and to determine the optimal dose of Prolanta™. We will also evaluate the effect of Prolanta™ on the ovarian cancer during the course of the study, including the effect on tumor size, blood chemistry, and tumor biochemistry.
Because of the unmet medical need in ovarian cancer, we will seek accelerated approval as soon as our studies demonstrate safety and substantial efficacy. This could be as early as the completion of two Phase II studies, but that is dependent on our clinical trial results as well as the then-current position of the FDA.
In connection with our IND filing, we conducted toxicity and pharmacokinetic studies of Prolanta™ in cynomolgus monkeys and rabbits at doses that exceeded our Phase I dose levels (adjusted for body weight of each species). These studies indicated that Prolanta™ was well tolerated, and there were no significant adverse events. The monkeys developed anti-drug antibodies, but these antibodies did not appear to have any effect on safety or the circulating life of Prolanta™ in the blood stream. It is common for biological-based drugs to result in antibody development by the subject, and the development of antibodies may have no clinical effect. Our Phase I study will also evaluate anti-Prolanta™ antibody levels in humans.
During the Phase I study, we intend to plan follow-on studies that will be conducted if the Phase I is successful. At this time, our development plan includes two Phase II studies. Because of the substantial scientific data that indicates Prolanta™ may be synergistic with current therapies, we intend to design these studies to use Prolanta™ in combination with platinum-based and taxane therapy. In the first Phase II study, one group of patients would receive Prolanta™ at the same time as other therapy, and the results will be compared to a second group receiving other therapy alone. The timing of this study will be dependent on the Phase I results and available resources.
We are currently evaluating improvements in our manufacturing process to increase the supply of available drug. This process may involve a change in manufacturing.
We will need additional capital to fund our development activities.
We may also in-license or acquire additional drug candidates, preferably in oncology, to expand our pipeline.
Intellectual Property
As part of our business strategy, we seek patent protection in the US and certain major foreign jurisdictions for the use of Prolanta™, including the use of Prolanta™ with certain modifications or in conjunction with other molecules, to treat various diseases. We also rely on trade secrets, internal know-how, technological innovations and agreements with third parties to develop, maintain and protect our competitive position. In addition, where applicable, we intend to rely on laws and regulations that provide opportunities for marketing exclusivity, such as the US Orphan Drug designation. Our ability to be competitive will depend on the success of this strategy.
Upon the commencement of operations in 2007, GHCRDC and certain scientists contributed intellectual property to Oncolix, including certain issued and pending patent applications. After this contribution, we also have filed additional patent applications related to Prolanta™. As of this date, we own eight US patents and several pending applications for which we pay no royalties. These patents and applications are in various stages of review in major foreign jurisdictions. The first US patent application for the use of Prolanta™ to treat ovarian cancer has been granted in the US and related US and foreign patent applications are currently under review by patent authorities. We have also licensed technology from Monsanto Company related to methods of manufacturing Prolanta™, and we may be required to pay a royalty of up to 2% upon the commercial sale of Prolanta™.
| 38 |
| Table of Contents |
Our goal is to obtain, maintain and enforce patent protection for the use of Prolanta™ and its formulations, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the US and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible through a combination of contractual arrangements and patents, both in the US and abroad. However, patent protection may not afford us with complete protection against competitors who seek to circumvent our patents.
Our patents and patent applications are subject to procedural or legal challenges by others.
We also depend upon the skills, knowledge, experience and know-how of our management, advisors, consultants and other contractors. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely and will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
The US Patent and Trademark Office (PTO) compensates patentees with additional patent in-force days if there are delays by the US PTO during prosecution. These additional days are called patent term adjustment (PTA) days and they allow a patentee to keep its patent in force after the expiration date of the patent for a period of time that the US PTO has provided as PTA.
In addition to patent protection, we plan to seek marketing exclusivity under other laws and regulations. Our success for preserving market exclusivity for our product candidate relies on our ability to obtain and maintain a regulatory period of data exclusivity for an approved biologic, currently 12 years from the date of marketing approval, obtaining orphan designation in the major markets and to preserve effective patent coverage. Once any regulatory period of data exclusivity expires, depending on the status of our patent coverage, we may not be able to prevent others from marketing and selling biosimilar versions of our product candidates. We are also dependent upon the diligence of third parties, which control the prosecution of pending domestic and foreign patent applications and maintain granted domestic and foreign patents.
We may be unable to obtain, maintain and protect the intellectual property rights necessary to conduct our business, and we may be subject to claims that we infringe or otherwise violate the intellectual property rights of others, which could materially harm our business. For more information, see “Risk Factors- Risks Related to Our Intellectual Property Rights.”
Manufacturing
We do not currently have nor do we plan to build the internal infrastructure or capability to manufacture Prolanta™ or any future product candidates for use in the conduct of our clinical trials or for commercial supply. We currently rely on a third-party contract manufacturer to manufacture clinical supplies of Prolanta™, but we are evaluating alternative manufacturers.
Prolanta™ is a recombinant protein that is produced by Escherichia coli (“E. coli”) bacteria that have been genetically modified to produce this protein. We licensed this bacterial strain, or “expression system”, from Monsanto Company. See “Intellectual Property”. Other manufacturers have proprietary and non-proprietary manufacturing systems.
The production of Prolanta™ is a multi-step process that involves the growth of bacteria in large containers (also known as fermentation), the inducement of production of the Prolanta™ protein by the bacteria, the extraction of the protein from the cells, the folding of the protein into the proper three-dimensional shape, and purification of the protein and removal of impurities and process residuals. The purified protein and additional excipients are then filled into vials and lyophilized for storage. The vials are kept refrigerated until use, and the protein is reconstituted with sterile water for injection beneath the skin. To treat cancer patients, we currently believe the protein will be injected daily under the skin during the treatment period. Our contract manufacturer currently performs the manufacturing steps prior to vial filling and lyophilization, and these latter steps are performed by a subcontractor.
Manufacturers of our products are required to comply with applicable FDA manufacturing requirements contained in the FDA’s current good manufacturing practice standards (“cGMP”) regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. Pharmaceutical product manufacturers and other entities involved in the manufacture and distribution of approved pharmaceutical products are required to register their establishments with governmental authorities, such as the FDA and/or Health Canada, and certain state agencies, and are subject to periodic unannounced inspections by the governmental authorities and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
| 39 |
| Table of Contents |
Facilities used by contract manufacturers and their subcontractors must be approved by the respective governmental agencies after we submit a request for marketing approval for Prolanta™. We do not control the manufacturing process and are completely dependent on contract manufacturers for compliance with the applicable regulatory requirements for the manufacture of Prolanta™. If a contract manufacturer cannot successfully manufacture material that conforms to the strict regulatory requirements of the FDA and any applicable foreign regulatory authority, they will not be able to secure the applicable approval for their manufacturing facilities. If these facilities are not approved for commercial manufacture, we may need to find alternative manufacturing facilities, which could result in delays in obtaining approval for the applicable product candidate as well as substantial additional expense.
In addition, contract manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. Failure to comply with applicable cGMPs or other regulatory requirements could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply and criminal prosecutions, any of which could significantly and adversely affect supplies of our Prolanta™ and have a material adverse impact on our business, financial condition and results of operations.
Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved marketing authorization, including withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA (or other governmental agency) approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further governmental review and approval.
In the event we develop or acquire additional product candidates, we expect to make similar arrangements with a other third-party contract manufacturers for such product candidates.
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical companies, generic drug companies and other public and private institutions developing oncology products. In addition, many universities and private and public research institutes are active in cancer research, some in direct competition with us. Many of our potential competitors have substantially greater financial, technical and human resources than we do, as well as greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. We also must compete with these institutions in recruiting highly qualified personnel. Accordingly, our potential competitors may be more successful than us in obtaining FDA approval for their drugs and achieving widespread market acceptance for their products. Our potential competitors’ drugs may be more effective, or more effectively marketed and sold, than any product candidate we may commercialize and may render Prolanta™ or any future product candidates obsolete or non-competitive before we can recover the expenses of their development and commercialization. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally, the development of new treatment methods for the diseases we are targeting could render Prolanta™ or any future product candidate non-competitive or obsolete.
We believe the existing products for the treatment of ovarian and other cancers are not completely effective and that there is a need for better therapies. Furthermore, most current products have significant side effects. We currently intend to develop Prolanta™ for the treatment of ovarian cancer and then evaluate the drug for other cancers, such as breast, prostate and endometrial cancer. A number of product candidates that are currently under development by others may become commercially available in the future for the treatment of ovarian cancer or other cancers we may target. We are aware that many competitors have product candidates in Phase 2 or later stage clinical trials. However, our competitors’ products may be more effective, have fewer side effects, have lower costs or be better marketed and sold than Prolanta™ or any of our future product candidates. Additionally, products that our competitors successfully develop for the treatment of ovarian cancer may be marketed prior to Prolanta™.
We believe the key competitive factors that will affect the development and commercial success of Prolanta™, if successfully developed and approved for marketing, will be Prolanta™’s efficacy compared to existing or future products, the extent to which such efficacy is synergistic with existing or future products, Prolanta™’s safety and tolerability profile, the extent to which Prolanta™ is effective in all patients or identifiable subsets of patients, the convenience of dosing, Prolanta™’s price, and the availability of reimbursement from governmental and other third-party payors.
| 40 |
| Table of Contents |
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as Prolanta™. Any pharmaceutical candidate that we develop must be approved by the FDA before it may be legally marketed in the United States and by the appropriate foreign regulatory agency before it may be legally marketed in foreign countries. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
United States Pharmaceutical Product Development Process
In the United States, the FDA regulates pharmaceutical (drug and biologic) products under the Federal Food, Drug and Cosmetic Act and its implementing regulations (“FDCA”). Pharmaceutical products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable US requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a pharmaceutical product may be marketed in the United States generally includes the following:
· Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices (“GLPs”) or other applicable regulations; · Submission to the FDA of an Investigational New Drug (“IND”) application, which must become effective before human clinical trials may begin in the United States; · Performance of adequate and well-controlled human clinical trials according to the FDA’s current Good Clinical Practices (“GCPs”), to establish the safety and efficacy of the proposed pharmaceutical product for its intended use; · Submission to the FDA of a New Drug Application (“NDA”) or Biological Licensing Application (“BLA”) for a new pharmaceutical product; · Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the pharmaceutical product is produced to assess compliance with the FDA’s current good manufacturing practices (“cGMP”), to assure that the facilities, methods and controls are adequate to preserve the pharmaceutical product’s identity, strength, quality and purity; · Potential FDA audit of the preclinical and clinical trial sites that generated the data in support of the New Drug Application or Biological Licensing Application (“NDA/BLA”); and · FDA review and approval of the NDA/BLA.
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources and approvals are inherently uncertain.
Before testing any compounds with potential therapeutic value in humans, the pharmaceutical product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the pharmaceutical product candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs. The sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the IND on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. An IND that has become effective may also be referred to as “receiving clearance” from the FDA. The FDA may also impose clinical holds on a pharmaceutical product candidate at any time before or during clinical trials due to safety concerns or non-compliance.
We currently have an effective IND to conduct a Phase I study in ovarian cancer, and may submit additional INDs in the future for other cancers or other products. We cannot be certain that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such clinical trial.
| 41 |
| Table of Contents |
Clinical trials involve the administration of the pharmaceutical product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by the sponsor. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA if conducted under a US IND. Clinical trials must be conducted in accordance with the FDA’s GCPs. Further, each clinical trial must be reviewed and approved by an independent institutional review board (“IRB”) or, if conducted outside of the US, an ethics committee at or servicing each institution at which the clinical trial will be conducted. An IRB or ethics committee is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB or ethics committee also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
We intend to use third party contract research organizations (“CRO”) to administer and conduct many aspects of our planned clinical trials and will rely upon such CROs, as well as medical institutions, clinical investigators and consultants, to conduct our trials in accordance with our clinical protocols and to play a significant role in the subsequent collection and analysis of data from these trials. The failure by any of such third parties to meet expected timelines, adhere to our protocols or meet regulatory standards could adversely impact the subject product development program.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
· Phase 1: The pharmaceutical product is initially introduced into healthy human subjects or diseased patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, such as cancer treatments, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. Our Phase I clinical trial will be conducted in ovarian cancer patients. · Phase 2: The pharmaceutical product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. · Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA/BLA or foreign authorities for approval of marketing applications.
The size, extent, length and nature of clinical trials may vary by the nature of the disease. Sponsors are required to obtain regulatory approval to conduct each clinical trial, and the FDA may require certain types of patients, numbers of patients, clinical procedures or tests, length of treatment, and other criteria that may be difficult, expensive or time consuming.
The FDA and IND sponsor may agree in writing on the design and size of clinical trials intended to form the primary basis of an effectiveness claim in an NDA application. This process is known as a special protocol assessment (SPA). These agreements may not be changed after the clinical trials begin, except in limited circumstances. The existence of a SPA, however, does not assure approval of a product candidate.
Post-approval studies, or Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and may be requested by the FDA as a condition of approval.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or, if used, its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an independent review board (IRB) or ethics committee can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s or ethics committee’s requirements or if the pharmaceutical product has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the pharmaceutical product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the pharmaceutical product candidate and, among other things, must develop methods for testing the identity, potency, quality and purity of the final pharmaceutical product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the pharmaceutical product candidate does not undergo unacceptable deterioration over its shelf life.
| 42 |
| Table of Contents |
United States Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the pharmaceutical product, proposed labeling and other relevant information are submitted to the FDA as part of an NDA/BLA requesting approval to market the product. The submission of an NDA/BLA is subject to the payment of substantial user fees by the applicant; a waiver of such fees may be obtained under certain limited circumstances, such as Orphan Drug Designation.
The FDA conducts a preliminary review of all NDAs/BLAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA/BLA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA/BLA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has twelve months in which to complete its initial review of a standard NDA/BLA and respond to the applicant, and eight months for a priority NDA/BLA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs/BLAs.
After the NDA/BLA submission is accepted for filing, the FDA reviews the NDA/BLA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. In addition to its own review, the FDA may refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the drug. If the FDA concludes that a REMS is needed, the sponsor of the NDA/BLA must submit a proposed REMS; the FDA will not approve the NDA/BLA without a REMS, if required.
Before granting regulatory approval of an NDA/BLA, the FDA will inspect the facilities at which the product is to be manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA/BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGMP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable it will outline the deficiencies in the submission and often will request additional testing or information.
The NDA/BLA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA/BLA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA/BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA will issue a “complete response” letter if the agency decides not to approve the NDA/BLA. The complete response letter usually describes all of the specific deficiencies in the NDA/BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA/BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific types of patients or diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing, which involves clinical trials designed to further assess a product’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Expedited Development and Review Programs
The FDA has a “Fast Track” program that is intended to expedite or facilitate the process for reviewing new drug products that meet certain criteria. Specifically, new drug products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor meets certain requirements and the FDA agrees to accept sections on a rolling basis.
Any product submitted to the FDA for marketing approval, including those submitted to a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval.
| 43 |
| Table of Contents |
Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or there is a significant improvement in the treatment, diagnosis or prevention of a disease compared with marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug product designated for priority review in an effort to facilitate the review.
Additionally, a product may be eligible for accelerated approval. Drug products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA generally requires that a sponsor of a drug product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies to establish safety and efficacy for the approved indication. Failure to conduct such studies, or conducting such studies that do not establish the required safety and efficacy may result in revocation of the original approval. In addition, the FDA currently requires as a condition for accelerated approval the pre-approval of promotional materials, which could adversely impact the timing of the commercial launch or subsequent marketing of the product.
Moreover, under the provisions of the new Food and Drug Administration Safety and Innovation Act, or FDASIA, enacted in 2012, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States there is no reasonable expectation that the cost of developing and making a drug product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA/BLA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not indicate an endorsement by the FDA and may not provide any benefit in the marketing approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. The designation of such drug also entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers.
Orphan drug status in the European Union has similar but not identical benefits in that jurisdiction. In the EU, orphan drug designation is provided for a drug that is intended to diagnose, prevent or treat a life-threatening or chronically debilitating condition which affects no more than 5 in 10,000 individuals in the EU (approximately 245,000 individuals) and for which no satisfactory method of diagnosis, prevention or treatment of the condition already exists, or if such method does exist, that the orphan product must be of significant benefit to the patient population over existing products. The company that obtains the first EU approval for a designated orphan drug indication receives marketing exclusivity for use of that drug for that indication for a period of 10 years for the 27 member states in the EU.
We currently have Orphan Drug Designation for Prolanta™ for the treatment of ovarian cancer in the United States. We intend to seek Orphan Drug designation from the EU for Prolanta™. Orphan Designation for ovarian cancer does not apply to the treatment of other cancers.
Orphan drug exclusivity may not prevent other market entrants. Competitors may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug or biological product as defined by the FDA or if our drug candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity.
| 44 |
| Table of Contents |
Post-Approval Requirements
Any pharmaceutical products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties.
We rely, and expect to continue to rely, on a third party and its subcontractors for the production of clinical and commercial quantities of Prolanta™. Manufacturers of our product candidates are required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. These regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of comprehensive records and documentation. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are also required to register their establishments and list any products they make with the FDA and to comply with related requirements in certain states or countries in which the manufacturer is located. These entities are further subject to periodic unannounced inspections by the FDA and certain state or foreign agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in serious and extensive restrictions on a product, manufacturer or holder of an approved NDA/BLA. These restrictions may include suspension of a product until the FDA is assured that quality standards can be met, continuing oversight of manufacturing by the FDA under a “consent decree,” which frequently includes the imposition of costs and continuing inspections over a period of many years, as well as possible withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of any FDA approval of our product candidate, some of our United States patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA/BLA plus the time between the submission date of an NDA/BLA and the approval of that application. Only one patent applicable to an approved product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
| 45 |
| Table of Contents |
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA or BLA. We believe that if Prolanta™ is approved as a biological product under a BLA, it should qualify for a 12-year period of exclusivity currently permitted by the Biologics Price Competition and Innovation Act of 2009, or BPCIA. Specifically, the BPCIA established an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on their similarity to existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator BLA holder. These periods apply only to biosimilar applications, and not products submitted under other regulatory applications. The BPCIA is complex and is only beginning to be interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning is subject to uncertainty. Orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances, independent of any protection offered by BPCIA.
U.S. Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act, to which we are subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity.
International Regulation
In addition to regulations in the United States, there are a variety of foreign regulations governing clinical trials and commercial sales and distribution of any product candidates. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trials may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with International Conference on Harmonization (ICH) / WHO Good Clinical Practice standards and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug under European Union regulatory systems, we must submit a marketing authorization application to the European Medicines Agency, or the EMA, where it will be evaluated by the Committee for Medicinal Products for Human Use. A favorable opinion typically results in the grant by the European Commission of a single marketing authorization that is valid for all European Union member states within 67 days of receipt of the opinion. The initial marketing authorization is valid for five years, but once renewed is usually valid for an unlimited period. The application used to file an NDA/BLA in the United States is similar to that required in the European Union, with the exception of, among other things, country-specific document requirements.
For countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCPs and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including The Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments.
| 46 |
| Table of Contents |
For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy and security provisions of the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Under the Veterans Health Care Act, or VHCA, drug companies are required to offer certain pharmaceutical products at a reduced price to a number of federal agencies including the United States Department of Veterans Affairs and United States Department of Defense, the Public Health Service and certain private Public Health Service - designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes purport to require that discounted prices be offered for certain United States Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/or register their sales representatives, as well as to prohibit pharmacies and other healthcare entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing, and to prohibit certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Pharmaceutical Coverage, Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors, including government health administrative authorities, managed care providers, private health insurers and other organizations. \These third-party payors are increasingly reducing reimbursement amounts for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
In the United States, a number of recent legislative reform measures have been passed to contain healthcare reimbursement for pharmaceuticals, including drugs such as our product candidates. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, known collectively as ACA, among other things, establishes annual fees to be paid by manufacturers for certain branded prescription drugs, requires manufacturers to participate in a discount program for certain outpatient drugs under Medicare Part D, increases manufacturer rebate responsibilities under the Medicaid Drug Rebate Program for outpatient drugs dispensed to Medicaid recipients, and expands oversight and support for the federal government’s comparative effectiveness research of services and products. We cannot predict the full impact of ACA or future reform measures on our operations and the current legal challenges and congressional efforts to repeal ACA add to the uncertainty.
| 47 |
| Table of Contents |
In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
Environmental Regulation
We and our contract manufacturer are subject to federal, state, local and international laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens and wastes. Although we believe that we have complied with these laws, regulations and policies in all material respects and have not been required to take any significant action to correct any noncompliance, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future.
