Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - ROCKWELL MEDICAL, INC. | rmti-20161231ex321c08810.htm |
| EX-31.2 - EX-31.2 - ROCKWELL MEDICAL, INC. | rmti-20161231ex312b37b8c.htm |
| EX-31.1 - EX-31.1 - ROCKWELL MEDICAL, INC. | rmti-20161231ex3115e9e2c.htm |
| EX-23.1 - EX-23.1 - ROCKWELL MEDICAL, INC. | rmti-20161231ex231c424b4.htm |
vh
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10‑K
|
(Mark One) |
|
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
|
|
For the fiscal year ended December 31, 2016 |
|
|
|
|
|
OR |
|
|
|
|
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
|
|
For the transition period from to |
|
Commission file number 000‑23661
ROCKWELL MEDICAL, INC.
(Exact name of registrant as specified in its charter)
|
Michigan |
38‑3317208 |
|
30142 Wixom Road Wixom, Michigan |
48393 |
(248) 960‑9009
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class: |
|
Name of each exchange on which registered: |
|
Common Stock, no par value |
|
Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act:
(None)
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S‑T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S‑K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10‑K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b‑2 of the Exchange Act. (Check one):
|
Large accelerated filer ☐ |
Accelerated filer ☒ |
Non‑accelerated filer ☐ |
Smaller reporting company ☐ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the registrant’s voting and non‑voting common equity held by non‑affiliates of the registrant on June 30, 2016 (computed by reference to the closing sales price of the registrant’s Common Stock as reported on the Nasdaq Global Market on such date) was $318,842,000. For purposes of this computation, shares of common stock held by our executive officers, directors and common shareholders with 10% or more of the outstanding shares of Common Stock were excluded. Such determination should not be deemed an admission that such officers, directors and beneficial owners are, in fact, affiliates.
Number of shares outstanding of the registrant’s Common Stock, no par value, as of February 17, 2017: 51,527,711 shares.
Documents Incorporated by Reference
Portions of the Registrant’s definitive Proxy Statement pertaining to the 2017 Annual Meeting of Shareholders (the “Proxy Statement”) to be filed pursuant to Regulation 14A are herein incorporated by reference in Part III of this Annual Report on Form 10‑K.
References to “Rockwell”, the “Company,” “we,” “us” and “our” are to Rockwell Medical, Inc. and its subsidiaries unless otherwise specified or the context otherwise requires.
Triferic®, CitraPure®, RenalPure® and SteriLyte® are registered trademarks of Rockwell.
Forward Looking Statements
We make forward‑looking statements in this report and may make such statements in future filings with the Securities and Exchange Commission, or SEC. We may also make forward‑looking statements in our press releases or other public or shareholder communications. Our forward‑looking statements are subject to risks and uncertainties and include information about our expectations and possible or assumed future results of our operations. When we use words such as “may,” might,” “will,” “should,” “believe,” “expect,” “anticipate,” “estimate,” “continue”, “predict”, “forecast”, “projected,” “intend” or similar expressions, or make statements regarding our intent, belief, or current expectations, we are making forward‑looking statements. Our forward looking statements also include, without limitation, statements about our competitors, statements regarding the commercialization of our new products, statements regarding our new products such as Triferic and Calcitriol, and statements regarding our anticipated future financial condition, operating results, cash flows and business and financing plans.
We claim the protection of the safe harbor for forward‑looking statements contained in the Private Securities Litigation Reform Act of 1995 for all of our forward‑looking statements. While we believe that our forward‑looking statements are reasonable, you should not place undue reliance on any such forward‑looking statements, which are based on information available to us on the date of this report or, if made elsewhere, as of the date made. Because these forward‑looking statements are based on estimates and assumptions that are subject to significant business, economic and competitive uncertainties, many of which are beyond our control or are subject to change, actual results could be materially different. Factors that might cause such a difference include, without limitation, the risks and uncertainties discussed in this report, including without limitation in “Item 1A—Risk Factors,” and from time to time in our other reports filed with the SEC. Other factors not currently anticipated may also materially and adversely affect our results of operations, cash flows and financial position. We do not undertake, and expressly disclaim, any obligation to update or alter any statements whether as a result of new information, future events or otherwise except as required by law.
1
PART I
Item 1. Business.
General
Rockwell Medical, Inc., incorporated in the state of Michigan in 1996, is a fully‑integrated biopharmaceutical company targeting end‑stage renal disease (“ESRD”) and chronic kidney disease with innovative products and services for the treatment of iron deficiency, secondary hyperparathyroidism and hemodialysis (also referred to as “dialysis”).
Triferic
Rockwell’s lead drug, Triferic was approved by United States Food and Drug Administration (“FDA”) in late January 2015. Triferic is the only FDA-approved therapy indicated to replace iron and maintain hemoglobin in adult hemodialysis patients with chronic kidney disease. Triferic is an innovative iron therapy that replaces the ongoing iron loss that occurs to patients during every hemodialysis treatment, via dialysate. Triferic’s unique mode-of-action enables it to bind to transferrin immediately and completely once entering the blood, where it then is transported to the bone marrow to make hemoglobin.
We are actively marketing and commercializing Triferic in the United States hemodialysis market which is currently the largest market in the world for dialysis products. Feedback from users in the commercial market has been positive and consistent with our clinical program, which demonstrated maintenance of hemoglobin concentration and significant reduction in ESA use.
Because Medicare pays for treatment of the vast majority of all dialysis patients, reimbursement status is important to Rockwell’s ability to successfully commercialize Triferic. Triferic has been approved for reimbursement by the Centers for Medicare & Medicaid Services (“CMS”) as part of the standard “bundled” payment received by dialysis service providers for providing treatment to patients. In late 2015, we requested clarification from CMS on whether Triferic qualified for transitional add-on reimbursement and CMS confirmed to us that Triferic was included as part of the bundle in January 2016. Since that time, we have pursued securing transitional add-on reimbursement status for Triferic which provides separate reimbursement for two years outside of the bundled payment. We believe add-on reimbursement status is warranted for new innovative therapies such as Triferic so that patients have access to them and so that companies will commit the time and monetary resources to innovation in the renal space. We also believe that there is legal support in the Protecting Access to Medicare Act of 2014 for our position, and there is precedent for CMS granting add-on reimbursement status to a recent therapy after initially placing it in the bundle payment. CMS itself has stated that add-on reimbursement status for new innovative therapies is important. Triferic has received strong Congressional support for transitional add-on reimbursement status from multiple members of Congress, who have requested in writing that CMS and the Secretary of the Department of Health and Human Services provide it. Triferic has also received support from patient advocacy groups and dialysis service providers. Management believes there is high likelihood that Triferic receives transitional add-on reimbursement status, and that Triferic will become the standard of care for iron maintenance therapy in both the United States and globally regardless of reimbursement status, but that transitional add-on reimbursement would accelerate sales and adoption in the United States commercial market.
We are working to commercialize Triferic globally and we intend to continue to out-license Triferic to partner companies who we believe are best suited to commercialize Triferic. We executed license agreements in 2016 for Triferic and Calcitriol for the Chinese market as well as the Kingdom of Saudi Arabia and several other Middle East markets. Both markets are large and China is expected to become the largest in the world over the next several years. Commercial sales activity in these markets will commence following regulatory or registration approval. Rockwell retains manufacturing responsibilities for both Triferic and Calcitriol. We have also executed a distribution agreement to market Triferic in Canada, where we anticipate commercial availability in 2019 after regulatory approval. Additionally, we have formed a wholly-owned subsidiary in India to market Triferic where we anticipate market availability in 2018. We remain actively engaged in licensing negotiations for Triferic in a number of other regions and countries. We intend to leverage the development, regulatory and commercial presence and expertise of potential business partners to accelerate sales of our products throughout the world.
We are currently executing development of Triferic for other clinical indications. These clinical applications include peritoneal dialysis (PD), total parenteral nutrition (TPN) and an orphan indication, that if successful may lead to
2
treating cancer patients. We are also developing an intravenous injection for use in other iron deficiency anemia indications. Rockwell’s drug product pipeline is summarized below by stage of development.
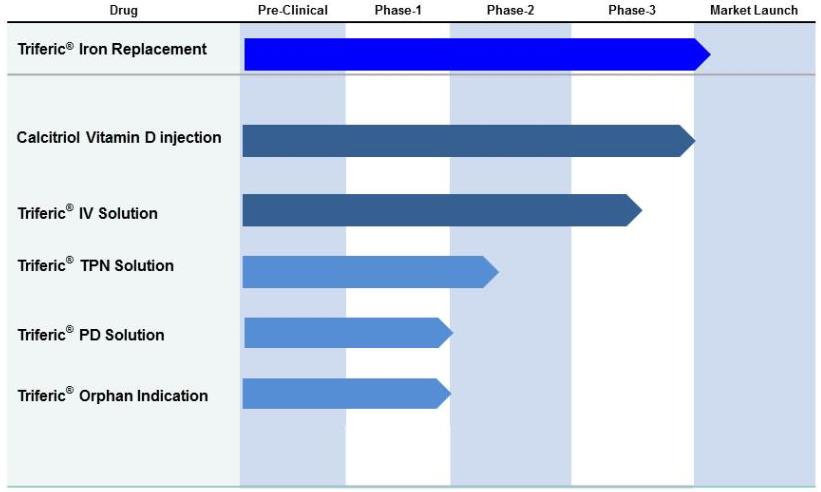
Calcitriol
Rockwell’s FDA approved generic drug, Calcitriol, is for treating secondary hyperparathyroidism in dialysis patients. Calcitriol (active vitamin D) injection is indicated in the management of hypocalcemia in patients undergoing chronic renal dialysis. It has been shown to significantly reduce elevated parathyroid hormone (“PTH”) levels. Reduction of PTH has been shown to result in an improvement in renal osteodystrophy. Based on industry estimates, we believe the United States market for vitamin D therapy for ESRD patients is about $200 million per year. We estimate that there are currently over 60,000,000 vitamin D treatments per year in the ESRD market in the United States. We recently were asked by the FDA to resubmit our manufacturing approval under a Prior Approval Supplement (“PAS”) and we intend to do so. As a result, we anticipate commercial availability during the second half of 2017. We intend to market Calcitriol to hemodialysis providers in the United States and elsewhere as soon as we have sufficient inventory.
Concentrate Business
Rockwell is an established manufacturer and leader in delivering high-quality hemodialysis concentrates/dialysates to dialysis providers and distributors in the United States and abroad. As one of the two major suppliers in the United States, Rockwell’s products are used to maintain human life by removing toxins and replacing critical nutrients in the dialysis patient’s bloodstream. Rockwell has three United States manufacturing/distribution facilities. Rockwell sells its concentrate products in the United States pursuant to an Exclusive Distribution Agreement (the “Distribution Agreement”) with Baxter Healthcare Corporation (“Baxter”) under which Baxter is our exclusive distributor. We have entered into an arbitration proceeding with Baxter related to the Distribution Agreement. See “Item 3 – Legal Proceedings.”
The Hemodialysis Market
3
The great majority of hemodialysis patients receive dialysis treatment three times per week, or approximately 156 times per year. Most patients have their dialysis treatment performed at a free-standing clinic for permanent loss of kidney function; these are called “chronic” patients. Some have their treatment performed at hospitals for temporary loss of kidney function; these are called “acute” patients. A small percent of chronic patients receive their treatment at home; these are called “home” patients. In each setting, a dialysis machine dilutes concentrated solution, such as Rockwell’s concentrate products, with purified water. The resulting solution is called dialysate. Dialysate is pumped through an artificial kidney or filter (called a dialyzer) while the patient's blood is pumped through a semi-permeable membrane inside the dialyzer in the opposite direction the dialysate is flowing. The dialysate infuses calcium, magnesium and bicarbonate into the patient’s blood while removing water and waste. Dialysate generally contains dextrose, sodium chloride, calcium, potassium, magnesium, sodium bicarbonate and citric acid or acetic acid. The patient's physician chooses the proper concentrations required for each patient based on each particular patient's needs.
In addition to using reusable concentrate products, a dialysis provider also uses other products such as blood tubing, fistula needles, dialyzers, drugs, specialized component kits, dressings, cleaning agents, filtration salts and other supplies, many of which we sell.
Dialysis Industry Trends
Hemodialysis is the primary treatment modality employed in the United States with over 90% of all dialysis patients receiving hemodialysis. The Company does not compete in the peritoneal or home dialysis segments. Hemodialysis treatments are primarily performed in freestanding clinics, as well as in some hospitals. The majority of dialysis services are performed by national and regional for profit dialysis chains. Based on data published by the United States Renal Data Systems (“USRDS”) we estimate that there are approximately 7,000 Medicare-certified treatment clinics in the United States. The two largest national for-profit dialysis chains service approximately 70% of the domestic hemodialysis market. According to the most recent statistics published by USRDS, there were approximately 460,000 dialysis patients in the United States as of the end of 2014.
Based on a global market study published by a major dialysis products manufacturer, the global ESRD population receiving some form of treatment was estimated to be over 2.8 million patients at the end of 2016 with the overall global patient population growing approximately 7-8% annually. According to the National Kidney Foundation, 10% of the worldwide population is affected by chronic kidney disease and millions die each year because they do not have access to affordable treatments. We have observed that the ESRD patient population in the United States has grown steadily over the past several decades and, coupled with data provided in that report, we expect the United States dialysis population to grow approximately 3-4% annually over the next several years. The Asia-Pacific market is projected to experience rapid growth in both the incidence of kidney disease and by total treatment in the ESRD population over the decade ahead.
Drug Products
Triferic (Ferric Pyrophosphate Citrate)
Iron deficiency anemia is pervasive for chronic patients receiving dialysis. Triferic is the only FDA approved drug indicated to replace iron and maintain hemoglobin in hemodialysis patients. We believe Triferic will become the standard of care in iron maintenance therapy for dialysis patients and address an important unmet need in the treatment of anemia in ESRD patients.
Triferic is an innovative iron therapy that replaces the iron lost by patients during every hemodialysis treatment. Triferic’s unique mode-of-action enables it to bind to transferrin immediately and completely once entering the blood via dialysate, where it then is transported to the bone marrow to make hemoglobin. Triferic delivers sufficient iron to the bone marrow and maintains hemoglobin without increasing iron stores (ferritin).
To address anemia associated with the ongoing blood losses associated with dialysis, long standing industry practice has been to inject patients with intravenous (“IV”) iron and erythropoiesis stimulating agents (“ESAs”). ESA is synthetic erythropoietin that acts in the bone marrow, together with iron, to increase the production of red blood cells, which carry oxygen throughout the body to nourish tissues and sustain life. Hemoglobin is an important constituent of red blood cells and is composed largely of iron and protein. IV iron was approved for use in hemodialysis patients in the 1990’s as there was no other iron product available that could work effectively with ESA. IV iron products metabolize
4
in the liver. Because of the constant inflammation levels present in hemodialysis patients the great majority of IV iron gets trapped in the patient’s liver, blocked by a protein called hepcidin. In contrast to Triferic, IV iron is unable to bind immediately to transferrin and travel to the bone marrow. IV iron is a repletion therapy, not an iron maintenance therapy.
Triferic is distinctly different from IV iron. Triferic is different in molecular structure, different in mode-of-action (bypassing liver storage) and different in FDA approved indication (to replace iron and maintain hemoglobin). Triferic is an iron maintenance therapy approved to be given to patients every treatment whereas IV iron is a repletion or “rescue” therapy approved to be given only when a patient experiences significant blood loss and has a ferritin level < 200 ng/mL. The current average ferritin level in dialysis patients in the U.S. has increased significantly to in excess of 750 ng/mL according to US-DOPPS Practice Monitor published December 2016. Triferic delivers iron and maintains hemoglobin without increasing iron stores (ferritin).
Triferic has demonstrated an excellent safety profile in its Phase 3 clinical program and has not been attributed to any anaphylaxis in an estimated 200,000 administrations. We received FDA approval to market Triferic in liquid form in 2015 and in powder form in 2016.
Calcitriol (Active Vitamin D) Injection
Calcitriol is a generic active vitamin D and is indicated for the treatment of secondary hyperparathyroidism in dialysis patients. The majority of ESRD patients receive vitamin D on a routine basis using primarily one of two branded drugs or in some cases oral drugs. Clinical data shows Calcitriol to be clinically equivalent in safety and efficacy to the two branded drugs as well as the most potent and physiological vitamin D therapy. We believe the lower cost of Calcitriol will entice dialysis providers to purchase it over current vitamin D options.
Out-Licensing Arrangements for Drug Products
We have made significant progress with our international business development effort for Triferic, including securing a licensing agreement with Wanbang Biopharmaceutical in the first quarter of 2016 for the rights to commercialize Triferic and Calcitriol for ESRD patients in the People’s Republic of China. Under the terms of the Wanbang Agreement, we received an upfront payment of $4 million, which we are recognizing over the term of the agreement. Rockwell may also receive milestone payments of up to an additional $35 million over the life of the agreement in regulatory and revenue milestone payments plus ongoing earnings on product sales.
In the third quarter of 2016, we entered into an exclusive license and manufacturing supply with ARAM Medical for the sale of Triferic and Calcitriol in the Kingdom of Saudi Arabia and a number of other countries in the Middle East for an initial term of 10 years. In consideration for the exclusive rights, ARAM Medical will pay us a $1 million licensing fee and a royalty on product sales, and has committed to annual minimum purchase quantities. ARAM Medical will also assume responsibility for all clinical and regulatory expenses for the countries covered by its agreement. Rockwell retains manufacturing responsibilities for both Triferic and Calcitriol.
Dialysis Concentrate Products
We manufacture, sell, deliver and distribute hemodialysis concentrates, along with a full line of ancillary products abroad. We use Baxter as our exclusive marketer and distributor in the United States and in select foreign markets. Dialysate concentrates accounted for over 95% of our 2016 revenue with ancillary products accounting for most of the remainder. All of our products are manufactured according to Association for the Advancement of Medical Instrumentation and current good manufacturing practices (“cGMP”) guidelines. Our concentrate products are diluted with clean water on-site at the clinic in the dialysis machine, creating dialysate, which works to clean the patient’s blood.
CitraPure Citric Acid Concentrate
Our CitraPure Concentrate is 100% acetate-free, in contrast to the acetate-based products used for many years. Acetate promotes inflammation so its removal is beneficial to the patient. Citrate has anticoagulant properties and has been shown in clinical studies to reduce the need for heparin during dialysis treatment (although CitraPureis not
5
indicated for heparin sparing). is packaged as a liquid and as a dry powder acid concentrate for use with our Dry Acid Concentrate Mixer. CitraPure contains citric acid, sodium chloride, dextrose, magnesium, potassium and calcium. CitraPure is packaged as dry acid concentrate in 25 gallon cases and liquid acid concentrate in 55 gallon drums and four one gallon jugs to a case.
Dri-Sate Dry Acid Concentrate
Our Dri-Sate Concentrate is our original acetate-based product. Dri-Sate is packaged as a dry powder acid concentrate for use with our Dry Acid Concentrate Mixer. Dri-Sate contains acetic acid, sodium chloride, dextrose, magnesium, potassium and calcium. Dri-Sate is packaged as dry acid concentrate in 25 gallon cases.
Renal Pure Liquid Acid Concentrate
Our RenalPure Liquid Concentrate is acetate-based and contains acetic acid, sodium chloride, dextrose, magnesium, potassium and calcium and packaged in 55 gallon drums and four one gallon jugs to a case.
Dry Acid Concentrate Mixer
Our Dry Acid Concentrate Mixer is designed for our CitraPureand Dri-Sate Dry Acid product and enables the clinic to mix acid concentrate on-site. Clinics using Rockwell’s Dry Acid Concentrate products realize numerous advantages, including lower cost per treatment, reduced storage space requirements, reduced number of deliveries and more flexibility in scheduling deliveries, while enabling the Company to reduce distribution and warehousing costs.
RenalPure and SteriLyte Bicarbonate Concentrate
RenalPure bicarbonate is a dry powder mixed on-site at the clinic and is packaged for bulk and individual treatment and SteriLyte bicarbonate is a liquid packaged in four one gallon jugs to a case and is used mainly in acute care settings.
Ancillary Products
We offer a wide range of ancillary products including blood tubing, fistula needles, specialized custom kits, dressings, cleaning agents, filtration salts and other supplies used by hemodialysis providers.
Distribution Agreement with Baxter
Pursuant to the Distribution Agreement, Baxter is our exclusive agent for commercializing our hemodialysis concentrate and ancillary products in the United States and various foreign countries for an initial term of 10 years. We retain sales, marketing and distribution rights for our hemodialysis concentrate products for our international customers and in those countries in which we have an established commercial presence. During the term of the Distribution Agreement, Baxter has agreed not to manufacture or sell any competitive concentrate products in the United States hemodialysis market, other than specified products. The Distribution Agreement does not include any of the Company’s drug products. We are currently involved in arbitration with Baxter regarding various disputes under the Distribution Agreement. See “Item 3 – Legal Proceedings.”
Under the Distribution Agreement, Baxter purchases concentrate-related products from us at pre-determined gross margin-based prices per unit adjusted each year during the term and subject to an annual true up. The Distribution Agreement also requires Baxter to meet minimum annual purchase levels, subject to a cure period and certain other relief, in order to maintain its exclusive distribution rights. The minimum purchase levels increase each year over the term of the Distribution Agreement. Purchases in any contract year that exceed the minimum may be carried forward and applied to future years’ minimum requirements. The Distribution Agreement also contains provisions governing the operating relationship between the parties, our obligations to maintain specified manufacturing capacity and quality levels, remedies, as well as representations, warranties and indemnification obligations of the parties. We will continue to manage customer service, transportation and certain other functions for our current customers through at least December 31, 2017: Baxter will pay us an amount equal to our related costs plus a slight mark-up for these services.
6
The Distribution Agreement also provides that Baxter will pay us, at our discretion, up to $10 million to build a new manufacturing facility in the Pacific time zone that will serve customers in the Western United States. The fee payable in connection with building the facility will be reduced to the extent that the facility is not operational within 12 months after the start of construction. Except for any leased components, we will own and operate the facility when completed. Baxter’s obligation to pay us this amount when we choose to build the facility is being contested in the Baxter arbitration.
Either party may terminate the Distribution Agreement upon the insolvency or material breach of the other party or in the event of a force majeure. In addition, Baxter may also terminate the Distribution Agreement at any time upon 270 days’ prior written notice to us or if (1) prices increase beyond certain thresholds and notice is provided within 45 days after the true up payment is due for the year in which the price threshold is exceeded, (2) a change of control of the Company occurs and 270 days’ notice is provided, or (3) upon written notice that Baxter has been enjoined by a court of competent jurisdiction from selling in the United States any product covered by the Distribution Agreement due to a claim of intellectual property infringement or misappropriation relating to such product. If Baxter terminates the Distribution Agreement under the discretionary termination or the price increase provisions, it would be subject to a limited non-compete obligation in the United States with respect to certain products for a period of two years.
If a “Refund Trigger Event” occurs, we would be obligated to repay a portion of the $20 million upfront fee and any paid portion of the facility fee. A “Refund Trigger Event” means any of the following: (1) a change of control of the Company involving any of certain specified companies; (2) a termination by Baxter due to the Company’s bankruptcy or breach, or due to price increases that exceed the stated thresholds; (3) a termination by either party due to a force majeure; (4) settlement or adjudication of any claim, action or litigation relating to a covered product that materially and adversely affects Baxter’s commercialization of the product; and (5) any regulatory action or ruling relating to a covered product that materially and adversely affects Baxter’s commercialization of the product. In addition, if Baxter terminates the Distribution Agreement because Baxter has been enjoined by a court of competent jurisdiction from selling in the United States any product covered by the Distribution Agreement due to a claim of intellectual property infringement or misappropriation relating to such product prior to the end of 2019, Baxter would be entitled to a partial refund. In no event would more than one refund be required to be paid.
The Distribution Agreement may be extended an additional five years by Baxter if Baxter achieves a specified sales target and pays an extension fee of $7.5 million. If the first extension occurs, the Distribution Agreement term may later be extended an additional five years at Baxter’s option at no additional cost.
