Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - HELIUS MEDICAL TECHNOLOGIES, INC. | exhibit32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - HELIUS MEDICAL TECHNOLOGIES, INC. | exhibit31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - HELIUS MEDICAL TECHNOLOGIES, INC. | exhibit31-1.htm |
| EX-23.1 - EXHIBIT 23.1 - HELIUS MEDICAL TECHNOLOGIES, INC. | exhibit23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K/A
(Amendment No. 1)
|
[X] |
Annual report pursuant to Section 13 or 15(d) of the
Securities Exchange Act of 1934. |
| [ ] | Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. |
For the transition period from _______to _______
Commission File No. 000-55364
![]()
HELIUS MEDICAL TECHNOLOGIES,
INC.
(Exact name of registrant as specified in its
charter)
| WYOMING | 36-4787690 |
| (State or other jurisdiction of | (I.R.S. Employer |
| incorporation or organization) | Identification No.) |
Suite 400, 41 University Drive
Newtown,
Pennsylvania, 18940
(Address of principal executive offices) (Zip
Code)
Registrant’s telephone number, including area code: (215) 809-2018
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered |
| None | Toronto Stock Exchange |
Securities registered pursuant to Section 12(g) of the Act: Class A Common Stock
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes
[ ] No [X]
Indicate by check mark if the
registrant is not required to file reports pursuant to Section 13 or Section
15(d) of the Act.
Yes [ ] No [X]
Indicate by check mark whether
the registrant (1) has filed all reports required to be filed by Section 13 or
15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports),
and (2) has been subject to such filing requirements for the past 90 days:
Yes [X] No [ ]
Indicate by check mark whether
the registrant has submitted electronically and posted on its corporate Web
site, if any, every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of Regulation S-T (Section §232.405 of this chapter) during
the preceding 12 months (or for such shorter period that the registrant was
required to submit and post such files).
Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer [ ] | Accelerated filer [ ] | Non-accelerated filer [ ] | Smaller reporting company |
| [X] | |||
| (Do not check if a smaller | |||
| reporting company) |
Indicate by check mark whether
the registrant is a shell company (as defined in Rule 12b-2 of the Exchange
Act).
Yes [ ] No [X]
The aggregate market value of the common equity held by non-affiliates of the registrant on September 30, 2015, based on the closing price on that date of CAD $0.84 (USD $0.61), was approximately $39,359,835. As of October 31, 2016 there were 84,534,684 shares of the registrant’s common stock outstanding.
Explanatory Note
This Amendment No. 1 to the Annual Report on Form 10-K (this “Amendment No. 1”) of Helius Medical Technologies, Inc. (the “Company”) amends the Company’s Annual Report on Form 10-K for the year ended March 31, 2016, which was filed with the Securities and Exchange Commission (“SEC”) on June 28, 2016 (the “Original Filing”). The Company is filing this Amendment No. 1 to include an audit report that is signed by our independent registered public accountants as required by Article 2 of Regulation S-X, and Section 302 and 906 certifications.
This Amendment No. 1 speaks as of the filing date of the Original Filing, does not reflect events which may have occurred since the filing date of the Original Filing, and does not amend or modify any disclosure made in the Original Filing except to incorporate changes to Form 10-K Page F-2, Exhibits 23.1, 31.1, 31.2, and 32.1.
This additional disclosure does not revise or alter the Company’s financial statements and any forward-looking statements contained in the Original Filing. As required by Rule 12b-15 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), this Amendment No. 1 contains new certifications by our principal executive officer and principal financial officer, which are being filed or furnished as exhibits to this Amendment No. 1.
TABLE OF CONTENTS
1
In this annual report on Form 10-K, unless otherwise specified, references to “we,” “us,” “our,” “Helius” or “the Company” mean Helius Medical Technologies, Inc. (formerly known as “0996445 B.C. Ltd.”) and its wholly-owned subsidiaries, NeuroHabilitation Corporation, or NHC, and Helius Medical Technologies (Canada), Inc., unless the context otherwise requires. All financial information is stated in U.S. dollars unless otherwise specified. Our financial statements are prepared in accordance with accounting principles generally accepted in the United States, or U.S. GAAP.
FORWARD-LOOKING STATEMENTS
This annual report on Form 10-K (“Annual Report”) includes certain statements that may constitute “forward-looking statements.” All statements contained in this Annual Report, other than statements of historical facts, that address events or developments that the Company expects to occur, are forward-looking statements. These statements are based on management’s expectations at the time the statements are made and are subject to risks, uncertainty, and changes in circumstances, which may cause actual results, performance, financial condition or achievements to differ materially from anticipated results, performance, financial condition or achievements. All statements contained herein that are not clearly historical in nature are forward-looking and the words “anticipate,” “believe,” “calls for,” “could” “depends,” “estimate,” “expect,” “extrapolate,” “foresee,” “goal,” “intend,” “likely,” “might,” “plan,” “project,” “propose,” “potential,” “target,” “think,” and similar expressions, or that events or conditions “may,” “should occur” “will,” “would,” or any similar expressions are generally intended to identify forward-looking statements.
The forward-looking statements in this Annual Report include but are not limited to statements relating to: enrollment and future plans for our clinical trials, progress of and reports of results from clinical studies, clinical development plans, product development activities, other products not yet developed or acquired, our product candidate success, plans for U.S. Food and Drug Administration (“FDA”) filings and their subsequent approvals, other foreign or domestic regulatory filings by us or our collaboration partners, our ability to commercialize the product(s), the safety and efficacy of our product candidate, the timeline for our improvement plans, our market awareness, our ability to compete effectively, the ability and limitation of our manufacturing source(s), our distribution network, the adequacy of our intellectual property protection, our future patent approvals, potential infringement of our intellectual property, future litigation waged against us and its outcome, any product liability we may incur, the sufficiency of our operating insurance, including sufficient product liability insurance, our limited operating history, our dependence on a small number of employees, employee conflicts of interest, our dependence on outside scientists and third party research institutions, our future expenses and cash flow, our ability to become profitable, our future financing arrangements, our ability to accurately report our financial position, our accountants’ future perspective including any going concerns, our ability to maintain effective internal controls, any future stock price, the potential dilution of the stock, future sales of the Company’s equity securities, any future Financial Industry Regulatory Authority sales practice requirements, the ability of a limited number of shareholders to take shareholder action without the involvement of the management or the Company’s other shareholders, future disclosure requirements, future regulatory risks, our relationship with the U.S. Army, our ability to use existing reimbursement codes or receive reimbursement codes from the American Medical Association and the U.S. Department of Health and Human Services, and our ability to receive reimbursement coverage under Medicare, Medicaid or under other insurance plans. These and additional risks and uncertainties are more fully described in this Annual Report and our other public filings with the Securities and Exchange Commission (“SEC”).
Although the Company believes the expectations expressed in such forward-looking statements are based on reasonable assumptions at the time they were made, they are subject to risks and uncertainties, known and unknown, which could cause actual results and developments to differ materially from those expressed or implied in such statements. Forward-looking statements are not guarantees of future performance and actual results may differ significantly from such forward-looking statements. Factors that could cause the actual results to differ materially from those in the forward-looking statements include future economic, competitive, reimbursement and regulatory conditions; new product introductions, demographic trends, the intellectual property landscape, litigation, financial market conditions, continued availability of capital and financing, and, future business decisions made by us and our competitors. All of these factors are difficult or impossible to predict accurately and many of them are beyond our control. Investors are cautioned that any such forward-looking statements are not guarantees of future performance and actual results or developments may differ materially from those projected in the forward-looking statements. Undue reliance should not be placed on forward looking statements which speak only as of the date they are made. Except as required by applicable securities laws, the Company undertakes no obligation to update or alter these forward-looking statements (and expressly disclaims any such intention or obligation to do so) in the event that management's beliefs, estimates, opinions, or other factors should change.
1
INDUSTRY AND MARKET DATA
In this Annual Report, we reference information, statistics and estimates regarding the medical devices and healthcare industries. We have obtained this information from various independent third-party sources, including independent industry publications, reports by market research firms and other independent sources. This information involves a number of assumptions and limitations, and we have not independently verified the accuracy or completeness of this information. Some data and other information are also based on the good faith estimates of management, which are derived from our review of internal surveys, general information discussed in the industry, and independent sources. We believe that these external sources and estimates are reliable but have not independently verified them. The industries in which we operate are subject to a high degree of uncertainty, change, and risk due to a variety of factors, including those described in “Item 1A. Risk Factors.” These and other factors could cause results to differ materially from those expressed in this report and other publications.
2
PART I
| ITEM 1. | BUSINESS |
Our Business
We are a medical technology company focused on neurological wellness and our mission statement is to:
“Develop, license and acquire unique, non-invasive treatments designed to help patients affected by neurological symptoms caused by disease or trauma.”
Our first product in development, the portable neuromodulation stimulator (“PoNS™”) device, exemplifies this mission as the device, when used in combination with physiotherapy, is designed to enhance the brain’s ability to compensate for damage due to trauma or disease.
Improving the process by which the brain can reorganize itself and compensate for damage, or even augment how we learn, has far-reaching implications for the treatment of disease as well as for the healthy population. We intend to pursue these opportunities using our platform PoNS™ technology. Neuroplasticity, the ability of the brain to reorganize a neural network or re-task neurons and form new synaptic connections, is core to all cerebral learning, training, and rehabilitation. Neuromodulation is the alteration of nerve activity in response to the delivery of electrical stimulation or chemical agents. Our proprietary PoNS™ device is designed to induce Cranial Nerve Non Invasive Neuromodulation through a dramatic increase in stimulation of the facial and trigeminal nerves which innervate the tongue. This appears to enhance neuroplasticity and benefit persons with neurological, cognitive, sensory, and motor disorders when combined with the rehabilitation process.
Traditional rehabilitation interventions have typically involved medication and various forms of therapies, including physical therapy. Our patented PoNS™ device has been developed to deliver to the tongue a non-invasive neurostimulation, in a form that induces neuromodulation. Published studies, suggest neurologic diseases and disorders such as Traumatic Brain Injury (TBI), Multiple Sclerosis (MS), Stroke, Parkinson’s disease, Alzheimer’s diseases, Depression, Attention Deficit Hyperactivity Disorder, and Autism, all have symptoms that may benefit from enhanced neuromodulation as a component of their rehabilitation therapy.
When the PoNS™ device is placed into and held in the patient’s mouth, it stimulates the trigeminal and facial nerves that innervate the anterior two-thirds of the human tongue using a sequenced pattern of superficial electrical stimulation. This stimulation excites a natural flow of neural impulses to the brainstem and cerebellum that is designed to effect changes in the function of these targeted brain structures. A series of studies, which are further described below, suggest that prolonged activation of 20 minutes or more of neuronal circuits, when combined with physical therapy, initiate a durable neuronal reorganization with a variety of positive results, including the correction of gait/balance impairments resulting from TBI. These results represent what we refer to as anecdotal evidence, and must be confirmed by a larger well-controlled, independently reviewed scientific study. Successful results from FDA approved and reviewed clinical trials are required prior to regulatory clearance for sale in the U.S.
The inventors of the PoNS™ device conducted a series of Institutional Review Board sanctioned feasibility studies, case studies, and one placebo-controlled study. In total, these studies involved approximately 260 patients using the first generation PoNS™ device in conjunction with physical or cognitive therapy at the University of Wisconsin-Madison. An Institutional Review Board is a scientific and patient advocacy board that reviews the validity and safety of clinical trials on behalf of patients. A “feasibility study” is a study with a small sample size that allows for early clinical evaluation for proof of principle and initial clinical safety data. A feasibility study may be appropriate early in device development when clinical experience is necessary because nonclinical testing methods are not available or adequate to provide the information needed to advance the developmental process. We use the term “case study” to mean a study of one patient that may support at most anecdotal evidence of efficacy. By “placebo-controlled study,” we mean a way of testing a medical therapy in which, in addition to a group of subjects that receives the treatment to be evaluated, a separate control group receives an artificial “placebo” treatment which is specifically designed to have no real effect. These studies were conducted primarily at the Tactile Communication and Neurorehabilitation Laboratory, or TCNL, at the University of Wisconsin-Madison with the approval and oversight by the university’s Institutional Review Board, which is required for scientific studies involving human subjects.
3
Based on the prior results in subjects with MS, a further controlled feasibility study in 14 MS subjects was independently performed at the Montreal Neurological Institute in 2015 using the next generation PoNS™ device to treat gait and balance disorder associated with MS. The results of this study were consistent with the previously completed studies on MS and further supported the device’s safety and efficacy in this cohort.
As described below, we have developed the PoNS™ device to secure FDA clearance for commercial use in treating balance disorder in TBI subjects. We are conducting a clinical trial of our PoNS™ device for the treatment of balance disorder in patients with TBI. Should the PoNS™ device be cleared by the FDA, we believe the addressable market for our product device to treat balance disorder associated with TBI is potentially over $5.0 billion. According to the U.S. Center for Disease Control and Prevention, approximately 5.3 million individuals in the U.S. were living with permanent TBI symptoms in 1999, and the incidence of new TBI diagnoses, as measured by hospitalizations and emergency department visits, has increased between 2001 and 2010. Additionally, the Brain Injury Association of America estimates that approximately 40% of patients diagnosed with TBI experience balance disturbance. Our addressable market estimate for TBI in the U.S. is based on the number of persons living with TBI (5.3 million) multiplied by the rate of balance disturbance in TBI patients (40%), and multiplied by the expected price per unit of our product.
In addition to the ongoing TBI study, we plan on conducting a registrational clinical trial of our PoNS™ device for the treatment of gait and balance disorder in patients with MS. Should the PoNS™ device be cleared by the FDA, we believe the addressable markets for our PoNS™ device to treat gait and balance disorder associated with MS is potentially over $500 million. According to the National Multiple Sclerosis Society, there are approximately 400,000 individuals in the U.S. with MS. Our addressable market estimate for MS is calculated by multiplying the estimated number of persons with MS by the rate of balance disturbance in MS patients (50%) and the expected price per unit of our product.
Supporting our three issued U.S. medical method patents, are a further twenty-one issued design and utility patents. Additional domestic and international patent filings for the PoNS™ device connected to both further medical methods and other designs and utility are a key component of the value of the Company and will continue to be expanded.
Our Principal Product
The predecessor to the current PoNS™ device was developed in 2008 at the TCNL. Since then, we have conducted a significant amount of experimentation, research, and development to arrive at the present-day PoNS™ device. We have completed the technical and product design phases of the PoNS™ device as well as the manufacturing development phase which will enable us to manufacture the device commercially. We are performing the registration clinical trials for FDA clearance with the device for use in treating balance disorder in TBI subjects and plan to launch a registrational clinical trial for MS subjects in late 2016.
We anticipate that the full commercial device will be ready for release in the fourth quarter of calendar year 2016, and we expect to produce the PoNS™ device in accordance with FDA’s Quality System Regulation, or QSR, current good manufacturing practices, or cGMPs, and in compliance with European and Canadian regulatory requirements. Previously, we disclosed that we anticipated that the full commercial device would be ready for release in the first quarter of calendar 2016. We delayed the commercial device build based on our revised forecast for the clinical trial completion.
The PoNS™ device is ergonomically designed for patient comfort, is relatively light, contains a replaceable hygienic mouthpiece, a rechargeable battery and allows for technical data logging and communications. See Figure 1.
4

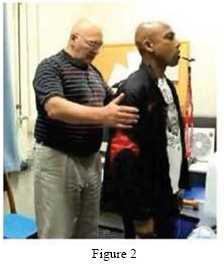
The PoNS™ device is an electrical pulse generator that delivers controlled electrical stimulation to the tongue. Pulses are generated and organized by a counter, timer, and wave-shaping electronic components. The device is held lightly in place by the lips and teeth around the neck of the tab that goes into the mouth and rests on the anterior and superior part of the tongue. See Figure 2.
The paddle-shaped tab of the device has a hexagonally patterned array of 143 gold-plated circular electrodes (1.50 mm diameter, on 2.34 mm centers) that is created by a photolithographic process used to make printed circuit boards. It is designed to use low-level electrical current to stimulate the lingual branch projections of at least two cranial nerves in the anterior tongue through the gold-plated electrodes. Device function is controlled by four buttons: On, Off, Intensity Up, and Intensity Down.
A rechargeable lithium- polymer battery with built-in charge safety circuitry provides power. While the voltage and pulse timing to each electrode are programmed into the device and cannot be altered, the stimulus intensity can be adjusted by the user. The sensation produced by the array is similar to the feeling of drinking a carbonated beverage. The waveform is specifically designed to minimize the potential for tissue irritation.
When the PoNS™ device is turned off, the intensity setting automatically resets to zero. Upon first introduction to the device stimulation, subjects are instructed to press the “Up” intensity button and hold it for approximately 4-5 seconds to reach sensation threshold. Subjects will frequently notice that the sensation intensity decreases 2-4 minutes after stimulation onset. Subjects are instructed to simply increase the sensation level to return to the predetermined perceptual midpoint of their individual perceptual dynamic range.
5
Ximedica
We have completed the design phase of the PoNS™ device and we will subcontract the building of commercial quantities of the device to Ximedica, LLC (“Ximedica”), a contract manufacturer based in Providence, Rhode Island, that we selected after an exhaustive procurement process. We place an emphasis on protecting our patented technology, trade secrets and know-how and only share confidential information on a need to know basis, even with our manufacturers. We expect that the PoNS™ device will require some very light assembly and labeling that will be performed by Ximedica. Ximedica is registered as a medical device manufacturer in good standing with the FDA, and is certified with International Organization for Standardization (ISO) 13485, a comprehensive quality management system for the design and manufacture of medical devices.
We are currently evaluating commercial-scale manufacturers for the PoNS™ device with the goal of building sufficient stock to warehouse and ship the product to our distributor, who will in turn manage customer distribution.
U.S. Army
We are designing the PoNS™ device with the cooperation of the U.S. Army pursuant to an agreement known as a cooperative research and development agreement, (the “CRADA”). The U.S. Army was interested in signing the CRADA because of the very high incidence of TBI in soldiers and the fact that there are very few proven, effective treatments available for those soldiers who suffer from chronic TBI symptoms. Department of Defense statistics show that incidence of TBI in the U.S. Army has numbered approximately 30,000 per year from 2012 to 2014 in active duty personnel, and over 300,000 U.S. military personnel have been diagnosed with TBI since 2000. Of the 30,000 active duty personnel who suffer from TBI annually, we estimate that approximately 20-30% will develop chronic symptoms related to their TBI. While the number of cases of TBI among active duty personnel may vary based on troop levels maintained by the federal government, our primary target market will be the large number of retired soldiers who suffer from chronic TBI symptoms as this population is less subject to material, year-to-year fluctuation. The Army has expressed its desire to distribute our PoNS™ device to service members who would benefit, should the device be cleared by the FDA. However, the U.S. Army is not under any obligation to purchase our product under the CRADA or any other agreement with us, and there is no assurance that the U.S. Army will ultimately purchase our product.
Pursuant to the CRADA, as amended, the laboratories of the U.S. Army Medical Material Agency (“USAMMA”) and the U.S. Army Medical Material Development Activity (“USAMMDA”) (collectively, the “Army Laboratories”), agreed to cooperate with NHC on research for the ongoing design and development to determine if the PoNS™ device can be developed for commercial use in assisting physical therapy in the treatment of soldiers and others with military relevant neurological manifestations of TBI, including but not limited to Tinnitus, post-traumatic stress disorder, or PTSD, pain and any subsequent indications identified by the parties. The CRADA may be terminated by NHC or the Army Laboratories unilaterally at any time by providing the other party written notice at least 30 days prior to the desired termination date. In addition, the CRADA automatically expires on December 31, 2017 unless modified in writing by the parties, provided that the CRADA is subject to a four-year automatic extension as required for both FDA clearance in the event that a pre-market approval application with the FDA is required for a PoNS™ indication in respect of aid to therapy for chronic balance deficits resulting from TBI as well as for commercialization of the PoNS™ device.
We will initially seek FDA clearance only for treatment of patients with chronic balance deficit due to TBI. The U.S. Army has expressed an interest in supplying PoNS™ devices to the military personnel who need it, subject to our ability to demonstrate its safety and effectiveness and our ability to obtain such FDA clearance. Based on this interest, we estimate that there is a sufficient potential market of active duty and retired soldiers who could potentially benefit from the PoNS™ device due to their chronic TBI symptoms. However, the U.S. Army has not made any guarantees and is not otherwise under any contractual obligations to purchase PoNS™ devices, even if we do demonstrate effectiveness and obtain FDA clearance.
If we are able to complete development of the PoNS™ device and obtain FDA clearance of the device to treat chronic balance deficit due to mild to moderate TBI, we plan to develop the PoNS™ device to treat other indications, or symptoms caused by neurological disorders. As set forth in the January 12, 2015 amendment of our CRADA, the U.S. Army has also expressed interest in our development of the PoNS™ device to treat other symptoms of TBI or any other indications caused by neurological disorders. We would be required to commit our own resources to sponsor the regulatory process for these additional indications. However, the Army Laboratories has agreed in the January 12, 2015 amendment to our CRADA to be responsible for supporting the execution of studies using the PoNS™ device as a treatment for mutually agreed-upon military relevant neurological disorders, which could include but not be limited to Tinnitus, PTSD, sleep regulation and pain (headache) and any subsequent indications identified by the parties. The amount of such support, if any, and the terms of such responsibility to support such studies are not yet negotiated and we have no assurance that we can ultimately reach agreement with the Army Laboratories on such amount or terms of support. There can be no assurance that the Army Laboratories will not otherwise attempt to renegotiate its responsibilities under the CRADA.
6
Food and Drug Administration
To date, no prior premarket notifications for clearance of the PoNS™ device have been submitted by NHC to the FDA, but the Army Laboratories, which previously was responsible as the regulatory sponsor until such role was assumed by NHC, submitted a request for information with the FDA with respect to the potential classification of the PoNS™ device through what is known as a 513(g) request for information. In response to a 513(g) request, the FDA provides information regarding the classification of the device or the requirements applicable to a device under the Federal Food, Drug, and Cosmetic Act, or the FD&C Act. Under the 513(g) request, the Army Laboratories sought guidance from the FDA regarding the classification of the PoNS™ device and the applicable requirements under the FD&C Act. As a result of this process, the FDA responded with guidance on pursuing de novo classification of the PoNS™ device as a Class II medical device.
We plan to utilize the de novo classification process to obtain Class II classification and 510(k) clearance from the FDA for the PoNS™ device. We have been deemed by the FDA through the pre-submission process a non-significant risk device in the context of the TBI clinical trial and thus do not need an Investigation Device Exemption to complete our clinical trials. We are seeking to complete a safety and effectiveness clinical trial by the first quarter of calendar year 2017 and will thereafter submit a request for de novo classification and the premarketing notification (i.e., 510(k)) to the FDA. Previously, we had sought to complete the safety and effectiveness clinical trial by the first quarter of calendar year 2016.
On a parallel path to our request for de novo classification and premarket notification to the FDA, we expect to submit an application for the clearances of the PoNS™ device for both TBI and MS indications to Europe for a CE Mark and to Health Canada (the department of the government of Canada with responsibility for national public health). Our goal is that the CE Mark and Canadian clearance for the PoNS™ device will be obtained in early 2017.
On April 29, 2014, NHC, as cooperator, entered into Notice of Modification No. 1 of the CRADA, with Advanced NeuroRehabilitation LLC (“ANR”), one of our significant shareholders, the inventors, and the Army Laboratories, whereby NHC will no longer provide expertise and training in the design of clinical study protocols or for U.S. Army and/or VA personnel in the physical therapy interventions required for clinical studies. In addition, pursuant to the amended CRADA, ANR will share all data with USAMMA and NHC will provide all data supporting clinical claims for regulatory approval.
On January 12, 2015, NHC, as cooperator, entered into Notice of Modification No. 2 of the CRADA, with ANR, the inventors, and the Army Laboratories. Under this amendment to the CRADA, the Army Laboratories agreed to transfer some of the CRADA responsibilities to NHC. We believe the Army Laboratories agreed to transfer certain responsibilities to us under the CRADA to enable us to accelerate development of the PoNS™ device for the eventual potential treatment of soldiers. One of the material changes reflected in the amendment to the CRADA is the shifting from the Army Laboratories to NHC of sole responsibility as the regulatory sponsor for all interactions with the FDA in order to gain approval and clearance from the FDA, including the initial 513(g) submission. As part of the amendments to the CRADA, NHC has agreed to be responsible to fund the FDA process as well as to provide the supply of all devices to support all studies governed by the CRADA. While under the amendments NHC gains control of the FDA regulatory process, the amendments materially increase the financial burden on NHC to meet these funding and supply obligations. The amendments also extend from two to four years both the time for regulatory approval in the event a pre-market approval application, or PMA, is required by the FDA as well as for commercialization of the PoNS™ device.
While NHC has sole responsibility as the regulatory sponsor under the CRADA, the U.S. Army Medical Research and Materiel Command (“USAMRMC”) has entered into a sole-source contractual agreement to support the execution of the registration trial for treatment of balance disorder associated with mild to moderate TBI. The objective of this contract is to defray the costs of the registration trial. The Army Laboratories also agreed in the January 12, 2015 amendment to our CRADA to be responsible for supporting the execution of studies using the PoNS™ device as a treatment for mutually agreed-upon military relevant neurological disorders, which could include but not be limited to Tinnitus, PTSD, and pain and any subsequent indications identified by the parties. The amount of such support, if any, and the terms of such responsibility to support such clinical studies are not yet negotiated and we have no assurance that we can ultimately reach agreement with the Army Laboratories on such amount or terms of support, and there can be no assurance that the Army Laboratories will not otherwise attempt to renegotiate its responsibilities under the CRADA. The Army Laboratories may terminate their obligations under the CRADA at any time upon 30 days prior written notice to us. If there are insufficient funds available to cover the necessary research and development costs for our product, the Army Laboratories could terminate the CRADA and cease research and development efforts which could jeopardize our ability to commercialize our PoNS™ device.
7
On July 7, 2015, the Company announced that NHC entered into a sole source cost sharing contract with the USAMRMC. The contract will support the Company’s registrational trial investigating the safety and effectiveness of the PoNS™ device. Under the contract, the USAMRMC will reimburse the Company for approximately 62% of the initially budgeted costs related to the registrational trial of up to a maximum amount of $2,996,244. The sole source cost sharing agreement expires December 31, 2016. As of March 31, 2016, the Company has received a total of $1,458,374 in respect of expenses reimbursed.
On December 28, 2015, NHC, as cooperator, entered into Notice of Modification No. 3 of the CRADA, with ANR, the inventors, and the Army Laboratories to extend the expiration date of the CRADA to December 31, 2017.
Our Market
NHC is in the neurostimulation market. According to a study by Grand View Research, the neurostimulation market was valued at $3.4 billion in 2013 and is expected to grow at a compounded annual growth rate of 14.4% from 2014 to 2020. The leading sectors in the industry are Spinal Cord Stimulation, Deep Brain Stimulation, Sacral Nerve Stimulation and Vagal Nerve Stimulation. We believe that due to the significant lack of non-invasive devices, non-invasive stimulation addresses only approximately 3% of the overall neurostimulation market today. This allows for a significant market opportunity for the Company.
Market Competition
The neurostimulation market is competitive and growing. Our competitors in the industry are predominantly large, publically-traded companies that have a history in the market, have significantly easier access to resources and have an established product pipeline. The combined clinical research and product development done by the industry, including by us and all of our competitors, is foundational, and neurostimulation has slowly become integrated into neurological therapy. This foundation has allowed for new and innovative neurostimulation companies to enter the market.
We believe that our technology, the PoNS™ device, introduces an innovative target and method of stimulation because targeting the tongue for neurostimulation provides several clear advantages, which are discussed below. While we believe that the factors described below competitively distinguish our technologies and provide the PoNS™ device a competitive advantage for non-invasive neuromodulation therapy, we note that these factors are only supported by anecdotal evidence of efficacy from the initial work done at the TCNL Laboratories. We believe that our pilot study on MS done at the Montreal Neurological Institute and Hospital and Concordia University’s PERFORM Center using functional MRI provides scientific evidence of efficacy, and from our Press Release dated November 2, 2015, we announced that our device met all of its study objectives and that the results suggested the device may be facilitating neural plasticity. We therefore are making the assumption that the results of our current TBI clinical trial program will be positive and further support these claims at that time.
