Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - Phio Pharmaceuticals Corp. | d271779dex231.htm |
Table of Contents
As filed with the Securities and Exchange Commission on November 10, 2016
Registration No. 333-214199
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 1
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
RXi PHARMACEUTICALS CORPORATION
(Exact name of Registrant as specified in its charter)
| Delaware | 2834 | 45-3215903 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
257 Simarano Drive, Suite 101
Marlborough, Massachusetts 01752
(508) 767-3861
(Address, including zip code, and telephone number, including area code, of Registrant’s principal executive offices)
Geert Cauwenbergh, Dr. Med. Sc.
President
RXi Pharmaceuticals Corporation
257 Simarano Drive, Suite 101
Marlborough, Massachusetts 01752
(508) 767-3861
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Ryan A. Murr
Christina Greenberg
Gibson, Dunn & Crutcher LLP
555 Mission Street, Suite 3000
San Francisco, CA 94105
Telephone: (415) 393-8373
Facsimile: (415) 374-8430
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||||
| Non-accelerated filer | ☐ | (Do not check if a smaller reporting company) | Smaller reporting company | ☒ |
CALCULATION OF REGISTRATION FEE
|
| ||||||
| Title of each class of securities to be registered(1) |
Proposed maximum offering price per unit |
Proposed offering price(1)(2) |
Amount of registration fee | |||
| Class A Units consisting of: |
$4,200,000 | $486.78 | ||||
| (i) Shares of common stock, par value $0.0001 per share |
||||||
| (ii) Warrants to purchase common stock(3) |
||||||
| Class B Units consisting of: |
$9,600,000 | $1,112.64 | ||||
| (i) Shares of Series B Convertible Preferred Stock, par value $0.0001 per share |
||||||
| (ii) Shares of common stock issuable on conversion of Series B Convertible Preferred Stock(3) |
||||||
| (iii) Warrants to purchase common stock(3) |
||||||
| Common stock issuable upon exercise of warrants |
$6,900,000 | $799.71 | ||||
| Total |
$20,700,000 | $2,399.13(4) | ||||
|
| ||||||
|
| ||||||
| (1) | Estimated solely for the purpose of computing the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended (the “Securities Act”). |
| (2) | Includes 1,512,150 additional units, consisting of one share of common stock and a warrant to purchase half of one share of common stock, that the underwriters have the option to purchase to cover over-allotments, if any. |
| (3) | No separate fee is required pursuant to Rule 457(g) under the Securities Act. |
| (4) | Previously paid. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities, and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to completion, dated November 10, 2016
Preliminary Prospectus

2,017,000 Class A Units consisting of Common Stock and warrants and
9,600 Class B Units consisting of shares of Series B Convertible Preferred Stock and warrants
(and 13,104,500 shares of Common Stock underlying shares of Series B Convertible Preferred Stock and warrants)
We are offering 2,017,000 Class A Units, with each Class A Unit consisting of one share of common stock, par value $0.0001 per share (the “common stock”) and a warrant to purchase half of one share of our common stock (based on an assumed offering price per common share of $1.19, which was the last reported sale price of our common stock on November 10, 2016, which assumption is used throughout this preliminary prospectus) (together with the shares of common stock underlying such warrants, the “Class A Units”) at a public offering price of $ per Class A Unit. Each warrant included in the Class A Units entitles its holder to purchase half of one share of common stock at an exercise price of $ .
We are also offering to those purchasers, whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 9.99% of our outstanding common stock following the consummation of this offering, the opportunity to purchase, in lieu of the number of Class A Units that would result in ownership in excess of 9.99%, 9,600 Class B Units. Each Class B Unit will consist of one share of Series B Convertible Preferred Stock, par value $0.0001 per share (the “Series B Convertible Preferred Stock”), convertible into 840 shares of common stock and warrants to purchase 420 shares of our common stock (based on an assumed offering price per common share of $1.19, which was the last reported sale price of our common stock on November 10, 2016, which assumption is used throughout this preliminary prospectus) (together with the shares of common stock underlying such shares of Series B Convertible Preferred Stock and such warrants, the “Class B Units” and, together with the Class A Units, the “units”) at a public offering price of $1,000 per Class B Unit. Each warrant included in the Class B Units entitles its holder to purchase half of one share of common stock at an exercise price of $ .
The Class A Units and Class B Units will not be certificated and the shares of common stock, Series B Convertible Preferred Stock and warrants comprising such units are immediately separable and will be issued separately in this offering. The underwriters have the option to purchase additional shares of common stock, and/or warrants to purchase shares of common stock solely to cover over-allotments, if any, at the price to the public less the underwriting discounts and commissions. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Convertible Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for 45 days from the date of this prospectus.
Our common stock is listed on The NASDAQ Capital Market under the symbol “RXII”. The closing price of our common stock on November 10, 2016, as reported by NASDAQ, was $1.19 per share. We do not intend to apply for listing of the shares of Series B Convertible Preferred Stock or the warrants on any securities exchange or other trading system.
Investing in the units involves a high degree of risk. Before making any investment in these securities, you should consider carefully the risks and uncertainties in the section entitled “Risk Factors” beginning on page 10 of this prospectus.
| Per Class A Unit(1) |
Per Class B Unit(1) |
Total | ||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discount(2)(3) |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to RXi Pharmaceuticals Corporation |
$ | $ | $ | |||||||||
| (1) | The public offering price and underwriting discount corresponds to (x) in respect of the Class A Units (i) an assumed public offering price per share of common stock of $ and (ii) an assumed public offering price per warrant of $ and (y) in respect of the Class B Units (i) an assumed public offering price per share of Series B Convertible Preferred Stock of $ and (ii) an assumed public offering price per warrant of $ . |
| (2) | We have also agreed to reimburse for certain expenses. See “Underwriting.” |
| (3) | We have granted a 45-day day option to the underwriter to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Convertible Preferred Stock) and warrants sold in the primary offering) solely to cover over-allotments, if any. |
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense. The securities are not being offered in any jurisdiction where the offer is not permitted.
Ladenburg Thalmann
Griffin Securities
The date of this prospectus is , 2016
Table of Contents
| 1 | ||||
| 10 | ||||
| 22 | ||||
| 23 | ||||
| 23 | ||||
| 24 | ||||
| 25 | ||||
| Management’s Discussion and Analysis of Financial Condition and Results of Operations |
26 | |||
| 36 | ||||
| 50 | ||||
| 53 | ||||
| 55 | ||||
| Certain Relationships, Related-Party Transactions and Director Independence |
56 | |||
| 57 | ||||
| 59 | ||||
| 65 | ||||
| 69 | ||||
| 69 | ||||
| 69 | ||||
| F-1 | ||||
We have not authorized anyone to provide you with information other than that contained in this prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give to you. We are offering to sell, and seeking offers to buy, securities only in jurisdictions where offers and sales are permitted. The information contained in this prospectus or any free writing prospectus is accurate only as of its date, regardless of its time of delivery or any sale of our securities. Our business, financial condition, results of operations, and prospects may have changed since that date.
No action is being taken in any jurisdiction outside the United States to permit an offering of our securities or possession or distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this prospectus applicable to that jurisdiction.
i
Table of Contents
The items in the following summary are described in more detail later in this prospectus. This summary provides an overview of selected information and does not contain all of the information you should consider in making your investment decision. Therefore, you should read the entire prospectus carefully before investing in our securities. Investors should carefully consider the information set forth under “Risk Factors” beginning on page 10 of this prospectus. In this prospectus, unless the context otherwise requires, references to “the Company,” “we,” “us,” “our,” or “RXi” refer to RXi Pharmaceuticals Corporation.
Overview
RXi Pharmaceuticals Corporation (“RXi,” “we,” “our” or the “Company”) is a clinical-stage RNAi company developing innovative therapeutics that address significant unmet medical needs. The Company’s development programs are based on our proprietary self-delivering RNAi (sd-rxRNA®) platform and Samcyprone™, a topical immunomodulator. Our clinical development programs include RXI-109, an sd-rxRNA for the treatment of dermal and ocular scarring, and Samcyprone™, for the treatment of such disorders as warts, alopecia areata, non-malignant skin tumors and cutaneous metastases of melanoma. In addition to these clinical programs, we have a pipeline of discovery and preclinical product candidates in our core therapeutic areas, as well as in other areas of interest. The Company’s pipeline, coupled with our extensive patent portfolio, provides for product development and business development opportunities across a broad spectrum of therapeutic areas.
1
Table of Contents
Our Pipeline
Our pipeline is focused on three areas: dermatology, ophthalmology and cosmetic product development. Our RNAi therapies are designed to “silence,” or down-regulate, the expression of a specific gene that may be over-expressed in a disease condition and our immunotherapy agent treats diseases by inducing, enhancing or suppressing an immune response. The following is a summary of our current product candidates and their development status:
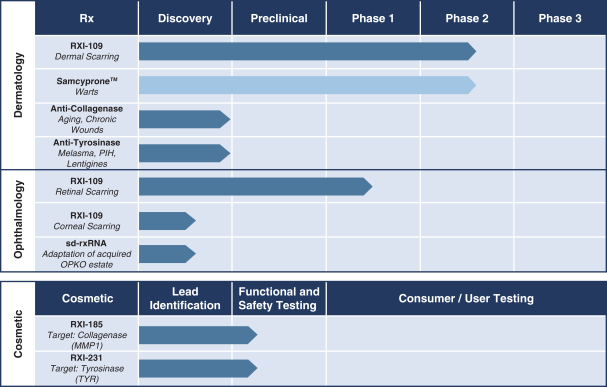
Dermatology Franchise
RXI-109 — Dermal Scarring
The Company’s lead product candidate and first RNAi clinical product candidate, RXI-109, is a self-delivering RNAi compound (sd-rxRNA) that commenced human clinical trials in 2012. RXI-109 is designed to reduce the expression of connective tissue growth factor (“CTGF”), a critical regulator of several biological pathways involved in fibrosis, including scar formation in the skin and eye. RXI-109 is currently being evaluated in a Phase 2 clinical trial, Study 1402, to prevent or reduce dermal scarring following scar revision surgery of an existing hypertrophic scar and a Phase 1/2 clinical trial, Study 1502, to evaluate the safety and clinical activity of RXI-109 to prevent the progression of retinal scarring in subjects with wet age-related macular degeneration (“AMD”).
The Company conducted two Phase 1 clinical trials evaluating RXI-109 in a surgical setting. Both trials demonstrated the safety and tolerability of RXI-109 in ascending single and multi-doses, and also provided the first evidence of clinical activity in a surgical setting. With the successful completion of the Phase 1 trials, the Company initiated its Phase 2 program for RXI-109 with Study 1301, a Phase 2a clinical trial evaluating the use
2
Table of Contents
of RXI-109 to prevent the recurrence of hypertrophic scars following scar revision surgery, in November 2013. This was followed by a second Phase 2 clinical trial in April 2014, Study 1401, which evaluated the use of RXI-109 to prevent the recurrence of keloids, raised and reddened or darkened scars resulting from increased collagen production, after surgical revision. Enrollment and dosing for both of these studies has been completed.
Preliminary data observations from Study 1301 were used in the design of the Company’s third Phase 2 clinical trial in hypertrophic scars, Study 1402, which commenced in July 2014. In October 2015, we reported that preliminary data from Study 1402 demonstrated that scars at revision sites were less visible at three months after a treatment regimen with intradermal administration of RXI-109 than scars at untreated revision sites in those same subjects. Based in part on this new information, two more cohorts were added to Study 1402 in November 2015. For these two cohorts, the number of doses was increased to either eight or nine doses of RXI-109 over a six-month period to better cover the extended wound healing/scarring profile of hypertrophic scars. Enrollment of subjects into these two new cohorts completed ahead of schedule during the third quarter of 2016.
Scarring represents a high unmet medical need as there are currently no U.S. Food and Drug Administration (“FDA”) approved therapies in the U.S. for the treatment and prevention of scars in the skin. Scar revision surgery is a common option, but often the scar recurs. If approved, RXI-109 could be a “first-in-class” RNAi treatment for the prevention or reduction of post-surgical dermal scarring. Given the large number of surgical procedures, there is a significant market for a scar prevention therapeutic such as RXI-109.
Samcyprone™ — Warts
In December 2014, the Company broadened its clinical pipeline with an exclusive, global license to Samcyprone™, our second clinical candidate. Samcyprone™ is a proprietary topical formulation of the small molecule diphenylcyclopropenone (“DPCP”), an immunomodulator that works by initiating a T-cell response. The use of Samcyprone™ allows sensitization using much lower concentrations of DPCP than are used with existing compounded DPCP solutions, avoiding hyper-sensitization to subsequent challenge doses. DPCP, the active ingredient in Samcyprone™, has long been used to treat warts and has also been used for several other indications, such as to stimulate hair re-growth in alopecia areata and to clear cutaneous metastases of melanoma. In March 2015, the FDA granted Orphan Drug Designation to the Company for Samcyprone™ for the treatment of malignant melanoma stage IIb to IV. Samcyprone™ is currently being evaluated in a Phase 2a clinical trial, Study 1502, for the clearance of common warts. Study 1502 was initiated in December 2015.
Study 1502 includes a sensitization phase in which a spot on the subject’s upper arm and one wart are treated with Samcyprone™. After being sensitized in this way, the subjects will enter into the treatment phase where up to four warts are treated on a once weekly basis for ten weeks with a ten-fold lower concentration of Samcyprone™ than in the sensitization phase. During the trial, the warts are scored, photographed and measured to monitor the level of clearance. The Company is currently enrolling subjects and is adding a second cohort to explore the opportunity to reduce the sensitization dose level and potentially reduce the treatment length. With this second cohort, enrollment is expected to be completed in the first quarter of 2017.
Cutaneous warts are extremely common, being experienced by most people at some time during their lives. Although most warts will spontaneously disappear without treatment, treatment of these lesions is sought for recalcitrant warts and to prevent recurrence. There are many different treatment modalities for warts, including physical destruction and immunomodulation. However, treatment of warts is complicated by low success rates, prolonged duration of therapy and the potential for recurrence. There is a clear unmet need for new therapies for warts and if approved, Samcyprone™ could be a more effective and convenient treatment than the currently available therapies.
3
Table of Contents
Additional Dermatology Programs
In addition to our dermal scarring and wart programs, we continue to advance our preclinical and discovery programs with our sd-rxRNA technology. The Company has selected tyrosinase (“TYR”) and collagenase (“MMP1”) as targets for our self-delivering platform because they are relevant for both consumer health and therapeutic development. TYR is a key enzyme in the synthesis of melanin. Melanin is produced by melanocytes and is the pigment that gives human skin, hair and eyes their color. The inhibition of TYR can play a key role in the management of diseases including cutaneous hyperpigmentation disorders such as lentigines (freckles, age spots and liver spots) and possibly melanoma. MMP1 is a key enzyme involved in the breakdown of extracellular matrix. Reduction of MMP1 may be beneficial in the treatment of skin aging disorders, arthritis, acne scarring, blistering skin disorders, corneal erosions, endometriosis and possible cancer metastasis. In addition to our cosmetic program (described below), the Company is actively evaluating similar sd-rxRNA compounds that target TYR and MMP1 to move forward on a separate therapeutic development path.
Ophthalmology Franchise
RXI-109 — Retinal Scarring
As in dermal scarring, CTGF is known to play a role in retinal scarring. RXI-109 can also be used to target CTGF in the eye, where it is known to be involved in retinal scarring. Building on the work in our dermal clinical program, the Company filed a new investigational drug application (“IND”) in July 2015 for RXI-109 as a potential therapeutic for the scarring component of retinal diseases in the eye, such as AMD. In November 2015, we initiated a Phase 1/2 clinical trial to evaluate the safety and clinical activity of RXI-109 in reducing the progression of retinal scarring.
Study 1501 is a multi-dose, dose escalation study conducted in subjects with AMD with evidence of subretinal fibrosis. Each subject will receive four doses of RXI-109 by intraocular injection at one month intervals for a total dosing period of three months. The safety and tolerability of RXI-109, as well as the potential for clinical activity, will be evaluated over the course of the study using numerous assessments to monitor the health in the retina and to assess visual acuity. The Company is currently enrolling subjects in Study 1501.
Currently, there is no effective way to prevent the formation or progression of retinal scars that may occur as a consequence of the number of debilitating ocular diseases. In advanced neo-vascular or wet-AMD, our first area of study, retinal scarring can result in continued vision loss even if the patient is being treated with an anti-vascular endothelial growth factor (“VEGF”) therapy. RXI-109 has the potential to fill this unmet medical need by reducing this continuing damage to the retina and in doing so help preserve these patients’ vision for a longer period of time.
Additional Ophthalmology Programs
In addition to the clinical trial for the use of RXI-109 as a potential therapeutic for retinal scarring, we are advancing other early-stage ophthalmology programs. Currently, the Company is directing its development efforts toward advancing RXI-109 for the treatment of corneal scarring. To date, preclinical studies have shown that CTGF protein levels are reduced in a dose-dependent manner in both the retina and cornea following an intravitreal injection of RXI-109 in monkeys. Elevated CTGF is implicated in the formation of corneal scarring that can occur after eye injury or after certain infections, and it has been proposed that a reduction of CTGF may be an important step towards reducing corneal scarring. Scarring of the cornea can impact the transparency of the cornea, and thus negatively impact vision. We are currently working towards a non-invasive delivery formulation of RXI-109 to reduce CTGF in the front of the eye. The Company also continues its exploratory efforts to identify potential sd-rxRNA lead compounds and targets from the RNAi-related assets acquired from OPKO Health Inc. (“OPKO”) in March 2013.
4
Table of Contents
Cosmetic Franchise
RXi’s cosmetic development program is based on our proprietary sd-rxRNA technology. Cosmetics are compounds that affect the appearance of the skin and make no preventative or therapeutic claims. These compounds may be developed more rapidly than therapeutics, therefore the path to market may be much shorter and less expensive.
In October 2015, we announced the selection of lead compounds targeting TYR and MMP1 for cosmetic development. RXI-231, an sd-rxRNA compound targeting TYR, is in development as a cosmetic ingredient that may improve the appearance of uneven skin tone and pigmentation. RXI-185, an sd-rxRNA compound targeting MMP1, is in development as a cosmetic ingredient that may improve the appearance of wrinkles or skin laxity. The Company is currently developing topical delivery application methods, including formulations and microneedling, for use with these compounds and completed functional and safety testing to support the initiation of human testing of one of these consumer health targets.
MirImmune Exclusive Option Agreement
In March 2015, RXi granted an exclusive license to MirImmune, Inc. (“MirImmune”), a private biopharmaceutical company, to utilize the Company’s novel and proprietary sd-rxRNA technology for use in developing ex vivo cell-based cancer immunotherapies. After obtaining the exclusive license from RXi, MirImmune has raised $500,000 in funding to date and used these proceeds on the advancement of their preclinical research, for patent prosecution and filing fees and for general corporate purposes.
MirImmune’s approach to immunotherapy builds on well-established methodologies of adoptive cell transfer. Immune cells, such as T-lymphocytes, are isolated from specific patients or retrieved from allogeneic immune cell banks and then expanded and possibly processed to express tumor-binding receptors. MirImmune’s method will introduce a new and important step in ex vivo processing of the immune cells. This step will reduce or eliminate the expression of immunosuppressive receptors or proteins by the therapeutic immune cells, potentially making them less sensitive to tumor resistance mechanisms and thus improving their ability to destroy the tumor cells.
MirImmune’s approach builds on current immunotherapy approaches, but provides some key advantages. One major advantage is that pre-treatment with MirImmune’s targeted compounds allow multiple immune checkpoints to be attenuated within the same therapeutic cell; an improvement which could dramatically increase their tumor cell killing capability. In addition, these therapeutic immune cells may lack some known side effects associated with the checkpoint inhibitor toxicity while potentially improving efficacy over current immunotherapy approaches.
Using RXi’s sd-rxRNA technology, MirImmune demonstrated in vitro that multiple sd-rxRNA compounds can be used alone or in combination to target and silence extracellular, as well as intracellular, checkpoints in immune cells. Additional in vitro data demonstrated that PD-1 silencing by sd-rxRNA in patient-derived tumor infiltrating lymphocytes (TILs) resulted in enhanced killing of melanoma tumor cells from the same patient in culture. MirImmune has also shown in a mouse model of human ovarian cancer that in vivo treatment with mesothelin CAR T-cells transfected with a PD-1 targeting sd-rxRNA significantly reduced the rate of tumor growth as compared to vehicle control. Furthermore, the silencing of PD-1 in the CAR T-cells isolated from these tumors persisted for at least one month.
MirImmune has identified lead sd-rxRNA compounds for each of six different checkpoints, both extracellular and intracellular. Since March 2015, MirImmune has been able to advance the potential of RXi’s sd-rxRNA platform for use in cell-based cancer immunotherapy with their preclinical research.
On October 7, 2016, RXi entered into an exclusive option agreement to acquire all outstanding capital stock of MirImmune in consideration for a number of shares equal to 19.99% of the then-outstanding shares of
5
Table of Contents
common stock of the Company, plus additional potential consideration contingent on MirImmune reaching certain milestones. The Company can exercise the option on the terms set forth in the option agreement at any time prior to April 5, 2017, but has no obligation to do so.
Market Opportunity
As there are currently no FDA-approved drugs to prevent scar formation, a therapeutic of this type could have great benefit for trauma and surgical patients, particularly as a treatment during the surgical revision of existing unsatisfactory scars. There are over 42 million medical procedures in the U.S. each year that could potentially benefit from a therapeutic treatment that could successfully reduce or prevent scarring; thus, the market potential is quite large. According to the American Society for Plastic Surgery, there are approximately 177,000 scar revision surgeries in the United States every year. In addition to cosmetic and reconstructive surgeries, medical interventions which could incorporate an anti-scarring agent include treatment of scarring that results from trauma, surgery or burns (especially relating to raised or hypertrophic scarring or contracture scarring), and surgical revision of existing unsatisfactory scars.
Overexpression of CTGF is implicated in dermal scarring, subretinal fibrosis and other fibrotic diseases. Because of this, we believe that RXI-109 or other CTGF-targeting RNAi compounds may be able to treat the fibrotic component of numerous indications. These indications are as wide ranging as acute spinal injury, endometriosis, organ fibrosis including liver and pulmonary fibrosis, cutaneous scleroderma and vascular restenosis, in addition to numerous ocular diseases that result in retinal scarring. If the current clinical trials of RXI-109 produce successful results, we may explore opportunities in these additional indications that can be accessed by local administration, starting with intradermal or intravitreal injection. Although the Company does not intend to develop systemic uses of RXI-109 at this time, the Company is open to business development and out-licensing opportunities for those applications.
DPCP, the active ingredient in Samcyprone™, is a small molecule that has been used since the late 1970s to stimulate regrowth of hair in patients with alopecia areata. Recent publications have supported its use as an immunomodulator for the treatment of alopecia areata, warts and cutaneous metastases of malignant melanoma, a combined market potential of over an estimated $1 billion. Although it has been used by physicians for several decades, it has never been reviewed or approved by a regulatory authority as a drug. If FDA approval is granted, Samcyprone™, RXi’s proprietary formulation of DPCP, is expected to achieve market exclusivity.
Risks Associated with Our Business
Our ability to implement our business strategy is subject to numerous risks that you should be aware of before making an investment decision. These risks are described more fully in the section entitled “Risk Factors” immediately following this prospectus summary. These risks include, among others:
| • | We are dependent on the success of our lead drug candidate, which may not receive regulatory approval or be successfully commercialized; |
| • | A number of different factors could prevent us from obtaining regulatory approval or commercializing our product candidates on a timely basis, or at all; |
| • | The approach we are taking to discover and develop novel therapeutics using RNAi is unproven and may never lead to marketable products; |
| • | The FDA could impose a unique regulatory regime for RNAi therapeutics; |
| • | Even if we receive regulatory approval to market our product candidates, our product candidates may not be accepted commercially, which may prevent us from becoming profitable; |
6
Table of Contents
| • | We are subject to significant competition and may not be able to compete successfully; |
| • | We are dependent on technologies we license, and if we lose the right to license such technologies or fail to license new technologies in the future, our ability to develop new products would be harmed; |
| • | We may be unable to protect our intellectual property rights licensed from other parties; our intellectual property rights may be inadequate to prevent third parties from using our technologies or developing competing products; and we may need to license additional intellectual property from others; |
| • | We may not be able to obtain sufficient financing and may not be able to develop our product candidates; |
| • | Future financing may be obtained through, and future development efforts may be paid for by, the issuance of debt or equity, which may have an adverse effect on our stockholders or may otherwise adversely affect our business; |
| • | We expect to continue to incur significant research and development expenses, which may make it difficult for us to attain profitability, and may lead to uncertainty as to our ability to continue as a going concern; |
| • | We will rely upon third parties for the manufacture of our clinical product candidates; |
| • | We may not be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our product candidates; |
| • | We are subject to potential liabilities from clinical testing and future product liability claims; |
| • | Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could have a material adverse effect on our business; |
| • | Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory review. If we fail to comply with continuing U.S. and foreign regulations, we could lose our approvals to market drugs and our business would be materially and adversely affected; |
| • | If we fail to attract, hire and retain qualified personnel, we may not be able to design, develop, market or sell our products or successfully manage our business; |
| • | We may decide not to exercise our option to acquire MirImmune; and |
| • | If we do acquire MirImmune, the acquisition may not create operational efficiencies or make commercial success more likely. |
Our Corporate Information
RXi is a Delaware corporation. Our principal executive offices are located at 257 Simarano Drive, Suite 101 Marlborough, Massachusetts 01752, and our telephone number is (508) 767-3861. Our Internet address is www.rxipharma.com. Our website and the information contained on that site, or connected to that site, is not part of or incorporated by reference into this prospectus.
7
Table of Contents
THE OFFERING
| Units offered by us |
We are offering 2,017,000 Class A Units and 9,600 Class B Units (based on an assumed offering price per common share of $1.19, which was the last reported sale price of our common stock on November 10, 2016). |
| Class A Units offered by us |
Each Class A Unit consists of one share of common stock and a warrant to purchase half of one share of our common stock, together with the shares of common stock underlying the warrants. The Class A Units will not be certificated and the shares of common stock and warrants part of such Unit are immediately separable and will be issued separately in this offering. |
| Public offering price per Class A Unit |
$ . |
| Class B Units offered by us |
We are also offering to those purchasers, whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 9.99% of our outstanding common stock following the consummation of this offering, the opportunity to purchase, in lieu of the number of Class A Units that would result in ownership in excess of 9.99%, 9,600 Class B Units. Each Class B Unit will consist of one share of Series B Convertible Preferred Stock, par value $0.0001 per share, convertible into 840 shares of common stock and warrants to purchase 420 shares of our common stock (together with the shares of common stock underlying such shares of Series B Convertible Preferred Stock and warrants). No shares of Series B Convertible Preferred Stock were outstanding prior to this offering. The Class B Units will not be certificated and the shares of Series B Convertible Preferred Stock and warrants part of such Unit are immediately separable and will be issued separately in this offering. |
| Public offering price per Class B Unit |
$ . |
| Over-allotment option |
The underwriters have the option to purchase additional shares of common stock, and/or warrants to purchase shares of common stock solely to cover over-allotments, if any, at the price to the public less the underwriting discounts and commissions. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters, but such purchases cannot exceed an aggregate of 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Convertible Preferred Stock) and warrants sold in the primary offering. The over-allotment option is exercisable for 45 days from the date of this prospectus. |
| Description of warrants |
The warrants will be exercisable beginning on the date of issuance and expire on the year anniversary of the date of issuance at an initial exercise price per share equal to . |
| Common stock outstanding prior to this |
6,599,846 shares, as of September 30, 2016. |
8
Table of Contents
| Series B Convertible Preferred Stock outstanding prior to this offering |
0. |
| Common stock outstanding after this offering |
8,616,846 shares(1)(2). |
| Series B Convertible Preferred Stock to be outstanding after this offering. |
9,600 shares. |
| Use of proceeds |
We intend to use the proceeds from this offering (i) to support our ongoing and future clinical trials, (ii) if the option agreement with MirImmune is exercised by the Company, to support the potential acquisition of MirImmune and the development of its immunotherapy pipeline and (iii) to support general corporate purposes and general and administrative expenses. See “Use of Proceeds.” |
| Risk factors |
You should read the “Risk Factors” section of this prospectus for a discussion of factors to consider carefully before deciding to invest in shares of our common stock. |
| NASDAQ Capital Market symbol |
Our common stock is listed on the Nasdaq Capital Market under the symbol “RXII.” On November 10, 2016, the closing price for our common stock was $1.19 per share. |
| Governing Law |
The offering will be governed by the laws of the State of Delaware. |
| (1) | The number of shares of common stock to be outstanding after this offering is based on 6,599,846 shares of common stock outstanding as of September 30, 2016. The number of shares of our common stock to be outstanding after this offering excludes the following: |
| • | Shares of our common stock that may be issued upon conversion of shares of Series B Convertible Preferred Stock and exercise of warrants issued in this offering; |
| • | 390,969 shares of common stock issuable upon the exercise of stock options outstanding, having a weighted average exercise price of $26.38 per share; |
| • | 1,300,464 shares of common stock issuable upon the exercise of warrants outstanding, having a weighted average exercise price of $5.21 per share; |
| • | An aggregate of 108,831 shares of common stock reserved for future issuance under our 2012 Long-Term Incentive Plan; |
| • | An aggregate of zero shares of common stock reserved for future issuance under our Employee Stock Purchase Plan. |
| (2) | Except as otherwise indicated, the number of shares of common stock presented in this prospectus excludes shares issuable pursuant to the exercise of the underwriters’ over-allotment option. |
9
Table of Contents
Investing in the units involves a high degree of risk. Before investing in the units, you should consider carefully the risks described below, together with the other information contained in this prospectus. If any of the risks set forth below occur, our business, financial condition, results of operations and future growth prospects could be materially and adversely affected. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment.
Risks Relating to Our Business and Industry
We are dependent on the success of our lead drug candidates, which may not receive regulatory approval or be successfully commercialized.
RXI-109, our lead product candidate and first RNAi-based product candidate, is designed to reduce the expression of CTGF, a critical regulator of several biological pathways involved in fibrosis. Samcyprone™, our second clinical product candidate, is a proprietary topical formulation of the small molecule diphenylcyclopropenone (“DPCP”), an immunomodulator that works by initiating a T-cell response. We began the clinical program to reduce the formation of hypertrophic scars with RXI-109 in June 2012, and are currently conducting a Phase 2 clinical trial for RXI-109 in this indication and a Phase 1/2 clinical trial in retinal scarring. We initiated our Phase 2 clinical trial for the treatment of cutaneous warts with Samcyprone™ in December 2015. The U.S. Food and Drug Administration (“FDA”) may require additional information from the Company regarding our current or planned trials at any time, and such information may be costly to provide or cause potentially significant delays in development. There is no assurance that we will be able to successfully develop RXI-109, Samcyprone™ or any other product candidate.
We have no commercial products and currently generate no revenue from commercial sales or collaborations and may never be able to develop marketable products. The FDA or similar foreign governmental agencies must approve our products in development before they can be marketed. The process for obtaining FDA approval is both time-consuming and costly, with no certainty of a successful outcome. Before obtaining regulatory approval for the sale of any drug candidate, we must conduct extensive preclinical tests and successful clinical trials to demonstrate the safety and efficacy of our product candidates in humans. For example, although the results of our Phase 1 clinical trials and preliminary results of our Phase 2 clinical trials of RXI-109 are promising, additional clinical trials will be required to establish the safety and efficacy of RXI-109. While DPCP has been used by physicians for decades, we have not yet shown safety or efficacy in humans for Samcyprone™ or for any of our other product candidates. A failure of any preclinical study or clinical trial can occur at any stage of testing. The results of preclinical and initial clinical testing of these products may not necessarily indicate the results that will be obtained from later or more extensive testing. Preliminary observations made in early stages of clinical trials with small numbers of subjects are inherently uncertain. Investors are cautioned that initial clinical trial results are not necessarily indicative of results that will be obtained when full data sets are analyzed or in subsequent clinical trials.
A number of different factors could prevent us from obtaining regulatory approval or commercializing our product candidates on a timely basis, or at all.
We, the FDA or other applicable regulatory authorities, or an Institutional Review Board (“IRB”) may suspend clinical trials of a drug candidate at any time for various reasons, including if we or they believe the subjects participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a drug candidate on subjects in a clinical trial could result in the FDA or other regulatory authorities suspending or terminating the trial and refusing to approve a particular drug candidate for any or all indications of use.
Clinical trials of a new drug candidate require the enrollment of a sufficient number of subjects, including subjects who are suffering from the disease or condition the drug candidate is intended to treat and who meet
10
Table of Contents
other eligibility criteria. Rates of subject enrollment are affected by many factors, and delays in subject enrollment can result in increased costs and longer development times.
Clinical trials also require the review and oversight of IRBs, which approve and continually review clinical investigations and protect the rights and welfare of human subjects. An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical trials, and the FDA may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB review and approval.
Numerous factors could affect the timing, cost or outcome of our drug development efforts, including the following:
| • | Delays in filing or acceptance of initial drug applications for our product candidates; |
| • | Difficulty in securing centers to conduct clinical trials; |
| • | Conditions imposed on us by the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
| • | Problems in engaging IRBs to oversee trials or problems in obtaining or maintaining IRB approval of studies; |
| • | Difficulty in enrolling subjects in conformity with required protocols or projected timelines; |
| • | Third-party contractors failing to comply with regulatory requirements or to meet their contractual obligations to us in a timely manner; |
| • | Our drug candidates having unexpected and different chemical and pharmacological properties in humans than in laboratory testing and interacting with human biological systems in unforeseen, ineffective or harmful ways; |
| • | The need to suspend or terminate clinical trials if the participants are being exposed to unacceptable health risks; |
| • | Insufficient or inadequate supply or quality of our drug candidates or other necessary materials necessary to conduct our clinical trials; |
| • | Effects of our drug candidates not being the desired effects or including undesirable side effects or the drug candidates having other unexpected characteristics; |
| • | The cost of our clinical trials being greater than we anticipate; |
| • | Negative or inconclusive results from our clinical trials or the clinical trials of others for similar drug candidates or inability to generate statistically significant data confirming the efficacy of the product being tested; |
| • | Changes in the FDA’s requirements for testing during the course of that testing; |
| • | Reallocation of our limited financial and other resources to other clinical programs; and |
| • | Adverse results obtained by other companies developing similar drugs. |
It is possible that none of the product candidates that we may attempt to develop will obtain the appropriate regulatory approvals necessary to begin selling them or that any regulatory approval to market a product may be subject to limitations on the indicated uses for which we may market the product. The time required to obtain FDA and other approvals is unpredictable, but often can take years following the commencement of clinical trials, depending upon the complexity of the drug candidate. Any analysis we perform of data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenue from the particular drug candidate.
