Attached files
As filed with the Securities and Exchange Commission on April
11, 2017
Registration
No. 333-216052
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment
No. 1
to
Form S-1
REGISTRATION STATEMENT UNDER
THE SECURITIES ACT OF 1933
Adgero Biopharmaceuticals Holdings, Inc.
(Exact Name of Registrant as Specified in its Charter)
|
Delaware
|
2834
|
47-5506831
|
|
|
|
|
|
(State
or other jurisdiction of incorporation or
organization)
|
(Primary
Standard Industrial
Classification
Code Number)
|
(I.R.S.
Employer Identification No.)
|
4365 US 1 South, Suite 211
Princeton, NJ 08540
Telephone: 609-917-9796
(Address, including zip code, and telephone number,
including area code, of principal executive offices)
Frank Pilkiewicz, PhD
Chief Executive Officer
Adgero Biopharmaceuticals Holdings, Inc.
4365 US 1 South, Suite 211
Princeton, NJ 08540
Telephone: 609-917-9796
(Address, including zip code, and telephone number,
including area code, of agent for service)
Copies to:
Michael J. Lerner, Esq.
Steven M. Skolnick, Esq.
Lowenstein Sandler LLP
1251 Avenue of the Americas
New York, New York 10020
Telephone: (212) 262-6700
Approximate
date of proposed sale to public: As soon as practicable on or after
the effective date of this registration statement.
If any
of the securities being registered on this Form are to be offered
on a delayed or continuous basis pursuant to Rule 415 under the
Securities Act of 1933 check the following box. ☒
If this
Form is filed to register additional securities for an offering
pursuant to Rule 462(b) under the Securities Act, please check the
following box and list the Securities Act registration statement
number of the earlier effective registration statement for the same
offering. ☐
If this
Form is a post-effective amendment filed pursuant to Rule 462(c)
under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier
effective registration statement for the same offering.
☐
If this
Form is a post-effective amendment filed pursuant to Rule 462(d)
under the Securities Act, check the following box and list the
Securities Act registration statement number of the earlier
effective registration statement for the same offering.
☐
Indicate
by check mark whether the registrant is a large accelerated filer,
an accelerated filer, a non-accelerated filer, or a smaller
reporting company. See definitions of “large accelerated
filer,” “accelerated filer,” and smaller
reporting company’ in Rule 12b-2 of the Exchange Act. (Check
one):
|
Large
accelerated filer ☐
|
Accelerated
filer ☐
|
|
|
|
|
Non-accelerated
filer ☐
|
Smaller
reporting company ☒
|
(Do not
check if a smaller reporting company)
The
Registrant hereby amends this Registration Statement on such date
or dates as may be necessary to delay its effective date until the
Registrant shall file a further amendment which specifically states
that this Registration Statement shall thereafter become effective
in accordance with Section 8(a) of the Securities Act of 1933, as
amended, or until this Registration Statement shall become
effective on such date as the Securities and Exchange Commission,
acting pursuant to such Section 8(a), may determine.
The information in this prospectus is not complete and may be
changed. We may not sell these securities until the Securities and
Exchange Commission declares our registration statement effective.
This prospectus is not an offer to sell nor does it seek an offer
to buy these securities in any state where the offer or sale is not
permitted.
|
Preliminary
Prospectus
|
|
Subject
to Completion, dated April 11, 2017.
|
Adgero Biopharmaceuticals Holdings, Inc.

3,467,680 Shares
Common Stock
This
prospectus relates to the offer for sale of up to an aggregate of
3,467,680 shares of common stock of Adgero Biopharmaceuticals
Holdings, Inc. by the selling stockholders named herein. We are not
offering any securities pursuant to this prospectus. The shares of
common stock offered by the selling stockholders include 1,749,272
shares of common stock underlying warrants with an exercise price
of $5.00 per share.
Our
common stock is not presently traded on any market or securities
exchange, and we have not applied for listing or quotation on any
exchange. We are seeking sponsorship for the trading of our common
stock on the Over-the-Counter, or OTC, Bulletin Board and/or OTCQB
Market operated by OTC Markets Group, Inc. (together, the
“OTCBB/OTCQB”) upon the effectiveness of the
registration statement of which this prospectus forms a part. The
3,467,680 shares of our common stock can be sold by selling
security holders at a fixed price of $5.00 per share until our
shares are quoted on the OTCBB/OTCQB and thereafter at prevailing
market prices or privately negotiated prices. There can be no
assurance that a market maker will agree to file the necessary
documents with the Financial Industry Regulatory Authority
(“FINRA”) nor can we provide assurance that our shares
will actually be quoted on the OTCBB/OTCQB or, if quoted, that a
viable public market will materialize or be sustained.
Following the
effectiveness of the registration statement of which this
prospectus forms a part, the sale and distribution of securities
offered hereby may be effected in one or more transactions that may
take place on the OTCBB/OTCQB, including ordinary brokers’
transactions, privately negotiated transactions or through sales to
one or more dealers for resale of such securities as principals, at
market prices prevailing at the time of sale, at prices related to
such prevailing market prices or at negotiated prices. Usual and
customary or specifically negotiated brokerage fees or commissions
may be paid by the selling stockholders. See “Plan of
Distribution.”
Certain
of the selling stockholders and intermediaries, who are identified
as broker-dealers in the footnotes to the selling stockholder table
contained in this prospectus, through whom such securities are sold
are deemed “underwriters” within the meaning of the
Securities Act of 1933, as amended (the “Securities
Act”), with respect to the securities offered hereby, and any
profits realized or commissions received may be deemed underwriting
compensation. We believe that all securities purchased by
broker-dealers or affiliates of broker-dealers were purchased by
such persons and entities in the ordinary course of business and at
the time of purchase, such purchasers did not have any agreements
or understandings, directly or indirectly, with any person to
distribute such securities.
We
are an “emerging growth company” under the federal
securities laws and, as such, we intend to comply with certain
reduced public company reporting requirements. Investing in our
common stock is highly speculative and involves a significant
degree of risk. See “Risk Factors” beginning on page 7
of this prospectus for a discussion of information that should be
considered before making a decision to purchase our common
stock.
Neither
the Securities and Exchange Commission nor any state securities
commission has approved or disapproved of these securities or
determined if this prospectus is truthful or complete. Any
representation to the contrary is a criminal offense.
The date of this prospectus
is
, 2017.
TABLE OF CONTENTS
|
PROSPECTUS
SUMMARY
|
1
|
|
|
|
|
THE
OFFERING
|
5
|
|
|
|
|
RISK
FACTORS
|
7
|
|
|
|
|
CAUTIONARY NOTE
REGARDING FORWARD-LOOKING STATEMENTS
|
37
|
|
|
|
|
USE OF
PROCEEDS
|
39
|
|
|
|
|
DIVIDEND
POLICY
|
40
|
|
|
|
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
41
|
|
|
|
|
BUSINESS
|
45
|
|
|
|
|
MANAGEMENT AND
BOARD OF DIRECTORS
|
67
|
|
|
|
|
EXECUTIVE
COMPENSATION
|
73
|
|
|
|
|
PRINCIPAL
STOCKHOLDERS
|
82
|
|
|
|
|
CERTAIN
RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
|
84
|
|
|
|
|
DESCRIPTION OF
SECURITIES
|
87
|
|
|
|
|
SELLING
STOCKHOLDERS
|
92
|
|
|
|
|
PLAN
OF DISTRIBUTION
|
99
|
|
|
|
|
MARKET
FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
|
101
|
|
|
|
|
LEGAL
MATTERS
|
101
|
|
|
|
|
EXPERTS
|
101
|
|
|
|
|
DISCLOSURE OF
COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES ACT
LIABILITIES
|
101
|
|
|
|
|
WHERE
YOU CAN FIND ADDITIONAL INFORMATION
|
102
|
|
|
|
|
SIGNATURES
|
II-9
|
You
should rely only on the information contained in this prospectus.
We have not authorized any other person to provide you with
information different from or in addition to that contained in this
prospectus. If anyone provides you with different or inconsistent
information, you should not rely on it. We are not making an offer
to sell these securities in any jurisdiction where an offer or sale
is not permitted. You should assume that the information appearing
in this prospectus is accurate only as of the date on the front
cover of this prospectus. Our business, financial condition,
results of operations and prospects may have changed since that
date.
Additional
risks and uncertainties not presently known or that are currently
deemed immaterial may also impair our business operations. The
risks and uncertainties described in this document and other risks
and uncertainties which we may face in the future will have a
greater impact on those who purchase our common stock. These
purchasers will purchase our common stock at the market price or at
a privately negotiated price and will run the risk of losing their
entire investments.
For investors outside the United States:
We have not done anything that would permit this offering or
possession or distribution of this prospectus in any jurisdiction
where action for that purpose is required, other than in the United
States. You are required to inform yourselves about and to observe
any restrictions relating to this offering and the distribution of
this prospectus.
In this
prospectus, we rely on and refer to information and statistics
regarding our industry. We obtained this statistical, market and
other industry data and forecasts from publicly available
information.
PROSPECTUS SUMMARY
This summary highlights information contained in other parts of
this prospectus. Because it is a summary, it does not contain all
of the information that you should consider in making your
investment decision. Before investing in our common stock,
you should read the entire
prospectus carefully, including our consolidated financial
statements and the related notes included in this prospectus and
the information set forth under the headings “Risk
Factors” on page 7 and “Management’s Discussion
and Analysis of Financial Condition and Results of
Operations” on page 41.
When used herein, unless the context requires otherwise, references
to the “Company,” “Holdings,”
“we,” “our” and “us” refer to
Adgero Biopharmaceuticals Holdings, Inc., a Delaware corporation,
collectively with its wholly-owned subsidiary, Adgero
Biopharmaceuticals, Inc., a Delaware corporation.
All trademarks or trade names referred to in this prospectus are
the property of their respective owners. Solely for convenience,
the trademarks and trade names in this prospectus are referred to
without the ® and ™ symbols, but such references should
not be construed as any indicator that their respective owners will
not assert, to the fullest extent under applicable law, their
rights thereto. We do not intend the use or display of other
companies’ trademarks and trade names to imply a relationship
with, or endorsement or sponsorship of us by, any other
companies.
Our Company
General
We are
a biopharmaceutical company, focused on the development of
photodynamic therapy (“PDT”) for the treatment of rare,
unmet medical needs. PDT is a treatment that uses light sensitive
compounds, or photosensitizers, that, when exposed to specific
wavelengths of light, act as a catalyst to produce a form of oxygen
that induces local tumor cell death. Our lead product candidate,
the REM-001 Therapy product, consists of three parts, the laser
light source, the light delivery device and the drug REM-001
(collectively, the “REM-001 Therapy”). REM-001 is a
second generation photosensitizer drug that has undergone late
stage clinical development and which we believe possesses multiple
advantages over earlier generation PDT compounds. Our lead
indication is unresectable cutaneous metastatic breast cancer
(“CMBC”), a disease that may strike individuals with
advanced breast cancer and for which effective treatment options
are limited. In four Phase 2 and/or Phase 3 clinical trials in CMBC
patients, primarily targeting patients who had
previously received chemotherapy
and failed radiation therapy, our REM-001 Therapy was able
to reduce or eliminate a substantial number of the treated CMBC
tumors. Specifically, our analysis of the data collected from these
trials indicates that in approximately 80% of evaluable tumor sites
treated with REM-001 Therapy, there was a complete response,
meaning that follow-up clinical assessments indicated no visible
evidence of the tumor remaining. We believe clinical data indicates
that REM-001 Therapy holds promise as a treatment to locally
eliminate or slow the growth of treated cutaneous cancerous tumors
in this difficult-to-treat patient population.
In
2012, we acquired certain assets and regulatory filings, including
REM-001 Therapy developed by Miravant Medical Technologies, and its
wholly-owned subsidiaries, a former public pharmaceutical and
research development company (collectively,
“Miravant”), and the associated technology, clinical
data and intellectual property, from a creditor of Miravant.
Between February 1996 and January 1999, Miravant, with support from
certain corporate partners, conducted the above-referenced four
Phase 2 and/or Phase 3 clinical trials for the treatment of CMBC
using REM-001 Therapy (collectively, the “Miravant CMBC
Trials”). The primary motivation behind our acquisition was
to secure the rights to the REM-001 Therapy and its associated
technology, proprietary processes and regulatory filings which have
already undergone substantial clinical development, which we
believe will help expedite the process of gaining regulatory
approval to market our REM-001 Therapy.
Our
initial product goal is to achieve marketing approval of REM-001
Therapy for the treatment of CMBC in the United States. We
conducted a first preliminary
analysis of all existing REM-001 Therapy clinical trial data for
CMBC, including data from the Miravant CMBC Trials. We then
conducted a more in-depth analysis that was overseen by regulatory experts who
have expertise in interacting with the Food and Drug Administration
(the “FDA”). The experts
we engaged were either former FDA employees with directly related
experience in reviewing similar oncology treatments or individuals
who have provided senior regulatory guidance to major
pharmaceutical or medical device companies in situations that led
to regulatory approval. The results of this second more in-depth
analysis were consistent with our original analysis. As a result of
our review, we submitted questions to FDA under a Type C
format to review the technology and results and determine
the anticipated requirements for regulatory approval. On
March 3, 2017, we received FDA’s written response to our
questions. Based on that response, we believe our plans to
manufacture REM-001 by revising the prior quality standards to meet
the currently recommended regulatory standards will be acceptable.
FDA also indicated our plans for utilizing light delivery devices
that have been shown to be functionally equivalent to the devices
used by Miravant will be acceptable. FDA also recognized that CMBC
represents an unmet clinical need and provided guidance on a number
of clinical pharmacology parameters they would like us to measure
in our planned clinical trial. Based on FDA’s responses, we
plan to conduct a clinical trial in CMBC to test the safety and
efficacy of REM-001 Therapy for marketing approval and we are
planning further discussions with FDA regarding the specifics of
such a trial.
1
We also
believe REM-001 Therapy holds promise as a treatment for cutaneous
metastatic cancers other than CMBC as well as locally advanced
basal cell cancer such as often occurs in patients with Basal Cell
Nevus Syndrome and cutaneously recurrent basal cell
cancer.
Formation of Holdings
We are
a Delaware corporation. In connection with our formation in October
2015, we sold an aggregate of 1,000,000 shares of common stock for
an aggregate of $50,000 ($0.05 per share), which includes 500,000
shares of common stock owned by an affiliate of Aegis Capital
Corporation (“Aegis Capital”), the placement agent in
our 2016 Private Placement described below.
Recent Developments
The Merger Transaction
On
January 11, 2016, Adgero Biopharmaceuticals, Inc.
(“Adgero”) entered into a merger agreement (the
“Merger Agreement”) by and among Adgero, Adgero
Biopharmaceuticals Holdings, Inc. (“Holdings”), and
Adgero Acquisition, Inc. a Delaware corporation and our
wholly-owned subsidiary (“Merger Sub”). Pursuant to the
terms of the Merger Agreement, as a condition of and
contemporaneously with the initial closing of the 2016 Private
Placement, described below, (the “Initial Closing”),
Merger Sub merged with and into Adgero and Adgero became a
wholly-owned subsidiary of us. In connection with the merger (the
“Merger”), stockholders of Adgero received an aggregate
of 2,000,000 shares of our common stock. In addition, the holders
of warrants to purchase common stock of Adgero prior to the Merger
received warrants (the “Replacement Warrants”), to
purchase 30,864 shares of our common stock with an exercise price
of $5.00. The terms of the Replacement Warrants are substantially
similar to the Investor Warrants, described below. At the closing
of the Merger, the board of directors of Holdings consisted of
Frank Pilkiewicz, PhD, the Chief Executive Officer of Holdings and
Chief Executive officer of Adgero, Allen Bloom, PhD, JD, Roman
Perez-Soler, MD, Tim McInerney, and David Hochman, a Board designee
of Aegis Capital.
The
Merger was treated as a reverse acquisition and recapitalization of
Adgero for financial accounting purposes and the historical
financial statements of Adgero are our financial statements as a
result of the Merger. The parties to the Merger Agreement have
agreed to take all actions necessary to ensure the Merger is
treated as a “plan of reorganization” under Section
368(a) of the Internal Revenue Code of 1986, as
amended.
2016 Private Placement
We
conducted a private placement offering from January to September
2016 (the “2016 Private Placement”). We issued an
aggregate 1,873,299 shares of our common stock for $5.00 per share,
inclusive of 87,099 shares of our common stock issued pursuant to
the conversion of promissory notes in connection with the 2016
Private Placement, and warrants (the “Investor
Warrants”), to purchase 1,873,299 shares of our common stock
at an exercise price of $5.00, inclusive of Investor Warrants to
purchase 87,099 shares of our common stock issued pursuant to the
conversion of promissory notes in connection with the 2016 Private
Placement. The Investor Warrants have a five year term. Gross
proceeds totaled $8,931,000, plus an additional $435,495 in
connection with the conversion of certain promissory notes included
in the 2016 Private Placement, and net proceeds were $7,271,904.
Aegis Capital acted as the placement agent (the “Placement
Agent”), for the 2016 Private Placement. Pursuant to the
registration statement of which this prospectus is a part, we are
registering those shares of common stock and shares of common stock
underlying the Investor Warrants issued in the 2016 Private
Placement as described in the “Selling Stockholders”
section on page 89, as well as (i) 5,154 shares of common stock and
5,154 shares of common stock underlying warrants issued pursuant to
the conversion of a promissory note not included in the 2016
Private Placement (see “Certain Relationships and Related
Party Transactions - Bridge Offering Affiliate
Participation”) and (ii) the shares of common stock
underlying the Replacement Warrants, for public resale by the
selling stockholders named herein and their assigns.
In
connection with the 2016 Private Placement, we paid the Placement
Agent and selected dealers an aggregate cash fee of $1,164,110,
inclusive of a non-accountable expense allowance equal to $275,564,
and we incurred approximately $494,986 of other expenses related to
the financing. In addition, as part of its compensation for acting
as placement agent for the 2016 Private Placement, we issued
warrants (the “Placement Agent Warrants”) to the
Placement Agent to purchase 367,418 shares of our common stock with
an exercise price of $5.00 per share. Such warrants contain a
“cashless exercise” feature and are exercisable at any
time prior to five years from the date of grant.
December 2016 Private Placement
We
conducted a private placement offering in December 2016 (the
“December 2016 Private Placement”), which closed
in January 2017. We issued an aggregate 400,000 shares of
our common stock for $5.00 per share, and a warrant (the
“December 2016 Investor Warrant”), to purchase 400,000
shares of our common stock at an exercise price of $5.00. The
December 2016 Investor Warrant has a five year term. Gross proceeds
totaled $2,000,000, and net proceeds were $1,784,145. Aegis Capital
acted as the placement agent, for the December 2016 Private
Placement.
2
In
connection with the December 2016 Private Placement, we paid the
Placement Agent and selected dealers an aggregate cash fee of
$200,000, and we incurred $15,855 of other expenses related to the
financing. In addition, as part of its compensation for acting as
placement agent for the December 2016 Private Placement, we issued
warrants (the “December 2016 Placement Agent Warrants”)
to the Placement Agent to purchase 80,000 shares of our common
stock with an exercise price of $5.00 per share. Such warrants
contain a “cashless exercise” feature and are
exercisable at any time prior to five years from the date of
grant.
Our Risks
An
investment in our common stock involves a high degree of risk. You
should carefully consider the risks summarized below. These risks
are discussed more fully in the “Risk Factors” section
of this prospectus on page 7 herein. These risks include, but are
not limited to, the following:
|
|
●
|
we have
a limited operating history and have incurred operating losses of
approximately $3,383,000 from inception through
December 31, 2016 and we expect to incur substantial
losses for the foreseeable future and may never achieve or maintain
profitability which could materially limit our ability to raise
additional funds through the issuance of new debt or equity
securities or otherwise;
|
|
|
●
|
we will
need to obtain additional financing to complete clinical
development of REM-001 Therapy;
|
|
|
●
|
clinical
trials for our product candidate, REM-001 Therapy, may not be
successful and we may not obtain approval from the FDA or other
regulatory bodies in different jurisdictions for REM-001
Therapy;
|
|
|
●
|
we are
highly dependent on the success of our product candidate, REM-001
Therapy, which is still in clinical development;
|
|
|
●
|
we
expect to rely on third parties to manufacture the components of
REM-001 Therapy and to conduct our clinical trials;
|
|
|
●
|
we
currently do not have the infrastructure to commercialize REM-001
Therapy should we be successful in obtaining FDA
approval;
|
|
|
●
|
we face
significant competition from other biotechnology and pharmaceutical
companies;
|
|
|
●
|
even if
we obtain marketing approval for REM-001 Therapy, we will be
subject to ongoing obligations and continued regulatory review;
and
|
|
|
●
|
we rely
on our key employees and executives and the loss of the services of
our key employees and executives would adversely impact our
business prospects.
|
Implications of Being an Emerging Growth Company
We are
an “emerging growth company,” as defined in the
Jumpstart Our Business Startups Act of 2012 (the “JOBS
Act”), and, for as long as we continue to be an
“emerging growth company,” we may choose to take
advantage of exemptions from various reporting requirements
applicable to other public companies but not to “emerging
growth companies,” including, but not limited to, not being
required to comply with the auditor attestation requirements of
Section 404 of the Sarbanes-Oxley Act of 2002, as amended, reduced
disclosure obligations regarding executive compensation in our
periodic reports, proxy statements, and exemptions from the
requirements of holding a nonbinding advisory vote on executive
compensation and stockholder approval of any golden parachute
payments not previously approved. We could be an “emerging
growth company” for up to five years, or until the earliest
of (i) the last day of the first fiscal year in which our annual
gross revenues exceed $1 billion, (ii) the date that we become a
“large accelerated filer” as defined in Rule 12b-2
under the Securities Exchange Act of 1934, as amended, which would
occur if the market value of our common stock that is held by
non-affiliates exceeds $700 million as of the last business day of
our most recently completed second fiscal quarter, or (iii) the
date on which we have issued more than $1 billion in
non-convertible debt during the preceding three-year period. We
intend to take advantage of these reporting exemptions described
above until we are no longer an “emerging growth
company.” Under the JOBS Act, “emerging growth
companies” can also delay adopting new or revised accounting
standards until such time as those standards apply to private
companies. We have irrevocably elected not to avail ourselves of
this exemption from new or revised accounting standards and,
therefore, we will be subject to the same new or revised accounting
standards as other public companies that are not “emerging
growth companies.”
3
Corporate Information
We are
a Delaware corporation formed in 2015 under the name Adgero
Biopharmaceuticals Holdings, Inc. We are the parent company of
Adgero Biopharmaceuticals, Inc., our operating subsidiary, a
Delaware corporation.
Our
principal offices are located at 4365 US 1 South, Suite 211,
Princeton, NJ 08540. Our web address is www.adgerobiopharm.com.
Information contained in or accessible through our web site is not,
and should not be deemed to be, part of this
prospectus.
We
currently do not own or license any United States federal trademark
registrations or applications. Some trademarks referred to in this
prospectus are referred to without the ® and ™ symbols,
but such references should not be construed as any indicator that
their respective owners will not assert, to the fullest extent
under applicable law, their rights thereto. We do not intend the
use or display of other companies’ trademarks and trade names
to imply a relationship with, or endorsement or sponsorship of us
by, any other companies.
4
THE OFFERING
|
Common Stock Outstanding
|
5,398,531
shares (1)
|
|
|
|
|
Common Stock Offered by Selling Stockholders
|
3,467,680
shares (2)
|
|
|
|
|
Use of Proceeds
|
We will
not receive any proceeds from the sale of the common stock by the
selling stockholders. We would, however, receive
proceeds upon the exercise of the warrants held by the selling
stockholders which, if such warrants are exercised in full, would
be approximately $8,746,360. Proceeds, if any, received
from the exercise of such warrants will be used for working capital
and general corporate purposes. No assurances can be given that any
of such warrants will be exercised.
|
|
|
|
|
Quotation of Common Stock
|
Our
common stock is not presently traded on any market or securities
exchange, and we have not at this time applied for listing or
quotation on any exchange. We are seeking sponsorship
for the trading of our common stock on the Over-the-Counter, or
OTC, Bulletin Board and/or OTCQB Market operated by OTC Markets
Group, Inc. (together, the “OTCBB/OTCQB”) upon the
effectiveness of the registration statement of which this
prospectus forms a part. The 3,467,680 shares of our
common stock can be sold by selling stockholders at a fixed price
of $5.00 per share until our shares are quoted on the OTCBB/OTCQB
and thereafter at prevailing market prices or privately negotiated
prices. There can be no assurance that a market maker
will agree to file the necessary documents with the Financial
Industry Regulatory Authority (“FINRA”), nor can we
provide any assurance that our shares will actually be quoted on
the OTCBB/OTCQB or, if quoted, that a viable public market will
materialize.
|
|
|
|
|
Risk Factors
|
An
investment in our company is highly speculative and involves a
significant degree of risk. See “Risk
Factors” and other information included in this prospectus
for a discussion of factors you should carefully consider before
deciding to invest in shares of our common stock.
|
|
(1)
|
|
Excludes: (i)
outstanding options to purchase 1,283,937 shares of
our common stock at an exercise price of $5.00 per share; (ii) up
to 1,437,393 shares of our common stock that are available for
issuance under our stock option plan (subject to stockholder
approval of an amendment to adjust the total number of shares
authorized under the 2016 Equity Incentive Plan to
2,756,330); (iii) Investor Warrants exercisable for
1,873,299 shares of common stock at an exercise price of $5.00 per
share issued in our 2016 Private Placement, inclusive of Investor
Warrants exercisable for 87,099 shares of our common stock issued
pursuant to the conversion of promissory notes in connection with
the 2016 Private Placement, (iv) December 2016 Investor Warrant
exercisable for 400,000 shares of common stock at an exercise price
of $5.00 per share issued in our December 2016 Private Placement,
(v) Replacement Warrants exercisable for 30,864 shares of our
common stock at an exercise price of $5.00 per share, (vi) the
Placement Agent Warrants exercisable for 367,418 shares of our
common stock at an exercise price of $5.00 per share, (vii) the
December 2016 Placement Agent Warrants exercisable for 80,000
shares of our common stock at exercise price of $5.00 per share,
(viii) warrants exercisable for 73,998 shares of our common stock
at an exercise price of $5.00 per share issued in connection with
the conversion of senior convertible notes on August 3, 2016, (ix)
warrants exercisable for 5,154 shares of common stock at an
exercise price of $5.00 per share issued in connection with the
conversion of promissory notes on April 8, 2016 not included in the
2016 Private Placement, and (x) 35,000 restricted shares of our
common stock issued under our stock option plan.
|
5
|
(2)
|
|
Includes: (i)
Investor Warrants exercisable for 1,713,254 shares of common stock
at an exercise price of $5.00 per share issued in our 2016 Private
Placement, inclusive of Investor Warrants exercisable for 2,054
shares of common stock issued pursuant to the conversion of a
promissory note in connection with the 2016 Private Placement, (ii)
warrants exercisable for 5,154 shares of common stock at an
exercise price of $5.00 per share issued in connection with the
conversion of a promissory note on April 8, 2016 not included in
the 2016 Private Placement, and (iii) Replacement Warrants
exercisable for 30,864 shares of our common stock at an exercise
price of $5.00 per share. Excludes (i) 160,045 shares of our common
stock issued in our 2016 Private Placement to affiliates of the
Placement Agent and holders of certain convertible promissory notes
in connection with the 2016 Private Placement, for which no
registration rights were granted, and (ii) Investor Warrants
exercisable for 160,045 shares of our common stock at an exercise
price of $5.00 per share issued in our 2016 Private Placement to
affiliates of the Placement Agent and holders of certain
convertible promissory notes in connection with the 2016 Private
Placement, for which no registration rights were
granted.
|
6
RISK FACTORS
An investment in our common stock is speculative and illiquid and
involves a high degree of risk including the risk of a loss of your
entire investment. You should carefully consider the risks and
uncertainties described below and the other information contained
in this prospectus before purchasing shares of our common stock.
The risks set forth below are not the only ones facing us.
Additional risks and uncertainties may exist that could also
adversely affect our business, operations and prospects. If any of
the following risks actually materialize, our business, financial
condition, prospects and/or operations could suffer. In such event,
the value of our common stock could decline, and you could lose all
or a substantial portion of the money that you pay for our common
stock.
Risks Related to Our Financial Position and Need for
Capital
We are a biopharmaceutical company with a limited operating
history.
We
are a biopharmaceutical company with a limited operating history.
Marketing approval of our therapeutic product candidate, the
REM-001 Therapy product, consisting of three parts, the laser light
source, the light delivery device and the drug REM-001
(collectively, the “REM-001 Therapy”), requires
extensive clinical testing data to support the safety and efficacy
requirements needed for regulatory approval. Although we believe
substantial clinical safety and efficacy data exists for our
REM-001 Therapy in cutaneous metastatic breast cancer
(“CMBC”), from the trials completed by Miravant Medical
Technologies, and its wholly-owned subsidiaries, a former public
pharmaceutical and research development company (collectively,
“Miravant”), our recent interaction with
the Food and Drug Administration (the “FDA”) indicated
that further clinical trials by us are needed prior to approval in
this indication. In any other indications we may pursue, we will
need to undertake extensive clinical testing to demonstrate the
safety and efficacy of our REM-001 Therapy or any other product
candidates we develop. In addition, since REM-001 was previously
manufactured and tested in the clinical studies conducted by
Miravant, we intend to use very similar or the same procedures to
manufacture our clinical drug supply and based on our
interaction with FDA we believe that Adgero’s manufacturing
plan is in alignment with the FDA expectations. However, the FDA
may later determine that our proposed approach is not
acceptable and may request more extensive approaches be employed.
Similarly, the other two components of our REM-001 Therapy, namely
the laser and light delivery device, or their equivalents, were
used previously by Miravant in certain clinical studies. We intend
to use the same lasers, or commercially available FDA cleared
lasers that are functionally equivalent to the previously used
devices, in our clinical trials and we intend to manufacture
our light delivery devices using the same design that Miravant
used. Based on our recent interactions with FDA, we believe
this approach should be acceptable, but FDA may later
determine this proposed approach is not acceptable and may request
that more extensive approaches be
employed.
The
likelihood of success of our business plan must be considered in
light of the problems, substantial expenses, difficulties,
complications and delays frequently encountered in connection with
developing and expanding clinical-stage businesses and the
regulatory and competitive environment in which we operate.
Biopharmaceutical product development is a highly speculative
undertaking, involves a substantial degree of risk and is a
capital-intensive business.
Accordingly, you
should consider our prospects in light of the costs, uncertainties,
delays and difficulties frequently encountered by companies in the
clinical stages of development. Potential investors should
carefully consider the risks and uncertainties that a company with
a limited operating history will face. In particular, potential
investors should consider that we cannot assure you that we will be
able to:
|
|
●
|
successfully
implement or execute our current business plan, or that our
business plan is sound;
|
|
|
●
|
receive
FDA approval of clinical trial protocols so that anticipated
additional clinical trials of REM-001 Therapy
commence;
|
|
|
●
|
successfully
complete clinical trials and obtain regulatory approval for the
marketing of REM-001 Therapy;
|
|
|
●
|
successfully
contract for the manufacture of clinical drug product and establish
a commercial drug supply;
|
|
|
●
|
successfully
contract for the manufacture of light delivery devices for clinical
trials;
|
7
|
|
●
|
receive
FDA approval to use our existing lasers (or lasers that are
functionally equivalent) or light delivery devices in
clinical trials or for commercial release
|
|
|
●
|
secure
market exclusivity and/or adequate intellectual property protection
for REM-001 Therapy;
|
|
|
●
|
attract
and retain an experienced management and advisory team;
and
|
|
|
●
|
raise
sufficient funds in the capital markets to effectuate our business
plan including clinical development, regulatory approval and
commercialization for REM-001Therapy.
|
If we
cannot successfully execute any one of the foregoing, our business
may not succeed and your investment will be adversely
affected.
We have incurred operating losses in each year since our inception
and expect to continue to incur substantial losses for the
foreseeable future. We may never become profitable or, if achieved,
be able to sustain profitability.
We
expect to incur substantial expenses without corresponding revenues
unless and until we are able to obtain regulatory approval and
successfully commercialize REM-001 Therapy. To date, we have not
generated any revenue from REM-001 Therapy and we expect to incur
significant expense to complete our clinical program for REM-001
Therapy in the United States and elsewhere. We may never be able to
obtain regulatory approval for the marketing of REM-001 Therapy in
any indication in the United States or internationally. Even if we
are able to commercialize REM-001 Therapy or any other product
candidate, there can be no assurance that we will generate
significant revenues or ever achieve profitability. Our net loss
for the years ended December 31, 2016 and 2015 was
approximately $2,578,000 and $142,000, respectively.
At December 31, 2016, our accumulated deficit since
inception was approximately $3,383,000.
Until
we obtain FDA approval for our REM-001 Therapy, which we do not
expect until 2019 at the earliest, we expect that our
research and development expenses will continue to increase as we
advance our clinical trials for indications for the treatment of
CMBC and potentially other cutaneous metastatic cancers and locally
advanced basal cell carcinomas. We may elect to pursue FDA approval
for REM-001 Therapy in other indications including cutaneous
metastatic cancers other than CMBC as well as locally advanced
basal cell carcinomas such as in patients with Basal Cell Nevus
Syndrome (“BCNS”), which will result in significant
additional research and development expenses. As a result, we
expect to continue to incur substantial losses for the foreseeable
future, and these losses will be increasing. We are uncertain when
or if we will be able to achieve or sustain profitability. If we
achieve profitability in the future, we may not be able to sustain
profitability in subsequent periods. Failure to become and remain
profitable would impair our ability to sustain operations and
adversely affect the price of our common stock and our ability to
raise capital.
Our cash or cash equivalent will only fund our operations for a
limited time and we will need to raise additional capital to
support our development and commercialization efforts.
We are
currently operating at a loss and expect our operating costs will
increase significantly as we incur costs related to the clinical
trials for REM-001 Therapy. At December 31, 2016, we
had a cash and cash equivalents balance of approximately
$3,165,000 and certificates of deposit of
$2,802,000. We believe that our cash and cash
equivalents and certificates of deposit as of December 31,
2016, and the $2,000,000 of gross proceeds received
subsequent to December 31, 2016 through the December
2016 Private Placement that closed in January 2017,
will be sufficient to fund our operations for at least through the
next twelve months from the date of this
filing.
We do
not currently have any arrangements or credit facilities in place
as a source of funds, and there can be no assurance that we will be
able to raise sufficient additional capital on acceptable terms, or
at all. We may seek additional capital through a combination of
private and public equity offerings, debt financings and strategic
collaborations. Debt financing, if obtained, may involve agreements
that include covenants limiting or restricting our ability to take
specific actions, such as incurring additional debt, and could
increase our expenses and require that our assets secure such
debt.
Moreover, any debt
we incur must be repaid regardless of our operating results. At
December 31, 2016, we had no debt outstanding. All
debt outstanding at December 31, 2015 was exchanged for common
stock and warrants on July 29, 2016 and August 3, 2016. Equity
financing, if obtained, could result in dilution to our then
existing stockholders and/or require such stockholders to waive
certain rights and preferences. If such financing is not available
on satisfactory terms, or is not available at all, we may be
required to delay, scale back or eliminate the development of
business opportunities and our operations and financial condition
may be materially adversely affected. We can provide no assurances
that any additional sources of financing will be available to us on
favorable terms, if at all. In addition, if we are unable to secure
sufficient capital to fund our operations, we might have to enter
into strategic collaborations that could require us to share
commercial rights to REM-001 Therapy with third parties in ways
that we currently do not intend or on terms that may not be
favorable to us. If we choose to pursue additional indications
and/or geographies for REM-001 Therapy or otherwise expand more
rapidly than we presently anticipate we may also need to raise
additional capital sooner than expected.
8
Risks
Related to Product Development, Regulatory Approval, Manufacturing
and Commercialization
Our plan to achieve marketing approval of REM-001 Therapy depends
partly on the accuracy of our preliminary efficacy analysis of
REM-001Therapy CMBC trial data. While we believe the results of our
preliminary efficacy analysis accurately reflect the actual
clinical trial results, a detailed analysis overseen by regulatory
experts may yield different results.
We plan
to utilize existing REM-001 Therapy clinical trial data as
supportive data when seeking marketing approval of REM-001
Therapy for the treatment of CMBC. Between February 1996 and
January 1999, Miravant, with support from certain corporate
partners, conducted four clinical trials for the treatment of CMBC
using REM-001 Therapy. As part of our review of REM-001
Therapy’s data package, we noted that while Miravant’s
investigators had done a safety analysis of all treated patients,
these reports indicated an efficacy analysis was only performed on
two of their four clinical trials. Notably, there had been no
efficacy analysis on the other two trials which constituted
approximately half of the CMBC patients who were treated with
REM-001 Therapy. We originally performed a preliminary efficacy
analysis on the data from all four CMBC trials, including the two
that had not previously been analyzed. We then engaged regulatory experts who were either
former FDA employees with directly related experience in reviewing
similar oncology treatments who are now acting as independent
consultants or individuals who have provided senior regulatory
guidance to major pharmaceutical or medical device companies in
situations that led to regulatory approval. These individuals
guided us in conducting a second more in-depth analysis that
yielded results consistent with our original analysis. Following
that, we compiled a briefing document and submitted questions
to FDA. While we believe the results of our
preliminary efficacy analysis, and
subsequent analysis conducted under the guidance of these experts
which was consistent with our original preliminary analysis,
accurately reflect the actual clinical trial results and that the
age of the underlying data from the clinical studies is not
material, a more in-depth review may yield different conclusions.
Such differing results may negatively impact our ability to pursue
or achieve, or result in delays to obtain, marketing approval of
REM-001 Therapy. There can be no certainty that results from our
analyses done to date or results from future analyses that we may
undertake will be sufficiently complete to satisfy FDA requests or
that any results will be favorable to us.
Our REM-001 Therapy clinical trial data may not be deemed
acceptable by the FDA to support our new drug
applications.
In
seeking regulatory approval for REM-001, we intend to rely at
least in part upon data gathered by Miravant Medical
Technologies in its initial Phase 1 studies and in four later Phase
2/3 clinical studies that were conducted approximately 20 years
ago. Based on our initial interactions with the FDA, we
believe the agency will accept these results as supportive data but
we cannot ultimately be certain that the FDA will accept
data that old to support our new drug applications. Also
based on our initial interactions with the FDA, we believe our
plans for manufacturing investigational test materials will lead to
investigational test materials that FDA will recognize as being
sufficiently comparable to Miravant’s materials and also
suitable for further investigational trials but FDA may
later raise questions about the similarity of
Miravant’s investigational testing material versus our
manufactured investigational testing material, or may
raise questions about the processes and methods under which this
old data was collected or may raise additional concerns regarding
the elapsed time period. If the FDA does not accept this
data, we will have to incur significant costs which may require
additional capital to redo some or all of the Miravant studies or
supplement these studies with additional studies.
We depend entirely on the success of REM-001 Therapy, which has not
received FDA approval for the treatment of CMBC, our lead
indication. If we are unable to obtain regulatory approval for and
generate revenues from REM-001 Therapy, our ability to create
stockholder value will be limited.
We do
not generate revenues from any FDA approved drug products. Our only
product candidate is REM-001 Therapy, which we believe has
previously demonstrated safety and efficacy results in patients
suffering from CMBC in four late stage (Phase 2/3) clinical trials
completed by Miravant. However, we are not currently actively
engaged in clinical trials for this or any other product candidate.
Moreover, there can be no assurance that any positive clinical
results obtained for REM-001 Therapy in prior clinical studies will
be repeated in future clinical studies, or that such results will
be sufficient to obtain regulatory approval. If we do not obtain
regulatory approval, or the approval process for REM-001 Therapy
takes longer than we anticipate, our expenses and need for
additional capital will increase. Most drug candidates that reach
clinical development have only a small chance of successfully
completing clinical development and gaining regulatory approval.
Therefore, our business currently depends entirely on the
successful development, regulatory approval and commercialization
of REM-001 Therapy, which may never occur.
If we are not able to obtain any required regulatory approvals for
REM-001 Therapy, we will not be able to commercialize our only
product candidate, REM-001 Therapy, and we will not be able to
generate revenue.
We will
need to successfully complete additional clinical trials for
REM-001 Therapy before we can apply for its marketing approval,
including conducting a pivotal Phase 3 clinical trial (or
equivalent) in CMBC patients with the goal of submitting a
New Drug Application (“NDA”) for REM-001 Therapy.
However, we may never commence this trial, and if we do, there can
be no assurance that the trial will be successful. Even if this
clinical trial is successful, we may be required to conduct
additional clinical trials to establish REM-001 Therapy’s
safety and efficacy before the NDA for REM-001 Therapy in CMBC can
be approved, if at all.
9
Clinical trials are
expensive, difficult to design and implement, can take many years
to complete and are uncertain as to outcome. Success in early
phases of pre-clinical and clinical trials does not ensure that
later clinical trials will be successful and interim results of a
clinical trial do not necessarily predict final results. A failure
of one or more of our clinical trials can occur at any stage of
testing. We may experience numerous unforeseen events during, or as
a result of, the clinical trial process that could delay or prevent
our ability to receive regulatory approval for or commercialize
REM-001 Therapy. The research, testing, manufacturing, labeling,
packaging, storage, approval, sale, marketing, advertising and
promotion, pricing, export, import and distribution of drug
products are subject to extensive regulation by the FDA and other
regulatory authorities in the United States and other countries,
which regulations differ from country to country. We are not
permitted to market REM-001 Therapy, including the REM-001 drug and
associated device components, as a prescription pharmaceutical
product in the United States until we receive approval of an NDA
from the FDA, or in any foreign countries until we receive the
requisite approval from such countries. In the United States, the
FDA generally requires the completion of clinical trials of each
drug to establish its safety and efficacy and extensive
pharmaceutical development to ensure its quality before an NDA is
approved. Regulatory authorities in other jurisdictions impose
similar requirements. Of the large number of drugs in development,
only a small percentage result in the submission of an NDA to the
FDA and even fewer are eventually approved for commercialization.
We have not submitted an NDA to the FDA or comparable applications
to other regulatory authorities. If our development efforts for
REM-001 Therapy, including regulatory approval, are not successful
for its planned indications, or if adequate demand for REM-001
Therapy is not generated, our business will be materially adversely
affected.
Our
success depends on the receipt of regulatory approval and the
issuance of such regulatory approvals is uncertain and subject to a
number of risks, including the following:
|
|
●
|
the FDA
or comparable foreign regulatory authorities or institutional
review boards (“IRBs”), may disagree with the design or
implementation of our clinical trials;
|
|
|
●
|
we may
not be able to provide acceptable evidence of our product
candidate’s safety and efficacy;
|
|
|
●
|
the
results of our clinical trials may not be satisfactory or may not
meet the level of statistical or clinical significance required by
the FDA, European Medicines Agency (“EMA”), or other
regulatory agencies for marketing approval;
|
|
|
●
|
the
dosing of REM-001 Therapy in a particular clinical trial may not be
at an optimal level;
|
|
|
●
|
patients
in our clinical trials may suffer adverse effects for reasons that
may or may not be related to REM-001 Therapy;
|
|
|
●
|
the
data collected from clinical trials may not be sufficient to
support the submission of an NDA or other submission or to obtain
regulatory approval in the United States or elsewhere;
|
|
|
●
|
the FDA
or comparable foreign regulatory authorities may fail to approve
the manufacturing processes or facilities of third-party
manufacturers with which we contract for clinical and commercial
supplies; and
|
|
|
●
|
the
approval policies or regulations of the FDA or comparable foreign
regulatory authorities may significantly change in a manner
rendering our clinical data insufficient for approval.
|
Failure
to obtain regulatory approval for REM-001 Therapy for the foregoing
or any other reasons will prevent us from commercializing this
product candidate as a prescription product and generating revenue.
We cannot guarantee that regulators will agree with our assessment
of the results of the clinical trials conducted by Miravant or that
we may conduct in the future or that such trials will be
successful. The FDA, EMA and other regulators have substantial
discretion in the approval process and may refuse to accept any
application or may decide that our data is insufficient for
approval and require additional clinical trials or other studies.
In addition, varying interpretations of the data obtained from
clinical testing could delay, limit or prevent regulatory approval
of our product candidate.
10
We are
a clinical-stage company and we have not submitted an NDA or
received regulatory approval to market REM-001 Therapy in any
jurisdiction. We have only limited experience in filing the
applications necessary to gain regulatory approvals and expect to
rely on consultants and clinical CROs with expertise in this area
to assist us in this process. Securing FDA approval requires the
submission of pre-clinical, clinical, and/or pharmacokinetic data,
information about product manufacturing processes and inspection of
facilities and supporting information to the FDA for each
therapeutic indication to establish a product candidate’s
safety and efficacy for each indication. REM-001 Therapy may prove
to have undesirable or unintended side effects, toxicities or other
characteristics that may preclude our obtaining regulatory approval
or prevent or limit commercial use with respect to one or all
intended indications.
The
process of obtaining regulatory approvals is expensive, often takes
many years, if approval is obtained at all, and can vary
substantially based upon, among other things, the type, complexity
and novelty of the product candidates involved, the jurisdiction in
which regulatory approval is sought and the substantial discretion
of the regulatory authorities. Changes in the regulatory approval
policy during the development period, changes in or the enactment
of additional statutes or regulations, or changes in regulatory
review for a submitted product application may cause delays in the
approval or rejection of an application. Regulatory approval
obtained in one jurisdiction does not necessarily mean that a
product candidate will receive regulatory approval in all
jurisdictions in which we may seek approval, but the failure to
obtain approval in one jurisdiction may negatively impact our
ability to seek approval in a different jurisdiction. Failure to
obtain regulatory marketing approval for REM-001 Therapy in any
indication will prevent us from commercializing the product
candidate, and generating revenue.
We do not have a clinical supply of REM-001. Moreover, we do not
have our own manufacturing facilities nor have we identified a
third-party to manufacture the product for us. If we are unable to
identify a third-party manufacturer, or if this third-party
manufacturer fails to meet applicable regulatory requirements or to
supply us for any reason, we will be unable to complete clinical
trials for REM-001 Therapy and our business will be materially
impaired.
We do
not have a clinical supply of REM-001. We will need to
engage a third party manufacturer to produce the product for us. If
and when approved, we intend to have a third-party manufacture
commercial supplies of the product as well. We have not yet
identified a third-party manufacturer and our failure to timely do
so will delay the commencement of our clinical trials and the
submission of our NDA for REM-001 Therapy.
We do not have a clinical supply of light delivery devices for use
with REM-001 Therapy. Moreover, we do not have our own
manufacturing facilities nor have we identified a third-party to
manufacture these devices for us. If we are unable to identify a
third-party manufacturer, or if this third-party manufacturer fails
to meet applicable regulatory requirements or to supply us for any
reason, we will be unable to complete clinical trials for REM-001
Therapy and our business will be materially impaired.
We do
not have a clinical supply of REM-001 Therapy light delivery
devices. We will need to engage a third party
manufacturer to produce these devices for us. If and when approved,
we intend to have a third-party manufacture commercial supplies of
these devices as well. We have not yet identified a third-party
manufacturer and our failure to timely do so will delay the
commencement of our clinical trials and the submission of our NDA
for REM-001 Therapy.
We are planning to use our existing laser light devices, some of
which were used in certain of Miravant’s clinical trials,
or laser light devices that are functionally equivalent to
the Miravant devices, in our clinical trials. We have not
confirmed that these laser designs are still acceptable for use by
the FDA or other local regulatory bodies. If we are unable to use
these lasers we may need to arrange for the manufacture of new
lasers or significantly modify our existing laser units. We do not
have our own manufacturing facilities for conducting these
activities nor have we identified a third-party to manufacture or
modify these devices for us. If we need to perform either of these
steps and are unable to identify a third-party manufacturer, or if
this third-party manufacturer fails to meet applicable regulatory
requirements or to supply us for any reason, we will be unable to
complete clinical trials for REM-001 Therapy and our business will
be materially impaired.
11
Our
plan relies on our using our existing laser supply, some of which
are the same units used by Miravant in certain of their clinical
trials or laser light devices that are functionally
equivalent to the Miravant devices. We will need to engage a
third party manufacturer to modify these devices or to produce new
laser devices for us. We have not yet identified such a third-party
and our failure to timely do so will delay the commencement of our
clinical trials and the submission of our NDA for REM-001
Therapy.
REM-001 Therapy is our only product candidate. If we fail to
successfully commercialize REM-001 Therapy, we may need to acquire
additional product candidates or our business will be materially
adversely affected.
We have
never commercialized any product candidates and do not have any
other compounds in pre-clinical testing, lead optimization or lead
identification stages beyond REM-001 Therapy. We cannot be certain
that REM-001 Therapy will prove to be sufficiently effective and
safe to meet applicable regulatory standards for any indication. If
we fail to successfully commercialize REM-001 Therapy as a
treatment for CMBC or any other indication, whether as a
stand-alone therapy or in combination with other treatments, our
business would be materially adversely affected.
Even if we receive regulatory approval for REM-001 Therapy, we
still may not be able to successfully commercialize it and the
revenue that we generate from its sales, if any, may be
limited.
If
approved for marketing, the commercial success of REM-001 Therapy
will depend upon its acceptance by the medical community, including
physicians, patients and health care payors. The degree of market
acceptance of REM-001 Therapy will depend on a number of factors,
including:
|
|
●
|
demonstration
of clinical safety and efficacy;
|
|
|
●
|
relative
convenience, burden and ease of administration;
|
|
|
●
|
the
prevalence and severity of any adverse effects;
|
|
|
●
|
the
willingness of physicians to prescribe REM-001 Therapy and of the
target patient population to try new therapies;
|
|
|
●
|
efficacy
of REM-001 Therapy compared to competing products;
|
|
|
●
|
the
introduction of any new products that may in the future become
available to treat indications for which REM-001 Therapy may be
approved;
|
|
|
●
|
new
procedures or methods of treatment that may reduce the incidences
of any of the indications in which REM-001 Therapy may show
utility;
|
|
|
●
|
pricing
and cost-effectiveness;
|
|
|
●
|
the
inclusion or omission of REM-001 Therapy in applicable treatment
guidelines;
|
|
|
●
|
the
effectiveness of our or any future collaborators’ sales and
marketing strategies;
|
|
|
●
|
limitations
or warnings contained in FDA-approved labeling;
|
12
|
|
●
|
our
ability to obtain and maintain sufficient third-party coverage or
reimbursement from government health care programs, including
Medicare and Medicaid, private health insurers and other
third-party payors; and
|
|
|
●
|
the
willingness of patients to pay out-of-pocket in the absence of
third-party coverage or reimbursement.
|
If
REM-001 Therapy is approved, but does not achieve an adequate level
of acceptance by physicians, health care payors and patients, we
may not generate sufficient revenue and we may not be able to
achieve or sustain profitability. Our efforts to educate the
medical community and third-party payors on the benefits of REM-001
Therapy may require significant resources and may never be
successful.
In
addition, even if we obtain regulatory approvals, the timing or
scope of any approvals may prohibit or reduce our ability to
commercialize REM-001 Therapy successfully. For example, if the
approval process takes too long, we may miss market opportunities
and give other companies the ability to develop competing products
or establish market dominance. Any regulatory approval we
ultimately obtain may be limited or subject to restrictions or
post-approval commitments that render REM-001 Therapy not
commercially viable. For example, regulatory authorities may
approve REM-001 Therapy for fewer or more limited indications than
we request, may grant approval contingent on the performance of
costly post-marketing clinical trials, or may approve REM-001
Therapy with a label that does not include the labeling claims
necessary or desirable for the successful commercialization of that
indication. Further, the FDA or comparable foreign regulatory
authorities may place conditions on approvals such as risk
management plans and the requirement for a Risk Evaluation and
Mitigation Strategy (“REMS”) to assure the safe use of
the drug. If the FDA concludes a REMS is needed, the sponsor of the
NDA must submit a proposed REMS; the FDA will not approve the NDA
without an approved REMS, if required. A REMS could include
medication guidelines, physician communication plans, or elements
to assure safe use, such as restricted distribution methods,
patient registries and other risk minimization tools. The FDA may
also require a REMS for an approved product when new safety
information emerges. Any of these limitations on approval or
marketing could restrict the commercial promotion, distribution,
prescription or dispensing of REM-001. Moreover, product approvals
may be withdrawn for non-compliance with regulatory standards or if
problems occur following the initial marketing of the product. Any
of the foregoing scenarios could materially harm the commercial
success of REM-001 Therapy.
To obtain regulatory approval to market REM-001 Therapy in
indications other than CMBC, costly and lengthy clinical trials
will be required, and the results of the studies and trials are
highly uncertain.
As part
of the regulatory approval process, we must conduct, at our own
expense, clinical trials on humans for each indication that we
intend to pursue. We expect the number of nonclinical studies and
clinical trials that the regulatory authorities will require will
vary depending on the disease or condition the drug is being
developed to address and regulations applicable to the particular
drug. Generally, the number and size of clinical trials required
for approval varies based on the nature of the disease and size of
the expected patient population that may be treated with a drug. We
must demonstrate that our drug products are safe and efficacious
for use in the targeted human patients in order to receive
regulatory approval for commercial sale, and regulatory approval
for one indication does not guaranty regulatory approval of the
same drug for another indication. If we do not obtain regulatory
approval for any indication for which it is sought, our business
may be materially adversely impacted.
Even if we obtain marketing approval for REM-001 Therapy, we will
be subject to ongoing obligations and continued regulatory review,
which will result in significant additional expense. Additionally,
REM-001 Therapy could be subject to labeling and other restrictions
and withdrawal from the market and we may be subject to penalties
if we fail to comply with regulatory requirements or if we
experience unanticipated problems with REM-001
Therapy.
Even if
we obtain United States regulatory approval of REM-001 Therapy for
an indication, the FDA may still impose significant restrictions on
its indicated uses or marketing or the conditions of approval, or
impose ongoing requirements for potentially costly and
time-consuming post-approval studies, including Phase 4
clinical trials, and post-market surveillance to monitor safety and
efficacy. REM-001 Therapy will also be subject to ongoing
regulatory requirements governing the manufacturing, labeling,
packaging, storage, distribution, safety surveillance, advertising,
promotion, recordkeeping and reporting of adverse events and other
post-market information. These requirements include registration
with the FDA, as well as continued compliance with current Good
Clinical Practices regulations (“cGCPs”) for any
clinical trials that we conduct post-approval. In addition,
manufacturers of drug products and their facilities are subject to
continual review and periodic inspections by the FDA and other
regulatory authorities for compliance with current Good
Manufacturing Practices (“cGMP”), requirements relating
to quality control, quality assurance and corresponding maintenance
of records and documents.
13
The FDA
has the authority to require a REMS as part of an NDA or after
approval, which may impose further requirements or restrictions on
the distribution or use of an approved drug, such as limiting
prescribing to certain physicians or medical centers that have
undergone specialized training, limiting treatment to patients who
meet certain safe-use criteria or requiring patient testing,
monitoring and/or enrollment in a registry.
With
respect to sales and marketing activities by us or any future
partner, advertising and promotional materials must comply with FDA
rules in addition to other applicable federal, state and local laws
in the United States and similar legal requirements in other
countries. In the United States, the distribution of product
samples to physicians must comply with the requirements of the U.S.
Prescription Drug Marketing Act. Application holders must obtain
FDA approval for product and manufacturing changes, depending on
the nature of the change. We may also be subject, directly or
indirectly through our customers and partners, to various fraud and
abuse laws, including, without limitation, the U.S. Anti-Kickback
Statute, U.S. False Claims Act, and similar state laws, which
impact, among other things, our proposed sales, marketing, and
scientific/educational grant programs. If we participate in the
U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of
the U.S. Department of Veterans Affairs, or other government drug
programs, we will be subject to complex laws and regulations
regarding reporting and payment obligations. All of these
activities are also potentially subject to U.S. federal and state
consumer protection and unfair competition laws. Similar
requirements exist in many of these areas in other
countries.
