Attached files
UNITED
STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): July 20, 2017
NeuroOne
Medical Technologies Corporation
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation) |
000-54716 (Commission File |
27-0863354 (IRS Employer Identification No.) |
10006
Liatris Lane, Eden Prairie, MN 55347
(Address of principal executive offices, including zip code)
952-237-7412
(Registrant's telephone number, including area code)
Original
Source Entertainment, Inc.
24 Turnberry Dr.
Williamsville, New York 14221
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company ¨
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
TABLE OF CONTENTS
| i |
| Item 5.03 Amendments to Certificate of Incorporation or Bylaws; Change in Fiscal Year | 104 |
| Item 5.06 Change in Shell Company Status | 104 |
| Item 9.01 Financial Statements and Exhibits | 104 |
| ii |
Upon the completion of the transactions contemplated by the Agreement and Plan of Merger and Reorganization, or the Merger Agreement, NeuroOne Medical Technologies Corporation became the Parent Company of NeuroOne, Inc., as more fully described below (the “Acquisition”).
We were originally incorporated as Original Source Entertainment, Inc. under the laws of the State of Nevada on August 20, 2009. We were originally formed to license songs to the television and movie industry. From our inception and prior to the Acquisition, our operations have been primarily limited to organizational, start-up, and capital formation activities.
As previously disclosed, prior to the Closing of the Acquisition, we completed a series of steps contemplated by a Plan of Conversion pursuant to which we, among other things, changed our name to NeuroOne Medical Technologies Corporation, increased our authorized number of shares of common stock from 45,000,000 to 100,000,000, increased our authorized number of shares of preferred stock from 5,000,000 to 10,000,000 and reincorporated in Delaware. Upon the closing of the Acquisition, all of the outstanding capital stock, options and warrants to purchase shares of NeuroOne, Inc. were converted into 6,291,994 shares of our common stock, options to purchase 365,716 shares of our common stock and warrants to purchase shares of our common stock, as a result of which NeuroOne, Inc. became our wholly-owned subsidiary. In connection with the Acquisition, we also assumed the outstanding convertible promissory notes of NeuroOne, Inc., and our largest stockholder pre-Acquisition, Mr. Samad, tendered for cancellation 3,500,000 shares of our common stock held by him.
As a result of the Acquisition, we acquired the business of NeuroOne, Inc., an early-stage medical technology company developing comprehensive neuromodulation cEEG and sEEG monitoring, ablation, and brain stimulation solutions to diagnose and treat patients with epilepsy, Parkinson’s disease, dystonia, essential tremors, and other brain related disorders.
Unless otherwise indicated in this Current Report on Form 8-K, or this Report, all references herein to “we,” “us,” “our Company,” “our,” “NeuroOne,” the “Company,” or the “Registrant” refers to NeuroOne Medical Technologies Corporation and the business of our subsidiary, NeuroOne, Inc., after giving effect to the Acquisition. Unless otherwise indicated in this Report, all references in this Report to our board of directors refer to our board of directors as reconstituted upon the closing of the Acquisition. Our business following the Acquisition consists solely of that of our subsidiary, NeuroOne, Inc.
This Report contains summaries of material terms of various agreements executed in connection with the transactions described herein. The summaries of these agreements are subject to, and are qualified in their entirety by reference to these agreements, which are filed as exhibits hereto and incorporated herein by reference.
CAUTIONARY STATEMENT CONCERNING FORWARD-LOOKING INFORMATION
This Report contains forward-looking statements that involve substantial risks and uncertainties. The forward-looking statements are contained principally in the sections entitled “Risk Factors,” “Management's Discussion and Analysis of Financial Condition and Results of Operations” and “Business,” but are also contained elsewhere in this Report. In some cases, you can identify forward-looking statements by the words “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “objective,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “target,” “seek,” “contemplate,” “continue” and “ongoing,” or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Report, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain. Forward-looking statements include statements about:
| · | our plans to develop and commercialize our cortical strip, grid and depth electrode technology; |
| 3 |
| · | our plans for and our expectations regarding the pre-clinical testing and clinical trials of our cortical strip, grid and depth electrode technology that will be required by the FDA or foreign regulatory bodies; |
| · | the timing and availability of data from pre-clinical tests or clinical trials; |
| · | the timing of our planned regulatory filings; |
| · | the timing of and our ability to obtain and maintain regulatory approval of our cortical strip, grid and depth electrode technology; |
| · | our expectations regarding international opportunities for commercializing our cortical strip, grid and depth electrode technology under development; |
| · | the clinical utility of our cortical strip, grid and depth electrode technology under development; |
| · | our ability to develop our cortical strip, grid and depth electrode technology with the benefits we hope to offer as compared to existing technology, or at all; |
| · | our ability to develop future generations of our cortical strip, grid and depth electrode technology; |
| · | our future development priorities; |
| · | our ability to obtain reimbursement coverage for our cortical strip, grid and depth electrode technology; |
| · | our expectations about the willingness of healthcare providers to recommend our cortical strip, grid and depth electrode technology to people with epilepsy, Parkinson’s disease, essential tremors, and other brain related disorders; |
| · | our future commercialization, marketing and manufacturing capabilities and strategy; |
| · | our ability to comply with applicable regulatory requirements; |
| · | our ability to maintain our intellectual property position; |
| · | our estimates regarding the size of, and future growth in, the market for our technology under development; and |
| · | our estimates regarding our future expenses and needs for additional financing. |
Forward-looking statements are based on management's current expectations, estimates, forecasts and projections about our business and the industry in which we operate, and management's beliefs and assumptions are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. You should refer to the “Risk Factors” section of this Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all.
These forward-looking statements speak only as of the date of this Report. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future. You should, however, review the factors and risks and other information we describe in the reports we will file from time to time with the SEC after the date of this Report.
Item 1.01 Entry into a Material Definitive Agreement.
The information contained in Item 2.01 below relating to the various agreements described therein is incorporated herein by reference.
Item 2.01 Completion of Acquisition or Disposition of Assets.
Merger with NeuroOne, Inc.
On July 20, 2017, we entered into the Merger Agreement with NeuroOne, Inc. and OSOK Acquisition Company to acquire NeuroOne, Inc. The transactions contemplated by the Merger Agreement were consummated on July 20, 2017 and, pursuant to the terms of the Merger Agreement, (i) all outstanding shares of common stock of NeuroOne, Inc., par value $0.0001 per share, or the NeuroOne Shares, were exchanged for shares of our common stock, par value $0.001 per share, or the Company Shares, based on the Exchange Ratio of 17.0103706 Company Shares for every one NeuroOne Share, (ii) all NeuroOne Options were replaced with Company Options based on the Exchange Ratio, with corresponding adjustments to their respective exercise prices, (iii) all NeuroOne Warrants were replaced with Company Warrants and (iv) we assumed the outstanding convertible promissory notes, or the Notes, of NeuroOne, Inc. We refer herein to the transactions described in clauses (i), (ii), (iii) and (iv), collectively, as the Acquisition. Accordingly, we acquired 100% of NeuroOne, Inc. in exchange for the issuance of shares of our common stock and NeuroOne, Inc. became our wholly-owned subsidiary. Also at the Closing, Mr. Samad tendered for cancellation 3,500,000 Company Shares held by him as part of the conditions to Closing. The Merger Agreement is filed as an exhibit to this Report and is incorporated herein by reference.
| 4 |
Issuance and Exchange of Company Shares for NeuroOne Shares
At the Closing, each NeuroOne Share outstanding immediately prior to the Closing was converted into the right to receive 17.0103706 Company Shares, or the Exchange Ratio, with all fractional shares rounded down to the nearest whole share. Accordingly, we issued an aggregate of 6,291,994 Company Shares for all of the then-outstanding NeuroOne Shares.
Issuance and Exchange of Company Options and Company Warrants for NeuroOne Options and NeuroOne Warrants
At the Closing, we issued Company Options in exchange for NeuroOne Options. The Company Options cover a number of Company Shares equal to the product (rounded down to the next whole number of Company Shares) of the number of NeuroOne Shares underlying each NeuroOne Option immediately prior to the Closing multiplied by the Exchange Ratio, and have an exercise price per Company Share equal to the per share exercise price of such NeuroOne Option immediately prior to the Closing divided by the Exchange Ratio. The Company Options continue to vest and become exercisable on the same vesting schedule. We similarly issued Company Warrants in exchange for NeuroOne Warrants. We reserved 365,716 Company Shares for issuance upon the exercise of Company Options, or the Reserved Shares. For a description of the warrants and information about the number of shares of common stock for which they can be exercised, see “Form 10 Information—Management's Discussion And Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Capital Resources”.
The NeuroOne Options had been issued pursuant to the NeuroOne, Inc. 2016 Equity Incentive Plan, or the 2016 Plan. Pursuant to the Merger Agreement, we assumed the 2016 Plan upon the Closing.
Reincorporation and Name Change
Prior to the Acquisition, we completed a series of steps contemplated by a Plan of Conversion pursuant to which we, among other things, changed our name to NeuroOne Medical Technologies Corporation, increased our authorized number of shares of common stock from 45,000,000 to 100,000,000, increased our authorized number of shares of preferred stock from 5,000,000 to 10,000,000 and reincorporated in Delaware. Our Certificate of Incorporation is filed as an exhibit to this Report and is incorporated herein by reference.
Change in Directors and Officers of the Company
In connection with the Acquisition, Amer Samad, formerly our sole director and officer, appointed the person designated by NeuroOne, Inc. to our board of directors, resigned from all officer positions and resigned as a director, subject to the filing by us of a Schedule 14F-1 providing notice of a change in the majority of our board of directors, or the Schedule 14F-1, and waiting the requisite time pursuant to the rules thereunder. Our newly constituted board of directors immediately appointed the officers designated by NeuroOne, Inc. Identification of our directors and officers, including biographical information for each of them, is included elsewhere in the “Management” section of this Report.
| 5 |
Aggregate Beneficial Ownership of our Common Stock After the Acquisition
Prior to the Closing, the former NeuroOne, Inc. stockholders owned no shares of our common stock and there were no material relationships between the management of NeuroOne, Inc. and our management. After the Closing, and after giving effect to the issuance of the Company Shares and the reservation of the Reserved Shares, the number of shares of our common stock issued and outstanding was 7,864,994 and an additional 365,716 shares of common stock were reserved for issuance upon the exercise of options. Following the Acquisition, the former holders of securities in NeuroOne, Inc. own approximately 80.0% of our outstanding common stock and the stockholders owning all of our common stock immediately prior to the Closing (other than Amer Samad, whose shares of common stock were cancelled upon the Closing of the Acquisition) own approximately 20.0% of our outstanding common stock.
The foregoing description is a summary of the material terms of the Acquisition and the terms of the Merger Agreement are not intended to modify or supplement any factual disclosures about us or NeuroOne, Inc. in any public reports filed by us with the Securities and Exchange Commission, or the SEC. The representations, warranties, and covenants contained in the Merger Agreement were made only for purposes of the Merger Agreement as of the specified dates set forth therein, were solely for the benefit of the parties to the Merger Agreement, and are subject to limitations agreed upon by the parties to the Merger Agreement, including being qualified by disclosure schedules. These disclosure schedules contain information that modifies, qualifies and creates exceptions to the representations and warranties set forth in the Merger Agreement. Moreover, certain representations and warranties in the Merger Agreement have been made for the purposes of allocating risk between the parties to the Merger Agreement instead of establishing matters of fact. Accordingly, the representations and warranties in the Merger Agreement may not constitute the actual state of facts about us or NeuroOne, Inc. The representations and warranties set forth in the Merger Agreement may also be subject to a contractual standard of materiality different from the actual state of facts. Moreover, information concerning the subject matter of the representations and warranties may change after the date of the Merger Agreement, which subsequent information may or may not be fully reflected in our public filings with the SEC.
Corporate Overview of NeuroOne Medical Technologies Corporation
We were originally incorporated as Original Source Entertainment, Inc. under the laws of the State of Nevada on August 20, 2009. Prior to the Closing of the Acquisition, we completed a series of steps contemplated by a Plan of Conversion pursuant to which we, among other things, changed our name to NeuroOne Medical Technologies Corporation, increased our authorized number of shares of common stock from 45,000,000 to 100,000,000, increased our authorized number of shares of preferred stock from 5,000,000 to 10,000,000 and reincorporated in Delaware. On July 20, 2017, we closed the Acquisition and acquired NeuroOne, Inc. Immediately following the Closing, the business of NeuroOne, Inc. became our sole focus. Unless otherwise stated or unless the context otherwise requires, the description of our business set forth below is provided on a combined basis, taking into account our wholly-owned subsidiary, NeuroOne, Inc.
Corporate Overview and History of NeuroOne, Inc.
NeuroOne, Inc. was incorporated under the laws of the State of Delaware on October 7, 2016. Its predecessor entity, NeuroOne LLC, or the LLC, was formed on December 13, 2013 and operated as a limited liability company until it was merged with and into NeuroOne, Inc. on October 27, 2016 with NeuroOne, Inc. as the surviving entity (the “Merger”). As a result of the Merger, all of the properties, rights, privileges, powers and franchises of the LLC vested in NeuroOne, Inc., and all debts, liabilities and duties of the LLC became the debts, liabilities and duties of NeuroOne, Inc., except for the WARF License, which was not legally transferred until May 2017. The purpose of the Merger was to change the jurisdiction of incorporation from Minnesota to Delaware, change of ownership of the LLC’s underlying assets and to convert from a limited liability company to a corporation.
We are headquartered in Eden Prairie, Minnesota.
| 6 |
We are a medical technology company focused on the development and commercialization of thin film electrode technology for cEEG and sEEG recording, brain stimulation and ablation solutions for patients suffering from Epilepsy, Parkinson’s Disease, Dystonia, Essential Tremors and other related brain related disorders. Members of our management team have held senior leadership positions at a number of medical technology and biopharmaceutical companies, including Boston Scientific, St. Jude Medical, Stryker Instruments, C.R. Bard, A-Med Systems, Sunshine Heart, Empi, Don-Joy and PMT Corporation.
We are developing our cortical and sheet and depth electrode technology to provide solutions for diagnosis through continuous electroencephalogram (cEEG) recording and stereoelectroencephalography (sEEG) recording and treatment through brain stimulation and ablation, all in one product. A cEEG is a continuous recording of the electrical activity of the brain that identifies the location of irregular brain activity, which information is required for proper treatment. cEEG recording involves an invasive surgical procedure, referred to as a craniotomy. sEEG involves a less invasive procedure whereby doctors place electrodes in targeted brain areas through drilling small holes through the skull. Both methods of seizure diagnosis are used to identify areas of the brain where epileptic seizures originate in order to precisely locate the seizure source for therapeutic treatment if possible.
Deep brain stimulation, or DBS, therapies involve activating or inhibiting the brain with electricity that can be given directly by electrodes on the surface or implanted deeper in the brain via depth electrodes. Introduced in 1987, this procedure involves implanting a power source referred to as a neurostimulator, which sends electrical impulses through implanted depth electrodes, to specific targets in the brain for the treatment of disorders such as Parkinson's disease, essential tremor, dystonia, and chronic pain. Alzheimer’s is another indication evaluating the effects of DBS. Unlike ablative technologies, the effects of DBS are reversible.
RF ablation is a procedure that uses radiofrequency under the electrode contacts that is directed to the site of the brain tissue that is targeted for removal. The process involves delivering energy to the contacts, thereby heating them and destroying the brain tissue. The ablation does not remove the tissue. Rather, it is left in place and typically scar tissue forms in the place where the ablation occurs. This procedure is also known as brain lesioning as it causes irreversible lesions.
Our cortical sheet electrode and depth electrode technology has been tested over the years at both Wisconsin Alumni Research Foundation, or WARF, the owners of our licensed patents, and Mayo Clinic located in Rochester Minnesota, in both pre-clinical models as well as through an IRB approval at Mayo Clinic for clinical research. These pre-clinical tests have demonstrated that the technology is capable of recording, ablation and acute stimulation, although our technology remains in product development for all of the therapeutic modalities. In addition, a great deal of bench top, pre-clinical and clinical testing remains for all therapeutic modalities as well as for the diagnostic technologies for our technology. Once the research and development has been completed, these devices will have to undergo both acute and long term testing to ensure the product’s long term viability. Bench top, electrical, sterility, aging and biocompability testing are some of the tests that will have to be conducted. After that, we expect that we will have to conduct a clinical trial for long term stimulation to achieve regulatory approval in any jurisdiction. The size and endpoints of these trials will require additional dialogue with U.S. Food and Drug Administration, or FDA, and other regulatory bodies in any foreign jurisdiction in which we seek to commercialize our technology.
Our Market Opportunity
Epilepsy Market
We expect to initially target the diagnosis and treatment of epilepsy. Epilepsy can be caused by a variety of conditions that affect a person’s brain, some of which are: stroke, brain tumor, traumatic brain injury and central nervous system infections. According to the Centers for Disease Control and Prevention, or CDC, and Citizens United for Research in Epilepsy, or CURE, there are approximately 3 million patients annually suffering with epilepsy in the U.S., with an additional 200,000 diagnosed every year. They also estimate that epilepsy costs the United States $15.5 billion per year. We believe the European market is similar. Approximately 720,000 of these patients are not receptive to pharmaceutical treatment and therefore are appropriate for surgical treatment of this disorder. In addition to poor quality of life, epilepsy also is associated with fairly high mortality rates, especially in children. CURE reported that Sudden Unexpected Death in Epilepsy, or SUDEP, accounts for 34% of all deaths in children. Such deaths have increased by close to 100% from 2005 to 2015 according to the CDC. Despite the large market opportunity, it is estimated that there are only 16,000 craniotomies performed for epilepsy cases each year in the United States with 18,000 performed in Europe.1 These numbers represent an underpenetrated market due to the invasiveness of a full craniotomy required just to perform the diagnostic procedure. After the diagnostic procedure, a second therapeutic procedure is required and at times even a third surgery if the seizures persist. We believe patients are unwilling to proceed due to the long diagnostic times (1-4 weeks in the hospital with a craniotomy), infection rates and 50% rate of success in the diagnosis and treatment of the disorder. As detailed above, after the diagnosis is completed, if successful, the patient must undergo an additional procedure to have the affected area of brain tissue removed. The average cost for the diagnostic technology per procedure is $10,000, with ablation devices costing $15,000 and brain stimulation devices costing $25,000 to $30,000. We believe our technology, once developed, will offer an all in one solution with diagnostic and therapeutic capabilities.
1 American Association of Neurological Surgeons (AANS) National Neurosurgical Procedural Statistics 2012.
| 7 |
We believe that many leading neurologists believe that the limits of today’s current technologies are the reason the exact affected area of the brain causing epileptic seizures is not determinable. We expect our technology under development, which has been developed to date by physicians at WARF and Mayo Clinic, will provide a number of significant advantages over the current technologies, including the following:
| · | Our proprietary thin film technology under development has a smaller footprint with many more electrodes. |
| · | We expect our technology eventually will be able to be implanted using a minimally invasive procedure utilizing a dime sized burr hole versus a full craniotomy required to implant the current technology. Our physician advisors are providing critical support in this endeavor. |
| · | Limited clinical testing to date by Mayo Clinic suggests that our proprietary thin film technology under development can detect single neuron brain activity. This could allow for more rapid detection of irregular brain activity versus an average of two and a half weeks with the currently available technology, during which time the patient remains hospitalized. In limited clinical testing, Doctors at Mayo Clinic have documented pre-seizure activity (micro-seizures) during their clinical research with their patients using this technology. |
| · | We expect our technology can ablate through the electrodes as well as perform brain stimulation, allowing for diagnosis and treatment through the same product and in the same procedure. |
Parkinson’s Disease
The Parkinson’s Disease Foundation estimates that as many as one million patients live with Parkinson’s disease with an additional 60,000 patients diagnosed per year. Over 10,000,000 patients worldwide are living with Parkinson’s disease. There have not been any drugs introduced that have been effective at treating Parkinson’s disease. The average onset is over 60 years old but some people have been diagnosed as young as 40 years old. Parkinson’s is a disorder of the central nervous system caused by loss of brain cells throughout various regions of the brain. It is attributed to the loss of dopamine production in the brain, a messenger in the brain that allows for movement and coordination. There are no objective tests to diagnose Parkinson’s disease, and misdiagnosis rates are still very high. Doctors look to find two or more signs to make a diagnosis, including balance problems, rigidity and tremors that occur during rest. In 2011, the FDA approved the first imaging device called a DaTscan that can capture images of the dopamine system in the brain. By itself, these scans cannot diagnose Parkinson’s but can help confirm a doctor’s diagnosis. Parkinson’s disease is typically not fatal; however, complications caused by the symptoms of Parkinson’s, such as difficulty swallowing causing food to travel to the lungs resulting in pulmonary issues or falls related to loss of balance, can be fatal.
| 8 |
Today’s primary treatment for Parkinson’s disease involves medications that have not proven to resolve symptoms but rather ease symptoms. Years ago, surgical procedures such as thalamotomy and pallidotomy targeted certain parts of the brain and involved destroying the tissue. More recently, these procedures have been replaced with DBS. A doctor evaluates the patient by reviewing the patient’s symptoms and medications taken and administering detailed memory, thinking and imaging tests to determine if they are appropriate for DBS. According to the Michael J. Fox Parkinson’s Disease Research Foundation website, patients that seem to do best with DBS are those that have had the disease for at least four years and have benefited from taking medications prescribed to control the disease. In addition, DBS seems to help with reducing the issues with motor functions such as tremors, stiffness and slowness but not for balance issues. Doctors are evaluating treatment to other parts of the brain in an effort to address more symptoms to treat walking or balance issues. In addition, research is being conducted to provide stimulation when the symptoms return as opposed to all of the time. We expect our technology under development will improve doctors’ ability to diagnose and treat Parkinson’s assuming our technology has the ability to detect single neuron activity. According to Mayo Clinic, detecting brain activity down to a sub-millimeter scale and detecting “microseizure activity” may allow for detection and thus prevention of a seizure before it occurs.
Essential Tremors
Essential tremors are thought to be due to electrical irregularities in the brain that send abnormal signals to the muscles. It is a progressive condition that worsens over time and is linked to genetic disorders that typically appear over 40 years old. Essential tremors usually occur alone and without any other neurological symptoms or signs. The tremors usually occur when the hands are raised and primarily affect the hands. Muscles in the trunk, face and neck may also experience symptoms. Sometimes misdiagnosed as Parkinson’s disease, essential tremors are an involuntary rhythmic shaking of the hands that is not present at rest. It is apparent during activities such as drinking, writing and eating. Symptoms can worsen due to stress, anxiety, smoking, caffeine, fatigue, etc. Genetics Home Reference estimates that as many as ten million people in the United States are affected by the disease. Treatments for the disease include medical therapy, weighting the limbs and DBS. Patients need to eliminate any medications they are taking that cause tremors as this can exacerbate the symptoms. For some patients, using wrist weights may ease symptoms allowing the patient to function. Other patients may also use relaxation techniques as stress can increase symptoms. Medical therapy is also used to treat patient’s symptoms. Primidone is typically the first drug prescribed as it has had success in some situations for epilepsy. Botox is also used at times to control head tremors. When these fail, surgery is the next alternative. A surgical procedure used years ago created lesions in the ventral intermediate thalamus and was highly successful with treating essential tremors but is no longer commonly used due to increased risk of developing speech problems. The latest therapy is DBS, which, unlike other therapies, is reversible and programmable, helping to adjust the settings to maximize patient benefit. Similar to Parkinson’s disease, the ability to detect this irregular brain activity before it causes a tremor is highly desirable. We expect our technology will detect brain activity to a single neuron, which we believe would be highly desirable by both physicians diagnosing and treating patients with essential tremors.
Dystonia
Dystonia is a neurological condition recognized as a motion disorder that involves over activity of a variety of different muscles simultaneously that work against each other. It presents itself in a variety of symptoms but typically involves repetitive, patterned and often twisting involuntary muscle contractions resembling tremors. According to the Dystonia Medical Research Foundation, over 300,000 people are affected in the United States and Canada alone. Dystonia is the third most common problem seen in movement disorder clinics. Because it has many different manifestations, it is often misdiagnosed. In addition, similar to Parkinson’s Disease, there are no specific tests that can positively diagnose dystonia. A doctor typically will evaluate patient and family history, potentially do genetic testing, EEG testing, blood and urine tests. There are also many treatment options for patients but depend on the type of dystonia. Botox and certain medications may be helpful or DBS may be used. As was described in previous sections, if our technology under development is able to detect single neuron activity as we expect it will be, our technology could be helpful in preventing or even minimizing these involuntary muscle contractions.
Limitations of Currently Available Therapies
There are a limited number of currently available products for diagnosis and treatment for people with neurological disorders such as epilepsy. Although the currently available systems provide diagnosis and treatment for patients, they have certain inherent limitations and shortcomings that we believe limit their use and validate the need for improved technology in the market. These limitations include:
| · | Lengthy diagnostic times: Patients spend one to four weeks in the hospital waiting to have seizures that will allow doctors to determine where the seizures are occurring. |
| 9 |
| · | Inability of diagnostic technologies to identify seizure location or micro seizures: Current technology does not have the ability to detect brain activity down to a single neuron, or what has been referred to as micro seizure activity. In addition, the spacing requirements between electrodes increase the potential for missing data that may be critical in the removal of brain tissue causing the irregular activity. Micro seizure activity could be a major predictor of where a future seizure will occur. |
| · | Need to perform a full craniotomy (invasiveness): Currently available cortical electrode technology is placed through a craniotomy, which requires removing the top part of the cranium and is a very painful and invasive procedure. Procedural times for a craniotomy range from a minimum of 4 to 8 hours. A variety of complications can occur when a full craniotomy is performed, including but not limited to: stroke, bleeding, infection, seizures, swelling of the brain (which may require a second craniotomy), nerve damage, which may cause muscle paralysis or weakness, cerebrospinal fluid (CSF) leak, which may require repair, loss of mental functions and permanent brain damage with associated disabilities. The invasiveness, procedural times and possible surgical complications have limited the growth of surgical treatment of epilepsy. |
| · | Multiple technologies required for diagnosis and treatment: Currently, a patient undergoes a craniotomy for implantation of diagnostic film technologies. The patient then waits in the hospital for one to four weeks waiting to have seizures that will allow doctors to pinpoint where the seizures are occurring in the brain. After this is complete, a patient has to undergo another lengthy procedure to have the brain tissue removed or undergo permanent implantation of depth electrodes for chronic stimulation. There is a need for an all in one technology that can potentially allow for diagnosis and treatment concurrently and potentially offer real time treatment without the need for surgery. |
Our Solution
As a result of the inherent limitations and inconvenience of existing systems, we believe that there is a significant unmet need among people with neurological disorders for cortical strip, grid and depth electrodes that provide diagnostic capabilities through cEEG and sEEG recording in addition to therapeutic modalities, such as brain stimulation and ablation, offered as an all in one product. In comparison to currently available technologies, we are currently developing our strip, grid and depth electrodes with the goal of providing the following expected advantages:
| · | Reduced time for diagnosis: If we are successful in identifying brain activity more quickly, in offering a minimally invasive procedure and developing an all in one solution, we expect our technology will reduce overall procedural times. While our pre-clinical and clinical experience to date is very limited, our cortical grid technology under development has, in some cases, demonstrated the ability to provide hi fidelity recordings that have allowed physicians to identify the affected brain tissue causing seizures in hours versus weeks. This represents the potential for meaningful cost savings for hospitals and patients and improved quality of life for patients. |
| · | Improved accuracy of diagnostic technologies: Because we believe our thin film technology will be capable of recording at higher fidelity than current technologies, we believe our technology may be able to more precisely determine the brain tissue causing seizures. In the limited clinical tests performed by Mayo Clinic with five patients to date, our technology under development has identified what clinicians refer to as pre-seizure activity (made possible by the ability to detect brain activity down to a single neuron and populations of neurons). We believe our technology under development may be able to improve outcomes compared to using other therapeutic technologies regardless of whether we are able to offer an all in one diagnostic and therapeutic solution. |
| 10 |
| · | Possibility to implant via minimally invasive procedure with fewer post-procedure complications: We are currently developing an approach to deliver the cortical electrodes, including minimizing the invasiveness of the procedure. We expect that patients who have qualified for this therapy will be more accepting of a minimally-invasive procedure. Such a procedure would potentially reduce the patient’s pain, bleeding and other adverse events associated with a full craniotomy. Our technology is expected to also have fewer wires exiting the patient’s head, which can also reduce the potential for infections. Furthermore, the material we currently use in our cortical electrodes has shown in pre-clinical evaluations to cause less inflammation than current electrode substrates as it appears more compatible with brain tissue. As discussed under “Strategy” below, our technology under development, if approved, will be implanted via a full craniotomy until such time, if ever, as we are able to develop our minimally invasive procedure. |
| · | All-in-one diagnostic and therapeutic technology solution: Due to the expected high fidelity recording capabilities of our technology under development, we have received feedback from physicians that they will attempt to perform the diagnosis and treatment in a single procedure, thereby eliminating the need for a second surgical procedure, reducing the likelihood of patient infection and minimizing the diagnostic, procedural and hospital costs. As discussed in under “Strategy” below, our initial product offering will offer diagnostic-only capabilities while we advance the development of our all in one approach. |
Our Strategy
Our goal is to be the global leader in cEEG and sEEG recording, deep brain stimulation and ablation, owning the procedure from diagnosis through treatment. The key elements of our strategy include:
| · | Introduce cortical strip and grid electrodes for the diagnosis of epilepsy in U.S. – In the first quarter of 2018, we intend to complete the development, testing and 510(k) device submission to the FDA for our cortical and strip electrodes for temporary (less than 30 day) use with recording, monitoring, and acute stimulation equipment for the recording, monitoring and stimulation of electrical signals on the surface of the brain. Our initial product offering will be placed through traditional surgical means involving a craniotomy until such time, if any, that we launch our minimally invasive procedure. We believe, due to physician feedback, that our technology under development would represent a major improvement over existing devices for the diagnosis of epilepsy. We are initially targeting epilepsy as we believe this is a clinical area of great need and a market that is underpenetrated with the fastest path to commercialization. We believe the largest and quickest-to-market geography for our cortical strip and grid technology under development is in the United States for a number of reasons, including the following: (i) many industry sources believe there is a large underserved U.S. market, (ii) healthy procedural reimbursement for centers and physicians, (ii) robust average selling prices, (iii) physician enthusiasm for our technology under development and (iv) that we may seek additional intellectual property protection and will be required to seek additional regulatory approvals to commercialize outside the U.S. We expect to hire direct experienced sales representatives to market our technology, if approved, in the U.S. |
| · | Evaluate international opportunities and initiate approval processes in targeted geographies – We are currently considering international opportunities to market our technology under development. We believe there is a market for our technology in Europe, but we are evaluating epilepsy surgery rates, reimbursement and growth opportunities, the regulatory pathway and intellectual property rights in each jurisdiction. Once we have compiled all this data, we will determine whether to seek to commercialize in any foreign jurisdictions, for which we would seek additional intellectual property protection and be required to obtain additional regulatory approvals. |
| · | Launch depth electrodes for sEEG recording – Given the reluctance of patients to undergo epilepsy surgery due to its invasiveness, a number of epilepsy centers have adopted the use of depth electrodes, which are placed by drilling small holes into the patient’s cranium, thereby avoiding a craniotomy. We believe our technology will offer advantages to current depth electrode technology and will enable us to offer a therapeutic solution using this technology in the future. As we develop our technology, we plan to release further information about the expected advantages of our technology over currently available therapies. |
| 11 |
| · | Introduce minimally invasive delivery system for cortical electrodes – Cortical electrodes generally require a craniotomy, which is a very invasive procedure that can cause patient complications. Because of this, many patients have opted to not have epilepsy surgery, instead accepting the consequences and risks associated with epilepsy. We intend to develop a procedure that may include a delivery system placed through a small circular incision in the skull for implantation of the cortical grid and strip electrodes. We believe this will increase patient willingness to accept the surgery and increase market penetration. Until we are able to develop this procedure, if at all, our initial product offering will be placed through traditional surgical means involving a craniotomy and may be less likely to be adopted by physicians and patients due to unwillingness of patients to undergo epilepsy surgery. |
| · | Utilize these core technologies to develop all in one diagnostic and therapeutic solutions – Patients currently undergo one surgical procedure for diagnosis (either to have a cortical electrode placed via a craniotomy or depth electrodes placed via holes drilled into the skull) and, hopefully after the brain recordings successfully indicate where the affected brain tissue is located, a second procedure or surgery is then required to treat the patient. There is strong physician interest in being able to perform both the diagnostic and therapeutic procedure concurrently. We are developing our technology with the goal of being able to offer this benefit although there can be no assurance that we will be able to do so. We are pursuing cortical grid, strip and depth electrode technology that can record brain activity (diagnose), ablate brain tissue and also provide both acute and long term stimulation. The technology has demonstrated these functions in acute and short term animal models; however, additional development is required to offer a device that has long term therapeutic application. These therapeutic technologies are expected to require more robust regulatory approvals for the United States, ranging from a 510(k) with human clinical data to pre-market approvals (PMAs). We will engage the FDA at the proper time to determine the most efficient clinical path. |
| · | Gain approval for other brain or motor related disorders such as Parkinson’s with the therapeutic technologies developed for epilepsy – While we are developing our technology for the diagnosis and treatment of epilepsy, we believe that our technology has strong application and utilization for other brain or motor related disorders such as Parkinson’s disease, dystonia, essential tremors and facial pain as these diseases are currently treated with DBS if medications are not effective. As previously mentioned, we are planning to offer electrodes that can be implanted for long term stimulation applications, but such use will require that we pursue additional approvals from the FDA and any international regulatory bodies where we seek to commercialize our technology. |
| · | Explore partnerships with other companies that leverage our core technology – Given that our technology enables, complements and/or competes with a number of companies that are in the market or attempting to enter the market with diagnostic or therapeutic technologies to treat brain related disorders, we believe there may be opportunities to establish mutually beneficial relationships. In addition, our technology may have application in cardiovascular, orthopedic and pain related indications that could benefit from a hi-fidelity thin film electrode product that can provide stimulation and/or ablation therapies. |
Our Technology
Epilepsy Mapping and Monitoring
Epileptic seizures occur when the neurons in the brain miscommunicate. This miscommunication typically results in involuntary muscle seizure activities and/or periods of perceptual disconnect where the individual appears frozen. Modern medical science has advanced the treatment of epileptic seizures by mapping the electrical communication activity of neurons and understanding their special orientation in the brain. This mapping is accomplished by access to the cranium (through a craniotomy) and placing conductive contacts on the brain directly. The craniotomy procedure is very invasive, traumatic to the surrounding tissue, results in high patient down time, and increases the risk of infection.
| 12 |
Our Technology
We seek to leverage scale-able technology and produce ultra-thin electrodes that allow for higher density contacts thus increasing the mapping resolution and signal acquisition. We also believe that the electrodes’ unique thinness and flexibility will afford a non-invasive approach to electrode placement to replace a craniotomy with placement and removal utilizing access via a small burr hole in the skull.
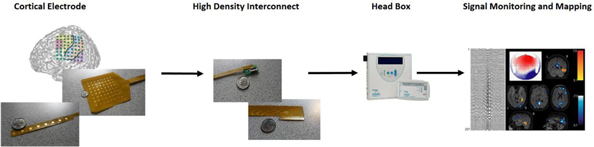
Our technology consists of three primary types of cortical electrodes: grid electrodes, strip electrodes and dual-sided electrodes. These electrodes have a patented design that utilizes proprietary processing and materials technology, which we believe will allow the electrodes to have improved features over the current industry standard recording electrodes.
What sets our technology apart from others is the integration of state of the art design leveraging the latest in flexible printed circuit technology. We believe our patented designs will provide the surgeon a higher tactile perspective on electrode placement allowing for ultra-precise neuron recording. We expect the benefits of our electrode designs to include the ability to detect better defined margins between healthy tissue and resect-able tissue, less immune-response from the brain and surrounding tissue, better signal acquisition due to superior conformability of the electrode over the brain, improved flexibility that physicians have been requested, which we expect will enable a minimally invasive approach and the electrodes unique thinness that is unmatched by current products being used.
The Future of Epilepsy Mapping with NeuroOne
We seek to develop superior “scale-able” technology for future product system iterations in higher density contact placement. This will open the doors to other brain related disease recording procedures by providing hi-fidelity, more accurate diagnostic capabilities and also the ability to provide an all in one therapy capable of diagnosis, ablation and/or stimulation. Beyond the brain, we believe our technology under development has applications in other neurological signal recording disease states related to voluntary or involuntary motor neuron abnormalities, understanding sensory neuro behavior (pain), limb prosthetics and degenerative muscle disease.
Clinical Development and Regulatory Pathway
Clinical Experience, Future Development and Clinical Trial Plans
Our technology under development has not been approved for commercialization by any U.S. or foreign regulatory body, and, prior to receiving such approval, our technology will need to undergo extensive pre-clinical testing and clinical trials. As disclosed in more detail below, our technology has been tested in very limited trials to date, and we have very limited clinical data to support our expectations regarding the performance of our technology, its safety, efficacy and anticipated benefits compared to currently available technology.
| 13 |
In parallel with the development and testing needed to launch our cortical strip and grid electrodes, we intend to expand our product offerings to include less invasive means and all in one solutions, thus providing both patients and physicians better options to treat epilepsy and other brain related disorders. While we expect to make modifications to this initial system, we believe that most of our future product development initiatives will involve unique and transformational next generation technology that should drive further appeal of our products with both physicians and patients.
We are utilizing a number of resources to develop these technologies. We license three critical patents from WARF that are the foundation of the technology we are developing and intend to commercialize and benefit from the thin film technology know-how of Mayo Clinic doctors through our license and development agreement. WARF and Mayo Clinic have been responsible for all pre-clinical studies of our technology under development to date. See “—WARF License” and “—The Mayo Foundation for Medical Education and Research License and Development Agreement” below.
