Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - Moleculin Biotech, Inc. | v462503_ex32-2.htm |
| EX-32.1 - EXHIBIT 32.1 - Moleculin Biotech, Inc. | v462503_ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - Moleculin Biotech, Inc. | v462503_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - Moleculin Biotech, Inc. | v462503_ex31-1.htm |
| EX-23.2 - EXHIBIT 23.2 - Moleculin Biotech, Inc. | v462503_ex23-2.htm |
| EX-23.1 - EXHIBIT 23.1 - Moleculin Biotech, Inc. | v462503_ex23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C., 20549
FORM 10-K
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
| For the fiscal year ended December 31, 2016 | Commission File Number: 001-37758 |

Moleculin Biotech, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 47-4671997 |
| (State or Other Jurisdiction of | (Primary Standard Industrial | (I.R.S. Employer |
| Incorporation or Organization) | Classification Code Number) | Identification Number) |
2575 West Bellfort, Suite 333
Houston, Texas 77054
(713) 300-5160
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant’s Principal Executive Offices)
Securities registered pursuant to Section
12(b) of the Act: Common Stock, par value $0.001
per share
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter periods as the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (check one)
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer ¨ | Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). YES ¨ NO x
The aggregate market value of the registrant’s voting equity held by non-affiliates of the registrant, computed by reference to the price at which the common stock was last sold as of the last business day of the registrant’s most recently completed second fiscal quarter, was $27,612,783. In determining the market value of the voting equity held by non-affiliates, securities of the registrant beneficially owned by directors, officers and 10% or greater shareholders of the registrant have been excluded. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The number of shares of the registrant’s common stock outstanding as of March 24, 2017 was 17,176,872.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of this registrant’s definitive proxy statement for its 2017 Annual Meeting of Stockholders to be filed with the SEC no later than 120 days after the end of the registrant’s fiscal year are incorporated herein by reference in Part III of this Annual Report on Form 10-K.
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
The Securities and Exchange Commission, referred to herein as the SEC, encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. Certain statements that we may make from time to time, including, without limitation, statements contained in this report constitute “forward- looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995.
We make forward-looking statements under the “Risk Factors,” “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in other sections of this report. In some cases, you can identify these statements by forward-looking words such as “may,” “might,” “should,” “would,” “could,” “expect,” “plan,” “anticipate,” “intend,” “believe,” “estimate,” “predict,” “potential” or “continue,” and the negative of these terms and other comparable terminology. These forward-looking statements, which are subject to known and unknown risks, uncertainties and assumptions about us, may include projections of our future financial performance based on our growth strategies and anticipated trends in our business. These statements are only predictions based on our current expectations and projections about future events. There are important factors that could cause our actual results, level of activity, performance or achievements to differ materially from the results, level of activity, performance or achievements expressed or implied by the forward-looking statements. In particular, you should consider the numerous risks and uncertainties described under “Risk Factors.”
While we believe we have identified material risks, these risks and uncertainties are not exhaustive. Other sections of this report describe additional factors that could adversely impact our business and financial performance. Moreover, we operate in a very highly regulated, competitive and rapidly changing environment. New risks and uncertainties emerge from time to time, and it is not possible to predict all risks and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. Moreover, neither we nor any other person assumes responsibility for the accuracy or completeness of any of these forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We are under no duty to update any of these forward-looking statements after the date of this report to conform our prior statements to actual results or revised expectations, and we do not intend to do so.
Forward-looking statements include, but are not limited to, statements about:
| · | our ability to obtain additional funding to develop our product candidates; |
| · | the need to obtain regulatory approval of our product candidates; |
| · | the success of our clinical trials through all phases of clinical development; |
| · | our ability to complete our clinical trials in a timely fashion and within our expected budget; |
| · | compliance with obligations under intellectual property licenses with third parties; |
| · | any delays in regulatory review and approval of product candidates in clinical development; |
| · | our ability to commercialize our product candidates; |
| · | market acceptance of our product candidates; |
| · | competition from existing products or new products that may emerge; |
| · | potential product liability claims; |
1
| · | our dependency on third-party manufacturers to supply or manufacture our products; |
| · | our ability to establish or maintain collaborations, licensing or other arrangements; |
| · | our ability and third parties’ abilities to protect intellectual property rights; |
| · | our ability to adequately support future growth; and |
| · | our ability to attract and retain key personnel to manage our business effectively. |
We caution you not to place undue reliance on the forward-looking statements, which speak only as of the date of this Form 10-K in the case of forward-looking statements contained in this Form 10-K.
References in this Annual Report on Form 10-K to “MBI” or “the Company”, “we”, “our” and “us” are used herein to refer to Moleculin Biotech, Inc.
Overview
MBI is a preclinical-stage pharmaceutical company organized as a Delaware corporation in July 2015 to focus on the development of anti-cancer drug candidates, some of which are based on license agreements with The University of Texas System on behalf of the M.D. Anderson Cancer Center, which we refer to as MD Anderson. Our lead drug candidate is liposomal Annamycin, which we refer to as Annamycin, an anthracycline being studied for the treatment of relapsed or refractory acute myeloid leukemia, or AML. Annamycin has been in clinical trials pursuant to an Investigational New Drug application, or IND, that had been filed with the FDA. Due to a lack of development activity by a prior drug developer, this IND was terminated. To permit the renewed investigation of Annamycin, we are submitting a new IND augmenting the data that supported the original IND, with subsequent clinical data and additional information. We have two other drug development projects in process, one involving a collection of small molecules, which we refer to as the WP1066 Portfolio, focused on the modulation of key regulatory transcription factors involved in the progression of cancer, and the WP1222 Portfolio, a suite of molecules targeting the metabolic processes involved in cancer in general and glioblastoma (the most common form of brain tumor) in particular. We also continue to sponsor ongoing research at MD Anderson in order to improve and expand our drug development pipeline.
We have been granted royalty-bearing, worldwide, exclusive licenses for the patent and technology rights related to our WP1066 Portfolio and WP1122 Portfolio drug technologies, as these patent rights are owned by MD Anderson. The Annamycin drug substance is no longer covered by any existing patent protection. We intend to submit patent applications for formulation, synthetic process and reconstitution related to our Annamycin drug product candidate, although there is no assurance that we will be successful in obtaining such patent protection. Independently from potential patent protection, we have received Orphan Drug designation from the FDA for Annamycin for the treatment of AML, which would entitle us to market exclusivity of 7 years from the date of approval of a New Drug Application (NDA) in the United States. If we submit and receive approval for a New Drug Application (NDA) for Annamycin for the treatment of AML, we may then benefit from Orphan Drug exclusivity, during which period FDA generally could not approve another Annamycin product for the same use. We also intend to apply for similar status in the European Union (EU) where market exclusivity extends to 10 years from the date of Marketing Authorization Application (MAA). Separately, the FDA may also grant market exclusivity of 5 years for newly approved new chemical entities (of which Annamycin would be one), but there can be no assurance that such exclusivity will be granted.
2
Corporate History
We were founded in 2015 by Walter Klemp (our chairman and CEO), Dr. Don Picker (our President and Chief Operating Officer) and Dr. Waldemar Priebe of MD Anderson (Chairman of our Scientific Advisory Board) in order to combine and consolidate development efforts that include several MD Anderson anti-cancer technologies. This effort began with the acquisition of the Annamycin development project from AnnaMed, Inc., or AnnaMed, followed by the acquisition of the license rights to the WP1122 Portfolio from IntertechBio Corporation, or IntertechBio. Further, we undertook an effort to gain control of the WP1066 Portfolio, which culminated with the merger of Moleculin, LLC and MBI and the establishment of a co-development agreement with Houston Pharmaceuticals, Inc., or HPI, coincident with our IPO.
AnnaMed was formed in 2012 to take over the development of Annamycin from a prior drug development company, Callisto Pharmaceuticals, Inc., or Callisto. Callisto ceased development work on Annamycin, leading to the termination of its IND by the FDA. Callisto disclosed publicly in its Form 10-K filing for the year ended 2009 that the clinical data relating to acute leukemia patients “did not support further clinical evaluation of L-Annamycin as a single agent to treat relapsed or refractory adult acute leukemia patients.” In order to satisfy unmet license obligations, Callisto agreed to transfer all available Annamycin data to AnnaMed, which data we are using and have augmented with additional data and information to apply for a new IND. As such, notwithstanding Callisto’s determination to terminate its development of Annamycin, we are utilizing the clinical data from Callisto’s trials to apply for a new IND. The basis for our decision to proceed notwithstanding Callisto’s determination is that we believe the actual clinical data as reported by Dr. Robert Shepard, our Chief Medical Officer and who was Callisto’s Chief Medical Officer at the time of the clinical trials, to the 2009 Annual Meeting of the American Society of Clinical Oncology, and as further reported by the Principal Investigators of the clinical trials in a peer-reviewed journal article (Clin Lymphoma Myeloma Leuk. 2013 August; 13(4): 430–434. doi:10.1016/j.clml.2013.03.015.), supports further clinical evaluation. In addition, the conclusion published in the 2013 Clinical Lymphoma, Myeloma & Leukemia journal article was that “Single agent nanomolecular liposomal annamycin appears to be well-tolerated and evidence of clinical activity as a single agent in refractory adult ALL.” As reported in both the ASCO presentation and the 2013 journal article referenced, the definition of efficacy is based on the following Response Criteria: “Response criteria were achievement of CR defined as ≤5% blasts, granulocyte count of ≥1×109/L, and a platelet count of ≥100×109/L. Partial remission was defined the same as CR, except for the presence of 6% to 25% blasts. Hematologic improvement was defined as for CR but platelet count <100×109/L.” The summary of patient response from the 2013 journal article reads: “After determining the MTD, a 10-patient phase IIA was conducted. Eight of the patients completed one cycle of the three days of treatment at the MTD. Of these, five (62%) demonstrated encouraging anti-leukemic activity with complete clearing of circulating peripheral blasts. Three of these subjects also cleared bone marrow blasts with one subsequently proceeding onto successful stem cell transplantation. The other two developed tumor lysis syndrome and unfortunately expired prior to response assessment.” In the Company’s review of these trials, it confirmed that the activity demonstrated in this summary corresponds with a “Partial remission” as described in the Response Criteria and that the three subjects who “cleared bone marrow blasts” correspond with “CR” (Complete Response).
In 2012, AnnaMed out-licensed development rights in a limited territory to a Polish special purpose drug development company called Dermin in exchange for Dermin’s development work based on its successful effort to obtain Polish government grant funding to assist in the development of Annamycin. Since that time, such grant funding has been used to produce Annamycin in preparation for future clinical trials. In August 2015, we entered into a rights transfer agreement with AnnaMed pursuant to which, in exchange for 1,431,000 shares of our common stock, AnnaMed agreed to transfer any and all data it had regarding the development of Annamycin and the Annamycin IND, including all trade secrets, know-how, confidential information and other intellectual property rights held by AnnaMed.
IntertechBio was formed in 2009 to license and begin development on the WP1122 Portfolio. The WP1122 Portfolio was also out-licensed to Dermin, which was awarded a Polish government grant to assist in drug development efforts. In August 2015, IntertechBio agreed to assign all license rights to us in exchange for our common stock.
3
Moleculin, LLC was formed in 2006 and has been working to develop the WP1066 Portfolio it licensed from MD Anderson. As a part of the formation of Moleculin, LLC, an agreement was reached with HPI to limit Moleculin, LLC’s development efforts to uses in dermatology only, leaving non-dermatology indications to HPI. From 2006 to 2014, Moleculin, LLC funded its operations through the sale of equity and debt securities and through fees received from an out-licensing agreement with a Japanese dermatology drug development company, Maruho, Ltd., or Maruho. Beginning in 2012, Moleculin, LLC conducted a series of clinical trials focused on the topical treatment of psoriasis. Although those trials demonstrated drug activity, the results were not conclusive enough to warrant full commercialization as a topical dermatology drug. Additional study was required to determine optimal dosing and scheduling regimens for the topical treatment of psoriatic plaques. As a result of this additional complexity with regard to the potential treatment of psoriasis, Maruho did not elect to continue its funding of the psoriasis drug development effort and, therefore, forfeited their license rights to the WP1066 Portfolio that had been granted to it by Moleculin, LLC. Dermin is planning to clinically evaluate a candidate from the WP1066 Portfolio for the topical treatment of cutaneous T-cell lymphoma, or CTCL. We do not have control of the clinical plan or timeline for Dermin’s development effort.
Prior to our IPO, Moleculin, LLC was merged with and into our company. The merger agreement contains mutual representations and warranties between the parties. Pursuant to the merger agreement, we agreed for a period of six years to indemnify and hold harmless each present and former director and/or officer of Moleculin, LLC whom Moleculin, LLC would have had the power to indemnify under Delaware law that is made a party or threatened to be made a party to any threatened, pending or completed proceeding or claim by reason of the fact that he or she was a director or officer of the Moleculin, LLC prior to the effective time of the merger and arising out of actions or omissions of the indemnified party in any such capacity occurring at or prior to the effective time of the merger against any losses or damages reasonably incurred in connection with any claim. To our knowledge, no such proceeding or claim exists or has been threatened on the date hereof and Moleculin, LLC made representations to this effect in the merger agreement as of the date of such agreement. As additional consideration payable to the Moleculin, LLC unit holders, we agreed pursuant to the merger agreement that if drugs for dermatology indications are successfully developed by us (or our successors) using any of the Existing IP Assets, then the Moleculin, LLC unit holders, in the aggregate, will be entitled to receive a 2.5% royalty on the net revenues generated by such drugs. Any such net revenues would include a deduction for license fees or royalty obligations payable to MD Anderson for such Existing IP Assets. The merger agreement defined “Existing IP Assets” to mean all intellectual property, licensed by us and Moleculin, LLC as of the time of the merger, including, without limitation, the intellectual property licensed from MD Anderson under the Patent and Technology License Agreement entered into by and between IntertechBio Corporation and MD Anderson dated April 2, 2012, as amended, and the Patent and Technology License Agreement dated June 21, 2010, as amended, between MD Anderson and Moleculin, LLC, but excluding any intellectual property relating to Annamycin. The right to receive the contingent royalty payments described herein are for drugs developed only for dermatology indications, and do not include drugs developed for any other indications. We have no obligation of any nature to pursue the development of any drugs for dermatology indications.
From 2006 through 2015, HPI was engaged in research related to the use of the WP1066 Portfolio for the treatment of non-dermatology cancers and received grant awards totaling approximately $4.9 million toward this effort. Prior to our IPO, we entered into a co-development agreement with HPI whereby HPI is continuing its grant-funded research and making all resulting data available for our use in exchange for a development fee. We may buy HPI out of its co-development rights in the WP1066 Portfolio at our option. Please see the section “Business – License Agreements” for a description of our agreement with HPI.
Neither our founders nor MBI have or will have any ownership stake in Dermin, our Polish licensee. No Dermin-related grant money is expected to be paid directly to us, but rather the sublicense agreements require Dermin to share resulting data, which we believe will reduce costs we might otherwise have to incur directly. There can be no assurance, however, that Dermin will continue to receive the funds they have been awarded or that such funds will be spent by Dermin in a manner that will benefit MBI. The sublicensed territories granted to Dermin for the WP1066 Portfolio are Poland, Ukraine, Czech Republic, Hungary, Romania, Slovakia, Belarus, Lithuania, Latvia and Estonia. For the WP1122 Portfolio, the territory expanded to include Russia and Kazakhstan. For Annamycin, the territory is further expanded to include Netherlands, Turkey, Belgium, Switzerland, Austria, Sweden, Greece, Portugal, Norway, Denmark, Ireland, Finland, Luxembourg, Iceland, Uzbekistan, Georgia, Armenia, Azerbaijan and Germany. However, in the case of Germany, this territory may be reclaimed by us for a payment of $500,000 to Dermin.
4
The following summarizes the transactional history and common relationships among HPI, IntertechBio, Moleculin, LLC, MBI, Callisto, AnnaMed, MD Anderson, and our officers and major shareholders:
· Moleculin, LLC: Prior to our IPO, Moleculin, LLC was merged with and into our company. Moleculin, LLC was the holder of the license agreement with MD Anderson covering our WP 1066 Portfolio. As a result of the merger, we issued the equity interests holders of Moleculin, LLC an aggregate of 999,931 shares of our common stock. Waldemar Priebe, chairman of our Scientific Advisory Board, Walter Klemp, our chairman and chief executive officer, and Don Picker, our president and chief operating officer, were members of Moleculin, LLC and received 6,046 shares, 22,795 shares and 6,046 shares, respectively, of our common stock as a result of the merger. In addition, Walter Klemp and Don Picker were members of the board of Moleculin, LLC.
· Houston Pharmaceuticals, Inc.: Prior to our IPO, MBI on Moleculin, LLC’s behalf, entered into a co-development agreement with HPI whereby HPI is continuing its grant-funded research and making all resulting data available for our use in exchange for a development fee. Waldemar Priebe, chairman of our Scientific Advisory Board, and Don Picker, our president and chief operating officer, are shareholders of HPI, and Dr. Priebe has the voting and dispositive power over the shares held by HPI.
· IntertechBio Corporation: In August 2015, in exchange for the issuance of 630,000 shares of common stock, we acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation. Waldemar Priebe, chairman of our Scientific Advisory Board, and Don Picker, our president and chief operating officer, are shareholders of IntertechBio and control the voting and dispositive power over the shares held by IntertechBio.
· AnnaMed, Inc.: In August 2015, in exchange for 1,431,000 shares of our common stock, we acquired the rights to the Annamycin data related to the original Annamycin IND and the development of Annamycin held by AnnaMed, Inc., a company controlled by Walter Klemp, our chairman and chief executive officer.
· Callisto Pharmaceuticals, Inc.: Our president and chief operating officer, Don Picker and our chief medical officer, Robert Shepard, were chief operating officer and chief medical officer, respectively, of Callisto. Since 2009, neither individual has had any relationship or ownership with Callisto.
· MD Anderson: Both Moleculin, LLC and IntertechBio entered into license agreements with MD Anderson. See “Business – Our Licenses Agreements” below. Waldemar Priebe, chairman of our Scientific Advisory Board, is a Professor of Medicinal Chemistry, Department of Experimental Therapeutics, Division of Cancer Medicine, at the University of Texas MD Anderson Cancer Center.
Our Drug Candidates
Annamycin
Our lead product candidate is Annamycin, for which FDA has granted Orphan Drug designation for the treatment of AML. We intend to conduct clinical trials for Annamycin as a monotherapy for the treatment of relapsed or refractory AML. If Annamycin is ultimately approved on this basis, it is possible, with the necessary subsequent testing and approval, that it could be used in the future in combination with other drugs.
Market for Annamycin
Leukemia is a cancer of the white blood cells and acute forms of leukemia can manifest quickly and leave patients with limited treatment options. AML is the most common type of acute leukemia in adults. It occurs when a clone of leukemic progenitor white blood cells proliferates in the bone marrow, suppressing the production of normal blood cells. Currently, the only viable option for acute leukemia patients is a bone marrow transplant, which is successful in a significant number of patients. However, in order to qualify for a bone marrow transplant, the patient’s leukemia cells must be decreased to a sufficiently low level. This usually begins with a therapy of combining two chemotherapeutic drugs, which almost always includes an anthracycline to induce remission (a complete response, or CR). This therapy has not improved since it was first used in the 1970s and we estimate that this induction therapy has a success rate of about 20%, as it has from the time of its initial use. Unfortunately, the current clinically approved anthracyclines are cardiotoxic (i.e., can damage the heart), which can limit the dosage amount that may be administered to patients. Additionally, the tumor cells often present de novo or develop resistance to the first line anthracycline, through what is called “multidrug resistance,” enabling the tumor cells to purge themselves of the available anthracyclines. Consequently, there remains no effective therapy for inducing remission in these patients sufficient to enable a curative bone marrow transplant and unfortunately most will succumb quickly to their leukemia. If a patient’s leukemia reappears before they can be prepared for a bone marrow transplant, they are considered to have “relapsed.” If a patient fails to achieve a sufficient response from the induction therapy to qualify for a bone marrow transplant, they are considered to be “refractory” (resistant to therapy). Together, this group of relapsed and refractory AML patients constitutes our primary focus for treatment with Annamycin and our intent is to pursue FDA approval for Annamycin as a second-line induction therapy for adult relapsed and refractory AML patients.
5
We believe that pursuing approval as a second line induction therapy for adult relapsed or refractory AML patients is the shortest path to regulatory approval, but we also believe that one of the most important potential uses of Annamycin is in the treatment of children with either AML or ALL (acute lymphoblastic leukemia, which is more common in children). Accordingly, we also intend to pursue approval for pediatric use in these conditions when practicable.
Of the estimated 18,860 U.S. cases of acute myeloid leukemia diagnosed in 2014, an estimated 97% were adult and although exact numbers are not available, we estimate that 70% to 80% (approximately 13,000 to 14,000 patients) were expected to relapse or be resistant to first-line therapy.
Prior Clinical Trials for Annamycin
Annamycin is a liposome formulated anthracycline (also referred to in literature as “L-Annamycin”). It has been tested in 6 clinical trials and 114 patients with little to no reported cardiotoxicity and, in the two clinical trials focused on leukemia, with fewer dose-limiting toxicities than are normally expected with doxorubicin (one of the leading first-line anthracyclines used for induction therapy). Each of these trials was conducted by a prior developer of Annamycin, and not by our company. Annamycin demonstrated significant activity in 8 of 16 patients in a Phase I study in adult relapsed or refractory AML and ALL patients, with 6 of 14 patients completely clearing leukemic bone marrow blasts. The reason only 14 (rather than 16) patients were tested for leukemic bone marrow blasts is that 2 of the 16 patients succumbed to their disease before bone marrow testing could be completed. In a 30 patient dose-ranging Phase I/II study in ALL, 3 of 8 patients treated with the maximum tolerable dose cleared their leukemic blasts to a level sufficient to qualify for a bone marrow transplant. One of these patients went on to receive a successful curative bone marrow transplant. The other two of these three patients died of tumor lysis syndrome, a condition resulting from the overloading of their system with the debris from leukemic blast cells destroyed by the induction therapy. Armed with the knowledge of this potential, prophylactic pretreatment intended to protect patients from the effects of tumor lysis syndrome will be deployed where appropriate in future trials. Based on the results of the above clinical trials, we believe Annamycin may be different from currently approved induction therapy drugs in four key ways: (i) it has demonstrated clinical activity in a patient population for whom there are currently no effective therapies, (ii) it appears to be capable of avoiding the “multi-drug resistance” mechanisms that have been associated with limiting the effectiveness of currently approved anthracyclines; (iii) it has been shown to be non-cardiotoxic in animal models and little to no cardiotoxicity has been reported from the use of Annamycin in 114 patients; and (iv) in certain AML cell lines, it has been shown to be more potent than one of the leading approved anthracyclines.
Based on initial conversations with the FDA, because of the serious unmet medical need, we believe Annamycin may qualify for accelerated approval based on our planned clinical trials. In order to facilitate our communication with the FDA, we requested access to and reviewed in detail the available data supporting the dose-ranging Phase I/II clinical trial discussed above, which was conducted by a previous developer of Annamycin. In October 2016, we announced that we had identified some positive findings from this review, which gave rise to a modification of our own clinical development plan. We had indicated that our plan was to conduct a detailed review of the clinical results generated by that prior developer, and then to use those results to reestablish an IND in order to continue clinical trials of Annamycin. However, in the course of our review, we identified that Annamycin may have greater potential for efficacy than we originally believed, based on an unexpected potential opportunity to increase the drug’s Maximum Tolerable Dose (“MTD”).
In particular, the Dose Limiting Toxicities (“DLTs”) reported in the previous trial that led to the establishment of the current MTD of 150 mg/m2 were all from patients who had an unusually high number of first-line induction therapy failures prior to being treated with Annamycin. Specifically, of the three patients in the last clinical trial who experienced these DLTs, one of them had failed nineteen prior induction therapy attempts, another had failed sixteen and the other had failed fifteen before being enrolled in the trial. We concluded from our review of this data that, if the heavily treated patients are excluded from the data set, the MTD could have been closer to 250 mg/m2, substantially higher than the level that was actually set by this previous trial.
6
We view this as an encouraging development because it means we may have an opportunity to increase the MTD for our next trial from 150 mg/m2 to 200 or even 250 mg/m2. If that turns out to be the case, we believe it could increase the chance for positive outcomes in our next trial.
With the discovery that we may be able to increase our MTD, we determined to adjust our clinical strategy by adding in a Phase I arm to our next Phase II trial, which will add expense to our development effort. We believe this change in strategy will add several months to the eventual final approval of the drug, if the drug is approved.
Because the prior developer of Annamycin allowed their IND to lapse, we are required to submit a new IND for continued clinical trials with Annamycin. We filed our IND application for Annamycin, with the clinical strategy of increasing the MTD mentioned above, on February 10, 2017. In subsequent discussions with us, FDA requested certain revisions to the protocol, additional information, and additional data related to Chemistry, Manufacturing and Controls (“CMC”). We have the additional information, have made the requested revisions to the protocol, and we are working on developing the CMC data. In the interim, we have withdrawn the IND application in order to resubmit it when the requested data are available. We believe that resubmission of the IND application will occur in time for the IND to go into effect and the announcement of the beginning of Phase I/II clinical trials in the first half of 2017. However, if we are unable to obtain the required CMC data on a timely basis, we will be delayed in resubmitting our IND application, which will delay the commencement of our clinical trials beyond the first half of 2017.
On March 21, 2017 we received notice that FDA had granted Orphan Drug designation for Annamycin for the treatment of AML, effective March 20, 2017.
Little to No Cardiotoxicity
One of the key dose-limiting toxicities associated with currently available anthracyclines is their propensity to induce life-threatening heart damage. This is especially problematic for pediatric leukemia patients whose life spans can be severely shortened by the very induction therapy designed to cure them of acute leukemia. In the animal model relied upon by the FDA as an indicator of human cardiotoxicity, the non-liposomal (free) form of Annamycin has been shown to be significantly less likely than doxorubicin to create heart lesions in mice, and the liposomal formulation (L-Annamycin) has been shown in these same models to have reduced cardiotoxicity to the point where it is unlikely to cause harm to human patients. This possible lack of human cardiotoxicity means L-Annamycin may be able to be used more aggressively in helping patients achieve remission. This would be especially valuable in the case of pediatric acute leukemia (both AML and ALL) where long-term survival can be greatly impacted by cardiotoxicity. Unless otherwise noted, all of our references to Annamycin refer to the liposomal form (L-Annamycin).
