Attached files
UNITED STATES
SECURITIES
AND EXCHANGE COMMISSION
WASHINGTON, DC
20549
_________________
FORM 10-K
☒
ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934
For the
fiscal year ended: December 31,
2016
OR
☐
TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934
For the
transition period from _________________ to
______________________
Commission
file number: 001-34673
CORMEDIX INC.
(Exact
name of Registrant as Specified in Its Charter)
|
Delaware
|
|
20-5894890
|
|
(State
or Other Jurisdiction ofIncorporation or Organization)
|
|
(I.R.S.
EmployerIdentification No.)
|
|
1430 US
Highway 206, Suite 200, Bedminster, NJ
|
|
07921
|
|
(Address
of Principal Executive Offices)
|
|
(Zip
Code)
|
Registrant’s
telephone number, including area code: (908) 517-9500
Securities
registered pursuant to Section 12(b) of the Act:
|
Title
of each class
|
|
Name of
each exchange on which registered
|
|
Common
Stock, $0.001 Par Value
|
|
NYSE
MKT LLC
|
Securities
registered pursuant to Section 12(g) of the Act: none
Indicate
by check mark if the registrant is a well-known seasoned issuer, as
defined in Rule 405 of the Securities Act.
Yes ☐
No ☒
Indicate
by check mark if the registrant is not required to file reports
pursuant to Section 13 or Section 15(d) of the Act.
Yes
☐ No ☒
Indicate
by check mark whether the registrant: (1) has filed all reports
required to be filed by Section 13 or 15(d) of the Securities
Exchange Act of 1934 during the preceding 12 months (or for such
shorter period that the registrant was required to file such
reports), and (2) has been subject to such filing requirements for
the past 90 days.
Yes
☒ No ☐
Indicate
by check mark whether the registrant has submitted electronically
and posted on its corporate Web site, if any, every Interactive
Data File required to be submitted and posted pursuant to Rule 405
of Regulation S-T during the preceding 12 months (or for such
shorter period that the registrant was required to submit and post
such files).
Yes
☒ No ☐
Indicate
by check mark if disclosure of delinquent filers pursuant to Item
405 of Regulations S-K is not contained herein, and will not be
contained, to the best of the registrant’s knowledge, in
definitive proxy or information statements incorporated by
reference in Part III of this Form 10-K or any amendment to this
Form 10-K.
☐
Indicate
by check mark whether the registrant is a large accelerated filer,
an accelerated filer, a non-accelerated filer or a smaller
reporting company. See the definitions of “large accelerated
filer,” “accelerated filer” and “smaller
reporting company” in Rule 12b-2 of the Exchange Act. (Check
one):
|
Large
accelerated filer ☐
|
Accelerated
filer ☒
|
|
|
|
|
Non-accelerated
filer ☐
|
Smaller
reporting company ☐
|
Indicate
by check mark whether the registrant is a shell company (as defined
in Rule 12b-2 of the Act).
Yes
☐ No ☒
The
aggregate market value of the registrant’s voting and
non-voting common equity held by non-affiliates of the registrant,
based upon the closing price of the registrant’s common stock
on the last business day of the registrant’s most recently
completed second fiscal quarter was approximately $72.6 million.
Solely for the purpose of this calculation, shares held by
directors and executive officers of the registrant have been
excluded. Such exclusion should not be deemed a determination or an
admission by the registrant that such individuals are, in fact,
affiliates of the registrant.
The
number of outstanding shares of the registrant’s common stock
was 40,720,838 as of March 14, 2017.
DOCUMENTS INCORPORATED BY REFERENCE
Certain
portions of the registrant’s definitive Proxy Statement for
its 2017 Annual Meeting of Stockholders are incorporated herein by
reference, as indicated in Part III.
CORMEDIX INC.
|
PART I
|
|
|
|
Item 1.
|
Business.
|
1
|
|
Item 1A.
|
Risk Factors.
|
16
|
|
Item 1B.
|
Unresolved Staff Comments.
|
33
|
|
Item 2.
|
Properties.
|
33
|
|
Item 3.
|
Legal Proceedings.
|
33
|
|
Item 4.
|
Mine Safety Disclosures.
|
35
|
|
PART II
|
|
|
|
Item 5.
|
Market for the Registrant’s Common Equity, Related
Stockholder Matters and Issuer Purchases of Equity
Securities.
|
35
|
|
Item 6.
|
Selected Financial Data.
|
37
|
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition
and Results of Operations.
|
38
|
|
Item 7A.
|
Quantitative and Qualitative Disclosures About Market
Risk.
|
50
|
|
Item 8.
|
Financial Statements and Supplementary Data.
|
50
|
|
Item 9.
|
Changes in and Disagreements With Accountants on Accounting and
Financial Disclosure.
|
50
|
|
Item 9A.
|
Controls and Procedures.
|
50
|
|
Item 9B.
|
Other Information.
|
53
|
|
PART III
|
|
|
|
Item 10.
|
Directors and Executive Officers and Corporate
Governance.
|
53
|
|
Item 11.
|
Executive Compensation.
|
53
|
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters.
|
53
|
|
Item 13.
|
Certain Relationships and Related Transactions and Director
Independence.
|
53
|
|
Item 14.
|
Principal Accountant Fees and Services.
|
53
|
|
PART IV
|
|
|
|
Item 15.
|
|
54
|
Neutrolin®
is our registered trademark. All other trade names, trademarks and
service marks appearing in this report are the property of their
respective owners. We have assumed that the reader understands that
all such terms are source-indicating. Accordingly, such terms, when
first mentioned in this report, appear with the trade name,
trademark or service mark notice and then throughout the remainder
of this report without trade name, trademark or service mark
notices for convenience only and should not be construed as being
used in a descriptive or generic sense.
PART I
Forward-Looking Statements
This report contains
“forward-looking statements” that involve risks and
uncertainties, as well as assumptions that, if they never
materialize or prove incorrect, could cause our results to differ
materially from those expressed or implied by such forward-looking
statements. The statements contained in this report that are not
purely historical are forward-looking statements within the meaning
of Section 27A of the Securities Act of 1933, as amended (the
“Securities Act”), and Section 21E of the
Securities Exchange Act of 1934, as amended (the “Exchange
Act”). Forward-looking statements are often identified by the
use of words such as, but not limited to, “anticipate,”
“believe,” “can,” “continue,”
“could,” “estimate,” “expect,”
“intend,” “may,” “will,”
“plan,” “project,” “seek,”
“should,” “target,” “will,”
“would,” and similar expressions or variations intended
to identify forward-looking statements. These statements are based
on the beliefs and assumptions of our management based on
information currently available to management. Such forward-looking
statements are subject to risks, uncertainties and other important
factors that could cause actual results and the timing of certain
events to differ materially from future results expressed or
implied by such forward-looking statements. Factors that could
cause or contribute to such differences include, but are not
limited to, those identified below in the section titled
“Item 1A. Risk Factors.” Furthermore, such
forward-looking statements speak only as of the date of this
report. Except as required by law, we undertake no obligation to
update any forward-looking statements to reflect events or
circumstances after the date of such statements.
Overview
We are
a biopharmaceutical company focused on developing and
commercializing therapeutic products for the prevention and
treatment of infectious and inflammatory diseases.
Our
primary focus is on the development of our lead product candidate,
Neutrolin® (also known as CRMD003), for potential
commercialization in the United States (“U.S.”) and
other key markets. We have in-licensed the worldwide rights to
develop and commercialize Neutrolin®. Neutrolin is a novel
anti-infective solution (a formulation of taurolidine, citrate and
heparin 1000 u/ml) for the reduction and prevention of
catheter-related infections and thrombosis in patients requiring
central venous catheters in clinical settings such as dialysis,
critical/intensive care, and oncology. Infection and thrombosis
represent key complications among critical care / intensive care
and cancer patients with central venous catheters. These
complications can lead to treatment delays and increased costs to
the healthcare system when they occur due to hospitalizations, need
for IV antibiotic treatment, long-term anticoagulation therapy,
removal/replacement of the central venous catheter, related
treatment costs and increased mortality. We believe Neutrolin
addresses a significant unmet medical need and a potential large
market opportunity.
Neutrolin – United States
The
U.S. Food and Drug Administration, or FDA, has designated Neutrolin
as a Qualified Infectious Disease Product, or QIDP, for prevention
of catheter related blood stream infections in patients with end
stage renal disease receiving hemodialysis through a central venous
catheter. Catheter-related blood stream infections and clotting can
be life-threatening. The QIDP designation provides an additional
five years of market exclusivity in addition to the five years
granted for a New Chemical Entity. In addition, in January 2015,
the FDA granted Fast Track designation to Neutrolin Catheter Lock
Solution, pursuant to the Food and Drug Administration Safety
Innovation Act, or FDASIA, highlighting the large unmet need to
prevent infections in the U.S. healthcare system. The Fast Track
designation of Neutrolin provides us with the opportunity to meet
with the FDA on a more frequent basis during the development
process, and also ensures eligibility to request priority review of
the marketing application.
In late
2013, we met with the FDA to determine the pathway for U.S.
marketing approval of Neutrolin. Based on those discussions, we
determined to conduct two pivotal trials to demonstrate safety and
effectiveness of Neutrolin to secure marketing approval in the U.S.
We initiated a Phase 3 clinical trial in hemodialysis patients with
a central venous catheter in December 2015 and are currently
planning to initiate a Phase 3 trial in oncology patients with
catheters.
1
We launched the
Phase 3 clinical trial in hemodialysis catheters in the U.S. in
December 2015. The clinical trial, named Catheter Lock Solution
Investigational Trial, or LOCK-IT-100, is a prospective,
multicenter, randomized, double-blind, placebo-controlled, active
control trial which aims to demonstrate the efficacy and safety of
Neutrolin in preventing catheter-related bloodstream infections, or
CRBSI, in subjects receiving hemodialysis therapy as treatment for
end stage renal disease. The primary
endpoint for the trial is time to CRBSI. The trial will evaluate
whether Neutrolin is superior to the active control heparin by
documenting the incidence of CRBSI and the time until the
occurrence of CRBSI. Key secondary endpoints are catheter patency
which is defined as required use of tissue plasminogen activating
factor (tPA) or removal of catheter due to dysfunction or for any
reason. We now project to complete enrollment in the fourth
quarter of 2017, subject to funding requirements.
We are
in discussions with the FDA to develop the design of a Phase 3
clinical trial in oncology patients with catheters, or LOCK-IT-200.
This trial also is subject to funding requirements (see
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations - Liquidity and Capital
Resources - Funding and Capital Requirements” in Part II,
Item 7 of this report).
Neutrolin – International
In July
2013, we received CE Mark approval for Neutrolin. As a result, in
December 2013, we commercially launched Neutrolin in Germany
for the prevention of catheter-related bloodstream infections
(“CRBSI”), and maintenance of catheter patency in
hemodialysis patients using a tunneled, cuffed central venous
catheter for vascular access. To date, Neutrolin is
registered and may be sold in certain European Union and Middle
Eastern countries for such treatment.
In
September 2014, the TUV-SUD and The Medicines Evaluation Board of
the Netherlands granted a label expansion for Neutrolin for these
same expanded indications for the European Union
(“EU”). In December 2014, we received approval from the
Hessian District President in Germany to expand the label to
include use in oncology patients receiving chemotherapy, IV
hydration and IV medications via central venous catheters. The
expansion also adds patients receiving medication and IV fluids via
central venous catheters in intensive or critical care units
(cardiac care unit, surgical care unit, neonatal critical care
unit, and urgent care centers). An indication for use in total
parenteral nutrition was also approved.
Additional Development Possibilities
We are
evaluating opportunities for the
possible expansion for taurolidine as a platform. Patent
applications have been filed in: wound closure, surgical meshes,
wound management, and osteoarthritis, including
visco-supplementation. There exists a need to
control and protect against surgical site infections upon wound
closure. We believe taurolidine can also offer benefits
not currently available in marketed antimicrobial medical
devices. It can also provide a significant advantage in
devices for burn victims and use in less sterile
environments. We are also involved in a pre-clinical
research collaboration for the use of taurolidine as a possible
treatment for rare orphan pediatric tumors.
Neutrolin
Market Opportunity
Central
venous catheters and peripherally inserted central catheters are an
important and frequently used method for accessing the vasculature
in hemodialysis (a form of dialysis where the patient’s blood
is circulated through a dialysis filter), administering
chemotherapy and basic fluids (total parenteral nutrition) in
cancer patients and for cancer chemotherapy, long term antibiotic
therapy, total parenteral nutrition (complete or partial dietary
support via intravenous nutrients) and critical care/intensive care
patients.
The
treatment of patients undergoing hemodialysis requires access to
their vascular system on a recurring basis. According to the 2015
United States Renal Disease System, there were 660,000 patients on
hemodialysis. It has been reported by Hemodialysis National
Kidney Foundation that patients requiring a catheter represent over
63 million catheter/dialysis treatment days per year. In the United
States, 5.7 million intensive care patients were admitted annually
according to the Society of Critical Care Medicine, which is
estimated to represent 28.5 million catheter days associated with
ICU stays alone. As of 2014, there were over 14.5 million
patients in the United States living with cancer, with an estimated
7.7 million having had a long-term central venous catheter. When
stages of disease and types of chemotherapy regime are considered,
the number of catheter days per year are 90 million. Infections and
thrombosis represent key complications among cancer patients with
central venous catheters. One of the major and common complications
for all patients requiring central venous catheters is catheter
related blood stream infections, or CRBSIs, and the clinical
complications associated with them. There are an estimated 250,000
CRBSIs each year. The U.S. Centers for Disease Control and
Prevention stated in the Journal of American Medicine that the
total annual cost in the United States of treating all CRBI
episodes and their complications would amount to approximately $6
billion.
2
Biofilm
build up is the pathogenesis of both infections and thrombotic
complications in central venous catheters. Prevention of CRBIs and
inflammatory complications requires both decontamination of the
internal surface of the catheter to prevent the systemic
dissemination of organisms contained within the biofilm as well as
an anticoagulant to retain patency. Biofilm forms when bacteria
adhere to surfaces in aqueous environments and begin to excrete a
slimy, glue-like substance that can anchor them to various types of
materials, including intravenous catheters. The presence of biofilm
has many adverse effects, including the ability to release bacteria
into the blood stream. The current standard of catheter care is to
instill a heparin lock solution at a concentration of 1,000 u/mL
into each catheter lumen immediately following treatment, in order
to prevent clotting between dialysis treatments. However, a heparin
lock solution provides no protection from the risk of
infection.
Currently, there
are no pharmacologic agents approved in the U.S. for the prevention
of CRBIs in central venous catheters. As noted above, we received
the CE Mark approval for Neutrolin from the Medical Evaluation
Board, or MEB, at the EU in July 2013.
We
believe there is a significant need for prevention of CRBIs in the
hemodialysis patient population as well as for other patient
populations utilizing central venous catheters and peripherally
inserted central catheters, such as oncology/chemotherapy, total
parenteral nutrition and intensive care unit patients.
Neutrolin is a
broad-spectrum antimicrobial/antifungal and anticoagulant
combination that is active against common microbes including
antibiotic-resistant strains and in addition may prevent biofilm
formation. We believe that using Neutrolin as an anti-infective
solution will significantly reduce the incidence of
catheter-related blood stream infections, thus reducing the need
for local and systemic antibiotics while prolonging catheter
life.
Initially, we
expect to sell Neutrolin directly to hospitals and also to key
dialysis center operators. We anticipate that Medicare
reimbursement could be available for Neutrolin in other catheter
indications in intensive care, oncology and total parenteral
nutrition through relevant hospital inpatient diagnosis-related
groups (DRGs) or outpatient ambulatory payment classifications
(APCs), the End-Stage Renal Disease Prospective Payment System
(ESRD PPS) base payment, or under the Durable Medical Equipment,
Prosthetics, Orthotics, and Supplies (DMEPOS) Fee Schedule,
depending on the setting of care. We also plan to seek separate
reimbursement as a drug, where available under Medicare, through
mechanisms such as pass-through status under the Hospital
Outpatient Prospective Payment System, the transitional drug add-on
payment adjustment under the ESRD PPS, or reimbursement as a drug
used with a DME infusion pump. We cannot fully anticipate changes
in reimbursement requirements and mechanisms in the coming years,
however, and we cannot be certain that Neutrolin will be granted
separate reimbursement under any of these mechanisms.
Furthermore, we
anticipate that the U.S. Centers for Medicare & Medicaid
Services (CMS), and private payers will increasingly demand that
manufacturers demonstrate the cost effectiveness of their product
as part of the reimbursement review and approval process. With this
in mind, we have incorporated health economic evaluations into our
ongoing clinical studies to support this review in the context of
the prospective use of Neutrolin in dialysis, the ICU and oncology
settings. Our studies might not be sufficient to support
coverage or reimbursement at levels that allow providers to use
Neutrolin.
Competitive Landscape
The
drug and medical device industries are highly competitive and
subject to rapid and significant technological change.
Neutrolin’s current and future competitors include large as
well as specialty pharmaceutical and biotechnology companies. Many
of our competitors have substantially greater financial, technical
and human resources than we do and significantly more experience in
the development and commercialization of drugs and medical devices.
Further, the development of new treatment methods could render
Neutrolin non-competitive or obsolete.
We
believe that the key competitive factors that will affect the
development and commercial success of Neutrolin are efficacy and
safety, as well as pricing and reimbursement.
Drug:
To the
best of our knowledge, the following product candidates have been
recognized for the prevention and treatment of catheter-related
blood stream infections.
3
●
TauroLock,
manufactured by Tauro-Implant (Winsen, Germany). TauroLock has
received a CE Mark and is distributed in 25 countries. It has
anti-microbial and anti-coagulant activity and contains a
combination of citrate 4% with (cyclo)-taurolidine and heparin or
urokinase TauroLock has four formulations: TauroLock,
Tauro_lock Heparin 100, TauroLock Heparin 500 and TauroLock
Urokinase 2500IU.
●
Zuragen, being
developed by Ash Access Technology (Lafayette,IN). It has
antimicrobial and anticoagulant activity and contains methylene
blue, parabens and 7% citrate.
●
B-Lock, being
developed by Great Lakes Pharmaceuticals Inc. (Cleveland,
OH). It has anti-microbial, anti-coagulant and anti-fungal
activity and contains trimethoprim, EDTA and ethanol combinations.
Initiated study in 2012 in Poland and Hungary to support CE Mark in
European Union.
●
DuraLock-C,
manufactured by Medical Components, Inc. (Harleysville,PA).
DuraLock-C received a CE Mark and is distributed in a number of
European Union countries. It has anti-microbial and
anti-thrombosis activity and contains trisodium citrate in 46.7%,
30% and 4% concentrations.
●
IntraLock,
manufactured by Fresenius Medical Care AG & Co. (Bad Homburg,
Germany). IntraLock received a CE Mark and is distributed in a
number of European Union countries. It is an anticoagulant solution
to prevent thrombus formation in catheters. IntraLock contains
citrate (4%) for anticoagulation and a small amount of polyhexanide
for preservation.
●
TauroSept,
manufactured by Geistlich Pharma (Wolhusen, Switzerland). TauroSept
received Class 3 CE Mark and is distributed in a number of European
Union countries. TauroSept contains 2% taurolidine solution, 5%
polyvinylpyrrolidone and traces of HCl and NaOH to adjust pH. It
contains no anticoagulant substances.
Medical Devices:
●
Tego®
Needlefree Connector, manufactured by ICU Medical Inc. (California,
USA) Tego Needlefree Connector received 510(k) clearance from the
FDA. The Tego connector creates a mechanical and microbiology
closed system when attached to the hub of the catheter and works
with all hemodialysis CVC related applications.
●
Curos®
(Luer-lock caps twist on, stay on) disinfecting port protectors
designed specifically for Tego Needlefree Connectors, manufactured
by Ivera Medical Corporation. Curos received 510 (k) clearance from
the FDA. Curos for Tego Needlefree Connectors contains 70%
isopropyl alcohol-saturated, sponge-like foam that disinfects ports
in three minutes and keeps ports clean for seven days.
●
ClearGuard®
HD End Caps for Hemodialysis Catheters, manufactured by Pursuit
Vascular, Inc. ClearGuard HD End Caps received 510 (k) clearance
from the FDA. The ClearGuard HD End Cap consists of 1) a
copolyester polymer plug, which has a rod extending from the tier
region that is coated with the antimicrobial agent chlorhexidine
acetate (CHA) and 2) a nylon lock ring with threads that are also
coated with CHA.
●
BioFlo DuraMax
Dialysis Catheter with Endexo Technology, manufactured by
AngioDynamics. The product received 510(k) clearance by the FDA.
The BioFlo DuraMax chronic dialysis catheter features Endexo
Technology, a catheter material more resistant to thrombus
accumulation. Endexo technology is permanent, non-eluting polymer
“blended” into the polyurethane from which the catheter
is made.
Some
device companies have launched antibiotic or antimicrobial-coated
catheters as short-term prevention of catheter infection. We
believe these are not effective for hemodialysis catheters due to
the long term use and high blood flow associated with
hemodialysis.
Manufacturing
All of
our manufacturing processes currently are, and we expect them to
continue to be, outsourced to third parties. We rely on third-party
manufacturers to produce sufficient quantities of drug product for
use both commercially and in clinical trials. We intend to continue
this practice in the future.
In
April 2015, we entered into a Preliminary Services Agreement with
[RC]2 Pharma Connect LLC
(“RC2”), pursuant to which RC2 coordinates certain
manufacturing services related to taurolidine, which is a key
ingredient in Neutrolin. Specifically, RC2 undertook a critical
parameters evaluation for our manufacturing needs and to coordinate
the cGMP processes set forth in the agreement that we believe are
necessary for the submission of our planned new drug application
for Neutrolin to the FDA, as well as any foreign regulatory
applications. The total cost for RC2’s services under the
preliminary services agreement is approximately $1.7 million which
is expected to be incurred under the terms of this agreement
through the first quarter of 2017. Since inception
through December 31, 2016, RC2 completed and we recognized expense
of approximately $1,505,000 for its services related to this
agreement.
4
We are
also working with RC2 under several service agreements for an
aggregate amount of $7.6 million for the manufacture of clinical
supplies to support our ongoing and planned Phase 3 clinical
trials. During the years ended December 31, 2016 and 2015, we
recognized research and development expense of approximately
$2,359,000 and $1,348,000, respectively, related to these
agreements. We may terminate these agreements upon 30 days written
notice and are only obligated for project costs and reasonable
project shut down costs provided through the date of
termination.
Navinta
LLC, a U.S.-based active pharmaceutical ingredient, or API,
developer, provides API manufacturing (manufactured in India at an
FDA-compliant facility) and a Drug Master File for Neutrolin,
pursuant to a supply agreement dated December 7, 2009 (the
“Navinta Agreement”). On March 24, 2015, we and Navinta
LLC entered into an amendment to the Navinta Agreement to extend
the term of the Navinta Agreement to March 31, 2016 and to lower
the price per kilogram of API that we purchase from Navinta LLC
under the Navinta Agreement. We also agreed to purchase a minimum
amount of product from Navinta LLC during 2015, which replaced the
prior minimum purchase requirement. The Navinta Agreement was
terminable by either party upon 30 days written notice. The Navinta
Agreement expired in accordance with its terms without delivery of
the minimum purchase requirement and without any further
obligations by either party.
We are
confident that there exist a sufficient number of potential
alternate sources for the drug substances required to produce our
products, as well as third-party manufacturers, that we will be
able to find alternate suppliers and third-party manufacturers in
the event that our relationship with any supplier or third-party
manufacturer deteriorates.
United States Government Regulation
The
research, development, testing, manufacture, labeling, promotion,
advertising, distribution, and marketing, among other things, of
our products are extensively regulated by governmental authorities
in the United States and other countries. Our products may be
classified by the FDA as a drug or a medical device depending upon
the indications for use or claims. Because certain of our product
candidates are considered as medical devices and others are
considered as drugs for regulatory purposes, we intend to submit
applications to regulatory agencies for approval or clearance of
both medical devices and pharmaceutical product
candidates.
In the
United States, the FDA regulates drugs and medical devices under
the Federal Food, Drug, and Cosmetic Act and the agency’s
implementing regulations. If we fail to comply with the applicable
United States requirements at any time during the product
development process, clinical testing, and the approval process or
after approval, we may become subject to administrative or judicial
sanctions. These sanctions could include the FDA’s refusal to
approve pending applications, license suspension or revocation,
withdrawal of an approval, warning letters, adverse publicity,
product recalls, product seizures, total or partial suspension of
production or distribution, injunctions, fines, civil penalties or
criminal prosecution, among other actions. Any agency enforcement
action could have a material adverse effect on us.
Drug Approval Process
The
research, development, and approval process in the United States
and elsewhere is intensive and rigorous and generally takes many
years to complete. The typical process required by the FDA before a
therapeutic drug may be marketed in the United States
includes:
●
preclinical
laboratory and animal tests performed under the FDA’s Good
Laboratory Practices, or GLP, regulations;
●
submission to the
FDA of an investigational new drug application, or IND, which must
become effective before human clinical trials may
commence;
●
human clinical
studies to evaluate the drug’s safety and effectiveness for
its intended uses;
●
FDA review of
whether the facility in which the drug is manufactured, processed,
packaged, or held meets standards designed to assure the
product’s continued quality and FDA review of clinical trial
sites to determine whether the clinical trials were conducted in
accordance with Good Clinical Practices, or GCPs; and
●
submission of a new
drug application, or NDA, to the FDA, and approval of the
application by the FDA to allow sales of the drug.
During
preclinical testing, studies are performed with respect to the
chemical and physical properties of candidate formulations. These
studies are subject to GLP requirements. Biological testing is
typically done in animal models to demonstrate the activity of the
compound against the targeted disease or condition and to assess
the apparent effects of the new product candidate on various organ
systems, as well as its relative therapeutic effectiveness and
safety. An IND application must be submitted to the FDA and become
effective before studies in humans may commence.
5
Clinical trial
programs in humans generally follow a three-phase process.
Typically, Phase 1 studies are conducted in small numbers of
healthy volunteers or, on occasion, in patients afflicted with the
target disease. Phase 1 studies are conducted to determine the
metabolic and pharmacological action of the product candidate in
humans and the side effects associated with increasing doses, and,
if possible, to gain early evidence of effectiveness. In Phase 2,
studies are generally conducted in larger groups of patients having
the target disease or condition in order to validate clinical
endpoints, and to obtain preliminary data on the effectiveness of
the product candidate and optimal dosing. This phase also helps
determine further the safety profile of the product candidate. In
Phase 3, large-scale clinical trials are generally conducted in
patients having the target disease or condition to provide
sufficient data for the statistical proof of effectiveness and
safety of the product candidate as required by United States and
foreign regulatory agencies. Typically two Phase 3 trials are
required for marketing approval.
In the
case of products for certain serious or life-threatening diseases,
the initial human testing may be done in patients with the disease
rather than in healthy volunteers. Because these patients are
already afflicted with the target disease or condition, it is
possible that such studies will also provide results traditionally
obtained in Phase 2 studies. These studies are often referred to as
“Phase 1/2” studies. However, even if patients
participate in initial human testing and a Phase 1/2 study is
carried out, the sponsor is still responsible for obtaining all the
data usually obtained in both Phase 1 and Phase 2
studies.
Before
proceeding with a study, sponsors may seek a written agreement
known as a Special Protocol Assessment, or SPA, from the FDA
regarding the design, size, and conduct of a clinical trial. Among
other things, SPAs can cover clinical studies for pivotal trials
whose data will form the primary basis to establish a
product’s efficacy. SPAs help establish up-front agreement
with the FDA about the adequacy of a clinical trial design to
support a regulatory approval, but the agreement is not binding on
the FDA if new circumstances arise. An
SPA may only be modified with the agreement of the FDA and the
trial sponsor or if the director of the FDA reviewing division
determines that a substantial scientific issue essential to
determining the safety or efficacy of the drug was identified after
the testing began. There is no guarantee that a study
will ultimately be adequate to support an approval even if the
study is subject to an SPA.
Additionally,
some clinical trials are overseen by an independent group of
qualified experts organized by the clinical trial sponsor, known as
a data safety monitoring board or committee. This group regularly
reviews accumulated data and advises the study sponsor regarding
the continuing safety of trial subjects, and the continuing
validity and scientific merit of the clinical trial. The data
safety monitoring board receives special access to unblinded data
during the clinical trial and may advise the sponsor to halt the
clinical trial if it determined there is an unacceptable safety
risk for subjects or on other grounds, such as no demonstration of
efficacy.
The
manufacture of investigational drugs for the conduct of human
clinical trials is subject to current Good Manufacturing Practice,
or cGMP, requirements. Investigational drugs and active
pharmaceutical ingredients imported into the United States are also
subject to regulation by the FDA relating to their labeling and
distribution. Further, the export of investigational drug products
outside of the United States is subject to regulatory requirements
of the receiving country as well as U.S. export requirements under
the Federal Food, Drug, and Cosmetic Act, or FDCA.
IND
sponsors are also required to submit a number of reports to the FDA
during the course of a development program. For instance, sponsors
are required to make annual reports to the FDA concerning the
progress of their clinical trial programs as well as more frequent
reports for certain serious adverse events. Sponsors must submit a protocol for each clinical trial,
and any subsequent protocol amendments to the FDA. Investigators
must also provide certain information to the clinical trial
sponsors to allow the sponsors to make certain financial
disclosures to the FDA. Information about certain clinical
trials, including a description of the study and study results,
must be submitted within specific timeframes to the National
Institutes of Health, or NIH, for public dissemination on their
clinicaltrials.gov website. Moreover, under the 21st Century Cures
Act, manufacturers or distributors of investigational drugs for the
diagnosis, monitoring, or treatment of one or more serious diseases
or conditions must have a publicly available policy concerning
expanded access to investigational drugs.
United
States law requires that studies conducted to support approval for
product marketing be “adequate and well controlled.” In
general, this means that either a placebo or a product already
approved for the treatment of the disease or condition under study
must be used as a reference control. The recently passed 21st
Century Cures Act, however, provides for FDA acceptance of new
kinds of data such as such as patient experience data, real world
evidence, and, for appropriate indications sought through
supplemental marketing applications, data summaries. Studies must
also be conducted in compliance with good clinical practice
requirements, and informed consent must be obtained from all study
subjects.
6
In
addition, under the Pediatric Research Equity Act, or PREA, an NDA
or supplement to an NDA for a new active ingredient, indication,
dosage form, dosage regimen, or route of administration must
contain data that are adequate to assess the safety and
effectiveness of the drug for the claimed indications in all
relevant pediatric subpopulations, and to support dosing and
administration for each pediatric subpopulation for which the
product is safe and effective. The FDA may, on its own initiative
or at the request of the applicant, grant deferrals for submission
of some or all pediatric data until after approval of the product
for use in adults, or full or partial waivers from the pediatric
data requirements.
The
FDA also may require submission of a risk evaluation and mitigation
strategy, or REMS, to ensure that the benefits of the drug outweigh
the risks of the drug. The REMS plan could include medication
guides, physician communication plans, and elements to assure safe
use, such as restricted distribution methods, patient registries,
or other risk minimization tools. An assessment of the REMS must
also be conducted at set intervals. Following product approval, a
REMS may also be required by the FDA if new safety information is
discovered and the FDA determines that a REMS is necessary to
ensure that the benefits of the drug outweigh the risks of the
drug.
The
clinical trial process for a new compound can take ten years or
more to complete. The FDA may prevent clinical trials from
beginning or may place clinical trials on hold at any point in this
process if, among other reasons, it concludes that study subjects
are being exposed to an unacceptable health risk. Trials may also
be prevented from beginning or may be terminated by institutional
review boards, or IRBs, who must review and approve all research
involving human subjects and amendments thereto. The IRB must continue to oversee the clinical
trial while it is being conducted. This includes the IRBs receiving
information concerning unanticipated problems involving risk to
subjects. Side effects or adverse events that are reported
during clinical trials can delay, impede, or prevent marketing
authorization. Similarly, adverse events that are reported after
marketing authorization can result in additional limitations being
placed on a product’s use and, potentially, withdrawal of the
product from the market.
Following the
completion of a clinical trial, the data are analyzed by the
sponsoring company to determine whether the trial successfully
demonstrated safety and effectiveness and whether a product
approval application may be submitted. In the United States, if the
product is regulated as a new drug, an NDA must be submitted and
approved before commercial marketing may begin. The NDA must
include a substantial amount of data and other information
concerning the safety and effectiveness of the compound from
laboratory, animal, and human clinical testing, as well as data and
information on manufacturing, product quality and stability, and
proposed product labeling.
Each
domestic and foreign manufacturing establishment, including any
contract manufacturers that we may decide to use, must be listed in
the NDA and must be registered with the FDA. The application
generally will not be approved until the FDA conducts a
manufacturing inspection, approves the applicable manufacturing
process for the drug product, and determines that the facility is
in compliance with current cGMP requirements. Moreover, FDA will
also typically inspect one or more clinical trial sites to confirm
that the applicable clinical trials were conducted in accordance
with GCPs.
Under
the Prescription Drug User Fee Act, as amended, the FDA receives
fees for reviewing an NDA, as well as annual fees for commercial
manufacturing establishments and for approved products. These fees
can be significant. Fee waivers or reductions are available in
certain circumstances. One basis for a waiver of the application
user fee is if the applicant employs fewer than 500 employees,
including employees of affiliates, the applicant does not have an
approved marketing application for a product that has been
introduced or delivered for introduction into interstate commerce,
and the applicant, including its affiliates, is submitting its
first marketing application. Product candidates that are designated
as orphan drugs, which are further described below, are also not
subject to application user fees unless the application includes an
indication other than the orphan indication. Under certain
circumstances, orphan products may also be exempt from product and
establishment fees.
Each
NDA submitted for FDA approval is usually reviewed for
administrative completeness and reviewability. Following this
review, the FDA may request additional information rather than
accept an NDA for filing. In this event, the application must be
resubmitted with the additional information. The resubmitted
application is also subject to review before the FDA accepts it for
filing.
Once
accepted for filing, the FDA’s review of an application may
involve review and recommendations by an independent FDA advisory
committee. The FDA must refer
applications for drugs that contain active ingredients, including
any ester or salt of the active ingredients that have not
previously been approved by the FDA to an advisory committee or
provide in an action letter a summary for not referring it to an
advisory committee. The FDA may also refer drugs to advisory
committees when it is determined that an advisory committee’s
expertise would be beneficial to the regulatory decision-making
process, including the evaluation of novel products and the use of
new technology. An advisory committee is typically a panel that
includes clinicians and other experts, which review, evaluate, and
make a recommendation as to whether the application should be
approved and under what conditions. The FDA is not bound by the
recommendations of an advisory committee, but it considers such
recommendations carefully when making
decisions.
7
After
evaluating the NDA and all related information, including the
advisory committee recommendation, if any, and inspection reports
regarding the manufacturing facilities and clinical trial sites,
the FDA may issue an approval letter, or, in some cases, a Complete
Response Letter, or CRL. If a CRL is issued, the applicant may
either resubmit the NDA, addressing all of the deficiencies
identified in the letter; withdraw the application; or request an
opportunity for a hearing. A CRL indicates that the review cycle of
the application is complete and the application is not ready for
approval and describes all of the specific deficiencies that the
FDA identified in the NDA. A CRL generally contains a statement of
specific conditions that must be met in order to secure final
approval of the NDA, and may require additional clinical or
preclinical testing in order for the FDA to reconsider the
application. The deficiencies identified may be minor, for example,
requiring labeling changes; or major, for example, requiring
additional clinical trials. Even with submission of this additional
information, the FDA ultimately may decide that the application
does not satisfy the regulatory criteria for approval. If and when
those conditions have been met to the FDA’s satisfaction, the
FDA may issue an approval letter. An approval letter authorizes
commercial marketing of the drug with specific prescribing
information for specific indications.
Even if
the FDA approves a product, it may limit the approved therapeutic
uses for the product as described in the product labeling, require
that warning statements be included in the product labeling,
require that additional studies be conducted following approval as
a condition of the approval, impose restrictions and conditions on
product distribution, prescribing, or dispensing in the form of a
REMS or otherwise limit the scope of any approval.
Special FDA Expedited Review and Approval Programs
The
FDA has various programs, including Fast Track designation,
priority review and breakthrough designation, that are intended to
expedite or simplify the process for the development and FDA review
of certain drug products that are intended for the treatment of
serious or life threatening diseases or conditions, and demonstrate
the potential to address unmet medical needs or present a
significant improvement over existing therapy. The purpose of these
programs is to provide important new drugs to patients earlier than
under standard FDA review procedures.
To
be eligible for a Fast Track designation, the FDA must determine,
based on the request of a sponsor, that a product is intended to
treat a serious or life threatening disease or condition and
demonstrates the potential to address an unmet medical need. The
FDA will determine that a product will fill an unmet medical need
if the product will provide a therapy where none exists or provide
a therapy that may be potentially superior to existing therapy
based on efficacy, safety, or public health factors. If Fast Track
designation is obtained, drug sponsors may be eligible for more
frequent development meetings and correspondence with the FDA. In
addition, the FDA may initiate review of sections of an NDA before
the application is complete. This “rolling review” is
available if the applicant provides and the FDA approves a schedule
for the remaining information. A Fast Track product is also
eligible to apply for accelerated approval and priority
review.
The
FDA may give a priority review designation to drugs that are
intended to treat serious conditions and, if approved, would
provide significant improvements in the safety or effectiveness of
the treatment, diagnosis, or prevention of serious conditions. A
priority review means that the goal for the FDA is to review an
application within six months, rather than the standard review of
ten months under current PDUFA guidelines, of the 60-day filing
date for new molecular entities.
Moreover,
under the provisions of the Food and Drug Administration Safety and
Innovation Act, or FDASIA, enacted in 2012, a sponsor can request
designation of a product candidate as a “breakthrough
therapy.” A breakthrough therapy is defined as a drug that is
intended, alone or in combination with one or more other drugs, to
treat a serious or life-threatening disease or condition, and
preliminary clinical evidence indicates that the drug may
demonstrate substantial improvement over existing therapies on one
or more clinically significant endpoints, such as substantial
treatment effects observed early in clinical development. Drugs
designated as breakthrough therapies are eligible for the Fast
Track designation features as described above, intensive guidance
on an efficient drug development program beginning as early as
Phase 1 trials, and a commitment from the FDA to involve
senior managers and experienced review staff in a proactive
collaborative, cross-disciplinary review.
Even
if a product qualifies for one or more of these programs, the FDA
may later decide that the product no longer meets the conditions
for qualification or decide that the time period for FDA review or
approval will not be shortened.
8
A
final new program to expedite the development of drug products is
the limited population pathway for antibacterial and antifungal
drugs, which was passed as part of the recent enacted 21st Century
Cures Act. Under this program, a sponsor can request drug approval
under this new pathway for an antibacterial or antifungal drug if
the drug is intended to treat a serious or life-threatening
infection in a limited population of patients with unmet needs.
Under this program, the FDA’s determination of safety and
effectiveness would reflect the risk-benefit profile of the drug in
the intended limited population, taking into account the severity,
rarity, or prevalence of the infection and the availability of
alternative treatments in the limited population. The drug may be
approved for the limited population notwithstanding a lack of
evidence to fully establish a favorable benefit-risk profile in a
broader population. Under this program, the FDA must provide prompt
advice to sponsors seeking approval under this pathway to enable
them to plan a development program. If approved under this pathway,
certain post-marketing requirements would apply, such as required
labeling and advertising statements and pre-distribution submission
of promotional materials to FDA. If after approval for a limited
population, a product receives a broader approval, the FDA may
remove such post-marketing restrictions. While a drug may only be
approved for a limited population under this program, the 21st
Century Cures Act states that it is not intended to restrict the
prescribing of antimicrobial drugs or other products by healthcare
professionals.
Exclusivity
For
approved drug products, market exclusivity provisions under the
FDCA provide periods of regulatory exclusivity, which gives the
holder of an approved NDA limited protection from new competition
in the marketplace for the innovation represented by its approved
drug.
Section 505
of the FDCA describes three types of marketing applications that
may be submitted to the FDA to request marketing authorization for
a new drug. A Section 505(b)(1) NDA is an application that
contains full reports of investigations of safety and efficacy. A
Section 505(b)(2) NDA is an application in which the
applicant, in part, relies on investigations that were not
conducted by or for the applicant and for which the applicant has
not obtained a right of reference or use from the person by or for
whom the investigations were conducted. Section 505(j)
establishes an abbreviated approval process for a generic version
of approved drug products through the submission of an Abbreviated
New Drug Application, or ANDA. An ANDA provides for marketing of a
generic drug product that has the same active ingredients, dosage
form, strength, route of administration, labeling, performance
characteristics, and intended use, among other things, to a
previously approved product. Limited changes must be pre-approved
by the FDA via a suitability petition.
Five
years of exclusivity are available to New Chemical Entities, or
NCEs. A NCE is a drug that contains no active moiety that has been
approved by the FDA in any other NDA. An active moiety is the
molecule or ion, excluding those appended portions of the molecule,
that cause the drug to be an ester, salt, including a salt with
hydrogen or coordination bonds, or other noncovalent derivatives,
such as a complex, chelate, or clathrate, of the molecule,
responsible for the therapeutic activity of the drug substance.
During the exclusivity period, the FDA may not accept for review
and make an ANDA or a 505(b)(2) NDA approval effective for an
application submitted by another company that contains the
previously approved active moiety. An ANDA or 505(b)(2)
application, however, may be submitted one year before NCE
exclusivity expires if the applicant submits a certification
stating that the patents listed by the NCE sponsor in FDA’s
list of Approved Drug Products with Therapeutic Equivalence
Evaluations, or Orange Book, are invalid or will not be infringed
by the manufacture, use, or sale of the drug product for which
approval is sought. Five-year exclusivity will also not delay the
submission or approval of a full NDA; however, an applicant
submitting a full NDA would be required to conduct or obtain a
right of reference to all of the preclinical studies and adequate
and well-controlled clinical trials necessary to demonstrate safety
and efficacy.
Pediatric
exclusivity is another type of non-patent marketing exclusivity in
the United States and, if granted, provides for the
attachment of an additional six months of marketing protection to
the term of any existing regulatory exclusivity, including the
non-patent exclusivity period described above. This six-month
exclusivity may be granted if an NDA sponsor submits pediatric data
that fairly respond to a written request from the FDA for such
data. The data do not need to show the product to be effective in
the pediatric population studied; rather, if the clinical trial is
deemed to fairly respond to the FDA’s request, the additional
protection is granted. If reports of requested pediatric studies
are submitted to and accepted by the FDA within the required time
frames, whatever statutory or regulatory periods of exclusivity or
Orange Book listed patent protection cover the drug are extended by
six months. Moreover, pediatric exclusivity attaches to all
formulations, dosage forms, and indications for products with
existing marketing exclusivity or patent life that contain the same
active moiety as that which was studied.
9
The
Orphan Drug Act also provides incentives for the development of
drugs intended to treat rare diseases or conditions, which
generally are diseases or conditions affecting fewer than 200,000
individuals annually in the United States, or affecting more than
200,000 in the United States and for which there is no reasonable
expectation that the cost of developing and making the drug
available in the United States will be recovered from sales in the
United States. Additionally, sponsors must present a plausible
hypothesis for clinical superiority to obtain orphan designation if
there is a drug already approved by the FDA that is intended for
the same indication and that is considered by the FDA to be the
same drug as the already approved drug. This hypothesis must be
demonstrated to obtain orphan drug exclusivity. If granted, prior
to product approval, Orphan Drug Designation entitles a party to
financial incentives such as opportunities for grant funding
towards clinical study costs, tax advantages, and user-fee waivers.