Advantages of the PoNS™ Device
|
|
• |
Other technologies stimulate other branches of the trigeminal nerve. We target the lowest branch of the trigeminal nerve, which is found in the tongue. It is also the largest branch, having the highest amount of nerve fibers of the three branches. |
|
|
• |
Stimulating the tongue also allows for the simultaneous stimulation of a second cranial nerve found in the tongue, the facial nerve. The ability to stimulate more than one nerve alone differentiates us from our competition. However, it has not been scientifically proven that stimulating additional nerves adds to the efficacy of the treatment. |
8
• |
The tongue has an anatomically unique surface with a high density of receptors, a consistently moist and conductive environment, constant pH, constant temperature and a direct connection to the brain through at least two cranial nerves. | |
| • | We believe that the trigeminal and facial cranial nerves offer a high-bandwidth pathway for impulses to directly affect the central nervous system. The trigeminal and facial nerves project directly onto several areas of the brain, primarily the brainstem (trigeminal and solitary nuclei), cerebellum, cochlear nuclei and spinal cord. Secondary targets include the limbic system, basal ganglia and thalamus. We believe that this range of projections allows impulses be sent through sites regulating dozens of functions. | |
| • | Unlike Deep Brain Stimulation devices, implantable vagal nerve devices and other invasive forms of electrical stimulation, the tongue allows for neurostimulation to be delivered non-invasively and portably. This opens the door for integration of neurostimulation with a wide range of therapies previously unexplored for neurological rehabilitation. |
Reimbursement
If we complete our clinical trials and obtain FDA clearance, and ultimately receive customer orders for the PoNS™ device, we plan to submit applications for appropriate reimbursement codes so that insurers, including Medicare and Medicaid, are able to pay for the device. We plan to seek coverage and reimbursement of the PoNS™ device from public payers, such as Medicare and Medicaid, as well as private payers. There are complex laws, regulations and guidance that set forth Medicare coverage and reimbursement policies. To help us navigate the regulatory complexities, we have engaged consultants to assist us with our reimbursement strategy.
From time to time, Congress enacts laws that impact Medicare coverage and reimbursement policy. In addition, the Centers for Medicare & Medicaid Services, or CMS, regularly engage in rulemaking activities and issues instructions and guidance that may affect Medicare coverage and reimbursement policy. Similarly, the federal and state governments may enact future laws or issue regulations or guidance that may impact Medicaid coverage and reimbursement policies, or the coverage and reimbursement policies of private insurers. We must ensure that we are in full compliance with all applicable requirements, and that we remain abreast of potential legislative or regulatory developments that could impact its business. For all payers, the PoNS™ device must fit within an identifiable coverage category and fully meet the requirements of such category.
Once we complete our clinical trials and obtain FDA clearance, and ultimately receive customer orders for the PoNS™ device, we intend to seek coverage for the PoNS™ device under the Medicare part B durable medical equipment benefit. This will involve ensuring that the PoNS™ device meets all of the criteria for coverage under that benefit. In addition, as part of the coverage process, we may have to submit an application request to CMS to revise the Healthcare Common Procedure Coding System, or HCPCS, level II national code set so that the PoNS™ device becomes eligible to be covered and reimbursed, not only by Medicare, but by other public and private payers. The HCPCS Level II Code Set is a standardized coding set used for claims submitted to public and private payers that identifies particular products, supplies and services. At present, we do not believe that the PoNS™ device would fit easily within an existing HCPCS code. Thus, we are considering submitting a request to CMS for a new HCPCS code and are evaluating our options with our consultants. An applicant can request that (1) a new permanent code be added to the HCPCS level II national code set; (2) the language used to describe an existing code be modified; or (3) an existing code be deleted. However, prior to submitting its coding request application, we must satisfy several criteria, including but not limited to receiving documentation of the FDA’s approval of the device and having sufficient claims activity or volume in the United States (evidenced by 3 months of marketing activity). The national codes are updated annually. Coding requests must be received by January 3 of the current year to be considered for the January update of the following year.
If we do submit such a request for a new HCPCS code, it will be reviewed by the CMS HCPCS Workgroup, which is comprised of representatives of CMS, Medicaid state agencies, and the Pricing, Data Analysis and Coding contractor. The HCPCS Workgroup meets monthly and determines whether each coding request warrants a change to the HCPCS national coding set.
In addition, Medicare and other insurers must find that the PoNS™ device is medically reasonable and necessary for the treatment of patients’ illness or injuries. If Medicare and other insurers find that the PoNS™ device does not meet their medical necessity criteria, it will not be reimbursed. Medicare and commercial insurers must also develop a payment amount for the PoNS™ device. If that amount is inadequate to cover the costs of the PoNS™ device, healthcare providers will be unlikely to use this device.
9
Deployment
Our PoNS™ device has a design feature that stops delivering therapy every fourteen weeks. This is expected to require patients to return to their physician or physical therapy center, or PTC, for assessment of their progress and reestablishment of challenging physical therapy to achieve higher goals. We currently expect the device to be inspected visually by the physical therapist, reset for another fourteen weeks of treatment, and the tongue array would be replaced by a new one to ensure no degradation of the electrodes occurs. We expect this business model feature to ensure proper support for patients in the early phase of their therapy.
We expect physicians will be informed to prescribe both the PoNS™ device and the “local” trained PTCs for their patients to receive the PoNS™ device and certified their training. We anticipate supporting the launch of the PoNS™ device with the development and implementation of a hub services center to help facilitate the healthcare transaction.
Upon discharge from the PTC, patients are expected to be monitored in their home therapy through the PTCs. At the end of their prescribed treatment, we expect patients to be directed back to their physician for assessment and then return to the PTC for additional treatment as well as replacement of the tongue array if the fourteen weeks have expired.
PoNS™ in the U.S. Army
If it ultimately decides to purchase PoNS™ devices from us, we expect that the U.S. Army would deploy the device to Active Duty Personnel through their rehabilitation centers under orders from the central medical command. All personnel are expected to be certified PoNS™ trainers supported by live, paper and video-based training materials developed through this project by the U.S. Army.
We have also approached the Canadian and United Kingdom Armed Forces to discuss their support of a similar program in Canada and discussions are ongoing. We also intend to pursue other military organizations in relevant countries based on need and size of potential deployment.
PoNS™ in Civilian Population
We believe that a key to deployment success will be to create a national framework of PoNS™-trained Physical Therapists (PTs). We have developed a training certification program where PTs can become certified PoNS™ therapists after on-line and in-person training. We expect there to be a strong financial incentive for the PT community to partner with us because PoNS™ training offers substantial opportunity for growth for the PTs. We anticipate that PTs will be able to use existing reimbursement codes for the physical therapy portion of the therapy. As discussed above, we plan to apply for reimbursement codes for the PoNS™ device.
We will concentrate our efforts in the United States, Canadian and UK marketplaces as first launch markets. We are currently uncertain which of these three markets will launch first, primarily due to the relative speed of the regulatory process, and there is no assurance that any will launch at all. Following the initial launch of marketplaces, we intend to commercialize the PoNS™ device in the rest of Europe, Australia and Japan as second phase countries (2018-2019) and Brazil, India and other markets as phase III countries (2019-2020). Previously, we disclosed that we intended to commercialize the PoNS™ device in the phase II countries in 2017 and the phase III countries in 2018. This change is based on the timing of our current clinical trial.
In November 2014 we signed a development and distribution agreement with Altair (a Russian company based in Moscow) to apply for registration and distribute the PoNS™ device in the territories of the former Soviet Union. Thus far the device has received a letter of conformity as an adjunct to physical therapy in Russia and Uzbekistan following regulatory applications to the health authorities of these two countries.
10
Licensed Intellectual Property
Pursuant to the Second Amended and Restated Patent Sub-License agreement dated as of June 6, 2014 entered into between ANR and NHC (the “Sublicense Agreement”), ANR has granted NHC a worldwide, exclusive license to make, have made, use, lease and sell devices utilizing certain patent applications, which are collectively referred to as the “Patent Pending Rights.” The Patent Pending Rights relate to the PoNSTM device and include the following patents and patent applications, which cover a device that noninvasively delivers neurostimulation through the skin or intra- orally to the brain stem via the trigeminal nerve, the facial nerve or both:
| U.S. Patent
Application No. |
Application Filing Date |
Status | U.S. Patent No. |
Issue Date | Subject Matter |
| 12/348,301 | 1/4/2009 | Issued | 8,849,407 | 9/30/2014 | non-invasive neurostimulation of the skin combined with simultaneous physical therapy to provide neurorehabilitation of a patient to treat various maladies including, e.g., TBI, stroke and Alzheimer’s disease |
| 14/340,144 | 7/24/2014 | Issued | 8,909,345 | 12/9/2014 | non- invasive neurostimulation within a patient’s mouth combined with physical therapy to provide neurorehabilitation of a patient to treat various maladies including, e.g., TBI, stroke, and Alzheimer’s disease |
| 14/341,141 | 7/25/2014 | Issued | 9,020,612 | 4/28/2015 | non- invasive neurostimulation within a patient’s mouth combined with cognitive therapy to provide neurorehabilitation of a patient resulting in improved reading comprehension and increased attention span as well as the treatment various maladies including, but not limited to, TBI, stroke, and Alzheimer’s disease |
| 14/615,766 | 2/6/2015 | Pending | N/A | N/A | non- invasive neurostimulation within a patient’s mouth combined with stimulation of the patient’s vision, hearing, vestibular systems, or somatosensory systems for the treatment of tinnitus |
| 14/689,462 | 4/17/2015 | Pending | N/A | N/A | non- invasive neurostimulation of a patient’s skin combined with cognitive therapy to provide neurorehabilitation of a patient resulting in improved reading comprehension and increased attention span as well as the treatment various maladies including, e.g., TBI, stroke, and Alzheimer’s disease |
| 14/815,171 | 7/31/2015 | Pending | N/A | N/A | non- invasive neurostimulation of a patient’s mouth combined with therapy to provide neurorehabilitation of a patient, with a focus on features of a neurostimulation device |
| 61/019,061 (Provisional) |
1/4/2008 | Expired | N/A | N/A | N/A |
| 61/020,265 (Provisional) |
1/10/2008 | Expired | N/A | N/A | N/A |
11
U.S. Patent Nos. 8,909,345 and 9,020,612 and U.S. Patent Application Nos. 14/615,766, 14/689,462 and 14/815,171 claim priority to U.S. Patent No. 8,849,407.
A U.S. provisional patent application provides the means to establish an early effective filing date for a later filed nonprovisional patent application. Therefore, though the two provisional applications have expired, they establish a priority date for U.S. Patent Nos. 8,849,407, 8,909,345, 9,020,612, and U.S. Patent Application Nos. 14/615,766, 14/689,462, 14/815,171 and any future filings that claim priority. We intend to file additional continuation applications in the USPTO claiming priority to U.S. Patent Application Nos. 14/615,766, 14/689,462, and 14/815,171 to protect other aspects of the PoNSTM device and related non- invasive neurostimulation techniques.
ANR, which is one of Helius’ significant shareholders, holds an interest in the Patent Pending Rights pursuant to an exclusive license from the inventors. U.S. Patent Application Nos. 14/615,766, 14/689,462, 14/815,171 are included in the exclusive license as the exclusive license agreement covers (i) U.S. Patent Application No. 12/348,301 and Provisional Application No. 61/019,061, (ii) any patents issuing therefrom and (iii) any patents claiming priority to U.S. Patent Application No. 12/348,301 or Provisional Application No. 61/019,061, which U.S. Patent Application Nos. 14/615,766, 14/689,462, 14/815,171 claim priority through such provisional application as well as through Provisional Application 61/020,265.
In addition, ANR has agreed that ownership of any improvements, enhancements or derivative works of the Patent Pending Rights that are developed by NHC or ANR shall be owned by NHC, provided that if NHC decides not to patent such improvements, ANR may choose to pursue patent rights independently. Pursuant to the Sublicense Agreement, NHC has agreed to pay ANR royalties equal to 4% of NHC’s revenues collected from the sale of devices covered by the Patent Pending Rights and services related to the therapy or use of devices covered by the Patent Pending Rights in therapy services. The Sublicense Agreement provides that the sublicense granted by ANR to NHC, if in good standing, shall not be cancelled, limited or impaired in any way should there be a termination of the master license granted by the inventors to ANR, which was acknowledged by the inventors in the Sublicense Agreement. On June 6, 2014, NHC and ANR entered into a second amended and restated sublicense agreement, or the Second Sublicense Agreement, which acknowledges the Reverse Merger (see “Our Corporate History - Acquisition of NeuroHabilitation Corporation and Concurrent Financing” below), and adds us as a party to the agreement.
The license of the Patent Pending Rights are subject to the right of the government of the United States, which funded certain research relating to the development of the PoNSTM device, to a nonexclusive, non- transferable, irrevocable, paid- up license to use the Patent Pending Rights for governmental purposes. In addition, NHC has granted a perpetual, royalty-free license to the Patent Pending Rights back to ANR for non- profit research and development activities which do not compete with NHC’s business and to produce and derive revenues from devices and services in connection with investigational uses of the PoNSTM device and related technology.
The license of the Patent Pending Rights is also subject to the terms of the CRADA. In the event that Helius is not willing or is unable to commercialize the PoNSTM technology within four years from the expiration of the CRADA, the Company is required to transfer possession, ownership and sponsorship/holdership of the regulation application, regulatory correspondence and supporting regulatory information related technology to USAMRMC and grant the U.S. Government a non-exclusive, irrevocable license to any patent, copyright, data rights, proprietary information or regulatory information for the U.S. Government to commercialize the technology.
12
Company Owned Intellectual Property
On July 17, 2015, the Company announced that the USPTO issued the Company its first patent related to the design of the current PoNSTM device. U.S. Patent No. 9,072,889, “Systems for Providing Non-Invasive Neurorehabilitation of a Patient,” issued on July 7, 2015, is the first patent Helius has received related specifically to the new device design.
The Company filed 27 U. S. patent applications related to various technical and ornamental aspects of the PoNSTM device. The Company filed eleven non-provisional patent applications that describe various technical features in the current version device and 16 design patent applications describing various ornamental designs. Helius is the sole assignee for these 27 U.S. patent filings. Prior to issuance, once the USPTO determines that a patent application meets all of the statutory requirements for patentability it provides a notice of allowance. In addition to the first issued patent (U.S. Patent No. 9,072,889), the USPTO has issued three utility patents, 16 design patents and provided notices of allowance for utility applications as summarized in the table below:
| U.S. Patent Application No. |
Application Filing Date |
Status | U.S. Patent No. |
Issue Date | Subject Matter |
| 14/558,768 | 12/3/2014 | Issued | 9,072,889 | 7/7/2015 | Utility application covering overall system design, including controller and mouthpiece |
| 14/559,123 | 12/3/2014 | Issued | 9,272,133 | 3/1/2016 | Utility application covering strain relief mechanisms for the connection between the mouthpiece and the controller |
| 14/558,787 | 12/3/2014 | Issued | 9,227,051 | 1/5/2016 | Utility application covering shape of the mouthpiece |
| 14/558,789 | 12/3/2014 | Issued | 9,283,377 | 3/15/2016 | Utility application covering center of gravity of the mouthpiece |
| 14/559,080 | 12/3/2014 | Allowed | TBD | TBD | Utility application covering structural support of the mouthpiece |
| 14/559,105 | 12/3/2014 | Allowed | TBD | TBD | Utility application covering glue wells of the mouthpiece |
| 29/510,741 | 12/3/2014 | Issued | D750264 | 2/23/2016 | Design application covering an alternative version of the current PoNS™ device (over-ear double boom design) |
| 29/510,742 | 12/3/2014 | Issued | D749746 | 2/16/2016 | Design application covering an alternative version of the current PoNS™ device (overhead minimal interference design) |
| 29/510,743 | 12/3/2014 | Issued | D752236 | 3/22/2016 | Design application covering system design used in the current PoNS™ device |
| 29/510,745 | 12/3/2014 | Issued | D750265 | 2/23/2016 | Design application covering an alternative mouthpiece not used in the current PoNS™ device |
13
| U.S. Patent Application No. |
Application Filing Date |
Status | U.S. Patent No. |
Issue Date | Subject Matter |
| 29/510,754 | 12/3/2014 | Issued | D750794 | 3/1/2016 | Design application covering the controller used in the PoNS™ device |
| 29/510,755 | 12/3/2014 | Issued | D751215 | 3/8/2016 | Design application covering an alternative controller not used in the current PoNS™ device |
| 29/510,746 | 12/3/2014 | Issued | D750266 | 2/23/2016 | Design application covering an alternative mouthpiece not used in the current PoNS™ device |
| 29/510,749 | 12/3/2014 | Issued | D750268 | 2/23/2016 | Design application covering an alternative mouthpiece not used in the current PoNS™ device |
| 29/510,747 | 12/3/2014 | Issued | D751213 | 3/8/2016 | Design application covering an alternative mouthpiece not used in the current PoNS™ device |
| 29/510,748 | 12/3/2014 | Issued | D750267 | 2/23/2016 | Design application covering an alternative mouthpiece not used in the current PoNS™ device |
| 29/510,750 | 12/3/2014 | Issued | D753315 | 4/5/2016 | Design application covering mouthpiece used in the current PoNS™ device |
| 29/510,751 | 12/3/2014 | Issued | D751722 | 3/15/2016 | Design application covering an alternative controller not used in the current PoNS™ device |
| 29/510,752 | 12/3/2014 | Issued | D752766 | 3/29/2016 | Design application covering an alternative controller not used in the current PoNS™ device |
| 29/510,753 | 12/3/2014 | Issued | D753316 | 4/5/2016 | Design application covering an alternative controller not used in the current PoNS™ device |
| 29/510,744 | 12/3/2014 | Issued | D760397 | 6/28/2016 | Design application covering system design used in the current PoNS™ device |
| 29/510,756 | 12/3/2014 | Issued | D759830 | 6/21/2016 | Design application covering system design used in the current PoNS™ device |
Additionally, Helius has filed three international applications, and 14 foreign design applications: seven in Canada, three in China, three in Russia, and one community design in Europe. The following three applications filed in China, which have been assigned to China Medical Systems Holdings LTD. pursuant to an asset purchase agreement (the “Strategic Agreement”) dated effective October 9, 2015 with A&B have issued:
14
| Chinese Patent Application No. |
Application Filing Date |
Status | Chinese Patent No. |
Issue Date | Subject Matter |
| 201530177804.4 | 6/3/2015 | Issued | CN303597712S | 2/24/2016 | Design application covering the system design currently used in the PoNSTM 4.0 device |
| 201530178171.9 | 6/3/2015 | Issued | CN303597713S | 2/24/2016 | Design application covering the mouthpiece design currently used in the PoNSTM 4.0 device |
| 201530177398.1 | 6/3/2015 | Issued | CN303597711S | 2/24/2016 | Design application covering the controller design currently used in the PoNSTM 4.0 device |
Further, the three design applications filed in Russia have been allowed, and the Canadian Design applications and European community design have issued:
| Russian Design Application No. |
Application Filing Date |
Status | Russian Patent No. |
Issue Date | Subject Matter |
| 2015501883 | 6/3/2015 | Allowed | TBD | TBD | Design application covering the system design currently used in the PoNSTM 4.0 device |
| 2015501882 | 6/3/2015 | Allowed | TBD | TBD | Design application covering the mouthpiece design currently used in the PoNSTM 4.0 device |
| 2015501881 | 6/3/2015 | Allowed | TBD | TBD | Design application covering the controller design currently used in the PoNSTM 4.0 device |
| Canadian Design Application No. |
Application Filing Date |
Status | Canadian Patent No. |
Issue Date | Subject Matter |
| 162676 | 6/2/2015 | Issued | 162676 | 2/29/2016 | Design application covering system design used in the current PoNSTM device |
| 162672 | 6/2/2015 | Issued | 162672 | 2/29/2016 | Design application covering an alternative mouthpiece not used in the current PoNSTM device |
| 162671 | 6/2/2015 | Issued | 162671 | 2/29/2016 | Design application covering an alternative mouthpiece not used in the current PoNSTM device |
| 162674 | 6/2/2015 | Issued | 162674 | 2/29/2016 | Design application covering mouthpiece used in the current PoNSTM device |
| 162675 | 6/2/2015 | Issued | 162675 | 2/29/2016 | Design application covering an alternative controller not used in the current PoNSTM device |
| 162670 | 6/2/2015 | Issued | 162670 | 2/29/2016 | Design application covering the controller used in the PoNSTM device |
| 162673 | 6/2/2015 | Issued | 162673 | 2/29/2016 | Design application covering system design used in the current PoNSTM device |
| EU Community Design Application No. |
Application Filing Date |
Status | EU Community Design Reg. No. |
Issue Date | Subject Matter |
| 002712026 | 6/3/2015 | Issued | 002712026 | 9/4/2015 | Design application covering several aspects of the system design currently used in the PoNSTM 4.0 device |
Currently, Helius uses four trademarks in connection with the operation of the business: PoNSTM, NeuroHabilitation, NHC and Helius Medical Technologies. Helius owns the rights to the PoNSTM mark by virtue of an assignment agreement having an effective date of October 27, 2014 and entered into with ANR and the inventors of the PoNSTM technology. Helius is the sole owner of the rights in the NeuroHabilitation and NHC trademarks, and Helius is the owner of the rights in the Helius Medical Technologies mark. On October 31, 2014, Helius filed trademark applications in the USPTO for these four trademarks.
On January 7, 2015, Helius filed trademark applications with the Canada Intellectual Property Office, claiming priority to the corresponding U.S. applications filed on October 31, 2014. The Company is the owner of the rights in the NeuroHabilitation, NHC, and PoNS marks in Canada, and Helius is the owner of the rights in the Helius Medical Technologies mark in Canada. The Company has also applied for the PoNS trademark in Canada, Europe, Russia and China.
We take precautions to safeguard our intellectual property, and it has been and may be the subject of lawsuits. See Part I Item 3, “Legal Proceedings.”
15
Government Regulation
Our products under development and our operations are subject to significant government regulation. In the United States, our products are regulated as medical devices by the FDA and other federal, state, and local regulatory authorities. The following is a general description of the review and clearance process of the FDA for medical devices.
FDA Regulation of Medical Devices
The FDA and other U.S. and foreign governmental agencies regulate, among other things, the following activities with respect to medical devices:
| • | design, development and manufacturing; | |
| • | testing, labeling, content and language of instructions for use and storage; | |
| • | clinical trials; | |
| • | product storage and safety; | |
| • | marketing, sales and distribution; | |
| • | pre-market clearance and approval; | |
| • | record keeping procedures; | |
| • | advertising and promotion; | |
| • | recalls and field safety corrective actions; | |
|
• |
post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; | |
| • | post-market approval studies; and | |
| • | product import and export. |
In the United States, numerous laws and regulations govern all the processes by which medical devices are brought to market and marketed. These include the FD&C Act and the FDA’s implementing regulations, among others.
The FDA Review, Clearance and Approval Process
Each medical device we seek to commercially distribute in the United States must first receive either clearance under Section 510(k) of the FD&C Act, receive de novo down-classification, or pre-market approval, or PMA, from the FDA, unless specifically exempted by the FDA. FDA review and approval is required for each application of a device, regardless of whether the device has been approved for other applications. The FDA classifies all medical devices into one of three classes. Devices deemed to pose the lowest risk are categorized as either Class I or II, which requires the manufacturer to submit to the FDA a 510(k) pre-market notification submission requesting clearance of the device for commercial distribution in the United States, unless the device is exempted from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously 510(k) cleared device are categorized as Class III and require submission and approval of a PMA application.
In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support a determination of substantial equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. However, some devices are automatically subject to the PMA pathway regardless of the level of risk they pose, because they have not previously been classified into a lower risk class by the FDA. Manufacturers of these devices may request that the FDA review such devices in accordance with the de novo classification procedure, which allows a manufacturer whose novel device would otherwise require the submission and approval of a PMA prior to marketing to request down-classification of the device on the basis that the device presents low or moderate risk. If the FDA agrees with the down-classification, the applicant will then receive approval to market the device. This device type can then be used as a predicate device for future 510(k) submissions.
16
We intend to utilize the de novo classification procedures to seek marketing authorization for the PoNS™ device, because there is currently no predicate cleared or approved by the FDA for commercial distribution and no existing classification decision by the FDA for such a device. The process of obtaining regulatory clearances or approvals, or completing the de novo classification process, to market a medical device can be costly and time consuming, and we may not be able to successfully obtain pre-market reviews on a timely basis, if at all.
If the FDA requires us to go through a lengthier, more rigorous examination for the PoNS™ device, introducing the product could be delayed or canceled, which could cause our launch to be delayed. In addition, the FDA may determine that the PoNS™ device requires the more costly, lengthy and uncertain PMA process. For example, if the FDA disagrees with our determination that the de novo classification procedures are the appropriate path to obtain marketing authorizations for the PoNS™ device, the FDA may require us to submit a PMA application, which is generally more costly and uncertain and can take from one to three years, or longer, from the time the application is submitted to the FDA until an approval is obtained. Further, even with respect to those future products where a PMA is not required, we cannot be certain that we will be able to obtain 510(k) clearances with respect to those products.
510(k) Clearance Process
To obtain 510(k) clearance, we must submit a pre-market notification to the FDA demonstrating that the proposed device is substantially equivalent to a previously-cleared 510(k) device or is a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of PMA applications. The FDA’s 510(k) clearance process usually takes from three to 12 months from the date the application is submitted and filed with the FDA, but may take significantly longer and clearance is never assured. Although many 510(k) pre-market notifications are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. In reviewing a pre-market notification submission, the FDA may request additional information, including clinical data, which may significantly prolong the review process.
After a device receives 510(k) clearance, any subsequent modification of the device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k) clearance or could require PMA. The FDA requires each manufacturer to make this determination initially, but the FDA may review any such decision and may disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA may require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA is obtained. Under these circumstances, the FDA may also subject a manufacturer to significant regulatory fines or other penalties. In addition, the FDA is currently evaluating the 510(k) process and may make substantial changes to industry requirements, including which devices are eligible for 510(k) clearance, the ability to rescind previously granted 510(k)s and additional requirements that may significantly impact the process.
De novo Classification Process
If a previously unclassified new medical device does not qualify for the 510(k) pre-market notification process because no predicate device to which it is substantially equivalent can be found, the device is automatically classified Class III regardless of the level of risk it poses. The Food and Drug Administration Modernization Act of 1997 established a new route to market for low to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act, or FDASIA, in July 2012, a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. The FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination. Under the FDASIA, the FDA is required to classify the device within 120 days following receipt of the de novo application. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed.
17
We plan to utilize the de novo classification process to obtain marketing authorization for the PoNS™ device under development, and we plan to seek Class II classification. In order to be placed in Class II, the FDA would need reasonable assurance of safety and effectiveness of the PoNS™ device. Under Class II, general controls (e.g., premarket notification) and special controls (e.g., specific performance testing) would be applicable. Our goal would be to complete in six months a safety and effectiveness clinical trial using the PoNS™ device, initially only for the treatment of balance disorder in patients with mild to moderate TBI and balance disorder associated with MS. Our overall goal for submission of the de novo application and FDA clearance of a 510(k) would be 24 months from December 2014. The application to the FDA will be made after the completion of the registration trial, which we anticipate will be completed in the first quarter of calendar year 2017. Originally, we anticipated that the registration trial would be completed at the end of 2015, but that timing was revised due to slower than expected enrollment. We have invested resources to expand recruiting to recoup for time lost. It will take us approximately four weeks to prepare the premarket notification to the FDA. We thus anticipate that we will be applying for clearance in the first half of calendar year 2017. To the extent the FDA completes its review in 90 days, we anticipate clearance by the third quarter of calendar year 2017.
Obtaining FDA clearance, de novo down-classification, or approval for medical devices can be expensive and uncertain, generally takes from several months to several years, and generally requires detailed and comprehensive scientific and clinical data. Notwithstanding the expense, these efforts may never result in FDA clearance. Even if we were to obtain regulatory clearance, it may not be for the uses we believe are important or commercially attractive, in which case we would not be permitted to market our product for those uses.