Our manufacturing activities involve the controlled use of hazardous materials, including, but not limited to, certain hazardous chemicals. Although we believe that our contract manufacturer has safety procedures for handling and disposing of such materials that comply with the standards prescribed by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. In the event of such an accident, we could be held liable, directly or indirectly through our contractual relationships, for any damages that result and any such liability could exceed our resources.
Legal Proceedings
We are not currently a party to any material legal proceedings.
Employees
As of this date, we have 2 employees, both of whom are executive officers of the Company. We utilize and plan to continue to utilize key consultants and contractors for certain operations, such as manufacturing.
Properties
Our principal executive offices at 14405 Walters Road, Suite 780, Houston, Texas 77014 are occupied under a two-year lease expiring in March 2019 for approximately 2,500 square feet of space providing for rental payments of approximately $3,300 per month. Our telephone number is (281) 402-3167.
General
The information contained on our website is not incorporated by reference into this prospectus, and you should not consider any information contained on, or that can be accessed through, our website as part of this prospectus or in deciding whether to purchase our Common Stock. You may access and read our SEC filings through the SEC’s web site (http:www.sec.gov). This site contains reports, proxy and information statements and other information regarding registrants, including us, that file electronically with the SEC.
| 48 |
| Table of Contents |
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with, and is qualified in its entirety by, the financial statements and the related notes thereto in this prospectus.
Basis of Presentation
On April 8, 2017, Oncolix, Inc. (“Oncolix”) acquired approximately 66% of the outstanding common stock of Advanced Environmental Petroleum Producers Inc. (“AEPP”), a public entity traded on the OTC Bulletin Board with limited or no operating activities and no indebtedness. Oncolix paid $315,000 in cash and paid a fee to an investment banker of $50,000 in connection with this transaction.
On August 3, 2017, Oncolix merged into a wholly-owned subsidiary of AEPP and AEPP acquired all of the outstanding equity interest of Oncolix and assumed responsibility for the issuance of equity securities under Oncolix’s options, warrants and convertible debt as described further herein. This transaction is treated as a reverse acquisition of AEPP for financial accounting and reporting purposes. As such, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes.
Accordingly, the financial statements of AEPP for all periods prior to August 3, 2017 are those of Oncolix. Because of our acquisition of a controlling interest on April 8, 2017, the financial statements of AEPP are consolidated with Oncolix since April 2017. The $365,000 purchase price for stock of AEPP by Oncolix is treated as an expense of the merger, and the shares of AEPP Common Stock not owned by Oncolix are treated as shares outstanding subsequent to April 2017. AEPP’s assets, liabilities and results of operations for periods after April 2017 are the consolidated assets, liabilities and results of operations of AEPP and Oncolix.
In the reverse merger, each stockholder of Oncolix received 20 shares of a substantially identical class of AEPP stock (Common or Series A Preferred, each with a par value of $0.0001) in exchange for each share of Oncolix (par value $0.001). In addition, each option or warrant became exercisable for 20 shares of the identical class of AEPP stock for each share of Oncolix, and the exercise price was adjusted by dividing the price per Oncolix share by 20. The shares of Common Stock owned by Oncolix were cancelled in the merger. After the merger, including shares issued by Oncolix in July 2017 as payment of accrued interest at June 30, 2017, AEPP had 103,536,703 shares of Common Stock, $0.0001 par value, issued and outstanding, of which 71,770,200 shares were issued to the former stockholders of Oncolix. In addition, AEPP had 62,138,680 shares of Series A Preferred Stock, $0.0001, issued and outstanding, all of which were owned by the former stockholders of Oncolix. The financial statements for all periods have been adjusted to reflect the adjusted number of shares resulting from the exchange ratio in the reverse merger (the 20:1 exchange ratio), effectively a stock split for accounting purposes, and the adjustment of the par value to $0.0001.
As used in the following discussion, “We,” “Us,” “Our,” “AEPP”, or the “Company” refers to Oncolix prior to April 2017 and the combined AEPP and Oncolix subsequent to such date.
| 49 |
| Table of Contents |
Overview
We are a clinical-stage biopharmaceutical company developing medical therapies for the treatment of cancer. We commenced operations on January 24, 2007, and since 2008, our activities have been focused on the development of Prolanta™, a biological drug for the treatment of cancer. Our initial focus is ovarian cancer, and we have commenced a Phase I clinical trial of Prolanta™ in the treatment of ovarian cancer after we received clearance from the FDA. We also believe that Prolanta™ may be effective in the treatment of breast, prostate and other cancers.
We believe our success primarily will be dependent on (i) the safety and efficacy of our drug candidate Prolanta™ in the treatment of ovarian and other cancers, (ii) our ability to retain and attract additional members of management, and (iii) our ability to raise sufficient capital to fund our development plans for Prolanta™ and/or acquire other product candidates. There are numerous other factors that will affect our success, such as competitive products, our ability to maintain a proprietary position, and the relative resources of others.
In early 2016, we commenced a Phase 1 clinical trial of Prolanta™ for the treatment of ovarian cancer. This trial will evaluate three dose-levels of Prolanta™ according to a protocol submitted to the FDA. We completed the first dosing group in June 2016, and our Data and Safety Monitoring Board (“DSMB”) has given us permission to dose a second group of patients with a higher dose of Prolanta™. Dosing of this second group will commence after we manufacture additional supplies of Prolanta™.
We have no revenues, and we will need to complete additional development and testing of Prolanta™ before we seek governmental approvals to sell the drug for patient use. Also, it is likely we will need to complete some additional development in order to seek a collaboration with a partner to fund development of Prolanta™. Accordingly, our liquidity and results of operations are dependent on our ability to obtain additional funds for our development activities.
We have experienced negative cash flows from operations since the commencement of operations, and we expect these losses to continue into the foreseeable future as we continue the development and clinical testing of Prolanta™ and seek regulatory approval to market products. In August 2017, we completed a debt financing through the issuance of the Convertible Notes that should fund operations into 2018. The Convertible Notes mature in seven installments during 2018. Accordingly, our continued operations are dependent on our ability to raise additional capital through the sale of equity or debt securities or through the licensing of our products. In the event we are unable to raise sufficient funds, we would have to substantially alter, or possibly even discontinue or curtail operations, or sell assets at distressed prices. This uncertainty raises substantial doubt about our ability to continue as a going concern, as noted in the opinion of our independent registered public accounting firm in the audited financial statements as of December 31, 2016. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
On August 3, 2017, the Company entered into the Purchase Agreement with various investors in connection with a $4,190,463 Convertible Note and Warrant financing. Pursuant to the Purchase Agreement, the Company issued Convertible Notes to the Purchasers in an aggregate principal amount of $4,190,463 for a purchase price of $3,561,894, consisting of gross cash proceeds of $2,000,000 and the exchange of existing convertible debt and related accrued interest of Oncolix totaling $1,561,894. The Convertible Notes were issued at a 15% discount as noted.
The Convertible Notes bear interest at a rate of 10% per annum, payable quarterly on November 1, 2017, February 1, 2018, and May 1, 2018 and thereafter, on a monthly basis until maturity. The interest is payable in cash or, at the option of the Company, subject to compliance with certain strict volume and price conditions, in fully tradable Common Stock at a 25% discount to the average of the five lowest closing bid prices for the 20 trading days prior to the interest payment date. The final maturity of the Convertible Notes is November 1, 2018, but commencing on May 1, 2018 and on the first day of each month thereafter until maturity, AEPP is required to redeem an amount of the Convertible Notes equal to 1/7th of the original principal amount, plus 10% of such monthly redemption amount as a bonus. The principal payments shall be made in cash or, subject to the satisfaction of with certain strict volume and price conditions, in Common Stock valued at a 25% discount to the average of the five lowest closing bid prices for the 20 trading days prior to the amortization date.
| 50 |
| Table of Contents |
The Convertible Notes are convertible at the option of the Purchasers at a conversion price equal to the lesser of (i) $0.075 per share of Common Stock, (ii) 75% of the 10-day average closing bid price of AEPP Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of the Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share). The Convertible Notes contain customary anti-dilution protection, including full-ratchet anti-dilution adjustments in the event of certain dilutive issuances (that adjust both price and share amounts).
In the event of a closing of an $8 million financing in connection with a change in listing of the Common Stock to Nasdaq or the NYSE, the Convertible Notes shall be subject to mandatory conversion into Common Stock at a 30% discount to the offering price. AEPP shall be required to offer to prepay in cash the aggregate principal amount of the Convertible Notes at 120% of the principal amount thereof plus any unpaid accrued interest to the date of repayment, at maturity, on the sale of substantially all of the assets of AEPP, upon a change of control, upon a qualified offering, or upon a “fundamental transaction” of the Company (such as a reverse merger); in such an event, the Purchasers shall have the right to convert the Convertible Notes prior to the date of any such prepayment.
The Convertible Notes contain standard negative covenants customary for transactions of this type. The events of default are also customary for transactions of this type, including default in timely payment of principal or interest, failure to observe or perform any covenant or agreement contained in the Convertible Note and Transaction Documents, the commencement of bankruptcy or insolvency proceedings, failure to timely deliver conversion shares underlying the Convertible Notes, failure to timely file Exchange Act filings, and failure to satisfy certain Equity Conditions.
Use of Estimates
The preparation of financial statements in conformity with US generally accepted accounting principles (“GAAP”) requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. In addition, significant estimates were made regarding realization of deferred tax assets, the calculation of the fair value of stock options awarded, the estimation of the value of warrants issued in conjunction with capital stock, and the projected realization of the carrying basis of our assets. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from the estimates and assumptions used.
Critical Accounting Policies
Our significant accounting policies are described in more detail in the notes to our financial statements appearing elsewhere.
Recent accounting standards. On February 25, 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. ASU No. 2016-02 requires lessees to recognize right-of-use assets and lease liabilities on the balance sheet for all leases of property, plant and equipment with lease terms greater than 12 months. Prior to this accounting standard, only capital leases were recognized on the balance sheet. ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2019, and interim periods within fiscal years beginning after December 15, 2020, and early adoption is permitted. This ASU must be applied using a retrospective approach upon adoption. The Company believes this ASU will not have a significant effect on its financial statements.
Results of Operations
General. We are in the early stage of development of our product candidate, Prolanta™, which will require extensive testing in humans and regulatory approvals prior to sale for medical use. To date, we have not generated any revenues from operations. While we may in the future generate revenue from a variety of sources, including license fees, milestone payments, research and development payments in connection with strategic partnerships and/or product sales, Prolanta™ may never be successfully developed or commercialized. At June 30, 2017, Oncolix had an accumulated deficit of $20.3 million primarily as a result of expenditures for research and development and general and administrative expenses. We expect to continue to incur substantial losses from operations for the foreseeable future and there can be no assurance that we will ever generate significant revenues or achieve profitability.
| 51 |
| Table of Contents |
Research and Development Expense. Research and development expense includes wages and associated employee benefits of employees engaged in research; stock-based compensation expense related to such employees, the cost of consultants engaged in our research activities; the cost of developing methods to manufacture our investigational drug products; the costs of production of investigation drugs for use in research, animal and human testing; the costs of the testing of our products in animal models and in human testing; the cost of research conducted on our behalf at academic institutions; the depreciation of equipment used in research; the cost of obtaining patents for the protection of our intellectual property; and other costs associated with these activities.
We expect to incur substantial research and development expenses in the foreseeable future as we continue product development of Prolanta™. The amount and timing of such costs will depend on various factors, including the availability of resources to fund these activities, the timing or regulatory approvals for stages of testing or commercialization, the results of testing at each stage of product development, the size and length of clinical studies required at each stage of development, the regulatory requirements for commercialization, and the costs of intellectual property protection, among other factors. In general, expenses increase substantially as a product progresses from early to later stages of development, and we expect our research and development expenses to increase in a similar manner. In the event that we license or acquire additional product candidates, research and development expense may increase as a result of the development of such product candidates.
General and Administrative Expense. General and administrative expense includes wages and associated employee benefits of employees engaged in administration; stock-based compensation expense related to such employees, professional fees for legal, consulting, audit and tax services, rent and other general operating expenses not otherwise included in research and development.
We expect general and administrative expenses to increase as we expand our activities to support our expanded research and development activities and incur costs related to the expanded infrastructure and increased professional fees associated with being a reporting company under the Exchange Act.
Comparison of Years Ended December 31, 2016 and 2015. The results of operations of Oncolix for the years ended December 31, 2016 and 2015 are as follows:
|
|
| Year Ended December 31, |
|
| Increase (Decrease) |
| ||||||||||
|
|
| 2016 |
|
| 2015 |
|
| $ |
|
| % |
| ||||
| Revenue |
| $ | - |
|
| $ | - |
|
| $ | - |
|
|
| - | % |
| Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Research and development expenses |
|
| 709,009 |
|
|
| 561,671 |
|
|
| 147,338 |
|
|
| 26 | % |
| General and administrative expenses |
|
| 620,835 |
|
|
| 777,499 |
|
|
| (156,664 | ) |
|
| -20 | % |
| Total operating expenses |
|
| 1,329,844 |
|
|
| 1,339,170 |
|
|
| (9,326 | ) |
|
| -1 | % |
| Interest expense on convertible notes |
|
| 55,912 |
|
|
| 7,812 |
|
|
| 48,100 |
|
|
| 616 | % |
| Net loss |
| $ | (1,385,756 | ) |
| $ | (1,346,982 | ) |
|
| (38,774 | ) |
|
| 3 | % |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Loss per Common Stock share - basic and diluted |
| $ | (0.019 | ) |
| $ | (0.019 | ) |
| $ | (0.001 | ) |
|
| 4 | % |
| Weighted average Common Stock shares outstanding - basic and diluted |
|
| 71,148,163 |
|
|
| 71,790,615 |
|
|
|
|
|
|
|
|
|
Research and development expense increased approximately $147,000 from 2015 to 2016 principally as a result of the costs of the Phase I clinical trial. Research costs in 2015 were principally comprised of patent related costs of approximately $192,000, Prolanta™ manufacturing costs of approximately $294,000, and development of clinical laboratory assays of approximately $47,000. Research costs in 2016 were principally comprised of patent related costs of approximately $204,000, Prolanta™ manufacturing costs of approximately $305,000, development of clinical laboratory assays of approximately $10,000 and clinical trial subject-related costs of approximately $168,000. As noted previously, the enrollment of subjects in the second dosing group in the clinical trial was approved in July 2016 but was not commenced, so only the first half of 2016 includes significant clinical trial costs.
General and administrative costs declined approximately $157,000 from 2015 to 2016, principally as a result of approximately $173,000 in stock-based compensation expense in 2015 related to grants in 2011. These grants were fully expensed in 2015, and no expense was recorded in 2016. In addition, Oncolix incurred approximately $35,000 in financial consultant costs in 2015, and the consultant was not engaged in 2016. These 2015 costs were partially offset by approximately $42,000 of legal and auditing costs in 2016 in preparation for financing activities.
| 52 |
| Table of Contents |
Interest expense in 2015 was attributable to certain bridge notes that converted into Oncolix Series A Preferred Stock in January 2015. Interest expense in 2016 was attributable to the secured notes sold in September and November 2016, and these notes included features that resulted in a higher recorded interest expense. Interest expense in 2016 includes approximately $51,000 of accretion attributable to the warrant to acquire Oncolix Series A Preferred Stock sold with the notes as well as a premium equal to 20% of the principal balance that may be payable at maturity.
The net loss for 2016 increased approximately $39,000, or 3%, as a result of these offsetting effects. The decline in stock-based compensation and financial consulting from 2015 to 2016 was offset by the higher clinical trial costs and interest expense in 2016.
Comparison of Six Months Ended June 30, 2017 and 2016. The results of operations of the Company for the six months ended June 30, 2017 and 2016 are as follows:
|
|
| Six Months Ended June 30, |
|
| Increase (Decrease) |
| ||||||||||
|
|
| 2017 |
|
| 2016 |
|
| $ |
|
| % |
| ||||
| Revenue |
| $ | - |
|
| $ | - |
|
| $ | - |
|
|
| - | % |
| Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Research and development expenses |
|
| 77,095 |
|
|
| 579,202 |
|
|
| (502,107 | ) |
|
| -87 | % |
| General and administrative expenses |
|
| 794,124 |
|
|
| 272,558 |
|
|
| 521,566 |
|
|
| 191 | % |
| Total operating expenses |
|
| 871,219 |
|
|
| 851,760 |
|
|
| 19,459 |
|
|
| 2 | % |
| Interest expense on convertible notes |
|
| 462,679 |
|
|
| - |
|
|
| 462,679 |
|
| * |
| |
| Net loss |
| $ | (1,333,898 | ) |
| $ | (851,760 | ) |
|
| (482,138 | ) |
|
| 57 | % |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Loss per Common Stock share - basic and diluted |
| $ | (0.016 | ) |
| $ | (0.012 | ) |
| $ | (0.004 | ) |
|
| 30 | % |
| Weighted average Common Stock shares outstanding - basic and diluted |
|
| 85,985,247 |
|
|
| 71,142,970 |
|
|
|
|
|
|
|
|
|
_______
* Not Meaningful
Research and development expense decreased $502,107 from the six months ended June 30, 2016 to the comparable period in 2017 principally as a result of the costs of the Phase I clinical trial conducted in 2016. Manufacturing activities were conducted in late 2015 and early 2016, and subject enrollment, treatment and monitoring occurred in the first half of 2016. Patent related costs decreased from approximately $82,000 in 2016 to approximately $45,000 in 2017, primarily as a result of the reduction in the number of matters being prosecuted as prior filings were granted. Prolanta™ manufacturing costs decreased from approximately $305,000 in 2016 to approximately $3,000 in 2017 because no drug was manufactured during the 2017 period. Clinical trial related costs and related assay-development costs decreased from approximately $180,000 in 2016 to approximately $22,000 in 2017 because of need for additional drug supplies to continue the clinical trial in 2017.
General and administrative costs increased $521,566 from the six months ended June 30, 2016 to the comparable period in 2017 principally as a result of (i) the expensing of the $365,000 cost to acquire control of the public shell and (ii) and legal costs incurred in 2017 as the Company prepared for its financing activities and the merger of Oncolix and AEPP. Audit costs increased from $0 in 2016 to approximately $37,000 in 2017. Legal costs for corporate activities increased from $0 in 2016 to $99,000 in 2017. Commencing with the acquisition of control of AEPP, the Company incurred approximately $26,000 of transfer agent, legal and other costs associated with the public trading of AEPP and the merger.
Interest expense in the six months ended June 30, 2017 was attributable to the notes sold in 2017 as well as the notes sold in September and November 2016. These notes included features that resulted in a higher recorded interest expense. Interest expense in 2017 includes approximately $407,000 of accretion attributable to the warrants sold with the notes as well as a premium equal to 20% of the principal balance that may be payable at maturity and the fees and expenses of the offering.
The net loss for the six months ended June 30, 2017 increased approximately $482,000, or 57%, compared to the 2016 period as a result of these offsetting effects. The decline in clinical trial and manufacturing costs from 2016 to 2017 was more than offset by the higher merger costs, including the cost of acquiring control of the public shell and the legal and auditing costs and interest expense in 2017. However, because of the higher number of outstanding shares of Common Stock from the consolidation of AEPP and Oncolix in April 2017, net loss per Common Share increased by30%, or $0.004 per Common share.
| 53 |
| Table of Contents |
Liquidity and Capital Resources
We are a clinical-stage biopharmaceutical company focused on the development and commercialization of Prolanta™ for the treatment of cancer. The FDA has granted clearance to conduct a Phase I clinical trial in subjects with ovarian cancer, and we commenced the first phase of this study in early 2016. To complete this study we require additional drug supplies. If this and other studies are successful, substantial additional resources will be necessary to complete the development of Prolanta™ and seek regulatory approval to market the drug. Based on our current expectations, we believe this development will require at least $25 million of additional funds.
The development of pharmaceutical products requires laboratory and preclinical studies before seeking regulatory approval for human testing, as well various stages of testing in humans. We have completed these laboratory and preclinical studies, but the results of our human studies or requests of regulatory authorities may require additional studies. In addition, the manufacturing of Prolanta™ must be conducted under controlled conditions, and we may seek to improve the process. After we complete the Phase I clinical trial, the human studies to determine the safety of Prolanta™ will be conducted in larger and larger groups of patients. All of these activities will require the expenditure of substantial funds. In the event that we obtain or develop additional product candidates in addition to Prolanta™, we will incur similar types of costs.
