Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Pulse Biosciences, Inc. | plse-20161231xex32_1.htm |
| EX-31.2 - EX-31.2 - Pulse Biosciences, Inc. | plse-20161231xex31_2.htm |
| EX-31.1 - EX-31.1 - Pulse Biosciences, Inc. | plse-20161231xex31_1.htm |
| EX-21.1 - EX-21.1 - Pulse Biosciences, Inc. | plse-20161231xex21_1.htm |
| EX-10.1 - EX-10.1 - Pulse Biosciences, Inc. | plse-20161231xex10_1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
____________________________________________
Form 10-K
____________________________________________
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-34899
____________________________________________
Pulse Biosciences, Inc.
(Exact name of registrant as specified in its charter)
____________________________________________
|
Nevada |
46-5696597 |
|
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
|
|
|
|
849 Mitten Road, Suite 104 Burlingame, CA |
94010 |
|
(Address of principal executive offices) |
(Zip Code) |
(Registrant’s telephone number, including area code): (650) 697-3939
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
Title of Each Class |
Name of Each Exchange on Which Registered |
|
Common Stock, par value $0.001 per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
☐ |
Accelerated filer |
☐ |
|
Non-accelerated filer |
☐ (Do not check if a smaller reporting company) |
Smaller reporting company |
☒ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
Aggregate market value of registrant’s common stock held by non-affiliates of the registrant on June 30, 2016, based upon the closing price of Common Stock on such date as reported by NASDAQ Capital Markets, was approximately $54,677,000. Shares of voting stock held by each officer and director have been excluded in that such persons may be deemed to be affiliates. This assumption regarding affiliate status is not necessarily a conclusive determination for other purposes.
Number of shares outstanding of the issuer’s common stock as of March 3, 2017: 14,136,220
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the registrant’s definitive Proxy Statement relating to its 2017 Annual Meeting of Stockholders to be held on May 16, 2017 are incorporated by reference into Part III of this Form 10-K where indicated. Such Proxy Statement will be filed with the U.S. Securities and Exchange Commission within 120 days after the end of the fiscal year to which this report relates.
|
|
Page |
|
|
PART I |
||
|
Item 1. |
3 | |
|
Item 1A. |
16 | |
|
Item 1B. |
37 | |
|
Item 2. |
37 | |
|
Item 3. |
37 | |
|
Item 4. |
37 | |
|
|
||
|
PART II |
||
|
Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and |
38 |
|
Item 6. |
41 | |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
42 |
|
Item 7A. |
50 | |
|
Item 8. |
51 | |
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
71 |
|
Item 9A. |
71 | |
|
Item 9B. |
72 | |
|
|
||
|
PART III |
||
|
Item 10. |
73 | |
|
Item 11. |
73 | |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
73 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
73 |
|
Item 14. |
73 | |
|
|
||
|
PART IV |
||
|
Item 15. |
74 | |
|
|
||
| 75 | ||
| 76 | ||
2
SPECIAL NOTE ABOUT FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, or Annual Report, contains “forward-looking statements” that involve substantial risks and uncertainties. In some cases, you can identify forward-looking statements by the following words: “may,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue,” “ongoing” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. These statements relate to future events or our future financial performance or condition and involve known and unknown risks, uncertainties and other factors that could cause our actual results, levels of activity, performance or achievement to differ materially from those expressed or implied by these forward-looking statements.
You should read this Annual Report and the documents that we reference elsewhere in this Annual Report completely and with the understanding that our actual results may differ materially from what we expect as expressed or implied by our forward-looking statements. In light of the significant risks and uncertainties to which our forward-looking statements are subject, you should not place undue reliance on or regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified timeframe, or at all. We discuss many of these risks and uncertainties in greater detail in this Annual Report, particularly in Part I. Item 1A. “Risk Factors.” These forward-looking statements represent our estimates and assumptions only as of the date of this Annual Report regardless of the time of delivery of this Annual Report. Except as required by law, we undertake no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise after the date of this Annual Report.
In this Annual Report on Form 10-K, references to “Pulse,” “Pulse Biosciences,” “we,” “us,” “our” and the “Company” refer to Pulse Biosciences, Inc. and its wholly owned subsidiaries, unless expressly indicated or the context otherwise requires.
Part I
Item 1. Business
General
We are a medical technology company developing a novel tissue treatment platform based upon our proprietary Nano-Pulse Stimulation (NPS) technology. We believe NPS will provide unique benefits to patients in a wide variety of medical applications. We are currently pursuing applications in immuno-oncology, dermatology, general tissue treatment and veterinary medicine, and we may pursue many others in the future
Nano-Pulse Stimulation is a non-thermal, precise, focal drug-free tissue treatment technology that initiates cell death within treated tissue. NPS utilizes nanosecond pulsed electric fields to induce cell signaling and the activation of cellular pathways by creating transient nanopores in cellular membranes and organelles. Once created, these transient nanopores allow ions to pass through these membranes, which disrupts cellular function and initiates cell death. NPS cell death eliminates treated tissue cells with a minimal inflammatory response, leading to a favorable healing process and the replacement of treated tissue cells with healthy tissue cells.
In pre-clinical models of cancerous lesions, NPS has been shown to induce immunogenic cell death (ICD), a process that leads to the exposure of the unique cancer cell antigens to the immune system, resulting in the generation of cytotoxic T-cells and the mounting of an adaptive immune response targeted against those cells, without any observed toxic side effects. Based on this pre-clinical research, we believe NPS can offer a novel tumor treatment therapy, as a monotherapy and in combination with other therapies.
The PulseTxTM System (PulseTx) is our proprietary NPS delivery system comprised of a tunable nanosecond pulse generation system and interchangeable tissue applicators, designed to enable the application of NPS across a variety of tissue treatment applications. We believe the unique biological response of cells to our novel NPS technology enables Pulse Biosciences the opportunity to pursue therapeutic applications across a wide array of tissues types, including benign and cancerous lesions. The favorable healing characteristics of NPS afford opportunities in applications such a as dermatology and aesthetics, while the ability to initiate ICD holds promise in key applications such as immune-oncology.
Pulse Biosciences strategy is to deploy the PulseTx System in pilot clinical studies to help identify those applications where NPS represents a high value opportunity. Based on these results and the market opportunity we will pursue the required regulatory clearances and ultimately commercialize these opportunities.
3
Our Proprietary Nano-Pulse Stimulation Technology
We are developing a therapeutic tissue treatment platform based upon our proprietary NPS technology. NPS is a local, non-thermal, and drug-free treatment that can physically stimulate cell death, a process in which cells systematically eliminate themselves to make way for new cells. The NPS cell death process triggers a cascade of cellular events that in preclinical models has been shown to induce a sustained adaptive immune response when employed in cancerous tumor cells, producing a durable systemic therapeutic benefit from a local treatment.
Programmed cell death is a normal process exhibited by many cells in the human body when they are no longer functioning properly. It involves a slow “digestion” of cellular proteins and DNA in the cells that are then recognized and removed by the immune system. When this process results in ICD, it stimulates the immune system to generate an immune response that actively seeks out and destroys any similar cells in the body. Pre-clinical studies suggest that NPS can stimulate programmed cell death and ICD in cancer cells.
We believe the unique characteristics of NPS cell death will translate to positive clinical outcomes and may establish NPS as a superior treatment modality across a variety of potential applications, including oncology, dermatology, and other minimally invasive applications where current treatment modalities do not provide benefits comparable to those afforded by NPS.
NPS exerts an electrical force on water molecules, driving them into the lipid bilayer, and producing transient, water-filled nanopores in cell and organelle membranes.
|
|
|
|
|
|
|
These transient pores allow ions to pass through them, and this can have several immediate effects including the release of calcium ions from the endoplasmic reticulum and the termination of the mitochondrial membrane potential. Downstream effects of NPS include initiating a signaling cascade that we believe can result in ICD in immunogenic lesions. ICD is a process by which cells are induced to die in a manner that activates the immune system to both clear the dying tumor cell and enroll cytotoxic T cells (CD8 +) and other immune system cells to recognize and eliminate cells of the same tumor type. Alternatively, in non-immunogenic lesions, we believe NPS can initiate a positive healing response with little inflammation. |
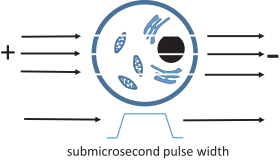
It is this nanoporation effect on internal cellular membranes that differentiates our technology from other energy -based therapies, such as irreversible electroporation (IRE) and radiofrequency ablation (RFA).
We believe we are a uniquely positioned medical technology company with the intellectual property, technology, and know-how to commercialize our NPS technology. Many other medical device companies produce products for treating and ablating tumors using a number of different modalities, including the use of extreme heat (e.g. RFA, microwave ablation or electrocauterization) or cold (e.g. cryoablation), or weaker electric fields with much longer pulses (e.g. IRE). The use of these modalities generally leads to immediate cellular necrosis. We believe that NPS differs significantly as it offers a non-thermal and non-ionizing technology that can be used to precisely treat a volume of tissue with minimal inflammatory response, potentially reducing collateral damage to surrounding tissue with a favorable healing profile. We believe these attributes, as well as the ability to induce ICD, will enable the use of NPS in a number of high value applications.
The cellular response to NPS is believed to occur in the following steps:
|
· |
Nanopores form within cellular membranes immediately and calcium increases in the cytoplasm within one second; |
|
· |
Phosphatidylserine (PS) is externalized to the cell surface within several seconds and is one marker used by the immune system to target and phagocytose unhealthy cells; |
|
· |
Calcium-dependent reactive oxygen species (ROS) generation occurs within approximately one minute; |
|
· |
Pyknosis and DNA fragmentation are stimulated within approximately 10 minutes (pyknosis is the shrinking of a cell nucleus along with condensation of the tissue in a cell that is undergoing necrosis or programmed cell death); |
4
|
· |
Calreticulin protein is externalized to the cell surface to become a second “eat me” signal within approximately two hours; |
|
· |
Caspase activation occurs within approximately three hours (caspases are a family of cellular proteins that are normally inactive but can be activated during programmed cell death to degrade other cellular proteins); and |
|
· |
We believe an adaptive immune response to the treated tumor can be triggered over a period of 14-28 days, as demonstrated in pre-clinical models showing the presence of CD8 + T cells, |
Potential Benefits of NPS Technology:
|
· |
Fast, precise, and focal treatment of tissue; |
|
· |
Induction of ICD, which can induce a targeted adaptive immune response; |
|
· |
Elimination of unwanted tissue with a favorable healing profile; |
|
· |
Reduction of potential damage to adjacent tissue because NPS does not rely on heat for ablation; and, |
|
· |
Utilization as a monotherapy or in combination with other therapeutics such as checkpoint inhibitor therapies (αPD-1, αPD-L1, αCTLA-4). |
Side Effects of NPS Technology
We recently completed a clinical study treating skin with NPS at multiple energy levels, utilizing multiple applicator configurations and applying up to 30 separate NPS treatments to each patient over the course of the 60-day treatment phase of the study. During the study, no adverse events were reported and no side effects were observed. Furthermore, subsequent histological review of the treated tissues did not reveal negative unintended tissue responses resulting from the NPS treatments.
During the course of conducting pre-clinical studies, over 1,000 tumors have been treated with NPS with no consistent side effects observed. Our predecessors and others carried out longer-term experiments in which a single tumor was ablated in each animal followed by observation for a period of 4 months for melanoma and 300 days for pancreatic cancer. No side effects were observed in these animals. The few reported problems observed over the past 10 years of animal studies resulted from improper placement of the applicators.
Our Proprietary NPS Delivery System
To deliver our NPS therapies and treatments we have developed our NPS platform, the PulseTx system. The PulseTx is a first-of-its-kind tunable nanosecond pulse generation and interchangeable applicator system designed to enable NPS treatments and therapies across multiple clinical and research application. The PulseTx is comprised of two primary components:
|
· |
The PulseTx Generator: a novel and proprietary tunable pulse generator capable of delivering treatments with varying pulse amplitude, duration, frequency and number. These are the key NPS parameters for stimulating cell death and ICD in tissue. The ability to tune these parameters with the PulseTx enables the development treatments of and therapies for a variety of applications and tissue types. |
|
· |
PulseTx Applicator Suite; a suite of interchangeable NPS treatment applicators that deliver NPS directly to the tissue being treated. We are designing a suite of single and multiple use PulseTx Applicators tailored to specific applications, including open or minimally invasive surface, surgery or solid tissue treatments. |
We believe that the design of the PulseTx system will allow the system to be deployed in a standard clinic or laboratory setting, without the need for special facilities or installation requirements.
We submitted a United States Food and Drug Administration (FDA) 510(k) for the PulseTx System for soft tissue ablation during the first quarter of 2017. We plan to conduct post-market studies that we believe will demonstrate improved clinical outcomes and drive specific indications. Although additional and broader indications may be cleared through the 510(k) process, we may be required to pursue Premarket Approval from the FDA to make labeling claims for specific indications.
5
Applications
Dermatology/Aesthetics
We believe NPS has high potential to offer improved clinical outcomes for a broad range of dermatology conditions and aesthetic improvement applications for which targeted removal of skin lesions is medically or cosmetically desirable. Current dermatology procedures to remove lesions or undesired skin tissue typically involve either excision (e.g. surgery) or the use of extreme heat (e.g. lasers or radiofrequency energy) or extreme cold (e.g. cryoablation). Patients are advised that these methods of tissue removal can be effective, but carry a risk of collateral damage to nearby healthy tissue and the associated inflammatory response to severe tissue injury, which can result in visible scars and pigment changes that can appear as cosmetically undesirable as the original lesion.
The novel NPS non-thermal mechanism for removing undesired skin lesions has the potential for both reducing collateral damage to surrounding healthy tissue and minimizing the inflammatory response as compared to standard treatment modalities, both of which can help contribute to cosmetically desired appearance of the skin when the lesion is eliminated and the skin is healed.
We recently completed our first NPS clinical study in dermatology, a dose response study in skin. This important skin safety and dose response study was designed to evaluate the tissue effects of a wide range of NPS energy “doses” on healthy skin. Over 170 individual sections of skin were tested during the course of the study. The interim analysis of investigator evaluations, clinical photographs, and microscopic examination of tissue samples consistently demonstrated a pattern of controlled destruction of targeted tissue in the upper layer of the skin (the epidermis) where many skin lesions are located, and a tissue sparing effect in the dermal skin layer. This finding of selective epidermal effects was consistent and apparent for all tested energy settings in the study. In addition, based on key microscopic indicators of long-term skin safety, a dermatopathologist concluded NPS has a low risk of undesired dermal damage and a restoration of a normal population of the melanocytes that produce skin pigment in the epidermis, when compared to untreated controls.
Based on the promising interim analysis from this recent NPS dose-response study, we have identified multiple potentially high value applications in dermatology for which the non-thermal effects of NPS in normal skin epidermis may lead to improved patient experiences and improved outcomes when compared to existing more destructive lesion removal methods. Potential application candidates in dermatology include Seborrheic Keratosis, warts, cherry angiomas, Basal Cell Carcinoma, Squamous Cell Carcinoma (SCC), and Actinic Keratosis, although we believe there are more skin conditions to consider for future studies. Key considerations in prioritizing our initial clinical application include, a substantial market size for skin treatments that are more typically paid directly by patients, a likelihood of favorable clinical outcomes based on the continued analysis of our dose response study data, comparisons to the current standards of care for lesion removal, and an indication for which there is a 510(k) pathway for clearance may be possible. Once we choose the initial application in dermatology, we will pursue a clinical study that we believe can be the basis for a 510(k) clearance for a specific indication.
We intend to pursue one or more clinical studies of NPS in dermatology applications during 2017 with the longer term plan of demonstrating the benefits of NPS in a range of dermatology conditions for which a non-thermal alternative to current methods may be preferred.
Oncology
We believe that NPS may afford a new paradigm in the treatment of certain cancers, either as a standalone therapy or in combination with other therapies currently available and in development. Immunotherapy continues to transform cancer care as the field advances with scientific discoveries in the laboratory and clinic.
It is well established that cancer cells can be recognized by the immune system. Under normal circumstances, these cancer cells will be detected by the immune surveillance system and eliminated. However, when cancer cells either evade or defeat the immune surveillance system, tumors grow and spread in an unregulated manner resulting in malignancies and, over time, may even cause the lack or loss of response to treatments.
We believe that NPS may offer a novel approach to treating tumors as a monotherapy and in combination with other therapies. Pre-clinical research demonstrates that NPS ablates treated tumors, modulates the tumor microenvironment of treated tumors and induces ICD in a drug-free manner. As a result of ICD, the patient’s immune system is able to detect the tumor cell antigens and mount an immune response specifically against those tumor cells that were treated with NPS.
Specifically, when tumors are treated with NPS, the cellular transient nanopores in individual tumor cells trigger endoplasmic reticulum stress and the release of intracellular calcium along with ROS and the emission of danger-associated
6
molecular patterns (DAMPs). As a result, these tumor cells undergo ICD, which is a unique form of cell death that is responsible for recruiting immune cells to the site of the NPS-treated tumors for antigen processing and presentation.
ICD is a desirable form of cell death in cancer therapy as it exhibits a preference for the cross-presentation of tumor cell antigens in the lymph nodes. The hallmark of ICD and cross-presentation is the generation of an adaptive immune response including CD4+ helper-T cells and, more important for killing cancer cells, CD8+ cytotoxic-T cells. In effect, NPS serves as an in situ personalized cancer vaccination against a patient’s own tumors. Furthermore, we believe the microenvironment in NPS-treated tumors is modified and that the protective mechanisms surrounding the tumor are reduced and possibly eliminated.
The ability of NPS to initiate an immune response against cancerous tumors and the related cancer microenvironment may lend itself more favorably to immunogenic cancers. Cancer immunogenicity reflects a tumor’s ability to stimulate an immune response. It has been hypothesized that cancer immunogenicity increases with mutation rate, meaning the more mutations a tumor has, the higher the chance the tumor antigens can trigger the immune response. Cancers with the highest mutation rates include melanoma, certain lung cancers, and bladder cancer.
Relative to existing immune therapies, we believe that NPS therapy will activate the adaptive immune response specific to an individual’s tumors in a non-toxic, targeted manner. Given the many different approaches to modulating an immune response and the ability of an immune response to access tumors, significant potential exists for NPS to be synergistic with existing therapies and therapies in development.
Our previously published pre-clinical work demonstrated the ability of NPS to treat and eliminate targeted melanoma tumors and that NPS-treated tumor cells can be used as a vaccine to protect mice against fibrosarcoma subdermal allografts. Additional pre-clinical studies conducted to date confirm the elimination of NPS treated tumors along with the generation of an adaptive immune response against various other tumor types. We continue to develop NPS, delineate the immune response and explore additional applications in oncology using pre-clinical animal models in preparation for upcoming clinical studies.
We are in the early stages of planning to commence our first clinical oncology study in patients with unresectable in-transit melanoma. We believe the high immunogenicity of melanoma lends itself well to our NPS therapy and we plan to initiate an open label study of in-transit melanoma in 2017.
Minimally Invasive Ablation Applications
We believe the use of NPS to treat and/or ablate tissue in a minimally invasive manner is an exciting opportunity for the technology. NPS may offer a new approach to eliminate unwanted tissue that we believe will be predictable and uniform and will result in minimal collateral damage to surrounding tissue. We believe that these benefits can be important to several minimally invasive high value applications, including atrial fibrillation and other cardiac applications, lung disease, Barret’s esophagus, ear, nose and throat papillomas and thyroid nodules.
Veterinary Applications
We believe that NPS can provide a novel treatment in veterinary medicine, one that can provide a minimally invasive approach for pets in this largely cash pay-for-service market. Although the FDA does have regulatory oversight over medical devices used in veterinary medicine and can take appropriate regulatory action if a device is misbranded, no regulatory clearance is necessary to introduce a therapy or device to the market.
We believe that addressing the veterinary oncology market is attractive because:
|
· |
animal data might yield information on the novel biological effects of NPS on tumors, especially confirmation of an adaptive immune response, which would help validate potential translational applications to humans; |
|
· |
it may offer a faster path to commercialization because of the less stringent regulatory pathway for veterinary medical devices; and |
|
· |
it is large and continues to grow. |
Strategy
We have consolidated several different entities working on nanosecond pulsed electric fields, and we now own or license 49 issued patents and 61 filed patent applications in the United States and worldwide. This novel platform technology with strong IP protection allows us to follow a broad, platform based approach to introducing our NPS technology and related
7
therapies and devices.
Our strategy is to:
|
· |
Develop a general purpose NPS platform for use across a broad array of applications. We are developing a versatile nanosecond pulse generation system, the PulseTx System, that can produce pulses of variable number, width, amplitude, and frequency and can be used with various applicator types and deployed into a wide range of applications; |
|
· |
Demonstrate the unique benefits of our NPS technology and the PulseTx System across a number of compelling treatment applications. We intend to conduct multiple clinical trials to demonstrate the unique ability of NPS to treat tissues across a number of applications with the highest value to clinicians and patients. We believe that a solid foundation of clinical data will provide the opportunity to pursue regulatory clearances and demonstrate to clinicians and patients the favorable treatment outcomes and patient experience afforded by our technology; |
|
· |
Pursue 510(k) clearance from the FDA for general soft tissue ablation followed by post-market studies to show improved clinical outcomes across a number of applications. We submitted an FDA 510(k) for the PulseTx System in the first quarter of 2017. We believe a 510(k) clearance for soft tissue ablation will provide a foundation for future clearances with specific indications that we will pursue with the addition of clinical data; and |
|
· |
Commercialize applications by focusing initially on those clinical uses requiring modest clinical data to demonstrate efficacy and safety and for which cash payment patterns or coded reimbursement arrangements are well-established. Conditions that require long-term evidence to support third-party reimbursement and/or a longer time horizon to evaluate efficacy and safety, including various skin cancers, are expected later in the commercial launch sequence. |
Intellectual Property
We believe that our current and any future patents and other proprietary rights we own or license are and will be essential to our business and create an important competitive advantage for us. We also rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect our intellectual property, in part, through confidentiality agreements with our employees, consultants and other parties, patent registration and access control to sensitive information. Our success also will depend on the ability of our licensors to obtain, maintain (including making periodic filings and payments) and enforce confidentiality agreements and patent protection for their intellectual property, in particular, those patents and other intellectual property to which we have secured rights.
We own or license 49 issued patents and 61 filed patent applications in the United States and worldwide to protect the intellectual property on which nanosecond pulsed electric field technology is based on. Our United States issued patents are set to expire between 2020 and 2034.
As we expand our business internationally, we will seek patent, trademark and copyright protections as appropriate and available and conduct our business with the protections of confidentiality and trade secrets. Depending on the jurisdiction, we may not be able to obtain the scope of protections we seek, in which event we will need to balance the available protections against the importance of such market to us.
Research and Development
We are currently conducting research and development activities in pursuit of commercial applications for our NPS technology, but have not yet commercialized or recognized revenue from our technology. Therefore, the majority of our business activities are devoted to research, including clinical trials, related to our core technologies and development of devices and products based on those technologies. Research and development expenses totaled $6.0 million, $2.6 million and $26,000 for the years ended December 31, 2016 and 2015 and for the period of May 19, 2014 (inception) through December 31, 2014, respectively. Research and development expenses are expected to increase substantially during the fiscal year ending 2017, reflecting the increased growth in our operational activities and expansion of our clinical trial programs.
Our R&D development team incorporates data and feedback obtained from our clinical and research programs into the development of the PulseTx NPS System and further inform our clinical strategies. We believe that developing and leveraging relationships among clinicians is a key element in driving the adoption of our technology in clinical studies in the
8
short-term and enhances the potential for possible commercial systems in the future.