In
addition, if REM-001 Therapy is approved for an indication, our
product labeling, advertising and promotion would be subject to
regulatory requirements and continuing regulatory review. The FDA
strictly regulates the promotional claims that may be made about
prescription products. In particular, a product may not be promoted
for uses that are not approved by the FDA as reflected in the
product’s approved labeling. If we receive marketing approval
for REM-001 Therapy, physicians may nevertheless legally prescribe
our products to their patients in a manner that is inconsistent
with the approved label. If we are found to have promoted such
off-label uses, we may become subject to significant liability and
government fines. The FDA and other agencies actively enforce the
laws and regulations prohibiting the promotion of off-label uses,
and a company that is found to have improperly promoted off-label
uses may be subject to significant sanctions. The federal
government has levied large civil and criminal fines against
companies for alleged improper promotion and has enjoined several
companies from engaging in off-label promotion. The FDA has also
requested that companies enter into consent decrees of permanent
injunctions under which specified promotional conduct is changed or
curtailed.
If we
or a regulatory agency discovers previously unknown problems with a
product, such as adverse events of unanticipated severity or
frequency, problems with the facility where the product is
manufactured, or we or our manufacturers fail to comply with
applicable regulatory requirements, we may be subject to the
following administrative or judicial sanctions:
|
|
●
|
restrictions
on the marketing or manufacturing of the product, withdrawal of the
product from the market, or voluntary or mandatory product
recalls;
|
|
|
●
|
issuance
of warning letters or untitled letters;
|
|
|
●
|
clinical
holds;
|
|
|
●
|
injunctions
or the imposition of civil or criminal penalties, monetary fines,
restitution, or disgorgement;
|
|
|
●
|
suspension
or withdrawal of regulatory approval;
|
|
|
●
|
refusals
of government contracts;
|
|
|
●
|
suspension
of any ongoing clinical trials;
|
14
|
|
●
|
refusal
to approve pending applications or supplements to approved
applications filed by us, or suspension or revocation of product
license approvals;
|
|
|
●
|
suspension
or imposition of restrictions on operations, including costly new
manufacturing requirements; or
|
|
|
●
|
product
seizure or detention or refusal to permit the import or export of
product.
|
The
occurrence of any event or penalty described above may inhibit our
ability to commercialize REM-001 Therapy and generate revenue.
Adverse regulatory action, whether pre- or post-approval, can also
potentially lead to product liability claims and increase our
product liability exposure.
We currently have no sales and marketing organization. If we are
unable to secure a sales and marketing partner or establish
satisfactory sales and marketing capabilities, we may not
successfully commercialize REM-001 Therapy.
At
present, we have no sales or marketing personnel. In order to
commercialize products that are approved for commercial sales, we
must either collaborate with third parties that have such
commercial infrastructure or develop our own sales and marketing
infrastructure. If we are not successful entering into
appropriate collaboration arrangements, or recruiting sales and
marketing personnel or in building a sales and marketing
infrastructure, we will have difficulty successfully
commercializing REM-001 Therapy, which would adversely affect our
business, operating results and financial condition.
We may
not be able to enter into collaboration agreements on terms
acceptable to us or at all. In addition, even if we enter into such
relationships, we may have limited or no control over the sales,
marketing and distribution activities of these third parties. Our
future revenues may depend heavily on the success of the efforts of
these third parties. If we elect to establish a sales and marketing
infrastructure we may not realize a positive return on this
investment. In addition, we will have to compete with established
and well-funded pharmaceutical and biotechnology companies to
recruit, hire, train and retain sales and marketing personnel.
Factors that may inhibit our efforts to commercialize REM-001
Therapy without strategic partners or licensees
include:
|
|
●
|
our
inability to recruit and retain adequate numbers of effective sales
and marketing personnel;
|
|
|
●
|
the
inability of sales personnel to obtain access to or persuade
adequate numbers of physicians to prescribe REM-001
Therapy;
|
|
|
●
|
the
lack of complementary products to be offered by sales personnel,
which may put us at a competitive disadvantage relative to
companies with more extensive product lines; and
|
|
|
●
|
unforeseen
costs and expenses associated with creating an independent sales
and marketing organization.
|
REM-001 Therapy may exhibit adverse side effects that prevent its
widespread adoption or that may necessitate its withdrawal from the
market.
Our REM-001 Therapy may exhibit undesirable and unintended side
effects that may prevent or limit its commercial adoption and use.
One such side effect upon the use of our REM-001 Therapy
drug and device components as
potential therapeutic agents may be a period of photosensitivity
for a certain period of time after receiving REM-001
Therapy. This period of
photosensitivity is generally dose dependent and typically declines
over time. A second such side effect is pain that arises from the
treatment or results from the treatment. Treatment related pain has
been experienced by some patients and it is often treated with
analgesics but in some cases more aggressive treatment can be
required. Even upon receiving approval by the FDA and other
regulatory authorities, our products may later exhibit adverse side
effects that prevent widespread use or necessitate withdrawal from
the market. The manifestation of such side effects could cause our
business to suffer.
15
We face competition from other biotechnology and pharmaceutical
companies and our operating results will suffer if we fail to
compete effectively.
The
biotechnology and pharmaceutical industries are intensely
competitive and subject to rapidly evolving technology and intense
research and development efforts. We have competitors in a number
of jurisdictions that have substantially greater name recognition,
commercial infrastructures and financial, technical and personnel
resources than we have. Established competitors may invest heavily
to quickly discover and develop novel compounds that could make
REM-001 Therapy obsolete or uneconomical. Any new product that
competes with an approved product may need to demonstrate
compelling advantages in efficacy, cost, convenience, tolerability
and safety to be commercially successful. Other competitive
factors, including generic competition, could force us to lower
prices or could result in reduced sales. In addition, new products
developed by others could emerge as competitors to REM-001 Therapy.
If we are not able to compete effectively against our current and
future competitors, our business will not grow and our financial
condition and operations will suffer.
Our
primary competitors in the CMBC field are Celsion Corporation
(NASDAQ: CLSN) in the United States and IGEA Medical S.p.A. in
Europe, both of which are developing alternative treatment
therapies for CMBC. Celsion Corporation is developing a drug
treatment for CMBC that is activated by a hyperthermia device and
has completed Phase 1/2 studies in CMBC. IGEA Medical S.p.A. is
developing an electro-chemotherapy treatment for CMBC. Pinnacle
Biologics Inc., a subsidiary of Concordia Healthcare Corp (NASDAQ:
CXRX), sells Photofrin, a first generation photodynamic therapy
(“PDT”) product for treatment of certain endobronchial
non-small-cell lung cancers and esophageal cancers. Photofrin is
currently in Phase 2 studies for the treatment of mesothelioma and
Phase 2 studies in recurrent glioma. To our knowledge, there is no
reported development program for Photofrin in CMBC.
There
are numerous therapies currently used to treat CMBC patients
including chemotherapy, radiation therapy, surgical excision,
hyperthermia, cryotherapy, electro-chemotherapy, topical drugs and
intra-lesional chemotherapy injections, but, to our knowledge,
there are no PDT therapies currently approved by the FDA for the
treatment of CMBC or similar cutaneous cancers. Some topical PDT
agents have been approved by FDA for actinic keratosis which is a
precancerous skin condition and they have been approved in some
other countries for some conditions that we believe pose low
medical risk such as basal cell cancer and acne.
Additionally, Rogers Sciences Inc. is a medical device
company that is developing a light delivery device for use with PDT
treatment of cutaneous cancers that they are currently clinically
testing in a Phase 2 study in CMBC patients.
Recently enacted and future legislation may increase the difficulty
and cost for us to obtain marketing approval of and
commercialization of REM-001 Therapy and may affect the prices we
may obtain.
In the
United States and some foreign jurisdictions, there have been a
number of legislative and regulatory changes and proposed changes
regarding the healthcare system that could prevent or delay
marketing approval for REM-001 Therapy, restrict or regulate
post-approval activities and affect our ability to profitably sell
REM-001 Therapy. Legislative and regulatory proposals have been
made to expand post-approval requirements and restrict sales and
promotional activities for pharmaceutical products. We do not know
whether additional legislative changes will be enacted, or whether
the FDA regulations, guidance or interpretations will be changed,
or what the impact of such changes on the marketing approvals of
REM-001 Therapy, if any, may be. In addition, increased scrutiny by
the U.S. Congress of the FDA’s approval process may
significantly delay or prevent marketing approval, as well as
subject us to more stringent product labeling and post-marketing
testing and other requirements.
In the
United States, the Medicare Modernization Act (the
“MMA”) changed the way Medicare covers and pays for
pharmaceutical products. The legislation expanded Medicare coverage
for drug purchases by the elderly and introduced a new
reimbursement methodology based on average sales prices for drugs.
In addition, this legislation authorized Medicare Part D
prescription drug plans to use formularies where they can limit the
number of drugs that will be covered in any therapeutic class. As a
result of this legislation and the expansion of federal coverage of
drug products, we expect that there will be additional pressure to
contain and reduce costs. These cost reduction initiatives and
other provisions of this legislation could decrease the coverage
and price that we receive for REM-001 Therapy and could seriously
harm our business. While the MMA applies only to drug benefits for
Medicare beneficiaries, private payors often follow Medicare
coverage policy and payment limitations in setting their own
reimbursement rates, and any reduction in reimbursement that
results from the MMA may result in a similar reduction in payments
from private payors.
16
In
March 2010, President Obama signed into law the Patient Protection
and Affordable Care Act, as amended by the Health Care and
Education Affordability Reconciliation Act of 2010
(collectively, the “Health Care Reform Law”), a
sweeping law intended to broaden access to health insurance, reduce
or constrain the growth of healthcare spending, enhance remedies
against fraud and abuse, add new transparency requirements for
healthcare and health insurance industries, impose new taxes and
fees on the health industry and impose additional health policy
reforms. Effective October 1, 2010, the Health Care Reform Law
revised the definition of “average manufacturer price”
for reporting purposes, which could increase the amount of Medicaid
drug rebates to states. Further, the law imposed a significant
annual fee on companies that manufacture or import branded
prescription drug products. Substantial provisions affecting
compliance have also been enacted, which may require us to modify
our business practices with healthcare practitioners and incur
substantial costs to ensure compliance. Despite initiatives to
repeal the Health Care Reform Law, at this time it
appears the implementation of the Health Care Reform Law will
continue. Although it is too early to determine the full effect of
the Health Care Reform Law, the law appears likely to continue the
pressure on pharmaceutical pricing, especially under the Medicare
program, and may also increase our regulatory burdens and operating
costs.
In
addition, other legislative changes have been proposed and adopted
in the United States since the Health Care Reform Law was enacted.
On August 2, 2011, the Budget Control Act of 2011 among other
things, created measures for spending reductions by Congress. A
Joint Select Committee on Deficit Reduction, tasked with
recommending a targeted deficit reduction of at least $1.2 trillion
for the years 2013 through 2021, was unable to reach required
goals, thereby triggering the legislation’s automatic
reduction to several government programs. This includes aggregate
reductions to Medicare payments to providers of up to 2% per fiscal
year, starting in 2013. On January 2, 2013, President Obama
signed into law the American Taxpayer Relief Act of 2012 (the
“ATRA”) which delayed for another two months the budget
cuts mandated by these sequestration provisions of the Budget
Control Act of 2011. The ATRA, among other things, also reduced
Medicare payments to several providers, including hospitals,
imaging centers and cancer treatment centers, and increased the
statute of limitations period for the government to recover
overpayments to providers from three to five years. We expect that
additional federal healthcare reform measures will be adopted in
the future, any of which could limit the amounts that federal and
state governments will pay for healthcare products and services,
and in turn could significantly reduce the projected value of
certain development projects and reduce any future
profitability.
Our future growth depends, in part, on our ability to penetrate
foreign markets, where we would be subject to additional regulatory
burdens and other risks and uncertainties.
Our
future profitability will depend, in part, on our ability to
commercialize REM-001 Therapy in foreign markets for which we
intend to rely on collaborations with third parties. If we
commercialize REM-001 Therapy in foreign markets, we would be
subject to additional risks and uncertainties,
including:
|
|
●
|
our
customers’ ability to obtain reimbursement for REM-001
Therapy in foreign markets;
|
|
|
●
|
our
inability to directly control commercial activities because we are
relying on third parties;
|
|
|
●
|
the
burden of complying with complex and changing foreign regulatory,
tax, accounting and legal requirements;
|
|
|
●
|
different
medical practices and customs in foreign countries affecting
acceptance in the marketplace;
|
|
|
●
|
import
or export licensing requirements;
|
17
|
|
●
|
longer
accounts receivable collection times;
|
|
|
●
|
longer
lead times for shipping;
|
|
|
●
|
language
barriers for technical training;
|
|
|
●
|
reduced
protection of intellectual property rights in some foreign
countries;
|
|
|
●
|
foreign
currency exchange rate fluctuations; and
|
|
|
●
|
the
interpretation of contractual provisions governed by foreign laws
in the event of a contract dispute.
|
Foreign
sales of REM-001 Therapy could also be adversely affected by the
imposition of governmental controls, political and economic
instability, trade restrictions and changes in tariffs, any of
which may adversely affect our results of operations.
If we market REM-001 Therapy in a manner that violates healthcare
fraud and abuse laws, or if we violate government price reporting
laws, we may be subject to civil or criminal
penalties.
The FDA
enforces laws and regulations which require that the promotion of
pharmaceutical products be consistent with the approved prescribing
information. While physicians may prescribe an approved product for
a so-called “off label” use, it is unlawful for a
pharmaceutical company to promote its products in a manner that is
inconsistent with its approved label and any company which engages
in such conduct can subject that company to significant liability.
Similarly, industry codes in the EU and other foreign jurisdictions
prohibit companies from engaging in off-label promotion and
regulatory agencies in various countries enforce violations of the
code with civil penalties. While we intend to ensure that our
promotional materials are consistent with our label, regulatory
agencies may disagree with our assessment and may issue untitled
letters, warning letters or may institute other civil or criminal
enforcement proceedings. In addition to FDA restrictions on
marketing of pharmaceutical products, several other types of state
and federal healthcare fraud and abuse laws have been applied in
recent years to restrict certain marketing practices in the
pharmaceutical industry. These laws include the U.S. Anti-Kickback
Statute, U.S. False Claims Act and similar state laws. Because of
the breadth of these laws and the narrowness of the safe harbors,
it is possible that some of our business activities could be
subject to challenge under one or more of these laws.
The
U.S. Anti-Kickback Statute prohibits, among other things, knowingly
and willfully offering, paying, soliciting or receiving
remuneration to induce, or in return for, purchasing, leasing,
ordering or arranging for the purchase, lease or order of any
healthcare item or service reimbursable under Medicare, Medicaid or
other federally financed healthcare programs. This statute has been
interpreted broadly to apply to arrangements between pharmaceutical
manufacturers on the one hand and prescribers, purchasers and
formulary managers on the other. Although there are several
statutory exemptions and regulatory safe harbors protecting certain
common activities from prosecution, the exemptions and safe harbors
are drawn narrowly, and practices that involve remuneration
intended to induce prescribing, purchasing or recommending may be
subject to scrutiny if they do not qualify for an exemption or safe
harbor. Our practices may not, in all cases, meet all of the
criteria for safe harbor protection from anti-kickback liability.
Moreover, recent health care reform legislation has strengthened
these laws. For example, the Health Care Reform Law, among other
things, amends the intent requirement of the U.S. Anti-Kickback
Statute and criminal health care fraud statutes; a person or entity
no longer needs to have actual knowledge of this statute or
specific intent to violate it. In addition, the Health Care Reform
Law provides that the government may assert that a claim including
items or services resulting from a violation of the U.S.
Anti-Kickback Statute constitutes a false or fraudulent claim for
purposes of the U.S. False Claims Act. Federal false claims laws
prohibit any person from knowingly presenting, or causing to be
presented, a false claim for payment to the federal government or
knowingly making, or causing to be made, a false statement to get a
false claim paid.
Over
the past few years, several pharmaceutical and other healthcare
companies have been prosecuted under these laws for a variety of
alleged promotional and marketing activities, such as: allegedly
providing free trips, free goods, sham consulting fees and grants
and other monetary benefits to prescribers; reporting to pricing
services inflated average wholesale prices that were then used by
federal programs to set reimbursement rates; engaging in off-label
promotion that caused claims to be submitted to Medicare or
Medicaid for non-covered, off-label uses; and submitting inflated
best price information to the Medicaid Rebate Program to reduce
liability for Medicaid rebates. Most states also have statutes or
regulations similar to the U.S. Anti-Kickback Statute and the U.S.
False Claims Act, which apply to items and services reimbursed
under Medicaid and other state programs, or, in several states,
apply regardless of the payor. Sanctions under these federal and
state laws may include substantial civil monetary penalties,
exclusion of a manufacturer’s products from reimbursement
under government programs, substantial criminal fines and
imprisonment.
18
We are, and will be, completely dependent on third parties to
manufacture REM-001 Therapy, and our commercialization of REM-001
Therapy could be halted, delayed or made less profitable if those
third parties fail to obtain manufacturing approval from the FDA or
comparable foreign regulatory authorities, fail to provide us with
sufficient quantities of REM-001or REM-001 Therapy device
components or fail to do so at acceptable quality levels or
prices.
We do
not currently have, nor do we plan to acquire, the capability or
infrastructure to manufacture the active pharmaceutical ingredient
(“API”) in REM-001, as well as the other related device
components, for use in our clinical trials, if required, or for
commercial product, if any. In addition, we do not have the
capability to produce REM-001 or the other related device
components for commercial distribution. As a result, we will be
obligated to rely on contract manufacturers for any clinical trials
we commence, and if and when REM-001 Therapy is approved we will
also rely on contract manufacturers for commercialization. We have
not entered into an agreement with any contract manufacturers for
clinical or commercial supply of REM-001 or the device components
and we may not be able to engage a contract manufacturer for such
supply on favorable terms to us, or at all.
The
facilities used by any future contract manufacturers, if any, to
manufacture REM-001 and its related device components must be
approved by the FDA pursuant to inspections that will be conducted
after we submit our NDA to the FDA. We do not control the
manufacturing process of, and are completely dependent on, any
future contract manufacturing partners, if any, for compliance with
cGMPs for manufacture of both active drug substances and finished
drug and device components. These cGMP regulations cover all
aspects of the manufacturing, testing, quality control and record
keeping relating to the drug and device components of REM-001
Therapy. If any future contract manufacturers cannot successfully
manufacture material that conforms to our specifications and the
strict regulatory requirements of the FDA or others, they will not
be able to secure and/or maintain regulatory approval for their
manufacturing facilities. If the FDA or a comparable foreign
regulatory authority does not approve these facilities for the
manufacture of the elements of REM-001 Therapy or if it withdraws
any such approval in the future, we may need to find alternative
manufacturing facilities, which would significantly impact our
ability to develop, obtain regulatory approval for or market
REM-001 Therapy, if approved.
Our
future contract manufacturers, if any, will be subject to ongoing
periodic unannounced inspections by the FDA and corresponding state
and foreign agencies for compliance with cGMPs and similar
regulatory requirements. We will not have control over our contract
manufacturers’ compliance with these regulations and
standards. Failure by any of our contract manufacturers to comply
with applicable regulations could result in sanctions being imposed
on us, including fines, injunctions, civil penalties, failure to
grant approval to market REM-001 Therapy, delays, suspensions or
withdrawals of approvals, operating restrictions and criminal
prosecutions, any of which could significantly and adversely affect
our business. In addition, we will not have control over the
ability of our future contract manufacturers, if any, to maintain
adequate quality control, quality assurance and qualified
personnel. Failure by our future contract manufacturers, if any, to
comply with or maintain any of these standards could adversely
affect our ability to develop, obtain regulatory approval for or
market REM-001 Therapy.
If, for
any reason, these third parties are unable or unwilling to perform,
we may not be able to terminate our agreements, if any, with them,
and we may not be able to locate alternative manufacturers or
formulators or enter into favorable agreements with them and we
cannot be certain that any such third parties will have the
manufacturing capacity to meet future requirements. If these future
manufacturers or any alternate manufacturer of finished drug
product experiences any significant difficulties in its respective
manufacturing processes for our API or finished REM-001 product and
the other device components or should cease doing business with us,
we could experience significant interruptions in the supply of
REM-001 and the other device components or may not be able to
create a supply of these items at all. Were we to encounter
manufacturing issues, our ability to produce a sufficient supply of
REM-001 or the other device components might be negatively
affected. Our inability to coordinate the efforts of our future
third party manufacturing partners, if any, or the lack of capacity
available at our third party manufacturing partners, could impair
our ability to supply REM-001 and the other device components at
required levels. Because of the significant regulatory requirements
that we would need to satisfy in order to qualify a new bulk or
finished product manufacturer, if we face these or other
difficulties with our future manufacturing partners, if any, we
could experience significant interruptions in the supply of REM-001
and the device components if we decided to transfer the manufacture
of them to one or more alternative manufacturers in an effort to
deal with the difficulties.
19
Any
manufacturing problem or the loss of a contract manufacturer could
be disruptive to our operations and result in lost sales.
Additionally, we will rely on third parties to supply the raw
materials needed to manufacture our potential products. Any
reliance on suppliers may involve several risks, including a
potential inability to obtain critical materials and reduced
control over production costs, delivery schedules, reliability and
quality. Any unanticipated disruption to a future contract
manufacturer caused by problems at suppliers could delay shipment
of REM-001 Therapy, increase our cost of goods sold and result in
lost sales.
We
cannot guarantee that our future manufacturing and supply partners,
if any, will be able to reduce the costs of commercial scale
manufacturing of REM-001 and the other device components over time.
If the commercial-scale manufacturing costs of these items are
higher than expected, these costs may significantly impact our
operating results. In order to reduce costs, our future
manufacturing partners, if any, will need to develop and implement
process improvements. However, in order to do so, such partners
will need, from time to time, to notify or make submissions with
regulatory authorities, and the improvements may be subject to
approval by such regulatory authorities. We cannot be sure that
these necessary approvals will be granted in a timely fashion or at
all. We also cannot guarantee that we will be able to enhance and
optimize output in our commercial manufacturing process. If we
cannot enhance and optimize output, we may not be able to reduce
our costs over time.
In the event the FDA requires us to conduct further clinical trials
with REM-001 Therapy, which we believe will be
required for approval in any indication, we expect that we will
rely on third parties to conduct clinical trials for REM-001
Therapy. If these third parties do not successfully carry out their
contractual duties or meet expected deadlines, we may not be able
to obtain regulatory approval for or commercialize REM-001 Therapy
and our business would be substantially harmed.
We
expect to enter into agreements with clinical CROs to conduct and
manage our clinical programs including contracting with clinical
sites to perform our clinical studies. We plan to rely heavily on
these parties for execution of clinical studies for REM-001 Therapy
and will control only certain aspects of their activities.
Nevertheless, we will be responsible for ensuring that each of our
studies is conducted in accordance with the applicable protocol,
legal, regulatory and scientific standards, and our reliance on
clinical CROs and clinical sites will not relieve us of our
regulatory responsibilities. We and our clinical CROs will be
required to comply with cGCPs, which are regulations and guidelines
enforced by the FDA, the Competent Authorities of the Member States
of the European Economic Area and comparable foreign regulatory
authorities for any products in clinical development. The FDA
enforces these cGCP regulations through periodic inspections of
trial sponsors, principal investigators and trial sites. If we or
our clinical CROs fail to comply with applicable cGCPs, the
clinical data generated in our clinical trials may be deemed
unreliable and the FDA or comparable foreign regulatory authorities
may require us to perform additional clinical trials before
approving our marketing applications. We cannot assure you that,
upon inspection, the FDA will determine that any of our clinical
trials comply with cGCPs. In addition, our clinical trials must be
conducted with products produced under cGMP regulations and will
require a large number of test subjects. Our failure or the failure
of our clinical CROs or clinical sites to comply with these
regulations may require us to repeat clinical trials, which would
delay the regulatory approval process and could also subject us to
enforcement action up to and including civil and criminal
penalties.
Although we intend
to design the clinical trials for REM-001 Therapy in consultation
with CROs, we expect that the clinical CROs will manage all of the
clinical trials conducted at contracted clinical sites. As a
result, many important aspects of our product development programs
would be outside of our direct control. In addition, the clinical
CROs and clinical sites may not perform all of their obligations
under arrangements with us or in compliance with regulatory
requirements. If the clinical CROs or clinical sites do not perform
clinical trials in a satisfactory manner, breach their obligations
to us or fail to comply with regulatory requirements, the
development and commercialization of REM-001 Therapy for the
subject indication may be delayed or our development program
materially and irreversibly harmed. We cannot control the amount
and timing of resources these clinical CROs and clinical sites will
devote to our program or REM-001 Therapy. If we are unable to rely
on clinical data collected by our CROs, we could be required to
repeat, extend the duration of, or increase the size of our
clinical trials, which could significantly delay commercialization
and require significantly greater expenditures.
20
If any
of our relationships with these clinical CROs or clinical sites
terminate, we may not be able to enter into arrangements with
alternative clinical CROs or clinical sites. If clinical CROs do
not successfully carry out their contractual duties or obligations
or meet expected deadlines, if they need to be replaced or if the
quality or accuracy of the clinical data they obtain is compromised
due to the failure to adhere to our clinical protocols, regulatory
requirements or for other reasons, any such clinical trials may be
extended, delayed or terminated, and we may not be able to obtain
regulatory approval for or successfully commercialize REM-001
Therapy. As a result, our financial results and the commercial
prospects for REM-001 Therapy would be harmed, our costs could
increase and our ability to generate revenue could be
delayed.
Any termination or suspension of, or delays in the commencement or
completion of, any necessary studies of REM-001 Therapy for any
indications could result in increased costs to us, delay or limit
our ability to generate revenue and adversely affect our commercial
prospects.
The
commencement and completion of clinical studies can be delayed for
a number of reasons, including delays related to:
|
|
●
|
the FDA
failing to grant permission to proceed and placing the clinical
study on hold;
|
|
|
●
|
subjects
failing to enroll or remain in our trials at the rate we
expect;
|
|
|
●
|
a
facility manufacturing any REM-001 Therapy component being ordered
by the FDA or other government or regulatory authorities to
temporarily or permanently shut down due to violations of cGMP
requirements or other applicable requirements, or
cross-contaminations of product candidates in the manufacturing
process;
|
|
|
●
|
any
changes to our manufacturing process that may be necessary or
desired;
|
|
|
●
|
subjects
choosing an alternative treatment for the indications for which we
are developing REM-001 Therapy, or participating in competing
clinical studies;
|
|
|
●
|
subjects
experiencing severe or unexpected drug-related adverse
effects;
|
|
|
●
|
reports
from clinical testing on similar technologies and products raising
safety and/or efficacy concerns;
|
|
|
●
|
third-party
clinical investigators losing their license or permits necessary to
perform our clinical trials, not performing our clinical trials on
our anticipated schedule or employing methods consistent with the
clinical trial protocol, cGMP requirements, or other third parties
not performing data collection and analysis in a timely or accurate
manner;
|
|
|
●
|
inspections
of clinical study sites by the FDA or IRBs finding regulatory
violations that require us to undertake corrective action, result
in suspension or termination of one or more sites or the imposition
of a clinical hold on the entire study, or that prohibit us from
using some or all of the data in support of our marketing
applications;
|
|
|
●
|
third-party
contractors becoming debarred or suspended or otherwise penalized
by the FDA or other government or regulatory authorities for
violations of regulatory requirements, in which case we may need to
find a substitute contractor, and we may not be able to use some or
any of the data produced by such contractors in support of our
marketing applications;
|
21
|
|
●
|
one or
more IRBs refusing to approve, suspending or terminating the study
at an investigational site, precluding enrollment of additional
subjects, or withdrawing its approval of the trial; reaching
agreement on acceptable terms with prospective clinical CROs and
clinical trial sites, the terms of which can be subject to
extensive negotiation and may vary significantly among different
clinical CROs and trial sites;
|
|
|
●
|
deviations
of the clinical sites from trial protocols or dropping out of a
trial;
|
|
|
●
|
adding
new clinical trial sites;
|
|
|
●
|
the
inability of the clinical CRO to execute any clinical trials for
any reason; and
|
|
|
●
|
government
or regulatory delays or “clinical holds” requiring
suspension or termination of a trial.
|
Product
development costs for REM-001 Therapy will increase if we have
delays in testing or approval or if we need to perform more or
larger clinical studies than planned. Additionally, changes in
regulatory requirements and policies may occur and we may need to
amend study protocols to reflect these changes. Amendments may
require us to resubmit our study protocols to the FDA and IRBs for
reexamination, which may impact the costs, timing or successful
completion of that study. If we experience delays in completion of,
or if we, the FDA or other regulatory authorities, an IRB, or other
reviewing entities, or any of our clinical study sites suspend or
terminate any of our clinical studies of REM-001 Therapy, its
commercial prospects may be materially harmed and our ability to
generate product revenues will be delayed. Any delays in completing
our clinical trials will increase our costs, slow down our
development and approval process and jeopardize our ability to
commence product sales and generate revenues. Any of these
occurrences may harm our business, financial condition and
prospects significantly. In addition, many of the factors that
cause, or lead to, termination or suspension of, or a delay in the
commencement or completion of, clinical studies may also ultimately
lead to the denial of regulatory approval of REM-001 Therapy. In
addition, if one or more clinical studies are delayed, our
competitors may be able to bring products to market before we do,
and the commercial viability of REM-001 Therapy could be
significantly reduced.
Clinical drug development involves a lengthy and expensive process
with an uncertain outcome, and results of earlier studies and
trials may not be predictive of future trial results.
Clinical testing is
expensive and can take many years to complete, and its outcome is
inherently uncertain. Failure can occur at any time during the
clinical trial process. The results of pre-clinical studies and
early clinical trials may not be predictive of the results of
later-stage clinical trials. The results of any clinical trials
conducted by us may not replicate those of earlier clinical trials
conducted by Miravant. We cannot assure you that the FDA will view
the results of the four Phase 2 and/or Phase 3 clinical trials for
the treatment of CMBC using REM-001 Therapy, conducted by Miravant,
with support from certain corporate partners, between February 1996
and January 1999 (collectively, the “Miravant CMBC
Trials”) as positively as we do or that any future trials of
REM-001 Therapy in any indication will achieve positive results.
Product candidates in later stages of clinical trials may fail to
show the desired safety and efficacy traits despite having
progressed through pre-clinical studies and initial clinical
trials. A number of companies in the biopharmaceutical industry
have suffered significant setbacks in advanced clinical trials due
to lack of efficacy or adverse safety profiles, notwithstanding
promising results in earlier trials. Any future clinical trial
results for REM-001 Therapy may not be successful.
In
addition, a number of factors could contribute to a lack of
favorable safety and efficacy results for REM-001 Therapy. For
example, such trials could result in increased variability due to
varying site characteristics, such as local standards of care,
differences in evaluation period and surgical technique and varying
patient characteristics including demographic factors and health
status.
We have not yet applied for and may not obtain or maintain the
benefits associated with orphan drug designation.
As a
key part of our business strategy, we intend to request orphan drug
designation in the United States for REM-001 for the treatment of
patients with CMBC. There is no guarantee that the FDA will grant
any future application for orphan drug designation for REM-001,
which would make us ineligible for the additional exclusivity and
other benefits of orphan drug designation.
22
Under
the Orphan Drug Act, the FDA may grant orphan drug designation to a
drug intended to treat a rare disease or condition, which is
generally a disease or condition that affects fewer than 200,000
individuals in the United States or for which there is no
reasonable expectation that the cost of developing and making a
drug available in the Unites States for this type of disease or
condition will be recovered from sales of the product. Orphan drug
designation must be requested before submitting an NDA. After the
FDA grants orphan drug designation, the identity of the therapeutic
agent and its potential orphan use are disclosed publicly by the
FDA. Orphan product designation does not convey any advantage in or
shorten the duration of regulatory review and approval
process.
If a
product that has orphan designation subsequently receives the first
FDA approval for the disease or condition for which it has such
designation, the product is entitled to orphan drug exclusivity,
which means the FDA may not approve any other applications to
market the same drug for the same indication for seven years,
except in limited circumstances, such as (i) the drug’s
orphan designation is revoked; (ii) its marketing approval is
withdrawn; (iii) the orphan exclusivity holder consents to the
approval of another applicant’s product; (iv) the orphan
exclusivity holder is unable to assure the availability of a
sufficient quantity of the drug; or (v) a showing of clinical
superiority to the product with orphan exclusivity by a competitor
product. If a drug designated as an orphan product receives
marketing approval for an indication broader than what is
designated, it may not be entitled to orphan drug exclusivity. In
addition to the potential period of exclusivity, orphan designation
makes a company eligible for tax credits for clinical research
expenses and potential exemption from the FDA application user fee.
There can be no assurance that we will receive orphan drug
designation for REM-001 in the indication of CMBC or in any other
indication, if we elect to seek such applications.
Although we intend to pursue expedited regulatory approval pathways
for REM-001 Therapy, it may not qualify for expedited development
or, if it does qualify for expedited development, it may not
actually lead to a faster development or regulatory review or
approval process.
Although we believe
there may be an opportunity to accelerate the development of
REM-001 Therapy through one or more of the FDA’s expedited
programs, such as special protocol assessment, fast track,
breakthrough therapy, accelerated approval or priority review, and
we intend to pursue one or more of these expedited programs, we
cannot be assured that REM-001 Therapy or any other product
candidates that we may develop will qualify for such
programs.
If we
apply for any expedited program for REM-001 Therapy, the FDA may
determine that REM-001 Therapy, our proposed target indication or
other aspects of our clinical development plans do not qualify for
such expedited program. Even if we are successful in obtaining a
designation or access to any expedited program, we may not
experience faster development timelines or achieve faster review or
approval compared to conventional FDA procedures. Access to an
expedited program may also be withdrawn by the FDA if it believes
that the designation is no longer supported by data from our
clinical development program. Additionally, qualification for any
expedited review procedure does not ensure that we will ultimately
obtain regulatory approval for REM-001 Therapy or any other product
candidate that we may develop.
The
Federal Food, Drug and Cosmetic Act directs the FDA to meet with
sponsors, pursuant to a sponsor’s written request, for the
purpose of reaching agreement on the design and size of clinical
trials intended to form the primary basis of an efficacy claim in
an NDA. If an agreement is reached, the FDA will reduce the
agreement to writing and make it part of the administrative record.
This agreement is called a special protocol assessment
(“SPA”). Our plan is to work closely with the FDA in
developing any Phase 3 trial we may run in CMBC to help ensure it
receives a SPA classification, but there is no assurance that this
classification will be granted. Furthermore, even if we receive an
SPA classification and we complete a Phase 3 trial that meets the
criteria defined in the clinical plan, the FDA may later add
additional requirements for approval or it may decline to grant
marketing approval altogether.
23
Third-party coverage and reimbursement and health care cost
containment initiatives and treatment guidelines may constrain our
future revenues.
Our
ability to successfully market REM-001 Therapy will depend in part
on the level of reimbursement that government health administration
authorities, private health coverage insurers and other
organizations provide for the cost of our products and related
treatments. Countries in which pharmaceutical or medical device
products are sold through reimbursement schemes under national
health insurance programs frequently require that manufacturers and
sellers of pharmaceutical products or medical devices obtain
governmental approval of initial prices and any subsequent price
increases. In certain countries, including the United States,
government-funded and private medical care plans can exert
significant indirect pressure on prices. We may not be able to sell
REM-001 Therapy profitably if adequate prices are not approved or
coverage and reimbursement is unavailable or limited in scope.
Increasingly, third-party payors attempt to contain health care
costs in ways that are likely to impact our development of products
including:
|
|
●
|
failing
to approve or challenging the prices charged for health care
products;
|
|
|
●
|
introducing
reimportation schemes from lower priced jurisdictions;
|
|
|
●
|
limiting
both coverage and the amount of reimbursement for new therapeutic
products;
|
|
|
●
|
denying
or limiting coverage for products that are approved by the
regulatory agencies but are considered to be experimental or
investigational by third-party payors; and
|
|
|
●
|
refusing
to provide coverage when an approved product is used in a way that
has not received regulatory marketing approval.
|
Risks Relating to Our Intellectual Property Rights
It is difficult and costly to protect our intellectual property
rights, and we cannot ensure the protection of these
rights.
Our
commercial success may depend, in part, on obtaining and
maintaining patent protection for our technologies, products and
processes, successfully defending these patents against third-party
challenges and successfully enforcing these patents against third
party competitors. The patent positions of biopharmaceutical
companies can be highly uncertain and involve complex legal,
scientific and factual questions for which important legal
principles remain unresolved. Changes in either the patent laws or
in interpretations of patent laws may diminish the value of our
intellectual property. Accordingly, we cannot predict the breadth
of claims that may be allowable or enforceable in our patents
(including patents owned by us). We currently have eight issued
patents and two patents we do not believe are relevant to our
current product development objectives. The existing patent and
related technologies may be challenged, invalidated or circumvented
by third parties and might not protect us against competitors with
similar products or technologies.
The
degree of future protection for our proprietary rights is
uncertain, because legal means afford only limited protection and
may not adequately protect our rights, permit us to gain or keep
our competitive advantage, or provide us with any competitive
advantage at all. For example, others have filed, and in the future
are likely to file, patent applications covering products and
technologies that are similar, identical or competitive to REM-001
Therapy, or important to our business. We cannot be certain that
any patent application owned by a third party will not have
priority over patent applications filed by us, or that we will not
be involved in interference, opposition or invalidity proceedings
before United States or foreign patent offices.
We also
rely on trade secrets to protect technology, especially in cases
when we believe patent protection is not appropriate or obtainable.
However, trade secrets are difficult to protect. While we require
employees, academic collaborators, consultants and other
contractors to enter into confidentiality agreements, we may not be
able to adequately protect our trade secrets or other proprietary
or licensed information. Typically, research collaborators and
scientific advisors have rights to publish data and information in
which we may have rights. If we cannot maintain the confidentiality
of our proprietary technology and other confidential information,
our ability to receive patent protection and our ability to protect
valuable information owned by us may be imperiled. Enforcing a
claim that a third-party entity illegally obtained and is using any
of our trade secrets is expensive and time consuming, and the
outcome is unpredictable. In addition, courts are sometimes less
willing to protect trade secrets than patents. Moreover, our
competitors may independently develop equivalent knowledge, methods
and know-how.
24
If we
fail to obtain or maintain patent protection or trade secret
protection for REM-001 Therapy or our technologies, third parties
may be able to use our proprietary information, which could impair
our ability to compete in the market and adversely affect our
ability to generate revenues and attain profitability.
We may
also rely on the trademarks we may develop to distinguish our
products from the products of our competitors. We cannot guarantee
that any trademark applications filed by us or our business
partners will be approved. Third parties may also oppose such
trademark applications, or otherwise challenge our use of the
trademarks. In the event that the trademarks we use are
successfully challenged, we could be forced to rebrand our
products, which could result in loss of brand recognition, and
could require us to devote resources to advertising and marketing
new brands. Further, we cannot provide assurance that competitors
will not infringe the trademarks we use, or that we will have
adequate resources to enforce these trademarks.
REM-001 Therapy may infringe the intellectual property rights of
others, which could increase our costs and delay or prevent our
development and commercialization efforts.
Our
success depends in part on avoiding infringement of the proprietary
technologies of others. The biopharmaceutical industry has been
characterized by frequent litigation regarding patent and other
intellectual property rights. Identification of third party patent
rights that may be relevant to our proprietary technology is
difficult because patent searching is imperfect due to differences
in terminology among patents, incomplete databases and the
difficulty in assessing the meaning of patent claims. Additionally,
because patent applications are maintained in secrecy until the
application is published, we may be unaware of third-party patents
that may be infringed by commercialization of REM-001 Therapy or
any future product candidate. There may be certain issued patents
and patent applications claiming subject matter that we may be
required to license in order to research, develop or commercialize
REM-001 Therapy, and we do not know if such patents and patent
applications would be available to license on commercially
reasonable terms, or at all. Any claims of patent infringement
asserted by third parties would be time-consuming and
may:
|
|
●
|
result
in costly litigation;
|
|
|
●
|
divert
the time and attention of our technical personnel and
management;
|
|
|
●
|
prevent
us from commercializing a product until the asserted patent expires
or is held finally invalid or not infringed in a court of
law;
|
|
|
●
|
require
us to cease or modify our use of the technology and/or develop
non-infringing technology; or
|
|
|
●
|
require
us to enter into royalty or licensing agreements.
|
We
acquired all of the rights, assets and technology related to
REM-001 Therapy from St. Cloud Investments, LLC (“St.
Cloud”), a creditor of Miravant, who acquired the same
through foreclosure, and we believe that St. Cloud owned all of
such rights prior to our acquisition. Although no third party has
asserted a claim of infringement or other claim against us, others
may hold or claim to hold proprietary or other rights that could
prevent REM-001 Therapy from being developed or marketed. Any legal
action against us claiming damages and seeking to enjoin commercial
activities relating to REM-001 Therapy or our processes could
subject us to potential liability for damages and require us to
obtain a license to continue to manufacture or market REM-001
Therapy or any future product candidates. We cannot predict whether
we would prevail in any such actions or that any license required
under any of these patents would be made available on commercially
acceptable terms, if at all. In addition, we cannot be sure that we
could redesign REM-001 Therapy or any future product candidates or
processes to avoid infringement, if necessary. Accordingly, an
adverse determination in a judicial or administrative proceeding,
or the failure to obtain necessary licenses, could prevent us from
developing and commercializing REM-001 Therapy or a future product
candidate, which could harm our business, financial condition and
operating results.
25
A
number of companies, including several major pharmaceutical
companies, have conducted research on PDT therapies which resulted
in the filing of many patent applications that could be interpreted
as pertaining to our planned applications. If we were to challenge
the validity of these or any issued United States patent in
court, we would need to overcome a statutory presumption of
validity that attaches to every issued United States patent.
This means that, in order to prevail, we would have to present
clear and convincing evidence as to the invalidity of the
patent’s claims. If we were to challenge the validity of
these or any issued United States patent in an administrative
trial before the Patent Trial and Appeal Board in the United
States Patent and Trademark Office, we would have to prove
that the claims are unpatentable by a preponderance of the
evidence. There is no assurance that a jury and/or court would find
in our favor on questions of infringement, validity or
enforceability.
We do not hold any patents covering the DD series laser light
source or the ML2-0400 light delivery device.
The DD
series laser light source and the ML2-0400 light delivery devices
are not currently covered by any patents; we have no patents
pending, and do not currently intend to seek patent protection for
these devices. As a result, competitors may be able to offer and
sell products or drug delivery technology, as the case may be,
using the same technology as our laser light source and/or light
delivery devices, so long as these competitors do not infringe any
other valid patents that we or third parties hold.
While
we plan to protect our proprietary information related to the DD
series laser light source and the ML2-0400 light delivery devices
as trade secrets through certain agreements with our employees,
consultants, agents and other organizations to which we disclose
our proprietary information, we cannot give any assurance that
these agreements will provide effective protection for our
proprietary information in the event of unauthorized use or
disclosure of such information. If other laser light sources or
light delivery devices are approved and marketed, we will be unable
to prevent them from competing with REM-001 Therapy in the
marketplace using a different drug molecule that is not encompassed
by any of our owned or licensed patents. We expect that the
presence of one or more competing products would reduce our market
share and could negatively impact price levels and third party
reimbursement policies for REM-001 Therapy, any of which would
materially affect our business.
We may be subject to claims that we have wrongfully hired an
employee from a competitor or that we or our employees have
wrongfully used or disclosed alleged confidential information or
trade secrets of their former employers.
As is
commonplace in our industry, we employ and plan to employ
individuals who were previously employed at other pharmaceutical
companies, including our competitors or potential competitors.
Although no claims against us are currently pending, we may be
subject in the future to claims that our employees or prospective
employees are subject to a continuing obligation to their former
employers (such as non-competition or non-solicitation obligations)
or claims that our employees or we have inadvertently or otherwise
used or disclosed trade secrets or other proprietary information of
their former employers. Litigation may be necessary to defend
against these claims. Even if we are successful in defending
against these claims, litigation could result in substantial costs
and be a distraction to management.
General Company-Related Risks
We will need to grow the size of our organization, and we may
experience difficulties in managing this growth.
We
currently have five full-time employees and one
part-time employee. As our development and commercialization plans
and strategies develop, we will need to expand the size of our
employee and consultant base for managerial, operational, sales,
marketing, financial and other resources. Future growth would
impose significant added responsibilities on members of management,
including the need to identify, recruit, maintain, motivate and
integrate additional employees. In addition, our management may
have to divert a disproportionate amount of its attention away from
our day-to-day activities and devote a substantial amount of time
to managing these growth activities. Our future financial
performance and our ability to commercialize REM-001 Therapy and
any other future product candidates and our ability to compete
effectively will depend, in part, on our ability to effectively
manage our future growth.
26
Our
success will depend in part on our ability to manage our operations
as we advance our product candidate through clinical trials and to
expand our development or regulatory capabilities or contract with
third parties to provide these capabilities for us. Failure to
achieve any of these goals could have a material adverse effect on
our business, financial condition or results of
operations.
Future capital raises may dilute our existing stockholders’
ownership and/or have other adverse effects on our
operations.
If we
raise additional capital by issuing equity securities, our existing
stockholders’ percentage ownership will be reduced and these
stockholders may experience substantial dilution. We may also issue
equity securities that provide for rights, preferences and
privileges senior to those of our common stock. If we raise
additional funds by issuing debt securities, these debt securities
would have rights senior to those of our common stock and the terms
of the debt securities issued could impose significant restrictions
on our operations, including liens on our assets. If we raise
additional funds through collaborations and licensing arrangements,
we may be required to relinquish some rights to our technologies or
candidate products, or to grant licenses on terms that are not
favorable to us.
If we are not successful in attracting and retaining highly
qualified personnel, we may not be able to successfully implement
our business strategy. In addition, the loss of the services of
certain key employees, including Frank Pilkiewicz, PhD, our
President and CEO, Jane Maida, our Chief Financial Officer and
Steven Rychnovsky, PhD, our Vice President of Operations and
Product Development, would adversely impact our business
prospects.
Our
ability to compete in the highly competitive pharmaceuticals
industry depends in large part upon our ability to attract highly
qualified managerial, scientific and medical personnel. In order to
induce valuable employees to remain with us, we intend to provide
employees with stock options that vest over time. The value to
employees of stock options that vest over time will be
significantly affected by movements in our stock price that we will
not be able to control and may at any time be insufficient to
counteract more lucrative offers from other companies.
Our
management team has expertise in many different aspects of drug
development and commercialization. However, we will need to hire
additional personnel as we further develop REM-001 Therapy.
Competition for skilled personnel in our market is intense and
competition for experienced scientists may limit our ability to
hire and retain highly qualified personnel on acceptable terms.
Despite our efforts to retain valuable employees, members of our
management, scientific and medical teams may terminate their
employment with us on short notice. In connection with the 2016
merger transaction (the “Merger”), we entered into
employment agreements with our Chief Executive Officer and Vice
President of Operations and Product Development. We entered into an
employment agreement with Jane Maida, our Chief Financial Officer,
in February 2017. While these arrangements provide for a term of
employment, our President and Chief Executive Officer, Chief
Financial Officer and Vice President of Operations and Product
Development still may leave our employment at any time, with
appropriate notice, which generally would be 60 days notice. The
loss of the services of any of our executive officers or other key
employees could potentially harm our business, operating results or
financial condition. In particular, we believe that the loss of the
services of Frank Pilkiewicz PhD, our President and Chief Executive
Officer, Jane Maida, our Chief Financial Officer and Steven
Rychnovsky, PhD, our Vice President of Operations and Product
Development, would have a material adverse effect on our business.
Our success also depends on our ability to continue to attract,
retain and motivate highly skilled junior, mid-level, and senior
managers as well as junior, mid-level, and senior scientific and
medical personnel.
Other
biopharmaceutical companies with which we compete for qualified
personnel have greater financial and other resources, different
risk profiles, and a longer history in the industry than we do.
They also may provide more diverse opportunities and better chances
for career advancement. Some of these characteristics may be more
appealing to high-quality candidates than what we have to offer. If
we are unable to continue to attract and retain high-quality
personnel, the rate and success at which we can develop and
commercialize product candidates would be limited.
27
If product liability lawsuits are brought against us, we may incur
substantial liabilities and may be required to limit
commercialization of REM-001 Therapy.
We face
a potential risk of product liability as a result of the clinical
testing of REM-001 Therapy and will face an even greater risk if we
commercialize REM-001 Therapy or any other future product. For
example, we may be sued if any product we develop, including
REM-001 Therapy, or any materials that we use in our products
allegedly causes injury or is found to be otherwise unsuitable
during product testing, manufacturing, marketing or sale. Any such
product liability claims may include allegations of defects in
manufacturing, defects in design, a failure to warn of dangers
inherent in the product, negligence, strict liability and a breach
of warranties. Claims could also be asserted under state consumer
protection acts. If we cannot successfully defend ourselves against
product liability claims, we may incur substantial liabilities or
be required to limit commercialization of REM-001 Therapy. Even
successful defense would require significant financial and
management resources. Regardless of the merits or eventual outcome,
liability claims may result in:
|
|
●
|
decreased
demand for REM-001 Therapy or any future products that we may
develop;
|
|
|
●
|
injury
to our reputation;
|
|
|
●
|
withdrawal
of clinical trial participants;
|
|
|
●
|
costs
to defend the related litigation;
|
|
|
●
|
a
diversion of management’s time and our
resources;
|
|
|
●
|
substantial
monetary awards to trial participants or patients;
|
|
|
●
|
product
recalls, withdrawals or labeling, marketing or promotional
restrictions;
|
|
|
●
|
the
inability to commercialize REM-001 Therapy; and
|
|
|
●
|
a
decline in the value of our stock.
|
Our
inability to obtain and retain sufficient product liability
insurance at an acceptable cost to protect against potential
product liability claims could prevent or inhibit the
commercialization of products we develop. We intend to obtain
product liability insurance covering our clinical trials. Although
we will maintain such insurance, any claim that may be brought
against us could result in a court judgment or settlement in an
amount that is not covered, in whole or in part, by our insurance
or that is in excess of the limits of our insurance coverage. Our
insurance policies will also have various exclusions, and we may be
subject to a product liability claim for which we have no coverage.
We may have to pay any amounts awarded by a court or negotiated in
a settlement that exceed our coverage limitations or that are not
covered by our insurance, and we may not have, or be able to
obtain, sufficient capital to pay such amounts.
We may acquire businesses or products, or form strategic alliances,
in the future, and we may not realize the benefits of such
acquisitions.
We may
acquire additional businesses or products, form strategic alliances
or create joint ventures with third parties that we believe will
complement or augment our existing business. If we acquire
businesses with promising markets or technologies, we may not be
able to realize the benefit of acquiring such businesses if we are
unable to successfully integrate them with our existing operations
and company culture. We may encounter numerous difficulties in
developing, manufacturing and marketing any new products resulting
from a strategic alliance or acquisition that delay or prevent us
from realizing their expected benefits or enhancing our business.
We cannot assure you that, following any such acquisition, we will
achieve the expected synergies to justify the
transaction.
28
Risks Related to our Common Stock
We have engaged in transactions with the Placement Agent and its
related parties that could present conflicts of
interest.
There
have been transactions between us and related parties of the
Placement Agent for our private placement, that could present
potential conflicts of interest. These transactions include, but
are not limited to, the engagement of the Placement Agent, the
issuance of founder shares of Adgero Biopharmaceuticals Holdings,
Inc. (“Holdings”) to affiliates of the Placement Agent,
the Placement Agent’s right to select a designee to our Board
of Directors and the payment of compensation in the form of cash,
warrants, and a non-accountable expense allowance to the Placement
Agent for securities sold in the private placement. Further, an
affiliate of the Placement Agent, purchased a bridge note in an
aggregate principal amount of $250,000 in 2015, which he
subsequently converted into the securities offered in the 2016
Private Placement. Each of these and other present and future
financial commitments or agreements could constitute potential
conflicts of interest.
Our majority stockholders will control our company for the
foreseeable future, including the outcome of matters requiring
stockholder approval.
Our
officers, directors, founders and affiliates of the Placement Agent
collectively own approximately 46.2% of our outstanding shares of
common stock. In addition, the stockholders of Adgero
Biopharmaceuticals, Inc. (“Adgero”) prior to the Merger
(the “Adgero Stockholders”), and the stockholders of
Holdings prior to the Merger (the “Holdings
Stockholders”), entered into a voting agreement in connection
with the Merger, whereby they have agreed to vote in favor of
nominees for directors selected by the parties to the voting
agreement as described herein. As a result, such entities and
individuals will have the ability, acting together, to control the
election of our directors and the outcome of corporate actions
requiring stockholder approval, such as: (i) a merger or a sale of
our company, (ii) a sale of all or substantially all of our assets,
and (iii) amendments to our certificate of incorporation and
bylaws. This concentration of voting power and control could have a
significant effect in delaying, deferring or preventing an action
that might otherwise be beneficial to our other stockholders and be
disadvantageous to our stockholders with interests different from
those entities and individuals. Certain of these individuals also
have significant control over our business, policies and affairs as
officers or directors of our company. Therefore, you should not
invest in reliance on your ability to have any control over our
company.
An investment in our company should be considered
illiquid.
An
investment in our company requires a long-term commitment, with no
certainty of return. Because we do not plan to become an SEC
reporting company by the traditional means of conducting an initial
public offering of our common stock, we may be unable to establish
a liquid market for our common stock. Moreover, we do not expect
security analysts of brokerage firms to provide coverage of our
company in the near future. In addition, investment banks may be
less likely to agree to underwrite primary or secondary offerings
on behalf of our company or its stockholders in the future than
they would if we were to become a public reporting company by means
of an initial public offering of common stock. If all or any of the
foregoing risks occur, it would have a material adverse effect on
our security holders.
29
If this resale registration statement is declared effective, we
will become subject to the reporting requirements of federal
securities laws, which will be expensive and require use of
resources that might otherwise go to develop our
business.
If we
become a public reporting company and, accordingly, subject to the
information and reporting requirements of the Exchange Act of 1934,
as amended (the “Exchange Act”), and other federal
securities laws, the costs of preparing and filing periodic and
other reports, proxy statements and other information with the
Securities and Exchange Commission, and furnishing audited reports
to stockholders, our expenses will be significantly higher than
they would be if we remained privately held. The cost of being a
public company will divert resources that might otherwise have been
used to develop our business, which could have a material adverse
effect on our company.
No public market for our common stock currently exists, and an
active trading market may not develop or be sustained.
As we
are in our early stages, an investment in our company will require
a long-term commitment, with no certainty of return. There is no
public market for our common stock, and even if we become a
publicly-listed company, of which no assurances can be given, we
cannot predict whether an active market for our common stock will
ever develop in the future. In the absence of an active trading
market:
|
|
●
|
investors
may have difficulty buying and selling or obtaining market
quotations;
|
|
|
●
|
market
visibility for shares of our common stock may be limited;
and
|
|
|
●
|
a lack
of visibility for shares of our common stock may have a depressive
effect on the market price for shares of our common
stock.
|
Assuming we can
find market makers to establish quotations for our common stock in
the future, we expect that our common stock will be quoted on the
Over-the-Counter, or OTC, Bulletin Board and/or OTCQB Market
operated by OTC Markets Group, Inc. (together, the
“OTCBB/OTCQB”). These markets are relatively
unorganized, inter-dealer, over-the-counter markets that provide
significantly less liquidity than NASDAQ or the NYSE MKT (formerly
known as the NYSE AMEX). No assurances can be given that our common
stock, even if quoted on such markets, will ever trade on such
markets, much less a senior market like NASDAQ or NYSE MKT. In this
event, there would be a highly illiquid market for our common stock
and you may be unable to dispose of your common stock at desirable
prices or at all. Moreover, there is a risk that our common stock
could be delisted from the OTCBB/OTCQB, in which case it might be
listed on the so called “Pink Sheets”, which is even
more illiquid than the OTCBB/OTCQB.