11
Table of Contents
We also are subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not assure approval by regulatory authorities outside of the United States.
The approach we are taking to discover and develop novel therapeutics using RNAi is unproven and may never lead to marketable products.
RNA interference is a relatively new scientific discovery. Our RNAi technologies have been subject to only limited clinical testing. To date, no company has received regulatory approval to market therapeutics utilizing RNAi, and a number of clinical trials of RNAi technologies by other companies have been unsuccessful. The scientific evidence to support the feasibility of developing drugs based on these discoveries is both preliminary and limited. To successfully develop RNAi-based products, we must resolve a number of issues, including stabilizing the RNAi material and delivering it into target cells in the human body. We may spend large amounts of money trying to resolve these issues and may never succeed in doing so. In addition, any compounds that we develop may not demonstrate in subjects the chemical and pharmacological properties ascribed to them in laboratory studies, and they may interact with human biological systems in unforeseen, ineffective or even harmful ways.
Samcyprone™ represents a novel approach, topical immunotherapy, to the treatment of skin disorders that presents development challenges to us and may never lead to marketable products.
Although DPCP, the active ingredient in Samcyprone™, has been used by physicians for several decades to stimulate regrowth of hair in patients with alopecia areata and to clear common warts, it has never been reviewed or approved by a regulatory authority as a drug. Other immunomodulatory compounds, such as Imiquimod and Picato®, have been approved for topical use in other indications by the FDA. Our formulation of DPCP, Samcyprone™, has been subject to only limited clinical testing. Further testing may show that Samcyprone™ may interact with human biological systems in unforeseen or ineffective ways. In addition, to successfully develop Samcyprone™ we must resolve a number of development challenges, including developing a consistent process for the safe administration of the product and establishing a consistent manufacturing process in line with the good manufacturing practice regulations. We may spend significant amounts of money to resolve these development challenges and to obtain regulatory approval for Samcyprone™ and may never succeed in doing so.
The FDA could impose a unique regulatory regime for our therapeutics.
The substances we intend to develop may represent a new class of drug, and the FDA has not yet established any definitive policies, practices or guidelines in relation to these drugs. While we expect any product candidates that we develop will be regulated as a new drug under the Federal Food, Drug, and Cosmetic Act, the FDA could decide to regulate them or other products we may develop as biologics under the Public Health Service Act. The lack of policies, practices or guidelines may hinder or slow review by the FDA of any regulatory filings that we may submit. Moreover, the FDA may respond to these submissions by defining requirements that we may not have anticipated.
Even if we receive regulatory approval to market our product candidates, our product candidates may not be accepted commercially, which may prevent us from becoming profitable.
The product candidates that we are developing are based on new technologies and therapeutic approaches. For example, RNAi products may be more expensive to manufacture than traditional small molecule drugs, which may make them costlier than competing small molecule drugs. Additionally, RNAi products do not readily cross the so-called blood brain barrier, are rapidly eliminated from circulating blood and, for various applications, are likely to require injection or implantation, which will make them less convenient to administer
12
Table of Contents
than drugs administered orally. Key participants in the pharmaceutical marketplace, such as physicians, medical professionals working in large reference laboratories, public health laboratories and hospitals, third-party payors and consumers may not accept products intended to improve therapeutic results based on our technologies. As a result, it may be more difficult for us to convince the medical community and third-party payors to accept and use our products or to provide favorable reimbursement. If medical professionals working with large reference laboratories, public health laboratories and hospitals choose not to adopt and use our technologies, our products may not achieve broader market acceptance.
Additionally, although we expect that we will have intellectual property protection for our technology, certain governments may elect to deny patent protection for drugs targeting diseases with high unmet medical need (e.g., as in the case of HIV) and allow in their country internationally unauthorized generic competition. If this were to happen, our commercial prospects for developing any such drugs would be substantially diminished in these countries.
We are subject to significant competition and may not be able to compete successfully.
We believe that numerous companies are investigating or plan to investigate a variety of proposed anti-scarring therapies in clinical trials or are working in the RNAi area generally. Many other companies are pursuing non-RNAi-based therapies for one or more fibrotic disease indications, including ocular scarring or other indications that we may seek to pursue. The companies include large and small pharmaceuticals, chemical and biotechnology companies, as well as universities, government agencies and other private and public research organizations.
We do not believe that there are any companies developing treatments directly competing with Samcyprone™ for warts, or for alopecia areata or cutaneous metastases of malignant melanoma. However, there are several treatments for each condition with which Samcyprone™ could potentially compete. For example, current topical medicinal treatments for common warts include salicylic acid, off label use of Imiquimod and Picato® and the most common ablative treatments include removal through medical procedures, such as cryotherapy, surgery or chemical peels.
Most of these competitors have substantially greater research and development capabilities and financial, scientific, technical, manufacturing, marketing, distribution and other resources than we have, and we may not be able to successfully compete with them. In addition, even if we are successful in developing our product candidates, in order to compete successfully we may need to be first to market or to demonstrate that our products are superior to therapies based on different technologies. A number of our competitors have already commenced clinical testing of product candidates and may be more advanced than we are in the process of developing products. If we are not first to market or are unable to demonstrate superiority, any products for which we are able to obtain approval may not be successful.
We are dependent on technologies we license, and if we lose the right to license such technologies or fail to license new technologies in the future, our ability to develop new products would be harmed.
Many patents in the fields we are pursuing have already been exclusively licensed to third parties, including our competitors. If any of our existing licenses are terminated, the development of the products contemplated by the licenses could be delayed or terminated and we may not be able to negotiate additional licenses on acceptable terms, if at all, which would have a material adverse effect on our business.
We may be unable to protect our intellectual property rights licensed from other parties; our intellectual property rights may be inadequate to prevent third parties from using our technologies or developing competing products; and we may need to license additional intellectual property from others.
Therapeutic applications of gene silencing technologies, formulations, delivery methods and other technologies that we license from third parties are claimed in a number of pending patent applications, but there
13
Table of Contents
is no assurance that these applications will result in any issued patents or that those patents would withstand possible legal challenges or protect our technologies from competition. The United States Patent and Trademark Office and patent granting authorities in other countries have upheld stringent standards for the RNAi patents that have been prosecuted so far. Consequently, pending patents that we have licensed and those that we own may continue to experience long and difficult prosecution challenges and may ultimately issue with much narrower claims than those in the pending applications. Third parties may hold or seek to obtain additional patents that could make it more difficult or impossible for us to develop products based on our technologies without obtaining a license to such patents, which licenses may not be available on attractive terms, or at all.
In addition, others may challenge the patents or patent applications that we currently license or may license in the future or that we own and, as a result, these patents could be narrowed, invalidated or rendered unenforceable, which would negatively affect our ability to exclude others from using the technologies described in these patents. There is no assurance that these patent or other pending applications or issued patents we license or that we own will withstand possible legal challenges. Moreover, the laws of some foreign countries may not protect our proprietary rights to the same extent as do the laws of the United States. Any patents issued to us or our licensors may not provide us with any competitive advantages, and there is no assurance that the patents of others will not have an adverse effect on our ability to do business or to continue to use our technologies freely. Our efforts to enforce and maintain our intellectual property rights may not be successful and may result in substantial costs and diversion of management time. Even if our rights are valid, enforceable and broad in scope, competitors may develop products based on technology that is not covered by our licenses or patents or patent applications that we own.
There is no guarantee that future licenses will be available from third parties for our product candidates on timely or satisfactory terms, or at all. To the extent that we are required and are able to obtain multiple licenses from third parties to develop or commercialize a product candidate, the aggregate licensing fees and milestones and royalty payments made to these parties may materially reduce our economic returns or even cause us to abandon development or commercialization of a product candidate.
Our success depends upon our ability to obtain and maintain intellectual property protection for our products and technologies.
The applications based on RNAi technologies claim many different methods, compositions and processes relating to the discovery, development, delivery and commercialization of RNAi therapeutics. Because this field is so new, very few of these patent applications have been fully processed by government patent offices around the world, and there is a great deal of uncertainty about which patents will issue, when, to whom and with what claims. Although we are not aware of any blocking patents or other proprietary rights, it is likely that there will be significant litigation and other proceedings, such as interference and opposition proceedings in various patent offices, relating to patent rights in the RNAi field. It is possible that we may become a party to such proceedings.
We may not be able to obtain sufficient financing and may not be able to develop our product candidates.
The Company’s current cash resources may not provide sufficient capital to fund our currently planned operations for at least the next twelve months. In the future, we may need to incur debt or issue equity in order to fund our planned expenditures as well as to make acquisitions and other investments. We cannot assure you that debt or equity financing will be available to us on acceptable terms or at all. If we cannot, or are limited in the ability to, incur debt, issue equity or enter into strategic collaborations, we may be unable to fund the discovery and development of our product candidates, address gaps in our product offerings or improve our technology.
We anticipate that we will need to raise substantial amounts of money to fund a variety of future activities integral to the development of our business, which may include but are not limited to the following:
| • | To conduct research and development to successfully develop our RNAi and immunotherapy technologies; |
14
Table of Contents
| • | To obtain regulatory approval for our products; |
| • | To file and prosecute patent applications and to defend and assess patents to protect our technologies; |
| • | To retain qualified employees, particularly in light of intense competition for qualified scientists; |
| • | To manufacture products ourselves or through third parties; |
| • | To market our products, either through building our own sales and distribution capabilities or relying on third parties; and |
| • | To acquire new technologies, licenses or products. |
We cannot assure you that any needed financing will be available to us on acceptable terms or at all. If we cannot obtain additional financing in the future, our operations may be restricted and we may ultimately be unable to continue to develop and potentially commercialize our product candidates.
Future financing may be obtained through, and future development efforts may be paid for by, the issuance of debt or equity, which may have an adverse effect on our stockholders or may otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity, any debt securities or preferred stock issued will have rights, preferences and privileges senior to those of holders of our common stock in the event of a liquidation. In such event, there is a possibility that once all senior claims are settled, there may be no assets remaining to pay out to the holders of common stock. In addition, if we raise funds through the issuance of additional equity, whether through private placements or public offerings, such an issuance would dilute your ownership in us.
The terms of debt securities may also impose restrictions on our operations, which may include limiting our ability to incur additional indebtedness, to pay dividends on or repurchase our capital stock, or to make certain acquisitions or investments. In addition, we may be subject to covenants requiring us to satisfy certain financial tests and ratios, and our ability to satisfy such covenants may be affected by events outside of our control.
We expect to continue to incur significant research and development expenses, which may make it difficult for us to attain profitability, and may lead to uncertainty as to our ability to continue as a going concern.
We expend substantial funds to develop our RNAi and immunotherapy technologies, and additional substantial funds will be required for further research and development, including preclinical testing and clinical trials of any product candidates, and to manufacture and market any products that are approved for commercial sale. Because the successful development of our products is uncertain, we are unable to precisely estimate the actual funds we will require to develop and potentially commercialize them. In addition, we may not be able to generate enough revenue, even if we are able to commercialize any of our product candidates, to become profitable.
If we are unable to achieve or sustain profitability or to secure additional financing, we may not be able to meet our obligations as they come due, raising substantial doubts as to our ability to continue as a going concern. Any such inability to continue as a going concern may result in our common stockholders losing their entire investment. There is no guarantee that we will become profitable or secure additional financing. Our financial statements contemplate that we will continue as a going concern and do not contain any adjustments that might result if we were unable to continue as a going concern. Changes in our operating plans, our existing and anticipated working capital needs, the acceleration or modification of our expansion plans, increased expenses, potential acquisitions or other events will all affect our ability to continue as a going concern.
15
Table of Contents
We will rely upon third parties for the manufacture of our clinical product candidates.
We do not have the facilities or expertise to manufacture supplies of any of our potential product candidates for clinical trials. Accordingly, we depend on a limited number of manufacturers to obtain supplies and we will need to either develop, contract for, or otherwise arrange for the necessary manufacturers for these supplies. If for any reason we are unable to obtain the supplies for our potential product candidates, we would have to seek to obtain it from another major manufacturer. There is no assurance that we will be able to timely secure needed supply arrangements on satisfactory terms, or at all. Our failure to secure these arrangements as needed could have a material adverse effect on our ability to complete the development of our product candidates or, if we obtain regulatory approval for our product candidates, to commercialize them.
We may not be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations and other third parties to support our discovery efforts, to formulate product candidates, to manufacture our product candidates and to conduct clinical trials for some or all of our product candidates. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, vendors and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential partners’ evaluation of the superiority of our technology over competing technologies, the quality of the preclinical and clinical data that we have generated and the perceived risks specific to developing our product candidates. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates. We cannot necessarily control the amount or timing of resources that our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill their obligations to us.
We are subject to potential liabilities from clinical testing and future product liability claims.
If any of our future products are alleged to be defective, they may expose us to claims for personal injury by subjects in clinical trials of our products or as a result of our distribution agreement with Ethicor Ltd. If our products are approved by the FDA, users may claim that such products caused unintended adverse effects. We will seek to obtain clinical trial insurance for clinical trials that we conduct, as well as liability insurance for any products that we market. There is no assurance that we will be able to obtain insurance in the amounts we seek, or at all. We anticipate that licensees who develop our products will carry liability insurance covering the clinical testing and marketing of those products. There is no assurance, however, that any insurance maintained by us or our licensees will prove adequate in the event of a claim against us. Even if claims asserted against us are unsuccessful, they may divert management’s attention from our operations and we may have to incur substantial costs to defend such claims.
Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could have a material adverse effect on our business.
If approved, we intend to sell our products to physicians, plastic surgeons and dermatologists, as well as hospitals, oncologists and clinics that receive reimbursement for the healthcare services they provide to their patients from third-party payors, such as Medicare, Medicaid and other domestic and international government programs, private insurance plans and managed care programs. Most third-party payors may deny reimbursement if they determine that a medical product was not used in accordance with cost-effective treatment methods, as determined by the third-party payor, was used for an unapproved indication or if they believe the cost of the product outweighs its benefits. Third-party payors also may refuse to reimburse for experimental procedures and
16
Table of Contents
devices. Furthermore, because our programs are still in development, we are unable at this time to determine their cost-effectiveness and the level or method of reimbursement for them. Increasingly, the third-party payors who reimburse patients are requiring that drug companies provide them with predetermined discounts from list prices, and are challenging the prices charged for medical products. If the price we are able to charge for any products we develop is inadequate in light of our development and other costs, our profitability could be adversely affected.
We currently expect that any drugs we develop may need to be administered under the supervision of a physician. Under currently applicable law, drugs that are not usually self-administered may be eligible for coverage by the Medicare program if:
| • | They are “incidental” to a physician’s services; |
| • | They are “reasonable and necessary” for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standard of medical practice; |
| • | They are not excluded as immunizations; and |
| • | They have been approved by the FDA. |
Insurers may refuse to provide insurance coverage for newly approved drugs, or insurance coverage may be delayed or be more limited than the purpose for which the drugs are approved by the FDA. Moreover, eligibility for insurance coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for new drugs that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to develop products and our overall financial condition.
Additionally, third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for medical products and services. Levels of reimbursement may decrease in the future, and future legislation, regulation or reimbursement policies of third-party payors may adversely affect the demand for and price levels of our products. If our customers are not reimbursed for our products, they may reduce or discontinue purchases of our products, which could have a material adverse effect on our business, financial condition and results of operations.
Comprehensive healthcare reform legislation, which became law in 2010, could adversely affect our business and financial condition. Among other provisions, the legislation provides that a “biosimilar” product may be approved by the FDA on the basis of analytical tests and certain clinical studies demonstrating that such product is highly similar to an existing, approved product and that switching between an existing product and the biosimilar product will not result in diminished safety or efficacy. This abbreviated regulatory approval process may result in increased competition if we are able to bring a product to market. The legislation also includes more stringent compliance programs for companies in various sectors of the life sciences industry with which we may need to comply and enhanced penalties for non-compliance with the new healthcare regulations. Complying with new regulations may divert management resources, and inadvertent failure to comply with new regulations may result in penalties being imposed on us.
Some states and localities have established drug importation programs for their citizens, and federal drug import legislation has been introduced in Congress. The Medicare Prescription Drug Plan legislation, which
17
Table of Contents
became law in 2003, required the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States. The Secretary, however, retained the discretion not to implement a drug reimportation plan if the Secretary finds that the benefits do not outweigh the costs, and has so far declined to approve a reimportation plan. Proponents of drug reimportation may attempt to pass legislation that would directly allow reimportation under certain circumstances. Legislation or regulations allowing the reimportation of drugs, if enacted, could decrease the price we receive for any products that we may develop and adversely affect our future revenues and prospects for profitability.
Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory review. If we fail to comply with continuing U.S. and foreign regulations, we could lose our approvals to market drugs and our business would be materially and adversely affected.
Following regulatory approval of any drugs we may develop, we will remain subject to continuing regulatory review, including the review of adverse drug experiences and clinical results that are reported after our drug products are made available to patients. This would include results from any post-marketing tests or vigilance required as a condition of approval. The manufacturer and manufacturing facilities we use to make any of our drug products will also be subject to periodic review and inspection by the FDA. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the drug or manufacturer or facility, including withdrawal of the drug from the market. We would continue to be subject to the FDA requirements governing the labeling, packaging, storage, advertising, promotion, recordkeeping and submission of safety and other post-market information for all of our product candidates, even those that the FDA had approved. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
If we fail to attract, hire and retain qualified personnel, we may not be able to design, develop, market or sell our products or successfully manage our business.
Our business prospects are dependent on our management team and all of our employees. The loss of any of our key employees, including Drs. Cauwenbergh and Pavco, who serve as our Chief Executive Officer and our Chief Development Officer, respectively, or our inability to identify, attract, retain and integrate additional qualified key personnel, could make it difficult for us to manage our business successfully and achieve our business objectives.
Competition for skilled research, product development, regulatory and technical personnel is intense, and we may not be able to recruit and retain the personnel we need. The loss of the services of any key research, product development, regulatory and technical personnel, or our inability to hire new personnel with the requisite skills, could restrict our ability to develop our product candidates.
Risks Relating to Our Securities
The price of our common stock has been and may continue to be volatile.
The stock markets, in general, and the markets for drug delivery and pharmaceutical company stocks, in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may adversely affect the trading price of our common stock. In addition, the limited trading volume of our stock may contribute to its volatility.
In the past, following periods of volatility in the market price of a particular company’s securities, litigation has often been brought against that company. If litigation of this type is brought against us, it could be extremely expensive and divert management’s attention and the Company’s resources.
18
Table of Contents
We are issuing shares of Series B Convertible Preferred Stock in this offering and possibly may issue more shares of Series B Convertible Preferred Stock or designate more classes of preferred stock in the future, and the terms of the preferred stock may reduce the value of our common stock.
We are authorized to issue up to 10,000,000 shares of preferred stock in one or more series. Our Board of Directors may determine the terms of future preferred stock offerings without further action by our stockholders. The issuance of our preferred stock could affect your rights or reduce the value of our outstanding preferred stock or common stock. In particular, rights granted to holders of certain series of preferred stock may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights and restrictions on our ability to merge with or sell our assets to a third party.
We may acquire other businesses or form joint ventures that may be unsuccessful and could dilute your ownership interest in the Company.
As part of our business strategy, we may pursue future acquisitions of other complementary businesses and technology licensing arrangements. We also may pursue strategic alliances. We have no experience with respect to acquiring other companies and limited experience with respect to the formation of collaborations, strategic alliances and joint ventures. We may not be able to integrate such acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. We also could experience adverse effects on our reported results of operations from acquisition related charges, amortization of acquired technology and other intangibles and impairment charges relating to write-offs of goodwill and other intangible assets from time to time following the acquisition. Integration of an acquired company requires management resources that otherwise would be available for ongoing development of our existing business. We may not realize the anticipated benefits of any acquisition, technology license or strategic alliance.
To finance future acquisitions, we may choose to issue shares of our common stock or preferred stock as consideration, which would dilute your ownership interest in us. Alternatively, it may be necessary for us to raise additional funds through public or private financings. Additional funds may not be available on terms that are favorable to us and, in the case of equity financings, may result in dilution to our stockholders. Any future acquisitions by us also could result in large and immediate write-offs, the incurrence of contingent liabilities or amortization of expenses related to acquired intangible assets, any of which could harm our operating results.
We may decide not to exercise our option to acquire MirImmune Inc., and if we do acquire MirImmune Inc., the acquisition may be unsuccessful and could dilute your ownership interest in the Company.
On October 7, 2016, we entered into an exclusive option agreement pursuant to which the Company has the exclusive option, but not the obligation, to purchase 100% of the outstanding capital stock of MirImmune Inc. (“MirImmune”) at any time prior to April 5, 2017, for 19.99% of the then-outstanding shares of common stock of the Company plus additional potential consideration contingent on MirImmune achieving certain milestones. As part of our business strategy, we may decide to exercise this option to acquire MirImmune. If we exercise this option, we may not be able to integrate the acquisition successfully into our existing business or may not realize the anticipated benefits from the acquisition. To finance the acquisition, we would issue shares of our common stock as consideration, which would dilute your ownership interest in the Company. Your ownership interest in the Company could be further diluted, should MirImmune achieve certain milestones and receive additional shares of our common stock. Alternatively, we may decide not to exercise this option to acquire MirImmune.
We do not anticipate paying cash dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings in the development and growth of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of potential gain for the foreseeable future.
19
Table of Contents
You may experience dilution of your ownership interests due to the future issuance of additional shares or other securities that are convertible into or exercisable for shares.
We may issue additional shares of our authorized and unissued securities in the future, resulting in the dilution of the ownership interests of our present stockholders and investors in this offering. We currently are authorized to issue up to 100,000,000 shares of common stock shares, of which we expect 8,616,846 shares of common stock will be outstanding after closing of this offering.
Provisions of our certificate of incorporation and bylaws and Delaware law might discourage, delay or prevent a change of control of the Company or changes in our management and, as a result, depress the trading price of our common stock.
Our certificate of incorporation and bylaws contain provisions that could discourage, delay or prevent a change of control of the Company or changes in our management that the stockholders of the Company may deem advantageous. These provisions:
| • | Authorize the issuance of “blank check” preferred stock that our Board of Directors could issue to increase the number of outstanding shares and to discourage a takeover attempt; |
| • | Prohibit stockholder action by written consent, which requires all stockholder actions to be taken at a meeting of our stockholders; |
| • | Provide that the Board of Directors is expressly authorized to adopt, alter or repeal our bylaws; and |
| • | Establish advance notice requirements for nominations for election to our Board of Directors or for proposing matters that can be acted upon by stockholders at stockholder meetings. |
Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our Board of Directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management team by making it more difficult for stockholders to replace members of our Board of Directors, which is responsible for appointing the members of our management.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Risks Related to this Offering
We have broad discretion in how we use the net proceeds of this offering, and we may not use these proceeds effectively or in ways with which you agree.
Our management will have broad discretion as to the application of the net proceeds of this offering and could use them for purposes other than those contemplated at the time of this offering. Our stockholders may not agree with the manner in which our management chooses to allocate and spend the net proceeds. Moreover, our management may use the net proceeds for corporate purposes that may not increase the market price of our common stock.
The offering price was set by our Board of Directors and does not necessarily indicate the actual or market value of our common stock.
Our Board of Directors approved the offering price and other terms of this offering after considering, among other things: the number of shares authorized in our certificate of incorporation; the current market price of our
20
Table of Contents
common stock; trading prices of our common stock over time; the volatility of our common stock; our current financial condition and the prospects for our future cash flows; the availability of and likely cost of capital of other potential sources of capital; and market and economic conditions at the time of the offering. The offering price is not intended to bear any relationship to the book value of our assets or our past operations, cash flows, losses, financial condition, net worth or any other established criteria used to value securities. The offering price may not be indicative of the fair value of the common stock.
Because the public offering price of our common stock will be substantially higher than the net tangible book value per share of our outstanding common stock following this offering, new investors will experience immediate and substantial dilution.
The public offering price of our common stock will be substantially higher than the net tangible book value per share of our common stock immediately following this offering based on the total value of our tangible assets less our total liabilities. Therefore, if you purchase shares of our common stock in this offering, you will experience immediate and substantial dilution. See the section entitled “Dilution” below for a more detailed discussion of the dilution you will incur if you purchase common stock in this offering.
The warrants and the Series B Convertible Preferred Stock are unlisted securities and there is no public market for them.
There is no established public trading market for the warrants or the Series B Convertible Preferred Stock, and we do not expect a market to develop. In addition, the warrants and Series B Convertible Preferred Stock are not listed, and we do not intend to apply for listing of the warrants or the Series B Convertible Preferred Stock on any securities exchange or trading system. Without an active market, the liquidity of the warrants and the Series B Convertible Preferred Stock is limited, and investors may be unable to liquidate their investments in the warrants and Series B Convertible Preferred Stock.
The warrants may not have any value.
The warrants will be exercisable for years from the closing date at an initial exercise price per share of $ . In the event that the price of a share of our common stock does not exceed the exercise price of the warrants during the period when the warrants are exercisable, the warrants may not have any value.
A warrant does not entitle the holder to any rights as common stockholders until the holder exercises the warrant for shares of our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, the warrants will not provide you any rights as a common stockholder. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs on or after the exercise date.
21
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements can be identified by words such as “intends,” “believes,” “anticipates,” “indicates,” “plans,” “expects,” “suggests,” “may,” “should,” “potential,” “designed to,” “will” and similar references. Such statements include, but are not limited to, statements about: our ability to successfully develop RXI-109 and our other product candidates; the future success of our clinical trials with RXI-109; the timing for the commencement and completion of clinical trials; the future success of our strategic partnerships; and our ability to implement cost-saving measures. Forward-looking statements are neither historical facts nor assurances of future performance. These statements are based only on our current beliefs, expectations and assumptions regarding the future of our business, future plans and strategies, projections, anticipated events and trends, the economy and other future conditions.
Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict and many of which are outside of our control. Our actual results and financial condition may differ materially from those indicated in the forward-looking statements. Therefore, you should not rely on any of these forward-looking statements. Important factors that could cause our actual results and financial condition to differ materially from those indicated in the forward-looking statements include, among others: the risk that our clinical trials with RXI-109 may not be successful in evaluating the safety and tolerability of RXI-109 or providing evidence of increased surgical scar reduction compared to placebo; the successful and timely completion of clinical trials; uncertainties regarding the regulatory process; the availability of funds and resources to pursue our research and development projects, including our clinical trials with RXI-109; the risk that we may not exercise our option to acquire MirImmune; and those identified in this prospectus under the heading “Risk Factors” and in other filings the Company periodically makes with the Securities and Exchange Commission. Forward-looking statements contained in this prospectus speak as of the date hereof and the Company does not undertake to update any of these forward-looking statements to reflect a change in its views or events or circumstances that occur after such date.
In evaluating our business, prospective investors should carefully consider these factors in addition to the other information set forth in this prospectus, including under the caption “Risk Factors.” All forward-looking statements included in this document are based on information available to us on the date hereof. We disclaim any intent to update any forward-looking statements.
22
Table of Contents
We intend to use the proceeds from this offering (i) to support our ongoing and future clinical trials, (ii) if the option agreement with MirImmune is exercised by the Company, to support the potential acquisition of MirImmune and the development of its immunotherapy pipeline and (iii) to support general corporate purposes and general and administrative expenses. We may also use a portion of the net proceeds to acquire or invest in complementary businesses, products and technologies or to fund the development of any such complementary businesses, products or technologies that we may acquire in a stock-based acquisition. Other than with respect to our option agreement with MirImmune, we have no current plans for any such acquisitions.
Our common stock is listed on the NASDAQ Capital Market under the symbol “RXII.” On April 14, 2016, we effected a 1-for-10 reverse stock split. The share prices in the table below are shown on a post-split basis. The following table shows the high and low per-share sale prices of our common stock for the periods indicated:
| High | Low | |||||||
| 2014 |
||||||||
| First Quarter |
$ | 68.40 | $ | 28.20 | ||||
| Second Quarter |
44.40 | 26.00 | ||||||
| Third Quarter |
39.80 | 19.30 | ||||||
| Fourth Quarter |
23.00 | 14.00 | ||||||
| 2015 |
||||||||
| First Quarter |
$ | 17.30 | $ | 6.90 | ||||
| Second Quarter |
8.70 | 3.42 | ||||||
| Third Quarter |
5.50 | 3.50 | ||||||
| Fourth Quarter |
6.49 | 3.56 | ||||||
| 2016 |
||||||||
| First Quarter |
$ | 4.00 | $ | 2.60 | ||||
| Second Quarter |
3.27 | 1.26 | ||||||
| Third Quarter |
2.67 | 1.70 | ||||||
| Fourth Quarter (through November 10, 2016) |
1.85 | 0.94 | ||||||
On November 10, 2016, the last reported sale price of our common stock on The NASDAQ Capital Market was $1.19 per share. As of September 30, 2016, we had approximately 107 holders of record of our common stock.
23
Table of Contents
We have never declared or paid any cash dividends and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We expect to retain future earnings, if any, for use in our development activities and the operation of our business. The payment of any future dividends will be subject to the discretion of our Board of Directors and will depend, among other things, upon our results of operations, financial condition, cash requirements, prospects and other factors that our Board of Directors may deem relevant.
24
Table of Contents
Our net tangible book value as of September 30, 2016 was $3.0 million or $0.46 per share of common stock. Net tangible book value per share represents total tangible assets less total liabilities, divided by the number of shares of common stock outstanding. Assuming that we issue only Class A Units (and no Class B Units) at a per share offering price of $1.19, which is the last reported sale of our common stock on the NASDAQ Capital Market on November 10, 2016, and excluding units that may be issued upon exercise of the underwriters’ over-allotment option and shares of common stock that may be issued and any proceeds received upon exercise of warrants and shares of common stock issuable upon exercise of warrants and after deduction of the underwriters’ fees and estimated offering expenses payable by us, our net tangible book value as of September 30, 2016, would have been $5.0 million, or $0.58 per share of common stock. This represents an immediate increase in net tangible book value of $0.12 per share to existing stockholders and an immediate dilution in net tangible book value of $0.61 per share to purchasers of common stock in this offering.
The following table illustrates this per-share dilution:
| Public offering price per share of common stock included in a Class A Unit |
$1.19 | |
|
| ||
| Net tangible book value per share as of September 30, 2016 |
$0.46 | |
| Increase per share attributable to this offering |
$0.12 | |
| As adjusted net tangible book value per share after giving effect to this offering |
$0.58 | |
| Dilution per share to investors in this offering |
$0.61 |
If the underwriters’ over-allotment option is exercised in full, dilution per share to new investors would be $0.54 per share of common stock.
The number of shares of common stock outstanding used in the calculations above is based on 6,599,846 shares outstanding as of September 30, 2016, and excludes:
| • | Shares of our common stock that may be issued upon conversion of shares of Series B Convertible Preferred Stock and exercise of warrants issued in this offering; |
| • | 390,969 shares of common stock issuable upon the exercise of stock options outstanding, having a weighted average exercise price of $26.38 per share; |
| • | 1,300,464 shares of common stock issuable upon the exercise of warrants outstanding, having a weighted average exercise price of $5.21 per share; |
| • | an aggregate of 108,831 shares of common stock reserved for future issuance under our 2012 Long-Term Incentive Plan; and |
| • | an aggregate of zero shares of common stock reserved for future issuance under our Employee Stock Purchase Plan. |
Furthermore, we may choose to raise additional capital through the sale of equity or convertible debt securities due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. New investors will experience further dilution if any of our outstanding options or warrants are exercised, new options are issued and exercised under our equity incentive plans or we issue additional shares of common stock, other equity securities or convertible debt securities in the future.
25
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion of our financial condition and results of operations should be read in conjunction with our financial statements and the notes to those financial statements included elsewhere in this prospectus. This discussion contains forward-looking statements that involve significant risks and uncertainties. As a result of many factors, such as those set forth under “Risk Factors” and elsewhere in this prospectus, our actual results may differ materially from those anticipated in these forward-looking statements. Please refer to the discussion under the heading “Forward-Looking Statements” above.
Overview
RXi Pharmaceuticals Corporation (“RXi,” “we,” “our” or the “Company”) is a clinical-stage RNAi company developing innovative therapeutics that address significant unmet medical needs. The Company’s development programs are based on our proprietary self-delivering RNAi (sd-rxRNA®) platform and Samcyprone™, a topical immunomodulator. Our clinical development programs include RXI-109, an sd-rxRNA for the treatment of dermal and ocular scarring, and Samcyprone™, for the treatment of such disorders as warts, alopecia areata, non-malignant skin tumors and cutaneous metastases of melanoma. In addition to these clinical programs, we have a pipeline of discovery and preclinical product candidates in our core therapeutic areas, as well as in other areas of interest. The Company’s pipeline, coupled with our extensive patent portfolio, provides for product and business development opportunities across a broad spectrum of therapeutic areas.
RNAi therapies are designed to “silence,” or down-regulate, the expression of a specific gene that may be over-expressed in a disease condition. The Company’s first RNAi clinical product candidate, RXI-109, is a self-delivering RNAi compound (sd-rxRNA) that commenced human clinical trials in 2012. RXI-109 is designed to reduce the expression of connective tissue growth factor (“CTGF”), a critical regulator of several biological pathways involved in fibrosis, including scar formation in the skin and eye. RXI-109 is currently being evaluated in a Phase 2 clinical trial, Study 1402, to prevent or reduce dermal scarring following scar revision surgery of an existing hypertrophic scar and a Phase 1/2 clinical trial, Study 1501, to evaluate the safety and clinical activity of RXI-109 to prevent the progression of retinal scarring in subjects with wet age-related macular degeneration (“AMD”).
Study 1402 commenced in July 2014. In October 2015, we reported that preliminary data from Study 1402 demonstrated that scars at revision sites were less visible at three months after a treatment regimen with intradermal administration of RXI-109 than scars at untreated revision sites in those same subjects. Based in part on this new information, two more cohorts were added to Study 1402 in November 2015. For these two cohorts, the number of doses was increased to either eight or nine doses of RXI-109 over a six-month period to better cover the extended wound healing/scarring profile of hypertrophic scars. Enrollment of subjects into these two new cohorts completed ahead of schedule during the third quarter of 2016.
Study 1501 commenced in November 2015, and is a multi-dose, dose escalation study conducted in subjects with AMD with evidence of subretinal fibrosis. Each subject will receive four doses of RXI-109 by intraocular injection at one month intervals for a total dosing period of three months. The safety and tolerability of RXI-109, as well as the potential for clinical activity, will be evaluated over the course of the study using numerous assessments to monitor the health in the retina and to assess visual acuity. The Company is currently enrolling subjects in Study 1501.