Below we have summarized, for each component of our technology under development, the current stage of development, the pre-clinical testing done to date by WARF or Mayo Clinic on such component, if any, our plans for further testing or clinical trials and our expectations regarding the requirements for regulatory approval and timing of regulatory submissions:
| Technology |
Stage of Development and Pre-Clinical Testing to Date |
Expected Requirements for Regulatory Approval | |||
|
Cortical strip and grid electrodes for the diagnosis of epilepsy
|
Design freeze complete
Pre-clinical testing has been conducted on the versions used for clinical research by The Mayo Clinic and WARF (described below) |
In vitro bench top and pre-clinical (bio compatibility and sterilization) testing expected to be required prior to human use and scheduled to be completed in the fourth quarter of 2017
There may be continued clinical evaluation of the technology under a pre-existing IRB research protocol approved by Mayo’s institutional review board, which will provide us with additional clinical evidence that may assist with product acceptance and launch
Planned U.S. commercial launch in early 2018 upon FDA clearance, if received | |||
| Depth electrodes for diagnostic purposes |
In development
Have not been tested in clinical or pre-clinical studies to date, although made of the same material and electrical contacts as our cortical grid and strip electrodes |
In vitro bench top, biocompatibility and sterilization tests expected to be required
Design testing to be determined in the second half of 2017 and expected to be complete in the fourth quarter of 2017
Expect to file for FDA 510(k) clearance in the second quarter of 2018 | |||
| Minimally invasive cortical electrode delivery system |
In development
No clinical testing to date |
Future clinical testing requirements for regulatory clearance currently unknown
Currently researching predicate devices and procedures to support position to file with the FDA as a 510(k) submission and to determine required testing |
| 14 |
| Technology |
Stage of Development and Pre-Clinical Testing to Date |
Expected Requirements for Regulatory Approval | |||
| Cortical and depth electrode ablation devices |
In development
No clinical testing to date
|
Future clinical testing requirements for regulatory clearance/approval currently unknown
Expect to perform clinical study in 2019 and then submit such clinical study to the FDA
No FDA feedback has been sought or received by us to date on the clinical process that may be required for an ablation indication, but we expect regulatory clearance/approval will require a more robust clinical process, which could range from 510(k) clearance with human clinical data to a PMA, depending on proposed indications for use
In-vitro bench top, pre-clinical and safety (animal) studies and FDA-approved human clinical studies will most likely be required | |||
| Cortical and depth electrode chronic stimulation devices |
In development
No clinical testing to date
|
Future clinical testing requirements for regulatory clearance/approval currently unknown
Expect to conduct clinical testing in 2019
No FDA feedback has been sought or received by us to date on the clinical process that will be required for chronic stimulation, but we expect regulatory clearance/approval for chronic stimulation may require a more robust clinical process, which could range from 510(k) clearance with human clinical data to a PMA
In-vitro bench-top, pre-clinical and safety (animal) studies and FDA-approved human clinical studies will most likely be required |
Our cortical technology for the diagnosis of epilepsy has been tested by doctors at Mayo Clinic in multiple pre-clinical tests conducted from 2012 to 2017. In pre-clinical models, doctors examined the biological impact on mammalian brains. Polyimide substrate electrodes (NeuroOne technology) were implanted on the pig’s brain for one week alongside standard competitive electrodes. The tissue underneath the two types of electrodes was removed, fixed, stained, and examined for immunological responses. Electrophysiological (brain neuron activity) properties were examined by recordings in pigs and from tissue to be removed in five patients undergoing surgery. Doctors examined the changes in local field potential activity of the brain with thin film electrodes (NeuroOne technology) and then compared to competitive electrodes. Intra-operative brain activity recordings were obtained and evaluated in a pig seizure model and in five human subjects undergoing surgery for drug resistant epilepsy.
The results of a histological (evaluation of brain tissue under a microscope) analysis showed reduced immunological reaction to prolonged polyimide substrate implants (NeuroOne technology) compared to standard silicone substrate competitive clinical electrodes. Electrophysiological recordings showed data obtained from polyimide electrodes showed feasibility of high fidelity multi-scale electrophysiology while also displaying easier deployment of polyimide electrodes (NeuroOne technology) through burr holes utilizing a minimally invasive approach.
Conclusions reached by the physicians at Mayo Clinic are that thin, flexible polyimide electrodes (NeuroOne technology) provide recordings similar to standard clinical electrodes with markedly reduced immunological response. In addition, the flexibility and reduced volume of polyimide electrodes should reduce pain and swelling associated with implantation of the device, and the single wire exiting the skull should reduce infection risk. Combined, these properties suggest that the replacement of current competitive silicone electrodes with polyimide substrate electrodes (NeuroOne technology) for recording brain activity for epilepsy could provide enhanced clinical value with reduced cost, reduced infection risk, and improved patient comfort.
| 15 |
In addition, our cortical implant technology has been tested by researchers at the University of Wisconsin-Madison in multiple pre-clinical studies conducted from 2006 to 2016. These studies, illustrated in a variety of pre-clinical animal models that included mice, rats and primates, have shown that thin film cortical implant technology can reliably record brain activity from different areas of the brain, can be implanted successfully in a minimally invasive fashion, can be safely implanted over long-time periods of up to five years, can electrically provide brain stimulation and tissue ablation, and has increased flexibility versus existing commercially available technology that allows the grids to conform to the brain surface.
Sales and Marketing
Based on the size and maturity of the U.S. market, our initial commercial focus, if our technology is approved for commercialization for the diagnosis of epilepsy in the U.S., will be to invest in developing a direct sales force and infrastructure to support the launch of the product in the United States and target what we estimate to be approximately 188 Level 4 epilepsy centers along with their respective epilepsy teams comprised of neurologists, neurosurgeons and technicians in the United States who are clinically active.
In parallel, we are evaluating the opportunity to commercialize our products in select European markets, which we would do through third-party distributors or country independent agents. We believe there is a market for our technology in Europe, but we are evaluating epilepsy surgery rates, reimbursement and growth opportunities, the regulatory pathway and intellectual property rights in each jurisdiction. If we plan to commercialize in foreign jurisdictions, we plan to initially target markets that have active epilepsy programs with adequate reimbursement for the procedures. Simultaneously, we have engaged European consultants to identify the requirements to gain acceptance in the European Union.
In addition, if our technology is approved for commercialization for the diagnosis of epilepsy in the U.S., we will look to educate neurologists, neurosurgeons and primary care physicians on the advantages to existing epilepsy approaches through a variety of targeted marketing tools and social media.
Reimbursement
Coverage in the United States
Reimbursement from private third-party healthcare payors and, to a lesser extent, Medicare will be an important element of our success. Although the Centers for Medicare and Medicaid, or CMS, and third-party payors have adopted coverage policies for our targeted indications, there is no guarantee this will continue at the same levels or at all in the future. Current Procedural Terminology, or CPT, is a medical code set that is used to report medical, surgical, and diagnostic procedures and services to entities such as physicians, health insurance companies and accreditation organizations.
Applicable diagnostic CPT codes for mapping (diagnosing) the brain for diagnostic procedures are as follows:
| · | 61531 Subdural implantation of strip electrodes through one or more burr or trephine (saw) hole(s) for long-term seizure monitoring |
| · | 61533 Craniotomy with elevation of bone flap: for subdural implantation of an electrode array, for long term seizure monitoring |
| · | 61535 Craniotomy with elevation of bone flap; for removal of epidural or subdural electrode array, without excision of cerebral tissue (separate procedure) |
| · | 61760 Stereotactic implantation of depth electrodes into the cerebrum for long term seizure monitoring |
| 16 |
Regarding ICD-10 codes, the International Classification of Diseases, Tenth Edition (ICD-10) is a clinical cataloging system that went into effect for the U.S. healthcare industry on Oct. 1, 2015, after a series of lengthy delays. Accounting for modern advances in clinical treatment and medical devices, ICD-10 codes offer many more classification options compared to those found in its predecessor, ICD-9. Within the healthcare industry, providers, coders, IT professionals, insurance carriers, government agencies and others use ICD codes to properly note diseases on health records, to track epidemiological trends and to assist in medical reimbursement decisions.
ICD-10 codes for epilepsy are as follows:
| · | G40.0 Localization-related (focal) (partial) idiopathic epilepsy and epileptic syndromes with seizures of localized onset |
| · | G40.1 Localization-related (focal) (partial) symptomatic epilepsy and epileptic syndromes with simple partial seizures |
| · | G40.2 Localization-related (focal) (partial) symptomatic epilepsy and epileptic syndromes with complex partial seizures |
| · | G40.3 Generalized idiopathic epilepsy and epileptic syndromes |
| · | G40.A Absence epileptic syndrome |
| · | G40.4 Other generalized epilepsy and epileptic syndromes |
| · | G40.50 Epileptic seizures related to external causes, not intractable |
| · | G40.80 Other epilepsy |
| · | G40.82 Epileptic spasms |
We believe that many of the indications we are pursuing with our technologies are currently reimbursed on a widespread basis by Medicare, Medicaid and private insurance companies.
Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medical devices, and, as a result, their coverage policies may be restrictive, or they may not cover or provide adequate payment for our products. In order to obtain reimbursement arrangements, we may have to agree to a net sales price lower than the net sales price we might charge in other sales channels. Our revenue may be limited by the continuing efforts of government and third-party payors to contain or reduce the costs of healthcare through various increasingly sophisticated means, such as requiring prospective reimbursement and second opinions, purchasing in groups, or redesigning benefits. Our future dependence on the commercial success of our technologies makes us particularly susceptible to any cost containment or reduction efforts. Accordingly, unless government and other third-party payors provide adequate coverage and reimbursement for our products and the related insertion and removal procedures, our financial performance may be limited.
Coverage Outside the United States
If we seek to commercialize in countries outside the United States, coverage for epilepsy surgical procedures are available from certain governmental authorities, private health insurance plans, and labor unions. Coverage systems in international markets vary significantly by country and, within some countries, by region. If we seek to commercialize our technology, if approved, outside the U.S., coverage approvals must be obtained on a country-by-country, region-by-region or, in some instances, a case-by case basis. Based on our ongoing evaluation, certain countries reimburse more highly than others.
| 17 |
Manufacturing, Supply and Quality Assurance
We currently outsource the supply and manufacture of all components of our prototypes of our technology under development. We plan to continue with an outsourced manufacturing arrangement for the foreseeable future. Our third-party manufacturers are recognized in their field for their competency to manufacture the respective portions of our system and have quality systems established that meet FDA requirements. We believe the manufacturers we currently utilize have sufficient capacity to meet our launch requirements if our technology under development is approved in the future and are able to scale up their capacity relatively quickly with minimal capital investment. We believe that, as we increase our demand in the future, our per unit costs will decrease materially. We have also identified capable second source manufacturers and suppliers in the event of disruption from any of our primary vendors.
Our suppliers meet the latest ISO 13485 certification, which includes design control requirements. As a medical device developer, the facilities of our sterilization and other critical suppliers are subject to periodic inspection by the FDA and corresponding state and foreign agencies. We believe that our quality systems and those of our suppliers are robust and achieve high product quality. We plan to audit our suppliers periodically to ensure conformity with the specifications, policies and procedures for our devices.
Research and Development
Our research and development team includes 10 employees and consultants who specialize in thin film technology, many of whom have considerable experience in brain related neurovascular technologies and related conditions. Our research and development team is focused on the development of thin film cortical grid and strip electrodes and depth electrodes for recording, ablation and chronic stimulation for brain related disorders. Our research and development expenses were $0 and $2,400 for the years ended December 31, 2016 and 2015, respectively, and $72,041 and $0 for the three months ended March 31, 2017 and 2016, respectively.
Competition
In the market for Epilepsy diagnosis, our cortical strip, sheet and depth electrode technology will likely compete with Integra Life Science’s Integra Epilepsy Strip, Grid and depth electrodes, which provide a similar function to our diagnostic technologies under development. These products are well established in the marketplace and Integra has greater resources than us, which could allow them to innovate faster. Ad-Tech Medical Instrument Corporation’s Epilepsy/LTM (subdural grid, strip and depth) electrodes, which have become the market leaders for diagnostic mapping in epilepsy, and PMT Corporation’s Cortac Strips and grid electrodes and Depthalon depth electrodes are used for recording brain activity similar to other competitive technologies. These technologies are very different from our thin film strip technology under development, which, if developed as expected and approved, would represent next generation recording technology that can be placed minimally invasively, allow for smaller footprints with increased number of electrodes, different shaped electrodes and much higher fidelity that may be able to detect microseizure activity, which would be helpful in improving clinical rates of eliminating seizure activity in patients. Today’s success rates for seizure free post-operative conditions remain at 50%, which has limited patient’s acceptance to undergo the currently highly invasive surgical procedure. We will also compete against other companies in early stages of development of thin film technologies.
In the neuro-ablation market, we expect to compete with Medtronic’s Visualase® guided-laser ablation technology and Monteris Medical’s NeuroBlate technology, which use MRI guided laser surgical ablation for use to ablate, necrotize or coagulate soft tissue through interstitial irradiation or thermal therapy in medicine and surgery in the discipline of neurosurgery with 1064 nm lasers. Their website claims it is used for ablation in the brain for soft tissue and tumors. We believe there are other laser based systems in development that will compete with these technologies.
In the neurostimulation market, we expect to compete with NeuroPace’s RNS system approved for epilepsy, Medtronic’s Activa system approved for Parkinson’s disease, Boston Scientific Vercise (indicated for Parkinson’s, Dystonia and Essential Tremors), Abbott/St. Jude Medical’s Infinity DBS system (approved for Parkinson’s disease and essential tremors), Liva Nova/Cyberonic’s VNS therapy intended for patient’s suffering with epilepsy. We believe there are additional companies pursuing this high growth space although none are expected to be commercially available in 2017 based on current reports.
| 18 |
Although we will face potential competition from many different sources, we believe that our technology, knowledge, experience and scientific resources will provide us with competitive advantages. We expect the key competitive factors affecting the success of our cortical strip and sheet electrodes under development, if successfully developed and approved, are likely to be: hi-fidelity recording that allows for detection of pre-seizure activity, ability to place the devices minimally invasively, deliverability of cortical grid, strip and depth electrode technology, ability to offer grid, strip and depth electrodes in various electrode shapes and sizes, potential reduction in infections and ability to record brain activity both on the surface using cortical grid and strip technology and deeper into the brain using depth electrodes concurrently.
Many of the companies against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and subject registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our development.
WARF License
We have entered into an Exclusive Start-Up Company License Agreement with Wisconsin Alumni Research Foundation, or WARF, pursuant to which WARF has granted us an exclusive license, or the WARF License, to make, use and sell, in the United States only, products that employ certain licensed patents for a neural probe array or thin-film micro electrode array and method. In exchange for the license, we have agreed to pay WARF a one-time fee of $120,000 (representing a license fee and reimbursement for costs incurred by WARF in maintaining the licensed patents) upon the earliest to occur of certain events related to the Company raising a minimum amount of financing, a change of control or our revenue reaching a threshold amount. We have also agreed to pay WARF a royalty equal to a single-digit percentage of our product sales pursuant to the WARF License, with a minimum annual royalty payment of $50,000 for 2019, $100,000 for 2020 and $150,000 for 2021 and each calendar year thereafter that the WARF License is in effect. If we or any of our sublicenses contest the validity of any licensed patent, the royalty rate will be doubled during the pendency of such contest and, if the contested patent is found to be valid and would be infringed by us if not for the WARF License, the royalty rate will be tripled for the remaining term of the WARF License.
We have agreed to diligently develop, manufacture, market and sell products under the WARF License in the United States during the term of the agreement and, specifically, that we will submit a business plan to WARF by February 1, 2018 and file an application for 510(k) marketing clearance with the FDA by February 1, 2019. WARF may terminate this license in the event that we fail to meet these milestones on 30 days’ written notice, if we default on the payments of amounts due to WARF or fail to timely submit development reports, actively pursue our development plan or breach any other covenant in the WARF License and fail to remedy such default in 90 days or in the event of certain bankruptcy events involving us. WARF may also terminate this license (i) on 90 days’ notice if we fail to have commercial sales of one or more FDA-approved products under the WARF License by March 31, 2019 or (ii) if, after royalties earned on sales begin to be paid, such earned royalties cease for more than four calendar quarters. The WARF License otherwise expires by its terms on the date that no valid claims on the patents licensed thereunder remain. We expect the latest expiration of a licensed patent to occur in 2030.
In addition, WARF reserves the right to grant non-profit research institutions and government agencies non-exclusive licenses to practice and use the inventions of the licensed patents for non-commercial research purposes, and we grant WARF a non-exclusive, sub licensable, royalty-free right and license for non-commercial research purposes to use improvements to the licensed patents. In the event that we discontinue use or commercialization of the licensed patents or improvements thereon, we must grant WARF an option to obtain a non-exclusive, sub-licensable royalty-bearing license to use the improvements for commercial purposes.
| 19 |
See “Risk Factors—We depend on intellectual property licensed from Wisconsin Alumni Research Foundation for our cortical strip, grid electrode and depth electrode technology under development, and the termination of this license would harm our business” for additional information regarding the WARF License and our past breach thereof.
Mayo Foundation for Medical Education and Research License and Development Agreement
We have entered into a license and development agreement, or the Mayo Development Agreement, with Mayo Foundation for Medical Education and Research (referred to herein as Mayo) to license worldwide (i) certain know how for the development and commercialization of products, methods and processes related to flexible circuit thin film technology for the recording of tissue and (ii) the products developed therefrom, and to partner with Mayo to assist the Company in the investigation, research application, development and improvement of such technology. Mayo has agreed to assist us by providing access to certain individuals at Mayo, or the Mayo Principal Investigators, in developing our cortical thin film flexible circuit technology, including prototype development, animal testing, protocol development for human and animal use, abstract development and presentation and access to and license of any intellectual property that the Mayo Principal Investigators develop relating to the procedure.
Whether or not any such technology, product, method, process, device or delivery system is developed, we agreed, in consideration for Mayo’s efforts under the Mayo Development Agreement, to pay Mayo a cash payment of approximately $92,000 on the earlier of September 30, 2017 or the date we raise a minimum amount of financing, and on May 25, 2017, prior to the closing of the Acquisition, NeuroOne, Inc. issued Mayo 50,556 NeuroOne Shares pursuant to a Subscription Agreement. Finally, we have agreed to pay Mayo a royalty equal to a single-digit percentage of our product sales pursuant to the Mayo Development Agreement. Mayo may purchase any developed products licensed under the Mayo Development Agreement at the best price offered by us to the end user in the prior year. The Mayo Development Agreement generally will expire in October 2034, unless the Mayo know-how and improvements under the Mayo Development Agreement remain in use, and the Mayo Development Agreement may be terminated by Mayo for cause or under certain circumstances.
For additional information regarding the Mayo Development Agreement and our past breach thereof, see “Risk Factors—We depend on our partnership with Mayo Foundation for Medical Education and Research to license certain know how for the development and commercialization of our technology. Termination of this partnership would harm our business, and even if this partnership continues, it may not be successful.”
Intellectual Property
Protection of our intellectual property is a strategic priority for our business. We rely on a combination of patents, trademarks, copyrights, trade secrets as well as nondisclosure and assignment of invention agreements, material transfer agreements, confidentiality agreements and other measures to protect our intellectual property and other proprietary rights.
Patents
As of June 19, 2017, our patent estate consists of 3 issued United States patents licensed from WARF covering a neural probe array and thin-film micro electrode array and method and one pending United States provisional patent application filed by us on March 31, 2017 covering our depth electrode technology and ancillary devices used during the diagnostic and therapeutic ablation and stimulation procedures. The licensed issued patents expire between 2025 and 2030, subject to any patent extensions that may be available for such patents. If a patent is issued on our pending patent application, the resulting patent is projected to expire in 2038.
Our patent application may not result in an issued patent, and any patents that have been issued or may be issued in the future may not protect the commercially important aspects of our technology. Furthermore, the validity and enforceability of our issued patents may be challenged by third parties and our patents could be invalidated or modified by the issuing governmental authority. Third parties may independently develop technology that is not covered by our patents that is similar to or competes with our technology. In addition, our intellectual property may be infringed or misappropriated by third parties, particularly in foreign countries where the laws and governmental authorities may not protect our proprietary rights as effectively as those in the United States.
| 20 |
The medical device industry in general, and the recording, ablation and neurostimulation sector of this industry in particular, are characterized by the existence of a large number of patents and frequent litigation based on assertions of patent infringement. We are aware of numerous patents issued to third parties that may relate to the technology used in our business, including the design and manufacture of electrodes and pulse generators, as well as methods for device placement. Each of these patents contains multiple claims, any one of which may be independently asserted against us. The owners of these patents may assert that the manufacture, use, sale or offer for sale of our cortical strip and sheet electrodes infringe one or more claims of their patents. Furthermore, there may be additional patents issued to third parties of which we are presently unaware that may relate to aspects of our technology that such third parties could assert against us and materially and adversely affect our business. In addition, because patent applications can take many years to issue, there may be patent applications that are currently pending and unknown to us, which may later result in issued patents that third parties could assert against us and materially and adversely affect our business.
Any adverse determination in litigations, post grant trial proceedings, including interference proceedings, at the Patent Office relating to intellectual property to which we are or may become a party could subject us to significant liabilities to third parties or require us to seek licenses from third parties, and result in the cancellation and/or invalidation of our intellectual property. Furthermore, if a court finds that we have willfully infringed a third party's intellectual property, we could be required to pay treble damages and/or attorney fees for the prevailing party, in addition to other penalties. Although intellectual property disputes in the medical device area are often settled through licensing or similar arrangements, costs associated with such arrangements can be substantial and often require ongoing royalty payments. We may be unable to obtain necessary licenses on satisfactory terms, if at all. If we do not obtain necessary licenses, we may not be able to redesign our products to avoid infringement; if we are able to redesign our products to avoid infringement, we may not receive FDA approval in a timely manner. Adverse determinations in a judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from manufacturing and selling our products, which could have a significant adverse impact on our business.
Trademarks
We have 1 pending U.S. trademark application for the “NeuroOne” trademark.
Trade Secrets
We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. We seek to protect such intellectual property and proprietary information by generally requiring our employees, consultants, contractors, scientific collaborators and other advisors to execute non-disclosure and assignment of invention agreements upon the commencement of their employment or engagement as the case may be. Our agreements with our employees prohibit them from providing us with any intellectual property or proprietary information of third parties. We also generally require confidentiality agreements or material transfer agreements with third parties that receive or have access to our confidential information, data or other materials. Notwithstanding the foregoing, there can be no assurance that our employees and third parties that have access to our confidential proprietary information will abide by the terms of their agreements. Despite the measures that we take to protect our intellectual property and confidential information, unauthorized third parties may copy aspects of our products or obtain and use our proprietary information.
Government Regulation
Our cortical strip, grid and depth electrodes are a medical device subject to extensive and ongoing regulation by the FDA, the U.S. Centers for Medicare & Medicaid Services, or CMS, the European Commission, and regulatory bodies in other countries. Regulations cover virtually every critical aspect of a medical device company's business operations, including research activities, product development, quality and risk management, contracting, reimbursement, medical communications, and sales and marketing. In the United States, the Federal Food, Drug and Cosmetic Act, or FDCA, and the implementing regulations of the FDA govern product design and development, pre-clinical and clinical testing, premarket clearance or approval, product manufacturing, quality systems, import and export, product labeling, product storage, recalls and field safety corrective actions, advertising and promotion, product sales and distribution, and post-market clinical surveillance. Our business is subject to federal, state, local, and foreign regulations, such as ISO 13485, ISO 14971, FDA's Quality System Regulation, or QSR, contained in 21 CFR Part 820, and the European Commission's Directive 93/42/EEC concerning medical devices and its amendments.
| 21 |
The FDA characterizes medical devices into one of three classes. Devices that are considered by the FDA to pose lower risk are classified as Class I or II. Class I devices and are subject to controls for labeling, pre-market notification and adherence to the FDA's QSR. This pertains to manufacturers' methods and documentation of the design, testing, production, control quality assurance, labeling, packaging, sterilization, storage and shipping of products, but are usually exempt from premarket notification requirements. Class II devices are subject to the same general controls but may be subject to special controls such as performance standards, post-market surveillance, FDA guidelines, or particularized labeling, and may also require clinical testing prior to clearance or approval. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general or special controls, including devices that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury.
Some Class I and Class II devices are exempted by regulation from the pre-market notification requirement under Section 510(k) of the FDCA, also referred to as a 510(k) clearance, and the requirement of compliance with substantially all of the QSR. However, a pre-market approval, or PMA application, is required for devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or certain implantable devices, or those that are "not substantially equivalent" either to a device previously cleared through the 510(k) process or to a "preamendment" Class III device in commercial distribution before May 28, 1976 when PMA applications were not required. The PMA approval process is more comprehensive than the 510(k) clearance process and typically takes several years to complete. Based on FDA definitions, we believe our diagnostic strip, grid and depth electrode technology will be categorized by the FDA as a Class II device that does not require clinical testing and can be filed as a 510(k), similar to existing competitive technology. The Company expects that indications for treating epilepsy, Parkinson’s and other patients suffering from motor related neurological deficiencies via a permanent implant for chronic treatment will require a PMA process to commercially distribute in the United States. While the 510(k) process is typically shorter than a PMA process, both the 510(k) clearance and PMA processes can be expensive and lengthy.
FDA review of a PMA application generally takes between one and three years, but may take significantly longer. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
| · | the device may not be safe, effective, reliable or accurate to the FDA's satisfaction; |
| · | the data from pre-clinical studies and clinical trials may be insufficient to support approval; |
| · | the manufacturing process or facilities may not meet applicable requirements; and |
| · | changes in FDA approval policies or adoption of new regulations may require additional data. |
If an FDA evaluation of a PMA application is favorable, the FDA will either issue an approval letter, or approvable letter, which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of a device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA's evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA. The PMA process can be expensive, uncertain and lengthy and a number of devices for which FDA approval has been sought by other companies have never been approved by the FDA for marketing.
New PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling, device specifications, materials or design of a device that has been approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive technical or clinical data or the convening of an advisory panel.
| 22 |
Clinical trials are typically required to support a PMA application and are sometimes required for a 510(k) clearance. These trials generally require submission of an application for an IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for abbreviated IDE requirements. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate institutional review boards at the clinical trial sites. The FDA's approval of an IDE allows clinical testing to go forward, but it does not bind the FDA to accept the results of the trial as sufficient to prove the product's safety and efficacy, even if the trial meets its intended success criteria. All clinical trials must be conducted in accordance with the FDA's IDE regulations that govern investigational device labeling, prohibit promotion, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with the FDA's regulations for institutional review board approval and for informed consent and other human subject protections. Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product. Clinical trials must be entered into the clinical trials registry at clintrials.gov.
The commencement or completion of any clinical trial may be delayed or halted, or be inadequate to support approval of a PMA application, for numerous reasons, including, but not limited to, the following:
| · | the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| · | patients do not enroll in clinical trials at the rate expected; |
| · | patients, sponsor (NeuroOne) or study sites do not comply with trial protocols; |
| · | patient follow-up is not at the rate expected; |
| · | patients experience adverse side effects; |
| · | patients die during a clinical trial, even though their death may not be related to the products that are part of our trial; |
| · | institutional review boards and third-party clinical investigators may delay or reject the trial protocol; |
| · | third-party clinical investigators decline to participate in a trial or do not perform a trial on the anticipated schedule or consistent with the clinical trial protocol, good clinical practices or other FDA requirements; |
| · | the sponsor (NeuroOne) or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans; |
| · | third-party clinical investigators have significant financial interests related to the sponsor (NeuroOne) or the study that the FDA deems to make the study results unreliable, or the company or investigators fail to disclose such interests; |
| · | regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; |
| · | changes in governmental regulations or administrative actions; |
| · | the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or efficacy; and |
| · | the FDA concludes that our trial design is inadequate to demonstrate safety and efficacy. |
International Regulation
International sales of medical devices are subject to local government regulations, which may vary substantially from country to country. The time required to obtain approval in another country may be longer or shorter than that required for FDA approval, and the requirements may differ. There is a trend towards harmonization of quality system standards among the European Union, United States, Canada and various other industrialized countries.
| 23 |
The primary regulatory body in Europe is that of the European Union, the European Commission, which includes most of the major countries in Europe. Other countries, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the European Union with respect to medical devices. The European Union has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices. Devices that comply with the requirements of these relevant directives will be entitled to bear the CE conformity marking, indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout Europe. The method of assessing conformity varies depending on the class of the product, but normally involves a combination of self-assessment by the manufacturer and a third party assessment by a "Notified Body." This third-party assessment may consist of an audit of the manufacturer's quality system and specific testing of the manufacturer's product. An assessment by a Notified Body of one country within the European Union is required in order for a manufacturer to commercially distribute the product throughout the European Union. Additional local requirements may apply on a country-by-country basis. Outside of the European Union, regulatory approval would need to be sought on a country-by-country basis in order for us to market our products.
Medical devices in Europe are classified into four primary categories. They are as follows:
| · | Non-invasive devices |
| · | Invasive medical devices |
| · | Active medical devices |
| · | Special Rules (including contraceptive, disinfectant, and radiological diagnostic medical devices) |
Devices are further segmented into the classes noted below. In Vitro Diagnostic devices(IVDs) have their own classification scheme and while active implantable devices do not follow the same classification system as provided by the Medical Device Directive(MDD), they are subject to similar requirements as Class III devices:
| · | Class I – Provided non-sterile or do not have a measuring function (low risk) |
| · | Class I – Provided sterile and/or have a measuring function (low/medium risk) |
| · | Class IIa (medium risk) |
| · | Class IIb (medium/high risk) |
| · | Class III (high risk) |
After a review of our technology, an international regulatory consultant advised us that our strip, grid and depth electrode diagnostic technology is likely a Class III device (since it comes into contact with the central nervous system) which will require a lengthy approval process as a design dossier including clinical data will be required for approval. The Company is currently investigating the market potential of the European Union before this process is initiated to gain European Union approval.
Other Regulatory Requirements
Even after a device receives clearance or approval and is placed in commercial distribution, numerous regulatory requirements apply. These include:
| · | establishment registration and device listing; |
| · | QSR, which requires manufacturers, including third party manufacturers, to follow stringent design, testing, risk management, production, control, supplier/contractor selection, complaint handling, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
| · | labeling regulations that prohibit the promotion of products for uncleared, unapproved or "off-label" uses, and impose other restrictions on labeling, advertising and promotion; |
| 24 |
| · | MDR regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
| · | voluntary and mandatory device recalls to address problems when a device is defective and could be a risk to health; and |
| · | corrections and removals reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health. |
Also, the FDA may require us to conduct post-market surveillance studies or establish and maintain a system for tracking our products through the chain of distribution to the patient level. The FDA enforces regulatory requirements by conducting periodic, unannounced inspections and market surveillance. Inspections may include the manufacturing facilities of our subcontractors.
Failure to comply with applicable regulatory requirements can result in enforcement actions by the FDA and other regulatory agencies. These may include any of the following sanctions or consequences:
| · | warning letters or untitled letters that require corrective action; |
| · | fines and civil penalties; |
| · | unanticipated expenditures; |
| · | delays in approving or refusal to approve future products; |
| · | FDA refusal to issue certificates to foreign governments needed to export products for sale in other countries; |
| · | suspension or withdrawal of FDA clearance or approval; |
| · | product recall or seizure; |
| · | interruption of production; |
| · | operating restrictions; |
| · | injunctions; and |
| · | criminal prosecution. |
Our contract manufacturers, specification developers and some suppliers of components or device accessories, also are required to manufacture our products in compliance with current good manufacturing practice requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and it includes extensive requirements with respect to quality management and organization, device design, buildings, equipment, purchase and handling of components or services, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing, and record keeping. The FDA evaluates compliance with the QSR through periodic unannounced inspections that may include the manufacturing facilities of our subcontractors. If the FDA believes that any of our contract manufacturers or regulated suppliers are not in compliance with these requirements, it can shut down such manufacturing operations, require recall of our products, refuse to approve new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations or assess civil and criminal penalties against us or our officers or other employees.
Health Insurance Portability and Accountability Act of 1996 and Similar Foreign and State Laws and Regulations Affecting the Transmission, Security and Privacy of Health Information
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA's security standards directly applicable to business associates, defined as service providers of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions. In addition, many state laws govern the privacy and security of health information in certain circumstances, many of which differ from HIPAA and each other in significant ways and may not have the same effect.
| 25 |
Foreign data privacy regulations, such as the EU Data Protection Directive (Directive 95/46/EC), the country-specific regulations that implement Directive 95/46/EC, and the EU General Data Protection Regulation (GDPR) also govern the processing of personally identifiable data, and may be stricter than U.S. laws.
Fraud and Abuse Laws
In addition to FDA restrictions, there are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback laws and physician self-referral laws. Our relationships with healthcare providers and other third parties are subject to scrutiny under these laws. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state healthcare programs, including the Medicare, Medicaid and Veterans Administration health programs.
Federal Anti-Kickback and Self-Referral Laws
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, receiving, offering or providing remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, to induce either the referral of an individual, or the furnishing, recommending, or arranging of a good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid or other federal healthcare programs. The term "remuneration" has been broadly interpreted to include anything of value, including such items as gifts, discounts, the furnishing of supplies or equipment, credit arrangements, waiver of payments and providing anything at less than its fair market value. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a review of all its relevant facts and circumstances. Several courts have interpreted the statute's intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of (or purchases, or recommendations related to) federal healthcare covered business, the Anti-Kickback Statute has been implicated and potentially violated.
The penalties for violating the federal Anti-Kickback Statute include imprisonment for up to five years, fines of up to $25,000 per violation and possible exclusion from federal healthcare programs such as Medicare and Medicaid. Many states have adopted prohibitions similar to the federal Anti-Kickback Statute, some of which do not have the same exceptions and apply to the referral of patients for healthcare services reimbursed by any source, not only by the Medicare and Medicaid programs. Further, the Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act, or PPACA. Specifically, as noted above, under the Anti-Kickback Statute, the government must prove the defendant acted "knowingly" to prove a violation occurred. The PPACA added a provision to clarify that with respect to violations of the Anti-Kickback Statute, "a person need not have actual knowledge" of the statute or specific intent to commit a violation of the statute. This change effectively overturns case law interpretations that set a higher standard under which prosecutors had to prove the specific intent to violate the law. In addition, the PPACA codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
We plan to provide the initial training to providers and patients necessary for appropriate use of our technology either through our own educators or by contracting with outside educators that have completed an appropriate training course. Outside educators are reimbursed for their services at fair market value.
Noncompliance with the federal anti-kickback legislation could result in our exclusion from Medicare, Medicaid or other governmental programs, restrictions on our ability to operate in certain jurisdictions, and civil and criminal penalties.
| 26 |
Federal law also includes a provision commonly known as the "Stark Law," which prohibits a physician from referring Medicare or Medicaid patients to an entity providing "designated health services," including a company that furnishes durable medical equipment, in which the physician has an ownership or investment interest or with which the physician has entered into a compensation arrangement. Violation of the Stark Law could result in denial of payment, disgorgement of reimbursements received under a noncompliant arrangement, civil penalties, and exclusion from Medicare, Medicaid or other governmental programs. We believe that we have structured our provider arrangements to comply with current Stark Law requirements.
Nevertheless, a determination of liability under such laws could result in fines and penalties and restrictions on our ability to operate in these jurisdictions.
Additionally, as some of these laws are still evolving, we lack definitive guidance as to the application of certain key aspects of these laws as they relate to our arrangements with providers with respect to patient training. We cannot predict the final form that these regulations will take or the effect that the final regulations will have on us. As a result, our provider and training arrangements may ultimately be found to be not in compliance with applicable federal law.
Federal False Claims Act
The Federal False Claims Act provides, in part, that the federal government may bring a lawsuit against any person whom it believes has knowingly presented, or caused to be presented, a false or fraudulent request for payment from the federal government, or who has made a false statement or used a false record to get a claim approved. In addition, amendments in 1986 to the Federal False Claims Act have made it easier for private parties to bring "qui tam" whistleblower lawsuits against companies under the Federal False Claims Act. Penalties include fines ranging from $5,500 to $11,000 for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person. Qui tam actions have increased significantly in recent years, causing greater numbers of healthcare companies to have to defend a false claim action, pay fines or be excluded from Medicare, Medicaid or other federal or state healthcare programs as a result of an investigation arising out of such action.
There are other federal anti-fraud laws that that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
Additionally, HIPAA established two federal crimes in the healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines or imprisonment.
Civil Monetary Penalties Law
In addition to the Anti-Kickback Statute and the civil and criminal False Claims Acts, the federal government has the authority to seek civil monetary penalties, or CMPs, assessments, and exclusion against an individual or entity based on a wide variety of prohibited conduct. For example, the Civil Monetary Penalties Law authorizes the imposition of substantial CMPs against an entity that engages in activities including, but not limited to: (1) knowingly presenting or causing to be presented, a claim for services not provided as claimed or which is otherwise false or fraudulent in any way; (2) knowingly giving or causing to be given false or misleading information reasonably expected to influence the decision to discharge a patient; (3) offering or giving remuneration to any beneficiary of a federal health care program likely to influence the receipt of reimbursable items or services; (4) arranging for reimbursable services with an entity which is excluded from participation from a federal health care program; (5) knowingly or willfully soliciting or receiving remuneration for a referral of a federal health care program beneficiary; or (6) using a payment intended for a federal health care program beneficiary for another use. Noncompliance can result in civil money penalties of up to $10,000 for each wrongful act, assessment of three times the amount claimed for each item or service and exclusion from the federal healthcare programs.
| 27 |
State Fraud and Abuse Provisions
Many states have also adopted some form of anti-kickback and anti-referral laws and a false claims act. We believe that we are in conformance to such laws. Nevertheless, a determination of liability under such laws could result in fines and penalties and restrictions on our ability to operate in these jurisdictions.