In addition, if a product receives FDA approval for the indication
for which it has orphan designation, the product is generally
entitled to orphan drug exclusivity, which means the FDA may not
approve any other application to market the same drug for the same
indication for a period of seven years, except in limited
circumstances, such as a showing of clinical superiority over the
product with orphan exclusivity.
For
certain infectious disease products, the above discussed
exclusivity periods may be further extended under the FDA’s
qualified infectious disease product program. A qualified
infections disease product, or QIDP, is an antibacterial or
antifungal drug for human use intended to treat serious or
life-threatening infections, including those caused by an
antibacterial or antifungal resistant pathogen, including novel or
emerging infectious pathogens; or qualifying pathogens designated
by the FDA that has the potential to pose a serious threat to
public health. If the FDA approves an NDA for a drug designated as
a QIDP, the NCE exclusivity period is extended to ten years and the
FDA may not accept applications for nine years. Moreover, if a
product is designated as a QIDP and an orphan product, the orphan
product exclusivity period is extended to twelve years. These
extensions are in addition to any extension that an application may
be entitled to under the pediatric exclusivity provisions. To
receive a QIDP designation, the sponsor must request that the FDA
designate the product as such prior to the submission of an NDA.
This designation may not be withdrawn except if the FDA finds that
the request for designation contained an untrue statement of
material fact. QIDPs are also eligible for fast track status and
priority review.
Post Approval Requirements
Significant legal
and regulatory requirements also apply after FDA approval to market
under an NDA. These include, among other things, requirements
related to adverse event and other reporting, product tracking and
tracing, suspect and illegitimate product investigations and
notifications, product advertising and promotion and ongoing
adherence to cGMPs, as well as the need to submit appropriate new
or supplemental applications and obtain FDA approval for certain
changes to the approved product, product labeling, or manufacturing
process. The FDA also enforces the requirements of the Prescription
Drug Marketing Act which, among other things, imposes various
requirements in connection with the distribution of product samples
to physicians. The FDA enforces these requirements through, among
other ways, periodic announced and unannounced facility
inspections.
The
FDA also strictly regulates marketing, labeling, advertising, and
promotion of products that are placed on the market. A company can
make only those claims relating to safety and efficacy that are
approved by the FDA. Physicians, in their independent professional
medical judgment, may prescribe legally available products for
unapproved indications that are not described in the
product’s labeling and that differ from those tested and
approved by the FDA. Pharmaceutical companies, however, are allowed
to promote their drug products only for the approved indications
and in accordance with the provisions of the approved label. The
FDA and other agencies actively enforce the laws and regulations
prohibiting the promotion of off-label uses, and a company that is
found to have improperly promoted off-label uses may be subject to
significant liability, including, but not limited to, criminal and
civil penalties under the FDCA and the civil False Claims Act, or
FCA, exclusion from participation in federal healthcare programs,
mandatory compliance programs under corporate integrity agreements,
debarment, and refusal of government contracts.
The
regulatory framework applicable to the production, distribution,
marketing, and/or sale, of our products may change significantly
from the current descriptions provided herein in the time that it
may take for any of our products to reach a point at which a NDA is
approved. Moreover, individual states may have laws and regulations
that we must comply with, such as laws and regulations concerning
licensing, promotion, sampling, distribution, and
reporting.
Overall
research, development, and approval times depend on a number of
factors, including the period of review at the FDA, the number of
questions posed by the FDA during review, how long it takes to
respond to the FDA’s questions, the severity or
life-threatening nature of the disease in question, the
availability of alternative treatments, the availability of
clinical investigators and eligible patients, the rate of
enrollment of patients in clinical trials, and the risks and
benefits demonstrated in the clinical trials.
10
Fraud and Abuse, Data Privacy and Security, and Transparency Laws
and Regulations
Our
business activities, including but not limited to research, sales,
promotion, distribution, medical education, and other activities
following product approval, if any, will be subject to regulation
by numerous federal and state regulatory and law enforcement
authorities in the United States in addition to the FDA, including
potentially the Department of Justice, the Department of Health and
Human Services and its various divisions, including the Centers for
Medicare & Medicaid Services, or CMS, and the Health
Resources and Services Administration, the Department of Veterans
Affairs, the Department of Defense, and state and local
governments. Our business activities must comply with numerous
healthcare laws, including but not limited to, anti-kickback and
false claims laws and regulations as well as data privacy and
security laws and regulations, which are described
below.
The
federal Anti-Kickback Statute prohibits, among other things, any
person or entity, from knowingly and willfully offering, paying,
soliciting, or receiving any remuneration, directly or indirectly,
overtly or covertly, in cash or in kind, to induce or in return for
purchasing, leasing, ordering, or arranging for or recommending the
purchase, lease, furnishing, or order of any item or service
reimbursable under Medicare, Medicaid, or other federal healthcare
programs, in whole or in part. The term “remuneration”
has been interpreted broadly to include anything of value. The
Anti-Kickback Statute has been interpreted to apply to arrangements
between pharmaceutical manufacturers on one hand and prescribers,
purchasers, formulary managers, and beneficiaries on the other.
There are certain statutory exceptions and regulatory safe harbors
protecting some common activities from prosecution. The exceptions
and safe harbors are drawn narrowly, and practices that involve
remuneration that may be alleged to be intended to induce
prescribing, purchases, reimbursement, or recommendations may be
subject to scrutiny if they do not qualify for an exception or safe
harbor. Failure to meet all of the requirements of a particular
applicable statutory exception or regulatory safe harbor does not
make the conduct per se illegal under the Anti-Kickback Statute.
Instead, the legality of the arrangement will be evaluated on a
case-by-case basis based on a cumulative review of all of its facts
and circumstances. Several courts have interpreted the
statute’s intent requirement to mean that if any one purpose
of an arrangement involving remuneration is to induce referrals of
federal healthcare covered business, the statute has been violated.
The Patient Protection and Affordable Care Act, or ACA, of 2010, as
amended, modified the intent requirement under the Anti-Kickback
Statute, such that a person or entity no longer needs to have
actual knowledge of the statute or specific intent to violate it in
order to have committed a violation. In addition, the ACA also
provided that a violation of the federal Anti-Kickback Statute is
grounds for the government or a whistleblower to assert that a
claim for payment of items or services resulting from such
violation constitutes a false or fraudulent claim for purposes of
the FCA.
The
federal FCA prohibits, among other things, any person or entity
from knowingly presenting, or causing to be presented, a false or
fraudulent claim for payment to, or approval by, the federal
government, knowingly making, using, or causing to be made or used
a false record or statement material to a false or fraudulent claim
to the federal government, or avoiding, decreasing, or concealing
an obligation to pay money to the federal government. The FCA
authorizes imposition of treble damages and a civil penalty for
each false claim submitted. For violations after November 2, 2015,
the penalty has increased from a minimum of $5,500 to $10,781, and
a maximum of $11,000 to $21,563. A claim includes “any
request or demand” for money or property presented to the
U.S. government, including an invoice. Accordingly, FCA penalties
for high claim volume products like pharmaceuticals have frequently
aggregated into millions of dollars. The FCA has been used to
assert liability on the basis of kickbacks and other improper
referrals, improperly reported government pricing metrics such as
Best Price or Average Manufacturer Price, improper use of Medicare
provider or supplier numbers when detailing a provider of services,
improper promotion of off-label uses not expressly approved by the
FDA in a drug’s label, and allegations as to
misrepresentations with respect to product quality or services
rendered. Several pharmaceutical and other healthcare companies
have further been sued under these laws for allegedly providing
free product to customers with the expectation that the customers
would bill federal programs for the product. Intent to deceive is
not required to establish liability under the FCA. Liability may be
predicated on reckless disregard for the truth. FCA actions may be
brought by the government or may be brought by private individuals
on behalf of the government, called “qui tam” actions.
If the government decides to intervene in a qui tam action and
prevails in the lawsuit, the individual will share in the proceeds
from any fines or settlement funds. If the government declines to
intervene, the individual may pursue the case alone. Since 2004,
these False Claims Act lawsuits against pharmaceutical companies
have increased significantly in volume and breadth, leading to
substantial civil and criminal settlements regarding certain sales
practices and promoting off label drug uses. Further, liability may
be predicated on non-compliance with federal laws and regulations
under the theory of implied certification. However, the Supreme
Court imposed a materiality standard for liability under this
theory – the non-compliance must be material to the
government’s payment decision. FCA liability may also be
imposed for Medicare or Medicaid overpayments, for example,
overpayments caused by understated rebate amounts that are not
refunded within 60 days of discovering the overpayment, even
if the overpayment was not caused by a false or fraudulent
act.
11
The
government may further prosecute conduct constituting a false claim
under the criminal False Claims Act. The criminal False Claims Act
prohibits the making or presenting of a claim to the government
knowing such claim to be false, fictitious, or fraudulent and,
unlike the civil False Claims Act, requires proof of intent to
submit a false claim.
The
civil monetary penalties statute is another potential statute under
which biopharmaceutical companies may be subject to enforcement.
Among other things, the civil monetary penalties statute imposes
fines against any person who is determined to have presented, or
caused to be presented, claims to a federal healthcare program that
the person knows, or should know, is for an item or service that
was not provided as claimed or is false or fraudulent. In 2016, the
Department of Justice doubled the amount of these penalties, which
are specified in the civil FCA.
Many
states have also adopted laws similar to each of the above federal
laws, which may be broader in scope. Certain states also require
implementation of commercial compliance programs and compliance
with the pharmaceutical industry’s voluntary compliance
guidelines and the applicable compliance guidance promulgated by
the federal government, or otherwise restrict payments or the
provision of other items of value that may be made to healthcare
providers and other potential referral sources; impose restrictions
on marketing practices; or require drug manufacturers to track and
report information related to payments, gifts, and other items of
value to physicians and other healthcare providers. These laws may
affect our future sales, marketing, and other promotional
activities by imposing administrative and compliance
burdens.
If our
operations are found to be in violation of any of the laws or
regulations described above or any other laws that apply to us, we
may be subject to penalties or other enforcement actions, including
criminal and significant civil monetary penalties, damages, fines,
disgorgement, imprisonment, exclusion from participation in
government healthcare programs, corporate integrity agreements,
debarment from receiving government contracts or refusal of new
orders under existing contracts, reputational harm, diminished
profits and future earnings, and the curtailment or restructuring
of our operations, any of which could adversely affect our ability
to operate our business and our results of operations. Enforcement
actions can be brought by federal or state governments, or as qui
tam actions brought by individual whistleblowers in the name of the
government under the civil False Claims Act if the violations are
alleged to have caused the government to pay a false or fraudulent
claim.
To the
extent that any of our products are sold in a foreign country, we
may be subject to similar foreign laws and regulations, which may
include, for instance, applicable post-marketing requirements,
including safety surveillance, anti-fraud and abuse laws, and
implementation of corporate compliance programs and reporting of
payments or transfers of value to healthcare
professionals.
Medical Device Approval Process
In
addition to our lead product candidate Neutrolin, which is subject
to regulation by the FDA as a drug, we are developing other
products that may be regulated as medical devices in the United
States. The FDA regulates the design, development, clinical
testing, manufacture, labeling, distribution, import and export,
sale and promotion of medical devices. Unless an exemption applies
or a product is a Class I device, all medical devices must receive
either 510(k) clearance or an approved pre-market application, or
PMA, from the FDA before they may be commercially distributed in
the U.S. In addition, certain modifications made to marketed
devices also may require 510(k) clearance or approval of a PMA
supplement.
To
obtain a 510(k) clearance for a device, a pre-market notification
to the FDA must be submitted demonstrating that the device is
substantially equivalent to a legally marketed predicate device.
The FDA attempts to respond to a 510(k) pre-market notification
within 90 days of submission, but as a practical matter,
pre-market clearance can take significantly longer, potentially up
to one year or more. The PMA process is much more demanding and
uncertain than the 510(k) pre-market notification process and must
be supported by extensive clinical, technical and other
information, including at least one adequate and well-controlled
clinical investigation. The FDA has 180 days to review an
accepted PMA, although the review generally occurs over a
significantly longer period of time, and can take up to several
years.
After a
device is placed on the market, numerous regulatory requirements
apply, including:
●
Quality System
Regulations, or QSRs, which require manufacturers to have a quality
system for the design, manufacture, packaging, labeling, storage,
installation, and servicing of finished medical
devices;
●
labeling
regulations, which govern product labels and labeling, prohibit the
promotion of products for unapproved, or off-label, uses and impose
other restrictions on labeling and promotional
activities;
●
medical device
listing and establishment registration;
●
post-approval
restrictions or conditions, including post-approval study
commitments;
12
●
post-market
surveillance requirements;
●
medical device
reporting, or MDR, regulations, which require that manufacturers
evaluate and investigate potential adverse events and malfunctions,
and report to the FDA if their device may have caused or
contributed to a death or serious injury or malfunctioned in a way
that would likely cause or contribute to a death or serious injury
if it were to recur;
●
regulations
requiring the reporting of any device corrections or removals if
the correction or removal was initiated to reduce a risk to health
posed by the device or remedy a violation of the FDCA which may
present a risk to health; and
●
the FDA’s
recall authority, whereby it can ask, or under certain conditions
order, device manufacturers to recall from the market a product
that is a risk to health.
Our
manufacturing facilities, as well as those of certain of our
suppliers, are subject to periodic and for-cause inspections by the
FDA and other governmental authorities to verify compliance with
the QSR and other regulatory requirements.
Reimbursement and Pricing Controls
In many
of the markets where we or our collaborative partners have targeted
or will target Neutrolin for sale, laws control the prices charged
to certain purchasers of pharmaceutical products and the prices
paid by drug reimbursement programs through varying price control
mechanisms. Public and private health care payors control costs and
influence drug pricing through a variety of mechanisms, including
through negotiating rebates with the manufacturers, limiting the
reimbursement rate paid to providers, and using tiered formularies,
co-payment structures that incentivize beneficiaries to request
lower cost alternatives, and other mechanisms that provide
preferential access to certain drugs over others within a
therapeutic class. Federal and commercial payors use competition
for health plan coverage and market share as leverage to obtain
rebates on products they reimburse, which impacts the
manufacturer’s net realization on the sale of the products.
These rebates may be paid on drugs sold at a mandatory discount.
Additionally, federal and commercial health plans may choose to
reimburse dialysis providers for dialysis services and drugs used
in the provision of those services through a single bundled payment
rate, which tends to make cost a more important factor for
providers when making drug purchase decisions than it would
otherwise be if the providers were reimbursed for drugs on a
stand-alone basis. Payors also set other criteria to govern the
uses of a drug that will be deemed medically appropriate and
therefore reimbursed or otherwise covered. In particular, many
public and private health care payors limit reimbursement and
coverage to the uses of a drug that are either approved by the FDA
or that are supported by other appropriate evidence (for example,
published medical literature) and appear in a recognized drug
compendium. Drug compendia are publications that summarize the
available medical evidence for particular drug products and
identify which uses of a drug are supported or not supported by the
available evidence, whether or not such uses have been approved by
the FDA.
Foreign
Regulatory Requirements
We and
our collaborative partners may be subject to widely varying foreign
regulations, which may be quite different from those of the FDA,
governing clinical trials, manufacture, product registration and
approval, and pharmaceutical sales. Whether or not FDA approval has
been obtained, we or our collaboration partners must obtain a
separate approval for a product by the comparable regulatory
authorities of foreign countries prior to the commencement of
product marketing in those countries. In certain countries,
regulatory authorities also establish pricing and reimbursement
criteria. The approval process varies from country to country, and
the time may be longer or shorter than that required for FDA
approval. In addition, under current United States law, there are
restrictions on the export of products not approved by the FDA,
depending on the country involved and the status of the product in
that country.
International sales
of medical devices manufactured in the U.S. that are not approved
by the FDA for use in the U.S., or are banned or deviate from
lawful performance standards, are subject to FDA export
requirements. Exported devices are subject to the regulatory
requirements of each country to which the device is exported. Some
countries do not have medical device regulations, but in most
foreign countries, medical devices are regulated. Frequently,
regulatory approval may first be obtained in a foreign country
prior to application in the U.S. to take advantage of differing
regulatory requirements. Most countries outside of the U.S. require
that product approvals be recertified on a regular basis, generally
every five years. The recertification process requires that we
evaluate any device changes and any new regulations or standards
relevant to the device and conduct appropriate testing to document
continued compliance. Where recertification applications are
required, they must be approved in order to continue selling our
products in those countries.
13
In the
European Union, in order for our product candidates to be marketed
and sold, we are required to comply with the Medical Devices
Directive and obtain CE Mark certification. The CE Mark
certification encompasses an extensive review of our quality
management system which is inspected by a notified body’s
auditor as part of a Stage 1 and 2 International Organization for
Standardization, or ISO, 13485:2003 audit, in accordance with
worldwide recognized ISO standards and applicable European Medical
Devices Directives for quality management systems for medical
device manufacturers. Once the quality management system and design
dossier has been successfully audited by a notified body and
reviewed and approved by a competent authority, a CE certificate
for the medical device will be issued. We are also required to
comply with other foreign regulations such as the requirement that
we obtain Ministry of Health, Labor and Welfare approval before we
can launch new products in Japan. The time required to obtain these
foreign approvals to market our products may vary from U.S.
approvals, and requirements for these approvals may differ from
those required by the FDA.
Medical
device laws and regulations are in effect in many of the countries
in which we may do business outside the United States. These laws
and regulations range from comprehensive device approval
requirements for our medical device product to requests for product
data or certifications. The number and scope of these requirements
can be complex and could increase. We may not be able to obtain or
maintain regulatory approvals in such countries and we may be
required to incur significant costs in obtaining or maintaining our
foreign regulatory approvals. In addition, the export of certain of
our products which have not yet been cleared for domestic
commercial distribution may be subject to FDA export restrictions.
Any failure to obtain product approvals in a timely fashion or to
comply with state or foreign medical device laws and regulations
may have a serious adverse effect on our business, financial
condition or results of operations.
Intellectual Property
CRMD003 and CRMD004
On
January 30, 2008, we entered into a License and Assignment
Agreement, or the NDP License Agreement, with ND Partners, LLC, or
NDP. Pursuant to the NDP License Agreement, NDP granted us
exclusive, worldwide licenses for certain antimicrobial catheter
lock solutions, processes for treating and inhibiting infections, a
biocidal lock system and a taurolidine delivery apparatus, and the
corresponding United States and foreign patents and applications
(the “NDP Technology”). We acquired such licenses and
patents through our assignment and assumption of NDP’s rights
under certain separate license agreements by and between NDP and
Dr. Hans-Dietrich Polaschegg, Dr. Klaus Sodemann, and Dr. Johannes
Reinmueller. NDP also granted us exclusive licenses, with the right
to grant sublicenses, to use and display certain trademarks in
connection with the NDP Technology. As consideration in part for
the rights to the NDP Technology, we paid NDP an initial licensing
fee of $325,000 and granted NDP an equity interest in our company
consisting of 365,534 shares of common stock as of December 31,
2010. In addition, we are required to make payments to NDP upon the
achievement of certain regulatory and sales-based milestones.
Certain of the milestone payments are to be made in the form of
shares of common stock currently held in escrow for NDP, and other
milestone payments are to be paid in cash. The maximum aggregate
number of shares issuable upon achievement of milestones and the
number of shares held in escrow is 145,543 shares of common stock.
The maximum aggregate amount of cash payments upon achievement of
milestones is $3,000,000 with $2,500,000 remaining at December 31,
2016. Events that trigger milestone payments include but are not
limited to the reaching of various stages of regulatory approval
processes and certain worldwide net sales
amounts.
On
April 11, 2013, we entered into an amendment to the NDP License
Agreement. Under Article 6 of the NDP License Agreement, we were
obligated to make a milestone payment of $500,000 to NDP upon the
first issuance of a CE Mark for a licensed product, which payment
was payable to NDP within 30 days after such issuance. Pursuant to
the terms of the amendment, we and NDP agreed to delay such
milestone payment to a time, to be chosen by us, anytime within
twelve months after the achievement of such issuance. As
consideration for the amendment, we issued NDP a warrant to
purchase 125,000 shares of our common stock at an exercise price of
$1.50 per share. The warrant is exercisable immediately upon
issuance and has a term of five years. The warrant contains a
cashless exercise feature and standard adjustment features in the
event of a stock split, stock dividend, recapitalization or similar
events.
During the year
ended December 31, 2013, a milestone payment of $500,000 was earned
by NDP upon the first issuance of the CE Mark for Neutrolin, which
was converted in January 2014 into 50,000 Series C-3 non-voting
preferred stock and 250,000 warrants at an exercise price of $1.50
per share. During the year ended December 31, 2014, a certain
milestone was achieved resulting in the release of 36,386 shares
held in escrow. The number of shares held in escrow as of December
31, 2016 is 109,157 shares of common stock. There were no
milestones achieved in 2015 or 2016.
14
The NDP
License Agreement will expire on a country-by-country basis upon
the earlier of (i) the expiration of the last patent claim under
the NDP License Agreement in a given country, or (ii) the payment
of all milestone payments and release of all shares of our common
stock held in escrow under the NDP License Agreement. Upon the
expiration of the NDP License Agreement in each country, we will
have an irrevocable, perpetual, fully paid-up, royalty-free
exclusive license to the NDP Technology in such country. The NDP
License Agreement also may be terminated by NDP if we materially
breach or default under the NDP License Agreement and that breach
is not cured within 60 days following the delivery of written
notice to us, or by us on a country-by-country basis upon 60 days
prior written notice. If the NDP License Agreement is terminated by
either party, our rights to the NDP Technology will revert back to
NDP.
On
January 30, 2008, we also entered into an Exclusive License and
Consulting Agreement with Dr. Polaschegg. Pursuant to the
Polaschegg License Agreement, Dr. Polaschegg granted us an
exclusive, worldwide license for a gel lock invention and certain
taurolidine treatments and the corresponding United States patent
applications (the “Polaschegg Technology”). The
Polaschegg Technology served as a basis for CRMD004, which is the
gel formation of Neutrolin. As consideration for the rights to the
Polaschegg Technology, in addition to an initial fee of $5,000, we
agreed to pay Dr. Polaschegg certain royalty payments ranging from
1% to 3% of the net sales of the Polaschegg Technology. The
Polaschegg License Agreement also set forth certain minimum royalty
payments (on an annual basis) to be made to Dr. Polaschegg in
connection with the Polaschegg Technology, which payments range
from $10,000 to $45,000. Additional minimum royalty payments would
become payable to Dr. Polaschegg if he developed new intellectual
property that was applied to the Polaschegg Technology. As of
December 31, 2015, we recorded an aggregate of approximately
$300,000 in licensing and minimum royalty payments under the
Polaschegg License Agreement.
We had
the right to terminate the Polaschegg License Agreement with
respect to the gel lock invention or taurolidine treatments
(individually or together) upon 60 days notice. Dr. Polaschegg had
the right to terminate the Polaschegg License Agreement with
respect to the gel lock invention and/or taurolidine treatments if
no product based on the particular portion of Polaschegg Technology
has been made available to the market by the later of eight years
after (i) the date of the Polaschegg License Agreement, and (ii)
the priority date of any new patent. If the Polaschegg License
Agreement is terminated with respect to any piece of Polaschegg
Technology by either party, all rights with respect to such portion
of Polaschegg Technology will revert to Dr. Polaschegg. In November
2015, we terminated the Polaschegg License Agreement.
We
believe that the patents and patent applications we have licensed
pursuant to the NDP License Agreement cover effective solutions to
the various problems discussed previously when using taurolidine in
clinical applications, and specifically in hemodialysis
applications. We intend to file additional patent applications to
cover any additional related subject matter we develop. We
currently have five non-provisional patent applications, five
international (PCT) patent applications and four provisional patent
applications pending which cover additional applications using
taurolidine in, among others, sutures, hydrogels, meshes,
transdermal and biofilm products.
Employees
As of
March 10, 2017, we had fourteen full time employees, including our
customer service representative and office manager in Germany. We
also engage various consultants and contractors for project
management and research and development, manufacturing and
regulatory development, marketing, financing, sales and marketing
and administrative activities.
Corporate Information
We were
organized as a Delaware corporation on July 28, 2006 under the name
“Picton Holding Company, Inc.” and we changed our
corporate name to “CorMedix Inc.” on January 18, 2007.
Our principal executive offices are located at 1430 US Highway 206,
Suite 200, Bedminster, New Jersey 07921. Our telephone number is
(908) 517-9500.
We
maintain a website at www.cormedix.com; however, the information
on, or that can be accessed through, our website is not part of
this report. This report and all of our filings under the
Exchange Act, including copies of annual reports on Form 10-K,
quarterly reports on Form 10-Q, current reports on Form 8-K, and
any amendments to those reports, are available free of charge
through our website on the date we file those materials with, or
furnish them to, the Securities and Exchange Commission (the
"SEC"). Such filings are also available to the public on the
internet at the SEC's website at www.sec.gov. The public may also read
and copy any document that we file at the SEC's Public Reference
Room located at 100 F Street, NE, Washington, DC 20549 on official
business days during the hours of 10 a.m. to 3 p.m. For
further information on the Public Reference Room, the public
is instructed to call the SEC at 1-800-SEC-0300.
15
Risks Related to Our Financial Position and Need for Additional
Capital
We have a history of operating losses, expect to incur additional
operating losses in the future and may never be
profitable.
Our
prospects must be considered in light of the uncertainties, risks,
expenses, and difficulties frequently encountered by companies in
the early stages of operation. We incurred net losses of
approximately $24.6 million, $18.2 million and $20.5 million for
the years ended December 31, 2016, 2015 and 2014, respectively. As
of December 31, 2016, we had an accumulated deficit of
approximately $119.2 million. We expect to incur substantial
additional operating expenses over the next several years as our
research, development, pre-clinical testing, clinical trial and
commercialization activities increase as we develop and
commercialize Neutrolin. As a result, we expect to experience
negative cash flow as we fund our operating losses and capital
expenditures. The amount of future losses and when, if ever, we
will achieve profitability are uncertain. Neutrolin was launched in
December 2013 and is currently available for distribution in
certain European Union and Middle East countries. We have not
generated any significant commercial revenue and do not expect to
generate substantial revenues from Neutrolin until it is approved
by the FDA and launched in the U.S. market, and might never
generate significant revenues from the sale of Neutrolin or any
other products. Our ability to generate revenue and achieve
profitability will depend on, among other things, the following:
successfully marketing Neutrolin in Germany and other countries in
which it is approved for sale; obtaining necessary regulatory
approvals for Neutrolin from the other applicable European and
Middle East agencies, other foreign agencies and the FDA and
international regulatory agencies for any other products;
establishing manufacturing, sales, and marketing arrangements,
either alone or with third parties; and raising sufficient funds to
finance our activities. We might not succeed at any of these
undertakings. If we are unsuccessful at some or all of these
undertakings, our business, prospects, and results of operations
may be materially adversely affected.
We have received a going concern opinion from our registered public
accounting firm.
Our
operations are subject to a number of factors that can affect our
operating results and financial condition. Such factors include,
but are not limited to: the results of clinical testing and trial
activities of our product candidates; the ability to obtain
regulatory approval to market our products; ability to manufacture
successfully; competition from products manufactured and sold or
being developed by other companies; the price of, and demand for,
our products; our ability to negotiate favorable licensing or other
manufacturing and marketing agreements for our products; and our
ability to raise capital to support our operations.
To
date, our commercial operations have not generated sufficient
revenues to enable profitability. As of December 31, 2016, we had
an accumulated deficit of $119.2 million, and incurred net losses
from operations of $24.6 million for the year then ended. Based on
the current development plans for Neutrolin in both the U.S. and
foreign markets (including the ongoing hemodialysis Phase 3
clinical trial in the U.S.) and our other operating requirements,
management believes that the existing cash at December 31, 2016
will not be sufficient to fund operations for at least the next 12
months following the filing of this annual report on Form 10-K.
These factors raise substantial doubt regarding our ability to
continue as a going concern. Additionally, we will need additional
funding to complete the hemodialysis clinical trial in the U.S.
which commenced in December 2015 as well as to initiate the planned
Phase 3 clinical trial in oncology patients with
catheters.
As a
result of the above matters, our independent auditors have
indicated in their report on our December 31, 2016 financial
statements that there is substantial doubt about our ability to
continue as a going concern. A “going concern” opinion
indicates that the financial statements have been prepared assuming
we will continue as a going concern and do not include any
adjustments to reflect the possible future effects on the
recoverability and classification of assets, or the amounts and
classification of liabilities that may result if we do not continue
as a going concern. Therefore, you should not rely on our
consolidated balance sheet as an indication of the amount of
proceeds that would be available to satisfy claims of our
creditors, and potentially be available for distribution to our
stockholders, in the event of liquidation.
Our
continued operations will ultimately depend on our ability to raise
additional capital through various potential sources, such as
equity and/or debt financings, strategic relationships, or
out-licensing of our products in order to complete our ongoing and
planned Phase 3 clinical trials and until we achieve profitability,
if ever. We can provide no assurances that such financing or
strategic relationships will be available on acceptable terms, or
at all. Without this funding, we could be required to delay, scale
back or eliminate some or all of our research and development
programs which would likely have a material adverse effect on our
business.
We will need to finance our future cash needs through public or
private equity offerings, debt financings or corporate
collaboration and licensing arrangements. Any additional funds that
we obtain may not be on terms favorable to us or our stockholders
and may require us to relinquish valuable
rights.
We
have launched Neutrolin in certain European Union and Middle East
countries, but to date have no other approved product on the market
and have not generated significant product revenue from Neutrolin
to date. Unless and until we receive applicable regulatory approval
for Neutrolin in the U.S., we cannot sell Neutrolin in the U.S.
Therefore, for the foreseeable future, we will have to fund all of
our operations and capital expenditures from Neutrolin sales in
Europe and other foreign markets, if approved, cash on hand,
additional financings, licensing fees and
grants.
16
We believe that our cash resources as of December
31, 2016, will not be sufficient to enable us to fund our projected
operating requirements for at least the next 12 months following
the balance sheet date. We will need additional funding thereafter
to complete our ongoing Phase 3 hemodialysis clinical trial
in the U.S., as well initiate as our planned Phase 3 clinical trial
in the U.S. in oncology patients with catheters. If we are unable
to raise additional funds when needed, we may not be able to
complete our ongoing Phase 3 clinical trial, commence and complete
our planned Phase 3 clinical trial or commercialize Neutrolin and
we could be required to delay, scale back or eliminate some or all
of our research and development programs. We can provide no
assurances that any financing or strategic relationships will be
available to us on acceptable terms, or at all. We expect to incur
increases in our cash used in operations as we continue to
commercialize Neutrolin in Europe and other markets, conduct our
ongoing Phase 3 clinical trial and prepare for our planned Phase 3
clinical trial in oncology patients, seek FDA approval of Neutrolin
in the U.S., commercialize Neutrolin in Europe and other markets,
pursue business development activities, and incur additional legal
costs to defend our intellectual property.
To
raise needed capital, we may sell additional equity or debt
securities, obtain a bank credit facility, or enter into a
corporate collaboration or licensing arrangement. The sale of
additional equity or debt securities, if convertible, could result
in dilution to our stockholders. The incurrence of indebtedness
would result in fixed obligations and could also result in
covenants that would restrict our operations. Raising additional
funds through collaboration or licensing arrangements with third
parties may require us to relinquish valuable rights to our
technologies, future revenue streams, research programs or product
candidates, or to grant licenses on terms that may not be favorable
to us or our stockholders.
Our efforts to explore strategic alternatives aimed at accelerating
Neutrolin’s development and commercialization and maximizing
shareholder value may not result in any definitive transaction or
deliver the expected benefits, and may create a distraction for our
management and uncertainty that may adversely affect our operating
results and business.
Strategic alternatives we may pursue could
include, but are not limited to, joint ventures or partnering or
other collaboration agreements, licensing arrangements, or another
transaction intended to maximize shareholder value, such as a
merger, a sale of the Company or some or all of its assets, or
another strategic transaction. In March 2015, the Board
commenced a process to evaluate our strategic alternatives in order
to accelerate the global development of Neutrolin and maximize
shareholder value. The Board previously engaged the
investment bank Evercore Group L.L.C. to provide financial advice
and assist the Board with its evaluation process. After
the process with Evercore, we announced in July 2015 that we expect
to continue to pursue product development and commercialization
opportunities as we move forward with the planned Phase 3 clinical
trials, rather than pursuing a possible sale of our company at that
time. No transaction materialized
pursuant to the Evercore engagement. We
terminated the arrangement with Evercore in July 2016. Although we
will remain open to and consider strategic partnerships,
there can be no assurance that any
transaction will present itself.
There
are various uncertainties and risks relating to our evaluation and
negotiation of possible strategic alternatives and our ability to
consummate a definitive transaction, including:
●
expected benefits
may not be successfully achieved;
●
evaluation and
negotiation of a proposed transaction may distract management from
focusing our time and resources on execution of our operating plan,
which could have a material adverse effect on our operating results
and business;
●
the process
of evaluating proposed transactions may be time consuming and
expensive and may result in the loss of business
opportunities;
●
perceived
uncertainties as to our future direction may result in increased
difficulties in retaining key employees and recruiting new
employees, particularly senior management;
●
even if our Board
of Directors negotiates a definitive agreement, successful
integration or execution of the strategic alternative will be
subject to additional risks;
●
the current market
price of our common stock may reflect a market assumption that a
transaction will occur, and during the period in which we are
considering a transaction, the market price of our common stock
could be highly volatile; and
●
a failure to
complete a transaction could result in a negative perception by
investors in the Company generally and could cause a decline in the
market price of our common stock, as well as lead to greater
volatility in the market price of our common stock, all of which
could adversely affect our ability to access the equity and
financial markets, as well as our ability to explore and enter into
different strategic alternatives.
17
Risks Related to the Development and Commercialization of Our
Product Candidates
Our only product is only approved in Europe and is still in
development in the U. S.
Neutrolin
currently and for at least the near future is our only current
product as well as product candidate. Neturolin has received CE
Mark approval in Europe, and we launched it in Germany in December
2013. We also are pursuing development of Neutrolin in the U.S. Our
product commercialization and development efforts may not lead to
commercially viable products for any of several reasons. For
example, our product candidates may fail to be proven safe and
effective in clinical trials, or we may have inadequate financial
or other resources to pursue development efforts for our product
candidates. Even if approved, our products may not be accepted in
the marketplace. Neutrolin will require significant additional
development, clinical trials, regulatory clearances and/or
investment by us or our collaborators as we continue its
commercialization, as will any of our other products. Specifically,
we plan to expand marketing of Neutrolin in other foreign countries
and to develop Neutrolin for sale in the U.S., which will take time
and capital.
We have
entered into agreements with a Saudi Arabian company to market and
sell Neutrolin in Saudi Arabia, and with a South Korean company to
market, sell and distribute Neutrolin in South Korea upon receipt
of regulatory approval in that country. We also have some
commercial presence in Germany and the United Arab Emirates.
Consequently, we will be dependent on these companies and
individuals for the success of sales in those countries and any
other countries in which we receive regulatory approval and in
which we contract with third parties for the marketing, sale and/or
distribution of Neutrolin. If these companies or individuals do not
perform for whatever reason, our business, prospects and results of
operations will me materially adversely affected. Finding a
suitable replacement organization or individual for these or any
other companies or individuals with whom we might contract could be
difficult, which would further harm our business, prospects and
results of operations.
Successful development and commercialization of our products is
uncertain.
Our development and commercialization of current and future product
candidates is subject to the risks of failure and delay inherent in
the development of new pharmaceutical products, including but not
limited to the following:
●
inability
to produce positive data in pre-clinical and clinical
trials;
●
delays
in product development, pre-clinical and clinical testing, or
manufacturing;
●
unplanned
expenditures in product development, clinical testing, or
manufacturing;
●
failure
to receive regulatory approvals;
●
emergence
of superior or equivalent products;
●
inability
to manufacture our product candidates on a commercial scale on our
own, or in collaboration with third parties; and
●
failure
to achieve market acceptance.
Because
of these risks, our development efforts may not result in any
commercially viable products. If a significant portion of these
development efforts are not successfully completed, required
regulatory approvals are not obtained or any approved products are
not commercialized successfully, our business, financial condition,
and results of operations will be materially harmed.
Clinical trials required for our product candidates are expensive
and time-consuming, and their outcome is uncertain.
In order to obtain FDA or foreign approval to market a new drug or
device product, we must demonstrate proof of safety and
effectiveness in humans. Foreign regulations and requirements are
similar to those of the FDA. To meet FDA requirements, we must
conduct “adequate and well-controlled” clinical trials.
Conducting clinical trials is a lengthy, time-consuming, and
expensive process. The length of time may vary substantially
according to the type, complexity, novelty, and intended use of the
product candidate, and often can be several years or more per
trial. Delays associated with products for which we are directly
conducting clinical trials may cause us to incur additional
operating expenses. The commencement and rate of completion of
clinical trials may be delayed by many factors, including, for
example:
●
inability
to manufacture sufficient quantities of qualified materials under
the FDA’s cGMP requirements for use in clinical
trials;
●
slower
than expected rates of patient recruitment;
●
failure
to recruit a sufficient number of patients;
●
modification
of clinical trial protocols;
●
changes
in regulatory requirements for clinical trials;
18
●
lack
of effectiveness during clinical trials;
●
emergence
of unforeseen safety issues;
●
delays,
suspension, or termination of clinical trials due to the
institutional review board responsible for overseeing the study at
a particular study site; and
●
government
or regulatory delays or “clinical holds” requiring
suspension or termination of the trials.
The
results from early pre-clinical and clinical trials are not
necessarily predictive of results to be obtained in later clinical
trials. Accordingly, even if we obtain positive results from early
pre-clinical or clinical trials, we may not achieve the same
success in later clinical trials.
Our
clinical trials may be conducted in patients with serious or
life-threatening diseases for whom conventional treatments have
been unsuccessful or for whom no conventional treatment exists, and
in some cases, our product is expected to be used in combination
with approved therapies that themselves have significant adverse
event profiles. During the course of treatment, these patients
could suffer adverse medical events or die for reasons that may or
may not be related to our products. We cannot ensure that safety
issues will not arise with respect to our products in clinical
development.
Clinical
trials may not demonstrate statistically significant safety and
effectiveness to obtain the requisite regulatory approvals for
product candidates. The failure of clinical trials to demonstrate
safety and effectiveness for the desired indications could harm the
development of our product candidates. Such a failure could cause
us to abandon a product candidate and could delay development of
other product candidates. Any delay in, or termination of, our
clinical trials would delay the filing of any NDA or any Premarket
Approval Application, or PMA, with the FDA and, ultimately, our
ability to commercialize our product candidates and generate
product revenues. Any change in, or termination of, our clinical
trials could materially harm our business, financial condition, and
results of operations.
If we fail to comply with international regulatory
requirements we could be subject to regulatory delays,
fines or other penalties.
Regulatory requirements in foreign countries for international
sales of medical devices often vary from country to country. The
occurrence and related impact of the following factors would harm
our business:
●
delays
in receipt of, or failure to receive, foreign regulatory approvals
or clearances;
●
the
loss of previously obtained approvals or clearances;
or
●
the
failure to comply with existing or future regulatory
requirements.
The
CE Mark is a mandatory conformity mark for products to be sold in
the European Economic Area. Currently, 28 countries in Europe
require products to bear CE Marking. To market in Europe,
a product must first obtain the certifications necessary to
affix the CE Mark. The CE Mark is an international symbol of
adherence to the Medical Device Directives and the
manufacturer’s declaration that the product complies with
essential requirements. Compliance with these requirements is
ascertained within a certified Quality Management System (QMS)
pursuant to ISO 13485. In order to obtain and to maintain a CE
Mark, a product must be in compliance with the applicable
quality assurance provisions of the aforementioned ISO and obtain
certification of its quality assurance systems by a recognized
European Union notified body. We received CE Mark approval for
Neutrolin on July 5, 2013. However, certain individual countries
within the European Union require further approval by their
national regulatory agencies. Failure to receive or
maintain these other requisite approvals could prohibit us
from marketing and selling Neutrolin in the entire
European Economic Area or elsewhere.
We do not have, and may never obtain, the regulatory approvals we
need to market our product candidates outside of the European
Union.
While
we have received the CE Mark approval for Neutrolin in Europe,
certain individual countries within the European Union require
further approval by their national regulatory
agencies. Failure to receive or maintain these other
requisite approvals could prohibit us from marketing and
selling Neutrolin in the entire European Economic Area. In
addition, we will need regulatory approval to market and sell
Neutrolin in foreign countries outside of Europe. We have received
regulatory approval in Saudi Arabia and Kuwait.
19
In
the United States, we have no current application for, and have not
received the regulatory approvals required for, the commercial sale
of any of our products. None of our product candidates has been
determined to be safe and effective in the United States, and we
have not submitted an NDA or PMA to the FDA for any product. We
have received approval from the FDA to proceed with our ongoing
Phase 3 clinical trial for Neutrolin in hemodialysis catheters and
our planned Phase 3 trial in oncology patients with catheters,
which is subject to finalization of the protocol with the FDA. In
December 2015, we initiated the Phase 3 trial in hemodialysis
catheters; however, we will not initiate the Phase 3 trial in
oncology patients with catheters until we receive sufficient
funding. We are seeking one or more strategic partners or other
sources of capital to complete the Phase 3 trial in hemodialysis
and to start the Phase 3 trial for oncology patients with
catheters. However, we might not obtain any commercial partner or
financing and may never start the Phase 3 clinical trial for
oncology patients with catheters.
It
is possible that Neutrolin will not receive any further approval or
that any of our other product candidates will be approved for
marketing. Failure to obtain regulatory approvals, or delays in
obtaining regulatory approvals, would adversely affect the
successful commercialization of Neutrolin or any other drugs or
products that we or our partners develop, impose additional costs
on us or our collaborators, diminish any competitive advantages
that we or our partners may attain, and/or adversely affect our
cash flow.
Even if approved, our products will be subject to extensive
post-approval regulation.
Once
a product is approved, numerous post-approval requirements apply in
the United States and abroad. Depending on the circumstances,
failure to meet these post-approval requirements can result in
criminal prosecution, fines, injunctions, recall or seizure of
products, total or partial suspension of production, denial or
withdrawal of pre-marketing product approvals, or refusal to allow
us to enter into supply contracts, including government contracts.
In addition, even if we comply with FDA, foreign and other
requirements, new information regarding the safety or effectiveness
of a product could lead the FDA or a foreign regulatory body to
modify or withdraw product approval.
The successful commercialization of Neutrolin will depend on
obtaining coverage and reimbursement for use of Neutrolin from
third-party payors.
Sales
of pharmaceutical products largely depend on the reimbursement of
patients’ medical expenses by government health care programs
and/or private health insurers, both in the U.S. and abroad.
Further, significant uncertainty exists as to the reimbursement
status of newly approved health care products. We initially
expect to sell Neutrolin directly to hospitals and key dialysis
center operators, but also plan to expand its usage into intensive
care, oncology and total parenteral nutrition patients needing
catheters, including Medicare patients. All of these potential
customers are healthcare providers who depend upon reimbursement by
government and commercial insurance payors for dialysis and other
treatments. Reimbursement is strictly governed by these insurance
payors. We believe that Neutrolin would be eligible for coverage
under various reimbursement programs, including hospital inpatient
diagnosis-related groups (DRGs), outpatient ambulatory payment
classification (APCs) and the End-Stage Renal Disease Prospective
Payment System (ESRD PPS) or under the Durable Medical Equipment,
Prosthetics, Orthotics and Supplies (DMEPOS) Fee Schedule,
depending on the treatment setting. However, coverage by any of
these reimbursement programs is not assured, and even if coverage
is granted it could later be revoked or modified under future
regulations. Further, the U.S. Centers for Medicare & Medicaid
Services (CMS), which administers Medicare and works with states to
administer Medicaid, has adopted and will continue to adopt and/or
amend rules governing reimbursement for specific treatments,
including those we intend to address such as dialysis and ESRD PPS.