The Company believes that it has sufficient cash on hand to fund its operations into 2018. The exact duration that our liquidity will be sufficient to fund operations depends upon many factors, some of which are outside the control of the Company, and is difficult to predict. We expect our research and development expenses to increase in connection with our ongoing activities relating to Prolanta™, particularly as we focus on and proceed with our clinical trials for our Prolanta™ product. In addition, our expenses could increase beyond current expectations if applicable regulatory authorities, including the FDA, require that we perform studies in addition to those that we currently anticipate, assuming we successfully complete our Phase I clinical trials, commence and complete our proposed Phase II clinical trials. We currently estimate that we require approximately $2 million to complete the Phase I clinical trial, depending on the cost and availability of drug supplies. We will still need substantial additional capital in the future in order to complete the clinical trials and obtain regulatory approval of Prolanta™. Accordingly, we will need to raise additional capital to fund future operations and we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Such funding may not be available on favorable terms, if at all. Failure to raise additional capital could cause us to curtail operations and result in the loss of your investment in our common stock.
If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or to grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
Our forecast of the period of time through which our current capital will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
As of June 30, 2017, we had cash of approximately $93,000 and secured notes payable with a balance due at maturity of approximately $1,705,000. In addition, we have liabilities to third parties and related parties totaling approximately $1,012,000 and deferred compensation to our employees of approximately $1,268,000 as of June 30, 2017.
We completed a debt financing subsequent to June 30, 2017 as described in “-Overview”. The proceeds from this debt financing will fund our operations into 2018, but also requires repayment in the event the holders do not convert the Convertible Notes into Common Stock. According, the Convertible Notes provide short-term liquidity but require us to raise additional funds promptly.
Our employees have agreed to continue to defer the payment of prior compensation until Oncolix has raised substantial additional equity. We will require additional resources to fund future activities and to complete our Phase I clinical study.
We expect to incur substantial expenditures in the foreseeable future for the research, development and potential commercialization of Prolanta™. We will require additional financing to develop, obtain regulatory approvals, fund operating losses, and, if deemed appropriate, establish additional manufacturing, sales and marketing capabilities.
| 54 |
| Table of Contents |
Accordingly, our continued operations are dependent on our ability to raise additional capital through the sale of equity or debt securities or through the licensing of our products. There can be no assurances that we will be able to obtain funding, or that such arrangements will be on terms attractive to us. In the event we are unable to raise sufficient funds, we would have to substantially alter, or possibly even discontinue, operations, or sell assets at distressed prices. This uncertainty raises substantial doubt about our ability to continue as a going concern, as noted in the opinion of our independent registered public accounting firm in the audited financial statements as of December 31, 2016.
The following table shows a summary of the Company’s cash flows for the periods presented.
|
|
| Year Ended December 31, |
|
|
|
|
| Six Months Ended June 30, |
| |||||||||||||||
|
|
| 2016 |
|
| 2015 |
|
| Change |
|
| 2017 |
|
| 2016 |
|
| Change |
| ||||||
| Cash provided by (used in): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
| Operating activities |
| $ | (445,269 | ) |
| $ | (907,584 | ) |
| $ | 462,315 |
|
| $ | (952,508 | ) |
| $ | (244,927 | ) |
| $ | (707,581 | ) |
| Financing activities |
|
| 186,470 |
|
|
| 1,176,690 |
|
|
| (990,220 | ) |
|
| 1,030,621 |
|
|
| 2,470 |
|
|
| 1,028,151 |
|
| Net increase (decrease) in cash |
|
| (258,799 | ) |
|
| 269,106 |
|
| $ | (527,905 | ) |
|
| 78,113 |
|
|
| (242,457 | ) |
| $ | 320,570 |
|
| Cash at beginning of period |
|
| 274,063 |
|
|
| 4,957 |
|
|
|
|
|
|
| 15,264 |
|
|
| 274,063 |
|
|
|
|
|
| Cash at end of period |
| $ | 15,264 |
|
| $ | 274,063 |
|
|
|
|
|
| $ | 93,377 |
|
| $ | 31,606 |
|
|
|
|
|
| Non-cash activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Stock issued for interest payable |
| $ | - |
|
| $ | - |
|
| $ | - |
|
| $ | 13,358 |
|
| $ | - |
|
| $ | 13,358 |
|
| Interest paid in cash |
| $ | - |
|
| $ | - |
|
| $ | - |
|
| $ | 8,362 |
|
| $ | - |
|
| $ | 8,362 |
|
Cash Provided by (Used in) the Years Ended December 31, 2016 and 2015
Cash used in operating activities decreased approximately $462,000 from the year ended December 31, 2015 to the year ended December 31, 2016, reflecting the reduced cash resources available to Oncolix in 2016. With the cash proceeds from the sale of Series A Preferred Stock and warrants in January 2015, Oncolix began the activities necessary to commence its Phase I clinical trial. This included commencing the manufacturing of supplies of Oncolix for the clinical trial. During 2015, trade accounts payable and amounts payable to related parties decreased approximately $123,000, reflecting that most operating activities were paid during the period. The enrollment and dosing of subjects in the clinical trial commenced in early 2016, and the first doing phase was completed in the first half of 2016. Oncolix began the sale of a unit of notes and warrants in September 2016 as noted below. However, as a result of reduced cash resources available, the deferral of payment of certain operating costs occurred in 2016, and accounts payable and amounts payable to related parties increased approximately $760,000 in total in 2016.
Cash provided by financing activities decreased approximately $990,000 from the year ended December 31, 2015 to the year ended December 31, 2016. In January 2015, Oncolix closed the sale of units of Series A Preferred Stock and a warrant to acquire Series A Preferred stock, with net proceeds of approximately $1,177,000. In addition, certain bridge loans totaling approximately $1,809,000 were also converted into these units. Oncolix also issued units to a clinical research firm in exchange for $450,000 of services to be provided in the clinical trial. These cash proceeds were used to commence the activities related to the Phase I clinical trial described above. During September and November 2016, Oncolix sold units of secured promissory notes and warrants to acquire Series A Preferred Stock for net proceeds of approximately $184,000. These proceeds would not fully fund 2016 activities, resulting in the increase in liabilities in 2016 described above.
Cash Provided by (Used in) the Six Months Ended June 30, 2017 and 2016
Cash used in operating activities increased approximately $708,000 from the six months ended March 31, 2016 as compared to the six months ended June 30, 2017, reflecting the increased cash resources available to the Company in 2017. During the 2016 period, the Company was conducting its Phase I clinical trial and incurred higher research and development expenses. However, a substantial portion of these costs were not paid during the period, resulting in an approximately $492,000 increase in accounts payable and amounts payable to related parties during the six months ended June 30, 2016. During the six months ended June 30, 2017, Oncolix received proceeds from the sale of units of a secured note and warrants, and these proceeds were used to fund current activities. As a result, expenses were funded by cash and there was no significant change in accounts payable and amounts payable to related parties during this period. During this period, we acquire control of a public shell for $365,000. As a result of the merger, these costs were expensed as acquisition costs, and additional accounting and legal costs were incurred, increasing the cash used by operating activities in this period.
Cash provided by financing activities increased approximately $1,028,000 from the six months ended June 30, 2016 as compared to the six months ended June 30, 2017. During the 2017 period Oncolix sold units of a secured promissory note and a warrant to acquire Common Stock with net proceeds of approximately $1,026,000. These cash proceeds were used to fund ongoing activities as described above, with no significant increase in liabilities.
| 55 |
| Table of Contents |
Off-Balance Sheet Arrangements
We do not currently have, nor have we ever had, any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts.
Quantitative and Qualitative Disclosures about Market Risks
We held no marketable securities at any time during the periods presented. Our existing short-term debt is at a fixed rate, but has a conversion feature based on the market price of our equity securities, which requires a period valuation adjustment. Because we intend to conduct activities globally, certain of our contracts either will require payment in foreign currencies or will be billed in US dollars based on a specified exchange rate. In particular, payments under our previous manufacturing services agreements were adjusted for certain changes in the exchange rate for Canadian dollars. Our exposure to changes in currency exchange rates will be dependent on the amounts of future billings by each service provider.
Net Operating Loss Tax Carry-Forwards
As of December 31, 2016, we had net operating loss carryforwards of approximately $8.4 million and an Orphan Drug Tax Credit carryforward of approximately $0.4 million to offset future federal income taxes. In addition, we have deferred the deduction of approximately $6.9 million in research and administrative costs as of December 31, 2016, and these may be deducted in future federal tax returns. We make decisions regarding tax attributes annually based on the results for the entire year, and such decisions include expensing for tax loss carryforwards or deferrals. As of June 30, 2017, we estimate the combined tax loss carryforward and deferral to increase by approximately $0.3 million for the period.
Current federal tax laws include substantial restrictions on the utilization of net operating loss and tax credits in the event of a change in the ownership, as such term is defined in the federal tax code. Even if the carryforwards are available, they may be subject to annual limitations, lack of future taxable income, or future ownership changes that could result in the expiration of the carryforwards before they are utilized. Our principal stockholder, GHC is considering a reorganization that may result in a change in ownership that would restrict our ability to utilize our prior accumulated tax benefits, but we do not expect such reorganization to have a significant effect on our operations.
At December 31, 2016, and June 30, 2017, we recorded a 100% valuation allowance against our deferred tax assets as our management believes it is uncertain that they will be fully realized. If we determine in the future that we will be able to realize all or a portion of our net operating loss carryforwards, an adjustment to our net operating loss carryforwards would increase net income in the period in which we make such a determination.
DETERMINATION OF OFFERING PRICE
We are not selling any of the Common Stock that we are registering. The Common Stock will be sold by the selling security holders as detailed in this prospectus. Such selling security holders may sell the Common Stock at the market price as of the date of sale or a price negotiated in a private sale. Our Common Stock is listed for quotation on the OTC Pink quotation systems under the symbol “AEPP.”
MARKET PRICE INFORMATION AND DIVIDEND POLICY
Market Information
Our Common Stock is quoted on the OTC Pink under the symbol “AEPP.” Shares of our Common Stock have historically been thinly traded, and currently there is a limited, sporadic and highly volatile trading market for our Common Stock. As a result, our stock price as quoted by the OTC Pink may not reflect an actual or perceived value. The following table sets forth the approximate high and low bid prices for our Common Stock for the last two fiscal years and interim periods. The Company Common Stock did not commence to trade until the first quarter of 2016. The quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions.
| 56 |
| Table of Contents |
|
|
| High |
|
| Low |
| ||
|
|
|
|
|
|
|
| ||
| YEAR 2017 |
|
|
|
|
|
|
|
|
| Quarter ended June 30, 2017 |
| $ | 0.07 |
|
| $ | 0.03 |
|
| Quarter ended March 31, 2017 |
| $ | 0.06 |
|
| $ | 0.01 |
|
|
|
|
|
|
|
|
|
|
|
| YEAR 2016 |
|
|
|
|
|
|
|
|
| Quarter ended December 31, 2016 |
| $ | 0.30 |
|
| $ | 0.01 |
|
| Quarter ended September 30, 2016 |
| $ | 0.50 |
|
| $ | 0.30 |
|
| Quarter ended June 30, 2016 |
| $ | 0.40 |
|
| $ | 0.36 |
|
| Quarter ended March 31, 2016 |
| $ | 0.40 |
|
| $ | 0.36 |
|
|
|
|
|
|
|
|
|
|
|
| YEAR 2015 |
|
|
|
|
|
|
|
|
| Quarter ended December 31, 2015 |
|
| -- |
|
|
| -- |
|
| Quarter ended September 30, 2015 |
|
| -- |
|
|
| -- |
|
| Quarter ended June 30, 2015 |
|
| -- |
|
|
| -- |
|
| Quarter ended March 31, 2015 |
|
| -- |
|
|
| -- |
|
The closing sales price of our Common Stock on August 24, 2017 was $0.11 per share.
Holders
The number of record holders of the Company’s Common Stock, as of August 21, 2017, is approximately 98.
Dividends
The Company has not declared any dividends with respect to its Common Stock and does not intend to declare any dividends in the foreseeable future. The future dividend policy of the Company cannot be ascertained with any certainty.
Securities Authorized for Issuance Under Equity Compensation Plans
The Company doesn’t maintain a shareholder approved equity compensation plan and disclosure with respect to existing options issued to our executive officers is set forth in “Management – Outstanding Option Awards at Fiscal Year End.”
| 57 |
| Table of Contents |
Our executive officers and directors and their respective ages, positions and biographical information are set forth below.
| Name |
| Age |
| Position |
| Michael T. Redman |
| 62 |
| Director, President and Chief Executive Officer |
| J. Donald Payne |
| 62 |
| Director, Secretary |
Michael T. Redman. Mr. Redman has served as the Chief Executive Officer and a Director of Oncolix since the inception of operations in January 2007, and as Chief Executive Officer and Director of AEPP since April 2017. Mr. Redman created and named Oncolix from technology acquired from GHS. Since joining Oncolix, Mr. Redman has prepared business plans, presented at major life science conferences and has been primarily responsible for eight issued US patents and additional foreign patents. Prior to forming Oncolix, Mr. Redman was the chief executive officer of Bone Medical in Australia and thereafter the chief executive officer and co-founder of Opexa Pharmaceuticals, which commenced operations in February of 2001 and was sold to PharmaFrontiers Corporation in November 2004. In addition to the above positions, Mr. Redman also has held key management positions with Zonagen (now Repros Therapeutics), Aronex Pharmaceuticals, Biovail, and American Home Products (now Pfizer). Mr. Redman is an active member of the Licensing Executives Society and formerly served on its Executive Committee. Mr. Redman earned a BA in Biology from the University of Missouri and a Masters in Business Administration from the University of Phoenix.
J. Donald Payne. Mr. Payne has served as the Senior Vice President - Administration and Secretary of Oncolix since 2011, and has served as a Director and as the Secretary of AEPP since April 2017. In such capacities, Mr. Payne has been responsible for assisting with managing the clinical and manufacturing operations of Oncolix as well as handling regulatory filings and intellectual property matters. Mr. Payne also currently is the co-founder, president and director of AuroRad, Inc., an unaffiliated start-up that intends to develop radiation oncology products. Until June 2012, Mr. Payne was a director and chairman of the audit committee of Pressure Biosciences, Inc., a Nasdaq-listed life science instrumentation company. Previously, Mr. Payne held various senior management positions at Nanospectra Biosciences, Sensus Drug Development, LifeCell Corporation, Aprogenex and Entex Energy. Mr. Payne has co-authored 9 scientific publications and is a co-inventor of 2 issued patents. Mr. Payne is a Summa Cum Laude graduate of Rice University with a Masters in Business Administration (1992) and a Summa Cum Laude graduate of Texas A&M University (1976) with a Bachelor in Business Administration. Mr. Payne is a licensed certified Public Accountant in Texas (1978).
| 58 |
| Table of Contents |
Compensation Committee Interlocks and Insider Participation; Board Committees
The Company does not presently have a separately designated audit committee, compensation committee, nominating committee, executive committee or any other committee of its Board of Directors. As such, the directors act in those capacities. The Company believes that committees of the Board are not necessary at this time given that the Company is a development stage company. However, the directors will continue to study this matter, and the Company plans to add Board members and/or committees of the Board as its business develops.
Messrs. Redman and Payne have not participated in deliberations concerning executive officer compensation. None of the Company’s executive officers serves on the board of directors or compensation committee of a company that has an executive officer that serves on the Company’s board of directors. No member of the Company’s board of directors is an executive officer of a company in which one of the Company’s executive officers serves as a member of the board of directors or compensation committee of that company.
Director Independence
Quotations for our Common Stock are entered on the OTC Pink inter-dealer quotation system, which does not have director independence requirements. For purposes of determining director independence, the Company applied the definitions set out in Nasdaq Rule 4200(a)(15). Under Nasdaq Rule 4200(a)(15), a director is not considered to be independent if he or she is also an executive officer or employee of the corporation. As a result, the Company does not have any independent directors. Our directors are executive officers of the Company.
Involvement in Certain Legal Proceedings
There are currently no material pending legal proceedings to which the Company is a party or of which any of its property is the subject, in which any of the above referenced directors or officers is a party adverse to the Company or has a material interest adverse to the Company. Furthermore, during the past ten years, none of the Company's officers or directors described above were involved in any legal proceedings that are material to an evaluation of the ability or integrity of such directors and officers.
Compensation to Officers of the Company
The following tables contain compensation data for our named executive officers as of the fiscal years ended December 31, 2016 and 2015:
Summary Compensation Table
The following table sets forth the cash and non-cash compensation for the last two fiscal years awarded to or earned by our named executive officers.
| Name and Principal Position |
| Year |
| Salary |
|
| Non-Equity Incentive |
|
| Total |
| |||
| Michael T. Redman, |
| 2016 |
| $ | 290,016 | (1) |
| $ | - |
|
| $ | 290,016 |
|
| President and Chief Executive Officer |
| 2015 |
| $ | 290,016 | (1) |
| $ | 20,000 | (2) |
| $ | 310,016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| J. Donald Payne, |
| 2016 |
| $ | 95,700 | (3) |
| $ | – |
|
| $ | 95,700 |
|
| Secretary |
| 2015 |
| $ | 99,600 | (3) |
| $ | – |
|
| $ | 99,600 |
|
____________
| (1) | The base salary of Mr. Redman was $290,016 for the periods noted. A variable percentage of salary was paid each period depending on the available cash resources of the Company, and the remainder was deferred. During 2016, $99,254 was paid in cash and $190,762 was deferred. During 2015, $145,008 was paid in cash, and $145,008 was deferred. Mr. Redman has agreed to defer payment of the unpaid amounts for these and prior years until such time as the Company has raised additional funds, and these unpaid amounts are reflected as deferred compensation in the Company’s financial statements. |
| (2) | The board of directors of Oncolix adopted an incentive cash compensation plan for Mr. Redman, pursuant to which he earned $20,000 in 2015. Mr. Redman has agreed to defer payment of this amount until such time as the Company has raised additional funds, and this unpaid amount is reflected as deferred compensation in the Company’s financial statements. |
| (3) | The base salary of Mr. Payne is $200,000 per year, and the amounts incurred by the Company vary based on the percentage of time devoted to the Company’s activities. During 2016 and 2015, approximately 50% and 48% of Mr. Payne’s time was devoted to Oncolix, but increased in 2017 to between 80% and 95%. A variable percentage of salary was paid during 2016 depending on the available cash resources of the Company, and the remainder was deferred. During 2016, $67,800 was paid in cash, and $27,900 was deferred. During 2015, all amounts were paid. Mr. Payne has agreed to defer payment of the unpaid amounts for these and prior years until such time as the Company has raised additional funds, and these unpaid amounts are reflected as deferred compensation in the Company’s financial statements. |
| 59 |
| Table of Contents |
Employment Arrangements
Mr. Redman is paid pursuant to a verbal arrangement, and his annual salary is $290,000. In 2016, Mr. Redman and the Company entered into a severance agreement providing for the payment of 18-months of his then current base salary in the event of termination without cause or upon certain change of control events.
Mr. Payne is paid pursuant to a verbal arrangement at an annual salary of $200,000, prorated for the time committed to the Company. Mr. Payne’s time commitment has varied, and during 2017 has committed between 80% and 95% of his time to the Company. In 2016, Mr. Payne and the Company entered into a severance agreement providing for the payment of 12-months of his then current base salary in the event of termination without cause or upon certain change of control events.
The Company believes that there are no material risks posed to the Company as a result of its employment arrangements with its named executive officers. As there are only two employees, the named executive officers, no pay ratio disclosure is set forth herein.
Director Compensation
During 2016 and 2015, the directors of the Company were not compensated for their services as directors and it is not expected that the directors will receive compensation in 2017. The Company has no formal arrangement pursuant to which directors are compensated for their services in their capacity as directors. Directors are reimbursed for reasonable out of pocket expenses incurred.
Grants of Plan-Based Equity Awards
No plan-based equity awards were granted to any of our named executive officers during the fiscal years ended December 31, 2016 and 2015, or during the interim period through June 30, 2017.
