Attached files
| file | filename |
|---|---|
| EX-32 - EX-32 - MYRIAD GENETICS INC | mygn-ex32_12.htm |
| EX-31.2 - EX-31.2 - MYRIAD GENETICS INC | mygn-ex312_14.htm |
| EX-31.1 - EX-31.1 - MYRIAD GENETICS INC | mygn-ex311_13.htm |
| EX-23.1 - EX-23.1 - MYRIAD GENETICS INC | mygn-ex231_15.htm |
| EX-21.1 - EX-21.1 - MYRIAD GENETICS INC | mygn-ex211_16.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2017
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 0-26642
MYRIAD GENETICS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
87-0494517 |
|
(State or other jurisdiction |
|
(I.R.S. Employer |
|
|
|
|
|
320 Wakara Way, Salt Lake City, UT (Address of principal executive offices) |
|
84108 (Zip Code) |
Registrant's telephone number, including area code: (801) 584-3600
Securities registered pursuant to Section 12(b) of the Exchange Act:
|
Title of each class |
|
Name of each exchange on which registered |
|
Common Stock, $.01 Par Value Per Share |
|
The NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer”, and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
☒ |
|
Accelerated filer |
☐ |
|
|
Non-accelerated filer |
☐ |
(do not check if a smaller reporting company) |
Smaller reporting company |
☐ |
|
|
Emerging growth company |
☒ |
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐ |
|||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the registrant's common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate), computed by reference to the price at which the common stock was last sold on December 31, 2016, the last business day of the registrant’s most recently completed second fiscal quarter, was $1,104,079,888.
As of August 3, 2017 the registrant had 68,437,971 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The following documents (or parts thereof) are incorporated by reference into the following parts of this Form 10-K: Certain information required in Part III of this Annual Report on Form 10-K is incorporated from the Registrant's Proxy Statement, to be filed no later than 120 days following June 30, 2017, for the Annual Meeting of Stockholders to be held on November 30, 2017.
|
|
|
|
|
Page |
|
PART I |
||||
|
|
|
|
|
|
|
Item 1. |
|
|
3 |
|
|
Item 1A. |
|
|
17 |
|
|
Item 1B. |
|
|
32 |
|
|
Item 2. |
|
|
32 |
|
|
Item 3. |
|
|
33 |
|
|
Item 4. |
|
|
33 |
|
|
|
|
|
|
|
|
PART II |
||||
|
|
|
|
|
|
|
Item 5. |
|
|
34 |
|
|
Item 6. |
|
|
36 |
|
|
Item 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
39 |
|
Item 7A. |
|
|
47 |
|
|
Item 8. |
|
|
48 |
|
|
Item 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
77 |
|
Item 9A. |
|
|
77 |
|
|
Item 9B. |
|
|
79 |
|
|
|
|
|
|
|
|
PART III |
||||
|
|
|
|
|
|
|
Item 10. |
|
|
80 |
|
|
Item 11. |
|
|
80 |
|
|
Item 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
80 |
|
Item 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
80 |
|
Item 14. |
|
|
80 |
|
|
|
|
|
|
|
|
PART IV |
||||
|
|
|
|
|
|
|
Item 15. |
|
|
81 |
|
|
|
|
|
|
|
|
|
82 |
|||
“We,” “us,” “Myriad” and the “Company” as used in this Annual Report on Form 10-K refer to Myriad Genetics, Inc., a Delaware corporation, and its subsidiaries.
“Myriad,” BRACAnalysis, BRACAnalysis CDx, BART, COLARIS, COLARIS AP, MELARIS, myPath, myPlan, myChoice, myRisk, Myriad myRisk, PANEXIA, PREZEON, Prolaris, myChoice HRD, Vectra, Vectraview, TruCulture, DiscoveryMAP, RodentMap, GeneSight, and EndoPredict are registered trademarks or trademarks of Myriad.
2
Overview
We are one of the largest specialty molecular diagnostic laboratories in the world and since our founding in 1992, have tested over 3.0 million patients. We are headquartered in Salt Lake City, Utah and generated worldwide revenues of approximately $771 million during our fiscal year ended June 30, 2017. We are a leading personalized medicine company acting as a trusted advisor to transform patient lives through pioneering molecular diagnostics. Through our proprietary technologies, we believe we are positioned to identify important disease genes, the proteins they produce, and the biological pathways in which they are involved to better understand the genetic basis of human disease. We believe that identifying these biomarkers (DNA, RNA and proteins) will enable us to develop novel molecular diagnostic tests that can provide important information to solve unmet medical needs.
Our Mission
Our goal is to provide physicians with critical information to guide the healthcare management of their patients by addressing four major questions a patient may have about their healthcare:
|
|
• |
What is the likelihood of my getting a disease? |
|
|
• |
Do I have a disease? |
|
|
• |
How aggressively should my disease be treated? |
|
|
• |
Which therapy will work best to treat my disease? |
Over time, we have developed and plan to develop additional products that answer these important questions in six medical specialties: oncology, preventive care, urology, dermatology, autoimmune and neuroscience. We believe that these product channels represent markets where there is a significant opportunity for high-value molecular diagnostic tests to positively impact patient care and drive value for the healthcare system.
Our Business Strategy
Our strategy is focused on executing the following five critical success factors:
|
|
1. |
Stabilize the hereditary cancer market - In fiscal year 2017, approximately 74 percent of our revenue was derived from the sale of products to assess a patient’s risk for hereditary cancer. Given that this is our most important market and that we are the worldwide leader in hereditary cancer testing, we are focused on maintaining this global leadership position. We are currently working on expanding professional guidelines for hereditary cancer testing to expand the addressable market, and have signed long-term contracts with commercial insurers to ensure pricing visibility going forward. |
|
|
2. |
Grow new product volume – In fiscal year 2017 volume from new products outside of hereditary cancer comprised greater than two-thirds of our overall volume. We are currently less than 10 percent penetrated in the U.S. market with our new products and see significant opportunity for future revenue growth. We are focused on further penetrating these markets and believe in the future our new products could represent the largest component of our revenue. |
|
|
3. |
Expand reimbursement coverage for new products – On average, our currently recognized average selling price for our pipeline tests is only about 25 percent of our target average selling price. This is because we currently do not have broad insurance coverage for our new products. We are actively working on demonstrating scientific evidence supporting both the clinical efficacy and utility of these products to commercial payers to broaden insurance coverage. |
|
|
4. |
Increase RNA kit sales internationally – Outside of the United States, we are primarily focused on selling kit-based versions of our RNA expression based tests. We currently market one RNA expression based test, EndoPredict, which we acquired through our acquisition of Sividon Diagnostics. In addition, we are working on kit based versions of Prolaris and myPath Melanoma which we also plan to sell in international markets. |
|
|
5. |
Improve profitability – In the fourth-quarter of fiscal year 2017 we launched a new operating margin improvement program called Elevate 2020. The goal of this program is to identify projects that can lead to $50 million in incremental operating income by fiscal year 2020 through leveraging centralized resources, implementing new technology solutions, executing strategic sourcing agreements, and focusing on laboratory efficiency. |
3
Our molecular diagnostic tests are designed to analyze genes, their expression levels and corresponding proteins to assess an individual’s risk for developing disease later in life, accurately diagnose disease, determine a patient’s likelihood of responding to a particular drug, or disease recurrence and assess a patient’s risk of disease progression. Provided with this valuable information, physicians may more effectively manage their patient’s healthcare.
Below are the descriptions of our molecular diagnostic tests:
|
|
• |
myRisk™ Hereditary Cancer: DNA sequencing test for assessing the risks for hereditary cancers. Our myRisk Hereditary Cancer test represents the next generation of our existing hereditary cancer testing franchise which we anticipate will eventually replace our current predictive medicine test offerings (BRACAnalysis, BART, Colaris and Colaris AP, and Melaris) with a single comprehensive test. myRisk Hereditary Cancer is designed to determine a patient’s hereditary cancer risk for breast cancer, ovarian cancer, colon cancer, uterine cancer, melanoma, pancreatic cancer, prostate cancer and gastric cancer. The test analyzes 28 separate genes to look for deleterious mutations that would put a patient at a substantially higher risk than the general population for developing one or more of the above cancers. All 28 genes in the panel are well documented in clinical literature for the role they play in hereditary cancer and have been shown to have actionable clinical interventions for the patient to lower disease risk or risk of cancer recurrence. The myRisk report presents the myRisk Genetic Test Result and myRisk Management Tool that summarizes published management guidelines related to the patient’s genetic mutation as well as their personal and family history of cancer. myRisk Hereditary Cancer testing identifies more mutation carriers than BRACAnalysis® and COLARIS® combined. We believe the global market for myRisk Hereditary Cancer and all of our hereditary cancer tests is approximately $5 billion annually. myRisk Hereditary Cancer was initially released through an early access launch that began in September 2013. |
|
|
• |
BRACAnalysis®: DNA sequencing test for assessing the risk of developing breast and ovarian cancer. Our BRACAnalysis test is an analysis of the BRCA1 and BRCA2 genes for assessing a woman’s risk of developing hereditary breast and ovarian cancer. A woman who tests positive for a deleterious mutation with the BRACAnalysis test has up to an 87% risk of developing breast cancer and up to a 44% risk of developing ovarian cancer by age 70. As published in the New England Journal of Medicine, researchers have shown that pre-symptomatic individuals who have a high risk of developing breast or ovarian cancer can reduce their risk by more than 90% with appropriate preventive therapies. Additionally, BRACAnalysis may be used to assist patients already diagnosed with breast or ovarian cancer and their physicians in determining the most appropriate therapeutic interventions to address their disease. |
|
|
• |
BART®: DNA sequencing test for hereditary breast and ovarian cancer. Our BART test is based on proprietary technology for detecting large genomic rearrangements in the genes involved in hereditary breast and ovarian cancer patients. |
|
|
• |
BRACAnalysis CDx TM: DNA sequencing test for use as a companion diagnostic with the PARP inhibitor LynparzaTM (olaparib) currently indicated for use in identifying ovarian cancer patients with deleterious or suspected deleterious germline BRCA variants eligible for treatment with LynparzaTM,and as a complementary diagnostic test in ovarian cancer patients associated with enhanced progression-free survival (PFS) from Tesaro’s PARP inhibitor Zejula™ (niraparib) maintenance therapy. Approximately 15% of patients with epithelial ovarian cancer are BRCA positive. |
|
|
• |
Tumor BRACAnalysis CDx TM: DNA sequencing test designed to be utilized to predict response to DNA damaging agents such as platinum based chemotherapy agents and poly ADP ribose (PARP) inhibitors. Tumor BRACAnalysis CDx evaluates both germline and somatic mutations in the BRCA1 and BRCA2 genes giving a more complete picture of potential loss of DNA repair ability within the tumor. Approximately 22% of epithelial ovarian cancer patients will test positive for Tumor BRACAnalysis CDx. Currently the test is only offered through our European laboratory in Munich Germany. |
|
|
• |
COLARIS®: DNA sequencing test for assessing the risk of colorectal and uterine cancer. Our COLARIS test is an analysis of the MLH1, MSH2, MSH6, PMS2, EPCAM and MYH genes for assessing a person’s risk of developing colorectal and uterine cancer. Individuals who carry a deleterious mutation in one of the colon cancer genes in the COLARIS test have a greater than 80% lifetime risk of developing colon cancer and women have up to a 71% lifetime chance of developing uterine cancer. Colon cancer is a preventable disease if high-risk individuals diligently have colonoscopies and remove any precancerous polyps. |
4
|
|
polyposis (FAP), a more common variation of the syndrome known as attenuated FAP, and the MYH-associated polyposis signature (MAP). Individuals who carry a deleterious mutation in the APC or MYH gene may have a greater than 90% lifetime risk of developing colon cancer. Effective preventive measures include colonoscopy and the removal of pre-cancerous polyps and prophylactic surgery. |
|
|
• |
Vectra®DA: protein quantification test for assessing the disease activity of rheumatoid arthritis. Our Vectra DA test is a quantitative, objective multi-biomarker blood test validated to measure rheumatoid arthritis (RA) disease activity. Vectra DA assesses multiple mechanisms and pathways associated with RA disease activity and integrates the concentrations of 12 serum proteins into a single score reported on a scale of 1 to 100. The test may be used throughout the course of a patient’s disease and provides clinicians with expanded insight on disease severity and the risk of radiographic progression. |
We believe the global market for Vectra DA is approximately $3 billion annually.
|
|
• |
Prolaris®: RNA expression test for assessing the aggressiveness of prostate cancer. Our Prolaris test is a gene expression assay that assesses whether a patient is likely to have a slow growing, indolent form of prostate cancer that can be safely monitored through active surveillance, or a more aggressive form of the disease that would warrant aggressive intervention such as a radical prostatectomy or radiation therapy. The Prolaris test was developed to improve physicians’ ability to predict disease outcome and to thereby optimize patient treatment. |
We believe the global market for Prolaris is approximately $1.5 billion annually.
|
|
• |
EndoPredict®: RNA expression test for assessing the aggressiveness of breast cancer. The EndoPredict test is a next-generation RNA expression test used to determine which women with breast cancer would benefit from chemotherapy. EndoPredict predicts the likelihood of metastases to help guide treatment decisions for chemotherapy and extended anti-hormonal therapy. EndoPredict has been shown to accurately predict recurrence in Her 2-, ER+, node negative and node positive breast cancer patients with no confusing intermediate results in 13 published clinical studies with more than 2,200 patients and is CE marked. We believe the global market opportunity for EndoPredict is greater than $600 million annually with the majority of that market existing in major European countries, Canada, and the United States. |
|
|
• |
myPath™ Melanoma: RNA expression test for diagnosing melanoma. Our myPath Melanoma test is a gene expression based profile that is performed on biopsy tissue for the purpose of aiding a dermatopathologist in the diagnosis of melanoma. Every year in the United States, there are approximately two million skin biopsies performed specifically for the diagnosis of melanoma. Approximately 14% of these biopsies are classified as indeterminate where a dermatopathologist cannot make a definitive call as to whether the biopsy is benign or malignant. Outcomes for patients are poor if melanoma is not caught in early stages with five year survival rates dropping from 98% for localized to less than 20% for distant stage disease cancer based upon data from the American Cancer Society. We believe myPath Melanoma may provide an accurate tool to assist physicians in correctly diagnosing indeterminate skin lesions. |
We believe the global market for myPath Melanoma is approximately $1 billion annually. myPath Melanoma was released through an early access launch that began in November 2013.
|
|
• |
myChoice® HRD: Companion diagnostic to measure three modes of homologous recombination deficiency (HRD) including loss of heterozygosity, telomeric allelic imbalance and large-scale state transitions in cancer cells. Our myChoice HRD test is the most comprehensive homologous recombination deficiency test to detect when a tumor has lost the ability to repair double-stranded DNA breaks, resulting in increased susceptibility to DNA-damaging drugs such as platinum drugs or PARP inhibitors. The myChoice HRD score is a composite of three proprietary technologies: loss of heterozygosity, telomeric allelic imbalance and large-scale state transitions. Positive myChoice HRD scores, reflective of DNA repair deficiencies, are prevalent in all breast cancer subtypes, ovarian and most other major cancers. In previously published data, Myriad showed that the myChoice HRD test predicted drug response to platinum therapy in certain patients with triple-negative breast and ovarian cancers. It is estimated that 1.4 million people in the United States and Europe who are diagnosed with cancers annually may be candidates for treatment with DNA-damaging agents. |
5
|
|
treatment decisions, patients were up to twice as likely to respond to the selected medication compared to standard of care. |
Pharmaceutical and Clinical Services
Our pharmaceutical and clinical services consist of the following:
|
|
• |
Through Myriad RBM, we provide biomarker discovery and pharmaceutical and clinical services to the pharmaceutical, biotechnology, and medical research industries utilizing our multiplexed immunoassay technology. Our technology enables us to efficiently screen large sets of well-characterized clinical samples from both diseased and non-diseased populations against our extensive menu of biomarkers. During the year ended June 30, 2017, Myriad RBM accounted for 3.6% of total revenue. In addition to the fees received from analyzing these samples, we also use this information to create and validate potential molecular diagnostic tests. |
|
|
• |
Privatklinik Dr. Robert Schindlbeck GmbH & Co. KG (the “Clinic”) located approximately 15 miles from the Company’s European laboratories in Munich, Germany. It is an internal medicine emergency hospital that is considered a specialized hospital for internal medicine and hemodialysis. |
The Molecular Diagnostic Industry and Competition
The markets in which we compete are rapidly evolving, and we face competition from multiple public companies, private companies, and academic/university laboratories for a number of our laboratory testing services.
In the hereditary cancer testing market we have faced increased competition since a landmark U.S. Supreme Court decision in June 2013 invalidated some of the key patent claims covering our hereditary cancer testing products. These patents were originally set to begin expiring in 2015 and beyond. Since this Supreme Court decision, numerous large reference laboratories, small private laboratories, and academic/university laboratories have launched competing hereditary cancer tests. Despite the impact from competition, we continue to be the world leader in hereditary cancer testing.
The market for hereditary cancer testing has evolved dramatically over time. Broad reimbursement coverage for hereditary cancer tests began emerging in the early 2000s and coupled with increased public awareness around genetics and Myriad’s marketing and promotional efforts, there has been significant growth in testing volumes. One of the largest drivers of growth has been increased testing in asymptomatic patients in the preventive care setting which now comprise nearly half of all tests performed in the United States. We are working to continue to expand awareness around hereditary cancer testing and expand the number of patients that qualify for hereditary cancer testing under medical guidelines and health insurance coverage policies.
Another factor influencing the marketplace has been the advent of next generation sequencing. This has allowed the transition from single syndrome tests to targeted pan-cancer panels in a cost effective manner without sacrificing test accuracy. We launched our first pan-cancer panel, myRisk Hereditary Cancer, in September, 2013, and we believe panel based tests will become standard of care in the marketplace based upon their greater sensitivity at finding cancer causing mutations. We have presented multiple studies showing that myRisk Hereditary Cancer can detect greater than 60 percent more deleterious mutations when compared to our legacy hereditary cancer tests.
Myriad competes in the hereditary cancer testing market based upon several factors including:
|
|
1) |
the analytical accuracy of our tests |
|
|
2) |
our ability to classify genetic variants in hereditary cancer genes |
|
|
3) |
the quality of our sales and marketing for our products |
|
|
4) |
the quality of our customer service and support |
|
|
5) |
turnaround time; and |
|
|
6) |
value |
We believe that we have substantial advantages in terms of our test accuracy and ability to classify variants. Based on our testing experience of over 2.0 million patients, and our substantial investments in our variant classification program, we have compiled a proprietary database of over 50,000 unique genetic variants in the genes tested by myRisk Hereditary Cancer. We believe this database allows us to provide more accurate results to patients and return a variant of unknown significance (VUS)
6
result to patients less frequently. We have demonstrated that this classification advantage leads to lower long-term healthcare costs and lower utilization of unnecessary healthcare services.
Given our scale relative to other laboratories in the hereditary cancer testing market, we believe we also have substantial competitive advantages in terms of cost efficiencies and laboratory automation, which leads to faster turnaround times for our tests.
In the urology market, we compete against a small number of public and private companies for our prostate cancer prognostic test, Prolaris. We compete in this market primarily based upon the quality of the clinical data supporting the test, its first mover advantage in the marketplace and the strength of its sales support and customer service.
In the autoimmune market, our Vectra DA test competes primarily against traditional methodologies for assessing rheumatoid arthritis disease activity such as a physician’s clinical assessment of the patient and single marker laboratory tests such as C-reactive protein (CRP). We believe we have the most predictive product on the market to assess rheumatoid arthritis disease activity.
In the neuroscience market, our GeneSight test meets a significant unmet clinical need and is the leading product for psychotropic drug selection. It is used by healthcare providers to help patients who are affected by neuropsychiatric conditions including depression, anxiety, ADHD, bipolar disorder, schizophrenia, post-traumatic stress disorder (PTSD) and other behavioral health conditions, as well as chronic pain. The test is clinically proven to enhance medication selection, helping healthcare providers get their patients on the right medication faster.
In the pharmaceutical and clinical services segment, our Myriad RBM division competes against other contract research organizations and academic laboratories for business from pharmaceutical and research customers.
Sales and Marketing
We sell our tests through our own direct sales force and marketing efforts in the United States, Europe, Australia and Canada. Our United States sales force is comprised of approximately 750 individuals across five separate sales channels. In connection with any additional tests that we may launch, we may expand our existing oncology, preventive care, urology, dermatology, and autoimmune care sales forces, or build new sales forces to address other physician specialty groups. In addition to our direct sales force, we have entered into distributor agreements with organizations in selected European, Latin American, Middle Eastern, Asian and African countries.
Research and Development
We plan to continue to use our proprietary DNA sequencing, RNA expression and protein analysis technologies, including our supporting bioinformatics and robotic technologies, in an effort to efficiently discover important genes and their proteins and to understand their role in human disease. Based on these biomarkers we plan to develop highly accurate, informative tests that may help physicians better manage their patients’ healthcare. We believe that our technologies provide us with a significant competitive advantage and the potential for numerous product opportunities. For the years ended June 30, 2017, 2016 and 2015, we had research and development expense of $74.4 million, $70.6 million, and $75.5 million, respectively.
Acquisitions
We intend to continue to take advantage of in-licensing or acquisition opportunities to augment our internal research and development programs. We recognize that we cannot meet all of our research discovery goals internally and can benefit from the research performed by other organizations. We hope to leverage our financial strength, product development expertise, and sales and marketing presence to acquire new product opportunities in our molecular diagnostic areas of focus.
In February 2014, we completed the acquisition of privately-held Crescendo Bioscience, Inc. (“Crescendo”) for $270 million in cash, which was reduced by the repayment of a loan made to Crescendo and other customary adjustments in accordance with the acquisition agreement. We believe that the acquisition of Crescendo facilitates our entry into the high growth autoimmune and inflammatory disease market, diversifies our product revenues and enhances our strength in protein-based diagnostics. The business of Crescendo, including its Vectra®DA blood test for rheumatoid arthritis disease management, is operated as a wholly owned subsidiary.
In February 2015, we completed the acquisition of the Clinic located approximately 15 miles from our European laboratories in Munich, Germany for total consideration of $20.1 million. We believe the acquisition of the Clinic should facilitate our
7
penetration into the German molecular diagnostic market. The Clinic’s MVZ designation allows us to directly negotiate reimbursement with government and private insurance providers in the German market and collaborate with hospitals and physician groups.
In May 2016, we completed the acquisition of Sividon Diagnostics GmbH (“Sividon”), a leading breast cancer prognostic company, for $39.0 million upfront with the potential for €15.0 million ($17.1 million converted at the June 30, 2017 period end exchange rate) in additional performance-based milestones. We believe the acquisition brings us the best-in-class breast cancer prognostic test and strengthens our market leading oncology portfolio of high value personalized medicine products.
In August 2016, the Company completed the acquisition of Assurex Health, Inc. (“Assurex) for total consideration of $351.6 million, net of cash acquired of $5.5 million, including a cash payment of $216.1 million, and two potential performance-based milestones totaling $185.0 million. We believe the acquisition establishes the foundation for our neuroscience business and leverages our existing preventative care business unit with the addition of a product, GeneSight, which has significant growth potential.
Seasonality
We experience seasonality in our testing business. The volume of testing is negatively impacted by the summer holiday season which is generally reflected in our fiscal first quarter. Our fiscal second quarter ending December 31 is generally strong as we see an increase in volume from patients who have met their annual insurance deductible. Conversely, fiscal third quarter ending March 31 is typically negatively impacted by the annual reset of patient deductibles.
Patents and Proprietary Rights
We own or have license rights to various issued patents as well as patent applications in the United States and foreign countries. These patents and patent applications relate to a variety of subject matter including, diagnostic biomarkers, gene expression signatures, antibodies, primers, probes, assays, disease-associated genetic mutations, methods for determining genetic predisposition, methods for disease diagnosis, methods for determining disease progression, methods for disease treatment, methods for determining disease treatment, and general molecular diagnostic techniques. For some of the patent assets, we hold rights through exclusive or non-exclusive license agreements. We also own additional patent assets and hold other non-exclusive license rights to patents which relate to various aspects of our tests or processes. Material patent assets relating to our tests that generate material revenue are described below.
Vectra DA. We hold an exclusive license to one issued U.S. patent and pending patent applications in the US and other jurisdictions relating to Vectra®DA testing. This issued U.S. patent has a term expected to expire in 2031 and these U.S. applications, if issued as patents and depending on term adjustments or terminal disclaimers if applicable, are expected to have similar expiration timeframes. These patents and applications contain multiple claims including but not limited to claims relating to biomarkers, kits, systems and methods for measuring and monitoring inflammatory disease activity.
Prolaris. We own or hold an exclusive license to issued patents and pending patent applications in the U.S. and other jurisdictions relating to Prolaris® testing. These issued U.S. patents will have terms to begin expiring in 2032 and these applications, if issued as patents and depending on term adjustments or terminal disclaimers if applicable, are expected to have similar expiration timeframes. These patents and applications contain multiple claims including but not limited to claims relating to biomarkers, kits, systems and methods for detecting, diagnosing, prognosing and selecting therapy for prostate cancer.
EndoPredict. We own or hold an exclusive license to two issued European patents and pending patent applications in the U.S. and other jurisdictions relating to EndoPredict® testing. These issued European patents have terms expected to begin expiring in 2031 and these applications, if issued as patents and depending on term adjustments or terminal disclaimers if applicable, are expected to have similar expiration timeframes. These patents and applications contain multiple claims including but not limited to claims relating to biomarkers, kits, systems and methods for prognosing and selecting therapy for breast cancer.
8
myChoice HRD. We own or hold an exclusive license to issued patents and pending patent applications in the U.S. and other jurisdictions relating to myChoice® HRD testing. These issued patents have terms expected to expire in 2032 and these applications, if issued as patents and depending on term adjustments or terminal disclaimers if applicable, are expected to have similar expiration timeframes. These patents contain multiple claims including but not limited to claims relating to biomarkers, kits, systems and methods for detecting homologous recombination deficiency and selecting therapy based on such detection.
GeneSight. We own or hold an exclusive license to issued patents and pending patent applications in the U.S. and other jurisdictions relation to GeneSight® testing. These issued patents have terms expected to begin expiring in 2024 and these applications, if issued as patents and depending on term adjustments or terminal disclaimers if applicable, are expected to have similar expiration timeframes. These patents contain multiple claims including but not limited to claims relating to biomarkers, kits, systems and methods for detecting single nucleotide polymorphisms and selecting and/or optimizing therapy based on such detection.
We intend to seek patent protection in the United States and major foreign jurisdictions for synthetic nucleic acids, antibodies, biomarker signatures, assays, probes, primers, technologies, methods, processes and other inventions which we believe are patentable and where we believe our interests would be best served by seeking patent protection. However, any patents issued to us or our licensors may not afford meaningful protection for our products or technology or may be subsequently circumvented, invalidated or narrowed or found unenforceable. Any patent applications which we have filed or will file or to which we have licensed or will license rights may not issue, and patents that do issue may not contain commercially valuable claims. In addition, others may obtain patents having claims which cover aspects of our tests or processes which are necessary for or useful to the development, use or performance of our diagnostic products. Should any other group obtain patent protection with respect to our discoveries, our commercialization of our molecular diagnostic tests could be limited or prohibited.
Others may offer clinical diagnostic genomic laboratory testing services which may infringe patents we control. We may seek to negotiate a license to use our patent rights or decide to seek enforcement of our patent rights through litigation. Patent litigation is expensive and the outcome is often uncertain and we may not be able to enforce our patent rights against others.
Our tests and processes may also conflict with patents which have been or may be granted to competitors, academic institutions or others. In addition, third parties could bring legal actions against us seeking to invalidate our owned or licensed patents, claiming damages, or seeking to enjoin clinical testing, developing and marketing of our tests or processes. If any of these actions are successful, in addition to any potential liability for damages, we could lose patent coverage for our tests, be required to cease the infringing activity or obtain a license in order to continue to develop or market the relevant test or process. We may not prevail in any such action, and any license required under any such patent may not be made available on acceptable terms, if at all. Our failure to maintain patent protection for our test and processes or to obtain a license to any technology that we may require to commercialize our tests and technologies could have a material adverse effect on our business.
We also rely upon unpatented proprietary technology, and in the future may determine in some cases that our interests would be better served by reliance on trade secrets or confidentiality agreements rather than patents or licenses. These include some of our genomic, proteomic, RNA expression, mutation analysis, robotic and bioinformatic technologies which may be used in discovering and characterizing new genes and proteins and ultimately used in the development or analysis of molecular diagnostic tests. We also maintain a database of gene mutations and their status as either harmful or benign for all of our hereditary cancer tests. To further protect our trade secrets and other proprietary information, we require that our employees and consultants enter into confidentiality and invention assignment agreements. However, those confidentiality and invention assignment agreements may not provide us with adequate protection. We may not be able to protect our rights to such unpatented proprietary technology and others may independently develop substantially equivalent technologies. If we are unable to obtain strong proprietary rights to our processes or tests, competitors may be able to market competing processes and tests.
License Agreements
We are a party to license agreements which give us the rights to use certain technologies in the research, development, testing processes, and commercialization of our molecular diagnostic tests and pharmaceutical and clinical services. We may not be able to continue to license these technologies on commercially reasonable terms, if at all. Additionally, patents underlying our license agreements may not afford meaningful protection for our technology or tests or may be subsequently circumvented, invalidated or narrowed, or found unenforceable. Our failure to maintain rights to this technology could have a material adverse effect on our business.
9
In 2006, Assurex Health, Inc. (now a wholly-owned subsidiary of the Company) entered into an agreement with the Mayo Foundation for Medical Education and Research (“Mayo”), which granted to Assurex an exclusive world-wide license to utilize certain rights of Mayo in intellectual property relating to what is now GeneSight testing. Under this license agreement we pay Mayo a royalty based on net sales of our GeneSight test. This license expires upon expiration of the last to expire patent covered by the Mayo agreement, which presently is not anticipated to expire until 2024. Mayo has the right to terminate the agreement for the uncured breach of any material term of the agreement.
In 2006, Assurex Health, Inc. entered into a license agreement with the Children’s Hospital Medical Center in Cincinnati (“CHMC”) for the exclusive world-wide right to utilize certain rights of CHMC in intellectual property relating to what is now GeneSight testing. Under this license agreement we pay CHMC a royalty based on net sales of our GeneSight test. This license agreement has no expiration, but CHMC has the right to terminate the agreement for the uncured breach of any material term of the license agreement.
In 2010, Crescendo Bioscience, Inc. (now a wholly-owned subsidiary of the Company) entered into a license agreement with the Oklahoma Medical Research Foundation (the “OMRF”), for the exclusive world-wide right to utilize certain intellectual property rights of OMRF including patent applications relating to what is now Vectra DA testing. Under this license agreement we pay OMRF a royalty based on net sales of our Vectra DA test. This license agreement ends on expiration of the last to expire patent covered by the license agreement, which presently is not anticipated to expire until 2031. OMRF has the right to terminate the license agreement for the uncured breach of any material term of the license agreement.
In 2012, we entered into a license agreement with the University of Texas M.D. Anderson Cancer Center (the “UTMDACC”), for the exclusive world-wide right to utilize certain rights of UTMDACC in intellectual property relating to what is now myChoice® HRD testing. Under this license agreement we will pay UTMDACC a royalty based on net sales of our myChoice® HRD test, if any. This license agreement ends on expiration of the last to expire patent covered by the license agreement, which presently is not anticipated to expire until 2032. UTMDACC has the right to terminate the license agreement for the uncured breach of any material term of the license agreement.
In 2012, we entered into a license agreement with Children’s Medical Center in Boston (“CMCC”) for the exclusive world-wide right to utilize certain rights of CMCC in intellectual property relating to what is now myChoice® HRD testing. Under this license agreement we expect to pay CMCC a royalty based on net sales of our myChoice® HRD test, if any. This license agreement ends on expiration of the last to expire patent covered by the license agreement, which presently is not anticipated to expire until 2032. CMCC has the right to terminate the license agreement for the uncured breach of any material term of the license agreement.
In 2013, we entered into a license agreement with Institut Curie and INSERM (“INSERM”) for the exclusive world-wide right to utilize certain rights of INSERM in intellectual property relating to what is now myChoice® HRD testing. Under this license agreement we expect to pay INSERM a royalty based on net sales of our myChoice® HRD test, if any. This license agreement ends on expiration of the last to expire patent covered by the license agreement, which presently is not anticipated to expire until 2032. INSERM has the right to terminate the license agreement for the uncured breach of any material term of the license agreement.
Governmental Regulation
The services that we provide are regulated by federal, state and foreign governmental authorities. Failure to comply with the applicable laws and regulations can subject us to repayment of amounts previously paid to us, significant civil and criminal penalties, loss of licensure, certification, or accreditation, or exclusion from state and federal health care programs. The significant areas of regulation are summarized below.