Physician Payment Sunshine Act
Transparency laws regarding payments or other items of value provided to healthcare providers and teaching hospitals may also impact our business practices. The federal Physician Payment Sunshine Act requires most medical device manufacturers to report annually to the Secretary of Human Health Services financial arrangements, payments, or other transfers of value made by that entity to physicians and teaching hospitals. The payment information is made publicly available in a searchable format on a CMS website. Over the next several years, we will need to dedicate significant resources to establish and maintain systems and processes in order to comply with these regulations. Failure to comply with the reporting requirements can result in significant civil monetary penalties. Similar laws have been enacted or are under consideration in foreign jurisdictions.
U.S. Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act, or FCPA, prohibits U.S. corporations and their representatives from offering, promising, authorizing or making corrupt payments, gifts or transfers to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. Activities that violate the FCPA, even if they occur wholly outside the United States, can result in criminal and civil fines, imprisonment, disgorgement, oversight, and debarment from government contracts.
Employees
As of June 19, 2017, we had 4 employees, all of whom are located in the United States. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Facilities
We currently have no leased or owned properties, including office space. To meet our current needs, we intend to lease office space near Eden Prairie, Minnesota.
Investing in our common stock involves a high degree of risk. Before you invest in our common stock, you should carefully consider the following risks, as well as general economic and business risks, and all of the other information contained in this Report. Any of the following risks could harm our business, operating results and financial condition and cause the trading price of our common stock to decline, which would cause you to lose all or part of your investment. When determining whether to invest, you should also refer to the other information contained in this Report including our financial statements and the related notes thereto.
| 28 |
Risks Relating to our Business
We have incurred significant operating losses since inception and cannot assure you that we will ever achieve or sustain profitability.
We have incurred losses since inception and had an accumulated deficit of $266,370 as December 31, 2016. The LLC, prior to the Merger, also incurred losses since its inception and had cumulative losses of $49,930 as of the date of the Merger. As of March 31, 2017, we had an accumulated deficit of $995,945, primarily as a result of expenses incurred in connection with our research and development programs and from general and administrative expenses associated with our operations. We expect to continue to incur significant expenses and increasing operating and net losses for the foreseeable future. To date, we have financed our operations primarily through debt and equity financings. We had very limited resources prior to our convertible note and warrant financing commencing in November 2016. To date, our primary activities have been limited to, and our limited resources have been dedicated to, performing business and financial planning, raising capital, recruiting personnel, negotiating with business partners and the licensors of our intellectual property and conducting development activities.
To implement our business strategy we need to, among other things, successfully complete the development, testing and 510(k) device submission to the FDA for our cortical and strip electrodes for the diagnosis of epilepsy, successfully complete the development, testing and all required steps for regulatory approval of our depth electrodes for sEEG recording in the U.S., develop and introduce a minimally invasive delivery system for our cortical electrodes, develop an all-in-one diagnostic and therapeutic solution, successfully complete the necessary testing and clinical trials required for regulatory approval of our technology for ablation and stimulation therapies, gain approval for other brain or motor related disorders such as Parkinson’s with the therapeutic technologies developed for epilepsy, convince physicians and patients that our technology, if approved, represents an improvement over existing diagnostic or treatment options, hire direct experienced sales representatives to market our technology, if approved, in the U.S., evaluate international opportunities and initiate and successfully complete the approval processes in targeted geographies and engage in beneficial partnerships that can leverage our core technology. We have never been profitable and do not expect to be profitable in the foreseeable future. We expect our expenses to increase significantly as we pursue our objectives. The extent of our future operating losses and the timing of profitability are highly uncertain, and we expect to continue incurring significant expenses and operating losses over the next several years. Our prior losses have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital. Any additional operating losses may have an adverse effect on our stockholders' equity, and we cannot assure you that we will ever be able to achieve profitability. Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our development efforts, obtain regulatory approvals or continue our operations.
We are a development stage company with a limited operating history, making it difficult for you to evaluate our business and your investment.
NeuroOne, Inc. was incorporated on October 7, 2016, and our predecessor, the LLC, had very limited operations. We are an early-stage medical technology company developing comprehensive neuromodulation cEEG and sEEG monitoring, ablation, and brain stimulation solutions to diagnose and treat patients with epilepsy, Parkinson’s disease, essential tremors, and other brain related disorders. Our cortical strip technology under development has only been used by Mayo in five patients for research purposes and has not been tested in any clinical trials. Our operations are subject to all of the risks inherent in the establishment of a new business enterprise, including but not limited to the absence of an operating history, lack of fully-developed or commercialized products, insufficient capital, expected substantial and continual losses for the foreseeable future, limited experience in dealing with regulatory issues, lack of manufacturing and marketing experience, need to rely on third parties for the development and commercialization of our proposed products, a competitive environment characterized by well-established and well-capitalized competitors and reliance on key personnel.
Since inception, we have not established any revenues or operations that will provide financial stability in the long term, and there can be no assurance that we will realize our plans on our projected timetable (or at all) in order to reach sustainable or profitable operations.
Investors are subject to all the risks incident to the creation and development of a new business and each investor should be prepared to withstand a complete loss of his, her or its investment. Furthermore, the accompanying financial statements have been prepared assuming that we will continue as a going concern. We have not emerged from the development stage, and may be unable to raise further equity. These factors raise substantial doubt about our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| 29 |
Our company has limited experience in medical device development and may not be able to successfully develop any device or therapy. Our ability to become profitable depends primarily on: our ability to develop our cortical strip, grid electrode and depth electrode technology, our successful completion of all necessary pre-clinical testing and clinical trials on such technology, our ability to obtain approval for such technology and, if approved, successfully commercialize such technology, our ongoing research and development efforts, the timing and cost of clinical trials, our ability to identify personnel with the necessary skill sets or enter into favorable alliances with third-parties who can provide substantial capabilities in clinical development, regulatory affairs, sales, marketing and distribution and our ability to obtain and maintain necessary intellectual property rights to such technology. Our limited experience in medical device development may make it more difficult for us to complete these tasks.
Even if we successfully develop and market such technology, we may not generate sufficient or sustainable revenue to achieve or sustain profitability, which could cause us to cease operations and cause you to lose all of your investment. Because we are subject to these risks, you may have a difficult time evaluating our business and your investment in our Company.
Our ability to continue our operations requires that we raise additional capital and our operations could be curtailed if we are unable to obtain the additional funding as or when needed. As a result, our registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements included in this Report. We will need to raise substantial additional funds in the future, and these funds may not be available on acceptable terms or at all. A failure to obtain this necessary capital when needed could force us to delay, limit, scale back or cease some or all operations.
Upon the completion of the audit of our financial statements for the year ended December 31, 2016, we concluded there was substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph regarding this uncertainty in its report on those financial statements.
At March 31, 2017, we had approximately $486,418 in cash deposits. We believe our existing cash and cash equivalents will be sufficient to fund our operating expenses only to August 2017. Our Board of Directors approved a convertible note and warrant financing for gross proceeds of up to $1.5 million in November 2016 and increased such financing authority to $2.5 million in June 2017. Our convertible notes, or the Notes, issued between November 2016 and June 2017 in aggregate principal amount of $1,625,120 pursuant to such authority, bear interest at a fixed rate of 8% per annum and require us to repay the principal and accrued and unpaid interest thereon at the earlier of November 21, 2017 or the consummation of the next equity or equity-linked round of financing resulting in more than $3 million in gross proceeds. If such a financing occurs before November 21, 2017, the outstanding principal and accrued and unpaid interest on the Notes shall automatically convert into the securities issued by us in such financing based on the greater number of such securities resulting from either (i) the outstanding principal and accrued interest on the Notes divided by $1.80 or (ii) the outstanding principal and accrued interest on the Notes multiplied by 1.25, divided by the price paid per security in such financing. If a change of control transaction or initial public offering occurs prior to such a financing, the Notes would, at the election of the holders of a majority of the outstanding principal of the Notes, either become payable on demand as of the closing date of such transaction or become convertible into shares of common stock immediately prior to such transaction at a price per share equal to the lesser of (i) the per share value as determined by our board of directors as if in connection with the granting of stock based compensation or in a private sale to a third party in an arms’ length transaction or (ii) at the per share consideration to be paid in such transaction. Change of control means a merger or consolidation with another entity in which our stockholders do not own more than 50% of the outstanding voting power of the surviving entity or the disposition of all or substantially all of our assets. The Notes are unsecured.
As of July 13, 2017, the outstanding principal and accrued and unpaid interest on the Notes totaled $1,677,345. If we fail to complete an equity financing by November 21, 2017, the notes will be immediately due and payable on such date and we will not have sufficient cash to pay the principal and accrued and unpaid interest thereon.
We will need to secure additional funding on or prior to August 2017. We plan to complete an equity financing in the third quarter of 2017.
| 30 |
The continued growth of our business, including the development, regulatory approval and commercialization of our cortical strip, grid electrode and depth electrode technology, will significantly increase our expenses going forward. As a result, we may be required to seek substantial additional funds in the future. Our future capital requirements will depend on many factors, including:
| · | the cost of developing our cortical strip, grid electrode and depth electrode technology; |
| · | obtaining and maintaining regulatory clearance or approval for our cortical strip, grid electrode and depth electrode technology; |
| · | the costs associated with commercializing our cortical strip, grid electrode and depth electrode technology; |
| · | any change in our development priorities; |
| · | the revenue generated by sales of our cortical strip, grid electrode and depth electrode technology, if approved; |
| · | the costs associated with expanding our sales and marketing infrastructure for commercialization of our cortical strip grid electrode and depth electrode technology, if approved; |
| · | any change in our plans regarding the manner in which we choose to commercialize any approved product in the United States or internationally; |
| · | the cost of ongoing compliance with regulatory requirements; |
| · | expenses we incur in connection with potential litigation or governmental investigations; |
| · | the costs to develop additional intellectual property: |
| · | anticipated or unanticipated capital expenditures; and |
| · | unanticipated general and administrative expenses. |
As a result of these and other factors, we do not know whether and the extent to which we may be required to raise additional capital. We may in the future seek additional capital from public or private offerings of our capital stock, borrowings under credit lines or other sources.
We may not be able to raise additional capital on terms acceptable to us, or at all. Any failure to raise additional capital could compromise our ability to execute on our business plan, and we may be forced to liquidate our assets. In such a scenario, the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements.
If we issue equity or debt securities to raise additional funds, our existing stockholders may experience dilution, and the new equity or debt securities may have rights, preferences and privileges senior to those of our existing stockholders. In addition, if we raise additional funds through collaborations, licensing, joint ventures, strategic alliances, partnership arrangements or other similar arrangements, it may be necessary to relinquish valuable rights to our potential future products or proprietary technologies, or grant licenses on terms that are not favorable to us.
Medical device development involves a lengthy and expensive process, with an uncertain outcome. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of any product.
Before obtaining marketing approval from regulatory authorities for the sale of our cortical strip, grid electrode and depth electrode technology under development in the United States or elsewhere, we must complete all pre-clinical testing, clinical trials and other regulatory requirements necessitated by the FDA and foreign regulatory bodies and demonstrate the performance and safety of our technology. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. Further, the outcomes of completed clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Clinical data is often susceptible to varying interpretations and analyses, and many companies that have believed their products performed satisfactorily in clinical trials have nonetheless failed to obtain marketing approval. We have limited resources to complete the expensive process of medical device development, pre-clinical testing and clinical trials, putting at a disadvantage, particularly compared to some of our larger and established competitors, and we may not have sufficient resources to commercialize our products under development in a timely fashion, if ever.
We may experience numerous unforeseen events during or as a result of clinical trials that could delay or prevent our ability to receive marketing approval or commercialize our products, including:
| 31 |
| · | regulators may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
| · | the failure to successfully complete pre-clinical testing requirements required by the FDA and international organizations; |
| · | we may experience delays in reaching, or fail to reach, agreement on acceptable clinical trial contracts with third parties or clinical trial protocols with prospective trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different trial sites; |
| · | clinical trials of our cortical strip, grid electrode and depth electrode technology may produce negative or inconclusive results, including failure to demonstrate statistical significance, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon our development programs; |
| · | the number of people with brain related disorders required for clinical trials may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or people may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
| · | our products may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or institutional review boards to suspend or terminate the trials; |
| · | our third-party contractors conducting the clinical trials may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| · | regulators may require that we or our investigators suspend or terminate clinical development for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
| · | the cost of clinical trials of our products may be greater than we anticipate; |
| · | the supply or quality of our products or other materials necessary to conduct clinical trials of our products may be insufficient or inadequate; and |
| · | delays from our suppliers and manufacturers could impact clinical trial completion and impact revenue. |
If we are required to conduct additional clinical trials or other testing of our cortical strip, grid electrode and depth electrode technology under development beyond those that we contemplate, if we are unable to successfully complete clinical trials of our cortical strip, grid electrode and depth electrode technology under development or other testing, if the results of these trials or tests are not favorable or if there are safety concerns, we may:
| · | not obtain marketing approval at all; |
| · | be delayed in obtaining marketing approval for our cortical strip, grid electrode and depth electrode technology under development in a jurisdiction; |
| · | be subject to additional post-marketing testing requirements; or |
| · | have our cortical strip, grid electrode and depth electrode technology removed from the market after obtaining marketing approval. |
Our development costs will also increase if we experience delays in testing or marketing approvals. We do not know whether any of our clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant clinical trial delays also could allow our competitors to bring innovative products to market before we do and impair our ability to successfully commercialize our products.
Changes in the configuration of our cortical strip, grid electrode and depth electrode technology under development may result in additional costs or delay.
As products are developed through pre-clinical testing and clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and configuration, are altered along the way in an effort to optimize processes and results. Any changes we make carry the risk that they will not achieve the intended objectives. Any of these changes could cause our products to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered device. Such changes may also require additional testing, regulatory notification or regulatory approval. This could delay completion of pre-clinical testing or clinical trials, increase costs, delay approval of our future products and jeopardize our ability to commence sales and generate revenue.
| 32 |
We have no products that are approved for commercial sale. If we are unable to successfully develop, receive regulatory approval for and commercialize our cortical strip, grid electrode and depth electrode technology under development, or if we experience significant delays in doing so, our business will be harmed.
We have no products that are approved for commercial sale. We initially plan to seek regulatory approval to commercialize our cortical strip, grid electrode and depth electrode technology under development in the U.S. and we may seek approval to commercialize in select international geographies. Our ability to generate revenue from our developed products, if any, will depend heavily on their successful development, regulatory approval and eventual commercialization. The success of any products that we develop will depend on several factors, including:
| · | FDA approval of our planned regulatory pathway (or approval of foreign regulatory body if we seek approval in any jurisdiction outside the U.S.); |
| · | successful completion of all necessary pre-clinical testing and clinical trials; |
| · | receipt of timely commercialization approvals from applicable regulatory authorities; |
| · | our ability to procure and maintain suppliers and manufacturers of the components of our current cortical strip, grid electrode and depth electrode technology and future versions; |
| · | launching commercial sales of our cortical strip, grid electrode and depth electrode technology, if approved for marketing; |
| · | market acceptance of our cortical strip, grid electrode and depth electrode technology, if approved, by people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders, the medical community and third-party payors; |
| · | our ability to obtain extensive coverage and reimbursement for our cortical strip, grid electrode and depth electrode technology and implantation procedures; |
| · | our success in educating healthcare providers and people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders about the benefits, administration and use of our cortical strip, grid electrode and depth electrode technology and future versions; |
| · | the prevalence and severity of adverse events; |
| · | the perceived advantages, cost, safety, convenience and accuracy of alternative therapies; |
| · | obtaining and maintaining patent, trademark and trade secret protection and regulatory exclusivity for our cortical strip, grid electrode and depth electrode technology and otherwise protecting our rights in our intellectual property portfolio; |
| · | maintaining compliance with regulatory requirements, including current good manufacturing practices; and |
| · | maintaining a continued acceptable performance and safety profile of our cortical strip, grid electrode and depth electrode technology following approval. |
Whether regulatory approval will be granted is unpredictable and depends upon numerous factors, including the substantial discretion of the regulatory authorities. Our success in clinical trials will not guarantee regulatory approval. The FDA and, if we seek to commercialize in select international geographies, other comparable foreign regulatory authorities may require that we conduct additional pre-clinical testing or clinical trials, provide additional data, take additional manufacturing steps, or require other conditions before they will grant us approval. If the FDA or other comparable foreign regulatory authorities require additional clinical trials or data, we would incur increased costs and delays in the marketing approval process, which may require us to expend more resources than we have available. In addition, the FDA or other comparable foreign regulatory authorities may not consider sufficient any additional required clinical trials, data or information that we perform and complete or generate.
In cases where we are successful in obtaining regulatory approval to market one or more of our products, our revenue will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to obtain coverage and reimbursement, and whether we own the commercial rights for that territory. If the number of people we target is not as significant as we estimate or the treatment population is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved.
| 33 |
Approval or clearance in the United States by the FDA or by a regulatory agency in another country does not guarantee approval by the regulatory authorities in other countries or jurisdictions or ensure approval for the same conditions of use. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. It is possible that no product we develop will never obtain regulatory approval in the United States or any other jurisdiction, even if we expend substantial time and resources seeking such approval. If we do not achieve one or more of these approvals in a timely manner or at all, we could experience significant delays or an inability to fully commercialize any product and achieve profitability.
Both before and after a product is commercially released, we will have ongoing responsibilities under U.S. and foreign regulations. We will also be subject to periodic inspections by the FDA and comparable foreign authorities to determine compliance with regulatory requirements, such as the Quality System Regulation, or QSR, of the FDA, medical device reporting regulations, vigilance in reporting of adverse events and regulations regarding notification, corrections, and recalls. These inspections can result in observations or reports, warning letters or other similar notices or forms of enforcement action. If the FDA or any comparable foreign authority concludes that we are not in compliance with applicable laws or regulations, or that any of our products are ineffective or pose an unreasonable health risk, such authority could ban these products, suspend or cancel our marketing authorizations, impose “stop-sale” and “stop-import” orders, refuse to issue export certificates, detain or seize adulterated or misbranded products, order a recall, repair, replacement, correction or refund of such products, or require us to notify health providers and others that the products present unreasonable risks of substantial harm to the public health. Discovery of previously unknown problems with our product's design or manufacture may result in restrictions on use, restrictions placed on us or our suppliers, or withdrawal of an existing regulatory clearance. The FDA or comparable foreign authorities may also impose operating restrictions, enjoin and restrain certain violations of applicable law pertaining to medical devices, assess civil or criminal penalties against our officers, employees or us, or recommend criminal prosecution of our company. Adverse regulatory action may restrict us from effectively marketing and selling our products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our business, financial condition, and operating results.
Foreign governmental regulations have become increasingly stringent and more extensive, and we may become subject to even more rigorous regulation by foreign governmental authorities in the future. Penalties for a company's noncompliance with foreign governmental regulation could be severe, including revocation or suspension of a company's business license and civil or criminal sanctions. In some jurisdictions, such as Germany, any violation of a law related to medical devices is also considered to be a violation of unfair competition law. In such cases, governmental authorities, our competitors and business or consumer associations may then file lawsuits to prohibit us from commercializing a product in such jurisdictions. Our competitors may also sue us for damages. Any domestic or foreign governmental law or regulation imposed in the future may have a material adverse effect on our business, financial condition and operating results.
Depending on the cost and market opportunity, we may never seek approval to commercialize our cortical strip, grid electrode and depth electrode technology in the European Union. We anticipate the cost to seek approval to commercialize in the EU will be significantly greater than the cost to seek approval to commercialize in the U.S. This is because we believe commercial approval by the corresponding Notified Body in the European Union and the European Economic Area, or EEA, even for diagnostic purposes, will require human clinical trials, which we do not believe will be required for regulatory approval by the FDA in the U.S. in order to seek approval of the use of our technology for diagnostic purposes.
Our success depends on our ability to continue to develop, commercialize and gain market acceptance for our product under development, our cortical strip, grid electrode and depth electrode technology.
Our current business strategy is highly dependent on developing and commercially launching one product, our cortical strip, grid electrode and depth electrode technology, and achieving and maintaining market acceptance. In order for us to sell cortical strip, grid electrode and depth electrode technology to people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders, we must convince them, their caregivers and healthcare providers that cortical strip, grid electrode and depth electrode technology is an attractive alternative to competitive products for neuromodulation cEEG and sEEG recording, ablation, and brain stimulation. Market acceptance and adoption of our cortical strip, grid electrode and depth electrode technology depends on educating people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders, as well as their caregivers and healthcare providers, and other perceived benefits of our cortical strip, grid electrode and depth electrode technology as compared to competitive products. We may face challenges convincing physicians, many of whom have extensive experience with competitors’ products and established relationships with other companies, to appreciate the benefits of our cortical strip, grid electrode and depth electrode technology and, in particular, its ability to successfully diagnose and treat epilepsy, Parkinson’s disease, and other brain related disorders in a way that is superior to and differentiated from currently available technology, and adopt it for treatment of their patients.
| 34 |
Achieving and maintaining market acceptance of cortical strip, grid electrode and depth electrode technology could be negatively impacted by many factors, including:
| · | the failure of our cortical strip, grid electrode and depth electrode technology to achieve wide acceptance among people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders, their caregivers, healthcare providers, third-party payors and key opinion leaders in the community; |
| · | lack of evidence supporting the performance criteria or other perceived benefits of our cortical strip, grid electrode and depth electrode technology over competitive products or other currently available technology; |
| · | perceived risks associated with the use of our cortical strip, grid electrode and depth electrode technology or similar products or technologies generally; |
| · | the introduction of competitive products and the rate of acceptance of those products as compared to our cortical strip, grid electrode and depth electrode technology; |
| · | adverse results of clinical trials relating to our cortical strip, grid electrode and depth electrode technology or similar competitive products; and |
| · | loss of regulatory approval for our cortical strip, grid electrode and depth electrode technology, adverse publicity or other adverse events including any product liability lawsuits. |
In addition, our cortical strip, grid electrode and depth electrode technology may be perceived by people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders, their caregivers or healthcare providers to be more complicated or less effective than current technology, and people may be unwilling to change their current regimens.
Moreover, we believe that healthcare providers tend to be slow to change their medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third-party reimbursement. Accordingly, healthcare providers may not recommend our cortical strip, grid electrode and depth electrode technology until, if ever, there is sufficient evidence to convince them to alter the treatment methods they typically recommend, such as receiving recommendations from prominent healthcare providers or other key opinion leaders in the community.
If we are not successful in convincing people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders of the benefits of our cortical strip, grid electrode and depth electrode technology, or if we are unable to achieve the support of caregivers and healthcare providers or widespread market acceptance for our cortical strip, grid electrode and depth electrode technology, then our sales potential, strategic objectives and profitability could be negatively impacted, which would adversely affect our business, financial condition and operating results.
We may fail to obtain regulatory approvals to market our products in the United States or in other countries.
Before we can market or sell a new regulated product in the United States, we must obtain either clearance under Section 510(k) of the FDCA or approval of a PMA application from the FDA, unless an exemption from pre-market review applies. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. Both the 510(k) and PMA processes can be expensive and lengthy and require the payment of significant fees, unless exempt. The FDA’s 510(k) clearance process usually takes from three to 12 months, but may last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is submitted to the FDA until an approval is obtained. The process of obtaining regulatory clearances or approvals to market a medical device can be costly and time-consuming, and we may not be able to obtain these clearances or approvals on a timely basis, if at all.
| 35 |
Even if we obtain clearance or approval by the FDA, said clearance or approval by the FDA does not ensure approval or certification by regulatory authorities in other countries or jurisdictions, and approval or certification by one foreign regulatory authority does not ensure approval or certification by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval or certification process may include all of the risks associated with obtaining FDA clearance or approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals or certifications and may not receive necessary approvals to commercialize our products in any market. If we fail to receive necessary approvals or certifications to commercialize our products in foreign jurisdictions on a timely basis, or at all, our business, results of operations and financial condition could be adversely affected.
Failure to secure or retain coverage or adequate reimbursement for our cortical strip, grid electrode and depth electrode technology or future versions of thereof, including the implantation procedures, by third-party payors could adversely affect our business, financial condition and operating results.
We plan to derive nearly all of our revenue from sales of our cortical strip, grid electrode and depth electrode technology under development, if approved, in the United States and potentially select international geographies and expect to do so for the next several years. We anticipate a substantial portion of the purchase price of our cortical strip, grid electrode and depth electrode technology will be paid for by third-party payors, including private insurance companies, preferred provider organizations and other managed care providers. Patients who receive treatment for their medical conditions and their healthcare providers generally rely on third-party payors to reimburse all or part of the costs associated with their medical treatment, including healthcare providers' services. Coverage and adequate reimbursement from third-party payors, including governmental healthcare programs, such as Medicare and Medicaid, and commercial payors, is critical to new product acceptance. Future sales of our cortical strip, grid electrode and depth electrode technology will be limited unless people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders can rely on third-party payors to pay for all or part of the cost to purchase our cortical strip, grid electrode and depth electrode technology. Access to adequate coverage and reimbursement for our cortical strip, grid electrode and depth electrode technology by third-party payors is essential to the acceptance of our products by people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders.
In the United States, a third-party payor's decision to provide coverage for our products does not imply that an adequate reimbursement rate will be obtained. Further, one third-party payor's decision to cover our products does not assure that other payors will also provide coverage for the products or will provide coverage at an adequate reimbursement rate. Healthcare providers may choose not to order a product unless third-party payors pay a substantial portion of the product. Within and outside the United States, reimbursement is obtained from a variety of sources, including government-sponsored and private health insurance plans. These third-party payors determine whether to provide coverage and reimbursement for specific products and procedures. Coverage determinations and reimbursement levels of both our products and the healthcare provider's performance of the insertion and removal procedures are critical to the commercial success of our product, and if we are not able to secure positive coverage determinations and reimbursement levels for our products or the insertion and removal procedures, our business would be materially adversely affected.
In addition, there may be significant delays in obtaining reimbursement, and coverage may be more limited than the purposes for which the product is cleared by the FDA or other foreign regulatory authorities. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower cost products that are already reimbursed, and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or third-party payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States.
| 36 |
Because there is generally no separate reimbursement for medical devices and other supplies used in such procedures, including our cortical strip, grid electrode and depth electrode technology, and because we believe that our cortical strip, grid electrode and depth electrode technology, if approved, would be adequately described by existing DRG and ICD-9 codes for epilepsy surgery, some of our target customers may be unwilling to adopt our cortical strip, grid electrode and depth electrode technology over more established or lower cost therapeutic alternatives already available or subsequently become available. Further, any decline in the amount payors are willing to reimburse our customers for procedures using our cortical strip, grid electrode and depth electrode technology could make it difficult for new customers to adopt our cortical strip, grid electrode and depth electrode technology and could create additional pricing pressure for us, which could adversely affect our ability to invest in and grow our business.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, no uniform policy of coverage and reimbursement for medical device products and services exists among third-party payors. Therefore, coverage and reimbursement for medical device products and services can differ significantly from payor to payor. In addition, payors continually review new technologies for possible coverage and can, without notice, deny coverage for these new products and procedures. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained, or maintained if obtained.
Reimbursement systems in international markets vary significantly by country and by region within some countries, and reimbursement approvals must be obtained on a country-by-country basis. In many international markets, a product must be approved for reimbursement before it can be approved for sale in that country. Further, many international markets have government-managed healthcare systems that control reimbursement for new devices and procedures. In most markets there are private insurance systems as well as government-managed systems. If sufficient coverage and reimbursement is not available for our any product we develop, in either the United States or internationally, the demand for our products and our revenues will be adversely affected.
Reimbursement by Medicare is highly regulated and subject to change.
The Medicare program is administered by CMS, which imposes extensive and detailed requirements on medical services providers, including, but not limited to, rules that govern how we structure our relationships with physicians, and how and where we provide our solutions. Our failure to comply with applicable Medicare rules could result in discontinuing the ability for physicians to receive reimbursement as they will likely utilize our cortical strip, grid electrode and depth electrode technology under the Medicare payment program, civil monetary penalties, and/or criminal penalties, any of which could have a material adverse effect on our business and revenues.
The impact of the Patient Protection and Affordable Care Act remains uncertain.
In 2010, significant reforms to the health care system were adopted as law in the United States. The law includes provisions that, among other things, reduce or limit Medicare reimbursement, require all individuals to have health insurance (with limited exceptions) and impose increased taxes. These factors, in turn, could result in reduced demand for our products, if approved, and increased downward pricing pressure. Because other parts of the 2010 health care law remain subject to implementation, the long-term impact on us is uncertain. The new law or any future legislation could reduce medical procedure volumes, lower reimbursement for our products, and impact the demand for our products or the prices at which we sell our products. Accordingly, while it is too early to understand and predict the ultimate impact of the law on our business, the legislation and resulting regulations could have a material adverse effect on our business, cash flows, financial condition and results of operations.
If our competitors are better able to develop and market products for the diagnosis and treatment of epilepsy, Parkinson’s disease, essential tremors and other brain related disorders that are safer, more effective, less costly, easier to use or otherwise more attractive than our cortical strip, grid electrode and depth electrode technology, our business will be adversely impacted.
The medical device industry is highly competitive and subject to technological change. Our success depends, in part, upon our ability to establish a competitive position in the market for the diagnosis and treatment of epilepsy, Parkinson’s disease, essential tremors and other brain related disorders by securing broad market acceptance of our cortical strip, grid electrode and depth electrode technology under development. Any product we develop that achieves regulatory clearance or approval will have to compete for market acceptance and market share. If developed as anticipated, we believe that the primary competitive factors of our cortical strip, grid electrode and depth electrode technology under development will be: product efficacy, reduced infections, ability to record additional brain activity, minimally invasive surgical procedure, ease of use and cost effectiveness. We face significant competition in the United States and internationally, which we believe will intensify. For example, our major competitors (i) in the market for diagnosis are PMT Corporation, Ad-Tec Medical and Integra Lifesciences, (ii) in the market for neuro-ablation are Medtronic and Monteris Medical and (iii) in the market for neurostimulation are Medtronic, Boston Scientific, NeuroPace Biotronik and Abbott. Each of the foregoing competitors has systems approved in the U.S. and certain foreign jurisdictions and has been established for several years. We face a particular challenge overcoming the long-standing practices by some physicians of using the existing technology of our larger, more established competitors. Physicians may be reluctant to try new products from a source with which they are less familiar. If these physicians do not try and subsequently adopt our product, then our revenue growth will slow or decline.
| 37 |
In addition to these major competitors, we may also face competition from other emerging competitors or smaller companies with active development programs that may emerge in the future.
Many of the companies developing or marketing competing products enjoy several advantages over us, including:
| • | more experienced sales forces; | |
| • | greater name recognition; | |
| • | more established sales and marketing programs and distribution networks; | |
| • | earlier regulatory approval in the U.S. or foreign jurisdictions; | |
| • | long established relationships with physicians and hospitals; | |
| • | significant patent portfolios, including issued U.S. and foreign patents and pending patent applications, as well as the resources to enforce patents against us or any of our third-party suppliers and distributors; | |
| • | the ability to acquire and integrate our competitors and/or their technology; | |
| • | demonstrated ability to develop product enhancements and new product offerings; | |
| • | established history of product reliability, safety and durability; | |
| • | the ability to offer rebates or bundle multiple product offerings to offer greater discounts or incentives; | |
| • | greater financial and human resources for product development, sales, and marketing; and | |
| • | greater experience in and resources for conducting research and development, clinical studies, manufacturing, preparing regulatory submissions, obtaining regulatory clearance or approval for products and marketing approved products. |
Our competitors may develop and patent processes or products earlier than us, obtain patents that may apply to us at any time, obtain regulatory clearance or approvals for competing products more rapidly than us or develop more effective or less expensive products or technologies that render our technology or products obsolete or less competitive. Furthermore, the frequent introduction by competitors of products that are, or claim to be, superior to our products may create market confusion that may make it difficult to differentiate the benefits of our products over competitive products. In addition, the entry of multiple new products may lead some of our competitors to employ pricing strategies that could adversely affect the pricing of any product we may develop and commercialize. We also face fierce competition in recruiting and retaining qualified sales, scientific, and management personnel, establishing clinical trial sites and enrolling patients in clinical studies. If our competitors are more successful than us in these matters, our business may be harmed.
| 38 |
The size and future growth in the market for our cortical strip, grid electrode and depth electrode technology under development has not been established with precision and may be smaller than we estimate, possibly materially. If our estimates and projections overestimate the size of this market, our sales growth may be adversely affected.
Our estimates of the size and future growth in the market for our cortical strip, grid electrode and depth electrode technology under development, including the number of people with epilepsy, Parkinson’s disease, essential tremors and other brain related disorders who may benefit from and be amenable to using cortical strip, grid electrode and depth electrode technology for diagnosis and treatment, is based on a number of internal and third-party studies, reports and estimates. In addition, our internal estimates are based in large part on current treatment patterns by healthcare providers using current generation technology and our belief is that the incidence of epilepsy, Parkinson’s disease, essential tremors and other brain related disorders in the United States and worldwide is increasing. While we believe these factors have historically provided and may continue to provide us with effective tools in estimating the total market for cortical strip, grid electrode and depth electrode technology, these estimates may not be correct and the conditions supporting our estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. The actual incidence of brain related disorders, and the actual demand for our products or competitive products, could differ materially from our projections if our assumptions are incorrect. As a result, our estimates of the size and future growth in the market for cortical strip, grid electrode and depth electrode technology may prove to be incorrect. If the actual number of people with brain related disorders who would benefit from cortical strip, grid electrode and depth electrode technology and the size and future growth in the market for cortical strip, grid electrode and depth electrode technology is smaller than we have estimated, it may impair our projected sales growth and have an adverse impact on our business.
We depend on intellectual property licensed from Wisconsin Alumni Research Foundation for our technology under development, and the termination of this license would harm our business.
Wisconsin Alumni Research Foundation, or WARF, has granted us an exclusive license, or the WARF License, to make, use and sell, in the United States only, products that employ certain licensed patents for a neural probe array or thin-film micro electrode array and method. See “Business — WARF License” for additional information regarding our license agreement with WARF.
We have agreed to diligently develop, manufacture, market and sell products under the WARF License in the United States during the term of the agreement and, specifically, that we will submit a business plan to WARF by February 1, 2018 and file an application for 510(k) marketing clearance with the FDA by February 1, 2019. WARF may terminate this license in the event that we fail to meet these milestones on 30 days’ written notice, if we default on the payments of amounts due to WARF or fail to timely submit development reports, actively pursue our development plan or breach any other covenant in the WARF License and fail to remedy such default in 90 days or in the event of certain bankruptcy events involving us. WARF may also terminate this license (i) on 90 days’ notice if we fail to have commercial sales of one or more FDA-approved products under the WARF License by March 31, 2019 or (ii) if, after royalties earned on sales begin to be paid, such earned royalties cease for more than four calendar quarters. The WARF License otherwise expires by its terms on the date that no valid claims on the patents licensed thereunder remain.
Disputes may arise between us and WARF regarding intellectual property subject to this agreement, including with respect to: the scope of rights granted under the WARF License and other interpretation-related issues; whether and the extent to which our technology and processes infringe on intellectual property of WARF that is not subject to the WARF License; the amount and timing of milestones and royalty payments; the rights of WARF under the license; our right to sublicense; and the ownership of inventions and know-how resulting from the WARF License. For example, if we or any of our sublicenses for any reason contest the validity of any patent licensed under the WARF License, the royalty rate will be doubled during the pendency of such contest and, if the contested patent is found to be valid and would be infringed by us if not for the WARF License, the royalty rate will be tripled for the remaining term of the WARF License.
Any disputes with WARF may prevent or impair our ability to maintain our current licensing arrangement. We depend on the intellectual property licensed from WARF to develop our cortical strip, grid electrode and depth electrode technology. We cannot assure you that we will be able to meet the milestones or commercialize a product under the WARF License by the dates required. In fact, the original license agreement entered into with WARF in 2014 required that we meet certain earlier milestones than set forth above and make certain payments to WARF. We failed to do so and were in default under the original license agreement. Furthermore, the LLC was not able to transfer the rights and obligations under the 2014 WARF Agreement to us at the time of the Merger without the consent of WARF. As a result, in February 2017, we signed an amendment to the WARF License which, among other things, modified and removed certain previous milestones and provided WARF’s consent to such transfer. Because of this past breach, WARF may be less likely to waive future defaults or breaches or further amend the WARF License in the future, to the extent we request any waiver or amendment. See “Note 4 – Intellectual Property” to the financial statements included in this Report.
| 39 |
Termination of our license could result in the loss of significant rights and would harm our ability to further develop our cortical strip, grid electrode and depth electrode technology. In addition, WARF reserves the right to grant non-profit research institutions and government agencies non-exclusive licenses to practice and use the inventions of the licensed patents for non-commercial research purposes, and we grant WARF a non-exclusive, sub licensable, royalty-free right and license for non-commercial research purposes to use improvements to the licensed patents. In the event that we discontinue use or commercialization of the licensed patents or improvements thereon, we must grant WARF an option to obtain a non-exclusive, sub-licensable royalty-bearing license to use the improvements for commercial purposes. Such rights, if exercised by WARF, could harm our ability to develop and commercialize our cortical strip, grid electrode and depth electrode technology.
We depend on our partnership with Mayo Foundation for Medical Education and Research to license certain know how for the development and commercialization of our technology. Termination of this partnership would harm our business, and even if this partnership continues, it may not be successful.