Circumventing Multidrug Resistance
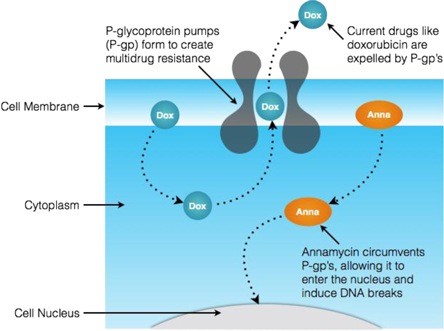
In addition to cardiotoxicity, the effectiveness of currently approved anthracyclines is limited by their propensity for succumbing to “multidrug resistance,” whereby transporters (one type of which is referred to as a “P-glycoprotein pump”) develop on the outer surface of cells to expel drugs like anthracyclines as a natural defense mechanism. The dosing of current therapies cannot be increased in an attempt to overcome multidrug resistance because of the likelihood of cardiotoxicity and other serious side effects. This limitation prevents adequate dosing of current anthracyclines to produce lasting remission. A laboratory study has suggested that Annamycin may resist being expelled by P-glycoprotein pumps and other similar tested multidrug resistance transporters, which may mean the drug circumvents multidrug resistance. This characteristic has been shown in pre-clinical testing to allow for higher drug uptake in diseased cells, which we believe could allow for more effective induction therapy with less risk to the patient.
7
In order for anthracyclines to provide effective induction therapy, they must be allowed to accumulate in leukemic cells sufficiently to enter the cell’s nucleus, where they damage the cell’s DNA and induce apoptosis (cell death). As induction therapy progresses, however, the targeted cells can develop a natural defense mechanism to prevent the anthracycline activity. The graphic below provides a simplified depiction of the formation of a P-glycoprotein pump on the outer surface (membrane) of a leukemic cell. As typical anthracyclines enter the cell, they are attracted to such pumps and are expelled (referred to as “efflux”) before they can accumulate sufficiently to serve their purpose. In contrast, Annamycin appears to avoid such pumps, thereby being allowed to accumulate sufficiently to destroy the leukemia cell despite the presence of the multidrug resistance mechanisms.
The WP1066 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to our WP1066 Portfolio and its close analogs, molecules targeting the modulation of key oncogenic transcription factors.
Clinical Testing of WP1066 Portfolio
In vitro testing has shown a high level of activity for WP1066 against a wide range of solid tumors, and in vivo testing has shown significant activity against head and neck, pancreatic, stomach, and ovarian cancers, as well as metastatic melanoma and glioblastoma, among others. In vivo testing in mouse tumor models has shown that WP1066 inhibits tumor growth, blocks angiogenesis (a process that provides necessary blood supply to tumors) and increases survival.
With respect to our WP1066 Portfolio, we must complete pre-clinical toxicology testing, along with additional chemistry, manufacturing and control work to fully characterize the drug, establish the desired formulation and develop reference standards for future drug release, among other things, prior to submitting an application for IND. A clinician at MD Anderson has advised us that she is proceeding with a physician-sponsored IND for WP1066 treatment of brain tumors. We are not participating in this IND process. The clinician has submitted an IND to the FDA and has indicated that this IND is on hold until documentation of Good Manufacturing Process, or GMP, production of WP1066 can be presented to the FDA.
8
An analog of WP1066, referred to as WP1220, was previously the subject of an IND (WP1220 was referred to as MOL4239 for purposes of this IND) related to use of the molecule in the topical treatment of psoriasis. Clinical trials were commenced on WP1220 in the US, but were terminated early due to limited efficacy in the topical treatment of psoriatic plaques. Notwithstanding its limitations in treating psoriasis, our pre-clinical research has suggested that WP1220 may be effective in inhibiting cutaneous T-cell lymphoma (CTCL) in multiple CTCL cell lines. Based on this data, we are collaborating with a Polish drug development company, Dermin, which has received Polish government grant money to develop WP1220 in Poland for the topical treatment of early stage CTCL patients. CTCL is a potentially deadly form of skin cancer for which there are limited treatment options.
We also conducted a Phase II clinical trial for WP1066 for the topical treatment of psoriasis, however this trial was terminated early as a significant number of patients experienced a non-permanent worsening of their psoriatic plaques after extended use of the drug, suggesting that its use as a topical agent for non-life threatening diseases such as psoriasis will require further study to optimize dosing and scheduling regimens.
Scientific Rationale for WP1066 Portfolio
Cellular biology depends upon signaling mechanisms to regulate functions such as cell growth, death and adaptation. Signal “transduction” is such a mechanism that converts an upstream stimulus to a cell into a specific cellular response. Signal transduction starts with a signal to a receptor or via a compound capable of passing through the cell membrane and ends with a change in cell function. The end result of this signal is often the activation of “transcription,” whereby genetic information is expressed and, in the case of oncogenic transcription, disease processes are initiated or maintained.
Receptors span the cell membrane, with part of the receptor outside and part inside the cell. When a chemical signal represented by a specific protein binds to the outer portion of the receptor, it conveys another signal inside the cell. Often there is a cascade of signals within the cell, wherein an upstream inducer starts a chain of events that resembles a domino effect. Collectively, this sequence is referred to as a “signaling pathway.” Eventually, the signal creates a change in the cell function by changing the expression of specific genes and production of specific proteins within the cell, and again, in the case of tumor development, such expression results in unwanted oncogenic processes.
Importantly, while normal healthy cell function relies on signaling mechanisms, diseases are capable of co-opting these mechanisms with negative consequences. Oncogenic processes (including inflammation and proliferation) depend upon signaling pathways that are responsible for coordinating functions such as cell growth, survival and cell differentiation. A particular class of proteins referred to as Signal Transducers and Activators of Transcription (such proteins are “STATs”) regulates the process of disease cell survival and proliferation, angiogenesis and immune system function and is persistently activated in a large number of human inflammatory processes and in hyper-proliferating diseases. Because certain of these proteins are known to be co-opted by tumor cells, we refer to them as “oncogenic transcription factors,” of which certain STATs are a subset.
Some STATs, such as STAT3, can be activated by any one of many different upstream inducers, making them very difficult to target by blocking just one or more of these upstream inducers. We believe that blocking a targeted STAT directly rather than via its multiple upstream inducers should result in greater efficacy with lower toxicity.
9
In the diagram shown below, any one of many different pathways (categorized as Growth Factor Receptors, Cytokine Receptors and Non-Receptor Tyrosine Kinases) triggers the activation of STAT3 proteins in a process called “phosphorylation”. In this process, phosphates attach to corresponding receptors on STAT3 and, eventually, two phosphorylated STAT3 proteins (“p-STAT3”) bind together in a pair referred to as a “dimer.” Once the dimer is formed, it enters the cell nucleus and triggers gene transcription. Conversely, if we reduce the presence of p-STAT3 before dimers can be formed, we can prevent the triggering of gene transcription and effectively inhibit the disease process.
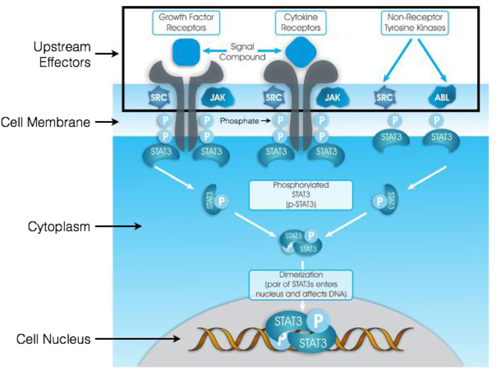
The upstream effectors shown in the above diagram (SRC, JAK and ABL) are just some of those capable of activating STAT3 once they themselves are activated by a variety of signal compounds. The complexity and diversity of pathways capable of activating STAT3 make it very difficult to develop effective drugs that attempt to target the upstream effectors. Furthermore, many of these upstream pathways are necessary for normal healthy cell function, so blocking them indiscriminately can lead to unwanted toxicities.
Published research has identified STAT3 as a master regulator of a wide range of tumors and linked STAT3 activation with the progression of these tumors. For this reason, it is believed that direct inhibition of p-STAT3 may be an effective way to reduce or eliminate the progression of these diseases.
Many research efforts have been directed toward development of specific methods to control activation of STAT3, but most have focused on targeting the upstream effectors of these pathways like growth factors, cytokines, and specific kinases including Janus kinases (JAKs). However, we believe that the multifactorial nature of the activation of STAT3 limits the effectiveness of such upstream approaches. Because the activity of p-STAT3 is a final and determinative step in triggering unwanted transcription, we believe it is preferable to inhibit p-STAT3 more directly and independently from upstream effectors.
We believe the WP1066 Portfolio represents a novel class of agents capable of hitting multiple targets, including p-STAT3, regardless of their upstream method of activation. Numerous preclinical tests involving a wide range of tumor cells suggest that by inhibiting the presence of p-STAT3, WP1066 directly attacks tumor cells. We believe the effectiveness of WP1066 is not only the result of attacking tumors directly, but also indirectly by stimulating the immune system, increasing the patient’s natural ability to fight off tumor development. STAT1 is believed to stimulate T-cell activity and thereby the immune system responsible for fighting tumors. WP1066 has been shown to increase the activity of STAT1 at the same time it inhibits the activity of p-STAT3. We believe this dual activity makes WP1066 a uniquely promising anticancer drug candidate, although we recognize that substantial additional preclinical and clinical research remains to be done, and may not bear out these early results and our optimism.
10
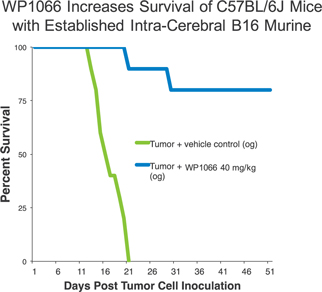
We believe the combination of the direct and indirect effects of WP1066 may ultimately be shown to provide significant tumor suppression and increased survival in a number of in vitro cancer models. Below is one example of an animal model suggesting an increase in survival by treating mice with metastatic melanoma with WP1066.
The WP1122 Portfolio
We have a license agreement with MD Anderson pursuant to which we have been granted a royalty-bearing, worldwide, exclusive license for the patent and technology rights related to our WP1122 Portfolio and similar molecules targeting the treatment of glioblastoma multiforme (GBM) and related central nervous system malignancies.
We believe this technology has the potential to target a wide variety of solid tumors, which eventually become resistant to all treatments, and thereby provide a large and important opportunity for novel drugs. Notwithstanding this potential, we are focused on the treatment of central nervous system malignancies and especially GBM. Although less prevalent than some larger categories of solid tumors, cancers of the central nervous system are particularly aggressive and resistant to treatment. The prognosis for such patients can be particularly grim and the treatment options available to their physicians are among the most limited of any cancer.
The American Cancer Society has estimated 23,770 new cases of brain and other nervous system cancers will occur in the United States in 2016, resulting in 16,050 deaths. Despite the severity and poor prognosis of these tumors, there are few FDA-approved drugs on the market.
We have preliminary preclinical data for WP1122, including in vitro activity against cancer cell lines, as well as data on survival of animals subjected to xenografts of human brain tumors, including data regarding biodistribution and pharmacokinetics. In non-optimal doses and treatment regimes, WP1122 performed equal to or better than the current market leader, temozolomide and provided for superior survival for animals treated in combination with temozolomide. Notwithstanding these early results, we recognize that substantial additional preclinical and clinical research remains to be done, and may not support these initial findings or their translation into activity in humans.
Scientific Rationale for WP1122
Science has recognized that many cancer cells have a unique metabolism, distinct from that of normal cells. Dubbed the “Warburg Effect” because of its discoverer, tumors rely preferentially on glycolysis for the metabolism of glucose, even in the presence of abundant oxygen, for energy (adenosine triphosphate (ATP)) production. This alternative form of energy production makes cancer cells as much as 17 times more dependent on glucose than normal cells.
The fundamental mechanism for imaging actively growing tumors using positron emission tomography (PET scans) is the Warburg Effect. A radiolabeled glucose decoy called F18DG accumulates disproportionately in tumors because of their dramatically increased rate of glucose uptake and accumulation.
11
Researchers have theorized that if a tumor’s access to glucose could be blocked, the tumor could be starved out of existence. Previous attempts at targeting the metabolism of tumor cells have failed due to the rapid metabolism and short half-life (minutes) of the drugs being investigated. Efforts to target tumor metabolism in the brain were further thwarted by the inability to get glycolytic inhibitors into the brain in sufficient/therapeutic amounts due to the presence of what is called the “blood brain barrier.”
We believe WP1122 has the potential for developing a technology platform for enabling increased cellular uptake, increased drug half-life and, importantly, an increased ability to cross the blood brain barrier, enabling greater uptake in the brain. Our approach was inspired by the same principle that distinguishes morphine from heroin. Heroin is chemically the diacetyl ester of morphine. While morphine has a limited ability to cross the blood brain barrier (making it a good candidate for pain killing without impairing mental function), its diacetyl form, heroin, has the ability to accumulate in the brain by 10 to 100 times more than morphine. Once across the blood brain barrier, the acetyl groups are cleaved off by natural enzyme esterases, leaving pure morphine to accumulate in the brain. Similarly, we believe, based on pre-clinical testing, that WP1122, the diacetyl form of a glucose analog and decoy known as “2-DG,” crosses the blood brain barrier where its acetyl groups are cleaved off, allowing the resulting 2-DG to accumulate in the brain at a much higher rate than free 2-DG can do by itself.
Adding to the difficulty in getting free 2-DG across the blood brain barrier in therapeutic quantities is the relatively short half-life of 2-DG. The free form of 2-DG is rapidly metabolized and rendered ineffective within minutes of entering the body. In contrast, WP1122 has a half-life of approximately 6 hours, making it much more feasible to deliver quantities adequate for a therapeutic effect. In addition, we believe WP1122 may represent an improvement to current PET scan diagnostics techniques because of its unique ability to cross the blood brain barrier. These observations are based only on preliminary data and significant additional development is required to determine if these findings are valid and if they will translate into the desired activity in humans.
On October 25, 2016, we announced promising initial results of the preclinical toxicology work on WP1122, our unique inhibitor of glucose metabolism, which is an important driver of glycolytic brain tumor progression and survival. We view this as an important step toward future clinical trials for WP1122. A similar chemical structure to that which turns morphine into heroine has been used to allow WP1122 to successfully enter the brain and increase circulation time. We indicated that preliminary escalating single dose toxicity testing in mice (oral administration) was successfully completed and even at the highest possible dose, no toxic death was observed. In multiple therapeutic doses, WP1122 was well tolerated during intense twice-daily oral dosing.
Overview of the market for our anti-cancer drugs
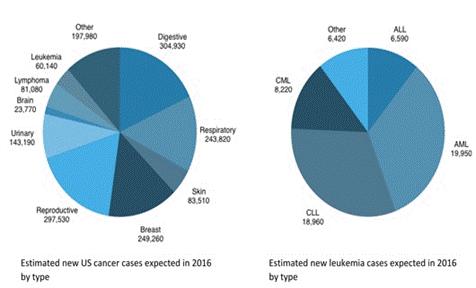
Cancer is the second leading cause of death in the United States behind heart disease. In 2014, an estimated 14.5 million people in the United States were living with a past or current diagnosis of cancer and, in 2016, the National Institutes of Health estimated that nearly 1.7 million new cases will be diagnosed and almost 600,000 Americans will die from cancer.
Source: American Cancer Society - Cancer Facts & Figures 2016
12
Digestive, reproductive, breast and respiratory cancers comprise 65% of expected cancer diagnoses in 2016, while cancers like leukemia and brain tumors are considered “rare diseases”. Leukemia in particular, can be divided into acute, chronic and other, with acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) comprising 26,540 of the estimated 60,140 new cases expected in the United States this year.
The worldwide cancer drug business has been estimated to represent approximately $100 billion in annual sales. Our lead drug, Annamycin, is in a class of drugs referred to as anthracyclines, which are chemotherapy drugs designed to destroy the DNA of targeted cancer cells. The most common approved anthracyclines are daunorubicin and doxorubicin and, prior to the expansion of their generic equivalents, annual revenues generated from anthracyclines have been estimated in the range of $600 million. Acute leukemia is one of a number of cancers that are treated with anthracyclines. One industry report estimates that annual drug revenues generated from the demand for AML-related therapies in the United States, United Kingdom, France, Germany, Italy and Spain were in the range of $151 million in 2012, and we believe that this number may increase if and when improved AML treatments are available.
Our other two active development projects have applications (among others) in the treatment of brain tumors, another rare disease for which there are few available treatments. The leading brain tumor drug is temozolomide, a drug introduced under the brand name Temodar. In 2012, one industry source reported annual revenues of approximately $882 million for Temodar before the expiration of its patent protection, at which point generic versions of the drug began to enter the market and reduce prices.
The Orphan Drug Act and other legislative initiatives provide incentives, including market exclusivity and accelerated approval pathways, for companies that pursue the development of treatments for rare diseases and serious diseases for which there are few or no acceptable available treatment alternatives. Over the last 10 years, an increasing number of companies have begun using these designations to obtain new drug approvals for drugs where patent coverage has expired and/or where accelerated approval appears possible. An IMS Health report estimated that, in 2013, the sale of drugs with full or partial Orphan Drug exclusivity represented approximately $29 billion in revenue. We consider obtaining Orphan Drug exclusivity and accelerated approval to be an important part of our development strategy for our drug candidates. Notwithstanding these potential opportunities, we can provide no assurance that our drugs will receive Orphan Drug designation (other than Annamycin, which recently received such designation) or, if approved, exclusivity or any other special designation that could, among other things, provide for accelerated approval.
13
Our License Agreements
We acquired the rights and obligations under the Patent and Technology License Agreement entered into by and between IntertechBio and MD Anderson dated April 2, 2012 (the “IntertechBio Agreement”). Pursuant to that license agreement and an October 2015 amendment, IntertechBio obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to our WP1122 Portfolio and to our drug product candidate, WP1122. In consideration, IntertechBio agreed to make payments to MD Anderson, including an up-front payment, license documentation fee, annual maintenance fee, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. Specifically, under the IntertechBio Agreement, IntertechBio agreed to pay a nonrefundable upfront documentation fee in the amount of $80,000; annual maintenance fee in the amount of $10,000 on the first anniversary of the effective date of the IntertechBio Agreement, $20,000 on the second anniversary of the effective date of the agreement, $40,000 on the third anniversary of the effective date of the agreement, $60,000 on the fourth anniversary of the effective date of the agreement, $80,000 on the fifth anniversary of the effective date of the agreement and $100,000 on the sixth anniversary of the effective date of the agreement and every anniversary thereafter, except that such payments will no longer be due upon the first sale of a licensed product. Under the IntertechBio Agreement, IntertechBio also agreed to make a minimum annual royalty in the amount of $200,000 for the first anniversary following the first sale of a licensed product, $400,000 for the second anniversary following the first sale of a licensed product, and $600,000 for the third year following the first sale of a licensed product and every anniversary thereafter. IntertechBio also agreed to make one-time milestone payments in the amount of $200,000 upon commencement of a Phase II study for a licensed product, $250,000 upon commencement of a Phase III study for a licensed product, $400,000 upon filing of a New Drug Application (“NDA”) for a licensed product and $500,000 upon receipt of market approval for sale of a licensed product. MD Anderson has the right to terminate the agreement upon advance notice in the event of a default by IntertechBio. The agreement will also be terminated immediately upon IntertechBio’s insolvency. Additionally, MD Anderson has the right to terminate the license agreement if (i) a preclinical toxicology program for a licensed product is not initiated within one year of the of effective date of the amendment, (ii) an investigational new drug application (IND) is not filed with the Food and Drug Administration (FDA) for a Phase I study for a licensed product within three years of the effective date of the amendment, or (iii) a Phase I study for a licensed product is not commenced within five years of the effective date of the amendment. The IntertechBio Agreement will expire upon the expiration of the licensed intellectual property. In August 2015, the IntertechBio Agreement and amendments thereto were assigned to MBI. Under the assignment, MBI has assumed the rights and obligations of IntertechBio including, without limitation, the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. However, the rights obtained pursuant to the assignment of the IntertechBio Agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by MBI. MBI is not required to issue MD Anderson any equity upon the completion of any milestones. On October 8, 2015, IntertechBio entered into a letter agreement with MD Anderson where MD Anderson agreed to receive past due maintenance fees and patent expenses of $98,108 owed by IntertechBio in four installments. The past due amount is related to certain metabolic inhibitor technology license that was assigned to MBI by IntertechBio and was owed by IntertechBio prior to MBI’s acquisition of the license. Pursuant to the letter, IntertechBio Corporation also agreed to pay $65,504 patent fees to a law firm. In order to have the license in good standing, MBI agreed to pay MD Anderson the $98,108 and the $65,504 to a law firm on behalf of IntertechBio Corporation; all such payments have been made in full by MBI.
Due to our acquisition of Moleculin, LLC, we obtained a royalty-bearing, worldwide, exclusive license to intellectual property rights, including patent rights related to our WP1066 drug product candidate from MD Anderson. Moleculin, LLC entered into a June 2010 Patent and Technology License Agreement with MD Anderson (the “Moleculin Agreement”). Under the Moleculin Agreement and an October 2015 amendment, Moleculin, LLC obtained the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. In consideration, Moleculin, LLC agreed to make payments to MD Anderson including an up-front payment, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. Specifically, under the Moleculin Agreement, Moleculin, LLC agreed to pay a nonrefundable upfront documentation fee in the amount of $223,585; annual maintenance fee in the amount of $20,000 on the first anniversary of the effective date of the Moleculin Agreement, which shall increase in $10,000 increments on an annual basis thereafter up to a maximum of $100,000, except that such payments will no longer be due upon marketing approval in any country of a licensed product. Under the Moleculin Agreement, Moleculin, LLC also agreed to make a minimum annual royalty payment to MD Anderson in the amount of $200,000 after the first sale of a licensed product. Moleculin, LLC also agreed to make one-time milestone payments in the amount of $150,000 upon commencement of the first Phase III study for a licensed product within the United States, Europe, China or Japan; $500,000 upon submission of the first NDA for a licensed product in the United States; and $600,000 upon receipt of the first marketing approval for sale of a licensed product in the United States. MD Anderson has the right to terminate the Moleculin Agreement upon advanced notice in the event of a default by Moleculin, LLC. Moleculin, LLC has the right to terminate the Moleculin Agreement for any reason upon advance written notice to MD Anderson. The Moleculin Agreement will also be terminated immediately upon Moleculin, LLC’s insolvency. Per the October 2015 amendment to the Moleculin Agreement, MD Anderson relinquished any rights to any equity previously due MD Anderson in Moleculin, LLC. Upon completion of our acquisition of Moleculin, LLC, we assumed the rights and obligations of Moleculin, LLC including, without limitation, the right to manufacture, have manufactured, use, import, offer to sell and/or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. However, the rights we have obtained pursuant to the assignment of the Moleculin Agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by us. MBI is not required to issue MD Anderson any equity in our company upon the completion of any milestones.
14
On October 8, 2015, Moleculin, LLC entered into a letter agreement with MD Anderson for Moleculin, LLC’s past due fees to MD Anderson in the amount of $691,186 of which $300,000 had been paid prior to the letter agreement. Pursuant to the letter agreement, MD Anderson agreed to receive the remaining past due fee in three installments, which payments have been made in full.
Moleculin, LLC out-licensed certain intellectual property rights including rights covering the WP1066 drug product candidate to Dermin (“Moleculin Out-License Agreement”). The licensed intellectual property includes rights obtained by Moleculin, LLC pursuant to a license agreement with MD Anderson (“Moleculin-MD Anderson Agreement”). Under the Moleculin Out-License Agreement, Moleculin, LLC granted Dermin a royalty-bearing, exclusive license to manufacture, have manufactured, use, import, offer to sell and/or sell products in the field of human therapeutics under the licensed intellectual property in the countries of Belarus, Czech Republic, Estonia, Hungary, Latvia, Lithuania, Poland, Romani, Slovakia and Ukraine (“licensed territories”). Additionally, Moleculin, LLC agreed to develop and provide a dossier containing data related to the licensed subject matter to Dermin. In consideration, Dermin agreed to make payments to Moleculin, LLC, including upfront development fees, annual royalty payments, sublicense fee, and milestone payments. Specifically, under the Moleculin Out-License Agreement, Dermin agreed to make a nonrefundable upfront dossier development payment in the amount of $100,000; a service fee in the amount of $100,000 for assistance provided to Dermin in securing additional funding; and royalty payments on sales of any licensed product at a rate of no less than the royalty rate due to MD Anderson under the Moleculin-MD Anderson Agreement plus 1%. Dermin also agreed to provide a percentage of certain consideration Dermin receives pursuant to sublicense agreements in the amount of 25% prior to completion of a Phase IIb clinical study in a licensed territory and 10% on or after completion of a Phase IIb clinical study in a licensed territory, provided, however, if the sublicense fee is less than the sublicense fee due to MD Anderson under the Moleculin-MD Anderson Agreement, then Dermin shall be obligated to pay no less than the amount due to MD Anderson plus 5%. Also under the Moleculin Out-License Agreement, Dermin agreed to pay all out-of-pocket expenses incurred by Moleculin, LLC in filing, prosecuting and maintaining the licensed patents for which the license has been granted. The parties to the Moleculin Out-License Agreement each have the right to terminate the agreement upon advance notice in the event of a default by the other party. Dermin has the right to terminate the agreement if (i) Moleculin, LLC fails to timely provide the dossier to Dermin after Moleculin, LLC’s filing of an NDA for a licensed product in the United States; or (ii) Moleculin, LLC does not cooperate in assisting Dermin to secure funds to develop the licensed subject matter. Upon completion of our acquisition of Moleculin, LLC, we assumed the rights and obligations of Moleculin, LLC under the Moleculin Out-License Agreement.