10
Clinical Laboratory Improvement Amendments of 1988 and State Regulation
Each of our clinical laboratories must hold certain federal, state and local licenses, certifications and permits to conduct our business. Laboratories in the United States that perform testing on human specimens for the purpose of providing information for the diagnosis, prevention, or treatment of disease are subject to the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”). CLIA requires such laboratories to be certified by the federal government and mandates compliance with various operational, personnel, facilities administration, quality and proficiency testing requirements intended to ensure that testing services are accurate, reliable and timely. CLIA certification also is a prerequisite to be eligible to bill state and federal health care programs, as well as many private insurers, for laboratory testing services. Our laboratories in Salt Lake City, Utah, Austin, Texas, Mason, Ohio, and South San Francisco, California are CLIA certified to perform high complexity tests.
In addition, CLIA requires each of our certified laboratories to enroll in an approved proficiency testing program if performing testing in any category for which proficiency testing is required. Each of our laboratories periodically tests specimens received from an outside proficiency testing organization and then submits the results back to that organization for evaluation. If one of our laboratories fails to achieve a passing score on a proficiency test, then it loses its right to perform testing. Further, failure to comply with other proficiency testing regulations, such as the prohibition on referral of a proficiency testing specimen to another laboratory for analysis, can result in revocation of the laboratory’s CLIA certification.
As a condition of CLIA certification, each of our laboratories is subject to survey and inspection every other year, in addition to being subject to additional random inspections. The biennial survey is conducted by the Centers for Medicare & Medicaid Services (“CMS”), a CMS agent (typically a state agency), or, a CMS-approved accreditation organization. Because our laboratories are accredited by the College of American Pathologists (“CAP”), which is a CMS-approved accreditation organization, they are typically subject to CAP inspections.
Our laboratories are licensed by the appropriate state agencies in the states in which they operate, if such licensure is required. In addition, our laboratories hold state licenses or permits, as applicable, from various states including, but not limited to, California, Florida, New York, Pennsylvania, Rhode Island and Maryland, to the extent that they accept specimens from one or more of these states, each of which requires out-of-state laboratories to obtain licensure.
If a laboratory is out of compliance with state laws or regulations governing licensed laboratories or with CLIA, may include suspension, limitation or revocation of the license or CLIA certificate, assessment of financial penalties or fines, or imprisonment. Loss of a laboratory’s CLIA certificate or state license may also result in the inability to receive payments from state and federal health care programs as well as private third party payors. We believe that we are in material compliance with CLIA and all applicable licensing laws and regulations.
Food and Drug Administration
Although the Food and Drug Administration (FDA) has consistently claimed that it has the authority to regulate laboratory-developed tests (“LDTs”) that are developed, validated and performed only by a CLIA certified laboratory, it has historically exercised enforcement discretion in not otherwise regulating most LDTs and has not required laboratories that furnish LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post-market controls). In recent years, however, the FDA has stated it intends to end its policy of general enforcement discretion and regulate certain LDTs as medical devices. To this end, on October 3, 2014, the FDA issued two draft guidance documents, entitled “Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs)” and “FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs)”, respectively, that set forth a proposed risk-based regulatory framework that would apply varying levels of FDA oversight to LDTs. The FDA has indicated that it does not intend to modify its policy of enforcement discretion until the draft guidance documents are finalized. It is unclear at this time when, or if, the draft guidance documents will be finalized, and even then, the new regulatory requirements are proposed to be phased-in consistent with the schedule set forth in the guidance (in as little as 12 months after the draft guidance is finalized for certain high-priority LDTs). Nevertheless, the FDA may decide to regulate certain LDTs on a case-by-case basis at any time. LDTs with the same intended use as a cleared or approved companion diagnostic are defined in FDA’s draft guidance as “high-risk LDTs (Class III medical devices)” for which premarket review would be first to occur.
We are developing companion diagnostic tests for use with drug products in development by pharmaceutical companies, such as our collaborations with pharmaceutical companies on PARP inhibitors for the treatment of ovarian, breast and other cancers. Companion diagnostic tests are currently subject to regulation by the FDA as medical devices. The FDA issued Guidance on In-Vitro Companion Diagnostic Devices in July 2014, which is intended to assist companies developing in vitro companion diagnostic devices and companies developing therapeutic products that depend on the use of a specific in-vitro companion
11
diagnostic for the safe and effective use of the product. The FDA defined an invitro companion diagnostic device (“IVD Companion Dx”) as a device that provides information that is essential for the safe and effective use of a corresponding therapeutic product. The FDA expects that the therapeutic sponsor will address the need for an approved or cleared IVD Companion Dx in its therapeutic product development plan and that, in most cases, the therapeutic product and its corresponding companion diagnostic will be developed contemporaneously. On July 15, 2016, FDA released a draft guidance entitled, “Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product.: This draft guidance document is intended to be a practical guide to assist therapeutic product sponsors and IVD sponsors in developing a therapeutic product and an accompanying IVD companion diagnostic.
The FDA has also introduced the concept of a complementary diagnostic that it defines as a test that is not required but which provides significant information about the use of a drug. A complementary test can help guide treatment strategy and identify which patients are likely to derive the greatest benefit from therapy, and if approved by the FDA information regarding the IVD will be included in the therapeutic product labelling. Although the FDA has not yet issued any written guidance regarding complementary diagnostics, it has already approved a couple of complementary diagnostics, including a supplementary premarket approval for BRACAnalysis CDx, which was approved in March 2017 as a complementary diagnostic test in ovarian cancer patients associated with enhanced progression-free survival (PFS) when used with Tesaro’s PARP inhibitor Zejula™ (niraparib) maintenance therapy.
In December 2014, we obtained premarket approval for BRACAnalysis CDx, which is used as a companion diagnostic test to identify ovarian cancer patients who may benefit from AstraZeneca’s PARP inhibitor Lynparza™ (olaparib). The premarket approval process is a complex, costly and time consuming procedure. Approvals must be supported by valid scientific evidence, submitted as part of a premarket approval application (“PMA”), which typically requires extensive data, including quality technical, preclinical, clinical and manufacturing data to demonstrate to the FDA’s satisfaction the safety and effectiveness of the companion diagnostic. We are currently collaborating with several pharmaceutical companies, including an expanded collaboration with AstraZeneca for an additional indication for BRACAnalysis CDx, to evaluate the use of several of our tests as companion diagnostics with other drugs.
After a medical device is placed on the market, numerous regulatory requirements apply. These include:
|
|
• |
compliance with the FDA’s Quality System Regulation (“QSR”), which requires manufacturers to follow stringent design, testing, control, documentation, record maintenance, including maintenance of complaint and related investigation files, and other quality assurance controls during the manufacturing process; |
|
|
• |
labeling regulations, which prohibit the promotion of products for uncleared, or unapproved uses, or “off-label” uses, and impose other restrictions on labeling; and |
|
|
• |
medical device reporting obligations, which require that manufacturers investigate and report to the FDA adverse events, including deaths, or serious injuries that may have been or were caused by a medical device and malfunctions in the device that would likely cause or contribute to a death or serious injury if it were to recur. |
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include sanctions, including but not limited to, warning letters; fines, injunctions, and civil penalties; recall or seizure of the device; operating restrictions, partial suspension or total shutdown of production; refusal to grant 510(k) clearance or approval of PMAs of new devices; withdrawal of clearance or approval; and civil or criminal prosecution. To ensure compliance with regulatory requirements, medical device manufacturers are subject to market surveillance and periodic, pre-scheduled and unannounced inspections by the FDA.
Other Regulatory Requirements
Our laboratories are subject to federal, state and local regulations relating to the handling and disposal of regulated medical waste, hazardous waste and biohazardous waste, including chemical, biological agents and compounds, blood and bone marrow samples and other human tissue. Typically, we use outside vendors who are contractually obligated to comply with applicable laws and regulations to dispose of such waste. These vendors are licensed or otherwise qualified to handle and dispose of such waste.
12
The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), established comprehensive federal standards for the privacy and security of health information. The HIPAA standards apply to three types of organizations: health plans, healthcare clearing houses, and healthcare providers that conduct certain healthcare transactions electronically (“Covered Entities”). Title II of HIPAA, the Administrative Simplification Act, contains provisions that address the privacy of health data, the security of health data, the standardization of identifying numbers used in the healthcare system and the standardization of certain healthcare transactions. The privacy regulations protect medical records and other protected health information by limiting their use and release, giving patients the right to access their medical records and limiting most disclosures of health information to the minimum amount necessary to accomplish an intended purpose. The HIPAA security standards require the adoption of administrative, physical, and technical safeguards and the adoption of written security policies and procedures.
On February 17, 2009, Congress enacted Subtitle D of the Health Information Technology for Economic and Clinical Health Act, or HITECH, provisions of the American Recovery and Reinvestment Act of 2009. HITECH expanded and strengthened HIPAA, created new targets for enforcement, imposed new penalties for noncompliance and established new breach notification requirements for Covered Entities. Regulations implementing major provisions of HITECH were finalized on January 25, 2013 through publication of the HIPAA Omnibus Rule (the “Omnibus Rule”).
Under HITECH's breach notification requirements, Covered Entities must report breaches of protected health information that has not been encrypted or otherwise secured in accordance with guidance from the Secretary of the U.S. Department of Health and Human Services (the “Secretary”). Required breach notices must be made as soon as is reasonably practicable, but no later than 60 days following discovery of the breach. Reports must be made to affected individuals and to the Secretary and, in some cases depending on the size of the breach; they must be reported through local and national media. Breach reports can lead to investigation, enforcement and civil litigation, including class action lawsuits.
We are currently subject to the HIPAA regulations and maintain an active compliance program that is designed to identify security incidents and other issues in a timely fashion and enable us to remediate, mitigate harm or report if required by law. We are subject to prosecution and/or administrative enforcement and increased civil and criminal penalties for non-compliance, including a new, four-tiered system of monetary penalties adopted under HITECH. We are also subject to enforcement by state attorneys general who were given authority to enforce HIPAA under HITECH. To avoid penalties under the HITECH breach notification provisions, we must ensure that breaches of protected health information are promptly detected and reported within the company, so that we can make all required notifications on a timely basis. However, even if we make required reports on a timely basis, we may still be subject to penalties for the underlying breach.
In May 2016, a complaint was filed with the Office for Civil Rights of the Department of Health and Human Services (“OCR”) by four patients alleging deficiencies in our policies regarding information that must be disclosed to patients as part of a “designated record set” under HIPAA. We proactively reached out to OCR on these issues to explain our compliance with all applicable regulations and OCR guidance. We are currently working with OCR to favorably resolve the concerns raised by the allegations.
A similar complaint was filed with the College of American Pathologists (“CAP”), which is the designated agency administering CLIA for Myriad Genetic Laboratories. We responded to CAP’s notice of the complaint and, on July 27, 2016, received a letter notifying us that the matter has been closed.
In addition to the federal privacy and security regulations, there are a number of state laws regarding the privacy and security of health information and personal data that are applicable to our clinical laboratories. Many states have also implemented genetic testing and privacy laws imposing specific patient consent requirements and protecting test results by strictly limiting the disclosure of those results. State requirements are particularly stringent regarding predictive genetic tests, due to the risk of genetic discrimination against healthy patients identified through testing as being at a high risk for disease. We believe that we have taken the steps required of us to comply with health information privacy and security statutes and regulations, including genetic testing and genetic information privacy laws in all jurisdictions, both state and federal. However, these laws constantly change and we may not be able to maintain compliance in all jurisdictions where we do business. Failure to maintain compliance, or changes in state or federal laws regarding privacy or security could result in civil and/or criminal penalties, significant reputational damage and could have a material adverse effect on our business.
We are subject to laws and regulations related to the protection of the environment, the health and safety of employees and the handling, transportation and disposal of medical specimens, infectious and hazardous waste and radioactive materials. For example, the U.S. Occupational Safety and Health Administration (“OSHA”) has established extensive requirements relating specifically to workplace safety for healthcare employers in the U.S. This includes requirements to develop and implement
13
multi-faceted programs to protect workers from exposure to blood-borne pathogens, including preventing or minimizing any exposure through needle stick injuries. For purposes of transportation, some biological materials and laboratory supplies are classified as hazardous materials and are subject to regulation by one or more of the following agencies: the U.S. Department of Transportation, the U.S. Public Health Service, the United States Postal Service and the International Air Transport Association. We generally use third-party vendors to dispose of regulated medical waste, hazardous waste and radioactive materials and contractually require them to comply with applicable laws and regulations.
Transparency Laws and Regulations
A federal law known as the Physician Payments Sunshine Act (the “Sunshine Act”) requires medical device manufacturers to track and report to the federal government certain payments and other transfers of value made to physicians and teaching hospitals and ownership or investment interests held by physicians and their immediate family members. Manufacturers must report data for the previous calendar year by the 90th day of the then-current calendar year. CMS then publishes the data on a publicly available website no later than June 30th. There are also state “sunshine” laws that require manufacturers to provide reports to state governments on pricing and marketing information. Several states have enacted legislation requiring medical device manufacturers to, among other things, establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales and marketing activities, and such laws may also prohibit or limit certain other sales and marketing practices. These laws may adversely affect our sales, marketing, and other activities by imposing administrative and compliance burdens on us. If we fail to track and report as required by these laws or to otherwise comply with these laws, we could be subject to the penalty provisions of the pertinent state and federal authorities.
Reimbursement and Billing
Reimbursement and billing for diagnostic services is highly complex. Laboratories must bill various payors, such as private third-party payors, including managed care organizations (“MCO”), and state and federal health care programs, such as Medicare and Medicaid, and each may have different billing requirements. Additionally, the audit requirements we must meet to ensure compliance with applicable laws and regulations, as well as our internal compliance policies and procedures, add further complexity to the billing process. Other factors that complicate billing include:
|
|
• |
variability in coverage and information requirements among various payors; |
|
|
• |
patient financial assistance programs; |
|
|
• |
missing, incomplete or inaccurate billing information provided by ordering physicians; |
|
|
• |
billings to payors with whom we do not have contracts; |
|
|
• |
disputes with payors as to which party is responsible for payment; and |
|
|
• |
disputes with payors as to the appropriate level of reimbursement. |
Depending on the reimbursement arrangement and applicable law, the party that reimburses us for our services may be:
|
|
• |
a third party who provides coverage to the patient, such as an insurance company or MCO; |
|
|
• |
a state or federal healthcare program; or |
|
|
• |
the patient. |
Presently, approximately 85% of our revenue comes from private third party payors.
14
Federal and State Fraud and Abuse Laws
A variety of federal laws prohibit fraud and abuse involving state and federal health care programs, such as Medicare and Medicaid. These laws are interpreted broadly and enforced aggressively by various state and federal agencies, including CMS, the Department of Justice, the Office of Inspector General for the Department of Health and Human Services (“OIG”), and various state agencies. In addition, the Medicare and Medicaid programs increasingly use a variety of contractors to review claims data and to identify improper payments as well as fraud and abuse. Any overpayments must be repaid within 60 days of identification unless a favorable decision is obtained on appeal. In some cases, these overpayments can be used as the basis for an extrapolation, by which the error rate is applied to a larger universe of claims, and which can result in even higher repayments.
Anti-Kickback Laws
The Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for or recommending of an item or service that is reimbursable, in whole or in part, by a federal health care program. “Remuneration” is broadly defined to include anything of monetary value, such as, for example, cash payments, gifts or gift certificates, discounts, or the furnishing of services, supplies or equipment. The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the health care industry.
Recognizing the breadth of the Anti-Kickback Statute and the fact that it may technically prohibit many innocuous or beneficial arrangements within the health care industry, the OIG has issued a series of regulations, or safe harbors intended to protect such arrangements. Compliance with all requirements of a safe harbor immunizes the parties to the business arrangement from prosecution under the Anti-Kickback Statute. The failure of a business arrangement to fit within a safe harbor does not necessarily mean that the arrangement is illegal or that the OIG will pursue prosecution. Still, in the absence of an applicable safe harbor, a violation of the Anti-Kickback Statute may occur even if only one purpose of an arrangement is to induce referrals. The penalties for violating the Anti-Kickback Statute can be severe. These sanctions include criminal and civil penalties, imprisonment and possible exclusion from the federal health care programs. Many states have adopted laws similar to the Anti-Kickback Statute, and some apply to items and services reimbursable by any payor, including private third-party payors.
Physician Self-Referral Bans
The federal ban on physician self-referrals, commonly known as the Stark Law, prohibits, subject to certain exceptions, physician referrals of Medicare patients to an entity providing certain designated health services, which include laboratory services, if the physician or an immediate family member of the physician has any financial relationship with the entity. Several Stark Law exceptions are relevant to arrangements involving clinical laboratories, including but not limited to: (1) fair market value compensation for the provision of items or services; (2) payments by physicians to a laboratory for clinical laboratory services; (3) certain space and equipment rental arrangements that satisfy certain requirements; and (4) personal services arrangements. Penalties for violating the Stark Law include the return of funds received for all prohibited referrals, fines, civil monetary penalties and possible exclusion from federal health care programs. In addition to the Stark Law, many states have their own self-referral bans, which may extend to all self-referrals, regardless of the payor.
State and Federal Prohibitions on False Claims
The federal False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government. Under the False Claims Act, a person acts knowingly if he has actual knowledge of the information or acts in deliberate ignorance or in reckless disregard of the truth or falsity of the information. Specific intent to defraud is not required. The qui tam provisions of the False Claims Act allow a private individual to bring an action on behalf of the federal government and to share in any amounts paid by the defendant to the government in connection with the action. Penalties include payment of up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 and $11,000 for each false claim, as well as possible exclusion from the federal health care programs. In addition, various states have enacted similar laws modeled after the False Claims Act that apply to items and services reimbursed under Medicaid and other state health care programs, and, in several states, such laws apply to claims submitted to any payor.
15
The federal Civil Monetary Penalties Law, or the CMP Law, prohibits, among other things, (1) the offering or transfer of remuneration to a Medicare or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies; (2) employing or contracting with an individual or entity that the provider knows or should know is excluded from participation in a federal health care program; (3) billing for services requested by an unlicensed physician or an excluded provider; and (4) billing for medically unnecessary services. The penalties for violating the CMP Law include exclusion, substantial fines, and payment of up to three times the amount billed, depending on the nature of the offense.
International regulations
We market some of our tests outside of the United States and are subject to foreign regulatory requirements governing laboratory licensure, human clinical testing, use of tissue, privacy and data security, and marketing approval for our tests. These requirements vary by jurisdiction, differ from those in the United States and may require us to implement additional compliance measures or perform additional pre-clinical or clinical testing. On September 26, 2012, the European Commission released the first drafts of the new European Union (“EU”) regulations for medical devices and IVDs that if finalized will impose additional regulatory requirements on IVDs used in the EU. In many countries outside of the United States, coverage, pricing and reimbursement approvals are also required. We are also required to maintain accurate information on and control over sales and distributors’ activities that may fall within the purview of the Foreign Corrupt Practices Act, its books and records provisions and its anti-bribery provisions.
Human Resources
As of June 30, 2017, we have over 2,400 full-time equivalent employees. Most of our employees are engaged directly in research, development, production, sales and marketing activities. We believe that the success of our business will depend, in part, on our ability to attract and retain qualified personnel. Our employees are not covered by a collective bargaining agreement, and we consider our relations with our employees to be good.
Available Information
We are a Delaware corporation with our principal executive offices located at 320 Wakara Way, Salt Lake City, Utah 84108. Our telephone number is (801) 584-3600 and our web site address is www.myriad.com. We make available free of charge through the Investor Relations section of our web site our Corporate Code of Conduct and Ethics, our Audit Committee and other committee charters and our other corporate governance policies, as well as our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission. We include our web site address in this Annual Report on Form 10-K only as an inactive textual reference and do not intend it to be an active link to our web site.
16
Risks Related to Our Business and Our Strategy
We may not be successful in transitioning from our existing product portfolio to our new products, such as our myRisk Hereditary Cancer test, which represents the next generation of our existing hereditary cancer franchise. We may not be able to generate sufficient revenue from our existing tests and our new tests or develop new tests to maintain profitability.
Although we have developed and marketed several molecular diagnostic tests to date, we believe our future success is dependent upon our ability to successfully market our existing molecular diagnostic tests to additional patients within the United States, to expand into new markets outside the United States, and to develop and commercialize new molecular diagnostic and companion diagnostic tests. Importantly, in 2014 we launched our myRisk Hereditary Cancer test, which represents the next generation of our existing hereditary cancer testing franchise. We anticipate that the myRisk Hereditary Cancer test will eventually replace our current predictive medicine test offerings (BRACAnalysis, BART, Colaris and Colaris AP and Melaris) with a single comprehensive test. However, we may not be successful in transitioning from our existing product portfolio to our new tests and in launching and commercializing our new tests. The demand for our existing molecular diagnostic tests may decrease or may not continue to increase at historical rates due to sales of the myRisk Hereditary Cancer test and our other new tests that are replacing our existing product portfolio, or for other reasons. For example, because most of our molecular diagnostic tests are only utilized once per patient, we will need to sell our services through physicians to new patients or develop new molecular diagnostic tests in order to continue to generate revenue. Our pipeline of new molecular diagnostic and companion diagnostic test candidates is in various stages of development and may take several more years to develop and must undergo extensive clinical validation. We may be unable to discover or develop any additional molecular diagnostic or companion diagnostic tests through the utilization of our technologies or technologies we license or acquire from others. Even if we develop tests or services for commercial use, we may not be able to develop tests or services that:
|
|
• |
meet applicable regulatory standards, in a timely manner or at all; |
|
|
• |
successfully compete with other technologies and tests; |
|
|
• |
avoid infringing the proprietary rights of others; |
|
|
• |
are adequately reimbursed by third-party payors; |
|
|
• |
can be performed at commercial levels or at reasonable cost; or |
|
|
• |
can be successfully marketed. |
We must generate significant revenue to maintain profitability. Even if we succeed in marketing myRisk Hereditary Cancer and our existing molecular diagnostic tests to physicians for use in new patients and in developing and commercializing any additional molecular diagnostic tests and companion diagnostic tests, we may not be able to generate sufficient revenue and we may not be able to maintain profitability.
We may not be able to sustain or increase profitability on a quarterly or annual basis.
In order to develop and commercialize our molecular diagnostic and companion diagnostic tests, we expect to incur significant expenses over the next several years as we increase our research and development activities, expand clinical validation trials for our molecular diagnostic tests and companion diagnostic tests currently in development, potentially license or acquire additional companies or technologies and engage in commercialization activities in anticipation of the launch of additional molecular diagnostic tests companion diagnostic tests. Because of the numerous risks and uncertainties associated with developing our tests and their potential for commercialization, we are unable to predict the extent of any future profits. If we are unable to sustain or increase profitability, the market value of our common stock will likely decline. Our ability to maintain profitability will depend upon numerous factors, including:
|
|
• |
our ability to transition from our existing product portfolio to our new products, such as our myRisk Hereditary Cancer test, and to commercialize these new tests; |
|
|
• |
successful outcomes of clinical trials (including but not limited to the GeneSight clinical trial); |
|
|
• |
our ability to obtain full or partial reimbursement for new products; |
|
|
• |
our ability to sell our other existing molecular diagnostic tests to new patients; |
|
|
• |
our ability to identify biomarkers that may lead to future molecular diagnostic tests and companion diagnostic tests; |
17
|
|
• |
our ability to successfully commercialize our tests in our existing markets and to extend into new markets outside the United States; |
|
|
• |
the approval and introduction of competitive tests; |
|
|
• |
reductions in reimbursement by third-party payors or their willingness to provide full or even partial reimbursement for our tests; |
|
|
• |
our ability to maintain and enforce our intellectual property rights covering our molecular diagnostic tests and companion diagnostic tests; |
|
|
• |
our ability to maintain and grow our sales force and marketing team to market our tests; |
|
|
• |
our ability to successfully integrate, develop and grow products and services and the business of any other companies or technologies that we may license or acquire; |
|
|
• |
our ability to increase commercial acceptance of our current molecular diagnostic tests; and |
|
|
• |
our ability to maintain or grow our current revenues. |
If we do not continue to generate sufficient revenue from sales of our molecular diagnostic tests and are unable to secure additional funding, we may have to reduce our operations.
As of June 30, 2017, we had $199.2 million in cash, cash equivalents and marketable securities. For the fiscal year ended June 30, 2017 our consolidated revenues were $771.4 million, and net cash from operating activities was $106.2 million. To develop and bring new molecular diagnostic tests and companion diagnostic tests to market, we must commit substantial resources to costly and time-consuming research, development testing and clinical testing. In addition, we entered into an unsecured revolving debt facility (the “Facility) in December 2016, pursuant to which we borrowed a principal amount of $205.0 million. The Facility is due on December 23, 2021. As of June 30, 2017, the balance due under our Facility was $99.1 million.
While we anticipate that our existing cash, cash equivalents and marketable securities and expected net cash to be generated from sales of our molecular diagnostic tests and pharmaceutical and clinical services will be sufficient to fund our current operations for the foreseeable future, changes could occur that would consume available capital resources more quickly than we currently expect and we may need or want to raise additional financing. If we are unable to secure additional funding, we may be unable to repay our Revolving Debt Facility when it becomes due, and be required to reduce research and development projects, limit sales and marketing activities, scale back our expansion efforts outside the United States, reduce headcount or potentially even discontinue operations. Our future capital requirements will depend on many factors that are currently unknown to us, including:
|
|
• |
the scope, progress, results and cost of development, clinical testing and pre-market studies of any new molecular diagnostic tests that we may discover or acquire; |
|
|
• |
the progress, results, and costs to develop additional molecular diagnostic tests; |
|
|
• |
the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our current issued patents, and defending intellectual property-related claims; |
|
|
• |
our ability to enter into collaborations, licensing or other arrangements favorable to us; |
|
|
• |
the costs of acquiring technologies or businesses, and our ability to successfully integrate and achieve the expected benefits of our business development activities and acquisitions; |
|
|
• |
the progress, cost and results of our international expansion efforts; |
|
|
• |
the costs of expanding our sales and marketing functions and commercial operation facilities in the United States and in new markets; |
|
|
• |
the costs, timing and outcome of any litigation against us; and |
|
|
• |
the costs to satisfy our current and future obligations. |
18
We are subject to debt covenants that impose operating and financial restrictions on us and could limit our ability to grow our business.
Covenants in the Facility, which went into effect during the quarter ending March 31, 2017, impose operating and financial restrictions on the Company. These restrictions may prohibit or place limitations on, among other things, the Company’s ability to incur additional indebtedness, create certain types of liens, mergers or consolidations, and/or change in control transactions. The Facility may also prohibit or place limitations on the Company’s ability to sell assets, pay dividends or provide other distributions to shareholders. These restrictions could also limit our ability to take advantage of business opportunities. The Company must maintain a specified leverage and interest ratios measured as of the end of each quarter as a financial covenant in the Facility. Our ability to comply with this ratio may be affected by events beyond our control, including prevailing economic, financial and industry conditions.
Under the Facility, a change in control in our Company, which means that a shareholder or a group of shareholders is or becomes the beneficial owner, directly or indirectly, of more than 35% of the total voting power of the voting stock of the Company would require mandatory prepayment of the outstanding debt.
If we are unable to comply with the covenants and ratio in the Facility in the future, we may be in default under the agreement. A default would result in an increase in the rate of interest and may cause the loan repayment to be accelerated. This could have a material adverse effect on our business.
We may acquire technologies, assets or other businesses that could cause us to incur significant expense and expose us to a number of unanticipated operational and financial risks.
In addition to organic growth, we intend to continue to pursue growth through the acquisition of technology, assets or other businesses that may enable us to enhance our technologies and capabilities, expand our geographic market, add experienced management personnel and increase our test offerings. For example, in May 2011, we completed the acquisition of Rules Based Medicine, Inc., which we renamed Myriad RBM, and are now offering pharmaceutical and clinical services and developing additional product candidates using the acquired technology. In February 2014, we completed the acquisition of Crescendo Bioscience, Inc., and are now offering molecular diagnostic tests for patients suffering from rheumatoid arthritis and developing additional product candidates in the inflammatory and autoimmune disease area. In February 2015, we acquired the Clinic and believe the acquisition may facilitate our penetration into the German molecular diagnostic market. In May 2016, we acquired Sividon Diagnostics GmbH. Now as a wholly-owned subsidiary, Sividon will continue to offer EndoPredict testing in the European market, which we offered under an exclusive distribution agreement with Sividon prior to the acquisition. Additionally, in August 2016, we acquired Assurex Health, Inc. and are now offering a molecular diagnostic test providing treatment decision support to healthcare providers for mental health patients. However, these acquisitions may not achieve profitability or generate a positive return on our investment. Additionally, we may be unable to implement our growth strategy if we cannot identify suitable acquisition candidates, reach agreement on potential acquisitions on acceptable terms, successfully integrate personnel or assets that we acquire or for other reasons. Our acquisition efforts may involve certain risks, including:
|
|
• |
we may have difficulty integrating operations and systems; |
|
|
• |
key personnel and customers of the acquired company may terminate their relationships with the acquired company as a result of the acquisition; |
|
|
• |
we may not be successful in launching new molecular diagnostic tests or companion diagnostic tests, or if those tests are launched they may not prove successful in the market place; |
|
|
• |
we may experience additional financial and accounting challenges and complexities in areas such as tax planning and financial reporting; |
|
|
• |
we may assume or be held liable for risks and liabilities, including for environmental-related costs, as a result of our acquisitions, some of which we may not discover during our due diligence; |
|
|
• |
we may incur significant additional operating expenses; |
|
|
• |
our ongoing business may be disrupted or receive insufficient management attention; and |
|
|
• |
we may not be able to realize synergies, the cost savings or other financial and operational benefits we anticipated, or such synergies, savings or benefits may take longer than we expected. |
The process of negotiating acquisitions and integrating acquired tests, services, technologies, personnel or businesses might result in operating difficulties and expenditures and might require significant management attention that would otherwise be
19
available for ongoing development of our business, whether or not any such transaction is ever consummated. Moreover, we might never realize the anticipated benefits of any acquisition. Future acquisitions could result in the use of our available cash and marketable securities, potentially dilutive issuances of equity securities, the incurrence of debt, contingent liabilities, or impairment expenses related to goodwill, and impairment or amortization expenses related to other intangible assets, which could harm our financial condition. In addition, if we are unable to integrate any acquired businesses, tests or technologies effectively, our business, financial condition and results of operations may be materially adversely affected.
We may not be able to successfully integrate the operations of businesses that we acquire with our own or realize the anticipated benefits of the acquisitions, which could adversely affect our financial condition, results of operations and business prospects.
There can be no assurance that we will be able to successfully integrate our recent acquisitions or develop or commercialize products based on recently acquired technologies, or that we will be able to successfully integrate any other companies, products or technologies that we acquire and may not realize all or any of the expected benefits of any acquisitions as and when planned. Additionally, we may experience increased expenses, distraction of our management, personnel and customer uncertainty.
The difficulties and risks associated with the integration of any other businesses that we may acquire include:
|
|
• |
possible inconsistencies in the standards, controls, procedures, policies and compensation structures; |
|
|
• |
the increased scope and complexity of the acquired company’s operations; |
|
|
• |
the potential loss of key employees and the costs associated to retain key employees; |
|
|
• |
risks and limitations on our ability to consolidate corporate and administrative infrastructures of the two companies; and |
|
|
• |
the possibility of unanticipated delays, costs or inefficiencies associated with the integration of our operations with the operations of any other companies that we may acquire. |
As a result of these difficulties and risks, we may not accomplish the integration of the business of any companies we may acquire smoothly, successfully or within our budgetary expectations and anticipated timetable. Accordingly, we may fail to realize some or all of the anticipated benefits of the acquisition, such as increase in our scale, diversification, cash flows and operational efficiency and meaningful accretion to our diluted earnings per share.
If we were successfully sued for product liability, we could face substantial liabilities that exceed our resources.
Our business exposes us to potential liability risks inherent in the testing, marketing and processing of molecular diagnostic products, including possible misdiagnoses. Although we are insured against such risks in amounts that we believe to be commercially reasonable, our present professional and product liability insurance may be inadequate. A successful product liability claim in excess of our insurance coverage could have a material adverse effect on our business. Any successful product liability claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable or reasonable terms. An inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our products.
We are dependent on our information technology and telecommunications systems, and any failure of these systems could harm our business.
We depend on information technology, or IT, and telecommunications systems for significant aspects of our business. These IT and telecommunications systems support a variety of functions, including sample processing, tracking, quality control, customer service and support, billing, research and development activities, and various general and administrative activities. Failures or significant downtime of our IT or telecommunications systems could prevent us from processing samples, providing test results to physicians, billing payors, addressing patient or physician inquiries, conducting research and development activities and conducting general and administrative elements of our business. Any disruption or loss of IT or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business, financial condition and results of operations.