In December 2014, the Company broadened its clinical pipeline with an exclusive, global license to Samcyprone™, our second clinical candidate. Samcyprone™ is a proprietary topical formulation of the small molecule diphenylcyclopropenone (“DPCP”), an immunomodulator that works by initiating a T-cell response. The use of Samcyprone™ allows sensitization using much lower concentrations of DPCP than are used with existing compounded DPCP solutions, avoiding hyper-sensitization to subsequent challenge doses. DPCP, the active ingredient in Samcyprone™, has long been used to treat warts and has also been used for several other
26
Table of Contents
indications, such as to stimulate hair re-growth in alopecia areata and to clear cutaneous metastases of melanoma. Although it has been used by physicians for several decades, it has never been reviewed or approved by a regulatory authority as a drug. If U.S. Food and Drug Administration approval is granted, Samcyprone™, RXi’s proprietary formulation of DPCP, is expected to achieve market exclusivity. Samcyprone™ is currently being evaluated in a Phase 2a clinical trial, Study 1502, for the clearance of common warts.
Study 1502 was initiated in December 2015. Study 1502 includes a sensitization phase in which a spot on the subject’s upper arm and one wart are treated with Samcyprone™. After being sensitized in this way, the subjects will enter into the treatment phase where up to four warts are treated on a once weekly basis for ten weeks with a ten-fold lower concentration of Samcyprone™ than in the sensitization phase. During the trial, the warts are scored, photographed and measured to monitor the level of clearance. The Company is currently enrolling subjects and is adding a second cohort to explore the opportunity to reduce the sensitization dose level and potentially reduce the treatment length. With this second cohort, enrollment is expected to be completed in the first quarter of 2017.
The Company continues to advance additional preclinical and discovery programs using our sd-rxRNA technology. Within our ophthalmology program, we are also directing our development efforts toward advancing RXI-109 for the treatment of corneal scarring. To date, we have shown that CTGF protein levels are reduced in a dose-dependent manner in both the retina and cornea following an intravitreal injection of RXI-109 in monkeys. Elevated CTGF is implicated in the formation of corneal scarring that can occur after eye injury or after certain infections. Scarring of the cornea can impact the transparency of the cornea, and thus negatively impact vision. We are currently working towards a non-invasive delivery formulation of RXI-109 to reduce CTGF in the front of the eye.
Within our dermatology franchise, the Company has selected tyrosinase (“TYR”) and collagenase (“MMP1”) as targets for our self-delivering RNAi platform because they are relevant for both consumer health and therapeutic development. TYR is a key enzyme involved in the synthesis of melanin. RXI-231, an sd-rxRNA compound targeting TYR, is in development as a cosmetic ingredient that may improve the appearance of uneven skin tone and pigmentation. MMP1 is a key enzyme involved in the breakdown of the extracellular matrix. RXI-185, an sd-rxRNA compound targeting MMP1, is in development as a cosmetic ingredient that may improve the appearance of wrinkles or skin laxity. The Company is currently developing topical delivery application methods, including formulations and microneedling, for use with these compounds and completed functional and safety testing to support the initiation of human testing of one of these consumer health targets.
Further, the Company has identified additional sd-rxRNA compounds like RXI-231 and RXI-185 that are available to move forward on a separate therapeutic development path. For example, selected reduction of MMP1 may be beneficial in the treatment of arthritis, corneal erosions, endometriosis and possible cancer metastasis and the inhibition of tyrosinase can play a key role in the management of diseases such as cutaneous hyperpigmentation disorders and possibly melanoma.
On April 14, 2016, the Board of Directors of the Company approved a 1-for-10 reverse stock split of the Company’s outstanding common stock, which was effected on April 18, 2016. The number of authorized shares of the Company remain unchanged. Stockholders who would have otherwise been entitled to fractional shares as a result of the reverse stock split received a cash payment in lieu of receiving fractional shares. Shares of common stock underlying outstanding stock options and other equity instruments were proportionately reduced and the respective exercise prices, if applicable, were proportionately increased in accordance with the terms of the agreements governing such securities. All share and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock split, including reclassifying an amount equal to the reduction in par value to additional paid-in capital.
Since inception, we have incurred significant losses. Substantially all of our losses to date have resulted from research and development expenses in connection with our clinical and research programs and from general administrative costs. We had an accumulated deficit of $63.8 million and $57.1 million at September 30, 2016
27
Table of Contents
and December 31, 2015 respectively. We expect to continue to incur significant losses for the foreseeable future, particularly as we advance our development programs for RXI-109 and Samcyprone™.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with United States generally accepted accounting principles (“GAAP”). The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to the impairment of long-lived assets, certain accrued expenses and stock-based compensation. We base our estimates on historical experience and various other assumptions that are believed to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions and could have a material impact on our reported results. While our significant accounting policies are more fully described in the notes to our financial statements included elsewhere in this Form S-1, we believe the following accounting policies to be the most critical in understanding the judgments and estimates we use in preparing our financial statements.
Research and Development Expenses
Research and development costs are charged to expense as incurred and relate to salaries, employee benefits, facility-related expenses, supplies, stock-based compensation related to employees and non-employees involved in the Company’s research and development, external services, other operating costs and overhead related to our research and development departments, costs to acquire technology licenses and expenses associated with preclinical activities and our clinical trials. Payments made by the Company in advance for research and development services not yet provided and/or for materials not yet received are recorded as prepaid expenses. Accrued liabilities are recorded related to those expenses for which vendors have not yet billed us with respect to services provided and/or materials that we have received.
Preclinical and clinical trial expenses relate to third-party services, subject-related fees at the sites where our clinical trials are being conducted, laboratory costs, analysis costs, toxicology studies and investigator fees. Costs associated with these expenses are generally payable on the passage of time or when certain milestones are achieved. Expense is recorded during the period incurred or in the period in which a milestone is achieved. In order to ensure that we have adequately provided for preclinical and clinical expenses during the proper period, we maintain an accrual to cover these expenses. These accruals are assessed on a quarterly basis and are based on such assumptions as expected total cost, the number of subjects and clinical trial sites and length of the study. Actual results may differ from these estimates and could have a material impact on our reported results. Our historical accrual estimates have not been materially different from our actual costs.
Stock-based Compensation
The Company follows the provisions of the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (“ASC”) Topic 718, “Compensation — Stock Compensation” (“ASC 718”), which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees, officers and non-employee directors, including stock options. Stock compensation expense based on the grant date fair value estimated in accordance with the provisions of ASC 718 is recognized as an expense over the requisite service period. Determining the amount of stock-based compensation to be recorded requires us to develop highly subjective estimates to be used in calculating the grant-date fair value of stock options. We use the Black-Scholes option pricing model to value our option grants and determine the related compensation expense. The use of the model requires us to make estimates of the following assumptions:
Expected volatility — Due to our limited trading history, we are responsible for estimating volatility and currently use the expected volatilities of similar entities. We have considered a number of factors in making our determination as to entities that are considered similar, such as the industry, stage of development, size of the company, and financial leverage.
28
Table of Contents
Expected term — We use the simplified method to estimate the expected term assumption. The simplified method is based on the vesting period and the contractual term for each grant, or for each vesting-tranche for awards with graded vesting.
Risk-free interest rate — The yield on zero-coupon U.S. Treasury securities for a period that is commensurate with the expected term assumption is used as the risk-free interest rate.
Dividend yield — We utilize a dividend yield of zero based on the fact that we have never paid cash dividends and currently have no intention to pay cash dividends.
For stock options granted as consideration for services rendered by non-employees, the Company recognizes compensation expense in accordance with the requirements of FASB ASC Topic 505-50, “Equity Based Payments to Non-Employees.” Non-employee option grants that do not vest immediately upon grant are recorded as an expense over the requisite service period of the underlying stock options. At the end of each financial reporting period prior to vesting, the value of these options, as calculated using the Black-Scholes option-pricing model, will be re-measured using the fair value of the Company’s common stock and the non-cash compensation recognized during the period will be adjusted accordingly. Since the fair market value of options granted to non-employees is subject to change in the future, the amount of the future compensation expense will include fair value re-measurements until the stock options are fully vested.
Derivative Financial Instruments
During the normal course of business we may issue warrants to vendors as consideration to perform services. We may also issue warrants as part of a debt of equity financing. Warrants and other derivative financial instruments are accounted for either as equity or as an asset or liability, depending on the characteristics of each derivative financial instrument. Warrants classified as equity are measured at fair value and recorded as additional paid in capital in stockholders’ equity at the date of issuance. No further adjustments to their valuation are made. Derivative financial instruments classified as an asset or liability are measured at fair value on the issuance date and are revalued on each subsequent balance sheet date. The changes in the fair value are recognized as current period income or loss.
Results of Operations
The following data summarizes the results of our operations for the periods indicated, in thousands:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Net revenues |
$ | — | $ | — | $ | 19 | $ | 34 | ||||||||
| Operating expenses |
(2,216 | ) | (2,504 | ) | (6,695 | ) | (7,649 | ) | ||||||||
| Operating loss |
(2,216 | ) | (2,504 | ) | (6,676 | ) | (7,615 | ) | ||||||||
| Net loss |
(2,212 | ) | (2,496 | ) | (6,655 | ) | (7,607 | ) | ||||||||
| Net loss applicable to common stockholders |
$ | (2,212 | ) | $ | (2,496 | ) | $ | (6,655 | ) | $ | (7,816 | ) | ||||
| For the Years Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Revenue |
$ | 34 | $ | 71 | ||||
| Operating expenses |
(10,271 | ) | (8,897 | ) | ||||
| Operating loss |
(10,237 | ) | (8,826 | ) | ||||
| Net loss |
(10,223 | ) | (8,800 | ) | ||||
| Net loss applicable to common stockholders |
(10,432 | ) | (12,930 | ) | ||||
29
Table of Contents
Comparison of the Three and Nine Months Ended September 30, 2016 and 2015
Net Revenues
To date, we have primarily generated revenues through government grants. The following table summarizes our total net revenues, for the periods indicated, in thousands:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Net revenues |
$ | — | $ | — | $ | 19 | $ | 34 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
There were no net revenues for the three months ended September 30, 2016 and September 30, 2015.
Net revenues were approximately $19,000 for the nine months ended September 30, 2016, as compared with $34,000 for the nine months ended September 30, 2015. The decrease of $15,000, or 44%, was due to the completion of a government grant from the National Cancer Institute, a division of the National Institutes of Health, in 2015. Net revenues for the nine months ended September 30, 2016 were due to the Company’s exclusive license agreements with MirImmune, Inc. and Thera Neuropharma, Inc.
Operating Expenses
The following table summarizes our total operating expenses, for the periods indicated, in thousands:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Research and development |
$ | 1,464 | $ | 1,734 | $ | 4,108 | $ | 5,202 | ||||||||
| General and administrative |
752 | 770 | 2,587 | 2,447 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
$ | 2,216 | $ | 2,504 | $ | 6,695 | $ | 7,649 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Research and Development Expenses
Research and development expenses consist of compensation-related costs for our employees dedicated to research and development activities, fees related to our Scientific Advisory Board members, expenses related to our ongoing research and development efforts primarily related to our clinical trials, drug manufacturing, outside contract services, licensing and patent fees and laboratory supplies and services for our research programs. We expect research and development expenses to increase as we expand our discovery, preclinical and clinical activities.
Research and development expenses were $1,464,000 for the three months ended September 30, 2016, compared with $1,734,000 for the three months ended September 30, 2015. The decrease of $270,000, or 16%, was primarily due to a decrease of $148,000 in research and development expenses, primarily due to manufacturing expenses for the RXI-109 drug product completed in the second half of 2015, offset by manufacturing and clinical trial-related costs for Samcyprone™, and a decrease of $122,000 in stock-based compensation expense due to the full vesting of stock options granted in 2012.
Research and development expenses were $4,108,000 for the nine months ended September 30, 2016, compared with $5,202,000 for the nine months ended September 30, 2015. The decrease of $1,094,000, or 21%, was primarily due to a decrease of $754,000 in research and development expenses related to the cash and equity fees payable to Hapten Pharmaceuticals, LLC upon the close of the Samcyprone™ license agreement, toxicology studies performed in connection with the Company’s investigational drug application for retinal scarring, both of which occurred in the first quarter of 2015, and manufacturing expenses for the RXI-109 drug product completed
30
Table of Contents
in the second half of 2015, offset by manufacturing and clinical trial-related costs for Samcyprone™. An additional decrease of $340,000 in stock-based compensation expense was due to the full vesting of stock options granted in 2012.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation-related costs for our employees dedicated to general and administrative activities, legal fees, audit and tax fees, consultants, professional services and general corporate expenses.
General and administrative expenses were $752,000 for the three months ended September 30, 2016, compared with $770,000 for the three months ended September 30, 2015. The decrease of $18,000, or 2%, was due to a decrease of $135,000 in stock-based compensation expense due to the full vesting of stock options granted in 2012, offset by an increase of $117,000 in general and administrative expenses primarily related to the use of outside professional services due to the Company’s increase in business development activities in line with its key corporate initiatives.
General and administrative expenses were $2,587,000 for the nine months ended September 30, 2016, compared with $2,447,000 for the nine months ended September 30, 2015. The increase of $140,000, or 6%, was primarily due to an increase of $394,000 in general and administrative expenses driven by the Company’s use of outside professional services due to the Company’s focus on business development activities in line with its key corporate initiatives and an increase in professional service fees primarily related to the Company’s special meeting and reverse stock split in April 2016, offset by a decrease of $254,000 in stock-based compensation expense due to the full vesting of stock options granted in 2012.
Series A and Series A-1 Convertible Preferred Stock Dividends
The following table summarizes our total Series A and Series A-1 convertible preferred stock (“Series A and Series A-1 Preferred Stock”) dividends for the periods indicated, in thousands:
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Series A and Series A-1 Preferred Stock dividends |
$ | — | $ | — | $ | — | $ | 209 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
On May 27, 2015, all shares of Series A and Series A-1 Preferred Stock were fully converted, with no shares remaining outstanding. Consequently, the Company has not paid any dividends on the Series A and Series A-1 Preferred Stock since the second quarter of 2015. Additionally, on November 6, 2015, the Company eliminated both series of preferred stock from its Certificate of Incorporation. As a result, the Company does not have any shares of Series A and Series A-1 Preferred Stock authorized, issued or outstanding.
Comparison of the Years Ended December 31, 2015 and 2014
Revenue
To date, we’ve generated revenue through government grants. The following table summarizes our total revenues from government grants, for the periods indicated, in thousands:
| For the Years Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Revenue |
$ | 34 | $ | 71 | ||||
|
|
|
|
|
|||||
31
Table of Contents
Total revenues were approximately $34,000 for the year ended December 31, 2015, compared with $71,000 for the year ended December 31, 2014. The decrease of $37,000, or 52%, was due to the completion of work on the Company’s outstanding government grant during the first quarter of 2015.
Operating Expenses
The following table summarizes our total operating expenses, for the periods indicated, in thousands:
| For the Years Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Research and development expenses |
$ | 6,925 | $ | 5,680 | ||||
| General and administrative expenses |
3,346 | 3,217 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
$ | 10,271 | $ | 8,897 | ||||
|
|
|
|
|
|||||
Research and Development Expenses
Total research and development expense was approximately $6,925,000 for the year ended December 31, 2015, compared with $5,680,000 for the year ended December 31, 2014. The increase of $1,245,000, or 22%, was due to an increase of $1,449,000 in research and development expense primarily related to the drug manufacturing expense related to the manufacture of RXI-109 and Samcyprone™ drug product during the year for use in the Company’s clinical trials as compared with the prior year. In addition, research and development expense increased due to research performed for the selection of our cosmetic targets and topical delivery applications for their use, as well as increases in employee-related expense due to the hire of new employees. The increase in research and development expense was offset by $204,000 in stock-based compensation due to a decrease in the valuation of stock options granted as compared to the prior year.
General and Administrative Expenses
General and administrative expense was approximately $3,346,000 for the year ended December 31, 2015, compared with $3,217,000 for the year ended December 31, 2014. The increase of $129,000, or 4%, was primarily due to an increase of $236,000 in general and administrative expenses primarily due to an increase in compensation expense, as well as professional services expense, due to the Company’s focus on business development activities as one of its key corporate initiatives, offset by a decrease of $107,000 in employee stock-based compensation expense.
Series A and Series A-1 Preferred Stock Dividends
The following table summarizes our total Series A and Series A-1 Preferred Stock dividends for the periods indicated, in thousands:
| For the Years Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Series A and Series A-1 Preferred Stock Dividends |
$ | 209 | $ | 4,130 | ||||
|
|
|
|
|
|||||
Total Series A and Series A-1 Preferred Stock dividends were approximately $209,000 for the year ended December 31, 2015, compared with $4,130,000 for the year ended December 31, 2014. The decrease of $3,921,000, or 95%, was due to a decrease in the Company’s common stock price on the dividend payment dates, the number of preferred shares earning dividends each quarter and the full conversion of the Series A and Series A-1 Preferred Stock during the quarter ended June 30, 2015, resulting in no further accumulation and payment of dividends on these series of preferred stock.
32
Table of Contents
No shares of the Series A and Series A-1 Preferred Stock remained outstanding at December 31, 2015, as all outstanding shares of Series A and Series A-1 Preferred Stock were fully converted into common stock on May 27, 2015, with no shares remaining outstanding. On November 6, 2015, the Company filed the Certificates of Elimination with respect to the Series A and Series A-1 Preferred Stock, as described further in Item 8 to our audited financial statements. As a result, the Company does not have any shares of Series A and Series A-1 Preferred Stock authorized, issued or outstanding.
Liquidity and Capital Resources
On June 2, 2015, we sold 2.6 million units in a public offering at a price of $4.00 per unit (the “Offering”). Each unit consists of one share of common stock, a 13-month overallotment purchase right to purchase one-half of one share of common stock at a price of $4.55 per full share of common stock (the “Overallotment Purchase Rights”) and a five-year warrant to purchase one-half of one share of common stock at a price of $5.20 per full share of common stock (the “2015 Warrants”). As a result of the Offering, the Company received net proceeds of approximately $9.2 million after placement agent fees and estimated Offering expenses, and assuming the Overallotment Purchase Rights and 2015 Warrants are not exercised.
Overallotment Purchase Rights totaling 1,300,002 were issued in connection with the Offering. During the year ended December 31, 2015, 43,500 Overallotment Purchase Rights were exercised for gross proceeds of $198,000. The Company’s remaining outstanding Overallotment Purchase Rights of 1,256,502 expired on July 2, 2016 and were not exercised. As of September 30, 2016, 1,300,002 2015 Warrants were issued and outstanding.
On December 18, 2014, the Company entered into a purchase agreement (the “Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“LPC”), pursuant to which the Company has the right to sell to LPC up to $10.8 million in shares of the Company’s common stock, subject to certain limitations and conditions set forth in the Purchase Agreement. To date, the Company has sold a total of 70,000 shares of common stock to LPC for net proceeds of approximately $216,000.
We had cash, cash equivalents and short-term investments of $4.4 million as of September 30, 2016, compared with $10.6 million as of December 31, 2015. Based on the Company’s operational spending rate to advance our clinical products through clinical trials, the Company’s current cash resources may not provide sufficient capital to fund operations for at least the next twelve months. The Company hopes to obtain additional funding through one or more of several options including strategic opportunities, such as a merger, acquisition or other business development transaction, and/or through sales of the Company’s securities, which could be dilutive to our stockholders. There can be no assurance that the Company will be successful in accomplishing these plans in order to continue as a going concern.
The following table summarizes our cash flows for the periods indicated, in thousands:
| Nine Months Ended September 30, |
||||||||
| 2016 | 2015 | |||||||
| Net cash used in operating activities |
$ | (6,388 | ) | $ | (5,735 | ) | ||
| Net cash provided by (used in) investing activities |
3,498 | (8,039 | ) | |||||
| Net cash provided by financing activities |
152 | 9,313 | ||||||
|
|
|
|
|
|||||
| Net decrease in cash and cash equivalents |
$ | (2,738 | ) | $ | (4,461 | ) | ||
33
Table of Contents
| For the Years Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Net cash used in operating activities |
$ | (7,317 | ) | $ | (7,758 | ) | ||
| Net cash provided by (used in) investing activities |
(5,557 | ) | 2,917 | |||||
| Net cash provided by financing activities |
9,495 | 1,947 | ||||||
|
|
|
|
|
|||||
| Net decrease in cash and cash equivalents |
$ | (3,379 | ) | $ | (2,894 | ) | ||
Net Cash Flow from Operating Activities
Net cash used in operating activities was $6,388,000 for the nine months ended September 30, 2016 compared with $5,735,000 for the nine months ended September 30, 2015. The increase in cash used in operating activities was due to changes in working capital items of $755,000, primarily due to payments related to the manufacturing of RXI-109 and Samcyprone™ drug product during the first quarter of 2016, and changes in non-cash expenses of $850,000 partially offset by a decrease in net loss of $952,000.
Net cash used in operating activities was $7,317,000 for the year ended December 31, 2015 compared with $7,758,000 for the year ended December 31, 2014. The decrease in cash used in operating activities was primarily due changes in working capital of $1,947,000 partially offset by an increase in net loss of $1,423,000.
Net Cash Flow from Investing Activities
Net cash provided by investing activities was $3,498,000 for the nine months ended September 30, 2016 compared with net cash used in investing activities of $8,039,000 for the nine months ended September 30, 2015. The increase in net cash provided by investing activities was primarily related to net purchases and maturities of short-term investments as compared with the same period in the prior year.
Net cash used in investing activities was $5,557,000 for the year ended December 31, 2015 compared with net cash provided by investing activities of $2,917,000 for the year ended December 31, 2014. Net cash used in and provided by investing activities primarily relates to net purchases of short-term investments and purchases of property and equipment.
Net Cash Flow from Financing Activities
Net cash provided by financing activities was $152,000 for the nine months ended September 30, 2016 compared with $9,313,000 for the nine months ended September 30, 2015. Net cash provided by financing activities in 2016 was due to net proceeds from the issuance of common stock to LPC under the Purchase Agreement. Net cash provided by financing activities in 2015 was primarily due to net proceeds received from the Offering and the issuance of common stock to LPC under the Purchase Agreement.
Net cash provided by financing activities was $9,495,000 for the year ended December 31, 2015 compared with $1,947,000 for the year ended December 31, 2014. Net cash provided by financing activities in 2015 was primarily due to net proceeds of $9,266,000 received in connection with the Offering and from the issuance of common stock to LPC pursuant to the Purchase Agreement and proceeds of $198,000 from the issuance of common stock upon the exercise of the 2015 Warrants. Net cash provided by financing activities in 2014 was primarily due to net proceeds of $1,886,000 from the issuance of common stock to LPC.
Off-Balance Sheet Arrangements
In connection with certain license agreements, we are required to indemnify the licensor for certain damages arising in connection with the intellectual property rights licensed under the agreement. In addition, we are a party to a number of agreements entered into in the ordinary course of business that contain typical provisions
34
Table of Contents
that obligate us to indemnify the other parties to such agreements upon the occurrence of certain events. These indemnification obligations are considered off-balance sheet arrangements in accordance with ASC Topic 460, “Guarantor’s Accounting and Disclosure Requirements for Guarantees, Including Indirect Guarantees of Indebtedness of Others.” To date, we have not encountered material costs as a result of such obligations and have not accrued any liabilities related to such obligations in our financial statements. See Note 6 to our financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2015, which was filed with the SEC on March 30, 2016, for further discussion of these indemnification agreements.
Recently Issued Accounting Standards
See Note 4 to our unaudited condensed financial statements and Note 3 to our audited financial statements in this prospectus for a description of recent accounting pronouncements applicable to our business.
35
Table of Contents
Overview
RXi Pharmaceuticals Corporation (“RXi,” “we,” “our” or the “Company”) is a clinical-stage RNAi company developing innovative therapeutics that address significant unmet medical needs. Our development programs are based on our proprietary self-delivering RNAi (sd-rxRNA®) platform and Samcyprone™, a topical immunomodulator. Our clinical development programs include RXI-109, an sd-rxRNA, for the treatment of dermal and ocular scarring, and Samcyprone™, for the treatment of such disorders as warts, alopecia areata, non-malignant skin tumors and cutaneous metastases of melanoma. In addition to these clinical programs, we have a pipeline of discovery and preclinical product candidates in our core therapeutic areas, as well as in other areas of interest. The Company’s pipeline, coupled with our extensive patent portfolio, provides for product and business development opportunities across a broad spectrum of therapeutic areas.
Our Pipeline
Our pipeline is focused on three areas: dermatology, ophthalmology and cosmetic product development. Our RNAi therapies are designed to “silence,” or down-regulate, the expression of a specific gene that may be over-expressed in a disease condition and our immunotherapy agent treats diseases by inducing, enhancing or suppressing an immune response. The following is a summary of our current product candidates and their development status:
Dermatology Franchise
RXI-109 — Dermal Scarring
The Company’s lead product candidate and first RNAi clinical product candidate, RXI-109, is a self-delivering RNAi compound (sd-rxRNA) that commenced human clinical trials in 2012. RXI-109 is designed to reduce the expression of connective tissue growth factor (“CTGF”), a critical regulator of several biological
36
Table of Contents
pathways involved in fibrosis, including scar formation in the skin and eye. RXI-109 is currently being evaluated in a Phase 2 clinical trial, Study 1402, to prevent or reduce dermal scarring following scar revision surgery of an existing hypertrophic scar and a Phase 1/2 clinical trial, Study 1502, to evaluate the safety and clinical activity of RXI-109 to prevent the progression of retinal scarring in subjects with wet age-related macular degeneration (“AMD”).
The Company conducted two Phase 1 clinical trials evaluating RXI-109 in a surgical setting. Both trials demonstrated the safety and tolerability of RXI-109 in ascending single and multi-doses, and also provided the first evidence of clinical activity in a surgical setting. With the successful completion of the Phase 1 trials, the Company initiated its Phase 2 program for RXI-109 with Study 1301, a Phase 2a clinical trial evaluating the use of RXI-109 to prevent the recurrence of hypertrophic scars following scar revision surgery, in November 2013. This was followed by a second Phase 2 clinical trial in April 2014, Study 1401, which evaluated the use of RXI-109 to prevent the recurrence of keloids, raised and reddened or darkened scars resulting from increased collagen production, after surgical revision. Enrollment and dosing for both of these studies has been completed.
Preliminary data observations from Study 1301 were used in the design of the Company’s third Phase 2 clinical trial in hypertrophic scars, Study 1402, which commenced in July 2014. In October 2015, we reported that preliminary data from Study 1402 demonstrated that scars at revision sites were less visible at three months after a treatment regimen with intradermal administration of RXI-109 than scars at untreated revision sites in those same subjects. Based in part on this new information, two more cohorts were added to Study 1402 in November 2015. For these two cohorts, the number of doses was increased to either eight or nine doses of RXI-109 over a six-month period to better cover the extended wound healing/scarring profile of hypertrophic scars. Enrollment of subjects into these two new cohorts completed ahead of schedule during the third quarter of 2016.
Scarring represents a high unmet medical need as there are currently no U.S. Food and Drug Administration (“FDA”) approved therapies in the U.S. for the treatment and prevention of scars in the skin. Scar revision surgery is a common option, but often the scar recurs. If approved, RXI-109 could be a “first-in-class” RNAi treatment for the prevention or reduction of post-surgical dermal scarring. Given the large number of surgical procedures, there is a significant market for a scar prevention therapeutic such as RXI-109.
Samcyprone ™ — Warts
In December 2014, the Company broadened its clinical pipeline with an exclusive, global license to Samcyprone™, our second clinical candidate. Samcyprone™ is a proprietary topical formulation of the small molecule diphenylcyclopropenone (“DPCP”), an immunomodulator that works by initiating a T-cell response. The use of Samcyprone™ allows sensitization using much lower concentrations of DPCP than are used with existing compounded DPCP solutions, avoiding hyper-sensitization to subsequent challenge doses. DPCP, the active ingredient in Samcyprone™, has long been used to treat warts and has also been used for several other indications, such as to stimulate hair re-growth in alopecia areata and to clear cutaneous metastases of melanoma. In March 2015, the FDA granted Orphan Drug Designation to the Company for Samcyprone™ for the treatment of malignant melanoma stage IIb to IV. Samcyprone™ is currently being evaluated in a Phase 2a clinical trial, Study 1502, for the clearance of common warts.
Study 1502 was initiated in December 2015. Study 1502 includes a sensitization phase in which a spot on the subject’s upper arm and one wart are treated with Samcyprone™. After being sensitized in this way, the subjects will enter into the treatment phase where up to four warts are treated on a once weekly basis for ten weeks with a ten-fold lower concentration of Samcyprone™ than in the sensitization phase. During the trial, the warts are be scored, photographed and measured to monitor the level of clearance. The Company is currently enrolling subjects and is adding a second cohort to explore the opportunity to reduce the sensitization dose level and potentially reduce the treatment length. With this second cohort, enrollment is expected to be completed in the first quarter of 2017.
37
Table of Contents
Cutaneous warts are extremely common, being experienced by most people at some time during their lives. Although most warts will spontaneously disappear without treatment, treatment of these lesions is sought for recalcitrant warts and to prevent recurrence. There are many different treatment modalities for warts, including physical destruction and immunomodulation. However, treatment of warts is complicated by low success rates, prolonged duration of therapy and the potential for recurrence. There is a clear unmet need for new therapies for warts and if approved, Samcyprone™ could be a more effective and convenient treatment than the currently available therapies.
Additional Dermatology Programs
In addition to our dermal scarring and wart programs, we continue to advance our preclinical and discovery programs with our sd-rxRNA technology. The Company has selected and tyrosinase (“TYR”) and collagenase (“MMP1”) as targets for our self-delivering platform because they are relevant for both consumer health and therapeutic development. TYR is a key enzyme in the synthesis of melanin. Melanin is produced by melanocytes and is the pigment that gives human skin, hair and eyes their color. The inhibition of TYR can play a key role in the management of diseases including cutaneous hyperpigmentation disorders such as lentigines (freckles, age spots and liver spots) and possibly melanoma. MMP1 is a key enzyme involved in the breakdown of extracellular matrix. Reduction of MMP1 may be beneficial in the treatment of skin aging disorders, arthritis, acne scarring, blistering skin disorders, corneal erosions, endometriosis and possible cancer metastasis. In addition to our cosmetic program (described below), the Company is actively evaluating similar sd-rxRNA compounds that target TYR and MMP1 to move forward on a separate therapeutic development path.
Ophthalmology Franchise
RXI-109 — Retinal Scarring
As in dermal scarring, CTGF is known to play a role in retinal scarring. RXI-109 can also be used to target CTGF in the eye, where it is known to be involved in retinal scarring. Building on the work in our dermal clinical program, the Company filed a new investigational drug application (“IND”) in July 2015 for RXI-109 as a potential therapeutic for the scarring component of retinal diseases in the eye, such as AMD. In November 2015, we initiated a Phase 1/2 clinical trial to evaluate the safety and clinical activity of RXI-109 in reducing the progression of retinal scarring.
Study 1501 is a multi-dose, dose escalation study conducted in subjects with AMD with evidence of subretinal fibrosis. Each subject will receive four doses of RXI-109 by intraocular injection at one month intervals for a total dosing period of three months. The safety and tolerability of RXI-109, as well as the potential for clinical activity, will be evaluated over the course of the study using numerous assessments to monitor the health in the retina and to assess visual acuity. The Company is currently enrolling subjects in Study 1501.
Currently, there is no effective way to prevent the formation or progression of retinal scars that may occur as a consequence of the number of debilitating ocular diseases. In advanced neo-vascular or wet-AMD, our first area of study, retinal scarring can result in continued vision loss even if the patient is being treated with an anti-vascular endothelial growth factor (“VEGF”) therapy. RXI-109 has the potential to fill this unmet medical need by reducing this continuing damage to the retina and in doing so help preserve these patients’ vision for a longer period of time.
Additional Ophthalmology Programs
In addition to the clinical trial for the use of RXI-109 as a potential therapeutic for retinal scarring, we are advancing other early-stage ophthalmology programs. Currently, the Company is directing its development efforts toward advancing RXI-109 for the treatment of corneal scarring. To date, preclinical studies have shown that CTGF protein levels are reduced in a dose-dependent manner in both the retina and cornea following an intravitreal injection of RXI-109 in monkeys. Elevated CTGF is implicated in the formation of corneal scarring
38
Table of Contents
that can occur after eye injury or after certain infections, and it has been proposed that a reduction of CTGF may be an important step towards reducing corneal scarring. Scarring of the cornea can impact the transparency of the cornea, and thus negatively impact vision. We are currently working towards a non-invasive delivery formulation of RXI-109 to reduce CTGF in the front of the eye.
The Company also continues its exploratory efforts to identify potential sd-rxRNA lead compounds and targets from the RNAi-related assets acquired from OPKO Health Inc. (“OPKO”) in March 2013.
Cosmetic Franchise
RXi’s cosmetic development program is based on our proprietary sd-rxRNA technology. Cosmetics are compounds that affect the appearance of the skin and make no preventative or therapeutic claims. These compounds may be developed more rapidly than therapeutics, therefore the path to market may be much shorter and less expensive.
In October 2015, we announced the selection of lead compounds targeting TYR and MMP1 for cosmetic development. RXI-231, an sd-rxRNA compound targeting TYR, is in development as a cosmetic ingredient that may improve the appearance of uneven skin tone and pigmentation. RXI-185, an sd-rxRNA compound targeting MMP1, is in development as a cosmetic ingredient that may improve the appearance of wrinkles or skin laxity. The Company is currently developing topical delivery application methods, including formulations and microneedling, for use with these compounds and completed functional and safety testing to support the initiation of human testing of one of these consumer health targets.
MirImmune Exclusive Option Agreement
In March 2015, RXi granted an exclusive license to MirImmune, Inc. (“MirImmune”), a private biopharmaceutical company, to utilize the Company’s novel and proprietary sd-rxRNA technology for use in developing ex vivo cell-based cancer immunotherapies. After obtaining the exclusive license from RXi, MirImmune has raised $500,000 in funding to date and used these proceeds on the advancement of their preclinical research, for patent prosecution and filing fees and for general corporate purposes.
MirImmune’s approach to immunotherapy builds on well-established methodologies of adoptive cell transfer. Immune cells, such as T-lymphocytes, are isolated from specific patients or retrieved from allogeneic immune cell banks and then expanded and possibly processed to express tumor-binding receptors. MirImmune’s method will introduce a new and important step in ex vivo processing of the immune cells. This step will reduce or eliminate the expression of immunosuppressive receptors or proteins by the therapeutic immune cells, potentially making them less sensitive to tumor resistance mechanisms and thus improving their ability to destroy the tumor cells.
MirImmune’s approach builds on current immunotherapy approaches, but provides some key advantages. One major advantage is that pre-treatment with MirImmune’s targeted compounds allow multiple immune checkpoints to be attenuated within the same therapeutic cell; an improvement which could dramatically increase their tumor cell killing capability. In addition, these therapeutic immune cells may lack some known side effects associated with the checkpoint inhibitor toxicity while potentially improving efficacy over current immunotherapy approaches.