We anticipate that CMS and private insurers will increasingly
demand that manufacturers demonstrate the cost effectiveness of
their products as part of the reimbursement review and approval
process. Rising healthcare costs have also lead many European and
other foreign countries to adopt healthcare reform proposals and
medical cost containment measures. Any measures affecting the
reimbursement programs of these governmental and private insurance
payors, including any uncertainty in the medical community
regarding their nature and effect on reimbursement programs, could
have an adverse effect on purchasing decisions regarding Neutrolin,
as well as limit the prices we may charge for Neutrolin.
The failure to obtain or maintain
reimbursement coverage for Neutrolin or any other products could
materially harm our operations.
In
anticipation that the U.S. Centers for Medicare & Medicaid
Services (CMS), and private payers will demand that we demonstrate
the cost effectiveness of Neutrolin as part of the reimbursement
review and approval process, we have incorporated health economic
evaluations into our ongoing clinical studies to support this
review in the context of the prospective use of Neutrolin in
dialysis, the ICU and oncology settings. However, our studies
might not be sufficient to support coverage or reimbursement at
levels that allow providers to use Neutrolin.
20
Physicians and patients may not accept and use our
products.
Even with the CE Mark approval of Neutrolin, and even if we receive
FDA or other foreign regulatory approval for Neutrolin or other
product candidates, physicians and patients may not accept and use
our products. Acceptance and use of our products will depend upon a
number of factors including the following:
●
perceptions
by members of the health care community, including physicians,
about the safety and effectiveness of our drug or device
product;
●
cost-effectiveness
of our product relative to competing products;
●
availability
of reimbursement for our product from government or other
healthcare payors; and
●
effectiveness
of marketing and distribution efforts by us and our licensees and
distributors, if any.
Because
we expect sales of Neutrolin to generate substantially all of our
product revenues for the foreseeable future, the failure of
Neutrolin to find market acceptance would harm our business and
would require us to seek additional
financing.
Risks Related to Our Business and Industry
We might not be able to access any of the $40.0 million available
to us under our new at-the-market common stock sales
program.
In
August 2016, we entered into a sales agreement with the FBR Capital
Markets & Co. whereby we can sell up to $40 million of shares
of our common stock. However, pursuant to a Consent and Exchange
Agreement between us and Manchester, dated September 15, 2014 (and
entered into as part of our removal of anti-dilution, price reset
and change of control provisions in various securities that had
caused those securities to be classified as derivative
liabilities), (as amended in April 2015), Manchester has a right of
participation in equity financings undertaken by us prior to
September 15, 2017. The participation right extends to the
at-the-market common stock sales program. Any common stock sold as
part of our new at-the-market common stock sales program would be
subject to Manchester’s participation right,
Manchester’s waiver of its participation right or another
arrangement with Manchester. In the event we are not able to obtain
such a waiver or reach an agreement with Manchester, we would not
be able to sell shares of common stock in our new at-the-market
program, which could have a material adverse effect on our
financial condition and operations.
Competition and technological change may make our product
candidates and technologies less attractive or
obsolete.
We compete with established pharmaceutical and medical device
companies that are pursuing other forms of prevention or treatment
for the same indications we are pursuing and that have greater
financial and other resources. Other companies may succeed in
developing products earlier than we do, obtaining FDA or any other
regulatory agency approval for products more rapidly, or developing
products that are more effective than our product candidates.
Research and development by others may render our technology or
product candidates obsolete or noncompetitive, or result in
processes, treatments or cures superior to any therapy we develop.
We face competition from companies that internally develop
competing technology or acquire competing technology from
universities and other research institutions. As these companies
develop their technologies, they may develop competitive positions
that may prevent, make futile, or limit our product
commercialization efforts, which would result in a decrease in the
revenue we would be able to derive from the sale of any
products.
There
can be no assurance that Neutrolin or any other product candidate
will be accepted by the marketplace as readily as these or other
competing treatments. Furthermore, if our competitors’
products are approved before ours, it could be more difficult for
us to obtain approval from the FDA or any other regulatory agency.
Even if our products are successfully developed and approved for
use by all governing regulatory bodies, there can be no assurance
that physicians and patients will accept any of our products as a
treatment of choice.
Furthermore,
the pharmaceutical and medical device industry is diverse, complex,
and rapidly changing. By its nature, the business risks associated
with the industry are numerous and significant. The effects of
competition, intellectual property disputes, market acceptance, and
FDA or other regulatory agency regulations preclude us from
forecasting revenues or income with certainty or even
confidence.
We face the risk of product liability claims and the amount of
insurance coverage we hold now or in the future may not be adequate
to cover all liabilities we might incur.
Our business exposes us to the risk of product liability claims
that are inherent in the development of drugs. If the use of one or
more of our or our collaborators’ drugs or devices harms
people, we may be subject to costly and damaging product liability
claims brought against us by clinical trial participants,
consumers, health care providers, pharmaceutical companies or
others selling our products.
21
We
currently carry product liability insurance that covers our
clinical trials. We cannot predict all of the possible harms or
side effects that may result and, therefore, the amount of
insurance coverage we hold may not be adequate to cover all
liabilities we might incur. Our insurance covers bodily injury and
property damage arising from our clinical trials, subject to
industry-standard terms, conditions and exclusions. This coverage
includes the sale of commercial products. We have expanded our
insurance coverage to include the sale of commercial products due
to the receipt of the CE Mark approval, but we may be unable to
maintain such coverage or obtain commercially reasonable product
liability insurance for any other products approved for
marketing.
If
we are unable to obtain insurance at an acceptable cost or
otherwise protect against potential product liability claims, we
may be exposed to significant liabilities, which may materially and
adversely affect our business and financial position. If we are
sued for any injury allegedly caused by our or our
collaborators’ products and do not have sufficient insurance
coverage, our liability could exceed our total assets and our
ability to pay the liability. A successful product liability claim
or series of claims brought against us would decrease our cash and
could cause the value of our capital stock to
decrease.
We may be exposed to liability claims associated with the use of
hazardous materials and chemicals.
Our research, development and manufacturing activities and/or those
of our third-party contractors may involve the controlled use of
hazardous materials and chemicals. Although we believe that our
safety procedures for using, storing, handling and disposing of
these materials comply with federal, state and local, as well as
foreign, laws and regulations, we cannot completely eliminate the
risk of accidental injury or contamination from these materials. In
the event of such an accident, we could be held liable for any
resulting damages and any liability could materially adversely
affect our business, financial condition and results of operations.
In addition, the federal, state and local, as well as foreign, laws
and regulations governing the use, manufacture, storage, handling
and disposal of hazardous or radioactive materials and waste
products may require us to incur substantial compliance costs that
could materially adversely affect our business, financial condition
and results of operations.
Healthcare policy changes, including reimbursement policies for
drugs and medical devices, may have an adverse effect on our
business, financial condition and results of
operations.
Market
acceptance and sales of Neutrolin or any other product candidates
that we develop will depend on reimbursement policies and may be
affected by health care reform measures in the United States and
abroad. Government authorities and other third-party payors, such
as private health insurers and health maintenance organizations,
decide which drugs they will pay for and establish reimbursement
levels. We cannot be sure that reimbursement will be available for
Neutrolin or any other product candidates that we develop. Also, we
cannot be sure that the amount of reimbursement available, if any,
will not reduce the demand for, or the price of, our products. If
reimbursement is not available or is available only at limited
levels, we may not be able to successfully commercialize Neutrolin
or any other product candidates that we develop.
In
the United States, there have been a number of legislative and
regulatory proposals to change the health care system in ways that
could affect our ability to sell our products profitably. The
Patient Protection and Affordable Care Act, as amended by the
Health Care and Education Reconciliation Act of 2010, or
collectively, the Healthcare Reform Act, substantially changes the
way healthcare is financed by both governmental and private
insurers, and significantly impacts the pharmaceutical industry.
The Healthcare Reform Act contains a number of provisions,
including those governing enrollment in federal healthcare
programs, reimbursement changes and fraud and abuse, which will
impact existing government healthcare programs and will result in
the development of new programs, including Medicare payment for
performance initiatives and improvements to the physician quality
reporting system and feedback program. We anticipate that if we
obtain approval for our products, some of our revenue may be
derived from U.S. government healthcare programs, including
Medicare. Furthermore, beginning in 2011, the Healthcare Reform Act
imposed a non-deductible excise tax on pharmaceutical manufacturers
or importers who sell “branded prescription drugs,”
which includes innovator drugs and biologics (excluding orphan
drugs or generics) to U.S. government programs. We expect that the
Healthcare Reform Act and other healthcare reform measures that may
be adopted in the future could have an adverse effect on our
industry generally and our products specifically.
In
addition to the Healthcare Reform Act, we expect that there will
continue to be proposals by legislators at both the federal and
state levels, regulators and third-party payors to keep healthcare
costs down while expanding individual healthcare benefits. Certain
of these changes could impose limitations on the prices we will be
able to charge for any products that are approved or the amounts of
reimbursement available for these products from governmental
agencies or other third-party payors or may increase the tax
requirements for life sciences companies such as ours. While it is
too early to predict what effect the Healthcare Reform Act or any
future legislation or regulation will have on us, such laws could
have an adverse effect on our business, financial condition and
results of operations.
22
Health
administration authorities in countries other than the United
States may not provide reimbursement for Neutrolin or any of our
other product candidates at rates sufficient for us to achieve
profitability, or at all. Like the United States, these countries
could adopt health care reform proposals and could materially alter
their government-sponsored health care programs by reducing
reimbursement rates.
Any
reduction in reimbursement rates under Medicare or private insurers
or foreign health care programs could negatively affect the pricing
of our products. If we are not able to charge a sufficient amount
for our products, then our margins and our profitability will be
adversely affected.
If we lose key management or scientific personnel, cannot recruit
qualified employees, directors, officers, or other personnel or
experience increases in compensation costs, our business may
materially suffer.
We are highly dependent on the principal members of our management
and scientific staff, specifically, Khoso Baluch, a director and
our Chief Executive Officer, Robert Cook, our Chief Financial
Officer, Judith Abrams, our Chief Medical Officer and John
Armstrong, our Executive Vice President for Technical Operations.
Our future success will depend in part on our ability to identify,
hire, and retain current and additional personnel. We experience
intense competition for qualified personnel and may be unable to
attract and retain the personnel necessary for the development of
our business. Moreover, our work force is located in the New Jersey
metropolitan area, where competition for personnel with the
scientific and technical skills that we seek is extremely high and
is likely to remain high. Because of this competition, our
compensation costs may increase significantly. In addition, we have
only limited ability to prevent former employees from competing
with us.
If we are unable to hire additional qualified personnel, our
ability to grow our business may be harmed.
Over time, we expect to hire additional qualified personnel with
expertise in clinical testing, clinical research and testing,
government regulation, formulation and manufacturing, and sales and
marketing. We compete for qualified individuals with numerous
pharmaceutical companies, universities and other research
institutions. Competition for such individuals is intense, and we
cannot be certain that our search for such personnel will be
successful. Attracting and retaining such qualified personnel will
be critical to our success.
We may not successfully manage our growth.
Our success will depend upon the expansion of our operations to
commercialize Neutrolin and the effective management of any growth,
which could place a significant strain on our management and our
administrative, operational and financial resources. To manage this
growth, we may need to expand our facilities, augment our
operational, financial and management systems and hire and train
additional qualified personnel. If we are unable to manage our
growth effectively, our business may be materially
harmed.
Risks Related to Our Intellectual Property
If we materially breach or default under any of our license
agreements, the licensor party to such agreement will have the
right to terminate the license agreement, which termination may
materially harm our business.
Our commercial success will depend in part on the maintenance of
our license agreements. Each of our license agreements provides the
licensor with a right to terminate the license agreement for our
material breach or default under the agreement, including the
failure to make any required milestone or other payments. Should
the licensor under any of our license agreements exercise such a
termination right, we would lose our right to the intellectual
property under the respective license agreement, which loss may
materially harm our business.
If we and our licensors do not obtain protection for and
successfully defend our respective intellectual property rights,
our competitors may be able to take advantage of our research and
development efforts to develop competing
products.
Our
commercial success will depend in part on obtaining further patent
protection for our products and other technologies and successfully
defending any patents that we currently have or will obtain against
third-party challenges. The patents which we currently believe are
most material to our business are as follows:
23
●
U.S.
Patent No. 8,541,393 (expiring in November 2024) (the “Prosl
Patent”) - use of Neutrolin for preventing infection and
maintenance of catheter patency in hemodialysis catheters (for
CRMD003);
●
U.S.
Patent No. 6,166,007 (expiring May 2019) (the “Sodemann
Patent”) - a method of inhibiting or preventing infection and
blood coagulation at a medical prosthetic device (for CRMD003);
and
●
European
Patent EP 1 814 562 B1 (expiring October 12, 2025) (the
“Prosl European Patent”) - a low heparin catheter lock
solution for maintaining and preventing infection in a hemodialysis
catheter.
We
are currently seeking further patent protection for our compounds
and methods of treating diseases. However, the patent process is
subject to numerous risks and uncertainties, and there can be no
assurance that we will be successful in protecting our products by
obtaining and defending patents. These risks and uncertainties
include the following:
●
patents
that may be issued or licensed may be challenged, invalidated, or
circumvented, or otherwise may not provide any competitive
advantage;
●
our
competitors, many of which have substantially greater resources
than we have and many of which have made significant investments in
competing technologies, may seek, or may already have obtained,
patents that will limit, interfere with, or eliminate our ability
to make, use, and sell our potential products either in the United
States or in international markets;
●
there
may be significant pressure on the United States government and
other international governmental bodies to limit the scope of
patent protection both inside and outside the United States for
treatments that prove successful as a matter of public policy
regarding worldwide health concerns; and
●
countries
other than the United States may have less restrictive patent laws
than those upheld by United States courts, allowing foreign
competitors the ability to exploit these laws to create, develop,
and market competing products.
In
addition, the United States Patent and Trademark Office, or PTO,
and patent offices in other jurisdictions have often required that
patent applications concerning pharmaceutical and/or
biotechnology-related inventions be limited or narrowed
substantially to cover only the specific innovations exemplified in
the patent application, thereby limiting the scope of protection
against competitive challenges. Thus, even if we or our licensors
are able to obtain patents, the patents may be substantially
narrower than anticipated.
The
above mentioned patents and patent applications are exclusively
licensed to us. To support our patent strategy, we have engaged in
a review of patentability and certain freedom to operate issues,
including performing certain searches. However, patentability and
certain freedom to operate issues are inherently complex, and we
cannot provide assurances that a relevant patent office and/or
relevant court will agree with our conclusions regarding
patentability issues or with our conclusions regarding freedom to
operate issues, which can involve subtle issues of claim
interpretation and/or claim liability. Furthermore, we may not be
aware of all patents, published applications or published
literature that may affect our business either by blocking our
ability to commercialize our product candidates, preventing the
patentability of our product candidates to us or our licensors, or
covering the same or similar technologies that may invalidate our
patents, limit the scope of our future patent claims or adversely
affect our ability to market our product
candidates.
In
addition to patents, we also rely on trade secrets and proprietary
know-how. Although we take measures to protect this information by
entering into confidentiality and inventions agreements with our
employees, scientific advisors, consultants, and collaborators, we
cannot provide any assurances that these agreements will not be
breached, that we will be able to protect ourselves from the
harmful effects of disclosure if they are breached, or that our
trade secrets will not otherwise become known or be independently
discovered by competitors. If any of these events occurs, or we
otherwise lose protection for our trade secrets or proprietary
know-how, the value of our intellectual property may be greatly
reduced.
Ongoing and future intellectual property disputes could require us
to spend time and money to address such disputes and could limit
our intellectual property rights.
The
biotechnology and pharmaceutical industries have been characterized
by extensive litigation regarding patents and other intellectual
property rights, and companies have employed intellectual property
litigation to gain a competitive advantage. We may initiate or
become subject to infringement claims or litigation arising out of
patents and pending applications of our competitors, or we may
become subject to proceedings initiated by our competitors or other
third parties or the PTO or applicable foreign bodies to reexamine
the patentability of our licensed or owned patents. In addition,
litigation may be necessary to enforce our issued patents, to
protect our trade secrets and know-how, or to determine the
enforceability, scope, and validity of the proprietary rights of
others.
24
We
initiated court proceedings in Germany for patent infringement and
unfair use of our proprietary information related to Neutrolin (as
described below). We also have had opposition proceedings brought
against the European Patent and the German utility model patent
which are the basis of our infringement proceedings (as described
below). The defense and prosecution of these ongoing and any future
intellectual property suits, PTO or foreign proceedings, and
related legal and administrative proceedings are costly and
time-consuming to pursue, and their outcome is uncertain. An
adverse determination in litigation or PTO or foreign proceedings
to which we may become a party could subject us to significant
liabilities, including damages, require us to obtain licenses from
third parties, restrict or prevent us from selling our products in
certain markets, or invalidate or render unenforceable our licensed
or owned patents. Although patent and intellectual property
disputes might be settled through licensing or similar
arrangements, the costs associated with such arrangements may be
substantial and could include our paying large fixed payments and
ongoing royalties. Furthermore, the necessary licenses may not be
available on satisfactory terms or at all.
In
February 2007, Geistlich Söhne AG für Chemische
Industrie, Switzerland (“Geistlich”) brought an action
against the European Sodemann Patent covering our Neutrolin product
candidate, which is owned by ND Partners, LLC (“NDP”)
and licensed to us pursuant to the License and Assignment Agreement
between us and NDP. This action was brought at the Board of the
European Patent Office (“EPO”) opposition division (the
“Opposition Board”) based upon alleged lack of
inventiveness in the use of citric acid and a pH value in the range
of 4.5 to 6.5 with having the aim to provide an alternative lock
solution through having improved anticoagulant characteristics
compared to the lock solutions of the prior art. The Opposition
Board rejected the opposition by Geistlich. In a letter dated
September 30, 2013, we were notified that the opposition division
of the EPO reopened the proceedings before the first instance and
gave their preliminary non-binding opinion that the patent as
amended during the appeal proceedings fulfills the requirements of
clarity, novelty, and inventive step, and invited the parties to
provide their comments and/or requests by February 10,
2014. We filed our response on February 3, 2014 to
request that the patent be maintained as amended during the appeal
proceedings. Geistlich did not file a further statement
within the required timeline. On November 5, 2014, the
Opposition Division at the EPO issued the interlocutory decision to
maintain the patent on the basis of the claims as amended during
the appeal proceedings. This decision became final as no further
appeal was lodged by Geistlich.
On
September 9, 2014, we filed in the District Court of Mannheim,
Germany a patent infringement action against TauroPharm GmbH and
Tauro-Implant GmbH as well as their respective CEOs (the
“Defendants”) claiming infringement of our European
Patent EP 1 814 562 B1, which was granted by the EPO on January 8,
2014 (the “Prosl European Patent”). The Prosl European
Patent covers a low dose heparin catheter lock solution for
maintaining patency and preventing infection in a hemodialysis
catheter. In this action, we claim that the Defendants
infringe on the Prosl European Patent by manufacturing and
distributing catheter locking solutions to the extent they are
covered by the claims of the Prosl European Patent. We
believe that our patent is sound, and are seeking injunctive relief
and raising claims for information, rendering of accounts, calling
back, destruction and damages. Separately, TauroPharm has filed an
opposition with the EPO against the Prosl European Patent alleging
that it lacks novelty and inventive step. We cannot
predict what other defenses the Defendants may raise, or the
ultimate outcome of either of these related matters.
In the
same complaint against the same Defendants, we also alleged an
infringement (requesting the same remedies) of NDP’s utility
model DE 20 2005 022 124 U1 (the “Utility Model”),
which we believe is fundamentally identical to the Prosl European
Patent in its main aspects and claims. The Court separated the two
proceedings and the Prosl European Patent and the Utility Model
claims are now being tried separately. TauroPharm has
filed a cancellation action against the Utility Model before the
German Patent and Trademark Office (the “German PTO”)
based on the similar arguments as those in the opposition against
the Prosl European Patent.
On
March 27, 2015, the District Court held a hearing to evaluate
whether the Utility Model has been infringed by TauroPharm in
connection with the manufacture, sale and distribution of its
TauroLock-HEP100TM and TauroLock-HEP500TM products. A hearing
before the same court was held on January 30, 2015 on the separate,
but related, question of infringement of the Prosl European Patent
by TauroPharm.
The
Court issued its decisions on May 8, 2015 staying both
proceedings. In its decisions, the Court found that the
commercialization by TauroPharm in Germany of its TauroLock
catheter lock solutions Hep100 and Hep500 infringes both the
Prosl European Patent and the Utility Model and further that there
is no prior use right that would allow TauroPharm to continue to
make, use or sell its product in Germany. However, the Court
declined to issue an injunction in favor of us that would preclude
the continued commercialization by TauroPharm based upon its
finding that there is a sufficient likelihood that the EPO, in the
case of the Prosl European Patent, or the German PTO, in the case
of the Utility Model, may find that such patent or utility model is
invalid. Specifically, the Court noted the
possible publication of certain instructions for product use
that may be deemed to constitute prior art. As such, the District
Court determined that it will defer any consideration of the
request by us for injunctive and other relief until such time as
the EPO or the German PTO has ruled on the underlying validity of
the Prosl European Patent and the Utility Model.
25
The
opposition proceeding against the Prosl European Patent before the
EPO is ongoing. In its preliminary consideration of the matter, the
EPO (and the German PTO) regarded the patent as not inventive or
novel due to publication of prior art. Oral proceedings
before the Opposition Division at the EPO were held on November 25,
2015, at which the three judge patent examiner panel considered
arguments related to the validity of the Prosl European Patent. The
hearing was adjourned due to the fact that the panel was of the
view that Claus Herdeis, one of the managing directors of
TauroPharm, has to be heard as a witness in a further hearing in
order to close some gaps in the documentation presented by
TauroPharm as regards the publication of prior art. In October
2016, TauroPharm submitted a further writ to the EPO requesting a
date for the hearing and bringing forward further arguments, in
particular in view of the recent decision of the German PTO on the
invalidity of the utility model. The EPO has scheduled a further
oral hearing for November 22 / 23, 2017. While we continue to
believe that the referenced publication and instructions for use do
not, in fact, constitute prior art and that the Prosl European
Patent will be found to be valid by the EPO, there can be no
assurance that we will prevail in this matter.
The
German PTO held a hearing in the validity proceedings relating to
the Utility Model on June 29, 2016, at which the panel affirmed its
preliminary finding that the Utility Model was invalid based upon
prior publication of a reference to the benefits that may be
associated with adding heparin to a taurolidine based solution. The
decision is subject to appeal and has only a declaratory effect, as
the Utility Model had expired in November 2015. Furthermore, it has
no bearing on the ongoing consideration of the validity and
possible infringement of the Prosl Patent by the EPO. We filed an
appeal against the ruling on September 7, 2016.
On
January 16, 2015, we filed a complaint against TauroPharm GmbH and
its managing directors in the District Court of Cologne,
Germany. In the complaint, we allege violation of the
German Unfair Competition Act by TauroPharm for the unauthorized
use of its proprietary information obtained in confidence by
TauroPharm. We allege that TauroPharm is improperly and
unfairly using its proprietary information relating to the
composition and manufacture of Neutrolin, in the manufacture and
sale of TauroPharm’s products TauroLockTM, TauroLock-HEP100
and TauroLock-HEP500. We seek a cease and desist order
against TauroPharm from continuing to manufacture and sell any
product containing taurolidine (the active pharmaceutical
ingredient (“API”) of Neutrolin) and citric acid in
addition to possible other components, damages for any sales in the
past and the removal of all such products from the market. An
initial hearing in the District Court of Cologne, Germany was held
on November 19, 2015 to consider our claims. The judge made no
decision on the merits of our complaint. On January 14, 2016,
the court issued an interim decision in the form of a court order
outlining several issues of concern that relate primarily to
court's interest in clarifying the facts and reviewing any and all
available documentation, in particular with regard to the question
which specific know-how was provided to TauroPharm by whom and
when. We have prepared the requested reply and produced the
respective documentation. TauroPharm has also filed another writ
within the same deadline and both parties have filed further writs
at the end of April setting out their respective argumentation in
more detail. A further oral hearing was held on November 15, 2016.
The court made no rulings from the bench, and indicated that it is
prepared to further examine the underlying facts of our
allegations. At this time, there are no further hearings scheduled.
We intend to continue to pursue this matter, and to provide
additional supplemental documentary and other evidence as may be
necessary to support our claims.
If we infringe the rights of third parties we could be prevented
from selling products and forced to pay damages and defend against
litigation.
If
our products, methods, processes and other technologies infringe
the proprietary rights of other parties, we could incur substantial
costs and we may have to do one or more of the
following:
●
obtain
licenses, which may not be available on commercially reasonable
terms, if at all;
●
abandon
an infringing product candidate;
●
redesign
our products or processes to avoid infringement;
●
stop
using the subject matter claimed in the patents held by
others;
●
pay
damages; or
●
defend
litigation or administrative proceedings, which may be costly
whether we win or lose, and which could result in a substantial
diversion of our financial and management resources.
26
Risks Related to Dependence on Third Parties
If we are not able to develop and maintain collaborative marketing
relationships with licensees or partners, or create an effective
sales, marketing, and distribution capability, we may be unable to
market our products or market them successfully.
Our
business strategy for Neutrolin relies on collaborating with larger
firms with experience in marketing and selling medical devices and
pharmaceutical products; for other products we may also rely on
such marketing collaborations or out-licensing of our product
candidates. Specifically, for Neutrolin, we have entered into an
agreement with a German company to market and sell Neutrolin in
Germany and a distributor agreement with each of a Saudi Arabian
and a South Korean company for sales and marketing in those two
countries (upon receipt of approval to market in South Korea). In
addition, we have an independent sales representative marketing and
selling in the Middle East. Assuming we receive applicable
regulatory approval for other markets, we plan to enter into
distribution agreements with one or more third parties for the sale
of Neutrolin in various European, Middle East and other markets.
However, there can be no assurance that we will be able to
successfully maintain those relationships or establish and maintain
additional marketing, sales, or distribution relationships. Nor can
there be assurance that such relationships will be successful, or
that we will be successful in gaining market acceptance for our
products. To the extent that we enter into any marketing, sales, or
distribution arrangements with third parties, our product revenues
will be lower than if we marketed and sold our products directly,
and any revenues we receive will depend upon the efforts of such
third-parties.
If
we are unable to establish and maintain such third-party sales and
marketing relationships, or choose not to do so, we will have to
establish our own in-house capabilities. We currently have no
sales, marketing, or distribution infrastructure. To market any of
our products directly, we would need to develop a marketing, sales,
and distribution force that has both technical expertise and the
ability to support a distribution capability. The establishment of
a marketing, sales, and distribution capability would take time and
significantly increase our costs, possibly requiring substantial
additional capital. In addition, there is intense competition for
proficient sales and marketing personnel, and we may not be able to
attract individuals who have the qualifications necessary to
market, sell, and distribute our products. There can be no
assurance that we will be able to establish internal marketing,
sales, or distribution capabilities. If we are unable to, or choose
not to establish these capabilities, or if the capabilities we
establish are not sufficient to meet our needs, we will be required
to establish collaborative marketing, sales, or distribution
relationships with third parties, which we might not be able to do
on acceptable terms or at all.
We currently have no internal marketing and sales organization and
currently rely and intend to continue to rely on third parties to
market and sell Neutrolin. If we are unable to enter into or
maintain agreements with third parties to market and sell Neutrolin
or any other product after approval or are unable to establish our
own marketing and sales capabilities, we may not be able to
generate significant or any product revenues.
We do
not have an internal sales organization. To date we have relied,
and intend to continue to rely, on third parties for the marketing,
sales and distribution of Neutrolin and any other product we might
develop. However, we may not be able to maintain current and future
arrangements or enter into new arrangements with third parties to
sell Neutrolin or any other product on favorable terms or at all.
In that event, we would have to develop our own marketing and sales
force. The establishment and development of our own sales force
would be expensive and time consuming and could delay any product
launch, and we cannot be certain that we would be able to
successfully develop this capability. In addition, the use of third
parties to commercialize our approved products reduces the revenues
that we would receive if we commercialized these products
ourselves.
We have
entered into agreements with independent companies to market
Neutrolin in Germany and in Saudi Arabia and, upon regulatory
approval, South Korea. We also have an independent sales
representative in the Middle East. We intend to seek a sales
partner in the U.S. if Neutrolin receives FDA approval.
Consequently, we will be dependent on these firms and individuals
for the success of sales in these and any other countries in which
approval is granted. If these firms or individuals do not perform
for whatever reason, our business, prospects and results of
operations will be materially adversely affected. Finding a new or
replacement organization for sales and marketing could be
difficult, which would further harm our business, prospects and
results of operations.
27
If we or our collaborators are unable to manufacture our products
in sufficient quantities or are unable to obtain regulatory
approvals for a manufacturing facility, we may be unable to meet
demand for our products and we may lose potential
revenues.
Completion
of our clinical trials and commercialization of Neutrolin and any
other product candidate require access to, or development of,
facilities to manufacture a sufficient supply of our product
candidates. All of our manufacturing processes currently are, and
we expect them to continue to be, outsourced to third parties.
Specifically, we will rely on one or more manufacturers to supply
us and/or our distribution partners with commercial quantities of
Neutrolin. If, for any reason, we become unable to rely on our
current sources for the manufacture of Neutrolin or any other
product candidates or for active pharmaceutical ingredient, or API,
either for clinical trials or for commercial quantities, then we
would need to identify and contract with additional or replacement
third-party manufacturers to manufacture compounds for
pre-clinical, clinical, and commercial purposes. We may not be
successful in identifying such additional or replacement
third-party manufacturers, or in negotiating acceptable terms with
any that we do identify. Such third-party manufacturers must
receive FDA or applicable foreign approval before they can produce
clinical material or commercial product, and any that are
identified may not receive such approval or may fail to maintain
such approval. In addition, we may be in competition with other
companies for access to these manufacturers’ facilities and
may be subject to delays in manufacturing if the manufacturers give
other clients higher priority than they give to us. If we are
unable to secure and maintain third-party manufacturing capacity,
the development and sales of our products and our financial
performance may be materially affected.
Before
we could begin to commercially manufacture Neutrolin or any other
product candidate on our own, we must obtain regulatory approval of
the manufacturing facility and process. The manufacture of drugs
for clinical and commercial purposes must comply with cGMP and
applicable non-U.S. regulatory requirements. The cGMP requirements
govern quality control and documentation policies and procedures.
Complying with cGMP and non-U.S. regulatory requirements would
require that we expend time, money, and effort in production,
recordkeeping, and quality control to assure that the product meets
applicable specifications and other requirements. We would also
have to pass a pre-approval inspection prior to FDA or non-U.S.
regulatory agency approval. Failure to pass a pre-approval
inspection may significantly delay regulatory approval of our
products. If we fail to comply with these requirements, we would be
subject to possible regulatory action and may be limited in the
jurisdictions in which we are permitted to sell our products. As a
result, our business, financial condition, and results of
operations could be materially adversely
affected.
Corporate
and academic collaborators may take actions that delay, prevent, or
undermine the success of our products.
Our
operating and financial strategy for the development, clinical
testing, manufacture, and commercialization of our product
candidates is heavily dependent on our entering into collaborations
with corporations, academic institutions, licensors, licensees, and
other parties. Our current strategy assumes that we will
successfully establish and maintain these collaborations or similar
relationships. However, there can be no assurance that we will be
successful establishing or maintaining such collaborations. Some of
our existing collaborations, such as our licensing agreements, are,
and future collaborations may be, terminable at the sole discretion
of the collaborator in certain circumstances. Replacement
collaborators might not be available on attractive terms, or at
all.
In addition, the activities of any collaborator will not be within
our control and may not be within our power to influence. There can
be no assurance that any collaborator will perform its obligations
to our satisfaction or at all, that we will derive any revenue or
profits from such collaborations, or that any collaborator will not
compete with us. If any collaboration is not pursued, we may
require substantially greater capital to undertake on our own the
development and marketing of our product candidates and may not be
able to develop and market such products successfully, if at all.
In addition, a lack of development and marketing collaborations may
lead to significant delays in introducing product candidates into
certain markets and/or reduced sales of products in such
markets.
Data provided by collaborators and others upon which we rely that
has not been independently verified could turn out to be false,
misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to
provide us with significant data and other information related to
our projects, clinical trials, and business. If such third parties
provide inaccurate, misleading, or incomplete data, our business,
prospects, and results of operations could be materially adversely
affected.
Risks Related to our Common Stock
Prior to fiscal 2015, we had identified a material weakness in our
internal control over financial reporting, and our current internal
control over financial reporting and our disclosure controls and
procedures may not prevent all possible errors that could
occur.
In the
several years prior to fiscal 2015, we had identified a material
weakness in our internal control over financial reporting that was
related to our limited finance staff and the resulting ineffective
management review over financial reporting, coupled with
increasingly complex accounting treatments associated with our
financing activities and European expansion. While we remediated
this material weakness in 2015, we cannot be assured that material
weaknesses will not arise again.
28
A
control system, no matter how well designed and operated, can
provide only reasonable, not absolute, assurance that the control
system’s objectives will be satisfied. Internal control over
financial reporting and disclosure controls and procedures are
designed to give a reasonable assurance that they are effective to
achieve their objectives. We cannot provide absolute assurance that
all of our possible future control issues will be detected. These
inherent limitations include the possibility that judgments in our
decision making can be faulty, and that isolated breakdowns can
occur because of simple human error or mistake. The design of our
system of controls is based in part upon assumptions about the
likelihood of future events, and there can be no assurance that any
design will succeed absolutely in achieving our stated goals under
all potential future or unforeseeable conditions. Because of the
inherent limitations in a cost effective control system,
misstatements due to error could occur and not be detected. This
and any future failures could cause investors to lose confidence in
our reported financial information, which could have a negative
impact on our financial condition and stock price.
Our common stock price has fluctuated considerably and is likely to
remain volatile, in part due to the limited market for our common
stock and you could lose all or a part of your
investment.
During the period from the completion of our initial public
offering, or IPO, on March 30, 2010 through February 28, 2017, the
high and low sales prices for our common stock were $10.40 and
$0.15, respectively. There is a limited public market for our
common stock and we cannot provide assurances that an active
trading market will develop. As a result of low trading volume in
our common stock, the purchase or sale of a relatively small number
of shares could result in significant share price
fluctuations.
Additionally,
the market price of our common stock may continue to fluctuate
significantly in response to a number of factors, some of which are
beyond our control, including the
following:
●
market
acceptance of Neutrolin in those markets in which it is approved
for sale;
●
our
need for additional capital;
●
the
receipt of or failure to obtain additional regulatory approvals for
Neutrolin, including FDA approval in the U.S.;
●
results
of clinical trials of our product candidates, including our planned
Phase 3 trial for Neutrolin in the U.S., or those of our
competitors;
●
our
entry into or the loss of a significant collaboration;
●
regulatory
or legal developments in the United States and other countries,
including changes in the healthcare payment systems;
●
changes
in financial estimates or investment recommendations by securities
analysts relating to our common stock;
●
announcements
by our competitors of significant developments, strategic
partnerships, joint ventures or capital commitments;
●
changes
in key personnel;
●
variations
in our financial results or those of companies that are perceived
to be similar to us;
●
market
conditions in the pharmaceutical and medical device sectors and
issuance of new or changed securities analysts’ reports or
recommendations;
●
general
economic, industry and market conditions;
●
developments
or disputes concerning patents or other proprietary
rights;
●
future
sales or anticipated sales of our securities by us or our
stockholders; and
●
any
other factors described in this “Risk Factors”
section.
In
addition, the stock markets in general, and the stock of
pharmaceutical and medical device companies in particular, have
experienced extreme price and volume fluctuations that have often
been unrelated or disproportionate to the operating performance of
these companies. Broad market and industry factors may negatively
affect the market price of our common stock, regardless of our
actual operating performance.
For
these reasons and others, an investment in our securities is risky
and invest only if you can withstand a significant loss and wide
fluctuations in the value of your investment.
29
A significant number of additional shares of our common stock may
be issued at a later date, and their sale could depress the market
price of our common stock.
As
of December 31, 2016, we had outstanding the following securities
that are convertible into or exercisable for shares of our common
stock:
●
warrants
for 227,273 shares of common stock issued in July 2013 with an
exercise price of $1.50 that expire on July 30, 2018;
●
warrants
for 500,000 shares of common stock issued in May 2013 with an
exercise price of $0.65 per share that expire on May 30,
2019;
●
warrants
for 125,000 shares issued to ND Partners in April 2013 in
connection with the amendment to the license and assignment
agreement with an exercise price of $1.50 per share that expire on
April 11, 2018;
●
options
to purchase an aggregate of 130,000 shares of our common stock
issued to our officers, directors, employees and non-employee
consultants under our Amended and Restated 2006 Stock Incentive
Plan, or the 2006 Stock Plan, with a weighted average exercise
price of $1.38 per share;
●
options
to purchase an aggregate of 4,479,755 shares of our common stock
issued to our officers, directors and non-employee consultants
under our 2013 Stock Plan, with a weighted average exercise price
of $2.31 per share;
●
warrants
issued to investors in our 2012 private placement to purchase an
aggregate of 312,500 shares of our common stock with an exercise
price of $0.40 per share, which expire on September 20,
2017;
●
a
warrant for 795 shares of our common stock issued to the placement
agent for our 2012 private placement with an exercise price of
$0.40 per share, which expires on September 20, 2017;
●
a
warrant to purchase 400,000 shares of our common stock issued on
February 19, 2013 with an exercise price of $1.50 that expire on
February 19, 2018;
●
warrants
for 750,000 shares of common stock with an exercise price of $0.90
that expire on October 22, 2019;
●
warrants
for 725,000 shares of common stock with an exercise price of $0.90
that expire on January 8, 2020;
●
Series C-2
Preferred Stock convertible into 1,500,000 shares of
common;
●
Series C-3
Preferred Stock convertible into 1,365,000 shares of common
stock;
●
Series D Preferred
Stock convertible into 1,479,240 shares of common
stock;
●
Series E Preferred
Stock convertible into 1,959,759 shares of common
stock;
●
warrants for
682,500 shares of common stock issued in March 2014 with an
exercise price of $2.50 per shares that expire on September 10,
2019;
●
warrants for
200,000 shares of common stock with an exercise price of $7.00 that
expire on March 3, 2020; and
●
warrants for 83,400
shares of common stock with an exercise price of $7.00 that expire
on March 25, 2020.
The
possibility of the issuance of these shares, as well as the actual
sale of such shares, could substantially reduce the market price
for our common stock and impede our ability to obtain future
financing.
We will need additional financing to fund our activities in the
future, which likely will dilute our stockholders.
To date, our commercial operations
have not generated sufficient revenues to enable profitability. As
of December 31, 2016, we had an accumulated deficit of $119.2
million, and incurred net losses from operations of $24.6 million
for the year then ended. Based on the current development plans for
Neutrolin in both the U.S. and foreign markets (including the
ongoing hemodialysis Phase 3 clinical trial in the U.S.) and our
other operating requirements, management believes that the existing
cash at December 31, 2016 will not be sufficient to fund operations
for at least the next 12 months following the balance sheet date.
Additionally, we will need additional funding to complete the
hemodialysis clinical trial in the U.S. which commenced in December
2015 as well as to initiate the planned Phase 3 clinical trial in
oncology patients with catheters. Further, we anticipate that we will incur
operating losses for the foreseeable future. Additionally, we will
require substantial funds in the future to support our operations.
We expect to seek equity or debt financings in the future to fund
our operations. The issuance of additional equity securities, or
convertible debt or other derivative securities, likely will dilute
some if not all of our then existing stockholders, depending on the
financing terms.
30
Future sales and issuances of our equity securities or rights to
purchase our equity securities, including pursuant to equity
incentive plans, would result in additional dilution of the
percentage ownership of our stockholders and could cause our stock
price to fall.
To the extent we raise additional capital by issuing equity
securities, our stockholders may experience substantial dilution.
We may, as we have in the past, sell common stock, convertible
securities or other equity securities in one or more transactions
at prices and in a manner we determine from time to time. If we
sell common stock, convertible securities or other equity
securities in more than one transaction, investors may be further
diluted by subsequent sales. Such sales may also result in material
dilution to our existing stockholders, and new investors could gain
rights superior to existing stockholders.
Pursuant
to our 2013 Stock Plan, our Board of Directors is authorized to
award up to a total of 11,000,000 shares of common stock or options
to purchase shares of common stock to our officers, directors,
employees and non-employee consultants. As of December 31, 2016,
options to purchase 130,000 shares of common stock issued under our
2006 Stock Plan at a weighted average exercise price of $1.38 per
share, and options to purchase 4,479,755 shares of common stock
issued under our 2013 Stock Plan at a weighted average exercise
price of $2.31 per share were outstanding. In addition, at December
31, 2016, there were outstanding warrants to purchase an aggregate
of 4,006,468 shares of our common stock at prices ranging from
$0.40 to $7.00, and shares of our outstanding Series C-2, C-3, D
and E preferred stock convertible into an aggregate of 6,303,999
shares of our common stock. Stockholders will experience dilution
in the event that additional shares of common stock are issued
under our 2006 Stock Plan or 2013 Stock Plan, or options issued
under our 2006 Stock Plan or 2013 Stock Plan are exercised, or any
warrants are exercised for, or preferred stock shares are converted
to, common stock.
Provisions in our corporate charter documents and under Delaware
law could make an acquisition of us, which may be beneficial to our
stockholders, more difficult.
Provisions in our Amended and Restated Certificate of
Incorporation, as amended, and our Amended and Restated Bylaws, as
well as provisions of the General Corporation Law of the State of
Delaware, or DGCL, may discourage, delay or prevent a merger,
acquisition or other change in control of our company, even if such
a change in control would be beneficial to our stockholders. These
provisions include the following:
●
authorizing
the issuance of “blank check” preferred stock, the
terms of which may be established and shares of which may be issued
without stockholder approval;
●
prohibiting
our stockholders from fixing the number of our directors;
and
●
establishing
advance notice requirements for stockholder proposals that can be
acted on at stockholder meetings and nominations to our Board of
Directors.
These
provisions may frustrate or prevent any attempts by our
stockholders to replace or remove our current management by making
it more difficult for stockholders to replace members of our board
of directors, which is responsible for appointing the members of
our management. In addition, we are subject to Section 203 of the
DGCL, which generally prohibits a Delaware corporation from
engaging in any of a broad range of business combinations with an
interested stockholder for a period of three years following the
date on which the stockholder became an interested stockholder,
unless such transactions are approved by the board of directors.
This provision could have the effect of discouraging, delaying or
preventing someone from acquiring us or merging with us, whether or
not it is desired by, or beneficial to, our stockholders. Any
provision of our Amended and Restated Certificate of Incorporation,
as amended, or Amended and Restated Bylaws or Delaware law that has
the effect of delaying or deterring a change in control could limit
the opportunity for our stockholders to receive a premium for their
shares of our common stock and could also affect the price that
some investors are willing to pay for our common
stock.