20
Security breaches, loss of data and other disruptions could compromise sensitive information related to our business, prevent us from accessing critical information or expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we collect and store sensitive data, including legally protected patient health information, credit card information, personally identifiable information about our employees, intellectual property, and proprietary business information. We manage and maintain our applications and data utilizing on-site systems. These applications and data encompass a wide variety of business critical information including research and development information, commercial information and business and financial information.
The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers, or viruses, breaches or interruptions due to employee error, malfeasance or other disruptions, or lapses in compliance with privacy and security mandates. Any such virus, breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. We have measures in place that are designed prevent, and if necessary to detect and respond to such security incidents and breaches of privacy and security mandates. While we have experienced unauthorized accesses to our information technology systems and infrastructure in the past, which may occur again in the future, our security measures have been able to detect, respond to and prevent any material adverse effect to our information systems and business operations from such breaches. However, in the future, any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA, government enforcement actions and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process samples, provide test results, bill payors or patients, provide customer support services, conduct research and development activities, process and prepare company financial information, manage various general and administrative aspects of our business and may damage our reputation, any of which could adversely affect our business, financial condition and results of operations.
If our current operating plan changes and we find that our existing capital resources will not meet our needs, we may find it necessary to raise additional funding, which may not be available.
We anticipate that our existing capital resources and expected net cash to be generated from sales of our molecular diagnostic tests will enable us to maintain our currently planned operations for the foreseeable future. However, we base this expectation on our current operating plan, which may change. We have incurred, and will continue to incur, significant costs in the discovery, development and marketing of current and prospective molecular diagnostic and companion diagnostic tests. Our ongoing efforts to develop tests and expand our business which may be through internally developed products, in licensing and mergers and acquisitions will require substantial cash resources. If, due to changes in our current operating plan, adequate funds are not available, we may be required to raise additional funds. Sources of potential additional capital resources may include, but are not limited to, public or private equity financings, establishing a credit facility, or selling convertible or non-convertible debt securities. This additional funding, if necessary, may not be available to us on reasonable terms, or at all. If we issue shares of stock or other securities to acquire new companies or technologies, the ownership interests of our existing stockholders may be significantly diluted.
Because of our potential long-term capital requirements, we may access the public or private equity or debt markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. Under SEC rules, we currently qualify as a well-known seasoned issuer, or WKSI, and can at any time file a registration statement registering securities to be sold to the public which would become effective upon filing. If additional funds are raised by issuing equity securities, existing shareholders may suffer significant dilution. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or tests, or grant licenses on terms that are not favorable to us.
Our business involves environmental risks that may result in liability for us.
In connection with our research and development activities, we are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens, chemicals and wastes. Although we believe that we have complied with the applicable laws, regulations and policies in all material respects and have not been required to correct any material noncompliance, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Although we believe that our safety procedures for handling and disposing of controlled materials comply with the standards prescribed by
21
state and federal regulations, accidental contamination or injury from these materials may occur. In the event of such an occurrence, we could be held liable for any damages that result and any such liability could exceed our resources.
Changes in health care policy could increase our costs, decrease our revenues and impact sales of and reimbursement for our tests.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the ACA became law. This law substantially changed the way health care is financed by both governmental and private insurers, and significantly impacts our industry. The ACA contains a number of provisions that are expected to impact our business and operations, some of which in ways we cannot currently predict, including those governing enrollment in state and federal health care programs, reimbursement changes and fraud and abuse, which will impact existing state and federal health care programs and will result in the development of new programs. The Trump administration supports a repeal of the ACA and an Executive Order was signed commanding federal agencies to try to waive or delay requirements of the ACA that impose economic or regulatory burdens on states, families, the health-care industry and others. The Executive Order also declares that the administration will seek the “prompt repeal” of the law and that the government should prepare to “afford the States more flexibility and control to create a more free and open healthcare market.” In addition, following the passage of the budget resolution for fiscal year 2017, the U.S. House of Representatives passed legislation known as the American Health Care Act, which, if enacted, would amend or repeal significant portions of the ACA. Although it seems unlikely, the U.S. Senate could adopt the American Health Care Act as passed by the U.S. House of Representatives or other legislation to amend or replace elements of the ACA. It is uncertain whether the American Health Care Act will become law. At this time, the immediate impact of the Executive Order is not clear, and we cannot know how any legislation that may be passed to amend or replace the ACA will impact our business in the United States.
In addition to the ACA, there will continue to be proposals by legislators at both the federal and state levels, regulators and private third-party payors to reduce costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge for our tests or the amounts of reimbursement available for our tests from governmental agencies or private third-party payors.
We face risks associated with currency exchange rate fluctuations, which could adversely affect our operating results.
We receive a portion of our revenues and pay a portion of our expenses in currencies other than the United States dollar, such as the Euro, the Swiss franc, the British pound, the Australian dollar and the Canadian dollar. As a result, we are at risk for exchange rate fluctuations between such foreign currencies and the United States dollar, which could affect the results of our operations. If the U.S. dollar strengthens against foreign currencies, the translation of these foreign currency denominated transactions will result in decreased revenues and operating expenses. We may not be able to offset adverse foreign currency impact with increased revenues. We do not currently utilize hedging strategies to mitigate foreign currency risk and even if we were to implement hedging strategies to mitigate foreign currency risk, these strategies might not eliminate our exposure to foreign exchange rate fluctuations and would involve costs and risks of their own, such as ongoing management time and expertise, external costs to implement the strategies and potential accounting implications.
Risks Related to Commercialization of Our Tests, Our Services and Test Candidates
We generate most of our revenues from three products and we may not be able to maintain revenue growth and profitability.
We may not be able to generate revenue growth or maintain existing revenue levels. Presently, our molecular diagnostic business operates profitably providing a cash contribution to our current funding and operational needs. We may not, however, be able to continue to operate our molecular diagnostic business on a profitable basis. We launched our first molecular diagnostic test, BRACAnalysis, our test for hereditary breast and ovarian cancer, in November 1996. BRACAnalysis test sales accounted for approximately 9% of our revenues for the year ended June 30, 2017, and this percentage has been declining in recent years as we transition to our myRisk Hereditary Cancer test. An interruption or cessation of BRACAnalysis sample flow would have a material impact on our revenues and future profitability. In 2014 we launched our myRisk Hereditary Cancer test, which represents the next generation of our existing hereditary cancer franchise. We may not be successful in transitioning from our existing product portfolio to our new products, such as myRisk Hereditary Cancer Test, and in commercializing these tests over time. Other potential events or factors that may have a significant impact on our ability to sustain revenue growth and profitability for our molecular diagnostic business include the following:
|
|
• |
increased costs of reagents and other consumables required for molecular diagnostic testing; |
|
|
• |
increased personnel and facility costs; |
22
|
|
• |
our inability to hire competent, trained staff, including laboratory directors required to review and approve all reports we issue in our molecular diagnostic business, and sales personnel; |
|
|
• |
our inability to obtain necessary equipment or reagents to perform molecular diagnostic testing; |
|
|
• |
our inability to increase production capacity as demand increases; |
|
|
• |
our inability to expand into new markets outside the United States; |
|
|
• |
the efforts of third party payors to limit or decrease the amounts that they are willing to pay for our tests; |
|
|
• |
increased licensing or royalty costs, and our ability to maintain and enforce the intellectual property rights underlying our tests and services; |
|
|
• |
changes in intellectual propriety law applicable to our patents or enforcement in the United States and foreign countries; |
|
|
• |
potential obsolescence of our tests; |
|
|
• |
our inability to increase commercial acceptance of our molecular diagnostic tests; |
|
|
• |
increased competition and loss of market share; and |
|
|
• |
increased regulatory requirements. |
Our international business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
As part of our business strategy, we have expanded into international markets. We have established sales offices in Germany, Switzerland, France, Spain, the United Kingdom, Italy, Canada and Australia; laboratory operations in Germany; and international headquarters in Switzerland. We may establish additional operations or acquire additional properties outside the United States in order to advance our international sales doing business internationally involves a number of risks, including:
|
|
• |
failure by us to obtain regulatory approvals or adequate reimbursement for the use of our tests in various countries; |
|
|
• |
difficulty in staffing and managing foreign operations; |
|
|
• |
managing multiple payor reimbursement and self-pay systems; |
|
|
• |
logistics and regulations associated with shipping patient samples, including infrastructure conditions and transportation delays; |
|
|
• |
limits in our ability to penetrate international markets if we are not able to process tests locally; |
|
|
• |
financial risks, such as longer payment cycles, difficulty collecting accounts receivable and exposure to foreign currency exchange rate fluctuations; |
|
|
• |
political and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; |
|
|
• |
multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, data and privacy laws, regulatory requirements and other governmental approvals, permits and licenses; and |
|
|
• |
regulatory and compliance risks that relate to maintaining accurate information and control over sales and distributors’ activities that may fall within the purview of the U.S. Foreign Corrupt Practice Act, anti-boycott and other laws. |
Any of these factors could significantly harm our international operations and, consequently, our revenues and results of operations. In addition, any failure to comply with applicable legal and regulatory obligations could impact us in a variety of ways that include, but are not limited to, significant criminal, civil and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, seizure of shipments, and restrictions on certain business activities. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our distribution and sales activities.
23
Our international operations could be affected by changes in laws, trade regulations, labor and employment regulations, and procedures and actions affecting approval, production, pricing, reimbursement and marketing of tests, as well as by inter-governmental disputes. Any of these changes could adversely affect our business.
Our success internationally will depend, in part, on our ability to develop and implement policies and strategies that are effective in anticipating and managing these and other risks in the countries in which we do business. Failure to manage these and other risks may have a material adverse effect on our operations in any particular country and on our business as a whole.
Foreign governments may impose reimbursement standards, which may adversely affect our future profitability.
We market our tests in foreign jurisdictions and as such may be subject to rules and regulations in those jurisdictions relating to our testing. In some foreign countries, including countries in the European Union, the reimbursement of diagnostic tests is subject to governmental control. In these countries, reimbursement negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a test candidate. If reimbursement of our future tests is unavailable or limited in scope or amount, or if reimbursement rates are set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
We may experience increased price competition and price erosion.
We may experience pricing pressures from managed care organizations and other private third-party payors in the future. Any declines in average selling prices of our products due to pricing pressures may have an adverse impact on our business, results of operations and financial condition.
Our pharmaceutical testing services customers may reduce the amount of testing they conduct through us.
If there is a change in the regulatory environment or intellectual property law, or our pharmaceutical testing services customers consolidate, our customers may divert resources from testing, resulting in a reduced demand for our laboratory testing services. Alternatively, customers may decide to perform their own laboratory testing services in-house.
We rely on a single laboratory facility to process each of our molecular diagnostic tests in the United States and Europe and a single laboratory facility to perform our pharmaceutical and clinical services. Failure to maintain the operations of these laboratories in compliance with applicable regulations would seriously harm our business.
We rely on a CLIA-certified and FDA approved laboratory facility in Salt Lake City, Utah to perform most of our molecular diagnostic tests; a CLIA-certified laboratory in South San Francisco, California to perform our VectraDA test; a single laboratory facility in Munich, Germany to perform our international molecular diagnostic tests; a CLIA-certified lab in Mason, Ohio to perform our GeneSight test; and a CLIA-certified laboratory facility in Austin, Texas to perform our pharmaceutical and clinical testing services. These facilities and certain pieces of laboratory equipment would be difficult to replace and may require significant replacement lead-time. In the event our clinical testing facilities were to lose their CLIA certification or other required certifications or licenses or were affected by a man-made or natural disaster, we would be unable to continue our molecular diagnostic and pharmaceutical and clinical services business at current levels to meet customer demands for a significant period of time. Although we maintain insurance on these facilities, including business interruption insurance, it may not be adequate to protect us from all potential losses if these facilities were damaged or destroyed. In addition, any interruption in our molecular diagnostic or pharmaceutical and clinical services business would result in a loss of goodwill, including damage to our reputation. If our molecular diagnostic or pharmaceutical and clinical services business were interrupted, it would seriously harm our business.
We depend on a limited number of third parties for some of our supplies of equipment and reagents. If these supplies become unavailable, then we may not be able to successfully perform our research or operate our business on a timely basis or at all.
We currently rely on a small number of suppliers to provide our gene sequencing equipment, content enrichment equipment, multiplex protein analysis equipment, robots, and specialty reagents and laboratory supplies required in connection with our testing and research. We believe that currently there are limited alternative suppliers of these equipment, robots, and reagents. The equipment, robots, or the reagents may not remain available in commercial quantities at acceptable costs. If we are unable to obtain when needed additional or alternative equipment, robots, or an adequate supply of reagents or other ingredients at commercially reasonable rates, our ability to continue to identify genes and perform molecular diagnostic testing and pharmaceutical and clinical services would be adversely affected.
24
Our molecular diagnostic and companion diagnostic tests in development may never achieve significant commercial market acceptance.
We may not succeed in achieving significant commercial market acceptance of our diagnostic test and clinical service offerings that we have launched in recent years or are currently developing. Our ability to successfully develop and commercialize our current molecular diagnostic and companion diagnostic tests, as well as any future molecular diagnostic and companion diagnostic tests that we may develop, will depend on several factors, including:
|
|
• |
our ability to convince the medical community of the clinical utility of our tests and their potential advantages over existing tests; |
|
|
• |
our ability to collaborate with biotechnology and pharmaceutical companies to develop and commercialize companion diagnostic tests for their therapeutic drugs and drug candidates; |
|
|
• |
the agreement by third-party payors to reimburse our tests, the scope and extent of which will affect patients’ willingness or ability to pay for our tests and will likely heavily influence physicians’ decisions to recommend our tests; and |
|
|
• |
the willingness of physicians to utilize our tests, which can be difficult to interpret. This difficulty is caused by the ability of our tests to predict only as to a probability, not certainty, that a tested individual will develop, have the disease, benefit from a particular therapy or has an aggressive form of the disease that the test is intended to predict. |
These factors present obstacles to commercial acceptance of our tests, which we would have to spend substantial time and money to overcome, if we can do so at all. Our inability to successfully do so would harm our business.
If we do not compete effectively with scientific and commercial competitors, we may not be able to successfully commercialize our tests.
The clinical laboratory and genetics testing fields are intense and highly competitive. Tests that are developed are characterized by rapid technological change. Our competitors in the United States and abroad are numerous and include, among others, major diagnostic companies, reference laboratories, molecular diagnostic firms, universities and other research institutions. Some of our potential competitors have considerably greater financial, technical, marketing and other resources than we do, which may allow these competitors to discover important genes and determine their function before we do. We could be adversely affected if we do not discover genes, proteins or biomarkers and characterize their function, develop molecular diagnostic and pharmaceutical and clinical services based on these discoveries, obtain required regulatory and other approvals and launch these tests and their related services before our competitors. We also expect to encounter significant competition with respect to any molecular diagnostic and companion diagnostic tests that we may develop or commercialize. Those companies that bring to market new molecular diagnostic and companion tests before we do may achieve a significant competitive advantage in marketing and commercializing their tests. We may not be able to develop additional molecular diagnostic tests successfully and we or our licensors may not obtain or enforce patents covering these tests that provide protection against our competitors. Moreover, our competitors may succeed in developing molecular diagnostic and companion diagnostic tests that circumvent our technologies or tests. Furthermore, our competitors may succeed in developing technologies or tests that are more effective or less costly than those developed by us or that would render our technologies or tests less competitive or obsolete. We expect competition to intensify in the fields in which we are involved as technical advances in these fields occur and become more widely known and changes in intellectual property laws generate challenges to our intellectual property position.
If our current research collaborators or scientific advisors terminate their relationships with us or develop relationships with a competitor, our ability to discover genes, proteins, and biomarkers, and to validate and commercialize molecular diagnostic and companion diagnostic tests could be adversely affected.
We have relationships with research collaborators at academic and other institutions who conduct research at our request. These research collaborators are not our employees. As a result, we have limited control over their activities and, except as otherwise required by our collaboration agreements, can expect only limited amounts of their time to be dedicated to our activities. Our ability to discover genes, proteins, and biomarkers involved in human disease and validate and commercialize molecular diagnostic and companion diagnostic tests will depend in part on the continuation of these collaborations. If any of these collaborations are terminated, we may not be able to enter into other acceptable collaborations. In addition, our existing collaborations may not be successful.
Our research collaborators and scientific advisors may have relationships with other commercial entities, some of which could compete with us. Our research collaborators and scientific advisors sign agreements which provide for the confidentiality of our
25
proprietary information and the results of studies conducted at our request. We may not, however, be able to maintain the confidentiality of our technology and other confidential information related to all collaborations. The dissemination of our confidential information could have a material adverse effect on our business.
If we fail to retain our key personnel and hire, train and retain qualified employees and consultants, we may not be able to successfully continue our business.
Because of the specialized scientific nature of our business, we are highly dependent upon our ability to attract and retain qualified management, scientific and technical personnel. We are currently recruiting additional qualified management, scientific and technical personnel. Competition for such personnel is intense. Loss of the services of or failure to recruit additional key management, scientific and technical personnel would adversely affect our research and development programs and molecular diagnostic and pharmaceutical and clinical services business and may have a material adverse effect on our business as a whole.
Our agreements with our employees generally provide for employment that can be terminated by either party without cause at any time, subject to specified notice requirements. Further, the non-competition provision to which each employee is subject expires for certain key employees on the applicable date of termination of employment.
As we expand our commercial tests we may be required to incur significant costs and devote significant efforts to expand our existing tests sales and marketing capabilities.
Our sales and marketing experience and capabilities consist primarily of our sales force that markets our molecular diagnostic tests to oncologists, obstetricians, gynecologists, urologists, dermatopathologists and rheumatologists in the United States. We are currently expanding our sales efforts outside the United States, which will require us to hire additional personnel and engage in additional sales and marketing efforts. We have limited sales and marketing experience outside the Unites States. As we expand our business operations internationally, we expect to face a number of additional costs and risks, including the need to recruit a large number of additional experienced marketing and sales personnel.
Risks Related to Our Intellectual Property
If we are not able to protect our proprietary technology, others could compete against us more directly, which would harm our business.
As of June 30, 2017, our patent portfolio included issued patents owned or licensed by us and numerous patent applications in the United States and other countries with claims protecting our intellectual property rights. Our commercial success will depend, in part, on our ability to obtain additional patents and licenses and protect our existing patent position, both in the United States and in other countries, for compositions, processes, methods and other inventions that we believe are patentable. Our ability to preserve our trade secrets, proprietary data bases and other intellectual property is also important to our long-term success. If our intellectual property is not adequately protected, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to maintain profitability. Patents may also issue to third parties which could interfere with our ability to bring our molecular diagnostic tests to market. The laws of some foreign countries do not protect our proprietary rights to the same extent as U.S. laws, and we may encounter significant problems in protecting our proprietary rights in these countries.
The patent positions of diagnostic companies, including our patent position, are generally highly uncertain and involve complex legal and factual questions, and, therefore, any patents issued to us may be challenged, deemed unenforceable, invalidated or circumvented. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies and any future tests are covered by valid and enforceable patents or are effectively maintained as trade secrets. Our patent applications may never issue as patents, and the claims of any issued patents may not afford meaningful protection for our technology or tests. In addition, any patents issued to us or our licensors may be challenged, and subsequently narrowed, invalidated or circumvented.
Where necessary, we may initiate litigation to enforce our patent or other intellectual property rights. Any such litigation may require us to spend a substantial amount of time and money and could distract management from our day-to-day operations. Moreover, there is no assurance that we will be successful in any such litigation.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
|
|
• |
we or our licensors were the first to make the inventions covered by each of our patent applications; |
26
|
|
• |
others will not independently develop similar or alternative technologies or duplicate any of our technologies; |
|
|
• |
any of our or our licensors’ patent applications will result in issued patents; |
|
|
• |
any of our or our licensors’ patents will be valid or enforceable; |
|
|
• |
any patents issued to us or our licensors and collaborators will provide a basis for commercially viable tests, will provide us with any competitive advantages or will not be challenged by third parties; |
|
|
• |
we will develop additional proprietary technologies or tests that are patentable; |
|
|
• |
the patents of others will not have an adverse effect on our business; or |
|
|
• |
our patents or patents that we license from others will survive legal challenges, and remain valid and enforceable. |
If a third party files a patent application with claims to subject matter we have invented, the PTO may declare interference between competing patent applications. If an interference is declared, we may not prevail in the interference. If the other party prevails in the interference, we may be precluded from commercializing services or tests based on the invention or may be required to seek a license. A license may not be available to us on commercially acceptable terms, if at all.
We also rely upon unpatented proprietary technologies and databases. Although we require employees, consultants and collaborators to sign confidentiality agreements, we may not be able to adequately protect our rights in such unpatented proprietary technologies and databases, which could have a material adverse effect on our business. For example, others may independently develop substantially equivalent proprietary information or techniques or otherwise gain access to our proprietary technologies or disclose our technologies to our competitors.
If we were sued for patent infringement by third parties, we might incur significant costs and delays in test introduction.
Our tests may also conflict with patents that have been or may be granted to others. Our industry includes many organizations that have or are seeking to discern biomarkers and develop genomic, proteomic and other technologies. To the extent any patents are issued or have been issued to those organizations, the risk increases that the sale of our molecular diagnostic and companion diagnostic tests currently being marketed or under development may give rise to claims of patent infringement. Others may have filed and in the future are likely to file patent applications covering biomarkers that are similar or identical to our tests. Any of these patent applications may have priority over our patent applications and these entities or persons could bring legal proceedings against us seeking damages or seeking to enjoin us from testing or marketing our tests. Patent litigation is costly, and even if we prevail, the cost of such litigation could have a material adverse effect on us. If the other parties in any such actions are successful, in addition to any liability for damages, we could be required to cease the infringing activity or obtain a license. Any license required may not be available to us on commercially acceptable terms, if at all. Our failure to obtain a license to any technology that we may require to commercialize our tests could have a material adverse effect on our business. We believe that there may be significant litigation in the industry regarding patent and other intellectual property rights. If we become involved in this litigation, it could consume a substantial portion of our managerial and financial resources.
We may be unable to adequately prevent disclosure of trade secrets, proprietary databases, and other proprietary information.
We rely on trade secrets to protect our proprietary technologies and databases, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and others to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy if unauthorized disclosure of confidential information occurs. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive position.
If we fail to comply with our obligations under license or technology agreements with third parties, we could lose license rights that are critical to our business.
We license intellectual property that is important to our business, including licenses underlying the technology in our molecular diagnostic and pharmaceutical and clinical services, and in the future we may enter into additional agreements that provide us
27
with licenses to valuable intellectual property or technology. These licenses impose various royalty payments, milestones, and other obligations on us. If we fail to comply with any of these obligations, the licensor may have the right to terminate the license. Termination by the licensor would cause us to lose valuable rights, and could prevent us from distributing our current tests, or inhibit our ability to commercialize future test candidates. Our business would suffer if any current or future licenses terminate, if the licensors fail to abide by the terms of the license, if the licensors fail to prevent infringement by third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms.
We may be subject to claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is commonplace in our industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Government Regulation
If we fail to comply with the complex federal, state, local and foreign laws and regulations that apply to our business, we could suffer severe consequences that could materially and adversely affect our operating results and financial condition.
Our operations are subject to extensive federal, state, local and foreign laws and regulations, all of which are subject to change. These laws and regulations currently include, among other things:
|
|
• |
CLIA, which requires that laboratories obtain certification from the federal government, and state licensure laws; |
|
|
• |
FDA laws and regulations; |
|
|
• |
HIPAA, which imposes comprehensive federal standards with respect to the privacy and security of protected health information and requirements for the use of certain standardized electronic transactions; amendments to HIPAA under HITECH, which strengthen and expand HIPAA privacy and security compliance requirements, increase penalties for violators, extend enforcement authority to state attorneys general and impose requirements for breach notification; |
|
|
• |
state laws regulating genetic testing and protecting the privacy of genetic test results, as well as state laws protecting the privacy and security of health information and personal data and mandating reporting of breaches to affected individuals and state regulators; |
|
|
• |
the federal anti-kickback law, or the Anti-Kickback Statute, which prohibits knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for, or recommending of an item or service that is reimbursable, in whole or in part, by a federal health care program; |
|
|
• |
the federal False Claims Act, which imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government; |
|
|
• |
the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies; |
|
|
• |
other federal and state fraud and abuse laws, such as anti-kickback laws, prohibitions on self-referral, and false claims acts, which may extend to services reimbursable by any third-party payor, including private insurers; |
|
|
• |
the federal Physician Payments Sunshine Act, which requires medical device manufactures to track and report to the federal government certain payments and other transfers of value made to physicians and teaching hospitals and ownership or investment interests held by physicians and their immediate family members; |
|
|
• |
Section 216 of the federal Protecting Access to Medicare Act of 2014 (“PAMA”), which requires applicable laboratories to report private payer data in a timely and accurate manner beginning in 2017 and every three years thereafter (and in some cases annually); |
28
|
|
• |
similar foreign laws and regulations that apply to us in the countries in which we operate. |
These laws and regulations are complex and are subject to interpretation by the courts and by government agencies. Our failure to comply could lead to civil or criminal penalties, exclusion from participation in state and federal health care programs, or prohibitions or restrictions on our laboratories’ ability to provide or receive payment for our services. We believe that we are in material compliance with all statutory and regulatory requirements, but there is a risk that one or more government agencies could take a contrary position, or that a private party could file suit under the qui tam provisions of the federal False Claims Act or a similar state law. Such occurrences, regardless of their outcome, could damage our reputation and adversely affect important business relationships with third parties, including managed care organizations, and other private third-party payors.
Failure to comply with government laws and regulations related to submission of claims for our services could result in significant monetary damages and penalties and exclusion from the Medicare and Medicaid programs and corresponding foreign reimbursement programs.
We are subject to laws and regulations governing the submission of claims for payment for our services, such as those relating to: coverage of our services under Medicare, Medicaid and other state, federal and foreign health care programs; the amounts that we may bill for our services; and the party to which we must submit claims. Our failure to comply with applicable laws and regulations could result in our inability to receive payment for our services or in attempts by state and federal healthcare programs, such as Medicare and Medicaid, to recover payments already made. Submission of claims in violation of these laws and regulations can result in recoupment of payments already received, substantial civil monetary penalties, and exclusion from state and federal health care programs, and can subject us to liability under the federal False Claims Act and similar laws. The failure to report and return an overpayment to the Medicare or Medicaid program within 60 days of identifying its existence can give rise to liability under the False Claims Act. Further, a government agency could attempt to hold us liable for causing the improper submission of claims by another entity for services that we performed if we were found to have knowingly participated in the arrangement at issue.
Our business could be harmed by the loss, suspension, or other restriction on a license, certification, or accreditation, or by the imposition of a fine or penalties, under CLIA, its implementing regulations, or other state, federal and foreign laws and regulations affecting licensure or certification, or by future changes in these laws or regulations.
The diagnostic testing industry is subject to extensive laws and regulations, many of which have not been interpreted by the courts. CLIA requires virtually all laboratories to be certified by the federal government and mandates compliance with various operational, personnel, facilities administration, quality and proficiency testing requirements intended to ensure that testing services are accurate, reliable and timely. CLIA certification is also a prerequisite to be eligible to bill state and federal health care programs, as well as many private third-party payors, for laboratory testing services. As a condition of CLIA certification, each of our laboratories is subject to survey and inspection every other year, in addition to being subject to additional random inspections. The biennial survey is conducted by CMS; a CMS agent (typically a state agency); or, if the laboratory holds a CLIA certificate of accreditation, a CMS-approved accreditation organization. Sanctions for failure to comply with CLIA requirements, including proficiency testing violations, may include suspension, revocation, or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as the imposition of significant fines or criminal penalties. In addition, we are subject to regulation under state laws and regulations governing laboratory licensure. Some states have enacted state licensure laws that are more stringent than CLIA. We are also subject to laws and regulations governing our reference laboratory in Germany. Changes in state or foreign licensure laws that affect our ability to offer and provide diagnostic services across state or foreign country lines could materially and adversely affect our business. In addition, state and foreign requirements for laboratory certification may be costly or difficult to meet and could affect our ability to receive specimens from certain states or foreign countries.
Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing licensure, or our failure to renew a CLIA certificate, a state or foreign license, or accreditation, could have a material adverse effect on our business. If the CLIA certificate of any one of our laboratories is revoked, CMS could seek revocation of the CLIA certificates of our other laboratories based on their common ownership or operation, even though they are separately certified.
29
Changes in the way that the FDA regulates tests performed by laboratories like ours could result in delay or additional expense in offering our tests and tests that we may develop in the future.
Historically, the FDA has exercised enforcement discretion with respect to most LDTs and has not required laboratories that furnish LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post-market controls). In recent years, however, the FDA publicly announced its intention to regulate certain LDTs and issued two draft guidance documents that set forth a proposed phased-in risk-based regulatory framework that would apply varying levels of FDA oversight to LDTs. However, these guidance documents were withdrawn at the end of the Obama administration and replaced by an informal discussion paper reflecting some of the feedback that FDA had received on LDT regulation. The FDA acknowledged that the discussion paper in January 2017 that the FDA stated does not represent the formal position of the FDA and is not enforceable. Nevertheless, the FDA wanted to share its synthesis of the feedback that it had received in the hope that it might advance public discussion on future LDT oversight. Notwithstanding the discussion paper, the FDA continues to exercise enforcement discretion and may decide to regulate certain LDTs on a case-by-case basis at any time, which could result in delay or additional expense in offering our tests and tests that we may develop in the future.
Companion and complementary diagnostic tests require FDA approval and we may not be able to secure such approval in a timely manner or at all.
Our companion and complementary diagnostic products, marketing, sales and development activities and manufacturing processes are subject to extensive and rigorous regulation by the FDA pursuant to the Federal Food, Drug, and Cosmetic Act (FDC Act), by comparable agencies in foreign countries, and by other regulatory agencies and governing bodies. Under the FDC Act, companion diagnostics must receive FDA clearance or approval before they can be commercially marketed in the U.S. The process of obtaining marketing approval or clearance from the FDA or by comparable agencies in foreign countries for new products could:
|
|
• |
take a significant period of time; |
|
|
• |
require the expenditure of substantial resources; |
|
|
• |
involve rigorous pre-clinical testing, as well as increased post-market surveillance; |
|
|
• |
require changes to products; and |
|
|
• |
result in limitations on the indicated uses of products. |
Although we obtained FDA approval for our BRACAnalysis CDx test, which is used as a companion diagnostic to identify ovarian cancer patients who may benefit from AstraZeneca’s PARP inhibitor Lynparza™ (olaparib) and as a complementary diagnostic in ovarian cancer patients associated with enhanced progression-free survival (PFS) from Tesaro’s PARP inhibitor Zejula™ (niraparib) maintenance therapy, we cannot predict whether or when we will be able to obtain FDA approval for other companion diagnostics that we are developing.
If the government and third-party payors fail to provide coverage and adequate payment for our tests and future tests, if any, our revenue and prospects for profitability will be harmed.
In both domestic and foreign markets, sales of our molecular diagnostic tests or any future diagnostic tests will depend in large part, upon the availability of reimbursement from third-party payors. Such third-party payors include state and federal health care programs such as Medicare, managed care providers, private health insurers and other organizations. These third-party payors are increasingly attempting to contain healthcare costs by demanding price discounts or rebates and limiting both coverage on which diagnostic tests they will pay for and the amounts that they will pay for new molecular diagnostic tests. We have recently experienced price reductions from CMS for some of our products and may experience future price reductions from managed care organizations and other third-party payors. The fact that a diagnostic test has been approved for reimbursement in the past, for any particular indication or in any particular jurisdiction, does not guarantee that such a diagnostic test will remain approved for reimbursement or that similar or additional diagnostic tests will be approved in the future. Moreover, there can be no assurance that any new tests we launch, such as myRisk Hereditary Cancer, myPath Melanoma and myPlan Lung Cancer, will be reimbursed at rates that are comparable to the rates that we historically obtained for our existing product portfolio. As a result, third-party payors may not cover or provide adequate payment for our current or future molecular diagnostic tests. Adequate third-party reimbursement might not be available to enable us to maintain price levels sufficient to realize an appropriate return on investment in product development. Further, beginning in 2018 under PAMA, Medicare reimbursement for any given diagnostic test will be based on the weighted-median of the payments made by private payors for such test, rendering private payor payment levels even more significant. As a result, future Medicare
30
payments may fluctuate more often and become subject to the willingness of private payors to recognize the value of diagnostic tests generally and any given test individually.
U.S. and foreign governments continue to propose and pass legislation designed to reduce the cost of health care. For example, in some foreign markets, the government controls the pricing of many health care products. We expect that there will continue to be federal and state proposals to implement governmental controls or impose health care requirements. In addition, the Medicare program and increasing emphasis on managed care in the United States will continue to put pressure on product pricing. Cost control initiatives could decrease the price that we would receive for any tests in the future, which would limit our revenue and profitability.