We acquired the rights and obligations to the Patent and Technology License Agreement entered into by and between IntertechBio and Dermin dated April 15, 2011 (the “IntertechBio Out-License Agreement”). Pursuant to that license agreement, IntertechBio exclusively out-licensed intellectual property rights to Dermin, including rights covering the WP1122 drug product candidate obtained from MD Anderson pursuant to the IntertechBio Agreement. Under the IntertechBio Out-License Agreement, IntertechBio granted Dermin a royalty-bearing, exclusive license to manufacture, have manufactured, use, import, offer to sell and/or sell products in the field of human therapeutics under the licensed intellectual property in the countries of Belarus, Russia, Kazakhstan, Uzbekistan, Turkmenistan, Czech Republic, Estonia, Hungary, Latvia, Lithuania, Poland, Romania, Slovakia and Ukraine (“licensed territories”). Additionally, IntertechBio agreed to develop and provide a dossier containing data related to the licensed subject matter to Dermin. In consideration, Dermin agreed to make payments to IntertechBio, including upfront development fees, annual royalty payments, sublicense fees, and milestone payments. Specifically, under the IntertechBio Out-licensing Agreement, Dermin agreed to make a nonrefundable upfront dossier development fee in the amount of $35,000; a service fee in the amount of $40,000 for assistance provided to Dermin in securing additional funding; and royalty payments on sales of any licensed product at a rate of no less than the royalty rate due to MD Anderson under the IntertechBio Agreement plus 2%. Dermin also agreed to provide a percentage of certain consideration Dermin receives pursuant to sublicense agreements in the amount of 25% prior to completion of a Phase IIb clinical study in a licensed territory and 10% on or after completion of a Phase IIb clinical study in the licensed territories, provided, if the sublicense fee is less than the sublicense fee due to MD Anderson under the IntertechBio Agreement, then Dermin shall be obligated to pay not less than the amount due to MD Anderson plus 5%. Also under the IntertechBio Out-Licensing Agreement, Dermin agreed to pay all out-of-pocket expenses incurred by IntertechBio in filing, prosecution and maintaining the licensed patents for which the license has been granted. The parties to the IntertechBio Out-licensing Agreement each have the right to terminate the agreement upon advanced notice in the event of a default by the other party. Dermin has the right to terminate the agreement if (i) IntertechBio fails to timely provide the dossier to Dermin after IntertechBio’s filing of an NDA for a licensed product in the United States; or (ii) IntertechBio does not cooperate in assisting Dermin to secure funds to develop the licensed subject matter.
15
We entered into a May 2016 out-licensing agreement with HPI, pursuant to which we granted HPI certain intellectual property rights, including rights covering the potential drug candidate, WP1066 (“HPI Out-Licensing Agreement”). Under the HPI Out-Licensing Agreement we are required to make an upfront $100,000 payment and quarterly payments in the amount of $37,500 for the first four quarters following the effective date of the HPI Out-Licensing Agreement and $75,000 per quarter for the following eight quarters thereafter in consideration for the right to development data related to the development of licensed products. Notwithstanding our obligation to make the foregoing payments, the HPI Out-Licensing Agreement does not obligate HPI to conduct any specific research or to meet any milestones. Upon payment in the amount of $1,000,000 to HPI within three years of the effective date of the HPI Out-Licensing Agreement we will regain all rights to the licensed subject matter and rights to any and all development data and any regulatory submissions including any IND, NDA or ANDA related to the licensed subject matter. In the event that we do not exercise our right to regain our rights to the licensed subject matter within three years of the effective date of the HPI Out-Licensing Agreement the license granted to HPI shall convert to an exclusive license upon HPI’s written notice and we shall be obligated to transfer all existing data relating to licensed subject matter including any development data and any IND to HPI.
Annamed out-licensed certain intellectual property rights including rights covering the potential drug product, Annamycin to Dermin pursuant to a Patent and Technology Development and License Agreement dated June 28, 2012 (the “Annamed Agreement”). The licensed intellectual property includes rights obtained by Annamed pursuant to a license agreement with MD Anderson (“Annamed-MD Anderson Agreement”). Under the Annamed Agreement, Annamed granted Dermin a royalty-bearing, exclusive license to manufacture, have manufactured, use, import, offer to sell and/or sell products in the field of human therapeutics under the licensed intellectual property in the countries of Poland, Ukraine, Czech Republic, Hungary, Romania, Slovakia, Belarus, Lithuania, Latvia, Estonia, Netherlands, Turkey, Belgium, Switzerland, Austria, Sweden, Greece, Portugal, Norway, Denmark, Ireland, Finland, Luxembourg, Iceland, Kazachstan, Russian Federation, Uzbekistan, Georgia, Armenia, Azerbaijan and Germany (“Annamed licensed territories”). Additionally, Annamed agreed to develop and provide a dossier containing data related to the licensed subject matter to Dermin. In consideration, Annamed agreed to provide a running royalty at a rate of no less than the royalty due to MD Anderson under the Annamed-MD Anderson Agreement plus 2.5% for the sale of any licensed product in the Annamed licensed territories excluding Germany and at a rate of 15% for the sale of any licensed product in Germany. Dermin also agreed to provide a percentage of certain consideration Dermin receives pursuant to sublicense agreements in the amount of 25% prior to completion of a Phase IIb clinical study in an Annamed licensed territory and 10% on or after completion of a Phase IIb clinical study in an Annamed licensed territory, provided, however, if the sublicense fee is less than the sublicense fee due to MD Anderson under the Annamed-MD Anderson Agreement, then Dermin shall be obligated to pay no less than the amount due to MD Anderson plus 5%. Also under the Annamed Agreement, Dermin agreed to pay all out-of-pocket expenses incurred by Annamed in filing, prosecuting and maintaining the licensed patents for which the license has been granted. Annamed has the right to terminate the Annamed Agreement as to any country within the Annamed licensed territories if Dermin fails to provide evidence of its use of commercially reasonable efforts to commercialize a licensed product in such country within ninety days of Annamed’s written request. The Annamed Agreement may also be terminated by Annamed upon advanced written notice in the event that Dermin defaults. Dermin has the right to terminate the agreement if (i) Annamed fails to timely provide the documents and information required for Dermin to prepare a dossier within 30 days of Annamed’s filing of an NDA for a licensed product in the United States; or (ii) Annamed does not cooperate in assisting Dermin to secure funds to develop the licensed subject matter. As of August 2015, we have obtained the rights and obligations of Annamed under the Annamed Agreement.
Competition
We operate in a highly competitive segment of the pharmaceutical market, which market is highly competitive as a whole. We face competition from numerous sources including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies, and private and public research institutions. Many of our competitors may have significantly greater financial, product development, manufacturing and marketing resources. Additionally, many universities and private and public research institutes are active in cancer research, and some may be in direct competition with us. We may also compete with these organizations to recruit scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
16
The unmet medical need for more effective cancer therapies is such that anticancer drugs are, by far, the leading class of drugs in development. These include a wide array of products against cancer targeting many of the same indications as our drug candidates. While the introduction of newer targeted agents may result in extended overall survival, induction therapy regimens are likely to remain a cornerstone of cancer treatment in the foreseeable future.
There are a number of established therapies that may be considered competitive for the cancer indications for which we intend to develop our lead product, Annamycin. A key consideration when treating AML patients is whether the patient is suitable for intensive therapy. The standard of care for the treatment of newly diagnosed AML patients who can tolerate intensive therapy is cytarabine in combination with an anthracycline (e.g., doxorubicin or daunorubicin). For some patients, primarily those less than 60 years of age, a stem cell transplant could also be considered if the induction regimen is effective in attaining a CR (Complete Response). The regimen of cytarabine in combination with an anthracycline has been the standard of care for decades. A patient not suitable for intensive therapy may be offered the option for low-intensity therapy such as low-dose cytarabine, azacitidine or decitabine. It should be noted that, in the US, these are not approved by the FDA for the treatment of AML patients and there remains no effective therapy for these patients or for relapsed or refractory AML. The initial focus for Annamycin development is in patients for whom the standard induction regimen has failed. Also, several major pharmaceutical companies and biotechnology companies are aggressively pursuing new cancer development programs for the treatment of AML.
A number of attempts have been made or are under way to provide an improved treatment for AML. Recently, Celator Pharmaceuticals reported Phase III clinical trial results for a new combined formulation of cytarabine and daunorubicin (commonly used induction therapy drugs) they call Vyxeos. This new liposome formulation provides a 5:1 ratio of cytarabine and daunorubicin in each of three injections. When compared with patients receiving 7 injections of cytarabine and 3 injections of daunorubicin (traditional 7+3 induction therapy), patients receiving Vyxeos achieved an average increase in overall survival of approximately 3.5 months (9.5 months compared with 6 months). Despite this extension of overall survival, Vyxeos did not reduce the toxic side effects of daunorubicin (including cardiotoxicity) and it failed to qualify a significant majority of patients for curative bone marrow transplant.
Drugs attempting to target a subset of AML patients who present with particular anomalies involving a gene referred to as FLT3 are currently in clinical trials, as well as other approaches that include immunotherapy relying on other biomarkers, other attempts at improved chemotherapy and alternative approaches to radiation therapy. Other approaches to improve the effectiveness of induction therapy are in early stage clinical trials and, although they do not appear to address the underlying problems with anthracyclines, we can provide no assurance that such improvements, if achieved, would not adversely impact the need for improved anthracyclines. A modified version of doxorubicin designed to reduce cardiotoxicity is in clinical trials for the treatment of sarcoma and, although this drug does not appear to address multidrug resistance and is not currently intended for the treatment of acute leukemia, we can provide no assurance that it will not become a competitive alternative to Annamycin. Although we are not aware of any other single agent therapies in clinical trials that would directly compete against Annamycin in the treatment of relapsed and refractory AML, we can provide no assurance that such therapies are not in development, will not receive regulatory approval and will reach market before our drug candidate Annamycin. In addition, any such competing therapy may be more effective and / or cost-effective than ours.
Government Regulation
Government authorities in the US, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. The pharmaceutical drug product candidates that we develop must be approved by the FDA before they may be marketed and distributed.
17
In the United States, the FDA regulates pharmaceutical products under the Federal Food, Drug, and Cosmetic Act, and implementing regulations. Pharmaceutical products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA and related enforcement activity could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a pharmaceutical product may be marketed in the US generally involves the following:
· Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations;
· Submission to the FDA of an Investigational New Drug application, or IND, which must become effective before human clinical studies may begin;
· Performance of adequate and well-controlled human clinical studies according to the FDA’s current good clinical practices (“GCP”), to establish the safety and efficacy of the proposed pharmaceutical product for its intended use;
· Submission to the FDA of an NDA for a new pharmaceutical product;
· Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the pharmaceutical product is produced, to assess compliance with current good manufacturing practices (“cGMP”), to assure that the facilities, methods and controls are adequate to preserve the pharmaceutical product’s identity, strength, quality and purity;
· Potential FDA audit of the preclinical and clinical study sites that generated the data in support of the NDA; and
· FDA review and approval of the NDA.
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources and approvals, and continued compliance are inherently uncertain.
Before testing any compounds with potential therapeutic value in humans, the pharmaceutical product candidate enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the pharmaceutical product candidate. These early proof-of-principle studies are done using sound scientific procedures and thorough documentation. The conduct of the single and repeat dose toxicology and toxicokinetic studies in animals must comply with federal regulations and requirements including good laboratory practices. The sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA has concerns and notifies the sponsor. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. If resolution cannot be reached within the 30-day review period, either the FDA places the IND on clinical hold or the sponsor withdraws the application. The FDA may also impose clinical holds on a pharmaceutical product candidate at any time before or during clinical studies for various reasons. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical studies to begin, or that, once begun, issues will not arise that suspend or terminate such clinical study.
18
Clinical studies involve the administration of the pharmaceutical product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the clinical study sponsor’s control. Clinical studies are conducted under protocols detailing, among other things, the objectives of the clinical study, dosing procedures, subject selection and exclusion criteria, how the results will be analyzed and presented and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA as part of the IND. Clinical studies must be conducted in accordance with GCP. Further, each clinical study must be reviewed and approved by an independent institutional review board (“IRB”) at, or servicing, each institution at which the clinical study will be conducted. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical studies are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical study subject or his or her legal representative and must monitor the clinical study until completed.
Human clinical studies are typically conducted in three sequential phases that may overlap or be combined:
· Phase 1: The pharmaceutical product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases such as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients, with a goal of characterizing the safety profile of the drug and establishing a maximum tolerable dose (“MTD”). Our pharmaceutical products fall into this latter category because its products are intended to treat cancer and contain cytotoxic agents. Hence, our Phase 1 studies are conducted in late-stage cancer patients whose disease has progressed after treatment with other agents.
· Phase 2: With the maximum tolerable dose established in a Phase 1 trial, the pharmaceutical product is evaluated in a limited patient population at the MTD to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases, to determine dosage tolerance, optimal dosage and dosing schedule and to identify patient populations with specific characteristics where the pharmaceutical product may be more effective.
· Phase 3: Clinical studies are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These clinical studies are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. The studies must be well controlled and usually include a control arm for comparison. One or two Phase 3 studies are usually required by the FDA for an NDA approval, depending on the disease severity and other available treatment options. In some instances, an NDA approval may be obtained based on Phase 2 clinical data with the understanding that the approved drug can be sold subject to a confirmatory trial to be conducted post-approval.
Post-approval studies, or Phase 4 clinical studies, may be conducted after initial marketing approval. These studies are often used to gain additional experience from the treatment of patients in the intended therapeutic indication. The FDA also may require Phase 4 studies, Risk Evaluation and Mitigation Strategies (“REMS”) and post-marketing surveillance, among other things, to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical studies may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical study at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the pharmaceutical product has been associated with unexpected serious harm to patients.
19
Concurrent with clinical studies, companies may complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the pharmaceutical product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the pharmaceutical product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final pharmaceutical product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the pharmaceutical product candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical studies, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the pharmaceutical product, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees. A waiver of such fees may be obtained under certain limited circumstances.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act (“PDUFA”), the FDA has 10 months after the 60-day filing date in which to complete its initial review of a standard review NDA and respond to the applicant, and six months after the 60-day filing date for a priority review NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs.
After the NDA submission is accepted for filing, the FDA reviews the NDA application to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel pharmaceutical products or pharmaceutical products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the pharmaceutical product approval process, the FDA also will determine whether a risk evaluation and mitigation strategy (“REMS”) is necessary to assure the safe use of the pharmaceutical product. If the FDA concludes that a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without a REMS, if required.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites as well as the site where the pharmaceutical product is manufactured to assure compliance with GCP and cGMP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. In addition, the FDA will require the review and approval of product labeling.
The NDA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical studies are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA. The complete response letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical studies. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical studies designed to further assess pharmaceutical product safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
20
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new pharmaceutical products that meet certain criteria. Specifically, new pharmaceutical products are eligible for Fast Track designation if they are intended to treat a serious condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, if the FDA determines that the schedule is acceptable and if the sponsor pays any required user fees upon submission of the first section of the NDA.
Any product submitted to the FDA for market, including a Fast Track program, may also be eligible for other FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it is intended to treate a serious condition and it offers a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new pharmaceutical product designated for priority review in an effort to facilitate the review. Additionally, accelerated approval may be available for a product intended to treat a serious condition that provides meaningful therapeutic benefit over existing treatments, which means the product may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on an intermediate clinical endpoint. As a condition of accelerated approval, the FDA may require the sponsor to perform adequate and well-controlled post-marketing clinical studies. In addition, the FDA currently requires pre-approval of promotional materials for products receiving accelerated approval, which could impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-Approval Requirements
Any pharmaceutical products for which the Company receives FDA approvals are subject to continuing regulation by the FDA, including, among other things, cGMP compliance, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, prohibitions on promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, actions by the U.S. Department of Justice and/or U.S. Department of Health and Human Services’ Office of Inspector General, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available pharmaceutical products for off-label uses, manufacturers may not directly or indirectly market or promote such off-label uses.
We rely and expect to continue to rely on third parties for the production of clinical and commercial quantities of our products. Manufacturers of our products are required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. cGMP regulations require, among other things, quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Pharmaceutical product manufacturers and other entities involved in the manufacture and distribution of approved pharmaceutical products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
21
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of the use of our pharmaceutical product candidates, some of our products covered by U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process for a product the approval of which is the first permitted commercial marketing of the active pharmaceutical ingredient. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved pharmaceutical product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent unless an extension is obtained. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and renders a decision on the application for any patent term extension or restoration. In the future, we may be able to apply for extension of patent term for one or more of our currently licensed patents or any future owned patents to add patent life beyond its current expiration date, depending upon the expected length of the clinical studies and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the U.S. Food, Drug, and Cosmetic Act can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA or seeking approval for a similar product. Pediatric exclusivity adds six months to existing exclusivity periods and patent terms and may be granted based on the completion of a pediatric clinical study that “fairly responds” to an FDA-issued “Written Request” for such a clinical study.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical product candidates for which we may obtain regulatory approval. In the United States and in markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part upon the availability of reimbursement from third-party payers. Third-party payers include government payers such as Medicare and Medicaid, managed care providers, private health insurers and other organizations. The process for determining whether a payer will provide coverage for a pharmaceutical product may be separate from the process for setting the price or reimbursement rate that the payer will pay for the pharmaceutical product. Third-party payers may limit coverage to specific pharmaceutical products on an approved list, or formulary, which might not, and frequently does not, include all of the FDA-approved pharmaceutical products for a particular indication. Third-party payers are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. A payer’s decision to provide coverage for a pharmaceutical product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. In addition, in the United States there is a growing emphasis on comparative effectiveness research, both by private payers and by government agencies. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our pharmaceutical product candidates may not be considered medically necessary or cost-effective. To the extent other drugs or therapies are found to be more effective than our products, payers may elect to cover such therapies in lieu of our products and/or reimburse our products at a lower rate.
Orphan Drug exclusivity prevents for seven years the approval of another product with the same active moiety for the same rare disease. If a product is a new chemical entity (i.e., generally that the moiety has not previously been approved), it may receive five years of exclusivity, during which period FDA may not accept for review certain NDAs for another product with the same moiety. If approval of a product required new clinical data, it may convey three years of exclusivity against approval of certain NDAs for similar products.
22
Different pricing and reimbursement schemes exist in other countries. In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed upon. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical studies that compare the cost-effectiveness of a particular pharmaceutical product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any pharmaceutical product candidates for which we may receive regulatory approval for commercial sale may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect this will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we may receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
International Regulation
In addition to regulations in the United States, there are a variety of foreign regulations governing clinical studies and commercial sales and distribution of our current and future product candidates. Whether or not FDA approval is obtained for a product, approval of a product must be obtained by the comparable regulatory authorities of foreign countries, or jurisdictions such as the EU, before clinical studies or marketing of the product can commence in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical studies, product licensing, pricing and reimbursement vary greatly from country to country. In addition, certain regulatory authorities may require us to repeat previously conducted preclinical and/or clinical studies under specific criteria for approval in their respective country or jurisdictions, which may delay and/or increase the cost of approval in certain markets targeted for approval by us.
Subsequent Event
On February 9, 2017, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with Roth Capital Partners, LLC, as representative of the several underwriters identified therein (collectively, the “Underwriters”), pursuant to which we sold in a registered public offering (the “Offering”), 3,710,000 units, priced at a public offering price of $1.35 per unit, with each unit consisting of: (i) one share of common stock, (ii) a five-year Series A warrant to purchase 0.50 of a share of common stock, (iii) a 90-day Series B warrant to purchase one share of common stock, and (iv) a five-year Series C warrant to purchase 0.50 of a share of common stock. The Series C warrants in a unit may only be exercised to the extent and in proportion to a holder of the Series C warrants exercising its Series B warrants included in the unit. The Series A and Series C warrant have an exercise price of $1.50 per share of common stock. The Series B warrant has an exercise price of $1.35 per share of common stock.
Under the terms of the Underwriting Agreement, we granted the Underwriters a 45-day option to purchase an additional 556,500 shares of common stock and/or an additional 556,500 warrants combinations (comprised of an aggregate of 278,250 Series A warrants, 556,500 Series B warrants and 278,250 Series C warrants), in any combinations thereof, from us to cover over-allotments at the public offering price per share of $1.349 and public offering price per warrant combination of $.001, respectively, less the underwriting discounts and commissions. Upon the closing of the Offering, the Underwriters exercised the over-allotment option with respect to 278,100 warrant combinations. We received approximately $4.4 million in net proceeds from the Offering, after deducting underwriting discounts and commissions and estimated offering expenses.
23
Subsequent to March 22, 2017, investors in the aforementioned offering on February 9, 2017, have excerised approximately 600,000 Series B warrants resulting in approximately gross proceeds to the Company of $800,000 and approximately 600,000 shares of the Company’s common stock being issued.
Employees
As of December 31, 2016, we had two full-time employees and four part-time employees, and accordingly, a high percentage of the work performed for our development projects is outsourced to qualified independent contractors.
Legal Proceedings
We are not subject to any litigation.
The following risks and uncertainties should be carefully considered. If any of the following occurs, our business, financial condition or operating results could be materially harmed. An investment in our securities is speculative in nature, involves a high degree of risk and should not be made by an investor who cannot bear the economic risk of its investment for an indefinite period of time and who cannot afford the loss of its entire investment.
Risks Relating to Our Business
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We intend to use the proceeds from our previous offerings, as well as the proceeds any possible future offerings, to, among other uses, advance Annamycin through clinical development. Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We will require substantial additional future capital in order to complete clinical development and commercialize Annamycin. If the FDA requires that we perform additional nonclinical studies or clinical trials, or if we determine, as we did in October 2016, that additional clinical trials are required for Annamycin, our expenses would further increase beyond what we currently expect and the anticipated timing of any potential approval of Annamycin would likely be delayed. In addition, if our current plan to obtain the required CMC data to be included with our IND application for Annamycin is unsuccessful, we will be required to incur unexpected costs and a delay in submiting our IND application. Further, there can be no assurance that the costs we will need to incur to obtain regulatory approval of Annamycin will not increase.
We will continue to require substantial additional capital to continue our clinical development and commercialization activities. Because successful development of our product candidates is uncertain, we are unable to estimate the actual amount of funding we will require to complete research and development and commercialize our products under development.
The amount and timing of our future funding requirements will depend on many factors, including but not limited to:
· whether our updated plan for clinical trials will be completed on a timely basis and, if completed, expect to publicly announce results from our phase I/II clinical trial until sometime in 2018;
· whether we are successful in obtaining the benefits of FDA’s expedited development and review programs related to Annamycin;
· the progress, costs, results of and timing of our clinical trials for Annamycin;
· the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals;
· the costs associated with securing and establishing commercialization and manufacturing capabilities;
· market acceptance of our product candidates;
24
· the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies;
· our ability to maintain, expand and enforce the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights;
· our need and ability to hire additional management and scientific and medical personnel;
· the effect of competing drug candidates and new product approvals;
· our need to implement additional internal systems and infrastructure, including financial and reporting systems; and
· the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future.
Some of these factors are outside of our control. Based upon our currently expected level of operating expenditures, we believe that we will be able to fund our operational plan into the first quarter of 2018, assuming not a significant amount of the warrants in our recent public offering are execised for cash. This period could be shortened if there are any significant increases in planned spending on development programs or more rapid progress of development programs than anticipated. We do not believe that our existing capital resources are sufficient to enable us to complete the development and commercialization of Annamycin, if approved, or to initiate any clinical trials or additional development work needed for any other drug candidates. Accordingly, we expect that we will need to raise additional funds in the future.
We may seek additional funding through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. Additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. In addition, the issuance of additional shares by us, or the possibility of such issuance, may cause the market price of our shares to decline.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our research or development programs. We also could be required to seek funds through arrangements with collaborative partners or otherwise that may require us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us.
We have in the past completed related party transactions that were not conducted on an arm’s length basis.
We acquired the rights to the license agreement with MD Anderson covering our WP1122 Portfolio held by IntertechBio Corporation, a company affiliated with certain members of our management and board of directors. We acquired the rights to all data related to the development of Annamycin held by AnnaMed, Inc., a company affiliated with certain members of our management and board of directors. Prior to our IPO, Moleculin, LLC merged with and into our company. Moleculin, LLC was affiliated with certain members of our management and board of directors. Prior to our IPO, we, on Moleculin, LLC’s behalf, entered into an agreement with HPI whereby HPI agreed to terminate its option to sublicense certain rights to the WP1066 Portfolio and entered into a co-development agreement with us. Our largest shareholder and a member of our management are shareholders of HPI. None of the foregoing transactions were conducted on an arm’s length basis. As such, it is possible that the terms were less favorable to us than in an arm’s length transaction.
25
Our ability to retain the development rights to the WP1066 Portfolio will require us to make up to a total of $1.75 million in future payments to HPI, in addition to payments of shares of our common stock and cash made in connection with our IPO, pursuant to the development agreement we entered into with HPI.
Our acquisition of Moleculin, LLC prior to our IPO provided us with the rights to the license agreement Moleculin, LLC had with MD Anderson covering the WP1066 Portfolio. However, Moleculin, LLC previously granted HPI an option to obtain an exclusive sub-license to develop the WP1066 Portfolio in all non-dermatological fields. Prior to our IPO, we, on Moleculin, LLC’s behalf, entered into two agreements with HPI. The first agreement terminated HPI’s option to obtain the aforementioned exclusive sublicense in exchange for a payment of $100,000 and the issuance of 629,000 shares of our common stock. The second agreement, the HPI Out-Licensing Agreement is a technology rights and development license agreement that provided HPI with a non-exclusive sublicense to develop the WP1066 Portfolio. Pursuant to this HPI Out-Licensing Agreement, we agreed to make payments to HPI of $750,000 over a three-year period commencing after our IPO in exchange for HPI allowing us to access any data, information or know-how resulting from the research and development conducted by HPI, which payments will be expensed when incurred. Notwithstanding our obligation to make the foregoing payments, the HPI Out-Licensing Agreement does not obligate HPI to conduct any specific research or to meet any milestones. Pursuant to the HPI Out-Licensing Agreement, we have the right within three years of the date we entered into the agreement to buy-out from HPI all rights granted to HPI under the agreement for a payment of $1.0 million. If we do not exercise the foregoing buy-out right within three years, the license granted to HPI shall convert into an exclusive license even as to our company. As such, if we do not exercise the buy-out right for any reason, we will no longer have access to the non-dermatology uses of the WP1066 Portfolio and all amounts paid to HPI prior to such date will have value only to the extent that the data, information and know-how may be applicable to dermatology applications of the WP1066 Portfolio. We do not expect to maintain a reserve of $1.0 million to exercise the buy-out payment and, as such, we will need to raise additional funds to make the buy-out payment. We cannot assure you that such additional funding, if required, will be available on satisfactory terms, or at all.
We have never been profitable, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to reduce our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have not yet submitted any drug candidates for approval by regulatory authorities in the United States or elsewhere. For the year ended December 31, 2016, we incurred a net loss of $3,926,361. We had an accumulated deficit of $4,674,721 as of December 31, 2016.