If we fail to comply with the continued listing standards of the
NYSE MKT, it may result in a delisting of our common stock from the
exchange.
Our
common stock is currently listed for trading on the NYSE MKT, and
the continued listing of our common stock on the NYSE MKT is
subject to our compliance with a number of listing standards. These
listing standards include the requirement for avoiding sustained
losses and maintaining a minimum level of stockholders’
equity. In 2012 and 2014, we received notices from the
NYSE MKT that we did not meet continued listing standards of the
NYSE MKT as set forth in Part 10 of the Company
Guide. Specifically, we were not in compliance with
Section 1003(a)(i) and Section 1003(a)(ii) of the Company Guide
because we reported stockholders’ equity of less than the
required amounts. As a result, we became subject to the
procedures and requirements of Section 1009 of the Company Guide
and were subject to possible delisting. In March 2015, we regained
compliance with the NYSE MKT listing requirements due to our market
capitalization, pursuant to Section 1003(a) of the Company Guide.
However, there can be no assurance that we will continue to meet
the continued listing standards of the NYSE MKT.
31
If our common stock were no longer listed on the
NYSE MKT, investors might only be able to trade on the OTC Bulletin
Board ®
or in the Pink Sheets
®
(a quotation medium operated by Pink
Sheets LLC). This would impair the liquidity of our common stock
not only in the number of shares that could be bought and sold at a
given price, which might be depressed by the relative illiquidity,
but also through delays in the timing of transactions and reduction
in media coverage.
Because the average daily trading volume of our common stock has
been low historically, the ability to sell our shares in the
secondary trading market may be limited.
Because the average daily trading
volume of our common stock on the NYSE MKT has been low
historically, the liquidity of our common stock may be impaired. As
a result, prices for shares of our common stock may be lower than
might otherwise prevail if the average daily trading volume of our
common stock was higher. The average daily trading volume of our
common stock may be low relative to the stocks of other
exchange-listed companies, which could limit investors’
ability to sell shares in the secondary trading
market.
Penny stock regulations may impose certain restrictions on
marketability of our securities.
The
SEC has adopted regulations which generally define a “penny
stock” to be any equity security that has a market price of
less than $5.00 per share or an exercise price of less than $5.00
per share, subject to certain exceptions. A security listed on a
national securities exchange is exempt from the definition of a
penny stock. Our common stock is listed on the NYSE MKT and so is
not considered a penny stock. However, if we fail to maintain our
common stock’s listing on the NYSE MKT, our common stock
would be considered a penny stock. In that event, our common stock
would be subject to rules that impose additional sales practice
requirements on broker-dealers who sell such securities to persons
other than established customers and accredited investors
(generally those with assets in excess of $1,000,000 or annual
income exceeding $200,000, or $300,000 together with their spouse).
For transactions covered by such rules, the broker-dealer must make
a special suitability determination for the purchase of such
securities and have received the purchaser’s written consent
to the transaction prior to the purchase. Additionally, for any
transaction involving a penny stock, unless exempt, the rules
require the delivery, prior to the transaction, of a risk
disclosure document mandated by the SEC relating to the penny stock
market. The broker-dealer must also disclose the commission payable
to both the broker-dealer and the registered representative,
current quotations for the securities and, if the broker-dealer is
the sole market maker, the broker-dealer must disclose this fact
and the broker-dealer’s presumed control over the market.
Finally, monthly statements must be sent disclosing recent price
information for the penny stock held in the account and information
on the limited market in penny stocks. Broker-dealers must wait two
business days after providing buyers with disclosure materials
regarding a security before effecting a transaction in such
security. Consequently, the “penny stock” rules
restrict the ability of broker-dealers to sell our securities and
affect the ability of investors to sell our securities in the
secondary market and the price at which such purchasers can sell
any such securities, thereby affecting the liquidity of the market
for our common stock.
Stockholders
should be aware that, according to the SEC, the market for penny
stocks has suffered in recent years from patterns of fraud and
abuse. Such patterns include:
●
control
of the market for the security by one or more broker-dealers that
are often related to the promoter or issuer;
●
manipulation
of prices through prearranged matching of purchases and sales and
false and misleading press releases;
●
“boiler
room” practices involving high pressure sales tactics and
unrealistic price projections by inexperienced sales
persons;
●
excessive
and undisclosed bid-ask differentials and markups by selling
broker-dealers; and
●
the
wholesale dumping of the same securities by promoters and
broker-dealers after prices have been manipulated to a desired
level, along with the inevitable collapse of those prices with
consequent investor losses.
We do not intend to pay dividends on our common stock so any
returns on our common stock will be limited to the value of our
common stock.
We
have never declared dividends on our common stock, and currently do
not plan to declare dividends on shares of our common stock in the
foreseeable future. Pursuant to the terms of our Series D and E
Non-Voting Convertible Preferred Stock, we may not declare or pay
any dividends or make any distributions on any of our shares or
other equity securities as long as any of those preferred shares
remain outstanding. We currently expect to retain future earnings,
if any, for use in the operation and expansion of our business. The
payment of cash dividends in the future, if any, will be at the
discretion of our board of directors and will depend upon such
factors as earnings levels, capital requirements, our overall
financial condition and any other factors deemed relevant by our
board of directors. Any return to holders of our common stock will
be limited to the value of their common stock.
32
None.
Our
principal executive offices are located in approximately 4,700
square feet of office space in Bedminster, New Jersey. We sublease
this office space pursuant to a sublease agreement dated December
2014 which runs from April 1, 2015 until March 31, 2018. Rent is
$5,000 per month plus occupancy costs such as utilities,
maintenance and taxes. The total lease obligation is approximately
$180,000. Our remaining sublease obligation is approximately
$75,000 as of December 31, 2016.
Our
subsidiary leases its offices in Fulda, Germany for a term of 36
months which commenced on September 1, 2013 for a base monthly
payment of €498. The total 36 month lease obligation was
approximately €17,900. The 36 month lease has terminated. A
new three-month lease agreement commenced on November 1, 2016 which
is renewable every three months for a base monthly payment of
€461.
We
believe that our existing facilities are adequate to meet our
current needs, and that suitable additional alternative spaces will
be available in the future on commercially reasonable
terms.
Item
3.
Legal
Proceedings
In
February 2007, Geistlich Söhne AG für Chemische
Industrie, Switzerland (“Geistlich”) brought an action
against the European Sodemann Patent covering our Neutrolin product
candidate, which is owned by ND Partners, LLC (“NDP”)
and licensed to us pursuant to the License and Assignment Agreement
between us and NDP. This action was brought at the Board of the
European Patent Office (“EPO”) opposition division (the
“Opposition Board”) based upon alleged lack of
inventiveness in the use of citric acid and a pH value in the range
of 4.5 to 6.5 with having the aim to provide an alternative lock
solution through having improved anticoagulant characteristics
compared to the lock solutions of the prior art. The Opposition
Board rejected the opposition by Geistlich. In a letter dated
September 30, 2013, we were notified that the opposition division
of the EPO reopened the proceedings before the first instance and
gave their preliminary non-binding opinion that the patent as
amended during the appeal proceedings fulfills the requirements of
clarity, novelty, and inventive step, and invited the parties to
provide their comments and/or requests by February 10, 2014. We
filed our response on February 3, 2014 to request that the patent
be maintained as amended during the appeal proceedings. Geistlich
did not file a further statement within the required timeline. On
November 5, 2014, the Opposition Division at the EPO issued the
interlocutory decision to maintain the patent on the basis of the
claims as amended during the appeal proceedings. This decision
became final as no further appeal was lodged by
Geistlich.
On
September 9, 2014, we filed in the District Court of Mannheim,
Germany a patent infringement action against TauroPharm GmbH and
Tauro-Implant GmbH as well as their respective CEOs (the
“Defendants”) claiming infringement of our European
Patent EP 1 814 562 B1, which was granted by the EPO on January 8,
2014 (the “Prosl European Patent”). The Prosl European
Patent covers a low dose heparin catheter lock solution for
maintaining patency and preventing infection in a hemodialysis
catheter. In this action, we claim that the Defendants infringe on
the Prosl European Patent by manufacturing and distributing
catheter locking solutions to the extent they are covered by the
claims of the Prosl European Patent. We believe that our patent is
sound, and are seeking injunctive relief and raising claims for
information, rendering of accounts, calling back, destruction and
damages. Separately, TauroPharm has filed an opposition with the
EPO against the Prosl European Patent alleging that it lacks
novelty and inventive step. We cannot predict what other defenses
the Defendants may raise, or the ultimate outcome of either of
these related matters.
In the
same complaint against the same Defendants, we also alleged an
infringement (requesting the same remedies) of NDP’s utility
model DE 20 2005 022 124 U1 (the “Utility Model”),
which we believe is fundamentally identical to the Prosl European
Patent in its main aspects and claims. The Court separated the two
proceedings and the Prosl European Patent and the Utility Model
claims are now being tried separately. TauroPharm has filed a
cancellation action against the Utility Model before the German
Patent and Trademark Office (the “German PTO”) based on
the similar arguments as those in the opposition against the Prosl
European Patent.
33
On
March 27, 2015, the District Court held a hearing to evaluate
whether the Utility Model has been infringed by TauroPharm in
connection with the manufacture, sale and distribution of its
TauroLock-HEP100TM and TauroLock-HEP500TM products. A hearing
before the same court was held on January 30, 2015 on the separate,
but related, question of infringement of the Prosl European Patent
by TauroPharm.
The
Court issued its decisions on May 8, 2015, staying both
proceedings. In its decisions, the Court found that the
commercialization by TauroPharm in Germany of its TauroLock
catheter lock solutions Hep100 and Hep500 infringes both the Prosl
European Patent and the Utility Model and further that there is no
prior use right that would allow TauroPharm to continue to make,
use or sell its product in Germany. However, the Court declined to
issue an injunction in favor of us that would preclude the
continued commercialization by TauroPharm based upon its finding
that there is a sufficient likelihood that the EPO, in the case of
the Prosl European Patent, or the German PTO, in the case of the
Utility Model, may find that such patent or utility model is
invalid. Specifically, the Court noted the possible publication of
certain instructions for product use that may be deemed to
constitute prior art. As such, the District Court determined that
it will defer any consideration of the request by us for injunctive
and other relief until such time as the EPO or the German PTO made
a final decision on the underlying validity of the Prosl European
Patent and the Utility Model.
The
opposition proceeding against the Prosl European Patent before the
EPO is ongoing. In its preliminary consideration of the matter, the
EPO (and the German PTO) regarded the patent as not inventive or
novel due to publication of prior art. Oral proceedings before the
Opposition Division at the EPO were held on November 25, 2015, at
which the three judge patent examiner panel considered arguments
related to the validity of the Prosl European Patent. The hearing
was adjourned due to the fact that the panel was of the view that
Claus Herdeis, one of the managing directors of TauroPharm, has to
be heard as a witness in a further hearing in order to close some
gaps in the documentation presented by TauroPharm as regards the
publication of prior art. In October 2016, TauroPharm submitted a
further writ to the EPO requesting a date for the hearing and
bringing forward further arguments, in particular in view of the
recent decision of the German PTO on the invalidity of the utility
model. The EPO has scheduled a further oral hearing for November 22
/ 23, 2017. While we continue to believe that the referenced
publication and instructions for use do not, in fact, constitute
prior art and that the Prosl European Patent will be found to be
valid by the EPO, there can be no assurance that we will prevail in
this matter.
The
German PTO held a hearing in the validity proceedings relating to
the Utility Model on June 29, 2016, at which the panel affirmed its
preliminary finding that the Utility Model was invalid based upon
prior publication of a reference to the benefits that may be
associated with adding heparin to a taurolidine based solution. The
decision has only a declaratory effect, as the Utility Model had
expired in November 2015. Furthermore, it has no bearing on the
ongoing consideration of the validity and possible infringement of
the Prosl European Patent by the EPO. We filed an appeal against
the ruling on September 7, 2016.
On
January 16, 2015, we filed a complaint against TauroPharm GmbH and
its managing directors in the District Court of Cologne, Germany.
In the complaint, we allege violation of the German Unfair
Competition Act by TauroPharm for the unauthorized use of its
proprietary information obtained in confidence by TauroPharm. We
allege that TauroPharm is improperly and unfairly using its
proprietary information relating to the composition and manufacture
of Neutrolin, in the manufacture and sale of TauroPharm’s
products TauroLockTM, TauroLock-HEP100 and TauroLock-HEP500. We
seek a cease and desist order against TauroPharm from continuing to
manufacture and sell any product containing taurolidine (the active
pharmaceutical ingredient (“API”) of Neutrolin) and
citric acid in addition to possible other components, damages for
any sales in the past and the removal of all such products from the
market. An initial hearing in the District Court of Cologne,
Germany was held on November 19, 2015 to consider our claims. The
judge made no decision on the merits of our complaint. On January
14, 2016, the court issued an interim decision in the form of a
court order outlining several issues of concern that relate
primarily to court's interest in clarifying the facts and reviewing
any and all available documentation, in particular with regard to
the question which specific know-how was provided to TauroPharm by
whom and when. We have prepared the requested reply and produced
the respective documentation. TauroPharm has also filed another
writ within the same deadline and both parties have filed further
writs at the end of April setting out their respective
argumentation in more detail. A further oral hearing in this matter
was held on November 15, 2016. In this hearing, the court heard
arguments from CorMedix and TauroPharm concerning the allegations
of unfair competition. The court made no rulings from the bench,
and indicated that it is prepared to further examine the underlying
facts of our allegations. At this time, there are no further
hearings scheduled. The Company intends to continue to pursue this
matter, and to provide additional supplemental documentary and
other evidence as may be necessary to support its
claims.
34
On July
7, 2015, a putative class action lawsuit was commenced against the
Company and certain of its current and former officers in the
United States District Court for the District of New Jersey,
captioned Li v. Cormedix Inc., et al., Case 3:15-cv-05264 (the
“Securities Class Action”). On September 4, 2015, two
individuals, Shahm Martini and Paul Chretien (the “Martini
Group”), filed a Motion to Appoint Lead Plaintiff. On that
same date, another individual, Elaine Wood, filed a competing
Motion to Appoint Lead Plaintiff. On September 18, 2015, the
Martini Group withdrew its motion. Thereafter, on September 22,
2015, the Court appointed Elaine Wood as Lead Plaintiff and, on
October 2, 2015, appointed the Rosen Law Firm as Lead
Counsel.
On
December 1, 2015, Lead Plaintiff filed an Amended Complaint
asserting claims that the Company and Steven Lefkowitz, Randy Milby
and Harry O’Grady (the “Cormedix Defendants”)
violated Section 10(b) of the Exchange Act and Rule 10b-5
promulgated thereunder and Section 20(a) of the Exchange Act. The
Amended Complaint also named as defendants several unrelated
entities that allegedly were paid stock promoters. Lead Plaintiff
alleged generally that the Cormedix Defendants made materially
false or misleading statements and omissions concerning, among
other things, the competitive landscape for the Company’s
Neutrolin product and the alleged use of stock promoters. The
Amended Complaint sought unspecified damages, interest,
attorneys’ fees, and other costs.
On
February 1, 2016, the Cormedix Defendants filed a motion to dismiss
all claims asserted against them in the Amended Complaint on the
grounds, among others, that the Amended Complaint fails to
adequately allege: (1) material misstatements or omissions; (2)
scienter by any of the Cormedix Defendants; or (3) loss causation.
The Court heard oral argument on this motion on July 18, 2016 and
in an order dated October 27, 2016, the Court granted the Cormedix
Defendants’ Motion to Dismiss and dismissed with prejudice
the Amended Complaint (the “Dismissal Order”). On
December 16, 2016, the parties filed a stipulation with the Court
in which the plaintiffs and their counsel agreed not to appeal,
move for reconsideration or otherwise challenge the Dismissal
Order. No settlement payment was made in exchange for the
stipulation.
On May
13, 2016, a putative shareholder derivative action was filed in the
Superior Court of New Jersey against the Company and certain
present and former directors and officers captioned Raval v. Milby,
et. al., Docket No. C-12034-6 (the “Derivative
Action”). The factual allegations of the Derivative Action
substantially overlap the factual allegations contained in the
Amended Complaint in the Securities Class Action. The plaintiff
purports to assert claims against the individual defendants on
behalf of the Company for breach of fiduciary duty, unjust
enrichment, abuse of control, gross mismanagement and waste of
corporate assets. The complaint in the Derivative Action seeks
unspecified damages, interest, attorneys’ fees and other
costs, and certain amendments to the Company’s
“corporate governance and internal procedures”. On June
30, 2016, the Court entered a stipulated order, among other things,
staying the Derivative Action until 30 days after either: (a) the
entry of any order denying any motion to dismiss the Derivative
Action in the Securities Class Action, or (b) the entry of a final
order dismissing the Securities Class Action with prejudice.
Following entry of the Dismissal Order in the Securities Class
Action, the parties entered into a stipulation, among other things,
staying the Derivative Action until 30 days after either (a)
November 30, 2016, in the event plaintiffs in the Securities Class
Action did not file an appeal of the Dismissal Order or (b) the
final disposition of any appeal in the event the plaintiffs in the
Securities Class Action filed a Notice of Appeal of the Dismissal
Order. On January 6, 2017, the parties filed a Stipulation of
Dismissal without Prejudice of the Derivative Action.
Item 4. Mine Safety Disclosures
Not
applicable.
PART II
Item
5.
Market
for the Registrant’s Common Equity, Related Stockholder
Matters and Issuer Purchases of Equity Securities
Market for Common Equity
Our
common stock trades on the NYSE MKT under the symbol
“CRMD.” The following table sets forth the high and low
sales prices for our common stock for the periods indicated as
reported by NYSE MKT:
|
|
2016
|
|
2015
|
||||
|
|
High
|
|
Low
|
|
High
|
|
Low
|
|
First
Quarter
|
$
2.88
|
|
$1.15
|
|
$
9.90
|
|
$1.63
|
|
Second
Quarter
|
$
4.54
|
|
$1.72
|
|
$10.40
|
|
$3.20
|
|
Third
Quarter
|
$
3.12
|
|
$1.35
|
|
$
4.31
|
|
$1.72
|
|
Fourth
Quarter
|
$
3.26
|
|
$1.46
|
|
$
2.96
|
|
$1.72
|
Based
upon information furnished by our transfer agent, at March 14,
2017, we had approximately 63 holders of record of our common
stock.
35
Stock Performance Graph
The
following performance graph shall not be deemed to be
“soliciting material” or “filed” or
incorporated by reference in future filings with the SEC, or
subject to the liabilities of Section 18 of the Exchange Act except
as shall be expressly set forth by specific reference in such
filing. The performance graph compares the performance of our
common stock to the NASDAQ Composite Index and the NASDAQ
Biotechnology Index. The graph covers the most recent five-year
period ended December 31, 2016. The graph assumes that the
value of the investment in our common stock and each index was
$100.00 at December 31, 2011, and that all dividends are
reinvested.
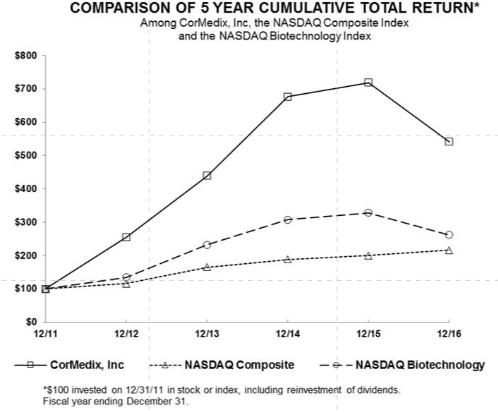
|
|
Cumulative Total
Return
|
|||||
|
|
12/2011
|
12/2012
|
12/2013
|
12/2014
|
12/2015
|
12/2016
|
|
CorMedix,
Inc.
|
$100
|
$255.03
|
$439.09
|
$676.35
|
$718.84
|
$541.78
|
|
NASDAQ
Composite
|
$100
|
$116.41
|
$165.47
|
$188.69
|
$200.32
|
$216.54
|
|
NASDAQ
Biotechnology
|
$100
|
$134.68
|
$232.37
|
$307.67
|
$328.76
|
$262.08
|
Dividend Policy
We have
never declared dividends on our equity securities, and currently do
not plan to declare dividends on shares of our common stock in the
foreseeable future. We expect to retain our future earnings, if
any, for use in the operation and expansion of our business.
Further, pursuant to the terms of our
Series D and Series E Non-Voting Convertible Preferred Stock, we
may not declare or pay any dividends or make any distributions on
any of our shares or other equity securities as long as any of
those preferred shares remain outstanding. Subject to the
foregoing, the payment of cash dividends in the future, if any,
will be at the discretion of our Board of Directors and will depend
upon such factors as earnings levels, capital requirements, our
overall financial condition and any other factors deemed relevant
by our Board of Directors.
Equity Compensation Plan Information
The
following table provides information as of December 31, 2016 about
our common stock that may be issued upon the exercise of options,
warrants and rights under all of our existing equity compensation
plans (including individual arrangements):
36
|
Plan
Category
|
Number of
securities to be issued upon exercise of outstanding options,
warrants and rights
(a)
|
Weighted-average
exercise price of outstanding options, warrants and
rights
(b)
|
Number
ofsecurities remaining available for future issuance under equity
compensation plans (excluding securities reflected in column
(a)
(c)
|
|
Equity compensation
plans approved by security holders (1)
|
4,609,755
|
$2.29
|
5,682,790
|
|
Equity compensation
plans not approved by security holders (2)
|
125,795
|
1.49
|
--
|
|
Total
|
4,735,550
|
$1.81
|
5,682,790
|
(1)
Our Amended and
Restated 2006 Stock Incentive Plan was approved by our stockholders
on February 19, 2010. Our 2013 Stock Incentive Plan was approved by
our stockholders on July 30, 2013.
(2) Consists
of 795 shares of common stock issuable pursuant to a warrant issued
to the placement agent of our convertible note financing in 2012
(with an exercise price of $0.40 per share) and 125,000 shares of
common stock issuable pursuant to a warrant issued to ND Partners
in April 2013 as consideration for the amendment of the ND Partners
License Agreement.
Item
6.
Selected
Consolidated Financial Data
The
consolidated statement of income data set forth below with respect
to the years ended December 31, 2016, December 31, 2015, and
December 31, 2014, and the consolidated balance sheet data at
December 31, 2016, December 31, 2015 and December 31, 2014 are
derived from the audited consolidated financial statements included
in Item 8 of this report and should be read in conjunction with
those financial statements and notes thereto. The consolidated
statement of income data for the years ended December 31, 2013 and
December 31, 2012 and the consolidated balance sheet data at
December 31, 2013 and December 31, 2012 are derived from audited
consolidated financial statements not included herein.
|
(amounts in
thousands, except for per share amounts)
|
2016
|
2015
|
2014
|
2013
|
2012
|
|
RESULTS OF
OPERATIONS
|
|
|
|
|
|
|
Net
sales
|
$224
|
$210
|
$189
|
$2
|
$-
|
|
Gross
(loss)
|
(143)
|
(109)
|
(257)
|
(200)
|
-
|
|
(Loss) from
operations
|
(24,761)
|
(16,654)
|
(8,903)
|
(4,915)
|
(3,000)
|
|
(Loss) before
income taxes
|
(24,644)
|
(18,187)
|
(20,453)
|
(9,133)
|
(3,381)
|
|
Net
(loss)
|
(24,644)
|
(18,187)
|
(20,453)
|
(9,133)
|
(3,381)
|
|
Comprehensive
income (loss)
|
19
|
(37)
|
108
|
(9)
|
-
|
|
Comprehensive
(loss)
|
(24,625)
|
(18,224)
|
(20,345)
|
(9,142)
|
(3,381)
|
|
(LOSS) PER
SHARE
|
|
|
|
|
|
|
Basic and
diluted
|
$(0.65)
|
(0.58)
|
(0.96)
|
(0.69)
|
$(0.30)
|
|
BALANCE SHEET
DATA
|
|
|
|
|
|
|
Total cash and
marketable securities
|
$20,165
|
$35,386
|
$4,340
|
$2,374
|
$835
|
|
Total
assets
|
21,906
|
37,102
|
5,098
|
2,968
|
1,153
|
|
Total
liabilities
|
4,092
|
3,090
|
1,463
|
6,990
|
1,489
|
|
Total
stockholders’ equity (deficiency)
|
17,815
|
34,011
|
3,634
|
(4,022)
|
(335)
|
37
Item
7.
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
You should read the following discussion and
analysis together with our audited financial statements and the
accompanying notes. This discussion contains forward-looking
statements, within the meaning of Section 27A of Securities Act,
Section 21E of the Exchange Act, and the Private Securities
Litigation Reform Act of 1995, including statements
regarding our expected financial
condition, business and financing plans. These statements involve
risks and uncertainties. Our actual results could differ materially
from the results described in or implied by these forward-looking
statements as a result of various factors, including those
discussed below and elsewhere in this report, particularly under
the heading “Risk Factors.”
Overview
We are
a biopharmaceutical company focused on developing and
commercializing therapeutic products for the prevention and
treatment of infectious and inflammatory diseases.
Our
primary focus is on the development of our lead product candidate,
Neutrolin, for potential commercialization in the U.S. and other
key markets. We have in-licensed the worldwide rights to develop
and commercialize Neutrolin®. Neutrolin is a
novel anti-infective solution (a formulation of taurolidine,
citrate and heparin 1000 u/ml) for the reduction and prevention of
catheter-related infections and thrombosis in patients requiring
central venous catheters in clinical settings such as dialysis,
critical/intensive care, and oncology. Infection and thrombosis
represent key complications among critical care / intensive care
and cancer patients with central venous catheters. These
complications can lead to treatment delays and increased costs to
the healthcare system when they occur due to hospitalizations, need
for IV antibiotic treatment, long-term anticoagulation therapy,
removal/replacement of the central venous catheter, related
treatment costs and increased mortality. We believe Neutrolin
addresses a significant unmet medical need and a potential large
market opportunity.
We plan
to conduct two pivotal trials to demonstrate safety and
effectiveness of Neutrolin to secure marketing approval in the U.S.
We initiated one Phase 3 clinical trial in hemodialysis patients
with a central venous catheter in December 2015 and plan to
initiate one Phase 3 trial in oncology patients with catheters,
subject to funding requirements.
In July
2013, we received CE Mark approval for Neutrolin. As a result, in
December 2013, we commercially launched Neutrolin in Germany
for the prevention of catheter-related bloodstream infections
(“CRBSI”), and maintenance of catheter patency in
hemodialysis patients using a tunneled, cuffed central venous
catheter for vascular access. To date, Neutrolin is
registered and may be sold in certain European Union and Middle
Eastern countries for such treatment.
Since
our inception, we have not generated sufficient revenue from
product sales to be profitable. Our operations to date
have been primarily limited to licensing product candidates,
developing clinical trials for our product candidates, establishing
manufacturing for our product candidates, performing business and
financial planning, performing research and development, seeking
regulatory approval for our products, initial commercialization
activities for Neutrolin, and maintaining and improving our patent
portfolio. We have funded our operations primarily
through debt and equity financings. We have generated
significant losses to date, and we expect to incur increases in our
cash use in operations as we continue to commercialize Neutrolin in
Europe and other markets, prepare for and undertake our ongoing and
planned Phase 3 clinical trials, pursue business development
activities, incur additional legal costs to defend our intellectual
property, and seek FDA approval of Neutrolin in the
U.S. As of December 31, 2016, we had an accumulated
deficit of approximately $119.2 million. We are unable
to predict the extent of any future losses or when we will become
profitable, if at all.
38
Financial Operations Overview
Revenue
We have
not generated substantial revenue since our inception. Through
December 31, 2016, we have funded our operations primarily through
debt and equity financings and our initial public offering, and to
a lesser extent, the receipt from Federal grants under the
Qualifying Therapeutic Discovery Project program and the sale of
our unused net operating losses through the State of New
Jersey’s Economic Development Authority Technology Business
Tax Certificate Transfer Program.
Research and Development Expense
Research and
development, or R&D, expense consists of: (i) internal costs
associated with our development activities; (ii) payments we make
to third-party contract research organizations, contract
manufacturers, investigative sites, and consultants; (iii)
technology and intellectual property license costs; (iv)
manufacturing development costs; (v) personnel related expenses,
including salaries, stock-based compensation, benefits, travel and
related costs for the personnel involved in drug development; (vi)
activities relating to regulatory filings and the advancement of
our product candidates through preclinical studies and clinical
trials; and (vii) facilities and other allocated expenses, which
include direct and allocated expenses for rent, facility
maintenance, as well as laboratory and other supplies. All R&D
is expensed as incurred.
Conducting a
significant amount of development is central to our business model.
Product candidates in later-stage clinical development generally
have higher development costs than those in earlier stages of
development, primarily due to the significantly increased size and
duration of the clinical trials. We plan to increase our R&D
expenses for the foreseeable future in order to complete
development of Neutrolin in the U.S., especially for the ongoing
Phase 3 trial in hemodialysis patients and the planned Phase 3
trial in oncology.
The
following table summarizes the percentages of our R&D payments
related to our sole product candidate Neutrolin and our former
product candidate CRMD0004 (we ceased development of and returned
the rights to CRMD004 in late 2015). The percentages summarized in
the following table reflect payments directly attributable to each
development candidate, which are tracked on a project basis. A
portion of our internal costs, including indirect costs relating to
our product candidates, are not tracked on a project basis and are
allocated based on management’s estimate.
|
|
Year
EndedDecember 31,
|
||
|
|
2016
|
2015
|
2014
|
|
CRMD003
|
100%
|
98%
|
98%
|
|
CRMD004
|
-
|
2%
|
2%
|
The
process of conducting pre-clinical studies and clinical trials
necessary to obtain regulatory approval is costly and time
consuming. The probability of success for each product candidate
and clinical trial may be affected by a variety of factors,
including, among others, the quality of the product
candidate’s early clinical data, investment in the program,
competition, manufacturing capabilities and commercial viability.
As a result of the uncertainties associated with clinical trial
enrollments and the risks inherent in the development process, we
are unable to determine the duration and completion costs of
current or future clinical stages of our product candidates or
when, or to what extent, we will generate revenues from the
commercialization and sale of any of our product
candidates.
Development
timelines, probability of success and development costs vary
widely. Our current focus is on clinical development efforts in the
U.S. and optimization of sales in markets where Neutrolin is
approved. We are seeking to develop Neutrolin in the U.S. Based on
our discussions with the FDA, we are conducting a Phase 3 clinical
trial in hemodialysis catheters and, subject to finalization of the
protocol, plan to conduct one Phase 3 clinical trial in oncology.
We plan to initiate the Phase 3 trial in oncology depending on our
ability to raise additional capital and our ability to complete the
hemodialysis catheters trial within our expected budget. We expect
that the ongoing Phase 3 trial for hemodialysis catheters will cost
approximately $26 million to $30 million and will take 30 months to
complete after initiation. We are still finalizing the details of
the protocol for the planned second Phase 3 trial for
oncology/total parenteral nutrition and are unable to provide a
cost estimate at this time.
We are seeking one or more strategic partners or other sources of
capital to help complete the development of Neutrolin in the
U.S.
39
On July
5, 2013, we received CE Mark approval for Neutrolin. As a result,
in late 2013, we commercially launched Neutrolin in Germany for the
prevention of catheter-related bloodstream infections, or CRBI, and
maintenance of catheter patency in hemodialysis patients using a
tunneled, cuffed central venous catheter for vascular access. To
date, Neutrolin is registered and may be sold in certain European
Union and Middle East countries for such treatment.
Selling, General and Administrative Expense
Selling, general
and administrative, or SG&A, expense includes costs related to
commercial personnel, medical education professionals, marketing
and advertising, salaries and other related costs, including
stock-based compensation expense, for persons serving in our
executive, sales, finance and accounting functions. Other SG&A
expense includes facility-related costs not included in R&D
expense, promotional expenses, costs associated with industry and
trade shows, and professional fees for legal services and
accounting services.
Loss on Issuance of Preferred Stock, Convertible Notes and
Warrants
We
issued preferred stock and related warrants during the year ended
December 31, 2014. The loss on the issuance of preferred stock and
related warrants represents the difference on the issuance date
between the combined derivative related fair value of the
conversion option and the warrants, and the proceeds that were
received net of all fees and expenses related to the
issuance.
Change in Fair Value of Derivative Liabilities
As
previously disclosed in our December 31, 2014 Form 10-K, we entered
into consent and exchange agreements with investors holding our
outstanding Series C-2, Series C-3, Series D, and Series E
non-voting convertible preferred stock. We modified certain terms
within the preferred stock which resulted in the reclassification
of the remaining derivative liability to equity in September
2014.
The
change in the fair value of derivative liabilities represents the
change in the fair value of the Series C, D and E preferred stock
conversion options and the change in the fair value of warrants
that were recorded at fair value on a recurring basis under
accounting principles generally accepted in the United States
(“GAAP”). This includes any changes in fair value
resulting from the re-measurement of the derivative liabilities in
connection with the redemption or conversion of the preferred stock
and the exercise of warrants.
Loss on Modification of Equity Instruments and Extinguishment of
Derivative Liabilities
As
discussed in Note 7 to the financial statements included in this
Report, the loss on modification of equity instruments and
extinguishment of derivative liabilities represents the change in
the fair value of the preferred stock hybrid instruments and
liability classified warrants resulting from the modifications made
to those instruments during the year ended December 31,
2014.
Foreign Currency Exchange Transaction Gain (Loss)
Foreign
currency exchange transaction gain (loss) is the result of
re-measuring transactions denominated in a currency other than our
functional currency and is reported in the consolidated statement
of operations as a separate line item within other income
(expense). In 2014, foreign currency exchange transaction gain
(loss) consists of foreign exchange transaction gains and losses on
intercompany loans that are in place between our company, which is
based in New Jersey, and our German subsidiary. Effective October
1, 2014, we determined that the intercompany loans outstanding are
not expected to be repaid in the foreseeable future and the nature
of the funding advanced is of a long-term investment nature. As
such, beginning October 1, 2014, unrealized foreign exchange
movements related to long-term intercompany loans are recorded in
other comprehensive income (loss).
Interest Income
Interest income
consists of interest earned on our cash and cash equivalents and
short-term investments.
Interest Expense
Interest expense
consists of interest incurred on financing of
expenditures.
40
Results of Operations
Comparison of the Years Ended December 31, 2016, 2015 and
2014
The
following is a tabular presentation of our consolidated operating
results (in
thousands):
|
|
For
the Year Ended December 31,
|
% of
Change Increase
(Decrease)
|
|||
|
|
2016
|
2015
|
2014
|
2016 versus
2015
|
2015 versus
2014
|
|
Revenue
|
$224
|
$210
|
$189
|
7%
|
11%
|
|
Cost of
sales
|
(367)
|
(319)
|
(446)
|
15%
|
(29)%
|
|
Gross profit
(loss)
|
(143)
|
(109)
|
(257)
|
31%
|
(58)%
|
|
Operating
Expenses:
|
|
|
|
|
|
|
Research and
development
|
(15,735)
|
(6,282)
|
(1,319)
|
150%
|
376%
|
|
Selling, general
and administrative
|
(8,883)
|
(10,263)
|
(7,327)
|
(13)%
|
40%
|
|
Total operating
expenses
|
(24,618)
|
(16,545)
|
(8,646)
|
49%
|
91%
|
|
Loss from
operations
|
(24,761)
|
(16,654)
|
(8,903)
|
49%
|
87%
|
|
Interest
income
|
127
|
61
|
3
|
108%
|
1933%
|
|
Foreign exchange
transaction loss
|
(8)
|
(7)
|
(151)
|
14%
|
(96)%
|
|
Value of warrants
issued in connection with backstop financing
|
-
|
(1,583)
|
-
|
(100%)
|
(100)%
|
|
Loss on issuance of
preferred stock, convertible notes and warrants
|
-
|
-
|
(89)
|
-
|
(100)%
|
|
Change in fair
value of derivative liabilities
|
-
|
-
|
(8,849)
|
-
|
(100)%
|
|
Loss on
modification of equity instruments and extinguishment of derivative
liabilities
|
-
|
-
|
(2,462)
|
-
|
(100)%
|
|
Interest
expense
|
(1)
|
(4)
|
(2)
|
(75)%
|
90%
|
|
Net
loss
|
(24,643)
|
(18,187)
|
(20,453)
|
35%
|
(12)%
|
|
Other comprehensive
income gain (loss)
|
19
|
(37)
|
108
|
(152)%
|
(134)%
|
|
Comprehensive
loss
|
$(24,624)
|
$(18,224)
|
$(20,345)
|
35%
|
(11)%
|
Revenue. Revenue for the year ended
December 31, 2016 was $224,000 as compared to $210,000 for the same
period in 2015, an increase of $14,000. The increase was due to the
recognition of deferred revenue of $106,000 for the products sold
with warranty, substantially offset by a decrease in sales of
Neutrolin in Germany and the Middle East of $92,000.
Revenue
for the year ended December 31, 2015 was $210,000 as compared to
$189,000 for the same period in 2014, an increase of $21,000. The
majority of the revenue is from sales of Neutrolin in Germany and
Middle East markets. In addition, we realized $8,000 associated
with the amortization of deferred revenue from a non-refundable
payment received from a distribution agreement in 2015 as compared
to $4,000 in 2014.
Cost of Sales. Cost of sales for the
year ended December 31, 2016 was $367,000 as compared to $319,000
for the same period in 2015, an increase of $48,000. The increase
was primarily due to the $63,000 recognition of cost of sales
associated with deferred revenue, an increase in write-off of
expired raw materials of $58,000, and an increase of inventory
reserve of $5,000, offset by decreases in ongoing stability studies
of $35,000 and decreased cost of materials of $43,000 due to lower
sales in 2016 excluding deferred revenue.
Cost of
sales for the year ended December 31, 2015 was $319,000 as compared
to $446,000 for the same period in 2014, a decrease of $127,000.
The decrease was primarily due to decreases in ongoing stability
studies and services performed in the management of manufacturing
of $131,000 and other manufacturing expenses mainly due to costs in
transitioning Neutrolin to new labels and packaging of $13,000,
offset by an increase in direct cost of materials of $67,000 due to
the use of new commercial batches as compared to the use of old
previously expensed research and development batches in 2014. In
addition, we recorded a charge of $125,000 associated with
pre-launch inventory build-up and start-up related manufacturing
inefficiencies in 2015 as compared to $175,000 in 2014, a decrease
of $50,000.
41
Research and Development Expense.
R&D expense for the year ended December 31, 2016 was
$15,735,000, an increase of $9,453,000 from $6,282,000 for the same
period in 2015. The increase was primarily attributable to
$5,317,000 of expenses related to the ongoing Phase 3 clinical
trial in hemodialysis catheters in the U.S.; cost of new studies
related to antimicrobial sutures, nanofiber webs, wound management
and osteoarthritis and visco-supplementation of $2,438,000; costs
to support the U.S. clinical trial drug supply consisting of
manufacturing process development activities of $1,928,000;
personnel cost of $627,000, mainly due to hiring employees and
consulting fees of $290,000. These increases were partially offset
by decreases in pharmacoeconomics and pricing and market research
studies conducted in 2015 of $401,000 and decreased non-cash stock
based compensation of $758,000.
R&D
expense for the year ended December 31, 2015 was $6,282,000, an
increase of $4,963,000 from $1,319,000 for the same period in 2014.
The increase was primarily attributable to $1,020,000 for the
initiation of the Phase 3 clinical trial in hemodialysis catheters
in the U.S.; higher costs to support the U.S. clinical trial drug
supply consisting of manufacturing process development activities
of $1,718,000 and pharmacoeconomics, and pricing and market
research studies of $203,000. Additionally, increases in non-cash
stock based compensation of $871,000, consulting fees pertaining
mainly to the clinical supply manufacturing development process and
regulatory activities of $996,000, and personnel costs of $120,000
were recognized.
Selling, General and Administrative
Expense. SG&A expense for the year ended December 31,
2016 was $8,883,000, a decrease of $1,380,000 from $10,263,000 for
the same period in 2015. The decrease was primarily attributable to
a $1,133,000 decrease in non-cash stock-based compensation expense,
mainly due to the modification of our former CEO’s stock
options recorded in 2015; a decrease in selling expenses in the EU
of $666,000, mainly due to reduction in sales force and a decrease
in expenses related to business development activities of $430,000.
These decreases, among others of lesser significance, were
partially offset by increased consulting fees of $587,000,
primarily due to interim CFO and executive search fees; increased
legal fees of $159,000; increased personnel expenses of $56,000 and
increased investor relations activities of $50,000.
SG&A expense
for the year ended December 31, 2015 was $10,263,000, an increase
of $2,936,000 from $7,327,000 for the same period in 2014. The
increase was attributable to increases in personnel expenses of
$650,000 due to employee benefits and the Release of Claims and
Severance Modification with our CEO; legal fees due mainly to
ongoing intellectual property and securities litigation, increased
SEC and clinical activities of $784,000; expenses related to
business development activities of $292,000 and marketing research
studies of $248,000; consulting fees of $554,000 primarily due to
an executive search fee, increased investor relations activities of
$193,000; and a non-cash charge of $187,000 for stock-based
compensation expense due to the modification of the stock options
of our CEO and $113,000 for modification of warrants. These
increases, among others of lesser significance, were partially
offset by a decrease in selling costs related to commercialization
of Neutrolin in the EU of $268,000.
Loss on Issuance of Preferred Stock,
Convertible Notes and Warrants. The loss on the issuance of
preferred stock and warrants of $90,000 for the year ended December
31, 2014 represents the difference on the issuance date between the
combined fair value of the conversion option and the warrants of
$2,054,000, and the combined proceeds received and liabilities
settled, net of all issuance-related fees and expenses of
$1,965,000. Due to the elimination of the downround protection of
these derivative liabilities through an agreement modification in
September 2014, which resulted in the reclassification of
derivative liabilities to equity, there was no charge to earnings
during the years ended December 31, 2016 and 2015.
Change in Fair Value of Derivative
Liabilities. The change in the value of derivative
liabilities for the year ended December 31, 2014 of $8,849,000
consists of increases in the fair value of preferred stock
conversion options and warrants between December 31, 2013 and
September 15, 2014 of $7,138,000 and $1,711,000, respectively. Due
to the modification of certain terms within the preferred stock
which resulted in the reclassification of the remaining derivative
liability to equity in September 2014, there was no charge to
earnings during the years ended December 31, 2016 and
2015.
Loss on Modification of Equity Instruments and
Extinguishment of Derivative Liabilities. The loss on
extinguishment of derivative liabilities for the year ended
December 31, 2014 of $2,463,000 represents the change in the fair
value of the preferred stock hybrid instruments of $2,119,000 and
liability classified warrants of $344,000 resulting from the
modifications made to those instruments on September 15, 2014 for
the purpose of changing the balance sheet classification from
liability to equity.
42
Foreign Exchange Transaction Gain
(Loss). Foreign exchange transaction losses for the years
ended December 31, 2016 and 2015 of $8,000 and $7,000,
respectively, were due to the foreign exchange rate fluctuations
for the payment of invoices paid in foreign currency. Foreign
exchange transaction loss for the year ended December 31, 2014 was
$151,000 due to additional funding to our German subsidiary through
September 30, 2014 and the corresponding fluctuation in the
exchange rates. Effective October 1, 2014, we considered the
intercompany loans to be of long-term investment nature. Foreign
exchange gains or losses subsequent to October 1, 2014 have been
recorded in other comprehensive income.