Our business could be adversely impacted by our failure or the failure of physicians to comply with the ICD-10-CM Code Set.
CMS adopted a new coding set for diagnoses, commonly known as ICD‑10-CM, which significantly expanded the previous coding set. Compliance with ICD-10-CM is required for all claims with dates of service on or after October 1, 2015. We believe we have fully implemented ICD-10-CM, however, our failure to implement and apply the new code set could adversely impact our business. In addition, if physicians fail to provide appropriate codes for desired tests, we may not be reimbursed for tests we perform.
Risks Related to Our Common Stock
Our stock price is highly volatile, and our stock may lose all or a significant part of its value.
The market prices for securities of molecular diagnostic companies have been volatile. This volatility has significantly affected the market prices for these securities for reasons frequently unrelated to the operating performance of the specific companies. These broad market fluctuations may adversely affect the market price of our common stock. The market price for our common stock has fluctuated significantly since public trading commenced in October 1995, and it is likely that the market price will continue to fluctuate in the future. In the two years ended June 30, 2017, our stock price has ranged from $15.15 per share to $46.24 per share. In addition, the stock market in general has experienced extreme price and volume fluctuations. Events or factors that may have a significant impact on our business and on the market price of our common stock include the following:
|
|
• |
failure of any of our recently launched tests and any new test candidates to achieve commercial success; |
|
|
• |
failure to sustain revenue growth or margins in our molecular diagnostic business; |
|
|
• |
changes in the structure of healthcare payment systems and changes in the governmental or private insurers reimbursement levels for our molecular diagnostic tests; |
|
|
• |
introduction of new commercial tests or technological innovations by competitors; |
|
|
• |
termination of the licenses underlying our molecular diagnostic and pharmaceutical and clinical services; |
|
|
• |
delays or other problems with operating our laboratory facilities; |
|
|
• |
failure of any of our research and development programs; |
|
|
• |
changes in intellectual property laws of our patents or enforcement in the United States and foreign countries; |
|
|
• |
developments or disputes concerning patents or other proprietary rights involving us directly or otherwise affecting the industry as a whole; |
|
|
• |
missing or changing the financial guidance we provide; |
|
|
• |
changes in estimates or recommendations by securities analysts relating to our common stock or the securities of our competitors; |
|
|
• |
changes in the governmental regulatory approved process for our existing and new tests: |
|
|
• |
failure to meet estimates or recommendations by securities analysts that cover our common stock; |
|
|
• |
public concern over our approved tests and any test candidates; |
|
|
• |
litigation; |
|
|
• |
future sales or anticipated sales of our common stock by us or our stockholders; |
31
|
|
• |
general market conditions; |
|
|
• |
seasonal slowness in sales, particularly in the quarters ending September 30 and March 31, the effects of which may be difficult to understand during periods of growth; |
|
|
• |
celebrity publicity; |
|
|
• |
economic, healthcare and diagnostic trends, disasters or crises and other external factors; and |
|
|
• |
period-to-period fluctuations in our financial results. |
These and other external factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, securities class action litigation against companies has been on the rise. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit regardless of the outcome. Such a lawsuit could also divert the time and attention of our management.
Anti-takeover provisions of Delaware law, provisions in our charter and bylaws and re-adoption of our stockholders’ rights plan, or poison pill, could make a third-party acquisition of us difficult.
Because we are a Delaware corporation, the anti-takeover provisions of Delaware law could make it more difficult for a third party to acquire control of us, even if the change in control would be beneficial to stockholders. We are subject to the provisions of Section 203 of the General Corporation Law of Delaware, which prohibits us from engaging in certain business combinations, unless the business combination is approved in a prescribed manner. In addition, our restated certificate of incorporation and restated bylaws also contain certain provisions that may make a third-party acquisition of us difficult, including:
|
|
• |
a classified board of directors, with three classes of directors each serving a staggered three-year term; |
|
|
• |
the ability of the board of directors to issue preferred stock; |
|
|
• |
a 70% super-majority shareholder vote to amend our bylaws and certain provisions of our certificate of incorporation; and |
|
|
• |
the inability of our stockholders to call a special meeting or act by written consent. |
In the past, we implemented a stockholders’ rights plan, also called a poison pill, which could make it uneconomical for a third party to acquire our company on a hostile basis. Although the plan expired in July 2011, our Board of Directors could adopt a new plan at any time. The provisions in a stockholders’ rights plan, as well as Section 203, may discourage certain types of transactions in which our stockholders might otherwise receive a premium for their shares over then current market price, and may limit the ability of our stockholders to approve transactions that they think may be in their best interests.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
Our corporate headquarters and facilities are located in Salt Lake City, Utah. We currently lease a total of 307,000 square feet of building space in Salt Lake City dedicated to research and development, administration and our laboratory that has received federal certification under CLIA. Activities related to our oncology, urology, dermatology and preventative care molecular diagnostic business are performed at this location. The leases on our existing Salt Lake City facilities have terms of fifteen years, expiring from 2022 through 2027, and provide for renewal options for up to ten additional years.
We also lease approximately 36,000 square feet in Austin, Texas under a lease that expires in June 2020. This space is dedicated to administration, research and development and the CLIA-certified laboratory. Activities related to our pharmaceutical and clinical services are performed at this location.
32
In addition, we lease approximately 54,000 square feet in South San Francisco, California under a lease that expires in February 2021. This space is dedicated to administration, research and development and the CLIA-certified laboratory for our Crescendo subsidiary. Activities related to our autoimmune molecular diagnostic business are performed at this location.
We also lease approximately 3,600 square feet in Munich, Germany under a lease expiring in March 2018. This space is used as a laboratory for our international molecular diagnostic businesses.
In December 2012, we entered into a lease expiring in November 2017 for approximately 5,000 square feet in Zurich, Switzerland. This space is used for the administration of our international operations. We also maintain lease agreements for our administrative offices in Paris, France; Milan, Italy; London, United Kingdom; Canada and Australia.
As part of our acquisition of Sividon in March 2016 we acquired a lease on an approximately 7,500 square foot facility with laboratory and office space in Cologne, Germany expiring in December 2017.
As part of our acquisition of the Clinic in February 2015, we acquired 20 buildings comprising 127,000 square feet in Herrsching, Germany. Activities related to our pharmaceutical and clinical services are performed at this location.
As part of our acquisition of Assurex in August 2016, we acquired leases of 2 spaces in Mason, OH and Toronto, ON Canada with a total square footage of approximately 11,250 expiring in March 2018 and August 2024.
We believe that our existing facilities and equipment are well maintained and in good working condition. We believe our current facilities and those planned will provide adequate capacity for at least the next two years. We continue to make investments in capital equipment as needed to meet the anticipated demand for our molecular diagnostic tests and our pharmaceutical and clinical services.
As previously reported in Part II, Item 1 of our Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2016, on September 7, 2016, Esoterix Genetic Laboratories, LLC (“EGL”) and The John Hopkins University (“JHU”) (collectively “Plaintiffs”) filed a complaint against Myriad Genetics, Inc. and Myriad Genetic Laboratories, Inc. (collectively “Myriad”) in the United States District Court for the Middle District of North Carolina, Greensboro Division (Civil Action No. 16-cv-1112), alleging that certain laboratory processes utilized by Myriad in conducting certain clinical diagnostic testing services infringe patent claims owned by JHU and exclusively licensed to EGL. The Plaintiffs are seeking a judgment of infringement, injunctive relief, compensatory damages, recovery of costs and legal fees, and other relief. On November 2, 2016, Myriad filed its Answer, Affirmative Defenses and Counterclaims to the Plaintiffs’ complaint wherein, amongst other things, Myriad requested declaratory rulings of non-infringement, invalidity and unenforceability of the asserted patent claims. On March 17, 2017, the Plaintiffs filed their answer to Myriad’s counterclaims. Myriad intends to vigorously defend the claims being asserted.
On March 16, 2017, Myriad filed four Petitions for Inter Partes Review with the United States Patent and Trademark Office before the Patent Trial and Appeal Board seeking the cancellation of patent claims being asserted against Myriad in the above litigation as unpatentable for anticipation and/or obviousness.
Other than as set forth above, we are not a party to any legal proceedings that we believe will have a material impact on our business, financial position or results of operations.
None.
33
|
Item 5. |
MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock is traded on The NASDAQ Global Select Market under the symbol "MYGN." The following table sets forth the high and low sales prices for our common stock, as reported by The NASDAQ Global Select Market for the last two fiscal years:
|
|
|
High |
|
|
Low |
|
||
|
Fiscal Year Ended June 30, 2017 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
26.99 |
|
|
$ |
17.50 |
|
|
Third Quarter |
|
$ |
19.90 |
|
|
$ |
15.15 |
|
|
Second Quarter |
|
$ |
21.53 |
|
|
$ |
15.86 |
|
|
First Quarter |
|
$ |
32.97 |
|
|
$ |
19.10 |
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal Year Ended June 30, 2016 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
39.74 |
|
|
$ |
28.82 |
|
|
Third Quarter |
|
$ |
43.68 |
|
|
$ |
33.87 |
|
|
Second Quarter |
|
$ |
46.24 |
|
|
$ |
36.47 |
|
|
First Quarter |
|
$ |
41.81 |
|
|
$ |
30.30 |
|
Stockholders
As of August 3, 2017, there were approximately 82 stockholders of record of our common stock and, according to our estimates, approximately 25,852 beneficial owners of our common stock.
Equity Compensation Plan Information
We incorporate information regarding the securities authorized for issuance under our equity compensation plans into this section by reference from the section entitled “Equity Compensation -- Equity Compensation Plan Information” to be included in the proxy statement for our 2017 Annual Meeting of Stockholders.
Unregistered Sales of Securities
None.
Issuer Purchases of Equity Securities
In June 2016, we announced that our board of directors had authorized us to repurchase an additional $200 million of our outstanding common stock increasing the cumulative share repurchase authorization since we first authorized the program in May 2010 to $1.4 billion. In connection with our most recent stock repurchase authorization, we have been authorized to complete the repurchase through open market transactions or through an accelerated share repurchase program, in each case to be executed at management’s discretion based on market conditions. As of the date of this report, we have not entered into an accelerated share repurchase agreement under our most recent stock repurchase program. The repurchase program may be suspended or discontinued at any time without prior notice. The transactions occurred in open market purchases and pursuant to a trading plan under Rule 10b5-1.
34
The details of the activity under our stock repurchase programs during the fiscal quarter ended June 30, 2017, were as follows:
Issuer Purchases of Equity Securities
(in millions, except per share data)
|
|
|
(a) |
|
|
(b) |
|
|
(c) |
|
|
(d) |
|
||||
|
|
|
|
|
|
|
|
|
|
|
Total Number of |
|
|
Approximate Dollar |
|
||
|
|
|
|
|
|
|
|
|
|
|
Shares Purchased as |
|
|
Value of Shares that |
|
||
|
|
|
Total Number |
|
|
Average Price |
|
|
Part of Publicly |
|
|
May Yet Be |
|
||||
|
|
|
of Shares |
|
|
Paid |
|
|
Announced Plans |
|
|
Purchased Under the |
|
||||
|
Period |
|
Purchased |
|
|
per Share |
|
|
or Programs |
|
|
Plans or Programs |
|
||||
|
April 1, 2017 to April 30, 2017 |
|
|
- |
|
|
$ |
- |
|
|
|
- |
|
|
|
160.7 |
|
|
May 1, 2017 to May 31, 2017 |
|
|
- |
|
|
$ |
- |
|
|
|
- |
|
|
|
160.7 |
|
|
June 1, 2017 to June 30, 2017 |
|
|
- |
|
|
$ |
- |
|
|
|
- |
|
|
|
160.7 |
|
|
Total |
|
|
- |
|
|
|
|
|
|
|
- |
|
|
$ |
160.7 |
|
Stock Performance Graph
The graph set forth below compares the annual percentage change in our cumulative total stockholder return on our common stock during a period commencing on June 29, 2012 and ending on June 30, 2017 (as measured by dividing (A) the difference between our share price at the end and the beginning of the measurement period; by (B) our share price at the beginning of the measurement period) with the cumulative total return of The NASDAQ Stock Market, Inc. and the NASDAQ Health Care Providers Stock Index during such period. We have not paid any cash dividends on our common stock, and we do not include cash dividends in the representation of our performance. The price of a share of common stock is based upon the closing price per share as quoted on The NASDAQ Global Select Market on the last trading day of the year shown. The graph lines merely connect year-end values and do not reflect fluctuations between those dates. The comparison assumes $100 was invested on June 29, 2012 in our common stock and in each of the foregoing indices. The comparisons shown in the graph below are based upon historical data. We caution that the stock price performance shown in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our common stock.
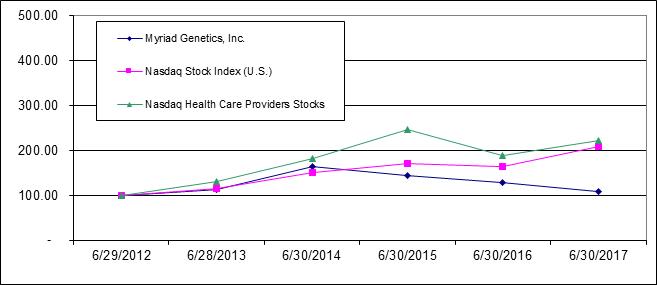
|
|
|
6/29/2012 |
|
|
6/28/2013 |
|
|
6/30/2014 |
|
|
6/30/2015 |
|
|
6/30/2016 |
|
|
6/30/2017 |
|
||||||
|
Myriad Genetics, Inc. |
|
|
100.00 |
|
|
|
113.04 |
|
|
|
163.74 |
|
|
|
143.00 |
|
|
|
128.73 |
|
|
|
108.71 |
|
|
NASDAQ Stock Index (U.S.) |
|
|
100.00 |
|
|
|
115.95 |
|
|
|
150.19 |
|
|
|
169.91 |
|
|
|
164.99 |
|
|
|
209.21 |
|
|
NASDAQ Health Care Providers Stocks |
|
|
100.00 |
|
|
|
131.58 |
|
|
|
181.64 |
|
|
|
247.05 |
|
|
|
187.69 |
|
|
|
221.90 |
|
35
Note: Information used on the graph was obtained from the CRSP Total Return Indexes, a source believed to be reliable, but we are not responsible for any errors or omission in such information.
The performance graph shall not be deemed to be incorporated by reference by means of any general statement incorporating by reference this Form 10-K into any filing under the Securities Act of 1933, as amended or the Securities Exchange Act of 1934, as amended, except to the extent that we specifically incorporate such information by reference, and shall not otherwise be deemed filed under such acts.
The following table sets forth our selected consolidated financial data and has been derived from our audited consolidated financial statements. Consolidated balance sheets as of June 30, 2017 and 2016, as well as consolidated statements of operations for the years ended June 30, 2017, 2016 and 2015 and the reports thereon are included elsewhere in this Annual Report on Form 10-K. The information below should be read in conjunction with our audited consolidated financial statements (and notes thereon) and "Management's Discussion and Analysis of Financial Condition and Results of Operations," included in Item 7.
|
In millions, except per share amounts |
|
Years Ended June 30, |
|
|||||||||||||||||
|
Consolidated Statement of Operations Data: |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Molecular diagnostic testing |
|
$ |
722.1 |
|
|
$ |
705.7 |
|
|
$ |
695.5 |
|
|
$ |
748.2 |
|
|
$ |
582.4 |
|
|
Pharmaceutical and clinical services |
|
|
49.3 |
|
|
|
48.1 |
|
|
|
27.6 |
|
|
|
30.0 |
|
|
|
30.8 |
|
|
Total Revenue |
|
|
771.4 |
|
|
|
753.8 |
|
|
|
723.1 |
|
|
|
778.2 |
|
|
|
613.2 |
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of molecular diagnostic testing |
|
|
145.2 |
|
|
|
132.8 |
|
|
|
132.8 |
|
|
|
96.1 |
|
|
|
64.4 |
|
|
Cost of pharmaceutical and clinical services |
|
|
26.0 |
|
|
|
24.5 |
|
|
|
14.6 |
|
|
|
13.1 |
|
|
|
15.3 |
|
|
Research and development expense |
|
|
74.4 |
|
|
|
70.6 |
|
|
|
75.5 |
|
|
|
67.5 |
|
|
|
53.7 |
|
|
Selling, general and administrative expense |
|
|
476.4 |
|
|
|
359.1 |
|
|
|
366.0 |
|
|
|
327.1 |
|
|
|
251.8 |
|
|
Total costs and expenses |
|
|
722.0 |
|
|
|
587.0 |
|
|
|
588.9 |
|
|
|
503.8 |
|
|
|
385.2 |
|
|
Operating income |
|
|
49.4 |
|
|
|
166.8 |
|
|
|
134.2 |
|
|
|
274.4 |
|
|
|
228.0 |
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
1.2 |
|
|
|
0.9 |
|
|
|
0.4 |
|
|
|
5.4 |
|
|
|
5.5 |
|
|
Interest expense |
|
|
(6.0 |
) |
|
|
(0.3 |
) |
|
|
(0.2 |
) |
|
|
- |
|
|
|
- |
|
|
Change in the fair value of contingent consideration |
|
|
0.8 |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Other |
|
|
(2.5 |
) |
|
|
1.5 |
|
|
|
0.5 |
|
|
|
(2.0 |
) |
|
|
(0.2 |
) |
|
Total other income (expense) |
|
|
(6.5 |
) |
|
|
2.1 |
|
|
|
0.7 |
|
|
|
3.4 |
|
|
|
5.3 |
|
|
Income before income taxes |
|
|
42.9 |
|
|
|
168.9 |
|
|
|
134.9 |
|
|
|
277.8 |
|
|
|
233.3 |
|
|
Income tax provision |
|
|
21.3 |
|
|
|
43.6 |
|
|
|
54.7 |
|
|
|
101.6 |
|
|
|
86.1 |
|
|
Net income |
|
|
21.6 |
|
|
|
125.3 |
|
|
|
80.2 |
|
|
|
176.2 |
|
|
|
147.2 |
|
|
Net loss attributable to non-controlling interest |
|
|
(0.2 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
21.8 |
|
|
$ |
125.3 |
|
|
$ |
80.2 |
|
|
$ |
176.2 |
|
|
$ |
147.2 |
|
|
Earnings per basic share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
0.32 |
|
|
$ |
1.79 |
|
|
$ |
1.12 |
|
|
$ |
2.33 |
|
|
$ |
1.82 |
|
|
Diluted |
|
$ |
0.32 |
|
|
$ |
1.71 |
|
|
$ |
1.08 |
|
|
$ |
2.25 |
|
|
$ |
1.77 |
|
|
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
68.3 |
|
|
|
70.0 |
|
|
|
71.3 |
|
|
|
75.7 |
|
|
|
80.9 |
|
|
Diluted |
|
|
68.8 |
|
|
|
73.4 |
|
|
|
74.5 |
|
|
|
78.2 |
|
|
|
83.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of June 30, |
|
|||||||||||||||||
|
Consolidated Balance Sheet Data: |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Cash, cash equivalents and marketable investment securities |
|
$ |
199.2 |
|
|
$ |
238.9 |
|
|
$ |
185.4 |
|
|
$ |
270.5 |
|
|
$ |
531.1 |
|
|
Working capital |
|
|
99.6 |
|
|
|
242.2 |
|
|
|
214.9 |
|
|
|
241.8 |
|
|
|
419.5 |
|
|
Total assets |
|
|
1,224.4 |
|
|
|
880.5 |
|
|
|
766.2 |
|
|
|
823.8 |
|
|
|
803.8 |
|
|
Stockholders’ equity |
|
|
777.8 |
|
|
|
748.1 |
|
|
|
662.1 |
|
|
|
719.0 |
|
|
|
728.6 |
|
36
Quarterly Financial Data (Unaudited)
|
In millions, except per share amounts |
|
Quarters Ended |
|
|||||||||||||
|
|
|
Jun 30, |
|
|
Mar 31, |
|
|
Dec 31, |
|
|
Sep 30, |
|
||||
|
Consolidated Statement of Operations Data: |
|
2017 |
|
|
2017 |
|
|
2016 |
|
|
2016 |
|
||||
|
Molecular diagnostic testing |
|
$ |
187.9 |
|
|
$ |
185.2 |
|
|
$ |
183.9 |
|
|
$ |
165.1 |
|
|
Pharmaceutical and clinical services |
|
|
12.6 |
|
|
|
11.7 |
|
|
|
12.6 |
|
|
|
12.4 |
|
|
Total Revenue |
|
|
200.5 |
|
|
|
196.9 |
|
|
|
196.5 |
|
|
|
177.5 |
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of molecular diagnostic testing |
|
|
35.6 |
|
|
|
37.9 |
|
|
|
37.4 |
|
|
|
34.3 |
|
|
Cost of pharmaceutical and clinical services |
|
|
6.9 |
|
|
|
6.4 |
|
|
|
7.0 |
|
|
|
5.7 |
|
|
Research and development expense |
|
|
18.8 |
|
|
|
17.6 |
|
|
|
18.6 |
|
|
|
19.4 |
|
|
Selling, general and administrative expense |
|
|
122.1 |
|
|
|
122.1 |
|
|
|
120.3 |
|
|
|
111.9 |
|
|
Total costs and expenses |
|
|
183.4 |
|
|
|
184.0 |
|
|
|
183.3 |
|
|
|
171.3 |
|
|
Operating income |
|
|
17.1 |
|
|
|
12.9 |
|
|
|
13.2 |
|
|
|
6.2 |
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
0.3 |
|
|
|
0.3 |
|
|
|
0.3 |
|
|
|
0.3 |
|
|
Interest expense |
|
|
(1.2 |
) |
|
|
(1.5 |
) |
|
|
(2.6 |
) |
|
|
(0.7 |
) |
|
Change in the fair value of contingent consideration |
|
|
2.7 |
|
|
|
(5.2 |
) |
|
|
3.8 |
|
|
|
(0.5 |
) |
|
Other |
|
|
(0.1 |
) |
|
|
1.5 |
|
|
|
(2.6 |
) |
|
|
(1.3 |
) |
|
Total other income (expense) |
|
|
1.7 |
|
|
|
(4.9 |
) |
|
|
(1.1 |
) |
|
|
(2.2 |
) |
|
Income before income taxes |
|
|
18.8 |
|
|
|
8.0 |
|
|
|
12.1 |
|
|
|
4.0 |
|
|
Income tax provision |
|
|
6.1 |
|
|
|
3.8 |
|
|
|
6.2 |
|
|
|
5.2 |
|
|
Net income |
|
|
12.7 |
|
|
|
4.2 |
|
|
|
5.9 |
|
|
|
(1.2 |
) |
|
Net loss attributable to non-controlling interest |
|
|
(0.2 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
12.9 |
|
|
$ |
4.2 |
|
|
$ |
5.9 |
|
|
$ |
(1.2 |
) |
|
Earnings per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
0.19 |
|
|
$ |
0.06 |
|
|
$ |
0.09 |
|
|
$ |
(0.02 |
) |
|
Diluted |
|
$ |
0.19 |
|
|
$ |
0.06 |
|
|
$ |
0.09 |
|
|
$ |
(0.02 |
) |
|
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
68.2 |
|
|
|
68.1 |
|
|
|
68.2 |
|
|
|
68.8 |
|
|
Diluted |
|
|
68.9 |
|
|
|
68.3 |
|
|
|
68.3 |
|
|
|
68.8 |
|
37
|
In millions, except per share amounts |
|
Quarters Ended |
|
|||||||||||||
|
|
|
Jun 30, |
|
|
Mar 31, |
|
|
Dec 31, |
|
|
Sep 30, |
|
||||
|
Consolidated Statement of Operations Data: |
|
2016 |
|
|
2016 |
|
|
2015 |
|
|
2015 |
|
||||
|
Molecular diagnostic testing |
|
$ |
173.8 |
|
|
$ |
177.4 |
|
|
$ |
182.6 |
|
|
$ |
171.9 |
|
|
Pharmaceutical and clinical services |
|
|
12.7 |
|
|
|
13.1 |
|
|
|
10.7 |
|
|
|
11.6 |
|
|
Total Revenue |
|
|
186.5 |
|
|
|
190.5 |
|
|
|
193.3 |
|
|
|
183.5 |
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of molecular diagnostic testing |
|
|
34.2 |
|
|
|
33.6 |
|
|
|
34.1 |
|
|
|
30.9 |
|
|
Cost of pharmaceutical and clinical services |
|
|
5.8 |
|
|
|
6.6 |
|
|
|
6.5 |
|
|
|
5.6 |
|
|
Research and development expense |
|
|
19.5 |
|
|
|
17.2 |
|
|
|
16.7 |
|
|
|
17.2 |
|
|
Selling, general and administrative expense |
|
|
91.3 |
|
|
|
90.5 |
|
|
|
90.8 |
|
|
|
86.5 |
|
|
Total costs and expenses |
|
|
150.8 |
|
|
|
147.9 |
|
|
|
148.1 |
|
|
|
140.2 |
|
|
Operating income |
|
|
35.7 |
|
|
|
42.6 |
|
|
|
45.2 |
|
|
|
43.3 |
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
0.4 |
|
|
|
0.3 |
|
|
|
0.1 |
|
|
|
0.1 |
|
|
Interest Expense |
|
|
(0.1 |
) |
|
|
(1.5 |
) |
|
|
(2.6 |
) |
|
|
(0.7 |
) |
|
Change in contingent consideration |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Other |
|
|
1.3 |
|
|
|
1.7 |
|
|
|
2.3 |
|
|
|
0.8 |
|
|
Total other income (expense) |
|
|
1.6 |
|
|
|
0.5 |
|
|
|
(0.2 |
) |
|
|
0.2 |
|
|
Income before income taxes |
|
|
37.3 |
|
|
|
43.1 |
|
|
|
45.0 |
|
|
|
43.5 |
|
|
Income tax provision |
|
|
13.9 |
|
|
|
8.6 |
|
|
|
7.9 |
|
|
|
13.2 |
|
|
Net income |
|
$ |
23.4 |
|
|
$ |
34.5 |
|
|
$ |
37.1 |
|
|
$ |
30.3 |
|
|
Earnings per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
0.33 |
|
|
$ |
0.49 |
|
|
$ |
0.53 |
|
|
$ |
0.44 |
|
|
Diluted |
|
$ |
0.32 |
|
|
$ |
0.47 |
|
|
$ |
0.50 |
|
|
$ |
0.42 |
|
|
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
70.0 |
|
|
|
70.9 |
|
|
|
70.5 |
|
|
|
68.7 |
|
|
Diluted |
|
|
72.4 |
|
|
|
73.8 |
|
|
|
74.6 |
|
|
|
72.6 |
|
38
Overview
Our consolidated revenues consist primarily of sales of molecular diagnostic tests and pharmaceutical and clinical services through our wholly-owned subsidiaries. During the year ended June 30, 2017, we reported total revenues of $771.4 million, net income of $21.8 million and diluted earnings per share of $0.32 that included income tax expense of $21.3 million.
In August 2016, the Company completed the acquisition of Assurex Health, Inc. (“Assurex) for total consideration of $351.6 million, net of cash acquired of $5.5 million, including a cash payment of $216.1 million, and two potential performance-based milestones totaling $185.0 million. We believe the acquisition establishes the foundation for our neuroscience business and leverages our existing preventative care business unit with the addition of a product, GeneSight, which has significant growth potential.
See Note 17 “Segment and Related Information” in the notes to our consolidated financial statements for information regarding our operating segments.
Our research and development expenses include costs incurred in formulating, improving, validating and creating alternative or modified processes related to and expanding the use of our current molecular diagnostic test offerings and costs incurred for the discovery, development and validation of our pipeline of molecular diagnostic and companion diagnostic candidates. In general, costs associated with research and development can fluctuate dramatically due to the timing of clinical studies, the staging of products in the pipeline and other factors.
Our selling, general and administrative expenses include costs associated with growing our businesses domestically and internationally. Selling, general and administrative expenses consist primarily of salaries, commissions and related personnel costs for sales, marketing, customer service, billing and collection, legal, finance and accounting, information technology, human resources, and allocated facilities expenses. We expect that our selling, general and administrative expenses may continue to increase and that such increases may be substantial, depending on the number and scope of any new molecular diagnostic test launches, our efforts in support of our existing molecular diagnostic tests and pharmaceutical and clinical services as well as our continued international expansion efforts.
Business Highlights
On August 31, 2016, we completed the acquisition of Assurex Health Inc. (“Assurex”). We believe the acquisition establishes the foundation for our neuroscience business and leverages our existing preventative care business unit with the addition of a neuropsychiatric pharmacogenomic test product, GeneSight, which has growth potential.
During the year the Medicare administrative contractor (MAC) in charge of the Mol Dx program published a draft non-coverage decision for Vectra DA based on data from the recently published AMPLE study. Myriad strongly disagrees with the conclusions of this study as published, which has significant limitations and shortcomings. The public comment period on the draft non-coverage decision has now closed. There was a significant support from patients and physicians for continued coverage of VectraDA. Feedback on the non-coverage decision is now under review by the MAC and we are awaiting a final decision.
Results of Operations
Years Ended June 30, 2017, 2016 and 2015
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
|
2017 |
|
|
|
2016 |
|
|
Revenue |
|
$ |
771.4 |
|
|
$ |
753.8 |
|
|
$ |
723.1 |
|
|
$ |
17.6 |
|
|
$ |
30.7 |
|
In 2017, the increase in revenue was primarily driven by an increase of $78.4 million in Genesight revenue from the acquisition of Assurex, and a $3.1 million increase in EndoPredict revenue from increased volumes. This increase was partially offset by a decrease of $63.6 million primarily due to reduced volumes and reimbursement for our hereditary cancer tests.
39
In 2016, the increase in revenue was primarily driven by an increase of $20.5 million in pharmaceutical and clinical services revenue mainly from the inclusion of $14.3 million in full-year revenue from the Clinic, which we acquired in February 2015 and a $9.2 million increase in Prolaris revenues from increased volumes and the initiation of Medicare coverage for a portion of the Medicare population.
The following table presents additional detail regarding the composition of our total revenue:
|
|
|
Years Ended June 30, |
|
|
Change |
|
|
% of Total Revenue |
|
|||||||||||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
||||||||
|
Molecular diagnostic revenues: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hereditary Cancer Testing |
|
$ |
568.7 |
|
|
$ |
632.3 |
|
|
$ |
638.3 |
|
|
$ |
(63.6 |
) |
|
$ |
(6.0 |
) |
|
|
74 |
% |
|
|
84 |
% |
|
|
88 |
% |
|
VectraDA |
|
|
43.7 |
|
|
|
47.8 |
|
|
|
43.7 |
|
|
|
(4.1 |
) |
|
|
4.1 |
|
|
|
5 |
% |
|
|
6 |
% |
|
|
6 |
% |
|
Prolaris |
|
|
12.1 |
|
|
|
11.3 |
|
|
|
2.1 |
|
|
|
0.8 |
|
|
|
9.2 |
|
|
|
2 |
% |
|
|
2 |
% |
|
|
1 |
% |
|
GeneSight |
|
|
78.4 |
|
|
|
- |
|
|
|
- |
|
|
|
78.4 |
|
|
|
- |
|
|
|
10 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
EndoPredict |
|
|
7.6 |
|
|
|
4.5 |
|
|
|
2.2 |
|
|
|
3.1 |
|
|
|
2.3 |
|
|
|
1 |
% |
|
|
1 |
% |
|
|
0 |
% |
|
Other |
|
|
11.6 |
|
|
|
9.8 |
|
|
|
9.2 |
|
|
|
1.8 |
|
|
|
0.6 |
|
|
|
2 |
% |
|
|
1 |
% |
|
|
1 |
% |
|
Total molecular diagnostic revenue |
|
|
722.1 |
|
|
|
705.7 |
|
|
|
695.5 |
|
|
|
16.4 |
|
|
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pharmaceutical and clinical service revenue |
|
|
49.3 |
|
|
|
48.1 |
|
|
|
27.6 |
|
|
|
1.2 |
|
|
|
20.5 |
|
|
|
6 |
% |
|
|
6 |
% |
|
|
4 |
% |
|
Total revenue |
|
$ |
771.4 |
|
|
$ |
753.8 |
|
|
$ |
723.1 |
|
|
$ |
17.6 |
|
|
$ |
30.7 |
|
|
|
100 |
% |
|
|
100 |
% |
|
|
100 |
% |
Cost of Sales
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
Cost of Sales |
|
$ |
171.2 |
|
|
$ |
157.3 |
|
|
$ |
147.4 |
|
|
$ |
13.9 |
|
|
$ |
9.9 |
|
|
Cost of sales as a % of Sales |
|
|
22.2 |
% |
|
|
20.9 |
% |
|
|
20.4 |
% |
|
|
|
|
|
|
|
|
Cost of sales as a percentage of revenues increased from 20.9% to 22.2% during fiscal 2017 compared to fiscal 2016. The increase was primarily driven by a change in existing product mix, lower fixed-cost absorption from lower hereditary cancer revenues and reduced hereditary cancer reimbursement.