Distribution and Delivery Operations
The majority of our domestic dialysis concentrate products are delivered through our subsidiary, Rockwell Transportation, Inc., which operates a fleet of trucks used to deliver products to our customers. Rockwell distribution and delivery will continue to operate under the Distribution Agreement on behalf of Baxter for domestic business. We perform delivery services that are generally not available from common carriers or our competitors, such as stock rotation, non-loading-dock delivery and drum pump-off service. As a result, we believe we offer a higher level of service than other providers. Our drug products are generally delivered by third party drug distributors in the United States.
Sales and Marketing
The top ten dialysis providers treat approximately 380,000 hemodialysis patients in their centers according to an article published by Nephrology News in 2016, which we believe constitutes approximately 83% of the hemodialysis patient population in the United States. Due to the concentrated nature of our customers, we will market our drug products using a small team of skilled salespeople. Our Chief Executive Officer leads and directs our sales team, and handles much of the sales effort with our major accounts.
We market and advertise through trade publications, journals, product literature, industry trade conferences, and the internet. We target our sales and marketing efforts to senior and operating management of dialysis companies, dialysis service providers, nephrologists, clinic administrators, nurses, medical directors and technical and purchasing personnel.
7
Our dialysis concentrate products are sold to customers in the United States through Baxter in accordance with the Distribution Agreement. Our dialysis concentrate products are sold to international customers through independent sales agents, distributors and direct.
Competition
Dialysis Concentrate Solutions and Dialysis Products Market Competition
In the United States, the principal competitor for our concentrate products is Fresenius Medical Care NA, a vertically integrated manufacturer and marketer of dialysis devices, drugs and supplies and dialysis clinic operator, which has substantially greater financial, technical, manufacturing, marketing, and research and development resources than us. Fresenius operates approximately 1,700 clinics and treats approximately 36% of the dialysis patients in the United States. Fresenius also manufactures and sells a full range of renal products, including dialysis machines, dialyzers (artificial kidneys), concentrates and other supplies used in hemodialysis. Fresenius also services clinics owned by others with its products where it commands a market leading position in its key product lines. Fresenius manufactures its concentrate in its own regional manufacturing facilities. Fresenius and Rockwell are the two major dialysis concentrate suppliers in the United States.
Iron Delivery Market Competition
We believe Triferic has potential to become the standard of care for iron maintenance therapy for hemodialysis patients due to its unique mode of action, clinical benefits, ability to lower treatment cost for providers, ease of administration and excellent safety profile. Presently, the IV iron drug Venofer® has the majority of the market for delivering iron to chronic dialysis patients in the United States, but is an iron repletion therapy and not an iron maintenance therapy. Venofer® is owned by Switzerland-based Galenica. Galenica also markets Ferinject® which is primarily used to treat anemia in a non-dialysis setting. Fresenius has a sublicense agreement that allows them to distribute Venofer® to the dialysis market in the United States and Canada. Other IV iron competitors include Sanofi with Ferrlecit® and Watson with a generic IV iron called Nulecit®.
The markets for drug products are highly competitive. Competition in drug delivery systems is generally based on marketing strength, product performance characteristics (i.e., reliability, safety, patient convenience) and product price. Acceptance by dialysis providers and nephrologists is also critical to the success of a product. The first product on the market in a particular therapeutic area typically is able to obtain and maintain a significant market share. In a highly competitive marketplace and with evolving technology, additional product introductions or developments by others could render our products or technologies noncompetitive or obsolete. In addition, pharmaceutical and medical device companies are largely dependent upon health care providers being reimbursed by private insurers and government payors. Drugs approved by the FDA might not receive reimbursement from private insurers or government payors.
Prior to 2011, CMS had historically paid providers for dialysis treatments under the Medicare program in two parts: the composite rate and separately reimbursed drugs and services. The composite rate is payment for the complete dialysis treatment except for physicians’ professional services, separately billed laboratory services and separately billed drugs. CMS began implementation of a fully bundled reimbursement rate in 2011. The bundled rate is a single payment per treatment, thereby eliminating reimbursement for individual drugs and services to providers. Regulations provide that the rate is recalculated each year. As a result, dialysis drugs are now viewed by providers as an additional cost rather than as a source of revenue. We believe Triferic, due to its potential for improved therapeutic response and lower cost of administration, is an attractive therapy under this reimbursement landscape. In addition, we are seeking transitional add-on reimbursement for Triferic which if gained would pay dialysis service providers an additional 6% above their reimbursed cost to cover the expense of new drug adoption.
Vitamin D Therapy Market Competition
We intend to market Calcitriol injection against two competitors with branded vitamin D products and against other generic drug competitors as well as oral forms of vitamin D. Abbott Laboratories markets Zemplar® and Sanofi-Aventis, through its Genzyme subsidiary, markets Hectorol®. Other companies offer oral forms of vitamin D. We believe the dialysis reimbursement law that went into effect in January 2011, along with Calcitriol being the lowest dose vitamin D injection available and our relationships with many dialysis providers gives us an advantage to sell Calcitriol against competitors in the market.
8
Quality Assurance and Control
Dialysis Concentrate Solutions Business
We operate under FDA and cGMP guidelines and place significant emphasis on providing quality products and services to our customers. Our quality management plays an essential role in meeting product quality requirements and FDA guidelines. We have implemented quality systems that involve control procedures that result in rigid conformance to specifications. Our quality systems also include assessments of suppliers of raw materials, packaging components and finished goods, and quality management reviews designed to inform management of key issues that may affect the quality of products, assess the effectiveness of our quality systems and identify areas for improvement.
Technically trained professionals at our production facilities maintain our quality system. To assure quality and consistency of our concentrates, we conduct specific analytical tests during the manufacturing process for each type of product that we manufacture. Prior to shipment, our quality control laboratory at each facility conducts analytical tests to verify that the chemical properties of the concentrates comply with the specifications required by industry standards. Each product is assigned a lot number for tracking purposes.
Drug Manufacturing
We will utilize CMOs to manufacture and package our drug products for sale. These contract manufacturers are FDA registered drug manufacturing establishments. We follow defined procedures to qualify manufacturers of our products and to review and approve all manufactured products to ensure compliance with FDA cGMP regulations.
Government Regulation
The testing, manufacture and sale of our hemodialysis concentrates and the ancillary products we distribute are subject to regulation by numerous governmental authorities, principally the FDA and corresponding state and foreign agencies. Under the Federal Food, Drug and Cosmetic Act, as amended (the “FD&C Act”), and FDA regulations, the FDA regulates the pre‑clinical and clinical testing, manufacture, labeling, distribution and marketing of medical devices. Noncompliance with applicable requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant pre‑market clearance or pre‑market approval for devices, withdrawal of marketing clearances or approvals and criminal prosecution.
We plan to develop and commercialize selected drug candidates, such as Triferic, other Triferic indications and Calcitriol. The development and regulatory approval process for new drugs and additional indications for approved drugs includes preclinical testing and human clinical trials and is lengthy and uncertain. Before marketing in the United States, any pharmaceutical or therapeutic product must undergo rigorous preclinical testing and clinical trials and an extensive regulatory approval process implemented by the FDA under the FD&C Act.
Moreover, the FDA imposes substantial requirements on new product research and the clinical development, manufacture and marketing of pharmaceutical products, including testing and clinical trials to establish the safety and effectiveness of these products.
Medical Device Approval and Regulation
A medical device may be marketed in the United States only with prior authorization from the FDA unless it is subject to a specific exemption. Devices classified as Class I devices (general controls) or Class II devices (general and special controls) are eligible to seek “510(k) clearance” from the FDA. Such clearance generally is granted when submitted information establishes that a proposed device is “substantially equivalent” in terms of safety and effectiveness to a legally marketed device that is not subject to premarket approval. A legally marketed device is a “pre‑amendment” device that was legally marketed prior to May 28, 1976 (for which a PMA is not required), a device that has been reclassified from Class III to Class I or II, or a device which has been found substantially equivalent through the 510(k) process. The FDA in recent years has been requiring a more rigorous demonstration of substantial equivalence than in the past, including requiring clinical trial data in some cases. For any devices that are cleared through the 510(k) process, modifications or enhancements that could significantly affect safety or effectiveness, or constitute a new or major change in the intended use of the device, will require new 510(k) submissions. We have been
9
advised that it usually takes from three to six months from the date of submission to obtain 510(k) clearance, and may take substantially longer. Our hemodialysis concentrates, liquid bicarbonate and other ancillary products are categorized as Class II devices.
A device which sustains or supports life, prevents impairment of human health or presents a potential unreasonable risk of illness or injury is categorized as a Class III device. A Class III device generally must receive approval through a pre‑market approval (“PMA”) application, which requires proving the safety and effectiveness of the device to the FDA. The process of obtaining PMA approval is expensive and uncertain. We have been advised that it usually takes approximately one year to obtain approval after filing the request, and may take substantially longer.
If human clinical trials of a device are required, whether for a 510(k) submission or a PMA application, and the device presents a “significant risk,” the sponsor of the trial (usually the manufacturer or the distributor of the device) will have to file an investigational device exemption (“IDE”) application prior to commencing human clinical trials. The IDE application must be supported by data, typically including the results of animal and laboratory testing. If the IDE application is approved by the FDA and one or more appropriate Institutional Review Boards (“IRBs”), the device may be shipped for the purpose of conducting the investigations without compliance with all of the requirements of the FD&C Act and human clinical trials may begin. The FDA will specify the number of investigational sites and the number of patients that may be included in the investigation. If the device does not present a “significant risk” to the patient, a sponsor may begin the clinical trial after obtaining approval for the study by one or more appropriate IRBs without the need for FDA approval.
Any devices manufactured or distributed by us pursuant to FDA clearances or approvals are subject to pervasive and continuing regulation by the FDA and certain state agencies. As a manufacturer of medical devices for marketing in the United States we are required to adhere to regulations setting forth detailed cGMP requirements, which include testing, control and documentation requirements. We must also comply with medical device reporting regulations which require that we report to the FDA any incident in which our products may have caused or contributed to a death or serious injury, or in which our products malfunctioned and, if the malfunction were to recur, it would be likely to cause or contribute to a death or serious injury. Under such a scenario, our products may be subject to voluntary recall by us or required recall by the FDA. Labeling and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. The FD&C Act prohibits the marketing of approved medical devices for unapproved uses.
We are subject to routine inspection by the FDA and certain state agencies for compliance with cGMP requirements and other applicable quality system regulations. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, transportation and disposal of hazardous or potentially hazardous substances.
We have 510(k) clearance from the FDA to market hemodialysis concentrates in both liquid and powder form. In addition, we have received 510(k) clearance for our Dry Acid Concentrate Mixer.
We must comply with the FD&C Act and related laws and regulations, including cGMP, to retain 510(k) clearances. We cannot assure you that we will be able to maintain our 510(k) clearances from the FDA to manufacture and distribute our products. If we fail to maintain our 510(k) clearances, we may be required to cease manufacturing and/or distributing our products, which would have a material adverse effect on our business, financial condition and results of operations. If any of our FDA clearances are denied or rescinded, sales of our products in the United States would be prohibited during the period we do not have such clearances.
Drug Approval and Regulation
The marketing of pharmaceutical products in the United States, such as Triferic, requires the approval of the FDA. We received FDA approval to market Triferic in January 2015. The FDA has established regulations, guidelines and safety standards which apply to the pre‑clinical evaluation, clinical testing, manufacturing and marketing of our new iron maintenance therapy product and other pharmaceutical products. The steps required before a pharmaceutical product can be produced and marketed for human use include: (i) pre‑clinical studies; (ii) submission to the FDA of an Investigational New Drug Application (“IND”), which must become effective before human clinical trials may commence in the United States; (iii) adequate and well controlled human clinical trials; (iv) submission to the FDA of a New Drug Application (“NDA”) or, in some cases, an Abbreviated New Drug Application (“ANDA”); and (v) review
10
and approval of the NDA or ANDA by the FDA. An NDA generally is required for products with new active ingredients, new indications, new routes of administration, new dosage forms or new strengths. An NDA requires that complete clinical studies of a product’s safety and efficacy be submitted to the FDA, the cost of which is substantial. The costs are often less, however, for new delivery systems which utilize already approved drugs than for drugs with new active ingredients.
An ANDA is a marketing application filed as part of an abbreviated approval process that is available for generic drug products that have been scientifically determined to be “bioequivalent” to an FDA‑approved drug. This requires that the generic drug product have the same amount of active ingredient(s) absorbed in the same amount of time, use indication, route of administration, dosage form and strength as an existing FDA‑approved product. In addition the generic drug product must be manufactured in accordance with cGMP and meet requirements for batch identity, strength, purity and quality. Under applicable regulations, companies that seek to introduce an ANDA product must also certify that the product does not infringe on the approved product’s patent or that such patent has expired. If the applicant certifies that its product does not infringe on the approved product’s patent, the patent holder may institute legal action to determine the relative rights of the parties and the application of the patent, and the FDA may not finally approve the ANDA until a court finally determines that the applicable patent is invalid or would not be infringed by the applicant’s product.
Pre‑clinical studies are conducted to obtain preliminary information on a pharmaceutical product’s efficacy and safety in animal or in vitro models. The results of these studies are submitted to the FDA as part of the IND and are reviewed by the FDA before human clinical trials begin. Human clinical trials may begin 30 days after receipt of the IND by the FDA unless the FDA objects to the commencement of clinical trials.
Human clinical trials are typically conducted in three sequential phases, but the phases may overlap. Phase 1 trials consist of testing the product primarily for safety, metabolism and pharmacologic action in a small number of patients or healthy volunteers at one or more doses. In Phase 2 trials, the safety and efficacy of the product are evaluated in a patient population somewhat larger than the Phase 1 trials with the primary intent of determining the effective dose range. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded population at a large number of test sites. A clinical plan, or protocol, accompanied by documentation from the institutions participating in the trials, must be received by the FDA prior to commencement of each of the clinical trials. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time.
The results of product development and pre‑clinical and clinical studies are submitted to the FDA as an NDA or an ANDA for approval. If an application is submitted, there can be no assurance that the FDA will review and approve the NDA or an ANDA in a timely manner. The FDA may deny an NDA or an ANDA if applicable regulatory criteria are not satisfied or it may require additional testing, including pre‑clinical, clinical and or product manufacturing tests. Even if such data are submitted, the FDA may ultimately deny approval of the product. Further, if there are any modifications to the drug, including changes in indication, manufacturing process, labeling, or a change in a manufacturing facility, an NDA or an ANDA supplement may be required to be submitted to the FDA. Product approvals may be withdrawn after the product reaches the market if compliance with regulatory standards is not maintained or if problems occur regarding the safety or efficacy of the product. The FDA may require testing and surveillance programs to monitor the effect of products which have been commercialized, and has the power to prevent or limit further marketing of these products based on the results of these post‑marketing programs.
Manufacturing facilities are subject to periodic inspections for compliance with regulations and each domestic drug manufacturing facility must be registered with the FDA. Foreign regulatory authorities may also have similar regulations. We expend significant time, money and effort in the area of quality assurance to fully comply with all applicable requirements. FDA approval to manufacture a drug is site specific. In the event an approved manufacturing facility for a particular drug becomes inoperable, obtaining the required FDA approval to manufacture such drug at a different manufacturing site could result in production delays, which could adversely affect our business and results of operations. Manufacturers and distributors must comply with various post‑market requirements, including adverse event reporting, re‑evaluation of approval decisions and notices of changes in the product or in the process or procedures used to manufacture a product.
11
Other Government Regulations
The federal and state governments in the United States, as well as many foreign governments, from time to time explore ways to reduce medical care costs through health care reform. Due to uncertainties regarding the ultimate features of reform initiatives and their enactment and implementation, we cannot predict what impact any reform proposal ultimately adopted may have on the pharmaceutical and medical device industry or on our business or operating results. The Patient Protection and Affordable Care Act (“PPACA”), which was enacted in 2010, imposes excise taxes on manufacturers on the sale of medical devices and pharmaceutical products and requires medical device and pharmaceutical manufacturers annually to report certain financial and ownership relationships they have with physicians and teaching hospitals. The medical device excise tax received a two year moratorium ending December 31, 2017 and its status subsequent to the expiration of the moratorium is unclear. There are various bills pending or that are expected to be introduced in Congress to repeal, replace or modify the PPACA. Most versions would eliminate the medical device tax as early as December 31, 2017. Our activities are subject to various federal, state and local laws and regulations regarding occupational safety, laboratory practices, and environmental protection and may be subject to other present and possible future local, state, federal and foreign regulations.
The approval procedures for the marketing of our products in foreign countries vary from country to country, and the time required for approval may be longer or shorter than that required for FDA approval. We generally depend on our foreign distributors or marketing partners to obtain the appropriate regulatory approvals to market our products in those countries which typically do not require additional testing for products that have received FDA approval.
However, since medical practice and governmental regulations differ across regions, further testing may be needed to support market introduction in some foreign countries. Some foreign regulatory agencies may require additional studies involving patients located in their countries. Even after foreign approvals are obtained, further delays may be encountered before products may be marketed. Issues related to import and export can delay product introduction. Many countries require additional governmental approval for price reimbursement under national health insurance systems.
Product License Agreements
We are party to an in‑license agreement for Triferic that covers issued patents in the United States, the European Union and Japan, as well as other foreign jurisdictions. We licensed the product from a company owned by Dr. Ajay Gupta who subsequently joined us as our Chief Scientific Officer. The license agreement continues for the duration of the underlying patents in each country plus a period of ten years. Patents were issued in the United States in 2004 and extended through 2016 and may be extended thereafter under the Hatch‑Waxman Act. Our request for a term extension is currently under review and is anticipated to be approved. In view of the pending review, the United States Patent and Trademark Office has granted an interim extension on these patents for the period of one year from the original expiration date and would extend through December 31, 2017. The European patent was issued in 2005 and extends through 2017. The Japanese patent was issued in 2007 and extends through 2017. We intend to apply for an extension of our patent exclusivity for up to five years in Europe and Japan. As noted below in “Trademarks and Patents,” the Company has also received patent protection on the pharmaceutical grade formulation of the active pharmaceutical ingredient in Triferic which extends patent protection until 2029.
Our Triferic license agreement requires us to obtain and pay the cost of obtaining FDA approval of the product and patent maintenance expenses in order to realize any benefit from commercialization of the product. In addition, we were obligated to make certain milestone payments during development of the product. As of December 31, 2016, there were no remaining milestones to be completed although we continue to be obligated to pay ongoing royalties.
Trademarks and Patents
We have several trademarks and service marks used on our products and in our advertising and promotion of our products, and we have applied for United States registration of such marks. Most such applications have resulted in registration of such trademarks and service marks.
We were issued a United States patent on the synthesis and formulation of our pharmaceutical grade formulation of Triferic. The United States patent expires on April 17, 2029. Patents have also been granted in Europe,
12
Japan and Canada. We have numerous other patents and patent applications connected to Triferic pending in various countries.
We also own patents in the United States and Canada for our Dry Acid Concentrate method and apparatus for preparing liquid dialysate which expire on September 17, 2019. Expiration of these patents is not expected to have a material impact on our business.
Suppliers
We believe the raw materials and packaging materials for our hemodialysis concentrates, the components for our hemodialysis kits and the ancillary hemodialysis products distributed by us are generally available from several potential suppliers. We intend to engage CMOs for the manufacture and packaging of our drug products. There are several potential CMOs that are able to manufacture and package our drug products and so it is unlikely we will be dependent on any particular CMO. However, the lead time to bring on an additional CMO could be lengthy.
Customers
We operate in one market segment, the hemodialysis market, which involves the manufacture, sale and distribution of hemodialysis products to hemodialysis clinics including pharmaceutical, dialysis concentrates, dialysis kits and other ancillary products used in the dialysis process. In October 2014, we entered into a Distribution Agreement with Baxter and under this agreement Baxter received exclusive distribution rights for our concentrate products in the United States. Rockwell domestic customer contracts for the supply of dialysis concentrate products that permitted assignment to Baxter without customer consent have been assigned to Baxter. As a result, for the years ended December 31, 2016 and 2015, our direct sales to Baxter aggregated approximately 24% and 28% of sales, respectively and we had a receivable from Baxter of $2,430,159 and $2,088,000 as of December 31, 2016 and 2015, respectively.
For the years ended December 31, 2016, 2015 and 2014, one customer, DaVita Healthcare Partners, Inc., accounted for 52% of our sales in 2016, 48% of our sales in 2015 and 49% of our sales in 2014. Our accounts receivable from this customer were $2,224,046 and $2,156,000 as of December 31, 2016 and 2015, respectively. DaVita and Baxter and the accounts administered by Baxter are important to our business, financial condition and results of operations. The loss of any significant accounts could have a material adverse effect on our business, financial condition and results of operations. No other customers accounted for more than 10% of our sales in any of the last three years.
The majority of our international sales in each of the last three years were sales to domestic distributors that were resold to end users outside the United States. Our sales to foreign customers and distributors were less than 5% of our total sales in 2016, 2015 and 2014. Our total international sales, including sales to domestic distributors for resale outside the United States, aggregated 12%, 13% and 13%, of overall sales in 2016, 2015 and 2014, respectively.
Employees
As of December 31, 2016, we had approximately 300 employees, substantially all of whom are full time employees. Our arrangements with our employees are not governed by any collective bargaining agreement. Our employees are employed on an “at‑will” basis.
Research & Development
Over the last several years we have invested heavily in the testing and development of Triferic. We completed human clinical trials and other testing in 2013, and submitted our NDA for Triferic to the FDA in 2014. We received FDA approval for Triferic in January 2015. Since approval of Triferic, we have conducted additional clinical studies of Triferic for other indications, presentation in IV formulation and for a pediatric study of Triferic.
We engaged outside service providers, contract research organizations, consultants and legal counsel to assist us with clinical trials, product development and obtaining regulatory approval. We incurred product development and research costs related to the commercial development, patent approval and regulatory approval of new products, including Triferic, aggregating approximately $5,840,000, $4,961,000 and $7,784,000, in 2016, 2015 and 2014, respectively.
13
Future research and product development spending on the Triferic platform may include clinical testing in connection with peritoneal dialysis, total parenteral nutrition, an orphan indication and a pediatric indication. Future spending on such indications is expected to be minor in relation to the Company’s cash resources.
Where You Can Get Information We File with the SEC
Our internet address is http://www.rockwellmed.com. Our internet address is included as an inactive textual reference only and nothing on the website is incorporated by reference into this Annual Report on Form 10‑K. You can access free of charge on our website all of our reports filed pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including our annual reports on Form 10‑K, quarterly reports on Form 10‑Q, current reports on Form 8‑K, and amendments to those reports. These reports are available as soon as practicable after they are electronically filed with the SEC.
The SEC also maintains a website on the internet that contains reports, proxy and information statements and other information regarding issuers, such as us, that file electronically with the SEC. The address of the SEC’s website is http://www.sec.gov.
Item 1A. Risk Factors.
Investing in our common stock involves a high degree of risk and there can be no assurance that future results will meet expectations. You should carefully consider the risks and uncertainties described below before purchasing our common stock. The risks and uncertainties described below are not the only ones facing our company. Additional risks and uncertainties may also impair our business operations. If any of these risks actually occur, our business, financial condition or results of operations would likely suffer. In that case, the trading price of our common stock could fall, and you may lose all or part of the money you paid to buy our common stock.
RISKS RELATED TO OUR DRUG BUSINESS
Although Triferic has been approved by the FDA, we may not be able to commercialize it successfully.
The commercial success of Triferic will depend on a number of factors, including the following:
|
· |
IV iron currently dominates treatment for iron deficiency and Triferic will have to compete against it and possibly other existing and future products; |
|
· |
It may be difficult to gain market acceptance from dialysis chains, anemia managers and nephrologists or such acceptance may be slower than expected. Market acceptance will depend on a number of factors, such as demonstration of Triferic’s safety and efficacy, cost-effectiveness, advantages over existing products, and the reimbursement policies of government and third party payers, including Medicare; |
|
· |
We are seeking transitional add-on reimbursement status from CMS to separately pay for Triferic outside of the standard dialysis bundled payment. Dialysis providers have been slow to adopt Triferic in the absence of such status. In the absence of a favorable determination from CMS, dialysis service providers are likely to adopt Triferic at a much slower rate than if Triferic is granted such status due to the cost of conversion and lack of an immediate financial incentive to adopt Triferic; |
|
· |
Maintaining compliance with ongoing regulatory requirements applicable to Triferic or which apply generally to the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping applicable to the product; |
|
· |
The effectiveness of our marketing, sales and distribution strategies and operations for development and commercialization, and our ability to execute our marketing strategy without significant additional expenditures; |
14
|
· |
Competitors may engage in anti-competitive practices and other tactics to retain their market share; |
|
· |
Our ability to avoid third party patent interference or patent infringement claims; |
|
· |
A continued acceptable safety profile of Triferic; and |
|
· |
Discovery of previously unknown problems with Triferic or with any third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements. |
An adverse development with respect to any of the foregoing may have a material adverse effect on our ability to manufacture and market Triferic. These factors are largely beyond our control. Accordingly, we cannot assure you that we will receive transitional add-on reimbursement status for Triferic from CMS or that we will be able to generate significant revenues through the sale of Triferic. If we are not successful in commercializing Triferic, or are significantly delayed in doing so, our entire investment in Triferic may be worthless, our licensing rights could be affected and the price of our common stock could substantially decline. If we are unable to successfully commercialize Triferic and achieve sufficient sales volumes over the next one to two years, we may have to write off a portion of our inventory investment in Triferic. Even if we were successful in commercializing Triferic, due to the highly concentrated nature of the market, our continued success may depend on adoption of Triferic by a few customers.