Interest Income. Interest income for
the year ended December 31, 2016 was $126,800, an increase of
$66,400 from $60,400 for the same period in 2015. The increase was
attributable to recording higher interest income on short-term
investments during 2016 as compared to the same period in
2015.
Interest income for
the year ended December 31, 2015 was $60,400, an increase of
$57,700 from $2,700 for the same period in 2014. The increase was
attributable to higher average interest-bearing cash balances
during the year ended December 31, 2015 as compared to the same
period in 2014.
Interest Expense. Interest expense for
the year ended December 31, 2016 was $1,300 as compared to $4,000
for the same period in 2015, a decrease of $2,700.
Interest expense
for the year ended December 31, 2015 was $4,000 as compared to
$2,000 for the same period in 2014, an increase of
$2,000.
Other Comprehensive Income (Loss).
Unrealized foreign exchange movements related to long-term
intercompany loans and the translation of the foreign affiliate
financial statements to U.S. dollars and unrealized movements
related to short-term investment are recorded in other
comprehensive income resulting in a $19,000 gain in 2016, $37,000
loss in 2015 and $108,000 gain in 2014.
Liquidity and Capital Resources
Sources of Liquidity
As a
result of our cost of sales, R&D and SG&A expenditures and
the lack of substantial product sales revenue, we have not been
profitable and have generated operating losses since we were
incorporated in July 2006. We received the following proceeds from
the issuance of our common stock during the years
ended:
|
|
December
31,
|
|||||
|
|
2016
|
2015
|
||||
|
|
Shares
Issued
|
Net
Proceeds
|
Wtd. Ave.
Price
|
Shares
Issued
|
Net
Proceeds
|
Wtd. Ave.
Price
|
|
At-the-market
program
|
3,360,037
|
$6,229,351
|
$1.92
|
5,310,037
|
$28,451,848
|
$5.55
|
|
Exercise of stock
options
|
1,087,500
|
863,101
|
$0.79
|
499,955
|
492,960
|
$0.99
|
|
Exercise of
warrants
|
-
|
-
|
$-
|
4,581,783
|
14,658,161
|
$3.20
|
|
Totals
|
4,447,537
|
$7,092,452
|
|
10,391,775
|
$43,602,969
|
|
During
the year ended December 31, 2014, we sold the
following:
●
200,000
shares of our Series C-3 non-voting convertible preferred stock and
warrants to purchase up to 1,000,000 shares of our common stock for
net cash proceeds of $1,319,000 and the settlement of accounts
payable and accrued expenses of $645,000; and
●
2,960,000
units, each unit consisted of one share of our common stock and
0.35 of a warrant to purchase one share of our common stock, for
gross proceeds of $7,400,000. We received net proceeds of
approximately $6,723,000.
43
Net Cash Used in Operating Activities
Net
cash used in operating activities for the year ended December 31,
2016 was $22,265,000 as compared to $12,527,000 in 2015, an
increase in net cash use of $9,738,000. The increase was primarily
attributable to an increase in net loss of $6,456,000 driven by
increased research and development expenses. The net loss of
$24,644,000 for the year ended December 31, 2016 was higher than
cash used in operating activities by $2,379,000. The difference is
primarily attributable to non-cash stock-based compensation of
$1,335,000, increase in accrued expenses of $1,122,000 and
decreases in trade receivables and inventory of $308,000 and
$80,000, respectively, offset by an increase in prepaid expenses of
$505,000 due to expenses associated with the Phase 3 clinical trial
in hemodialysis catheters in the U.S and decrease in accounts
payable of $64,000.
Net
cash used in operating activities for the year ended December 31,
2015 was $12,527,000 as compared to $6,321,000 in 2014. The net
loss of $18,187,000 for the year ended December 31, 2015 was higher
than cash used in operating activities by $5,660,000. The
difference is attributable primarily to a non-cash stock-based
compensation of $3,226,000, non-cash charge for warrants issued in
connection with the March 2015 backstop agreement of $1,583,000 and
value of warrants related to the extension of the expiration date
of $113,000. In comparison for the same period last year, the net
loss of $20,453,000 for the year ended December 31, 2014 was higher
than cash used in operating activities by $14,132,000. The
difference is attributable primarily to revaluation of derivative
liabilities of $8,849,000, non-cash loss on extinguishment of
derivative liabilities of $2,463,000, non-cash stock-based
compensation of $2,168,000, and losses on foreign currency
transactions and issuance of preferred stock of $151,000 and
$90,000, respectively.
Net Cash Provided by (Used in) Investing Activities
Cash
provided by (used in) investing activities for the year ended
December 31, 2016 was $11,420,000, attributable to the proceeds on
the sale of short-term investments as compared to cash used in
investing activities of $23,608,000 for the same period in 2015 due
to the purchase of short-term investments.
Cash
used in investing activities for the year ended December 31, 2015
was $23,608,000 attributable to the purchase of short-term
investments as compared to $25,000 for the same period in 2014 due
to the purchase of software for our German subsidiary.
Net Cash Provided by Financing Activities
Net
cash provided by financing activities for the year ended December
31, 2016 was $7,092,000 as compared to $43,629,000 for the same
period in 2015. During 2016, we generated net proceeds of
$6,229,000 from the sale of our common stock in the at-the-market
program and received net proceeds of $863,000 from the exercise of
stock options.
Net
cash provided by financing activities for the year ended December
31, 2015 was $43,629,000 as compared to $8,358,000 for the same
period in 2014. During 2015, we generated $28,452,000 net proceeds
from the sale of our common stock in an at-the-market program;
received $14,658,000 and $493,000 net proceeds from the exercise of
warrants and stock options, respectively. In comparison to the same
period in 2014, we generated net proceeds of $6,723,000 and
$1,319,000 from the sale of our common stock and Series C-3
preferred stock, respectively, and received net proceeds of
$318,000 from the exercise of stock options.
Funding Requirements and Liquidity
Our
total cash on hand and short-term investments as of December 31,
2016 was $20.2 million, excluding restricted cash of $0.2 million,
compared with $35.4 million and $4.3 million at December 31, 2015
and 2014, respectively. In addition, we have approximately $4.1
million available under our current at-the-market program at
December 31, 2016. In
August 2016, we entered into a new At-the-Market Issuance Sales
Agreement (the "ATM program") with FBR Capital Markets & Co.
(“FBR”), the successor in interest to MLV, on terms
identical to the Sales Agreement for our current at-the-market
program. This new agreement allows us to sell up to $40 million of
shares of our common stock; however, we cannot access the
at-the-market program under the new sales agreement unless and
until (i) we and Manchester Securities Corp.
(“Manchester”) agree as to the exercise or waiver of
Manchester’s participation rights in the new ATM program,
which rights were granted in a Consent and Exchange Agreement dated
September 15, 2014, and apply to any equity financing we undertake
until September 15, 2017, and (ii) we amend our existing shelf
registration statement or file a new registration statement that
includes the prospectus for the new at-the-market program. (See
Note 4).
44
Because
our business has not currently generated positive operating cash
flow, we will need to raise additional capital in order to continue
to fund our research and development activities and our business
development activities, as well as to fund operations generally.
Our continued operations and completion of our ongoing Phase 3
clinical trial for Neutrolin in hemodialysis catheters in the U.S.,
which was initiated in December 2015, will depend on our ability to
raise sufficient additional funds through various potential
sources, such as equity, debt financings, and/or strategic
relationships. We plan to conduct a Phase 3 clinical trial in
oncology patients with catheters in the U.S., for which additional
funds over and above the funds needed for the ongoing hemodialysis
Phase 3 clinical trial will be required before such trial can
commence. We can provide no assurances that financing or strategic
relationships will be available on acceptable terms, or at all,
that may enable us to complete our hemodialysis study and commence
our oncology study.
We
expect to continue to fund operations from cash on hand and through
capital raising sources as previously described, which may be
dilutive to existing stockholders, through revenues from the
licensing of our products, or through strategic alliances. At
December 31, 2016, we have
approximately $4.1 million available under our current at-the
market program. We also have $60.0 million available under our
current shelf registration for the issuance of equity, debt or
equity-linked securities. We may utilize our ATM program, if
conditions allow, to support our ongoing Phase 3 clinical trial for
Neutrolin in hemodialysis catheters in the U.S. Additionally, we
may seek to sell additional equity or debt securities through one
or more discrete transactions, or enter into a strategic alliance
arrangement, but can provide no assurances that any such
financing or strategic alliance arrangement will be available on
acceptable terms, or at all. Moreover, the incurrence of
indebtedness in connection with a debt financing would result in
increased fixed obligations and could contain covenants that would
restrict our operations. Raising
additional funds through strategic alliance arrangements with third
parties may require significant time to complete and force us to
relinquish valuable rights to our technologies, future revenue
streams, research programs or product candidates, or to grant
licenses on terms that may not be favorable to us or our
stockholders. Our actual cash requirements may vary
materially from those now planned due to a number of factors,
including any change in the focus and direction of our research and
development programs, any acquisition or pursuit of development of
new product candidates, competitive and technical advances, the
costs of commercializing any of our product candidates, and costs
of filing, prosecuting, defending and enforcing any patent claims
and any other intellectual property rights.
While
we expect to grow product sales, we do not anticipate that we will
generate significant product revenues in the foreseeable future. In
the absence of such revenue, we are likely to continue generating
operating cash flow deficits. We expect to incur increases in our
cash used in operations as we continue our ongoing and planned
Phase 3 clinical trials, pursue business development activities,
incur additional legal costs to defend our intellectual property
and seek FDA approval of Neutrolin in the U.S.
Based on our cash resources at December 31,
2016 and the expected cost of the Phase 3 clinical trial in
hemodialysis catheters in the U.S., we believe that our existing
cash and short-term investments will be insufficient to fund our
operations through 2017 and will not be sufficient to complete the
ongoing Phase 3 trial for hemodialysis catheters in the U.S. or to
begin the planned Phase 3 trial for oncology patients with
catheters. If we are unable to raise additional funds when needed,
we may be forced to slow or discontinue our ongoing Phase 3
clinical trial, and will be unable to commence our planned Phase 3
clinical trial for oncology patients with catheters. We could also
be required to delay, scale back or eliminate some or all of our
research and development programs. Each of these alternatives would
likely have a material adverse effect on our business.
Contractual Obligations
We
entered into a sublease for 4,700 square feet of office space in
Bedminster, New Jersey, which runs from April 1, 2015 until March
31, 2018. Rent is $5,000 per month plus occupancy costs such as
utilities, maintenance and taxes. In accordance with the lease
agreement, we deposited $5,000 with the landlord, the equivalent of
one month rent.
Our
German subsidiary entered into a lease agreement for its offices in
Fulda, Germany. The lease initially had a term of 36 months which
commenced on September 1, 2013 for a base monthly payment of
€498. A new lease agreement commenced on November 1, 2016
which is renewable every three months for a base monthly payment of
€461.
Under
our current lease agreements, the total estimated remaining lease
obligation as of December 31, 2016 is set forth below:
|
2017
|
$62,237
|
|
2018
|
15,000
|
|
Total
|
$77,237
|
45
Critical Accounting Estimates
Our
management’s discussion and analysis of our financial
condition and results of operations is based on our financial
statements, which have been prepared in accordance with accounting
principles generally accepted in the United States, or GAAP. The
preparation of these financial statements requires us to make
estimates and judgments that affect the reported amounts of assets,
liabilities and expenses. On an ongoing basis, we evaluate these
estimates and judgments, including those described below. We base
our estimates on our historical experience and on various other
assumptions that we believe to be reasonable under the
circumstances. These estimates and assumptions form the basis for
making judgments about the carrying values of assets and
liabilities that are not readily apparent from other sources.
Actual results and experiences may differ materially from these
estimates.
While
our significant accounting policies are more fully described in
Note 3 to our financial statements included with this report, we
believe that the following accounting policies are the most
critical to aid you in fully understanding and evaluating our
reported financial results and affect the more significant
judgments and estimates that we use in the preparation of our
financial statements.
Stock-Based Compensation
We
account for stock options according to the Financial Accounting
Standards Board (“FASB”) Accounting Standards
Codification (“ASC”) No. 718, “Compensation
— Stock Compensation” (“ASC 718”).
Share-based compensation cost is measured at grant date, based on
the estimated fair value of the award using a Black-Scholes option
pricing model for options with service or performance based
conditions and a Monte Carlo option pricing model for options with
market vesting conditions. Stock-based compensation cost is
recognized as expense, over the employee’s requisite service
period on a straight-line basis.
Effective October
1, 2016, we adopted Accounting Standards Update (“ASU”)
2016-09, Improvements to Employee Share-Based Payment Accounting,
which was applied retroactively effective January 1, 2016, to
account for forfeitures as they occur. Under ASU 2016-09, all
share-based awards will be recognized on a straight-line method,
assuming all awards granted will vest. Forfeitures of share-based
awards will be recognized in the period in which they occur. Prior
to the adoption of ASU 2016-09, share-based compensation cost was
measured at grant date, based on the estimated fair value of the
award, and was recognized as expense net of expected forfeitures,
over the employee’s requisite service period on a
straight-line basis.
We
account for stock options granted to non-employees on a fair value
basis using the Black-Scholes option pricing model in accordance
with ASC 718 and ASC No. 505-50, “Equity-Based Payments to
Non-Employees” (“ASC 505”). The non-cash
charge to operations for non-employee options with time based
vesting provisions is based on the fair value of the options
remeasured each reporting period and amortized to expense over the
related vesting period. The non-cash charge to operations for
non-employee options with performance based vesting provisions is
recorded when the achievement of the performance condition is
probable and remeasured each reporting period until the performance
condition is achieved.
Valuations
incorporate several variables, including expected term, expected
volatility, expected dividend yield and a risk-free interest rate.
We estimate the expected term of the options granted based on
anticipated exercises in future periods. Prior to 2015, the
expected volatility used in the valuation of our stock options was
based on the historical volatility of publicly traded peer group
companies due to the limited trading history of our common stock.
Beginning in the first quarter of 2015, the expected stock price
volatility for our stock options is calculated based on the
historical volatility since the initial public offering of our
common stock in March 2010, weighted pre and post CE Mark approval
in the European Union. The expected dividend yield reflects our
current and expected future policy for dividends on our common
stock. To determine the risk-free interest rate, we utilize
the U.S. Treasury yield curve in effect at the time of grant with a
term consistent with the expected term of our
awards.
Revenue Recognition
We
recognize revenue in accordance with SEC SAB No. 101,
“Revenue Recognition in Financial Statements”
(“SAB 101”), as amended by SAB No. 104,
“Revenue Recognition” (“SAB 104”) and FASB
ASC 605, “Revenue Recognition” (“ASC
605”). Our product
Neutrolin received its CE Mark in Europe in July 2013 and shipment
of product to the dialysis centers began in December 2013. In
accordance with SAB 101 and SAB 104, we recognize revenue from
product sales when the following four revenue recognition criteria
are met: persuasive evidence of an arrangement exists, delivery has
occurred, the selling price is fixed or determinable, and
collectability is reasonably assured. We recognize revenue upon
shipment of product to the dialysis centers because the four
revenue recognition criteria are met at that time. For an upfront
payment related to an exclusive distribution agreement, we record
it as deferred revenue and recognize revenue on a straight-line
basis over the contractual term of the agreement
46
In
October 2015, we shipped product with less than 75% of its
remaining shelf life to a customer and issued a guarantee that any
product shipped with less than 75% of its shelf life remaining
would be replaced by us if the customer was not able to sell the
product before it expired. As a result of this warranty, we may
have an additional performance obligation (i.e. accept returned
product and deliver new product to the customer) if the customer is
unable to sell the short-dated product. Due to limited sales
experience with the customer, we were unable to estimate the amount
of the warranty obligation that may be incurred as a result of this
shipment. Therefore, we deferred the revenue and related cost of
sales associated with the original shipment of this product. Since
we will be unable to resell the expired product if returned by the
customer, the deferred revenue and related cost of sales is
presented net as “Deferred revenue” on the consolidated
balance sheet. During the year ended December 31, 2016, we
recognized 35.6% of deferred revenue and related cost of sales
amounting to $106,000 and $63,000, respectively. At December 31,
2016 and 2015, deferred revenue on shipment to distributor was
$75,000 and $121,000, respectively.
During
the year ended December 31, 2014, we entered into a distribution
agreement with Wonik Corporation, a South Korean company, to
market, sell and distribute Neutrolin for hemodialysis and
oncolytic patients upon receipt of regulatory approval in Korea.
Upon execution of the agreement, Wonik paid to us a non-refundable
$50,000 payment and will pay an additional $50,000 upon receipt of
the product registration necessary to sell Neutrolin in the
Republic of Korea. Revenue associated with the non-refundable
up-front payment under this arrangement is deferred and recognized
as revenue on a straight-line basis over the contractual term of
our agreement. Deferred revenue
related to this agreement at December 31, 2016 and 2015 amounted to
approximately $29,000 and $38,000,
respectively.
Inventory Valuation
We
engage third parties to manufacture and package inventory held for
sale and warehouse such goods until packaged for final distribution
and sale. Inventories are stated at the lower of cost or market
price with cost determined on a first-in, first-out basis.
Inventories are reviewed periodically to identify slow-moving or
obsolete inventory based on sales activity, both projected and
historical, as well as product shelf-life. In evaluating the
recoverability of our inventories, we consider the probability that
revenue will be obtained from the future sale of the related
inventory and, if required, will write down inventory quantities in
excess of expected requirements. Expired inventory is disposed of
and the related costs are recognized as cost of product sales in
our consolidated statements of operations.
We
analyze our inventory levels to identify inventory that may expire
prior to sale, inventory that has a cost basis in excess of its
estimated realizable value, or inventory in excess of expected
sales requirements. Although the manufacturing of our products is
subject to strict quality controls, certain batches or units of
product may no longer meet quality specifications or may expire,
which would require adjustments to our inventory
values.
In the
future, reduced demand, quality issues or excess supply beyond
those anticipated by management may result in an adjustment to
inventory levels, which would be recorded as an increase to cost of
product sales. The determination of whether or not inventory costs
will be realizable requires estimates by our management. A critical
input in this determination is future expected inventory
requirements based on our internal sales forecasts which we then
compare to the expiry dates of inventory on hand. To the extent
that inventory is expected to expire prior to being sold, we will
write down the value of inventory. If actual results differ from
those estimates, additional inventory write-offs may be
required.
Short-Term Investments
We
determine the appropriate classification of marketable securities
at the time of purchase and reevaluate such designation as of each
balance sheet date. Investments in marketable debt and equity
securities classified as available-for-sale are reported at fair
value. Fair values of our investments are determined using quoted
market prices in active markets for identical assets or liabilities
or quoted prices for similar assets or liabilities or other inputs
that are observable or can be corroborated by observable market
data for substantially the full term of the assets or liabilities.
Our marketable securities are highly liquid and consist of U.S.
government agency securities, high-grade corporate obligations and
commercial paper with maturities of more than 90 days but less than
12 months. Changes in fair value that are considered temporary are
reported net of tax in other comprehensive income (loss). Realized
gains and losses, amortization of premiums and discounts and
interest and dividends earned are included in income (expense) on
the condensed consolidated statements of operations and
comprehensive income (loss). The cost of investments for purposes
of computing realized and unrealized gains and losses is based on
the specific identification method. Investments with maturities
beyond one year, if any, are classified as short-term based on
management’s intent to fund current operations with these
securities or to make them available for current operations. For
declines, if any, in the fair value of equity securities that are
considered other-than-temporary, impairment losses are charged to
other (income) expense, net. We consider available evidence in
evaluating potential impairments of our investments, including the
duration and extent to which fair value is less than cost and, for
equity securities, our ability and intent to hold the
investments.
47
Fair Value Measurements
We
categorize our financial instruments into a three-level fair value
hierarchy that prioritize the inputs to valuation techniques used
to measure fair value. The fair value hierarchy gives the highest
priority to quoted prices in active markets for identical assets
(Level 1) and the lowest priority to unobservable inputs (Level 3).
If the inputs used to measure fair value fall within different
levels of the hierarchy, the category level is based on the lowest
priority level input that is significant to the fair value
measurement of the instrument. Financial assets recorded at fair
value on our condensed consolidated balance sheets are categorized
as follows:
●
Level 1
inputs—Observable inputs that reflect quoted prices
(unadjusted) for identical assets or liabilities in active
markets.
●
Level 2
inputs— Significant other observable inputs (e.g., quoted
prices for similar items in active markets, quoted prices for
identical or similar items in markets that are not active, inputs
other than quoted prices that are observable such as interest rate
and yield curves, and market-corroborated inputs).
●
Level 3
inputs—Unobservable inputs for the asset or liability, which
are supported by little or no market activity and are valued based on management’s
estimates of assumptions that market participants would use in
pricing the asset or liability.
Embedded Derivative Liabilities:
We
have previously had several series of preferred stock and warrants
that contained embedded derivatives. We evaluate all our
financial instruments to determine if those instruments or any
embedded components of those instruments qualify as derivatives
that need to be separately accounted for in accordance with FASB
ASC 815, “Derivatives and Hedging”.
Embedded derivatives satisfying certain criteria are recorded at
fair value at issuance and marked-to-market at each balance sheet
date with the change in the fair value recorded as income or
expense. In addition, upon the occurrence of an event that
requires the derivative liability to be reclassified to equity, the
derivative liability is revalued to fair value at that
date.
We
account for stock warrants as either equity instruments or
derivative liabilities depending on the specific terms of the
warrant agreement. Stock warrants that allow for cash
settlement or provide for certain modifications of the warrant
exercise price are accounted for as derivative liabilities.
For those liability-classified warrants that have down-round
provisions which allow the exercise price to be adjusted as a
result of certain future financing transactions, we use level 3
inputs to value those warrants. The estimated fair values of
the warrant liabilities with downround protection were determined
using a Monte Carlo option pricing model which takes into account
the probabilities of certain events occurring over the life of the
warrants. The derivative liabilities are adjusted to
their estimated fair values at each reporting period, with any
decrease or increase in the estimated fair value being recorded in
other income (expense).
Recent Authoritative Pronouncements:
In
May 2014, the FASB issued new guidance related to how an entity
should recognize revenue. The guidance specifies that an entity
should recognize revenue to depict the transfer of promised goods
or services to customers in an amount that reflects the
consideration to which the entity expects to be entitled in
exchange for those goods and services. In addition, the guidance
expands the required disclosures related to revenue and cash flows
from contracts with customers. The guidance is effective for us
beginning in the first quarter of 2017. Early adoption is not
permitted and retrospective application is required. We are
currently evaluating the impact of adopting this guidance on our
consolidated financial statements.
In
July 2015, the FASB issued an accounting standard that requires
inventory be measured at the lower of cost and net realizable value
and options that currently exist for market value be eliminated.
The standard defines net realizable value as estimated selling
prices in the ordinary course of business, less reasonably
predictable costs of completion, disposal, and transportation and
is effective for reporting periods beginning after December 15,
2016 and interim periods within those fiscal years with early
adoption permitted. The guidance should be applied prospectively.
We are evaluating the impact the adoption of this guidance will
have on the determination or reporting of our consolidated
financial statements.
48
In
November 2015, the FASB issued guidance that requires that deferred
tax liabilities and assets be classified as noncurrent in a
classified statement of financial position. The current requirement
that deferred tax liabilities and assets of a tax-paying component
of an entity be offset and presented as a single amount is not
affected by this amendment. The new guidance is effective for
fiscal years, and interim periods within those years, beginning
after December 15, 2016. Early adoption is permitted and the
standard may be applied either retrospectively or on a prospective
basis to all deferred tax assets and liabilities. We are evaluating the impact the adoption of this
guidance will have on the determination or reporting of our
consolidated financial statements.
In
January 2016, the FASB issued a new standard that modifies certain
aspects of the recognition, measurement, presentation, and
disclosure of financial instruments. The accounting standard update
is effective for fiscal years, and interim periods within those
years, beginning after December 15, 2017, and early adoption is
permitted. We are currently assessing the impact that adopting this
new accounting guidance will have on our consolidated financial
statements.
In February 2016, the FASB issued new guidance
related to how an entity should lease assets and lease liabilities.
The guidance specifies that an entity who is a lessee under lease
agreements should recognize lease assets and lease
liabilities for those leases classified as operating leases under
previous FASB guidance. Accounting for
leases by lessors is largely unchanged under the new guidance. The
guidance is effective for us beginning in the first quarter of
2019. Early adoption is permitted. In transition, lessees
and lessors are required to recognize and measure leases at the
beginning of the earliest period presented using a modified
retrospective approach. We are
evaluating the impact of adopting this guidance on our consolidated
financial statements.
In
April 2016, the FASB issued an update which clarifies two aspects
of the new revenue guidance by providing guidance on how to
identify performance obligations and providing implementation
guidance surrounding licensing. The amendments in this update do
not change the core principle of the new revenue guidance. The
guidance is effective for us beginning in the first quarter of
2017. Early adoption is not permitted and retrospective application
is required. We are currently evaluating the impact of adopting
this guidance on our consolidated financial
statements.
In June
2016, the FASB issued new guidance which replaces the incurred loss
impairment methodology in current GAAP with a methodology that
reflects expected credit losses and requires consideration of a
broader range of reasonable and supportable information to inform
credit loss estimates. The guidance is effective for us beginning
in the first quarter of fiscal year 2020. Early adoption is
permitted beginning in the first quarter of fiscal year 2019. We
are evaluating the impact of adopting this guidance on our
consolidated financial statements.
In
August 2016, the FASB issued new guidance which clarifies how
certain cash receipts and cash payments are presented and
classified in the statement of cash flows in order to reduce
diversity in practice. The guidance is effective for us beginning
in the first quarter of fiscal year 2018. Early adoption is
permitted. We are evaluating the impact of adopting this guidance
on our consolidated financial statements.
In
November 2016, the FASB issued new guidance which clarifies how
restricted cash is presented and classified in the statement of
cash flows. The guidance is effective for us beginning in the first
quarter of fiscal year 2018. Early adoption is permitted. We are
evaluating the impact of adopting this guidance on our consolidated
financial statements.
In
January 2017, the FASB issued new guidance which clarifies the
definition of a business in a business combination. The guidance is
effective for us beginning in the first quarter of fiscal year
2018. Early adoption is permitted. We are evaluating the impact of
adopting this guidance on our consolidated financial
statements.
In
January 2017, the FASB issued new guidance which simplifies the
test for goodwill impairment. The guidance is effective for us
beginning in the first quarter of fiscal year 2020. Early adoption
is permitted for interim or annual goodwill impairments tests after
January 1, 2017. We are evaluating the impact of adopting this
guidance on our consolidated financial statements.
Recently Adopted Authoritative Pronouncements:
In
June 2014, the FASB issued an accounting standard that clarifies
the accounting for share-based payments when the terms of an award
provide that a performance target could be achieved after the
requisite service period. The standard requires that a
performance target that affects vesting and that could be achieved
after the requisite service period be treated as a performance
condition. The amendments are effective for
interim and annual reporting periods beginning after December 15,
2015. This adoption did not have a material impact on
our consolidated financial statements.
49
In
August 2014, the FASB issued new guidance related to disclosures of
uncertainties about an entity’s ability to continue as a
going concern. The ASU requires management to evaluate whether
there are conditions and events that raise substantial doubt about
the entity’s ability to continue as a going concern within
one year after the financial statements are issued and if
management’s plans will alleviate that doubt. Management will
be required to make this evaluation for both annual and interim
reporting periods. We adopted this
guidance as of the fiscal year ended December 31,
2016.
In
April 2015, the FASB issued new guidance which requires that debt
issuance costs related to a recognized debt liability be presented
in the balance sheet as a direct deduction from the carrying amount
of that debt liability, consistent with debt discounts. This
guidance requires retrospective adoption and was effective for us
beginning in the first quarter of 2016. This adoption
did not have a material impact on our consolidated financial
statements.
In March 2016, the FASB issued new guidance which
simplifies several aspects of the accounting for share-based
payment transactions, including the income tax consequences,
classification of awards as either equity or liabilities, and
classification on the statement of cash flows. We adopted this guidance in the fourth quarter of
fiscal year 2016. This adoption did not have a material impact on
our consolidated financial statements.
Off-Balance Sheet Arrangements
We do
not have any off-balance sheet arrangements.
Item
7A.
Quantitative
and Qualitative Disclosures About Market Risk
Not
applicable.
Item
8.
Financial
Statements and Supplementary Data
Item
9.
Changes
in and Disagreements With Accountants on Accounting and Financial
Disclosure
Not
applicable.
Item
9A.
Controls
and Procedures
As of
the end of the period covered by this Annual Report on Form 10-K,
we carried out an evaluation, under the supervision and with the
participation of our management, including our Chief Executive
Officer and our Chief Financial Officer, of the effectiveness of
the design and operation of our disclosure controls and procedures
(as defined in the Exchange Act Rules 13a-15(e) and 15d-15(e)) (the
“Exchange Act”). Based on the foregoing evaluation, our
Chief Executive Officer and Chief Financial Officer have concluded
that our disclosure controls and procedures are effective to ensure
that information required to be disclosed by us in the reports we
file or submit under the Exchange Act is recorded, processed,
summarized and reported within the time periods specified in the
rules and forms of the SEC, and that such information is
accumulated and communicated to our management, including our Chief
Executive Officer and Chief Financial Officer, to allow timely
decisions regarding required disclosures.
Changes
in Internal Control Over Financial Reporting
There
were no changes in our internal control over financial reporting
during our fourth quarter ended December 31, 2016, or in other
factors that could significantly affect these controls, that
materially affected, or are reasonably likely to materially affect,
our internal control over financial reporting.
50
Management’s
Annual Report on Internal Controls Over Financial
Reporting
Our
management is responsible for establishing and maintaining adequate
internal control over financial reporting and for the assessment of
the effectiveness of internal control over financial reporting. As
defined by the Securities and Exchange Commission, internal control
over financial reporting is a process designed by, or under the
supervision of, our principal executive and principal financial
officers and effected by our Board of Directors, management and
other personnel, to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of the
consolidated financial statements in accordance with U.S. generally
accepted accounting principles.
Our
internal control over financial reporting includes those policies
and procedures that (1) pertain to the maintenance of records that,
in reasonable detail, accurately and fairly reflect our
transactions and dispositions of our assets; (2) provide reasonable
assurance that transactions are recorded as necessary to permit
preparation of the consolidated financial statements in accordance
with generally accepted accounting principles, and that our
receipts and expenditures are being made only in accordance with
authorizations of our management and directors; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use or disposition of our assets that
could have a material effect on the consolidated financial
statements.
Because
of its inherent limitations, internal control over financial
reporting may not prevent or detect misstatements. Also,
projections of any evaluation of effectiveness to future periods
are subject to the risk that controls may become inadequate because
of changes in conditions, or that the degree of compliance with the
policies or procedures may deteriorate.
In
connection with the preparation of our annual consolidated
financial statements, management, including, our Chief Executive
Officer and Chief Financial Officer, have undertaken an assessment
of the effectiveness of our internal control over financial
reporting as of December 31, 2016, based on the criterial
established in Internal Control—Integrated Framework (2013)
issued by the Committee of Sponsoring Organizations of the Treadway
Commission (COSO). Management’s assessment included an
evaluation of the design of our internal control over financial
reporting and testing of the operational effectiveness of those
controls.
Based
on this evaluation, management has concluded that our internal
control over financial reporting was effective as of
December 31, 2016.
Friedman LLP, the
independent registered public accounting firm that audited our
consolidated financial statements included in this report, has
issued their report on the effectiveness of internal control over
financial reporting as of December 31, 2016, a copy of which is
included herein.
51
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the
Board of Directors and Stockholders of CorMedix, Inc.
We have
audited CorMedix, Inc. and subsidiary’s (the
“Company”) internal control over financial reporting as
of December 31, 2016, based on criteria established in Internal
Control-Integrated Framework (2013) issued by the Committee of
Sponsoring Organizations of the Treadway Commission (COSO). The
Company’s management is responsible for maintaining effective
internal control over financial reporting and for its assessment of
the effectiveness of internal control over financial reporting,
included in the accompanying Management’s Report on Internal
Control Over Financial Reporting. Our responsibility is to express
an opinion on the effectiveness of the Company’s internal
control over financial reporting based on our audit.
We
conducted our audit in accordance with the standards of the Public
Company Accounting Oversight Board (United States). Those standards
require that we plan and perform the audit to obtain reasonable
assurance about whether effective internal control over financial
reporting was maintained in all material respects. Our audit
included obtaining an understanding of internal control over
financial reporting, assessing the risk that a material weakness
exists, and testing and evaluating the design and operating
effectiveness of internal control based on the assessed risk. Our
audit also included performing such other procedures as we
considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A
company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with generally
accepted accounting principles. A company’s internal control
over financial reporting includes those policies and procedures
that (1) pertain to the maintenance of records that, in reasonable
detail, accurately and fairly reflect the transactions and
dispositions of the assets of the company; (2) provide reasonable
assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally
accepted accounting principles, and that receipts and expenditures
of the company are being made only in accordance with
authorizations of management and directors of the company; and (3)
provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because
of its inherent limitations, internal control over financial
reporting may not prevent or detect misstatements. Also,
projections of any evaluation of effectiveness to future periods
are subject to the risk that controls may become inadequate because
of changes in conditions, or that the degree of compliance with the
policies or procedures may deteriorate.
In our
opinion, CorMedix, Inc. and subsidiary maintained, in all material
respects, effective internal control over financial reporting as of
December 31, 2016, based on criteria established in Internal
Control-Integrated Framework issued by COSO in 2013.
We also
have audited, in accordance with the standards of the Public
Company Accounting Oversight Board (United States), the
consolidated balance sheets of CorMedix, Inc. and subsidiary as of
December 31, 2016, and 2015, and the related consolidated
statements of operations, comprehensive income (loss), cash flows,
and stockholder’s equity for each of the years in the
three-year period ended December 31, 2016, and our report dated
March 16, 2017 expressed an unqualified opinion thereon that
included an explanatory paragraph regarding CorMedix, Inc.’s
ability to continue as a going concern.
|
/s/ Friedman LLP
|
|
|
East Hanover, NJ
|
|
|
March 16, 2017
|
|
52
Item
9B.
Other
Information
None.
PART III
Item 10.
Directors,
Executive Officers, and Corporate Governance
We have
adopted a written Code of Conduct and Ethics that applies to our
directors, executive officers and all employees. We intend to
disclose any amendments to, or waivers from, our code of ethics and
business conduct that are required to be publicly disclosed
pursuant to rules of the SEC by filing such amendment or waiver
with the SEC. This code of ethics and business conduct can be found
in the “Investors - Corporate Governance” section of
our website, www.cormedix.com.
The
other information required by this Item concerning our directors
and executive officers is incorporated by reference from the
section captioned “Proposal No. 1—Election of
Directors” and “Corporate Governance” contained
in our proxy statement related to the 2017 Annual Meeting of
Stockholders scheduled to be held on June 6, 2017 which we intend
to file with the SEC within 120 days of the end of our fiscal year
pursuant to General Instruction G(3) of Form 10-K. The information
required by this Item concerning compliance with Section 16(a) of
the Exchange Act by our directors, executive officers and persons
who own more than 10% of our outstanding common stock is
incorporated by reference from the section captioned “Section
16(a) Beneficial Ownership Reporting Compliance” contained in
the proxy statement.
Item 11.
Executive
Compensation
The
information required by this Item concerning directors and
executive compensation is incorporated by reference from the
section captioned “Director Compensation,”
“Executive Compensation – Summary Compensation
Table” “Executive Compensation – Compensation
Discussion and Analysis,” “Executive Compensation
– Grants of Plan Based Awards,” “Executive
Compensation – Option Exercises and Stock Vested,”
“Executive Compensation – Outstanding Equity Awards at
Fiscal Year End 2015” “Executive Compensation –
Compensation Committee Interlocks and Insider Participation,”
and “Executive Compensation – Compensation Committee
Report” contained in the proxy statement.
Item 12.
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
The
information regarding our equity compensation plans required by
this Item is found in Item 5 of this report. The other information
required by this Item is incorporated by reference to the
information under the section captioned “Security Ownership
of Certain Beneficial Owners and Management” contained in the
proxy statement.
Item 13.
Certain
Relationships and Related Transactions and Director
Independence
The
information required by this Item is incorporated by reference to
the information under the section captioned “Certain
Relationships and Related Transactions” and “Proposal
No. 1—Election of Directors” contained in the
proxy statement.
Item 14.
Principal
Accountant Fees and Services
The
information required by this Item is incorporated by reference to
the information under the section captioned “Auditor and
Audit Committee Matters” contained in the proxy
statement.
53
PART IV
Item
15.
Exhibits
and Financial Statement Schedules
(a)
List of documents
filed as part of this report:
1.
Financial
Statements:
The
financial statements of the Company and the related reports of the
Company’s independent registered public accounting firms
thereon have been filed under Item 8 hereof.
2.
Financial Statement
Schedules:
None.
3.
Exhibit
Index
The
following is a list of exhibits filed as part of this Form
10-K:
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
3.1
|
|
Form of
Amended and Restated Certificate of Incorporation.
|
|
S-1/A
|
|
3/01/2010
|
|
3.3
|
|
|
|
3.2
|
|
Form of
Amended and Restated Bylaws.
|
|
S-1/A
|
|
3/01/2010
|
|
3.4
|
|
|
|
3.3
|
|
Certificate
of Amendment to Amended and Restated Certificate of Incorporation,
dated December 3, 2012.
|
|
10-K
|
|
3/27/2013
|
|
3.3
|
|
|
|
3.4
|
|
Certificate
of Designation of Series A Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
February 18, 2013, as corrected on February 19, 2013.
|
|
8-K
|
|
2/19/2013
|
|
3.3
|
|
|
|
3.5
|
|
Certificate
of Designation of Series B Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
July 26, 2013.
|
|
8-K
|
|
7/26/2013
|
|
3.4
|
|
|
|
3.6
|
|
Certificate
of Designation of Series C-1 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.5
|
|
|
|
3.7
|
|
Certificate
of Amendment to Certificate of Designation of Series C-1 Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.10
|
|
|
|
3.8
|
|
Certificate
of Designation of Series C-2 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.6
|
|
|
|
3.9
|
|
Certificate
of Amendment to Certificate of Designation of Series C-2 Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.11
|
|
|
|
3.10
|
|
Certificate
of Designation of Series C-3 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.9
|
|
|
|
3.11
|
|
Certificate
of Designation of Series D Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.7
|
|
|
|
3.12
|
|
Certificate
of Amendment to Certificate of Designation of Series D Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.12
|
|
|
54
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
3.13
|
|
Certificate
of Designation of Series E Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.8
|
|
|
|
3.14
|
|
Certificate
of Amendment to Certificate of Designation of Series E Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.13
|
|
|
|
4.1
|
|
Specimen
of Common Stock Certificate.
|
|
S-1/A
|
|
3/19/2010
|
|
4.1
|
|
|
|
4.2
|
|
Stockholder
Agreement, dated as of January 30, 2008, between CorMedix Inc. and
ND Partners LLC.
|
|
S-1
|
|
11/25/2009
|
|
4.7
|
|
|
|
4.3
|
|
Form of
Warrant Issued in September/November 2012.
|
|
10-Q
|
|
11/13/2012
|
|
4.2
|
|
|
|
4.4
|
|
Form of
Sales Agent Warrant issued in September 2012.
|
|
10-Q
|
|
11/13/2012
|
|
4.3
|
|
|
|
4.5
|
|
Warrant
issued to ND Partners in April 2013.
|
|
10-Q
|
|
5/15/2013
|
|
4.18
|
|
|
|
4.6
|
|
Form of
Warrant issued on February 19, 2013.
|
|
8-K
|
|
2/19/2013
|
|
4.13
|
|
|
|
4.7
|
|
Form of
Warrant issued on July 30, 2013.
|
|
8-K
|
|
7/26/2013
|
|
4.21
|
|
|
|
4.8
|
|
Form of
Warrant issued on October 22, 2013.
|
|
8-K
|
|
10/18/2013
|
|
4.22
|
|
|
|
4.9
|
|
Form of
Warrant issued on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
4.23
|
|
|
|
4.10
|
|
Form of
Warrant issued on March 10, 2014.
|
|
8-K
|
|
3/05/2014
|
|
4.24
|
|
|
|
4.11
|
|
Form of
Warrant issued on March 3, 2015.
|
|
8-K
|
|
3/04/2015
|
|
4.1
|
|
|
|
4.12
|
|
Amended
and Restated Warrant originally issued May 30, 2013.
|
|
8-K
|
|
3/04/2015
|
|
4.3
|
|
|
|
4.13
|
|
Amended
and Restated Warrant originally issued March 24, 2010.
|
|
8-K
|
|
3/04/2015
|
|
4.2
|
|
|
|
4.14
|
|
Registration
Rights Agreement, dated March 3, 2015, by and between CorMedix Inc.
and Manchester Securities Corp.
|
|
8-K
|
|
3/04/2015
|
|
4.5
|
|
|
|
10.1*
|
|
License
and Assignment Agreement, dated as of January 30, 2008, between the
Company and ND Partners LLC.
|
|
S-1/A
|
|
12/312009
|
|
10.5
|
|
|
|
10.2
|
|
Escrow
Agreement, dated as of January 30, 2008, among the Company, ND
Partners LLC and the Secretary of the Company, as Escrow
Agent.
|
|
S-1
|
|
11/25/2009
|
|
10.6
|
|
|
|
10.3
|
|
Consulting
Agreement, dated as of January 30, 2008, between the Company and
Frank Prosl.
|
|
S-1
|
|
11/25/2009
|
|
10.12
|
|
|
|
10.4*
|
|
Manufacture
and Development Agreement, dated as of March 5, 2007, by and
between the Company and Emcure Pharmaceuticals USA,
Inc.
|
|
S-1/A
|
|
12/31/2009
|
|
10.14
|
|
|
|
10.5
|
|
Amended
and Restated 2006 Stock Incentive Plan.
|
|
S-1/A
|
|
3/01/2010
|
|
10.8
|
|
|
|
10.6
|
|
Form of
Indemnification Agreement between the Company and each of its
directors and executive officers.
|
|
S-1/A
|
|
3/01/2010
|
|
10.17
|
|
|
|
10.7
|
|
Agreement
for Work on Pharmaceutical Advertising dated January 10, 2013 by
and between MKM Co-Pharma GmbH and CorMedix Inc.
|
|
8-K
|
|
1/16/2013
|
|
10.22
|
|
|
|
10.8
|
|
2013
Stock Incentive Plan
|
|
10-K
|
|
3/27/2013
|
|
10.27
|
|
|
|
10.9
|
|
Form of
Securities Purchase Agreement, dated January 7, 2014, between
CorMedix Inc. and the investors named therein.
|
|
8-K
|
|
1/09/2014
|
|
10.36
|
|
|
|
10.10
|
|
Preliminary
Services Agreement dated April 8, 2015, between CorMedix Inc. and
[RC]2 Pharma Connect
LLC.
|
|
10-Q
|
|
8/06/2015
|
|
10.1
|
|
|
|
10.11
|
|
Release
of Claims and Severance Modification, dated July 17, 2015, between
Randy Milby and CorMedix Inc.
|
|
|
|
|
|
|
|
X
|
55
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
10.12
|
|
Employment
Agreement, effective February 1, 2017, between CorMedix Inc. and
Robert Cook.**
|
|
|
|
|
|
|
|
X
|
|
10.13
|
|
Employment
Agreement, effective February 1, 2017, between CorMedix Inc. and
Judith Abrams.**
|
|
|
|
|
|
|
|
X
|
|
10.14
|
|
Employment
Agreement, effective March 1, 2017, between CorMedix Inc. and John
Armstrong.
|
|
|
|
|
|
|
|
X
|
|
21.1
|
|
List of
Subsidiaries
|
|
10-K
|
|
3/27/2013
|
|
21.1
|
|
|
|
23.1
|
|
Consent
of Independent Registered Public Accounting Firm.
|
|
|
|
|
|
|
|
X
|
|
31.1
|
|
Certification
of Principal Executive Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
31.2
|
|
Certification
of Principal Financial Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
32.1
|
|
Certification
of Principal Executive Officer pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
32.2
|
|
Certification
of Principal Financial Officer pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
101
|
|
The
following materials from CorMedix Inc. Form 10-K for the year ended
December 31, 2016, formatted in Extensible Business Reporting
Language (XBRL): (i) Balance Sheets at December 31, 2016 and 2015,
(ii) Statements of Operations for the years ended December 31,
2016, 2015 and 2014, (iii) Statements of Changes in
Stockholders’ Equity for the years ended December 31, 2016,
2015 and 2014, (iv) Statements of Cash Flows for the years ended
December 31, 2016, 2015 and 2014 and (v) Notes to the Financial
Statements.***
|
|
|
|
|
|
|
|
X
|
_____________
|
*
|
Confidential
treatment has been granted for portions of this document. The
omitted portions of this document have been filed separately with
the SEC.
|
|
**
|
Confidential
treatment has been requested with respect to certain portions of
this exhibit. Omitted portions have been filed
separately with the SEC.
|
|
***
|
Pursuant
to Rule 406T of Regulation S-T, the Interactive Data Files in
Exhibit 101 hereto are deemed not filed or part of a registration
statement or prospectus for purposes of Sections 11 or 12 of the
Securities Act of 1933, as amended, are deemed not filed for
purposes of Section 18 of the Securities Exchange Act of 1934, as
amended, and otherwise are not subject to liability under those
sections.
|
56
SIGNATURES
Pursuant to the
requirements of the Securities Exchange Act of 1934, the Registrant
has duly caused this report to be signed on its behalf by the
undersigned thereunto duly authorized.
|
|
|
CORMEDIX
INC.
|
|
|
|
|
|
|
|
|
|
|
|
|
March
16, 2017
|
|
By: /s/
Khoso Baluch
|
|
|
|
|
Khoso
Baluch
|
|
|
|
|
Chief
Executive Officer
(Principal
Executive Officer)
|
|
|
March
16, 2017
|
|
By: /s/
Robert Cook
|
|
|
|
|
Robert
Cook
|
|
|
|
|
Chief
Financial Officer
(Principal
Financial and Accounting Officer)
|
|
Pursuant to the
requirements of the Securities Exchange Act of 1934, this report
has been signed below by the following persons on behalf of the
Registrant and in the capacities and on the dates
indicated:
|
Signature
|
Title
|
Date
|
|
|
|
|
|
/s/
Khoso Baluch
|
Chief
Executive Officer and Director
|
March
16, 2017
|
|
Khoso
Baluch
|
(Principal
Executive Officer)
|
|
|
|
|
|
|
/s/
Robert Cook
|
Chief
Financial Officer
|
March
16, 2017
|
|
Robert
Cook
|
(Principal
Financial and Accounting Officer)
|
|
|
|
|
|
|
/s/
Cora Tellez
|
Chairman of the
Board and Director
|
March
16, 2017
|
|
Cora
Tellez
|
|
|
|
|
|
|
|
/s/
Janet Dillione
|
Director
|
March
16, 2017
|
|
Janet
Dillione
|
|
|
|
|
|
|
|
/s/
Michael George
|
Director
|
March
16, 2017
|
|
Michael
George
|
|
|
|
|
|
|
|
/s/
Myron Kaplan
|
Director
|
March
16, 2017
|
|
Myron
Kaplan
|
|
|
|
|
|
|
|
/s/
Taunia Markvicka
|
Director
|
March
16, 2017
|
|
Taunia
Markvicka
|
|
|
57
CORMEDIX
INC. AND SUBSIDIARY
FINANCIAL
STATEMENTS
Financial
Statements Index
|
|
|
|
Report
of Independent Registered Public Accounting Firm
|
F-2
|
|
|
|
|
Consolidated
Balance Sheets December 31, 2016
and 2015
|
F-3
|
|
|
|
|
Consolidated
Statements of Operations and Comprehensive Income (Loss)
Years
Ended December 31, 2016, 2015 and 2014
|
F-4
|
|
|
|
|
Consolidated
Statements of Changes in Stockholders’ Equity
Years
Ended December 31, 2016, 2015 and 2014
|
F-5
|
|
|
|
|
Consolidated
Statements of Cash Flows Years Ended
December 31, 2016, 2015 and 2014
|
F-6
|
|
|
|
|
Notes
to Consolidated Financial Statements
|
F-8
|
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the
Board of Directors and Stockholders
CorMedix,
Inc.