We have entered into the Mayo Development Agreement to (i) exclusively license worldwide certain Mayo improvements for the development and commercialization of products, methods and processes related to flexible circuit technology for the recording and stimulation of tissue and (ii) license, on a non-exclusive basis, worldwide Mayo thin film electrode technology know-how for the development and commercialization of products, methods and processes related to flexible circuit technology for the recording and stimulation of tissue. Mayo has agreed to assist the Company by providing access to the Mayo Principal Investigators in developing a minimally invasive device/delivery system and procedure for a minimally invasive approach for the implantation of any flexible circuit technology developed by the Company, including prototype development, animal testing, protocol development for human and animal use, abstract development and presentation and access to and license of any intellectual property that the Mayo Principal Investigators develop relating to the procedure. See “Business—Mayo Foundation for Medical Education and Research License and Development Agreement” for additional information regarding our agreement with Mayo.
Mayo Development Agreement generally will expire in October 2034, unless the Mayo know-how and improvements under the Mayo Development Agreement remain in use, and the Mayo Development Agreement may be terminated by Mayo for cause or under certain circumstances. Mayo and the Company may not be successful in their efforts to develop any product, method, process, device, delivery system or minimally invasive approach by such expiration date or termination, if at all. If no such minimally invasive device or delivery system and procedure for minimally invasive approach is developed, the Company may never receive regulatory approval of its cortical strip, grid electrode and depth electrode technology under development or the market may never accept such technology, if approved.
Disputes may arise between us and Mayo regarding intellectual property subject to the Mayo Development Agreement or other matters, including with respect to: the scope of rights granted under the agreement and other interpretation-related issues; the amount and timing of payments; the rights and obligations of Mayo under the license agreement; and the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by Mayo and us.
Any disputes with Mayo may prevent or impair our ability to maintain our current arrangement. We depend on the intellectual property licensed from and development assistance from Mayo to develop our cortical strip, grid electrode and depth electrode technology. We cannot assure you that we will be able to continue to comply with the Mayo Development Agreement. In fact, the original license and development agreement entered into with Mayo in 2014 required that, upon the Merger with the LLC, we make certain payments and issue shares of common stock to Mayo, which we failed to do at such time. We signed the Mayo Development Agreement in May 2017, which, among other things, modified or removed certain provisions of the original agreement, including those we breached. Because of this past breach, Mayo may be less likely to waive future defaults or breaches or further amend the Mayo Development Agreement in the future, to the extent we request any waiver or amendment. See “Note 4 – Intellectual Property” to the financial statements included in this Report. Termination of the Mayo Development Agreement could result in the loss of significant rights and would harm our ability to further develop our technology.
We do not have the sales and marketing personnel necessary to sell any products we may develop, if approved for commercialization. Even if we have our cortical strip, grid electrode and depth electrode technology approved for commercial sale, if we are unable to establish a sales and marketing infrastructure, we may not be successful in commercializing our cortical strip, grid electrode and depth electrode technology in the United States.
We are an early stage development company with limited resources. Even if we had products available for sale, which we currently do not, we have not secured sales and marketing staff at this early stage of operations to sell products. To achieve commercial success in the United States for our cortical strip, grid electrode and depth electrode technology, we will need to establish and expand our sales and marketing infrastructure to drive adoption of our products, which will include a team of educators that will train healthcare providers and people with brain related disorders on the benefits and use of our cortical strip, grid electrode and depth electrode technology. There is significant competition for sales personnel experienced in relevant medical device sales. We expect that we will face significant challenges as we recruit and subsequently grow our sales and marketing infrastructure. If we are unable to attract and retain sufficient, and skilled, sales and marketing representatives, our sales could be adversely affected. If one of our sales or marketing representatives were to depart and be retained by one of our competitors, they could help competitors solicit business from customers, if any, which could further harm our sales. In addition, if our sales and marketing representatives or educators fail to achieve their objectives or if we are not able to recruit and retain a network of educators, we may not be able to successfully train healthcare providers on the use of our cortical strip, grid electrode and depth electrode technology, which could delay new sales and harm our reputation.
| 40 |
As we increase our sales and marketing expenditures with respect to our cortical strip, grid electrode and depth electrode technology under development, if approved, or future versions thereof, we will need to hire, train, retain and motivate skilled sales and marketing representatives with significant industry-specific knowledge in various areas. Our success will depend largely on the competitive landscape for our products and the ability of our sales personnel to obtain access to healthcare providers and persuade those healthcare providers to recommend our cortical strip, grid electrode and depth electrode technology. Recently hired sales representatives require training and take time to achieve full productivity. If we fail to train new hires adequately, or if we experience high turnover in our sales force in the future, we cannot be certain that new hires will become as productive as may be necessary to maintain or increase our sales. In addition, the expansion of our sales and marketing personnel will place significant burdens on our management team.
If approved for sale, we anticipate that we will derive nearly all of our U.S. revenue from the sales of our cortical strip, grid electrode and depth electrode technology or future versions thereof. As a result, our financial condition and operating results will be highly dependent on the ability of our sales representatives to adequately promote, market and sell our cortical strip, grid electrode and depth electrode technology and the ability of our educators to train healthcare providers on the use of our cortical strip, grid electrode and depth electrode technology. If we are unable to establish and expand our sales and marketing capabilities, we may not be able to effectively commercialize our existing or planned products, or enhance the strength of our brand, either of which could impair our projected sales growth and have an adverse impact on our business.
We will depend on a limited number of third-party suppliers for the components of our cortical strip, grid electrode and depth electrode technology under development and the loss of any of these suppliers, or their inability to provide us with an adequate supply of materials, could harm our business.
We will rely on third-party suppliers to supply and manufacture the components of our cortical strip, grid electrode and depth electrode technology. For our business strategy to be successful, our suppliers must be able to provide us with components in sufficient quantities, in compliance with regulatory requirements and quality control standards, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. Future increases in sales of our cortical strip and sheet electrode technology, if approved, whether expected or unanticipated, could strain the ability of our suppliers to deliver an increasingly large supply of components and our cortical strip, grid electrode and depth electrode technology in a manner that meets these various requirements.
We will likely use a small number of suppliers of components for our products. Depending on a limited number of suppliers exposes us to risks, including limited control over pricing, availability, quality and delivery schedules. We may not have long-term supply agreements with our suppliers and, in many cases, we may make our purchases on a purchase order basis. Our ability to purchase adequate quantities of components or our products may be limited and we may not be able to convince suppliers to make components and products available to us. Additionally, our suppliers may encounter problems that limit their ability to supply components or manufacture products for us, including financial difficulties, damage to their manufacturing equipment or facilities, or product discontinuations. As a result, there is a risk that certain components could be discontinued and no longer available to us. We may be required to make significant “last time” purchases of component inventory that is being discontinued by the supplier to ensure supply continuity. If we fail to obtain sufficient quantities of high quality components to meet demand for our products in a timely manner or on terms acceptable to us, we would have to seek alternative sources of supply. Because of factors such as the proprietary nature of our products, our quality control standards and regulatory requirements, we may not be able to quickly engage additional or replacement suppliers for some of our critical components. Failure of any supplier to deliver components at the level our business requires could disrupt the manufacturing of our products and, if approved, limit our ability to meet our sales commitments, which could harm our reputation and adversely affect our business.
| 41 |
Furthermore, vandalism, terrorism or a natural or other disaster, such as an earthquake, fire or flood, could damage or destroy equipment or our inventory of component supplies or finished products, cause substantial delays in development or our operations, result in the loss of key information, and cause us to incur additional expenses. We do not currently have insurance to cover such losses or expenses and, once we obtain such insurance, it may not cover our losses in any particular case. In addition, regardless of the level of insurance coverage, damage to our or our suppliers' facilities could harm our business, financial condition and operating results.
We may also have difficulty obtaining similar components from other suppliers that are acceptable to the FDA or other regulatory agencies, and the failure of any supplier to comply with strictly enforced regulatory requirements could expose us to regulatory action including warning letters, product recalls, and termination of distribution, product seizures or civil penalties. It could also require us to cease using the components, seek alternative components or technologies and modify our products to incorporate alternative components or technologies, which could result in a requirement to seek additional regulatory approvals. Any disruption of this nature or increased expenses could harm our development, approval or commercialization efforts and adversely affect our operating results.
We plan to contract with third parties for the manufacture of our cortical strip, grid electrode and depth electrode technology under development and expect to continue to do so for clinical trials and commercialization. Risks associated with the manufacturing of our products could reduce our gross margins and negatively affect our operating results.
We currently rely, and expect to continue to rely, on third parties for the manufacture of our cortical strip, grid electrode and depth electrode technology during development, for clinical testing, as well as for commercial manufacture if our cortical strip, grid electrode and depth electrode technology receives regulatory approval. Therefore, our business strategy depends on our third-party manufacturers' ability to manufacture our cortical strip, grid electrode and depth electrode technology and future generations thereof in sufficient quantities and on a timely basis so as to meet consumer demand, while adhering to product quality standards, complying with regulatory requirements and managing manufacturing costs. To date, we have only had an initial supply of our product manufactured. As a result, we currently have limited data and experience regarding the quality, reliability and timeliness of our third-party manufacturers.
We are subject to numerous risks relating to the manufacturing capabilities of our third-party manufacturers, including:
| · | quality or reliability defects; |
| · | inability to secure product components in a timely manner, in sufficient quantities or on commercially reasonable terms; |
| · | failure to increase production to meet demand; |
| · | inability to modify production lines to enable us to efficiently produce future products or implement changes in current products in response to regulatory requirements; |
| · | difficulty identifying and qualifying alternative manufacturers in a timely manner; |
| · | inability to manufacture product components cost-effectively; |
| · | inability to establish agreements with future third-party manufacturers or to do so on acceptable terms; or |
| · | potential damage to or destruction of our manufacturers' equipment or facilities. |
These risks are likely to be exacerbated by our limited experience with our cortical strip, grid electrode and depth electrode technology and its manufacturing process. As demand for our products increases, our third-party suppliers will need to invest additional resources to purchase components, hire and train employees, and enhance their manufacturing processes. If our manufacturers fail to increase production capacity efficiently, our sales may not increase in line with our expectations and our operating margins could fluctuate or decline. In addition, manufacturing any future versions of our cortical strip, grid electrode and depth electrode technology may require the modification of production lines, the identification of new manufacturers for specific components, or the development of new manufacturing technologies. It may not be possible for us to manufacture these products at a cost or in quantities sufficient to make any future versions of our cortical strip, grid electrode and depth electrode technology commercially viable.
| 42 |
If we or our third-party suppliers or manufacturers fail to comply with the FDA's good manufacturing practice regulations, this could impair our ability to market our products in a cost-effective and timely manner.
We and our third-party suppliers are required to comply with the FDA's QSR, which covers the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. The FDA audits compliance with the QSR through periodic announced and unannounced inspections of manufacturing and other facilities. The FDA may impose inspections or audits at any time. If we or our suppliers or manufacturers have significant non-compliance issues or if any corrective action plan that we or our suppliers propose in response to observed deficiencies is not sufficient, the FDA could take enforcement action against us. Any of the foregoing actions could impair our reputation, business, financial condition and operating results.
Various factors outside our direct control may adversely affect manufacturing, sterilization and distribution of our products.
The manufacture, sterilization and distribution of our products is challenging. Changes that our suppliers may make outside the purview of our direct control can have an impact on our processes, quality of our products and the successful delivery of products to our customers. Necessary materials for our product under development may not be available from our third-party suppliers in a timely fashion or at all. Mistakes and mishandling are not uncommon and can affect supply and delivery. Some of these risks include:
| · | failure to complete sterilization on time or in compliance with the required regulatory standards; |
| · | transportation and import and export risk; |
| · | delays in analytical results or failure of analytical techniques that we will depend on for quality control and release of products; |
| · | natural disasters, labor disputes, financial distress, raw material availability, issues with facilities and equipment or other forms of disruption to business operations affecting our manufacturers or suppliers; and |
| · | latent defects that may become apparent after products have been released and that may result in a recall of such products. |
If any of these risks were to materialize, our ability to develop products, conduct clinical trials or provide our products to customers on a timely basis, if approved, would be adversely impacted.
Potential complications from our cortical strip, grid electrode and depth electrode technology may come to light or may not be revealed by our clinical experience.
Based on our industry experience and the experience of the physicians that use products similar to our cortical strip, grid electrode and depth electrode technology, complications from use of our cortical strip, grid electrode and depth electrode technology may include post-operative hemorrhage, infection, brain inflammation, brain tissue necrosis, inability to accurately localize the epileptogenic focus (the area of the cerebral cortex responsible for causing epileptic seizures), neurologic deficit (abnormal function of a body area due to weaker function of the brain, spinal cord, muscles or nerves, such as abnormal reflexes, inability to speak and decreased sensation) and extra axial fluid collections (fluid that occurs in the brain after surgery). If these or unanticipated complications or side-effects result from the use of our cortical strip, grid electrode and depth electrode technology, our product development may be delayed, we may not be able to obtain regulatory approval for any product, we could be subject to liability and, even if approved, our technology would not be widely adopted. Additionally, we have no clinical experience with use of our cortical strip, grid electrode and depth electrode technology. We cannot assure you that use, even for a limited time, would not result in unanticipated complications, even after the device is removed.
Undetected errors or defects in our cortical strip, grid electrode and depth electrode technology under development or future versions thereof could harm our reputation, decrease the market acceptance of our cortical strip, grid electrode and depth electrode technology or expose us to product liability claims.
Our cortical strip, grid electrode and depth electrode technology may contain undetected errors or defects. Disruptions or other performance problems with our cortical strip, grid electrode and depth electrode technology may delay development, prevent regulatory approval or harm our reputation. If that occurs, we may incur significant costs, the attention of our key personnel could be diverted or other significant customer relations problems may arise. We may also be subject to warranty and liability claims for damages related to errors or defects in our cortical strip and sheet electrode technology or future versions thereof. A material liability claim or other occurrence that harms our reputation or decreases market acceptance of our cortical strip, grid electrode and depth electrode technology could harm our business and operating results. This risk exists even if a device is cleared or approved for commercial sale and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority. Our products are designed to affect, and any future products will be designed to affect, important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our cortical strip, grid electrode and depth electrode technology or future versions thereof could result in patient injury or death. The medical device industry has historically been subject to extensive litigation over product liability claims, and we cannot offer any assurance that we will not face product liability lawsuits.
| 43 |
The sale and use of our cortical strip, grid electrode and depth electrode technology or future versions thereof could lead to the filing of product liability claims if someone were to allege that our cortical strip, grid electrode and depth electrode technology or one of our products contained a design or manufacturing defect. A product liability claim could result in substantial damages and be costly and time consuming to defend, either of which could materially harm our business or financial condition. Product liability claims may be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our products, among others. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| · | costs of litigation; |
| · | distraction of management's attention from our primary business; |
| · | the inability to commercialize our cortical strip, grid electrode and depth electrode technology; |
| · | decreased demand; |
| · | damage to our business reputation; |
| · | product recalls or withdrawals from the market; |
| · | withdrawal of clinical trial participants; |
| · | substantial monetary awards to patients or other claimants; or |
| · | loss of revenue. |
Product liability lawsuits and claims, safety alerts or product recalls, with or without merit, could cause us to incur substantial costs, delay our product development efforts, place a significant strain on our financial resources, divert the attention of management from our core business, harm our reputation, increase our product liability insurance rates, once we obtain such insurance, or prevent us from securing such insurance coverage in the future and adversely affect our ability to attract and retain customers, if approved, any of which could harm our business, financial condition and operating results.
We do not currently maintain any product liability insurance and do not anticipate obtaining product liability insurance until we commence clinical trials. Once we obtain such insurance, we cannot assure you that such insurance would adequately protect our assets from the financial impact of defending a product liability claim. Even if any product liability loss is covered by an insurance policy, these policies typically have substantial deductibles for which we are responsible. Product liability claims in excess of applicable insurance coverage would negatively impact our business, financial condition and operating results. Insurance coverage varies in cost and can be difficult to obtain, and we cannot guarantee that we will be able to obtain insurance coverage in the future on terms acceptable to us or at all.
If there are significant disruptions in our information technology systems, our business, financial condition and operating results could be adversely affected.
The efficient operation of our business depends on our information technology systems. We rely on our information technology systems to effectively manage product development tasks, research and development data and accounting and financial functions. We expect in the future we will rely on our information technology systems for inventory management and technical support functions, if and once implemented. Our information technology systems are vulnerable to damage or interruption from earthquakes, fires, floods and other natural disasters, terrorist attacks, attacks by computer viruses or hackers, power losses, and computer system or data network failures. In addition, our data management application and a variety of our software systems are hosted by third-party service providers whose security and information technology systems are subject to similar risks, which could be subject to computer viruses or hacker attacks or other failures. If our or our third-party service provider's security systems are breached or fail, unauthorized persons may be able to obtain access to sensitive data.
| 44 |
The failure of our or our service providers' information technology systems or our transmitter's software to perform as we anticipate or our failure to effectively implement new information technology systems could disrupt our entire operation or adversely affect our products and could delay our product development, clinical trial or commercialization efforts, result in increased overhead costs and damage our reputation, all of which could negatively affect our business, financial condition and operating results.
We have no business insurance; any unanticipated events or expenses may hurt our business substantially.
We have no general liability or umbrella liability insurance, no business liability or disruption insurance and no criminal insurance. Any unanticipated events or expenses may result in our incurring substantial costs and the diversion of our resources and hurt our business substantially.
We may enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances or partnerships with third-parties that may not result in the development of commercially viable products or the generation of significant future revenues.
In the ordinary course of our business, we may enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances, partnerships or other arrangements to develop products and to pursue new markets. Proposing, negotiating and implementing collaborations, in-licensing arrangements, joint ventures, strategic alliances or partnerships may be a lengthy and complex process. Other companies, including those with substantially greater financial, marketing, sales, technology or other business resources, may compete with us for these opportunities or arrangements. We may not identify, secure, or complete any such transactions or arrangements in a timely manner, on a cost-effective basis, on acceptable terms or at all. We have limited institutional knowledge and experience with respect to these business development activities, and we may also not realize the anticipated benefits of any such transaction or arrangement. In particular, these collaborations may not result in the development of products that achieve commercial success or result in significant revenues and could be terminated prior to developing any products.
Additionally, we may not be in a position to exercise sole decision making authority regarding the transaction or arrangement, which could create the potential risk of creating impasses on decisions, and our future collaborators may have economic or business interests or goals that are, or that may become, inconsistent with our business interests or goals. It is possible that conflicts may arise with our collaborators, such as conflicts concerning the achievement of performance milestones, or the interpretation of significant terms under any agreement, such as those related to financial obligations or the ownership or control of intellectual property developed during the collaboration. If any conflicts arise with any future collaborators, they may act in their self-interest, which may be adverse to our best interest, and they may breach their obligations to us. In addition, we may have limited control over the amount and timing of resources that any future collaborators devote to our or their future products. Disputes between us and our collaborators may result in litigation or arbitration which would increase our expenses and divert the attention of our management. Further, these transactions and arrangements will be contractual in nature and will generally be terminable under the terms of the applicable agreements and, in such event, we may not continue to have rights to the products relating to such transaction or arrangement or may need to purchase such rights at a premium.
If we enter into in-bound intellectual property license agreements, we may not be able to fully protect the licensed intellectual property rights or maintain those licenses. Future licensors could retain the right to prosecute and defend the intellectual property rights licensed to us, in which case we would depend on the ability of our licensors to obtain, maintain and enforce intellectual property protection for the licensed intellectual property. These licensors may determine not to pursue litigation against other companies or may pursue such litigation less aggressively than we would. Further, entering into such license agreements could impose various diligence, commercialization, royalty or other obligations on us. Future licensors may allege that we have breached our license agreement with them, and accordingly seek to terminate our license, which could adversely affect our competitive business position and harm our business prospects.
| 45 |
We may seek to grow our business through acquisitions of complementary products or technologies, and the failure to manage acquisitions, or the failure to integrate them with our existing business, could harm our business, financial condition and operating results.
From time to time, we may consider opportunities to acquire other companies, products or technologies that may enhance our product platform or technology, expand the breadth of our markets or customer base, or advance our business strategies. Potential acquisitions involve numerous risks, including:
| · | problems assimilating the acquired products or technologies; |
| · | issues maintaining uniform standards, procedures, controls and policies; |
| · | unanticipated costs associated with acquisitions; |
| · | diversion of management's attention from our existing business; |
| · | risks associated with entering new markets in which we have limited or no experience; |
| · | increased legal and accounting costs relating to the acquisitions or compliance with regulatory matters; and |
| · | unanticipated or undisclosed liabilities of any target. |
We have no current commitments with respect to any acquisition. We do not know if we will be able to identify acquisitions we deem suitable, whether we will be able to successfully complete any such acquisitions on favorable terms or at all, or whether we will be able to successfully integrate any acquired products or technologies. Our potential inability to integrate any acquired products or technologies effectively may adversely affect our business, operating results and financial condition.
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on the management, research and development, clinical, financial and business development expertise of David Rosa, Mark Christianson and Thomas Bachinski, as well as our scientific and physician advisory board members. Although we have an employment agreement with David Rosa, he (and each of our other key employees) may terminate his employment with us at any time and will continue to be able to do so. We do not maintain “key person” insurance for any of our executives or employees.
Recruiting and retaining qualified scientific and clinical personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize our products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous medical device companies for similar personnel, many of which have greater financial and other resources dedicated to attracting and retaining personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
Prolonged negative economic conditions could adversely affect us, our customers and third-party partners, manufactures or suppliers, if any, which could harm our financial condition.
We are subject to the risks arising from adverse changes in general economic and market conditions. Uncertainty about future economic conditions could negatively impact our existing and potential customers, adversely affect the financial ability of health insurers to pay claims, adversely impact our expenses and ability to obtain financing of our operations, and cause delays or other problems with key suppliers.
Healthcare spending in Europe and the United States has been, and is expected to continue to be, under significant pressure and there are many initiatives to reduce healthcare costs. As a result, we believe that some insurers are scrutinizing insurance claims more rigorously and delaying or denying coverage and reimbursement more often. Because the sale, if approved, of our cortical strip, grid electrode and depth electrode technology under development will generally depend on the availability of third-party coverage and reimbursement, any delay or decline in coverage and reimbursement will adversely affect our sales.
| 46 |
Risks Related to our Intellectual Property
Our ability to protect our intellectual property and proprietary technology is uncertain.
We rely primarily on patent, trademark and trade secret laws, as well as confidentiality and non-disclosure agreements, to protect our proprietary technologies. Our patent estate consists of 3 issued United States patents licensed from WARF covering a neural probe array and thin-film micro electrode array and method, and one pending United States provisional patent application filed by us covering a wide variety of concepts, ranging from accessories for brain surgery to ablation and stimulation concepts for both cortical and depth electrodes. The licensed issued patents expire between 2025 and 2030, subject to any patent extensions that may be available for such patents. If a patent is issued on our pending patent application, the resulting patent is projected to expire in 2038. We continue to review new technological developments in order to make decisions about what additional filings would be the most appropriate for us. We also plan to seek patent protection for our proprietary technology in select countries internationally. We also have one pending U.S. trademark application and one pending foreign trademark application, as well as one foreign trademark registration. We have applied for patent protection relating to certain existing and proposed products and processes. Currently, several of our issued U.S. patents licensed from WARF as well as our pending U.S. patent application relate to our cortical and depth electrode technologies and are therefore important to the functionality of our products. If we fail to timely file a patent application in any jurisdiction, we may be precluded from doing so at a later date. Furthermore, we cannot assure you that any patent application will be approved in a timely manner or at all. The rights granted to us under our patents, and the rights we are seeking to have granted in our pending patent applications, may not be meaningful or provide us with any commercial advantage. In addition, those rights could be opposed, contested or circumvented by our competitors, or be declared invalid or unenforceable in judicial or administrative proceedings. The failure of our patents to adequately protect our technology might make it easier for our competitors to offer the same or similar products or technologies. Even if we are successful in receiving patent protection for certain products and processes, our competitors may be able to design around our patents or develop products that provide outcomes which are comparable to ours without infringing on our intellectual property rights. Due to differences between foreign and U.S. patent laws, our patented intellectual property rights may not receive the same degree of protection in foreign countries as they would in the United States. Even if patents are granted outside the United States, effective enforcement in those countries may not be available.
We rely on our trademarks and trade names to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks. For example, we have one pending application in the United States for the “NeuroOne” trademark. We cannot assure you that our trademark applications will be approved in a timely manner or at all. Third parties also may oppose our trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote additional resources to marketing new brands. Further, we cannot assure you that competitors will not infringe upon our trademarks, or that we will have adequate resources to enforce our trademarks.
We also rely on trade secrets, know-how and technology, which are not protectable by patents, to maintain our competitive position. We try to protect this information by entering into confidentiality agreements and intellectual property assignment agreements with our officers, employees, temporary employees and consultants regarding our intellectual property and proprietary technology. In the event of unauthorized use or disclosure or other breaches of those agreements, we may not be provided with meaningful protection for our trade secrets or other proprietary information. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in the related or resulting know-how and inventions. If any of our trade secrets, know-how or other technologies not protected by a patent were to be disclosed to or independently developed by a competitor, our business, financial condition and results of operations could be materially adversely affected.
If a competitor infringes upon one of our patents, trademarks or other intellectual property rights, enforcing those patents, trademarks and other rights may be difficult and time-consuming. Patent law relating to the scope of claims in the industry in which we operate is subject to rapid change and constant evolution and, consequently, patent positions in our industry can be uncertain. Even if successful, litigation to defend our patents and trademarks against challenges or to enforce our intellectual property rights could be expensive and time consuming and could divert management's attention from managing our business. Moreover, we may not have sufficient resources or desire to defend our patents or trademarks against challenges or to enforce our intellectual property rights. Litigation also puts our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing. Additionally, we may provoke third-parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially valuable. The occurrence of any of these events may harm our business, financial condition and operating results.
| 47 |
We may not be able to establish or strengthen our brand.
We believe that establishing and strengthening our brand is critical to achieving widespread acceptance of our cortical strip, grid electrode and depth electrode technology. Promoting and positioning our brand will depend largely on the success of our marketing efforts and our ability to provide physicians with a reliable product for successful treatment of brain-related disorders. Additionally, we believe the quality and reliability of our product is critical to building physician support in the United States, and any negative publicity regarding the quality or reliability of our cortical strip, grid electrode and depth electrode technology could significantly damage our reputation in the market. Further, given the established nature of our competitors, it is likely that our future marketing efforts will require us to incur significant additional expenses. These brand promotion activities may not yield increased sales and, even if they do, any sales increases may not offset the expenses we incur to promote our brand. If we fail to successfully promote and maintain our brand, or if we incur substantial expenses in an unsuccessful attempt to promote and maintain our brand, our cortical strip, grid electrode and depth electrode technology may not be accepted by physicians, which would adversely affect our business, results of operations and financial condition.
The medical device industry is characterized by patent litigation, and we could become subject to litigation that could be costly, result in the diversion of management's time and efforts, stop our development and commercialization measures or require us to pay damages.
Our success will depend in part on not infringing the patents or violating the other proprietary rights of third-parties. Significant litigation regarding patent rights exists in our industry. Our competitors in both the United States and abroad, many of which have substantially greater resources and have made substantial investments in competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with our ability to make and sell our products. The large number of patents, the rapid rate of new patent issuances, and the complexities of the technology involved increase the risk of patent litigation.
In the future, we could receive communications from various industry participants alleging our infringement of their intellectual property rights. Any potential intellectual property litigation could force us to do one or more of the following:
| · | stop selling our products or using technology that contains the allegedly infringing intellectual property; |
| · | incur significant legal expenses; |
| · | pay substantial damages to the party whose intellectual property rights we are allegedly infringing; |
| · | redesign those products that contain the allegedly infringing intellectual property; or |
| · | attempt to obtain a license to the relevant intellectual property from third-parties, which may not be available on reasonable terms or at all, and if available, may be non-exclusive, thereby giving our competitors access to the same technology. |
Patent litigation can involve complex factual and legal questions, and its outcome is uncertain. Any litigation or claim against us, even those without merit, may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. Further, as the number of participants in the neurostimulation market increases, the possibility of intellectual property infringement claims against us increases.
We may be subject to damages resulting from claims that we, or our employees, have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
Some of our current or future employees may have previously been employed at other medical device companies, including those that are our direct competitors or could potentially be our direct competitors. We may be subject to claims that we, or our employees, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of these former employers or competitors. In addition, we may in the future be subject to allegations that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims.
| 48 |
We received a letter in May 2017 from the former employer of Mark Christianson and Wade Fredrickson claiming, among other things, that such officers are breaching their non-competition obligations with such employer, by virtue of such officers’ current work for us and such officer’s prior work during employment with the prior employer, and their confidentiality and non-disclosure obligations to such prior employer and federal and state law by misappropriating confidential and trade secret information, and claiming that the Company is responsible for tortious interference with contract. The letter seeks that Mr. Fredrickson, Mr. Christianson and the Company cease and desist all competitive activities against the former employer, that Mr. Fredrickson step down from his position and that Mr. Christianson and the Company provide the former employer access to our systems to demonstrate that we are not using trade secrets or proprietary information nor competing with the former employer. We have no insurance coverage to protect against any losses we may experience due to this claim. Furthermore, Mr. Fredrickson or Mr. Christianson are key officers and the loss of either or both of them would be detrimental to our operations and prospects. The Company, Mr. Fredrickson and Mr. Christianson intend to vigorously defend themselves.
Even if we successfully defend against these claims, litigation could cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. If our defense to those claims fails, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. There can be no assurance that this type of litigation will not occur, and any future litigation or the threat thereof may adversely affect our ability to hire additional employees. A loss of key personnel or their work product could hamper or prevent our ability to develop or commercialize our cortical strip, grid electrode and depth electrode technology or future versions thereof, which could have an adverse effect on our business, financial condition and operating results.
We are subject to the patent laws of countries other than the United States, which may not offer the same level of patent protection and whose rules could seriously affect how we draft, file, prosecute and maintain patents, trademarks and patent and trademark applications.
Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, the patent owner has failed to “work” the invention in that country, or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patent and other intellectual property protection which makes it difficult to stop infringement.
We cannot be certain that the patent or trademark offices of countries outside the United States will not implement new rules that increase costs for drafting, filing, prosecuting and maintaining patents, trademarks and patent and trademark applications or that any such new rules will not restrict our ability to file for patent protection. For example, we may elect not to seek patent protection in some jurisdictions in order to save costs. We may be forced to abandon or return the rights to specific patents due to a lack of financial resources.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| · | others may be able to make devices that are the same as or similar to our cortical strip, grid electrode and depth electrode technology but that are not covered by the claims of the patents that we own; |
| · | we or any collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own; |
| · | we might not have been the first to file patent applications covering certain of our inventions; |
| · | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| · | it is possible that our pending patent applications will not lead to issued patents; |
| · | issued patents that we own may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
| 49 |
| · | we might enforce our patent rights or defend a challenge to our issued patents or pending application, putting the patents and patent applications at risk of being invalidated or interpreted narrowly; |
| · | our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; and |
| · | we may not develop additional proprietary technologies that are patentable. |
Risks Related to our Legal and Regulatory Environment
Our products and operations are subject to extensive governmental regulation, and failure to comply with applicable requirements could cause our business to suffer.
The medical device industry is regulated extensively by governmental authorities, principally the FDA and corresponding state regulatory agencies in the United States and the European Commission and corresponding Notified Body in the European Union and the EEA. The regulations are very complex and are subject to rapid change and varying interpretations. Regulatory restrictions or changes could limit our ability to carry on or expand our operations or result in higher than anticipated costs or lower than anticipated sales. These governmental authorities enforce laws and regulations that are meant to assure product safety and effectiveness, including the regulation of, among other things:
| · | product design and development; |
| · | pre-clinical studies and clinical trials; |
| · | product safety; |
| · | establishment registration and product listing; |
| · | labeling, content and language of instructions for use and storage; |
| · | marketing, manufacturing, sales and distribution; |
| · | pre-market clearance or approval; |
| · | servicing and post-market surveillance; |
| · | record-keeping procedures; |
| · | product import and export; |
| · | advertising and promotion; and |
| · | recalls and field safety corrective actions. |
The regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated revenues.
Failure to comply with applicable regulations could jeopardize our ability to sell our products and result in enforcement actions such as fines, civil penalties, injunctions, warning letters, recalls of products, delays in the introduction of products into the market, refusal of the regulatory agency or other regulators to grant future clearances or approvals, and the suspension or withdrawal of existing approvals by such regulatory agencies. Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales, and harm our reputation, business, financial condition and operating results.
The FDA regulatory clearance process is expensive, time-consuming and uncertain, and the failure to obtain and maintain required regulatory clearances and approvals could prevent us from commercializing our cortical strip, grid electrode and depth electrode technology under development and future versions thereof.
Our products and operations are subject to extensive and rigorous regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FFDCA, and its implementing regulations, guidances, and standards. The FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, promotion, distribution, and production of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. The FDA also regulates the export of medical devices manufactured in the United States to international markets. Any violations of these laws and regulations could result in a material adverse effect on our business, financial condition and results of operations. In addition, if there is a change in law, regulation or judicial interpretation, we may be required to change our business practices, which could have a material adverse effect on our business, financial condition and results of operations.
| 50 |
Under the FFDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to ensure safety and effectiveness.
Class I devices are those for which safety and effectiveness can be assured by adherence to FDA’s “general controls” for medical devices, which include compliance with the applicable portions of the QSR facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising, and promotional materials. Some Class I devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below.
Class II devices are subject to FDA’s general controls, and any other “special controls” deemed necessary by FDA to ensure the safety and effectiveness of the device. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification procedure, though certain Class II devices are exempt from this premarket review process. When a 510(k) is required, the manufacturer must submit to the FDA a premarket notification submission demonstrating that the device is “substantially equivalent” to a legally marketed device, which in some cases may require submission of clinical data. A legally marketed device is defined by statute to mean a device that was legally marketed prior to May 28, 1976, the date upon which the Medical Device Amendments of 1976 were enacted, or another commercially available, similar device that was cleared through the 510(k) process. Unless a specific exemption applies, 510(k) premarket notification submissions are subject to user fees. If the FDA determines that the device, or its intended use, is not substantially equivalent to a legally marketed device, the FDA will place the device, or the particular use of the device, into Class III, and the device sponsor must then fulfill much more rigorous premarketing requirements in the form of a premarket approval, or PMA.
A Class III device includes devices deemed by the FDA to pose the greatest risk such as life-supporting or life-sustaining devices, or implantable devices, in addition to a device that has a new intended use or utilizes advanced technology that is not substantially equivalent to that of a legally marketed device. The safety and effectiveness of Class III devices cannot be assured solely by general and special controls. These devices almost always require formal clinical studies to demonstrate safety and effectiveness. Submission and FDA approval of a PMA application is required before marketing of a Class III device can proceed.
We believe our cortical strip, grid electrode and depth electrode technology under development will be a Class II medical device. The FDA has not made any determination about whether our specific technology is a Class II medical device. While such a determination is not necessary in order for us to list a device with the FDA and bring that device to the U.S. market, we may decide to get clarification from the FDA prior to introducing a product into the market. From time to time, the FDA may disagree with the classification and require us to apply for approval as a Class III medical device. In the event that the FDA determines that our technology should be classified as Class III, we could be precluded from marketing the devices for clinical use within the United States for months, years or longer, depending on the specific change in the classification. Reclassification of our technology as Class III could significantly increase our regulatory costs, including the timing and expense associated with required clinical trials and other costs.
If the FDA requires us to go through more costly, lengthy and uncertain PMA process for our cortical strip, grid electrode and depth electrode technology, future products or modifications to existing products than we had expected, we may be less likely to receive approval for our cortical strip, grid electrode and depth electrode technology or such approval may take longer and be more costly.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| · | we may not be able to demonstrate that our products are safe and effective for their intended users; |
| · | the data from our clinical trials may be insufficient to support clearance or approval; and |
| · | the manufacturing process or facilities we use may not meet applicable requirements. |
| 51 |
When FDA approval of a device requires human clinical trials, and if the device presents a “significant risk” to human health, the device sponsor is required to file an investigational device exemption, or IDE, application with the FDA and obtain IDE approval prior to commencing the human clinical trial. If the device is considered a “non-significant risk,” IDE submission to FDA is not required. Instead, only approval from the Institutional Review Board, or IRB, overseeing the investigation at each clinical trial site is required. Human clinical studies are generally required in connection with approval of Class III devices and may be required for Class I and II devices. The FDA or the IRB at each institution at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk. We believe that we will need to complete human clinical trials and submit an application for an IDE in order to seek approval to use of our cortical strip, grid electrode and depth electrode technology for stimulation and ablation but not for diagnostic purposes. Because any IDE, if required, must be cleared by the FDA prior to the start of a clinical investigation, this requirement may delay our product development or clinical trial efforts. Any delay in, or failure to receive or maintain, clearance or approval for our products under development could prevent us from generating revenue from these products or achieving profitability.
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently cleared or approved products on a timely basis.
After the FDA permits a device to enter commercial distribution, numerous regulatory requirements apply. These include: compliance with the QSR, which requires manufacturers to follow elaborate design, testing, control, documentation and other quality assurance procedures during the manufacturing process; labeling regulations; the FDA’s general prohibition against promoting products for unapproved or “off-label” uses; the reports of Corrections and Removals regulation, which requires manufacturers to report recalls and field actions to the FDA if initiated to reduce a risk of health posed by the device or to remedy a violation of the Federal Food, Drug and Cosmetic Act; and the Medical Device Reporting regulation, which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to reoccur. Manufacturers are also required to register and list their devices with the FDA, based on which the FDA will conduct inspections to ensure continued compliance with applicable regulatory requirements.
