Attached files
| file | filename |
|---|---|
| EX-32.2 - Immune Therapeutics, Inc. | ex32-2.htm |
| EX-32.1 - Immune Therapeutics, Inc. | ex32-1.htm |
| EX-31.2 - Immune Therapeutics, Inc. | ex31-2.htm |
| EX-31.1 - Immune Therapeutics, Inc. | ex31-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 2016
OR
[ ] TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
For the Transition Period From ____________ to ____________
Commission File Number: 001-34918
IMMUNE THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| Florida | 59-3226705 | |
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) |
37
North Orange Ave, Suite 607,
Orlando, Florida 32801
(Address of principal executive offices)
(888) 613-8802
Registrant’s telephone number, including area code:
None
Securities Registered Pursuant to Section 12(b) of the Act:
Common Stock, par value $0.0001 per share
Securities Registered Pursuant to Section 12(g) of the Act:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter time that the registrant was required to submit and post such files). Yes [ ] No [X]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. Yes [X] No [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | [ ] | Accelerated filer | [ ] |
| Non-accelerated filer | [ ] (Do not check if a smaller reporting company) | Smaller reporting company | [X] |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes [ ] No [X]
The aggregate market value of voting stock held by non-affiliates of the registrant on June 30, 2016, the last business day of the registrant’s most recently completed second quarter, was $31,415,587, based on the last reported sale price of the registrant’s Common Stock on the OTC Markets on that date.
As of March 31, 2017, the registrant had outstanding 273,640,164 shares of common stock, $0.0001 par value per share.
IMMUNE THERAPEUTICS, INC.
2016 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
| 2 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Certain statements contained or incorporated by reference in this Annual Report on Form 10-K are considered forward-looking statements (within the meaning of the Private Securities Litigation Reform Act of 1995) concerning our business, results of operations, economic performance and/or financial condition, based on management’s current expectations, plans, estimates, assumptions and projections. Forward-looking statements are included, for example, in the discussions about:
| ● | strategy; | |
| ● | new product discovery and development; | |
| ● | current or pending clinical trials; | |
| ● | our products’ ability to demonstrate efficacy or an acceptable safety profile; | |
| ● | actions by the FDA and other regulatory authorities; | |
| ● | product manufacturing, including our arrangements with third-party suppliers; | |
| ● | product introduction and sales; | |
| ● | royalties and contract revenues; | |
| ● | expenses and net income; | |
| ● | credit and foreign exchange risk management; | |
| ● | liquidity; | |
| ● | asset and liability risk management; | |
| ● | the outcome of litigation and other proceedings; | |
| ● | intellectual property rights and protection; | |
| ● | economic factors; | |
| ● | competition; and | |
| ● | legal risks. |
Any statements contained in this report that are not statements of historical fact may be deemed forward-looking statements. Forward-looking statements generally are identified by the words “expects,” “anticipates,” “believes,” “intends,” “estimates,” “aims,” “plans,” “may,” “could,” “will,” “will continue,” “seeks,” “should,” “predict,” “potential,” “outlook,” “guidance,” “target,” “forecast,” “probable,” “possible” or the negative of such terms and similar expressions. Forward-looking statements are subject to change and may be affected by risks and uncertainties, most of which are difficult to predict and are generally beyond our control. Forward-looking statements speak only as of the date they are made, and we undertake no obligation to update any forward-looking statement in light of new information or future events, except as required by law, although we intend to continue to meet our ongoing disclosure obligations under the U.S. securities laws and other applicable laws.
We caution you that a number of important factors could cause actual results or outcomes to differ materially from those expressed in, or implied by, the forward-looking statements, and therefore you should not place too much reliance on them. These factors include, among others, those described herein, under “Risk Factors” and elsewhere in this Annual Report and in our other public reports filed with the Securities and Exchange Commission. It is not possible to predict or identify all such factors, and therefore the factors that are noted are not intended to be a complete discussion of all potential risks or uncertainties that may affect forward-looking statements. If these or other risks and uncertainties materialize, or if the assumptions underlying any of the forward-looking statements prove incorrect, our actual performance and future actions may be materially different from those expressed in, or implied by, such forward-looking statements. We can offer no assurance that our estimates or expectations will prove accurate or that we will be able to achieve our strategic and operational goals.
Forward-looking statements are based on information we have when those statements are made or management’s good faith belief as of that time with respect to future events, and are subject to significant risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to:
| ● | our lack of operating history; | |
| ● | our current and future capital requirements and our ability to satisfy our capital needs; | |
| ● | our inability to keep up with industry competition; | |
| ● | interpretations of current laws and the passages of future laws; | |
| ● | acceptance of our business model by investors and our ability to raise capital; | |
| ● | our drug discovery and development activities may not result in products that are approved by the applicable regulatory authorities. Even if our drug candidates do obtain regulatory approval they may never achieve market acceptance or commercial success; | |
| ● | our reliance on key personnel, including our ability to attract and retain scientists; | |
| ● | our reliance on third party manufacturing to supply drugs for clinical trials and sales; | |
| ● | our limited distribution organization with no sales and marketing staff; | |
| ● | our being subject to product liability claims; | |
| ● | our reliance on key personnel, including our ability to attract and retain scientists; | |
| ● | legislation or regulation that may increase the cost of our business or limit our service and product offerings; | |
| ● | risks related to our intellectual property, including our ability to adequately protect intellectual property rights; | |
| ● | risks related to government regulation, including our ability to obtain approvals for the commercialization of some or all of our drug candidates, and ongoing regulatory obligations and continued regulatory review which may result in significant additional expense and subject us to penalties if we fail to comply with applicable regulatory requirements; and | |
| ● | our ability to obtain regulatory approvals in foreign jurisdictions to allow us to market our products internationally. |
Moreover, new risks regularly emerge and it is not possible for our management to predict or articulate all risks we face, nor can we assess the impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from those contained in any forward-looking statements. All forward-looking statements included in this prospectus are based on information available to us on the date of this Annual Report. Except to the extent required by applicable laws or rules, we undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise. All subsequent written and oral forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements contained above and throughout this Annual Report.
| 3 |
JUMPSTART OUR BUSINESS STARTUPS ACT
We qualify as an “emerging growth company” as defined in Section 101 of the Jumpstart our Business Startups Act (“JOBS Act”) as we do not have more than $1,000,000,000 in annual gross revenue and did not have such amount as of December 31, 2016, the last day of our last fiscal year. We are electing to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act.
As an emerging growth company, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
| ● | being permitted to present only two years of audited financial statements and only two years of related “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this annual report; | |
| ● | not being requested to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended (“Sarbanes-Oxley Act”); | |
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and | |
| ● | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We will remain an emerging growth company until the earliest to occur of: (i) our reporting $1 billion or more in annual gross revenues; (ii) the end of fiscal year 2019; (iii) our issuance, in a three year period, of more than $1 billion in non-convertible debt; and (iv) the end of the fiscal year in which the market value of our common stock held by non-affiliates exceeded $700 million on the last business day of our second fiscal quarter.
| 4 |
Company Overview
Immune Therapeutics, Inc. (the “Company”) was initially incorporated in Florida on December 2, 1993 as Resort Clubs International, Inc. (“Resort Clubs”). It was formed to manage and market golf course properties in resort markets throughout the United States. Galliano International Ltd. (“Galliano”) was incorporated in Delaware on May 27, 1998 and began trading in November 1999 through the filing of a 15C-211. On November 10, 2004, Galliano merged with Resort Clubs. Resort Clubs was the surviving corporation. On August 23, 2010, Resort Clubs changed its name to pH Environmental Inc. (“pH Environmental”).
On April 23, 2012, pH Environmental completed a name change to TNI BioTech, Inc., and on April 24, 2012, we executed a share exchange agreement for the acquisition of all of the outstanding shares of TNI BioTech IP, Inc. On September 4, 2014, a majority of our shareholders approved an amendment to our Amended and Restated Articles of Incorporation, as amended, to change our name to Immune Therapeutics, Inc. We filed our name change amendment with the Secretary of State of Florida on October 27, 2014 changing our name to Immune Therapeutics, Inc.
The Company currently operates out of Orlando, Florida. In July 2012, the Company’s focus turned to acquiring patents that would protect and advance the development of new uses of opioid-related immune- therapies, such as low dose naltrexone (“LDN”) and Methionine [Met5]-enkephalin (“MENK”). The Company’s therapies are believed to stimulate and/or regulate the immune system in such a way that they provide the potential to treat a variety of diseases. We believe our therapies may be able to correct abnormalities or deficiencies in the immune system in diseases such as HIV infection, autoimmune disease, immune disorders, or cancer; all of which can lead to disease progression and life-threatening situations when the immune system is not functioning optimally.
In October 2012, the Company formed TNI BioTech International, Ltd., a BVI company in Tortola, British Virgin Islands, which was set up to allow the Company to market and sell LDN in those countries outside the U.S. in which we have been able to obtain approval to sell the Company’s products.
In August 2013, the Company formed its United Kingdom subsidiary, TNI BioTech, LTD (the “UK Subsidiary”). The UK Subsidiary received approval to be considered a micro, small or medium-sized enterprise (“SME”) with the European Medicines Agency (“EMA”) on August 21, 2013. The designation provides the UK Subsidiary with significant discounts when holding meetings or submitting filings to the EMA. On September 19, 2013, the UK Subsidiary submitted a pre-submission package to the EMA regarding Crohn’s Disease. The EMA granted the UK Subsidiary a meeting that took place on September 27, 2013. The UK Subsidiary is eligible to benefit from the provisions for administrative and financial assistance for SMEs set out in Regulation (EC) No 2049/2005. The Company will apply to obtain EMA benefits once funding becomes available.
In December 2013, the Company formed a new subsidiary, Cytocom Inc., to focus on conducting LDN and MENK clinical trials in the United States. In December 2014, the Company finalized the distribution of common stock of Cytocom Inc. to its shareholders. As part of the transaction, the Company retained exclusive rights to all international patents, in-country approvals, formulations, trademarks, manufacturing, marketing, sales, and distributions rights in emerging nations, including Africa, Central America, South America, Russia, India, China, Far East, and The Commonwealth of Independent States (former Soviet Union). The Company will continue to have access to existing clinical data as well as any new data generated by Cytocom Inc. during drug development. On December 8, 2014, the number of Cytocom Inc. shares of common stock that were issued to our shareholders totaled 113,242,522 shares. In connection with the transaction, Cytocom Inc. issued 140,100,000 shares of its common stock to the Company, which gave the Company a 55.3% stake in Cytocom Inc. on that date. In April 2016, the Board of Directors and a majority of shareholders of Cytocom approved a reverse stock split of Cytocom’s outstanding common stock with one new share of stock for each twenty old shares of common stock. Cytocom effectuated and finalized the reverse split in June 2016. At December 31, 2016, the Company’s equity interest had been further reduced to 13%, by subsequent issuances of Cytocom common stock to shareholders in settlement of notes payable.
| 5 |
In March 2014, the Company incorporated Airmed Biopharma Limited, an Irish corporation with an address in Dublin, Ireland, and Airmed Holdings Limited, an Irish company domiciled in Bermuda. The Irish companies were set up to benefit from incentives granted by the Irish government for the establishment of pharmaceutical companies (many of the world’s leading pharmaceutical companies have located in Ireland), and so that the Company could take advantage of Ireland’s status as a member of the European Union and the European Economic Area. An Irish limited liability company enjoys a low corporate income tax rate of 12.5%, one of the lowest in the world. The Irish-domiciled company hopes to qualify for tax incentives for Irish holding/headquartered companies and to benefit from the network of double tax treaties that reduce withholding taxes. TNI BioTech International, Ltd. will manage our international distribution, using product that is manufactured in Ireland and elsewhere.
Today, Immune Therapeutics is focused on the commercialization of affordable non-toxic immunotherapies focused on the activation and rebalancing of the body’s immune system. Stimulating the body’s immune system remains one of the most promising approaches in the treatment of Cancers, HIV, Autoimmune Diseases, inflammatory conditions and other opportunistic infections for chronic often life-threatening diseases through the mobilization of the body’s immune system in Emerging Nations using existing clinical data.
Cytocom Inc, is a clinical-stage pharmaceutical company focused on the development of the first affordable non-toxic immunodulator for the treatment of inflammatory diseases, immune-related disorders, and cancer and is responsible for the development of our patented therapies with the FDA and EMA.
As of this date, neither we nor our collaboration partners are permitted to market our drug candidates in the United States until we receive approval of a New Drug Application from the FDA. Neither we nor our collaboration partners have submitted an application for or received marketing approval for any of our drug candidates. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process.
Some of the Company’s more substantial risks include, but are not limited to, its lack of operating history, its high needs for capital, strict government regulation, risk of law suits from trial participants and otherwise, requirement for drug approvals which may never occur, changes in the industry, failure of the Company’s products to make it through trials, reliance on third parties to conduct trials and manufacture and distribute the Company’s drugs, and fierce competition. All of these factors and more could affect investors’ investments in the Company.
The Company’s technology platform is built surrounding two different immune therapies, IRT-103, low dose naltrexone herein sometimes referred to as “LDN” or “Lodonal™” or IRT-101, herein sometimes referred to as Methionine-Enkephalin or “MENK.” Both therapies have been decades in the making, over forty clinical trials run by institutions such as the Pennsylvania State University Medical School at Hershey, University of Chicago, State University of New York, and Multiple Sclerosis Center at UCSF. When the Company acquired the assets from the either the patent holder or licensee we also acquired the rights to the clinical data, orphan drug designations and IRB. The Company has completed one trial in Nigeria for Lodonal™ and has a second trial underway for cervical cancer in Malawi. The Company has also submitted briefing packaged to the FDA for a phase IIB/III trial for IRT-103 for Crohn’s Disease and is preparing a new briefing package for IRT-103 for pancreatic cancer. In addition to the work in the United States we have been completing pre-clinical and clinical work for IRT-013 with Professor Shan at the School of Immunology in China.
LDN
The Company has branded our Immediate Release Low Dose Naltrexone as IRT-103TM when working with the FDA and EMA and trademarked our product in Africa as LodonalTM. Lodonal™ with an immediate release 4.5mg formulation of Naltrexone is the brand name for our HIV treatment. LDN can be any formulation between .05 and to 10mg and may or may not be immediate release and may be an oral or liquid formulation.
The FDA approved naltrexone HCl in 1984 for the treatment of opioid addiction. The typical daily dosage for opioid addiction is 50mg to 100mg, and 50mg tablets are available commercial. There is no FDA-approved use for naltrexone at any lower dosage then 50mg for the treatment of any other medical conditions or treatments.
| 6 |
Where high dose Naltrexone at 50mg to 100mg and Slow release Naltrexone between .01mg and 10mg and Immediate Release Naltrexone between .01mg and 10mg share commonality” in categories of genes and are considered the same drug the difference in dosing and delivery method (immediate release) difference in the overall response to the immune system. There is a difference in the cell patterns of genes that are altered by the treatment of immediate release naltrexone verses high does naltrexone and slow release low naltrexone between .01mg and 10mg. The differences are important because immediate release naltrexone between .01 and 10mg acts as an immunomodulator.
Since immediate release naltrexone blocks the opiate receptors only for a few hours before it is naturally excreted, what results is a rebound effect; in which both the production and utilization of methionine-enkephalin or opiate growth factor are increased. Once the immediate release naltrexone has been metabolized, the elevated endorphins produced as a result of the rebound effect can now interact with the more sensitive and more-plentiful receptors and assist in regulating cell growth and immunity. There is no rebound effect with either high dose naltrexone or slow release naltrexone and it is the rebound immunomodulatory effect that effects the treatment of treating patients suffering from human immunodeficiency virus (HIV) acquired immune deficiency syndrome (AIDS, autoimmune disease, opportunistic infections, cancer, inflammation, and neurodegenerative diseases
It has been demonstrated in trials that in the presence of LDN, the numbers of T-cells, both CD4+ helper T cells and CD8+ cytotoxic T cells, may increase by more than 300%.
The Mechanism of Action of immediate release LDN is not fully understood at this time, but based on clinical work there are three current theoretical models for how immediate release LDN works in autoimmune disease, inflammatory disease, cancer and HIV/AIDS.
Immediate Release LDN, which is different from either slow or extended release LDN, works by triggering a number of receptors including (1) the opioid and T Cell receptors on immune cells which activate or balance various cells of the immune system, and (2) tolling receptors to shift Th1 (pro-inflammatory) to Th2 (anti-inflammatory) which is critical when dealing with autoimmune and inflammatory disease. (1) Increases the production of cytokines specifically an endorphin referred to as Methionine-enkephaline or OGF.
These compounds then produce pain relief similar to opiates. The body responds to these compounds by inhibition of cell growth, promoting healing, and reducing inflammation, all in an effort to restore homeostasis. IRLDN also causes increase in OGF receptor.
Regulatory applications submitted, if any, to commence clinical trials and the current status of such applications.
Clinical trials under the INDs are anticipated to be conducted in the United States and the EU.
| IND # | Indication | Product Name | Sponsor | Status | ||||
| 34,442 | HIV/AIDS | MENK (IRT-101) | Dr. Ronald Herberman and Dr. Bernard Bihari | Inactive; filed in 1997 (1) | ||||
| 67442 | Crohn’s Disease | Naltrexone HCL (IRT-103) | Dr. Jill Smith | Active; filed May 31, 2003 and filed with the FDA in March, 2013 | ||||
| 50987 | Pancreatic Cancer | MENK (IRT-101) | Dr. Jill Smith | Active; was filed with the FDA in March, 2013 |
| (1) | Currently inactive but will soon be reactivated and transferred to TNIB. |
Immune Therapeutics, Inc. Drug Development Plan
Nigeria
Immune Therapeutics Inc., through its wholly owned subsidiary TNI BioTech Intl., completed a 90-day bridging trial for the treatment of patients with HIV/AIDS in Nigeria. The National Agency for Food and Drug Administration and Control (NAFDAC) approval is based on previous clinical data and the Nigeria Bridging Trial was a single center, open labeled, randomized, bridging study. The trial consisted of a total of one hundred and fifty [150] patients of both genders between the ages of 18-60, each of whom was infected with the human immunodeficiency virus (HIV).
| 7 |
The 90-Day Bridging Trial was undertaken at the State Specialist Hospital in Asubiaro, Osogbo, Osun State, Nigeria and the primary objective of this Bridging Trial was to confirm that LodonalTM has a beneficial effect on the immune system of immune deficient patients and that it is safe. The trial separated the patients into a Control (placebo) Group and a Treatment Group (which was administered LodonalTM). The efficacy of increasing CD4 count [cell/mm3] between Day-1 and Day-90 by at least 25% was set as the criteria for demonstrating beneficial effect on the immune system. Safety was demonstrated through quality of life assessment and vitals both of which were not adversely affected. Treatment Group patients were given a daily dose of 4.5-mg/kg of Lodonal™.
The results yielded an average increase of 44% increase in CD4 count in the Lodonal Treatment Group compared to an 11% increase in the Control Group. Additionally, there were no reported opportunistic infections and no toxicity levels uncovered. Liver function remained normal and there was no negative impact on other systems based on blood results. No sleep disturbance or vivid dreams were present enough to justify trial discontinuation. No appreciable adverse CNS, renal, cardiac, hepatic, musculoskeletal, hematopoietic side effects were present.
NAFDAC has issued approval of Lodonal™as an immune system regulator in the management of HIV patients and the company is now in the process of completing the registration to import the drug into Nigeria.
Malawi
The Company, through its wholly owned subsidiary TNI BioTech International, received permission from the Pharmacy, Medicines and Poisons Board (PMPB) and The College of Medicine; University of Malawi to initiate a clinical trial for a Single Visit Approach to Cervical Cancer Prevention in the Republic of Malawi. The PMPB issued drug approval to import the drug in 2015.
The Malawi Clinical Trial’s primary endpoint includes Safety, Acceptability, and Feasibility of a Single Visit Approach to Cervical Cancer Prevention in patients. (Trial number: VIA-LDN-401 -0 I). The secondary objectives’ is to determine life extension; to improve the immune system of HIV and Cancer positive patients by starting treatment with LDN (“LodonalTM”) and to ensure marked improvement in Clinical benefit based upon parameters that reflect the overall well- being of the patient, including Pain control, performance status, and body weight under the supervision of Dr. Frank Taulo, Dr. Gladys Gadama, Dr. Effie Chipeta as Principal Investigators. The recruitment of study participants, testing and follow up is still on-going. The first evaluation report from the doctors involved is expected by the end of April 2017 as per the study protocol.
The Company intends to initiate a number of additional trials in Africa in the next 6 months, which will include trials as an adjunct to chemotherapy in Kenya and Ghana and HIV/AIDS in Malawi.
FDA and EMA Development Plan for Cytocom, Inc.
After the completion of the spin-off of our subsidiary, Cytocom, Inc., to shareholders, all work with the FDA, EMA or any of the G7 countries will be under the supervision of Cytocom Inc. However, the Company plans to submit all trials and fees on Cytocom Inc.’s behalf until such time as Cytocom, Inc. is sufficiently funded, as all funding is currently being provided through the Company. Nonetheless, the trials will be supervised by Cytocom, Inc. following submission by the Company. At this time the INDs have not been transferred to Cytocom; however they will be transferred before the trials begin. Studies and trials in countries that are not part of the G7 are being conducted directly by the Company.
Our lead product candidate is IRT-103 ™, a first-in-class, proprietary fixed-dose therapy entering Phase 2b clinical trials for the treatment of adult and pediatric Crohn’s disease where we have orphan drug designation. The Company acquired all of the clinical data from Dr. Jill Smith in conjunction with the acquisition of the license. The Company had no direct involvement in the trials.
Crohn’s disease affects over 1.6 million adults in the United States and an estimated 80,000 children. Data from the Phase 2a clinical trial indicate that Lodonal™ was generally well-tolerated therapy that can provide promise for the treatment of Crohn’s disease. The Department of Medicine at The Pennsylvania State University, (PSU) College of Medicine, GI Medicine were responsible for three of the Crohn’s trials that were part of our briefing package to the FDA.
| 8 |
PSU researchers have demonstrated in a mouse model of Crohn’s disease from with dextran sodium sulfate (a chemically induced colitis) that opioid receptor blockade by a low dose of naltrexone resulted in less weight loss, lower disease activity index scores and less histological evidence of inflammation when compared to controls (Matters, GL et al., 2008). Furthermore, the researchers demonstrated that tissue inflammatory cytokine mRNA was reversed to baseline levels in the colons of mice treated with naltrexone.
A Phase 2 trial with low-dose naltrexone was completed on patients with Crohn’s Disease and the objective of the trial was to determine the role of endogenous opioids and opioid antagonists in healing and repair of tissues. Eligible subjects with histologically and endoscopically confirmed active Crohn’s disease activity index (CDAI) score of 220–450 were enrolled in a study using 4.5 mg naltrexone per day. Infliximab was not allowed for a minimum of 8 weeks prior to study initiation. Other therapy for Crohn’s disease that was at a stable dose for 4 weeks prior to enrollment was continued at the same doses. Patients completed the inflammatory bowel disease questionnaire (IBDQ) and the short-form (SF-36) quality of life surveys and CDAI scores were assessed pre-treatment, every 4 weeks while on therapy and 4 weeks after completion of the study. The drug was administered by mouth each evening for a 12-week period. Seventeen patients with a mean CDAI score of 356 °æ 27 were enrolled. CDAI scores decreased significantly (P = 0.01) with LDN, and remained lower than baseline 4 weeks after completing therapy. Eighty-nine percent of patients exhibited a response to therapy and 67% achieved a remission (P < 0.001). Improvement was recorded in both quality of life surveys with LDN compared with baseline. No laboratory abnormalities were noted. The most common side effect was sleep disturbances, occurring in seven patients. The trial concluded that LDN therapy appears effective and safe in subjects with active Crohn’s disease.
Another Phase 2 trial therapy was conducted, entitled, Therapy with the Opioid Antagonist Naltrexone Promotes Mucosal Healing in Active Crohn’s Disease: A Randomized Placebo-Controlled Phase 11 trial at the Department of Medicine, The Pennsylvania State University, College of Medicine, GI Medicine [Smith, JP. et al, 2011]. Aims – A randomized double-blind placebo-controlled study was designed to test the efficacy and safety of an opioid antagonist for 12 weeks in adults with active Crohn’s disease. The phase 2A trial had 3 major endpoints for the study including: 1) Clinical improvement based upon the Crohn’s Disease Activity Index (CDAI) Score, 2) Mucosal healing by colonoscopy, and 3) Safety. The human studies were done with FDA approval under IND 67442. Forty subjects with active Crohn’s disease were enrolled in the study. Randomized patients received daily oral administration of 4.5-mg naltrexone or placebo. Providers and patients were masked to treatment assignment. The primary outcome was the proportion of subjects in each arm with a 70-point decline in Crohn’s Disease Activity Index (CDAI) score. The secondary outcome included mucosal healing based upon colonoscopy appearance and histology. Eighty-eight percent of those treated with naltrexone had at least a 70-point decline in CDAI scores compared to 40% of placebo-treated patients (p = 0.009). After 12 weeks, 78% of subjects treated with naltrexone exhibited an endoscopic response as indicated by a 5-point decline in the Crohn’s disease endoscopy index severity score (CDEIS) from baseline compared to 28% response in placebo-treated controls (p = 0.008). 33% achieved remission with a CDEIS score <6, whereas only 8% of those on placebo, showed the same change. Fatigue was the only side effect reported that was significantly greater in subjects receiving placebo. The study concluded that Naltrexone improves clinical and inflammatory activity of subjects with moderate to severe Crohn’s Disease compared to placebo-treated controls. Strategies to alter the endogenous opioid system provide promise for the treatment of Crohn’s Disease.
A pilot study was completed entitled Safety and Tolerability of Low-dose Naltrexone Therapy in Children With Moderate to Severe Crohn’s Disease trial at the Department of Medicine, The Pennsylvania State University, College of Medicine, GI Medicine Smith, (JP. et al, 2013). The aims of this study were to evaluate the safety and tolerability of an opioid antagonist, naltrexone, in children with moderate to severe Crohn’s disease. The pilot clinical trial was conducted in children with moderate to severe Crohn’s disease. Fourteen subjects with a mean age of 12.3 years (range, 8 to 17 y) were enrolled. Children were randomized to placebo or naltrexone (0.1 mg/kg) orally for 8 weeks followed by open-labeled treatment with 8 additional weeks of naltrexone. Safety and toxicity were monitored by physical examinations and blood chemistries. Clinical activity was assessed by the Pediatric Crohn’s Disease Activity Index (PCDAI) and Quality of life was monitored by the Impact III survey. The results indicated that oral naltrexone was well tolerated without any serious adverse events in children with moderate to severe Crohn’s disease. PCDAI scores decreased from pre-treatment values (34.2Å 3.3) with an 8-week course of naltrexone therapy (21.7Å 3.9) (P=0.005). Twenty-five percent of those treated with naltrexone were considered in remission (score r10) and 67% had improved with mild disease activity (decrease in PCDAI score by at least 10 points) at the end of the study. Systemic and social quality of life improved with naltrexone treatment (P=0.035). The study concluded that naltrexone therapy seems safe with limited toxicity when given to children with Crohn’s disease and may reduce disease activity.
| 9 |
Food And Drug Administration
There are three types of meetings with the FDA: Type A Meeting – is a meeting that is “immediately necessary for an otherwise stalled drug development program to proceed.” This type of meeting refers to meetings to resolve disputes, talk about clinical holds, special protocols. Type B Meetings are identified as (1) pre-IND meetings, (2) certain end of Phase I meetings, (3) end of Phase 2/pre-Phase 3 meetings and (4) pre-NDA/BLA meetings. A type C Meeting is any other kind of meeting.
The Company attended a Type C meeting with the FDA June 26, 2013 with the Division of Gastroenterology and Inborn Errors Products regarding the clinical and regulatory aspects of the proposed Phase IIB/III development program and future 505(b)(2) application for Low Dose Naltrexone (LDN) in the treatment of adults and pediatric patients with Crohn’s Disease. In principal the FDA agreed that a 505(b)(2) application would be an acceptable approach at FDA recommends that sponsors considering the submission of an application through the 505(b)(2) pathway consult FDA’s regulations at 21 CFR 314.54, and the draft guidance for industry Applications Covered by Section 505(b)(2) (October 1999). The Company is planning to submit a request for “breakthrough technology” designation. If this request is granted, what impact could it have on minimizing the Phase 3 study design(s) and the package needed for filing the 505(b)(2) application.
The Company retained the services of Cote Orphan to provide a new briefing package and work with the Division for Breakthrough Therapy Designation is granted to determine the most efficient development program for your product and proposed indication. The Company notes that a 505(b)(2) NDA will need to be submitted with all the same components that a regular NDA requires. The Company hopes to submit our briefing package to the FDA because CDAI was previously been accepted by the FDA for phase 3 trials in CD, FDA has reconsidered the use of the CDAI as a measure of clinical response to therapeutic intervention and we our new trial design endpoint will include endoscopies to show true mescal healing. The Company delayed submitting our new briefing package to the FDA as a number of changes were under consideration at the FDA in connection with 505(2)(b) pathway drug development. The Company has recently begun to make final changes to the briefing package, and expects to present its new briefing package to the FDA before the end of the third quarter 2017, with a meeting shortly thereafter.
Anticipated developmental timelines for Cytocom, Inc.
Guidance meeting held with FDA regarding pediatric and adult studies provided feedback and based on the new submission of a briefing package we anticipate presenting final protocols for both adult and pediatric trial.
| ● | Current development plan includes: |
| ● | Two adult studies planned: |
| ● | Double-blind, randomization, 24-week study of LDN vs. Placebo at 4.5mg |
| ● | Double-blind, randomization 12-week multi-dose study including 4.5mg |
| ● | Both studies will roll-over to an open label |
| ● | Studies to commence Q1 2017 |
| ● | Two pediatric studies |
| ● | Double-blind randomization, 24 week study of LDN vs. Placebo at |
| ● | Both studies will roll-over to open label |
| ● | Studies to commence Q1 of 2017 |
Competitive Advantage
The Company believes many of the same advantages of our therapies apply to both the US market as well as the African market. LodonalTM could provide the first affordable, non-toxic approach for treatment of immune dysfunction, cancer and d chronic inflammatory state.
| 10 |
Some of the Competitive Advantages and Benefits of LodonalTM include the following:
Lower production costs and sales price of treatments
Today the majority of the drugs under development are both more expensive and more toxic. We do not believe this is the right way to move forward. Biologic agents cost between $12,000 and $150,000 a year.
LodonalTM/ IRT-103 can be manufactured and delivered in Emerging Nations for under $.90 cents a day and we estimate a price of $3,600 dollars a year in developed country underwritten to $10 to $15 dollars per month and when not underwritten the company will provide to patients for $30 dollars a month.
Lodonal™ should be able to substantially reduce health care costs for a number of reasons:
| ● | IRT-103 and Lodonal™ can provides a new, non-toxic inexpensive method of medical treatment by mobilizing the natural defenses of one’s own immune system. It can be used as a stand-alone therapy or an adjunct to existing immunosuppressive therapies by reducing the toxic side effects of immunosuppressive drugs. |
| o | Patients who are taking immunosuppressant drugs should see their doctor on a regular basis to monitor the patient for unwanted side effects. |
| ● | Lodonal™ does not require the medical supervision of antiretroviral or immunosuppressive therapies. |
| o | Immunosuppressive drugs are very powerful and can cause such serious side effects as high blood pressure, kidney problems, malignancies and liver disorder. Immunosuppressant drugs lower a person’s resistance to infection and can make infections harder to treat. The drugs can also increase the chance of uncontrolled bleeding. |
| ● | Lodonal™ has no toxic side effects as it is an immunomodulator and activates and re-balances the immune system. |
| o | HIV as a Chronic Disease: For reasons that remain to be elucidated, antiretroviral-treated HIV disease is associated with a new constellation of problems, generally referred to as “non-AIDS morbidity”, and, in the popular press, “premature aging” |
| ● | Health care systems in those regions where most people with HIV reside (e.g., sub-Saharan Africa) were designed to provide acute care only and are ill equipped to provide the chronic care, which is now required to manage HIV. |
| ● | Lodonal as an immunomodulator has a way of helping what is now referred to as “non-AIDS morbidity”, and, in the popular press, “premature aging”. |
| ● | Blocks release of proinflammatory cytokines including Interleukins IL6 and IL12, TNFα, NF-ĸB (nuclear factor kappa light chain enhancer of activated B cells) |
| ● | Modulates T and B lymphocyte production and cause Shift from Th1 (pro-inflammatory) to Th2 (anti-inflammatory) |
| ● | Affects microglia – macrophages/ 1st line of immune defense in CNS; normally quiescent; |
| ● | Cell death, inflammation, infection à Activated microglia à increase in proinflammatory cytokines, excitatory amino acids, and nitric oxide |
| ● | (NO); Increased NFkB à additional proinflammatory cytokines that act on neurons to create pain, fatigue, etc.; Naltrexone suppresses microglial activation; |
| ● | Low dose naltrexone (LDN) enhances maturation of bone marrow dendritic cells (BMDCs) |
| ● | Reduces inducible nitric oxide synthase activity à decreased peroxynitrite formation à glutamate transporters inhibited |
| ● | Excitatory neurotoxicity of glutamate on neuronal cells and oligodendrocytes is prevented |
| ● | Apoptosis of oligodendrocytes reduced |
| ● | Downregulates NFk2, inflammatory cytokines (TNF, IL-1, IL-6) |
| 11 |
| ● | These are all immune system changes associated with aging, which potentially can be improved by Lodonal, which will improve the quality of life for HIV/AIDS patients and reduce the health care burden on the medical system. |
| o | Avoiding contact with people who have infections is also important. In addition, people who are taking or have been taking immunosuppressant drugs should not have such immunizations as smallpox vaccinations without consulting their physician. Because their resistance to infection has been lowered, people taking these drugs might get the disease that the vaccine is designed to prevent. People taking immunosuppressant drugs should avoid contact with anyone who has had a recent dose of oral polio vaccine, as there is a chance that the virus used to make the vaccine could be passed on to them. |
| ● | Lodonal™ has no toxic side effects. |
| ● | LodonalTM does not compromise the immune system. |
| o | Indirect Cost of Immunosuppressive drugs |
| ● | HIV/AIDS still ranks 5th among the 14 diseases, with indirect per person costs ranging from $890 to $2663 in Zaire and from $2425 to $5903 in Tanzania indirect costs are roughly 95% of the total costs which are not covered by donor dollars. |
| ● | The total economic burden of CD was $10.9-15.5 billion in the United States and euro 2.1-16.7 billion of which is approximately 30% to 50% are indirect cost. |
| ● | Lodonal™ has been shown to reduce the number of opportunistic infections with HIV/AIDS |
Lodonal™ can improve compliance. When used correctly, antiretroviral therapy (ART) is effective. However, according to recent studies, ART regimens require 70–90% adherence in order to be effective. Sustaining adherence to ART over the long term requires accurate and consistent monitoring, and this is a particular challenge for countries in sub-Saharan Africa.
| o | Lodonal is a simple, once-daily regimen, which can be taken with or without a food, |
| o | Occasional non-compliance will not affect the overall success therapy. |
| o | Does not require the medical supervision of antiretroviral or immunosuppressive therapies (people will not lose time from work). |
| o | Compliance is further challenged by various social and clinical obstacles, where inadequate suppression of viral replication by ART are resulting due to poor adherence to therapy, low potency of the antiretroviral regimens and viral resistance to antiretroviral medications. |
| o | Estimates suggest that the average rates of non-adherence to antiretroviral therapy range from 50% to 70%. |
| o | Due to lack of compliance, the transmissibility of the antiretroviral resistant viruses from person to person further compounds the problem as a clinical and public health challenge. Which continues to be one of the major problems with treatment around the world. |
Our lead product candidate with the FDA is IRT-103 and is posed to initiate a pivotal phase IIB/III clinical trial for moderate-to-severe adult Crohn’s disease as well for pediatric Crohn’s disease, an orphan indication. We will need to complete both a Phase 2B and Phase 3 clinical trial for both Pediatric and Adult Crohn’s Disease for IRT-103 before we can obtain final approval to market the drug in the U.S. from the FDA.
The Company seeks to benefit patients with chronic and often life-threatening diseases through the stimulation and/or regulation of the body’s immune system. Using our patented immunotherapy, management believes that the Company’s products, technologies and patents will harness the power of the immune system to improve the treatment of cancer, HIV/AIDS, autoimmune diseases, opportunistic infections and inflammatory disorders.
Recent Accomplishments
The Company obtained drug approval from NAFDAC (National Agency for Food and Drug Administration and Control) in April of 2016. In May the Company began the regulatory process with NAFDAC for marketing authorization. There were a number of steps the Company is required to complete before marketing approval will be granted despite the current approval of Lodonal™ as a treatment of immune deficiency by NAFDAC.
| 12 |
The Company is currently completing the regulatory process to begin marketing the drug in Nigeria. Beginning in July of 2016 NAFDAC began requiring a manufacturing site inspection before authorizing the marketing of any new products, including Lodonal™.
On October 3, 2016 the Company received a letter from NAFDAC requesting a site visit of our manufacturing facility as the final step in regulatory marketing approval. Upon completion of the site visit the Company expects to receive marketing approval shortly thereafter.
The Company has been informed that the Nigerian approval allows the company to use the approval to be “fast tracked” for approval in the Economic Community of West African States, a regional group of sixteen countries, of which Nigeria is a member. The countries include Benin, Burkina, Faso, Cote d’Ivoire, Gambia, Ghana, Guinea-Bissau, Liberia, Mali, Nigeria, Senegal, Sierra Leone, and Togo. These countries play a major role in our Africa development strategy, as they are part of the “Test and Treat Program” for West and Central Africa. While the trend in international health funding for HIV/AIDS and ART (Antiretroviral Therapy) and the policies that drive the funding, has been to focus on high-burden countries and HIV ‘hotspots’ in Sub-Saharan Africa, most countries in the region classified by the UN as the West and Central Africa (WCA) region have been neglected. In the WCA region, 76% of those who need antiretroviral therapy – a total of five million people – are still awaiting treatment. The unmet needs in most of these countries are slipping further out of focus and we believe that Lodonal™ could help the people of West and Central Arica.
In September of 2016, the company retained the services of GLOBALMEDLINE SARL in Senegal to assist the drug registration of Lodonal™ in Senegal for HIV/AIDS and Cancer. The filing in Senegal is part of the company’s program to register Lodonal™ throughout the francophone (French-speaking) countries in Africa using our approval in Senegal to fast track the process.
The Company is currently in the process of filing in Kenya, Ghana, Liberia, Mali and Uganda. We will be adding additional countries in 2017.
In September of 2016 the Company signed a consulting agreement with the Honorable Joyce Banda as a member of the Champions for an AIDS Free Generation. His Excellency Festus Mogae, the Former President of the Republic of Botswana, first launched The Champions in 2008. The Champions program works to ensure that all children are born free from HIV in Africa and that all people have access to quality HIV prevention and treatment services.
The Company has been assigned three provisional patent applications: No. 62/296,759, a Method for Inducing a Sustained Immune Response; No. 62/379,272, a Method for Treating and Preventing Protozoan Infection; and No. 62/450,635, Methods and Compositions Useful for Treating Cancer Application. The Company expects to file additional patent applications in the coming months. Our intellectual property portfolio includes biotech assets acquired either through acquisition or exclusive licensing. The Company converted the provisional patent for method for inducing immune response into a US patent, and a PCT application was filed on February 17, 2017 under number PCT/IB2017/000124.
The Company has retained the services Coté Orphan to assist in obtaining FDA and EMEA approvals. Cote Orphan is a boutique, full-service, lab-to-market regulatory group focused on Orphan Drugs. Team Coté is led by Dr. Tim Coté, the former Director of the FDA’s Office of Orphan Products Development (OOPD). Dr. Cote is currently working with the Company to file a briefing package and final protocols for adult and pediatric Crohn’s Disease. The Company expects to finish this process by the end of the second quarter of 2017. The Company has delayed its request for a Type B meeting with the FDA on Crohn’s Disease, as the FDA is in the process of making changes in a number of areas as it applies to both 505(2)(b) pathway development and paediatric trials. The Company is preparing its final submission and expects to have the briefing package ready in the next 90 days.
The Company recently submitted its application for drug approval in Senegal and Kenya, and expects to have responses from the regulatory authorities by the end of the fourth quarter of 2017.
The Company’s Board has authorized continued discussions with a number of potential partners in Far East as well as discussion with drug development partners in both the US and EU.
| 13 |
MENK
MENK, also herein referred to as IRT-101, opiate growth factor or OGF, and Methionine-enkephalin is a synthetic peptide that activates natural killer (“NK”) cells of the immune system to seek and destroy cancer cells of the immune system to seek and destroy cancer cells. IRT-101 is a small peptide normally made by nerve cells and immune cells.
Further work in the laboratories of Plotnikoff, et al. has shown that daily injection of enkephalins into mice for 1 week resulted in increases in the size and weight of the thymus gland and a concomitant decrease in size and weight of the spleen. These observations led the same workers to study the antitumor action of the enkephalins. Mice carrying L 1210 tl.~mor cells were treated with methionine and leucine enkephalins, and their survival was compared to placebo treated controls.
The survival of the enkephalin-treated mice was longer than that of controls. Such observations in the animal model prompted these and other researchers to evaluate the immune potentiating effects of enkephalins and endorphins in humans. Plotnikoff and his group found that enkephalins and endorphins stimulated active rosettes in humans. Similar results were obtained by Gilman, et al., and Wybran, et al. This work was expanded further to study the effect of enkephalins on an immunosuppressed population of individuals. Lymphocytes from a group of lymphoma patients were treated with the enkephalins in vitro and then evaluated for rosetting with sheep red blood cells (SRBC’s.) It was found that both methionine and leucine enkephalins enhanced the ability of these cells to form T cell rosettes when compared to controls. Methionine enkephalin exhibited this immunopotentiation at a concentration as low as 10-14 mg/ml.
Studies have been undertaken in laboratories to investigate the effects of enkephalins on natural killer (NK) cell activity. The NK cells are a population of cells that can selectively lyse certain tumor cells in vitro without prior sensitization. This is a heterogeneous population of cells present in a variety of animals and humans. It is presently believed that these cells play a major role in protective surveillance against cancer. Anticancer potential of many of the immune activators has been measured by their ability to boost the (NK) defenses of the host. Results to date indicate enkephalins are capable of enhancing NK activity in vitro from normal volunteers and more importantly from cancer patients, some of whom have been heavily pretreated with chemotherapy.
Three separate groups have now reported in vivo clinical findings with the use of methionine enkephalin in patients with AIDS and symptomatic HIV infection. A Low Dose Study (10 micrograms/kg l.V.3 times/wk. for 12 weeks) Zunich and Kirkpatrick administered 10 μg/kg MEK intravenously three times weekly for up to 12 weeks to seven patients with various stages of HIV infection. This trial was conducted prior to the advent of potent antiretroviral therapy. In evaluating cellular immunity, the authors stated that MEK appears to temporarily enhance selected immune responses in patients infected with HIV. However, in this study, the results were neither clinically, nor statistically significant. There were no adverse reactions or evidence of toxicity.
Moderate-High Dose Studies (20, 25, 40, 50, 60, 80, and 100 micrograms/kg 1.V. 1 to 3 times/week for one to 24 months) have also been conducted. The preliminary clinical studies of MEK in symptomatic HIV infection, AIDS and cancer patients (30, 32, 45, 46, 47, 48 and 49) involved individualized treatment schedules with doses ranging up to 100 μg/kg 3x/week and durations extending beyond one year. There were no serious adverse reactions attributed to treatment for any of the patients studied. All reactions were transitory and appear to have been directly related to the infusion.
Eight Kaposi’s Sarcoma Patients were administered MEK (10-100 μg/kg 1-3 x week for 1-24 months). There were no serious adverse reactions attributed to treatment for any of the patients studied.
Symptomatic HIV Infection (Treatment one month up to 24 months, MEK administered at doses of 20-100 μg/kg 1-3x week). In a pilot study in these patients, Dr. J. Wybran in Brussels, Belgium indicated that there may be immunological improvements with MEK treatment. No adverse reactions attributable to MEK treatment were observed.
| 14 |
Asymptomatic HIV+ patients. Four asymptomatic HIV+ patients were treated with methionine enkephalin (60 μg/kg i.v.) once a week for 1-4 months. The monoclonal marker Leu 19 (CD56) for natural killer and killer cells was found to be increased in all four patients (18/24 discrete measures). No adverse reactions to methionine enkephalin were reported. It appears, based on the above data, that a new group of clinically useful immune modulators called enkephalins are emerging. Their in vitro magnitude of potency is at least 106 - 107 greater than that of interferon, interleukins and thymus hormones. Stimulatory effects were observed at concentrations as low as 1012- 10-14 mg/ml (33-37). Such characteristics make endogenous peptides ideal for therapeutic trials. The following additional facts are of interest in this regard: a) Methionine enkephalin stimulates humoral immunity (at low doses) and chemotaxis; b) Methionine enkephalin selectively stimulates production of cytotoxic T cells. Cytotoxic T cells (CD3, CD8) have been reported to inhibit reverse transcriptase of HIV; c) Subacute safety studies in rats and dogs at doses up to 25 mg/kg resulted in no toxicity; d) Human safety studies using doses up to 250 micrograms/kg resulted in no toxicity; e) Enkephalins may in fact be the natural mediators for the endogenous release of both interleukin 2 (32, 44), gamma interferon (32, 44) and IL-12.
Prior clinical studies
Eight (8) Phase I and four (4) Phase II clinical trials completed in cancer and HIV/AIDS between the years 1997-2015.
Notably promising data in HIV/AIDS and Cancer with strong efficacy signals:
123 adults in Phase I/II trials in cancer Between the years 1997-2014
250 adults in Phase I/II for HIV/AIDS Between the years 2000 -2014
Our first acquisition was the patents and intellectual property of Dr. Nicholas P. Plotnikoff and Professor Fengping Shan in 2012. While Dr. Plotnikoff was with Oral Roberts University, he was a member of the team that developed and patented the specific application of MENK as a treatment for cancer, HIV/AIDS, and infectious diseases. All of the clinical data generated by Dr. Plotnikoff has been included in our briefing package to the FDA.
Dr. Nicholas Plotnikoff initiated and completed Phase I and Phase II clinical studies of MENK under Investigational New Drug (“IND”) protocols filed with the U.S. Food and Drug Administration (“FDA”). In these clinical trials, MENK has been shown to reduce the symptoms of early AIDS and AIDS Related Complex (“ARC”), a condition also known as pre-AIDS which includes symptoms such as fever, diarrhea, weight loss, swollen lymph nodes and herpes. In addition to the therapeutic effects of the treatments, trial reports indicated an elevation in mood of the patients treated (Bihari, B., Plotnikoff, N., Freeman, K., Dowling, J., Duguid, C., and Altmann, E., ’‘Methionine Enkephalin in the Treatment of ARC,’’ Seventh Int. Conf. on AIDS, Florence, Italy, 1991).