Cost of sales as a percentage of revenues increased from 20.4% to 20.9% during fiscal 2016 compared to fiscal 2015. The increase was primarily driven by our acquisition of the Clinic in February 2015, which has a higher cost of sales than our molecular diagnostic testing business, and a change in existing product mix. This increase was partially offset by improved efficiencies in the laboratory performing molecular diagnostic tests. We expect to continue to see improved efficiencies in our laboratories.
Research and Development Expenses
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
R&D expense |
|
$ |
74.4 |
|
|
$ |
70.6 |
|
|
$ |
75.5 |
|
|
$ |
3.8 |
|
|
$ |
(4.9 |
) |
|
R&D expense as a % of Sales |
|
|
9.6 |
% |
|
|
9.4 |
% |
|
|
10.4 |
% |
|
|
|
|
|
|
|
|
In 2017, R&D expense increased compared to the same period in the prior year primarily due to the inclusion of Assurex spend of $8.5 million. This increase was partially offset by reductions of $2.0 million in spend related to internal development of new products, $1.3 million in share-based compensation expense and $0.9 million in development of pharmaceutical CDx development.
In 2016, R&D expense decreased compared to the same period in the prior year driven by a reduction in the cost of formulating, improving, validating and creating alternative or modified processes relating to the myRisk production process and the timing of clinical studies. These decreases were partially offset by increase in costs related to the development of new products.
40
Selling, General and Administrative Expenses
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
SG&A expense |
|
$ |
476.4 |
|
|
$ |
359.1 |
|
|
$ |
366.0 |
|
|
$ |
117.3 |
|
|
$ |
(6.9 |
) |
|
SG&A expense as a % of Sales |
|
|
61.8 |
% |
|
|
47.6 |
% |
|
|
50.6 |
% |
|
|
|
|
|
|
|
|
In 2017, the increase in SG&A expense compared to the prior year is primarily due to the inclusion of Assurex spend of $62.2 million, and $34.6 million in costs related to amortization and acquisition/integration activities for Assurex and Sividon. There were also increases of $9.5 million increase in other various administrative costs, $4.6 million increase in sales and marketing efforts, $4.3 million increase related to sales force compensation and $4.5 million in bad debt expense related to reduced collections. These increases were partially offset by reduction of $1.9 million in share-based compensation and $1.1 million reduction in international administrative costs.
In 2016, the decrease in SG&A expense was primarily driven by a $15.0 million reduction in share based compensation expense, as well as a $5.1 million reduction in administrative costs related to improved efficiencies. The decrease in share based compensation was due to reduction of $15.9 million related to the acceleration of vesting of certain options for executive transitions in the prior year offset by a $0.9 million increase in share based compensation excluding the executive transitions. These decreases were partially offset by an increase of $6.7 million in commissions and employee benefits, $4.4 million increase in costs relating to the Clinic acquisition and $1.1 million increase in sales and marketing efforts.
Other Income
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
Other Income (expense) |
|
$ |
(6.5 |
) |
|
$ |
2.1 |
|
|
$ |
0.7 |
|
|
$ |
(8.6 |
) |
|
$ |
1.4 |
|
In 2017, the decrease in other income (expense) was primarily driven by $5.7 million in increased interest expense, $1.9 million increase in Sividon’s contingent consideration, $1.3 million loss on extinguishment of debt, $2.4 million impairment of our investment in RainDance, and a $0.9 million indirect tax expense. These were partially offset by a decrease in Assurex’s contingent consideration of $2.7 million.
In 2016, the increase in other income was primarily driven by a $2.6 million increase in foreign exchange gains and the disposition of assets and a $0.4 million increase in interest income from marketable investment securities. This increase was partially offset by a $1.7 million increase in other taxes and interest expense.
Income Tax Expense
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
Income tax expense |
|
$ |
21.3 |
|
|
$ |
43.6 |
|
|
$ |
54.7 |
|
|
$ |
(22.3 |
) |
|
$ |
(11.1 |
) |
|
Effective tax rate |
|
|
49.7 |
% |
|
|
25.8 |
% |
|
|
40.6 |
% |
|
|
|
|
|
|
|
|
Our tax rate is a product of a U.S. federal effective rate of 35% and a blended state income tax rate of 2.6%. Certain significant or unusual items are separately recognized during the period in which they occur and can be a source of variability in the effective tax rates from period to period. The increase in the effective rate for the year ended June 30, 2017 as compared to the years ended June 30, 2016 and 2015 is due to an increase in our liability for unrecognized tax benefits, acquisition related transaction costs, an increase in the valuation allowance attributable to state research credits, foreign losses for which benefit was not recognized and the prior year adoption of ASU 2016-09 (see Note 10 “Income Taxes” in the Notes to Consolidated Financial Statements). The fiscal 2015 foreign losses, and the resulting impact on the effective rate, were larger in proportion to consolidated income as compared to later years.
Liquidity and Capital Resources
We believe that our existing capital resources and the cash to be generated from future sales will be sufficient to meet our projected operating requirements, including repayment of the outstanding Facility which matures on December 23, 2021, for
41
the foreseeable future. There are no scheduled principal payments of the Facility prior to its maturity date; however, our available capital resources may be consumed more rapidly than currently expected and we may need or want to raise additional financing. We may not be able to secure such financing in a timely manner or on favorable terms, if at all. Without additional funds, we may be forced to delay, scale back or eliminate some of our sales and marketing efforts, research and development activities, or other operations and potentially delay development of our diagnostic tests in an effort to provide sufficient funds to continue our operations. If any of these events occur, our ability to achieve our development and commercialization goals would be adversely affected.
Our capital deployment strategy focuses on use of resources in four key areas: research and development, acquisitions, debt repayment and the repurchase of our common stock. We believe that research and development provides the best return on invested capital. We also allocate capital for acquisitions that support our business strategy and share repurchases based on business and market conditions.
The following table represents the balances of cash, cash equivalents and marketable investment securities:
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
Cash and cash equivalents |
|
$ |
102.4 |
|
|
$ |
68.5 |
|
|
$ |
64.1 |
|
|
$ |
33.9 |
|
|
$ |
4.4 |
|
|
Marketable investment securities |
|
|
48.3 |
|
|
|
90.5 |
|
|
|
80.7 |
|
|
|
(42.2 |
) |
|
|
9.8 |
|
|
Long-term marketable investment securities |
|
|
48.5 |
|
|
|
79.9 |
|
|
|
40.6 |
|
|
|
(31.4 |
) |
|
|
39.3 |
|
|
Cash, cash equivalents and marketable investment securities |
|
$ |
199.2 |
|
|
$ |
238.9 |
|
|
$ |
185.4 |
|
|
$ |
(39.7 |
) |
|
$ |
53.5 |
|
In 2017, the decrease in cash, cash equivalents and marketable investment securities was primarily driven by the $216.1 million used for the acquisition of Assurex, the $105.0 million used to pay down the principal on our long-term debt facility, the $31.6 million used on share repurchase and $5.9 million used to purchase capital equipment. These decreases were partially offset by $203.0 million in net cash received from the issuance of debt, $106.0 million in cash flows from operations and $73.3 million in net proceeds from the maturities and sales of marketable investment securities.
In 2016, the increase in cash, cash equivalents and marketable investment securities was primarily driven by $166.3 million in cash flows from operations and $94.3 million in net proceeds from common stock issued under share-based compensation plans. These increases were partially offset by $162.6 million used for the repurchase and retirement of common stock and $49.4 million in net purchases of marketable investment securities.
The following table represents the condensed cash flow statement:
|
|
|
Years Ended June 30, |
|
|
Change |
|
||||||||||||||
|
(In millions) |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 |
|
|
2016 |
|
|||||
|
Cash flows from operating activities |
|
$ |
106.2 |
|
|
$ |
166.3 |
|
|
$ |
140.5 |
|
|
$ |
(60.1 |
) |
|
$ |
25.8 |
|
|
Cash flows from investing activities |
|
|
(146.3 |
) |
|
|
(91.4 |
) |
|
|
40.9 |
|
|
|
(54.9 |
) |
|
|
(132.3 |
) |
|
Cash flows from financing activities |
|
|
71.8 |
|
|
|
(68.3 |
) |
|
|
(177.3 |
) |
|
|
140.1 |
|
|
|
109.0 |
|
|
Effect of foreign exchange rates on cash and cash equivalents |
|
|
2.2 |
|
|
|
(2.2 |
) |
|
|
(4.8 |
) |
|
|
4.4 |
|
|
|
2.6 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
33.9 |
|
|
|
4.4 |
|
|
|
(0.7 |
) |
|
|
29.5 |
|
|
|
5.1 |
|
|
Cash and cash equivalents at the beginning of the year |
|
|
68.5 |
|
|
|
64.1 |
|
|
|
64.8 |
|
|
|
4.4 |
|
|
|
(0.7 |
) |
|
Cash and cash equivalents at the end of the year |
|
$ |
102.4 |
|
|
$ |
68.5 |
|
|
$ |
64.1 |
|
|
$ |
33.9 |
|
|
$ |
4.4 |
|
Cash Flows from Operating Activities
In 2017, the primary driver of the decrease in cash flows from operating activities was the $103.3 million decrease in net income partially offset by a $28.6 million increase due to changes in assets and liabilities associated with operating activities and $14.4 million increase in non-cash charges.
In 2016, the primary driver of the increase of cash flows from operating activities was the $45.1 million increase in net income partially offset by a $24.7 million decrease due to changes in assets and liabilities associated with operating activities. The use of cash was primarily driven by an increase in inventory related to the Company’s unconditional purchase obligation as well as growth in trade accounts receivable driven by increased revenues.
42
Cash Flows from Investing Activities
In 2017, the decrease in cash flows from investing activities was primarily related to the $179.1 million increase in cash outlays related to acquisition activity partially offset by a $122.7 million increase in the net proceeds from the liquidation of marketable investment securities.
In 2016, the decrease in cash flows from investing activities was primarily related to the $134.3 million reduction in net proceeds from the liquidation of marketable investment securities and an increase of $16.9 million in cash outlays related to acquisition activity partially offset by an $18.9 million decrease in capital expenditures.
Cash Flows from Financing Activities
In 2017, the increase in cash flows from financing activities was driven primarily by $203.0 million in net cash received from the issuance of debt and $131.0 million reduction in cash spent on share repurchase. These were partially offset by $105.0 million used to pay down the principal on our long-term debt facility and $88.3 million reduction in cash proceeds from common stock issued under share-based compensation plans.
In 2016, the increase in cash flows from financing activities was driven primarily by $64.3 million increase in net proceeds from common stock issued under share-based compensation plans and $48.1 million reduction in repurchases and retirement of common stock from the prior year.
Contractual Obligations
The following table represents our contractual obligations as of June 30, 2017:
|
|
|
|
|
|
|
Less Than |
|
|
1-3 |
|
|
4-5 |
|
|
More Than |
|
||||
|
(In millions) |
|
Total |
|
|
1 Year |
|
|
Years |
|
|
Years |
|
|
5 Years |
|
|||||
|
Purchase obligations |
|
$ |
11.2 |
|
|
$ |
9.0 |
|
|
$ |
1.5 |
|
|
$ |
0.7 |
|
|
$ |
— |
|
|
Operating Leases |
|
|
89.1 |
|
|
|
12.8 |
|
|
|
24.6 |
|
|
|
19.3 |
|
|
|
32.4 |
|
|
Total |
|
|
100.3 |
|
|
|
21.8 |
|
|
|
26.1 |
|
|
|
20.0 |
|
|
|
32.4 |
|
The expected timing of payment for the obligations listed above is estimated based on current information. Actual payment timing and amounts may differ depending on the timing of goods or services received or other changes. The table above only includes payment obligations that are fixed or determinable. The table excludes royalties to third parties based on future sales of any of our product candidates that are approved for sale, as the amounts, timing, and likelihood of any such payments are based on the level of future sales of tests and are unknown.
Effects of Inflation
We do not believe that inflation has had a material impact on our business, revenues, or operating results during the periods presented.
Off-Balance Sheet Arrangements
None.
Certain Factors That May Affect Future Results of Operations
The Securities and Exchange Commission encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. This Annual Report on Form 10-K contains such “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995.
Words such as “may,” “anticipate,” “estimate,” “expects,” “projects,” “intends,” “plans,” “believes,” “seek,” “could,” continue,” “likely,” “will,” “strategy, “goal” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, identify forward-looking statements. All forward-looking statements are management’s present expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those described in the forward-looking statements. These risks include, but
43
are not limited to: the risk that sales and profit margins of our existing molecular diagnostic tests and pharmaceutical and clinical services may decline or will not continue to increase at historical rates; risks related to our ability to successfully transition from our existing product portfolio to our new tests; risks related to changes in governmental or private insurers reimbursement levels for our tests or our ability to obtain reimbursement for our new tests at comparable levels to our existing tests; risks related to increased competition and the development of new competing tests and services; the risk that we may be unable to develop or achieve commercial success for additional molecular diagnostic tests and pharmaceutical and clinical services in a timely manner, or at all; the risk that we may not successfully develop new markets for our molecular diagnostic tests and pharmaceutical and clinical services, including our ability to successfully generate revenue outside the United States; the risk that licenses to the technology underlying our molecular diagnostic tests and pharmaceutical and clinical services tests and any future tests are terminated or cannot be maintained on satisfactory terms; risks related to delays or other problems with operating our laboratory testing facilities; risks related to public concern over genetic testing in general or our tests in particular; risks related to regulatory requirements or enforcement in the United States and foreign countries and changes in the structure of the healthcare system or healthcare payment systems; risks related to our ability to obtain new corporate collaborations or licenses and acquire new technologies or businesses on satisfactory terms, if at all; risks related to our ability to successfully integrate and derive benefits from any technologies or businesses that we license or acquire; risks related to our projections about the potential market opportunity for our products; the risk that we or our licensors may be unable to protect or that third parties will infringe the proprietary technologies underlying our tests; the risk of patent-infringement claims or challenges to the validity of our patents; risks related to changes in intellectual property laws covering our molecular diagnostic tests and pharmaceutical and clinical services and patents or enforcement in the United States and foreign countries, such as the Supreme Court decision in the lawsuit brought against us by the Association for Molecular Pathology et al; risks of new, changing and competitive technologies and regulations in the United States and internationally; the risk that we may be unable to comply with financial operating covenants under our credit or lending agreements: the risk that we will be unable to pay, when due, amounts due under our credit or lending agreements: and other factors discussed under the heading “Risk Factors” contained in Item 1A of this Annual Report.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this Annual Report or in any document incorporated by reference might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this Annual Report. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to us or to any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
Market, Industry and Other Data
This Annual Report on Form 10-K contains estimates, projections and other information concerning our industry, our business and relevant molecular diagnostics markets, including data regarding the estimated size of relevant molecular diagnostic markets, patient populations, and the perceptions and preferences of patients and physicians regarding certain therapies, as well as data regarding market research and estimates. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances that are assumed in this information. Unless otherwise expressly stated, we obtained this industry, business, market and other data from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources that we believe to be reliable. In some cases, we do not expressly refer to the sources from which this data is derived. In that regard, when we refer to one or more sources of this type of data in any paragraph, you should assume that other data of this type appearing in the same paragraph is derived from the same sources, unless otherwise expressly stated or the context otherwise requires.
Critical Accounting Policies
Critical accounting policies are those policies which are both important to the portrayal of a company’s financial condition and results and require management’s most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Our critical accounting policies are as follows:
|
|
• |
revenue recognition; |
|
|
• |
allowance for doubtful accounts; |
|
|
• |
goodwill; and |
|
|
• |
income taxes. |
44
Revenue Recognition. Revenue includes the sale of our molecular diagnostic tests and of our pharmaceutical and clinical services. Revenue is recorded at the invoiced amount net of any discounts or allowances and is recognized when persuasive evidence of an agreement exists, delivery has occurred, the fee is fixed or determinable, and collection is reasonably assured. Revenue is recognized upon completion of the test or service, communication of results, and when collectability is reasonably assured.
Allowance for Doubtful Accounts. The preparation of our financial statements in accordance with U.S. GAAP requires us to make estimates and assumptions that affect the reported amount of assets at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Trade accounts receivable are comprised of amounts due from sales of our molecular diagnostic tests, which are recorded net of any discounts or contractual allowances. We analyze trade accounts receivable and consider historic experience, customer creditworthiness, facts and circumstances specific to outstanding balances, and payment terms when evaluating the adequacy of the allowance for doubtful accounts.
We periodically evaluate and adjust the allowance for doubtful accounts when trends or significant events indicate that a change in estimate is appropriate. Such changes in estimate could materially affect our results of operations or financial position; however, to date these changes have not been material. It is possible that we may need to adjust our estimates in future periods.
After a review of our allowance for doubtful accounts as of June 30, 2017 and 2016, we have determined that a hypothetical ten percent increase in our allowance for doubtful accounts would result in additional bad debt expense and an increase to our allowance for doubtful accounts of $0.8 million and $0.7 million, respectively.
Goodwill. We test goodwill for impairment on an annual basis and in the interim by reporting unit if events and circumstances indicate that goodwill may be impaired. The events and circumstances that are considered include business climate and market conditions, legal factors, operating performance indicators and competition. Impairment of goodwill is evaluated on a qualitative basis to determine if using a two-step process is necessary. If the qualitative assessment suggests that impairment is more likely than not, a two-step impairment analysis is performed. The first step involves comparison of the fair value of a reporting unit with its carrying amount. The valuation of a reporting unit requires judgment in estimating future cash flows, discount rates and other factors. In making these judgments, we evaluate the financial health of our business, including such factors as industry performance, market saturation and opportunity, changes in technology and operating cash flows. Changes in our forecasts or decreases in the value of our common stock could cause book value of reporting units to exceed their fair values. If the carrying amount of a reporting unit exceeds its fair value, the second step of the process involves a comparison of the fair value and the carrying amount of the goodwill of that reporting unit. If the carrying amount of the goodwill of the reporting unit exceeds the fair value of that goodwill, an impairment loss would be recognized in an amount equal to the excess of carrying value over fair value. If an event occurs that would cause a revision to the estimates and assumptions used in analyzing the value of the goodwill, the revision could result in a non-cash impairment charge that could have a material impact on the financial results.
We have recorded goodwill of $316.1 million from the acquisitions of Assurex that was completed on August 31, 2016, Sividon that was completed on May 31, 2016, the Clinic that was completed on February 27, 2015, Crescendo that was completed on February 28, 2014 and Myriad RBM that was completed on May 31, 2011. Of this goodwill, $250.5 million related to our molecular diagnostic segment for Crescendo, Sividon and Assurex and $65.6 million for Myriad RBM and the Clinic related to our other segment (see note 15 for segment descriptions). We qualitatively evaluated the Assurex, Sividon and the Clinic reporting units for impairment noting no indicators of impairment from the date of acquisition. We measured the fair value of Myriad RBM utilizing the income approach using the discounted cash flow method. This considered management’s plans and projections as the basis for expected cash flows for the next fifteen years using a 4% long term growth rate. We also used a discount rate of 30%. We noted the fair value of the Myriad RBM reporting unit exceeded its carrying value by 88%. We measured the fair value of Crescendo utilizing income and market approaches. The income approach considered management’s business plans and projections as the basis for expected cash flows for the next fifteen years and a 2.0% residual growth rate. We also used a weighted average discount rate of 16%. Other significant estimates used in the analysis include the profitability of the respective reporting unit and working capital effects of each unit. We noted the fair value of the Crescendo reporting unit exceeded its carrying value by 47% using these assumptions described above.
Income Taxes. Our income tax provision is based on income before taxes and is computed using the liability method in accordance with Accounting Standards Codification (“ASC”) 740 – Income Taxes. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using tax rates projected to be in effect for the year in which the differences are expected to reverse. Significant estimates are required in determining our provision for income taxes. Some of these estimates are based on interpretations of existing tax laws or regulations, or the expected results from any future tax examinations. Various internal and external factors may have favorable
45
or unfavorable effects on our future provision for income taxes. Those factors include, but are not limited to, changes in tax laws, regulations and/or rates, the results of any future tax examinations, changing interpretations of existing tax laws or regulations, changes in estimates of prior years’ items, past levels of research and development spending, acquisitions, changes in our corporate structure, and changes in overall levels of income before taxes all of which may result in periodic revisions to our provision for income taxes.
Developing our provision for income taxes, including our effective tax rate and analysis of potential uncertain tax positions, if any, requires significant judgment and expertise in federal and state income tax laws, regulations and strategies, including the determination of deferred tax assets and liabilities and any estimated valuation allowance we deem necessary to offset deferred tax assets. If we do not maintain taxable income from operations in future periods, we may increase the valuation allowance for our deferred tax assets and record material adjustments to our income tax expense. Our judgment and tax strategies are subject to audit by various taxing authorities. While we believe we have provided adequately for our uncertain income tax positions in our consolidated financial statements, adverse determination by these taxing authorities could have a material adverse effect on our consolidated financial condition, results of operations or cash flows. Interest and penalties on income tax items are included as a component of overall income tax expense.
Recent Accounting Pronouncements
Stock Compensation
In March 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-09, Compensation – Stock Compensation (Topic 718) (“ASU 2016-09”), which simplified certain aspects of the accounting for share-based payment transactions, including income taxes, classification of awards and classification in the statement of cash flows. ASU 2016-09 will be effective for the Company beginning in its first quarter of 2018. We adopted this guidance during the prior year. For discussion of impact see note 10 to the financial statements.
Leases
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”), which modified lease accounting for both lessees and lessors to increase transparency and comparability by recognizing lease assets and lease liabilities by lessees for those leases classified as operating leases under previous accounting standards and disclosing key information about leasing arrangements. ASU 2016-02 will be effective for the Company beginning in its first quarter of 2020 and early adoption is permitted. We are currently evaluating the timing of its adoption and the impact of adopting the new lease standard on its consolidated financial statements.
Revenue Recognition
In May 2014, the Financial Accounting Standards Board (FASB) issued the converged standard on revenue recognition with the objective of providing a single, comprehensive model for all contracts with customers to improve comparability in the financial statements of companies reporting using International Financial Reporting Standards and U.S. GAAP. The standard contains principles that an entity must apply to determine the measurement of revenue and timing of when it is recognized. The underlying principle is that an entity must recognize revenue to depict the transfer of goods or services to customers at an amount that the entity expects to be entitled to in exchange for those goods or services. An entity can apply the revenue standard retrospectively to each prior reporting period presented (full retrospective method) or retrospectively with the cumulative effect of initially applying the standard recognized at the date of initial application in retained earnings (modified retrospective method). The standard will be effective for the Company beginning July 1, 2018, with early adoption permitted for annual periods beginning after December 16, 2016.
The Company plans to adopt the standard July 1, 2018 using the full retrospective method. The Company continues to assess the impact of this standard on its results of operations, financial position and cash flows. Based on its preliminary assessment, the Company expects the majority of the amounts that have historically been classified as bad debt expense, primarily related to patient responsibility, will be reflected as a reduction of the transaction price and therefore as a reduction in revenue. The Company anticipates an increase in the level of required financial statement disclosures due to the standard.
46
We maintain an investment portfolio in accordance with our written investment policy. The primary objectives of our investment policy are to preserve principal, maintain proper liquidity to meet operating needs and maximize yields. Our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure to any single issue, issuer or type of investment.
Our investments consist of securities of various types and maturities of five years or less, with a maximum average maturity of three years. These securities are classified as available for sale. Available-for-sale securities are recorded on the balance sheet at fair market value with unrealized gains or losses reported as part of accumulated other comprehensive income/loss. Realized gains and losses on investment security transactions are reported on the specific-identification method. Dividend and interest income are recognized when earned. A decline in the market value of any available-for-sale security below cost that is deemed other-than-temporary results in a charge to earnings and establishes a new cost basis for the security.
Although our investment policy guidelines are intended to ensure the preservation of principal, market conditions can result in high levels of uncertainty. Our ability to trade or redeem the marketable investment securities in which we invest, including certain corporate bonds, may become difficult. Valuation and pricing of these securities can also become variable and subject to uncertainty.
As of June 30, 2017 we have $0.1 million in unrealized losses in our investment portfolio. For the year ended June 30, 2017 we have experienced fluctuations in our portfolio value primarily from our investments in bonds of various municipalities. If interest rates rise, the market value of our investments may decline, which could result in a realized loss if we are forced to sell an investment before its scheduled maturity. A hypothetical increase in interest rates by 25 basis points would have resulted in a decrease in the fair value of our net investment position of approximately $0.3 million and $0.5 million as of June 30, 2017 and 2016, respectively. We do not utilize derivative financial instruments to manage our interest rate risks.
47
48
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Myriad Genetics, Inc.
We have audited the accompanying consolidated balance sheets of Myriad Genetics, Inc. and subsidiaries as of June 30, 2017 and 2016, and the related consolidated statements of operations, comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended June 30, 2017. Our audits also included the financial statement schedule listed in the Index at Item 15(a). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Myriad Genetics, Inc. and subsidiaries at June 30, 2017 and 2016, and the consolidated results of their operations and their cash flows for each of the three years in the period ended June 30, 2017, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Myriad Genetics, Inc. and subsidiaries’ internal control over financial reporting as of June 30, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated August 9, 2017 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Salt Lake City, UT
August 9, 2017
49
AND SUBSIDIARIES
(In millions)
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
102.4 |
|
|
$ |
68.5 |
|
|
Marketable investment securities |
|
|
48.3 |
|
|
|
90.5 |
|
|
Prepaid expenses |
|
|
12.7 |
|
|
|
18.4 |
|
|
Inventory |
|
|
42.2 |
|
|
|
38.3 |
|
|
Trade accounts receivable, less allowance for doubtful accounts of $8.2 in 2017 and $6.8 in 2016 |
|
|
105.6 |
|
|
|
91.7 |
|
|
Prepaid taxes |
|
|
0.2 |
|
|
|
3.8 |
|
|
Other receivables |
|
|
5.7 |
|
|
|
3.3 |
|
|
Total current assets |
|
|
317.1 |
|
|
|
314.5 |
|
|
Property, plant and equipment, net |
|
|
51.1 |
|
|
|
58.3 |
|
|
Long-term marketable investment securities |
|
|
48.5 |
|
|
|
79.9 |
|
|
Intangibles, net |
|
|
491.6 |
|
|
|
227.5 |
|
|
Goodwill |
|
|
316.1 |
|
|
|
195.3 |
|
|
Other assets |
|
|
- |
|
|
|
5.0 |
|
|
Total assets |
|
$ |
1,224.4 |
|
|
$ |
880.5 |
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
22.0 |
|
|
$ |
21.1 |
|
|
Accrued liabilities |
|
|
65.6 |
|
|
|
49.5 |
|
|
Short-term contingent consideration |
|
|
127.3 |
|
|
|
- |
|
|
Deferred revenue |
|
|
2.6 |
|
|
|
1.7 |
|
|
Total current liabilities |
|
|
217.5 |
|
|
|
72.3 |
|
|
Unrecognized tax benefits |
|
|
25.2 |
|
|
|
24.0 |
|
|
Other long-term liabilities |
|
|
7.2 |
|
|
|
7.8 |
|
|
Contingent consideration |
|
|
13.2 |
|
|
|
10.4 |
|
|
Long-term debt |
|
|
99.1 |
|
|
|
- |
|
|
Long-term deferred taxes |
|
|
84.4 |
|
|
|
17.9 |
|
|
Total liabilities |
|
|
446.6 |
|
|
|
132.4 |
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Common stock, 68.4 and 69.1 shares outstanding at June 30, 2017 and 2016 respectively |
|
|
0.7 |
|
|
|
0.7 |
|
|
Additional paid-in capital |
|
|
851.4 |
|
|
|
830.1 |
|
|
Accumulated other comprehensive loss |
|
|
(5.5 |
) |
|
|
(9.5 |
) |
|
Accumulated deficit |
|
|
(68.4 |
) |
|
|
(73.2 |
) |
|
Total Myriad Genetics, Inc. stockholders' equity |
|
|
778.2 |
|
|
|
748.1 |
|
|
Non-controlling interest |
|
|
(0.4 |
) |
|
|
- |
|
|
Total stockholders' equity |
|
|
777.8 |
|
|
|
748.1 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
1,224.4 |
|
|
$ |
880.5 |
|
See accompanying notes to consolidated financial statements.
50
AND SUBSIDIARIES
Consolidated Statements of Operations
(In millions, except per share amounts)
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Molecular diagnostic testing |
|
$ |
722.1 |
|
|
$ |
705.7 |
|
|
$ |
695.5 |
|
|
Pharmaceutical and clinical services |
|
|
49.3 |
|
|
|
48.1 |
|
|
|
27.6 |
|
|
Total revenue |
|
|
771.4 |
|
|
|
753.8 |
|
|
|
723.1 |
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of molecular diagnostic testing |
|
|
145.2 |
|
|
|
132.8 |
|
|
|
132.8 |
|
|
Cost of pharmaceutical and clinical services |
|
|
26.0 |
|
|
|
24.5 |
|
|
|
14.6 |
|
|
Research and development expense |
|
|
74.4 |
|
|
|
70.6 |
|
|
|
75.5 |
|
|
Selling, general, and administrative expense |
|
|
476.4 |
|
|
|
359.1 |
|
|
|
366.0 |
|
|
Total costs and expenses |
|
|
722.0 |
|
|
|
587.0 |
|
|
|
588.9 |
|
|
Operating income |
|
|
49.4 |
|
|
|
166.8 |
|
|
|
134.2 |
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
1.2 |
|
|
|
0.9 |
|
|
|
0.4 |
|
|
Interest expense |
|
|
(6.0 |
) |
|
|
(0.3 |
) |
|
|
(0.2 |
) |
|
Change in the fair value of contingent consideration |
|
|
0.8 |
|
|
|
- |
|
|
|
- |
|
|
Other |
|
|
(2.5 |
) |
|
|
1.5 |
|
|
|
0.5 |
|
|
Total other income (expense): |
|
|
(6.5 |
) |
|
|
2.1 |
|
|
|
0.7 |
|
|
Income before income tax |
|
|
42.9 |
|
|
|
168.9 |
|
|
|
134.9 |
|
|
Income tax provision |
|
|
21.3 |
|
|
|
43.6 |
|
|
|
54.7 |
|
|
Net income |
|
|
21.6 |
|
|
|
125.3 |
|
|
|
80.2 |
|
|
Net loss attributable to non-controlling interest |
|
|
(0.2 |
) |
|
|
- |
|
|
|
- |
|
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
21.8 |
|
|
$ |
125.3 |
|
|
$ |
80.2 |
|
|
Earnings per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
0.32 |
|
|
$ |
1.79 |
|
|
$ |
1.12 |
|
|
Diluted |
|
$ |
0.32 |
|
|
$ |
1.71 |
|
|
$ |
1.08 |
|
|
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
68.3 |
|
|
|
70.0 |
|
|
|
71.3 |
|
|
Diluted |
|
|
68.8 |
|
|
|
73.4 |
|
|
|
74.5 |
|
See accompanying notes to consolidated financial statements.
51
AND SUBSIDIARIES
Consolidated Statements of Comprehensive Income
(In millions)
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
21.8 |
|
|
$ |
125.3 |
|
|
$ |
80.2 |
|
|
Unrealized gain (loss) on available-for-sale securities, net of tax |
|
|
(0.6 |
) |
|
|
0.3 |
|
|
|
(0.3 |
) |
|
Change in pension liability |
|
|
0.2 |
|
|
|
(0.2 |
) |
|
|
— |
|
|
Change in foreign currency translation adjustment |
|
|
4.4 |
|
|
|
(2.6 |
) |
|
|
(5.2 |
) |
|
Comprehensive income |
|
$ |
25.8 |
|
|
$ |
122.8 |
|
|
$ |
74.7 |
|
See accompanying notes to consolidated financial statements.