To date, we have devoted most of our financial resources to research and development, including our drug discovery research, preclinical development activities and clinical trial preparation, as well as corporate overhead. We have not generated any revenues from product sales. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for Annamycin, prepare for and begin the commercialization of any approved products, and add infrastructure and personnel to support our continuing product development efforts. We anticipate that any such losses could be significant for the next several years. If Annamycin or any of our other drug candidates fail in clinical trials or does not gain regulatory approval, or if our drug candidates do not achieve market acceptance, we may never become profitable. As a result of the foregoing, we expect to continue to experience net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders' equity and working capital.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. In addition, our expenses could increase if we are required by the FDA to perform studies or trials in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our drug candidates. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues.
Our financial condition would be adversely impacted if our intangible assets become impaired.
As a result of the accounting for our acquisition of Moleculin, LLC and the agreement we, on Moleculin, LLC’s behalf, entered into with Houston Pharmaceuticals, Inc., we have carried on our balance sheet within intangible assets in-process research and development (“IPR&D”) of $11,147,540 as of December 31, 2016. Intangibles are evaluated quarterly and are tested for impairment at least annually or when events or changes in circumstances indicate the carrying value of each segment, and collectively our company taken as a whole, might exceed its fair value. We have retained a third party valuation firm to provide us with a valuation of these intangible assets and have incorporated their final report results in this filing of our Form 10-K for the year ended December 31, 2016.
26
Intangible assets related to IPR&D are considered indefinite-lived intangible assets and are assessed for impairment annually or more frequently if impairment indicators exist. If the associated research and development effort is abandoned, the related assets will be written-off and the Company will record a noncash impairment loss on its statement of operations. For those compounds that reach commercialization, if any, the IPR&D assets will be amortized over their estimated useful lives.
If we determine that the value of our intangible assets is less than the amounts reflected on our balance sheet, we will be required to reflect an impairment of our intangible assets in the period in which such determination is made. An impairment of our intangible assets would result in our recognizing an expense in the amount of the impairment in the relevant period, which would also result in the reduction of our intangible assets and a corresponding reduction in our stockholders’ equity in the relevant period. As the transactions discussed above were related party transactions and were not conducted on an arm’s length basis, it is possible that the terms were less favorable to us than what we would have received in an arm’s length transaction.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on an annual basis, which may make it difficult to predict our future performance.
We are a preclinical pharmaceutical company with a limited operating history. Our operations to date have been limited to acquiring our technology portfolio. We have not yet commenced any clinical trials or obtained any regulatory approvals for any of our drug candidates. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or approved products on the market. Our operating results are expected to significantly fluctuate from quarter-to-quarter or year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include:
· any delays in regulatory review and approval of our product candidates in clinical development, including our ability to receive approval from the FDA for Annamycin;
· delays in the commencement, enrollment and timing of clinical trials;
· difficulties in identifying patients suffering from our target indications;
· the success of our clinical trials through all phases of clinical development;
· potential side effects of our product candidate that could delay or prevent approval or cause an approved drug to be taken off the market;
· our ability to obtain additional funding to develop drug candidates;
· our ability to identify and develop additional drug candidates beyond Annamycin and our WP1066 and WP1122 Portfolios;
· competition from existing products or new products that continue to emerge;
· the ability of patients or healthcare providers to obtain coverage or sufficient reimbursement for our products;
· our ability to adhere to clinical trial requirements directly or with third parties such as contract research organizations (CROs);
27
· our dependency on third-party manufacturers to manufacture our products and key ingredients;
· our ability to establish or maintain collaborations, licensing or other arrangements, particularly with MD Anderson;
· our ability to defend against any challenges to our intellectual property including, claims of patent infringement;
· our ability to enforce our intellectual property rights against potential competitors;
· our ability to secure additional intellectual property protection for our developing drug candidates and associated technologies;
· our ability to attract and retain key personnel to manage our business effectively; and
· potential product liability claims.
Accordingly, the results of any historical quarterly or annual periods should not be relied upon as indications of future operating performance.
We cannot be certain that Annamycin will receive regulatory approval, and without regulatory approval we will not be able to market Annamycin.
Our business currently depends largely on the successful development and commercialization of Annamycin. Our ability to generate revenue related to product sales, if ever, will depend on the successful development and regulatory approval of Annamycin for the treatment of relapsed or refractory acute myeloid leukemia, or AML.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the United States and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the United States until we receive approval of a NDA from the FDA. We have not submitted any marketing applications for any of our product candidates.
NDAs must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. NDAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a NDA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA review processes can take years to complete and approval is never guaranteed. If we submit a NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators in other jurisdictions have their own procedures for approval of product candidates. Even if a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and Europe also have requirements for approval of drug candidates with which we must comply with prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. In addition, delays in approvals or rejections of marketing applications in the United States, Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding our product candidates or other products. Also, regulatory approval for any of our product candidates may be withdrawn.
28
If we are unable to obtain approval from the FDA, or other regulatory agencies, for Annamycin and our other product candidates, or if, subsequent to approval, we are unable to successfully commercialize Annamycin or our other product candidates, we will not be able to generate sufficient revenue to become profitable or to continue our operations.
Any statements in this report indicating that Annamycin has demonstrated preliminary evidence of efficacy are our own and are not based on the FDA’s or any other comparable governmental agency’s assessment of Annamycin and do not indicate that Annamycin will achieve favorable efficacy results in any later stage trials or that the FDA or any comparable agency will ultimately determine that Annamycin is effective for purposes of granting marketing approval.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for Annamycin and our other product candidates.
Delays in the commencement, enrollment and completion of clinical trials could increase our product development costs or limit the regulatory approval of our product candidates. We do not know whether any future trials or studies of our other product candidates will begin on time or will be completed on schedule, if at all. The start or end of a clinical study is often delayed or halted due to changing regulatory requirements, manufacturing challenges, including delays or shortages in available drug product, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparative drug or required prior therapy, clinical outcomes or financial constraints. For instance, delays or difficulties in patient enrollment or difficulties in retaining trial participants can result in increased costs, longer development times or termination of a clinical trial. Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the eligibility criteria for the clinical trial, that include the age and condition of the patients and the stage and severity of disease, the nature of the protocol, the proximity of patients to clinical sites and the availability of effective treatments and/or availability of investigational treatment options for the relevant disease.
A product candidate can unexpectedly fail at any stage of preclinical and clinical development. The historical failure rate for product candidates is high due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables. The results from preclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in later phase clinical trials of the product candidate. We, the FDA or other applicable regulatory authorities may suspend clinical trials of a product candidate at any time for various reasons, including, but not limited to, a belief that subjects participating in such trials are being exposed to unacceptable health risks or adverse side effects, or other adverse initial experiences or findings. We may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
· inability to obtain sufficient funds required for a clinical trial;
· inability to reach agreements on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
· negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program;
· serious and unexpected drug-related side effects experienced by subjects in our clinical trials or by individuals using drugs similar to our product candidates;
· conditions imposed by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials;
29
· delays in enrolling research subjects in clinical trials;
· high drop-out rates and high fail rates of research subjects;
· inadequate supply or quality of product candidate components or materials or other supplies necessary for the conduct of our clinical trials;
· greater than anticipated clinical trial costs;
· poor effectiveness of our product candidates during clinical trials; or
· unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or vendor.
We have never conducted a clinical trial or submitted an NDA before, and any product candidate we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of our clinical development. Clinical trials may produce negative or inconclusive results, and our collaborators or we may decide, or regulators may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Success in preclinical studies and early clinical trials does not ensure that subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier clinical trials.
In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts.
If Annamycin is found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for it and our business would be harmed.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether any clinical trials we or any of our potential future collaborators may conduct will demonstrate the consistent or adequate efficacy and safety that would be required to obtain regulatory approval and market any products. If we are unable to bring Annamycin to market, or to acquire other products that are on the market or can be developed, our ability to create long-term stockholder value will be limited.
Our product candidates may have undesirable side effects that may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Unforeseen side effects from any of our product candidates could arise either during clinical development or, if Annamycin is approved, after the approved product has been marketed. For example, in the most recent Phase I/II dose-ranging clinical trial of Annamycin, two patients succumbed to tumor lysis syndrome (TLS) resulting from the debris created by Annamycin killing the targeted leukemic blasts more rapidly than anticipated. Now that this potential has been identified, prophylactic measures intended to protect patients from TLS will be deployed in future clinical trials, but there can be no assurance that such measures will be effective or that other adverse events may not emerge related to our drug. As another example, we intend to attempt to increase the maximum tolerable dose (MTD) for Annamycin by conducting another Phase I dose-ranging trial, however, unforeseen side effects could prevent us from increasing the MTD from the one established in the prior Phase I/II trial. Additional or unforeseen side effects from Annamycin or any of our other product candidates could arise either during clinical development or, if approved, after the approved product has been marketed.
30
The range and potential severity of possible side effects from therapies such as Annamycin are significant. If Annamycin causes undesirable or unacceptable side effects in the future, this could interrupt, delay or halt clinical trials and result in the failure to obtain or suspension or termination of marketing approval from the FDA and other regulatory authorities, or result in marketing approval from the FDA and other regulatory authorities only with restrictive label warnings or other limitations.
If any of our product candidates receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by such products:
· regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies;
· we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product;
· we may be subject to limitations on how we may promote the product;
· sales of the product may decrease significantly;
· regulatory authorities may require us to take our approved product off the market;
· we may be subject to litigation or product liability claims; and
· our reputation may suffer.
Any of these events could prevent us or our potential future collaborators from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
If the FDA does not find the manufacturing facilities of our future contract manufacturers acceptable for commercial production, we may not be able to commercialize any of our product candidates.
We do not intend to manufacture the pharmaceutical products that we plan to sell. We are currently utilizing contract manufacturers for the production of the active pharmaceutical ingredients and the formulation of drug product for our trials of Annamycin that we will need to conduct prior to seeking regulatory approval. However, we do not have agreements for supplies of Annamycin or any of our other product candidates and we may not be able to reach agreements with these or other contract manufacturers for sufficient supplies to commercialize Annamycin if it is approved. Additionally, the facilities used by any contract manufacturer to manufacture Annamycin or any of our other product candidates must be the subject of a satisfactory inspection before the FDA approves the product candidate manufactured at that facility. We are completely dependent on these third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture material that conform to our specifications and the FDA’s current good manufacturing practice standards, or cGMP, and other requirements of any governmental agency whose jurisdiction to which we are subject, our product candidates will not be approved or, if already approved, may be subject to recalls or other negative actions. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured our product candidates, including:
· the possibility that we are unable to enter into a manufacturing agreement with a third party to manufacture our product candidates;
31
· the possible breach of the manufacturing agreements by the third parties because of factors beyond our control; and
· the possibility of termination or nonrenewal of the agreements by the third parties before we are able to arrange for a qualified replacement third-party manufacturer.
Any of these factors could cause the delay of approval or commercialization of our product candidates, cause us to incur higher costs or prevent us from commercializing our product candidates successfully. Furthermore, if any of our product candidates are approved and contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis at commercially reasonable prices and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the government agencies that regulate our products.
We have no sales, marketing or distribution experience and we will have to invest significant resources to develop those capabilities or enter into acceptable third-party sales and marketing arrangements.
We have no sales, marketing or distribution experience. To develop sales, distribution and marketing capabilities, we will have to invest significant amounts of financial and management resources, some of which will need to be committed prior to any confirmation that Annamycin or any of our other product candidates will be approved by the FDA. For product candidates where we decide to perform sales, marketing and distribution functions ourselves or through third parties, we could face a number of additional risks, including that we or our third-party sales collaborators may not be able to build and maintain an effective marketing or sales force. If we use third parties to market and sell our products, we may have limited or no control over their sales, marketing and distribution activities on which our future revenues may depend.
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop certain of our product candidates and our financial condition and operating results.
Because developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products are expensive, we may seek to enter into collaborations with companies that have more experience. Additionally, if any of our product candidates receives marketing approval, we may enter into sales and marketing arrangements with third parties with respect to our unlicensed territories. If we are unable to enter into arrangements on acceptable terms, if at all, we may be unable to effectively market and sell our products in our target markets. We expect to face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement and they may require substantial resources to maintain. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of our product candidates.
When we collaborate with a third party for development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the future success of that product candidate to the third party. For example, we have formed a collaboration with a Polish drug development company called Dermin, where we have provided them with sub-license rights to our technologies for use in limited territories in exchange for their use of Polish government grant funding to pay for development costs we would otherwise have to fund ourselves. With the exception of Annamycin, Dermin’s territories are primarily Poland and lesser surrounding countries, but not including any of the major European markets (UK, Germany, France, Spain and Italy). In the case of Annamycin, Dermin’s territories also include Germany, but we retain the right to repurchase that territory for $500,000 at any time in the future.
We announced in October 2016 that we had secured an agreement with Dermin to utilize Dermin’s supply of Annamycin for our upcoming clinical trial. If Dermin is unable to provide us with sufficient supply for clinical trials, or if the drug product supplied is unacceptable for use in our clinical trials, our clinical trials may be delayed or the outcomes may be adversely affected.
32
One or more of our collaboration partners may not devote sufficient resources to the commercialization of our product candidates or may otherwise fail in their commercialization. The terms of any collaboration or other arrangement that we establish may contain provisions that are not favorable to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of our product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a product candidate or research program under a collaboration arrangement, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development. If we are unable to reach agreements with suitable collaborators for our product candidates, we would face increased costs, we may be forced to limit the number of our product candidates we can commercially develop or the territories in which we commercialize them. As a result, we might fail to commercialize products or programs for which a suitable collaborator cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition could be materially and adversely affected.
Our success depends greatly on the success of Annamycin’s development for the treatment of relapsed or refractory AML, and our pipeline of product candidates beyond this lead indication is extremely early stage and limited.
Other than Annamycin, our other two drug candidates, our WP1066 Portfolio and our WP1122 Portfolio, are each in the early stages of development. In addition, our current plan is to use a significant portion of our available funds to support our clinical plan for Annamycin, although this cash utilization may change in the future. As such, we are dependent on the success of Annamycin in the near term. We cannot provide you any assurance that we will be able to successfully advance any of these indications through the development process.
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have competitors in the United States, Europe and other jurisdictions, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies and universities and other research institutions. Many of our competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing drugs for the diseases that we are targeting before we do or may develop drugs that are deemed to be more effective or gain greater market acceptance than ours. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. In addition, many universities and private and public research institutes may become active in our target disease areas. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, technologies and drug products that are more effective or less costly than any of our product candidates that we are currently developing or that we may develop, which could render our products obsolete or noncompetitive.
A number of attempts have been made or are under way to provide an improved treatment for AML. Drugs attempting to target a subset of AML patients who present with particular anomalies involving a gene referred to as FLT3 are currently in clinical trials. Other approaches to improve the effectiveness of induction therapy are in early stage clinical trials and, although they do not appear to address the underlying problems with anthracyclines, we can provide no assurance that such improvements, if achieved, would not adversely impact the need for improved anthracyclines. A modified version of doxorubicin designed to reduce cardiotoxicity is in clinical trials for the treatment of sarcoma and, although this drug does not appear to address multidrug resistance and is not currently intended for the treatment of acute leukemia, we can provide no assurance that it will not become a competitive alternative to Annamycin. Although we are not aware of any other single agent therapies in clinical trials that would directly compete against Annamycin in the treatment of relapsed and refractory AML, we can provide no assurance that such therapies are not in development, will not receive regulatory approval and will reach market before our drug candidate Annamycin. In addition, any such competing therapy may be more effective and / or cost-effective than ours.
33
If our competitors market products that are more effective, safer or less expensive or that reach the market sooner than our future products, if any, we may not achieve commercial success. In addition, because of our limited resources, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
Annamycin does not have any patent protections, including composition of matter patent protection.
On March 21, 2017, we received notice that we have obtained Orphan Drug designation for Annamycin with the FDA for the treatment of AML. Additionally, we intend to pursue patents with claims directed to Annamycin drug product formulations and the methods of use of Annamycin to treat relapsed or refractory AML and other conditions, and methods for its synthesis, as the composition of matter patent protection for Annamycin has expired. As a result, competitors may be able to offer and sell products so long as these competitors do not infringe any other patents that third parties or we hold, including formulation, synthesis and method of use patents. However, method of use patents, in particular, are more difficult to enforce than composition of matter patents because of the risk of off-label sale or use of the subject compounds. Physicians are permitted to prescribe an approved product for uses that are not described in the product's labeling. Although off-label prescriptions may infringe our method of use patents, the practice is common across medical specialties and such infringement is difficult to prevent or prosecute. Off-label sales would limit our ability to generate revenue from the sale of Annamycin, if approved for commercial sale.
The intellectual property rights we have licensed from MD Anderson are subject to the rights of the U.S. government.
We have obtained a royalty-bearing, worldwide, exclusive license to intellectual property rights, including patent rights related to our WP1066 Portfolio and WP1122 Portfolio drug product candidates from MD Anderson. Some of our licensed intellectual property rights from MD Anderson have been developed in the course of research funded by the U.S. government. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future products pursuant to the Bayh-Dole Act of 1980. Government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us, or an assignee or exclusive licensee to such inventions, to grant licenses to any of these inventions to a third party if they determine that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; (iii) government action is necessary to meet requirements for public use under federal regulations; or (iv) the right to use or sell such inventions is exclusively licensed to an entity within the U.S. and substantially manufactured outside the U.S. without the U.S. government’s prior approval. Additionally, we may be restricted from granting exclusive licenses for the right to use or sell our inventions created pursuant to such agreements unless the licensee agrees to additional restrictions (e.g., manufacturing substantially all of the invention in the U.S.). The U.S. government also has the right to take title to these inventions if we fail to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. In addition, the U.S. government may acquire title in any country in which a patent application is not filed within specified time limits. Additionally, certain inventions are subject to transfer restrictions during the term of these agreements and for a period thereafter, including sales of products or components, transfers to foreign subsidiaries for the purpose of the relevant agreements, and transfers to certain foreign third parties. If any of our intellectual property becomes subject to any of the rights or remedies available to the U.S. government or third parties pursuant to the Bayh-Dole Act of 1980, this could impair the value of our intellectual property and could adversely affect our business.
34
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
We may from time to time seek to enforce our intellectual property rights against infringers when we determine that a successful outcome is probable and may lead to an increase in the value of the intellectual property. If we choose to enforce our patent rights against a party, then that individual or company has the right to ask the court to rule that such patents are invalid or should not be enforced. Additionally, the validity of our patents and the patents we have licensed may be challenged if a petition for post grant proceedings such as inter-partes review and post grant review is filed within the statutorily applicable time with the U.S. Patent and Trademark Office (USPTO). These lawsuits and proceedings are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party's activities do not infringe our intellectual property rights. In addition, in recent years the U.S. Supreme Court modified some tests used by the USPTO in granting patents over the past 20 years, which may decrease the likelihood that we will be able to obtain patents and increase the likelihood of a challenge of any patents we obtain or license.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As of December 31, 2016, we had two full-time and four part-time employees. As we advance our product candidates through preclinical studies and clinical trials, we will need to increase our product development, scientific and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we may need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel and consultants due to the intense competition for qualified personnel and consultants among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel and consultants to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
35
We are highly dependent on the development, regulatory, commercialization and business development expertise of our management team, key employees and consultants. If we lose one or more of our executive officers or key employees or consultants, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers or key employees or consultants may terminate their employment at any time. Replacing executive officers, key employees and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire and retain employees and consultants from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel and consultants. Our failure to retain key personnel or consultants could materially harm our business.
In addition, we have scientific and clinical advisors and consultants who assist us in formulating our research, development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us and typically they will not enter into non-compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
Our chief executive officer and our chief operating officer are currently working for us on a part-time basis.
Three of our key employees are currently part-time and provide services for other biotechnology development efforts. Specifically, Walter Klemp, our chairman and chief executive officer is also the chief executive officer of Soliton, Inc., a medical device development company whose business operations we do not believe conflict with those of our company, and Donald Picker, our president and chief operating officer, is currently also serving as chief operating officer of two other biotechnology companies, whose business operations we do not believe conflict with those of our company. As we progress, if the full-time services of a CEO or COO are required and the current officers cannot provide that level of commitment, we will need to identify a suitable CEO or COO who can dedicate such time to our company. We can provide no assurance that we will be able to successfully identify and retain a qualified candidate for this position.
We do not expect that our insurance policies will cover all of our business exposures thus leaving us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. In particular, we do not carry product liability insurance covering any clinical trials liability that we may incur. Although we intend to obtain such insurance before we commence any clinical trials, there can be no assurance that we will secure adequate insurance coverage or that any such insurance coverage will be sufficient to protect our operations to significant potential liability in the future. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
Risks Relating to Our Common Stock
Our stock price has been and may continue to be volatile, which could result in substantial losses for investors.
Since our IPO in June 2016, our stock price has ranged from a high of $9.58 to a low of $0.95, and the market price of our common stock is likely to continue to be highly volatile and could fluctuate widely in response to various factors, many of which are beyond our control. In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also significantly affect the market price of our common stock.
36
Your ownership may be diluted if additional capital stock is issued to raise capital, to finance acquisitions or in connection with strategic transactions.
We intend to seek to raise additional funds, finance acquisitions or develop strategic relationships by issuing equity or convertible debt securities, which would reduce the percentage ownership of our existing stockholders. Our board of directors has the authority, without action or vote of the stockholders, to issue all or any part of our authorized but unissued shares of common or preferred stock. Our certificate of incorporation authorizes us to issue up to 75,000,000 shares of common stock and 5,000,000 shares of preferred stock. Future issuances of common or preferred stock would reduce your influence over matters on which stockholders vote and would be dilutive to earnings per share. In addition, any newly issued preferred stock could have rights, preferences and privileges senior to those of the common stock. Those rights, preferences and privileges could include, among other things, the establishment of dividends that must be paid prior to declaring or paying dividends or other distributions to holders of our common stock or providing for preferential liquidation rights. These rights, preferences and privileges could negatively affect the rights of holders of our common stock, and the right to convert such preferred stock into shares of our common stock at a rate or price that would have a dilutive effect on the outstanding shares of our common stock.
The concentration of our common stock ownership by our current management will limit your ability to influence corporate matters.
Our founders, directors and executive officers beneficially own and are able to vote in the aggregate 48.3% of our outstanding common stock. As such, our founders, directors and executive officers, as stockholders, will continue to have the ability to exert significant influence over all corporate activities, including the election or removal of directors and the outcome of tender offers, mergers, proxy contests or other purchases of common stock that could give our stockholders the opportunity to realize a premium over the then-prevailing market price for their shares of common stock. This concentrated control will limit your ability to influence corporate matters and, as a result, we may take actions that our stockholders do not view as beneficial. In addition, such concentrated control could discourage others from initiating changes of control. In such cases, the perception of our prospects in the market may be adversely affected and the market price of our common stock may decline.
Certain provisions in our organizational documents could enable our board of directors to prevent or delay a change of control.
Our organizational documents contain provisions that may have the effect of discouraging, delaying or preventing a change of control of, or unsolicited acquisition proposals, that a stockholder might consider favorable. These include provisions:
· prohibiting the stockholders from acting by written consent;
· requiring advance notice of director nominations and of business to be brought before a meeting of stockholders;
· requiring a majority vote of the outstanding shares of common stock to amend the bylaws; and
· limiting the persons who may call special stockholders’ meetings.
Furthermore, our board of directors has the authority to issue shares of preferred stock in one or more series and to fix the rights and preferences of these shares without stockholder approval. Any series of preferred stock is likely to be senior to our common stock with respect to dividends, liquidation rights and, possibly, voting rights. The ability of our board of directors to issue preferred stock also could have the effect of discouraging unsolicited acquisition proposals, thus adversely affecting the market price of our common stock.
In addition, Delaware law makes it difficult for stockholders that recently have acquired a large interest in a corporation to cause the merger or acquisition of the corporation against the directors’ wishes. Under Section 203 of the Delaware General Corporation Law, a Delaware corporation may not engage in any merger or other business combination with an interested stockholder for a period of three years following the date that the stockholder became an interested stockholder except in limited circumstances, including by approval of the corporation’s board of directors.
37
We have no intention of declaring dividends in the foreseeable future.
The decision to pay cash dividends on our common stock rests with our board of directors and will depend on our earnings, unencumbered cash, capital requirements and financial condition. We do not anticipate declaring any dividends in the foreseeable future, as we intend to use any excess cash to fund our operations. Investors in our common stock should not expect to receive dividend income on their investment, and investors will be dependent on the appreciation of our common stock to earn a return on their investment.
Failure to maintain effective internal control over our financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could cause our financial reports to be inaccurate.
We are required pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to maintain internal control over financial reporting and to assess and report on the effectiveness of those controls. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. Our management concluded that our internal controls over financial reporting were, and continue to be ineffective, and as of the year ended December 31, 2016, identified a material weakness in our internal controls due to the lack of segregation of duties. While management is working to remediate the material weakness, there is no assurance that such changes, when economically feasible and sustainable, will remediate the identified material weaknesses or that the controls will prevent or detect future material weaknesses. If we are not able to maintain effective internal control over financial reporting, our financial statements, including related disclosures, may be inaccurate, which could have a material adverse effect on our business.
Failure to continue improving our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with the Sarbanes-Oxley Act of 2002, and the related rules and regulations of the SEC. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
Management did elect to perform an annual assessment as of December 31, 2016 of the effectiveness of our internal control over financial reporting for its first annual report. Our management concluded that our internal control over financial reporting was, and continues to be, ineffective and as of the year ended December 31, 2016, due to a material weakness in our internal controls due to the lack of segregation of duties. This annual assessment was performed earlier than required. Section 404(a) of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC. However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we have and intend to consider to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. We may continue to take advantage of these reporting exemptions until we are no longer an “emerging growth company.” If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
On November 14, 2016, our Audit Committee, after discussion with management and our previous independent registered public accountants, determined that our unaudited financial statements for the quarter ended June 30, 2016, as reported in our Quarterly Report on Form 10-Q filed on August 15, 2016 should no longer be relied upon due to an error identified therein, and that a restatement of these financial statements is required. We identified certain non-cash errors due to an error in the accounting for the business combination of Moleculin, LLC. We filed a Form 10-Q/A for the quarter ended June 30, 2016 on November 21, 2016 reflecting such corrections in errors.
38
As an “emerging growth company” under the Jumpstart Our Business Startups Act, or JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements.
As an “emerging growth company” under the JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements. We are an emerging growth company until the earliest of:
· the last day of the fiscal year during which we have total annual gross revenues of $1 billion or more;
· the last day of the fiscal year following the fifth anniversary of our IPO, or December 31, 2021;
· the date on which we have, during the previous 3-year period, issued more than $1 billion in non-convertible debt; or
· the date on which we are deemed a “large accelerated issuer” as defined under the federal securities laws.