The
lack of an active market impairs your ability to sell your shares
at the time you wish to sell them or at a price that you consider
reasonable. The lack of an active market may also reduce the fair
market value of your shares. An inactive market may also impair our
ability to raise capital to continue to fund operations by selling
shares and may impair our ability to acquire additional
intellectual property assets by using our shares as
consideration.
30
We may not qualify for OTCBB/OTCQB inclusion, and therefore you may
be unable to sell your shares.
We
believe that, at some time following the effectiveness of this
registration statement of which this prospectus forms a part our
common stock will become eligible for quotation on the OTCBB/OTCQB.
No assurances can be given, however, that this eligibility will be
granted. OTCBB/OTCQB eligible securities include securities not
listed on a registered national securities exchange in the United
States and that are also required to file reports pursuant to
Section 13 or 15(d) of the Securities Act of 1933, as amended (the
“Securities Act”), and require that the company be
current in its periodic securities reporting
obligations.
Among
other matters, in order for our common stock to become OTCBB/OTCQB
eligible, a broker/dealer member of the Financial Industry
Regulatory Authority (“FINRA”), must file a Form 211
with FINRA and commit to make a market in our securities once the
Form 211 is approved by FINRA. As of the date of this offering
memorandum, a Form 211 has not been filed with FINRA by any
broker/dealer. If for any reason our common stock does not become
eligible for quotation on the OTCBB/OTCQB or a public trading
market does not develop, purchasers of shares of our common stock
may have difficulty selling their shares should they desire to do
so. If we are unable to satisfy the requirements for quotation on
the OTCBB/OTCQB, any quotation of our common stock would be
conducted in the “Pink Sheets” market. As a result, a
purchaser of our common stock may find it more difficult to dispose
of, or to obtain accurate quotations as to the price of their
shares.
Even if
our securities become listed on a registered national securities
exchange such as the NYSE MKT or NASDAQ, we may not be able to
continue to meet such exchange’s minimum listing requirements
or those of any other national exchange. In addition, a liquid
market may not develop for our common stock. If we are unable to
maintain listing on such a registered national securities exchange
or if a liquid market for our common stock does not develop, our
common stock may remain thinly traded. The listing rules of
registered national securities exchanges require listing issuers to
comply with certain standards in order to remain listed on such
exchanges. Our stockholders may suffer a material adverse effect
if, for any reason, we should fail to maintain compliance with
these listing standards and such exchange should delist our
securities from trading on its exchange and we are unable to obtain
listing on another national securities exchange.
Even if our common stock becomes publicly-traded and an active
trading market develops, the market price our common stock may be
significantly volatile.
Even if
our securities become publicly-traded and even if an active market
for our common stock develops, of which no assurances can be given,
the market price for our common stock may be volatile and subject
to wide fluctuations in response to factors including the
following:
|
|
●
|
actual
or anticipated fluctuations in our quarterly or annual operating
results;
|
|
|
●
|
changes
in financial or operational estimates or projections;
|
|
|
●
|
conditions
in markets generally;
|
|
|
●
|
changes
in the economic performance or market valuations of companies
similar to ours; and
|
|
|
●
|
general
economic or political conditions in the United States or
elsewhere.
|
In
particular, the market prices of biotechnology companies like ours
have been highly volatile due to factors, including, but not
limited to:
|
|
●
|
any
delay or failure to conduct a clinical trial for our product or
receive approval from the FDA and other regulatory
agents;
|
31
|
|
●
|
developments
or disputes concerning our product’s intellectual property
rights;
|
|
|
●
|
our or
our competitors’ technological innovations;
|
|
|
●
|
changes
in market valuations of similar companies;
|
|
|
●
|
announcements
by us or our competitors of significant contracts, acquisitions,
strategic partnerships, joint ventures, capital commitments, new
technologies, or patents;
|
|
|
●
|
failure
to complete significant transactions or collaborate with vendors in
manufacturing our product; and
|
|
|
●
|
proposals
for legislation that would place restrictions on the price of
medical therapies.
|
The
securities market has from time to time experienced significant
price and volume fluctuations that are not related to the operating
performance of particular companies. These market fluctuations may
also materially and adversely affect the market price of shares of
our common stock.
The registration for resale of a significant portion of our
outstanding shares of common stock in this registration statement
may have a depressive effect on our stock price.
We are
registering for resale 1,718,408 shares of our common stock plus
1,749,272 shares of common stock underlying outstanding warrants.
If our existing stockholders sell substantial amounts of our common
stock in the public market, or if the public perceives that such
sales could occur, this could have an adverse impact on the market
price of our common stock, even if there is no relationship between
such sales and the performance of our business.
Our common stock may be considered a “penny stock,” and
thereby be subject to additional sale and trading regulations that
may make it more difficult to sell. Further, if our common stock is
considered a “penny stock,” the protection provided by
the federal securities laws relating to forward looking statements
would not apply to us.
The SEC
has adopted rules that regulate broker-dealer practices in
connection with transactions in penny stocks. Penny stocks are
generally equity securities with a price of less than $5.00 (other
than securities registered on certain national securities exchanges
or authorized for quotation on certain automated quotation systems,
provided that current price and volume information with respect to
transactions in such securities is provided by the exchange or
system). The OTCBB/OTCQB does not meet such requirements and if the
price of our common stock is less than $5.00, our common stock may
be deemed a penny stock. The penny stock rules require a
broker-dealer, prior to a transaction in a penny stock not
otherwise exempt from those rules, to deliver a standardized risk
disclosure document containing specified information. In addition,
the penny stock rules require that prior to effecting any
transaction in a penny stock not otherwise exempt from those rules,
a broker-dealer must make a special written determination that the
penny stock is a suitable investment for the purchaser and receive
(i) the purchaser’s written acknowledgment of the receipt of
a risk disclosure statement; (ii) a written agreement to
transactions involving penny stocks; and (iii) a signed and dated
copy of a written suitability statement. These disclosure
requirements may have the effect of reducing the trading activity
in the secondary market for our common stock, and therefore stock
holders may have difficulty selling their shares once our common
stock is publicly traded.
Although federal
securities laws provide a safe harbor for forward-looking
statements made by a public company that files reports under the
federal securities laws, this safe harbor is not available to
issuers of penny stocks. As a result, we may not have the benefit
of this safe harbor protection in the event of any legal action
based upon a claim that the material provided by us contained a
material misstatement of fact or was misleading in any material
respect because of our failure to include any statements necessary
to make the statements not misleading. Such an action could hurt
our financial condition.
32
FINRA sales practice requirements may also limit your ability to
buy and sell our common stock, which could depress the price of our
shares.
FINRA
rules require broker-dealers to have reasonable grounds for
believing that an investment is suitable for a customer before
recommending that investment to the customer. Prior to recommending
speculative low-priced securities to their non-institutional
customers, broker-dealers must make reasonable efforts to obtain
information about the customer’s financial status, tax status
and investment objectives, among other things. Under
interpretations of these rules, FINRA believes that there is a high
probability such speculative low-priced securities will not be
suitable for at least some customers. Thus, FINRA requirements make
it more difficult for broker-dealers to recommend that their
customers buy our common stock, which may limit your ability to buy
and sell our shares once publicly traded, have an adverse effect on
the market for our shares, and thereby depress our share
price.
You may face significant restrictions on the resale of your shares
due to state “blue sky” laws.
Each
state has its own securities laws, often called “blue
sky” laws, which (1) limit sales of securities to a
state’s residents unless the securities are registered in
that state or qualify for an exemption from registration, and
(2) govern the reporting requirements for broker-dealers doing
business directly or indirectly in the state. Before a security is
sold in a state, there must be a registration in place to cover the
transaction, or it must be exempt from registration. The applicable
broker-dealer must also be registered in that state.
We do
not know whether our securities will be registered or exempt from
registration under the laws of any state. A determination regarding
registration will be made by those broker-dealers, if any, who
agree to serve as market makers for our common stock. We have not
yet applied to have our securities registered in any state and will
not do so until we receive expressions of interest from investors
resident in specific states in the future. There may be significant
state blue sky law restrictions on the ability of investors to
sell, and on purchasers to buy, our securities. You should
therefore consider the resale market for our common stock to be
limited, as you may be unable to resell your shares without the
significant expense of state registration or
qualification.
Shareholders will experience dilution by exercises of outstanding
warrants and options.
There are currently 2,830,733 shares of common
stock issuable upon the exercise of our outstanding warrants, as
well as options to purchase an aggregate of up to
1,283,937 shares of our common stock pursuant to
employment agreements with our officers, and issuances to our
directors and strategic advisory board members, each at an exercise
price of $5.00. The exercise of such options and warrants will
result in dilution of your investment. As a result of this
dilution, you may receive significantly less than the full purchase
price you paid for securities of the Company in the event of
liquidation.
We are an “emerging growth company,” and will be able
take advantage of reduced disclosure requirements applicable to
“emerging growth companies,” which could make our
common stock less attractive to investors.
We are
an “emerging growth company,” as defined in the
Jumpstart Our Business Startups Act of 2012 (the “JOBS
Act”) and, for as long as we continue to be an
“emerging growth company,” we intend to take advantage
of certain exemptions from various reporting requirements
applicable to other public companies but not to “emerging
growth companies,” including, but not limited to, not being
required to comply with the auditor attestation requirements of
Section 404 of the Sarbanes-Oxley Act, reduced disclosure
obligations regarding executive compensation in our periodic
reports and proxy statements, and exemptions from the requirements
of holding a nonbinding advisory vote on executive compensation and
stockholder approval of any golden parachute payments not
previously approved. We could be an “emerging growth
company” for up to five years, or until the earliest of (i)
the last day of the first fiscal year in which our annual gross
revenues exceed $1 billion, (ii) the date that we become a
“large accelerated filer” as defined in Rule 12b-2
under the Exchange Act, which would occur if the market value of
our common stock that is held by non-affiliates exceeds $700
million as of the last business day of our most recently completed
second fiscal quarter, or (iii) the date on which we have issued
more than $1 billion in non-convertible debt during the preceding
three year period.
33
We
intend to take advantage of these reporting exemptions described
above until we are no longer an “emerging growth
company.” Under the JOBS Act, “emerging growth
companies” can also delay adopting new or revised accounting
standards until such time as those standards apply to private
companies. We have irrevocably elected not to avail ourselves of
this exemption from new or revised accounting standards and,
therefore, we will be subject to the same new or revised accounting
standards as other public companies that are not “emerging
growth companies.”
We
cannot predict if investors will find our common stock less
attractive if we choose to rely on these exemptions. If some
investors find our common stock less attractive as a result of any
choices to reduce future disclosure, there may be a less active
trading market for our common stock and our stock price may be more
volatile.
We will incur significantly increased costs and devote substantial
management time as a result of operating as a public company
particularly after we are no longer an “emerging growth
company.”
As a
public company, we will incur significant legal, accounting and
other expenses that we did not incur as a private company. For
example, we will be required to comply with certain of the
requirements of the Sarbanes-Oxley Act and the Dodd-Frank Wall
Street Reform and Consumer Protection Act, as well as rules and
regulations subsequently implemented by the SEC, including the
establishment and maintenance of effective disclosure and financial
controls and changes in corporate governance practices. We expect
that compliance with these requirements will increase our legal and
financial compliance costs and will make some activities more time
consuming and costly. In addition, we expect that our management
and other personnel will need to divert attention from operational
and other business matters to devote substantial time to these
public company requirements. In particular, we expect to incur
significant expenses and devote substantial management effort
toward ensuring compliance with the requirements of Section 404 of
the Sarbanes-Oxley Act. In addition, after we no longer qualify as
an “emerging growth company,” as defined under the JOBS
ACT we expect to incur additional management time and cost to
comply with the more stringent reporting requirements applicable to
companies that are deemed accelerated filers or large accelerated
filers, including complying with the auditor attestation
requirements of Section 404 of the Sarbanes-Oxley Act. We are just
beginning the process of compiling the system and processing
documentation needed to comply with such requirements. We may not
be able to complete our evaluation, testing and any required
remediation in a timely fashion. In that regard, we currently do
not have an internal audit function, and we will need to hire or
contract for additional accounting and financial staff with
appropriate public company experience and technical accounting
knowledge.
We
cannot predict or estimate the amount of additional costs we may
incur as a result of becoming a public company or the timing of
such costs.
There may be limitations on the effectiveness of our internal
controls, and a failure of our control systems to prevent error or
fraud may materially harm our company.
Proper
systems of internal controls over financial accounting and
disclosure controls and procedures are critical to the operation of
a public company. As we are a start-up company, we only have
five full-time employees and one part-time employee,
which results in a lack of segregation of duties and are at the
very early stages of establishing, and we may be unable to
effectively establish such systems, especially in light of the fact
that we expect to operate as a publicly reporting company. This
would leave us without the ability to reliably assimilate and
compile financial information about our company and significantly
impair our ability to prevent error and detect fraud, all of which
would have a negative impact on our company from many
perspectives.
Moreover, we do not
expect that disclosure controls or internal control over financial
reporting, even if established, will prevent all error and all
fraud. A control system, no matter how well designed and operated,
can provide only reasonable, not absolute, assurance that the
control system’s objectives will be met. Further, the design
of a control system must reflect the fact that there are resource
constraints and the benefits of controls must be considered
relative to their costs. Because of the inherent limitations in all
control systems, no evaluation of controls can provide absolute
assurance that all control issues and instances of fraud, if any,
have been detected. Failure of our control systems to prevent error
or fraud could materially adversely impact us.
34
We have a material weakness in our internal control over
financial reporting. In addition, because of our status as an
emerging growth company, our independent registered public
accountants are not required to provide an attestation report as to
our internal control over financial reporting for the foreseeable
future.
We may
be required, pursuant to Section 404 of the Sarbanes-Oxley Act, to
furnish a report by our management on, among other things, the
effectiveness of our internal control over financial reporting for
the first fiscal year beginning after the effective date of the
registration statement of which this prospectus is a part. This
assessment will need to include disclosure of any material
weaknesses identified by our management in our internal control
over financial reporting, as well as a statement that our
independent registered public accounting firm has issued an opinion
on our internal control over financial reporting. We are in the
very early stages of the costly and challenging process of
compiling the system and processing documentation necessary to
perform the evaluation needed to comply with Section
404.
A
“material weakness” is a deficiency, or a combination
of deficiencies, in internal control over financial reporting such
that there is a reasonable possibility that a material misstatement
of our annual or interim financial statements will not be prevented
or detected on a timely basis. In connection with the audit of our
financial statements for the year ended December 31, 2015, we
determined that our disclosure controls and procedures were
ineffective, and that there was a material weakness in our internal
controls over financial reporting, due to insufficient segregation
of duties in our finance and accounting function because of our
limited personnel. We currently have five full-time
employees and one part-time employee. This resulted in not ensuring
appropriate segregation of duties between incompatible functions,
and made it more difficult to ensure that financial information is
adequately analyzed and reviewed on a timely basis to detect
misstatements. These above deficiencies represent a material
weakness in our internal control over financial reporting given
that they result in a reasonable possibility that a material
misstatement to the annual or interim financial statements would
not have been prevented or detected.
We have
begun evaluating and implementing additional procedures to improve
the segregation of duties, however, because of our limited
resources we cannot assure that these or other measures will fully
remediate the deficiencies or material weakness described above in
a timely manner. We intend to address the weakness identified above
by increasing the oversight and review procedures of the board of
directors with regard to financial reporting, financial processes
and procedures and internal control procedures; and when funding is
available, hiring additional finance and accounting personnel.
Nevertheless, there can be no assurances that we will have enough
financial resources to remedy our current material weaknesses and
significant deficiencies. If we are unable to remediate the
material weakness, or otherwise maintain effective internal control
over financial reporting, we may not be able to report our
financial results accurately, prevent fraud or file our periodic
reports in a timely manner. We cannot assure you that we have
identified all of our existing significant deficiencies and
material weaknesses, or that we will not in the future have
additional significant deficiencies or material
weaknesses.
Our
independent registered public accounting firm will not be required
to formally attest to the effectiveness of our internal control
over financial reporting pursuant to Section 404 until the later of
the year following our first annual report required to be filed
with the SEC, or the date we are no longer an “emerging
growth company” as defined in the recently enacted JOBS Act,
if we take advantage (as we expect to do) of the exemptions
contained in the JOBS Act. We will remain an “emerging growth
company” for up to five years, although if the market value
of our common stock that is held by non-affiliates exceeds $700
million as of any June 30th before that time, we would cease to be
an “emerging growth company” as of the following
December 31st. At such time, our independent registered public
accounting firm may issue a report that is adverse in the event it
is not satisfied with the level at which our controls are
documented, designed or operating. Our remediation efforts may not
enable us to avoid a material weakness in our internal control over
financial reporting in the future.
Any of
the foregoing occurrences, should they come to pass, could
negatively impact the public perception of our company, which could
have a negative impact on our stock price.
We do not currently intend to pay dividends on our common stock in
the foreseeable future, and consequently, your ability to achieve a
return on your investment will depend on appreciation in the price
of our common stock.
35
We have
never declared or paid cash dividends on our common stock and do
not anticipate paying any cash dividends to holders of our common
stock in the foreseeable future. Consequently, investors must rely
on sales of their common stock after price appreciation, which may
never occur, as the only way to realize any future gains on their
investments. There is no guarantee that shares of our common stock
will appreciate in value or even maintain the price at which our
stockholders have purchased their shares.
Upon dissolution of our company, you may not recoup all or any
portion of your investment.
In the
event of a liquidation, dissolution or winding-up of our company,
whether voluntary or involuntary, the proceeds and/or assets of our
company remaining after giving effect to such transaction, and the
payment of all of our debts and liabilities and distributions
required to be made to holders of any outstanding preferred stock
will then be distributed to the stockholders of common stock on a
pro rata basis. There can be no assurance that we will have
available assets to pay to the holders of common stock, or any
amounts, upon such a liquidation, dissolution or winding-up of our
Company. In this event, you could lose some or all of your
investment.
Our ability to use our net operating loss carryforwards and certain
other tax attributes may be limited.
As a
result of the Merger, our ability to utilize our federal net
operating loss, carryforwards and federal tax credit may be limited
under Sections 382 of the Internal Revenue Code of 1986, as
amended. The limitations apply if an “ownership
change,” as defined by Section 382, occurs. Generally, an
ownership change occurs if the percentage of the value of the stock
that is owned by one or more direct or indirect “five percent
shareholders” increases by more than 50 percentage points
over their lowest ownership percentage at any time during the
applicable testing period (typically three years). In addition,
future changes in our stock ownership, which may be outside of our
control, may trigger an “ownership change” and,
consequently, Section 382 limitations. As a result, if we earn net
taxable income, our ability to use our pre-change net operating
loss carryforwards and other tax attributes to offset United States
federal taxable income may be subject to limitations, which could
potentially result in increased future tax liability to
us.
Our certificate of incorporation allows for our board to create new
series of preferred stock without further approval by our
stockholders, which could adversely affect the rights of the
holders of our common stock.
Our
board of directors has the authority to fix and determine the
relative rights and preferences of preferred stock. We anticipate
that our board of directors will have the authority to issue up to
10 million shares of our preferred stock without further
stockholder approval. As a result, our board of directors could
authorize the issuance of a series of preferred stock that would
grant to holders the preferred right to our assets upon liquidation
and the right to receive dividend payments before dividends are
distributed to the holders of common stock. In addition, our board
of directors could authorize the issuance of a series of preferred
stock that has greater voting power than our common stock or that
is convertible into our common stock, which could decrease the
relative voting power of our common stock or result in dilution to
our existing stockholders.
Our certificate of incorporation, as amended, designates the Court
of Chancery of the State of Delaware as the sole and exclusive
forum for certain types of actions and proceedings that may be
initiated by our stockholders, which could discourage lawsuits
against us, and our directors and officers.
Our
certificate of incorporation, as amended, provides that unless we
consent in writing to the selection of an alternative forum, the
Court of Chancery of the State of Delaware will be the sole and
exclusive forum for (i) any derivative action or proceeding brought
on our behalf; (ii) any action asserting a claim of breach of a
fiduciary duty; (iii) any action asserting a claim against us, or
any of our officers or directors, arising pursuant to, or a claim
against us, or any of our officers or directors, with respect to
the interpretation or application of any provision of the Delaware
General Corporation Law, our certificate of incorporation, as
amended, or our bylaws; or (iv) any action asserting a claim
governed by the internal affairs doctrine. However, if the Court of
Chancery of the State of Delaware dismisses any such action for
lack of subject matter jurisdiction, the action may be brought in
another state court sitting in the State of Delaware.
Although
our certificate of incorporation, as amended, includes this
exclusive forum provision, it is possible that a court could rule
that this provision is inapplicable or unenforceable. This
exclusive forum provision may limit the ability of our stockholders
to bring a claim in a judicial forum that such stockholders find
favorable for the disputes listed above, which may discourage such
lawsuits against us and our officers and directors. Alternatively,
if a court were to find this exclusive forum provision inapplicable
to, or unenforceable in respect of, one or more of the specified
types of actions or proceedings described above, we may incur
additional costs associated with resolving such matters in other
jurisdictions, which could negatively affect our business, results
of operations and financial condition.
IN
ADDITION TO THE ABOVE RISKS, BUSINESSES ARE OFTEN SUBJECT TO RISKS
NOT FORESEEN OR FULLY APPRECIATED BY OUR MANAGEMENT. IN REVIEWING
THIS PROSPECTUS, POTENTIAL INVESTORS SHOULD KEEP IN MIND THAT THERE
MAY BE OTHER POSSIBLE RISKS THAT COULD BE IMPORTANT.
36
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This
prospectus contains, and our officers and representatives may from
time to time make, “forward-looking statements,” which
include information relating to future events, future financial
performance, financial projections, strategies, expectations,
competitive environment and regulation. Words such as
“may,” “should,” “could,”
“would,” “predicts,”
“potential,” “continue,”
“expects,” “anticipates,”
“future,” “intends,” “plans,”
“believes,” “estimates,”
“goal,” “seek,” “project,”
“strategy,” “likely,” and similar
expressions, as well as statements in future tense, identify
forward-looking statements. Forward-looking statements are neither
historical facts, nor should they be read as a guarantee of future
performance or results and may not be accurate indications of when
such performance or results will be achieved. Forward-looking
statements are based on information we have when those statements
are made or management’s good faith belief as of that time
with respect to future events, and are subject to risks and
uncertainties that could cause actual performance or results to
differ materially from those expressed in or suggested by the
forward-looking statements. Important factors that could cause such
differences include, but are not limited to:
|
|
●
|
our
limited operating history;
|
|
|
●
|
our
history of operating losses in each year since inception and
expectation that we will continue to incur operating losses for the
foreseeable future;
|
|
|
●
|
our
current and future capital requirements to support our development
and commercialization efforts for our lead product candidate, the
REM-001 Therapy product, consisting of three parts, the laser light
source, the light delivery device and the drug REM-001
(collectively, the “REM-001 Therapy”) and our ability
to satisfy our capital needs;
|
|
|
●
|
our
dependence on the REM-001 Therapy, our sole product candidate,
which is still in development,
|
|
|
●
|
our
ability to obtain approval from the Food and Drug Administration
(the “FDA”) or other regulatory agents in different
jurisdictions for REM-001 Therapy;
|
|
|
●
|
our
lack of a sales and marketing organization and our ability to
commercialize REM-001 Therapy, if we obtain regulatory
approval;
|
|
|
●
|
our
dependence on third-parties to manufacture REM-001 and the related
device components;
|
|
|
●
|
our
ability to maintain or protect the validity of our patents and
other intellectual property;
|
|
|
●
|
our
ability to retain key executives and medical and science
personnel;
|
|
|
●
|
our
ability to internally develop new inventions and intellectual
property;
|
|
|
●
|
interpretations
of current laws and the passages of future laws;
|
|
|
●
|
acceptance
of our business model by investors;
|
|
|
●
|
the
accuracy of our estimates regarding expenses and capital
requirements; and
|
|
|
●
|
our
ability to adequately support growth.
|
The
foregoing does not represent an exhaustive list of matters that may
be covered by the forward-looking statements contained herein or
risk factors that we are faced with that may cause our actual
results to differ from those anticipate in our forward-looking
statements. Please see “Risk Factors” for additional
risks which could adversely impact our business and financial
performance.
37
Moreover, new risks
regularly emerge and it is not possible for our management to
predict or articulate all risks we face, nor can we assess the
impact of all risks on our business or the extent to which any
risk, or combination of risks, may cause actual results to differ
from those contained in any forward-looking statements. All
forward-looking statements included in this prospectus are based on
information available to us on the date of this prospectus. Except
to the extent required by applicable laws or rules, we undertake no
obligation to publicly update or revise any forward-looking
statement, whether written or oral, that may be made from time to
time, whether as a result of new information, future events or
otherwise. All subsequent written and oral forward-looking
statements attributable to us or persons acting on our behalf are
expressly qualified in their entirety by the cautionary statements
contained above and throughout this prospectus.
IN
ADDITION TO THE ABOVE RISKS, BUSINESSES ARE OFTEN SUBJECT TO RISKS
NOT FORESEEN OR FULLY APPRECIATED BY OUR MANAGEMENT. IN REVIEWING
THIS PROSPECTUS, POTENTIAL INVESTORS SHOULD KEEP IN MIND THAT THERE
MAY BE OTHER POSSIBLE RISKS THAT COULD BE IMPORTANT.
38
USE OF PROCEEDS
We will
not receive any of the proceeds from the sale of the common stock
by the selling stockholders named in this prospectus. All proceeds
from the sale of the common stock will be paid directly to the
selling stockholders.
We
would, however, receive proceeds upon the exercise of the warrants
held by the selling stockholders which, if such warrants are
exercised in full (and assuming no “cashless” exercise
features are utilized), would be approximately $8,746,360.
Proceeds, if any, received from the exercise of such warrants will
be used for working capital and general corporate purposes. No
assurances can be given that any of such warrants will be
exercised.
39
DIVIDEND POLICY
We have
never paid any cash dividends on our common stock. We anticipate
that we will retain funds and future earnings to support operations
and to finance the growth and development of our business.
Therefore, we do not expect to pay cash dividends in the
foreseeable future following this offering. Any future
determination to pay dividends will be at the discretion of our
board of directors and will depend on our financial condition,
results of operations, capital requirements and other factors that
our board of directors deems relevant. In addition, the terms of
any future debt or credit financings may preclude us from paying
dividends.
40
MANAGEMENT’S DISCUSSION AND
ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Prospective investors should read the following discussion and
analysis of our financial condition and results of operations
together with our consolidated financial statements and the related
notes and other financial information included elsewhere in this
prospectus. Some of the information contained in this discussion
and analysis or set forth elsewhere in this prospectus, including
information with respect to our plans and strategy for our
business, includes forward-looking statements that involve risks
and uncertainties. See “Cautionary Note Regarding
Forward-Looking Statements.” You should review the
“Risk Factors” section of this prospectus for a
discussion of important factors that could cause actual results to
differ materially from the results described in or implied by the
forward-looking statements contained in the following discussion
and analysis.
Overview
We are
a biopharmaceutical company, focused on the development of
photodynamic therapy (“PDT”) for the treatment of rare,
unmet medical needs. PDT is a treatment that uses light sensitive
compounds, or photosensitizers, that, when exposed to specific
wavelengths of light, act as a catalyst to produce a form of oxygen
that induces local tumor cell death. Our lead product candidate,
the REM-001 Therapy product, consists of three parts, the laser
light source, the light delivery device and the drug REM-001
(collectively, the “REM-001 Therapy”). REM-001 is a
second generation photosensitizer drug that has undergone late
stage clinical development and which we believe possesses multiple
advantages over earlier generation PDT compounds. Our lead
indication is unresectable cutaneous metastatic breast cancer
(“CMBC”), a disease that may strike individuals with
advanced breast cancer and for which effective treatment options
are limited. In four Phase 2 and/or Phase 3 clinical trials in CMBC
patients, primarily targeting patients who had previously received
chemotherapy and failed radiation therapy, our REM-001 Therapy was
able to reduce or eliminate a substantial number of the treated
CMBC tumors. Specifically, our analysis of the data collected from
these trials indicates that in approximately 80% of evaluable tumor
sites treated with REM-001 Therapy, there was a complete response,
meaning that follow-up clinical assessments indicated no visible
evidence of the tumor remaining. We believe clinical data indicates
that REM-001 Therapy holds promise as a treatment to
locally eliminate or slow the growth of treated cutaneous cancerous
tumors in this difficult-to-treat patient population.
In
2012, we acquired certain assets and regulatory filings, including
REM-001 Therapy developed by Miravant Medical Technologies, and its
wholly-owned subsidiaries, a former public pharmaceutical and
research development company (collectively,
“Miravant”), and the associated technology, clinical
data and intellectual property, from a creditor of Miravant.
Between February 1996 and January 1999, Miravant, with support from
certain corporate partners, conducted the above-referenced four
Phase 2 and/or Phase 3 clinical trials for the treatment of CMBC
using REM-001 Therapy (collectively, the “Miravant CMBC
Trials”). The primary motivation behind our acquisition was
to secure the rights to the REM-001 Therapy and its associated
technology, proprietary processes and regulatory filings which have
already undergone substantial clinical development, which we
believe will help expedite the process of gaining regulatory
approval to market our REM-001 Therapy.
Our
initial product goal is to achieve marketing approval of REM-001
Therapy for the treatment of CMBC in the United States. We
conducted a first preliminary analysis of all existing REM-001
Therapy clinical trial data for CMBC, including data from the
Miravant CMBC Trials. We then conducted a more in-depth analysis
that was overseen by regulatory experts who have expertise in
interacting with the Food and Drug Administration (the
“FDA”). The experts
we engaged were either former FDA employees with directly related
experience in reviewing similar oncology treatments or individuals
who have provided senior regulatory guidance to major
pharmaceutical or medical device companies in situations that led
to regulatory approval. The results of this second more in-depth
analysis were consistent with our original analysis. As a result of
our review, we submitted questions to FDA under
a Type C format to review the technology and results
and determine the anticipated requirements for regulatory approval.
On March 3, 2017, we received FDA’s written response to
our questions. Based on that response, we believe our plans to
manufacture REM-001 by revising the prior quality standards to meet
the currently recommended regulatory standards will be acceptable.
FDA also indicated our plans for utilizing light delivery devices
that have been shown to be functionally equivalent to the devices
used by Miravant will be acceptable. FDA also recognized that CMBC
represents an unmet clinical need and provided guidance on a number
of clinical pharmacology parameters they would like us to measure
in our planned clinical trial. Based on FDA’s responses, we
plan to conduct a clinical trial in CMBC to test the safety and
efficacy of REM-001 Therapy for marketing approval and we are
planning further discussions with FDA regarding the specifics of
such a trial.
We also
believe REM-001 Therapy holds promise as a treatment for cutaneous
metastatic cancers other than CMBC as well as locally advanced
basal cell cancer such as often occurs in patients with Basal Cell
Nevus Syndrome and cutaneously recurrent basal cell
cancer.
41
Financial Operations Overview
We are
a development stage company and have not generated any revenues
from the sale of products. We have never been profitable and, from
inception through December 31, 2016, our losses from
operations have been approximately $3,383,000. Our net
loss for the years ended December 31, 2016 and 2015
were approximately $2,578,000 and $142,000. We expect
to incur significant expenses and increasing operating losses for
the foreseeable future. We expect our expenses to increase
significantly in connection with our ongoing activities to develop
and seek regulatory approval and commercialization of REM-001
Therapy. Furthermore, we expect to incur additional costs
associated with operating as a public company. Accordingly, we will
need additional financing to support our continuing operations. We
will seek to fund our operations through public or private equity
or debt financings or other sources, which may include
collaborations with third parties. Adequate additional financing
may not be available to us on acceptable terms, or at all. Our
failure to raise capital as and when needed would have a negative
impact on our financial condition and our ability to pursue our
business strategy. We will need to generate significant revenues to
achieve profitability, and we may never do so.
We
expect to continue to incur significant expenses and increasing
operating losses for at least the next several years. We expect our
expenses will increase substantially in 2017 and in
the future in connection with our ongoing activities, as
we:
|
|
●
|
conduct
clinical trials and obtain regulatory approval for the marketing of
REM0-001 Therapy;
|
|
|
●
|
contract
for the manufacture of our clinical drug product and establish a
commercial drug supply;
|
|
|
●
|
contract
for the manufacture of light delivery devices for clinical
trials;
|
|
|
●
|
attract
and retain an experienced management and advisory
team;
|
|
|
●
|
raise
sufficient funds in the capital market to effectuate our business
plan including clinical development, regulatory approval and
commercialization for REM-001 Therapy, and
|
|
|
●
|
operate
as a public company.
|
Critical Accounting Policies and Estimates
Our
consolidated financial statements have been prepared in accordance
with accounting principles generally accepted in the United States
of America. The preparation of these financial statements requires
management to make estimates and assumptions that affect the
reported amounts of assets and liabilities and the disclosure of
contingent assets and liabilities at the date of the financial
statements and the reported amounts of revenues and expenses during
the reporting period.
On an
ongoing basis, we evaluate our estimates and judgments for all
assets and liabilities, including those related to fair value
calculations for equity securities, assessing contingent
liabilities, establishing valuation allowances for deferred taxes,
and the recovery of deferred costs. We base our estimates and
judgments on historical experience, current economic and industry
conditions and on various other factors that are believed to be
reasonable under the circumstances. This forms the basis for making
judgments about the carrying values of assets and liabilities that
are not readily apparent from other sources. Actual results may
differ from these estimates under different assumptions or
conditions.
We
believe that full consideration has been given to all relevant
circumstances that we may be subject to, and the consolidated
financial statements accurately reflect our best estimate of the
results of operations, financial position and cash flows for the
periods presented.
Revenue
To
date, we have not generated any revenues from the sales of
products. We do not expect to generate revenue from product sales
unless and until we successfully complete development and obtain
regulatory approval for the marketing of REM-001 Therapy which we
expect will take a number of years and is subject to significant
uncertainty.
42
Research and Development
Research and
development expenses are incurred for the development of REM-001
Therapy and consist primarily of compensation costs (including
stock-based compensation), and payments to contract research and
development companies (consulting costs). To date, these costs are
related to regulatory and clinical consulting services and
acquiring and analyzing our pre-clinical and clinical data. These
costs are expected to increase significantly in the future as
REM-001 Therapy undergoes additional regulatory review and is
evaluated in clinical trials.
Total Other Income (Expense)
Other
income (expense) consists primarily of interest income we earn on
interest-bearing accounts and interest expense incurred on our
outstanding debt.
General and Administrative Expenses
General
and administrative expenses consist primarily of payroll and
professional services. Other general and administrative expenses
include accounting and legal services and expenses associated with
obtaining and maintaining patents. We anticipate that our general
and administrative expenses will increase significantly during
2017 and in the future as we increase our headcount to
support our continued research and development and the potential
commercialization of our product candidates. We also anticipate
increased expenses related to audit, legal, regulatory, and
tax-related services associated with maintaining compliance with
exchange listing and SEC requirements, director and officer
insurance premiums, and investor relations costs associated with
being a public company. Additionally, commencing in 2016, we began
to compensate our outside directors.
Stock-Based Compensation
We
account for equity awards in accordance with FASB ASC Topic No.
718, Compensation-Stock Compensation. Under FASB ASC Topic No. 718,
compensation expense related to stock-based payments to employees
and directors is recorded over the requisite service period based
on the grant date fair value of the awards. The grant date value of
performance-based equity awards is recognized over the service
period, so long as completion of the performance criteria is deemed
to be probable. Compensation previously recorded for unvested
equity awards that are forfeited is reversed upon forfeiture. The
Company uses the Black-Scholes option pricing model for determining
the estimated fair value for stock-based awards. The Black-Scholes
model requires the use of assumptions which determine the fair
value of stock-based awards, including the option’s expected
term and the price volatility of the underlying
stock.
Our accounting
policy for equity instruments issued to consultants and vendors in
exchange for goods and services follows the provisions of FASB ASC
Topic No. 505-50, Equity Based Payments to Non-Employees.
Accordingly, the measurement date for the fair value of the equity
instruments issued is determined at the earlier of (i) the date at
which a commitment for performance by the consultant or vendor is
reached or (ii) the date at which the consultant or vendor’s
performance is complete. In the case of equity instruments issued
to consultants, the fair value of the award is generally
re-measured on vesting dates and interim financial reporting dates
until the service period is complete. The fair value amount is then
recognized over the period during which services are required to be
provided in exchange for the award, usually the vesting period.
Performance-based equity awards without a performance commitment
are recognized upon completion of the performance criteria at their
then market value.
Emerging Growth Company Status
Under
Section 107(b) of the Jumpstart Our Business Startups Act of 2012,
emerging growth companies can delay adopting new or revised
accounting standards until such time as those standards apply to
private companies. We have irrevocably elected not to avail
ourselves of this exemption from new or revised accounting
standards and, therefore, we will be subject to the same new or
revised accounting standards as other public companies that are not
emerging growth companies.
Results of Operations
Comparison
of Year Ended December 31, 2016 to 2015
Research and Development. Research and
development expenses for the year ended December 31, 2016 totaled
$783,656, an increase of $769,886 over the $13,770 recorded for the
year ended December 31, 2015. The increase was primarily
attributable to the milestone payment of $100,000, $474,000 in
compensation costs (including stock-based compensation), and
$144,000 in payments to contract research and development companies
(consulting costs).
General and Administrative. General and
administrative expense for the year ended December 31, 2016 totaled
$1,787,013 an increase of $1,678,892 over the $108,121 recorded for
the year ended December 31, 2015. The increase was primarily
attributable to an increase of $879,000 for compensation costs
(including stock-based compensation), $499,000 for professional
fees, $84,000 for Board fees, $26,000 for marketing, $26,000 for
insurance and $194,000 for consulting costs.
43
Liquidity and Capital Resources
Since
inception, we have experienced negative cash flows from operations.
We have financed our operations primarily through sales of
equity-related securities, convertible notes and loans from
stockholders. At December 31, 2016, our accumulated deficit since
inception was approximately $3,383,000.
At December 31,
2016, we had total current assets of approximately $6,017,000 and
current liabilities of approximately $927,000 resulting in working
capital of approximately $5,091,000. At December 31, 2016, we had
total assets of approximately $6,060,000 and total liabilities of
approximately $927,000, resulting in a stockholders’ equity
of $5,133,000.
Net cash used in
operating activities for the year ended December 31, 2016 was
approximately $1,652,000, which includes cash used from a net loss
of approximately $2,578,000, reduced by approximately $475,000 of
non-cash stock-based compensation, plus cash used from an increase
in prepaid expenses totaling approximately $51,000 and
approximately $502,000 of cash provided from a net increase in
accounts payable and accrued expenses.
Net cash used in
investing activities for the year ended December 31, 2016 totaled
approximately $2,814,000, primarily related to the purchase of
certificates of deposit of $5,302,000, partially offset by
$2,500,000 related to proceeds from maturing certificates of
deposit.
Cash provided from
financing activities for the year ended December 31, 2016 totaled
approximately $7,403,000, primarily from the issuance of common
stock and warrants in the 2016 Private
Placement.
At December 31,
2016, we had no debt outstanding. All of the debt outstanding as of
December 31, 2015 was exchanged for shares of our common stock and
warrants on July 29, 2016 and August 3, 2016.
At
December 31, 2016, we had a cash and cash equivalents
balance of approximately $3,165,000 and certificates
of deposits of $2,802,000. We expect our current cash
on hand, our certificates of deposit and the $2,000,000 of gross
proceeds received subsequent to December 31, 2016
through the December 2016 Private Placement, to be sufficient to
meet our operating and capital requirements until at least March
2018. We will need to raise significant additional capital to fund
the clinical trials for REM-001 Therapy. The source, timing and
availability of any future financing will depend principally upon
market conditions, and, more specifically, on the progress of our
clinical development programs. Funding may not be available when
needed, at all, or on terms acceptable to us. Lack of necessary
funds may require us, among other things, to delay, scale back or
eliminate some or all of our planned clinical trials.
Off-Balance Sheet Arrangements
We did
not have during the periods presented, and we do not currently
have, any off-balance sheet arrangements, as defined under SEC
rules, such as relationships with unconsolidated entities or
financial partnerships, which are often referred to as structured
finance or special purpose entities, established for the purpose of
facilitating financing transactions that are not required to be
reflected on our balance sheets.
Quantitative and Qualitative Disclosures about Market
Risk
Our
exposure to market risk is limited to our cash and cash
equivalents, all of which have maturities of three months or less.
The primary objectives of our investment activities are to preserve
principal, provide liquidity and maximize income without
significantly increasing risk. Our primary exposure to market risk
is interest income sensitivity, which is affected by changes in the
general level of U.S. interest rates. However, because of the
short-term nature of the instruments in our portfolio, a sudden
change in market interest rates would not be expected to have a
material impact on our financial condition and/or results of
operation. We do not have any foreign currency or other derivative
financial instruments.
44
BUSINESS
Overview
We are a biopharmaceutical
company, focused on the development of photodynamic therapy
(“PDT”) for the treatment of rare, unmet medical needs.
PDT is a treatment that uses light sensitive compounds, or
photosensitizers, that, when exposed to specific wavelengths of
light, act as a catalyst to produce a form of oxygen that induces
local tumor cell death. Our lead product candidate, the REM-001
Therapy product, consists of three parts, the laser light source,
the light delivery device and the drug REM-001 (collectively, the
“REM-001 Therapy”). REM-001 is a second generation
photosensitizer drug that has undergone late stage clinical
development and which we believe possesses multiple advantages over
earlier generation PDT compounds. Our lead indication is
unresectable cutaneous metastatic breast cancer
(“CMBC”), a disease that may strike individuals with
advanced breast cancer and for which effective treatment options
are limited. In four Phase 2 and/or Phase 3 clinical trials in CMBC
patients, primarily targeting patients who had previously received
chemotherapy and failed radiation therapy, our REM-001 Therapy was
able to reduce or eliminate a substantial number of the treated
CMBC tumors. Specifically, our analysis of the data collected from
these trials indicates that in approximately 80% of evaluable tumor
sites treated with REM-001 Therapy, there was a complete response;
meaning that follow-up clinical assessments indicated no visible
evidence of the tumor remaining. We believe clinical data indicates
that REM-001 Therapy holds promise as a treatment to locally
eliminate or slow the growth of treated cutaneous cancerous tumors
in this difficult-to-treat patient population.
In
2012, we acquired certain assets and regulatory filings, including
REM-001 Therapy developed by Miravant Medical Technologies, and its
wholly-owned subsidiaries, a former public pharmaceutical and
research development company (collectively,
“Miravant”), and the associated technology, clinical
data and intellectual property, from a creditor of Miravant.
Between February 1996 and January 1999, Miravant, with support from
certain corporate partners, conducted the above-referenced four
Phase 2 and/or Phase 3 clinical trials for the treatment of CMBC
using REM-001 Therapy (collectively, the “Miravant CMBC
Trials”). The primary motivation behind our acquisition was
to secure the rights to the REM-001 Therapy and its associated
technology, proprietary processes and regulatory filings which have
already undergone substantial clinical development, which we
believe will help expedite the process of gaining regulatory
approval to market our REM-001 Therapy.
Numerous approaches have been utilized
to treat CMBC patients, including various forms of
chemotherapy, radiation therapy, surgical excision, hyperthermia,
cryotherapy, electro-chemotherapy, topical drugs, and
intra-lesional chemotherapy injections; however, for the most part,
we believe that these therapies are often inadequate given the
limited efficacy, toxicities and/or side effects of each. We
believe our REM-001 Therapy has several advantages for this
indication: it can be highly directed to the tumor site, has
minimal systemic effects or normal tissue toxicities, can be used
in conjunction with other therapies, and can be periodically
repeated. Our analysis of the data collected from the Miravant CMBC
Trials indicates that in approximately 80% of evaluable tumor sites
treated with REM-001 Therapy, there was a complete response,
meaning that follow-up clinical assessments indicated no visible
evidence of the tumor remaining. Based on these results, we believe
that REM-001 Therapy also holds promise as a treatment for other
cutaneous metastatic cancers and locally advanced basal cell
carcinomas.
Our
initial product goal is to achieve marketing approval of REM001
Therapy for the treatment of CMBC in the United States. We
conducted a first preliminary analysis of all existing REM001
Therapy clinical trial data for CMBC, including data from the
Miravant CMBC Trials. We then conducted a more in-depth analysis
that was overseen by regulatory experts who have expertise in
interacting with the Food and Drug Administration (the
“FDA”). The experts we engaged were either former FDA
employees with directly related experience in reviewing similar
oncology treatments or individuals who have provided senior
regulatory guidance to major pharmaceutical or medical device
companies in situations that led to regulatory approval. The
results of this second more in-depth analysis were consistent with
our original analysis. As a result of our review, we
submitted questions to FDA under a Type C format to
review the technology and results and determine the anticipated
requirements for regulatory approval. On March 3, 2017, we
received FDA’s written response to our questions. Based on
that response, we believe our plans to manufacture REM-001 by
revising the prior quality standards to meet the currently
recommended regulatory standards will be acceptable. FDA also
indicated our plans for utilizing light delivery devices that have
been shown to be functionally equivalent to the devices used by
Miravant will be acceptable. FDA also recognized that CMBC
represents an unmet clinical need and provided guidance on a number
of clinical pharmacology parameters they would like us to measure
in our planned clinical trial. Based on FDA’s responses, we
plan to conduct a clinical trial in CMBC to test the safety and
efficacy of REM-001 Therapy for marketing approval and we are
planning further discussions with FDA regarding the specifics of
such a trial.
We also
believe REM-001 Therapy holds promise as a treatment for cutaneous
metastatic cancers other than CMBC as well as locally advanced
basal cell cancer such as often occurs in patients with Basal Cell
Nevus Syndrome and cutaneously recurrent basal cell
cancer.
45
Our History and REM-001 Background
Adgero
Biopharmaceuticals, Inc. (“Adgero”) was formed in 2007
in the State of Delaware for the purposes of acquiring
pharmaceutical technology. In 2012, we acquired certain
assets and regulatory filings, including the REM-001 Therapy
developed by Miravant and the associated technology, clinical data
and intellectual property, from a creditor of Miravant.
Following the acquisition, the FDA acknowledged the ownership
transfer of Miravant’s oncology and ophthalmology
investigational new drug applications (“INDs”) for
REM-001 to Adgero.
Miravant initiated
commercial development of REM-001 and its associated device
components in the 1990s. This led to late stage clinical
trials in CMBC and also in an aspect of “wet”
age-related macular degeneration (“AMD”) a disease that
affects over 1.5 million people in the United States and is a cause
of vision loss in older individuals. Of these two
indications, AMD represented a much larger market, and in 1998, for
what we believe were business reasons, Miravant discontinued its
CMBC program and, together with or through its corporate partners,
ultimately focused its REM-001 development efforts on AMD.
That program remained Miravant’s main clinical focus until it
ran out of funding and ceased operations in 2006.
While
Miravant did not pursue the CMBC indication through to approval, it
did compile substantial clinical data in the four Miravant CMBC
Trials. The first two of these trials were Phase 2/3 trials
that treated 67 CMBC patients who, for the most part, previously
failed radiation therapy, and were then treated with REM-001
Therapy. Miravant compiled both safety and efficacy data for
these two studies. At the time Miravant discontinued its CMBC
program, REM-001 Therapy was also being tested in two additional
Phase 2 or 3 clinical trials that treated a total of 81
patients. Our review of internal Miravant records indicates
that data was collected in all four trials generally in accordance
with Good Clinical Practice and the data was analyzed for safety,
and reports were filed with the FDA. Our review also
indicates that Miravant never conducted an efficacy analysis of the
81 patients in the last two studies which were not yet complete
when Miravant discontinued its CMBC program.
In
2004, Miravant submitted a new drug application (“NDA”)
to the FDA for the use of REM-001 to treat an aspect of AMD.
The FDA reviewed this submission and granted Miravant an approvable
letter for REM-001 in the treatment of AMD, with final approval
contingent on, among other things, the successful completion of a
Phase 3 trial. Miravant ceased operations prior to completing
this trial.
Since
acquiring the rights to REM-001 Therapy, we recently performed a
preliminary analysis of the data collected from the 81 patients
that Miravant never analyzed for efficacy. Based on our
analysis of both that data, and data collected from the initial 67
patients, we believe REM-001 Therapy provided promising safety and
efficacy in CMBC patients and that, taken together, these results
provide strong support for REM-001 Therapy as a potential therapy
for this disease. Furthermore, we believe the approvable
letter previously granted to Miravant with respect to its New Drug
Application (“NDA”) for REM-001 in an aspect of AMD may
indicate that many of the elements required for approval have
already been completed for REM-001.
Overview of Key Regulatory Filings
The
initial investigational new drug (“IND”) filing for
REM-001 Therapy was IND 39,940 which was filed in June 1992 with
the FDA’s Division of Oncology and Pulmonary Drug Products.
This IND is now under the purview of the FDA’s Division of
Oncology Products. All CMBC trials were conducted under this IND.
Miravant kept this IND in place but in 2005 they placed it on
inactive status since they had focused their REM-001 development
efforts on ophthalmology. In 2012, following St Cloud’s
foreclosure action on Miravant and Adgero’s subsequent
purchase of the Miravant assets, St. Cloud transferred ownership of
this IND to Adgero. This transfer was formally recognized by the
FDA with a Change of Sponsor letter dated December 14, 2012.
Adgero’s interactions with the FDA for CMBC are under the
auspices of this IND. It is our expectation, based on input from
regulatory consultants, that clinical development in CMBC, non-CMBC
cutaneous metastatic cancer and basal cell nevus syndrome would be
conducted under this IND. Recent FDA approvals in locally advanced
basal cell cancers, including basal nevus syndrome, have been under
the purview of the FDA’s Division of Oncology
Products.
As part
of its purchase agreement with St Cloud, sponsorship of two other
IND’s was transferred to us. On February 25, 2013, the
FDA’s Division of Dermatology and Dental Products notified us
with a Change of Sponsor letter that it recognized us as the
sponsor of IND 50,116. On May 8, 2013 the FDA’s Division of
Transplant and Ophthalmology Products notified us with a Change of
Sponsor letter that it recognized us as the sponsor of IND 49,648.
At this time, we do not anticipate any of our planned or
contemplated clinical development activities would be under either
of these IND’s.
Our REM-001 Therapy
Overview
Our
REM-001 Therapy product consists of three parts, the DD series
laser light source (or equivalent), the ML2-0400 light
delivery device (or equivalent) and the drug REM-001. Pursuant to
the Miravant oncology IND, the FDA previously approved all three
components to be used together in certain Miravant CMBC Trials. In
use, the drug REM-001 is first administered by intravenous infusion
and allowed to distribute within the body and be taken up by the
tumors. Tumors are then illuminated with light using the light
delivery device, which is attached to the laser light source, so
that the accumulated drug REM-001 can be activated for the desired
clinical effect. Our analysis of clinical data collected in the
Miravant CMBC Trials shows that REM-001 Therapy provides a stronger
reaction in tumor tissues than in healthy tissues, which was a goal
with REM-001’s nanoparticle liposomal
formulation.
46
We plan
to use either the Miravant DD2 (or equivalent), which
was used in certain prior Miravant clinical trials, or DD4 laser
light source in any clinical trials for CMBC. The lasers are
capable of delivering of 1 watt (DD4) or 2 watts (DD2) of optical
power centered at a wavelength of 664 nanometers. We believe we
acquired a sufficient number of them to conduct currently
anticipated CMBC clinical trials. Alternatively, we may
manufacture new laser units for future clinical trials that have
been determined to be functionally equivalent to the DD2/DD4
lasers. Based on our interactions with FDA, we believe that use of
such equivalent lasers will be acceptable to
FDA.
The
light delivery devices we plan to use in our CMBC program are the
same basic proprietary design developed and used previously by
Miravant in its clinical trials. In the case of cutaneous
treatment, such as with CMBC, the light delivery device consists of
an optical fiber which has a modified end to allow it to deliver a
uniform light treatment field to the tumor. Our plan is to have
clinical light delivery devices built by a contract medical device
manufacturer using the basic Miravant design and tested to
the same performance specifications as used
previously.
The REM-001 Drug
REM-001
is a light activated photosensitizer drug used in PDT. During light
activation, photosensitizer drugs act as a catalyst and absorb
light energy which they transfer to surrounding oxygen-containing
molecules to create reactive oxygen species (“ROS”).
ROS can initiate various biological mechanisms of
action:
|
|
●
|
Apoptosis
– Certain photosensitizer drugs associate with the
cells’ mitochondria. When light activated, these drugs
generate ROS that alter mitochondria membrane permeability to allow
the release of activators that initiate a programmed cell death
process known as apoptosis. Apoptosis is a desirable means of
inducing tumor cell death as it is the body’s natural mode
for eliminating damaged cells.
|
|
|
●
|
Necrosis
– At higher doses these photosensitizer-generated ROS can
overwhelm a cell and induce cellular necrosis.
|
|
|
●
|
Anti-angiogenesis
– As they grow, tumors develop their own micro-vasculature
network. ROS can be used to create permeability in these
micro-vessels which reduces their effectiveness and cuts off the
tumor’s blood supply.
|
|
|
●
|
Immune
Response – PDT is known to induce an immune response
including activation of CD8+ T
cells to attack tumor cells. Such T cells provide one of the key
mechanisms making up the body’s immune response system, which
response may enhance anti-tumor immunity. Therapeutic drugs that
produce such an immune response are known as immunotherapies. We
believe that immunotherapies are promising areas of cancer
treatment and are being developed as either monotherapies or in
combination with other treatments.
|
REM-001 has been shown to induce apoptosis and, in treating an
aspect of AMD, have anti-angiogenesis properties. REM-001 is a
second generation photosensitizer drug designed with the following
attributes to overcome several of the shortcomings of earlier,
first generation photosensitizer drugs such as
Photofrin:
|
|
●
|
It is
activated with longer wavelength, deeper penetrating
light;
|
|
|
●
|
It has
a stronger light absorption coefficient;
|
|
|
●
|
It is a
synthetic single molecule; and
|
|
|
●
|
It
causes transient photosensitivity of shorter duration.
|
Photofrin, which is
sold by Pinnacle Biologics Inc. (“Pinnacle”), a
subsidiary of Concordia Healthcare Corp (NASDAQ: CXRX), is the only
PDT compound that we are aware of which is approved by the FDA for
the treatment of cancer. Specifically, it is approved in the United
States for certain non-small-cell lung cancers and esophageal
cancers. Currently, Photofrin it is not approved for treatment of
CMBC or similar cutaneous tumors and we are not aware of any
efforts to get approval in these indications.
47
REM-001’s
chemical structure is designed to allow the use of longer
wavelength, deeper penetrating light than is used in Photofrin.
Deeper penetrating light means the treatment effect can reach
deeper into the tumor which we believe should allow for the
treatment of larger tumor volumes. REM-001 also has a stronger
light absorption coefficient than Photofrin, which we believe
should allow it to generate ROS more efficiently. In addition,
REM-001 is an easily synthesized single molecule meaning that its
manufacturing process is consistent with modern drug manufacturing
strategies; we believe this will make REM-001 better suited for
today’s rigorous regulatory environment. Unlike REM-001,
Photofrin is a polymer mixture derived from naturally occurring
substances. Polymer mixtures can present challenges in achieving a
consistent drug product in line with modern regulatory
requirements. An additional advantage provided by REM-001 is the
rate at which it clears from the skin. Clinical data from a Phase 1
clinical trial conducted by Miravant in healthy volunteers showed
that, at the 1.2 mg/kg dose, there was no measurable
photosensitivity when patients were exposed 15 days after drug
administration to light equivalent to one half hour of midday
sunlight. Further data indicates this effect is present for longer
periods if higher drug doses or more extended periods of light
exposure are used. Based on our review of limited published data
(Wagnieres, et. al., Photochemistry and Photobiology, 1998, 68(3):
382-87), we believe that, when used under similar conditions, the
photosensitivity of REM-001 is of shorter duration and is less
intense than that seen with Photofrin. In the Wagnieres paper, the
authors describe photosensitivity measurements on a human subject
that was done using test conditions that were virtually identical
to a study conducted by Miravant using REM-001. All patients in the
Miravant study had photosensitivity reactions that were much less
intense and of much shorter duration than that seen in the
Wagnieres paper.