A double-blind, randomized controlled Phase II study of 46 patients was performed with ARC (Bihari B, Plotnikoff NP. Methionine Enkephalin in the Treatment of AIDS-Related Complex. CRC Press, LLC; Cytokines: Stress and Immunity. 1999; 77-91) that was designed to measure the effect of a regular weekly dosing schedule of MENK at two different dose levels. The study involved randomized assignment to three arms: (i) patients on the first arm received weekly doses of 60µg/kg of MENK (low dose) for 12 weeks; (ii) patients on the second arm received a weekly infusion of 60µg/kg of MENK (low dose) for 2 weeks, followed by 10 weekly doses of MENK at 125µg/kg (high dose); and (iii) the patients on the third arm received a placebo intravenously for 12 weeks.
Product Development Status
| ● | Phase I Trial Pancreatic was completed in 2007 – Department of Medicine, The Pennsylvania State University, College of Medicine, GI Medicine. |
| ● | Dose is 250-300 μg/kg IV weekly. |
| ● | Showed minimal side effects. |
| ● | Determine the maximum tolerated dose standard 3+3 regimen starting at 25 μg/kg. |
| ● | Safety and toxicity of OGF (paresthesia, hypotension at 250μg/kg over 30 min). |
| ● | Compare route of administration (IV vs. SC); solubility issues with sc in low volume. |
| ● | Pharmacokinetic assays; [Met5]-enkephalin by RIA. |
| ● | Examine safety of chronic administration. |
| 15 |
Phase I combination trial OGF & Gemcitabine
| ● | Primary |
Evaluate the safety and toxicity of the combination of OGF biotherapy and gemcitabine chemotherapy in patients with advanced pancreatic cancer / pharmacokinetics. N =20
| ● | Secondary |
Examine the role of OGF given with gemcitabine on, patient survival.
Patients were treatment naïve.
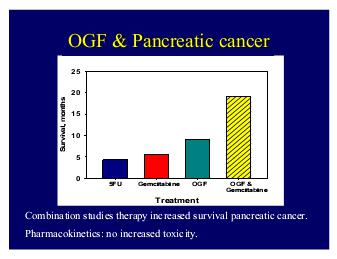
All of the symptoms and possible side effects of MENK/OGF therapy such as nausea, constipation, dry mouth, flushing, diarrhea and abdominal pain thought to be due to the advanced cancer rather than the treatment.
The two side effects thought to be due to the MENK/OGF therapy include the transient paresthesia or tingling at the beginning of the infusion and the hypotension.
The survival of patients with metastatic pancreatic cancer was increased to almost 9 months compared to standard historical medications used.5 FU only lengthens the survival to 4.5 months and gemcitabine to 5.8 months.
Phase II Trial Pancreatic Cancer
In our larger Phase 2 study when we compared survival of OGF patients to 266 untreated control subjects, the survival was increased. (Department of Medicine, The Pennsylvania State University, College of Medicine, GI Medicine)
| ● | Funded by FDA Orphan Drug Program | |
| ● | Treatment OGF 250mg/kg over 45 min weekly | |
| ● | Open-labeled, untreated controls | |
| ● | Primary endpoint: Survival | |
| ● | Secondary endpoints: efficacy, QOL | |
| ● | 25 OGF-treated subjects & 166 controls | |
| ● | Eligible patients: |
| ● | Unresectable pancreatic cancer | |
| ● | Failed standard therapy | |
| ● | Karnofsky status 50% |
Most importantly, there were no changes in the blood laboratory tests with OGF. Compared to standard chemotherapy that reduces the blood count from bone marrow toxicity, the blood count remained stable with OGF.
| 16 |
One of the most important features of OGF therapy in cancer patients with advanced metastatic disease is the marked improvement in Clinical Benefit. Clinical benefit is based upon parameters that reflect the overall well-being of the patient, including Pain control, performance status, and body weight. 53% of those receiving OGF experienced a clinical benefit compared to patients treated with standard chemotherapy.
The Company is planning to rely on the following available information and historical data to support the initiation of the Phase 3 study and filing of the NDA:
1. Clinical safety data from studies conducted under this IND (50,987 studies NIH# R03
CA80646, NCT00109941, and IRB Protocol No. 26336;
2. Clinical safety data from studies conducted under IND 34,442 (MENK for the treatment of AIDS/ARC and cancer patients and normal healthy volunteers
Toxicology Studies completed by Baxter and Travenol 1984 and 1985
Published literature as summarized in this package (to provide nonclinical pharmacology and additional safety data). To support the NDA filing, we are also planning to conduct the following additional study:
Phase 2b study will run in parallel with the Phase 1 PK and 3 month GLP toxicology studies.
| ● | Population to be studied: Patients on first line therapy with local advanced or metastatic disease |
| ● | The Company is running the GLP toxicology at an FDA approved facility in China and those studies will be completed before the end of Q4 2017. |
| ● | The company anticipates a database of 300 – 600 patients at the time of NDA filing. |
| ● | The FDA stated that a genotoxicity study will not need to be conducted. |
| ● | The MTD has been established previously under this IND in pancreatic cancer patients |
| ● | Due to safety data collected to date and established MTD, TNI BioTech believes that a Phase 1 PK study in healthy volunteers can be run concurrently with a Phase 2 clinical trial in pancreatic cancer patients |
| ● | Doses will not exceed the previously established MTD (250 µg/kg) |
Sponsor, which in this case is the Company, will submit the Phase 2b clinical protocol for FDA review. The FDA confirmed that the Phase 2b study could be conducted as an open-label randomized study with two doses of MENK in combination with nap-paclitaxel + gemcitabine versus nap-paclitaxel + gemcitabine. Exact study design will be determined by the Sponsor, and submitted to the FDA for review.
After completion of the Phase 2b study, the Agency would grant the Sponsor an End-of-Phase 2 meeting.
Hubei Qianjiang and Immune Therapeutics have a signed agreement for the development of MENK and have moved forward on that project since 2014-present.
PK and Toxicology Studies were delayed in 2015 because the China Food and Drug Administration (CFDA) required Chemistry, Manufacturing and Controls to be completed before starting the toxicology study. They have been started and will be completed by the end of 2017.
Hubei Qianjiang signed Pharmaceutical Development Agreement for Formulation Development and CTM Manufacturing of Methionine–Enkephalin for Quinjiang and Immune Therapeutics with China Peptide Company (“CPC”) in Q3 2015
CPC perform analytical, pre-formulation, formulation development, clinical trial manufacturing, release testing and ICH1 stability for Methione- Enkephalin
CPC is among only a handful of companies in the world that can claim both ISO Certification and cGMP licensing. In February 2012, CPC became the first peptide company to successfully pass US FDA inspection outside of US and Europe regions.
CMC and formulations are required for mass production of MENK, which is also required for pivotal trials with the FDA for this work has been ongoing since our meeting with the FDA.
| i. | Qianjiang will provide Cote Orphan the CMC data which is required as part of filing MENK protocols. |
| 17 |
In addition Hubei has completed pre-clinical studies using MENK on various cancers in the lab using mouse models and has shown to be successful in a number of cancers including colon, pancreatic and hepatic. The data will be translated and provided as part of the briefing package for both pancreatic and liver cancer.
The MENK treatment was generally well tolerated with no appreciable toxicity observed. The high dose of MENK increased adaptive cell immunity resulting in increased activity of the body’s immune system (e.g. increased IL-2 receptors, CD56 NK and LAK cells, CD3, CD4 and CD8 cells) and a reduction in the size of lymph nodes. One patient in the high dose group administered by rapid intravenous infusion experienced dizziness, diaphoresis, elevated blood pressure and decreased pulse rate. These signs and symptoms were responded to with supportive measures.
Recently, Professor Fengping Shan and Dr. Plotnikoff have published, in a number of peer-reviewed international journals, that MENK inhibited regulatory T-cells, increasing the functional activities of T cells and NK cells and, thus, is a key to improved cancer therapy. They additionally published results showing that MENK alone or in combination with Interleukin-2 (“IL -2”) or Interferon-γ (“IFN-γ”) can enhance the production of interferon- γ or IL-2 from CD4+T cells, respectively (Shan F, Yanjie Xia, Ning Wang, Jingjuan Meng, Changlong Lu, Yiming Meng, Nicolas P. Plotnikoff. Functional modulation of the pathway between dendritic cells (DCs) and CD4+T cells by the neuropeptide: Methionine enkephalin (MENK). Peptides 32. 2011; 929–937). MENK also appeared to be more potent than IL -2 or IFN-γ, alone (Hua H, Changlong Lu, Weiwei Li, Jingjuan Meng, Danan Wang, Nicolas Plotnikoff, Enhua Wang and Fengping Shan. Comparison of stimulating effect on subpopulations of lymphocytes in human peripheral blood by methionine enkephalin with IL-2 and IFN-γ. Human Vaccines & Immunotherapeutics 8:8, 2012; 1082-1089), two widely known cytokines that have been approved by the FDA for marketing.
Plotnikoff & Shan History
| ● | 1983 Baxter takes license Conducts sub-chronic multiple dose pathology and toxicology studies |
| ● | Beginning 1984 Open Label clinical studies started in cancer and AIDs patients |
| ● | 1989 NIH AID’s Committee recommends Methionine-enkephalin for inclusion in AIDs clinical trials (low priority). Methionine-enkephalin discovered to activate LAK cells which destroy AIDs virus |
| ● | 1990-1995 Double Blind placebo controlled study in HIV patients begun at C.R.L in New York |
| ● | 1995-2000 Open Label Tulsa, Belgium, Denver and New York |
| ● | 2012-2014 Open Label 1Department of Cord Blood Bank, Shengjing Hospital; China Medical University; Heping District, Shenyang, PR China; Department of dermatology; No.1hospital; China Medical University; Heping District, Shenyang, PR China; Department of Immunology; School of Basic Medical Science; China Medical University; Heping District, Shenyang, PR China and TNI Bio. Tech. Inc.; Orlando, FL USA |
| ● | In 1984 Nicholas P Plotnikoff and Gerald C miller and Joseph Wybran (etc) initiated a trial in 14 healthy volunteers and 8 cancer patients the Clinical pharmacology of methionine-enkephalin was studied in normal volunteers at doses of 1, 10, 50, 100, 150, 200, and 250 pg/kg. Immunologically, increases in total lymphocytes, B lymphocytes, active rosette-forming cells, T lymphocytes (OKTl 1), T-helper lymphocytes (OKT4), and T-suppressor lymphocytes (OKT8) were seen after infusion with methionine-enkephalin. In addition, increased mitogen-stimulated blastogenesis with PHA, Con A, and pokeweed were also seen with methionine-enkephalin treatment. No material changes were seen in EKG, blood pressure, heart rate, respiratory rate, temperature, or neurologic reflexes of normal volunteers receiving methionine enkephalin in doses of 1 to 200 pg/kg. |
Our human (in vivo) studies have demonstrated that methionine-enkephalin is an activator of T-cell subsets, NK cells, and potentiator of blastogenesis in the presence of PHA, Con A, pokeweed, or Staph A. All of the clinical pharmacologic variables were normal, including EKG, heart rate, blood pressure, respiration, temperature, and neurologic reflexes, as well as urinalysis and SMAC 26. Transient side effects such as vasodilation and/or gastrointestinal cramps were seen only at high doses (100, 150, 200, and 250 pg/kg). Thus, in this study, methionine-enkephalin, a natural hormone, was administered without significant adverse effect in a dose range of 1 to 250 pg/kg (by intravenous infusion).
| 18 |
In our studies in cancer patients with Kaposi’s sarcoma (due to AIDS), melanoma, lung cancer, and hypernephroma increases in T-cell subsets were also observed. Increased levels of blastogenesis with the mitogens PHA, Con A, and pokeweed were also observed. An increased expression of interleukin-2 receptors was also observed, while Wybran reported increased blood levels of interleukin-2 in patients receiving methionine-enkephalin. In addition, Wybran et al. 28 have reported that methionine enkephalin elevates T-cell subset numbers in pre-AIDS or ARC patients. Methionine enkephalin may well be useful as an immunomodulator in the treatment of patient in the early stages of their illness and/or following surgery, radiation, or chemotherapy treatment.
Cancer Patients
Methionine-enkephalin was administrated to seven patients with lung cancer. These patients were newly diagnosed and had not yet received prior treatment such as surgery, chemotherapy, radiotherapy, or immunotherapy. Immunologic tests were performed before Methionine-enkephalin injection, and 2 hours, 24 hours, 6 days after Methionine-enkephalin perfusion. The results can be summarized as follows: in four patients active T-cells were increased in the blood; in five patients the percentage of OKTlO increased in the blood; and in seven the cells with the Leu 11 phenotype increased (to more than twice the initial value in four of these patients). More interestingly, NK activity increased in five of seven patients (by more than 100 in three of them, in whom NK activity increased from 30 to 60, 7 to 15%, and 18 to 36%).
Once again, the absence of subjective or objective side effects should be stressed in this single-injection study.
ARC Patients
Seven patients with AIDS-related complex received Methionine-enkephalin three times a week intravenously for a minimum of 21 days; the concentrations varied from 20 to 100 pg/kg at each injection. Some patients have already been treated for 130 days. The immunologic results can be summarized as follows after 21 days of treatment: increase in the numbers of blood OKT3 and OKT4 lymphocytes, no increase in the absolute count of lymphocytes, borderline increase in NK activity (p < 0.10), and increase in IL-2 production as well as in PHA response. The most striking result is the enhancement of the PHA response. Some patients were treated for a longer period of time and one patient has shown a remarkable immunologic and clinical course. This 32-year-old Caucasian homosexual male had a prior history of an 11-kg weight loss, night sweats, recurrent scrota infections, and lymphadenopathy for a period of 2 years. Testing of his immunologic status showed a reduced OKT4 percentage (20%) and count 336/mm3), low NK activity (22%), low ILL-2 production (0.5 units), and low PHA response (147,000 cpm). He was started on Methionine-enkephalin and followed immunologically as well as clinically. At day 130, his OKT4 percentage is 38% and the OKT4 count 730/mm3. The NK activity has increased to 43% and the IL-2 production is also normalized to 2.1 units. Finally, his PHA response has presently reached 414,000 cpm. Clinically, this patient has no more scrota1 infections, he has gained 8 kg, the night sweats have disappeared, and the lymph nodes have completely regressed within 3 months. In summary, this patient shows an almost complete immunologic nonspecific functional reconstitution, except with respect to the OKT4 subset. Clinically, he is in complete remission. All the data indicate that Methionine-enkephalin can enhance some immunologic functions in ARC patients, and preliminary data suggest some therapeutic beneficial effect in some patients. These results have to be confirmed in larger series in double-blind randomized trials.
AIDS Patients
One AIDS patient received a single injection of 20 pg/kg of Methionine-enkephalin and his PHA response increased from 4300 cpm to 14,000 cpm! Another AIDS patient with Kaposi’s Sarcoma is now receiving chronic treatment with Methionine-enkephalin. His lesions have remained stable for 4 months. More interestingly, an AIDS patient (with Hodgkin’s lymphoma, Toxoplasma cerebral abscess, atypical mycobacteria) was diagnosed as having Kaposi’s Sarcoma lesions (biopsy-proven). He was started on Methionine-enkephalin treatment 1 month after the Kaposi’s Sarcoma lesions appeared, and after 4 weeks of treatment these lesions have flattened and are in the process of disappearing (as determined by biopsy). In the case, Methionine-enkephalin administration was associated with the regression of the Kaposi’s Sarcoma.
Clinical Studies finished with 178 patients in Tulsa, Brussels the Belgium Medical School, New York SUNY, Denver Colorado Medical School and Chicago to increase number of cytotoxic cells CD4, CD8 and NK Cells in AIDS and Cancer patients. The studies demonstrated that Methionine-enkephalin is an effective and potent immunomodulator in HIV/AIDS patients. Methionine-enkephalin exhibited a dose response between the dose of 10, 20, 50 and 100 micrograms/kg in terms of increasing number of CD3, CD4, CD8 and NK cells in these patients (average increase of 50%). Higher doses of Methionine-enkephalin (150, 200, 250 and 300 micrograms/kg) exhibited a plateau effect. No toxicity was seen in these patients. Methionine-enkephalin increases the number of cytotoxic cells (subsets CD4, CD8 and NK) that are known to specifically destroy HIV. This “antiviral” effect was recorded by marked reduction of p24 both in vitro and in vivo.
| 19 |
A phase II clinical study under IND was completed. The study, was a double-blind and placebo controlled, involved randomized assignment to three arms receiving a weekly intravenous infusion of 60mpg/km (low Dose) or receiving a weekly infusion of 125 micrograms/kg and the third receiving a placebo infusion of normal saline. Twenty subjects completed 12 weeks of the trial, twenty-six patients completed 8 weeks, and 33 patients completed 4 weeks. Eligibility for this clinical study included a positive HIV serology, CD4 level between 200 and 500.
Substantial differences from baseline were observed (at eight weeks in patients receiving 125 micrograms/km) for the following parameters: CD4, DC8, DC35, CD38, CD56, NK, PHA, PWM and CMV. The most important findings of the study were the increase in CD4 (T helper cells) and CD56 (cytotoxic cells NK-K-LAK). There were no serious adverse reactions.
Phase I and Phase II studies have been conducted under the Company’s IND (previously held by Penn State University) that have demonstrated that MENK can be delivered to patients suffering from advanced pancreatic cancer, hepatocellular carcinoma (PI: Eric Kimchi, M.D. at Penn State University) and advanced head and neck cancer (PI: David Goldenberg, M.D., FACS at Penn State University). The maximum tolerated dose has been found to be 250 µg/kg using the intravenous route of administration over a 30-min infusion time in a Phase I trial in fourteen normal volunteers and eight cancer patients. This maximum tolerated dose was later confirmed in Phase I and Phase II trials in patients with advanced unresectable pancreatic cancer.
Patients with Kaposi’s Sarcoma (9 patients), lung cancer (12 patients), melanoma (3 patients), hypernephroma (1 patient), or pancreatic cancer (1 patient) were treated with MENK for one week to 12 months at doses of 10 µg/kg three times per week up to 80 µg/kg 3 times per week. After 1-2 weeks increases in T cell subsets (CD3, CD4, CD8, and CD2 positive cells) were observed. An increase also occurred in IL-2 receptor expression. NK cell activity was measured in 14 patients and an increased NK activity was present in 12/14 patients. No toxicity attributable to treatment with MENK was observed in any patient.
Two case studies have been reported in an infant and a 20-month old child who were treated with MENK. The infant was diagnosed with hepatoblastoma and was treated with one course of neoadjuvant chemotherapy at approximately one week of age. Due to complications from the chemotherapy (neutropenic fever, pneumonia and sepsis), the patient’s parents declined further chemotherapy, and the infant was treated with surgical resection and MENK/LDN. She is currently close to ten years disease–free survival. The 20-month-old child was diagnosed with hepatoblastoma. Due to existing comorbidities (including autosomal recessive polycystic kidney disease and hypertension), and biopsy results that indicated the tumor might be insensitive to chemotherapy, the parents elected not to proceed with neoadjuvant chemotherapy. The patient was treated with surgical resection and MENK/LDN, and is currently at more than five years disease-free survival.
2013- 2014 Open Label China Medical School
2012-Present: We have been running trials both in-vitro and in-vino and have published on some of the work that was completed.
MENK, a penta-peptide is considered as being involved in the regulatory feedback loop between the immune and neuroendocrine systems, with marked modulation of various functions of human immune cells. The aim of the present work was to investigate change of lymphocyte subpopulations in peripheral blood of 50 cancer patients before and after treatment with MENK. Peripheral blood mononuclear cells (PBMCs) of peripheral blood from 50 cancer patients were isolated by density gradient centrifugation using Ficoll-Paque solution and cultured with MENK. We measured proliferation of total nucleated cells, subpopulations of individual CD4+T cells, CD8+T cells, CD4+CD25+ regulatory T cells (Treg), natural killer cells (NK) before and after treatment with 10-12M MENK in cell culture by flow cytometry (FCM).
| 20 |
Our results indicated that MENK showed a strong inhibiting effect on Treg cells while it stimulated marked proliferation of other lymphocyte subpopulations. All data obtained were of significance statistically. It was therefore concluded that MENK could work as a strong immune booster with great potential in restoring damaged human immune system and we could consider MENK as a drug to treat cancer patients, whose immune systems are damaged by chemotherapy or radiotherapy. Furthermore we could consider MENK as a chemotherapy additive, which would sustain immune system of cancer patients during the process of chemotherapy to get maximized efficacy with minimized side effect.
Methionine enkephalin (MENK) improves lymphocyte subpopulations in human peripheral blood of 50 cancer patients by inhibiting regulatory T cells (Tregs). The patients recruited for this study were all with terminal cancers with broad metastasis, underwent chemotherapy and their immune system were damaged severely. They failed to respond to any therapy available and were desperate. After they signed informed consent we began to give treatment. The concrete cases distribution was as following: Rectal cancer: 4, Colon cancer: 6, Stomach cancer: 3, Hepanocellular cancer: 2, Lung cancer: 7, Oval cancer: 3, Pancreatic cancer: 2, Breast cancer: 3, Urinary bladder cancer: 1
This approach helps us learn more knowledge about MENK’s action in rehabilitating human immune system and the conclusion of MENK as an immune enhancer, drawn from present study is fully supported by the data obtained. We believe that this is the first time that published data based on large samples show that MENK could stimulate proliferation of lymphocyte subpopulations by inhibiting Tregs in peripheral blood of cancer patients.
Research results indicate that MENK, at suitable doses, boosts the immune system through the following possible mechanisms:
| ● | increasing proliferation and functional activities of CD4+T-cells and CD8+T-cells which will play a role in anti-virus and anti-tumor activities; | |
| ● | increasing maturation of dendritic cells which will initiate and intensify T-cell responses; | |
| ● | increasing secretion of cytokines such as IL-2, TNF, IL-12 and IFN-γ which will amplify the T-cell response and mediate interaction among immune cells, forming a modulated and balanced immunity; | |
| ● | increasing functions of macrophages, resulting in enhanced cellular immunity through secreting a set of cytokines; and | |
| ● | increasing activity of NK cells which have the ability to kill cancer cells and virus-infected cells. |
Based upon published literature, the Company believes that, in oncology in particular, MENK has two possible mechanisms of actions:
1. Immune stimulation and regulation effects; and
2. Direct anti-cancer inhibitory effects.
Based upon data from multiple in vivo and in vitro studies conducted by Zagon et al. over the past 15 years, the onset and/or progression of some cancers may be related to defects in MENK and/or OGFr, which would promote or exacerbate tumorigenesis. These findings show there may be an advantage in up-regulating the peptide (e.g., MENK administration) to enhance anti-cancer activity (Zagon IS, Donahue RN, McLaughlin PJ. Opioid growth factor-opioid growth factor receptor axis is a physiological determinant of cell proliferation in diverse human cancers. Am J Physiol Regul Integr Comp Physiol. 2009; 297: R1154–R1161). Further exploration and clinical trials are needed to confirm MENK’s mechanism of action and its ability to stop the growth of cancerous cells in human subjects with advanced cancer; however, supportive literature around the possible mechanism of actions for MENK are provided below.
MENK as an immune stimulator/regulator
While Dr. Nicholas Plotnikoff was a faculty member at Oral Roberts University, he discovered that all three of the classical opioid receptors are expressed on most subsets of immune cells, and that either in vitro incubation with MENK or parenteral administration of and humans, especially those with immunodeficiencies associated with cancer or HIV/AIDS, MENK in vivo increased the number and functional activities of T cells, including cytotoxic CD8+ T cells, and also natural killer (NK) cells (Plotnikoff NP, Faith RE, Murgo AJ, Herberman RB, Good RA. Methionine Enkephalin: A New Cytokine – Human Studies. Clinical Immunology and Immunopathology. February 1997; 82(2): 93-101). Following those pioneering studies, several other investigators observed that administration of MENK to mice increased CD4+ and CD+ T cells, and increased various immune functions, including cytotoxic activities of both T cells and NK cells (Wybran J, Schandené L, Van Vooren JP, Vandermoten G, Latinne D, Sonnet J, De Bruyère M, Taelman H, Plotnikoff NP. Immunologic properties of methionine-enkephalin, and therapeutic implications in AIDS, ARC, and cancer. Ann N Y Acad Sci. 1987; 496:108-14). Recently, Drs. Fengping Shan and Nicholas Plotnikoff have reported that MENK treatment of mice stimulates the cytotoxic activities of T cells and NK cells and reduces levels of T regulatory cells, and augments therapeutic effects in tumor-bearing immunocompetent mice.
| 21 |
MENK as an inhibitor of cancer cell growth
MENK has been found to exert a profound inhibition on the initiation and progression of human pancreatic cancer in vitro and in vivo (Zagon IS, Smith JP, McLaughlin PJ. Opioid Growth Factor (OFG) Inhibits Human Pancreatic Cancer Transplanted into Nude Mice. Cancer Letters. 1997 Jan 30; 112(2):167-175. Zagon IS, Smith JP, Conter R, McLaughlin PJ. Identification and Characterization of Opioid Growth Factor Receptor in Human Pancreatic Adenocarcinoma. International J of Molecular Med. 2000 Jan; 5(1):77-84.). This led to the discovery that MENK interacted with a novel opioid receptor (OGFr) on human cancer cells (ovarian, SCCHN, pancreatic, colorectal and others) creating a competitive inhibition profile and subcellular location that is different from other well-known “classic” opioid receptors [mu (µ), delta (δ) and kappa (κ)]. The other “classic” opioid receptors have not been found to have any impact on cell growth; thus there is specificity in the MENK-OGFr interaction which regulates cell proliferation. In an extensive number of experiments that have been conducted on human pancreatic cancer cells in tissue culture exposed to a variety of opioid-related compounds, MENK was the only compound that inhibited cell proliferation. Based upon data from multiple in vivo and in vitro studies conducted by Zagon et al., the onset and/or progression of some cancers may be related to defects in MENK and/or OGFr, which would promote or exacerbate tumorigenesis. These findings show there may be an advantage in up-regulating the peptide (e.g., MENK administration) to enhance anti-cancer activity.
Therefore, as opioid receptors are not only found on cancer cells, but also on most subsets of immune cells, MENK has the ability to not only inhibit cancer cell growth, but also have a direct impact on the patient’s immune system by increasing the number and functional activities of T cells and NK cells.
Zagon and McLaughlin have not recorded in any of their animal studies any side effects of MENK for non-oncological indications such as experimental autoimmune encephalomyelitis (EAE, the mouse model of multiple sclerosis) or relapse-remitting EAE (RR-EAE) with MENK being administered daily. Treatment with MENK or LDN did not exacerbate EAE and was able to halt progression of disease, reverse neurological deficits, and prevent the onset of neurological dysfunction over time (Rahn KA, McLaughlin PJ, Zagon IS. Prevention and diminished expression of experimental autoimmune encephalomyelitis by low dose naltrexone (LDN) or opioid growth factor (OGF) for an extended period: Therapeutic implications for multiple sclerosis).
Management believes clinical trials involving LDN hold great promise for the millions of people worldwide for the treatment of autoimmune diseases or disorders, central nervous system disorders or those who face cancer. Management also believes it could be the first low-cost, easy to administer therapy with minimal to no side-effects for the treatment of HIV/AIDS, autoimmune diseases and immune disorders, in particular Crohn’s disease, multiple sclerosis, and/or fibromyalgia.
Naltrexone is an orally effective opioid receptor antagonist, used as a treatment for opiate addiction. Naltrexone was originally synthesized in 1963 and patented in 1967. In 1984, the FDA approved naltrexone in a 50 mg dose as a treatment for heroin addiction. Naltrexone 50 mg film-coated tablets have been approved in Europe since at least 1989 for the treatment of opiate addiction and more recently alcohol dependency. At lower doses (approximately 4.5 mg/day), it has been gaining popularity as a treatment for signs and symptoms of autoimmune diseases and immune disorders, HIV/AIDS and cancer. Research studies by others have indicated that the short-term blockage of opioid receptors on circulating and tissue cells by LDN was followed by a substantial rebound in opioid receptor expression and increased levels of β-endorphin and methionine-enkephalin (Zagon IS, McLaughlin PJ. Opioid antagonist modulation of murine neuroblastoma: A profile of cell proliferation and opioid peptides and receptors. Brain Res. 1989; 480:16–28.)
| 22 |
Oral administration of LDN has been shown to transiently (approximately 4 hours) inhibit opioid receptors which in turn provides a remaining window of approximately 20 hours for the unregulated opioids and receptors to interact (Donahue RN, McLaughlin PJ, Zagon IS. The opioid growth factor (OGF) and low dose naltrexone (LDN) suppress human ovarian cancer progression in mice. Gynecol Oncol. 2011 Aug; 122(2):382-8).
LDN has been shown to increase the levels of endogenous opioid activity, thereby having the ability to play a direct role in enhancing the human body’s stress resilience, improving psychiatric problems such as autism, in addition to being able to have a direct impact on the immune system and regulation of how the immune system works when faced with disease. LDN is believed to facilitate the body’s own resources to slow down or combat cancers, autoimmune diseases and HIV/AIDS; thus reducing the overall impact and load on the body (Brown N, and Panksepp J. Low dose naltrexone for disease prevention and quality of life. Med Hypotheses. 2008 Mar: 72(3):293-6.).
Naltrexone was originally patented in 1967 by the specialty pharmaceutical company Endo Health Solutions Inc. At the time, it seemed unlikely that naltrexone would be developed because the experimental drug had relatively low market potential, and naltrexone’s patent protection would likely expire before the completion of clinical trials. With the assistance of DuPont, a division of Merck & Co. that acquired Endo in 1969, the US government’s National Institute on Drug Abuse (“NIDA”) advanced naltrexone through the FDA approval process, leading to approval for marketing as a treatment for heroin addiction in a 50 mg dose in 1984. Although its patent expired that same year, naltrexone gained seven additional years of marketing exclusivity for DuPont when the FDA designated it (trademarked as Trexan) an Orphan Drug. Marketing exclusivity provides a pharmaceutical company the right to sell its drug for a certain length of time free of competition from generic versions of the drug and is often granted to encourage companies to develop a use for a drug whose patent has expired or to encourage a company to develop an already approved drug for a new use. With market exclusivity, the anticipated returns on investment are higher, improving the profitability of a drug. With funding provided by the US National Institute on Alcohol Abuse and Alcoholism (“NIAAA”) and the potential to gain three additional years of post-approval market exclusivity for naltrexone, DuPont advanced naltrexone through additional clinical trials, and gained FDA approval for a 50 mg dose (trademarked as ReVia) as a treatment for alcohol abuse in 1995. As naltrexone had already been on the market for 10 years as a treatment for heroin addiction, the FDA’s confidence in its safety resulted in approval only six months after naltrexone’s regulatory application was submitted.
Naltrexone has a black box warning for liver toxicity, which was included based on liver enzyme elevations reported with daily dosing at 100 mg-300 mg. These doses were evaluated in clinical trials for obesity; however, they have not been approved for this use. Our review of the literature and adverse effect reports in naltrexone clinical trials did not demonstrate a risk for liver damage with daily dosing at 50 mg. Although the black box warning does remain, the FDA has stated that naltrexone does not appear to be a hepatotoxin at the recommended doses for the currently approved indications. Recently, naltrexone at 4 mg and 8 mg, in combination with the anti-depressant drug, bupropion at 90 mg, was evaluated as an anti-obesity drug by Orexigen Therapeutics Inc. and submitted for FDA approval in 2010. Data from studies with the drug combination (trademarked Contrave) showed potential hepatotoxicity in 1.2% of subjects treated with Contrave (n = 3,239). [Note: After the FDA requested a long-term study to demonstrate the daily recommended dose of the drug combination (two tablets each with 8 mg naltrexone plus 90 mg bupropion taken twice daily) does not raise the risk of heart attacks, Orexigen initiated a Phase III trial to evaluate Contrave in a study expected to enroll more than 9,000 subjects and is anticipated to be completed in 2017.] Other than its small potential association with liver toxicity at high doses, the most common adverse effects reported with naltrexone are non-specific gastrointestinal complaints such as diarrhea and abdominal cramping.
Notable published clinical trial evidence indicates that LDN, particularly daily dosing at 3mg - 4.5 mg, stimulates the immune system and is effective in the treatment of some immunodeficiency diseases, such as HIV/AIDS diseases, and advanced cancer as shown in the studies referenced herein. The first clinical trial results with LDN for immune disorders, however, were published only recently in a peer-reviewed medical journal in 2007 which evaluated LDN treatment in a pilot phase II study of 17 patients with Crohn’s disease, a form of inflammatory bowel disease that most commonly affects the ileum and the beginning of the colon. Two-Thirds of patients in this study went into remission after 4.5 mg daily LDN treatment (p < 0.001), with 89% of patients overall showing some degree of response.
| 23 |
An open-label pilot study was conducted by Pennsylvania State University with LDN to evaluate response, safety and toxicity in adult subjects with moderate to severe, active Crohn’s disease. Patients were treated with LDN orally each evening at a dose of 4.5 mg for 3 months. A total of 17 patients were enrolled, 16 of whom completed the study. No laboratory abnormalities were noted. The most common side effect was sleep disturbances (occurred when dosing at night, at about bed-time), occurring in seven patients (41%).
A second clinical study was conducted by Pennsylvania State University as a randomized double-blind, placebo-controlled study to test the efficacy and safety of LDN for 12 weeks in adults with moderate to severe active Crohn’s disease. Forty subjects with moderate to severe active Crohn’s disease were enrolled in the study. Randomized patients received daily oral administration LDN (4.5 mg/day) or placebo. Fatigue was the only side effect reported of statistical significance, and it was greater in subjects receiving placebo.
A pilot Phase II clinical trial was conducted by Pennsylvania State University in children with moderate to severe active Crohn’s disease. Fourteen subjects were enrolled, 12 subjects were randomized and treated with a mean age of 12.3 years (range 8-17 years). Children were randomized to placebo or LDN (0.1 mg/kg or a maximum dose of 4.5 mg) orally for 8 weeks followed by open-label treatment for an additional 8 weeks of LDN at the same dose of 0.1 mg/kg or 4.5 mg. Oral LDN was well tolerated without any serious adverse events.
Fourteen (14) subjects were enrolled, 12 subjects were randomized and treated (Smith J. et al., 2007; Smith J. et al., 2011; Smith J. et al., 2013).
Pennsylvania State University researchers have also demonstrated in a mouse model of Crohn’s disease (chemically induced colitis with dextran sodium sulfate) that opioid receptor blockade by LDN resulted in less weight loss, lower disease activity index scores and less histological evidence of inflammation when compared to controls. Furthermore, the researchers demonstrated that tissue inflammatory cytokine mRNA was reversed to baseline levels in the colons of mice treated with LDN.
Management believes in LDN’s potential treatment effects for Crohn’s disease, as the treatments currently available for Crohn’s disease are expensive and carry black box warning due to the toxic side effects associated with virtually all of the currently used drugs. Management believes that LDN provides an attractive alternative. Three published clinical trials in patients with moderate to severe disease, two in adults and one in children, have shown notable disease and quality of life improvement by 12-weeks. LDN was able to reverse the inflammatory activity, promote mucosal healing, and decrease histologic inflammation when compared to placebo-treated controls (Smith J. et al, 2011; Smith J. et al, 2013). Based on these results, the Company has placed a very high priority on implementing a pivotal Phase IIB/III study. By using the 505(b)(2) pathway, confirmation of efficacy in our Phase III study is expected to result in approval by the FDA.
With its increasing recognition in children and adolescents, Crohn’s disease has become one of the chronic diseases that affect young people. Pediatric Crohn’s disease affects approximately 80,000 patients in the United States, and thus has led to orphan drug designation with the FDA. In addition to the common GI symptoms due to inflammation in the small and/or large intestine, children often experience growth failure, malnutrition, pubertal delay, bone demineralization, and psychological issues. Crohn’s tends to be both severe and extensive in the pediatric population with a relatively high proportion of pediatric Crohn’s patients having involvement of their small intestine, proximal to the ileum.
| 24 |
The following table provides a summary of clinical trials for LDN that have recently been or are being conducted by Pennsylvania State University:
| Title | Indication(s) | Dose | ClinicalTrials.gov Identifier / Status | |||
| Low Dose Naltrexone for Metastatic Melanoma, Castrate Resistant Prostate Cancer and Renal Cancer | Metastatic Melanoma, Castrate Resistant Prostate Cancer and Renal Cancer | 5 mg/day | NCT01650350 / Currently Recruiting (verified May 2013) | |||
| Effects of Low Dose Naltrexone in Fibromyalgia | Fibromyalgia | 3-4.5 mg/day | NCT00568555 / Completed June 2012 (verified June 2012) | |||
| Low Dose Naltrexone in Symptomatic Inflammatory Bowel Disease | Inflammatory Bowel Disease | 4.5 mg/day | NCT01810185 / Not yet recruiting (verified June 2013) | |||
| Low dose Naltrexone for Glioma Patients | Malignant Glioma | 4.5 mg/day | NCT01303835 / Active, not recruiting (verified January 2014) | |||
| Low dose Naltrexone for Depression Relapse and Recurrence | Major Depressive Disorder Depression, Unipolar Recurrence Relapse |
1 mg/day | NCT01874951 / Recruiting (verified September 2013) |
The 505(b)(2) Regulatory Pathway
Traditionally, pharmaceutical drugs had to be approved by the FDA under the standard 505(b)(1) regulatory pathway, which could take as long as 15 years. Now, drugs approved under 505(b)(2) may rely in part on data from existing reference drugs meaning they can be developed and achieve FDA approval in as little as 30 months with only a fraction of the number of required clinical trials and at a much lower cost.
Developing LDN using the 505(b)(2) regulatory pathway decreases the amount of development time and cost in order to obtain FDA approval. As naltrexone is an FDA-approved product for alcohol or opiate dependence, prescriptions are currently being filled for naltrexone in 50 mg doses by hundreds of local pharmacies and mail-order pharmacies around the United States.
The FDA’s 505(b)(2) pathway for approving drugs opens the door for the Company to gain FDA approval of LDN (which is used at doses of approximately 1/10th the approved dose) for new diseases. A 505(b)(2) drug application for LDN will contain full reports of clinical investigations to support the safety and effectiveness in the new indication(s); however, at least some of the information required for approval will come from studies not conducted by or for the applicant, and for which the applicant has not obtained a right of reference or use, as many of these drugs are now off-patent. With the opportunity to use previous findings of safety, the Company intends to use the 505(b)(2) pathway to study and gain approval for LDN in other diseases, with Crohn’s disease slated as its first therapeutic indication.
As there is a sufficient database of information around the safety of this product, and the reference listed drug (NDA 018932 REVIA) is being used, the FDA agreed that a 505(b)(2) application would be an acceptable approach at this time.
The Company, through its subsidiary, will need to complete both a Phase 2B and Phase 3 clinical trial for both Pediatric and Adult Crohn’s Disease for IRT-103 before final approval to market the drug in the U.S. from the FDA. Similarly Phase 2 and Phase 3 clinical trials will need to be completed in the U.S. for IRT-101 before we can obtain final approval to market the drug in the U.S. from the FDA. All clinical trials in the U.S. are being completed under the supervision of our subsidiary Cytocom, Inc.
| 25 |
Intellectual Property
The Company has been developing active forms of immunotherapies through the acquisition of patents, IND Applications, clinical data and all proprietary technical information, know-how, procedures, protocols, methods, prototypes, designs, data and reports which are not readily available to others through public means, and which were owned, generated or developed through experiments or testing by Dr. Plotnikoff, Professor Shan, Dr. Bernard Bihari, Dr. Ian S. Zagon, Dr. Jill Smith, Dr. Patricia J. McLaughlin and Moshe Rogosnitzky.
The Company has been able to acquire many of the patents and intellectual property it was seeking, and has also been able to team up with some of the leaders in the field of immunology, experts such as the late Dr. Ronald Herberman (1940-2013), Dr. Fenping Shan, Dr. Jill Smith and Dr. Terry Grossman.
Dr. Plotnikoff is the inventor behind a number of patents granted for cancer treatments and an adjunct to patents for autoimmune diseases including: European Patent United Kingdom, Germany, France, Ireland EP 1401471 BI Methods for inducing sustained immune response; Russian Patent Russian Federation patent number 2313364; The Patent Office of the People’s Republic of China, Application No.: 200810165784.8 China Patent CN1015113407 A The Patent Office of the People’s Republic of China ISSN: 1006-2858 CN 21-1349/R; Patent Agencies Government of India Patent, Application number 1627/KOLNP/2003 number 220265 an Enkephalin Peptide Composition; and the US Patent Pending, US Patent Application 10/146.999 which was approved in January 2014 US patent number 20140024588 (the “Plotnikoff Patents”). The Patent Cooperation Treaty (“PCT”) enables a U.S. applicant to file a single application, known as “an international application,” in a standardized format in English in the U.S. Receiving Office (the U.S. Patent and Trademark Office) that is acknowledged as a regular national or regional filing in any state or region that is party to the PCT.
The Company entered into a Sale of Technology Agreement with Dr. Nicholas P. Plotnikoff on March 4, 2012, wherein Dr. Plotnikoff agreed to transfer and assign all of his rights, title and interest in the Plotnikoff Patents to the Company. The Company received all the production formulations and technology designs from Dr. Plotnikoff necessary for the manufacturing, formulation, production and protocols of the MENK treatment of cancer and HIV/AIDS. As consideration for entering into the Sale of Technology Agreement, Dr. Plotnikoff received 6,000,000 shares of common stock, a royalty of a one percent on all sales of MENK in perpetuity and was granted the position of Non-Executive Chairman of the Board of Directors. The Sale of Technology Agreement was filed with Amendment No. 6 to the Form 10 Registration and incorporated herein by reference.
In addition to the above patents, we also signed an exclusive licensing agreement for all of the intellectual property developed at Pennsylvania State University by Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Dr. Jill P. Smith for the treatment of cancer. The patents cover methods and formulations related to the treatment and prevention of cancers. More specifically, the present inventions describe the use of drugs that interact with opioid receptors (naltrexone, naloxone and the pentapeptide MENK) to inhibit and arrest the growth of cancer. Such efficacy has been discovered to be partially due to the functional manipulation of the zeta opioid receptor through exogenous and endogenous MENK. This receptor has been determined to be present in a variety of cancers, including pancreatic, ovarian, liver, head and neck, and colon cancer. US Patent Numbers 6,737,397, CA 2,557,504, US 20010046968 , US 6737397 , US 6136780 , US 20080015211 , US 20070053838 , US 8003630 , US 20110123437 , US 7807368 , US 7576180 , US 7517649 , US 20080146512 , US 7122651 , US 20060073565, US 20050191241 , Patent No 8,003,630. In addition to the approved patents we have four other patents pending: U.S. Patent Application No. 11/061,932, U.S. Application No. 13/660,129; Israeli Patent Application No. 194734; Chinese Patent Application No.: 200810165784.8 and US Application No. 62/296,759. The licensing agreement referenced in this paragraph was previously filed with Amendment No. 7 to the Form 10 Registration Statement filed January 22, 2014 and is incorporated herewith.
We also acquired the licensing rights to the patent portfolio and intellectual property developed by Dr. Bernard Bihari relating to treatments with drugs that interact with opioid receptors such as LDN and MENK for a variety of diseases and conditions including malignant lymphoma, chronic lymphocyctic leukemia, Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma, chronic herpes virus infections, and chronic infections due to the Epstein-Barr virus and a treatment method for humans infected with HTLV-III (AIDS) virus including patients clinically diagnosed as suffering from HIV/AIDS and those suffering from ARC. The licensed rights include all reissues or modifications, reexaminations, or other related U.S. patent filings directed to the same subject matter and the use of U.S. Patent Number 6,586,443, U.S. Patent Number 6,384,044, U.S. Patent Number 6,288,074, U.S. Patent Number 5,356,900, U.S. Patent Number 5,013,739, U.S. Patent Number 4,888,346. The license agreement with Dr. Bihari was previously filed with Amendment No. 1 to the Form 10 Registration Statement filed on June 7, 2013 and is incorporated herewith.
| 26 |
Once the Company acquired the above patents, it was then able to sign a licensing agreement to acquire the exclusive patent rights for the intellectual property of the licensors, Dr. Jill Smith and LDN Research Group, LLC, whose members include Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Moshe Rogosnitzky. The patents cover methods and formulations for the treatment of the inflammatory and ulcerative diseases of the bowel, using naltrexone in low doses as an opioid antagonist. Endogenous opioids and opioid antagonists at low doses have been shown to play a role in stimulating and rebalancing the immune system and the healing and repair of tissues. US Patent No. 6,136,780, Patent No. US 7879870. The Company then negotiated with Dr. Jill Smith to arrange the transfer of the Orphan Drug Designation for the use of naltrexone for the treatment of pediatric Crohn’s disease with the FDA. Dr. Smith has since transferred the IND to the Company, and the FDA acknowledged that the Company is now the sponsor for this IND. In September 2014, the Company and the licensors jointly agreed to terminate the license agreement, and in place thereof, have the licensors grant a similar license in their patent rights to Cytocom Inc. pursuant to a Patent License Agreement between the licensors, Cytocom Inc. and the Company with substantially similar terms as set forth in the original license agreement. Pursuant to this agreement, the Company issued 1,000,000 shares of its common stock to the licensors and the Company guaranteed the obligations of Cytocom Inc. to the licensors under the agreement.
The Company originally acquired the patents and intellectual property from Dr. Smith and LDN Research Group, LLC because management believed clinical trials involving LDN held great promise for the millions of people worldwide with autoimmune diseases or disorders, central nervous system disorders or those who face cancer. Management also believed it could be the first low-cost, easy to administer therapy with minimal to no side-effects for the treatment of HIV/AIDS, autoimmune diseases and immune disorders, in particular Crohn’s disease, multiple sclerosis, and/or fibromyalgia.
Dr. Nicholas Plotnikoff, Professor Fenping Shan and Noreen Griffin recently filed a Provisional Application for a Utility Patent US Application No. 62/296,759 Method for Inducing a Sustained Immune Response, which was assigned to the Company in March 2016. U.S. Patent and Trademark Office has issued the Official Filing Receipt in connection with this application, and accorded a filing date of February 18, 2016 during its pendency in the USPTO.
Summaries of the Company’s agreements are as follows:
Dr. Nicholas Plotnikoff
In connection with the share exchange, the following is the scope of the intellectual property transfer: we entered into a Sale of Technology Agreement with Dr. Nicholas P. Plotnikoff on March 4, 2012, wherein Dr. Plotnikoff agreed to a 100% transfer and assign all of his rights, title and interest in: European Patent United Kingdom, Germany, France, Ireland EP 1401471 BI Methods for inducing sustained immune response; Russian Patent Russian Federation patent number 2313364; The Patent Office of the People’s Republic of China, Application No.: 200810165784.8 China Patent CN1015113407 A The Patent Office of the People’s Republic of China ISSN: 1006-2858 CN 21-1349/R; Patent Agencies Government of India Patent, Application number 1627/KOLNP/2003 number 220265 an Enkephalin Peptide Composition; and the US Patent Pending, US Patent Application 10/146.999 e. The Company received all the production formulations and technology designs from Dr. Plotnikoff necessary for the manufacturing, formulation, production and protocols of the MENK treatment of cancer and HIV/AIDS. As consideration for entering into the Sale of Technology Agreement, Dr. Plotnikoff received 8,000,000 shares of common stock, a royalty of a single-digit percentage on all sales of MENK and was granted the position of Non-Executive Chairman of the Board of Directors. There were no other payment provisions included in the termination provisions.