Competition
The applications we intend to target are subject to intense competition from rapidly evolving companies and new scientific discoveries. We compete against well-established incumbent technologies offering products in oncology, dermatology and aesthetics, minimally invasive treatments, and veterinary applications. Given the broad scope of our technology, we face competition ranging from large manufacturers with multiple business lines to small companies with focused products, as well as providers of other medical therapies and therapeutics for conditions that we intend to treat. Our future success will depend on our ability to establish and maintain a competitive position in current and future technologies.
In immuno-oncology, we compete with multiple new technologies stimulating the immune system to target cancer. An increased understanding of the multiple mechanisms by which cancer or precancerous cells can evade the immune system has helped researchers develop drugs including those that target immune inhibitors or stimulate T-cell production. For example, approved checkpoint inhibitor therapeutics are administered systemically and modulate the immune system in a more global way, which can lead to significant side effects including autoimmune diseases. Companies with approved checkpoint inhibitors include: Bristol-Myers Squibb and Merck. CAR-T cell therapy has gained attention recently; which refers to a therapy where T cells are removed from a patient and modified to express receptors on its surface that are specific to a cancer type. These cells are then cultured and infused back into the body. Companies developing CAR-T cell therapies include Juno Therapeutics and Kite Pharma.
We compete with multiple tissue removal technologies. These technologies cause immediate cell necrosis, killing cells within seconds to hours following exposure and triggering inflammation. Our technology is unique and differentiated in that NPS stimulates primarily intracellular cell death which we believe would be less traumatic to treated tissue and would result in less scarring or collateral damage to surrounding tissues. Tissue removal technologies include RFA, microwave ablation, cryoablation, laser therapies and IRE.
IRE uses pulsed electric fields at a high voltage in hundreds of microsecond pulse widths. These pulses cause irreversible damage to cell membranes, resulting in necrosis (death) of the tumor cells. However, this technology stimulates nerves and muscles in a manner that makes it common for clinicians to use general anesthesia and muscle blockade during treatment. In contrast, NPS utilizes pulses 1,000 times shorter and in pre-clinical and limited clinical studies has not required the use of muscle blockade. Moreover, our technology transiently permeabilizes internal organelles which can lead to a signaling cascade ending in immunogenic cell death.
Tissue ablation companies for therapeutic applications include: Medtronic, Boston Scientific, AngioDynamics and St. Jude Medical. Ablation companies for dermatologic and aesthetic applications include: Alma Lasers, Cutera and Syneron Medical.
Government Regulation
In general, medical device companies must navigate a challenging regulatory environment. The FDA regulates the medical device market to ensure the safety and efficacy of these products. The FDA allows for two primary pathways for a medical device to gain approval for commercialization: a successful pre-market approval, or PMA application or 510(k) clearance. A completely novel product must go through the more rigorous premarket approval, or PMA, process if it cannot receive authorization through a 510(k). The FDA has established three different classes of medical devices that indicate the level of risk associated with using a device and consequent degree of regulatory controls needed to govern its safety and efficacy. Level I and Level II devices are considered lower risk and often can gain approval for commercial distribution by submitting a notification request to the FDA, generally known as the 510(k) process. The devices regarded as the highest risk by the FDA are designated Class III status and generally require the submission of a PMA application for approval to commercialize a product. These generally include life-sustaining, life-supporting, or implantable devices or devices without a known predicate technology already approved by the FDA.
The 510(k) clearance path can be significantly less time-consuming than PMA approval, making this route preferable for a medical device company. Through a 510(k), a company must provide documentation that its device is safe and effective by showing it is substantially equivalent to a device already cleared through a 510(k) or in distribution before May 28, 1976 for which the FDA has not yet required a PMA submission. The FDA has a 90 calendar day review goal from the date of the 510(k) submission to authorize or decline commercial distribution of the device. However, similar to the PMA
9
process, approval may take longer than this 90 day goal. If the FDA resolves that the product is not substantially equivalent to a predicate device, a clearance will not be granted.
A PMA application must be accompanied by substantial data that supports the safety and efficacy of the device, which includes the provision of preclinical, clinical, technical, manufacturing and labeling information. If the FDA deems the application acceptable to pass through the first level of scrutiny, it has 180 days to review the submission, but it can typically take longer (up to several years) as this regulatory body can request additional information or clarifications. The FDA may also impose additional regulatory hurdles for a PMA, including the institution of an outside advisory panel of experts to assess the application or provide recommendations as to whether to approve the device. Although the FDA in the end approves or disapproves the device, in nearly all cases the FDA follows the recommendation from the independent panel concerning approvability of the new device. As part of this process, the FDA will also inspect the manufacturing operations of the company requesting approval to verify compliance with quality control regulations. Significant changes in the fabrication of a device, or alterations in the labeling or design of a product require new PMA applications or PMA supplements for a product originally approved under a PMA. This creates substantial regulatory risk for devices undergoing the PMA route.
Pervasive and Continuing Regulation
After a device is placed on the market, numerous regulatory requirements continue to apply. These include:
The FDA’s QSR which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process;
labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label uses;
clearance or approval of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use;
medical device reporting, or MDR, regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; and
post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device.
After a device receives 510(k) clearance or PMA approval, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new clearance or approval. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with the determination not to seek a new 510(k) clearance or PMA, the FDA may retroactively require a new 510(k) clearance or premarket approval. The FDA could also require a manufacturer to cease marketing and distribution and/or recall the modified device until 510(k) clearance or premarket approval is obtained. Also, in these circumstances, it may be subject to significant regulatory fines, penalties, and warning letters.
The MDR regulations require that we report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury.
The FDA has broad post-market and regulatory enforcement powers. We may be subject to unannounced inspections by the FDA and the Food and Drug Branch of CDHS to determine our compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of our suppliers.
Failure to comply with applicable regulatory requirements can result in enforcement action by FDA, which may include any of the following sanctions:
warning letters, fines, injunctions, consent decrees and civil penalties;
repair, replacement, refunds, recall or seizure of our products;
10
operating restrictions, partial suspension or total shutdown of production;
refusing our requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products;
withdrawing 510(k) clearance or premarket approvals that have already been granted; and
criminal prosecution.
Regulatory System for Medical Devices in Europe
The European Union consists of 28 member states and has a coordinated system for the authorization of medical devices. The E.U. Medical Devices Directive, or MDD, sets out the basic regulatory framework for medical devices in the European Union. This directive has been separately enacted in more detail in the national legislation of the individual member states of the European Union.
The system of regulating medical devices operates by way of a certification for each medical device. Each certificated device is marked with CE mark which shows that the device has a Certificat de Conformité. There are national bodies known as Competent Authorities in each member state which oversee the implementation of the MDD within their jurisdiction. The means for achieving the requirements for CE mark varies according to the nature of the device. Devices are classified in accordance with their perceived risks, similarly to the U.S. system. The class of a product determines the requirements to be fulfilled before CE mark can be placed on a product, known as a conformity assessment. Conformity assessments for our products are carried out as required by the MDD. Each member state can appoint Notified Bodies within its jurisdiction. If a Notified Body of one member state has issued a Certificat de Conformité, the device can be sold throughout the European Union without further conformance tests being required in other member states.
Health Insurance Portability and Accountability Act
The Health Insurance Portability and Accountability Act of 1996, or HIPAA, established for the first time comprehensive federal protection for the privacy and security of health information. The HIPAA standards apply to three types of organizations, or Covered Entities: health plans, healthcare clearing houses, and healthcare providers which conduct certain healthcare transactions electronically. Title II of HIPAA, the Administrative Simplification Act, contains provisions that address the privacy of health data, the security of health data, the standardization of identifying numbers used in the healthcare system and the standardization of certain healthcare transactions. The privacy regulations protect medical records and other protected health information by limiting their use and release, giving patients the right to access their medical records and limiting most disclosures of health information to the minimum amount necessary to accomplish an intended purpose. The HIPAA security standards require the adoption of administrative, physical, and technical safeguards and the adoption of written security policies and procedures. HIPAA requires Covered Entities to obtain a written assurance of compliance from individuals or organizations who provide services to Covered Entities involving the use or disclosure of protected health information (“Business Associates”).
On February 17, 2009, Congress enacted Subtitle D of the Health Information Technology for Economic and Clinical Health Act, or HITECH, provisions of the American Recovery and Reinvestment Act of 2009. HITECH amends HIPAA and, among other things, expands and strengthens HIPAA, creates new targets for enforcement, imposes new penalties for noncompliance and establishes new breach notification requirements for Covered Entities and Business Associates. Regulations implementing major provisions of HITECH were finalized on January 25, 2013 through publication of the HIPAA Omnibus Rule, or the Omnibus Rule. The Omnibus Rule contained significant changes for Covered Entities and Business Associates with respect to permitted uses and disclosures of Protected Health Information.
Under HITECH’s new breach notification requirements, Covered Entities must report breaches of protected health information that has not been encrypted or otherwise secured in accordance with guidance from the Secretary of the U.S. Department of Health and Human Services, or the Secretary. Required breach notices must be made as soon as is reasonably practicable, but no later than 60 days following discovery of the breach. Reports must be made to affected individuals and to the Secretary and in some cases, they must be reported through local and national media, depending on the size of the breach. We are currently subject to the HIPAA regulations. We are subject to audit under the U.S. Department of Health and Human Services, or HHS, HITECH-mandated audit program. We may also be audited in connection with a privacy complaint. We are subject to prosecution and/or administrative enforcement and increased civil and criminal penalties for non-compliance, including a new, four-tiered system of monetary penalties adopted under HITECH. We are also subject to enforcement by
11
state attorneys general who were given authority to enforce HIPAA under HITECH. To avoid penalties under the HITECH breach notification provisions, we must ensure that breaches of protected health information are promptly detected and reported within the company, so that we can make all required notifications on a timely basis. However, even if we make required reports on a timely basis, we may still be subject to penalties for the underlying breach.
In addition to the federal privacy regulations, there are a number of state laws regarding the privacy and security of health information and personal data that are applicable to clinical laboratories. The compliance requirements of these laws, including additional breach reporting requirements, and the penalties for violation vary widely and new privacy and security laws in this area are evolving. Requirements of these laws and penalties for violations vary widely. We believe that we have taken the steps required of us to comply with health information privacy and security statutes and regulations in all jurisdictions, both state and federal. However, we may not be able to maintain compliance in all jurisdictions where we do business. Failure to maintain compliance, or changes in state or federal laws regarding privacy or security, could result in civil and/or criminal penalties and could have a material adverse effect on our business.
If we or our operations are found to be in violation of HIPAA, HITECH or their implementing regulations, we may be subject to penalties, including civil and criminal penalties, fines, and exclusion from participation in U.S. federal or state health care programs, and the curtailment or restructuring of our operations. HITECH increased the civil and criminal penalties that may be imposed against Covered Entities, their Business Associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions.
In addition to federal privacy regulations, there are a number of state laws governing confidentiality of health information that are applicable to our operations. New laws governing privacy may be adopted in the future as well. We have taken steps to comply with health information privacy requirements that are applicable to us.
Federal, State and Foreign Fraud and Abuse Laws
Because of the significant federal funding involved in Medicare and Medicaid, Congress and the states have enacted, and actively enforce, a number of laws to eliminate fraud and abuse in federal healthcare programs. Our business is subject to compliance with these laws. In March 2010, the Recipient Protection and Affordable Care Act, as amended by the Healthcare and Education Affordability Reconciliation Act, which we refer to collectively as the Affordable Care Act, was enacted in the United States. The provisions of the Affordable Care Act are effective on various dates. The Affordable Care Act expands the government’s investigative and enforcement authority and increases the penalties for fraud and abuse, including amendments to both the Anti-Kickback Statute and the False Claims Act, to make it easier to bring suit under these statutes. The Affordable Care Act also allocates additional resources and tools for the government to police healthcare fraud, with expanded subpoena power for HHS, additional funding to investigate fraud and abuse across the healthcare system and expanded use of recovery audit contractors for enforcement.
Anti-Kickback Statutes. The federal healthcare programs’ Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid.
The definition of “remuneration” has been broadly interpreted to include anything of value, including, for example, gifts, certain discounts, the furnishing of free supplies, equipment or services, credit arrangements, payment of cash and waivers of payments. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered businesses, the statute has been violated. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. In addition some kickback allegations have been claimed to violate the Federal False Claims Act, discussed in more detail below.
The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are otherwise lawful in businesses outside of the healthcare industry. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the Office of Inspector General, or OIG, of HHS to issue a series of regulations known as “safe harbors.” These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do
12
not fully satisfy an applicable safe harbor may result in increased scrutiny by government enforcement authorities such as OIG.
Many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to referral of recipients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
Government officials have focused their enforcement efforts on the marketing of healthcare services and products, among other activities, and recently have brought cases against companies, and certain individual sales, marketing and executive personnel, for allegedly offering unlawful inducements to potential or existing customers in an attempt to procure their business.
Federal False Claims Act. Another development affecting the healthcare industry is the increased use of the federal False Claims Act, and in particular, action brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has violated the False Claims Act and to share in any monetary recovery. In recent years, the number of suits brought against healthcare providers by private individuals has increased dramatically. In addition, various states have enacted false claims laws analogous to the False Claims Act, and many of these state laws apply where a claim is submitted to any third-party payor and not just a federal healthcare program.
When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 and $11,000 for each separate instance of false claim. As part of any settlement, the government may ask the entity to enter into a corporate integrity agreement, which imposes certain compliance, certification and reporting obligations. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The federal government has used the False Claims Act to assert liability on the basis of inadequate care, kickbacks and other improper referrals, and improper use of Medicare numbers when detailing the provider of services, in addition to the more predictable allegations as to misrepresentations with respect to the services rendered. In addition, the federal government has prosecuted companies under the False Claims Act in connection with off-label promotion of products. Our future activities relating to the reporting of wholesale or estimated retail prices of our products, the reporting of discount and rebate information and other information affecting federal, state and third-party reimbursement of our products and the sale and marketing of our products may be subject to scrutiny under these laws.
While we are unaware of any current matters, we are unable to predict whether we will be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the costs of defending such claims, as well as any sanctions imposed, could significantly affect our financial performance.
The Sunshine Act. The Physician Payment Sunshine Act, or the Sunshine Act, which was enacted as part of the Affordable Care Act, requires all entities that operate in the United States and manufacturers of a drug, device, biologic or other medical supply that is covered by Medicare, Medicaid or the Children’s Health Insurance Program to report annually to the Secretary of HHS: (i) payments or other transfers of value made by that entity, or by a third-party as directed by that entity, to physicians and teaching hospitals or to third parties on behalf of physicians or teaching hospitals; and (ii) physician ownership and investment interests in the entity. The payments required to be reported include the cost of meals provided to a physician, travel reimbursements and other transfers of value, including those provided as part of contracted services such as speaker programs, advisory boards, consultation services and clinical trial services. The final rule implementing the Sunshine Act required data collection on payments to begin on August 1, 2013. The first annual report, comprised of data collected from August 1, 2013 to December 31, 2013, was due March 31, 2014. The statute requires the federal government to make reported information available to the public starting September 2014, which it has. Failure to comply with the reporting requirements can result in significant civil monetary penalties ranging from $1,000 to $10,000 for each payment or other transfer of value that is not reported (up to a maximum per annual report of $150,000) and from $10,000 to $100,000 for each knowing failure to report (up to a maximum per annual report of $1.0 million). Additionally, there are criminal penalties if an entity intentionally makes false statements in such reports. We are subject to the Sunshine Act and the information we disclose may lead to greater scrutiny, which may result in modifications to established practices and additional costs. Additionally, similar reporting requirements have also been enacted on the state level domestically, and an increasing number of countries worldwide either have adopted or are considering similar laws requiring transparency of interactions with healthcare professionals.
13
Foreign Corrupt Practices Act. The Foreign Corrupt Practices Act, or FCPA, prohibits any United States individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, if any, and to devise and maintain an adequate system of internal accounting controls for international operations.
International Laws. In Europe, various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines, for individuals and/or companies committing a bribery offense. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United Kingdom, under the Bribery Act 2010, which went into effect in July 2011, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act of 2010, faces imprisonment of up to 10 years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
There are also international privacy laws that impose restrictions on the access, use, and disclosure of health information. All of these laws may impact our business. Our failure to comply with these privacy laws or significant changes in the laws restricting our ability to obtain required patient information could significantly impact our business and our future business plans.
U.S. Healthcare Reform
Changes in healthcare policy could increase our costs and subject us to additional regulatory requirements that may interrupt commercialization of our current and future solutions. Changes in healthcare policy could increase our costs, decrease our revenues and impact sales of and reimbursement for our current and future solutions. The Affordable Care Act substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts our industry. The Act contains a number of provisions that impact our business and operations, some of which in ways we cannot currently predict, including those governing enrollment in federal healthcare programs and reimbursement changes.
There will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to reduce costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge for our current and future solutions or the amounts of reimbursement available for our current and future solutions from governmental agencies or third-party payors. While in general it is too early to predict specifically what effect the Affordable Care Act and its implementation or any future healthcare reform legislation or policies will have on our business, current and future healthcare reform legislation and policies could have a material adverse effect on our business and financial condition.
Third-Party Reimbursement
Payment for patient care in the United States is generally made by third-party payors, including private insurers and government insurance programs, such as Medicare and Medicaid. The Medicare program, the largest single payor in the United States, is a federal governmental health insurance program administered by the Centers for Medicare and Medicaid Services, or CMS, and covers certain medical care expenses for eligible elderly and disabled individuals. Because a large percentage of the population with PAD includes Medicare beneficiaries, and private insurers may follow the coverage and payment policies of Medicare, Medicare’s coverage and payment policies are significant to our operations
Environmental
We are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain hazardous and potentially hazardous substances used in connection with our operations. Although we believe that we have complied with these laws and regulations in all material respects and, to date, have not been required to take any action to correct any noncompliance, there can be no assurance that we will not be required to incur significant costs to comply with environmental regulations in the future.
14
Insurance
We maintain product and clinical trial liability insurance coverage which includes a maximum of per claim and an annual aggregate policy limits, subject to self-insured retentions. The policy covers, subject to policy conditions and exclusions, claims of bodily injury and property damage from any product manufactured by us or from trial-related adverse events.
There is no assurance that our level of coverage is adequate. We may not be able to sustain or maintain our current level of coverage and cannot assure you that adequate insurance coverage will continue to be available on commercially reasonable terms, or at all. A successful product liability claim may exceed our existing coverages and may make future coverages significantly more expensive, if available at all.
Employees
As of December 31, 2016, we had 13 full-time employees. Of these employees, nine were in research and development and four were in general and administration. Substantially all of our employees are located at our headquarters in Burlingame, California. None of our employees are represented by labor unions or are covered by a collective bargaining agreement with respect to their employment. We have not experienced any work stoppages, and we consider our relationship with our employees to be good.
Available Information
We were incorporated in Nevada on May 19, 2014 under the name Electroblate, Inc. Electroblate, Inc. changed its name to Pulse Biosciences, Inc. effective December 8, 2015. Our corporate offices are located at 849 Mitten Road, Suite 104, Burlingame, California. Our telephone number is (650) 697-3939.
Our website is located at www.pulsebiosciences.com. The information that can be accessed through our website is not incorporated into this Annual Report on Form 10-K, and the inclusion of our website address is an inactive textual reference only. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge through the “Investor Relations” section of our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission (SEC).
Additionally, we use our website as a channel for distribution for important company information. Important information, including press releases, analyst presentations and financial information regarding us, as well as corporate governance information, is routinely posted and accessible on the “Investor Relations” section of the website, which is accessible by clicking “Investors” on the menu tab labeled “About Us” on our website home page.
15
Investing in our common stock involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report on Form 10-K, including our financial statements and related notes, which could have a material adverse effect on our business, financial condition, results of operations and prospects. The risks described below are not the only risks facing us. Risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially affect our business, financial condition, results of operations and prospects.
Risks Relating to Our Business, Industry and Financial Condition
Since we have a limited operating history and have not commenced any revenue producing operations, it is difficult for potential investors to evaluate the future of our business.
We are a clinical-stage medical technology company and have not yet commenced revenue-producing operations. To date, our operations on a consolidated basis have consisted of the continued development of our technologies and implementation of the early parts of our business plan. We have incurred significant operating losses in each year since our inception and we expect to continue to incur additional loses for the next several years. In addition, a high percentage of our expenses will continue to be fixed; accordingly, our losses may be greater than expected and our operating results may suffer. We have limited historical financial data upon which we may base our projected revenue and base our planned operating expenses. Our limited operating history makes it difficult for potential investors to evaluate our technology or prospective operations and business prospects.
We currently have no commercial products or product revenue and may never become profitable.
To date, we have not generated revenue and have relied on equity-based financing from the sale of securities to fund our operations. We expect that our future financial results will depend primarily on our success in obtaining approval for, launching, selling and supporting our PulseTxTM System or other products based on Nano Pulse Stimulation, or NPS; however, our technology is still in development. We expect to expend significant resources on hiring of personnel, continued scientific and product research and development, potential product testing and preclinical and clinical investigation, intellectual property development and prosecution, marketing and promotion, capital expenditures, working capital, general and administrative expenses, and fees and expenses associated with our capital raising efforts. We expect to incur costs and expenses related to consulting costs, laboratory development costs, hiring of scientists, engineers, and other operational personnel, and the continued development of relationships with potential partners. We are incurring significant operating losses, we expect to continue to incur additional loses for the next several years, and we cannot assure you that we will generate revenue or be profitable in the future. Our future products may never be approved or become commercially viable or accepted for use. Even if we find commercially viable applications for our technology, which may include licensing, we may never recover our research and development expenses.
Investment in medical technology is highly speculative, because it entails substantial upfront capital expenditures and significant risk that any potential planned product will fail to demonstrate adequate efficacy or clinical utility. Investors should evaluate an investment in us in light of the uncertainties encountered by developing medical technology companies in a competitive environment. There can be no assurance that our efforts will be successful or that we will ultimately be able to achieve profitability. Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable could adversely affect the market price of our common stock and could significantly impair our ability to raise capital, expand our business or continue to implement our business plan.
We anticipate needing additional financing to execute our business plan and fund operations, which additional financing may not be available on reasonable terms or at all.
Our ability to continue as a going concern ultimately is dependent upon our generating cash flow from sales that are sufficient to fund operations or finding adequate financing to support our operations. Currently, we have no product revenue, and we do not have arrangements in place for all the anticipated, required financing to be able to fully implement our business plan.
We cannot give any assurance that we will be able to obtain all the necessary funding that we may need. We believe we will require additional capital in the future to fully develop our technologies and planned products to the stage of a commercial launch. We may pursue additional funding through various financing sources, including the private sale of our equity and debt securities, licensing fees for our technology, joint ventures with capital partners and project type financing. If we raise funds by issuing equity or equity-linked securities, dilution to our stockholders will result. Any equity securities
16
issued may also provide for rights, preferences or privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings could impose significant restrictions on our operations. We also may seek government based financing, such as development and research grants. There can be no assurance that funds will be available on commercially reasonable terms, if at all.
The incurrence of additional indebtedness or the issuance of certain equity securities could result in increased fixed payment obligations and could also result in restrictive covenants, such as limitations on our ability to incur additional debt or issue additional equity, limitations on our ability to acquire or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business. In addition, the issuance of additional equity securities by us, or the possibility of such issuance, may cause the market price of our common stock to decline. In the event that we enter into collaborations or licensing arrangements to raise capital, we may be required to accept unfavorable terms. These agreements may require that we relinquish, or license to a third party on unfavorable terms, our rights to technologies or product candidates that we otherwise would seek to develop or commercialize ourselves, or reserve certain opportunities for future potential arrangements when we might otherwise be able to achieve more favorable terms. In addition, we may be forced to work with a partner on one or more of our products or market development programs, which could lower the economic value of those programs to us.