Given
what we believe are its potential multiple mechanisms of action,
efficacy results to date and substantial development, we believe
REM-001Therapy is a promising platform therapy for the treatment of
CMBC and other cutaneous metastatic cancers.
Safety and Toxicology
PDT
carries what we believe is an inherent safety advantage since it
uses photosensitizer compounds that are largely inactive except
when they are being illuminated by intense light at specific
wavelengths. Nevertheless, drug molecules, including
photosensitizer molecules, can carry safety or toxicology risks on
their own. REM-001 has previously undergone preclinical and
clinical studies throughout its development cycle and has undergone
certain tests typically required for FDA drug approval. REM-001 has
been safely administered to over 1000 patients in prior clinical
trials. Most significantly, REM-001 has been previously reviewed by
the FDA as part of the NDA submitted by Miravant for the use of
REM-001 to treat an aspect of AMD, a non-CMBC indication. Following
that review, the FDA granted an approvable letter for REM-001 in an
aspect of AMD in 2004, with final approval contingent on, among
other things, the successful completion of a Phase 3 trial. While
not definitive, we believe this letter, along with feedback
we received from FDA on March 3, 2017, indicates that it is
unlikely that there will be significant safety or toxicology issues
associated with REM-001 that would ultimately prevent marketing
approval.
Based
on our review of the clinical data of the Miravant CMBC Trials, we
believe pain was the most common treatment-related adverse event
experienced by patients in these Trials. The second most common
safety issue experienced with REM-001 was a transient
photosensitivity, meaning extended exposure in bright light and
direct sunlight should be avoided, which may occur with all
photosensitizers to some degree. We believe this issue
can be addressed by minimizing one’s exposure to bright light
and sunlight for two to four weeks after treatment. In general, the
potentially treatment-related adverse events observed were expected
in nature (pain, edema, skin photosensitivity) and severity, and
mostly resolved during the course of the studies.
Markets for REM-001 Therapy
Our
development plan for REM-001 Therapy is focused on the treatment of
rare unmet needs in cancer, particularly those where
the tumor can be accessed with a light delivery fiber
device.
Cutaneous Metastatic Breast Cancer (CMBC)
While
most internal cancers can metastasize to the skin, the internal
cancer where this most commonly occurs is breast cancer.
Radiotherapy is often used as an adjunctive therapy in breast
cancer, in part to help prevent the development of local
recurrences including CMBC. However, breast cancer survivors may
still develop CMBC lesions, even over a decade after their original
cancer treatment. In fact, physicians often watch for cutaneous
(skin surface) metastases as a sign of breast cancer recurrence. A
2003 meta-analysis of approximately 20,000 cancer patients found
that 24% of metastatic breast cancer patients included in the
analysis had developed cutaneous metastases, which was the highest
rate of any cancer type. Given that approximately 160,000 women
suffer from metastatic breast cancer, we believe the prevalence of
CMBC may approach 40,000 in the United States. In many
cases of CMBC, surgical excision is not possible, so various
standard cancer therapies, particularly radiotherapy or
chemotherapy, are the first course of treatment. We believe these
therapies are inadequate given the well-known dose limiting
toxicities, limited efficacy, and/or side effects of each. We are
not aware of any prospective clinical trials that have led to FDA
approval of a therapy specifically for the treatment of CMBC and we
do not expect any to be approved in the near future.
48
Cutaneous Metastatic Cancers
A
meta-analysis has shown that approximately five percent of people
with internal (non-melanoma, non-lymphatic, non-leukemic) cancers
develop cutaneous metastatic tumors in their skin. Based on an
estimated incidence of 1,500,000 such internal cancers in the
United States, this means that the incidence of such cutaneous
metastases is approximately 75,000 with a substantially higher
prevalence given the fact that individuals often live with
metastatic cancer for years. Regardless of the primary source of
the cancer, these cutaneous metastatic tumors often begin as small
skin nodules but, as the cancer spreads, more nodules form and can
eventually cover large areas of skin. With progression, the tumor
field generally becomes more painful as tumors may grow larger and
more numerous, ulcerate, bleed and carry a strong odor. Part of our
goal is to treat these cutaneous tumors as early as possible to
either cause them to be locally eliminated or to slow their growth
sufficiently to reduce their late stage development.
Adjuvant and Neo-adjuvant Applications in Oncology
REM-001 Therapy possesses a number of
characteristics that we believe make it well suited as a potential
therapy in adjuvant and neo-adjuvant applications in oncology.
Adjuvant therapy in cancer is any treatment given after
primary therapy, commonly surgery, to increase the chance of
long-term disease-free survival. Neo-adjuvant therapy is treatment given before
primary therapy with the same long-term goal.
We
believe there are a number of potential applications for REM-001
Therapy in the areas of adjuvant and neo-adjuvant
therapy in oncology. One of these is the treatment of
breast cancer, which has approximately 232,000 new cases in the U.S
each year. The standard of primary care for most
patients with breast cancer includes surgery via
a mastectomy or a lumpectomy, a
more breast-conserving surgery, to remove the tumor. In
the United States, lumpectomy surgery is currently used over
mastectomy by an approximately two-to-one ratio, although the
number and rate of mastectomies has been increasing. Given its
potential to selectively induce anti-angiogenesis as well as
an immune response, we believe REM-001 Therapy could help to
prevent breast cancer recurrence when used in a neo-adjuvant
manner prior to surgery. If effective in this mode, we
believe it could even help to increase the use of lumpectomies
relative to mastectomies in the treatment of primary breast
cancer.
49
Beyond
this potential application, PDT and its mechanisms of action have
been investigated in preclinical research studies as an adjuvant
therapy for potential use in combination with standard cancer
therapies and in a number of cancers (e.g., lung, colon, ovarian,
brain, etc.). We believe that as this work progresses, REM-001
Therapy may be well positioned for clinical development as an
adjuvant or neo-adjuvant therapy in one or more significant
indications in cancer.
Basal Cell Nevus Syndrome (BCNS)
In
addition to the clinical studies that Miravant conducted with
REM-001 Therapy in CMBC, it also generated clinical data for
patients with Basal Cell Nevus Syndrome (“BCNS”) who
developed extensive basal cell carcinoma. BCNS is a rare but
serious condition that is often characterized by the formation of
multiple and recurring cutaneous basal cell carcinoma lesions.
Estimates of its prevalence range from 1 in 57,000 to 1 in 256,000
which indicates it could qualify as an orphan disease indication.
In a Miravant Phase 1/2 clinical trial (CA001B), 14 patients with
BCNS were enrolled and treated with REM-001 Therapy using the same
dosing conditions as were used in the CMBC trials. A total of 157
lesions were treated in these patients and showed a 91% overall
response rate. This was composed of a 68% complete response rate
(no remaining visible evidence of a lesion) and a 23% partial
response rate (lesion was reduced in size by more than 50%). In
addition, 7% of lesions had stable disease (any increase in lesion
size was less than 25%). The various response rates are shown in
the graph below and are similar to the results seen in CMBC
patients as we would expect.
Until
the recent FDA approval of the drugs Odomzo and Erivedge, treatment
options for these BCNS patients were very limited. However, we
believe that, based on their package inserts, Odomzo and Erivedge
have dose limiting toxicity profiles which are broader in scope
than the primarily transient adverse effects observed with REM-001
Therapy. We believe that the potential toxicity limitations related
to the existing therapies for BCNS, plus the positive initial Phase
1/2 data generated in clinical trials with REM-001 Therapy, suggest
that REM-001 Therapy could be a viable alternative in treating
recurrent basal cell carcinoma in BCNS patients.
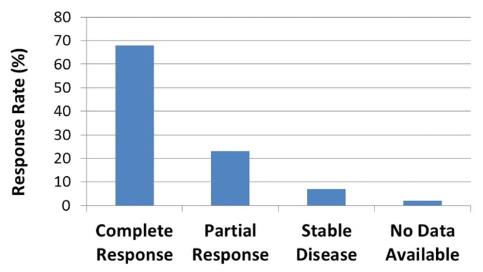
50
Our Pipeline
The following table summarizes
the most advanced stage of development of our technology platform.
As noted above, our REM-001 Therapy has already been tested on 149
patients with CMBC in Phase 2/3 clinical trials. In addition, in a
Phase 2 trial, four patients with non-CMBC cutaneous metastatic
cancer were treated with REM-001 Therapy. Lesion response rates on
those four non-CMBC patients were similar to those achieved with
REM-001 Therapy in other cutaneous cancers tested.
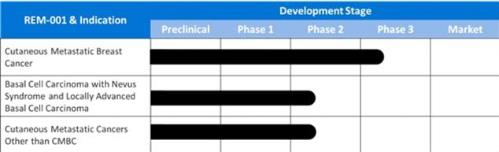
CMBC: Our Lead Indication
Current and Experimental Treatments for CMBC
As with
many cancers, the current standard treatment for CMBC is surgical
excision. However, this is often not feasible due to the extent of
the tumor field or the condition of the skin, particularly in
patients who had radiation therapy. A number of other therapies
have been used on patients with CMBC, including various forms of
chemotherapy, radiation therapy, hyperthermia, cryotherapy,
electro-chemotherapy, topical drugs and intra-lesional chemotherapy
injections. Researchers have also attempted to combine therapies in
an effort to improve efficacy. However, we believe that these
therapies are often inadequate given the limited efficacy,
toxicities and/or side effects of each. The side effects associated
with therapies may be particularly difficult for patients who may
have already experienced extensive surgery along with a full course
of radiation and/or systemic chemotherapy. Also, the fact that CMBC
tumors continue to develop following these therapies is a signal
that the tumor cells may have developed a resistance to some of
these approaches. Based on the above our discussions with
clinicians and literature reviews, and our March 3, 2017
response from FDA, we believe that treatment of unresectable
CMBC tumors is a largely unmet medical need, particularly in
patients who have already received extensive radiation and
chemotherapy.
Key Clinical Results in CMBC
We have
conducted an analysis of the Phase 1 and four Phase 2 and/or Phase
3 CMBC clinical trials done previously with REM-001 Therapy by
Miravant (the “Miravant CMBC Trials”) and have
concluded that, in these studies, REM-001 Therapy provided higher
tumor response rates than are generally seen with alternative CMBC
treatments but this program was discontinued in 1998.
Our review of Miravant’s records further indicates that,
following this decision, Miravant continued to monitor patients in
the CMBC trials and collected data as required by protocol, but
they conducted no further treatment of CMBC patients with REM-001
Therapy. We believe that Miravant primarily chose to discontinue
this program in order to focus its REM-001 development efforts on
an aspect of “wet” age-related macular degeneration
(“AMD”).
Phase 1 Clinical Trial
A Phase
1 dose escalation clinical trial was initially conducted by
Miravant to establish the REM-001 dosimetry to be used in
subsequent safety and efficacy trials. The trial was initiated in
1993 and enrolled 22 patients with 213 treated cutaneous cancer
lesions who received escalating REM-001 drug and light doses. This
study used earlier generation light delivery devices than those
used in later trials but these devices provided
equivalent light output to those units used in later trials. In
these studies REM-001 drug doses ranged from 0.1 mg/kg to 1.2
mg/kg, light doses ranged from 100 to 200 J/cm2 and treatment
time-points ranged from 24 to 72 hours. This study indicated that a
drug dose in excess of 0.8 mg/kg and a light dose of 200 J/cm2
administered at 24 hours provided a high overall response rate when
delivered in a variety of cutaneous cancer lesions. The previously
tested dose of 1.2mg/kg was then tested further in a second Phase 1
trial, where it was administered to 27 cutaneous tumor lesions and
provided a 66% complete response rate and a 90% overall response
rate. Based on these results, this dosimetry was used in subsequent
CMBC trials, including the Miravant CMBC Trials described
below.
51
Phase 2/3 Studies
Miravant conducted
four Phase 2/3 trials with REM-001 Therapy for the treatment of
CMBC as summarized below. These trials all used the same dosimetry
as described above and most of the patients had been previously
treated with radiation therapy and chemotherapy. The light delivery
devices used in these trials were the ML1-0400 or the functionally
equivalent ML2-0400. The laser light source used in three of the
trials was the Miravant DD2 laser and one trial used the KTP model
laser manufactured by LaserScope. Each trial was conducted under
Miravant’s REM-001 cancer Investigational New Drug
Application (“IND”) using Good Clinical Practices with
safety and efficacy data collected accordingly. In connection with
our acquisition of the Miravant assets, ownership of that IND has
been transferred to us.
The
table below summarizes the Miravant CMBC Trials. Trials CA008,
CA009 and CA019 required that the patients enrolled had received
prior radiation therapy. Trial CA013 did not have this specific
inclusion requirement but our review of the data indicates that at
least 50 of the 56 patients in CA013 had received prior radiation
therapy. A second difference across the trials is that trials
CA008, CA009 and CA019 had a 24 week follow-up period while trial
CA013 had a 52 week follow-up period. Also in studies CA008 and
CA009 two tumor lesions on each patient were randomly selected as
controls and did not receive light activation. CA013 was conducted
in Europe by a corporate partner of Miravant. Beyond these
differences and those device differences noted above. We believe
there were no other substantive differences between the trials and
that all trials enrolled similar patients.
Table
of Phase 2 and/or 3 Miravant CMBC Trials
(Note:
SnET2 is now called REM-001.)
|
Trial Title
|
Phase
|
Location
|
Total Patients
|
Total Patients Previously Treated with Radiotherapy
|
Included Randomly Selected Control Tumors
|
|
CA008: Open-Label Randomized No
Treatment Concurrent Controlled Study of Single Dose Tin Ethyl
Etiopurpurin (SnET2) Photodynamic Therapy (PDT) in Patients with
Advanced Breast Cancer Who Have Failed Radiation Therapy for the
Management of Cutaneous Metastatic Breast Carcinoma (24 Week Follow
Up)
|
2/3
|
U.S.
|
32
|
32
|
Yes
|
|
CA009: Open-Label Randomized No
Treatment Concurrent Controlled Study of Single Dose Tin Ethyl
Etiopurpurin (SnET2) Photodynamic Therapy (PDT) in Patients with
Advanced Breast Cancer Who Have Failed Radiation Therapy for the
Management of Cutaneous Metastatic Breast Carcinoma (24 Week Follow
Up)
|
2/3
|
U.S.
|
36
|
36
|
Yes
|
|
CA013: Multinational, Open-Label Study
of Single Dose Tin Ethyl Etiopurpurin (SnET2) Photodynamic Therapy
(PDT) in Patients with Advanced Breast Cancer for the Management of
Cutaneous Metastases of Breast Carcinoma (52 Week Follow
Up)
|
2
|
Europe
|
56
|
50
|
No
|
|
CA019: Open-Label Study of Single Dose
Tin Ethyl Etiopurpurin (SnET2) Photodynamic Therapy (PDT) in
Patients with Advanced Breast Cancer Who Have Failed Radiation
Therapy for the Management of Cutaneous Metastatic Breast Carcinoma
(24 Week Follow Up)
|
3
|
U.S.
|
25
|
25
|
No
|
52
The
primary endpoints for studies CA008 and CA009 were objective tumor
response rate, quality-of-life change, device performance and
patient safety. Adgero’s review of the tumor response rate
and quality-of-life endpoints indicated they were defined as
follows:
|
|
●
|
Tumor Response: Measured as paired response difference, as
calculated by the percentage of a patient’s evaluable lesions
that respond minus the percentage of the patient’s control
lesions that respond with this difference averaged over all treated
patients.
|
|
|
●
|
Quality of Life Change: Measured using the Dermatologic Life
Quality Index (DLQI, A.Y. Finlay and O.K. Khan, "Dermatology Life
Quality Index (DLQI - a simple practical measure for routine
clinical use". Clinical and Experimental Dermatology 1994; 19:
210-2 16) with change measured from baseline
measurements.
|
The
following table shows the results of these two endpoints for
studies CA008 and CA009 as calculated by Miravant. In some cases
patients dropped out of the study before lesion responses could be
assessed or they did not complete their quality of life
questionnaires. The Eligible Patients column in this and the
following tables refers to the number of patients in each case for
which sufficient data is available to calculate the relevant
endpoint.
|
|
Tumor Response as Measured by Paired Response Endpoint
|
24 Week Quality of Life Change
|
||||
|
Study
|
Eligible Patients
(N)
|
Mean ± SD (%)
|
P value
|
Eligible Patients
(N)
|
Mean ± SD
|
P value
|
|
CA008
|
18
|
33%
± 37%
|
<
0.001
|
7
|
0.4
± 4.8
|
0.813
|
|
CA009
|
19
|
39%
± 47%
|
<
0.001
|
10
|
-0.3
± 4.1
|
0.554
|
FDA
typically requires a p value of 0.05 or less for approval. Based on
the above results, it appears that the Paired Response endpoint
achieved statistical significance in both the CA008 and CA009
studies. However, it is Adgero’s understanding that FDA
questions the strength of this data, in part due to the small
number of patients involved as well as the fact that each patient
had only two control lesions.
Following
discussions with the FDA, an endpoint called Clinical Success was
added as an additional measure of tumor response. This was defined
as follows:
|
|
●
|
Clinical Success: Clinical success is determined by a
two-step process. First, for each patient, clinical success occurs
when the fraction of evaluable lesions that respond minus the
fraction of evaluable lesions that progress is greater than 0.5.
Second, for the entire study, an average rate of clinical success
is determined, simply by taking the ratio of individual patients
who are clinical successes to the total number of eligible
patients. Note this endpoint does not involve the control lesions
or any other control, so a p-value is not appropriate since
p-values refer to the difference between a treated and a control
group. In such uncontrolled settings, the statistical measure
commonly used by regulatory agencies instead of a p-value is the
confidence interval, which is provided in the charts
below.
|
The
clinical success rates for studies CA008 and CA009 as calculated by
Miravant are provided in the following table:
|
|
Tumor Response as Measured by Clinical Success
|
||
|
Study
|
Eligible Patients
(N)
|
Average Rate of Clinical Success (%)
|
95% Confidence Interval
|
|
CA008
|
20
|
60%
|
39% -
81%
|
|
CA009
|
20
|
50%
|
28% -
72%
|
No
significant device failures were observed in either study.
Secondary endpoints in CA008 and CA009 were patient disease burden,
duration of response and patient pain assessment. Miravant?s
analysis indicated, for patients for which data was available,
there was a treatment benefit in disease burden (p = 0.0017 for
CA008, p = 0.0020 for CA009) and duration of response (p < 0.001
for CA008, not significant in CA009) when comparing treated and
control lesions. In terms of pain, there was no significant change
in pain in CA008 and a treatment related increase in pain at 4
Weeks post-treatment in CA009. Treatment related pain, particularly
during the first month after treatment, was the most commonly
reported adverse event and was often treated with
analgesics.
53
Studies
CA013 and CA019 used similar endpoints with one notable exception.
Tumor Response as Measured by Paired Response was not possible in
these studies since this measurement relies on control lesions and
CA013 and CA019 did not include controls. Miravant did not conduct
an efficacy analysis of these two studies but Adgero has conducted
an analysis of the Quality of Life and Clinical Success endpoints
used in the pivotal CA008 and CA009 trials. Results from that
analysis are shown in the following table:
|
|
Clinical Success
|
24 Week Quality of Life Change
|
||||
|
Study
|
Eligible Patients
(N)
|
Average Rate of Clinical Success (%)
|
95% Confidence Interval
|
Eligible Patients
(N)
|
Mean ± SD
|
P value
|
|
CA013
|
32
|
88%
|
71% -
97%
|
16
|
1.3
± 3.6
|
1.00
|
|
CA019
|
18
|
83%
|
45% -
86%
|
11
|
2.5
± 4.7
|
1.00
|
Adgero
has not attempted any further analysis of the endpoints included in
these two studies.
The
most common adverse events seen in these four studies (CA008,
CA009, CA013, CA019) were pain and photosensitivity, both of which
are expected with this therapy. In the four studies there were a
total of 17 serious adverse events (SAE’s) that were judged
by investigators to be possibly, probably or definitely related to
treatment. None of these were classified by the investigator as
life threatening and none resulted in death. Of these 17
SAE’s, 8 were related to necrosis of the treated lesions, 3
were related to treatment field infection, 4 were treatment related
pain, 1 was a photosensitivity skin reaction and 1 was an allergic
reaction.
Adgero believes
that the data from these studies show that REM-001 Treatment is a
promising therapy for CMBC. However, because there are no approved
therapies for CMBC, Adgero has no basis for comparing these results
to existing therapies. Based on FDA’s March 3, 2017 response
we believe FDA will view these results as supportive data and will
require us to conduct a new pivotal Phase 3 trial using endpoints
that will be negotiated with the agency and that will reflect
current standards for development of oncology
drugs.
Miravant
discontinued its CMBC program in 1998 and clinical study reports
for the Miravant CMBC Trials were not completed until 2001, by
which time we believe Miravant’s clinical development efforts
for REM-001 were focused entirely on their ophthalmology program.
These clinical study reports were focused primarily on the safety
aspects of REM-001 Therapy; they include only an efficacy analysis
of the CA008 and CA009 trials and state no efficacy analysis was
done for the CA013 and CA019 studies. Based on this observation, we
believe that trials CA013 and CA019 may not have been analyzed for
efficacy. Notably, CA013 and CA019 comprise over half of the CMBC
patients treated in the Miravant CMBC Trials. While we cannot state
with certainty, it is our understanding that the lack of an
in-depth efficacy analysis of the Miravant CMBC Trial data in CA013
and CA019 was due to the fact that the study reports were prepared
well after the CMBC program had been discontinued and the main goal
in preparing these reports was to ensure that REM-001 safety data
was properly reported in preparation for Miravant’s NDA in
the ophthalmology AMD program.
After
acquiring Miravant’s assets we first performed a
preliminary efficacy analysis of all of the Miravant CMBC Trials.
This analysis was conducted by us using clinical data stored in
digital backup form on Miravant’s server. Our
initial review was based on a
last-observation-carried-forward (“LOCF”) analysis of
recorded lesion measurements of evaluable tumor lesions from these
electronic records. According to the Miravant clinical protocol,
tumor lesions were evaluable, meaning they could be measured and
scored for a treatment response, when the REM-001 Therapy
post-treatment redness or swelling had resolved so that any
underlying tumor could be visually identified. To minimize the
likelihood of error, our initial preliminary analysis
considered only the complete response rate, or the
fraction of tumor lesion sites where there is no remaining visible
evidence of a tumor. An analysis that also considers tumor lesions
with a partial response would yield a higher response rate; thus,
we believe our initial preliminary analysis utilized a
more rigorous standard than the overall response rate that is the
benchmark often used in clinical cancer trials involving cutaneous
tumors.
The
figure below shows the results of this initial
preliminary analysis of Miravant clinical data and depicts
the percentage of evaluable lesions in each Miravant CMBC Trial for
which there was a complete response; i.e. where all visible
clinical evidence of the tumor is gone after treatment with REM-001
Therapy.
54
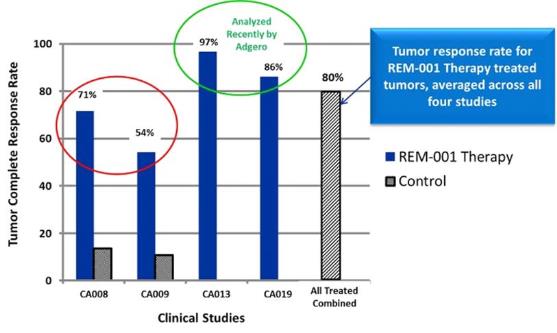
Clinical Development Plans
CMBC
Our
initial product goal is to achieve marketing approval of REM-001
Therapy for the treatment of CMBC. We conducted a first in-depth
analysis of all existing REM001 Therapy clinical trial data for
CMBC, including data from the Miravant CMBC Trials. This analysis
was overseen by regulatory experts who have expertise in
interacting with the Food and Drug Administration (the
“FDA”). The experts we have engaged are either former
FDA employees with directly related experience in reviewing similar
oncology treatments who are now acting as independent consultants
or individuals who have provided senior regulatory guidance to
major pharmaceutical or medical device companies in situations that
led to regulatory approval. For this first analysis, we have
submitted questions to FDA under a Type C
format to review the technology and results and
determine the anticipated requirements for regulatory approval.
On March 3, 2017, we received FDA’s written response to
our questions. Based on that response, we believe our plans to
manufacture REM-001 by revising the prior quality standards to meet
the currently recommended regulatory standards will be acceptable.
FDA also indicated our plans for utilizing light delivery devices
that have been shown to be functionally equivalent to the devices
used by Miravant will be acceptable. FDA also recognized that CMBC
represents an unmet clinical need and provided guidance on a number
of clinical pharmacology parameters they would like us to measure
in our planned clinical trial. Based on FDA’s responses, we
plan to conduct a clinical trial in CMBC to test the safety and
efficacy of REM-001 Therapy for marketing approval and we are
planning further discussions with FDA regarding the specifics of
such a trial.
At this time
we estimate the necessary trial design will be a pivotal
Phase 3 multi-center trial that would enroll approximately 100-150
CMBC patients who have received prior radiation therapy and
chemotherapy. This study will be designed with input
from the FDA with the goal of gaining expedited development and
review under the following FDA programs: Fast Track, Breakthrough
Therapy, Accelerated Approval and Priority Review designations. We
would also plan to coordinate closely with the FDA to attempt to
receive a special protocol assessment (“SPA”) based on
what we believe is the substantial body of existing clinical data
generated with REM-001 Therapy in CMBC. We also plan to seek an
orphan drug designation for REM-001 in CMBC from the FDA. Our
research indicates that CMBC prevalence is less than 200,000 in the
United States, thus we believe it should qualify for an orphan drug
designation. Our request will be based on our existing clinical
data in CMBC patients. The FDA also grants five years of
exclusivity to the first applicant to obtain approval of an NDA for
a new chemical entity (“NCE”). A drug is an NCE if the
FDA has not previously approved any other new drug containing the
same active ingredient. We believe that REM-001 would also qualify
for this form of exclusivity. There is no guarantee that we will
receive an orphan drug designation or NCE exclusivity for REM-001
or any of our product candidates. See “Regulatory
Matters.”
55
St. Cloud Asset Purchase Agreement
We
acquired certain Miravant assets, including the REM-001 Therapy and
the associated technology and intellectual property, through an
asset purchase agreement with St. Cloud Investments, LLC
(“St. Cloud”), dated November 26, 2012, as amended (the
“St. Cloud Agreement”). St. Cloud was previously a
Miravant creditor and acquired these Miravant assets pursuant to a
foreclosure process St. Cloud completed under California law.
Pursuant to the terms of the St. Cloud Agreement, we are obligated
to make certain payments to St. Cloud and Steven Rychnovsky, PhD,
who became our Vice President of Operations and Product Development
after the consummation of the St. Cloud Agreement, in consideration
of his services to St. Cloud in helping to negotiate the St. Cloud
Agreement, as St. Cloud’s designee. The amounts paid or owed
under that agreement are as follows:
●
Thirteen thousand
dollars ($13,000) was paid to Steven Rychnovsky, PhD upon the
Initial Closing of the 2016
Private Placement .
●
Forty thousand dollars ($40,000) was paid
to St. Cloud upon the Initial Closing of the 2016 Private
Placement.
●
Fifty
thousand dollars ($50,000) was paid to Steven Rychnovsky, PhDduring
the 2016 Private Placement, because the 2016 PrivatePlacement was
completed for an amount that exceeded four milliondollars
($4,000,000).
●
Fifty
thousand dollars ($50,000) was paid to St. Cloud during the2016
Private Placement, because the 2016 Private Placement wascompleted
for an amount that exceeded four million
dollars($4,000,000).
●
Upon
the earlier of (i) a subsequent equity financing to take placeafter
we conduct a Phase 2B clinical trial in which fifty
patientscomplete the trial and their clinical data can be evaluated
or (ii)the commencement of a clinical trial intended to be used as
adefinitive study for market approval in any country, we
areobligated to pay an aggregate amount of three hundred
thousanddollars ($300,000) in cash or an equivalent amount of
common stock,with two hundred forty thousand dollars ($240,000) to
St. Cloud andsixty thousand dollars ($60,000) to Steven
Rychnovsky,PhD.
●
Upon
receipt of regulatory approval of REM-001 Therapy, we are
obligatedto pay an aggregate amount of seven hundred thousand
dollars($700,000) in cash or an equivalent amount of common stock,
withfive hundred and sixty thousand dollars ($560,000) to St. Cloud
andone hundred forty thousand dollars ($140,000) to Steven
Rychnovsky,PhD.
With
respect to the $300,000 and $700,000 potential milestone payments
referenced above (each a "Milestone Payment"), if either such
Milestone Payment becomes payable, and in the event we elect to pay
either such Milestone Payment in shares of our common stock, the
value of the common stock will equal the price per share of the
most recent financing, or, if we are considered to be a
publicly-traded company, the average of the closing price per share
of our common stock over the twenty (20) trading days following the
first public announcement of the applicable event described
above.
In
addition, we must pay to St. Cloud and Steven Rychnovsky, PhD, in
the aggregate, a royalty fee of six percent (6%) of net sales
during the royalty term on a country-by-country and
product-by-product basis with St. Cloud receiving a royalty rate of
four and eight tenths percent (4.8%) and Steven Rychnovsky, PhD,
receiving a royalty of one and two tenths percent (1.2%). The
royalty term for a product commences on the first commercial sale
of the product, such as REM-001 Therapy, in any country, and the
royalty fee must be paid within 30 days of each calendar quarter
during which revenue is collected. The royalty term terminates on
the later of (i) the invalidation, revocation, lapse or expiration
of the last to expire valid claim on any patent acquired in the St.
Cloud Agreement that would be infringed by the sale of the product
in the country where the commercial sale takes place or (ii) the
expiration of the period for which we hold exclusive marketing
rights of the product in the country, if we were granted those
rights under the St. Cloud Agreement.
56
In
connection with and pursuant to the St. Cloud Agreement, on
November 26, 2012, we issued a senior convertible note to each of
St. Cloud and Steven Rychnovsky, PhD. The notes had an aggregate
principal amount of two hundred thousand dollars ($200,000) and
accrued interest at a rate of eight percent (8%) per annum.
Pursuant to the terms of the notes, because our 2016 Private
Placement raised an amount in excess of five million dollars
($5,000,000) in the aggregate, on August 3, 2016, the notes
converted into an aggregate of 73,998 shares of our common stock
and 73,998 warrants to purchase shares of our common stock at an
exercise price of $5.00 per share, is the quantity of such
securities being equal to the outstanding balance of such notes,
plus interest accrued thereon but unpaid, divided by seventy
percent (70%) of the purchase price per share paid by the investors
participating the financing.
Competition
The
biotechnology and pharmaceutical industries are highly competitive
and characterized by rapidly evolving technology and intense
research and development efforts. We expect to compete with
companies, including major international pharmaceutical companies
that have substantially greater financial, research and
development, marketing and sales capabilities and have
substantially greater experience in undertaking preclinical and
clinical testing of products, obtaining regulatory approvals and
marketing and selling biopharmaceutical products. We will
face competition based on, among other things, product efficacy and
safety, the timing and scope of regulatory approvals, product ease
of use and price.
Our
primary competitors in the CMBC field are Celsion Corporation
(NASDAQ: CLSN) in the United States and IGEA Medical S.p.A. in
Europe, both of which are developing alternative treatment
therapies for CMBC. Celsion Corporation is developing a drug
treatment for CMBC that is activated by a hyperthermia device and
has completed Phase 1/2 studies in CMBC. IGEA Medical S.p.A. is
developing an electro-chemotherapy treatment for CMBC. Pinnacle
Biologics Inc., a subsidiary of Concordia Healthcare Corp (NASDAQ:
CXRX), sells Photofrin, a first generation PDT product for
treatment of certain endobronchial non-small-cell lung cancers and
esophageal cancers. Photofrin is currently in Phase 2 studies in
recurrent glioma. To our knowledge, there is no reported
development program for Photofrin in CMBC. Rogers Sciences Inc. is
a medical device company that is developing a light delivery device
for use with PDT treatment of cutaneous cancers that they are
currently clinically testing in a Phase 2 study in CMBC
patients.
There
are numerous therapies currently used to treat CMBC patients
including chemotherapy, radiation therapy, surgical excision,
hyperthermia, cryotherapy, electro-chemotherapy, topical drugs and
intra-lesional chemotherapy injections, but, to our knowledge,
there are no PDT therapies currently approved by the FDA for the
treatment of CMBC or similar cutaneous cancers. Some topical PDT
agents have been approved by the FDA for actinic keratosis which is
a precancerous skin condition and they have been approved in some
other countries for some conditions that we believe pose low
medical risk such as basal cell cancer and acne.
Research and Development
We
intend to use a portion of the net proceeds from our recently
completed 2016 Private Placement and December 2016 Private
Placement for research and development activities, which we expect
to include compiling a complete audited database of all REM-001
CMBC data from the Miravant CMBC Trials, updating our oncology IND,
manufacturing REM-001, preparing materials and conducting a meeting
with the FDA to discuss regulatory approval options and contracting
for the manufacture of light delivery devices suitable for use in
CMBC. Prior to our acquisition of the assets of Miravant pursuant
to the St. Cloud Agreement, Miravant, with support from certain
corporate partners, conducted preclinical and clinical development
work on REM-001, including the development of a manufacturing
process, preclinical safety, pharmacokinetic and toxicology studies
and human clinical trials.
57
Manufacturing and Supply
The
manufacturing process for the active pharmaceutical ingredient in
REM-001 was developed over a ten year period and we believe it is
now well established and suitable for commercial scale production.
This process was also included as part of Miravant’s prior
NDA for the use of REM-001 to treat an aspect of AMD, which
underwent a FDA review where an approvable letter was granted. The
final REM-001 drug product uses a straightforward lipid formulation
and has already been produced at commercial scale by a large
contract manufacturer for use in Miravant’s past clinical
trials and commercial scale-up activities. We do not own or operate
manufacturing facilities for the production of REM-001, nor the
laser light source, light delivery device or catheters required for
use with REM-001 Therapy. We will depend on third-party suppliers
and manufacturing organizations for both commercial and clinical
trial supplies of all of our raw materials, our REM-001 drug
substance, drug product and the REM-001 Therapy light delivery
device. We believe our existing supply of laser systems should be
sufficient to conduct our currently anticipated CMBC clinical
trials. If additional laser units are needed for either commercial
products or clinical trials, we plan to use third party contract
medical product manufacturers to build those additional
units. In the case of the light delivery device, we will need
to obtain these from third party contract medical device
manufacturers. We believe there are readily available supplies of
all raw materials needed for the manufacture of REM-001 and the
related required REM-001 Therapy components.
Intellectual Property
Our
intellectual property and product pipeline is based on technology
we acquired that was originally developed by Miravant. We acquired
this intellectual property through an asset purchase agreement and
our retention of the intellectual property is dependent on our
meeting the terms of that agreement, most of which are milestone
and royalty based payments. The acquired intellectual property
includes scientific and regulatory data, product know-how and eight
issued US Utility patents. Two of the patents contain process
claims that pertain specifically to REM-001 and its production with
one of these set to expire in August 2020 and the other set to
expire in March 2021. Two of the patents are for light delivery
devices intended to deliver light to internal body surfaces; the
first of these has device and process claims and is set to expire
in August 2020 and the other has device claims and is set to expire
in September 2024. Of the other four patents, two contain method of
use claims that pertain to cardiovascular PDT with one of these set
to expire in November 2021 and the other set to expire in May 2021.
The two remaining patents are composition of matter patients for
next generation photosensitizer drugs that we believe may be useful
in a range of diseases. The first of these is set to expire in
March 2020 and the second is set to expire in November 2021. The
proprietary regulatory data we own includes two INDs for use of
REM-001 in oncology and ophthalmology, and one NDA for use of
REM-001 to treat an aspect of AMD. We do not hold any patents
covering the DD series laser light source or the ML2-0400 light
delivery device.
Adgero Assigned Patents
Our
success will depend on our ability to obtain and maintain patent
and other proprietary rights in commercially important technology,
inventions and know-how related to our business, the validity and
enforceability of our patents, the continued confidentiality of our
trade secrets as well as our ability to operate without infringing
the valid and enforceable patents and proprietary rights of third
parties. We also plan to rely on continuing technological
innovation and in-licensing opportunities to develop and maintain
our proprietary position.
There
is no guarantee that patents will be granted with respect to any
patent applications we may submit, own or license in the future,
nor can we be sure that any of our existing patents or any patents
we may own or license in the future will be useful in protecting
our technology.
In
addition to patents, we rely on trade secrets and know-how to
develop and maintain our competitive position. For example,
significant aspects of our proprietary technology platform are
based on unpatented trade secrets and know-how. Trade secrets and
know-how can be difficult to protect. We seek to protect our
proprietary technology and processes, in part, by confidentiality
agreements and invention assignment agreements with our current and
future employees, consultants, scientific advisors, contractors and
commercial partners. These agreements are designed to protect our
proprietary information and, in the case of the invention
assignment agreements, to grant us ownership of technologies that
are developed through a relationship with a third party. We will
also seek to preserve the integrity and confidentiality of our data
and trade secrets by maintaining physical security of our premises
and physical and electronic security of our information technology
systems. While we have confidence in these individuals,
organizations and systems, agreements or security measures may be
breached, and we may not have adequate remedies for any breach. In
addition, our trade secrets may otherwise become known or be
independently discovered by competitors. To the extent that our
future contractors use intellectual property owned by others in
their work for us, disputes may arise as to the rights in related
or resulting know-how and inventions.
58
We also
plan to seek trademark protection in the United States and outside
of the United States where available and when appropriate. We
intend to use these registered marks in connection with our
pharmaceutical research and development as well as our product
candidates.
Regulatory Matters
Government Regulation
Any
product development activities related to REM-001 Therapy or
products that we may develop or acquire in the future will be
subject to extensive regulation by various government authorities,
including the FDA and other federal, state and local statutes and
regulations and comparable regulatory authorities in
other countries, which regulate the design, research, clinical and
non-clinical development, testing, manufacturing, storage,
distribution, import, export, labeling, advertising and marketing
of pharmaceutical products and devices. Generally, before a new
drug can be sold, considerable data demonstrating its quality,
safety and efficacy must be obtained, organized into a format
specific to each regulatory authority, submitted for review and
approved by the regulatory authority. The data is often generated
in two distinct development states: pre-clinical and clinical.
REM-001 Therapy or other products that we may develop or acquire in
the future must be approved by the FDA through the IND process
before they may be legally marketed in the United States. For new
chemical entities, the pre-clinical development stage generally
involves synthesizing the active component, developing the
formulation and determining the manufacturing process, as well as
carrying out non-human toxicology, pharmacology and drug metabolism
studies which support subsequent clinical testing.
The
clinical stages of development can generally be divided into three
sequential phases that may overlap, Phase 1, Phase 2 and Phase 3
clinical trials. In Phase 1, generally, small numbers of healthy
volunteers are initially exposed to single escalating doses and
then multiple escalating doses of the product candidate. The
primary purpose of these studies is to assess the metabolism,
pharmacologic action, side effect tolerability and safety of the
drug. Phase 2A trials typically involve studies in disease-affected
patients to determine the dose required to produce the desired
benefits, while Phase 2B trials are designed to determine efficacy.
At the same time, safety and further pharmacokinetic and
pharmacodynamic information is collected. In most cases a drug
therapy only gains marketing approval after successful completion
of a Phase 3 study. In such a study the therapy is given to a large
group of people to confirm its effectiveness, monitor side effects
and collect information that the therapy is safe.
Post-approval
studies may be conducted after initial marketing approval.
Sometimes, these studies are used to gain additional experience
from the treatment of patients in the intended therapeutic
condition, and are then often referred to as Phase 4 clinical
trials. In certain instances, the FDA may mandate the performance
of Phase 4 studies. In other situations, post-approval studies aim
to gain additional indications for a medication, often indicated as
Phase 3B studies.
Development of Drugs in the United States
In the
United States, the process of obtaining regulatory approvals and
the subsequent compliance with appropriate federal, state, local
and foreign statutes and regulations require the expenditure of
substantial time and financial resources. Failure to comply with
the applicable United States requirements at any time during the
product development process, approval process or after approval,
may subject an applicant to administrative or judicial sanctions.
These sanctions could include the FDA’s refusal to approve
pending applications, withdrawal of an approval, a clinical hold,
warning letters, product recalls or withdrawals from the market,
product seizures, total or partial suspension of production or
distribution, injunctions, fines, refusals of government contracts,
restitution, disgorgement, or civil or criminal penalties. Any
agency or judicial enforcement action could have a material adverse
effect on us.
Prior
to the start of human clinical studies for a new drug in the United
States, pre-clinical laboratory and animal tests are often
performed under the FDA’s Good Laboratory Practices
regulations. The sponsor must submit the result of the pre-clinical
tests, together with manufacturing information, analytical data and
any available clinical data or literature and a proposed clinical
protocol to the FDA as part of an IND application, which is a
request for authorization from the FDA to administer an
investigational drug or biological product to humans. Similar
filings are required in other countries. The amount of data that
must be supplied in the IND depends on the phase of the study.
Phase 1 studies typically require less data than larger Phase 2 and
3 studies. A clinical plan must be submitted to the FDA prior to
commencement of a clinical trial. If the FDA has concerns about the
clinical plan or the safety of the proposed studies, they may
suspend or terminate the study at any time. Studies must be
conducted in accordance with Good Clinical Practices and regulator
reporting of study progress and any adverse experiences is
required. Studies are also subject to review by independent
institutional review boards responsible for overseeing studies at
particular sites andprotecting human research study subjects. An
independent institutional review board may also suspend or
terminate a study once initiated. Accordingly, we cannot be sure
that submission of an IND will result in the FDA allowing clinical
trials to begin, or that once begun, issues will not arise that
could cause the trial to be suspended or terminated.
59
Review and Approval in the United States
Following pivotal
or Phase 3 trial completion, data is analyzed to determine safety
and efficacy. Data is then filed with the FDA in a NDA, along with
proposed labeling for the product and information about the
manufacturing and testing processes and facilities that will be
used to ensure product quality. In the United States, FDA approval
of a NDA must be obtained before marketing a pharmaceutical
product. The NDA must contain proof of safety, purity, potency and
efficacy, which entails extensive pre-clinical and clinical
testing.
The FDA
will likely re-analyze the clinical trial data, which could result
in extensive discussions between the FDA and us during the review
process. The review and evaluation of applications by the FDA is
extensive and time consuming and may take several years to
complete. The FDA may conduct a pre-approval inspection of the
manufacturing facilities for the new product to determine whether
they comply with current good manufacturing practice requirements
and may also audit data from clinical and pre-clinical
trials.
There
is no assurance that the FDA will act favorably or quickly in
making such reviews and significant difficulties or costs may be
encountered in our efforts to obtain FDA approval. The FDA may
require that certain contraindications, warnings or precautions be
included in the product labeling, or may condition the approval of
the NDA on other changes to the proposed labeling, development of
adequate controls and specifications, or a commitment to conduct
post-marketing testing or clinical trials and surveillance programs
to monitor the safety of approved products that have been
commercialized. Further, the FDA may place conditions on approvals
including the requirement for a risk evaluation and mitigation
strategy (“REMS”) to assure the safe use of the drug.
If the FDA concludes a REMS is needed, the sponsor of the NDA must
submit a proposed REMS; the FDA will not approve the NDA without an
approved REMS, if required. A REMS could include medication guides,
physician communication plans, or elements to assure safe use, such
as restricted distribution methods, patient registries andother
risk minimization tools. Any of these limitations on approval or
marketing could restrict the commercial promotion, distribution,
prescription or dispensing of products. Product approval may be
withdrawn for non-compliance with regulatory standards or if
problems occur, following the initial marketing of the
product.
Orphan Drug Designation
Under
the Orphan Drug Act, the FDA may grant orphan drug designation to a
drug intended to treat a rare disease or condition, which is
generally a disease or condition that affects fewer than 200,000
individuals in the United States or for which there is no
reasonable expectation that the cost of developing and making a
drug available in the United States for this type of disease or
condition will be recovered from sales of the product. Orphan
product designation must be requested before submitting an NDA.
After the FDA grants orphan drug designation, the identity of the
therapeutic agent and its potential orphan use are disclosed
publicly by the FDA. Orphan product designation does not convey any
advantage in or shorten the duration of regulatory review and
approval process.
If a
product that has orphan designation subsequently receives the first
FDA approval for the disease or condition for which it has such
designation, the product is entitled to orphan drug exclusivity,
which means the FDA may not approve any other applications to
market the same drug for the same indication for seven years,
except in limited circumstances, such as (i) the drug’s
orphan designation is revoked; (ii) its marketing approval is
withdrawn; (iii) the orphan exclusivity holder consents to the
approval of another applicant’s product; (iv) the orphan
exclusivity holder is unable to assure the availability of a
sufficient quantity of the drug; or (v) a showing of clinical
superiority to the product with orphan exclusivity by a competitor
product. If a drug designated as an orphan product receives
marketing approval for an indication broader than what is
designated, it may not be entitled to orphan drug exclusivity. In
addition to the period of exclusivity, orphan designation makes a
company eligible for tax credits for clinical research expenses and
potential exemption from the normal prescription drug user fee
(“PDUFA”) required with an NDA submission. There can be
no assurance that we will receive orphan drug designation for
REM-001in the indication of CMBC or any other
indication.
60
New Chemical Entity Exclusivity
The FDA
grants five years of exclusivity to the first applicant to obtain
approval of an NDA for a NCE. A drug is an NCE if the FDA has not
previously approved any other new drug containing the same active
ingredient. During the exclusivity period, the FDA may not accept
for review an abbreviated new drug application
(“ANDA”), or a 505(b)(2) NDA submitted by another
company for a drug based on the same active ingredient, regardless
of whether the drug is intended for the same indication as the
original innovative drug or for another indication, where the
applicant does not own orhave a legal right of reference to all the
data required for approval. However, an application may be
submitted after four years if it contains a certification of patent
invalidity or non-infringement to one of the patents listed with
the FDA by the innovator NDA holder. As in these cases, the FDA can
only accept and begin to review new applications after the
exclusivity period has expired, so the data exclusivity can
effectively expand the protection period by another one and a half
years to a total of six and a half years, before competitors with
the same active ingredient can reach the market. We believe REM-001
is a novel mixture different from any other FDA approved active
drug ingredient, and, therefore, should be regarded as an NCE,
although there can be no guarantee that the FDA will take the same
position. Analogous data and market exclusivity provisions, of
varying duration, may be available in Europe and other
countries.
FDA Programs
The FDA
expedited programs and designations for serious conditions, like
Fast Track, Breakthrough Therapy, Accelerated Approval and Priority
Review, are intended to make certain drugs available as rapidly as
possible. We must request Fast Track designation from the FDA,
which would provide access to a process to facilitate the
development and expedite the review of a drug intended to treat
serious conditions and fill an unmet need. The request can be
initiated at any time during the drug development process. The FDA
will review the request and make a decision within 60 days based on
whether the drug fulfills an unmet medical need in a serious
condition. A drug that receives Fast Track designation is eligible
for some or all of the following: (i) more frequent meetings with
FDA to discuss the drug’s development plan and ensure
collection of appropriate data needed to support drug approval;
(ii) more frequent written correspondence from FDA about things
such as the design of the proposed clinical trial and the use of
biomarkers; and (iii) eligibility for Accelerated Approval and
Priority Review, if relevant criteria are met. Under the
Breakthrough Therapy authority, a drug may be eligible for
designation as a Breakthrough Therapy if the drug is intended,
alone or in combination with one or more other drugs, to treat a
serious or life-threatening condition and preliminary
clinical evidence indicates that the drug may demonstrate
substantial improvement over existing therapies on one or more
clinically significant endpoints. If a drug is designated a
Breakthrough Therapy, the FDA will expedite the development and
review of the drug. Under its Accelerated Approval authority, the
FDA may approve a product for a serious disease or condition that
fills an unmet need, including a Fast Track product, if it is found
to have an effect on a surrogate endpoint – a marker that is
thought to predict a clinical benefit. The FDA may also approve a
product under Accelerated Approval authority if it is found to have
an effect on an intermediate endpoint – a measure of
therapeutic effect that is considered reasonably likely to predict
a clinical benefit. The endpoint evidence to support Accelerated
Approval may be epidemiological, pathophysiological, therapeutic,
and pharmacologic or based on the use of biomarkers. Accelerated
Approval can be withdrawn or the labeled indication of the drug
changed if trials fail to verify clinical benefit or do not
demonstrate sufficient clinical benefit to justify the risk
associated with the drug. Every drug application submitted to the
FDA is subject to consideration for Priority Review designation,
even if the applicant does not request it. The FDA informs the
applicant of a Priority Review designation within 60 days of the
receipt of the original NDA. Designation of a drug as
“Priority” does not alter the scientific/medical
standard for approval or the quality of evidence necessary for
approval. Priority Review does shorten the planned time period for
review of an NDA (from six months compared with the ten month
standard review) by the FDA. A SPA request can be requested after a
pre-Phase 3 meeting with the FDA. It allows the FDA and sponsor to
agree on the study design for a Phase 3 trial whose efficacy
results will be the basis of an NDA. There is no guarantee that we
will receive Fast Track designation, Accelerated Approval
designation, Special Protocol Assessment or Priority Review
designation for REM-001 Therapy or any of our product
candidates.
61
Special Protocol Assessment
The
Federal Food, Drug and Cosmetic Act directs the FDA to meet with
sponsors, pursuant to a sponsor’s written request, for the
purpose of reaching agreement on the design and size of clinical
trials intended to form the primary basis of an efficacy claim in
an NDA. If an agreement is reached, the FDA will reduce the
agreement to writing and make it part of the administrative record.
This agreement is called a special protocol assessment or SPA.
While the FDA’s guidance on SPAs states that documented SPAs
should be considered binding on the review division, the FDA has
latitude to change its assessment if certain exceptions apply.
Exceptions include public health concerns emerging that were
unrecognized at the time of the protocol assessment, identification
of a substantial scientific issue essential to the safety or
efficacy testing that later comes to light, a sponsor’s
failure to follow the protocol agreed upon, or the FDA’s
reliance on data, assumptions or information that are determined to
be wrong.
Drug Development in Europe
In the
European Union, our future products may also be subject to
extensive regulatory requirements. Similar to the United States,
the marketing of medicinal products has been subject to the
granting of marketing authorizations by regulatory agencies.
Particular emphasis is also being placed on more sophisticated and
faster procedures for reporting of adverse events to the competent
authorities.
As in
the United States, the various phases of pre-clinical and clinical
research in the European Union are subject to significant
regulatory controls. Although the regulatory controls on clinical
research are currently undergoing a harmonization process following
the adoption of the Clinical Trials Directive 2001/20/EC, there are
currently significant variations in the member state regimes. All
member states, however, currently require independent institutional
review board approval of interventional clinical trials. Generally,
all clinical trials require either prior governmental notification
or approval. Most regulators also require the submission of adverse
event reports during a study and a copy of the final study
report.
Review and Approval in the European Union
In the
European Union, approval of new medicinal products can be obtained
through one of three processes: the mutual recognition procedure,
the centralized procedure and the decentralized procedure. We
intend to determine which process we will follow, if any, in the
future.
Mutual
Recognition Procedure: An applicant submits an application in one
European Union member state, known as the reference member state.
Once the reference member state has granted the marketing
authorization, the applicant may choose to submit applications in
other concerned member states, requesting them to mutually
recognize the marketing authorizations already granted. Under this
mutual recognition process, authorities in other concerned member
states have 55 days to raise objections, which must then be
resolved by discussion among the concerned member states, the
reference member state and the applicant within 90 days of the
commencement of the mutual recognition procedure. If any
disagreement remains, all considerations by authorities in the
concerned member states are suspended and the disagreement is
resolved through an arbitration process. The mutual recognition
procedure results in separate national marketing authorizations in
the reference member state.
Centralized
Procedure: This procedure is currently mandatory for, among other
things, products developed by means of a biotechnological process,
products that target cancer and orphan medicines, and optional for
new active substances and other “innovative medicinal
products with novel characteristics.” Under this procedure,
an application is submitted to the European Agency for the
Evaluation of Medical Products. Two European Union member states
are appointed to conduct an initial evaluation of each application.
These countries each prepare an assessment report that is then used
as the basis of a scientific opinion of the Committee on
Proprietary Medical Products. If this opinion is favorable, it is
sent to the European Commission, which drafts a decision. After
consulting with the member states, the European Commission adopts a
decision and grants a marketing authorization, which is valid
throughout the European Union and confers the same rights and
obligations in each of the member states as a marketing
authorization granted by that member state.
62
Decentralized
Procedure: The most recently introduced of the three processes for
obtaining approval of new medicinal processes in the European
Union, the decentralized procedure is similar to the Mutual
Recognition procedure described above, but with differences in the
timing that key documents are provided to concerned member states
by the reference member state, the overall timing of the procedure
and the possibility of, among other things, “clock
stops” during the procedure.
Post-Marketing Requirements
Following approval
of a new product, a pharmaceutical company and the approved product
are subject to continuing regulation by the FDA and other federal
and state regulatory authorities, including, among other things,
monitoring and recordkeeping activities, reporting to applicable
regulatory authorities of adverse experiences with the product,
providing the regulatory authorities with updated safety and
efficacy information, product sampling and distribution
requirements, and complying with promotion and advertising
requirements, which include, among others, standards for
direct-to-consumer advertising, restrictions on promoting drugs for
uses or in patient populations not described in the drug’s
approved labeling (known as “off-label use”),
limitations on industry-sponsored scientific and educational
activities, and requirements for promotional activities involving
the internet. Although physicians may prescribe legally available
drugs for off-label uses, manufacturers may not market or promote
such off-label uses. Modifications or enhancements to the products
or labeling or changes of site of manufacture are often subject to
the approval of the FDA and other regulators, which may or may not
be received or may result in a lengthy review process.
Prescription drug
advertising is subject to federal, state and foreign regulations.
In the United States, the FDA regulates prescription drug
promotion, including direct-to-consumer advertising. Prescription
drug promotion materials must be submitted to the FDA in
conjunction with their first use. Any distribution of prescription
drug products and pharmaceutical samples must comply with the U.S.
Prescription Drug Marketing Act, a part of the U.S. Federal Food,
Drug and Cosmetic Act. Once a product is approved, its manufacture
is subject to comprehensive and continuing regulations by the FDA.
The FDA regulations require the products be manufactured in
specific approved facilities and in accordance with current Good
Manufacturing Practices (“cGMP”), and NDA holders must
list their products and register their manufacturing establishments
with the FDA. These regulations also impose certain organizational,
procedural and documentation requirements with respect to
manufacturing and quality assurance activities. Drug manufacturers
and other entities involved in the manufacture and distribution of
approved drugs are subject to periodic unannounced inspections by
the FDA and certain state agencies for compliance with cGMP and
other laws. NDA holders using contract manufacturers’
laboratories or packagers are responsible for the selection and
monitoring of qualified firms. These firms are subject to
inspections by the FDA at any time, and the discovery of violative
conditions could result in enforcement actions that interrupt the
operation of any such facilities or the ability to distribute
products manufactured, processed or tested by them.