Using RXi’s sd-rxRNA technology, MirImmune demonstrated in vitro that multiple sd-rxRNA compounds can be used alone or in combination to target and silence extracellular, as well as intracellular, checkpoints in immune cells. Additional in vitro data demonstrated that PD-1 silencing by sd-rxRNA in patient-derived tumor infiltrating lymphocytes (TILs) resulted in enhanced killing of melanoma tumor cells from the same patient in culture. MirImmune has also shown in a mouse model of human ovarian cancer that in vivo treatment with mesothelin CAR T-cells transfected with a PD-1 targeting sd-rxRNA significantly reduced the rate of tumor
39
Table of Contents
growth as compared to vehicle control. Furthermore, the silencing of PD-1 in the CAR T-cells isolated from these tumors persisted for at least one month.
MirImmune has identified lead sd-rxRNA compounds for each of six different checkpoints, both extracellular and intracellular. Since March 2015, MirImmune has been able to advance the potential of RXi’s sd-rxRNA platform for use in cell-based cancer immunotherapy with their preclinical research.
On October 7, 2016, RXi entered into an exclusive option agreement to acquire all outstanding capital stock of MirImmune in consideration for a number of shares equal to 19.99% of the then-outstanding shares of common stock of the Company, plus additional potential consideration contingent on MirImmune reaching certain milestones. The Company can exercise the option on the terms set forth in the option agreement at any time prior to April 5, 2017, but has no obligation to do so.
Market Opportunity
As there are currently no FDA-approved drugs to prevent scar formation, a therapeutic of this type could have great benefit for trauma and surgical patients, particularly as a treatment during the surgical revision of existing unsatisfactory scars. There are over 42 million medical procedures in the U.S. each year that could potentially benefit from a therapeutic treatment that could successfully reduce or prevent scarring; thus, the market potential is quite large. According to the American Society for Plastic Surgery, there are approximately 177,000 scar revision surgeries in the United States every year. In addition to cosmetic and reconstructive surgeries, medical interventions which could incorporate an anti-scarring agent include treatment of scarring that results from trauma, surgery or burns (especially relating to raised or hypertrophic scarring or contracture scarring), and surgical revision of existing unsatisfactory scars.
Overexpression of CTGF is implicated in dermal scarring, subretinal fibrosis and other fibrotic diseases. Because of this, we believe that RXI-109 or other CTGF-targeting RNAi compounds may be able to treat the fibrotic component of numerous indications. These indications are as wide ranging as acute spinal injury, endometriosis, organ fibrosis including liver and pulmonary fibrosis, cutaneous scleroderma and vascular restenosis, in addition to numerous ocular diseases that result in retinal scarring. If the current clinical trials of RXI-109 produce successful results, we may explore opportunities in these additional indications that can be accessed by local administration, starting with intradermal or intravitreal injection. Although the Company does not intend to develop systemic uses of RXI-109 at this time, the Company is open to business development and out-licensing opportunities for those applications.
DPCP, the active ingredient in Samcyprone™, is a small molecule that has been used since the late 1970s to stimulate regrowth of hair in patients with alopecia areata. Recent publications have supported its use as an immunomodulator for the treatment of alopecia areata, warts and cutaneous metastases of malignant melanoma, a combined market potential of over an estimated $1 billion. Although it has been used by physicians for several decades, it has never been reviewed or approved by a regulatory authority as a drug. If FDA approval is granted, Samcyprone™, RXi’s proprietary formulation of DPCP, is expected to achieve market exclusivity.
Introduction to RNAi
RNAi is a naturally occurring phenomenon where short, double-stranded RNA molecules interfere with the expression of targeted genes. The discovery of RNAi is regarded as a significant advancement in the scientific community, as evidenced by the 2006 Nobel Prize in Medicine awarded to the co-discoverers of RNAi, including Dr. Craig Mello, one of the founders of RXi and the Chairman of our Scientific Advisory Board.
RNAi offers a novel approach to the drug development process because RNAi compounds can potentially be designed to target any one of the thousands of human genes, many of which are “undruggable” by other modalities. The specificity of RNAi is achieved by an intrinsic, well-understood biological mechanism based on
40
Table of Contents
designing the sequence of an RNAi compound to match the sequence of the targeted gene. The sequence of the entire human genome is now known, and the mRNA coding sequence for many proteins is already available. Supported by numerous gene-silencing reports and our own research, we believe that this sequence information can be used to design RNAi compounds to interfere with the expression of almost any specific gene.
Our RNAi Therapeutic Platform
The first design of RNAi compounds to be pursued for the development of human therapeutics were short, double-stranded RNAs that included at least one overhanging single-stranded region and limited modifications, known as small-interfering RNA, or siRNA, which we will also refer to as classic siRNA.
We believe that classic siRNAs have drawbacks that may limit the usefulness of those agents as human therapeutics, and that we may be able to utilize the technologies we have licensed and developed internally to optimize RNAi compounds for use as human therapeutic agents. It is the combination of the length, the nucleotide sequence and the configuration of chemical modifications that are important for effective RNAi therapeutics.
Drug delivery has been the primary challenge in developing RNAi therapeutics since its initial discovery. One conventional solution to the delivery problem involves encapsulation into a lipid-based particle, such as a liposome, to improve circulation time and cellular uptake. Scientists at RXi have used an alternative approach to delivery in which drug-like properties were built into the RNAi compound itself. These novel compounds are termed ‘self-delivering’ RNAi compounds or sd-rxRNA.
sd-rxRNAs are hybrid oligonucleotide compounds that the Company believes combine the beneficial properties of both conventional RNAi and antisense technologies. Traditional, single-stranded antisense compounds have favorable tissue distribution and cellular uptake properties. However, they do not have the intracellular potency that is a hallmark of double-stranded RNAi compounds. Conversely, the duplex structure and hydrophilic character of traditional RNAi compounds results in poor tissue distribution and cellular uptake. In an attempt to combine the best properties of both technologies, sd-rxRNA have a single-stranded phosphorothioate region, a short duplex region, and contain a variety of nuclease-stabilizing and lipophilic chemical modifications. The combination of these features allows sd-rxRNA to achieve efficient spontaneous cellular uptake and potent, long-lasting intracellular activity.
We believe that our next generation sd-rxRNA compounds offer significant advantages over classic siRNA used by other companies developing RNAi therapeutics, highlighted by the following characteristics:
| • | Potent RNAi activity in the absence of a delivery vehicle; |
| • | More resistant to nuclease degradation; |
| • | Readily manufactured; |
| • | Potentially more specific for the target gene; and |
| • | More reliable at blocking immune side effects than classic siRNA. |
Our Route of Administration
The route by which an RNAi therapeutic is brought into contact with the body depends on the intended organ or tissue to be treated. Delivery routes can be simplified into two major categories: (1) local (when a drug is delivered directly to the tissue of interest); and (2) systemic (when a drug accesses the tissue of interest through the circulatory system). Local delivery may avoid some hurdles associated with systemic approaches such as rapid clearance from circulation and inefficient tissue extravasation (crossing the endothelial barrier from the blood stream). However, the local delivery approach can only be applied to a limited number of organs or tissues (e.g., skin, eye, lung and potentially the central nervous system).
41
Table of Contents
The key to therapeutic success with RNAi lies in delivering intact RNAi compounds to the target tissue and the interior of the target cells. To accomplish this, we have developed a comprehensive platform that includes chemically synthesized RNAi compounds that are optimized for stability and efficacy and combine delivery at the site of action with a local delivery approach.
Our sd-rxRNA molecules have unique properties that improve tissue and cell uptake. We have studied sd-rxRNA molecules in animal models for dermal and ocular delivery. Direct administration of sd-rxRNA via intradermal injection with no additional delivery vehicle to the skin or to the eye demonstrates that target gene silencing can be measured after local administration. The dose levels required for these direct-injection methods are small and suitable for clinical development. The Company has a number of clinical trials currently ongoing with RXI-109, an sd-rxRNA compound, for local delivery in the skin and the eye. Other target tissues that are potentially accessible for local delivery using sd-rxRNA compounds include the lung, the central nervous system, mucosal tissues and sites of inflammation and tumor (direct administration).
The built-in drug-like properties of sd-rxRNAs, including an extended circulation time and better tissue distribution, may make them amenable for system delivery. Target tissues that are potentially accessible using sd-rxRNA compounds by systemic delivery include kidney, fat, heart, lung, sites of inflammation, tumors and vascular endothelium. Further optimization efforts are required to expand this technology to systemic applications.
Introduction to Immunomodulators
Immunotherapy is the treatment of disease by inducing, enhancing or suppressing an immune response. Active agents in immunotherapy are collectively called immunomodulators. They are a diverse array of recombinant, synthetic and natural preparations that help to regulate or normalize the immune system.
Our Immunomodulator Therapeutic Pipeline
Samcyprone™, licensed by the Company in 2014, is a proprietary topical formulation of the small molecule DPCP. DPCP has been used for decades as a treatment to stimulate hair re-growth in patients with alopecia areata and more recently as a treatment for recalcitrant wart removal and as an aid in the reduction of cutaneous metastases of melanoma. As it is currently used, a doctor must prescribe DPCP to be formulated by a compounding facility, generally in acetone. There are no standardized methods of formulation or procedures for use. Because it works by causing an immune response, the level of response can vary greatly from person to person. Moreover, some pharmacies will not even compound it, even if it is prescribed.
The Company’s formulation of DPCP, called Samcyprone™, works by initiating a T-cell response. T-cells or T lymphocytes are a type of white blood cell that play a key role in cell-mediated immunity. The use of Samcyprone™ will allow for lower sensitizing and challenge doses than in current use and should result in an improved safety margin and a more consistent immune response.
There will be several advantages to using an FDA regulated formulation like the one we are developing. First, the amount of DPCP used in our own ointment formulation will be lower than that generally used in acetone formulation. This should result in reduced side effects that happen due to accidental over-sensitization when a higher than necessary concentration is used. Second, we are developing an optimized dosing regimen so that a standardized response can be expected. And third, the ointment formulation will be easier to prescribe and to use than an acetone formulation, allowing for ease of application at the appropriate site on the skin.
The mechanism of action of Samcyprone™ is linked to DPCP’s ability to alter the expression of multiple genes involved in the immune response. Research with Samcyprone™ may also allow us to discover specific targets and develop new sd-rxRNA compounds for the potential treatment of immunological disorders that are relevant to the skin, as well as various systemic diseases.
42
Table of Contents
Intellectual Property
We protect our proprietary information by means of United States and foreign patents, trademarks and copyrights. In addition, we rely upon trade secret protection and contractual arrangements to protect certain of our proprietary information and products. We have pending patent applications that relate to potential drug targets, compounds we are developing to modulate those targets, methods of making or using those compounds and proprietary elements of our drug discovery platform.
Much of our technology and many of our processes depend upon the knowledge, experience and skills of key scientific and technical personnel. To protect our rights to our proprietary know-how and technology, we require all employees, as well as our consultants and advisors when feasible, to enter into confidentiality agreements that require disclosure and assignment to us of ideas, developments, discoveries and inventions made by these employees, consultants and advisors in the course of their service to us, and we vigorously defend that position with partners, as well as with employees who leave the Company.
We have also obtained rights to various patents and patent applications under licenses with third parties, which require us to pay royalties, milestone payments, or both. The degree of patent protection for biotechnology products and processes, including ours, remains uncertain, both in the United States and in other important markets, because the scope of protection depends on decisions of patent offices, courts and lawmakers in these countries. There is no certainty that our existing patents or others, if obtained, will afford us substantial protection or commercial benefit. Similarly, there is no assurance that our pending patent applications or patent applications licensed from third parties will ultimately be granted as patents or that those patents that have been issued or are issued in the future will stand if they are challenged in court. We assess our license agreements on an ongoing basis, and may from time to time terminate licenses to technology that we do not intend to employ in our technology platforms, or in our product discovery or development activities.
Patents and Patent Applications
We are actively prosecuting twenty-nine patent families covering our compounds and technologies, including RXI-109 and Samcyprone™. A combined summary of these patents and patent applications is set forth below in the following table:
| Pending Applications |
Issued Patents |
|||||||
| United States |
19 | 31 | ||||||
| Canada |
6 | 1 | ||||||
| Europe |
8 | 31 | ||||||
| Japan |
4 | 5 | ||||||
| Other Markets |
10 | 8 | ||||||
Patents and Patent Applications Relating to RNAi
Our portfolio includes 76 issued patents, thirteen of which cover our self-delivering RNAi platform. These thirteen patents broadly cover both the composition and methods of use of our self-delivering platform technology and uses of our sd-rxRNAs targeting CTGF for the treatment of fibrotic disorders, including RXI-109 for the treatment of dermal and ocular fibrosis. These patents are scheduled to expire between 2029 and 2031. Furthermore, there are 44 patent applications, encompassing what we believe to be important new RNAi compounds and their use as therapeutics and/or cosmetics, chemical modifications of RNAi compounds that improve the compounds’ suitability for therapeutic uses (including delivery) and compounds directed to specific targets (i.e., that address specific disease states).
The patents and any patents that may issue from these pending patent applications will, if issued, be set to expire between 2022 and 2035, not including any patent term extensions that may be afforded under the Federal
43
Table of Contents
Food, Drug, and Cosmetic Act (and the equivalent provisions in foreign jurisdictions) for any delays incurred during the regulatory approval process relating to human drug products (or processes for making or using human drug products).
Patent and Patent Applications Relating to Samcyprone™
The Samcyprone™ portfolio includes one issued patent and three patent applications. The patent and patent applications cover both the compositions and methods of use of Samcyprone™ for the treatment of warts, human papilloma virus (HPV) skin infections, skin cancer (including melanoma) and immunocompromised patients.
The patent and any patents that may issue from the pending applications will be set to expire between 2019 and 2031, not including any patent term extensions that may be afforded under the Federal Food, Drug, and Cosmetic Act (and the equivalent provisions in foreign jurisdictions) for any delays incurred during the regulatory approval process relating to human drug products (or processed for making or using human drug products).
Intellectual Property License Agreements
We have secured exclusive and non-exclusive rights to develop therapeutics by licensing key RNAi technologies, Samcyprone™ and patent rights from third parties. These rights relate to chemistry and configuration of compounds, delivery technologies of compounds to cells and therapeutic targets. As we continue to develop our own proprietary compounds, we continue to evaluate both our in-licensed portfolio as well as the field for new technologies that could be in-licensed to further enhance our intellectual property portfolio and unique position in the RNAi and immunotherapy space.
Advirna. In September 2011, we entered into agreements with Advirna, LLC (“Advirna”) pursuant to which Advirna assigned to us its existing patent and technology rights related to sd-rxRNA technology in exchange for our agreement to issue 5% of the Company’s fully-diluted shares, pay an annual maintenance fee of $100,000 and pay a one-time milestone payment of $350,000 upon the issuance of the first patent with valid claims covering the assigned technology. The common shares of the Company were issued to Advirna in 2012 and the one-time milestone payment was paid in 2014. Additionally, we will be required to pay a 1% royalty to Advirna on any license revenue received by us with respect to future licensing of the assigned Advirna patent and technology rights. We also granted back to Advirna a license under the assigned patent and technology rights for fields of use outside human therapeutics and diagnostics.
Our rights under the Advirna agreement will expire upon the later of: (i) the expiration of the last-to-expire of the “patent rights” (as defined therein) or (ii) the abandonment of the last-to-be abandoned of such patents, unless earlier terminated in accordance with the provisions of the agreement.
We may terminate the Advirna agreement at any time upon 90 days’ written notice to Advirna, and Advirna may terminate the agreement upon 90 days’ prior written notice in the event that we cease using commercially reasonable efforts to research, develop, license or otherwise commercialize the patent rights or “royalty-bearing products” (as defined therein), provided that we may refute such claim within such 90-day period by showing budgeted expenditures for the research, development, licensing or other commercialization consistent with other technologies of similar stage of development and commercial potential as the patent rights or royalty-bearing products. Further, either party at any time may provide to the other party written notice of a material breach of the agreement. If the other party fails to cure the identified breach within 90 days after the date of the notice, the aggrieved party may terminate the agreement by written notice to the party in breach.
Hapten. In December 2014, the Company entered into an Assignment and License Agreement with Hapten Pharmaceuticals, LLC (“Hapten”) under which Hapten agreed, effective at a closing that was subject to the satisfaction of certain closing conditions which occurred in February 2015, to sell and assign to us certain patent rights and related assets and rights, including an investigational new drug application and clinical data, for
44
Table of Contents
Hapten’s Samcyprone™ products for therapeutic and prophylactic use. Under the Assignment and License Agreement and upon the closing, Hapten received a one-time upfront cash payment of $100,000 and we issued to Hapten 200,000 shares of Company common stock. Pursuant to the Assignment and License Agreement, Hapten will be entitled to receive: (i) future milestone payments tied to the achievement of certain clinical and commercial objectives (all of which payments may be made at our option in cash or through the issuance of common stock) and (ii) escalating royalties based on product sales by us and any sublicensees.
We have certain customary diligence obligations under the Assignment and License Agreement requiring us to use commercially reasonable efforts to develop and commercialize one or more products covered by the Assignment and License Agreement, which obligations, if not performed, could result in rights assigned or licensed to us reverting back to Hapten.
In addition to the license agreements listed above, the Company has entered into and may enter into other license agreements that may benefit us as we develop our RNAi and Samcyprone™ pipelines.
Other Strategic Agreements
OPKO. In March 2013, the Company entered into an Asset Purchase Agreement with OPKO, in which we acquired substantially all of its RNAi-related assets, which included patents and patent applications, licenses, clinical and preclinical data and other related assets. In exchange for the assets that we purchased from OPKO, we issued 1,666,666 shares of our common stock and agreed to pay, if applicable: (i) up to $50 million in development and commercialization milestones for the successful development and commercialization of each “Qualified Drug” (as defined therein) and (ii) royalty payments equal to: (a) a mid-single-digit percentage of “Net Sales” (as defined therein) with respect to each Qualified Drug sold for an ophthalmologic use during the applicable “Royalty Period” (as defined therein) and (b) a low-single-digit percentage of Net Sales with respect to each Qualified Drug sold for a non-ophthalmologic use during the applicable Royalty Period.
We have certain customary diligence obligations under the Asset Purchase Agreement requiring us to use commercially reasonable efforts to develop and commercialize one or more products covered by the Asset Purchase Agreement, which obligations, if not performed, could result in assets transferred and rights assigned or licensed to us reverting back to OPKO.
MirImmune. In March 2015, RXi granted an exclusive license to MirImmune, Inc. (“MirImmune”) to utilize the Company’s novel and proprietary sd-rxRNA technology for MirImmune’s use in developing ex-vivo cell-based cancer immunotherapies. Under the terms of the agreement, MirImmune will be responsible for all research, development, manufacturing, regulatory and commercialization activities for the licensed products. MirImmune will develop ex-vivo cell-based therapeutics utilizing our sd-rxRNA technology to target immune inhibitory pathways (checkpoints) which are responsible for limiting the efficacy of cancer immunotherapies. The Company is eligible to receive an annual licensing fee, clinical milestone payments and royalties on sales from MirImmune. Further, upon the achievement of gating milestones, the Company will have the right to acquire a double-digit equity stake in MirImmune.
The Company does not expect to realize any significant milestone payments or royalties under this agreement in the near term. However, if successful, this collaboration has the potential to result in novel, more effective and patient friendly cancer treatments that could contribute to developments in personalized medicine.
On October 7, 2016, RXi entered into an exclusive option agreement pursuant to which the Company has the exclusive option, but not the obligation, to purchase 100% of the outstanding capital stock of MirImmune within 180 days of October 7, 2016, for 19.99% of the then-outstanding shares of common stock of the Company plus additional potential consideration contingent on MirImmune achieving certain milestones.
Thera Neuropharma, Inc. In May 2016, RXi granted an exclusive license to Thera Neuropharma, Inc. (“Thera”) to the Company’s novel and proprietary sd-rxRNA platform to develop therapeutics for
45
Table of Contents
neurodegenerative diseases. Under the terms of the agreement, Thera will be responsible for all research, development, manufacturing, regulatory and commercialization activities for the licensed products. Thera’s initial focus will be on sd-rxRNA compounds targeting superoxide dismutase 1 (SOD1) for use in developing innovative treatments for amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig’s disease. Upon execution of the license agreement, RXi was issued shares of common stock of Thera and was granted a five year warrant to purchase additional shares of common stock of Thera pursuant to the terms of the license agreement. The Company is eligible to receive future cash, additional equity and royalties based on the achievement of certain milestones.
Research and Development
To date, our research programs have primarily focused on developing technology necessary to make RNAi compounds available by local administration for diseases for which we intend to develop an RNAi therapeutic, identifying and testing RNAi compounds against therapeutically relevant targets in the fields of dermatology and ophthalmology and identifying lead product candidates and moving those product candidates into the clinic. Since we commenced operations, research and development has comprised a significant proportion of our total operating expenses and is expected to comprise the majority of our spending for the foreseeable future.
There are risks in any new field of drug discovery that preclude certainty regarding the successful development of a product. We cannot reasonably estimate or know the nature, timing and costs of the efforts necessary to complete the development of, or the period in which material net cash inflows are expected to commence from, any product candidate. Our inability to make these estimates results from the uncertainty of numerous factors, including but not limited to:
| • | Our ability to advance product candidates into preclinical research and clinical trials; |
| • | The scope and rate of progress of our preclinical program and other research and development activities; |
| • | The scope, rate of progress and cost of any clinical trials we commence; |
| • | The cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
| • | Clinical trial results; |
| • | The terms and timing of any collaborative, licensing and other arrangements that we may establish; |
| • | The cost and timing of regulatory approvals; |
| • | The cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop; |
| • | The cost and timing of establishing sales, marketing and distribution capabilities; |
| • | The effect of competing technological and market developments; and |
| • | The effect of government regulation and insurance industry efforts to control healthcare costs through reimbursement policy and other cost management strategies. |
Failure to complete any stage of the development of our product candidates in a timely manner could have a material adverse effect on our operations, financial position and liquidity.
Research and Development Expenses
Research and development expenses consist of compensation-related costs for our employees dedicated to research and development activities, fees related to our Scientific Advisory Board members, expenses related to our ongoing research and development efforts primarily related to our clinical trials, drug manufacturing, outside
46
Table of Contents
contract services, licensing and patent fees and laboratory supplies and services for our research programs. We expect research and development expenses to increase as we expand our discovery, preclinical and clinical activities.
Total research and development expenses for the nine months ended September 30, 2016 and 2015 was $4,108,000 and $5,202,000, respectively. Total research and development expenses for the years ended December 31, 2015 and 2014 was $6,925,000 and $5,680,000, respectively.
Competition
We believe that numerous companies are investigating or plan to investigate a variety of proposed anti-scarring therapies in clinical trials or are working in the RNAi area generally. Many other companies are pursuing non-RNAi-based therapies for one or more fibrotic disease indications, including ocular scarring or other indications that we may seek to pursue. The companies include large and small pharmaceuticals, chemical and biotechnology companies, as well as universities, government agencies and other private and public research organizations.
We believe that other companies currently developing anti-scarring therapies, both dermal and ocular, include CoDa Therapeutics, Inc., Sirnaomics, Inc., FirstString Research, Inc., Promedior, Inc., FibroGen, Inc., MiRagen Therapeutics, Inc., Ophthotech Corporation, Vascular BioSciences, Allergan plc, and Suneva Medical, Inc.
We believe that other companies working in the RNAi area, generally, include Alnylam Pharmaceuticals, Inc., Benitec Limited, Silence Therapeutics plc, Quark Pharmaceuticals, Inc., Arbutus Biopharma, Arrowhead Research Corporation, Dicerna Pharmaceuticals, Inc., Sylentis, S.A. and Roche Innovation Center Copenhagen A/S, as well as a number of large pharmaceutical companies.
We do not believe that there are any companies developing treatments directly competing with Samcyprone™ for warts or for alopecia areata or cutaneous metastases of malignant melanoma. However, there are several existing treatments for each condition with which Samcyprone™ could potentially compete. Current topical medicinal treatments for warts include salicylic acid, off label use of Imiquimod and Picato® and the most common ablative treatments include removal through medical procedures, such as cryotherapy, surgery or chemical peels. There currently are no FDA-approved treatments specifically for alopecia areata and the most common treatments used by medical professionals include cortisone injections or pills, topical ointments, such as minoxidil or anthralin, and topical immunotherapy with the use of chemicals such as DPCP or dinitrochlorobenzene. Finally, common treatment therapies for cutaneous metastases of malignant melanoma include cryotherapy, photodynamic therapy, laser therapy, chemotherapy and immunotherapy, such as the use of Imiquimod.
Government Regulation
The United States and many other countries extensively regulate the preclinical and clinical testing, manufacturing, labeling, storage, record-keeping, advertising, promotion, export, marketing and distribution of drugs and biologic products. The FDA regulates pharmaceutical and biologic products under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal statutes and regulations.
To obtain approval of our future product candidates from the FDA, we must, among other requirements, submit data supporting safety and efficacy for the intended indication as well as detailed information on the manufacture and composition of the product candidate. In most cases, this will require extensive laboratory tests and preclinical and clinical trials. The collection of these data, as well as the preparation of applications for review by the FDA involve significant time and expense. The FDA also may require post-marketing testing to monitor the safety and efficacy of approved products or place conditions on any approvals that could restrict the
47
Table of Contents
therapeutic claims and commercial applications of these products. Regulatory authorities may withdraw product approvals if we fail to comply with regulatory standards or if we encounter problems at any time following initial marketing of our products.
The first stage of the FDA approval process for a new biologic or drug involves completion of preclinical studies and the submission of the results of these studies to the FDA. These data, together with proposed clinical protocols, manufacturing information, analytical data and other information submitted to the FDA in an IND application, must become effective before human clinical trials may commence. Preclinical studies generally involve FDA regulated laboratory evaluation of product characteristics and animal studies to assess the efficacy and safety of the product candidate.
After the IND becomes effective, a company may commence human clinical trials. These are typically conducted in three sequential phases, but the phases may overlap. Phase 1 trials consist of testing the product candidate in a small number of patients or healthy volunteers, primarily for safety at one or more doses. Phase 2 trials, in addition to safety, evaluate the efficacy of the product candidate in a patient population somewhat larger than Phase 1 trials. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded population at multiple test sites. A company must submit to the FDA a clinical protocol, accompanied by the approval of the Institutional Review Board (“IRB”) at the institutions participating in the trials, prior to commencement of each clinical trial.
To obtain FDA marketing authorization, a company must submit to the FDA the results of the preclinical and clinical testing, together with, among other things, detailed information on the manufacture and composition of the product candidate, in the form of a new drug application (an “NDA”), or, in the case of a biologic, a biologics license application (a “BLA”).
The amount of time taken by the FDA for approval of an NDA or BLA will depend upon a number of factors, including whether the product candidate has received priority review, the quality of the submission and studies presented, the potential contribution that the compound will make in improving the treatment of the disease in question and the workload at the FDA.
The FDA may, in some cases, confer upon an investigational product the status of a fast track product. A fast track product is defined as a new drug or biologic intended for the treatment of a serious or life threatening condition that demonstrates the potential to address unmet medical needs for this condition. The FDA can base approval of an NDA or BLA for a fast track product on an effect on a surrogate endpoint, or on another endpoint that is reasonably likely to predict clinical benefit. If a preliminary review of clinical data suggests that a fast track product may be effective, the FDA may initiate review of entire sections of a marketing application for a fast track product before the sponsor completes the application.
We anticipate that our products will be manufactured by our strategic partners, licensees or other third parties. Before approving an NDA or BLA, the FDA will inspect the facilities at which the product is manufactured and will not approve the product unless the manufacturing facilities are in compliance with the FDA’s current good manufacturing practices (“cGMP”), which are regulations that govern the manufacture, holding and distribution of a product. Manufacturers of biologics also must comply with the FDA’s general biological product standards. Our manufacturers also will be subject to regulation under the Occupational Safety and Health Act, the Nuclear Energy and Radiation Control Act, the Toxic Substance Control Act and the Resource Conservation and Recovery Act and other applicable environmental statutes. Following approval, the FDA periodically inspects drug and biologic manufacturing facilities to ensure continued compliance with the cGMP. Our manufacturers will have to continue to comply with those requirements. Failure to comply with these requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing or recall or seizure of product. Adverse patient experiences with the product must be reported to the FDA and could result in the imposition of marketing restrictions through labeling changes or market removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
48
Table of Contents
The labeling, advertising, promotion, marketing and distribution of a drug or biologic product also must be in compliance with FDA and Federal Trade Commission requirements which include, among others, standards and regulations for off-label promotion, industry sponsored scientific and educational activities, promotional activities involving the internet, and direct-to-consumer advertising. We also will be subject to a variety of federal, state and local regulations relating to the use, handling, storage and disposal of hazardous materials, including chemicals and radioactive and biological materials. In addition, we will be subject to various laws and regulations governing laboratory practices and the experimental use of animals. In each of these areas, as above, the FDA has broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of product approvals, seize or recall products and deny or withdraw approvals.
We will also be subject to a variety of regulations governing clinical trials and sales of our products outside the United States. Whether or not FDA approval has been obtained, approval of a product candidate by the comparable regulatory authorities of foreign countries and regions must be obtained prior to the commencement of marketing the product in those countries. The approval process varies from one regulatory authority to another and the time may be longer or shorter than that required for FDA approval. In the European Union, Canada and Australia, regulatory requirements and approval processes are similar, in principle, to those in the United States.
Environmental Compliance
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specific waste products. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of bio-hazardous materials. The cost of compliance with these laws and regulations could be significant and may adversely affect capital expenditures to the extent we are required to procure expensive capital equipment to meet regulatory requirements.
Employees
As of October 15, 2016, we had fifteen full-time employees, nine of whom were engaged in research and development, and six of whom were engaged in management, administration and finance. None of our employees are represented by a labor union or covered by a collective bargaining agreement, nor have we experienced any work stoppages.
Corporate Information
RXi was incorporated in the state of Delaware in 2011. Our executive offices are located at 257 Simarano Drive, Suite 101, Marlborough, MA 01752, and our telephone number is (508) 767-3861.
Investor Information
The Company’s website address is http://www.rxipharma.com. We make available on our website, free of charge, copies of our annual reports on Form 10-K, our quarterly reports on Form 10-Q and our current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the Securities and Exchange Commission (the “SEC”).
You may read and copy any materials the Company files with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding RXi and other issuers that file electronically with the SEC. The SEC’s website address is http://www.sec.gov.
49
Table of Contents
Directors and Executive Officers of RXi
The following table sets forth our directors and executive officers, their ages and the positions currently held by each person:
| Name |
Age | Position | ||||
| Geert Cauwenbergh, Dr. Med. Sc. |
62 | President, Chief Executive Officer, acting Chief Financial Officer and Director | ||||
| Pamela Pavco, Ph.D. |
60 | Chief Development Officer | ||||
| Robert J. Bitterman |
65 | Chairman of the Board of Directors | ||||
| Keith L. Brownlie |
64 | Director | ||||
| H. Paul Dorman |
79 | Director | ||||
| Curtis A. Lockshin, Ph.D. |
56 | Director | ||||
Geert Cauwenbergh, Dr. Med. Sc. was appointed to the Board and was elected as President and Chief Executive Officer of the Company in April 2012. Prior to joining us, from June 2011 to April 2012, Dr. Cauwenbergh was active, through his consulting company Phases123 LLC, in advising various small biotech and healthcare companies. From July 2008 to June 2011, Dr. Cauwenbergh was the Chief Executive Officer of Rhei Pharmaceuticals HK Ltd, a Chinese company that licenses western drugs for development and commercialization in China, and Managing Director of the Center for Medical Innovation, a government subsidized center for translational medicine for the Belgian Region of Flanders. In February 2008 and May 2009, Dr. Cauwenbergh founded Phases123 LLC and Aramis LLC, a consulting company and a dermatology company, respectively. From September 2008 to March 2010, Dr. Cauwenbergh served as a member of the board of directors of DARA Biosciences, Inc., a publicly-traded biopharmaceutical company. From 2002 to 2008, Dr. Cauwenbergh served as Chief Executive Officer of Barrier Therapeutics, Inc., a publicly traded biopharmaceutical company that he founded in 2001 and where he also served as Chairman of the board of directors from 2002 to 2006. Barrier, which focused on dermatology drug development and commercialization, was acquired by Stiefel Laboratories, Inc. in 2008. Prior to founding Barrier, Dr. Cauwenbergh held a number of ascending senior management positions at Johnson & Johnson, where he was employed for 23 years. As Vice President of Research and Development for Johnson & Johnson’s Skin Research Center, he was responsible for the worldwide research and development of all skin care products for the Johnson & Johnson consumer companies. He is a member of the board of directors of Moberg Derma AB, a Swedish pharmaceutical company. In February 2014, Dr. Cauwenbergh joined the board of directors of Phosphagenics Ltd., an Australian company focused on developing new transdermal delivery systems for pharmaceutical products. Dr. Cauwenbergh currently serves as chairman of the nominating and governance committee of Phosphagenics. In September 2011, Dr. Cauwenbergh also joined the board of directors of Cutanea Life Sciences, Inc., a wholly owned subsidiary of Maruho Company, LTD., which focuses on the development and commercialization of proprietary technologies to treat diseased and aging skin. In 2005, Dr. Cauwenbergh was inducted into the New Jersey High-Tech Hall of Fame, and, from 2009 to 2010, he served as Chairman of the Board of Trustees of BioNJ. He has authored more than 100 publications and has been a guest editor for numerous books addressing mycology and infectious diseases. Dr. Cauwenbergh received his Doctorate in Medical Sciences from the Catholic University of Leuven, Faculty of Medicine (Belgium), where he also completed his masters and undergraduate work. Based on Dr. Cauwenbergh’s understanding of the business through his role as our Chief Executive Officer and as an incumbent member of the Board, as well as his extensive experience in dermatology and company-building, the Nominating Committee concluded that Dr. Cauwenbergh has the requisite experience, qualifications, attributes and skill necessary to serve as a member of the Board.
Pamela Pavco, Ph.D. currently serves as our Chief Development Officer. Prior to this, Dr. Pavco served as our Senior Vice President of Pharmaceutical Development from September 24, 2011 until April 2012. From March 2007 to September 24, 2011, she served as the Vice President of Pharmaceutical Development of Galena
50
Table of Contents
Biopharma, Inc. Dr. Pavco has over 25 years of research and development experience in oligonucleotides. Dr. Pavco was Senior Director of Research and Development Project Management at Sirna Therapeutics, Inc., from 2002 until 2006, when it was acquired by Merck & Co., Inc. for $1.1 billion. While at Sirna, she was responsible for the discovery research and development of Sirna-027, the first chemically modified siRNA to enter clinical trials. Dr. Pavco also managed Sirna’s alliance with Allergan, Inc. that was initiated to continue discovery research in the area of ophthalmology and take Sirna-027 forward into Phase 2 clinical studies. While at Sirna, Dr. Pavco served in various additional capacities, including Director of Biology Research and Director of Pharmacology and she also managed numerous corporate collaborations and internal programs focusing on the development of therapeutic oligonucleotides in the fields of oncology, antiangiogenesis, hepatitis, respiratory disease and Huntington’s disease. Dr. Pavco has authored numerous scientific articles and contributed to approximately 60 patents and patent applications in the oligonucleotide therapeutics field. Dr. Pavco received a Ph.D. in Biochemistry from Virginia Commonwealth University and did her post-doctoral work at Duke University. She is a member of the American Association of Cancer Research and the Association for Research and Vision in Ophthalmology.