For so long as we remain an emerging growth company, we will not be required to:
· have an auditor report on our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002;
· comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis);
· submit certain executive compensation matters to shareholders advisory votes pursuant to the “say on frequency” and “say on pay” provisions (requiring a non-binding shareholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding shareholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010;
· include detailed compensation discussion and analysis in our filings under the Securities Exchange Act of 1934, as amended, and instead may provide a reduced level of disclosure concerning executive compensation; and
· may present only two years of audited financial statements and only two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations, or MD&A.
We intend to take advantage of all of these reduced reporting requirements and exemptions. Certain of these reduced reporting requirements and exemptions were already available to us due to the fact that we also qualify as a “smaller reporting company” under SEC rules. For instance, smaller reporting companies are not required to obtain an auditor attestation and report regarding management’s assessment of internal control over financial reporting; are not required to provide a compensation discussion and analysis; are not required to provide a pay-for-performance graph or CEO pay ratio disclosure; and may present only two years of audited financial statements and related MD&A disclosure.
Under the JOBS Act, we may take advantage of the above-described reduced reporting requirements and exemptions until December 31, 2021, or such earlier time that we no longer meet the definition of an emerging growth company. In this regard, the JOBS Act provides that we would cease to be an “emerging growth company” if we have more than $1.0 billion in annual revenues, have more than $700 million in market value of our common stock held by non-affiliates, or issue more than $1.0 billion in principal amount of non-convertible debt over a three-year period. Further, under current SEC rules, we will continue to qualify as a “smaller reporting company” for so long as we have a public float (i.e., the market value of common equity held by non-affiliates) of less than $75 million as of the last business day of our most recently completed second fiscal quarter.
We cannot predict if investors will find our securities less attractive due to our reliance on these exemptions. If investors were to find our common stock less attractive as a result of our election, we may have difficulty raising capital.
39
ITEM 1B. UNRESOLVED STAFF COMMENTS
There are no unresolved comments from the staff of the SEC.
Our corporate and executive offices are in located in a leased facility in Houston, Texas. The current lease is month-to-month but is expected to be renegotiated in the next six months. We believe our facilities are sufficient to meet our current needs and that suitable space will be available as and when needed. We do not own any real property.
We are not a party to any pending legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURE
Not applicable.
40
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock has been listed on the NASDAQ Capital Market since June 1, 2016, the date of our initial public offering, under the symbol “MBRX”. On March 24, 2017, the closing price reported on the NASDAQ Capital Market for our common stock was $$1.13.
| Year-Ended December 31, 2016 | High | Low | ||||||
| First Quarter | n/a | n/a | ||||||
| Second Quarter (commencing June 1, 2016) | $ | 9.58 | $ | 6.24 | ||||
| Third Quarter | $ | 7.02 | $ | 5.50 | ||||
| Fourth Quarter | $ | 5.90 | $ | 1.46 | ||||
Holders
As of February 23, 2017 there were approximately 229 holders of record of our common stock. In addition, we believe that a significant number of beneficial owners of our common stock hold their shares in nominee or in “street name” accounts through brokers.
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain any future earnings to finance the growth and development of our business. Therefore, we do not anticipate declaring any cash dividends in the foreseeable future. Any future determination as to the payment of dividends, if any, will be at the discretion of our board of directors and will depend on then existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
Securities Authorized for Issuance under Equity Compensation Plans
The following table sets forth information regarding our equity compensation plans at December 31, 2016:
| Plan category | Number
of securities to (a) | Weighted-average (b) | Number
of securities (by (c) | |||||||||
| Equity compensation plans approved by security holders (1) | 510,000 | $ | 5.28 | 1,990,000 | ||||||||
| Equity compensation plans not approved by security holders (2) | 107,802 | $ | 7.50 | — | ||||||||
| Total | 617,802 | $ | 5.67 | 1,990,000 | ||||||||
| (1) | Represents shares of common stock issuable upon exercise of outstanding stock options under our 2015 Stock Plan, as amended. Our 2015 Stock Plan has been approved by our stockholders. |
| (2) | Consists of a five-year warrant issued to the underwriters in our initial public offering. |
41
Recent Sales of Unregistered Securities
On various dates from August 31, 2015 through January 19, 2016, each as amended on March 10, 2016, we issued certain 8% unsecured promissory notes in aggregate principal amount of $615,000 to certain accredited investors. Upon the completion of the IPO, these notes provided that they be automatically converted into shares of our common stock at their applicable conversion prices, which were $0.1299 with respect to $250,000 in notes and $0.20 per share with respect to the remaining $365,000, to the extent and provided that no holder of these notes was or will be permitted to convert such notes to the extent that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock after such conversion. Due to this 4.99% limitation, the remaining principal and accrued interest amounts of the effected notes will remain outstanding and will be converted into shares of our common stock at such time as the 4.99% limitation continues to be met. The IPO was completed on May 31, 2016. On May 31, 2016, pursuant to the conversion feature of the foregoing notes and with restriction of the 4.99% beneficially owned condition limitation, we issued 1,166,503 common shares in total, reducing convertible debt principal by $183,356 and accrued interest by $17,699. During the three months ended December 31, 2016, an additional 110,038 common shares were issued due to the number of common shares outstanding allowing for conversion of additional shares under the 4.99% beneficially owned condition limitation. This reduced the convertible debt principal by $21,577 and accrued interest by $430. The remaining convertible debt without consideration of accrued interest as of December 31, 2016, if converted on December 31, 2016, would result in an additional 1,821,013 common shares to be issued. See Note 3 of our financial statements for more information regarding the issuance of the notes.
We believe that the issuances were exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act regarding transactions not involving a public offering.
On May 31, 2016, we completed our initial public offering, which commenced on May 2, 2016, pursuant to which we sold 1,540,026 shares of our common stock at $6.00 per share with gross proceeds of $9,240,156 and net proceeds of $8,464,183 after deducting underwriting discounts and commissions and offering expenses payable by us. The offer and sale of all of the shares in the offering were registered under the Securities Act pursuant to a registration statement on Form S-1 (File No. 333-209323), which was declared effective by the SEC on May 2, 2016. Bonwick Capital Partners LLC and Network 1 Financial Securities, Inc. acted as underwriters for the offering.
There has been no material change in the planned use of proceeds from our IPO as described in our final prospectus filed with the SEC on May 3, 2016 pursuant to Rule 424(b). No direct or indirect payments were made by us to any of our directors or officers or their associates, to persons owning ten percent or more of our common stock or to their associates, or to our affiliates, other than payments in the ordinary course of business to officers for salaries and those payments disclosed in “Item 1. Business” with regard to the license arrangements with HPI. Pending the uses described, we intend to invest the net proceeds in short-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the U.S. government.
42
Subsequent Event
On February 9, 2017, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with Roth Capital Partners, LLC, as representative of the several underwriters identified therein (collectively, the “Underwriters”), pursuant to which we sold in a registered public offering (the “Offering”), 3,710,000 units, priced at a public offering price of $1.35 per unit, with each unit consisting of: (i) one share of common stock, (ii) a five-year Series A warrant to purchase 0.50 of a share of common stock, (iii) a 90-day Series B warrant to purchase one share of common stock, and (iv) a five-year Series C warrant to purchase 0.50 of a share of common stock. The Series C warrants in a unit may only be exercised to the extent and in proportion to a holder of the Series C warrants exercising its Series B warrants included in the unit. The Series A and Series C warrant have an exercise price of $1.50 per share of common stock. The Series B warrant has an exercise price of $1.35 per share of common stock.
Under the terms of the Underwriting Agreement, we granted the Underwriters a 45-day option to purchase an additional 556,500 shares of common stock and/or an additional 556,500 warrants combinations (comprised of an aggregate of 278,250 Series A warrants, 556,500 Series B warrants and 278,250 Series C warrants), in any combinations thereof, from us to cover over-allotments at the public offering price per share of $1.349 and public offering price per warrant combination of $.001, respectively, less the underwriting discounts and commissions. Upon the closing of the Offering, the Underwriters exercised the over-allotment option with respect to 278,100 warrant combinations. We received approximately $4.4 million in net proceeds from the Offering, after deducting underwriting discounts and commissions and estimated offering expenses.
Subsequent to March 22, 2017, investors in the aforementioned offering on February 9, 2017, have excerised approximately 600,000 Series B warrants resulting in approximately gross proceeds to the Company of $800,000 and approximately 600,000 shares of the Company’s common stock being issued.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
We did not repurchase any of our equity securities during the year ended December 31, 2016.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable to the Company, which is a smaller reporting company.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) contains certain forward-looking statements. Historical results may not indicate future performance. Our forward-looking statements reflect our current views about future events, are based on assumptions and are subject to known and unknown risks and uncertainties that could cause actual results to differ materially from those contemplated by these statements. Factors that may cause differences between actual results and those contemplated by forward-looking statements include, but are not limited to, those set forth at the beginning of this report under the section titled “Cautionary Note Regarding Forward-Looking Statements” and discussed in “Item 1A. Risk Factors.” We undertake no obligation to publicly update or revise any forward-looking statements, including any changes that might result from any facts, events, or circumstances after the date hereof that may bear upon forward-looking statements. Furthermore, we cannot guarantee future results, events, levels of activity, performance, or achievements.
Highlights
We are a preclinical-stage pharmaceutical development company organized as a Delaware corporation in July 2015 to focus on the development of anti-cancer drug candidates. We have three drug development projects. Our lead drug candidate is liposomal Annamycin, which is referred to as Annamycin, an anthracycline intended for the treatment of relapsed or refractory acute myeloid leukemia, or AML. Annamycin has been in clinical trials pursuant to an investigational new drug application, or IND, that had been filed with the U.S. Food and Drug Administration, or FDA. Due to a lack of development activity by a prior drug developer, this IND was terminated.
43
We filed our IND application, with a Phase I/II approach with the intent of increasing the MTD, for Annamycin on February 10, 2017. In subsequent discussions, FDA requested certain revisions to the protocol, additional information, and additional data related to Chemistry, Manufacturing and Controls (“CMC”). We have the additional information, have made the requested revisions to the protocol, and we are working on developing the CMC data. In the interim, we have withdrawn the IND application, in order to resubmit it when the requested data are available. We believe that resubmission of the IND application will occur in time for the IND to go into effect in the first half of 2017. However, if we are unable to obtain the required CMC data on a timely basis, we will be delayed in resubmitting our IND application, which will delay the commencement of our clinical trials beyond the first half of 2017. Furthermore on March 21, 2017, we received notice that we have obtained Orphan Drug designation for Annamycin with the FDA for the treatment of AML.
We have two other drug development projects in progress. One of them involves a collection of small molecules we refer to as the WP1066 Portfolio that was obtained via our acquisition of Moleculin, LLC and is focused on the modulation of key regulatory transcription factors involved in the progression of cancer. The other, which we call the WP1122 Portfolio, is a suite of molecules targeting the metabolic processes involved in cancer in general, and glioblastoma in particular that we acquired from IntertechBio Corporation. Both of these technologies are licensed on a worldwide exclusive basis from The University of Texas M.D. Anderson Cancer Center, or MD Anderson.
Overview
MBI was founded in 2015 in order to combine and consolidate the development efforts involving several anti-cancer technologies, some of which are based on license agreements with MD Anderson. This effort began with the acquisition of the Annamycin development project from AnnaMed, Inc., or AnnaMed, followed by the acquisition of the license rights to the WP1122 Portfolio from IntertechBio Corporation, or IntertechBio. Further, we, on Moleculin, LLC’s behalf, entered into a co-development agreement with Houston Pharmaceuticals, Inc., or HPI, which culminated with the merger of Moleculin, LLC into MBI coincident with our initial public offering allowing us to gain control of the WP1066 Portfolio.
AnnaMed was formed in 2012 to take over the development of Annamycin from a prior drug development company, Callisto Pharmaceuticals, Inc., or Callisto. Callisto ceased development work on Annamycin leading to the termination of its IND by the FDA. In order to satisfy unmet license obligations, Callisto agreed to transfer all available Annamycin data to AnnaMed, which data we used in our initial filing of an IND.
IntertechBio was formed in 2009 to license and begin development on the WP1122 Portfolio. In August 2015, IntertechBio agreed to assign all license rights to us in exchange for 630,000 shares of our common stock.
Moleculin, LLC was formed in 2006 and was working to develop the WP1066 Portfolio it licensed from MD Anderson. On May 2, 2016, Moleculin, LLC was merged with and into MBI. As a result of the merger, we issued the holders of Moleculin, LLC equity interests and convertible notes an aggregate of approximately 999,931 shares of our common stock.
Since Moleculin, LLC commenced operations in 2006, substantially all of its efforts had been focused on research, development and the advancement of the WP1066 Portfolio. Moleculin, LLC did not generate any revenue from product sales and, as a result, incurred significant losses.
Neither Moleculin, LLC nor MBI has manufacturing facilities and all manufacturing activities are contracted out to third parties. Additionally, Moleculin, LLC utilized third-party clinical research organizations to carry out clinical trials. Neither Moleculin, LLC nor MBI have a sales organization.
Recent Business Developments
Accelerated Plan for Clinical Drug Production – We announced on October 7, 2016, that we had secured an agreement with Dermin Sp. Zo. O. (“Dermin”) to utilize Dermin’s supply of Annamycin for our upcoming clinical trial, substantially reducing the expenditures required for drug product and shortening the time required to produce clinical supplies. We believe that this is an important agreement and key milestone to be reached and that it reduces the potential for drug production to negatively impact our clinical timeline. With this agreement in place, our drug product expense for upcoming clinical trials should be below previous estimates.
44
Moleculin, LLC previously licensed Annamycin to Dermin within a limited region in Europe, enabling Dermin to deploy Polish grant funds toward producing Annamycin. The agreement reached between the two companies allows us to utilize this Annamycin in our upcoming clinical trials rather than having to produce new Annamycin for its own use. We believe Dermin benefits from a data sharing arrangement giving it access to our clinical data on a faster timeline than it would be able develop on its own.
Updated Plan for Clinical Trials for Annamycin – On October 20, 2016, we announced in a conference call that we had identified some significantly positive findings from our detailed review of the last clinical trial for Annamycin by a prior developer, which has given rise to a modification of our own clinical development plan.
As we previously disclosed, a prior developer had conducted a clinical trial with Annamycin, but then subsequently failed to maintain their IND with the FDA. We had previously indicated that our plan was to conduct a detailed review of the clinical results generated by that prior developer. We would then use those results to reestablish an IND in order to continue clinical trials of Annamycin. However, we announced recently that, in the course of our review, we identified that Annamycin may have greater potential for efficacy than we originally believed, based on an unexpected potential opportunity to increase the drug’s Maximum Tolerable Dose (“MTD”).
In particular, the Dose Limiting Toxicities (“DLTs”) reported in that previous trial that led to the establishment of the current MTD of 150 mg/m2 were all from patients who had an unusually high number of first-line induction therapy failures prior to being treated with Annamycin. Specifically, of the three patients in the last clinical trial who experienced these DLTs, one of them had failed nineteen prior induction therapy attempts, another had failed sixteen and the other had failed fifteen before being enrolled in the trial. We concluded from our review of this data that, if the heavily treated patients are excluded from the data set, the MTD could have been closer to 250 mg/m2, substantially higher than the level that was actually set by this previous trial.
We view this as an encouraging development because it means we may have an opportunity to increase the MTD for our next trial from 150 mg/m2 to 200 or even 250 mg/m2. If that turns out to be the case, we believe it could increase the chance for positive outcomes in our next trial.
With the discovery that we may be able to increase our MTD, we determined to adjust our clinical strategy by adding in a Phase I arm to our next Phase II trial, which will add some expense to our development effort. We believe this change in strategy will add several months to the eventual final approval of the drug, however, we believe that we will publicly announce results from our phase I/II clinical trial sometime in 2018.
Filing of an IND for Annamycin – We filed our IND application, with the clinical strategy of increasing the MTD mentioned above, for Annamycin on February 10, 2017. In subsequent discussions, FDA requested certain revisions to the protocol, additional information, and additional data related to Chemistry, Manufacturing and Controls (“CMC”). We have the additional information, we have made the requested revisions to the protocol, and we are developing the CMC data. In the interim, we have withdrawn the IND application, in order to resubmit it when the requested data are available. We believe that resubmission of the IND application will occur in time for the IND to go into effect and the announcement of the beginning of phase I/II clinical trials in the first half of 2017. However, if we are unable to obtain the required CMC data on a timely basis, we will be delayed in resubmitting our IND application, which will delay the commencement of our clinical trials beyond the first half of 2017.
Filing for Orphan Drug Status for Annamycin – On March 21, 2017 we received notice that we had obtained Orphan Drug designation for Annamycin for the treatment of AML with the FDA effective March 20, 2017.
Update on WP1066 – A clinician at MD Anderson has advised us that she is proceeding with a physician-sponsored IND for WP1066 for the treatment of brain tumors. We are not participating in nor have influence on this IND process. The clinician has submitted an IND to the FDA and has indicated that this IND is on hold until documentation of Good Manufacturing Process or GMP production of WP1066 can be presented to the FDA. We expect that this IND will eventually move forward in 2017 and will produce publishable clinical results in 2018.
45
Advancement of Preclinical Testing for Brain Tumors with WP1122 – On October 25, 2016, we announced promising initial results of the preclinical toxicology work on WP1122, our unique inhibitor of glucose metabolism, which is an important driver of glycolytic brain tumor progression and survival. We view this as an important step toward future clinical trials for WP1122. A similar chemical structure to that which turns morphine into heroine has been used to allow WP1122 to successfully enter the brain and increase circulation time. We indicated that preliminary escalating single dose toxicity testing in mice (oral administration) was successfully completed and even at the highest possible dose, no toxic death was observed. In multiple therapeutic doses, WP1122 was well tolerated during intense twice-daily oral dosing.
We believe moving forward with preclinical toxicology is the key to our ability to generate proof of concept in humans. We had previously announced the presentation of promising preclinical data in July 2016, supporting the potential for using WP1122 as a treatment for glioblastoma.
Subsequent Event
On February 9, 2017, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with Roth Capital Partners, LLC, as representative of the several underwriters identified therein (collectively, the “Underwriters”), pursuant to which we sold in a registered public offering (the “Offering”), 3,710,000 units, priced at a public offering price of $1.35 per unit, with each unit consisting of: (i) one share of common stock, (ii) a five-year Series A warrant to purchase 0.50 of a share of common stock, (iii) a 90-day Series B warrant to purchase one share of common stock, and (iv) a five-year Series C warrant to purchase 0.50 of a share of common stock. The Series C warrants in a unit may only be exercised to the extent and in proportion to a holder of the Series C warrants exercising its Series B warrants included in the unit. The Series A and Series C warrant have an exercise price of $1.50 per share of common stock. The Series B warrant has an exercise price of $1.35 per share of common stock.
Under the terms of the Underwriting Agreement, we granted the Underwriters a 45-day option to purchase an additional 556,500 shares of common stock and/or an additional 556,500 warrants combinations (comprised of an aggregate of 278,250 Series A warrants, 556,500 Series B warrants and 278,250 Series C warrants), in any combinations thereof, from us to cover over-allotments at the public offering price per share of $1.349 and public offering price per warrant combination of $.001, respectively, less the underwriting discounts and commissions. Upon the closing of the Offering, the Underwriters exercised the over-allotment option with respect to 278,100 warrant combinations. We received approximately $4.4 million in net proceeds from the Offering, after deducting underwriting discounts and commissions and estimated offering expenses.
Subsequent to March 22, 2017, investors in the aforementioned offering on February 9, 2017, have excerised approximately 600,000 Series B warrants resulting in approximately gross proceeds to the Company of $800,000 and approximately 600,000 shares of the Company’s common stock being issued.
46
Results of Operations
We were formed on July 28, 2015; therefore, the financial information for 2015 is not comparable to the financial results of the year ended December 31, 2016. The following table sets forth, for the periods indicated, data derived from our statement of operations:
| Year Ended December 31, | From Inception through December 31, | |||||||
| 2016 | 2015 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 1,495,561 | 260,418 | ||||||
| General and administrative | 2,381,424 | 477,810 | ||||||
| Depreciation | 6,162 | - | ||||||
| Total operating expense | 3,883,147 | 738,228 | ||||||
| Loss from operations | (3,883,147 | ) | (738,228 | ) | ||||
| Other expense: | ||||||||
| Interest expense | (43,214 | ) | (10,132 | ) | ||||
| Net loss | $ | (3,926,361 | ) | $ | (748,360 | ) | ||
Research and Development Expense. Research and development (R&D) expense was $1,495,561 and $260,418 for the twelve months ended December 31, 2016 and for the period from July 28, 2015 (inception) to December 31, 2015, respectively. The increase of approximately $1,235,000 mainly represents an increase of approximately: $253,000 related to an increase in R&D headcount and associated payroll costs, $111,000 for sponsored research and related expenses; approximately $660,000 associated with developing and testing drug product as we prepare for clinical trials; and, $245,000 related to travel, consultants, and other research costs associated in preparing our IND and Orphan Drug applications with the FDA. This was all offset by an approximate $34,000 reduction in patent prosecution and other expenses.
General and Administrative Expense. General and administrative expense was $2,381,424 and $477,810 for the twelve months ended December 31, 2016 and for the period from July 28, 2015 (inception) to December 31, 2015, respectively. The expense increase of approximately $1,904,000 was mainly attributable to: (a) the increase in headcount and associated payroll costs of $648,000 including severance of $118,000 and roughly $254,000 of stock based compensation and deferred salary; (b) approximately $584,000 in legal, accounting, consulting, and other professional expenses; (c) approximately $389,000 in public company costs; (d) approximately $183,000 in insurance expense; (e) roughly $50,000 in travel expenses; and (f) approximately $50,000 in occupancy, office and other costs. All of these increases are directly related to our being fully operational versus this period a year ago.
We utilize outside consultants. Total wages paid to our employees, including our CEO, CFO, COO, CMO plus two other employees, were approximately $549,000, predominately in the second half of the year.
Interest Expense. Interest expense included expense accrued on our convertible promissory notes issued in 2015 and 2016 bearing interest at the rate of 8% per annum.
Net Loss. The net loss for the twelve months ended December 31, 2016 was $3,926,361 which included non-cash expenses of approximately $450,000 which included $6,000 for depreciation, $324,000 for stock based compensation and other stock based expenses and a one-time expense of $120,000 related to the severance of our former Chief Financial Officer. This loss for the period is a significant increase from the loss for the period from July 28, 2015 (inception) to December 31, 2015 of $748,360 as we had, at that time, just begun operations.
Liquidity and Capital Resources
As of December 31, 2016, we had $5,007,216 in cash. During the period from January 1, 2016 through May 2, 2016, we sold 234,296 of common stock for $702,894. On May 31, 2016, we completed our initial public offering, pursuant to which we sold 1,540,026 shares of our common stock at $6.00 per share for net proceeds of $8,464,183 after deducting underwriting discounts and commissions and direct offering expenses payable by us. In February 2017, we completed a public offering of our common stock and warrants, pursuant to which we received approximately $4.4 million in net proceeds, after deducting underwriting discounts and commissions and estimated offering expenses. We believe that our existing cash and cash equivalents as of December 31, 2016 along with the cash generated by the February 2017 offering described above will be sufficient to fund our planned operations into the first quarter of 2018.
47
We will not generate revenue from product sales unless and until we successfully complete development of, obtain regulatory approval for and begin to commercialize one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Accordingly, we anticipate that we will need to raise additional capital to fund our future operations. Until such time that we can generate substantial revenue from product sales, if ever, we expect to finance our operating activities through a combination of equity offerings and debt financings and we may seek to raise additional capital through strategic collaborations. However, we may be unable to raise additional funds or enter into such arrangements when needed on favorable terms, or at all, which would have a negative impact on our financial condition and could force us to delay, limit, reduce or terminate our development programs or commercialization efforts or grant to others rights to develop or market product candidates that we would otherwise prefer to develop and market ourselves. Failure to receive additional funding could cause us to cease operations, in part or in full. Furthermore, even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital due to favorable market conditions or strategic considerations, which may cause dilution to our existing stockholders.
The following table sets forth the primary sources and uses of cash of MBI for the period indicated:
| For the Twelve Months Ended December 31, 2016 | From Inception through December 31, 2015 | |||||||
| Net cash used in operating activities | $ | (3,764,905 | ) | $ | (423,509 | ) | ||
| Net cash used in investing activities | (121,108 | ) | - | |||||
| Net cash provided by financing activities | 8,865,138 | 451,600 | ||||||
| Net increase in cash and cash equivalents | $ | 4,979,125 | $ | 28,091 | ||||
Cash used in operating activities
Net cash used in operating activities was $3,764,905 for the twelve months ended December 31, 2016 compared to $423,509 for the period from inception to December 31, 2015. This increase in use of cash for operations is due to our becoming operational post IPO in mid-2016. This mainly included payments made for payroll, travel, insurance and professional fees to our consultants, attorneys and accountants for services related to our becoming a publicly traded company and related filing fees, along with payments made to MD Anderson for license and maintenance fees.
Cash used in investing activities
Net cash used in investing activities was $121,108 for the twelve months ended December 31, 2016 and primarily represents the cash paid as part of the acquisition of Moleculin, LLC. No investing activities were done in the prior period.
48
Cash provided by financing activities
Net cash provided by financing activities was $8,865,138 for the twelve months ended December 31, 2016 compared to the prior period of $451,600. We received $8,464,183 net proceeds from our IPO stock issuance, $705,894 from issuance of common stock at $3.00 per share, and $165,000 from issuance of convertible notes. The prior period financing activities mainly consisted of the issuance of convertible notes payable. Net cash used in financing activities included approximately $470,000 for payments of notes payable.
Since Moleculin, LLC’s inception and through December 31, 2015, Moleculin, LLC funded its operations primarily through the sale and issuance of convertible preferred units and convertible and non-convertible promissory notes. From May to November 2014, Moleculin, LLC issued various convertible notes to its creditors. The note proceeds were $1.5 million. These notes bore interest at 8% per annum and were due on the earlier of June 30, 2016 or the consummation of a liquidation event (which event included the merger between MBI and Moleculin, LLC that occurred on May 2, 2016), unless earlier converted.
Off-Balance Sheet Transactions
We do not engage in off-balance sheet transactions.
JOBS Act and Recent Accounting Pronouncements
The recently enacted JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
We have implemented all new accounting pronouncements that are in effect and may impact our financial statements and we do not believe that there are any other new accounting pronouncements that have been issued that might have a material impact on our financial position or results of operations.