If we are unable to obtain adequate financing or financing on terms satisfactory to us when we require it, we may terminate or delay the development of one or more of our products, delay clinical trials necessary to market our products, or delay establishment of sales and marketing capabilities or other activities necessary to commercialize our products. If this were to occur, our ability to grow and support our business and to respond to market challenges could be significantly limited or we may be unable to continue operations, in which case you could lose your entire investment.
If we lose key management personnel, our ability to identify, develop and commercialize new or next generation product candidates will be impaired, could result in loss of markets or market share and could make us less competitive.
We are highly dependent upon the principal members of our management team and the members of our scientific team, including our Chief Executive Officer, Darrin Uecker. These persons have significant experience and knowledge with sub-microsecond pulsed electric fields and more broadly in life sciences and medical technologies. The loss of any team member could impair our ability to design, identify, and develop new intellectual property and new scientific or product ideas. The loss of a key employee, the failure of a key employee to perform in his or her current position or our inability to attract and retain skilled employees could result in our inability to continue to grow our business or to implement our business strategy. We compete for qualified management and scientific personnel with other life science companies, academic institutions and research institutions. Our employees could leave our company with little or no prior notice and may be free to work for a competitor. If one or more of our senior executives or other key personnel were unable or unwilling to continue in their present positions, we may not be able to replace them easily or at all, and other senior management may be required to divert attention from other aspects of the business. In addition, we do not have “key person” life insurance policies covering any member of our management team or other key personnel. The loss of any of these individuals or any inability to attract or retain qualified personnel, including scientists, engineers and others, could prevent us from pursuing collaborations and materially and adversely affect our product development and introductions, business growth prospects, results of operations and financial condition.
There is a limited talent pool of experienced professionals in our industry. If we are not able to retain and recruit personnel with the requisite technical skills, we may be unable to successfully execute our business strategy.
The specialized nature of our industry results in an inherent scarcity of experienced personnel in the field. Our future success depends upon our ability to attract and retain highly skilled personnel, including scientific, technical, commercial, business, regulatory and administrative personnel, necessary to support our anticipated growth, develop our business and perform certain contractual obligations. Given the scarcity of professionals with the scientific knowledge that we require and the competition for qualified personnel among life science businesses, we may not succeed in attracting or retaining the personnel we require to continue and grow our operations.
17
We expect to operate in a highly competitive market, we may face competition from large, well-established medical device and product manufacturers with significant resources, and we may not be able to compete effectively.
The medical technology, medical device, biotechnology and pharmaceutical industries are characterized by intense and dynamic competition to develop new technologies and proprietary therapies. We may find ourselves in competition with companies that have competitive advantages over us, such as:
|
· |
additional lines of products, and the ability to offer rebates, higher discounts or incentives to gain a competitive advantage; |
|
· |
greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products, and marketing approved products; and |
|
· |
greater financial and human resources for product development, sales and marketing, and patent litigation. |
We may also face increased competition in the future as new companies enter our markets and as scientific developments surrounding electro-signaling therapeutics continue to accelerate. While we will seek to expand our technological capabilities to remain competitive, research and development by others may render our technology or product candidates obsolete or noncompetitive or result in treatments or cures superior to any therapy developed by us. As a result, we may not be able to compete effectively against current and potential future competitors or their devices and products.
We may rely on third parties for our sales, marketing, manufacturing and distribution, and these third parties may not perform satisfactorily.
We do not currently conduct many aspects of sales, marketing, manufacturing or distribution. To be able to commercialize our planned products, we may elect to internally develop all of the foregoing or utilize third parties with respect to one or more of these items. Our reliance on these third parties may reduce our control over these activities; however, reliance on third parties does not relieve us of our responsibility to ensure compliance with all required legal, regulatory and scientific standards. Any failure of these third parties to perform satisfactorily and in compliance with relevant laws and regulations could lead to delays in the development of our planned products, including delays in our clinical trials, or failure to obtain regulatory approval for our planned products, or failure to successfully commercialize our planned products or other future products. Some of these events could be the basis for FDA or other regulatory action, including injunction, recall, seizure or total or partial suspension of production.
We do not have any corporate experience in establishing these capabilities, and therefore, we may be unsuccessful in achieving commercialization and earning revenues. We believe that setting up the commercialization aspects of a company will take a substantial amount of capital and commitment of time and effort. We may seek development and marketing partners and license our technology to others in order to avoid our having to provide the marketing, manufacturing and distribution capabilities within our organization. There can be no assurance that we will find any development and marketing partners or companies that are interested in licensing our technology. If we are unable to establish and maintain adequate sales, marketing, manufacturing and distribution capabilities, independently or with others, we will not be able to generate product revenue, and may not become profitable.
If we are unable to manage the anticipated growth of our business, our future revenue and operating results may be harmed.
Any growth that we experience in the future could provide challenges to our organization, requiring us to expand our sales personnel and manufacturing operations and general and administrative infrastructure. Future growth will impose significant added responsibilities on management, including the need to identify, recruit, train and integrate additional employees. Rapid expansion in personnel could mean that less experienced people carry out our research and development activities, manufacture our PulseTx System devices and market and sell our NPS therapies and treatments, which could result in inefficiencies and unanticipated costs, reduced quality and disruptions to our operations. In addition, rapid and significant growth may strain our administrative and operational infrastructure, and the failure to continue to upgrade our technical, administrative, operating and financial control systems or the occurrence of unexpected expansion difficulties could have a material adverse effect on our business, financial condition and results of operations and our ability to timely execute our business plan. If we are unable to manage our growth effectively, it may be difficult for us to execute our business strategy and our business could be harmed.
18
Rapidly changing technology in life sciences could make the products we are developing obsolete.
The medical technology industry is characterized by rapid and significant technological changes, frequent new product introductions and enhancements and evolving industry standards. Our future success will depend on our ability to continually develop and then improve the products that we design and to develop and introduce new products that address the evolving needs of our customers on a timely and cost-effective basis. We also will need to pursue new market opportunities that develop as a result of technological and scientific advances. These new market opportunities may be outside the scope of our proven expertise or in areas which have unproven market demand. Any new products developed by us may not be accepted in the intended markets. Our inability to gain market acceptance of new products could harm our future operating results.
Security breaches, loss of data and other disruptions to us or our third-party service providers could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we, and our third-party service providers may collect and store sensitive data, including legally protected health information, personally identifiable information about our patients, information related to our trials, intellectual property, and our proprietary business and financial information. We manage and maintain our applications and data utilizing a combination of on-site and vendor-owned systems. We face a number of risks related to our protection of, and our service providers’ protection of, this critical information, including loss of access, unauthorized disclosure and unauthorized access, as well as risks associated with our ability to identify and audit such events.
Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers or viruses or otherwise breached due to employee error, malfeasance or other activities. While we have not experienced any such attack or breach, if such an event were to occur, our networks would be compromised and the information we store on those networks could be accessed by unauthorized parties, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process tests, provide test results, provide services, conduct research and development activities, collect, process and prepare company financial information, provide information about our product candidates and manage the administrative aspects of our business and could damage our reputation, any of which could adversely affect our business.
In addition, the interpretation and application of federal and state consumer, health-related and data protection laws in the United States are often uncertain, contradictory and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. Complying with these various laws could cause us to incur substantial costs or require us to change our business practices, systems and compliance procedures in a manner adverse to our business.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the future sale of planned products and the use of planned products in human clinical studies. For example, we may be sued if any of our product candidates, including any that are developed in combination therapies, allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. We may also be subject to liability for a misunderstanding of, or inappropriate reliance upon, the information we provide. If we cannot successfully defend ourselves against claims that our planned products caused injuries, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
19
For example, if we pursue clinical trials in the field of oncology, patients with the types and stages of cancer targeted by our NPS technology may already be in severe and advanced stages of disease, may have worsened conditions despite traditional therapies, may not be surgical candidates, and/or may have both known and unknown significant pre-existing and potentially life-threatening conditions. During the course of treatment, patients may suffer adverse events, including death, for reasons that may be related to our NPS Technology or our PulseTx System. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market those products, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to our product, the investigation into the circumstance may be time-consuming or inconclusive. These investigations may interrupt our sales efforts, delay our regulatory approval processes, or impact and limit the type of regulatory approvals our products could receive or maintain. As a result of these factors, a product liability claim, even if successfully defended, could harm our business.
We currently maintain product liability insurance coverage, which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
We have incurred net losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. If not utilized, the federal and state NOL carryforwards will begin to expire in various years beginning after 2032. Under the Internal Revenue Code of 1986, as amended, or the Code, and certain similar state tax provisions, a corporation is generally allowed a deduction for net operating losses, or NOLs, carried over from a prior taxable year. Under those provisions, we can carry forward our NOLs to offset our future taxable income, if any, until such NOLs are used or expire. The same is true of other unused tax attributes, such as tax credits.
In addition, under Section 382 of the Internal Revenue Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change net operating losses (“NOLs”) to offset future taxable income. We believe that we may have had one or more ownership changes, and, as a result, a portion of our existing NOLs may be subject to limitation. Future changes in our stock ownership could result in additional limitations. We may not be able to utilize a material portion of our NOLs even if we attain profitability.
We have a substantial amount of goodwill and intangible assets which over time may have to be written down as we make the required periodic assessments as to their value as reflected on our financial statements.
A significant portion of our total assets are comprised of goodwill and intangibles that arose from our 2014 business acquisitions. We review goodwill for impairment at least annually or whenever changes in circumstances indicate that the carrying value of the goodwill may not be recoverable. We also review our intangible assets for impairment at each fiscal year end or when events or changes in circumstances indicate the carrying value of these assets may exceed their current fair values. If we take an impairment charge for either goodwill or intangible assets, the overall assets will be reduced. Such an impairment charge may result in a change in the perceived value of the company and ultimately may be reflected as a reduction in the market price of our securities. Additionally, an impairment charge may also adversely influence our ability to raise capital in the future.
We have identified a material weakness in our internal control over financial reporting. If our remediation of this material weakness is not effective, or if we experience additional material weaknesses in the future or otherwise fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. Section 404 of the Sarbanes-Oxley Act requires that we evaluate and determine the effectiveness of our internal control over financial reporting and, beginning with our second annual report following our initial public offering, which will be the year ending December 31, 2017, provide a management report on internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis.
20
In connection with the audit of our financial statements as of and for the year ended December 31, 2016, we identified a material weakness in our internal control over financial reporting. The material weakness related to a lack of effective controls to adequately restrict access and segregate duties. Specifically, due to the limited number of staff in our accounting function, certain personnel had the ability to prepare and post journal entries without a qualified independent review performed by someone without this ability. Upon identifying this material weakness, we performed additional procedures to evaluate the impact on the financial statements. Based on these procedures, we believe the material weakness did not result in any material misstatements to our financial statements. However, this material weakness could result in a misstatement of our accounts or disclosures that would result in a material misstatement of our financial statements that would not be prevented or detected. We are implementing measures designed to improve our internal control over financial reporting to remediate this material weakness, including the following:
|
· |
We are evaluating our accounting system access rights so that there are accounting personnel without journal entry access who can perform review activities. |
|
· |
We are formalizing our internal control documentation and strengthening supervisory reviews by our management. |
|
· |
We are in the process of adding additional accounting personnel and will be segregating duties amongst accounting personnel. |
We cannot assure you that the measures we have taken to date, and are continuing to implement, will be sufficient to remediate the material weakness we have identified or avoid potential future material weaknesses. If the steps we take do not correct the material weakness in a timely manner, we will be unable to conclude that we maintain effective internal control over financial reporting. Accordingly, there could continue to be a reasonable possibility that a material misstatement of our financial statements would not be prevented or detected on a timely basis.
In addition to the remediation efforts related to the material weakness described above, we are in the process of designing and implementing the internal control over financial reporting required to comply with Section 404 of the Sarbanes Oxley Act. This process will be time consuming, costly and complicated. If during the evaluation and testing process, we identify one or more other material weaknesses in our internal control over financial reporting, our management will be unable to assert that our internal control over financial reporting is effective. Even if our management concludes that our internal control over financial reporting is effective, our independent registered public accounting firm may conclude that there are material weaknesses with respect to our internal controls or the level at which our internal controls are documented, designed, implemented or reviewed. If we are unable to assert that our internal control over financial reporting is effective, or when required in the future, if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could be adversely affected, and we could become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, which could require additional financial and management resources.
Our facilities in California are located near known earthquake faults, and the occurrence of an earthquake or other catastrophic disaster could cause damage to our facilities and equipment, which could require us to cease or curtail operations.
Our facilities in the San Francisco Bay Area are located near known earthquake fault zones and are vulnerable to damage from earthquakes. We are also vulnerable to damage from other types of disasters, including fire, floods, power loss, communications failures and similar events. If any disaster were to occur, our ability to operate our business at our facilities would be seriously, or potentially completely, impaired. In addition, the nature of our activities could cause significant delays in our research programs and commercial activities and make it difficult for us to recover from a disaster. The insurance we maintain may not be adequate to cover our losses resulting from disasters or other business interruptions. Accordingly, an earthquake or other disaster could materially and adversely harm our ability to conduct business.
Risks Related to Product Development
We currently do not have any products approved by the FDA or other similar foreign regulatory authorities for commercial sale or any commercialized products.
To date, we have invested a substantial amount of time and capital to research and develop the foundations of our technology and potential applications. For us to develop any products that might ultimately be commercialized, we will have to invest further time and capital in research and product development, medical and other regulatory compliance, and market development. Therefore, we may never develop any products that can be commercialized. All of our development efforts will
21
require substantial additional investment, which may never result in any revenue. Our efforts may not lead to approved or commercially successful products for a number of reasons, including:
|
· |
we may not be able to obtain regulatory approvals for our planned products, or the approved indications may be narrower than we seek; |
|
· |
we may experience delays in our development program, clinical trials and the regulatory approval process; |
|
· |
our NPS technology may not prove to be safe or effective in clinical trials; |
|
· |
physicians may not receive any reimbursement from third-party payers, or the level of reimbursement may be insufficient to support widespread adoption of any of our products; |
|
· |
any products that are approved may not be accepted in the marketplace by physicians or patients; |
|
· |
we may not be able to manufacture our products in commercial quantities or at an acceptable cost; and |
|
· |
rapid technological change or the appearance of a new competitive technology may make our technology and products obsolete. |
Our efforts may never demonstrate the feasibility of our technology.
Our research and development efforts remain subject to all of the risks associated with the development of new therapies, devices, treatment modalities, and related products based on our NPS technology. NPS applications are not yet fully developed. Development of the underlying technology, including the development of the PulseTx System, may be affected by unanticipated technical or other problems, among other development and research issues, and the possible insufficiency of funds needed in order to complete development of these products or devices. Safety, regulatory and efficacy issues, clinical hurdles or challenges also may result in delays and cause us to incur additional expenses that may increase our need for capital and result in additional losses. In addition, the potential indications for NPS are numerous, and we may fail to pursue the most optimal indications. If we cannot complete, or if we experience significant delays in developing our technology, applications or products for use in potential commercial applications, particularly after incurring significant expenditures, our business may fail and investors may lose the entirety of their investment.
The mechanism of action of NPS has not been fully determined or validated.
The exact mechanism(s) of action(s) of NPS is not fully understood, and data is still being gathered regarding its use. Furthermore, there are only a relatively small number of scientists and researchers who can be considered experts in the use of this emerging technology. A full understanding of a future product’s mechanism of action and a large stable of scientific experts are typically believed to make product development less risky. The FDA or similar foreign regulatory authorities may view this as increasing the potential risks, and diminishing the potential benefits, of products based on NPS technology. In addition, potential partners may view this as a limitation of the program, and it may be more challenging for us to obtain a partnership on favorable terms as a result.
NPS or our planned products may cause serious adverse side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial desirability of an approved label or result in significant negative consequences following any marketing approval.
The risk of failure of clinical development is high. For example, the vast majority of our in vivo data has been a result of animal testing, and we have only recently begun our first pilot study in humans. It is impossible to predict when or if this or any planned products will prove safe enough to receive regulatory approval. Undesirable side effects caused by NPS or any of our planned products could cause us or regulatory authorities to interrupt, delay or halt clinical trials. They could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authority.
Additionally, if NPS or any of our planned products receive marketing approval but, we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including:
|
· |
regulatory authorities may require additional warnings on the label that could diminish the usage or otherwise limit the commercial success of such products; |
22
|
· |
the FDA or other regulatory authorities may issue safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings about such product; |
|
· |
we may be required to change the way the product is administered or conduct additional clinical trials; |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular planned product, if approved.
Our business is dependent upon physicians adopting our NPS technology, and if we fail to obtain broad adoption, our business would be adversely affected.
If we obtain regulatory approval for an NPS application or product, our success will depend on our ability to educate physicians regarding the benefits of NPS, such as our PulseTx System, over existing treatment modalities and to persuade them to prescribe PulseTx System treatments for their patients. We do not know if NPS will be successful over the long term, and market acceptance may be hindered if physicians are not presented with compelling data demonstrating the efficacy of our service compared to alternative treatments. Any studies we, or third parties, may conduct comparing our NPS technology with alternative treatments may be expensive, time consuming or may not yield positive results. Additionally, adoption will be directly influenced by a number of financial factors, including the ability of providers to attract cash reimbursement from patients or to obtain sufficient reimbursement from third party commercial payors, and the Centers for Medicare & Medicaid Services, or CMS, for the professional services they provide in administering NPS treatments. The efficacy, safety, performance and cost-effectiveness of our NPS technology, PulseTx System or other potential products based on NPS technology, on a stand-alone basis and relative to competing services, will determine the availability and level of reimbursement received by us and providers. If physicians do not adopt and prescribe our future products, we may never become profitable.
We may find it difficult to enroll patients in our clinical trials. If we cannot enroll a sufficient number of eligible patients to participate in the clinical trials, we may not be able to initiate or continue clinical trials, which could delay or prevent development of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our product candidates as well as completion of required follow-up periods. In general, if patients are unwilling to participate in our trials because of negative publicity from adverse events in the life sciences industry or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting trials and obtaining regulatory approval of planned products may be delayed. If there are delays in accumulating the required patients and patient data, there may be delays in completing the trial. Further, if any of our clinical trial sites fail to comply with FDA-approved good clinical practices, we may be unable to use the data gathered at those sites. If our clinical investigators fail to carry out their contractual duties or regulatory obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of the clinical trials altogether.
Laboratory conditions differ from commercial conditions and field conditions, and the safety and effectiveness of our planned products may depend on the technique of the user.
Observations and developments that may be achievable under laboratory circumstances may not be able to be replicated in broader research and development phases, in commercial settings, or in the use of any of the planned products in the field. Furthermore, if commercialized, NPS will be administered by healthcare professionals and will require a degree of training and practice to administer correctly. Treatment results achieved during the laboratory or in clinical trials conducted by us or other investigators may not be representative of the results actually encountered during commercial use of our products due to variability in administration technique. The training and skills of investigators in our clinical trials may not be representative of the training and skills of future product users, which could negatively affect treatment results. In addition,
23
there may be a selection bias in the patients and/or sites of administration chosen for any clinical trials that would positively affect treatment results.
Issues with our firmware and software may negatively affect the function of our devices.
The safety and effectiveness of NPS-based treatments and therapies may depend, in part, on the function of firmware run by the microprocessors embedded in the device and associated software. This firmware and software is proprietary to us. While we have made efforts to test the firmware and software extensively, it is potentially subject to malfunction which in turn may harm a patient. Further, it may be vulnerable to physical break-ins, hackers, improper employee or contractor access, computer viruses, programming errors, or similar problems. Any of these might result in harm to a patient or the unauthorized release of confidential medical, business or other information of other persons or of ourselves.
We may encounter manufacturing problems or delays that could result in lost revenue. Additionally, we currently rely on third-party suppliers for critical materials needed to manufacture NPS devices such as the PulseTx System and related applicators and, if we obtain regulatory approval, our planned products. Any problems experienced by these suppliers could result in a delay or interruption of their supply to us, and as a result, we may face delays in the development and commercialization of planned products.
We perform final assembly of our devices to support our current research and development activities at our facility in Burlingame, California and will be moving to a larger facility in Hayward, California. We believe that we currently have, and at our new facility, will continue to have, adequate manufacturing capacity for these purposes. However, if demand for our planned products increases significantly, we will need to either expand our manufacturing capabilities or outsource to other manufacturers. We have no corporate experience in commercial-scale manufacturing of our planned products, and we currently rely upon third-party suppliers to manufacture and supply components for our NPS devices. The manufacture of these products in compliance with the FDA’s regulations requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medical device products often encounter difficulties in production, including difficulties with production costs and yields, quality control, quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced FDA requirements, other federal and state regulatory requirements, and foreign regulations.
We currently purchase components for our NPS devices under purchase orders and do not have long-term contracts with most of the suppliers of these materials. If suppliers were to delay or stop producing our components, or if the prices they charge us were to increase significantly, or if they elected not to sell to us, we would need to identify other suppliers. We could experience delays in manufacturing the devices while finding another acceptable supplier, which could impact our results of operations.
We may not become commercially viable if our ultimate commercialized products or related treatments fail to obtain an adequate level of reimbursement by Medicare and other third party payers.
We believe that the commercial viability of our potential devices and products and related treatments, and therefore our commercial success as a company, will be affected by the availability of government reimbursement and medical insurance coverage and reimbursement for newly approved medical therapies, technologies and devices. Insurance coverage and reimbursement is not assured. It typically takes a period of use in the market place before coverage and reimbursement is granted, if it is granted at all. In the United States and other jurisdictions in Europe and other regions, physicians and other healthcare providers generally rely on insurance coverage and reimbursement for their revenues, therefore this is an important factor in the overall commercialization plans of a proposed product and whether it will be accepted for use in the marketplace. Without insurance coverage and reimbursement for our planned products, we would expect to earn only diminished revenues, if any revenues are earned.
Medicare, Medicaid, health maintenance organizations and other third-party payers are increasingly attempting to contain healthcare costs by limiting both the scope of coverage and the level of reimbursement of new medical technologies and products, and as a result, they may not cover or provide adequate payment for the use of our planned products. In order to obtain satisfactory reimbursement arrangements, we may have to agree to a fee or sales price lower than the fee or sales price we might otherwise charge. Even if Medicare and other third-party payers decide to cover procedures involving our proposed devices and products, we cannot be certain that the reimbursement levels will be adequate. Accordingly, even if our planned products are approved for commercial sale, unless government and other third-party payers provide adequate coverage and reimbursement for our devices and products, some physicians may be discouraged from using them, and our sales would suffer.
24
Medicare reimburses for medical technologies and products in a variety of ways, depending on where and how the item is used. However, Medicare only provides reimbursement if CMS determines that the item should be covered and that the use of the device or product is consistent with the coverage criteria. A coverage determination can be made at the local level by the Medicare administrative contractor, a private contractor that processes and pays claims on behalf of CMS for the geographic area where the services were rendered, or at the national level by CMS through a national coverage determination. There are statutory provisions intended to facilitate coverage determinations for new technologies, but it is unclear how these new provisions will be implemented and it is not possible to indicate how they might apply to any of our proposed devices and products, as they are still in the development stages. Coverage presupposes that the technology, device, or product has been cleared or approved by the FDA and further, that the coverage will be no broader than the approved intended uses of the device or product as approved or cleared by the FDA, but coverage can be narrower. A coverage determination may be so limited that relatively few patients will qualify for a covered use of a device or product.