We have
audited the accompanying consolidated balance sheets of CorMedix,
Inc. and Subsidiary (the “Company”) as of December 31,
2016 and 2015, and the related consolidated statements of
operations and comprehensive income (loss), stockholders’
equity, and cash flows for each of the years in the three-year
period ended 2016. The Company’s management is responsible
for these financial statements. Our responsibility is to express an
opinion on these financial statements based on our
audits.
We
conducted our audits in accordance with the standards of the Public
Company Accounting Oversight Board (United States). Those standards
require that we plan and perform the audit to obtain reasonable
assurance about whether the financial statements are free of
material misstatement. An audit includes examining, on a test
basis, evidence supporting the amounts and disclosures in the
financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by
management, as well as evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable basis
for our opinion.
In our
opinion, the financial statements referred to above present fairly,
in all material respects, the financial position of the Company as
of 2016 and 2015, and the results of its operations and its cash
flows for each of the years in the three-year period ended December
31, 2016, in conformity with accounting principles generally
accepted in the United States of America.
The
accompanying consolidated financial statements have been prepared
assuming the Company will continue as a going concern. As described
in Note 2, the Company has an accumulated deficit of $119.2 million
as of December 31, 2016, has recurring losses, and negative cash
flow from operations. These conditions, among others, raise
substantial doubt about the Company’s ability to continue as
a going concern. Management’s plans in regard to these
matters are also described in Note 2. The consolidated financial
statements do not include any adjustments that may result from the
outcome of this uncertainty. If the Company is unable to obtain
additional financing, there could be a material adverse effect on
the Company.
We also
have audited, in accordance with the standards of the Public
Company Accounting Oversight Board (United States), the
Company’s internal control over financial reporting as of the
year ended December 31, 2016, based on criteria established in
Internal Control—Integrated Framework (2013) issued by the
Committee of Sponsoring Organizations of the Treadway Commission
(COSO), and our report dated March 16, 2017, expressed an
unqualified opinion.
|
/s/ Friedman LLP
|
|
|
East Hanover, NJ
|
|
|
March 16, 2017
|
|
F-2
CORMEDIX INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
December
31, 2016 and 2015
|
|
December
31,
|
|
|
|
2016
|
2015
|
|
ASSETS
|
|
|
|
Current
assets
|
|
|
|
Cash and cash
equivalents
|
$8,064,490
|
$11,817,418
|
|
Restricted
cash
|
171,553
|
171,553
|
|
Short-term
investments
|
12,100,920
|
23,568,386
|
|
Trade
receivables
|
12,014
|
315,771
|
|
Inventories,
net
|
166,733
|
376,569
|
|
Prepaid research
and development expenses
|
943,924
|
430,162
|
|
Other prepaid
expenses and current assets
|
372,057
|
379,004
|
|
Total current
assets
|
21,831,691
|
37,058,863
|
|
Property and
equipment, net
|
69,695
|
37,866
|
|
Other
assets
|
5,000
|
5,000
|
|
TOTAL
ASSETS
|
$21,906,386
|
$37,101,729
|
|
|
|
|
|
LIABILITIES
AND STOCKHOLDERS’ EQUITY
|
|
|
|
Current
liabilities
|
|
|
|
Accounts
payable
|
$1,645,298
|
$1,709,397
|
|
Accrued
expenses
|
2,342,352
|
1,221,557
|
|
Deferred
revenue
|
104,210
|
130,409
|
|
Total current liabilities
|
4,091,860
|
3,061,363
|
|
Deferred revenue,
long term
|
-
|
28,878
|
|
TOTAL
LIABILITIES
|
4,091,860
|
3,090,241
|
|
|
|
|
|
COMMITMENTS
AND CONTINGENCIES (Note 6)
|
|
|
|
|
|
|
|
STOCKHOLDERS’
EQUITY
|
|
|
|
Preferred stock -
$0.001 par value: 2,000,000 shares authorized; 450,085 shares
issued and outstanding at December 31, 2016 and 2015,
respectively
|
450
|
450
|
|
Common stock -
$0.001 par value: 80,000,000 shares authorized; 40,432,339 and
35,963,348 shares issued and outstanding at December 31, 2016 and
2015, respectively
|
40,433
|
35,964
|
|
Accumulated other
comprehensive gain
|
81,186
|
62,130
|
|
Additional paid-in
capital
|
136,857,409
|
128,304,539
|
|
Accumulated
deficit
|
(119,164,952)
|
(94,391,595)
|
|
TOTAL
STOCKHOLDERS’ EQUITY
|
17,814,526
|
34,011,488
|
|
TOTAL
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
$21,906,386
|
$37,101,729
|
The
accompanying notes are integral part of these consolidated
financial statements.
F-3
CORMEDIX INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME
(LOSS)
Years
Ended December 31, 2016, 2015 and 2014
|
|
December
31,
|
||
|
|
2016
|
2015
|
2014
|
|
Revenue
|
|
|
|
|
Net
sales
|
$224,105
|
$210,130
|
$189,274
|
|
Cost
of sales
|
(366,673)
|
(318,718)
|
(445,799)
|
|
Gross
loss
|
(142,568)
|
(108,588)
|
(256,525)
|
|
Operating
Expenses
|
|
|
|
|
Research and
development
|
(15,735,300)
|
(6,281,823)
|
(1,318,734)
|
|
Selling, general
and administrative
|
(8,883,050)
|
(10,263,560)
|
(7,326,861)
|
|
Total operating
expenses
|
(24,618,350)
|
(16,545,383)
|
(8,645,595)
|
|
Loss
From Operations
|
(24,760,918)
|
(16,653,971)
|
(8,902,120)
|
|
Other
Income (Expense)
|
|
|
|
|
Interest
income
|
126,774
|
60,393
|
2,714
|
|
Foreign exchange
transaction loss
|
(8,172)
|
(6,735)
|
(150,803)
|
|
Loss on issuance of
preferred stock, convertible notes and warrants
|
-
|
-
|
(89,590)
|
|
Value of warrants
issued in connection with backstop financing
|
-
|
(1,583,252)
|
-
|
|
Change in fair
value of derivative liabilities
|
-
|
-
|
(8,848,953)
|
|
Loss on
modification of equity instruments and extinguishment of derivative
liabilities
|
-
|
-
|
(2,462,588)
|
|
Interest
expense
|
(1,311)
|
(3,964)
|
(2,087)
|
|
Total income
(expense)
|
117,291
|
(1,533,558)
|
(11,551,307)
|
|
Net
Loss
|
(24,643,627)
|
(18,187,529)
|
(20,453,427)
|
|
Other
Comprehensive Income Gain (Loss)
|
|
|
|
|
Unrealized gain
(loss) from investments
|
11,027
|
(24,239)
|
-
|
|
Foreign currency
translation gain (loss)
|
8,029
|
(12,603)
|
108,295
|
|
Total other
comprehensive income gain (loss)
|
19,056
|
(36,842)
|
108,295
|
|
Comprehensive
Loss
|
$(24,624,571)
|
$(18,224,371)
|
$(20,345,132)
|
|
Net
Loss
|
$(24,643,627)
|
$(18,187,529)
|
$(20,453,427)
|
|
Dividends,
including deemed dividends
|
-
|
(33,121)
|
(82,899)
|
|
Net
Loss Attributable To Common Shareholders
|
$(24,643,627)
|
$(18,220,650)
|
$(20,536,326)
|
|
Net
Loss Per Common Share – Basic and Diluted
|
$(0.65)
|
$(0.58)
|
$(0.96)
|
|
Weighted
Average Common Shares Outstanding – Basic and
Diluted
|
37,967,373
|
31,343,545
|
21,441,906
|
The
accompanying notes are integral part of these consolidated
financial statements.
F-4
CORMEDIX INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERSí
EQUITY
Years Ended December 31, 2016, 2015 and 2014
|
|
Common
Stock
|
Non-Voting
Preferred Stock – Series A, Series B, Series C-1, Series C-2,
C-3, Series D and Series E
|
Accumulated
Other Comprehen-sive Gain (Loss)
|
AdditionalPaid-inCapital
|
Accumulated
Deficit
|
Total
Stockholders’ Equity (Deficiency)
|
||
|
|
Shares
|
Amount
|
Shares
|
Amount
|
|
|
|
|
|
Balance at
December 31, 2013
|
16,606,695
|
$16,606
|
857,160
|
$857
|
$(9,323)
|
$51,720,156
|
$(55,750,639)
|
$(4,022,343)
|
|
Series C-3 non-voting preferred
stock issued in January 2014 financing at $10 per share,
net
|
-
|
-
|
200,000
|
200
|
-
|
-
|
-
|
200
|
|
Conversion of Series C-1 non-voting
preferred stock to common stock
|
1,400,000
|
1,400
|
(140,000)
|
(140)
|
-
|
2,446,124
|
-
|
2,447,384
|
|
Stock issued in connection with
March 2014 public offering at $2.50 per unit,
net
|
2,960,000
|
2,960
|
-
|
-
|
-
|
4,991,838
|
-
|
4,994,798
|
|
Reclassification of Series C-2 and
Series C-3 preferred stock conversion option derivative liability
to equity
|
-
|
-
|
-
|
-
|
-
|
6,235,398
|
-
|
6,235,398
|
|
Reclassification of derivative
liabilities to equity from modification of various equity
instruments including payment-in-kind dividends
|
-
|
-
|
53,788
|
54
|
-
|
11,740,809
|
-
|
11,740,863
|
|
Shares held in escrow upon
achievement of certain milestone
|
-
|
-
|
-
|
-
|
-
|
(36)
|
-
|
-
|
|
Stock-based
compensation
|
-
|
-
|
-
|
-
|
-
|
2,168,303
|
-
|
2,168,303
|
|
Stock issued in connection with
warrants cashless exercised
|
772,589
|
773
|
-
|
-
|
-
|
(773)
|
-
|
-
|
|
Stock issued in connection with
stock options exercised
|
455,000
|
455
|
-
|
-
|
-
|
317,695
|
-
|
318,150
|
|
Conversion of wages and fees to
common stock
|
57,384
|
57
|
-
|
-
|
-
|
96,794
|
-
|
96,851
|
|
Conversion of Series C-3 non-voting
preferred stock to common stock
|
210,000
|
210
|
(21,000)
|
(21)
|
-
|
(189)
|
-
|
-
|
|
Other comprehensive
gain
|
-
|
-
|
-
|
-
|
108,295
|
-
|
-
|
108,295
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(20,453,427)
|
(20,453,427)
|
|
Balance at
December 31, 2014
|
22,461,668
|
22,461
|
949,948
|
950
|
98,972
|
79,716,155
|
(76,204,066)
|
3,634,472
|
|
Conversion of Series B non-voting
preferred stock to common stock
|
454,546
|
455
|
(454,546)
|
(455)
|
-
|
-
|
-
|
-
|
|
Conversion of Series C-3 non-voting
preferred stock to common stock
|
425,000
|
425
|
(42,500)
|
(42)
|
-
|
(383)
|
-
|
-
|
|
Conversion of Series E non-voting
preferred stock to common stock
|
61,598
|
62
|
(2,817)
|
(3)
|
-
|
(59)
|
-
|
-
|
|
Stock issued in connection with
warrants exercised
|
4,581,783
|
4,582
|
-
|
-
|
-
|
14,653,579
|
-
|
14,658,161
|
|
Stock issued in connection with
warrants cashless exercised
|
2,158,033
|
2,158
|
-
|
-
|
-
|
(2,158)
|
-
|
-
|
|
Stock issued in connection with
stock options exercised
|
499,955
|
500
|
-
|
-
|
-
|
492,460
|
-
|
492,960
|
|
Stock issued in connection with
sale of common stock
|
5,310,037
|
5,310
|
-
|
-
|
-
|
28,446,538
|
-
|
28,451,848
|
|
Stock issued in connection with
conversion of wages
|
10,728
|
11
|
-
|
-
|
-
|
49,989
|
-
|
50,000
|
|
Value of warrants in connection
with backstop financing
|
-
|
-
|
-
|
-
|
-
|
1,583,252
|
-
|
1,583,252
|
|
Modification of warrant
agreement
|
-
|
-
|
-
|
-
|
-
|
112,982
|
-
|
112,982
|
|
Short swing profit
recovery
|
-
|
-
|
-
|
-
|
-
|
26,525
|
-
|
26,525
|
|
Stock-based
compensation
|
-
|
-
|
-
|
-
|
-
|
3,225,659
|
-
|
3,225,659
|
|
Other comprehensive
loss
|
-
|
-
|
-
|
-
|
(36,842)
|
-
|
-
|
(36,842)
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(18,187,529)
|
(18,187,529)
|
|
Balance at
December 31, 2015
|
35,963,348
|
35,964
|
450,085
|
450
|
62,130
|
128,304,539
|
(94,391,595)
|
34,011,488
|
|
Cumulative
effect of change in accounting principle (Note
8)
|
-
|
-
|
-
|
-
|
-
|
129,730
|
(129,730)
|
-
|
|
Stock issued in connection with
sale of common stock, net
|
3,360,037
|
3,360
|
-
|
-
|
-
|
6,225,991
|
-
|
6,229,351
|
|
Stock issued in connection with
warrants cashless exercised
|
21,454
|
21
|
-
|
-
|
-
|
(21)
|
-
|
-
|
|
Stock issued in connection with
stock options exercised
|
1,087,500
|
1,088
|
-
|
-
|
-
|
862,013
|
-
|
863,101
|
|
Stock-based
compensation
|
-
|
-
|
-
|
-
|
-
|
1,335,157
|
-
|
1,335,157
|
|
Other comprehensive
gain
|
-
|
-
|
-
|
-
|
19,056
|
-
|
-
|
19,056
|
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(24,643,627)
|
(24,643,627)
|
|
Balance at
December 31, 2016
|
40,432,339
|
$40,433
|
450,085
|
$450
|
$81,186
|
$136,857,409
|
$(119,164,952)
|
$17,814,526
|
The
accompanying notes are integral part of these consolidated
financial statements.
F-5
CORMEDIX
INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF CASH FLOWS
Years Ended December 31, 2016, 2015 and
2014
|
|
December
31,
|
||
|
|
2016
|
2015
|
2014
|
|
CASH
FLOWS FROM OPERATING ACTIVITIES:
|
|
|
|
|
Net
loss
|
$(24,643,627)
|
$(18,187,529)
|
$(20,453,427)
|
|
Adjustments to
reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
Stock-based
compensation
|
1,335,157
|
3,225,659
|
2,168,303
|
|
Value of warrants
issued in connection with backstop financing
|
-
|
1,583,252
|
-
|
|
Modification of
warrant agreement
|
-
|
112,982
|
-
|
|
Loss on foreign
exchange transactions
|
-
|
-
|
150,803
|
|
Loss on issuance of
preferred stock, convertible notes and warrants
|
-
|
-
|
89,590
|
|
Loss on
modification of equity instruments and extinguishment of derivative
liabilities
|
-
|
-
|
2,462,588
|
|
Inventory
reserve
|
130,000
|
125,000
|
175,000
|
|
Revaluation of
derivative liabilities
|
-
|
-
|
8,848,953
|
|
Depreciation
|
25,596
|
15,076
|
15,074
|
|
Changes in
operating assets and liabilities:
|
|
|
|
|
Restricted
cash
|
-
|
(171,553)
|
220,586
|
|
Trade
receivables
|
307,774
|
(248,186)
|
(85,412)
|
|
Inventory
|
79,836
|
(38,540)
|
(558,008)
|
|
Prepaid expenses
and other current assets
|
(505,492)
|
(645,356)
|
72,958
|
|
Accounts
payable
|
(63,586)
|
825,105
|
8,055
|
|
Accrued
expenses
|
1,121,863
|
764,114
|
522,995
|
|
Deferred
revenue
|
(52,916)
|
113,078
|
41,123
|
|
Net cash used in
operating activities
|
(22,265,395)
|
(12,526,898)
|
(6,320,819)
|
|
CASH
FLOWS FROM INVESTING ACTIVITIES:
|
|
|
|
|
Sale (purchase) of
short-term investments
|
11,478,493
|
(23,592,625)
|
-
|
|
Purchase of
equipment
|
(58,723)
|
(15,446)
|
(25,402)
|
|
Net cash provided
by (used in) investing activities
|
11,419,770
|
(23,608,071)
|
(25,402)
|
|
CASH
FLOWS FROM FINANCING ACTIVITIES:
|
|
|
|
|
Proceeds from sale
of common stock from at-the-market program
|
6,229,351
|
28,451,848
|
-
|
|
Proceeds from
Series C-3 preferred stock, net
|
-
|
-
|
743,884
|
|
Proceeds from
Series C-3 preferred stock, related party
|
-
|
-
|
575,000
|
|
Proceeds from
exercise of warrants
|
-
|
14,658,161
|
-
|
|
Proceeds from
exercise of stock options
|
863,101
|
492,960
|
318,150
|
|
Payment of deferred
financing costs
|
-
|
-
|
(2,366)
|
|
Proceeds from sale
of equity securities
|
-
|
-
|
6,723,248
|
|
Proceeds from short
swing profit recovery
|
-
|
26,525
|
-
|
|
Net cash provided
by financing activities
|
7,092,452
|
43,629,494
|
8,357,916
|
|
Foreign exchange
effects on cash
|
245
|
(16,647)
|
(46,048)
|
|
NET
INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS
|
(3,752,928)
|
7,477,878
|
1,965,647
|
|
CASH
AND CASH EQUIVALENTS – BEGINNING OF YEAR
|
11,817,418
|
4,339,540
|
2,373,893
|
|
CASH
AND CASH EQUIVALENTS – END OF YEAR
|
$8,064,490
|
$11,817,418
|
$4,339,540
|
The
accompanying notes are integral part of these consolidated
financial statements.
F-6
CORMEDIX INC. AND SUBSIDIARY
CONSOLIDATED
STATEMENTS OF CASH FLOWS
Years Ended
December 31, 2016, 2015 and 2014
|
|
December
31,
|
||
|
|
2016
|
2015
|
2014
|
|
Cash paid for
interest
|
$1,197
|
$3,964
|
$2,074
|
|
|
|
|
|
|
Supplemental
Disclosure of Non Cash Financing Activities:
|
|
|
|
|
Unrealized gain
(loss) from investments
|
$11,027
|
$(24,239)
|
$-
|
|
Conversion of
preferred stock to common stock
|
$-
|
$500
|
$2,447,384
|
|
Conversion of
accounts payable and accrued expenses to preferred
stock
|
$-
|
$-
|
$645,458
|
|
Reclassification of
derivative liabilities to equity
|
$-
|
$-
|
$17,955,143
|
|
Settlement of
accrued dividends with issuance of preferred stock
|
$-
|
$-
|
$102,845
|
|
Conversion of wages
and fees to common stock
|
$-
|
$50,000
|
$96,851
|
|
Dividend, including
deemed dividends
|
$-
|
$33,121
|
$82,899
|
The
accompanying notes are integral part of these consolidated
financial statements.
F-7
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 — Organization, Business and Basis of
Presentation:
Organization and Business:
CorMedix Inc.
(“CorMedix” or the “Company”) was
incorporated in the State of Delaware on July 28, 2006. The Company
is a biopharmaceutical company focused on developing and
commercializing therapeutic products for the prevention and
treatment of infectious and inflammatory diseases. In 2013, the
Company formed a wholly-owned subsidiary, CorMedix Europe
GmbH.
The
Company’s primary activities have been acquiring licenses for
its pharmaceutical product candidates, performing research and
development, seeking regulatory approval for its products and
developing its lead product Neutrolin® (also known as
CRMD003) for potential commercialization in the United States and
other key markets. The Company has in-licensed the worldwide rights
to develop and commercialize Neutrolin®. The Company
had also previously in-licensed CRMD004 which ceased development in
late 2015.
The
Company received CE Mark approval for Neutrolin in 2013 and
commercially launched Neutrolin in Germany for the prevention of
catheter-related bloodstream infections and maintenance of catheter
patency in hemodialysis patients using a tunneled, cuffed central
venous catheter for vascular access. To date, Neutrolin is
registered and may be sold in certain European Union and Middle
Eastern countries for such treatment.
In
September 2014, the TUV-SUD and The Medicines Evaluation Board of
the Netherlands granted labeling for expanded indications for
Neutrolin in the European Union (“EU”). In December
2014, the Company received approval from the Hessian District
President in Germany to expand the label to include use in oncology
patients receiving chemotherapy, intravenous (“IV”),
hydration and IV medications via central venous catheters. The
expansion also adds patients receiving medication and IV fluids via
central venous catheters in intensive or critical care units
(cardiac care unit, surgical care unit, neonatal critical care
unit, and urgent care centers). An indication for use in total
parenteral, or IV, nutrition was also approved.
The
Company launched its Phase 3 clinical trial in hemodialysis
catheters in the U.S. in December 2015 and is currently planning to
conduct a Phase 3 clinical trial in oncology, subject to funding
requirements.
Note 2 — Liquidity, Going Concern and
Uncertainties:
The
financial statements have been prepared in conformity with
generally accepted accounting principles which contemplate
continuation of the Company as a going concern. To date, the
Company’s commercial operations have not generated sufficient
revenues to enable profitability. As of December 31, 2016, the
Company had an accumulated deficit of $119.2 million, and incurred
losses from operations of $24.8 million, $16.7 million and $8.9
million for the years ended December 31, 2016, 2015 and 2014,
respectively. Based on the current development plans for Neutrolin
in both the U.S. and foreign markets (including the ongoing
hemodialysis Phase 3 clinical trial in the U.S.) and the
Company’s other operating requirements, management believes
that the existing cash at December 31, 2016 will not be sufficient
to fund operations for at least the next twelve months following
the filing date of the Company’s Annual Report on Form 10-K
for the year ended December 31, 2016 (the “Form 10-K”).
These factors raise substantial doubt regarding the Company’s
ability to continue as a going concern.
The
Company’s continued operations will depend on its ability to
raise additional capital through various potential sources, such as
equity and/or debt financings, strategic relationships, or
out-licensing of its products in order to complete its ongoing and
planned Phase 3 clinical trials and until it achieves
profitability, if ever. Management is actively pursuing financing
plans but can provide no assurances that such financing or
strategic relationships will be available on acceptable terms, or
at all. Without this funding, the Company could be required to
delay, scale back or eliminate some or all of its research and
development programs which would likely have a material adverse
effect on the Company.
F-8
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
At
December 31, 2016, approximately $4.1 million remained available
for sale under an April 2015 At-the-Market Issuance Sales Agreement
(the “ATM program”) with MLV & Co. LLC, now a
subsidiary of FBR Capital Markets & Co. (“FBR”),
pursuant to which the Company is able to issue and sell up to $40
million of shares of its common stock from time to time (See Note
8). In August 2016, the Company entered into a new ATM program with
FBR, which allows the Company to sell up to another $40 million of
shares of its common stock. However, the Company cannot access the
August 2016 ATM program unless and until (i) the Company and
Manchester Securities Corp. (“Manchester”) agree as to
the exercise or waiver of Manchester’s participation rights
in the new ATM program, which rights were granted in a Consent and
Exchange Agreement dated September 15, 2014, and apply to any
equity financing we undertake until September 15, 2017 (See Note
4), and (ii) the registration statement that includes the
prospectus for the new ATM program that the Company filed with the
Securities and Exchange Commission is declared effective. As of the
filing date of the Form 10-K, Manchester has not agreed to exercise
or waive its rights nor has the registration statement that the
Company filed in August 2016 been declared effective.
The
financial statements do not include any adjustments relating to the
recoverability and classification of asset carrying amounts or the
amount and classification of liabilities that might result should
the Company be unable to continue as a going concern.
The
Company’s operations are subject to a number of factors that
can affect its operating results and financial condition. Such
factors include, but are not limited to: the results of clinical
testing and trial activities of the Company’s product
candidates; the ability to obtain regulatory approval to market the
Company’s products; ability to manufacture successfully;
competition from products manufactured and sold or being developed
by other companies; the price of, and demand for, Company products;
the Company’s ability to negotiate favorable licensing or
other manufacturing and marketing agreements for its products; and
the Company’s ability to raise capital to support its
operations.
Note 3 — Summary of Significant Accounting
Policies:
Use of Estimates
The
preparation of financial statements in conformity with accounting
principles generally accepted in the United States of America
requires management to make estimates and assumptions that affect
the reported amounts of assets and liabilities and disclosure of
contingent assets and liabilities at the date of the financial
statements and reported amounts of revenue and expenses during the
reporting period. Actual results could differ from those
estimates.
Basis of Consolidation
The
consolidated financial statements include the accounts of the
Company and CorMedix Europe GmbH, a wholly owned subsidiary. All
significant intercompany accounts and transactions have been
eliminated in consolidation.
Financial Instruments
Financial
instruments that potentially subject the Company to concentrations
of credit risk consist principally of cash and cash equivalents and
short-term investments. The Company maintains its cash and cash
equivalents in bank deposit and other interest bearing accounts,
the balances of which, at times, may exceed federally insured
limits.
The
appropriate classification of marketable securities is determined
at the time of purchase and reevaluated as of each balance sheet
date. Investments in marketable debt and equity securities
classified as available-for-sale are reported at fair value. Fair
value is determined using quoted market prices in active markets
for identical assets or liabilities or quoted prices for similar
assets or liabilities or other inputs that are observable or can be
corroborated by observable market data for substantially the full
term of the assets or liabilities. Changes in fair value that are
considered temporary are reported net of tax in other comprehensive
income (loss). Realized gains and losses, amortization of premiums
and discounts and interest and dividends earned are included in
income (expense). For declines in the fair value of equity
securities that are considered other-than-temporary, impairment
losses are charged to other (income) expense, net. The Company
considers available evidence in evaluating potential impairments of
its investments, including the duration and extent to which fair
value is less than cost. There were no deemed permanent impairments
at December 31, 2016 or 2015.
F-9
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
Company’s marketable securities are highly liquid and consist
of U.S. government agency securities, high-grade corporate
obligations and commercial paper with original maturities of more
than 90 days. As of December 31, 2016 and 2015, all of the
Company’s investments had contractual maturities which were
less than one year. The following table summarizes the amortized
cost, unrealized gains and losses and the fair value at December
31, 2016 and 2015:
|
December 31, 2016:
|
Amortized Cost
|
Gross Unrealized Losses
|
Gross Unrealized Gains
|
Fair Value
|
|
Money
Market Funds included in Cash Equivalents
|
$95,949
|
$-
|
$-
|
$95,949
|
|
Corporate
Securities
|
10,619,583
|
(13,212)
|
-
|
10,606,371
|
|
Commercial
Paper
|
1,494,549
|
-
|
-
|
1,494,549
|
|
Subtotal
|
12,114,132
|
(13,212)
|
-
|
12,100,920
|
|
Total
December 31, 2016
|
$12,210,081
|
$(13,212)
|
$-
|
$12,196,869
|
|
December 31, 2015:
|
|
|
|
|
|
Money
Market Funds included in Cash Equivalents
|
$3,353,067
|
$-
|
$-
|
$3,353,067
|
|
U.S.
Government Agency Securities
|
6,531,914
|
(3,014)
|
-
|
6,528,900
|
|
Corporate
Securities
|
15,065,595
|
(21,637)
|
412
|
15,044,370
|
|
Commercial
Paper
|
1,995,116
|
-
|
-
|
1,995,116
|
|
Subtotal
|
23,592,625
|
(24,651)
|
412
|
23,568,386
|
|
Total
December 31, 2015
|
$26,945,692
|
$(24,651)
|
$412
|
$26,921,453
|
Fair Value Measurements
The
Company’s financial instruments recorded in the consolidated
balance sheets include cash and cash equivalents, accounts
receivable, investment securities, accounts payable and accrued
expenses. The carrying value of certain financial
instruments, primarily cash and cash equivalents, accounts
receivable, accounts payable, and accrued expenses approximate
their estimated fair values based upon the short-term nature of
their maturity dates.
The
Company categorizes its financial instruments into a three-level
fair value hierarchy that prioritizes the inputs to valuation
techniques used to measure fair value, which is set out below. The
fair value hierarchy gives the highest priority to quoted prices in
active markets for identical assets (Level 1) and the lowest
priority to unobservable inputs (Level 3). If the inputs used to
measure fair value fall within different levels of the hierarchy,
the category level is based on the lowest priority level input that
is significant to the fair value measurement of the
instrument.
●
Level 1
inputs—Observable inputs that reflect quoted prices
(unadjusted) for identical assets or liabilities in active
markets.
●
Level 2
inputs— Significant other observable inputs (e.g., quoted
prices for similar items in active markets, quoted prices for
identical or similar items in markets that are not active, inputs
other than quoted prices that are observable such as interest rate
and yield curves, and market-corroborated inputs).
●
Level 3
inputs—Unobservable inputs for the asset or liability, which
are supported by little or no market activity and are valued based on management’s
estimates of assumptions that market participants would use in
pricing the asset or liability.
F-10
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
following table provides the carrying value and fair value of the
Company’s financial assets measured at fair value as of
December 31, 2016 and 2015:
|
December 31, 2016:
|
Carrying Value
|
Level 1
|
Level 2
|
Level 3
|
|
Money
Market Funds
|
$95,949
|
$95,949
|
$-
|
$-
|
|
Available
for sale securities:
|
|
|
|
|
|
Corporate
Securities
|
10,606,371
|
-
|
10,606,371
|
-
|
|
Commercial
Paper
|
1,494,549
|
-
|
1,494,549
|
-
|
|
Subtotal
|
12,100,920
|
-
|
12,100,920
|
-
|
|
Total
December 31, 2016
|
$12,196,869
|
$95,949
|
$12,100,920
|
$-
|
|
December 31,
2015:
|
|
|
|
|
|
Money
Market Funds
|
$3,353,067
|
$3,353,067
|
$-
|
$-
|
|
Available
for sale securities:
|
|
|
|
|
|
US
Government Agency Securities
|
6,528,900
|
-
|
6,528,900
|
-
|
|
Corporate
Securities
|
15,044,370
|
-
|
15,044,370
|
-
|
|
Commercial
Paper
|
1,995,116
|
-
|
1,995,116
|
-
|
|
Subtotal
|
23,568,386
|
-
|
23,568,386
|
-
|
|
Total
December 31, 2015
|
$26,921,453
|
$3,353,067
|
$23,568,386
|
$-
|
Foreign Currency Translation and Transactions
The
consolidated financial statements are presented in U.S. Dollars
(USD), the reporting currency of the Company. For the financial
statements of the Company’s foreign subsidiary, whose
functional currency is the EURO, foreign currency asset and
liability amounts, if any, are translated into USD at end-of-period
exchange rates. Foreign currency income and expenses are translated
at average exchange rates in effect during the year. Translation
gains and losses are included in other comprehensive loss. The
Company had foreign currency translation gain of $8,029 in 2016, a
loss of $12,603 in 2015 and a gain of $108,295 in
2014.
Foreign
currency exchange transaction gain (loss) is the result of
re-measuring transactions denominated in a currency other than the
functional currency of the entity recording the
transaction.
Segment and Geographic Information
The
Company reported revenues of $224,105, $210,130 and $189,274 for
the years ended December 31, 2016, 2015 and 2014, respectively. Of
the Company’s 2016, 2015 and 2014 revenues, $215,282,
$201,306 and $185,598, respectively, were attributable to its
European and Mideast operations, which are based in Germany. Total
assets at December 31, 2016 and 2015 were $22,643,953 and
$37,101,729, respectively, of which $22,333,139 and $36,190,835
were located in the United States at December 31, 2016 and 2015,
respectively, with the remainder in Germany.
Restricted Cash
As of
December 31, 2016 and 2015, the Company’s restricted cash is
in connection with the patent and utility model infringement
proceedings against TauroPharm (see Note 6). The Company was
required by the District Court Mannheim to provide a security
deposit of approximately $132,000 to cover legal fees in the event
TauroPharm is entitled to reimbursement of these costs. The
Company furthermore had to provide a deposit in the amount of
$40,000 in connection with the unfair competition proceedings in
Cologne.
Prepaid Research and Development and Other Prepaid
Expenses
Prepaid
expenses consist of payments made in advance to vendors relating to
service contracts for clinical trial development, manufacturing,
preclinical development and insurance policies. These advanced
payments are amortized to expense either as services are performed
or over the relevant service period using the straight-line
method.
F-11
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Inventories, net
Inventories are
valued at the lower of cost or market on a first in, first out
basis. Inventories consist of raw materials (including labeling and
packaging), work-in-process, and finished goods, if any, for the
Neutrolin product. Inventories consist of the
following:
|
|
December
31,
|
|
|
|
2016
|
2015
|
|
Raw
materials
|
$79,900
|
$244,459
|
|
Work in
process
|
463,897
|
424,622
|
|
Finished
goods
|
52,936
|
7,488
|
|
Inventory
reserve
|
(430,000)
|
(300,000)
|
|
Total
|
$166,733
|
$376,569
|
Property and Equipment
Property and
equipment consist primarily of furnishings, fixtures, leasehold
improvements, office equipment and computer equipment which are
recorded at cost. Depreciation is provided for by the straight-line
method over the estimated useful lives of the related
assets. Leasehold improvements are amortized using the
straight-line method over the remaining lease term or the life of
the asset, whichever is shorter. Property and equipment,
as of December 31, 2016 and 2015 were 69,695 and $37,866,
respectively, net of accumulated depreciation of $117,949 and
$92,353, respectively. Depreciation and amortization of property
and equipment is included in selling, general and administrative
expenses.
|
Description
|
|
Estimated Useful
Life
|
|
Office
equipment and furniture
|
|
5
years
|
|
Leasehold
improvements
|
|
5
years
|
|
Computer
equipment
|
|
5
years
|
|
Computer
software
|
|
3
years
|
Accrued Expenses
Accrued
expenses consist of the following at December 31:
|
|
2016
|
2015
|
|
Professional and
consulting fees
|
$335,198
|
$282,975
|
|
Accrued payroll and
payroll taxes
|
737,607
|
532,084
|
|
Clinical trial and
manufacturing development
|
875,500
|
226,042
|
|
Product
development
|
374,839
|
-
|
|
Monitoring program
fees
|
-
|
65,076
|
|
Statutory
taxes
|
1,833
|
67,236
|
|
Other
|
17,375
|
48,144
|
|
Total
|
$2,342,352
|
$1,221,557
|
Revenue Recognition
Revenue
is recognized from product sales when the following four revenue
recognition criteria are met: persuasive evidence of an arrangement
exists, delivery has occurred, the selling price is fixed or
determinable, and collectability is reasonably
assured.
Neutrolin
received its CE Mark in Europe in July 2013 and product shipments
to dialysis centers began in December 2013. Orders are processed
through a distributor; however, Neutrolin is drop-shipped via a
pharmacy directly to the Company’s customer, the dialysis
center. The Company recognizes net sales upon shipment of product
to the dialysis centers.
F-12
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Deferred Revenue
In
October 2015, the Company shipped product with less than 75% of its
remaining shelf life to a customer and issued a guarantee that the
specific product shipped would be replaced by the Company if the
customer was not able to sell the product before it expired. As a
result of this warranty, the Company may have an additional
performance obligation (i.e. accept returned product and deliver
new product to the customer) if the customer is unable to sell the
short-dated product. Due to limited sales experience with the
customer, the Company is unable to estimate the amount of the
warranty obligation that may be incurred as a result of this
shipment. Therefore, the Company has deferred the revenue and
related cost of sales associated with the shipment of this product.
During the year ended December 31, 2016, the Company recognized
35.6% of deferred revenue and related cost of sales amounting to
$106,000 and $63,000, respectively. Deferred revenue at December
31, 2016 and 2015 amounted to approximately $75,000 and $121,000,
respectively.
In August 2014, the Company entered into an
exclusive distribution agreement (the “Wonik
Agreement”) with Wonik Corporation, a South Korean company,
to market, sell and distribute Neutrolin for hemodialysis and
oncolytic patients upon receipt of regulatory approval in Korea.
Upon execution, Wonik paid the Company a non-refundable $50,000
payment and will pay an additional $50,000 upon receipt of
the product registration necessary to sell Neutrolin in the
Republic of Korea (the
“Territory”). The term of the Wonik Agreement commenced
on August 8, 2014 and will continue for three years after the first
commercial sale of Neutrolin in the Territory. The non-refundable
up-front payment has been recorded as deferred revenue and will be
recognized as revenue on a straight-line basis over the contractual
term of the Agreement. Deferred revenue related to this agreement
at December 31, 2016 and 2015 amounted to approximately $29,000 and
$38,000, respectively.
Loss Per Common Share
Basic
loss per common share excludes dilution and is computed by dividing
net loss by the weighted average number of common shares
outstanding during the period. Diluted loss per common share
reflects the potential dilution that could occur if securities or
other contracts to issue common stock were exercised or converted
into common stock or resulted in the issuance of common stock that
then shared in the earnings of the entity. Since the Company has
only incurred losses, basic and diluted loss per share are the same
as potentially dilutive shares have been excluded from the
calculation of diluted net loss per share as their effect would be
anti-dilutive.
The
shares outstanding at the end of the respective periods presented
below were excluded from the calculation of diluted net loss per
shares due to their anti-dilutive effect:
|
|
December
31,
|
||
|
|
2016
|
2015
|
2014
|
|
Series B non-voting
preferred stock
|
-
|
-
|
454,546
|
|
Series C non-voting
preferred stock
|
2,865,000
|
2,865,000
|
3,290,000
|
|
Series D non-voting
preferred stock
|
1,479,240
|
1,479,240
|
1,479,240
|
|
Series E non-voting
preferred stock
|
1,959,759
|
1,959,759
|
2,021,358
|
|
Shares underlying
outstanding warrants
|
4,006,468
|
4,422,188
|
11,520,762
|
|
Shares underlying
outstanding stock options
|
4,609,755
|
3,600,045
|
3,664,500
|
|
Total Potentially
Dilutive Shares
|
14,920,222
|
14,326,232
|
22,430,406
|
Stock-Based Compensation
The
Company accounts for stock options granted to employees, officers
and directors according to ASC No. 718, “Compensation — Stock
Compensation” (“ASC 718”).
Share-based compensation cost is measured at grant date, based on
the estimated fair value of the award using a Black-Scholes option
pricing model for options with service or performance based
conditions and a Monte Carlo option pricing model for options with
market vesting conditions. Stock-based compensation cost is
recognized as expense over the employee’s requisite service
period on a straight-line basis.
F-13
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Effective October
1, 2016, the Company adopted Accounting Standards Update
(“ASU”) 2016-09, Compensation — Stock Compensation
(“Topic 718”), Improvements to Employee Share-Based Payment
Accounting to account for forfeitures as they occur. All
share-based awards will be recognized on a straight-line method,
assuming all awards granted will vest. Forfeitures of share-based
awards will be recognized in the period in which they occur. Prior
to the adoption of ASU 2016-09, share-based compensation expense
was recognized by applying the expected forfeiture rate during the
vesting period to the fair value of the award. As of January
1, 2016, a cumulative effect adjustment of $129,730 was recognized
to reflect the forfeiture rate that had been applied to unvested
option awards prior to fiscal year 2016.