Triferic is currently limited to use in adult patients receiving hemodialysis treatments and has not been approved for other indications. Regulatory approval for any approved product is limited by the FDA to those specific indications and conditions for which clinical safety and efficacy have been demonstrated, which may limit our ability to market our drug products.
While physicians may choose to prescribe drugs for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical studies and approved by regulatory authorities, our ability to promote the products or encourage our customers to use the products is limited to those indications that are specifically approved by the FDA as safe and effective. Any new indication for an approved product also requires FDA approval. If we are not able to obtain FDA approval for any other indications for Triferic, our ability to effectively market and sell Triferic may be reduced and our business may be adversely affected. Moreover, if our promotional activities fail to comply with the FDA’s regulations or guidelines, we may be subject to warnings from, or enforcement action by, the FDA that may include penalties, fines, injunctions, recall or seizure of products, suspension of production, denial of future regulatory approvals, withdrawal or suspension of existing regulatory approvals, operating restrictions, debarment, exclusion and criminal prosecution, any of which could materially harm our business.
If we do not obtain protection under the Hatch-Waxman Act to extend patent protection for Triferic, our business may be harmed.
The United States Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as the “Hatch-Waxman Act,” provides that patent holders may apply for an extension of patent term for drugs for a period of up to five years to compensate for time spent in development and regulatory approval. We have applied for an extension, and received a temporary one year extension but there can be no assurance that we will receive the full extension of the patent term provided under the Hatch-Waxman Act for either of the licensed Triferic patents that expired at the end of 2016. If we fail to receive the full extension, our ability to prevent competitors from manufacturing, marketing and selling generic versions of Triferic could be impaired and we would have to rely on the protection afforded us by the United States patent we hold on the synthesis and formulation of our pharmaceutical grade formulation of Triferic which expires in 2029 or on other patents related to Triferic that may be issued to us in the future.
Our Calcitriol contract manufacturer has made changes to the manufacturing process for Calcitriol that require FDA approval prior to commercial sale of Calcitriol. The FDA review process has delayed our launch of Calcitriol and, even if approved, we may not be able to commercialize Calcitriol successfully.
While we have received FDA approval to manufacture a generic version of Calcitriol, our CMO made changes to the process that requires prior FDA approval before the commercial products can be sold that were produced under the process change. Even if approved, we must also meet certain ongoing regulatory requirements for product testing and stability of our commercially marketed products. If our testing does not meet approvable standards, if our CMO cannot
15
make the product in sufficient quantities and on a consistent basis or if we experience operational issues with our CMO, we may not be able to market Calcitriol successfully or the launch may be further delayed.
The market for generic drugs such as Calcitriol is generally very competitive, which may make it difficult for us to capture significant market share. If we have success in capturing market share with Calcitriol, it may attract other entrants to market their own Calcitriol product, which could have a material adverse effect on our future revenues and results of operations. Branded competitors may aggressively lower their prices to maintain market share. Dialysis service providers may seek alternative forms of treatment for this indication.
We may not be successful in obtaining foreign regulatory approvals or in arranging an out-licensing or other venture to realize commercialization of our drug products outside of the United States. Even if we are successful in out-licensing our drug products, the licensees or partners may not be effective at marketing our products in certain markets or at all.
The approval procedures for marketing our new drug products, such as Triferic, outside the United States vary from country to country, can be difficult to obtain and carry the same risks as FDA approval. In particular, regulatory approval in foreign countries may require additional testing and may otherwise be expensive and time consuming to undertake. Even after foreign approvals are obtained, further delays may be encountered before products may be marketed. Many countries require additional government approval for price reimbursement under national health insurance systems.
Even if we obtain the necessary foreign approval in a particular market, we do not have substantial expertise selling and marketing on an international level and therefore may not be successful in realizing commercial value from our products should we attempt to develop international markets ourselves. Our strategy for addressing the need for expertise in obtaining foreign approvals and marketing in foreign markets is to out-license rights to our drugs in markets outside the United States. However, we may not be successful in finding partners in addition to Wanbang, our Chinese market partner, or ARAM Medical, our distributor in the Middle East, who will be willing to invest in our drugs outside the United States or our partners may be unable to obtain the necessary regulatory approvals. If we are not successful in out-licensing our drugs outside of the United States or entering into some other business development arrangement to obtain the necessary approvals to commercialize them, or if our partners are unable to obtain the necessary regulatory approvals, we may be forced to seek regulatory approval and market these products ourselves. If we elect to seek approval ourselves, it may take longer than expected to obtain regulatory approval and to market and manufacture our drugs, and we may decide to delay or abandon development efforts in certain markets.
Any such delay or abandonment, or any failure to receive one or more foreign approvals, may have an adverse effect on the benefits otherwise expected from marketing in foreign countries.
If we are successful in obtaining other business partners to commercialize our products in foreign markets, we will be dependent upon their effectiveness in selling and marketing our products in those foreign markets. These partners may face stiff competition, government price regulations, generic versions of our drug products, violations of our intellectual property rights and other negative events or may otherwise be ineffective in commercializing our products, any of which could reduce the market potential for our products and our success in those markets.
We will rely on third party suppliers for raw materials, packaging components and manufacturing of our drug products. We may not be able to obtain the raw materials, proper components or manufacturing capacity we need, or the cost of the materials, components or manufacturing capacity may be higher than expected, any of which could have a material adverse effect on our expected results of operations, financial position and cash flows.
We may not be able to obtain needed raw materials, packaging components and manufacturing capacity for a variety of reasons, including among others:
|
· |
We may be required to purchase certain raw materials and packaging components from unaffiliated third-party suppliers who may not be able to supply us consistently or at all; |
|
· |
Regulatory requirements or action by regulatory agencies or others, including delays in receiving necessary approvals; |
16
|
· |
Adverse financial or other strategic developments at or affecting a supplier or contract manufacturer; |
|
· |
Unexpected demand for or shortage of raw materials or packaging components; |
|
· |
Failure to comply with cGMP standards which results in quality or product failures, adulteration, contamination and/or recall; |
|
· |
Changes made to manufacturing processes by our contract manufacturers may result in regulatory delays until such changes are approved regulatory authorities; |
|
· |
Limitations in capacity of contract manufacturers; and |
|
· |
Changes in product demand. |
If we are unable to obtain the raw materials, components and manufacturing capacity we require, or if we are charged more than expected for these items, we may not be able to produce the desired quantities of our drug products or our expected gross profit margins may be materially adversely affected.
Before it can be marketed, an investigational drug requires FDA approval, which is a long, expensive process with no guarantee of success.
Performing clinical trials and obtaining FDA approval for any drug can take a long time. Clinical trials typically take many months or years to complete. Once trials are completed and the NDA, is submitted to the FDA, the FDA may find deficiencies in our NDA, may raise safety or efficacy concerns or may otherwise require additional clinical testing or impose other requirements before granting approval, which could significantly delay approval or result in us not receiving approval at all.
Clinical trials and the NDA approval process for any new drug candidate are expensive. If we were to develop new drug candidates and we experience delays, or additional testing or other unplanned requirements are imposed on us, we may need to raise additional capital, which may not be available when needed or may be available only on terms that are not in the best interests of the Company and its shareholders, or which result in substantial dilution of shareholders’ voting power and ownership. New indications of Triferic when submitted to the FDA for approval will require us to pay review fees. If approval is not granted for any new products submitted, our entire investment in the related products may be worthless, any licensing rights could be forfeited and the price of our common stock could substantially decline.
Our drug business will depend on government funding of health care, and changes could impact our ability to be paid in full for our products, increase prices or cause consolidation in the dialysis provider market.
Many dialysis providers receive the majority of their funding from the government and are supplemented by payments from private health care insurers. These providers depend on Medicare and Medicaid funding to be viable businesses. Congress continuously enacts a variety of changes to health insurance and reimbursement, some of which could have a negative impact on Medicare and Medicaid funding, which fund the majority of dialysis costs in the United States, and on reimbursement protocols. If Medicare and Medicaid funding were to be materially decreased, these providers would be severely impacted, increasing our risk of not being paid in full. An increase in our exposure to uncollectible accounts could have a material adverse effect on our financial position, results of operations and cash flows.
Since 2011, CMS has continued to modify reimbursement policies for dialysis under the ESRD prospective payment system generally resulting in lower payment to dialysis providers. We anticipate that dialysis providers will continue to seek ways to reduce their costs per treatment due to this change in reimbursement practice which could reduce our sales and profitability and have a material adverse effect on our business, financial condition and results of operations.
CMS continues to make changes to the ESRD Quality Incentive Program, or QIP, which pays dialysis providers an incentive to improve the quality of care. Final ESRD regulations published in October 2015 include changes to QIP for CYs 2017-2019. Each facility’s total performance score is posted on the CMS website. Low performance scores at
17
our customers could result in a reduction in patient volume, a reduction in payment rates and a decrease in sales for those customers.
PPACA also proposes changes to Medicaid, including decreasing the amount of federal dollars available to the states and eliminating the Medicaid expansion program, resulting in fewer people qualifying for Medicaid benefits. If implemented, these changes could impact reimbursement by the Medicaid program for our drug products and dialysis.
As a result of these changes to Medicare and Medicaid reimbursement, the dialysis provider industry may continue to consolidate. This may result in increased purchasing leverage for providers across all dialysis product categories and increased pricing pressure on all suppliers to the industry.
Our efforts to obtain transitional add on reimbursement status for Triferic may not be successful.
We have worked to obtain broad based support for transitional add-on reimbursement status from CMS for Triferic and we believe that there is a strong likelihood that Triferic will receive transitional add-on reimbursement status. However, we cannot predict when or even if CMS will grant such status. If CMS does not grant transitional add-on reimbursement status for Triferic we believe the pace of adoption for Triferic will be much slower than if such status is granted due to cost considerations by dialysis service providers. While we believe that Triferic’s attributes make it a better choice for patients and that it will become the standard of care in iron maintenance therapy for hemodialysis patients, we cannot predict future usage of Triferic. We have invested over $10,000,000 in Triferic inventory, including approximately $9 million in Triferic API, which generally has a three year shelf life, and $1.3 million in finished goods inventory with shelf lives ranging from 1-3 years. If we are unable to utilize some or all of the Triferic inventory before its shelf life expires, some or all of our investment in Triferic inventory may not be saleable, reducing the inventory we have available for sale and requiring us to reserve for the reduction in value. We may also need to reserve for inventory that we estimate will not be sold before such inventory expires. Any such inventory reserve could have a material adverse effect on our results of operations and financial condition.
Health care reform could adversely affect our business.
The federal and state governments in the United States, as well as many foreign governments, from time to time explore ways to reduce medical care costs through health care reform. The federal Medicare and Medicaid programs are facing financial challenges and are looking at ways to reduce the costs of the Medicare and Medicaid programs. Similarly, many states have large deficits which may prove unsustainable, resulting in defaults on state debt obligations which may ultimately result in the reduction or curtailment of health care benefits or state Medicaid reimbursement.
The United States government faces structural deficits that may require changes to government funded healthcare programs such as Medicare and Medicaid which may negatively impact customers of our products. Our financial position, results of operations, and cash flows and ability to commercialize our drug products could be materially impacted by the PPACA, future health care reform or reduced Medicare and Medicaid spending by the federal government. In addition, legislative and administrative efforts to repeal or modify the PPACA are underway. We cannot predict how reform or replacement of PPACA or other health care reform will affect our business and any such changes could substantially modify the methodology for reimbursing medical services, drugs and devices or the number of patients eligible for reimbursement.
Device and pharmaceutical manufacturers are required to report annually to the Department of Health and Human Services regarding certain financial relationships they have with physicians and teaching hospitals. This reporting requirement will increase governmental scrutiny on our contractual relationships with physicians and teaching hospitals and will increase the risk that inadvertent violations result in liability under the federal fraud and abuse laws, which could have a material adverse effect on our results of operations, financial position and cash flows.
RISKS RELATED TO OUR CONCENTRATE BUSINESS
We are in arbitration to resolve disputes with Baxter regarding the Distribution Agreement, which could result in termination of the Distribution Agreement or have other material adverse consequences for us.
Under the Distribution Agreement, Baxter is our exclusive agent for commercializing our hemodialysis concentrate and ancillary products in the United States and various foreign countries. The Agreement does not involve
18
Rockwell’s drug products. In September 2016, Baxter initiated an arbitration proceeding against us, alleging that we have materially breached the Distribution Agreement in various respects. Baxter seeks declaratory relief giving Baxter the right to terminate the Distribution Agreement and recover a portion of the upfront fee, injunctive relief to prevent us from establishing a West Coast facility, and unspecified damages. We filed a response denying all of Baxter’s claims of breach and wrongdoing, and have counterclaimed that Baxter is itself in material breach of the Distribution Agreement for failing to pay certain accounts receivable and for repudiating its obligation to pay the West Coast facility fee. We are seeking damages, declaratory, injunctive and other equitable relief, as well as interest, costs and attorney fees. In addition, in October 2016, we gave notice to Baxter that it breached the minimum purchase requirement for the contract year ending October 2, 2016 and that we intended to cause its distribution rights to become non-exclusive unless it cured the shortfall within the 30-day period specified in the Distribution Agreement. Baxter disputed the existence of a breach and failed to cure the shortfall. Rockwell subsequently provided Baxter with notice of loss of exclusivity due to its failure to cure as provided in the Distribution Agreement. The determination of whether a breach occurred resulting in a loss of exclusivity and the outcome of the other pending disputes with Baxter will be determined through the arbitration process.
There can be no assurance that our disputes with Baxter will be resolved in the arbitration in our favor. If resolution of the dispute permits Baxter to not pay us the amounts owed to us, requires us to pay amounts to Baxter in excess of any reserve we may establish in connection with this matter or requires us to refund a portion of the upfront payment made by Baxter to us, it could have a material adverse effect on our results of operations, cash flows and financial condition. Moreover, the arbitration process will be expensive and may divert management’s attention away from operation of our business.
In addition, given the possibility that either the Distribution Agreement will be terminated by us or Baxter upon completion of the arbitration or that Baxter’s distribution rights will become non-exclusive, we believe that Baxter is unlikely to commit material financial and other resources to the marketing and distribution of our products while the arbitration process continues. As a result, unit sales of our concentrate products may fall and we may lose customers to our competitors during the arbitration process, resulting in lower revenues and gross margin for us. If the Distribution Agreement were to terminate or if Baxter’s distribution rights were to become non-exclusive, we may have to compete directly for customers with competitors. Such competition could have the effect of further reducing our sales and adversely affecting our results of operations, cash flows and financial condition.
We may be required to repay a portion of the fees received from Baxter, which could materially and adversely affect our financial position and cash reserves.
Pursuant to the terms of the Distribution Agreement, we may be required to repay a portion of the upfront fee and a portion of the facility fee to Baxter upon the occurrence of a "Refund Trigger Event." A "Refund Trigger Event" includes, among other events, termination due to an uncured material breach by us. Occurrence of a Refund Trigger Event would obligate us to refund 50% of the $20 million upfront fee (and any portion of the facility fee paid by Baxter) if the event occurs prior to December 31, 2016, 33% of the $20 million upfront fee if the event occurs in 2017 or 2018, and 25% of the $20 million upfront fee if the event occurs in 2019, 2020 or 2021.
In September 2016, Baxter initiated an arbitration proceeding against us alleging material breaches of the Distribution Agreement and seeking various forms of relief, including the right to terminate the Distribution Agreement and receive a refund of a portion of the upfront fee. We have filed a response denying all of Baxter’s claims and a counterclaim alleging that Baxter is in material breach of the Distribution Agreement.
In addition, if Baxter terminates the Distribution Agreement because it has been enjoined by a court of competent jurisdiction from selling in the United States any product covered by the Distribution Agreement due to a claim of intellectual property infringement or misappropriation relating to such product prior to the end of 2018, Baxter would be entitled to a refund of up to $10 million, or $6.6 million if the termination occurs in 2019.
If we are required to make any such refund payment, we may need to reallocate funds from other parts of our business, which could force us to change or delay plans for use of that capital. In any such event, our financial condition, results of operations, and cash reserves could be materially and adversely affected.
19
A few customers account for a substantial portion of the end user sales of our concentrate products. The loss of any of these customers could have a material adverse effect on our results of operations and cash flow from our concentrate business.
A substantial portion of our concentrate and ancillary products are primarily sold to or through Baxter. Its sales of our products are highly concentrated in a few customers and Baxter’s loss of any of those customers could adversely affect our results of operations for one year or less depending on when Baxter would lose exclusivity for not meeting the minimum order threshold; one customer accounted for nearly half of our sales in each of the last three years and for a substantial number of the clinics we serve.
The concentrate market is very competitive and has a large competitor with substantial resources.
The primary competitor in the market for our concentrate products is a large diversified company which has substantial financial, technical, manufacturing, marketing, research and management resources. We and our distributor, Baxter, may not be able to successfully compete with them or other companies. The primary competitor has historically used product bundling and low pricing as marketing techniques to capture market share of the concentrate products we sell. We and Baxter may be at a disadvantage in competing against their marketing strategies to sell our products. Furthermore, the primary competitor is vertically integrated and is the largest provider of dialysis services in the United States, treating approximately 36% of all U.S. patients through its clinics. This competitor has routinely acquired smaller clinic chain operations which we supply through Baxter. This competitor may acquire more of the customers we service in the future.
We may be affected materially and adversely by increases in raw material costs.
A significant portion of our costs relates to chemicals and other raw materials, which are subject to price volatility based on demand and are highly influenced by the overall level of economic activity in the United States and abroad. These costs have tended to rise from year to year and are likely to continue to rise in the future. Under our Distribution Agreement with Baxter, such cost inflation may result in increases in the prices we charge Baxter. If these increases exceed specified levels in the Distribution Agreement, Baxter is permitted to terminate the Distribution Agreement and obtain a refund of a portion of the fees we received from Baxter. Any such termination or refund could have a material adverse effect on our business, results of operations, financial position and cash flows.
Our concentrate business is highly regulated, which increases our costs and the risk and consequence of noncompliance.
Although our hemodialysis concentrates have been cleared by the FDA, it could rescind these clearances and any new products or modifications to our current products that we develop could fail to receive FDA clearance. If the FDA rescinds or denies any current or future clearances or approvals for our products, we would be prohibited from selling those products in the United States until we obtain such clearances or approvals. Our business would be adversely affected by any such prohibition, any delay in obtaining necessary regulatory approvals, and any limits placed by the FDA on our intended use. Our products are also subject to federal regulations regarding good manufacturing practices and quality. Our failure to comply with these regulations could result in FDA action or product liability litigation adverse to us. Any of these events could constitute a breach by us of the Distribution Agreement, providing Baxter with various remedies that would be material and adverse to us, including without limitation, termination of the Distribution Agreement. Moreover, changes in applicable regulatory requirements could significantly increase the costs of our operations and, if such higher costs result in price increases that exceed the thresholds specified in the Distribution Agreement, could give Baxter the right to terminate the Distribution Agreement and obtain a partial refund of certain fees paid to us pursuant to that agreement.
RISKS RELATED TO OUR BUSINESS AS A WHOLE
We may not be successful in expanding our product portfolio or in our business development efforts related to in-licensing, acquisitions or other business collaborations. Even if we are able to enter into business development arrangements, they could have a negative impact on our business and our profitability.
As part of our business strategy to expand our product portfolio, we are seeking to acquire or in-license other drug products that we believe are a complementary fit with our current product portfolio as well as other products that
20
we believe have substantial development potential. Our experience with respect to these business development activities is limited. The negotiation of such arrangements can be a lengthy and complex process and there can be no assurance that any such negotiations will be completed on a timely basis or on terms that are cost-effective and acceptable to us or, if they are completed, that we will be able to effectively integrate, develop and launch such products effectively.
In addition, the market potential for new products is highly uncertain and evaluation of such potential requires significant judgment and assumptions. There is a significant risk that any new product may not be able to be brought to market as profitably as expected or at all. If the results of any new product initiative were materially worse than expected, it could have a material adverse effect on our financial results and condition.
Our drug and concentrate businesses are highly regulated, resulting in additional expense and risk of noncompliance that can materially and adversely affect our business, financial condition and results of operations.
Our businesses are highly regulated. The testing, manufacture and sale of the products we manufacture directly or through third party contractors are subject to extensive regulation by the FDA and by other federal, state and foreign authorities. Before drugs or medical devices, such as our concentrate products, can be commercially marketed in the United States, the FDA must give either premarket approval or 510(k) clearance. Even after a product is approved, regulatory authorities may still impose significant restrictions on a product’s indicated uses or marketing or impose requirements for potentially costly post-marketing studies. In addition, our products are subject to ongoing regulatory requirements for labeling, packaging, storage, advertising, promotion, sampling, record-keeping and reporting of safety and other post-market information, including both federal and state requirements in the United States and in other jurisdictions where they are marketed. In addition, manufacturers and their facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to current cGMP and applicable state laws. As such, we and our contract manufacturers are subject to continual review and periodic inspections to assess compliance with cGMP and state laws. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas to achieve and maintain regulatory compliance. We are also required to report certain adverse reactions and production problems, if any, to the FDA, state agencies and foreign regulatory authorities, when applicable, and to comply with requirements concerning advertising and promotion for our products.
If a regulatory agency determines that we do not comply with any applicable regulatory requirements, we may be subject to warnings from, or enforcement action by, state and federal government authorities that may include penalties, fines, injunctions, recall or seizure of products, suspension of production, denial of future regulatory approvals, withdrawal or suspension of existing regulatory approvals, operating restrictions, injunctions and criminal prosecution. If regulatory sanctions are applied, the value of our Company and our operating results could be materially and adversely affected.
We depend on key personnel, the loss of which could harm our ability to operate.
Our success depends heavily on the efforts of Robert L. Chioini, our founder and Chief Executive Officer, Dr. Ajay Gupta, our Chief Scientific Officer, Dr. Raymond D. Pratt, our Chief Medical Officer, and Thomas E. Klema, our Chief Financial Officer, Secretary and Treasurer. Mr. Chioini is primarily responsible for the strategic direction of the Company and for managing our sales and marketing efforts. Dr. Gupta is primarily responsible for discovery and development of new technologies. Dr. Pratt is primarily responsible for the clinical development, testing and regulatory approval of our products. No member of our executive management team has an employment agreement with the Company. If we lose the services of Mr. Chioini, Dr. Gupta, Dr. Pratt or Mr. Klema, our business, product development efforts, financial condition and results of operations could be adversely affected.
We could be prevented from selling products, forced to pay damages and compelled to defend against litigation if we infringe the rights of a third party.
Our success, competitive position and future revenues will depend in part on our ability to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties.
21
We could incur substantial costs in seeking enforcement of our patent rights against infringement, and we cannot guarantee that such patents will successfully preclude others from using technology that we rely upon. We have no knowledge of any infringement or patent litigation, threatened or filed at this time. It is possible that we may infringe on intellectual property rights of others without being aware of the infringement. If a third party believes that one of our products infringes on the third party’s patent, it may sue us even if we have received our own patent protection for the technology. If we infringe the rights of a third party, we could be prevented from selling products, forced to pay damages and compelled to defend against litigation. If Baxter is prevented from selling from any of our concentrate or ancillary products due to a patent infringement or if its ability to sell any of our concentrate or ancillary products due to a patent infringement is materially and adversely affected, Baxter may be entitled to terminate our Distribution Agreement and obtain a refund of a portion of the upfront fee and facility fee.