Obtaining a coverage determination, whether local or national, is a time-consuming, expensive and highly uncertain proposition, especially for a new technology, and inconsistent local determinations are possible. On average, Medicare coverage determinations for medical devices and products lag behind FDA approval. The Medicare statutory framework is also subject to administrative rulings, interpretations and discretion that affect the amount and timing of reimbursement made under Medicare. Medicaid coverage determinations and reimbursement levels are determined on a state by state basis, because Medicaid, unlike Medicare, is administered by the states under a state plan filed with the Secretary of the United States Department of Health and Human Services (HHS). Medicaid generally reimburses at lower levels than Medicare. Moreover, Medicaid programs and private insurers are frequently influenced by Medicare coverage determinations.
We work with outside scientists and their institutions in developing product candidates. These scientists may have other commitments or conflicts of interest, which could limit our access to their expertise and harm our ability to leverage our discovery platforms.
We work with scientific advisors and collaborators at academic research institutions in connection with our product development. These scientists and collaborators are not our employees, but they serve as either independent contractors or researchers under research agreements that we have with their sponsoring clinic, academic institution or research institution. Such scientists and collaborators may have other commitments that would limit their availability to us. Although our scientific advisors generally agree not to do competing work, if an actual or potential conflict of interest between their work for us and their work for another entity arises, we may lose their services. It is also possible that some of our valuable proprietary knowledge may become publicly known through these scientific advisors if they breach their confidentiality agreements with us, which would cause competitive harm to our business.
Risks Related to Intellectual Property
If we or our licensors are unable to protect our/their intellectual property, then our financial condition, results of operations and the value of our technology and products could be adversely affected.
Patents and other proprietary rights are essential to our business, and our ability to compete effectively with other companies is dependent upon the proprietary nature of our technologies. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect these, in part, through confidentiality agreements with certain employees, consultants and other parties. Our success will depend in part on the ability of our licensors and us to obtain, to maintain (including making periodic filings and payments) and to enforce patent protection for the licensed intellectual property, in particular, those patents to which we have secured rights. We, and our licensors, may not successfully prosecute or continue to prosecute the patent applications which we have licensed. Even if patents are issued in respect of these patent applications, we or our licensors may fail to maintain these patents, may determine not to pursue litigation against entities that are infringing upon these patents, or may pursue such enforcement less aggressively than we ordinarily would for our own patents. Without adequate protection for the intellectual property that we own or license, other companies might be able to offer substantially identical products for sale, which could unfavorably affect our competitive business position and harm our business prospects. Even if issued, patents may be challenged, invalidated, or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of term of patent protection that we may have for our products.
25
Litigation or third-party claims of intellectual property infringement or challenges to the validity of our patents would require us to use resources to protect our technology and may prevent or delay our development, regulatory approval or commercialization of our product candidates.
If we are the target of claims by third parties asserting that our products or intellectual property infringe upon the rights of others we may be forced to incur substantial expenses or divert substantial employee resources from our business. If successful, those claims could result in our having to pay substantial damages or could prevent us from developing one or more product candidates. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we or our collaborators experience patent infringement claims, or if we elect to avoid potential claims others may be able to assert, we or our collaborators may choose to seek, or be required to seek, a license from the third-party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly. The cost to us of any litigation or other proceeding, regardless of its merit, even if resolved in our favor, could be substantial. Some of our competitors may be able to bear the costs of such litigation or proceedings more effectively than we can because of their having greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Intellectual property litigation and other proceedings may, regardless of their merit, also absorb significant management time and employee resources.
If we fail to comply with our obligations in the agreements under which we license development or commercialization rights to products or technology from third-parties, we could lose license rights that are important to our business.
We hold licenses from ODURF and EVMS and from AMI-USC to intellectual property relating to the sub-microsecond electric field technology, as well as applicator design and configuration, and pulse generators in addition to the intellectual property that we own for these things. For the continuance of the license with ODURF and EVMS, Pulse Biosciences needs to commence pursuing one or more applications with the FDA by December 15, 2018 and continue to comply with the various obligations set forth in the license. If we fail to meet these obligations, the licensor will have the right to terminate the applicable license or modify certain terms of the license agreement. Generally, the loss of any one of our current licenses, or any other license we may acquire in the future, could harm our business, prospects, financial condition and results of operation.
Our intellectual property rights will not necessarily provide us with competitive advantages.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
|
· |
others may be able to make products that are similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed; |
|
· |
others may independently develop similar or alternative technologies without infringing our intellectual property rights; |
26
|
· |
the patents of others may have an adverse effect on our business, for example by preventing us from marketing one or more of our product candidates for one or more indications. |
Any of the aforementioned threats to our competitive advantage could harm our business.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely upon, among other things, unpatented proprietary technology, processes, trade secrets and know-how. Any involuntary disclosure to or misappropriation by third-parties of our confidential or proprietary information could enable competitors to duplicate or surpass our technological achievements, potentially eroding our competitive position in our market. We seek to protect confidential or proprietary information in part by confidentiality agreements with our employees, consultants and third-parties. While we require all of our employees, consultants, advisors and any third-parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure. To the extent that any of our staff were previously employed by other pharmaceutical, medical technology or biotechnology companies, those employers may allege violations of trade secrets and other similar claims in relation to their medical device development activities for us.
If we are unable to protect the intellectual property used in our products, others may be able to copy our innovations which may impair our ability to compete effectively in our markets.
The strength of our patents involves complex legal and scientific questions and can be uncertain. Our patents or patent applications may be challenged or our patent applications may fail to result in issued patents and our existing or future patents may be too narrow to prevent third-parties from developing or designing around our intellectual property and in that event we may lose competitive advantage and our business may suffer. Further, the patent applications that we license or have filed may fail to result in issued patents. The claims may need to be amended. Even after amendment, a patent may not issue and in that event we may not obtain the use of the intellectual property that we seek and may lose competitive advantage which could result in harm to our business.
We may become involved in future lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we or our licensors may file infringement claims, which can be expensive and time consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or of our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. If we or any current licensors or future licensees or licensors with rights to prosecute, assert or defend patents related to our product candidates fail to appropriately prosecute and maintain patent protection for patents covering any of our product candidates, or if patents covering any of our product candidates are asserted against infringers or defended against claims of invalidity or unenforceability in a manner which adversely affects such coverage, our ability to develop and commercialize any such product candidate may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
The United States Patent and Trademark Office may initiate interference proceedings to determine the priority of inventions described in or otherwise affecting our patents and patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if a prevailing party does not offer us a license on terms that are acceptable to us. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction of our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
27
Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of trade secrets and other proprietary information, which would harm our competitive position.
In addition to patents, we rely on trade secrets, technical know-how and proprietary information concerning our business strategy and product candidates in order to protect our competitive position, which are difficult to protect. As we collaborate with various third parties on the research and development of our planned products, we must, at times, share trade secrets with them. In the course of our research and development activities and our business activities, we rely on confidentiality agreements to protect our proprietary information. Such confidentiality agreements are used, for example, when we talk to vendors or potential strategic collaborators. In addition, each of our employees and consultants is required to sign a confidentiality agreement and invention assignment agreement upon joining our company. Our employees, consultants, contractors, business partners or outside scientific collaborators might intentionally or inadvertently disclose our trade secret information in breach of these confidentiality agreements or our trade secrets may otherwise be misappropriated. Our collaborators might also have rights to publish data, and we might fail to apply for patent protection prior to such publication. It is possible that a competitor will make use of such information, and that our competitive position will be compromised. In addition, to the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States sometimes are less willing than U.S. courts to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how, and our trade secrets cannot be enforced against such independently developed knowledge. If we cannot maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information would be jeopardized, which would adversely affect our competitive position.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Although we seek to protect our ownership of intellectual property rights by ensuring that our agreements with our independent contractors, collaborators and other third parties with whom we do business include provisions requiring such parties to assign rights in inventions to us, we may also be subject to claims that former employees, collaborators or other third parties have an ownership interest in our patents or other intellectual property. We may be subject to ownership disputes in the future arising, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could harm our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our current or future product candidates, if any, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. We believe this is caused by both the technical nature of the subject matter and a general enthusiasm for generic competition in developing countries, and is not a concern that is specific to any particular foreign jurisdiction. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against
28
us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We have not yet registered some of our trademarks in all of our potential markets, and failure to secure those registrations could adversely affect our business.
If we apply to register our trademarks in all of our potential markets, our applications may not be allowed for registration, and our registered trademarks may not be maintained or enforced. In addition, in the U.S. Patent and Trademark Office and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our existing and future products.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. In 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and also may affect patent litigation. These also include provisions that switched the United States from a “first-to-invent” system to a “first-to-file” system, allow third party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack the validity of a patent by the USPTO administered post grant proceedings. Under a first-to-file system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective in 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, patent reform legislation may pass in the future that could lead to additional uncertainties and increased costs surrounding the prosecution, enforcement and defense of our patents and applications. Furthermore, the U.S. Supreme Court and the U.S. Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the patent laws of the United States are interpreted. Similarly, foreign courts have made, and will likely continue to make, changes in how the patent laws in their respective jurisdictions are interpreted. We cannot predict future changes in the interpretation of patent laws or changes to patent laws that might be enacted into law by U.S. and foreign legislative bodies. Those changes may materially affect our patents or patent applications and our ability to obtain additional patent protection in the future.
Risks Related to Government Regulation
We may never receive regulatory approval, including that from the FDA, for any of our planned products.
We may never receive regulatory approvals, including from the FDA, for any potential therapies, devices or products in the United States or in any foreign market. Therefore, it is highly speculative as to any timing for our planned products to be commercialized. Investors need to take a long-term approach to an investment in our securities, as the commercial realization of our technology is speculative and well in the future.
29
We will be subject to stringent domestic and foreign regulation in respect of any potential therapies, devices and products. Any unfavorable regulatory action may materially and adversely affect our future financial condition and business operations and prospects.
Our potential therapies, devices and products, further development activities and manufacturing and distribution, once developed and determined, will be subject to extensive, rigorous and ongoing regulation by numerous government agencies, including the FDA and similar foreign regulatory authorities. To varying degrees, each of these agencies monitors and enforces our compliance with laws and regulations governing the development, testing, manufacturing, labeling, marketing, distribution, and the safety and effectiveness of our medical technology. The process of obtaining and maintaining marketing approval or clearance from the FDA and similar foreign regulatory authorities for new therapies, devices and products, or for enhancements, expansion of the indications or modifications to existing products, could:
If we experience any of these occurrences, our operations may suffer, we might experience harm to our competitive standing and result in further losses that adversely affect our financial condition. We will have ongoing responsibilities under FDA and international regulations, both before and after a product is approved and commercially released. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections. If an inspection were to conclude that we are not in compliance with applicable laws or regulations, or that any of our therapies or devices are ineffective or pose an unreasonable health risk, the FDA or similar foreign regulatory authorities could ban such medical therapies, devices or products, detain or seize such devices or products, order a recall, repair, replacement, or refund of such devices or products, or require us to notify health professionals and others that the therapies, devices or products present unreasonable risks of substantial harm to the public health. Additionally, the FDA or similar foreign regulatory authorities may impose other operating restrictions, enjoin and restrain certain violations of applicable law pertaining to therapies, devices and products and assess civil or criminal penalties against our officers, employees, or us. The FDA and similar foreign regulatory authorities have been increasing its scrutiny of the industry and the government is expected to continue to scrutinize the industry closely with inspections and possibly enforcement actions. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively manufacturing, marketing and selling our therapies, devices and products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our financial condition and results of operations.
We will have to comply with complex statutes prohibiting fraud and abuse, and both we and physicians utilizing our planned products could be subject to significant penalties for noncompliance.
There are many federal and state laws and regulations prohibiting fraud and abuse in the healthcare industry that can result in significant criminal and civil penalties. These federal laws include: the anti-kickback statutes which prohibit certain business practices and relationships, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs; the physician self-referral prohibition, commonly referred to as the Stark Law; the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; the Civil False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs and the Civil Monetary Penalties Law, which authorizes the imposition of civil penalties administratively for fraudulent or abusive acts.
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. As federal and state budget pressures continue, federal and state administrative agencies may also continue to escalate investigation and enforcement efforts to root out waste and to control fraud and abuse in governmental healthcare programs. Private enforcement of healthcare fraud has also increased, due in large part to amendments to the Civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. A violation of any of these federal and state fraud and abuse laws and regulations could have a material adverse effect on our liquidity and financial condition.
30
To obtain the necessary device and marketing and manufacturing clearance or approval, as a pre-condition, we will have to conduct various preclinical and clinical tests, which may be costly and time consuming, and may not provide results that will allow us to seek regulatory approval.
The number of preclinical and clinical tests that will be required for regulatory clearance or approval varies depending on the disease or condition to be treated, the method of treatment, the nature of the device, the jurisdiction in which we are seeking approval and the applicable regulations. Regulatory agencies, including those in the United States, Canada, Europe and other countries where medical devices and products are regulated, can delay, limit or deny approval of a product for many reasons. For example, regulatory agencies:
|
· |
may conclude that our device does not meet quality standards for durability, long-term reliability, biocompatibility, electromagnetic compatibility, or electrical safety; and |
The FDA may make requests or suggestions regarding conduct of our clinical trials, resulting in an increased risk of difficulties or delays in obtaining regulatory approval in the US. Any of these occurrences could prove materially harmful to our operations and business.
Even if a potential device or product ultimately is cleared or approved by the different regulatory authorities, it may be cleared or approved only for narrow indications which may render it commercially less viable.
Even if a potential device or product of ours is cleared or approved, it may not be cleared or approved for the indications that are necessary or desirable for a successful commercialization. Our preference will be to obtain as broad an indication as possible for use in connection with the particular disease or treatment for which it is designed. However, the final classification may be more limited than we originally seek. The limitation on use may make the device or product commercially less viable and more difficult, if not impractical, to market. Therefore we may not obtain the revenues that we seek in respect of the proposed product, and we will not be able to become profitable and provide an investment return to our investors.
Even if we obtain clearance or approval to sell a potential product, we will be subject to ongoing requirements and inspections that could lead to the restriction, suspension or revocation of our clearance.
We, as well as any potential collaborative partners such as manufacturers and distributors, will be required to adhere to applicable FDA regulations regarding good manufacturing practice, which include testing, control, and documentation requirements. We will be subject to similar regulations in foreign countries. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Ongoing compliance with good manufacturing practice and other applicable regulatory requirements is strictly enforced in the United States through periodic inspections by state and federal agencies, including the FDA, and in international jurisdictions by comparable agencies. Failure to comply with regulatory requirements could result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure to obtain premarket clearance or premarket approval for devices, withdrawal of approvals previously obtained, and criminal prosecution. The restriction, suspension or revocation of regulatory approvals or any other failure to comply with regulatory requirements will limit our ability to operate and could increase our costs.
Any failure or delay in completing clinical trials or studies for our therapies, devices and products and the expense of those trials may adversely affect our business.
Preclinical studies, clinical trials and post-clinical monitoring and trials required to demonstrate the safety and efficacy of our potential devices and products will be time consuming and expensive. If we must conduct additional clinical trials or other studies with respect to any of our proposed product candidates to those that are initially contemplated, if we are unable to successfully complete any clinical trials or other studies, or if the results of these trials or studies are not positive or are only modestly positive, we may be delayed in obtaining marketing approval for the planned products, we may not be able to obtain marketing approval, or we may obtain approval for indications that are not as broad as we seek. Our research and product development costs also will increase if we experience delays in testing or approvals. The completion of clinical trials for our proposed therapies, devices and products could be delayed because of our inability to manufacture or obtain from
31
third-parties materials sufficient for use in preclinical studies and clinical trials; delays in patient enrollment and variability in the number and types of patients available for clinical trials; difficulty in maintaining contact with patients after treatment, resulting in incomplete data; poor effectiveness of proposed devices and products during clinical trials; unforeseen safety issues or side effects; and governmental or regulatory delays and changes in regulatory requirements and guidelines. If we incur significant delays in our clinical trials, our competitors may be able to bring their products to market before we do, which could result in harming our ability to commercialize our planned products. If we experience any of these occurrences our business will be materially harmed.
Because we and one of our licensors have used federal funding in the development of certain aspects of our technology, the federal government retains ‘march-in’ rights in connection with results derived from these grants.
March-in rights give the federal government the right to grant to other entities, which may include competitors, licenses or to take a license for itself if the government funded the development of a patent. The march-in right applies to patents that have been issued. The march-in right is intended to be used only if there is a threat to public health and safety that the owner of the patent is not equipped to handle. The march-in right may also be used to remove the exclusive rights belonging to a patent holder if the patent for which the government provided funding is not suitable for public use. If march-in rights are used by the government, the entities using the patent are required to pay royalties to the patent holder, which amount would be subject to negotiation. Because federal funding was used for some aspects of the company’s technology that will be the subject of some of our patents, the company could be subject to the march-in right and lose its exclusivity of those patents, and may suffer direct competition if any license is granted by the government under the march-in right to a competitor.
Our employees, collaborators and other personnel may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, collaborators, vendors, principal investigators, consultants and commercial partners. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA and similar foreign regulatory authorities, provide accurate information to the FDA and similar foreign regulatory authorities, comply with data privacy and security and healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. Additionally, laws regarding data privacy and security, including HIPAA, as amended by HITECH, as well as comparable laws in non-U.S. jurisdictions, may impose obligations with respect to safeguarding the privacy, use, security and transmission of individually identifiable health information such as genetic material.
Various laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Any misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause harm to our reputation. We adopted a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Risks Related to Owning Our Common Stock
The price of our common stock is expected to be volatile, and you may be unable to sell your shares at or above the price you paid to acquire them.
The market price of our common stock has been volatile, and we expect it to continue to be volatile for the foreseeable future in response to many risk factors listed in this section, and others beyond our control, including:
32
|
· |
actual or anticipated fluctuations in our financial condition and operating results; |
|
· |
announcements by our customers, partners or suppliers relating directly or indirectly to our products, services or technologies; |
|
· |
announcements of technological innovations by us or our competitors; |
|
· |
overall conditions in our industry and market; |
|
· |
changes in laws or regulations applicable to our planned products; |
|
· |
announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, capital commitments or achievement of significant milestones; |
|
· |
additions or departures of key personnel; |
|
· |
competition from existing products or new products that may emerge; |
|
· |
fluctuations in the valuation of companies perceived by investors to be comparable to us; |
|
· |
disputes or other developments related to proprietary rights, including patents, litigation matters or our ability to obtain intellectual property protection for our technologies; |
|
· |
announcement or expectation of additional financing efforts; |
|
· |
sales of our common stock by us or our stockholders; |
|
· |
stock price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
|
· |
reports, guidance and ratings issued by securities or industry analysts; and |
|
· |
general economic and market conditions. |
If any of the foregoing occurs, it may cause our stock price or trading volume to decline. Stock markets in general and the market for companies in our industry in particular have experienced price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of our common stock. Investors may not realize any return on their investment in us and may lose some or all of their investment. In the past, companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
We do not know whether an active, liquid and orderly trading market will develop or be maintained for our common stock or what the market price of our common stock will be and as a result it may be difficult for you to sell your common stock.
Prior to our initial public offering in May 2016, there was no public market for our common stock. Although our common stock is listed on The NASDAQ Capital Market, the market for our shares has demonstrated varying levels of trading activity. As a result of these and other factors, you may not be able to sell your common stock quickly or at or above the price paid to acquire the stock or at all. Further, an inactive market may also impair our ability to raise capital by selling additional common stock and may impair our ability to enter into strategic collaborations or acquire companies or products by using our common stock as consideration.
Control by our principal stockholders may limit your ability to influence the outcome of director elections and other transactions requiring stockholder approval.
A significant percentage of our outstanding stock is held by a limited number of investors. As a result, such persons will have significant influence over corporate actions requiring stockholder approval, including the following actions:
|
· |
to elect or defeat the election of our directors; |
|
· |
to amend or prevent amendment of our articles of incorporation or bylaws; |
|
· |
to effect or prevent a merger, sale of assets or other corporate transaction; and |
|
· |
to control the outcome of any other matter submitted to our stockholders for vote. |
33
Such persons’ stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of our company, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price.
Management currently beneficially holds a small percentage of our common stock. Other than their positions as directors and officers, and the restriction on the stockholders being able to call a special meeting limited to a majority of the outstanding shares, our management will not be able to greatly influence corporate actions requiring stockholder approval.
We have incurred and will continue to incur costs as a result of operating as a public company and our management has been and will be required to devote substantial time to public company compliance initiatives.
As a public company, listed in the United States, we have incurred and will continue to incur significant legal, accounting and other expenses due to our compliance with regulations and disclosure obligations applicable to us, including compliance with the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC and the NASDAQ. The SEC and other regulators have continued to adopt new rules and regulations and make additional changes to existing regulations that require our compliance.
Stockholder activism, the current political environment, and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact, in ways we cannot currently anticipate, the manner in which we operate our business. Our management and other personnel have and will continue to devote a substantial amount of time to these compliance programs and monitoring of public company reporting obligations and, as a result of the new corporate governance and executive compensation related rules, regulations, and guidelines prompted by the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, and further regulations and disclosure obligations expected in the future, we will likely need to devote additional time and costs to comply with such compliance programs and rules. New laws and regulations as well as changes to existing laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002, the Dodd-Frank Act, and rules adopted by the SEC and NASDAQ, will likely result in increased costs to us as we respond to their requirements. We are currently evaluating and monitoring developments with respect to these rules and regulations, and we cannot predict or estimate the amount of additional costs we may incur or the timing of such costs.
Furthermore, these and future rules and regulations could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as our executive officers.
We agreed in the underwriting agreement for our initial public offering to conduct a rights offering as a pre-condition to certain future offers and sales of our common stock, which may hinder our ability to raise capital during the term of the provision and because of the exceptions stockholders may not be offered the right to participate in future offerings.
We agreed with one of the underwriters of our May 2016 initial public offering that for a period of up to five years after completion of our initial public offering, we will conduct offerings of our common stock so as to give the holders of our common stock the ability to participate through a rights offering. There are several exceptions to this obligation, including (i) stock dividends and splits, (ii) exercises and conversions of outstanding securities, (iii) equity awards under a stockholder approved plan which are authorized by the board of directors, (iv) merger, consolidation and combination transactions and business and asset acquisition transactions, (v) equity financings in any 12-month period that do not exceed both $2,500,000 in gross proceeds and 5% of the then issued and outstanding shares of our common stock, and (vi) transactions which are approved by MDB Capital Group, LLC, one of the underwriters of the offering. Should any one of these exceptions be applicable to an offering, we would be able to proceed with the offering without first giving our current stockholders the right to participate. Although a rights offering may provide to the current stockholders the opportunity to maintain their ownership percentage, it may delay or disrupt an offering of common stock or securities related to common stock. Rights offerings are typically held open for a period of 16 to 30 days, after the required corporate actions and documentation, including a registration statement, are completed, which may range from a few weeks to several months. Because a rights offering may not raise all the capital sought by a company, the company may have to structure the offering with over-subscription rights, standby purchasers, private placement agents and/or underwriters in order to sell the offered and any additional securities in order to obtain the sought amount of capital.
34
We are an “emerging growth company” under the JOBS Act of 2012 and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We will remain an “emerging growth company” for up to five years, although we will lose that status sooner if our revenues exceed $1 billion, if we issue more than $1 billion in non-convertible debt in a three-year period, or if the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30.