The
Company accounts for stock options granted to non-employees on a
fair value basis using the Black-Scholes option pricing model in
accordance with ASC 718 and ASC No. 505-50, “Equity-Based Payments to
Non-Employees” (“ASC 505”). The
non-cash charge to operations for non-employee options with time
based vesting provisions is based on the fair value of the options
remeasured each reporting period and amortized to expense over the
related vesting period. The non-cash charge to operations for
non-employee options with performance based vesting provisions is
recorded when the achievement of the performance condition is
probable and remeasured each reporting period until the performance
condition is achieved.
Research and Development
Research and
development costs are charged to expense as incurred. Research and
development includes fees associated with operational consultants,
contract clinical research organizations, contract manufacturing
organizations, clinical site fees, contract laboratory research
organizations, contract central testing laboratories, licensing
activities, and allocated executive, human resources and facilities
expenses. The Company accrues for costs incurred as the services
are being provided by monitoring the status of the trial and the
invoices received from its external service providers. As actual
costs become known, the Company adjusts its accruals in the period
when actual costs become known. Costs related to the acquisition of
technology rights and patents for which development work is still
in process are charged to operations as incurred and considered a
component of research and development expense.
Income Taxes
Deferred tax assets
and liabilities are recognized for the future tax consequences
attributable to temporary differences between the financial
statement carrying amounts of existing assets and liabilities and
their respective tax bases. Deferred tax assets and liabilities are
measured using enacted tax rates expected to apply to taxable
income in the years in which those temporary differences are
expected to be recovered or settled. The effect on deferred tax
assets and liabilities of a change in tax rates is recognized in
income in the period that includes the enactment date. Valuation
allowances are established when it is more likely than not that
some or all of the deferred tax assets will not be
realized.
Embedded Derivative Liabilities and Warrant
Liabilities
The Company has several series of preferred stock
and warrants and evaluates all its financial instruments to
determine if those instruments or any potential embedded components
of those instruments qualify as derivatives that need to be
separately accounted for in accordance with FASB ASC 815,
“Derivatives
and Hedging”. Embedded
derivatives satisfying certain criteria are recorded at fair value
at issuance and marked-to-market at each balance sheet date with
the change in the fair value recorded as income or expense.
In addition, upon the occurrence of an event that requires
the derivative liability to be reclassified to equity, the
derivative liability is revalued to fair value at that date. (See
Note 7).
F-14
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
Company accounts for stock warrants as either equity instruments or
derivative liabilities depending on the specific terms of the
warrant agreement. Stock warrants that allow for cash
settlement or provide for certain modifications of the warrant
exercise price were accounted for as derivative liabilities.
For those liability-classified warrants that have down-round
provisions which allow the exercise price to be adjusted as a
result of certain future financing transactions, the Company uses
level 3 inputs to value those warrants. The estimated fair
values of the warrant liabilities with downround protection were
determined using a Monte Carlo option pricing model which takes
into account the probabilities of certain events occurring over the
life of the warrants. The derivative liabilities were
adjusted to their estimated fair values at each reporting period,
with any decrease or increase in the estimated fair value being
recorded in other income (expense). The significant inputs and
assumptions are as follows:
Stock price –
Due to the historical volatility of
the Company’s common stock price, a one month volume-weighted
average stock price was used as of each valuation
date.
Conversion/redemption strike
price – These assumptions
incorporate both the initial contractual conversion price as well
as subsequent downward adjustments (wherever applicable) based on
management’s estimate of the probabilities of additional
future financings that would include a stock price or conversion
price that is lower than the then existing conversion
price.
Volatility – The Company used a weighted average of (i)
the historical volatility of the Company’s common stock for
approximately five years, (ii) the volatility used for prior period
valuations, and (iii) the volatilities of comparable companies
(provided by the Company’s management) from the date product
approval is received to the various valuation dates. Then,
appropriate weights were applied to these data points to arrive at
the weighted average historical volatility.
Term – Although the Series C, D and E preferred
stocks do not have a specified contracted life, the Company has
assumed a five year life from the date of inception for the purpose
of the valuations.
Risk-free Rate
– The U.S. Treasury Bond Rate
with a term approximating the term of the instrument was used as
the risk-free interest rate in the valuation.
Credit adjusted discount
rate – Management
believes that its debt, if rated, would be equivalent to
Moody’s C rated bonds or lower.
Dividend rate
- Management does not expect to pay
any dividends during the term of the hybrid
instrument.
Recently Adopted Authoritative Pronouncements
In
June 2014, the FASB issued an accounting standard that clarifies
the accounting for share-based payments when the terms of an award
provide that a performance target could be achieved after the
requisite service period. The standard requires that a
performance target that affects vesting and that could be achieved
after the requisite service period be treated as a performance
condition. The amendments are effective for
interim and annual reporting periods beginning after December 15,
2015. This adoption did not have a material impact on
the Company’s consolidated financial statements.
In
August 2014, the FASB issued new guidance related to disclosures of
uncertainties about an entity’s ability to continue as a
going concern. The ASU requires management to evaluate whether
there are conditions and events that raise substantial doubt about
the entity’s ability to continue as a going concern within
one year after the financial statements are issued and if
management’s plans will alleviate that doubt. Management will
be required to make this evaluation for both annual and interim
reporting periods. The Company adopted
this guidance for the fiscal year ended December 31, 2016. This
adoption did not have a material impact on the Company’s
consolidated financial statements.
In
April 2015, the FASB issued new guidance which requires that debt
issuance costs related to a recognized debt liability be presented
in the balance sheet as a direct deduction from the carrying amount
of that debt liability, consistent with debt discounts. This
guidance was effective for the Company beginning in the first
quarter of 2016. This adoption did not have a material
impact on the Company’s consolidated financial
statements.
In March 2016, the FASB issued new guidance which
simplifies several aspects of the accounting for share-based
payment transactions, including the income tax consequences,
classification of awards as either equity or liabilities, and
classification on the statement of cash flows. The Company adopted this guidance in the fourth
quarter of fiscal year 2016, which was applied retroactively
effective January 1, 2016. This adoption did not have a material
impact on the Company’s consolidated financial
statements.
F-15
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Recent Authoritative Pronouncements
In
May 2014, the FASB issued new guidance related to how an entity
should recognize revenue. The guidance specifies that an entity
should recognize revenue to depict the transfer of promised goods
or services to customers in an amount that reflects the
consideration to which the entity expects to be entitled in
exchange for those goods and services. In addition, the guidance
expands the required disclosures related to revenue and cash flows
from contracts with customers. The guidance is effective for the
Company beginning in the first quarter of 2017. Early adoption is
not permitted and retrospective application is required. The
Company is currently evaluating the impact of adopting this
guidance on its consolidated financial statements.
In
July 2015, the FASB issued an accounting standard that requires
inventory be measured at the lower of cost and net realizable value
and options that currently exist for market value be eliminated.
The standard defines net realizable value as estimated selling
prices in the ordinary course of business, less reasonably
predictable costs of completion, disposal, and transportation and
is effective for reporting periods beginning after December 15,
2016 and interim periods within those fiscal years with early
adoption permitted. The guidance should be applied prospectively.
The Company is evaluating the impact the adoption of this guidance
will have on the determination or reporting of its consolidated
financial statements.
In
November 2015, the FASB issued guidance that requires that deferred
tax liabilities and assets be classified as noncurrent in a
classified statement of financial position. The current requirement
that deferred tax liabilities and assets of a tax-paying component
of an entity be offset and presented as a single amount is not
affected by this amendment. The new guidance is effective for
fiscal years, and interim periods within those years, beginning
after December 15, 2016. Early adoption is permitted and the
standard may be applied either retrospectively or on a prospective
basis to all deferred tax assets and liabilities. The Company is evaluating the impact the adoption
of this guidance will have on the determination or reporting of its
consolidated financial statements.
In
January 2016, the FASB issued a new standard that modifies certain
aspects of the recognition, measurement, presentation, and
disclosure of financial instruments. The accounting standard update
is effective for fiscal years, and interim periods within those
years, beginning after December 15, 2017, and early adoption is
permitted. The Company is currently assessing the impact that
adopting this new accounting guidance will have on its consolidated financial
statements.
In February 2016, the FASB issued new guidance
related to how an entity should lease assets and lease liabilities.
The guidance specifies that an entity who is a lessee under lease
agreements should recognize lease assets and lease
liabilities for those leases classified as operating leases under
previous FASB guidance. Accounting for
leases by lessors is largely unchanged under the new guidance. The
guidance is effective for the Company beginning in the first
quarter of 2019. Early adoption is permitted. In transition,
lessees and lessors are required to recognize and measure leases at
the beginning of the earliest period presented using a modified
retrospective approach. The Company is
evaluating the impact of adopting this guidance on its consolidated
financial statements.
In
April 2016, the FASB issued an update which clarifies two aspects
of the new revenue guidance by providing guidance on how to
identify performance obligations and providing implementation
guidance surrounding licensing. The amendments in this update do
not change the core principle of the new revenue guidance. The
guidance is effective for the Company beginning in the first
quarter of 2017. Early adoption is not permitted and retrospective
application is required. The Company is currently evaluating the
impact of adopting this guidance on its consolidated financial
statements.
In June 2016,
the FASB issued new guidance which replaces the incurred loss
impairment methodology in current GAAP with a methodology that
reflects expected credit losses and requires consideration of a
broader range of reasonable and supportable information to inform
credit loss estimates. The guidance is effective for the Company
beginning in the first quarter of fiscal year 2020. Early adoption
is permitted beginning in the first quarter of fiscal year 2019.
The Company is evaluating the impact of adopting this guidance on
its consolidated financial
statements.
F-16
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In
August 2016, the FASB issued new guidance which clarifies how
certain cash receipts and cash payments are presented and
classified in the statement of cash flows in order to reduce
diversity in practice. The guidance is effective for the Company
beginning in the first quarter of fiscal year 2018. Early adoption
is permitted. The Company is evaluating the impact of adopting this
guidance on its consolidated financial statements.
In
November 2016, the FASB issued new guidance which clarifies how
restricted cash is presented and classified in the statement of
cash flows. The guidance is effective for the Company beginning in
the first quarter of fiscal year 2018. Early adoption is permitted.
The Company is evaluating the impact of adopting this guidance on
its consolidated financial statements.
Note 4 — Related Party Transactions:
In
September 2014, as part of the removal of anti-dilution, price
reset and change of control provisions in various securities that
had caused those securities to be classified as derivative
liabilities, the Company entered into a Consent and Exchange
Agreement dated September 15, 2014, with Manchester Securities
Corp., or Manchester, pursuant to which Manchester has a right of
participation in equity financings undertaken by the Company prior
to September 15, 2017.
On
March 3, 2015, the Company entered into a backstop agreement with
an existing institutional investor, Manchester Securities Corp., a
wholly owned subsidiary of Elliott Associates, L.P., and a
beneficial holder of more than 5% of the Company’s
outstanding common stock. Pursuant to the backstop agreement,
Manchester agreed to lend the Company, at its request, up to
$4,500,000 on or before April 30, 2015, provided that the loan
could not exceed $3,000,000. The Company issued two
warrants exercisable for an aggregate of up to 283,400 common
shares with an exercise price of $7.00 per share and a term of five
years as a result of entering into the backstop agreement. The
Company did not access the loan and the loan expired on April 30,
2015. Additionally, the Company granted Manchester the right for as
long as it or its affiliates hold any of the Company’s common
stock or securities convertible into its common stock the right to
appoint up to two members to the Company’s board of directors
and/or to have up to two observers attend board meetings in a
non-voting capacity. As of December 31, 2016, two board members had
been appointed to the Company’s board of directors under this
provision.
On
April 7, 2015, the Company entered into a one year agreement with a
consultant to advise management with their investment banking
relationships and assist in the negotiations with potential
external parties, if applicable. The consultant is a member of the
board of directors of Sterling HSA which was founded by the
Chairman of the Board of Directors of the Company. The arrangement
called for a $30,000 retainer, a monthly fee of $6,000, and a
multiple of the price per share upon a merger or acquisition or a
percentage of any strategic partnership. This agreement was
terminated at the end of August 2015.
In
January 2014, the following related parties participated in the
private placement of Series C-3 preferred stock and warrants to purchase the
Company’s common stock at an exercise price of $1.25 per
share, which was reduced to $0.90 in September 2014. Each
share of Series C-3 preferred stock is convertible into 10 shares
of common stock. All terms were the
same as the Series C-1 and C-2 preferred stock issued in the
October 2013 private placement:
|
|
|
Amount
|
Number of Series
C-3 Preferred Stock
|
Number of
Warrants
|
|
Gary A. Gelbfish
(1)
|
Former Chairman of
the Board
|
$500,000
|
50,000
|
250,000
|
|
Randy Milby and
affiliate
|
CEO and
Director
|
$250,000
|
25,000
|
125,000
|
|
Steven W. Lefkowitz
and affiliate
|
Director and Former
Interim CFO
|
$75,000
|
7,500
|
37,500
|
(1) Gary A. Gelbfish
resigned effective June 13, 2014 and ceased to be a related party
90 days thereafter.
In each
instance, the purchase was on the same terms as all other
purchasers in the offerings. The Audit Committee of the Board of
Directors approved the purchase by these insiders.
F-17
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 5 — Income Taxes:
The
Company’s U.S. and foreign loss before income taxes are set
forth below:
|
|
December 31,
|
||
|
|
2016
|
2015
|
2014
|
|
United
States
|
$(23,743,682)
|
$(16,690,084)
|
$(18,653,576)
|
|
Foreign
|
(899,945)
|
(1,497,445)
|
(1,799,851)
|
|
Total
|
$(24,643,627)
|
$(18,187,529)
|
$(20,453,427)
|
There
were no current or deferred income tax provision for the years
ended December 31, 2016, 2015 or 2014 because the Company has
incurred operating losses since inception.
The
Company’s deferred tax assets consist of the
following:
|
|
December 31,
|
|
|
|
2016
|
2015
|
|
Net operating loss
carryforwards – Federal
|
$27,798,000
|
$18,282,000
|
|
Net operating loss
carryforwards – State
|
4,082,000
|
2,522,000
|
|
Net operating loss
carryforwards – Foreign
|
1,373,000
|
1,103,000
|
|
Capitalized
licensing fees
|
1,734,000
|
1,915,000
|
|
Stock-based
compensation
|
2,511,000
|
2,349,000
|
|
Accrued
compensation
|
132,000
|
206,000
|
|
Other
|
181,000
|
150,000
|
|
Totals
|
37,811,000
|
26,527,000
|
|
Less valuation
allowance
|
(37,811,000)
|
(26,527,000)
|
|
Deferred tax
assets
|
$-
|
$-
|
The
Company had approximately the following potentially utilizable net
operating loss tax carryforwards:
|
|
December 31,
|
||
|
|
2016
|
2015
|
2014
|
|
Federal
|
$81,759,000
|
$56,429,000
|
$38,023,000
|
|
State
|
$68,713,000
|
$45,113,000
|
$25,772,000
|
|
Foreign
|
$4,577,000
|
$3,678,000
|
$2,183,000
|
The net
operating loss tax carryforwards will start to expire in 2026 for
Federal purposes and 2016 for state purposes. The foreign net
operating loss tax carryforwards do not expire. Our federal and
state operating loss carryforwards include windfall tax deductions
from stock option exercises.
The
utilization of the Company’s federal and state net operating
losses may be subject to a substantial limitation due to the
“change of ownership provisions” under Section 382 of
the Internal Revenue Code and similar state provisions. Such
limitation may result in the expiration of the net operating loss
carryforwards before their utilization.
The
Company’s foreign earnings are derived from its German
subsidiary. The Company does not expect any foreign earnings to be
repatriated in the U.S. in the near future.
F-18
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
effective tax rate varied from the statutory rate as
follows:
|
|
December
31,
|
||
|
|
2016
|
2015
|
2014
|
|
Statutory Federal
tax rate
|
(34.0)%
|
(34.0)%
|
(34.0)%
|
|
State income tax
rate (net of Federal)
|
(5.7)%
|
(6.2)%
|
(0.6)%
|
|
Effect of foreign
operations
|
(1.1)%
|
(2.5)%
|
0.4%
|
|
Non-deductible
expenses associated with derivative liabilities
|
0.0%
|
0.0%
|
23.5%
|
|
Warrant related
expenses
|
0.0%
|
3.2%
|
0.0%
|
|
Prior year return
to provision adjustment
|
0.0%
|
(3.1)%
|
0.0%
|
|
Other permanent
differences
|
(0.7)%
|
(0.1)%
|
(0.1)%
|
|
Effect of valuation
allowance
|
41.5%
|
42.7%
|
10.8%
|
|
Effective tax
rate
|
0.0%
|
0.0%
|
0.0%
|
In
assessing the realizability of deferred tax assets, management
considers whether it is more-likely-than-not that some portion or
all of the deferred tax assets will not be realized. The ultimate
realization of deferred tax assets is dependent upon the generation
of future taxable income of the appropriate character during the
periods in which those temporary differences become deductible and
the loss carryforwards are available to reduce taxable income. In
making its assessment, the Company considered all sources of
taxable income including carryback potential, future reversals of
existing deferred tax liabilities, prudent and feasible tax
planning strategies, and lastly, objectively verifiable projections
of future taxable income exclusive of reversing temporary
differences and carryforwards. At December 31, 2016 and 2015, the
Company maintained a full valuation allowance against its net
deferred tax assets. The Company will continue to assess all
available evidence during future periods to evaluate the
realization of its deferred tax assets.
The
following table presents the changes in the deferred tax asset
valuation allowance for the periods indicated:
|
Year Ended
|
Balance at Beginning of Year
|
Increase (Decrease) Charged (Credited) to Income Taxes
(Benefit)
|
Increase (Decrease) Charged (Credited) to OCI
|
Balance at End of Year
|
|
December
31, 2016
|
$26,527,000
|
$11,309,800
|
$(25,800)
|
$37,811,000
|
|
December
31, 2015
|
$18,744,000
|
$7,770,000
|
$13,000
|
$26,527,000
|
|
December
31, 2014
|
$16,564,000
|
$2,212,000
|
$(32,000)
|
$18,744,000
|
Accounting
for uncertainty in income taxes requires uncertain tax positions to
be classified as non-current income tax liabilities unless they are
expected to be paid within one year. The Company has concluded that
there are no uncertain tax positions requiring recognition in its
consolidated financial statements as of December 31, 2016, 2015 and
2014. The Company recognizes interest and penalties related to
uncertain tax positions if any as a component of income tax
expense.
The
Company files income tax returns in the U.S. federal, state and
foreign jurisdictions. Tax years 2012 to 2016 remain open to
examination for both the U.S. federal and state jurisdictions. Tax
years 2013 to 2015 remain open for Germany.
F-19
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 6 — Commitments and
Contingencies:
Contingency Matters
In
February 2007, Geistlich Söhne AG für Chemische
Industrie, Switzerland (“Geistlich”) brought an action
against the European Sodemann Patent covering the Company’s
Neutrolin product candidate, which is owned by ND Partners, LLC
(“NDP”) and licensed to the Company pursuant to the
License and Assignment Agreement between the Company and NDP. This
action was brought at the Board of the European Patent Office
(“EPO”) opposition division (the “Opposition
Board”) based upon alleged lack of inventiveness in the use
of citric acid and a pH value in the range of 4.5 to 6.5 with
having the aim to provide an alternative lock solution through
having improved anticoagulant characteristics compared to the lock
solutions of the prior art. The Opposition Board rejected the
opposition by Geistlich. In a letter dated September 30, 2013, the
Company was notified that the opposition division of the EPO
reopened the proceedings before the first instance and gave their
preliminary non-binding opinion that the patent as amended during
the appeal proceedings fulfills the requirements of clarity,
novelty, and inventive step, and invited the parties to provide
their comments and/or requests by February 10, 2014. The Company
filed its response on February 3, 2014 to request that the patent
be maintained as amended during the appeal proceedings. Geistlich
did not file a further statement within the required
timeline. On November 5, 2014, the Opposition Division
at the EPO issued the interlocutory decision to maintain the patent
on the basis of the claims as amended during the appeal
proceedings. This decision became final as no further appeal was
lodged by Geistlich.
On
September 9, 2014, the Company filed in the District Court of
Mannheim, Germany a patent infringement action against TauroPharm
GmbH and Tauro-Implant GmbH as well as their respective CEOs (the
“Defendants”) claiming infringement of the
Company’s European Patent EP 1 814 562 B1, which was granted
by the European Patent Office (the “EPO”) on January 8,
2014 (the “Prosl European Patent”). The Prosl
European Patent covers a low dose heparin catheter lock solution
for maintaining patency and preventing infection in a hemodialysis
catheter. In this action, the Company claims that the
Defendants infringe on the Prosl European Patent by manufacturing
and distributing catheter locking solutions to the extent they are
covered by the claims of the Prosl European Patent. The
Company believes that its patent is sound, and is seeking
injunctive relief and raising claims for information, rendering of
accounts, calling back, destruction and damages. Separately,
TauroPharm has filed an opposition with the EPO against the Prosl
European Patent alleging that it lacks novelty and inventive
step. The Company cannot predict what other defenses the
Defendants may raise, or the ultimate outcome of either of these
related matters.
In the
same complaint against the same Defendants, the Company also
alleged an infringement (requesting the same remedies) of
NDP’s utility model DE 20 2005 022 124 U1 (the “Utility
Model”), which the Company believes is fundamentally
identical to the Prosl European Patent in its main aspects and
claims. The Court separated the two proceedings and the Prosl
European Patent and the Utility Model claims are now being tried
separately. TauroPharm has filed a cancellation action against the
Utility Model before the German Patent and Trademark Office (the
“German PTO”) based on the similar arguments as those
in the opposition against the Prosl European Patent.
On
March 27, 2015, the District Court held a hearing to evaluate
whether the Utility Model has been infringed by TauroPharm in
connection with the manufacture, sale and distribution of its
TauroLock-HEP100TM and
TauroLock-HEP500TM products. A hearing
before the same court was held on January 30, 2015 on the separate,
but related, question of infringement of the Prosl European Patent
by TauroPharm.
The
Court issued its decisions on May 8, 2015, staying both
proceedings. In its decisions, the Court found that the
commercialization by TauroPharm in Germany of its TauroLock
catheter lock solutions Hep100 and Hep500 infringes both the
Prosl European Patent and the Utility Model and further that there
is no prior use right that would allow TauroPharm to continue to
make, use or sell its product in Germany. However, the Court
declined to issue an injunction in favor of the Company that would
preclude the continued commercialization by TauroPharm based upon
its finding that there is a sufficient likelihood that the EPO, in
the case of the Prosl European Patent, or the German PTO, in the
case of the Utility Model, may find that such patent or utility
model is invalid. Specifically, the Court noted the possible
publication of certain instructions for product use that may be
deemed to constitute prior art. As such, the District Court
determined that it will defer any consideration of the request by
the Company for injunctive and other relief until such time as the
EPO or the German PTO has made a final decision on the underlying
validity of the Prosl European Patent and the Utility
Model.
F-20
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
opposition proceeding against the Prosl European Patent before the
EPO is ongoing. The EPO held a hearing in the opposition proceeding
on November 25, 2015. In its preliminary consideration of the
matter, the EPO (and the German PTO) had regarded the patent as not
inventive or novel due to publication of prior art. However, the
EPO did not issue a decision at the end of the hearing but
adjourned the matter due to the fact that the panel was of the view
that Claus Herdeis, one of the managing directors of TauroPharm,
has to be heard as a witness in a further hearing in order to close
some gaps in the documentation presented by TauroPharm as regards
the publication of the prior art. In October 2016, TauroPharm
submitted a further writ to the EPO requesting a date for the
hearing and bringing forward further arguments, in particular in
view of the recent decision of the German PTO on the invalidity of
the utility model. The EPO has issued a further oral hearing for
November 22 / 23, 2017. While the Company continues to believe that
the referenced publication and instructions for use do not, in
fact, constitute prior art and that the Prosl European Patent will
be found to be valid by the EPO, there can be no assurance that the
Company will prevail in this matter.
The
German PTO held a hearing in the validity proceedings relating to
the Utility Model on June 29, 2016, at which the panel affirmed its
preliminary finding that the Utility Model was invalid based upon
prior publication of a reference to the benefits that may be
associated with adding heparin to a taurolidine based solution. The
decision has only a declaratory effect, as the Utility Model had
expired in November 2015. Furthermore, it has no bearing on the
ongoing consideration of the validity and possible infringement of
the Prosl European Patent by the EPO. The Company filed an appeal
against the ruling on September 7, 2016.
On
January 16, 2015, the Company filed a complaint against TauroPharm
GmbH and its managing directors in the District Court of
Cologne, Germany. In the complaint, the Company alleges
violation of the German Unfair Competition Act by TauroPharm for
the unauthorized use of its proprietary information obtained in
confidence by TauroPharm. The Company alleges that
TauroPharm is improperly and unfairly using its proprietary
information relating to the composition and manufacture of
Neutrolin, in the manufacture and sale of TauroPharm’s
products TauroLockTM, TauroLock-HEP100
and TauroLock-HEP500. The Company seeks a cease and
desist order against TauroPharm from continuing to manufacture and
sell any product containing taurolidine (the active pharmaceutical
ingredient (“API”) of Neutrolin) and citric acid in
addition to possible other components, damages for any sales in the
past and the removal of all such products from the market. An
initial hearing in the District Court of Cologne, Germany was held
on November 19, 2015 to consider the Company’s claims. In
this hearing, the presiding judge explained that the court needed
more information with regard to several aspects of the case. As a
consequence, the court issued an interim decision in the form of a
court order outlining several issues of concern that relate
primarily to the court's interest in clarifying the facts and
reviewing any and all available documentation, in particular with
regard to the question which specific know-how was provided to
TauroPharm by whom and when. The Company's legal team has prepared
the requested reply and produced the respective documentation.
TauroPharm has also filed another writ within the same deadline and
both parties have filed further writs at the end of April setting
out their respective argumentation in more detail. A further oral
hearing in this matter was held November 15, 2016. In this hearing,
the court heard arguments from CorMedix and TauroPharm concerning
the allegations of unfair competition. The court made no rulings
from the bench, and indicated that it is prepared to further
examine the underlying facts of our allegations. At this time,
there are no further hearings scheduled. The Company intends to
continue to pursue this matter, and to provide additional
supplemental documentary and other evidence as may be necessary to
support its claims.
In
connection with the aforementioned patent and utility model
infringement proceedings against TauroPharm, the Company was
required by the District Court Mannheim to provide a security
deposit of approximately $132,000 to cover legal fees in the event
TauroPharm is entitled to reimbursement of these costs. The
Company recorded the deposit as restricted cash for the year ended
December 31, 2015. The Company furthermore had to provide a deposit
in the amount of $40,000 in connection with the unfair competition
proceedings in Cologne.
On July 7, 2015, a putative class action lawsuit
was commenced against the Company and certain of its current and
former officers in the United States District Court for the
District of New Jersey, captioned Li v. Cormedix Inc., et
al., Case 3:15-cv-05264 (the
“Securities Class Action”). On September 4, 2015, two
individuals, Shahm Martini and Paul Chretien (the “Martini
Group”), filed a Motion to Appoint Lead Plaintiff. On that
same date, another individual, Elaine Wood, filed a competing
Motion to Appoint Lead Plaintiff. On September 18, 2015, the
Martini Group withdrew its motion. Thereafter, on September 22,
2015, the Court appointed Elaine Wood as Lead Plaintiff and, on
October 2, 2015, appointed the Rosen Law Firm as Lead Counsel.
F-21
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On December 1, 2015, Lead Plaintiff filed an
Amended Complaint asserting claims that the Company and Steven
Lefkowitz, Randy Milby and Harry O’Grady (the “Cormedix
Defendants”) violated Section 10(b) of the Securities
Exchange Act of 1934, as amended (the “Exchange Act”)
and Rule 10b-5 promulgated thereunder and Section 20(a) of the
Exchange Act. The Amended Complaint also named as defendants
several unrelated entities that allegedly were paid stock
promoters. Lead Plaintiff alleged generally that the Cormedix
Defendants made materially false or misleading statements and
omissions concerning, among other things, the competitive landscape
for the Company’s Neutrolin product and the alleged use of
stock promoters. The Amended Complaint sought unspecified
damages, interest, attorneys’ fees, and other costs. The
Court heard oral argument on this motion on July 18, 2016 and in an
order dated October 27, 2016, the Court granted the Cormedix
Defendants’ Motion to Dismiss and dismissed with prejudice
the Amended Complaint (the “Dismissal Order”).
On December 16, 2016, the parties filed a stipulation with the
Court in which the plaintiffs and their counsel agreed not to
appeal, move for reconsideration or otherwise challenge the
Dismissal Order. No settlement payment was made in exchange for the
stipulation.
On May 13, 2016, a putative shareholder derivative
action was filed in the Superior Court of New Jersey against the
Company and certain present and former directors and officers
captioned Raval v. Milby, et.
al., Docket No. C-12034-6 (the
“Derivative Action”). The factual allegations of the
Derivative Action substantially overlap the factual allegations
contained in the Amended Complaint in the Securities Class Action.
The complaint in the Derivative Action seeks unspecified damages,
interest, attorneys’ fees and other costs, and certain
amendments to the Company’s “corporate governance and
internal procedures”. On June 30, 2016, the Court entered a
stipulated order, among other things, staying the Derivative Action
until 30 days after either: (a) the entry of any order denying any
motion to dismiss the Derivative Action in the Securities Class
Action, or (b) the entry of a final order dismissing the Securities
Class Action with prejudice. Following entry of the
Dismissal Order in the Securities Class Action, the parties entered
into a stipulation, among other things, staying the Derivative
Action until 30 days after either (a) November 30, 2016, in the
event plaintiffs in the Securities Class Action did not file an
appeal of the Dismissal Order or (b) the final disposition of any
appeal in the event the plaintiffs in the Securities Class Action
filed a Notice of Appeal of the Dismissal Order. On January 6,
2017, the parties filed a Stipulation of Dismissal without
Prejudice of the Derivative Action.
Commitments
Manufacturing
Navinta
LLC, a U.S.-based API developer, provides API manufacturing
(manufactured in India at an FDA-compliant facility) and a Drug
Master File for CRMD003, pursuant to an original supply agreement
dated December 7, 2009 (the “Navinta Agreement”). The
Navinta Agreement provided that Navinta will supply taurolidine
(the API for CRMD003) to the Company on an exclusive worldwide
basis in the field of the prevention and treatment of human
infection and/or dialysis so long as the Company purchases a
minimum of $2,250,000 of product on an annual basis for five years
following the Company’s first commercial sale of a product
incorporating taurolidine. The Company did not purchase the
required amounts and as a result, lost its exclusive manufacturing
rights. The Company is also required to make certain cash payments
to Navinta upon the achievement of certain sales-based milestones
which is based on a tiered approach and does not commence until the
Company achieves a designated net sales threshold. The maximum
aggregate amount of such payments, assuming achievement of all
milestones, is $1,975,000 over five years. There were no milestones
achieved in 2016, 2015 and 2014.
On
March 24, 2015, the Company and Navinta LLC entered into an
amendment to the Navinta Agreement to extend the term of the
Navinta Agreement to March 31, 2016 and to lower the price per
kilogram of API that the Company purchases from Navinta LLC under
the Navinta Agreement. The Company also agreed to purchase a
minimum amount of product from Navinta LLC during 2015, which
replaced the prior minimum purchase requirement. The Navinta
Agreement expired on March 31, 2016, was not renewed and there are
no further purchase obligations or milestone payments.
The
Company has developed a program aimed at reducing the cost of goods
of Neutrolin through a more efficient, custom synthesis of the
active ingredient taurolidine. As part of that program, on April 8,
2015, the Company entered into a Preliminary Services Agreement
with [RC]2
Pharma Connect LLC (“RC2”), pursuant to which RC2 will
coordinate certain manufacturing services related to taurolidine
that the Company believes are necessary for the submission of its
planned new drug application for Neutrolin to the FDA, as well as
any foreign regulatory applications. The total cost for RC2’s
services under the preliminary services agreement was expected to
approximate $1.7 million through the first quarter of 2017. During
the years ended December 31, 2016 and 2015, the Company recognized
research and development expense of $1,278,000 and $227,000,
respectively, for its services related to the
agreement.
F-22
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
Company also has several service agreements with RC2 for the
manufacture of clinical supplies to support its ongoing and planned
Phase 3 clinical trials for an aggregate amount of $7.6
million. During the years ended December 31, 2016 and 2015,
the Company recognized research and development expense of
approximately $2,359,000 and $1,348,000, respectively, related to
these agreements. The Company may terminate these agreements upon
30 days written notice and is only obligated for project costs and
reasonable project shut down costs provided through the date of
termination.
Clinical and Regulatory
On
December 3, 2015 CorMedix signed a Master Service Agreement and
Work Order (the “Master Service Agreement”) with PPD
Development, LP (“PPD”) for a $19.5 million
(originally, $19.2 million) Phase 3 multicenter, double-blind,
randomized active control study (the “Phase 3 Clinical
Trial”) to demonstrate the safety and effectiveness of
Neutrolin in preventing catheter-related bloodstream infections and
blood clotting in subjects receiving hemodialysis therapy as
treatment for end stage renal disease. The Phase 3 Clinical Trial
is expected to accrue up to 632 patients in 70 sites in the US.
Prior to the signing of the Master Service Agreement, CorMedix
signed a Letter of Agreement (“LOA”) with PPD in July
2015. During the years ended December 31, 2016 and 2015, the
Company recognized $5,283,000 and $1,019,000 research and
development expense related to this agreement,
respectively.
In-Licensing
In
2008, the Company entered into a License and Assignment Agreement
(the “NDP License Agreement”) with NDP. Pursuant to the
NDP License Agreement, NDP granted the Company exclusive, worldwide
licenses for certain antimicrobial catheter lock solutions,
processes for treating and inhibiting infections, a biocidal lock
system and a taurolidine delivery apparatus, and the corresponding
United States and foreign patents and applications (the “NDP
Technology”). The Company acquired such licenses and patents
through its assignment and assumption of NDP’s rights under
certain separate license agreements by and between NDP and Dr.
Hans-Dietrich Polaschegg, Dr. Klaus Sodemann and Dr. Johannes
Reinmueller. As consideration in part for the rights to the NDP
Technology, the Company paid NDP an initial licensing fee of
$325,000 and granted NDP a 5% equity interest in the Company,
consisting of 39,980 shares of the Company’s common
stock.
In
addition, the Company is required to make payments to NDP upon the
achievement of certain regulatory and sales-based milestones.
Certain of the milestone payments are to be made in the form of
shares of common stock currently held in escrow for NDP, and other
milestone payments are to be paid in cash. The maximum aggregate
number of shares issuable upon achievement of milestones is 145,543
shares. During the year ended December 31, 2014, a certain
milestone was achieved resulting in the release of 36,386 shares
held in escrow. The number of shares held in escrow as of December
31, 2016 and 2015 is 109,157 shares of common stock. The maximum
aggregate amount of cash payments upon achievement of milestones is
$3,000,000 with $2,500,000 remaining at December 31, 2016 and 2015.
Events that trigger milestone payments include but are not limited
to the reaching of various stages of regulatory approval and upon
achieving certain worldwide net sales amounts. There were no milestones achieved in 2016 and
2015.
The NDP
License Agreement may be terminated by the Company on a
country-by-country basis upon 60 days prior written notice. If the
NDP License Agreement is terminated by either party, the
Company’s rights to the NDP Technology will revert back to
NDP.
F-23
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On
January 30, 2008, the Company also entered into an Exclusive
License and Consulting Agreement with Dr. Polaschegg (the
“Polaschegg License Agreement”). Pursuant to the
Polaschegg License Agreement, Dr. Polaschegg granted the Company an
exclusive, worldwide license for a certain antimicrobial solution
and certain taurolidine treatments and the corresponding United
States patent applications (the “Polaschegg
Technology”), and agreed to provide the Company with certain
consulting services. As consideration for the rights to the
Polaschegg Technology, the Company paid Dr. Polaschegg an initial
payment of $5,000 and agreed to pay Dr. Polaschegg certain royalty
payments ranging from 1% to 3% of the net sales of the Polaschegg
Technology. The
Polaschegg License Agreement also set forth certain minimum royalty
payments (on an annual basis) to be made to Dr. Polaschegg in
connection with the Polaschegg Technology, which payments range
from $10,000 to $45,000. The Company could terminate the Polaschegg
License Agreement with respect to any piece of the Polaschegg
Technology upon 60 days notice. If the Polaschegg License Agreement
is terminated with respect to any piece of the Polaschegg
Technology by either party, all rights with respect to such portion
of the Polaschegg Technology revert to Dr. Polaschegg. On November
5, 2015, the Company gave notice of its termination of the
Polaschegg License Agreement. During the years ended December 31,
2015 and 2014, the Company expensed $30,000 and $40,000,
respectively, in connection with the Polaschegg License
Agreement.
Other
On
September 27, 2016, the Company entered into an employment
agreement, effective October 3, 2016, with Khoso Baluch, its Chief
Executive Officer. Unless renewed pursuant to the terms thereof,
the agreement will expire on October 3, 2019. After the initial
three-year term of Mr. Baluch’s employment agreement, the
agreement will automatically renew for additional successive
one-year periods, unless either party notifies the other in writing
at least 90 days before the expiration of the then current term
that the agreement will not be renewed. Mr. Baluch also was
appointed to the Company’s Board of Directors (the
“Board”) on October 3, 2016. Mr. Baluch was granted stock options to purchase
1,850,000 shares of common stock, subject to four-year vesting for
some of these options and subject to performance and market based
vesting requirements for the rest of the
options.
If the
Company terminates Mr. Baluch’s employment other than for Cause
(as defined in the agreement), death or disability, other than by
notice of nonrenewal, or if Mr. Baluch resigns for Good Reason (as
defined in the agreement), Mr. Baluch will receive his base salary
and benefits for a period of 12 months following the effective date
of the termination of his employment and all restricted shares and
unvested stock options that are scheduled to vest on or before the
next succeeding anniversary of the date of termination shall be
accelerated and deemed to have vested as of the termination
date.
On
August 3, 2015, the Company entered into a Release of Claims and
Severance Modification with Randy Milby, its former Chief Executive
Officer, that Mr. Milby may not compete against the Company by
engaging in any business involving the development or
commercialization of (i) a preventive anti-infective product that
would be a direct competitor of Neutrolin or (ii) a product
containing taurolidine. The non-compete term did not change and
remains at twelve months following termination of his employment.
The employment agreement was also amended to allow Mr. Milby a
period in which to exercise all vested options and warrants until
the later of 60 months following the termination date of his
employment, provided in no event shall he be able to exercise after
the respective expiration date of any stock option or warrant.
During the year ended December 31, 2015, the Company recorded
non-cash expense of $507,341 as a result of this
modification.
Mr.
Milby resigned as the Company’s Interim Chief Executive
Officer on October 2, 2016. Pursuant to the terms of his employment
agreement, Mr. Milby will be entitled to receive his base salary
and benefits for a period of twelve months following the effective
date of the termination of his employment.
The
Company entered into a sublease for 4,700 square feet of office
space in Bedminster, New Jersey, which sublease runs from April 1,
2015 until March 31, 2018. Rent is $5,000 per month plus occupancy
costs such as utilities, maintenance and taxes. In accordance with
the lease agreement, the Company has deposited $5,000 with the
landlord, the equivalent of one month rent.
The
Company’s subsidiary entered into a lease agreement for its
offices in Fulda, Germany. The lease has a term of 36 months which
commenced on September 1, 2013 for a base monthly payment of
€498. The total 36 month lease obligation is approximately
€17,900 ($20,000). The 36 month lease has terminated. A new
lease agreement commenced on November 1, 2017 which is renewable
every three months for a base monthly payment of
€461.
Rent
expense for the years ended December 31, 2016, 2015 and 2014, was
$68,096, $72,119 and $70,337, respectively.
Under
the Company’s current lease agreements, the total remaining
lease obligation as of December 31, 2016 is set forth
below:
F-24
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
2017
|
$62,237
|
|
2018
|
15,000
|
|
Total
|
$77,237
|
Note 7 — Equity Instruments Modification and Fair
Value Measurements:
The
following table presents the fair value hierarchy and the change in
fair values of the Company’s derivative liabilities measured
at fair value on a recurring basis during the year ended December
31, 2014.
|
|
Fair Value
Hierarchy Level
|
Change in Fair
Value From Jan. 1 to Sept. 15, 2014 (Modification
Date)
|
|
Series C-1, C-2 and
C-3 non-voting preferred stock conversion option issued in October
2013 and January 2014
|
3
|
$599,814
|
|
Series D non-voting
preferred stock conversion option issued in October
2013
|
3
|
2,017,960
|
|
Series E non-voting
preferred stock conversion option issued in October
2013
|
3
|
1,786,902
|
|
Warrants issued in
connection with convertible debt issued in May 2013
|
3
|
1,566,444
|
|
Warrants issued in
connection with Series C-1, C-2 and C-3 non-voting preferred stock
issued in October 2013 and January 2014
|
3
|
3,732,962
|
|
Warrants issued in
March 2014 in connection with the private placement of common stock
and warrants
|
3
|
(855,129)
|
|
Total
|
|
$8,848,953
|
On
September 15, 2014, the Company entered into consent and exchange
agreements with the investors holding its outstanding Series C-2
preferred stock and related warrants, Series C-3 preferred stock
and related warrants, Series D preferred stock and Series E
preferred stock, and the investors holding warrants issued in March
2014. Pursuant to those agreements, the Company and the investors
agreed to amend and restate the Series C-2 preferred stock and
related warrants, Series C-3 preferred stock and related warrants,
Series D preferred stock and Series E preferred stock and the
warrants, to remove anti-dilution, price reset, cash settlement
features and certain change of control provisions that caused those
instruments to be classified as derivative
liabilities. The Company also eliminated the preferred
dividends on the Series D preferred stock and Series E preferred
stock.
In
exchange for the removal of the anti-dilution, price reset, cash
settlement, change of control and dividend provisions, the Company
agreed to the following:
1.
Decrease the
exercise price of the warrants issued in May 2013 from $1.00 to
$0.65, decrease the exercise price of the warrants issued in
October 2013 from $1.25 to $0.90, decrease the exercise price of
the warrants issued in January 2014 from $1.25 to $0.90, and
decrease the exercise price of the warrants issued in March 2014
from $3.10 to $2.50;
2.
Extend the existing
right of the two institutional investors in our May and October
2013 financings to participate in future financings to the later of
two years (subsequently extended by an additional year) after
September 15, 2014 or the date on which the respective
holder holds less than 5% of the Company’s common stock on a
fully diluted basis;
3.
Increase the
conversion ratio of the Series E preferred stock from 20 shares to
21.8667 shares of common stock for every share of Series
E preferred stock;
4.
Issue 16,562 shares
of the Company’s Series D preferred stock to the investor
holding all of the outstanding shares of the Series D preferred
stock in satisfaction of the 9.0% payment-in-kind dividend on that
stock; and
F-25
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
5.
Issue an aggregate
of 37,226 shares of Series E preferred stock to the two investors
holding all of the outstanding shares of Series E preferred stock
in satisfaction of the 8.0% payment-in-kind dividend on that
stock.