The FDA has broad post-market and regulatory and enforcement powers. Failure to comply with the applicable U.S. medical device regulatory requirements could result in, among other things, warning letters; fines; injunctions; consent decrees; civil penalties; repairs, replacements or refunds; recalls, corrections or seizures of products; total or partial suspension of production; the FDA’s refusal to grant future premarket clearances or approvals; withdrawals or suspensions of current product applications; and criminal prosecution. Regulatory enforcement or inquiries, or other increased scrutiny on us, could dissuade some people with brain related disorders from using our products and adversely affect our reputation and the perceived accuracy and safety of our products. If any of these events were to occur, they could have a material adverse effect on our business, financial condition and results of operations.
International sales are subject to regulatory requirements in the countries in which our products are sold. The regulatory review process varies from country to country and may in some cases require the submission of clinical data. In addition, the FDA must be notified of, or approve the export to certain countries of devices that require a PMA, and are not yet approved in the United States.
A recall of our products, or the discovery of serious safety issues with our products, could have a significant negative impact on us.
The FDA has the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture or in the event that a product poses an unacceptable risk to health. Our third-party suppliers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our third-party distributors, if any, could occur as a result of an unacceptable risk to health, component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, financial condition and operating results, which could impair our ability to produce our products in a cost-effective and timely manner.
| 52 |
Further, under the FDA's medical device reporting regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Repeated product malfunctions may result in a voluntary or involuntary product recall, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner and have an adverse effect on our reputation, financial condition and operating results.
Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or regulatory agency action, which could include inspection, mandatory recall or other enforcement action. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
We will be subject to the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act and other anti-corruption and anti-money-laundering laws, as well as export control laws, customs laws, sanctions laws and other laws governing our future global operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our future global operations will expose us to trade and economic sanctions and other restrictions imposed by the United States, the European Union and other governments and organizations. The U.S. Departments of Justice, Commerce, State and Treasury and other federal agencies and authorities have a broad range of civil and criminal penalties they may seek to impose against corporations and individuals for violations of economic sanctions laws, export control laws, the Foreign Corrupt Practices Act, or the FCPA, and other federal statutes and regulations, including those established by the Office of Foreign Assets Control, or OFAC. In addition, the U.K. Bribery Act of 2010, or the Bribery Act, prohibits both domestic and international bribery, as well as bribery across both private and public sectors. An organization that “fails to prevent bribery” by anyone associated with the organization can be charged under the Bribery Act unless the organization can establish the defense of having implemented “adequate procedures” to prevent bribery. Under these laws and regulations, as well as other anti-corruption laws, anti-money-laundering laws, export control laws, customs laws, sanctions laws and other laws governing our operations, various government agencies may require export licenses, may seek to impose modifications to business practices, including cessation of business activities in sanctioned countries or with sanctioned persons or entities and modifications to compliance programs, which may increase compliance costs, and may subject us to fines, penalties and other sanctions. A violation of these laws or regulations could adversely impact our business, results of operations and financial condition.
We will implement and maintain policies and procedures designed to ensure compliance by us, and our directors, officers, employees, representatives, third-party distributors, if any, consultants and agents with the FCPA, OFAC restrictions, the Bribery Act and other export control, anticorruption, anti-money-laundering and anti-terrorism laws and regulations. We cannot assure you, however, that our policies and procedures will be sufficient or that directors, officers, employees, representatives, third-party distributors, if any, consultants and agents have not engaged and will not engage in conduct for which we may be held responsible, nor can we assure you that our business partners have not engaged and will not engage in conduct that could materially affect their ability to perform their contractual obligations to us or even result in our being held liable for such conduct. Violations of the FCPA, OFAC restrictions, the Bribery Act or other export control, anti-corruption, anti-money-laundering and anti-terrorism laws or regulations may result in severe criminal or civil sanctions, and we may be subject to other liabilities, which could have a material adverse effect on our business, financial condition, cash flows and results of operations.
We are subject to additional federal, state and foreign laws and regulations relating to our healthcare business; our failure to comply with those laws could have an adverse impact on our business.
Although we will not provide healthcare services, submit claims for third-party reimbursement, or receive payments directly from government health insurance programs or other third-party payors for our cortical strip, grid electrode and depth electrode technology, we are subject to healthcare fraud and abuse regulation and enforcement by federal, state and foreign governments, which could adversely impact our business. Healthcare fraud and abuse and health information privacy and security laws potentially applicable to our operations include, but are not limited to:
| 53 |
| · | the federal Anti-Kickback Statute, which will apply to our marketing practices, educational programs, pricing policies and relationships with healthcare providers, by prohibiting, among other things, soliciting, receiving, offering or providing remuneration intended to induce the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare or Medicaid programs. A person or entity does not need to have actual knowledge of this statute or specific intent to violate it to have committed a violation; |
| · | federal civil and criminal false claims laws and civil monetary penalty laws, including civil whistleblower or qui tam actions that prohibit, among other things, knowingly presenting, or causing to be presented, claims for payment or approval to the federal government that are false or fraudulent, knowingly making a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay or transmit money or property to the federal government. The government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes; |
| · | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, and its implementing regulations, which created federal criminal laws that prohibit, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. A person or entity does not need to have actual knowledge of these statutes or specific intent to violate them; |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their implementing regulations, also imposes certain regulatory and contractual requirements regarding the privacy, security and transmission of individually identifiable health information; |
| · | federal “sunshine” requirements imposed by the Patent Protection and Affordable Care Act, or PPACA, on device manufacturers regarding any “transfer of value” made or distributed to physicians and teaching hospitals. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission. Manufacturers must submit reports by the 90th day of each subsequent calendar year; |
| · | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| · | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require device companies to comply with the industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state laws that require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of certain health information, many of which differ from each other in significant ways and often are not preempted by HIPAA; and |
| · | foreign data privacy regulations, such as the EU Data Protection Directive (Directive 95/46/EC), and the country-specific regulations that implement Directive 95/46/EC, which impose strict obligations and restrictions on the ability to collect, analyze and transfer personal data, including health data from clinical trials and adverse event reporting, and may be stricter than U.S. laws. |
The risk of our being found in violation of these laws and regulations is increased by the fact that the scope and enforcement of these laws is uncertain, many of them have not been fully interpreted by the regulatory authorities or the courts, their provisions are open to a variety of interpretations, or they vary country by country. We are unable to predict what additional federal, state or foreign legislation or regulatory initiatives may be enacted in the future regarding our business or the healthcare industry in general, or what effect such legislation or regulations may have on us. Federal, state or foreign governments may (i) impose additional restrictions or adopt interpretations of existing laws that could have a material adverse effect on us or (ii) challenge our current or future activities under these laws. Any of these challenges could impact our reputation, business, financial condition and operating results.
| 54 |
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us now or in the future, we may be subject to penalties, including civil and criminal penalties, damages, fines, disgorgement of profits, exclusion from governmental health care programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results. Any federal, state or foreign regulatory review to which we may become subject, regardless of the outcome, would be costly and time-consuming.
For example, to enforce compliance with the federal laws, the U.S. Department of Justice, or DOJ, has recently increased its scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Dealing with investigations can be time and resource consuming and can divert management's attention from our core business. Additionally, if we settle an investigation with law enforcement or other regulatory agencies, we may be forced to agree to additional onerous compliance and reporting requirements as part of a consent decree or corporate integrity agreement. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
We may be liable if the FDA or another regulatory agency concludes that we have engaged in the off-label promotion of our products.
Our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of the off-label use of our products. Healthcare providers may use our products, if approved, off-label, as the FDA does not restrict or regulate a physician's choice of treatment within the practice of medicine. However, if the FDA determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties. Although we intend to train our marketing and direct sales force to not promote our products for uses outside of their cleared uses and our policy will be to refrain from statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion. In addition, the off-label use of our products may increase the risk of product liability claims. Product liability claims are expensive to defend and could result in substantial damage awards against us and harm our reputation.
Further, if we seek commercial approval in Europe, the advertising and promotion of our products is subject to the laws of EEA Member States implementing Directive 93/42/EEC concerning medical devices, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other EEA Member State legislation governing the advertising and promotion of medical devices. EEA Member State legislation may also restrict or impose limitations on our ability to advertise our products directly to the general public. In addition, voluntary EU and national codes of conduct provide guidelines on the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare providers harming our business, operating results and financial condition.
Legislative or regulatory healthcare reforms may make it more difficult and costly for us to obtain regulatory clearance or approval of our products.
Recent political, economic and regulatory influences are subjecting the healthcare industry to fundamental changes. The sales of our products depend in part on the availability of coverage and reimbursement from third-party payors such as government health administration authorities, private health insurers, health maintenance organizations and other healthcare-related organizations. Both the federal and state governments in the United States continue to propose and pass new legislation and regulations designed to contain or reduce the cost of healthcare. This legislation and regulation may result in decreased reimbursement for medical devices, which may further exacerbate industry-wide pressure to reduce the prices charged for medical devices. This could harm our ability to market our products and generate sales.
In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of or failure to receive regulatory clearances or approvals for our products would harm our business, financial condition and operating results.
| 55 |
While the goal of healthcare reform is to expand coverage to more individuals, it also involves increased government price controls, additional regulatory mandates and other measures designed to constrain medical costs. For example, the PPACA was enacted in March 2010. The PPACA substantially changes the way healthcare is financed by both governmental and private insurers, encourages improvements in the quality of healthcare items and services and significantly impacts the medical device industries. Among other things, the PPACA:
| · | establishes a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research; |
| · | implements payment system reforms including value-based payment programs, increased funding for comparative effectiveness research, reduced hospital payments for avoidable readmissions and hospital acquired conditions, and pilot programs to evaluate alternative payment methodologies that promote care coordination (such as bundled physician and hospital payments); and |
| · | creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
At this time, we cannot predict which, if any, additional healthcare reform proposals will be adopted, when they may be adopted or what impact they, or the PPACA, may have on our business and operations, and any of these impacts may be adverse on our operating results and financial condition.
Our financial performance may be adversely affected by medical device tax provisions in the healthcare reform laws.
The PPACA imposes, among other things, an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States beginning in 2013. Due to subsequent legislative amendments, the excise tax has been suspended from January 1, 2016 to December 31, 2017, and, absent further legislative action, will be reinstated starting January 1, 2018. We do not believe that our cortical strip, grid electrode and depth electrode technology under development is currently subject to this tax based on the retail exemption under applicable Treasury Regulations. However, the availability of this exemption is subject to interpretation by the Internal Revenue Service, or IRS, and the IRS may disagree with our analysis. In addition, future products that we manufacture, produce or import may be subject to this tax. The financial impact this tax may have on our business is unclear and there can be no assurance that our business will not be materially adversely affected by it.
Risks Related to our Common Stock
There is not now, and there may never be, an active market for our common stock and we cannot assure you that our common stock will become liquid or that it will be listed on a securities exchange.
There currently is no liquid market for our common stock. An investor may find it difficult to obtain accurate quotations as to the market value of the common stock and trading of our common stock may be extremely sporadic. For example, several days may pass before any shares may be traded. A more active market for our common stock may never develop. In addition, if we failed to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling the common stock, which may further affect its liquidity. This would also make it more difficult for us to raise additional capital.
The price of our common stock might fluctuate significantly, and you could lose all or part of your investment.
Volatility in the market price of our common stock may prevent you from being able to sell your shares of our common stock at or above the price you paid for your shares. The trading price of our common stock may be volatile and subject to wide price fluctuations in response to various factors, including:
| · | actual or anticipated fluctuations in our quarterly financial and operating results; |
| · | our progress toward developing our cortical strip and sheet electrode technology; |
| · | the commencement, enrollment and results of our future clinical trials; |
| · | adverse results from, delays in or termination of our clinical trials; |
| · | adverse regulatory decisions, including failure to receive regulatory approval; |
| 56 |
| · | publication of research reports about us or our industry or positive or negative recommendations or withdrawal of research coverage by securities analysts, if any; |
| · | perceptions about the market acceptance of our products and the recognition of our brand; |
| · | adverse publicity about our products or industry in general; |
| · | overall performance of the equity markets; |
| · | introduction of products, or announcements of significant contracts, licenses or acquisitions, by us or our competitors; |
| · | legislative, political or regulatory developments; |
| · | additions or departures of key personnel; |
| · | threatened or actual litigation and government investigations; |
| · | sale of shares of our common stock by us or members of our management; and |
| · | general economic conditions. |
These and other factors might cause the market price of our common stock to fluctuate substantially, which may negatively affect the liquidity of our common stock. In addition, in recent years, the stock market has experienced significant price and volume fluctuations. This volatility has had a significant impact on the market price of securities issued by many companies across many industries. The changes frequently appear to occur without regard to the operating performance of the affected companies. Accordingly, the price of our common stock could fluctuate based upon factors that have little or nothing to do with our company, and these fluctuations could materially reduce our share price.
Securities class action litigation has often been instituted against companies following periods of volatility in the overall market and in the market price of a company's securities. This litigation, if instituted against us, could result in substantial costs, divert our management's attention and resources, and harm our business, operating results and financial condition.
We are a smaller reporting company, and the reduced reporting requirements applicable to smaller reporting companies may make our common stock less attractive to investors.
We are a “smaller reporting company” as defined in Section 12 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). For as long as we continue to be a smaller reporting company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not smaller reporting companies, including not being required to comply with the auditor attestation requirements of Section 404 of Sarbanes-Oxley, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding nonbinding advisory votes on executive compensation, and stockholder approval of any golden parachute payments not previously approved. We could remain a smaller reporting company until the last day of the fiscal year when the aggregate worldwide market value of the voting and nonvoting common equity held by our nonaffiliates is $75 million or more on the last business day of our most recently completed second fiscal quarter. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Our common stock is subject to the “penny stock” rules of the SEC, which makes transactions in our stock cumbersome and may reduce the value of an investment in our stock.
The SEC has adopted regulations which generally define a "penny stock" as an equity security that has a market price of less than $5.00 per share, subject to specific exemptions. The SEC's penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document that provides information about penny stocks and the risks in the penny stock market. The broker-dealer must also provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and the salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer's account. In addition, the penny stock rules generally require that before a transaction in a penny stock occurs, the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser's agreement to the transaction. If applicable in the future, these rules may restrict the ability of brokers-dealers to sell our common stock and may affect the ability of investors to sell their shares, until our common stock no longer is considered a penny stock.
| 57 |
Concentration of ownership of our common stock among our existing executive officers, directors and principal stockholders may prevent new investors from influencing significant corporate decisions.
Our executive officers, directors and current beneficial owners of 5% or more of our common stock and their respective affiliates, in the aggregate, beneficially own approximately 95.0% of our outstanding common stock. As a result, these persons, acting together, would be able to significantly influence all matters requiring stockholder approval, including the election and removal of directors, any merger, consolidation, sale of all or substantially all of our assets, or other significant corporate transactions.
Some of these persons or entities may have interests different than yours. For example, they may be more interested in selling our company to an acquirer than other investors, or they may want us to pursue strategies that deviate from the interests of other stockholders.
We intend to issue more shares to raise capital, which will result in substantial dilution.
Our certificate of incorporation authorizes the issuance of a maximum of 100,000,000 shares of common stock and 10,000,000 shares of preferred stock. Any additional financings effected by us may result in the issuance of additional securities without stockholder approval and the substantial dilution in the percentage of common stock held by our then existing stockholders. Moreover, the common stock issued in any such transaction may be valued on an arbitrary or non-arm's-length basis by our management, resulting in an additional reduction in the percentage of common stock held by our current stockholders. Our board of directors has the power to issue any or all of such authorized but unissued shares without stockholder approval. To the extent that additional shares of common stock are issued in connection with a financing, dilution to the interests of our stockholders will occur and the rights of the holder of common stock might be materially and adversely affected.
In addition, as of the Closing, we had outstanding options to purchase an aggregate of 365,716 shares of common stock at a weighted average exercise price of $0.035 per share and Notes and warrants convertible into or exercisable for shares of our common stock upon their terms. As of July 13, 2017, the outstanding principal and accrued and unpaid interest on the Notes totaled $1,677,345. For a description of the Notes and warrants and information about the number of shares of common stock for which they are convertible or exercisable, see “Management's Discussion And Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Capital Resources”. To the extent these outstanding options or warrants are exercised or the Notes are converted, there will be further dilution to investors.
Anti-takeover provisions that may be in our charter and bylaws may prevent or frustrate attempts by stockholders to change the board of directors or current management and could make a third-party acquisition of us difficult.
Our certificate of incorporation and bylaws may contain provisions that may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could limit the price that investors might be willing to pay in the future for shares of our common stock.
We do not intend to pay cash dividends in the foreseeable future.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings for use in the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. Accordingly, you may have to sell some or all of your shares of our common stock in order to generate cash flow from your investment. You may not receive a gain on your investment when you sell shares and you may lose the entire amount of the investment.
We expect to incur increased costs and demands upon management as a result of being a public company.
As a public company in the United States, we expect to incur significant additional legal, accounting and other costs. These additional costs could negatively affect our financial results. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including regulations implemented by the SEC and the stock exchange on which we may list our common stock, may increase legal and financial compliance costs and make some activities more time-consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management's time and attention from revenue-generating activities to compliance activities. If, notwithstanding our efforts to comply with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
| 58 |
Failure to comply with these rules might also make it more difficult for us to obtain some types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors or as members of senior management.
Failure to establish and maintain an effective system of internal controls could result in material misstatements of our financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud in which case, our stockholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our stock. Furthermore, our management and our independent auditors have identified certain internal control deficiencies, which management and our independent auditors believe constitute material weaknesses.
Prior to the Acquisition, NeuroOne, Inc. was a private company with limited accounting personnel and other resources with which to address our internal controls and procedures. We review and update our internal controls, disclosure controls and procedures, and corporate governance policies as our Company continues to evolve. In addition, in connection with the Acquisition and becoming a company required to file reports with the SEC, we are required to comply with the internal control evaluation and certification requirements of Section 404 of SOX and management is required to report annually on our internal control over financial reporting. Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal control over financial reporting pursuant to Section 404 of SOX until the date we are no longer a "smaller reporting company" as defined by applicable SEC rules. We will remain a "smaller reporting company" as long as our public float remains less than $75 million as of the last business day of our most recently-completed second fiscal quarter.
During the 2016 audit, it was determined that our internal control over financial reporting is not effective. Such shortcoming could have an adverse effect on our business and financial results and the price of our common stock could be negatively affected once we become a Registrant. This reporting requirement could also make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. Any system of internal controls, however well designed and operated, is based in part on certain assumptions and can provide only reasonable, not absolute, assurances that the objectives of the system are met. Any failure or circumvention of the controls and procedures or failure to comply with regulation concerning control and procedures could have a material effect on our business, results of operation and financial condition. Any of these events could result in an adverse reaction in the financial marketplace due to a loss of investor confidence in the reliability of our financial statements, which ultimately could negatively affect the market price of our shares, increase the volatility of our stock price and adversely affect our ability to raise additional funding. The effect of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors and as executive officers.
Our management’s evaluation of the effectiveness of our internal controls over financial reporting as of December 31, 2016 and March 31, 2017 concluded that our controls were not effective, due to material weaknesses resulting from:
| · | Management did not maintain effective internal controls relating to the accounting closing and financial reporting process pertaining to certain stock transactions and complicated convertible debt instruments; |
| · | The Company has insufficient internal personnel resources and technical accounting and reporting expertise within the Company’s financial closing and reporting functions; and |
| · | Due to our small size, the Company did not maintain effective internal controls to assure proper segregation of duties as the same employee was responsible for initiating and recording of transactions, thereby creating a segregation of duties weakness. |
| 59 |
Management believes there is a reasonable possibility that these control deficiencies, if uncorrected, could result in material misstatements in the annual or interim financial statements that would not be prevented or detected in a timely manner. Accordingly, we have determined that these control deficiencies constitute material weaknesses. Although the Company is taking steps to remediate the material weaknesses, there can be no assurance that similar incidents can be prevented in the future if the internal controls are not followed by senior management and our Board of Directors.
We will need to evaluate our existing internal controls over financial reporting against the criteria set forth in Internal Control – Integrated Framework (2013) (the “Framework”) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). During the course of our ongoing evaluation of the internal controls, we may identify other areas requiring improvement, and may have to design enhanced processes and controls to address issues identified through this review. Remediating any deficiencies, significant deficiencies or material weaknesses that we or our independent registered public accounting firm may identify may require us to incur significant costs and expend significant time and management resources. We cannot assure you that any of the measures we implement to remedy any such deficiencies will effectively mitigate or remedy such deficiencies. The existence of one or more material weaknesses could affect the accuracy and timing of our financial reporting. Investors could lose confidence in our financial reports, and the value of our common stock may be harmed, if our internal controls over financial reporting are found not to be effective by management or by an independent registered public accounting firm or if we make disclosure of existing or potential material weaknesses in those controls.
Even if we conclude that our internal control over financial reporting provides reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our future reporting obligations.
Our reporting obligations as a public company will place a significant strain on our management, operational and financial resources and systems for the foreseeable future. If we fail to timely achieve and maintain the adequacy of our internal control over financial reporting, we may not be able to produce reliable financial reports or help prevent fraud. Our failure to achieve and maintain effective internal control over financial reporting could prevent us from filing our periodic reports on a timely basis which could result in the loss of investor confidence in the reliability of our financial statements, harm our business and negatively impact the trading price of our common stock.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. If our stockholders sell, or the market perceives that our stockholders intend to sell, substantial amounts of our common stock in the public market, the market price of our common stock could decline significantly.
Of the 7,864,994 shares of our common stock issued and outstanding after the closing of the Acquisition, 224,151 shares are freely tradable without restriction by stockholders who are not our affiliates. Of our outstanding shares, 1,348,849 shares that were outstanding before the Acquisition are “restricted securities” as defined in Rule 144. We issued an aggregate of 6,291,994 shares of our common stock to the former NeuroOne, Inc. stockholders pursuant to an exemption from the registration requirements of the Securities Act of 1933, as amended, or the Securities Act, and such shares are also “restricted securities” as defined in Rule 144. These restricted securities may be publicly resold under Rule 144 beginning one year following the date of the filing of this Report with the SEC.
In addition, in the future, we intend to file one or more registration statements on Form S-8 registering the issuance of approximately 365,716 shares of common stock subject to options or other equity awards issued as well as the 1,300,000 additional shares reserved for future issuance, under our 2017 Equity Incentive Plan, subject to adjustment as set forth in such plan. Shares registered under these registration statements on Form S-8 will be available for sale in the public market subject to vesting arrangements and exercise of options and the restrictions of Rule 144 in the case of our affiliates.
| 60 |
If securities or industry analysts do not publish research or reports, or publish unfavorable research or reports, about us, our business or our market, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that securities or industry analysts publish about us and our business. Securities or industry analysts may elect not to provide coverage of our common stock, and such lack of coverage may adversely affect the market price of our common stock. In the event we do not secure additional securities or industry analyst coverage, we will not have any control over the analysts or the content and opinions included in their reports. The price of our stock could decline if one or more securities or industry analysts downgrade our stock or issue other unfavorable commentary or research. If one or more securities or industry analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our stock could decrease, which in turn could cause our stock price or trading volume to decline.
Risks Related to our Acquisition by Original Source Entertainment, Inc.
We may be subject to unknown risks as a result of our recently completed acquisition by Original Source Entertainment, Inc.
Original Source Entertainment, Inc., which was renamed NeuroOne Medical Technologies Corporation in connection with the Acquisition, was formed to license songs to the television and movie industry and has generated very little revenues. Prior to the Acquisition, its operations have been primarily limited to organizational, start-up, and capital formation activities, with no employees other than the former officers. On February 5, 2014, the board of directors of Original Source Entertainment, Inc. authorized the spin-off of Original Source Music, Inc., a wholly-owned subsidiary which then held all of our operations and assets, to our stockholders of record as of February 25, 2014. Under the terms of the spin-off, the common stock, par value $0.001 per share, of Original Source Music was distributed on a pro-rata basis to each holder of our common stock on the February 25, 2014 record date without any consideration or action on the part of such holders, and the holders of our common stock as of the February 25, 2014 record date became owners of 100% of the common stock of Original Source Music. The spin-off of Original Source Music was effective as of May 13, 2016, due to the satisfactory resolution of all comments from the Securities and Exchange Commission to the Registration Statement on Form 10 of Original Source Music and the Form 10’s effectiveness. Therefore, upon the spinoff of Original Source Music, which held all of our operations and assets at the time, on May 13, 2016, Original Source Entertainment, Inc. ceased having a specific business plan and purpose.
In connection with the Acquisition, the liabilities existing in Original Source Entertainment, Inc. at the time of the Acquisition were cancelled or paid by a related party, as required by the Merger Agreement. Despite this requirement and the representations and warranties of Original Source Entertainment, Inc. in the Merger Agreement, there may be unknown liabilities, or liabilities that were known but believed to be immaterial, related to the business of Original Source Entertainment, Inc. that may become material liabilities we are subject to in the future. If we are subject to material liability as a result of the conduct of Original Source Entertainment, Inc., we may have limited recourse for such liabilities, which could have a material impact on our business and stock price.
Management's Discussion And Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of financial condition and results of operations of NeuroOne, Inc. together with our financial statements and the related notes included elsewhere in this Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Report, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” section of this Report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
| 61 |
Overview
On July 20, 2017, we entered into the Merger Agreement with NeuroOne, Inc. and OSOK Acquisition Company to acquire NeuroOne, Inc. The transactions contemplated by the Merger Agreement were consummated on July 20, 2017 and, pursuant to the terms of the Merger Agreement, (i) all outstanding NeuroOne Shares were exchanged for Company Shares based on the Exchange Ratio of 17.0103706 Company Shares for every one NeuroOne Share, (ii) all NeuroOne Options were replaced with Company Options based on the Exchange Ratio, with corresponding adjustments to their respective exercise prices, (iii) all NeuroOne Warrants were replaced with Company Warrants and (iv) we assumed the outstanding convertible promissory notes, or the Notes, of NeuroOne, Inc. Accordingly, we acquired 100% of NeuroOne, Inc. in exchange for the issuance of shares of our common stock and NeuroOne, Inc. became our wholly-owned subsidiary. Our sole business is the business of NeuroOne, Inc. Our management's discussion and analysis below is based on the financial results of NeuroOne, Inc. Except as otherwise indicated herein, all share and per share information in this “Management's Discussion and Analysis of Financial Condition and Results of Operations” section gives retroactive effect to the exchange of NeuroOne Shares, NeuroOne Options and NeuroOne Warrants for Company Shares, Company Options and Company Warrants, respectively, in the Acquisition, as well as the corresponding exercise price adjustments for such Company Options. The following discussion and analysis provides information which we believe to be relevant to an assessment and understanding of the results of operations and financial condition of NeuroOne, Inc.
We are an early-stage medical technology company developing comprehensive neuromodulation cEEG and sEEG monitoring, ablation, and brain stimulation solutions to diagnose and treat patients with epilepsy, Parkinson’s disease, essential tremors, and other brain related disorders. Our cortical strip technology under development has only been used by Mayo in five patients for research purposes and has not been tested in any clinical trials.
The LLC was formed on December 13, 2013 and operated as a limited liability company until it was merged with and into NeuroOne, Inc. on October 27, 2016 with NeuroOne, Inc. as the surviving entity. As a result of the Merger, all of the properties, rights, privileges, powers and franchises of the LLC vested in NeuroOne, Inc., and all debts, liabilities and duties of the LLC became the debts, liabilities and duties of NeuroOne, Inc., except for the WARF License, which was not legally transferred until May 2017. The purpose of the Merger was to change the jurisdiction of incorporation from Minnesota to Delaware, change of ownership of the LLC’s underlying assets and to convert from a limited liability company to a corporation. All financial results prior to October 27, 2016 are from the operations of the LLC. The LLC and NeuroOne, Inc. are collectively referred to as the Company. The sole member of the LLC received, upon the effectiveness of the Merger, in consideration for the cancellation of his membership interests in the LLC, 5,000 shares of NeuroOne, Inc. The holders of shares of common stock of NeuroOne, Inc. received, upon the effectiveness of the Merger, one share of common stock of NeuroOne, Inc. for every three shares of common stock of NeuroOne, Inc. held pre-merger. Except as otherwise indicated herein, all share and per share information in this Report gives retroactive effect to the stock combination completed in connection with the Merger.
NeuroOne, Inc. and the LLC were not entities under common control. As the LLC did not have an integrated set of activities that contained the required complement of inputs, processes and outputs to be considered a business the 2016 Merger was accounted for as an asset acquisition as prescribed under Accounting Standards Codification (ASC) 805 – Business Combinations. As such, the activities of NeuroOne, Inc. and the LLC were not combined and are shown separately in the accompanying financial statements included in this Report. For purposes of the management discussion and analysis the 2016 results for NeuroOne, Inc and the LLC have been combined for comparative purposes.
We had very limited resources prior to our convertible note and warrant financing commencing in November 2016. To date, our primary activities have been limited to, and our limited resources have been dedicated to, performing business and financial planning, raising capital, recruiting personnel, negotiating with business partners and the licensors of our intellectual property and conducting development activities. Our cortical strip, grid electrode and depth electrode technology is still under development, we do not yet have regulatory approval in any jurisdiction to sell any products and, to date, we have not generated any revenue.
We have incurred losses since inception and had an accumulated deficit of $266,370 as December 31, 2016. The LLC, prior to the Merger, also incurred losses since its inception and had cumulative losses of $49,930 as of the date of the Merger. As of March 31, 2017, we had an accumulated deficit of $995,945, primarily as a result of expenses incurred in connection with our research and development programs and from general and administrative expenses associated with our operations. We expect to continue to incur significant expenses and increasing operating and net losses for the foreseeable future.
| 62 |
We do not expect to generate revenue from product sales unless and until we obtain marketing authorization to sell our cortical strip, grid electrode and depth electrode technology from applicable regulatory authorities. See “Business—Clinical Experience, Future Development and Clinical Trial Plans” for an explanation of our plans for pre-clinical testing, clinical trials and regulatory submissions in the U.S.
Our primary source of cash has been proceeds from the issuance of Notes and common stock purchase warrants. From November 2016 to June 2017, we issued Notes (in aggregate principal amount of $1,625,120) and common stock purchase warrants, or the Warrants, for aggregate net proceeds of $1,511,510 to fund our operations.
We need to obtain substantial additional funding in connection with our continuing operations through public or private equity or debt financings or other sources, which may include collaborations with third parties. However, we may be unable to raise additional funds when needed on favorable terms or at all. Our failure to raise such capital as and when needed would have a negative impact on our financial condition and our ability to develop and commercialize our cortical strip, grid electrode and depth electrode technology and future products and our ability to pursue our business strategy. See “–Liquidity and Capital Requirements” below.
Financial Overview
Revenue
To date, we have not generated any revenue. We do not expect to generate revenue unless or until we develop, obtain regulatory approval for and commercialize our cortical strip, grid electrode and depth electrode technology. If we fail to complete the development of our cortical strip, grid electrode and depth electrode technology, or any other product candidate we may pursue in the future, in a timely manner, or fail to obtain regulatory approval, we may never be able to generate any revenue.
General and Administrative
General and administrative expenses consist primarily of personnel-related costs for personnel in functions not directly associated with research and development activities. Other significant costs include legal fees relating to corporate matters, intellectual property costs, professional fees for consultants assisting with regulatory, clinical, product development and financial matters, and product costs. We anticipate that our general and administrative expenses will significantly increase in the future to support our continued research and development activities, potential commercialization of our cortical strip, grid electrode and depth electrode technology, if approved, and the increased costs of operating as a public company. These increases will include increased costs related to the hiring of additional personnel and fees for legal and professional services, as well as other public-company related costs.
Research and Development
Research and development expenses consist of expenses incurred in performing research and development activities in developing our cortical strip, grid electrode and depth electrode technology. Research and development expenses include compensation and benefits for research and development employees, overhead expenses, cost of laboratory supplies, clinical trial and related clinical manufacturing expenses, costs related to regulatory operations, fees paid to consultants, and other outside expenses. Research and development costs are expensed as incurred and costs incurred by third parties are expensed as the contracted work is performed.
We expect our research and development expenses to significantly increase over the next several years as we develop our cortical strip, grid electrode and depth electrode technology and conduct preclinical testing and clinical trials and will depend on the duration, costs and timing to complete our preclinical programs and clinical trials.
| 63 |
Interest Expense
Interest expense primarily consists of amortized Note issuance costs and interest costs related to the Notes we issued between November 2016 and June 2017. The Notes bear interest at a fixed rate of 8% per annum, compounding annually.
Interest expense also includes the change in the fair value of warrant liability and the premium conversion derivative during the particular period. The change in fair value of the warrant liability results from the marking to market at the end of every reporting period of the fair value of the warrant liability related to the warrants to purchase shares of common stock issued in connection with the issuance of the Notes. The fair value of this warrant liability will fluctuate based on the change in the price of our common stock in the public markets until these warrants are exercised or expire.
Results of Operations
For comparison purposes, the results of operations for the LLC for the three-month period ended March 31, 2016 is compared to the operating results for NeuroOne, Inc. for the three-month period ended March 31, 2017. For the fiscal year ended December 31, 2016, the results of operations for the LLC from January 1, 2016 to October 26, 2016 have been combined with the operating results for NeuroOne, Inc. from October 7, 2016 to December 31, 2016 and such combined results have been compared to the operating results of the LLC for the year ended December 31, 2015.
Comparison of the Three Months Ended March 31, 2016 and 2017
The following table sets forth the results of operations of NeuroOne, LLC and NeuroOne, Inc. for the three-months ended March 31, 2016 and 2017, respectively.
| Three Months Ended March 31, | ||||||||||||
NeuroOne LLC 2016 | NeuroOne, Inc. 2017 | Period-to- Period Change | ||||||||||
| Expenses: | ||||||||||||
| General and administrative | $ | 2,001 | $ | 444,016 | $ | (442,015 | ) | |||||
| Research and development | — | 72,041 | (72,041 | ) | ||||||||
| Operating Loss | (2,001 | ) | (516,057 | ) | (514,056 | ) | ||||||
| Other expense: | ||||||||||||
| Interest expense | (3,498 | ) | (213,518 | ) | (210,020 | ) | ||||||
| Net loss and comprehensive loss | $ | (5,499 | ) | $ | (729,575 | ) | $ | (724,076 | ) | |||
General and administrative expenses
General and administrative expenses were $444,016 for the three months ended March 31, 2017, compared to $2,001 for the three months ended March 31, 2016. The increase was primarily due to an increase in salary related expenses to support the increased level of commercialization and development activities. The increase in spending was primarily attributable to salary and related expenses for additional staffing $300,000, legal and accounting expenses of $63,500, and investment banker fees of $50,000.
Research and development expenses
Research and development expenses were $72,041 for the three months ended March 31, 2017, compared to $0 for the three months ended March 31, 2016. The increase was primarily due to an increase in salary-related expenses and development materials and supplies to support the increased level of development activities.
| 64 |
Interest expense
Interest expense, for the three months ended March 31, 2017 was $213,518, consisting of interest expense and amortization of debt issuance costs of $212,200 related to the Notes and other interest expense of $1,318. There were no outstanding Notes in the prior year.
Comparison of the Years Ended December 31, 2015 and 2016
The following table sets forth our results of operations for the years ended December 31, 2015 and 2016.
| NeuroOne LLC | NeuroOne, Inc. | NeuroOne, Inc. and NeuroOne LLC 2016 Combined | ||||||||||||||||||
For the year ended December 31, 2015 | For the period January 1, 2016 to October 26, 2016 | For the period October 7, 2016 to December 31, 2016 | For the period January 1, 2016 to December 31, 2016 | Fiscal Year Period to Period Change | ||||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| General and administrative | $ | 7,936 | $ | 6,657 | $ | 182,667 | $ | 189,324 | $ | (181,388 | ) | |||||||||
| Research and development | 2,400 | — | — | — | 2,400 | |||||||||||||||
| Total operating expenses | 10,336 | 6,657 | 182,667 | 189,324 | (178,988 | ) | ||||||||||||||
| Loss from operations | (10,336 | ) | (6,657 | ) | (182,667 | ) | (189,324 | ) | (178,988 | ) | ||||||||||
| Interest expense | (4,187 | ) | (11,947 | ) | (83,703 | ) | (95,650 | ) | (91,463 | ) | ||||||||||
| Net loss | $ | (14,523 | ) | $ | (18,604 | ) | $ | (266,370 | ) | $ | (284,974 | ) | $ | (270,451 | ) | |||||
General and administrative expenses
General and administrative expenses were $189,324 for the year ended December 31, 2016, compared to $7,936 for the year ended December 31, 2015, an increase of $181,388. The increase was primarily due to increases in compensation expenses from additional personnel of $124,634 and professional fees for legal and accounting services of $48,687.
Research and development expenses
Research and development expenses were $0 for the year ended December 31, 2016, compared to $2,400 for the year ended December 31, 2015. Development activities were limited in 2016 while our focus was on laying the groundwork for future growth by creating our corporate structure and obtaining funding needed to support an increased level of future operations.
Interest expense
Interest expense for the year ended December 31, 2016 was $95,650 compared to $4,187 for the year ended December 31, 2015. The $91,463 increase in interest expense was primarily due to the amortization of $78,466 of debt issuance costs for the Notes in the fourth quarter of 2016.