At the time of the acquisition, the valuation of goodwill and other intangible assets were determined using the fair market price for the Company’s common stock, which were exchanged for shares of TNI IP. In the fourth quarter of 2012, the Company performed an annual valuation to determine whether any goodwill or intangible assets that had been acquired by the Company were impaired. The result of this valuation was that material impairments were identified. The Company recognized an impairment of the goodwill arising on the acquisition of TNI IP of $98,000,000.
| 27 |
Jacqueline Young
On August 13, 2012, the Company signed an exclusive License Agreement with Ms. Jacqueline Young (the “Young Agreement”) for the intellectual property developed by Dr. Bernard Bihari relating to treatments with opioid antagonists such as naltrexone and Met-enkephalin for a variety of diseases and conditions including malignant lymphoma, chronic lymphocytic leukemia, Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma, chronic herpes virus infections, chronic herpes viral infections such as chronic genital herpes caused by the herpes simplex virus Type 2 and chronic infections due to the Epstein-Barr virus and a treatment method for humans infected with HTLV-III (AIDS) virus, including patients clinically diagnosed as suffering from AIDS and those suffering from AIDS-related complex (ARC).
The Young Agreement is valid for the life of the patents and expires on a country by country basis in each country where patent rights exist, upon the expiration of the last to expire patent in each country or in the event the patent in such country is held to be invalid and/or unenforceable (by a court or government body of competent jurisdiction) or admitted to be invalid or unenforceable.
The termination provision states that, additionally, we can cancel the Young Agreement upon 120 days’ written notice and shall pay all royalties and fees that have accrued. We have the exclusive rights to the intellectual property; however, Ms. Young retains a right to practice the patents licensed under the Young Agreement solely for non-commercial, academic research purposes.
The Bihari patents were acquired in exchange for 540,000 shares of the Company’s common stock with a fair value of $972,000 and assumed liabilities of $400,000, which is payable to Ms. Young over a twenty-four month period in equal installments to reimburse her for the costs of a New York City office in accordance with the Young Agreement. The patent liability at December 31, 2013 totaled $118,333. The cost of the patent totaled $1,372,000. Additionally, the Company will pay the licensor a royalty payment of 1% of gross MENK sales and provide the licensor a position as non-executive chairman of the Company.
Dr. Jill Smith
On December 24, 2012, the Company signed an agreement for the acquisition of patent rights (the “Smith Agreement”) for the intellectual property of Dr. Jill Smith and LDN Research Group, LLC (collectively, the “Licensor Parties”), whose members are Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Moshe Rogosnitzky and orphan drug designation by the FDA to a novel late-stage drug, trademarked “LDN,” for the treatment of Pediatric Crohn’s disease. The patent covers methods and formulations for treatment of the inflammatory and ulcerative diseases of the bowel, using naltrexone in low doses as an opioid antagonist. These patents were acquired in exchange for the purchase of 300,000 shares of our common stock with a fair market value of $2,715,000 and an up-front payment of $165,384 (consisting of a $100,000 initial license fee and payment of $65,384 of expenses), which totaled $2,880,384.
The Smith Agreement requires the Company to (i) use commercially reasonable efforts to develop, commercialize, market and sell licensed products in a manner consistent with a business plan, (ii) expend a minimum amount of funds per annum to develop and commercialize licensed products as soon as practicable, (iii) obtain all requisite regulatory approvals needed to use or sell licensed products in the field of use, and (iv) make the first commercial sale of a licensed product by March of 2017.
In addition to the above stated 300,000 shares with a fair market value of $2,715,000 at the time of signing with the Company, it was agreed that 1,000,000 shares of common stock at the time of signing of a licensing agreement with Cytocom Inc, with all fees paid by the Company, on behalf of the contract until such time as Cytocom is funded.
| 28 |
The aggregate amounts paid to date under the agreement are: $100,000 in 2014, $100,000 in 2015, and $100,000 set to be paid in December of 2016. The aggregate future potential milestone payments to be paid are: $250,000 upon the initiation of a phase III trial by the FDA, $250,000 upon acceptance of a NDA by the FDA, and $750,000 upon marketing approval by the FDA. The royalty rates for the transactions are 4% on Crohn’s Disease in the United States and 1% on sales in Emerging Markets.
Unless terminated sooner pursuant to the Agreement, the Agreement will terminate upon the later of: (a) the expiration or abandonment of the last patent to expire or become abandoned of the Patent Rights; or (b) Ten (10) years after the first Sale of the first Licensed Product. The Company may terminate the Smith Agreement upon 90 days’ written notice, provided all sublicenses are terminated and all amounts due and owing are paid to the Licensor Parties. The Licensor Parties may terminate the agreement ten days’ after notice to the Company if the Company is ten days late in payment or there is a breach that remains uncured for ten days after written notice of such breach.
The Company is required to pay an annual license fee, an annual running royalty on net sales of each licensed product or a minimum royalty, whichever is greater, and a sublicense fee on payments received by the Company from sublicensees. The Company has an exclusive, worldwide license to make, have made, use, lease, import, offer for sale and sell licensed products and to use the method under the patent rights.
The Company is also required to pay milestone payments after substantial achievement of certain milestone events for each licensed product including payment: upon initiation of each Phase III trial; upon positive completion of each Phase III clinical trial of the therapeutic use of an LDN compound in the field of use; when a New Drug Application (“NDA”) is accepted for review by the FDA; and when FDA approval to market the NDA is approved. The Company will issue shares upon reaching certain milestones including upon the first dosing of the first patient in a Phase III clinical trial for each licensed product, upon the first sale of each licensed product, and upon the achievement of a set dollar amount in cumulative sales for each licensed product covered by NDAs.
As part of the Smith Agreement, the Company has the right to apply to the FDA for the transfer of the orphan drug status for the use of naltrexone for the treatment of pediatric Crohn’s disease and ulcerative colitis, the Investigation New Drug Application (“IND”), and the right to acquire the relevant clinical data set from Dr. Jill Smith. Dr. Jill Smith made arrangements to transfer the IND to the Company as well as the relevant clinical data set, and the FDA has acknowledged that the Company is now the sponsor for this IND.
On September 24, 2014, the Company and the Licensor Parties jointly agreed to terminate the Smith Agreement, and in place thereof, have the Licensor Parties grant a similar license in their patent rights to Cytocom Inc. pursuant to a Patent License Agreement between the Licensor Parties, Cytocom Inc. and the Company with substantially similar terms as set forth in the Smith Agreement. Pursuant to this agreement, the Company issued 1,000,000 shares of its common stock valued at $270,000, upon execution to the Licensor Parties and the Company guaranteed the obligations of Cytocom Inc. to the Licensor Parties under the agreement.
The Penn State Research Foundation
On January 18, 2013, the Company signed an exclusive licensing agreement with The Penn State Research Foundation to license all of the intellectual property developed by Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Dr. Jill P. Smith for the treatment of cancer titled “Opioid Growth Factor and Cancer” and “Combination Therapy with Opioid Growth Factor and Taxanes for the Treatment of Cancer” (the “Foundation Agreement”).
The Foundation Agreement requires the Company to: (a) use commercially reasonable efforts to develop, commercialize, market and sell licensed products in a manner consistent with a business plan; (b) expend a minimum amount of funds per annum to develop and commercialize licensed products as soon as practicable; (c) obtain all requisite regulatory approvals needed to use or sell licensed products in the field of use; and (d) make the first commercial sale of a licensed product by December 31, 2016.
The Foundation Agreement provides that the Company must pay to the licensor an initial license fee, a license maintenance fee on each anniversary of the effective date of the Foundation Agreement, and an annual running royalty on net sales for each licensed product or a minimum royalty, whichever is greater. In addition, the Company must pay a sublicense fee on payments received by the Company from sublicensees.
| 29 |
The Foundation Agreement also requires the Company to make payments upon the achievement of certain milestone events including: initiation of each Phase II trial; initiation of each Phase III trial; when the NDA is accepted for review by the FDA; and when FDA approval to market is approved. The Company must also issue shares upon certain milestones including upon the first dosing of the first patient in a Phase II clinical trial for each licensed product, upon the first dosing of the first patient in a Phase III clinical trial for each licensed product, upon the first sale of each licensed product, and upon the achievement of a set dollar amount of cumulative sales for each licensed product covered by NDAs.
The Foundation Agreement terminates on the expiration or abandonment of the last patent to expire or become abandoned. The Company may terminate the Foundation Agreement at any time upon 60 days’ prior written notice and ceasing to make and sell all licensed products, the termination of all sublicenses and payment of all monies owed under the Foundation Agreement. The licensor may terminate the agreement 30 days after notice to the Company if the Company is 30 days late in payment or a breach that remains uncured for 45 days after written notice of such breach
The Penn State Agreement included an upfront execution payment of $100,000 that was previously paid.
Each year the Company incurs $10,000 in annual payments and maintenance fees on the patents as well as the legal fees in connection with the pending patents.
The aggregate future potential milestone payments to be paid are as follows:
| ● | One Hundred Thousand Dollars ($100,000) upon initiation of each Phase II trial. | |
| Two Hundred Fifty Thousand Dollars ($250,000) upon initiation of each Phase III trial. | ||
| ● | One Million Dollars ($1,000,000) to be paid as follows: Two Hundred Fifty Thousand ($250,000) when the NDA is accepted for review by the FDA and Seven Hundred Fifty Thousand ($750,000) when FDA approval to market is approved. | |
| ● | First dosing of first patient in a Phase II trial for each Licensed Product 250,000 shares | |
| ● | First dosing of first patient in Phase III trial 50,000 shares for each Licensed Product. | |
| ● | First Sale of each Licensed Product in the United States and/or Canada 100,000 shares. | |
| ● | Achievement of Twenty Million Dollars ($20,000,000) in cumulative Sales for each Licensed Product covered by NDA’s 200,000 shares of common stock. |
Royalty rates are equal to four percent (4%) of Net Sales of US and Canadian sales.
Professor Fengping Shan
In May of 2013, the Company executed a Patent License Agreement with Professor Fengping Shan (the “Shan Agreement”) pursuant to which it obtained exclusive rights to develop and commercialize the licensed technology. The licensed technology is the intellectual property developed and owned by Professor Shan (i) relating to the treatment of a variety of diseases and conditions with MENK including multiple forms of lymphoma and cancer and (ii) a treatment method for humans infected with the HLTV-III (AIDS) virus including AIDS and AIDS related complex (ARC). The licensed technology includes the methods and formulations for these treatments including all INDs, communications with regulatory agencies, patient data, and letters relating to these treatments. The licensed technology also includes certain patents developed by Professor Shan.
Under the Shan Agreement, the Company must issue 500,000 shares to Professor Shan upon final transfer of the licenses, and reimburse Professor Shan for all out of pocket expenses in connection with the patents. The Company will pay Professor Shan a running royalty on gross sales subject to decreases if third party intellectual property is needed to complete such sale or product. The Shan Agreement lasts for the duration of each of the licensed patents however the Company may terminate the Shan Agreement on 120 days’ written notice to Professor Shan.
| 30 |
The payment provisions under the Shan Agreement do not provide any up-front or executions payments and no aggregate amounts have been paid or received under the agreement. No future potential milestone payments will be paid or received either. The royalty rate under the agreement is .5% and there are no profit or revenue-sharing provisions to be followed.
On August 6, 2014, Professor Fengping Shan executed an Assignment pursuant to which he transferred to the Company his entire right, title and interest in and to the licensed patents under the Shan Agreement and CN 201210302259 Application of combination of low-dose naltrexone and methionine-enkephalin to preparation of anti-cancer drug for the consideration of 500,000 shares of common stock valued at $140,000. Patents Overview:
| Patent: | Title: | Expiration: | License/Assigned: | Product or Use: | ||||
U.S. Patent Number 6,586,443 (Related to US 5,356,900, 5,013,739 and 4,888,346 – all expired) (No related foreign patents) |
Multiple sclerosis in a human patient is treated by the administration preferably via a pharmacologically effective route of an essentially pure opiate receptor antagonist. | January 3, 2019 | Exclusive License from Jacqueline Young. | IRT-103 (LDN) | ||||
U.S. Patent Number 6,384,044 (No related foreign patents) |
Cancer of the prostate in human male patients even at an advanced state with metastasis to other organs is preferably treated by administration. | November 8, 2019 | Exclusive License from Jacqueline Young. | IRT-103 (LDN) | ||||
U.S. Patent Number 6,288,074 (No related foreign patents) |
Lymphoproliferative syndrome, including such diseases as malignant lymphoma, chronic lymphocytic leukemia, Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma, are treated in human patients via administration. | November 15, 2019 | Exclusive License from Jacqueline Young. | IRT-103 (LDN) | ||||
U.S. Patent Number 6,136,780 (Related to US 6,737,397) (No related foreign applications) |
Control of cancer growth through the interaction of [Met5] - Enkephalin and the zeta (s) receptor. | May 17, 2021 | Exclusive License: Penn State University. | IRT-101 (MENK) and IRT-103 (LDN) | ||||
U.S. Patent No. 6,737,397 (Related to US 6,136,780) (No related foreign applications) |
Control of cancer growth through the interaction of [Met5]-Enkephalin and the zeta receptor. | May 17, 2021
|
Exclusive license: Penn State University. | IRT-101 (MENK) and IRT-103 (LDN) |
| 31 |
U.S. Patent No. 7,879,870 (US PgPub 2008/0015211) (No related foreign patents) |
Treatment of inflammatory and ulcerative diseases of the bowel with opioid antagonists. | February 1, 2028 | License to Cytocom Inc.: Dr. Jill Smith and LDN Research Group, LLC. | IRT-103 (LDN) | ||||
Israeli Patent mentioned in license
|
Treatment of inflammatory and ulcerative diseases of the bowel with opioid antagonists. | Pending | License to Cytocom Inc.: Dr. Jill Smith and LDN Research Group, LLC.
|
Treatment of Crohn’s disease | ||||
U.S. Application Number: 11/061,932 (Claims Priority to US60/548,021) Canadian Application Number: 2,557,504 (Pending) |
Combinatorial therapies for the treatment of neoplasias using the opioid growth factor receptor. | Pending application
|
Exclusive license: Penn State University. | IRT-101 (MENK) | ||||
U.S. Patent No. 8,003,630 (Application Number: 11/510,682) (US PgPub 2007/0053838) (Claims Priority to US60/548,021) |
Combinatorial therapies for the treatment of neoplasias using the opioid growth factor receptor. | May 22, 2028 | Exclusive license: Penn State University. | IRT-101 (MENK) | ||||
U.S. PgPub 2013/0084242 A1 (Application Number: 13/660,129) (Claims Priority to US60/548,021)
Patent Cooperation Treaty (PCT) application: PCT/US2010/030967 (Claims priority to US61/173,351) |
Combinatorial therapies for the treatment of neoplasias using the opioid growth factor receptor. | Pending | Exclusive license: Penn State University. | IRT-101 (MENK) | ||||
US 7,807,368 (US PgPub 2008-0146512 A1) (No related foreign applications) |
Cyclin-dependent kinase inhibitors as targets for opioid growth factor treatment. | October 4, 2027 | Exclusive license: Penn State University. | IRT-101 (MENK) |
| 32 |
US 7,576,180 (Claims priority to US60/106,879) (There is a related PCT application PCT/US1999/025802, claiming priority to the US60/106,879, but no National Phase applications were filed) |
Opioid growth factor receptors. | August 17, 2026 | Exclusive license: Penn State University. | IRT-101 (MENK) | ||||
US 7,122,651 (No related foreign applications)
|
Novel nucleic acid molecules encoding opioid growth factor receptors. | October 17, 2023 | Exclusive license: Penn State University. | Treatment of cancer |
US 7,517,649 (US PgPub 20060073565) (No related foreign applications) |
Methods of detecting opioid growth factor receptor (OGFr) in tissue. | April 13, 2026 | Exclusive license: Penn State University. | IRT-101 (MENK) | ||||
CN 200910011030 (No related U.S. applications) |
Shan Fengping, Nikola Polonikov, Lu Changlong: Application of naloxone and composition thereof in preparing drug for treating cancer. Shan Fengping: August 26, 2009. | August 23, 2026 | Assigned by Fengping Shan. | IRT-101 (MENK) and IRT-103 (LDN) | ||||
CN 200710051586 (No related U.S. applications) |
Huang Jianyin, Zhang Ding, Shan Fengping, Luo Zhinong: Application of methionine enkephalin in preparing human or animal vaccination. Huang Jianyin: August, 20 2008. | August 20, 2025 | Assigned by Fengping Shan. | IRT-101 (MENK) | ||||
CN 200710158742 (No related U.S. applications) |
Shan Fengping, Lv Changlong, Nikola Polonikov, Huang Jianyin: Application of compounds Methionine Enkephalin for preparing medicine for curing blood medulla hematopoietic system cancer. Dan Fengping: May 14, 2008. | May 13, 2025 | Assigned by Fengping Shan. | IRT-101 (MENK) |
| 33 |
CN 200610046249 (No related U.S. applications) |
Shan Fengping, Lv Changlong, Huang Jianyin, Zhang Ding, Luo Zhinong: Aerosol containing Met-Enkephalin. Shan Fengping: November 15, 2006. | November 14, 2023 | Assigned by Fengping Shan. | IRT-101 (MENK) | ||||
CN 200310120896 (No related U.S. applications) |
Shan Fengping, Li Li: Integrated health food for regulating human body immune balance. Liaoning Academy of Microorganism Sciences: July 6, 2005. | July 5, 2022 | Exclusive license: Nicholas Plotnikoff and Fengping Shan. | Oncology treatments and cancer treatment | ||||
WO 2007/067753 (PCT/US2006/046925 |
Huang John, Chang Ding, Lo Shi-Lung, Shan Fengping: Methods of reducing side effects in cancer therapy. Penta Biotech: June 14, 2007. | Pending | Exclusive license: Fengping Shan. | IRT-101 (MENK) |
| CN 200510019964 | Huang Jianyin, Zhang Ding, Luo Zhinong, Shan Fengping: Use of Methionine Enkephalin in preparation of medicine for reducing toxic side effects of chemical or radioactive therapy. Huang Jianyin: August 9, 2006. | August 8, 2023 | Exclusive license: Fengping Shan. | IRT-101 (MENK) | ||||
US PgPub 2003/0148942 A1 (Application Number: 10/146,999) Our Docket #6463-0101PUS1 (Claims Priority to US60/291,237) |
Methods for inducing sustained immune response. | May 16, 2022 | Assigned and licensed: Nicholas Plotnikoff. | IRT-101 (MENK) | ||||
Russian Application 2003136161/14
(Claims Priority to US60/291,237) |
Methods for inducing sustained immune response. | May 16, 2022 | Assigned and licensed: Nicholas Plotnikoff. | IRT-101 (MENK) | ||||
PCT application PCT/US2002/018529 (Claims priority to the US60/291,237) |
Methods for inducing sustained immune response. | May 16, 2022 | Assigned and licensed: Nicholas Plotnikoff. | IRT-101 (MENK) |
| 34 |
National Phase entries filed off the PCT/US2002/018529 China 02814327.2 (Pending) EP App 2002746503 Granted: November 29, 2006
India Patent No. 220265 (App 01627/KOLNP/2003)
Japan App Withdrawn |
Methods for inducing sustained immune response. | May 16, 2022
|
Assigned and licensed: Nicholas Plotnikoff. | IRT-101 (MENK) | ||||
| China Patent 200810229085 | The invention belongs to the technical field of treating tumors by immunization therapy. In particular, a method for treating intestinal cancer and pancreatic cancer cells by Methionine Enkephalin under conditions of in-vivo injection and in-vitro cell culture so as to achieve the treating aim. | March 21, 2026 | Assigned by Fengping Shan. | IRT-101 (MENK) |
Employees
As of December 31, 2016, the Company had 5 full time employees.
Reports to Security Holders
Our common stock is registered under the Securities Exchange Act of 1934 and we are required to file current, quarterly and annual reports and other information with the SEC. You may read and copy any document that we file at the SEC’s public reference facilities at 100 F. Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-732-0330 for more information about its public reference facilities. Our SEC filings are available to you free of charge at the SEC’s web site at www.sec.gov. We are an electronic filer with the SEC and, as such, our information is available through the Internet site maintained by the SEC that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC. This information may be found at www.sec.gov and posted on our website at www.immunetherapeutics.com.
Research and Development
Our research and development (“R&D”) organization focuses primarily on new uses for the opioid-related immuno-therapies, such as LDN and MENK. These therapies stimulate the immune system in such a way that provides the potential to treat a variety of diseases that have abnormalities in the immune system.
| 35 |
Our R&D priorities include development of MENK IRT-101, a small synthetic pentapeptide that is naturally occurring in the body, and LDN IRT-103, an opioid receptor antagonist. Our pipeline provides two therapies with an extremely wide range of indications that can be pursued. Both molecules have the ability to stimulate and/or regulate the immune system in order to treat a variety of autoimmune diseases including multiple sclerosis, immune disorders such as Crohn’s disease, cancer, and viral infections such as HIV/AIDS.
Our R&D is overseen and managed internally, working with individuals, universities, and Contract Research Organizations (“CROs”) in order to utilize patents that we have licensed or acquired since our inception. We continue to seek to expand our pipeline by reviewing other compounds, technologies or capabilities. We also seek out promising compounds and innovative technologies developed by third parties to incorporate into our discovery and development processes or projects.
Drug discovery and development is time-consuming, expensive and unpredictable. According to the Pharmaceutical Research and Manufacturers of America (PhRMA), out of 5,000-10,000 screened compounds, only 250 enter preclinical testing, five enter human clinical trials and one is approved by the FDA. The process from early discovery or design to development to regulatory approval can take more than 10 years. Drug candidates can fail at any stage of the process, and candidates may not receive regulatory approval even after many years of research.
As of December 31, 2015, we had two compounds (IRT-101 and IRT-103) in research and development. In 2015 our development programs focused on both compounds, one in oncology and one in Crohn’s disease; which we are expecting to move into Phase II clinical trials.
The following table provides information about notable regulatory actions by, and filings pending with the FDA and regulatory authorities in the EU, as well as additional indications and new drug candidates in late-stage development.
| NEW DRUG CANDIDATES IN LATE-STAGE DEVELOPMENT | ||||
| CANDIDATE | INDICATION | REGULATORY ACTIONS | ||
| IRT-101 | Pancreatic Cancer | End-of-Phase 1 Meeting with FDA Complete 3Q 2013 | ||
| IRT-103 | Crohn’s Disease | Type C Meeting with FDA Complete 2Q 2013 Scientific Advice with EMA Complete 1Q 2014 | ||
The Company expects it will incur future research and development expenditures in the next 12 months through Cytocom. Cytocom plans to conduct Phase II and Phase IIB trials for the treatment of IRT-103 Crohn’s disease, at an estimated cost of $3,900,000 and $7,500,000 respectively for each phase. If the trials do not commence before the end of 2017, the Company will be required to make a payment of $100,000 in December 2017 under its license agreements. In prior years, the Company has been able to raise funds through sales of notes payable, and it expects to do the same for the payment due in 2017. With funding Cytocom will be responsible for the development of IRT-101 MENK for pancreatic cancer.
Government Regulations
United States
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries with regulations differing from country to country. Neither we nor our collaboration partners are permitted to market our drug candidates in the United States until we receive approval of a New Drug Application (“NDA”) from the FDA. Neither we nor our collaboration partners have submitted an application for or received marketing approval for any of our drug candidates. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process.
Prior to receiving approval to commercialize any of our drug candidates in the United States or abroad, we and our collaboration partners must demonstrate with substantial evidence from well controlled clinical trials, and to the satisfaction of the FDA and other regulatory authorities abroad, that such drug candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Regulatory approval of an NDA or NDA supplement is not guaranteed, and the approval process is expensive and may take several years.
| 36 |
Before a drug can be tested in people, the sponsor (in this case the Company) performs laboratory and animal tests to discover how the drug works and whether it’s likely to be safe and effective in humans. As LDN and MENK have previously been used in clinical trials, this phase of development was not required by the Company to initiate clinical trials under its applications.
Next, a series of tests in people (i.e. clinical trials) is begun to determine whether the drug is safe when used to treat a disease and whether it provides a real health benefit. The clinical phase typically starts at Phase 1 and progresses to Phase 3. The Company will have an abbreviated list of clinical trials that need to be conducted due to published literature on previously conducted studies, as well as utilizing the approval of naltrexone previously at 50 mg by the FDA.
Upon completion of the clinical trials, the Company will send the FDA and/or the EMA the evidence from these tests to prove the drug is safe and effective for its intended use (New Drug Application (NDA) in the US or Marketing Authorization Application in the EU). The regulatory bodies will review these data and determine if the sponsor has approval to market the product at the specified dose(s) and formulation(s) for the specified indication(s) (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/default.htm).
Once regulatory approval has been granted, the approved product and its manufacturer are subject to continual review by the FDA and/or non-U.S. regulatory authorities. Any regulatory approval that we or our collaboration partners receive for our drug candidates may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies to monitor the safety and efficacy of the product. In addition, if the FDA and/or non-U.S. regulatory authorities approve any of our drug candidates, we will be subject to extensive and ongoing regulatory requirements by the FDA and other regulatory authorities with regard to the labeling, packaging, adverse event reporting, storage, advertising, promotion and recordkeeping for our products. In addition, manufacturers of our drug products are required to comply with current cGMP regulations which include requirements related to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Further, regulatory authorities must approve these manufacturing facilities before they can be used to manufacture our drug products, and these facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations.
European Union
We intend to seek distribution and marketing partners for IRT-101 (MENK) and IRT-103 (LDN) in the European Union (“EU”). To market our future products in the European Economic Area (“EEA”) (which is comprised of the 27 member states of the EU plus Norway, Iceland and Liechtenstein) and many other foreign jurisdictions, we must obtain separate regulatory approvals. More concretely, in the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization (“MA”).
| ● | The Community MA is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use of the EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. | |
| ● | National MAs, which are issued by the competent authorities of the member states of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a member state of the EEA, this National MA can be recognized in another member state through the Mutual Recognition Procedure. If the product has not received a National MA in any member state at the time of application, it can be approved simultaneously in various member states through the Decentralized Procedure. |
| 37 |
Under the procedures described above, before granting the MA, the EMA or the competent authorities of the member states of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Our IND is being conducted per 21 Code of Federal Regulations Title 21, Part 312. In addition, we follow ICH guidelines, including good clinical practices (ICH E6) and current good manufacturing practice (ICH Q7) throughout the development process. After completion of Phase III clinical trials, the Company will file our NDA for LDN (IRT-103) as a 505(b)(2) application. IRT-103 products will follow the 505(b)(2) pathway relying on the Reference Listed Drug (RLD) REVIA to support the safety of the product. Efficacy will be submitted by the Company directly to the LDN NDA. IRT-101 products will follow the traditional approval pathway as a RLD is not available for MENK. However, published literature will support this program.
Nigeria
NAFDAC is the equivalent in Nigeria of the FDA. It undertakes registration of food, drugs, medical devices, cosmetics, agrochemicals and other similar products in Nigeria. At the end of the process, a registration number is given to the product and a registration certificate is issued to the applicant.
Many of the African countries do not have a local FDA equivalent organization or agency. We plan to use the NAFDAC Registration as the guideline for submission in Africa for countries that do not have their own application and approval procedures.
The Company has partnered with AHAR Pharma, a company duly incorporated and existing under the laws of the Republic of Nigeria to distribute LDN in the Republic of Nigeria. The distribution agreement was originally signed in 2013 and extended in 2016 for 5 years with an optional 5 year extension. AHAR Pharma is responsible for securing all required governmental or regulatory approvals, registrations, permits and licenses necessary to market, promote, offer for sale, sell, supply and distribute LDN in the Republic of Nigeria. Pursuant to the distribution agreement, the Company shall pay AHAR Pharma twenty-five U.S. cents for each tablet sold. Of the twenty-five cents paid, twelve and a half cents may be credited against the Company’s invoices and twelve and a half U.S. cents shall be paid to AHAR Pharma’s affiliate GB Pharma.
In addition, the Company has worked with GB Pharma Holdings since its formation in 2012. Over the last two years GB Pharma has worked directly with the NAFDAC and Minister of Health’s in Africa and the African Union on the approval of Lodonal™ for HIV/AIDS in Africa. GB Pharma has also worked to introduce LDN to the UNAIDS and World Health Organization. Dr. Gloria Herndon, the President of GB Pharma Holdings has traveled extensively to Nigeria, Kenya, Angola and South Africa in the past two years meeting with and holding discussion about the Company’s immunotherapies. Dr. Herndon has spoken on behalf of the Company at a number of conferences and was responsible for the introduction of the therapy to the Africa Union and the Champions, a group of former presidents of Africa to help promote the approval through the various in-country agencies.
The Company’s wholly owned subsidiary, Airmed Biopharma Limited, entered into an Exclusive Agency Agreement with GB Parma Holdings, Inc. on June 12, 2014 to distribute Lodonal™ in various emerging markets including Equatorial Guinea. Pursuant to the distribution agreement, GB Pharma shall be entitled to a commission of 5% of the net product sales. The Company’s relationships with the GB Pharma and its subsidiaries and affiliates are contractual, with the rights and obligations of the parties dictated by their respective agreements. GB Pharma and certain of its affiliates may also now own, or in the past owned, minority share positions in the Company via their investments or shares granted for services.
Malawi
No formal governmental agency is in place in Malawi to govern the application of a new drug. Malawi is a member of the Southern Africa Development Community (“SADC”). The SADC has been making efforts to synchronize the regulation of medication in the SADC countries.
| 38 |
The guidelines require filing an application prior to approval of registration. However, these guidelines are preliminary. The Regional Indicative Strategic Development Plan (“RISDP”) is a comprehensive development and implementation framework guiding the Regional Integration agenda of the SADC over a period of fifteen years (2005-2020). It is designed to provide clear strategic direction with respect to SADC programs, projects and activities in line with the SADC Common Agenda and strategic priorities, as enshrined in the SADC Treaty of 1992.
In July 2014, the Republic of Malawi approved Lodonal™ as an adjunct for the treatment of cancer. Protocols for a Lodonal™ trial were approved in November 2015. The Brewer Group, Inc. has paid for production of the first shipment to Malawi of Lodonal™, which was delivered to Malawi from Nicaragua in 2015.
The Company and the Brewer Foundation arranged for the donation of the Wallach LL100 Cryosurgical system that were necessary to run the trial and treat patients with cervical cancer. In July 2014, the Republic of Malawi approved Lodonal™ as an adjunct for the treatment of cancer. Protocols for a Lodonal™ trial were approved in November 2015.
In 2012, the Company started its collaboration with the Brewer Foundation and the Brewer Group. Over the last two years The Brewer Group and Foundation has worked directly with the Government of Malawi on the approval of the clinical trials and protocols. Once the protocols were approved the Brewer Group help to arrange funding for the trials.
The Brewer Group has entered into a distribution agreement with Airmed Bioparhma Limited, a wholly owned subsidiary of the Company, to distribute Lodonal™ in emerging markets. The distribution agreement was entered into in 2014 and has a term of 5 years. Pursuant to the distribution agreement the Company shall sell Lodonal™ to The Brewer Group at a 25% discount to the list price. The Brewer Group has also arranged a number of meetings with various hedge funds in New York to assist with the funding of the company.
The Company’s relationships with the Brewer Group and Brewer Foundation are contractual, with the rights and obligations of the parties dictated by their respective agreements. The Brewer Group, Brewer Foundation and certain of their affiliates may also now own, or in the past owned, minority share positions in the Company via their investments or shares granted for services.
Equatorial Guinea
Equatorial Guinea does not have procedures for the official approval of traditional medical practices or remedies. Accordingly, the Company was requested to make a presentation to the Health Sector and Minister on the use of naltrexone in the treatment of certain indications. After due discussion, the Government approved the following:
| 1. | Drug use naltrexone (4.5 mg) in the treatment of diseases requiring immune system stimulating cancer, HIV infection, multiple sclerosis, etc., as demonstrated by the Company, at a cost of $1/day or 450 x F.CFA/day. | |
| 2. | Management – laboratory for quality control and to analyze drugs imported in to Equatorial Guinea. | |
| 3. | The implementation of local production of quality essential medicines. |
The Company’s wholly owned subsidiary Airmed Biopharma Limited, entered into an Exclusive Agency Agreement with GB Parma Holdings, Inc. on June 12, 2014 to distribute Lodonal™ in various emerging markets including Equatorial Guinea. Pursuant to the distribution agreement, GB Pharma shall be entitled to a commission of 5% of the net product sales.
China
On October 18, 2012, the Company and Hubei Qianjiang Pharmaceutical Co., Ltd. (“Qianjiang Pharmaceutical”), signed a Venture Cooperation Agreement on New Drug Methionine Enkephalin (the “Venture Agreement”) pursuant to which Qianjiang Pharmaceutical acquired an exclusive license for the production of MENK in China. The Venture Agreement requires that Qianjiang Pharmaceutical conduct drug research and pilot testing for MENK, organize pre-clinical studies, and apply for clinical trials for MENK with the Chinese State Food and Drug Administration. Under the Venture Agreement, Qianjiang Pharmaceutical must open a co-administration account for the development of MENK in China. Qianjiang Pharmaceutical must pay the Company, upon the marketing of MENK products, a half-year amount equaling 6% of its gross sales from MENK of the preceding half year.
| 39 |
Qianjiang Pharmaceutical is required to obtain all approvals and permits required for the importation and sale of the Company’s products in China.
The Company may cancel the Venture Agreement if Qianjiang Pharmaceutical does not pay expenses for a period exceeding nine months or does not commence clinical trials within 12-months after receiving certain approvals. Qianjiang Pharmaceutical may cancel the Venture Agreement if the Company fails to perform its obligations for a period of nine months or the failure to receive approval of clinical trials is due to the Company’s MENK technologies.
On August 6, 2014, the Company entered into a Supplementary Agreement on New Drug Methionine – Enkephalin Cooperation (the “Amendment”) with Qianjiang Pharmaceutical, amending the Venture Agreement, as amended. The Company and Qianjiang Pharmaceutical executed the Amendment to accelerate clinical trials in both the United States and China, and agreed to immediately initiate three month Good Laboratory Practice (“GLP”) Toxicology Studies (rat and dog) within 30 days of signing the Amendment. The Amendment requires that the GLP Toxicology Studies Trials are conducted in China in accordance with international standards and standards acceptable to the FDA.
The Company modified the agreement one more time due to the fact toxicology studies were not completed by the end of 2015 as others steps were required before the studies could start.
Qianjiang has completed the formulation and required Chemistry, Manufacturing, and Controls (“CMC”) for Methionine–Enkephalin. All work was completed in China in accordance with international cGMP standards acceptable to the U.S. Food and Drug Administration with Chinese Peptide Company (“CPC”). CPC is among only a handful of companies in the world that can claim both ISO Certification and cGMP licensing. In February 2012, we became the first peptide company to successfully pass US FDA inspection outside of US and Europe regions.
Nicaragua and Dominican Republic
Laboratorios Ramos in Managua, Nicaragua has been issued approval from the Minster of Health to manufacture Naltrexone for the Company under the trademark name Lodonal™. The certificate of free sale allows Naltrexone to be exported from the Managua facility to other jurisdictions where the Company is approved to market and sell Naltrexone in satisfaction of the import requirements of such jurisdictions. The free sale certificate is not a license for export, which is issued separately for a specific product in both the country of export as well as the country of import.
Due to political developments in Nicaragua in the second half of 2016, the Company decided it would be prudent to obtain a second source of LDN product supply for sales outside of North America. In October 2016, the Company and Acromax Dominicana, SA (“Acromax”) entered into a contract for the manufacture of LDN tablets, capsules and/or creams (“Agreement”). Acromax is located in Santo Domingo; in the Dominican Republic. It is both cGMP certified and ISO 9001: 2008 certified. Acromax exports over 160 products and has sold throughout the Caribbean and Central and South America. More information about Acromax is available at http://acromaxdominicana.com/.
The Agreement has an initial term of five years unless terminated by either party in accordance with the terms. Subject to the terms and conditions of the Agreement, Acromax will obtain all necessary licenses and permits to carry out the manufacturing and packaging of LDN in exchange for a fixed fee per tablet plus an additional fee for packaging, shipping and customs clearance. In 2017, Acromax received approval from the Ministry of Public Health and Social Assistance to manufacture and sell Naltrexone both in the Dominican Republic as well as for export on behalf of TNI BioTech International under the trademark name Lodonal™. The company was granted and issued a Certificate of Pharmaceutical Product (“COPP”) for the following countries: Nigeria, Senegal, Kenya and Malawi. We expect to add additional countries to the COPP in 2017.
| 40 |
Business Strategy
The Company’s short-term business strategy focuses on several key areas described below, all of which are being undertaken simultaneously.
International Regulatory Approval in 2017: The Company has been in discussions with drug regulators regarding the regulatory and approvals process for sale of its products in 2017 in a number of countries: South Africa, Nigeria, Malawi, Kenya, Angola, Niger, Gabon, Egypt, Sri Lanka, and China. The process can take between four to 12 months depending on the local authorities. In December 2016, the Company presented the final results of a bridging trial in Nigeria. As of March 31, 2017, we are still awaiting receipt of final approvals for sale of Lodonal™ in Nigeria. The Company has begun discussions for a 90-day Lodonal™ bridging trial as an adjunct to chemotherapy in Kenya. If we complete the Kenya trial and if we meet the primary and secondary end points, the Company expects to receive approval for Lodonal™ as an adjunct treatment for cancer in Kenya. An approval in Nigeria would allow the Company to fast track approvals in other countries.
While working on regulatory approval throughout Africa, The Company has begun discussions with a number of international organizations including the World Health Organization, the Joint United Nations Programme on HIV/AIDS (UNAIDS), and PATH, an international nonprofit organization dedicated to saving lives and improving health, especially among women and children. The Company expects to reach agreements with them in the coming year.
Establish Partnerships in Africa: Receipt of the recent approvals in Nigeria will allow the Company to establish partnerships in Africa with large employers that maintain onsite clinics; there is increasing recognition that health creates wealth and advances GDP. HIV and AIDS have had a significant negative impact on labor and productivity. The vast majority of people living with HIV in Africa are of working age 15-49 years old. The Company believes that Lodonal™ can be used as a prophylactic to avoid many of the standard opportunistic infections accompanied with HIV and cancer. The Company will now move forward on this program if it receives approval for Lodonal™ sales in Nigeria.
Funding Cytocom Inc. Clinical Trials: If we generate significant revenue from sales to Africa and under the licensing of our LDN formulation to., which is described in detail below under “Agreements to Promote Development and Sale of Company Products,” we believe that in 2017 we will be able to commit financial resources to help fund Cytocom Inc.’s clinical trials in the United States to validate the use of Lodonal LDN in a number of indications. Unless other funding becomes available, the Company expects to use revenue from the sale of Lodonal™ in Nigeria to help underwrite clinical trials in the U.S. and Europe.
Clinical Studies:
In 2016, the Company focused on receipt of approvals in Nigeria and Malawi to conduct clinical development programs for Lodonal™. We completed a trial in Nigeria in December 2016, and we have final NAFDAC approval to commence sales in Nigeria. The Company also expects to commence a bridging trial for cancer in 2017 in the Republic of Malawi for Lodonal as a stand-along therapy. The recruitment of study participants, VIA testing and follow up of patients is still on-going. The first evaluation report from the doctors involved is expected by the end of April 2017 as per the study protocol. Since we have a cancer trial with Lodonal as an adjunct, we decided to hold off requesting final approval for the second trial until we complete our first trial, which is expected to occur in the third quarter of 2017.
On the regulatory front, Cytocom Inc. has held constructive dialogue with the FDA with respect to appropriate trial designs and study protocols for Lodonal™ and IRT-101. The Company has received approval for a phase IIB/III to be run in conjunction with this trial, but as of the date of this Report the FDA remains in the process of determining the primary endpoint for the trial for Crohn’s Disease. The Company has retained Cote Orphan to submit a final package to the FDA during the third quarter of 2017.
Cytocom Inc.’s immediate focus will be on pediatric and adult Crohn’s Disease. The Company expects recruitment in both pharmacokinetic (PK) and Dosing Trials for Lodonal™ to begin in 2017 as long as funds are available.
| 41 |
In addition to the trial design as described immediately above, the Company intends to undertake a Toxicologist and PK study in China with its partner, Hubei Qianjiang Pharmaceutical Co., Ltd, for IRT-101 for the purpose of furthering its understanding of the effect of the pharmacokinetics of IRT-101. The trial has been delayed until 2017 because the State Food and Drug Administration of the People’s Republic of China on the Safety of Drugs and Medical Devices (SFDA) has required chemistry, manufacturing and controls to be completed before the start of the toxicology study. Quinjiang expects to have the PK and Toxicology studies completed before the end of the year. This will allow the Company to better understand the potential for drug interactions to further optimize treatment for Phase 3 development of IRT-101 for cancer in both the United States and China. These trials are required before Hubei can file for a phase III trial for liver cancer with the SFDA.
The Company recently received the pharmacology and toxicology reports for MENK completed at GLP Lab by Hubei Qianjiang on. In addition to the pharmacology and toxicology, China Peptide Company has completed the Chemistry, Manufacturing, and Controls (CMC) required to file with both the SFDA and the FDA, as no pivotal trial can be started without CMC.
Strategy for Growth:
Since 2015, the Company has worked aggressively to build the business both upward and outward. It has worked to create a multi-faceted leader in immunotherapies by exploring opportunities beyond HIV/AIDS and cancer in emerging nations.
The goal and strategic vision is to build a diverse pipeline and to develop and commercialize novel drug treatments to improve the lives of patients suffering from chronic often life-threating disease.
To fulfill these goals, the Company received funding from the sale of stock, the exercise of stock warrants and issuance of notes payable totaling approximately $3.0 million in 2016. The Company anticipates generating revenues in 2017 from a number of sources, including revenues from the use of its formulation and patents and the sale of Lodonal™ in Nigeria, Malawi, Kenya, and Equatorial Guinea.
Company Growth:
To build visibility and positive awareness of the Company, management has made presentations at key investor and industry conferences internationally.
If the Company receives approvals for the sale of LDN in Nigeria following the 2016 bridging trial there, the Company believes it will be able to “fast track” the approval process in a number of African nations that will accept NAFDAC Nigeria’s approval of Lodonal™ for the treatment of HIV/AIDS.
Focus on HIV/AIDS
HIV/AIDS remains one of the three global public health threats. This disease results in substantial morbidity, mortality, negative socioeconomic consequences, and human suffering. Despite the significant increase in financial support and recent progress in addressing HIV/AIDS, many obstacles and unmet priorities remain.
Disease-specific interventions must be developed to ensure successful treatments of this disease. Apart from human suffering, the associated high adult mortality caused by HIV/AIDS, negatively impacts the socioeconomic development in some countries, especially in Nigeria and South Africa.
Adopting a treatment regimen for life that involves taking daily medication with potential side effects, presents many challenges that must be overcome if patients are to successfully remain on treatment. If drug resistance occurs through failure to adhere to antiretroviral treatments (ARVs), far more expensive second line therapy may be necessary. In some cases drug-resistant strains of HIV are transmitted, which can impact national treatment programs. Drug resistance has been found to be more prevalent the longer a country has provided antiretroviral therapies.
| 42 |
The Company believes Lodonal™ can provide an alternative in areas where patients have stopped taking their therapies due to side effects, or cost; Lodonal™ has been shown in trials to lessen the toxic side effects of ARVs. In those countries where people must travel monthly for a medical check-up (and lose a day’s pay), Lodonal™ can cost-effectively provide the ability to slow the progression of the disease without any toxic side effects.
Nigeria: In Nigeria, it is estimated that more than 3.6 million people are living with HIV/AIDS, with the annual number of new infections for adults at 323,000 and 57,000 for children. Currently, only 600,000 people receive HIV treatment (see http://www.unaids.org/en/regionscountries/).
South Africa: Based on a wide range of data including the household and antenatal studies, UNAIDS (see http://www.unaids.org/en/regionscountries) estimated that HIV prevalence was 17.3% among 15-49 year olds at the end of 2011. The high and low estimates were 16.6% and 18.1%, respectively. This implies that approximately 5.6 million South Africans were living with HIV at the end of 2011, including 460,000 children under 15 years old. By October 2012, only two million people were receiving ARVs. The biggest problem is compliance with prescribed treatment, as people are required to take pills 2 to 3 times a day at specific times and many times with food with considerable toxic side effects. In contrast, Lodonal™ is taken only once per day, does not have to be taken with food, and has no toxic side effects.
Agreements to Promote Development and Sale of Company Products
The Company is focused on its lead therapies designed for the treatment of cancer, HIV/AIDS, Crohn’s disease, fibromyalgia and MS. Management believes the pharmaceutical industry is eager to acquire advanced clinical-phase and approved products. However, despite the strong demand for advanced clinical-phase products, nearly 4,000 known compounds have had their development suspended in Phase II or earlier. Many of these are promising therapeutic drug candidates, but their development was discontinued because of strategic or financial constraints rather than for clinical reasons. Therefore, management believes there are clear market opportunities with a significant amount of unmet needs and a robust potential for partnering activities.
To further the business strategy, the Company has entered into relationships with a number of groups to promote the sale of its products outside the U.S., focusing initially on countries in Africa. They include: The Brewer Group, Inc.; GB Oncology & Imaging Group, LLC; American Hospitals and Resorts Limited (“AHAR”), an advanced surgical and medical facility, as well as a number of U.S. doctors that own and operate clinics in the U.S. and Nigeria.
The Brewer Group, Inc. is an international business advisory firm engaged in the business of identifying and capitalizing on opportunities with international governments, non-government organizations and professional athletes. The CEO of The Brewer Group is also the founder and Executive Director of The Jack Brewer Foundation. The Jack Brewer Foundation seeks to provide the Company with medical equipment where it is needed. Under the Engagement Agreement for Corporate Advisory Services dated February 5, 2013, the Brewer Group agreed to evaluate the Company’s options for expansion and growth into certain international markets, including Africa and, upon request, markets in Haiti, the Dominican Republic and/or Panama. Pursuant to the Engagement Agreement, the Brewer Group agreed to endorse the Company publicly and assist the Company in securing strategic partnership deals to enhance brand and market awareness. The initial term of the Engagement Agreement was 12 months, with an option for either party to terminate upon 30 calendar days with written notice to the other party. The agreement has been extended through 2017. The Company has issued the Brewer Group a total of 6,650,000 shares of its common stock since the signing of the Engagement Agreement.