Critical Accounting Policies and Significant Judgments and Estimates
The financial statements have been prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that the following accounting policies are the most critical to aid in fully understanding and evaluating our reported financial results, and they require our most difficult, subjective or complex judgments, resulting from the need to make estimates about the effect of matters that are inherently uncertain.
Acquisition
We acquired Moleculin, LLC on May 2, 2016, and, going forward our financial statements will include the operations of Moleculin, LLC. We account for acquired businesses using the acquisition method of accounting, which requires, among other things, that most assets acquired and liabilities assumed be recognized at their estimated fair values as of the acquisition date and that the fair value of acquired in-process research and development (“IPR&D”) be recorded on the balance sheet. Transaction costs are expensed as incurred. Any excess of the consideration transferred over the assigned values of the net assets acquired will be recorded as goodwill. The Company obtained input from third-parties regarding its tangible and intangible assets and other information necessary to measure the fair value of the assets acquired and liabilities assumed in connection with the acquisition of Moleculin, LLC.
49
Beneficial Conversion Feature
From time to time, we may issue convertible notes that have conversion prices that create an embedded beneficial conversion feature on the issuance date. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of any attached equity instruments, if any related equity instruments were granted with the debt. We estimate the fair value of our common stock using the most recent selling price available. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid-in capital. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
Research and Development Costs
We record accrued expenses for estimated costs of our research and development activities conducted by third-party service providers, which include the conduct of pre-clinical studies and preparation for clinical trials and contract manufacturing activities. We record the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced, and we include these costs in accrued liabilities in the balance sheets and within research and development expense in the statement of operations. These costs are a significant component of our research and development expenses. We record accrued expenses for these costs based on the estimated amount of work completed and in accordance with agreements established with these third parties.
We estimate the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. We make significant judgments and estimates in determining the accrued balance in each reporting period. As actual costs become known, we adjust our accrued estimates. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed may vary from our estimates and could result in us reporting amounts that are too high or too low in any particular period. Our accrued expenses are dependent, in part, upon the receipt of timely and accurate reporting from clinical research organizations and other third-party service providers. To date, there have been no material differences from our accrued expenses to actual expenses.
Impairment of Long-Lived Assets
Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable or at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its carrying value.
Components of our Results of Operations and Financial Condition
Operating expenses
We classify our operating expenses into three categories: research and development, general and administrative and depreciation.
50
Research and development. Research and development expenses consist primarily of:
· costs incurred to conduct research, such as the discovery and development of our product candidates;
· costs related to production of clinical supplies, including fees paid to contract manufacturers;
· fees paid to clinical consultants, clinical trial sites and vendors, including clinical research organizations, in preparation for clinical trials and our IND and Orphan Drug applications with the FDA; and
· costs related to compliance with drug development regulatory requirements.
We recognize all research and development costs as they are incurred. Pre-clinical costs, contract manufacturing and other development costs incurred by third parties are expensed as the contracted work is performed.
We expect our research and development expenses to increase in the future as we advance our product candidates into and through clinical trials and pursue regulatory approval of our product candidates in the United States and Europe. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming. The actual probability of success for our product candidates may be affected by a variety of factors including: the quality of our product candidates, early clinical data, investment in our clinical program, competition, manufacturing capability and commercial viability. We may never succeed in achieving regulatory approval for any of our product candidates. As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent, if any, we will generate revenue from the commercialization and sale of our product candidates.
General and administrative. General and administrative expense consists of personnel related costs, which include salaries, as well as the costs of professional services, such as accounting and legal, facilities, information technology and other administrative expenses. We expect our general and administrative expense to increase due to the anticipated growth of our business and related infrastructure as well as accounting, insurance, investor relations and other costs associated with becoming a public company.
Depreciation. Depreciation expense consists of depreciation on our property and equipment. We depreciate our assets over their estimated useful life. We estimate leasehold improvements to have a 1-year life; computer equipment to have a 2-year life; machinery and equipment to have a 5-year life and furniture and office equipment to have a 7-year life. Property and equipment assets acquired as a result of the acquisition of Moleculin, LLC were given a 2-year life given the assessment at acquisition of their age and condition and expected useful remaining life.
Other income (expense), net
Other income (expense), net consists of interest expense associated with our notes payable and interest income.
51
Accounting for warrants
We issued warrants to purchase shares of common stock related to equity transactions in 2016. We account for our warrants issued in accordance with Accounting Standards Codification (ASC) Topic 815, Derivatives and Hedging, guidance applicable to derivative instruments, which requires every derivative instrument within its scope to be recorded on the balance sheet as either an asset or liability measured at its fair value, with changes in fair value recognized in earnings for liability classified warrants. Based on this guidance, we determined that our warrants meet the criteria for classification as equity. Accordingly, the warrants were classified as equity and are not subject to remeasurement at each balance sheet date. The fair value was estimated using the Black-Scholes option pricing model, based on the market value of the underlying common stock at the measurement date, the contractual term of the warrant, risk-free interest rates, expected dividends and expected volatility of the price of the underlying common stock.
Stock-based compensation
Stock based compensation transactions are recognized as compensation expense in the statement of operations based on their fair values on the date of the grant, with the compensation expense recognized over the period in which a grantee is required to provide service in exchange for the award. We estimate the fair value of options granted using the Black-Scholes option valuation model. This estimate uses assumptions regarding a number of inputs that require us to make significant estimates and judgments.
Because we are a relatively new publicly traded common stock the expected volatility assumption was based on industry peer information.
Income taxes
We account for income taxes using ASC 740 Income Taxes. ASC 740 Income Taxes is an asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in our financial statements or tax returns. In estimating future tax consequences, ASC 740 generally considers all expected future events other than enactments of and changes in the tax law or rates. The measurement of deferred tax assets is reduced, if necessary, by the amount of any tax benefits that, based on available evidence, are not expected to be realized. Valuation allowances are provided if, considering available evidence, it is more likely than not that the deferred tax assets will not be realized. ASC 740 clarifies the criteria that must be met prior to recognition of the financial statement benefit of a position taken in a tax return. ASC 740 provides a benefit recognition model with a two-step approach consisting of “more-likely-than-not” recognition criteria, and a measurement attribute that measures a given tax position as the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate settlement. ASC 740 also requires the recognition of liabilities created by differences between tax positions taken in a tax return and amounts recognized in the financial statements.
Recent accounting pronouncements
In May 2014, the FASB issued Accounting Standard Update ("ASU") 2014-09, Revenue from Contracts with Customers (Topic 606), which will replace numerous requirements in U.S. GAAP, including industry-specific requirements, and provide companies with a single revenue recognition model for recognizing revenue from contracts with customers. The core principle of the new standard is that a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In August 2015, the FASB approved a proposal to defer the effective date of the guidance until annual and interim reporting periods beginning after December 15, 2017. The Company is currently evaluating the impact that this standard will have on its financial statements at the time the Company starts to generate revenue or enters into other contractual arrangements, which the Company does not expect in the near term.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. Under the new guidance, management will be required to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures in certain circumstances. The provisions of this ASU are effective for annual periods ending after December 15, 2016, and for annual and interim periods thereafter; early adoption is permitted. This disclosure is effective within these financial statements for the year ended December 31, 2016.
In January 2016, the FASB issued ASU No. 2016-01, Financial Instruments – Overall: Recognition and Measurement of Financial Assets and Financial Liabilities (“ASU 2016-01”). ASU 2016-01 affects the accounting for equity investments, financial liabilities under the fair value option and the presentation and disclosure requirements of financial instruments. ASU 2016-01 is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. The Company is currently evaluating the impact that this standard will have on its financial statements.
52
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”). Under ASU 2016-02, an entity will be required to recognize right-of-use assets and lease liabilities on its balance sheet and disclose key information about leasing arrangements. ASU 2016-02 offers specific accounting guidance for a lessee, a lessor and sale and leaseback transactions. Lessees and lessors are required to disclose qualitative and quantitative information about leasing arrangements to enable a user of the financial statements to assess the amount, timing and uncertainty of cash flows arising from leases. For public companies, ASU 2016-02 is effective for annual reporting periods beginning after December 15, 2018, including interim periods within that reporting period, and requires a modified retrospective adoption, with early adoption permitted. The Company is currently evaluating the impact that this standard will have on its financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation (Topic 718). The new guidance changes the accounting and simplifies various aspects of the accounting for share-based payments to employees. The guidance allows for a policy election to account for forfeitures as they occur or based on an estimated number of awards that are expected to vest. ASU 2016-09 is effective for annual periods beginning after December 15, 2016, with early adoption permitted. The adoption of this standard on January 1, 2017, did not have a significant impact on the Company’s financial statements.
In August 2016, the FASB issued ASU, Statement of Cash Flows (Topic 230). This ASU applies to all entities that are required to present a statement of cash flows under Topic 230. The amendments provide guidance on eight specific cash flow issues and includes clarification on how these items should be classified in the statement of cash flows and is designed to help eliminate diversity in practice as to where items are classified in the cash flow statement. Furthermore in November 2016, the FASB issued additional guidance on this Topic that requires amounts generally described as restricted cash and restricted cash equivalents to be included with cash and cash equivalents when reconciling the statement of cash flows. This ASU is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years, with earlier application permitted for all entities. We plan to adopt the provisions of this ASU for our fiscal year beginning January 1, 2018 and are currently evaluating the impact the adoption of this new accounting standard will have on our financial statements.
On November 20, 2015, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update ("ASU") 2015-17, "Balance Sheet Classification of Deferred Taxes", requiring all deferred tax assets and liabilities, and any related valuation allowance, to be classified as non-current on the balance sheet. The classification change for all deferred taxes as non-current simplifies entities’ processes as it eliminates the need to separately identify the net current and net non-current deferred tax asset or liability in each jurisdiction and allocate valuation allowances. The Company elected to adopt the accounting in the fourth quarter of 2016.
The Company does not believe that any other recently issued effective pronouncements, or pronouncements issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISKS
Not applicable to us, as we are a smaller reporting company.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required by this item are set forth beginning in Item 15 of this report and are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
There have been no disagreements with our independent registered public accountants on accounting or financial disclosure matters during our two most recent fiscal years.
53
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures.
Our management, including our principal executive officer and principal financial officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of the end of the period covered by this Form 10-K. Based on this evaluation, our Chief Executive Officer (“CEO”) and our Chief Financial Officer (“CFO”), concluded that as a result of the material weakness in our internal controls over financial reporting discussed below, our disclosure controls and procedures were not effective at ensuring that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission's rules and forms and that such information is accumulated and communicated to our management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding disclosure.
Attestation Report of the Registered Public Accounting Firm
Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal controls over financial reporting for as long as we are an “emerging growth company” pursuant to the provisions of the Jumpstart Our Business Startups Act.
Management's Report on Internal Control Over Financial Reporting
Our principal executive officer and our principal accounting and financial officer, are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Management conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2016. In making this assessment, management used the criteria described in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Our management concluded that our internal controls over financial reporting were, and continue to be ineffective, as of December 31, 2016 due to a material weakness in our internal controls due to the lack of segregation of duties.
It should be noted that any system of controls, however well designed and operated, can provide only reasonable and not absolute assurance that the objectives of the system are met. In addition, the design of any control system is based in part upon certain assumptions about the likelihood of certain events. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote.
In light of the material weakness described below, we performed additional analysis and other post-closing procedures to ensure our financial statements were prepared in accordance with generally accepted accounting principles. Accordingly, we believe that the financial statements included in this report fairly present, in all material respects, our financial condition, results of operations and cash flows for the periods presented.
A material weakness is a control deficiency (within the meaning of the Public Company Accounting Oversight Board (PCAOB) Auditing Standard 1305) or combination of control deficiencies that result in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected.
During the last quarter of fiscal 2016, and as our operational activities increased, management determined that it does not have sufficient segregation of duties within its accounting functions, which is a basic internal control. Due to our size and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Management evaluated the impact of our failure to maintain effective segregation of duties on our assessment of our internal control over financial reporting and has concluded that the control deficiency represents a material weakness. Management intends to increase its accounting staff, as soon as economically feasible and sustainable, to remediate this material weakness.
In reporting its third quarter results, we identified a material weakness that since has been remediated. Our accounting for business combinations resulted in an error that caused a restatement of one of our quarterly reports. To remediate this material weakness, management engaged an outside firm to assist management with such accounting and will continue to use outside firms as a resource to deal with other non-recurring or unusual transactions that have occurred since the business combination.
54
Changes in Internal Control over Financial Reporting
During the last quarter of fiscal 2016, and as our operational activities increased, management determined that it does not have sufficient segregation of duties within its accounting functions, which is a basic internal control. Due to our size and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Management evaluated the impact of our failure to maintain effective segregation of duties on our assessment of our internal control over financial reporting and has concluded that the control deficiency represents a material weakness. Management intends to increase its accounting staff, as soon as economically feasible and sustainable, to remediate this material weakness.
In reporting its third quarter results, we identified a material weakness that since has been remediated. Our accounting for business combinations resulted in an error that caused a restatement of one of our quarterly reports. To remediate this material weakness, management engaged an outside firm to assist management with such accounting and has continued to use this outside firm as a resource to deal with other non-recurring or unusual transactions that have occurred since the business combination.
ITEM 9B. OTHER INFORMATION.
None.
55
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference to our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2016.
Our Board of Directors has adopted a written Code of Business Conduct and Ethics applicable to all officers, directors and employees, which is available on our website (www.moleculin.com) under “Governance Documents” within the “Corporate Governance” section. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding amendment to, or waiver from, a provision of this Code and by posting such information on the website address and location specified above.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference to our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2016.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference to our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2016.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference to our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2016.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated by reference to our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of the fiscal year ended December 31, 2016.
56
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENTS
| (a) | Documents filed as part of this Report |
| 1. | Financial Statements. |
The following financial statements and notes thereto which are attached hereto beginning on page F-1 have been included by reference into Item 8 of this part of the annual report on Form 10-K:
| Page | ||
| 1. | Nature of Business and Liquidity | F-8 |
| 2. | Summary of Significant Accounting Policies | F-9 |
| 3. | – Intangible Assets | F-12 |
| 4. | – Convertible Notes Payable | F-15 |
| 5. | – Equity | F-16 |
| 6. | – Income Taxes | F-18 |
| 7. | Commitments and Contingencies | F-20 |
| 8. | Subsequent Events | F-24 |
2. Financial Statement Schedules
All schedules are omitted because they are inapplicable or not required or the required information is shown in the financial statements or notes thereto.
3. Exhibits
The information required by this Item is set forth in the Exhibit Index that follows the signature page of this Annual Report.
57
Moleculin Biotech, Inc.
Index to Financial Statements
F-1
| REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM |
Board of Directors and Stockholders
Moleculin Biotech, Inc.
We have audited the accompanying balance sheet of Moleculin Biotech, Inc. (a Delaware corporation) (the “Company”) as of December 31, 2016, and the related statements of operations, stockholders’ equity, and cash flows for the year ended December 31, 2016. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Moleculin Biotech, Inc. as of December 31, 2016, and the results of its operations and its cash flows for the year ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred an accumulated deficit of $4,674,721 since inception and has not yet generated any revenue from operations. These conditions, along with other matters as set forth in Note 2, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ GRANT THORNTON LLP
Houston, Texas
April 3, 2017
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Moleculin Biotech, Inc.
Houston, Texas
We have audited the accompanying balance sheet of Moleculin Biotech, Inc. as of December 31, 2015 and the related statements of operations, stockholders’ equity (deficit), and cash flows for the period from July 28, 2015 (Inception) to December 31, 2015. Moleculin Biotech, Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Moleculin Biotech, Inc. as of December 31, 2015 and the results of its operations and cash flows for the period from July 28, 2015 (Inception) to December 31, 2015 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that Moleculin Biotech, Inc. will continue as a going concern. As discussed in Note 2 to the financial statements, Moleculin Biotech, Inc. incurred an accumulated loss and has not yet generated any revenue from operations that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/GBH CPAs, PC
GBH CPAs, PC
www.gbhcpas.com
Houston, Texas
March 21, 2016
F-3
Balance Sheets
| December 31, 2016 | December 31, 2015 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 5,007,216 | $ | 28,091 | ||||
| Prepaid expenses | 215,052 | – | ||||||
| Total current assets | 5,222,268 | 28,091 | ||||||
| Long-Term Assets: | ||||||||
| Furniture and equipment, net of accumulated depreciation of $6,162 and $0, respectively | 23,128 | – | ||||||
| Intangible assets | 11,147,540 | – | ||||||
| Total Assets | $ | 16,392,936 | $ | 28,091 | ||||
| Liabilities and Stockholders' Equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | 1,048,655 | $ | 322,790 | ||||
| Convertible notes payable | 296,412 | 450,000 | ||||||
| Total current liabilities | 1,345,067 | 772,790 | ||||||
| Long-term deferred compensation – related party | 87,500 | – | ||||||
| Total Liabilities | 1,432,567 | 772,790 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders' Equity: | ||||||||
| Preferred stock, $0.001 par value; 5,000,000 authorized, no shares issued and outstanding | – | – | ||||||
| Common stock, $0.001 par value; 75,000,000 authorized, 12,164,852 and 6,661,000 shares issued and outstanding, respectively | 12,165 | 6,661 | ||||||
| Subscription receivable | - | (3,000 | ) | |||||
| Additional paid-in capital | 19,622,925 | – | ||||||
| Accumulated deficit | (4,674,721 | ) | (748,360 | ) | ||||
| Total Stockholders' Equity (Deficit) | 14,960,369 | (744,699 | ) | |||||
| Total Liabilities and Stockholders' Equity (Deficit) | $ | 16,392,936 | $ | 28,091 | ||||
See accompanying notes to the financial statements.
F-4
Statements of Operations
| Year Ended December 31, | From July 28, 2015 (Inception) Through December 31, | |||||||
| 2016 | 2015 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 1,495,561 | 260,418 | ||||||
| General and administrative | 2,381,424 | 477,810 | ||||||
| Depreciation | 6,162 | - | ||||||
| Total Operating Expenses | 3,883,147 | 738,228 | ||||||
| Loss from operations | (3,883,147 | ) | (738,228 | ) | ||||
| Other expense: | ||||||||
| Interest expense | (43,214 | ) | (10,132 | ) | ||||
| Net loss | $ | (3,926,361 | ) | $ | (748,360 | ) | ||
| Net loss per common share - basic and diluted | $ | (0.40 | ) | $ | (0.13 | ) | ||
| Weighted average common shares outstanding - basic and diluted | 9,827,510 | 5,691,803 | ||||||
See accompanying notes to the financial statements.
F-5
Statements of Cash Flows
| Year Ended December 31, | From July 28, 2015 (Inception) Through December 31, | |||||||
| 2016 | 2015 | |||||||
| Cash Flows from Operating Activities: | ||||||||
| Net loss | $ | (3,926,361 | ) | $ | (748,360 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 6,162 | - | ||||||
| Stock-based compensation | 323,974 | - | ||||||
| Deferred CEO compensation | 87,500 | |||||||
| Stock issued for licenses used for research and development | - | 2,061 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses | (215,052 | ) | ||||||
| Accounts payable and accrued expenses | (41,128 | ) | 322,790 | |||||
| Net Cash Used in Operating Activities | (3,764,905 | ) | (423,509 | ) | ||||
| Cash Flows from Investing Activities: | ||||||||
| Cash paid for purchase of fixed assets | (21,470 | ) | - | |||||
| Cash paid for acquisition of Moleculin, LLC, net with cash acquired | (99,638 | ) | - | |||||
| Net Cash Used in Investing Activities | (121,108 | ) | - | |||||
| Cash Flows from Financing Activities: | ||||||||
| Proceeds from notes payable | 165,000 | 450,000 | ||||||
| Payments on notes payable | (469,939 | ) | - | |||||
| Proceeds from sale of common stock, net of cash stock issuance costs | 9,170,077 | 1,600 | ||||||
| Net Cash Provided by Financing Activities | 8,865,138 | 451,600 | ||||||
| Net change in cash and cash equivalents | 4,979,125 | 28,091 | ||||||
| Cash and cash equivalents, at beginning of period | 28,091 | - | ||||||
| Cash and cash equivalents, at end of period | $ | 5,007,216 | $ | 28,091 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for interest | $ | 4,585 | $ | - | ||||
| Cash paid for income taxes | $ | - | $ | - | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Common stock issued for the Acquisition of Moleculin, LLC | $ | 9,773,586 | $ | - | ||||
| Common stock issued for conversion of debt | $ | 363,792 | $ | - | ||||
| Warrants issued for services provided | $ | 374,763 | $ | - | ||||
| Common stock issued for services provided | $ | 157,680 | $ | - | ||||
| Shares subscribed | $ | - | $ | 3,000 | ||||
See accompanying notes to the financial statements.
F-6
Statements of Stockholders’ Equity
| Common Stock | ||||||||||||||||||||||||
| Number | Amount | Additional Paid-In-Capital | Subscriptions Receivable | Accumulated Loss | Stockholders' Equity (Deficit) | |||||||||||||||||||
| Stock issued for cash and subscription receivable | 4,600,000 | $ | 4,600 | $ | $ | (3,000 | ) | $ | $ | 1,600 | ||||||||||||||
| Stock issued for licenses | 2,061,000 | 2,061 | 2,061 | |||||||||||||||||||||
| Net loss | (748,360 | ) | (748,360 | ) | ||||||||||||||||||||
| Balance at December 31, 2015 | 6,661,000 | 6,661 | - | (3,000 | ) | (748,360 | ) | (744,699 | ) | |||||||||||||||
| Private issuance @ $3.00 / share | 234,297 | 234 | 702,660 | 702,894 | ||||||||||||||||||||
| Issued for Moleculin acquisition | 999,931 | 1,000 | 5,998,586 | 5,999,586 | ||||||||||||||||||||
| Issued for technology | 629,000 | 629 | 3,773,371 | 3,774,000 | ||||||||||||||||||||
| Issued for cash - IPO, net of stock issuance costs of $1,150, 736 | 1,540,026 | 1,540 | 8,087,880 | 8,089,420 | ||||||||||||||||||||
| Warrants issued for services | 374,763 | 374,763 | ||||||||||||||||||||||
| Stock granted for services | 24,000 | 24 | 157,656 | 157,680 | ||||||||||||||||||||
| Stock option expense | 166,294 | 166,294 | ||||||||||||||||||||||
| Issued for convertible debt | 2,076,598 | 2,077 | 361,715 | 363,792 | ||||||||||||||||||||
| Subscription agreement settled for cash | 3,000 | 3,000 | ||||||||||||||||||||||
| Net loss | (3,926,361 | ) | (3,926,361 | ) | ||||||||||||||||||||
| Balance at December 31, 2016 | 12,164,852 | $ | 12,165 | $ | 19,622,925 | $ | 0 | $ | (4,674,721 | ) | $ | 14,960,369 | ||||||||||||
See accompanying notes to the financial statements.
F-7
Notes to the Financial Statements
| 1. | Nature of Business and Liquidity |
The terms “MBI” or “the Company”, “we”, “our” and “us” are used herein to refer to Moleculin Biotech, Inc. MBI is a preclinical pharmaceutical company organized as a Delaware corporation in July 2015 to focus on the development of anti-cancer drug candidates, some of which are based on license agreements with The University of Texas System on behalf of the M.D. Anderson Cancer Center, which we refer to as MD Anderson.
Our lead drug candidate is liposomal Annamycin, which we refer to as Annamycin, an anthracycline being studied for the treatment of relapsed or refractory acute myeloid leukemia, or AML. In August 2015, the Company entered into a rights transfer agreement with AnnaMed, Inc. (“AnnaMed”), a company affiliated with certain members of the Company’s management and board of directors, pursuant to which, in exchange for 1,431,000 shares of the Company’s common stock, AnnaMed agreed to transfer to MBI any and all data it had regarding the development of Annamycin and the Annamycin investigative new drug application (“IND”) with the U.S. Food and Drug Administration (“FDA”) , including all trade secrets, know-how, confidential information and other intellectual property rights held by AnnaMed. Annamycin has been in clinical trials pursuant to an IND that had been filed with the FDA. This IND was terminated due to a lack of activity by a prior drug developer. However, in the course of our review of that data, we identified that Annamycin may have greater potential for efficacy than we originally believed, based on an unexpected potential opportunity to increase the drug’s Maximum Tolerable Dose (“MTD”). We determined to adjust our clinical strategy by adding in a Phase I arm to our next Phase II trial.
Because the prior developer of Annamycin allowed their IND to lapse, we are required to submit a new IND for continued clinical trials with Annamycin. We filed our IND application, with the clinical strategy of increasing the MTD mentioned above, for Annamycin on February 10, 2017. In subsequent discussions with us, FDA requested certain revisions to the protocol, additional information, and additional data related to Chemistry, Manufacturing and Controls (“CMC”). We have the additional information, have made the requested revisions to the protocol, and we are working on developing the CMC data. In the interim, we have withdrawn the IND application in order to resubmit it when the requested data are available. We believe that resubmission of the IND application will occur in time for the IND to go into effect and the announcement of the beginning of Phase I/II clinical trials in the first half of 2017. However, if we are unable to obtain the required CMC data on a timely basis, we will be delayed in resubmitting our IND application, which will delay the commencement of our clinical trials beyond the first half of 2017.
The Annamycin drug substance is no longer covered by any existing patent protection. We intend to submit patent applications for formulation, synthetic process and reconstitution related to our Annamycin drug product candidate, although there is no assurance that we will be successful in obtaining such patent protection. On March 21, 2017, we received notice that FDA had granted us Orphan Drug designation for Annamycin for the treatment of AML . Orphan Drug status could entitle us to market exclusivity of up to 7 and 10 years from the date of approval of a New Drug Application (“NDA”) or Marketing Authorization Application (“MAA”), in the US and the European Union (“EU”), respectively. Separately, the FDA may also grant market exclusivity of up to five years for newly approved new chemical entities (of which Annamycin would be one), but there can be no assurance that such exclusivity will be granted.
F-8
We have two other drug development projects in progress, one involving a portfolio of small molecules, which we refer to as the WP1066 Portfolio, focused on the modulation of key oncogenic transcription factors involved in the progression of cancer, and the WP1122 Portfolio, a suite of molecules targeting the metabolic processes involved in cancer in general, and glioblastoma (the most common form of brain tumor) in particular. We have been granted royalty-bearing, worldwide, exclusive licenses for the patent and technology rights related to our WP1066 Portfolio and WP1122 Portfolio drug technologies, as these patent rights are owned by MD Anderson.