Pre-market Approval Process
A PMA application must be submitted if the medical device is in Class III (although the FDA has the discretion to continue to allow certain pre-amendment Class III devices to use the 510(k) process) or cannot be cleared through the 510(k) process. A PMA application must be supported by, among other things, extensive technical, preclinical, clinical trial, manufacturing and labeling data to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use.
After a PMA application is submitted and filed, the FDA begins an in-depth review of the submitted information, which typically takes between one and three years, but may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with Quality System Regulations, or QSR, which impose elaborate design development, testing, control, documentation and other quality assurance procedures in the design and manufacturing process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution and collection of long-term follow-up data from patients in the clinical study that supported approval. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the loss or withdrawal of the approval. New PMA applications or supplements are required for significant modifications to the manufacturing process, labeling of the product and design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an original pre-market approval application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
Clinical Trials
A clinical trial is typically required to support a PMA application and is sometimes required for a 510(k) pre-market notification. After a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to an unacceptable health risk. Any trials we conduct must be conducted in accordance with FDA regulations as well as other federal regulations and state laws concerning human subject protection and privacy. Moreover, the results of a clinical trial may not be sufficient to obtain clearance or approval of the product, and separate clinical trials will be necessary to obtain clearance for multiple uses of one device.
18
Risks of Delay from the FDA Clearance Process and Regulatory Compliance Risks
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
|
• |
we may not be able to demonstrate to the FDA’s satisfaction that our product candidate is safe and effective, sensitive and specific diagnostic tests, for its intended users; | |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and | |
| • | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis. For example, in response to industry and healthcare provider concerns regarding the predictability, consistency and rigor of the 510(k) regulatory pathway, the FDA initiated an evaluation of the program, and in January 2011, announced several proposed actions intended to reform the review process governing the clearance of medical devices. The FDA intends these reform actions to improve the efficiency and transparency of the clearance process, as well as bolster patient safety. In addition, as part of the FDASIA the U.S. Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms that are further intended to clarify and improve medical device regulation both pre- and post-approval. Any delay in, or failure to receive or maintain, clearance or approval for our product candidate could prevent us from generating revenue from our product candidate and adversely affect our business operations and financial results.
Even if we obtain FDA clearance for our PoNS™ device, we will still be required to pursue a 510(k) clearance, de novo down-classification, or PMA for any future product which will delay future product launches and would likely place substantial restrictions on how our device is manufactured, marketed and sold. For example, the manufacture of medical devices must comply with FDA’s QSR. In addition, manufacturers must register their manufacturing facilities, list the products with FDA, and comply with requirements relating to labeling, marketing, complaint handling, adverse event and medical device reporting, reporting of corrections and removals, and import and export. FDA monitors compliance with the QSR and these other requirements through periodic inspections. If our facilities or those of our manufacturers or suppliers are found to be in violation of applicable laws and regulations, or if we or our manufacturers or suppliers fail to take satisfactory corrective action in response to an adverse inspection, the regulatory authority could take enforcement action, including any of the following sanctions:
|
|
• |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
|
• |
customer notifications or repair, replacement, refunds, detention or seizure of our products; |
|
|
• |
operating restrictions or partial suspension or total shutdown of production; |
|
|
• |
refusing or delaying requests for 510(k) marketing clearance or pre-market approvals of new products or modified products; |
|
|
• |
withdrawing 510(k) marketing clearances or pre-market approvals that have already been granted; |
|
|
• |
refusing to provide Certificates for Foreign Government; |
|
|
• |
refusing to grant export approval for our products; or |
|
|
• |
pursuing criminal prosecution. |
19
Additionally, FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could affect the perceived safety and efficacy of our product candidate and dissuade our customers from using our product candidate, if and when they are authorized for marketing.
Pervasive and Continuing U.S. Food and Drug Administration Regulation
After a medical device is placed on the market, numerous FDA regulatory requirements apply, including, but not limited to the following:
|
|
• |
the QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures during the manufacturing process; |
|
|
|
|
|
|
• |
establishment registration, which requires establishments involved in the production and distribution of medical devices, intended for commercial distribution in the United States, to register with the FDA; |
|
|
|
|
|
|
• |
medical device listing, which requires manufacturers to list the devices they have in commercial distribution with the FDA; |
|
|
|
|
|
|
• |
correction and removal reporting regulations which require that manufacturers report to the FDA field corrections and product recalls or removals undertaken to reduce a risk to health posed by the device or remedy a violation of the FD&C Act that may present a risk to health; |
|
|
|
|
|
|
• |
labeling regulations, which prohibit “misbranded” devices from entering the market, as well as prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; |
|
|
|
|
|
|
• |
clearance or approval of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use; |
|
|
|
|
|
|
• |
post-market surveillance including Medical Device Reporting, which requires manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury, or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and |
|
|
|
|
|
|
• |
other post-approval restrictions or conditions. |
Our Corporate History Highlights
Formation and Arrangement with Boomerang Oil, Inc.
We were incorporated on March 13, 2014 under the British Columbia Business Corporations Act, or the BCBCA, as “0996445 B.C. Ltd.” On March 25, 2014, and amended on April 8, 2014, we entered into an arrangement agreement with Boomerang Oil, Inc. (formerly known as 0922327 B.C. Ltd.) and 0995162 B.C. Ltd. to reorganize the business structure of such three entities in such a manner which would allow Boomerang Oil, Inc. to spin us out to become an independent entity that is a reporting issuer in Canada and for us to complete a reverse take-over of 0995162 B.C. Ltd. As a result of the arrangement agreement, we became a reporting issuer in the provinces of British Columbia and Alberta. In addition, the arrangement resulted in 0995162 B.C. Ltd. becoming our wholly-owned subsidiary. The assets of 0995162 B.C. Ltd. consisted of cash and 0995162 B.C. Ltd.’s interest in a letter agreement pursuant to which it had agreed to acquire all of the outstanding shares of NHC, a Delaware corporation, and to seek a listing on a recognized stock exchange.
Reincorporation in Wyoming
On May 23, 2014, we changed our name to “Helius Medical Technologies, Inc.” and filed articles of continuation with the Wyoming Secretary of State office to reincorporate from being a corporation governed by the BCBCA to a corporation governed by the Wyoming Business Corporation Act, or WBCA.
Acquisition of NeuroHabilitation Corporation and Concurrent Financing
On June 13, 2014, we completed the acquisition of NHC by way of an agreement and plan of merger. We refer to this transaction as the Reverse Merger. Pursuant to the agreement and plan of merger, HMT Mergersub, Inc., our wholly-owned subsidiary, merged with and into NHC with NHC as the surviving corporation. In connection with the Reverse Merger, we issued an aggregate of 35,300,083 shares of our Class A common stock, or our common stock, to the former shareholders of NHC. The Reverse Merger was deemed to be a capital transaction in substance and recorded as a reverse recapitalization of NeuroHabilitation Corporation whereby NeuroHabilitation Corporation is deemed to be the continuing, surviving entity for accounting purposes, but through reorganization, has deemed to have adopted the capital structure of Helius.
20
In connection with the Reverse Merger, we completed a non-brokered private placement financing of $7.016 million (CAD$7.62 million) by issuing 15.24 million subscription receipts. Pursuant to its terms, each subscription receipt automatically converted into one unit upon satisfaction of certain escrow release conditions, which had been satisfied. Each unit consisted of one share of our common stock and one-half of one share purchase warrant with each whole warrant being exercisable at CAD$1.00 per share for a period of two years. In connection with the concurrent private placement financing, we paid aggregate finders’ fees of $379,806 (CAD $412,200) and issued 824,000 finder’s warrants. Each finder warrant is exercisable at CAD$1.00 per share for a period of two years.
General Development of the Business of NeuroHabilitation Corporation
Our primary operations are conducted through our wholly-owned subsidiary NHC. On January 22 2013, NHC entered into a patent sub-license agreement whereby ANR granted NHC exclusive worldwide rights to ANR’s trade secrets, knowhow, and patent pending technology for a non-invasive means for delivering neurostimulation through the oral cavity, or the PoNS™ device. NHC obtained these rights in exchange for 50% of the outstanding equity in NHC and an obligation to pay ANR a royalty equal to 4% of any revenue collected by NHC from (1) the sale of products covered by any claim of the patent rights to end users and (2) services related to the therapy or use of such products in therapy services. This agreement was subsequently amended by the Amended and Restated Patent Sub-License and again by the Sublicense Agreement described above.
Listing of our Common Stock on the CSE, TSX and OTCQB
Following our Reverse Merger, we obtained approval of the listing of our common stock on the Canadian Securities Exchange (the “CSE”).
On April 18, 2016, our common stock was listed on the Toronto Stock Exchange (“TSX”) under the symbol “HSM.” At the same time, we delisted our common stock from the CSE. Our Warrants were also approved for listing on the TSX on April 18, 2016.
Our common stock is currently quoted on the OTCQB under the symbol “HSDT.”
Our shares of Class A common stock were approved for listing on the TSX on April 18, 2016. However, some of our shares of common stock were issued in an offshore offering in April and May of 2016 (the “Offshore Offering”) in transactions exempt from the registration requirements of the Securities Act of 1933, as amended (the “Securities Act”) and are listed under a separate ticker symbol for trading on the TSX. These shares of common stock are subject to restrictions on their resale to a U.S. person (as that term is defined in Regulation S), or to a person in the United States, unless in a registered transaction or pursuant to an applicable safe harbor or exemption from registration. Certain of our warrants were also approved for listing on the TSX on April 18, 2016. However, because only warrants issued in the Offshore Offering in transactions exempt from the registration requirements of the Securities Act were approved for listing on the TSX, the Warrants listed on the TSX may not be purchased by or on behalf of a U.S. person, or by a person in the United States, unless in a registered transaction or pursuant to an applicable safe harbor or exemption from registration.
Employees
As of June 21, 2016, we have six full time employees and 20 full-time equivalent independent contractors.
Business Uncertainties and Going Concern Risk
To date we have not generated any revenue from the sales of products or services. There are a number of conditions that we must satisfy before we will be able to generate revenue, including but not limited to successful completion of the TBI, FDA, CE Mark or Health Canada clearance of the PoNS™ device for balance disorder associated with TBI, manufacturing of a commercially-viable version of the PoNS™ device and demonstration of safety and effectiveness sufficient to generate commercial orders by customers for our product. In addition, given the importance of the U.S. Army to our early commercial plans, if the U.S. Army were to eventually decide not to purchase our product, we would need to replace those sales in the civilian market which will lower our early commercialization forecast. To date, we have not achieved many of these conditions, and the successful achievement of such conditions will require significant expenditures. Because we have not generated any revenues, we are dependent entirely on funding from outside investors. There is no guarantee that such funding will be available at all or in sufficient amounts to satisfy our required expenditures. Furthermore, even if we were able to raise sufficient capital to successfully design and manufacture a commercially-viable version of the PoNS™ device and to receive FDA, CE Mark or Health Canada clearance, we do not currently have any contract or other arrangement to sell the PoNS™ device. Accordingly, we cannot know for certain that we will ever be able to generate any revenue from the sales of products or services.
21
Additionally, based on management’s assessment that there is substantial doubt about the Company’s ability to continue as a going concern. This means that there is substantial doubt that we can continue as an on-going business for the next twelve months. While we had $2,643,937 of cash as of March 31, 2016 and we have raised an additional $7,199,781 (CAD$9,215,000) in the Offshore Offering and private placement, and $1,409,947 (CAD$1,825,600) from the exercise of certain warrants issued in 2014, we do not currently have sufficient resources to accomplish all of the above conditions necessary for us to generate revenue.
In reviewing this filing, you should carefully consider the risks described in the section entitled “Risk Factors” and other risks described throughout this Annual Report.
| ITEM 1A. | RISK FACTORS |
RISK FACTORS
An investment in our common stock involves a number of very significant risks. You should carefully consider the following risks and uncertainties in addition to other information in this Annual Report in evaluating our company and its business before purchasing shares of our common stock. Our business, operating results and financial condition could be seriously harmed due to any of the following risks. The risks described below may not be all of the risks facing our company. Additional risks not presently known to us or that we currently consider immaterial may also impair our business operations. You could lose all or part of your investment due to any of these risks.
Risks Related to Our Company
We have a very limited operating history and have a history of operating losses.
Helius Medical Technologies, Inc. is our holding company and it has no material assets other than cash and cash equivalents and its ownership of all of the outstanding shares of NHC, which is our wholly owned subsidiary. NHC was incorporated in Delaware on January 22, 2013 and has had limited operations to date. Since our inception, we have incurred significant net losses. As of March 31, 2016, our accumulated deficit was approximately $26,305,263.
We are heavily dependent upon the ability and expertise of our CEO and a very limited number of employees and the loss of such individuals could have a material adverse effect on our business, operating results or financial condition.
We currently have a very small management team and almost no other employees. Our success is dependent upon the ability, expertise, judgment, discretion and good faith of our senior management, and in particular Mr. Philippe Deschamps, our President and Chief Executive Officer. Currently, Mr. Deschamps is joined by Jonathan Sackier, our Chief Medical Officer, Joyce LaViscount, our Chief Financial Officer and Chief Operating Officer, Brian Bapty, our Vice President of Strategy and Business Development, and two others as our only full-time employees. We also have engaged 20 full-time equivalent persons as independent contractors. While employment agreements are customarily used as a primary method of retaining the services of key employees, these agreements cannot assure the continued services of such employees. Any loss of the services of such individuals could have a material adverse effect on our business, operating results or financial condition.
22
Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements. We may be unable to continue to operate without the threat of liquidation for the foreseeable future.
In connection with our management’s assessment, our report from our independent registered public accounting firm for the fiscal year ended March 31, 2016 includes an explanatory paragraph stating that our recurring losses from operations and net capital deficiency raise substantial doubt about our ability to continue as a going concern. If we are unable to obtain sufficient funding, our business, prospects, financial condition and results of operations will be materially and adversely affected and we may be unable to continue as a going concern. For example, our existing capital resources will be insufficient to fund our operations beyond the end of the fourth quarter of calendar 2016. If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our consolidated financial statements, and investors will likely lose all or a part of their investment. Future reports from our independent registered public accounting firm may also contain statements expressing doubt about our ability to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all.
We have identified a material weakness in our internal controls over financial reporting. If we do not maintain effective internal controls over financial reporting, we could fail to report our financial results accurately.
We have identified material weaknesses in our internal control over financial reporting. Under the direction of our Chief Executive Officer and our Chief Financial Officer, our management evaluated our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and concluded that our disclosure controls and procedures were ineffective as of March 31, 2015, June 30, 2015, September 30, 2015, December 31, 2015, and March 31, 2016.
In April of 2016, the Board, after consulting with the Company’s management, determined that it was necessary to reevaluate the Company’s accounting relating to warrants issued as part of its private placements conducted in April, June and July of 2015 (the “2015 Warrants”), and to restate the Company’s previously reviewed, unaudited, condensed consolidated financial statements for the three months ended June 30, 2015, the three months and six months ended September 30, 2015, and the three and nine months ended December 31, 2015, as a result of an error in the classification of the 2015 Warrants. The Company previously recorded the issuance of the 2015 Warrants as equity instruments instead of liabilities. The warrant exercise prices are denominated in U.S. dollars whereas the functional currency of the Company is the Canadian dollar; as such, the settlement of the warrants fails the fixed for fixed criteria of ASC 815 and they are required to be recorded as a liability at their fair value on inception. The warrant liability is required to be re-measured at its fair value on each reporting date with the changes in fair value recorded in the Company’s Statement of Comprehensive Loss.
The Company had previously restated its consolidated financial statements for the periods as of and for the twelve months ended March 31, 2015 and the quarters therein and its interim condensed consolidated financial statements for the three months ended June 30, 2015 and the three months and six months ended September 30, 2015, as a result of the Company not previously re-measuring the fair value of stock options awarded to non-employees that had not yet vested. The Company’s management has determined that the improper design of controls with respect to the calculation of the fair value of the Company’s share based compensation was a deficiency in its internal control over financial reporting. It is possible that other control deficiencies could be identified in the future or may exist or occur without being identified. In the event additional material weaknesses in our internal controls are discovered in the future, they may adversely affect our ability to record, process, summarize and report financial information timely and accurately and, as a result, our financial statements may contain material misstatements or omissions.
We have incurred substantial net losses since our inception and anticipate that we will continue to incur substantial net losses for the foreseeable future. We may never achieve or sustain profitability.
We have incurred substantial net losses since our inception. For our fiscal years ending March 31, 2016 and March 31, 2015, we incurred a net loss of $6,881,812 and $9,838,317, respectively, and used cash in operations of $7,816,536 and $6,321,285 respectively. We have an accumulated deficit of $26,305,263 and $19,423,451 as of March 31, 2016 and March 31, 2015, respectively. Our losses have resulted primarily from costs incurred in connection with our design, manufacturing and development, research and development activities, stock based compensation, legal, advertising, marketing and investor relations, and general and administrative expenses associated with our operations. Even if we are successful in obtaining clearance from the FDA and launching our PoNS™ device into the market, we expect to continue to incur substantial losses for the foreseeable future as we continue to sell and market our current product and research and develop, and seek regulatory approvals for, other potential product candidates.
23
We are subject to all of the business risks and uncertainties associated with any new business enterprise, including under-capitalization, cash shortages, limitations with respect to personnel, financial and other resources, lack of revenue and the risk that we will not achieve our growth objective. If sales revenue from our current product or any potential product candidates that receive marketing clearance from the FDA or other regulatory body is insufficient, if we are unable to develop and commercialize any of our potential product candidates, or if our product development is delayed, we may never achieve or sustain profitability.
We will require additional financing to carry out our plan of operations and if we are unable to obtain such financing, our business may fail.
We currently have limited working capital and liquid assets. We held cash totaling $2,643,937 as at March 31, 2016. To date we have not generated any revenue from the sales of products or of services. There are a number of conditions that we must satisfy before we will be able to generate revenue, including but not limited to completion of our clinical trial for the treatment of balance disorder in subjects with mild to moderate TBI, FDA clearance of the PoNS™ device for treating balance disorder in patients with mild to moderate TBI, manufacturing of a commercially-viable version of the PoNS™ device and demonstration of effectiveness sufficient to generate commercial orders by customers for our product. While we are currently seeking additional funding, we do not currently have sufficient resources to accomplish any of these conditions necessary for us to generate revenue, and we do not expect to generate revenue in an amount sufficient to fund our operations for the foreseeable future. We will therefore require substantial additional funds in order to continue to conduct the research and development and regulatory clearance and approval activities necessary to bring our product to market, to establish effective marketing and sales capabilities and to develop other product candidates. Our existing capital resources will not be sufficient to enable us to fund the completion of the development and commercialization of our current product and our product candidates. We cannot determine with certainty the duration and completion costs of the current or future development and commercialization of our product candidate or if, when, or to what extent we will generate revenues from the commercialization and sale of our current product candidate or potential future product candidates for which we obtain regulatory approval. We may never succeed in achieving regulatory approval for our current product candidate and any potential future product candidates. We may be unable to raise the additional funding to finance our business on commercially reasonable terms, or at all. If we are unable to obtain additional financing as needed, we may be required to reduce the scope of our operations and pursue only those projects that can be funded through cash flows generated from its existing operations, if any.
Raising additional capital by issuing securities or through debt financings or licensing arrangements may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidate on terms unfavorable to us.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through strategic partnerships with third parties, we may have to relinquish valuable rights to our technologies or product candidate, future revenue streams, research programs or product candidate, or otherwise grant licenses on terms that are not favorable to us. If we are unable to raise additional capital when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts for our product candidate or our preclinical product candidates, or grant rights to develop and market potential future product candidates that we would otherwise prefer to develop and market ourselves. Any of these events could adversely affect our ability to achieve our product development and commercialization goals and have a material adverse effect on our business, financial condition and results of operations.
We currently only have one product candidate, which is still in development, and we have not obtained clearance from the FDA to commercially distribute the device in the United States or clearance from Health Canada to commercially distribute the device in Canada, and we may never obtain such clearances.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of public and private equity offerings, debt financings, and license and development agreements through strategic partnerships with third parties. For example, we recently completed a license agreement and debt and equity financing arrangement with A&B. Under the agreements with A&B, we licensed the use of our intellectual property in Asia, and arranged for financing through the issuance of significant amounts of our common stock. We currently have no products approved for commercial distribution. We currently are dependent on the potential development of a single product which is our PoNS™ device for use in the neuromodulation market. We are still developing this product, and we cannot begin marketing and selling the device in the United States or Canada until we obtain clearances from the FDA or Health Canada, respectively. We have not yet submitted applications for regulatory clearance in the United States, Europe, or Canada. The process of obtaining regulatory clearance is expensive and time-consuming and can vary substantially based upon, among other things, the type, complexity and novelty of a product. Changes in regulatory policy, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application may cause delays in the clearance of a product candidate or rejection of a regulatory application altogether. The FDA has substantial discretion in the de novo review and clearance processes and may refuse to accept any application or may decide that our data are insufficient for clearance and require additional pre-clinical, clinical, or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit, or prevent marketing authorization from the FDA or regulatory clearance of a product candidate. Any marketing authorization from the FDA or regulatory clearance we ultimately obtain may be limited or subject to restrictions or post-market commitments that render the product candidate not commercially viable. If our attempts to obtain marketing authorization are unsuccessful, we may be unable to generate sufficient revenue to sustain and grow our business, and our business, financial condition, and results of operations will be materially adversely affected.
24
If we are able to complete development of the PoNS™ device and obtain clearance of the PoNS™ device for treatment of chronic balance deficit in patients with mild to moderate TBI or chronic gait and balance deficit associated with MS in the United States, Europe, or Canada, we plan to develop the PoNS™ device to treat other indications, or symptoms caused by neurological disorders. We would be required to commit our own resources to fund development of any other indications and each would require separate FDA clearance. The costs of such development efforts and FDA clearances would be substantial and would likely require additional funding, and each such indication would be subject to the same foregoing risks and uncertainties for FDA clearance.
We are and will continue to be dependent in significant part on outside scientists and third-party research institutions for our research and development in order to be able to commercialize our product candidate.
We currently have a limited number of employees and resources available to perform the research and development necessary to commercialize our PoNS™ device and potential future product candidates. We therefore rely at present, and will continue to rely on third-party research institution collaborators for this capability.
Our subsidiary, NHC, is currently party to the CRADA (as defined below) with the inventors, background patent owners and the Army Laboratories. Pursuant to the CRADA, the Army Laboratories agree to cooperate with NHC on research for the ongoing design and development to determine if the PoNS™ device can be developed for commercial use in assisting physical therapy in the treatment of soldiers and others with military relevant neurological disorders, including but not limited to Tinnitus, post-traumatic stress disorder, or PTSD, pain and any subsequent indications identified by the parties. Under the terms of the initial CRADA, we are solely responsible to fund and oversee clinical studies for the PoNS™ device and seek FDA clearance and approval of the PoNS™ device. We are also solely responsible to complete the research and development efforts necessary to commercialize our PoNS™ device. However, on July 7, 2015, we announced that NHC entered into a sole-source contractual agreement with USAMRMC to support the execution of the registration trial for treatment of balance disorder associated with mild to moderate TBI. The objective of this contract is to defray the costs of the registration trial. The Army Laboratories also agreed in the January 12, 2015 amendment to our CRADA to be responsible to support the execution of clinical studies for the PoNS™ device as a treatment for mutually agreed upon military relevant neurological disorders, which could include but not be limited to Tinnitus, PTSD, and pain and any subsequent indications identified by the parties. The amount of such support, if any, and the terms of such responsibility to support such clinical studies are not yet negotiated and we have no assurance that we can ultimately reach agreement with the Army Laboratories on such amount or terms of support, and there can be no assurance that the Army Laboratories will not otherwise attempt to renegotiate its responsibilities under the CRADA. The Army Laboratories may terminate their obligations under the CRADA at any time upon 30 days prior written notice to us. If there are insufficient funds available to cover the necessary research and development costs for our product, the Army Laboratories could terminate the CRADA and cease research and development efforts which could jeopardize our ability to commercialize our PoNS™ device.
25
If we fail to obtain FDA clearance for commercialization of or otherwise fail to ensure that the PoNS™ device is available for purchase by the U.S. Government by December 31, 2017, we are subject to significant risk of loss of data, proprietary rights and to certain contractual penalties.
Under the CRADA if we fail to obtain FDA clearance of the PoNS™ device or otherwise fail to ensure that the PoNS™ device is available for purchase by the U.S. Government, in each case by the expiration date under the CRADA of December 31, 2017, we may forfeit the right to pursue commercialization on our own. Specifically, in either such case, we will be required to (i) transfer possession, ownership and sponsorship of any regulatory application, and correspondence supporting the PoNS™ technology to the USAMRMC and (ii) provide the U.S. Government with a non-exclusive, irrevocable license to any patent, copyright, data rights, proprietary information and regulatory information, in order to permit the U.S. Government to pursue commercialization on its own. Any such loss of our ability to exclusively market and sell the PoNS™ device would have a material adverse effect on our business.
Additionally, under our Strategic Agreement with A&B if we fail to obtain FDA clearance for commercialization of or otherwise fail to ensure that the PoNS™ device is available for purchase by the U.S. Government by December 31, 2017, we are subject to a US$2,000,000 contract penalty payable to A&B.
We may encounter substantial delays in our clinical trials, or our clinical trials may fail to demonstrate the safety and efficacy of our product candidate to the satisfaction of applicable regulatory authorities.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidate, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidate. Clinical testing is expensive, time consuming and uncertain as to outcome. We cannot guarantee that clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Delays can be costly and could negatively affect our ability to complete a clinical trial.
There is limited market awareness of our product and the neuromodulation market is new and uncertain.
There is currently limited market awareness of our product. In order to succeed, we must among other things increase market awareness of our PoNS™ product and implement a sales and marketing strategy. If we fail in any of these endeavors or experience delays in pursuing them, we will not generate revenues as planned and will need to curtail operations or seek additional financing earlier than otherwise anticipated. In addition, should the neuromodulation market fail to expand, it could have a materially adverse effect on our business and financial position.
Our PoNS™ technology is a new “untested” form of neurostimulation therapy and the medical community tends not to adopt new therapies very rapidly, which may have a material adverse effect on our business and financial position.
The effectiveness of our PoNS™ technology to treat TBI or any other neurological disorder has not been established in studies conducted in a controlled environment designed to produce scientifically significant results. Accordingly, our PoNS™ technology is a new “untested”, and therefore unproven, therapy. Unproven and untested technologies are usually more slowly adopted by the medical community as the medical community tends to be very conservative and does not adopt new “untested” therapies very rapidly. Physicians may elect not to use our products for a variety of reasons, including:
| • | lack or perceived lack of evidence supporting the beneficial characteristics of our technology; | |
| • | limited long-term data on the use of PoNS™ technology for therapy; | |
| • | physicians’ perception that there are insufficient advantages of our product relative to currently available products; | |
| • | hospitals may choose not to purchase our product; | |
| • | group purchasing organizations may choose not to contract for our product, thus limiting availability of our products to hospital purchasers; | |
| • | lack of coverage or adequate payment from managed care plans and other third-party payers for our product; | |
| • | Medicare, Medicaid or other third-party payers may limit or not permit reimbursement for our product; and | |
| • | the development of or improvement of competitive products. |
If the medical community reacts in a similar fashion to adopting our PoNS™ device for neurostimulation therapy, we will not be able to generate significant revenues, if any.
26
In order to be successful, we must expand our products beyond our single product by commercializing new potential product candidates, but we may not be able to do so in a timely fashion and at expected costs, or at all.
In order to be successful, we will need to expand our product lines beyond our PoNS™ device which is currently our only product. To succeed in our commercialization efforts, we must effectively continue product development and testing, obtain regulatory clearances and approvals, and enhance our sales and marketing capabilities. There is no assurance that we will succeed in bringing any of our current or potential future product candidates to market. If we fail in bringing our product candidate to market, or experience delays in doing so, we will not generate revenues as planned and will need to curtail operations or seek additional financing earlier than otherwise anticipated.