GB Pharma lead by Dr. Gloria B. Herndon, a former director and current consultant of the Company has been committed to sourcing sustainable solutions in the field of health care in Africa, and has been involved since the 1990s on health-care related issues in Africa. The Company and GB Pharma have continued working with the ministries of health in African countries to provide better access to and public awareness of the prevention, diagnosis and treatment of cancer and chronic infectious diseases.
| 43 |
In 2015, the Company submitted protocols seeking permission from the Pharmacy, Medicines and Poisons Board of Malawi (“PMPB”) to conduct two trials involving Lodonal™ in Malawi:
| a. | The first protocol, submitted jointly with The Jack Brewer Foundation (“JBF Worldwide”), received PMPB approval on November 11, 2016. The protocol covers a 12-month trial for a “Single Visit Approach to Cervical Cancer Prevention.” The approach is designed to deliver a preventive and simple procedure that can be performed in a clinical setting without the use of a laboratory and to allow for immediate treatment of any precancerous lesions utilizing Wallach LL100 Cryosurgical systems. The protocol provides for 50% of the patient group to be put on Lodonal™ to determine if the drug lowers the number of opportunistic infections during the year, and if it can be shown that LDN increases CD4, CE8, NK and T cell count, which would show that the incidence rates of opportunistic infection could decrease with Lodonal™ and that Lodonal™ could be used as a prophylaxis to prevent substantial HIV-related morbidity in Malawi. The Company expects the final trial agreement with PMPB to be signed by the end of the third quarter of 2017 as there was a delay in training and recruitment. |
| b. | The second protocol, which has not yet been approved, covers a trial using Lodonal™ for the treatment of cancer. The protocol is still under discussion with the PMPB. The Company has held off on this trial as the approval of Lodonal™ in Nigeria and the pending approval in Kenya and Senegal for cancer treatment will allow it to file for drug approval without the need for a trial. |
In September 2013, TNI BioTech International, Ltd., a wholly-owned subsidiary of the Company, signed a Distribution Agreement with AHAR Pharma, a Nigerian company, to market Lodonal™, in Nigeria for the treatment of autoimmune diseases and cancer. AHAR intends to distribute Lodonal™ through a local distributor network, an Internet client base and directly to hospitals, pharmacists and doctors in Nigeria. The agreement gives AHAR exclusive rights to sell to customers in the private sector and non-exclusive rights to public-sector companies. The Company may terminate the exclusivity if AHAR fails to meet minimum purchase targets. Unless terminated earlier, the agreement is valid for five years, subject to the right of the parties to extend for one additional five-year term. The Company had hoped to implement the agreement in 2015 but did not finish the trial until December 2015. We now anticipate launch of the product in the third quarter of 2017. This delay was caused by the change in manufacturing from Nicaragua to the Dominican Republic. Under the agreement, the Company is obligated to provide delivery of an initial supply of between 1 million and 1.5 million doses of Lodonal™ product to cover AHAR Pharma’s first-year purchase commitment.
In August 2015, the Company announced the signing of a letter of interest with GB Pharma/AHAR and Fidson Healthcare of Nigeria to enable Fidson to market and sell LodonalTM in Nigeria. The agreement will become effective if we receive NAFDAC approval to distribute LodonalTM in Nigeria, based on the 2015 trial evaluating the efficacy and safety of Lodonal™. The agreement will require an amendment to the agreement signed in September 2013 with AHAR, so that both contracts conform. On January 21, 2016 the company submitted the initial results of the clinical trial to NAFDAC, and on February 22, 2016 AHAR Pharma submitted final document to NAFDAC requesting the issuance for marketing approval. As of March 31, 2017 the Company is still awaiting marketing approval for distribution of Lodonal™ in Nigeria under this agreement.
In September 2014, Airmed Biopharma Limited (“Airmed”), a wholly owned subsidiary of the Company, signed an exclusive agency agreement with GB Pharma Holdings Inc., to market and promote Lodonal™, and to solicit purchase orders for Lodonal™, in various counties in Africa. Airmed is required to pay GB Pharma Holdings Inc. a commission based on payments actually received on purchase orders procured by the GB Pharma Holdings Inc. from customers in Africa during the term of the agreement, after deduction for certain costs incurred by Airmed for product supply. The agreement has an initial term of five years, with automatic renewals for additional one-year periods unless terminated by either party.
On October 18, 2012, the Company and Hubei Qianjiang Pharmaceutical Co., Ltd. (“Qianjiang Pharmaceutical”), signed a Venture Cooperation Agreement on New Drug Methionine Enkephalin (the “Venture Agreement”) pursuant to which Qianjiang Pharmaceutical acquired an exclusive license for the production of MENK in China. The Venture Agreement requires that Qianjiang Pharmaceutical conduct drug research and pilot testing for MENK, organize pre-clinical studies, and apply for clinical trials for MENK with the Chinese State Food and Drug Administration. Under the Venture Agreement, Qianjiang Pharmaceutical must open a co-administration account for the development of MENK in China. Qianjiang Pharmaceutical must pay the Company, upon the marketing of MENK products, a half-year amount equaling 6% of its gross sales from MENK of the preceding half year. The Company may cancel the Venture Agreement if Qianjiang Pharmaceutical does not pay expenses for a period exceeding nine months or does not commence clinical trials within 12-months after receiving certain approvals. Qianjiang Pharmaceutical may cancel the Venture Agreement if the Company fails to perform its obligations for a period of nine months or the failure to receive approval of clinical trials is due to the Company’s MENK technologies. The Venture Agreement was amended on February 24, 2013 to expand the clinical trials from pancreatic to both pancreatic and liver cancer and amended on March 6, 2014 to require Qianjiang Pharmaceutical to commence studies and clinical trials in China and place funds in the co-administration account.
| 44 |
On August 6, 2014, the Company entered into a Supplementary Agreement on New Drug Methionine – Enkephalin Cooperation (the “Amendment”) with Qianjiang Pharmaceutical, amending the Venture Agreement, as amended. The Company and Qianjiang Pharmaceutical executed the Amendment to accelerate clinical trials in both the United States and China, and agreed to immediately initiate three month Good Laboratory Practice (“GLP”) Toxicology Studies (rat and dog) within 30 days of signing the Amendment. The Amendment requires that the GLP Toxicology Studies Trials are conducted in China in accordance with international standards and standards acceptable to the FDA.
In February 2013, the Company signed a Strategic Framework Agreement for Cooperation with Qianjiang Pharmaceutical. Under the agreement, the parties will work together to further the development of new products and conduct research and development on the Company’s licensed patented technology. Specifically, the parties aim to co-invest to develop and market products focusing on HIV, cancer and related autoimmune system therapies, develop co-ventured manufacturing facilities in China, and develop co-ventured distribution of the developed products in China and Africa. The agreement does not have a definitive term, as each new agreement resulting from the cooperation will set forth the material terms, including, but not limited to, fees, duration and termination therein.
In accordance with these agreements, Qianjiang Pharmaceutical has acquired MENK material for the preclinical and clinical trial. MENK toxicology studies are in process under the trial, including a six-month toxicology study in animals. Other studies, including stability and general pharmacology (on normal animals to determine the effect to heart, blood pressure, etc.) have commenced. All FDA-required tests, including formulation and quality control tests, are in process in China.
The Company recently received the pharmacology and toxicology reports for MENK at GLP lab completed by Hubei Qianjiang. In addition to the pharmacology and toxicology, China Peptide has completed the Chemistry, Manufacturing, and Controls (CMC) required to file with both the SFDA and the FDA, as no pivotal trial can be started without CMC.
Production
On April 23, 2013, the Company signed a Contract with ViPharma for the Supervision and Inspection of Manufacturing Processes as part of its negotiations for a contract for the manufacturing of LDN in a tablet, capsule and/or cream. The contract sets out the terms and conditions under which ViPharma will carry out the services of inspecting and supervising the manufacturing and packaging processes of LDN and ensure compliance with the FDA’s Current Good Manufacturing Practice regulations (“cGMP”) and the Company’s specifications. ViPharma will carry out its obligations in whatever Latin American country the Company ultimately decides to manufacture LDN. Under the contract, ViPharma has the exclusive rights to supervise and inspect all manufacturing processes of LDN in Latin America. The initial term of the agreement is ten years commencing in September 2013, with automatic five-year renewal terms provided neither party is in breach. The agreement may be terminated by (i) mutual agreement, (ii) in the event of a breach after a 45 day cure period or (iii) by either party upon provision of written notice at least 90 days before the end of the agreement, provided however that if the Company terminates the contract without cause it will be required to pay ViPharma a $10 million penalty.
The Company executed a manufacturing agreement with Laboratorios Ramos, a current good manufacturing practice (“cGMP”) facility for Lodonal™ effective August 16, 2013. Under the agreement, Laboratorios Ramos will produce LDN tablets, capsules and/or cream in accordance with the technical specifications we provided, cGMPs and the practices of Nicaragua and of any other regulatory body of the countries where the products will be exported. Laboratorios Ramos has obtained all permits and licenses necessary to carry out the manufacturing of LDN. The manufacturing agreement has a five-year term, renewable by a signed agreement by the parties at least 60 days before the expiration of the agreement. The agreement may be terminated earlier through mutual agreement or upon expiration of a 30-day cure period following notice from the non-breaching party to the breaching party of a material failure of the obligations under the agreement. Additionally, we may terminate the agreement upon at least 30 days written notice if Laboratorios Ramos does not act in strict accordance with the technical specifications we provided and with cGMPs or those of any regulatory body of the importing countries. We will pay Laboratorios Ramos a low single digit cent amount per tablet or capsule and a low double-digit cent amount per each cream produced.
| 45 |
Raw Materials and Principal Suppliers
The Company has decided to enter into third-party manufacturing agreements; accordingly, we rely on third parties for clinical production of our products and product candidates.
The active pharmaceutical ingredient (“API”) for initiating clinical trials in the United States has been and will continue to be sourced from a cGMP-established vendor that has filed or will file a Type II Drug Master File in the United States. Prior to sourcing, a quality due diligence/vendor qualification will be completed that will include, but is not limited to, a review of the vendor’s inspection and compliance history with the FDA and, as relevant, the vendor’s inspection and compliance history with other regulatory bodies (i.e. the European Medicines Agency, or EMA).
The Company expects that the Finished Pharmaceutical Product (“FPP”) for initiating the proposed clinical trials will be prepared by a U.S. vendor with extensive cGMP experience, a strong record of compliance with FDA regulations as evidenced by a site Quality Audit, and an extensive history of manufacturing products administered to humans in the U.S.
American Peptide Company is the Company’s supplier of the API in MENK. S.A.L.A.R.S SpA supplies the API in LDN.
Competition
The pharmaceutical industry is characterized by rapidly evolving technology and intense competition. A large number of companies of all sizes, including major pharmaceutical companies and specialized biotechnology companies, engage in activities similar to ours. Many of our competitors have substantially greater financial and other resources available to them. In addition, colleges, universities, governmental agencies and other public and private research organizations continue to conduct research and are becoming more active in seeking patent protection and licensing arrangements to collect royalties for use of technologies that they have developed. Some of our competitors’ current or future products and technologies may be in direct competition with ours. We also must compete with these institutions in recruiting highly qualified personnel.
Established pharmaceutical companies that currently sell or are developing drugs in our markets of interest include, for example; Abbott Laboratories, Amgen, AstraZeneca, Biogen Idec, Bayer, Elan, Johnson & Johnson, Merck, Merck Serono, Takeda, Novartis, Pfizer, Reata, Sanofi-Aventis and Teva. Many or all of these established competitors are also involved in research and drug development regarding various OGF receptors. Pharmaceutical and biotechnology companies which are known to be involved in immuno therapy research and related drug development include Pfizer, Bristol-Myers Squibb, Merck, Takeda, Sanofi-Aventis, Incyte, and UCB Pharma, among others.
Protalex, Inc. is a clinical stage biopharmaceutical company that is developing a class of biopharmaceutical drugs for treating autoimmune and inflammatory diseases and rtax Biopharma is a clinical stage biopharmaceutical company developing breakthrough therapies for autoimmune diseases. Its pipeline of drugs is based on novel science that targets the interaction between T-cell receptors (TCR) and Nck, a key protein involved in T-cell activation.
There are also comparable companies focused on advancing drugs for various diseases using the FDA’s 505(b)(2) pathway including Akorn Inc., Chelsea Therapeutics Inc., MAP Pharmaceuticals Inc., Pain Therapeutics Inc., Rexahn Pharmaceuticals Inc., Santarus Inc., Ventrus Biosciences Inc., and XenoPort Inc.
| 46 |
The current lack of safe, effective, low cost oral treatments for autoimmune diseases such as multiple sclerosis and Crohn’s disease provides an attractive opportunity for our product. Treatment of cancer is an unmet medical need in developing countries, and the need for a safe, effective, low-cost oral treatment provides an opportunity for our Company. Clinical results with LDN trials support further evaluation of our products in the treatment of Crohn’s disease.
Customers
In 2016, the Company recorded Lodonal™ sales totaling $3,463 to individuals in the USA under its agreement with KRS. The Company had $16,197 of sales to customers in 2015.
The Company has made final applications for the importation of Lodonal™ into the Republic of Nigeria and the Republic of Equatorial Guinea. In Equatorial Guinea, the Company has obtained approval for the sale of the drug, but the government has not allocated any funds to initiate purchases. In December 2015, NAFDAC granted permission to the Company and AHAR Pharma for a Lodonal™ trial for HIV/AIDS in Nigeria. The objective of this bridging trial was to assure safety and tolerability as well as efficacy with the local population. The Company completed this trial in December 2016, and as of March 31, 2017 had not yet received approval for the importation of Lodonal™ in Nigeria.
The Company, with approval from the Minister of Health in Nicaragua and countries that approve and register the product inclusive of the Certificate of Pharmaceutical Products issued for that jurisdiction, will look to open distribution channels in Central and South America in 2017.
Lodonal’s commercialization in Nigeria was delayed by about three quarters as we replaced our Lodonal manufacturer in Nicaragua in favor of Dominican Republic. Through our political contacts, we determined the political landscape in Nicaragua was shifting in the wrong direction and, rather than risk a major, costly supply chain disruption after beginning distribution, our Board decided to secure a relationship with a second manufacturer.
The Company signed an agreement with Acromax Dominicana SA October 25, 2016 one of the premier pharmaceutical companies in the Dominican Republic. Acromax is both cGMP certified but has received ISO 9001: 2008 certification as evidence that they fully meet the requirements dictated by the said rule and, as a result, is able to meet national and international regulations related to their activities ensuring quality, safety and reliability of their medicines. Acromax exports over 160 products and has shipped into Nigeria in the past. This required a new round of regulatory approvals in both Nigeria with NAFDAC and with the Dominican Republic with the Minister of Public Health and Social Assistance.
The Dominican Minister of Public Health and Social Assistance granted manufacturing and export approval for LodonalTM on January 23, 2017, registration number 2017-0068. As of March 16, 2017, the Company had submitted all of the required documents and samples to NAFDAC to apply for final marketing approval in Nigeria of Dominican-manufactured LodonalTM. The company is hopes to receive marketing approval by the third quarter of 2017.
Once the Company obtained drug approval for Lodonal in the Dominican Republic, we were able to begin the new regulatory process in Nigeria for marketing approval. The Company was required to provide the following documents as part of marketing approval: Acromax Dossier, Drug Approval issued by the relevant health/regulatory body Dominican Republic, Certificate of Pharmaceutical Products (COPP – WHO FORMAT); Current Good Manufacturing Practice (GMP) Certificate of the manufacturing facility; Certificate of Registration of Brand Name with the trademark Registry in the Ministry of Commerce in Nigeria, Clinical Trial Data, Comprehensive Certificate of Analysis; Issued by the manufacturer. Current Annual License to Practice for the Superintendent Pharmacists issued by the Pharmacists Council of Nigeria; Current Certificate of Registration Retention of Premise issued by the Pharmacists Council of Nigeria, 100 Samples including packaging, Patient Data Sheet and A letter of Invitation to inspect the factory abroad.
The Company has received approval from the Republic of Malawi for the importation of Lodonal™, subject to completion of a clinical trial as an adjunct treatment for cancer.
| 47 |
Research and Development
Our research and development (“R&D”) organization focuses primarily on new uses for the opioid-related immuno-therapies, such as LDN and MENK. These therapies stimulate the immune system in such a way that provides the potential to treat a variety of diseases that have abnormalities in the immune system.
Our R&D priorities include development of MENK IRT-101, a small synthetic pentapeptide that is naturally occurring in the body, and Lodonal™, an opioid receptor antagonist. Our pipeline provides two therapies with an extremely wide range of indications that can be pursued. Both molecules have the ability to stimulate and/or regulate the immune system in order to treat a variety of autoimmune diseases including multiple sclerosis, immune disorders such as Crohn’s disease, cancer, and viral infections such as HIV/AIDS.
Our R&D is overseen and managed internally, working with individuals, universities, and Contract Research Organizations (“CROs”) in order to utilize patents that we have licensed or acquired since our inception. We continue to seek to expand our pipeline by reviewing other compounds, technologies or capabilities. We also seek out promising compounds and innovative technologies developed by third parties to incorporate into our discovery and development processes or projects.
Drug discovery and development is time-consuming, expensive and unpredictable. According to the Pharmaceutical Research and Manufacturers of America (PhRMA), out of 5,000-10,000 screened compounds, only 250 enter preclinical testing, five enter human clinical trials and one is approved by the FDA. The process from early discovery or design to development to regulatory approval can take more than 10 years. Drug candidates can fail at any stage of the process, and candidates may not receive regulatory approval even after many years of research.
As of December 31, 2016, we had two compounds (IRT-101 and Lodonal™) in research and development. In 2016 our development programs focused on both compounds, one in oncology and one in Crohn’s disease; which we are expecting to move into Phase II clinical trials.
The following table provides information about significant regulatory actions by, and filings pending with the FDA and regulatory authorities in the EU, as well as additional indications and new drug candidates in late-stage development.
| NEW DRUG CANDIDATES IN LATE-STAGE DEVELOPMENT | ||||
| CANDIDATE | INDICATION | REGULATORY ACTIONS | ||
| IRT-101 | Pancreatic Cancer | End-of-Phase 1 Meeting with FDA Complete 3Q 2013 | ||
| IRT-103 Lodonal™ | Crohn’s Disease | Type C Meeting with FDA Complete 2Q 2013 Scientific Advice with EMA Complete 1Q 2015 | ||
The Company has incurred, and expects to continue incurring, substantial research, development and other costs in connection with compound partnering agreements. In 2016, the Company incurred cash expenses of $444,165 for research and development. The Company estimates it could spend approximately $15,000,000 in cash for research and development costs in 2017.
Employees
As of December 31, 2016, the Company had 5 full time employees.
Available Information
Our Current Reports on Form 8-K, and Quarterly Reports are electronically filed with or furnished to the Securities and Exchange Commission (SEC), and all such reports and amendments to such reports have been and will be made available, free of charge, through our website (http://www.immunetherapeutics.com) as soon as reasonably practicable after such submission to the SEC. Such reports will remain available on our website for at least 12 months. The contents of our website are not incorporated by reference into this Annual Report on Form 10-K. The public may read and copy any materials filed by us with the SEC at the SEC’s Public Reference Room at 100 F Street, NW, Washington, D.C. 20549.
| 48 |
You should carefully consider the following factors and other information in this Annual Report and our other SEC filings before making a decision to invest in our common stock. Additional risks and uncertainties that we are unaware of may become important factors that affect us. If any of the following events occur, our business, financial conditions and operating results may be materially and adversely affected. In that event, the trading price of our common stock may decline, and you could lose all or part of your investment.
Risks Related to our Business
We have a limited operating history and are expected to incur significant operating losses during the early stage of our corporate development.
We have a limited operating history. Our historical financial information consists only of an audit of our financial results at and for the years ended December 31, 2016, 2015, 2014, 2013 and 2012. There is limited historical financial information upon which to base an evaluation of our performance. We are an emerging company, and thus our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operation, particularly in the pharmaceutical industry.
Since inception, we have invested a substantial portion of our time and financial resources in the acquisition and development of our most advanced drug candidate, LDN. We have generated cumulative losses of approximately $xxx million and $xxx stockholders’ deficit since inception, and we expect to continue to incur losses until IRT-103 (LDN) is approved by the FDA and foreign regulatory authorities. Even if regulatory approval is obtained, there is a risk that we will not be able to generate material sales of IRT-103 (LDN), which would cause us to continue to incur losses.
We may never generate revenue, are not profitable and may never become profitable.
We expect to incur substantial losses and negative operating cash flow for the foreseeable future, and we may never achieve or maintain profitability. Even if we are able to launch IRT-103 (LDN) we expect to incur substantial losses for the foreseeable future and may never become profitable.
We do not anticipate that we will generate revenue from the sale of our products for the foreseeable future. In addition, if approved, we expect to incur significant costs to commercialize our drug candidates and our drugs may never gain market acceptance. If our drug candidates fail to demonstrate safety and efficacy in clinical trials, do not gain regulatory approval, or do not achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. If we are unable to achieve and sustain profitability, the market value of our common stock will likely decline. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or whether we will become profitable.
We will see losses from our clinical trials conducted either directly or through our subsidiaries for the foreseeable future, and if we fail at one or more of our clinical trials, it could affect the value of the Company’s stock.
We rely on financings to fund and conduct clinical trials directly or through our subsidiaries needed for NDA submission with respect to IRT-103 (LDN). Any of the following events or factors could have a material adverse effect on our ability to generate revenue from the commercialization of IRT-103 (LDN):
| ● | The Company may be unable to successfully complete the clinical development of IRT-103 (LDN); | |
| ● | The Company must comply with any possible additional requests and recommendations from the FDA, including additional clinical trials; | |
| ● | The Company may not obtain all necessary approvals from the FDA and similar foreign regulatory agencies; | |
| ● | The Company may not commit sufficient resources to the development, regulatory approval, marketing and distribution of IRT-103 (LDN); | |
| ● | IRT-103 (LDN) must be manufactured in compliance with requirements of the FDA and similar foreign regulatory agencies and in commercial quantities sufficient to meet market demand; |
| 49 |
| ● | IRT-103 (LDN) may not achieve market acceptance by physicians, patients and third party payers; | |
| ● | IRT-103 (LDN) may not successfully compete against alternative products and therapies; and | |
| ● | The Company or any other pharmaceutical organization may independently develop products that compete with IRT-103 (LDN). |
To obtain approval from the FDA of an NDA, for IRT-103 (LDN), The Company will need to demonstrate through evidence of adequate and well-controlled clinical trials that IRT-103 (LDN) is safe and effective for each proposed indication. However, IRT-103 (LDN) may not be approved even though it achieved its specified endpoints in future Phase III clinical trials intended to support an NDA, which may be conducted by the Company. The FDA may disagree with the trial design and the interpretation of data from clinical trials, may ask the Company to conduct additional costly and time consuming clinical trials in order to obtain marketing approval or approval to enter into an advanced phase of development, or may change the requirements for approval even after it has reviewed and commented on the design for our future clinical trials. The FDA may also approve IRT-103 (LDN) for fewer or more limited indications than the Company may request, or may grant approval contingent on the performance of costly post-approval clinical trials. In addition, the FDA may not approve the labeling claims that we believe are necessary or desirable for the successful commercialization of IRT-103 (LDN).
The Company anticipates that if Cytocom initiates a clinical trial in the next 12 months, Cytocom would need approximately $7-$15 million to fully develop products and for Phase III clinical trials for Crohn’s disease. We expect that two-thirds of this amount will be spent by Cytocom in the USA, the balance by Immune Therapeutics, Inc. and/or its subsidiaries for international trials. Cytocom trials are expected to be split evenly between LDN and MENK. The international trials will focus the use of MENK for treatment of cancer in Africa.
The development of new drugs is a highly risky undertaking which involves a lengthy process, and therefore our drug discovery and development activities may not result in products that are approved by the applicable regulatory authorities on the time schedule we have planned, or at all.
Our drug candidates are in early stages of drug discovery or clinical trials and are prone to the risks of failure inherent in drug development. As of the date of this Form 10-K, both of our current drug candidates, IRT-101 (MENK) and IRT-103 (LDN) have been tested on human beings. We will need to conduct additional clinical trials before we can demonstrate that our drug candidates are safe and effective to the satisfaction of the FDA and other regulatory authorities. Clinical trials are expensive and uncertain processes that can take multiple years to complete. We cannot assure you that our ongoing clinical trials or any future clinical trial of any of our other drug candidates, will be completed on schedule, or at all, or whether our planned clinical trials will start in a timely manner. The commencement of our planned clinical trials could be substantially delayed or prevented by a number of factors, including:
| ● | delays or failures in obtaining sufficient quantities of the API and/or drug product; | |
| ● | delays or failures in reaching an agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites and with the FDA or other foreign regulatory bodies; | |
| ● | delays or failures in obtaining Institutional Review Board (“IRB”) or Ethics Committee (“EC”) approvals to conduct a clinical trial at a prospective site; | |
| ● | the need to successfully complete, on a timely basis, preclinical safety pharmacology studies (for IRT-101 (MENK)); | |
| ● | the limited number of, and competition for, suitable sites to conduct the clinical trials; | |
| ● | the limited number of, and competition for, suitable patients for enrollment in the clinical trials; and | |
| ● | delays or failures in obtaining regulatory approval to commence a clinical trial. |
The completion of our clinical trials could also be substantially delayed or prevented by a number of factors, including:
| ● | slower than expected rates of patient recruitment and enrollment; | |
| ● | failure of patients to complete the clinical trials; | |
| ● | failure of our third party vendors to timely or adequately perform their contractual obligations relating to the clinical trials; |
| 50 |
| ● | inability or unwillingness of patients or medical investigators to follow our clinical trial protocols; | |
| ● | inability to monitor patients adequately during or after treatment; | |
| ● | termination of the clinical trials by one or more clinical trial sites; | |
| ● | unforeseen safety issues; | |
| ● | lack of efficacy demonstrated during clinical trial results; | |
| ● | lack of adequate funding to continue the clinical trials; | |
| ● | the need for unexpected discussions with the FDA or other foreign regulatory agencies regarding the scope or design of our clinical trials or the need to conduct additional trials; | |
| ● | unforeseen delays by the FDA or other foreign regulatory agencies after submission of our results; | |
| ● | an unfavorable FDA inspection of our contract manufacturers of APIs or drug products; and/or | |
| ● | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold. |
Any failure or significant delay in completing clinical trials for our drug candidates will harm the commercial prospects for our drug candidates and adversely affect our financial results.
Additionally, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs or ECs for reexamination, which may impact the costs, timing or successful completion of a clinical trial. If we experience delays in completion of a clinical trial, or if we terminate any of our clinical trials, the commercial prospects for our drug candidates may be harmed and our ability to generate product revenues will be delayed. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a drug candidate.
If we are required to suspend or discontinue clinical trials due to side effects or other safety risks, or if we are required to conduct studies on the long-term effects associated with the use of our drug candidates, our efforts to commercialize our products could be delayed or halted.
Our clinical trials may be suspended or terminated at any time for a number of safety-related reasons. For example, administering any drug candidate to humans may produce undesirable side effects. We may voluntarily suspend or terminate our clinical trials if at any time we believe that our drug candidates present an unacceptable safety risk to the clinical trial patients. In addition, IRBs, ECs or regulatory agencies may order the temporary discontinuation or termination of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, including if they present an unacceptable safety risk to patients. The existence of undesirable side effects resulting from our drug candidates could cause us or regulatory authorities, such as the FDA, to interrupt, delay or halt clinical trials of our drug candidates and could result in the FDA or other regulatory agencies denying further development or approval of our drug candidates for any or all targeted indications.
Further, cytokine receptors and opiate growth factor receptors are a novel class of targets. As a result, we may experience unforeseen adverse side effects with our existing and future drug candidates, including IRT-101 (MENK) and IRT-103 (LDN). As of the date of this registration statement, although we have not observed harmful side effects in prior studies of LDN or MENK, later trials could reveal such side effects. The pharmacokinetic profile and results of preclinical studies may not be indicative of results in any clinical trial.
We have not conducted studies on the long-term effects associated with the use of our drug candidates. Studies of long-term effects and chronic dosing (approximately 1 year of dosing); will be required for regulatory approval and may delay introduction of our therapies or our other drug candidates into the market. Additional studies could also be required at any time after regulatory approval of any of our drug candidates. Some or all of our drug candidates may prove to be unsafe for human use.
| 51 |
Even if our drug candidates do obtain regulatory approval they may never achieve market acceptance or commercial success.
Even if we obtain FDA or other regulatory approval, our drug candidates may not achieve market acceptance among physicians, patients and/or third party payers or they may be used only in applications more restricted than we anticipate, and ultimately, may not be commercially successful. Our treatments, if successfully developed, will compete with a number of traditional products manufactured and marketed by major pharmaceutical and biotechnology companies. Our treatments may also compete with new products currently under development by such companies and others. Physicians will prescribe a product only if they determine, based on experience, clinical data, side effect profiles and other factors, that it is beneficial as compared to other products currently available and in use. Physicians also will prescribe a product based on their traditional preferences. Market acceptance of our drug candidates for which we receive approval depend on a number of factors, including:
| ● | the efficacy and safety of our drug candidates as demonstrated in clinical trials; | |
| ● | the clinical indications for which the drug is approved; | |
| ● | acceptance by physicians, major operators of clinics and patients of the drug as a safe and effective treatment; | |
| ● | the potential and perceived advantages of our drug candidates over alternative treatments; | |
| ● | the safety of drug candidates seen in a broader patient group, including its use outside the approved indications; | |
| ● | the cost of treatment in relation to alternative treatments; | |
| ● | the availability of adequate reimbursement and pricing by third parties and government authorities; | |
| ● | relative convenience and ease of administration; | |
| ● | the prevalence and severity of adverse side effects; and | |
| ● | the effectiveness of our sales and marketing efforts. |
If our drug candidates that obtain regulatory approval fail to achieve market acceptance or commercial success, the Company’s financial results will be adversely affected.
The commercial success of IRT-103 depends, in part, on Cytocom’s ability to develop and market the drug in North America, and if we fail in these initiatives, our ability to generate future revenue in the United States could be reduced.
If Cytocom successfully completes the clinical development program in the U.S. for our lead independent drug candidate, IRT-103 (LDN), we plan to retain commercial rights to IRT-103 as we have exclusive licensing rights. Any of the following events or factors could have a material adverse effect on both the ability to generate revenue in the U.S. from the commercialization of IRT-103:
| ● | we may be unable to successfully complete the clinical development of IRT-103; | |
| ● | our lack of experience in commercializing and marketing drug products; | |
| ● | we may not have or be able to obtain sufficient financial resources to develop and commercialize IRT-103; | |
| ● | we may not be able to identify a suitable co-development partner; | |
| ● | we, or any of our future partners, may fail to fulfill our responsibilities in a timely manner or fail to commit sufficient resources to the development, regulatory approval, and commercialization efforts related to IRT-103; | |
| ● | we, or any of our future partners, must comply with additional requests and recommendations from the FDA, including additional clinical trials; | |
| ● | we, or any of our future partners, may not obtain all necessary approvals from the FDA and similar foreign regulatory agencies; | |
| ● | IRT-103 must be manufactured in compliance with requirements of the FDA and similar foreign regulatory agencies and in commercial quantities sufficient to meet market demand; | |
| ● | IRT-103 may not achieve market acceptance by physicians, patients and third party payers; | |
| ● | IRT-103 may not compete successfully against alternative products and therapies; and | |
| ● | we, or any pharmaceutical company, may independently develop products that compete with IRT-103. |
| 52 |
Changes in pharmaceutical and biotechnology industry trends could adversely affect the Company’s operating results.
Industry trends, economic and political factors that affect pharmaceutical, biotechnology, medical device companies and academic/government entities sponsoring clinical research directly affect the Company’s business. For example, many companies in such industries and government organizations have been hiring companies (like the Company) to conduct large development projects. The Company’s operations, financial condition and growth rate could be materially and adversely affected if these industries reduce outsourcing of such projects. In the past, mergers, product withdrawals, liability lawsuits and other factors in the pharmaceutical industry have slowed decision making by pharmaceutical companies and correlating government bodies significantly delaying and/or halting drug development projects. Continuation or increases in such trends could have an adverse effect on the Company’s business. Additionally, numerous government agencies have undertaken efforts to control growing healthcare costs through legislation, regulation and voluntary agreements with medical care providers and pharmaceutical companies. If future regulatory cost-containment efforts limit potential profits derived from new drugs, the Company’s clients may reduce their drug discovery and development spending. A reduction in drug discovery and development spending could have a material adverse effect on the Company’s results and operations creating a significant reduction of the Company’s revenue.
We currently rely on third parties to conduct all our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize any of our drug candidates.
We currently do not have the ability to independently conduct clinical trials. We rely on medical institutions, clinical investigators, contract laboratories, collaborative partners and other third parties, such as contract research organizations, to conduct clinical trials on our drug candidates. The third parties with whom we contract for execution of our clinical trials play a significant role in the conduct of these trials and the subsequent collection and analysis of data. These third parties are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs. Our IND is being conducted per 21 Code of Federal Regulations Title 21, Part 312. In addition, we follow ICH guidelines, including good clinical practices (ICH E6) and current good manufacturing practice (ICH Q7) throughout the development process. After completion of Phase III clinical trials, the Company will file our NDA for LDN (IRT-103) as a 505(b)(2) application. Although we rely on these third parties to conduct our clinical trials, we remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as current Good Clinical Practices (“cGCPs”) for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials.
In addition, the execution of preclinical studies and clinical trials, and the subsequent compilation and analysis of the data produced, requires coordination among various parties. In order for these functions to be carried out effectively and efficiently, it is imperative that these parties communicate and coordinate with one another. Moreover, these third parties may also have relationships with other commercial entities, some of which may compete with us. In most cases, these third parties may terminate their agreements upon a material breach by us that is not cured within 30 days by providing us with 30 days’ prior written notice. Many of these agreements may also be terminated by such third parties under certain other circumstances, including our insolvency or our failure to comply with applicable laws. In general, these agreements require such third parties to reasonably cooperate with us at our expense for an orderly winding down of services of such third parties under the agreements. If the third parties conducting our clinical trials do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical trial protocols or cGCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties, which could be costly, and our clinical trials may be extended, delayed or terminated or may need to be repeated, and we may not be able to obtain regulatory approval for or commercialize the drug candidate being tested in such trials.
| 53 |
If any of our drug candidates receive marketing approval, and the Company or others later identify undesirable side effects caused by the drug candidate, our ability to market and derive revenue from the drugs could be compromised.
If the Company or others identify undesirable side effects caused by one of our drug candidates, any of the following adverse events could occur:
| ● | regulatory authorities may withdraw approval of the drug or seize the drug; | |
| ● | we may be required to recall the drug or change the way the drug is administered; | |
| ● | additional restrictions may be imposed on the marketing or the manufacturing processes of the particular drug; | |
| ● | we may be subject to fines, injunctions or the imposition of civil or criminal penalties; | |
| ● | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; | |
| ● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; | |
| ● | we could be sued and held liable for harm caused to patients; | |
| ● | the drug may become less competitive; and | |
| ● | our reputation may suffer. |
Any of these could result in the loss of significant revenues, which would materially and adversely affect our results of operations and business.
We may need additional financing and may be unable to raise capital on acceptable terms, or at all, when needed, which could force us to delay, reduce or eliminate our research and development programs and other operations or commercialization efforts.
We are advancing multiple drug candidates through discovery and development and will require substantial funds to conduct development, including preclinical studies and clinical trials, of our drug candidates. Commercialization of any drug candidate will also require substantial expenditures. To further the development and commercialization efforts of our drug candidates, we may need additional financing to hire additional employees to co-promote drug candidates or to commercialize drug candidates that may not be covered by our current collaboration agreements.
At December 31, 2016, we had $74,389 in cash and cash equivalents. We do not believe that our available cash and cash equivalents will be sufficient to fund our anticipated level of operations for the next 12 months and we will likely need to seek outside sources of funding. Assuming that anticipated investment and revenue does not materialize business operations would not be able to continue more than 30 days. We believe we require at least $2,000,000 for our operations over the next 12 months. Our future financing requirements will depend on many factors, some of which are beyond our control, including:
| ● | the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities; | |
| ● | the timing of, and costs involved in, seeking and obtaining FDA and other regulatory approvals; | |
| ● | the continuation and success of our strategic alliances and future collaboration partners; | |
| ● | the exercise of remaining options under current collaborative agreements; | |
| ● | the costs of preparing, filing, prosecuting, maintaining and enforcing any patent claims and other intellectual property rights, including litigation costs and the results of such litigation; | |
| ● | our ability to enter into additional collaboration, licensing, government or other arrangements and the terms and timing of such arrangements; | |
| ● | potential acquisition or in-licensing of other products or technologies; and | |
| ● | the technologies or other adverse market developments. |
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies. We currently have no understandings, commitments or agreements relating to any of these types of transactions.
| 54 |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity offerings, debt financings, government grants and contracts and/or strategic collaborations. Additional financing may not be available to us when we need it or it may not be available on favorable terms, if at all. If we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or our commercialization efforts. We may be required to enter into collaborative partnerships for one or more of our drug candidate programs at an earlier stage of development or on less favorable terms, which may require us to relinquish rights to some of our drug candidates that we would otherwise have pursued on our own. We may also be required to pursue strategic alternatives that may affect our business or corporate structure in order to make ourselves more attractive to investors.
In addition, If the Company or any of its future collaboration partners does not perform in the manner we expect or fulfill its responsibilities in a timely manner, or at all, the clinical development, regulatory approval, and commercialization efforts related to IRT-103 (LDN) could be delayed or terminated. It may be necessary for us to assume the responsibility at our own expense for the development of IRT-103 (LDN). In that event, we would likely be required to seek additional funding.
We may form additional strategic alliances in the future with respect to our independent programs, and we may not realize any benefits of such alliances.
We may form strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties with respect to our independent programs that we believe will complement or augment our existing business. For example, we plan to find a partner to co-develop and commercialize IRT-101 (MENK) and IRT-103 (LDN) outside North America upon completion of clinical development of IRT-103 (LDN) for the treatment of pediatric and adult patients with Crohn’s disease. We face significant competition in seeking appropriate strategic partners. The negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for any future product candidates and programs because our research and development pipeline may be insufficient, our product candidates and programs may be deemed to be at too early a stage of development for collaborative effort and/or third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy. We cannot be certain that, following a strategic transaction or license, we will achieve the revenues or specific net income that justifies such transactions. Any delays in entering into new strategic partnership agreements related to our product candidates could also delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market.
We do not currently manufacture IRT-103 Low Dose Naltrexone (LDN) and therefore must rely on third-party manufacturing to supply the drug for clinical trials. If one of our suppliers or manufacturers fails to perform adequately or fulfill our needs, we may be required to incur significant costs and devote significant efforts to find new suppliers or manufacturers, which would cause delays in the development and commercialization of our drug candidates.
The manufacture of pharmaceutical products in compliance with cGMPs requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, including difficulties with production costs and yields, quality control, including stability of the drug candidate and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced FDA cGMP requirements, other federal and state regulatory requirements, and foreign regulations. If our manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations to us or under applicable regulations, our ability to provide study drugs in our preclinical studies and clinical trials would be jeopardized. Any delay or interruption in the supply of preclinical study or clinical trial materials could delay the completion of our preclinical studies and clinical trials, increase the costs associated with maintaining our preclinical study and clinical trial programs and, depending upon the period of delay, require us to commence new trials at significant additional expense or terminate the studies and trials completely.
All manufacturers of our drug candidates must comply with cGMP requirements enforced by the FDA through its facilities inspection program. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. Manufacturers of our component materials may be unable to comply with these cGMP requirements and with other FDA, state and foreign, regulatory requirements. The FDA or similar foreign regulatory agencies at any time may also implement new standards, or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. We have little control over our manufacturers’ compliance with these regulations and standards. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any product supplied is compromised due to our manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products, and we may be held liable for any injuries sustained as a result. Any of these factors could cause a delay of clinical trials, regulatory submissions, approvals or commercialization of our drug candidates or entail higher costs or impair our reputation.
| 55 |
We source the API for IRT-103 (LDN) from a third-party manufacturing vendor. Another pharmaceutical company manufactures the API for IRT-101. Our current agreements with our suppliers provide for the entire supply of the API necessary for additional clinical trials or for full-scale commercialization. In the event that we and our suppliers cannot agree to the terms and conditions for them to continue to provide some or all of our API clinical and commercial supply needs, or if any single source supplier terminates the agreement in response to a breach by us, we would not be able to manufacture the API on a commercial scale until a qualified alternative supplier is identified, which could also delay the development of, and impair our ability to commercialize, our drug candidates.
Although alternative sources of supplies exist, the number of third party suppliers with the necessary manufacturing and regulatory expertise and facilities are limited, and it could be expensive and take a significant amount of time to arrange for alternative suppliers, which could have a material adverse effect on our business. New suppliers of any API would be required to qualify under applicable regulatory requirements and would need to have sufficient rights to the method of manufacturing such ingredients under applicable intellectual property laws. Obtaining the necessary FDA approvals or other qualifications under applicable regulatory requirements and ensuring non-infringement of third party intellectual property rights could result in a significant interruption of supply and could require the new manufacturer to bear significant additional costs which may be passed on to us.
We currently have only a limited distribution organization with no sales and marketing staff. If we are unable to develop sales and marketing and expand distribution capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our future products.
We currently have only a limited distribution organization with no sales or marketing staff. If our products are approved for sale in the United States we will need to execute a number of sales and marketing agreements, but there can be no assurance that the Company will be able to sign an agreement to market and distribute our products. To the extent we rely on third parties for marketing and distributing our approved products, any revenue we receive will depend upon the efforts of third parties, which may not be successful, and are only partially within our control. Our reliance on third parties makes it likely that our product revenue is likely to be lower than if we directly marketed or sold our products. If we are unable to enter into arrangements with third parties to commercialize the approved products on acceptable terms or at all, we may not be able to successfully commercialize our future products or we will have to market these products ourselves, which will be expensive and require us to build our own sales force, which we do not have experience doing. We cannot assure you we will be successful in any of these initiatives. If we are not successful in commercializing our future products, either on our own or through collaborations with one or more third parties, our future product revenue will be materially adversely affected.
We are dependent on market acceptance of compounding pharmacies and compounded formulations, and physicians may be unwilling to prescribe, and patients may be unwilling to use, our proprietary LDN compounded formulation.
We are currently distributing our proprietary LDN formulation through Complete Pharmacy and Medical Solutions, LLC and expect to distribute such formulation through other compounding pharmacies outside of the U.S. Formulations prepared and dispensed by compounding pharmacies contain FDA-approved ingredients, but are not themselves approved by the FDA. As a result, our formulation has not undergone the FDA approval process and only limited data, if any, may be available with respect to the safety and efficiency of our formulation for any particular indication. Some physicians may be hesitant to prescribe, and some patients may be hesitant to purchase and use, this non-FDA approved compounded formulation. In addition, certain compounding pharmacies have been the subject of widespread negative media coverage in recent years, and the actions of these pharmacies have resulted in increased scrutiny of compounding pharmacy activities from the FDA and state governmental agencies. As a result, physicians may be unwilling to prescribe a compounded formulation when an FDA-approved alternative is available, even if they believe the compounded formulation to be superior and less expensive. Other reasons physicians may be unwilling to prescribe or patients may be unwilling to use our proprietary LDN compounded formulation could include the following, among others: our proprietary formulation is not required to be, and has not been, approved for marketing and sale by the FDA; there may be limited or no data available with respect to the clinical efficacy or safety of our compounded formulation the physician is prescribing; and to the extent there is such data available, we are limited in our ability to discuss the efficacy or safety of our formulation with potential purchasers of our formulation.
| 56 |
Additionally, some third-party payors, including the government Medicare and Medicaid programs, may not provide reimbursement for compounded formulations. Physicians who may otherwise be interested in prescribing our formulation or utilizing our compounding pharmacy services may be unwilling to do so if third party payor reimbursement, including Medicare and Medicaid reimbursement, is not available for our compounded formulation. Any failure by physicians, patients and/or third-party payors to accept and embrace compounded formulations could substantially limit our market and cause our operations to suffer.
We aim to generate revenue from our proprietary LDN formulation through our licensing arrangement with Complete Pharmacy and Medical Solutions, LLC and potentially other compounding pharmacies outside of the United States, but we may not be successful in our efforts to generate revenue from such formulation.
One aspect of our business strategy is to continue to develop our licensing arrangement with Complete Pharmacy and Medical Solutions, LLC and potentially enter into other licensing arrangements with other compounding pharmacies outside of the U.S., through which we can generate revenue from the sale of our proprietary LDN formulation. On December 8, 2014, we entered into a Contract for the Compounding of Pharmaceutical Products with Complete Pharmacy and Medical Solutions, LLC pursuant to which Complete Pharmacy and Medical Solutions, LLC will carry out the services of compounding, packaging and distributing tablets of our LDN formulation in the U.S. We have limited experience commercializing our formulation through licensing arrangements with compounding pharmacies. Even if we are successful, we may be unable to generate sufficient revenue to recover our costs.
We have minimal experience licensing products to pharmacies and outsourcing facilities and we may not be successful in our efforts to develop our licensing arrangements. If we elect to license our proprietary LDN formulation to one or more pharmacies or outsourcing facilities outside of the U.S., we may not be able to enter into licensing agreements when desired, on acceptable terms, or at all. Establishing licensing or other relationships with pharmacies and outsourcing facilities could be expensive and time consuming, disrupt our other operations, require significant capital expenditures and distract management and our other employees from other aspects of our business.
Failure to achieve and maintain effective internal controls could have a material adverse effect on our business.
Effective internal controls are necessary for us to safeguard our assets and provide reliable financial reports. If we cannot provide reliable financial reports, our operating results could be harmed. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
While we continue to evaluate and improve our internal controls, we are a small company with limited staff, and we cannot be certain that the measures we implement will ensure that we design, undertake and maintain adequate controls over our financial processes and reporting in the future. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations.
Failure to achieve and maintain an effective internal control environment could cause investors to lose confidence in our reported financial information, which could have a material adverse effect on our stock price. In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
| 57 |
Our independent registered public accounting firm has identified material weaknesses in our financial reporting process.
Our independent registered public accounting firm has identified two material weaknesses in our financial reporting process. Specifically, our independent registered public accounting firm identified material weaknesses with respect to:
| ● | currently inadequate segregation of duties by management in the financial reporting area; and | |
| ● | the lack of an audit committee to oversee the financial reporting process. |
We intend to remediate this weakness by increasing the size of our accounting staff in 2016 and by appointing an audit committee with membership that is qualified to oversee the Company’s financial reporting. However, there can be no assurance that we will be able to successfully implement our plans to remediate the material weaknesses in our financial reporting process. Our failure to successfully implement our plans to remediate these material weaknesses could cause us to fail to meet our reporting obligations, to produce timely and reliable financial information, and to effectively prevent fraud. Additionally, such failure, or other weaknesses that we may experience in our financial reporting process or other internal controls, could cause investors to lose confidence in our reported financial information, which could have a negative impact on our financial condition and stock price.