Outstanding Option Awards at Fiscal Year End
These options were granted between 2009 and 2011.
| Name |
| Number of Share of Common Stock Underlying Unexercised Options (Exercisable) |
|
| Number of Share of Common Stock Underlying Unexercised Options (Unexercisable) |
|
| Option Exercise Price |
|
| Option Expiration Date | ||||
| Michael T. Redman |
|
| 2,000,000 |
|
|
| -- |
|
| $ | 0.005 |
|
| April 2021 | |
| Michael T. Redman |
|
| 800,000 |
|
|
| -- |
|
| $ | 0.005 |
|
| November 2021 | |
| Michael T. Redman |
|
| 2,000,000 |
|
|
| -- |
|
| $ | 0.015 |
|
| November 2021 | |
| J. Donald Payne |
|
| 500,000 |
|
|
| -- |
|
| $ | 0.005 |
|
| April 2021 | |
| J. Donald Payne |
|
| 1,100,000 |
|
|
| -- |
|
| $ | 0.015 |
|
| November 2021 | |
| 60 |
| Table of Contents |
Certain Relationships and Related Party Transactions
GHC Research Development Corporation owns 48,000,000 shares of Company Common Stock, 19,938,500 shares of Company Series A Preferred Stock, and warrants to purchase 19,938,500 shares of Company Series A Preferred Stock at an exercise price of $0.0825 that expire in January 2020, which securities were acquired between 2007 and 2015 in consideration for the assignment of certain intellectual property to Oncolix, for cash investments of $2,400,000, and the conversion of indebtedness of $1,495,388. GHC retains the rights to the contributed intellectual property in the event of a bankruptcy or liquidation of the Company, although GHC has subordinated these rights to the holders of the Convertible Notes. Biovectra LLC owns 10,000,000 shares of Company Common Stock, which shares were acquired pursuant to a 2011 contract for services valued at $1,500,000. Integrium LLC owns 6,000,000 shares of Company Series A Preferred Stock, and a warrant to purchase 6,000,000 shares of Company Series A Preferred Stock at an exercise price of $0.0825 that expire in January 2020, which securities were acquired in 2015 pursuant to contracted services rendered valued at $450,000. Texas Treasury Safekeeping Trust Company, Emerging Technology Program (“TTSTC”) owns 36,172,180 shares of Company Series A Preferred Stock and warrants to purchase 20,177,520 shares of Company Series A Preferred Stock at an exercise price of $0.0825 that expire in January 2020, which securities were acquired between 2012 and 2015 in consideration for $3,600,000 in cash and the conversion of $313,314 of indebtedness. TTSTC holds shares acquired by an economic development affiliate of the State of Texas, and in connection with such investment, we agreed that if we do not maintain our principal place of business or principal executive offices in Texas through September 30, 2020, TTSTC has the right to require us to repay the $3,913,314 investment plus eight percent interest per annum, compounded annually, from the date of the investment (a stipulated damage provision) and TTSTC will retain its shares of Series A Preferred Stock. We do not intend to relocate our business or principal executive office outside of the State of Texas. Mr. Redman acquired (i) 4,000,000 shares of Company Common Stock in 2007 for nominal consideration, (ii) 14,000 shares of Company Series A Preferred Stock and warrants to acquire 14,000 shares of Series A Preferred Stock at an exercise price of $0.0825 that expire in January 2020 for $1,050 paid in 2015 and 2016, and (iii) 400,000 shares of Common Stock pursuant to stock options previously granted for a nominal consideration paid in 2013. Mr. Payne acquired (i) 14,000 shares of Company Series A Preferred Stock and warrants to acquire 14,000 shares of Series A Preferred Stock at an exercise price of $0.0825 that expire in January 2020, for $1,050 paid in 2015 and 2016 and (ii) 400,000 shares of Common Stock pursuant to stock options previously granted for a nominal consideration paid in 2013.
An affiliate of GHC Research Development Corporation provided services for the Company’s clinical trial, and is owed $61,935 as of December 31, 2016 and June 30, 2017 (none of such costs were incurred in 2016 or 2017). Biovectra provided contract manufacturing services, and the Company incurred an estimated net liability of $328,217 as of December 31, 2016, substantially all of which was incurred in 2016 and such amount remains outstanding as of June 30, 2017. Integrium LLC provided clinical trial services under a 2015 agreement in which it was issued equity for such services, and the Company incurred approximately $135,000 of expenses pursuant to this arrangement in 2016, which amount was applied to the prepayment related to shares issued as of December 31, 2016 and June 30, 2017.
The Company has not adopted a policy or procedure for approval with respect to any related party.
The following table sets forth, as of August 28, 2017, certain information concerning the beneficial ownership of our Common Stock and Series A Preferred Stock by (i) each stockholder known by us to own beneficially five percent or more of either our outstanding Common Stock or our Series A Preferred Stock; (ii) each director; (iii) each named executive officer, as defined in Item 402 of Registration S-K; and (iv) all of our executive officers and directors as a group, and their percentage ownership and voting power. As of August 28, 2017, there were 103,536,703 shares of Common Stock issued and outstanding and 62,138,680 shares of Series A Preferred Stock issued and outstanding that are voting and convertible into 63,038,284 shares of Common Stock, for an aggregate of 166,574,987 shares of voting capital stock issued and outstanding.
Unless otherwise stated, beneficial ownership has been determined in accordance with Rule 13d-3 under the Exchange Act. Under this rule, certain shares may be deemed to be beneficially owned by more than one person (if, for example, persons share the power to vote or the power to dispose of the shares). In addition, shares are deemed to be beneficially owned by a person if the person has the right to acquire shares (for example, upon exercise of an option or warrant) within 60 days of the date as of which the information is provided. In computing the percentage ownership of any person or group of persons, the number of shares beneficially owned by such person or group of persons is deemed to include the number of shares beneficially owned by such person or the members of such group by reason of such acquisition rights, and the total number of shares outstanding is also deemed to include such shares (but not shares subject to similar acquisition rights held by any other person or group) for purposes of that calculation. As a result, the percentage of outstanding shares of any person as shown in the following table does not necessarily reflect the person’s actual voting power at any particular date. To our knowledge, except as indicated in the footnotes to this table and pursuant to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of Common Stock shown as beneficially owned by them.
| 61 |
| Table of Contents |
The address for each of the beneficial owners is the Company’s address, unless otherwise indicated.
| Amount and Nature of Beneficial Ownership (Shares) |
|
|
|
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
|
|
| Common Stock |
|
| Series A Preferred Stock |
|
| % of Total |
| |||||||||||
| Name and Address of Beneficial Owner |
| Shares |
|
| Percent |
|
| Shares |
|
| Percent |
|
| Voting Power |
| |||||
| GHC Research Development Corporation, 300 East McBee Avenue, Suite 302, Greenville, South Carolina 29601 |
|
| 87,877,000 | (2) |
|
| 71.2 | % |
|
| 39,877,000 | (3) |
|
| 48.1 | % |
|
| 47.1 | %(2) |
|
| ||||||||||||||||||||
| Biovectra Inc., 11 Aviation Avenue Charlottetown, PEI Canada C1E 0A |
| 10,000,000 |
| 9.7 | % |
| - |
| 0.0 | % |
| 6.0 | % | |||||||
|
| ||||||||||||||||||||
| Texas Treasury Safekeeping Trust Company, Emerging Technology Program, Rusk State Office Building 208 East 10th Street, 4th Floor, Austin, Texas 78701 |
| 56,349,700 | (4) |
| 45.5 | % |
| 56,349,700 | (5) |
| 67.7 | % |
| 30.2 | %(4) | |||||
|
| ||||||||||||||||||||
| Integrium LLC, 100 E. Hanover Ave., Suite 401 Cedar Knolls, NJ 07927 |
| 12,000,000 | (6) |
| 11.0 | % |
| 12,000,000 | (7) |
| 17.4 | % |
| 7.0 | %(6) | |||||
|
| ||||||||||||||||||||
| Michael T Redman, Director and CEO, 14405 Walters Road, Ste , Houston, TX 77014 |
| 9,228,000 | (8) |
| 8.5 | % |
| 28,000 | (9) |
| 0.0 | % |
| 5.5 | %(8) | |||||
|
| ||||||||||||||||||||
| J. Donald Payne, Director and Secretary, 14405 Walters Road, Ste , Houston, TX 77014 |
| 2,028,000 | (10) |
| 1.9 | % |
| 28,000 | (11) |
| 0.0 | % |
| 1.2 | %(10) | |||||
|
| ||||||||||||||||||||
| All officers and directors as a group (2 persons) |
| 11,256,000 | (12) |
| 10.2 | % |
| 56,000 | (13) |
| 0.1 | % |
| 6.8 | %(12) | |||||
____________
| (1) | Based upon an aggregate of 103,536,703 shares of Common Stock issued and outstanding and 62,138,680 shares of Series A Preferred Stock issued and outstanding as of August 28, 2017. All options and warrants are immediately exercisable. |
| (2) | Includes 19,938,500 shares of Common Stock issuable upon conversion of 19,938,500 shares of Series A Preferred Stock, and 19,938,500 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 19,938,500 shares of Series A Preferred Stock. |
| (3) | Includes 19,938,500 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 19,938,500 shares of Series A Preferred Stock. |
| (4) | Includes 36,172,180 shares of Common Stock issuable upon conversion of 36,172,180 shares of Series A Preferred Stock, and 20,177,520 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 20,177,520 shares of Series A Preferred Stock. |
| (5) | Includes 20,177,520 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 20,177,520 shares of Series A Preferred Stock. |
| (6) | Includes 6,000,000 shares of Common Stock issuable upon conversion of 6,000,000 shares of Series A Preferred Stock, and 6,000,000 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 26,000,000 shares of Series A Preferred Stock. |
| (7) | Includes 6,000,000 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 6,000,000 shares of Series A Preferred Stock. |
| (8) | Includes options to acquire 4,800,000 shares of Common Stock and 14,000 shares of Common Stock issuable upon conversion of 14,000 shares of Series A Preferred Stock, and 14,000 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 14,000 shares of Series A Preferred Stock. |
| (9) | Includes 14,000 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 14,000 shares of Series A Preferred Stock. |
| (10) | Includes options to acquire 1,600,000 shares of Common Stock and 14,000 shares of Common Stock issuable upon conversion of 14,000 shares of Series A Preferred Stock, and 14,000 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 14,000 shares of Series A Preferred Stock. |
| (11) | Includes 14,000 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 14,000 shares of Series A Preferred Stock. |
| (12) | Includes options to acquire 6,400,000 shares of Common Stock and 28,000 shares of Common Stock issuable upon conversion of 28,000 shares of Series A Preferred Stock, and 28,000 shares of Common Stock that may be acquired upon the exercise and conversion of warrants to acquire 28,000 shares of Series A Preferred Stock. |
| (13) | Includes 28,000 shares of Series A Preferred Stock that may be acquired upon the exercise of warrants to acquire 28,000 shares of Series A Preferred Stock |
| 62 |
| Table of Contents |
Contractual Lock-Up
The officers and directors of the Company, Messrs. Redman and Payne, as well as GHC, agreed for a period of fifteen months from August 3, 2017, not to sell, assign or otherwise transfer any of their shares of our Common Stock or Series A Preferred Stock.
DISCLOSURE OF COMMISSION POSITION OF INDEMNIFICATION FOR SECURITIES ACT LIABILITIES
Our directors and officers are indemnified to the fullest extent permitted under the Florida Business Corporations Act.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing, or otherwise, we have been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by us of expenses incurred or paid by a director, officer or controlling person of ours in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
As a result of the Merger as previously described, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes. Accordingly, the financial statements of the Company are the historical financial statements of Oncolix prior to the Merger. Accordingly, the Company financial statements as of December 31, 2015 and 2016 have been audited by Pannell Kerr Forster of Texas, P.C., an independent registered public accounting firm (“PKF”) to the extent and for the periods set forth in their report thereon, appearing elsewhere in this registration statement, and are included in reliance upon such report given on the authority of PKF as experts in auditing and accounting.
The financial statements of AEPP prior to the Merger as of December 31, 2016 were audited by B F Borgers CPA PC, an independent public accounting firm (“Borgers”) and the AEPP financial statements as of December 31, 2015 were audited by Stevenson & Company CPAS LLC (S&C), an independent public accounting firm (“Stevenson”).
On August 4, 2017, the Board of Directors of the Company approved engagement of PKF as the independent registered public accounting firm for the Company. PKF has previously conducted audits of Oncolix, and the historical financial statements of Oncolix are presented as the historical financial statements of AEPP after the Merger. The previous independent registered public accounting firm for AEPP was Borgers, which conducted the audit of the financial statements for the year ended December 31, 2016. On August 17, 2016, AEPP’s Board of Directors approved the engagement of Borgers as its principal accountant to audit the Company's financial statements. The report of Borgers on the AEPP financial statements did not contain an adverse opinion or disclaimer of opinion, nor was it qualified or modified as to uncertainty, audit scope or accounting principles, except that the report contains an explanatory paragraph stating that there was substantial doubt about the company's ability to continue as a going concern.
There were no disagreements between the Company and Borgers for 2016 and the March 31, 2017 interim filing on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which, if not resolved to the satisfaction of Borger would have caused it to make reference to the subject matter of the disagreement in connection with its report. On August 16, 2016, Stevenson resigned as AEPP’s principal accountant. None of the reports of Stevenson on AEPP’s financial statements for either 2015 or 2014 contained an adverse opinion or disclaimer of opinion, or were qualified or modified as to uncertainty, audit scope or accounting principles, except that the report contains an explanatory paragraph stating that there was substantial doubt about the company's ability to continue as a going concern. There were no disagreements between AEPP and Stevenson for the two most recent fiscal years and the March 31, 2016 interim filing on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which, if not resolved to the satisfaction of Stevenson, would have caused to make reference to the subject matter of the disagreement in connection with its report.
| 63 |
| Table of Contents |
The validity of the shares of Common Stock and the shares of Common Stock to be sold in this offering will be passed upon by Thomas C. Pritchard, P.C., Houston, Texas.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC under the Securities Act, a registration statement on Form S-1 relating to the securities offered hereby. This prospectus does not contain all of the information set forth in the registration statement and the exhibits and schedules thereto. For further information with respect to our company and the securities we are offering by this prospectus you should refer to the registration statement, including the exhibits and schedules thereto. You may inspect a copy of the registration statement without charge at the Public Reference Room of the SEC at 100 F Street, NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an internet site that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. The SEC’s internet address is http://www.sec.gov. We maintain a website at http://www.oncolixbio.com. You may access our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not part of this prospectus.
| 64 |
|
|
| Page(s) |
|
| Audited Financial Statements of Advanced Environmental Petroleum Producers Inc. |
|
|
|
|
| F - 2 |
| |
|
| F - 3 |
| |
| Statements of Operations for the years ended December 31, 2016 and 2015 |
| F - 4 |
|
| Statements of Stockholders’ Deficit for the years ended December 31, 2016 and 2015 |
| F - 5 |
|
| Statements of Cash Flows for the years ended December 31, 2016 and 2015 |
| F - 6 |
|
|
| F - 7 |
| |
|
|
|
|
|
| Unaudited Financial Statements of Advanced Environmental Petroleum Producers Inc. |
|
|
|
|
| F - 16 |
| |
| Statements of Operations for the six months ended June 30, 2017 and 2016 |
| F - 17 |
|
| Statements of Cash Flows for the six months ended June 30, 2017 and 2016 |
| F - 18 |
|
|
| F - 19 |
|
| F-1 |
| Table of Contents |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Advanced Environmental Petroleum Producers Inc.
We have audited the accompanying balance sheets of Advanced Environmental Petroleum Producers Inc. (the “Company”), as of December 31, 2016 and 2015, and the related statements of operations, stockholders’ deficit, and cash flows for each of the years then ended, and the related notes to the financial statements. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2016 and 2015, and the results of its operations and cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has experienced negative cash flows from operations since the commencement of operations and expects these losses to continue into the foreseeable future as the Company continues the development and clinical testing of Prolanta™ and seeking regulatory approval to market products. These conditions raise substantial doubt about its ability to continue as a going concern. Management’s plans regarding those matters are described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to that matter.
/s/ Pannell Kerr Forster of Texas, P.C.
Houston, Texas
May 25, 2017, except for Note 1, as to which the date is August 29, 2017
| F-2 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Balance Sheets
|
|
| December 31, |
|
| December 31, |
| ||
|
|
| 2016 |
|
| 2015 |
| ||
| Current assets |
|
|
|
|
|
| ||
| Cash and cash equivalents |
| $ | 15,264 |
|
| $ | 274,063 |
|
| Prepaid services |
|
| 363,089 |
|
|
| 493,090 |
|
| Prepaid expenses |
|
| 3,802 |
|
|
| 3,802 |
|
| Total current assets |
|
| 382,155 |
|
|
| 770,955 |
|
| Deferred offering costs |
|
| 38,687 |
|
|
| - |
|
| Total assets |
| $ | 420,842 |
|
| $ | 770,955 |
|
|
|
|
|
|
|
|
|
|
|
| Current liabilities |
|
|
|
|
|
|
|
|
| Accounts payable and accrued expenses |
| $ | 643,473 |
|
| $ | 480,412 |
|
| Related party payable and accrued expenses |
|
| 375,217 |
|
|
| 58,119 |
|
| Convertible notes and accrued interest payable |
|
| 198,547 |
|
|
| - |
|
| Accounts payable to Greenville Health System and affiliates |
|
| 61,935 |
|
|
| 22,911 |
|
| Total current liabilities |
|
| 1,279,172 |
|
|
| 561,442 |
|
| Deferred compensation |
|
| 1,268,013 |
|
|
| 1,032,623 |
|
| Total liabilities |
|
| 2,547,185 |
|
|
| 1,594,065 |
|
| Commitments and contingencies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Stockholders' deficit |
|
|
|
|
|
|
|
|
| Common stock, $0.0001 par value; 500,000,000 shares authorized, 71,153,300 and 71,133,300 shares issued and outstanding at December 31, 2016 and 2015, respectively |
|
| 7,115 |
|
|
| 7,113 |
|
| Series A preferred stock, $0.001 par value, 100,000,000 shares authorized, 62,138,680 and 62,119,080 shares issued and outstanding at December 31, 2016 and 2015, respectively |
|
| 6,214 |
|
|
| 6,212 |
|
| Additional paid-in capital |
|
| 16,840,328 |
|
|
| 16,757,809 |
|
| Accumulated deficit |
|
| (18,980,000 | ) |
|
| (17,594,244 | ) |
| Total stockholders' deficit |
|
| (2,126,343 | ) |
|
| (823,110 | ) |
| Total liabilities and stockholders' deficit |
| $ | 420,842 |
|
| $ | 770,955 |
|
See accompanying notes to financial statements.
| F-3 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Statements of Operations
|
|
| Year Ended December 31, |
| |||||
|
|
| 2016 |
|
| 2015 |
| ||
|
|
|
|
|
|
|
|
|
|
| Revenue |
| $ | - |
|
| $ | - |
|
| Expenses |
|
|
|
|
|
|
|
|
| Research and development expenses |
|
| 709,009 |
|
|
| 561,671 |
|
| General and administrative expenses |
|
| 620,835 |
|
|
| 777,499 |
|
| Total operating expenses |
|
| 1,329,844 |
|
|
| 1,339,170 |
|
| Interest expense on convertible notes |
|
| 55,912 |
|
|
| 7,812 |
|
| Net loss |
| $ | (1,385,756 | ) |
| $ | (1,346,982 | ) |
|
|
|
|
|
|
|
|
|
|
| Loss per common share - basic and diluted |
| $ | (0.019 | ) |
| $ | (0.019 | ) |
| Weighted average common shares outstanding - basic and diluted |
|
| 71,148,163 |
|
|
| 71,790,615 |
|
See accompanying notes to financial statements.