The development of additional products is subject to the risks of failure inherent in the development of new, state of the art products, laboratory devices and products based on new technologies. These risks include: (a) delays in product development or manufacturing; (b) unplanned expenditures for product development or manufacturing; (c) failure of new products to have the desired effect or an acceptable accuracy profile; (d) emergence of superior or equivalent products; (e) failure by any potential collaborative partners to successfully develop products; and (f) the dependence on third parties for the manufacture, development and sale of our products. Because of these risks, our research and development efforts or those of potential collaborative partners may not result in any commercially viable products. If a significant portion of these development efforts is not successfully completed, or any products are not commercially successful, we are less likely to generate significant revenues, or become profitable. The failure to perform such activities could have a material adverse effect on our business, financial condition and results of its operations.
We can provide no assurance that the development by others of new or improved devices or products will not result in our present and future products from becoming obsolete.
The areas in which we plan to commercialize, distribute, and/or sell products involves rapidly developing technology. There can be no assurance that we will be able to establish ourselves in such fields, or, if established, that we will be able to maintain our market position, if any. There can be no assurance that the development by others of new or improved products will not make our present and future products, if any, superfluous or obsolete.
Our future success depends on our ability to obtain approval on the patent for the PoNS™ technology, failing which we may be unable to protect our proprietary information and any competitive advantage which may have a material adverse effect on our business and financial condition.
Our future success will depend, in part, on our ability to obtain patent approval for the PoNS™ technology. There can be no assurance that the patent applications made will result in the issuance of patents or that the term of the patents will be extendable after they expire in due course, which would prevent us from being able to protect our proprietary information and may have a material adverse effect on our business and financial condition.
Much of our know-how and technology may not be patentable, though they may constitute trade secrets. There can be no assurance, however, that we will be able to meaningfully protect our trade secrets. To help protect our intellectual property rights and proprietary technology, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance that these agreements will provide meaningful protection for our trade secrets, knowhow or other proprietary information in the event of any unauthorized use or disclosure.
Our intellectual property has been and may be the subject of lawsuits. See Part I Item 3,“Legal Proceedings.”
If our intellectual property protection is inadequate, competitors may gain access to our technology and undermine our competitive position.
We regard our intended and future intellectual property as important to our success, and we intend to rely on patent law to protect our proprietary rights. Despite our precautions, unauthorized third parties may copy certain portions of our devices or products or reverse engineer or obtain and use information that we regard as proprietary. We may seek additional patents in the future. We do not know if any future patent application will be issued with the scope of the claims we seek, if at all, or whether any patents we receive will be challenged or invalidated. Thus, we cannot assure you that any intellectual property rights that we may receive can be successfully asserted in the future or that they will not be invalidated, circumvented or challenged. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as do the laws of the United States. Our means of protecting any proprietary rights we may receive in the United States or abroad may not be adequate and competitors may independently develop a similar technology. Any failure to protect our proprietary information and any successful intellectual property challenges or infringement proceedings against us could have a material adverse effect on our business, financial condition, or results of operations.
27
We may be subject to various litigation claims and legal proceedings, including intellectual property litigation, such as patent infringement claims, which could adversely affect our business.
We, as well as certain of our directors and officers, may be subject to claims or lawsuits. These lawsuits may result in significant legal fees and expenses and could divert management’s time and other resources. If the claims contained in these lawsuits are successfully asserted against us, we could be liable for damages and be required to alter or cease certain of our business practices or product lines. Any of these outcomes could cause our business, financial performance and cash position to be negatively impacted.
Additionally, our commercial success will also depend, in part, on not infringing on the patents or proprietary rights of others. There can be no assurance that the technologies and products used or developed by us will not infringe such rights. If such infringement occurs and we are not able to obtain a license from the relevant third party, we will not be able to continue the development, manufacture, use, or sale of any such infringing technology or product. There can be no assurance that necessary licenses to third-party technology will be available at all or on commercially reasonable term. In some cases, litigation or other proceedings may be necessary to defend against or assert claims of infringement or to determine the scope and validity of the proprietary rights of third parties. Any potential litigation could result in substantial costs to, and diversion of, our resources and could have a material and adverse impact on us.
An adverse outcome in any such litigation or proceeding could subject us to significant liabilities, require us to cease using the subject technology or require us to license the subject technology from the third party, all of which could have a material adverse effect on our business.
On January 5, 2015, Wicab sued the Company, NHC, Mitch Tyler, a director of the Company and NHC and Yuri Danilov, a former director of the Company and a director of NHC, and ANR, in the U.S. District Court for the Western District of Wisconsin. ANR is the licensor to the Company of three issued patents (U.S. Patent Nos. 8,849,407 and 8,909,345 and 9,020,612) and other patents pending related to neurostimulation methods and devices. The complaint contained various state and common law claims arising from Messrs. Danilov’s and Tyler’s prior employment with Wicab and relating to ownership of two of the issued patents (U.S. Patent Nos. 8,849,407 and 8,909,345). U.S. Patent No. 9,020,612 was not included in the Wicab complaint. The complaint alleged, among other things, that following their departure from Wicab, Danilov and Tyler knowingly filed patent applications for and used ideas and inventions developed at Wicab in violation of various non-competition and confidentiality agreements, and that the two issued patents are therefore rightfully the property of Wicab. The complaint sought an unspecified amount of monetary damages, an injunction preventing NHC from using the ideas and inventions in the two patents, an order transferring ownership of the patents from ANR to Wicab, and recovery of costs and attorneys’ fees. The Company conducted an internal investigation and determined that Wicab expressly waived all rights in the two issued patents and, additionally, that Wicab’s claims were barred by the six year statute of limitations in Wisconsin. On January 14, 2015, the Company informed Wicab of its belief that the claims were barred due to the express waiver and the statute of limitations. On the same day, Wicab dismissed the complaint without prejudice.
On October 12, 2015, the Company received a letter from Wicab alleging that the two issued patents were invalid in view of prior art cited in the letter, including scientific publications and patent applications, and that Paul Bach-y-Rita, Wicab’s founder, should have been named as an inventor on these two issued patents. Wicab indicated in the letter that it may file reexamination or inter partes review proceedings with the U.S. Patent Office to attempt to invalidate the claims in the two issued patents. Wicab also stated that it would consider an unspecified “business solution” to resolve this matter. On December 10, 2015, representatives of each of the Company and Wicab met to discuss the parameters of a potential settlement. As at the date of this filing, these discussions are ongoing and there can be no guarantee that a settlement will be reached. In the event that a settlement with Wicab is not reached, Wicab may file reexamination or inter partes review proceedings with the U.S. Patent Office to challenge the validity of the two issued patents. If the Company receives an adverse decision from the U.S. Patent Office in connection with these proceedings, some or all of the claims in the two patents may be invalidated or otherwise impaired, which could prevent the Company from bringing an infringement suit against a future competitor for making use of the PoNS™ technology for neurorehabilitation, and could have a material adverse effect on the Company’s business, operating results and financial condition. Wicab may also take other actions against the Company, its assets, intellectual property rights, officers, directors, employees, agents or other persons or entities which may also have a material on business, operating results and financial condition.
28
See Part I Item 3, “Legal Proceedings.”
If our expenses are greater than anticipated, then we will have fewer funds with which to pursue our plan of operations and our financing requirements will be greater than anticipated.
We may find that the costs of carrying out our plan of operations are greater than we anticipate. Increased operating costs may cause the amount of financing that we require to increase. Investors may be more reluctant to provide additional financing if we cannot demonstrate that we can control our operating costs. There is no assurance that additional financing required as a result of our operating costs being greater than anticipated will be available to us. If we do not control our operating expenses, then we will have fewer funds with which to carry out our plan of operations with the result that our business may fail.
Our ability to use net operating losses to offset future taxable income may be subject to certain limitations.
Under Section 382 of the Internal Revenue Code of 1986, as amended, substantial changes in a corporation’s ownership may limit the amount of net operating losses (“NOL”s) that can be utilized annually in the future to offset the corporation’s (and the corporation’s affiliates’) U.S. federal and state taxable income. Specifically, this limitation may arise in the event of a cumulative change in ownership of more than 50% within any three-year period. The amount of the annual limitation is determined based on the value of the corporation that underwent the ownership change, immediately before the ownership change. Subsequent ownership changes may further affect any limitation in future years (including by the way of exercising of warrants). We plan to undertake a study to analyze and determine if any historical ownership changes of us or our subsidiary NHC have occurred to determine if there are any permanent limitations on our ability to utilize NOLs in the future. If we determine that an ownership change has occurred, the limitations on the use of our NOLs could increase our U.S. federal and state tax liability and reduce the amount of cash available for distribution to shareholders or otherwise adversely affect the value of an investment in our common stock or Warrants.
We may not be able to build an effective distribution network for our products.
We currently have very few employees and will likely need to rely on third party distributors to sell our product. We cannot assure you that we will succeed in entering into and maintaining productive arrangements with an adequate number of distributors that are sufficiently committed to selling our products. The establishment of a distribution network is expensive and time consuming. As we launch new products and increase our marketing effort with respect to existing products, we will need to continue to hire, train, retain and motivate skilled independent distributors with significant technical knowledge. In addition, the commissions we pay our distributors could increase over time which would result in higher sales and marketing expenses. Furthermore, current and potential distributors may market and sell the products of our competitors. Even if the distributors market and sell our products, our competitors may be able, by offering higher commission payments or other incentives, to persuade these distributors to reduce or terminate their sales and marketing efforts related to our products. The distributors may also help competitors solicit business from our existing customers. Some of our independent distributors will likely account for a significant portion of our sales volume, and, if we were to lose them, our sales could be adversely affected. Even if we engage and maintain suitable relationships with an adequate number of distributors, they may not generate revenue as quickly as we expect them to, commit the necessary resources to effectively market and sell our products, or ultimately succeed in selling our products.
We depend on a single source for the manufacture of our product and the loss of this third-party manufacture could harm our business.
We will be dependent on a single third-party to manufacture and supply our PoNS™ device. This manufacturer will also hold some inventory and ship our products to our distribution center who will hold the bulk of our inventory, warehouse and ship our products to customers as well as handle customer service related tasks. Our reliance on a single third-party manufacturer to supply us with our PoNS™ device and a separate vendor to provide such other distribution and warranty services exposes us to risks that could delay our sales, or result in higher costs or lost product revenues. In particular, our manufacturer could:
| • | encounter difficulties in achieving volume production, quality control and quality assurance or suffer shortages of qualified personnel, which could result in their inability to manufacture sufficient quantities of our commercially available product to meet market demand, or it could experience similar problems that result in the manufacture of insufficient quantities of our product candidate; and |
29
| • | fail to follow and remain in compliance with the FDA-mandated QSRs, compliance which is required for all medical devices, or fail to document their compliance to QSRs, either of which could lead to significant delays in the availability of materials for our product. |
If we are unable to obtain adequate supplies of our product that meet our specifications and quality standards, it will be difficult for us to compete effectively. We have no supply agreements in place with our manufacturer and it may change the terms of our future orders or choose not to supply us with products in the future. Furthermore, if such manufacturer fails to perform its obligations, we may be forced to purchase our product from other third-party manufacturers, which we may not be able to do on reasonable terms or in sufficient time, if at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer or the reverification of an existing manufacturer could negatively affect our ability to produce and distribute our product in a timely manner.
If the U.S. Army were to decide not to purchase our product or chose to no longer provide financial support for our clinical testing through the sole-source cost sharing contract we would face risks related to finding new partners or customers.
The U.S. Army is under no obligation to purchase the PoNS™ device from us and there is no assurance that the U.S. Army will ultimately purchase the Company’s product. Given the importance of the U.S. Army to our commercial plans, if the U.S. Army were to eventually decide not to purchase our product, we would need to find other buyers for our product. If the U.S. Army were to decline to purchase our product, we may have more difficulty persuading other third parties to purchase our product. Additionally, through our subsidiary NHC, we are party to a sole source cost sharing contract with the USAMRMC. Under the contract, the USAMRMC will reimburse the Company for costs related to a registrational trial investigating the safety and effectiveness of the PoNS™ device up to a maximum amount of $2,996,244. The contract expires on December 31, 2016, however, the Company is working with the USAMRMC to extend the contract into 2017 based on the current trial forecast timelines . If we fail to complete the registrational trial or renew the contract by that time we face the risk of needing to find additional financial support for the trial.
If and when we sell our products, we may be liable for product liability claims and we may not carry sufficient product liability insurance.
The devices and products that we intend to develop may expose us to potential liability from personal injury claims by end-users of the product. We intend to carry product liability insurance to protect us against the risk that in the future a product liability claim or product recall could materially and adversely affect our business. Inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our intended products. We cannot assure you that if and when we commence distribution of our product that we will be able to obtain or maintain adequate coverage on acceptable terms, or that such insurance will provide adequate coverage against all potential claims. Moreover, even if we maintain adequate insurance, any successful claim could materially and adversely affect our reputation and prospects, and divert management’s time and attention. If we are sued for any injury allegedly caused by our future products our liability could exceed our total assets and our ability to pay the liability.
We are an “emerging growth company” under the Jumpstart Our Business Startups Act of 2012, or JOBS Act, and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act. As an “emerging growth company”, we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of section 404 of the Sarbanes-Oxley Act of 2002, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation, shareholder approval of any golden parachute payments not previously approved and presenting the relationship between executive compensation actually paid and our financial performance. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. Additionally, we have irrevocably elected to comply with new or revised accounting standards even though we are an emerging growth company.
30
We will remain an “emerging growth company” for up to five years after our first sale of common stock pursuant to a Securities Act of 1933, as amended, or the Securities Act, registration statement, although we will lose that status sooner if our revenues exceed $1 billion, if we issue more than $1 billion in non-convertible debt in a three year period, or if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the end of our third quarter in any calendar year.
Our status as an “emerging growth company” under the JOBS Act may make it more difficult to raise capital as and when we need it. Because of the exemptions from various reporting requirements provided to us as an “emerging growth company”, we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
We are a small company with limited resources compared to some of our current and potential competitors and we may not be able to compete effectively and increase market share.
There is potential that we will face intense competition from other companies, some of which can be expected to have longer operating histories and more financial resources and manufacturing and marketing experience than us. Increased competition by larger and better financed competitors could materially and adversely affect our business, financial condition and our results of operations.
Because of the early stage of the industry in which we intend to operate, we expect to face additional competition from new entrants. To be competitive, we will require a continued high level of investment in research and development, marketing, sales and client support. We may not have sufficient resources to maintain research and development, marketing, sales and client support efforts on a competitive basis which could materially and adversely affect our business, financial condition and our results of operations.
We have incurred increased costs and have become subject to additional regulations and requirements as a result of becoming a public company, which could lower our profits, if any, or make it more difficult to run our business.
As a public company, we have incurred significant legal, accounting and other expenses that we did not incur as a private company, including costs associated with public company reporting requirements. We will continue to incur costs associated with the rules implemented by the SEC, the TSX, the OTCQB, and any other exchange on which our common stock may become listed. The expenses incurred by public companies for reporting and corporate governance purposes have generally been increasing. These rules and regulations have increased our legal and financial compliance costs and have made some activities more time-consuming and costly. These laws and regulations also could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. These laws and regulations could also make it more difficult for us to attract and retain qualified persons to serve on our Board of Directors, our board committees or as our executive officers. Furthermore, if we are unable to satisfy our obligations as a public company, we could be subject to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
Several people who work for us on a part-time consulting basis may be subject to conflicts of interest.
Several people who provide services to us do so on a part-time consulting basis. Each may devote part of his working time to other business endeavors, including consulting relationships with other corporate entities, and may have responsibilities to these other entities. Because of these relationships, some of the persons who provide services to us may be subject to conflicts of interest. Such conflicts may include deciding how much time to devote to our affairs, as well as what business opportunities should be presented to us.
31
Risks Related to Government Regulation
Before we can market and sell our products, we will be required to obtain approval and clearance by the FDA and foreign regulatory authorities. These approvals and clearances will take significant time and require significant research, development, and clinical study expenditures, and ultimately may not succeed.
Before we begin to label and market the PoNS™ device for use in the United States, we are required to obtain clearance from the FDA under Section 510(k) of the FD&C Act, approval of a de novo reclassification petition for our product, or approval of pre-market approval application from the FDA, unless an exemption from pre-market review applies. We intend to utilize the de novo classification procedures to seek marketing authorization for the PoNS™ device, because there is currently no predicate cleared or approved by the FDA for commercial distribution and no existing classification decision by the FDA for such a device. We will also be required to comply with costly and time-consuming compliance by foreign regulatory authorities if we want to sell our products outside of the United States. The process of obtaining regulatory clearances or approvals, or completing the de novo classification process, to market a medical device can be costly and time consuming, and we may not be able to successfully obtain pre-market reviews on a timely basis, if at all.
If the FDA requires us to go through a lengthier, more rigorous examination for the PoNS™ device, introducing the product could be delayed or canceled, which could cause our launch to be delayed. In addition, the FDA may determine that the PoNS™ device requires the more costly, lengthy and uncertain pre-market approval process. For example, if the FDA disagrees with our determination that the de novo classification procedures are the appropriate path to obtain marketing authorizations for the PoNS™ device, the FDA may require us to submit a PMA application, which is generally more costly and uncertain and can take from one to three years, or longer, from the time the application is submitted to the FDA until an approval is obtained.
Further, even with respect to those future products where a PMA is not required, we cannot be certain that we will be able to obtain 510(k) clearances with respect to those products.
Obtaining FDA clearance will be costly, may result in time-consuming delays and will subject us to ongoing compliance costs and regulatory risk for non-compliance.
Obtaining FDA clearance, de novo down-classification, or approval for medical devices can be expensive and uncertain, and generally takes from several months to several years, and generally requires detailed and comprehensive scientific and clinical data. Notwithstanding the expense, these efforts may never result in FDA clearance. Even if we were to obtain regulatory clearance, it may not be for the uses we believe are important or commercially attractive, in which case we would not be permitted to market our product for those uses.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| • | we may not be able to demonstrate to the FDA’s satisfaction that our product candidate is safe and effective, sensitive and specific diagnostic tests, for its intended users; | |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and | |
| • | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis. For example, in response to industry and healthcare provider concerns regarding the predictability, consistency and rigor of the 510(k) regulatory pathway, the FDA initiated an evaluation of the program, and in January 2011, announced several proposed actions intended to reform the review process governing the clearance of medical devices. The FDA intends these reform actions to improve the efficiency and transparency of the clearance process, as well as bolster patient safety. In addition, as part of the FDASIA the U.S. Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms which are further intended to clarify and improve medical device regulation both pre- and post-approval. Any delay in, or failure to receive or maintain, clearance or approval for our product candidate could prevent us from generating revenue from our product candidate and adversely affect our business operations and financial results.
Even if granted, a 510(k) clearance, de novo down-classification, or pre-market approval for any future product would likely place substantial restrictions on how our device is marketed or sold, and FDA will continue to place considerable restrictions on our products and operations. For example, the manufacture of medical devices must comply with FDA’s QSR. In addition, manufacturers must register their manufacturing facilities, list the products with FDA, and comply with requirements relating to labeling, marketing, complaint handling, adverse event and medical device reporting, reporting of corrections and removals, and import and export. FDA monitors compliance with the QSR and these other requirements through periodic inspections. If our facilities or those of our manufacturers or suppliers are found to be in violation of applicable laws and regulations, or if we or our manufacturers or suppliers fail to take satisfactory corrective action in response to an adverse inspection, the regulatory authority could take enforcement action, including any of the following sanctions:
32
|
|
• |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
|
• |
customer notifications of repair, replacement, refunds, detention or seizure of our products |
|
|
• |
operating restrictions or partial suspension or total shutdown of production; |
|
|
• |
refusing or delaying requests for 510(k) marketing clearance or pre-market approvals of new products or modified products; |
|
|
• |
withdrawing 510(k) marketing clearances or pre-market approvals that have already been granted; |
|
|
• |
refusing to provide Certificates for Foreign Government; |
|
|
• |
refusing to grant export approval for our products; or |
|
|
• |
pursuing criminal prosecution |
Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could affect the perceived safety and efficacy of our product candidate and dissuade our customers from using our product candidate, if and when it is authorized for marketing.
We expect to be required to conduct clinical trials to support regulatory approval of some of our potential future product candidates. We have limited experience in the clinical trials process, they may proceed more slowly than anticipated, and we cannot be certain that our product candidate will be shown to be safe and effective for human use.
In order to commercialize our product candidate in the United States, we may be required by the FDA to submit an application for PMA for review and approval by the FDA. A PMA application must be submitted to the FDA if our device cannot be cleared through the 510(k) clearance process or is not exempt from premarket review by the FDA. We could also be required to submit a PMA application for other potential future product candidates. If we are required by the FDA to submit a PMA application, the FDA will also require us to conduct clinical trials. The FDA could also require us to provide the FDA with clinical trial data to support some of our 510(k) premarket notifications. We will receive approval or clearance from the FDA to commercialize products requiring a clinical trial only if we can demonstrate to the satisfaction of the FDA, through well-designed and properly conducted clinical trials, that our product candidate is safe and effective and otherwise meet the appropriate standards required for approval or clearance for specified indications.
Clinical trials are complex, expensive, time consuming, uncertain and are subject to substantial and unanticipated delays. Before we may begin clinical trials, we must submit and obtain approval for an investigational device exemption, or IDE, that describes, among other things, the manufacture of, and controls for, the device and a complete investigational plan. Clinical trials generally involve a substantial number of patients in a multi-year study. Because we do not have the experience or the infrastructure necessary to conduct clinical trials, we will have to hire one or more contract research organizations, or CROs, to conduct trials on our behalf. CRO contract negotiations may be costly and time consuming and we will rely heavily on the CRO to ensure that our trials are conducted in accordance with regulatory and industry standards. We may encounter problems with our clinical trials and any of those problems could cause us or the FDA to suspend those trials, or delay the analysis of the data derived from them.
A number of events or factors, including any of the following, could delay the completion of our clinical trials in the future and negatively impact our ability to obtain FDA approval for, and to introduce our product candidate:
| • |
failure to obtain financing necessary to bear the cost of designing and conducting clinical trials; | |
| • |
failure to obtain approval from the FDA or foreign regulatory authorities to commence investigational studies; | |
| • |
conditions imposed on us by the FDA or foreign regulatory authorities regarding the scope or design of our clinical trials; | |
| • |
failure to find a qualified CRO to conduct our clinical trials or to negotiate a CRO services agreement on favorable terms; | |
| • |
delays in obtaining or in our maintaining required approvals from institutional review boards or other reviewing entities at clinical sites selected for participation in our clinical trials; | |
| • |
insufficient supply of our product candidate or other materials necessary to conduct our clinical trials; | |
| • |
difficulties in enrolling patients in our clinical trials; |
33
|
|
• |
negative or inconclusive results from clinical trials, or results that are inconsistent with earlier results, that necessitate additional clinical studies; |
|
|
• |
failure on the part of the CRO to conduct the clinical trial in accordance with regulatory requirements; |
|
|
• |
our failure to maintain a successful relationship with the CRO or termination of our contractual relationship with the CRO before completion of the clinical trials; |
|
|
• |
serious or unexpected side effects experienced by patients in whom our product candidate are implanted; or |
|
|
• |
failure by any of our third-party contractors or investigators to comply with regulatory requirements or meet other contractual obligations in a timely manner. |
Our clinical trials may need to be redesigned or may not be completed on schedule, if at all. Delays in our clinical trials may result in increased development costs for our product candidate, which could cause our stock price to decline and limit our ability to obtain additional financing. In addition, if one or more of our clinical trials are delayed, competitors may be able to bring products to market before we do, and the commercial viability of our product candidate could be significantly reduced.
We will be substantially dependent on third parties to conduct clinical trials.
As we are required to conduct clinical trials to obtain FDA clearance, we need to rely heavily on third parties over the course of our clinical trials, and as a result will have limited control over the clinical investigators and limited visibility into their day-to-day activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol and legal, regulatory, and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and these third parties are required to comply with current good clinical practices, or cGCPs, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for product candidates in clinical development. Regulatory authorities enforce these cGCPs through periodic inspections of trial sponsors, principal investigators, and trial sites. If we or any of these third parties fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional nonclinical or clinical trials before approving our marketing applications. We cannot be certain that, upon inspection, such regulatory authorities will determine that any of our clinical trials comply with the cGCP regulations. In addition, our clinical trials may be required to be conducted with a large number of test patients. Our failure or any failure by these third parties to comply with these regulations or to recruit a sufficient number of patients may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, our business may be implicated if any of these third parties violates federal or state fraud and abuse or false claims laws and regulations or healthcare privacy and security laws.
Any third parties conducting our clinical trials are not and will not be our employees and, except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our ongoing preclinical, clinical, and nonclinical programs. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other drug development activities, which could affect their performance on our behalf. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed, or terminated and we may not be able to complete development of, obtain regulatory approval of or successfully commercialize our product candidate. As a result, our financial results and the commercial prospects for our product candidate would be harmed, our costs could increase, and our ability to generate revenue could be delayed.
If any of our relationships terminate with these third-party CROs, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO begins work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition, and prospects.
34
If we are unable to obtain a reimbursement code from the U.S. Department of Health and Human Services so that the PoNS™ device is covered under Medicare and Medicaid, this would have a negative impact on our intended sales and would have a material adverse effect on our business, financial condition and operating results.
We plan to submit an application to the U.S. Department of Health and Human Services for reimbursement code so that the PoNS™ device is covered under Medicare and Medicaid. There can be no assurance that our application will be successful, or that we will be able to obtain a reimbursement code in a timely manner. In the event that we do not obtain a reimbursement code for the PoNS™ device, our customers may be unable to obtain reimbursement for their purchases under private or government-sponsored insurance plans which would have a negative impact on sales and have a material adverse effect on our business, financial condition and operating results. In addition, Medicare and its administrative contractors as well as other insurers must find that the PoNS™ device meets their medical necessity requirements for the treatment of patients or they will not pay for the device. In addition, there is a risk that the payment amount for the PoNS™ device is too low to incentivize customer adoption.
If hospitals and other healthcare providers are unable to obtain coverage or adequate reimbursement for procedures performed with our products, our product will not likely be widely used.
In the United States, the commercial success of our existing product and any future products will depend, in part, on the extent to which governmental payers at the federal and state levels, including Medicare and Medicaid, private health insurers and other third-party payers provide coverage for and establish adequate reimbursement levels for procedures utilizing our products. Hospitals and other healthcare providers that purchase our product for treatment of their patients generally rely on third-party payers to pay for all or part of the costs and fees associated with our products as part of a “bundled” rate for the associated procedures. The existence of coverage and adequate reimbursement for our products and the procedures performed with them by government and private payers critical to market acceptance of our existing and future products. Neither hospitals nor physicians are likely to use our product and any future products if they do not receive adequate reimbursement for the procedures utilizing our products.
Many private payers currently base their reimbursement policies on the coverage decisions and payment amounts determined by the CMS, which administers the Medicare program. Others may adopt different coverage or reimbursement policies for procedures performed with our products, while some governmental programs, such as Medicaid, have reimbursement policies that vary from state to state, some of which may not pay for the procedures performed with our products in an adequate amount, if at all. A Medicare national or local coverage decision denying coverage for one or more of our products could result in private and other third-party payers also denying coverage for our products. Third-party payers also may deny reimbursement for our products if they determine that a product used in a procedure was not medically necessary, was not used in accordance with cost-effective treatment methods, as determined by the third-party payer, or was used for an unapproved use. Unfavorable coverage or reimbursement decisions by government programs or private payers underscore the uncertainty that our products face in the market and could have a material adverse effect on our business.
Many hospitals and clinics in the United States belong to group purchasing organizations, which typically incentivize their hospital members to make a relatively large proportion of purchases from a limited number of vendors of similar products that have contracted to offer discounted prices. Such contracts often include exceptions for purchasing certain innovative new technologies, however. Accordingly, the commercial success of our products may also depend to some extent on our ability to either negotiate favorable purchase contracts with key group purchasing organizations and/or persuade hospitals and clinics to purchase our product “off contract.”