We will need to increase the size of our organization, but we may experience difficulties in managing growth.
We will need to continue to expand our managerial, operational, financial and other resources in order to manage our operations and clinical trials, continue our development activities and commercialize our drug candidates. Our current management, personnel systems and facilities may not be adequate to support this future growth. Our need to effectively execute our growth strategy requires that we:
| ● | manage our clinical trials effectively, including our clinical trials for IRT-103 (LDN) which will be conducted at numerous trial sites throughout the world; | |
| ● | manage our internal development efforts effectively while carrying out our contractual obligations to licensors, contractors, collaborators, government agencies and other third parties; | |
| ● | manage operations in both regulated and unregulated businesses; | |
| ● | continue to improve our operational, financial and management controls and reporting systems and procedures; and | |
| ● | identify, recruit, maintain, motivate and integrate additional employees. |
If we are unable to expand our managerial, operational, financial and other resources to the extent required to manage our development and commercialization activities, our business will be materially adversely affected.
We face substantial competition. Our competitors may discover, develop or commercialize products faster or more successfully than us.
The biotechnology and pharmaceutical industries are highly competitive. We face significant competition from companies in the pharmaceutical, biotechnology and other related markets that are researching and marketing products designed to address Crohn’s Disease, multiple sclerosis, other autoimmune diseases or immune disorders, inflammatory disorders, HIV/AIDS and cancer. Established pharmaceutical companies that currently sell or are developing drugs in our markets of interest include, for example; Abbott Laboratories, Amgen, AstraZeneca, Biogen Idec, Bayer, Elan, Johnson & Johnson, Merck, Merck Serono, Takeda, Novartis, Pfizer, Reata, Sanofi-Aventis and Teva. Many or all of these established competitors are also involved in research and drug development regarding various OGF receptors. Pharmaceutical and biotechnology companies which are known to be involved in immunotherapy research and related drug development include Pfizer, Bristol-Myers Squibb, Merck, Takeda, Sanofi-Aventis, Incyte, and UCB Pharma among others.
We are developing small molecule therapeutics that will compete with other drugs and alternative therapies that are currently marketed or are being developed to treat Crohn’s Disease, HIV/AIDS, other autoimmune diseases and inflammatory disorders, HIV/AIDS and cancer. If approved for marketing by the FDA, IRT-103 (LDN), our lead Inflammatory Bowel Disease (“IBD”) drug candidate, would compete against existing IBD treatments such as Sulfasalazine (Azulfidine); Mesalamine (Asacol, Rowasa) Corticosteroids; Azathioprine (Imuran) and mercaptopurine (Purinethol); Infliximab (Remicade); Adalimumab (Humira); Certolizumab pegol (Cimzia); Methotrexate (Rheumatrex); Cyclosporine (Gengraf, Neoral, Sandimmune) and Natalizumab (Tysabri). Similarly, other future drug candidates we are pursuing would compete against numerous existing and established drugs and potentially against other novel drugs and therapies that are currently in development. We also anticipate that we will face increased competition in the future as new companies enter our target markets and scientific developments surrounding the chemokine system continue to develop.
| 58 |
Many of our competitors have greater name recognition and financial, manufacturing, marketing, research and drug development resources than we do. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Large pharmaceutical companies in particular have extensive expertise in preclinical and clinical testing and in obtaining regulatory approvals for drugs. In addition, academic institutions, government agencies, and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies. These organizations may also establish exclusive collaborative or licensing relationships with our competitors, thus giving our competitors a significant advantage. We may be unable to respond to competitive forces presently in the marketplace, which would severely impact our business.
In addition, in terms of the licensing of our LDN formulation to Complete Pharmacy and Medical Solutions, LLC, we compete against branded drug companies, generic drug companies, outsourcing facilities and other compounding pharmacies. We are currently and expect to continue our efforts on making available our proprietary compounded formulation through Complete Pharmacy and Medical Solutions, LLC and other compounding pharmacies outside of the U.S. The drug products available through branded and generic drug companies with which our formulation competes have been approved for marketing and sale by the FDA and are required to be manufactured in facilities compliant with cGMP standards. As a result, some physicians may be unwilling to prescribe them. Because our proprietary LDN formulation is compounded in accordance with The U.S. Federal Food, Drug, and Cosmetic Act Section 503B and is not required to be, and has not been, approved for marketing and sale by the FDA, our business may be subject to limitations our competitors with FDA-approved drugs may not face.
Under state and federal laws applicable to compounding pharmacies, Complete Pharmacy and Medical Solutions, LLC is not permitted to prepare significant amounts of a specific formulation in advance of a prescription, compound quantities for office use or utilize a wholesaler for distribution for our formulation; instead, our compounded formulation must be prepared and dispensed in connection with a physician prescription for an individually identified patient. Pharmaceutical companies typically sell most of their FDA-approved products to large pharmaceutical wholesalers, who in turn sell to and supply hospitals and retail pharmacies. As a result, the sale of our formulation by Complete Pharmacy and Medical Solutions, LLC is not scalable on the scope available to our competitors with FDA-approved drugs, which may limit our potential for revenue.
We may be subject to costly product liability claims related to our clinical trials and drug candidates and, if we are unable to obtain adequate insurance or are required to pay for liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage, a material liability claim could adversely affect our financial condition.
Because we conduct clinical trials with human patients, we face the risk that the use of our drug candidates may result in adverse side effects to patients and to otherwise healthy volunteers in our clinical trials. We face even greater risks upon any commercialization of our drug candidates. Although we will maintain product liability insurance for clinical trials, our insurance may be insufficient to reimburse us for any expenses or losses we may suffer, and we will be required to increase our product liability insurance coverage for the advanced clinical trials that we plan to initiate. We do not know whether we will be able to continue to obtain product liability coverage and obtain expanded coverage on acceptable terms, or at all. We may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage. There is also a risk that third parties that we have agreed to indemnify could incur liability. An individual may bring a product liability claim against us if one of our drug candidates, products or compounded formulations cause, or is claimed to have caused, an injury or is found to be unsuitable for consumer use. Any product liability claim brought against us, with or without merit, could result in:
| 59 |
| ● | withdrawal of clinical trial volunteers, investigators, patients or trial sites; | |
| ● | the inability to commercialize our drug candidates; | |
| ● | decreased demand for our drug candidates; | |
| ● | regulatory investigations that could require costly recalls or product modifications; | |
| ● | loss of revenues; | |
| ● | substantial costs of litigation; | |
| ● | liabilities that substantially exceed our product liability insurance, which we would then be required to pay ourselves; | |
| ● | an increase in our product liability insurance rates or the inability to maintain insurance coverage in the future on acceptable terms, if at all; | |
| ● | the diversion of management’s attention from our business; and | |
| ● | damage to our reputation and the reputation of our products. |
Our business involves the use of hazardous materials. As a result, we, including our third party manufacturers, must comply with environmental laws and regulations, which may be expensive and restrict how we do business.
Our third party manufacturers’ activities and our own activities involve the controlled storage, use and disposal of hazardous materials, including the components of our pharmaceutical products, test samples and reagents, biological materials and other hazardous compounds. We and our manufacturers are subject to federal, state and local, and foreign laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these hazardous materials. We currently carry no insurance specifically covering environmental claims relating to the use of hazardous materials. Although we believe that our safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of an accident, state or federal or other applicable authorities may curtail our use of these materials and/or interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. The substantial unexpected costs we may incur could significantly harm our financial condition and results of operations.
Future financings may adversely affect our stockholders or impose restrictions on our assets or operations, which may harm our business.
If we raise additional capital by issuing equity securities or convertible debt securities, our existing stockholders’ ownership will be diluted and the terms of any new equity securities may have preferences over our common stock. If we raise additional capital through the issuance of debt securities, the debt will have rights senior to the holders of our common stock and may contain covenants that restrict our operational flexibility or impose liens or other restrictions on our assets. In addition, the terms of future financings may restrict our ability to raise additional capital, which would delay or prevent the further development or commercialization of our drug candidates.
If we raise additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish potentially valuable rights to our current drug candidates, potential products or proprietary technologies, or grant licenses on terms that are not favorable to us. Additionally, we may consider pursuing strategic opportunity for our business and corporate structure that may make us a more attractive investment candidate. If adequate funds are not available, our ability to achieve profitability or to respond to competitive pressures would be significantly limited and we may be required to delay, significantly curtail or eliminate the development of one or more of our drug candidates.
We may be adversely affected by the current economic environment.
Our ability to attract and retain collaboration partners or customers, invest in and grow our business and meet our financial obligations depends on our operating and financial performance, which, in turn, is subject to numerous factors, including the prevailing economic conditions and financial, business and other factors beyond our control, such as the rate of unemployment, the number of uninsured persons in the United States and inflationary pressures. We cannot anticipate all the ways in which the current economic climate and financial market conditions could adversely impact our business.
| 60 |
We are exposed to risks associated with reduced profitability and potential financial instability of our collaboration partners or customers, many of which may be adversely affected by volatile conditions in the financial markets. For example, unemployment and underemployment, and the resultant loss of insurance, may decrease the demand for healthcare services and pharmaceuticals. If fewer patients are seeking medical care because they do not have insurance coverage, our collaboration partners or customers may experience reductions in revenues, profitability and/or cash flow that could lead them to reduce their support of our programs or financing activities. If collaboration partners or customers are not successful in generating sufficient revenue or are precluded from securing financing, they may not be able to pay, or may delay payment of, accounts receivable that are owed to us. This, in turn, could adversely affect our financial condition and liquidity. To the extent economic challenges result in fewer individuals pursuing or being able to afford our products once commercialized, our business, results of operations, financial condition and cash flows could be adversely affected.
Our internal computer systems, or the computer systems of our contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our drug development programs.
Despite the implementation of security measures, our internal computer systems and the computer systems of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for any of our drug candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our drug candidates could be delayed.
Our current and future operations substantially depend on our management team and our ability to have other key personnel, the loss of any of whom could disrupt our business operations.
The Company’s future success depends on the efforts and abilities of principal members of its senior management and scientific staff to provide strategic direction, business development, operations management and maintenance of a cohesive and stable work environment. The Company relies on the services of Dr. Nicholas P. Plotnikoff and Professor Fengping Shan. If we lost their services or the services of any other key member of management, it could be impossible to replace them.
Additionally, the Company’s ability to maintain, expand and renew existing business with its clients and maximize potential business opportunities from new clients (in both the drug development and the drug discovery areas) depends on its ability to hire and retain scientists with necessary skills. The scientists working for the Company must have the ability to lead ahead of continuing changes and trends in drug discovery and development technologies to create the most innovative products on the market in order to remain competitive within the drug development industry. The Company faces risks, challenges and competition attracting and retaining experienced scientists and healthcare providers.
The Company’s inability to hire qualified personnel may increase the workload for both existing and new personnel. The Company may not be successful in attracting new healthcare providers, scientists and management or in retaining/motivating existing personnel. The shortage of experienced healthcare providers and scientists or other factors may lead to increased recruiting, relocation and compensation costs for the Company. Such increased costs may reduce profit margins or make hiring necessary experts (i.e. healthcare providers or scientists) impracticable. If the Company is unable to attract or retain any of its key personnel its ability to execute a competitive and profitable business plan will be adversely affected. Services and products will be less competitive if not obsolete. If competing companies introduce superior technologies that compete with the Company’s services and products, the Company may not be able to make the necessary enhancements to its services and products that will maintain a competitive position in the marketplace. The Company’s competitive position, business, revenues and financial condition will be materially and adversely affected.
| 61 |
Any failure by the Company to comply with existing health care and drug regulations could harm its reputation, operating results, the quality of the Company’s business strategy and the quality of the Company’s products.
The Company has not experienced any failure to comply and has not received any notice or violation of either good clinical practices, laboratory practices or good manufacturing practices. Any future failure by the Company to comply with existing health care and drug regulations could result in the termination of ongoing research and/or the disqualification of data for submission to regulatory authorities. Failure to comply with existing regulations will harm the Company’s reputation, brand name, its prospects for immediate and future work and its operating results. For example, if the Company fails to verify that informed consent is obtained from patient participants in connection with a particular clinical trial or grant deviations from the inclusion/exclusion criteria in a study protocol, the data collected from that trial could be disqualified at which point the Company may be required to conduct the trial again at no further cost to its client. Furthermore, the issuance of a FDA notice based on a finding of a material violation of good clinical practice, good laboratory practice or good manufacturing practice requirements could materially and adversely affect the Company.
Proposed and future legislation or regulation may increase the cost of the Company’s business or limit its service and product offerings.
Federal, state, and/or international authorities might adopt healthcare legislation or regulations that are more burdensome than existing regulations. For example, recent product safety concerns and the creation of the Drug Safety Oversight Board could change the regulatory environment for drug products including the process for FDA product approval and post-approval safety surveillance. Such changes and other possible changes in regulation could increase the Company’s expenses or limit its ability to offer some of its services or products. For example, the confidentiality of patient-specific information and the circumstances under which it may be released for inclusion in the Company’s databases or used in other aspects of business are subject to substantial government regulation. Additional legislation or regulation governing the possession, use and dissemination of medical record information or other personal health information may require the Company to implement new security measures requiring substantial expenditures or limiting the ability to offer services and products. These regulations might also increase costs by creating new privacy requirements for the Company’s business mandating additional privacy procedures for its clinical research business.
Requirements associated with being a public company will increase our costs significantly, as well as divert significant company resources and management attention.
Prior to June 2013, we were not subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). We are working with our legal, independent accounting and financial advisors to identify those areas in which changes should be made to our financial and management control systems to manage our growth and our obligations as a public company. These areas include corporate governance, corporate control, internal audit, disclosure controls and procedures and financial reporting and accounting systems. We have made, and will continue to make, changes in these and other areas. However, the expenses that will be required in order to adequately prepare for being a public company could be material.
Compliance with the various reporting and other requirements applicable to public companies will also require considerable time and attention of management. As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and NASDAQ, has imposed various requirements on public companies. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. In addition, the changes we make may not be sufficient to allow us to satisfy our obligations as a public company on a timely basis.
Moreover, we anticipate that compliance with these rules and regulations will increase our legal, accounting and financial compliance costs substantially. In addition, these rules and regulations may make our activities related to legal, accounting and financial compliance more difficult, time-consuming and costly and may also place undue strain on our personnel, systems and resources. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations.
| 62 |
In addition, being a public company could make it more difficult or more costly for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
We estimate the additional costs we may incur to respond to these requirements to range from $100 to $500 thousand annually, although unforeseen circumstances could increase actual costs.
As an “emerging growth company” under applicable law, we will be subject to lessened disclosure requirements, which could leave our stockholders without information or rights available to stockholders of more mature companies.
For as long as we remain an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (which we refer to herein as the JOBS Act), we have elected to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to:
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; |
| ● | taking advantage of an extension of time to comply with new or revised financial accounting standards; |
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements; and |
| ● | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We expect to take advantage of these reporting exemptions until we are no longer an “emerging growth company.” Because of these lessened regulatory requirements, our stockholders would be left without information or rights available to stockholders of more mature companies.
If we are unable to attract suitable and willing investigators and volunteers for clinical trials and product development, business may suffer.
Our clinical research studies rely on the accessibility and participation of physician investigators and volunteer subjects. Investigators are typically located at hospitals, clinics or other sites and supervise administration of the study drug to patients during the course of a clinical trial. Volunteer subjects generally include individuals from the locale where the studies are conducted. Our clinical research development business could be adversely affected if it is unable to attract suitable and willing investigators or volunteers on a consistent basis.
We may not obtain government approval for our products and/or uses.
The development and commercialization of pharmaceutical products are subject to extensive governmental regulation in the United States and foreign countries. Government approvals are required to develop, market and sell potential drug candidates. Obtaining government approval to develop, market and sell drug candidates is time-consuming and expensive. The clinical trial results for a particular drug candidate might not satisfy necessary requirements to obtain government approvals. Even if we are successful in obtaining all required approvals to market and sell a drug candidate, post-approval requirements and the failure to comply with other regulations could result in suspension or limitation of government approvals.
In connection with drug discovery activities outside of the United States, we and our strategic partners will be subject to foreign regulatory requirements governing testing, approval, manufacturing, labeling, marketing and sale of pharmaceutical products. These requirements vary with location. Even if approval has been obtained for a product in the United States, approval in a foreign country must be obtained prior to marketing the product. The approval process in foreign countries may be more or less rigorous than the United States and the time required for approval may be longer or shorter. Clinical studies conducted outside of a specific country may not be acceptable. The approval of a pharmaceutical product in one country does not guarantee approval in another.
| 63 |
Even if approved, the products that we may develop and market may be later withdrawn from the market or subject to promotional limitations.
We may not be able to obtain the labeling claims necessary or desirable for the promotion of our treatments if approved. We may also be required to undertake post-marketing clinical trials. If the results of such post-marketing studies are not satisfactory or if adverse events or other safety issues arise after approval, the FDA or a comparable regulatory agency in another country may withdraw marketing authorization or may condition continued marketing on commitments from us that may be expensive and/or time consuming to complete. In addition, if we or others identify adverse side effects after any of our products are on the market, or if manufacturing problems occur, regulatory approval may be withdrawn and reformulation of our products, additional clinical trials, changes in labeling of our products and additional marketing applications may be required. Any reformulation or labeling changes may limit the marketability of our products if approved.
Florida Law and our Articles of Incorporation may protect our Directors and Officers from certain types of lawsuits.
Florida law provides that our officers and directors will not be liable to us or our stockholders for monetary damages for all but certain types of conduct as officers and directors. Our Bylaws permit us broad indemnification powers to all persons against all damages incurred in connection with our business to the fullest extent provided or allowed by law. The exculpation provisions may have the effect of preventing stockholders from recovering damages against our officers and directors caused by their negligence, poor judgment or other circumstances. The indemnification provisions may require us to use our limited assets to defend our officers and directors against claims, including claims arising out of their negligence, poor judgment or other circumstances.
We may seek to grow our business through acquisitions of or investments in new or complementary businesses, products or technologies, and the failure to manage acquisitions or investments, or the failure to integrate them with our existing business, could have a material adverse effect on us.
From time to time we expect to consider opportunities to acquire or make investments in other technologies, products and businesses that may enhance our capabilities, complement our current products or expand the breadth of our markets or customer base. Potential and completed acquisitions and strategic investments involve numerous risks, including:
| ● | problems assimilating the purchased technologies, products or business operations; | |
| ● | issues maintaining uniform standards, procedures, controls and policies; | |
| ● | unanticipated costs associated with acquisitions; | |
| ● | diversion of management’s attention from our core business; | |
| ● | adverse effects on existing business relationships with suppliers and customers; | |
| ● | risks associated with entering new markets in which we have limited or no experience; | |
| ● | potential loss of key employees of acquired businesses; and | |
| ● | increased legal and accounting compliance costs. |
We have no current commitments with respect to any acquisition or investment. We do not know if we will be able to identify acquisitions we deem suitable, whether we will be able to successfully complete any such acquisitions on favorable terms or at all, or whether we will be able to successfully integrate any acquired business, product or technology into our business or retain any key personnel, suppliers or distributors. Our ability to successfully grow through acquisitions depends upon our ability to identify, negotiate, complete and integrate suitable target businesses and to obtain any necessary financing. These efforts could be expensive and time-consuming, and may disrupt our ongoing business and prevent management from focusing on our operations. If we are unable to integrate any acquired businesses, products or technologies effectively, our business, results of operations and financial condition will be materially adversely affected.
We may expend our limited resources to pursue a particular opportunity and fail to capitalize on current research and products that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we have focused on specific research programs, treatments, and products. As a result, we may forego or delay pursuit of opportunities with other products or research that later may prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial treatments or profitable market opportunities. Our spending on current and future research and development programs may not yield any commercially viable treatments.
| 64 |
We are subject to risks associated with our non-U.S. operations.
The Foreign Corrupt Practices Act (“FCPA”) and similar worldwide anti-bribery laws in non-U.S. jurisdictions generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. The FCPA also imposes accounting standards and requirements on publicly traded U.S. corporations and their foreign affiliates which are intended to prevent the diversion of corporate funds to the payment of bribes and other improper payments, and to prevent the establishment of “off books” slush funds from which such improper payments can be made. Because of the predominance of government-sponsored healthcare systems around the world, many of our customer relationships outside of the United States are with governmental entities and are therefore subject to such anti-bribery laws. Our internal control policies and procedures may not always protect us from reckless or criminal acts committed by our employees or agents. Violations of these laws, or allegations of such violations, could disrupt our operations, involve significant management distraction and result in a material adverse effect on our business, results of operations and financial condition. We also could suffer severe penalties, including criminal and civil penalties, disgorgement and other remedial measures, including further changes or enhancements to our procedures, policies and controls, as well as potential personnel changes and disciplinary actions.
Furthermore, we are subject to the export controls and economic embargo rules and regulations of the United States, including, but not limited to, the Export Administration Regulations and trade sanctions against embargoed countries, which are administered by the Office of Foreign Assets Control within the Department of the Treasury, as well as the laws and regulations administered by the Department of Commerce. These regulations limit our ability to market, sell, distribute or otherwise transfer our products or technology to prohibited countries or persons. A determination that we have failed to comply, whether knowingly or inadvertently, may result in substantial penalties, including fines and enforcement actions and civil and/or criminal sanctions, the disgorgement of profits, the imposition of a court-appointed monitor, the denial of export privileges and/or an adverse effect on our reputation.
These and other factors may have a material adverse effect on our international operations or on our business, results of operations and financial condition generally.
Because our some of our manufacturing activities occur in Nicaragua, which is subject to political, economic and other uncertainties, situations may arise that could have a material adverse effect on our business.
The status of Nicaragua as a developing country may make it difficult for us to obtain additional financing for our projects. Notwithstanding the progress achieved in recent years in political institutions and revitalizing the Nicaraguan economy, the present administration, or any successor government, may not be able to sustain the progress achieved. While the Nicaraguan economy has experienced growth in recent years, such growth may not continue in the future at similar rates or at all. If the economy of Nicaragua fails to continue its growth or suffers a recession, our manufacturing efforts may be affected.
Further, Nicaragua has in the past experienced a difficult security environment as well as political instability. In particular, various illegal groups that may be active in and around regions in which we are present may pose a credible threat of terrorism, extortion and kidnapping, which could have an adverse effect on our operations in such regions. In the event that continued operations in these regions compromise our security or business principles, we may withdraw from these regions on a temporary or permanent basis, which in turn, could have an adverse impact on our results of operations and financial condition. Any changes in regulations or shifts in political attitudes are beyond our control and may adversely affect our business.
Our independent auditors have expressed substantial doubt about our ability to continue as a going concern.
Due to our net losses, negative cash flow and negative working capital, in their report on our audited financial statements for the years ended December 31, 2015 and 2014, our independent auditors included an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern.
| 65 |
We have incurred substantial losses since inception. Because of these losses, we will require additional working capital to develop our business operations. We intend to raise additional working capital through private placements, public offerings, bank financing and/or advances from related parties or shareholder loans.
There are no assurances that we will be able to achieve a level of revenues adequate to generate sufficient cash flow from operations. To the extent that funds generated from operations and any private placements, public offerings and/or bank financing are insufficient, we will have to raise additional working capital. No assurance can be given that additional financing will be available, or, if available, will be on terms acceptable to us. If adequate working capital is not available we may not increase our operations.
These conditions raise substantial doubt about our ability to continue as a going concern. The financial statements do not include any adjustments relating to the recoverability and classification of asset carrying amounts or the amount and classification of liabilities that might be necessary should we be unable to continue as a going concern.
Risks Related to Intellectual Property
Our inability to adequately protect our intellectual property rights could hurt business.
Our commercial success will depend in part on obtaining and maintaining intellectual property protection for our products, formulations, processes, methods and other technologies. We will only be able to protect these technologies and products from unauthorized use by third parties to the extent that valid and enforceable intellectual property rights, including patents or other market exclusionary rights apply.
The patent positions of pharmaceutical companies, like ours, can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States. The general environment for pharmaceutical patents outside the United States also involves significant uncertainty. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced, or that the scope of these patent rights could provide a sufficient degree of future protection that could permit us to gain or keep our competitive advantage with respect to these products and technologies. For example, we cannot predict:
| ● | the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to make, use, sell, offer to sell or import competitive products without infringing our patents; | |
| ● | if and when patents will issue; | |
| ● | whether or not others will obtain patents claiming inventions similar to those covered by our patents and patent applications; or | |
| ● | whether we will need to initiate litigation or administrative proceedings in connection with patent rights, which may be costly whether we win or lose. |
Some of our patents we have licensed may be subject to challenge and possibly invalidated or rendered unenforceable by third parties. Changes in either the patent laws or in the interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property.
In addition, others may independently develop similar or alternative products and technologies that may be outside the scope of our intellectual property. Furthermore, others may have invented technology claimed by our patents before our licensors or we did so, and they may have filed patents claiming such technology before we did so, weakening our ability to obtain and maintain patent protection for such technology. Should third parties obtain patent rights to similar products or technology, this may have an adverse effect on our business.
We may also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. Trade secrets, however, are difficult to protect. While we believe that we will use reasonable efforts to protect our trade secrets, our own or our strategic partners’ employees, consultants, contractors or advisors may unintentionally or willfully disclose our information to competitors. We seek to protect this information, in part, through the use of non-disclosure and confidentiality agreements with employees, consultants, advisors and others. These agreements may be breached, and we may not have adequate remedies for a breach. In addition, we cannot ensure that those agreements will provide adequate protection for our trade secrets, know-how or other proprietary information or prevent their unauthorized use or disclosure.
| 66 |
If competitors that have greater experience and financial resources learn our trade secrets, the competitors may copy or use our trade secrets and other proprietary information in the advancement of their products, methods or technologies. If we were to prosecute a claim that a third party had illegally obtained and was using our trade secrets, it could be expensive and time consuming and the outcome could be unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets than courts in the United States. Moreover, if our competitors independently develop equivalent knowledge, we would lack any legal or contractual claim to prevent them from using such information, and our business could be harmed.
The Company’s most important intellectual property includes:
| ● | For IRT - 103 for Crohn’s disease, Patent Number 7879870, filed April 16, 2007, issued February 1, 2011, Methods for the treatment of inflammatory and ulcerative diseases of the bowel (e.g., Crohn’s disease and ulcerative colitis) with low dose opioid antagonists (e.g., naltrexone, nalmefene or naloxone), pharmaceutical compositions for use in such methods, and methods for the manufacture of such pharmaceutical compositions. | |
| ● | We depend extensively on our license agreement with Pennsylvania State University for the development of IRT-101 for pancreatic cancer covered by patents US Patent Numbers 6,737,397, CA 2,557,504, US 20010046968 , US 6737397 , US 6136780 , US 20080015211 , US 20070053838 , US 8003630 , US 20110123437 , US 7807368 , US 7576180 , US 7517649, US 20080146512 , US 7122651 , US 20060073565 , US 20050191241 , Patent No 8,003,630 issued between 2001 and 2011. Our license agreement with Pennsylvania State University may be terminated if we materially breach the agreement and fail to cure our breach during an applicable cure period. Our failure to use commercially reasonable efforts to develop and commercialize OGF sometimes referred to as MENK (intravenous) and IRT-101 in the United States and certain other specified countries or to perform our other diligence obligations under the license agreement would constitute a material breach of the license agreement. In the event our license agreement with Pennsylvania State University is terminated, we will lose all of our rights to develop and commercialize the drug candidates covered by such license, which would harm our business and future prospects. We own a number of other patents having to do with the development of MENK which would allow us to continue our development of those indications. |
We may become subject to intellectual property suits that could cause us to incur significant costs or pay significant damages or that could prohibit us from selling its products.
The Company’s competitors also seek to obtain patents or other protection of their proprietary rights. Third parties may claim in the future that the Company’s products infringe upon their proprietary rights. To date, there have been no claims of infringement. However, in the future, intellectual property claims could force the Company to alter its existing products or withdraw them from the market or could delay the introduction of new products.
Various patents have been issued to the Company’s competitors and these competitors may assert that the Company’s products infringe their patent or other proprietary rights. If the Company’s products are found to infringe third-party intellectual property rights, the Company may be unable to obtain a license to use such technology, and it could incur substantial costs to redesign its products or to defend legal actions.
The drug discovery and development industry has a history of patent and other intellectual property litigation; thus, we may be involved in costly intellectual property lawsuits.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. We or one of our collaborators may be subject to third party claims in the future that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing a third party’s patents. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or drug candidate that is the subject of the suit. As a result of patent infringement claims, or in order to avoid potential claims, we or our collaborators may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or forced to redesign it, or to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly.
| 67 |
In addition to infringement claims against us, if third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings with the United States Patent and Trademark Office (“USPTO”) to determine the priority of invention. We may also become involved in similar opposition proceedings in the European Patent Office regarding our intellectual property rights with respect to our products and technology.
The failure to obtain or maintain patents, licensing agreements and other intellectual property could impact our ability to compete effectively.
Our success will depend, in part, on our ability to obtain and maintain patent protection for our drug candidates, preserve our trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others. While the patents we own have been issued, pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents. We cannot be certain that patents will be issued as a result of any of our pending applications, and we cannot be certain that any of our issued patents, whether issued pursuant to our pending applications or licensed from third parties, will give us adequate protection from competing products.
Composition of Matter patents on APIs are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, as they apply without regard to any method of use. Entirely new individual chemical compounds, often referred to as new chemical entities, are typically entitled to Composition of Matter coverage. However, we cannot be certain that the current law will remain the same, or that our drug candidates will be considered novel and non-obvious by the USPTO and courts.
In addition to Composition of Matter patents and patent applications, we also have filed Method of Use patent applications. This type of patent protects the use of the product only for the specified method. However, this type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if these competitors do not actively promote their product for our targeted indication, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of Method of Use patents, the practice is common and such infringement is difficult to prevent or prosecute.
Patent applications in the United States and most other countries are confidential for a period of time until they are published. The publication of discoveries in scientific or patent literature typically lags actual discoveries by several months or more. As a result, we cannot be certain whether the Company or another inventor were the inventors of the issued patents and applications or that the Company or another inventor were the first to conceive of the inventions covered by such patents and pending patent applications or that the Company or another inventor were the first to file patent applications covering such inventions.
Others may obtain issued patents that could prevent us from commercializing our product candidates or require us to obtain licenses requiring the payment of considerable fees or royalties in order to enable us to conduct our business. As to those patents that we have licensed, our rights depend on maintaining our obligations to the licensor under the applicable license agreement, and we may be unable to do so.
We have numerous issued patents and some patent applications pending before the USPTO. The protection may lapse before we manage to obtain commercial value from the patents, which might result in increased competition and materially affect our position in the market.
| 68 |
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Many of our employees were previously employed at universities, biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper our ability to commercialize, or prevent us from commercializing our drug candidates, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Some of our intellectual property that was discovered through government funded programs may be subject to federal regulation such as “march-in” rights, certain reporting requirements, and a preference for United States industry. Compliance with such regulations may limit our exclusive rights, subject us to expenditure of resources with respect to reporting requirements, and limit our ability to contract with foreign manufacturers.
Some of our existing drug candidates, including LDN and MENK, and some of the research and development work conducted before we had licensing rights may have been funded, at least in part, by the U.S. government and therefore would be subject to certain federal regulations. Under the “march-in” provisions of the Bayh-Dole Act, the government may have the right under limited circumstances to require the patent owners to grant exclusive, partially exclusive or non-exclusive rights to third parties for intellectual property discovered through the government-funded program. The government can exercise its march-in rights if it determines that action is necessary because the patent owner fails to achieve practical application of the new invention or because action is necessary to alleviate health concerns or address the safety needs of the public. Intellectual property discovered under the government-funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. Such intellectual property is also subject to a preference for U.S. industry, which may limit our ability to contract with foreign product manufacturers for products covered by such intellectual property. We may apply for additional U.S. government funding, and it is possible that we may discover compounds or drug candidates as a result of such funding. Intellectual property under such discoveries would be subject to the applicable provisions of the Bayh-Dole Act.
Risks Related to Government Regulation
The regulatory approval process is expensive, time consuming and uncertain and may prevent us or our collaboration partners from obtaining approvals for the commercialization of some or all of our drug candidates.
The research, testing, manufacturing, labeling, approval, selling, import, export, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries which regulations differ from country to country. Neither we nor our collaboration partners are permitted to market our drug candidates in the United States until we receive approval of a NDA from the FDA. Neither we nor our collaboration partners have submitted an application for or received marketing approval for any of our drug candidates. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including:
| ● | warning letters; | |
| ● | civil and criminal penalties; | |
| ● | injunctions; | |
| ● | withdrawal of approved products; | |
| ● | product seizure or detention; | |
| ● | product recalls; | |
| ● | total or partial suspension of production; and | |
| ● | refusal to approve pending NDAs or supplements to approved NDAs. |
| 69 |
Regulatory approval of an NDA or NDA supplement is not guaranteed, and the approval process is expensive and may take several years. The FDA also has substantial discretion in the approval process. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon or repeat clinical trials, or perform additional preclinical studies and clinical trials. The number of preclinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including, but not limited to, the following:
| ● | a drug candidate may not be deemed safe or effective; | |
| ● | FDA officials may not find the data from preclinical studies and clinical trials sufficient; | |
| ● | the FDA might not approve our or our third party manufacturer’s processes or facilities; or | |
| ● | the FDA may change its approval policies or adopt new regulations. |
If any of our drug candidates fail to demonstrate safety and efficacy in clinical trials or do not gain regulatory approval, our business and results of operations will be materially and adversely harmed.
Even if we receive regulatory approval for a drug candidate, we will be subject to ongoing regulatory obligations and continued regulatory review which may result in considerable additional expense and subject us to penalties if we fail to comply with applicable regulatory requirements.
Once regulatory approval has been granted, the approved product and its manufacturer are subject to continual review by the FDA and/or non-U.S. regulatory authorities. Any regulatory approval that we or our collaboration partners receive for our drug candidates may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies to monitor the safety and efficacy of the product. In addition, if the FDA and/or non-U.S. regulatory authorities approve any of our drug candidates, we will be subject to extensive and ongoing regulatory requirements by the FDA and other regulatory authorities with regard to the labeling, packaging, adverse event reporting, storage, advertising, promotion and recordkeeping for our products. In addition, manufacturers of our drug products are required to comply with current cGMP regulations which include requirements related to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory authorities must approve these manufacturing facilities before they can be used to manufacture our drug products, and these facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory authority discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory authority may impose restrictions on that product, the manufacturer or us, including requiring withdrawal of the product from the market or suspension of manufacturing. If we, our drug candidates or the manufacturing facilities for our drug candidates fail to comply with regulatory requirements of the FDA and/or other non-U.S. regulatory authorities, we could be subject to administrative or judicially imposed sanctions, including:
| ● | warning letters; | |
| ● | civil or criminal penalties; | |
| ● | injunctions; | |
| ● | suspension of or withdrawal of regulatory approval; | |
| ● | suspension of any ongoing clinical trials; | |
| ● | voluntary or mandatory product recalls and publicity requirements; | |
| ● | refusal to approve pending applications for marketing approval of new drugs or supplements to approved applications filed by us; | |
| ● | restrictions on operations, including costly new manufacturing requirements; or | |
| ● | seizure or detention of our products or import bans. |
| 70 |
The regulatory requirements and policies may change and additional government regulations may be enacted for which we may also be required to comply. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or in other countries. If we are not able to maintain regulatory compliance, we will not be permitted to market our future products and our business will suffer.
The availability of adequate third-party coverage and reimbursement for newly approved drugs is uncertain, and failure to obtain adequate coverage and reimbursement from third-party payers could impede our ability to market any future products we may develop and could limit our ability to generate revenue.
There is significant uncertainty related to the third-party payor coverage and reimbursement of newly approved drugs. The commercial success of our future products in both domestic and international markets depends on whether such third-party coverage and reimbursement is available for our future products. Governmental payers, including Medicare and Medicaid, health maintenance organizations and other third-party payers are increasingly attempting to manage their healthcare expenditures by limiting both coverage and the level of reimbursement of new drugs and, as a result, they may not cover or provide adequate reimbursement for our future products. These payers may not view our future products as cost-effective, and coverage and reimbursement may not be available to our customers or may not be sufficient to allow our future products to be marketed on a competitive basis. Third-party payers are exerting increasing influence on decisions regarding the use of, and coverage and reimbursement levels for, particular treatments. Such third-party payers, including Medicare, are challenging the prices charged for medical products and services, and many third-party payers limit or delay coverage and reimbursement for newly approved healthcare products. In particular, third-party payers may limit the covered indications. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated product revenues. If the prices for our drug candidates decrease or if governmental and other third-party payers do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
Even if we obtain FDA approval of any product candidate we may develop or acquire in the future, we may never obtain approval or commercialize our products outside of the U.S., which would limit our ability to realize their full market potential. If foreign approval is obtained, there are risks in conducting business in international markets.
We have and continue to seek other distribution and marketing partners for IRT-101 and IRT-103 (LDN) outside North America that will and may market future products in international markets. In order to market any of our products we may develop or acquire outside of the U.S., we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and require additional preclinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in the U.S. or any foreign country may delay or have negative effects on the process for regulatory approval in other countries. If we fail to comply with regulatory requirements in a foreign country or to obtain and maintain required approvals, our potential market for our products will be reduced and our ability to realize the full market potential of our products will be harmed.
The Company, either directly or through its collaborating partners, is working with drug regulatory authorities in Nicaragua, China and in those African Nations where an FDA equivalent exists. The Company is working with the agencies to obtain local approval for the therapies for each modality that we intend to market for. We believe this will reduce our risk due to The Agreement on Trade Related Aspects of Intellectual Property Rights (“TRIPS”) which is an international agreement administered by the World Trade Organization (“WTO”). TRIPS allows emerging nations to manufacture drugs around existing patents.
| 71 |
If approved for commercialization in a foreign country, we intend to enter into agreements with third parties to market our products whenever they may be approved and wherever we have the right to market them. Consequently, we expect that we will be subject to additional risks relating to entering into international business relationships, including:
| ● | lack of adequate protection from intellectual property rights in foreign countries, which could occur if we do not have issued patents in force in such foreign countries covering our products, their methods of use and methods of manufacture; | |
| ● | the potential for so-called parallel importing, which is what happens when a local seller, faced with high or higher local prices (for instance, because the goods have patent protection in such country), opts to import goods from a foreign market (with low or lower prices) rather than buy them locally; | |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements | |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
| ● | compliance with laws for employees traveling abroad; | |
| ● | foreign taxes, including withholding of payroll taxes; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues; | |
| ● | workforce uncertainty in countries where labor unrest is more common than in the U.S.; | |
| ● | production shortages resulting from any events affecting the API and/or finished drug product supply or manufacturing capabilities abroad; | |
| ● | business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires; and | |
| ● | failure to comply with Office of Foreign Asset Control rules and regulations and the Foreign Corrupt Practices Act |
These and other risks may materially adversely affect our ability to attain or sustain revenue from international markets.
Healthcare policy changes may have a material adverse effect on us.
Our business may be affected by the efforts of government and third-party payers to contain or reduce the cost of healthcare through various means. For example, the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the Affordable Care Act or ACA), enacted in March 2010, substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacted the pharmaceutical industry. With regard to pharmaceutical products, among other things, ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare D program. ACA has been held constitutional. This adds to the uncertainty of the legislative changes enacted as part of ACA, and we cannot predict the impact that ACA or any other legislative or regulatory proposals will have on our business. We expect both government and private health plans to continue to require healthcare providers, including healthcare providers that may one day purchase our products, to contain costs and demonstrate the value of the therapies they provide.
If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected.
Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payers, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The regulations that may affect our ability to operate include, without limitation:
| ● | the federal healthcare program Anti-Kickback Statute, which prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; | |
| ● | the federal False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, false claims, or knowingly using false statements to obtain payment from the federal government, and which may apply to entities like us which may provide coding and billing advice to customers; | |
| ● | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; and | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; and state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
| 72 |
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert Management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
The FDA may not accept the results of clinical trials conducted outside of the United States.
It is possible that the FDA may not accept the results of our clinical trials; and this risk can increase when a clinical trial is conducted outside of the United States. All clinical trials and clinical trial sites that are outside of the United States but will be used to support a FDA application will be run in accordance with all US guidelines and regulations; however, this does not guarantee the FDA’s acceptance of the clinical trial results. Clinical studies to support US licensure will only be conducted in countries that are typically used to support a US licensure such as Canada, Australia, and countries within the EU. We would need to obtain approval from the FDA to conduct the trial outside of the United States and/or to allow clinical sites outside of the US, prior to initiation of such study. We would also need to ensure that the study is conducted in accordance with local legal and regulatory requirements and all applicable United States federal regulations, European Union regulations, International Conference on Harmonisation of Good Clinical Practice guidelines and any other applicable regulatory requirements for the overall conduct of the clinical investigation.
Risks Related to our Common Stock
Because of their significant stock ownership, our chief executive officer, our other executive officers, and our directors and principal stockholders may be able to exert control over us and our significant corporate decisions. Our other stockholders will have limited ability to influence corporate actions or decisions.
This concentration of ownership may harm the value of our common stock by, among other things:
| ● | delaying, deferring or preventing a change in control of our company; | |
| ● | impeding a merger, consolidation, takeover or other business combination involving our company; or | |
| ● | causing us to enter into transactions or agreements that are not in the best interests of all stockholders |
As a group, our officers and directors own 14.0% of the outstanding common stock of the Company. Our other stockholders will have limited ability to influence corporate actions or decisions.
The price of our common stock may be volatile, and you may not be able to resell your shares.
An active and liquid trading market for our common stock may not develop or be sustainable. Shareholders may be unable to sell shares of common stock at or above their purchase price due to fluctuations in the market price of our common stock. The market price of our common stock may fluctuate significantly in response to factors, some of which are beyond our control. Factors that could cause volatility in the market price of our common stock include, but are not limited to:
| ● | results from, and delays in, clinical trial programs relating to our drug candidates, including the ongoing and planned clinical trials for IRT-103 (LDN), IRT-101 (MENK) and other drug candidates; |
| 73 |
| ● | announcements of regulatory approvals or disapprovals of our drug candidates including IRT-103 (LDN) and IRT-101 (MENK) or delays in any regulatory agency review or approval processes; | |
| ● | failure or discontinuation of any of our research programs; | |
| ● | loss of significant clients or customers; | |
| ● | loss of significant strategic relationships; | |
| ● | announcements relating to future collaborations or our existing collaborations; | |
| ● | our failure to achieve and maintain profitability; | |
| ● | changes in earnings estimates and recommendations by financial analysts; | |
| ● | changes in market valuations of similar companies; | |
| ● | wholesalers’ buying patterns; | |
| ● | addition or termination of clinical trials or funding support; | |
| ● | regulatory developments affecting our drug candidates or those of our competitors; | |
| ● | the Company’s sales decrease internationally; | |
| ● | variations in the level of expenses related to our drug candidates or future development programs; | |
| ● | ability to secure new government contracts and allocation of our resources to or away from performing work under government contracts; | |
| ● | general economic conditions in the United States and abroad; | |
| ● | acquisitions and sales of new products, technologies or business; | |
| ● | market conditions in the pharmaceutical, biopharmaceutical and biotechnology sectors; | |
| ● | the issuance of new or changed securities analysts’ reports or recommendations regarding us, our competitors or our industry in general; | |
| ● | actual and anticipated fluctuations in our quarterly operating results; | |
| ● | disputes concerning our intellectual property or other proprietary rights; | |
| ● | introduction of technological innovations or new products by us or our competitors; | |
| ● | manufacturing issues related to our drug candidates for clinical trials or future products for commercialization; | |
| ● | market acceptance of our future products; | |
| ● | deviations in our operating results from the estimates of analysts; | |
| ● | third party payor coverage and reimbursement policies; | |
| ● | new legislation in the United States relating to the sale or pricing of pharmaceuticals; | |
| ● | FDA or other U.S. or foreign regulatory actions affecting us or our industry; | |
| ● | product liability claims or other litigation or public concern about the safety of our drug candidates or future drugs; | |
| ● | our ability to obtain necessary intellectual property licenses including, if necessary, those relating to IRT-103 (LDN) and other drug candidates; | |
| ● | the outcome of any future legal actions to which we are a party; | |
| ● | sales of our common stock by our officers, directors or significant stockholders; | |
| ● | frequent, irregular, under market, or large sales of shares of our common stock by any shareholder; | |
| ● | additions or departures of key personnel; and | |
| ● | external factors, including natural disasters and other crises. |
In addition, the stock markets in general, and the markets for pharmaceutical, biopharmaceutical and biotechnology stocks in particular, have experienced extreme volatility that has often been unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation suits against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
Future sales of our common stock or securities convertible or exchangeable for our common stock may depress our stock price.
If our existing stockholders or holders of our convertible notes, options or warrants sell, or indicate an intention to sell substantial amounts of our common stock in the public market, the trading price of our common stock could decline. The perception in the market place that these sales may occur could also cause the trading price of our common stock to decline.
| 74 |
Certain holders of shares of our common stock, warrants to purchase our common stock, and shares of common stock issuable upon exercise of warrants will be entitled to rights with respect to the registration of their shares under the Securities Act. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates. In addition, our directors may, and we expect that our executive officers will establish programmed selling plans under Rule 10b5-1 of the Exchange Act, for the purpose of effecting sales of our common stock. Any sales of securities by these stockholders, or the perception that those sales may occur, including the entry into such programmed selling plans, could have a material adverse effect on the trading price of our common stock.
If we sell shares of our common stock in future financings, common stockholders may experience immediate dilution and, as a result, our stock price may decline.
We may from time to time issue additional shares of common stock at a discount from the current trading price of our common stock. As a result, our common stockholders would experience immediate dilution upon the purchase of any shares of our common stock sold at such a discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock.
Provisions of our charter documents or Florida law could delay or prevent an acquisition of the Company, even if the acquisition would be beneficial to our stockholders, and could make it more difficult for stockholders to change management.
Provisions of our amended and restated articles of incorporation, as amended, and amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. In addition, these provisions may frustrate or prevent any attempt by our stockholders to replace or remove our current management by making it more difficult to replace or remove our board of directors.
We do not anticipate paying any cash dividends on our capital stock in the foreseeable future, therefore capital appreciation, if any, of our common stock will be our shareholders sole source of gain for the foreseeable future.
We have never declared or paid cash dividends on our capital stock. We do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of the common stock will be our shareholders sole source of gain for the foreseeable future.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend, in part, on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on our Company. If no securities or industry analysts commence coverage of our Company, the trading price for our stock would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. In addition, if our operating results fail to meet the forecast of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of our Company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
| 75 |
Our board of directors is authorized to issue and designate shares of our preferred stock in additional series without stockholder approval.