Other Regulatory Matters
Manufacturing,
sales, promotion and other activities following product approval
are also subject to regulation by numerous regulatory authorities
in addition to the FDA, including, in the United States, the
Centers for Medicare & Medicaid Services (?CMS?), other
divisions of the Department of Health and Human Services, the Drug
Enforcement Administration, the Consumer Product Safety Commission,
the Federal Trade Commission, the Occupational Safety & Health
Administration, the Environmental Protection Agency, and state and
local governments. Sales, marketing and scientific/educational
programs must also comply with federal and state fraud and abuse
laws. Pricing and rebate programs must comply with the Medicaid
rebate requirements of the U.S. Omnibus Budget Reconciliation Act
of 1990. If products are made available to authorized users of the
Federal Supply Schedule of the General Services Administration,
additional laws and requirements apply. The handling of any
controlled substances must comply with the U.S. Controlled
Substances Act and Controlled Substances Import and Export Act.
Products must meet applicable child-resistant packaging
requirements under the U.S. Poison Prevention Packaging Act.
Manufacturing, sales, promotion and other activities are also
potentially subject to federal and state consumer protection and
unfair completion laws.
63
The
distribution of pharmaceutical products is subject to additional
requirements and regulations, including extensive record-keeping,
licensing, storage and security requirements intended to prevent
the unauthorized sale of pharmaceutical products.
Third-Party Payer Coverage and Reimbursement
Significant
uncertainty exists as to the coverage and reimbursement status of
REM-001 Therapy should it ultimately obtain regulatory approval. In
both the United States and foreign markets, our ability to
commercialize REM-001 Therapy successfully, and to attract
commercialization partners for REM-001 Therapy, depends in
significant part on the availability of adequate financial coverage
and reimbursement from third-party payers, including, in the United
States, governmental payers such as the Medicare and Medicaid
programs, managed care organizations, and private health insurers.
Medicare is a federally funded program managed by the CMS, through
local fiscal intermediaries and carriers that administer coverage
and reimbursement for certain healthcare items and services
furnished to the elderly and disabled. Medicaid is an insurance
program for certain categories of patients whose income and assets
fall below state defined levels and who are otherwise uninsured
that is both federally and state funded and managed by each state.
The federal government sets general guidelines for Medicaid and
each state creates specific regulations that govern its individual
program. Each payer has its own process and standards for
determining whether it will cover and reimburse a procedure or
particular product. Private payers often rely on the lead of the
governmental payers in rendering coverage and
reimbursement determinations. Therefore, achieving favorable CMS
coverage and reimbursement is usually a significant gating issue
for successful introduction of a new product. The competitive
position of REM-001 Therapy will depend in part, upon the extent of
coverage and adequate reimbursement for such product and for the
procedures in which such product is used. Prices at which we or our
customers seek reimbursement for REM-001 Therapy can be subject to
challenge, reduction or denial by the government and other
payers.
The
United States Congress and state legislatures may, from time to
time, propose and adopt initiatives aimed at cost containment,
which could impact our ability to sell REM-001 Therapy profitably.
For example, in March 2010, President Obama signed into law the
Patient Protection and Affordable Care Act, as amended by the
Health Care and Education Affordability Reconciliation Act of 2010
(collectively, the “Health Care Reform Law”), a
sweeping law intended to broaden access to health insurance, reduce
or constrain the growth of healthcare spending, enhance remedies
against fraud and abuse, add new transparency requirements for
healthcare and health insurance industries, impose new taxes and
fees on the health industry and impose additional health policy
reforms. Effective October 1, 2010, the Health Care Reform Law
revised the definition of “average manufacturer price”
for reporting purposes, which could increase the amount of Medicaid
drug rebates to states once the provision is effective. Further,
the law imposes a significant annual fee on companies that
manufacture or import branded prescription drug products.
Substantial new provisions affecting compliance have also been
enacted, which may require us to modify our business practices with
healthcare practitioners. Although it is too early to determine the
full effect of the Health Care Reform Law, the law appears likely
to continue the pressure on pharmaceutical pricing, especially
under the Medicare program, and may also increase our regulatory
burdens and operating costs. Moreover, in the coming years,
additional changes could be made to governmental healthcare
programs that could significantly impact the success of REM-001
Therapy.
The
cost of pharmaceuticals continues to generate substantial
governmental and third-party payer interest. We expect that the
pharmaceutical industry will experience pricing pressures due to
the trend toward managed healthcare, the increasing influence of
managed care organizations and additional legislative proposals.
Our results of operations could be adversely affected by current
and future healthcare reforms.
Some
third-party payers also require pre-approval of coverage for new or
innovative devices or drug therapies before they will reimburse
healthcare providers that use such therapies. While we cannot
predict whether any proposed cost-containment measures will be
adopted or otherwise implemented in the future, the announcement or
adoption of these proposals could have a material adverse effect on
our ability to obtain adequate prices REM-001 Therapy and operate
profitably.
In
addition, in some foreign countries, the proposed pricing for a
drug must be approved before it may be lawfully marketed. The
requirements governing drug pricing vary widely from country to
country. For example, the European Union provides options for its
member states to restrict the range of medicinal products for which
their national health insurance systems provide reimbursement and
to control the prices of medicinal products for human
use. A member state may approve a specific price for the medicinal
product or it may instead adopt a system of direct or indirect
controls on the profitability of the company placing the medicinal
product on the market. There can be no assurance that any country
that has price controls or reimbursement limitations for
pharmaceutical products will allow favorable reimbursement and
pricing arrangements for any of our products. Historically,
products launched in the European Union do not follow price
structures of the United States and generally tend to be
significantly lower.
64
Other Healthcare Laws and Compliance Requirements
In the
United States, our activities are potentially subject to regulation
by various federal, state and local authorities in addition to the
FDA, including the CMS, other divisions of the United States
Department of Health and Human Services (e.g., the Office of
Inspector General), the United States Department of Justice and
individual United States Attorney offices within the Department of
Justice, and state and local governments. These regulations
include:
|
|
●
|
the
federal healthcare program anti-kickback law which prohibits, among
other things, persons from soliciting, receiving or providing
remuneration, directly or indirectly, to induce either the referral
of an individual, for an item or service or the purchasing or
ordering of a good or service, for which payment may be made under
federal healthcare programs such as the Medicare and Medicaid
programs;
|
|
|
●
|
federal
false claims laws which prohibit, among other things, individuals
or entities from knowingly presenting, or causing to be presented,
claims for payment from Medicare, Medicaid, or other government
reimbursement programs that are false or fraudulent. The government
may assert that a claim including items or services resulting from
a violation of the federal healthcare program anti-kickback law or
related to off-label promotion constitutes a false or fraudulent
claim for purposes of the federal false claims laws;
|
|
|
●
|
the
federal Health Insurance Portability and Accountability Act of 1996
(“HIPPA”) which prohibits executing a scheme to defraud
any healthcare benefit program or making false statements relating
to healthcare matters and which also imposes certain requirements
relating to the privacy, security and transmission of individually
identifiable health information;
|
|
|
●
|
the
federal transparency requirements under the Health Care Reform Law
requires manufacturers of drugs, devices, biologics, and medical
supplies to report to the Department of Health and Human Services
information related to physician payments and other transfers of
value and physician ownership and investment
interests;
|
|
|
●
|
the
Federal Physician Payments Sunshine Act within the Patient
Protection and Affordable Care Act, and its implementing
regulations, require that certain manufacturers of drugs, devices,
biological and medical supplies for which payment is available
under Medicare, Medicaid or the Children’s Health Insurance
Program (with certain exceptions) to report information related to
certain payments or other transfers of value made or distributed to
physicians and teaching hospitals, or to entities or individuals at
the request of, or designated on behalf of, the physicians and
teaching hospitals and to report annually certain ownership and
investment interests held by physicians and their immediate family
members; and
|
|
|
●
|
state
law equivalents of each of the above federal laws, such as
anti-kickback and false claims laws, which may apply to items or
services reimbursed by any third party payer, including commercial
insurers, and state laws governing the privacy and security of
health information in certain circumstances, many of which differ
from each other in significant ways and often are not preempted by
federal laws, thus complicating compliance efforts.
|
In
addition, we may be subject to data privacy and security regulation
by both the federal government and the states in which we conduct
our business. HIPAA, as amended by the Health Information
Technology for Economic and Clinical Health Act
(“HITECH”), and its implementing regulations, imposes
certain requirements relating to the privacy, security and
transmission of individually identifiable health information. Among
other things, HITECH makes HIPAA’s privacy and security
standards directly applicable to “business
associates”—independent contractors or agents of
covered entities that receive or obtain protected health
information in connection with providing a service on behalf of a
covered entity. HITECH also created four new tiers of civil
monetary penalties, amended HIPAA to make civil and criminal
penalties directly applicable to business associates and possibly
other persons, and gave state attorneys general new authority to
file civil actions for damages or injunctions in federal courts to
enforce the federal HIPAA laws and seek attorneys’ fees and
costs associated with pursuing federal civil actions. In addition,
state laws govern the privacy and security of health information in
certain circumstances, many of which differ from each other in
significant ways and may not have the same effect, thus
complicating compliance efforts.
65
Employees
We
currently have five full-time employees and one
part-time employee, and plan to hire additional personnel over the
next year, as needed, to perform administration, finance, clinical,
regulatory and business development functions. We believe our
relations with our employees are good. We anticipate that the
number of employees may grow as we continue to develop our current
product or if we develop new product candidates in the future. In
addition, we utilize and will continue to utilize consultants,
clinical research organizations and third parties to perform our
pre-clinical studies, clinical studies, manufacturing and
regulatory functions.
Facilities
Due to the stage of
development of our REM-001 Therapy, we do not currently have need
for laboratory space. Our principal offices are 4365 US 1 South,
Suite 211, Princeton, NJ 08540. We have signed a three year lease,
commencing December 2016, and entered into an amendment to expand
the leased office space effective April 1, 2017. Basic rent in
connection with the amended lease is $6,026 per month.We believe
our facilities are suitable and adequate for our foreseeable
needs.
Legal Matters
We are
not currently subject to any material legal proceedings; however,
we may from time to time become a party to various legal
proceedings arising in the ordinary course of our
business.
66
MANAGEMENT AND BOARD OF DIRECTORS
The
following sets forth certain information with respect to our
executive officers and directors.
|
Name
|
Age
|
Position(s)
|
|
Frank
Pilkiewicz, PhD
|
70
|
President,
Chief Executive Officer, Director, Chairman of the
Board
|
|
Steven
Rychnovsky, PhD
|
58
|
Vice
President of Operations and Product Development
|
|
Jane
Maida
|
62
|
Chief
Financial Officer and Vice President of Finance
|
|
Allen
Bloom, PhD, JD
|
73
|
Director
|
|
Roman
Perez-Soler, MD
|
63
|
Director
|
|
David
P. Hochman
|
41
|
Director
|
|
Tim
McInerney
|
56
|
Director
|
________________________
Management
Frank Pilkiewicz, PhD, President, Chief Executive Officer,
Director, Chairman of the Board
Dr.
Pilkiewicz has served as our President and Chief Executive
Officer since the 2016 merger transaction (the ?Merger?). Dr.
Pilkiewicz formed Adgero Biopharmaceuticals, Inc. (?Adgero?), our
operating subsidiary since the Merger, in 2007 for the purposes of
acquiring biotech and pharmaceutical technologies with high
commercial potential and has served as its President and Chief
Executive Officer and Chairman since its founding. From 2010 to
2014, Dr. Pilkiewicz was the Principal at Pilkiewicz Consulting and
Development where he provided business and technical consulting
services, primarily to the pharmaceutical and biotechnology
industry and venture and early stage investment groups focused on
those areas. Dr. Pilkiewicz was the President and Chief Executive
Officer of CellXplore, Inc. from 2008 to 2010, a biotechnology
company focused on the treatment and diagnosis of breast cancer.
Prior to joining CellXplore, Inc., from 2000 to 2006 Dr. Pilkiewicz
was the President and Chief Executive Officer of Transave, Inc., a
pharmaceutical company that he founded following his invention of
Transave, Inc.?s inhalation drug delivery technology for treating
serious pulmonary diseases. Transave, Inc. was acquired by Insmed
in 2010 and its lead product, Arykace, developed by the inhalation
drug delivery technology invented by Dr. Pilkiewicz, is currently
awaiting approval. Previously, from 1986 to 1992 Dr. Pilkiewicz
served as Vice President of Research & Development at The
Liposome Company, Inc. where his team developed a number of
commercial liposomal products including its lead products;
Myocet® for metastatic
breast cancer and Abelcet® for serious fungal infections. In
2000, with these lead products in late stage development, the
Liposome Company was acquired by Elan. Dr. Pilkiewicz served as a
member of the board of directors of CellXplore, Inc. from 2008 to
2010, Charis Pharmaceutical, Inc. from 2007 to 2010, Transave, Inc.
from 1999 to 2006, and OncoTherapeutics, Inc. from 1992 to 1995. He
is currently serving on the Life Sciences Advisory Board for the
New Jersey Economic Development Authority. Dr. Pilkiewicz earned a
B.S. in chemistry from Saint Peter’s University, an M.S. in
organic chemistry from Rutgers University, and has a PhD in organic
chemistry from Rutgers University. He was a Postdoctoral Research
Fellow at Columbia University in organic chemistry. Dr. Pilkiewicz was
selected as a director because of his business and leadership
experience in the biopharmaceutical sector, as well as a result of
having served as a director since our inception; his broad
scientific background is also seen as an asset to
us.
Steven Rychnovsky, PhD, Vice President of Operations and Product
Development
Dr.
Rychnovsky has served as our Vice President of Operations and
Product Development since the Merger, and has held identical
positions with Adgero since 2012. Dr. Rychnovsky is experienced in
all aspects of the photodynamic therapy (“PDT”)
developed by Miravant Medical Technologies, and its wholly-owned
subsidiaries, a former public pharmaceutical and research
development company (collectively, “Miravant”), and,
since 2012, Dr. Rychnovsky has worked with Dr. Pilkiewicz to
develop Adgero’s business strategy and plans for
commercialization of the REM-001 Therapy product, consisting of
three parts, the laser light source, the light delivery device and
the drug REM-001 (collectively, the “REM-001 Therapy”).
From 2008 to 2012 Dr. Rychnovsky served as a consultant to St.
Cloud Investments where his role was maintaining the Miravant
assets and identifying a party to license or purchase those assets
and pursue commercial development. In 2012, Dr. Rychnovsky was a
co-founder of Endocole, LLC, a medical device company. He worked
with Endocole from 2012 to 2015, where he focused on raising
initial grant financing and worked in the development and
preclinical testing of its proof-of-concept device and was a
co-inventor of Endocole, LLC’s key intellectual property.
EndoCole has completed its initial preclinical studies and is
currently raising private funding to initiate a clinical study.
From 2008 to 2012, Dr. Rychnovsky was a Senior Research Physicist
at Sotera Defense Solutions, Inc., a Naval Research Laboratory
optical nanotechnology group focused on applied research in optical
materials and devices. From 2003 to 2008, Dr. Rychnovsky served as
the Cardiovascular Program Manager at Miravant, where he invented
key elements of Miravant’s cardiovascular technology. Dr.
Rychnovsky also served as the Director of Systems and Engineering
at Miravant where he managed the team responsible for development
of Miravant’s PDT light delivery technology. During his time
at Miravant, Dr. Rychnovsky was involved in new product development
for its cancer, ophthalmology and cardiovascular programs,
including clinical development of REM-001 and related PDT
technology. Dr. Rychnovsky earned a B.S. in electrical engineering
from Iowa State University, an M.S. in electrical engineering from
the University of Minnesota, and has a PhD in photonics from the
University of Iowa.
67
Jane M. Maida, Chief Financial Officer and Vice President of
Finance
Ms.
Maida has served as our Chief Financial Officer since February
2017. Ms. Maida has over 20 years of senior financial experience
with emerging biotechnology companies. From March 2011 to February
2017, Ms. Maida was the Chief Financial Officer at Signum
Biosciences, Inc. From September 2004 through August 2008, Ms.
Maida served at Abeille Pharmaceuticals, Inc., as Vice President
and Chief Financial Officer. From February 2002 to August 2004 she
worked as a consultant providing confidential financial services,
assisting private companies with funding opportunities, financial
and corporate structures, and managerial issues, as well as serving
as acting Chief Financial Officer. From May 2000 to February 2002,
Ms. Maida served at Physiome Sciences, Inc. (which merged into
Predix Pharmaceuticals, Inc. in 2003), as Vice President, Chief
Financial Officer, and Treasurer. From March 1997 to May 2000, Ms.
Maida served at Cytogen Corporation (which merged into EUSA in
2008), as Vice President of Finance and Administration, and Chief
Accounting Officer. Ms. Maida holds CPA certificates from New York
and New Jersey. Ms. Maida earned a B.S.
in Education from University of Pennsylvania and a Master of
Science in Accountancy from State University of New York at
Albany.
Significant Employees
Laura Edgerly-Pflug, Vice President, Manufacturing Operations and
Quality Control
Ms.
Edgerly-Pflug has served as our Vice President of Manufacturing
Operations and Quality Control since October 2016. From October
2013 to October 2016, Ms. Edgerly-Pflug was the Owner at Pflug
BioPharm Solutions where she provided strategic direction and
implementation to clients in the areas of manufacturing, quality
assurance, new technologies, new products and life cycle
initiatives. From 2012 through 2013, Ms. Edgerly-Pflug served at
Insmed Incorporated, formerly Transave Inc., as Vice President of
Technical Operations and Chemistry, Manufacturing and Controls
where she was responsible for product development and manufacturing
of sterile liposomal products from preclinical development through
commercialization. From 2006 through 2012 she served at Insmed as
Executive Director of Technical Operations and Chemistry,
Manufacturing and Controls. Ms. Edgerly-Pflug
earned a B.S. in Chemistry from Kean College of New
Jersey.
Directors
Frank Pilkiewicz, PhD, President, Chief Executive Officer,
Director, Chairman
See
description under Management.
Allen Bloom, PhD, JD, Director
Dr.
Bloom became one of our directors in April 2016, in connection with
the Merger. Since 2004, Dr. Bloom has been an independent business
consultant in the life sciences sector specializing in business
development, strategic partnering and licensing and evaluation of
early stage investment opportunities. He has over forty years of
experience in the biotechnology and pharmaceutical sector
specializing primarily in business development and intellectual
property. He has served as a consultant to Adgero since its
inception. From 1994 until 2003, Dr. Bloom was a partner at the law
firm of Dechert LLP, where he served as Co-Chair of the
Intellectual Property Group and headed the patent practice group
focusing on biotechnology, pharmaceuticals and medical devices.
From 1985 until 1994, Dr. Bloom was Vice President, General Counsel
and Secretary of The Liposome Company, Inc., a pharmaceutical
company that developed and commercialized liposomal and other
lipid-based drug delivery systems. As General Counsel, his
responsibilities included managing corporate and securities
matters, patent, regulatory and licensing activities. Prior to his
position at The Liposome Company, Inc., Dr. Bloom served as Patent
Attorney and Patent Counsel for Pfizer, Inc. and RCA Corporation,
respectively. Dr. Bloom has served as a director of Redpoint Bio
Corporation, Unigene Laboratories, Inc. and Cytogen Corporation.
Dr. Bloom served as Lead Director and Chair of the Audit Committee
at Unigene Laboratories, Inc. and also served as a member of the
Audit Committee at Redpoint Bio Corporation. Dr. Bloom earned a
B.S. in chemistry from Brooklyn College, a JD degree from New York
Law School and a PhD in organic chemistry from Iowa State
University. Dr. Bloom was selected
as a director because of his background in the biotechnology and
pharmaceutical sector, and his legal background is seen as an asset
to us.
Roman Perez-Soler, MD, Director
Dr.
Perez-Soler became one of our directors in April 2016, in
connection with the Merger. Since July 2001, Dr. Perez-Soler has
been the Gutman Professor of Oncology and Chairman of the
Department of Oncology at Montefiore
Medical Center, and Professor
of Medicine and Molecular Pharmacology and Chief of the Division of
Medical Oncology at the Albert Einstein College of Medicine. He is
currently Deputy Director of the Albert Einstein Cancer Center. Dr.
Perez-Soler is currently a member of the Board of Scientific
Advisors of the National Cancer Institute. Dr. Perez-Soler
is an inventor in 17 patents, and is well known in cancer research
with over 200 scientific publications and several book chapters
including in the fields of drug delivery, target therapies for
solid tumors and his work in the field of anti-epidermal growth
factor receptor therapies has received myriad of citations. He
has been the co-founder of two
pharmaceutical companies, Argus Pharmaceuticals, Inc. and Transave,
Inc. and has served as a consultant to a large number of
pharmaceutical companies. He served as a director for Transave,
Inc. from 2000 to 2006. Dr. Perez-Soler earned his MD from
Universidad Autonoma in Barcelona, where he also completed a
residency in internal medicine. He completed his fellowship in
Medical Oncology at the University of Texas, MD Anderson Hospital
Cancer Center and is a past Fulbright
Scholar. Dr. Roman Perez-Soler
was selected as a director because of his experience in oncology
and business experience in other biopharmaceutical
companies.
68
David P. Hochman, Director
Mr.
Hochman has been one of our directors since November 2015. Since
June 2006, Mr. Hochman has been Managing Partner of Orchestra
Medical Ventures, LLC, an investment firm that employs a strategy
to create, build and invest in medical technology companies
intended to generate substantial clinical value and superior
investor returns. He is also President of Accelerated Technologies,
Inc., a medical device accelerator company managed by Orchestra.
Mr. Hochman is the Vice Chairman and a Director of Naked Brand
Group Inc. He has over seventeen years of venture capital and
investment banking experience. Mr. Hochman is the Chairman of Vital
Access Corp. and serves as a director of MOTUS GI Medical
Technologies Ltd., Caliber Therapeutics, BackBeat Medical (where he
is also President), FreeHold Surgical, Maternity Neighborhood, and
Prescient Medical, Inc., an interventional cardiovascular device
company Mr. Hochman co-founded in 2004. Prior to joining Orchestra,
Mr. Hochman was Chief Executive Officer of Spencer Trask Edison
Partners, LLC, an investment partnership focused on early stage
healthcare companies. He was also Managing Director of Spencer
Trask Ventures, Inc. during which time he led financing
transactions for over twenty early-stage companies. Mr. Hochman was
a board advisor of Health Dialog Services Corporation, a leader in
collaborative healthcare management that was acquired in 2008 by
the British United Provident Association for $750 million. From
2005 to 2007, he was a co-founder and director of PROLOR Biotech,
Inc., a biopharmaceutical company developing longer-lasting
versions of approved therapeutic proteins, which was purchased by
Opko Health (NYSE: OPK) in 2013 for over $600 million. Mr. Hochman
also currently serves as a board member of Corbus Pharmaceuticals
Holdings, Inc. (NASDAQ: CRBP), a clinical stage biopharmaceutical
company focused on the development and commercialization of novel
therapeutics to treat rare, life-threating inflammatory-fibrotic
diseases with clear unmet medical needs, and two non-profit
organizations: the Citizens Committee for New York City and the
Mollie Parnis Livingston Foundation. He has a B.A. degree with
honors from the University of Michigan. Mr. Hochman was
selected as a director due to his leadership experience at other
public companies, including pharmaceutical companies, his financial
experience and his expertise in governance
matters.
Tim McInerney, Director
Mr.
McInerney became one of our directors in April 2016, in connection
with the Merger. Since 2007, Mr. McInerney has been a principal at
Two River Group, a merchant bank involved in founding, financing or
managing companies that are focused on developing preventative and
therapeutic technologies for a broad spectrum of disease areas
including oncology, cardiovascular disease and neurological
disorders, and a Partner of Riverbank Capital Securities, Inc., a
Financial Industry Regulatory Authority
(“FINRA”)/Securities Investor
Protection Corporation (“SIPC”)
member broker dealer that provides capital raising and advisory
services to companies primarily in the life science industry,
including Two River portfolio companies. From 1992 to March 2007,
Mr. McInerney was a Managing Director of Paramount BioCapital, Inc.
where he oversaw the overall distribution of Paramount's private
equity product. Prior to 1992, Mr. McInerney was a research analyst
focusing on the biotechnology industry at Ladenburg, Thalmann &
Co., Inc. Prior to that, Mr. McInerney held equity sales positions
at Bear Stearns & Co. and Shearson Lehman Brothers, Inc. and
worked in sales and marketing for Bristol-Myers Squibb Co.
(NYSE:BMY). Mr. McInerney currently serves on the boards of
Emisphere Technologies, Inc. (OTCBB:EMIS), a specialty
pharmaceutical company partnered with global pharmaceutical
companies for the development of new orally delivered therapeutics,
and Edgemont Pharmaceuticals, LLC, a specialty pharmaceutical
company focused on developing, acquiring, and commercializing
improved versions of widely-prescribed products that provide
meaningful clinical benefits over the current standard of care in
treating psychiatric disorders. From 2008 to 2015, Mr. McInerney
served as Chairman of the Board of Insite Vision, Inc. and from
2005 to 2015, he served as a director of Ziopharm Oncology Inc.
(NASDAQ: ZIOP). Mr. McInerney received his B.S. in pharmacy from
St. John's University. He also completed a post-graduate residency
at the New York University Medical Center in drug information
systems. Mr. McInerney was
selected as a director due to his leadership experience at other
public companies, and his financial and accounting experience and
his expertise in governance matters.
Committees of the Board
Our
board of directors has established an Audit Committee, a
Compensation Committee, and a Nominating and Corporate Governance
Committee. Our board of directors may establish other committees to
facilitate the management of our business. The composition and
functions of each committee are described below. Members serve on
these committees until their resignation or until otherwise
determined by our board of directors. Each of these committees
operate under a charter that has been approved by our board of
directors, which will be available on our website.
Audit Committee. Our Audit Committee
consists of Mr. McInerney, Dr. Bloom, and Dr. Perez-Soler, with Mr.
McInerney serving as the Chairman of the Audit Committee. Our board
of directors has determined that the three directors currently
serving on our Audit Committee are independent within the meaning
of the NASDAQ Marketplace Rules and Rule 10A-3 under the Exchange
Act. In addition, our board of directors has determined that Mr.
McInerney qualifies as an audit committee financial expert within
the meaning of SEC regulations and The NASDAQ Marketplace
Rules.
69
The
Audit Committee oversees and monitors our financial reporting
process and internal control system, reviews and evaluates the
audit performed by our registered independent public accountants
and reports to the Board any substantive issues found during the
audit. The Audit Committee is directly responsible for the
appointment, compensation and oversight of the work of our
registered independent public accountants. The Audit Committee
reviews and approves all transactions with affiliated
parties.
Compensation Committee. Our Compensation
Committee consists of Mr. McInerney, Dr. Bloom, and Dr.
Perez-Soler, with Dr. Perez-Soler serving as the Chairman of the
Compensation Committee. Our board of directors has determined that
the three directors currently servicing on our Compensation
Committee are independent under the listing standards, are
“non-employee directors” as defined in rule 16b-3
promulgated under the Exchange Act and are “outside
directors” as that term is defined in Section 162(m) of the
Internal Revenue Code of 1986, as amended.
The
Compensation Committee provides advice and makes recommendations to
the Board in the areas of employee salaries, benefit programs and
director compensation. The Compensation Committee also reviews and
approves corporate goals and objectives relevant to the
compensation of our President, Chief Executive Officer, and other
officers and makes recommendations in that regard to the Board as a
whole.
Nominating and Corporate Governance
Committee. Our Nominating and Corporate Governance Committee
consists of Mr. McInerney, Dr. Bloom, and Dr. Perez-Soler, with Mr.
Bloom serving as the Chairman of the Nominating and Corporate
Governance Committee. The Nominating and Corporate Governance
Committee nominates individuals to be elected to the Board by our
stockholders. The Nominating and Corporate Governance Committee
considers recommendations from stockholders if submitted in a
timely manner in accordance with the procedures set forth in our
Bylaws and will apply the same criteria to all persons being
considered. All members of the Nominating and Corporate Governance
Committee are independent directors as defined under the NASDAQ
listing standards.
Director Independence
Our
board of directors undertook a review of its composition, the
composition of its committees and the independence of each
director. Based upon information requested from and provided by
each director concerning his or her background, employment and
affiliations, including family relationships, our board of
directors has determined that Mr. McInerney, Dr. Bloom, and Dr.
Perez-Soler do not have a relationship that would interfere with
the exercise of independent judgment in carrying out the
responsibilities of a director and that each of these directors is
“independent” as that term is defined under the Rules
of the NASDAQ Stock Market and the SEC.
Scientific Advisory Board
We
believe in seeking and attracting scientific and clinical leaders
in the field of oncology to provide counsel and support our growth.
We intend for our scientific advisory board to include the
following individuals, and we expect to add additional members in
the future.
Ron R. Allison, MD
Dr.
Allison is a board certified Radiation Oncologist with
expertise in PDT. Dr. Allison retired as Professor and the Chair of
Radiation Oncology at the Brody School of Medicine at the
University of North Carolina and the Director of the Leo W. Jenkins
Cancer Center. Dr. Allison has authored over 100 oncology
publications and has delivered over 200 presentations at oncology
meetings and forums worldwide. Dr. Allison's area of expertise is
PDT, and has been a principle investigator for multiple PDT
clinical trials, treated numerous patients with PDT and
served as a reviewer and editor of grants and articles in this
domain. Dr. Allison has entered into a Scientific Advisory Board
agreement with us as of June 23, 2016, for an initial term of one
year. The agreement provides for automatic term extensions for
consecutive periods of one year each, unless a notice of
non-renewal is given by either Dr. Allison or us at least 30 days
prior to any renewal date.
70
Thomas S. Mang, PhD
Dr.
Mang is an Associate Professor and Director of Research for the
Oral and Maxillofacial Surgery Department at the University at
Buffalo, School of Dental Medicine. He also is the Director of the
Great Lakes Biomedical Laser Center at the UB School of Dental
Medicine. Dr. Mang previously served as Director of the PDT Clinic
at Roswell Park Cancer Institute, in Buffalo NY. Dr. Mang is an
expert in PDT with over 30 years of experience, and has experience
in the corporate aspects of PDT having previously held the position
of Clinical Manager for PDT at Axcan Pharma Inc. He was also
engaged in prior clinical work with our lead product candidate, the
REM-001 Therapy. Dr. Mang has entered into a Scientific Advisory
Board agreement with us as of August 12, 2016, for an initial term
of one year. The agreement provides for automatic term extensions
for consecutive periods of one year each, unless a notice of
non-renewal is given by either Dr. Mang or us at least 30
days prior to any renewal date.
Roman Perez-Soler, MD
See
description under Management.
Stephen B. Solomon, MD
Dr.
Solomon is Chief of Interventional Radiology in the Department of
Radiology at Memorial Sloan Kettering and is the Co-Director of
Memorial Sloan Kettering’s Center for Image-Guided
Intervention. As an interventional radiologist, Dr. Solomon
specializes in minimally invasive treatments carried out using
image guidance, including x-ray, CT, ultrasound, and MRI and has
expertise in robotics and other image-guided therapies. During his
career, he has authored over 200 articles in peer-reviewed
scientific journals and has been the principal investigator on
multiple clinical trials in cancer. Dr. Solomon has entered into a Scientific Advisory
Board agreement with us as of February 3, 2017, for an initial term
of one year. The agreement provides for automatic term extensions
for consecutive periods of one year each, unless a notice of
non-renewal is given by either Dr. Solomon or us at least 30 days
prior to any renewal date.
Leonard A. Farber, MD
Dr.
Farber is a board certified physician in Radiation Oncology and an
Assistant Professor in the Department of Radiation Oncology at
Weill- Cornell Hospital. Dr. Farber previously served as the
Chairman of Radiation Oncology at Staten Island University
Hospital. Dr. Farber specializes in adult radiation oncology, with
particular focus on breast and gastrointestinal malignancies,
lymphomas, lung cancers, and head and neck cancer and has expertise
in high dose rate brachytherapy for various types of cancers,
including breast, skin, and gynecologic malignancies, and
intracranial and body stereotactic radiation therapy. Dr. Farber
has entered into a Scientific Advisory Board agreement with us as
of June 15, 2016, for an initial term of one year. The agreement
provides for automatic term extensions for consecutive periods of
one year each, unless a notice of non-renewal is given by either
Dr. Farber or us at least 30 days prior to any renewal
date.
Code of Business Conduct and Ethics
We have
adopted a written code of business conduct and ethics that applies
to our employees, officers and directors. A current copy of the
code will be posted on the Corporate Governance section of our
website, which will be located at www.adgerobiopharm.com. We intend
to disclose future amendments to certain provisions of our code of
business conduct and ethics, or waivers of such provisions
applicable to any principal executive officer, principal financial
officer, principal accounting officer or controller, or persons
performing similar functions, and our directors, on our website
identified above or in a current report on Form 8-K.
71
Limitation of Directors Liability and Indemnification
The
Delaware General Corporation Law authorizes corporations to limit
or eliminate, subject to certain conditions, the personal liability
of directors to corporations and their stockholders for monetary
damages for breach of their fiduciary duties. Our certificate of
incorporation limits the liability of our directors to the fullest
extent permitted by Delaware law. In addition, we have entered into
indemnification agreements with certain of our directors and
officers whereby we have agreed to indemnify those directors and
officers to the fullest extent permitted by law, including
indemnification against expenses and liabilities incurred in legal
proceedings to which the director or officer was, or is threatened
to be made, a party by reason of the fact that such director or
officer is or was a director, officer, employee or agent of the
Company, provided that such director or officer acted in good faith
and in a manner that the director or officer reasonably believed to
be in, or not opposed to, the best interests of the
Company.
We have
director and officer liability insurance to cover liabilities our
directors and officers may incur in connection with their services
to us, including matters arising under the Securities Act of 1933,
as amended (the “Securities Act”). Our certificate of
incorporation and bylaws also provide that we will indemnify our
directors and officers who, by reason of the fact that he or she is
or was one of our officers or directors of our company, is involved
in any action, suit or proceeding, whether civil, criminal,
administrative or investigative related to their board role with
the company.
There
is no pending litigation or proceeding involving any of our
directors, officers, employees or agents in which indemnification
will be required or permitted. We are not aware of any threatened
litigation or proceeding that may result in a claim for such
indemnification.
72
EXECUTIVE COMPENSATION
Summary Compensation Table
No
named executive officers received compensation during the fiscal
year ended December 31, 2015. The following table presents
information regarding the total compensation awarded to, earned by,
or paid to our chief executive officer and the most
highly-compensated executive officer (other than the chief
executive officer) who was serving as an executive officer as of
December 31, 2016 for services rendered in all capacities to us for
the year ended December 31, 2016. These individuals
are our named executive officers for 2016.
|
Name and Principal Position
|
Year
|
Salary
($) (1)
|
Bonus
($)
|
Option Awards
($) (2)
|
Total
($)
|
|
Frank
Pilkiewicz, PhD
|
2016
|
254,479
|
261,750
|
776,446
|
1,292,675
|
|
Chief Executive Officer and Chief Financial Officer
|
|||||
|
Steven
Rychnovsky, PhD
|
2016
|
181,563
|
74,700
|
388,223
|
644,486
|
|
Vice President of Operations and Product Development
|
|||||
(1)
Each
of the named executive officers began receiving salary on April 8,
2016.
(2)
Amounts reflect
the grant date fair value of option awards granted in 2016 in
accordance with Accounting Standards Codification Topic 718. For
information regarding assumptions underlying the valuation of
equity awards, see Note 2 and Note 4 to our Condensed Consolidated
Financial Statements. These amounts do not correspond to the actual
value that may be received by the named executive officers if the
stock options are exercised.
Employment Agreements
In
connection with the Merger, we entered into an employment agreement
with Frank Pilkiewicz, PhD, which is effective for a period of
three years, and a Non-Disclosure and Invention Assignment
Agreement (“NDIAA”). Under the terms of Dr.
Pilkiewicz’s employment agreement, he holds the position of
Chief Executive Officer, President and Chairman of the Board and
Treasurer and receives a base salary of $349,000 annually, subject
to adjustments in the discretion of the Board of Directors;
provided, however, that the base salary shall be increased upon the
achievement of certain milestones, specifically, an increase of
$50,000 upon a future closing of an additional underwritten round
of financing of at least $20 million, exclusive of proceeds from
exercise of investor warrants, that results in listing of the
Company’s shares on a major exchange such as Nasdaq or the
New York Stock Exchange if the shares are not already so listed,
and an increase of $150,000 upon approval of a Company New Drug
Application (“NDA”). On March 3, 2017, our
Board of Directors approved an adjustment to the base salary for
Dr. Pilkiewicz to $400,000. In addition, Dr. Pilkiewicz is
eligible to receive an annual bonus, which is targeted at up to 75%
of his base salary but which may be adjusted by our Board of
Directors based on his individual performance and our performance
as a whole. On July 29, 2016, Dr. Pilkiewicz received a grant
of options covering 335,958 shares of common stock at an exercise
price of $5.00 per share. These options vest in three (3) equal
annual installments, beginning on the first anniversary of the date
of grant, and continuing on the second and third anniversaries,
provided that Dr. Pilkiewicz remains employed by the Company
through each applicable vesting date. In addition,
pursuant to the terms of his employment agreement, Dr. Pilkiewicz
is eligible to receive, from time to time, equity awards under our
existing equity incentive plan, or any other equity incentive plan
we may adopt in the future, and the terms and conditions of such
awards, if any, will be determined by our Board of Directors or
Compensation Committee, in their discretion. If we terminate
Dr. Pilkiewicz’s employment without cause or he terminates
his employment for good reason, we are required to provide him
severance including (i) continued payments of twelve (12) months of
his annual base salary, paid in installments in accordance with the
Company’s regular payroll practices, (ii) payment for his
health care coverage costs at such rate as in effect as of the
termination date for a period of twelve (12) months, and (iii) an
additional twelve (12) months of service vesting credit for each of
his stock options outstanding at the time of his termination, and
all of his vested options will remain exercisable for up to a
twelve (12)-month period measured from his termination date (or
earlier expiration of the options term). Additionally, in the
event that we terminate Dr. Pilkiewicz’s employment without
cause or he terminates his employment for good reason within
twenty-four (24) months following a change in control, Dr.
Pilkiewicz will be entitled to (i) continued payments of eighteen
(18) months of his annual base salary, paid in installments in
accordance with the Company’s regular payroll practices, (ii)
payment for his health care coverage costs at such rate as in
effect as of the termination date for a period of twelve (12)
months, and (iii) an additional eighteen (18) months of service
vesting credit for each of his stock options outstanding at the
time of his termination, and all of his vested options will remain
exercisable for a period of eighteen (18) months following
termination (or earlier expiration of the options term).
Notwithstanding the foregoing, Dr. Pilkiewicz’s
post-severance salary continuation and healthcare coverage payments
as described herein will cease at such time as Dr. Pilkiewicz
becomes gainfully employed prior to the expiration of his receipt
of such severance benefits. Dr. Pilkiewicz’s severance
benefits will be subject to reduction to the extent doing so would
put him in a better after-tax position after taking into account
any excise tax he may incur under Internal Revenue Code
(“Code”) Section 4999 in connection with any change in
control of us or his subsequent termination of employment.
Dr. Pilkiewicz is also subject to non-compete and non-solicitation
provisions, which will apply during the term of his employment and
for a period of twelve months following termination of his
employment. In addition, the NDIAA contains confidentiality
provisions. In February 2017, we entered into an amendment to
the employment agreement with Dr. Pilkiewicz that provides for
accelerated vesting of all unvested equity awards granted to Dr.
Pilkiewicz pursuant to his employment agreement upon certain
terminations of employment following a change in
control.
73
In
February 2017, we entered into an employment agreement with Jane M.
Maida and a Non-Disclosure and Invention Assignment Agreement
(“NDIAA”). Ms. Maida’s employment agreement is
not for a specified term. Under the terms of Ms. Maida’s
employment agreement, she holds the position of Chief Financial
Officer and Vice President of Finance and receives a base salary of
$275,000 annually, subject to adjustments in the discretion of our
Board of Directors and/or Compensation Committee. In addition, Ms.
Maida is eligible to receive an annual bonus, which is targeted at
up to 35% of her base salary but which may be adjusted by our Board
of Directors and/or Compensation Committee based on her individual
performance and our performance as a whole. At the commencement of
Ms. Maida's employment, Ms. Maida received a grant of options
covering 100,000 shares of our common stock at an exercise price of
$5.00 per share. These options will vest in three (3) equal annual
installments, beginning on the first anniversary of the date of
grant, and continuing on the second and third anniversaries,
provided that Ms. Maida remains employed by us through each
applicable vesting date. In addition, pursuant to the terms
of her employment agreement, Ms. Maida is eligible to receive, from
time to time, equity awards under our existing equity incentive
plan, or any other equity incentive plan we may adopt in the
future, and the terms and conditions of such awards, if any, will
be determined by our Board of Directors or Compensation Committee,
in their discretion. Ms. Maida’s employment agreement
provides for accelerated vesting of all unvested equity awards
granted to Ms. Maida upon certain terminations of employment
following a change in control. If we terminate Ms. Maida’s
employment without cause or she terminates her employment for good
reason, we are required to provide her severance, provided her
termination date is at least six months after she began employment
with us, including (i) continued payments of six (6) months of her
annual base salary, paid in installments in accordance with our
regular payroll practices, (ii) reimbursement of healthcare
continuation payments under COBRA for a period of six (6) months,
and (iii) an additional three (3) months of service vesting credit
for each of her stock options outstanding at the time of her
termination, and all of her vested options will remain exercisable
for up to a six (6)-month period measured from her termination date
(or earlier expiration of the options term). If we terminate Ms.
Maida’s employment without cause or she terminates her
employment for good reason within twenty-four (24) months following
a change in control, provided her termination date is at least six
months after she began employment with us, Ms. Maida shall receive
full vesting of all unvested options upon her date of termination,
and all of her vested options will remain exercisable for a period
of six (6) months following her termination date (or earlier
expiration of the options term). Notwithstanding the foregoing, Ms.
Maida’s post-employment healthcare coverage payments as
described herein will cease at such time as Ms. Maida becomes
otherwise eligible to obtain alternative healthcare coverage from a
new employer if such event occurs prior to the expiration of her
receipt of such benefit. Ms. Maida’s severance benefits will
be subject to reduction to the extent doing so would put her in a
better after-tax position after taking into account any excise tax
she may incur under Internal Revenue Code (“Code”)
Section 4999 in connection with any change in control of us or her
subsequent termination of employment. Ms. Maida is also subject to
non-compete and non-solicitation provisions, which will apply
during the term of her employment and for a period of twelve months
following termination of her employment. In addition, the NDIAA
contains confidentiality provisions.
In
connection with the Merger, we entered into an employment agreement
with Steven Rychnovsky, PhD, which is effective for a period of two
years, and a NDIAA. Under the terms of Dr. Rychnovsky’s
employment agreement, he holds the position of Vice President of
Operations and Product Development and receives a base salary of
$249,000 annually subject to adjustments in the discretion of the
Board of Directors; provided, however, that the base salary shall
be increased upon the achievement of certain milestones,
specifically an increase of $25,000 upon a future closing of an
additional underwritten round of financing of at least $20 million,
exclusive of proceeds from exercise of investor warrants, that
results in listing of the Company’s shares on a major
exchange such as Nasdaq or the New York Stock Exchange if the
shares are not already so listed, and an increase of $75,000 upon
approval of an NDA. On March 3, 2017, our Board of Directors
approved an adjustment to the base salary for Dr. Rychnovsky to
$275,000. In addition, Dr. Rychnovsky is also eligible to
receive an annual bonus, which is targeted at up to 30% of his base
salary but which may be adjusted by our Board of Directors based on
his individual performance and our performance as a whole. On July
29, 2016, Dr. Rychnovsky received a grant of options covering
167,979 shares of common stock at an exercise price of $5.00 per
share. These options vest in three (3) equal annual installments,
beginning on the first anniversary of the date of grant, and
continuing on the second and third anniversaries, provided that Dr.
Rychnovsky remains employed by the Company through each applicable
vesting date. In addition, pursuant to the terms of his
employment agreement, Dr. Rychnovsky is eligible to receive, from
time to time, equity awards under our existing equity incentive
plan, or any other equity incentive plan we may adopt in the
future, and the terms and conditions of such awards, if any, will
be determined by our Board of Directors or Compensation Committee,
in their discretion. If we terminate Dr. Rychnovsky’s
employment without cause or he terminates his employment for good
reason, we are required to provide him severance including (i)
continued payments of six (6) months of his annual base salary,
paid in installments in accordance with the Company’s regular
payroll practices, (ii) reimbursement of his health care coverage
costs under the Consolidated Omnibus Budget Reconciliation Act
(“COBRA”) for a period of six (6) months, and (iii) an
additional six (6) months of service vesting credit for each of his
stock options outstanding at the time of his termination, and all
of his vested options will remain exercisable for up to six (6)
months measured from his termination date (or earlier expiration of
the options term). Additionally, in the event that we terminate Dr.
Rychnovsky’s employment without cause or he terminates his
employment for good reason within twenty-four (24) months following
a change in control, Dr. Rychnovsky will be entitled to an
additional nine (9) months of service vesting credit for each of
his stock options outstanding at the time of the date his
termination, and all of his vested options will remain exercisable
for a period of nine (9) months following his termination (or
earlier expiration of the term). Dr. Rychnovsky’s severance
benefits will be subject to reduction to the extent doing so would
put him in a better after-tax position after taking into account
any excise tax he may incur under Code Section 4999 in connection
with any change in control of us or his subsequent termination of
employment. Notwithstanding the foregoing, Dr. Rychnovsky’s
post-severance salary continuation and COBRA coverage as described
herein will cease at such time as Dr. Rychnovsky becomes gainfully
employed prior to the expiration of his receipt of such severance
benefits. Dr. Rychnovsky is also subject to non-compete and
non-solicitation provisions, which will apply during the term of
his employment and for a period of twelve (12) months following
termination of his employment. In addition, the NDIAA contains
certain confidentiality provisions. In February 2017, we
entered into an amendment to the employment agreement with Dr.
Rychnovsky that provides for accelerated vesting of all unvested
equity awards granted to Dr. Rychnovsky pursuant to his employment
agreement upon certain terminations of employment following a
change in control.
74
In
October 2016, we entered into an employment agreement with Laura
Edgerly-Pflug and a Non-Disclosure and Invention Assignment
Agreement (“NDIAA”). Ms. Edgerly-Pflug’s
employment agreement is not for a specified term. Under the terms
of Ms. Edgerly-Pflug’s employment agreement, she holds the
position of Vice President of Manufacturing Operations and Quality
Control and receives a base salary of $275,000 annually, subject to
adjustments in the discretion of our Board of Directors and/or
Compensation Committee. In addition, Ms. Edgerly-Pflug is eligible
to receive an annual bonus, which is targeted at up to 35% of her
base salary but which may be adjusted by our Board of Directors
and/or Compensation Committee based on her individual performance
and our performance as a whole. On October 21, 2016, Ms.
Edgerly-Pflug received a grant of options covering 150,000 shares
of common stock at an exercise price of $5.00 per share. These
options vest in three (3) equal annual installments, beginning on
the first anniversary of the date of grant, and continuing on the
second and third anniversaries, provided that Ms. Edgerly-Pflug
remains employed by us through each applicable vesting date.
In addition, pursuant to the terms of her employment
agreement, Ms. Edgerly-Pflug is eligible to receive, from time to
time, equity awards under our existing equity incentive plan, or
any other equity incentive plan we may adopt in the future, and the
terms and conditions of such awards, if any, will be determined by
our Board of Directors or Compensation Committee, in their
discretion. If we terminate Ms. Edgerly-Pflug’s
employment without cause or she terminates her employment for good
reason, we are required to provide her severance including (i)
continued payments of three (3) months of her annual base salary,
paid in installments in accordance with our regular payroll
practices, (ii) reimbursement of healthcare continuation payments
under COBRA for a period of three (3) months, and (iii) an
additional three (3) months of service vesting credit for each of
her stock options outstanding at the time of her termination, and
all of her vested options will remain exercisable for up to a six
(6)-month period measured from her termination date (or earlier
expiration of the options term). If we terminate Ms.
Edgerly-Pflug’s employment without cause or she terminates
her employment for good reason within twenty-four (24) months
following a change in control, Ms. Edgerly-Pflug shall receive an
additional six (6) months of service vesting credit for each of her
stock options outstanding at the time of her termination (instead
of three (3) months), and all of her vested options will remain
exercisable for a period of six (6) months following her
termination date (or earlier expiration of the options term).
Notwithstanding the foregoing, Ms. Edgerly-Pflug’s
post-employment healthcare coverage payments as described herein
will cease at such time as Ms. Edgerly-Pflug becomes otherwise
eligible to obtain alternative healthcare coverage from a new
employer if such event occurs prior to the expiration of her
receipt of such benefit. Ms. Edgerly-Pflug’s severance
benefits will be subject to reduction to the extent doing so would
put her in a better after-tax position after taking into account
any excise tax she may incur under Internal Revenue Code
(“Code”) Section 4999 in connection with any change in
control of us or her subsequent termination of employment. Ms.
Edgerly-Pflug is also subject to non-compete and non-solicitation
provisions, which will apply during the term of her employment and
for a period of twelve months following termination of her
employment. In addition, the NDIAA contains confidentiality
provisions. In February 2017, we entered into an amendment to
the employment agreement with Ms. Pflug that provides for
accelerated vesting of all unvested equity awards granted to Ms.
Pflug pursuant to her employment agreement upon certain
terminations of employment following a change in
control.
Outstanding Equity Awards at Fiscal Year-End
The
following table summarizes, for each of the named executive
officers, the number of shares of common stock underlying
outstanding stock options held as of December 31,
2016.
|
|
Number of securities underlying unexercised
options (#)
|
|
|
|
|
Name
|
Exercisable
|
Unexercisable
|
Option Exercise Price
($)
|
Option Expiration Date
|
|
Frank
Pilkiewicz, PhD
|
0
|
335,958
|
5.00
|
July
28, 2026
|
|
|
|
|
|
|
|
Steven
Rychnovsky, PhD
|
0
|
167,979
|
5.00
|
July
28, 2026
|
(1)
Represents
options to purchase shares of our common stock granted on July 29,
2016. The shares underlying the option vest in 3 equal annual
installments beginning on the first anniversary of the date of
grant.
75
Director Compensation
The
following table sets forth information concerning the compensation
paid to our non-employee directors during the fiscal year ended
December 31, 2016.
|
Name
|
Fees earned or paid in cash
($) (1)
|
Option awards
($) (2)
|
Total
($)
|
|
Allen
Bloom, PhD, JD (3)
|
21,923
|
89,871
|
111,794
|
|
Roman
Perez--Soler, MD (4)
|
21,923
|
89,871
|
111,794
|
|
David
P. Hochman (5)
|
18,269
|
89,871
|
108,140
|
|
Tim
McInerney (6)
|
21,923
|
89,871
|
111,794
|
(1)
Represents
fees earned for 2016 commencing on April 8, 2016
.
(2)
Amounts
reflect the aggregate grant date fair value of each stock option
granted in 2016, in accordance with the Accounting Standards
Codification Topic 718. For information regarding assumptions
underlying the valuation of equity awards, see Note 2 and Note 4 to
our Condensed Consolidated Financial Statements. These amounts do
not correspond to the actual value that may be received by the
directors if the stock options are exercised.
(3)
The
aggregate number of shares of common stock underlying stock options
outstanding as of December 31, 2016 held by Dr. Bloom was
40,000.
(4)
The
aggregate number of shares of common stock underlying stock options
outstanding as of December 31, 2016 held by Dr. Perez-Soler was
40,000.
(5)
The
aggregate number of shares of common stock underlying stock options
outstanding as of December 31, 2016 held by Mr. Hochman was
40,000.
(6)
The
aggregate number of shares of common stock underlying stock options
outstanding as of December 31, 2016 held by Mr. McInerney was
40,000.
Non-Employee Director Compensation and Advisory Board
Compensation
Our
Board of Directors approved a director compensation policy for our
non-employee directors. This policy provides for the following cash
compensation:
|
|
●
|
each
non-employee director is entitled to receive an annual fee from us
of $25,000; and
|
|
|
●
|
each
chair of a Board of Director committee will receive an annual fee
from us of $5,000.
|
Each
non-employee director on our board of directors receives an annual
option grant to purchase shares of our common stock under our
existing equity incentive plan, or any other equity incentive plan
we may adopt in the future, which shall be fully vested on the one
year anniversary of the grant date. For the fiscal year ending
December 31, 2016, our non-employee directors received a grant of
40,000 options to purchase shares of our common stock.
All
fees under the director compensation policy will be paid on a
quarterly basis in arrears and no per meeting fees will be paid. We
will also reimburse non-employee directors for reasonable expenses
incurred in connection with attending board of director and
committee meetings.
76
Scientific Advisory Board Compensation
Our
Board of Directors approved a Scientific Advisory Board (the
“SAB”), compensation policy for appointed members of
the SAB. Prior to receiving compensation, each member of the SAB
will enter into an agreement with the Company providing for the
following cash compensation:
|
|
●
|
each
SAB member is entitled to receive reimbursement for all reasonable
and necessary out-of-pocket expenses incurred by the SAB member,
provided that such expenses are approved in advance by an officer
of the Company;
|
|
|
●
|
each
SAB member is entitled to receive a per diem fee from us of $2,500
to attend each formal face-to-face SAB meeting; and
|
|
|
●
|
each
SAB member is entitled to receive an hourly fee from us of $350 for
non-meeting services performed at our request.
|
With
the exception of Dr. Perez-Soler, a member of our Board of
Directors, each SAB member who enters into an engagement agreement
will receive an initial option grant to purchase 25,000 shares of
our common stock under our existing equity incentive plan, or any
other equity incentive plan we may adopt in the future, which shall
vest in 36 equal monthly installments, the first vesting date to
occur on the date of the grant.
Business Advisory Board Compensation
On May
20, 2016, our Board of Directors approved a Business Advisory Board
(the “BAB”), compensation policy for appointed members
of the BAB. This policy provides for the following cash
compensation:
|
|
●
|
each
BAB member is entitled to receive reimbursement for all reasonable
and necessary out-of-pocket expenses incurred by the BAB member,
provided that such expenses are approved in advance by an officer
of the Company.
|
In
addition, each BAB member will be entitled to receive an initial
option grant to purchase 25,000 shares of our common stock under
our existing equity incentive plan, or any other equity incentive
plan we may adopt in the future.
Board Leadership Structure and Role in Risk Oversight
Risk
is inherent with every business, and how well a business manages
risk can ultimately determine its success. While the Board of
Directors oversees risk management, our management is responsible
for our day-to-day risk management process. Our board of directors
has an active role, directly and through its committee structure,
in the oversight of our risk management efforts.
Our
board of directors satisfies this responsibility through full
reports by each committee chair regarding the committee's
considerations and actions, as well as through regular reports
directly from officers responsible for oversight of particular
risks within our company. Our Audit Committee assists the board in
performing its oversight responsibilities relating to our processes
and policies with respect to identifying, monitoring, assessing,
reporting on, managing and controlling our business and financial
risk. The Audit committee oversees, reviews, monitors and assesses
(including through regular reports by, and discussions with,
management), our processes and policies for risk identification,
risk assessment, reporting on risk, risk management and risk
control (including with respect to risks arising from our
compensation policies and practices and in connection with the
business and operations of its subsidiaries), and the steps that
management has taken to identify, assess, monitor, report on,
manage and control risks. The Audit committee also discusses with
management the balancing of risk versus reward for us and areas of
specific risk identified by management and/or the Audit
committee.
Our
board of directors believes that full and open communication
between management and the board of directors is essential for
effective risk management and oversight.
77
2016 Equity Compensation Plan
General
On
January 8, 2016, our Board of Directors adopted the 2016 Equity
Incentive Plan (the “2016 Plan”), subject to
stockholder approval, which was received on February 4, 2016,
pursuant to the terms described herein. On March 3, 2017, our Board
of Directors approved the First Amendment to the 2016 Plan (the
“First Amendment to the 2016 Plan”) which adjusted the
total number of shares authorized under the 2016 Plan to an
aggregate of 2,756,330, and remains subject to stockholder approval
within 12 months after the date the First Amendment to the 2016
Plan was approved by our Board of Directors.