The healthcare industry in the United States has experienced a trend toward cost containment as government and private payers seek to control healthcare costs by paying service providers lower rates. While we believe that hospitals will be able to obtain coverage for procedures using our products, the level of payment available to them for such procedures may change over time. State and federal healthcare programs, such as Medicare and Medicaid, closely regulate provider payment levels and have sought to contain, and sometimes reduce, payment levels. Private payers frequently follow government payment policies and are likewise interested in controlling increases in the cost of medical care. In addition, some payers are adopting pay-for-performance programs that differentiate payments to healthcare providers based on the achievement of documented quality-of-care metrics, cost efficiencies, or patient outcomes. These programs are intended to provide incentives to providers to deliver the same or better results while consuming fewer resources. As a result of these programs, and related payer efforts to reduce payment levels, hospitals and other providers are seeking ways to reduce their costs, including the amounts they pay to medical device manufacturers. We may not be able to sell our implants profitably if third-party payers deny or discontinue coverage or reduce their levels of payment below that which we project, or if our production costs increase at a greater rate than payment levels. Adverse changes in payment rates by payers to hospitals could adversely impact our ability to market and sell our products and negatively affect our financial performance.
35
In international markets, medical device regulatory requirements and healthcare payment systems vary significantly from country to country, and many countries have instituted price ceilings on specific product lines. We cannot assure you that our products will be considered cost-effective by international third-party payers, that reimbursement will be available or, if available, that the third-party payers’ reimbursement policies will not adversely affect our ability to sell our products profitably. Any failure to receive regulatory or reimbursement approvals would negatively impact market acceptance of our products in any international markets in which those approvals are sought.
Risks Related to Our Common Stock
A decline in the price of our common stock could affect our ability to raise any required working capital and adversely impact our operations.
A decline in the price of our common stock could result in a reduction in the liquidity of our common stock and a reduction in our ability to raise any required capital for our operations. Because our operations to date have been principally financed through the sale of equity securities, a decline in the price of our common stock could have an adverse effect upon our liquidity and our continued operations. A reduction in our ability to raise equity capital in the future may have a material adverse effect upon our business plan and operations. If our stock price declines, we may not be able to raise additional capital or generate funds from operations sufficient to meet our obligations.
Our common stock does not have a well-established trading market in the United States. Trading of our common stock is sporadic, and the price of our common stock may be volatile; we caution you as to the highly illiquid nature of an investment in our shares.
Our common stock is currently periodically quoted on the OTCQB electronic quotation service operated by OTC Markets Group Inc. A well-established market for our common stock may never develop in the United States. Trading in stock quoted on the OTCQB is often thin and characterized by wide fluctuations in trading prices, due to many factors that may have little to do with our operations or business prospects. This volatility could depress the market price of our common stock for reasons unrelated to operating performance or future prospects of our business. Moreover, the OTCQB is not a stock exchange, and trading of securities on the OTCQB is often more sporadic than the trading of securities listed on a quotation system like NASDAQ or a stock exchange like Amex. Accordingly, shareholders may have difficulty reselling any of the shares.
Our common stock has been listed on the TSX since April 18, 2016. Certain shares of our common stock are also restricted for immediate resale to U.S. persons or to anyone for the account or on behalf of any U.S. person, pursuant to the requirements of Regulation S. These shares are traded separately on the TSX under a separate ticker symbol. To date, trading on the TSX in our common stock has been extremely limited and sporadic. Trading in our common stock on the CSE was also extremely limited.
Our Warrants were also approved for listing on the TSX on April 18, 2016. However, because only the Warrants issued in the Offshore Offering in transactions exempt from the registration requirements of the Securities Act were approved for listing on the TSX, the Warrants listed on the TSX may not be purchased by or on behalf of a U.S. person, or by a person in the United States, unless in a registered transaction or pursuant to an applicable safe harbor or exemption from registration.
Securities of microcap and small-cap companies have experienced substantial volatility in the past, often based on factors unrelated to the companies’ financial performance or prospects. We believe that trading in our stock, if it occurs at all, will likely be subject to significant volatility since, among other reasons, we do not have nor will we have in the foreseeable future an active trading market in our stock. These factors include macroeconomic developments in North America and globally and market perceptions of the attractiveness of particular industries. Factors unrelated to our performance that may affect the price of our common stock include the following: the extent of analytical coverage available to investors concerning our business may be limited if investment banks with research capabilities do not follow us, a reduction in trading volume and general market interest in our common stock may affect an investor’s ability to trade significant numbers of shares of our common stock; the size of our public float may limit the ability of some institutions to invest in our common stock; and a substantial decline in the price of shares of our common stock that persists for a significant period of time could cause our common stock, if listed on an exchange, to be delisted from such exchange, further reducing market liquidity. As a result of any of these factors, the market price of our common stock at any given point in time may not accurately reflect our long-term value. The price of our common shares may increase or decrease in response to a number of events and factors, including: changes in financial estimates; our acquisitions and financings; quarterly variations in our operating results; the operating and share price performance of other companies that investors may deem comparable; and purchase or sale of blocks of our common stock. These factors, or any of them, may materially adversely affect the prices of our common shares regardless of our operating performance. We caution you as to the highly illiquid nature of an investment in our shares.
36
The market price of our common stock is affected by many other variables which are not directly related to our success and are, therefore, not within our control. These include other developments that affect the breadth of the public market for shares of our common stock and the attractiveness of alternative investments. The effect of these and other factors on the market price of our common stock is expected to make our common stock price volatile in the future, which may result in losses to investors.
We have not voluntarily implemented various corporate governance measures, in the absence of which, shareholders may have more limited protections against interested director transactions, conflicts of interest and similar matters.
Federal legislation, including the Sarbanes-Oxley Act of 2002, has resulted in the adoption of various corporate governance measures designed to promote the integrity of the corporate management and the securities markets. Some of these measures have been adopted in response to legal requirements. Others have been adopted by companies in response to the requirements of national securities exchanges, such as the NYSE or the Nasdaq Stock Market, on which their securities are listed. Among the corporate governance measures that are required under the rules of national securities exchanges are those that address board of directors’ independence, and audit committee oversight. We have not yet adopted many of these corporate governance measures, including
|
|
• |
the requirement that our board of directors be composed of a majority of independent directors; and |
|
|
• |
the requirement that we have a nominating and corporate governance committee, a compensation committee and an audit committee composed entirely of independent directors, with written charters addressing the committees’ purpose and responsibilities. |
It is possible that if we were to adopt some or all of these corporate governance measures, stockholders would benefit from somewhat greater assurances that internal corporate decisions were being made by disinterested directors and that policies had been implemented to define responsible conduct. Investors should bear in mind our current lack of corporate governance measures in formulating their investment decisions.
Our shares are subject to potential delisting if we do not meet or continue to maintain the listing requirements of the TSX.
The TSX rules for continued listing include minimum market capitalization and other requirements. Failure to maintain our listing on the TSX or being de-listed from the TSX would make it more difficult for shareholders to dispose of our common stock and more difficult to obtain accurate quotations on our common stock. This could have an adverse effect on the price of our common stock. Our ability to issue additional securities for financing or other purposes, or to otherwise arrange for any financing we may need in the future, may also be materially and adversely affected if our common stock is not traded on a national securities exchange.
The market price of our common stock is likely to be highly volatile and subject to wide fluctuations, and you may be unable to resell your shares at or above the price at which you acquired them, or at all.
The market price of our common stock is likely to be highly volatile and could be subject to wide fluctuations in response to a number of factors that are beyond our control, including, but not limited to:
| • | quarterly variations in our revenues and operating expenses; | |
| • | developments in the financial markets and worldwide or regional economies; | |
| • | announcements of innovations or new products or services by us or our competitors; | |
| • | announcements by the government relating to regulations that govern our industry; | |
| • | significant sales of our common stock or other securities in the open market; | |
| • | variations in interest rates; | |
| • | changes in the market valuations of other comparable companies; and | |
| • | changes in accounting principles. |
37
In the past, stockholders have often instituted securities class action litigation after periods of volatility in the market price of a company’s securities. If a stockholder were to file any such class action suit against us, we would incur substantial legal fees and our management’s attention and resources would be diverted from operating our business to respond to the litigation, which could harm our business.
Our two major shareholders have the ability to take shareholder action without the involvement of our other shareholders.
In accordance with our governing documents, any action required to be taken at a shareholders' meeting may be taken without a meeting if consents in writing setting forth the action so taken are signed by the holders of our outstanding shares having not less than the minimum number of votes that would be required to authorize or take the action at a meeting at which all shares entitled to vote on the action were present and voted. Currently, our two major shareholders, MPJ Healthcare, LLC (“MPJ”) and ANR, hold approximately 39% of our outstanding shares of common stock. Philippe Deschamps, our Chief Executive Officer, and Jonathan Sackier, our Chief Medical Officer, each serve on the board of members of MPJ.
Our two major shareholders may have the ability to take shareholder action at a shareholders' meeting even if they do not hold a majority of our outstanding common stock.
As long as our two major shareholders, MPJ and ANR, collectively hold at least 33 1/3% of our outstanding common stock, they may be able to effect a vote requiring shareholder approval. In accordance with our governing documents, shareholders holding at least five percent of all the votes entitled to be cast on a proposal may call a special meeting to vote on the proposal. Also in accordance with our governing documents, quorum for a shareholders' meeting is at least 33 1/3% of our outstanding common stock entitled to vote and, where quorum is present, shareholder action may be taken by the affirmative vote of a majority of the shares represented at the meeting and entitled to vote. Accordingly, if our two major shareholders call a meeting and establish quorum, they can effect shareholder approval on a proposal unless other shareholders holding a greater number of shares than our two major shareholders were present at the meeting, either in person or by proxy, and vote against the proposal. There is no guarantee that such other shareholders will be present at any such meeting or, even if they were present at such meeting, will vote against the proposal.
We are authorized to issue an unlimited number of Class A common stock, and we intend to issue significantly more shares to raise capital, which would result in substantial dilution to your investment in our shares.
Our Articles of Incorporation authorize the issuance of an unlimited number of Class A common shares, that can be issued for such consideration and on such terms and conditions as are established by our board of directors without the approval of any of our shareholders. Any additional financings effected by us may result in the issuance of additional securities without stockholder approval and the substantial dilution in the percentage of common stock held by our then existing stockholders. Moreover, the common stock issued in any such transaction may be valued on an arbitrary or non-arm’s-length basis by our management, resulting in an additional reduction in the percentage of common stock held by our current stockholders. Our board of directors has the power to issue any or all of such authorized but unissued shares without stockholder approval. To the extent that additional shares of common stock or preferred stock are issued in connection with a financing, dilution to the interests of our stockholders will occur and the rights of the holders of common stock might be materially and adversely affected. We may issue additional common shares in connection with a future financing or acquisition. The issuance of additional common shares may dilute an investor’s investment in us and reduce cash available for distribution per common share, if any dividends are declared by the board of directors in the future.
We have not paid any dividends and do not foresee paying dividends in the future.
We intend to retain earnings, if any, to finance the growth and development of our business and do not intend to pay cash dividends on shares of our common stock in the foreseeable future. The payment of future cash dividends, if any, will be reviewed periodically by the board of directors and will depend upon, among other things, conditions then existing including earnings, financial condition and capital requirements, restrictions in financing agreements, business opportunities and other factors.
38
A significant portion of our outstanding common stock may be sold into the public market in the future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur in the future. These sales, or the market perception that the holders of a large number of shares of our common stock intend to sell shares, could reduce the market price of our common stock.
Our stock is a penny stock. Trading of our stock may be restricted by the SEC’s penny stock regulations which may limit a stockholder’s ability to buy and sell our stock.
Our stock is a penny stock. The SEC has adopted Rule 15g-9 which generally defines “penny stock” to be any equity security that has a market price (as defined) less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. Our securities are covered by the penny stock rules, which impose additional sales practice requirements on broker-dealers who sell to persons other than established customers and “accredited investors”. The term “accredited investor” refers generally to institutions with assets in excess of $5,000,000 or individuals with a net worth in excess of $1,000,000, not including any equity in that person’s or person’s spouse’s primary residence, or annual income exceeding $200,000 or $300,000 jointly with their spouse. The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document in a form prepared by the SEC which provides information about penny stocks and the nature and level of risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction and monthly account statements showing the market value of each penny stock held in the customer’s account. The bid and offer quotations, and the broker-dealer and salesperson compensation information, must be given to the customer orally or in writing prior to effecting the transaction and must be given to the customer in writing before or with the customer’s confirmation. In addition, the penny stock rules require that prior to a transaction in a penny stock not otherwise exempt from these rules, the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for the stock that is subject to these penny stock rules. Consequently, these penny stock rules may affect the ability of broker-dealers to trade our securities. We believe that the penny stock rules discourage investor interest in and limit the marketability of our common stock.
FINRA sales practice requirements may also limit a stockholder’s ability to buy and sell our stock.
In addition to the “penny stock” rules promulgated by the SEC, the Financial Industry Regulatory Authority (FINRA) has adopted rules that require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. The FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our stock.
Any future sales of our equity securities will dilute the ownership percentage of our existing stockholders and may decrease the market price for our common stock.
Future sales or issuances of equity securities could decrease the value of our common stock, dilute stockholders’ voting power and reduce future potential earnings per share. We intend to sell additional equity securities in future offerings (including through the sale of securities convertible into shares of our common stock) and may issue additional equity securities to finance our operations, development, acquisitions or other projects. We cannot predict the size of future sales and issuances of equity securities or the effect, if any, that future sales and issuances of equity securities will have on the market price of our common stock. Sales or issuances of a substantial number of equity securities, or the perception that such sales could occur, may adversely affect prevailing market prices for our common stock. With any additional sale or issuance of equity securities, investors will suffer dilution of their voting power and may experience dilution in our earnings per share.
Anti-takeover provisions may limit the ability of another party to acquire us, which could cause our stock price to decline.
Though not now, we may be or in the future we may become subject to Wyoming’s control share law. The law focuses on the acquisition of a “controlling interest” which means the ownership of outstanding voting shares sufficient, but for the control share law, to enable the acquiring person to exercise the following proportions of the voting power of the corporation in the election of directors: (i) one-fifth or more but less than one-third, (ii) one-third or more but less than a majority, or (iii) a majority or more. The ability to exercise such voting power may be direct or indirect, as well as individual or in association with others. The effect of the control share law is that the acquiring person, and those acting in association with it, obtains only such voting rights in the control shares as are conferred by a resolution of the stockholders of the corporation, approved at a special or annual meeting of stockholders. The control share law contemplates that voting rights will be considered only once by the other stockholders. Thus, there is no authority to strip voting rights from the control shares of an acquiring person once those rights have been approved. If the stockholders do not grant voting rights to the control shares acquired by an acquiring person, those shares do not become permanent non-voting shares. The acquiring person is free to sell its shares to others. If the buyers of those shares themselves do not acquire a controlling interest, their shares do not become governed by the control share law. If control shares are accorded full voting rights and the acquiring person has acquired control shares with a majority or more of the voting power, any stockholder of record, other than an acquiring person, who has not voted in favor of approval of voting rights is entitled to demand fair value for such stockholder’s shares.
39
Wyoming’s control share law may have the effect of discouraging takeovers of the corporation. In addition to the control share law, Wyoming has a business combination law which prohibits certain business combinations between Wyoming corporations and “interested stockholders” for three years after the “interested stockholder” first becomes an “interested stockholder,” unless the corporation’s board of directors approves the combination in advance. For purposes of Wyoming law, an “interested stockholder” is any person who is (i) the beneficial owner, directly or indirectly, of ten percent or more of the voting power of the outstanding voting shares of the corporation, or (ii) an affiliate or associate of the corporation and at any time within the three previous years was the beneficial owner, directly or indirectly, of ten percent or more of the voting power of the then outstanding shares of the corporation. The definition of the term “business combination” is sufficiently broad to cover virtually any kind of transaction that would allow a potential acquirer to use the corporation’s assets to finance the acquisition or otherwise to benefit its own interests rather than the interests of the corporation and its other stockholders. The effect of Wyoming’s business combination law is to potentially discourage parties interested in taking control of the Company from doing so if it cannot obtain the approval of our board of directors.
In addition, our Articles of Incorporation provide for unlimited authorized shares of our Class A common stock. Our authorized but unissued shares of common stock will be available for future issuance without stockholder approval. These additional shares may be utilized for a variety of corporate purposes, including future public offerings to raise additional capital, corporate acquisitions and employee benefit plans. The existence of unlimited authorized but unissued shares of common stock could render more difficult or discourage an attempt to obtain control of a majority of our Class A common stock by means of a proxy contest, tender offer, merger or otherwise.
Holders of our Warrants will have no rights as shareholders until such holders exercise their Warrants and acquire our common shares.
Until holders of Warrants acquire common shares upon exercise of the Warrants, holders of Warrants will have no rights with respect to the common shares underlying such Warrants. Upon exercise of the Warrants, the holders thereof will be entitled to exercise the rights of common shareholders only as to matters for which the record date occurs after the exercise date.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. If any of the analysts who may cover us change their recommendation regarding our stock adversely, or provide more favorable relative recommendations about our competitors, our stock price would likely decline. If any analyst who may cover us were to cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
40
| ITEM 2. | PROPERTIES |
Our head office is located at Suite 400, 41 University Drive, Newtown, PA 18940. We currently lease three office rooms from Regus for approximately $3,896 per month. The lease is for one year expiring on April 30, 2017, at which time we will determine whether we should have a dedicated office space. Currently, we do not have any other material physical properties as we seek to contract out all the non-core functions such as research and development, human resources and investor relations in order to maintain a low fixed cost business model. Our registered office and registered agent is located at CT Corporation System, 1712 Pioneer Ave., Ste. 120, Cheyenne, Wyoming 82001.
| ITEM 3. | LEGAL PROCEEDINGS |
From time to time, we are subject to various legal proceedings and claims that arise in the ordinary course of our business activities. Although the results of litigation and claims cannot be predicted with certainty, as of the date of this filing, we do not believe we are party to any claim or litigation, the outcome of which, if determined adversely to us, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business, other than as set forth below in respect of the matters described below. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Intellectual Property Litigation
On January 27, 2015 we received a demand letter containing allegations that we had entered into a consulting arrangement with the complainants and breached certain of its terms, and used certain intellectual property in the form of business and marketing plans allegedly prepared by the complainants, and seeking damages. On May 7, 2015, Mr. Rainier Maas and Dr. Jochen Scheld filed a complaint in the U.S. District Court for the Eastern District of Pennsylvania seeking monetary damages in excess of $225,000. On December 22, 2015, the Company entered into a settlement agreement with the plaintiffs for an amount of €57,000, which was paid on January 12, 2016. The parties have since executed the settlement agreement for the aforementioned amount and the case has been dismissed without prejudice.
On January 5, 2015, Wicab sued the Company, NHC, Mitch Tyler, a director of the Company and NHC, Yuri Danilov, a former director of the Company and a director of NHC, and ANR, in the U.S. District Court for the Western District of Wisconsin. ANR is the licensor to the Company of three issued patents (U.S. Patent Nos. 8,849,407 and 8,909,345 and 9,020,612) and other patents pending related to neurostimulation methods and devices. The complaint contained various state and common law claims arising from Messrs. Danilov’s and Tyler’s prior employment with Wicab and relating to ownership of two of the issued patents (U.S. Patent Nos. 8,849,407 and 8,909,345). U.S. Patent No. 9,020,612 was not included in the Wicab complaint. The complaint alleged, among other things, that following their departure from Wicab, Danilov and Tyler knowingly filed patent applications for and used ideas and inventions developed at Wicab in violation of various non-competition and confidentiality agreements, and that the two issued patents are therefore rightfully the property of Wicab. The complaint sought an unspecified amount of monetary damages, an injunction preventing NHC from using the ideas and inventions in the two patents, an order transferring ownership of the patents from ANR to Wicab, and recovery of costs and attorneys’ fees. The Company conducted an internal investigation and determined that Wicab expressly waived all rights in the two issued patents and, additionally, that Wicab’s claims were barred by the six year statute of limitations in Wisconsin. On January 14, 2015, the Company informed Wicab of its belief that the claims were barred due to the express waiver and the statute of limitations. On the same day, Wicab dismissed the complaint without prejudice.
On October 12, 2015, the Company received a letter from Wicab alleging that the two issued patents were invalid in view of prior art cited in the letter, including scientific publications and patent applications, and that Paul Bach-y-Rita, Wicab’s founder, should have been named as an inventor on these two issued patents. Wicab indicated in the letter that it may file reexamination or inter partes review proceedings with the U.S. Patent Office to attempt to invalidate the claims in the two issued patents. Wicab also stated that it would consider an unspecified “business solution” to resolve this matter. On December 10, 2015, representatives of each of the Company and Wicab met to discuss the parameters of a potential settlement. There can be no guarantee that a settlement will be reached. In the event that a settlement with Wicab is not reached, Wicab may file reexamination or inter partes review proceedings with the U.S. Patent Office to challenge the validity of the two issued patents. If the Company receives an adverse decision from the U.S. Patent Office in connection with these proceedings, some or all of the claims in the two patents may be invalidated or otherwise impaired, which could prevent the Company from bringing an infringement suit against a future competitor for making use of the PoNS™ technology for neurorehabilitation, and could have a material adverse effect on the Company’s business, operating results and financial condition. Wicab may also take other actions against the Company, its assets, intellectual property rights, officers, directors, employees, agents or other persons or entities which may also have a material on business, operating results and financial condition.
41
Except as described above, we are not aware of any legal proceedings contemplated by any governmental authority or any other party involving us or our properties. As of March 31, 2016, no director, officer or affiliate is: (i) a party adverse to us in any legal proceeding, or (ii) has an adverse interest to us in any legal proceedings. We are not aware of any other legal proceedings pending or that have been threatened against us or our properties.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
42
PART II
|
ITEM 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our shares of common stock commenced trading on the TSX under the symbol “HSM” on April 18, 2016. Our Warrants were also approved for listing on the TSX on April 18, 2016. See Part I Item 1, “Listing of our Common Stock on the CSE, TSX and OTCQB.”
Our common stock is currently quoted on the OTCQB under the symbol “HSDT.”
The following table sets forth, for the periods indicated, the high and low prices relating to our common stock for the periods indicated, as provided by the CSE, the TSX and the OTCQB. OTC prices in the table below prior to February 10, 2015 reflect pricing on the OTC’s Grey Market. The Company’s common stock was delisted from the CSE concurrently with the TSX listing. These quotations reflect inter-dealer prices without retail mark-up, mark-down, or commissions, and may not reflect actual transactions.
| OTC (US$) | CSE / TSX (CAD$) | |||||||||||
|
Period |
High | Low | High | Low | ||||||||
| Fiscal Year Ended March 31, 2015 | ||||||||||||
| First Quarter | - | - | CAD$ 2.37 | CAD$ 1.00 | ||||||||
| Second Quarter | $2.49 | $2.03 | CAD$ 2.72 | CAD$ 2.27 | ||||||||
| Third Quarter | $2.79 | $1.90 | CAD$ 3.00 | CAD$ 2.25 | ||||||||
| Fourth Quarter | $2.70 | $1.80 | CAD$ 3.40 | CAD$ 2.24 | ||||||||
| Fiscal Year Ended March 31, 2016 | ||||||||||||
| First Quarter | $2.60 | $1.90 | CAD$ 3.28 | CAD$ 2.30 | ||||||||
| Second Quarter | $2.10 | $0.62 | CAD$ 2.55 | CAD$ 0.80 | ||||||||
| Third Quarter | $1.15 | $0.58 | CAD$ 1.55 | CAD$ 0.75 | ||||||||
| Fourth Quarter | $0.86 | $0.68 | CAD$ 1.24 | CAD$ 0.95 | ||||||||
| Fiscal Year Ended March 31, 2017 | ||||||||||||
| First Quarter(1) | $1.50 | $0.70 | CAD$ 1.95 | CAD$ 1.01 | ||||||||
(1) Through June 21, 2016.
As of June 21, 2016, the last reported sales price of our common stock on the TSX was CAD$1.30 per share. As of June 21, 2016, the last reported sales price of our common stock on the OTCQB was US$0.99 per share.
The exchange rate in effect on June 21, 2016 as reported by Bank of Canada was US$1.00 = CAD$1.28.
Holders
On June 21, 2016, there were approximately 213 record holders of our common stock. The number of holders of record is based on the actual number of holders registered on the books of our transfer agent and does not reflect holders of shares in “street name” or persons, partnerships, associations, corporations or other entities identified in security position listings maintained by depository trust companies.
43
Compensation Options
As of June 21, 2016 we had compensation options outstanding which are exercisable to purchase a total of 501,457 units, each unit consisting of one share of our Class A common stock and one half of one common share purchase warrant. Each compensation option will entitle the holder thereof to acquire one unit at a price of CAD$1.00 per unit until April 18, 2018.
Options
As of June 21, 2016, options to purchase 6,675,360 shares of our common stock with a weighted average exercise price of $1.08 per share, were outstanding.
Warrants
As of June 21, 2016, we had 9,681,171 common share purchase warrants outstanding which are exercisable into 9,681,171 shares of common stock.
Dividend Policy
We have not paid any cash dividends on our common shares since our inception and do not anticipate paying any cash dividends in the foreseeable future. We plan to retain our earnings, if any, to provide funds for the expansion of our business.
RECENT SALES OF UNREGISTERED SECURITIES.
Other than as previously disclosed in our Quarterly Reports on Form 10-Q, as amended, and our Current Reports on Form 8-K, there were no sales of equity securities by the Company that were not registered under the Securities Act during the fiscal year ended March 31, 2016.
| ITEM 6. | SELECTED FINANCIAL DATA |
As a smaller reporting company, we have elected not to provide selected financial data in reliance on Item 301(c) of Regulation S-K.
|
ITEM 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results of operations together with the section entitled “Selected Financial Data” and our consolidated financial statements and related notes appearing in this 10-K filing. Some of the information contained in this discussion and analysis or set forth elsewhere in this 10-K filing, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors” section of this annual filing, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Restatements
Our Management’s Discussion and Analysis of Financial Condition and Results of Operations gives effect to the restatements of our consolidated financial statements on January 11, 2016 and again on April 26, 2016.
Overview
We are a medical technology company focused on neurological wellness. We seek to develop, license or acquire unique and non-invasive platform technologies that amplify the brain’s ability to heal itself.
Our mission is to develop, license and acquire non-invasive treatments designed to help patients affected by neurological symptoms caused by disease or trauma. Applying the principles of neuroplasticity, our patented PoNS™ device induces Cranial Nerve Non Invasive Neuromodulation that utilizes the brain’s innate ability to achieve neuroplastic change to aid persons with neurological, cognitive, sensory, and motor disorders when combined with the rehabilitation process.
44
Since our inception we have incurred significant operating losses. Our net loss was $6,881,812 and $9,838,317 for the fiscal years ended March 31, 2016 and 2015, respectively. As of March 31, 2016, we had an accumulated deficit of $26,305,263. We expect to incur significant expenses and operating losses for the foreseeable future as we continue to advance our products through clinical trials, and seek regulatory approval and pursue commercialization of such products. In addition, if we obtain marketing approval for any of our products, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. In addition, we may incur expenses in connection with the in-license or acquisition of other potential products.
As a result, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through the sale of equity, debt financings, or other capital sources, including potential collaborations with other companies or other strategic transactions. We may be unable to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements, as, and when, needed, we may have to reduce the scope of our operations and planned capital expenditures or sell certain assets, including intellectual property assets.
As of March 31, 2016, we had cash, cash equivalents and short term investments of $2,643,937. As discussed in more detail below, we recently raised additional capital in an unregistered offering of common stock and warrants and we intend to seek additional funding. However, we do not currently have sufficient resources to accomplish all of the conditions necessary for us to generate revenue. For this reason, there is substantial doubt that we can continue as a going concern for the next twelve months unless we obtain additional capital to pay our expenditures.
Components of Our Results of Operations
Revenue
We have not generated any revenue since our inception and do not expect to generate any revenue from the sale of products in the near future.
Research and Development Expenses
Research and development expenses consists of expenses incurred in connection with the discovery and development of our product candidates. We expense research and development costs as incurred. These expenses include:
| • | expenses incurred under agreements with consultants that conduct our clinical trials; | |
| • | outsourced professional scientific development services; | |
| • | employee-related expenses, which include salaries, benefits and stock-based compensation; | |
| • | expenses relating to regulatory activities, including filing fees paid to regulatory agencies; | |
| • | laboratory materials and supplies used to support our research activities; and | |
| • | allocated expenses for utilities and other facility-related costs |
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect our research and development expenses to increase significantly over the next several years as we increase personnel costs, conduct clinical trials and prepare regulatory filings for our product candidates.