Our amended and restated articles of incorporation, as amended, authorize our board of directors, without the approval of our stockholders, to issue shares of our preferred stock, subject to limitations prescribed by applicable law, rules and regulations and the provisions of our amended and restated articles of incorporation, as amended, as shares of preferred stock in series, and to establish from time to time the number of shares to be included in each such series, and to fix the designation, powers, preferences and rights of the shares of each such series and the qualifications, limitations or restrictions thereof. The powers, preferences and rights of these additional series of preferred stock may be senior to or on parity with our common stock, which may reduce its value. We do not currently have any class of preferred stock authorized.
Our shares may be subject to the “penny stock” rules, which may subject you to restrictions on marketability and limit your ability to sell your shares.
Broker-dealer practices in connection with transactions in “Penny Stocks” are regulated by certain penny stock rules adopted by the SEC. Penny stocks generally are equity securities with a price of less than $5.00 per share (other than securities registered on certain national securities exchanges or quoted on the NASDAQ system). The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document that provides information about penny stocks and the risk associated with the penny stock market. The broker-dealer must also provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer’s account. In addition, the penny stock rules generally require that prior to a transaction in a penny stock, the broker-dealer must make a written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for a stock that becomes subject to the penny stock rules. The Company’s securities may be subject to the penny stock rules, and investors may find it more difficult to sell their securities.
An active and visible trading market for our common stock may not develop.
We cannot predict whether an active market for our common stock will develop in the future. In the absence of an active trading market:
| ● | Investors may have difficulty buying and selling or obtaining market quotations; | |
| ● | Market visibility for our common stock may be limited; and | |
| ● | A lack of visibility for our common stock may have a depressive effect on the market price for our common stock |
Our common stock is currently quoted on the OTC Market under the trading symbol “IMUN”. The OTC Market is unorganized, inter-dealers, over-the-counter markets that provides significantly less liquidity than the New York Stock Exchange or NASDAQ. No assurances can be given that we will ever obtain a listing for our securities on a senior exchange. The trading price of our common stock is therefore expected to be subject to significant fluctuations in response to variations in quarterly operating results, changes in analysts’ earnings estimates, announcements of innovations by us or our competitors, general conditions in the industry in which we operate and other factors. These fluctuations, as well as general economic and market conditions, may have a material or adverse effect on the market price of our common stock.
Because we have elected to use the extended transition period for complying with new or revised accounting standards for an “emerging growth company” our financial statements may not be comparable to companies that comply with public company effective dates.
We have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates, and thus investors may have difficulty evaluating or comparing our business, performance or prospects in comparison to other public companies, which may have a negative impact on the value and liquidity of our common stock.
| 76 |
We may not register or qualify our securities with any state agency pursuant to blue sky regulations.
The holders of our shares of common stock and persons who desire to purchase them in the future should be aware that there may be significant state law restrictions upon the ability of investors to resell our shares. We currently do not intend to and may not be able to qualify securities for resale in other states which require shares to be qualified before they can be resold by our shareholders.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Certain statements contained or incorporated by reference in this Form S-1 are considered forward-looking statements (within the meaning of the Private Securities Litigation Reform Act of 1995) concerning our business, results of operations, economic performance and/or financial condition, based on management’s current expectations, plans, estimates, assumptions and projections. Forward-looking statements are included, for example, in the discussions about:
| ● | strategy; | |
| ● | new product discovery and development; | |
| ● | current or pending clinical trials; | |
| ● | our products’ ability to demonstrate efficacy or an acceptable safety profile; | |
| ● | actions by the FDA and other regulatory authorities; | |
| ● | product manufacturing, including our arrangements with third-party suppliers; | |
| ● | product introduction and sales; | |
| ● | royalties and contract revenues; | |
| ● | expenses and net income; | |
| ● | credit and foreign exchange risk management; | |
| ● | liquidity; | |
| ● | asset and liability risk management; | |
| ● | the outcome of litigation and other proceedings; | |
| ● | intellectual property rights and protection; | |
| ● | economic factors; | |
| ● | competition; and | |
| ● | legal risks. |
Any statements contained in this report that are not statements of historical fact may be deemed forward-looking statements. Forward-looking statements generally are identified by the words “expects,” “anticipates,” “believes,” “intends,” “estimates,” “aims,” “plans,” “may,” “could,” “will,” “will continue,” “seeks,” “should,” “predict,” “potential,” “outlook,” “guidance,” “target,” “forecast,” “probable,” “possible” or the negative of such terms and similar expressions. Forward-looking statements are subject to change and may be affected by risks and uncertainties, most of which are difficult to predict and are generally beyond our control. Forward-looking statements speak only as of the date they are made, and we undertake no obligation to update any forward-looking statement in light of new information or future events, except as required by law, although we intend to continue to meet our ongoing disclosure obligations under the U.S. securities laws and other applicable laws.
We caution you that a number of important factors could cause actual results or outcomes to differ materially from those expressed in, or implied by, the forward-looking statements, and therefore you should not place too much reliance on them. These factors include, among others, those described herein, under “Risk Factors” and elsewhere in this Annual Report and in our other public reports filed with the Securities and Exchange Commission. It is not possible to predict or identify all such factors, and therefore the factors that are noted are not intended to be a complete discussion of all potential risks or uncertainties that may affect forward-looking statements. If these or other risks and uncertainties materialize, or if the assumptions underlying any of the forward-looking statements prove incorrect, our actual performance and future actions may be materially different from those expressed in, or implied by, such forward-looking statements. We can offer no assurance that our estimates or expectations will prove accurate or that we will be able to achieve our strategic and operational goals.
| 77 |
Forward-looking statements are based on information we have when those statements are made or management’s good faith belief as of that time with respect to future events, and are subject to significant risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to:
| ● | our lack of operating history; | |
| ● | our current and future capital requirements and our ability to satisfy our capital needs; | |
| ● | our inability to keep up with industry competition; | |
| ● | interpretations of current laws and the passages of future laws; | |
| ● | acceptance of our business model by investors and our ability to raise capital; | |
| ● | our drug discovery and development activities may not result in products that are approved by the applicable regulatory authorities. Even if our drug candidates do obtain regulatory approval they may never achieve market acceptance or commercial success; | |
| ● | our reliance on key personnel, including our ability to attract and retain scientists; | |
| ● | our reliance on third party manufacturing to supply drugs for clinical trials and sales; | |
| ● | our limited distribution organization with no sales and marketing staff; | |
| ● | our being subject to product liability claims; | |
| ● | our reliance on key personnel, including our ability to attract and retain scientists; | |
| ● | legislation or regulation that may increase the cost of our business or limit our service and product offerings; | |
| ● | risks related to our intellectual property, including our ability to adequately protect intellectual property rights; | |
| ● | risks related to government regulation, including our ability to obtain approvals for the commercialization of some or all of our drug candidates, and ongoing regulatory obligations and continued regulatory review which may result in significant additional expense and subject us to penalties if we fail to comply with applicable regulatory requirements; and | |
| ● | our ability to obtain regulatory approvals in foreign jurisdictions to allow us to market our products internationally. |
Moreover, new risks regularly emerge and it is not possible for our management to predict or articulate all risks we face, nor can we assess the impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from those contained in any forward-looking statements. All forward-looking statements included in this prospectus are based on information available to us on the date of this registration statement. Except to the extent required by applicable laws or rules, we undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise. All subsequent written and oral forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements contained above and throughout this registration statement.
Item 1B. Unresolved Staff Comments
Not applicable.
We maintain our headquarters at 37 North Orange Ave, Suite 607, Orlando, Florida 32801. The Company leases approximately 800 sq. feet at a monthly cost of approximately $1,350. The lease expires on December 31, 2018.
On November 11, 2016, the Company received a fax containing a copy of a lawsuit supposedly filed against the Company on November 10, 2016. Public records reflect Kacem Enterprises, Inc. sued the Company on November 10, 2016 in the Circuit Court of Arlington County, Virginia, Case Number CL-16-2870. Kacem Enterprise, Inc. alleges it served the Company by mail on January 23, 2017. The Company was never personally served with the lawsuit in accordance with Florida law and is unaware of the alleged service by mail. Public records further reflect a default judgment was entered against the Company on March 10, 2017. Kacem Enterprise, Inc. sued the Company for $21,777 plus interest, costs and attorneys’ fees for fees Kacem Enterprise claims it is owed for travel related expenses incurred by certain consultants of the Company. The Company has not accrued any amount for these claims, on the basis that the claims should have been filed only against the consultants.
Item 4. Mine Safety Disclosures
Not applicable.
| 78 |
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed for quotation on the OTCQB marketplace under the symbol IMUN. Our common stock began trading in November 1999 on OTC under the name Galliano International Ltd. Trading under the name of TNI BioTech, Inc. commenced in March 2012 under the symbol TNIB. The symbol was changed to IMUN on December 11, 2014. The following table sets forth the high and low sales prices per share of our common stock for the periods indicated as reported by the OTC Markets Group, Inc.
Period
| Price Range | ||||||||
| High | Low | |||||||
| Quarter Ended December 31, 2016: | $ | 0.09 | $ | 0.04 | ||||
| Quarter Ended: | ||||||||
| September 30, 2016 | $ | 0.18 | $ | 0.12 | ||||
| June 30, 2016 | $ | 0.19 | $ | 0.11 | ||||
| March 31, 2016 | $ | 0.22 | $ | 0.12 | ||||
| Quarter Ended December 31, 2015: | $ | 0.28 | $ | 0.08 | ||||
| Quarter Ended | ||||||||
| September 30, 2015 | $ | 0.36 | $ | 0.05 | ||||
| June 30, 2015 | $ | 0.11 | $ | 0.05 | ||||
| March 31, 2015 | $ | 0.28 | $ | 0.07 | ||||
Record Holders
As of March 29, 2017, we have approximately 773 stockholders of record of our common stock.
Dividends
The Company paid no dividends in 2016 or 2015.
We do not anticipate paying any cash dividends in the foreseeable future.
The payment of dividends is within the discretion of our Board of Directors and will depend on our earnings, capital requirements, financial condition, and other relevant factors. There are no restrictions that currently limit our ability to pay dividends on its common stock other than those generally imposed by applicable state law.
Securities Authorized for Issuance Under Equity Compensation Plans
On September 4, 2014, the Company’s shareholders approved the Company’s 2014 Stock Incentive Plan (“2014 Plan). The 2014 Plan is an important incentive for our employees and is critical to the Company’s ongoing effort to build shareholder value and align the interests of employees and directors with those of the Company’s shareholders. Equity awards are a significant part of the Company’s ability to attract, retain, and motivate our executives and employees whose skills and performance are critical to our success.
A total of 5,000,000 shares of the Company’s common stock has been reserved for issuance under the 2014 Plan. Awards under the 2014 Plan may be made to employees, directors, consultants and advisors of the Company and any successor entity that adopts the 2014 Plan. Awards under the 2014 Plan may be made in the form of incentive stock options, nonqualified stock options, stock appreciation rights, restricted stock awards and restricted stock unit awards. Vesting of awards will be determined by our Board. No more than 500,000 shares may be issued to a single participant pursuant to stock options and stock appreciation rights in a calendar year. Re-pricing of outstanding stock awards is not permitted under the 2014 Plan. The 2014 Plan will terminate upon the earlier of the adoption of a resolution of the Board terminating the 2014 Plan, or September 3, 2024.
| 79 |
Item 6. Selected Financial Data
Not required for smaller reporting companies.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation
General
Immune Therapeutics, Inc. (the “Company”) was initially incorporated in Florida on December 2, 1993 as Resort Clubs International, Inc. (“Resort Clubs”). It was formed to manage and market golf course properties in resort markets throughout the United States. Galliano International Ltd. (“Galliano”) was incorporated in Delaware on May 27, 1998 and began trading in November 1999 through the filing of a 15C-211. On November 10, 2004, Galliano merged with Resort Clubs. Resort Clubs was the surviving corporation. On August 23, 2010, Resort Clubs changed its name to pH Environmental Inc. (“pH Environmental”).
On April 23, 2012, pH Environmental completed a name change to TNI BioTech, Inc., and on April 24, 2012, we executed a share exchange agreement for the acquisition of all of the outstanding shares of TNI BioTech IP, Inc. On September 4, 2014, a majority of our shareholders approved an amendment to our Amended and Restated Articles of Incorporation, as amended, to change our name to Immune Therapeutics, Inc. We filed our name change amendment with the Secretary of State of Florida on October 27, 2014 changing our name to Immune Therapeutics, Inc.
The Company currently operates out of Orlando, Florida. In July 2012, the Company’s focus turned to acquiring patents that would protect and advance the development of new uses of opioid-related immune- therapies, such as low dose naltrexone (“LDN”) and Methionine [Met5]-enkephalin (“MENK”). The Company’s therapies are believed to stimulate and/or regulate the immune system in such a way that they provide the potential to treat a variety of diseases. We believe our therapies may be able to correct abnormalities or deficiencies in the immune system in diseases such as HIV infection, autoimmune disease, immune disorders, or cancer; all of which can lead to disease progression and life-threatening situations when the immune system is not functioning optimally.
Subsidiaries
In October 2012, the Company formed TNI BioTech International, Ltd., a BVI company in Tortola, British Virgin Islands, which was set up to allow the Company to market and sell LDN in those countries outside the U.S. in which we have been able to obtain approval to sell the Company’s products.
In August 2013, the Company formed its United Kingdom subsidiary, TNI BioTech, LTD (the “UK Subsidiary”). The UK Subsidiary received approval to be considered a micro, small or medium-sized enterprise (“SME”) with the European Medicines Agency (“EMA”) on August 21, 2013. The designation provides the UK Subsidiary with significant discounts when holding meetings or submitting filings to the EMA. On September 19, 2013, the UK Subsidiary submitted a pre-submission package to the EMA regarding Crohn’s Disease. The EMA granted the UK Subsidiary a meeting that took place on September 27, 2013. The UK Subsidiary is eligible to benefit from the provisions for administrative and financial assistance for SMEs set out in Regulation (EC) No 2049/2005. The Company will apply to obtain EMA benefits once funding becomes available.
In December 2013, the Company formed a new subsidiary, Cytocom Inc., to focus on conducting LDN and MENK clinical trials in the United States. In December 2014, the Company finalized the distribution of common stock of Cytocom Inc. to its shareholders. As part of the transaction, the Company retained exclusive rights to all international patents, in-country approvals, formulations, trademarks, manufacturing, marketing, sales, and distributions rights in emerging nations, including Africa, Central America, South America, Russia, India, China, Far East, and The Commonwealth of Independent States (former Soviet Union). The Company will continue to have access to existing clinical data as well as any new data generated by Cytocom Inc. during drug development. On December 8, 2014, the number of Cytocom Inc. shares of common stock that were issued to our shareholders totaled 113,242,522. In connection with the transaction, Cytocom Inc. issued 140,100,000 shares of its common stock to the Company, which gave the Company a 55.3% stake in Cytocom Inc. on that date. In April 2016, the Board of Directors and a majority of shareholders of Cytocom approved a reverse stock split of Cytocom’s outstanding common stock with one new share of stock for each twenty old shares of common stock. Cytocom effectuated and finalized the reverse split in June 2016. At December 31, 2016, the Company’s equity interest had been further reduced to 13%, by subsequent issuances of Cytocom common stock to shareholders in settlement of notes payable.
| 80 |
In March 2014, the Company incorporated Airmed Biopharma Limited, an Irish corporation with an address in Dublin, Ireland, and Airmed Holdings Limited, an Irish company domiciled in Bermuda. The Irish companies were set up to benefit from incentives granted by the Irish government for the establishment of pharmaceutical companies (many of the world’s leading pharmaceutical companies have located in Ireland), and so that the Company could take advantage of Ireland’s status as a member of the European Union and the European Economic Area. An Irish limited liability company enjoys a low corporate income tax rate of 12.5%, one of the lowest in the world. The Irish-domiciled company hopes to qualify for tax incentives for Irish holding/headquartered companies and to benefit from the network of double tax treaties that reduce withholding taxes. TNI BioTech International, Ltd. will manage our international distribution, using product that is manufactured in Ireland and elsewhere.
We are focused on the development and commercialization of therapeutic treatments for cancer, HIV/AIDS and autoimmune diseases and immune disorders by combating these severe and fatal diseases through the stimulation and/or regulation of the body’s immune system. TNI’s growth strategy includes the near-term commercialization of its existing immunotherapies targeting cancer, Crohn’s disease and/or HIV/AIDS.
Financial Operations Overview
Revenue
The Company reported revenues of $3,463 in the year ending December 31, 2016, compared to revenues of $16,197 in 2015. The decrease in revenues was primarily attributable to the cancellation of the Company’s manufacturing agreement with KRS in the second quarter of 2016. As of December 31, 2016, the Company had not signed an agreement with another contract manufacturer or distributor for its products in the United States.
The Company expects to derive revenues from the sale of its products in Nigeria in the second quarter of 2017 under distribution and partnership agreements announced in 2012 and 2013.
Direct Expenses and Gross Margin
The Company incurred no direct expenses in 2016 and 2015. In future, direct expenses will include the cost of finished product to be purchased from the Company’s contract manufacturers, sales incentives, associated travel, and inventory return charges.
Gross margin, which is gross profit as a percent of revenue, will be affected by a number of factors, including the type of product sold and the geographic region in which the sale is made.
Research and Development
The Company makes investments in research and development (“R&D) in support of ongoing proprietary product development programs to support its pipeline of new drug candidates. See Item 1. “Business” for a summary the current stage of development of our new drug candidates. Expenses related to both direct research and to the use of outsourced research arrangements decreased in 2016 compared to 2015, as the Company was not able to raise the cash required to fund its planned research program in 2016.
R&D expenses consist of salaries and benefits paid to employees directly engaged in research, payments made in cash or in kind to contractors for services directly related to research and product development, product development costs, payments made for patents and licenses to which the Company has acquired rights to use, and travel, telecommunications, facilities and external legal costs incurred in relation to research activities.
\
| 81 |
We do not collect costs on a product basis or for any category of product involved in carrying out research projects. While we do perform cost calculations to facilitate our internal evaluation of individual products, these calculations include significant estimations and allocations that are not relevant to, or included in, our external financial reporting mechanisms. As a consequence, we do not report research and development costs at the product level.
Operating Expenses
Selling, general and administrative expenses primarily include salary and benefit costs for employees and contractors included in our sales, marketing, finance, legal and administrative organizations, professional services, insurance, unallocated travel expenses, telecommunications, impairment of intangibles, and office expenses. Professional services consist principally of recruiting costs, external legal, audit, tax and other consulting services.
Other Expenses, Net
Other expenses, net consists primarily of interest income on cash balances, and interest expense on borrowings. Interest expense will vary periodically depending on prevailing short-term interest rates.
Loss on settlement of debt
Loss on settlement of debt comprises the cost of issuance of common stock for the retirement of principal and accrued interest on promissory notes.
Critical Accounting Policies
The preparation of financial statements and related disclosures in conformity with U.S. generally accepted accounting principles (“GAAP”) and the Company’s discussion and analysis of its financial condition and operating results require the Company’s management to make judgments, assumptions, and estimates that affect the amounts reported in its consolidated financial statements and accompanying notes. Note 1, “Summary of Significant Accounting Policies” of the Notes to Consolidated Financial Statements in Part II, Item 8 of this Form 10-K describes the significant accounting policies and methods used in the preparation of the Company’s consolidated financial statements. Management bases its estimates on historical experience and on various other assumptions it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities.
We have identified the policies below as critical to our business operations and the understanding of its results of operations. The Company’s senior management has reviewed these critical accounting policies and related disclosures with the Company’s Board of Directors. The impact and any associated risks related to these policies on our business operations are discussed throughout this section where such policies affect our reported and expected financial results. Our preparation of financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of our financial statements, and the reported amounts of revenues and expenses during the reporting period. There can be no assurance that actual results will not differ from those estimates and such differences may be material.
Cash and Cash Equivalents
We consider all highly liquid debt instruments and other short-term investments with a maturity of three months or less at the time of purchase to be cash equivalents.
Net Loss Per Share of Common Stock
The basic net loss per common share is computed by dividing the net loss by the weighted average number of common shares outstanding. Diluted net loss per share gives effect to all dilutive potential common shares outstanding during the period using the “as if converted” basis.
| 82 |
Uncertainty in Income Taxes
Management considers the likelihood of changes by taxing authorities in its filed income tax returns, and recognizes a liability for or discloses potential changes that management believes are more likely than not to occur upon examination by tax authorities. Management has not identified any uncertain tax positions in filed income tax returns that require recognition or disclosure. The Company’s income tax returns for the past three years are subject to examination by tax authorities, and may change upon examination.
We follow Accounting Standards Codification (“ASC”) 740-10, Accounting for Uncertainty in Income Taxes (“ASC 740-10”). This interpretation requires recognition and measurement of uncertain income tax positions using a “more-likely-than-not” approach. ASC 740-10 is effective for fiscal years beginning after December 15, 2006. The Company has adopted ASC 740-10 and evaluates its tax positions on an annual basis.
Results of Operations
Year Ended December 31, 2016 Compared to Year Ended December 31, 2015
Revenues
We had revenues of $3,463 from operations for the year ended December 31, 2016, compared to revenues of $16,197 for the year ended December 31, 2015. All revenues were earned under the agreement signed on December 8, 2015 with KRS Global Biotechnology, Inc. (“KRS”). The decrease in revenues resulted from the cancellation in the second quarter of 2016 of the contract with KRS. As of December 31, 2016, the Company had not entered into a contract to replace KRS as its contract manufacturer and distributor in the United States.
Operating Expenses
Selling, general and administrative
Selling, general and administrative expenses and related percentages for the years ended December 31, 2016 and 2015 were as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Selling, general and administrative | $ | 3,887 | $ | 2,734 | ||||
| Increase / (decrease) from prior year | $ | 1,153 | $ | (1,338 | ) | |||
| Percent increase / (decrease) from prior year | 42 | % | (33 | )% | ||||
In 2016, total cash spent on in selling, general and administrative expense was $3,887, compared to $2,734 for 2015, an increase of $1,153 or 42%. For the years ended December 31, 2016 and 2015, selling, general and administrative expenses were made up as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Stock listing and investor relations expenses | $ | 187 | $ | 44 | ||||
| Consulting and contractors | 1,052 | 1,033 | ||||||
| Payroll | 1,719 | 833 | ||||||
| Professional fees | 196 | 265 | ||||||
| Travel | 228 | 174 | ||||||
| Insurance | - | (107 | ) | |||||
| Other expenses | 505 | 492 | ||||||
| $ | 3,887 | $ | 2,734 | |||||
The increase year over year in selling, general and administrative expense was attributable primarily to an $866 increase year over year in payroll (due to higher headcount at the Company for the year, increases in officer compensation in accordance with their contracts, and accruals for payroll owed to Cytocom employees), a $143 increase year over year in investor relations expenses, a $107 increase year over year in insurance expenses (in 2015, the Company reversed accruals taken in 2014 on insurance policies that lapsed in 2015), and to a $54 increase in travel activities to promote the manufacture and sale of the Company’s products in South America, the Caribbean and Africa. These increases were offset by a $69 reduction in professional fees (mostly related to the incorporation and spin-out of Cytocom in 2015).
| 83 |
Significant cash spending included:
| ● | consulting services obtained to assist the Company in raising capital, manage investor relations, and develop business in new markets, in the amount of $1,052 in 2016, an increase of $19 or 2% of the $1,033 spent in 2015; | |
| ● | professional fees for legal, tax and accounting services in the amount of $196 in 2016, a decrease of $69, or 26% over the $265 spent in 2015; | |
| ● | payroll in the amount of $1,719 in 2016, an increase of $886 or 106% over the $833 spent in 2015. $658 of the increase was attributable to payroll owed to officers under new contracts signed with Cytocom and agreed-upon pay increases under their employment contracts, $85 was caused by an increase in headcount at the Company, and $143 arose from payroll taxes arising from the aforementioned increases.; and | |
| ● | travel in the amount of $228 in 2016, an increase of $54 or 31% of the $174 spent in 2015, the result of travel to Africa to market the Company’s products and to obtain regulatory approvals for their sale, and to Nicaragua and Dominican Republic to meet with contract manufacturers and government regulatory agencies. |
Research and development
R&D expenses and related percentages for the years ended December 31, 2016 and 2015 were as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Research and development | $ | 444 | $ | 977 | ||||
| Increase/ (decrease) from prior year | $ | (533 | ) | $ | (1,436 | ) | ||
| Percent increase/ (decrease) from prior year | (55 | )% | (60 | )% | ||||
R&D is overseen and managed internally, working with individuals, universities, and CROs in order to utilize patents that we have licensed or acquired since our inception. We continue to seek to expand our pipeline of patents by reviewing other compounds, technologies or capabilities. We also seek out promising compounds and innovative technologies developed by third parties to incorporate into our discovery and development processes or projects.
Drug discovery and development is time-consuming, expensive and unpredictable. According to PhRMA, out of 5,000-10,000 screened compounds, only 250 enter preclinical testing, five enter human clinical trials and one is approved by the FDA. The process from early discovery or design to development to regulatory approval can take more than 10 years. Drug candidates can fail at any stage of the process, and candidates may not receive regulatory approval even after many years of research.
As of December 31, 2016, we had two compounds (IRT-101, and IRT-103 or Lodonal™) in research and development in oncology and Crohn’s disease, both of which are expected to move into Phase 3 clinical trials in the United States in 2017.
For the years ended December 31, 2016 and 2015, research and development expenses were made up as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Contracted and consulting services | $ | 50 | $ | 205 | ||||
| Patent expenses | 185 | 156 | ||||||
| Payroll | (5 | ) | 5 | |||||
| Trials | 177 | 541 | ||||||
| Professional fees | 100 | 32 | ||||||
| Supplies and materials | - | 14 | ||||||
| Rent | (84 | ) | 22 | |||||
| Travel | 12 | - | ||||||
| Other | 9 | 2 | ||||||
| $ | 444 | $ | 977 | |||||
| 84 |
Expenses for research and development in 2016 decreased by 55% compared to expenses in the same period 2015. Most of the R&D spending in both years focused on the development of LDN, primarily under trials in Africa. The overall decrease was the result of lower funding available to conduct research and development.
In 2016, total cash spent on R&D was $444, a decrease of $533 or 55% over the $977 spent in 2015. Significant cash items included:
| ● | payments for contracted services ($50 in 2016, a decrease of $155 or 76% over the $205 spent in 2015). A significant portion of the difference ($100) arose from the settlement of amounts that had been accrued in prior years for services owed to a vendor. There was also a decrease in use of contractors to perform research activities in the United States; | |
| ● | patent expenses ($185 in 2016, compared to $156 for 2015, an increase of $29 or 19% over 2015), reflecting increased costs incurred in 2016 to maintain licenses and patents acquired in 2015 and 2016, and the registration of patents in new countries; | |
| ● | legal fees ($100 in 2016, compared to $32 in 2015, an increase of $68, or 212% over 2015), incurred for the support of patents and licenses and to register patents in new jurisdictions; | |
| ● | rent $(84) in 2016, compared to $22 in 2015, a decrease $106, or 477% over 2015), reflecting mediated settlements in 2016 of past-due amounts for rent for premises in Maryland); | |
| ● | payroll ($(5) in 2016, a decrease $10, or 200% over the $5 accrued in 2015), reflecting reversals in 2016 of accruals for payroll taxes; and | |
| ● | travel ($12 in 2016, an increase of $12 or 100% over the $0 in 2015), reflecting the need for travel to monitor trials in Africa. |
Stock issued for services
The cost of stock issued for services G&A and related percentages for the years ended December 31, 2016 and 2015 were as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Consulting services G&A | $ | 5,212 | $ | 6,240 | ||||
| Percentage increase/(decrease) from prior year | (16 | )% | (67 | )% | ||||
The decrease in expense reflects the decrease in the price of the Company’s stock year over year.
The number of shares issued for prepaid consulting services G&A in 2016 was 32,633,910 (16,672,504 in 2015).
Prepaid consulting services G&A 2016 consisted of the following:
| Amortization of cost of stock issued prior to 2016 under G&A consulting contracts | $ | 564 | ||
| Amortization of cost incurred for new stock issued in 2016 under G&A consulting contracts entered into in 2016 | $ | 4,648 |
| 85 |
Warrant valuation expense
When the Company sells its stock to stockholders for cash, it periodically issues warrants to those stockholders to acquire additional stock at prices agreed at the date of the original sale. The Company incurs a cost for the rights attached to the warrants, which is calculated using the Black-Scholes Model (see above 4. Capital Structure—Common Stock and Common Stock Purchase Warrants). This expense is reported in the Consolidated Statements of Operations above as the Warrant valuation expense.
In 2016, the Company issued 50,222,904 warrants to stockholders at exercise prices ranging between $0.03 and $2.00, for which it recorded an expense of $4,731. In 2015, the Company issued 435,000 warrants to stockholders at exercise prices ranging between $0.07 and $0.50, for which it recorded an expense of $46.
Depreciation and amortization
The Company amortizes the costs incurred to acquire patents and licenses over the period of the related agreements. The decrease year over year in depreciation and amortization expense reflects the fact that all of the Company’s patents and licenses had been impaired by the end of 2015.
Depreciation and amortization expenses for the years ended December 31, 2016 and 2015 were as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Depreciation expense | $ | 2 | $ | 3 | ||||
| Amortization expense | $ | 0 | $ | 592 | ||||
| Increase/ (decrease) from prior year | $ | (593 | ) | $ | (2,285 | ) | ||
| Percentage increase (decrease) from prior year | (99 | )% | (79 | )% | ||||
Interest Expense
Interest expense for the years ended December 31, 2016 and 2015 were as follows (dollar amounts in thousands):
| 2016 | 2015 | |||||||
| Interest expense | $ | 3,447 | $ | 271 | ||||
| Increase/(decrease) from prior year | $ | 3,176 | $ | (117 | ) | |||
| Percentage increase/(decrease) from prior year | 1,172 | % | (30 | )% | ||||
Interest expenses are comprised of loan origination fees and interest owed by the Company. The significant increase in interest expense reflects the fact that the debt levels were higher in 2016 than in 2015, and the Company incurred default penalties of $1,000 in 2016.
Loss on settlement of debt (dollar amounts in thousands):
In 2016, certain lenders to the Company exercised their rights to convert all or a portion of their notes payable to equity. The Company also settled certain obligations in 2016 through the issuance of its common stock. The Company recorded an expense of $2,101, reflecting the fair value of the 36,235,380 shares of common stock issued in exchange for notes and other obligations. The corresponding expense in 2015 was $844, reflecting the fair value of the 14,058,833 shares of common stock issued in exchange for notes.
Liquidity:
Liquidity is measured by the Company’s ability to secure enough cash to meet its contractual and operating needs as they arise. The Company had cash of $74,389 at December 31, 2016, compared to $23,149 at December 31, 2015. For the years ended December 31, 2016 and 2015, net cash used in operating activities was $2,839,739 and $2,832,313, respectively. $1,725 cash was used in investing activities for the year ended December 31, 2016 ($0 was used in 2015).
In May 2016, the Company announced that it had received approval for sale of LodonalTM in Nigeria. The Company expects to commence sales to Nigeria by the end of the second quarter of 2017. The Company believes that those sales will generate sufficient cash flows for the next 12 months to pay for operating expenses and to pay off current and past-due obligations. Until such sales commence, the Company expects to fund operations through sales of equity and notes payable, and conversions of exiting obligations into equity. Over the next 12 months, the Company believes it will require between $300,000 and $350,000 monthly to meet its ongoing expenses and obligations.
| 86 |
If the Company is unable to generate sufficient cash flows from sales, or if it does not raise additional working capital to meet all of its operating obligations and expenditures, the Company may have to modify its business plan.
In addition to the cost of its ongoing operations, the Company expects it will incur future research and development expenditures in the next 12 months through Cytocom. Cytocom plans to conduct Phase II and Phase IIB trials for the treatment of Crohn’s disease, at an estimated cost of $3,900,000 and $7,500,000 respectively for each phase. If the trials do not commence before the end of 2017, the Company will be required to make a payment of $100,000 in December 2017 under its license agreements. In prior years, the Company has been able to raise funds through sales of notes payable, and it expects to do the same if the payment becomes due in December 2017.
During the year ended December 31, 2016 proceeds from the sale of stock and exercise of stock warrants totaled $200,000, compared to $605,500 for the corresponding period in 2015. The Company also received $2,768,631 from the issuance of notes payable in year ended December 31, 2016, compared to $2,057,975 in 2015.Loan repayments made in cash in the year ended December 31, 2016 totaled $75,927 ($0 in 2015).
Off-Balance Sheet Arrangements
During the years ended December 31, 2016 and 2015, we did not engage in any off balance sheet arrangements as defined in item 303(a)(4) of the SEC’s Regulation S-K.
| Item 7A. | Quantitative and Qualitative Disclosure About Market Risk |
Not applicable.
| Item 8. | Financial Statements and Supplementary Data |
Our Consolidated Financial Statements and Notes thereto, for the fiscal years ended December 31, 2016 and 2015 and the report of Turner, Stone & Company, L.L.P. (“Turner”), our independent registered public accounting firm, are set forth on pages F-1 through F-23 of this Annual Report.
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported, within the time period specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports filed under the Exchange Act is accumulated and communicated to management, including the Chief Executive Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on the evaluation, the CEO and CFO have concluded that our disclosure controls and procedures are ineffective to ensure that information disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms. This determination was based on the small size of our accounting staff, the lack of segregation of duties and the lack of an audit committee which creates a material weakness. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. A material weakness means there is a risk that our financial reports or other filings may contain an error or inaccuracy or not submitted timely. The Company plans to remediate this weakness by increasing the size of its accounting staff in 2016 and by appointing an audit committee with membership that is qualified to oversee financial reporting.
| 87 |
Management Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control system was designed to provide reasonable assurance to our management and board of directors regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Any internal control system, no matter how well designed, has inherent limitations and may not prevent or detect misstatements. Accordingly, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management, with the participation of our Chief Executive Officer and Chief Financial Officer, have evaluated the effectiveness of our internal control over financial reporting as of December 31, 2016 based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, because of the Company’s limited resources and limited number of employees, and the absence of an audit committee, management concluded that, as of December 31, 2016, our internal control over financial reporting is not effective in providing reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles.
This annual report does not include an attestation report of the Company’s independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s independent registered public accounting firm pursuant to permanent rules of the SEC that permit the Company to provide only management’s report in this Annual Report.
Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Securities and Exchange Act of 1934) during the year ended December 31, 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
| 88 |
| Item 10. | Directors, Executive Officers and Corporate Governance |
Directors
As of March 31, 2017, the number of voting members of our Board of Directors was 4. Our Non-Executive Chairman of the Board is a non-voting member of our Board. The members of our Board of Directors as of March 31, 2017 are as follows:
| Name | Age | Director Since | Position | |||
| Noreen Griffin | 64 | March 2012 | Chief Executive Officer and Director | |||
| Dr. Nicholas Plotnikoff | 89 | March 2012 | Non-Executive Chairman of the Board (non-voting director) | |||
| Edward Teraskiewicz | 70 | January 2015 | Director | |||
| Dr. Clifford Selsky | 68 | February 2016 | Director | |||
| Paul Akin | 58 | February 2016 | Director |
The biographies of each director below contains information regarding the person’s service as a director, business experience, director positions held currently or at any time during the last five years, and information regarding involvement in certain legal or administrative proceedings, if applicable.
Noreen Griffin – Ms. Griffin is one of our Founders and has served as our Chief Executive Officer and as a member of our Board of Directors since March 2012. Ms. Griffin was a vital part of the acquisition of our patents and therapies involving MENK and LDN. She was involved with the inventors and patent holders for over five years before deciding to join the team that formed the Company. Prior to joining our Board, Ms. Griffin was the sole officer and director of Supertrail Manufacturing Co., Inc., a private company in Aberdeen, Mississippi from 1998 to 2013. Ms. Griffin was Chief Financial Officer of Environmental Remediation Holdings, Inc., a public reporting company in Lafayette, Louisiana from 1997 to 2014. From 2004 to 2009, she was an advisor to Global Environmental Energy Corp, a public company, and assumed the role of sole officer for the purpose of the company’s bankruptcy. From 2007 to 2013, Ms. Griffin was Chief Executive Officer of pH Solutions, a private company in Boston, Massachusetts. Since 2008, Ms. Griffin has been a partner of Griffin Enterprises Group. The firm provides chief financial officer services to its clients on a part- time basis. In that capacity, Ms. Griffin acted as sole officer and director for the bankruptcy of James M. Jost and Company Inc. and Avalon from 2010 through 2013. Ms. Griffin has over 25 years of industry experience, having founded and led a number of startup companies. She has played an integral role in raising multiple rounds of private and venture capital funds on behalf of clients. Ms. Griffin has served as Chief Financial Officer and Vice President of a number of small public companies over the last 10 years. In addition, Ms. Griffin has significant experience in the administration of companies in bankruptcies.
Dr. Nicholas Plotnikoff – Dr. Plotnikoff has served as our non-voting, Non-Executive Chairman of the Board since March 2012. Dr. Plotnikoff has a Ph.D. in Pharmacology and over 20 years’ experience in the pharmaceutical industry working in Pharmacology, Toxicology, and Clinical Research. Prior to joining our Board, Dr. Plotnikoff was a Professor of Pharmacology at the University of Illinois Medical Center in Chicago from December 1987 until his retirement in May 2008. Dr. Plotnikoff formed TNI Pharmaceutical, Inc. in 1987, which became a public company in 1991. Dr. Plotnikoff served as its Chief Executive Officer and President for 20 years where he helped develop the immunological effects of MENK. He successfully managed the project teams that developed the new drug applications for Traxene (a Valium-like tranquilizer) and Cylert (a non-amphetamine psychostimulant). Dr. Plotnikoff was responsible for the Phase I and Phase II trials for MENK in HIV/AIDS. In basic research, he was the first to identify the central nervous system effects of hypothalamic releasing factors (brain hormones), resulting in clinical development for treatment of depression and Parkinson’s disease. Dr. Plotnikoff has co-authored 25 publications with Dr. Andrew Schally, covering basic research in the field of depression and Parkinson’s disease. Dr. Plotnikoff was issued a number of international patents for the use of MENK in HIV/AIDS and cancer, which we acquired in 2012.
| 89 |
Edward Teraskiewicz – Mr. Teraskiewicz has served on our Board since January 2015. He has over 35 years of financial services experience. From 1992 to 2004, Mr. Teraskiewicz was 50% owner Co-Founder, Chief Executive Officer and Director of Prebon Yamane International Limited, one of the world’s pre-eminent money brokerage firms, with over 1700 employees in 17 cities around the world with revenue over $500,000,000 annually. Having retired from daily operations at the end of 1994, he continued his involvement with Prebon Yamane as a member of the Board of Directors of Fulton Prebon Group U.K., the holding company which owns the money brokerage business, until October 2004, at which time he resigned from the Board upon a merger with Collins, Steward, Tullett. Since 2004, Mr. Teraskiewicz has been overseeing and managing various residential real estate development projects and has been an investor in numerous other projects in the music and financial industry. Mr. Teraskiewicz is a graduate of the American Institute of Banking, having commenced his career as a trainee at Citibank in 1964. In 1970, he joined Mabon, Nugent & Co., a New York Stock Exchange member firm, where he advanced to the rank of Senior General Partner. Mr. Teraskiewicz is an experienced investor, having developed several residential sub-divisions and luxury estate home projects in the United States, as well as pursuing other transactions around the world.
Dr. Clifford Selsky— Dr. Selsky has served on our Board since February 2016. He has been a practicing pediatrician in Central Florida for the past twenty years. He is the founder of the Children’s Center for Cancer and Blood Disease at Florida Hospital cancer institute, which he established after training in pediatrics at Yale New Haven hospital and completing a pediatric hematology and oncology fellowship at Yale University School of Medicine. Dr. Selsky is board certified in Pediatrics, Pediatric hematology and oncology and Palliative medicine. Currently, he is a pediatrician at Family First Pediatrics which he established in 2013.
Also an accomplished scientist, Dr. Selsky obtained his PH.D. in Microbiology and Molecular Genetics at the University of Miami School of Medicine. He then did DNA repair research studies at the radiobiology laboratory at Harvard School of Public Health and the biophysics laboratory at Stanford University. Dr. Selsky has numerous publications in peer reviewed journals relating to DNA repair and clinical conditions such as angiocentric lymphoma and chemotherapy related neurological disorders. As a toxicologist for Stauffer Chemical Company, he designed and implemented research on molecular dosimetry and genetic risk estimation, including DNA adduct separation and quantitation.
Over the course of his career, Dr. Selsky has served as principal investigator for both the Pediatric Oncology Group at Florida Hospital Cancer Institute and the Children’s Oncology group at Florida Hospital overseeing more than 140 cooperative group protocols. He was department chair for Pediatrics at Florida Hospital for Children for seven years. Additionally, he has served on numerous committees including Florida Hospital Cancer Center Medical Advisory committee, Florida Hospital Ethics committee, Florida Hospital Quality Assurance committee and Florida Hospital Pharmacy and Therapeutics committee. Dr. Selsky was elected president of the Orange County Medical Society in 2016 and has received numerous awards including the Florida Hospital Medical Staff recognition Award for Excellence 2008 and being named Top Doctor, Orlando Magazine in 2001, 2005, 2006, 2007, 2008, 2009, 2010, 2011 and 2015.
Paul Akin— Mr. Akin has been a member of our Board since February 2016. Mr. Akin is a significant investor and active strategist for Immune Therapeutics. As an early investor, he was instrumental in the strategic planning and coordination of market penetration, expansion and capitalization. In 2015, he drove the research, development and subsequent creation of LDN Information Management, Inc. (“LDNIM”), whose goal is twofold: to educate the general population on the benefits of LDN, and to distribute LDN domestically through partnership with the Company. LDNIM also serves as an adjunct marketing team with the goal of accelerating consumer and physician education and distributing LDN in certain states in the USA over the next 12 months.
Mr. Akin currently serves as Chief Executive Officer and Executive Chairman of Collier Warehouse Inc., which sells residential and commercial products and services for architects, contractors, homeowners, and developers. Outside of the day-to-day operations of Collier and LDNIM, Mr. Akin is an active investor in a variety of emerging growth companies, having served in a number of business roles including Executive Chairman, Independent Board Director, Strategic Advisor, Venture Capital Limited Partner, market pundit, guest speaker/moderator, private investor and trustee. An avid sportsman, Mr. Akin is an active member at the San Francisco Olympic Club and competes on the basketball and triathlon teams, as well as a distance runner and golfer. He currently resides in San Francisco.
| 90 |
Executive Officers
Biographical information concerning Noreen Griffin, our Chief Executive Officer is set forth above. Biographical information concerning our other executive officers is set forth below.
Peter Aronstam – Mr. Aronstam, age 64, has been our Chief Financial Officer since January 2013. Mr. Aronstam brings more than 30 years of experience in accounting, finance, banking, international trade and law to his clients. His career is marked by a progression of senior finance roles with growth and performance-driven enterprises, from start-up technology and internet companies to chief financial officer roles with small service providers to very large international manufacturers to global banks. Mr. Aronstam has advised publicly-held and privately-owned businesses since 1978, providing both full-time and part-time CFO services. His background includes start-up VC-backed entrepreneurial companies, manufacturing technology and service companies, serving as a public-company CFO, corporate and international banking in major multinational banks, managing HR and IT functions, and raising more than $500,000,000 in debt and equity for his companies and their customers. From 2001 to 2006, Mr. Aronstam was the Chief Financial Officer of Airspan Networks, Inc., a public reporting company in Boca Raton, Florida. He also was the CFO of private company Mainstream Holdings, LLC in West Palm Beach, Florida from 2007 to 2008 and private company The Neptune Society in Plantation, Florida from 2008 to 2009. Since 2010, Mr. Aronstam has been a partner of B2B CFO Partners, LLC, doing business as B2B CFO©. The firm provides CFO services to its clients on a part time basis. Born and educated in South Africa, Mr. Aronstam earned his Bachelor of Commerce, Bachelor of Law and PhD from the University of the Witwatersrand in South Africa. Mr. Aronstam has worked in South Africa, Canada, and Florida.
Dr. Fengping Shan – Dr. Shan, age 58, has been our Chief Science Officer since March 2012 and was a member of our Board from March 2012 until September 2014. Dr. Shan has a Ph.D. in Microbiology and Tumor Immunology. Dr. Shan is Professor of Immunology and Vice Director of the Institute of Immunology, China Medical University, in Shenyang, China. He has been with the University from 2006 to present. Dr. Shan was the Senior Scientist for Penta Biotech from 2000 to 2006, Chief Scientist for the China Liaoning Institute of Microbiological Science from 1995 to 2000 and studied at the National Cancer Research in Paris, France from 1990 to 1994. Dr. Shan has authored 90 publications, been issued 11 patents, and is the unique inventor of a thrombolytic enzyme from the earthworm. From 2000 to present, Dr. Shan has worked both in the United States and China on the clinical trials with Dr. Nicholas Plotnikoff involving new immunotherapies for the treatment of cancer. Based on the trials, a number of patents were filed in China beginning in 2009 and 2010, and approved in 2011.
Dr. Joseph M. Fortunak – Dr. Fortunak, age 62, has been our Vice President of Global Research and Development and Chemical Development since April 2013. He graduated from the University of Wisconsin-Madison with a PhD in Organic Chemistry. After spending time holding a postdoctoral position at Cambridge University, he spent over 20 years in the pharmaceutical industry, most recently as Director and Head of Global Process Research and Development at Abbott Laboratories, from 2000 to 2004. Before taking the position at Abbott Laboratories, from 1983 to 1993, Dr. Fortunak worked as Associate Senior Research Investigator, Senior Research Investigator, and Assistant Director for Smith Kline Beechman (GlaxoSmithKline), and from 1993 to 2000 was the Associate Director, Director, Senior Director, and Executive Director of DuPont Pharmaceutical Company. In 2004, Dr. Fortunak assumed the position of Associate Professor of Chemistry and Pharmaceutical Sciences at Howard University in Washington, D.C., with a goal of developing an international program for cGMP practices worldwide. Dr. Fortunak has extensive experience dealing with the FDA and other regulatory agencies and has worked on industry initiatives (PQRI, BACPAC I & II) as a member of the PhRMA API Technical Group. He has significantly contributed to new drug development for illnesses including Malaria, Parkinson’s disease, Ovarian, Breast and Small-Cell Lung cancer, Hypertension and Congestive Heart Failure, AIDS (NNRTI), Breast and Ovarian cancer and Karposi’s sarcoma and AIDS (NRTI). In addition to his other work, Dr. Fortunak is a consultant for the Clinton Foundation’s HIV/AIDS Initiative (CHAI) and the World Health Organization; advising these organizations on pricing and production of antiretroviral drugs as well as advising generic manufacturers of antiretroviral drugs on the requirements for cGMPs, general strategies for process development and route discovery for the production of API.
| 91 |
Board Committees
At December 31, 2016, we had not yet established an audit committee, compensation committee, or nominating committee. During 2016, the functions ordinarily handled by these committees were handled by our entire Board. In February of 2015, the Board authorized formation of and adopted charters for an audit committee, compensation committee and nominating committee. As of the date of filing this annual report, no members were appointed to the audit committee, compensation committee or nominating committee. The audit committee, compensation committee and nominating committee have not yet held any meetings.