52
AND SUBSIDIARIES
Consolidated Statements of Stockholders’ Equity
(In millions)
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
Retained |
|
|
Myriad |
|
|||
|
|
|
|
|
|
|
Additional |
|
|
other |
|
|
earnings |
|
|
Genetics, Inc. |
|
||||
|
|
|
Common |
|
|
paid-in |
|
|
comprehensive |
|
|
(accumulated |
|
|
Stockholders’ |
|
|||||
|
|
|
stock |
|
|
capital |
|
|
loss |
|
|
deficit) |
|
|
equity |
|
|||||
|
BALANCES AT JUNE 30, 2014 |
|
$ |
0.7 |
|
|
$ |
717.8 |
|
|
$ |
(1.5 |
) |
|
$ |
2.0 |
|
|
$ |
719.0 |
|
|
Issuance of common stock under share-based compensation plans |
|
|
— |
|
|
|
30.0 |
|
|
|
— |
|
|
|
— |
|
|
|
30.0 |
|
|
Share-based payment expense |
|
|
— |
|
|
|
45.7 |
|
|
|
— |
|
|
|
— |
|
|
|
45.7 |
|
|
Share-based compensation tax benefits |
|
|
— |
|
|
|
3.4 |
|
|
|
— |
|
|
|
— |
|
|
|
3.4 |
|
|
Repurchase and retirement of common stock |
|
|
— |
|
|
|
(51.5 |
) |
|
|
— |
|
|
|
(159.2 |
) |
|
|
(210.7 |
) |
|
Net income |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
80.2 |
|
|
|
80.2 |
|
|
Other comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
(5.5 |
) |
|
|
— |
|
|
|
(5.5 |
) |
|
BALANCES AT JUNE 30, 2015 |
|
$ |
0.7 |
|
|
$ |
745.4 |
|
|
$ |
(7.0 |
) |
|
$ |
(77.0 |
) |
|
$ |
662.1 |
|
|
Issuance of common stock under share-based compensation plans |
|
|
— |
|
|
|
94.3 |
|
|
|
— |
|
|
|
— |
|
|
|
94.3 |
|
|
Share-based payment expense |
|
|
— |
|
|
|
31.6 |
|
|
|
— |
|
|
|
— |
|
|
|
31.6 |
|
|
Repurchase and retirement of common stock |
|
|
— |
|
|
|
(41.2 |
) |
|
|
— |
|
|
|
(121.5 |
) |
|
|
(162.7 |
) |
|
Net income |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
125.3 |
|
|
|
125.3 |
|
|
Other comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
(2.5 |
) |
|
|
— |
|
|
|
(2.5 |
) |
|
BALANCES AT JUNE 30, 2016 |
|
$ |
0.7 |
|
|
$ |
830.1 |
|
|
$ |
(9.5 |
) |
|
$ |
(73.2 |
) |
|
$ |
748.1 |
|
|
Issuance of common stock under share-based compensation plans |
|
|
— |
|
|
|
6.0 |
|
|
|
— |
|
|
|
— |
|
|
|
6.0 |
|
|
Share-based payment expense |
|
|
— |
|
|
|
29.9 |
|
|
|
— |
|
|
|
— |
|
|
|
29.9 |
|
|
Repurchase and retirement of common stock |
|
|
— |
|
|
|
(14.6 |
) |
|
|
— |
|
|
|
(17.0 |
) |
|
|
(31.6 |
) |
|
Net income |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
21.8 |
|
|
|
21.8 |
|
|
Other comprehensive income, net of tax |
|
|
— |
|
|
|
— |
|
|
|
4.0 |
|
|
|
— |
|
|
|
4.0 |
|
|
BALANCES AT JUNE 30, 2017 |
|
$ |
0.7 |
|
|
$ |
851.4 |
|
|
$ |
(5.5 |
) |
|
$ |
(68.4 |
) |
|
$ |
778.2 |
|
See accompanying notes to consolidated financial statements.
53
AND SUBSIDIARIES
Consolidated Statements of Cash Flows
(In millions)
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
21.8 |
|
|
$ |
125.3 |
|
|
$ |
80.2 |
|
|
Adjustments to reconcile net income to net cash provided by operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
48.3 |
|
|
|
26.7 |
|
|
|
25.0 |
|
|
Non-cash interest expense |
|
|
0.4 |
|
|
|
— |
|
|
|
— |
|
|
Loss (gain) on disposition of assets |
|
|
(0.3 |
) |
|
|
(0.9 |
) |
|
|
0.5 |
|
|
Share-based compensation expense |
|
|
29.9 |
|
|
|
31.6 |
|
|
|
45.7 |
|
|
Impairment of cost basis investment |
|
|
2.4 |
|
|
|
— |
|
|
|
— |
|
|
Bad debt expense |
|
|
37.3 |
|
|
|
33.3 |
|
|
|
31.5 |
|
|
Loss on extinguishment of debt |
|
|
1.3 |
|
|
|
— |
|
|
|
— |
|
|
Deferred income taxes |
|
|
1.3 |
|
|
|
18.1 |
|
|
|
(0.4 |
) |
|
Unrecognized tax benefits |
|
|
1.2 |
|
|
|
(2.4 |
) |
|
|
2.1 |
|
|
Change in fair value of contingent consideration |
|
|
(0.8 |
) |
|
|
— |
|
|
|
— |
|
|
Excess tax benefit from share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
(3.4 |
) |
|
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses |
|
|
7.8 |
|
|
|
(7.2 |
) |
|
|
(5.5 |
) |
|
Trade accounts receivable |
|
|
(41.4 |
) |
|
|
(39.2 |
) |
|
|
(34.4 |
) |
|
Other receivables |
|
|
(4.0 |
) |
|
|
(0.9 |
) |
|
|
2.5 |
|
|
Inventory |
|
|
(1.2 |
) |
|
|
(14.6 |
) |
|
|
(0.8 |
) |
|
Prepaid taxes |
|
|
3.6 |
|
|
|
(3.8 |
) |
|
|
13.6 |
|
|
Accounts payable |
|
|
(3.0 |
) |
|
|
— |
|
|
|
(3.1 |
) |
|
Accrued liabilities |
|
|
0.7 |
|
|
|
0.5 |
|
|
|
(13.4 |
) |
|
Deferred revenue |
|
|
0.9 |
|
|
|
(0.2 |
) |
|
|
0.4 |
|
|
Net cash provided by operating activities |
|
|
106.2 |
|
|
|
166.3 |
|
|
|
140.5 |
|
|
CASH FLOWS FROM INVESTING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
|
|
|
Capital expenditures |
|
|
(6.1 |
) |
|
|
(5.0 |
) |
|
|
(23.9 |
) |
|
Acquisitions, net of cash acquired |
|
|
(216.1 |
) |
|
|
(37.0 |
) |
|
|
(20.1 |
) |
|
Sale of cost basis investment |
|
|
2.6 |
|
|
|
- |
|
|
|
- |
|
|
Purchases of marketable investment securities |
|
|
(87.5 |
) |
|
|
(164.5 |
) |
|
|
(80.7 |
) |
|
Proceeds from maturities and sales of marketable investment securities |
|
|
160.8 |
|
|
|
115.1 |
|
|
|
165.6 |
|
|
Net cash provided by (used in) investing activities |
|
|
(146.3 |
) |
|
|
(91.4 |
) |
|
|
40.9 |
|
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net proceeds from common stock issued under share-based compensation plans |
|
|
6.0 |
|
|
|
94.3 |
|
|
|
30.0 |
|
|
Net proceeds from revolving credit facility |
|
|
204.0 |
|
|
|
— |
|
|
|
— |
|
|
Net proceeds from term loan |
|
|
199.0 |
|
|
|
— |
|
|
|
— |
|
|
Repayment of term loan |
|
|
(200.0 |
) |
|
|
— |
|
|
|
— |
|
|
Repayment of revolving credit facility |
|
|
(105.0 |
) |
|
|
— |
|
|
|
— |
|
|
Fees paid for extinguishment of debt |
|
|
(0.6 |
) |
|
|
— |
|
|
|
— |
|
|
Excess tax benefit from share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
3.4 |
|
|
Repurchase and retirement of common stock |
|
|
(31.6 |
) |
|
|
(162.6 |
) |
|
|
(210.7 |
) |
|
Net cash provided by (used in) financing activities |
|
|
71.8 |
|
|
|
(68.3 |
) |
|
|
(177.3 |
) |
|
Effect of foreign exchange rates on cash and cash equivalents |
|
|
2.2 |
|
|
|
(2.2 |
) |
|
|
(4.8 |
) |
|
Net (decrease) increase in cash and cash equivalents |
|
|
33.9 |
|
|
|
4.4 |
|
|
|
(0.7 |
) |
|
Cash and cash equivalents at beginning of year |
|
|
68.5 |
|
|
|
64.1 |
|
|
|
64.8 |
|
|
Cash and cash equivalents at end of year |
|
$ |
102.4 |
|
|
$ |
68.5 |
|
|
$ |
64.1 |
|
See accompanying notes to consolidated financial statements.
54
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(in millions, except per share data)
|
1. |
ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Basis of Financial Statement Presentation
Myriad Genetics, Inc. and subsidiaries (collectively, the Company) is a leading molecular diagnostic company focused on developing and marketing novel predictive medicine, personalized medicine and prognostic medicine tests. The Company employs a number of proprietary technologies, including DNA, RNA and protein analysis, that help it to understand the genetic basis of human disease and the role that genes and their related proteins may play in the onset and progression of disease. The Company uses this information to guide the development of new molecular diagnostic and companion diagnostic tests that are designed to assess an individual’s risk for developing disease later in life (predictive medicine), identify a patient’s likelihood of responding to drug therapy and guide a patient’s dosing to ensure optimal treatment (personalized medicine), or assess a patient’s risk of disease progression and disease recurrence (prognostic medicine). The Company generates revenue by performing molecular diagnostic tests as well as by providing pharmaceutical and clinical services to the pharmaceutical and biotechnology industries and medical research institutions utilizing its multiplexed immunoassay technology. The Company’s corporate headquarters are located in Salt Lake City, Utah.
The accompanying condensed consolidated financial statements have been prepared by Myriad Genetics, Inc. (the “Company” or “Myriad”) in accordance with U.S. generally accepted accounting principles (“GAAP”) for interim financial information and pursuant to the applicable rules and regulations of the Securities and Exchange Commission (“SEC”). The condensed consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation. In the opinion of management, the accompanying financial statements contain all adjustments (consisting of normal and recurring accruals) necessary to present fairly all financial statements in accordance with U.S. GAAP. A reclassification of ($0.3) and ($0.2) from other to interest expense in the consolidated statements of operations for fiscal years ended June 30, 2016 and 2015 and a reclassification of $10.4 from other long-term liabilities to contingent consideration in the consolidated balance sheet for year ended June 30, 2016 in order to conform with the current-year presentation.
Marketable Investment Securities
The Company has classified its marketable investment securities as available-for-sale. Available-for-sale investment securities with remaining maturities of greater than one year are classified as long-term. Available-for-sale investment securities with remaining maturities of one year or less are classified as short-term. Available-for-sale investment securities with remaining maturities of less than three months at the time of purchase are classified as cash equivalents. Marketable securities are carried at estimated fair value with unrealized holding gains and losses, net of the related tax effect, included in accumulated other comprehensive loss in stockholders’ equity until realized. Gains and losses on investment security transactions are reported using the specific-identification method. Dividend and interest income are recognized when earned.
A decline in the market value of any available-for-sale security below cost that is deemed other than temporary results in a charge to earnings and establishes a new cost basis for the security. Losses are charged against “Other income” when a decline in fair value is determined to be other than temporary. We review several factors to determine whether a loss is other than temporary. These factors include but are not limited to: (i) the extent to which the fair value is less than cost and the cause for the fair value decline, (ii) the financial condition and near term prospects of the issuer, (iii) the length of time a security is in an unrealized loss position and (iv) our ability to hold the security for a period of time sufficient to allow for any anticipated recovery in fair value. There were no other-than-temporary impairments recognized during the fiscal years ended June 30, 2017, 2016 and 2015.
Inventories consist of reagents, plates and testing kits. Inventories are stated at the lower of cost or market on a first-in, first-out basis. In order to assess the ultimate realization of inventories, the Company is required to make judgments as to future demand requirements compared to current or committed inventory levels.
The Company evaluates its inventories for excess quantities and obsolescence. Inventories that are considered obsolete are expensed. The valuation of inventories requires the use of estimates as to the amounts of current inventories that will be sold. These estimates are dependent on management’s assessment of current and expected orders from the Company’s customers.
55
Trade Accounts Receivable and Allowance for Doubtful Accounts
Trade accounts receivable are comprised of amounts due from sales of the Company’s molecular diagnostic tests and pharmaceutical and clinical services and are recorded at the invoiced amount, net of discounts and contractual allowances. The allowance for doubtful accounts is based on the Company’s best estimate of the amount of probable losses in the Company’s existing accounts receivable, which is based on historical write-off experience, customer creditworthiness, facts and circumstances specific to outstanding balances, and payment terms. Account balances are charged against the allowance after all means of collection have been exhausted and the potential for recovery is considered remote. The Company does not have any off-balance-sheet credit exposure related to its customers and does not require collateral.
Property, Plant and Equipment
Equipment and leasehold improvements are stated at cost less accumulated depreciation. Depreciation and amortization are computed using the straight-line method based on the lesser of estimated useful lives of the related assets or lease terms. Equipment items have depreciable lives of five to seven years. Leasehold improvements are depreciated over the shorter of the estimated useful lives or the associated lease terms, which range from three to ten years. Repairs and maintenance costs are charged to expense as incurred.
Intangible Assets and Other Long-Lived Assets
Intangible and other long-lived assets are comprised of acquired licenses, technology and intellectual property and purchased in-process research and development. Acquired intangible assets are recorded at fair value and amortized over the shorter of the contractual life or the estimated useful life. The estimated useful life of acquired in-process research and development was also evaluated in conjunction with the annual impairment analysis of intangible assets. The classification of the acquired in-process research and development as an indefinite lived asset was deemed appropriate as the related research and development was not yet complete nor had it been abandoned.
The Company continually reviews and monitors long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future undiscounted net cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future undiscounted cash flows, an impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. Assets to be disposed of are reported at the lower of the carrying amount or fair value less costs to sell.
Goodwill
Goodwill is tested for impairment on an annual basis as of April 1 and in the interim by reporting unit if events and circumstances indicate that goodwill may be impaired. The events and circumstances that are considered include business climate and market conditions, legal factors, operating performance indicators and competition. Impairment of goodwill was first assessed using a qualitative approach. If the qualitative assessment suggests that impairment is more likely than not, a two-step impairment analysis is performed. The first step involves a comparison of the fair value of the reporting unit with its carrying amount. If the carrying amount of the reporting unit exceeds its fair value, the second step of the process involves a comparison of the fair value and the carrying amount of the goodwill of that reporting unit. If the carrying amount of the goodwill of the reporting unit exceeds the fair value of that goodwill, an impairment loss would be recognized in an amount equal to the excess of carrying value over fair value. If an event occurs that would cause a revision to the estimates and assumptions used in analyzing the value of the goodwill, the revision could result in a non-cash impairment charge that could have a material impact on the financial results.
Revenue Recognition
Molecular diagnostic testing revenue is recognized when persuasive evidence of an agreement exists, results have been communicated to the patient, the fee is fixed or determinable, and collection is reasonably assured. Revenue from the sale of molecular diagnostic tests and related marketing agreements is recorded at the invoiced amount net of any discounts or contractual allowances.
56
Pharmaceutical and clinical service revenue is recognized when persuasive evidence of an agreement exists, the fee is fixed and or determinable, when the service has been completed and the results of the tests/service are provided to the customer, and collectability is reasonably assured. In addition, the Company’s wholly owned subsidiary, Myriad RBM, has received national, state, foreign government and private foundation grants and contracts. Revenue associated with these grants and contracts is recognized in the period in which qualifying costs for the services by the grants and contracts are incurred and the related grant or contract fee is earned.
Income Taxes
The Company recognizes income taxes under the asset and liability method. This approach requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the carrying amounts and the tax bases of assets and liabilities.
The provision for income taxes, including the effective tax rate and analysis of potential tax exposure items, if any, requires significant judgment and expertise in federal and state income tax laws, regulations and strategies, including the determination of deferred tax assets and liabilities and any estimated valuation allowances deemed necessary to recognize deferred tax assets at an amount that is more likely than not to be realized. The Company’s filings, including the positions taken therein, are subject to audit by various taxing authorities. While the Company believes it has provided adequately for its income tax liabilities in the consolidated financial statements, adverse determinations by these taxing authorities could have a material adverse effect on the consolidated financial condition, results of operations or cash flows.
Earnings Per Share
Basic earnings per share is computed based on the weighted-average number of shares of common stock outstanding. Diluted earnings per share is computed based on the weighted-average number of shares of common stock, including the dilutive effect of common stock equivalents, outstanding.
The following is a reconciliation of the denominators of the basic and diluted earnings per share computations:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
Denominator: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average shares outstanding used to compute basic EPS |
|
|
68.3 |
|
|
|
70.0 |
|
|
|
71.3 |
|
|
Effect of dilutive stock options |
|
|
0.5 |
|
|
|
3.4 |
|
|
|
3.2 |
|
|
Weighted-average shares outstanding and dilutive securities used to compute diluted EPS |
|
|
68.8 |
|
|
|
73.4 |
|
|
|
74.5 |
|
Certain outstanding options and RSUs were excluded from the computation of diluted earnings per share because the effect would have been anti-dilutive. These potential dilutive common shares, which may be dilutive to future diluted earnings per share, are as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
Anti-dilutive options and RSUs excluded from EPS computation |
|
|
7.0 |
|
|
|
— |
|
|
|
— |
|
Foreign Currency
The functional currency of the Company’s international subsidiaries is the local currency. For those subsidiaries, expenses denominated in the functional currency are translated into U.S. dollars using average exchange rates in effect during the period and assets and liabilities are translated using period-end exchange rates. The foreign currency translation adjustments are included in accumulated other comprehensive loss as a separate component of stockholders’ (deficit) equity.
57
The following table shows the cumulative translation adjustments included in other comprehensive income:
|
Ending balance June 30, 2016 |
|
$ |
(10.1 |
) |
|
Period translation adjustments |
|
|
4.4 |
|
|
Ending balance June 30, 2017 |
|
|
(5.7 |
) |
Use of Estimates
The preparation of the consolidated financial statements in accordance with U.S. GAAP requires Company management to make estimates and assumptions relating to the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the period. Significant items subject to such estimates and assumptions include the carrying amount of fixed assets, valuation allowances for receivables and deferred income tax assets, certain accrued liabilities, share-based compensation and impairment analysis of goodwill and intangible assets. Actual results could differ from those estimates.
Recent Accounting Pronouncements
In March 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-09, Compensation – Stock Compensation (Topic 718) (“ASU 2016-09”), which simplified certain aspects of the accounting for share-based payment transactions, including income taxes, classification of awards and classification in the statement of cash flows. We adopted this guidance during the prior fiscal year. For discussion of impact see note 10 to the financial statements.
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”), which modified lease accounting for both lessees and lessors to increase transparency and comparability by recognizing lease assets and lease liabilities by lessees for those leases classified as operating leases under previous accounting standards and disclosing key information about leasing arrangements. ASU 2016-02 will be effective for the Company beginning in its first quarter of 2020 and early adoption is permitted. We are currently evaluating the timing of its adoption and the impact of adopting the new lease standard on our consolidated financial statements.
In May 2014, the Financial Accounting Standards Board (FASB) issued the converged standard on revenue recognition with the objective of providing a single, comprehensive model for all contracts with customers to improve comparability in the financial statements of companies reporting using International Financial Reporting Standards and U.S. GAAP. The standard contains principles that an entity must apply to determine the measurement of revenue and timing of when it is recognized. The underlying principle is that an entity must recognize revenue to depict the transfer of goods or services to customers at an amount that the entity expects to be entitled to in exchange for those goods or services. An entity can apply the revenue standard retrospectively to each prior reporting period presented (full retrospective method) or retrospectively with the cumulative effect of initially applying the standard recognized at the date of initial application in retained earnings (modified retrospective method). The standard will be effective for the Company beginning July 1, 2018, with early adoption permitted for annual periods beginning after December 16, 2016.
The Company plans to adopt the standard July 1, 2018 using the modified retrospective method. The Company continues to assess the impact of this standard on its results of operations, financial position and cash flows. Based on its preliminary assessment, the Company expects the majority of the amounts that have historically been classified as bad debt expense, primarily related to patient responsibility, will be reflected as a reduction of the transaction price and therefore as a reduction in revenue. The Company anticipates an increase in the level of required financial statement disclosures due to the standard.
|
2. |
BUSINESS ACQUISITIONS |
Assurex
On August 31, 2016, the Company completed the acquisition of Assurex, pursuant to the Agreement and Plan of Merger (as amended, the “Merger Agreement”), dated August 3, 2016. Pursuant to the terms of the Merger Agreement, Myriad Merger Sub, Inc., a wholly owned subsidiary of the Company was merged with and into Assurex, with Assurex continuing as the surviving corporation, and wholly owned subsidiary of Myriad. The Company acquired Assurex for total consideration of $351.6, net of cash acquired of $5.5, including a cash payment of $216.1, and two potential
58
performance-based milestones totaling $185.0 with a fair value of $130.0. The fair value of the performance-based milestones was determined by using the Monte Carlo method.
Of the cash consideration, $19.1 was deposited into an escrow account to fund (i) any post-closing adjustments payable to Myriad based upon differences between the estimated working capital and the actual working capital of Assurex at closing, and (ii) any indemnification claims made by Myriad against Assurex within 18 months following closing.
Total consideration transferred was allocated to tangible assets acquired and liabilities assumed based on their fair values as of the acquisition date including current adjustments as set forth below. The Company believes the acquisition establishes the foundation for our neuroscience business and leverages our existing preventative care business unit with the addition of a product, GeneSight, which has growth potential. These factors contributed to consideration transferred in excess of the fair value of Assurex’s net tangible and intangible assets acquired, resulting in the Company recording $119.1 in goodwill in connection with the transaction. During the year ended June 30, 2017 there were fair value reductions as of the date of the acquisition to intangible assets and equipment totaling $1.7 due to adoption of our capitalization policy which increased goodwill by that amount. During the period there was an adjustment to working capital as of the date of acquisition which required an additional $3.1 cash payment and increased goodwill by that amount, $2.0 decrease in the deferred tax liability due to a change in the blended state tax rate as well as changes in the pre-acquisition deferred tax liabilities which decreased goodwill by the same amount.
Management estimated the fair value of tangible and intangible assets and liabilities in accordance with the applicable accounting guidance for business combinations and utilized the services of third-party valuation consultants. The preliminary allocation of the consideration transferred is based on a preliminary valuation and is subject to potential adjustments. Balances subject to adjustment primarily include the valuations of acquired assets (tangible and intangible), the fair value of equipment, non-controlling interest, as well as tax-related matters, including tax basis of acquired assets and liabilities in a foreign jurisdiction. During the measurement period, the Company may record adjustments to the provisional amounts recognized in the Company’s initial accounting for the acquisition. The Company expects the allocation of the consideration transferred to be final within the measurement period (up to one year from the acquisition date). The preliminary purchase price allocation as of June 30, 2017 is as follows:
|
|
|
Estimated Fair Value |
|
|
|
Current assets |
|
$ |
18.2 |
|
|
Intangible assets |
|
|
295.6 |
|
|
Equipment |
|
|
1.8 |
|
|
Goodwill |
|
|
119.1 |
|
|
Current liabilities |
|
|
(18.6 |
) |
|
Deferred tax liability |
|
|
(64.5 |
) |
|
Total fair value purchase price |
|
$ |
351.6 |
|
|
Less: Contingent consideration |
|
|
(130.0 |
) |
|
Less: Cash acquired |
|
|
(5.5 |
) |
|
Total cash consideration transferred |
|
$ |
216.1 |
|
Identifiable Intangible Assets
The Company acquired intangible assets that consisted of developed technology which had an estimated fair value of $256.5 and a database with an estimated fair value of $39.1. The fair value of the developed technology was determined using a probability-weighted income approach that discounts expected future cash flows to present value. The fair value of the database was determined using a combination of the lost profits and replacement cost methods. The estimated net cash flows were discounted using a discount rate of 16% which is based on the estimated internal rate of return for the acquisition and represents the rate that market participants might use to value the intangible assets. The projected cash flows were based on key assumptions such as: estimates of revenues and operating profits; the time and resources needed to recreate databases and product and commercial development and approval; the life of the commercialized product; and associated risks related to viability and product alternatives. The Company will amortize the intangible assets on a straight-line basis over their estimated useful lives of 17 years for the developed technology and 5 years for the database. This amortization is not deductible for income tax purposes. During the year the internally developed software was written off as it was already included in the fair value of the developed technology.
59
The $119.1 of goodwill represents the excess of consideration transferred over the fair value of assets acquired and liabilities assumed and is attributable to the benefits expected from combining the Company’s research and commercial operations with Assurex’s. This goodwill is not deductible for income tax purposes. As discussed above, the change in goodwill from the date of acquisition is shown below:
|
|
|
Carrying |
|
|
|
|
|
amount |
|
|
|
Balance August 31, 2016 |
|
$ |
116.3 |
|
|
Fair value adjustment to equipment and intangibles |
|
|
1.7 |
|
|
Working capital adjustment |
|
|
3.1 |
|
|
Change in deferred tax liability |
|
|
(2.0 |
) |
|
Ending balance June 30, 2017 |
|
$ |
119.1 |
|
Pro Forma Information
The unaudited pro-forma results presented below include the effects of the Assurex acquisition as if it had been consummated as of July 1, 2015, with adjustments to give effect to pro forma events that are directly attributable to the acquisition which includes adjustments related to the amortization of acquired intangible assets, interest income and expense, and depreciation. The unaudited pro forma results do not reflect any operating efficiency or potential cost savings which may result from the consolidation of Assurex. Accordingly, these unaudited pro forma results are presented for informational purposes only and are not necessarily indicative of what the actual results of operation of the combined company would have been if the acquisition had occurred at the beginning of the period presented nor are they indicative of future results of operations and are not necessarily indicative of results that might have been achieved had the acquisition been consummated as of July 1, 2015.
|
|
|
Years Ended |
|
|||||
|
|
|
June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Revenue |
|
$ |
782.8 |
|
|
$ |
819.7 |
|
|
Income from operations |
|
|
41.9 |
|
|
|
102.2 |
|
|
Net income |
|
|
2.5 |
|
|
|
71.8 |
|
|
Net income per share, basic |
|
$ |
0.04 |
|
|
$ |
1.03 |
|
|
Net income per share, diluted |
|
$ |
0.04 |
|
|
$ |
0.98 |
|
To complete the purchase transaction, we incurred approximately $5.0 of acquisition costs, which were recorded as selling, general and administrative expenses. For the year ended June 30, 2017, Assurex contributed revenue of approximately $78.4. For year ended June 30, 2017 operating expenses related to Assurex were approximately $96.4.
Sividon
On May 31, 2016 we completed the acquisition of Sividon Diagnostics GmbH (“Sividon”), a leading breast cancer prognostic company with cash paid and total cash consideration transferred of $39.0 upfront and the potential for €15.0 ($17.1 converted at the June 30, 2017 period end exchange rate) in additional performance-based milestones.
Total consideration transferred was allocated to tangible assets acquired and liabilities assumed based on their fair values at the acquisition date as set forth below. We believe the acquisition brings us the best-in-class breast cancer prognostic test and strengthens our market leading oncology portfolio of high value personalized medicine products which can be expanded internationally as well as brought to the US market. These factors contributed to consideration transferred in excess of the fair value of Sividon’s net tangible and intangible assets acquired, resulting in the Company recording goodwill in connection with the transaction. The goodwill related to the purchase is not tax deductible.
60
Management estimated the fair value of tangible and intangible assets and liabilities in accordance with the applicable accounting guidance for business combinations and utilized the services of third-party valuation consultants. During the measurement period, the Company recorded adjustments to the provisional amounts recognized in the Company’s initial accounting for the acquisition. During the year ended June 30, 2017 there was a decrease in contingent consideration of $0.4 and intangibles of $0.4 which increased goodwill by $0.8 based upon updated fair value calculations as of the purchase date. The allocation of consideration transferred is now final.
|
|
|
Estimated Fair |
|
|
|
|
|
Value |
|
|
|
Current assets |
|
$ |
2.7 |
|
|
Intangible assets |
|
|
45.8 |
|
|
Equipment |
|
|
0.3 |
|
|
Goodwill |
|
|
18.5 |
|
|
Current liabilities |
|
|
(15.4 |
) |
|
Total fair value purchase price |
|
$ |
51.9 |
|
|
Less: Contingent consideration |
|
|
(10.9 |
) |
|
Less: Cash acquired |
|
|
(2.0 |
) |
|
Total cash consideration transferred |
|
$ |
39.0 |
|
The acquisition of Sividon has been deemed insignificant in relation to the consolidated financials. As such, pro forma financial information has not been provided.
|
3. |
MARKETABLE INVESTMENT SECURITIES |
The amortized cost, gross unrealized holding gains, gross unrealized holding losses, and fair value for available-for-sale securities by major security type and class of security at June 30, 2017 and 2016 were as follows:
|
|
|
|
|
|
|
Gross |
|
|
Gross |
|
|
|
|
|
||
|
|
|
|
|
|
|
unrealized |
|
|
unrealized |
|
|
|
|
|
||
|
|
|
Amortized |
|
|
holding |
|
|
holding |
|
|
Estimated |
|
||||
|
|
|
cost |
|
|
gains |
|
|
losses |
|
|
fair value |
|
||||
|
At June 30, 2017: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash |
|
$ |
83.5 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
83.5 |
|
|
Cash equivalents |
|
|
18.9 |
|
|
|
— |
|
|
|
— |
|
|
|
18.9 |
|
|
Total cash and cash equivalents |
|
|
102.4 |
|
|
|
— |
|
|
|
— |
|
|
|
102.4 |
|
|
Available-for-sale: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Corporate bonds and notes |
|
|
45.4 |
|
|
|
0.1 |
|
|
|
(0.1 |
) |
|
|
45.4 |
|
|
Municipal bonds |
|
|
32.7 |
|
|
|
— |
|
|
|
— |
|
|
|
32.7 |
|
|
Federal agency issues |
|
|
11.6 |
|
|
|
— |
|
|
|
(0.1 |
) |
|
|
11.5 |
|
|
US government securities |
|
|
7.2 |
|
|
|
— |
|
|
|
— |
|
|
|
7.2 |
|
|
Total |
|
$ |
199.3 |
|
|
$ |
0.1 |
|
|
$ |
(0.2 |
) |
|
$ |
199.2 |
|
61
|
|
|
|
|
|
|
Gross |
|
|
Gross |
|
|
|
|
|
||
|
|
|
|
|
|
|
unrealized |
|
|
unrealized |
|
|
|
|
|
||
|
|
|
Amortized |
|
|
holding |
|
|
holding |
|
|
Estimated |
|
||||
|
|
|
cost |
|
|
gains |
|
|
losses |
|
|
fair value |
|
||||
|
At June 30, 2016: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash |
|
$ |
66.1 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
66.1 |
|
|
Cash equivalents |
|
|
2.4 |
|
|
|
— |
|
|
|
— |
|
|
|
2.4 |
|
|
Total cash and cash equivalents |
|
|
68.5 |
|
|
|
— |
|
|
|
— |
|
|
|
68.5 |
|
|
Available-for-sale: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Corporate bonds and notes |
|
|
50.8 |
|
|
|
0.2 |
|
|
|
— |
|
|
|
51.0 |
|
|
Municipal bonds |
|
|
85.4 |
|
|
|
0.2 |
|
|
|
— |
|
|
|
85.6 |
|
|
Federal agency issues |
|
|
25.5 |
|
|
|
— |
|
|
|
— |
|
|
|
25.5 |
|
|
US government securities |
|
|
8.2 |
|
|
|
0.1 |
|
|
|
— |
|
|
|
8.3 |
|
|
Total |
|
$ |
238.4 |
|
|
$ |
0.5 |
|
|
$ |
— |
|
|
$ |
238.9 |
|
Cash, cash equivalents, and maturities of debt securities classified as available-for-sale are as follows at June 30, 2017:
|
|
|
Amortized |
|
|
Estimated |
|
||
|
|
|
cost |
|
|
fair value |
|
||
|
Cash |
|
$ |
83.5 |
|
|
$ |
83.5 |
|
|
Cash equivalents |
|
|
18.9 |
|
|
|
18.9 |
|
|
Available-for-sale: |
|
|
|
|
|
|
|
|
|
Due within one year |
|
|
48.3 |
|
|
|
48.3 |
|
|
Due after one year through five years |
|
|
48.6 |
|
|
|
48.5 |
|
|
Due after five years |
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
199.3 |
|
|
$ |
199.2 |
|
Debt securities in an unrealized loss position as of June 30, 2017 were not impaired at acquisition and the declines in fair value are not attributed to declines in credit quality. Management believes that it is more likely than not that the securities will be held until a recovery of par value. All securities in an unrealized loss position as of June 30, 2017 and 2016 are debt securities. Debt securities available-for-sale in a gross unrealized loss position as of June 30, 2017 and 2016 are summarized as follows:
|
|
|
Less than 12 months |
|
|
More than 12 months |
|
|
Total |
|
|||||||||||||||
|
|
|
Fair |
|
|
Unrealized |
|
|
Fair |
|
|
Unrealized |
|
|
Fair |
|
|
Unrealized |
|
||||||
|
|
|
value |
|
|
losses |
|
|
value |
|
|
losses |
|
|
value |
|
|
losses |
|
||||||
|
At June 30, 2017: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Debt securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Corporate bonds and notes |
|
|
29.9 |
|
|
|
0.1 |
|
|
|
— |
|
|
|
— |
|
|
|
29.9 |
|
|
|
0.1 |
|
|
Municipal bonds |
|
|
13.3 |
|
|
|
— |
|
|
|
0.7 |
|
|
|
— |
|
|
|
14.0 |
|
|
|
— |
|
|
Federal agency issues |
|
|
11.5 |
|
|
|
0.1 |
|
|
|
— |
|
|
|
— |
|
|
|
11.5 |
|
|
|
0.1 |
|
|
US government securities |
|
|
5.3 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
5.3 |
|
|
|
— |
|
|
|
|
$ |
60.0 |
|
|
$ |
0.2 |
|
|
$ |
0.7 |
|
|
$ |
— |
|
|
$ |
60.7 |
|
|
$ |
0.2 |
|
|
|
|
Less than 12 months |
|
|
More than 12 months |
|
|
Total |
|
|||||||||||||||
|
|
|
Fair |
|
|
Unrealized |
|
|
Fair |
|
|
Unrealized |
|
|
Fair |
|
|
Unrealized |
|
||||||
|
|
|
value |
|
|
losses |
|
|
value |
|
|
losses |
|
|
value |
|
|
losses |
|
||||||
|
At June 30, 2016: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Debt securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Corporate bonds and notes |
|
|
5.0 |
|
|
|
— |
|
|
|
1.5 |
|
|
|
— |
|
|
|
6.5 |
|
|
|
— |
|
|
Municipal bonds |
|
|
7.6 |
|
|
|
— |
|
|
|
6.3 |
|
|
|
— |
|
|
|
13.9 |
|
|
|
— |
|
|
Federal agency issues |
|
|
6.0 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
6.0 |
|
|
|
— |
|
|
US government securities |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
$ |
18.6 |
|
|
$ |
— |
|
|
$ |
7.8 |
|
|
$ |
— |
|
|
$ |
26.4 |
|
|
$ |
— |
|
62
Additional information relating to fair value of marketable investment securities can be found in Note 12.