Our products may have undesirable side effects and our product liability insurance may not be sufficient to protect us from material liability or harm to our business.
If concerns are raised regarding the safety of a new drug as a result of undesirable side effects identified during clinical testing, the FDA may decline to approve the drug at the end of the NDA review period or issue a letter requesting additional data or information prior to making a final decision regarding whether or not to approve the drug. Following FDA approval, if we or others later identify previously unknown undesirable side effects caused by our drug or concentrate products, if known side effects are more frequent or severe than in the past, or if we or others detect unexpected safety signals for such products or any products perceived to be similar to such products, the FDA or other applicable regulatory authorities may require the addition of unfavorable labeling statements, specific warnings or contraindications, may suspend or withdraw their approval of the product, may require it to be removed from the market or may impose restrictions on the distribution or use of the product. Such side effects may also result in litigation against the Company by private litigants.
We maintain products liability insurance in the amount of $10 million per occurrence and $10 million in the aggregate. We cannot be sure that such insurance would be sufficient to protect us against liabilities associated with any of these events in view of our expanding business or that such insurance will remain available at economical levels. We may have significant legal expenses that are not covered by insurance. In addition, our reputation could be damaged by such sanctions or product liability litigation and that could harm our marketing ability. Any such sanctions or litigation could also hurt our ability to retain products liability insurance or make such insurance more expensive. In any such event, our business, financial condition and results of operations could be materially adversely affected.
We may be unable to obtain certain debt financing in the future as a result of our arrangement with Baxter.
The Distribution Agreement prohibits us from entering into a contract encumbering the assets used in our concentrate business without the prior written consent of Baxter, and Baxter would be under no obligation to provide us with consent. The assets used in our concentrate business currently constitute a substantial portion of the tangible assets we own. If our development activities require substantial cash resources in the future in excess of our liquid resources on hand and if our cash flows are not sufficient to support financing through unsecured indebtedness, we may not be able to obtain debt financing and our capital financing options may become limited. If we are unable to obtain this type of debt financing, our business and our future development and expansion strategies may be adversely affected.
RISKS RELATED TO OUR COMMON STOCK
Shares eligible for future sale may affect the market price of our common shares.
Any future sales by us of substantial amounts of our common shares, or the possibility of such sales, could adversely affect the market price of our common shares and also impair our ability to raise capital through an offering of our equity securities in the future. In the future, we may issue additional shares or warrants in connection with investments or for other purposes considered advisable by our Board of Directors. Any substantial sale of our common shares may have an adverse effect on the market price of our common shares and may dilute the economic value and voting rights of existing shareholders.
22
In addition, as of December 31, 2016, there were 5,989,335 shares issuable upon the exercise of outstanding and exercisable stock options, 1,702,166 shares issuable upon the exercise of outstanding stock options that are not yet exercisable and 545,694 additional shares available for future grant under our 2007 Long Term Incentive Plan. The market price of the common shares may be depressed by the potential exercise of these options. The holders of these options are likely to exercise them when we would otherwise be able to obtain additional capital on more favorable terms than those provided by the options.
The market price for our common stock is volatile.
Our stock price, like the market price of many stocks in the biotechnology and pharmaceutical industries, is volatile. Events such as announcements around clinical testing results or regulatory approval of a product, as well as the reporting of sales, operating results and cash resources, may cause significant fluctuations in our share price. In addition, third parties may engage in trading strategies that result in intentional volatility to and control over our share price.
We could have a material weakness in our internal control over financial reporting, which, until remedied, could result in errors in our financial statements requiring restatement of our financial statements. As a result, investors may lose confidence in our reported financial information, which could lead to a decline in our stock price.
SEC rules require us to evaluate the effectiveness of our internal control over financial reporting as of the end of each year, and to include a management report assessing the effectiveness of our internal control over financial reporting in each Annual Report on Form 10-K. It is possible, due to the small size of our accounting staff, that we may identify control deficiencies in the future that constitute one or more material weaknesses. If our internal control over financial reporting or disclosure controls and procedures are not effective, there may be errors in our financial statements and in our disclosure that could require restatements. Investors may lose confidence in our reported financial information and in our disclosure, which could lead to a decline in our stock price.
No system of internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. Over time, controls may become inadequate because changes in conditions or deterioration in the degree of compliance with policies or procedures may occur. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. As a result, we cannot assure you that significant deficiencies or material weaknesses in our internal control over financial reporting will not be identified in the future.
Structural and anti-takeover provisions reduce the likelihood that you will receive a takeover premium.
The Board of Directors has the authority, without shareholder approval, to issue shares of preferred stock having such rights, preferences and privileges as the Board of Directors may determine. Any such issuance of preferred stock could, under certain circumstances, have the effect of delaying or preventing a change in control and may adversely affect the rights of holders of common shares, including by decreasing the amount of earnings and assets available for distribution to holders of common shares and adversely affect the relative voting power or other rights of the holders of the common shares. In addition, we may become subject to Michigan statutes regulating business combinations or our Board may take other actions which might also hinder or delay a change in control. Any such actions can have a depressive effect on the market price of our common shares and can limit shareholders’ ability to receive a premium on their shares by discouraging takeover and tender offers.
Our shareholders do not have the right to cumulative voting in the election of directors. Moreover, our directors serve staggered three-year terms, and directors may not be removed without cause. These provisions could have an anti-takeover effect by making it more difficult to acquire us by means of a tender offer, a proxy contest or otherwise, or to remove incumbent directors. These provisions could delay, deter or prevent a tender offer or takeover attempt that a shareholder might consider in his or her best interests, including those attempts that might result in a premium over the market price for the common shares.
23
We do not anticipate paying dividends in the foreseeable future.
Since inception, we have not paid any cash dividend on our common shares and do not anticipate paying such dividends in the foreseeable future. The payment of dividends is within the discretion of our Board of Directors and depends upon our earnings, capital requirements, financial condition and requirements, future prospects, restrictions in future financing agreements, business conditions and other factors deemed relevant by the Board. We intend to retain earnings and any cash resources to finance our operations. Therefore, it is highly unlikely we will pay cash dividends.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 2. Properties.
We occupy a 51,000 square foot facility and a 17,500 square foot facility in Wixom, Michigan under a lease expiring in August 2018. We also occupy a 51,000 square foot facility in Grapevine, Texas under a lease expiring in December 2020. In addition, we lease a 57,000 square foot facility in Greer, South Carolina under a lease expiring in February 2018.
We intend to use each of our facilities to manufacture and warehouse our products. All such facilities and their contents are covered under various insurance policies which management believes provide adequate coverage. We also use the office space in Wixom, Michigan as our principal administrative office. With our continued growth we expect that we will require additional office space, manufacturing capacity and distribution facilities to meet our business requirements.
Item 3. Legal Proceedings.
Baxter Arbitration
On September 12, 2016, Baxter initiated an arbitration proceeding against Rockwell in accordance with the International Institute for Conflict Prevention and Resolution, Inc.’s Rules for Non-Administered Arbitration under the Distribution Agreement. Baxter alleges that Rockwell has breached the Distribution Agreement in various respects associated with its dealings with customers, its allocation of expenses and its true-up notices, and by improperly threatening to build a West Coast facility. Baxter seeks declaratory relief giving Baxter the right to terminate the Distribution Agreement and recover up to $10 million of the upfront fee, injunctive relief to prevent Rockwell from establishing a West Coast facility, and unspecified damages.
Rockwell filed a response denying all of Baxter’s claims of breach and wrongdoing, and has counterclaimed that Baxter is itself in breach of the Distribution Agreement for failing to pay substantial accounts receivable and for repudiating its obligation to pay the West Coast facility fee of up to $10 million. Rockwell is seeking damages, declaratory, injunctive and other equitable relief, as well as interest, costs and attorney fees.
In addition, in October 2016, Rockwell gave notice to Baxter that it breached the minimum purchase requirement for the contract year ended October 2, 2016 and that Rockwell intended to cause its distribution rights to become non-exclusive unless it cured the shortfall within the 30-day period specified in the Distribution Agreement. Baxter disputed the existence of a breach and failed to cure the deficiency. Rockwell subsequently provided Baxter with notice of loss of exclusivity due to its failure to cure as provided in the Distribution Agreement. The determination of whether a breach occurred resulting in a loss of exclusivity and the outcome of the other pending disputes with Baxter will be determined through the arbitration process. Such arbitration process is anticipated to conclude during the third quarter of 2017.
Richmond/Ravich Litigation
On March 8, 2017, Rockwell filed suit in the United States District Court for the Eastern District of Michigan against Richmond Brothers, Inc. and certain related entities, David S. Richmond, Mark H. Ravich and certain related trusts, Matthew J. Curfman, and certain other individual investors seeking declaratory and injunctive relief relating to
24
alleged violations of Section 13(d) of the Securities Exchange Act of 1934, as amended, and the rules promulgated thereunder by the Securities and Exchange Commission. The complaint alleges that the defendants failed to file required Schedules 13D and made various material misstatements in a Schedule 13G and Schedule 13D that they filed.
Other Proceedings
In addition to the Baxter arbitration, we are involved in certain other legal proceedings from time to time before various courts and governmental agencies. We cannot predict the final disposition of such proceedings. We regularly review legal matters and record provisions for claims that are considered probable of loss. The resolution of these pending proceedings is not expected to have a material effect on our operations or consolidated financial statements in the period in which they are resolved.
Item 4. Mine Safety Disclosures.
Not applicable.
25
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common shares trade on the Nasdaq Global Market under the trading symbol “RMTI”. The prices below are the high and low sale prices as reported by the Nasdaq Global Market in each quarter during 2016 and 2015.
|
|
|
Price Range |
|
||||
|
|
|
High |
|
Low |
|
||
|
2016 |
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
8.37 |
|
$ |
3.55 |
|
|
Third Quarter |
|
|
8.45 |
|
|
6.27 |
|
|
Second Quarter |
|
|
10.58 |
|
|
6.86 |
|
|
First Quarter |
|
|
10.50 |
|
|
5.47 |
|
|
2015 |
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
13.50 |
|
$ |
7.46 |
|
|
Third Quarter |
|
|
18.90 |
|
|
7.09 |
|
|
Second Quarter |
|
|
18.04 |
|
|
9.01 |
|
|
First Quarter |
|
|
12.47 |
|
|
9.11 |
|
As of February 28, 2017, there were 23 holders of record of our common shares.
Dividends
Our Board of Directors has discretion whether or not to pay dividends. Among the factors our Board of Directors considers when determining whether or not to pay dividends are our earnings, capital requirements, financial condition, future business prospects and business conditions. We have never paid any cash dividends on our common shares and do not anticipate paying dividends in the foreseeable future. We intend to retain earnings, if any, to finance the development and expansion of our operations.
Securities Authorized for Issuance Under Equity Compensation Plans
The information contained under “Item 12—Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” of this Annual Report on Form 10‑K under the heading “Securities Authorized for Issuance Under Equity Compensation Plans” is incorporated herein by reference.
26
Performance Graph
The following graph compares the cumulative 5‑year total return of holders of the Company’s common stock with the cumulative total returns of the Russell 2000 index and the Nasdaq Biotechnology index. The graph tracks the performance of a $100 investment in our common stock and in each of the indexes (with reinvestment of all dividends, if any) on December 31, 2011 with relative performance tracked through December 31, 2016. The stock price performance included in this graph is not necessarily indicative of future stock price performance.
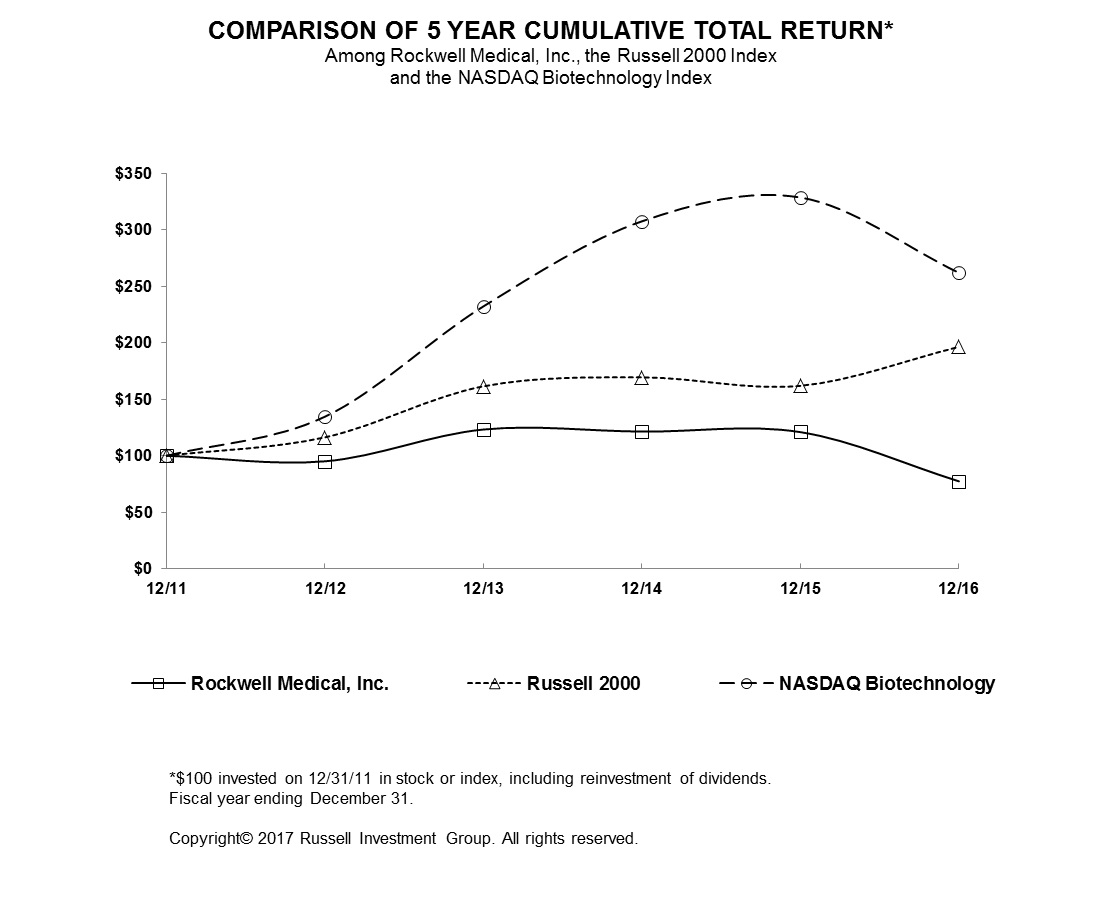
|
|
|
12/31/2011 |
|
12/31/2012 |
|
12/31/2013 |
|
12/31/2014 |
|
12/31/2015 |
|
12/31/2016 |
|
|
Rockwell Medical, Inc. |
|
100.00 |
|
95.04 |
|
123.26 |
|
121.37 |
|
120.90 |
|
77.33 |
|
|
Russell 2000 |
|
100.00 |
|
116.35 |
|
161.52 |
|
169.43 |
|
161.95 |
|
196.45 |
|
|
NASDAQ Biotechnology |
|
100.00 |
|
134.68 |
|
232.37 |
|
307.67 |
|
328.76 |
|
262.08 |
|
The information furnished under the heading “Stock Performance Graph” shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liabilities of that Section, and such information shall not be deemed incorporated by reference in any filing under the Securities Act of 1933, as amended.
27
Item 6. Selected Financial Data.
The financial data in the following tables should be read in conjunction with the consolidated financial statements and notes thereto and Management’s Discussion and Analysis of Financial Condition and Results of Operations included in this Form 10‑K.
|
|
|
For the Year Ended December 31, |
|
|||||||||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|
2012 |
|
|||||
|
Net sales |
|
$ |
53,284,166 |
|
$ |
55,350,702 |
|
$ |
54,188,444 |
|
$ |
52,379,543 |
|
$ |
49,842,392 |
|
|
Cost of sales |
|
|
46,531,648 |
|
|
46,412,848 |
|
|
45,643,231 |
|
|
45,720,323 |
|
|
43,148,965 |
|
|
Gross profit |
|
|
6,752,518 |
|
|
8,937,854 |
|
|
8,545,213 |
|
|
6,659,220 |
|
|
6,693,427 |
|
|
Income from continuing operations before interest expense and income taxes |
|
|
(20,208,729) |
|
|
(15,102,326) |
|
|
(17,559,101) |
|
|
(47,059,266) |
|
|
(54,262,082) |
|
|
Interest (expense) and Investment Income, net |
|
|
810,340 |
|
|
681,876 |
|
|
(3,768,056) |
|
|
(1,724,046) |
|
|
240,567 |
|
|
Income from continuing operations before income taxes(1) |
|
|
(19,398,389) |
|
|
(14,420,450) |
|
|
(21,327,157) |
|
|
(48,783,312) |
|
|
(54,021,515) |
|
|
Income taxes |
|
|
404,527 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Net income |
|
|
(19,802,916) |
|
|
(14,420,450) |
|
|
(21,327,157) |
|
|
(48,783,312) |
|
|
(54,021,515) |
|
|
Earnings per common share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
(0.39) |
|
$ |
(0.29) |
|
$ |
(0.52) |
|
$ |
(1.48) |
|
$ |
(2.65) |
|
|
Diluted |
|
$ |
(0.39) |
|
$ |
(0.29) |
|
$ |
(0.52) |
|
$ |
(1.48) |
|
$ |
(2.65) |
|
|
Weighted average number of common shares and common share equivalents |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
50,676,180 |
|
|
50,068,129 |
|
|
41,404,999 |
|
|
32,882,333 |
|
|
20,395,889 |
|
|
Diluted |
|
|
50,676,180 |
|
|
50,068,129 |
|
|
41,404,999 |
|
|
32,882,333 |
|
|
20,395,889 |
|
|
|
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|
2012 |
|
|||||
|
Total assets |
|
$ |
83,153,638 |
|
$ |
87,822,125 |
|
$ |
97,999,716 |
|
$ |
36,362,124 |
|
$ |
17,025,086 |
|
|
Current assets |
|
|
78,509,195 |
|
|
84,626,316 |
|
|
94,707,149 |
|
|
31,917,774 |
|
|
13,149,432 |
|
|
Current liabilities |
|
|
10,145,602 |
|
|
8,091,451 |
|
|
9,804,402 |
|
|
17,849,671 |
|
|
26,986,956 |
|
|
Working capital |
|
|
68,363,593 |
|
|
76,534,865 |
|
|
84,902,747 |
|
|
14,068,103 |
|
|
(13,837,524) |
|
|
Long term debt |
|
|
— |
|
|
— |
|
|
— |
|
|
17,916,914 |
|
|
— |
|
|
Shareholders’ equity(2) |
|
|
52,956,299 |
|
|
62,319,822 |
|
|
68,702,794 |
|
|
595,539 |
|
|
(9,961,870) |
|
|
Book value per outstanding common share |
|
$ |
1.03 |
|
$ |
1.21 |
|
$ |
1.37 |
|
$ |
0.01 |
|
$ |
(0.46) |
|
|
Common shares outstanding |
|
|
51,527,711 |
|
|
51,501,877 |
|
|
50,284,007 |
|
|
40,110,661 |
|
|
21,494,696 |
|
|
(1) |
Decrease in loss in since 2014 reflects a significant decrease in research and development expenses associated with completion of clinical trials for Triferic and elimination of interest expense in 2015 following repayment of our long term indebtedness. |
|
(2) |
There were no cash dividends paid during the periods presented. Shareholders’ equity reflects the proceeds of public and private offerings in 2014, 2013 and 2012. |
28
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Overview and Recent Developments
Rockwell is a fully-integrated pharmaceutical company targeting end-stage renal disease and chronic kidney disease with innovative products and services for the treatment of iron deficiency, secondary hyperparathyroidism and hemodialysis. We are also an established manufacturer and leader in delivering high-quality hemodialysis concentrates/dialysates to dialysis providers and distributors in the United States and abroad.
Our business focus is on developing unique, proprietary renal drug therapies. These novel renal drug therapies support disease management initiatives to improve the quality of life and care of dialysis patients and are designed to deliver safe and effective therapy, while decreasing drug administration costs and improving patient convenience and outcome.
Our strategy is to develop high potential drugs while expanding our dialysis products business. In January 2015, we received FDA approval to market Triferic our lead branded drug. Based on our clinical trial results, we believe Triferic has the potential to capture significant market share due to its unique attributes and clinical benefits, including savings on nursing administration time, potential to reduce expensive ESA treatments and excellent safety profile. Triferic has a unique mode of action and has proven to be both safe and effective in iron replacement and maintenance of hemoglobin.
In 2016, we had revenue of $53.3 million, a decrease of $2.1 million or 3.7% less than 2015. To date, nearly all of our revenue has been from our dialysis concentrate business. We supply approximately 25% of the United States domestic market with dialysis concentrates and we also supply dialysis concentrates to distributors serving a number of foreign countries, primarily in the Americas and the Pacific Rim.
Although we are actively marketing Triferic, sales of Triferic to date have not been material as we seek transitional add-on reimbursement status from CMS as discussed in “Item 1 – Business – General - Triferic.” Transitional add-on reimbursement status would offset the costs incurred by providers who adopt Triferic in place of current IV iron treatment alternatives and we believe will cause providers to more readily adopt Triferic and make it widely available to dialysis patients. Although CMS stated that Triferic reimbursement was included within the standard “bundled” reimbursement for the dialysis treatment, we anticipate that CMS will ultimately grant Triferic transitional add-on reimbursement status for the reasons noted in “Item 1 – Business – General - Triferic.” This transitional status, if granted, would last for two years to provide CMS with an opportunity to evaluate utilization of the new drug. Future bundled reimbursement base rates could be impacted by the changes in utilization of Triferic used during the transitional period.
We have built up a significant inventory of Triferic and the active pharmaceutical ingredient of Triferic in anticipation of receiving transitional add-on reimbursement status. However, there is no assurance when, if ever, it will be received. Until the reimbursement issue is finally resolved by CMS, we do not anticipate realizing significant revenues from Triferic. If we are not successful in obtaining transitional add-on reimbursement status for Triferic, we still believe it will be adopted by providers, but expect its adoption to occur at a slower rate. If we are unable to successfully commercialize Triferic and achieve sufficient sales volumes over the next one to two years, we may have to write off a portion of our inventory investment in Triferic, which would have an adverse effect on our results of operations.
We also intend to develop Triferic to address other clinical needs. We are continuing our development work on peritoneal dialysis, total parenteral nutrition and other indications. In addition, we initiated a clinical study on an orphan indication. We intend to license our other Triferic indications to partners who can optimize the commercial opportunities. We also continue to evaluate opportunities to in-license other products that will complement our product portfolio. We are also working to produce sufficient inventory to begin marketing Calcitriol, our generic injectable vitamin-D analogue. We are dependent upon CMOs to manufacture Calcitriol within specification and for the product to remain within specifications over its shelf life. Early batches produced by our CMO were initially within specified tolerances for all components but later, certain inactive ingredients (“excipients”) were determined to be out of specification at later testing dates. The stability issue was not related to the active pharmaceutical ingredient in Calcitriol, which is supplied by a different manufacturer. Subsequent manufacturing procedural modifications were expected to address the stability issue for these ingredients that were unrelated to drug efficacy. The FDA has advised
29
us that the manufacturing changes made by our vendor require FDA review under a process called a Prior Approval Supplement (“PAS”) that is used when manufacturing processes are significantly modified and the product manufactured using this process cannot be sold until the change is approved. While we believe our CMO has successfully addressed the earlier stability issues with Calcitriol, our CMO has had limited experience to date in producing stable batches that remain within specification over prescribed periods. Assuming we receive FDA approval of our PAS in the second half of 2017, we expect to begin marketing Calcitriol as soon as we have sufficient inventory that meets specifications.
Our global strategy is to out-license Triferic and Calcitriol for key international markets. We are actively pursuing the international development and licensing of Triferic in targeted foreign markets. In the first quarter of 2016, we entered into a licensing agreement with Wanbang Biopharmaceutical for the rights to commercialize our Triferic and Calcitriol products for ESRD patients in the People’s Republic of China. Under the terms of the Wanbang Agreement, we received an upfront payment of $4 million, which we are recognizing over the term of the agreement. We may also receive milestone payments of up to an additional $35 million over the life of the agreement in regulatory and revenue milestone payments plus ongoing earnings on product sales. We believe that China will ultimately become a significant market for the Company due to its large and growing dialysis population with a hemodialysis market projected by some industry participants to become the largest in the world over the next several years. It is a market that we also expect will provide an ideal opportunity for our other Triferic therapeutic indications.