Family Relationships
There are no familial relationships between any of our officers and directors.
Director or Officer Involvement in Certain Legal Proceedings
Our directors and executive officers were not involved in any legal proceedings as described in Item 401(f) of Regulation S-K in the past ten years.
Director Independence
The Company is not currently listed on any national securities exchange that has a requirement that the board of directors be independent. At this time, Mr. Teraskiewicz, Dr. Selsky and Mr. Akin are “independent directors” as that term is defined under the rules of the NASDAQ Capital Market.
Code of Ethics
We have adopted a Code of Ethics that applies to all of our employees and officers, and the members of our Board of Directors. The Code of Ethics is available on our corporate website at www.immunetherapeutics.com. You can access the Code of Ethics on our website by first clicking “About Us” and then “Corporate Governance.” Any amendment to or waiver of the Code of Ethics will be disclosed on our website promptly following the date of such amendment or waiver.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than 10% of a registered class of our equity securities, to file reports of ownership and changes in ownership with the SEC. Officers, directors and greater than 10% shareholders are required by SEC regulation to furnish the Company with copies of all Section 16(a) forms they file. Based on our review of the reports filed by Reporting Persons, we believe that, during the year ended December 31, 2016, the Reporting Persons met all applicable Section 16(a) filing requirements, with the following exceptions: (i) Ms. Griffin was not timely with respect to Statement of Changes in Beneficial Ownership of Securities on Form 4 necessitated by the issuance of shares in error that were later cancelled; and (ii) Ms. Griffin who was not timely with respect to a Statement of Changes in Beneficial Ownership of Securities on Form 4 necessitated by the issuance of shares in error, the majority of which were later cancelled.
| Item 11. | Executive Compensation |
The following table summarizes the annual compensation of our Named Executive Officers (defined below), as of December 31, 2016. “Named Executive Officers,” consistent with Item 402(m) of Regulation S-K promulgated under the Exchange Act, include: (i) the Company’s Principal Executive Officer and individuals acting in a similar capacity during fiscal year 2016, regardless of compensation level; (ii) the Company’s two most highly compensated executive officers other than the Principal Executive Officer who were serving as executive officers at the end of fiscal year 2016; and (iii) up to two additional individuals who would have been included under (ii) above but for the fact that the applicable individual was not serving as an executive officer of the Company at the end of fiscal year 2016.
| 92 |
The Officers receive an annual salary from the Company as described in the table below for the services rendered on behalf of the Company.
| Name and Principal Position | Year | Salary | Bonus | Stock Awards |
Option Awards |
All
Other Compensation |
Total($) | |||||||||||||||||||
| Noreen Griffin | 2016 | $ | 401,953 | (1) | $ | — | $ | — | $ | 150,140 | $ | 37,180 | (2) | $ | 589,273 | |||||||||||
| Chief Executive Officer | 2015 | $ | 381,281 | (1) | $ | — | $ | — | $ | — | $ | 18,000 | (2) | $ | 399,281 | |||||||||||
| Christopher Pearce | 2016 | $ | 292,508 | (1) | $ | — | $ | — | $ | — | $ | 24,105 | (3) | $ | 316,713 | |||||||||||
| Chief Operating Officer, Director(*) | 2015 | $ | 285,961 | (1) | $ | — | $ | — | $ | — | $ | 18,000 | (3) | $ | 303,961 | |||||||||||
| Peter Aronstam | 2016 | $ | 90,000 | $ | — | $ | — | (4) | $ | 14,972 | $ | — | $ | 104,972 | ||||||||||||
| Chief Financial Officer | 2015 | $ | 90,000 | $ | — | $ | — | (4) | $ | — | $ | — | $ | 90,000 | ||||||||||||
| Dr. Fengping Shan | 2016 | $ | 20,000 | $ | — | $ | 230,000 |
(5) | $ | — | $ | — | $ | 250,000 | ||||||||||||
| Chief Science Officer | 2015 | $ | 20,000 | $ | — | $ | — | $ | — | $ | — | $ | 20,000 | |||||||||||||
(*) Mr. Pearce passed away on December 12, 2016.
(1) In 2015 and 2016, Ms. Griffin and Mr. Pearce agreed to defer a portion of their salaries until the Company had sufficient income to pay them in cash in full. In April 2015, Mr. Pearce agreed to waive payment of $282,252 of his deferred compensation in return for a royalty payable in perpetuity in an amount equal to (a) $0.01 per tablet or capsule of LDN sold by the Company outside of the United States, and (b) $0.005 per tablet or capsule of LDN sold by the Company in the United States. At December 31, 2016, the amounts of salaries deferred for Ms. Griffin and owed to Mr. Pearce’s estate were $856,812 and $230,471 respectively ($613,109 and $126,562, respectively, at December 31, 2015).
(2) The Company pays Ms. Griffin $1,500 per month for unaccountable expenses during each month of the term of her employment agreement. Unaccountable expenses refer to costs incurred by Ms. Griffin in the course of business that are not required to be reported on a monthly expense report. These costs include expenses incurred related to international travel. The Company also reimburses Ms. Griffin for the cost of health insurance premiums.
(3) The Company paid Mr. Pearce $1,500 per month for unaccountable expenses during each month of the term. Unaccountable expenses refer to costs incurred by Mr. Pearce in the course of business that are not required to be reported on a monthly expense report. These costs include expenses incurred related to international travel. The Company also reimbursed Mr. Pearce for the cost of health insurance premiums.
(4) In 2016, warrants were granted to Mr. Aronstam to acquire 100,000 shares of common stock of the Company and at a price of $0.14 each. The warrants were recorded at $14,972, which was determined to be the fair value as of the date of the warrant grant. The warrants expire on June 29, 2021. In 2015, there were no awards to Mr. Aronstam of Company common stock. In 2015, Mr. Aronstam was awarded 50,000 shares of common stock of Cytocom Inc.
(5) 1,437,500 shares were issued to Dr. Shan, representing 1 million shares for past services ($160,000) and 437,500 for past-due compensation ($70,000). No cash payments were made to Dr. Shan in 2016; the Company recorded a liability of $20,000 owed to Dr. Shan for services provided in 2016. In 2015, the Company recorded a liability of $20,000 owed to Dr. Shan for services provided.
Employment and Related Agreements
Noreen Griffin, the Company’s Chief Executive Officer, entered into an employment agreement with the Company in March 2012. The agreement has an initial two-year term and automatically renews for additional one-year periods at the election of our Board, unless sooner terminated in accordance with the agreement. The agreement provides for payment of an annual salary of $275,000, increasing by 5% per year during the term, payment of a monthly auto allowance of $1,500, payment for medical and insurance benefits, and payment of annual bonuses as determined by the Company. Ms. Griffin is also entitled to receive 2 million shares of the Company’s common stock when the Company commences shipments of its LDN product, in addition to the 3 million founder shares to which Ms. Griffin was already entitled. The agreement entitles Ms. Griffin to certain payments if more than 50.1% of the Company’s issued stock is acquired in a merger. The agreement was amended in March 2013, increasing Ms. Griffin’s annual salary to $350,000. The agreement was further amended as of March 14, 2016 extending the term to March 25, 2017. The amendment to the agreement also entitles Ms. Griffin warrants to purchase 1,000,000 shares of the Company’s common stock at $0.20 cents. The options were recorded at $150,140, which was determined to be the fair value as of the date of the option grant.
| 93 |
Peter Aronstam, our Chief Financial Officer, entered into a services agreement with the Company on December 15, 2014. The agreement expired on December 14, 2015, unless sooner terminated in accordance with the agreement. Pursuant to the agreement, Mr. Aronstam is required to devote twenty-five hours per month to carry out the chief financial officer services identified in the agreement. The agreement provides for a monthly payment of $7,500 and requires the Company to issue Mr. Aronstam a warrant, which is exercisable for five years, to purchase 250,000 shares of our common stock at an exercise price of $0.14 per share. On June 30, 2016, Mr. Aronstam’s agreement was extended to June 30, 2017. Under the extension, Mr. Aronstam received a warrant, which is exercisable for five years, to purchase 100,000 shares of the Company’s common stock at an exercise price of $0.17 per share. The warrant was recorded at $14,972, which was determined to be the fair value as of the date of the warrant grant.
Outstanding Equity Awards at Fiscal Year-End
Warrants were granted to Mr. Aronstam in December 2014 to acquire 250,000 shares of common stock of the Company and 250,000 shares of common stock of Cytocom Inc. at a price of $0.14 each. The warrants expire on December 14, 2019. Warrants were granted to Mr. Aronstam in June 2016 to acquire 100,000 shares of common stock of the Company at a price of $0.17 each. The warrants expire on June 29, 2021.
An option was granted to Ms. Griffin in March 2016 to acquire 1,000,000 shares of common stock of the Company at a price of $0.20 each. The options expire on March 13, 2021.
Summary Director Compensation Table
The following table shows information regarding the compensation earned or paid during 2016 to Non-Employee Directors who served on the Board during the year. The compensation paid to Ms. Griffin, Mr. Pearce, and Dr. Shan is shown under “Executive Compensation” and the related explanatory tables. Ms. Griffin, Mr. Pearce, and Dr. Shan did not receive any compensation for their service as members of the Board.
| Name
and Principal Position |
Year | Fees
Paid or Earned in Cash ($) |
Stock Awards ($) |
Option Awards ($) |
Non-equity incentive plan compensation |
Non-qualified incentive plan compensation |
All
Other Compensation ($) |
Total ($) | ||||||||||||||||||||||
| Dr. Nicholas Plotnikoff | 2016 | — | 16,202 | — | — | — | 16,202 | |||||||||||||||||||||||
| Non-Executive Chairman of the Board | 2015 | — | — | — | — | — | — | — | ||||||||||||||||||||||
| Edward Teraskiewicz, | 2016 | 60,000 | (1) | 42,500 | (1) | 339,077 | — | — | — | 441,577 | ||||||||||||||||||||
| Director | 2015 | 50,000 | (1) | 53,990 | (1) | — | — | — | — | 103,990 | ||||||||||||||||||||
| Dr. Clifford | 2016 | 55,000 | (2) | 42,500 | (2) | 339,077 | — | — | — | 436,577 | ||||||||||||||||||||
| Selsky | 2015 | — | — | — | — | — | — | |||||||||||||||||||||||
| Paul | 2016 | 55,000 | (3) | 42,500 | (3) | 309,539 | — | — | — | 407,039 | ||||||||||||||||||||
| Akin | 2015 | — | — | — | — | — | — | |||||||||||||||||||||||
| 94 |
(1) For services as a director in 2016, Mr. Teraskiewicz was entitled to receive payment of $60,000. Mr. Teraskiewicz agreed to defer payment until 2017. In 2016, Mr. Teraskiewic was awarded 250,000 shares of common stock the Company for services as director. The stock was recorded at $42,500. In 2016, he also received 2,000,000 warrants, the cost of which was recorded at $339,077. In 2015, Mr. Teraskiewic was awarded 350,000 shares of common stock the Company for services as director. The stock was recorded at $53,990.
(2) For services as a director in 2016, Dr. Selsky was entitled to receive payment of $55,000. Dr. Selsky agreed to defer $27,500 of this amount until 2017. In 2016, Dr. Selsky was awarded 250,000 shares of common stock the Company for services as director. The stock was recorded at $42,500. In 2016, he also received 2,000,000 warrants, the cost of which was recorded at $339,077
(3) For services as a director in 2016, Mr. Akin was entitled to receive payment of $55,000. Mr. Akin agreed to defer $55,000 of this amount until 2017. In 2016, Mr. Akin was awarded 250,000 shares of common stock the Company for services as director. The stock was recorded at $42,500. In 2016, he also received 2,000,000 warrants, the cost of which was recorded at $309,539.
For 2016, all members of the Board of Directors who are not our employees, or Non-Employee Directors, currently receive an annual retainer of $60,000 per year, payable monthly in arrears. In addition, Non-Employee Directors are eligible to receive stock upon their appointment to the Board. Certain members of the Board of Directors are also entitled to receive an annual payment of Company shares for their services. We do not currently have minimum stock ownership guidelines for Non-Employee Directors.
We reimburse Non-Employee Directors for actual out-of-pocket costs incurred to attend board meetings. No additional compensation is paid for attendance in person or by telephone at board meetings.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The following table sets forth certain information regarding the ownership of our common stock as of March 16, 2017 (the “Determination Date”) by: (i) each current director of our company; (ii) each of our named executive officers; (iii) all current executive officers and directors of our company as a group; and (iv) all those known by us to be beneficial owners of more than five percent (5%) of our common stock.
Beneficial ownership and percentage ownership are determined in accordance with the rules of the SEC. Under these rules, beneficial ownership generally includes any shares as to which the individual or entity has sole or shared voting power or investment power and includes any shares that an individual or entity has the right to acquire beneficial ownership of within 60 days of the Determination Date, through the exercise of any option, warrant or similar right (such instruments being deemed to be “presently exercisable”). In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of our common stock that could be issued upon the exercise of presently exercisable options and warrants are considered to be outstanding. These shares, however, are not considered outstanding as of the Determination Date when computing the percentage ownership of each other person.
| 95 |
To our knowledge, except as indicated in the footnotes to the following table, and subject to state community property laws where applicable, all beneficial owners named in the following table have sole voting and investment power with respect to all shares shown as beneficially owned by them. Percentage of ownership is based on 326,615,497 shares of common stock and warrants outstanding as of the March 27, 2017. Unless otherwise indicated, the business address of each person in the table below is c/o Immune Therapeutics, Inc., 37 North Orange Avenue, Suite 607, Orlando, Florida 32801.
| Name and Address | Amount
of Beneficial Ownership | Percentage
of Class % | ||||||
| Dr. Nicholas Plotnikoff, Non-Executive Chairman of the Board | 6,650,000 | (1) | 2.0 | |||||
| Noreen Griffin, Chief Executive Officer and Director | 4,817,710 | (2) | 1.5 | |||||
| Edward Teraskiewicz, Director (5) | 3,600,000 | (3) | 1.1 | |||||
| Paul Akin, Director (5) | 6,885,712 | 2.1 | ||||||
| Dr. Clifford Selsky, Director (5) | 2,250,000 | 0.7 | ||||||
| Dr. Fengping Shan, Chief Science Officer | 3,702,499 | 1.1 | ||||||
| Peter Aronstam, Chief Financial Officer | 425,000 | (4) | 0.2 | |||||
| Dr. Joseph M. Fortunak, VP of Global R&D and Chemical Development | 1,000,000 | 0.3 | ||||||
| Robert J. Dailey (6) | 16,496,667 | 5.0 | ||||||
| All directors and officers as a group (8 persons) | 45,827,588 | 14.0 | ||||||
| (1) | These shares are held by the Plotnikoff Family Trust. An additional 700,000 shares are in the process of being transferred from the Plotnikoff Family Trust to a trustee in the TNI Pharma bankruptcy. The trust does not have any beneficial ownership of these shares, nor does it control or direct their voting interests in any manner or have dispositive control. |
| (2) | Represents 704,009 shares held in the name of Griffin Enterprises Group, Inc., which is 50% owned and managed by Robert Wilson, Ms. Griffin’s son; 2,685,000 shares held by the Griffin Family Trust, an irrevocable trust that is not managed by Ms. Griffin; and 1,428,701 shares held by Noreen Griffin, individually. |
| (3) | 1,600,000 of these shares are held by the Raster Investments, Inc., an irrevocable trust that is not managed by Mr. Teraskiewicz. Warrants for 2,000,000 shares were issued to Mr. Teraskiewicz. |
| (4) | Represents warrants to purchase 425,000 shares of common stock, as follows: (i) for price of $1.00 (75,000 shares until December 2018); for price of $0.14 (250,000 shares until December 2019); and for price of $0.17 (100,000 shares until June 2021). In addition, Mr. Aronstam holds a warrant to purchase 250,000 shares of Cytocom Inc. at $0.14 per share until December 2019. |
| (5) | As compensation for their appointment as directors, the Company has agreed to issue Dr. Selsky and Mr. Akin each a total of 500,000 shares of restricted common as follows: 250,000 shares of stock will be issued and fully earned upon their agreement to serve as directors, and 250,000 shares will be issued and fully earned after each serves as director for 12 consecutive months. Warrants for 2,000,000 shares each were issued to Mr. Akin and Dr. Selsky. |
| (6) | Mr. Dailey’s address is 401 First Street, Los Altos, California 94002. |
(*) Beneficial ownership of less than 1.0 percent is omitted.
| 96 |
| Item 13. | Certain Relationships and Related Party Transactions |
In 2012, Webfoot, Inc. provided a loan to the Company in the amount of $121,128. Webfoot, Inc. is owned by the son of Noreen Griffin, the Company’s Chief Executive Officer. On February 21, 2013, the Company entered into a formal loan agreement to evidence the amount owed by the Company. In the nine months ended September 30, 2014, the Company repaid $40,000 of the loan. On September 30, 2014, the remaining balance of $71,128 owed under the agreement, together with accrued and unpaid interest totaling $10,212, was converted into 813,404 shares of the Company’s common stock in full and final settlement of the loan. The loss on conversion was $162,681.
In 2012, Noreen Griffin made payments totaling $30,000 on the Company’s behalf covering the costs of incorporation and merger-related expenses. On February 13, 2013, the Company entered into a formal loan agreement to evidence repayment of the amount owed on December 31, 2012. On September 30, 2014, the full balance of the loan, together with accrued and unpaid interest totaling $2,870, was converted into 328,701 shares of the Company’s common stock in full and final settlement of the loan. The loss on conversion was $65,740.
In 2012, Griffin Enterprises, Inc. made payments totaling $46,000 on the Company’s behalf covering the cost of incorporation and merger-related expenses. Griffin Enterprises, Inc. is wholly owned by Noreen Griffin. On February 13, 2013, the Company entered into a formal loan agreement to evidence repayment of the amount owed on December 31, 2012. On September 30, 2014, the full balance of the loan, together with accrued and unpaid interest totaling $4,401, was converted into 504,009 shares of the Company’s common stock in full and final settlement of the loan. The loss on conversion was $100,802.
On May 1, 2016, the Company formalized the terms under which Robert Wilson, the son of Noreen Griffin, the Company’s Chief Executive Officer, is employed. The terms of the agreement define Mr. Wilson’s base salary and health insurance coverage. In the year ended December 31, 2016, the Company paid compensation to Mr. Wilson totaling $70,000 ($nil in the year ended December 31, 2015).
On January 3, 2013, the Company formalized the terms under which Kelly O’Brien Wilson, the daughter-in-law of Noreen Griffin, the Company’s Chief Executive Officer, is employed. Ms. Wilson had been working with the Company since 2012 and her three-year employment agreement is effective as of December 1, 2012. The terms of the agreement define her base salary, a grant of a common stock, and health insurance coverage. Ms. Wilson was issued 500,000 shares of common stock of the Company in January 2014. In October 2015, the expiration date of the agreement was extended to October 1, 2018. In the year ended December 31, 2016, the Company paid compensation to Ms. Wilson totaling $156,582 ($163,954 in the year ended December 31, 2015).
In May 2013, the Company executed a Patent License Agreement with Professor Fengping Shan. The Company obtained exclusive rights to develop and commercialize the licensed technology. The licensed technology is the intellectual property developed and owned by Professor Shan (i) relating to the treatment of a variety of diseases and conditions with MENK including multiple forms of lymphoma and cancer and (ii) a treatment method for humans infected with the HLTV-III (AIDS) virus including AIDS and AIDS related complex (ARC). The licensed technology includes the methods and formulations for these treatments including but not limited to all INDs, communications with regulatory agencies, patient data, and letters relating to these treatments. The licensed technology also includes the following patents: 200710158742.7 MENK, its application is in treating leukemia and other blood cancers; No. 200710051586.4 MENK, its application is in preparation of human and animal vaccines; No. 200610046249.1, a nasal spray formulation containing MENK; No. 201210290150.1 LDN, combined with MENK, its application is in preparation of an anticancer drug (Pending); No. 201210302259.2 LDN, combined with MENK, its application is in preparation of leukophoresis for anticancer (Pending); No. 200810229085.5 Compound MENK as a drug for colon cancer and pancreatic cancer; No. 200910011030.1, Naltrexone as well as analogues being anticancer drug. In August 2014, Professor Shan executed an Assignment under which he transferred to the Company his entire right, title and interest in and to the licensed patents under the Patent License Agreement and to CN 201210302259 Application of combination of LDN and MENK to preparation of anti-cancer drug for the consideration of 500,000 shares of our common stock.
The Company entered into a Sale of Technology Agreement with Dr. Nicholas P. Plotnikoff on March 4, 2012, wherein Dr. Plotnikoff agreed to transfer and assign all of his rights, title and interest in: European Patent United Kingdom, Germany, France, Ireland EP 1401471 BI Methods for inducing sustained immune response; Russian Patent Russian Federation patent number 2313364; The Patent Office of the People’s Republic of China, Application No.: 200810165784.8 China Patent CN1015113407 A The Patent Office of the People’s Republic of China ISSN: 1006-2858 CN 21-1349/R; Patent Agencies Government of India Patent, Application number 1627/KOLNP/2003 number 220265 an Enkephalin Peptide Composition; and the US Patent Pending, US Patent Application 10/146.999 e. The Company received all the production formulations and technology designs from Dr. Plotnikoff necessary for the manufacturing, formulation, production and protocols of the MENK treatment of cancer and HIV/AIDS. As consideration for entering into the Sale of Technology Agreement, Dr. Plotnikoff received 8,000,000 shares of common stock, a royalty of a single-digit percentage on all sales of MENK and was granted the position of Non-Executive Chairman of the Board of Directors.
| 97 |
On April 23, 2012, the Company acquired TNI BioTech IP, Inc. (“TNI IP”), its wholly-owned subsidiary, in exchange for 20,250,000 shares of the Company’s common stock, of which 8,000,000 shares were issued to Dr. Plotnikoff for TNI IP’s acquisition of the patents and the remaining 12,250,000 shares were issued to the founders of TNI IP in exchange for all of their right, title and interest in their TNI IP shares.
In April 2015, the Company entered into an agreement with LDN Information Management, LLC, (“LDNIM”), a California limited liability company. Paul Akin, a member of the Company’s board of directors, is the managing member of LDNIM. Under the contract, LDNIM is required to provide outreach and educational services to physicians relating to the Company’s LDN formulation in Arizona, California, Hawaii, Nevada, Oregon and Washington (the Territory”). This includes attending seminars and conferences and otherwise providing educational information about the Company’s licensing of its LDN formulation to KRS Global Biotechnology, Inc. (“KRS”).
LDNIM has agreed that its outreach efforts should result in a threshold set of patients filling prescriptions at KRS, reaching 60,000 patients by December 31, 2017 (excluding patients who purchase from KRS via prescriptions from the specified physicians). As compensation, the Company has agreed to pay LDNIM monthly an amount equal to ten percent (10%) of the licensing revenue paid by KRS for revenues generated in the Territory. Revenues generated from patients that have placed an order with KRS via prescriptions written by specified physicians are not included. In a related agreement, in May 2015, Mr. Akin also received 190,000 shares of common stock of the Company, for which the Company recorded an expense of $12,350.
| Item 14. | Principal Accounting Fees and Services |
The following table sets forth fees billed to us by Turner, Stone & Company, L.L.P., our independent registered public accounting firm, during the fiscal years ended December 31, 2016 and December 31, 2015 for: (i) services rendered for the audit of our annual financial statements and the review of our quarterly financial statements; (ii) services by our independent registered public accounting firms that are reasonably related to the performance of the audit or review of our financial statements and that are not reported as audit fees; (iii) services rendered in connection with tax compliance, tax advice and tax planning; and (iv) all other fees for services rendered.
| December 31, 2016 | December 31, 2015 | |||||||
| Audit Fees | $ | 78,553 | $ | 66,167 | ||||
| Audited Related Fees | $ | — | $ | — | ||||
| Tax Fees | $ | 1,841 | $ | 4,000 | ||||
| All Other Fees | $ | 7,120 | $ | 14,095 | ||||
| 98 |
| Item 15. | Exhibits, Financial Statement Schedules. |
Exhibits Schedule
The following exhibits are filed with this Annual Report:
| Exhibit | Description | |
| 3.1 | Restated Articles of Incorporation* | |
| 3.2 | Bylaws* | |
| 10.1 | Sale and Assignment of Patent and Transfer of Technology Agreement with Nicholas Plotnikoff †> | |
| 10.2 | Agreement with Professor Shan †> | |
| 10.3 | Patent License Agreement with Penn State Research Foundation r† | |
| 10.4 | Patent License Agreement Between Immune Therapeutics, Inc. and Jacqueline Young for the intellectual property of Dr. Bernard Bihari# | |
| 10.5 | Strategic Framework Agreement for Cooperation with Hubei Qianjiang Pharmaceutical Company, and Commissioned Processing Contract, and Addendum to Venture Cooperation †> | |
| 10.6 | Malawi Memorandum of Agreement with GB Oncology & Imaging Group Ltd.# | |
| 10.7 | Letter of Intent between GB Oncology & Imaging Group Ltd. and G-Ex Technologies St. Maris Pharma Limited # | |
| 10.8 | Distribution Agreement in Nigeria with GB Pharma Holdings Inc. †> | |
| 10.9 | ViPharma Agreement †> | |
| 10.10 | Strategic Framework Agreement, Addendum to Venture Cooperation and Supplementary Agreement with Hubei Qianjiang Pharmaceutical Company (MENK) (filed as Exhibit 10.11 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference). | |
| 10.11 | Manufacturing Agreement with Laboratorios Ramos (and English translation)> | |
| 10.12 | Engagement Agreement for Corporate Advisory Services by the Brewer Group? | |
| 10.13 | Employment Agreement with Noreen Griffin (filed as Exhibit 10.14 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference). | |
| 10.14 | Master Service Agreement with American Peptide Company (filed as Exhibit 10.17 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference). | |
| 10.15 | Agreement with AHAR Pharma (filed as Exhibit 10.18 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference). | |
| 10.16 | Consulting agreement with Dr. Graham Burton (filed as Exhibit 10.19 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference). | |
| 10.17 | Promissory note to Robert J. Dailey, issued February 6, 2014, for $200,000 (filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, and incorporated herein by reference). | |
| 10.18 | Promissory note to Robert J. Dailey, issued March 7, 2014, for $200,000 (filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, and incorporated herein by reference). | |
| 10.19 | Promissory note to Robert J. Dailey, issued March 28, 2014, for $200,000 (filed as Exhibit 10.3 to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, and incorporated herein by reference). | |
| 10.20 | Promissory Note Settlement Agreement, dated September 26, 2014, between Immune Therapeutics, Inc. and Robert Dailey for notes equaling $399,000 µ | |
| 10.21 | Promissory Note Settlement Agreement, dated September 26, 2014, between Immune Therapeutics, Inc. and Robert Dailey for notes equaling $400,000 µ | |
| 10.22 | Distribution Agreement, effective June 11, 2014, between Airmed Biopharma Limited and PanAm Global Logistics, Inc. (filed as Exhibit 1.1 to our Current Report on Form 8-K dated June 25, 2014, and incorporated herein by reference). | |
| 10.23 | Immune Therapeutics, Inc. 2014 Stock Incentive Plan (filed as Appendix A to our Definitive Proxy Statement on Schedule 14A filed on July 16, 2014, and incorporated herein by reference). | |
| 10.24 | Supplementary Agreement on New Drug Methionine – Enkephalin Cooperation, dated August 6, 2014, between Immune Therapeutics, Inc. and Hubei Qianjiang Pharmaceutical Co., Ltd. (filed as Exhibit 1.1 to our Current Report on Form 8-K dated August 13, 2014, and incorporated herein by reference). |
| 99 |
| 10.25 | Assignment by Professor Fengping Shan Ph.D. to Immune Therapeutics, Inc., executed August 6, 2014 (filed as Exhibit 1.2 to our Current Report on Form 8-K dated August 13, 2014, and incorporated herein by reference). | |
| 10.26 | Clinical Trial Agreement, dated October 20, 2014, between TNI BioTech International Ltd. and American Hospital and Resorts (filed as Exhibit 10.1 to our Current Report on Form 8-K dated October 30, 2014, and incorporated herein by reference). | |
| 10.27 | Contract for the Compounding of Pharmaceutical Products, dated December 8, 2014, between Immune Therapeutics, Inc. and KRS Global Biotechnology, Inc. (filed as Exhibit 10.1 to our Current Report on Form 8-K dated December 15, 2014, and incorporated herein by reference). | |
| 10.28 | Services Agreement, dated December 15, 2014, between Immune Therapeutics, Inc. and Aronstam Management Services, Inc. µ | |
| 10.29 | Royalty Agreement (Filed as Exhibit 10.1 to our Current Report on Form 8-K dated May 21, 2015, and incorporated herein by reference.) | |
| 10.30 | Change of Transfer Agent Agreement (Filed as Exhibit 99.1 to our Current Report on Form 8-K dated January 26, 2015, and incorporated herein by reference.) | |
| 10.31 | Real Property Lease for Florida Office and Extension | |
| 10.32 | Compounding Contract with Complete Pharmacy and Medical Solutions, LLC (filed as Exhibit 10.1 to our Current Report 8-K dated May 20, 2016, and incorporated herein by reference.) | |
| 10.33 | Employment Agreement with Robert Wilson (filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q for the quarter ended June 30, 2016, and incorporated herein by reference). | |
| 10.34 | Letter of Intent with Super T-Cell Cancer Co., (filed as Exhibit 10.1 to our Current Report on Form 8-K dated April 21, 2016, and incorporated herein by reference.) | |
| 10.35 | Extension of Employment Agreement with Noreen Griffin | |
| 10.36 | Extension of AHAR Agreement | |
| 10.37 | Extension of Employment Agreement with Peter Aronstam | |
| 10.38 | Agreement with Joyce Banda | |
| 10.39 | Patent License Agreement with Cytocom | |
| 10.40 | Agreement with Cote Orphan | |
| 10.41 | Securities Purchase Agreement with JMJ Financial | |
| 10.42 | Agreements with Jill Smith | |
| 21.1 | List of Subsidiaries µ | |
| 31.1 | Chief Executive Officer certification under Section 302 of the Sarbanes-Oxley Act of 2002 µ | |
| 31.2 | Chief Financial Officer certification under Section 302 of the Sarbanes-Oxley Act of 2002 µ | |
| 32.1 | Chief Executive Officer certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 µ |
| * | filed with the Form 10 Registration Statement filed with the SEC on April 22, 2013 and the Amendment No. 1 to the Form 10 Registration Statement filed with the SEC on June 7, 2013 and incorporated herein by reference. |
| # | filed with the Amendment No. 1 to the Form 10 Registration Statement filed with the SEC on June 7, 2013 and incorporated herein by reference. |
| + | filed with the Amendment No. 2 to the Form 10 Registration Statement filed with the SEC on July 18, 2013 and incorporated herein by reference. |
| ^ | filed with the Amendment No. 3 to the Form 10 Registration Statement filed with the SEC on August 23, 2013 and incorporated herein by reference. |
| ? | filed with the Amendment No. 4 to the Form 10 Registration Statement filed with the SEC on September 25, 2013 and incorporated herein by reference. |
| Î | filed with the Amendment No. 5 to the Form 10 Registration Statement filed with the SEC on October 11, 2013 and incorporated herein by reference. |
| † | Portions of this exhibit have been redacted pursuant to a confidential treatment order granted by the Securities and Exchange Commission. |
| > | Filed with the Amendment No. 6 to the Form 10 Registration Statement filed with the SEC on November 21, 2013 and incorporated hereby by reference. |
| R | Filed with the Amendment No. 7 to the Form 10 Registration Statement filed with the SEC on January 22, 2015 and incorporated hereby by reference. |
| µ | Filed herewith. |
| 100 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Immune Therapeutics, Inc.
Orlando, Florida
We have audited the accompanying consolidated balance sheets of Immune Therapeutics, Inc. and its subsidiaries (the “Company”) as of December 31, 2016 and 2015 and the related consolidated statements of operations, stockholders’ deficit and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Immune Therapeutics, Inc. and its subsidiaries as of December 31, 2016 and 2015, and the results of their consolidated operations and their cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations since inception and has a working capital deficiency both of which raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| /s/ Turner, Stone & Company, L.L.P. | |
| Dallas, Texas | |
March 31, 2017 |
| F-1 |
IMMUNE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
DECEMBER 31, 2016 AND 2015
| 2016 | 2015 | |||||||
| ASSETS | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 74,389 | $ | 23,149 | ||||
| Accounts receivable | - | 16,197 | ||||||
| Total current assets | 74,389 | 39,346 | ||||||
| Fixed Assets: | ||||||||
| Computer equipment, net of accumulated depreciation of $7,888 and $6,331 respectively | 1,850 | 1,682 | ||||||
| Deposits | 200 | 200 | ||||||
| Total assets | $ | 76,439 | $ | 41,228 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 1,818,605 | $ | 1,924,672 | ||||
| Accrued liabilities | 3,156,759 | 1,281,039 | ||||||
| Current portion of notes payable | 4,225,419 | 2,793,701 | ||||||
| Total current liabilities | 9,200,783 | 5,999,412 | ||||||
| Total liabilities | 9,200,783 | 5,999,412 | ||||||
| Commitments and Contingencies (Note 10) | ||||||||
| Stockholders’ Deficit: | ||||||||
| Common stock - par value $0.0001; 500,000,000 shares authorized; 250,428,133 and 174,850,047 shares issued and outstanding respectively | 25,043 | 17,485 | ||||||
| Additional paid in capital | 360,420,026 | 343,434,786 | ||||||
| Stock issuances due | 962,429 | 1,140,303 | ||||||
| Prepaid services | (822,500 | ) | (660,417 | ) | ||||
| Accumulated deficit | (365,718,976 | ) | (349,861,173 | ) | ||||
| Deficit attributable to common stockholders | (5,133,978 | ) | (3,857,732 | ) | ||||
| Non-controlling interest | (3,990,366 | ) | (2,100,452 | ) | ||||
| Total stockholders’ deficit | (9,124,344 | ) | (5,958,184 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 76,439 | $ | 41,228 | ||||
The accompanying footnotes are an integral part of these consolidated financial statements
| F-2 |
IMMUNE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2016 AND 2015
| 2016 | 2015 | |||||||
| Revenues, net | $ | 3,463 | $ | 16,197 | ||||
| Operating expenses: | ||||||||
| Selling, general and administrative | 3,886,599 | 2,734,414 | ||||||
| Research and development expense | 444,165 | 977,203 | ||||||
| Stock issued for services general and administrative | 5,212,304 | 6,240,143 | ||||||
| Warrant valuation | 4,730,726 | 46,189 | ||||||
| Depreciation and amortization expense | 1,557 | 594,785 | ||||||
| Impairment of intangible assets | - | 5,226,352 | ||||||
| Total operating expenses | 14,275,351 | 15,819,086 | ||||||
| Loss from operations | (14,271,888 | ) | (15,802,889 | ) | ||||
| Other expense: | ||||||||
| Interest expense | (3,446,564 | ) | (270,989 | ) | ||||
| Impairment of investment | - | (32,000 | ) | |||||
| Loss on settlement of debt | (2,100,549 | ) | (843,573 | ) | ||||
| Total other expense | (5,547,113 | ) | (1,146,562 | ) | ||||
| Net loss | $ | (19,819,001 | ) | $ | (16,949,451 | ) | ||
| Net loss attributable to non-controlling interest | (297,600 | ) | (2,706,939 | ) | ||||
| Net loss attributable to common shareholders | (19,521,401 | ) | (14,242,512 | ) | ||||
| Basic loss per share attributed to common shareholders | $ | (0.09 | ) | $ | (0.09 | ) | ||
| Weighted average number of shares outstanding | 216,687,993 | 153,247,023 | ||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-3 |
IMMUNE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY/ (DEFICIT)
FOR THE YEARS ENDED DECEMBER 31, 2016 AND 2015
| Common Stock | Additional Paid-in |
Stock To | Prepaid | Accumulated | Non-Controlling | |||||||||||||||||||||||||||
| Shares | Amount | Capital | Be Issued | Services | Deficit | Interest | Total | |||||||||||||||||||||||||
| Balance December 31, 2014 | 134,417,210 | $ | 13,442 | $ | 337,985,787 | $ | 847,279 | $ | (3,553,185 | ) | $ | (333,547,378 | ) | $ | 606,487 | $ | 2,352,432 | |||||||||||||||
| Issuance of common stock for prepaid services | 16,672,504 | 1,667 | 2,730,708 | 535,000 | (1,436,000 | ) | - | - | 1,831,376 | |||||||||||||||||||||||
| Amortization of prepaid services | - | - | - | - | 4,328,768 | - | - | 4,328,768 | ||||||||||||||||||||||||
| Issuance of common stock for investment | 400,000 | 40 | 31,960 | - | - | - | - | 32,000 | ||||||||||||||||||||||||
| Issuance of common stock for legal settlement | 1,000,000 | 100 | 79,900 | - | - | - | - | 80,000 | ||||||||||||||||||||||||
| Issuance of common stock for interest expense | 62,500 | 6 | 15,619 | - | - | - | - | 15,625 | ||||||||||||||||||||||||
| Issuance of common stock in exchange for debt | 14,058,833 | 1,406 | 1,954,971 | (257,000 | ) | - | - | 1,699,377 | ||||||||||||||||||||||||
| Issuance of common stock for cash and exercise of warrants | 8,239,000 | 824 | 589,651 | 15,025 | - | - | 605,500 | |||||||||||||||||||||||||
| Issuance and modification of common stock warrants | - | 46,189 | - | - | 46,189 | |||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (14,242,512 | ) | (2,706,939 | ) | (16,949,451 | ) | |||||||||||||||||||||
| Balance December 31, 2015 | 174,850,047 | $ | 17,485 | $ | 343,434,786 | $ | 1,140,303 | $ | (660,417 | ) | $ | (347,789,889 | ) | $ | (2,100,452 | ) | $ | (5,958,184 | ) | |||||||||||||
| Issuance of common stock for prepaid services | 32,633,910 | 3,262 | 5,678,998 | (307,875 | ) | (3,072,535 | ) | - | - | 2,301,850 | ||||||||||||||||||||||
| Amortization of prepaid services | - | - | - | - | 2,910,452 | - | - | 2,910,452 | ||||||||||||||||||||||||
| Discount on warrants issued in connection with notes payable | - | - | 682,665 | - | - | - | - | 682,665 | ||||||||||||||||||||||||
| Issuance of common stock for legal expenses | 150,000 | 15 | 22,485 | - | - | - | - | 22,500 | ||||||||||||||||||||||||
| Issuance of common stock for interest expense | 4,621,296 | 463 | 403,287 | - | - | - | - | 403,750 | ||||||||||||||||||||||||
| Issuance of common stock in exchange for debt | 36,235,380 | 3,625 | 5,242,272 | 155,001 | - | - | - | 5,400,898 | ||||||||||||||||||||||||
| Issuance of common stock for cash and exercise of warrants | 1,937,500 | 193 | 224,807 | (25,000 | ) | - | - | - | 200,000 | |||||||||||||||||||||||
| Issuance and modification of common stock warrants | - | - | 4,730,726 | - | - | - | - | 4,730,726 | ||||||||||||||||||||||||
| Issuance of stock of Cytocom | 1,592,314 | (1,592,314 | ) | - | ||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (19,521,401 | ) | (297,600 | ) | (19,819,001 | ) | |||||||||||||||||||||
| Balance December 31, 2016 | 250,428,133 | $ | 25,043 | $ | 360,420,026 | $ | 962,429 | $ | (822,500 | ) | $ | (365,718,976 | ) | $ | (3,990,366 | ) | $ | (9,124,344 | ) | |||||||||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-4 |
IMMUNE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2016 AND 2015
| 2016 | 2015 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net loss including non-controlling interest | $ | (19,819,001 | ) | $ | (16,949,451 | ) | ||
| Adjustments to reconcile net loss to net cash flows used in operating activities: | ||||||||
| Depreciation | 1,557 | 2,552 | ||||||
| Amortization | - | 592,233 | ||||||
| Stock issued for services | 2,324,350 | 1,911,375 | ||||||
| Amortization of stock issued for prepaid services | 2,910,452 | 4,328,768 | ||||||
| Impairment of intangible assets | - | 5,226,352 | ||||||
| Impairment of investment | - | 32,000 | ||||||
| Loss on settlement of debt and accounts payable | 2,100,549 | 843,573 | ||||||
| Stock warrant expense | 4,730,726 | 46,189 | ||||||
| Stock issued for loan expenses and interest | 403,750 | 15,625 | ||||||
| Amortization of debt discount | 989,035 | |||||||
| Expenses paid by lenders | 115,202 | |||||||
| Accounts receivable write off | 2,661 | |||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | 13,536 | (16,197 | ) | |||||
| Prepaid expenses and other current assets | - | 39,983 | ||||||
| Accrued liabilities | 2,846,461 | 194,201 | ||||||
| Accounts payable | 540,983 | 900,484 | ||||||
| Net cash used in operating activities | (2,839,739 | ) | (2,832,313 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||
| Purchase of computer equipment | (1,725 | ) | - | |||||
| Net cash provided used in investing activities | (1,725 | ) | - | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
| Proceeds from sale of common stock and exercise of warrants | 200,000 | 605,500 | ||||||
| Proceeds from notes payable | 2,768,631 | 2,057,975 | ||||||
| Repayment of notes payable | (75,927 | ) | - | |||||
| Net cash provided by financing activities | 2,892,704 | 2,663,475 | ||||||
| Increase/(decrease) in cash | 51,240 | (168,838 | ) | |||||
| Cash and cash equivalents, beginning of year | 23,149 | 191,987 | ||||||
| Cash and cash equivalents, end of year | $ | 74,389 | $ | 23,149 | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-5 |
IMMUNE THERAPEUTICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2016 and 2015
| 2016 | 2015 | |||||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION: | ||||||||
| Cash paid for interest | $ | 29,567 | $ | 20,500 | ||||
| SUPPLEMENTAL DISCLOSURES OF NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||
| Conversion of notes payable, accrued liabilities and accounts payable to common stock | $ | 3,363,731 | $ | 855,804 | ||||
| Accrued liabilities converted to note payable | $ | 388,143 | $ | - | ||||
| Settlement of note payable by issuance of new note payable | $ | - | $ | 50,000 | ||||
| Notes payable for expenses paid by lender | $ | 115,201 | $ | 46,933 | ||||
| Debt discounts on notes payable | $ | 1,007,716 | $ | - | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-6 |
1. Organization and Description of Business
Immune Therapeutics, Inc. (the “Company,” “we,” or “our”) was initially incorporated in Florida on December 2, 1993 as Resort Clubs International, Inc. (“Resort Clubs”). It was formed to manage and market golf course properties in resort markets throughout the United States. Galliano International Ltd. (“Galliano”) was incorporated in Delaware on May 27, 1998 and began trading in November 1999 through the filing of a 15C-211. On November 10, 2004, Galliano merged with Resort Clubs. Resort Clubs was the surviving corporation. On August 23, 2010, Resort Clubs changed its name to pH Environmental Inc. (“pH Environmental”).
On April 23, 2012, pH Environmental completed a name change to TNI BioTech, Inc., and on April 24, 2012, we executed a share exchange agreement for the acquisition of all of the outstanding shares of TNI BioTech IP, Inc. On September 4, 2014, a majority of our shareholders approved an amendment to our Amended and Restated Articles of Incorporation, as amended, to change our name to Immune Therapeutics, Inc. We filed our name change amendment with the Secretary of State of Florida on October 27, 2014 changing our name to Immune Therapeutics, Inc.
The Company currently has an office in Orlando, Florida. In July 2012, the Company’s focus turned to acquiring patents that would protect and advance the development of new uses of opioid-related immune- therapies, such as low dose naltrexone (“LDN”) and Methionine [Met5]-enkephalin (“MENK”).
In October 2012, the Company formed TNI BioTech International, Ltd., a BVI company in Tortola, British Virgin Islands, which was set up to allow the Company to market and sell LDN in those countries outside the U.S. in which we have been able to obtain approval to sell the Company’s products.
In August 2013, the Company formed its United Kingdom subsidiary, TNI BioTech, LTD (the “UK Subsidiary”). The UK Subsidiary received approval to be considered a micro, small or medium-sized enterprise (“SME”) with the European Medicines Agency (“EMA”) on August 21, 2013. The designation provides the UK Subsidiary with significant discounts when holding meetings or submitting filings to the EMA. On September 19, 2013, the UK Subsidiary submitted a pre-submission package to the EMA regarding Crohn’s Disease. The EMA granted the UK Subsidiary a meeting that took place on September 27, 2013. The UK Subsidiary is eligible to benefit from the provisions for administrative and financial assistance for SMEs set out in Regulation (EC) No 2049/2005. The Company will apply to obtain EMA benefits once funding becomes available.
In December 2013, the Company formed a new subsidiary, Cytocom Inc., to focus on conducting LDN and MENK clinical trials in the United States. In December 2014, the Company completed the distribution of common stock of Cytocom Inc. to its shareholders. As part of the transaction, the Company retained exclusive rights to all international patents, in-country approvals, formulations, trademarks, manufacturing, marketing, sales, and distributions rights in emerging nations, including Africa, Central America, South America, Russia, India, China, Far East, and The Commonwealth of Independent States (former Soviet Union). The Company will continue to have access to existing clinical data as well as any new data generated by Cytocom Inc. during drug development. On December 8, 2014, the number of Cytocom Inc. shares of common stock that were issued to our shareholders totaled 113,242,522 shares. In connection with the transaction, Cytocom Inc. issued 140,100,000 shares of its common stock to the Company, which gave the Company a 55.3% stake in Cytocom Inc. on that date. In April 2016, the Board of Directors and a majority of shareholders of Cytocom approved a reverse stock split of Cytocom’s outstanding common stock with one new share of stock for each twenty old shares of common stock. Cytocom effectuated and finalized the reverse split in June 2016. At December 31, 2016, the Company’s equity interest had been further reduced to 13%, by subsequent issuances of Cytocom common stock to shareholders in settlement of notes payable.
| F-7 |
In March 2014, the Company incorporated Airmed Biopharma Limited, an Irish corporation with an address in Dublin, Ireland, and Airmed Holdings Limited, an Irish company domiciled in Bermuda. The Irish companies were set up to benefit from incentives granted by the Irish government for the establishment of pharmaceutical companies (many of the world’s leading pharmaceutical companies have located in Ireland), and so that the Company could take advantage of Ireland’s status as a member of the European Union and the European Economic Area. An Irish limited liability company enjoys a low corporate income tax rate of 12.5%, one of the lowest in the world. The Irish-domiciled company hopes to qualify for tax incentives for Irish holding/headquartered companies and to benefit from the network of double tax treaties that reduce withholding taxes. TNI BioTech International, Ltd. will manage our international distribution, using product that is manufactured in Ireland and elsewhere.