| F-4 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Statements of Stockholders’ Deficit
|
|
| Shares of Common Stock |
|
| Common Stock Amount |
|
| Shares of Series A Preferred Stock |
|
| Series A Preferred Stock Amount |
|
| Additional Paid-In Capital |
|
| Accumulated Deficit |
|
| Total Stockholders' Deficit |
| |||||||
| Balance, December 31, 2014 |
|
| 87,127,960 |
|
| $ | 8,712 |
|
|
| - |
|
| $ | - |
|
| $ | 13,154,222 |
|
| $ | (16,247,262 | ) |
| $ | (3,084,328 | ) |
| Stock-based compensation |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 172,808 |
|
|
| - |
|
|
| 172,808 |
|
| Sale of Series A Preferred stock and warrants for cash, net of offering costs |
|
| - |
|
|
| - |
|
|
| 16,008,400 |
|
|
| 1,601 |
|
|
| 1,175,088 |
|
|
| - |
|
|
| 1,176,689 |
|
| Exchange of Series A Preferred for Common stock |
|
| (15,994,660 | ) |
|
| (1,599 | ) |
|
| 15,994,660 |
|
|
| 1,600 |
|
|
| - |
|
|
| - |
|
|
| 1 |
|
| Conversion of indebtedness for Series A Preferred and warrants |
|
| - |
|
|
| - |
|
|
| 24,116,020 |
|
|
| 2,411 |
|
|
| 1,806,291 |
|
|
| - |
|
|
| 1,808,702 |
|
| Sale of Series A Preferred and warrants for services |
|
| - |
|
|
| - |
|
|
| 6,000,000 |
|
|
| 600 |
|
|
| 449,400 |
|
|
| - |
|
|
| 450,000 |
|
| Net loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (1,346,982 | ) |
|
| (1,346,982 | ) |
| Balance, December 31, 2015 |
|
| 71,133,300 |
|
|
| 7,113 |
|
|
| 62,119,080 |
|
|
| 6,212 |
|
|
| 16,757,809 |
|
|
| (17,594,244 | ) |
|
| (823,110 | ) |
| Sale of Series A Preferred stock and warrants for cash |
|
| - |
|
|
| - |
|
|
| 19,600 |
|
|
| 2 |
|
|
| 1,468 |
|
|
| - |
|
|
| 1,470 |
|
| Warrants issued with indebtedness |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 41,366 |
|
|
| - |
|
|
| 41,366 |
|
| Warrants issued to Placement Agent |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| 38,687 |
|
|
| - |
|
|
| 38,687 |
|
| Exercise of stock option for cash |
|
| 20,000 |
|
|
| 2 |
|
|
| - |
|
|
| - |
|
|
| 998 |
|
|
| - |
|
|
| 1,000 |
|
| Net loss |
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| - |
|
|
| (1,385,756 | ) |
|
| (1,385,756 | ) |
| Balance, December 31, 2016 |
|
| 71,153,300 |
|
| $ | 7,115 |
|
|
| 62,138,680 |
|
| $ | 6,214 |
|
| $ | 16,840,328 |
|
| $ | (18,980,000 | ) |
| $ | (2,126,343 | ) |
See accompanying notes to financial statements.
| F-5 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Statements of Cash Flows
|
|
| Year Ended December 31, |
| |||||
|
|
| 2016 |
|
| 2015 |
| ||
| Cash flows from operating activitities |
|
|
|
|
|
| ||
| Net loss |
| $ | (1,385,756 | ) |
| $ | (1,346,982 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities |
|
|
|
|
|
|
|
|
| Stock based compensation |
|
| - |
|
|
| 172,808 |
|
| Accretion of discount on notes payable |
|
| 50,749 |
|
|
| - |
|
| Changes in operating assets and liabilities |
|
|
|
|
|
|
|
|
| Prepaid and other current assets |
|
| 130,001 |
|
|
| 143,292 |
|
| Related party accounts payable and accrued expenses |
|
| 591,512 |
|
|
| 123,604 |
|
| Accounts payable and accrued expenses |
|
| 168,225 |
|
|
| (306 | ) |
| Net cash used in operating activities |
|
| (445,269 | ) |
|
| (907,584 | ) |
| Cash flows from financing activities |
|
|
|
|
|
|
|
|
| Proceeds from exercise of stock options |
|
| 1,000 |
|
|
| - |
|
| Proceeds from sale of convertible notes and warrants |
|
| 184,000 |
|
|
| - |
|
| Proceeds from sale of Series A preferred stock and warrants |
|
| 1,470 |
|
|
| 1,176,690 |
|
| Net cash provided by financing activities |
|
| 186,470 |
|
|
| 1,176,690 |
|
| Net increase (decrease) in cash and cash equivalents |
|
| (258,799 | ) |
|
| 269,106 |
|
| Cash and cash equivalents at beginning of period |
|
| 274,063 |
|
|
| 4,957 |
|
| Cash and cash equivalents at end of period |
| $ | 15,264 |
|
| $ | 274,063 |
|
| Non-cash financing activities |
|
|
|
|
|
|
|
|
| Stock issued for prepaid services |
| $ | 38,687 |
|
| $ | 450,000 |
|
| Stock issued upon conversion of indebtedness |
| $ | - |
|
| $ | 1,808,702 |
|
| Interest accrued on related party debt |
| $ | - |
|
| $ | 7,812 |
|
See accompanying notes to financial statements.
| F-6 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Notes to Financial Statements
Note 1 – Basis of Presentation
On April 8, 2017, Oncolix, Inc. (“Oncolix”) acquired approximately 66% of the outstanding common stock of Advanced Environmental Petroleum Producers Inc. (“AEPP”), a public entity traded on the OTC pink sheets with limited or no operating activities and no indebtedness. On August 3, 2017, Oncolix merged into a wholly-owned subsidiary of AEPP and AEPP acquired all of the outstanding equity interest of Oncolix and assumed responsibility for the issuance of equity securities under Oncolix’s options, warrants and convertible debt as described further herein. This transaction is treated as a reverse acquisition of AEPP for financial accounting and reporting purposes. As such, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes. Accordingly, these financial statements of AEPP for are those of Oncolix and reflect the operations, cash flows and stockholders’ equity of Oncolix during the periods indicated, but do not reflect the minority interest held by the remaining shareholders of AEPP until April 2017, when Oncolix acquired control of AEPP.
In the reverse merger, each stockholder of Oncolix received 20 shares of a substantially identical class of AEPP stock (Common or Series A Preferred, each with a par value of $0.0001) in exchange for each share of Oncolix (par value $0.001). In addition, each option or warrant became exercisable for 20 shares of the identical class of AEPP stock for each share of Oncolix, and the exercise price was adjusted by dividing the price per Oncolix share by 20. The shares of AEPP Common Stock owned by Oncolix were cancelled in the reverse merger. These financial statements have been adjusted to reflect the adjusted number of shares resulting from the exchange ratio in the reverse merger (the 20:1 exchange ratio), effectively a stock split for accounting purposes, and the adjustment of the par value to $0.0001. The authorized shares of capital stock are the authorized shares of AEPP.
AEPP has 500,000,000 authorized shares of Common Stock, $0.0001 par value, and 100,000,000 authorized shares of Series A Preferred Stock, $0.0001 par value.
As used in the following notes, “We,” “Us,” “Our,” “AEPP”, or the “Company” refers to Oncolix prior to April 2017 and the combined AEPP and Oncolix subsequent to such date.
Note 2 – Background
Description of business
We are developing medical therapies for the treatment of cancer. The Company’s activities are focused on the development of Prolanta™ for the treatment of gynecological cancers.
The Company is also in the process of raising additional equity capital to support the completion of its development and commercialization activities. The Company’s business is subject to significant risks and uncertainties, including the risk of failure to secure additional funding for the Company’s business.
We commenced operations on January 24, 2007. At formation, certain intellectual property was transferred to us by a subsidiary of Greenville Health Corporation (collectively, “GHC”), a component unit of Greenville Health System (“GHS”) of South Carolina. As of December 31, 2016 and 2015, GHC was the majority stockholder of Oncolix. See Notes 4 and 7.
Going concern
We have experienced negative cash flows from operations since the commencement of operations, and we expect these losses to continue into the foreseeable future as we continue the development and clinical testing of Prolanta™ and seek regulatory approval to market products. We are currently seeking financing that would fund operations beyond the end of 2017. Accordingly, our continued operations are dependent on our ability to raise additional capital through the sale of equity or debt securities or through the licensing of our products. In the event we are unable to raise sufficient funds, we would have to substantially alter, or possibly even discontinue, operations, or sell assets at distressed prices. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| F-7 |
| Table of Contents |
Note 3 - Summary of Significant Accounting Policies
Use of estimates
To prepare our financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”), we are required to make significant estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. In addition, significant estimates were made regarding the realization of deferred tax assets, the calculation of the fair value of stock options awarded, the estimation of the value of warrants issued in conjunction with capital stock, and the projected realization of the carrying basis of our assets. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from the estimates and assumptions used.
Cash and cash equivalents
Our cash balances are not restricted from use and we have no investments or marketable securities during the periods reported. Marketable securities with maturities of three months or less are considered to be cash equivalents.
Research and development
Research and development costs, which are comprised of costs incurred in performing research and development activities including wages and associated employee benefits of employees engaged in research; the cost of consultants engaged in our research activities; the cost of developing methods to manufacture our investigational drug products; the costs of production of investigation drugs for use in research, animal and human testing; the costs of the testing of our products in animal models and in human testing; the cost of research conducted on our behalf at academic institutions; the depreciation of equipment used in research; the cost of obtaining patents for the protection of our intellectual property; and other costs associated with these activities. These costs are accounted for in accordance with Accounting Standards Codification (“ASC”) subtopic 730-10, “Research and Development,” which requires all research and development costs to be charged to expense as incurred.
Our agreements with both the manufacturer of our drug Prolanta™ and the organizations that will provide monitoring and regulatory services for our clinical trials provide that Oncolix capital stock will be issued for part of the value of the total services rendered. The value of the stock issued is expensed as the service is rendered, and any prepayment of future services is reflected as prepaid services in the accompanying balance sheets until the services are rendered.
Property and equipment
Property and equipment are stated at cost, less accumulated depreciation. For financial reporting purposes, depreciation is recognized using the straight-line method, allocating the cost of the assets over their estimated useful lives of five years for laboratory equipment and three years for office furniture and equipment. As of December 31, 2016 and 2015, property and equipment was fully depreciated, and there was no depreciation expense recorded in either year.
Intangible assets
The costs of obtaining patent protection for our intellectual property are expensed as incurred given the uncertainty of successful development, ultimate regulatory approval and commercialization of products developed using this technology.
Deferred offering costs
Oncolix defers legal and certain other costs related to financing activities until the completion or termination of the offering. If the financing to which such costs are related is terminated, such costs are expensed. If the costs are related to a debt financing, the costs are amortized as additional interest expense over the period prior to maturity.
Concentrations of risk
We have engaged a contract manufacturer to develop methods of production of Prolanta™ and to produce quantities necessary for our animal and human testing. Accordingly, our operations are dependent upon the manufacturer’s ability to manufacture suitable quantities of Prolanta™ in compliance with regulatory requirements and within acceptable timeframes, and to maintain regulatory licenses for its operations. Failure to maintain such regulatory requirements would have a material impact on our operations. We incurred contract manufacturing costs of $305,098 and $293,807 during the years ended December 31, 2016 and 2015, respectively.
Financial instruments
Our financial instruments consist of cash and cash equivalents and accounts payable. We believe that the carrying amounts of the financial instruments classified as current assets and liabilities approximate their fair values because of their short-term nature.
Income taxes
We account for income taxes under the asset and liability method, which requires recognition of deferred tax assets, subject to valuation allowances, and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and income tax purposes. A valuation allowance is established if it is more likely than not that all or a portion of the net deferred tax assets will not be realized.
| F-8 |
| Table of Contents |
If substantial changes in our ownership should occur, as defined in Section 382 of the Internal Revenue Code, there could be limitations on the amount of net loss carry forwards that could be used to offset future taxable income.
We will recognize the impact of an uncertain tax position only if that position is more likely than not of being sustained upon examination by the taxing authority based on the technical merits of the position. We will account for interest and penalties relating to an uncertain tax position in the current period statement of operations, as necessary.
Stock-based compensation
Through December 31, 2016, we maintained an equity compensation plan under which incentive stock options and non-qualified stock options were granted to employees, independent members of our Board of Directors and outside consultants. We expect a new plan will be adopted in 2017. We recognize stock-based compensation in accordance with ASC topic 718, “Compensation.”
Deferred compensation
The officers of the Company have voluntarily deferred the payment of certain compensation since 2012. During 2015, these officers agreed that the deferred compensation would be paid only from the proceeds of future equity offerings or the sale of assets, and not be paid using the existing current assets of the Company. Accordingly, the cumulative unpaid compensation is included in the accompanying balance sheets as a non-current liability, deferred compensation. As of December 31, 2016 and 2015, respectively, $1,268,013 and $1,032,623 are reflected as deferred compensation in the accompanying balance sheets.
Recent accounting standards
On February 25, 2016, the Financial Accounting Standards Board (“FASB”) issued ASU No. 2016-02, Leases. ASU No. 2016-02 requires lessees to recognize right-of-use assets and lease liabilities on the balance sheet for all leases of property, plant and equipment with lease terms greater than 12 months. Prior to this accounting standard, only capital leases were recognized on the balance sheet. ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2019, and interim periods within fiscal years beginning after December 15, 2020, and early adoption is permitted. This ASU must be applied using a retrospective approach upon adoption. The Company is currently evaluating the requirements of this ASU and has not determined the impact this ASU may have on its financial statements.
Rent expense
We lease administrative offices in Houston, Texas under a lease which expires on March 31, 2019. Rent expense for the years ended December 31, 2016 and 2015 was $42,083 and $40,431, respectively. The remaining rental obligation under the lease is $40,083 in 2017, $40,680 in 2018 and $10,248 in 2019.
Note 4 - Stockholders’ Deficit
As of December 31, 2016, 71,153,300 shares of Common Stock and 62,138,680 shares of Series A Preferred Stock were issued and outstanding. In addition, as of December 31, 2016, we have issued options to acquire up to 7,200,000 shares of Common Stock and warrants to acquire up to 50,810,720 shares of Series A Preferred Stock.
Series A Preferred Stock
Each share of Series A Preferred Stock is currently convertible into one share of Common Stock. This conversion ratio is subject to adjustment for certain dilutive events. If a liquidation event occurs, each share of Series A Preferred Stock is entitled to a liquidation preference of $0.075 per share, and then each share will receive distributions ratably with the Common Stock based on the then-existing conversion ratio. As of December 31, 2016, the liquidation preference of the outstanding shares of Series A Preferred Stock is $4,660,401. The holders of Series A Preferred Stock have voting rights with the Common Stock based on the then-existing conversion ratio, and also have certain separate voting rights. As long as at least 14,000,000 shares of Series A Preferred Stock are outstanding, the holders of the Series A Preferred Stock have certain protective rights to nominate a director to the Board of Directors, who shall have the right to approve certain transactions. In addition, as long as at least 14,000,000 shares of Series A Preferred Stock are outstanding, the holders of the Series A Preferred Stock have the right to vote separately as a class to approve certain amendments to the certificate of incorporation, any liquidation event, and certain issuances of capital stock. The shares of Series A Preferred Stock may be converted into Common Stock at any time, but will automatically convert upon either the written consent of the holders of the majority of such shares or the closing of a firm-commitment underwritten public offering with gross proceeds of at least $10 million.
Common Stock
On January 24, 2007, Oncolix issued 24,000,000 shares of Common Stock to GHC in exchange for certain intellectual property and a commitment by GHC to invest $3.0 million to fund Oncolix’s operations. GHC retains the rights to the contributed intellectual property in the event of a bankruptcy or liquidation of the Company. Also on January 24, 2007, Oncolix issued 7,999,960 shares of Common Stock for $0.01 per share in cash to four individuals (the “Scientific Founders”) in exchange for future services and sold 4,000,000 shares of Common Stock for $0.0005 per share to the Chief Executive Officer (“CEO”). Oncolix retains the right to repurchase 1,000,000 shares until such time as the Company has received $10 million in capital contributions or income from licensing or sales of our intellectual property.
| F-9 |
| Table of Contents |
In connection with this purchase of stock at formation, the Scientific Founders and CEO agreed to certain conditions, including the obligation to offer to other holders on a pro rata basis any offer the individual receives to sell his/her Common Stock, the right to require other holders to sell their pro rata shares in any sale of Common Stock by the individual. Additionally, in the event of a proposed sale of Common Stock by the individuals, GHC was granted the right of first refusal to acquire the shares from the individuals. The individuals were also granted certain rights to include their shares in registered public offerings, and agreed to certain restrictions on sales of Common Stock following certain public offerings.
Contract manufacturing arrangements and payment in equity
During August 2009, we engaged a non-US manufacturer for the production of Prolanta™ and to provide certain quality assurance services. Our agreement ended in August 2016. Of amounts earned by the manufacturer, approximately 58%, or $1.5 million, was payable in Oncolix Common Stock at $0.15 per share. The remaining amounts due are payable in cash. As of December 31, 2016 and 2015, there are no prepaid services on the accompanying balance sheets.
Clinical research arrangements and payment in equity
During August 2011, we agreed to pay 10% of the cost of services rendered by a non-US based clinical research organization through the issuance of 16,667 shares of Common Stock at $0.15 per share. The $50,000 value of the Common Stock was recorded as a prepayment of future services. As of December 31, 2016 and 2015, the unearned portion of the value of the equity issued, $43,090, is reflected as prepaid services in the accompanying balance sheets.
During January 2015, as part of a financing transaction described below, we agreed to pay the cost of services by a US-based clinical research organization through the issuance of Series A Preferred Stock and warrants to acquire Series A Preferred Stock. The $450,000 value of the initial issuance of Series A Preferred Stock was recorded as a prepayment of future services. As of December 31,2016 and 2015, respectively, the unearned portion of the value of the equity issued reflected as a prepayment in the accompanying balance sheets was $319,999 and $450,000. The clinical research organization may perform an additional $1,050,000 in services, and receive an additional $1,050,000 of Series A Preferred Stock.
Texas Emerging Technology Fund Investment
During 2014, an agreement with an existing investor, the Texas Emerging Technology Fund (“TETF”), an economic development affiliate of the State of Texas, was amended and the TETF invested $300,000 through a bridge loan, which was converted into Series A Preferred Stock in 2015. Additionally, the TETF purchased an additional $1,200,000 of Series A Preferred Stock in 2015. See the description of the sale of convertible notes and the sale of Series A Preferred Stock herein.
We agreed to certain conditions related to the TETF investment, including certain representations and warranties. Our obligations to TETF generally expire on September 30, 2020. In the event that we do not maintain our principal place of business or principal executive offices in Texas, TETF may require us to repay the $3.9 million investment plus eight percent interest per annum, compounded annually, from the date of the investment, essentially as a stipulated damage or penalty, and TETF will retain the TETF Warrant or any stock acquired through the exercise of the Warrant. In the event of a breach of other conditions of the investment or representations and warranties, TETF may seek any remedies it may have in law or equity. We can terminate our obligations to TETF at any time by the repayment of the amount invested plus 8% interest calculated as described above.
Sale of convertible notes to affiliates
Prior to 2015, GHC funded our continuing operations by purchasing units consisting of convertible promissory notes (“Bridge Notes”) and warrants to acquire Common Stock (“Bridge Warrants”). As of January 1, 2015, GHC had advanced $750,000 under such Bridge Notes.
Prior to 2015, GHC and TETF also advanced funds by purchasing units consisting of convertible promissory notes secured by the assets of Oncolix (“Secured Bridge Notes”) and warrants to acquire Common Stock (“Secured Bridge Warrants”). As of January 1, 2015, GHC had acquired $435,000 and TETF had acquired $300,000 of such Secured Bridge Notes.
The Bridge Notes and Secured Bridge Notes accrued interest at the rate of 12% per annum, simple interest, until maturity. During the year ended December 31, 2015, we incurred $7,812 of interest under these notes.
Pursuant to the terms of the notes, the principal amount of the Bridge Notes and Secured Bridge Notes and accrued interest automatically converted into shares of Series A Preferred Stock and warrants to acquire Series A Preferred Stock in January 2015. In addition, the holder of the notes received a warrant to acquire additional shares equal to 30% of the shares received upon conversion of the principal amount of the notes. These additional warrants expired in June 2015.
| F-10 |
| Table of Contents |
Sale of Series A Preferred Stock and Warrants in 2015
During January 2015, we issued 62,119,080 shares of Series A Preferred Stock and warrants to acquire 62,119,080 shares of Series A Preferred Stock (the “Preferred Warrants”) in exchange for $1,200,630 of cash, the conversion of $1,808,702 of indebtedness and accrued interest, and the agreement to provide $450,000 of future clinical services. In connection with this transaction, 15,994,660 shares of Common Stock previously issued to TETF were exchanged for 15,994,660 shares of Series A Preferred Stock. The stated price of the unit of one share of Series A Preferred Stock and the Preferred Warrant was $0.075. The expenses related to the transaction were $23,941 and are deducted from the proceeds of the offering in the accompanying Statements of Stockholders’ Deficit.
The Preferred Warrants expire on January 16, 2020 if not otherwise exercised. The exercise price is $0.0825 per share, subject to adjustment for certain dilutive transactions. The Preferred Warrants may be exercised on a cash-less basis.
The purchase price of the units of Series A Preferred Stock and the Preferred Warrant were allocated between the stock and warrant using a Black-Scholes pricing model. As a result, the Preferred Warrants were ascribed a value of $854,455 at the date of the offering.