Because of the exemptions from various reporting requirements provided to us as an “emerging growth company,” we may be less attractive to investors as an investment opportunity and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our reporting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our common stock will depend on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. We currently have no analysts covering us and there can be no assurance that analysts will cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our share price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our share price or trading volume to decline.
Sales of shares of our common stock may adversely affect the market for our common stock.
If our stockholders, particularly our directors, executive officers and significant stockholders, sell, register for sale, or indicate an intent to sell, shares our common stock in the public market after the contractual lock-up agreements executed as part of our initial public offering and other legal restrictions on resale lapse, it may have a material adverse effect on the market price of our common stock.
We have not paid dividends in the past and have no plans to pay dividends.
We plan to reinvest all of our earnings, to the extent we have earnings, into our product research and development. We do not plan to pay any cash dividends with respect to our securities in the foreseeable future. We cannot assure you that we would, at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common stock as a dividend. Therefore, you should not expect to receive cash dividends on our outstanding common stock.
Our charter documents and Nevada law may inhibit a takeover that stockholders consider favorable.
Provisions of our articles of incorporation and bylaws and applicable provisions of Nevada law may delay or discourage transactions involving an actual or potential change in control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Some of the following provisions in our articles and bylaws that implement these are:
|
· |
5,000,000 shares of “blank check” preferred stock, which may be issued at the discretion of the board of directors, without further approval of the stockholders; |
|
· |
no cumulative voting rights for the holders of common stock in the election of directors; and |
|
· |
vacancies in the board of directors may be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum. |
35
The Revised Nevada Statutes also provide for restrictions on voting our equity securities in connection with unapproved business combinations and control shares, which we have not opted out of.
These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to potential acquirers at a premium over prevailing prices.
36
Item 1B. Unresolved Staff Comments
None.
As of December 31, 2016, we leased approximately 4,300 square feet in Burlingame, California, where we house our executive offices and research and development facilities.
On January 25, 2017, we entered into a lease agreement for premises consisting of approximately 15,697 square feet in Hayward, California. The premises will be used for our corporate headquarters and principal operating facility. The term of this lease is sixty-two months, commencing on the date that is the earlier of (1) the date upon which we commence business in the premises or (2) the date upon which the premises is ready for occupancy as defined in the lease. We anticipate commencing business in the premises in the third quarter of 2017. We have the right to extend this lease for five years upon written notice not more than twelve months nor less than nine months prior to the expiration of the original lease term. We have the right to terminate this lease if the landlord is unable to deliver the facility to us by December 1, 2017.
We believe that our existing facilities, together with the new premises under the lease agreement entered into in January 2017, will be sufficient to meet our needs in the foreseeable future.
From time to time, we may be involved in a variety of claims, lawsuits, investigations and proceedings relating to securities laws, product liability, patent infringement, contract disputes and other matters relating to various claims that arise in the normal course of our business. In addition, third parties may, from time to time, assert claims against us in the form of letters and other communications. We currently believe that these ordinary course matters will not have a material adverse effect on our business; however, the results of litigation and claims are inherently unpredictable. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Item 4. Mine Safety Disclosures
Not applicable.
37
Part II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed on The Nasdaq Capital Market and has been traded under the symbol “PLSE” since May 18, 2016. Prior to that date, there was no established public trading market for our common stock. As a result, we have not set forth quarterly information with respect to the high and low prices for our common stock for the two most recent fiscal years. The following table sets forth the range of high and low closing sales prices per share for our common stock as reported on the NASDAQ from May 18, 2016 through December 31, 2016:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2016 |
||||
|
|
|
High |
|
Low |
||
|
2nd Quarter |
|
$ |
4.54 |
|
$ |
4.08 |
|
3rd Quarter |
|
$ |
6.43 |
|
$ |
4.40 |
|
4th Quarter |
|
$ |
6.50 |
|
$ |
5.21 |
Holders of Record
As of March 3, 2017, there were approximately 63 stockholders of record of our common stock. We believe the actual number of stockholders is greater than this number of record holders and includes stockholders who are beneficial owners, but whose shares are held in “street” name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Dividend Policy
We have never declared or paid any cash dividend on our common stock and have no present plans to do so. We intend to retain earnings for use in the operation and expansion of our business.
Sales of Unregistered Securities
Warrants
During the year ended December 31, 2016, in connection with the closing of our IPO, we issued warrants as compensation to the underwriters of our IPO to purchase a total 574,985 shares of our common stock at a price of $5.00 per share. The warrants became exercisable 180 days after issuance and are exercisable for a period of five years. This issuance was undertaken in reliance upon the exemption that registration requirements available under Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”).
Stock Options
During the year ended December 31, 2016, we issued 405,132 stock options at a weighted average exercise price of $4.44 under our 2015 Stock Incentive Plan which was adopted in August 2015. These issuances were undertaken in reliance upon the exemptions from registration requirements available under Rule 701 and Section 4(a)(2) of the Securities Act.
Private Placement
On February 7, 2017, we entered into a securities purchase agreement (the “Purchase Agreement”) with Robert W. Duggan and Maky Zanganeh (the “Investors”), pursuant to which we, in a private placement, agreed to issue and sell to the Investors an aggregate of 819,673 shares of our common stock, par value $0.001 per share, at a price per share of $6.10 (the “Shares”), for gross proceeds of approximately $5 million (the “Private Placement”).
Pursuant to this Private Placement we sold the Shares to “accredited investors,” as that term is defined in the Securities Act of 1933, in reliance on the exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated under the Securities Act and corresponding provisions of state
38
securities or “blue sky” laws. The Investors represented that they were acquiring the Shares for investment only and not with a view towards, or for resale in connection with, the public sale or distribution thereof.
Use of Proceeds from Public Offerings of Common Stock
Our IPO of common stock was effected through a Registration Statement on Form S-1 (File No. 333-208694), as amended, which was declared effective on May 13, 2016. Our IPO closed on May 23, 2016 and resulted in net proceeds of approximately $20.3 million, including proceeds from the exercise of the overallotment option granted to the underwriters, net of underwriting discounts and commissions and other offering costs. There has been no material change in the planned use of proceeds from our IPO as described in our final prospectuses filed with the SEC on May 17, 2016 pursuant to Rule 424(b).
39
Performance Graph
The performance graph included in this Annual Report on Form 10-K shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or incorporated by reference into any filing of Pulse Biosciences, Inc. under the Securities Act of 1933, as amended, or the Exchange Act, except as shall be expressly set forth by specific reference in such filing.
The following graph shows a comparison from May 18, 2016 (the date our common stock commenced trading on the Nasdaq Capital Market through December 31, 2016 of the cumulative total return for our common stock, the Nasdaq Composite Index and the Nasdaq Biotechnology Index. Such returns are based on historical results and are not intended to suggest future performance. Data for The Nasdaq Composite Index and the Nasdaq Biotechnology Index assume reinvestment of dividends.
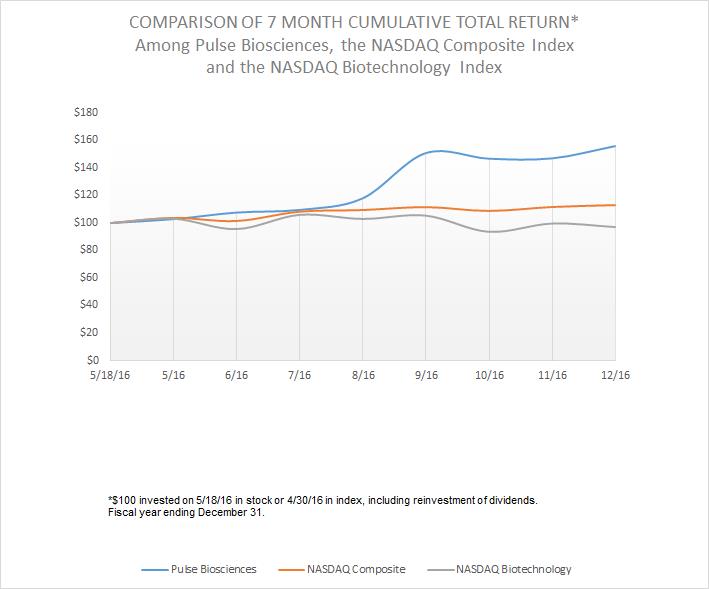
40
Item 6. Selected Financial Data
The following table sets forth selected financial data as of and for the periods indicated. The selected consolidated balance sheets as of December 31, 2016 and 2015, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for the year ended December 31, 2016 and 2015, and for the period from May 19, 2014 (inception) through December 31, 2014 are derived from our consolidated audited financial statements appearing in Item 8, “Financial Statements and Supplementary Data,” of this Annual Report on Form 10-K. Our historical results are not necessarily indicative of the results to be expected for any future period. The following selected financial data should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
(in thousands, except per share amounts) |
|
2016 |
|
2015 |
|
2014 |
|||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
2,933 |
|
|
1,224 |
|
|
43 |
|
Research and development |
|
|
5,988 |
|
|
2,578 |
|
|
26 |
|
Amortization of intangible assets |
|
|
665 |
|
|
666 |
|
|
111 |
|
Costs of business acquisitions |
|
|
— |
|
|
— |
|
|
120 |
|
Total operating expenses |
|
|
9,586 |
|
|
4,468 |
|
|
300 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
68 |
|
|
— |
|
|
— |
|
Total other income |
|
|
68 |
|
|
— |
|
|
— |
|
Loss from operations, before income taxes |
|
|
(9,518) |
|
|
(4,468) |
|
|
(300) |
|
Income tax benefit |
|
|
— |
|
|
(1,657) |
|
|
(23) |
|
Net loss |
|
$ |
(9,518) |
|
$ |
(2,811) |
|
$ |
(277) |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share: |
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share |
|
$ |
(0.86) |
|
$ |
(0.37) |
|
$ |
(0.11) |
|
Weighted average shares used to compute net loss per common share — basic and diluted |
|
|
11,009 |
|
|
7,565 |
|
|
2,511 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, |
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Cash, cash equivalents and marketable investments |
|
$ |
16,395 |
|
$ |
3,606 |
|
$ |
7,009 |
|
Working capital |
|
|
15,647 |
|
|
3,337 |
|
|
6,866 |
|
Total assets |
|
|
26,314 |
|
|
14,325 |
|
|
17,896 |
|
Total liabilities |
|
|
1,016 |
|
|
660 |
|
|
1,821 |
|
Total stockholders' equity |
|
|
25,298 |
|
|
13,665 |
|
|
16,074 |
|
|
|
|
|
|
|
|
|
|
|
41
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes thereto included in Item 8 under the heading “Financial Statements and Supplementary Data”. Some of the information contained in this discussion and analysis or set forth elsewhere in this Form 10-K contains forward-looking statements that involve risks and uncertainties, including statements regarding our expected financial results in future periods. The words “anticipates,” “believes,” “could,” “estimates,” “expects,” “intends,” “may,” “might,” “plans,” “projects,” “will,” “would,” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements that we make. You should read the “Risk Factors” section of this Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. We do not assume any obligation to update any forward-looking statements.
Overview
We are a clinical-stage medical technology company developing a non-thermal tissue treatment platform technology based upon our proprietary Nano-Pulse Stimulation (“NPS”) technology and pursuing applications in oncology, dermatology, general tissue treatment and veterinary medicine. NPS is a novel patented technology which leverages nano-second duration energy pulses that have demonstrated effective local tumor control and the initiation of an adaptive immune response in pre-clinical studies. We are pursuing a number of clinical applications for NPS, including oncology, dermatology, aesthetics and other minimally invasive applications where we believe NPS has the potential to compare favorably with current therapies and treatments. We are currently conducting research and development activities in pursuit of commercial applications for our NPS technology, but we have not yet commercialized or recognized revenue from our technology.
Plan of Operation
We plan to establish ourselves as a medical technology company with a local, non-thermal, and drug-free treatment platform that initiates cell death in targeted tissue by a process of cell signaling and also induces a systemic adaptive immune response to the targeted tissue. In order to accomplish this, we plan to:
|
· |
Improve our technology by continuing our research and product development efforts. We expect to develop different devices to target different tissue types that will leverage the novel characteristics of our technology platform. |
|
· |
Further explore and understand the benefits of NPS with the objectives of broadening the currently planned cosmetic and therapeutic applications and identifying new applications. We anticipate that results of our clinical studies will enable us to recognize certain unmet medical needs that may be addressed by our technology. |
|
· |
Continue to protect and expand our intellectual property portfolio with respect to NPS technology, which we expect will increase our ability to deter competitors and position our company for favorable licensing and partnering opportunities. |
|
· |
Partner with medical or biomedical device companies for certain applications which we anticipate may accelerate product development and acceptance into target market areas and allow us to gain the sales and marketing advantages of the distribution infrastructure. |
Critical Accounting Policies and Use of Estimates
The following discussion and analysis of financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in conformity with accounting principles generally accepted in the United States of America. Certain accounting policies and estimates are particularly important to the understanding of our financial position and results of operations and require the application of significant judgment by management or can be materially affected by changes from period to period in economic factors or conditions that are outside of the company’s control. As a result, these issues are subject to an inherent degree of uncertainty. In applying these policies, management uses its judgment to determine the appropriate assumptions to be used in the determination of certain estimates. Those estimates are based on our historical operations, future business plans and the projected financial results, the terms of existing contracts, trends in the industry and information available from other outside sources.
42
Long-Lived Assets
We review long-lived assets, consisting of equipment and intangible assets, for impairment at each fiscal year end or when events or changes in circumstances indicate the carrying value of these assets may exceed their current fair values. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the assets. Assets to be disposed of are separately presented in the consolidated balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated.
Goodwill
We record goodwill when the consideration paid in a business acquisition exceeds the fair value of the net tangible assets and the identified intangible assets acquired. We review goodwill for impairment at least annually or whenever changes in circumstances indicate that the carrying value of the goodwill may not be recoverable.
Stock-Based Compensation
We periodically issue stock options to officers, directors, employees and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date. Stock-based payments to officers, directors and employees, including grants of employee stock options, are recognized in the financial statements based on their fair values. Stock option grants, which are generally time vested, are measured at the grant date fair value and charged to operations on a straight-line basis over the vesting period. We estimate the grant date fair value of stock options, using the Black-Scholes option-pricing model on a straight-line basis over the requisite service period of the award.
The Black-Scholes option-pricing model requires the use of highly subjective assumptions which determine the fair value of stock-based awards. The assumptions used in our option-pricing model represent management’s best estimates. These estimates are complex, involve a number of variables, uncertainties and assumptions and the application of management’s judgment, so that they are inherently subjective. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the future. These assumptions are estimated as follows:
Risk-Free Interest Rate. We base the risk-free interest rate used in the Black-Scholes valuation model on the implied yield based on the U.S. Treasury yield curve in effect at the time of grant.
Expected Term. The expected term represents the period that our stock-based awards are expected to be outstanding. Because of the limitations on the sale or transfer of our common stock as a privately held company before our IPO, we did not believe our historical exercise pattern was indicative of the pattern we would experience as a publicly traded company post IPO. We have consequently used the Staff Accounting Bulletin No. 110 (“SAB 110”) simplified method to calculate expected term, which is the average of the contractual term and vesting period. We plan to continue using the SAB 110 simplified method until we have sufficient trading history as a publicly traded company.
Volatility. We determine the price volatility factor based on the historical volatilities of comparable public companies in a similar industry. We intend to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of our own common stock share price becomes available, or unless circumstances change such that the identified companies are no longer similar to us, in which case, more suitable companies whose share prices are publicly available would be utilized in the calculation.
Dividend Yield. The expected dividend assumption is based on our current expectations about our anticipated dividend policy. We currently do not expect to issue any dividends.
In addition to assumptions used in the Black-Scholes option-pricing model, we must also estimate a forfeiture rate to calculate stock-based compensation for our awards. We will continue to use judgment in evaluating the assumptions related to our stock-based compensation on a prospective basis. As we continue to accumulate additional data, we may have refinements to our estimates, which could materially impact our future stock-based compensation expense.
Income Taxes
We account for income taxes using the asset and liability method, whereby deferred tax assets and liability account balances are determined based on differences between the financial reporting and tax bases of assets and liabilities, and are measured using the enacted rates and laws that will be in effect when the differences are expected to reverse.
43
We provide a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. If we determine that we would be able to realize deferred tax assets in the future in excess of the recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should we determine that we would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
We account for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by FASB Accounting Standards Codification (“ASC”) 740-10, “Accounting for Uncertainty in Income Taxes.” The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized.
We are subject to U.S. federal income taxes and income taxes of various state tax jurisdictions. As our net operating losses have yet to be utilized, previous tax years remain open to examination by federal authorities and other jurisdictions in which we currently operates or have operated in the past. We are not currently under examination by any tax authority.
During 2014, we recorded deferred tax liabilities totaling $1.7 million reflecting the book and tax basis difference of the recorded intangible assets as the underlying intangible assets will be amortized to expense over their estimated useful lives but will not be expensed for tax purposes. During 2015, operating losses incurred resulted in the realization of deferred tax assets that exceeded deferred tax liabilities. The tax benefit recorded during 2015 reflects the benefit resulting from the deferred tax assets, partially offset by the net difference between the deferred tax liabilities and the valuation allowance recorded. The effect of this treatment in 2015 resulted in the realization of a $1.7 million tax benefit and the elimination of the deferred tax liabilities.
Segment and Geographical Information
We operate and manage our business as one reportable and operating segment. Our Chief Executive Officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for purposes of allocating resources and evaluating financial performance. Primarily all of our long-lived assets are based in the United States.
Components of Results of Operations
Operating Expenses
We generally recognize operating expenses as they are incurred in two general categories, general and administrative costs and research and development costs, as well as non-cash amortization of intangible assets. Our operating expenses also include non-cash components related to depreciation of equipment and stock-based compensation costs, which are allocated, as appropriate, to general and administrative costs and research and development costs.
|
· |
General and administrative expenses consist of salaries and related expenses for executive, finance, legal, human resources, information technology and administrative personnel, professional fees, insurance costs and other general corporate expenses. We expect general and administrative expenses to increase in the future as we hire personnel and incur additional costs to support the expansion of our research and development activities and our operation as a public company, including higher legal, accounting, insurance, compliance, compensation and other costs. |
|
· |
Research and development expenses consist of salaries and related expenses and consulting costs related to the design, development and enhancement of our potential future products, prototypes material and devices, patent filing fees and costs and rent, partially offset by grants received in support of specific research projects. We expect research and development costs to increase in the future as we develop next generation PulseTXTM systems and pursue commercial applications of our NPS technology and expand our clinical programs. |
44
Results of Operations
Comparison of the Years ended December 31, 2016 and 2015
Our consolidated statements of operations as discussed herein are presented below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended |
|
|
|
||||
|
|
|
December 31, |
|
|
|
||||
|
(in thousands) |
|
2016 |
|
2015 |
|
$ Change |
|||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
2,933 |
|
|
1,224 |
|
|
1,709 |
|
Research and development |
|
|
5,988 |
|
|
2,578 |
|
|
3,410 |
|
Amortization of intangible assets |
|
|
665 |
|
|
666 |
|
|
(1) |
|
Total operating expenses |
|
|
9,586 |
|
|
4,468 |
|
|
5,118 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
68 |
|
|
— |
|
|
68 |
|
Total other income |
|
|
68 |
|
|
— |
|
|
68 |
|
Loss from operations, before income taxes |
|
|
(9,518) |
|
|
(4,468) |
|
|
5,050 |
|
Income tax benefit |
|
|
— |
|
|
(1,657) |
|
|
(1,657) |
|
Net loss |
|
$ |
(9,518) |
|
$ |
(2,811) |
|
$ |
6,707 |
General and Administrative
General and administrative expenses increased by $1,709,000 to $2,933,000 in 2016, from $1,224,000 in 2015. The increase was due primarily to $908,000 of increased compensation costs, $277,000 of increased stock-based compensation expense, $219,000 of increased insurance costs, $151,000 of increased professional and consulting services, $58,000 of increased travel costs and $62,000 of increased supplies. Compensation, including stock-based compensation costs, increased due to increased headcount. Insurance, professional and consulting services, travel costs and supplies increased primarily as a result of fees incurred in preparation for and operating as a public company. General and administrative expenses are expected to increase substantially during 2017 reflecting the buildout of additional operational infrastructure to support the increased level of clinical and development activities, in addition to increased operational compliance activities.
Research and Development
Research and development expenses increased by $3,410,000 to $5,988,000 in 2016, from $2,578,000 in 2015. The increase was due primarily to $1,194,000 of increased consulting and outside services, $1,157,000 of increased prototype and development supplies, $813,000 of increased compensation costs, $192,000 of increased stock-based compensation expense, $84,000 of increased intellectual property-related legal costs and $67,000 of increased facility costs, partially offset by $123,000 of decreased sponsored research expenses. Consultant and outside services and prototype and development supplies increased due to increased product development activities and the costs associated with manufacturing pre-production prototypes. Compensation, including stock-based compensation costs, increased due to increased headcount. Facility costs increased as we expanded our research facility to support increased research and development activities during 2016 compared to the prior year. Sponsored research expenses decreased primarily due to higher grant funding provided in 2015 compared to 2016 related to the sponsored research agreement entered into with Old Dominion University Research Foundation (“ODURF”). Intellectual property-related legal costs increased as a result of ongoing research and development. Research and development expenses are expected to increase substantially during 2017 compared to 2016, as we expand our clinical study activities, continue development of our PulseTx systems towards commercialization, and pursue regulatory clearance for our technology in one or more indications.
Income Tax Benefit
We recognized an income tax benefit of $1,657,000 in 2015, due primarily to income tax benefit realized from deferred tax assets stemming from the net operating losses generated during 2015, net of the deferred tax liabilities as of December 31, 2015.
45
Comparison of the period from May 19, 2014 through December 31, 2014 and the year ended December 31, 2015
Our consolidated statements of operations as discussed herein are presented below.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
|
|
|
Year Ended |
|
through |
|
|
|
||
|
|
|
December 31, |
|
December 31, |
|
|
|
||
|
(in thousands) |
|
2015 |
|
2014 |
|
$ Change |
|||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
1,224 |
|
|
43 |
|
|
1,181 |
|
Research and development |
|
|
2,578 |
|
|
26 |
|
|
2,552 |
|
Amortization of intangible assets |
|
|
666 |
|
|
111 |
|
|
555 |
|
Costs of business acquisitions |
|
|
— |
|
|
120 |
|
|
120 |
|
Total operating expenses |
|
|
4,468 |
|
|
300 |
|
|
4,168 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
— |
|
|
— |
|
|
— |
|
Total other income |
|
|
— |
|
|
— |
|
|
— |
|
Loss from operations, before income taxes |
|
|
(4,468) |
|
|
(300) |
|
|
4,168 |
|
Income tax benefit |
|
|
(1,657) |
|
|
(23) |
|
|
1,634 |
|
Net loss |
|
$ |
(2,811) |
|
$ |
(277) |
|
$ |
2,534 |
The period from May 19, 2014 (inception) through November 6, 2014, was a period of limited activity for us, as we were in the formation stage and capital raising stage, until we completed the acquisition of the businesses and the license of the intellectual property rights necessary for our business in November 2014.
Year ended December 31, 2015
The operating results for the year ended December 31, 2015, reflect the first full year of operational activities and the increased development activities involving our proprietary technology, including sponsored research costs, in combination with the establishment of general and administrative functions during the year. We expect both research and development and general and administrative expenses to increase during 2016 reflecting continuation and expansion of activities commencing during 2015.