On August 11, 2015, the Company entered into a rights transfer agreement for WP1122 with IntertechBio Corporation (“IntertechBio”), a company affiliated with certain members of our management, whereby IntertechBio agreed to assign its license or sublicense its license to certain metabolic inhibitor technology owned by MD Anderson. In consideration, the Company issued 630,000 common shares to IntertechBio. IntertechBio agreed to make payments to MD Anderson including an up-front payment, license documentation fee, annual maintenance fee, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. The Company has assumed the rights and obligations of IntertechBio under the license agreement with MD Anderson. Therefore, all out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by the Company.
In accordance with FASB ASC Topic 280, Segment Reporting, we view our operations and manage our business as principally one segment. As a result, the financial information disclosed herein represents all of the material financial information related to our principal operating segment.
The Company filed a registration statement on Form S-1 (which was declared effective on May 2, 2016) with respect to the Company’s initial public offering of shares of its common stock (“IPO”) to fund the development of its technologies. Prior to the declaration of effectiveness of the registration statement on Form S-1, we acquired Moleculin, LLC which was merged with and into MBI. Moleculin, LLC was the holder of a license agreement with MD Anderson covering technology referred to as the WP1066 Portfolio, which is focused on the modulation of key oncogenic transcription factors.
| 2. | Summary of Significant Accounting Policies |
The accompanying audited financial statements and related notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for financial information, and in accordance with the rules and regulations of the United States Securities and Exchange Commission (the “SEC”).
Use of Estimates in Financial Statement Presentation - The preparation of these financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Acquisition – We acquired Moleculin, LLC (“Moleculin”) on May 2, 2016, and, going forward our financial statements include the operations of Moleculin, LLC. We account for acquired businesses using the acquisition method of accounting, which requires, among other things, that assets acquired and liabilities assumed be recognized at their estimated fair values as of the acquisition date and that the fair value of acquired in-process research and development (“IPR&D”) be recorded on the balance sheet. Transaction costs are expensed as incurred. Any excess of the consideration transferred over the assigned values of the net assets acquired will be recorded as goodwill. The Company obtained input from third-parties regarding its tangible and intangible assets and other information necessary to measure the fair value of the assets acquired and liabilities assumed in connection with the acquisition of Moleculin, LLC.
Going Concern - These financial statements have been prepared on a going concern basis, which assumes the Company will continue to realize its assets and discharge its liabilities in the normal course of business. The continuation of the Company as a going concern is dependent upon the ability of the Company to obtain continued financial support from its stockholders’, necessary equity financing to continue operations and the attainment of profitable operations. As of December 31, 2016, the Company has incurred an accumulated deficit of $4,674,721 since inception, and had not yet generated any revenue from operations. Additionally, management anticipates that its cash on hand as of December 31, 2016 plus the additional cash generated from its equity offering subsequent to year-end, discussed further within these notes to the financial statements, is sufficient to fund its planned operations into but not beyond the near term. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern. The Company may seek additional funding through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements, other collaborations, strategic alliances and licensing arrangements and delay planned cash outlays or a combination thereof. Management cannot be certain that such events or a combination thereof can be achieved.
F-9
Cash and Cash Equivalents - The Company considers all highly liquid accounts with original maturities of three months or less at the date of acquisition to be cash equivalents. Periodically, the Company may carry cash balances at financial institutions in excess of the federally insured limit of $250,000. The amount in excess of the FDIC insurance at December 31, 2016 was $256,836 and $0 as of December 31, 2015. Compensation expense is recognized only for share based payments expected to vest.
Property and equipment - Property and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line depreciation method as follows:
| Leasehold improvement | Shorter of estimated useful lives or the term of the lease |
| Computer equipment | 2 years* |
| Machinery and equipment | 5 years* |
| Furniture and office equipment | 7 years* |
*Property and equipment assets acquired in the merger with Moleculin, LLC are being depreciated over a 2 year useful life due to their age and condition and expected remaining life assessed at merger date.
Intangible assets - Intangible assets with finite lives are amortized using the straight-line method over their estimated period of benefit. If an intangible asset is identified as an in-process research & development asset, then no amortization will occur until the development is complete. If the associated research and development effort is abandoned, the related assets will be written-off and the Company will record a noncash impairment loss on its statements of operations. For those compounds that reach commercialization, the IPR&D assets will be amortized over their estimated useful lives.
We evaluate the recoverability of intangible assets periodically and take into account events or circumstances that warrant revised estimates of useful lives or that indicate that impairment exists. No material impairments of intangible assets have been identified during any of the periods presented. Intangible assets are tested for impairment on an annual basis, and between annual tests if indicators of potential impairment exist, using a fair-value-based approach.
Beneficial Conversion Feature - From time to time, the Company may issue convertible notes that have conversion prices that create an embedded beneficial conversion feature on the issuance date. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of any attached equity instruments, if any related equity instruments were granted with the debt. The Company estimated the fair value of its common stock on the dates issued. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid-in capital, if any. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
F-10
Income Taxes - The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of reported assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company must then assess the likelihood that the resulting deferred tax assets will be realized. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
The Company accounts for uncertain tax positions in accordance with the provisions of ASC 740-10 which prescribes a recognition threshold and measurement attribute for financial statement disclosure of tax positions taken, or expected to be taken, on its tax return. The Company evaluates and records any uncertain tax positions based on the amount that management deems is more likely than not to be sustained upon examination and ultimate settlement with the tax authorities in the tax jurisdictions in which it operates.
Stock-based Compensation - Stock-based compensation expense includes the estimated fair value of equity awards vested during the reporting period. The expense for equity awards vested during the reporting period is determined based upon the grant date fair value of the award and is recognized as expense over the applicable vesting period of the stock award using the straight-line method.
Loss Per Common Share - Basic net loss per common share are computed by dividing net loss available to common shareholders by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is determined using the weighted-average number of common shares outstanding during the period, adjusted for the dilutive effect of common stock equivalents. In periods when losses are reported, the weighted-average number of common shares outstanding excludes common stock equivalents, because their inclusion would be anti-dilutive. As of December 31, 2016, the Company’s potentially dilutive shares, which were not included in the calculation of net loss per share, included notes convertible to 1,821,013 common shares, options to purchase 510,000 common shares and warrants to purchase 107,802 common shares.
Research and Development Costs - Research and development costs are expensed as incurred.
Subsequent Events - The Company’s management reviewed all material events through the date these financial statements were issued for subsequent event disclosure consideration and has noted such events as described in Note 8.
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standard Update ("ASU") 2014-09, Revenue from Contracts with Customers (Topic 606), which will replace numerous requirements in U.S. GAAP, including industry-specific requirements, and provide companies with a single revenue recognition model for recognizing revenue from contracts with customers. The core principle of the new standard is that a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In August 2015, the FASB approved a proposal to defer the effective date of the guidance until annual and interim reporting periods beginning after December 15, 2017. The Company is currently evaluating the impact that this standard will have on its financial statements at the time the Company starts to generate revenue or enters into other contractual arrangements, which the Company does not expect in the near term.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. Under the new guidance, management will be required to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures in certain circumstances. The provisions of this ASU are effective for annual periods ending after December 15, 2016, and for annual and interim periods thereafter; early adoption is permitted. This disclosure is effective within these financial statements for the year ended December 31, 2016.
In January 2016, the FASB issued ASU No. 2016-01, Financial Instruments – Overall: Recognition and Measurement of Financial Assets and Financial Liabilities (“ASU 2016-01”). ASU 2016-01 affects the accounting for equity investments, financial liabilities under the fair value option and the presentation and disclosure requirements of financial instruments. ASU 2016-01 is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. The Company is currently evaluating the impact that this standard will have on its financial statements.
F-11
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”). Under ASU 2016-02, an entity will be required to recognize right-of-use assets and lease liabilities on its balance sheet and disclose key information about leasing arrangements. ASU 2016-02 offers specific accounting guidance for a lessee, a lessor and sale and leaseback transactions. Lessees and lessors are required to disclose qualitative and quantitative information about leasing arrangements to enable a user of the financial statements to assess the amount, timing and uncertainty of cash flows arising from leases. For public companies, ASU 2016-02 is effective for annual reporting periods beginning after December 15, 2018, including interim periods within that reporting period, and requires a modified retrospective adoption, with early adoption permitted. The Company is currently evaluating the impact that this standard will have on its financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation-Stock Compensation (Topic 718). The new guidance changes the accounting and simplifies various aspects of the accounting for share-based payments to employees. The guidance allows for a policy election to account for forfeitures as they occur or based on an estimated number of awards that are expected to vest. ASU 2016-09 is effective for annual periods beginning after December 15, 2016, with early adoption permitted. The adoption of this standard on January 1, 2017, did not have a significant impact on the Company’s financial statements.
In August 2016, the FASB issued ASU, Statement of Cash Flows (Topic 230). This ASU applies to all entities that are required to present a statement of cash flows under Topic 230. The amendments provide guidance on eight specific cash flow issues and includes clarification on how these items should be classified in the statement of cash flows and is designed to help eliminate diversity in practice as to where items are classified in the cash flow statement. Furthermore in November 2016, the FASB issued additional guidance on this Topic that requires amounts generally described as restricted cash and restricted cash equivalents to be included with cash and cash equivalents when reconciling the statement of cash flows. This ASU is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years, with earlier application permitted for all entities. We plan to adopt the provisions of this ASU for our fiscal year beginning January 1, 2018 and are currently evaluating the impact the adoption of this new accounting standard will have on our financial statements.
On November 20, 2015, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update ("ASU") 2015-17, "Balance Sheet Classification of Deferred Taxes", requiring all deferred tax assets and liabilities, and any related valuation allowance, to be classified as non-current on the balance sheet. The classification change for all deferred taxes as non-current simplifies entities’ processes as it eliminates the need to separately identify the net current and net non-current deferred tax asset or liability in each jurisdiction and allocate valuation allowances. The Company elected to adopt the accounting in the fourth quarter of 2016.
The Company does not believe that any other recently issued effective pronouncements, or pronouncements issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
| 3. | Intangible Assets |
The Acquisition of Moleculin, LLC
On May 2, 2016, Moleculin, LLC, a Texas limited liability company, was merged with and into the Company. As a result of the merger, the Company issued to the holders of Moleculin equity interests an aggregate of 999,931 shares of the Company’s common stock valued at $5,999,586, based on the estimated acquisition-date fair value of our common stock of $6.00 per share, equal to the IPO price announced in our prospectus filed on that date. These shares contain certain trading restrictions. Prior to the Company’s acquisition of Moleculin, the Company had loaned $57,822 to Moleculin which was treated as part of the consideration paid to acquire Moleculin.
As additional consideration payable to the Moleculin unit holders, we agreed pursuant to the merger agreement that if drugs for dermatology indications are successfully developed by us using any of the Existing IP Assets, then the Moleculin, LLC unit holders, in the aggregate, will be entitled to receive a 2.5% royalty on the net revenues generated by such drugs. Any such net revenues would include a deduction for license fees or royalty obligations payable to MD Anderson for such Existing IP Assets. The merger agreement defined “Existing IP Assets” to mean all intellectual property, licensed by us and Moleculin as of the time of the merger, including, without limitation, the intellectual property licensed from MD Anderson under the Patent and Technology License Agreement entered into by and between IntertechBio Corporation and MD Anderson dated April 2, 2012, as amended, and the Patent and Technology License Agreement dated June 21, 2010, as amended, between MD Anderson and Moleculin, LLC, but excluding any intellectual property relating to Annamycin. The right to receive the contingent royalty payments described herein is limited to drugs developed only for dermatology indications, and does not include drugs developed for any other indications. We have no obligation of any nature to pursue the development of any drugs for dermatology indications.
Our acquisition of Moleculin, LLC, occurring prior to our IPO offering, provided us with the rights to the license agreement that Moleculin, LLC had with MD Anderson covering the WP1066 Portfolio. However, Moleculin, LLC had previously granted Houston Pharmaceuticals, Inc. (“HPI”), a related party, an option, which could be exercised at any time, to obtain an exclusive sub-license to develop the WP1066 Portfolio in all non-dermatological fields. Moleculin, LLC had previously pursued development of the WP1066 Portfolio for treatment of psoriasis, however, psoriasis related clinical trials had been terminated. Because WP1066 has shown significant activity against a wide range of tumors, Moleculin, LLC’s focus prior to the acquisition included the development of drugs for cancer treatment. However, the exclusive sub-license option held by HPI precluded Moleculin, LLC from pursuing drug development related to non-skin cancers, in addition to potentially creating significant intellectual property, clinical and commercialization risks associated with drug development for skin cancers. Re-acquisition of the HPI option was therefore essential for the values of both the WP1066 Portfolio and Moleculin, LLC.
F-12
Additionally, the merger agreement contained mutual representations and warranties between the parties. Pursuant to the merger agreement, we agreed for a period of six years to indemnify and hold harmless each present and former director and/or officer of Moleculin, LLC whom Moleculin, LLC would have had the power to indemnify under Delaware law that is made a party or threatened to be made a party to any threatened, pending or completed proceeding or claim by reason of the fact that he or she was a director or officer of the Moleculin, LLC prior to the effective time of the merger and arising out of actions or omissions of the indemnified party in any such capacity occurring at or prior to the effective time of the merger against any losses or damages reasonably incurred in connection with any claim. To our knowledge, no such proceeding or claim exists or has been threatened on the date hereof.
In connection with the acquisition of Moleculin, LLC, we also negotiated on behalf of Moleculin, LLC two agreements with HPI. Under the first agreement, HPI’s option to obtain the aforementioned exclusive sublicense was terminated in exchange for a payment of $100,000 and the issuance of 629,000 shares of our common stock, valued at $6 per share. Under the second agreement (HPI Out-Licensing Agreement), HPI has received a non-exclusive technology rights and development sublicense under which it may continue its ongoing work to develop the WP1066 Portfolio related to treatment of non-skin cancer. Pursuant to this HPI Out-Licensing Agreement, we agreed to make payments to HPI totaling $750,000 over a three-year period, of which $75,000 was paid in calendar year 2016. The Company expenses such costs as incurred as research and development expense, commencing after the IPO offering in exchange for HPI allowing us to access any data, information or know-how resulting from the research and development conducted by HPI. As of December 31, 2016, notwithstanding our obligation to make the foregoing payments, the HPI Out-Licensing Agreement does not obligate HPI to conduct any specific research or to meet any milestones. Pursuant to the HPI Out-Licensing Agreement, we have the right within three years of the effective date to buy-out from HPI all rights granted to HPI under the agreement for a payment of $1.0 million. Upon our exercise of the buy-out we will no longer be obligated to make any payments to HPI remaining from the $750,000 obligation discussed above. If we do not exercise the foregoing buy-out right within three years, the license granted to HPI shall convert into an exclusive license. As such, if we do not exercise the buy-out right for any reason, we will no longer have access to the non-skin cancer uses of the WP1066 Portfolio. As noted above, this will also potentially create risks for the development of skin cancer drugs. We do not intend to set aside and designate cash and cash equivalents in the amount of $1.0 million to make the buy-out payment. If we ultimately decide to exercise the buy-out right from HPI, we will need to raise additional funds to make the buy-out payment. We cannot assure that such additional funding will be available on satisfactory terms, or at all.
The agreements with HPI were executed on May 2, 2016, simultaneously with the closing of the Moleculin, LLC acquisition, and were non-cancelable but contingent on the Company’s ability to complete the IPO by June 30, 2016. They became effective on May 31, 2016.
The termination of the HPI option was completed on behalf of Moleculin, LLC, which was required to enable the sale of Moleculin, LLC by materializing the value of its most significant asset, and was non-cancelable by either party. Further, the HPI option termination price was determined simultaneously with the acquisition on May 2, 2016 as our IPO price was established at that time. Accordingly, we concluded that this transaction was primarily for the benefit of Moleculin, LLC and its former owners, resulting in control of the underlying intellectual property and thereby increasing the value of Moleculin, LLC intangible assets immediately prior to the closing of its acquisition by us.
The HPI option termination price amounted to $3,874,000, consisting of 629,000 shares of our common stock valued at the IPO price of $6.00 per share, and $100,000 paid in cash in July 2016, and was included in acquisition-date liabilities assumed.
Purchase Price Allocation
The acquisition price was allocated to the assets acquired and liabilities assumed based upon their estimated fair values and the information available to management. The following table summarizes the estimated fair values of the assets acquired and liabilities assumed as of the acquisition date.
| Cash | $ | 362 | ||
| Property and equipment | 7,820 | |||
| Intangibles | 11,147,540 | |||
| Total assets acquired | $ | 11,155,722 | ||
| Liability assumed (HPI) | (3,874,000 | ) | ||
| Other liabilities assumed, including $469,939 of notes payable | (1,224,314 | ) | ||
| Net assets acquired/total consideration transferred | $ | 6,057,408 |
F-13
The Company obtained input from third-parties regarding its tangible and intangible assets and other information necessary to measure the fair value of the assets acquired and liabilities assumed in connection with the acquisition of Moleculin, LLC. Management believes all or most of the intangible assets are IPR&D related to the WP1066 Portfolio, and, as such, no amortization has been recorded to date.
Intangible assets consisted of the following at December 31, 2016 and December 31, 2015:
| December 31, 2016 | December 31, 2015 | |||||||
| Intangibles acquired from Moleculin, LLC | $ | 11,147,540 | $ | – | ||||
F-14
Unaudited Pro Forma Results of Operations
The following comparative table presents the unaudited condensed pro forma results of operations that reflect the acquisition of Moleculin as if the acquisition had occurred as of the first day of each period presented, adjusted for items that are directly attributable to the acquisition. This information has been compiled from historical financial statements and is not necessarily indicative of the results that actually would have been achieved had the transaction already occurred or that may be achieved in the future.
| Pro Forma For the Year Ended December 31, 2016 | Pro Forma For Year Ended December 31, 2015 | |||||||
| Total operating expenses | $ | (3,979,316 | ) | (688,101 | ) | |||
| Net loss | $ | (3,962,997 | ) | (164,128 | ) | |||
| Net loss per common share – basic and diluted | $ | (0.42 | ) | (0.04 | ) | |||
| Weighted average outstanding common shares – basic and diluted | 9,401,028 | 4,273,239 | ||||||
The years ended December 31, 2016 and December 31, 2015 are adjusted on a pro forma basis to exclude $145,078 and $422,874, respectively, in net interest expense related to the amortization of deferred financing costs and debt discount amortization for Moleculin, LLC’s convertible notes. The holders of the convertible notes were issued the Company’s common shares upon the Company’s acquisition of Moleculin, LLC.
| 4. | Convertible Notes Payable |
On various dates from August 31, 2015 through January 19, 2016, each as amended on March 10, 2016, the Company entered into seven unsecured promissory notes with three separate third party investors. Each note bears interest at 8.0% per annum and was to mature on the earlier of June 30, 2016 or the completion of an IPO of the Company’s securities.
Since the completion of the IPO occurred prior to June 30, 2016, these notes were to be automatically converted according to their terms into shares of the Company’s common stock at applicable conversion price upon the Company’s IPO to the extent and provided that no holder of these notes was or will be permitted to convert such notes to the extent that the holder or any of its affiliates would beneficially own in excess of 4.99% of our common stock after such conversion. Due to this 4.99% limitation, a portion of these notes was not converted at the time of the IPO and the remaining unconverted principal and accrued interest amounts of the effected notes will remain outstanding and will be converted into shares of our common stock at such time as the 4.99% limitation continues to be met. Until such time as the notes are converted into shares of common stock, the maturity date of the notes will automatically be extended and we will not be required to repay the notes or the accrued interest relating to the notes in cash.
F-15
The IPO was completed on May 31, 2016. On May 31, 2016, pursuant to the conversion feature of the foregoing notes and with restriction of the 4.99% beneficially owned condition limitation, discussed above, the Company issued 1,166,503 common shares in total, reducing convertible debt principal by $183,356 and accrued interest by $17,699. Subsequent to these transactions and through December 31, 2016, an additional 910,095 common shares were issued due to the number of common shares outstanding allowing for conversion of additional shares under the 4.99% beneficially owned condition limitation. This reduced the convertible debt principal by $155,565 and accrued interest by $7,172. The remaining convertible debt without consideration of accrued interest as of December 31, 2016, if converted on December 31, 2016, would result in an additional 1,821,013 common shares to be issued.
The convertible notes were analyzed for a beneficial conversion feature on various issuance dates, at which time it was concluded that a beneficial conversion feature did not exist.
The table below represents the shares that are convertible at December 31, 2016 relating to the principal amounts of these convertible notes payable and excludes any shares that are convertible relating to the associated accrued interest:
| Issuance Date | December 31, 2016 (e) | December 31, 2015 | Conversion Rate | Shares Convertible at Dec 31, 2016 | ||||||||||||
| August 31, 2015(a) | $ | 38,299 | $ | 125,000 | $ | 0.1299 | 294,832 | |||||||||
| September 3, 2015 | 125,000 | 125,000 | 0.1299 | 962,279 | ||||||||||||
| October 6, 2015(a)(c) | 30,280 | 147,000 | 0.20 | 151,402 | ||||||||||||
| October 6, 2015(b) | – | 3,000 | 0.20 | – | ||||||||||||
| October 28, 2015(b) | – | 50,000 | 0.20 | – | ||||||||||||
| January 14, 2016(d) | – | – | 0.20 | – | ||||||||||||
| January 19, 2016 | 82,500 | – | 0.20 | 412,500 | ||||||||||||
| Total | $ | 276,079 | $ | 450,000 | 1,821,013 | |||||||||||
(a) Debt partially converted on May 31, 2016 and on August 19, 2016.
(b) Debt fully converted to common shares on May 31, 2016.
(c) Debt partially converted on September 1, 2016.
(d) Debt fully converted to common shares effective November 30, 2016 .
(e) Excluded from the $276,079 is $20,333 in accrued interest on the convertible notes payable which is included in the accompanying balance sheet.
The common shares relating to the above mentioned convertible notes payable contain the following trading restrictions: (a) beginning 90 days after the initial closing of our IPO and until the one-year anniversary of the initial closing of the IPO, the holder of the note will be able to sell 1% of the number of shares of common stock underlying the note on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; (b) if the common stock price is over $7.00 per share for five consecutive trading days then the holder of the note can sell up to 3% of the number of shares of common stock underlying the note on a monthly basis, subject to a maximum sale on any trading day of 4% of the daily volume; (c) if the common stock price is over $10.00 per share for five consecutive trading days then the holder of the note can sell up to an additional 5% of the number of shares of common stock underlying the note on a monthly basis, subject to a maximum sale on any trading day of 7% of the daily volume; and (d) if the common stock price is over $14.00 per share then the holder of the note is not restricted from making any sales until such time as the common stock price falls back below $14.00 per share; and (b) thereafter, until the two-year anniversary of the initial closing of IPO, the holder of the note can sell on any trading day 10% of the daily volume; provided that if the common stock price is over $10.00 per share then the holder of the note is not restricted from making any sales until such time as the common stock falls back below $10.00 per share. The foregoing lock-up restrictions relate to public sales and do not restrict the transfer of the shares privately, if permitted by applicable law, provided the acquirer of the shares agrees to comply with the above restrictions with respect to any public sales.
| 5. | Equity |
On May 2, 2016, the Company amended and restated its certificate of incorporation to increase the number of shares authorized to 80,000,000 of which 5,000,000 shares of preferred stock are authorized and 75,000,000 shares of common stock are authorized.
F-16
Preferred Stock
We are authorized to issue up to 5,000,000 shares of preferred stock. Our certificate of incorporation authorizes the board to issue these shares in one or more series, to determine the designations and the powers, preferences and relative, participating, optional or other special rights and the qualifications, limitations and restrictions thereof, including the dividend rights, conversion or exchange rights, voting rights (including the number of votes per share), redemption rights and terms, liquidation preferences, sinking fund provisions and the number of shares constituting the series. As of December 31, 2016, there was no issued preferred stock.
Common Stock
On May 31, 2016, the Company completed its IPO and sold 1,540,026 shares of the Company’s common stock. The IPO price per share was $6.00. The Company received net cash proceeds of $8,464,183 after deducting underwriting discounts, commissions and direct offering expenses payable by us. Pursuant to our agreement with our underwriters, as additional compensation, we issued the underwriters warrants to purchase 107,802 shares of common stock exercisable for a period of 5 years from date of issuance at an exercise price of $7.50 per share. The relative fair value of these warrants was $374,763 and is included in stock issuance costs calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) discount rate of 1.39% (2) expected life of 5 years, (3) expected volatility of 80.61%, and (4) zero expected dividends.
In August 2015, the Company agreed to issue 4,600,000 shares of common stock to its directors, officers and founders for subscriptions of $4,600 cash to be received of which $1,600 was received in 2015.
On August 11, 2015, the Company was granted an assignment of a license agreement between MD Anderson and IntertechBio in exchange for 630,000 shares of the Company's common stock. The sublicense gives the Company rights to access certain metabolic inhibitor technology owned by MD Anderson that had been licensed to IntertechBio. The shares were valued at a total of $630 and the related expense included in research and development costs.
On August 21, 2015, the Company acquired the right to the intellectual property of AnnaMed in exchange for 1,431,000 shares of the Company's common stock. The license gives the Company full ownership rights to the data package supporting the FDA IND Number 46869, allowing the Company to resubmit a request for IND to the FDA to begin development work on Annamycin. The shares were valued at a total of $1,431 and the related expense included in research and development costs.
During the period from January 1, 2016 through May 2, 2016, the Company sold 234,297 common shares for $702,894. These shares are subject to the following lock-up agreement, from and after the later of six months after issuance or 90 days from the effective date of our IPO registration statement until the one-year anniversary thereof, (a) the holder of the shares can sell up to 10% of the purchased shares per month, subject to a maximum sale on any trading day of 8% of the daily volume of the common stock; (b) if the common stock price is over $7.00 per share for five consecutive trading days then the holder of the shares can sell up to 20% of the purchased shares per month, subject to a maximum sale on any trading day of 10% of the daily volume of the common stock; and (c) if the common stock price is over $12.00 per share then the holder of the shares is not restricted from making any sales until such time as the common stock price falls back below $12.00 per share.
On June 20, 2016, the Company agreed to issue 24,000 shares of common stock to PCG Advisory Group, the Company’s investor relations firm, for services provided. The fair value of these shares was $157,680 based on the market price on the grant date.