The successful development of our product candidates is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the remainder of the development of, or when, if ever, material net cash inflows may commence from any of our other product candidates. This uncertainty is due to the numerous risks and uncertainties associated with the duration and cost of clinical trials, which vary significantly over the life of a project as a result of many factors, including:
45
| • | the number of clinical sites included in the trials; | |
| • | the length of time required to enroll suitable patients; | |
| • | the number of patients that ultimately participate in the trials; | |
| • | the number of doses patients receive; | |
| • | the duration of patient follow-up; and | |
| • | the results of our clinical trials. |
Our expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals, and the expense of filing, prosecuting, defending and enforcing any patent claims or other intellectual property rights. We may never succeed in achieving regulatory approval for any of our product candidates. We may obtain unexpected results from our clinical trials. We may elect to discontinue, delay or modify clinical trials of some product candidates or focus on others. A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or other regulatory authorities were to require us to conduct clinical trials beyond those that we currently anticipate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development. Drug commercialization will take several years and millions of dollars in development costs.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive, finance and legal functions, including stock-based compensation, and travel expenses. Other general and administrative expenses include facility related costs, professional fees for legal, auditing and tax services, consulting, and insurance costs.
We anticipate that our general and administrative expenses will increase as a result of increased personnel costs, including stock-based compensation, expanded infrastructure and higher consulting, legal and tax-related services associated with maintaining compliance with stock exchange listing and Securities and Exchange Commission, or SEC, requirements, accounting and investor relations costs, and director and officer insurance premiums associated with being a public company. Additionally, if and when we believe a regulatory approval of a drug candidate appears likely, we anticipate an increase in payroll and expense as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of that drug candidate.
Interest Expense, net
Interest expense, net consists of accretion of convertible debenture note discounts and interest income from cash held in interest-bearing accounts.
Other Income
Other income primarily stems from the distribution of prototype devices into approved territories in Russia through Altair distribution agreement. Distribution amounts have been immaterial to date and will continue to be immaterial until the PoNSTM device becomes commercially available.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based upon our financial statements that have been prepared in accordance with U.S. GAAP. This preparation requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, and the disclosure of contingent assets and liabilities. U.S. GAAP provides the framework from which to make these estimates, assumption and disclosures. We choose accounting policies within U.S. GAAP that management believes are appropriate to accurately and fairly report our operating results and financial position in a consistent manner. Management regularly assesses these policies in light of current and forecasted economic conditions. Actual results could differ from those estimates made by management. While there are a number of significant accounting policies affecting our financial statements, we believe the critical accounting policies involving the most complex, difficult and subjective estimates and judgments are: valuation of non-monetary transactions, stock compensation for services, valuation of options and valuation of income taxes.
46
Stock-Based Compensation
We account for all of our stock-based payments and awards under the fair value based method. We recognize our stock-based compensation using the straight-line method.
Stock-based payments to non-employees are measured at the fair value of the consideration received, or the fair value of the equity instruments issued, or liabilities incurred, whichever is more reliably measurable. The fair value of stock-based payments to non-employees is periodically re-measured until the counterparty performance is complete, and any change therein is recognized over the vesting period of the award and in the same manner as if we had paid cash instead of paying with or using equity based instruments. The fair value of the stock-based payments to non-employees that is fully vested and non-forfeitable as at the grant date is measured and recognized at that date.
We account for the granting of share purchase options to employees using the fair value method whereby all awards to employees will be recorded at fair value on the date of the grant. The fair value of all share purchase options are expensed over their vesting period with a corresponding increase to additional capital surplus. Upon exercise of share purchase options, the consideration paid by the option holder, together with the amount previously recognized in additional paid-in capital is recorded as an increase to share capital. Share purchase options granted to employees are accounted for as liabilities when they contain conditions or other features that are indexed to other than a market, performance or service condition.
We use the Black-Scholes option pricing model to calculate the fair value of our share purchase options. We lack historical and implied volatility information. Therefore, we estimate our expected stock volatility based on the historical volatility of a publicly traded set of peer companies and expect to continue to do so until such time as we have adequate historical data regarding the volatility of our own traded stock price. The expected term of our stock options has been determined utilizing the “simplified” method for awards that qualify as “plain vanilla” options. The expected term of stock options granted to non-employees is equal to the contractual term of the option award. The risk free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that we have never paid cash dividends and do not expect to pay any cash dividends in the foreseeable future.
Derivative Liabilities
We evaluate our financial instruments and other contracts to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for in accordance with ASC 815. The result of this accounting treatment is that the fair value of the embedded derivative is marked-to-market at each balance sheet date and recorded as a liability and the change in fair value is recorded in the consolidated statements of operations and comprehensive loss. Upon conversion or exercise of a derivative instrument, the instrument is marked to fair value at the conversion date and then that fair value is reclassified to equity.
The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period. Derivative instruments that become subject to reclassification are reclassified at the fair value of the instrument on the reclassification date. Derivative instrument liabilities will be classified in the balance sheet as current or non-current based on whether or not settlement of the derivative instrument is expected within 12 months of the balance sheet date.
We use the Black-Scholes option valuation model to value derivative liabilities. This model uses Level 3 inputs in the fair value hierarchy established by ASC 820 - Fair Value Measurement.
Results of Operations
Comparison of Fiscal Year Ended March 31, 2015 and 2014
The following table summarizes our results of operations for the fiscal years ended March 31, 2016 and 2015:
47
| Year Ended March 31, | |||||||||
| 2016 | 2015 | Change | |||||||
| Revenue | $ | — | $ | — | $ | — | |||
| Operating expenses: | |||||||||
| Research and development | 3,645,796 | 4,500,073 | (854,277 | ) | |||||
| General and administrative | 5,671,598 | 5,308,371 | 363,227 | ||||||
| Total operating expenses | (9,317,394 | ) | (9,808,444 | ) | (491,050 | ) | |||
| Loss from operations | |||||||||
| Other items: | |||||||||
| Interest expense, net | (46,920 | ) | (176,488 | ) | (129,568 | ) | |||
| Other income | 150,250 | 20,074 | 130,176 | ||||||
| Change in fair value of derivative liability | 2,082,703 | (739,375 | ) | 2,822,078 | |||||
| Foreign exchange | (18,785 | ) | 865,916 | (884,701 | ) | ||||
| Gain on extinguishment of debt | 268,334 | - | 268,334 | ||||||
| Net loss | $ | (6,881,812 | ) | $ | (9,838,317 | ) | $ | (2,956,505 | ) |
Revenues
During the fiscal years ended March 31, 2016 and 2015, we did not generate any revenues.
Research and Development Expenses
Research and development expenses were $3,645,796 for the fiscal year ended March 31, 2016, compared to $4,500,073 for the fiscal year ended March 31, 2015. The decrease of $854,277 was primarily attributable to research and development reimbursements received from the USAMRC totaling $596,547 (March 31, 2015 - $nil) which were credited directly to research and development expenses.
General and Administrative Expenses
General and administrative expenses were $5,671,598 for the fiscal year ended March 31, 2016, compared to $5,308,371 for the fiscal year ended March 31, 2015. The increase of $363,227 was primarily attributable to a general increase in business activities.
Interest Expense, net
Interest expense, net was $46,920 for the fiscal year ended March 31, 2016, compared to $176,488 for the fiscal year ended March 31, 2015. The decrease of $129,568 was primarily attributable to the fact that the convertible debenture was settled in fiscal 2015 and the accompanying accreted interest was recorded in fiscal 2015.
48
Other Income
Other income was $150,250 for the fiscal year ended March 31, 2016, compared to $20,074 for the fiscal year ended March 31, 2015. The increase of $130,176 was primarily attributable to distribution of prototype devices into approved territories in Russia through the Altair distribution agreement.
Change in fair value of derivative liability
The change in fair value of derivative liability was $2,082,703 for the fiscal year ended March 31, 2016, compared to ($739,375) for the fiscal year ended March 31, 2015. The change in fair value of derivative liability is mostly attributable to the change in our stock price during the year, as well as the fair value of warrants issued in private placements. The derivative liabilities do not represent cash liabilities
Foreign exchange
Foreign exchange loss was ($18,785) for the fiscal year ended March 31, 2016, compared to a gain of $865,916 for the fiscal year ended March 31, 2015.
Gain on extinguishment of debt
Gain on extinguishment of debt relating to the A&B promissory note was $268,334 for the fiscal year ended March 31, 2016, compared to a nil amount for the fiscal year ended March 31, 2015. As a result of the bifurcation of the embedded conversion option, for accounting purposes, two instruments were considered outstanding and, upon exercise of the contractual conversion option, extinguishment accounting has been applied. Consequently, the shares issued pursuant to the conversion are recorded at their fair value on the date of issuance, determined with reference to their quoted market price on the date of conversion. The resulting difference between the fair value of the shares issued, less the fair value of the related conversion feature and the carrying value of the related debt, is recorded as a gain or loss on the consolidated statement of operations.
Statement of Cash Flows
Fiscal Year Ended March 31, 2016 Compared to the Fiscal Year Ended March 31, 2015
The following table summarizes our cash flows for each of the periods presented:
| Year Ended March 31, | ||||||
| 2016 | 2015 | |||||
| Cash used in/provided by operating activities | $ | (7,937,412 | ) | $ | (6,321,285 | ) |
| Cash used in/provided by investing activities | 378,000 | (378,000 | ) | |||
| Cash used in/provided by financing activities | 9,691,336 | 7,482,728 | ||||
| Net increase/decrease in cash and cash equivalents | 2,225,044 | 402,925 | ||||
During the fiscal year ended March 31, 2016, our net cash increased by $2,225,044 (March 31, 2015 – increase of $402,925), which included net cash used in operating activities of $7,937,412 (March 31, 2015 - $6,321,285) stemming from our increase in operations, net cash provided by investing activities of $378,000 (March 31, 2016 – ($378,000)) stemming from the redemption of a short-term investment and net cash provided by financing activities of $9,691,336 (March 31, 2015 - $7,482,728) stemming mainly from the closing of multiple private placements and drawing down of the A&B convertible promissory note and credit facility.
Cash Used in Operating Activities
Operating activities for the fiscal year ended March 31, 2016 used cash of $7,937,412 (March 31, 2015 - $6,321,285). This was made up of a net loss of $6,881,812 (March 31, 2015 - $9,838,317) less adjustments for non-cash items such as change in fair value of derivative liability of ($2,082,703) (March 31, 2015 – $739,375), interest accretion of $29,045 (March 31, 2015 - $176,488), stock based compensation of $1,231,250 (March 31, 2015 - $2,340,876), a gain on extinguishment of debt of $268,334 (March 31, 2015 - $nil), receivables of ($390,273) (March 31, 2015 – ($8,945)), accounts payable of $637,935 (March 31, 2015 – $979,040), prepaid expenses and other current assets of ($91,644) (March 31, 2015 – ($110,873)) and unrealized foreign exchange gain of $120,876 (March 31, 2015 – ($598,929)). Receivables increased due to the higher amount of refundable Canadian commodity tax, the Company’s reimbursements from the USAMRC, and the sale of some prototype devices. Prepaid expenses increased due to our increase in operations, while payables remained relatively unchanged.
49
Cash Provided by Investing Activities
During the fiscal year ended March 31, 2016, cash provided by investing activities totaled $378,000 (March 31, 2015 - ($378,000)). This was comprised of the redemption of a short-term investment.
Cash Provided by Financing Activities
During the fiscal year ended March 31, 2016, financing activities provided cash of $9,691,336 (March 31, 2015 - $7,482,728). Financing activities during the fiscal year ended March 31, 2016, consisted of: issuance of share capital of $7,636,910 (March 31, 2015 - $6,637,203) stemming from multiple private placements and the A&B credit facility draw-down, proceeds from shares to be issued of $nil (March 31, 2015 - $39,545), exercise of warrants and stock options of $54,426 (March 31, 2015 - $nil), proceeds from the issuance of convertible debt of $2,000,000 (March 31, 2015 - $nil), proceeds from the issuance of a promissory note of $200,000 (March 31, 2015 - $632,076), the repayment of said promissory note of ($200,000) (March 31, 2015 - $nil), proceeds from a bridge loan of $nil (March 31, 2015 - $150,000), and cash acquired on recapitalization of $nil (March 31, 2015 - $23,904).
Liquidity and Capital Resources
Our financial statements have been prepared assuming that we will continue as a going concern and, accordingly, does not include adjustments relating to the recoverability and realization of assets and classification of liabilities that might be necessary should we be unable to continue in operation.
The following table sets out our cash and working capital as of March 31, 2016 and 2015:
| March 31, 2016 | March 31, 2015 | |||||
| Cash and cash equivalents | $ | 2,643,937 | $ | 418,893 | ||
| Working capital (deficit) | $ | 1,409,568 | $ | 18,543 |
We currently have limited working capital and liquid assets. Our cash and cash equivalents as of March 31, 2016 were $2,643,937. To date we have not generated any revenue from the commercial sales of products or services. There are a number of conditions that we must satisfy before we will be able to generate revenue, including but not limited to successful completion of the clinical trial, FDA clearance of the PoNS™ device for treating balance disorder associated with mild to moderate TBI, manufacturing of a commercially-viable version of the PoNS™ device and demonstration of effectiveness sufficient to generate commercial orders by customers for our product. While we are currently seeking additional funding, we do not currently have sufficient resources to accomplish any of these conditions necessary for us to generate revenue. We will therefore require substantial additional funds in order to continue to conduct the research and development and regulatory clearance and approval activities necessary to bring our product to market, to establish effective marketing and sales capabilities and to develop other product candidates.
We will have to continue to rely on equity and debt financing. There can be no assurance that financing, whether debt or equity, will always be available to us in the amount required at any particular time or for any particular period or, if available, that it can be obtained on terms satisfactory to us. Without additional financing, we do not believe our resources will be sufficient to meet our operating and capital needs through the fourth quarter of calendar 2016.
50
Off Balance Sheet Arrangements
To the best of management’s knowledge, there are no off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our results of operations or financial condition.
Tabular Disclosure of Contractual Obligations
As of March 31, 2016, we did not have any contractual obligations required to be disclosed by Item 303(a)(5) of Regulation S-K during the fiscal year ended March 31, 2016.
Recently Issued Accounting Pronouncements
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). The new standard establishes a ROU model that requires a lessee to record a ROU asset and a lease liability on the consolidated balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the consolidated income statement. ASU 2016-02 is effective for annual periods beginning after December 15, 2018, including interim periods within those annual periods, with early adoption permitted. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. We are currently evaluating the potential impact of the adoption of this standard.
In January 2016, the FASB issued ASU 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities. The amendments in this update revise the accounting related to the classification and measurement of investments in equity securities and the presentation of certain fair value changes for financial liabilities measured at fair value. The amendments are effective for annual reporting periods after December 15, 2017, including interim periods within those fiscal years. Early adoption is permitted. We are currently evaluating the potential impact of the adoption of this standard.
In November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes. The amendments in this update simplify the presentation of deferred income taxes to require that deferred tax liabilities and assets are classified as noncurrent in a statement of financial position. The amendments are effective for annual reporting periods beginning after December 15, 2016 and interim reporting periods within those annual periods. Early adoption is permitted. We have adopted the provisions of this standard early, the impact of which on our consolidated financial statements was not significant.
In April 2015, the FASB issued ASU 2015-03, Interest- Imputation of Interest (Subtopic 835-30). This guidance is to simplify the presentation of debt issuance costs by recognizing a debt liability in the balance sheet as a direct deduction from that debt liability consistent with the presentation of a debt discount. The amendments in this update are effective for financial statements issued for fiscal years beginning after December 15, 2015, and interim periods within those fiscal years. We have adopted this standard and the adoption did not have a material impact on our financial position.
JOBS Act
In April 2012, the JOBS Act was enacted in the United States. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth public companies.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Not applicable
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
See the Index to Financial Statements included in this Annual Report.
51
|
ITEM 9. |
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not applicable
| ITEM 9A. | CONTROLS AND PROCEDURES |
(a) Evaluation of Disclosure Controls and Procedures
As required by Rule 13(a)-15 under the Exchange Act, in connection with this Annual Report, under the direction of the Chief Executive Officer and the Chief Financial Officer, the Company has evaluated its disclosure controls and procedures as of March 31, 2016, and has concluded the disclosure controls and procedures were ineffective as discussed in greater detail below. As of the date of this filing, the Company is still in the process of remediating such material weaknesses in its internal controls and procedures.
(b) Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Securities Exchange Act of 1934. Our management evaluated, under the supervision and with the participation of the Chief Executive Officer and Chief Financial Officer, the effectiveness of its internal control over financial reporting as of March 31, 2016.
Based on its evaluation under the framework in Internal Control—Integrated Framework (2013), issued by the Committee of Sponsoring Organizations of the Treadway Commission, management with the participation of our Chief Executive Officer and our Chief Financial Officer concluded that the Company’s internal control over financial reporting was not effective as of March 31, 2016, due to the existence of a material weakness, as described in greater detail below. A material weakness is a control deficiency, or combination of control deficiencies, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis.
In light of this material weakness, the Company hired an independent firm to provide technical accounting services for the Company and they performed additional post-closing procedures and analyses in order to prepare the consolidated financial statements included in this report. As a result of these procedures, the Company believes its consolidated financial statements included in this report present fairly, in all material respects, the financial position, results of operations and cash flows for the fiscal year ended March 31, 2016.
Limitations on Effectiveness of Controls
The Company’s Chief Executive Officer and Chief Financial Officer do not expect that disclosure controls or internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additional controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Material Weaknesses Identified
In connection with the preparation of the consolidated financial statements for the fiscal year ended March 31, 2016, management identified the following material weakness in internal control:
Our company’s accounting staff does not have sufficient technical accounting knowledge relating to accounting for income taxes and complex US GAAP matters.
52
Our Plans for Remediation of the Material Weakness
Subsequent to March 31, 2016, the Company engaged the services of a consulting group with expertise in US GAAP and SEC compliance matters to assist the Company with its financial reporting and SEC filings. In the ensuing fiscal year, we intend to monitor the progress of our remediation of our identified material weakness.
| ITEM 9B. | OTHER INFORMATION |
None.
53
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Our directors and executive officers and their respective ages as of the date of June 21, 2016 are as follows:
| Name | Age | Position |
| Philippe Deschamps | 54 | President, Chief Executive Officer, and Director |
| Joyce LaViscount | 54 | Chief Financial Officer and Chief Operating Officer |
| Jonathan Sackier | 58 | Chief Medical Officer |
| Brian Bapty | 47 | Vice President, Strategy and Business Development |
| Savio Chiu | 34 | Director |
| Huaizheng Peng | 54 | Director |
| Mitch Tyler | 63 | Director |
| Edward M. Straw | 77 | Director |
| Blane Walter | 46 | Director |
The following describes the business experience of each of our directors and executive officers, including other directorships held in reporting companies:
Philippe Deschamps, Chief Executive Officer, President and a Director
Mr. Deschamps has served as our CEO, President and a Director since June 13, 2014. Mr. Deschamps has extensive experience in pharmaceutical and healthcare commercialization. The depth of his expertise stems from his 30 years in the health sciences industry, approximately half spent at Bristol Myers Squibb (NYSE: BMY), and approximately half on the service side as CEO of GSW Worldwide, a healthcare commercialization company. From 1986 to 1998, Mr. Deschamps served as director of neuroscience marketing at Bristol Myers Squibb in Princeton, N.J., where he participated on several pre-launch global marketing teams in the neuroscience and pain therapeutic areas. Mr. Deschamps started at GSW Worldwide in February 1998 as a Vice President and Account Director and became President and CEO of GSW Worldwide in January 2002, serving in that role until September 2011. Mr. Deschamps was responsible for the GSW Worldwide operations which includes offices in 15 major markets around the world. He primarily consulted on global marketing, commercialization and new business model development for pharmaceutical, device and diagnostics companies. In February 2012, Mr. Deschamps joined MediMedia Health, a marketing services company as CEO where he served until October 2013. At MediMedia Health, he was responsible for the evaluating the different businesses of the company and developing recommendations for the sale of the company to the private equity company that owned it. In October 2013, he became President of NHC. Mr. Deschamps has a BSc. from the University of Ottawa in Canada which he obtained in 1985.
Joyce LaViscount, Chief Financial Officer and Chief Operating Officer
Ms. LaViscount has served as our Chief Financial Officer and Chief Operating Officer since October 19, 2015 and she previously served as one of our directors from March 2, 2015 until December 29, 2015. Ms. LaViscount was at MM Health Solutions (formerly MediMedia Health), a marketing services company, from July 2012 until August 2015 where she served as Chief Operating Officer and Chief Financial Officer. Concurrent with her role at MediMedia Health, Ms. LaViscount also served as the CFO for MediMedia Pharmaceutical Solutions from January 2014 until February 2015. Prior to joining MM Health Solutions, Ms. LaViscount was Executive Director/Group Controller North America for Aptalis Pharmaceuticals (2010 to 2012). From 2004 to 2009 Ms. LaViscount worked for Endo Pharmaceuticals in a variety of roles, including Chief Accounting Officer, VP-Investor Relations and Corporate Communications, and VP Finance Operations, as well as holding operational roles in Sales Operations, Training and Corporate Strategy Development. Ms. LaViscount’s pharmaceutical industry experience also includes more than 15 years in finance at Bristol-Myers Squibb and Pharmacia. Ms. LaViscount began her career with Ernst & Young and is a New Jersey Certified Public Accountant and has Bachelor of Arts in Business with a concentration in Accounting from Franklin and Marshall College.
54
Jonathan Sackier, Chief Medical Officer
Dr. Sackier joined the Company in December of 2014 as Chief Medical Officer and brings to his role extensive experience in new technologies and treatment methodologies gained over more than 30 years in the healthcare industry. Since 2014, Dr. Sackier has been a Visiting Professor of Surgery at the Nuffield Department of Surgical Sciences at Oxford University. From 2005 to 2014, Dr. Sackier was a Visiting Professor of Surgery at the University of Virginia and prior to that a served as a Clinical Professor at George Washington University in Washington, DC from 1995 to 1999. In 1995, while at George Washington University, Dr. Sackier founded and funded the Washington Institute of Surgical Endoscopy, a center for education, research, innovation and technology transfer.
He is widely recognized as one of the leaders of the laparoscopic surgery revolution. In addition to his academic work, Dr. Sackier has helped build several companies including medical technology, research and product-design and medical contract sales organizations. He has also collaborated with pharmaceutical and medical device technology partners including ConvaTec, Pfizer, Karl Storz, Applied Medical, Stryker, Siemens, Bayer and Novartis. Dr. Sackier served as Chairman of Adenosine Therapeutics from 1992 to 1998, which became part of Clinical Data and then Forest Laboratories. Dr. Sackier also worked to develop and market the AESOP robot with Computer Motion from 1992 to 1998. He also founded Genethics in 1985, which patented and licensed amniotic stem cell technology.
Dr. Sackier sits on several boards of directors, he has served as a member of Kypha’s board since 2014, a director of Clinvue since 2010, and a director of Brandon Medical since 2009. Dr. Sackier was also director for Hemoshear from 2008 to 2015 and served as Chairman of Adenosine Therapeutics which became part of Clinical Data and then Forest Laboratories from 2002 to 2008. He is a Trustee of First Star and previously chaired The Larry King Cardiac Foundation Board of Governors. He has also served as a board member of The American College of Surgeons Foundation, The Surgical Fellowship Foundation and Rex Bionics.
A keen pilot, Jonathan advises the Aircraft Owners & Pilots Association (AOPA) on medical issues germane to pilots and authors the “Fly Well” column in AOPA Pilot magazine.
Brian Bapty, Vice President, Strategy and Business Development
Dr. Bapty joined Helius as a consultant in July 2014, and full time as the Company’s Vice President, Strategy and Business Development in October 2015. His sixteen years of experience in capital markets and public companies began in 2000, when he Joined Raymond James as an equity analyst for Canadian healthcare companies. In 2008, still with Raymond James he moved to the London desk supporting institutional equity sales. Early in 2009, Dr. Bapty joined Northland Bancorp Private Equity as a partner and held management positions in investee companies. These positions included Director of Research at Galileo Equity Advisors (a small to midcap focused asset management company) and CEO of Northland Securities (in institutional focused brokerage firm). In March 2012, Dr. Bapty left Northland Bancorp to join Confederation Minerals as President and Director where he served until November 2014.
Dr. Bapty has Ph.D. (Research Medicine, Nephrology) from the University of British Columbia (UBC), and B.Sc. (UBC) in Cell and Developmental Biology.
Savio Chiu, Director
Mr. Chiu has served as one of our Directors since June 13, 2014. From June 2009 to present, Mr. Chiu has been the Senior Manager, Corporate Finance of V Baron Global Financial Canada Ltd. (“V Baron”), which provides us with corporate advisory services pursuant to the terms of a management agreement. Since April 2011, Mr. Chiu has served as the Chief Financial Officer and Corporate Secretary of Confederation Minerals Ltd. (TSXV: CFM). From December 2010 to August 2014, Mr. Chiu served as a director of Finore Mining Inc. (CSE: FIN). From October 2010 to August 2013, Mr. Chiu served as the Chief Financial Officer of Pan American Fertilizer Corp. (formerly Golden Fame Resources Corp.) (TSXV: PFE). From July 2010 to June 2011, he served as the Chief Financial Officer of Cassius Ventures Ltd. (TSXV: CZ).
Mr. Chiu is a Chartered Accountant and holds a Bachelor of Commerce degree in Accounting from the University of British Columbia which he obtained in 2005. Mr. Chiu’s accounting and financial expertise brings a valuable oversight role to the board.
55
Mitch Tyler, Director
Mr. Tyler has served as one of our Directors since June 13, 2014. Mr. Tyler is a co-inventor of the PoNS™ device and co-owner of ANR and Clinical Director of ANR(2009 to present). Mr. Tyler is also the Clinical Director of the Tactile Communication and NeuroRehabilitation Laboratory, University of Wisconsin - Madison (1998 to present), and a Senior Lecturer in Biomedical Engineering. From 1998 through 2005, Mr. Tyler was the Vice President and Principal Investigator for Wicab Inc. He received his M.S. in Bioengineering from University of California, Berkeley in 1985 and is currently working on his Ph.D. in Biomedical Engineering at the UW-Madison. Mr. Tyler’s extensive knowledge of our principal product and history in the medical device industry brings invaluable experience to the board.
Edward M. Straw, Director
Vice Admiral Edward Straw has served as one of our Directors since November 18, 2014. He founded Osprey Venture Partners, a firm that mentors young entrepreneurs seeking investment capital and assists with business development, in 2011 and serves as the Managing Director. Previously he was President, Global Operations of The Estée Lauder Companies from 2000 to 2005, SVP, Global Operations of the Compaq Computer Corporation from 1998 to 2000, and former President of Ryder Integrated Logistics from 1996 to 1998. Prior to joining the private sector, he had a distinguished 35 year career in the U.S. Navy and retired as a three-star admiral. During his military service, Vice Admiral Straw was Chief Executive Officer of the Defense Logistics Agency, the largest military logistics command supporting the American armed forces. Vice Admiral Straw holds an MBA from The George Washington University, a Bachelor of Science degree from Annapolis, and is a graduate of the National War College. He has been a member of the Defense Science Board, Chairman of Odyssey Logistics and currently sits on the boards of: The Boston Consulting Federal Group, Performance Equity Management, and Capital Teas. He was a board member of: Eddie Bauer, MeadWestvaco, Ply Gem Industries and Panther Logistics. Vice Admiral Straw is an “audit committee financial expert” as that term is defined in Item 407(d)(5)(ii) of Regulation S-K. Vice Admiral Straw brings extensive leadership experience to our board.