Robert J. Bitterman has served as a member and the Chairman of our Board of Directors since 2012. Prior to joining the Company, Mr. Bitterman founded Cutanea Life Sciences, Inc. in September 2005 as its President, Chief Executive Officer and Board Director. Cutanea Life Sciences, Inc. focuses on the development and commercialization of proprietary technologies to treat diseased and aging skin and was successfully acquired by Maruho Company, LTD. in February 2012, where Mr. Bitterman has continued his role as President and Chief Executive Officer. Mr. Bitterman also served as President and General Manager of Dermik Laboratories, the global dermatology strategic business unit of Aventis S.A. from 1994 to 2004. Prior to assuming senior operational leadership positions, Mr. Bitterman held various positions of increasing responsibility in financial and commercial capacities within Aventis S.A. From September 2004 until April 2005, Mr. Bitterman also held the position of President and Chief Executive Officer of Isolagen, Inc., a publicly traded bioscience technology company which developed and commercialized autologous human fibroblasts targeting soft tissue enhancement. Mr. Bitterman holds an A.B. degree in Economics from The College of the Holy Cross and a Master of Business Administration degree from Boston University. He also holds a Doctor of Human Letters (Honoris Causa) from the New York College of Podiatric Medicine and is a member of the Philadelphia Business Leaders Network. Based on Mr. Bitterman’s significant leadership roles in other bioscience companies, including the role of chief executive officer, his experience with development stage organizations, and his knowledge of dermatology and the pharmaceutical industry, the Nominating Committee concluded that Mr. Bitterman has the requisite experience, qualifications, attributes and skill necessary to serve as a member of the Board.
Keith L. Brownlie has served as a member of our Board of Directors since June 2012. Prior to joining us, Mr. Brownlie was employed by the accounting firm Ernst & Young LLP from 1974 to 2010. At Ernst & Young, he served as audit partner for numerous public companies and was the Life Sciences Industry Leader for the New York Metro Area. Mr. Brownlie co-founded the New Jersey Entrepreneur of the Year Program and was co-chair of the BIONJ/PABIO Annual Symposium. Since his retirement from Ernst & Young in 2010, Mr. Brownlie currently serves as a member of the board of directors and chairman of the audit committee of Soligenix, Inc., which develops products to treat life-threatening side effects of cancer treatments and serious gastrointestinal diseases and vaccines for certain bioterrorism agents. From 2011 to 2013, Mr. Brownlie also served as a member of the board of directors and served as the chairman of the audit committee of EpiCept Corporation, which focused on the development and commercialization of pharmaceutical products for the treatment of pain and cancer and merged with Immune Pharmaceuticals in August 2013. From 2013 to 2014, Mr. Brownlie was a member of the board of directors and served as the chairman of the audit committee of Cancer Genetics, Inc., an emerging leader in DNA-based cancer diagnostics that personalizes the clinical management of difficult-to-diagnose cancers. Mr. Brownlie received a B.S. in Accounting from Lehigh University and is a Certified Public Accountant. Based on Mr. Brownlie’s experience in the area of public company financial reporting, his responsibilities as an audit partner, which qualify him as a financial expert, and his membership on the board of directors of other public companies, the Nominating Committee concluded that Mr. Brownlie has the requisite experience, qualifications, attributes and skill necessary to serve as a member of the Board.
51
Table of Contents
H. Paul Dorman has served as a member of our Board of Directors since April 2013. Mr. Dorman currently serves as the Chairman and CEO of DFB Pharmaceuticals, a holdings company specializing in investing in and operating pharmaceutical businesses. From 1990 to 2012, Mr. Dorman also served as the Chairman and CEO of DPT Laboratories, a contract manufacturer and developer of pharmaceutical products. During that time, Mr. Dorman expanded DPT into a portfolio of healthcare companies that provides services and proprietary branded pharmaceutical products to the global market. Prior to acquiring DPT, Mr. Dorman was employed by Johnson & Johnson for 12 years, where he served in various positions, including Vice President and as a member of the board of directors. Prior to Johnson & Johnson, Mr. Dorman was employed by Baxter-Travenol, a large pharmaceuticals company. Mr. Dorman holds a B.S. degree in Mechanical Engineering from Tulane University and a Juris Doctor of Law from Loyola University. Based on Mr. Dorman’s experience through his roles as Chairman and CEO and his deep understanding of the pharmaceutical industry in holding executive positions at large public companies, the Nominating Committee concluded that Mr. Dorman has the requisite experience, qualifications, attributes and skill necessary to serve as a member of the Board.
Curtis A. Lockshin, Ph.D. has served as a member of our Board of Directors since April 2013. Since July 2015, Dr. Lockshin has served as Chief Executive Officer and Director of SciVac Therapeutics Inc., and its subsidiary SciVac, Ltd., a biologics and vaccine company in Rehovot, Israel, where he has been serving as CEO and Director since September 2014. Dr. Lockshin has also served as the Vice President of Research and Operations of Xenetic Biosciences, Inc., a biopharmaceutical company focused on developing biologic drugs and novel oncology therapeutics since March 2014 and as the President and CEO of Guardum Pharmaceuticals, LLC, a private pharmaceutical company, since May 2013. From October 2011 to February 2013, Dr. Lockshin served as Vice President of Corporate R&D Initiatives for OPKO Health, Inc., a multinational pharmaceutical and diagnostics company, at which time he then assumed the position of consultant to OPKO until December 2013. From March 2011 until December 2013, Dr. Lockshin served as a member of the board of directors for ChromaDex, Inc., a natural products company engaged in the dietary supplement, food & beverage, cosmetic and pharmaceutical industries. From October 2009 to September 2012, Dr. Lockshin served as a member of the board of directors for Sorrento Therapeutics, Inc., a development-stage biopharmaceutical company. Since April 2004, Dr. Lockshin has also served as a member of the board of directors of the Ruth K. Broad Biomedical Research Foundation. The foundation is a Duke University Support Corporation that supports basic research related to Alzheimer’s disease and neurodegeneration via intramural, extramural and international grants. Since 2003, Dr. Lockshin has worked as an independent consultant, focusing on small private companies in the healthcare, biotechnology and security sectors. From August 2002 to March 2003, Dr. Lockshin held the position of Director of Discovery Biology at Beyond Genomics, Inc. (now BG Medicine, Inc.), a company engaged in the discovery of disease-associated biomarkers and identification of therapeutic targets. Dr. Lockshin held various positions from June 1998 to July 2002 at Sepracor, Inc. (now Sunovion Pharmaceuticals, Inc.), a pharmaceutical company that develops therapeutic products for the central nervous system and respiratory disorders. Dr. Lockshin holds a S.B. degree in Life Sciences and a Ph.D. in Biological Chemistry from the Massachusetts Institute of Technology. Based on Dr. Lockshin’s industry knowledge in the biotechnology and pharmaceutical fields and his membership on the board of directors of other public companies, the Nominating Committee concluded that Dr. Lockshin has the requisite experience, qualifications, attributes and skill necessary to serve as a member of the Board.
52
Table of Contents
The following describes the compensation earned in fiscal 2015 and 2014 by each of the executive officers identified below in the Summary Compensation Table, who are referred to collectively as our “named executive officers.” Our named executive officers with respect to the fiscal year that ended on December 31, 2015 are Geert Cauwenbergh, Dr. Med. Sc., President, Chief Executive Officer, acting Chief Financial Officer and Director, and Pamela Pavco, Ph.D., Chief Development Officer.
Summary Compensation Table
| Name and principal position |
Year | Salary ($) |
Option awards ($)(1) |
Non-equity incentive plan compensation ($)(2) |
All other compensation ($)(3) |
Total ($) |
||||||||||||||||||
| Geert Cauwenbergh, Dr. Med. Sc. |
2015 | 398,361 | 37,240 | 190,000 | 300 | 625,901 | ||||||||||||||||||
| President, Chief Executive Officer and acting Chief Financial Officer |
2014 | 381,274 | 305,900 | 114,000 | 552 | (4) | 801,726 | |||||||||||||||||
| Pamela Pavco, Ph.D. |
2015 | 363,808 | 18,480 | 104,025 | 300 | 486,613 | ||||||||||||||||||
| Chief Development Officer |
2014 | 350,577 | 150,480 | 63,000 | 300 | 564,357 | ||||||||||||||||||
| (1) | The amounts shown reflect the grant date fair value computed in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 718 for the indicated year, adjusted to disregard the effects of any estimate of forfeitures related to service-based vesting. The assumptions we used in valuing options are described more fully in the “Management’s Discussion and Analysis” section and the Notes to Financial Statements included in this prospectus. |
| (2) | The amounts shown reflect the annual cash bonus earned for performance for each respective year under the Company’s Incentive Bonus Program. The annual cash bonuses were paid in February of the calendar year following the year to which the bonus relates. |
| (3) | Represents amounts for the dollar value of life insurance premiums paid. |
| (4) | Represents amounts for the dollar value of life insurance premiums paid and a gross-up for the related tax liability in connection with Dr. Cauwenbergh’s health insurance premiums. |
Outstanding Equity Awards at Fiscal Year-End
The following table shows information regarding outstanding equity awards as of December 31, 2015 for our named executive officers. None of the named executive officers held any outstanding stock awards as of that date.
| Option Awards | ||||||||||||||||
| Name |
Number of Securities Underlying Unexercised Options Exercisable (#) |
Number of Securities Underlying Unexercised Options Unexercisable (#) |
Option Exercise Price ($) |
Option Expiration Date |
||||||||||||
| Geert Cauwenbergh, Dr. Med. Sc.(1) |
104,363 | 9,489 | 25.50 | 06/08/2022 | ||||||||||||
| 8,334 | 5,000 | 60.00 | 06/07/2023 | |||||||||||||
| 4,988 | 8,313 | 28.50 | 06/02/2024 | |||||||||||||
| — | 13.301 | 3.80 | 06/01/2025 | |||||||||||||
| Pamela Pavco, Ph.D.(2) |
55,810 | — | 39.00 | 05/04/2022 | ||||||||||||
| 4,167 | 2,500 | 60.00 | 06/07/2023 | |||||||||||||
| 2,475 | 4,125 | 28.50 | 06/02/2024 | |||||||||||||
| 825 | 5,776 | 3.80 | 06/01/2025 | |||||||||||||
53
Table of Contents
| (1) | The option awards granted to Dr. Cauwenbergh vest as to 25% of the award on the first anniversary of the grant date and as to the remaining 75% of the option in equal monthly installments over a three year period thereafter. |
| (2) | The option awards granted to Dr. Pavco vest in equal monthly installments over a four year period. |
Nonqualified Deferred Compensation
We do not have any nonqualified deferred compensation plans.
Employment and Change of Control Agreements
Geert Cauwenbergh, Dr. Med. Sc.
Dr. Cauwenbergh was appointed Chief Executive Officer pursuant to an employment agreement, dated April 27, 2012, pursuant to which he is entitled to receive an initial base salary of $360,000 per annum, as well as a performance bonus of up to 50% of his base salary, subject to the achievement of performance goals to be established annually. On June 8, 2012, Dr. Cauwenbergh received an option entitling him to purchase 113,852 shares of Company common stock at an exercise price equal to the fair value of the underlying common stock on the date of grant. The option vested with respect to one quarter of the underlying shares on April 27, 2013, and then vested on a ratable basis monthly thereafter over the next three years such that the option became fully vested and exercisable on April 27, 2016.
Dr. Cauwenbergh’s employment agreement provides that, upon termination of Dr. Cauwenbergh’s employment without “cause” (as defined therein) by us or by Dr. Cauwenbergh for “good reason” (as defined therein), he will be entitled to payment of: (1) any accrued but unpaid salary, business expenses and unused vacation as of the date of his termination as well as any unpaid bonus compensation awarded for the prior year; (2) six months of salary from the date of termination; and (3) continued participation, at our expense, during the applicable six-month severance period in our sponsored group medical and dental plans. In the event his employment is terminated within twelve months following a “change of control” of RXi, he will be entitled to: (x) twelve months of salary from the date of termination; (y) accelerated vesting of any unvested RXi stock options held by him; and (z) continued participation, at our expense, during the twelve-month severance period in our sponsored group medical and dental plans.
Pamela Pavco, Ph.D.
Dr. Pavco serves as our Chief Development Officer. Under her employment agreement dated September 24, 2011, Dr. Pavco is entitled to receive an initial annual salary of $300,000. She also received an option to purchase up to 55,810 shares of common stock at an exercise price equal to the fair value of the underlying common stock on the date of grant. The option vested in equal monthly installments over four years, beginning on October 24, 2011, such that the option became fully vested and exercisable on September 24, 2015.
Dr. Pavco’s employment agreement provides that, upon termination of Dr. Pavco’s employment without “cause” (as defined therein) by us or by Dr. Pavco for “good reason” (as defined therein), she will be entitled to payment of: (1) any accrued but unpaid salary and unused vacation as of the date of her termination; (2) six months of salary from the date of termination; and (3) continued participation, at our expense, during the applicable six-month severance period in our sponsored group medical and dental plans. In the event her employment is terminated within twelve months following a “change of control” of RXi, she will be entitled to: (x) twelve months of salary from the date of termination; (y) accelerated vesting of any unvested RXi stock options held by her as to 50% of the unvested option shares or the portion of the unvested option shares that would have vested over the following twenty-four months, whichever is greater; and (z) continued participation, at our expense, during the twelve-month severance period in our sponsored group medical and dental plans.
54
Table of Contents
The following table shows the compensation paid in fiscal year 2015 to the Company’s non-employee directors.
| Name |
Fees Earned or Paid in Cash ($) | Option Awards ($)(1)(2) |
Total ($) | |||||||||
| Robert J. Bitterman |
35,000 | 4,333 | 39,333 | |||||||||
| Keith L. Brownlie |
35,000 | 4,333 | 39,333 | |||||||||
| H. Paul Dorman |
27,500 | 4,333 | 31,833 | |||||||||
| Curtis A. Lockshin, Ph.D. |
30,000 | 4,333 | 34,333 | |||||||||
| (1) | The amounts shown reflect the grant date fair value computed in accordance with FASB ASC 718 for the indicated year, adjusted to disregard the effects of any estimate of forfeitures related to service-based vesting. The assumptions we used in valuing options are described more fully in the “Management’s Discussion and Analysis” section and the Notes to Financial Statements included in this prospectus. |
| (2) | Since their service on the Board, the aggregate number of shares underlying stock options outstanding at fiscal yearend to our non-employee directors is as follows: Robert J. Bitterman — 8,335 option awards, Keith L. Brownlie 8,335 option awards, H. Paul Dorman — 6,668 option awards and Curtis A. Lockshin, Ph.D. 6,668 option awards. |
We compensate our non-employee directors for their service as a member of our Board of Directors. As our only director who is also an employee, Dr. Cauwenbergh receives no separate compensation for Board service. Dr. Cauwenbergh’s compensation is set forth above in the Summary Compensation Table.
Each non-employee director is entitled to receive an annual cash retainer of $25,000. The chairs of our Board and the Audit Committee are entitled to receive an additional annual cash retainer of $10,000 and the chair of the Nominating and Governance Committee is entitled to receive an additional annual cash retainer of $5,000.
Each non-employee director is entitled to receive an option award for 3,334 shares of the Company’s common stock, vesting in equal quarterly installments over one year, upon initial election to our Board of Directors. In addition, each non-employee director is also entitled to receive an additional annual option award for 1,667 shares of the Company’s common stock, vesting in equal quarterly installments over one year.
Non-employee directors are also reimbursed for their travel and reasonable out-of-pocket expenses incurred in connection with attending Board and committee meetings and in attending continuing education seminars, to the extent that attendance is required by the Board or the committee(s) on which that director serves.
THE COMPENSATION COMMITTEE AND THE BOARD OF DIRECTORS REASSESS THE APPROPRIATE LEVEL OF EQUITY COMPENSATION FOR NON-EMPLOYEE DIRECTORS ON AN ANNUAL BASIS. FUTURE EQUITY COMPENSATION PAYMENTS WILL BE DETERMINED ON A YEAR-BY-YEAR BASIS FOR THE FORESEEABLE FUTURE DUE TO THE VOLATILITY OF THE COMPANY’S STOCK PRICE.
55
Table of Contents
CERTAIN RELATIONSHIPS, RELATED-PARTY TRANSACTIONS AND DIRECTOR INDEPENDENCE
Since the past two years, there has not been, nor is there currently proposed, any transaction or series of related transactions to which we were or will be a party in which the amount involved exceeded or will exceed $120,000 and in which the other parties included or will include any of our directors, executive officers, holders of 5% or more of our voting securities, or any member of the immediate family of any of the foregoing persons, other than compensation arrangements with directors and executive officers, which are described where required in “Management,” “Executive Compensation,” and the transactions described below.
Procedures for Review, Approval or Ratification of Transactions with Related Persons
Our Board of Directors has a policy to review and approve all transactions with directors, officers and holders of more than 5% of our voting securities and their affiliates. The policy provides that, prior to Board consideration of a transaction with such a related party, the material facts as to the related party’s relationship or interest in the transaction must be disclosed to the Board, and the transaction will not be considered approved by the Board unless a majority of the directors who are not interested in the transaction (if applicable) approve the transaction. Furthermore, when stockholders are entitled to vote on a transaction with a related party, the material facts of the related party’s relationship or interest in the transaction must be disclosed to the stockholders, who must approve the transaction in good faith.
Indemnification Agreements
We have entered into indemnification agreements with each of our executive officers and directors. These agreements provide that, subject to limited exceptions and among other things, we will indemnify each of our executive officers and directors to the fullest extent permitted by law and advance expenses to each indemnitee in connection with any proceeding in which a right to indemnification is available.
Director Independence
We believe that the Company benefits from having a strong and independent Board. For a director to be considered independent, the Board must determine that the director does not have any direct or indirect material relationship with the Company that would affect his or her exercise of independent judgment. On an annual basis, the Board reviews the independence of all directors under the applicable NASDAQ listing standards. The Company also considers each director’s affiliations with the Company and members of management, as well as significant holdings of Company securities. This review considers all known relevant facts and circumstances in making an independence determination. Based on this review, the Board has made an affirmative determination that all directors, other than Dr. Cauwenbergh, are independent. It was determined that Dr. Cauwenbergh lacks independence because of his status as the Company’s President and Chief Executive Officer.
In addition, NASDAQ listing standards require that, subject to specified exceptions, each member of our Audit, Compensation and Nominating and Governance Committees be independent and that our Audit Committee members also satisfy independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Our Board of Directors has determined that Messrs. Brownlie and Dorman and Dr. Lockshin, members of the Audit Committee, Messrs. Bitterman and Brownlie and Dr. Lockshin, members of the Compensation Committee, and Dr. Lockshin and Messrs. Brownlie and Dorman, members of the Nominating and Governance Committee, are independent under the applicable NASDAQ listing standards and the Exchange Act.
56
Table of Contents
SECURITY OWNERSHIP OF BENEFICIAL OWNERS AND MANAGEMENT
Based on information available to us and filings with the Securities and Exchange Commission (the “SEC”), the following table sets forth certain information regarding the beneficial ownership (as defined by Rule 13d-3 under the Securities Exchange Act of 1934) of our outstanding common stock for (i) each of our directors, (ii) each of our “named executive officers,” as defined in the Executive Compensation section above, (iii) all of our directors and executive officers as a group and (iv) persons known to us to beneficially own more than 5% of our outstanding common stock. The following information is presented as of September 30, 2016 or such other date as may be reflected below.
Beneficial ownership and percentage ownership are determined in accordance with the rules of the SEC and include voting or investment power with respect to shares of stock. This information does not necessarily indicate beneficial ownership for any other purpose. Under these rules, shares of common stock issuable under stock options or warrants that are exercisable within 60 days of September 30, 2016 are deemed outstanding for the purpose of computing the percentage ownership of the person holding the options or warrants, but are not deemed outstanding for the purpose of computing the percentage ownership of any other person.
Unless otherwise indicated and subject to applicable community property laws, to our knowledge, each stockholder named in the following table possesses sole voting and investment power over their shares of common stock, except for those jointly owned with that person’s spouse. Unless otherwise indicated below, the address of each person listed on the table is c/o RXi Pharmaceuticals Corporation, 257 Simarano Drive, Suite 101, Marlborough, MA 01752.
| Shares Beneficially Owned | ||||||||
| Name and Address of Beneficial Owner |
Number(1) | Percent of Class(2) |
||||||
| Greater than 5% Holders |
||||||||
| Broadfin Capital, LLC(3) |
||||||||
| 300 Park Avenue, 25th Floor New York, NY 10022 |
661,713 | 9.90 | % | |||||
| Directors, Officers and Named Executive Officers: |
||||||||
| Geert Cauwenbergh, Dr. Med. Sc.(4) |
160,738 | 2.38 | % | |||||
| Robert J. Bitterman(5) |
15,860 | * | ||||||
| Keith L. Brownlie(6) |
9,585 | * | ||||||
| H. Paul Dorman(7) |
13,543 | * | ||||||
| Curtis A. Lockshin, Ph.D.(8) |
9,818 | * | ||||||
| Pamela Pavco, Ph.D.(9) |
75,756 | 1.14 | % | |||||
| All current directors and executive officers as a group (six persons) |
285,300 | 4.13 | % | |||||
| * | Indicates less than 1%. |
| (1) | Represents shares of common stock and shares of restricted stock held as of September 30, 2016 plus shares of common stock that may be acquired upon exercise of options, warrants and other rights exercisable within 60 days of September 30, 2016. |
| (2) | Based on 6,599,846 shares of the registrant’s common stock that were issued and outstanding as of September 30, 2016. The percentage ownership and voting power for each person (or all directors and executive officers as a group) is calculated by assuming the exercise or conversion of all options, warrants and convertible securities exercisable or convertible within 60 days of September 30, 2016 held by such person and the non-exercise and non-conversion of all outstanding warrants, options and convertible securities held by all other persons. |
| (3) | Based solely on information set forth in a 13G/A filed with the SEC on February 12, 2016. Voting and dispositive power with respect to the shares is shared with Broadfin Healthcare Master Fund, Ltd. and Kevin Kotler. Kevin Kotler is the Managing Member of Broadfin Capital, LLC and Director of Broadfin Healthcare Master Fund, Ltd. |
57
Table of Contents
| (4) | Consists of (a) 18,750 shares of common stock and (b) 137,988 shares of common stock issuable upon the exercise of options and 4,000 shares of common stock issuable upon the exercise of warrants exercisable within 60 days of September 30, 2016. |
| (5) | Consists of (a) 4,400 shares of common stock and (b) 9,585 shares of common stock issuable upon the exercise of options and 1,875 shares of common stock issuable upon the exercise of warrants exercisable within 60 days of September 30, 2016. |
| (6) | Consists of 9,585 shares of common stock issuable upon the exercise of options exercisable within 60 days of September 30, 2016. |
| (7) | Consists of (a) 3,750 shares of common stock and (b) 7,918 shares of common stock issuable upon the exercise of options and 1,875 shares of common stock issuable upon the exercise of warrants exercisable within 60 days of September 30, 2016. |
| (8) | Consists of (a) 1,300 shares of common stock and (b) 7,918 shares of common stock issuable upon the exercise of options and 600 shares of common stock issuable upon the exercise of warrants exercisable within 60 days of September 30, 2016. |
| (9) | Consists of (a) 6,062 shares of common stock and (b) 69,069 shares of common stock issuable upon the exercise of options and 625 shares of common stock issuable upon the exercise of warrants exercisable within 60 days of September 30, 2016. |
Securities Authorized for Issuance Under Equity Compensation Plans
The following tables provides information, as of September 30, 2016, about the securities authorized for issuance under our equity compensation plans, which consisted of our 2012 Long Term Incentive Plan and our 2013 Employee Stock Purchase Plan:
| Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in First Column) |
|||||||||
| Equity compensation plans approved by security holders |
390,969 | $ | 26.38 | 108,831 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
390,969 | $ | 26.38 | 108,831 | ||||||||
58
Table of Contents
Authorized Capital Stock
Our authorized capital stock consists of 100,000,000 shares of common stock, par value $0.0001 per share, and 10,000,000 shares of preferred stock, par value $0.0001 per share. As of September 30, 2016, 6,599,846 shares of our common stock were outstanding and zero shares of our preferred stock were outstanding.
Common Stock
The holders of our common stock are entitled to one vote for each share on all matters voted on by stockholders, including elections of directors, and, except as otherwise required by law or provided in any resolution adopted by our Board with respect to any series of preferred stock, the holders of such shares will possess all voting power. Our certificate of incorporation does not provide for cumulative voting in the election of directors. The shares of common stock have no conversion rights or sinking fund provisions and are not liable for further call or assessment. Subject to any preferential rights of any outstanding series of our preferred stock created by our board from time to time, the holders of common stock are entitled to such dividends as may be declared from time to time by our board from funds available therefor and upon liquidation are entitled to receive their pro rata share of all assets available for distribution to such holders. Our common stock is not redeemable.
The holders of our common stock have no preemptive rights.
The rights, preferences and privileges of holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate and issue in the future.
On April 14, 2016, the Board of Directors of the Company approved a 1-for-10 reverse stock split of the Company’s outstanding common stock, which was effected on April 18, 2016. The number of authorized shares of the Company remain unchanged. Stockholders who would have otherwise been entitled to fractional shares as a result of the reverse stock split received a cash payment in lieu of receiving fractional shares. Shares of common stock underlying outstanding stock options and other equity instruments were proportionately reduced and the respective exercise prices, if applicable, were proportionately increased in accordance with the terms of the agreements governing such securities. Following the implementation of the reverse stock split, the Company regained compliance with the minimum bid price requirement set forth in Nasdaq Listing Rule 5550(a)(2) for continued listing on The Nasdaq Capital Market.
Preferred Stock
Our board of directors, without further action by the holders of our common stock, may issue shares of our preferred stock in one or more series. Our board is vested with the authority to fix by resolution the designations, preferences and relative, participating, optional or other special rights, and such qualifications, limitations or restrictions thereof, including, without limitation, redemption rights, dividend rights, liquidation preferences and conversion or exchange rights of any class or series of preferred stock, and to fix the number of classes or series of preferred stock, the number of shares constituting any such class or series and the voting powers for each class or series.
The authority possessed by our board to issue preferred stock could potentially be used to discourage attempts by third parties to obtain control of us through a merger, tender offer, proxy contest or otherwise by making such attempts more difficult or more costly. Our board may issue preferred stock with voting rights or conversion rights that, if exercised, could adversely affect the voting power of the holders of common stock. Other than with respect to the Series B Preferred Stock, there are no current agreements or understandings with respect to the future issuance of preferred stock and our board has no present intention to issue any additional shares of preferred stock.
59
Table of Contents
Outstanding Warrants
As of September 30, 2016, we had outstanding warrants to purchase 1,300,464 shares of common stock at a weighted-average exercise price of $5.21 per share, which expire on April 27, 2017 and June 2, 2020.
Units
We are offering 2,017,000 Class A Units, with each Class A Unit consisting of one share of common stock and a warrant to purchase half of one share of our common stock (based on an assumed offering price per common share of $1.19, which was the last reported sale price of our common stock on November 10, 2016), together with the shares of common stock underlying such warrants, at a public offering price of $ per Class A Unit. Each warrant included in the Class A Units entitles its holder to purchase half of one share of common stock at an exercise price of $ . The Class A Units will not be certificated and the shares of common stock and warrants part of such Unit are immediately separable and will be issued separately in this offering.
We are also offering to those purchasers, whose purchase of Class A Units in this offering would result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 9.99% of our outstanding common stock following the consummation of this offering, the opportunity to purchase, in lieu of Class A Units that would result in ownership in excess of 9.99%, 9,600 Class B Units, with each Class B Unit consisting of one share of Series B Convertible Preferred Stock, par value $0.0001 per share, convertible into 840 shares of common stock and warrants to purchase 420 shares of our common stock (based on an assumed offering price per common share of $1.19, which was the last reported sale price of our common stock on November 10, 2016), together with the shares of common stock underlying such shares of Series B Convertible Preferred Stock and warrants, at a public offering price of $1,000 per Class B Unit. Each warrant included in the Class B Units entitles its holder to purchase half of one share of common stock at an exercise price of $ . The Class B Units will not be certificated and the shares of Series B Convertible Preferred Stock and warrants part of such unit are immediately separable and will be issued separately in this offering.
Description of Warrants Included in the Units
The material terms and provisions of the warrants being issued in this offering are summarized below. The following description is subject to, and qualified in its entirety by, the form of warrant, which has been filed as an exhibit to the registration statement of which this prospectus is a part. Prospective investors should carefully review the terms and provisions set forth in the form of warrant.
Form. Pursuant to a warrant agency agreement between us and Computershare Trust Company, N.A., as warrant agent, the warrants will be issued in book-entry form and shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian on behalf of The Depositary Trust Company, or DTC, and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC.
Exercisability. The warrants are exercisable at any time after the date of issuance, and at any time up to the date that is years from the date of issuance, at which time any unexercised warrants will expire and cease to be exercisable. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and by payment in full in immediately available funds for the number of shares of common stock purchased upon such exercise. If a registration statement registering the issuance of the shares of common stock underlying the warrants under the Securities Act of 1933, as amended, is not then effective or available, the holder may only exercise the warrant through a cashless exercise, in whole or in part, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the warrant. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will either pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price or round up to the next whole share.
Exercise Limitation. A holder will not have the right to exercise any portion of the warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% of the number of shares of our stock
60
Table of Contents
outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days after such notice to us.
Exercise Price. The initial exercise price is $ per share of common stock. The exercise price is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock.
Transferability. Subject to applicable laws, the warrants may not be offered for sale, sold, transferred or assigned without our consent. There is currently no trading market for the warrants and a trading market is not expected to develop.
Exchange Listing. We do not plan to apply to list the warrants on the NASDAQ Capital Market, any other national securities exchange or any other nationally recognized trading system.
Fundamental Transactions. In the event of a fundamental transaction, as described in the warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, or our consolidation or merger with or into another person, the holders of the warrants will be entitled to receive upon exercise of the warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the warrants immediately prior to such fundamental transaction.
Rights as a Stockholder. Except as otherwise provided in the warrants or by virtue of such holder’s ownership of shares of our common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant.
Description of Series B Convertible Preferred Stock Included in the Class B Units
In connection with this offering our board of directors will authorize the issuance of shares of Series B Convertible Preferred Stock (“Series B Convertible Preferred Shares”). The Series B Convertible Preferred Shares preferences and rights will be as set forth in a Certificate of Designation (the “Series B Convertible Preferred Stock Certificate of Designation.”)
Holders of Series B Convertible Preferred Shares are entitled to be paid a liquidation preference equal to $0.01 per share. The Series B Convertible Preferred Stock Certificate of Designation provides, among other things, that we shall not pay any dividends on shares of common stock (other than dividends in the form of common stock) unless and until such time as we pay dividends on each Series B Convertible Preferred Share on an as-converted basis. Other than as set forth in the previous sentence, the Series B Convertible Preferred Stock Certificate of Designation provides that no other dividends shall be paid on Series B Convertible Preferred Shares and that we shall pay no dividends (other than dividends in the form of common stock) on shares of common stock unless we simultaneously comply with the previous sentence. The Series B Convertible Preferred Stock Certificate of Designation does not provide for any restriction on the repurchase of Series B Convertible Preferred Shares by us while there is any arrearage in the payment of dividends on the Series B Convertible Preferred Shares. There are no sinking fund provisions applicable to the Series B Convertible Preferred Shares.
With certain exceptions, as described in the Series B Convertible Preferred Stock Certificate of Designation, the Series B Convertible Preferred Shares have no voting rights. However, as long as any shares of Series B Convertible Preferred Shares remain outstanding, the Series B Convertible Preferred Stock Certificate of Designation provides that we shall not, without the affirmative vote of holders of a majority of the then-outstanding Series B Convertible Preferred Shares, (a) alter or change adversely the powers, preferences or rights given to the Series B Convertible Preferred Shares or alter or amend the Series B Convertible Preferred Stock Certificate of Designation, (b) increase the number of authorized shares of Series B Convertible Preferred Shares or (c) effect a stock split or reverse stock split of the Series B Convertible Preferred Shares or any like event.
61
Table of Contents
Each Series B Convertible Preferred Share is convertible at any time at the holder’s option into a number of shares of common stock equal to $1,000 per share divided by the Series B Convertible Preferred Share Conversion Price. The “Series B Convertible Preferred Share Conversion Price” is initially $ and is subject to adjustment for stock splits, stock dividends, distributions, subdivisions and combinations. Notwithstanding the foregoing, the Series B Convertible Preferred Stock Certificate of Designation further provides that we shall not effect any conversion of Series B Convertible Preferred Shares, with certain exceptions, to the extent that, after giving effect to an attempted conversion, the holder of Series B Convertible Preferred Shares (together with such holder’s affiliates, and any other person whose beneficial ownership of common stock would be aggregated with the holder’s for purposes of Section 13(d) of the Exchange Act and the applicable regulations thereunder, including any “group” of which the holder is a member) would beneficially own a number of shares of common stock in excess of 9.99% of the shares of our common stock then outstanding.
We do not intend to apply for listing of the Series B Convertible Preferred Shares on any securities exchange or other trading system.
The Series B Convertible Preferred Stock will be issued in book-entry form and shall initially be represented only by one or more global certificates deposited with The Depositary Trust Company, or DTC, and registered in the name of Cede & Co., a nominee or DTC, or as otherwise directed by DTC.
Anti-Takeover Effects of Provisions of the Certificate of Incorporation and Bylaws
Certificate of Incorporation and Bylaw Provisions. Our certificate of incorporation and bylaws include a number of provisions that may have the effect of encouraging persons considering unsolicited tender offers or other unilateral takeover proposals to negotiate with our board of directors rather than pursue non-negotiated takeover attempts. They are intended to enhance long-term value to our stockholders by increasing the likelihood of continued stability in the composition of our board of directors and its policies and discouraging certain types of transactions that may involve an actual or threatened acquisition of us. These provisions are also designed to reduce our vulnerability to an unsolicited acquisition proposal and to discourage certain tactics that may be used in proxy fights. However, such provisions could have the effect of discouraging others from making tender offers for our shares and, as a consequence, they also may inhibit fluctuations in the market price of our stock that could result from actual or rumored takeover attempts. These provisions include the items described below.