F-17
Adoption of 2015 Stock Plan
On December 5, 2015, the Board of Directors of the Company approved the Company’s 2015 Stock Plan, which was amended on April 22, 2016. The expiration date of the plan is December 5, 2025 and the total number of underlying shares of the Company’s common stock available for grant to employees, directors and consultants under the plan is 2,500,000 shares. The awards under the 2015 Stock Plan can be in the form of stock options, stock awards or stock unit awards. The following is a summary of option activities for the periods ended December 31, 2015 and 2016:
| Number of Shares | Weighted Average Grant Date Fair Value | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value | ||||||||||||||||
| Granted - 2015 | 200,000 | $ | 0.14 | $ | 0.20 | |||||||||||||||
| Outstanding, December 31, 2015 | 200,000 | 0.14 | $ | 0.20 | ||||||||||||||||
| Granted - 2016 | 460,000 | 3.75 | 5.83 | |||||||||||||||||
| Cancelled | (150,000 | ) | 0.14 | 0.20 | ||||||||||||||||
| Outstanding, December 31, 2016 | 510,000 | $ | 3.40 | $ | 5.28 | 9.29 | $ | 275,500 | ||||||||||||
| Exercisable, December 31, 2016 | 50,000 | $ | 0.14 | $ | 0.20 | 3.67 | $ | 275,500 | ||||||||||||
During the year ended December 31, 2016, the Company granted an officer and its board of directors’ options, in the aggregate, to purchase 460,000 shares of the Company’s common stock with an exercise price ranging from $5.71 per share to $5.85 per share, a term of 10 years, and a vesting period of 3 to 4 years. The exercise price was based upon the closing price of the stock on the day of the grant. The options have an aggregated fair value of $1,772,422 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) discount of 1.30% (2) expected lives of 6 to 6.25 years, (3) expected volatility of 70.18% to 70.44%, and (4) zero expected dividends. The Company, due to the limited number of participants in the plan and their positions within the Company, uses a 0% estimated forfeiture rate. During the year ended December 31, 2016, the Company recorded $166,294 in stock-based compensation in relation to these options. As of December 31, 2016, there was $1,565,599 of unrecognized compensation cost, net of estimated forfeitures, related to the Company’s non-vested equity awards, which is expected to be recognized over a weighted average period of 3.7 years.
The fair value of each stock option is estimated on the date of grant using the Black-Scholes option valuation model that uses the assumptions noted in the above paragraph and table. The expected term of the options was computed using the “plain vanilla” method as prescribed by the Securities and Exhange Commission Staff Accounting Bulletin 107 because we do not have suffient data regarding employee exercise behavior to estimate the expected term. The volatility was determined by referring to the average historical volatility of a peer group of public companies because we do not have sufficient trading history to determine our historical volatility. The risk-free rate for periods within the contractual life of the option is based on the U.S. Treasury yield curve in effect at the time of grant.
The Company entered into a separation agreement with its former Chief Financial Officer in October 2016 and as part of the agreement, options to purchase 150,000 shares of common stock issued to the former Chief Financial Officer were cancelled and the vesting was accelerated on the remaining options to purchase 50,000 shares of common stock.
| 6. | Income Taxes |
The provision for income taxes consists of the following components (in thousands):
| December 31, 2016 | December 31, 2015 | |||||||
| Current Expense (Benefit): | ||||||||
| Federal | $ | 0 | $ | 0 | ||||
| State | 0 | 0 | ||||||
| Current Income Tax Expense | $ | 0 | $ | 0 | ||||
| Deferred Expense (Benefit): | ||||||||
| Federal | $ | 0 | $ | 0 | ||||
| State | 0 | 0 | ||||||
| Deferred Income Tax Expense | $ | 0 | $ | 0 | ||||
| Net Deferred Taxes | $ | 0 | $ | 0 | ||||
F-18
The following summarizes activity related to the Company’s valuation allowance (in thousands):
| December 31, 2016 | December 31, 2015 | |||||||
| Valuation Allowance at Beginning of Period | $ | 155 | $ | 0 | ||||
| Income Tax Benefit | 1,242 | 155 | ||||||
| Release of Valuation Allowance | 0 | 0 | ||||||
| Valuation Allowance at End of Period | $ | 1,397 | $ | 155 | ||||
A reconciliation of the income tax benefit computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows:
| December 31, 2016 | December 31, 2015 | |||||||||||||||
| Amount | Percent | Amount | Percent | |||||||||||||
| Federal Tax Benefit at Statutory Rate | $ | 1,335 | 34.00 | % | $ | 254 | 34.00 | % | ||||||||
| State Tax Benefit Net of Federal | 137 | 3.48 | % | 0 | 0.00 | % | ||||||||||
| IPO Costs | (227 | ) | (5.78 | )% | (99 | ) | (13.24 | )% | ||||||||
| Other Permanent Differences | (3 | ) | (0.08 | )% | 0 | 0.00 | % | |||||||||
| Increase in Valuation Allowance | (1,242 | ) | (31.62 | )% | (155 | ) | (20.76 | )% | ||||||||
| Total Tax (Expense) / Benefit | $ | 0 | 0.00 | % | $ | 0 | 0.00 | % | ||||||||
The principal components of the Company’s deferred tax assets and liabilities consist of the following at December 31, 2016 and 2015 (in thousands):
| December 31, 2016 | December 31, 2015 | |||||||
| Deferred Tax Assets: | ||||||||
| Start Up Costs | $ | 798 | $ | 67 | ||||
| Federal Net Operating Loss Carryforwards | 520 | 89 | ||||||
| State Tax Loss Carryforwards | 50 | 0 | ||||||
| Deferred Compensation | 33 | 0 | ||||||
| Total Deferred Tax Assets | $ | 1,401 | $ | 155 | ||||
| Less Valuation Allowance | (1,397 | ) | (155 | ) | ||||
| Net Deferred Tax Assets | $ | 4 | $ | 0 | ||||
| Deferred Tax Liabilities: | ||||||||
| Fixed Assets | (4 | ) | 0 | |||||
| Total Deferred Tax Liabilities | $ | (4 | ) | $ | 0 | |||
| Net Deferred Taxes | $ | 0 | $ | 0 | ||||
The Company has incurred net operating losses since inception. As of December 31, 2016, the Company had total federal operating loss carry forwards of approximately $1.53 million which expire commencing in 2035. The value of these carryforwards depends on the Company’s ability to generate taxable income. Additionally, because federal tax laws limit the time during which the net operating loss carryforwards may be applied against future taxes, if the Company fails to generate taxable income prior to the expiration dates of the carry forwards the Company may not be able to fully utilize the net operating loss carryforwards to reduce future income taxes. Finally, the Company has not undertaken a detailed analysis of the application of IRC Section 382 with respect to limitations on the utilization of net operating loss carryforwards and other deferred tax assets. However, the Company believes that this matter is not material to the overall tax position within the financial statements due to the full valuation allowance against the net operating losses and the lack of utilization of the net operating losses during tax years open under statute.
F-19
The Company conducts business in various locations and, as a result, files income tax returns in the United States Federal jurisdiction and in multiple state jurisdictions. As of December 31, 2016, the Company had state operating losses of approximately $1.27 million which expire commencing in 2036. Since the Company is in a loss carryforward position, the Company is generally subject to examination by the U.S. federal, state and local income tax authorities for all tax years in which a loss carryforward is available.
Management has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets. The Company has cumulative losses and there is no assurance of future taxable income, therefore, valuation allowances have been recorded to fully offset the deferred tax asset at December 31, 2016. Management has determined that it is more likely than not that the Company will not recognize the benefits of its federal and state deferred tax assets, and as a result, a valuation allowance of $1.40 million and $0.16 million has been established at December 31, 2016 and 2015, respectively. The change in the valuation allowance for the year ended December 31, 2016 was primary due to additional operating losses and capitalized research costs. The Company may be eligible to claim research and development tax credits in the future, but has not conducted a study to date.
There are no unrecognized tax benefits from any federal, state or foreign jurisdictions. The only tax year open under statute for the Company is December 31, 2015.
The Company’s policy is to recognize interest and penalties related to any unrecognized tax liabilities as additional tax expense. No interest or penalties have been accrued at December 31, 2016 and 2015, as the Company has not recorded any uncertain tax positions. The Company believes it has appropriate and adequate support for the income tax positions taken and to be taken on its tax returns and that its accruals for tax liabilities are adequate for all open years based on an assessment of many factors including past experience and interpretations of tax law applied to the facts of each matter.
Although the Company believes its recorded assets and liabilities are reasonable, tax regulations are subject to interpretation and tax litigation is inherently uncertain; therefore the Company’s assessments can involve both a series of complex judgments about future events and rely heavily on estimates and assumptions. Although the Company believes that the estimates and assumptions supporting its assessments are reasonable, the final determination of tax audit settlements and any related litigation could be materially different from that which is reflected in historical income tax provisions and recorded assets and liabilities. If the Company were to settle an audit or a matter under litigation, it could have a material effect on the income tax provision, net income, or cash flows in the period or periods for which that determination is made. Any accruals for tax contingencies are provided for in accordance with U.S. GAAP.
The Company does not believe that its tax positions will significantly change due to any settlement and/or expiration of statutes of limitations prior to December 31, 2017.
| 7. | Commitments and Contingencies |
MD Anderson – IntertechBio Agreement
On August 11, 2015, the Company acquired the rights and obligations under the Patent and Technology License Agreement entered into between IntertechBio and MD Anderson dated April 2, 2012. Pursuant to the agreement, IntertechBio obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to the Company’s drug product candidate, WP1122. Under the agreement, IntertechBio agreed to pay annual maintenance fees in the amount of $10,000 on the first anniversary of the effective date of the agreement, $20,000 on the second anniversary of the effective date of the agreement, $40,000 on the third anniversary of the effective date of the agreement, $60,000 on the fourth anniversary of the effective date of the agreement, $80,000 on the fifth anniversary of the effective date of the agreement and $100,000 on the sixth anniversary of the effective date of the agreement, except that such payments will no longer be due upon the first sale of a licensed product. Under the agreement, IntertechBio also agreed to make a minimum annual royalty payment in the amount of $200,000 for the first anniversary following the first sale of a licensed product, $400,000 for the second anniversary following the first sale of a licensed product, and $600,000 for the third year following the first sale of a licensed product. IntertechBio also agreed to make certain milestone payments. Pursuant to an amendment on October 19, 2015, the Company will pay milestone payments as follows:
| Phase | Amount | |||
| Commencement of Phase II Study for a licensed product | $ | 200,000 | ||
| Commencement of Phase III Study for a licensed product | $ | 250,000 | ||
| Filing of a New Drug Application for a licensed product | $ | 400,000 | ||
| Receipt of market approval for a licensed product | $ | 500,000 | ||
Per the October 2015 amendment to the agreement, MD Anderson has the right to terminate the license agreement if (i) a preclinical toxicology program for a licensed product is not initiated within one year of the effective date of the amendment (which has occurred), (ii) an investigational new drug application is not filed with the Food and Drug Administration for a Phase I study for a licensed product within three years of the effective date of the amendment, or (iii) a Phase I study for a licensed product is not commenced within five years of the effective date of the amendment. The agreement will expire upon the expiration of the licensed intellectual property. The rights obtained by the Company pursuant to the agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by the Company.
F-20
On October 8, 2015, IntertechBio Corporation entered into a letter agreement with MD Anderson wherein MD Anderson agreed to receive past due maintenance fees and patent expenses of $98,108 owed by IntertechBio Corporation in four installments. The past due amount is related to certain metabolic inhibitor technology license that was assigned to the Company by IntertechBio Corporation and was owed by IntertechBio Corporation prior to the Company’s acquisition of the license. Pursuant to the letter, IntertechBio Corporation also agreed to pay $65,504 in patent fees to a law firm. In order to have the license in good standing, the Company agreed to pay MD Anderson the $98,108 and the $65,504 in patent fees to a patent law firm on behalf of IntertechBio Corporation. As of December 31, 2015, $45,000 of the past due amount to MD Anderson and $42,504 in patent fees to a patent law firm were still outstanding and were included in accounts payable and accrued liabilities. On April 15, 2016, the Company entered into a letter agreement with MD Anderson where MD Anderson agreed to receive the remaining outstanding amount on or before the earlier of a) May 31, 2016 or b) four days after the Company’s completion of the IPO. These amounts were paid prior to or on May 31, 2016.
MD Anderson – Patent & Technology License Agreement
Upon the Company’s acquisition of Moleculin, LLC on May 2, 2016, we obtained a royalty-bearing, worldwide, exclusive license to intellectual property rights, including patent rights related to our WP1066 drug product candidate from MD Anderson through a Patent and Technology License Agreement Moleculin, LLC entered with MD Anderson on June 21, 2010 (the “Moleculin License Agreement”). Under the Moleculin License Agreement, Moleculin, LLC obtained the right to manufacture, have manufactured, use, import, offer to sell or sell products worldwide for any indication under the licensed intellectual property with the right to sublicense. In consideration, Moleculin, LLC agreed to make payments to MD Anderson including an up-front payment, milestone payments and minimum annual royalty payments for sales of products developed under the license agreement. Specifically, under the Moleculin License Agreement, Moleculin, LLC agreed to pay a nonrefundable upfront documentation fee and an annual maintenance fee in the amount of $20,000 on June 21, 2011, which has and shall increase in $10,000 increments on an annual basis thereafter up to a maximum of $100,000, except that such payments will no longer be due upon marketing approval in any country of a licensed product. Under the Moleculin License Agreement, Moleculin, LLC also agreed to make a minimum annual royalty payment.
Upon completion of our acquisition of Moleculin, LLC, we assumed the rights and obligations of Moleculin, LLC. However, the rights we have obtained pursuant to the assignment of the Moleculin License Agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by us.
On October 8, 2015, Moleculin, LLC entered into a letter agreement with MD Anderson for Moleculin, LLC’s past due fees to MD Anderson in the amount of $691,186 of which $300,000 had been paid prior to the letter agreement. Pursuant to the letter agreement, MD Anderson agreed to receive the remaining past due fee in three installments: a) $125,000 on October 31, 2015; b) $175,000 on January 31, 2016; and c) $91,186 on April 30, 2016. Moleculin, LLC paid $125,000 to MD Anderson on November 2, 2015.
On October 19, 2015, the agreement was amended for the milestone payments. The amended milestone payments are as follows: (i) commencement of Phase III Study for first licensed drug/product within the United States, Europe, China or Japan - $150,000; (ii) submission of the first NDA within the United States - $500,000; and (iii) receipt of first marketing approval for sale of a license product in the United States - $600,000.
On January 28, 2016, the Company and Moleculin, LLC entered into a letter agreement with MD Anderson where MD Anderson agreed to receive the remaining outstanding amount on or before the earlier of April 30, 2016 or four days after our IPO. This date was amended and per the amended agreement, the Company paid the outstanding Moleculin, LLC fees on May 31, 2016 in the amount of $306,186 in cash.
F-21
Bonwick Capital Partners LLC
On January 22, 2016, as amended on February 15, 2016, the Company entered into a letter agreement with Bonwick Capital Partners LLC. (“Bonwick”) to engage Bonwick as an exclusive financial advisor of the Company. Pursuant to the agreement, the Company agreed to: a) pay success fees equal to 7% of the gross proceeds from any form of financing; and b) issue five-year warrants to purchase 7% of the Company’s equity securities sold with a cashless exercise provision, exercisable at 125% of the price per share of the Company’s common stock paid by investors in the transaction. In addition, the Company agreed to reimburse Bonwick for all of its out-of-pocket expenses incurred in connection with the offering, not to exceed $25,000, and fees and expenses of their counsel not to exceed $100,000. Upon completion of the Company’s IPO, the Company paid Bonwick a $50,000 advisory fee. In connection with the Company’s IPO, Bonwick received a success fee of $646,872, warrants to purchase 107,802 shares of common stock at an exercise price of $7.50 per share, and $6,266 for reimbursement of expenses.
Houston Pharmaceuticals, Inc.
Our acquisition of Moleculin, LLC, occurring prior to our IPO offering, provided us with the rights of the license agreement that Moleculin, LLC had with MD Anderson covering the WP1066 Portfolio. As discussed in Note 2, we are obligated to make payments to HPI totaling $750,000 over a three-year period commencing after the IPO offering in exchange for HPI allowing us to access any data, information or know-how resulting from the research and development conducted by HPI. Pursuant to the HPI Out-Licensing Agreement, we have the right within three years of the date we enter into the agreement to buy-out from HPI all rights granted to HPI under the agreement for a payment of $1.0 million. Upon our exercise of the buy-out we will no longer be obligated to make any payments to HPI remaining from the $750,000 obligation discussed above. We will need to raise additional funds to make the buy-out payment. We cannot assure that such additional funding will be available on satisfactory terms, or at all.
Employment Agreements
On August 19, 2016, the Company entered into an employment agreement with Mr. Jonathan P. Foster pursuant to which Mr. Foster agreed to serve as Chief Financial Officer and Executive Vice President of the Company commencing on such date for an initial term of three years, which will be automatically renewed for additional one-year terms unless either party chooses not to renew the agreement. The agreement provides for an annual salary of $250,000, a potential annual bonus, ten-year options to purchase 400,000 shares at an exercise price per share equal to the closing price of the Company’s common stock on the date of execution of his employment agreement, which was $5.85. The options vest in four equal installments. Termination payments may be made under certain conditions.
The Company accepted the resignation of Mr. Louis Ploth from his position as Chief Financial Officer, effective on August 19, 2016. On October 7, 2016, the Company entered into a separation agreement with Mr. Louis Ploth (the “Separation Agreement”), pursuant to which, among other items, Mr. Ploth generally released the Company from any claims he may have against the Company or its affiliates, and the Company agreed to pay Mr. Ploth a severance payment of $100,000 over a 12-month period and to pay his medical insurance premiums until August 31, 2017. Pursuant to the Separation Agreement, Mr. Ploth will be permitted to exercise 25% of the options to purchase common stock granted to him in December 2015, or 50,000 shares, until May 2020.
On October 13, 2016, the Company and its Chief Executive Officer, Walter Klemp, entered into an employment agreement which provides for an annual salary of $300,000, of which some is deferred, and a potential annual bonus. During the period commencing June 1, 2016 and ending June 1, 2017, $12,500 per month of the compensation is deferred. The deferred compensation shall be payable in a lump sum on the earlier of the termination of the employment agreement or June 1, 2019. As of December 31, 2016, deferred compensation of $87,500 was recorded in the accompanying balance sheet. If Mr. Klemp’s employment is terminated, his status reverts to a consultant and payments to him would continue until June 1, 2019.
F-22
| 8. | Subsequent Events |
On February 9, 2017, we entered into an Underwriting Agreement (the “Underwriting Agreement”) with Roth Capital Partners, LLC, as representative of the several underwriters identified therein (collectively, the “Underwriters”), pursuant to which we sold in a registered public offering (the “Offering”), 3,710,000 units, priced at a public offering price of $1.35 per unit, with each unit consisting of: (i) one share of common stock, (ii) a five-year Series A warrant to purchase 0.50 of a share of common stock, (iii) a 90-day Series B warrant to purchase one share of common stock, and (iv) a five-year Series C warrant to purchase 0.50 of a share of common stock. The Series C warrants in a unit may only be exercised to the extent and in proportion to a holder of the Series C warrants exercising its Series B warrants included in the unit. The Series A and Series C warrant have an exercise price of $1.50 per share of common stock. The Series B warrant has an exercise price of $1.35 per share of common stock.
Under the terms of the Underwriting Agreement, we granted the Underwriters a 45-day option to purchase an additional 556,500 shares of common stock and/or an additional 556,500 warrant combinations (comprised of an aggregate of 278,250 Series A warrants, 556,500 Series B warrants and 278,250 Series C warrants), in any combinations thereof, from us to cover over-allotments at the public offering price per share of $1.349 and public offering price per warrant combination of $.001, respectively, less the underwriting discounts and commissions. Upon the closing of the Offering, the Underwriters exercised the over-allotment option with respect to 278,100 warrant combinations. We received approximately $4.4 million in net proceeds from the Offering, after deducting underwriting discounts and commissions and estimated offering expenses.
On March 21, 2017, we received notice that FDA had granted us Orphan Drug designation for Annamycin for the treatment of AML.
Subsequent to March 22, 2017, investors in the aforementioned offering on February 9, 2017, have excerised approximately 600,000 Series B warrants resulting in approximately gross proceeds to the Company of $800,000 and approximately 600,000 shares of the Company’s common stock being issued.
On January 13, 2017, the Company issued to two of its Science Advisory Board (“SAB”) members 10,000 options each with an excerise price of $2.31 with 3-year annual vesting and 79,167 shares to a former SAB member to settle an accounts payable of $237,500 in past due amounts.
F-24
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| MOLECULIN BIOTECH, INC. | ||
| By: | /s/ Walter V. Klemp | |
| Walter V. Klemp, | ||
| Chief Executive Officer and Chairman | ||
Date: April 3, 2017
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Walter V. Klemp | Chief Executive Officer and Chairman | April 3, 2017 | ||
| Walter Klemp | (Principal Executive Officer) | |||
| /s/ Jonathan P. Foster | Executive Vice President Officer and | April 3, 2017 | ||
| Jonathan P. Foster | Chief Financial Officer | |||
| (Principal Financial and Accounting Officer) | ||||
| /s/ Donald Picker | President and Chief Operating Officer | April 3, 2017 | ||
| Donald Picker | ||||
| /s/ Robert George | Director | April 3, 2017 | ||
| Robert George | ||||
| /s/ Michael Cannon | Director | April 3, 2017 | ||
| Michael Cannon | ||||
| /s/ Jacqueline Northcut | Director | April 3, 2017 | ||
| Jacqueline Northcut |
58
EXHIBIT INDEX
| Exhibit Number |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation of Moleculin Biotech, Inc. (incorporated by reference to exhibit 3.1 of the Form S-1/A filed March 21, 2016) | |
| 3.2 | Amended and Restated Bylaws of Moleculin Biotech, Inc. (incorporated by reference to exhibit 3.2 of the Form S-1/A filed March 21, 2016) | |
| 4.1 | Form of Series A/B/C Warrant Agreement issued in February 2017 offering (incorporated by reference to Exhibit 4.1 of the Form 8-K filed February 9, 2017) | |
| 10.1 ** | Moleculin Biotech, Inc. 2015 Incentive Plan (incorporated by reference to exhibit 10.1 of the Form S-1/A filed March 21, 2016) | |
| 10.2 | Rights Transfer Agreement between Moleculin Biotech, Inc. and AnnaMed, Inc. (incorporated by reference to exhibit 10.2 of the Form S-1/A filed March 21, 2016) | |
| 10.3 | Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and Moleculin, LLC (incorporated by reference to exhibit 10.3 of the Form S-1/A filed March 21, 2016) | |
| 10.4 | Amendment No. 1 to the Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and Moleculin, LLC (incorporated by reference to exhibit 10.4 of the Form S-1/A filed March 21, 2016) | |
| 10.5 | Patent and Technology License Agreement dated April 2, 2012 by and between The Board of Regents of the University of Texas System and IntertechBio Corporation (incorporated by reference to exhibit 10.5 of the Form S-1/A filed March 21, 2016) | |
| 10.6 | Amendment No. 1 to the Patent and Technology License Agreement dated June 21, 2010 by and between The Board of Regents of the University of Texas System and IntertechBio Corporation (incorporated by reference to exhibit 10.6 of the Form S-1/A filed March 21, 2016) | |
| 10.7 | Patent and Technology Development and License Agreement June 28, 2012 by and between Annamed, Inc. and Dermin Sp. z.o.o (incorporated by reference to exhibit 10.7 of the Form S-1/A filed April 15, 2016) | |
| 10.8 | Patent and Technology Development and License Agreement dated April 15, 2011 by and between IntertechBio Corporation and Dermin Sp. z.o.o (incorporated by reference to exhibit 10.8 of the Form S-1/A filed March 21, 2016) | |
| 10.9 | Patent and Technology Development and License Agreement dated October 27, 2010 by and between Moleculin, LLC and Dermin Sp. z.o.o (incorporated by reference to exhibit 10.9 of the Form S-1/A filed March 21, 2016) | |
| 10.10 | Rights Transfer Agreement dated between Moleculin Biotech, Inc. and IntertechBio Corporation dated August 11, 2015 (incorporated by reference to exhibit 10.10 of the Form S-1/A filed March 21, 2016) | |
| 10.11 | Agreement and Plan of Merger between Moleculin Biotech, Inc. and Moleculin, LLC (incorporated by reference to exhibit 10.11 of the Form S-1/A filed March 21, 2016) | |
| 10.12 | Technology Rights and Development License Agreement to be entered into by Moleculin Biotech, Inc. and Houston Pharmaceuticals, Inc. (incorporated by reference to exhibit 10.13 of the Form S-1/A filed April 15, 2016) | |
| 10.13 ** | Employment Agreement between Moleculin Biotech, Inc. and Jonathan P. Foster dated August 19, 2016 (incorporated by reference to Exhibit 10.1 of the Form 8-K filed August 25, 2016) |
59
| 10.14 ** | Executive Employment Agreement between Moleculin Biotech, Inc. and Walter Klemp dated October 13, 2016 (incorporated by reference to Exhibit 10.1 of the Form 8-K filed October 13, 2016) | |
| 10.15 ** | General Release and Separation Agreement between Moleculin Biotech, Inc. and Louis Ploth dated October 7, 2016 (incorporated by reference to Exhibit 10.2 of the Form 8-K filed October 13, 2016) | |
| 10.16 | Development Collaboration Agreement between Moleculin Biotech, Inc. and Dermin Sp. Z o. o. dated September 30, 2016 (incorporated by reference to Exhibit 10.4 of the Form 10-Q filed November 21, 2016) | |
| 21 | Subsidiaries of the Registrant (incorporated by reference to exhibit 21 of the Form S-1/A filed April 15, 2016) | |
| 23.1* | Consent of GBH CPAs, PC | |
| 23.2* | Consent of Grant Thornton, LLP | |
| 31.1* | Certification of Principal Executive Officer Pursuant to Section 302 of Sarbanes- Oxley Act of 2002 | |
| 31.2* | Certification of Principal Financial Officer Pursuant to Section 302 of Sarbanes-Oxley Act of 2002 | |
| 32.1* | Certification of Principal Executive Officer Pursuant to Section 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32.2* | Certification of Principal Financial Officer Pursuant to Section 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101.INS * | XBRL Instance Document | |
| 101.SCH * | XBRL Taxonomy Extension Schema Document | |
| 101.CAL * | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF * | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB * | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE * | XBRL Taxonomy Extension Presentation Linkbase Document |
| * | Filed herewith. |
| ** | Denotes a management contract or compensatory plan or arrangement. |
60