Placement Agent Compensation Agreement
In connection with the sale of convertible notes in 2017 (see Note 9), we executed an agreement with a placement agent in 2016. The agreement provides that the agent will receive a cash payment equal to 8 to 10% of the cash proceeds received, as well as 4% of the net exercise value upon exercise of any warrants issued. The agent may also receive a warrant equal to 10% of the equity, including warrants, issued. In addition, the agent and its employees were granted a warrant to acquire 2,000,000 shares of Series A Preferred Stock at $0.0825 per share. The warrant expires if not exercised in January 2020, and has cashless exercise provisions. The warrant was issued in December 2016, and the estimated value of the warrant, $38,687, is reflected as a deferred offering cost in the accompanying balance sheets as of December 31, 2016.
2007 Stock Option Plan
The 2007 Stock Option Plan (the “2007 Plan”) authorized the Company to award options to acquire up to 10,880,000 shares of Common Stock to employees, Directors and consultants through December 31, 2016. There were no option awards granted, exercised or forfeited in 2015 as noted in the following table.
|
|
| Number of Shares |
|
| Weighted Average Exercise Price Per Share |
|
| Weighted average remaining life in years |
| |||
| Balance, December 31, 2014 |
|
| 7,820,000 |
|
| $ | 0.01 |
|
|
| 6.76 |
|
| Balance, December 31, 2015 |
|
| 7,820,000 |
|
| $ | 0.01 |
|
|
| 5.76 |
|
| Exercises |
|
| (20,000 | ) |
| $ | 0.05 |
|
|
|
|
|
| Forfeitures |
|
| (600,000 | ) |
| $ | 0.015 |
|
|
|
|
|
| Balance, December 31, 2016 |
|
| 7,200,000 |
|
| $ | 0.0095 |
|
|
| 4.40 |
|
| Exercisable December 31, 2016 |
|
| 7,200,000 |
|
| $ | 0.0095 |
|
|
| 4.40 |
|
The authority to make grants under the 2007 plan expired on December 31, 2016, and no further grants may be made. We expect to adopt a new plan in 2017.
We recognize stock-based compensation expense for equity awards over the requisite service period of each grant using the Black-Scholes option pricing model to estimate the fair value of the stock options on the date of grant. We do not currently have sufficient historical exercise or forfeiture data on which to base an estimate of the expected term of each option, so we use the life of the grant and assume no forfeitures. Additionally, we utilize an expected volatility of 200% because we do not have any trading experience. We base the risk-free interest rate used in the Black-Scholes option pricing model on the implied yield currently available on U.S. Treasury securities.
| F-11 |
| Table of Contents |
The following table summarizes the assumptions we utilized for grants of options to acquire Common Stock:
| Assumptions |
| Employees |
|
| Non-Employee Director |
|
| Consultants |
| |
|
|
|
|
|
|
|
|
|
|
| |
| Expected life (years) |
| 10 |
|
| 5-10 |
|
| 10 |
| |
| Expected volatility (%) |
| 200 |
|
| 200 |
|
| 200 |
| |
| Risk-free interest rate (%) |
| 2.0-3.39 |
|
| 0.93-3.39 |
|
| 2.0 |
| |
| Forfeiture rate (%) |
| 0.0 |
|
| 0.0 |
|
| 0.0 |
| |
| Expected dividend yield |
| 0.0 |
|
| 0.0 |
|
| 0.0 |
| |
| Assumed value per share of common stock on grant date |
| $0.15 |
|
| $0.15 |
|
| $0.125 to $0.15 |
| |
There were no independent determinations of the valuation of our Common Stock on the date of each option grant. For accounting purposes, the price of the most recent sale of Common Stock, regardless of the contractual date of such sale, was utilized, resulting in an assumed value of $0.15 per share. Since the exercise price was substantially less than the assumed value per share of Common Stock on the grant date, the value of each option granted was the approximate intrinsic value of each option. The most recent sales price of stock, Series A Preferred Stock, was at $0.075 per unit of one share and a warrant to acquire one share as described above.
There was no unamortized stock-based compensation expense recognized related to stock options during the year ended December 31, 2016. We recognized stock-based compensation expense of $172,808 in 2015.
Note 5 – Notes Payable Sold in 2016
During 2016, we sold a series of units of a 10% secured note payable and a warrant to acquire shares of Series A Preferred Stock. As of December 31, 2016, we issued $200,000 in principal amount of the notes and warrants to acquire 2,666,700 shares of Series A Preferred Stock at $0.0825 per share. The notes have maturities between March 2017 and May 2017 and are secured by the assets of Oncolix. The warrants are exercisable through January 2022, and were assigned a value of $41,365 using a Black-Scholes formula, which was recorded as a discount to the notes. Upon maturity, if the noteholders are not offered an option to convert the notes into a new equity offering by the Company, the noteholder will also be entitled to receive an additional 20% of the principal amount of the notes. The discount attributable to the value of the warrants, the 20% premium, and an additional discount related to the fees and expenses of the issuance of the notes of $16,000 are being accreted as additional interest expense over the life of the notes. During 2016, we recorded $55,912 of interest expense attributable to these notes, of which $50,749 is accretion.
During March 2017, $100,000 of principal amount of notes were exchanged for a new series of notes in the 2017 note offering. The remaining $100,000 of notes extended the maturity through May 2017 and received an additional warrant to acquire 500,000 shares of Series A Preferred Stock. See Note 9.
Note 6 - Federal Income Tax
At December 31, 2016, the Company has net operating loss carryforwards (“NOLs”) of approximately $8.4 million and an Orphan Drug Tax Credit of approximately $431,000 which are available to reduce future Federal income taxes. The future utilization of these carryforwards is restricted by various tax regulations. These carryforwards begin to expire in 2027, if not utilized. The following table the components of our net deferred tax asset as of December 31, 2016 and 2015. The amounts for 2015 have been adjusted for changes made in the actual tax return as filed.
| F-12 |
| Table of Contents |
|
|
| December 31, |
| |||||
|
|
| 2016 |
|
| 2015 |
| ||
|
|
|
|
|
|
|
| ||
| Deferred tax assets (liabilities): |
|
|
|
|
|
| ||
| Net operating losses |
| $ | 2,946,115 |
|
| $ | 2,635,788 |
|
| Unamortized R&D expenses |
|
| 689,178 |
|
|
| 839,122 |
|
| Unamortized startup costs |
|
| 1,737,633 |
|
|
| 1,520,860 |
|
| Orphan Drug Credit |
|
| 431,238 |
|
|
| 179,389 |
|
| Other, net |
|
| 108,104 |
|
|
| 88,912 |
|
|
|
|
|
|
|
|
|
|
|
| Total deferred tax assets |
|
| 5,912,268 |
|
|
| 5,264,071 |
|
|
|
|
|
|
|
|
|
|
|
| Valuation allowance |
|
| (5,912,268 | ) |
|
| (5,264,071 | ) |
|
|
|
|
|
|
|
|
|
|
| Net deferred tax asset |
| $ | - |
|
| $ | - |
|
Our effective income tax (benefit) provision rate was different than the statutory federal income tax (benefit) provision rate as follows:
|
|
| 2016 |
|
| 2015 |
| ||
|
|
|
|
|
|
|
| ||
| Federal tax benefit rate |
|
| 35.0 | % |
|
| 35.0 | % |
| Permanent differences |
|
| (6.4 | ) |
|
| (9.3 | ) |
| Valuation allowance |
|
| (28.6 | ) |
|
| (25.7 | ) |
| Effective income tax benefit (provision) rate |
|
| - | % |
|
| - | % |
There is no assurance that sufficient taxable income will be generated in the future to utilize the net operating loss carryforward prior to its expiration, a valuation allowance has been recorded to fully offset the deferred tax assets. For the year ended December 31, 2016, the valuation allowance increased by approximately $648,000 primarily to offset the net operating loss generated in 2016 and other deferred tax assets generated.
Note 7 - Related-Party Transactions
As of December 31, 2016, GHC was the majority stockholder of Oncolix, and GHC is an affiliate of GHS. An affiliate of GHS is the site for clinical trial activities. Accounts payable to GHC and GHS for services approximated $61,935 and $22,911 at December 31, 2016 and 2015, respectively.
We have engaged a contract manufacturer and clinical research firms and certain of the costs incurred have been paid or will be paid through the issuance of capital stock. See Note 3.
Note 8 - Commitments and Contingencies
Licenses
We have licensed certain manufacturing technology and will pay a royalty of up to 2% of future sales.
Incentive compensation for equity sales
Our Chief Executive Officer receives 1% of the gross proceeds from certain sales of equity securities, including services paid through the issuance of stock. During the years ended December 31, 2016 and 2015, we incurred $0 and $20,000 additional compensation expense pursuant to these arrangements.
Engagement of placement agent
In connection with Oncolix’s financing activities, we have engaged a placement agent for various offerings of securities. If successful, the agent will receive a fee based on the proceeds received by Oncolix and a warrant to acquire stock in Oncolix. During 2016, the agent received a warrant to acquire 2,000,000 shares of Series A Preferred Stock and $16,000 in fees related to notes and warrants issued. The agent will receive fees related to the sale of Notes in 2017 – See Note 9.
| F-13 |
| Table of Contents |
Contingent obligations to Texas Emerging Technology Fund
In connection with the investments by the TETF, we agreed to certain conditions, including certain representations and warranties, and the breach of which may result in penalties payable to TETF. See Note 4.
Obligation to GHC Upon Bankruptcy or Liquidation
In connection with the contribution of intellectual property upon the formation of Oncolix, GHC retains the rights to the contributed intellectual property in the event of the bankruptcy or liquidation of Oncolix.
Severance Arrangements for Officers
The Company has entered into employment agreements with its officers that provide for a severance payment of 12 to 18 months of the officer’s salary in the event of termination without cause or under certain other conditions.
Note 9 - Subsequent Events
Sale of Convertible Notes in 2017
During 2017, we commenced the sale of units consisting of a convertible promissory note (the “2017 Notes”) and a warrant to acquire shares of Common Stock at $0.085 per share. The 2017 Notes are secured by the assets of the Company, but are subordinated to the secured notes sold in 2016 (see Note 4). As stated in Note 5, one holder of notes sold in 2016 has exchanged such notes for a new unit. As of May 25, 2017, we have issued approximately $1,321,000 in principal amount of 2017 Notes and warrants to acquire approximately 17,600,000 shares of Common Stock. These warrants are exercisable through May 2022, and were assigned a value of $347,160 using a Black-Scholes formula, which was recorded as a discount to the notes.
The maturity of the 2017 Notes is January 2018. At maturity, if we have not offered the noteholders an option to exchange the 2017 Notes for equity securities in an offering with at least $5 million in gross proceeds, each noteholder may also receive an additional payment equal to 20% of the principal of the note. The discount attributable to the value of the warrants, the 20% premium, and an additional discount related to the fees and expenses of the issuance of the notes of $197,389 are being accreted as additional interest expense over the life of the notes.
The 2017 Notes and accrued interest are exchangeable at a 30% discount into such new equity offering. Interest of 10% is payable quarterly in arrears, and may be paid in our Common Stock at $0.075 per share. On March 31, 2017, we elected to pay accrued interest as of such date by issuing 178,100 shares of our Common Stock.
Acquisition of Voting Control of a Public Entity
On April 8, 2017, as discussed in Note 1, Oncolix acquired approximately 66% of the outstanding common stock of Advanced Environmental Petroleum Producers Inc. (“AEPP”), a public entity traded on the OTC pink sheets with limited or no operating activities and no indebtedness. On August 3, 2017, Oncolix merged into a wholly-owned subsidiary of AEPP and AEPP acquired all of the outstanding equity interest of Oncolix and assumed responsibility for the issuance of equity securities under Oncolix’s options, warrants and convertible debt as described further herein. This transaction is treated as a reverse acquisition of AEPP for financial accounting and reporting purposes. As such, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes. Accordingly, these financial statements of AEPP for are those of Oncolix and reflect the operations, cash flows and stockholders’ equity of Oncolix during the periods indicated, but do not reflect the minority interest held by the remaining shareholders of AEPP until April 2017, when Oncolix acquired control of AEPP.
In the reverse merger, each stockholder of Oncolix received 20 shares of a substantially identical class of AEPP stock (Common or Series A Preferred, each with a par value of $0.0001) in exchange for each share of Oncolix (par value $0.001). In addition, each option or warrant became exercisable for 20 shares of the identical class of AEPP stock for each share of Oncolix, and the exercise price was adjusted by dividing the price per Oncolix share by 20. The shares of AEPP Common Stock owned by Oncolix were cancelled in the reverse merger. These financial statements have been adjusted to reflect the adjusted number of shares resulting from the exchange ratio in the reverse merger (the 20:1 exchange ratio), effectively a stock split for accounting purposes, and the adjustment of the par value to $0.0001. The authorized shares of capital stock are the authorized shares of AEPP.
Sale of July Notes
During July 2017, we sold an additional $160,000 in principal of secured notes the (“July Notes”) and warrants to acquire 3,200,000 shares of Common Stock at $0.0825 per share. The July Notes are due in February 2018 and the warrants are exercisable through July 2022.
| F-14 |
| Table of Contents |
Issuance of New Debt
During August 2017, in conjunction with a new financing, all of the 2017 Notes, the July Notes and amounts due under a consulting agreement were exchanged for new AEPP Notes (the “August Notes”) with aggregate face amount of $1,837,522. In connection with this exchange, warrants to acquire 17,608,280 shares of Common Stock were exchanged for new warrants to acquire 24,500,290 shares of Common Stock (the “August Common Warrant”). Additionally, we sold for cash of $2,000,000 additional Notes with a face amount of $2,352,941 and August Common Warrants to acquire 31,372,547 shares of Common Stock at $0.09 per share.
The conversion rate of the August Notes and the exercise price of the August Common Warrant is subject to adjustment based on the trading price of our Common Stock at certain timepoints. Each holder of the August Notes may convert the principal balance of the note into Common Stock at the lower of (i) $0.075 per share, (ii) 75% of the 10-day average closing bid price of Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of the Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share).
In addition, we will be obligated to file one or more registration statements to register the shares of Common Stock issuable upon the conversion of the August Notes. The August Notes are due in seven installments commencing in February 2018, and bear interest at the rate of 10% per annum, payable quarterly. An additional 10% of the principal balance is due with each monthly payment.
Adjustments to the conversion price of the August Notes and exercise price of the August Common Warrants may result in an anti-dilution adjustment to our Series A Preferred Stock.
| F-15 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Condensed Balance Sheets
(Unaudited)
|
|
| June 30, |
|
| December 31, |
| ||
|
|
| 2017 |
|
| 2016 |
| ||
| Current assets |
|
|
|
|
|
| ||
| Cash and cash equivalents |
| $ | 93,377 |
|
| $ | 15,264 |
|
| Prepaid services |
|
| 363,089 |
|
|
| 363,089 |
|
| Prepaid expenses |
|
| 3,802 |
|
|
| 3,802 |
|
| Total current assets |
|
| 460,268 |
|
|
| 382,155 |
|
| Deferred offering costs |
|
| - |
|
|
| 38,687 |
|
| Total assets |
| $ | 460,268 |
|
| $ | 420,842 |
|
|
|
|
|
|
|
|
|
|
|
| Current liabilities |
|
|
|
|
|
|
|
|
| Accounts payable and accrued expenses |
| $ | 643,393 |
|
| $ | 643,473 |
|
| Related party payable and accrued expenses |
|
| 306,693 |
|
|
| 375,217 |
|
| Convertible notes and accrued interest payable |
|
| 1,217,797 |
|
|
| 198,547 |
|
| Accounts payable to Greenville Health System and affiliates |
|
| 61,935 |
|
|
| 61,935 |
|
| Total current liabilities |
|
| 2,229,818 |
|
|
| 1,279,172 |
|
| Deferred compensation |
|
| 1,268,013 |
|
|
| 1,268,013 |
|
| Total liabilities |
|
| 3,497,831 |
|
|
| 2,547,185 |
|
| Commitments and contingencies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Stockholders' deficit |
|
|
|
|
|
|
|
|
| Common stock, $0.0001 par value; 500,000,000 shares authorized, 103,097,783 and 71,153,300 shares issued and outstanding at June 30, 2017 and December 31, 2016, respectively |
|
| 10,310 |
|
|
| 7,115 |
|
| Series A preferred stock, $0.001 par value, 100,000,000 shares authorized, 62,138,680 shares issued and outstanding at June 30, 2017 and December 31, 2016 |
|
| 6,214 |
|
|
| 6,214 |
|
| Additional paid-in capital |
|
| 17,259,811 |
|
|
| 16,840,328 |
|
| Accumulated deficit |
|
| (20,313,898 | ) |
|
| (18,980,000 | ) |
| Total stockholders' deficit |
|
| (3,037,563 | ) |
|
| (2,126,343 | ) |
| Total liabilities and stockholders' deficit |
| $ | 460,268 |
|
| $ | 420,842 |
|
See accompanying condensed notes to condensed financial statements.
| F-16 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Condensed Statements of Operations
(Unaudited)
|
|
| Six Months Ended June 30, |
| |||||
|
|
| 2017 |
|
| 2016 |
| ||
| Revenue |
| $ | - |
|
| $ | - |
|
| Expenses |
|
|
|
|
|
|
|
|
| Research and development expenses |
|
| 77,095 |
|
|
| 579,202 |
|
| General and administrative expenses |
|
| 794,124 |
|
|
| 272,558 |
|
| Total operating expenses |
|
| 871,219 |
|
|
| 851,760 |
|
| Interest expense on convertible notes |
|
| 462,679 |
|
|
| - |
|
| Net loss |
| $ | (1,333,898 | ) |
| $ | (851,760 | ) |
|
|
|
|
|
|
|
|
|
|
| Loss per common share - basic and diluted |
| $ | (0.016 | ) |
| $ | (0.012 | ) |
| Weighted average common shares outstanding - basic and diluted |
|
| 85,985,247 |
|
|
| 71,142,970 |
|
See accompanying condensed notes to condensed financial statements.
| F-17 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Condensed Statements of Cash Flows
(Unaudited)
|
|
| Six Months Ended June 30, |
| |||||
|
|
| June 30, |
| |||||
|
|
| 2017 |
|
| 2016 |
| ||
| Cash flows from operating activitities |
|
|
|
|
|
| ||
| Net loss |
| $ | (1,333,898 | ) |
| $ | (851,760 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities |
|
|
|
|
|
|
|
|
| Accretion of discount on notes payable |
|
| 406,952 |
|
|
| - |
|
| Changes in operating assets and liabilities |
|
|
|
|
|
|
|
|
| Prepaid and other current assets |
|
| - |
|
|
| 114,355 |
|
| Related party accounts payable and accrued expenses |
|
| (68,524 | ) |
|
| 392,417 |
|
| Accounts payable and accrued expenses |
|
| 42,962 |
|
|
| 61,037 |
|
| Accounts payable to Greenville Health System and affiliates |
|
| - |
|
|
| 39,024 |
|
| Net cash used in operating activities |
|
| (952,508 | ) |
|
| (244,927 | ) |
| Cash flows from financing activities |
|
|
|
|
|
|
|
|
| Proceeds from exercise of stock options |
|
| - |
|
|
| 1,000 |
|
| Proceeds from sale of convertible notes and warrants |
|
| 1,030,621 |
|
|
| - |
|
| Proceeds from sale of Series A preferred stock and warrants |
|
| - |
|
|
| 1,470 |
|
| Net cash provided by financing activities |
|
| 1,030,621 |
|
|
| 2,470 |
|
| Net increase (decrease) in cash and cash equivalents |
|
| 78,113 |
|
|
| (242,457 | ) |
| Cash and cash equivalents at beginning of period |
|
| 15,264 |
|
|
| 274,063 |
|
| Cash and cash equivalents at end of period |
| $ | 93,377 |
|
| $ | 31,606 |
|
| Non-cash financing activities |
|
|
|
|
|
|
|
|
| Stock issued for interest payable |
| $ | 13,358 |
|
| $ | - |
|
| Interest paid in cash |
| $ | 8,362 |
|
| $ | - |
|
See accompanying condensed notes to condensed financial statements.
| F-18 |
| Table of Contents |
Advanced Environmental Petroleum Producers Inc.
Condensed Notes to Financial Statements
(Unaudited)
Note 1 - Background
Basis of presentation
On April 8, 2017, Oncolix, Inc. (“Oncolix”) acquired approximately 66% of the outstanding common stock of Advanced Environmental Petroleum Producers Inc. (“AEPP”), a public entity traded on the OTC Bulletin Board with limited or no operating activities and no indebtedness. Oncolix paid $315,000 in cash and paid a fee to an investment banker of $50,000 in connection with this transaction.