As a
result of these modifications, all of the outstanding derivative
liabilities were reclassified to equity on September 15, 2014. The
fair value was re-measured immediately prior to the modification
date with the original terms and immediately after the modification
date with the amended terms. The change in fair value resulting
from the modifications made to those instruments on September 15,
2014 was recorded as loss on modification of equity instruments and
extinguishment of derivative liabilities in the amount of
approximately $2,463,000.
The
table below sets forth a summary of changes in the fair value of
the Company’s Level 3 derivative liabilities related to the
non-voting preferred stock embedded derivatives and the liability
classified warrants.
|
|
December 31,
2014
|
|
Balance at
beginning of year
|
$5,308,804
|
|
Additions to
derivative liabilities
|
3,782,182
|
|
Conversion of
convertible preferred stock to common stock
|
(2,447,384)
|
|
Loss from
modification of preferred stock and warrant
instruments
|
2,462,588
|
|
Change in fair
value of derivative liabilities
|
8,848,953
|
|
Reclassification of
derivative liabilities to equity (excluding $21,117 dividends
issued in 2013)
|
(17,955,143)
|
|
Balance at December
31, 2014
|
$-
|
Note 8 — Stockholders’ Equity:
Common Stock:
During
2015 the Company entered into a Sales Agreement with MLV under
which the Company may issue and sell up to $40.0 million of shares
of its common stock from time to time through MLV acting as agent,
subject to limitations imposed by the Company, such as the number
or dollar amount of shares registered under the registration
statement to which the offering relates. When the Company wishes to
issue and sell common stock under the Sales Agreement, it notifies
MLV of the number of shares to be issued, the dates on which such
sales are anticipated to be made, any minimum price below which
sales may not be made and other sales parameters as the Company
deems appropriate. MLV is entitled to a commission of up to 3% of
the gross proceeds from the sale of common stock sold under the
Sales Agreement. The shares of common stock to be sold under the
Sales Agreement are registered under an effective registration
statement filed with the SEC. At December 31, 2016, approximately
$4.1 million was available for sale under this Sales Agreement.
During the years ended December 31, 2016 and 2015, the shares of
common stock under the Sales Agreement issued by the Company were
3,360,037 and 5,310,037, respectively, and realized net proceeds of
$6,229,000 in 2016 and $28,452,000 in 2015.
In
August 2016, the Company entered into a new sales agreement with
the same bank, which is identical in terms to the Sales Agreement
for the Company’s current ATM program and allows the Company
to sell up to $40 million of shares of its common stock. The
Company will not and cannot access the ATM program under the new
sales agreement unless and until (i) the Company and Manchester
agree as to the exercise or waiver of Manchester’s
participation rights in the new ATM program, which rights were
granted in a Consent and Exchange Agreement dated September 15,
2014, and apply to any equity financing we undertake until
September 15, 2017, and (ii) the registration statement that
includes the prospectus for the new ATM program that the Company
filed with the Securities and Exchange Commission is declared
effective. At such time, the Company will be able to access the new
ATM program and will terminate the current ATM program and the
related Sales Agreement. There is no assurance that conditions will
allow the Company to raise additional funds available under its
at-the-market program.
F-26
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During the year ended December 31, 2016,
the Company also issued the following
shares of common stock:
●
21,454
shares upon cashless exercise of 25,000 warrants;
●
1,087,500
shares upon exercise of stock options at a weighted average
exercise price of $0.79 per share, resulting in gross proceeds of
$863,101.
During
the year ended December 31, 2015, the Company also issued the
following shares of common stock:
●
4,581,783 shares of
common stock upon exercise of warrants with a weighted average
exercise price of $3.20 per share resulting in gross proceeds of
$14,658,161;
●
2,158,033
shares upon cashless exercise of 2,597,591 warrants;
●
499,955
shares upon exercise of stock options at a weighted average
exercise price of $0.99 per share, resulting in gross proceeds of
$492,960;
●
454,546
shares upon conversion of 454,546 shares of the Series B non-voting
preferred stock;
●
425,000
shares upon conversion of an aggregate of 42,500 shares of the
Series C-3 non-voting preferred stock;
●
61,598
shares upon conversion of 2,817 shares of the Series E non-voting
preferred stock; and
●
10,728
shares upon conversion of wages by an officer of the Company in an
aggregate amount of $50,000 at prices per share of $3.10 -
$8.55.
During
the year ended December 31, 2014, the Company issued the following
shares of common stock:
●
455,000
shares upon exercise of stock options resulting in gross proceeds
of $318,150;
●
1,400,000
shares upon conversion of an aggregate of 140,000 shares of the
Series C-1 non-voting preferred stock;
●
210,000
shares upon conversion of 21,000 shares of the Series C-3
non-voting preferred stock;
●
772,589
shares upon exercise of warrants to purchase 919,513 shares of the
Company’s common stock on a cashless basis; and
●
57,384
shares upon conversion of wages and board fees by an officer and
board member in an aggregate amount of $96,851 at prices of $1.32 -
$2.00 per share.
In
March 2014, the Company sold an aggregate of 2,960,000 units in a
registered direct offering at a purchase price of $2.50 per unit,
for net proceeds of $6,723,248. Each unit consisted of
one share of the Company’s common stock and 0.35 of a
warrant, each to purchase one share of the Company’s common
stock. Upon issuance, the warrants had an exercise
price of $3.10 per share, are exercisable commencing six months
from the date of issuance, and have a term of five years from the
date of exercisability. Under certain circumstances, the warrants
may be settled in cash and were therefore were initially classified
as derivative liabilities (See Note 7). The Company used the Black
Scholes option pricing model to value the warrants, of which
$1,728,450 was the ascribed value calculated at the issuance date.
These warrants were revalued at each balance sheet date and the
resulting changes were recorded in other income (expense) in the
statement of operations. On September 15, 2014, the exercise price
of these warrants was decreased to $2.50 in exchange for the
removal of the cash settlement provisions of the warrant. During
2014, the Company used the following assumptions in calculating the
Black Scholes values of these warrants:
|
|
At Issuance
Date
|
At September 15,
2014
|
|
Expected term
(years)
|
5.5
|
5
|
|
Volatility
|
75%
|
75%
|
|
Dividend
yield
|
0.0%
|
0.0%
|
|
Risk-free interest
rate
|
1.63%
|
1.8%
|
F-27
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Preferred Stock and Warrants
The
Company is authorized to issue up to 2,000,000 shares of preferred
stock in one or more series without stockholder approval. Our board
of directors has the discretion to determine the rights,
preferences, privileges and restrictions, including voting rights,
dividend rights, conversion rights, redemption privileges and
liquidation preferences, of each series of preferred stock. Of the
2,000,000 shares of preferred stock authorized, our board of
directors has designated (all with par value of $0.001 per share)
the following:
|
|
As of December 31, 2016 and 2015
|
As of December 31, 2014
|
||||
|
|
Preferred Shares
Outstanding
|
Liquidation
Preference (Per
Share)
|
Total Liquidation
Preference
|
Preferred Shares
Outstanding
|
Liquidation
Preference (Per
Share)
|
Total Liquidation
Preference
|
|
Series
B
|
-
|
$0.001
|
$-
|
454,546
|
$0.001
|
$455
|
|
Series
C-2
|
150,000
|
10.000
|
1,500,000
|
150,000
|
10.000
|
1,500,000
|
|
Series
C-3
|
136,500
|
10.000
|
1,365,000
|
179,000
|
10.000
|
1,790,000
|
|
Series
D
|
73,962
|
21.000
|
1,553,202
|
73,962
|
21.000
|
1,553,202
|
|
Series
E
|
89,623
|
49.200
|
4,409,452
|
92,440
|
49.200
|
4,548,048
|
|
Total
|
450,085
|
|
$8,827,654
|
949,948
|
|
$9,391,705
|
The following terms and conditions apply to
all of the non-voting convertible preferred stock outstanding at
December 31, 2016, 2015 and 2014:
Dividends - Holders of the Series
B, Series C, Series D and Series E non-voting preferred stock are
entitled to receive, and the Company is required to pay, dividends
on shares of the Series B, Series C, Series D and Series E
non-voting preferred stock equal to (on an
as-if-converted-to-common-stock basis) and in the same form as
dividends (other than dividends in the form of common stock)
actually paid on shares of the common stock when, as and if such
dividends (other than dividends in the form of common stock) are
paid on shares of the common stock.
Fundamental Transactions- If, at
any time that shares of Series B, Series C, Series D or Series E
non-voting preferred stock are outstanding, the Company effects a
merger or other change of control transaction, as described in the
certificate of designation and referred to as a fundamental
transaction, then, upon each and every fundamental transaction, a
holder will have the right to receive, upon any subsequent
conversion of a share of Series B, Series C, Series D or Series E
non-voting preferred stock (in lieu of conversion shares) for each
issuable conversion share, the same kind and amount of securities,
cash or property as such holder would have been entitled to receive
upon the occurrence of such fundamental transaction if such holder
had been, immediately prior to such fundamental transaction, the
holder of a share of common stock.
Redemption – The Company is not
obligated to redeem or repurchase any shares of Series B, Series C,
Series D or Series E non-voting preferred stock. Shares of Series
B, Series C, series D and Series E Preferred Stock are not
otherwise entitled to any redemption rights.
Listing- There is no established public
trading market for the Series B, Series C, Series D or Series E
non-voting preferred stock, and the Company does not expect a
market to develop. In addition, the Company does not intend to
apply for listing of the Series B, Series C, Series D or Series E
non-voting preferred stock on any national securities exchange or
trading system.
Series B Non-Voting Convertible Preferred Stock and
Warrants
On July
30, 2013, the Company sold 454,546 shares of its Series B
non-voting convertible preferred stock and a warrant to purchase up
to 227,273 shares of the Company’s common stock, for gross
proceeds of $500,000. Each share of Series B stock was convertible
into one share of the Company’s common stock at any time at
the holder’s option. All shares of Series B stock were
converted into 454,546 shares of common stock in 2015.
The
warrant was exercisable immediately upon issuance and has an
exercise price of $1.50 per share and a term of five
years.
F-28
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Series C-1, Series C-2 and Series C-3 Non-Voting Convertible
Preferred Stock and Warrants
On October 22, 2013, the Company issued 150,000
shares of Series C-1 non-voting convertible preferred stock and 150,000 shares of Series
C-2 non-voting convertible preferred stock, together with warrants to
purchase up to an aggregate of 1,500,000 shares of common
stock. As a condition to the closing, the Company
simultaneously exchanged a convertible note held by one of the
investors in the principal amount of $400,000 for 57,400 shares of
Series D non-voting convertible preferred stock and exchanged another convertible
note held by the same investor in the principal amount of $750,000
for 53,537 shares of Series E non-voting convertible
preferred stock. The
Company also issued 1,677 shares of Series E preferred stock to the
other investor in the offering.
In January 2014, the Company sold to various
investors 200,000 shares of Series C-3 non-voting
convertible preferred stock, together
with warrants to purchase up to an aggregate of 1,000,000 shares of
common stock, for aggregate gross proceeds of
$2,000,000. The Series C-3 non-voting convertible
preferred stock and the related warrants were sold together at a
price of $10.00 per share. The warrants are exercisable
one year after issuance, had an exercise price of $1.25 per share
(decreased to $0.90 per share in September 2014 – See Note
7), subject to adjustment, and a term of five years from the date
they are first exercisable. The Company received net proceeds of
$1,318,884.
The
Series C-1 non-voting preferred stock, Series C-2 non-voting
preferred stock and Series C-3 non-voting preferred stock have
identical rights, privileges and terms and are referred to
collectively as the “Series C Stock.” Each
share of Series C Stock is convertible into 10 shares of common
stock at any time at the holder’s option at a conversion
price of $1.00 per share. In the event of the
Company’s liquidation, dissolution, or winding up, holders of
the Series C Stock will receive a payment equal to $10.00 per share
of Series C Stock, subject to adjustment, before any proceeds are
distributed to the holders of common stock. Shares of
the Series C Stock will not be entitled to receive any dividends,
unless and until specifically declared by the Company’s board
of directors.
In
January 2014, all 140,000 outstanding shares of Series C-1
non-voting preferred stock were converted into 1,400,000 shares of
the Company’s common stock which resulted in the
reclassification of the derivative liability to equity in the
amount of $2,447,384 for the year ended December 31,
2014.
The Series C Preferred Stock ranks senior to the
Company’s common stock; senior to any class or series of capital stock created
after the issuance of the Series C Preferred Stock; and junior to
the Series D non-voting convertible preferred stock and Series E
non-voting convertible preferred stock. As long as any of the
Series C-2 Preferred Stock is outstanding, the Company cannot incur
any indebtedness other than indebtedness existing prior to
September 15, 2014, trade payables incurred in the ordinary course
of business consistent with past practice, and letters of credit
incurred in an aggregate amount of $3.0 million at any point in
time.
Due to the existence of downround provisions, the
conversion features of the Series C-3 non-voting convertible
preferred stock and the associated
warrants were initially classified as derivative liabilities upon
issuance and were valued using a Monte Carlo simulation model. On
the issuance date, the estimated value of the conversion features
and warrants was $1,398,158 and $655,574,
respectively.
In February 2014, the downround protection of
Series C-2 and Series C-3 non-voting convertible
preferred stock was eliminated as,
pursuant to their terms, the closing price of the Company’s
common stock was greater than $2.00 for a period of twenty trading
days for a consecutive thirty trading day period subsequent to the
closing (See Note 7).
The Series C-1 non-voting convertible
preferred stock, Series C-2
non-voting convertible preferred
stock, Series D non-voting convertible preferred stock and Series E non-voting
convertible preferred stock all
contained a prohibition on its respective conversion (in the case
of the Series C-1 and Series C-2, in the aggregate for both series)
if, as a result of such conversion, the Company would have issued
in each case shares of its common stock in an aggregate amount
equal to 3,190,221 shares, which is 20% of the shares of common
stock outstanding on October 17, 2013, unless the Company received
the approval of its stockholders for such overage. On February 28,
2014, the shareholders approved the issuance of such
overage.
F-29
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Series D Non-Voting Convertible Preferred Stock
Each share of Series D non-voting
convertible preferred stock is
convertible into 20 shares of common stock (subject to adjustment)
at a per share price of $0.35 at any time at the option of the
holder, except that a holder will be prohibited from converting
shares of Series D non-voting convertible preferred stock into shares of common stock if, as
a result of such conversion, such holder, together with its
affiliates, would beneficially own more than 9.99% of the total
number of shares of the Company’s common stock then issued
and outstanding. In the event of the Company’s liquidation,
dissolution or winding up, holders of Series D non-voting
convertible preferred stock originally
were to receive a payment equal to $7.00 per share (increased to
$21.00 per share in September 2014 – See Note 7) of Series
D non-voting convertible preferred stock on parity with the payment of the
liquidation preference due the Series E non-voting
convertible preferred stock, but
before any proceeds are distributed to the holders of common stock,
the Series C-1 non-voting convertible preferred stock and the Series C-2
non-voting convertible preferred
stock. Shares of Series D non-voting convertible
preferred stock received a dividend of
9% per annum through September 15, 2014 (See Note
7).
The Series D non-voting convertible preferred
stock ranks senior to the Company’s common stock; senior to
any class or series of capital stock created after the issuance of
the Series D non-voting convertible preferred stock; senior to the
Series C-2 non-voting convertible preferred stock and the Series
C-3 non-voting convertible preferred stock; and on parity with the
Series E non-voting convertible preferred stock. As long as any of
the Series D non-voting convertible preferred stock is outstanding, the Company cannot
incur any indebtedness other than indebtedness existing prior to
September 15, 2014, trade payables incurred in the ordinary course
of business consistent with past practice, and letters of credit
incurred in an aggregate amount of $3.0 million at any point in
time.
In
addition to the debt restrictions above, as long as any shares of
the Series D non-voting convertible preferred stock are
outstanding, the Company cannot, among others things: create,
incur, assume or suffer to exist any encumbrances on any of its
assets or property; or redeem, purchase or otherwise acquire or pay
or declare any dividend or other distribution on any junior
securities.
Series E Non-Voting Convertible Preferred Stock
Each share of Series E non-voting convertible
preferred stock was originally convertible into 20 shares
(increased to 21.8667 per share in September 2014 – See Note
7) of the Company’s common stock (subject to adjustment) at a
per share price of $0.82 (reduced to $0.75 per share in September
2014 – See Note 7) at any time at the option of the holder,
except that a holder will be prohibited from converting shares of
Series E non-voting convertible preferred stock into shares of
common stock if, as a result of such conversion, such holder,
together with its affiliates, would beneficially own more than
9.99% of the total number of shares of the Company’s common
stock then issued and outstanding. In the event of the
Company’s liquidation, dissolution or winding up, holders of
Series E preferred stock originally was to receive a payment equal
to $16.40 per share (increased to $49.20 per share in September
2014 – See Note 7) of Series E non-voting convertible
preferred stock on parity with the payment of the liquidation
preference due the Series D non-voting convertible preferred stock,
but before any proceeds are distributed to the holders of common
stock, the Series C-2 non-voting convertible preferred stock. Shares of Series E
non-voting convertible preferred stock
received a dividend of 8% per annum through September 15, 2014 (See
Note 7).
The Series E non-voting convertible
preferred stock ranks senior to the
Company’s common stock; senior to any class or series of
capital stock created after the issuance of the Series E non-voting
preferred stock; senior to the Series C-2 and the Series C-3
non-voting convertible preferred stock; and on parity with the
Series D non-voting convertible preferred stock. As long as any of
the Series E non-voting convertible preferred stock is outstanding, the Company cannot
incur any indebtedness other than indebtedness existing prior to
September 15, 2014, trade payables incurred in the ordinary course
of business consistent with past practice, and letters of credit
incurred in an aggregate amount of $3.0 million at any point in
time.
In addition to the debt restrictions above, as
long as any the Series E non-voting convertible
preferred stock is outstanding
, the Company cannot, among others things: create, incur, assume or
suffer to exist any encumbrances on any of our assets or property;
redeem, repurchase or pay any cash dividend or distribution on any
of our capital stock (other than as permitted); redeem, repurchase
or prepay any indebtedness; or engage in any material line of
business substantially different from our current lines of
business.
In the
event the Company issues any options, convertible securities or
rights to purchase stock or other securities pro rata to the
holders of common stock, then holders of Series E non-voting
convertible preferred stock will be entitled to acquire, upon the
same terms a pro rata amount of such stock or securities as if the
Series E non-voting convertible preferred stock had been converted
to common stock.
F-30
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
Company used a Monte Carlo model to separately value the Series
C-1, C-2, C-3, D and E preferred stock, the conversion options
associated with the those preferred stock instruments and the
warrants issued in connection with the Series C-1 and C-2 preferred
stock. A summary of the key assumptions used in the
Monte Carlo models are as follows:
Stock price – Due to the
historical volatility of the stock price, a 30-day volume-weighted
average stock price was used as of each valuation
date.
Conversion/redemption strike price
– These assumptions incorporate both the initial contractual
conversion price as well as subsequent downward adjustments based
on management’s estimate of the probabilities of additional
future financings that would include a stock price or conversion
price that is lower than the then existing conversion
price.
Volatility – The Company used a
weighted average of 1) the historical volatility of the stock of
CorMedix for approximately three years, 2) the volatility of the
stock of CorMedix after receiving product approval and 3) the
volatilities of comparable companies (provided by the management)
from the date product approval is received to the various valuation
dates. Then, appropriate weights were applied to these data points
to arrive at the weighted average historical volatility. The
concluded volatility is assumed to remain constant for all the
valuation dates.
Term – Although the preferred
Series C, D and E instruments do not have a specified contracted
life, the Company has assumed a five year life from the date of
inception for the purpose of the valuations, indicating that these
instruments would expire in October 2018 at which point the holder
would convert the investments into equity.
Risk-free Rate – The U.S.
Treasury Bond Rate with a term approximating the term of the
instrument was used as the risk-free interest rate in the
valuation.
Credit adjusted discount rate –
Management believes that its debt, if rated, would be equivalent to
Moody’s C rated bonds or lower.
Dividend rate - Management does not
expect to pay any dividends during the term of the hybrid
instrument.
A
summary of the assumptions used in the Monte Carlo models are as
follows:
|
|
At September 15,
2014
|
At Issuance
Date
|
|
Expected term
(months)
|
49 - 64
|
56 - 60
|
|
Volatility
|
75%
|
75%
|
|
Dividend
yield
|
0.0%
|
0.0%
|
|
Risk-free interest
rate
|
1.63 - 1.8%
|
1.3 - 1.5%
|
Stock Options:
In
2013, the Company’s board of directors approved the 2013
Stock Incentive Plan (the “2013 Plan”). The 2013 Plan
provides for the issuance of equity grants in the form of options,
restricted stock, stock awards and other forms of equity
compensation. Awards may be made to directors, officers, employees
and consultants under the 2013 Plan. An aggregate of 5,000,000
shares of the Company’s common stock is reserved for issuance
under the 2013 Plan. On January 19, 2016, the shareholders approved
the increase of the shares issuable under the 2013 Plan from
5,000,000 to 8,000,000 and on June 13, 2016 from 8,000,000 to
11,000,000.
F-31
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During
the year ended December 31, 2016, the Company granted ten-year
non-qualified stock options under the 2013 Plan covering an
aggregate of 2,891,000 shares of the Company’s common stock
to its officers, directors, employees and consultants. Of these
options, 1,850,000 were granted on September 30, 2016 to the
Company’s new CEO in connection with his employment.
1,250,000 of the options will vest in four equal annual
installments on the first four anniversaries of the grant date. Of
the remaining options, 300,000, split into three equal tranches,
become exercisable upon the achievement of specified performance
milestones, provided that these options will be forfeited if the
milestones are not achieved within four years of grant date and
provided further that these options will not vest before December
18, 2018. The remaining 300,000 options become exercisable upon the
achievement of a specified average closing stock price, provided
that these options will not vest before December 31, 2018 and if
the closing price per share of the Company’s common stock is
below the specified average closing stock price on December 31,
2018, the options will be forfeited. In each case, the new CEO must
be an employee of the Company or consultant to the Company on the
applicable vesting date. The total fair value of the 1,850,000
stock options issued to the Company’s CEO on the date of
grant was $3,186,450 which will be amortized to expense over the
related vesting periods.
During
the year ended December 31, 2015, the Company granted ten-year
service based non-qualified stock options under the 2013 Plan
covering an aggregate of 640,000 shares of the Company’s
common stock to its officers, directors and
consultants.
During
the years ended December 31, 2016, 2015 and 2014, total
compensation expense for stock options issued to employees,
directors, officers and consultants was $1,335,157, $3,225,659 and
$2,168,303, respectively.
As of
December 31, 2016, there was $3,375,165 total unrecognized
compensation expense related to stock options granted which expense
will be recognized over an expected remaining weighted average
period of 1.8 years.
Effective October
1, 2016, the Company adopted Accounting Standards Update
(“ASU”) 2016-09, Compensation — Stock Compensation
(“Topic 718”), Improvements to Employee Share-Based Payment
Accounting to account for forfeitures as they occur. All
share-based awards will be recognized on a straight-line method,
assuming all awards granted will vest. Forfeitures of share-based
awards will be recognized in the period in which they occur. Prior
to the adoption of ASU 2016-09, share-based compensation expense
was recognized by applying the expected forfeiture rate during the
vesting period to the fair value of the award. As of January
1, 2016, a cumulative effect adjustment of $129,730 was recognized
to reflect the forfeiture rate that had been applied to unvested
option awards prior to fiscal year 2016.
The
fair value at grants dates of the grants issued subject to service
and performance based vesting conditions were determined using the
Black-Scholes option pricing model with the following
assumptions:
|
|
|
Year Ended
December 31,
|
||||
|
|
|
2016
|
|
2015
|
|
2014
|
|
Risk-free
interest rate
|
|
1.14% -
1.94%
|
|
1.47% -
2.26%
|
|
1.5% -
2.9%
|
|
Expected
volatility
|
|
96% -
98%
|
|
93% -
94%
|
|
74% -
113%
|
|
Expected
term (years)
|
|
5 - 10
years
|
|
5 - 10
years
|
|
5 - 10
years
|
|
Expected
dividend yield
|
|
0.0%
|
|
0.0%
|
|
0.0%
|
|
Weighted-average
grant date fair value of options granted during the
period
|
|
$
1.76
|
|
$
3.46
|
|
$
1.50
|
The
Company estimated the expected term of the stock options granted to
employees based on anticipated exercises in future periods. The
expected term of the stock options granted to consultants is based
upon the full term of the respective option agreements. The
expected volatility is calculated based on the historical
volatility since the initial public offering of the Company’s
common stock in March 2010, weighted pre and post CE Mark approval
in the European Union. The expected dividend yield of 0.0% reflects
the Company’s current and expected future policy for
dividends on the Company’s common stock. To determine the
risk-free interest rate, the Company utilized the U.S. Treasury
yield curve in effect at the time of grant with a term consistent
with the expected term of the Company’s awards.
F-32
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
fair value of the grant issued subject to a market based vesting
condition was determined using the onte Carlo option pricing model
which values financial instruments whose value is dependent on
share price by sampling random paths for share price. The key
inputs for the simulation included the closing stock price of the
Company on the date of grant, the expected term of the stock
options granted was based on anticipated exercises in future
periods, the expected stock price volatility for the
Company’s stock options was calculated based on the
historical volatility since the initial public offering of the
Company’s common stock in March 2010, weighted pre and post
CE Mark, the expected dividend yield of 0.0% reflects the
Company’s current and expected future policy for dividends on
the Company’s common stock and the risk-free interest rate
which was determined by utilizing the U.S. Treasury yield curve in
effect at the time of grant with a term consistent with the
expected term of the Company’s awards. The table below
summarizes the key inputs used in the Monte Carlo
simulation:
|
Expected
Term
|
5
years
|
|
Volatility
|
97%
|
|
Dividend
yield
|
0.0%
|
|
Risk-free interest
rate
|
1.13%
|
|
Weighted-average
grant date fair value of options granted during the
period
|
$1.12
|
The
following table summarizes the Company’s stock options
activity and related information for the year ended December 31,
2016:
|
|
Shares
|
Weighted-Average
Exercise Price
|
Weighted-Average
Remaining Contractual Term (Years)
|
Aggregate
Intrinsic Value
|
|
Outstanding at
beginning of year
|
3,600,045
|
$1.82
|
7.6
|
$2,405,321
|
|
Granted
|
2,891,000
|
$2.38
|
|
|
|
Exercised
|
(1,087,500)
|
$0.79
|
|
|
|
Expired/Cancelled
|
(510,000)
|
$2.73
|
|
|
|
Forfeited
|
(283,790)
|
$2.28
|
|
|
|
Outstanding at end
of year
|
4,609,755
|
$2.29
|
8.2
|
$581,823
|
|
Vested at end of
year
|
2,136,458
|
$2.13
|
6.6
|
$581,823
|
|
Expected to vest in
the future
|
2,462,797
|
$2.42
|
9.6
|
$-
|
The
total intrinsic value of stock options exercised during the years
ended December 31, 2016, 2015 and 2014 was $1,497,506, $3,260,728
and $636,250, respectively. The aggregate intrinsic value is
calculated as the difference between the exercise prices of the
underlying options and the quoted closing price of the common stock
of the Company at the end of the reporting period for those options
that have an exercise price below the quoted closing
price.
In July
2015, the Company entered into a Release of Claims and Severance
Modification Agreement with Randy Milby (See Note 6), the
Company’s CEO, due to Mr. Milby’s anticipated
termination of employment. As a result, the Company recorded a
total of $507,341 compensation expense for the incremental value of
an aggregate of 762,500 options during the year ended December 31,
2015 using the Black-Scholes option pricing model with the
following assumptions:
|
Expected
term (years)
|
0.25
– 5
|
|
Volatility
|
94% -
97%
|
|
Dividend
yield
|
0.0%
|
|
Risk-free
interest rate
|
0.05% -
1.61%
|
F-33
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Warrants:
The
following table is the summary of warrant activities:
|
|
Shares
|
Weighted Average
Exercise Price
|
Weighted Average
Remaining Contractual Life
|
|
Outstanding at
December 31, 2015
|
4,422,188
|
$1.80
|
3.07
|
|
Expired
|
(390,720)
|
$3.44
|
-
|
|
Exercised
|
(25,000)
|
$0.40
|
-
|
|
Outstanding at
December 31, 2016
|
4,006,468
|
$1.65
|
2.36
|
During
the year ended December 31, 2015, the Company extended the
expiration date for an aggregate of 38,400 warrants with an
exercise price of $3.4375. The Company accounted for this
transaction as a modification of warrants and recorded additional
paid in capital and non-cash general and administrative expense in
the amount of $112,982. The warrants were valued using the
Black-Scholes option pricing model with the following
assumptions:
|
Expected term
(days)
|
5
|
|
Volatility
|
88.17%
|
|
Dividend
yield
|
0.0%
|
|
Risk-free interest
rate
|
.003%
|
On
March 2, 2015, the Company’s board of directors approved an
extension to April 30, 2015 of the expiration date of the
Company’s publicly traded warrants which resulted in deemed
dividend of $33,121.
In
March 2015, the Company issued two warrants exercisable for an
aggregate of up to 283,400 common shares with an exercise price of
$7.00 per share and a term of five years as a result of entering
into a backstop agreement with Manchester Securities Corp.
(“Manchester”) (See Note 4). Additionally, the
expiration date of March 24, 2015 of warrants to purchase 390,720
shares of common stock issued to Manchester in connection with the
Company’s initial public offering (“IPO”) was
extended by one year to March 24, 2016. The Company recorded
non-cash other expense of $1,583,252 for these warrants using the
Black-Scholes option pricing model with the following
assumptions:
|
Expected
term (years)
|
1 -
5
|
|
Volatility
|
75.81%
- 104.08%
|
|
Dividend
yield
|
0.0%
|
|
Risk-free
interest rate
|
0.01% -
1.61%
|
Stock-based Deferred Compensation Plan for Non-Employee
Directors
During
the third quarter of 2014, the Company established an unfunded
stock-based deferred compensation plan, providing non-employee
directors the opportunity to defer up to one hundred percent of
fees and compensation, including restricted stock units. The
amount of fees and compensation deferred by a non-employee director
is converted into stock units, the number of which is determined
based on the closing price of the Company’s common stock on
the date such compensation would have otherwise been payable.
At all times, the plan participants are one hundred percent vested
in their respective deferred compensation accounts. On the
tenth business day of January in the year following a
director’s termination of service, the director will receive
a number of common shares equal to the number of stock units
accumulated in the director’s deferred compensation
account. The Company accounts for this plan as stock based
compensation under ASC 718. During the year ended December
31, 2016, 2015 and 2014, the amount of compensation that was
deferred under this plan was $95,666, $79,200 and $21,826,
respectively.
Note 9 — Concentrations:
During
the years ended December 31, 2015 and 2014, the Company had
revenues of $100,000 and $55,000 from one customer which
represented 48% and 29% of the Company’s total revenue,
respectively. At December 31, 2015, approximately 93% of net
accounts receivable was due from this same customer. During the
year ended December 31, 2016, the Company did not have individual
sales in excess of 10% of its total sales.
Note 10 — Quarterly Results of Operations
(Unaudited):
F-34
CORMEDIX INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
following table sets forth certain of the Company's unaudited
quarterly consolidated results of operations for the years ended
December 31, 2016 and 2015 (amounts in thousands, except for per
share amounts):
|
|
First
Quarter
|
Second
Quarter
|
Third
Quarter
|
Fourth
Quarter
|
|
December
31, 2016:
|
|
|
|
|
|
Net
sales
|
$41
|
$16
|
$44
|
$121
|
|
Cost of
sales
|
(50)
|
(187)
|
(44)
|
(85)
|
|
Gross profit
(loss)
|
(9)
|
(171)
|
-
|
36
|
|
Research and
development
|
(2,089)
|
(2,773)
|
(6,840)
|
(4,032)
|
|
Selling, general
and administrative
|
(2,163)
|
(1,968)
|
(2,318)
|
(2,433)
|
|
(Loss) from
operations
|
(4,261)
|
(4,912)
|
(9,158)
|
(6,429)
|
|
Total other income
(expenses)
|
30
|
25
|
32
|
30
|
|
Net
loss
|
(4,231)
|
(4,887)
|
(9,126)
|
(6,399)
|
|
Other comprehensive
income (loss)
|
31
|
(3)
|
(7)
|
(2)
|
|
Comprehensive net
(loss)
|
$(4,200)
|
$(4,890)
|
$(9,133)
|
$(6,401)
|
|
Net loss
attributable to common shareholders
|
$(4,231)
|
$(4,887)
|
$(9,126)
|
$(6,429)
|
|
Net loss per common
shares - basic and diluted
|
$(0.12)
|
$(0.13)
|
$(0.23)
|
$(0.16)
|
|
Weighted Average
common shares outstanding – basic and diluted
|
36,013
|
36,447
|
39,054
|
40,381
|
|
|
|
|
|
|
|
December
31, 2015:
|
|
|
|
|
|
Net
sales
|
$31
|
$120
|
$36
|
$23
|
|
Cost of
sales
|
(17)
|
(102)
|
(35)
|
(164)
|
|
Gross
(loss)
|
14
|
18
|
1
|
(141)
|
|
Research and
development
|
(1,235)
|
(1,798)
|
(1,764)
|
(1,485)
|
|
Selling, general
and administrative
|
(2,693)
|
(2,355)
|
(2,949)
|
(2,267)
|
|
(Loss) from
operations
|
(3,914)
|
(4,135)
|
(4,712)
|
(3,893)
|
|
Total other income
(expenses)
|
(1,584)
|
-
|
24
|
27
|
|
Net
loss
|
(5,498)
|
(4,135)
|
(4,688)
|
(3,866)
|
|
Other comprehensive
income (loss)
|
3
|
(9)
|
7
|
(38)
|
|
Comprehensive net
(loss)
|
$(5,495)
|
$(4,144)
|
$(4,681)
|
$(3,904)
|
|
Net loss
attributable to common shareholders
|
$(5,498)
|
$(4,135)
|
$(4,688)
|
$(3,866)
|
|
Net loss per common
shares - basic and diluted
|
$(0.23)
|
$(0.13)
|
$(0.14)
|
$(0.11)
|
|
Weighted Average
common shares outstanding – basic and diluted
|
23,922
|
31,623
|
34,586
|
35,378
|
Note 11 — Subsequent Event:
During
January 2017, the Company issued an
aggregate of 198,630 shares under its current at-the-market sales
agreement with a weighted average sale price of $1.80 per share,
resulting in net proceeds of approximately
$347,000.
F-35
EXHIBIT INDEX
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
3.1
|
|
Form of
Amended and Restated Certificate of Incorporation.
|
|
S-1/A
|
|
3/01/2010
|
|
3.3
|
|
|
|
3.2
|
|
Form of
Amended and Restated Bylaws.
|
|
S-1/A
|
|
3/01/2010
|
|
3.4
|
|
|
|
3.3
|
|
Certificate
of Amendment to Amended and Restated Certificate of Incorporation,
dated December 3, 2012.
|
|
10-K
|
|
2/27/2013
|
|
3.3
|
|
|
|
3.4
|
|
Certificate
of Designation of Series A Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
February 18, 2013, as corrected on February 19, 2013.
|
|
8-K
|
|
2/19/2013
|
|
3.3
|
|
|
|
3.5
|
|
Certificate
of Designation of Series B Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
July 26, 2013.
|
|
8-K
|
|
7/26/2013
|
|
3.4
|
|
|
|
3.6
|
|
Certificate
of Designation of Series C-1 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.5
|
|
|
|
3.7
|
|
Certificate
of Amendment to Certificate of Designation of Series C-1 Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.10
|
|
|
|
3.8
|
|
Certificate
of Designation of Series C-2 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.6
|
|
|
|
3.9
|
|
Certificate
of Amendment to Certificate of Designation of Series C-2 Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.11
|
|
|
|
3.10
|
|
Certificate
of Designation of Series C-3 Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.9
|
|
|
|
3.11
|
|
Certificate
of Designation of Series D Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.7
|
|
|
|
3.12
|
|
Certificate
of Amendment to Certificate of Designation of Series D Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.12
|
|
|
|
3.13
|
|
Certificate
of Designation of Series E Non-Voting Convertible Preferred Stock
of CorMedix Inc., filed with the Delaware Secretary of State on
October 21, 2013.
|
|
8-K
|
|
10/23/2013
|
|
3.8
|
|
|
|
3.14
|
|
Certificate
of Amendment to Certificate of Designation of Series E Non-Voting
Convertible Preferred Stock of CorMedix Inc., filed with the
Delaware Secretary of State on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
3.13
|
|
|
|
4.1
|
|
Specimen
of Common Stock Certificate.
|
|
S-1/A
|
|
3/19/2010
|
|
4.1
|
|
|
|
4.2
|
|
Stockholder
Agreement, dated as of January 30, 2008, between CorMedix Inc. and
ND Partners LLC.
|
|
S-1
|
|
11/25/2009
|
|
4.7
|
|
|
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
4.3
|
|
Form of
Registration Rights Agreement.
|
|
10-Q
|
|
11/13/2012
|
|
4.5
|
|
|
|
4.4
|
|
Form of
Sales Agent Warrant issued in September 2012.
|
|
10-Q
|
|
11/13/2012
|
|
4.3
|
|
|
|
4.5
|
|
Warrant
issued to ND Partners in April 2013.
|
|
10-Q
|
|
5/15/2013
|
|
4.18
|
|
|
|
4.6
|
|
Form of
Warrant issued on February 19, 2013.
|
|
8-K
|
|
2/19/2013
|
|
4.13
|
|
|
|
4.7
|
|
Form of
Warrant issued on July 30, 2013.
|
|
8-K
|
|
7/26/2013
|
|
4.21
|
|
|
|
4.8
|
|
Form of
Warrant issued on October 22, 2013.
|
|
8-K
|
|
10/18/2013
|
|
4.22
|
|
|
|
4.9
|
|
Form of
Warrant issued on January 8, 2014.
|
|
8-K
|
|
1/09/2014
|
|
4.23
|
|
|
|
4.10
|
|
Form of
Warrant issued on March 4, 2014.
|
|
8-K
|
|
3/05/2014
|
|
4.24
|
|
|
|
4.11
|
|
Form of
Warrant issued on March 3, 2015.
|
|
8-K
|
|
3/04/2015
|
|
4.1
|
|
|
|
4.12
|
|
Amended
and Restated Warrant originally issued May 30, 2013.
|
|
8-K
|
|
3/04/2015
|
|
4.3
|
|
|
|
4.13
|
|
Amended
and Restated Warrant originally issued March 24, 2010.
|
|
8-K
|
|
3/04/2015
|
|
4.2
|
|
|
|
4.14
|
|
Registration
Rights Agreement, dated March 3, 2015, by and between CorMedix Inc.
and Manchester Securities Corp.
|
|
8-K
|
|
3/04/2015
|
|
4.5
|
|
|
|
10.1*
|
|
License
and Assignment Agreement, dated as of January 30, 2008, between the
Company and ND Partners LLC.
|
|
S-1/A
|
|
12/31/2009
|
|
10.5
|
|
|
|
10.2
|
|
Escrow
Agreement, dated as of January 30, 2008, among the Company, ND
Partners LLC and the Secretary of the Company, as Escrow
Agent.
|
|
S-1
|
|
11/25/2009
|
|
10.6
|
|
|
|
10.3
|
|
Consulting
Agreement, dated as of January 30, 2008, between the Company and
Frank Prosl.
|
|
S-1
|
|
11/25/2009
|
|
10.12
|
|
|
|
10.4*
|
|
Manufacture
and Development Agreement, dated as of March 5, 2007, by and
between the Company and Emcure Pharmaceuticals USA,
Inc.
|
|
S-1/A
|
|
12/31/2009
|
|
10.14
|
|
|
|
10.5
|
|
Amended
and Restated 2006 Stock Incentive Plan.
|
|
S-1/A
|
|
3/01/2010
|
|
10.8
|
|
|
|
10.6
|
|
Form of
Indemnification Agreement between the Company and each of its
directors and executive officers.
|
|
S-1/A
|
|
3/01/2010
|
|
10.17
|
|
|
|
10.7
|
|
Agreement
for Work on Pharmaceutical Advertising dated January 10, 2013 by
and between MKM Co-Pharma GmbH and CorMedix Inc.
|
|
8-K
|
|
1/16/2013
|
|
10.22
|
|
|
|
10.8
|
|
2013
Stock Incentive Plan
|
|
10-K
|
|
3/27/2013
|
|
10.27
|
|
|
|
10.9
|
|
Form of
Securities Purchase Agreement, dated January 7, 2014, between
CorMedix Inc. and the investors named therein.
|
|
8-K
|
|
1/09/2014
|
|
10.36
|
|
|
|
10.10
|
|
Preliminary
Services Agreement dated April 8, 2015, between CorMedix Inc. and
[RC]2 Pharma Connect
LLC.
|
|
10-Q
|
|
8/06/2015
|
|
10.1
|
|
|
|
10.11
|
|
Release
of Claims and Severance Modification, dated July 17, 2015, between
Randy Milby and CorMedix Inc.
|
|
|
|
|
|
|
|
X
|
|
10.12
|
|
Employment
Agreement, effective February 1, 2017, between CorMedix Inc. and
Robert Cook.**
|
|
|
|
|
|
|
|
X
|
|
10.13
|
|
Employment
Agreement, effective February 1, 2017, between CorMedix Inc. and
Judith Abrams.**
|
|
|
|
|
|
|
|
X
|
|
10.14
|
|
Employment
Agreement, effective March 1, 2017, between CorMedix Inc. and John
Armstrong.
|
|
|
|
|
|
|
|
X
|
|
21.1
|
|
List of
Subsidiaries
|
|
10-K
|
|
3/27/2013
|
|
21.1
|
|
|
|
23.1
|
|
Consent
of Independent Registered Public Accounting Firm.
|
|
|
|
|
|
|
|
X
|
|
Exhibit
Number
|
|
Description of Document
|
|
Registrant’s
Form
|
|
Dated
|
|
Exhibit Number
|
|
Filed Herewith
|
|
31.1
|
|
Certification
of Principal Executive Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
31.2
|
|
Certification
of Principal Financial Officer pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
32.1
|
|
Certification
of Principal Executive Officer pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
32.2
|
|
Certification
of Principal Financial Officer pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
X
|
|
101
|
|
The
following materials from CorMedix Inc. Form 10-K for the year ended
December 31, 2016, formatted in Extensible Business Reporting
Language (XBRL): (i) Balance Sheets at December 31, 2016 and 2015,
(ii) Statements of Operations for the years ended December 31,
2016, 2015 and 2014, (iii) Statements of Changes in
Stockholders’ Equity (Deficiency) for the years ended
December 31, 2016, 2015 and 2014, (iv) Statements of Cash Flows for
the years ended December 31, 2016, 2015 and 2014 and (v) Notes to
the Financial Statements.**
|
|
|
|
|
|
|
|
X
|
_____________
|
*
|
Confidential
treatment has been granted for portions of this document. The
omitted portions of this document have been filed separately with
the SEC.
|
|
**
|
Confidential
treatment has been requested with respect to certain portions of
this exhibit. Omitted portions have been filed
separately with the SEC.
|
|
***
|
Pursuant
to Rule 406T of Regulation S-T, the Interactive Data Files in
Exhibit 101 hereto are deemed not filed or part of a registration
statement or prospectus for purposes of Sections 11 or 12 of the
Securities Act of 1933, as amended, are deemed not filed for
purposes of Section 18 of the Securities Exchange Act of 1934, as
amended, and otherwise are not subject to liability under those
sections.
|
|
|
|