General and Administrative
General and administrative expenses totaled $43,000 and $1,224,000 for the period from May 19, 2014 (inception) to December 31, 2014 and the year ended December 31, 2015, respectively. Expenses incurred during 2014 consisted primarily of normal corporate formation and start-up legal costs, while in 2015 expenses reflect employee and board compensation expense of $733,000, professional and consulting services of $390,000 and other general expenses of $101,000. 2015 general and administration expenses include $398,000 of stock-based compensation expense compared to no expense in 2014.
Research and Development
Research and development expenses totaled $26,000 and $2,578,000 for the period from May 19, 2014 (inception to December 31, 2014 and the year ended December 31, 2015, respectively. Included in the expenses are National Institutes of Health (“NIH”) research grant revenues of $178,000 and $340,000 for 2014 and 2015, respectively. Expenses incurred during 2014 reflect compensation, patent expense and sponsored research expenses, while in 2015 expenses reflect compensation of $962,000, sponsored research and related expenses of $697,000, patent related expenses of $397,000, lab supplies and equipment of $243,000, and $279,000 of general research and development expenses. 2015 research and development expense include $5,000 of stock-based compensation compared to no expense in 2014.
Amortization of Intangible Assets
Amortization of intangible assets reflects for the presented periods the respective portion of the twelve-year amortization of the acquired technology and licensed intangible assets.
46
Costs of Business Acquisitions
Costs of business acquisitions reflect relevant legal fees of $120,000 for the period from May 19, 2014 (inception) through December 31, 2014.
Income Tax Benefit
We recognized an income tax benefit of $23,000 and $1,657,000 for the period from May 19, 2014 (inception) to December 31, 2014 and the year ended December 31, 2015 respectively. The income tax benefit realized during 2015 primarily reflects the deferred tax assets stemming from the net operating losses generated during 2015, net of the deferred tax liabilities as of December 31, 2015.
Liquidity and Capital Resources
To date, we have not generated any revenues from product sales, and management does not expect to generate revenues from product sales for the next few years. Since inception, we have funded our business plan through the issuance of equity securities and grants from governmental agencies. Over the next few years, we intend to invest in research and development to develop commercially viable products and to assess the feasibility of potential future products. Additionally, we expect that our general and administrative expenses will increase as we incur substantial incremental costs associated with being a public company.
In May and June 2016, we completed our IPO from which we received total net proceeds of $20.3 million, including proceeds from the exercise of the overallotment option granted to the underwriters, net of underwriting discounts and commissions and other offering costs.
In February 2017, we entered into a securities purchase agreement with Robert W. Duggan and Maky Zanganeh (the “Investors”), pursuant to which we, in this private placement, agreed to issue and sell to the Investors an aggregate of 819,673 shares of our common stock at a price per share of $6.10 (the “Shares”) for gross proceeds of approximately $5 million (the “Private Placement”).
Our condensed consolidated statements of cash flows as discussed herein are presented below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Year Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
(in thousands) |
|
2016 |
|
2015 |
|
2014 |
|||
|
Net cash used in operating activities |
|
$ |
(8,051) |
|
$ |
(3,317) |
|
$ |
(101) |
|
Net cash used in investing activities |
|
$ |
(14,381) |
|
$ |
(86) |
|
$ |
(45) |
|
Net cash provided by financing activities |
|
$ |
20,915 |
|
$ |
— |
|
$ |
7,155 |
|
Net increase (decrease) in cash |
|
$ |
(1,517) |
|
$ |
(3,403) |
|
$ |
7,009 |
At December 31, 2016, we had cash, cash equivalents and investments of $16.4 million. We believe that the net proceeds from our IPO and the Private Placement in February 2017 will be sufficient to fund our projected operating requirements for at least the next 12 months; however, we plan to raise additional capital in the future. These expectations are based on our current operating and financing plans which are subject to change. Until we are able to generate sustainable product revenues at profitable levels, we expect to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Such additional funds may not be available on terms acceptable to us or at all, particularly in light of recent market conditions. If we raise funds by issuing equity or equity-linked securities, the ownership of our stockholders will be diluted and the holders of new equity securities may have priority rights over our existing stockholders. If adequate funds are not available, we may be required to curtail operations significantly or to obtain funds by entering into agreements on unattractive terms. Our inability to raise capital could have a material adverse effect on our business, financial condition and results of operations.
Operating Activities
During 2016, we used cash of $8.1 million in operating activities. The difference between cash used in operating activities and net loss consisted primarily of stock-based compensation and depreciation and amortization, increased accrued expenses, partially offset by increased prepaid expenses and other current assets and deferred offering costs.
47
During 2015, we used cash of $3.3 million in operating activities. The difference between cash used in operating activities and net loss consisted primarily of depreciation and amortization and stock-based compensation, and changes in deferred income taxes.
Investing Activities
During 2016, we used cash of $14.4 million for investing activities for the purchase of investments and office and laboratory equipment.
During 2015, we used cash of $86,000 for investing activities for the purchase of office and laboratory equipment.
Financing Activities
During 2016, cash provided from financing activities was $20.9 million due to the net proceeds received from our IPO, after deducting underwriting discounts and commissions and other offering costs.
During 2015, we did not have any cash flows from financing activities.
During the period from May 19, 2014 (inception) through December 31, 2014, we generated cash from financing activities of $8,000 from the issuance of our common stock to our founders and net proceeds of $7.1 million from the November 6, 2014 common stock private placement, and $1,000 from the issuance of warrants to the placement agent.
Contractual Obligations
Frank Reidy Research Center Agreement
As provided for in the license agreement with ODURF and EVMS, both of which are stockholders of our company, effective on November 6, 2014, we sponsored certain approved research activities at ODURF’s Frank Reidy Research Center under a sponsored research agreement. In April 2016, the Company agreed to sponsor additional research under the SRA from April 2016 to March 2017, for an aggregate amount of $1.0 million. As of December 31, 2016, there was $0.25 million of approved budget remaining under this research agreement.
Operating Lease
We lease corporate offices and research facilities in Burlingame, California, under a lease expiring March 31, 2017, at a monthly cost of approximately $19,000.
In January 2017, we entered into a new lease agreement (the “Lease”) for premises consisting of approximately 15,697 rentable square feet located in Hayward, California (the “Premises”). The Premises will be used for our corporate headquarters and principal operating facility. The term of the Lease is sixty-two (62) months, commencing on the date that is the earlier of (i) the date upon which we commence business in the Premises or (ii) the date upon which the Premises is “Ready for Occupancy” as defined in the lease. Base monthly rent shall be abated for the first two (2) months of the Lease term and thereafter will be $42,400 per month during the first year of the Lease term, with specified annual increases thereafter until reaching approximately $50,300 per month during the last two (2) months of the Lease term. We are required to pay a refundable security deposit of approximately $101,000. The Landlord is obligated to provide us with improvement allowances in the amount of approximately $135.00 per rentable square foot of the Premises, which may be applied towards the costs of construction of the initial improvements in the Premises. We will be responsible for any such improvement costs in excess of the foregoing allowances. We may also be required to reimburse Landlord for certain expenses during the Lease term. We have the right to extend the Lease term by five (5) years upon written notice not more than twelve (12) months nor less than nine (9) months prior to the expiration of the original Lease term, with monthly payments equal to the “Fair Rental Value” as defined in the Lease. We also reserved the right to terminate the Lease if the Landlord is unable to deliver the Premises to us by December 1, 2017. Assuming the Landlord delivers the Premises to us by December 1, 2017, the lease obligations as of December 31, 2016 for less than one year, one to three years, three to five years and more than five years is approximately $0.3 million, $1.1 million, $1.1 million and $0.3 million, respectively.
Off-Balance Sheet Arrangements
At December 31, 2016, we did not have any transactions, obligations or relationships that could be considered off-balance sheet arrangements.
48
JOBS Act Accounting Election
Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Trends, Events and Uncertainties
Research and development of new technologies are, by their nature, unpredictable. Although we undertake development efforts with commercially reasonable diligence, there can be no assurance that the net proceeds from our financings will be sufficient to enable us to develop our technology to the extent needed to generate future sales to sustain our operations. If we do not continue to have enough funds to sustain our operations, we will consider other options to continue our path to commercialization of NPS, including, but not limited to, additional financing through follow-on stock offerings, debt financings, or co-development agreements and /or other alternatives.
We cannot assure investors that our technology will be adopted or that we will ever achieve sustainable revenues sufficient to support our operations. Even if we are able to generate revenues, there can be no assurances that we will be able to achieve profitability or positive operating cash flows. There can be no assurances that we will be able to secure additional financing in the future on acceptable terms or at all. If cash resources are insufficient to satisfy our ongoing cash needs, we would be required to scale back or discontinue our technology and product development programs, or obtain funds, if available, although there can be no assurances, through the sale, licensing or strategic alliances that could require us to relinquish rights to our technology and intellectual property, or to curtail, suspend or discontinue our operations entirely.
Other than as discussed above and elsewhere in this Annual Report on Form 10-K, we are not currently aware of any trends, events or uncertainties that are likely to have a material effect on our financial condition in the near term, although it is possible that new trends or events may develop in the future that could have a material effect on our financial condition.
49
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to market risk in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates.
Interest Rate and Market Risk
Our exposure to interest rate and market risk is confined to our cash, cash equivalents and investments, all of which have maturities of less than three years. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of our cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. The securities in our investment portfolio are not leveraged, are classified as available-for-sale, and are, due to their relatively short-term nature, subject to minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that a hypothetical 10% change in market interest rates would have any material negative impact on the value of our investment portfolio.
Foreign Exchange Risk
The majority of our expense and capital purchasing activities are transacted in U.S. dollars. We do not have any international operations. We may incur foreign exchange gains or losses in the future.
50
Item 8. Financial Statements and Supplementary Data
PULSE BIOSCIENCES, INC.
Index to Consolidated Financial Statements
|
|
|
|
|
|
|
Page |
|
|
|
Number |
|
CONSOLIDATED FINANCIAL STATEMENTS |
|
|
|
|
52 |
|
|
|
53 |
|
|
Consolidated Statements of Operations and Comprehensive Loss |
|
54 |
|
|
55 |
|
|
|
56 |
|
|
57 |
51
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Pulse Biosciences, Inc.
We have audited the accompanying consolidated balance sheets of Pulse Biosciences, Inc. (as defined in Note 2 to the consolidated financial statements) (the “Company”) as of December 31, 2016 and 2015, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for the years ended December 31, 2016 and 2015, and for the period from May 19, 2014 (inception) through December 31, 2014. The Company’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of their internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2016 and 2015, and the results of their operations and their cash flows for the years ended December 31, 2016 and 2015, and for the period from May 19, 2014 (inception) through December 31, 2014, in conformity with accounting principles generally accepted in the United States of America.
/s/ Gumbiner Savett Inc.
March 20, 2017
Santa Monica, California
52
Consolidated Balance Sheets
(in thousands, except per share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
ASSETS |
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
2,089 |
|
$ |
3,606 |
|
Marketable investments |
|
|
14,306 |
|
|
— |
|
Prepaid expenses and other current assets |
|
|
268 |
|
|
44 |
|
Deferred offering costs |
|
|
— |
|
|
347 |
|
Total current assets |
|
|
16,663 |
|
|
3,997 |
|
Equipment, net of accumulated depreciation |
|
|
317 |
|
|
329 |
|
Intangible assets, net of accumulated amortization |
|
|
6,543 |
|
|
7,208 |
|
Goodwill |
|
|
2,791 |
|
|
2,791 |
|
Total assets |
|
$ |
26,314 |
|
$ |
14,325 |
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
Accounts payable |
|
$ |
265 |
|
$ |
262 |
|
Accrued expenses |
|
|
751 |
|
|
398 |
|
Total current liabilities |
|
|
1,016 |
|
|
660 |
|
|
|
|
|
|
|
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
Preferred stock, $0.001 par value; |
|
|
— |
|
|
— |
|
Common stock, $0.001 par value; |
|
|
13 |
|
|
8 |
|
Additional paid-in capital |
|
|
37,898 |
|
|
16,745 |
|
Accumulated other comprehensive loss |
|
|
(7) |
|
|
— |
|
Accumulated deficit |
|
|
(12,606) |
|
|
(3,088) |
|
Total stockholders’ equity |
|
|
25,298 |
|
|
13,665 |
|
Total liabilities and stockholders’ equity |
|
$ |
26,314 |
|
$ |
14,325 |
See accompanying notes to the consolidated financial statements.
53
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except per share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
2,933 |
|
|
1,224 |
|
|
43 |
|
Research and development |
|
|
5,988 |
|
|
2,578 |
|
|
26 |
|
Amortization of intangible assets |
|
|
665 |
|
|
666 |
|
|
111 |
|
Costs of business acquisitions |
|
|
— |
|
|
— |
|
|
120 |
|
Total operating expenses |
|
|
9,586 |
|
|
4,468 |
|
|
300 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
68 |
|
|
— |
|
|
— |
|
Total other income |
|
|
68 |
|
|
— |
|
|
— |
|
Loss from operations, before income taxes |
|
|
(9,518) |
|
|
(4,468) |
|
|
(300) |
|
Income tax benefit |
|
|
— |
|
|
(1,657) |
|
|
(23) |
|
Net loss |
|
|
(9,518) |
|
|
(2,811) |
|
|
(277) |
|
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
Unrealized loss on available-for-sale securities, net of tax |
|
|
(7) |
|
|
— |
|
|
— |
|
Comprehensive loss |
|
$ |
(9,525) |
|
$ |
(2,811) |
|
$ |
(277) |
|
Net loss per share |
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share |
|
$ |
(0.86) |
|
$ |
(0.37) |
|
$ |
(0.11) |
|
Weighted average shares used to compute net loss per common share — basic and diluted |
|
|
11,009 |
|
|
7,565 |
|
|
2,511 |
See accompanying notes to the consolidated financial statements.
54
Consolidated Statements of Stockholders’ Equity
PERIOD FROM MAY 19, 2014 (INCEPTION) THROUGH DECEMBER 31, 2014,
AND THE YEARS ENDED DECEMBER 31, 2015 AND 2016
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
Accumulated Other |
|
|
|
|
Total |
|||
|
|
|
Common Stock |
|
Paid-in |
|
Comprehensive |
|
Accumulated |
|
Stockholders’ |
|||||||
|
|
|
Shares |
|
Amount |
|
Capital |
|
Loss |
|
Deficit |
|
Equity |
|||||
|
Common stock issued to founders |
|
1,125 |
|
$ |
1 |
|
$ |
7 |
|
$ |
— |
|
$ |
— |
|
$ |
8 |
|
Issuance of common stock in private placement, net of issuance costs of $853 |
|
2,996 |
|
|
3 |
|
|
7,144 |
|
|
— |
|
|
— |
|
|
7,147 |
|
Common stock issued in connection with |
|
2,027 |
|
|
2 |
|
|
5,409 |
|
|
— |
|
|
— |
|
|
5,411 |
|
Common stock issued in connection with |
|
1,417 |
|
|
2 |
|
|
3,783 |
|
|
— |
|
|
— |
|
|
3,785 |
|
Net loss for the period from May 19, 2014 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(277) |
|
|
(277) |
|
Balance, December 31, 2014 |
|
7,565 |
|
$ |
8 |
|
$ |
16,343 |
|
$ |
— |
|
$ |
(277) |
|
$ |
16,074 |
|
Stock-based compensation expense |
|
— |
|
|
— |
|
|
402 |
|
|
— |
|
|
— |
|
|
402 |
|
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(2,811) |
|
|
(2,811) |
|
Balance, December 31, 2015 |
|
7,565 |
|
$ |
8 |
|
$ |
16,745 |
|
$ |
— |
|
$ |
(3,088) |
|
$ |
13,665 |
|
Issuance of common stock upon initial public offering, net of issuance costs of $2,711 |
|
5,750 |
|
|
5 |
|
|
20,283 |
|
|
— |
|
|
— |
|
|
20,288 |
|
Stock-based compensation expense |
|
— |
|
|
— |
|
|
870 |
|
|
— |
|
|
— |
|
|
870 |
|
Unrealized loss on marketable investments, net of tax |
|
— |
|
|
— |
|
|
— |
|
|
(7) |
|
|
— |
|
|
(7) |
|
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(9,518) |
|
|
(9,518) |
|
Balance, December 31, 2016 |
|
13,315 |
|
$ |
13 |
|
$ |
37,898 |
|
$ |
(7) |
|
$ |
(12,606) |
|
$ |
25,298 |
See accompanying notes to the consolidated financial statements.
55
Consolidated Statements of Cash Flows
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(9,518) |
|
$ |
(2,811) |
|
$ |
(277) |
|
Adjustments to reconcile net loss to net cash used in |
|
|
|
|
|
|
|
|
|
|
Depreciation of equipment |
|
|
94 |
|
|
51 |
|
|
6 |
|
Amortization of intangible assets |
|
|
665 |
|
|
666 |
|
|
111 |
|
Stock-based compensation |
|
|
870 |
|
|
402 |
|
|
— |
|
Net premium amortization on marketable investments |
|
|
4 |
|
|
— |
|
|
— |
|
Change in deferred income taxes |
|
|
— |
|
|
(1,657) |
|
|
(23) |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
Prepaid expenses and other current assets |
|
|
(135) |
|
|
(12) |
|
|
12 |
|
Deferred offering costs |
|
|
(280) |
|
|
(347) |
|
|
— |
|
Accounts payable |
|
|
3 |
|
|
125 |
|
|
29 |
|
Accrued expenses |
|
|
246 |
|
|
305 |
|
|
58 |
|
Deferred grant revenue |
|
|
— |
|
|
(39) |
|
|
(17) |
|
Net cash used in operating activities |
|
|
(8,051) |
|
|
(3,317) |
|
|
(101) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
Purchase of equipment |
|
|
(64) |
|
|
(86) |
|
|
(46) |
|
Cash acquired in connection with acquisition of businesses |
|
|
— |
|
|
— |
|
|
1 |
|
Purchase of marketable investments |
|
|
(19,067) |
|
|
— |
|
|
— |
|
Maturities of marketable investments |
|
|
4,750 |
|
|
— |
|
|
— |
|
Net cash used in investing activities |
|
|
(14,381) |
|
|
(86) |
|
|
(45) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
Common stock issued to founders |
|
|
— |
|
|
— |
|
|
8 |
|
Net proceeds from private placement |
|
|
— |
|
|
— |
|
|
7,147 |
|
Proceeds from issuance of common stock from initial public offering, net of issuance costs |
|
|
20,915 |
|
|
— |
|
|
— |
|
Net cash provided by financing activities |
|
|
20,915 |
|
|
— |
|
|
7,155 |
|
Net increase (decrease) in cash |
|
|
(1,517) |
|
|
(3,403) |
|
|
7,009 |
|
Cash and cash equivalents at beginning of period |
|
|
3,606 |
|
|
7,009 |
|
|
— |
|
Cash and cash equivalents at end of period |
|
$ |
2,089 |
|
$ |
3,606 |
|
$ |
7,009 |
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of noncash investing |
|
|
|
|
|
|
|
|
|
|
Reclassification of deferred offering costs to additional paid-in capital upon initial public offering |
|
$ |
627 |
|
$ |
— |
|
$ |
— |
|
Fair value of common stock issued in connection |
|
$ |
— |
|
$ |
— |
|
$ |
3,785 |
|
Fair value of common stock issued in connection |
|
$ |
— |
|
$ |
— |
|
$ |
5,411 |
|
Equipment purchased in accrued expenses |
|
$ |
18 |
|
$ |
104 |
|
$ |
— |
See accompanying notes to the consolidated financial statements.
56
Notes to Consolidated Financial Statements
1. Organization and Description of Business
Business and Basis of Presentation
Pulse Biosciences, Inc., incorporated in Nevada on May 19, 2014, is a clinical-stage medical technologies company developing commercial clinical applications for its proprietary Nano-Pulse Stimulation (“NPS”) technology. NPS is a novel patented technology which leverages nano-second duration energy pulses that have demonstrated effective local tumor control and the initiation of an adaptive immune response in pre-clinical studies. The Company is pursuing a number of clinical applications for NPS, including human oncology, dermatology, aesthetics and other minimally invasive applications where NPS is believed to provide greater benefits compared to current therapies and treatments. Pulse Biosciences, Inc. is referred to individually as “Pulse” and collectively with its wholly-owned subsidiaries as described below as the “Company” or “Pulse Biosciences, Inc.” The Company’s corporate office and research facility are located in Burlingame, California.
The Company’s activities are subject to significant risks and uncertainties, including the need for additional capital. The Company has not yet commenced any revenue-generating operations, does not have any cash flows from operations, and will need to raise additional capital to finance its operations. However, there can be no assurances that the Company will be able to obtain additional financing on acceptable terms and in the amounts necessary to fully fund its operating requirements.
Initial Public Offering
On May 13, 2016, the Company’s registration statement on Form S-1 (File No. 333-208694), as amended (the “Registration Statement”), relating to its initial public offering (“IPO”) was declared effective by the Securities and Exchange Commission (“SEC”) and closed on May 23, 2016, whereby the Company sold 5,000,000 shares of common stock at $4.00 per share. The shares began trading on the NASDAQ Capital Market under the trading symbol “PLSE” on May 18, 2016. On June 21, 2016, the underwriters exercised their overallotment option to purchase an additional 749,846 shares of the Company’s common stock at $4.00 per share. The Company received net proceeds of approximately $20.3 million from the IPO, including proceeds from the exercise of the overallotment option granted to the underwriters, net of underwriting discounts and commissions and other offering costs.
2. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements are prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”) and include the financial statements of Pulse and its wholly-owned subsidiaries, BioElectroMed Corp. (“BEM”) and NanoBlate Corp. (“NB”), since their date of acquisition on November 6, 2014. ThelioPulse, Inc. (“TPI”), which was acquired on November 6, 2014, was merged into Pulse subsequent to its acquisition and ceased to exist as a separate entity. Intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions. These estimates and assumptions affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual amounts could differ from those estimates.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist of cash and cash equivalents and investments. The Company places its cash equivalents and investments with high credit quality financial institutions and, by policy, limits the amounts invested with any one financial institution or issuer. Deposits held with banks may exceed the amount of insurance provided on such deposits. The Company has not experienced any losses since inception.
57
Fair Value of Financial Instruments
The Company believes the carrying amounts of its financial instruments, including cash equivalents, prepaid expenses and other current assets, accounts payable and accrued expenses, approximate fair value due to the short-term nature of such instruments.
Cash, Cash Equivalents and Marketable Investments
The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents. The Company has designated all marketable investments as available-for-sale and therefore, such marketable investments are reported at fair value, with unrealized gains and losses recognized in accumulated other comprehensive income (loss) (“OCI”) in stockholders’ equity. The cost of marketable securities is adjusted for the amortization of premiums and discounts to expected maturity. Premium and discount amortization is included in other income, net. Realized gains and losses, as well as interest income, on available-for-sale securities are also included in other income, net. The Company includes all of its available-for-sale securities in current assets.
All of the Company’s marketable investments are subject to a periodic impairment review. The Company recognizes an impairment charge when a decline in the fair value of its marketable investments below the cost basis is judged to be other-than-temporary. Factors considered in determining whether a loss is temporary include the length of time and extent to which the marketable investments fair value has been less than the cost basis, the financial condition and near-term prospects of the investee, extent of the loss related to credit of the issuer, the expected cash flows from the security, the Company’s intent to sell the security and whether or not the Company will be required to sell the security before the recovery of its amortized cost. During the year ended December 31 2016, the Company did not recognize any impairment charges on its investments as it is more likely than not that the Company will recover their amortized cost basis upon sale or maturity. The Company did not hold investment securities at any time during the prior year.