The
general purpose of the 2016 Plan is to provide a means whereby
eligible employees, officers, non-employee directors and other
individual service providers develop a sense of proprietorship and
personal involvement in our development and financial success, and
to encourage them to devote their best efforts to our business,
thereby advancing our interests and the interests of our
stockholders. By means of the 2016 Plan, we seek to retain the
services of such eligible persons and to provide incentives for
such persons to exert maximum efforts for our success and the
success of our subsidiaries.
Description of the 2016 Equity Incentive Plan
The
following description of the principal terms of the 2016 Plan is a
summary and is qualified in its entirety by the full text of the
2016 Plan.
Administration. The
2016 Plan is administered by the Compensation Committee of our
Board of Directors. The Compensation Committee may grant options to
purchase shares of our common stock, stock appreciation rights,
restricted stock, stock units, performance shares, performance
units, incentive bonus awards, other cash-based awards and other
stock-based awards. The Compensation Committee also has authority
to determine the terms and conditions of each award, prescribe,
amend and rescind rules and regulations relating to the 2016 Plan,
and amend the terms of awards in any manner not inconsistent with
the 2016 Plan (provided that no amendment may adversely affect the
rights of a participant without consent). The Compensation
Committee may delegate authority to officers and employees to grant
options and other awards to employees (other than themselves),
subject to applicable law and restrictions in the 2016 Plan. No
award may be granted under the 2016 Plan on or after the ten year
anniversary of the adoption of the 2016 Plan by our Board of
Directors, but awards granted prior to the ten year anniversary may
extend beyond that date.
Eligibility. Persons
eligible to receive awards under the 2016 Plan include any person
who is an employee, officer, director, consultant, advisor or other
individual service provider of Adgero Biopharmaceuticals Holdings,
Inc. (“Holdings”) or any subsidiary, or any person who
is determined by the Compensation Committee to be a prospective
employee, officer, director, consultant, advisor or other
individual service provider of Holdings or any
subsidiary.
Shares Subject to the 2016 Plan. As of
March 3, 2017, the aggregate number of shares of common stock
authorized for issuance in connection with options and awards
granted under the 2016 Plan is 2,756,330 (subject to stockholder approval of
the First Amendment to the 2016 Plan). Incentive Stock
Options may, but need not be, granted with respect to all of the
shares available for issuance under the 2016 Plan. If any award
granted under the 2016 Plan payable in shares of common
stock is forfeited,
cancelled, returned for failure to satisfy vesting requirements, is
otherwise forfeited, otherwise terminates
without payment being made, or if
shares of common stock are withheld to cover withholding taxes on
options or other awards, the number of shares of common stock as to
which such option or award was forfeited, or which were withheld,
will be available for future grants under the 2016
Plan.
In
addition, the 2016 Plan contains an “evergreen”
provision allowing for an annual increase in the number of shares
of our common stock available for issuance under the 2016 Plan on
January 1 of each year during the period beginning January 1, 2017,
and ending on (and including) January 1, 2026. The annual increase
in the number of shares shall be equal to six percent (6%) of the
total number of shares of our common stock outstanding on December
31st of the preceding calendar year; provided, however, that our
board of directors may act prior to the first day of any calendar
year to provide that there shall be no increase such calendar year,
or that the increase shall be a lesser number of shares of common
stock than would otherwise occur.
Terms and Conditions of Options. Options granted under the 2016 Plan
may be either “incentive stock options” that are
intended to meet the requirements of Section 422 of the Code or
“nonqualified stock options” that do not meet the
requirements of Section 422 of the Code. The Compensation Committee
will determine the exercise price of options granted under the 2016
Plan. The exercise price of stock options may not be less than the
fair market value per share of our common stock on the date of
grant (or 110% of fair market value in the case of incentive stock
options granted to a ten-percent stockholder).
If on
the date of grant the common stock is listed on a stock exchange or
national market system, the fair market value will generally be the
closing sale price on the date of grant. If the common stock is not
traded on a stock exchange or national market system on the date of
grant, the fair market value will generally be the average of the
closing bid and asked prices for the common stock on the date of
grant. If no such prices are available, the fair market value shall
be determined in good faith by the Compensation Committee based on
the reasonable application of a reasonable valuation method.
Notwithstanding the foregoing, if the date for which fair market
value is determined is the date on which the final prospectus
relating to an initial public offering of the Company is filed, the
fair market value for such date will be the “Price to the
Public” (or equivalent) set forth on the cover page for the
final prospectus.
78
No option may be
exercisable for more than ten years from the date of grant (five
years in the case of an incentive stock option granted to a
ten-percent stockholder). Options granted under the 2016 Plan will
be exercisable at such time or times as the Compensation Committee
prescribes at the time of grant. No employee may receive incentive
stock options that first become exercisable in any calendar year in
an amount exceeding $100,000. The Compensation Committee may, in
its discretion, permit a holder of a nonqualified stock option to
exercise the option before it has otherwise become exercisable, in
which case the shares of our common stock issued to the recipient
will be restricted stock subject to vesting requirements analogous
to those that applied to the option before
exercise.
Generally, the
exercise price of an option may be paid (a) in cash or by certified
bank check, (b) through delivery of shares of our common stock
having a fair market value equal to the purchase price, or (c) such
other method as approved by the Compensation Committee and set
forth in an award agreement. The Compensation Committee is also
authorized to establish a cashless exercise program and to permit
the exercise price to be satisfied by reducing from the shares
otherwise issuable upon exercise a number of shares having a fair
market value equal to the exercise price.
No
option may be transferred other than by will or by the laws of
descent and distribution, and during a recipient’s lifetime
an option may be exercised only by the recipient. However, the
Compensation Committee may permit the holder of nonqualified stock
options, share-settled stock appreciation rights, restricted stock,
performance shares or other share-settled stock based awards to
transfer the option, right or other award to immediate family
members, to a trust for estate planning purposes, or by gift to
charitable institutions. The Compensation Committee will determine
the extent to which a holder of a stock option may exercise the
option following termination of service with us.
Stock Appreciation Rights. The Compensation Committee may grant
stock appreciation rights (“SAR”) independent of or in
connection with an option. The Compensation Committee will
determine the other terms applicable to SAR. The base price of a
SAR will be determined by the Compensation Committee, but will not
be less than 100% of the fair market value of a share of our common
stock on the date of grant. The maximum term of any SAR granted
under the 2016 Plan is ten years from the date of grant. Generally,
each SAR will entitle a participant upon exercise to an amount
equal to:
|
|
●
|
the
excess of the fair market value on the exercise date of one share
of our common stock over the base price, multiplied by
|
|
|
●
|
the
number of shares of common stock as to which the stock appreciation
right is exercised.
|
Payment
may be made in shares of our common stock, in cash, or partly in
common stock and partly in cash, all as determined by the
Compensation Committee.
Restricted Stock and Stock Units. The Compensation Committee may award
restricted common stock and/or stock units under the 2016 Plan.
Restricted stock awards consist of shares of stock that are
transferred to a participant subject to restrictions that may
result in forfeiture if specified conditions are not satisfied.
Stock units confer the right to receive shares of our common stock,
cash, or a combination of shares and cash, at a future date upon or
following the attainment of certain conditions specified by the
Compensation Committee. The Compensation Committee will determine
the restrictions and conditions applicable to each award of
restricted stock or stock units, which may include
performance-based conditions. Dividends with respect to restricted
stock may be paid to the holder of the shares as and when dividends
are paid to stockholders or at the time that the restricted stock
vests, as determined by the Compensation Committee. Dividend
equivalent amounts may be paid with respect to stock units, and
will be subject to the same restrictions on transferability as the
stock units with respect to which they were paid. Unless the
Compensation Committee determines otherwise, holders of restricted
stock will have the right to vote the shares.
Performance Shares and Performance Units. The Compensation Committee may award
performance shares and/or performance units under the 2016 Plan.
Performance shares and performance units are awards, denominated in
either shares or U.S. dollars, which are earned during a specified
performance period subject to the attainment of performance
criteria, as established by the Compensation Committee. The
Compensation Committee will determine the restrictions and
conditions applicable to each award of performance shares and
performance units.
Incentive Bonus Awards. The Compensation Committee may award
incentive bonus awards payable in cash or common stock, as set
forth in an award agreement. The Compensation Committee will
determine the terms and conditions applicable to each incentive
bonus award.
Other Stock-Based and Cash-Based Awards. The Compensation Committee may award
other types of equity-based or cash-based awards under the 2016
Plan, including the grant or offer for sale of shares of our common
stock that do not have vesting requirements and the right to
receive one or more cash payments subject to satisfaction of such
conditions as the Compensation Committee may impose.
79
Section 162(m) Compliance. If stock or cash-based awards are
intended to satisfy the conditions for deductibility under Section
162(m) of the Code as “performance-based compensation,”
the performance criteria will be selected from among the following,
which may be applied to our Company as a whole, or to any
subsidiary or any division or operating unit thereof: (a) pre-tax
income; (b) after-tax income; (c) net income; (d) operating income
or profit; (e) cash flow, free cash flow, cash flow return on
investment (discounted or otherwise), net cash provided by
operations, or cash flow in excess of cost of capital; (f) earnings
per share (basic or diluted); (g) return on equity; (h) returns on
sales or revenues; (i) return on invested capital or assets (gross
or net); (j) cash, funds or earnings available for distribution;
(k) appreciation in the fair market value of our common stock; (l)
operating expenses; (m) implementation or completion of critical
projects or processes; (n) return on investment; (o) total return
to stockholders (meaning the aggregate common stock price
appreciation and dividends paid (assuming full reinvestment of
dividends) during the applicable period); (p) net earnings growth;
(q) return measures (including but not limited to return on assets,
capital, equity, or sales); (r) increase in revenues; (s) the
Company’s published ranking against its peer group of
pharmaceutical companies based on total stockholder return; (t) net
earnings; (u) changes (or the absence of changes) in the per share
price of the Company’s common stock; (v) preclinical,
clinical or regulatory milestones; (w) earnings before or after any
one or more of the following items: interest, taxes, depreciation
or amortization, as reflected in the Company’s financial
reports for the applicable period; (x) total revenue growth
(meaning the increase in total revenues after the date of grant of
an award and during the applicable period, as reflected in the
Company’s financial reports for the applicable period); (y)
economic value created; (z) operating margin or profit margin; (aa)
share price or total shareholder return; (bb) cost targets,
reductions and savings, productivity and efficiencies; (cc)
strategic business criteria, consisting of one or more objectives
based on meeting objectively determinable criteria: specified
market penetration, geographic business expansion, investor
satisfaction, employee satisfaction, human resources management,
supervision of litigation, information technology, and goals
relating to acquisitions, divestitures, joint ventures and similar
transactions, and budget comparisons; (dd) objectively determinable
personal or professional objectives, including any of the following
performance goals: the implementation of policies and plans, the
negotiation of transactions, the development of long term business
goals, formation of joint ventures, research or development
collaborations, and the completion of other corporate transactions;
and (ee) any combination of, or a specified increase or improvement
in, any of the foregoing.
At the
end of the performance period established in connection with any
award, the Compensation Committee will determine the extent to
which the performance goal or goals established for such award have
been attained, and shall determine, on that basis, the shares or,
if applicable, the cash or other property that has been earned and
as to which payment will be made. The Compensation Committee will
certify in writing the extent to which it has determined that the
performance goal or goals established by it for such award have
been attained.
The
maximum number of shares of common stock with respect to which any
one participant may be granted stock options or stock appreciation
rights during any calendar year is 500,000 shares. With respect to
awards intended to be exempt from the deductibility limitation in
Section 162(m) of the Code (other than stock options and stock
appreciation rights), (i) the maximum number of shares of common
stock that may be paid to any one individual in respect of any
calendar year if the applicable performance goals are attained is
500,000 shares, and (ii) the maximum cash amount that may be paid
to any one participant in respect of any calendar year if the
applicable performance goals are attained is $1,000,000. Each such
maximum number of shares is subject to adjustment in the event of a
recapitalization, stock split, merger, reorganization or similar
corporate change affecting the common stock. If the performance
period for certain performance goals spans more than one calendar
year, the shares or cash paid in respect of each calendar year will
be determined by pro rating the shares or cash paid for the
performance period based on the number of performance period days
that fall in each respective calendar year.
Effect of Certain Corporate Transactions. The Compensation Committee may, at the
time of the grant of an award, provide for the effect of a change
in control (as defined in the 2016 Plan) on any award, including
(i) accelerating or extending the time periods for exercising,
vesting in, or realizing gain from any award, (ii) eliminating or
modifying the performance or other conditions of an award, (iii)
providing for the cash settlement of an award for an equivalent
cash value, as determined by the Compensation Committee, or (iv)
such other modification or adjustment to an award as the
Compensation Committee deems appropriate to maintain and protect
the rights and interests of participants following a change in
control. The Compensation Committee may, in its discretion and
without the need for the consent of any recipient of an award, also
take one or more of the following actions contingent upon the
occurrence of a change in control: (a) cause any or all outstanding
options and stock appreciation rights to become immediately
exercisable, in whole or in part; (b) cause any other
awards to become
non-forfeitable, in whole or in part; (c) cancel any option or
stock appreciation right in exchange for a substitute option; (d)
cancel any award of restricted stock, stock units, performance
shares or performance units in exchange for a similar award of the
capital stock of any successor corporation; (e) redeem any
restricted stock for cash and/or other substitute consideration
with a value equal to the fair market value of an unrestricted
share of our common stock on the date of the change in control; (f)
cancel any option or stock appreciation right in exchange for cash
and/or other substitute consideration based on the value of our
common stock on the date of the change in control, and cancel any option or stock
appreciation right without any payment if its exercise price
exceeds the value of our common stock on the date of the change in
control; or (g) make such other modifications, adjustments or
amendments to outstanding awards as the Compensation Committee
deems necessary or appropriate.
80
Amendment, Termination. The Compensation Committee may amend
the terms of awards in any manner not inconsistent with the 2016
Plan, provided that no amendment shall adversely affect the rights
of a participant with respect to an outstanding award without the
participant’s consent. In addition, our board of directors
may at any time amend, suspend, or terminate the 2016 Plan,
provided that (i) no such amendment, suspension or termination
shall materially and adversely affect the rights of any participant
under any outstanding award without the consent of such participant
and (ii) to the extent necessary to comply with any applicable law,
regulation, or stock exchange rule, the 2016 Plan requires us to
obtain stockholder consent. Stockholder approval is required for
any plan amendment that increases the number of shares of common
stock available for issuance under the 2016 Plan or changes the
persons or classes of persons eligible to receive
awards.
Tax Withholding
As and
when appropriate, we shall have the right to require each optionee
purchasing shares of common stock and each grantee receiving an
award of shares of common stock under the 2016 Plan to pay any
federal, state or local taxes required by law to be
withheld.
Option Grants and Stock Awards
The
grant of options and other awards under the 2016 Plan will be
discretionary and we cannot determine now the specific number or
type of options or awards to be granted in the future to any
particular person or group.
Indemnification Agreements
We have
entered into Indemnification Agreements with certain of our current
directors and executive officers. The Indemnification Agreements
provide for indemnification against expenses, judgments, fines and
penalties actually and reasonably incurred by an indemnitee in
connection with threatened, pending or completed actions, suits or
other proceedings, subject to certain limitations. The
Indemnification Agreements also provide for the advancement of
expenses in connection with a proceeding prior to a final,
nonappealable judgment or other adjudication, provided that the
indemnitee provides an undertaking to repay to us any amounts
advanced if the indemnitee is ultimately found not to be entitled
to indemnification by us. The Indemnification Agreement sets forth
procedures for making and responding to a request for
indemnification or advancement of expenses, as well as dispute
resolution procedures that will apply to any dispute between us and
an indemnitee arising under the Indemnification
Agreements.
81
PRINCIPAL STOCKHOLDERS
The
following table sets forth information regarding the beneficial
ownership of our common stock as of the date of this prospectus
by:
|
|
●
|
each
person known by us to be the beneficial owner of more than 5% of
our outstanding shares of common stock;
|
|
|
●
|
each of
our named executive officers;
|
|
|
●
|
each of
our directors; and
|
|
|
●
|
all of
our directors and current executive officers as a
group.
|
Beneficial
ownership is determined based on the rules and regulations of the
Commission. A person has beneficial ownership of shares if such
individual has the power to vote and/or dispose of shares. This
power may be sole or shared and direct or indirect. In computing
the number of shares beneficially owned by a person and the
percentage ownership of that person, shares of common stock that
are subject to options or warrants held by that person and
exercisable as of, or within 60 days of, April 4, 2017 are counted
as outstanding. These shares, however, are not counted as
outstanding for the purposes of computing the percentage ownership
of any other person(s). Except as may be indicated in the footnotes
to this table and pursuant to applicable community property laws,
each person named in the table has sole voting and dispositive
power with respect to the shares of common stock set forth opposite
that person’s name. Unless indicated below, the address of
each individual listed below is c/o Adgero Biopharmaceuticals,
Holdings Inc., 4365 US 1 South, Suite 211, Princeton, NJ
08540.
Applicable
percentage ownership in the following table is based on 5,398,531
shares of common stock outstanding as of April 4, 2017. Beneficial
ownership representing less than 1% is denoted with an asterisk
(*).
|
Name of Beneficial Owner
|
Number of Shares Beneficially Owned
|
Percentage of Shares Beneficially Owned
|
|
Officers and Directors
|
|
|
|
Frank Pilkiewicz, PhD (1)
|
1,468,863
|
26.5%
|
|
Steven Rychnovsky, PhD (2)
|
419,677
|
7.7%
|
|
Jane Maida (3)
|
0
|
*
|
|
Allen Bloom, PhD, JD (4)
|
76,085
|
1.4%
|
|
Roman Perez-Soler, MD (5)
|
44,108
|
*
|
|
David Hochman (6)
|
112,000
|
2.1%
|
|
Tim McInerney (7)
|
80,000
|
1.5%
|
|
|
|
|
|
Directors and Executive Officers as a Group (7
persons)
|
2,040,733
|
36.6%
|
|
|
|
|
|
5% Stockholders
|
|
|
|
Adam Stern (8)
|
761,784
|
13.6%
|
|
Fernovelty (Hong Kong)
Holding Co., Ltd (9)
|
800,000
|
13.8%
|
|
GJG Life Sciences, LLC (10)
|
842,800
|
14.5%
|
82
* Less
than 1%
(1)
Includes (i)
11,670 shares of common stock owned by his spouse, (ii) 6,173
shares of common stock issuable upon exercise of warrants owned by
his spouse, (iii) 4,369
shares of common stock owned jointly with his spouse, (iv) an
aggregate of 29,784 shares of common stock owned by his children
and, (v) 34,153 shares of common stock issuable upon exercise of
warrants owned jointly with his spouse. Does not include 335,958
shares of common stock issuable upon the exercise of stock options
granted to Frank Pilkiewicz, PhD in connection with his employment
agreement that are not exercisable within sixty days of April
4,
2017.
(2)
Includes 36,999
shares of common stock issuable upon exercise of warrants owned by
Steven Rychnovsky, PhD. Does not include 167,979 shares of common
stock issuable upon the exercise of stock options granted to Steve
Rychnovsky, PhD in connection with his employment agreement that
are not exercisable within sixty days of April 4,
2017.
(3)
Does
not include 100,000 shares of common stock issuable upon the
exercise of stock options granted to Jane Maida that are not
exercisable with sixty days of April 4,
2017.
(4)
Includes
40,000 shares of common stock issuable upon exercise of options
owned by Allen Bloom, PhD, JD. Does not include 45,000
shares of common stock issuable upon the exercise of stock options
granted to Allen Bloom, PhD, JD that are not exercisable within
sixty days of April 4, 2017.
(5)
Includes (i) 2,054 shares of
common stock issuable upon exercise of warrants
and (ii) 40,000 shares of common
stock issuable upon exercise of options owned by Roman
Perez-Soler. Does not include 45,000
shares of common stock issuable upon the exercise
of stock options granted to Roman Perez-Soler that are not
exercisable within sixty days of April 4,
2017.
(6)
Includes
40,000 shares of common stock issuable upon exercise of options
owned by David Hochman. Does not include 45,000
shares of common stock issuable
upon the exercise of stock options granted to David Hochman that
are not exercisable within sixty days of April
4, 2017.
(7)
Includes (i) 20,000 shares of
common stock issuable upon exercise of warrants
and (ii) 40,000
shares of common stock issuable upon exercise of
options owned by Tim McInerney. Does not
include 45,000 shares of common stock issuable upon the exercise
of stock options granted to Tim McInerney that are not exercisable
within sixty days of April 4,
2017.
(8)
Includes (i) 50,892 shares of common stock issuable upon exercise
of warrants and (ii) 160,000 shares of common stock issuable upon
exercise of Placement Agent Warrants owned by Adam Stern. Adam
Stern is an affiliate of the Placement Agent. Does not include up
to 80,000 shares of common stock issuable upon exercise of warrants
issued to the Placement Agent in the December 2016 Private
Placement. Adam Stern disclaims ownership of such shares except to
the extent of his pecuniary interest therein. The address of Adam
Stern is 810 Seventh Ave, 18th FL, New York, NY 10019.
(9)
Includes 400,000
shares of common stock issuable upon exercise of warrants owned by
Fernovelty (Hong Kong) Holding Co., Ltd. Haibo Wang is a
natural person with voting and dispositive power over the shares
held by Fernovelty (Hong Kong) Holding Co., Ltd. The address
of Fernovelty (Hong Kong) Holding Co., Ltd is No. 308, Cailun Road,
Zhangjiang Hi-Tech Park, Pudong, Shanghai, China
201210.
(10)
Includes 421,400 shares of common stock issuable upon exercise of
warrants owned by GJG Life Sciences, LLC. Jennifer Lorenzo is
a natural person with voting and dispositive power over the shares
held by GJG Life Sciences LLC. The address of GJG Life
Sciences, LLC is 107 Circle Road, Staten Island, NY
10301.
83
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Other
than compensation arrangements for our named executive officers and
directors, we describe below each transaction or series of similar
transactions, since January 1, 2014, to which we were a party or
will be a party, in which:
|
|
●
|
the
amounts involved exceeded or will exceed the lesser of (i) $120,000
or (ii) 1% of the average total assets of the Company at year end
for the last two completed fiscal years.; and
|
|
|
●
|
any of
our directors, executive officers, promoters or holders of more
than 5% of our capital stock, or any member of the immediate family
of the foregoing persons, had or will have a direct or indirect
material interest.
|
Compensation
arrangements for our named executive officers and directors are
described in the section entitled “executive
compensation.” Frank Pilkiewicz, PhD and Steven Rychnovsky,
PhD are our founders and, therefore, may be considered promoters,
as that term is defined in Rule 405 of Regulation C of the
Securities Act of 1933, as amended (the “Securities
Act”).
The Placement Agent and Related Parties
In
connection with the formation of Adgero Biopharmaceuticals
Holdings, Inc. (“Holdings”) in October, 2015 certain
affiliates of Aegis Capital Corporation (the “Placement
Agent”) and certain other parties not affiliated with us or
the Placement Agent subscribed for an aggregate of 1,000,000 shares
of common stock of Holdings for which they paid an aggregate of
$50,000 ($0.05 per share), including David Hochman, one of our
directors who purchased 72,000 shares. In addition, an affiliate of
the Placement Agent also purchased a Bridge Note in the amount of
$250,000 in the Bridge Offering, described below, which converted, together with accrued interest
thereon calculated through April 8, 2016, the date of the initial
closing (the “Initial Closing”) of our 2016 private
placement, for which closings occurred April 8, 2016 through
September 9, 2016 (the “2016 Private Placement”), at a
rate of 6% per annum, into 50,892 shares of common stock
and Investor Warrants to purchase
50,892 shares of our common stock with an exercise price of
$5.00 per share, in connection with the 2016 Private Placement.
Affiliates of the Placement Agent have not received registration
rights with respect to (i) shares of common stock held in Holdings
prior to the 2016 merger transaction (the “Merger”) or
(ii) the securities issued upon conversion of the Bridge Note.
Shareholders of Holdings pre-Merger (other than affiliates of the
Placement Agent), including, without limitation, David Hochman,
have not received registration rights with respect to the shares of
common stock held in Holdings pre-Merger.
Following
the Initial Closing of the 2016 Private Placement, the Placement
Agent has a right to appoint one member of our Board of Directors
for a two-year term (the “Aegis Nominee”). Upon the
Initial Closing, on April 8, 2016, David Hochman was appointed as
the Aegis Nominee and his successor, if any, will be chosen by the
Placement Agent, subject to the reasonable approval of the Company
and the terms of the Voting Agreement described below.
Voting Agreement
In
connection with the Initial Closing, the stockholders of Adgero
Biopharmaceuticals, Inc. (“Adgero”) prior to the Merger
(the “Adgero Stockholders”), and the stockholders of
Holdings prior to the Merger (the “Holdings
Stockholders”), entered into a voting agreement (the
“Voting Agreement”). Pursuant to the terms of the
Voting Agreement, (i) the Adgero Stockholders have the right to
nominate three (3) members to our Board (“the Adgero
Stockholders’ Nominees”), (ii) the Holdings
Stockholders shall vote in favor of the election of the Adgero
Stockholders’ Nominees, (iii) the Holdings Stockholders shall
vote in favor of the election of one Aegis Nominee to our Board,
(iv) the Adgero Stockholders shall vote in favor of the election of
the Aegis Nominee, and (v) the Adgero Stockholders and the Holdings
Stockholders will vote in favor of one independent candidate to the
Board of Directors acceptable to the Aegis Designee and the Adgero
Stockholders’ Nominees, which is currently Tim McInerney. The
Voting Agreement expires upon the earlier of (i) the approval of at
least 75% of the Adgero Stockholders and the Holdings Stockholders
voting together based upon their ownership of our common stock,
(ii) the closing of a firm commitment underwritten public offering
of shares of our common stock resulting in gross proceeds of at
least $10 million or (iii) the listing of our common stock on
Nasdaq or New York Stock Exchange.
84
Indemnification Agreements
We have
entered into indemnification agreements with our directors and
officers. For more information, see the description of the
indemnification agreements under “Management and Board of
Directors - Limitation of Directors Liability and
Indemnification.”
Debt to our Officers and Payments to our Vice President of
Operations and Product Development
On
July 9, 2014, we entered into a
promissory note with Frank Pilkiewicz, PhD, our President and Chief
Executive Officer, in the principal amount of $163,934. The
promissory note accrued interest at a rate of two percent (2%) per
annum. On July 29, 2016, the note was converted into 34,153 shares
of common stock owned jointly by Frank Pilkiewicz, PhD and Carolyn
Pilkiewicz, and Investor Warrants to purchase 34,153 shares of our
common stock with an exercise price of $5.00 per share owned
jointly by Frank Pilkiewicz, PhD and Carolyn Pilkiewicz, in
connection with the 2016 Private Placement.
Prior
to August 1, 2016, we paid Frank
Pilkiewicz, PhD and Steven Rychnovsky, PhD, approximately $46,000
and $13,000, respectively, for advancement of expenses each
previously made to the Company.
We
acquired certain assets from Miravant Medical Technologies, and its
wholly-owned subsidiaries, a former public pharmaceutical and
research development company (collectively,
“Miravant”), including the REM-001 Therapy product,
consisting of three parts, the laser light source, the light
delivery device and the drug REM-001 (collectively, the
“REM-001 Therapy”), through an asset purchase agreement
with St. Cloud Investments, LLC (“St. Cloud,” a
previous Miravant creditor who acquired these Miravant assets
pursuant to a foreclosure process St. Cloud completed under
California law) dated November 26, 2012, as amended (the “St.
Cloud Agreement”). Pursuant to the terms of the St. Cloud
Agreement, we are obligated to make certain payments to Steven
Rychnovsky, PhD, who was recruited to become our Vice President of
Operations and Product Development after consummation of the St.
Cloud Agreement. From 2008 to 2012 Steven Rychnovsky, PhD, served
as a consultant to St. Cloud where he maintained the assets of
Miravant, and worked to identify parties to license or purchase the
Miravant assets and initiate commercial development. As
compensation for those services, he would receive 50% of any
up-front payments and 20% of all subsequent payments from any
license or purchase agreement for those assets. This compensation
is detailed in the St. Cloud Agreement where Steven Rychnovsky,
PhD, is identified as the Seller’s Designee. The amounts paid
or owed under that agreement are as follows:
|
|
●
|
Thirteen
thousand dollars ($13,000) was paid to Steven Rychnovsky, PhD upon
the Initial Closing of the 2016
Private Placement.
|
|
|
●
|
Fiftythousand
dollars ($50,000) was paid to Steven Rychnovsky, PhDduring the 2016
Private Placement, because the 2016 PrivatePlacement was completed
for an amount that exceeded four milliondollars
($4,000,000).
|
|
|
●
|
Upon
the earlier of (i) a subsequent equity financing to take place
after we conduct a Phase 2B clinical trial in which fifty patients
complete the trial and their clinical data can be evaluated or (ii)
the commencement of a clinical trial intended to be used as a
definitive study for market approval in any country, we are
obligated to pay an amount of sixty thousand dollars ($60,000) in
cash or an equivalent amount of common stock to Steven Rychnovsky,
PhD.
|
|
|
●
|
Upon
receipt of regulatory approval of our lead product candidate, the
REM-001 Therapy, we are obligated to pay an aggregate amount of one
hundred forty thousand dollars ($140,000) in cash or an equivalent
amount of common stock to Steven Rychnovsky, PhD.
|
With
respect to the $60,000 and $140,000 potential milestone payments
referenced above (each a "Rychnovsky Milestone Payment"), if either
such Rychnovsky Milestone Payment becomes payable, and in the event
we elect to pay either such Rychnovsky Milestone Payment in shares
of our common stock, the value of the common stock will equal the
price per share of the most recent financing, or, if we are
considered to be a publicly-traded company, the average of the
closing price per share of our common stock over the twenty (20)
trading days following the first public announcement of the
applicable event described above.
85
In
addition, we must pay to Steven Rychnovsky, PhD, a royalty fee of
one and one fifth percent (1.2%) of net sales during the royalty
term on a country-by-country and product-by-product basis. The
royalty term for a product commences on the first commercial sale
of the product, such as REM-001 Therapy, in any country, and the
royalty fee must be paid within 30 days of each calendar quarter
during which revenue is collected. The royalty term terminates on
the later of (i) the invalidation, revocation, lapse or expiration
of the last to expire valid claim on any patent acquired in the St.
Cloud Agreement that would be infringed by the sale of the product
in the country where the commercial sale takes place or (ii) the
expiration of the period for which we hold exclusive marketing
rights of the product in the country, if we were granted those
rights under the St. Cloud Agreement.
In
connection with and pursuant to the St. Cloud Agreement, on
November 26, 2012, we issued a senior convertible note to Steven
Rychnovsky, PhD. The note had an aggregate principal amount of one
hundred thousand dollars ($100,000) and accrued interest at a rate
of eight percent (8%) per annum. During the 2016 Private Placement,
the note converted into 36,999 shares
of common stock and warrants
exercisable for 36,999 shares of common stock with an
exercise price of $5.00 per share, representing the number of
securities equal to the outstanding balance of the note, plus
interest accrued thereon but unpaid, divided by seventy percent
(70%) of the $5.00 purchase price per share paid by the investors
participating in such financing. In 2014, we entered into a
promissory note and pledge with Steven Rychnovsky, PhD in the
initial principal amount of $7,887. Such principal amount and the
interest accrued thereon was forgiven in 2015 in exchange for
services provided to us by Steven Rychnovsky, PhD.
Bridge Offering Affiliate Participation
In 2015, Adgero sold promissory notes (the
“Bridge Offering”) in an aggregate principal amount of
$285,000, to three individuals (the “Bridge Holders”).
One of our directors, Roman Perez-Soler, MD, purchased a Bridge
Note in an aggregate principal amount of $10,000. Upon the Initial
Closing of the 2016 Private Placement, Dr. Perez-Soler’s
Bridge Note automatically converted, together with accrued interest
thereon at a rate of 6% per annum, calculated through April 8,
2016, the date of the Initial Closing, into 2,054 shares of
common stock and Investor Warrants to
purchase 2,054 shares of our common stock with an exercise price of $5.00 per share,
in connection with the 2016 Private Placement. In addition an affiliate of the Placement Agent,
purchased a Bridge Note in an aggregate principal amount of
$250,000. Upon the Initial Closing of the 2016 Private Placement,
the Bridge Note purchased by the Affiliate of the Placement Agent
automatically converted, together with accrued interest thereon at
a rate of 6% per annum, calculated through April 8, 2016, the date
of the Initial Closing, into 50,892 shares of common stock
and Investor Warrants to purchase
50,892 shares of our common stock with an exercise price of $5.00 per share,
in connection with the 2016 Private Placement. The affiliate of the Placement Agent will not
receive registration rights for the shares of common stock
issued pursuant to the conversion of
his Bridge Note or the shares of common stock underlying the warrants issued pursuant to the
conversion of his Bridge Note.
Merger Transaction
On
January 11, 2016, Adgero entered into the merger agreement (the
“Merger Agreement”) with Adgero Acquisition, Inc. a
Delaware corporation (“Merger Sub”), a wholly owned
subsidiary of Holdings. Pursuant to the terms of the Merger
Agreement, as a condition of and contemporaneously with the closing
of the Merger on April 8, 2016, Merger Sub merged with and into
Adgero and Adgero became a wholly owned subsidiary of
Holdings.
While
we believe that all of these agreements and arrangements are in the
best interests of our Company, related parties of the Placement
Agent may derive material benefits as the result of these
transactions. In addition, related parties of the Placement Agent
will have a continuing substantial interest in our Company and will
derive substantial benefits from any success of our
Company.
Policies and Procedures for Related Party Transactions
Our
board of directors has adopted a policy that our executive
officers, directors, nominees for election as a director,
beneficial owners of more than 5% of any class of our common stock,
any members of the immediate family of any of the foregoing persons
and any firms, corporations or other entities in which any of the
foregoing persons is employed or is a partner or principal or in a
similar position or in which such person has a 5% or greater
beneficial ownership interest (collectively “related
parties”), are not permitted to enter into a transaction with
us without the prior consent of our board of directors acting
through the audit committee or, in certain circumstances, the
chairman of the audit committee. Any request for us to enter into a
transaction with a related party, in which the amount involved
exceeds $100,000 and such related party would have a direct or
indirect interest must first be presented to our audit committee,
or in certain circumstances the chairman of our audit committee,
for review, consideration and approval. In approving or rejecting
any such proposal, our audit committee, or the chairman of our
audit committee, is to consider the material facts of the
transaction, including, but not limited to, whether the transaction
is on terms no less favorable than terms generally available to an
unaffiliated third party under the same or similar circumstances,
the extent of the benefits to us, the availability of other sources
of comparable products or services and the extent of the related
party’s interest in the transaction.
86
DESCRIPTION OF SECURITIES
Our
current certificate of incorporation, as amended, authorizes us to
issue:
|
|
●
|
50,000,000
shares of common stock, par value $0.0001 per share;
and
|
|
|
●
|
10,000,000
shares of preferred stock, par value $0.0001 per
share.
|
As of
April 4, 2017, there were 5,398,531 shares of our common stock
outstanding, and no shares of preferred stock
outstanding.
The
following statements are summaries only of provisions of our
authorized capital stock and are qualified in their entirety by our
Certificate of Incorporation, as amended. You should review these
documents for a description of the rights, restrictions and
obligations relating to our capital stock. Copies of our
Certificate of Incorporation may be obtained from the Company upon
written request.
Common stock
Voting. The holders of our common stock
are entitled to one vote for each share held of record on all
matters on which the holders are entitled to vote (or consent
to).
Dividends. The holders of our common
stock are entitled to receive, ratably, dividends only if, when and
as declared by our Board of Directors out of funds legally
available therefor and after provision is made for each class of
capital stock having preference over the common stock.
Liquidation Rights. In the event of our
liquidation, dissolution or winding-up, the holders of our common
stock are entitled to share, ratably, in all assets remaining
available for distribution after payment of all liabilities and
after provision is made for each class of capital stock having
preference over the common stock.
Conversion Rights. The holders of our
common stock have no conversion rights.
Preemptive and Similar Rights. The
holders of our common stock have no preemptive or similar
rights.
Redemption/Put Rights. There are no
redemption or sinking fund provisions applicable to the common
stock. All of the outstanding shares of our common stock are
fully-paid and non-assessable.
Transfer Restrictions. Shares of our
common stock are subject to transfer restrictions. Holders of our
common stock may not transfer their securities unless (a) a
registration statement is in effect under the Securities Act of
1933, as amended (the “Securities Act”), covering the
proposed transfer and such transfer is made in accordance with such
registration statement or (b) the securities are transferred in a
transaction exempt from the registration requirements of the
Securities Act and any related requirements imposed by applicable
state securities laws. In the case of any transfer permitted under
clause (b), the holder must notify us in writing of the proposed
transfer and furnish us with an opinion of counsel, reasonably
satisfactory to us, that the transfer will not require registration
under the Securities Act or any applicable state securities laws.
Each certificate representing a security contains a legend
referring to this restriction on transfer and any legends required
by state securities laws. The securities are also subject to other
restrictions on transfer as provided in the Registration Rights
Agreement, described below.
Preferred Stock
We are
authorized to issue up to 10,000,000 shares of “blank
check” preferred stock, par value $0.0001 per share, with
such designations, rights, and preferences as may be determined
from time to time by our Board of Directors. Accordingly, our Board
of Directors is empowered, without stockholder approval, to issue
preferred stock with dividend, liquidation, conversion, voting, or
other rights that could adversely affect the voting power or other
rights of the holders of our common stock. The issuance of
preferred stock could have the effect of restricting dividends on
our common stock, diluting the voting power of our common stock,
impairing the liquidation rights of our common stock, or delaying
or preventing a change in control of our company, all without
further action by our stockholders. We currently have no shares of
preferred stock outstanding.
87
Warrants
Investor Warrants. In connection with
our private placement, for which closings occurred April 8, 2016
through September 9, 2016 (the “2016 Private
Placement”), we issued warrants to investors to purchase an
aggregate 1,873,299 shares of our common stock (the “Investor
Warrants”), inclusive of Investor Warrants to purchase 87,099
shares of our common stock issued pursuant to the conversion of
certain notes in connection with the 2016 Private Placement. The
Investor Warrants are exercisable for our common stock at an
exercise price equal to $5.00 per share (the “Exercise
Price”). The Investor Warrants are exercisable immediately
upon issuance and have a five year term. The Investor Warrants may
be exercised at any time in whole or in part at the applicable
exercise price until expiration of the Investor Warrants. No
fractional shares will be issued upon the exercise of the Investor
Warrants. Prior to the expiration date of the Investor Warrants,
the Company has the option to redeem all of the Investor Warrants
then outstanding upon not less than thirty (30) days nor more than
sixty (60) days prior written notice to the Investor Warrant
holders at any time provided that, at the time of delivery of such
notice (i) there is an effective registration statement covering
the resale of the Investor Warrant shares, and (ii) the closing
price of the Company’s common stock for each of the twenty
(20) consecutive trading days prior to the date of the notice of
redemption is at least $12.50, as proportionately adjusted to
reflect any stock splits, stock dividends, combination of shares or
like events. The redemption price to be paid to the holders of the
Investor Warrants will be $0.0001 for each share of common stock to
which the holder of the Investor Warrant would then be entitled
upon exercise of the Investor Warrant being redeemed. It is
contemplated that Aegis Capital Corporation (“Aegis
Capital”) will be retained as solicitation agent in the event
the Company elects to redeem the Investor Warrants and shall be
paid a fee of 5% of the gross proceeds derived from the exercise of
the Investor Warrants in such event.
In
connection with the December 2016 Private Placement, we issued the
December 2016 Private Placement Warrant to purchase 400,000 shares
of our common stock at an exercise price of $5.00 per share. The
December 2016 Private Placement Warrant is on substantially similar
terms as the Investor Warrants, except that registration rights
were not granted in connection with the December 2016 Private
Placement Warrant.
In
connection with the conversion of a note issued in the Bridge
Offering, which was not included in the 2016 Private Placement, we
issued a warrant on substantially similar terms as the Investor
Warrants to a Bridge Holder to purchase 5,154 shares of our common
stock at an exercise price of $5.00 per share. In connection with
the conversion of the notes issued pursuant to the St. Cloud
Agreement, we issued warrants to purchase 73,998 shares of our
common stock at an exercise price of $5.00 per share, to holders of
notes issued pursuant to the St. Cloud Agreement. The warrants
issued pursuant to the conversion of the convertible notes issued
pursuant to the St. Cloud Agreement are on substantially similar
terms as the Investor Warrants. See “Business - St. Cloud
Asset Purchase Agreement.”
Replacement Warrants. In connection
with the 2016 merger transaction (the “Merger”), we
issued warrants to purchase 30,864 shares of our common stock at an
exercise price of $5.00 per share to holders of warrants to
purchase an aggregate of 250,000 shares of common stock of Adgero
Biopharmaceuticals, Inc. common stock (the “Replacement
Warrants”). The Replacement Warrants are on substantially
similar terms as the Investor Warrants.
Placement Agent Warrants. In connection
with completion of the 2016 Private Placement, we issued Aegis
Capital (the “Placement Agent”) warrants to purchase
367,418 shares of our common stock at an exercise price of $5.00 as
partial compensation (the “Placement Agent Warrants”).
These warrants have a five year term and provide cashless
exercise.
In
connection with the completion of the December 2016 Private
Placement, we issued the Placement Agent warrants to purchase
80,000 shares of our common stock at an exercise price of $5.00 as
partial compensation (the “Placement Agent Warrants”).
These warrants have a five year term and provide cashless
exercise.
Registration Rights
In
connection with the 2016 Private Placement, we entered into a
registration rights agreement (as amended, the “Registration
Rights Agreement”) with the 2016 Private Placement investors,
a Bridge Holder whose bridge note was not included in the 2016
Private Placement, the Placement Agent and the holders of certain
of our outstanding warrants (collectively, the
“Investors”). We were required to file with the SEC
after the date of the final closing of the 2016 Private Placement
(the “Registration Filing Date”), a registration
statement covering the resale of the shares of common stock held by
the Investors (the “Investor Shares”) and certain of
the Investor Warrants, issued in the 2016 Private Placement, as
well as the shares of common stock underlying the Replacement
Warrants and the warrant issued to a Bridge Holder whose bridge
note was not included in the 2016 Private Placement (together with
the Investor Shares and the Investor Warrants, the
“Registrable Securities”). We are also required to use
commercially reasonable efforts to have the registration statement
declared effective within one hundred and fifty (150) days after
the registration statement is filed (the “Effectiveness
Deadline”); provided however, that if the Company signs a
letter of intent or comparable agreement with an underwriter which
contemplates an Initial Public Offering (“IPO”) or
holds an organizational meeting for an IPO, or otherwise orally
engages an underwriter to begin working with the Company towards an
IPO prior to the Effectiveness Deadline (the “IPO Process
Commencement Date”), then the Company shall file a joint
registration statement covering the primary shares to be issued in
the IPO and the resale of the Registrable Securities, and in such
event the Registration Filing Date shall be extended to a date that
is seventy five (75) calendar days after the IPO Process
Commencement Date and the Effectiveness Deadline shall be extended
to a date that is one hundred twenty (120) calendar days after the
initial filing of the Registration Statement with the Commission.
If the IPO is abandoned at any time, then the Registration Filing
Date will be 60 calendar days from the actual date of abandonment
and the Effectiveness Deadline will be one hundred and fifty (150)
calendar days after the date of abandonment. We are also required
to keep the registration statement continuously effective under the
Securities Act for a period of one year or for such shorter period
ending on the earlier to occur of the date when all the Registrable
Securities covered by the registration statement have been sold or
such time as all of the Registrable Securities covered by the
registration statement can be sold under Rule 144 without any
volume limitations.
88
If this
registration statement is not declared effective on or before the
Effectiveness Deadline, we will be required to pay to each holder
of Registrable Securities purchased in the 2016 Private Placement
an amount in cash equal to one-half of one percent (0.5%) of such
holder’s investment amount on every thirty (30) day
anniversary of such Effectiveness Deadline until such failure is
cured. The payment amount shall be prorated for partial thirty (30)
day periods. The maximum aggregate amount of payments to be made by
us as the result of such failure, shall be an amount equal to 6% of
each holder’s investment amount. Notwithstanding the
foregoing, no payments shall be owed with respect to any period
during which all of the holder’s Registrable Securities may
be sold by such holder without restriction under Rule
144.
We
shall keep the registration statement “evergreen” for
one (1) year from the date it is declared effective by the SEC or
until Rule 144 of the Securities Act is available to the holders of
Registrable Securities purchased in the 2016 Private Placement with
respect to all of their shares, whichever is earlier.
We will
pay all costs and expenses incurred by us in complying with our
obligations to file registration statements pursuant to the
Registration Rights Agreement, except that the selling holders will
be responsible for their shares of the attorney’s fees and
expenses and any commissions or other compensation to selling
agents and similar persons; provided, however, that, in any
registration, each party will pay for its own underwriting
discounts and commissions and transfer taxes.
Lock-Up Agreements
Each of
our directors and officers and the holders of substantially all of
5% or more of our common stock have agreed that they will not (a)
offer, sell, contract to sell, grant any option to purchase,
hypothecate, pledge or otherwise dispose of or (b) transfer title
to any shares of common stock acquired prior to the 2016 Private
Placement, which includes any shares of common stock acquired upon
the exercise of any warrants acquired prior to the 2016 Private
Placement, for a period beginning on April 8, 2016 and ending
twelve (12) months following the effective date of the registration
statement of which this prospectus is a part, without the prior
written consent of both the Company and Aegis Capital.
In
connection with the formation of Adgero Biopharmaceuticals
Holdings, Inc. (“Holdings”) in October, 2015, certain
affiliates of the Placement Agent and certain other parties not
affiliated with us or the Placement Agent subscribed for an
aggregate of 1,000,000 shares of common stock (the “Formation
Shares”), for which they paid an aggregate of $50,000 ($0.05
per share). Each of the holders of the Formation Shares have agreed
that they will not (a) offer, sell, contract to sell, grant any
option to purchase, hypothecate, pledge or otherwise dispose of or
(b) transfer title to any shares of common stock acquired prior to
the 2016 Private Placement, which includes any shares of common
stock acquired upon the exercise of any warrants acquired prior to
the 2016 Private Placement, for a period beginning on April 8, 2016
and ending fifteen (15) months following the effective date of the
registration statement of which this prospectus is a part, without
the prior written consent of both the Company and Aegis
Capital.
From
and after the effective date of the registration statement of which
this prospectus forms a part (the “Effective Date”),
the investors in the 2016 Private Placement have agreed that they
will not sell, offer, pledge, contract to sell, grant any option or
contract to purchase, lend or otherwise transfer or encumber,
directly or indirectly, any shares acquired in the 2016 Private
Placement nor will they enter into any swap, hedging or other
arrangement that transfers to another, in whole or in part, any of
the economic consequences of ownership of any shares acquired in
the 2016 Private Placement until 270 days from the Effective Date
(the “Release Date”), provided that each investor may
transfer up to one quarter of their Investor Shares held as of the
Effective Date, and following such date, a holder shall be entitled
to transfer up to an additional 25% of its Investor Shares
beginning at the end of each successive three month period
thereafter. Following the Release Date, the investors may sell the
remaining Registrable Securities that they hold. The foregoing
holdback provisions shall be terminated in the event the closing
price of our common stock is $15.00 or above for 20 consecutive
trading days. In addition, the investor in the December 2016
Private Placement has agreed to be subject to the same lock-up
provisions as applicable to the investors in the 2016 Private
Placement, described herein.
89
Transfer Agent and Registrar
Continental Stock
Transfer and Trust, located at 17 Battery Place, New York, NY
10004, is the transfer agent and registrar for our common
stock.
Quotation of Securities
We
intend to have a broker-dealer file a Form 211 in order to have our
common stock quoted on the Over-the-Counter, or OTC, Bulletin Board
and/or OTCQB Market operated by OTC Markets Group, Inc. (together,
the “OTCBB/OTCQB”). It is anticipated that our common
stock will be quoted on the OTCBB/OTCQB on or promptly after the
date of this prospectus, provided, however, that is no assurance
that our common stock will actually be approved and quoted on the
OTCBB/OTCQB.
Anti-Takeover Effect of Delaware Law, Certain Charter and Bylaw
Provisions
Our
certificate of incorporation and bylaws contain provisions that
could have the effect of discouraging potential acquisition
proposals or tender offers or delaying or preventing a change of
control of our company. These provisions are as
follows:
|
|
●
|
they
provide that special meetings of stockholders may be called by the
board of directors or at the request in writing by stockholders of
record owning at least twenty (20%) percent of the issued and
outstanding voting shares of common stock;
|
|
|
●
|
they do
not include a provision for cumulative voting in the election of
directors. Under cumulative voting, a minority stockholder holding
a sufficient number of shares may be able to ensure the election of
one or more directors. The absence of cumulative voting may have
the effect of limiting the ability of minority stockholders to
effect changes in our board of directors; and
|
|
|
●
|
they
allow us to issue, without stockholder approval, up to 10,000,000
shares of preferred stock that could adversely affect the rights
and powers of the holders of our common stock.
|
We are
subject to the provisions of Section 203 of the General Corporation
Law of the State of Delaware, an anti-takeover law. In general,
Section 203 prohibits a publicly held Delaware corporation from
engaging in a “business combination” with an
“interested stockholder” for a period of three years
after the date of the transaction in which the person became an
interested stockholder, unless the business combination is approved
in the following prescribed manner:
|
|
●
|
prior
to the time of the transaction, the board of directors of the
corporation approved either the business combination or the
transaction which resulted in the stockholder becoming an
interested stockholder;
|
|
|
●
|
upon
completion of the transaction that resulted in the stockholder
becoming an interested stockholder, the stockholder owned at least
85% of the voting stock of the corporation outstanding at the time
the transaction commenced, excluding for purposes of determining
the number of shares outstanding (1) shares owned by persons who
are directors and also officers and (2) shares owned by employee
stock plans in which employee participants do not have the right to
determine confidentially whether shares held subject to the plan
will be tendered in a tender or exchange offer; and
|
|
|
●
|
on or
subsequent to the time of the transaction, the business combination
is approved by the board and authorized at an annual or special
meeting of stockholders, and not by written consent, by the
affirmative vote of at least 66 2/3% of the outstanding voting
stock which is not owned by the interested stockholder.
|
Generally, for
purposes of Section 203, a “business combination”
includes a merger, asset or stock sale, or other transaction
resulting in a financial benefit to the interested stockholder. An
“interested stockholder” is a person who, together with
affiliates and associates, owns or, within three years prior to the
determination of interested stockholder status, owned 15% or more
of a corporation’s outstanding voting
securities.
90
Stockholder Action by Written Consent
Our
Certificate of Incorporation, as amended, provides that any action
required by law to be taken at any annual or special meeting of the
stockholders or any action which may be taken at such a meeting may
be taken without a meeting by written consent of the stockholders
in lieu of a meeting.
Choice of Forum
Our
Certificate of Incorporation, as amended, provides that, unless we
consent in writing to the selection of an alternative forum, the
Court of Chancery of the State of Delaware will be the exclusive
forum for any derivative action or proceeding brought on our
behalf; any action asserting a breach of fiduciary duty; any action
asserting a claim against us, or any of our officers or directors,
arising pursuant to the Delaware General Corporation Law, our
Certificate of Incorporation, as amended, or our bylaws; or any
action asserting a claim against us that is governed by the
internal affairs doctrine. This exclusive forum provision may limit
the ability of our stockholders to bring a claim in a judicial
forum that such stockholders find favorable for the disputes listed
above, which may discourage such lawsuits against us, or any of our
officers or directors.
91
SELLING STOCKHOLDERS
The
following table sets forth information as of the date of this
prospectus, to our knowledge, about the beneficial ownership of our
common stock by the selling stockholders prior to this offering,
the amount to be offered for the selling stockholder’s
account, and the amount to be owned by such selling stockholder
after completion of this offering.
All of
the selling stockholders received their securities: (i) as a result
of the 2016 merger transaction (the “Merger”), (ii) in
our private placement, for which closings occurred April 8, 2016
through September 9, 2016 (the “2016 Private
Placement”), and (iii) in our Bridge Offering; in each case
prior to the initial filing date of the registration statement of
which this prospectus is a part. We believe that the selling
stockholders have sole voting and investment power with respect to
all of the shares of common stock beneficially owned by them unless
otherwise indicated.
During
the 2016 Private Placement, we issued an aggregate 1,873,299 shares
of our common stock, inclusive of 87,099 shares of our common stock
issued pursuant to the conversion of promissory notes in connection
with the 2016 Private Placement, and Investor Warrants to purchase
1,873,299 shares of our common stock at an exercise price of $5.00,
inclusive of Investor Warrants to purchase 87,099 shares of our
common stock issued pursuant to the conversion of promissory notes
in connection with the 2016 Private Placement. Of those shares and
Investor Warrants issued in the 2016 Private Placement, only
1,713,254 shares of our common stock, and Investor Warrants to
purchase 1,713,254 shares of our common stock are being registered
as part of this registration statement. 160,045 shares of our
common stock and Investor Warrants to purchase 160,045 shares of
our common stock, at an exercise price of $5.00 per share, are
excluded from the below selling stockholder table as they were
issued in our 2016 Private Placement to affiliates of the Placement
Agent and holders of certain convertible promissory notes in
connection with the 2016 Private Placement, for which no
registration rights were granted.
Certain
of the selling stockholders and intermediaries, who are identified
as broker-dealers in the footnotes to the selling stockholder
table, through whom such securities are sold are deemed
“underwriters” within the meaning of the Securities Act
of 1933, as amended (the “Securities Act”), with
respect to the securities offered hereby, and any profits realized
or commissions received may be deemed underwriting compensation. We
believe that all securities purchased by broker-dealers or
affiliates of broker-dealers were purchased by such persons and
entities in the ordinary course of business and at the time of
purchase, such purchasers did not have any agreements or
understandings, directly or indirectly, with any person to
distribute such securities.
The
percent of beneficial ownership for the selling stockholders is
based on 5,398,531 shares of common stock outstanding as of the
date of this prospectus. Warrants to purchase shares of our common
stock held by certain investors that are currently exercisable or
exercisable within 60 days of the date of this prospectus are
considered outstanding and beneficially owned by such investors for
the purpose of computing the percentage ownership of their
respective percentage ownership but are not treated as outstanding
for the purpose of computing the percentage ownership of any other
stockholder. Unless otherwise stated below, to our knowledge, none
of the selling stockholders has had a material relationship with us
other than as a stockholder at any time within the past three years
or has ever been one of our officers or directors.
Pursuant to Rules
13d-3 and 13d-5 of the Securities Exchange Act of 1934, as amended
(the “Exchange Act”), beneficial ownership includes any
shares of our common stock as to which a stockholder has sole or
shared voting power or investment power, and also any shares of our
common stock which the stockholder has the right to acquire within
60 days, including upon exercise of warrants to purchase shares of
our common stock.
The
shares of common stock being offered pursuant to this prospectus
may be offered for sale from time to time during the period the
registration statement of which this prospectus is a part remains
effective, by or for the account of the selling stockholders. After
the date of effectiveness, the selling stockholders may have sold
or transferred, in transactions covered by this prospectus or in
transactions exempt from the registration requirements of the
Securities Act, some or all of their common stock.
Information about
the selling stockholders may change over time. Any changed
information will be set forth in an amendment to the registration
statement or supplement to this prospectus, to the extent required
by law.