In the third quarter of 2016, we entered into an exclusive license and manufacturing supply with ARAM Medical for the sale of Triferic and Calcitriol in the Kingdom of Saudi Arabia and a number of other countries in the Middle East for an initial term of 10 years. In consideration for the exclusive rights, ARAM Medical will pay us a licensing fee and a royalty on product sales, and has committed to annual minimum purchase quantities. ARAM Medical will also assume responsibility for all clinical and regulatory expenses for the countries covered by its agreement. Commercial sales activity will commence following regulatory approval. Rockwell retains manufacturing responsibilities for both Triferic and Calcitriol.
Under our Distribution Agreement with Baxter, Baxter holds the exclusive distribution rights for of our dialysis concentrates in the United States and certain foreign markets. The Distribution Agreement does not include our drug products. Rockwell receives a pre-defined gross profit margin on its concentrate products sold pursuant to the Distribution Agreement, which adjusts each year over the term of the agreement and is subject to an annual true-up. The Distribution Agreement requires Baxter to achieve certain minimum purchase requirements to maintain its exclusivity under the agreement.
In October 2016, we notified Baxter that they did not meet these minimum purchase requirements. Baxter disputed our assertion of a breach of the minimum purchase requirements and Baxter did not subsequently cure the deficiency in the prescribed period. The dispute regarding whether Baxter maintains exclusive distribution rights and the various other allegations of breach under the Distribution Agreement by Rockwell and by Baxter are the subject of a pending arbitration proceeding described under “Item 3 – Legal Proceedings.” For a more detailed description of the Distribution Agreement, see “Item 1—Business—Distribution Agreement with Baxter.”
Results of Operations
For the year ended December 31, 2016 compared to the year ended December 31, 2015
Sales
In 2016, our sales were $53.3 million compared to $55.4 million in 2015 a decrease of $2.1 million or 3.7%. Our domestic concentrate revenue increased $0.1 million or 0.2 % compared to 2015 due to increased concentrate product sales. Our international concentrate revenue for 2016 decreased $0.6 million or 8.0% compared to 2015 due to lower order volume. Third party contract manufacturing sales decreased $1.6 million compared to 2015 due to the completion of a manufacturing contract in 2015. We recognized deferred license revenue related to the Baxter Distribution Agreement of $2.1 million in both 2016 and 2015.
30
We continue to market to and educate our customers on Triferic while seeking to obtain transitional add-on reimbursement status for Triferic. Until the reimbursement issue is resolved with CMS, however, we expect Triferic sales will not be significant. Our drug business revenue was not significant in 2016. We recognized $0.3 million in deferred revenue related to Triferic licensing agreements in 2016.
Gross Profit
Our gross profit was $6.8 million in 2016, a decrease of $2.2 million compared to 2015. Gross profit margins were 12.7% in 2016 compared to 16.1% in 2015. Gross profit decreased by $1.7 million primarily due to approximately $1.5 million related to direct operating, regulatory, material, inventory and finished product expenses associated with our drug products. We also expensed $0.2 million in value added taxes paid on the $4 million in licensing payments received in connection with the Wanbang Agreement. The remainder of the decrease in gross profit was due to lower unit volumes on contract manufacturing sales and on international business.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $21.1 million in 2016 compared to $19.1 million in 2015 an increase of $2.0 million. The increase was primarily due to an increase in non-cash equity compensation charges of $1.5 million related to equity grants in prior years as no equity compensation plan grants were made to directors and officers in 2016 other than a grant to a new director. Other significant cost increases included increased legal fees of $0.7 million related to litigation expenses and increased marketing costs related to Triferic of $0.2 million. The increase was partially offset by the moratorium on medical device taxes for 2016 and 2017, resulting in a decrease of $0.4 million in 2016 compared to 2015.
Research and Development
We incurred product development and research costs related to the commercial development, patent approval and regulatory approval of new products, primarily Triferic, aggregating approximately $5.8 million and $5.0 million in 2016 and 2015, respectively. Costs incurred in 2016 and 2015 were largely related to testing of Triferic and included pharmacokinetic testing of Triferic for use in other indications, pediatric indications of Triferic, peritoneal dialysis, an orphan indication for Triferic, additional presentations of Triferic as well as other testing and development costs.
Interest Income, Net
Our net interest income was $0.8 million compared to net interest income of $0.7 million in 2015.
Income Tax Expense
We recognized approximately $0.4 million in income tax expense in 2016 compared to no income tax expense in 2015. Our income tax expense pertained to foreign income taxes paid related to license payments received under the Wanbang Agreement. The amount of foreign income tax paid can be credited against future United States tax liabilities and carried forward to offset future United States income tax liabilities.
We have substantial tax loss carryforwards from our losses in previous years. We have not recorded a federal income tax benefit from either our prior losses or our current year losses because we might not realize the carryforward benefit of the remaining losses.
For the year ended December 31, 2015 compared to the year ended December 31, 2014
Sales
In 2015, our sales were $55.4 million compared to $54.2 million in 2014 an increase of $1.2 million or 2.1%. Our domestic concentrate business sales increased 4.2% or $1.9 million in 2015 compared to 2014. We launched Triferic in late September 2015 and our net sales of Triferic were $0.2 million for 2015. Our international concentrate sales increased 1.3% or $0.1 million in 2015 over 2014. Our net revenue from third party contract manufacturing decreased
31
$1.0 million in 2015 compared to 2014 following cessation of contract manufacturing for a certain non‑hemodialysis customer.
As a result of our Distribution Agreement with Baxter, all domestic customer contracts for concentrate products that permitted assignment to Baxter without consent have been assigned to Baxter throughout 2015. Baxter subsequently began to invoice those customers following assignment. Our 2015 sales largely reflect the lower distributor prices paid by Baxter, such that our sales are lower on those accounts billed by Baxter than they were historically. Our 2015 sales were favorably impacted by the recognition of deferred license revenue under the Distribution Agreement of $2.1 million in 2015 compared to $0.5 million in 2014.
Gross Profit
Our gross profit was $8.9 million in 2015, an increase of $0.4 million or 4.6% compared to 2014. Gross profit margins were 16.1% in 2015 compared to 15.8% in 2014. Gross profit was favorably impacted by recognition of deferred license revenue under the Distribution Agreement of $2.1 million in 2015 compared to $0.5 million in 2014. Gross Profit was negatively impacted by lower sales on those accounts billed by Baxter following assumption of billing by Baxter.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $19.0 million in 2015 compared to $18.3 million in 2014. The increase of $0.7 million was primarily due to an increase in marketing expenses related to Triferic of $1.0 million. Total compensation including direct pay and equity compensation decreased $0.4 million.
Research and Development
We incurred product development and research costs related to the commercial development, patent approval and regulatory approval of new products, primarily Triferic, aggregating approximately $5.0 million and $7.8 million in 2015 and 2014, respectively. Costs incurred in 2014 were mostly related to Triferic and primarily for regulatory approval of Triferic while spending in 2015 included costs related to peritoneal dialysis, an orphan indication for Triferic, pediatric indications of Triferic, additional presentations of Triferic and other testing and development costs.
Interest Income, Net
Our net interest income was $0.7 million compared to a net interest expense of $3.8 million in 2014. The $4.5 million net increase in net interest income over interest expense was due to the repayment of all of our outstanding loan balance in the fourth quarter of 2014. We did not have any long term debt or loans outstanding as of December 31, 2015 or December 31, 2014.
Income Tax Expense
We have substantial tax loss carryforwards from our losses in earlier years. We have not recorded a federal income tax benefit from either our prior losses or our current year losses because we might not realize the carryforward benefit of the remaining losses.
Critical Accounting Estimates and Judgments
Our consolidated financial statements and accompanying notes are prepared in accordance with accounting principles generally accepted in the United States of America. These accounting principles require us to make estimates, judgments and assumptions that affect the reported amounts of revenues, expenses, assets, liabilities, and contingencies. All significant estimates, judgments and assumptions are developed based on the best information available to us at the time made and are regularly reviewed and updated when necessary. Actual results will generally differ from these estimates. Changes in estimates are reflected in our financial statements in the period of change based upon on‑going actual experience, trends, or subsequent realization depending on the nature and predictability of the estimates and contingencies.
32
Interim changes in estimates are generally applied prospectively within annual periods. Certain accounting estimates, including those concerning revenue recognition, allowance for doubtful accounts, inventory reserves, share based compensation, impairments of long‑lived assets, and accounting for income taxes, are considered to be critical in evaluating and understanding our financial results because they involve inherently uncertain matters and their application requires the most difficult and complex judgments and estimates. These are described below. For further information on our accounting policies, see Note 2 to our Consolidated Financial Statements.
Revenue recognition
Our policy is to recognize revenue consistent with authoritative guidance for revenue recognition including the provisions of the Financial Accounting Standards Board Accounting Standards Codification. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured.
Consistent with these guidelines we recognize revenue at the time we transfer title to our products to our customers which generally occurs when our products are delivered to our customer’s location consistent with our terms of sale. We recognize revenue for international shipments when title has transferred consistent with standard terms of sale.
We apply judgment as we analyze each element of our contractual agreements to determine appropriate revenue recognition. The terms of our contractual agreements may include milestone payments if specified research and development objectives are achieved, non-refundable licensing fees, milestone payments on sales or royalties from product sales.
When entering into an arrangement, we first determine whether the arrangement includes multiple deliverables and is subject to the accounting guidance in ASC subtopic 605-25, Multiple-Element Arrangements. If we determine that an arrangement includes multiple elements, we determine whether the arrangement should be divided into separate units of accounting and how the arrangement consideration should be measured and allocated among the separate units of accounting. An element qualifies as a separate unit of accounting when the delivered element has standalone value to the customer. Our arrangements do not include a general right of return relative to delivered elements. Any delivered elements that do not qualify as separate units of accounting are combined with other undelivered elements within the arrangement as a single unit of accounting. If the arrangement constitutes a single combined unit of accounting, we determine the revenue recognition method for the combined unit of accounting and recognize the revenue either on a straight-line basis or on a modified proportional performance method over the period from inception through the date the last deliverable within the single unit of accounting is delivered.
Non-refundable upfront license fees are recorded as deferred revenue and recognized into revenue over the estimated period of our substantive performance obligations. If we do not have substantive performance obligations, we recognize non-refundable upfront fees into revenue through the date the deliverable is satisfied. Analyzing the arrangement to identify deliverables requires the use of judgment and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation. In arrangements that include license rights and other non-contingent deliverables, such as participation in a steering committee, these deliverables do not have standalone value because the non-contingent deliverables are dependent on the license rights. That is, the non-contingent deliverables would not have value without the license rights, and only we can perform the related services. Upfront license rights and non-contingent deliverables, such as participation in a steering committee, do not have standalone value as they are not sold separately and they cannot be resold. In addition, when non-contingent deliverables are sold with upfront license rights, the license rights do not represent the culmination of a separate earnings process. As such, we account for the license and the non-contingent deliverables as a single combined unit of accounting. In such instances, the license revenue in the form of non-refundable upfront payments is deferred and recognized over the applicable relationship period.
For milestone payments based on sales and for royalties based on sales, we recognize revenue in the quarter that the information related to the sales becomes available and collectability is reasonably assured.
33
We generally recognize licensing fees over the term of the related license agreement. We received an upfront payment of $4 million pursuant to our License Agreement with Wanbang Biopharmaceutical Co., Ltd. in February 2016. We also executed a license agreement covering Saudi Arabia and a number of other countries in the Middle East. Deferred license revenue for our license agreements is being recognized over the term of the license agreements.
The initial payment of $20 million received pursuant to our Distribution Agreement with Baxter in October 2014 has been accounted for as deferred license revenue. Deferred license revenue is being recognized based on the proportion of product shipments to Baxter in each period to total expected sales volume for the term of the agreement.
We recognize other revenues at the time the related fees and or payments are earned.
Allowance for doubtful accounts
Accounts receivable are stated at invoice amounts. The carrying amount of trade accounts receivable is reduced by an allowance for doubtful accounts that reflects our best estimate of accounts that may not be collected. We review outstanding trade account receivable balances and based on our assessment of expected collections, we estimate the portion, if any, of the balance that may not be collected as well as a general valuation allowance for other accounts receivable based primarily on historical experience. All accounts or portions thereof deemed to be uncollectible are written off to the allowance for doubtful accounts. If we underestimate the allowance, we would incur a current period expense which could have a material adverse effect on earnings.
Inventories
Inventory is stated at the lower of cost or net realizable value. Cost is determined on the first‑in first‑out (FIFO) method. Our policy is to reserve for our drug product inventory that we determine is unlikely to be sold to, or if sold, unlikely to be utilized by our customers on or before its expiration date.
We evaluate how much of our current inventory we are likely to convert into cash over the next twelve months. If we have inventory that is in excess of our expectations to convert into cash over the next twelve months we will classify such inventory as non-current. We will evaluate such inventory for its net realizable value considering such factors as potential future product sales, marketability and future shelf life.
Share Based Compensation
We measure the cost of employee services received in exchange for equity awards, including stock options, based on the grant date fair value of the awards in accordance with ASC 718‑10, Compensation—Stock Compensation. The cost of equity based compensation is recognized as compensation expense over the vesting period of the awards.
We estimate the fair value of compensation involving stock options utilizing the Black‑Scholes option pricing model. This model requires the input of several factors such as the expected option term, expected volatility of our stock price over the expected option term, and an expected forfeiture rate, and is subject to various assumptions. We believe the valuation methodology is appropriate for estimating the fair value of stock options we grant to employees and directors which are subject to ASC 718‑10 requirements. These amounts are estimates and thus may not be reflective of actual future results or amounts ultimately realized by recipients of these grants.
Impairments of long‑lived assets
We account for impairment of long‑lived assets, which include property and equipment, amortizable and non‑amortizable intangible assets and goodwill, in accordance with authoritative accounting pronouncements. An impairment review is performed annually or whenever a change in condition occurs which indicates that the carrying amounts of assets may not be recoverable. Such changes may include changes in our business strategies and plans, changes to our customer contracts, changes to our product lines and changes in our operating practices. We use a variety of factors to assess the realizable value of long‑lived assets depending on their nature and use.
Goodwill is not amortized; however, it must be tested for impairment at least annually. The goodwill impairment analysis is based on the fair market value of our common shares. Amortization continues to be recorded for
34
other intangible assets with definite lives over the estimated useful lives. Intangible assets subject to amortization are reviewed for potential impairment whenever events or circumstances indicate that carrying amounts may not be recoverable based on future cash flows. If we determine that goodwill has been impaired, the change in value will be accounted for as a current period expense and could have a material adverse effect on earnings.
Accounting for income taxes
We estimate our income tax provision to recognize our tax expense and our deferred tax liabilities and assets for future tax consequences of events that have been recognized in our financial statements using current enacted tax laws. Deferred tax assets must be assessed based upon the likelihood of recoverability from future taxable income and to the extent that recovery is not likely, a valuation allowance is established. The allowance is regularly reviewed and updated for changes in circumstances that would cause a change in judgment about whether the related deferred tax asset may be realized. These calculations and assessments involve complex estimates and judgments because the ultimate tax outcome can be uncertain and future events unpredictable. If we determine that the deferred tax asset will be realized in the future, it may result in a material beneficial effect on earnings.
New Accounting Pronouncements
New accounting pronouncements are issued by the Financial Accounting Standards Board or other standard setting bodies that are adopted by us as of the specified effective date. Unless otherwise discussed, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption. For further discussion on recent accounting pronouncements, please see Note 2, “New Accounting Pronouncements,” to our consolidated financial statements included in this Annual Report on Form 10‑K for additional information.
Liquidity and Capital Resources
We believe we have adequate capital resources and substantial liquidity to pursue our business strategy. In addition to operating our concentrate business, our strategy is centered on developing, marketing and licensing high potential drug candidates including Triferic.
As of December 31, 2016, we had current assets of $78.5 million and net working capital of $68.4 million. We have approximately $57.9 million in cash and investments as of December 31, 2016. Our uses of cash have primarily been for research and product development, investments in inventory to support our drug product launches and for operating expenses. Operating activities used $12.5 million of cash in 2016, which included research and development expenses of $5.8 million and an increase of $6.1 million in inventory levels. We significantly increased our Triferic inventory in preparation of commercializing Triferic and believe we have adequate inventory to meet anticipated requirements. We received a net of $3.2 million in cash following execution of the Wanbang Agreement. Our capital expenditures were $0.4 million in 2016.
We anticipate that we will increase our accounts receivable as we increase our drug product sales and we may also increase inventories to a more modest degree as we commercialize Triferic and Calcitriol. We also expect to invest in research and product development in 2017 as we work to expand potential uses for Triferic. We believe that we have adequate capital resources to make these investments in accounts receivable, inventory and research and product development. We expect to generate positive cash flow from operations upon increased sales of our drug products.
We have no long term debt as of December 31, 2016 and do not expect to incur interest expense in 2017. Capital expenditures on our current facilities are not expected to materially exceed depreciation expense. We continue to plan to source our drug products from contract manufacturing organizations.
Our research and development expenses were reduced significantly following the completion of the clinical program for Triferic and FDA approval of Triferic. Future research and product development spending on the Triferic platform is expected to include clinical testing in connection with peritoneal dialysis, an orphan drug indication, pediatric indications and certain other indications. Future spending on such indications is expected to be minor in
35
relation to the Company’s cash resources. Our expected future cash investment for product launches is expected to be primarily related to accounts receivables in the near term.
The Company is in discussions with multiple potential business development partners to out‑license rights to Rockwell’s drug products outside the United States. Such licensing arrangements often include upfront fees, developmental milestone payments and royalties. If such licensing arrangements are negotiated for certain markets, we may receive such consideration in the future in addition to that which we are already entitled to receive under existing agreements. We are also considering other business development arrangements including joint ventures, partnerships and other transactions related to our products or other future products that we may develop or license.
Contractual Obligations
The following table details our contractual obligations as of December 31, 2016:
|
|
|
Payments due by period |
|
|||||||||||||
|
|
|
|
|
|
Less than |
|
|
|
|
|
|
|
More than |
|
||
|
Contractual Obligations |
|
Total |
|
1 year |
|
1 ‑ 3 years |
|
3 ‑ 5 years |
|
5 years |
|
|||||
|
Operating leases |
|
$ |
6,024,634 |
|
|
1,956,338 |
|
|
3,202,302 |
|
|
811,267 |
|
|
54,726 |
|
|
Purchase obligations |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
All other long term liabilities |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Total |
|
$ |
6,024,634 |
|
$ |
1,956,338 |
|
$ |
3,202,302 |
|
$ |
811,267 |
|
$ |
54,726 |
|
Off‑Balance Sheet Arrangements
We do not have any off‑balance sheet arrangements that have or are reasonably likely to have a material effect on our financial condition.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk.
Interest Rate Risk
We have invested $40.8 million in available for sale securities that are invested in short term bond funds which typically yield higher returns than the interest realized in money market funds. While these funds hold bonds of short duration, their market value is affected by changes in interest rates. Increases in interest rates will reduce the market value of bonds held in these funds and we may incur unrealized losses from the reduction in market value of the fund. If we liquidate our position in these funds, those unrealized losses may result in realized losses which may or may not exceed the interest and dividends earned from those funds. However, due to the short duration of these short term bond fund portfolios, we do not believe that a hypothetical 100 basis point increase or decrease in interest rates will have a material impact on the value of our investments.
Foreign Currency Exchange Rate Risk
Our international business is conducted in U.S. dollars with the exception of transactions by our subsidiary in India, which conducts business in Indian rupees and has had only minor transactional activity to date. It has not been our practice to hedge the risk of appreciation of the U.S. dollar against the predominant currencies of our trading partners. We have no significant foreign currency exposure to foreign supplied materials, and an immediate 10% strengthening or weakening of the U.S. dollar would not have a material impact on our shareholders’ equity or net income.
Item 8. Financial Statements.
The Consolidated Financial Statements of the Registrant and other information required by this item are set forth on pages F‑1 through F‑24 and incorporated herein by reference.
36
Item 9. Changes In and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure material information required to be disclosed in our reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required financial disclosure. In designing and evaluating the disclosure controls and procedures, we recognized that a control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. Management necessarily was required to apply its judgment in evaluating the cost‑benefit relationship of possible controls and procedures.
As of the end of the period covered by this report, we carried out an evaluation under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. We maintain internal control over financial reporting designed to provide reasonable, but not absolute, assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Therefore, internal control over financial reporting determined to be effective provides only reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, management evaluated the effectiveness of our internal control over financial reporting as of December 31, 2016. In making its assessment of internal control over financial reporting, management used the criteria described in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Our evaluation included documenting, evaluating and testing of the design and operating effectiveness of our internal control over financial reporting. Based on this evaluation, we concluded that the Company’s internal control over financial reporting was effective as of December 31, 2016.
Plante & Moran, PLLC, an independent registered public accounting firm, as auditors of our consolidated financial statements, has issued an attestation report on the effectiveness of our internal control over financial reporting as of December 31, 2016. Plante & Moran, PLLC’s report, which expresses an unqualified opinion on the effectiveness of our internal control over financial reporting, is included herein.
Changes in Internal Controls
There was no change in our internal control over financial reporting identified in connection with the Company’s evaluation of such internal controls that occurred during our fiscal quarter ended December 31, 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
37
Item 9B. Other Information.
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The required information will be contained in the Proxy Statement under the captions “Election of Directors” and “Section 16(a) Beneficial Ownership Reporting Compliance” and (excluding the Report of the Audit Committee) is incorporated herein by reference.
Item 11. Executive Compensation.
The required information will be contained in the Proxy Statement under the captions “Compensation of Executive Officers and Directors,” and “Compensation Committee” and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The required information will be contained in the Proxy Statement under the caption “Voting Securities and Principal Holders” and is incorporated herein by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table summarizes our compensation plans, including individual compensation arrangements, under which our equity securities are authorized for issuance as of December 31, 2016:
|
|
|
|
|
|
|
|
Number of securities |
|
|
|
|
Number of securities |
|
|
|
|
remaining available for |
|
|
|
|
to be issued upon |
|
Weighted‑average |
|
future issuance under |
|
|
|
|
|
exercise of |
|
exercise price of |
|
equity compensation plans |
|
|
|
|
|
outstanding options, |
|
outstanding options, |
|
(excluding securities |
|
|
|
Plan Category |
|
warrants and rights |
|
warrants and rights |
|
reflected in column (a)) |
|
|
|
|
|
(a) |
|
|
(b) |
|
(c) |
|
|
Equity compensation plans approved by security holders |
|
7,691,501 |
|
$ |
7.83 |
|
545,694 |
|
|
Equity compensation plans not approved by security holders |
|
— |
|
|
— |
|
— |
|
|
Total |
|
7,691,501 |
|
$ |
7.83 |
|
545,694 |
|
Item 13. Certain Relationships and Related Transactions and Director Independence.
The required information will be contained in the Proxy Statement under the captions “Independence” and “Related Party Transactions” and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services.
The required information will be contained in the Proxy Statement under the caption “Independent Accountants” and is incorporated herein by reference.
Item 15. Exhibits and Financial Statement Schedules.
(a) The financial statements and schedule filed herewith are set forth on the Index to Financial Statements and Schedule of the separate financial section of this annual report, which is incorporated herein by reference.