Filling Vacancies. Any vacancy on our board of directors, however occurring, including a vacancy resulting from an increase in the size of our board of directors, may only be filled by the affirmative vote of a majority of our directors then in office even if less than a quorum.
No Written Consent of Stockholders. Our certificate of incorporation provides that all stockholder actions are required to be taken by a vote of the stockholders at an annual or special meeting, and that stockholders may not take any action by written consent in lieu of a meeting.
Meetings of Stockholders. Our certificate of incorporation provides that only our board of directors or holders of 5% or more of our outstanding shares of common stock may call special meetings of stockholders and only those matters set forth in the notice of the special meeting may be considered or acted upon at a special meeting of stockholders. Our bylaws limit the business that may be conducted at an annual meeting of stockholders to those matters properly brought before the meeting.
Advance Notice Requirements. Our bylaws include advance notice procedures with regard to stockholder proposals relating to the nomination of candidates for election as directors or new business to be brought before meetings of our stockholders. These procedures provide that notice of stockholder proposals must be timely given in writing to our corporate secretary prior to the meeting at which the action is to be taken. Generally, to be timely, notice must be received at our principal executive offices not less than 90 days nor more than 120 days prior to the first anniversary date of the annual meeting for the preceding year. The notice must contain certain information specified in the amended and restated bylaws.
Amendment to Bylaws and Certificate of Incorporation. As required by the Delaware General Corporation Law (the “DGCL”) any amendment of our certificate of incorporation must first be approved by a majority of
62
Table of Contents
our board of directors and, if required by law or our certificate of incorporation, thereafter be approved by a majority of the outstanding shares entitled to vote on the amendment, and a majority of the outstanding shares of each class entitled to vote thereon as a class. Our bylaws may be amended by the affirmative vote of a majority vote of the directors then in office, subject to any limitations set forth in the bylaws.
Blank Check Preferred Stock. Our amended and restated certificate of incorporation provides for 10,000,000 authorized shares of preferred stock. The existence of authorized but unissued shares of preferred stock may enable our board of directors to render more difficult or to discourage an attempt to obtain control of us by means of a merger, tender offer, proxy contest, or otherwise. For example, if in the due exercise of its fiduciary obligations, our board of directors were to determine that a takeover proposal is not in the best interests of our company or our stockholders, our board of directors could cause shares of preferred stock to be issued without stockholder approval in one or more private offerings or other transactions that might dilute the voting or other rights of the proposed acquirer or insurgent stockholder or stockholder group. In this regard, our amended and restated certificate of incorporation grants our board of directors broad power to establish the rights and preferences of authorized and unissued shares of preferred stock. The issuance of shares of preferred stock could decrease the amount of earnings and assets available for distribution to holders of shares of common stock. The issuance may also adversely affect the rights and powers, including voting rights, of these holders and may have the effect of delaying, deterring, or preventing a change of control of us.
Delaware Business Combination Statute
Section 203 of the DGCL provides that, subject to exceptions set forth therein, an “interested stockholder” of a Delaware corporation shall not engage in any business combination, including mergers or consolidations or acquisitions of additional shares of the corporation, with the corporation for a three-year period following the date that such stockholder becomes an interested stockholder unless:
| • | Prior to such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
| • | Upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, other than statutorily excluded shares; or |
| • | On or subsequent to such date, the business combination is approved by the board of directors of the corporation and authorized at an annual or special meeting of the stockholders by the affirmative vote of at least 66-2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Except as otherwise set forth in Section 203, an “interested stockholder” is defined to include:
| • | Any person that is the owner of 15% or more of the outstanding voting stock of the corporation, or is an affiliate or associate of the corporation and was the owner of 15% or more of the outstanding voting stock of the corporation at any time within three years immediately prior to the date of determination; and |
| • | The affiliates and associates of any such person. |
The restrictions contained in Section 203 are not applicable to any of our existing stockholders that owned 15% or more of our outstanding common stock upon the completion of our spin-off from Galena.
Section 203 may make it more difficult for a person who would be an interested stockholder to effect various business combinations with a corporation for a three-year period. We have not elected to be exempt from the restrictions imposed under Section 203. The provisions of Section 203 may encourage persons interested in acquiring us to negotiate in advance with our board, since the stockholder approval requirement would be avoided if a majority of the directors then in office approves either the business combination or the transaction which results in any such person becoming an interested stockholder. Such provisions also may have the effect of preventing changes in our management. It is possible that such provisions could make it more difficult to accomplish transactions, which our stockholders may otherwise deem to be in their best interests.
63
Table of Contents
Exchange Listing
Our common stock is listed on the Nasdaq Capital Market under the trading symbol “RXII.”
Transfer Agent and Registrar
Computershare Trust Company, N.A. is the transfer agent and registrar for our common stock.
64
Table of Contents
We have entered into an underwriting agreement dated , 2016, with Ladenburg Thalmann & Co. Inc. as representative of the underwriters in the table below (the “Representative”). The underwriting agreement provides for the purchase of a specific number of Class A Units and Class B Units. Each Class A Unit consists of one share of common stock and a warrant to purchase half of one share of our common stock. Each Class B Unit consists of one share of Series B Convertible Preferred Stock, par value $0.0001 per share, convertible into 840 shares of common stock and warrants to purchase 420 shares of our common stock. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriters and the underwriters have agreed to purchase from us, at the public offering prices less the underwriting discounts set forth on the cover page of this prospectus, the number of our securities set forth opposite their name below.
| Underwriter | Class A Units | Class B Units | ||||||
| Ladenburg Thalmann & Co. Inc. |
||||||||
| Griffin Securities, Inc. |
||||||||
|
|
|
|
|
|||||
| Total |
||||||||
|
|
|
|
|
|||||
A copy of the underwriting agreement will be filed as an exhibit to the registration statement of which this prospectus is part. The securities we are offering are being offered by the underwriter subject to certain conditions specified in the underwriting agreement.
We have been advised by the underwriters that they propose to offer the units directly to the public at the applicable public offering price set forth on the cover page of this prospectus.
The underwriting agreement provides that the underwriters’ obligation to purchase the securities we are offering is subject to conditions contained in the underwriting agreement. Subject to the satisfaction or waiver by the Representative of such conditions, the underwriters are obligated to purchase and pay for all of the units offered by this prospectus.
No action has been taken by us or the underwriters that would permit a public offering of the units, or the common stock, Series B Convertible Preferred Stock and warrants to purchase common stock included in the units, or the common stock underlying the warrants and the Series B Convertible Preferred Stock in any jurisdiction outside the United States where action for that purpose is required. None of our securities included in this offering may be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sales of any of the units, the common stock, Series B Convertible Preferred Stock and warrants to purchase common stock, or the common stock underlying the warrants and the Series B Convertible Preferred Stock be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons who receive this prospectus are advised to inform themselves about and to observe any restrictions relating to this offering of units, the common stock, Series B Convertible Preferred Stock and warrants to purchase common stock, or the common stock underlying the warrants and the Series B Convertible Preferred Stock and the distribution of this prospectus. This prospectus is neither an offer to sell nor a solicitation of any offer to buy the units, the common stock, Series B Convertible Preferred Stock and warrants to purchase common stock, or the common stock underlying the warrants and the Series B Convertible Preferred Stock in any jurisdiction where that would not be permitted or legal.
The underwriters have advised us that they do not intend to confirm sales to any accounts over which they exercise discretionary authority.
65
Table of Contents
Underwriting Discount and Expenses
The following table summarizes the underwriting discount and commission to be paid to the underwriters by us.
| Per Class A Unit(1) | Per Class B Unit(1) | Total | ||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discount(2)(3) |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to RXi Pharmaceuticals Corporation |
$ | $ | $ | |||||||||
| (1) | The public offering price and underwriting discount corresponds to (x) in respect of the Class A Units (i) an assumed public offering price per share of common stock of $ and (ii) an assumed public offering price per warrant of $ and (y) in respect of the Class B Units (i) an assumed public offering price per share of Series B Convertible Preferred Stock of $ and (ii) an assumed public offering price per warrant of $ . |
| (2) | We have also agreed to reimburse the Representative for certain expenses. |
| (3) | We have granted a 45-day option to the underwriters to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Convertible Preferred Stock) and warrants sold in the primary offering) solely to cover over-allotments, if any. |
We estimate the total expenses payable by us for this offering to be approximately $1,076,997, which amount includes (i) the underwriting discount of $840,000 ($984,000 if the underwriters’ over-allotment option is exercised in full), (ii) reimbursement of the accountable expenses of the Representative up to $75,000 ($15,000 of which has been paid in advance, subject to FINRA Rule 5110(f)(2)(C)), including the legal fees of the Representative being paid by us, and (iii) other estimated expenses of approximately $236,997 which includes legal, accounting, printing costs and various fees associated with the registration and listing of our shares.
We have also agreed, subject to certain conditions, to give the Representative a twelve month right of first refusal from the closing date of any securities offering consummated during the term of our engagement with the Representative, to act as a sole bookrunner or exclusive placement agent in any financing involving an underwriter or placement agent following the closing date of this offering. In addition, we will pay the Representative a cash fee as provided under our engagement agreement with them equal to the underwriting discount percentage in this offering in the event that the offering contemplated hereby does not close and any investor contacted by the Representative in connection with this offering (other than certain insider investors) purchases securities from us at any time between the termination of the offering and six months after the date of termination or expiration of the offering.
Over-allotment Option
We have granted a 45 day option to the underwriters to purchase additional shares of common stock and/or warrants to purchase shares of common stock (up to 15% of the number of shares of common stock (including the number of shares of common stock issuable upon conversion of shares of Series B Convertible Preferred Stock) and warrants sold in the primary offering) solely to cover over-allotments, if any at the public offering price, less the underwriting discount, set forth on the cover page of this prospectus. The underwriters may exercise the option solely to cover over-allotments, if any, made in connection with this offering. If any additional shares of common stock and/or warrants are purchased pursuant to the over-allotment option, the underwriters will offer these shares of common stock and/or warrants on the same terms as those on which the other units are being offered hereby. The over-allotment option may be used to purchase shares of common stock, or warrants, or any combination thereof, as determined by the underwriters.
66
Table of Contents
Determination of Offering Price
Our common stock is listed on The NASDAQ Capital Market under the symbol “RXII”. The closing price of our common stock on November 10, 2016, as reported by NASDAQ, was $1.19 per share. We do not intend to apply for listing of the shares of Series B Convertible Preferred Stock or the warrants on any securities exchange or other trading system.
The public offering price of the units offered by this prospectus has been determined by negotiation between us and the Representative. Among the factors considered in determining the public offering price of the shares were:
| • | the number of shares authorized in our certificate of incorporation; |
| • | the current market price of our common stock; |
| • | trading prices of our common stock over time; |
| • | the volatility of our common stock; |
| • | our current financial condition and the prospects for our future cash flows; |
| • | the availability of and likely cost of capital of other potential sources of capital; and |
| • | market and economic conditions at the time of the offering. |
The offering price stated on the cover page of this prospectus should not be considered an indication of the actual value of the units. That price is subject to change as a result of market conditions and other factors, and we cannot assure you that the units can be resold at or above the public offering price.
Lock-up Agreements
Pursuant to certain “lock-up” agreements, our executive officers, directors and certain stockholders have agreed, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, any shares or any securities convertible into or exercisable or exchangeable for shares, whether currently owned or subsequently acquired, without the prior written consent of the Representative, for a period of 90 days from the date of effectiveness of the registration statement of which this prospectus is a part. Pursuant to the underwriting agreement, we and our subsidiaries have agreed, subject to certain exceptions, not to issue, enter into any agreement to issue or announce the issuance or proposed issuance of any shares of common stock or common stock equivalents for a period of 90 trading days from the date of effectiveness of the registration statement of which this prospectus is a part.
Stabilization, Short Positions and Penalty Bids
The underwriters may engage in syndicate covering transactions, stabilizing transactions and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of our common stock:
| • | Stabilizing transactions permit bids to purchase shares of common stock so long as the stabilizing bids do not exceed a specified maximum, and are engaged in for the purpose of preventing or retarding a decline in the market price of the shares of common stock while this offering is in progress. |
| • | Over-allotment transactions involve sales by the underwriters of shares of common stock in excess of the number of shares the underwriter is obligated to purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing shares and/or warrants in the open market. |
67
Table of Contents
| • | Syndicate covering transactions involve purchases of shares of common stock in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares and/or warrants available for purchase in the open market as compared with the price at which they may purchase shares and/or warrants through exercise of the over-allotment option. If the underwriters sells more shares than could be covered by exercise of the over-allotment option and, therefore, has a naked short position, the position can be closed out only by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could be downward pressure on the price of the shares in the open market that could adversely affect investors who purchase in this offering. |
| • | Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the securities originally sold by the syndicate member are purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These syndicate covering transactions, stabilizing transactions and penalty bids may have the effect of raising or maintaining the market prices of our securities or preventing or retarding a decline in the market prices of our securities. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of our common stock. These transactions may be effected on The NASDAQ Capital Market, in the over-the-counter market or on any other trading market and, if commenced, may be discontinued at any time.
In connection with this offering, the underwriters also may engage in passive market making transactions in our common stock in accordance with Regulation M during a period before the commencement of offers or sales of shares of our common stock in this offering and extending through the completion of the distribution. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for that security. However, if all independent bids are lowered below the passive market maker’s bid, that bid must then be lowered when specific purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Neither we nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the prices of our securities. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that any transactions, once commenced, will not be discontinued without notice.
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including certain liabilities arising under the Securities Act, or to contribute to payments that the underwriters may be required to make for these liabilities.
Other Relationships
The underwriters and its affiliates have engaged in, and may in the future engage in, investment banking and other commercial dealings in the ordinary course of business with us or our affiliates. They have received, or may in the future receive, customary fees and commissions for these transactions. We have not paid the underwriters any compensation in the 180 days prior to the date of this prospectus supplement, and we have no current arrangements or expectation to pay the underwriters any compensation (other than in connection with this offering) within the next 90 days. Affiliates of the Representative currently own approximately 232,000 shares of our common stock. These shares were acquired more than 180 days prior to this offering.
68
Table of Contents
Certain legal matters relating to the issuance of the securities offered by this prospectus will be passed upon for us by Gibson, Dunn & Crutcher LLP, San Francisco, California. Certain legal matters in connection with this offering will be passed upon for the underwriter by Ellenoff Grossman & Schole LLP.
The financial statements of RXi Pharmaceuticals Corporation as of December 31, 2015 and 2014 and for the years then ended included in this Prospectus have been so included in reliance on the report of BDO USA, LLP, an independent registered public accounting firm, appearing elsewhere herein and in the Registration Statement, given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We are required to file annual, quarterly and special reports, proxy statements and other information with the SEC. You may read and copy any document filed by us at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. Our filings with the SEC are also available to the public at the SEC’s Internet web site at http://www.sec.gov.
We have filed a registration statement, of which this prospectus is a part, covering the securities offered hereby. As allowed by SEC rules, this prospectus does not include all of the information contained in the registration statement and the included exhibits, financial statements and schedules. You are referred to the registration statement, the included exhibits, financial statements and schedules for further information. This prospectus is qualified in its entirety by such other information.
We are subject to the information and periodic reporting requirements of the Exchange Act and, in accordance therewith, file periodic reports, proxy statements and other information with the SEC. Such periodic reports, proxy statements and other information are available for inspection and copying at the public reference room and website of the SEC referred to above. We maintain a website at www.rxipharma.com. The reference to our website address does not constitute incorporation by reference of the information contained on our website, and you should not consider the contents of our website in making an investment decision with respect to our common stock.
69
Table of Contents
Unaudited Condensed Financial Statements
F-1
Table of Contents
RXi PHARMACEUTICALS CORPORATION
(Amounts in thousands, except share and per share data)
(Unaudited)
| September 30, 2016 |
December 31, 2015 |
|||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 2,379 | $ | 5,117 | ||||
| Restricted cash |
50 | 50 | ||||||
| Short-term investments |
2,000 | 5,500 | ||||||
| Prepaid expenses |
321 | 311 | ||||||
|
|
|
|
|
|||||
| Total current assets |
4,750 | 10,978 | ||||||
| Property and equipment, net |
124 | 163 | ||||||
| Other assets |
27 | 18 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 4,901 | $ | 11,159 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 457 | $ | 1,163 | ||||
| Accrued expenses |
1,408 | 1,106 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
1,865 | 2,269 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.0001 par value; 10,000,000 authorized; no shares issued or outstanding |
— | — | ||||||
| Common stock, $0.0001 par value, 100,000,000 shares authorized; 6,599,846 and 6,534,846 shares issued and outstanding at September 30, 2016 and December 31, 2015, respectively |
1 | 1 | ||||||
| Additional paid-in capital |
66,795 | 65,994 | ||||||
| Accumulated deficit |
(63,760 | ) | (57,105 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
3,036 | 8,890 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 4,901 | $ | 11,159 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
F-2
Table of Contents
RXi PHARMACEUTICALS CORPORATION
CONDENSED STATEMENTS OF OPERATIONS
(Amounts in thousands, except share and per share data)
(Unaudited)
| Three Months Ended September 30, |
Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Net revenues |
$ | — | $ | — | $ | 19 | $ | 34 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development(1) |
1,464 | 1,734 | 4,108 | 5,202 | ||||||||||||
| General and administrative(1) |
752 | 770 | 2,587 | 2,447 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
2,216 | 2,504 | 6,695 | 7,649 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating loss |
(2,216 | ) | (2,504 | ) | (6,676 | ) | (7,615 | ) | ||||||||
| Other income (expense): |
||||||||||||||||
| Interest income, net |
4 | 8 | 15 | 10 | ||||||||||||
| Other income (expense), net |
— | — | 6 | (2 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total other income (expense) |
4 | 8 | 21 | 8 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
(2,212 | ) | (2,496 | ) | (6,655 | ) | (7,607 | ) | ||||||||
| Series A and Series A-1 convertible preferred stock dividends |
— | — | — | (209 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss applicable to common stockholders |
$ | (2,212 | ) | $ | (2,496 | ) | $ | (6,655 | ) | $ | (7,816 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per common share applicable to common stockholders: |
||||||||||||||||
| Basic and diluted |
$ | (0.34 | ) | $ | (0.38 | ) | $ | (1.02 | ) | $ | (1.76 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted average common shares: basic and diluted |
6,576,096 | 6,494,912 | 6,548,696 | 4,445,192 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| (1) Non-cash stock-based compensation expenses included in operating expenses are as follows: |
||||||||||||||||
| Research and development |
$ | 52 | $ | 174 | $ | 212 | $ | 552 | ||||||||
| General and administrative |
76 | 211 | 437 | 691 | ||||||||||||
The accompanying notes are an integral part of these financial statements.
F-3
Table of Contents
RXi PHARMACEUTICALS CORPORATION
CONDENSED STATEMENTS OF CASH FLOWS
(Amounts in thousands)
(Unaudited)
| Nine Months Ended September 30, |
||||||||
| 2016 | 2015 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | (6,655 | ) | $ | (7,607 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
41 | 60 | ||||||
| Non-cash stock-based compensation |
649 | 1,243 | ||||||
| Value of non-marketable equity securities recognized as revenue |
(9 | ) | — | |||||
| Fair value of common stock issued in exchange for patent and technology rights |
— | 228 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Prepaid expenses and other assets |
(10 | ) | 10 | |||||
| Accounts payable |
(706 | ) | (63 | ) | ||||
| Accrued expenses |
302 | 441 | ||||||
| Deferred revenue |
— | (47 | ) | |||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(6,388 | ) | (5,735 | ) | ||||
| Cash flows from investing activities: |
||||||||
| Purchase of short-term investments |
(2,000 | ) | (8,000 | ) | ||||
| Maturities of short-term investments |
5,500 | — | ||||||
| Cash paid for purchase of property and equipment |
(2 | ) | (39 | ) | ||||
|
|
|
|
|
|||||
| Net cash provided by (used in) investing activities |
3,498 | (8,039 | ) | |||||
| Cash flows from financing activities: |
||||||||
| Net proceeds from the issuance of common stock |
152 | 9,266 | ||||||
| Proceeds from the issuance of common stock upon the exercise of warrants |
— | 16 | ||||||
| Proceeds from the issuance of common stock in connection with the employee stock purchase plan |
— | 31 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
152 | 9,313 | ||||||
| Net decrease in cash and cash equivalents |
(2,738 | ) | (4,461 | ) | ||||
| Cash and cash equivalents at the beginning of period |
5,117 | 8,496 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at the end of period |
$ | 2,379 | $ | 4,035 | ||||
|
|
|
|
|
|||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||
| Series A and Series A-1 convertible preferred stock dividends |
$ | — | $ | 126 | ||||
|
|
|
|
|
|||||
| Fair value of Series A and Series A-1 convertible preferred stock dividends |
$ | — | $ | 209 | ||||
|
|
|
|
|
|||||
| Exchange of Series A convertible preferred stock into Series A-1 convertible preferred stock |
$ | — | $ | 2,000 | ||||
|
|
|
|
|
|||||
| Conversion of Series A and Series A-1 convertible preferred stock into common stock |
$ | — | $ | 6,814 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
F-4
Table of Contents
RXi PHARMACEUTICALS CORPORATION
NOTES TO CONDENSED FINANCIAL STATEMENTS
(Unaudited)
1. Nature of Operations
Description of Business
RXi Pharmaceuticals Corporation (“RXi,” “we,” “our” or the “Company”) is a clinical-stage RNAi company developing innovative therapeutics that address significant unmet medical needs. The Company’s development programs are based on our proprietary self-delivering RNAi (sd-rxRNA®) platform and Samcyprone™, a topical immunomodulator. Our clinical development programs include RXI-109, an sd-rxRNA for the treatment of dermal and ocular scarring, and Samcyprone™, for the treatment of such disorders as warts, alopecia areata, non-malignant skin tumors and cutaneous metastases of melanoma. In addition to these clinical programs, we have a pipeline of discovery and preclinical product candidates in our core therapeutic areas, as well as in other areas of interest. The Company’s pipeline, coupled with our extensive patent portfolio, provides for product and business development opportunities across a broad spectrum of therapeutic areas.
On April 14, 2016, the Board of Directors of the Company approved a 1-for-10 reverse stock split of the Company’s outstanding common stock, which was effected on April 18, 2016. All share and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock split, including reclassifying an amount equal to the reduction in par value to additional paid-in capital.
2. Liquidity and Going Concern
The Company has limited cash resources, has reported recurring losses from operations since inception and has not yet received revenues from sales of products. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern, and the Company’s current cash resources may not provide sufficient capital to fund operations for at least the next twelve months. Historically, the Company’s primary source of financing has been through the sale of its securities. The continuation of the Company as a going concern depends upon the Company’s ability to raise additional capital through an equity offering, debt offering or strategic opportunity to fund its operations. There can be no assurance that the Company will be successful in accomplishing these plans in order to continue as a going concern. These financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
3. Significant Accounting Policies
Basis of Presentation
The accompanying condensed financial statements are unaudited and have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). Certain information and footnote disclosures included in the Company’s annual financial statements have been condensed or omitted. The year-end condensed balance sheet data was derived from audited financial statements, but does not include all disclosures required by GAAP. In the opinion of management, all adjustments (including normal recurring accruals) considered necessary for a fair presentation of the condensed financial statements have been included. Interim results are not necessarily indicative of results for a full year.
Uses of Estimates in Preparation of Financial Statements
The preparation of financial statements in accordance with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from these estimates.
F-5
Table of Contents
Cash Equivalents
The Company considers all highly liquid instruments with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of amounts invested in certificates of deposit.
Restricted Cash
Restricted cash consists of certificates of deposit held by financial institutions as collateral for the Company’s corporate credit cards.
Short-term Investments
Short-term investments consist of certificates of deposit with original maturities ranging from over three months to one year.
Investments in Non-marketable Equity Securities
The Company’s investments in non-marketable equity securities are accounted for under the cost method because the Company does not have the ability to exercise significant influence over the investee and the securities do not have readily determinable fair values. Our investments are carried at cost less any impairment write-downs. Annually, the Company’s cost method investments are assessed for impairment. The Company does not reassess the fair value of cost method investments if there are no identified events or changes in circumstances that may have a significant adverse effect on the fair value of the investments.
Derivative Financial Instruments
The Company follows the provisions of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 815, “Derivatives and Hedging” (“ASC 815”). Financial instruments that meet the definition of a derivative are classified as an asset or liability and measured at fair value on the issuance date and are revalued on each subsequent balance sheet date. The changes in fair value are recognized as current period income or loss.
Revenue Recognition
Revenue is recognized when there is persuasive evidence of an arrangement, the fee is fixed or determinable, delivery has occurred or services have been rendered and collection of the related receivable is reasonably assured. The Company may generate revenue from product sales, license agreements, collaborative research and development arrangements and government grants. Payments received prior to the recognition of revenue are recorded as deferred revenue.
The Company has entered into license agreements for its proprietary sd-rxRNA technology during the ordinary course of business with start-up biotechnology and pharmaceutical companies. Under these agreements, the Company has granted exclusive licenses to the Company’s technology in exchange for potential future equity, cash and royalty payments. For each agreement, the Company determines whether the agreement includes multiple deliverables, and if so, whether they should be considered separate or a single unit of accounting and whether the delivered items have standalone value. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition guidance is applied to each of the separate units.
Upfront fees are recognized on a straight-line basis over the contracted or estimated period of performance if they do not have standalone value. If upfront fees are determined to have standalone value from other identified deliverables, the Company recognizes revenue upon delivery.
F-6
Table of Contents
Substantive milestone payments are recognized upon achievement of the milestone. In evaluating whether a milestone has substance, the consideration earned from the achievement of a milestone is considered if the milestone is commensurate with the entity’s performance to achieve the milestone or the enhancement of value of the delivered item, if it relates solely to past performance and if it is reasonable relative to all the deliverables and payment terms within the arrangement. When a substantive milestone is achieved, revenue related to the milestone will be recognized in full. If a milestone is not considered substantive, revenue is recognized over the period of performance.
If the Company is entitled to reimbursement or payments for specific research and development services, the Company determines whether the funding would result in collaborative revenues or an offset to research and development expenses in accordance with the provisions of gross or net revenue presentation.
Research and Development Expenses
Research and development costs are charged to expense as incurred and relate to salaries, employee benefits, facility-related expenses, supplies, stock-based compensation related to employees and non-employees involved in the Company’s research and development, external services, other operating costs and overhead related to our research and development departments, costs to acquire technology licenses and expenses associated with preclinical activities and our clinical trials. Payments made by the Company in advance for research and development services not yet provided and/or for materials not yet received are recorded as prepaid expenses. Accrued liabilities are recorded related to those expenses for which vendors have not yet billed us with respect to services provided and/or materials that we have received.
Preclinical and clinical trial expenses relate to third-party services, subject-related fees at the sites where our clinical trials are being conducted, laboratory costs, analysis costs, toxicology studies and investigator fees. Costs associated with these expenses are generally payable on the passage of time or when certain milestones are achieved. Expense is recorded during the period incurred or in the period in which a milestone is achieved. In order to ensure that we have adequately provided for preclinical and clinical expenses during the proper period, we maintain an accrual to cover these expenses. These accruals are assessed on a quarterly basis and are based on such assumptions as expected total cost, the number of subjects and clinical trial sites and length of the study. Actual results may differ from these estimates and could have a material impact on our reported results. Our historical accrual estimates have not been materially different from our actual costs.
Stock-based Compensation
The Company follows the provisions of the FASB ASC Topic 718, “Compensation — Stock Compensation” (“ASC 718”), which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees, officers and non-employee directors, including stock options. Stock compensation expense based on the grant date fair value estimated in accordance with the provisions of ASC 718 is recognized as an expense over the requisite service period.
For stock options granted as consideration for services rendered by non-employees, the Company recognizes compensation expense in accordance with the requirements of FASB ASC Topic 505-50, “Equity Based Payments to Non-Employees.” Non-employee option grants that do not vest immediately upon grant are recorded as an expense over the requisite service period of the underlying stock options. At the end of each financial reporting period prior to vesting, the value of these options, as calculated using the Black-Scholes option-pricing model, will be re-measured using the fair value of the Company’s common stock and the non-cash compensation recognized during the period will be adjusted accordingly. Since the fair market value of options granted to non-employees is subject to change in the future, the amount of the future compensation expense will include fair value re-measurements until the stock options are fully vested.
Comprehensive Loss
The Company’s comprehensive loss is equal to its net loss for all periods presented.
F-7
Table of Contents
Net Loss per Share Attributable to Common Stockholders
The Company accounts for and discloses net loss per share attributable to common stockholders in accordance with FASB ASC Topic 260, “Earnings per Share.” Basic and diluted net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding. When the effects are not dilutive, diluted earnings per share is computed by dividing the Company’s net earnings by the weighted average number of common shares outstanding and the impact of all dilutive potential common shares.
4. Recent Accounting Pronouncements
In February 2016, the FASB issued Accounting Standards Update (“ASU”) 2016-02, “Leases (Topic 842),” which requires companies that are lessees to recognize a right-of-use asset and lease liability for most leases that do not meet the definition of a short-term lease. For income statement purposes, leases will continue to be classified as either operating or financing. Classification will be based on criteria that are largely similar to those applied in current lease accounting. This standard will result in extensive qualitative and quantitative disclosure changes. This standard will be effective for annual reporting periods beginning after December 15, 2018, including interim periods within that reporting period. The Company is currently evaluating the impact of this ASU on its financial position and results of operations.
In March 2016, the FASB issued ASU 2016-09, “Compensation — Stock Compensation (Topic 718),” which simplifies several aspects of accounting for share-based payment transactions, including the income tax consequences, classifications of awards as either equity or liabilities and classification on the statement of cash flows. This standard will be effective for annual reporting periods beginning after December 15, 2016 and interim periods within that reporting period. Early adoption is permitted. The Company is currently evaluating the impact of this ASU on its financial statements and related disclosures.
In April 2016, the FASB issued ASU 2016-10, “Revenue from Contracts with Customers (Topic 606),” which clarifies two aspects of the guidance on accounting for revenue contracts with customers: identifying performance obligations and the licensing implementation guidance. The amendments in this ASU do not change the core principles for those areas. This standard will be effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period. Early adoption is not permitted. The Company is currently evaluating the potential impact the update may have on its financial position and results of operations.
In August 2016, the FASB issued ASU 2016-12, “Statement of Cash Flows (Topic 230),” which clarifies how certain cash receipts and payments are presented and classified in the statement of cash flows. This standard will be effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period. Early adoption is permitted. The amendments in ASU 2016-12 should be applied using a retrospective transition method to each period presented. The Company is currently evaluating the potential impact the update may have on its statement of cash flows.
5. Other Assets
In May 2016, the Company entered into an exclusive license agreement with Thera Neuropharma, Inc. (“Thera”), a privately held company, pursuant to which the Company granted certain rights to its sd-rxRNA platform for neurodegenerative diseases in exchange for an upfront equity ownership interest and the potential to receive future cash, additional equity and royalties based on the achievement of certain milestones. The Company was issued shares of common stock in Thera upon execution of the license agreement. Due to the Company’s inability to exercise significant influence over Thera and the Company owning less than 20% of the voting equity of Thera’s stock, the Company accounted for this investment using the cost method. As of September 30, 2016, the carrying value of the investment in Thera of $4,500 was included in other assets on the balance sheet and no impairment has been recognized for this investment through September 30, 2016.
F-8
Table of Contents
The Company was also granted a five year warrant to purchase additional shares of common stock of Thera at a price of $0.001 per share of common stock (the “Thera Warrant”) pursuant to the terms of the license agreement. The Company first assessed the Thera Warrant under ASC 815. Under the related guidance, a financial instrument shall be considered a derivative when it includes an underlying and notional amount or payment provision, an initial net investment and a net settlement. The Company determined that the Thera Warrant met all of the characteristics of a derivative. Per ASC 815, the Thera Warrant is recognized at fair value on the balance sheet and gains and losses from changes in the fair value of the Thera Warrant are recognized in the statement of operations. The fair value of the Thera Warrant at the date of issuance totaled $4,500 and was included in other assets on the balance sheet. There have been no changes to the fair value since the date of issuance.
6. Fair Value Measurements
The Company follows the provisions of FASB ASC Topic 820, “Fair Value Measurements and Disclosures,” for the Company’s financial assets and liabilities that are re-measured and reported at fair value at each reporting period and are re-measured and reported at fair value at least annually using a fair value hierarchy that is broken down into three levels. Level inputs are defined as follows:
Level 1 — quoted prices in active markets for identical assets or liabilities.
Level 2 — other significant observable inputs for the assets or liabilities through corroboration with market data at the measurement date.
Level 3 — significant unobservable inputs that reflect management’s best estimate of what market participants would use to price the assets or liabilities at the measurement date.
The Company categorized its restricted cash, cash equivalents and short-term investments as Level 2 hierarchy. The assets classified as Level 2 have initially been valued at the applicable transaction price and subsequently valued, at the end of each reporting period, using other market observable data. Observable market data points include quoted prices, interest rates, reportable trades and other industry and economic events. The Company’s Thera Warrant is categorized as Level 3 hierarchy. The estimated fair value inputs utilizing the asset-based approach include the stage of enterprise development, terms of existing contractual arrangements of the entity’s equity securities, the achievement of milestones and other unobservable inputs. Financial assets measured at fair value on a recurring basis are summarized as follows, in thousands:
| Description |
At September 30, 2016 | Quoted Prices in Active Markets (Level 1) |
Other Significant Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
| Assets: |
||||||||||||||||
| Restricted cash |
$ | 50 | $ | — | $ | 50 | $ | — | ||||||||
| Short-term investments |
2,000 | — | 2,000 | — | ||||||||||||
| Thera Warrant |
5 | — | — | 5 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 2,055 | $ | — | $ | 2,050 | $ | 5 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Description |
At December 31, 2015 | Quoted Prices in Active Markets (Level 1) |
Other Significant Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
| Assets: |
||||||||||||||||
| Restricted cash |
$ | 50 | $ | — | $ | 50 | $ | — | ||||||||
| Cash equivalents |
2,500 | — | 2,500 | — | ||||||||||||
| Short-term investments |
5,500 | — | 5,500 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 8,050 | $ | — | $ | 8,050 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
F-9
Table of Contents
The following table presents additional information about assets measured at fair value on a recurring basis and for which the Company utilizes Level 3 inputs to determine fair value, in thousands:
| Nine Months Ended September 30, 2016 |
||||
| Balance, beginning of period |
$ | — | ||
| Fair value of Thera Warrant |
5 | |||
|
|
|
|||
| Balance, end of period |
$ | 5 | ||
|
|
|
|||
Fair Value of Financial Instruments
The carrying amounts reported in the balance sheet for restricted cash, cash equivalents, short-term investments, accounts payable and accrued expenses approximate their fair values due to their short-term nature.