Liquidity and Capital Resources
Capital Resources
As of December 31, 2016 and March 31, 2017, our principal source of liquidity consisted of cash deposits of $522,217 and $486,418, respectively. We have not generated any revenue, and we anticipate that we will continue to incur losses for the foreseeable future. We anticipate that our expenses will increase substantially as we develop our cortical strip, grid electrode and depth electrode technology and pursue pre-clinical testing and clinical trials, seek regulatory approvals, contract to manufacture any products, establish our own sales, marketing and distribution infrastructure to commercialize our cortical strip, grid electrode and depth electrode technology under development, if approved, hire additional staff, add operational, financial and management systems and operate as a public company.
| 65 |
Our primary source of cash has been proceeds from the issuance of the Notes and Warrants. From November 2016 to June 2017, we issued Notes (in aggregate principal amount of $1,625,120) and Warrants for aggregate net proceeds of $1,511,510.
The Notes bear interest at a fixed rate of 8% per annum and require us to repay the principal and accrued and unpaid interest thereon at the earlier of November 21, 2017 or the consummation of the next equity or equity-linked round of financing resulting in more than $3 million in gross proceeds. If such a financing occurs before November 21, 2017, the outstanding principal and accrued and unpaid interest on the Notes shall automatically convert into the securities issued by us in such financing based on the greater number of such securities resulting from either (i) the outstanding principal and accrued interest on the Notes divided by $1.80 or (ii) the outstanding principal and accrued interest on the Notes multiplied by 1.25, divided by the price paid per security in such financing. If a change of control transaction or initial public offering occurs prior to such a financing, the Note would, at the election of the holders of a majority of the outstanding principal of the Notes, either become payable on demand as of the closing date of such transaction or become convertible into shares of common stock immediately prior to such transaction at a price per share equal to the lesser of (i) the per share value as determined by our board of directors as if in connection with the granting of stock based compensation or in a private sale to a third party in an arms’ length transaction or (ii) at the per share consideration to be paid in such transaction. Change of control means a merger or consolidation with another entity in which our stockholders do not own more than 50% of the outstanding voting power of the surviving entity or the disposition of all or substantially all of our assets. The Notes are unsecured. As of July 13, 2017, the outstanding principal and accrued and unpaid interest on the Notes totaled $1,677,345. If we fail to complete a qualified equity financing by November 21, 2017, the Notes will be immediately due and payable on such date.
Each Warrant grants the holder the option to purchase the number of shares issuable upon the conversion of the Note held by such holder.
The terms of the Warrants were amended in June 2017 to be exercisable only in the event of conversion of the outstanding principal and accrued interest on the related Notes. Prior to such amendment, the Warrants were immediately exercisable into common stock and the number of shares were not fixed and determinable at the issuance date. No Warrants were exercised prior to the amendment. The Warrants have been accounted for as a liability at fair value. Following such amendment, the amount of warrant shares to be issued are now fixed to the number of shares of common stock to be received by the Holder upon conversion of such holder’s Note, and to an exercise price equal to the price at which the Notes convert into common shares. However, the warrants still have price protections that result in the accounting for these Warrants as a liability at fair value. Since the Warrants were no longer immediately exercisable as of the date of the amendment and the terms were modified, we will adjust the fair value of the underlying warrant liability in the second quarter of 2017.
In connection with the private placement of Notes and Warrants, we owe the placement agent a cash fee of $113,610 (8% of the gross proceeds received to date from certain Note and Warrant investors) and are obligated to issue to the placement agent a Warrant to purchase shares of common stock (or common stock equivalents) in an amount equal to 8% of the common stock purchased by certain investors in the private placement, which Warrant is expected to have an exercise price of $2.00 per Company Share.
The Warrants (other than the placement agent Warrant), after the amendment in June 2017, are exercisable following the conversion of the Notes and expire on November 21, 2021. The placement agent Warrants will be immediately exercisable and expire five years from the date of issuance. The placement agent Warrant is issuable upon the closing of the Company’s Note and Warrant financing. The exercise price and number of the shares of our common stock issuable upon exercising the Warrants will be subject to adjustment in the event of any stock dividends and splits, reverse stock split, recapitalization, reorganization, business combination or similar transaction, as described therein. In addition, the exercise price of the Warrants (but not those issued to the placement agent) is subject to reduction of the exercise price if we subsequently issue common stock or equivalents at an effective price less than the current exercise price of such Warrants.
We also received a short-term unsecured loan for $50,000 in November 2016 from the placement agent of the Notes and Warrants. We incurred no fees or interest costs related to such temporary loan and repaid it in full in February 2017.
The placement agent is entitled to receive warrants to purchase common stock in an amount equal to 10% of the common stock (or common stock equivalents) purchased by certain investors in subsequent equity financing rounds. Such warrants will have an exercise price determined in relation to the pricing of the subsequent financing, will be immediately exercisable once issued and have a term of 5 years.
| 66 |
Funding Requirements and Outlook
We have no current source of revenue to sustain our present activities, and we do not expect to generate revenue until, and unless, the FDA or other regulatory authorities approve our cortical strip, grid electrode and depth electrode technology under development and we successfully commercialize our cortical strip, grid electrode and depth electrode technology. Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of equity and debt financings as well as collaborations, strategic alliances and licensing arrangements. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or licensing arrangements with third-party partners, we may have to relinquish valuable rights to our technologies, future revenue streams or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or through collaborations, strategic alliances or licensing arrangements when needed, we may be required to delay, limit, reduce or terminate our product development, future commercialization efforts, or grant rights to develop and market our cortical strip, grid electrode and depth electrode technology that we would otherwise prefer to develop and market ourselves.
Our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the years ended December 31, 2015 and 2016, noting the existence of substantial doubt about our ability to continue as a going concern. This uncertainty arose from management's review of our results of operations and financial condition and its conclusion that, based on our operating plans, we did not have sufficient existing working capital to sustain operations through December 31, 2017.
We believe our existing cash and cash equivalents will be sufficient to fund our operating expenses only to August 2017. Furthermore, as of July 13, 2017, the outstanding principal and accrued and unpaid interest on the Notes totaled $1,677,345. If we fail to complete an equity financing by November 21, 2017, the Notes will be immediately due and payable on such date and we will not have sufficient cash to pay the principal and accrued and unpaid interest thereon.
We have agreements with WARF and Mayo that require us to make certain milestone and royalty payments. See “Business—WARF License” and “Business—Mayo Foundation for Medical Education and Research License and Development Agreement”.
To continue to fund operations, we will need to secure additional funding on or prior to August 2017. We plan to complete an equity financing in the third quarter of 2017. We may obtain additional financing in the future through the issuance of our common stock, through other equity or debt financings or through collaborations or partnerships with other companies. We may not be able to raise additional capital on terms acceptable to us, or at all, and any failure to raise capital as and when needed could compromise our ability to execute on our business plan.
The development of our cortical strip, grid electrode and depth electrode technology is subject to numerous uncertainties, and we have based these estimates on assumptions that may prove to be substantially different than we currently anticipate and could use our cash resources sooner than we expect. Additionally, the process of developing medical devices is costly, and the timing of progress in pre-clinical tests and clinical trials is uncertain. Our ability to successfully transition to profitability will be dependent upon achieving a level of product sales adequate to support our cost structure. We cannot assure you that we will ever be profitable or generate positive cash flow from operating activities.
| 67 |
Cash Flows
The following is a summary of cash flows for each of the periods set forth below.
| NeuroOne LLC | NeuroOne, Inc. | NeuroOne, Inc. and NeuroOne LLC 2016 Combined | NeuroOne LLC | NeuroOne, Inc. | ||||||||||||||||||||
For the year ended December 31, 2015 | For the period January 1, 2016 to October 26, 2016 | For the period October7, 2016 to December 31, 2016 | For the period January 1, 2016 to December 31, 2016 | For the period Ended March 31, 2016 | For the period Ended March 31, 2017 | |||||||||||||||||||
| Net cash used in operating activities | $ | (2,400 | ) | $ | — | $ | (175,783 | ) | $ | (175,783 | ) | $ | — | $ | (350,919 | ) | ||||||||
| Net cash provided by financing activities | 2,400 | — | 698,000 | 698,000 | — | 315,120 | ||||||||||||||||||
| Net increase(decrease) in cash | $ | — | $ | — | $ | 522,217 | $ | 522,217 | $ | — | $ | (35,799 | ) | |||||||||||
Net cash used in operating activities
Net cash used in operating activities was $350,919 for the three months ended March 31, 2017, which consisted of a net loss of $729,575 partially offset by non-cash interest, discount amortization, and warrant issuance costs on the convertible promissory notes of $212,231, an increase in accrued expenses of $107,627, and an increase in prepaid expenses of $53,823.
Net cash used in operating activities was $0 for the three months ended March 31, 2016. There was limited activity during the three month period ended March 31, 2016.
Net cash used in operating activities was $175,783 for the year ended December 31, 2016, which consisted of a net loss of $284,974 combined with a decrease in prepaid expenses of $53,823, partially offset by non-cash interest, discount amortization, and warrant issuance costs on the convertible promissory notes of $82,416, combined with an increase in accrued expenses of $72,266.
Net cash used in operating activities was $2,400 for the year ended December 31, 2015, which consisted primarily of a net loss of $14,523, partially offset by amortization expense of $7,765 combined with an increase in accrued expenses and accounts payable of $4,358.
Net cash provided by financing activities
Net cash provided by financing activities was $315,120 for the three months ended March 31, 2017, which consisted of $365,120 in net proceeds received upon the issuance of the Notes and Warrants during the quarter partially offset by the $50,000 repayment of a short-term unsecured loan.
We had no cash provided by financing activities in the three month period ended March 31, 2016.
Net cash provided by financing activities was $698,000 for the year ended December 31, 2016, which consisted of $648,000 in net proceeds received upon the issuance of the Notes and Warrants during the fourth quarter of 2016 and proceeds of $50,000 received from a short-term unsecured loan.
Net cash provided by financing activities was $2,400 for the year ended December 31, 2015, and consisted of LLC member contributions.
| 68 |
Critical Accounting Policies and Significant Judgments and Estimates
Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP. The preparation of these financial statements requires us to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities as of the dates of the balance sheets and the reported amounts of revenue and expenses during the reporting periods. In accordance with GAAP, we base our estimates on historical experience and on various other assumptions that we believe are reasonable under the circumstances at the time such estimates are made. Actual results may differ materially from our estimates and judgments under different assumptions or conditions. We periodically review our estimates in light of changes in circumstances, facts and experience. The effects of material revisions in estimates are reflected in our financial statements prospectively from the date of the change in estimate.
While our significant accounting policies are more fully described in the notes to our financial statements appearing elsewhere in this Report, we believe the following are the critical accounting policies used in the preparation of our financial statements that require significant estimates and judgments.
Management's Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Fair Value of Financial Instruments
We account for fair value measurements of assets and liabilities that are recognized or disclosed at fair value in the financial statements on a recurring or nonrecurring basis adhering to the Financial Accounting Standards Board (FASB) fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to measurements involving significant unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are as follows:
| · | Level 1 Inputs: Unadjusted quoted prices in active markets for identical assets or liabilities accessible to us at the measurement date. |
| · | Level 2 Inputs: Other than quoted prices included in Level 1 inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the asset or liability. |
| · | Level 3 Inputs: Unobservable inputs for the asset or liability used to measure fair value to the extent that observable inputs are not available, thereby allowing for situations in which there is little, if any, market activity for the asset or liability at measurement date. |
As of December 31, 2016 and 2015, the fair values of cash, other assets, accounts payable, accrued expenses and the unsecured loan approximated their carrying values because of the short-term nature of these assets or liabilities. The estimated fair value of the convertible promissory notes was based on amortized cost which was deemed to approximate fair value. The fair value of the warrant liability and the premium conversion derivative associated with the Notes were based on cash flow models discounted at current implied market rates evidenced in recent arms-length transactions representing expected returns by market participants for similar instruments which were based on Level 3 inputs. There were no transfers between fair value hierarchy levels during the year ended December 31, 2016 or the three-month period ended March 31, 2017.
Intellectual Property
We entered into two licensing agreements with major research institutions, which allow for access to certain patented technology and know-how. Milestone payments under those agreements are capitalized and amortized to general and administrative expense over the expected useful life of the acquired technology.
| 69 |
Impairment of Long-Lived Assets
We evaluate long-lived assets, which consists entirely of licensed intellectual property for impairment whenever events or changes in circumstances indicate that the carrying value of these assets may not be recoverable. We assess the recoverability of long-lived assets by determining whether or not the carrying value of such assets will be recovered through undiscounted expected future cash flows. If the asset is considered to be impaired, the amount of any impairment is measured as the difference between the carrying value and the fair value of the impaired asset. Through March 31, 2017, we had not impaired any long-lived assets.
Debt Issuance Costs
Debt issuance costs are recorded as a reduction of the Notes. Amortization of debt issuance costs is calculated using the straight-line method over the term of the Notes, which approximates the effective interest method, and is recorded in interest expense in the statement of operations.
Research and Development Costs
Research and development costs are charged to expense as incurred. Research and development expenses may comprise costs incurred in performing research and development activities, including clinical trial costs, manufacturing costs for both clinical and pre-clinical materials as well as other contracted services, license fees, and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity is performed or when the goods have been received, rather than when payment is made, in accordance with ASC 730, Research and Development.
Warrant Liability
We issued warrants to purchase equity securities in connection with the issuance of the Notes. We account for these warrants as a liability at fair value for each reporting period as the number of shares were not fixed and determinable at the issuance date. Additionally, issuance costs associated with the warrants are expensed as incurred and reflected as interest expense in the accompanying statement of operations. We will continue to adjust the warrant liability for changes in fair value until the earlier of the exercise or expiration of the warrants, or until such time, if any, as the number of shares to be exercised becomes fixed, at which point the warrants will be classified in stockholders’ (deficit) equity provided that there are sufficient authorized and unissued shares of common stock to settle the warrants and redeem any other contracts that may require settlement in shares of common stock. Any future change in fair value of the warrant liability will be recognized as a component of interest expense in the statement of operations.
Premium Debt Conversion Derivative
We evaluate all conversion and redemption features contained in a debt instrument to determine if there are any embedded derivative(s) that require separation from the host debt instrument. An embedded derivative that requires separation is bifurcated from its host debt instrument and a corresponding discount to the host debt instrument is recorded. The discount is amortized and recorded to interest expense over the term of the host debt instrument using the straight line method which approximates the effective interest method. The separated embedded derivative is accounted for separately on a fair market value basis. We record the fair value changes of a separated embedded derivative to interest expense at each reporting period. We issued Notes that contained a 125% conversion premium in the event that a qualified financing occurs at a price under $2.25 per common share (see Note 8 – Convertible Promissory Notes and Warrant Agreements). We determined that the redemption feature under the Notes qualified as an embedded derivative and was separated from its debt host.
Income Taxes
For NeuroOne, Inc., income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax base and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Deferred tax assets are reduced by a valuation allowance if it is more likely than not that some portion of all of the deferred tax asset will not be realized.
| 70 |
The LLC operated as a single-member LLC from formation on December 12, 2013 until it was merged into NeuroOne, Inc. on October 27, 2016. As such, it was a disregarded legal entity for income tax purposes. Accordingly, no provision for income taxes was included in the financial statements for the period from January 1, 2016 through October 26, 2016 and for the year ended December 31, 2015.
Net Loss Per Share
The LLC was a single-member LLC for which no units were outstanding. Accordingly, earnings per share is not presented for the LLC.
For NeuroOne, Inc., basic loss per share of common stock is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the period.
Diluted earnings or loss per share of common stock is computed similarly to basic earnings or loss per share except the weighted average shares outstanding are increased to include additional shares from the assumed exercise of any common stock equivalents, if dilutive. Our convertible promissory notes and warrants are considered common stock equivalents for this purpose. Diluted earnings is computed utilizing the treasury method for the warrants. Diluted earnings with respect to the convertible promissory notes utilizing the if-converted method was not applicable during the period from October 7, 2016 to December 31, 2016 as no conditions required for conversion had occurred during this period. No incremental common stock equivalents were included in calculating diluted loss per share because such inclusion would be anti-dilutive given the net loss reported for the period from October 7, 2016 to December 31, 2016.
Fair Value of Common Stock on Grant Dates
Prior to the Acquisition, we were a private company with no active public market for our common stock. Therefore, we have historically periodically determined for financial reporting purposes the estimated per share fair value of our common stock at various dates using contemporaneous valuations performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held Company Equity Securities Issued as Compensation, also known as the Practice Aid. We performed these contemporaneous valuations on an as-needed basis. In conducting the contemporaneous valuations, we considered all objective and subjective factors that we believed to be relevant for each valuation conducted, including our best estimate of our business condition, prospects and operating performance at each valuation date.
Common Stock Valuation Methodology
The contemporaneous valuations that we conducted were prepared in accordance with the guidelines in the Practice Aid, which prescribes several valuation approaches for setting the value of an enterprise, such as the asset, market and income approaches, and various methodologies for allocating the value of an enterprise to its common stock. In determining the fair value of the common stock underlying the stock options granted, our board of directors has historically considered, among other things, the most recent estimate of fair value provided by an independent third-party valuation specialist and our assessment of additional objective and subjective factors to determine the common stock fair market value each valuation date. The following factors, among others, were considered:
| · | our financial condition and operating results, including our projected results; |
| · | our stage of development and business strategy; |
| · | the financial condition and operating results of comparable publicly owned companies; |
| · | worldwide economic conditions; |
| · | our recent securities transactions; and |
| · | the likelihood of a liquidity event such as an initial public offering, a merger or the sale of our company. |
| 71 |
There are significant judgments and estimates inherent in the determination of fair value of our common stock, including the contemporaneous valuations. These judgments and estimates include assumptions regarding our future operating performance, and the determination of the appropriate valuation methods. If we had made different assumptions, our stock-based compensation expense, net loss and net loss per common share could have been significantly different.
In each of our contemporaneous valuations, we generally used the asset and market approaches to derive an estimated enterprise value. The asset approach establishes value based on the cost of reproducing or replacing the property, less economic depreciation due to physical deterioration, and functional or economic obsolescence, if present and measurable. The particular market approach utilizes the option pricing method, or OPM, backsolve method to determine our enterprise value. Under this method, an implied equity value of the company is derived from recent transactions involving the company's securities in arms-length transactions. Under the option pricing method, shares are valued by creating a series of hypothetical call options using exercise prices based on the liquidation preferences and conversion terms of each equity class. The values of the multiple classes of equity are inferred by analyzing these options and determining the option value attributable to each respective security. We applied a discount to the valuations due to the lack of marketability of the ordinary shares at the time of issuance. We calculated the discount for lack of marketability and applied it as appropriate to each allocation.
Recent Accounting Pronouncements
In November 2015, the Financial Accounting Standards Board (FASB) issued Accounting Standard Update (ASU) 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes (ASU 2015-17). The new guidance simplifies the presentation of deferred income taxes by requiring that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. ASU 2015-17 applies to all entities that present a classified statement of financial position. The current requirement that deferred tax liabilities and assets of a tax-paying component of an entity be offset and presented as a single amount is not affected by this ASU. For public entities, ASU 2015-17 is effective for financial statements issued for annual periods beginning after December 15, 2016 and for other entities beginning after December 15, 2017 with earlier application permitted. The new guidance may be applied either prospectively or retrospectively to all periods presented. We have adopted this standard for all periods presented. The adoption of this standard did not have a material impact on the our financial statements.
In January 2016, the FASB issued ASU No. 2016-01, Financial Instruments — Overall: Recognition and Measurement of Financial Assets and Financial Liabilities. The guidance affects the accounting for equity investments, financial liabilities under the fair value option and the presentation and disclosure requirements of financial instruments. The guidance is effective in the first quarter of fiscal 2018 for public entities and for all other entities in the first quarter of fiscal 2019. Early adoption is permitted for the accounting guidance on financial liabilities under the fair value option. We are currently evaluating the impact of the new guidance on our financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation – Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. This ASU simplifies the accounting for share-based payment award transactions including: income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. This ASU is effective for fiscal years beginning after December 15, 2016 for public entities and after December 15, 2017 for all other entities, including interim periods within those fiscal years. Early adoption is permitted. We are currently evaluating the requirements of this new guidance and have not yet determined its impact on our financial statements.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules.
We currently have no leased or owned properties, including office space. To meet our current needs, we intend to lease office space near Eden Prairie, Minnesota.
| 72 |
ITEM 4. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth the beneficial ownership of our common stock as of the Closing, taking into account the conversion of all NeuroOne Shares and NeuroOne Options into Company Shares and Company Options, respectively, in the Acquisition for:
| · | each person, or group of affiliated persons, who is known by us to beneficially own more than 5% of our common stock; |
| · | each of our named executive officers; |
| · | each of our directors; and |
| · | all of our current executive officers and directors as a group. |
We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options that are either immediately exercisable or exercisable within 60 days after the Closing. These shares are deemed to be outstanding and beneficially owned by the person holding those options for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws. Except as otherwise noted below, the address for persons listed in the table is c/o NeuroOne Medical Technologies Corporation, c/o David Rosa, 10006 Liatris Lane, Eden Prairie, Minnesota 55347.
| Name and Address of Beneficial Owner | Number of Shares of Common Stock Beneficially Owned | Percentage of Ownership(1) | ||||||
| Greater than 5% stockholders | ||||||||
| Wade Fredrickson(2) 4825 Suburban Drive Shorewood, Minnesota 55331 | 2,840,731 | 36.12 | % | |||||
Chromium 24 LLC(3) 135 East 18th Street New York, New York 10003 | 790,499 | 10.05 | % | |||||
| Mayo Foundation for Medical Education and Research (4) 200 First Street SW Rochester, Minnesota 55905 | 859,976 | 10.93 | % | |||||
Lifestyle Healthcare LLC(5) 404 East 79th Street, Apt 28G New York, New York 10075 | 542,622 | 6.90 | % | |||||
Named Executive Officers and Directors | ||||||||
| David Rosa | 793,822 | 10.09 | % | |||||
| Amer Samad(6) | 15,728 | * | ||||||
| Paul Buckman(7) | 34,020 | (8) | * | |||||
| Suraj Kalia(7) | 34,020 | (8) | * | |||||
| Jeffrey S. Mathiesen(7) | 34,020 | (8) | * | |||||
| Thomas Bachinski | 215,453 | (9) | 2.74 | % | ||||
| Mark Christianson | 1,423,206 | 18.10 | % | |||||
| All Directors and Officers as a Group (4 persons) (10) | 2,448,209 | 31.1 | % | |||||
* Less than one percent.
| (1) | Based on 7,864,994 shares of common stock outstanding as of the Closing. |
| 73 |
| (2) | Mr. Fredrickson is our former Vice President of Therapy and Product Development. |
| (3) | Chromium 24, LLC is a Delaware limited liability company that is controlled by John Kalem. Mr. Kalem has sole voting and investment power over these shares. |
| (4) | Mayo Clinic, a Minnesota corporation, is the controlling corporation of Mayo Foundation for Medical Education and Research. Mayo Clinic disclaims beneficial ownership of these shares. |
| (5) | Lifestyle Healthcare LLC is a Delaware limited liability company. Dmitri Saprikyn is a partner of Lifestyle Healthcare LLC and has voting and investment control over these shares. |
| (6) | Mr. Samad has resigned from the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. At the Closing, Mr. Samad tendered for cancellation 3,500,000 shares of our common stock held by him, as part of the conditions to Closing. |
| (7) | Messrs Buckman, Kalia and Mathiesen, who are directors of NeuroOne, Inc., have been appointed to the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. |
| (8) | Consists of an option to purchase 34,020 shares of our common stock issued pursuant to the 2016 Plan. |
| (9) | Consists of shares of restricted stock issued pursuant to the 2016 Plan and a subscription agreement. Pursuant to the subscription agreement, the shares of restricted stock were subject to a repurchase option until the vesting milestones had been achieved. All such vesting milestones were achieved as of the Closing of the Acquisition and, as such, the shares are held outright and no longer remain subject to the repurchase option. |
| (10) | Includes shares beneficially owned by Messrs. Rosa, Samad, Bachinski and Christianson, our directors and officers at the Closing of the Acquisition. The number of shares beneficially owned by Messrs. Rosa, Buckman, Kalia, Mathiesen, Bachinski and Christianson (6 persons), our directors and officers effective on the 10th day following our filing of the Schedule 14F-1, is 2,534,541 (31.8%). |
| 74 |
ITEM 5. DIRECTORS AND EXECUTIVE OFFICERS
The following table sets forth information concerning our directors and executive officers, including their ages as of the Closing of the Acquisition:
|
Name |
Age | Position | ||
| Executive Officers: | ||||
| David Rosa | 53 | Chief Executive Officer, President and Director | ||
| Mark Christianson | 50 | Vice President, Business Development and Marketing | ||
| Thomas Bachinski | 56 | Chief Development Officer | ||
| Non-management Directors: | ||||
| Amer Samad | 33 | Director (1) | ||
| Paul Buckman | 61 | Chairman of the Board of Directors(2) | ||
| Suraj Kalia | 44 | Director(2) | ||
| Jeffrey Mathiesen | 56 | Director(2) |
| (1) | Mr. Samad has resigned from the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. |
| (2) | Messrs Buckman, Kalia and Mathiesen, who are directors of NeuroOne, Inc., have been appointed to the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. |
Executive Officers
David Rosa
Mr. Rosa has served as the Chief Executive Officer and as a director of NeuroOne, Inc. since October 2016 and was appointed as Chief Executive Officer, President and a director of the Company upon the Closing of the Acquisition. From November 2009 to November 2015, Mr. Rosa served as the chief executive officer and president of Sunshine Heart, Inc., a publicly-held early-stage medical device company. From 2008 to November 2009, Mr. Rosa served as chief executive officer of Milksmart, Inc., a company that specializes in medical devices for animals. From 2004 to 2008, Mr. Rosa served as the vice president of global marketing for cardiac surgery and cardiology at St. Jude Medical.
Mr. Rosa’s qualifications to serve on the Board include his senior leadership experience in the medical device industry. In addition, his day-to-day leadership of the Company gives him critical insights into the Company’s operations, strategy and competition, and he facilitates the Board’s ability to perform its oversight function. Throughout his career at the Company and his former positions, he has demonstrated strong technical, strategic, and operational expertise, and he possesses in-depth knowledge of the medical device industry on a global basis.
Mark Christianson
Mr. Christianson has served as Vice President of Business Development and Marketing of NeuroOne, Inc. since December 2016 and of the Company since the Closing of the Acquisition. From May 2013 to December 2016 Mr. Christianson served as North American sales manager for Cortec Corporation. From February 2012 to May 2013 Mr. Christianson held the position of business development executive for Robert Half International. From May 2009 to February 2012 Mr. Christianson held the position of regional sales manager for PMT Corporation. Mr. Christianson studied accounting at Augsburg College in Minneapolis.
Mr. Christianson brings 15 years of high performing sales, sales management, and project management experience to the team. In addition, he also has contributed to the development and corporate strategy of the Company given his experience in the neurological field and his close relationships with key epilepsy opinion leaders.
| 75 |
Thomas Bachinski
Mr. Bachinski has served as Chief Development Officer of NeuroOne, Inc. since January 2017 and of the Company since the Closing of the Acquisition. From July 2015 to January 2017, Mr. Bachinski served as the vice president of research and development / engineering at Caerus Corporation, a privately held medical device company developing products for both the human medical and animal veterinarian industries. From December 2014 to the present Mr. Bachinski founded FutureWorks Innovative Engineering where technology commercialization strategies are developed and executed for a variety of clients across the medical device industry. Prior to FutureWorks, Mr. Bachinski was Vice President of research and development and director of the advanced concept development group at Empi, Inc., developing and commercializing a portfolio of neuro-stimulation devices and technologies.
We believe that Mr. Bachinski’s extensive engineering background and accomplishments in technology commercialization in the neurological market space will bring key engineering solutions and insight into our future product portfolio under development and future commercialization efforts, if approved. He holds a Master’s degree in Engineering and an MBA.
Non-Management Directors
Amer Samad
Amer Samad served as our Chief Executive Officer and a director since March 6, 2014. Mr. Samad resigned as our Chief Executive Officer upon the Closing of the Acquisition and tendered his resignation as a director effective on the 10th day following our filing of the Schedule 14F-1. Mr. Samad completed his undergraduate degree at the State University of New York at Buffalo with concentrations in Financial Analysis and Marketing. He later studied Real Estate Development and Real Estate Finance at the Massachusetts Institute of Technology and went on to found in November 2007, Samad Holdings & Construction Corp., a real estate development company based in Buffalo, New York that specializes in medical office and multi-family development in the United States and Canada. Mr. Samad earned a Masters in Business Administration degree from Columbia Business School in 2014.
Paul Buckman
Mr. Buckman has served as the chairman of the board of directors of NeuroOne, Inc. since October 2016 and has been appointed chairman of the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. He is currently the General Manager of TMVR for LivaNova PLC. Prior to joining LivaNova, Mr. Buckman served as chief executive officer of Conventus Orthopaedics, a Minnesota-based company specializing in peri-articular bone fracture fixation, from September 2013 until March 2017. Mr. Buckman was chief executive officer of Sentreheart, Inc., a medical technology company focused on closure of various anatomic structures, from February 2012 to September 2013. Previously, Mr. Buckman served as chief executive officer and chairman of Pathway Medical Technologies, Inc., a medical device company focused on treatment of peripheral arterial disease, from September 2008 to February 2012; as chief executive officer of Devax, Inc., a developer and manufacturer of drug eluting stents, from December 2006 to September 2008; as president of the cardiology division of St. Jude Medical, Inc., a publicly traded diversified medical products company, from August 2004 to December 2006; and as chairman of the board of directors and chief executive officer of ev3, LLC, a Minnesota-based medical device company focused on endovascular therapies that Mr. Buckman founded and developed into an $80 million business, from January 2001 to January 2004. Mr. Buckman has worked in the medical device industry for over 30 years, including 10 years at Scimed Life Systems, Inc. and Boston Scientific Corporation, a publicly traded medical device manufacturer, where he held several executive positions before becoming president of the cardiology division of Boston Scientific in January 2000. Mr. Buckman also currently serves as a business advisory board member for Bio Star Ventures, and as a director for Ablative Solutions, Inc., Aortica, Inc., Miromatrix, Inc. and Xtant Medical. He previously served as a director of Conventus Orthopaedics, Caisson Interventional LLC, Velocimed, Inc., where he was a co-founder, EndiCor, Inc., Microvena, Inc., Sunshine Heart, Inc., a publicly-held early-stage medical device company, and Micro Therapeutics, Inc. Mr. Buckman received a Master’s degree in Business Administration and Finance and a B.A. degree in Business Administration from Western Michigan University.
| 76 |
We believe that Mr. Buckman’s strong executive experience in medical device companies provides the Company with valuable guidance on product development and operational matters.
Suraj Kalia
Mr. Kalia has served as a member of the board of directors of NeuroOne, Inc. since March 2017 and has been appointed to the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. He currently serves as a Managing Director and Senior Research Analyst at Northland Capital Markets, where he covers the medical technology sector, after rejoining the firm in August 2012. His previous positions include Managing Director and Senior Medical Device Analyst at Rodman & Renshaw Capital Group, Senior Vice President at Madison Williams and Company LLC, Senior Research Analyst at Piper Jaffray Companies, and Project Manager at Entegris, Inc. as part of the Business Development & Engineering groups.
Mr. Kalia has served as a member of the Advisory Board of Levitronix, LLC from 2009 till its acquisition by Thoratec Corp in 2011. He has more than 18 years of experience working in the financial and health care industries. Mr. Kalia has also served as an Adjunct Professor of Finance and taught MBA level courses on Investment Theory and Mergers & Acquisitions at University of St. Thomas.
Mr. Kalia holds the Chartered Financial Analyst (CFA) designation. He received a Master's of Business Administration degree in Finance from the University of St. Thomas in Minneapolis, a Bachelor's degree in Chemical Engineering from the Indian Institute of Technology in Bombay, India and a Master's degree in Chemical Engineering from Stevens Institute of Technology in Hoboken, N.J.
We believe that Mr. Kalia’s extensive background in the financial and medtech industry will bring important practical experience from a financial and strategic perspective. In addition, his engineering background will enable him to provide key insights with respect to the Company’s product development strategy.
Jeffrey Mathiesen
Mr. Mathiesen has served as a member of the board of directors of NeuroOne, Inc. since April 2017 and has been appointed to the board of directors of the Company effective on the 10th day following our filing of the Schedule 14F-1. He currently serves as the Chief Financial Officer of Gemphire Therapeutics Inc., a publicly-held clinical-stage biopharmaceutical company developing therapies for patients with cardiometabolic disorders, a position he has held since September 2015. From August 2015 to September 2015 he was a consultant to Gemphire. Prior to joining Gemphire, Mr. Mathiesen served as Chief Financial Officer of Sunshine Heart, Inc., a publicly traded medical device company, from March 2011 to January 2015. From December 2005 to April 2010, Mr. Mathiesen served as Vice President and Chief Financial Officer of Zareba Systems, Inc., a manufacturer and marketer of medical products, perimeter fencing and security systems, which was purchased by Woodstream Corporation in April 2010. Mr. Mathiesen has held executive positions with publicly traded companies dating back to 1993, including vice president and chief financial officer positions. Mr. Mathiesen also serves as a director, audit committee chairman and nominating and governance committee member of Sun BioPharma, Inc., a publicly traded clinical stage biopharmaceutical company that develops therapies for pancreatic diseases. Mr. Mathiesen received a B.S. in Accounting from the University of South Dakota and is also a Certified Public Accountant.
We believe that Mr. Mathiesen brings financial insight and leadership and a wealth of experience in capital markets to the Board, as well as knowledge of public company accounting and financial reporting requirements and familiarity with the life sciences industry.
Family Relationships
There are no familial relationships between any of our officers and directors.
| 77 |
Structure and Operation of the Board
We do not have standing audit, compensation or nominating committees of our Board. However, the full Board performs all of the functions of a standing audit committee, compensation committee and nominating committee. The following is a brief description of these functions of the Board:
Nomination of Directors
The Board does not currently have a standing nominating committee, and thus we do not have a nominating committee charter. Due to our small size and limited operations to date, the Board determined that it was appropriate for the entire Board to act as the nominating committee. The full Board currently has the responsibility of selecting individuals to be nominated for election to the Board. Board candidates are typically identified by existing directors or members of management. The Board will consider director candidates recommended by stockholders. Any such candidates will be evaluated on the same basis as other candidates being evaluated by the Board. Information with respect to such candidates should be sent to NeuroOne Medical Technologies Corporation, c/o Mr. David Rosa, 10006 Liatris Lane, Eden Prairie, Minnesota 55347. The Board considers the needs for the Board as a whole when identifying and evaluating nominees and, among other things, considers diversity in background, age, experience, qualifications, attributes and skills in identifying nominees, although it does not have a formal policy regarding the consideration of diversity.
Audit Committee Related Function
We do not have a standing audit committee, and thus we do not have an audit committee charter. Due to our small size and limited operations to date, the Board determined that it was appropriate for the entire Board to act as the audit committee. The Board reviews with management and the Company’s independent public accountants the Company’s financial statements, the accounting principles applied in their preparation, the scope of the audit, any comments made by the independent accountants upon the financial condition of the Company and its accounting controls and procedures and such other matters as the Board deems appropriate. During fiscal year 2016, the Board met one time with respect to audit committee related matters. Because the Company’s common stock is traded on the Over the Counter Pink Open Market, the Company is not subject to the listing requirements of any securities exchange regarding audit committee related matters.
The Board currently consists of four directors: Mr. Rosa, Mr. Buckman, Mr. Kalia and Mr. Mathiesen.
Report of Board on Audit Related Matters
In discharging its responsibility for oversight of the audit process, the Board obtained from the Company’s newly appointed independent auditors, BDO USA, LLP (“BDO”), a formal written statement describing any relationships between the auditors and the Company that might bear on the auditors’ independence, consistent with the Independence Standards Board Standard No. 1, “Independence Discussions with Audit Committees.” In addition, the Board discussed with the auditors any relationships that might impact the auditors’ objectivity and independence. The Board is satisfied as to the auditors’ independence.
Audit Committee Financial Expert
We do not have an audit committee financial expert, because we do not have an audit committee.
Risk Oversight
The Board’s risk oversight is administered primarily through the following:
| · | review and approval of an annual business plan; |
| · | review of a summary of risks and opportunities at meetings of the Board; |
| · | review of business developments, business plan implementation and financial results; |
| · | oversight of internal controls over financial reporting; and |
| · | review of employee compensation and its relationship to our business plans. |
Due to the small size and early stage of the Company, we have not adopted a formal policy on whether the Chairman and Chief Executive Officer positions should be separate or combined.
| 78 |
Compensation Committee Related Function
The Board does not currently have a standing compensation committee, and thus we do not have a compensation committee charter. Due to our small size and limited operations to date, the Board determined that it was appropriate for the entire Board to act as the compensation committee. The full Board currently has the responsibility for reviewing and establishing compensation for executive officers and making policy decisions concerning salaries and incentive compensation for executive officers of the Company.
The Company’s executive compensation program is administered by the Board, which determines the compensation of the Chief Executive Officer and other executive officers of the Company. In reviewing the compensation of the individual executive officers (other than the Chief Executive Officer), the Board considers the recommendations of the Chief Executive Officer, published compensation surveys and current market conditions.
Communication with Stockholders
Stockholders wishing to communicate with the Board can send an email to daver@neurooneinc.com or write or telephone David Rosa at the Company’s corporate offices:
NeuroOne Medical Technologies Corporation
c/o David Rosa
10006 Liatris Lane
Eden Prairie, Minnesota 55347
Telephone: 952-237-7412
All such communication must state the type and amount of Company securities held by the stockholder and must clearly state that the communication is intended to be shared with the Board. Mr. Rosa will forward all such communications to the members of the Board.