Blane Walter, Director
Mr. Walter has served as one of our Directors since December 29, 2015. Mr. Walter has been a Partner at Talisman Capital Partners, a private investment partnership located in Columbus, Ohio, since 2011. He founded inChord Communications, Inc. in 1994, which he built into the largest independently-owned, healthcare communications company in the world. In 2005, inChord was acquired by Ventiv Health, the largest provider of outsourced sales and clinical services serving the pharmaceutical industry to create inVentiv Health. In 2008, Mr. Walter became CEO of the combined public company, a role in which he served until 2011. Mr. Walter’s background in the healthcare and pharmaceutical industries lends important perspective to our board.
Huaizheng Peng, Director
Dr. Peng has served as one of our Directors since December 29, 2015. Since 2013 Dr. Peng has served as the General Manager, and non-executive Director of China Medical System Holdings (“CMS”) where he is in charge of international operations, prior to becoming General Manager, Dr. Peng served on the CMS board of directors for a period of three years. Prior to joining CMS, Dr. Peng was a partner in a private equity firm, Northland Bancorp, from 2010 to 2012, head of global life sciences and a director of corporate finance at Seymour Pierce from 2007 to 2010, and served as a non-executive Director of China Medstar, an AIM listed medical service company from 2006 to 2008. Dr. Peng also worked as a senior portfolio manager, specializing in global life science and Asian technology investment at Reabourne Technology Investment Management Limited from 1999 to 2006. Dr. Peng was nominated to our board of directors by A&B pursuant to the terms of the A&B Credit Facility.
Dr. Peng received his Bachelor’s and Masters’ degree in medicine from Hunan Medical College, China. Dr. Peng was awarded his PhD in molecular pathology from University College London (UCL) Medical School where he subsequently worked as a clinical lecturer. We believe that Dr. Peng’s leadership experience in international contexts, knowledge of medicine and investment experience will help our board in its oversight role.
56
Director Independence
Our Board of Directors has determined that two of our directors, Blane Walter and Edward Straw, qualify as independent directors under the listing standards of the TSX and the NYSE MKT.
Term of Office
Our directors are appointed to hold office until the next annual general meeting of our stockholders or until they resign or are removed from the board in accordance with our bylaws. Our officers are appointed by our Board of Directors and hold office until they resign or are removed from office by the Board of Directors.
Committees of the Board of Directors
Our Board of Directors has the authority to appoint committees to perform certain management and administration functions. Our Board of Directors currently has an audit committee. The charter for the audit committee is available on our website.
Our audit committee is comprised of Edward Straw and Blane Walter each of whom are independent directors under the rules of the NYSE MKT and the SEC. The purpose of the audit committee is to assist our Board of Directors with oversight of: (i) the quality and integrity of our financial statements and its related internal controls over financial reporting, (ii) our compliance with legal and regulatory compliance, (iii) the independent registered public accounting firm’s qualifications and independence, and (iv) the performance of our independent registered public accounting firm. The audit committee’s primary function is to provide advice with respect to our financial matters and to assist our Board of Directors in fulfilling its oversight responsibilities regarding finance, accounting, and legal compliance. Vice Admiral Straw is an “audit committee financial expert” as that term is defined in Item 407(d)(5)(ii) of Regulation S-K.
Family Relationships
There are no family relationships among our directors and officers.
Code of Ethics
The Company has adopted a code of business conduct and ethics that applies to its directors, officers, and employees, including its principal executive officers, principal financial officer, principal accounting officer, controller or persons performing similar functions. Our code of business conduct and ethics (“Code of Ethics”) can be found on the Investor Relations page of our website. If we make substantive amendments to the Code of Ethics for or grant any waiver, including any implicit waiver, we will disclose the nature of such amendment or waiver on our website or in a report on Form 8-K within four days of such amendment or waiver.
| ITEM 11. | EXECUTIVE COMPENSATION |
During the fiscal year ended March 31, 2016, our named executive officers consisted of Philippe Deschamps, our Chief Executive Officer, Jonathan Sackier, our Chief Medical Officer, and Joyce LaViscount, our Chief Financial Officer. Ms. LaViscount joined us as a director on February 27, 2015, and became our Chief Financial Officer on October 19, 2015.
Summary Compensation Table
| Name and | All other | |||||||||||||||||
| principal | Fiscal | Option awards | Compensation ($) | |||||||||||||||
| position | Year | Salary ($) | ($) | Bonus ($) | Total ($) | |||||||||||||
| Philippe Deschamps | 2016 | 400,000 | - | (1) | 120,000 | 15,000 | 535,000 | |||||||||||
| Chief Executive Officer | 2015 | 360,417 | 432,198 | - | 5,000 | 797,615 | ||||||||||||
| Joyce LaViscount | 2016 | 137,500 | 205,848 | (3) | - | 5,500 | 348,848 | |||||||||||
| Chief Financial Officer and Chief Operating Officer(2) | ||||||||||||||||||
| Jonathan Sackier; | 2016 | 300,000 | - | (4) | - | - | 300,000 | |||||||||||
| Chief Medical Officer | 2015 | 100,000 | 449,797 | - | - | 549,797 |
57
| (1) |
The grant date fair value was denominated in Canadian dollars and converted into U.S. Dollars using the Bank of Canada nominal noon exchange rate on June 19, 2014 (the grant date) of CAD$1.00 = USD$0.9235. |
| (2) |
Ms. LaViscount was appointed as Chief Financial Officer and Chief Operating Officer on October 19, 2015, and resigned from our Board of Directors on December 29, 2015. The compensation reflected in the Summary Compensation Table reflects her compensation in connection with her role as an executive officer of the Company. Ms. LaViscount was not awarded any compensation in connection with her role as a director of the Company during the fiscal year ended March 31, 2016. |
| (3) |
The grant date fair value was denominated in Canadian dollars and converted into U.S. Dollars using the Bank of Canada nominal noon exchange rate on October 21, 2015 (the grant date) of CAD$1.00 = USD$0.7624. |
| (4) |
The grant date fair value was denominated in Canadian dollars and converted into U.S. Dollars using the Bank of Canada nominal noon exchange rate on December 8, 2015 (the grant date) of CAD$1.00 = USD$0.8717. |
Narrative Disclosure to Summary Compensation Table
Employment Agreement with Philippe Deschamps
On June 13, 2014, we entered into an employment agreement with Philippe Deschamps to serve as our President and CEO. This employment agreement was amended on September 1, 2014. Pursuant to the employment agreement, Mr. Deschamps received a base salary at an annualized rate of $250,000 until investments reached a level of $5 million, or the Financing Threshold, and after such Financing Threshold was met, on August 14, 2014, the Board approved the increase of his base salary to $400,000. In addition to Mr. Deschamps’ base salary, he has the opportunity to receive a target annual bonus of 30% of the base salary, conditional upon, and subject to upward or downward adjustment based upon, achievements and individual goals to be established in good faith by the Board of Directors and Mr. Deschamps. For the fiscal year ended March 31, 2016, Mr. Deschamps was granted a cash bonus of $120,000. If Mr. Deschamps is terminated without cause or if Mr. Deschamps resigns for good reason, we shall pay Mr. Deschamps an aggregate amount equal to the sum of his base salary and the earned portion of the annual bonus paid for the year preceding the year of his termination of which such amount is to be paid in equal monthly installments during the twelve month period following such termination of employment.
Employment Agreement with Joyce LaViscount
On October 19, 2015, we entered into an employment agreement with Joyce LaViscount to serve as our Chief Financial Officer and Chief Operating Officer. Pursuant to the employment agreement, Ms. LaViscount will receive a base salary at an annualized rate of $300,000 for her employment term, which is at-will. In addition to Ms. LaViscount’s base salary, she shall have the opportunity to receive a target annual bonus of 25% of the base salary, conditional upon, and subject to upward or downward adjustment based upon achievements and individual goals to be established in good faith by our CEO and Ms. LaViscount. If Ms. LaViscount is terminated without cause or if Ms. LaViscount resigns for good reason, we will pay Ms. LaViscount an aggregate amount equal to the sum of her base salary and the earned portion of the annual bonus paid for the year of her termination of which such amount is to be paid in equal monthly installments during the twelve month period following such termination of employment.
Employment Agreement with Jonathan Sackier, MD
On December 1, 2014, we entered into an employment agreement with Dr. Jonathan Sackier to serve as our Chief Medical Officer. Pursuant to the employment agreement, Dr. Sackier will receive a base salary at an annualized rate of $300,000 for his employment term, which is at-will. In addition to Dr. Sackier’s base salary, he shall have the opportunity to receive a target annual bonus of 25% of the base salary, conditional upon, and subject to upward or downward adjustment based on upon, achievements and individual goals to be established in good faith by our CEO and Dr. Sackier. If Dr. Sackier is terminated without cause, or if he resigns for good reason, we will pay Dr. Sackier an aggregate amount equal to the sum of his base salary and the earned portion of the annual bonus paid for the year of his termination of which such amount is to be paid in equal monthly installments during the twelve month period following such termination of employment.
58
Employment Agreement with Brian Bapty, PhD
On November 2, 2015, we entered into an employment agreement with Mr. Brian Bapty to serve as our Vice President of Strategy and Business Development. Pursuant to the employment agreement, Mr. Bapty will receive a base salary at an annualized rate of CAN $220,000 for his employment term, which is at-will. In addition to Mr. Bapty’s base salary, he shall have the opportunity to receive a target annual bonus of 25% of the base salary, conditional upon, and subject to upward or downward adjustment based on upon, achievements and individual goals to be established in good faith by our CEO and Mr. Bapty. If Mr. Bapty is terminated without cause, or if he resigns for good reason, we will pay Mr. Bapty an aggregate amount equal to the sum of his base salary of which such amount is to be paid in equal monthly installments during the twelve-month period following such termination of employment.
Option Grants during Fiscal Year 2016
During the fiscal year ended March 31, 2016, we granted 750,000 options to Joyce LaViscount. The grant was made pursuant to the June 2014 Stock Incentive Plan, which is further described below. Twenty five percent of Ms. LaViscount’s options vested upon grant, and the remaining seventy five percent will vest at a rate of twenty five percent annually from the grant date. Ms. LaViscount’s options have an exercise price of CAD$0.87 and expire on October 21, 2020.
Management Contract with V Baron Global Financial Canada Ltd.
Effective July 1, 2014, V Baron has been engaged as an advisor to provide corporate advisory and CFO services to the Company. V Baron was initially engaged for a period of 12 months ending on July 1, 2015. Once the 12 month period passed, V Baron continued to provide advisory services on a month-to-month basis. The corporate advisory services include advising on corporate governance, assisting in compliance with the standards and policies of stock exchanges and regulators, advising on continuous disclosure requirements, assisting in compilation of financial statements, liaising with legal counsel, auditors and the Company’s transfer agent, and assisting/advising on corporate finance related matters. During the duration of the agreement, each party may terminate the agreement by providing the other party with 60 days written notice. V Baron will receive CAD$12,500 per month for the services provided. Until her resignation in October of 2015, our CFO services were provided by Amanda Tseng, who is an employee of V Baron. On October 19, 2015, we appointed Joyce LaViscount to act as our Chief Financial Officer. During the fiscal year ended March 31, 2016, the Company incurred charges totaling CAD$150,000 (US$114,623) in respect of this agreement.
Savio Chiu, a member of our Board of Directors, is a Senior Manager, Corporate Finance of V Baron.
June 2014 Stock Incentive Plan
On June 18, 2014, our Board of Directors authorized and approved the adoption of the 2014 Plan, effective June 18, 2014, under which an aggregate of 12,108,016 shares of our common stock may be issued. The purpose of the 2014 Plan is to enhance our long-term stockholder value by offering opportunities to our directors, officers, employees and eligible consultants to acquire and maintain stock ownership in order to give these persons the opportunity to participate in our growth and success, and to encourage them to remain in our service. Pursuant to the terms of the 2014 Plan, we are authorized to grant stock options, as well as awards of stock appreciation rights, restricted stock, unrestricted shares, restricted stock units and deferred stock units.
The foregoing summary of the 2014 Plan is not complete and is qualified in its entirety by reference to the 2014 Plan.
59
Securities Authorized For Issuance Under Compensation Plans
The following table sets forth the securities to be issued under the Stock Option Plan as at March 31, 2016:
| Number of securities | |||||||||
| remaining available for | |||||||||
| Number of securities to | Weighted-average exercise | future issuance under | |||||||
| be | |||||||||
| issued upon exercise of | price of outstanding | equity compensation plans | |||||||
| outstanding options, | options, warrants and | (excluding securities | |||||||
| warrants and rights | rights | reflected in column (a)) | |||||||
| (a) | (b) | (c) | |||||||
|
Equity compensation plans approved by security holders |
- | - | - | ||||||
|
Equity compensation plans not approved by security holders(1) |
4,920,000 | $ | 0.8989(2) | 7,188,016 | |||||
| Total | 4,920,000 | $ | 0.8989(2) | 7,188,016 |
| (1) |
Represents grants of stock options pursuant to the 2014 Plan. See “Item 11. Executive Compensation— June 2014 Stock Incentive Plan” for a description of the material features of the 2014 Plan. |
| (2) |
The weighted-average exercise price was denominated in Canadian dollars and converted into U.S. dollars based on the Bank of Canada nominal noon exchange rate on March 31, 2016 of CAD$1.00 = USD $0.7710. |
Outstanding Equity Awards at Fiscal Year-End
| Number of | Number of | |||||||||||
| Securities | Securities | |||||||||||
| Underlying | Underlying | |||||||||||
| Unexercised | Unexercised | |||||||||||
| Options | Options | Option | ||||||||||
| (#) | (#) | Exercise Price | Option Expiration | |||||||||
|
Name |
Exercisable | Unexercisable | ($) | Date | ||||||||
| Philippe Deschamps | 1,200,000 | 600,000 | (1) | 0.55 | (2) | 06/18/2019 | ||||||
| Joyce LaViscount | 66,667 | 33,333 | (3) | 2.51 | (4) | 03/16/2020 | ||||||
| 250,000 | 500,000 | (5) | 0.66 | (6) | 10/21/2020 | |||||||
| Jonathan Sackier | 300,000 | 100,000 | (7) | 2.58 | (8) | 12/08/2019 |
| (1) |
600,000 options will vest on June 19, 2016. |
| (2) |
The option exercise price of CAD$0.60 was converted from Canadian dollars to U.S. dollars based on the Bank of Canada nominal noon exchange rate on June 19, 2014 (the grant date) of CAD$1.00 = USD$0.9235. |
| (3) |
33,333 options will vest on March 16, 2017. These options were awarded in connection with Ms. LaViscount’s role as a member of our Board of Directors. |
| (4) |
The option exercise price of CAD$3.20 was converted from Canadian dollars to U.S. dollars based on the Bank of Canada nominal noon exchange rate on March 16, 2015 (the grant date) of CAD$1.00 = USD$0.7834. |
| (5) |
250,000 options will vest on each of October 21, 2017 and October 21, 2018. |
| (6) |
The option exercise price of CAD$0.87 was converted from Canadian dollars to U.S. dollars based on the Bank of Canada nominal noon exchange rate on March 16, 2015 (the grant date) of CAD$1.00 = USD$0.7624. |
60
| (7) |
100,000 options will vest on June 8, 2016. |
| (8) |
The option exercise price of CAD$2.96 was converted from Canadian dollars to U.S. dollars based on the Bank of Canada nominal noon exchange rate on December 8, 2014 (the grant date) of CAD$1.00 = USD$0.8717. |
Director Compensation
| Option | All Other | Total | |||||||
| Awards | Compensation | Compensation | |||||||
|
Name(1) |
($) | ($) | ($) | ||||||
| Savio Chiu | -(2) | - | - | ||||||
| Yuri Danilov(3) | - | 12,350(8)(9) | - | ||||||
| Mitch Tyler | -(4) | 58,410(8)(9) | 58,410 | ||||||
| Edward Straw | -(5) | - | - | ||||||
| Blane Walter | 18,063(6) | - | 18,063 | ||||||
| Huaizheng Peng | 18,063(7) | - | 18,063 |
| (1) |
Ms. LaViscount resigned from our Board of Directors on December 29, 2015. The compensation awarded to Ms. LaViscount in connection with her role as a member of our Board of Directors during the fiscal year ended March 31, 2016 is reflected above in the Summary Compensation Table. |
| (2) |
Mr. Chiu had 60,000 options outstanding as of March 31, 2016, of which 20,000 were not vested. |
| (3) |
Mr. Danilov resigned from our Board of Directors on December 29, 2015. |
| (4) |
Mr. Tyler had 400,000 options outstanding as of March 31, 2016, of which 133,333 were not vested. |
| (5) |
Mr. Straw had 100,000 options outstanding as of March 31, 2016, of which 33,333 were not vested. |
| (6) |
Mr. Walter had 50,000 options outstanding as of March 31, 2016, of which 33,333 were not vested. The grant date fair value was denominated in Canadian dollars and converted into U.S. dollars using the Bank of Canada nominal noon exchange rate on December 31, 2015 (the grant date) of CAD$1.00 = USD$0.7225. |
| (7) |
Dr. Peng had 50,000 options outstanding as of March 31, 2016, of which 33,333 were not vested. The grant date fair value was denominated in Canadian dollars and converted into U.S. dollars using the Bank of Canada nominal noon exchange rate on December 31, 2015 (the grant date) of CAD$1.00 = USD$0.7225. |
| (8) |
These amounts were paid pursuant to a consulting agreement between each of Messrs. Danilov and Tyler and us. See “Certain Relationships and Related Transactions, and Director Independence—Related Party Transactions” for a description of the agreement. |
| (9) |
These awards were issued to Messrs. Danilov and Tyler as part of their compensation for services rendered as non- employee consultants. |
Narrative Disclosure to Director Compensation Table
During the fiscal year ended March 31, 2016, our directors did not receive any fees for their service. Instead, we granted stock options to two of our directors. We granted 50,000 options to Messrs. Walter and Peng, respectively. Messrs. Walter and Peng’s options expire on December 31, 2020 and have an exercise price of CAD$1.24.
|
ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The following table sets forth information relating to the beneficial ownership of our common stock as of June 21, 2016, by:
|
|
• |
Each of our directors and named executive officers; |
|
|
|
|
|
|
• |
All of our directors and executive officers as a group; |
|
|
|
|
|
|
• |
each person, or group of affiliated persons, known by us to beneficially own more than 5% of our outstanding shares of common stock; |
The number of shares beneficially owned by each entity, person, director or executive officer is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any shares over which the individual has sole or shared voting power or investment power as well as any shares that the individual has the right to acquire within 60 days of June 21, 2016 through the exercise of any stock options, warrants or other rights. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of common stock held by that person.
61
Shares of our common stock that a person has the right to acquire within 60 days of June 21, 2016 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and executive officers as a group. Unless otherwise indicated in the footnotes to the table, the information presented in this table is based on 84,323,934 shares of our Class A common stock outstanding on June 21, 2016. Unless otherwise indicated below, the address for each beneficial owner listed is c/o Helius Medical Technologies, Suite 400, 41 University Drive, Newtown, PA 18940.
| Name and Address of Beneficial Owner | Amount and Nature of Beneficial | |
| Ownership | ||
| Directors and Named Executive Officers: | Shares | % |
| Philippe Deschamps | 17,917,355(1) | 21.7% |
| President, Director, and Chief Executive Officer | ||
| Joyce LaViscount | 517,003(2) | (*)% |
| Chief Financial Officer and Chief Operating Officer | ||
| Jonathan Sackier | 16,435,026(3) | 19.9% |
| Chief Medical Officer | ||
| Savio Chiu | 60,000(4) | (*)% |
| Director | ||
| Mitch Tyler | 400,000(5) | (*)% |
| Director | ||
| Edward Straw | 79,167(6) | (*)% |
| Director | ||
| Blane Walter | 16,667(7) | (*)% |
| Director | ||
| Huaizheng Peng | 16,667(8) | (*)% |
| Director | ||
| All executive officers and directors as a group (9 persons): | 4.9% | |
| 5% or greater stockholders: | Shares | % |
| MPJ Healthcare, LLC | 16,035,026(9) | 19.4% |
| 208 Palmer Aly | ||
| Newtown, PA 18940 | ||
| Advanced NeuroRehabilitation, LLC | 16,035,026(10) | 19.4% |
| 510 Charmany Dr., Suite 175F | ||
| Madison, WI 53719 | ||
| A&B (HK) Company Limited | 11,458,334(11) | 13.9% |
| Unit A, 11th Floor, Chung Pont Commercial Building, 300 | ||
| Hennessy Road, Wanchai, Hong Kong, P.R.C. | ||
*Represents beneficial ownership of less than one percent of our outstanding common stock.
| (1) |
Includes 1,800,000 stock options which are immediately exercisable or which will become exercisable within 60 days, warrants to purchase 25,093 shares, and 16,917,355 shares held by MPJ Healthcare, LLC. Investment and voting decisions for the shares held by MPJ Healthcare, LLC are made by a board of three members, each holding one vote. The three board members are Philippe Deschamps, Jonathan Sackier and Montel Williams. This amount includes 7,215,762 shares held in escrow. The holder has only voting power and no investment power with respect to the escrowed shares. |
62
| (2) |
Includes 441,667 stock options which are immediately exercisable or which will become exercisable within 60 days and warrants to purchase 25,112 shares. | |
| (3) |
Includes 400,000 stock options which are immediately exercisable or which will become exercisable within 60 days and 16,917,355 shares held by MPJ Healthcare, LLC. Investment and voting decisions for the shares held by MPJ Healthcare, LLC are made by a board of three members, each holding one vote. The three board members are Philippe Deschamps, Jonathan Sackier and Montel Williams. This amount includes 7,215,762 shares held in escrow. The holder has only voting power and no investment power with respect to the escrowed shares. | |
| (4) |
Includes 60,000 stock options which are immediately exercisable or which will become exercisable within 60 days. | |
| (5) |
Includes 400,000 stock options which are immediately exercisable or which will become exercisable within 60 days. | |
| (6) |
Includes 66,667 stock options which are immediately exercisable or which will become exercisable within 60 days. | |
| (7) |
Include 16,667 stock options which are immediately exercisable or which will become exercisable within 60 days. | |
| (8) |
Includes 16,667 stock options which are immediately exercisable or which will become exercisable within 60 days. | |
| (9) |
Investment and voting decisions for the shares held by MPJ Healthcare, LLC are made by a board of three members, each holding one vote. The three board members are Philippe Deschamps, Jonathan Sackier and Montel Williams. This amount includes 7,215,762 shares held in escrow. The holder has only voting power and no investment power with respect to the escrowed shares. | |
| (10) |
Investment and voting decisions for shares held by Advanced NeuroRehabilitation, LLC are made by Kurt Kaczmarek, as the managing member. This amount includes 7,215,762 shares held in escrow. The holder has only voting power and no investment power with respect to the escrowed shares. | |
| (11) |
In a Schedule 13D filed March 4, 2016, each of A&B, A&B Brother Limited (“A&B BVI”), and Dr. Lam Kong disclosed shared investment and dispositive power over 11,458,334 shares. Based solely upon the disclosure in the Schedule 13D, Dr. Lam Kong is the sole officer and director of each of A&B and A&B BVI. The business address of A&B BVI is Trident Chambers, P.O. Box 146, Road Town, Tortola, British Virgin Islands. The business address of Dr. Lam Kong is 8/F Bldg. A, Tongfang Information Harbor, No. 11 Langshan Road, Shenzhen Hi-tech Industrial Park, Nanshan District, Shenzhen, P.R.C. |
Shares of our Common Stock that are owned by ANR and MPJ are subject to the terms of a Lock-Up Agreement as discussed herein below. Under Rule 144 promulgated under the Securities Act, our officers, directors and beneficial shareholders may sell, subject to the terms of the Lock-Up Agreement, up to one percent (1%) of the total outstanding shares (or an amount of shares equal to the average weekly reported volume of trading during the four calendar weeks preceding the sale) every three months provided that (i) current public information is available about our Company, (ii) the shares have been held for at least one year, (iii) the shares are sold in a broker’s transaction or through a market-maker, and (iv) the seller files a Form 144 with the SEC.
|
ITEM 13. |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Except as described below and in “Executive Compensation” above, there are no transactions since our inception, or any currently proposed transactions, in which we were or are to be a participant and in which any “related person” had or will have a direct or indirect material interest. “Related person” includes:
63
| (a) |
Any of our directors or executive officers; | |
| (b) |
Any person proposed as a nominee for election as a director; | |
| (c) |
Any person who beneficially owns more than 5% of our common stock; or | |
| (d) |
Any child, stepchild, parent, stepparent, spouse, sibling, mother-in-law, father-in-law, son-in-law, daughter- in- law, brother-in-law, sister-in-law or person (other than a tenant or employee) sharing the same household of any person enumerated in paragraph (a), (b), or (c). |
Related Party Transactions
Consulting and Employment Agreements with Brian Bapty
On November 2, 2015, we entered into an employment agreement with Dr. Bapty to serve as the Vice President of Strategy and Business Development of the Company. Pursuant to the employment agreement, Dr. Bapty will receive a base salary at an annualized rate of CAD$220,000 for his employment term, which is at-will. In addition to Dr. Bapty’s base salary, he shall have the opportunity to receive a target annual bonus of 25% of the base salary, conditional upon, and subject to upward or downward adjustment based upon achievements and individual goals to be established in good faith by the Company’s CEO and Dr. Bapty, which goals have not yet been established. If Dr. Bapty is terminated without cause or if Dr. Bapty resigns for good reason, the Company will pay Dr. Bapty an aggregate amount equal to the sum of his base salary and there will be accelerated vesting of the options described in the immediately preceding paragraph.
Strategic Agreement with A&B and A&B Credit Facility
On October 13, 2015, the Company announced that it, through its wholly owned subsidiary NHC, entered into the Strategic Agreement with A&B for the development and commercialization of the PoNS™ therapy in China, Hong Kong, Macau, Taiwan and Singapore (collectively, the “Territories”). A&B is an investment and development company owned by Dr. Kong Lam and based in Hong Kong. The Strategic Agreement transfers ownership of certain Asian patents, patent applications, and product support material for the PoNS™ device from NHC to A&B and grants to A&B, among other things, an exclusive, perpetual, irrevocable and royalty-free license, with the right to sublicense, to certain NHC technology, as more particularly described in the Strategic Agreement, to market, promote, distribute and sell PoNS™ devices solely within the Territories. Pursuant to the Strategic Agreement, A&B has assumed all development, patent (both application and defense), future manufacturing, clinical trial, and regulatory clearance costs for the Territories. The Company and A&B will share and transfer ownership of any intellectual property or support material (developed by either party) for their respective geographies. In connection with the Strategic Agreement, A&B agreed to provide a credit facility to the Company.
On October 9, 2015, the Company had issued a convertible promissory note (the “Note”) to A&B in connection with the drawdown of US$2.0 million under the Company’s US$7.0 million credit facility with A&B (the “A&B Credit Facility”). The Company also received notice of conversion on October 9, 2015and immediately satisfied the terms of the Note by issuing to A&B: (i) 2,083,333 common shares at a deemed price of US$0.96 per common share; and (ii) 1,041,667 common share purchase warrants, with each warrant entitling A&B to purchase an additional common share at a price of US$1.44 for a period of three years expiring on November 10, 2018.
On December 29, 2015, the Company drew down the remaining US$5.0 million from the A&B Credit Facility in exchange for the issuance to A&B of 5,555,556 common shares at a price of US$0.90 per common share and warrants to purchase 2,777,778 commons shares for a period of three years having an exercise price of US$1.35 per common share. Additionally, pursuant to the terms of the funding commitment from A&B, the Company granted A&B the right to nominate one person to serve on the Board. A&B nominated Dr. Peng and the Board appointed Dr. Peng to fill the new vacancy. The common shares and warrants issued to A&B, and the common shares underlying such warrants, are subject to a four-month statutory hold period.
Pursuant to the terms of the A&B Credit Facility, we have agreed to register the shares of common stock issued under the terms of the Credit Facility upon the request of A&B. A&B currently has beneficial ownership over 11,458,334 shares of our common stock.
64
Consulting Agreement with Montel Media, Inc.
On April 13, 2016, Montel Media, Inc. (“Montel Media”) entered into a consulting agreement, or the Montel Media Consulting Agreement, with the Company to provide consulting services in relation to the promotion of clinical trials as well as ongoing media/marketing strategy. Montel Media is owned by Montel Williams. Mr. Williams is one of three board members of MPJ. The Montel Media Consulting Agreement is valid for a period of 12 months and Montel Media will charge a monthly fee of $15,000. The total projected dollar value of the contract is $180,000. Pursuant to the Montel Media Consulting Agreement, Montel Media will be an independent contractor and subject to the confidentiality provisions contained in the Montel Media Consulting Agreement.