7. Stockholders’ Equity
The Company currently has authorized for issuance 100,000,000 shares of common stock, par value $0.0001 per share, and 10,000,000 shares of preferred stock, par value $0.0001 per share.
Common Stock
On April 14, 2016, the Board of Directors of the Company approved a 1-for-10 reverse stock split of the Company’s outstanding common stock, which was effected on April 18, 2016. The number of authorized shares of the Company remain unchanged. Stockholders who would have otherwise been entitled to fractional shares as a result of the reverse stock split received a cash payment in lieu of receiving fractional shares. Shares of common stock underlying outstanding stock options and other equity instruments were proportionately reduced and the respective exercise prices, if applicable, were proportionately increased in accordance with the terms of the agreements governing such securities. All share and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock split, including reclassifying an amount equal to the reduction in par value to additional paid-in capital. Following the implementation of the reverse stock split, the Company regained compliance with the minimum bid price requirement set forth in Nasdaq Listing Rule 5550(a)(2) for continued listing on The Nasdaq Capital Market.
During the three months ended September 30, 2016, the Company sold 65,000 shares of common stock to Lincoln Park Capital Fund, LLC (“LPC”) pursuant to a purchase agreement dated December 18, 2014 between the Company and LPC. The net proceeds to the Company totaled approximately $152,000.
Common Stock Warrants
The following table summarizes the Company’s warrants outstanding at September 30, 2016:
| Exercise prices |
Number of Shares Underlying Warrants |
Expiration | ||||||
| $39.00 |
462 | April 27, 2017 | ||||||
| $5.20 |
1,300,002 | June 2, 2020 | ||||||
|
|
|
|||||||
| Total warrants outstanding |
1,300,464 | |||||||
|
|
|
|||||||
On July 2, 2016, 1,256,502 of the Company’s outstanding warrants with an exercise price of $4.55 expired.
F-10
Table of Contents
8. Stock-based Compensation
The Company uses the Black-Scholes option-pricing model to determine the fair value of all its option grants. For valuing options granted during the three and nine months ended September 30, 2016 and 2015, the following assumptions were used:
| For the Three Months Ended September 30, |
For the Nine Months Ended September 30, |
|||||||||||||||
| 2016 | 2015 | 2016 | 2015 | |||||||||||||
| Risk-free interest rate |
1.46 | % | 1.82 – 2.43 | % | 1.18 – 2.02 | % | 1.47 – 2.43 | % | ||||||||
| Expected volatility |
116.88 | % | 85.29 – 116.81 | % | 79.42 – 116.88 | % | 85.29 – 116.81 | % | ||||||||
| Weighted average expected volatility |
116.88 | % | 107.49 | % | 89.12 | % | 89.43 | % | ||||||||
| Expected lives (in years) |
10.00 | 6.25 – 10.0 | 5.20 – 10.00 | 5.20 – 10.0 | ||||||||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | 0.00 | % | ||||||||
The weighted average fair value of options granted during the three month periods ended September 30, 2016 and 2015 was $2.27 and $4.40, respectively. The weighted average fair value of options granted during the nine month periods ended September 30, 2016 and 2015 was $2.15 and $4.10, respectively.
The risk-free interest rate used for each grant was based upon the yield on zero-coupon U.S. Treasury securities with a term similar to the expected life of the related option. The Company’s expected stock price volatility assumption is based upon the volatility of a composition of comparable companies. The expected life assumption for employee grants was based upon the simplified method provided for under ASC 718, and the expected life assumption for non-employees was based upon the contractual term of the option. The dividend yield assumption of zero is based upon the fact that the Company has never paid cash dividends and presently has no intention of paying cash dividends.
The following table summarizes the activity of Company’s stock option plan:
| Total Number of Shares |
Weighted- Average Exercise Price Per Share |
Aggregate Intrinsic Value |
||||||||||
| Balance at December 31, 2015 |
332,400 | $ | 30.50 | |||||||||
| Granted |
58,569 | 2.91 | ||||||||||
| Exercised |
— | — | ||||||||||
| Cancelled |
— | — | ||||||||||
|
|
|
|
|
|
|
|||||||
| Balance at September 30, 2016 |
390,969 | $ | 26.38 | $ | — | |||||||
|
|
|
|
|
|
|
|||||||
| Exercisable at September 30, 2016 |
298,347 | $ | 31.18 | $ | — | |||||||
|
|
|
|
|
|
|
|||||||
Stock-based compensation expense for the three months ended September 30, 2016 and 2015 was $128,000 and $385,000, respectively. Of this amount, the Company recognized non-employee stock-based compensation expense of $1,000 and $3,100 for the same respective periods.
Stock-based compensation expense for the nine months ended September 30, 2016 and 2015 was $649,000 and $1,243,000, respectively. Of this amount, the Company recognized non-employee stock-based compensation expense of $3,200 and a credit to non-employee stock-based compensation expense of $18,700 for the same respective periods.
F-11
Table of Contents
9. Net Loss per Share Attributable to Common Stockholders
The following table sets forth the potential common shares excluded from the calculation of net loss per common share attributable to common stockholders because their inclusion would be anti-dilutive:
| September 30, | ||||||||
| 2016 | 2015 | |||||||
| Options to purchase common stock |
390,969 | 330,876 | ||||||
| Warrants to purchase common stock |
1,300,464 | 2,596,966 | ||||||
|
|
|
|
|
|||||
| Total |
1,691,433 | 2,927,842 | ||||||
|
|
|
|
|
|||||
10. Subsequent Events
On October 7, 2016, the Company entered into an exclusive option agreement to acquire all outstanding capital stock of MirImmune, Inc. (“MirImmune”) in consideration for a number of shares equal to 19.99% of the then-outstanding shares of common stock of the Company, plus additional potential consideration contingent on MirImmune reaching certain milestones. RXi can exercise the option to acquire MirImmune on the terms set forth in the option agreement at any time prior to April 5, 2017, but has no obligation to do so. For accounting purposes, if the option is exercised, the transaction will be accounted for as an asset acquisition per the guidance in FASB ASC Topic 805, “Business Combinations.”
F-12
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
RXi Pharmaceuticals Corporation
Marlborough, Massachusetts
We have audited the accompanying balance sheets of RXi Pharmaceuticals Corporation (the “Company”) as of December 31, 2015 and 2014, and the related statements of operations, convertible preferred stock and stockholders’ equity and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of the Company at December 31, 2015 and 2014 and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
/s/ BDO USA, LLP
Boston, Massachusetts
March 30, 2016 (except for the stock split discussed in Note 13, as to which the date is April 18, 2016)
F-13
Table of Contents
RXi PHARMACEUTICALS CORPORATION
(Amounts in thousands, except share data)
| Years Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 5,117 | $ | 8,496 | ||||
| Restricted cash |
50 | 50 | ||||||
| Short-term investments |
5,500 | — | ||||||
| Prepaid expenses |
311 | 442 | ||||||
|
|
|
|
|
|||||
| Total current assets |
10,978 | 8,988 | ||||||
| Property and equipment, net of accumulated depreciation of $778 and $702, in 2015 and 2014, respectively |
163 | 183 | ||||||
| Other assets |
18 | 18 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 11,159 | $ | 9,189 | ||||
|
|
|
|
|
|||||
| LIABILITIES, CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 1,163 | $ | 285 | ||||
| Accrued expenses |
1,106 | 1,002 | ||||||
| Deferred revenue |
— | 47 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
2,269 | 1,334 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Note 6) |
||||||||
| Convertible preferred stock: |
||||||||
| Series A convertible preferred stock, $0.0001 par value, no shares authorized, issued or outstanding at December 31, 2015; 15,000 shares authorized and 5,110 shares issued and outstanding at December 31, 2014 (at liquidation value) |
— | 5,110 | ||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.0001 par value; 10,000,000 authorized |
||||||||
| Series A-1 convertible preferred stock, $0.0001 par value, no shares authorized, issued or outstanding at December 31, 2015; 10,000 shares authorized and 1,578 shares issued and outstanding at December 31, 2014 (at liquidation value) |
— | 1,578 | ||||||
| Common stock, $0.0001 par value, 100,000,000 shares authorized; 6,534,846 and 2,198,427 shares issued and outstanding at December 31, 2015 and 2014, respectively |
1 | — | ||||||
| Additional paid-in capital |
65,994 | 48,049 | ||||||
| Accumulated deficit |
(57,105 | ) | (46,882 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
8,890 | 2,745 | ||||||
|
|
|
|
|
|||||
| Total liabilities, convertible preferred stock and stockholders’ equity |
$ | 11,159 | $ | 9,189 | ||||
|
|
|
|
|
|||||
See accompanying notes to financial statements.
F-14
Table of Contents
RXi PHARMACEUTICALS CORPORATION
(Amounts in thousands, except share and per share data)
| Years Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Revenue |
$ | 34 | $ | 71 | ||||
| Operating expenses: |
||||||||
| Research and development expenses(1) |
6,925 | 5,680 | ||||||
| General and administrative expenses(1) |
3,346 | 3,217 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
10,271 | 8,897 | ||||||
| Operating loss |
(10,237 | ) | (8,826 | ) | ||||
| Other income: |
||||||||
| Interest income, net |
16 | 17 | ||||||
| Other income (expense), net |
(2 | ) | 9 | |||||
|
|
|
|
|
|||||
| Total other income |
14 | 26 | ||||||
|
|
|
|
|
|||||
| Loss before income taxes |
(10,223 | ) | (8,800 | ) | ||||
| Provision for income taxes |
— | — | ||||||
|
|
|
|
|
|||||
| Net loss |
(10,223 | ) | (8,800 | ) | ||||
| Series A and A-1 convertible preferred stock dividends |
(209 | ) | (4,130 | ) | ||||
|
|
|
|
|
|||||
| Net loss applicable to common stockholders |
$ | (10,432 | ) | $ | (12,930 | ) | ||
|
|
|
|
|
|||||
| Net loss per common share applicable to common stockholders: |
||||||||
| Basic and diluted |
$ | (2.10 | ) | $ | (7.90 | ) | ||
|
|
|
|
|
|||||
| Weighted average common shares: basic and diluted |
4,970,382 | 1,636,291 | ||||||
|
|
|
|
|
|||||
| (1) Non-cash stock-based compensation expenses included in operating expenses are as follows: |
| |||||||
| Research and development |
$ | 632 | $ | 836 | ||||
| General and administrative |
903 | 1,010 | ||||||
See accompanying notes to financial statements.
F-15
Table of Contents
RXi PHARMACEUTICALS CORPORATION
STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY
(Amounts in thousands, except share data)
| Series A Convertible Preferred Stock |
Series A-1 Convertible Preferred Stock |
Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||
| Shares Issued |
Amount | Shares Issued |
Amount | Shares Issued |
Amount | |||||||||||||||||||||||||||||||
| Balance at December 31, 2013 |
7,920 | $ | 7,920 | 2,054 | $ | 2,054 | 1,178,804 | $ | — | $ | 40,970 | $ | (38,082 | ) | $ | 4,942 | ||||||||||||||||||||
| Issuance of common stock under Lincoln Park Capital, LLC purchase agreement, net of offering costs of $114 |
— | — | — | — | 70,000 | — | 1,886 | — | 1,886 | |||||||||||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
— | — | — | — | 3,252 | — | 61 | — | 61 | |||||||||||||||||||||||||||
| Stock-based compensation expense |
— | — | — | — | — | — | 1,846 | — | 1,846 | |||||||||||||||||||||||||||
| Dividends issued on Series A and Series A-1 convertible preferred stock |
356 | 356 | 240 | 240 | — | — | 3,534 | — | 3,774 | |||||||||||||||||||||||||||
| Fair value of Series A and Series A-1 convertible preferred stock dividends |
— | — | — | — | — | — | (4,130 | ) | — | (4,130 | ) | |||||||||||||||||||||||||
| Exchange of Series A convertible preferred stock into Series A-1 convertible preferred stock |
(3,000 | ) | (3,000 | ) | 3,000 | 3,000 | — | — | — | — | 3,000 | |||||||||||||||||||||||||
| Conversions of Series A and Series A-1 convertible preferred stock into common stock |
(166 | ) | (166 | ) | (3,716 | ) | (3,716 | ) | 946,371 | — | 3,882 | — | 166 | |||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (8,800 | ) | (8,800 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2014 |
5,110 | 5,110 | 1,578 | 1,578 | 2,198,427 | — | 48,049 | (46,882 | ) | 2,745 | ||||||||||||||||||||||||||
| Issuance of common stock under Lincoln Park Capital, LLC purchase agreement |
— | — | — | — | 5,000 | — | 64 | — | 64 | |||||||||||||||||||||||||||
| Issuance of common stock in exchange for patent and technology rights |
— | — | — | — | 20,000 | — | 228 | — | 228 | |||||||||||||||||||||||||||
| Issuance of common stock in connection with public offering, net of offering costs of $1,198 |
— | — | — | — | 2,600,000 | 1 | 9,201 | — | 9,202 | |||||||||||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
— | — | — | — | 6,889 | — | 31 | — | 31 | |||||||||||||||||||||||||||
| Issuance of common stock upon exercise of warrants in connection with public offering |
— | — | — | — | 43,500 | — | 198 | — | 198 | |||||||||||||||||||||||||||
| Stock-based compensation expense |
— | — | — | — | — | — | 1,535 | — | 1,535 | |||||||||||||||||||||||||||
| Dividends issued on Series A and Series A-1 convertible preferred stock |
105 | 105 | 21 | 21 | — | — | 83 | — | 104 | |||||||||||||||||||||||||||
| Fair value of Series A and Series A-1 convertible preferred stock dividends |
— | — | — | — | — | — | (209 | ) | — | (209 | ) | |||||||||||||||||||||||||
| Exchange of Series A convertible preferred stock into Series A-1 convertible preferred stock |
(2,000 | ) | (2,000 | ) | 2,000 | 2,000 | — | — | — | — | 2,000 | |||||||||||||||||||||||||
| Conversions of Series A and Series A-1 convertible preferred stock into common stock |
(3,215 | ) | (3,215 | ) | (3,599 | ) | (3,599 | ) | 1,661,030 | — | 6,814 | — | 3,215 | |||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (10,223 | ) | (10,223 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2015 |
— | $ | — | — | $ | — | 6,534,846 | $ | 1 | $ | 65,994 | $ | (57,105 | ) | $ | 8,890 | ||||||||||||||||||||
See accompanying notes to financial statements.
F-16
Table of Contents
RXi PHARMACEUTICALS CORPORATION
(Amounts in thousands)
| Years Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | (10,223 | ) | $ | (8,800 | ) | ||
| Adjustment to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization expense |
77 | 87 | ||||||
| Gain on disposal of property and equipment |
— | (10 | ) | |||||
| Non-cash share-based compensation expense |
1,535 | 1,846 | ||||||
| Fair value of common stock issued in exchange for patent and technology rights |
228 | — | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Prepaid expenses |
131 | (139 | ) | |||||
| Accounts payable |
878 | 122 | ||||||
| Accrued expenses |
104 | (793 | ) | |||||
| Deferred revenue |
(47 | ) | (71 | ) | ||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(7,317 | ) | (7,758 | ) | ||||
|
|
|
|
|
|||||
| Cash flows from investing activities: |
||||||||
| Purchase of short-term investments |
(8,000 | ) | (5,000 | ) | ||||
| Maturities of short-term investments |
2,500 | 8,000 | ||||||
| Cash paid for purchase of property and equipment |
(57 | ) | (95 | ) | ||||
| Proceeds from disposal of property and equipment |
— | 12 | ||||||
|
|
|
|
|
|||||
| Net cash (used in) provided by investing activities |
(5,557 | ) | 2,917 | |||||
|
|
|
|
|
|||||
| Cash flows from financing activities: |
||||||||
| Net proceeds from the issuance of common stock |
9,266 | 1,886 | ||||||
| Proceeds from the issuance of common stock upon the exercise of warrants |
198 | — | ||||||
| Proceeds from the issuance of common stock in connection with the employee stock purchase plan |
31 | 61 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
9,495 | 1,947 | ||||||
|
|
|
|
|
|||||
| Net decrease in cash and cash equivalents |
(3,379 | ) | (2,894 | ) | ||||
| Cash and cash equivalents at the beginning of period |
8,496 | 11,390 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at the end of period |
$ | 5,117 | $ | 8,496 | ||||
|
|
|
|
|
|||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||
| Exchange of Series A convertible preferred stock into Series A-1 convertible preferred stock |
$ | 2,000 | $ | 3,000 | ||||
|
|
|
|
|
|||||
| Conversion of Series A and Series A-1 convertible preferred stock into common stock |
$ | 6,814 | $ | 3,882 | ||||
|
|
|
|
|
|||||
| Fair value of Series A and Series A-1 convertible preferred stock dividends |
$ | 209 | $ | 4,130 | ||||
|
|
|
|
|
|||||
| Series A and Series A-1 convertible preferred stock dividends |
$ | 126 | $ | 596 | ||||
|
|
|
|
|
|||||
See accompanying notes to financial statements.
F-17
Table of Contents
RXi PHARMACEUTICALS CORPORATION
1. Nature of Business
RXi Pharmaceuticals Corporation (“RXi,” “we,” “our” or the “Company”) is a clinical-stage RNAi company developing innovative therapeutics in dermatology and ophthalmology that address significant unmet medical needs. Our development programs are based on our proprietary self-delivering RNAi (sd-rxRNA®) platform and Samcyprone™, a topical immunomodulator. Our clinical development programs include, but are not limited to, RXI-109, an sd-rxRNA for the treatment of dermal and ocular scarring, and Samcyprone™ for the treatment of such disorders as warts, alopecia areata, non-malignant skin tumors and cutaneous metastases of melanoma. In addition to these clinical programs, we have a pipeline of discovery and preclinical product candidates in our core therapeutic areas, as well as in other areas of interest. The Company’s pipeline, coupled with our extensive patent portfolio, provides for product development and business development opportunities across a broad spectrum of therapeutic areas.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”).
Uses of Estimates in Preparation of Financial Statements
The preparation of financial statements in accordance with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ materially from these estimates.
Cash and Cash Equivalents
The Company considers all highly liquid debt instruments with an original maturity of three months or less to be cash equivalents. Cash equivalents consist primarily of amounts invested in certificates of deposit.
Restricted Cash
Restricted cash consists of certificates of deposit held by financial institutions as collateral for the Company’s corporate credit cards.
Short-term Investments
Short-term investments consist of certificates of deposit with original maturities ranging from three months to one year.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents and short-term investments. The Company maintains cash balances in several accounts with one bank, which at times are in excess of federally insured limits. The Company has established guidelines related to credit ratings and maturities intended to safeguard principal balances and
F-18
Table of Contents
maintain liquidity. The Company’s investments are maintained in accordance with the Company’s investment policy, which defines allowable investments, specifies credit quality standards and limits the exposure of any single issuer.
Fair Value of Financial Instruments
The carrying amounts reported in the balance sheet for cash equivalents, restricted cash, short-term investments and accounts payable approximate their fair values due to their short-term nature or market rates of interest.
Property and Equipment
Property and equipment are stated at cost and depreciated using the straight-line method based on the estimated useful lives of the related assets. The Company provides for depreciation over the assets’ estimated useful lives as follows:
| Computer equipment |
3 years | |||
| Machinery & equipment |
5 years | |||
| Office furniture |
5 years |
Depreciation and amortization expense for the years ended December 31, 2015 and 2014 was approximately $77,000 and $87,000, respectively.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment on an interim basis if an event occurs that might reduce the fair value of such assets below their carrying values. An impairment loss would be recognized based on the difference between the carrying value of the asset and its estimated fair value, which would be determined based on either discounted future cash flows or other appropriate fair value methods. The Company believes no impairment existed as of December 31, 2015 and 2014.
Revenue Recognition
Revenue is recognized when there is persuasive evidence of an arrangement, the fee is fixed or determinable, delivery has occurred or services have been rendered and collection of the related receivable is reasonably assured. The Company may generate revenue from product sales, license agreements, collaborative research and development arrangements and government grants. The Company’s principal source of revenue consists of government research grants. Revenue from a government grant is recognized over the respective contract periods as the services are performed. Payments received prior to the recognition of revenue are recorded as deferred revenue.
Research and Development Expenses
Research and development costs are charged to expense as incurred and relate to salaries, employee benefits, facility-related expenses, supplies, stock-based compensation related to employees and non-employees involved in the Company’s research and development, external services, other operating costs and overhead related to our research and development departments, costs to acquire technology licenses and expenses associated with preclinical activities and our clinical trials. Payments made by the Company in advance for research and development services not yet provided and/or for materials not yet received are recorded as prepaid expenses. Accrued liabilities are recorded related to those expenses for which vendors have not yet billed us with respect to services provided and/or materials that we have received.
F-19
Table of Contents
Preclinical and clinical trial expenses relate to third-party services, subject-related fees at the sites where our clinical trials are being conducted, laboratory costs, analysis costs, toxicology studies and investigator fees. Costs associated with these expenses are generally payable on the passage of time or when certain milestones are achieved. Expense is recorded during the period incurred or in the period in which a milestone is achieved. In order to ensure that we have adequately provided for preclinical and clinical expenses during the proper period, we maintain an accrual to cover these expenses. These accruals are assessed on a quarterly basis and are based on such assumptions as expected total cost, the number of subjects and clinical trial sites and length of the study. Actual results may differ from these estimates and could have a material impact on our reported results. Our historical accrual estimates have not been materially different from our actual costs.
Although the Company believes that its patents and underlying technology have continuing value, the amount of future benefits to be derived from the patents is uncertain. Patent costs are, therefore, expensed as research and development costs as incurred.
Stock-based Compensation
The Company follows the provisions of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 718, “Compensation — Stock Compensation” (“ASC 718”), which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees, officers and non-employee directors, including stock options. Stock compensation expense based on the grant date fair value estimated in accordance with the provisions of ASC 718 is recognized as an expense over the requisite service period.
For stock options granted as consideration for services rendered by non-employees, the Company recognizes compensation expense in accordance with the requirements of FASB ASC Topic 505-50, “Equity Based Payments to Non-Employees.” Non-employee option grants that do not vest immediately upon grant are recorded as an expense over the requisite service period of the underlying stock options. At the end of each financial reporting period prior to vesting, the value of these options, as calculated using the Black-Scholes option-pricing model, will be re-measured using the fair value of the Company’s common stock and the non-cash compensation recognized during the period will be adjusted accordingly. Since the fair market value of options granted to non-employees is subject to change in the future, the amount of the future compensation expense will include fair value re-measurements until the stock options are fully vested.
Income Taxes
The Company recognizes assets or liabilities for the deferred tax consequences of temporary differences between the tax basis of assets or liabilities and their reported amounts in the financial statements in accordance with FASB ASC Topic 740, “Accounting for Income Taxes” (“ASC 740”). These temporary differences will result in taxable or deductible amounts in future years when the reported amounts of the assets or liabilities are recovered or settled. ASC 740 requires that a valuation allowance be established when management determines that it is more likely than not that all or a portion of a deferred asset will not be realized. The Company evaluates the realizability of its net deferred income tax assets and valuation allowances as necessary, at least on an annual basis. During this evaluation, the Company reviews its forecasts of income in conjunction with other positive and negative evidence surrounding the realizability of its deferred income tax assets to determine if a valuation allowance is required. Adjustments to the valuation allowance will increase or decrease the Company’s income tax provision or benefit. The recognition and measurement of benefits related to the Company’s tax positions requires significant judgment, as uncertainties often exist with respect to new laws, new interpretations of existing laws, and rulings by taxing authorities. Differences between actual results and the Company’s assumptions or changes in the Company’s assumptions in future periods are recorded in the period they become known.
Comprehensive Loss
The Company’s comprehensive loss is equal to its net loss for all periods presented.
F-20
Table of Contents
Net loss per Share
The Company accounts for and discloses net loss per share attributable to common stockholders in accordance with FASB ASC Topic 260, “Earnings per Share.” Basic and diluted net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding. When the effects are not anti-dilutive, diluted earnings per share is computed by dividing the Company’s net earnings by the weighted average number of common shares outstanding and the impact of all dilutive potential common shares.
3. Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update (“ASU”) 2014-09, “Revenue from Contracts with Customers (Topic 606).” ASU 2014-09 states that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. On July 9, 2015, the FASB voted to delay the effective date of the new revenue standard by one year but to permit entities to choose to adopt the standard as of the original effective date. The new standard will be effective for the Company on January 1, 2018. The Company is currently evaluating the method of adoption and the potential impact the update may have on its financial position and results of operations.
In September 2015, the FASB issued ASU 2015-16, “Simplifying the Accounting for Measurement-Period Adjustments (Topic 805).” ASU 2015-16 states that an entity must recognize adjustments to provisional amounts that are identified during the measurement period in the reporting period in which the adjustment amounts are determined. ASU 2015-16 will be effective for annual periods beginning after December 15, 2015, including interim periods within those fiscal years. The new standard will be effective for the Company on January 1, 2016. The adoption of ASU 2015-16 is not expected to have a material impact on our financial statements.
In November 2015, the FASB issued ASU 2015-17, “Balance Sheet Classification of Deferred Taxes (Topic 740).” ASU 2015-17 simplifies the presentation of deferred income taxes by requiring that deferred tax assets and liabilities be classified as noncurrent on the balance sheet instead of separating into current and noncurrent amounts. ASU 2015-17 will be effective for annual periods beginning on or after December 15, 2016 and may be applied either prospectively or retrospectively. Early adoption is permitted. The Company has elected to early adopt this guidance on a prospective basis. The adoption of this update did not have an effect on our financial statements.
In February 2016, the FASB issued ASU 2016-02, “Leases (Topic 842)”, which requires companies that are lessees to recognize a right-of-use asset and lease liability for most leases that do not meet the definition of a short-term lease. For income statement purposes, leases will continue to be classified as either operating or financing. Classification will be based on criteria that are largely similar to those applied in current lease accounting. This standard will result in extensive qualitative and quantitative disclosure changes. This standard will be effective for annual reporting periods beginning after December 15, 2018, including interim periods within that reporting period. The Company is currently evaluating the impact of this ASU on its financial position and results of operations.
4. Fair Value Measurements
The Company follows the provisions of FASB ASC Topic 820, “Fair Value Measurements and Disclosures,” for the Company’s financial assets and liabilities that are re-measured and reported at fair value at each reporting period and are re-measured and reported at fair value at least annually using a fair value hierarchy that is broken down into three levels. Level inputs are defined as follows:
| Level 1 |
— | quoted prices in active markets for identical assets or liabilities. | ||
| Level 2 |
— | other significant observable inputs for the assets or liabilities through corroboration with market data at the measurement date. | ||
| Level 3 |
— | significant unobservable inputs that reflect management’s best estimate of what market participants would use to price the assets or liabilities at the measurement date. | ||
F-21
Table of Contents
The Company categorized its restricted cash, cash equivalents and short-term investments as Level 2 hierarchy. The assets classified as Level 2 have initially been valued at the applicable transaction price and subsequently valued, at the end of each reporting period, using other market observable data. Observable market data points include quoted prices, interest rates, reportable trades and other industry and economic events. Financial assets measured at fair value on a recurring basis are summarized as follows, in thousands:
| Description |
December 31, 2015 | Quoted Prices In Active Markets (Level 1) |
Other Significant Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
| Assets: |
||||||||||||||||
| Restricted cash |
$ | 50 | $ | — | $ | 50 | $ | — | ||||||||
| Cash equivalents |
2,500 | — | 2,500 | — | ||||||||||||
| Short-term investments |
5,500 | — | 5,500 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 8,050 | $ | — | $ | 8,050 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Description |
December 31, 2014 | Quoted Prices In Active Markets (Level 1) |
Other Significant Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
| Assets: |
||||||||||||||||
| Restricted cash |
$ | 50 | $ | — | $ | 50 | $ | — | ||||||||
| Cash equivalents |
4,000 | — | 4,000 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 4,050 | $ | — | $ | 4,050 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
5. Accrued Expenses
Accrued expenses consist of the following, in thousands:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Employee compensation and benefits |
$ | 725 | $ | 528 | ||||
| Clinical development expenses |
225 | 186 | ||||||
| Professional fees |
126 | 165 | ||||||
| Research and development costs |
20 | 118 | ||||||
| Other |
10 | 5 | ||||||
|
|
|
|
|
|||||
| Total accrued expenses |
$ | 1,106 | $ | 1,002 | ||||
|
|
|
|
|
|||||
6. Commitments and Contingencies
License Commitments
The Company acquires assets under development and enters into research and development arrangements with third parties that often require milestone and royalty payments based on the progress of the asset through development stages. Milestone payments may be required, for example, upon approval of the product for marketing by a regulatory agency. In certain agreements, the Company is required to make royalty payments based upon a percentage of the sales of the products licensed pursuant to such agreements. Because of the contingent nature of these payments, they are not included in the table of contractual obligations shown below (see also Note 12).
These arrangements may be material individually, and in the unlikely event that milestones for multiple products covered by these arrangements were reached in the same period, the aggregate charge to expense could be material to the results of operations. In addition, these arrangements often give the Company the discretion to
F-22
Table of Contents
unilaterally terminate development of the product, which would allow the Company to avoid making the contingent payments; however, the Company is unlikely to cease development if the compound successfully achieves clinical testing objectives.
The Company’s contractual license obligations that will require future cash payments as of December 31, 2015 are as follows, in thousands:
| Year Ending December 31, |
||||
| 2016 |
$ | 200 | ||
| 2017 |
200 | |||
| 2018 |
200 | |||
| 2019 |
165 | |||
| 2020 |
165 | |||
| Thereafter |
966 | |||
|
|
|
|||
| Total |
$ | 1,896 | ||
|
|
|
|||
Operating Leases
The Company leases office and laboratory space for its corporate headquarters and primary research facility in Marlborough, Massachusetts. The lease for the office and lab space will expire in March 2019. Monthly rental expense is approximately $9,500, which includes the Company’s pro rata share of annual real estate taxes and operating expenses.
Total rent expense under the Company’s operating leases was $118,000 and $107,500 for the years ended December 31, 2015 and 2014, respectively.
At December 31, 2015, the Company’s future minimum payments required under operating leases are as follows, in thousands:
| Year Ending December 31, |
||||
| 2016 |
$ | 120 | ||
| 2017 |
117 | |||
| 2018 |
120 | |||
| 2019 |
30 | |||
|
|
|
|||
| Total |
$ | 387 | ||
|
|
|
|||
The Company applies the disclosure provisions of FASB ASC Topic 460, “Guarantor’s Accounting and Disclosure Requirements for Guarantees, Including Indirect Guarantees of Indebtedness of Others” (“ASC 460”), to its agreements that contain guarantee or indemnification clauses. The Company provides: (i) indemnifications of varying scope and size to certain investors and other parties for certain losses suffered or incurred by the indemnified party in connection with various types of third-party claims; and (ii) indemnifications of varying scope and size to officers and directors against third-party claims arising from the services they provide to us. These indemnifications give rise only to the disclosure provisions of ASC 460. To date, the Company has not incurred costs as a result of these obligations and does not expect to incur material costs in the future. Accordingly, the Company has not accrued any liabilities in its financial statements related to these indemnifications.
7. Convertible Preferred Stock
Dividends
On May 22, 2015, the Company entered into an agreement (the “Acceleration and Conversion Agreement”) with Tang Capital Partners, L.P. (“TCP”) pursuant to which the Company and TCP agreed to accelerate the next dividend payment date from June 30, 2015 to no later than May 29, 2015, and upon payment
F-23
Table of Contents
of such dividend immediately convert the dividend shares into common stock. In connection therewith, the dividend payment date was accelerated to May 27, 2015. There were no shares of Series A convertible preferred stock (“Series A Preferred Stock”) outstanding after such date.
The Company paid dividends in additional shares of Series A Preferred Stock of 105 and 356 shares for the years ended December 31, 2015 and 2014, respectively. No dividends were paid on the Series A Preferred Stock after May 27, 2015, as all outstanding shares were converted into common stock on this date. Included in the Company’s net loss applicable to common stockholders related to the fair value of the Series A Preferred Stock dividends was $172,000 and $2,399,000 for the years ended December 31, 2015 and 2014, respectively.
Conversion
During the year ended December 31, 2015, 3,215 shares of Series A Preferred Stock were converted into 783,740 shares of common stock. There were no conversions of the Series A Preferred Stock after May 27, 2015, as all outstanding shares were converted into common stock on this date. During the year ended December 31, 2014, 166 shares of Series A Preferred Stock were converted into 40,572 shares of common stock.
Elimination
On November 6, 2015, the Company filed a Certificate Eliminating the Series A Preferred Stock from the Certificate of Incorporation of the Company with the Secretary of State of the State of Delaware, in order to eliminate from the Charter all matters set forth in the Charter, including the related certificates of designation and increase, relating to the previously issued series of preferred stock of the Company. As a result, the 15,000 shares of unissued Series A Preferred Stock were returned to the status of authorized but unissued shares of preferred stock of the Company, without designation as to series or preferences or rights. As of December 31, 2015, there were no shares of Series A Preferred Stock authorized, outstanding or issued.
8. Stockholders’ Equity
The Company currently has authorized for issuance 100,000,000 shares of common stock, par value $0.0001 per share, and 10,000,000 shares of preferred stock, par value $0.0001 per share.
Series A-1 Preferred Stock
Dividends
On May 22, 2015, the Company entered into the Acceleration and Conversion Agreement with TCP pursuant to which the Company and TCP agreed to accelerate the next dividend payment date from June 30, 2015 to no later than May 29, 2015, and upon payment of such dividend immediately convert the dividend shares into common stock. In connection therewith, the dividend payment date was accelerated to May 27, 2015. There were no shares of Series A-1 convertible preferred stock (“Series A-1 Preferred Stock”) outstanding after such date.
The Company paid dividends in additional shares of Series A-1 Preferred Stock of 21 and 240 shares for the years ended December 31, 2015 and 2014, respectively. No dividends were paid on the Series A-1 Preferred Stock after May 27, 2015, as all outstanding shares were converted into common stock on this date. Included in the Company’s net loss applicable to common stockholders related to the fair value of the Series A-1 Preferred Stock dividends was $37,000 and $1,731,000 for the years ended December 31, 2015 and 2014, respectively.
Conversion
During the year ended December 31, 2015, 3,599 shares of Series A-1 Preferred Stock were converted into 877,290 shares of common stock. There were no conversions of the Series A-1 Preferred Stock after May 27, 2015, as all outstanding shares were converted into common stock on this date. During the year ended December 31, 2014, 3,716 shares of Series A-1 Preferred Stock were converted into 905,799 shares of common stock.
F-24
Table of Contents
Elimination
On November 6, 2015, the Company filed a Certificate Eliminating the Series A-1 Preferred Stock from the Certificate of Incorporation of the Company with the Secretary of State of the State of Delaware, in order to eliminate from the Charter all matters set forth in the Charter, including the related certificates of designation and increase, relating to the previously issued series of preferred stock of the Company. As a result, the 10,000 shares of unissued Series A-1 Preferred Stock were returned to the status of authorized but unissued shares of preferred stock of the Company, without designation as to series or preferences or rights. As of December 31, 2015, there were no shares of Series A-1 Preferred Stock authorized, outstanding or issued.