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
Land |
|
$ |
2.3 |
|
|
$ |
2.3 |
|
|
Buildings and improvements |
|
|
17.1 |
|
|
|
17.3 |
|
|
Leasehold improvements |
|
|
22.1 |
|
|
|
18.7 |
|
|
Equipment |
|
|
106.9 |
|
|
|
103.4 |
|
|
|
|
|
148.4 |
|
|
|
141.7 |
|
|
Less accumulated depreciation |
|
|
(97.3 |
) |
|
|
(83.4 |
) |
|
Property, plant and equipment, net |
|
$ |
51.1 |
|
|
$ |
58.3 |
|
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
Depreciation expense |
|
$ |
15.0 |
|
|
$ |
14.1 |
|
|
$ |
12.3 |
|
|
5. |
GOODWILL AND INTANGIBLE ASSETS |
Goodwill
The Company has recorded goodwill of $316.1 from the acquisitions of Sividon Diagnostics that was completed on May 31, 2016, GmbH, Privatklinik Dr. Robert Schindlbeck GmbH & Co. KG that was completed on February 27, 2015, Crescendo Bioscience, Inc. that was completed on February 28, 2014 and Rules-Based Medicine, Inc. that was completed on May 31, 2011. Of this goodwill, $250.5 relates to the Company’s diagnostic segment and $65.6 related to the other segment. The Company assessed goodwill for impairment in accordance with the appropriate guidance (see Note 1) and recorded no impairment of goodwill for the period ended June 30, 2017, 2016 and 2015. Included in the diagnostic segment is $119.1 in goodwill associated with the acquisition of Assurex which is preliminary and subject to adjustment within the measurement period (see Note 2).
The following summarizes changes to the goodwill balance for the years ended June 30, 2017 and 2016:
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
Beginning balance |
|
$ |
195.3 |
|
|
$ |
177.2 |
|
|
Acquisitions (see note 2) |
|
|
116.3 |
|
|
|
17.7 |
|
|
Adjustments to acquisitions (see note 2) |
|
|
3.6 |
|
|
|
0.6 |
|
|
Translation adjustments |
|
|
0.9 |
|
|
|
(0.2 |
) |
|
Ending balance |
|
$ |
316.1 |
|
|
$ |
195.3 |
|
Intangible Assets
Intangible assets primarily consist of amortizable assets of purchased licenses and technologies, developed technology, a laboratory database, trademarks, and customer relationships as well as non-amortizable intangible assets of in-process technologies, research and development. Certain of these intangible assets were recorded as part of the Company’s purchase of Assurex on August, 31, 2016, Sividon on March 31, 2016, Crescendo on February 28, 2014 and Myriad RBM on May 31, 2011. The Company’s developed technology and database acquired have estimated remaining useful lives between14 and 19 years, trademarks acquired have an estimated remaining useful life of approximately 11 years and customer relationships have an estimated remaining useful life of approximately 4 years. The estimated useful life of acquired in-process research and development was also evaluated in conjunction with the annual impairment analysis of intangible assets. The classification of the acquired in-process research and development as an indefinite lived asset was deemed appropriate as the related research and development was not yet complete nor had it been abandoned. The Company concluded there was no impairment of long-lived assets for the years ended June 30, 2017, 2016 and 2015.
63
The following summarizes the amounts reported as intangible assets:
|
|
|
Gross |
|
|
|
|
|
|
|
|
|
|
|
|
|
Carrying |
|
|
Accumulated |
|
|
|
|
|
||
|
At June 30, 2017: |
|
Amount |
|
|
Amortization |
|
|
Net |
|
|||
|
Purchased licenses and technologies |
|
$ |
525.7 |
|
|
$ |
(61.2 |
) |
|
$ |
464.5 |
|
|
Customer relationships |
|
|
4.6 |
|
|
|
(2.8 |
) |
|
|
1.8 |
|
|
Trademarks |
|
|
3.0 |
|
|
|
(0.8 |
) |
|
|
2.2 |
|
|
Total amortizable intangible assets |
|
|
533.3 |
|
|
|
(64.8 |
) |
|
|
468.5 |
|
|
In-process research and development |
|
|
23.1 |
|
|
|
— |
|
|
|
23.1 |
|
|
Total unamortized intangible assets |
|
|
23.1 |
|
|
|
— |
|
|
|
23.1 |
|
|
Total intangible assets |
|
$ |
556.4 |
|
|
$ |
(64.8 |
) |
|
$ |
491.6 |
|
|
|
|
Gross |
|
|
|
|
|
|
|
|
|
|
|
|
|
Carrying |
|
|
Accumulated |
|
|
|
|
|
||
|
At June 30, 2016: |
|
Amount |
|
|
Amortization |
|
|
Net |
|
|||
|
Purchased licenses and technologies |
|
$ |
228.7 |
|
|
$ |
(28.5 |
) |
|
$ |
200.2 |
|
|
Customer relationships |
|
|
4.7 |
|
|
|
(2.4 |
) |
|
|
2.3 |
|
|
Trademarks |
|
|
3.0 |
|
|
|
(0.6 |
) |
|
|
2.4 |
|
|
Total amortizable intangible assets |
|
|
236.4 |
|
|
|
(31.5 |
) |
|
|
204.9 |
|
|
In-process research and development |
|
|
22.6 |
|
|
|
— |
|
|
|
22.6 |
|
|
Total unamortized intangible assets |
|
|
22.6 |
|
|
|
— |
|
|
|
22.6 |
|
|
Total intangible assets |
|
$ |
259.0 |
|
|
$ |
(31.5 |
) |
|
$ |
227.5 |
|
As of June 30, 2017 the weighted average remaining amortization period for purchased licenses and technologies, trademarks, and customer relationships is approximately 14 years.
The Company recorded amortization during the respective periods for these intangible assets as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
Amortization of intangible assets |
|
$ |
33.3 |
|
|
$ |
12.6 |
|
|
$ |
12.7 |
|
Amortization expense of intangible assets is estimated to be $37.2 in 2018, $37.2 in 2019, $36.9 in 2020, $36.9 in 2021 and $36.9 in 2022 and $283.4 thereafter.
|
6. |
COST BASIS INVESTMENT |
In April 2013, the Company acquired approximately 28 shares of Series E preferred stock of RainDance Technologies, Inc. (“RainDance”) of Lexington, Massachusetts, for $5.0. RainDance provides high-throughput picodroplet-based technology that can encapsulate a single molecule, cell or reaction and be digitally analyzed and sorted one at a time. The Series E shares purchased by the Company represented less than 5% of the total shares outstanding of RainDance’s capital stock. Subsequent to the investment the Company evaluated its relationship with RainDance and determined it did not have significant influence over the operations of RainDance. During the year ended June 30, 2017, the Company sold its investment in RainDance Technologies, Inc., which had been recorded under the cost method as an “Other Asset” on the Company’s condensed consolidated balance sheet for $2.6 and recognized a $2.4 impairment.
|
7. |
ACCRUED LIABILITIES |
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
Employee compensation and benefits |
|
$ |
44.4 |
|
|
$ |
37.3 |
|
|
Accrued taxes payable |
|
|
7.1 |
|
|
|
2.8 |
|
|
Other |
|
|
14.1 |
|
|
|
9.4 |
|
|
Total accrued liabilities |
|
$ |
65.6 |
|
|
$ |
49.5 |
|
64
On August 31, 2016, we entered into a Credit Agreement pursuant to which it borrowed term loans in an aggregate principal amount of $200.0 (the “Term Loan”). The Term Loan was to mature on August 31, 2017. There were no scheduled principal payments of the Term Loan prior to its maturity date.
The proceeds of the Term Loan were used to (i) finance the acquisition of Assurex, (ii) refinance certain existing indebtedness of Assurex and its subsidiaries, (iii) pay fees, commissions, transactions costs and expenses incurred in connection with the foregoing, and (iv) for working capital and other general corporate purposes.
On December 23, 2016, we entered into a senior secured revolving credit facility (the “Facility”). A portion of the proceeds of the Facility were used to extinguish in full the obligations under the Term Loan. We recognized a $1.3 loss on the extinguishment reflected which is presented as a component of interest expense on the consolidated statement of operations.
|
9. |
LONG-TERM DEBT |
On December 23, 2016, The Company entered into the Facility by and among Myriad, as borrower, the lenders from time to time party thereto, providing for the Facility in an aggregate principal amount of up to $300.0, which amount shall include $10.0 sublimits, in each case, for swingline loans and letters of credit. Pursuant to the Facility, Myriad borrowed revolving loans in an aggregate principal amount of $205.0 with $0.7 upfront fees and $0.3 debt issuance costs recorded as a debt discount to be amortized over the term of the Facility resulting in current net long-term debt of $204.0. The Facility matures on December 23, 2021. There are no scheduled principal payments of the Facility prior to its maturity date.
The proceeds of the Facility were used (i) to refinance in full the obligations under the Term Loan, (ii) to pay any fees and expenses related thereto, and (iii) for working capital and general corporate purposes.
The Facility contains customary loan terms, interest rates, representations and warranties, affirmative and negative covenants, in each case, subject to customary limitations, exceptions and exclusions. The Credit Agreement also contains certain customary events of default.
Covenants in the Facility, which went into effect during the quarter ending March 31, 2017, impose operating and financial restrictions on the Company. These restrictions may prohibit or place limitations on, among other things, the Company’s ability to incur additional indebtedness, create certain types of liens, mergers or consolidations, and/or change in control transactions. The Facility may also prohibit or place limitations on the Company’s ability to sell assets, pay dividends or provide other distributions to shareholders. The Company must maintain a specified leverage and interest ratios measured as of the end of each quarter as a financial covenant in the Facility. The Company in compliance with all financial covenants at June 30, 2017.
During the year ended June 30, 2017 the Company made $105.0 in principal repayments.
The Facility is secured by a first-lien security interest in substantially all of the assets of Myriad and certain of its domestic subsidiaries and each such domestic subsidiary of Myriad has guaranteed the repayment of the Facility. Amounts outstanding under the Facility were as follows:
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
Long-term debt |
|
$ |
100.0 |
|
|
$ |
- |
|
|
Long-term debt discount |
|
|
(0.9 |
) |
|
|
- |
|
|
Net long-term debt |
|
$ |
99.1 |
|
|
$ |
- |
|
|
10. |
OTHER LONG TERM LIABILITIES |
|
|
|
Years Ended June 30, |
|
|||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
Pension obligation |
|
$ |
5.9 |
|
|
$ |
5.9 |
|
|
Other |
|
|
1.3 |
|
|
|
1.9 |
|
|
Total other long term liabilities |
|
$ |
7.2 |
|
|
$ |
7.8 |
|
65
The Company has two non-contributory defined benefit pension plans for its current and former Clinic employees. The Company has closed participation in the plans to exclude those employees hired after 2002. As of June 30, 2017 the fair value of the plan assets were approximately $0.1 resulting in a net pension liability of $5.9.
|
11. |
PREFERRED AND COMMON STOCKHOLDER’S EQUITY |
The Company is authorized to issue up to 5.0 shares of preferred stock, par value $0.01 per share. There were no preferred shares outstanding at June 30, 2017, 2016 and 2015.
The Company is authorized to issue up to 150.0 shares of common stock, par value $0.01 per share. There were 68.4, 69.1, and 68.9 shares issued and outstanding at June 30, 2017, 2016 and 2015 respectively.
Common shares issued and outstanding
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|
Common stock issued and outstanding at July 1 |
|
|
69.1 |
|
|
|
68.9 |
|
|
|
73.5 |
|
|
Common stock issued upon exercise of options and employee stock plans |
|
|
0.9 |
|
|
|
4.7 |
|
|
|
1.4 |
|
|
Repurchase and retirement of common stock |
|
|
(1.6 |
) |
|
|
(4.5 |
) |
|
|
(6.0 |
) |
|
Common stock issued and outstanding at June 30 |
|
|
68.4 |
|
|
|
69.1 |
|
|
|
68.9 |
|
Stock Repurchase Program
In June 2016, the Company’s Board of Directors authorized an eighth share repurchase program of $200.0 of the Company’s outstanding common stock. The Company plans to repurchase its common stock from time to time or on an accelerated basis through open market transactions or privately negotiated transactions as determined by the Company’s management. The amount and timing of stock repurchases under the program will depend on business and market conditions, stock price, trading restrictions, acquisition activity and other factors. As of June 30, 2017, the Company has $160.7 remaining on its current share repurchase authorization.
The Company uses the par value method of accounting for its stock repurchases. As a result of the stock repurchases, the Company reduced common stock and additional paid-in capital and recorded charges to accumulated deficit. The shares retired, aggregate common stock and additional paid-in capital reductions, and related charges to accumulated deficit for the repurchases for periods ended June 30, 2017, 2016 and 2015 were as follows:
|
|
|
Year ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Shares purchased and retired |
|
|
1.6 |
|
|
|
4.5 |
|
|
|
6.0 |
|
|
Common stock and additional paid-in-capital reductions |
|
$ |
14.6 |
|
|
$ |
41.2 |
|
|
$ |
51.5 |
|
|
Charges to retained earnings |
|
$ |
17.0 |
|
|
$ |
121.4 |
|
|
$ |
159.2 |
|
66
Income tax expense consists of the following:
|
|
|
Year ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
$ |
17.2 |
|
|
$ |
22.2 |
|
|
$ |
53.7 |
|
|
State |
|
|
2.4 |
|
|
|
3.5 |
|
|
|
4.7 |
|
|
Total Current |
|
|
19.6 |
|
|
|
25.7 |
|
|
|
58.4 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
2.0 |
|
|
|
15.4 |
|
|
|
(2.1 |
) |
|
State |
|
|
(1.7 |
) |
|
|
(3.2 |
) |
|
|
8.7 |
|
|
Foreign |
|
|
(1.7 |
) |
|
|
3.5 |
|
|
|
(2.3 |
) |
|
Change in valuation allowance |
|
|
3.1 |
|
|
|
2.2 |
|
|
|
(8.0 |
) |
|
Total Deferred |
|
|
1.7 |
|
|
|
17.9 |
|
|
|
(3.7 |
) |
|
Total income tax expense |
|
$ |
21.3 |
|
|
$ |
43.6 |
|
|
$ |
54.7 |
|
Income (loss) before income taxes consists of the following:
|
|
|
Year ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
45.9 |
|
|
$ |
169.5 |
|
|
$ |
147.0 |
|
|
Foreign |
|
|
(3.0 |
) |
|
|
(0.6 |
) |
|
|
(12.1 |
) |
|
Total |
|
$ |
42.9 |
|
|
$ |
168.9 |
|
|
$ |
134.9 |
|
The differences between income taxes at the statutory federal income tax rate and income taxes reported in the consolidated statements of operations were as follows:
|
|
|
Year ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Federal income tax expense at the statutory rate |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
State income taxes, net of federal benefit |
|
|
1.7 |
|
|
|
2.0 |
|
|
|
1.7 |
|
|
Research and development credits, net of the federal tax on state credits |
|
|
(7.1 |
) |
|
|
(1.3 |
) |
|
|
(2.5 |
) |
|
Uncertain tax positions, net of federal benefit |
|
|
3.8 |
|
|
|
0.6 |
|
|
|
1.2 |
|
|
Uncertain tax benefits statute expired, net of federal benefit |
|
|
(0.9 |
) |
|
|
(4.4 |
) |
|
|
0.0 |
|
|
Incentive stock option and employee stock purchase plan expense |
|
|
1.9 |
|
|
|
(0.3 |
) |
|
|
0.2 |
|
|
Adjustment to deferred tax liability attributable to acquired intangible assets |
|
|
0.0 |
|
|
|
0.0 |
|
|
|
1.6 |
|
|
Foreign rate differential |
|
|
(1.2 |
) |
|
|
0.0 |
|
|
|
1.6 |
|
|
Change in valuation allowance |
|
|
7.1 |
|
|
|
1.2 |
|
|
|
2.6 |
|
|
California basis step-up election, net of related valuation allowance impact |
|
|
0.0 |
|
|
|
0.0 |
|
|
|
(1.2 |
) |
|
Early adoption of ASU 2016-09 |
|
|
7.2 |
|
|
|
(7.5 |
) |
|
|
0.0 |
|
|
Acquisition related transaction costs |
|
|
1.8 |
|
|
|
0.0 |
|
|
|
0.0 |
|
|
Other, net |
|
|
0.4 |
|
|
|
0.5 |
|
|
|
0.4 |
|
|
|
|
|
49.7 |
% |
|
|
25.8 |
% |
|
|
40.6 |
% |
The Company’s effective tax rate for the year ended June 30, 2017 was higher than the years ended June 30, 2016 and 2015. This increase was primarily due a valuation allowance relating to capital losses, the early adoption of ASU 2016-09 regarding stock compensation and an increase in the Company’s liability for unrecognized tax expense.
67
The significant components of the Company’s deferred tax assets and liabilities were comprised of the following at June 30, 2017 and 2016:
|
|
|
Year ended June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
75.5 |
|
|
$ |
54.4 |
|
|
Property, plant and equipment |
|
|
2.1 |
|
|
|
1.8 |
|
|
Accrued vacation |
|
|
3.0 |
|
|
|
2.0 |
|
|
Allowance for doubtful accounts |
|
|
9.7 |
|
|
|
2.4 |
|
|
Stock compensation expense |
|
|
29.9 |
|
|
|
27.0 |
|
|
Research and development credits |
|
|
9.9 |
|
|
|
9.4 |
|
|
Uncertain state tax positions |
|
|
1.4 |
|
|
|
1.1 |
|
|
Other, net |
|
|
2.3 |
|
|
|
0.7 |
|
|
Total gross deferred tax assets |
|
|
133.8 |
|
|
|
98.8 |
|
|
Less valuation allowance |
|
|
(40.5 |
) |
|
|
(35.6 |
) |
|
Total deferred tax assets |
|
|
93.3 |
|
|
|
63.2 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Intangible assets |
|
|
177.7 |
|
|
|
81.1 |
|
|
Total deferred tax liabilities |
|
|
177.7 |
|
|
|
81.1 |
|
|
Net deferred tax liability |
|
|
(84.4 |
) |
|
|
(17.9 |
) |
Due to sustained positive operating performance and the availability of expected future taxable income, the Company concluded that it is more likely than not that the benefits of the majority of its deferred income tax assets will be realized. However, for certain deferred tax assets, a valuation allowance has been established. For the years ended June 30, 2017 and 2016, the Company’s valuation allowance increased by $4.9 and decreased by $2.2, respectively. The net increase of the valuation allowance in the year ended June 30, 2017 consisted of an increase of $1.4 related to capital losses, an increase of $1.9 related to the acquisition of Assurex Health, Inc., an increase of $1.2 related to foreign losses and an increase of $0.4 related to Utah Research Activities Credits. The net increase of the valuation allowance in the year ended June 30, 2016 consisted of an increase of $3.8 related to the adoption of ASU 2016-09 with regard to state excess tax benefits, an increase of $1.9 related to additional state research credit carry-forwards, for which the company concluded it was more likely than not that the benefits of the losses and credits will not be realized. In addition, there was an offsetting decrease in the valuation allowance on foreign net operating losses in the amount of $3.5 which related to a corresponding decrease in the foreign net operating losses.
The Company acquired Sividon Diagnostics GmbH on May 31, 2016 (see Note 2). As part of the purchase accounting for this acquisition a net deferred tax liability of approximately $13.2 was recorded, consisting primarily of intangible assets for which the book basis exceeds the tax basis. The Company Acquired Assurex Health, Inc. on August 31, 2016 (see Note 2). As part of the purchase accounting for the acquisition, a net deferred tax liability of approximately $64.5 was recorded, consisting primarily of intangible assets for which the book basis exceeds the tax basis.
During the fiscal year ended June 30, 2016 the Company elected to early adopt ASU 2015-17, Balance Sheet Classification of Deferred Taxes, and ASU 2016-09, Improvements to Employee Share-Based Payment Accounting, both provisions were adopted prospectively. Prior to the adoption of ASU 2016-09, excess tax benefits from stock based compensation were credited directly to additional paid-in-capital. With the early adoption of ASU 2016-09, all excess tax benefits and tax deficiencies are recognized as income tax expense or benefit in the income statement. The Company realized a tax deficiency of $3.1 in the year ended June 30, 2017 and an excess tax benefit of $12.7 in the year ended June 30, 2016.
68
At June 30, 2017, the Company had the following net operating loss and research credit carryforwards, with their respective expiration periods. Certain carryforwards are subject to the limitations of Section 382 and 383 of the Internal Revenue Code as indicated.
|
|
|
|
|
|
|
Subject to |
|
Expires |
|
|
|
|
|
|
|
Carryforwards |
|
Amount |
|
|
sections 382, 383 |
|
beginning in year |
|
|
Through |
|
|||
|
Federal net operating loss |
|
$ |
160.9 |
|
|
Yes |
|
|
2027 |
|
|
|
2033 |
|
|
Utah net operating loss |
|
|
209.5 |
|
|
No |
|
|
2016 |
|
|
|
2024 |
|
|
Oklahoma net operating loss |
|
|
14.1 |
|
|
Yes |
|
|
2023 |
|
|
|
2033 |
|
|
Foreign net operating losses (various jurisdictions) |
|
|
26.0 |
|
|
No |
|
Various |
|
|
Various |
|
||
|
Federal research credit |
|
|
3.2 |
|
|
Yes |
|
|
2025 |
|
|
|
2032 |
|
|
Utah research credit |
|
|
10.1 |
|
|
No |
|
|
2021 |
|
|
|
2031 |
|
All of the Utah net operating loss carryforwards are ‘excess tax benefits’ as defined by ASC guidance and, if realized in future years, will be recognized as a credit to tax benefit, pursuant to the guidance of ASU 2016-09. The Company’s deferred tax asset for the Utah net operating loss ‘excess tax benefits’ is approximately $6.8 and is offset by a full valuation allowance at June 30, 2017.
Consistent with the indefinite reversal criteria of ASC 740-30-25-17, the Company intends to invest undistributed earnings of its foreign subsidiaries indefinitely. Due to the cumulative losses that have been incurred to date in its foreign operations, the amount of unrecorded deferred liability resulting from the indefinite reversal criteria at June 30, 2017 is $0.
In July 2006, the FASB issued ASC Topic 740 Subtopic 10 Section 05, which clarifies the accounting for uncertainty in tax positions. Accounting guidance requires that the impact of a tax position be recognized in the financial statements if that position is more likely than not of being sustained on audit, based on the technical merits of the position. The Company adopted the guidance on July 1, 2007 and recorded $0 cumulative effect. A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
|
|
|
Year ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Unrecognized tax benefits at the beginning of year |
|
$ |
24.2 |
|
|
$ |
26.3 |
|
|
$ |
24.2 |
|
|
Gross increases - current year tax positions |
|
|
0.7 |
|
|
|
0.6 |
|
|
|
1.1 |
|
|
Gross increases - prior year tax positions |
|
|
0.7 |
|
|
|
5.4 |
|
|
|
1.0 |
|
|
Gross increases - acquisitions |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Gross decreases - prior year tax positions |
|
|
(0.4 |
) |
|
|
(8.1 |
) |
|
|
— |
|
|
Unrecognized tax benefits at end of year |
|
$ |
25.2 |
|
|
$ |
24.2 |
|
|
$ |
26.3 |
|
|
Interest and penalties in year-end balance |
|
$ |
(0.9 |
) |
|
$ |
0.5 |
|
|
$ |
0.6 |
|
Interest and penalties related to uncertain tax positions are included as a component of income tax expense all other interest and penalties are included as a component of other income (expense).
The Company files U.S., foreign and state income tax returns in jurisdictions with various statutes of limitations. The years ended June 30, 2013 through June 30, 2017 remain subject to examination at June 30, 2017. The Company’s income tax returns for the following jurisdictions are currently under examination: California State income tax returns for the years ended June 30, 2013 through 2014; New Jersey State income tax returns for the years ended June 30, 2007 through 2013; and U.S. income tax returns for the years ended June 30, 2014 through 2015. The Company recently was subject to an examination of its payroll reporting by the Internal Revenue Service. The payroll audit concluded during the year ended June 30, 2017 and resulted in a penalty of $0.9, which is included in the Company’s results for that year. Annual tax provisions include amounts considered necessary to pay assessments that may result from examination of prior year tax returns; however, the amount ultimately paid upon resolution of issues may differ materially from the amount accrued. The Company’s foreign income tax returns and all other state tax returns are not currently under examination.
69
The Company maintains a share-based compensation plan, the 2010 Employee, Director and Consultant Equity Incentive Plan, as amended (the “2010 Plan”), that has been approved by the Company’s shareholders. The 2010 Plan allows the Company, under the direction of the Compensation Committee of the Board of Directors, to make grants of stock options, restricted and unrestricted stock awards and other stock-based awards to employees, consultants and directors. On December 1, 2016, the shareholders approved an amendment to the 2010 Plan to add 2.5 to the number of shares of common stock available for grant. As of June 30, 2017, a total of 2.8 shares of common stock are available for issuance under the 2010 Plan. In addition, as of June 30, 2017, the Company may grant up to 2.3 additional shares under the 2010 Plan if options previously granted under the Company’s terminated 2003 Employee, Director and Consultant Option Plan are cancelled or expire in the future without the issuance of shares of common stock by the Company. The exercise price of options granted in 2017, 2016 and 2015 was equivalent to the fair market value of the stock at the date of grant. The number of shares, terms, and vesting periods are determined by the Company’s board of directors or a committee thereof on an option-by-option basis. Options generally vest ratably over service periods of four years. Options granted after December 5, 2012 expire eight years from the date of grant, and options granted prior to that date generally expire ten years from the date of grant. In September 2014, the Company began issuing restricted stock units (“RSUs”) in lieu of stock options. RSUs generally vest ratably over four years on the anniversary date of the grant to all employees. The number of RSUs awarded to certain executive officers may be reduced if certain additional performance metrics are not met. Options and restricted stock units granted to our non-employee directors vest in full upon the earlier of (i) one full year of service on the Board following date of grant or (ii) the date of the next annual meeting of stockholders.
Stock Options
A summary of option activity is as follows for the fiscal years ended June 30:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
|||
|
|
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
||||||
|
|
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
|
of |
|
|
exercise |
|
||||||
|
|
|
shares |
|
|
price |
|
|
shares |
|
|
price |
|
|
shares |
|
|
price |
|
||||||
|
Options outstanding at beginning of year |
|
|
8.2 |
|
|
$ |
24.52 |
|
|
|
12.5 |
|
|
$ |
23.49 |
|
|
|
14.2 |
|
|
$ |
23.30 |
|
|
Options granted |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
Less: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercised |
|
|
(0.1 |
) |
|
$ |
14.81 |
|
|
|
(4.3 |
) |
|
$ |
21.51 |
|
|
|
(1.2 |
) |
|
$ |
20.17 |
|
|
Options canceled or expired |
|
|
(0.1 |
) |
|
$ |
26.55 |
|
|
|
— |
|
|
$ |
— |
|
|
|
(0.5 |
) |
|
$ |
25.98 |
|
|
Options outstanding at end of year |
|
|
8.0 |
|
|
$ |
24.67 |
|
|
|
8.2 |
|
|
$ |
24.52 |
|
|
|
12.5 |
|
|
$ |
23.49 |
|
|
Options exercisable at end of year |
|
|
7.5 |
|
|
$ |
24.55 |
|
|
|
6.7 |
|
|
$ |
24.04 |
|
|
|
9.6 |
|
|
$ |
22.80 |
|
|
Options vested and expected to vest |
|
|
8.0 |
|
|
$ |
24.67 |
|
|
|
8.2 |
|
|
$ |
24.52 |
|
|
|
12.5 |
|
|
$ |
23.49 |
|
|
Weighted average fair value of options granted during the year |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
The following table summarizes information about stock options outstanding at June 30, 2017:
|
|
|
Options outstanding |
|
|
Options exercisable |
|
||||||||||||||
|
|
|
Number |
|
|
Weighted |
|
|
|
|
|
|
Number |
|
|
|
|
|
|||
|
|
|
outstanding |
|
|
average |
|
|
Weighted |
|
|
exercisable |
|
|
Weighted |
|
|||||
|
Range of |
|
at |
|
|
remaining |
|
|
average |
|
|
at |
|
|
average |
|
|||||
|
exercise |
|
June 30, |
|
|
contractual |
|
|
exercise |
|
|
June 30, |
|
|
exercise |
|
|||||
|
prices |
|
2017 |
|
|
life (years) |
|
|
price |
|
|
2017 |
|
|
price |
|
|||||
|
13.28 - 21.66 |
|
|
2.1 |
|
|
|
3.73 |
|
|
$ |
18.66 |
|
|
|
2.1 |
|
|
$ |
18.66 |
|
|
21.73 - 26.49 |
|
|
3.2 |
|
|
|
3.72 |
|
|
|
25.42 |
|
|
|
2.7 |
|
|
|
25.23 |
|
|
27.07 - 30.12 |
|
|
2.2 |
|
|
|
4.39 |
|
|
|
27.81 |
|
|
|
2.2 |
|
|
|
27.81 |
|
|
30.34 - 37.73 |
|
|
0.5 |
|
|
|
2.27 |
|
|
|
30.46 |
|
|
|
0.5 |
|
|
|
30.44 |
|
|
|
|
|
8.0 |
|
|
|
3.81 |
|
|
$ |
24.67 |
|
|
|
7.5 |
|
|
$ |
24.55 |
|
Options exercisable at June 30, 2017 had a weighted average remaining contractual life of 3.8 years.
As of June 30, 2017, there was $1.0 of total unrecognized share-based compensation expense related to stock options that will be recognized over a weighted-average period of 0.2 years.
70
A summary of RSU activity is as follows:
|
|
|
2017 |
|
|
2016 |
|
||||||||||
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
Weighted |
|
||
|
|
|
Number |
|
|
average |
|
|
Number |
|
|
average |
|
||||
|
|
|
of |
|
|
grant date |
|
|
of |
|
|
grant date |
|
||||
|
|
|
shares |
|
|
fair value |
|
|
shares |
|
|
fair value |
|
||||
|
RSUs outstanding at the beginning of year |
|
|
1.4 |
|
|
$ |
38.76 |
|
|
|
1.0 |
|
|
$ |
37.63 |
|
|
RSUs granted |
|
|
1.1 |
|
|
|
21.47 |
|
|
|
0.8 |
|
|
|
40.62 |
|
|
Less: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
RSUs released |
|
|
(0.4 |
) |
|
|
22.20 |
|
|
|
(0.4 |
) |
|
|
39.74 |
|
|
RSUs canceled |
|
|
(0.1 |
) |
|
|
33.00 |
|
|
|
— |
|
|
|
— |
|
|
RSUs outstanding at end of year |
|
|
2.0 |
|
|
$ |
33.02 |
|
|
|
1.4 |
|
|
$ |
38.76 |
|
As of June 30, 2017, there was $31.3 of total unrecognized share-based compensation expense related to RSUs that will be recognized over a weighted-average period of 2.0 years.