We are focused on the development and commercialization of therapeutic treatments for cancer, HIV/AIDS and autoimmune diseases and immune disorders by combating these severe and fatal diseases through the stimulation and/or regulation of the body’s immune system. Our growth strategy includes the near-term commercialization of our existing immunotherapies targeting cancer, Crohn’s disease and/or HIV/AIDS.
Going Concern
The Company has incurred significant net losses since inception and has relied on its ability to fund its operations through private equity financings. Management expects operating losses and negative cash flows to continue at more significant levels in the future. As the Company continues to incur losses, transition to profitability is dependent upon the successful development, approval, and commercialization of its product candidate and the achievement of a level of revenues adequate to support the Company’s cost structure. The Company may never achieve profitability, and unless and until it does, the Company will continue to need to raise additional cash. Management intends to fund future operations through additional private or public debt or equity offerings, and may seek additional capital through arrangements with strategic partners or from other sources. Based on the Company’s operating plan, existing working capital at December 31, 2016 was not sufficient to meet the cash requirements to fund planned operations through December 31, 2017 without additional sources of cash. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements have been prepared assuming that the Company will continue as a going concern and do not include adjustments that might result from the outcome of this uncertainty. This basis of accounting contemplates the recovery of the Company’s assets and the satisfaction of liabilities in the normal course of business.
The Company experienced a net loss from operations of $19,819,001 and used cash and cash equivalents for operations in the amount of $2,839,739 during the year ended December 31, 2016, resulting in a stockholders’ deficit of $9,124,344.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with United States (U.S.) generally accepted accounting principles (U.S. GAAP) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented.
The Company qualifies as an “emerging growth company” as defined in Section 101 of the Jumpstart our Business Startups Act (“JOBS Act”) as we do not have more than $1,000,000,000 in annual gross revenue for the year ended December 31, 2016. We are electing to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act.
Revenue Recognition
Revenue from product sales is recognized upon passage of title and risk of loss to customers. Provisions for discounts, rebates and sales incentives to customers, and returns and other adjustments are provided for in the period the related sales are recorded. Historical data are not yet readily available and reliable for use in estimating the amount of the reduction in gross sales. Revenue from the launch of a new product, from an improved version of an existing product, or for shipments in excess of a customer’s normal requirements will be recorded when the conditions noted above are met. In those situations, management will record a returns reserve for such revenue, if necessary. If in future the Company participates in selling arrangements that include multiple deliverables (e.g., instruments, reagents, procedures, and service agreements), under these arrangements, the Company will recognize revenue upon delivery of the product or performance of the service and will allocate the revenue based on the relative selling price of each deliverable, which will be based primarily on vendor specific objective evidence. Revenue from license of product rights is recorded over the periods earned.
| F-8 |
In May 2014, the Financial Accounting Standards Board issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers, which provides a single comprehensive model for accounting for revenue from contracts with customers and will supersede most existing revenue recognition guidance. Early adoption is not permitted. The standard becomes effective for the Company in the first quarter of 2018. The Company is currently evaluating the effect, if any, that the standard will have on its consolidated financial statements and related disclosures.
Use of Estimates
The preparation of the Company’s financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from such estimates.
Cash, Cash Equivalents, and Short-Term Investments
The Company considers all highly liquid investments with original maturities at the date of purchase of three months or less to be cash equivalents. Cash and cash equivalents include bank demand deposits, marketable securities with maturities of three months or less at purchase, and money market funds that invest primarily in certificates of deposits, commercial paper and U.S. government and U.S. government agency obligations. Cash equivalents are reported at fair value.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk are primarily cash and cash equivalents. The Company is exposed to credit risk, subject to federal deposit insurance, in the event of a default by the financial institutions holding its cash and cash equivalents to the extent of amounts recorded on the balance sheets. The cash accounts are insured by the Federal Deposit Insurance Corporation up to $250,000. At December 31, 2016, the Company has no cash balances in excess of insured limits.
Segment and Geographic Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating segment and does not segment the business for internal reporting or decision making.
Fair Value of Financial Instruments
In accordance with the reporting requirements of Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 825, “Financial Instruments”, the Company calculates the fair value of its assets and liabilities which qualify as financial instruments under this standard and includes this additional information in the notes to the financial statements when the fair value is different than the carrying value of those financial instruments. Cash and accounts payable are accounted for at cost which approximates fair value due to the relatively short maturity of these instruments. The carrying value of notes payable also approximate fair value since they bear market rates of interest and other terms. None of these instruments are held for trading purposes.
Fair Value Measurements
The ASC Topic 820, Fair Value Measurement, defines fair value, establishes a framework for measuring fair value in accordance with U.S. generally accepted accounting principles, and requires certain disclosures about fair value measurements. In general, fair values of financial instruments are based upon quoted market prices, where available. If such quoted market prices are not available, fair value is based upon internally developed models that primarily use, as inputs, observable market-based parameters. Valuation adjustments may be made to ensure that financial instruments are recorded at fair value. These adjustments may include amounts to reflect counterparty credit quality and the customer’s creditworthiness, among other things, as well as unobservable parameters. Any such valuation adjustments are applied consistently over time.
| F-9 |
Fixed Assets
Fixed assets are stated at cost, less accumulated depreciation. Depreciation is determined on a straight-line basis over the estimated useful lives of the assets, which generally range from three to five years. Maintenance and repairs are charged against expense as incurred. Depreciation expense for the years ended December 31, 2016 and December 31, 2015 was $1,557 and $2,552, respectively.
Intangible Assets
Costs incurred to acquire and/or develop the Company’s product licenses and patents are capitalized and amortized by straight-line methods over estimated useful lives of ten to sixteen years. Intangible assets are stated at the lower of cost or estimated fair value. During the years ended December 31, 2016 and December 31, 2015, the Company capitalized $nil and $nil respectively, of such costs incurred for the Company’s acquisition of licenses for the patents. (See Note 10). Amortization expense for the years ended December 31, 2016 and December 31, 2015 was $0 and $592,233, respectively.
Impairment of Long-Lived Assets
The Company evaluates long-lived assets for impairment whenever events or change in circumstances indicate that the carrying amount of an asset may not be recoverable as prescribed by ASC Topic 360-10-05, “Property, Plant and Equipment.” If the carrying amount of the asset, including any intangible assets associated with that asset, exceeds its estimated undiscounted net cash flow, before interest, the Company will recognize an impairment loss equal to the difference between its carrying amount and its estimated fair value. In 2015, the Company determined that the carrying amount recorded for the acquisition of licenses and patents related to LDN were impaired, and recorded an impairment loss of $5,226,352. No such impairment charges were recorded in 2016.
Research and Development Costs
Research and development costs are charged to expense as incurred and are typically comprised of salaries and benefits, pre-clinical studies, clinical trial activities, drug development and manufacturing, fees paid to consultants and other entities that conduct certain research and development activities on the Company’s behalf and third-party service fees, including clinical research organizations and investigative sites. Costs for certain development activities, such as clinical trials are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the financial statements as operating expenses.
Income Taxes
The Company follows ASC Topic 740, Income Taxes, which requires recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are based on the differences between the financial statements and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Deferred tax assets are reduced by a valuation allowance to the extent management concludes it is more likely than not that the asset will not be realized. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled.
The standard addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC Topic 740, the Company may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the tax authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position should be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate settlement. ASC Topic 740 also provides guidance on de-recognition, classification, interest and penalties on income taxes, accounting in interim periods and requires increased disclosures. At the date of adoption, and as of December 31, 2016 and 2015, the Company does not have a liability for unrecognized tax uncertainties.
| F-10 |
The Company’s policy is to record interest and penalties on uncertain tax positions as income tax expense. As of December 31, 2016, and 2015, the Company has not accrued any interest or penalties related to uncertain tax positions.
Stock-Based Compensation and Issuance of Stock for Non-Cash Consideration
The Company measures and recognizes compensation expense for all share-based payment awards made to employees and directors, including employee stock options, based on estimated fair values equaling either the market value of the shares issued or the value of consideration received, whichever is more readily determinable. The majority of the non-cash consideration pertains to services rendered by consultants and others and has been valued at the fair value of the Company’s common stock at the date of the agreement.
The Company’s accounting policy for equity instruments issued to consultants and vendors in exchange for goods and services follows the provisions of ASC Topic 505-50, “Equity-Based Payments to Non-Employees.” The measurement date for the fair value of the equity instruments issued is determined at the earlier of (i) the date at which a commitment for performance by the consultant or vendor is reached or (ii) the date at which the consultant or vendor’s performance is complete.
Non-controlling Interest
In accordance with ASC Topic 810, Consolidation, the Company consolidates Cytocom, Inc. The non-controlling interests in Cytocom represent the interests of outside shareholders in the equity and results of operations of Cytocom.
Net Loss per Share
Basic net loss per share is calculated by dividing the net loss attributable to common stockholders by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by dividing the net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method and the if-converted method. Dilutive common stock equivalents are comprised of common stock purchase warrants outstanding. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding due to the Company’s net loss position.
A calculation of basic and diluted net loss per share follows:
| For
the year ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Historical net loss per share: | ||||||||
| Numerator | ||||||||
| Net loss | $ | (19,819,001 | ) | $ | (16,949,451 | ) | ||
| Non-controlling interest | (297,600 | ) | (2,706,939 | ) | ||||
| Net loss attributed to common stockholders | $ | (19,521,401 | ) | $ | (14,242,512 | ) | ||
| Denominator | ||||||||
| Weighted-average common shares outstanding— | ||||||||
| Denominator for basic and diluted net loss per share | 216,687,993 | 153,247,023 | ||||||
| Basic and diluted net loss per share attributed to common stockholders | $ | (0.09 | ) | $ | (0.09 | ) | ||
| F-11 |
The Company’s potential dilutive securities which include stock and warrants have been excluded from the computation of diluted net loss per share as the effect would be to reduce the net loss per share. Therefore, the weighted-average Common stock outstanding used to calculate both basic and diluted net loss per share is the same.
The following shares of potentially dilutive securities have been excluded from the computations of diluted weighted average shares outstanding as the effect of including such securities would be antidilutive:
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Common Stock Purchase Warrants | 59,191,904 | 9,131,500 | ||||||
Recent Accounting Standards
During the year ended December 31, 2016 and through March 31, 2017, there were several new accounting pronouncements issued by the Financial Accounting Standards Board. Each of these pronouncements, as applicable, has been or will be adopted by the Company. Management does not believe the adoption of any of these accounting pronouncements has had or will have a material impact on the Company’s consolidated financial statements.
The Company qualifies as an “emerging growth company” as defined in Section 101 of the Jumpstart our Business Startups Act (“JOBS Act”) as we do not have more than $1,000,000,000 in annual gross revenue and did not have such amount as of December 31, 2016, our last fiscal year. We are electing to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act.
3. Fixed Assets
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Fixed assets: | ||||||||
| Computer equipment | $ | 9,738 | $ | 8,013 | ||||
| Less accumulated depreciation | (7,888 | ) | (6,331 | ) | ||||
| Property and equipment, net | $ | 1,850 | $ | 1,682 | ||||
The Company utilizes the straight-line method for depreciation, using three to five-year depreciable asset lives. Depreciation expense was not material for all periods presented.
4. Accrued Liabilities
Accrued expenses and other liabilities consist of the following:
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Accrued payroll to officers and others | 1,126,261 | 758,342 | ||||||
| Accrued interest and penalties - notes payable | 1,902,018 | 236,671 | ||||||
| Estimated legal settlement | 128,087 | 282,136 | ||||||
| Other accrued liabilities | 393 | 323 | ||||||
| State payroll taxes | - | 3,567 | ||||||
| Total accrued expenses and other liabilities | $ | 3,156,759 | $ | 1,281,039 | ||||
| F-12 |
5. Notes Payable
Notes payable consist of the following:
| December 31, 2016 | December
31, 2015 | |||||||
| Promissory note issued July 29, 2014 to Ira Gaines. In 2016, the maturity date on the note was extended to December 1, 2017. The note earns interest at a rate of 18% per annum. | $ | 100,000 | $ | 100,000 | ||||
| Promissory notes issued between November 26, 2014 and September 30, 2015, to raise up to $2,000,000 in debt. Lenders earn interest at a rate of 10% per annum, plus a pro-rata share of two percent of the Company’s gross receipts for sales of IRT-103-LDN in perpetuity. Notes will be repaid in 36 monthly installments of principal and interest commencing no later than October 15, 2015. Principal of $425,500 and accrued interest of $37,427 were converted to 5,309,092 shares of common stock in the year ended December 31, 2016, Notes aggregating $286,000 were in default at December 31, 2016, as the Company was unable to pay installments on those notes on their due dates. No demands for repayment have been made by the lenders. | 286,000 | 711,500 | ||||||
| Promissory note issued October 17, 2014 to Roger Bozarth. The note matures on October 17, 2015 and earns interest at a rate of 2% per annum. The lender converted this note and $2,139 in accrued interest to 114,237 shares of common stock in January 2016. | - | 7,000 | ||||||
| Promissory notes issued between May 1, 2015 and December 31, 2016, and maturing between June 14, 2015 and September 30, 2017. Lenders on loans aggregating $505,994 earn interest at rates between 2% and 18% per annum. On loans aggregating $198,500, interest is payable in a fixed amount not tied to a specific interest rate. Notes aggregating $100,000 were in default at December 31, 2016, as the Company was unable to repay those notes on their due dates. No demands for repayment have been made by the lenders. | 704,494 | 669,933 | ||||||
| Promissory note issued January 26, 2015 to Robert J. Dailey. The note is senior to, and has priority in right of payment over, all indebtedness of the Company. The note earns interest at a rate of 2% per annum and was due on July 30, 2015. Principal of $200,000 and accrued interest of $4,778 was converted to 3,722,015 shares of common stock in 2016. | - | 200,000 | ||||||
| Promissory notes issued by Cytocom Inc. between April 29, 2015 and December 31, 2015. Lenders earn interest at rates between 5% and 10% per annum. These notes mature on September 30, 2016. $375,000 of principal and $12,036 in accrued interest was converted to 4,837,960 shares of common stock in 2016. The Company was unable to repay the remaining notes at maturity and the notes are in default, although no demand for repayment has been made by the lenders. | 425,000 | 800,000 | ||||||
| Promissory notes issued in December 2015. Lenders earn interest at a rate of 10% per month. Notes are repayable on March 9, 2016. $30,000 of principal and $49,000 of interest and penalties were converted to 987,500 shares of common stock in 2016. The Company was unable to repay the remaining note at maturity and the note is in default. The Company is obligated to pay late-payment penalties totaling $5,000 per day on the remaining obligation. | 100,000 | 130,000 | ||||||
| F-13 |
| Promissory note issued November 24, 2015 as settlement of amounts owing to a law firm. The Lender earns interest at the rate of 10% per annum. The note and $10,036 in interest was converted to 1,235,536 shares of common stock in July 2016. | - | 175,268 | ||||||
| Promissory notes issued between May 5, 2016 and June 2, 2016 that mature between October 1, 2016 and January 31, 2017, and include stock conversion features, warrants and original issue debt discounts. Notes aggregating $304,882 were paid on February 8, 2017. The remaining note's maturity date was extended and is in default on March 31, 2017. No demand for repayment has been made by the lender. | 554,882 | - | ||||||
| Promissory notes issued to an officer of the Company effective November 3, 2015 and maturing November 3, 2016 for settlement of accrued payroll, bearing interest at 10% per annum and including a stock conversion feature. | 112,737 | - | ||||||
| Promissory note issued in July 2016. The note was repayable on October 5, 2016 but was extended to December 31, 2016. The note earns interest at 6% per month. The Company was unable to repay the note at maturity and the note is in default. | 50,000 | - | ||||||
| Promissory note issued in July 2016 with an original issuance discount of $30,000. Net proceeds were $150,000. The note is repayable on April 7, 2017. | 180,000 | - | ||||||
| Promissory notes issued in August 2016 for $149,854 as a settlement of amounts owed to a law firm. The notes accrue interest at 5% per annum and are payable in 18 equal monthly installments of $8,641.88. The note was in default on December 31, 2016. | 120,987 | - | ||||||
| Promissory notes issued between July 1, 2016 and December 31, 2016. Lenders earn interest at 2% per annum. The notes mature on September 30, 2017. Notes aggregating $239,000 were converted in the fourth quarter of 2016. | 256,000 | - | ||||||
| Notes aggregating $1,354,000 were issued in the fourth quarter of 2016. The notes accrue interest at 2% per annum and mature between November 1, 2017 and December 31, 2017. | 1,354,000 | - | ||||||
| Less: Original issue discounts on notes payable and warrants issued with notes. | (18,681 | ) | - | |||||
| Total | 4,225,419 | 2,793,701 | ||||||
| Less: Current Portion | $ | (4,225,419 | ) | $ | (2,793,701 | ) | ||
| Long-Term debt, less current portion | $ | - | $ | - |
| F-14 |
As of December 31, 2016, the Company had accrued $399,271 in unpaid interest and $1,502,747 in unpaid default penalties. During the year ended December 31, 2016, 4,621,296 shares with a fair value of $403,750 were issued by the Company for interest expense under promissory notes.
As of December 31, 2015, the Company had accrued $236,671 in unpaid interest. During the year ended December 31, 2015, 62,500 shares with a fair value of $15,625 were issued by the Company for interest expense under promissory notes.
6. Capital Structure—Common Stock and Common Stock Purchase Warrants
Each holder of common stock is entitled to vote on all matters and is entitled to one vote for each share held. No holder of shares of stock of any class shall be entitled as a matter of right to subscribe for or purchase or receive any part of any new or additional issue of shares of stock of any class, or of securities convertible into shares of stock or any class, whether now hereafter authorized or whether issued for money, for consideration other than money, or by way of dividend.
As of December 31, 2016 and 2015, the Company was authorized to issue 500,000,000 common shares at a par value of $0.0001 per share.
As of December 31, 2016, the Company had 250,428,133 shares of common stock outstanding and 174,850,047 outstanding as of December 31, 2015.
Stock Warrants
In 2016, the Company issued 50,222,904 warrants, exercisable into one share of common stock of the Company for each warrant at prices between $0.03 and $2.00 per share. The warrants expire between Oct 2017 and Dec 2021.
When the Company sells its stock to stockholders for cash, it periodically issues warrants to those stockholders to acquire additional stock at prices agreed at the date of the original sale. The Company incurs a cost for the rights attached to the warrants, which is calculated using the Black-Scholes Model. This expense is reported in the Statements of Operations above as the Warrant valuation expense.
During 2016, there were no modifications of the terms of any warrants issued by the Company.
Following is a summary of outstanding stock warrants at December 31, 2016 and 2015 and activity during the years then ended:
| Number
of Shares |
Exercise Price |
Weighted Average Price |
||||||||||
| Warrants as of December 31, 2015 | 9,131,500 | $ | 0.07-15 | $ | 1.47 | |||||||
| Issued | 50,222,904 | $ | 0.03-2.00 | $ | 0.16 | |||||||
| Expired | (162,500 | ) | $ | 5.00 | $ | 5.00 | ||||||
| Exercised | 0 | $ | 0 | $ | 0 | |||||||
| Warrants as of December 31, 2016 | 59,191,904 | $ | 0.03-15.00 | $ | 0.35 | |||||||
| F-15 |
Summary of outstanding warrants as of December 31, 2016:
| Expiration Date | Number
of Shares |
Exercise Price |
Remaining Life (years) |
|||||||||
| Fourth Quarter 2017 | 350,000 | $ | 1.50-9.00 | 1.00 | ||||||||
| First Quarter 2018 | 127,500 | $ | 15.00 | 1.25 | ||||||||
| Second Quarter 2018 | 33,334 | $ | 15.00 | 1.50 | ||||||||
| Third Quarter 2018 | 250,000 | $ | 1.50 | 1.75 | ||||||||
| Fourth Quarter 2018 | 6,089,166 | $ | 1.00-1.50 | 2.00 | ||||||||
| First Quarter 2019 | 4,024,000 | $ | 0.50-2.00 | 2.25 | ||||||||
| Second Quarter 2019 | 135,000 | $ | 0.07-0.23 | 2.50 | ||||||||
| Third Quarter 2019 | 260,000 | $ | 0.50-1.50 | 2.75 | ||||||||
| Fourth Quarter 2019 | 400,000 | $ | 0.14 | 3.00 | ||||||||
| Second Quarter 2020 | 300,000 | $ | 0.50 | 3.50 | ||||||||
| Fourth Quarter 2020 | 1,000,000 | $ | 0.20 | 4.00 | ||||||||
| First Quarter 2021 | 12,600,000 | $ | 0.20 | 4.25 | ||||||||
| Second Quarter 2021 | 23,806,237 | $ | 0.03-0.20 | 4.50 | ||||||||
| Third Quarter 2021 | 9,166,667 |
$ | 0.03-0.20 | 4.75 | ||||||||
7. Stock Compensation
Shares Issued for Services
During the years ended December 31, 2016 and 2015, the Company issued 32,633,910 and 16,672,504 shares of common stock respectively for consulting fees. The Company valued these shares based upon the fair value of the common stock at the dates of the agreements. The consulting fees are amortized over the contract periods which are typically between 12 and 24 months. The amortization of prepaid services totaled $2,910,452 and $4,328,768 for the years ended December 31, 2016 and 2015.
8. Income Taxes - Results of Operations
There was no income tax expense reflected in the results of operations for the years ended December 31, 2016 and 2015 because the Company incurred a net loss in both years.
Deferred tax assets:
| 2016 | 2015 | ||||||
| Net operating losses | $ | 27,153,000 |
$ | 22,611,000 | |||
| Stock based compensation | 63,108,000 | 60,914,000 | |||||
| Amortization, depreciation, and impairment | 6,765,000 | 6,765,000 | |||||
| Capitalization of start-up costs for tax purposes | 1,854,000 | 1,854,000 | |||||
| Loss on debt conversion of debt | 1,216,000 | 502,000 | |||||
| Total deferred tax assets | 99,382,000 | 92,646,000 | |||||
| Valuation allowance | (99,382,000 | ) | (92,646,000 | ) | |||
| Total deferred tax assets, net | $ | - | $ | - | |||
The Company has recognized no tax benefit for the losses generated for the periods through December 31, 2016. ASC Topic 740 requires that a valuation allowance be provided if it is more likely than not that some portion or all of a deferred tax asset will not be realized. The Company’s ability to realize the benefit of its deferred tax asset will depend on the generation of future taxable income. Because the Company has yet to recognize revenue, we believe that the full valuation allowance should be provided.
| F-16 |
Our effective tax rate for fiscal years 2016 and 2015 was 0%. Our tax rate can be affected by recurring items, such as tax rates in foreign jurisdictions and the relative amount of income we earn in jurisdictions. It may also be affected by discrete items that may occur in any given year, but are not consistent from year to year.
As of December 31, 2016, we have estimated federal and state income tax net operating loss (“NOL”) carry-forwards of approximately $79,900,000, which will expire in 2032-2035.
| 2016 | 2015 | |||||||||||||||
| Amount | Percent | Amount | Percent | |||||||||||||
| Benefits for income tax at federal statutory rate | $ | 6,738,000 | 34 | % | $ | 5,763,000 | 34 | % | ||||||||
| Change in valuation allowance | (6,983,000 | ) | (34 | )% | $ | (5,762,000 | ) | (34 | ) | |||||||
| Permanent differences | (2,000 | ) | - | (1,000 | ) | - | ||||||||||
| Change in estimates | 247,000 | - | - | - | ||||||||||||
| $ | - | - | $ | - | - | % | ||||||||||
9. Licenses and Supply Agreements
Patent and Subsidiary Acquisition
The Company entered into a share exchange agreement on April 24, 2012 to acquire all of the outstanding shares of TNI BioTech IP, Inc. (“TNI IP”), a biotechnology firm incorporated in Florida and formed to acquire patents related to the treatment of cancer and HIV/AIDS and autoimmune diseases, using Met-enkephalin (“MENK”) and Naltrexone (“LDN”). The goal of TNI IP’s management was to enable mankind and civilization to combat fatal diseases by activating and mobilizing the body’s own immune system using TNI IP’s patented use of MENK. The first patents acquired by TNI IP were acquired from Dr. Nicholas P. Plotnikoff and Professor Fengping Shan in 2012. TNI IP was acquired in exchange for 20,250,000 shares of the Company’s common stock, of which 8,000,000 shares were issued to Dr. Plotnikoff for the acquisition of patents and the remaining 12,250,000 shares were issued to the founders of TNI IP in exchange for all of their right, title and interest in their TNI IP shares. The goodwill arising on the acquisition of TNI BioTech IP, Inc. was valued at $98,000,000 and license agreements arising from the acquisition of TNI IP were valued at $16,006,000.
In connection with the share exchange, we entered into a Sale of Technology Agreement with Dr. Nicholas P. Plotnikoff on March 4, 2012, wherein Dr. Plotnikoff agreed to transfer and assign all of his rights, title and interest in: European Patent United Kingdom, Germany, France, Ireland EP 1401471 BI Methods for inducing sustained immune response; Russian Patent Russian Federation patent number 2313364; The Patent Office of the People’s Republic of China, Application No.: 200810165784.8 China Patent CN1015113407 A The Patent Office of the People’s Republic of China ISSN: 1006-2858 CN 21-1349/R; Patent Agencies Government of India Patent, Application number 1627/KOLNP/2003 number 220265 an Enkephalin Peptide Composition; and the US Patent Pending, US Patent Application 10/146.999 e. The Company received all the production formulations and technology designs from Dr. Plotnikoff necessary for the manufacturing, formulation, production and protocols of the MENK treatment of cancer and HIV/AIDS. As consideration for entering into the Sale of Technology Agreement, Dr. Plotnikoff received 8,000,000 shares of common stock, a royalty of a single-digit percentage on all sales of MENK and was granted the position of Non-Executive Chairman of the Board of Directors.
At the time of the acquisition, the valuation of goodwill and other intangible assets were determined using the fair market price for the Company’s common stock, which were exchanged for shares of TNI IP. In the fourth quarter of 2012, the Company performed an annual valuation to determine whether any goodwill or intangible assets that had been acquired by the Company were impaired. The result of this valuation was that material impairments were identified. The Company recognized an impairment of the goodwill arising on the acquisition of TNI IP of $98,000,000.
| F-17 |
Patent License Agreements
On August 13, 2012, the Company signed an exclusive License Agreement with Ms. Jacqueline Young (the “Young Agreement”) for the intellectual property developed by Dr. Bernard Bihari relating to treatments with opioid antagonists such as naltrexone and Met-enkephalin for a variety of diseases and conditions including malignant lymphoma, chronic lymphocytic leukemia, Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma, chronic herpes virus infections, chronic herpes viral infections such as chronic genital herpes caused by the herpes simplex virus Type 2 and chronic infections due to the Epstein-Barr virus and a treatment method for humans infected with HTLV-III (AIDS) virus, including patients clinically diagnosed as suffering from AIDS and those suffering from AIDS-related complex (ARC). The Bihari patents were acquired in exchange for 540,000 shares of the Company’s common stock with a fair value of $972,000 and assumed liabilities of $400,000, which is payable to Ms. Young over a twenty-four month period in equal installments to reimburse her for the costs of a New York City office in accordance with the Young Agreement. The patent liability at December 31, 2013 totaled $118,333. The cost of the patent totaled $1,372,000. Additionally, the Company will pay the licensor a royalty payment of 1% of gross MENK sales and provide the licensor a position as non-executive chairman of the Company. The Young Agreement is valid for the life of the patents and expires on a country by country basis in each country where patent rights exist, upon the expiration of the last to expire patent in each country or in the event the patent in such country is held to be invalid and/or unenforceable (by a court or government body of competent jurisdiction) or admitted to be invalid or unenforceable. Additionally, we can cancel the Young Agreement upon 120 days’ written notice and shall pay all royalties and fees that have accrued under the Young Agreement. We have the exclusive rights to the intellectual property; however, Ms. Young retains a right to practice the patents licensed under the Young Agreement solely for noncommercial, academic research purposes.
On December 24, 2012, the Company signed an agreement for the acquisition of patent rights (the “Smith Agreement”) for the intellectual property of Dr. Jill Smith and LDN Research Group, LLC (collectively, the “Licensor Parties”), whose members are Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Moshe Rogosnitzky and orphan drug designation by the FDA to a novel late-stage drug, trademarked “LDN,” for the treatment of Pediatric Crohn’s disease. The patent covers methods and formulations for treatment of the inflammatory and ulcerative diseases of the bowel, using naltrexone in low doses as an opioid antagonist. These patents were acquired in exchange for 300,000 shares of our common stock with a fair value of $2,715,000 and payment of $165,384 (consisting of a $100,000 initial license fee and payment of $65,384 of expenses), which totaled $2,880,384.
The Smith Agreement requires the Company to (i) use commercially reasonable efforts to develop, commercialize, market and sell licensed products in a manner consistent with a business plan, (ii) expend a minimum amount of funds per annum to develop and commercialize licensed products as soon as practicable, (iii) obtain all requisite regulatory approvals needed to use or sell licensed products in the field of use, and (iv) make the first commercial sale of a licensed product by March of 2017.
The Company is required to pay an annual license fee, an annual running royalty on net sales of each licensed product or a minimum royalty, whichever is greater, and a sublicense fee on payments received by the Company from sublicensees. The Company has an exclusive, worldwide license to make, have made, use, lease, import, offer for sale and sell licensed products and to use the method under the patent rights. The Smith Agreement will terminate on the expiration or abandonment of the last patent to expire or ten years after the sale of the first licensed product. The Company may terminate the Smith Agreement upon 90 days’ written notice, provided all sublicenses are terminated and all amounts due and owing are paid to the Licensor Parties. The Licensor Parties may terminate the agreement ten days’ after notice to the Company if the Company is ten days late in payment or there is a breach that remains uncured for ten days after written notice of such breach.
The Company is also required to pay milestone payments after substantial achievement of certain milestone events for each licensed product including payment: upon initiation of each Phase III trial; upon positive completion of each Phase III clinical trial of the therapeutic use of an LDN compound in the field of use; when a New Drug Application (“NDA”) is accepted for review by the FDA; and when FDA approval to market the NDA is approved. The Company will issue shares upon reaching certain milestones including upon the first dosing of the first patient in a Phase III clinical trial for each licensed product, upon the first sale of each licensed product, and upon the achievement of a set dollar amount in cumulative sales for each licensed product covered by NDAs.
As part of the Smith Agreement, the Company has the right to apply to the FDA for the transfer of the orphan drug status for the use of naltrexone for the treatment of pediatric Crohn’s disease and ulcerative colitis, the Investigation New Drug Application (“IND”), and the right to acquire the relevant clinical data set from Dr. Jill Smith. Dr. Jill Smith made arrangements to transfer the IND to the Company as well as the relevant clinical data set, and the FDA has acknowledged that the Company is now the sponsor for this IND.
| F-18 |
On September 24, 2014, the Company and the Licensor Parties jointly agreed to terminate the Smith Agreement, and in place thereof, have the Licensor Parties grant a similar license in their patent rights to Cytocom Inc. pursuant to a Patent License Agreement between the Licensor Parties, Cytocom Inc. and the Company with substantially similar terms as set forth in the Smith Agreement. Pursuant to this agreement, the Company issued 1,000,000 shares of its common stock valued at $270,000, upon execution to the Licensor Parties and the Company guaranteed the obligations of Cytocom Inc. to the Licensor Parties under the agreement.
On January 18, 2013, the Company signed an exclusive licensing agreement with The Penn State Research Foundation to license all of the intellectual property developed by Dr. Ian S. Zagon, Dr. Patricia J. McLaughlin and Dr. Jill P. Smith for the treatment of cancer titled “Opioid Growth Factor and Cancer” and “Combination Therapy with Opioid Growth Factor and Taxanes for the Treatment of Cancer” (the “Foundation Agreement”).
The Foundation Agreement requires the Company to: (a) use commercially reasonable efforts to develop, commercialize, market and sell licensed products in a manner consistent with a business plan; (b) expend a minimum amount of funds per annum to develop and commercialize licensed products as soon as practicable; (c) obtain all requisite regulatory approvals needed to use or sell licensed products in the field of use; and (d) make the first commercial sale of a licensed product by December 31, 2016.
The Foundation Agreement provides that the Company must pay to the licensor an initial license fee, a license maintenance fee on each anniversary of the effective date of the Foundation Agreement, and an annual running royalty on net sales for each licensed product or a minimum royalty, whichever is greater. In addition, the Company must pay a sublicense fee on payments received by the Company from sublicensees.
The Foundation Agreement also requires the Company to make payments upon the achievement of certain milestone events including: initiation of each Phase II trial; initiation of each Phase III trial; when the NDA is accepted for review by the FDA; and when FDA approval to market is approved. The Company must also issue shares upon certain milestones including upon the first dosing of the first patient in a Phase II clinical trial for each licensed product, upon the first dosing of the first patient in a Phase III clinical trial for each licensed product, upon the first sale of each licensed product, and upon the achievement of a set dollar amount of cumulative sales for each licensed product covered by NDAs.
The Foundation Agreement terminates on the expiration or abandonment of the last patent to expire or become abandoned. The Company may terminate the Foundation Agreement at any time upon 60 days’ prior written notice and ceasing to make and sell all licensed products, the termination of all sublicenses and payment of all monies owed under the Foundation Agreement. The licensor may terminate the agreement 30 days after notice to the Company if the Company is 30 days late in payment or a breach that remains uncured for 45 days after written notice of such breach.
In May of 2013, the Company executed a Patent License Agreement with Professor Fengping Shan (the “Shan Agreement”) pursuant to which it obtained exclusive rights to develop and commercialize the licensed technology. The licensed technology is the intellectual property developed and owned by Professor Shan (i) relating to the treatment of a variety of diseases and conditions with MENK including multiple forms of lymphoma and cancer and (ii) a treatment method for humans infected with the HLTV-III (AIDS) virus including AIDS and AIDS related complex (ARC). The licensed technology includes the methods and formulations for these treatments including all INDs, communications with regulatory agencies, patient data, and letters relating to these treatments. The licensed technology also includes certain patents developed by Professor Shan. Under the Shan Agreement, the Company must issue 500,000 shares to Professor Shan upon final transfer of the licenses, and reimburse Professor Shan for all out of pocket expenses in connection with the patents. The Company will pay Professor Shan a running royalty on gross sales subject to decreases if third party intellectual property is needed to complete such sale or product. The Shan Agreement lasts for the duration of each of the licensed patents however the Company may terminate the Shan Agreement on 120 days’ written notice to Professor Shan.
| F-19 |
On August 6, 2014, Professor Fengping Shan executed an Assignment pursuant to which he transferred to the Company his entire right, title and interest in and to the licensed patents under the Shan Agreement and CN 201210302259 Application of combination of low-dose naltrexone and methionine-enkephalin to preparation of anti-cancer drug for the consideration of 500,000 shares of common stock valued at $140,000.
10. Commitments and Contingencies
Malawi Treatment Facilities
On July 14, 2012, GB Oncology and Imaging Group LTD (“GBOIG”) in partnership with the Company signed a letter of intent agreement to collaborate with the Government of Malawi to assist in expanding the treatment of cancer, HIV/AIDS and other infectious diseases.
In December of 2014, the Government of Malawi completed an oncology clinic at the Queen Elizabeth Central Hospital in Blantyre, Malawi for the treatment of cancer and infectious diseases. In 2015, the Company submitted protocols seeking permission from the Pharmacy, Medicines and Poisons Board of Malawi (“PMPB”) to conduct two trials involving Lodonal™ in Malawi:
| a. | The first protocol, submitted jointly with The Jack Brewer Foundation (“JBF Worldwide”), received PMPB approval on November 11, 2015. The protocol covers a 12-month trial for a “Single Visit Approach to Cervical Cancer Prevention.” The approach is designed to deliver a preventive and simple procedure that can be performed in a clinical setting without the use of a laboratory and to allow for immediate treatment of any precancerous lesions utilizing Wallach LL100 Cryosurgical systems. The protocol provides for 50% of the patient group to be put on Lodonal™ to determine if the drug lowers the number of opportunistic infections during the year, and if it can be shown that LDN increases CD4, CE8, NK and T cell count, which would show that the incidence rates of opportunistic infection could decrease with Lodonal™ and that Lodonal™ could be used as a prophylaxis to prevent substantial HIV-related morbidity in Malawi. The PMPB approved the trial in late 2016 and recruitment began in late 2016 and continued through first quarter of 2017, with the trial now ongoing. |
| b. | The second protocol, which has not yet been approved, covers a trial using Lodonal™ for the treatment of cancer. The Company has put this trial on hold as it may not be necessary with the approval in Nigeria in addition to the pending approval in Kenya and Senegal for Lodonal™ for the treatment of cancer. |
Distribution Agreements in Nigeria
Effective November 9, 2012, we signed an exclusive Distribution Agreement with G-Ex Technologies/St. Maris Pharma and GB Pharma Holdings, LLC for the Federal Republic of Nigeria.
The parties have been unable to perform under the agreement because a certificate of free sale was not obtained by the Company until November of 2013, and no extension has been granted.
In October 2013, the Company announced the signing of a Distribution Agreement with AHAR Pharma, a Nigerian company, to market Lodonal™, in Nigeria for the treatment of autoimmune diseases and cancer. AHAR intends to distribute Lodonal™ through a local distributor network, an Internet client base and directly to hospitals, pharmacists and doctors in Nigeria. The Company expects to implement the agreement in 2016. Under the agreement, the Company is obligated to provide delivery of an initial supply of between 1 million and 1.5 million doses of Lodonal™ product to cover AHAR Pharma’s first-year purchase commitment.
In August 2015, the Company announced the signing of a letter of intent with GB Pharma/AHAR and Fidson Healthcare Plc., in terms of which Fidson will promote LodonalTM upon execution of a definitive agreement between the companies and receipt of NAFDAC and other approvals to distribute LodonalTM in Nigeria.
| F-20 |
Agreements with Hubei Qianjiang Pharmaceutical Company
On October 18, 2012, the Company and Hubei Qianjiang Pharmaceutical Co., Ltd. (“Qianjiang Pharmaceutical”), signed a Venture Cooperation Agreement on New Drug Methionine Enkephalin (the “Venture Agreement”) pursuant to which Qianjiang Pharmaceutical acquired an exclusive license for the production of MENK in China. The Venture Agreement requires that Qianjiang Pharmaceutical conduct drug research and pilot testing for MENK, organize pre-clinical studies, and apply for clinical trials for MENK with the Chinese State Food and Drug Administration. Under the Venture Agreement, Qianjiang Pharmaceutical must open a co-administration account for the development of MENK in China. Qianjiang Pharmaceutical must pay the Company, upon the marketing of MENK products, a half-year amount equaling 6% of its gross sales from MENK of the preceding half year. The Company may cancel the Venture Agreement if Qianjiang Pharmaceutical does not pay expenses for a period exceeding six months or does not commence clinical trials within 12-months after receiving certain approvals. Qianjiang Pharmaceutical may cancel the Venture Agreement if the Company fails to perform its obligations for a period of six months or the failure to receive approval of clinical trials is due to the Company’s MENK technologies. The Venture Agreement was amended on February 24, 2013 to expand the clinical trials from pancreatic to both pancreatic and liver cancer and amended on March 6, 2014 to require Qianjiang Pharmaceutical to commence studies and clinical trials in China and place funds in the co-administration account.
On August 6, 2014, the Company entered into a Supplementary Agreement on New Drug Methionine – Enkephalin Cooperation (the “Amendment”) with Qianjiang Pharmaceutical, amending the Venture Agreement, as amended. The Company and Qianjiang Pharmaceutical executed the Amendment to accelerate clinical trials in both the United States and China, and agreed to immediately initiate three month Good Laboratory Practice (“GLP”) Toxicology Studies (rat and dog) within 30 days of signing the Amendment. The Amendment requires that the GLP Toxicology Studies Trials are conducted in China in accordance with international standards and standards acceptable to the FDA and that the studies include the following:
Exploratory Toxicology (nGLP)
| ● | Dose range finding studies | |
| ● | Different species and methods of administration | |
| ● | Multiple dosing regimens | |
| ● | Estimate the response vs. dose given |
Definitive Toxicology (GLP)
| ● | Performed in collaboration with Calvert Laboratories (USA) and MPI/Medicillon (China) | |
| ● | General toxicology studies | |
| ● | Different species and methods of administration | |
| ● | Immunogenicity study with NHPs |
Special Toxicology Studies (planned)
Pursuant to the Amendment, Qianjiang Pharmaceutical has made certain funds available from the co-administrative account opened by Qianjiang Pharmaceutical under the Venture Agreement, in accordance with an approved budget and timeline set forth in the Amendment. A portion of these funds are expected to be used by Cytocom to run PK and Dosing trials for MENK in the United States in 2016. The Amendment requires Cytocom and Qianjiang Pharmaceutical to meet with the China State Food and Drug Administration to determine that PK and Dosing Trials completed in the United States will be acceptable. All developments and trials run by Cytocom in the U.S. or the European Union will be used for requesting registration approval in China.
In February 2013, the Company signed a Strategic Framework Agreement for Cooperation with Qianjiang Pharmaceutical. Under the agreement, the parties will work together to further the development of new products and conduct research and development on the Company’s licensed patented technology. Specifically, the parties aim to co-invest to develop and market products focusing on HIV, cancer and related autoimmune system therapies, develop co-ventured manufacturing facilities in China, and develop co-ventured distribution of the developed products in China and Africa. The agreement does not have a definitive term, as each new agreement resulting from the cooperation will set forth the material terms, including, but not limited to, fees, duration and termination therein.
In December of 2016 Qianjiang Pharmaceutical completed the following documents:
| F-21 |
Exploratory Toxicology (nGLP)
| ● | Dose range finding studies | |
| ● | Different species and methods of administration | |
| ● | Multiple dosing regimens | |
| ● | Estimate the response vs. dose given |
Definitive Toxicology (GLP)
| ● | Performed in collaboration with Calvert Laboratories (USA) and MPI/Medicillon (China) | |
| ● | General toxicology studies | |
| ● | Different species and methods of administration | |
| ● | Immunogenicity study with NHPs |
In addition to the pharmacology and toxicology studies, Qianjiang Pharmaceutical and China Peptide completed the formulation and CMC necessary to scale up manufacturing of MENK.
Contract Manufacturing Agreements
On May 16, 2016, the Company entered into an agreement with Complete Pharmacy and Medical Solutions, LLC (“CPMS”) to compound, package and distribute the LDN tablets, capsules and/or creams in the United States. The initial term of the agreement is three years, with the option to renew for an additional year. The agreement may be terminated by (i) mutual agreement, (ii) in the event of a breach, provided however that if the Company terminates the agreement, the Company will be required to reimburse CPMS for all unused packaging materials for the LDN, which unused packaging materials CPMS will provide to IMUN. If CPMS does not receive and ship at least 1,000 orders (prescriptions) during the term of the agreement, the Company will be required to reimburse CPMS for 100% of the “ramp up costs” (defined as all costs and expenses of labor and materials related to the testing, and required FDA and other governmental documentation/approvals of test data) of providing and producing the LDN, even where the Company cancels/terminates the agreement, which provision shall survive the cancellation/termination of the agreement.
On October 25, 2016, the Company and Acromax Dominicana, SA ("Acromax") entered into a contract for manufacturing of LDN tablets, capsules and/or creams ("Agreement"). Subject to the terms and conditions of the Agreement, Acromax will obtain all necessary licenses and permits to carry out the manufacturing and packaging of LDN in exchange for a fixed fee per tablet plus an additional fee for packaging, shipping and customs clearance. The Agreement has an initial term of five years unless terminated by either party in accordance with the terms.
In January of 2017, Acromax obtained from the Ministry of Public Health and Social Assistance a Medications and Specialized Pharmaceuticals Registration Certification for LodonalTM, which allows for the manufacture and sale of LodonalTM in the Dominican Republic and for export. The Ministry also issued a Certificate of Pharmaceutical Product for Nigeria, Kenya, Senegal and Malawi, which will allow for the export of LodonalTM to those countries where we have drug and marketing approval.
Operating Leases
At December 31, 2016, the Company was a party to agreements to lease office space in Orlando, Florida. Cash rental expense for the years ended December 31, 2016 and 2015 was $16,041 and $53,205 respectively.
Legal Proceedings
On November 11, 2016, the Company received a fax containing a copy of a lawsuit supposedly filed against the Company on November 10, 2016. The Company has not been served and has not been provided a case number or evidence that the suit has been filed and is relying on the information in the facsimile received as the basis for this disclosure. Apparently, Kacem Enterprise, Inc. is suing the Company and certain of its consultants and their related entities for $21,777 plus interest, costs and attorneys’ fees for fees Kacem Enterprise claims it is owed for travel related expenses incurred by certain consultants of the Company. The suit was supposedly filed in the Circuit Court of Arlington, Virginia. The Company has not accrued any amount for these claims, on the basis that the claims should have been filed only against the consultants.
| F-22 |
11. Related Party Transactions
None.
12. Subsequent Events
Between January 1, 2017 and March 31, 2017, the Company issued shares as follows:
| Number
of Shares | ||||
| Issuance of common stock to employees and consultants | 6,045,460 | |||
| Issuance of common stock in exchange for debt | 10,010,638 | |||
| Issuance of common stock pursuant to a court order and settlement agreement | 3,000,000 | |||
| Issuance of common stock for the exercise of a warrant | 1,656,447 | |||
As of March 29, 2017, the Company had outstanding 273,640,638 shares of common stock.
Between January 1, 2017 and March 23, 2017, the Company issued 150,000 warrants for services and employment, exercisable into one share of common stock of the Company for each warrant at a price of $0.20 per share. The warrants expire in February 2022.
Between January 1, 2017 and March 23, 2017, the Company borrowed $735,000.
On February 8, 2017, the Company paid $321,845 in principal and interest to repay a note payable.
| F-23 |
In accordance with Section 13 or 15(d) of the Exchange Act, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Immune Therapeutics, Inc. | ||
| Date: March 31, 2017 | By: | /s/ Noreen Griffin |
| Name: | Noreen Griffin | |
| Title: | Chief Executive Officer | |
| (Principal Executive Officer) | ||
| By: | /s/ Peter Aronstam | |
| Name: | Peter Aronstam | |
| Title: | Chief Financial Officer | |
| (Principal Accounting Officer) | ||
In accordance with the Exchange Act, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Person | Capacity | Date | ||
| /s/ Nicholas Plotnikoff | Chairman of the Board | March 31, 2017 | ||
| Nicholas Plotnikoff | ||||
| /s/ Noreen Griffin | Director | March 31, 2017 | ||
| Noreen Griffin | ||||
| /s/ Paul Akin | Director | March 31, 2017 | ||
| Paul Akin | ||||
| /s/ Clifford Selsky | Director | March 31, 2017 | ||
| Clifford Selsky | ||||
| /s/ Edward Teraskiewicz | Director | March 31, 2017 | ||
| Edward Teraskiewicz |
| 101 |