Code of Business Conduct and Ethics
Our Board has not yet adopted a code of business conduct and ethics that applies to all of our employees, officers and directors, including our Chief Executive Officer and other executive officers, but we intend to adopt a code of business conduct and ethics following the Closing of the Acquisition.
| 79 |
ITEM 6. EXECUTIVE COMPENSATION
All NeuroOne Shares were converted into Company Shares and all NeuroOne Options were converted into Company Options in connection with the Closing of the Acquisition pursuant to the Exchange Ratio. The share and per share information included in this "Executive Compensation" section gives effect to the conversion of such shares and options in the Acquisition, except as otherwise reflected below.
NeuroOne, Inc.’s named executive officers for the year ended December 31, 2016 were:
| · | Dave Rosa, Chief Executive Officer and President |
| · | Wade Fredrickson, former Vice President of Therapy and Product Development |
| · | Mark Christianson, Vice President of Marketing and Sales |
Amer Samad, the Company’s sole named executive officer and director for the years ended December 31, 2016 and 2015 did not receive any compensation for services rendered to us.
Summary Compensation Table
The following table presents the compensation awarded to, earned by or paid to each of our named executive officers of NeuroOne, Inc. for the year ended December 31, 2016.
| Name and Principal Position | Salary ($) | Stock Awards(1) ($) | All Other Compensation ($) | Total ($) | ||||||||||||
| Dave Rosa, Chief Executive Officer and President | 75,000 | (2) | — | 2,898 | (3) | 79,298 | ||||||||||
| Wade Fredrickson, former Vice President, Market Development(4) | 16,667 | (5) | — | 204 | (6) | 21,881 | ||||||||||
| Mark Christianson, Vice President, Business Development and Marketing | 16,667 | (7) | — | 830 | (8) | 20,007 | ||||||||||
(1) In October 2016, NeuroOne, Inc. issued 301,670 NeuroOne Shares as founders’ shares to seven individuals, including the three named executive officers above. The investors did not pay the purchase price for the shares. The purchase price owed by the seven individuals for the founders’ shares under their respective subscription agreements totaling $9,050 was recorded as share subscription receivable in 2016. The receivable was forgiven in June 2017 and will be reportable as compensation in 2017.
(2) Represents the salary earned by Mr. Rosa for his service
from October 5, 2016, the effective date of his employment, through the end of 2016.
(3) Represents an $800 per month car allowance commencing in October 2016 and a $498 life insurance premium paid in 2016.
(4) Mr. Fredrickson was no longer an officer of the Company as of June 28, 2017.
(5) Represents the salary earned by Mr. Fredrickson for his service from December 1, 2016, the effective date of his employment, through December 31, 2016.
(6) Represents a $204 life insurance premium paid in 2016.
(7) Represents the salary earned by Mr. Christianson for his service from December 1, 2016, the effective date of his employment, through December 31, 2016.
(8) Represents a $500 per month car allowance, which was received for December 2016, and a $330 life insurance premium paid in 2016.
| 80 |
Narrative to Summary Compensation Table
Our board of directors sets the annual compensation for our named executive officers. As we are a development-stage company and commenced our operational efforts in 2016, we have not hired a compensation consultant and do not currently target a specific competitive position or a specific mix of compensation among base salary, bonus or long-term incentives.
Our subsidiary, NeuroOne, Inc., entered into an employment agreement with Mr. Rosa in October 2016 that establishes his base salary and target bonus opportunity. Mr. Fredrickson’s and Mr. Christianson’s base salaries and bonus opportunities were established in offer letters negotiated in connection with their hiring. In connection with the Acquisition, the named executive officers of NeuroOne, Inc. were appointed as officers of the Company, and we assumed those employment arrangements and entered into an amended and restated employment agreement with Mr. Rosa.
Except for car allowances provided to Mr. Rosa and Mr. Christianson and payment of a portion of the premiums for life, medical and disability insurance for all employees, NeuroOne, Inc. did not, and we do not, provide perquisites or personal benefits to our named executive officers.
Employment Agreements
Our subsidiary, NeuroOne, Inc. entered into a written employment agreement with Mr. Rosa, as described below. Each of our named executive officers has also executed our standard form of proprietary information, inventions assignment and non-competition agreement.
In October 2016, NeuroOne, Inc. entered into an employment agreement with Mr. Rosa that governs the terms of his employment with us. Pursuant to the agreement, Mr. Rosa is entitled to an annual base salary of $300,000 per year, and is eligible to earn an annual performance bonus of up to 40% of his base salary, as determined by our board of directors. Mr. Rosa was also entitled to an equity award equal to 14% of the fully diluted equity of Neuro One, Inc.
The employment agreement provides for an at-will arrangement and may be terminated by Mr. Rosa or NeuroOne, Inc. at any time, with or without cause. If NeuroOne, Inc. terminates Mr. Rosa’s employment without cause or if Mr. Rosa resigns for good reason, Mr. Rosa is eligible to receive a continuation of his base salary in effect on the termination date for 12 months and a prorated portion of his annual performance bonus, if any, based upon the number of full months during the year of termination in which Mr. Rosa was employed. All severance payments related to his annual base salary will be paid in equal monthly installments in accordance with our regular payroll practices; however, the initial payment will not be made until the first regular monthly payroll date that is more than 60 days after the termination date. Any severance payment related to his annual performance bonus will be made at the same time annual bonuses are paid to other employees.
If Mr. Rosa’s employment is terminated without cause or Mr. Rosa resigns for good reason within two years of a change in control, the annual base salary severance payments will increase from 12 months to 18 months and will be paid on the first payroll date that is more than 60 days after Mr. Rosa’s last date of employment.
In order to receive any severance payments, Mr. Rosa must deliver a general release to NeuroOne, Inc. and must not be in material breach of any other agreement between Mr. Rosa and NeuroOne, Inc. In the event Mr. Rosa materially breaches any agreement while still receiving severance payments, all benefits will cease being paid.
NeuroOne, Inc. 2016 Equity Incentive Plan
In October 2016 NeuroOne, Inc.’s board of directors adopted and its stockholders approved the 2016 Equity Incentive Plan, or the 2016 Plan. In connection with the Acquisition, we assumed the 2016 Plan. As of the Closing of the Acquisition, there were outstanding Company Options to purchase a total of 365,716 Company Shares issued and outstanding under the 2016 Plan and 215,453 restricted Company Shares had been granted under the 2016 Plan. Upon the closing of the Acquisition, we anticipate that no additional awards will be granted under the 2016 Plan, and all awards will be granted under the 2017 Plan.
| 81 |
Description of the Plan
The 2016 Plan authorizes the board of directors to provide incentive compensation in the form of stock options, stock appreciation rights and restricted stock and stock units. Under the 2016 Plan, the board of directors is authorized to issue up to 58,333 shares (pre-Acquisition).
Our board of directors, or a duly authorized committee thereof, has the authority to administer the 2016 Plan. Our board of directors may also delegate to one or more of its officers the authority to (1) designate officers and employees to be recipients of certain stock awards, and (2) determine the number of shares of common stock to be subject to such stock awards. Subject to the terms of the 2016 Plan, our board of directors or the authorized committee, referred to therein as the plan administrator, determines recipients, dates of grant, the numbers and types or combination of types of stock awards to be granted and the terms and conditions of the stock awards, including the vesting schedule applicable to a stock award. Subject to the limitations set forth below, the plan administrator also determines the exercise price, strike price or purchase price of awards granted and the types of consideration to be paid for the award.
The plan administrator has the authority to modify outstanding awards under the 2016 Plan. Subject to the terms of the 2016 Plan, the plan administrator has the authority to reduce the exercise, purchase or strike price of any outstanding stock award, cancel any outstanding stock award in exchange for new stock awards, cash or other consideration, or take any other action that is treated as a repricing under generally accepted accounting principles, with the consent of any adversely affected participant.
Stock Options
The plan administrator determines the term of stock options granted under the 2016 Plan, up to a maximum of ten years. Unless the terms of an optionholder’s stock option agreement provides otherwise, if an optionholder’s service relationship with the Company, or any of its affiliates, ceases for any reason other than disability, death or cause, the optionholder may generally exercise any vested options for a period of three months following the cessation of service. The option term may be extended in the event that exercise of the option following such a termination of service is prohibited by applicable securities laws. If an optionholder’s service relationship with the Company or any of its affiliates ceases due to disability or death, or an optionholder dies within a certain period following cessation of service, the optionholder or a beneficiary may generally exercise his or her options for a period of 12 months in the event of disability and 18 months in the event of death. In the event of a termination for cause, options generally terminate upon the termination date of the individual. In no event may an option be exercised beyond the expiration of its term.
Acceptable consideration for the purchase of common stock issued upon the exercise of a stock option is determined by the plan administrator and may include (1) cash, check, bank draft or money order, (2) the tender of shares of the Company’s common stock previously owned by the optionholder, (3) a net exercise of the option if it is a non-qualified stock option, or NSO, (4) a deferred payment or similar arrangement with the optionholder, and (5) other legal consideration approved by the plan administrator.
Unless the plan administrator provides otherwise, options generally are not transferable except by will, the laws of descent and distribution, or pursuant to a domestic relations order. An optionholder may designate a beneficiary, however, who may exercise the option following the optionholder’s death. The board of directors may also permit transfer of options to a trust provided certain conditions are met.
The aggregate fair market value, determined at the time of grant, of the Company’s common stock with respect to Incentive Stock Options, or ISOs, that are exercisable for the first time by an optionholder during any calendar year under all of the Company’s stock plans may not exceed $100,000. Options or portions thereof that exceed such limit are generally treated as NSOs. No ISO may be granted to any person who, at the time of the grant, owns or is deemed to own stock possessing more than 10% of the Company’s total combined voting power or that of any of the Company’s affiliates unless (1) the option exercise price is at least 110% of the fair market value of the stock subject to the option on the date of grant, and (2) the term of the ISO does not exceed five years from the date of grant.
| 82 |
Changes in Capital Structure
In the event that there is a specified type of change in the Company’s capital structure, such as a stock split or recapitalization, appropriate adjustments will be made to (1) the class and maximum number of shares reserved for issuance under the 2016 Plan, (2) the class and maximum number of shares by which the share reserve may increase automatically each year, (3) the class and maximum number of shares that may be issued upon the exercise of ISOs, (4) the class and number of shares and exercise price, strike price, or purchase price, if applicable, of all outstanding stock awards.
Other Corporate Transactions
In the event of certain specified significant corporate transactions, the plan administrator has the discretion to take any of the following actions with respect to stock awards:
| · | arrange for the assumption, continuation or substitution of stock awards outstanding under the 2016 Plan by a surviving or acquiring entity or parent company; |
| · | arrange for the reacquisition or repurchase rights held by the Company to the surviving or acquiring entity or parent company; |
| · | accelerate the vesting of the stock award and provide for its termination prior to the effective time of the corporate transaction; |
| · | terminate unexercised stock awards held by participants no longer employed by the Company and that have not been assumed, continued or substituted; or |
| · | make a payment equal to the excess of (1) the value of the property the participant would have received upon exercise of the stock award over (2) the exercise price applicable to the stock award. |
The Company’s plan administrator is not obligated to treat all stock awards, even those that are of the same type, in the same manner.
Under the 2016 Plan, a corporate transaction is generally the consummation of (1) a sale or other disposition of all or substantially all of the Company’s consolidated assets, (2) a sale or other disposition of at least 90% of the Company’s outstanding securities, (3) a merger, consolidation or similar transaction following which the Company is not the surviving corporation, or (4) a merger, consolidation or similar transaction following which the Company is the surviving corporation but the Company Shares outstanding immediately prior to such transaction are converted or exchanged into other property by virtue of the transaction.
The plan administrator may provide, in an individual award agreement or in any other written agreement between a participant and the Company that the stock award will be subject to additional acceleration of vesting and exercisability in the event of a change of control transaction. For example, certain of the Company’s employees may receive an award agreement that provides for vesting acceleration upon the individual’s termination without cause or resignation for good reason (including a material reduction in the individual’s base salary, duties, responsibilities or authority, or a material relocation of the individual’s principal place of employment with the Company) in connection with a change of control. Under the 2016 Plan, a change of control is generally (1) the acquisition by a person or entity of more than 50% of the Company’s combined voting power other than by merger, consolidation or similar transaction; (2) a consummated merger, consolidation or similar transaction immediately after which the Company’s stockholders cease to own more than 50% of the combined voting power of the surviving entity; or (3) a consummated sale, lease or license or other disposition of all or substantially of the Company’s consolidated assets.
| 83 |
2017 Equity Incentive Plan
In April 2017, our board of directors adopted and our stockholders approved the 2017 equity incentive plan, or the 2017 Plan. The 2017 Plan is designed to provide a vehicle under which a variety of stock-based and other awards can be granted to the Company’s employees, consultants and directors, which will align the interests of award recipients with those of our stockholders, reinforce key goals and objectives that help drive stockholder value, and attract, motivate and retain experienced and highly qualified individuals who will contribute to the Company’s financial success. The board of directors believes that the 2017 Plan will serve a critical role in attracting and retaining high caliber employees, consultants and directors essential to our success and in motivating these individuals to strive to meet our goals.
Shares Available Under the Plan
The 2017 Plan authorizes the compensation committee or board of directors to provide incentive compensation in the form of stock options, stock appreciation rights, restricted stock and stock units, performance shares and units and other stock-based awards. Under the 2017 Plan, the compensation committee is authorized to issue up to 1,300,000 shares, subject to adjustment as provided below.
Section 162(m) Tax Considerations
The 2017 Plan is designed to help us comply with the rules relating to our ability to deduct in full for federal income tax purposes the compensation recognized by our executive officers in connection with certain types of awards. Section 162(m) of the Internal Revenue Code generally denies a corporate tax deduction for annual compensation exceeding $1 million paid to the chief executive officer or any of the three other most highly compensated officers of a publicly held company other than the chief financial officer. However, qualified performance-based compensation is excluded from this limit. To enable compensation in connection with stock options, stock appreciation rights, certain restricted stock and restricted stock unit awards, performance shares, performance units and certain other stock-based awards granted under the 2017 Plan that are intended to qualify as “performance-based” within the meaning of Section 162(m) of the Code to be deductible, the stockholders were asked to approve certain material terms of the 2017 Plan. By approving the 2017 Plan, the Consenting Stockholder specifically approved, among other things:
| · | the eligibility requirements for participation in the 2017 Plan; |
| · | the maximum number of shares for which stock-based awards may be granted to an employee in any fiscal year; and |
| · | the performance measures that may be used by the compensation committee to establish the performance goals applicable to the grant or vesting of awards of restricted stock, restricted stock units, performance shares, performance units and other stock-based awards that are intended to result in qualified performance-based compensation. |
While we believe that compensation provided by such awards under the 2017 Plan generally will be deductible by us for federal income tax purposes, under certain circumstances, such as a change in control, compensation paid in settlement of certain awards may not qualify as performance-based. In addition, Section 162(m) of the Code imposes a number of other requirements that must be met in order for awards to qualify for deduction under the Code. Accordingly, there can be no assurance that awards under the 2017 Plan will be fully deductible under all circumstances. In addition, other awards under the 2017 Plan which are not intended to satisfy these “performance-based” compensation requirements generally will not so qualify. To the extent that such compensation, when added to other non-exempt compensation, exceeds $1 million in any given year paid to certain executives, then the amount in excess of $1 million will be subject to the deduction limitations of Section 162(m) of the Code.
Description of the 2017 Plan
Authorized Shares. The maximum number of shares of our common stock that may be issued under the 2017 Plan, is 1,300,000 shares. In addition, the number of shares of our common stock reserved for issuance under our 2017 Plan will automatically increase on January 1st of each calendar year, starting on January 1, 2018 through January 1, 2027, in an amount equal to 13.0% of the total number of fully-diluted shares of our common stock as of December 31 of the preceding calendar year, or a lesser number of shares determined by our board of directors. The maximum number of shares of our common stock that may be issued on the exercise of incentive stock options under the 2017 Plan is 1,300,000.
| 84 |
Shares subject to awards granted under the 2017 Plan that expire or terminate without being exercised in full, or that are paid out in cash rather than in shares, do not reduce the number of shares available for issuance under the 2017 Plan. Additionally, shares become available for future grant under the 2017 Plan if they were issued under stock awards under the 2017 Plan and if we repurchase them or they are forfeited. This includes shares used to pay the exercise price of a stock award or to satisfy the tax withholding obligations related to a stock award.
Plan Administration. Our board of directors, or a duly authorized committee of our board of directors, will administer the 2017 Plan. Our board of directors may also delegate to one or more of our officers the authority to (1) designate employees (other than officers) to receive specified stock awards and (2) determine the number of shares subject to such stock awards. Under the 2017 Plan, our board of directors has the authority to determine and amend the terms of awards and underlying agreements, including:
| · | recipients; |
| · | the exercise, purchase, or strike price of stock awards, if any; |
| · | the number of shares subject to each stock award; |
| · | the vesting schedule applicable to the awards, together with any vesting acceleration; and |
| · | the form of consideration, if any, payable on exercise or settlement of the award. |
Under the 2017 Plan, the board of directors also generally has the authority to effect, with the consent of any adversely affected participant:
| · | the reduction of the exercise, purchase, or strike price of any outstanding award; |
| · | the cancellation of any outstanding award and the grant in substitution therefore of other awards, cash, or other consideration; or |
| · | any other action that is treated as a repricing under generally accepted accounting principles. |
Section 162(m) Limits. At such time as necessary for compliance with Section 162(m) of the Code, no participant may be granted stock awards covering more than 1,300,000 shares of our common stock under the 2017 Plan during any calendar year pursuant to stock options, stock appreciation rights, and other stock awards whose value is determined by reference to an increase over an exercise price or strike price of at least 100% of the fair market value of our common stock on the date of grant. Additionally, under the 2017 Plan, in a calendar year, no participant may be granted a performance stock award covering more than 1,300,000 shares of our common stock or a performance cash award having a maximum value in excess of $1 million. These limitations are designed to allow us to grant compensation that will not be subject to the $1,000,000 annual limitation on the income tax deductibility of compensation paid to a covered executive officer imposed by Section 162(m) of the Code.
Stock Options. Incentive stock options and nonstatutory stock options are granted under stock option agreements adopted by the plan administrator. The plan administrator determines the exercise price for stock options, within the terms and conditions of the 2017 Plan, provided that the exercise price of a stock option generally cannot be less than 100% of the fair market value of our common stock on the date of grant. Options granted under the 2017 Plan vest at the rate specified in the stock option agreement as determined by the plan administrator.
Restricted Stock Unit Awards. Restricted Stock Units (“RSUs”) are granted under restricted stock unit award agreements adopted by the plan administrator. RSUs may be granted in consideration for any form of legal consideration that may be acceptable to our board of directors and permissible under applicable law. An RSU may be settled by cash, delivery of stock, a combination of cash and stock as deemed appropriate by the plan administrator, or in any other form of consideration set forth in the RSU agreement. Additionally, dividend equivalents may be credited in respect of shares covered by an RSU. Except as otherwise provided in the applicable award agreement, RSUs that have not vested will be forfeited once the participant’s continuous service ends for any reason.
| 85 |
Restricted Stock Awards. Restricted stock awards are granted under restricted stock award agreements adopted by the plan administrator. A restricted stock award may be awarded in consideration for cash, check, bank draft or money order, past services to us, or any other form of legal consideration that may be acceptable to our board of directors and permissible under applicable law. The plan administrator determines the terms and conditions of restricted stock awards, including vesting and forfeiture terms. If a participant’s service relationship with us ends for any reason, we may receive any or all of the shares of common stock held by the participant that have not vested as of the date the participant terminates service with us through a forfeiture condition or a repurchase right.
Stock Appreciation Rights. Stock appreciation rights are granted under stock appreciation grant agreements adopted by the plan administrator. The plan administrator determines the purchase price or strike price for a stock appreciation right, which generally cannot be less than 100% of the fair market value of our common stock on the date of grant. A stock appreciation right granted under the 2017 Plan vests at the rate specified in the stock appreciation right agreement as determined by the plan administrator.
Performance Awards. The 2017 Plan permits the grant of performance-based stock and cash awards that may qualify as performance-based compensation that is not subject to the $1,000,000 limitation on the income tax deductibility imposed by Section 162(m) of the Code. Our compensation committee may structure awards so that the stock or cash will be issued or paid only following the achievement of certain pre-established performance goals during a designated performance period.
The performance goals that may be selected include one or more of the following: (i) earnings (including earnings per share and net earnings); (ii) earnings before interest, taxes and depreciation; (iii) earnings before interest, taxes, depreciation and amortization; (iv) earnings before interest, taxes, depreciation, amortization and legal settlements; (v) earnings before interest, taxes, depreciation, amortization, legal settlements and other income (expense); (vi) earnings before interest, taxes, depreciation, amortization, legal settlements, other income (expense) and stock-based compensation; (vii) earnings before interest, taxes, depreciation, amortization, legal settlements, other income (expense), stock-based compensation and changes in deferred revenue; (viii) earnings before interest, taxes, depreciation, amortization, legal settlements, other income (expense), stock-based compensation, other non-cash expenses and changes in deferred revenue; (ix) total stockholder return; (x) return on equity or average stockholder’s equity; (xi) return on assets, investment, or capital employed; (xii) stock price; (xiii) margin (including gross margin); (xiv) income (before or after taxes); (xv) operating income; (xvi) operating income after taxes; (xvii) pre-tax profit; (xviii) operating cash flow; (xix) sales or revenue targets; (xx) increases in revenue or product revenue; (xxi) expenses and cost reduction goals; (xxii) improvement in or attainment of working capital levels; (xxiii) economic value added (or an equivalent metric); (xxiv) market share; (xxv) cash flow; (xxvi) cash flow per share; (xxvii) cash balance; (xxviii) cash burn; (xxix) cash collections; (xxx) share price performance; (xxxi) debt reduction; (xxxii) implementation or completion of projects or processes (including, without limitation, clinical trial initiation, new and supplemental indications for existing products, and product supply); (xxxiii) stockholders’ equity; (xxxiv) capital expenditures; (xxxv) debt levels; (xxxvi) operating profit or net operating profit; (xxxvii) workforce diversity; (xxxviii) growth of net income or operating income; (xxxix) billings; (xl) bookings; (xli) employee retention; (xlii) initiation of phases of clinical trials and/or studies by specific dates; (xliii) acquisition of new customers, including institutional accounts; (xliv) customer retention and/or repeat order rate; (xlv) number of institutional customer accounts; (xlvi) budget management; (xlvii) improvements in sample and test processing times; (xlviii) regulatory milestones; (xlix) progress of internal research or clinical programs; (l) progress of partnered programs; (li) partner satisfaction; (lii) milestones related to samples received and/or tests run; (liii) expansion of sales in additional geographies or markets; (liv) research progress, including the development of programs; (lv) patient samples processed and billed; (lvi) sample processing operating metrics (including, without limitation, failure rate maximums and reduction of repeat rates); (lvii) strategic partnerships or transactions (including in-licensing and out-licensing of intellectual property); and (lviii) and to the extent that an award is not intended to constitute “qualified performance-based compensation” under Section 162(m) of the Code, other measures of performance selected by the board of directors.
| 86 |
The performance goals may be based on company-wide performance or performance of one or more business units, divisions, affiliates, or business segments, and may be either absolute or relative to the performance of one or more comparable companies or the performance of one or more relevant indices. Unless specified otherwise by the board of directors or committee (as applicable) (i) in the award agreement at the time the award is granted or (ii) in such other document setting forth the performance goals at the time the performance goals are established, the board of directors or committee (as applicable) will appropriately make adjustments in the method of calculating the attainment of performance goals for a performance period as follows: (1) to exclude restructuring and/or other nonrecurring charges; (2) to exclude exchange rate effects; (3) to exclude the effects of changes to generally accepted accounting principles; (4) to exclude the effects of any statutory adjustments to corporate tax rates; (5) to exclude the effects of any “extraordinary items” as determined under generally accepted accounting principles; (6) to exclude the dilutive effects of acquisitions or joint ventures; (7) to assume that any business divested by the Company achieved performance objectives at targeted levels during the balance of a performance period following such divestiture; (8) to exclude the effect of any change in the outstanding shares of Common Stock of the Company by reason of any stock dividend or split, stock repurchase, reorganization, recapitalization, merger, consolidation, spin-off, combination or exchange of shares or other similar corporate change, or any distributions to common stockholders other than regular cash dividends; (9) to exclude the effects of stock based compensation and the award of bonuses under the Company’s bonus plans; (10) to exclude costs incurred in connection with potential acquisitions or divestitures that are required to be expensed under generally accepted accounting principles; (11) to exclude the goodwill and intangible asset impairment charges that are required to be recorded under generally accepted accounting principles; (12) to exclude the effect of any other unusual, non-recurring gain or loss or other extraordinary item; and (13) to exclude the effects of the timing of acceptance for review and/or approval of submissions to any regulatory body. In addition, subject to certain limitations, the board of directors or committee (as applicable) retains the discretion to reduce or eliminate the compensation or economic benefit due on attainment of performance goals and to define the manner of calculating the performance criteria it selects to use for such performance period. Partial achievement of the specified criteria may result in the payment or vesting corresponding to the degree of achievement as specified in the award agreement or the written terms of a performance cash award.
Other Stock Awards. The plan administrator may grant other awards based in whole or in part by reference to our Common Stock. The plan administrator will set the number of shares under the stock award and all other terms and conditions of such awards.
Changes to Capital Structure. In the event there is a specified type of change in our capital structure, such as a stock split, reverse stock split, or recapitalization, appropriate adjustments will be made to (1) the class and maximum number of shares reserved for issuance under the 2017 Plan, (2) the class and maximum number of shares that may be issued on the exercise of incentive stock options, (3) the class and maximum number of shares subject to stock awards that can be granted to a person in a calendar year (as established under the 2017 Plan under Section 162(m) of the Code), and (4) the class and number of shares and exercise price, strike price, or purchase price, if applicable, of all outstanding stock awards.
Corporate Transactions. The 2017 Plan provides that in the event of certain specified significant corporate transactions, including: (1) a sale of all or substantially all of our assets, (2) the sale or disposition of more than 90% of our outstanding securities, (3) the consummation of a merger or consolidation where we do not survive the transaction, and (4) the consummation of a merger or consolidation where we do survive the transaction but the shares of our Common Stock outstanding before such transaction are converted or exchanged into other property by virtue of the transaction, unless otherwise provided in an award agreement or other written agreement between us and the award holder, the administrator may take one or more of the following actions with respect to such stock awards:
| · | arrange for the assumption, continuation, or substitution of a stock award by a successor corporation; |
| · | arrange for the assignment of any reacquisition or repurchase rights held by us to a successor corporation; |
| · | accelerate the vesting, in whole or in part, of the stock award and provide for its termination before the transaction; |
| · | arrange for the lapse, in whole or in part, of any reacquisition or repurchase rights held by us; |
| · | cancel or arrange for the cancellation of the stock award before the transaction in exchange for a cash payment, or no payment, as determined by the board; or |
| · | make a payment, in the form determined by our board of directors, equal to the excess, if any, of the value of the property the participant would have received on exercise of the awards before the transaction over any exercise price payable by the participant in connection with the exercise. |
| 87 |
The plan administrator is not obligated to treat all stock awards or portions of stock awards, even those that are of the same type, in the same manner and is not obligated to treat all participants in the same manner.
In the event of a change in control, awards granted under the 2017 Plan will not receive automatic acceleration of vesting and exercisability, although this treatment may be provided for in an award agreement. Under the 2017 Plan, a change in control is defined to include (1) the acquisition by any person or company of more than 50% of the combined voting power of our then outstanding stock, (2) a merger, consolidation, or similar transaction in which our stockholders immediately before the transaction do not own, directly or indirectly, more than 50% of the combined voting power of the surviving entity (or the parent of the surviving entity), (3) a sale, lease, exclusive license, or other disposition of all or substantially all of our assets other than to an entity more than 50% of the combined voting power of which is owned by our stockholders, and (4) an unapproved change in the majority of the board of directors.
Transferability. A participant may not transfer stock awards under the 2017 Plan other than by will, the laws of descent and distribution, or as otherwise provided under the 2017 Plan.
Plan Amendment or Termination. Our board of directors has the authority to amend, suspend, or terminate the 2017 Plan, provided that such action does not materially impair the existing rights of any participant without such participant’s written consent. Certain material amendments also require the approval of our stockholders. No incentive stock options may be granted after the tenth anniversary of the date our board of directors adopted the 2017 Plan. No stock awards may be granted under the 2017 Plan while it is suspended or after it is terminated.
New Plan Benefits
A new plan benefits table for the 2017 Plan and the benefits or amounts that would have been received by or allocated to participants for the last completed fiscal year under the 2017 Plan if the 2017 Plan was then in effect, are not provided because all awards made under the 2017 Plan will be made at the board of directors or compensation committee’s discretion, subject to the terms of the 2017 Plan. Therefore, the benefits and amounts that will be received or allocated under the 2017 Plan are not determinable at this time.
Dissenters’ Rights
Our stockholders are not entitled to dissenters’ rights with respect to the 2017 Plan.
Rule 10b5-1 Sales Plans
Our directors and executive officers may adopt written plans, known as Rule 10b5-1 plans, in which they will contract with a broker to buy or sell shares of our common stock on a periodic basis. Under a Rule 10b5-1 plan, a broker executes trades pursuant to parameters established by the director or officer when entering into the plan, without further direction from them. The director or officer may amend a Rule 10b5-1 plan in some circumstances and may terminate a plan at any time. Our directors and executive officers also may buy or sell additional shares outside of a Rule 10b5-1 plan when they are not in possession of material nonpublic information subject to compliance with the terms of our insider trading policy.
Limitation on Liability and Indemnification of Directors and Officers
Our certificate of incorporation and bylaws limit our directors' liability to the fullest extent permitted under Delaware corporate law. Delaware corporate law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability:
| · | for any transaction from which the director derives an improper personal benefit; |
| · | for any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| 88 |
| · | under Section 174 of the Delaware General Corporation Law (unlawful payment of dividends or redemption of shares); or |
| · | for any breach of a director's duty of loyalty to the corporation or its stockholders. |
If the Delaware General Corporation Law is amended to authorize corporate action further eliminating or limiting the personal liability of directors, then the liability of our directors shall be eliminated or limited to the fullest extent permitted by the Delaware General Corporation Law, as so amended.
Delaware law and our certificate of incorporation and our bylaws provide that we will, in certain situations, indemnify our directors and officers and may indemnify other employees and other agents, to the fullest extent permitted by law. Any indemnified person is also entitled, subject to certain limitations, to payment or reimbursement of reasonable expenses (including attorneys' fees and disbursements) in advance of the final disposition of the proceeding.
In addition, we have entered, and intend to continue to enter, into separate indemnification agreements with our directors and officers. These agreements, among other things, require us to indemnify our directors and officers for certain expenses, including attorneys' fees, judgments, fines and settlement amounts incurred by a director or officer in any action or proceeding arising out of their services as one of our directors or officers or any other company or enterprise to which the person provides services at our request.
We are procuring a directors' and officers' insurance policy pursuant to which our directors and officers are insured against certain liability for actions taken in their capacities as directors and officers. We believe that these provisions in our certificate of incorporation and bylaws and these indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or control persons, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Non-Employee Director Compensation
All NeuroOne Shares were converted into Company Shares and all NeuroOne Options were converted into Company Options in connection with the Closing of the Acquisition pursuant to the Exchange Ratio. The share and per share information included in this "—Non-Employee Director Compensation " section gives effect to the conversion of such shares and options in the Acquisition.
We have not historically paid cash retainers or other cash compensation with respect to service on the Board, except for reimbursement of direct expenses incurred in connection with the business of the Company. The Company grants stock options to its non-employee directors in connection with their appointment to the Board under the Company’s 2016 Equity Incentive Plan. In April 2017, NeuroOne, Inc. granted an option to purchase 2,000 NeuroOne Shares (34,020 Company Shares) to each of Mr. Buckman, Mr. Kalia and Mr. Mathiesen, at an exercise price of $0.59 per NeuroOne Share ($0.035 per Company Share).
Director Compensation Table
Paul Buckman was the only non-employee director of NeuroOne, Inc. during the year ended December 31, 2016; no compensation was paid to Mr. Buckman during the year ended December 31, 2016. David Rosa, our Chief Executive Officer, was a NeuroOne, Inc. director during the year ended December 31, 2016, but did not receive any additional compensation for his service as a director. Mr. Rosa’s compensation as an executive officer is set forth below under "Executive Compensation—Summary Compensation Table."
| 89 |
ITEM 7. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
There have been no transactions since January 1, 2015 to which NeuroOne, Inc. has been a participant in which the amount involved exceeded or will exceed the lesser of $120,000 or 1% of the average of the Company’s total assets as of December 31, 2016, and in which any of our directors, executive officers or holders of more than five percent of our capital stock, or any members of their immediate family, had or will have a direct or indirect material interest, other than compensation arrangements which are described under "Executive Compensation."
NeuroOne LLC Merger into NeuroOne, Inc.
NeuroOne, Inc.’s predecessor company, NeuroOne LLC (the “LLC”), was formed as a limited liability company in Minnesota on December 13, 2013 and merged with and into NeuroOne, Inc. on October 27, 2016. The founder and sole member of the LLC received, upon the effectiveness of the merger, in consideration for the cancellation of his membership interests in the LLC, 5,000 shares of common stock of NeuroOne, Inc. The holders of shares of common stock of NeuroOne, Inc., including David Rosa, our Chief Executive Officer and Director, Mark Christianson, our Vice President of Marketing and Sales, and Wade Fredrickson, our former Vice President of Therapy and Product Development, received, upon the effectiveness of the Merger, one share of common stock of NeuroOne, Inc. for every three shares of common stock of NeuroOne, Inc. held pre-Merger.
NeuroOne, Inc. Founders Shares
In October 2016, prior to the Merger, NeuroOne, Inc. issued 301,670 NeuroOne Shares as founders’ shares in a private placement to seven individuals (who were all accredited investors), including David Rosa, our Chief Executive Officer and Director, Mark Christianson, our Vice President of Marketing and Sales, and Wade Fredrickson, our former Vice President of Therapy and Product Development. The value applied to the shares issued was $0.03 per NeuroOne Share based on a valuation utilizing a weighted average market value of invested capital methodology. In June 2017, the purchase price owed by the seven individuals for the founders’ shares under their respective subscription agreements totaling $9,050 was forgiven by the Company in its entirety. For more information, see “Item 3.02 –Unregistered Sales of Equity Securities.”
Stockholders Agreement
In connection with the issuance of founders’ shares described above, NeuroOne, Inc. entered into a stockholders agreement with the recipients of founders’ shares, which was also executed by the recipients of NeuroOne Shares subsequent to the execution of the stockholders agreement.
The stockholders agreement, among other things, grants certain of our stockholders drag-along rights and preemptive rights with respect to NeuroOne Shares issued in subsequent offerings. The parties to the stockholders agreement agreed to vote their NeuroOne Shares to ensure that the board size and composition is as directed by the then current NeuroOne Board and designated David A. Rosa and Paul Buckman as the initial directors of NeuroOne, Inc. The stockholders agreement, by its terms, terminated at the Closing of the Acquisition. The stockholders agreement is filed as an exhibit to this Report and is incorporated herein by reference.
Issuance of NeuroOne, Inc. Notes and Warrants
Our Board of Directors approved a Note and Warrant financing for gross proceeds of up to $1.5 million in November 2016 and increased such financing authority to $2.5 million in June 2017. Between November 2016 and June 2017, NeuroOne, Inc. issued Notes and Warrants to investors in a private placement, including, in June 2017, a Note for $50,000 and Warrants to the founder and sole owner of the LLC. For more information, see “Item 11. Description of Capital Stock—Convertible Promissory Notes” and “Item 11. Description of Capital Stock—Warrants”.
| 90 |
Mayo Development Agreement
Pursuant to the Mayo Development Agreement, we have agreed to license worldwide (i) certain know how for the development and commercialization of products, methods and processes related to flexible circuit thin film technology for the recording of tissue and (ii) the products developed therefrom, and to partner with Mayo to assist the Company in the investigation, research application, development and improvement of such technology. Mayo has agreed to assist us by providing access to the Mayo Principal Investigators in developing a minimally invasive device/delivery system and procedure for a minimally invasive approach for the implantation of our cortical thin film flexible circuit technology developed by the Company, including prototype development, animal testing, protocol development for human and animal use, abstract development and presentation and access to and license of any intellectual property that the Mayo Principal Investigators develop relating to the procedure.
Whether or not any such technology, product, method, process, device, delivery system or minimally invasive approach is developed or approved, we agreed, in consideration for Mayo’s efforts under the Mayo Development Agreement, to pay Mayo a cash payment of approximately $92,000 on the earlier of September 30, 2017 or the date we raise a minimum amount of financing, and on May 25, 2017, NeuroOne, Inc. issued Mayo 50,556 NeuroOne Shares pursuant to a Subscription Agreement, pursuant to which Mayo became a holder of over 5% of NeuroOne, Inc.’s outstanding common stock. Finally, we have agreed to pay Mayo a royalty equal to a single-digit percentage of our product sales pursuant to the Mayo Development Agreement. Mayo may purchase any developed products licensed under the Mayo Development Agreement at the best price offered by us to the end user in the prior year. The Mayo Development Agreement will expire on May 25, 2037 and may be terminated by Mayo for cause or under certain circumstances.
For additional information regarding the Mayo Development Agreement and our past breach thereof, see “Risk Factors—We depend on our partnership with Mayo Foundation for Medical Education and Research to license certain know how for the development and commercialization of our technology. Termination of this partnership would harm our business, and even if this partnership continues, it may not be successful.”