Share-based compensation expense recognized and included in the consolidated statements of operations for the fiscal years ended June 30, 2017, 2016 and 2015 were as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cost of molecular diagnostic testing |
|
$ |
0.9 |
|
|
$ |
0.9 |
|
|
$ |
0.9 |
|
|
Cost of pharmaceutical and clinical services |
|
|
0.3 |
|
|
|
0.4 |
|
|
|
0.5 |
|
|
Research and development expense |
|
|
5.8 |
|
|
|
5.4 |
|
|
|
4.3 |
|
|
Selling, general, and administrative expense |
|
|
22.9 |
|
|
|
24.9 |
|
|
|
40.0 |
|
|
Total share-based compensation expense |
|
$ |
29.9 |
|
|
$ |
31.6 |
|
|
$ |
45.7 |
|
In February 2015, the Company and its former Chief Executive Officer entered into a resignation agreement under which the vesting of certain awards were modified such that the specified awards were accelerated. As a result of this award modification the Company recognized approximately $12.8 in share-based compensation expense for year ended June 30, 2015.
The Company has unrecognized share-based compensation cost related to share-based compensation granted under its current plans. The estimated unrecognized share-based compensation cost and related weighted average recognition period, aggregate intrinsic value of options outstanding, aggregate intrinsic value of options that are fully vested and aggregate intrinsic value of RSUs vested and expected to vest is as follows:
|
|
|
As of |
|
|
|
|
|
June 30, 2017 |
|
|
|
Unrecognized share-based compensation cost |
|
$ |
32.3 |
|
|
Aggregate intrinsic value of options outstanding |
|
|
17.4 |
|
|
Aggregate intrinsic value of options fully vested |
|
|
17.4 |
|
|
Aggregate intrinsic value of RSUs outstanding |
|
$ |
52.5 |
|
The total intrinsic value of options exercised during 2017, 2016 and 2015 was as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Total intrinsic value of options exercised |
|
$ |
0.9 |
|
|
$ |
86.1 |
|
|
$ |
20.5 |
|
71
The Company had an Employee Stock Purchase Plan that was approved by shareholders in 1995 (the “1995 Purchase Plan”), and subsequently amended, under which 2.0 shares of common stock had been authorized. As of December 5, 2012, a total of 2.0 shares of common stock had been issued under the 1995 Purchase Plan when it was terminated. On December 5, 2012, following shareholder approval, the Company adopted the 2012 Employee Stock Purchase Plan (the “2012 Purchase Plan”), under which 2.0 shares of common stock have been authorized. Shares are issued under the 2012 Purchase Plan twice yearly at the end of each offering period. At June 30, 2017, a total of 0.7 shares of common stock had been purchased under the 2012 Plan. Shares purchased under and compensation expense associated with the 1995 and 2012 Plans for the years reported are as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Shares purchased under the plans |
|
|
0.5 |
|
|
|
0.2 |
|
|
|
0.2 |
|
|
Plan compensation expense |
|
$ |
2.3 |
|
|
$ |
1.9 |
|
|
$ |
1.8 |
|
On June 1, 2017 there was an amendment to the 2012 Purchase Plan such that the plan is non-compensatory. As of June 30, 2017, there is no unrecognized share-based compensation expense related to the 2012 Purchase Plan.
The fair value of shares issued under the Plan that was in effect for each period reported was calculated using the Black‑Scholes option-pricing model using the following weighted-average assumptions:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Risk-free interest rate |
|
|
0.6% |
|
|
|
0.4% |
|
|
|
0.1% |
|
|
Expected dividend yield |
|
|
0% |
|
|
|
0% |
|
|
|
0% |
|
|
Expected lives (in years) |
|
|
0.5 |
|
|
|
0.5 |
|
|
|
0.5 |
|
|
Expected volatility |
|
|
72% |
|
|
|
31% |
|
|
|
33% |
|
|
14. |
FAIR VALUE MEASUREMENTS |
The fair value of the Company’s financial instruments reflects the amounts that the Company estimates to receive in connection with the sale of an asset or paid in connection with the transfer of a liability in an orderly transaction between market participants at the measurement date (exit price). The fair value of contingent consideration related to the Sividon and Assurex acquisitions as well as the long-term debt were categorized as a level 3 liability, as the measurement amount is based primarily on significant inputs not observable in the market. For more information about the Sividon and Assurex acquisitions, see Note 2 "Acquisitions". The fair value hierarchy prioritizes the use of inputs used in valuation techniques into the following three levels:
Level 1—quoted prices in active markets for identical assets and liabilities.
Level 2— observable inputs other than quoted prices in active markets for identical assets and liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. Some of the Company’s marketable securities primarily utilize broker quotes in a non-active market for valuation of these securities.
Level 3—unobservable inputs.
All of the Company’s financial instruments are valued using quoted prices in active markets or based on other observable inputs. For Level 2 securities, the Company uses a third party pricing service which provides documentation on an ongoing basis that includes, among other things, pricing information with respect to reference data, methodology, inputs summarized by asset class, pricing application and corroborative information. For Level 3 contingent consideration, we reassess the fair value of expected contingent consideration and the corresponding liability each reporting period using the Monte Carlo Method, which is consistent with the initial measurement of the expected earn out liability. This fair value measurement is considered a Level 3 measurement because we estimate projections during the earn out period utilizing various potential pay-out scenarios. Probabilities were applied to each potential scenario and the resulting values were discounted using a rate that considers weighted average cost of capital as well as a specific risk premium associated with the riskiness of the earn out itself, the related projections, and the overall business. The contingent earn out liabilities are classified as a component of long-term and short-term contingent consideration in our consolidated balance sheets. Changes to the earn out liabilities are reflected in other income (expense) in our consolidated statements of operations. Changes to the unobservable inputs could have a material impact on our financial statements.
72
The fair value of our long-term debt, which we consider a Level 3 measurement, is estimated using discounted cash flow analyses, based on the Company’s current estimated incremental borrowing rates for similar borrowing arrangements. The fair value of long-term debt is estimated to be $87.3 at June 30, 2017.
The following tables set forth the fair value of the Company’s financial assets that are re-measured on a regular basis:
|
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
|
June 30, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds (a) |
|
$ |
7.4 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
7.4 |
|
|
Corporate bonds and notes |
|
|
— |
|
|
|
50.4 |
|
|
|
— |
|
|
|
50.4 |
|
|
Municipal bonds |
|
|
— |
|
|
|
36.9 |
|
|
|
— |
|
|
|
36.9 |
|
|
Federal agency issues |
|
|
— |
|
|
|
13.8 |
|
|
|
— |
|
|
|
13.8 |
|
|
US government securities |
|
|
— |
|
|
|
7.2 |
|
|
|
— |
|
|
|
7.2 |
|
|
Contingent consideration |
|
|
— |
|
|
|
— |
|
|
|
(140.5 |
) |
|
|
(140.5 |
) |
|
Total |
|
$ |
7.4 |
|
|
$ |
108.3 |
|
|
$ |
(140.5 |
) |
|
$ |
(24.8 |
) |
|
(a) |
Money market funds are primarily comprised of exchange traded funds and accrued interest |
|
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
|
June 30, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds (a) |
|
$ |
2.4 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
2.4 |
|
|
Corporate bonds and notes |
|
|
— |
|
|
|
51.0 |
|
|
|
— |
|
|
|
51.0 |
|
|
Municipal bonds |
|
|
— |
|
|
|
85.6 |
|
|
|
— |
|
|
|
85.6 |
|
|
Federal agency issues |
|
|
— |
|
|
|
25.5 |
|
|
|
— |
|
|
|
25.5 |
|
|
US government securities |
|
|
— |
|
|
|
8.3 |
|
|
|
— |
|
|
|
8.3 |
|
|
Contingent consideration |
|
|
— |
|
|
|
— |
|
|
$ |
(10.4 |
) |
|
|
(10.4 |
) |
|
Total |
|
$ |
2.4 |
|
|
$ |
170.4 |
|
|
$ |
(10.4 |
) |
|
$ |
162.4 |
|
|
(a) |
Money market funds are primarily comprised of exchange traded funds and accrued interest |
The following table reconciles the change in the fair value of the contingent consideration during the periods presented:
|
|
Carrying Amount |
|
|
|
Balance June 30, 2016 |
$ |
10.4 |
|
|
Purchases (see note 2) |
|
130.0 |
|
|
Adjustments to purchase accounting |
|
0.5 |
|
|
Change in fair value recognized in the statement of operations |
|
(0.8 |
) |
|
Translation adjustments recognized in other comprehensive income |
|
0.4 |
|
|
Balance June 30, 2017 |
$ |
140.5 |
|
|
15. |
COMMITMENT AND CONTINGENCIES |
The Company is subject to various claims and legal proceedings covering matters that arise in the ordinary course of its business activities. As of June 30, 2017, management of the Company believes any liability that may ultimately result from the resolution of these matters will not have a material adverse effect on the Company’s consolidated financial position, operating results, or cash flows.
The Company leases office and laboratory space under five non-cancelable operating leases, with terms that expire between 2022 and 2027 in Salt Lake City, Utah, one cancelable lease for office and laboratory space with a term that expires in 2018 in Munich, Germany, a non-cancelable operating lease for Myriad RBM for office and laboratory space that expires in 2020 in Austin, Texas, and a non-cancelable lease for office and laboratory space that expires in 2017 in Cologne, Germany. The Company also leases office and laboratory space under one non-cancellable operating lease that expires in 2021 in South San Francisco, California for Crescendo. The Company also leases office and laboratory space under two non-cancelable leases that expire between 2018 and 2024 in Mason, Ohio and Toronto, Canada for Assurex. In addition, the Company maintains lease agreements that expire between 2018 and 2022 for administrative offices in
73
Zurich, Switzerland; Paris, France; Madrid, Spain; Milan, Italy; London, UK and Munich, Germany. Furthermore, the Company leases information technology equipment under four non-cancelable leases, with terms that expire in 2018 and 2019.
The following is a summary of the Company’s rental expense for the fiscal years reported:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Rental expense |
|
$ |
15.2 |
|
|
$ |
14.3 |
|
|
$ |
13.8 |
|
Future minimum lease payments under the Company’s current leases as of June 30, 2017 are as follows:
|
Fiscal year ending: |
|
|
|
|
|
2018 |
|
$ |
12.8 |
|
|
2019 |
|
|
12.1 |
|
|
2020 |
|
|
12.4 |
|
|
2021 |
|
|
10.6 |
|
|
2022 |
|
|
8.7 |
|
|
Thereafter |
|
|
32.5 |
|
|
|
|
$ |
89.1 |
|
The Company has a deferred savings plan which qualifies under Section 401(k) of the Internal Revenue Code. Substantially all of the Company’s U.S. employees are covered by the plan. The Company makes matching contributions of 50% of each employee’s contribution with the employer’s contribution not to exceed 4% of the employee’s compensation.
The Company’s recorded contributions to the plan as follows:
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Deferred savings plan Company contributions |
|
$ |
6.6 |
|
|
$ |
5.5 |
|
|
$ |
5.1 |
|
|
17. |
SEGMENT AND RELATED INFORMATION |
The Company’s business units have been changed to align with how its Chief Operating Decision Maker (“CODM”) reviews performance and makes decisions in managing the Company. The business units were historically comprised of three reportable segments: (i) research, (ii) molecular diagnostics and (iii) pharmaceutical and clinical services. Beginning in fiscal 2015 the business units have been aggregated into two reportable segments: (i) diagnostics and (ii) other, which was formerly the research and pharmaceutical and clinical services segments. The diagnostics segment primarily provides testing and collaborative development of testing that is designed to assess an individual’s risk for developing disease later in life, identify a patient’s likelihood of responding to drug therapy and guide a patient’s dosing to ensure optimal treatment, or assess a patient’s risk of disease progression and disease recurrence. The other segment provides testing products and services to the pharmaceutical, biotechnology and medical research industries, research and development, and clinical services for patients, and includes corporate services such as finance, human resources, legal and information technology. Prior periods presented have been recast to conform to the current presentation.
The accounting policies of the segments are the same as those described in the summary of significant accounting policies (Note 1). The Company evaluates segment performance based on income (loss) before interest income and other income and expense.
74
|
|
|
Diagnostics |
|
|
Other |
|
|
Total |
|
|||
|
Year ended June 30, 2017: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
722.1 |
|
|
$ |
49.3 |
|
|
$ |
771.4 |
|
|
Depreciation and amortization |
|
|
42.8 |
|
|
|
5.5 |
|
|
|
48.3 |
|
|
Segment operating income (loss) |
|
|
130.9 |
|
|
|
(81.5 |
) |
|
|
49.4 |
|
|
Year ended June 30, 2016: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
705.7 |
|
|
$ |
48.1 |
|
|
$ |
753.8 |
|
|
Depreciation and amortization |
|
|
21.5 |
|
|
|
5.2 |
|
|
|
26.7 |
|
|
Segment operating income (loss) |
|
|
239.3 |
|
|
|
(72.5 |
) |
|
|
166.8 |
|
|
Year ended June 30, 2015: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
695.5 |
|
|
$ |
27.6 |
|
|
$ |
723.1 |
|
|
Depreciation and amortization |
|
|
20.5 |
|
|
|
4.5 |
|
|
|
25.0 |
|
|
Segment operating income (loss) |
|
|
217.1 |
|
|
|
(82.9 |
) |
|
|
134.2 |
|
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Total operating income for reportable segments |
|
$ |
49.4 |
|
|
$ |
166.8 |
|
|
$ |
134.2 |
|
|
Unallocated amounts: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
1.2 |
|
|
|
0.9 |
|
|
|
0.4 |
|
|
Interest Expense |
|
|
(6.0 |
) |
|
|
(0.3 |
) |
|
|
(0.2 |
) |
|
Change in the fair value of contingent consideration |
|
|
0.8 |
|
|
|
— |
|
|
|
— |
|
|
Other |
|
|
(2.5 |
) |
|
|
1.5 |
|
|
|
0.5 |
|
|
Income from operations before income taxes |
|
|
42.9 |
|
|
|
168.9 |
|
|
|
134.9 |
|
|
Income tax provision |
|
|
21.3 |
|
|
|
43.6 |
|
|
|
54.7 |
|
|
Net income |
|
|
21.6 |
|
|
|
125.3 |
|
|
|
80.2 |
|
|
Net loss attributable to non-controlling interest |
|
|
(0.2 |
) |
|
|
— |
|
|
|
— |
|
|
Net income attributable to Myriad Genetics, Inc. stockholders |
|
$ |
21.8 |
|
|
$ |
125.3 |
|
|
$ |
80.2 |
|
The following table sets forth a comparison of balance sheet assets by operating segment:
|
|
|
June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net equipment, leasehold improvements and property: |
|
|
|
|
|
|
|
|
|
Diagnostics |
|
|
20.2 |
|
|
|
26.3 |
|
|
Other |
|
|
30.9 |
|
|
|
32.0 |
|
|
Total |
|
$ |
51.1 |
|
|
$ |
58.3 |
|
|
Total Assets: |
|
|
|
|
|
|
|
|
|
Diagnostics |
|
|
900.9 |
|
|
|
512.4 |
|
|
Other |
|
|
124.3 |
|
|
|
129.2 |
|
|
Total |
|
$ |
1,025.2 |
|
|
$ |
641.6 |
|
The following table reconciles assets by geographical region:
|
|
|
June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net equipment, leasehold improvements and property: |
|
|
|
|
|
|
|
|
|
United States |
|
|
27.6 |
|
|
|
33.6 |
|
|
Rest of world |
|
|
23.5 |
|
|
|
24.7 |
|
|
Total |
|
$ |
51.1 |
|
|
$ |
58.3 |
|
|
Total Assets: |
|
|
|
|
|
|
|
|
|
United States |
|
|
912.8 |
|
|
|
532.6 |
|
|
Rest of world |
|
|
112.4 |
|
|
|
109.0 |
|
|
Total |
|
$ |
1,025.2 |
|
|
$ |
641.6 |
|
75
The following table reconciles assets by operating segment and geographic region to total assets:
|
|
|
June 30, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Total assets by segment and geographical region |
|
$ |
1,025.2 |
|
|
$ |
641.6 |
|
|
Cash, cash equivalents, and marketable investment securities (1) |
|
|
199.2 |
|
|
|
238.9 |
|
|
Total |
|
$ |
1,224.4 |
|
|
$ |
880.5 |
|
|
(1) |
The Company manages cash, cash equivalents, and marketable investment securities at the consolidated level for all segments |
The majority of the Company’s revenues were derived from the sale of diagnostic tests in the United States. There were no customers that accounted for greater than 10% of revenue in the years ended June 30, 2017, 2016 and 2015.
|
|
|
Years Ended June 30, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash paid during the year for income taxes |
|
$ |
12.3 |
|
|
$ |
32.0 |
|
|
$ |
39.1 |
|
|
Non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair value adjustment on marketable investment securities recorded to stockholders' equity |
|
|
(0.6 |
) |
|
|
0.3 |
|
|
|
— |
|
76
Not applicable.
1. Disclosure Controls and Procedures
We maintain disclosure controls and procedures (Disclosure Controls) within the meaning of Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Our Disclosure Controls are designed to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act, such as this Annual Report on Form 10-K, is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. Our Disclosure Controls are also designed to ensure that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating our Disclosure Controls, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily applied its judgment in evaluating and implementing possible controls and procedures.
As of the end of the period covered by this Annual Report on Form 10-K, we evaluated the effectiveness of the design and operation of the Company’s Disclosure Controls, which was done under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer. Based on the evaluation of our Disclosure Controls, our Chief Executive Officer and Chief Financial Officer have concluded that, as of June 30, 2017, our Disclosure Controls were effective to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
2. Internal Control Over Financial Reporting
a. Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, a company's principal executive and principal financial officers and effected by the company's board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that:
|
|
• |
pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Our management assessed the effectiveness of our internal control over financial reporting as of June 30, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (COSO) in Internal Control—Integrated Framework. Based on our assessment, management believes that, as of June 30, 2017, our internal control over financial reporting is effective based on those criteria.
77
Management has excluded Assurex from its assessment of internal control over financial reporting as of June 30, 2017, because we acquired Assurex in a business combination on August 31, 2016. Assurex is a wholly-owned subsidiary whose total assets and net assets represent 33.8% and 42.8%, respectively, and revenues represent 10.2% with a negative impact on net income as of June 30, 2017.
The effectiveness of Myriad Genetics, Inc.’s internal control over financial reporting as of June 30, 2017, has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report as follows:
b. Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of Myriad Genetics, Inc. and subsidiaries
We have audited Myriad Genetics, Inc. and subsidiaries’ internal control over financial reporting as of June 30, 2017, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Myriad Genetics, Inc. and subsidiaries’ management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
As indicated in the accompanying Management’s Report on Internal Control over Financial Reporting, management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of Assurex Health, Inc., which is included in the fiscal 2017 consolidated financial statements of Myriad Genetics, Inc. and subsidiaries and constituted 33.8% and 42.8% of total and net assets, respectively, as of June 30, 2017 and 10.2% of revenues for the year then ended. Our audit of internal control over financial reporting of Myriad Genetics, Inc. and subsidiaries also did not include an evaluation of the internal control over financial reporting of Assurex Health, Inc.
In our opinion, Myriad Genetics, Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of June 30, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Myriad Genetics, Inc. and subsidiaries as of June 30, 2017 and 2016, and the related consolidated statements of operations, comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended June 30, 2017 of Myriad Genetics, Inc. and subsidiaries and our report dated August 9, 2017 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Salt Lake City, UT
August 9, 2017
78
c. Change in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting, identified in connection with the evaluation of such internal control that occurred during the fourth quarter of our last fiscal year that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
79
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management and Corporate Governance,” “Section 16(a) Beneficial Ownership Reporting Compliance” and “Corporate Code of Conduct and Ethics” in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be held on November 30, 2017.
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Executive Compensation,” “Management and Corporate Governance – Committees of the Board of Directors and Meetings – Compensation Committee Interlocks and Insider Participation,” “Compensation Committee Report” and “Management and Corporate Governance – Board’s Role in the Oversight of Risk Management” in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be held on November 30, 2017.
|
Item 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Executive Compensation - Equity Compensation Plan Information” in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be held on November 30, 2017.
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance – Director Independence” in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be held on November 30, 2017.
The response to this item is incorporated by reference from the discussion responsive thereto in the proposal entitled “Independent Public Accountants” in our Proxy Statement for the 2017 Annual Meeting of the Stockholders to be held on November 30, 2017.
80
|
(a) |
The following documents are included as part of this Annual Report on Form 10-K. |
|
|
1. |
Financial Statements |
See “Index to Consolidated Financial Statements” at Item 8 to this Annual Report on Form 10-K.
|
|
2. |
Financial Statement Schedule |
The following schedule is filed as part of this Annual Report on Form 10-K:
Schedule II - Schedule of Valuation and Qualifying Accounts for the Years Ended June 30, 2017, 2016 and 2015.
Other financial statement schedules have not been included because they are not applicable or the information is included in the financial statements or notes thereto.
|
|
3. |
Exhibits |
The exhibits which are filed with or incorporated by reference into this Annual Report on Form 10-K are set forth in the Exhibit Index beginning on page A-1, which is incorporated herein by reference.
81
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on August 9, 2017.
|
|
|
MYRIAD GENETICS, INC. |
|
|
|
|
|
By: |
|
/s/ Mark C. Capone |
|
|
|
Mark C. Capone |
|
|
|
President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated below and on the dates indicated.
|
Signatures |
|
Title |
|
Date |
||
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Mark C. Capone |
|
President, Chief Executive |
|
August 9, 2017 |
|
|
|
Mark C. Capone |
|
Officer and Director |
|
|
|
|
|
|
|
(principal executive officer) |
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ R. Bryan Riggsbee |
|
Chief Financial Officer |
|
August 9, 2017 |
|
|
|
R. Bryan Riggsbee |
|
(principal financial and accounting officer) |
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ John T. Henderson |
|
Chairman of the Board |
|
August 9, 2017 |
|
|
|
John T. Henderson, M.D. |
|
|
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Walter Gilbert |
|
Vice Chairman of the Board |
|
August 9, 2017 |
|
|
|
Walter Gilbert, Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Lawrence C. Best |
|
Director |
|
August 9, 2017 |
|
|
|
Lawrence C. Best |
|
|
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Heiner Dreismann |
|
Director |
|
August 9, 2017 |
|
|
|
Heiner Dreismann, Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Dennis Langer |
|
Director |
|
August 9, 2017 |
|
|
|
Dennis Langer, M.D., J.D. |
|
|
|
|
|
|
|
|
|
|
|
|
|
By: |
|
/s/ S. Louise Phanstiel |
|
Director |
|
August 9, 2017 |
|
|
|
S. Louise Phanstiel |
|
|
|
|
82
MYRIAD GENETICS, INC.
Schedule of Valuation and Qualifying Accounts
Years Ended June 30, 2017, 2016 and 2015
(In millions)
|
|
|
Balance at Beginning of Period |
|
|
Addition Charged to Cost and Expenses |
|
|
Net Deductions and Other (1) |
|
|
Balance at End of Period |
|
||||
|
Allowance for doubtful accounts: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended June 30, 2017 |
|
$ |
6.8 |
|
|
$ |
37.3 |
|
|
$ |
(35.9 |
) |
|
$ |
8.2 |
|
|
Year ended June 30, 2016 |
|
$ |
7.6 |
|
|
$ |
33.3 |
|
|
$ |
(34.1 |
) |
|
$ |
6.8 |
|
|
Year ended June 30, 2015 |
|
$ |
9.0 |
|
|
$ |
31.5 |
|
|
$ |
(32.9 |
) |
|
$ |
7.6 |
|
|
(1) |
Primarily represents the write-off of accounts receivables net of recoveries. |
See report of independent registered public accounting firm.
|
Exhibit Number |
|
|
Exhibit Description |
|
Filed with this Report |
|
Incorporated by Reference herein from Form or Schedule |
|
Filing Date |
|
SEC File/ Registration Number |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
|
Restated Certificate of Incorporation, as amended |
|
|
|
10-K (Exhibit 3.1) |
|
08/15/11 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
|
Restated By-Laws |
|
|
|
8-K (Exhibit 3.1) |
|
09/24/14 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
|
Specimen common stock certificate |
|
|
|
10-K (Exhibit 4.1) |
|
08/15/11 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lease Agreements |
|||||||||||
|
|
|||||||||||
|
10.1 |
.1 |
|
Lease Agreement, dated October 12, 1995, between the Registrant and Boyer Research Park Associates V, by its general partner, the Boyer Company |
|
|
|
10-Q (Exhibit 10.2) |
|
11/08/96 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Amendment to Phase I Lease Agreement, dated February 3, 2016, between the Registrant and HCPI/UTAHII, LLC. |
|
|
|
10-Q (Exhibit 10.1) |
|
05/04/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2 |
.1 |
|
Lease Agreement-Research Park Building Phase II, dated March 6, 1998, between the Registrant and Research Park Associated VI, by its general partner, the Boyer Company, L.C. |
|
|
|
10-K (Exhibit 10.44) |
|
09/24/98 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Amendment to Phase II Lease Agreement, dated February 3, 2016, between Myriad Genetics, Inc. and HCPI/UTAH II, LLC. |
|
|
|
10-Q (Exhibit 10.2) |
|
05/04/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
.1 |
|
Lease Agreement, dated March 31, 2001, between the Registrant and Boyer Research Park Associates VI, by its general partner, The Boyer Company, L.C. |
|
|
|
10-Q (Exhibit 10.1) |
|
05/15/01 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Amendment to Phase III Lease Agreement, dated February 3, 2016, between Myriad Genetics, Inc. and HCPI/UTAH II, LLC. |
|
|
|
10-Q (Exhibit 10.3) |
|
05/04/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4 |
.1 |
|
Lease Agreement, effective as of May 31, 2005, dated June 29, 2005, between the Registrant and Boyer Research Park Associates VIII, by its general partner, The Boyer Company, L.C. |
|
|
|
8-K (Exhibit 99.1) |
|
07/05/05 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Letter of Understanding regarding Lease, dated June 29, 2005, between the Registrant and Boyer Research Park Associates VIII, by its general partner, The Boyer Company, L.C. |
|
|
|
8-K (Exhibit 99.2) |
|
07/05/05 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.3 |
|
Amendment to Phase IV Lease Agreement, dated February 16, 2007, between Myriad Genetics, Inc. and Boyer Research Park Associates VIII, L.C.. |
|
|
|
10-Q (Exhibit 10.4) |
|
05/04/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5 |
.1 |
|
Lease Agreement, dated March 11, 2008, between the Registrant and Boyer Research Park Associates IX, by its general partner, The Boyer Company, L.C. |
|
|
|
10-K (Exhibit 10.32) |
|
08/28/08 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Amendment to Lease Agreement, dated February 12, 2010 between the Registrant and Boyer Research Park Associates IX, L.C.. |
|
|
|
10-Q (Exhibit 10.4) |
|
05/05/10 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit Number |
|
|
Exhibit Description |
|
Filed with this Report |
|
Incorporated by Reference herein from Form or Schedule |
|
Filing Date |
|
SEC File/ Registration Number |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
|
Executive Retention Agreement, dated September 29, 2015, between the Registrant and Richard M. Marsh+ |
|
|
|
8-K (Exhibit 10.1) |
|
10/02/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
|
Executive Retention Agreement, dated September 29, 2015, between the Registrant and Jerry S. Lanchbury, Ph.D.+ |
|
|
|
8-K (Exhibit 10.1) |
|
10/02/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
.1 |
|
Form of Executive Retention Agreement+@ |
|
|
|
10-Q (Exhibit 10.1) |
|
05/05/10 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Form of Amendment to Form of Executive Retention Agreement+@ |
|
|
|
10-Q (Exhibit 10.2) |
|
05/05/10 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.3 |
|
Form of Executive Retention Agreement, as amended.+@ |
|
|
|
10-Q (Exhibit 10.1) |
|
11/04/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
|
Executive Retention Agreement, dated September 29, 2015, between the Registrant and Mark. C. Capone+ |
|
|
|
8-K (Exhibit 10.1) |
|
10/02/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10 |
|
|
Executive Retention Agreement between Myriad Genetics Inc. and R. Bryan Riggsbee dated September 29, 2015+ |
|
|
|
8-K (Exhibit 10.1) |
|
10/02/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11 |
|
|
Compensatory Arrangements of Certain Officers+ |
|
|
|
8-K |
|
06/02/15 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12 |
|
|
Non-Employee Director Compensation Policy+ |
|
|
|
10-K (Exhibit 10.15) |
|
08/10/17 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
|
Form of director and executive officer indemnification agreement+ |
|
|
|
10-K (Exhibit 10.34) |
|
08/25/09 |
|
000-26642
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
|
Indemnification Agreement by and between the Registrant and Virginia C. Drosos, dated September 26, 2016+ |
|
|
|
10-Q (Exhibit 10.3) |
|
11/02/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15 |
|
|
Executive Retention Agreement by and between the Registrant and Virginia C. Drosos, dated September 26, 2016+ |
|
|
|
10-Q (Exhibit 10.4) |
|
11/02/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity Compensation Plans |
|||||||||||
|
|
|||||||||||
|
10.16 |
.1 |
|
2003 Employee, Director and Consultant Stock Option Plan, as amended (the “2003 Plan”)+ |
|
|
|
10-Q (Exhibit 10.1) |
|
02/3/10 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Form of Incentive Stock Option Agreement under the 2003 Plan+ |
|
|
|
10-Q (Exhibit 10.7) |
|
11/01/07 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.3 |
|
Form of Non-Qualified Stock Option Agreement under the 2003 Plan+ |
|
|
|
10-Q (Exhibit 10.8) |
|
11/01/07 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit Number |
|
|
Exhibit Description |
|
Filed with this Report |
|
Incorporated by Reference herein from Form or Schedule |
|
Filing Date |
|
SEC File/ Registration Number |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.1 |
|
Myriad Genetics, Inc. 2010 Employee, Director and Consultant Equity Incentive Plan, as amended (the “2010 Plan”)+ |
|
|
|
8-K (Exhibit 10.1) |
|
12/06/13 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.2 |
|
Form of Stock Option Agreement under the 2010 Equity Incentive Plan+ |
|
|
|
10-Q (Exhibit 10.3) |
|
02/01/11 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.3 |
|
Form of Director Stock Option Agreement under the 2010 Equity Incentive Plan+ |
|
|
|
10-Q (Exhibit 10.4) |
|
02/01/11 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.4 |
|
Form of Restricted Stock Unit Agreement for Executive Officers under the 2010 Plan+ |
|
|
|
10-Q (Exhibit 10.1) |
|
11/05/14 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.5 |
|
Form of Restricted Stock Unit Agreement for Directors under the 2010 Plan+ |
|
|
|
10-Q (Exhibit 10.2) |
|
11/05/14 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
.6 |
|
Myriad Genetics, Inc. 2010 Employee, Director and Consultant Equity Incentive Plan, as amended+ |
|
|
|
8-K (Exhibit 10.1) |
|
12/02/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18 |
|
|
2012 Employee Stock Purchase Plan+ |
|
|
|
8-K (Exhibit 10.2) |
|
12/07/12 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
|
2013 Executive Incentive Plan+ |
|
|
|
8-K (Exhibit 10.3) |
|
12/07/12 |
|
000-26642 |
|
Credit Agreement |
|||||||||||
|
|
|||||||||||
|
10.20 |
|
|
Credit Agreement, dated December 23, 2016, among the Registrant and the lenders from time to time party thereto.
|
|
|
|
10-Q (Exhibit 10.1) |
|
02/08/17 |
|
000-26642 |
|
Merger Agreements |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
|
Agreement and Plan of Merger among the Registrant, Myriad Merger Sub, Inc., Assurex Health, Inc. and Fortis Advisors LLC, dated as of August 3, 2016. |
|
|
|
10-Q (Exhibit 10.1) |
|
11/02/16 |
|
000-26642 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other |
|||||||||||
|
|
|||||||||||
|
21.1 |
|
|
List of Subsidiaries of the Registrant |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
|
Consent of Independent Registered Public Accounting Firm (Ernst & Young LLP) |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
|
Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
|
Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32 |
|
|
Certification pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit Number |
|
|
Exhibit Description |
|
Filed with this Report |
|
Incorporated by Reference herein from Form or Schedule |
|
Filing Date |
|
SEC File/ Registration Number |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following materials from Myriad Genetics, Inc.’s Annual Report on Form 10-K for the fiscal year ended June 30, 2017, formatted in XBRL (Xtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Comprehensive Income, (iii) Consolidated Statements of Stockholders’ Equity, (iv) Consolidated Statements of Cash Flows, and (v) Notes to Consolidated Financial Statements. |
|
X |
|
|
|
|
|
|
|
(+) |
Management contract or compensatory plan arrangement. |
|
(@) |
The agreements with all executives are identical except for the executive who is a party to the agreement and the date of execution, which are listed at the end of the exhibit |