38
(b)Exhibits
The following documents are filed as part of this report or were previously filed and incorporated herein by reference to the filing indicated. Exhibits not required for this report have been omitted. Our Commission file number is 000‑23661.
| 3.1 |
|
Restated Articles of Incorporation, as amended as of May 1, 2013. (Company’s Form 10‑Q filed May 8, 2013). |
| 3.2 |
|
Amended and Restated Bylaws (Company’s Form 8‑K filed November 25, 2008). |
| 10.4 |
|
Licensing Agreement between the Company and Charak LLC and Dr. Ajay Gupta dated January 7, 2002 (with certain portions of the exhibit redacted pursuant to a confidential treatment order) (Company’s Form 10‑KSB filed April 1, 2002). |
| 10.11 |
|
Amending Agreement made the 16th day of January, 2006, by and between Dr. Ajay Gupta, Charak LLC and Rockwell Medical, Inc. (Company’s Form 10‑KSB filed March 31, 2006). |
|
*10.20 |
|
Form of Nonqualified Stock Option Agreement (Director Version) (Company’s Form 8‑K filed December 20, 2007). |
|
*10.21 |
|
Form of Nonqualified Stock Option Agreement (Employee Version) (Company’s Form 8‑K filed December 20, 2007). |
|
*10.54 |
|
Form of Restricted Stock Award Agreement June 2013 (Executive Version) (Company’s Form 10‑Q filed May 12, 2014). |
| 10.55 |
|
First Amended and Restated Products Purchase Agreement dated May 8, 2013, by and between Rockwell Medical, Inc. and DaVita Healthcare Partners, Inc. (with certain portions redacted pursuant to a confidential treatment order) (Company’s Form 10‑Q filed August 1, 2013). |
| 10.57 |
|
Exclusive Distribution Agreement, dated as of October 2, 2014, between the Company and Baxter Healthcare Corporation (with certain portions redacted pursuant to a confidential treatment order) (Company’s Form 10‑K filed March 3, 2015). |
| 10.58 |
|
Investment Agreement, dated as of October 2, 2014, between the Company and Baxter Healthcare Corporation (Company’s Form 10‑K filed March 3, 2015). |
|
*10.59 |
|
Amendment to October 1, 2014 Stock Option Agreement with Robert L. Chioini (Company’s Form 10‑K filed March 3, 2015). |
|
*10.60 |
|
Rockwell Medical, Inc. Amended and Restated 2007 Long Term Incentive Plan, as amended effective May 21, 2015 (Company’s Proxy Statement for the 2015 Annual Meeting of Shareholders filed on April 13, 2015). |
|
*10.61 |
|
Amendment to October 2, 2015 Stock Option Agreement with Robert L. Chioini (Company’s Form 10 K filed February 29, 2016). |
|
*10.62 |
|
Form of Restricted Stock Award Agreement October 2015 (Director Version) (Company’s Form 10 K filed February 29, 2016). |
| 14.1 |
|
Rockwell Medical, Inc. Code of Ethics (Company’s Proxy Statement filed April 23, 2004). |
| 21.1 |
|
List of Subsidiaries (Company’s Form SB‑2 (file No. 333‑31991)). |
| 23.1 |
|
Consent of Plante & Moran, PLLC. |
| 31.1 |
|
Certification of Chief Executive Officer Pursuant to Rule 13a‑14(a). |
| 31.2 |
|
Certification of Chief Financial Officer Pursuant to Rule 13a‑14(a). |
| 32.1 |
|
Certification of the Chief Executive Officer and Chief Financial Officer, Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes‑Oxley Act of 2002. |
|
101. INS |
|
XBRL Instance Document |
|
101. SCH |
|
XBRL Taxonomy Extension Schema |
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase |
39
|
101.DEF |
|
XBRL Taxonomy Extension Definition Database |
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase |
|
101.PRE |
|
XBRL Taxonomy Extension Presentation Linkbase |
*Current management contracts or compensatory plans or arrangements.
Item 16. Form 10-K Summary.
Not Applicable
40
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
ROCKWELL MEDICAL, INC. (Registrant) |
|
|
|
|
|
|
|
|
|
By: |
/s/ Robert L. Chioini |
|
|
|
|
Robert L. Chioini |
|
|
|
|
President and Chief Executive Officer |
|
|
|
Date: |
March 15, 2017 |
41
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of registrant and in the capacities and on the dates indicated.
|
SIGNATURE |
|
TITLE |
|
DATE |
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Robert L. Chioini |
|
President, Chief Executive Officer and |
|
March 15, 2017 |
|
Robert L. Chioini |
|
Director (Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vice President of Finance, Chief Financial |
|
|
|
/s/ Thomas E. Klema |
|
Officer, Treasurer and Secretary (Principal |
|
March 15, 2017 |
|
Thomas E. Klema |
|
Financial Officer and Principal Accounting |
|
|
|
|
|
Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Patrick J. Bagley |
|
Director |
|
March 15, 2017 |
|
Patrick J. Bagley |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Ronald D. Boyd |
|
Director |
|
March 15, 2017 |
|
Ronald D. Boyd |
|
|
||
|
|
|
|
|
|
|
/s/ Kenneth L. HOLT |
|
Director |
|
March 15, 2017 |
|
Kenneth L. Holt
|
|
|
||
|
|
|
|
|
|
|
/s/ Robin L. Smith |
|
Director |
|
March 15, 2017 |
|
Robin L. Smith
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
42
|
|
|
|
|
PAGE |
|
I. |
|
Consolidated Financial Statements for Rockwell Medical, Inc. and Subsidiaries |
|
|
|
|
|
|
F 2 - F 3 |
|
|
|
|
|
F-4 |
|
|
|
|
Consolidated Income Statements for the years ended December 31, 2016, 2015 and 2014 |
|
F-5 |
|
|
|
Consolidated Statements of Comprehensive Income for the years ended December 31, 2016, 2015 and 2014 |
|
F-6 |
|
|
|
|
F-7 |
|
|
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2016, 2015 and 2014 |
|
F-8 |
|
|
|
|
F-9 – F-24 |
|
|
|
|
S-1 |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders
Rockwell Medical, Inc. and Subsidiaries
We have audited the accompanying consolidated balance sheets of Rockwell Medical, Inc. and Subsidiaries (the Company) as of December 31, 2016 and 2015, and the related consolidated statements of income, comprehensive income, changes in shareholders’ equity, and cash flows for each of the years in the three‑year period ended December 31, 2016. Our audits of the basic financial statements also included the related financial statement Schedule II—Valuation and Qualifying Accounts. These financial statements and related schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and related schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Rockwell Medical, Inc. and Subsidiaries at December 31, 2016 and 2015, and the consolidated results of its operations and its cash flows for each of the years in the three‑year period ended December 31, 2016, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Rockwell Medical, Inc. and Subsidiaries’ internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated March 15, 2017 expressed an unqualified opinion on the effectiveness of internal control over financial reporting.
/s/ Plante & Moran, PLLC
Clinton Township, Michigan
March 15, 2017
F-2
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders
Rockwell Medical, Inc. and Subsidiaries
We have audited Rockwell Medical, Inc. and Subsidiaries’ (the Company) internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Rockwell Medical, Inc. and Subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Rockwell Medical, Inc. and Subsidiaries (the Company) as of December 31, 2016 and 2015, and the related consolidated statements of income, comprehensive income, changes in shareholders’ equity, and cash flows for each of the years in the three‑year period ended December 31, 2016 and related schedule and our report dated March 15, 2017 expressed an unqualified opinion thereon.
/s/ Plante & Moran, PLLC
Clinton Township, Michigan
March 15, 2017
F-3
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
As of December 31, 2016 and 2015
|
|
|
December 31, |
|
December 31, |
|
||
|
|
|
2016 |
|
2015 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
Cash and Cash Equivalents |
|
$ |
17,180,594 |
|
$ |
31,198,182 |
|
|
Investments Available for Sale |
|
|
40,759,703 |
|
|
39,482,732 |
|
|
Accounts Receivable, net of a reserve of $5,000 in 2016 and $75,000 in 2015 |
|
|
6,393,228 |
|
|
5,046,733 |
|
|
Inventory |
|
|
12,141,072 |
|
|
7,871,780 |
|
|
Other Current Assets |
|
|
2,034,598 |
|
|
1,026,889 |
|
|
Total Current Assets |
|
|
78,509,195 |
|
|
84,626,316 |
|
|
Property and Equipment, net |
|
|
1,391,575 |
|
|
1,646,568 |
|
|
Inventory, Non-Current |
|
|
1,826,554 |
|
|
— |
|
|
Intangible Assets |
|
|
4,382 |
|
|
165,657 |
|
|
Goodwill |
|
|
920,745 |
|
|
920,745 |
|
|
Other Non-current Assets |
|
|
501,187 |
|
|
462,839 |
|
|
Total Assets |
|
$ |
83,153,638 |
|
$ |
87,822,125 |
|
|
LIABILITIES AND SHAREHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
Accounts Payable |
|
$ |
5,858,234 |
|
$ |
3,995,216 |
|
|
Accrued Liabilities |
|
|
4,210,151 |
|
|
3,831,356 |
|
|
Customer Deposits |
|
|
77,217 |
|
|
264,879 |
|
|
Total Current Liabilities |
|
|
10,145,602 |
|
|
8,091,451 |
|
|
|
|
|
|
|
|
|
|
|
Deferred License Revenue |
|
|
20,051,737 |
|
|
17,410,852 |
|
|
|
|
|
|
|
|
|
|
|
Shareholders’ Equity: |
|
|
|
|
|
|
|
|
Common Shares, no par value, 51,527,711 and 51,501,877 shares issued and outstanding |
|
|
268,199,939 |
|
|
257,773,494 |
|
|
Accumulated Deficit |
|
|
(214,341,092) |
|
|
(194,538,176) |
|
|
Accumulated Other Comprehensive Income |
|
|
(902,548) |
|
|
(915,496) |
|
|
Total Shareholders’ Equity |
|
|
52,956,299 |
|
|
62,319,822 |
|
|
Total Liabilities And Shareholders’ Equity |
|
$ |
83,153,638 |
|
$ |
87,822,125 |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-4
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
CONSOLIDATED INCOME STATEMENTS
For The Years Ended December 31, 2016, 2015 and 2014
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Sales |
|
$ |
53,284,166 |
|
$ |
55,350,702 |
|
$ |
54,188,444 |
|
|
Cost of Sales |
|
|
46,531,648 |
|
|
46,412,848 |
|
|
45,643,231 |
|
|
Gross Profit |
|
|
6,752,518 |
|
|
8,937,854 |
|
|
8,545,213 |
|
|
Selling, General and Administrative |
|
|
21,120,901 |
|
|
19,078,867 |
|
|
18,320,720 |
|
|
Research and Product Development |
|
|
5,840,346 |
|
|
4,961,313 |
|
|
7,783,594 |
|
|
Operating Income (Loss) |
|
|
(20,208,729) |
|
|
(15,102,326) |
|
|
(17,559,101) |
|
|
Interest and Investment Income |
|
|
810,340 |
|
|
681,876 |
|
|
386,257 |
|
|
Interest (Expense) |
|
|
— |
|
|
— |
|
|
(4,154,313) |
|
|
Income (Loss) Before Income Taxes |
|
|
(19,398,389) |
|
|
(14,420,450) |
|
|
(21,327,157) |
|
|
Income Tax Expense |
|
|
(404,527) |
|
|
— |
|
|
— |
|
|
Net Income (Loss) |
|
$ |
(19,802,916) |
|
$ |
(14,420,450) |
|
$ |
(21,327,157) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic Earnings (Loss) per Share |
|
$ |
(0.39) |
|
$ |
(0.29) |
|
$ |
(0.52) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diluted Earnings (Loss) per Share |
|
$ |
(0.39) |
|
$ |
(0.29) |
|
$ |
(0.52) |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-5
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
For The Years Ended December 31, 2016, 2015 and 2014
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Net Income (Loss) |
|
$ |
(19,802,916) |
|
$ |
(14,420,450) |
|
$ |
(21,327,157) |
|
|
Unrealized Gain (Loss) on Available-for-Sale Investments |
|
|
13,619 |
|
|
(717,827) |
|
|
(230,088) |
|
|
Foreign Currency Translation Adjustments |
|
|
(671) |
|
|
— |
|
|
— |
|
|
Comprehensive Income (Loss) |
|
$ |
(19,789,968) |
|
$ |
(15,138,277) |
|
$ |
(21,557,245) |
|
The accompanying notes are an integral part of the consolidated financial statements.
F-6
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
For The Years Ended December 31, 2016, 2015 and 2014
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ACCUMULATED |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OTHER |
|
TOTAL |
|
||||||
|
|
|
COMMON SHARES |
|
PURCHASE WARRANTS |
|
ACCUMULATED |
|
COMPREHENSIVE |
|
SHAREHOLDER’S |
|
|||||||||||||
|
|
|
SHARES |
|
AMOUNT |
|
WARRANTS |
|
AMOUNT |
|
DEFICIT |
|
INCOME (LOSS) |
|
EQUITY |
|
|||||||||
|
Balance as of December 31, 2013 |
|
40,110,661 |
|
$ |
154,457,878 |
|
983,071 |
|
$ |
4,895,811 |
|
$ |
(158,790,569) |
|
$ |
32,419 |
|
$ |
595,539 |
|
||||
|
Net Loss |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
(21,327,157) |
|
|
— |
|
|
(21,327,157) |
|
||||
|
Unrealized Gain on Available-for-Sale Investments |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
(230,088) |
|
|
(230,088) |
|
||||
|
Issuance of Common Shares |
|
9,268,460 |
|
|
71,136,487 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
71,136,487 |
|
||||
|
Exercise of Purchase Warrants |
|
904,886 |
|
|
13,329,138 |
|
(983,071) |
|
|
(4,895,811) |
|
|
— |
|
|
— |
|
|
8,433,327 |
|
||||
|
Stock Option Based Expense |
|
— |
|
|
4,597,412 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
4,597,412 |
|
||||
|
Restricted Stock Amortization |
|
— |
|
|
5,497,274 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
5,497,274 |
|
||||
|
Balance as of December 31, 2014 |
|
50,284,007 |
|
$ |
249,018,189 |
|
— |
|
$ |
— |
|
$ |
(180,117,726) |
|
$ |
(197,669) |
|
$ |
68,702,794 |
|
||||
|
Net Loss |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
(14,420,450) |
|
|
— |
|
|
(14,420,450) |
|
||||
|
Unrealized Gain on Available-for-Sale Investments |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
(717,827) |
|
|
(717,827) |
|
||||
|
Issuance of Common Shares |
|
1,644,248 |
|
|
4,132,250 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
4,132,250 |
|
||||
|
Stock Tendered in Satisfaction of Tax Liabilities |
|
(426,378) |
|
|
(4,264,922) |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(4,264,922) |
|
||||
|
Stock Option Based Expense |
|
— |
|
|
5,193,481 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
5,193,481 |
|
||||
|
Restricted Stock Amortization |
|
— |
|
|
3,694,496 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
3,694,496 |
|
||||
|
Balance as of December 31, 2015 |
|
51,501,877 |
|
|
257,773,494 |
|
— |
|
|
— |
|
|
(194,538,176) |
|
|
(915,496) |
|
|
62,319,822 |
|
||||
|
Net Loss |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
(19,802,916) |
|
|
— |
|
|
(19,802,916) |
|
||||
|
Unrealized Gain on Available-for-Sale Investments |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
13,619 |
|
|
13,619 |
|
||||
|
Foreign Currency Rate Changes |
|
— |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
(671) |
|
|
(671) |
|
||||
|
Issuance of Common Shares |
|
25,834 |
|
|
80,161 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
80,161 |
|
||||
|
Stock Option Based Expense |
|
— |
|
|
5,984,524 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
5,984,524 |
|
||||
|
Restricted Stock Amortization |
|
— |
|
|
4,361,760 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
4,361,760 |
|
||||
|
Balance as of December 31, 2016 |
|
51,527,711 |
|
$ |
268,199,939 |
|
— |
|
$ |
— |
|
$ |
(214,341,092) |
|
$ |
(902,548) |
|
$ |
52,956,299 |
|
||||
The accompanying notes are an integral part of the consolidated financial statements.
F-7
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31, 2016, 2015 and 2014
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Cash Flows From Operating Activities: |
|
|
|
|
|
|
|
|
|
|
|
Net (Loss) |
|
$ |
(19,802,916) |
|
$ |
(14,420,450) |
|
$ |
(21,327,157) |
|
|
Adjustments To Reconcile Net Loss To Net Cash Used In Operating Activities: |
|
|
|
|
|
|
|
|
|
|
|
Depreciation and Amortization |
|
|
762,368 |
|
|
822,294 |
|
|
996,321 |
|
|
Share Based Compensation—Employees |
|
|
10,346,284 |
|
|
8,887,977 |
|
|
10,094,685 |
|
|
Restricted Stock Retained in Satisfaction of Tax Liabilities |
|
|
— |
|
|
(2,912,859) |
|
|
— |
|
|
Loss on Disposal of Assets |
|
|
8,168 |
|
|
5,281 |
|
|
7,338 |
|
|
Loss on Sale of Investments Available for Sale |
|
|
26,820 |
|
|
58,095 |
|
|
1,223 |
|
|
Amortization of Debt Issuance Costs |
|
|
— |
|
|
— |
|
|
882,716 |
|
|
Non-Cash Interest Expense |
|
|
— |
|
|
— |
|
|
874,942 |
|
|
Changes in Assets and Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
(Increase) in Accounts Receivable |
|
|
(1,162,469) |
|
|
(574,731) |
|
|
106,317 |
|
|
(Increase) in Inventory |
|
|
(6,095,846) |
|
|
(3,951,595) |
|
|
(1,120,537) |
|
|
(Increase) in Other Assets |
|
|
(1,230,084) |
|
|
(360,303) |
|
|
(13,466) |
|
|
(Decrease) in Accounts Payable |
|
|
1,863,018 |
|
|
(1,299,299) |
|
|
(3,391,638) |
|
|
(Decrease) in Other Liabilities |
|
|
191,134 |
|
|
(413,652) |
|
|
(2,345,486) |
|
|
Increase (decrease) in Deferred License Revenue |
|
|
(2,065,785) |
|
|
(2,081,668) |
|
|
19,492,520 |
|
|
Increase (decrease) in Deferred Drug License Revenue |
|
|
4,706,670 |
|
|
— |
|
|
— |
|
|
Changes in Assets and Liabilities |
|
|
(3,793,362) |
|
|
(8,681,248) |
|
|
12,727,710 |
|
|
Cash (Used) In Provided By Operating Activities |
|
|
(12,452,638) |
|
|
(16,240,910) |
|
|
4,257,778 |
|
|
Cash Flows From Investing Activities: |
|
|
|
|
|
|
|
|
|
|
|
Purchase of Investments Available for Sale |
|
|
(25,781,853) |
|
|
(21,800,000) |
|
|
(13,100,000) |
|
|
Sale of Investments Available for Sale |
|
|
24,491,677 |
|
|
1,468,656 |
|
|
4,976,000 |
|
|
Purchase of Equipment |
|
|
(355,264) |
|
|
(815,002) |
|
|
(684,593) |
|
|
Proceeds on Sale of Assets |
|
|
1,000 |
|
|
4,800 |
|
|
— |
|
|
Cash (Used In) Investing Activities |
|
|
(1,644,440) |
|
|
(21,141,546) |
|
|
(8,808,593) |
|
|
Cash Flows From Financing Activities: |
|
|
|
|
|
|
|
|
|
|
|
Proceeds from Issuance of Common Shares and Purchase Warrants |
|
|
80,161 |
|
|
2,780,187 |
|
|
79,569,815 |
|
|
Payments on Notes Payable and Capital Lease Obligations |
|
|
— |
|
|
— |
|
|
(21,100,000) |
|
|
Cash Provided By Financing Activities |
|
|
80,161 |
|
|
2,780,187 |
|
|
58,469,815 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effects of exchange rate changes |
|
|
(671) |
|
|
— |
|
|
— |
|
|
(Decrease) Increase In Cash |
|
|
(14,017,588) |
|
|
(34,602,269) |
|
|
53,919,000 |
|
|
Cash At Beginning Of Period |
|
|
31,198,182 |
|
|
65,800,451 |
|
|
11,881,451 |
|
|
Cash At End Of Period |
|
$ |
17,180,594 |
|
$ |
31,198,182 |
|
$ |
65,800,451 |
|
Supplemental Cash Flow Information:
|
|
|
2016 |
|
2015 |
|
2014 |
|
||||||||||
|
Interest Paid |
|
$ |
— |
|
$ |
— |
|
$ |
3,518,168 |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
Income Taxes Paid |
|
$ |
404,527 |
|
$ |
— |
|
$ |
— |
|
|||||||
The accompanying notes are an integral part of the consolidated financial statements
F-8
ROCKWELL MEDICAL, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS
Rockwell Medical, Inc. and Subsidiaries (collectively, “we”, “our”, “us”, or the “Company”) is a fully-integrated pharmaceutical company targeting end-stage renal disease and chronic kidney disease with innovative products for the treatment of iron deficiency, secondary hyperparathyroidism and hemodialysis. We are also an established manufacturer and leader in delivering high-quality hemodialysis concentrates/dialysates to dialysis providers and distributors in the United States and abroad.
We are currently developing unique, proprietary renal drug therapies. These novel renal drug therapies support disease management initiatives to improve the quality of life and care of dialysis patients and are designed to deliver safe and effective therapy, while decreasing drug administration costs and improving patient convenience and outcome. We have obtained global licenses for certain dialysis related drugs which we are developing and planning to market.
We manufacture, sell and distribute hemodialysis concentrates and other ancillary medical products and supplies used in the treatment of patients with End Stage Renal Disease, or “ESRD”. We supply our products to dialysis providers and distributors who treat patients with kidney disease. Our concentrate products are used to remove waste and replace needed nutrients in the blood of dialysis patients during their hemodialysis treatment. We primarily sell our products in the United States.
We are regulated by the Federal Food and Drug Administration (“FDA”) under the Federal Drug and Cosmetics Act, as well as by other federal, state and local agencies. We hold several FDA product approvals including both drugs and medical devices.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
Our consolidated financial statements include our accounts and the accounts for our wholly owned subsidiaries, Rockwell Transportation, Inc. and Rockwell Medical India Private Limited. Rockwell Medical India Private Limited was formed in 2016 for the purpose of conducting certain commercial activities in India.
All intercompany balances and transactions have been eliminated in consolidation.
Revenue Recognition
Our policy is to recognize revenue consistent with authoritative guidance for revenue recognition including the provisions of the Financial Accounting Standards Board Accounting Standards Codification. We recognize revenue when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller's price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured.
Consistent with these guidelines we recognize revenue at the time we transfer title to our products to our customers which generally occurs when our products are delivered to our customer’s location consistent with our terms of sale. We recognize revenue for international shipments when title has transferred consistent with standard terms of sale.
We apply judgment as we analyze each element of our contractual agreements to determine appropriate revenue recognition. The terms of our contractual agreements may include milestone payments if specified research and development objectives are achieved, non-refundable licensing fees, milestone payments on sales or royalties from product sales.
F-9
When entering into an arrangement, we first determine whether the arrangement includes multiple deliverables and is subject to the accounting guidance in ASC subtopic 605-25, Multiple-Element Arrangements. If we determine that an arrangement includes multiple elements, we determine whether the arrangement should be divided into separate units of accounting and how the arrangement consideration should be measured and allocated among the separate units of accounting. An element qualifies as a separate unit of accounting when the delivered element has standalone value to the customer. Our arrangements do not include a general right of return relative to delivered elements. Any delivered elements that do not qualify as separate units of accounting are combined with other undelivered elements within the arrangement as a single unit of accounting. If the arrangement constitutes a single combined unit of accounting, we determine the revenue recognition method for the combined unit of accounting and recognize the revenue either on a straight-line basis or on a modified proportional performance method over the period from inception through the date the last deliverable within the single unit of accounting is delivered.
Non-refundable upfront license fees are recorded as deferred revenue and recognized into revenue over the estimated period of our substantive performance obligations. If we do not have substantive performance obligations, we recognize non-refundable upfront fees into revenue through the date the deliverable is satisfied. Analyzing the arrangement to identify deliverables requires the use of judgment and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation. In arrangements that include license rights and other non-contingent deliverables, such as participation in a steering committee, these deliverables do not have standalone value because the non-contingent deliverables are dependent on the license rights. That is, the non-contingent deliverables would not have value without the license rights, and only we can perform the related services. Upfront license rights and non-contingent deliverables, such as participation in a steering committee, do not have standalone value as they are not sold separately and they cannot be resold. In addition, when non-contingent deliverables are sold with upfront license rights, the license rights do not represent the culmination of a separate earnings process. As such, we account for the license and the non-contingent deliverables as a single combined unit of accounting. In such instances, the license revenue in the form of non-refundable upfront payments is deferred and recognized over the applicable relationship period.