The Placement Agent
The placement agent of NeuroOne, Inc. for the private placement of the Notes and Warrants, HRA Capital, is affiliated with two of our greater than 5% stockholders, Chromium 24 LLC and Lifestyle Healthcare LLC. Pursuant to the engagement letter with such placement agent, NeuroOne, Inc. owes the placement agent a cash fee of $113,610 (8% of the gross proceeds received from certain Note and Warrant investors) and is obligated to issue to the placement agent a Warrant to purchase shares of common stock (or common stock equivalents) in an amount equal to 8% of the common stock purchased by investors in the private placement, which Warrants are expected to have an exercise price of $2.00 per Company Share. The placement agent Warrants are immediately exercisable and expire five years from the date of issuance. The exercise price and number of the shares of our common stock issuable upon exercising the placement agent Warrant will be subject to adjustment in the event of any stock dividends and splits, reverse stock split, recapitalization, reorganization, business combination or similar transaction, as described therein. Under the engagement letter, the placement agent is further entitled to receive warrants to purchase common stock in an amount equal to 10% of the common stock (or common stock equivalents) purchased by certain investors in subsequent equity financing rounds. Such warrants will have an exercise price determined in relation to the pricing of the subsequent financing and will be immediately exercisable once issued and have a term of 5 years.
We also received a short-term unsecured loan for $50,000 in November 2016 from the placement agent. We incurred no fees or interest costs related to such temporary loan and repaid it in full in February 2017.
The Acquisition
Pursuant to the Merger Agreement for the Acquisition whereby NeuroOne, Inc. became a wholly-owned subsidiary of the Company, each holder of NeuroOne Shares outstanding immediately prior to the Closing received Company Shares in exchange therefore based on the Exchange Ratio, with all fractional shares rounded down to the nearest whole share. Accordingly, we issued 793,822, 1,423,206 and 2,840,731 Company Shares to Messrs. Rosa, Christianson and Fredrickson, respectively, 34,020 Company Options to each of Messrs. Buckman, Kalia and Mathiesen and 859,976 Company Shares to Mayo, a holder of over 5% of our outstanding common stock. All of the Company Shares held by Mr. Samad prior to the Closing of the Acquisition were cancelled by us upon the Closing pursuant to the terms of the Merger Agreement. The Merger Agreement also provides that Mr. Rosa be appointed as a director of the Company upon the Closing of the Acquisition.
| 91 |
Indemnification Agreements
Our certificate of incorporation contains provisions limiting the liability of directors, and our bylaws provides that we indemnify each of our directors to the fullest extent permitted under Delaware law. Our certificate of incorporation and bylaws also provide our board of directors with discretion to indemnify our officers and employees when determined appropriate by the board.
In addition, we have entered into an indemnification agreement with our directors and our executive officers. For more information regarding these agreements, see "Executive Compensation—Limitations on Liability and Indemnification Matters."
Related Person Transaction Policy
The Board reviews, approves and oversees any transaction between us and any related person and any other potential conflict of interest situations on an ongoing basis, in accordance with our policies and procedures, and develops policies and procedures for the approval of related party transactions. Prior to consideration of a transaction with a related person, the material facts as to the related person's relationship or interest in the transaction are disclosed to the disinterested directors. The transaction is not approved unless a majority of the members of the Board who are not interested in the transaction approve the transaction. The Board takes into account, among other factors that it deems appropriate, whether the related person transaction is on terms no less favorable to us than terms generally available in a transaction with an unrelated third-party under the same or similar circumstances and the extent of the related person's interest in the related person transaction. Our current policy with respect to approval of related person transactions is not set forth in writing.
Director Independence
We are not currently listed on a national securities exchange or in an inter-dealer quotation system that has requirements that a majority of the board of directors be independent. However, our board of directors has undertaken a review of the independence of the directors and considered whether any director has a material relationship with us that could compromise his ability to exercise independent judgment in carrying out his or her responsibilities. As a result of this review, our board of directors has determined that, following the effectiveness of their appointments, Messrs. Buckman, Kalia and Mathiesen, representing three of our four directors, are "independent directors" as defined under the rules of the NASDAQ Global Market. Mr. Samad, the sole director of the Company for the year ended December 31, 2016, and Mr. Rosa, one of our current directors, are not considered independent due to their service as executive officers of the Company.
| 92 |
From time to time, we are subject to litigation and claims arising in the ordinary course of business. Other than the letter received in May 2017 from the former employer of Mark Christianson and Wade Fredrickson claiming, among other things, certain breaches of non-competition obligations and confidentiality and non-disclosure obligations to such prior employer and federal and state law by virtue of such officers’ work for us, we are not currently a party to any material legal proceedings and we are not aware of any pending or threatened legal proceeding against us that we believe could have a material adverse effect on our business, operating results or financial condition. See “Risk Factors—We may be subject to damages resulting from claims that we, or our employees, have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors”.
ITEM 9. MARKET PRICE OF AND DIVIDENDS ON COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Information
As of the date of the Closing of the Acquisition, 21,500 NeuroOne Shares (365,716 Company Shares giving effect to the Acquisition), each with an exercise price of $0.59 per NeuroOne Share ($0.035 per Company Share giving effect to the Acquisition) were issuable upon the exercise of Company Options issued under our 2016 Plan.
There has been no trading market for our common stock since inception. There can be no assurance that a trading market will ever develop or, if such a market does develop, that it will continue.
Holders
As of the date of the Report, after giving effect to the Closing of the Acquisition and the issuance of shares required thereunder, there are approximately 51 holders of record of our common stock.
Dividends
We have never declared or paid any cash dividend. We do not anticipate that we will declare or pay any dividends in the foreseeable future. Our current policy is to retain earnings, if any, to fund operations, and the development and growth of our business. Any future determination to pay cash dividends will be at the discretion of our Board and will be dependent upon our financial condition, operation results, capital requirements, applicable contractual restrictions, restrictions in our organizational documents, and any other factors that our Board deems relevant.
Securities Authorized for Issuance under Equity Compensation Plans
2016 Equity Incentive Plan
In October 2016, the NeuroOne, Inc. board of directors adopted and its stockholders approved the 2016 Plan. All NeuroOne Options were converted into Company Options in connection with the Closing of the Acquisition.
2017 Equity Incentive Plan
In April 2017, our board of directors adopted and our stockholders approved the 2017 Plan. The 2017 Plan is designed to provide a vehicle under which a variety of stock-based and other awards can be granted to the Company’s employees, consultants and directors, which will align the interests of award recipients with those of our stockholders, reinforce key goals and objectives that help drive stockholder value, and attract, motivate and retain experienced and highly qualified individuals who will contribute to the Company’s financial success. The board of directors believes that the 2017 Plan will serve a critical role in attracting and retaining high caliber employees, consultants and directors essential to our success and in motivating these individuals to strive to meet our goals.
| 93 |
Shares Available Under the Plan
At the Closing of the Acquisition, we assumed the 2016 Plan, under which the NeuroOne Options were exchanged for Company Options, based on the Exchange Ratio. The following table shows the number of securities issuable upon exercise of outstanding Company Options as of the date hereof.
| Plan Category | Number of Securities to be issued upon exercise of outstanding options (a) | Weighted-average exercise price of outstanding options (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans not approved by security holders | 0 | $ | 0 | 0 | ||||||||
| Equity compensation plan approved by security holders | 365,716 | 0.035 | 1,711,090 | (1) | ||||||||
| Total | 365,716 | 0.035 | 1,711,090 | (1) | ||||||||
| (1) | Consists of 1,300,000 shares reserved under the 2017 Plan and 411,090 shares remaining available for issuance under the 2016 Plan. Upon the closing of the Acquisition, we anticipate that no additional awards will be granted under the 2016 Plan, and all awards will be granted under the 2017 Plan. |
ITEM 10. RECENT SALES OF UNREGISTERED SECURITIES
See information contained in Item 3.02 below.
ITEM 11. DESCRIPTION OF CAPITAL STOCK
The following is a summary of the rights of holders of our capital stock and some of the provisions of our certificate of incorporation and bylaws and of the Delaware General Corporation Law. This summary is not complete. For more detailed information, please see our certificate of incorporation and bylaws, which are filed as exhibits to the registration statement of which this prospectus is a part, as well as the relevant provisions of the Delaware General Corporation Law.
Authorized Capital Stock
We have authorized capital stock consisting of 100,000,000 shares of common stock, $0.001 par value per share, and 10,000,000 shares of preferred stock, $0.001 par value per share. As of the date of this Report, all of our authorized preferred stock is undesignated.
Common Stock
As of the date of this Report, we had 7,864,994 shares of common stock issued and outstanding. Each outstanding share of our common stock is duly and validly issued, fully paid and non-assessable.
Voting Rights
Our common stock is entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, including the election of directors, and does not have cumulative voting rights. Accordingly, the holders of a plurality of the shares of our common stock present at the meeting and entitled to vote in any election of directors can elect all of the directors standing for election. For most other matters, the approval of a majority of the shares voting at an annual or special meeting of stockholders will be required. Exceptions to this include removing directors for cause and amending certain sections of our certificate of incorporation and bylaws, each of which will require the approval of the holders of at least 66 2/3% of the voting power of all of our then outstanding capital stock.
| 94 |
Dividends
Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Rights and Preferences
Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
Preferred Stock
As of the date of this Report, we had no shares of preferred stock issued and outstanding.
Under the certificate of incorporation, our board of directors will have the authority, without further action by the stockholders, to issue up to 10,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in our control that may otherwise benefit holders of our common stock and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock. We have no current plans to issue any shares of preferred stock.
Stockholders Agreement
In connection with the issuance of founders’ shares described above, NeuroOne, Inc. entered into a stockholders agreement with the recipients of founders’ shares, which was also executed by the recipients of NeuroOne Shares subsequent to the execution of the stockholders agreement.
The stockholders agreement, among other things, grants certain of our stockholders drag-along rights and preemptive rights with respect to NeuroOne Shares issued in subsequent offerings. The parties to the stockholders agreement agreed to vote their NeuroOne Shares to ensure that the board size and composition is as directed by the then current NeuroOne Board and designated David A. Rosa and Paul Buckman as the initial directors of NeuroOne, Inc. The stockholders agreement, by its terms, terminated at the Closing of the Acquisition. The stockholders agreement is filed as an exhibit to this Report and is incorporated herein by reference.
| 95 |
Options
As of the date of the Closing of the Acquisition, 365,716 Company Shares were issuable upon the exercise of outstanding Company Options issued under our 2016 Plan, each with an exercise price of $0.035 per share.
Convertible Promissory Notes
Between November 2016 and June 2017, we issued Notes and Warrants for net proceeds of approximately $1.5 million. The Notes bear interest at a fixed rate of 8% per annum and require us to repay the principal and accrued and unpaid interest thereon at the earlier of November 21, 2017 or the consummation of the next equity or equity-linked round of financing resulting in more than $3 million in gross proceeds. If such a financing occurs before November 21, 2017, the outstanding principal and accrued and unpaid interest on the Notes shall automatically convert into the securities issued by us in such financing based on the greater number of such securities resulting from either (i) the outstanding principal and accrued interest on the Notes divided by $1.80 or (ii) the outstanding principal and accrued interest on the Notes multiplied by 1.25, divided by the price paid per security in such financing. If a change of control transaction or initial public offering occurs prior to such a financing, the Note would, at the election of the holders of a majority of the outstanding principal of the Notes, either become payable on demand as of the closing date of such transaction or become convertible into shares of common stock immediately prior to such transaction at a price per share equal to the lesser of (i) the per share value as determined by our board of directors as if in connection with the granting of stock based compensation or in a private sale to a third party in an arms’ length transaction or (ii) at the per share consideration to be paid in such transaction. Change of control means a merger or consolidation with another entity in which our stockholders do not own more than 50% of the outstanding voting power of the surviving entity or the disposition of all or substantially all of our assets.
Pursuant to the Subscription Agreement governing the issuance of the Notes, the Company agreed to use commercially reasonable efforts to effect a reverse merger transaction with a public company within three months of the closing of the Notes and Warrants issuance at which time the Company had sold and issued at least $750,000 in aggregate original principal amount of the Notes. In addition, pursuant to the Subscription Agreement, the Company is entitled to notice in the event a holder receives a bona fide offer for any portion of the Notes or Warrants and the right to purchase the Notes or Warrants on the same terms as the bona fide offer as long as it exercises that right within 15 days of receiving written notice.
As of July 13, 2017, the outstanding principal and accrued and unpaid interest on the Notes totaled $1,677,345. If we fail to complete an equity financing by November 21, 2017, the notes will be immediately due and payable on such date.
Warrants
Each Warrant grants the holder the option to purchase the number of shares issuable upon the conversion of the Note held by such holder. In connection with the private placement of Notes and Warrants, we are obligated to issue a Warrant to purchase shares of common stock (or common stock equivalents) in an amount equal to 8% of the common stock purchased by investors in the private placement, which Warrants have an exercise price of $2.00 per Company Share. The Warrants (other than the placement agent Warrants) are exercisable following the conversion of the Notes, as amended in June 2017, and expire on November 21, 2021. The placement agent Warrants are immediately exercisable and expire five years from the date of issuance. The exercise price and number of the shares of our common stock issuable upon exercising the Warrants will be subject to adjustment in the event of any stock dividends and splits, reverse stock split, recapitalization, reorganization or similar transaction, as described therein. In addition, the Warrants (but not those issued to the placement agent) is subject to reduction of the exercise price if we subsequently issue common stock or equivalents at an effective price less than the current exercise price of such Warrants. For more information, see “Item 11. Description of Capital Stock—Convertible Promissory Notes.”
The placement agent Warrants have piggyback registration rights.
| 96 |
The placement agent engagement letter also provides that we will issue to the placement agent warrants to purchase shares of the Company’s common stock equal to 10% of the shares of common stock (or common stock equivalents) sold in the subsequent private placement transaction.
Anti-Takeover Provisions
We are subject to Section 203 of the Delaware General Corporation Law, or Section 203. Section 203 generally prohibits a public Delaware corporation from engaging in a "business combination" with an "interested stockholder" for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
| · | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
| · | the interested stockholder owned at least 85% of the voting stock of the corporation outstanding upon consummation of the transaction, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| · | on or subsequent to the consummation of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Section 203 defines a business combination to include:
| · | any merger or consolidation involving the corporation and the interested stockholder; |
| · | any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; |
| · | subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; |
| · | subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; and |
| · | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Certificate of Incorporation and Bylaws
Provisions of our certificate of incorporation and bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our certificate of incorporation and bylaws:
| 97 |
| · | permit our board of directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate; |
| · | provide that the authorized number of directors may be changed only by resolution adopted by a majority of the board of directors; |
| · | provide that the board of directors or any individual director may only be removed with cause and the affirmative vote of the holders of at least 66 2/3% of the voting power of all of our then outstanding capital stock; |
| · | provide that all vacancies, including newly created directorships, may, except as otherwise required by law or subject to the rights of holders of preferred stock as designated from time to time, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| · | divide our board of directors into three classes; |
| · | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent or electronic transmission; |
| · | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder's notice; |
| · | do not provide for cumulative voting rights, which means that holders of a majority of the shares of common stock entitled to vote in any election of directors can elect all of the directors standing for election; |
| · | provide that special meetings of our stockholders may only be called by the chairman of the board of directors, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors (whether or not any vacancies exist); and |
| · | provide that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors or officers to us or our stockholders, (iii) any action asserting a claim against us arising pursuant to any provision of the DGCL, or (iv) any action asserting a claim against us governed by the internal affairs doctrine. |
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require the affirmative vote of the holders of at least 66 2/3% of the voting power of all of our then outstanding capital stock.
Transfer Agent and Registrar
The Company’s registrar and transfer agent for the Common Stock is Action Stock Transfer and may be contacted at 2469 E. Fort Union Blvd., Suite 214, Salt Lake City, UT 84121. Their telephone number is 801-274-1088.
| 98 |
ITEM 12. INDEMNIFICATION OF DIRECTORS AND OFFICERS
The Company is incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law provides that a Delaware corporation may indemnify any persons who were, are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation, or is or was serving at the request of such corporation as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation's best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who were, are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys' fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation's best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses (including attorneys' fees) actually and reasonably incurred.
The Company’s certificate of incorporation provides for the indemnification of its directors to the fullest extent permitted under the Delaware General Corporation Law. The Company’s bylaws provide for the indemnification of its directors and officers to the fullest extent permitted under the Delaware General Corporation Law.
Section 102(b)(7) of the Delaware General Corporation Law permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| · | transaction from which the director derives an improper personal benefit; |
| · | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| · | unlawful payment of dividends or redemption of shares; or |
| · | breach of a director's duty of loyalty to the corporation or its stockholders. |
The Company’s certificate of incorporation includes such a provision. Under the Company’s bylaws, expenses incurred by any director or officers in defending any such action, suit or proceeding in advance of its final disposition shall be paid by the Company upon delivery to it of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by the Company, as long as such undertaking remains required by the Delaware General Corporation Law.
Section 174 of the Delaware General Corporation Law provides, among other things, that a director who willfully or negligently approves of an unlawful payment of dividends or an unlawful stock purchase or redemption, may be held liable for such actions. A director who was either absent when the unlawful actions were approved or dissented at the time may avoid liability by causing his or her dissent to such actions to be entered in the books containing minutes of the meetings of the board of directors at the time such action occurred or immediately after such absent director receives notice of the unlawful acts.
| 99 |
As permitted by the Delaware General Corporation Law, we have entered into indemnity agreements with each of our directors and executive officers, that require us to indemnify such persons against any and all expenses (including reasonable attorneys' fees), witness fees, damages, judgments, fines, settlements and other amounts incurred (including expenses of a derivative action) in connection with any action, suit or proceeding, whether actual or threatened, to which any such person may be made a party by reason of the fact that such person is or was a director, an officer or an employee of the Company or any of its affiliated enterprises, provided that such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to our best interests and, with respect to any criminal proceeding, had no reasonable cause to believe his or her conduct was unlawful. The indemnification agreements also set forth certain procedures that will apply in the event of a claim for indemnification thereunder.
There is at present no pending litigation or proceeding involving any of the Company’s directors or executive officers as to which indemnification is required or permitted, and the Company is not aware of any threatened litigation or proceeding that may result in a claim for indemnification, other than the letter received by the Company in May 2017 from the former employer of Mark Christianson and Wade Fredrickson claiming, among other things, certain breaches of non-competition obligations and confidentiality and non-disclosure obligations to such prior employer and federal and state law by virtue of such officers’ work for the Company. See “Risk Factors—We may be subject to damages resulting from claims that we, or our employees, have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors”.
The Company is procuring an insurance policy that covers its officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act or otherwise.
See information contained in Item 9.01 below.
ITEM 14. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING OR FINANCIAL DISCLOSURE
See information contained in Item 4.01 below.
ITEM 15. FINANCIAL STATEMENTS AND EXHIBITS
See information contained in Item 9.01 below.
Item 3.02 Unregistered Sales of Equity Securities.
Issuance of NeuroOne, Inc. Common Stock, Stock Awards and Options
Prior to the Merger, between October 16 and October 20, 2016, NeuroOne, Inc. issued 301,670 NeuroOne Shares (5,131,514 Company Shares giving effect to the Acquisition) as founders’ shares in a private placement to seven individuals (who were all accredited investors), including three officers of the Company. The value applied was $0.03 per NeuroOne Share based on a valuation utilizing a weighted average market value of invested capital methodology. In June 2017, the purchase price owed by the seven individuals for the founders’ shares under their respective subscription agreements totaling $9,050 was forgiven by the Company in its entirety.
The issuances of the NeuroOne Shares were exempt from the registration requirements of the Securities Act in reliance upon Section 4(a)(2) of the Securities Act and Regulation D promulgated under the Securities Act. Each of the purchasers represented to NeuroOne, Inc. that they acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. The purchasers also represented to NeuroOne, Inc. that they were accredited investors as defined in Rule 501 promulgated under the Securities Act.
In April 2017, NeuroOne, Inc. issued 12,666 restricted NeuroOne Shares (215,453 Company Shares giving effect to the Acquisition), subject to vesting, to one of our executive officers under the 2016 Plan. Between January 2017 and June 2017, NeuroOne, Inc. granted NeuroOne Options under the 2016 Plan to purchase an aggregate of 21,500 NeuroOne Shares (365,716 Company Shares giving effect to the Acquisition), each with an exercise price of $0.59 per NeuroOne Share ($0.035 per Company Share giving effect to the Acquisition), to certain of its consultants and directors.
| 100 |
The issuance of restricted NeuroOne Shares and NeuroOne Options was exempt from the registration requirements of the Securities Act in reliance upon Section 4(a)(2) of the Securities Act or Rule 701 promulgated under the Securities Act.
Issuance of NeuroOne, Inc. Notes and Warrants
Between November 2016 and June 2017, NeuroOne, Inc. issued Notes and Warrants to 16 accredited investors in a private placement for gross proceeds of $1,625,120. The offers, sales and issuances of the Notes and Warrants were exempt from the registration requirements of the Securities Act in reliance upon Section 4(a)(2) of the Securities Act and Regulation D promulgated under the Securities Act. Each of the purchasers represented to NeuroOne, Inc. that they acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. The purchasers also represented to NeuroOne, Inc. that they were accredited investors as defined in Rule 501 promulgated under the Securities Act. For more information, see “Item 11. Description of Capital Stock—Convertible Promissory Notes” and “Item 11. Description of Capital Stock—Warrants”.
Issuance of NeuroOne Shares Pursuant to Mayo Development Agreement
In May 2017, NeuroOne, Inc. issued to Mayo 50,556 NeuroOne Shares pursuant to a Subscription Agreement, as required by the terms of the Mayo Development Agreement. The issuance of the NeuroOne Shares was exempt from the registration requirements of the Securities Act in reliance upon Section 4(a)(2) of the Securities Act and Regulation D promulgated under the Securities Act. Mayo represented to NeuroOne, Inc. that it acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Mayo also represented to NeuroOne, Inc. that they were accredited investors as defined in Rule 501 promulgated under the Securities Act.
Securities Issued Pursuant to Acquisition
At the Closing, pursuant to and in connection with the Acquisition, we issued:
| · | 6,291,994 Company Shares to the former stockholders of NeuroOne, Inc.; |
| · | Company Options for the purchase of an aggregate of 365,716 Company Shares; and |
| · | Company Warrants, which are exercisable for Company Shares following conversion of the Notes on their terms. |
We also assumed the Notes of NeuroOne, Inc., pursuant to which $1,677,345 in principal and accrued and unpaid interest was outstanding in the aggregate as of July 13, 2017, which Notes are convertible into Company Shares on their terms. For a description of the Notes and Company Warrants and information about the number of shares of common stock for which they are convertible or exercisable, see “Form 10 Information—Management's Discussion And Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—Capital Resources”.
The 6,291,994 shares issued to the former NeuroOne, Inc. stockholders were issued with a restrictive legend that the shares had not been registered under the Securities Act. For more information, see “Item 2.01—Completion of Acquisition or Disposition of Assets.”
The issuance of the Company Shares and Company Warrants in conjunction with the Acquisition was exempt from the registration requirements of the Securities Act in reliance upon Section 4(a)(2) of the Securities Act and Regulation D promulgated under the Securities Act as an offering not involving a public offering. Each of the recipients of the Company Shares and Company Warrants represented that they were accredited investors and/or sophisticated, with fewer than 35 non-accredited investors. The issuance of the Company Options were exempt under Rule 701 under the Securities Act.
| 101 |
None of the Company Shares, Company Options or Company Warrants issued in connection with the Acquisition, nor the underlying shares of common stock issuable upon exercise, have been registered under the Securities Act, and all documents have been issued with a restrictive legend prohibiting further transfer of the securities without such securities either being first registered or otherwise exempt from registration in any further resale or disposition.
Item 4.01 Changes In Accountants.
Change in Independent Registered Public Accountants Resulting from the Acquisition
The financial statements of NeuroOne, Inc. as of December 31, 2016 and for the period from October 7, 2016 to December 31, 2016 and NeuroOne LLC for the period from January 1, 2016 to October 26, 2016 and as of and for the year ended December 31, 2015 were audited by BDO USA, LLP, or BDO. Our financial statements as of and for the fiscal years ended December 31, 2016 and 2015 were audited by, and our financial statements as of and for the first quarter ended March 31, 2017 were reviewed by, Pritchett, Siler & Hardy, P.C., or Pritchett, which served as our independent registered public accountant prior to the Acquisition.
Effective as of the Closing of the Acquisition, BDO will act as our auditor until its resignation or removal. Pursuant to Item 304 of Regulation S-K, we report as follows:
| · | The dismissal of Pritchett and the appointment of the new independent registered public accounting firm, BDO, each of which took effect on the Closing of the Acquisition, were related solely to the change of control resulting from the Acquisition, and were not related in any way to any disagreement with Pritchett. |
| · | Pritchett’s reports on the financial statements of Original Source Entertainment, Inc. have not contained any adverse opinion or disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope, or accounting principle, except for Pritchett’s audit reports dated April 17, 2017 and April 14, 2016, each of which contained a statement that cited certain conditions that raised substantial doubt about our ability to continue as a going concern. |
| · | The decision to utilize BDO was approved by our board of directors. |
| · | During our last two fiscal years and through the date of this Report, there were no disagreements between us and Pritchett on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreements, if not resolved to the satisfaction of Pritchett, would have caused it to make reference to the subject matter of the disagreements in connection with its report; and there were no reportable events with respect to the Company as defined in Item 304(a)(1)(v) of Regulation S-K (other than the material weaknesses in the internal controls over financial reporting of NeuroOne, Inc. as described under “Item 1A. Risk Factors—Failure to establish and maintain an effective system of internal controls could result in material misstatements of our financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud in which case, our stockholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our stock. Furthermore, our management and our independent auditors have identified certain internal control deficiencies, which management and our independent auditors believe constitute material weaknesses.” |
We have provided Pritchett a copy of the above disclosures in response to Item 304 of Regulation S-K in conjunction with the filing of this Report and requested that Pritchett provide us with a letter addressed to the SEC stating whether it agrees with the statements we have made in response to Item 304(a) of Regulation S-K. A copy of such letter is filed as an exhibit to this Report and is incorporated by reference herein.
During our two most recent fiscal years and through the date of this Report, we did not consult BDO with respect to the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company's consolidated financial statements, or any other matters or reportable events as set forth in Items 304(a)(2) of Regulation S-K.
| 102 |
Previous independent registered public accounting firm
On November 10, 2015, Cutler & Co., LLC (“Cutler”) resigned as the independent registered public accounting firm for the Company, as Cutler merged its SEC auditing practice with Pritchett. On November 10, 2015, the Company engaged Pritchett as its new independent registered public accounting firm. The change of the Company’s independent registered public accounting firm from Cutler to Pritchett was approved by our board of directors.
During the year ended December 31, 2015, there were (i) no disagreements between the Company and Cutler on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreement, if not resolved to the satisfaction of Cutler, would have caused Cutler to make reference thereto in their reports on the consolidated financial statements for such year, and (ii) no “reportable events” as that term is defined in Item 304(a)(1)(v) of Regulation S-K.
The Company provided Cutler with a copy of its Current Report on Form 8-K dated November 12, 2015 (the “Cutler 8-K”) and requested that Cutler furnish it with a letter addressed to the SEC stating whether or not Cutler agreed with the above statements. A copy of such letter, dated November 12, 2015, was attached as Exhibit 16.1 to the Cutler 8-K.
New independent registered public accounting firm
The Company engaged BDO as its new independent registered public accounting firm as of July 20, 2017. During the fiscal years ended December 31, 2016 and 2015 and the subsequent interim period through March 31, 2017 we have not consulted with BDO regarding either (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements, and neither a written report was provided to us or oral advice was provided that BDO concluded was an important factor considered by us in reaching a decision as to the accounting, auditing or financial reporting issue; or (ii) any matter that was either the subject of a disagreement (as defined in Regulation S-K, Item 304(a)(1)(iv)) and the related instructions to Regulation S-K 304, or a reportable event (as defined in Regulation S-K, Item 304(a)(1)(v)).
Item 5.01 Changes in Control of Registrant.
The information regarding the Acquisition set forth in Item 2.01, “Completion of Acquisition or Disposition of Assets” is incorporated herein by reference.
At the closing of the Acquisition, we issued 6,291,994 Company Shares to former NeuroOne, Inc. stockholders in exchange for all of their ownership of NeuroOne, Inc. and all of the shares held by our largest stockholder, Amer Samad, were cancelled. Prior to the Acquisition, the NeuroOne, Inc. stockholders did not own any of our shares of common stock.
After giving effect to the issuance of the Company Shares, the number of shares of our common stock issued and outstanding is 7,864,994 of which the former NeuroOne, Inc. stockholders own approximately 80.0%. Assuming the exercise of all outstanding Company Options as of the Closing, the number of shares of our common stock issued and outstanding would be 8,230,710, of which the former NeuroOne, Inc. stockholders would own approximately 81.0%. Stockholders beneficially owning all of our common stock immediately prior to the closing of the Acquisition were diluted to an aggregate ownership of 1,573,000 shares, or approximately 20.0% of our issued and outstanding shares. For information on arrangements with respect to the election of directors, see “Item 5.02—Departure of Directors or Principal Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.”
| 103 |
Item 5.02 Departure of Directors or Principal Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.
Change in the Directors Serving on our Board
In connection with the Closing of the Acquisition, the sole member of the Company’s Board, Amer Samad, increased the size of the Board to two members and appointed David Rosa as a director of the Company as of the Closing Date pursuant to the Merger Agreement. On the Closing, the Company’s Board accepted the resignation of Amer Samad, to be effective on the 10th day following our filing of the Schedule 14F-1, and, subject to the expiration of such 10 days, increased its size to four members and appointed Paul Buckman, Suraj Kalia and Jeffrey Mathiesen to serve on the Board with Mr. Rosa. The resignation of Mr. Samad was not in connection with any known disagreement with us on any matter.
Change in Executive Officers
In connection with the Closing of the Acquisition, Mr. Samad, the former sole officer, resigned and the following individuals were named as executive officers of the Company as of the Closing Date:
| Name | Position |
| Executive Officers: | |
| David Rosa | Chief Executive Officer and Director |
| Mark Christianson | Vice President, Business Development and Marketing |
| Thomas Bachinski | Chief Development Officer |
Our executive officers serve at the pleasure of our board of directors.
See “Item 5. Directors and Executive Officers” for information on each of our new directors and executive officers, “Item 6. Executive Compensation” for information on compensatory arrangements with our executive officers and “Item 7. Certain Relationships and Related Transactions, and Director Independence” for information on related party transactions.
Item 5.03 Amendments to Certificate of Incorporation or Bylaws; Change in Fiscal Year.
As previously disclosed, prior to the Closing of the Acquisition, we completed a series of steps contemplated by a Plan of Conversion pursuant to which we, among other things, changed our name to NeuroOne Medical Technologies Corporation, increased our authorized number of shares of common stock from 45,000,000 to 100,000,000, increased our authorized number of shares of preferred stock from 5,000,000 to 10,000,000 and reincorporated in Delaware. In connection with the foregoing, we adopted a new Certificate of Incorporation and By-Laws. The Certificate of Incorporation and By-Laws are annexed hereto as Exhibits 3.1 and 3.2 and incorporated by reference herein.
Item 5.06 Change in Shell Company Status.
As a result of the Acquisition, we are no longer a “shell company” within the meaning of Rule 12b-2 under the Exchange Act. For more information, see “Item 2.01 – Completion of Acquisition or Disposition of Assets.” In addition, the information contained in this Report is intended to provide “Form 10 information” within the meaning of Rule 144(i)(3) under the Securities Act.
Item 9.01 Financial Statements and Exhibits.
(a) Financial Statements of Business Acquired
See Financial Statements for NeuroOne, Inc. as of December 31, 2016 and for the period from October 7, 2016 to December 31, 2016 and NeuroOne LLC for the period from January 1, 2016 to October 26, 2016 and as of and for the year ended December 31, 2015, which are filed as Exhibit 99.1 to this Report and are incorporated herein by reference.
| 104 |
See Financial Statements for NeuroOne, Inc. for the three months ended March 31, 2017 and NeuroOne LLC for the three months ended March 31, 2016, which are filed as Exhibit 99.2 to this Report and are incorporated herein by reference.
(b) Pro Forma Financial Information
See Unaudited Pro Forma Condensed Combined Balance Sheets as of March 31, 2017 and Condensed Combined Statement of Operations for the three months ended March 31, 2017 and the year ended December 31, 2016, which is filed as Exhibit 99.3 to this Report and is incorporated herein by reference.
| 105 |
(d) Exhibits
| Exhibit No. | Document | |||
| 2.1 | * | Agreement and Plan of Merger and Reorganization by and among NeuroOne Medical Technologies Corporation, OSOK Acquisition Company and NeuroOne, Inc. dated as of July 20, 2017 | ||
| 3.1 | Certificate of Incorporation of NeuroOne Medical Technologies Corporation (incorporated by reference to Exhibit 3.4 on the Registrant’s Current Report on Form 8-K filed on June, 29, 2017) | |||
| 3.2 | Bylaws of NeuroOne Medical Technologies Corporation (incorporated by reference to Exhibit 3.5 on the Registrant’s Current Report on Form 8-K filed on June 29, 2017) | |||
| 4.1 | Form of Common Stock Certificate | |||
| 10.1 | # | Exclusive Start-Up Company License Agreement; WARF Agreement No. 14-00333 by and between Wisconsin Alumni Research Foundation and Neuro One LLC, dated October 1, 2014 | ||
| 10.2 | # | Amendment to Exclusive Start-Up Company License Agreement by and between Wisconsin Alumni Research Foundation, Neuro One LLC, and NeuroOne, Inc. dated as of February 22, 2017 | ||
| 10.3 | # | Mayo Foundation for Medical Education and Research Amended and Restated License and Development Agreement by and between Mayo Foundation for Medical Education and Research, and NeuroOne LLC dated as of May 25, 2017 | ||
| 10.4 | Form of October 2016 Common Stock Subscription Agreement | |||
| 10.5 | Form of Promissory Note and Warrant Subscription Agreement | |||
| 10.6 | Form of Promissory Note issued pursuant to Promissory Note and Warrant Subscription Agreement | |||
| 10.7 | First Amendment to Promissory Note issued pursuant to Promissory Note and Warrant Subscription Agreement | |||
| 10.8 | Form of Capital Stock Purchase Warrant issued pursuant to Promissory Note and Warrant Subscription Agreement | |||
| 10.9 | Form of First Amendment to Capital Stock Purchase Warrant issued pursuant to Promissory Note and Warrant Subscription Agreement | |||
| 10.10 | Stockholders Agreement by and among NeuroOne, Inc., and the stockholders party thereto dated as of October 20, 2016 | |||
| 10.11 | + | 2016 Equity Incentive Plan of NeuroOne, Inc. | ||
| 10.12 | Form of Stock Option Award Agreement pursuant to 2016 Equity Incentive Plan of NeuroOne, Inc. | |||
| 10.13 | Restricted Stock Purchase Agreement by and between NeuroOne, Inc. and Thomas Bachinski, dated as of April 10, 2017 | |||
| 10.14 | + | 2017 Equity Incentive Plan of the Company (incorporated by reference to Appendix G to Schedule 14C filed on April 20, 2017) | ||
| 10.15 | + | NeuroOne Medical Technologies Corporation 2017 Equity Incentive Plan Option Agreement | ||
| 106 |
| 10.16 | + | NeuroOne Medical Technologies Corporation 2017 Equity Incentive Plan Restricted Stock Unit Agreement | ||
| 10.17 | + | Employment Agreement by and between NeuroOne LLC and Dave Rosa, dated as of October 5, 2016 | ||
| 10.18 | + | Offer Letter to Mark Christianson from NeuroOne, Inc. dated December 1, 2016 | ||
| 10.19 | + | Offer Letter to Thomas Bachinski from NeuroOne, Inc. dated January 9, 2017 | ||
| 10.20 | + | Offer Letter to Wade Fredrickson from NeuroOne, Inc. dated December 1, 2016 | ||
| 10.21 | + | Form of Indemnification Agreement with the Company’s Officers and Directors (incorporated by reference to Appendix E to Schedule 14C filed on April 20, 2017) | ||
| 10.22 | Resignation Letter of Amer Samad | |||
| 10.23 | + | Release Agreement of Wade Fredrickson dated June 28, 2017. | ||
| 16.1 | Letter from Cutler & Co., LLC dated November 12, 2015 (incorporated by reference to Exhibit 16.1 on the Registrant’s Current Report on Form 8-K filed on November 12, 2015) | |||
| 16.2 | Letter from Pritchett, Siler & Hardy, P.C dated July 11, 2017 | |||
| 21.1 | Subsidiaries of the Registrant | |||
| 99.1 | Financial Statements for NeuroOne, Inc. as of December 31, 2016 and for the period from October 7, 2016 to December 31, 2016 and for NeuroOne LLC for the period from January 1, 2016 to October 26, 2016 and as of and for the year ended December 31, 2015 | |||
| 99.2 | Financial Statements for NeuroOne, Inc. for the three months ended March 31, 2017 and NeuroOne LLC for the three months ended March 31, 2016 | |||
| 99.3 | Unaudited Pro Forma Condensed Combined Balance Sheets as of March 31, 2017 and Condensed Combined Statement of Operations for the three months ended March 31, 2017 and the year ended December 31, 2016 |
*Pursuant to Item 601(b)(2) of Regulation S-K, the Registrant agrees to furnish supplementally a copy of any omitted schedule or exhibit to the Agreement and Plan of Merger to the Securities and Exchange Commission upon request.
#Portions of this exhibit have been omitted pursuant to a request for confidential treatment and have been separately filed with the Securities and Exchange Commission.
+Indicates management contract or compensatory plan.
| 107 |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
Date: July 20, 2017
| NEUROONE MEDICAL TECHNOLOGIES CORPORATION | |||
| By: | /s/ DAVID ROSA | ||
| David Rosa Chief Executive Officer | |||
| 108 |