On August 3, 2017, Oncolix merged into a wholly-owned subsidiary of AEPP and AEPP acquired all of the outstanding equity interest of Oncolix and assumed responsibility for the issuance of equity securities under Oncolix’s options, warrants and convertible debt as described further herein. This transaction is treated as a reverse acquisition of AEPP for financial accounting and reporting purposes. As such, Oncolix is treated as the acquirer for accounting and financial reporting purposes while AEPP is treated as the acquired entity for accounting and financial reporting purposes. Accordingly, the financial statements of AEPP for all periods prior to August 3, 2017 are those of Oncolix. Because of the acquisition of a controlling interest on April 8, 2017, the financial statements of AEPP are consolidated with Oncolix since April 2017. The $365,000 purchase price for stock of AEPP by Oncolix is treated as an expense of the merger, and the shares of AEPP Common Stock not owned by Oncolix are treated as shares outstanding subsequent to April 2017. AEPP’s assets, liabilities and results of operations for periods after April 2017 are the consolidated assets, liabilities and results of operations of AEPP and Oncolix.
In the reverse merger, each stockholder of Oncolix received 20 shares of a substantially identical class of AEPP stock (Common or Series A Preferred, each with a par value of $0.0001) in exchange for each share of Oncolix (par value $0.001). In addition, each option or warrant became exercisable for 20 shares of the identical class of AEPP stock for each share of Oncolix, and the exercise price was adjusted by dividing the price per Oncolix share by 20. The shares of AEPP Common Stock owned by Oncolix were cancelled in the reverse merger. After the reverse merger, including shares issued by Oncolix in July 2017 as payment of accrued interest at June 30, 2017, AEPP had 103,536,703 shares of Common Stock, $0.0001 par value, issued and outstanding, of which 71,770,200 shares were issued to the former stockholders of Oncolix. In addition, AEPP had 62,138,680 shares of Series A Preferred Stock, $0.0001, issued and outstanding, all of which were owned by the former stockholders of Oncolix. The financial statements for all periods have been adjusted to reflect the adjusted number of shares resulting from the exchange ratio in the reverse merger (the 20:1 exchange ratio), effectively a stock split for accounting purposes, and the adjustment of the par value to $0.0001.
As used in the following discussion, “We,” “Us,” “Our,” “AEPP”, or the “Company” refers to Oncolix prior to April 2017 and the combined AEPP and Oncolix subsequent to such date.
These condensed financial statements are unaudited; however, in the opinion of management, these condensed statements reflect all adjustments necessary for a fair statement of the results for the periods reported. All such adjustments are of a normal recurring nature unless disclosed otherwise. These condensed financial statements, including notes, have been prepared in accordance with the applicable rules of the SEC and do not include all of the information and disclosures required by U.S. GAAP for complete financial statements. These interim condensed financial statements should be read in conjunction with the financial statements and notes thereto included in our Current Report on Form 8-K filed with the Securities and Exchange Commission on August 9, 2017. The results of operations for the three and six month periods of 2017 are not necessarily indicative of the results to be expected for the full year.
Description of business
We are developing medical therapies for the treatment of cancer. The Company’s activities are focused on the development of Prolanta™ for the treatment of gynecological cancers. AEPP is incorporated in Florida, and we commenced operations on January 24, 2007. At formation, certain intellectual property was transferred to us by a subsidiary of Greenville Health Corporation (collectively, “GHC”), a component unit of Greenville Health System (“GHS”) of South Carolina. Prior to the merger in August 2017, and in all prior periods, GHC was the majority stockholder of Oncolix. See Notes 3 and 6.
As a clinical-stage biotechnology company, we have no revenues and must raise substantial additional capital to support the completion of its development and commercialization activities. The Company’s business is subject to significant risks and uncertainties, including the risk of failure to secure additional funding for the Company’s business.
| F-19 |
| Table of Contents |
Going concern
We have experienced negative cash flows from operations since the commencement of operations, and we expect these losses to continue into the foreseeable future as we continue the development and clinical testing of Prolanta™ and seek regulatory approval to market products. We have raised capital during 2017 through the sale of promissory notes secured by all of the assets of AEPP. See Note 5. Substantially all of these notes were exchanged for new notes and additional debt was sold in August 2017. See Note 8. We expect these funds to support our operations into 2018, and we are currently seeking financing that would fund repayment of this debt and fund operations through at least 2019. Accordingly, our continued operations are dependent on our ability to raise additional capital through the sale of equity or debt securities or through the licensing of our products. In the event we are unable to raise sufficient funds, we would have to substantially alter, or possibly even discontinue, operations, or sell assets at distressed prices. The accompanying condensed financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Note 2 - Summary of Significant Accounting Policies
Use of estimates
To prepare our condensed financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”), we are required to make significant estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed financial statements and the reported amounts of revenues and expenses during the reporting period. In addition, significant estimates were made regarding the realization of deferred tax assets, the calculation of the fair value of stock options awarded, the estimation of the value of warrants issued in conjunction with capital stock, and the projected realization of the carrying basis of our assets. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from the estimates and assumptions used.
Cash and cash equivalents
Our cash balances are not restricted from use and we have no investments or marketable securities during the periods reported. Marketable securities with maturities of three months or less are considered to be cash equivalents.
Research and development
Research and development costs, which are comprised of costs incurred in performing research and development activities including wages and associated employee benefits of employees engaged in research; the cost of consultants engaged in our research activities; the cost of developing methods to manufacture our investigational drug products; the costs of production of investigation drugs for use in research, animal and human testing; the costs of the testing of our products in animal models and in human testing; the cost of research conducted on our behalf at academic institutions; the depreciation of equipment used in research; the cost of obtaining patents for the protection of our intellectual property; and other costs associated with these activities. These costs are accounted for in accordance with Accounting Standards Codification (“ASC”) subtopic 730-10, “Research and Development,” which requires all research and development costs to be charged to expense as incurred.
Our agreements with both the manufacturer of our drug Prolanta™ and the organizations that will provide monitoring and regulatory services for our clinical trials provide that our capital stock was issued for part of the value of the total services rendered. The original value of the stock issued was recorded as a prepayment of future services and is expensed as the services are rendered. The remaining balance is reflected as prepaid services in the accompanying condensed balance sheets.
Property and equipment
Property and equipment are stated at cost, less accumulated depreciation. For financial reporting purposes, depreciation is recognized using the straight-line method, allocating the cost of the assets over their estimated useful lives of five years for laboratory equipment and three years for office furniture and equipment. Property and equipment is fully depreciated, and there was no depreciation expense recorded in any period presented.
| F-20 |
| Table of Contents |
Intangible assets
The costs of obtaining patent protection for our intellectual property are expensed as incurred given the uncertainty of successful development, ultimate regulatory approval and commercialization of products developed using this technology.
Deferred offering costs
Oncolix defers legal and certain other costs related to financing activities until the completion or termination of the offering. If the financing to which such costs are related is terminated, such costs are expensed. If the costs are related to a debt financing, the costs are amortized as additional interest expense over the period prior to maturity.
Concentrations of risk
We have engaged a contract manufacturer to develop methods of production of Prolanta™ and to produce quantities necessary for our animal and human testing. Accordingly, our operations are dependent upon the manufacturer’s ability to manufacture suitable quantities of Prolanta™ in compliance with regulatory requirements and within acceptable timeframes, and to maintain regulatory licenses for its operations. Failure to maintain such regulatory requirements would have a material impact on our operations. We incurred contract manufacturing costs of $0 and $2,418 during the three months ended June 30, 2017 and 2016 and $3,476 and $305,098 during the six months ended June 30, 2017 and 2016, respectively.
Financial instruments
Our financial instruments consist of cash and cash equivalents, accounts payable and notes payable. We believe that the carrying amounts of the financial instruments classified as current assets and liabilities approximate their fair values because of their short-term nature. As discussed in Note 5, certain of our notes payable have a conversion feature at a discount to future equity offerings. We adjust the carrying value of these notes payable at the end of each period based on the market value of the equity securities. As of June 30, 2017, no adjustments were required.
Income taxes
We account for income taxes under the asset and liability method, which requires recognition of deferred tax assets, subject to valuation allowances, and liabilities for the expected future tax consequences of events that have been included in the condensed financial statements or tax returns. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and income tax purposes. A valuation allowance is established if it is more likely than not that all or a portion of the net deferred tax assets will not be realized.
If substantial changes in our ownership should occur, as defined in Section 382 of the Internal Revenue Code, there could be limitations on the amount of net loss carry forwards that could be used to offset future taxable income.
We will recognize the impact of an uncertain tax position only if that position is more likely than not of being sustained upon examination by the taxing authority based on the technical merits of the position. We will account for interest and penalties relating to an uncertain tax position in the current period statement of operations, as necessary.
| F-21 |
| Table of Contents |
Stock-based compensation
We maintained an equity compensation plan under which incentive stock options and non-qualified stock options were granted to employees, independent members of our Board of Directors and outside consultants. The right to make grants under this plan expired on December 31, 2016, and we expect a new plan will be adopted in 2017. We recognize stock-based compensation in accordance with ASC topic 718, “Compensation.”
Deferred compensation
The officers of the Company have voluntarily deferred the payment of certain compensation since 2012, and such deferral has varied with the financial resources available to Oncolix. During 2015, these officers agreed that the deferred compensation would be paid only from the proceeds of future equity offerings or the sale of assets, and not be paid using the existing current assets of the Company. Accordingly, the cumulative unpaid compensation is included in the accompanying condensed balance sheets as a non-current liability, deferred compensation. As of June 30, 2017, and December 31, 2016, $1,268,013 is reflected as deferred compensation in the accompanying condensed balance sheets.
Recent accounting standards
On February 25, 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, Leases. ASU No. 2016-02 requires lessees to recognize right-of-use assets and lease liabilities on the balance sheet for all leases of property, plant and equipment with lease terms greater than 12 months. Prior to this accounting standard, only capital leases were recognized on the balance sheet. ASU No. 2016-02 is effective for fiscal years beginning after December 15, 2019, and interim periods within fiscal years beginning after December 15, 2020, and early adoption is permitted. This ASU must be applied using a retrospective approach upon adoption. The Company believes this ASU will not have a significant effect on its financial statements.
Rent expense
We lease administrative offices in Houston, Texas under a lease which expires on March 31, 2019. Rent expense for the three months ended June 30, 2017 and 2016 was $10,705 and $9,958, and for the six months ended June 30, 2017 and 2016 was $20,980 and $19,891, respectively. The remaining rental obligation under the lease is $19,872 in the remainder of 2017, $40,680 in 2018 and $10,248 in 2019.
Note 3 - Stockholders’ Deficit
We have 500,000,000 authorized shares of Common Stock, $0.0001 par value, and 100,000,000 authorized shares of Series A Preferred Stock, $0.0001 par value. As of June 30, 2017, 103,097,783 shares of Common Stock and 62,138,680 shares of Series A Preferred Stock were issued and outstanding. The shares of Common Stock include the 31,766,503 shares held by the remaining shareholders of AEPP prior to our acquisition of control on April 8, 2017. In addition, as of June 30, 2017, we have issued options to acquire up to 7,200,000 shares of Common Stock, warrants to acquire up to 52,077,380 shares of Series A Preferred Stock and warrants to acquire up to 19,188,280 shares of Common Stock. In addition, we may be obligated to issue an additional 14,000,000 shares of Series A Preferred Stock in exchange for future clinical research services, and we may issue an additional 18,941,615 shares of Common Stock if holders of promissory notes voluntarily convert.
Series A Preferred Stock
Each share of Series A Preferred Stock is currently convertible into one share of Common Stock. This conversion ratio is subject to adjustment for certain dilutive events. If a liquidation event occurs, each share of Series A Preferred Stock is entitled to a liquidation preference of $0.075 per share, and then each share will receive distributions ratably with the Common Stock based on the then-existing conversion ratio. As of June 30, 2017, the liquidation preference of the outstanding shares of Series A Preferred Stock is $4,660,401. The holders of Series A Preferred Stock have voting rights with the Common Stock based on the then-existing conversion ratio, and also have certain separate voting rights. As long as at least 14,000,000 shares of Series A Preferred Stock are outstanding, the holders of the Series A Preferred Stock have certain protective rights to nominate a director to the Board of Directors, who shall have the right to approve certain transactions. In addition, as long as at least 14,000,000 shares of Series A Preferred Stock are outstanding, the holders of the Series A Preferred Stock have the right to vote separately as a class to approve certain amendments to the certificate of incorporation, any liquidation event, and certain issuances of capital stock. The shares of Series A Preferred Stock may be converted into Common Stock at any time, but will automatically convert upon either the written consent of the holders of the majority of such shares or the closing of a firm-commitment underwritten public offering with gross proceeds of at least $10 million.
| F-22 |
| Table of Contents |
GHC liquidation rights
On January 24, 2007, the Company issued 24,000,000 shares of Common Stock to GHC in exchange for certain intellectual property and a commitment by GHC to invest $3.0 million to fund the Company’s operations. GHC retains the rights to the contributed intellectual property in the event of a bankruptcy or liquidation of the Company. GHC has subordinated these rights in connection with the Company’s borrowings.
Clinical research arrangements and payment in equity
During August 2011, we agreed to pay 10% of the cost of services rendered by a non-US based clinical research organization through the issuance of 333,340 shares of Common Stock at $0.15 per share. The $50,000 value of the Common Stock was recorded as a prepayment of future services. As of June 30, 2017, and December 31, 2016, the unearned portion of the value of the equity issued, $43,090, is reflected as prepaid services in the accompanying condensed balance sheets.
During January 2015, as part of a financing transaction described below, we agreed to pay the cost of services by a US-based clinical research organization through the issuance of Series A Preferred Stock and warrants to acquire Series A Preferred Stock. The $450,000 value of the initial issuance of Series A Preferred Stock was recorded as a prepayment of future services. As of June 30, 2017, and December 31, 2016, respectively, the unearned portion of the value of the equity issued reflected as a prepayment in the accompanying condensed balance sheets was $319,999. Additionally, the clinical research organization may perform an additional $1,050,000 in services, and receive an additional 14,000,000 shares of Series A Preferred Stock.
Texas Emerging Technology Fund Investment
The Texas Emerging Technology Fund (“TETF”), an economic development affiliate of the State of Texas, is the majority holder of the Company’s Series A Preferred Stock as of June 30, 2017 and December 31, 2016. We agreed to certain conditions related to the TETF investment, including certain representations and warranties. Our obligations to TETF generally expire on September 30, 2020. In the event that we do not maintain our principal place of business or principal executive offices in Texas, TETF may require us to repay the $3.9 million investment plus eight percent interest per annum, compounded annually, from the date of the investment, essentially as a stipulated damage or penalty, and TETF will retain its shares of Series A Preferred Stock. In the event of a breach of other conditions of the investment or representations and warranties, TETF may seek any remedies it may have in law or equity. We can terminate our obligations to TETF at any time by the repayment of the amount invested plus 8% interest calculated as described above.
Warrants to Acquire Series A Preferred Stock
As of June 30, 2017, and December 31, 2016, respectively, we have issued warrants to acquire up to 52,077,380 and 50,810,720 shares of Series A Preferred Stock (the “Preferred Warrants”). The Preferred Warrants expire if not otherwise exercised at various dates commencing on January 16, 2020 and ending May 12, 2022. The exercise price is $0.0825 per share, subject to adjustment for certain dilutive transactions. The Preferred Warrants may be exercised on a cash-less basis.
Warrants to Acquire Common Stock
As of June 30, 2017, and December 31, 2016, respectively, we have issued warrants to acquire up to 19,188,280 and 0 shares of Common Stock (the “Common Warrants”). The Common Warrants expire if not otherwise exercised on or prior to July 31, 2022. The exercise price is $0.0825 per share, subject to adjustment for certain dilutive transactions. The Common Warrants may be exercised on a cash-less basis. These Common Warrants were exchanged for a new class of warrants in August 2017. See Note 8.
2007 Stock Option Plan
The authority to make grants under the 2007 Stock Option Plan expired on December 31, 2016, and no further grants may be made. We expect to adopt a new plan in 2017. As of June 30, 2017, and December 31, 2016, options to acquire 7,200,000 shares of Common Stock were outstanding. These options are fully vested and are exercisable at an average price of $0.0093 per share.
We recognize stock-based compensation expense for equity awards over the requisite service period of each grant using the Black-Scholes option pricing model to estimate the fair value of the stock options on the date of grant. There was no unamortized stock-based compensation expense recognized related to stock options during any period in 2016 and 2017.
| F-23 |
| Table of Contents |
Note 4 – Loss Per Common Share
Basic loss per Common share is computed by dividing net loss by the weighted average number of shares of Common Stock outstanding during the period. Diluted loss per share is computed using the average shares outstanding for the period and applying the treasury stock method to potentially dilutive outstanding options and warrants. In all applicable periods, all potential Common Stock equivalents were anti-dilutive and, accordingly, were not included in the computation of diluted loss per share.
The following potential shares of Common Stock, including shares underlying warrants and options and issuable upon conversion of shares of Series A Preferred Stock, were excluded from the calculation of diluted loss per Common share because they were anti-dilutive: 62,138,680 shares underlying the 62,138,680 shares of Series A Preferred Stock outstanding during each period; 7,200,000 shares of Common Stock under outstanding stock options; 52,077,380 shares of Common Stock underlying the warrants to acquire 52,077,380 shares of Series A Preferred Stock; 19,199,280 shares of Common Stock underlying warrants to acquire shares of Common Stock.
Note 5 – Notes Payable
2016 Secured Notes
During the second half of 2016, we sold units of a 10% secured note payable (the “2016 Notes”) and a warrant to acquire shares of Series A Preferred Stock. As of December 31, 2016, we issued $200,000 in principal amount of the 2016 Notes and warrants to acquire 2,666,700 shares of Series A Preferred Stock at $0.0825 per share. The original maturities of the 2016 Notes were between March 2017 and May 2017 and are secured by the assets of Oncolix. The warrants are exercisable through January 2022, and were assigned a value of $41,365 using a Black-Scholes formula, which was recorded as a discount to the notes. Upon maturity, if the noteholders are not offered an option to convert the notes into a new equity offering by the Company, the noteholder will also be entitled to receive an additional 20% of the principal amount of the 2016 Notes. The discount attributable to the value of the warrants, the 20% premium, and an additional discount related to the fees and expenses of the issuance of the 2016 Notes of $16,000 was accreted as additional interest expense over the life of the notes.
The 2016 Notes have the right to voluntarily convert into the units sold in 2017, as described in the section below. As a result, the principal of the 2016 Notes may be indirectly converted into Common Stock at the price of $0.075 per Common share.
During March 2017, $100,000 of principal amount of the 2016 Notes was exchanged for a new series of notes being sold in 2017 – see the following section. The maturity date of the remaining $100,000 of principal of 2016 Notes was extended through May 12, 2017, in exchange for an increase in the interest rate to 18% and the issuance of a warrant to acquire 500,000 shares of Series A Preferred Stock. The warrant is exercisable at $0.0825 per share and expires if not exercised on or prior to March 14, 2022. The discount attributable to the value of this additional warrant ($8,465) was being accreted as additional interest expense over the extended maturity period of the 2016 Note. In May 2017, the maturity of this note was further extended in July 2017, and we issued a second warrant to acquire 500,000 shares of Series A Preferred Stock. The warrant is exercisable at $0.0825 per share and expires if not exercised on or prior to May 12, 2022. The discount attributable to the value of this additional warrant ($11,435) was being accreted as additional interest expense over the extended maturity period of the 2016 Note. See Note 8.
During August 2017, the balance of this debt was paid. See Note 8.
During the three months and six months ended June 30, 2017, respectively, we recorded $20,774 and $85,966 of interest expense attributable to the 2016 Notes, of which $16,286 and $76,521is accretion.
| F-24 |
| Table of Contents |
Sale of Convertible Notes in 2017
During 2017 we sold units consisting of a convertible promissory note (the “2017 Notes”) and a warrant to acquire shares of Common Stock at $0.0825 per share. The 2017 Notes are secured by the assets of the Company, but are subordinated to the 2016 Notes. As of June 30, 2017, we have issued units for $1,185,000 in cash and the exchange for 2016 Notes, with a principal balance of $1,320,621 and warrants to acquire 17,608,280 shares of Common Stock. We paid fees to the placement agent of $103,500 in cash and issued warrants to acquire 1,580,000 shares of Common Stock (in addition to the warrants to acquire 2,000,000 shares of Series A Preferred Stock issued in 2016). In addition, we incurred cash expenses of $93,889.