For milestone payments based on sales and for royalties based on sales, we recognize revenue in the quarter that the information related to the sales becomes available and collectability is reasonably assured.
We recognize drug licensing fees over the term of the related license agreement. We received an upfront payment of $4 million pursuant to our License Agreement with Wanbang Biopharmaceutical Co., Ltd. (“Wanbang”), a subsidiary of Shanghai Fosun Pharmaceutical (Group) Co., Ltd. and we also recorded deferred drug license revenue related to a license for the Middle East in 2016 with ARAM Medical. Deferred drug license revenue for our drug license agreements is being recognized over the term of those license agreements and we recognized drug license revenue of $0.3 million in 2016.
The initial payment of $20 million received pursuant to our long-term Exclusive Distribution Agreement (the “Distribution Agreement”) with Baxter Healthcare Corporation (“Baxter”) in October 2014 has been accounted for as deferred license revenue. Deferred license revenue is being recognized based on the proportion of product shipments to Baxter in each period to total expected sales volume for the term of the agreement.
We recognize other revenues at the time the related fees and or payments are earned.
Shipping and Handling Revenue and Costs
Our products are generally priced on a delivered basis with the price of delivery included in the overall price of our products which is reported as sales. Separately identified freight and handling charges are also included in sales.
We include shipping and handling costs, including expenses of Rockwell Transportation, Inc., in cost of sales.
Cash and Cash Equivalents
We consider cash on hand, money market funds and unrestricted certificates of deposit with an original maturity of 90 days or less as cash and cash equivalents.
F-10
Investments Available for Sale
Investments Available for Sale are short-term investments, consisting principally of investments in short term duration bond funds, and are stated at fair value based upon observed market prices (Level 1 in the fair value hierarchy). Unrealized holding gains or losses on these securities are included in accumulated other comprehensive income (loss). Realized gains and losses, including declines in value judged to be other-than-temporary on available-for-sale securities are included as a component of other income or expense.
Management evaluates securities for other-than-temporary impairment (“OTTI”) on a quarterly basis, and more frequently when conditions warrant such an evaluation. When evaluating investment securities, consideration is given to the length of time and the extent to which the fair value has been less than cost, the financial condition and near-term prospects of the issuer, and whether the Company has the intent to sell the security or more likely than not will be required to sell the security before its anticipated recovery. The assessment of whether an OTTI exists involves a high degree of subjectivity and judgment and is based on the information available to management at a point in time.
Accounts Receivable
Accounts receivable are stated at invoice amounts. The carrying amount of trade accounts receivable is reduced by an allowance for doubtful accounts that reflects our best estimate of accounts that may not be collected. We review outstanding trade accounts receivable balances and based on our assessment of expected collections, we estimate the portion, if any, of the balance that may not be collected as well as a general valuation allowance for other accounts receivable based primarily on historical experience. All accounts or portions thereof deemed to be uncollectible are written off to the allowance for doubtful accounts.
Inventory
Inventory is stated at the lower of cost or net realizable value. Cost is determined on the first‑in first‑out (FIFO) method. Inventory that is not expected to be converted to cash over the next year is classified as non-current. Our policy is to reserve for our drug product inventory that we determine is unlikely to be sold to, or if sold, unlikely to be utilized by our customers on or before its expiration date.
Property and Equipment
Property and equipment are recorded at cost. Expenditures for normal maintenance and repairs are charged to expense as incurred. Property and equipment are depreciated using the straight‑line method over their useful lives, which range from three to ten years. Leasehold improvements are amortized using the straight‑line method over the shorter of their useful lives or the related lease term.
Licensing Fees
License fees related to the technology, intellectual property and marketing rights for Triferic covered under certain issued patents have been capitalized and are being amortized over the life of the related patents which is generally 17 years.
Goodwill, Intangible Assets and Long Lived Assets
The recorded amounts of goodwill and other intangibles from prior business combinations are based on management’s best estimates of the fair values of assets acquired and liabilities assumed at the date of acquisition. Goodwill is not amortized; however, it must be tested for impairment at least annually. Amortization continues to be recorded for other intangible assets with definite lives over their estimated useful lives. Intangible assets subject to amortization are reviewed for potential impairment whenever events or circumstances indicate that carrying amounts may not be recoverable.
An impairment review of goodwill, intangible assets, and property and equipment is performed annually or whenever a change in condition occurs which indicates that the carrying amounts of assets may not be recoverable. Such changes may include changes in our business strategies and plans, changes to our customer contracts, changes to our
F-11
product lines and changes in our operating practices. We use a variety of factors to assess the realizable value of long‑lived assets depending on their nature and use.
The useful lives of other intangible assets are based on management’s best estimates of the period over which the assets are expected to contribute directly or indirectly to our future cash flows. Management annually evaluates the remaining useful lives of intangible assets with finite useful lives to determine whether events and circumstances warrant a revision to the remaining amortization periods. It is reasonably possible that management’s estimates of the carrying amount of goodwill and the remaining useful lives of other intangible assets may change in the near term.
Income Taxes
We account for income taxes in accordance with the provisions of ASC 740‑10, Income Taxes. A current tax liability or asset is recognized for the estimated taxes payable or refundable on tax returns for the year. Deferred tax liabilities or assets are recognized for the estimated future tax effects of temporary differences between book and tax accounting and operating loss and tax credit carryforwards. A valuation allowance is established for deferred tax assets if we determine it to be more likely than not that the deferred tax asset will not be realized.
The effects of tax positions are generally recognized in the financial statements consistent with amounts reflected in returns filed, or expected to be filed, with taxing authorities. For tax positions that the Company considers to be uncertain, current and deferred tax liabilities are recognized, or assets derecognized, when it is probable that an income tax liability has been incurred and the amount of the liability is reasonably estimable, or when it is probable that a tax benefit, such as a tax credit or loss carryforward, will be disallowed by a taxing authority. The amount of unrecognized tax benefits related to current tax positions is insignificant. The Company recognizes interest and penalties accrued related to unrecognized tax benefits as income tax expense.
Research and Product Development
We recognize research and product development costs as expenses as incurred. We incurred product development and research costs related to the commercial development, patent approval and regulatory approval of new products, including Triferic and for other indications of Triferic, aggregating approximately $5,840,000, $4,961,000 and $7,784,000 in 2016, 2015 and 2014, respectively.
Share Based Compensation
We measure the cost of employee services received in exchange for equity awards, including stock options, based on the grant date fair value of the awards in accordance with ASC 718‑10, Compensation — Stock Compensation. The cost of equity based compensation is recognized as compensation expense over the vesting period of the awards.
We estimate the fair value of compensation involving stock options utilizing the Black‑Scholes option pricing model. This model requires the input of several factors such as the expected option term, expected volatility of our stock price over the expected option term, and an expected forfeiture rate, and is subject to various assumptions. We believe the valuation methodology is appropriate for estimating the fair value of stock options we grant to employees and directors which are subject to ASC 718‑10 requirements. These amounts are estimates and thus may not be reflective of actual future results or amounts ultimately realized by recipients of these grants.
Employee Retirement Plans
We are the sponsor of a non‑contributory 401(k) Employee Savings Plan.
Earnings per Share
We compute our basic earnings (loss) per share using weighted average shares outstanding for each respective period. Diluted earnings per share also reflect the weighted average impact from the date of issuance of all potentially dilutive securities, consisting of stock options and common share purchase warrants, unless inclusion would have had an
F-12
anti‑dilutive effect. Actual weighted average shares outstanding used in calculating basic and diluted earnings per share were:
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Basic Weighted Average Shares Outstanding |
|
50,676,180 |
|
50,068,129 |
|
41,404,999 |
|
|
Effect of Dilutive Securities |
|
— |
|
— |
|
— |
|
|
Diluted Weighted Average Shares Outstanding |
|
50,676,180 |
|
50,068,129 |
|
41,404,999 |
|
For 2016, 2015 and 2014, the dilutive effect of stock options, unvested restricted share grants and common share purchase warrants have not been included in the average shares outstanding for the calculation of diluted loss per share as the effect would be anti-dilutive as a result of our net loss in these periods. The table below summarizes potentially dilutive securities.
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Stock Options |
|
7,691,501 |
|
7,759,002 |
|
6,885,083 |
|
|
Range of Exercise Prices of Stock Options |
|
$3.09 - $11.49 |
|
$3.09 - $11.49 |
|
$3.09 - $10.20 |
|
|
Unvested Restricted Common Shares |
|
850,000 |
|
850,000 |
|
740,000 |
|
|
Common Share Purchase Warrants |
|
None |
|
None |
|
None |
|
|
Range of Exercise Prices of Warrants |
|
n/a |
|
n/a |
|
n/a |
|
Other Comprehensive Income (Loss)
Accounting principles generally require that recognized revenue, expenses, gains, and losses be included in net income. Certain changes in assets and liabilities, however, such as unrealized gains and losses on available for sale securities, are reported as a direct adjustment to the equity section of the balance sheet. Such items, along with net income (loss), are considered components of comprehensive income (loss). Accumulated Other Comprehensive Income (Loss) consists almost entirely of unrealized gains and losses on available‑for‑sale investment securities. We also record foreign currency translation adjustments to Other Comprehensive Income (Loss). However, such amounts were insignificant in 2016.
Estimates in Preparation of Financial Statements
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and reported amounts of revenues and expenses during the period. Actual results could differ from those estimates.
New Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606), which will supersede the current revenue recognition requirements in Topic 605, Revenue Recognition. The standard’s core principle is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. The new guidance initially was effective for years beginning January 1, 2017, but on July 9, 2015 the FASB deferred the effective date and, as a result, the new guidance is effective for the year beginning January 1, 2018. The standard permits the use of either the retrospective or cumulative effect transition method. The Company is in the process of evaluating how the new revenue recognition standard could impact the financial statements and disclosures. For the majority of our sales transactions, the new standard is not expected to significantly change the timing of revenue recognition; however, we are still analyzing our licensing arrangements to determine the impact of the new standard. The new standard will also require expanded disclosures surrounding revenue in the notes to the financial statements.
F-13
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). The amendments in this ASU revise the accounting related to lessee accounting. Under the new guidance, lessees will be required to recognize a lease liability and a right-of-use asset for all leases. The amendments in this ASU are effective for the Company beginning on January 1, 2019 and should be applied through a modified retrospective transition approach for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. Early adoption is permitted. We anticipate this standard will have a material impact on our consolidated balance sheets. However, we do not believe adoption will have a material impact on our consolidated income statements. Upon implementation, the Company's lease payment obligations will be recognized at their estimated present value along with a corresponding right-of-use asset. Lease expense recognition will be generally consistent with current practice.
In July 2015, the FASB issued ASU 2015-11, Inventory (Topic 330) Related to Simplifying the Measurement of Inventory, which applies to all inventory except inventory that is measured using last-in, first-out (LIFO) or the retail inventory method. Inventory measured using first-in, first-out (FIFO) or average cost is covered by the new guidance and should be measured at the lower of cost and net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. The amendments will be applied prospectively for fiscal years beginning after December 15, 2016. The Company does not expect a significant change upon implementation as cost is expected to be lower than net realizable value.
3. FAIR MARKET VALUE MEASUREMENTS
Accounting standards require certain assets and liabilities be reported at fair value in the financial statements and provides a framework for establishing that fair value. The framework for determining fair value is based on a hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
|
Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
|
|
Level 2: |
Observable prices that are based on inputs not quoted in active markets, but corroborated by market data. |
|
Level 3: |
Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
In determining fair value, we utilize valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible as well as considering counterparty credit risk in its assessment of fair value. The following methods, assumptions, and valuation techniques were used to measure different financial assets and liabilities at fair value and in estimating its fair value disclosures for financial instruments.
Cash and Cash Equivalents: The carrying amounts reported in the consolidated balance sheets for cash and cash equivalents are deemed to approximate fair value
Investment Securities: Fair values for investment securities are determined by quoted market prices if available.
Accounts Receivable, Accounts Payable and Accrued Liabilities: The fair value of trade receivables and payables approximate their carrying amounts due to the short duration before collection or payment.
F-14
Based on the foregoing methods and assumptions, the carrying value and fair value of the Company’s financial instruments other than trade receivables and payables are as follows (in thousands):
|
|
|
Carrying |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
value |
|
Fair value |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
|||||
|
As of December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Financial assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
17,181 |
|
$ |
17,181 |
|
$ |
17,181 |
|
$ |
— |
|
$ |
— |
|
|
Investment securities available for sale |
|
|
40,760 |
|
|
40,760 |
|
|
40,760 |
|
|
— |
|
|
— |
|
|
As of December 31, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Financial assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
31,198 |
|
$ |
31,198 |
|
$ |
31,198 |
|
$ |
— |
|
$ |
— |
|
|
Investment securities available for sale |
|
|
39,483 |
|
|
39,483 |
|
|
39,483 |
|
|
— |
|
|
— |
|
The Company also has certain non‑financial assets that under certain conditions are subject to measurement at fair value on a non‑recurring basis. No such measurements were required in 2016 or 2015.
4. INVESTMENTS IN AVAILABLE FOR SALE SECURITIES
As of December 31, 2016, we held investments in available for sale securities in several short term bond funds. These funds generally held high credit quality short term debt instruments. These debt instruments were subject to changes in fair market value due to changes in interest rates. The market value of these investments was $40,759,703 as of December 31, 2016. In 2016, we purchased securities with a market value of $25,781,853 and had unrealized losses of $901,877 as of December 31, 2016. In 2016, we sold securities with a market value of $24,491,677 with an average cost basis of $24,518,497. We had realized gains of $156,461 and realized losses of $183,281.
As of December 31, 2015, we held investments in available for sale securities in several short term bond funds. These funds generally held high credit quality short term debt instruments. These debt instruments were subject to changes in fair market value due to changes in interest rates. As of December 31, 2015, the market value of investments in available for sale securities was $39,482,732. In 2015, we purchased securities with a market value of $21,800,000 and had unrealized gains of $69,877 and unrealized losses of $985,374 as of December 31, 2015. In 2015, we sold securities with a market value of $1,469,000 with an average cost basis of $1,527,095. We realized losses of $58,095 from sales of available for sale securities.
5. SIGNIFICANT MARKET SEGMENTS AND CUSTOMERS
We operate in one market segment, the hemodialysis market, which involves the manufacture, sale and distribution of hemodialysis products to hemodialysis clinics including pharmaceutical, dialysis concentrates, dialysis kits and other ancillary products used in the dialysis process. In October 2014, we entered into the Distribution Agreement with Baxter pursuant to which Baxter received exclusive distribution rights for our concentrate products in the United States. Rockwell domestic customer contracts for the supply of dialysis concentrate products that permitted assignment to Baxter without consent have been assigned to Baxter. As a result, for the years ended December 31, 2016 and 2015, our direct sales to Baxter aggregated approximately 24% and 28% of sales, respectively and we had a receivable from Baxter of $2,430,159 and $2,088,000 as of December 31, 2016 and 2015, respectively.
For the years ended December 31, 2016, 2015 and 2014, one customer, DaVita Healthcare Partners, Inc., accounted for 52% of our sales in 2016, 48% of our sales in 2015 and 49% of our sales in 2014. Our accounts receivable from this customer were $2,224,046 and $2,156,000 as of December 31, 2016 and 2015, respectively. DaVita and Baxter and the accounts administered by Baxter are important to our business, financial condition and results of operations. The loss of any significant accounts could have a material adverse effect on our business, financial condition and results of operations. No other customers accounted for more than 10% of our sales in any of the last three years.
The majority of our international sales in each of the last three years were sales to domestic distributors that were resold to end users outside the United States. Our sales to foreign customers and distributors were less than 5% of our total sales in 2016, 2015 and 2014. Our total international sales, including sales to domestic distributors for resale outside the United States, aggregated 12%, 13% and 13%, of overall sales in 2016, 2015 and 2014, respectively.
F-15
6. DISTRIBUTION AGREEMENT
As of October 2, 2014, we entered into the Distribution Agreement with Baxter, pursuant to which Baxter became the Company’s exclusive agent for sales, marketing and distribution activities for the Company’s hemodialysis concentrate and ancillary products in the United States and various foreign countries for an initial term of 10 years. The Distribution Agreement does not include any of the Company’s drug products. The Company will retain sales, marketing and distribution rights for its hemodialysis concentrate products in specified foreign countries in which the Company has an established commercial presence. During the term of the Distribution Agreement, Baxter has agreed not to manufacture or sell any competitive concentrate products in the United States hemodialysis market, other than specified products.
Pursuant to the Distribution Agreement, Baxter paid the Company $20 million in cash in October 2014 (the “Upfront Fee”). The Upfront Fee has been deferred and is being recognized as revenue based on the proportion of product shipments to Baxter in each period to total expected sales volume over the term of the Distribution Agreement. The Company recognized revenue associated with the Upfront Fee totaling $2,065,785 for the year ended December 31, 2016, $2,081,668 for the year ended December 31, 2015 and $507,480 for the year ended December 31, 2014.
Under the Distribution Agreement, Baxter purchases products from the Company at established gross margin-based prices per unit, adjusted each year during the term. The Company continues to manage customer service, transportation and certain other functions for its current customers on Baxter’s behalf through at least December 31, 2017, in exchange for which Baxter will pay the Company an amount equal to the Company’s related costs to provide such functions plus a slight mark-up.
The Distribution Agreement also requires Baxter to meet minimum annual gallon-equivalent purchase levels, subject to a cure period and certain other relief, in order to maintain its exclusive distribution rights. The minimum purchase levels increase each year over the term of the Distribution Agreement. Orders in any contract year that exceed the minimum will be carried forward and applied to future years’ minimum requirements. The Distribution Agreement also contains provisions governing the operating relationship between the parties, the Company’s obligations to maintain specified manufacturing capacity and quality levels, remedies, as well as representations, warranties and indemnification obligations of the parties.
Either party may terminate the Distribution Agreement upon the insolvency or material breach of the other party or in the event of a force majeure. In addition, Baxter may also terminate the Distribution Agreement at any time upon 270 days’ prior written notice to the Company or if (1) prices increase beyond certain thresholds and notice is provided within 45 days after the true up payment is due for the year in which the price threshold is exceeded, (2) a change of control of the Company occurs and 270 days’ notice is provided, or (3) upon written notice that Baxter has been enjoined by a court of competent jurisdiction from selling in the United States any product covered by the Distribution Agreement due to a claim of intellectual property infringement or misappropriation relating to such product. If Baxter terminates the Distribution Agreement under the discretionary termination or the price increase provisions, it would be subject to a limited non-compete obligation in the United States with respect to certain products for a period of two years.
If a “Refund Trigger Event” occurs, the Company would be obligated to repay a portion of the Upfront Fee and Facility Fee (described below) as follows: 50% if the event occurs prior to December 31, 2016, 33% if the event occurs in 2017 or 2018, and 25% if the event occurs in 2019, 2020 or 2021. A “Refund Trigger Event” means any of the following: (1) a change of control of the Company involving any of certain specified companies; (2) a termination by Baxter due to the Company’s bankruptcy or breach, or due to price increases that exceed the stated thresholds; (3) a termination by either party due to a force majeure; (4) settlement or adjudication of any claim, action or litigation relating to a covered product that materially and adversely affects Baxter’s commercialization of the product; and (5) any regulatory action or ruling relating to a covered product that materially and adversely affects Baxter’s commercialization of the product. In addition, if Baxter terminates the Distribution Agreement because Baxter has been enjoined by a court of competent jurisdiction from selling in the United States any product covered by the Distribution Agreement due to a claim of intellectual property infringement or misappropriation relating to such product prior to the end of 2018, Baxter would be entitled to a refund of up to $10 million, or $6.6 million if the termination occurs in 2019. In no event would Baxter be entitled to more than one refund payment
F-16
The Distribution Agreement also required the Company to prepay its outstanding secured long-term indebtedness within 180 days and prohibits the Company from entering into a subsequent contract encumbering the assets used in the Company’s concentrate business without the prior written consent of Baxter.
Baxter has also agreed to pay the Company up to $10 million (the “West Coast Facility Fee”) to build and operate a new manufacturing facility located in the Pacific time zone to service customers in the Western United States. The West Coast Facility Fee will be reduced to the extent that the facility is not operational within 12 months after the start of construction. Except for any leased components, the Company will own the facility when completed.
The Distribution Agreement may be extended an additional five years by Baxter if Baxter achieves a specified sales target and pays an extension fee of $7.5 million. If the first extension occurs, the Distribution Agreement term may later be extended an additional five years at Baxter’s option at no additional cost.
On September 12, 2016, Baxter initiated an arbitration proceeding against Rockwell under the Distribution Agreement. Baxter alleges that Rockwell has breached the Distribution Agreement in various respects associated with its dealings with customers, its allocation of expenses and its true-up notices, and by improperly threatening to build a West Coast facility. Baxter seeks declaratory relief giving Baxter the right to terminate the Distribution Agreement and recover up to $10 million of the upfront fee, injunctive relief to prevent Rockwell from establishing a West Coast facility, and unspecified damages.
Rockwell filed a response denying all of Baxter’s claims of breach and wrongdoing, and has counterclaimed that Baxter is itself in breach of the Distribution Agreement for failing to pay substantial accounts receivable and for repudiating its obligation to pay the West Coast facility fee of up to $10 million. Rockwell is seeking damages, declaratory, injunctive and other equitable relief, as well as interest, costs and attorney fees.
In addition, in October 2016, Rockwell gave notice to Baxter that it breached the minimum purchase requirement for the contract year ended October 2, 2016 and that we intended to cause its distribution rights to become non-exclusive unless it cured the shortfall within the 30-day period specified in the Distribution Agreement. Baxter disputed the existence of a breach and failed to cure the deficiency. Rockwell subsequently provided Baxter with notice of loss of exclusivity due to its failure to cure as provided in the Distribution Agreement. The determination of whether a breach occurred resulting in a loss of exclusivity and the outcome of the other pending disputes with Baxter will be determined through the arbitration process.
7. INVENTORY
Components of inventory as of December 31, 2016 and 2015 are as follows:
|
|
|
December 31, |
|
December 31, |
|
||
|
|
|
2016 |
|
2015 |
|
||
|
Raw Materials |
|
$ |
10,903,084 |
|
$ |
5,504,915 |
|
|
Work in Process |
|
|
86,452 |
|
|
165,910 |
|
|
Finished Goods |
|
|
2,978,090 |
|
|
2,200,955 |
|
|
Total |
|
$ |
13,967,626 |
|
$ |
7,871,780 |
|
As of December 31, 2016, we classified $1,826,554 of inventory as non-current all of which related to the active pharmaceutical ingredient for Triferic.
F-17
8. PROPERTY AND EQUIPMENT
Major classes of property and equipment, stated at cost, as of December 31, 2016 and 2015 are as follows:
|
|
|
2016 |
|
2015 |
|
||
|
Leasehold Improvements |
|
$ |
728,151 |
|
$ |
682,992 |
|
|
Machinery and Equipment |
|
|
7,169,223 |
|
|
7,042,836 |
|
|
Information Technology & Office Equipment |
|
|
2,293,587 |
|
|
2,273,289 |
|
|
Laboratory Equipment |
|
|
610,767 |
|
|
668,607 |
|
|
Transportation Equipment |
|
|
265,198 |
|
|
273,109 |
|
|
|
|
|
11,066,926 |
|
|
10,940,833 |
|
|
Accumulated Depreciation |
|
|
(9,675,351) |
|
|
(9,294,265) |
|
|
Net Property and Equipment |
|
$ |
1,391,575 |
|
$ |
1,646,568 |
|
Below is a summary of depreciation expense by period:
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Depreciation expense |
|
$ |
601,093 |
|
$ |
655,265 |
|
$ |
829,292 |
|
9. GOODWILL AND INTANGIBLE ASSETS
Total goodwill was $920,745 at December 31, 2016 and 2015. We completed our annual impairment tests as of November 30, 2016 and 2015, and determined that no adjustment for impairment of goodwill was required.
We have entered into a global licensing agreement for certain patents covering Triferic, a therapeutic drug compound to be delivered using our dialysate product lines. We received FDA approval for this product in January 2015. We have capitalized the licensing fees paid for the rights to use this patented technology as an intangible asset. We have capitalized certain patent approval costs.
During 2011, we acquired an abbreviated new drug application (“ANDA”) for a generic version of an intravenous vitamin‑D analogue, Calcitriol. Total capitalized costs related to this ANDA were approximately $695,000. These were amortized over a five year period ending December 31, 2016.