Deferred and Capitalized Offering Costs
Costs incurred in connection with ongoing equity financing activities, consisting primarily of legal, accounting and other professional fees, are deferred until the related financing is either completed or abandoned. Costs related to completed equity financings are charged directly to additional paid-in capital. Costs related to abandoned financings are charged to operations.
Equipment
Equipment is recorded at cost and depreciated on a straight-line basis over their estimated useful lives, ranging from three to five years.
Intangible Assets
The Company’s intangible assets consist of acquired patents and licenses, which are being amortized over their estimated useful lives of twelve years.
Long-Lived Assets
The Company reviews long-lived assets, consisting of equipment and intangible assets, for impairment during each fiscal year or when events or changes in circumstances indicate the carrying value of these assets may exceed their current fair values. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the assets. Assets to be disposed of are separately presented in the consolidated balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated. The Company has not historically recorded any impairment to its long-lived assets. In the future, if events or market conditions affect the estimated fair value to the extent that a long-lived asset is impaired, the Company will adjust the carrying value of these long-lived assets in the period in which the impairment occurs. For the years ended December 31, 2016, 2015 and the period from May 19, 2014 (inception) through December 31, 2014, the Company had not deemed any long-lived assets as impaired, and was not aware of the existence of any indicators of impairment at such dates.
58
Goodwill
The Company records goodwill when the consideration paid in a business acquisition exceeds the fair value of the net tangible assets and the identified intangible assets acquired. The Company reviews goodwill for impairment at least annually or whenever changes in circumstances indicate that the carrying value of the goodwill may not be recoverable. For the years ended December 31, 2016, 2015 and the period from May 19, 2014 (inception) through December 31, 2014, the Company had not deemed the value of goodwill as impaired, and was not aware of the existence of any indicators of impairment at such dates.
Stock-Based Compensation
The Company periodically issues stock options to officers, directors, employees and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date.
Stock-based payments to officers, directors and employees, including grants of employee stock options, are recognized in the financial statements based on their grant date fair values. Stock option grants, which are generally time vested, are measured at the grant date fair value and charged to operations on a straight-line basis over the vesting period. The fair value of stock options is determined utilizing the Black-Scholes option-pricing model, which is affected by several variables, including the risk-free interest rate, the expected dividend yield, the life of the equity award, the exercise price of the stock option as compared to the fair value of the common stock on the grant date, and the estimated volatility of the common stock over the term of the equity award.
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The Company estimates volatility based on the average historical volatilities of comparable public companies in a similar industry. The expected dividend yield is based on the current yield at the grant date; the Company has never declared or paid dividends and has no plans to do so for the foreseeable future. As permitted by Staff Accounting Bulletin No. 107, due to the Company’s lack of history and option activity, management utilizes the simplified method to estimate the expected term of options at the date of grant. Prior to the Company’s IPO, the fair value of common stock was determined by reference to either recent or anticipated cash transactions involving the sale of the Company’s common stock.
Stock options issued to non-employees as compensation for services provided to the Company are accounted for based upon the estimated fair value of the stock option. Management utilizes the Black-Scholes option-pricing model to determine the fair value of the stock options issued by the Company. The Company recognizes this expense over the period in which the services are provided.
The Company recognizes the fair value of stock-based compensation costs in general and administrative costs and in research and development costs, as appropriate, in the Company’s consolidated statements of operations. The Company issues new shares to satisfy stock option exercises.
Research Grants
Research grants are generally funded and paid through governmental, institutional, educational or research organizations. Grants received from agencies of the federal government are subject to federal regulation as to how the Company conducts its research activities, and the Company is required to comply with the respective research agreement terms relating to those grants. Amounts received under research grants are nonrefundable, regardless of the success of the underlying research project, to the extent that such amounts are expended in accordance with the approved grant project. The Company is permitted to draw down the research grants after incurring the related expenses. Amounts received under research grants are offset against the related research and development costs in the Company’s consolidated statement of operations as the costs are incurred.
Research and Development Costs
Research and development costs consist primarily of compensation costs, fees paid to consultants and outside service providers and organizations (including research institutes at universities), development prototypes, patent fees and costs, and other expenses relating to the acquisition, design, development and testing of the Company’s product candidates. Research and development costs incurred by the Company are expensed as incurred, unless the achievement of milestones, the completion of contracted work, or other information indicates that a different expensing schedule is more appropriate.
59
Patent Costs
The Company is the owner of numerous domestic and foreign patents. Due to the significant uncertainty associated with the successful development of one or more commercially viable products based on the Company’s research efforts and any related patent applications, patent costs not related to acquired patents, including patent-related legal fees, filing fees and other costs, including internally generated costs, are expensed as incurred. During the years ended December 31, 2016 and 2015 patent costs totaled $0.5 million and $0.4 million, respectively. During the period from May 19, 2014 (inception) through December 31, 2014, patent costs were $26,000. Patent costs are included in research and development costs in the consolidated statements of operations and comprehensive loss.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company determines that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
The Company is subject to U.S. federal income taxes and income taxes of various state tax jurisdictions. As the Company’s net operating losses have yet to be utilized, previous tax years remain open to examination by federal authorities and other jurisdictions in which the Company currently operates or has operated in the past. The Company is not currently under examination by any tax authority.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by U.S. GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized. At December 31, 2016 and 2015, the Company had not recorded any liability for uncertain tax positions. The Company includes interest and penalties related to uncertain tax positions as a component of income tax expense.
Comprehensive Loss
Comprehensive loss consists of net loss and unrealized gains or losses on available-for-sale investments. The Company displays comprehensive loss and its components as part of the consolidated statements of operations and comprehensive loss.
Net Loss per Share
The Company’s basic net loss per share is calculated by dividing the net loss by the weighted average number of shares of common stock outstanding for the period. Diluted net loss per share is computed by giving effect to all potential dilutive common stock equivalents outstanding for the period. For purposes of this calculation, options to purchase common stock and common stock warrants are considered common stock equivalents. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted net loss per share.
Basic and diluted net loss per common share is the same for all periods presented because all warrants and stock options outstanding are anti-dilutive.
The following outstanding stock options and warrants to purchase common stock were excluded from the computation of diluted net loss per share for the periods presented because including them would have had an anti-dilutive effect:
60
|
|
|||||||||
|
|
|||||||||
|
|
May 19, 2014 |
||||||||
|
|
(inception) |
||||||||
|
|
Years Ended |
through |
|||||||
|
|
December 31, |
December 31, |
|||||||
|
|
2016 |
2015 |
2014 |
||||||
|
Common stock warrants |
874,610 | 299,625 | 299,625 | ||||||
|
Common stock options |
1,229,355 | 875,221 |
— |
||||||
|
Total |
2,103,965 | 1,174,846 | 299,625 | ||||||
Segment and Geographical Information
The Company operates and manages its business as one reportable and operating segment. The Company’s Chief Executive Officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for purposes of allocating resources and evaluating financial performance. All of the Company’s long-lived assets are based in the United States.
Recently Adopted Standards
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Updates (“ASU”) No. 2014-15, Presentation of Financial Statements – Going Concern Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern, which requires management to evaluate whether there are conditions or events that raise substantial doubt about an entity’s ability to continue as a going concern and to provide disclosures when certain criteria are met. The guidance is effective for annual periods beginning after December 15, 2016 and interim reporting periods starting in the first quarter of 2017. The Company adopted this standard as of December 31, 2016.
Recent Accounting Pronouncements
During May 2014, the Financial Accounting Standards Board (“FASB”) issued (“ASU”) No. 2014-09, Revenue from Contracts with Customers. This updated standard will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective and permits the use of either the retrospective or cumulative effect transition method. In August 2015, the FASB issued an update to defer the effective date of this update by one year. This updated standard becomes effective for the Company in the first quarter of fiscal year 2018, but allows the Company to adopt the standard one year earlier if it so chooses. In March 2016, the FASB issued updates to this guidance to clarify the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued guidance to clarify aspects related to identifying performance obligations and the licensing implementation guidance. In May 2016, the FASB issued guidance to clarify the implementation on narrow scope improvements and practical expedients. The Company is currently evaluating the impact of adopting these standards.
During February 2016, the FASB issued ASU No. 2016-02, Leases, which amends the existing accounting standards for leases. The new standard requires lessees to record a right-of-use asset and a corresponding lease liability on the balance sheet (with the exception of short-term leases). For lessees, leases will continue to be classified as either operating or financing in the income statement. This ASU becomes effective for the Company in the first quarter of fiscal year 2019 and early adoption is permitted. This ASU is required to be applied with a modified retrospective approach and requires application of the new standard at the beginning of the earliest comparative period presented. The Company generally does not finance purchases of equipment or other capital, but does lease its facilities. While the Company is continuing to assess all potential impacts of this standard, it expects that most of its lease commitments will be subject to the updated standard and recognized as lease liabilities and right-of-use assets upon adoption.
During March 2016, the FASB issued ASU No. 2016-09, Improvements to Employee Share-based Payment Accounting. This ASU simplifies the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. This ASU requires that excess tax benefits and deficiencies be recognized as income tax benefit or expense in the income statement. The Company currently plans to implement this ASU as required in the first quarter of fiscal year 2017. The Company does not expect the adoption of this ASU to have a significant impact on its financial position.
61
During June 2016, the FASB issued ASU 2016-13, Financial Instruments — Measurement of Credit Losses on Financial Instruments, which requires measurement and recognition of expected credit losses for financial assets held. ASU 2016-13 is effective for the Company beginning January 1, 2020. The Company is currently evaluating the impact of adopting this standard.
During January 2017, the FASB issued ASU 2017-04, Intangibles — Goodwill and Other (Topic 350): Simplifying the Accounting for Goodwill Impairment, which simplifies the accounting for goodwill impairment. This ASU removes Step 2 of the goodwill impairment test, which requires hypothetical purchase price allocation. A goodwill impairment will now be the amount by which a reporting unit’s carrying value exceeds its fair value, not to exceed the carrying amount of goodwill. The new guidance also requires disclosure of the amount of goodwill at reporting units with zero or negative carrying amounts. ASU 2017-04 is effective for the Company beginning January 1, 2020. Early adoption is permitted for any impairment tests performed after January 1, 2017. The Company is currently evaluating the impact of adopting this standard.
3. Fair Value of Financial Instruments
The authoritative guidance with respect to fair value established a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels, and requires that assets and liabilities carried at fair value be classified and disclosed in one of three categories, as presented below.
Level 1 - Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2 - Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange based derivatives, mutual funds, and fair-value hedges.
Level 3 - Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently-traded, non-exchange-based derivatives and commingled investment funds, and are measured using present value pricing models.
The Company determines the level in the fair value hierarchy within which each fair value measurement falls in its entirety, based on the lowest level input that is significant to the fair value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
The Company classifies its investments in money market funds within Level 1 of the fair value hierarchy because they are valued using bank balances or quoted market prices. The Company classifies its Level 2 instruments based on market pricing and other observable inputs. The Company did not classify any of its investments within Level 3 of the fair value hierarchy.
The following table sets forth the fair value of the Company’s financial assets measured on a recurring basis as of December 31, 2016 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2016 |
||||||||||
|
Assets |
|
Classification |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
Money market funds |
|
Cash and cash equivalents |
|
$ |
1,726 |
|
$ |
— |
|
$ |
— |
|
$ |
1,726 |
|
Corporate bonds |
|
Investments |
|
|
— |
|
|
13,289 |
|
|
— |
|
|
13,289 |
|
Asset-backed security |
|
Investments |
|
|
— |
|
|
1,017 |
|
|
— |
|
|
1,017 |
|
Total assets measured at fair value |
|
|
|
$ |
1,726 |
|
$ |
14,306 |
|
$ |
— |
|
$ |
16,032 |
The Company did not have any financial assets measured on a recurring basis as of December 31, 2015. The Company did not have any financial liabilities measured on a recurring basis as of December 31, 2016 and 2015.
During the year ended December 31, 2016, the Company did not record any impairment charges related to its marketable investments. There were no transfers between Level 1, Level 2 or Level 3 assets reported at fair value on a recurring basis and the valuation techniques used did not change compared to the Company’s established practice.
62
4. Equipment
Equipment consisted of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
Laboratory equipment |
|
$ |
425 |
|
$ |
356 |
|
Software |
|
|
20 |
|
|
10 |
|
Furniture, fixtures and equipment |
|
|
17 |
|
|
20 |
|
|
|
|
462 |
|
|
386 |
|
Less: Accumulated depreciation |
|
|
(145) |
|
|
(57) |
|
|
|
$ |
317 |
|
$ |
329 |
Depreciation expense for the years ended December 31, 2016, 2015 and the period from May 19, 2014 (inception) to December 31, 2014 was $94,000, $51,000 and $6,000, respectively.
5. Intangible Assets
Intangible assets consisted of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
Acquired patents and licenses |
|
$ |
7,985 |
|
$ |
7,985 |
|
Less: Accumulated amortization |
|
|
(1,442) |
|
|
(777) |
|
|
|
$ |
6,543 |
|
$ |
7,208 |
A schedule of the amortization of intangible assets for the five years ending December 31, 2017 through 2021 and thereafter is as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
Year Ending December 31: |
|
|
|
|
2017 |
|
$ |
665 |
|
2018 |
|
|
666 |
|
2019 |
|
|
665 |
|
2020 |
|
|
666 |
|
2021 |
|
|
665 |
|
Thereafter |
|
|
3,216 |
|
|
|
$ |
6,543 |
6. Accrued Expenses
Accrued expenses consisted of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
Professional fees |
|
$ |
285 |
|
$ |
84 |
|
Compensation expense |
|
|
261 |
|
|
34 |
|
Other |
|
|
205 |
|
|
40 |
|
Offering costs |
|
|
— |
|
|
240 |
|
|
|
$ |
751 |
|
$ |
398 |
63
7. Stockholders’ Equity and Stock-Based Compensation
Preferred Stock
The Company has authorized a total of 5,000,000 shares of preferred stock, par value $0.001 per share, none of which were outstanding at December 31, 2016 and 2015. The Company’s Board of Directors has the authority to issue preferred stock and to determine the rights, preferences, privileges, and restrictions, including voting rights, without any further vote or action by the Company’s stockholders.
Common Stock
The Company has authorized a total of 45,000,000 shares of common stock, par value $0.001 per share.
In conjunction with the incorporation of the Company, 1,125,000 shares of common stock were issued to its founding stockholders, which consisted of MDB Capital Group, LLC and its affiliated persons, for cash consideration of $8,000.
During November 2014, the Company sold 2,996,253 shares of common stock in a private placement to accredited investors for $2.67 per share, resulting in gross cash proceeds of $8.0 million. Direct costs of the private placement consisted of a 10% placement agent fee to MDB Capital Group, LLC and its designees, of $0.8 million and related legal fees and reimbursable expenses of $54,000. In conjunction with this private placement, the Company issued warrants to MDB Capital Group, LLC and its designees for a consideration of $1,000. The placement agent warrants had a fair value of $0.6 million, calculated pursuant to the Black-Scholes option-pricing model, as described below. Issuance costs of the private placement, net of the consideration received from MDB Capital Group, LLC of $1,000, aggregated $0.9 million and were charged directly to additional paid-in capital.
During November 2014, the Company issued an aggregate of 3,444,198 shares of common stock valued at $9.2 million to acquire businesses and intellectual property license rights.
During May 2016, the Company completed an IPO whereby it sold 5,749,846 shares of the Company’s common stock at $4.00 as described in Note 1.
Common Stock Warrants
During the year ended December 31, 2016, in connection with the closing of the Company’s initial public offering, the Company issued warrants as compensation to the underwriters of its initial public offering to purchase a total 574,985 shares of is common stock at a price of $5.00 per share. The warrants became exercisable 180 days after issuance and are exercisable for a period of five years. These warrants were valued pursuant to the Black-Scholes option-pricing model at $1.4 million, based on the following assumptions: fair value of common stock – $4.12 to $4.27 per share; risk free interest rate: 1.22% – 1.38%; expected volatility – 80%; expected dividend yield – 0%; and expected term – 5 years.
In conjunction with the private placement of common stock during November 2014 at $2.67 per share, the Company issued warrants to the placement agent to purchase 299,625 shares of common stock, exercisable at issuance for a period of seven years at $2.67 per share. The placement agent warrants have standard anti-dilution protections and cashless exercise rights. The placement agent warrants have piggy-back registration rights commencing on the date that the Company became a reporting company under the Securities Exchange Act of 1934, as amended, and for a period of seven years thereafter, and a one-time demand registration right commencing six months after the date that the Company became a reporting company under the Securities Exchange Act of 1934, as amended, and for a period of 54 months thereafter.
64
A summary of warrant activity for the year ended December 31, 2016 is presented below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
Weighted |
|
Remaining |
|
|
|
|
Number of |
|
Average |
|
Contractual |
|
|
|
|
Shares |
|
Exercise Price |
|
Life (in Years) |
|
|
Warrants outstanding at December 31, 2015 |
|
299,625 |
|
$ |
2.67 |
|
5.85 |
|
Issued |
|
574,985 |
|
|
5.00 |
|
|
|
Exercised |
|
— |
|
|
— |
|
|
|
Expired/terminated |
|
— |
|
|
— |
|
|
|
Warrants outstanding and exercisable at December 31, 2016 |
|
874,610 |
|
$ |
4.20 |
|
4.53 |
The intrinsic value of exercisable in-the-money stock warrants was approximately $2.0 million as of December 31, 2016.
Stock Options
During August 2015, the Company adopted the 2015 Stock Incentive Plan (the “2015 Plan”) and reserved 1,134,818 shares of common stock for issuance under the 2015 Plan, including stock options and restricted or performance stock awards. The 2015 Plan is administered by the Compensation Committee of the Company’s Board of Directors. Eligible participants in the 2015 Plan include the Company’s employees, officers and directors, and any person who has a business relationship with the Company. Options issued under the 2015 Plan may have a term of up to ten years and may have variable vesting provisions. New hire grants generally vest 25% upon the first anniversary of the grant and 1/12 quarterly thereafter, over the subsequent twelve quarters. Grants issued to existing employees generally vest quarterly over four years.
During the year ended December 31, 2015, prior to the adoption of the 2015 Plan, all of the stock options granted to directors were at an exercise price of $2.67 per share, the same price as the common shares sold in the November 2014 private placement. Pursuant to an employment agreement with Darrin Uecker, the Company’s President and Chief Executive Officer (the “Executive”), the Company agreed to issue to the Executive, upon consummation of the proposed IPO, an additional stock option equal to 3% of the Company’s fully diluted shares of common stock outstanding after taking into account the shares to be issued in the IPO exercisable at the IPO per share price. On October 5, 2016, the Executive offered, and the Company accepted, to forgo receipt of such grant until such time the Company’s shareholders approve a new equity incentive plan or an increase in the number of shares available under the existing plan. In exchange for the Executive’s forgoing receipt of the post-IPO option grant, the Company agreed to amend the Executive’s employment agreement (the “Amendment”) so that the Executive will receive (i) an option grant to purchase 187,286 shares of the Company’s common stock, which is a number of shares equal to the number of shares he would have been entitled to receive upon completion of the IPO, and (ii) a restricted stock grant equal in value to (A) the difference between the exercise price previously agreed to for the post-IPO option grant, which was $4.00 per share, and the exercise price on the date of the deferral grant, multiplied by (B) 187,286. In the event of a change of control that precedes the aforementioned option grant while the Executive is still an employee of the Company, the Executive would be entitled to receive a cash bonus equal to the consideration the Executive would have received as a holder of 187,286 vested options to purchase the Company’s common stock at an exercise price of $4.00 per share.
A summary of stock option activity for the year ended December 31, 2016 is presented below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
Weighted |
|
Average |
|
|
|
|
|
|
Average |
|
Remaining |
|
|
|
|
Number of |
|
Exercise |
|
Contractual |
|
|
|
|
Shares |
|
Price |
|
Life (in Years) |
|
|
Stock options outstanding at December 31, 2015 |
|
875,221 |
|
$ |
3.51 |
|
7.8 |
|
Issued |
|
405,132 |
|
|
4.44 |
|
|
|
Exercised |
|
— |
|
|
— |
|
|
|
Expired/terminated |
|
(50,998) |
|
|
3.57 |
|
|
|
Stock options outstanding at December 31, 2016 |
|
1,229,355 |
|
$ |
3.82 |
|
|
|
Vested and expected to vest at December 31, 2016 |
|
1,229,355 |
|
$ |
3.82 |
|
7.6 |
|
Stock options exercisable at December 31, 2016 |
|
354,855 |
|
$ |
3.34 |
|
6.0 |
65
The exercise prices of stock options outstanding and exercisable are as follows at December 31, 2016:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding |
|
Options Exercisable |
||||
|
|
|
Weighted average |
|
|
|
|
|
|
Number |
remaining contractual |
Weighted average |
|
Number |
Weighted average |
|
Exercise Price |
outstanding |
life (in years) |
exercise price |
|
vested |
exercise price |
|
$2.67 |
302,620 |
3.3 |
$ 2.67 |
|
176,528 |
$ 2.67 |
|
$4.00 - $4.28 |
692,335 |
8.9 |
4.02 |
|
178,327 | 4.00 |
|
$4.67 - $4.68 |
234,400 |
9.6 |
4.68 |
|
— |
- |
|
|
1,229,355 |
7.6 |
$ 3.82 |
|
354,855 |
$ 3.34 |
The intrinsic value of exercisable in-the-money stock options at December 31, 2016 was approximately $1.1 million.
The fair value of option awards was estimated using the Black-Scholes option-pricing model utilizing the following assumptions:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Expected term in years |
|
|
6.08 |
|
|
3.5 - 6.25 |
|
|
— |
|
Expected volatility |
|
|
80% |
|
|
89% - 90% |
|
|
— |
|
Risk-free interest rate |
|
|
1.16%-1.45% |
|
|
0.88% - 1.89% |
|
|
— |
|
Dividend yield |
|
|
— |
|
|
— |
|
|
— |
The fair value of the stock options granted during the years ended December 31, 2016 and 2015, calculated pursuant to the Black-Scholes option-pricing model, was $1.2 million and $3.1 million, respectively.
Total stock-based compensation expense consisted of the following (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
General and administrative |
|
$ |
674 |
|
$ |
397 |
|
$ |
— |
|
Research and development |
|
|
196 |
|
|
5 |
|
|
— |
|
Total stock-based compensation expense |
|
$ |
870 |
|
$ |
402 |
|
$ |
— |
At December 31, 2016, there was $2.4 million of unrecognized compensation cost related to unvested stock-based compensation arrangements, which is expected to be recognized over a weighted average period of 2.84 years.
8. Research Grants and Agreements
Research Grants
The Company’s subsidiary, BEM, was acquired by Pulse in November 2014. BEM had received grants from the National Cancer Institute of the National Institutes of Health (the “NIH”), including grants from the NIH Small Business Innovation Research (“SBIR”) Program, to conduct research and develop devices that will provide health benefits utilizing bioelectric technology. At the time of the acquisition, BEM had an active research grant under the SBIR Program for a project entitled “EndoPulse System for Endoscopic Ultrasound-Guided Therapy of Pancreatic Carcinoma.” This research project was completed during the year ended December 31, 2015. During the period from May 19, 2014 (inception) through December 31, 2014 and for the year ended December 31, 2015, the Company received research grant funding of $0.2 million and $0.3 million, respectively. The Company has not subsequently pursued additional grants.