These warrants are exercisable through July 2022, and were assigned a value of $389,421 using a Black-Scholes formula.
The maturity of the 2017 Notes is July 2018. At maturity, if we have not offered the noteholders an option to exchange the 2017 Notes for equity securities in an offering with at least $5 million in gross proceeds, each noteholder may also receive an additional payment equal to 20% of the principal of the 2017 Note. The discount attributable to the value of the warrants, the 20% premium, and an additional discount related to the fees and expenses of the issuance of the notes of $197,389 are being accreted as additional interest expense over the life of the notes.
The 2017 Notes may also be converted by each holder into shares of Common Stock at a price of $0.075 per Common Shares.
The 2017 Notes and accrued interest are exchangeable at a discount into a new equity offering. Interest of 10% is payable quarterly in arrears, and may be paid in our Common Stock at $0.075 per share. On March 31, 2017, we elected to pay accrued interest as of such date by issuing 178,100 shares of our Common Stock. Such Common Stock was issued in April 2017.
During August 2017, all of the 2017 Notes and related warrants were exchanged for a new series of units of convertible debt and warrants. See Note 8.
During the three months and six months ended June 30, 2017, respectively, we recorded $260,511 and $376,713 of interest expense attributable to the 2017 Notes, of which $227,585 and $330,431 is accretion.
Summary of Notes as of June 30, 2017
|
|
| As of June 30, 2017 |
| |||||||||
|
|
| 2016 Notes |
|
| 2017 Notes |
|
| Total |
| |||
|
|
|
|
|
|
|
|
|
| ||||
| Principal due at maturity, including 20% premium |
| $ | 120,000 |
|
| $ | 1,584,745 |
|
| $ | 1,704,745 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| Unamortized issue costs and discount |
|
| (1,293 | ) |
|
| (520,503 | ) |
|
| (521,796 | ) |
|
|
|
|
|
|
|
|
|
|
|
|
| |
| Carrying amount in the accompanying Balance Sheet |
| $ | 118,707 |
|
| $ | 1,064,242 |
|
| $ | 1,182,949 |
|
Note 6 - Related-Party Transactions
As of June 30, 2017, GHC was the largest stockholder of the Company, and GHC is an affiliate of GHS. An affiliate of GHS is the site for clinical trial activities. Accounts payable to GHC and GHS for services approximated $61,935 at June 30, 2017 and December 31, 2016.
We have engaged clinical research firms and certain of the costs incurred have been paid or will be paid through the issuance of capital stock. See Note 3.
| F-25 |
| Table of Contents |
Note 7 - Commitments and Contingencies
Licenses
We have licensed certain manufacturing technology and will pay a royalty of up to 2% of future sales of products that utilize such technology.
Engagement of placement agent
In connection with our financing activities, we have engaged a placement agent for various offerings of securities. If successful, the agent will receive a fee based on the proceeds received by us and a warrant to acquire our Common Stock. During the six months ended June 30, 2017, the agent received $103,500 in fees and we issued warrants to acquire 1,580,000 shares of Common Stock (in addition to the warrants to acquire 2,000,000 shares of Series A Preferred Stock issued in 2016) and warrants to acquire 266,660 shares of Series A Preferred Stock. There were no fees in the three months ended June 30, 2017. The placement agent will also receive a fee equal to 4% of cash received by Oncolix on the exercise of warrants.
Contingent obligations to Texas Emerging Technology Fund
In connection with the investments by the TETF, we agreed to certain conditions, including certain representations and warranties, and the breach of which may result in penalties payable to TETF. See Note 3.
Obligation to GHC Upon Bankruptcy or Liquidation
In connection with the contribution of intellectual property upon the formation of the Company, GHC retains the rights to the contributed intellectual property in the event of the bankruptcy or liquidation of Oncolix.
Severance Arrangements for Officers
The Company has entered into employment agreements with its officers that provide for a severance payment of 12 to 18 months of the officer’s salary in the event of termination without cause or under certain other conditions.
Note 8 - Subsequent Events
Sale of July Notes
During July 2017, we sold an additional $160,000 in principal of secured notes the (“July Notes”) and warrants to acquire 3,200,000 shares of Common Stock at $0.0825 per share. The July Notes are due in February 2018 and the warrants are exercisable through July 2022.
Issuance of New Debt
During August 2017, in conjunction with a new financing, all of the 2017 Notes, the July Notes and amounts due under a consulting agreement were exchanged for new AEPP Notes (the “August Notes”) with aggregate face amount of $1,837,522. In connection with this exchange, warrants to acquire 17,608,280 shares of Common Stock were exchanged for new warrants to acquire 24,500,290 shares of Common Stock (the “August Common Warrant”). Additionally, we sold for cash of $2,000,000 additional Notes with a face amount of $2,352,941 and August Common Warrants to acquire 31,372,547 shares of Common Stock at $0.09 per share.
The conversion rate of the August Notes and the exercise price of the August Common Warrant is subject to adjustment based on the trading price of our Common Stock at certain timepoints. Each holder of the August Notes may convert the principal balance of the note into Common Stock at the lower of (i) $0.075 per share, (ii) 75% of the 10-day average closing bid price of Common Stock for the period prior to the filing of a registration statement as described below (subject to a floor of $0.0375 per share), and (iii) 75% of the 10-day average closing bid price of the Common Stock for the 10-day period prior to the effective date of the registration statement (subject to a floor of $0.0375 per share).
In addition, we will be obligated to file one or more registration statements to register the shares of Common Stock issuable upon the conversion of the August Notes. The August Notes are due in seven installments commencing in February 2018, and bear interest at the rate of 10% per annum, payable quarterly. An additional 10% of the principal balance is due with each monthly payment.
Adjustments to the conversion price of the August Notes and exercise price of the August Common Warrants may result in an anti-dilution adjustment to our Series A Preferred Stock.
| F-26 |
ADVANCED ENVIRONMENTAL PETROLEUM PRODUCERS INC.
___________ Shares
of Common Stock
PROSPECTUS
_____________, 2017
Until________________, 2017 all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers’ obligation to deliver a prospectus when acting as an underwriter and with respect to their unsold allotments or subscriptions.
BACK COVER PAGE
Part II
Information not required in prospectus
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the estimated expenses to be incurred in connection with the distribution of the securities being registered. The expenses shall be paid by the Company.
| SEC registration fees |
| $ | 1,542 |
|
| Legal fees |
| $ | 20,000 |
|
| Accounting fees |
| $ | 10,000 |
|
| EDGAR/financial printing |
| $ | 5,000 |
|
| Misc. |
| $ | 5,000 |
|
| Total |
| $ | 41,542 |
|
Item 14. Indemnification of directors and officers
Section 607.0850 of the Florida Business Corporations Act permits a corporation to have power to indemnify any person who was or is a party to any proceeding (other than an action by, or in the right of, the corporation), by reason of the fact that he or she is or was a director, officer, employee, or agent of the corporation or is or was serving at the request of the corporation as a director, officer, employee, or agent of another corporation, partnership, joint venture, trust, or other enterprise against liability incurred in connection with such proceeding, including any appeal thereof, if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful. The termination of any proceeding by judgment, order, settlement, or conviction or upon a plea of nolo contendere or its equivalent shall not, of itself, create a presumption that the person did not act in good faith and in a manner which he or she reasonably believed to be in, or not opposed to, the best interests of the corporation or, with respect to any criminal action or proceeding, had reasonable cause to believe that his or her conduct was unlawful.
A corporation shall have power to indemnify any person, who was or is a party to any proceeding by or in the right of the corporation to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee, or agent of the corporation or is or was serving at the request of the corporation as a director, officer, employee, or agent of another corporation, partnership, joint venture, trust, or other enterprise, against expenses and amounts paid in settlement not exceeding, in the judgment of the board of directors, the estimated expense of litigating the proceeding to conclusion, actually and reasonably incurred in connection with the defense or settlement of such proceeding, including any appeal thereof. Such indemnification shall be authorized if such person acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be made under this subsection in respect of any claim, issue, or matter as to which such person shall have been adjudged to be liable unless, and only to the extent that, the court in which such proceeding was brought, or any other court of competent jurisdiction, shall determine upon application that, despite the adjudication of liability but in view of all circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which such court shall deem proper.
To the extent that a director, officer, employee, or agent of a corporation has been successful on the merits or otherwise in defense of any indemnification pursuant to Section 607.0850, or in defense of any claim, issue, or matter therein, he or she shall be indemnified against expenses actually and reasonably incurred by him or her in connection therewith.
Any indemnification, unless pursuant to a determination by a court, shall be made by the corporation only as authorized in the specific case upon a determination that indemnification of the director, officer, employee, or agent is proper in the circumstances because he or she has met the applicable standard of conduct set forth in Section 607.0850.
Expenses incurred by an officer or director in defending a civil or criminal proceeding may be paid by the corporation in advance of the final disposition of such proceeding upon receipt of an undertaking by or on behalf of such director or officer to repay such amount if he or she is ultimately found not to be entitled to indemnification by the corporation pursuant to this section. Expenses incurred by other employees and agents may be paid in advance upon such terms or conditions that the board of directors deems appropriate.
| II-1 |
The indemnification and advancement of expenses provided pursuant to this section are not exclusive, and a corporation may make any other or further indemnification or advancement of expenses of any of its directors, officers, employees, or agents, under any bylaw, agreement, vote of shareholders or disinterested directors, or otherwise, both as to action in his or her official capacity and as to action in another capacity while holding such office. However, indemnification or advancement of expenses shall not be made to or on behalf of any director, officer, employee, or agent if a judgment or other final adjudication establishes that his or her actions, or omissions to act, were material to the cause of action so adjudicated.
Our Articles of Incorporation and bylaws contain provisions that provide for indemnification of officers and directors to the full extent permitted by, and in the manner permissible under, Florida law.
Item 15. Recent Sales of Unregistered Securities
Unless otherwise indicated below, the following securities were offered and sold in reliance upon exemptions from registration pursuant to Section 4(a)(2) of the Securities Act and/or Regulation D (“Regulation D”) promulgated under the Securities Act.
In connection with transactions below issued in reliance on Section 4(a)(2) of the Securities Act or Regulation D, other than issuances to the Company’s officers, directors, employees and consultants for services, the Company made the determination that Section 4(a)(2) or Regulation D applied based on the representations of the investors which included, in pertinent part, that each such investor was an “accredited investor” within the meaning of Rule 501 of Regulation D (there were no non-accredited investors in any of the offerings) and upon such further representations from each investor that (i) such investor is acquiring the securities for its own account for investment and not for the account of any other person and not with a view to or for distribution, assignment or resale in connection with any distribution within the meaning of the Securities Act, (ii) such investor agrees not to sell or otherwise transfer the purchased securities or shares underlying such securities unless they are registered under the Securities Act and any applicable state securities laws, or an exemption or exemptions from such registration are available, (iii) such investor has knowledge and experience in financial and business matters such that such investor is capable of evaluating the merits and risks of an investment in us, (iv) such investor had access to all of the Company’s documents, records, and books pertaining to the investment and was provided the opportunity to ask questions and receive answers regarding the terms and conditions of the offerings and to obtain any additional information which the Company possessed or was able to acquire without unreasonable effort and expense, and (v) such investor was acquiring the securities not with a view to sale or distribution and could afford the complete loss of such investment. In addition, there was no general solicitation or advertising for securities issued in reliance upon Regulation D and the investors received written disclosures that the securities had not been registered under the Securities Act and that any resale must be made pursuant to a registration or an available exemption from such registration.
| II-2 |
| Name of Security Holder |
| Securities Received |
|
| Date Securities Sold |
| Consideration ($) |
| ||
|
|
|
|
|
|
|
|
|
| ||
| Theresa Kearl |
| 3,500,000 shares |
|
| November 9, 2015 |
|
| 17,500 |
| |
| Devin Kyle Lavalle |
| 3,000,000 shares |
|
| November 9, 2015 |
|
| 15,000 |
| |
| Shivon Michelle Lavalle |
| 1,000,000 shares |
|
| November 9, 2015 |
|
| 5,000 |
| |
| New Opportunity Business Solutions, Inc. |
| 7,200,000 shares |
|
| November 19, 2015 |
|
| 36,000 |
| |
| Island Capital Management |
| 30,000 shares |
|
| January 6, 2016 |
|
| 4,000 |
| |
| Ricky Gatt |
| 500,000 shares |
|
| June 6, 2016 |
|
| 2,500 |
| |
| Bert Lavallee |
| 3,500,000 shares |
|
| June 6, 2016 |
|
| 17,500 |
| |
| Robert W. Harris |
| 1,000,000 shares |
|
| June 24, 2016 |
|
| 5,000 |
| |
| Robert W. Harris |
| 2,000,000 shares |
|
| June 27, 2016 |
|
| 10,000 |
| |
| Ricky Gatt |
| 500,000 shares |
|
| June 27, 2016 |
|
| 2,500 |
| |
| Tom Buggs |
| 1,600,000 shares |
|
| June 8, 2016 |
|
| 8,000 |
| |
| Margaret Studer |
| 75,000 shares |
|
| August 19, 2016 |
|
| 375 |
| |
| Kathleen Dinneen |
| 75,000 shares |
|
| August 19, 2016 |
|
| 375 |
| |
| Amy Bugg |
| 200,000 shares |
|
| August 19, 2016 |
|
| 1,000 |
| |
| Adam Hedayat |
| 4,000,000 shares |
|
| August 19, 2016 |
|
| 20,000 |
| |
| Rozalia Mako |
| 100,000 shares |
|
| August 19, 2016 |
|
| 500 |
| |
| Oncolix shareholders and option holders |
|
| (1) |
| August 3, 2017 |
| Exchange |
| ||
| Investors |
|
| (2) |
| August 3, 2017 |
| Cash and exchange |
| ||
___________
| (1) | On July 18, 2017, the Company entered into an agreement and plan of merger with AEPP Merger Sub, Inc., a wholly-owned subsidiary of the Company (“Merger Sub”) and Oncolix that closed on August 3, 2017, pursuant to which Merger Sub merged into Oncolix, the surviving entity of the Merger was Oncolix, and Oncolix became a wholly-owned subsidiary of the Company. Pursuant to the terms of the Merger, a total of 71,770,200 shares of Company Common Stock were issued to 19 former common stock holders of Oncolix and 62,138,680 shares of Company Series A Preferred Stock were issued to five former Series A Preferred Stock holders of Oncolix. Each share of Series A Preferred Stock issued by the Company is convertible into one share of Company Common Stock, has voting rights, a liquidation preference, and other customary rights and preferences. As a result of the Merger, the shares of Company Common Stock owned by Oncolix were cancelled and there were 103,536,703 shares of Company Common Stock issued and outstanding and 62,138,680 shares of Series A Preferred Stock issued and outstanding immediately after the Merger. Subsequent to the Merger, a warrant to purchase shares of Series A Preferred Stock was exercised and 899,064 shares of Series A Preferred Stock was issued. |
|
|
|
| (2) | On August 3, 2017, AEPP and Oncolix entered into agreements with 18 investors in connection with a $4,190,463 convertible debt and warrant financing. Such investment was effected through the issuance and sale of Convertible Notes and Warrants for (i) $2,000,000 in cash for Convertible Notes in the principal amount of $2,352,942 and Warrants to acquire 31,372,547 shares of Common Stock and (ii) (A) the exchange of outstanding Oncolix secured indebtedness in the amount of $1,561,894 and warrants to acquire Oncolix common stock, for (B) the issuance and sale of Convertible Notes issued by the Company in the aggregate principal amount of $1,837,522 and Warrants to acquire 27,700,290 shares of Company Common Stock. In connection with this issuance, the Company paid a sales commission to a FINRA-registered broker-dealer in the amount of $164,800. In addition, the placement agent received warrants to acquire 5,587,285 shares of Company Common Stock. |
| II-3 |
Item 16. Exhibits
| Exhibit No. |
| Description |
| 2.1 |
| Agreement and Plan of Merger (incorporated by reference to Exhibit 2.1 filed with the SEC in the Form 8-K filed on July 24, 2017) |
| 3.1 |
| Articles of Incorporation (incorporated by reference to Exhibit 3.1 filed with the SEC in the Form S-1 Registration Statement on November 19, 2013) |
| 3.2 |
| Amendment to Articles of Incorporation to designate the Series A Preferred Stock (incorporated by reference to Exhibit 3.1 filed with the SEC in the Form 8-K on August 9, 2017) |
| 3.3 |
| Bylaws (incorporated by reference to Exhibit 3.1 filed with the SEC in the Form S-1 Registration Statement on November 19, 2013) |
|
| ||
|
| ||
| 10.1 |
| Securities Purchase Agreement, dated as of August 3, 2017, by and among the Company, Purchasers and Oncolix, Inc. (incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.3 |
| Registration Rights Agreement, dated as of August 3, 2017, by and between the Company and the Purchasers (incorporated by reference to Exhibit 10.3 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.4 |
| Security Agreement, dated as of August 3, 2017, by and among the Company, Oncolix and Purchasers (incorporated by reference to Exhibit 10.4 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.5 |
| IP Security Agreement, dated as of August 3, 2017, by and among the Company, Oncolix and Collateral Agent (incorporated by reference to Exhibit 10.5 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.6 |
| Common Stock Purchase Warrants, dated as of August 3, 2017, issued by the Company to the Purchasers (incorporated by reference to Exhibit 10.6 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.7 |
| Lock-Up Agreement, dated as of August 3, 2017, by and among the Company, Oncolix and certain existing equity holders (incorporated by reference to Exhibit 10.7 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.8 |
| Subsidiary Guarantee, dated as of August 3, 2017, by and between the Company and Purchasers (incorporated by reference to Exhibit 10.8 of the Company’s Form 8-K filed on August 9, 2017) |
| 10.9 |
| Form of Series A Preferred Stock Warrant (incorporated by reference to Exhibit 10.8 of the Company’s Form 8-K filed on August 9, 2017) |
|
| ||
|
| ||
|
| ||
| 10.13 |
| Form of Series A Preferred Stock Warrant (incorporated by reference to Exhibit 10.9 of the Company’s Form 8-K filed on August 9, 2017) |
| 14.1 |
| Code of Ethics (incorporated by reference to Exhibit 14 filed with the SEC in the Form S-1 Registration Statement on November 19, 2013) |
| 16.1 |
| Letter from B. F. Borgers CPA PC (incorporated by reference to Exhibit 16.1 of the Company’s Form 8-K filed on August 9, 2017) |
| 16.2 |
| Letter from Stevenson & Co. CPAS LLC (incorporated by reference to Exhibit 16.2 of the Company’s Form 8-K filed on August 19, 2016) |
|
| ||
|
| ||
| 101(2) |
| The following financial information for the fiscal years ended December 31, 2015 and 2016 formatted in Extensible Business Reporting language (XBRL); (i) balance sheets, (ii) statements of operations, (iii) statements of cash flows and (iv) notes to the financial statements |
___________
* Management contract or compensatory plan or arrangement.
(1) Filed herewith (2) Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections.
II-4
ITEM 17. UNDERTAKINGS
The undersigned registrant hereby undertakes:
| 1. | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: | |
|
| ||
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933; | |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; | |
|
| ||
| 2. | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. | |
|
| ||
| 3. | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. | |
|
| ||
| 4. | For determining liability of the undersigned registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: | |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
Each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
| II-5 |
SIGNATURES
In accordance with the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and authorized this registration statement to be signed on its behalf by the undersigned, in the City of Houston, State of Texas, on August 29, 2017.
|
| ADVANCED ENVIRONMENTAL PETROLEUM PRODUCERS INC. |
| |
|
| |||
|
| By: | /S/ Michael T. Redman |
|
|
| Michael T. Redman | ||
|
| Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons in the capacities indicated below on August 29, 2017.
| Signature |
| Title |
| Date |
|
|
| |||
|
|
|
| ||
| /S/ Michael T. Redman |
| Chief Executive Officer and Director |
| August 29, 2017 |
| Michael T. Redman |
| (Principal Executive Officer) |
| |
|
|
| |||
|
|
|
| ||
| /S/ Michael T. Redman |
| Chief Financial Officer and |
| August 29, 2017 |
| Michael T. Redman |
| (Principal Financial Officer) |
| |
|
|
| |||
|
|
|
| ||
| /S/ J. Donald Payne |
| Director |
| August 29, 2017 |
| J. Donald Payne |
|
|
| II-6 |