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Capitalized Licensing Fees |
|
$ |
1,070,126 |
|
$ |
1,070,126 |
|
$ |
1,070,126 |
|
|
Accumulated Amortization |
|
|
(1,065,744) |
|
|
(904,469) |
|
|
(737,440) |
|
|
Capitalized Licensing Fees, Net of Amortization |
|
$ |
4,382 |
|
$ |
165,657 |
|
$ |
332,686 |
|
|
Amortization Expense |
|
$ |
161,275 |
|
$ |
167,029 |
|
$ |
167,029 |
|
Our policy is to amortize licensing fees over the life of the patents pertaining to certain licensing agreements and to amortize patent costs over the life of the patent. Estimated amortization expense for amortization of capitalized patent costs in 2017 is approximately $353. In 2015, we recognized expenses related to milestone achievements of $275,000.
10. ACCRUED LIABILITIES
We had the following accrued liabilities as of December 31, 2016 and 2015:
|
|
|
2016 |
|
2015 |
|
||
|
Accrued Research & Development Expense |
|
$ |
193,638 |
|
$ |
219,070 |
|
|
Accrued Compensation and Benefits |
|
|
2,002,767 |
|
|
1,893,144 |
|
|
Other Accrued Liabilities |
|
|
2,013,746 |
|
|
1,719,142 |
|
|
Total Accrued Liabilities |
|
$ |
4,210,151 |
|
$ |
3,831,356 |
|
F-18
11. OPERATING LEASES
We lease our production facilities and administrative offices as well as certain equipment used in our operations including leases on transportation equipment used in the delivery of our products. The lease terms range from monthly to seven years. We occupy a 51,000 square foot facility and a 17,500 square foot facility in Wixom, Michigan under a lease expiring in August 2018. We also occupy a 51,000 square foot facility in Grapevine, Texas under a lease expiring in December 2020. In addition, we lease a 57,000 square foot facility in Greer, South Carolina under a lease expiring February 2018.
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Rent Expense Recognized Under Operating Leases |
|
$ |
2,369,101 |
|
$ |
2,301,930 |
|
$ |
2,075,919 |
|
Future minimum rental payments under operating lease agreements are as follows:
|
Year ending December 31, 2017 |
|
$ |
1,956,338 |
|
|
Year ending December 31, 2018 |
|
|
1,827,752 |
|
|
Year ending December 31, 2019 |
|
|
1,374,550 |
|
|
Year ending December 31, 2020 |
|
|
614,292 |
|
|
Year ending December 31, 2021 |
|
|
196,975 |
|
|
Year ending December 31, 2022 and thereafter |
|
|
54,726 |
|
|
Total |
|
$ |
6,024,633 |
|
12. INCOME TAXES
A reconciliation of income tax expense at the statutory rate to income tax expense at our effective tax rate is as follows:
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Tax Expense (Benefit) Computed at 34 % of Pretax Income (Loss) |
|
$ |
(6,595,452) |
|
$ |
(4,903,000) |
|
$ |
(7,251,000) |
|
|
Foreign Income Tax Expense |
|
|
(404,527) |
|
|
— |
|
|
— |
|
|
Effect of Change in Valuation Allowance |
|
|
(6,595,452) |
|
|
(4,903,000) |
|
|
(7,251,000) |
|
|
Total Income Tax Expense |
|
$ |
404,527 |
|
$ |
— |
|
$ |
— |
|
The details of the net deferred tax asset are as follows:
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
Net Operating Loss Carryforward |
|
$ |
51,463,000 |
|
$ |
52,669,000 |
|
|
Stock Based Compensation |
|
|
7,704,000 |
|
|
4,223,000 |
|
|
Deferred Revenue |
|
|
6,478,000 |
|
|
5,920,000 |
|
|
General Business Credit |
|
|
6,146,000 |
|
|
5,741,000 |
|
|
Accrued Expenses |
|
|
605,000 |
|
|
511,000 |
|
|
Inventories |
|
|
494,000 |
|
|
163,000 |
|
|
Book over Tax Depreciation |
|
|
61,000 |
|
|
47,000 |
|
|
Allowance for Doubtful Accounts |
|
|
2,000 |
|
|
26,000 |
|
|
Total Deferred Tax Assets |
|
|
72,953,000 |
|
|
69,300,000 |
|
|
Deferred Tax Liabilities: |
|
|
|
|
|
|
|
|
Goodwill & Intangible Assets |
|
|
137,000 |
|
|
167,000 |
|
|
Prepaid Expenses |
|
|
182,000 |
|
|
119,000 |
|
|
Total Deferred Tax Liabilities |
|
|
319,000 |
|
|
286,000 |
|
|
Subtotal |
|
|
72,634,000 |
|
|
69,014,000 |
|
|
Valuation Allowance |
|
|
(72,634,000) |
|
|
(69,014,000) |
|
|
Net Deferred Tax Asset |
|
$ |
— |
|
$ |
— |
|
Deferred tax assets result primarily from net operating loss carryforwards. For tax purposes, we have net operating loss carryforwards of approximately $151,400,000 that expire between 2018 and 2036.
F-19
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized upon the generation of future taxable income during the periods in which those temporary differences become deductible. We recognized $404,527 in foreign income taxes paid for the year ended December 31, 2016. We recognized no income tax expense or benefit for the years ended December 31, 2015 and 2014. While we anticipate generating income within the next year or two, we expect to incur operating losses until our drug products are marketed and generating sufficient profits to offset our operating expenses. Considered together with our limited history of operating income and our net losses in 2016, 2015 and 2014, management has placed a full valuation allowance against the net deferred tax assets as of December 31, 2016 and 2015. The portion of the valuation allowance resulting from excess tax benefits on share based compensation that would be credited directly to contributed capital if recognized in subsequent periods is $5.4 million.
The Company accounts for its uncertain tax positions in accordance with ASC 740‑10, Income Taxes and the amount of unrecognized tax benefits related to tax positions is not significant at December 31, 2016 and 2015.
13. CAPITAL STOCK
Our authorized capital stock consists of 2,000,000 preferred shares, none of which were issued or outstanding at December 31, 2016, 2015 and 2014, and 120,000,000 common shares, no par value per share, of which the following shares were outstanding:
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Shares outstanding as of December 31, |
|
|
51,527,711 |
|
|
51,501,877 |
|
|
50,284,007 |
|
|
Summary of Share Issuances: |
|
|
|
|
|
|
|
|
|
|
|
Share Issuances related to Equity Compensation: |
|
|
|
|
|
|
|
|
|
|
|
Shares issued upon exercise of stock options by employees |
|
|
25,834 |
|
|
657,998 |
|
|
711,516 |
|
|
Proceeds realized from stock option exercises |
|
$ |
80,161 |
|
$ |
2,780,188 |
|
$ |
2,964,445 |
|
|
Average exercise price of options exercised |
|
$ |
3.10 |
|
$ |
4.23 |
|
$ |
4.17 |
|
|
Restricted Stock Grants |
|
|
— |
|
|
850,000 |
|
|
740,000 |
|
|
Share issuances related to Warrant Exercises |
|
|
|
|
|
|
|
|
|
|
|
Shares issued upon the exercise of warrants |
|
|
— |
|
|
|
|
|
904,886 |
|
|
Proceeds realized from warrant exercises |
|
|
— |
|
|
|
|
$ |
8,433,045 |
|
|
Share issuances related to Equity Offerings |
|
|
|
|
|
|
|
|
|
|
|
Shares issued pursuant to equity offerings |
|
|
— |
|
|
|
|
|
7,816,944 |
|
|
Proceeds realized from equity offerings |
|
|
— |
|
|
|
|
$ |
69,780,967 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common Shares
Holders of the common shares are entitled to one vote per share on all matters submitted to a vote of our shareholders and are entitled to receive dividends when and if declared by the Board of Directors. The Board is authorized to issue additional common shares within the limits of the Company’s Articles of Incorporation without further shareholder action, subject to applicable stock exchange rules.
Warrants
As of December 31, 2016 and 2015, we had no outstanding warrants. During 2014, we realized $8,433,045 in net proceeds from the exercise of 904,886 warrants at an average exercise price of $9.32.
14. LONG TERM INCENTIVE PLAN & STOCK OPTIONS
Long Term Incentive Plan & Stock Options
The Board of Directors adopted the Rockwell Medical, Inc., 2007 Long Term Incentive Plan (“LTIP”) on April 11, 2007 as a replacement for the 1997 Stock Option Plan (the “Old Plan”) which was terminated as to future grants. No options were granted under the Old Plan after 2006 and no options remain outstanding as of December 31,
F-20
2016. There are 11,500,000 common shares reserved for issuance under the LTIP. The Compensation Committee of the Board of Directors (the “Committee”) is responsible for the administration of the LTIP including the grant of stock based awards and other financial incentives including performance based incentives to employees, non‑employee directors and consultants.
The Committee determines the terms and conditions of options and other equity based incentives including, but not limited to, the number of shares, the exercise price, term of option and vesting requirements. The Committee approved stock option grants during 2016, 2015 and 2014 and restricted stock grants in 2015 and 2014. The stock option awards were granted with an exercise price equal to the market price of the Company’s stock on the date of the grant. The options expire 10 years from the date of grant or upon termination of employment and generally vest in three equal annual installments beginning on the first anniversary of the date of grant.
Restricted Stock Grants
There were no grants of restricted stock during 2016. We granted 850,000 and 740,000 restricted shares in 2015 and 2014, respectively under the LTIP. These restricted stock grants were valued at the market price on the date of grant.
During 2015, restricted stock grants aggregating 850,000 common shares were granted in October 2015 with a vesting date of approximately twenty months following the grant date. Vesting is conditioned upon continued employment with the Company.
During 2014, restricted stock grants aggregating 320,000 shares were granted in January 2014 with a vesting date of approximately fourteen months after the grant date and an additional 420,000 common shares were granted in October 2014 with a vesting date of approximately seven months following the grant date with vesting conditioned upon continued employment with the Company.
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Restricted Shares Granted |
|
|
- |
|
|
850,000 |
|
|
740,000 |
|
|
Average Market Value Per Share on Grant Date |
|
|
- |
|
$ |
8.23 |
|
$ |
9.42 |
|
|
Expense related to All Restricted Shares |
|
$ |
4,361,760 |
|
$ |
3,694,496 |
|
$ |
5,497,274 |
|
|
Unearned Stock Based Compensation for All Restricted Stock Awards Attributable to Future Periods. |
|
$ |
1,549,259 |
|
|
|
|
|
|
|
Stock Option Grants
Our standard stock option agreement under the 2007 Plan allows for the payment of the exercise price of vested stock options either through cash remittance in exchange for newly issued shares, or through non‑cash exchange of previously issued shares held by the recipient for at least six months in exchange for our newly issued shares. The 1997 Plan also allows for the retention of shares in payment of the exercise price and income tax withholding. The latter method results in no cash being received by us, but also results in a lower number of total shares being outstanding subsequently as a direct result of this exchange of shares. Shares returned to us in this manner would be retired.
In 2016, 2015 and 2014, the Company received cash proceeds of $80,161, $2,780,188 and $2,964,445 respectively, in exchange for shares issued upon the exercise of options during the year. No income tax benefits were recognized during 2016, 2015 and 2014 related to stock option activity as the Company has a full valuation allowance recorded against its deferred tax assets. However, tax benefits (expense) for the excess of the value of the shares issued over the price paid of $20,000, ($943,000) and $2,009,000 were created in 2016, 2015, and 2014. The cumulative excess tax benefit at December 31, 2016 is $5.4 million, which when realized, will be credited directly to shareholders' equity.
F-21
A summary of the status of the LTIP and the Old Plan is as follows:
|
|
|
|
|
WEIGHTED |
|
|
|
|
|
|
|
|
|
AVERAGE |
|
AGGREGATE |
|
|
|
|
|
|
|
EXERCISE |
|
INTRINSIC |
|
|
|
|
|
SHARES |
|
PRICE |
|
VALUE |
|
|
|
Outstanding at December 31, 2013 |
|
6,228,000 |
|
6.27 |
|
$ |
25,956,880 |
|
|
Granted |
|
1,731,500 |
|
9.43 |
|
|
|
|
|
Exercised |
|
(1,029,016) |
|
2.88 |
|
$ |
2,964,445 |
|
|
Forfeited |
|
(45,401) |
|
6.19 |
|
|
|
|
|
Outstanding at December 31, 2014 |
|
6,885,083 |
|
7.41 |
|
$ |
19,730,211 |
|
|
Granted |
|
1,697,500 |
|
8.30 |
|
|
|
|
|
Exercised |
|
(794,248) |
|
3.50 |
|
$ |
2,780,188 |
|
|
Forfeited |
|
(29,333) |
|
6.91 |
|
|
|
|
|
Outstanding at December 31, 2015 |
|
7,759,002 |
|
7.84 |
|
$ |
18,648,477 |
|
|
Granted |
|
30,000 |
|
6.54 |
|
|
|
|
|
Exercised |
|
(25,834) |
|
4.35 |
|
$ |
112,280 |
|
|
Forfeited |
|
(71,667) |
|
9.33 |
|
|
|
|
|
Outstanding at December 31, 2016 |
|
7,691,501 |
|
7.83 |
|
$ |
1,821,384 |
|
|
OPTIONS OUTSTANDING |
|
OPTIONS EXERCISABLE |
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
WEIGHTED |
|
|
|
|
|
|
|
|
REMAINING |
|
WEIGHTED |
|
|
|
|
AVERAGE |
|
||
|
|
|
NUMBER OF |
|
CONTRACTUAL |
|
EXERCISE |
|
NUMBER OF |
|
EXERCISE |
|
||||
|
RANGE OF EXERCISE PRICES |
|
OPTIONS |
|
LIFE |
|
PRICE |
|
OPTIONS |
|
PRICE |
|
||||
|
$3.09 to $4.93 |
|
|
392,500 |
|
2.0-6.5 yrs. |
|
$ |
3.31 |
|
|
392,500 |
|
$ |
3.31 |
|
|
$5.86 to $7.13 |
|
|
2,513,000 |
|
.9-9.7 yrs. |
|
$ |
6.44 |
|
|
2,483,000 |
|
$ |
6.44 |
|
|
$8.23 to 11.49 |
|
|
4,786,001 |
|
2.8-8.8 yrs. |
|
$ |
8.94 |
|
|
3,113,835 |
|
$ |
9.09 |
|
|
Total |
|
|
7,691,501 |
|
5.7 yrs. |
|
$ |
7.83 |
|
|
5,989,335 |
|
$ |
7.61 |
|
|
Intrinsic Value |
|
$ |
1,821,384 |
|
|
|
|
|
|
$ |
1,821,134 |
|
|
|
|
|
|
|
|
|
WEIGHTED |
|
|
|
|
|
|
|
AVERAGE |
|
|
|
|
|
NUMBER OF |
|
FAIR MARKET |
|
|
|
|
|
UNVESTED |
|
VALUE AT |
|
|
|
|
|
OPTIONS |
|
GRANT DATE |
|
|
|
As of December 31, 2013 |
|
1,714,433 |
|
|
|
|
|
Granted |
|
1,731,500 |
|
$ |
5.91 |
|
|
Forfeited |
|
(45,401) |
|
|
|
|
|
Vested |
|
(820,032) |
|
|
|
|
|
As of December 31, 2014 |
|
2,580,500 |
|
|
|
|
|
Granted |
|
1,697,500 |
|
$ |
4.56 |
|
|
Forfeited |
|
(28,333) |
|
|
|
|
|
Vested |
|
(1,135,056) |
|
|
|
|
|
As of December 31, 2015 |
|
3,114,611 |
|
|
|
|
|
Granted |
|
30,000 |
|
$ |
3.85 |
|
|
Forfeited |
|
(71,667) |
|
|
|
|
|
Vested |
|
(1,370,778) |
|
|
|
|
|
As of December 31, 2016 |
|
1,702,166 |
|
|
|
|
F-22
The Company values stock options awarded using the Black‑Scholes method. Assumptions used in the stock option valuations were:
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Volatility of share price |
|
64 - 65 % |
|
58 - 61 % |
|
69 - 70 % |
|
|
Risk free interest rate |
|
1.3-1.6 % |
|
1.5 - 1.7 % |
|
1.9 - 2.0 % |
|
|
Expected option life |
|
6 yrs. |
|
6 yrs. |
|
6 yrs. |
|
|
Dividend Yield |
|
0.0% |
|
0.0% |
|
0.0% |
|
We believe this valuation methodology is appropriate for estimating the fair value of stock options we grant to employees and directors which are subject to ASC 718‑10 requirements. We primarily base our determination of expected volatility through our assessment of the historical volatility of our common shares. We do not believe that we are able to rely on our historical stock option exercise and post‑vested termination activity to provide accurate data for estimating our expected term for use in determining the fair value of these options. Therefore, as allowed by Staff Accounting Bulletin (SAB) No. 107, Share‑Based Payment, we have opted to use the simplified method for estimating the expected option term equal to the midpoint between the vesting period and the contractual term. The contractual term of the option is 10 years from the date of grant and the vesting term of the option is three years from date of grant. Risk free interest rates utilized are based upon published U.S. Treasury yield curves at the date of the grant for the expected option term.
For the years ended December 31, 2016, 2015 and 2014, we recognized compensation expense of $5,984,524, $5,193,481 and $4,597,412 respectively related to options granted to employees under the LTIP with a corresponding credit to common stock. At December 31, 2016, the amount of unrecorded stock-based compensation expense for stock options attributable to future periods was approximately $5,834,369 which is expected to be amortized to expense over the remaining vesting periods of the options of 1 to 30 months.
As of December 31, 2016, the remaining number of common shares available for equity awards under the LTIP was 545,694.
15. RISK MANAGEMENT
Insurance
We evaluate various kinds of risk that we are exposed to in our business. In our evaluation of risk, we evaluate options and alternatives to mitigating such risks. For certain insurable risks we may acquire insurance policies to protect against potential losses or to partially insure against certain risks. For our subsidiary, Rockwell Transportation, Inc., we maintain a partially uninsured workers' compensation plan. Under the policy, the Company's self‑insurance retention is $350,000 per occurrence and $580,388 in aggregate coverage for the policy year ending July 1, 2017. The total amount at December 31, 2016 by which retention limits exceed the claims paid and accrued is approximately $298,000 for the policy year ending July 1, 2017. Estimated additional future claims subject to payment by the Company of approximately $196,537 have been accrued for the year ended December 31, 2016.
At December 31, 2016, approximately $300,000 was held in cash collateral and escrow by the insurance carrier for workers’ compensation insurance. At December 31, 2016 amounts held in cash collateral and escrow are included in prepaid expenses and other non-current assets in the consolidated financial statements.
F-23
16. QUARTERLY RESULTS OF OPERATIONS
The following is a summary of the quarterly results of operations for the years ended December 31, 2016 and 2015.
|
|
|
First Quarter |
|
Second Quarter |
|
Third Quarter |
|
Fourth Quarter |
|
||||
|
2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sales |
|
$ |
13,627,048 |
|
$ |
13,452,517 |
|
$ |
12,814,815 |
|
$ |
13,389,786 |
|
|
Cost of Sales |
|
|
11,932,122 |
|
|
11,962,989 |
|
|
11,234,934 |
|
|
11,401,603 |
|
|
Gross Profit |
|
|
1,694,926 |
|
|
1,489,528 |
|
|
1,579,881 |
|
|
1,988,183 |
|
|
Selling, General and Administrative |
|
|
4,986,741 |
|
|
5,014,370 |
|
|
5,070,127 |
|
|
6,049,663 |
|
|
Research and Product Development |
|
|
1,314,430 |
|
|
2,063,324 |
|
|
1,261,863 |
|
|
1,200,729 |
|
|
Operating Income (Loss) |
|
|
(4,606,245) |
|
|
(5,588,166) |
|
|
(4,752,109) |
|
|
(5,262,209) |
|
|
Interest and Investment Income, net |
|
|
186,562 |
|
|
227,020 |
|
|
188,847 |
|
|
207,911 |
|
|
Interest Expense |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Income (Loss) Before Income Taxes |
|
|
(4,419,683) |
|
|
(5,361,146) |
|
|
(4,563,262) |
|
|
(5,054,298) |
|
|
Income Tax Expense |
|
|
(404,527) |
|
|
— |
|
|
— |
|
|
— |
|
|
Net Income (Loss) |
|
$ |
(4,824,210) |
|
$ |
(5,361,146) |
|
$ |
(4,563,262) |
|
$ |
(5,054,298) |
|
|
Basic And Diluted Earnings (Loss) Per Share |
|
$ |
(0.10) |
|
$ |
(0.11) |
|
$ |
(0.09) |
|
$ |
(0.10) |
|
|
|
|
First Quarter |
|
Second Quarter |
|
Third Quarter |
|
Fourth Quarter |
|
||||
|
2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sales |
|
$ |
13,883,961 |
|
$ |
12,955,576 |
|
$ |
14,378,528 |
|
$ |
14,132,637 |
|
|
Cost of Sales |
|
|
11,571,618 |
|
|
10,889,619 |
|
|
11,875,122 |
|
|
12,076,489 |
|
|
Gross Profit |
|
|
2,312,343 |
|
|
2,065,957 |
|
|
2,503,406 |
|
|
2,056,148 |
|
|
Selling, General and Administrative |
|
|
5,325,761 |
|
|
3,835,596 |
|
|
3,827,904 |
|
|
6,089,606 |
|
|
Research and Product Development |
|
|
799,591 |
|
|
885,259 |
|
|
1,246,727 |
|
|
2,029,736 |
|
|
Operating Income (Loss) |
|
|
(3,813,009) |
|
|
(2,654,898) |
|
|
(2,571,225) |
|
|
(6,063,194) |
|
|
Interest and Investment Income, net |
|
|
113,815 |
|
|
118,151 |
|
|
156,672 |
|
|
293,238 |
|
|
Interest Expense |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Income (Loss) Before Income Taxes |
|
|
(3,699,194) |
|
|
(2,536,747) |
|
|
(2,414,553) |
|
|
(5,769,956) |
|
|
Income Tax Expense |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Net Income (Loss) |
|
$ |
(3,699,194) |
|
$ |
(2,536,747) |
|
$ |
(2,414,553) |
|
$ |
(5,769,956) |
|
|
Basic And Diluted Earnings (Loss) Per Share |
|
$ |
(0.07) |
|
$ |
(0.05) |
|
$ |
(0.05) |
|
$ |
(0.12) |
|
F-24
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS
|
|
|
Balance at |
|
|
|
|
|
|
|
Balance |
|
||
|
|
|
Beginning |
|
|
|
|
|
|
|
at End |
|
||
|
|
|
of Period |
|
Additions |
|
(Deductions) |
|
of Period |
|
||||
|
Allowance for Doubtful Accounts: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2016 |
|
$ |
75,160 |
|
$ |
13,348 |
|
$ |
(83,277) |
|
$ |
5,231 |
|
|
Year ended December 31, 2015 |
|
$ |
52,213 |
|
$ |
72,877 |
|
$ |
(49,930) |
|
$ |
75,160 |
|
|
Year ended December 31, 2014 |
|
$ |
37,392 |
|
$ |
40,714 |
|
$ |
(25,893) |
|
$ |
52,213 |
|
|
Inventory Reserve: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2016 |
|
$ |
10,662 |
|
$ |
564,451 |
|
$ |
(11,298) |
|
$ |
563,815 |
|
|
Year ended December 31, 2015 |
|
$ |
27,274 |
|
$ |
59,581 |
|
$ |
(76,193) |
|
$ |
10,662 |
|
|
Year ended December 31, 2014 |
|
$ |
35,009 |
|
$ |
19,701 |
|
$ |
(27,435) |
|
$ |
27,274 |
|
|
Deferred Tax Asset Valuation Allowance: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, 2016 |
|
$ |
69,014,000 |
|
$ |
3,620,000 |
|
$ |
— |
|
$ |
72,634,000 |
|
|
Year ended December 31, 2015 |
|
$ |
69,307,000 |
|
$ |
— |
|
$ |
(293,000) |
|
$ |
69,014,000 |
|
|
Year ended December 31, 2014 |
|
$ |
56,272,000 |
|
$ |
13,035,000 |
|
$ |
— |
|
$ |
69,307,000 |
|
Allowances and reserves are deducted from the accounts to which they apply
S-1