92
|
|
Shares Beneficially
|
Shares
|
Shares Beneficially
|
||
|
|
Owned as of the date of
|
Offered by
|
Owned After the
|
||
|
|
this Prospectus (1)
|
this
|
Offering (1) (2)
|
||
|
Name of Selling Stockholder
|
Number
|
Percent
|
Prospectus
|
Number
|
Percent
|
|
AI International Corporate Holdings, Ltd. (3)
|
140,000
|
2.6%
|
140,000
|
-
|
-
|
|
Alchemy Ventures Group, LLC (4)
|
70,000
|
1.3%
|
70,000
|
-
|
-
|
|
Alexander J. Brown Rev Trust (5)
|
30,000
|
*
|
30,000
|
-
|
-
|
|
Andrew & Melissa Fisher
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Anthony S. Quinto
|
2,000
|
*
|
2,000
|
-
|
-
|
|
AT Media Corp. (6)
|
162,000
|
3.0%
|
162,000
|
-
|
-
|
|
Barbara E. Arendash
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Barry Warner
|
7,043
|
*
|
2,469
|
4,574
|
*
|
|
Ben Reuben
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Carolyn Pilkiewicz
|
1,468,863
|
27.2%
|
6,172
|
1,462,691
|
27.1%
|
|
Charles S. and Beth A. Hall - Tenants by the Entirety
(7)
|
8,000
|
*
|
8,000
|
-
|
-
|
|
Christopher Meyer & Mary Rivet
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Christopher R. Ellis
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Christopher Reynolds and Linda Seyfert
|
110,000
|
2.0%
|
110,000
|
-
|
-
|
|
Christopher Spinello
|
7,043
|
*
|
2,469
|
4,574
|
*
|
|
Clayton A. Struve
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Daniel & Natalie Cohen
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Daniel Fagin
|
4,000
|
*
|
4,000
|
-
|
-
|
|
David Reimer (8)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Douglas Cohen
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Dr. Yacov Geva
|
100,000
|
1.8%
|
100,000
|
-
|
-
|
|
Eugene Bauer
|
10,000
|
*
|
10,000
|
-
|
-
|
|
First Riverside Investors, LP (9)
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Four JR Investments Ltd (10)
|
40,000
|
*
|
40,000
|
-
|
-
|
93
|
|
Shares Beneficially
Owned as of the date
of
this Prospectus
(1)
|
Shares
Offered by
this
|
Shares Beneficially
Owned After the
Offering (1)(2)
|
||
|
Name of Selling Stockholder
|
Number
|
Percent
|
Prospectus
|
Number
|
Percent
|
|
Frederick B. Polak
|
10,000
|
*
|
10,000
|
-
|
-
|
|
GJG Life Sciences, LLC (11)
|
842,800
|
14.5%
|
842,800
|
-
|
-
|
|
Greg Blackfelner
|
50,000
|
*
|
50,000
|
-
|
-
|
|
Growth Ventures, Inc. Pension Plan & Trust (12)
|
20,000
|
*
|
20,000
|
-
|
-
|
|
James G. Kelley
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Jeffrey Tarrand
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Joel B. Portnoff (13)
|
26,438
|
*
|
4,938
|
21,500
|
*
|
|
Joel Kovacs
|
6,000
|
*
|
6,000
|
-
|
-
|
|
John Burke
|
10,000
|
*
|
10,000
|
-
|
-
|
|
John V. Boulger
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Joseph A. Di Vito Jr. Personal Trust (14)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Joshua Movtady
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Keith Murphy
|
40,000
|
*
|
40,000
|
-
|
-
|
|
L.N.R. Family Trust (15)
|
8,000
|
*
|
8,000
|
-
|
-
|
|
Law Offices of Kenneth E. Chyten Defined Benefit Pension Plan
(16)
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Lawrence Scott Pierce
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Lester Petracca
|
100,000
|
1.8%
|
100,000
|
-
|
-
|
|
LGA Investment Family, L.P. (17)
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Ligi Investments LLLP (18)
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Linda Korey and Harvey Schliowitz
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Ludwig Bravmann (19)
|
100,000
|
1.8%
|
100,000
|
-
|
-
|
|
M.A. Patterson and Barbara McShane, Trustees of the Patterson and
McShane Trust (20)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Margrit Polak
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Mark Harger
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Martin Feinberg
|
56,000
|
1.0%
|
56,000
|
-
|
-
|
|
Martin Westerman
|
4,000
|
*
|
4,000
|
-
|
-
|
94
|
|
Shares Beneficially
Owned as of the date ofthis
Prospectus (1)
|
Shares
offered by
this
|
Shares Beneficially
Owned After the
Offering (1)(2)
|
||
|
Name of Selling Stockholder
|
Number
|
Percent
|
Prospectus
|
Number
|
Percent
|
|
Meekavi Revocable Trust (21)
|
14,000
|
*
|
14,000
|
-
|
-
|
|
Michael DeGidio
|
3,523
|
*
|
1,235
|
2,288
|
*
|
|
Michael J. Pierce
|
80,000
|
1.5%
|
80,000
|
-
|
-
|
|
Michelle Jaigobind
|
30,000
|
*
|
30,000
|
-
|
-
|
|
Northlea Partners, LLLP (22)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Oppenheimer & Co. Inc. Cust FBO Uri Kaufthal IRA
(23)
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Orchestra Medical Ventures, LLC (24)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Paul Kilgallon
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Ramnarain Jaigobind
|
30,000
|
*
|
30,000
|
-
|
-
|
|
RBC Capital Markets LLC Cust. FBO Laurence G. Allen, IRA
(25)
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Rexford Capital, LLC (26)
|
60,000
|
1.1%
|
60,000
|
-
|
-
|
|
Richard Brown
|
150,000
|
2.7%
|
150,000
|
-
|
-
|
|
Richard Roth
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Robert DiVincenzo
|
3,507
|
*
|
1,235
|
2,272
|
*
|
|
Robert H. Cohen
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Robert Hendrickson
|
10,308
|
*
|
10,308
|
-
|
-
|
|
Robert Herbst
|
80,000
|
1.5%
|
80,000
|
-
|
-
|
|
Robert Montgomery
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Roman Perez-Soler (27)
|
4,108
|
*
|
4,108
|
-
|
-
|
|
Russell Linderman & Diane Linderman
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Russell Wilson & Marilyn Wilson
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Souheil Haddad
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Sunny Wong
|
30,000
|
*
|
30,000
|
-
|
-
|
|
Terence R. Oi and Patricia M. Meehan
|
8,000
|
*
|
8,000
|
-
|
-
|
|
Thomas Mang
|
48,566
|
*
|
6,173
|
42,393
|
*
|
|
Thomas Newman and Irene Newman
|
17,572
|
*
|
6,173
|
11,399
|
*
|
|
Tim Elmes
|
10,000
|
*
|
10,000
|
-
|
-
|
95
|
|
Shares Beneficially
Owned as of the date ofthis Prospectus (1)
|
Shares
offered by
this
|
Shares Beneficially
Owned After the
Offering (1)(2)
|
||
|
Name of Selling Stockholder
|
Number
|
Percent
|
Prospectus
|
Number
|
Percent
|
|
Timothy McInerney (28)
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Vantgage fbo Laurence E Lof Roth IRA (29)
|
10,000
|
*
|
10,000
|
-
|
-
|
|
William Strawbridge
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Arnie Ross
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Chad Ekroth
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Cody Bennett
|
6,000
|
*
|
6,000
|
-
|
-
|
|
Ferdinand Smith
|
8,000
|
*
|
8,000
|
-
|
-
|
|
IDH Capital LLC (30)
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Ingram Tynes
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Jeremy Rosen
|
4,000
|
*
|
4,000
|
-
|
-
|
|
John Arcell
|
20,000
|
*
|
20,000
|
-
|
-
|
|
John B. Chambers
|
20,000
|
*
|
20,000
|
-
|
-
|
|
John Kacperski
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Johnny Armstead
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Megan N. Williams
|
6,000
|
*
|
6,000
|
-
|
-
|
|
Nelson Herrera
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Raymond J. Stowell
|
2,000
|
*
|
2,000
|
-
|
-
|
|
RBC Capital Markets LLC Cust. FBO Frank Mirchin, IRA
(31)
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Salvatore Pucci
|
3,200
|
*
|
3,200
|
-
|
-
|
|
Stephen Andrews
|
2,400
|
*
|
2,400
|
-
|
-
|
|
Stephen Papajan
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Steven Sinclair
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Steven Wietsma
|
10,000
|
*
|
10,000
|
-
|
-
|
|
William Dennis
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Jeffrey Funk
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Richard Kempski
|
10,000
|
*
|
10,000
|
-
|
-
|
96
|
|
Shares Beneficially
Owned as of the date ofthis Prospectus (1)
|
Shares
offered by
this
|
Shares Beneficially
Owned After the
Offering (1)(2)
|
||
|
Name of Selling Stockholder
|
Number
|
Percent
|
Prospectus
|
Number
|
Percent
|
|
Ralph Darling
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Todd Zahnow
|
10,000
|
*
|
10,000
|
-
|
-
|
|
Ron Gress Jr
|
6,000
|
*
|
6,000
|
-
|
-
|
|
Armondo Gallotta
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Daniel Waldman
|
20,000
|
*
|
20,000
|
-
|
-
|
|
Alexander J. Brown
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Diane A. Brown
|
2,000
|
*
|
2,000
|
-
|
-
|
|
David Bilan
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Allaeddin Jallad
|
2,000
|
*
|
2,000
|
-
|
-
|
|
Nancy Ganz
|
2,000
|
*
|
2,000
|
-
|
-
|
|
James A. Studdiford
|
40,000
|
*
|
40,000
|
-
|
-
|
|
Ann S. Totten
|
8,000
|
*
|
8,000
|
-
|
-
|
|
Michael Cohen
|
4,000
|
*
|
4,000
|
-
|
-
|
|
Robert Urs
|
4,000
|
*
|
4,000
|
-
|
-
|
* Less
than 1%.
(1)
Share
numbers include shares underlying warrants held by the selling
stockholder.
(2)
Assumes the sale
of all shares offered pursuant to this prospectus.
(3)
Ezzat
Jallad is a natural person with voting and dispositive power over
the shares held by AI International Corporate Holdings,
Ltd.
(4)
Michael Ross is a
natural person with voting and dispositive power over the shares
held by Alchemy Ventures Group, LLC. Mr. Ross is affiliated with a
FINRA member broker-dealer.
(5)
Richard A. Brown
is a trustee with voting and dispositive power over the shares held
by the Alexander J. Brown Rev Trust, dated April 11,
1996.
(6)
Radha
Freese is a natural person with voting and dispositive power over
the shares held by AT Media Corp.
(7)
Charles S. and
Beth A. Hall are affiliated with a FINRA member
broker-dealer.
(8)
David
Reimer is a broker-dealer. Mr. Reimer is affiliated with a FINRA
member broker-dealer.
97
(9)
Stephen Bolduc is
a natural person with voting and dispositive power over the shares
held by First Riverside Investors, LP.
(10)
Robert
D. Burke is a natural person with voting and dispositive power over
the shares held by Four JR Investments Ltd.
(11)
Jennifer Lorenzo
is a natural person with voting and dispositive power over the
shares held by GJG Life Sciences, LLC.
(12)
Gary
J. McAdam is a natural person with voting and dispositive power
over the shares held by Growth Ventures, Inc. Pension Plan &
Trust, dated February 1, 1981.
(13)
Subscription
documents for Mr. Portnoff were completed by Barbara Portnoff,
Executor of the Estate of Joel B. Portnoff. Shares held in Mr.
Portnoff's name will be transferred to the appropriate party upon
final settlement of his Estate.
(14)
Joseph
A. Di Vito is a trustee with voting and dispositive power over the
shares held by the Joseph A. Divito Jr. Personal Trust, dated
September 13, 2000. Mr. Di Vito is affiliated with a FINRA member
broker-dealer.
(15)
Laurence Rivkin is
a co-trustee with voting and dispositive power over the shares held
by the L.N.R. Family Trust, dated August 2, 1987.
(16)
Kenneth E. Chyten
is a trustee with voting and dispositive power over the shares held
by the Law Offices of Kenneth E. Chyten Defined Benefit Pension
Plan, dated December 1, 2001.
(17)
Laurence G. Allen
is a natural person with voting and dispositive power over the
shares held by LGA Investment Family, L.P. Mr. Allen is affiliated
with a FINRA member broker-dealer.
(18)
Jennifer Ligeti is
a natural person with voting and dispositive power over the shares
held by LIGI Investments LLLP.
(19)
Ludwig
Bravmann is affiliated with a FINRA member
broker-dealer.
(20)
Barbara McShane
and Michael Patterson are co-trustees with voting and dispositive
power over the shares held by the Patterson and McShane Trust,
dated July 7, 2005.
(21)
Meenakshi V
Bhavsar is a trustee with voting and dispositive power over the
shares held by the Meekavi Revocable Trust, dated July 15,
2005.
(22)
John
H. Abeles MD is a natural person with voting and dispositive power
over the shares held by Northlea Partners LLLP.
(23)
Uri
Kaufthal is a natural person with voting and dispositive power over
the shares held by Oppenheimer & Co. Inc. Cust FBO Uri Kaufthal
IRA. Mr. Kaufthal is affiliated with a FINRA member
broker-dealer.
(24)
David
Hochman and Darren Sherman are natural persons with voting and
dispositive power over the shares held by Orchestra Medical
Ventures, LLC. Mr. Hochman is a Director of the
Company.
(25)
Laurence G. Allen
is a natural person with voting and dispositive power over the
shares held by RBC Capital Markets LLC Cust. FBO Laurence G. Allen,
IRA. Mr. Allen is affiliated with a FINRA member
broker-dealer.
(26)
Kimberly Langston
is a natural person with voting and dispositive power over the
shares held by Rexford Capital, LLC.
(27)
Roman
Perez-Soler is a Director of the Company.
(28)
Timothy McInerney
is a Director of the Company. Mr. McInerney is affiliated with a
FINRA member broker-dealer.
(29)
Laurence E. Lof is a natural person with voting and dispositive
power over the shares held by Vantage fbo Laurence E. Lof Roth IRA.
Mr. Lof is affiliated with a FINRA member
broker-dealer.
(30)
Issakhar Daniell
is a natural person with voting and dispositive power over the
shares held by IDH Capital LLC.
(31)
Frank
Mirchin is a natural person with voting and dispositive power over
the shares held by RBC Capital Markets LLC Cust. FBO Frank Mirchin,
IRA.
98
PLAN OF DISTRIBUTION
The
selling stockholders, which as used herein includes donees,
pledgees, transferees or other successors in-interest selling
shares of common stock or interests in shares of common stock
received after the date of this prospectus from a selling
stockholder as a gift, pledge, partnership distribution or other
transfer, may, from time to time, sell, transfer or otherwise
dispose of any or all of their shares of common stock or interests
in shares of common stock on any stock exchange, market or trading
facility on which the shares are traded or in private
transactions.
The
selling security holders may sell some or all of their shares at a
fixed price of $5.00 per share until our shares are quoted on the
Over-the-Counter, or OTC, Bulletin Board and/or OTCQB Market
operated by OTC Markets Group, Inc. (together, the
“OTCBB/OTCQB”) and thereafter at prevailing market
prices or privately negotiated prices. Prior to being quoted on the
OTCBB/OTCQB, stockholders may sell their shares in private
transactions to other individuals.
Our
common stock is not listed or traded on any public exchange, and we
have not applied for listing or quotation on any exchange. We are
seeking sponsorship for the quotation of our common stock on the
OTCBB/OTCQB. In order to be quoted on the OTCBB/OTCQB, a market
maker must file an application on our behalf in order to make a
market for our common stock. There can be no assurance that a
market maker will agree to file the necessary documents with the
Financial Industry Regulatory Authority (“FINRA”), nor
can there be any assurance that such an application for quotation
will be approved. There is further no assurance that an active
trading market for our shares will develop, or, if developed, that
it will be sustained. In the absence of a trading market or an
active trading market, investors may be unable to liquidate their
investment.
The
selling stockholders may use any one or more of the following
methods when disposing of shares or interests therein:
|
|
●
|
ordinary
brokerage transactions and transactions in which the broker-dealer
solicits purchasers;
|
|
|
●
|
block
trades in which the broker-dealer will attempt to sell the shares
as agent, but may position and resell a portion of the block as
principal to facilitate the transaction;
|
|
|
●
|
purchases
by a broker-dealer as principal and resale by the broker-dealer for
its account;
|
|
|
●
|
privately
negotiated transactions;
|
|
|
●
|
short
sales;
|
|
|
●
|
through
the writing or settlement of options or other hedging transactions,
whether through an options exchange or otherwise;
|
|
|
●
|
broker-dealers
may agree with the selling stockholders to sell a specified number
of such shares at a stipulated price per share;
|
|
|
●
|
a
combination of any such methods of sale; and
|
|
|
●
|
any
other method permitted pursuant to applicable law.
|
The
selling stockholders may, from time to time, pledge or grant a
security interest in some or all of the shares of common stock
owned by them and, if they default in the performance of their
secured obligations, the pledgees or secured parties may offer and
sell the shares of common stock, from time to time, under this
prospectus, or under an amendment to this prospectus under Rule
424(b)(3) or other applicable provision of the Securities Act of
1933, as amended (the “Securities Act”), amending the
list of selling stockholders to include the pledgee, transferee or
other successors in interest as selling stockholders under this
prospectus. The selling stockholders also may transfer the shares
of common stock in other circumstances, in which case the
transferees, pledgees or other successors in interest will be the
selling beneficial owners for purposes of this prospectus;
provided, however, that prior to any such transfer the following
information (or such other information as may be required by the
federal securities laws from time to time) with respect to each
such selling beneficial owner must be added to the prospectus by
way of a prospectus supplement or post-effective amendment, as
appropriate: (1) the name of the selling beneficial owner; (2) any
material relationship the selling beneficial owner has had within
the past three years with us or any of our predecessors or
affiliates; (3) the amount of securities of the class owned by such
beneficial owner before the offering; (4) the amount to be offered
for the beneficial owner’s account; and (5) the amount and
(if one percent or more) the percentage of the class to be owned by
such beneficial owner after the offering is complete.
99
In
connection with the sale of our common stock or interests therein,
the selling stockholders may enter into hedging transactions with
broker-dealers or other financial institutions, which may in turn
engage in short sales of the common stock in the course of hedging
the positions they assume. The selling stockholders may also sell
shares of our common stock short and deliver these securities to
close out their short positions, or loan or pledge the common stock
to broker-dealers that in turn may sell these securities. The
selling stockholders may also enter into option or other
transactions with broker-dealers or other financial institutions or
the creation of one or more derivative securities which require the
delivery to such broker-dealer or other financial institution of
shares offered by this prospectus, which shares such broker-dealer
or other financial institution may resell pursuant to this
prospectus (as supplemented or amended to reflect such
transaction).
The
aggregate proceeds to the selling stockholders from the sale of the
common stock offered by them will be the purchase price of the
common stock less discounts or commissions, if any. Each of the
selling stockholders reserves the right to accept and, together
with their agents from time to time, to reject, in whole or in
part, any proposed purchase of common stock to be made directly or
through agents. We will not receive any of the proceeds from this
offering, provided, however, we will receive proceeds from the
exercise of the warrants held by certain investors.
The
selling stockholders also may resell all or a portion of the shares
in open market transactions in reliance upon Rule 144 under the
Securities Act, provided that they meet the criteria and conform to
the requirements of that rule.
The
selling stockholders and any underwriters, broker-dealers or agents
that participate in the sale of the common stock or interests
therein may be “underwriters” within the meaning of
Section 2(11) of the Securities Act. Any discounts, commissions,
concessions or profit they earn on any resale of the shares may be
underwriting discounts and commissions under the Securities Act.
Selling stockholders who are “underwriters” within the
meaning of Section 2(11) of the Securities Act will be subject to
the prospectus delivery requirements of the Securities
Act.
To the
extent required, the shares of our common stock to be sold, the
names of the selling stockholders, the respective purchase prices
and public offering prices, the names of any agents, dealer or
underwriter, any applicable commissions or discounts with respect
to a particular offer will be set forth in an accompanying
prospectus supplement or, if appropriate, a post-effective
amendment to the registration statement that includes this
prospectus.
The
maximum amount of compensation to be received by any FINRA member
or independent broker-dealer for the sale of any securities
registered under this prospectus will not be greater than 8.0% of
the gross proceeds from the sale of such securities.
In
order to comply with the securities laws of some states, if
applicable, the common stock may be sold in these jurisdictions
only through registered or licensed brokers or dealers. In
addition, in some states the common stock may not be sold unless it
has been registered or qualified for sale or an exemption from
registration or qualification requirements is available and is
complied with.
We have
advised the selling stockholders that the anti-manipulation rules
of Regulation M under the Securities Exchange Act of 1934, as
amended (the “Exchange Act”) may apply to sales of
shares in the market and to the activities of the selling
stockholders and their affiliates. In addition, we will make copies
of this prospectus (as it may be supplemented or amended from time
to time) available to the selling stockholders for the purpose of
satisfying the prospectus delivery requirements of the Securities
Act. The selling stockholders may indemnify any broker-dealer that
participates in transactions involving the sale of the shares
against certain liabilities, including liabilities arising under
the Securities Act.
100
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER
MATTERS
Market Information
There
is no public trading market on which our common stock is traded.
Among other matters, in order for our common stock to become
Over-the-Counter, or OTC, Bulletin Board and/or OTCQB Market
operated by OTC Markets Group, Inc. (together, the
“OTCBB/OTCQB”) eligible, a Financial Industry
Regulatory Authority (“FINRA”) member broker/dealer
must file a Form 211 with FINRA and commit to make a market in our
securities once the Form 211 is approved by FINRA. As of the date
of this prospectus, the Form 211 has not been filed with FINRA.
There is no assurance that our common stock will be included on the
OTCBB/OTCQB.
The
shares of common stock registered hereby can be sold by selling
stockholders at a fixed price of $5.00 per share until our shares
are quoted on the OTCBB/OTCQB and thereafter at prevailing market
prices or privately negotiated prices. We determined such fixed
price based on the highest price at which shares of our common
stock were sold in our private placement, for which closings
occurred April 8, 2016 through September 9, 2016 (the “2016
Private Placement”).
We can
offer no assurance that an active public market in our shares will
develop or be sustained. Future sales of substantial amounts of our
shares in the public market could adversely affect market prices
prevailing from time to time and could impair our ability to raise
capital through the sale of our equity securities.
Holders
As of
the date of this prospectus, there are approximately 162 record holders of our common
stock.
LEGAL MATTERS
The
validity of the securities offered in this prospectus is being
passed upon for us by Lowenstein Sandler LLP, New York, New York.
Members of the firm beneficially own 11,080 shares of our common
stock.
EXPERTS
The
consolidated balance sheets of Adgero Biopharmaceuticals, Inc., and
the related statements of operations, stockholders’
(deficit) equity, and cash flows for each of the years
in the two-year period ended December 31, 2016, have
been audited by Marcum LLP, an independent registered public
accounting firm, as stated in their report which is included in
this prospectus herein. Such financial statements have been
included herein in reliance on the report of such firm given upon
their authority as experts in accounting and auditing.
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION FOR SECURITIES
ACT LIABILITIES
Our
directors and officers are indemnified to the fullest extent
permitted under Delaware law. We also maintain insurance which
protects our officers and directors against any liabilities
incurred in connection with their service in such a
capacity.
Insofar
as indemnification for liabilities arising under the Securities Act
of 1933, as amended (the “Securities Act”), may be
permitted to our directors, officers and controlling persons
pursuant to the foregoing, or otherwise, we have been advised that
in the opinion of the Securities and Exchange Commission (the
“SEC”) such indemnification is against public policy as
expressed in the Securities Act and is, therefore, unenforceable.
In the event that a claim for indemnification against such
liabilities (other than the payment by us of expenses incurred or
paid by a director, officer or controlling person of ours in the
successful defense of any action, suit or proceeding) is asserted
by such director, officer or controlling person in connection with
the securities being registered, we will, unless in the opinion of
our counsel the matter has been settled by controlling precedent,
submit to a court of appropriate jurisdiction the question whether
such indemnification by it is against public policy as expressed in
the Securities Act and will be governed by the final adjudication
of such issue.
101
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have
filed with the Securities and Exchange Commission (the
“SEC”) a registration statement on Form S-1 under the
Securities Act of 1933, as amended (the “Securities
Act”), with respect to the common stock offered by this
prospectus. This prospectus, which is part of the registration
statement, omits certain information, exhibits, schedules and
undertakings set forth in the registration statement. For further
information pertaining to us and our common stock, reference is
made to the registration statement and the exhibits and schedules
to the registration statement. Statements contained in this
prospectus as to the contents or provisions of any documents
referred to in this prospectus are not necessarily complete, and in
each instance where a copy of the document has been filed as an
exhibit to the registration statement, reference is made to the
exhibit for a more complete description of the matters
involved.
You may
read and copy all or any portion of the registration statement
without charge at the office of the SEC at the Public Reference
Room at Station Place, 100 F Street, N.E., Washington, D.C. 20549.
Copies of the registration statement may be obtained from the SEC
at prescribed rates from the Public Reference Section of the SEC at
such address. In addition, registration statements and certain
other filings made with the SEC electronically are publicly
available through the SEC’s web site at http://www.sec.gov.
The registration statement, including all exhibits and amendments
to the registration statement, has been filed electronically with
the SEC.
Following
the effectiveness of the registration statement, of which this
prospectus is a part, we will become subject to the information and
periodic reporting requirements of the Securities Exchange Act of
1934, as amended (the “Exchange Act”) and, accordingly,
will file annual reports containing financial statements audited by
an independent registered public accounting firm, quarterly reports
containing unaudited financial data, current reports, proxy
statements and other information with the SEC. You will be able to
inspect and copy such periodic reports, proxy statements and other
information at the SEC’s public reference room, and the web
site of the SEC referred to above.
102
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
INDEX TO FINANCIAL STATEMENTS
|
|
Page Number
|
|
Report
of Independent Registered Public Accounting
Firm
|
F-2
|
|
Consolidated
Balance Sheets as of December 31, 2016 and 2015
|
F-3
|
|
Consolidated
Statements of Operations for the years ended December 31, 2016 and
2015
|
F-4
|
|
Consolidated
Statements of Stockholders’ (Deficit) Equity for the years
ended December 31, 2016 and 2015
|
F-5
|
|
Consolidated
Statements of Cash Flows for the years ended December 31, 2016 and
2015
|
F-6
|
|
Notes
to Consolidated Financial Statements
|
F-7
|
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the Audit
Committee of the
Board of Directors
and Shareholders
of Adgero
Biopharmaceuticals Holdings, Inc.
We
have audited the accompanying consolidated balance sheets of Adgero
Biopharmaceuticals Holdings, Inc. (the “Company”) as of
December 31, 2016 and 2015, and the related consolidated statements
of operations, changes in stockholders’ (deficit) equity and
cash flows for the years then ended. These consolidated financial
statements are the responsibility of the Company’s
management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We conducted our
audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards
require that we plan and perform the audit to obtain reasonable
assurance about whether the financial statements are free of
material misstatement. The
Company is not required to have, nor were we engaged to perform, an
audit of its internal control over financial reporting. Our audit
included consideration of internal control over financial reporting
as a basis for designing audit procedures that are appropriate in
the circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such
opinion. An audit also includes examining, on a test
basis, evidence supporting the amounts and disclosures in the
financial statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating the
overall financial statement presentation. We believe that our
audits provide a reasonable basis for our
opinion.
In our opinion, the financial statements referred
to above present fairly, in all material respects, the consolidated
financial position of Adgero Biopharmaceuticals Holdings, Inc., as
of December 31, 2016 and 2015, and the
consolidated results of its operations and its cash flows for
the years then ended in conformity with accounting principles
generally accepted in the United States of
America.
/s/ Marcum LLP
Marcum llp
New York,
NY
April
11,
2017
F-2
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Consolidated
Statements of Operations
|
|
December
31,
|
|
|
|
2016
|
2015
|
|
Assets
|
|
|
|
Current
assets:
|
|
|
|
Cash and cash
equivalents
|
$3,165,225
|
$228,737
|
|
Certificates of
deposit
|
2,802,000
|
-
|
|
Prepaid
expenses
|
50,659
|
-
|
|
Total current
assets
|
6,017,884
|
228,737
|
|
Property and
equipment, net
|
26,313
|
-
|
|
Deferred offering
costs
|
15,855
|
195,000
|
|
Total
Assets
|
$6,060,052
|
$423,737
|
|
|
|
|
|
Liabilities
and Stockholders' Equity (Deficit)
|
|
|
|
Current
Liabilities:
|
|
|
|
Accounts
payable
|
$524,883
|
$370,169
|
|
Accrued
interest
|
-
|
55,231
|
|
Accrued
expenses
|
400,825
|
54,000
|
|
Advances from
stockholder
|
962
|
29,108
|
|
Total current
liabilities
|
926,670
|
508,508
|
|
Note payable to
stockholder
|
-
|
163,934
|
|
Convertible
notes
|
-
|
485,000
|
|
Total
Liabilities
|
926,670
|
1,157,442
|
|
|
|
|
|
Commitments
and Contingencies (Note 5)
|
|
|
|
|
|
|
|
Stockholders'
Equity (Deficit):
|
|
|
|
Preferred stock,
$0.0001 par value, 10,000,000 shares authorized and no shares
issued or outstanding
|
-
|
-
|
|
Common stock,
$0.0001 par value, 50,000,000 shares authorized and 4,987,451 and
2,000,000 shares issued and outstanding at December 31, 2016 and
December 31, 2015, respectively
|
499
|
200
|
|
Additional paid in
capital
|
8,516,111
|
70,960
|
|
Accumulated
deficit
|
(3,383,228)
|
(804,865)
|
|
Total Stockholders'
Equity (Deficit)
|
5,133,382
|
(733,705)
|
|
Total Liabilities
and Stockholders' Equity (Deficit)
|
$6,060,052
|
$423,737
|
See Notes to
Consolidated Financial Statements
F-3
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Consolidated
Statements of Operations
|
|
Years Ended December 31,
|
|
|
|
2016
|
2015
|
|
|
|
|
|
Operating
expenses:
|
|
|
|
Research and
development
|
$783,656
|
$13,770
|
|
General and
administrative
|
1,787,013
|
108,121
|
|
Total operating
expenses
|
2,570,669
|
121,891
|
|
Loss
from operations
|
(2,570,669)
|
(121,891)
|
|
|
|
|
|
Other
income (expense):
|
|
|
|
Interest
income
|
8,400
|
271
|
|
Interest
expense
|
(16,094)
|
(20,100)
|
|
Total other
expense, net
|
(7,694)
|
(19,829)
|
|
Net
loss
|
$(2,578,363)
|
$(141,720)
|
|
Net
loss per common share:
|
|
|
|
Basic and
diluted
|
$(0.68)
|
$(0.07)
|
|
Weighted
average common shares:
|
|
|
|
Basic and
diluted
|
3,803,293
|
1,957,551
|
|
|
|
|
See Notes to
Consolidated Financial Statements
F-4
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Consolidated
Statements of Changes in Stockholders’ (Deficit)
Equity
|
|
|
|
|
|
|
|
|
|
Common Stock
|
|
|
|
|
|
|
|
Number of Shares
|
Amount
|
Additional Paid-In Capital
|
Accumulated Deficit
|
Subscription
Receivable
|
Total Stockholder's (Deficit) Equity
|
|
Balance at December 31, 2014
|
1,938,272
|
$194
|
$40,966
|
$(663,145)
|
$(7,887)
|
$(629,872)
|
|
Issuance
of restricted common stock for cash
|
61,728
|
6
|
29,994
|
-
|
-
|
30,000
|
|
Services
received in lieu of cash for settlement of the subscription
receivable
|
-
|
-
|
-
|
-
|
7,887
|
7,887
|
|
Net
loss
|
-
|
-
|
-
|
(141,720)
|
-
|
(141,720)
|
|
Balance at December 31, 2015
|
2,000,000
|
200
|
70,960
|
(804,865)
|
-
|
(733,705)
|
|
Equity
of Holdings at time of Reverse Merger
|
1,000,000
|
100
|
14,471
|
-
|
-
|
14,571
|
|
Conversion
of convertible notes and accrued interest for
Units
|
132,098
|
13
|
549,481
|
-
|
-
|
549,494
|
|
Exchange
of shareholder note and accrued interest for
Units
|
34,153
|
3
|
170,762
|
-
|
-
|
170,765
|
|
Issuance
of Units, net of offering costs
|
1,786,200
|
179
|
7,235,665
|
-
|
-
|
7,235,844
|
|
Issuance
of restricted stock and options
|
35,000
|
4
|
474,772
|
-
|
-
|
474,776
|
|
Net
loss
|
-
|
-
|
-
|
(2,578,363)
|
-
|
(2,578,363)
|
|
Balance at December 31, 2016
|
4,987,451
|
$499
|
$8,516,111
|
$(3,383,228)
|
$-
|
$5,133,382
|
See Notes to
Consolidated Financial Statements
F-5
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Consolidated
Statements of Cash Flows
|
|
Years Ended
December 31,
|
|
|
|
2016
|
2015
|
|
Cash
flows from operating activities
|
|
|
|
Net
loss
|
$(2,578,363)
|
$(141,720)
|
|
Adjustments to
reconcile net loss to net cash used in operating
activities:
|
|
|
|
Services received
in lieu of cash for settlement of the subscription
receivable
|
-
|
7,887
|
|
Depreciation
expense
|
306
|
-
|
|
Stock-based
compensation
|
474,776
|
-
|
|
Interest
expense
|
16,094
|
-
|
|
Changes in
operating assets and liabilities:
|
|
|
|
Interest
receivable
|
-
|
104
|
|
Prepaid expenses
and other current assets
|
(50,659)
|
-
|
|
Accounts
payable
|
138,859
|
50,654
|
|
Accrued
expenses
|
346,825
|
20,100
|
|
Total
adjustments
|
926,201
|
78,745
|
|
Net
cash used in operating activities
|
(1,652,162)
|
(62,975)
|
|
Cash
flows from investing activities
|
|
|
|
Cash acquired in
Reverse Merger
|
14,571
|
-
|
|
Purchase of
certificate of deposit
|
(5,302,000)
|
-
|
|
Proceeds from
certificate of deposit
|
2,500,000
|
-
|
|
Purchase of
property and equipment
|
(26,619)
|
-
|
|
Net
cash used in investing activitivies
|
(2,814,048)
|
-
|
|
Cash
flows from financing activities
|
|
|
|
Proceeds from
subscription receivable
|
-
|
1,105
|
|
Proceeds from
private placement, net of offering costs
|
7,430,844
|
-
|
|
Deferred offering
costs
|
-
|
(45,000)
|
|
Issuance of
restricted common stock
|
-
|
30,000
|
|
Proceeds from
convertible notes
|
-
|
285,000
|
|
(Repayment) advance
to/from stockholder
|
(28,146)
|
20,253
|
|
Net
cash provided by financing activitivies
|
7,402,698
|
291,358
|
|
Net
increase in cash and cash equivalents
|
2,936,488
|
228,383
|
|
Cash
and cash equivalents, beginning of year
|
228,737
|
354
|
|
Cash
and cash equivalents, end of year
|
$3,165,225
|
$228,737
|
|
Supplemental
disclosures of non-cash financing activities
|
|
|
|
Deferred
offering costs in accounts payable
|
$15,855
|
$150,000
|
|
Conversion
of convertible notes and accrued interest to
Units
|
$549,494
|
$-
|
|
Exchange
of note payable and accrued interest for Units
|
$170,765
|
$-
|
|
Deferred
issuance costs reclassified to paid-in capital
|
$195,000
|
$-
|
See Notes to
Consolidated Financial Statements
F-6
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
1.
Organization
Nature
of Business and Liquidity
Adgero
Biopharmaceuticals Holdings, Inc. (“Holdings”),
incorporated in Delaware on October 26, 2015, wholly owns Adgero
Biopharmaceuticals, Inc. (“Adgero”) which was
incorporated in Delaware on November 16, 2007 and wholly owns
Remelux Biopharmaceuticals, Inc (“Remelux”) which was
incorporated in Delaware on July 18, 2014 (collectively the
“Company”) and Remelux was dissolved on January 19,
2016. The Company is a biopharmaceutical company, focused on the
development of photodynamic therapy, for the treatment of rare,
unmet medical needs, specifically orphan cancer indications. The
Company is headquartered in Princeton, New
Jersey.
The Company is
devoting substantially all of its efforts towards research and
development of its photodynamic therapy and raising capital. The
Company has not generated any product revenue to
date.
The Company has
financed its operations to date primarily through the issuance of
its common stock and convertible notes, loans from stockholders and
through the 2016 Units Offering (see Note 8 – Equity –
2016 Units Offering). The Company expects to continue to incur net
losses in the foreseeable future.
As of December 31,
2016, the Company had cash and cash equivalents of $3,165,225 and
holds certificates of deposit totaling, in the aggregate,
$2,802,000 as of December 31, 2016. Although the Company has
incurred recurring losses, the Company expects its cash and cash
equivalents and certificates of deposit as of December 31, 2016,
plus the $1,784, 145 of net cash proceeds received from a January
2017 closing of Investor Units (see Note 9 – Subsequent
Events – Sale of Investor Units) will be sufficient to fund
operations for at least the next twelve months from the date of
this filing.
The Company will
need to continue to raise funds until it is able to generate
revenues from operations sufficient to fund its development and
commercial operations. The Company cannot be certain that
additional funding will be available on acceptable terms, or at
all, in which case it may have to significantly delay, scale back
or discontinue the development and/or commercialization of its
product. The Company may also be required to (a) seek collaborators
for its product at an earlier stage than otherwise would be
desirable and on terms that are less favorable than might otherwise
be available; or (b) relinquish or otherwise dispose of rights to
technology or its product that the Company would otherwise seek to
deploy or commercialize.
2.
Summary of Significant Accounting Policies
Basis
of Presentation and Consolidation
The consolidated
financial statements include the accounts of Holdings, Adgero and
Remelux. Significant inter-company account balances and
transactions have been eliminated in
consolidation.
On January 11,
2016, Adgero entered into a merger agreement (the ”Merger
Agreement”) by and among Holdings, the legal acquirer, which
had 1,000,000 shares of common stock outstanding (50% of which were
owned by a principal of the 2016 Units Offering placement agent),
Adgero Acquisition, Inc., a Delaware corporation and wholly-owned
subsidiary of Holdings (“Merger Sub”) and Adgero.
Pursuant to the terms of the Merger Agreement, as a condition of
and contemporaneously with the April 8, 2016 initial closing of the
2016 Units Offering (the “Initial Closing”), Merger Sub
merged with and into Adgero and Adgero became a wholly–owned
subsidiary of Holdings. In connection with the merger (the
“Merger”), the stockholders of Adgero received an
aggregate of 2,000,000 shares of Holdings’ common stock in
exchange for their outstanding shares of Adgero common stock,
utilizing an exchange ratio of 0.12345679. Immediately following
the Merger, the Adgero stockholders controlled 66.7% of Holdings.
In addition, the holders of warrants to purchase 30,864 shares of
common stock of Adgero with an exercise price of $4.05 per share
prior to the Merger received replacement warrants (the
“Replacement Warrants”) to purchase 30,864 shares of
Holdings’ common stock with an exercise price of $5.00 per
share.
For financial
reporting purposes, this was a capital transaction of Adgero or a
"Reverse Merger" rather than a business combination, because the
shareholders of Adgero controlled the combined company immediately
following the completion of the transaction. Adgero was deemed to
be the accounting acquirer in the transaction and, consequently,
the transaction was treated as a recapitalization of
Adgero. Accordingly, the assets and liabilities and the
historical operations that are reflected in these consolidated
financial statements are those of Adgero and are recorded at the
historical cost basis of Adgero. Holding’s net assets and
results of operations were consolidated with those of Adgero
effective with the April 8, 2016 consummation of the Reverse
Merger.
F-7
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Basis
of Presentation and Consolidation, continued
In the accompanying
financial statements, all share and per share amounts, of Adgero
were retroactively adjusted to reflect the 0.12345679 exchange
ratio effective with the consummation of the Reverse Merger for all
periods presented. All costs attributable to the Reverse Merger
were expensed.
Use
of Estimates
The preparation of
the Company’s financial statements in conformity with U.S.
GAAP requires management to make estimates and assessments that
affect the amounts reported in the consolidated financial
statements and accompanying notes. The Company bases its estimates
and judgments on historical experience and on various other
assumptions that it believes are reasonable under the
circumstances. The amounts of assets and liabilities reported in
the Company’s balance sheets and the amounts of expenses
reported for each of the periods presented are effected by
estimates and assumptions, which are used for, but not limited to,
fair value calculations for equity securities, establishing
valuation allowances for deferred taxes, and the recovery of
deferred costs. Actual results could differ from those
estimates.
Cash
and Cash Equivalents
The Company
considers all highly-liquid investments with an original maturity
of three months or less when purchased to be cash equivalents. All
cash and cash equivalents are held in United States financial
institutions.
Certificates
of Deposit
As of December 31,
2016, the Company holds certificates of deposit (“CDs”)
totaling, in the aggregate, $2,802,000, with original maturities
ranging from five months to nine months. The CDs earn a weighted
average interest rate of 0.64% per annum.
Property
and Equipment, net
Property and
equipment are stated at cost, net of accumulated depreciation,
which is recorded commencing at the in-service date using the
straight-line method at rates sufficient to charge the cost of
depreciable assets to operations over their estimated useful lives,
which is 7 years.
F-8
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Stock-Based
Compensation
The Company
accounts for equity awards in accordance with FASB ASC Topic No.
718, Compensation-Stock
Compensation. Under FASB ASC Topic No. 718, compensation
expense related to stock-based payments to employees and directors
is recorded over the requisite service period based on the grant
date fair value of the awards. The grant date value of
performance-based equity awards is recognized over the service
period, so long as completion of the performance criteria is deemed
to be probable. Compensation previously recorded for unvested
equity awards that are forfeited is reversed upon forfeiture. The
Company uses the Black-Scholes option pricing model for determining
the estimated fair value for stock-based awards. The Black-Scholes
model requires the use of assumptions which determine the fair
value of stock-based awards, including the option’s expected
term and the price volatility of the underlying
stock.
The Company’s
accounting policy for equity instruments issued to consultants and
vendors in exchange for goods and services follows the provisions
of FASB ASC Topic No. 505-50, Equity Based Payments to Non-Employees.
Accordingly, the measurement date for the fair value of the equity
instruments issued is determined at the earlier of (i) the date at
which a commitment for performance by the consultant or vendor is
reached or (ii) the date at which the consultant or vendor’s
performance is complete. In the case of equity instruments issued
to consultants, the fair value of the award is generally
re-measured on vesting dates and interim financial reporting dates
until the service period is complete. The fair value amount is then
recognized over the period during which services are required to be
provided in exchange for the award, usually the vesting
period.
Performance-based equity awards without a performance commitment
are recognized upon completion of the performance criteria at their
then market value.
Advertising
Costs
Advertising costs
are expensed as they are incurred. There were no advertising costs
incurred for the years ended December 31, 2016 and
2015.
F-9
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Research
and Development Expenses
Research and
development costs are expensed as incurred in accordance with FASB
ASC Topic No. 730, Research and
Development. Research and development expenses of $783,656
and $13,770 during the years ended December 31, 2016 and 2015,
respectively, consist primarily of (i) $100,000 paid in 2016 for achieving a
milestone relating to a 2012 asset purchase agreement; (ii)
compensation expense (including stock-based compensation), and
(iii) costs related to consulting fees and support
services.
Concentrations
of Credit Risk
Financial
instruments which potentially subject the Company to credit risk
consist principally of cash and cash equivalents and certificates
of deposit. All cash and cash equivalents are held in United States
financial institutions which, at times, exceed federally insured
limits. The Company manages their purchases of certificates of
deposit such that it keeps its exposure to any single bank to be
under the federally insured limits. The Company has not recognized
any losses from credit risks on such accounts. The Company believes
it is not exposed to significant credit risk on cash and cash
equivalents.
Deferred
Offering Costs
Deferred offering
costs in the amount of $15,855 at December 31, 2016, which
primarily consisted of direct, incremental professional fees
incurred in 2016 relating to the January 2017 closing of Investor
Units, were capitalized and included as non-current assets on the
balance sheet as of December 31, 2016 (see Note 9 –
Subsequent Events – Sale of Investor Units). Deferred
offering costs in the amount of $195,000 at December 31, 2015,
which primarily consisted of direct, incremental professional fees
incurred in 2015 relating to our 2016 Units Offering, were
capitalized and included as non-current assets on the balance sheet
as of December 31, 2015 and offset against the equity offering
proceeds upon the consummation of the 2016 Units Offering for the
year ended December 31, 2016 (see Note 8 – Equity –
2016 Units Offering).
F-10
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Income
Taxes
The Company
accounts for deferred taxes using the asset and liability method as
set specified in FASB ASC Topic No. 740-10, Income Taxes. Deferred income tax
assets and liabilities are determined based on differences between
the financial statement reporting and the tax basis of assets and
liabilities, operating losses and tax credit carryforwards.
Deferred income taxes are measured using the enacted tax rates and
the laws that are anticipated to be in effect when the differences
are expected to reverse. The measurement of deferred income tax
assets is reduced, if necessary, by a valuation allowance for any
tax benefits which are not expected to be realized. The effect on
deferred income tax assets and liabilities of a change in tax rates
is recognized in the period that such tax changes are
enacted.
The Company has
adopted the authoritative guidance on accounting for disclosure of
uncertain tax positions which prescribes a comprehensive model for
the financial statement recognition, measurement, presentation and
disclosure of uncertain tax positions taken or expected to be taken
in income tax returns. The Company has no uncertain tax positions
as of December 31, 2016 and 2015 that qualify for either
recognition or disclosure in the financial statements under this
guidance.
The Company’s
policy is to classify assessments, if any, for tax related interest
as interest expense and penalties as general and administrative
expenses in the consolidated statements of operations. There were
no amounts accrued for interest or penalties for the years ended
December 31, 2016 and 2015.
Financial
Instruments
Financial
instruments, which include cash and cash equivalents and accounts
payable are carried at cost, which management believes approximates
fair value due to the short-term nature of these instruments. The
Company’s other financial instruments have included
convertible and other notes, the carrying value of which
approximates their fair value as the notes bear terms and
conditions comparable to market for obligations with similar terms
and conditions.
F-11
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Financial
Instruments, continued
Fair value is
defined as the exchange price that would be received for an asset
or paid to transfer a liability (an exit price) in the principal or
most advantageous market for the asset or liability in an orderly
transaction between market participants at the measurement date.
Assets and liabilities that are measured at fair value are reported
using a three-level fair value hierarchy that prioritizes the
inputs used to measure fair value. This hierarchy maximizes the use
of observable inputs and minimizes the use of unobservable
inputs.
The three levels of
inputs used to measure fair value are as
follows:
●
Level 1 –
Unadjusted quoted prices in active markets that are assessable at
the measurement date of identical, unrestricted assets or
liabilities.
●
Level 2 –
Quoted prices in markets that are not active, or inputs that are
observable, either directly or indirectly, for substantially the
full term of the asset or liability; and
●
Level 3 –
Prices or valuation techniques that require inputs that are both
significant to the fair value measurement and unobservable
(supported by little or no market activity).
Prior to the first
closing of the 2016 Units Offering (see Note 8 – Equity
– 2016 Units Offering), the Company determined that the fair
value of its common stock was immaterial because it was a private
company with no market for its securities that (a) had not yet been
successful in accomplishing a significant capital raise; (b) had
negative net worth; and (c) had not yet commenced
operations.
Loss
per Common Share
The Company
utilizes FASB ASC Topic No. 260, Earnings per Share. Basic loss per
share is computed by dividing net loss available to common
shareholders by the weighted-average number of common shares
outstanding during the year. Diluted earnings per share gives
effect to all potentially dilutive common shares outstanding during
the period, including convertible notes, non-vested restricted
stock, stock options and warrants using the treasury
stock method and the if-converted method. Dilutive loss per share
excludes all potential common shares if their effect is
anti-dilutive.
F-12
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Loss
per Common Share, continued
The potentially
dilutive securities shown below have been excluded from the
computation of diluted loss per share for the years ended December
31, 2016 and 2015 since their inclusion would have been
anti-dilutive.
The following
securities are excluded from the calculation of weighted average
dilutive common shares:
|
|
As of December 31,
|
|
|
|
2016
|
2015
|
|
Convertible
notes
|
-
|
121,378
|
|
Options
|
933,937
|
-
|
|
Warrants
|
2,350,733
|
30,846
|
|
Non-vested
restricted stock
|
25,000
|
-
|
|
|
3,309,670
|
152,224
|
Recently
Issued Accounting Standards
In August 2014, the
Financial Accounting Standards Board (‘‘FASB”)
issued Accounting Standards Update (“ASU”) 2014-15,
Presentation of Financial
Statements-Going Concern (Subtopic 205-40): Disclosure of
Uncertainties about an Entity’s Ability to Continue as a
Going Concern, which defines management’s
responsibility to assess an entity’s ability to continue as a
going concern, and to provide related footnote disclosures if there
is substantial doubt about its ability to continue as a going
concern. The pronouncement is effective for annual reporting
periods ending after December 15, 2016 with early adoption
permitted. The adoption of this guidance did not have a material
impact on the Company’s financial
statements
In February 2016,
the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU
2016-02”). ASU 2016-02 requires an entity to recognize assets
and liabilities arising from a lease for both financing and
operating leases. ASU 2016-02 will also require new qualitative and
quantitative disclosures to help investors and other financial
statement users better understand the amount, timing, and
uncertainty of cash flows arising from leases. ASU 2016-02 is
effective for fiscal years beginning after December 15, 2018, with
early adoption permitted. The Company is currently evaluating ASU
2016-02 and its impact on the Company's consolidated financial
statements.
In March 2016, the
FASB issued ASU No. 2016-09, Compensation—Stock Compensation (Topic
718): Improvements to Employee Share-Based Payment
Accounting (“ASU 2016-09”). ASU 2016-09 aimed at
simplifying the accounting for share-based payment transactions.
Included in the update are modifications to the accounting for
income taxes upon vesting or settlement of awards, employer tax
withholding on share-based compensation, forfeitures, and financial
statement presentation of excess tax benefits. ASU 2016-09 is
effective for annual periods beginning after December 15, 2016 and
interim periods within those periods. The Company is currently
evaluating ASU 2016-09 and its impact on the Company's consolidated
financial statements.
F-13
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
2.
Summary
of Significant Accounting Policies (continued)
Recently
Issued Accounting Standards, continued
In August 2016, the
FASB issued ASU 2016-15, Statement
of Cash Flows (Topic 230): Classification of Certain Cash Receipts
and Cash Payments (“ASU 2016-15”). The new standard will make eight
targeted changes to how cash receipts and cash payments are
presented and classified in the statement of cash flows. The new
standard is effective for fiscal years beginning after December 15,
2017. We will require adoption on a retrospective basis unless it
is impracticable to apply, in which case we would be required to
apply the amendments prospectively as of the earliest date
practicable. The Company is currently evaluating ASU 2016-15 and
its impact on our consolidated financial statements or
disclosures.
3.
Property and Equipment
As of December 31,
2016, property and equipment, net consisted of office furniture in
the amount of $26,619, net of accumulated depreciation in the
amount of $306. Depreciation expense amounted to $306 and $0 for
the years ended December 31, 2016 and 2015, respectively.
Depreciation expense is reflected in general and administrative
expenses in the consolidated statements of
operations.
4.
Convertible Notes
Convertible notes
consisted of:
|
|
December 31, 2015
|
|
2015
Convertible Notes, bearing interest at
|
|
|
6%
per annum, maturing June 9, 2016
|
$285,000
|
|
|
|
|
2012
Senior Convertible Equity Securities, bearing
interest
|
|
|
at
8% per annum, maturing November 26, 2019
|
200,000
|
|
|
$485,000
|
As of December 31,
2015, the accrued interest on the outstanding convertible notes was
$49,556.
For the years ended
December 31, 2016 and 2015, the Company recorded interest expense
related to the outstanding convertible notes of $14,162 and
$16,781, respectively.
F-14
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
4.
Convertible
Notes (continued)
2015
Convertible Notes
During 2015, the
Company issued three (3) 6% convertible notes for $285,000 ($25,000
on October 6th, $10,000 on October
28th and
$250,000 on December 23rd). The principal
amounts outstanding under these notes bear interest at 6%. These
notes were scheduled to mature on March 31, 2016, then were
subsequently extended to June 9, 2016 and then were subsequently
converted to Units (the conversion of the 2015 Convertible Note
with a principal amount of $25,000 was not part of the 2016 Units
Offering) on April 8, 2016 (see Note 8 – Equity – 2016
Unit Offering). The extension did not qualify as a debt
modification as the net present value of the cash flows was
unchanged.
Prior to maturity,
the notes were automatically convertible into a Qualified
Financing, as defined, with minimum gross proceeds of $3,000,000,
at the price paid by investors in a Qualified
Financing.
At maturity, if the
notes were not converted into a Qualified Financing, the investors
had the sole option to be repaid in cash, including interest
accrued, or to convert at a conversion price of $5.00 per unit,
wherein a unit would be comprised of one (1) share of Adgero common
stock and one (1) five-year cashless warrant, exercisable for one
(1) share of Adgero common stock at an exercise price of $5.00 per
share. Furthermore, the warrants would have been callable at the
Company’s option at a price of $12.55 per
share.
The Company
determined that the conversion options embedded in the convertible
notes did not meet the defined criteria of a derivative in that the
net settlement requirement of delivery of common shares does not
meet the “readily convertible to cash” criteria
described in Accounting Standards Codifications 815 and therefore
bifurcating of the embedded conversion option was not required.
There is no established market for the Company’s common
stock. There was no beneficial conversion feature associated with
these convertible notes as the conversion price of $5.00 per Unit
of Holdings was equivalent to the fair value of a Unit on the
commitment date.
On April 8, 2016,
the $285,000 of principal and $5,501 of accrued interest due on the
2015 Convertible Notes, were automatically converted into 58,100
Units at $5.00 per Unit. No beneficial conversion feature was
recognized in connection with the 2015 Convertible Notes, since the
conversion price for a Unit in Holdings was equivalent to the fair
value of a Unit on the commitment date.
F-15
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
4.
Convertible
Notes (continued)
2012
Senior Convertible Equity Securities
In November 2012,
the Company issued two (2) 8% Senior Convertible Equity Securities
(the “Senior Notes”) with an aggregate principal amount
of $200,000 and a maturity of November 26, 2019 in conjunction with
an asset purchase agreement (see Note 5 - Commitments). Upon
completion of a Payment Equity Financing (as defined), with minimum
gross proceeds of $5,000,000, the holders could elect to convert
the Senior Notes into the securities sold in the Payment Equity
Financing at a conversion price equal to seventy percent (70%) of
the price paid by the investors in the Payment Equity
Financing.
The Company
determined that the conversion options embedded in the convertible
notes did not meet the defined criteria of a derivative in that the
net settlement requirement of delivery of common shares does not
meet the “readily convertible to cash” criteria
described in Accounting Standards Codifications 815 and therefore
bifurcating of the embedded conversion option was not required.
There is no established market for the Company’s common
stock. There was no measurement of the potential beneficial
conversion feature as of the commitment date because the qualifying
event had not occurred.
On August 3, 2016,
the $200,000 principal and $59,000 accrued interest due on the 2012
Senior Convertible Equity Securities, was converted into 73,998
Units (see Note 8 – Equity – 2016 Units Offering). No
beneficial conversion feature was recognized in connection with the
conversion of the 2012 Senior Convertible Equity Securities because
the fair value of the underlying stock at the 2012 commitment date
was negligible.
5.
Commitments
Legal
Matters
The Company is not
currently subject to any material legal proceedings; however, the
Company may from time to time become a party to various legal
proceedings arising in the ordinary course of the Company’s
business.
F-16
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
5.