Sponsored Research Agreement
The Company sponsors research activities performed by ODURF’s Frank Reidy Center. ODURF is compensated by the Company for its conduct of each study in accordance with the budget and payment terms set forth in the applicable task
66
order. During the years ended December 31, 2016 and 2015, the Company agreed to sponsor $1.0 million and $1.2 million, respectively, during the subsequent 12-month period. The principal investigator may transfer funds with the budget as needed without the Company’s approval so long as the obligations of ODURF under the task order and statement of work remain unchanged and unimpaired. During the year ended December 31, 2016 and 2015, the Company incurred $0.9 million and $1.0 million, respectively, of costs pursuant to various task orders.
9. Income Taxes
The income tax provision for the year ended December 31, 2016 was $0. The income tax provision for the year ended December 31, 2015 and the period from May 19, 2014 (inception) through December 31, 2014 was a benefit of $1.7 million and $23,000, respectively. The prior years’ tax benefits resulted from the realization of deferred tax assets related principally to the Company’s net operating loss for the year ended December 31, 2015, offset by deferred tax liability created based upon the difference in the value for book and tax purposes of certain acquired technology assets, which are considered temporary income tax differences under purchase accounting. A full valuation allowance is provided against the Company’s remaining deferred tax assets.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets at December 31, 2016 and 2015 are summarized below (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
Technology |
|
$ |
(1,449) |
|
$ |
(1,630) |
|
Temporary differences |
|
|
312 |
|
|
103 |
|
Credits |
|
|
639 |
|
|
198 |
|
Net operating loss carryforwards |
|
|
5,121 |
|
|
1,590 |
|
Total deferred tax assets |
|
|
4,623 |
|
|
261 |
|
Valuation allowance |
|
|
(4,623) |
|
|
(261) |
|
Net deferred tax assets |
|
$ |
— |
|
$ |
— |
In assessing the potential realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the Company attaining future taxable income during the periods in which those temporary differences become deductible. At December 31, 2016 and 2015, management was unable to determine that it was more likely than not that the Company’s deferred tax assets will be realized, and has therefore recorded an appropriate valuation allowance against deferred tax assets at such dates.
The Company’s effective tax rate is different from the federal statutory tax rate of 35% due primarily to net losses that receive no tax benefit as a result of a valuation allowance recorded for such losses.
Presented below is the reconcilement of the difference between the tax rate computed by applying the U.S. federal statutory tax rate and the effective tax rate for the years ended December 31, 2016, 2015 and for the period from May 19, 2014 through December 31, 2014:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
May 19, 2014 |
|
|
|
|
|
|
|
|
|
|
(inception) |
|
|
|
|
Years Ended |
|
through |
|||||
|
|
|
December 31, |
|
December 31, |
|||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
U.S. federal statutory tax rate |
|
(35.0) |
% |
|
(35.0) |
% |
|
(35.0) |
% |
|
Valuation allowance |
|
42.0 |
% |
|
4.0 |
% |
|
16.0 |
% |
|
Permanent differences |
|
2.0 |
% |
|
1.0 |
% |
|
15.0 |
% |
|
State tax benefit and other |
|
(9.0) |
% |
|
(7.0) |
% |
|
(4.0) |
% |
|
Effective tax rate |
|
— |
% |
|
(37.0) |
% |
|
(8.0) |
% |
At December 31, 2016, the Company had federal and California state net operating loss carryforwards of approximately $11.7 million and $11.5 million, respectively. The federal and state net operating loss carryforwards will begin to expire after 2032. At December 31, 2016, the Company had approximately $0.4 million and $0.4 million of federal and
67
California R&D credits, respectively. The federal R&D credits begin to expire after 2035 and the California R&D credits have an indefinite carryforward period.
These net operating loss carryforward and research and development credit amounts have full valuation allowances against them due to the remoteness of their expected utilization. While the Company has not performed a formal analysis of the availability of these operating loss carryforwards at December 31, 2016 under Internal Revenue Code Sections 382 and 383, management expects that the Company’s ability to use its net operating loss carryforwards may be limited in future periods.
The Company’s activity related to unrecognized tax benefits are summarized below (in thousands):
|
|
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
|
2016 |
|
2015 |
||
|
Balance, January 1, 2016 |
|
$ |
66 |
|
$ |
— |
|
Gross increases - tax positions in prior periods |
|
|
— |
|
|
— |
|
Gross decreases - tax positions in prior periods |
|
|
— |
|
|
— |
|
Gross increases - tax position in current period |
|
|
147 |
|
|
66 |
|
Settlements |
|
|
— |
|
|
— |
|
Lapses in statutes of limitations |
|
|
— |
|
|
— |
|
Balance, December 31, 2016 |
|
$ |
213 |
|
$ |
66 |
|
|
|
|
|
|
|
|
Although it is reasonably possible that certain unrecognized tax benefits may increase or decrease within the next twelve months due to tax examination changes, settlement activities, expirations of statute of limitations, or the impact on recognition and measurement considerations related to the results of published tax cases or other similar activities, the Company does not anticipate any significant changes to unrecognized tax benefits over the next twelve months. During the years ended December 31, 2016, 2015 and the period from May 19, 2014 to December 31, 2014, no interest or penalties were required to be recognized related to unrecognized tax benefits. Although the Company is not under examination, the tax years for 2014 and forward are subject to examination by United States tax authorities.
10. Related Party Transactions
MDB Capital Group, LLC (“MDB”) provided investment banking, executive recruiting and intellectual property management services to the Company. Furthermore, provisions of the Company’s investment banking agreement with MDB survive the June 2016 termination of the agreement for a period of two years and MDB retains significant rights under the 2016 IPO underwriting agreement with respect to the Company’s future financing alternatives for up to five years. The Company’s Chairman, Robert Levande, is a Senior Managing Director of MDB.
In connection with the Company’s 2016 IPO (Note 1), the underwriting syndicate led by MDB received $1.8 million in underwriting discounts, $0.2 million in unaccountable expense reimbursements and warrants valued in the aggregate of $1.4 million.
In addition, during the period from May 19, 2014 (inception) to December 31, 2014 (Note 7), MDB received cash placement agent fees of $0.8 million and the Company issued warrants to purchase 299,625 shares of common stock for a consideration of $1,000, exercisable for seven years at $2.67 per share, to MDB Capital Group, LLC and its designees.
During the year ended December 31, 2016, the Company incurred non-financing related expenses charged by MDB of $100,000 for services rendered with respect to intellectual property related services. Similarly, during the year ended December 31, 2015, the Company incurred expenses charged by MDB comprised of: $49,000 for services rendered with respect to executive search activities related to the hiring of the Company’s Chief Executive Officer and the appointment of one director, $42,000 for offering related expenses and $26,000 for intellectual property related services.
Gary Schuman, the Chief Financial Officer of MDB, was also the acting Chief Financial Officer of the Company and was compensated at a monthly rate of $4,000 from November 1, 2014 to December 31, 2015, reflecting an aggregate charge to general and administrative expenses of $48,000 and $8,000 for the year ended December 31, 2015 and the period from May 19, 2014 (inception) through December 31, 2014, respectively.
At December 31, 2016, accounts payable and accrued expenses did not include any amounts payable to MDB. At December 31, 2015, included in accounts payable and accrued expenses is an amount of $58,000 payable to MDB for their
68
expenses incurred relating to the Company’s planned IPO, which were recorded as deferred offering costs, and patent related services.
11. Commitments and Contingencies
Operating Leases
The Company leases its corporate offices and research facilities in Burlingame, California, under an operating lease expiring March 31, 2017 at a monthly cost of approximately $19,000.
In January 2017, the Company entered into a new lease agreement for premises consisting of approximately fifteen thousand six hundred ninety-seven (15,697) rentable square feet located in Hayward, California. See Note 14.
During the years ended December 31, 2016, 2015 and the period from May 19, 2014 (inception) through December 31, 2014, rent expense, including common area maintenance charges, was $0.2 million, $0.2 million and $19,000, respectively.
12. Employee Benefit Plans
The Company sponsors a defined contribution plan under which it may make discretionary contributions. The Company did not make any employer matching contributions to this plan during the years ended December 31, 2016, 2015 or the period from May 19, 2014 (inception) through December 31, 2014.
13. Selected Quarterly Financial Data (Unaudited)
The following table provides the selected quarterly financial data for the years ended December 31, 2016 and 2015 (in thousands, except per share data):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended |
||||||||||||||||||||||
|
|
|
2016 |
|
2015 |
||||||||||||||||||||
|
|
|
December 31, |
|
September 30, |
|
June 30, |
|
March 31, |
|
December 31, |
|
September 30, |
|
June 30, |
|
March 31, |
||||||||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
870 |
|
|
893 |
|
|
642 |
|
|
528 |
|
|
417 |
|
|
408 |
|
|
243 |
|
|
156 |
|
Research and development |
|
|
1,806 |
|
|
1,739 |
|
|
1,453 |
|
|
990 |
|
|
951 |
|
|
659 |
|
|
547 |
|
|
421 |
|
Amortization of intangible assets |
|
|
167 |
|
|
166 |
|
|
166 |
|
|
166 |
|
|
167 |
|
|
167 |
|
|
166 |
|
|
166 |
|
Total operating expenses |
|
|
2,843 |
|
|
2,798 |
|
|
2,261 |
|
|
1,684 |
|
|
1,535 |
|
|
1,234 |
|
|
956 |
|
|
743 |
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
34 |
|
|
31 |
|
|
3 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Total other income |
|
|
34 |
|
|
31 |
|
|
3 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Loss from operations, before income taxes |
|
|
(2,809) |
|
|
(2,767) |
|
|
(2,258) |
|
|
(1,684) |
|
|
(1,535) |
|
|
(1,234) |
|
|
(956) |
|
|
(743) |
|
Income tax benefit |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(518) |
|
|
(483) |
|
|
(357) |
|
|
(299) |
|
Net loss |
|
|
(2,809) |
|
|
(2,767) |
|
|
(2,258) |
|
|
(1,684) |
|
|
(1,017) |
|
|
(751) |
|
|
(599) |
|
|
(444) |
|
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unrealized gain (loss) on available-for-sale securities, net of tax: |
|
|
1 |
|
|
(8) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Comprehensive loss |
|
$ |
(2,808) |
|
$ |
(2,775) |
|
$ |
(2,258) |
|
$ |
(1,684) |
|
$ |
(1,017) |
|
$ |
(751) |
|
$ |
(599) |
|
$ |
(444) |
|
Net loss per share |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share |
|
$ |
(0.21) |
|
$ |
(0.21) |
|
$ |
(0.23) |
|
$ |
(0.22) |
|
$ |
(0.13) |
|
$ |
(0.10) |
|
$ |
(0.08) |
|
$ |
(0.06) |
|
Weighted average shares used to compute net loss per common share — basic and diluted |
|
|
13,315 |
|
|
13,315 |
|
|
9,791 |
|
|
7,565 |
|
|
7,565 |
|
|
7,565 |
|
|
7,565 |
|
|
7,565 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
69
14. Subsequent Events
Lease Agreement
In January 2017, the Company entered into a new lease agreement (the “Lease”) for premises consisting of approximately fifteen thousand six hundred ninety-seven (15,697) rentable square feet located in Hayward, California (the “Premises”).
The Premises will be used for the Company’s corporate headquarters and principal operating facility. The term of the Lease is sixty-two (62) months, commencing on the date that is the earlier of (i) the date upon which the Company commences business in the Premises or (ii) the date upon which the Premises is “Ready for Occupancy” as defined in the Lease. Base monthly rent shall be abated for the first two (2) months of the Lease term and thereafter will be $42,400 per month during the first year of the Lease term, with specified annual increases thereafter until reaching approximately $50,300 per month during the last two (2) months of the Lease term. The Company is required to pay a refundable security deposit of approximately $101,000. The Landlord is obligated to provide the Company with improvement allowances in the amount of approximately $135.00 per rentable square foot of the Premises, which may be applied towards the costs of construction of the initial improvements in the Premises. The Company will be responsible for any such improvement costs in excess of the foregoing allowances. The Company is required to reimburse Landlord for certain expenses during the Lease term.
The Company has the right to extend the Lease term by five (5) years upon written notice not more than twelve (12) months nor less than nine (9) months prior to the expiration of the original Lease term, with monthly payments equal to the “Fair Rental Value” as defined in the Lease. The Company has also reserved the right to terminate the Lease if the Landlord is unable to deliver the facility to the Company by December 1, 2017. Assuming the Landlord delivers the facility to us by December 1, 2017, as of December 31, 2016, the lease obligations for less than one year, one to three years, three to five years and more than five years is approximately $0.3 million, $1.1 million, $1.1 million and $0.3 million, respectively.
Private Placement
On February 7, 2017, the Company entered into a securities purchase agreement (the “Purchase Agreement”) with Robert W. Duggan and Maky Zanganeh (the “Investors”), pursuant to which the Company, in a private placement, agreed to issue and sell to the Investors an aggregate of 819,673 shares of the Company’s common stock, par value $0.001 per share (the “Common Stock”), at a price per share of $6.10 (the “Shares”), for gross proceeds of approximately $5 million (the “Private Placement”).
In connection with the Private Placement, the Company has granted certain registration rights to the Investors, pursuant to which, among other things, the Company will prepare and file with the Securities and Exchange Commission a registration statement to register for resale the Shares on or prior to July 31, 2017.
70
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management, including our Chief Executive Officer and our Chief Financial Officer, conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act of 1934, as amended, as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures were not effective at the reasonable level of assurance due to a material weakness in internal control over financial reporting.
In connection with the audit of our financial statements as of and for the year ended December 31, 2016, we identified a material weakness in our internal control over financial reporting. The material weakness related to a lack of effective controls to adequately restrict access and segregate duties. Specifically, due to the limited number of staff in our accounting function, certain personnel had the ability to prepare and post journal entries without a qualified independent review performed by someone without this ability. Upon identifying this material weakness, we performed additional procedures to evaluate the impact on the financial statements. Based on these procedures, we believe the material weakness did not result in any material misstatements to our financial statements. However, this material weakness could result in a misstatement of our accounts or disclosures that would result in a material misstatement of our financial statements that would not be prevented or detected. We are implementing measures designed to improve our internal control over financial reporting to remediate this material weakness, including the following:
|
· |
We are evaluating our accounting system access rights so that there are accounting personnel without journal entry access who can perform review activities. |
|
· |
We are formalizing our internal control documentation and strengthening supervisory reviews by our management. |
|
· |
We are in the process of adding additional accounting personnel and will be segregating duties amongst accounting personnel. |
We cannot assure you that the measures we have taken to date, and are continuing to implement, will be sufficient to remediate the material weakness we have identified or avoid potential future material weaknesses. If the steps we take do not correct the material weakness in a timely manner, we will be unable to conclude that we maintain effective internal control over financial reporting. Accordingly, there could continue to be a reasonable possibility that a material misstatement of our financial statements would not be prevented or detected on a timely basis.
Management’s Annual Report on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management's assessment regarding internal control over financial reporting or an attestation report from our independent registered public accounting firm due to a transition period established by rules of the SEC for newly public companies.
Changes in Internal Control over Financial Reporting
There were no changes in our internal controls over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the quarter ended December 31, 2016 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
71
Inherent Limitations on Effectiveness of Controls
Our management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
None.
72
Part III
Item 10. Directors, Executive Officers and Corporate Governance
Information responsive to this item is incorporated herein by reference to our definitive proxy statement with respect to our 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 11. Executive Compensation
Information responsive to this item is incorporated herein by reference to our definitive proxy statement with respect to our 2017 Annual Meeting of Stockholder to be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information responsive to this item is incorporated herein by reference to our definitive proxy statement with respect to our 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information responsive to this item is incorporated herein by reference to our definitive proxy statement with respect to our 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 14. Principal Accounting Fees and Services
Information responsive to this item is incorporated herein by reference to our definitive proxy statement with respect to our 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
73
Part IV
Item 15. Exhibits, Financial Statement Schedules
(a) The following documents are filed as part of, or incorporated by reference into, this Annual Report on Form 10-K:
1. Financial Statements: See Item 8 of this Annual Report on Form 10-K.
2. Financial Statement Schedules: All schedules are omitted because they are not required, are not applicable or the information is included in the consolidated financial statements or notes thereto.
3. Exhibits: We have filed, or incorporated by reference into this Annual Report on Form 10-K, the exhibits listed on the accompanying Exhibit Index immediately following the signature page of this Annual Report on Form 10-K.
(b) Exhibits: See Item 15(a)(3) above.
(c) Financial Statement Schedules: See Item 15(a)(2) above.
74
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
|
|
|
|
|
|
|
PULSE BIOSCIENCES, INC. |
|
||
|
|
|
|
|
|
|
|
Date: March 20, 2017 |
|
By: |
/s/ BRIAN B. DOW |
|
|
|
|
|
|
|
Brian B. Dow |
|
|
|
|
|
|
Chief Financial Officer, Senior Vice President of Finance and Administration, Secretary and Treasurer (Principal Financial and Principal Accounting Officer) |
|
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Darrin R. Uecker and Brian B. Dow, jointly and severally, as his true and lawful attorney-in-fact and agent, with full power of substitution, each with power to act alone, to sign and execute on behalf of the undersigned any and all amendments to this Annual Report on Form 10-K, and to perform any acts necessary in order to file the same, with all exhibits thereto and other documents in connection therewith with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requested and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or their or his or her substitutes, shall do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
|
/s/ Darrin R. Uecker
Darrin R. Uecker |
President, Chief Executive Officer and Director (Principal Executive Officer) |
March 20, 2017 |
|
/s/ Brian B. Dow
Brian B. Dow |
Chief Financial Officer, Senior Vice President of Finance and Administration, Secretary and Treasurer (Principal Financial and Principal Accounting Officer) |
March 20, 2017 |
|
/s/ Robert M. Levande Robert M. Levande |
Director |
March 20, 2017 |
|
|
|
|
|
/s/ Robert Greenberg, M.D.
Robert Greenberg, M.D. |
Director |
March 20, 2017 |
|
/s/ Mitchell Levinson
Mitchell Levinson |
Director |
March 20, 2017 |
|
/s/ Maky Zanganeh
Maky Zanganeh |
Director |
March 20, 2017 |
75
|
Exhibit |
Incorporation by Reference |
|||||||||
|
Number |
Exhibit Description |
Form |
File No. |
Exhibit(s) |
Filing Date |
|||||
|
1.1 |
Form of Underwriting Agreement |
S-1 |
333-208694 |
1.1 |
May 3, 2016 |
|||||
|
3.1 |
Articles of Incorporation of the Registrant, as amended on December 8, 2015 |
S-1 |
333-208694 |
3.1 |
December 22, 2015 |
|||||
|
3.2 |
Amended and Restated Bylaws of Pulse Biosciences, Inc. |
8-K |
001-37744 |
3.1 |
February 10, 2017 |
|||||
|
4.1 |
Specimen Certificate representing shares of common stock of Registrant |
S-1 |
333-208694 |
4.1 |
March 7, 2016 |
|||||
|
4.2 |
Form of Warrant dated November 9, 2014 issued to MDB Capital Group, LLC |
S-1 |
333-208694 |
4.2 |
December 22, 2015 |
|||||
|
4.3 |
Form of Underwriters' Warrant |
S-1 |
333-208694 |
4.3 |
March 28, 2016 |
|||||
|
10.1* |
Lease for facilities at 3955 Point Eden Way, Hayward, California, dated January 26, 2017 |
|||||||||
|
10.2 |
Eighth Amendment to Lease Agreement - ARE-819/863 Mitten Road, LLC and the Registrant |
10-Q |
001-37744 |
10.1 |
August 10, 2016 |
|||||
|
10.3# |
License Agreement among Old Dominion University Research Foundation, Eastern Virginia Medical School and the Registrant |
S-1 |
333-208694 |
10.12 |
May 3, 2016 |
|||||
|
10.4 |
Amendments No. 1 to License Agreement among Old Dominion University Research Foundation, Eastern Virgina Medical School and the Registrant |
S-1 |
333-208694 |
10.13 |
March 7, 2016 |
|||||
|
10.5# |
License Agreement among Univeristy of Southern California, The Alfred Mann Institute and the Registrant |
S-1 |
333-208694 |
10.14 |
May 3, 2016 |
|||||
|
10.6# |
Amendment No. 1 to the License Agreement among University of Southern California, The Alfred Mann Institute and the Registrant |
S-1 |
333-208694 |
10.15 |
May 3, 2016 |
|||||
|
10.7 |
Securities Purchase Agreement, dated February 7, 2017, by and between Pulse Biosciences, Inc. and certain purchasers |
8-K |
001-37744 |
10.1 |
February 10, 2017 |
|||||
|
10.8 |
2015 Stock Incentive Plan |
S-1 |
333-208694 |
10.2 |
December 22, 2015 |
|||||
|
10.9 |
Form of Director Option Agreement, not issued under the 2015 Stock Incentive Plan |
S-1 |
333-208694 |
10.3 |
December 22, 2015 |
|||||
|
10.10+ |
Employment Agreement between Dr. Richard Nuccitelli and the Registrant |
S-1 |
333-208694 |
10.8 |
December 22, 2015 |
|||||
|
10.11+ |
Executive Employment Agreement between Darrin R. Uecker and the Registrant |
S-1 |
333-208694 |
10.9 |
December 22, 2015 |
|||||
|
10.12+ |
Amendment to Employment Agreement between Darrin R. Uecker and Pulse Biosciences, Inc. dated October 5, 2016 |
8-K |
001-37744 |
10.10 |
October 11, 2016 |
|||||
|
10.13+ |
Executive Employment Agreement between Brian B. Dow and the Registrant |
S-1 |
333-208694 |
10.14 |
December 22, 2015 |
|||||
|
10.14+ |
Form of At-Will Employment, Confidential Information, Invention Assignment, and Arbitration Agreement for Employees |
S-1 |
333-208694 |
10.10 |
December 22, 2015 |
|||||
|
10.15 |
Engagement Agreement dated September 15, 2014 between MDB Capital Group LLC and the Registrant |
S-1 |
333-208694 |
10.4 |
December 22, 2015 |
|||||
|
10.16 |
Form of Securities Purchase Agreement dated November 6, 2014, among the purchasers of common stock and the Registrant |
S-1 |
333-208694 |
10.5 |
December 22, 2015 |
|||||
|
10.17 |
Form of Registration Rights Agreement dated November 6, 2014, among the purchasers of common stock and the Registrant |
S-1 |
333-208694 |
10.6 |
December 22, 2015 |
|||||
|
10.18 |
Form of Registration Rights Agreement dated November 6, 2014, among the holders of placement warrants and the Registrant |
S-1 |
333-208694 |
10.7 |
December 22, 2015 |
|||||
|
21.1* |
List of Subsidiaries |
|||||||||
76
|
31.1 |
Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|||||||||
|
31.2 |
Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|||||||||
|
32.1* |
Certification of the Chief Executive and Chief Financial Officers pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (18 U.S.C. Section 1350). |
|||||||||
|
101.INS |
XBRL Instance Document |
|||||||||
|
101.SCH |
XBRL Taxonomy Extension Schema Document |
|||||||||
|
101.CAL |
XBRL Taxonomy Extension Calculation Linkbase Document |
|||||||||
|
101.DEF |
XBRL Taxonomy Extension Definition Linkbase Document |
|||||||||
|
101.LAB |
XBRL Taxonomy Extension Label Linkbase Document |
|||||||||
|
101.PRE |
XBRL Taxonomy Extension Presentation Linkbase Document |
|||||||||
|
|
||||||||||
|
|
* Filed herewith |
|||||||||
|
|
+ Indicates a management contract or compensatory plan or arrangement. |
|||||||||
|
|
# Portions of this exhibit (indicated by asterisks) have been omitted pusuant to a grant of confidential treatment. |
77