Common Stock
Hapten License Agreement
On December 17, 2014, the Company entered into an assignment and exclusive license agreement, (the “Assignment and License Agreement”) with Hapten Pharmaceuticals, LLC (“Hapten”) under which Hapten agreed, effective at a closing that occurred on February 4, 2015, to sell and assign to the Company certain patent rights and related assets and rights, including an investigational new drug application and clinical data, for Hapten’s Samcyprone™ products for therapeutic and prophylactic use. Upon the closing of the Hapten Assignment and License Agreement on February 4, 2015, the Company paid to Hapten a one-time upfront cash payment of $100,000 and issued 20,000 shares of common stock, the fair value of which was determined using the quoted market price of the Company’s common stock on the date of issuance. Accordingly, the cash payment of $100,000 and the fair value of the common stock of $228,000 were recorded as research and development expense during the year ended December 31, 2015.
Lincoln Park Capital Equity Line
On April 22, 2014, the Company entered into a purchase agreement (the “Prior Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“LPC”), pursuant to which and subject to the terms and conditions contained in the Prior Purchase Agreement, the Company had the right to sell to LPC up to $20,000,000 in shares of the Company’s common stock over a 30-month term. The Prior Purchase Agreement was terminable, among other circumstances, by mutual agreement of LPC and the Company at any time. The Company and LPC executed a termination agreement dated December 18, 2014, whereby the parties mutually agreed to terminate the Prior Purchase Agreement effective immediately. During the year ended December 31, 2014, the Company sold a total of 50,000 shares of common stock for net proceeds of $1,900,000 and issued 10,000 shares of common stock at price per share of $40.00 as a commitment fee, recorded as a cost of capital, under the Prior Purchase Agreement.
On December 18, 2014, the Company entered into a purchase agreement (the “Purchase Agreement”) with LPC, pursuant to which the Company has the right to sell to LPC up to $10,800,000 in shares of the Company’s common stock, subject to certain limitations and conditions set forth in the Purchase Agreement. Pursuant to the Purchase Agreement, the Company issued 10,000 shares of common stock at price per share of $19.30 as a commitment fee, which was recorded as a cost of capital during the year ended December 31, 2014.
During the year ended December 31, 2015, the Company sold a total of 5,000 shares of common stock to LPC for net proceeds of $64,000 pursuant to and subject to the limitations and conditions set forth in the Purchase Agreement. There have been no other sales made to date under the Purchase Agreement. Per the terms of the public offering (see below), the Company cannot access the equity line until the expiration of the 13-month Overallotment Purchase Rights (defined below).
June 2015 Public Offering
On June 2, 2015, the Company sold a total of 2,600,000 units at a price of $4.00 per unit in a public offering (the “Offering”). Each unit consists of one share of common stock, a 13-month overallotment purchase right to
F-25
Table of Contents
purchase one-half of one share of common stock at a price of $4.55 per full share of common stock (the “Overallotment Purchase Rights”) and a five-year warrant to purchase one-half of one share of common stock at a price of $5.20 per full share of common stock (the “Warrants”). As a result of the Offering, the Company received gross proceeds of approximately $10,400,000 and net proceeds of approximately $9,200,000 after placement agent fees and estimated Offering expenses, and assuming the Overallotment Purchase Rights and Warrants are not exercised.
The Company first assessed the Overallotment Purchase Rights and the Warrants under FASB ASC Topic 480, “Distinguishing Liabilities and Equity” (“ASC 480”), and determined that the Overallotment Purchase Rights and the Warrants were outside the scope of ASC 480. The Company next assessed the Overallotment Purchase Rights and Warrants under FASB ASC Topic 815, “Derivatives and Hedging” (“ASC 815”). Under the related guidance, a reporting entity shall not consider a contract to be a derivative instrument if the contract is both (1) indexed to the entity’s own stock and (2) classified in stockholders’ equity. The Company determined that the warrant contracts are indexed to the Company’s stock, as the agreements do not contain any exercise contingencies and the warrants’ settlement amount equals the difference between the fair value of the Company’s common stock price and the warrant contract strike price, and the only variables which could affect the settlement amount would be inputs to the fair value for a fixed-for-fixed option on equity shares. The Company also assessed the classification in stockholders’ equity and determined the warrant contracts meet all of the criteria for classification as equity under ASC 815. Based on this analysis, the Company determined that the Overallotment Purchase Rights and the Warrants should be classified as equity.
During the year ended December 31, 2015, 43,500 Overallotment Purchase Rights were exercised for gross proceeds of $198,000.
Refer to the Series A Preferred Stock and Series A-1 Preferred Stock conversions described above in this Note and Note 7 for shares issued as a result of the conversions of Series A and Series A-1 Preferred Stock during the years ended December 31, 2015 and 2014, respectively.
9. Net Loss per Share Attributable to Common Stockholders
The following table sets forth the potential common shares excluded from the calculation of net loss per common share attributable to common stockholders because their inclusion would be anti-dilutive:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Options to purchase common stock |
332,400 | 300,026 | ||||||
| Common stock underlying Series A and Series A-1 convertible preferred stock |
— | 1,630,097 | ||||||
| Warrants to purchase common stock |
2,556,966 | 462 | ||||||
|
|
|
|
|
|||||
| Total |
2,889,366 | 1,930,585 | ||||||
|
|
|
|
|
|||||
10. Stock-based Compensation
Stock Plans
On January 23, 2012, the Company’s Board of Directors and sole stockholder adopted the RXi Pharmaceuticals Corporation 2012 Long-Term Incentive Plan (the “2012 Incentive Plan”). Under the 2012 Incentive Plan, the Company may grant incentive stock options, nonqualified stock options, cash awards, stock appreciation rights, restricted and unrestricted stock and stock unit awards and other stock-based awards. The Company’s Board of Directors currently acts as the administrator of the Company’s 2012 Incentive Plan. The administrator has the power to select participants from among the key employees, directors and consultants of and advisors to the Company, establish the terms, conditions and vesting schedule, if applicable, of each award and to accelerate vesting or exercisability of any award.
F-26
Table of Contents
As of December 31, 2015, an aggregate of 500,000 shares of common stock were reserved for issuance under the Company’s 2012 Incentive Plan, including 332,400 shares subject to outstanding common stock options granted under the 2012 Incentive Plan and 167,400 shares available for future grants. Stock options granted by the Company to employees may have different vesting parameters, but generally vest within 48 months after the option grant date and expire within ten years of issuance.
Stock-based Compensation
The Company uses the Black-Scholes option-pricing model to determine the fair value of all its option grants. For valuing options granted for the years ended December 31, 2015 and 2014, the following assumptions were used:
| Year Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Risk-free interest rate |
1.47 – 2.43 | % | 1.60 – 2.73 | % | ||||
| Expected volatility |
84.93 – 116.81 | % | 97.91 – 107.01 | % | ||||
| Weighted average expected volatility |
89.26 | % | 101.52 | % | ||||
| Expected lives (in years) |
5.20 – 10.00 | 5.20 – 10.00 | ||||||
| Expected dividend yield |
0.00 | % | 0.00 | % | ||||
The weighted-average fair value of options granted during the years ended December 31, 2015 and 2014 was $4.10 and $24.30 per share, respectively.
The risk-free interest rate used for each grant was based upon the yield on zero-coupon U.S. Treasury securities with a term similar to the expected life of the related option. The Company’s expected stock price volatility assumption is based upon the volatility of a composition of comparable companies. The expected life assumption for employee grants was based upon the simplified method provided for under ASC 718 and the expected life assumption for non-employees was based upon the contractual term of the option. The dividend yield assumption of zero is based upon the fact that the Company has never paid cash dividends and presently has no intention of paying cash dividends.
The following table summarizes the activity of the Company’s stock option plan for the period from January 1, 2015 to December 31, 2015:
| Total Number of Shares |
Weighted- Average Exercise Price Per Share |
Weighted- Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| Balance at January 1, 2015 |
300,026 | $ | 33.90 | |||||||||||||
| Granted |
40,516 | 5.50 | ||||||||||||||
| Exercised |
— | — | ||||||||||||||
| Cancelled |
(8,142 | ) | 30.70 | |||||||||||||
|
|
|
|||||||||||||||
| Balance at December 31, 2015 |
332,400 | $ | 30.50 | 7.15 years | $ | — | ||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2015 |
250,181 | $ | 32.90 | 6.78 years | $ | — | ||||||||||
|
|
|
|||||||||||||||
Stock-based compensation expense for the years ended December 31, 2015 and 2014 was approximately $1,535,000 and $1,846,000, respectively. Of this, the Company recognized approximately $16,800 of income and $81,000 of expense related to non-employee stock options for the same period. There is no income tax benefit as the Company is currently operating at a loss and an actual income tax benefit may not be realized.
As of December 31, 2015, the compensation expense for all unvested stock options in the amount of approximately $1,200,000 will be recognized in the Company’s results of operations over a weighted average period of 1.68 years.
F-27
Table of Contents
11. Income Taxes
For the years ending December 31, 2015 and 2014, all of the Company’s loss before income taxes was generated in the United States. The components of federal and state income tax expense are as follows (in thousands):
| Year Ended December 31, |
||||||||
| 2015 | 2014 | |||||||
| Current |
||||||||
| Federal |
$ | — | $ | — | ||||
| State |
— | — | ||||||
|
|
|
|
|
|||||
| Total current |
— | — | ||||||
| Deferred |
||||||||
| Federal |
(3,157 | ) | (2,727 | ) | ||||
| State |
(803 | ) | (583 | ) | ||||
|
|
|
|
|
|||||
| Total deferred |
(3,960 | ) | (3,310 | ) | ||||
| Valuation allowance |
3,960 | 3,310 | ||||||
|
|
|
|
|
|||||
| Total income tax expense |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The differences between the income taxes expected at the Federal statutory income tax rate and the reported income tax (benefit) expense is as follows:
| 2015 | 2014 | |||||||
| Federal statutory rate |
34.0 | % | 34.0 | % | ||||
| State income taxes, net of federal benefit |
4.8 | 4.2 | ||||||
| Non-deductible expenses |
(1.9 | ) | (2.1 | ) | ||||
| Income tax credits |
1.9 | 1.6 | ||||||
| Valuation allowance |
(38.8 | ) | (37.7 | ) | ||||
|
|
|
|
|
|||||
| Effective tax rate |
0.0 | % | 0.0 | % | ||||
|
|
|
|
|
|||||
As of December 31, 2015, the Company elected to early adopt ASU 2015-17 issued by the FASB in November 2015. ASU 2015-17 simplifies the presentation of deferred income taxes by requiring that deferred tax assets and liabilities be classified as noncurrent on the balance sheet. The Company early adopted the guidance on a prospective basis. There was no impact to the financial statements as a result of this change. The components of net deferred tax assets are as follows (in thousands):
| As of December 31, | ||||||||
| 2015 | 2014 | |||||||
| Net operating loss carryforwards |
$ | 13,552 | $ | 9,805 | ||||
| Tax credit carryforwards |
442 | 261 | ||||||
| Stock-based compensation |
1,744 | 1,392 | ||||||
| Licensing deduction deferral |
5,979 | 6,367 | ||||||
| Other timing differences |
178 | 110 | ||||||
|
|
|
|
|
|||||
| Gross deferred tax assets |
21,895 | 17,935 | ||||||
| Valuation allowance |
(21,895 | ) | (17,935 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax asset |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The Company’s deferred tax assets at December 31, 2015 and 2014 consisted primarily of its net operating loss carryforwards, deferred compensation, tax credit carryforwards, intangible assets capitalized for federal
F-28
Table of Contents
income tax purposes and certain accruals that for tax purposes are not deductible until future payment is made. The valuation allowance increased $3,960,000 and $3,310,000 for the years ended December 31, 2015 and 2014, respectively, and is primarily attributable to an increase in net operating losses, tax credits and stock-based compensation in 2015.
The Company has incurred net operating losses since inception. At December 31, 2015, the Company had federal and state net operating loss carryforwards of approximately $35,300,000 and $29,700,000, respectively, which are available to reduce future taxable income through 2035. In addition, the Company has federal and state research credits of $331,000 and $168,000, respectively, to offset future tax expense through 2035. Based on an assessment of all available evidence including, but not limited to the Company’s limited operating history in its core business and lack of profitability, uncertainties of the commercial viability of its technology, the impact of government regulation and healthcare reform initiatives, and other risks normally associated with biotechnology companies, the Company has concluded that it is more likely than not that these net operating loss carryforwards and credits will not be realized and, as a result, a full deferred income tax valuation allowance has been recorded against these assets.
Under the provisions of the Internal Revenue Code, certain substantial changes in the Company’s ownership may result in a limitation on the amount of net operating loss carryforwards and research and development credit carryforwards which could be utilized annually to offset future taxable income and taxes payable. The Company has not yet performed an analysis to determine if one or multiple ownership changes may have occurred in the past.
The Company files income tax returns in the United States, Massachusetts and New Jersey. The Company is subject to tax examinations for Federal and state purposes tax years 2012 through 2015. The Company has not recorded any uncertain tax positions as of December 31, 2015 or 2014. The Company does not believe there will be any material changes in its unrecognized tax positions over the next 12 months. The Company has not incurred any interest or penalties. In the event that the Company is assessed interest or penalties at some point in the future, they will be classified in the financial statements as general and administrative expenses.
12. License Agreements
As part of its business, the Company enters into licensing agreements with third parties that often require milestone and royalty payments based on the progress of the asset through development stages. Milestone payments may be required, for example, upon approval of the product for marketing by a regulatory agency. In certain agreements, the Company is required to make royalty payments based upon a percentage of the sales of the products licensed pursuant to such agreements.
The expenditures required under these arrangements may be material individually in the event that the Company develops product candidates covered by the intellectual property licensed under any such arrangement, and in the unlikely event that milestones for multiple products covered by these arrangements were reached in the same period, the aggregate charge to expense could be material to the results of operations. In addition, these arrangements often give the Company discretion to unilaterally terminate development of the product, which would allow the Company to avoid making the contingent payments; however, the Company is unlikely to cease development if the compound successfully achieves clinical testing objectives.
Advirna. We have entered into agreements with Advirna, pursuant to which Advirna assigned to us its existing patent and technology rights related to sd-rxRNA technology in exchange for our agreement to pay Advirna an annual maintenance fee of $100,000. Pursuant to the terms of the agreement, during the year ended December 31, 2014, we paid to Advirna and recorded research and development expense of $350,000 for a one-time milestone payment upon the issuance of the first patent with valid claims covering the assigned technology. Additionally, we will be required to pay a 1% royalty to Advirna on any licensing revenue received by us with respect to future licensing of the assigned Advirna patent and technology rights. We also granted back to Advirna a license under the assigned patent and technology rights for fields of use outside human therapeutics.
F-29
Table of Contents
Our rights under the Advirna agreement will expire upon the later of: (i) the expiration of the last-to-expire of the “patent rights” (as defined) included in the Advirna agreement; or (ii) the abandonment of the last-to-be abandoned of such patents, unless earlier terminated in accordance with the provisions of the agreement.
We may terminate the Advirna agreement at any time upon 90 days’ written notice to Advirna, and Advirna may terminate the agreement upon 90 days’ prior written notice in the event that we cease using commercially reasonable efforts to research, develop, license or otherwise commercialize the patent rights or “royalty-bearing products” (as defined), provided that we may refute such claim within such 90-day period by showing budgeted expenditures for the research, development, licensing or other commercialization consistent with other technologies of similar stage of development and commercial potential as the patent rights or royalty-bearing products. Further, either party at any time may provide to the other party written notice of a material breach of the agreement. If the other party fails to cure the identified breach within 90 days after the date of the notice, the aggrieved party may terminate the agreement by written notice to the party in breach.
Hapten. On December 17, 2014, the Company entered into the Assignment and License Agreement with Hapten under which Hapten agreed, effective at a closing that was subject to the satisfaction of certain closing conditions which occurred in February 2015, to sell and assign to us certain patent rights and related assets and rights, including an investigational new drug application and clinical data, for Hapten’s Samcyprone™ products for therapeutic and prophylactic use. Samcyprone™ is a proprietary formulation of diphenylcyclopropenone (“DPCP”), an immunomodulation agent that works by initiating a T-cell response. Hapten has been developing Samcyprone™ for the treatment of warts, alopecia areata and cutaneous metastases of malignant melanoma.
Under the Assignment and License Agreement, Hapten received at closing an upfront payment from us, paid in cash and stock, and will be entitled to receive: (i) future milestone payments tied to the achievement of certain clinical and commercial objectives (all of which payments may be made at our option in cash or through the issuance of common stock); and (ii) escalating royalties based on product sales by us and any sublicensees. The Assignment and License Agreement with Hapten is described further in Note 8.
13. Subsequent Events
On April 14, 2016, the Board of Directors of the Company approved a 1-for-10 reverse stock split of the Company’s outstanding common stock, which was effected on April 18, 2016. The number of authorized shares of the Company remain unchanged. Stockholders who would have otherwise been entitled to fractional shares as a result of the reverse stock split received a cash payment in lieu of receiving fractional shares. Shares of common stock underlying outstanding stock options and other equity instruments were proportionately reduced and the respective exercise prices, if applicable, were proportionately increased in accordance with the terms of the agreements governing such securities. All share and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock split, including reclassifying an amount equal to the reduction in par value to additional paid-in capital.
F-30
Table of Contents
RXi Pharmaceuticals Corporation
2,017,000 Class A Units consisting of Common Stock and warrants
and 9,600 Class B Units consisting of shares of Series B Convertible Preferred Stock and warrants
(and 13,104,500 shares of Common Stock underlying shares of Series B Convertible Preferred Stock and warrants)
PROSPECTUS
, 2016
Table of Contents
PART II
Information Not Required in Prospectus
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION
The following table sets forth the fees and expenses, other than placement agent fees and expenses, payable in connection with the registration of the common stock hereunder. All amounts are estimates except the SEC registration fee and the FINRA filing fee.
| Item |
Amount to be paid |
|||
| SEC registration fee |
$ | 2,399.13 | ||
| FINRA filing fee |
3,605.00 | |||
| Printing and engraving expenses |
40,000.00 | |||
| Legal fees and expenses |
120,000.00 | |||
| Accounting fees and expenses |
50,000.00 | |||
| Blue Sky, qualification fees and expenses |
— | |||
| Transfer Agent fees and expenses |
15,000.00 | |||
| Miscellaneous expenses |
5,992.87 | |||
|
|
|
|||
| Total |
$ | 236,997.00 | ||
| * | To be provided in amendment. |
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS
Section 145 of the Delaware General Corporation Law (“DGCL”) authorizes a corporation to indemnify its directors and officers against liabilities arising out of actions, suits and proceedings to which they are made or threatened to be made a party by reason of the fact that they have served or are currently serving as a director or officer to a corporation. The indemnity may cover expenses (including attorneys’ fees) judgments, fines and amounts paid in settlement actually and reasonably incurred by the director or officer in connection with any such action, suit or proceeding. Section 145 permits corporations to pay expenses (including attorneys’ fees) incurred by directors and officers in advance of the final disposition of such action, suit or proceeding. In addition, Section 145 provides that a corporation has the power to purchase and maintain insurance on behalf of its directors and officers against any liability asserted against them and incurred by them in their capacity as a director or officer, or arising out of their status as such, whether or not the corporation would have the power to indemnify the director or officer against such liability under Section 145.
Our certificate of incorporation provides that we will indemnify to the fullest extent authorized or permitted by the DGCL or any other applicable law as now or hereafter in effect any person made, or threatened to be made, a defendant or witness to any action, suit or proceeding (whether civil, criminal or otherwise) by reason of the fact that he is or was a director of our corporation or by reason of the fact that such director, at our request, is or was serving any other corporation, partnership, joint venture, trust, employee benefit plan or other enterprise in any capacity. Our certificate of incorporation also provides that no amendment or repeal of the certificate of incorporation will apply to or have any effect on any right to indemnification provided in the certificate of incorporation with respect to any acts or omissions occurring prior to such amendment or repeal.
As permitted by the DGCL, our bylaws, as amended, provide that we will indemnify to the fullest extent authorized or permitted by applicable law as now or hereafter in effect any person who was or is made, or is threatened to be made, a party or is otherwise involved in any action, suit or proceeding (whether civil, criminal, administrative or investigative), by reason of the fact that he (or a person for whom he is the legal representative)
II-1
Table of Contents
is or was a director or officer of our corporation, is or was serving at our request as a director, officer, employee, member, trustee or agent of another corporation or of a partnership, joint venture, trust, nonprofit entity or other enterprise.
Consequently, no director of the corporation will be personally liable to the corporation or its stockholders for monetary damages for any breach of fiduciary duty by such a director as a director. However, notwithstanding the preceding sentence, a director will be liable to the extent provided by Delaware law (1) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (2) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (3) for payments of unlawful dividends or for unlawful stock repurchases or redemption, or (4) for any transaction from which the director derived an improper personal benefit.
We have entered into indemnification agreements with each of our executive officers and directors. These agreements provide that, subject to limited exceptions and among other things, we will indemnify each of our executive officers and directors to the fullest extent permitted by law and advance expenses to each indemnitee in connection with any proceeding in which a right to indemnification is available.
We also maintain insurance on behalf of any person who is or was our director, officer, trustee, employee or agent or serving at our request as a director, officer, trustee, employee or agent of another corporation, partnership, joint venture, trust, non-profit entity or other enterprise against any liability asserted against the person and incurred by the person in any such capacity, or arising out of his or her status as such.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted for directors, officers, or persons who control us, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES
In the three years preceding the filing of this registration statement, we have issued the following securities that were not registered in the Securities Act.
On January 24, 2014, the Company entered into an exchange agreement with TCP pursuant to which TCP exchanged a total of 3,000 shares of Series A Preferred Stock for a like number of shares of Series A-1 Preferred Stock.
On March 20, 2015, the Company entered into an exchange agreement with TCP pursuant to which TCP exchanged a total of 2,000 shares of Series A Preferred Stock for a like number of shares of Series A-1 Preferred Stock.
With respect to the foregoing exchange agreements, the Company issued the Series A-1 Preferred Stock in transactions exempt from the registration requirements of the Securities Act by virtue of the exemption provided for in Section 3(a)(9) of the Act for securities exchanged by the issuer with an existing security holder.
On February 4, 2015, the Company issued 20,000 shares of common stock to Hapten Pharmaceuticals, LLC, pursuant to that certain Assignment and Exclusive License Agreement dated as of December 17, 2014, in partial consideration for certain patent rights and related assets and rights, including an investigational new drug application and clinical data for Hapten’s Samcyprone™ gel products for therapeutic and prophylactic use.
Holders of Series A Preferred Stock received dividends payable in shares of Series A Preferred Stock of 105, 356 and 628 during the years ended December 31, 2015, 2014 and 2013, respectively.
Holders of Series A-1 Preferred Stock received dividends payable in shares of Series A-1 Preferred Stock of 21, 240 and 54 during the years ended December 31, 2015, 2014 and 2013, respectively.
II-2
Table of Contents
On June 7, 2013, the Compensation Committee approved an employee stock purchase plan (“ESPP”), which was subsequently approved by the Company’s stockholders at the Company’s 2014 Annual Meeting of Stockholders. The ESPP allows employees to contribute a percentage of their cash earnings, subject to certain maximum amounts, to be used to purchase shares of the Company’s common stock on each of two semi-annual purchase dates. The purchase price is equal to 90% of the market value per share on either (a) the date of grant of a purchase right under the ESPP or (b) the date on which such purchase right is deemed exercised, whichever is lower.
As of September 30, 2016, an aggregate of zero shares of common stock were reserved for issuance under the Company’s ESPP, of which 11,333 shares of common stock have been issued under the ESPP and no shares are available for future issuances.
As of September 30, 2016, we have sold an aggregate of 11,333 shares of common stock to employees, directors, and consultants for cash consideration in the aggregate amount of approximately $121,000 upon the exercise of stock options and stock awards.
Unless otherwise noted, all of the transactions described in Item 15 were exempt from registration under the Securities Act pursuant to Section 4(a)(2) of the Securities Act in that such sales did not involve a public offering or under Rule 701 promulgated under the Securities Act, in that they were offered and sold either pursuant to written compensatory plans or pursuant to a written contract relating to compensation, as provided by Rule 701.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
See the Exhibit Index set forth on page II-6 to this Registration Statement, which is incorporated herein by reference.
ITEM 17. UNDERTAKINGS
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, or the Act, may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The Registrant hereby undertakes that:
(a) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the
II-3
Table of Contents
estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(b) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof;
(c) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering;
(d) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
(i) Each prospectus filed by the Registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
(ii) Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date;
(e) That, for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933 shall be deemed to be part of this registration statement as of the time it was declared effective; and
(f) That, for the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
Table of Contents
Signatures
Pursuant to the requirements of the Securities Act of 1933, as amended, the Registrant has duly caused this Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in Marlborough, Massachusetts, on November 10, 2016.
| RXi PHARMACEUTICALS CORPORATION | ||
| By: | /s/ Geert Cauwenbergh | |
| Geert Cauwenbergh, Dr. Med. Sc. | ||
| President, Chief Executive Officer and Chief Financial Officer | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Geert Cauwenbergh Geert Cauwenbergh, Dr. Med. Sc. |
President, Chief Executive Officer, Chief Financial Officer and Director (Principal Executive Officer and Principal Financial Officer) |
November 10, 2016 | ||
| /s/ Caitlin Kontulis Caitlin Kontulis |
Director of Finance and Secretary (Principal Accounting Officer) |
November 10, 2016 | ||
| * Robert J. Bitterman |
Director | November 10, 2016 | ||
| * Keith L. Brownlie |
Director | November 10, 2016 | ||
| * H. Paul Dorman |
Director | November 10, 2016 | ||
| * Curtis A. Lockshin, Ph.D. |
Director | November 10, 2016 | ||
| * By: | /s/ Geert Cauwenbergh |
|||
| Geert Cauwenbergh, | ||||
| Attorney-in-Fact | ||||
| November 10, 2016 |
II-5
Table of Contents
EXHIBIT INDEX
| Incorporated by Reference Herein | ||||||
| Exhibit Number |
Description | Form | Date | |||
| 1.1 | Form of Underwriting Agreement* | |||||
| 2.1 | Contribution Agreement, dated as of September 24, 2011, between RXi Pharmaceuticals Corporation (formerly RNCS, Inc.) and Galena Biopharma, Inc. (formerly RXi Pharmaceuticals Corporation). | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-177498) | December 8, 2011 | |||
| 2.2 | Securities Purchase Agreement, dated as of September 24, 2011, among RXi Pharmaceuticals Corporation (formerly RNCS, Inc.), Galena Biopharma, Inc. (formerly RXi Pharmaceuticals Corporation), Tang Capital Partners, LP and RTW Investments, LLC. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-177498) | December 8, 2011 | |||
| 2.3 | Asset Purchase Agreement, dated March 1, 2013, between RXi Pharmaceuticals Corporation and OPKO Health, Inc. + | Quarterly Report on Form 10-Q (File No. 000-54910) | March 15, 2013 | |||
| 3.1 | Amended and Restated Certificate of Incorporation of RXi Pharmaceuticals Corporation. | Amendment No. 4 to the Registration Statement on Form S-1 (File No. 333-177498) | February 7, 2012 | |||
| 3.2 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of RXi Pharmaceuticals Corporation. | Current Report on Form 8-K (File No. 000-54910) | July 22, 2013 | |||
| 3.3 | Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock of RXi Pharmaceuticals Corporation. | Amendment No. 4 to Registration Statement Form S-1 (File No. 333-177498) | February 7, 2012 | |||
| 3.4 | Certificate of Designations, Preferences and Rights of Series A-1 Convertible Preferred Stock of RXi Pharmaceuticals Corporation. | Quarterly Report on Form 10-Q (File No. 000-54910) | August 14, 2013 | |||
| 3.5 | Certificate of Increase, filed with the Secretary of State of the State of Delaware on January 24, 2014. | Current Report on Form 8-K (File No. 000-54910) | January 24, 2014 | |||
| 3.6 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of RXi Pharmaceuticals Corporation. | Registration Statement on Form S-1 (File No. 333-203389) | April 13, 2015 | |||
| 3.7 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of RXi Pharmaceuticals Corporation. | Current Report on Form 8-K (File No. 000-54910) | April 14, 2016 | |||
II-6
Table of Contents
| Incorporated by Reference Herein | ||||||
| Exhibit Number |
Description | Form | Date | |||
| 3.8 | Amended and Restated Bylaws of RXi Pharmaceuticals Corporation. | Quarterly Report on Form 10-Q (File No. 333-177498) | May 14, 2012 | |||
| 3.9 | Certificate Eliminating the Series A Convertible Preferred Stock from the Certificate of Incorporation of RXi Pharmaceuticals Corporation | Quarterly Report on Form 10-Q (File No. 001-36304) | November 12, 2015 | |||
| 3.10 | Certificate Eliminating the Series A-1 Convertible Preferred Stock from the Certificate of Incorporation of RXi Pharmaceuticals Corporation | Quarterly Report on Form 10-Q (File No. 001-36304) | November 12, 2015 | |||
| 3.11 | Form of Certificate of Designation of the Series B Convertible Preferred Stock* | |||||
| 4.1 | Form of Overallotment Purchase Right. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
| 4.2 | Form of Warrant | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
| 4.3 | Form of Warrant* | |||||
| 5.1 | Opinion of Gibson, Dunn & Crutcher LLP regarding the securities being registered.* | |||||
| 10.1 | Employment Agreement, dated September 24, 2011, between RXi Pharmaceuticals Corporation (formerly, RNCS, Inc.) and Pamela Pavco, Ph.D.** | Current Report on Form 8-K of Galena Biopharma, Inc. (File No. 001-33958) | September 26, 2011 | |||
| 10.2 | Patent and Technology Assignment Agreement between RXi Pharmaceuticals Corporation (formerly RNCS, Inc.) and Advirna, LLC, effective as of September 24, 2011. | Registration Statement on Form S-1 (FileNo. 333-177498) | October 25, 2011 | |||
| 10.3 | RXi Pharmaceuticals Corporation 2012 Long Term Incentive Plan.** | Amendment No. 3 to the Registration Statement on Form S-1 (FileNo. 333-177498) | January 23, 2012 | |||
| 10.4 | Form of Incentive Stock Option Award under the Company’s 2012 Long Term Incentive Plan, as amended.** | Registration Statement on Form S-1 (File No. 333-191236 | September 18, 2013 | |||
| 10.5 | Form of Non-qualified Stock Option Award under the Company’s 2012 Long Term Incentive Plan, as amended.** | Registration Statement on Form S-1 (File No. 333-191236 | September 18, 2013 | |||
II-7
Table of Contents
| Incorporated by Reference Herein | ||||||
| Exhibit Number |
Description | Form | Date | |||
| 10.6 | Form of Restricted Stock Unit Award under the Company’s 2012 Long Term Incentive Plan, as amended.** | Registration Statement on Form S-1 (File No. 333-191236 | September 18, 2013 | |||
| 10.7 | Amendment to RXi Pharmaceuticals Corporation Long-Term Incentive Plan.** | Registration Statement on Form S-1 (File No. 333-191236 | September 18, 2013 | |||
| 10.8 | RXi Pharmaceuticals Corporation Employee Stock Purchase Plan.** | Registration Statement on Form S-1 (File No. 333-191236 | September 18, 2013 | |||
| 10.9 | Form of Indemnification Agreement.** | Amendment No. 3 to the Registration Statement on Form S-1 (File No. 333-177498) | January 23, 2012 | |||
| 10.10 | Employment Agreement, dated April 27, 2012, between RXi Pharmaceuticals Corporation and Geert Cauwenbergh, Dr. Med. Sc.** | Current Report on Form 8-K (File No. 333-177498) | May 3, 2012 | |||
| 10.11 | Securities Purchase Agreement, dated as of March 6, 2013, among RXi Pharmaceuticals Corporation and the purchasers named therein. | Current Report on Form 8-K (File No. 000-54910) | March 7, 2013 | |||
| 10.12 | Lease Agreement dated December 17, 2013 between RXi Pharmaceuticals Corporation and 257 Simarano Drive, LLC, Brighton Properties, LLC, Robert Stubblebine 1, LLC and Robert Stubblebine 2, LLC | Current Report on Form 8-K (File No. 000-54910) | December 20, 2013 | |||
| 10.13 | Purchase Agreement, dated as of April 22, 2014, between RXi Pharmaceuticals Corporation and Lincoln Park Capital Fund, LLC | Current Report on Form 8-K (File No. 001-36304) | April 23, 2014 | |||
| 10.14 | Purchase Agreement, dated as of December 18, 2014, between RXi Pharmaceuticals Corporation and Lincoln Park Capital Fund, LLC | Current Report on Form 8-K (File No. 001-36304) | December 19, 2014 | |||
| 10.15 | Manufacturing and Distribution Agreement, dated November 14, 2013 between RXi Pharmaceuticals Corporation and Ethicor Pharmaceuticals Ltd. + | Annual Report on Form 10-K (File No. 000-54910) | March 28, 2014 | |||
| 10.16 | Engagement Agreement, dated February 25, 2015 between RXi pharmaceuticals Corporation and H.C. Wainwright & Co., L.L.C. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
II-8
Table of Contents
| Incorporated by Reference Herein | ||||||
| Exhibit Number |
Description | Form | Date | |||
| 10.17 | Amendment to Engagement Agreement, dated April 20, 2015 between RXi Pharmaceuticals Corporation and H.C. Wainwright & Co., L.L.C. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
| 10.18 | Amendment to Engagement Agreement, dated May 19, 2015 between RXi Pharmaceuticals Corporation and H.C. Wainwright & Co., L.L.C. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
| 10.19 | Form of Securities Purchase Agreement. | Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-203389) | May 21, 2015 | |||
| 23.1 | Consent of BDO USA, LLP, Independent Registered Public Accounting Firm*** | |||||
| 23.2 | Consent of Gibson, Dunn & Crutcher LLP (included in Exhibit 5.1)* | |||||
| 24.1 | Powers of Attorney (included on the signature page of Part II of prior filing) | |||||
| 101.INS | XBRL Instance Document**** | |||||
| 101.SCH | XBRL Taxonomy Extension Schema**** | |||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document**** | |||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document**** | |||||
| 101.LAB | XBRL Taxonomy Extension Labels Linkbase Document**** | |||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document**** | |||||
| * | To be filed by amendment. |
| ** | Indicates a management contract or compensatory plan or arrangement. |
| *** | Filed herewith. |
| **** | Filed herewith. Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files on Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
| + | Confidential treatment has been requested or granted for certain portions which have been blanked out in the copy of the exhibit filed with the Securities and Exchange Commission. The omitted information has been filed separately with the Securities and Exchange Commission. |
II-9