Commitments
(continued)
Asset
Purchase Agreement
In connection with
an asset purchase agreement with Miravant Medical Technologies
(“Miravant”) dated November 26, 2012, the Company
issued $200,000 of Senior Convertible Equity Securities (see Note 3
– Convertible Notes). In addition, the Company was required
to reimburse St. Cloud Investments (“St. Cloud”) and
its respective agents $69,000 for expenses associated with the
sale. As of December 31, 2016 and 2015, the balance due was $0 and
$53,000, respectively.
On May 12, 2014,
the Company amended the asset purchase agreement. Under the amended
agreement, ten percent (10%) of any monies raised through an equity
financing are to be paid to the seller and its agents until the
unpaid balance of $53,000 for expenses associated with sale as
described above, is paid in full. The Company raised sufficient
funds through the 2016 Unit offering and the balance of $53,000 was
paid in full.
In addition, the
milestone payments were amended as follows:
a)
Payment in cash of
the initial milestone of $100,000 if the equity financing exceeds
$4,000,000;
b)
Payments of
$300,000 in cash or an equivalent amount of stock, at the
Company’s sole discretion, upon the sooner of (1) the next
equity financing after a “non-exploratory” clinical
trial or (2) the commencement of a clinical trial intended to be
used as a definitive study for market approval in any
country;
c)
Payment of $700,000
in cash or an equivalent amount of stock, at the Company’s
sole discretion, upon the grant of the first regulatory approval of
a product; and
d)
Royalty equity to
six percent (6%) on net sales during the Royalty
Term.
The purchase was
accounted for as an asset acquisition. Contingent consideration in
an asset acquisition is generally recognized when it is probable
that a liability has incurred and the amount can be reasonably
estimated. None of the milestone payments were accrued for at the
time of acquisition as it was not probable that a liability had
been incurred.
After the May 31,
2016 closing of the 2016 Units Offering, the Company exceeded
equity financing in excess of $4,000,000 and made payment of the
$100,000 milestone which was recognized as research and development
costs. The remaining milestones payments have not yet been achieved
as of the date of the financial statements.
F-17
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
5.
Commitments
(continued)
Employment
Agreements
Chief Executive Officer
On April 8, 2016, the Company and the CEO agreed
to the terms of a three-year employment agreement. Pursuant to the
agreement, the CEO is entitled to receive a salary of $349,000 per
annum, subject to automatic increases as follows: (1) upon the
closing of an underwritten round of financing with gross proceeds
of $20 million or more, the CEO’s salary increases by
$50,000, and (2) upon acceptance of the Company’s first new
drug application (“NDA”), the CEO’s salary
increases by $150,000. As of the date of this filing, neither of
these milestones have been met. The CEO is also entitled to
receive annual bonuses of up to 75% of his annual base salary, in
the event certain performance goals, as determined by the
Company’s Compensation Committee, are satisfied. For the year
ended December 31, 2016, the Company accrued $261,750 of
bonus expense for the satisfaction of
the performance goals pursuant to the agreement, which is included
in accrued expenses on the accompanying consolidated balance sheet
at December 31, 2016. Pursuant to the agreement, upon the July 29,
2016 completion of the 2016 Units Offering, the CEO was awarded an
option grant for the equivalent of 5.0% of the Company’s
fully-diluted shares of common stock, which resulted in an option
grant to purchase 335,958 shares of common stock at an exercise
price of $5.00 per share (see Note 8 – Equity – Equity
Awards). The option vests ratably on each of the subsequent three
annual anniversary dates.
Also
pursuant to the agreement, in the event that (a) the CEO’s
employment is terminated by the Company without cause (with 60 days
notice), or (b) the CEO terminates his employment for “good
reason” (each as defined in the employment agreement), or (c)
the term of the CEO’s employment agreement is not extended
beyond the expiration date, the CEO would be entitled to receive
severance in an amount equal to his then annual base salary and
health insurance continuation benefits for 12 months and an
additional 12 months of service would be credited toward the
accelerated vesting of his outstanding options and would be
permitted to exercise all vested options for a period of 12
months.
Further,
in the event that the CEO’s employment is terminated by the
Company without cause, or the CEO terminates his employment for
“good reason”, within 24 months following a
“change in control” (as defined in the employment
agreement), the CEO would be entitled to receive severance in an
amount equal to his then annual base salary and health insurance
continuation benefits for 18 months and an additional 18 months of
service would be credited toward the accelerated vesting of his
outstanding options and would be permitted to exercise all vested
options for a period of 18 months.
F-18
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
5.
Commitments
(continued)
Employment
Agreements, continued
Chief Executive Officer, continued
In
February 2017, the Company entered into an amendment to the
employment agreement with the CEO that provides for accelerated
vesting of all unvested option awards granted to the CEO pursuant
to his employment agreement upon certain terminations of employment
following a change in control.
Vice President of Operations and Product
Development
On April 8, 2016, the Company and its Vice
President of Operations and Product Development (“VPO”) agreed to the terms of a
two-year employment agreement. Pursuant to the agreement, the VPO
is entitled to receive a salary of $249,000 per annum, subject to
automatic increases as follows: (1) upon the closing of an
underwritten round of financing with gross proceeds of $20 million
or more, the VPO’s salary increases by $25,000, and (2) upon
acceptance of the Company’s first new drug application
(“NDA”), the VPO’s salary increases by $75,000.
As of the date of this filing, neither of these milestones have
been met. The VPO is also entitled to receive annual bonuses
of up to 30% of his annual base salary, in the event certain
performance goals, as determined by the Company’s
Compensation Committee, are satisfied. For the year ended December
31, 2016, the Company accrued $74,700 of bonus expense for the
satisfaction of the performance goals pursuant to the agreement,
which is included in accrued expenses on the accompanying
consolidated balance sheet at December 31, 2016. Pursuant to the
agreement, upon the July 29, 2016 completion of the 2016 Units
Offering, the VPO was awarded an option grant for the equivalent of
2.5% of the Company’s fully-diluted shares of common stock,
which resulted in an option grant to purchase 167,979 shares of
common stock at an exercise price of $5.00 per share (see Note 8
– Equity – Stock Options). The option vests ratably on
each of the subsequent three annual anniversary
dates.
Also
pursuant to the agreement, in the event that (a) the VPO’s
employment is terminated by the Company without cause (with 60 days
notice), or (b) the VPO terminates his employment for “good
reason” (each as defined in the employment agreement), or (c)
the term of the VPO’s employment agreement is not extended
beyond the expiration date, the VPO would be entitled to receive
severance in an amount equal to his then annual base salary and
health insurance continuation benefits for 6 months and an
additional 6 months of service would be credited toward the
accelerated vesting of his outstanding options and he’d be
permitted to exercise all vested options for a period of 6
months.
F-19
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
5.
Commitments
(continued)
Employment
Agreements, continued
Vice President of Operations and Product Development,
continued
In
February 2017, the Company entered into an amendment to the
employment agreement with the VPO that provides for accelerated
vesting of all unvested option awards granted to the VPO pursuant
to his employment agreement upon certain terminations of employment
following a change in control.
Vice President of Manufacturing Operations and Quality
Control
On October 3, 2016, the Company and its
Vice President of Manufacturing Operations and Quality
Control (“VPQC”) agreed to the terms of an
open ended employment agreement. Pursuant to the agreement, the
VPQC is entitled to receive a salary of $275,000 per annum.
The VPQC is also entitled to receive annual bonuses targeted at 35%
of her annual base salary, in the event certain performance goals,
as determined by the Company’s Compensation Committee, are
satisfied. For the year ended December 31, 2016, the Company did
not accrue any bonus expense pursuant to the agreement. Pursuant to
the agreement, on October 21, 2016, the VPQC was awarded a stock
option for the purchase of 150,000 shares of the Company’s
common stock at an exercise price of $5.00 per share (provided that
such exercise price is not less than fair market value) which will
vest in three annual installments (see Note 8 – Equity
– Stock Options).
The
employment agreement may be terminated by the Company without cause
or by the VPQC with good reason, upon sixty days notice. Also
pursuant to the agreement, in the event that (a) the VPQC’s
employment is terminated by the Company without cause (with 60
days’ notice), or (b) the VPQC terminates her employment for
“good reason” (each as defined in the employment
agreement), the VPQC would be entitled to receive an additional 3
months of service credited toward the accelerated vesting of her
outstanding options and she would be permitted to exercise all
vested options for a period of 6 months and, subject to the
execution of a Release Agreement, as defined, severance in an
amount equal to three months of her then annual base salary and
health insurance continuation benefits for up to 3
months.
In
February 2017, the Company entered into an amendment to the
employment agreement with the VPQC that provides for accelerated
vesting of all unvested option awards granted to the VPQC pursuant
to her employment agreement upon certain terminations of employment
following a change in control.
F-20
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
5.
Commitments
(continued)
Consulting
Agreement
On
October 12, 2016, the Company entered into a one-year consulting
agreement with a consultant (the “Consultant”) for
regulatory for product development advice and services, at a rate
of $150 per hour. The agreement may be renewed or extended upon the
expiration of the one-year term upon mutual written agreement of
the parties, and may be terminated by either the Company or the
Consultant for any reason upon 30 days prior written notice. On
December 16, 2016, the Consultant was awarded an option to purchase
5,000 shares of the Company’s common stock at an exercise
price of $5.00 per share which vests on December 16, 2017 and
expires on December 15, 2026 (see Note 8 – Equity –
Stock Options).
Lease
Agreement
In
December 2016, a three-year lease commenced on office space in
Princeton. New Jersey. Basic rent in connection with the lease is
$3,962 per month. The Company entered into an amendment to expand
the leased office space which will become effective April 1, 2017.
Basic rent in connection with the lease amendment is $6,026 per
month.
Future
minimum payments under this lease agreement, as amended, is as
follows:
|
For the Years Ending,
|
|
|
December 31,
|
Amount
|
|
2017
|
$66,250
|
|
2018
|
74,015
|
|
2019
|
75,587
|
|
2020
|
25,676
|
|
|
$241,528
|
F-21
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
6.
Related Party Transactions
Since inception,
the CEO had advanced funds to the Company. On July 9, 2014, the CEO
exchanged his advances of $163,934 for a Note due July 1, 2019
bearing interest at two percent.
On July 29, 2016,
the CEO exchanged the Note and the related accrued interest which
totaled $170,765 ($163,934 plus accrued interest $6,831) for Units
consisting of 34,153 shares of Holdings’ common stock and
warrants to purchase 34,153 shares of Holdings’ common stock
at an exercise price of $5.00 per share. The carrying value of the
Note equaled the fair value of the Holdings’ equity
securities.
See Note 2 –
Summary of Significant Accounting Policies – Basis of
Presentation and Consolidation for details related to the ownership
interest of a principal of the placement agent responsible for the
2016 Units Offering.
7.
Income Taxes
As of December 31,
2016 and 2015, the Company had available federal net operating loss
carryforwards (“NOLs”) of approximately $3,001,000 and
$889,000, respectively, and state NOLs of approximately $2,903,000
and $791,000, respectively, which are available to offset future
federal and state taxable income, if any, and which expire between
2027 and 2036. The utilization of
these NOLs is subject to limitations based on past and future
changes in ownership of the Company pursuant to Internal Revenue
Code Section 382. The Company has determined that an ownership
change occurred for Internal Revenue Code Section 382 purposes. As
a result of this ownership change, the ability of the Company to
utilize its NOLs may be limited. Federal tax returns
for the years 2013, 2014 and 2015 remain subject to
audit.
The tax effects of
temporary differences that give rise to significant portions of the
deferred tax asset is presented below:
|
|
December 31,
|
|
|
|
2016
|
2015
|
|
Deferred
tax assets
|
|
|
|
Net
operating loss carryforwards
|
$1,194,402
|
$349,553
|
|
Stock-based
compensation
|
178,630
|
-
|
|
|
|
|
|
Total
gross deferred tax asset
|
1,373,032
|
349,553
|
|
Less:
Valuation allowance
|
(1,373,032)
|
(349,553)
|
|
|
|
|
|
Net
deferred tax asset
|
$-
|
$-
|
F-22
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
7.
Income Taxes (continued)
ASC 740,
“Income Taxes” requires that a valuation allowance be
established when it is “more likely than not” that all,
or a portion of, deferred tax assets will not be realized. A review
of all available positive and negative evidence needs to be
considered, including the scheduled reversal of deferred tax
liabilities, projected future taxable income, and tax planning
strategies. After consideration of all the information available,
management believes that uncertainty exists with respect to future
realization of its deferred tax assets and has, therefore,
established a full valuation allowance as of December 31, 2016 and
2015. The net change in valuation allowance for the years ended
December 31, 2016 and 2015 was an increase of approximately
$1,023,000 and $57,000, respectively.
The income tax
(provision) benefit consists of the following:
|
|
For The Years Ended
|
|
|
|
December 31,
|
|
|
|
2016
|
2015
|
|
Federal:
|
|
|
|
Current
|
$-
|
$-
|
|
Deferred
|
869,957
|
48,185
|
|
|
|
|
|
State
and local:
|
|
|
|
Current
|
-
|
-
|
|
Deferred
|
153,522
|
8,503
|
|
|
1,023,479
|
56,688
|
|
Change
in valuation allowance
|
(1,023,479)
|
(56,688)
|
|
Income
tax (provision) benefit
|
$-
|
$-
|
F-23
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
7.
Income Taxes (continued)
A
reconciliation of the U.S. Statutory income tax rate to the
Company’s effective tax rate is as
follows:
|
|
For The
Years Ended
|
|
|
|
December
31,
|
|
|
|
2016
|
2015
|
|
|
|
|
|
Federal
income tax at statutory rate
|
34.0%
|
34.0%
|
|
State
income tax benefit, net of federal benefit
|
6.0%
|
6.0%
|
|
Permanent
differences
|
(0.1%)
|
0.0%
|
|
Change
in valuation allowance
|
(39.9%)
|
(40.0%)
|
|
|
|
|
|
Effective
income tax rate
|
0.0%
|
0.0%
|
8.
Equity
Authorized
Capital
Holdings has
60,000,000 shares of authorized capital stock, including 50,000,000
shares of common stock and 10,000,000 shares of preferred stock. No
preferred stock has been designated to date.
Equity
Incentive Plan
On January 8, 2016,
the Holdings’ Board and stockholders adopted the 2016 Equity
Incentive Plan (“2016 Plan”), which has a ten-year life
for granting awards and initially reserved 750,000 shares of common
stock for awards, which increased to 15% of the quantity of the
Company’s outstanding common stock, on a fully diluted basis,
immediately following the final closing of the 2016 Units Offering,
up to a maximum of 2,000,000 shares. Beginning January 1, 2017 and
annually thereafter, the maximum shares will be increased by 6% of
the Holdings’ common stock outstanding at that time. Shares
of common stock issued under the 2016 Plan may either be authorized
but unissued Holdings’ common stock or shares held in
Holdings’ treasury. As of December 31, 2016, (i) the Company
has 1,007,875 shares of common stock authorized for awards granted
under the 2016 Plan; (ii) options to purchase an aggregate of
933,937 shares of common stock were outstanding under the 2016
Plan; and (iii) 35,000 shares of common stock were issued as
restricted stock grants pursuant to the 2016 Plan. Accordingly, as
of December 31, 2016, 38,938 shares are reserved and currently
available for future grants under the 2016 Plan. See Note 9 –
Subsequent Events – Equity Incentive Plan for details
regarding the amendment of the Company’s 2016 Plan and Note 9
– Subsequent Events – Other Equity Awards for details
regarding additional issuances under the plan.
F-24
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Equity
Incentive Plan, continued
Awards granted
under the 2016 Plan may be incentive stock options (they must meet
all statutory requirements), non-qualified stock options, stock
appreciation rights, restricted stock, stock units, performance
shares, performance units, incentive bonus awards, and other
cash-based or stock-based awards. Pursuant to the 2016 Plan, stock
options must expire within 10 years and must be granted with
exercise prices of no less than the fair value of the common stock
on the grant date, as determined by the Board of
Directors.
Sale
of Common Stock
On September 25,
2014, Adgero sold 411,676 shares of common stock for $9,393. Adgero
ultimately received cash proceeds of $1,506. On December 30, 2015,
Adgero settled the remaining $7,887 receivable in exchange for
services provided to Adgero for the year ended December 31,
2015.
On September 9,
2015, Adgero sold 61,728 shares of common stock for aggregate
proceeds of $30,000.
Placement
Agent Agreement
On January 11,
2016, Holdings entered into a placement agent’s agreement to
offer Units for sale in a private placement (the “2016 Units
Offering”) and agreed to pay the Placement Agent (i) a cash
fee equal to 10% of the gross proceeds and (ii) a non-accountable
expenses allowance equal to 3% of the gross proceeds raised in the
offering. In addition, the Placement Agent is entitled to five-year
warrants to purchase Holdings’ common stock at an exercise
price equal to $5.00 per share in a quantity equal to 10% of the
number of shares of common stock and warrants sold in this offering
(the “Placement Agent Warrants”).
2016
Units Offering
Each Unit in the
2016 Units Offering was sold at a price of $5.00 per Unit. Each
Unit consisted of (i) one share of Holdings’ common stock,
par value $0.0001 per share and (ii) one five-year warrant to
purchase one share of Holdings’ common stock at an exercise
price of $5.00 per share (“Investor
Warrant”).
F-25
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
2016
Units Offering, continued
The 2016 Units
Offering consisted of (a) the Initial Offering (the first six
closings through July 29, 2016). and (b) the Follow-on Offering
(the final two closings through September 9, 2016). In connection
with the 2016 Units Offering, the Company raised aggregate gross
proceeds of $8,931,000 and issued 1,786,200 Units consisting of
1,786,200 shares of common stock and Investor Warrants to purchase
1,786,200 shares of common stock. In addition, for the year ended
December 31, 2016, the Placement Agent has earned (a) a $888,546
cash fee. (b) a $275,564 allowance for nonaccountable expenses and
(c) Placement Agent Warrants to purchase 367,418 shares of
Holdings’ common stock, which have the same terms as the
Investor Warrants. The Company determined that the Investor and
Placement Agent Warrants were equity instruments. Through December
31, 2016, Holdings incurred an additional $531,046 of offering
costs (primarily legal costs), which were charged against the
proceeds of the 2016 Units Offering.
On April 8, 2016,
at the initial closing of the 2016 Units Offering, principal and
accrued interest due related to the 2015 Convertible notes,
totaling, in the aggregate, $290,501 ($285,000 plus accrued
interest of $5,501) was automatically converted into 58,100 Units
(5,154 of which were not included in the 2016 Units Offering)
consisting of an aggregate of 58,100 shares of Holdings’
common stock and Investor Warrants to purchase an aggregate of
58,100 shares of Holdings’ common stock at an exercise price
of $5.00 per share. No beneficial conversion feature was recognized
because the $5.00 conversion price for a Unit in Holdings was
equivalent to the fair value of a Unit on the commitment date. See
Note 4 – Convertible Notes for additional details related to
issuances upon the conversions of convertible
stock.
On July 29, 2016,
the CEO exchanged his Note and accrued interest which totaled
$170,765 ($163,934 plus accrued interest of $6,831) for Units
consisting of 34,153 shares of Holdings’ common stock and
five-year warrants to purchase 34,153 shares of Holdings’
common stock at an exercise price of $5.00 per
share.
F-26
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
2016
Units Offering, continued
On August 3, 2016,
the note holders of the 2012 Senior Convertible Equity Securities
exchanged the principal and accrued interest which totaled $258,993
($200,000 and accrued interest $58,993) for 73,998 Units (which
were not included in the 2016 Units Offering) consisting of 73,998
shares of Holdings’ common stock and five-year warrants to
purchase 73,998 shares of Holdings’ common stock at an
exercise price of $5.00 per share; representing the number of
securities equal to the outstanding balance of the 2012 Senior
Convertible Equity Securities, plus interest accrued thereon but
unpaid, divided by seventy percent (70%) of the $5.00 purchase
price per unit paid by the investors participating in the 2016
Units Offering, pursuant to the terms of the 2012 Senior
Convertible Equity Securities. See Note 4 – Convertible Notes
for additional details related to issuances upon the conversions of
convertible stock.
Registration
Rights Agreement
In connection with
the 2016 Units Offering, the Company entered into a registration
rights agreement (as amended, the “Registration Rights
Agreement”) with the 2016 Units Offering investors, a 2015
Convertible Note holder whose note was not included in the 2016
Units Offering, the Placement Agent and the holders of certain of
our outstanding warrants (collectively, the
“Investors”). The Company is required to file with the
SEC after the date of the final closing of the 2016 Units Offering
(the “Registration Filing Date”), a registration
statement covering the resale of the shares of common stock held by
the Investors (the “Investor Shares”) and certain of
the Investor Warrants, issued in the 2016 Units Offering, as well
as the shares of common stock underlying the Replacement Warrants
and the warrant issued to a 2015 Convertible Note holder whose note
was not included in the 2016 Units Offering (together with the
Investor Shares and the Investor Warrants, the “Registrable
Securities”).
F-27
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Registration
Rights Agreement, continued
The Company is also
required to use commercially reasonable efforts to have the
registration statement declared effective within one hundred and
fifty (150) days after the registration statement is filed (the
“Effectiveness Deadline”). provided however, that if
the Company signs a letter of intent or comparable agreement with
an underwriter which contemplates an Initial Public Offering
(“IPO”) or holds an organizational meeting for an IPO,
or otherwise orally engages an underwriter to begin working with
the Company towards an IPO prior to the Effectiveness Deadline (the
“IPO Process Commencement Date”), then the Company
shall file a joint registration statement covering the primary
shares to be issued in the IPO and the resale of the Registrable
Securities, and in such event the Registration Filing Date shall be
extended to a date that is seventy five (75) calendar days after
the IPO Process Commencement Date and the Effectiveness Deadline
shall be extended to a date that is one hundred twenty (120)
calendar days after the initial filing of the Registration
Statement with the Commission. If the IPO is abandoned at any time,
then the Registration Filing Date will be 60 calendar days from the
actual date of abandonment and the Effectiveness Deadline will be
one hundred and fifty (150) calendar days after the date of
abandonment. The Company is also required to keep the registration
statement continuously effective under the Securities Act for a
period of one year or for such shorter period ending on the earlier
to occur of the date when all the Registrable Securities covered by
the registration statement have been sold or such time as all of
the Registrable Securities covered by the registration statement
can be sold under Rule 144 without any volume
limitations.
If the registration
statement is not declared effective on or before the Effectiveness
Deadline, the Company will be required to pay to each holder of
Registrable Securities purchased in the 2016 Units Offering an
amount in cash equal to one-half of one percent (0.5%) of such
holder's investment amount on every thirty (30) day anniversary of
such Effectiveness Deadline until such failure is cured. The
payment amount shall be prorated for partial thirty (30) day
periods. The maximum aggregate amount of payments to be made by us
as the result of such failure, shall be an amount equal to 6% of
each holder's investment amount. Notwithstanding the foregoing, no
payments shall be owed with respect to any period during which all
of the holder's Registrable Securities may be sold by such holder
without restriction under Rule 144.
F-28
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Restricted
Stock
During July 2016,
Holdings granted an aggregate of 25,000 shares of restricted stock
with a grant date value of $70,500 to consultants. Of these awards
(a) 25% of the shares vest upon effectiveness of the
Company’s registration statement on Form S-1; while (b) the
remaining shares vest ratably at the end of each 90-day period
subsequent to the registration statement effectiveness. To date,
this restricted stock award is not vested. No stock based
compensation has been recognized in connection with this restricted
stock.
On the June 23,
2016, effective date of a consulting agreement, the Company became
contractually obligated to deliver 10,000 vested shares of the
Company’s restricted common stock and the Company recognized
the $28,200 fair value of these shares as stock-based compensation
expense for the year ended December 31, 2016. There is no
unrecognized stock-based compensation related to the restricted
common stock as of December 31, 2016.
A
summary of restricted stock activity for the year ended December
31, 2016 is presented below:
|
|
Number of Restricted Shares
|
Weighted Average Fair Value
|
|
Non-vested,
December 31, 2015
|
-
|
$-
|
|
Granted
|
35,000
|
2.82
|
|
Vested
|
(10,000)
|
2.82
|
|
Forfeited
|
-
|
-
|
|
Non-vested,
December 31, 2016
|
25,000
|
$2.82
|
Stock
Options
On July 22, 2016,
the Company granted options for the purchase of an aggregate
160,000 shares of the Company’s common stock to four members
of the Board of Directors (the “Director Options”). The
Director Options have a ten-year life, are exercisable at $5.00 per
share and vest on April 8, 2017. The Director Options have an
aggregate grant date value of $359,484 or $2.25 per share, which is
recognized ratably over the vesting period.
F-29
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Stock
Options, continued
On July 29, 2016,
the Company granted options for the purchase of 90,000 shares to
consultants (the “Consultant Options”) The Consultant
Options have a ten-year life and have an exercise price of $5.00
per share. Options for the purchase of 5,000 shares vested
immediately and the remainder vest ratably on each of the three
anniversary dates following the date of grant. The Consultant
Options had an aggregate grant date value of $208,003 or $2.31 per
share, which is re-measured on each financial reporting date and
vesting date until the service period is
complete.
On July 29, 2016,
the Company granted options for the purchase of an aggregate
503,937 shares to three officers of the Company (the
“Executive Options”). The Executive Options have a
ten-year life and have an exercise price of $5.00 per share. The
options vest ratably on each of the three anniversary dates
following the date of grant. The Executive Options had an aggregate
grant date value of $1,164,671 or $2.31 per share, which is
recognized over the vesting period.
On October 21,
2016, Holdings granted to a Scientific Advisory Board member an
option to purchase 25,000 shares of Holdings’ common stock at
an exercise price of $5.00 per share. The option vests
one-third on each of the next three anniversary dates, subject to
the grantee’s continued service through each such vesting
date. The options expire on the tenth anniversary of the grant
date. The option had a grant date value of $57,908 or $2.32 per
share, which is re-measured on each financial reporting date and
vesting date until the service period is
complete.
On October 21,
2016, Holdings awarded to its Vice President of Manufacturing
Operations and Quality Control a ten-year stock option to purchase
150,000 shares of Holdings' common stock at an exercise price of
$5.00 per share. The option vests one-third on each of the next
three anniversary dates. The option had a grant
date value of $347,447 or $2.32 per share, which is recognized over
the vesting period.
On December 16,
2016, a consultant was awarded an option to purchase 5,000 shares
of the Company’s common stock at an exercise price of $5.00
per share which vests on December 16, 2017 and expires on December
15, 2026. The option had a grant date value of $11,294 or $2.26 per
share, which is re-measured on each financial reporting date and
vesting date until the service period is
complete.
F-30
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Stock
Options, continued
In applying the
Black Scholes option pricing model, the Company used the following
assumptions:
|
|
For The Year Ended December 31, 2016
|
|
Risk
free interest rate
|
1.16% - 2.04%
|
|
Expected
term
|
5.5-6.0
years
|
|
Expected
volatility
|
120.00%
|
|
Expected
dividends
|
0.00%
|
For the year ended
December 31, 2016, the Company recognized stock based compensation
of $446,576 related to stock option grants, of which $104,378 is
included in research and development expenses, and $342,198 is
included in general and administrative expenses in the consolidated
statement of operations. As of December 31, 2016, there was
$1,702,233 of unrecognized stock-based compensation related to
stock option grants that will be amortized over a weighted average
period of 2.4 years, of which $233,726 of unrecognized expense is
subject to non-employee mark-to-market
adjustments.
A
summary of stock options activity for the year ended December 31,
2016 is presented below:
|
|
Number of Options
|
Weighted Average Exercise Price
|
Weighted Average Remaining LifeIn Years
|
Intrinsic Value
|
|
Outstanding,
December 31, 2015
|
-
|
$-
|
|
|
|
Granted
|
933,937
|
5.00
|
|
|
|
Exercised
|
-
|
-
|
|
|
|
Expired
|
-
|
-
|
|
|
|
Forfeited
|
-
|
-
|
|
|
|
Outstanding,
December 31, 2016
|
933,937
|
$5.00
|
9.6
|
$-
|
|
|
|
|
|
|
|
Exercisable,
December 31, 2016
|
5,000
|
$5.00
|
9.6
|
$-
|
F-31
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
8.
Equity
(continued)
Warrants
For the year ended
December 31, 2016 the Company issued an aggregate of 2,350,733
five-year warrants with an exercise price of $5.00, consisting of
30,864 Replacement Warrants (See Note 2 – Summary of
Significant Accounting Policies: Basis of Presentation and
Consolidation), 1,786,200 warrants issued as part of the 2016 Unit
Offering, 166,251 warrants issued upon the exchange or conversion
of debt and 367,418 Placement Agent Warrants issued in connection
with the 2016 Units Offering (see Note 8 – Equity –
2016 Units Offering).
A
summary of warrant activity for the year ended December 31, 2016 is
presented below:
|
|
Number of Warrants
|
Weighted Average Exercise Price
|
Weighted Average Remaining Life in
Years
|
Intrinsic Value
|
|
Outstanding,
December 31, 2015
|
-
|
$-
|
|
|
|
Issued
|
2,350,733
|
5.00
|
|
|
|
Exercised
|
-
|
-
|
|
|
|
Cancelled
|
-
|
-
|
|
|
|
Outstanding,
December 31, 2016
|
2,350,733
|
$5.00
|
4.5
|
$-
|
|
|
|
|
|
|
|
Exercisable,
December 31, 2016
|
2,350,733
|
$5.00
|
4.5
|
$-
|
F-32
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
9.
Subsequent Events
Management has
evaluated subsequent events through the date the consolidated
financial statements were available to be
issued.
Employment
Agreements
Chief Executive Officer
On
March 3, 2017, the Board of Directors of the Company approved an
amendment to the CEO’s employment agreement such that the
CEO’s base salary was increased from $349,000 per annum to
$400,000 per annum.
Chief Financial Officer
On
February 13, 2017, the Company entered into an employment agreement
with its new Chief Financial Officer (“CFO”). Pursuant
to the terms of the agreement, the CFO’s initial base salary
is $275,000, subject to annual review and adjustment at the
discretion of the Board or the Compensation Committee, and the CFO
is eligible to receive an annual bonus amount targeted at 35% of
her annual base salary, based upon the achievement of certain goals
and objectives as set by the Board or the Compensation Committee.
In connection with her employment, the CFO was granted options for
the purchase of 100,000 shares of the Company’s common stock
at an exercise price of $5.00 per share which vest in three annual
installments. The option expiration date is February 12,
2027.
The
CFO’s employment agreement can be terminated by the Company
without cause or by the CFO with good reason, upon sixty days
notice. In such case, after completing six months of employment,
the CFO is entitled to severance equal to six months of her base
salary and health insurance continuation benefits for up to six
months.
The
employment agreement with the CFO provides for accelerated vesting
of all unvested option awards granted to the CFO pursuant to her
employment agreement upon certain terminations of employment
following a change in control.
Vice President of Operations and Product
Development
On
March 3, 2017, the Board of Directors of the Company approved an
amendment to the VPO’s employment agreement such that the
VPO’s base salary was increased from $249,000 per annum to
$275,000 per annum.
F-33
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
9.
Subsequent Events (continued)
Advisory
Agreement
On
February 3, 2017, the Company entered into a one-year agreement
with an advisor (the “Advisor”) for services as a
member of the Advisory Board. Upon expiration, the agreement
automatically extends for one year, unless 30 days written notice
of non-renewal is given by either party. Pursuant to the terms of
the agreement, the Advisor will receive $2,500 to attend each
formal face-to-face meeting and $350 per hour for non-meeting
services performed at the request of the Company. On February 8,
2017, the Advisor was granted options for the purchase of 25,000
shares of the Company’s common stock at an exercise price of
$5.00 per share, which vest in three equal annual installments and
expire on February 7, 2027.
Equity
Incentive Plan
On January 1, 2017,
pursuant to the terms of the 2016 Plan, the authorized number of
shares of common stock for awards granted under the 2016 Plan
automatically increased from 1,007,875 to 1,307,122 shares. The
increase represents 6% of Holdings’ common stock as of
January 1, 2017. Furthermore, in March 2017, the Company’s
Board of Directors amended the 2016 Plan such that the authorized
number of shares of common stock for awards granted under the 2016
Plan was increased from 1,307,122 to 2,756,330 shares, subject to
shareholder approval.
Other
Equity Awards
In January 2017,
the Company issued 11,080 shares of immediately vested common stock
to an advisor as partial payment of services
rendered.
In March 2017, the
Company granted options for the purchase of an aggregate of 180,000
shares of the Company’s common stock at an exercise price of
$5.00 per share to four members of the Board of Directors. The
options have a ten-year life and vest on the one year anniversary
from the date of grant.
In March 2017, the
Company granted two employees and a consultant options for the
purchase of an aggregate of 45,000 shares of the Company’s
common stock at an exercise price of $5.00 per share. The options
have a ten-year life and vest in three equal annual
installments.
F-34
ADGERO
BIOPHARMACEUTICALS HOLDINGS, INC.
AND
SUBSIDIARIES
Notes to the
Consolidated Financial Statements
Years Ended
December 31, 2016 and 2015
9.
Subsequent Events (continued)
Sale
of Investor Units
On January 17,
2017, the Company closed on a subscription agreement with an
investor (the “Investor”), pursuant to which the
Investor agreed to purchase 400,000 Units (the “Investor
Units”) at a price of $5.00 per Investor Unit, or an
aggregate of $2,000,000. Each Investor Unit consists of (i) one
share of Holdings’ common stock, par value $0.0001 per share
and (ii) and one five-year warrant to purchase one share of
Holdings’ common stock at an exercise price of $5.00 per
share. The subscription agreement places certain restrictions on
the sale or transfer of the Investor Units, until 270 days after a
registration statement becomes effective. In connection with the
subscription agreement, the Company issued the placement agent a
five-year warrant to purchase 80,000 shares of common stock at an
exercise price of $5.00 per share. The Company paid a cash
compensation fee of $200,000 to the placement agent and incurred
legal fees of $15,855 in connection with this closing, for net
proceeds of $1,784,145.
F-35
ADGERO BIOPHARMACEUTICALS HOLDINGS, INC.
3,467,680 Shares
Common Stock
PROSPECTUS
, 2017
II-1
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
|
ITEM 13.
|
OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
|
Our
estimated expenses in connection with the issuance and distribution
of the securities being registered are:
|
SEC Registration
Fee
|
$2,009.52
|
|
|
|
|
Accounting Fees and
Expenses
|
$30,000
|
|
|
|
|
Legal Fees and
Expenses
|
$80,000
|
|
|
|
|
Miscellaneous Fees
and Expenses
|
$12,990.48
|
|
|
|
|
Total
|
$125,000
|
|
ITEM 14.
|
INDEMNIFICATION OF OFFICERS AND DIRECTORS
|
Section
145 of the Delaware General Corporation Law (the
“DGCL”) provides, in general, that a corporation
incorporated under the laws of the State of Delaware, as we are,
may indemnify any person who was or is a party or is threatened to
be made a party to any threatened, pending or completed action,
suit or proceeding (other than a derivative action by or in the
right of the corporation) by reason of the fact that such person is
or was a director, officer, employee or agent of the corporation,
or is or was serving at the request of the corporation as a
director, officer, employee or agent of another enterprise, against
expenses (including attorneys’ fees), judgments, fines and
amounts paid in settlement actually and reasonably incurred by such
person in connection with such action, suit or proceeding if such
person acted in good faith and in a manner such person reasonably
believed to be in or not opposed to the best interests of the
corporation and, with respect to any criminal action or proceeding,
had no reasonable cause to believe such person’s conduct was
unlawful. In the case of a derivative action, a Delaware
corporation may indemnify any such person against expenses
(including attorneys’ fees) actually and reasonably incurred
by such person in connection with the defense or settlement of such
action or suit if such person acted in good faith and in a manner
such person reasonably believed to be in or not opposed to the best
interests of the corporation, except that no indemnification will
be made in respect of any claim, issue or matter as to which such
person will have been adjudged to be liable to the corporation
unless and only to the extent that the Court of Chancery of the
State of Delaware or any other court in which such action was
brought determines such person is fairly and reasonably entitled to
indemnity for such expenses.
Our
certificate of incorporation and bylaws provide that we will
indemnify our directors, officers, employees and agents to the
extent and in the manner permitted by the provisions of the DGCL,
as amended from time to time, subject to any permissible expansion
or limitation of such indemnification, as may be set forth in any
amendment by stockholders or directors resolution.
II-2
Any
repeal or modification of these provisions approved by our
stockholders will be prospective only and will not adversely affect
any limitation on the liability of any of our directors or officers
existing as of the time of such repeal or
modification.
We have
director and officer liability insurance to cover liabilities our
directors and officers may incur in connection with their services
to us, including matters arising under the Securities Act of 1933,
as amended (the “Securities Act”).
We have
entered into indemnification agreements with certain of our
directors and officers whereby we have agreed to indemnify those
directors and officers to the fullest extent permitted by law,
including indemnification against expenses and liabilities incurred
in legal proceedings to which the director or officer was, or is
threatened to be made, a party by reason of the fact that such
director or officer is or was a director, officer, employee or
agent of the Company, provided that such director or officer acted
in good faith and in a manner that the director or officer
reasonably believed to be in, or not opposed to, the best interests
of the Company.
|
ITEM 15.
|
RECENT SALES OF UNREGISTERED SECURITIES
|
Since
January 1, 2013, the Company made sales of the following
unregistered securities:
Original Issuances of Stock and Warrants
Formation of Holdings
In
connection with our formation in October 2015, we sold an aggregate
of 1,000,000 shares of common stock for an aggregate of $50,000
($0.05 per share), which includes 500,000 shares of common stock
owned by an affiliate of Aegis Capital Corp., the placement agent
(“Placement Agent”) for our private placement, for
which closings occurred April 8, 2016 through September 9, 2016
(the “2016 Private Placement”), described
below.
2016 Private Placement
From
January through September of 2016, we sold an aggregate of
1,873,299 shares of our common stock, inclusive of 87,099 shares of
our common stock issued pursuant to the conversion of promissory
notes in connection with the 2016 Private Placement, and warrants
to purchase an aggregate of 1,873,299 shares of our common stock
with an exercise price of $5.00 per share, inclusive of Investor
Warrants to purchase 87,099 shares of our common stock issued
pursuant to the conversion of promissory notes in connection with
the 2016 Private Placement, to 124 accredited
investors.
In
connection with the 2016 Private Placement, we issued warrants to
the Placement Agent to purchase 367,418 shares of our common stock
with an exercise price of $5.00 per share.
December 2016 Private Placement
In
connection with the December 2016 Private
Placement, which closed in January 2017, we (i)
sold an aggregate of 400,000 shares of our common stock, and
warrants to purchase an aggregate of 400,000 shares of our common
stock with an exercise price of $5.00 per share, to 1 accredited
investor, and (ii) issued warrants to the Placement
Agent to purchase 80,000 shares of our common stock with an
exercise price of $5.00 per share.
Merger Transaction
On
April 8, 2016, pursuant to the terms of the merger agreement (the
“Merger Agreement”) by and among Adgero
Biopharmaceuticals, Inc. (“Adgero”), Adgero
Biopharmaceuticals Holdings, Inc. (“Holdings”) and
Adgero Acquisition, Inc. (“Merger Sub”), a wholly-owned
subsidiary of Holdings, Adgero merged with and into Merger Sub and
became a wholly-owned subsidiary of Holdings. In connection with
the merger (the “Merger”), we issued an aggregate of
2,000,000 shares of our common stock, and issued warrants
(“Replacement Warrants”), to purchase 30,864 shares of
our common stock at an exercise price of $5.00 per share to 18
stockholders of Adgero.
Bridge Note Conversion
In
2015, Adgero sold promissory notes (the “Bridge
Offering”) in an aggregate principal amount of $285,000
(“Bridge Notes”), of which a Bridge Note in an amount
of $250,000 was purchased by an affiliate of the Placement Agent.
On April 8, 2016, the initial closing (the “Initial
Closing”) of the 2016 Private Placement, these Bridge Notes,
together with accrued interest thereon at a rate of 6% per annum,
converted into 58,100 shares of our common stock, 5,154 of which
were not included in the 2016 Private Placement, and 58,100 shares
of our common stock underlying warrants with an exercise price of
$5.00 per share, 5,154 of which were not included in the 2016
Private Placement.
II-3
St. Cloud Note Conversion
In
2012, Adgero issued a senior convertible note to each of St. Cloud
and Steven Rychnovsky, PhD, in an aggregate principal amount of two
hundred thousand dollars ($200,000). On August 3, 2016 these notes,
together with accrued interest thereon at a rate of 8% per annum,
converted into 73,998 shares of our common stock and 73,998 shares
of our common stock underlying warrants with an exercise price of
$5.00 per share.
Lowenstein Sandler LLP
In
January 2017, we issued 11,080 shares of our common stock to
Lowenstein Sandler LLP as partial payment for services
rendered.
Stock Options
Since
January 1, 2016, we have granted stock options under our 2016
Equity Incentive Plan to purchase an aggregate of
1,283,937 shares of our common stock at an exercise
price of $5.00 per share.
Restricted Share Awards
Since
January 1, 2016, we have granted restricted stock awards under our
2016 Equity Incentive Plan for an aggregate of 35,000
shares of our common stock.
Securities Act Exemptions
We
deemed the offers, sales and issuances of the securities described
above under “—Original Issuances of Stock and
Warrants” to be exempt from registration under the Securities
Act of 1933, as amended (the “Securities Act”), in
reliance on Section 4(a)(2) of the Securities Act, including
Regulation D and Rule 506 promulgated thereunder, relative to
transactions by an issuer not involving a public offering. All
purchasers of securities in transactions exempt from registration
pursuant to Regulation D represented to us that they were
accredited investors and were acquiring the shares for investment
purposes only and not with a view to, or for sale in connection
with, any distribution thereof and that they could bear the risks
of the investment and could hold the securities for an indefinite
period of time. The purchasers received written disclosures that
the securities had not been registered under the Securities Act and
that any resale must be made pursuant to a registration statement
or an available exemption from such registration.
We
deemed the grants of stock options and issuances of common stock
upon exercise of such options, and the restricted share awards,
described above under “Stock Options” and "Restricted
Share Awards" to be exempt from registration under the Securities
Act in reliance on section 4(a)(2) of the Securities Act,
including Regulation D and Rule 506 promulgated thereunder
and Rule 701 of the Securities Act as offers and sales of
securities under compensatory benefit plans and contracts relating
to compensation in compliance with Rule 701. Each of the recipients
of securities in any transaction exempt from registration either
received or had adequate access, through employment, business or
other relationships, to information about us.
All
certificates representing the securities issued in the transactions
described in this Item 15 included appropriate legends setting
forth that the securities had not been offered or sold pursuant to
a registration statement and describing the applicable restrictions
on transfer of the securities. There were no underwriters employed
in connection with any of the transactions set forth in this Item
15.
|
ITEM 16.
|
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
|
Exhibit No.
|
|
Description
|
|
|
|
|
|
2.1*
+
|
|
Agreement and Plan
of Merger, dated January 11, 2016, by and among the Company, Adgero
Acquisition, Inc. and Adgero Biopharmaceuticals, Inc.
|
|
|
|
|
|
3.1*
|
|
Certificate of
Incorporation
|
|
|
|
|
|
3.2*
|
|
Certificate of
Amendment to the Certificate of Incorporation
|
|
|
|
|
|
3.3*
|
|
Bylaws
|
|
|
|
|
|
4.1*
|
|
Form
of Replacement Warrant
|
|
|
|
|
|
4.2*
|
|
Form
of Investor Warrant
|
|
|
|
|
II-4
|
4.3*
|
|
Form
of Placement Agent Warrant
|
|
|
|
|
|
4.4*
|
|
Registration
Rights Agreement
|
|
|
|
|
|
4.5*
|
|
Form
of August Investor Warrant
|
|
|
|
|
|
4.6*
|
|
Form
of Common Stock Certificate
|
|
|
|
|
|
4.7*
|
|
Form
of December 2016 Investor Warrant
|
|
|
|
|
|
5.1*
|
|
Opinion of
Lowenstein Sandler LLP
|
|
|
|
|
|
10.1*
|
|
Placement Agency
Agreement, dated January 11, 2016, between the Company and Aegis
Capital Corp.
|
|
|
|
|
|
10.2*
|
|
Form
of Subscription Agreement for the Company’s 2016 private
placement
|
|
|
|
|
|
10.3*
|
|
Form
of Voting Agreement, dated April 8, 2016, by and among the Company
and the stockholders named therein
|
|
|
|
|
|
10.4*
|
|
2016
Equity Incentive Plan
|
|
|
|
|
|
10.5*
|
|
Form
of Incentive Stock Option Agreement
|
|
|
|
|
|
10.6*
|
|
Form
of Non-Qualified Stock Option Agreement
|
|
|
|
|
|
10.7*
|
|
Form
of Restricted Stock Agreement
|
|
|
|
|
|
10.8*
|
|
Employment
Agreement, dated April 8, 2016, between the Company and Frank
Pilkiewicz, PhD
|
|
|
|
|
|
10.9*
|
|
Employment
Agreement, dated April 8, 2016, between the Company and Steven
Rychnovsky, PhD
|
|
10.10*
|
|
Form
of Subscription Agreement for the Company’s August private
placement
|
|
|
|
|
|
10.11*
|
|
Promissory Note,
dated July 9, 2014, among Adgero Biopharmaceuticals, Inc. and Frank
Pilkiewicz, PhD
|
|
|
|
|
|
10.12*
|
|
Amendment to
Promissory Note, dated December 30, 2015, among Adgero
Biopharmaceuticals, Inc. and Frank Pilkiewicz, PhD
|
|
|
|
|
|
10.13*
|
|
Asset
Purchase Agreement, dated as of November 26, 2012, among Adgero
Biopharmaceuticals, Inc. and St. Cloud Investments,
LLC
|
|
|
|
|
|
10.14*
|
|
Amendment to
Asset Purchase Agreement, dated May 12, 2014, among Adgero
Biopharmaceuticals, Inc. and St. Cloud Investments,
LLC
|
|
|
|
|
|
10.15*
|
|
6%
Convertible Note, dated October 6, 2015, among Adgero
Biopharmaceuticals, Inc. and Robert F. Hendrickson
|
|
|
|
|
|
10.16*
|
|
Amendment to 6%
Convertible Note, dated March 25, 2016, among Adgero
Biopharmaceuticals, Inc. and Robert F. Hendrickson
|
|
|
|
|
|
10.17*
|
|
6%
Convertible Note, dated October 28, 2015, among Adgero
Biopharmaceuticals, Inc. and Roman Perez-Soler, MD
|
|
|
|
|
|
10.18*
|
|
Amendment to 6%
Convertible Note, dated March 28, 2016, among Adgero
Biopharmaceuticals, Inc. and Roman Perez-Soler, MD
|
|
|
|
|
II-5
|
10.19*
|
|
6%
Convertible Note, dated December 23, 2015, among Adgero
Biopharmaceuticals, Inc. and Adam Stern
|
|
|
|
|
|
10.20*
|
|
Amendment to 6%
Convertible Note, dated March 21, 2016, among Adgero
Biopharmaceuticals, Inc. and Adam Stern
|
|
|
|
|
|
10.21*
|
|
Engagement Letter,
dated August 8, 2016, between the Company and Aegis Capital
Corporation.
|
|
|
|
|
|
10.22*
|
|
8%
Convertible Note, dated November 26, 2012, among Adgero
Biopharmaceuticals, Inc. and St. Cloud Investments,
LLC
|
|
|
|
|
|
10.23*
|
|
8%
Convertible Note, dated November 26, 2012, among Adgero
Biopharmaceuticals, Inc. and Steven Rychnovsky, PhD
|
|
|
|
|
|
10.24*
|
|
Form
of Indemnification Agreement
|
|
|
|
|
|
10.25*
|
|
Employment
Agreement, dated October 3, 2016, between the Company and Laura
Pflug
|
|
|
|
|
|
10.26*
|
|
Agreement of
Lease, dated November 8, 2016, between the Company and Sutman
Princeton Associates, L.P.
|
|
|
|
|
|
10.27*
|
|
Lease
Modification Agreement, dated November 8, 2016, between the Company
and Sutman Princeton Associates, L.P.
|
|
|
|
|
|
10.28*
|
|
Employment
Agreement, dated April 8, 2016, between the Company and Jane
Maida
|
|
|
|
|
|
10.29*
|
|
Amendment No. 1 to
Employment Agreement, dated February 8, 2017, between the Company
and Frank Pilkiewicz, PhD
|
|
|
|
|
|
10.30*
|
|
Amendment No. 1 to
Employment Agreement, dated February 8, 2017, between the Company
and Steven Rychnovsky, PhD
|
|
|
|
|
|
10.31*
|
|
Amendment No. 1 to
Employment Agreement, dated February 8, 2017, between the Company
and Laura Pflug
|
|
|
|
|
|
10.32*
|
|
Form
of Subscription Agreement for the Company's December 2016 Private
Placement
|
|
|
|
|
|
10.33
|
|
First Amendment to
the 2016 Equity Incentive Plan
|
|
|
|
|
|
21.1*
|
|
List
of Subsidiaries of the Company
|
|
|
|
|
|
23.1
|
|
Consent of Marcum
LLP
|
|
|
|
|
|
23.2*
|
|
Consent of
Lowenstein Sandler LLP (included in Exhibit 5.1)
|
|
|
|
|
|
24.1*
|
|
Power
of Attorney (included on the signature page)
|
|
|
*
|
Previously
filed.
|
|
|
|
|
|
|
+
|
As
permitted by Item 601(b)(2) of Regulation S-K, certain schedules to
this agreement have not been filed herewith. The company will
furnish supplementally a copy of any omitted schedule to the
Commission upon request.
|
|
|
|
|
II-6
|
ITEM 17.
|
UNDERTAKINGS
|
The
undersigned registrant hereby undertakes:
(1)
To file, during any period in which offers or sales are being made,
a post-effective amendment to this registration
statement:
(i)
To include
any prospectus required by Section 10(a)(3) of the Securities Act
of 1933;
(ii)
To reflect in the
prospectus any facts or events arising after the effective date of
the registration statement (or the most recent post-effective
amendment thereof) which, individually or in the aggregate,
represent a fundamental change in the information set forth in the
registration statement. Notwithstanding the foregoing, any increase
or decrease in volume of securities offered (if the total dollar
value of securities offered would not exceed that which was
registered) and any deviation from the low or high end of the
estimated maximum offering range may be reflected in the form of
prospectus filed with the Securities and Exchange Commission
pursuant to Rule 424(b) if, in the aggregate, the changes in volume
and price represent no more than a 20% change in the maximum
aggregate offering price set forth in the “Calculation of
Registration Fee” table in the effective registration
statement; and
(iii)
To include any material
information with respect to the plan of distribution not previously
disclosed in the registration statement or any material change to
such information in the registration statement;
(2)
That, for the purpose of
determining any liability under the Securities Act of 1933, each
such post-effective amendment shall be deemed to be a new
registration statement relating to the securities offered therein,
and the offering of such securities at that time shall be deemed to
be the initial bona fide offering thereof.
(3)
To remove from registration by means of a post-effective amendment
any of the securities being registered which remain unsold at the
termination of the offering.
(4)
That, for the purpose of
determining liability under the Securities Act of 1933 to any
purchaser, each prospectus filed pursuant to Rule 424(b) as part of
a registration statement relating to an offering, other than
registration statements relying on Rule 430B or other than
prospectuses filed in reliance on Rule 430A, shall be deemed to be
part of and included in the registration statement as of the date
it is first used after effectiveness. Provided, however, that no statement
made in a registration statement or prospectus that is part of the
registration statement or made in a document incorporated or deemed
incorporated by reference into the registration statement or
prospectus that is part of the registration statement will, as to a
purchaser with a time of contract of sale prior to such first use,
supersede or modify any statement that was made in the registration
statement or prospectus that was part of the registration statement
or made in any such document immediately prior to such date of
first use.
(5)
That, for the purpose of
determining liability of the registrant under the Securities Act of
1933 to any purchaser in the initial distribution of the
securities:
The
undersigned registrant undertakes that in a primary offering of
securities of the undersigned registrant pursuant to this
registration statement, regardless of the underwriting method used
to sell the securities to the purchaser, if the securities are
offered or sold to such purchaser by means of any of the following
communications, the undersigned registrant will be a seller to the
purchaser and will be considered to offer or sell such securities
to such purchaser:
(i)
Any
preliminary prospectus or prospectus of the undersigned registrant
relating to the offering required to be filed pursuant to Rule
424;
(ii)
Any free writing
prospectus relating to the offering prepared by or on behalf of the
undersigned registrant or used or referred to by the undersigned
registrant;
II-7
(iii)
The portion of any other
free writing prospectus relating to the offering containing
material information about the undersigned registrant or its
securities provided by or on behalf of the undersigned registrant;
and
(iv)
Any other communication that is an
offer in the offering made by the undersigned registrant to the
purchaser.
(6)
Insofar as
indemnification for liabilities arising under the Securities Act of
1933 may be permitted to directors, officers and controlling
persons of the registrant pursuant to the foregoing provisions, or
otherwise, the registrant has been advised that in the opinion of
the Securities and Exchange Commission such indemnification is
against public policy as expressed in the Securities Act of 1933
and is, therefore, unenforceable. In the event that a claim for
indemnification against such liabilities (other than the payment by
the registrant of expenses incurred or paid by a director, officer
or controlling person of the registrant in the successful defense
of any action, suit or proceeding) is asserted by such director,
officer or controlling person in connection with the securities
being registered, the registrant will, unless in the opinion of its
counsel the matter has been settled by controlling precedent,
submit to a court of appropriate jurisdiction the question whether
such indemnification by it is against public policy as expressed in
the Securities Act of 1933 and will be governed by the final
adjudication of such issue.
II-8
SIGNATURES
Pursuant to the
requirements of the Securities Act of 1933, as amended, the
registrant has duly caused this registration statement to be signed
on its behalf by the undersigned, thereunto duly authorized, in the
City of Princeton, State of New Jersey on April
11,
2017.
|
|
ADGERO BIOPHARMACEUTICALS HOLDINGS, INC.
|
|
|
|
|
|
|
|
|
|
By:
|
/s/
Frank Pilkiewicz,
PhD
|
|
|
|
Name:
Frank Pilkiewicz, PhD
|
|
|
|
|
Title:
Chief Executive Officer
|
|
|
Pursuant to the
requirements of the Securities Act of 1933, as amended, this
registration statement has been signed below by the following
persons in the capacities and on the dates
indicated.
|
Person
|
|
Capacity
|
|
Date
|
|
|
|
|
|
|
|
/s/ Frank
Pilkiewicz, PhD
|
|
Chief Executive
Officer, and Director
|
|
|
|
Frank Pilkiewicz,
PhD
|
|
(Principal
Executive Officer)
|
|
April
11, 2017
|
|
|
|
|
|
|
|
/s/
*
|
|
Chief Financial
Officer
|
|
|
|
Jane
Maida
|
|
(Principal
Financial and Accounting Officer)
|
|
April 11,
2017
|
|
|
|
|
|
|
|
/s/ *
|
|
|
|
|
|
Allen Bloom, PhD,
JD
|
|
Director
|
|
April 11,
2017
|
|
|
|
|
|
|
|
/s/
*
|
|
|
|
|
|
David
Hochman
|
|
Director
|
|
April 11,
2017
|
|
|
|
|
|
|
|
/s/
*
|
|
|
|
|
|
Roman
Perez-Soler, MD
|
|
Director
|
|
April 11,
2017
|
|
|
|
|
|
|
|
/s/
*
|
|
|
|
|
|
Tim
McInerney
|
|
Director
|
|
April
11, 2017
|
|
*By:
|
|
/s/ Frank
Pilkiewicz, PhD
|
|
|
|
|
|
|
Frank Pilkiewicz,
PhD
|
|
|
|
|
|
|
Attorney-in-fact
|
|
|
|
II-9