Review, Approval and Ratification of Related Party Transactions
Our Board of Directors has responsibility for establishing and maintaining guidelines relating to any related party transactions between us and any of our officers or directors. Any conflict of interest between a related party and us must be referred to the non-interested directors, if any, for approval. We intend to adopt written guidelines for the board of directors which will set forth the requirements for review and approval of any related party transactions.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The following are aggregate fees billed to us by BDO Canada LLP during the fiscal years ended March 31, 2016 and 2015:
| Fiscal Year Ended | Fiscal Year Ended | |||||
| March 31, 2016 | March 31, 2015 | |||||
| Audit Fees | $ | 155,000 | 86,715 | |||
| Audit-Related Fees | Nil | Nil | ||||
| Tax Fees | $ | 61,550 | 5,090 | |||
| All Other Fees | - | Nil | ||||
| Total Fees | $ | 216,550 | 91,805 |
Audit Fees
Audit fees consist of fees billed for professional services rendered for the audit of our consolidated financial statements and review of the interim consolidated financial statements included in quarterly reports and services that are normally provided by BDO Canada LLP in connection with statutory and regulatory filings, our registration statements and securities offerings.
Tax Fees
Tax fees consist of fees billed for professional services for tax compliance, tax advice and tax planning. These services include assistance regarding federal, state and tax compliance, customs and duties, mergers and acquisitions and tax planning.
All Other Fees
This was zero for 2016.
A majority of our independent directors, or the independent director to whom such authority was delegated by the independent directors, must pre-approve all services provided by the independent registered public accounting firm.
65
PART IV
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
The following documents are filed as part of this Annual Report:
| 1. |
Financial Statements—See the Index to Consolidated Financial Statements on Page F-1. |
| 2. |
Financial Statement Schedules—None. We have omitted financial statement schedules because they are not required or are not applicable, or the required information is shown in the consolidated financial statements or notes to the consolidated financial statements. |
| 3. |
Exhibits. |
|
Exhibit Number |
Exhibit |
|
2.2 |
Agreement and Plan of Merger among Helius Medical Technologies, Inc., HMT Mergersub, Inc. and NeuroHabilitation Corporation, dated June 6, 2014 (incorporated by reference to Exhibit 10.6 to the Form S-1 filed with the SEC on July 14, 2014) |
|
3.1 |
Articles of Continuation (incorporated by reference to Exhibit 3.1 to the Form S-1 filed with the SEC on July 14, 2014) |
|
3.2 |
Articles of Amendment filed with the Wyoming Secretary of State on July 3, 2014 (incorporated by reference to Exhibit 3.2 to the Form S-1 filed with the SEC on July 14, 2014) |
|
3.3 |
Articles of Amendment filed with the Wyoming Secretary of State on April 27, 2015 (incorporated by reference to Exhibit 3.3 to amendment no. 1 to the Form 10 filed with the SEC on May 4, 2015) |
|
3.4 |
Bylaws as amended and restated (incorporated by reference to Exhibit 3.1 to the Form 8-K filed with the SEC on March 23, 2016) |
|
4.1 |
Form of Warrant (included in Exhibit 4.2) |
|
4.2 |
Warrant Indenture dated April 18, 2016 by and between Helius Medical Technologies, Inc. and Computershare Investor Services Inc. (incorporated by reference to Exhibit 4.1 to amendment no. 1 to the Form 8-K filed April 18, 2016 and amended on April 20, 2016) |
|
10.1† |
Employment Agreement between Helius Medical Technologies, Inc. and Philippe Deschamps, dated June 13, 2014 (incorporated by reference to Exhibit 99.1 to the Form S-1 filed with the SEC on July 14, 2014) |
|
10.2† |
Amendment Agreement to the Employment Agreement between Helius Medical Technologies, Inc. and Philippe Deschamps, dated September 1, 2014 (incorporated by reference to Exhibit 99.5 to the Amendment to Form S-1 filed with the SEC on September 23, 2014) |
|
10.3† |
Employment Agreement between Helius Medical Technologies, Inc. and Jonathan Sackier, dated December 1, 2014 (incorporated by reference to Exhibit 10.4 to the Form 10-12G filed with the SEC on April 15, 2015) |
|
10.4† |
Consulting Agreement between NeuroHabilitation Corporation and Yuri Danilov, dated July 1, 2014 (incorporated by reference to Exhibit 99.4 to the Amendment to Form S-1 filed with the SEC on September 23, 2014) |
66
|
Exhibit Number |
Exhibit |
|
|
|
|
10.5† |
Consulting Agreement between NeuroHabilitation Corporation and Mitch Tyler, dated December 10, 2014 (incorporated by reference to Exhibit 10.5 to the Form 10-12G filed with the SEC on February 6, 2015) |
|
|
|
|
10.6† |
Advisory Agreement between Helius Medical Technologies, Inc. and V Baron Global Financial Canada Ltd., dated June 13, 2014 (incorporated by reference to Exhibit 99.2 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.7 |
License Agreement between Advanced NeuroRehabilitation, LLC and Yuri Danilov, Mitchell Tyler, Kurt Kaczmarek and John Klus, dated June 29, 2011 (incorporated by reference to Exhibit 10.8 to the Amendment to Form S-1 filed with the SEC on September 23, 2014) |
|
|
|
|
10.8 |
Amended and Restated Patent Sub-License Agreement between Advanced NeuroRehabilitation, LLC and NeuroHabilitation Corporation, having an effective date of January 22, 2013 (incorporated by reference to Exhibit 10.1 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.9 |
Second Amended and Restated Patent Sub-License Agreement between Advanced NeuroRehabilitation, LLC and NeuroHabilitation Corporation, dated June 6, 2014, but having an effective date of January 22, 2013 (incorporated by reference to Exhibit 10.7 to the Form S- 1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.10 |
Master Cooperative Research and Development Agreement between NeuroHabilitation Corporation, Advanced NeuroRehabilitation, LLC, Yuri Danilov, Mitchell Tyler, Kurt Kaczmarek and U.S. Army Medical Material Agency and U.S. Army Medical Material Development Activity, dated effective February 1, 2013 (incorporated by reference to Exhibit 10.2 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.11 |
Notice of Modification No. 1 to Cooperative Research and Development Agreement between NeuroHabilitation Corporation, Advanced NeuroRehabilitation, LLC, Yuri Danilov, Mitchell Tyler, Kurt Kaczmarek and U.S. Army Medical Material Agency and U.S. Army Medical Material Development Activity, dated April 29, 2014 (incorporated by reference to Exhibit 10.5 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.12 |
Notice of Modification No. 2 to Cooperative Research and Development Agreement between NeuroHabilitation Corporation, Advanced NeuroRehabilitation, LLC, Yuri Danilov, Mitchell Tyler, Kurt Kaczmarek and U.S. Army Medical Material Agency and U.S. Army Medical Material Development Activity, dated January 12, 2015 (incorporated by reference to Exhibit 10.12 to the Form 10-12G filed with the SEC on February 6, 2015) |
|
|
|
|
10.13 |
Design and Manufacturing Consultant Agreement between NeuroHabilitation Corporation and Clinvue, LLC, dated January 30, 2013 (incorporated by reference to Exhibit 10.3 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.14 |
Commercial Development-to-Supply Program between NeuroHabilitation Corporation and Ximedica, dated October 25, 2013 (incorporated by reference to Exhibit 10.4 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.15 |
Amendment No. 1 to the Commercial Development-to-Supply Program between NeuroHabilitation Corporation and Ximedica, dated October 25, 2013, amended January 15, 2016 (incorporated by reference to Exhibit 10.15 to the Form S-1 filed with the SEC on May 4, 2016) |
|
|
|
|
10.16† |
Employment Agreement between Helius Medical Technologies, Inc. and Joyce LaViscount, dated October 19, 2015 (incorporated by reference to Exhibit 10.3 to the Form 10-Q filed with |
67
|
Exhibit Number |
Exhibit |
|
|
|
|
10.17† |
Employment Agreement between Helius Medical Technologies, Inc. and Brian Bapty, dated November 2, 2015 (incorporated by reference to Exhibit 10.4 to the Form 10-Q filed with the SEC on February 16, 2016) |
|
|
|
|
10.18‡ |
Asset Purchase Agreement between the Company and A&B (HK) Company Limited, dated as of October 9, 2015 (incorporated by reference to Exhibit 2.1 to the Form 8-K filed with the SEC on October 13, 2015) |
|
|
|
|
10.19 |
Convertible Promissory Note between the Company and A&B (HK) Company Limited, dated as of October 9, 2015 (incorporated by reference to Exhibit 10.1 to the Form 8-K filed with the SEC on October 13, 2015) |
|
|
|
|
10.20 |
Notice of Modification No. 3 to Cooperative Research and Development Agreement between NeuroHabilitation Corporation, Advanced NeuroRehabilitation, LLC, Yuri Danilov, Mitchell Tyler, Kurt Kaczmarek and U.S. Army Medical Material Agency and U.S. Army Medical Material Development Activity, dated December 28, 2016 (incorporated by reference to Exhibit 2.1 to the Form 8-K filed with the SEC on December 31, 2015) |
|
|
|
|
10.21 |
Agency Agreement between the Company and Mackie Research Capital Corporation, dated as of March 23, 2016 (incorporated by reference to Exhibit 10.21 to the Form S-1 filed with the SEC on May 4, 2016) |
|
|
|
|
10.22 |
Sole-source cost sharing contract by and between NeuroHabilitation Corporation and the U.S. Army Medical Research and Materiel Command (USAMRMC) dated as of July 7, 2015 (incorporated by reference to Exhibit 10.22 to the Form S-1 filed with the SEC on May 4, 2016) |
|
|
|
|
10.23† |
2014 Stock Incentive Plan (incorporated by reference to Exhibit 4.1 to the Form S-1 filed with the SEC on July 14, 2014) |
|
|
|
|
10.24 |
Consulting Agreement between Helius Medical Technologies, Inc. and Montel Media, Inc., dated April 13, 2016 (incorporated by reference to Exhibit 10.24 to the Form S-1 filed with the SEC on May 4, 2016) |
|
|
|
|
16.1 |
Letter from Davidson & Company LLP, dated April 15, 2015 (incorporated by reference to Exhibit 16.1 to the Form 10-12G filed with the SEC on April 15, 2015) |
|
|
|
|
21.1* |
Subsidiaries of Helius Medical Technologies, Inc.: |
|
|
|
|
1. NeuroHabilitation Corporation is a wholly owned subsidiary of Helius Medical Technologies, Inc. | |
|
|
|
|
2. Helius Medical Technologies (Canada), Inc. is a wholly owned subsidiary of Helius Medical Technologies, Inc. | |
|
|
|
|
|
|
|
101.INS* |
XBRL Instance Document |
|
|
|
|
101.SCH* |
XBRL Taxonomy Extension Schema Document |
|
|
|
|
101.CAL* |
XBRL Taxonomy Extension Calculation Linkbase Document |
68
| Exhibit Number | Exhibit |
| 101.LAB* | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE* | XBRL Taxonomy Extension Presentation Linkbase Document |
| 101.DEF* | XBRL Taxonomy Extension Definition Linkbase Document |
* Filed herewith.
†Indicates a management contract or compensatory plan.
‡Confidential information has been omitted and filed separately with the Securities and Exchange Commission. Confidential treatment has been granted with respect to this omitted information.
69
INDEX TO FINANCIAL STATEMENTS
70
HELIUS MEDICAL TECHNOLOGIES, INC.
CONSOLIDATED FINANCIAL
STATEMENTS
March 31, 2016 and 2015
(Expressed in United States Dollars)
| Tel: 604 688 5421 Fax: 604 688 5132 www.bdo.ca |
BDO Canada LLP 600 Cathedral Place 925 West Georgia Street Vancouver BC V6C 3L2 Canada |
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors
Helius Medical
Technologies Inc.
We have audited the accompanying consolidated balance sheets of Helius Medical Technologies Inc. as of March 31, 2016 and 2015, and the related consolidated statements of operations and comprehensive loss, stockholders’ deficit, and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Helius Medical Technologies Inc. at March 31, 2016 and 2015, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As described in Note 1 to the consolidated financial statements, the Company incurred a net loss of $6,881,812 for the year ended March 31, 2016, had an accumulated deficit of $26,305,263 at March 31, 2016 and the Company expects to incur further losses in the development of its business. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ BDO Canada LLP
Chartered Professional Accountants
Vancouver, British Columbia
June 27, 2016
BDO Canada LLP, a Canadian limited liability partnership, is a member of BDO International Limited, a UK company limited by guarantee, and forms part of the international BDO network of independent member firms.
| Helius Medical Technologies, Inc. |
| Consolidated Balance Sheets |
| March 31, 2016 and 2015 |
| (Expressed in United States Dollars) |
| March 31, | March 31, | |||||
| 2016 | 2015 | |||||
| $ | $ | |||||
| ASSETS | ||||||
| Current assets | ||||||
| Cash and cash equivalents | 2,643,937 | 418,893 | ||||
| Short-term investment | - | 378,000 | ||||
| Receivables (Note 2) | 399,106 | 8,833 | ||||
| Prepaid expenses | 502,264 | 410,621 | ||||
| Other current assets (Note 14) | 495,415 | - | ||||
| Total current assets | 4,040,722 | 1,216,347 | ||||
| TOTAL ASSETS | 4,040,722 | 1,216,347 | ||||
| LIABILITIES | ||||||
| Current liabilities | ||||||
| Accounts payable and accrued liabilities | 2,181,154 | 1,197,804 | ||||
| Shares to be issued (Note 14) | 150,000 | - | ||||
| Total current liabilities | 2,331,154 | 1,197,804 | ||||
| Derivative liability (Note 7 and Note 8) | 1,725,760 | 1,581,444 | ||||
| TOTAL LIABILITIES | 4,056,914 | 2,779,248 | ||||
| Commitments and contingencies (Note 10) | ||||||
| STOCKHOLDERS’ DEFICIT | ||||||
| Common stock
(Unlimited Class A common shares
authorized); (72,193,209 shares issued and outstanding at March 31, 2016 and 63,104,788 shares issued and outstanding at March 31, 2015) (Note 7) |
24,347,930 | 16,358,093 | ||||
| Additional paid-in capital | 2,940,539 | 2,434,552 | ||||
| Shares to be issued | - | 39,545 | ||||
| Accumulated other comprehensive loss | (999,398 | ) | (971,640 | ) | ||
| Accumulated deficit | (26,305,263 | ) | (19,423,451 | ) | ||
| TOTAL STOCKHOLDERS’ DEFICIT | (16,192 | ) | (1,562,901 | ) | ||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT | 4,040,722 | 1,216,347 |
(The accompanying notes are an integral part of these
consolidated financial statements.)
F-5
| Helius Medical Technologies Inc. |
| Consolidated Statements of Operations and Comprehensive Loss |
| for the fiscal years ended March 31, 2016 and 2015 |
| (Expressed in United States Dollars) |
| Year Ended March 31, | ||||||
| 2016 | 2015 | |||||
| $ | $ | |||||
| Operating Expenses | ||||||
| General and administrative | 5,671,598 | 5,308,371 | ||||
| Research and development | 3,645,796 | 4,500,073 | ||||
| Loss from operations | 9,317,394 | (9,808,444 | ) | |||
| Other Income (expense) | ||||||
| Interest expense, net | (46,920 | ) | (176,488 | ) | ||
| Other income | 150,250 | 20,074 | ||||
| Change in fair value of derivative liability | 2,082,703 | (739,375 | ) | |||
| Foreign exchange gain (loss) | (18,785 | ) | 865,916 | |||
| Gain on extinguishment of debt | 268,334 | - | ||||
| 2,435,582 | (29,873 | ) | ||||
| Net loss | (6,881,812 | ) | (9,838,317 | ) | ||
| Other comprehensive income (loss) | ||||||
| Foreign currency translation adjustments | (27,758 | ) | (971,640 | ) | ||
| Comprehensive loss | (6,909,570 | ) | (10,809,957 | ) | ||
| Net loss per share | ||||||
| Basic | $ | (0.10 | ) | $ | (0.17 | ) |
| Diluted | $ | (0.12 | ) | $ | (0.17 | ) |
| Weighted average shares outstanding | ||||||
| Basic | 66,522,564 | 57,048,406 | ||||
| Diluted | 67,026,545 | 57,048,406 | ||||
(The accompanying notes are an integral part of these consolidated financial statements.)
F-6
| Helius Medical Technologies Inc. |
| Consolidated Statements of Stockholders’ Deficit |
| for the fiscal years ended March 31, 2016 and 2015 |
| (Expressed in United States Dollars) |
| Accumulated other | |||||||||||||||||||||
| Additional Paid-In | Shares to be | Accumulated | comprehensive income | ||||||||||||||||||
| Common Stock | Amount | Capital | Issued | Deficit | (loss) | Capital (Deficit) | |||||||||||||||
| $ | $ | $ | $ | $ | $ | ||||||||||||||||
| Balance – March 31, 2014 | 32,070,052 | 8,510,000 | 807,157 | - | (9,585,134 | ) | - | (267,977 | ) | ||||||||||||
| Stock-based compensation on 2,300,000 options granted | 50,303 | - | - | - | 50,303 | ||||||||||||||||
| Shares issued to consultant for option exercise (Note 6) | 2,300,000 | 717 | - | - | - | - | 717 | ||||||||||||||
| Shares issued to consultant for option exercise (Note 6) | 930,031 | 290 | - | - | - | - | 290 | ||||||||||||||
| Fair value of options allocated to share capital on exercise of options | - | 857,460 | (857,460 | ) | - | - | - | - | |||||||||||||
| Recapitalization of Helius Medical Technologies, Inc. (Note 3) | 10,000,000 | - | 162,890 | - | - | - | 162,890 | ||||||||||||||
| Issuance of common stock for private placement (Note 5) | 15,240,000 | 6,437,041 | 578,961 | - | - | - | 7,016,002 | ||||||||||||||
| Share issuance cost (Note 5) | - | (447,515 | ) | 67,709 | - | - | - | (379,806 | ) | ||||||||||||
| Beneficial conversion feature (Note 4) | - | - | 176,488 | - | - | - | 176,488 | ||||||||||||||
| Stock-based compensation on
3,370,000 options granted (Note 6) (Restated) |
- | - | 1,227,724 | - | - | - | 1,227,724 | ||||||||||||||
| Conversion of debenture (Note 4) | 2,564,705 | 1,000,100 | - | - | - | - | 1,000,100 | ||||||||||||||
| Stock-based compensation on 100,000 options granted (Note 6) | - | - | 74,190 | - | - | - | 74,190 | ||||||||||||||
| Stock-based compensation on 100,000 options granted (Note 6) | - | - | 43,229 | - | - | - | 43,229 | ||||||||||||||
| Stock-based compensation on 400,000 options granted (Note 6) | - | - | 135,564 | - | - | - | 135,564 | ||||||||||||||
| Stock-based compensation on 100,000 options granted (Note 6) | - | - | 41,987 | - | - | - | 41,987 | ||||||||||||||
| Fair value of vested non-employee options reallocated to derivative liability | - | - | (74,190 | ) | - | - | - | (74,190 | ) | ||||||||||||
| Private placement proceeds | - | - | - | 39,545 | - | - | 39,545 | ||||||||||||||
| Net loss for the year | - | - | - | - | (9,838,317 | ) | - | (9,838,317 | ) | ||||||||||||
| Translation adjustments | - | - | - | - | - | (971,640 | ) | (971,640 | ) | ||||||||||||
| Balance – March 31, 2015 | 63,104,788 | 16,358,093 | 2,434,552 | 39,545 | (19,423,451 | ) | (971,640 | ) | (1,562,901 | ) |
F-7
| Helius Medical Technologies Inc. |
| Consolidated Statements of Stockholders’ Deficit |
| for the fiscal years ended March 31, 2016 and 2015 |
| (Expressed in United States Dollars) |
| Accumulated other | |||||||||||||||||||||
| Additional Paid-In | Shares to be | Accumulated | comprehensive income | ||||||||||||||||||
| Common Stock | Amount | Capital | Issued | Deficit | (loss) | Capital (Deficit) | |||||||||||||||
| $ | $ | $ | $ | $ | $ | ||||||||||||||||
| Balance – March 31, 2015 | 63,104,788 | 16,358,093 | 2,434,552 | 39,545 | (19,423,451 | ) | (971,640 | ) | (1,562,901 | ) | |||||||||||
| Exercise of finder’s warrants | 14,400 | 11,926 | - | - | - | - | 11,926 | ||||||||||||||
| Issuance of common stock for private placement | 849,273 | 1,825,937 | - | - | - | - | 1,825,937 | ||||||||||||||
| Fair value of warrants issued in connection with private placement, classified to derivative liability | (360,413 | ) | (360,413 | ) | |||||||||||||||||
| Issuance of common stock for private placement | 335,463 | 721,243 | - | (39,545 | ) | - | - | 681,698 | |||||||||||||
| Fair value of warrants issued in connection with private placement, classified to derivative liability | (135,540 | ) | (135,540 | ) | |||||||||||||||||
| Issuance of common stock for private placement | 125,756 | 270,375 | - | - | - | - | 270,375 | ||||||||||||||
| Fair value of warrants issued in connection with private placement, classified to derivative liability | (36,569 | ) | (36,569 | ) | |||||||||||||||||
| Stock option exercise | 94,640 | 42,500 | - | - | - | - | 42,500 | ||||||||||||||
| Fair value of options exercised | - | 34,378 | (34,378 | ) | - | - | - | - | |||||||||||||
| Shares issued as a debt discount | 30,000 | 29,045 | - | - | - | - | 29,045 | ||||||||||||||
| Issuance of common stock upon conversion of convertible note | 2,083,333 | 1,731,667 | - | - | - | - | 1,731,667 | ||||||||||||||
| Fair value of warrants issued in connection with initial borrowing under credit facility, classified to derivative liability | (206,667 | ) | (206,667 | ) | |||||||||||||||||
| Issuance of common stock for draw down of remaining credit facility | 5,555,556 | 5,000,000 | - | - | - | - | 5,000,000 | ||||||||||||||
| Fair value of warrants issued in connection with draw down of remaining credit facility, classified to derivative liability | (796,945 | ) | (796,945 | ) | |||||||||||||||||
| Share issuance cost | - | (141,100 | ) | - | - | - | - | (141,100 | ) | ||||||||||||
| Stock-based compensation on 3,370,000 options granted | - | - | 656,512 | - | - | - | 656,511 | ||||||||||||||
| Stock-based compensation on 400,000 options granted | - | - | 197,637 | - | - | - | 197,637 | ||||||||||||||
| Stock-based compensation on 100,000 options granted | - | - | 33,483 | - | - | - | 33,483 | ||||||||||||||
| Stock-based compensation on 100,000 options granted | - | - | 36,140 | - | - | - | 36,140 | ||||||||||||||
| Stock-based compensation on 50,000 options granted | - | - | 7,967 | - | - | - | 7,967 | ||||||||||||||
| Stock-based compensation on 750,000 options granted | - | - | 72,792 | - | - | - | 72,792 | ||||||||||||||
| Stock-based compensation on 950,000 options granted | - | - | 211,274 | - | - | - | 211,274 | ||||||||||||||
| Stock-based compensation on 100,000 options granted | - | - | 15,445 | - | - | - | 15,445 | ||||||||||||||
| Fair value of non-employee vested options reallocated to derivative liability | - | - | (690,885 | ) | - | - | - | (690,885 | ) | ||||||||||||
| Net loss for the year | - | - | - | - | (6,881,812 | ) | - | (6,881,812 | ) | ||||||||||||
| Translation adjustments | - | - | - | - | - | (27,758 | ) | (27,758 | ) | ||||||||||||
| Balance – March 31, 2016 | 72,193,209 | 24,347,930 | 2,940,539 | - | (26,305,263 | ) | (999,398 | ) | (16,192 | ) |
(The accompanying notes are an integral part of these consolidated financial statements. )
F-8
| Helius Medical Technologies, Inc. |
| Consolidated Statements of Cash Flows |
| for the fiscal years ended March 31, 2016 and 2015 |
| (Expressed in United States Dollars) |
| March 31, 2016 | March 31, 2015 | |||||
| $ | $ | |||||
| Cash flows from operating activities | ||||||
| Net loss | (6,881,812 | ) | (9,838,317 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||
| Change in fair value of derivative liability | (2,082,703 | ) | 739,375 | |||
| Interest accretion | 29,045 | 176,488 | ||||
| Stock-based compensation | 1,231,250 | 2,340,876 | ||||
| Gain on extinguishment of debt | (268,334 | ) | - | |||
| Unrealized foreign exchange loss (gain) |
(120,876 |
) |
(598,929 |
) | ||
| Changes in operating assets and liabilities: | ||||||
| Receivables | (390,273 | ) | (8,945 | ) | ||
| Prepaids and other current assets | (91,644 | ) | (110,873 | ) | ||
| Accounts payable and accrued liabilities | 487,935 | 979,040 | ||||
| Shares to be issued | 150,000 | - | ||||
| Net cash used in operating activities | (7,937,412 | ) | (6,321,285 | ) | ||
| Cash flows from investing activities | ||||||
| Proceeds from (purchase of) the sale of short term investment | 378,000 | (378,000 | ) | |||
| Net cash provided by (used in) investing activities | 378,000 | (378,000 | ) | |||
| Cash flows from financing activities | ||||||
| Cash acquired on recapitalization | - | 23,904 | ||||
| Proceeds from the issuance of common stock, net | 7,636,910 | 6,637,203 | ||||
| Proceeds from shares to be issued | - | 39,545 | ||||
| Exercise of warrants and stock options | 54,426 | - | ||||
| Proceeds from issuance of convertible debt | 2,000,000 | 632,076 | ||||
| Proceeds from issuance of promissory note | 200,000 | - | ||||
| Repayment of promissory note | (200,000 | ) | - | |||
| Proceeds from bridge loan | - | 150,000 | ||||
| Net cash provided by financing activities | 9,691,336 | 7,482,728 | ||||
| Effect of foreign exchange rate changes on cash | 93,120 | (380,518 | ) | |||
| Net change in cash and cash equivalents | 2,225,044 | 402,925 | ||||
| Cash and cash equivalents, beginning of the period | 418,893 | 15,968 | ||||
| Cash and cash equivalents, end of the period | 2,643,937 | 418,893 | ||||
| Supplemental cash flow information | ||||||
| Interest paid in cash | $ | 1,651 | $ | 11,144 | ||
| Income taxes paid in cash | - | - |
(The accompanying notes are an integral part of these consolidated financial statements.)
F-9
Helius Medical Technologies, Inc.
Notes to the
Consolidated Financial Statements
Years ended March 31, 2016 and 2015
| 1. |
DESCRIPTION OF BUSINESS AND BASIS OF PRESENTATION |
Helius Medical Technologies, Inc. (the “Company”) is engaged primarily in the medical technology industry focused on neurological wellness. The Company’s planned principal operations include the development, licensing and acquisition of unique and non-invasive platform technologies to amplify the brain’s ability to heal itself. To date the Company has not generated any revenue.
The Company was incorporated in British Columbia, Canada, on March 13, 2014. On May 28, 2014, the Company completed a continuation via a plan of arrangement whereby the Company moved from being a corporation governed by the British Columbia Corporations Act to a corporation governed by the Wyoming Business Corporations Act. The Company’s head office is located in Newtown, Pennsylvania.
The Company is currently listed on the Toronto Stock Exchange (the “TSX”). The Company began trading on the Canadian Securities Exchange on June 23, 2014, under the ticker symbol “HSM”, and subsequently moved to the TSX on April 18, 2016. The Company also began trading on the OTCQB under the ticker symbol “HSDT” on February 10, 2015.
On June 13, 2014, the Company completed its acquisition of 100% of the issued and outstanding shares of Neurohabilitation Corporation (“Neuro”), a private company incorporated in Delaware, USA, on January 22, 2013. Prior to the transaction, the Company was a non-operating public shell company. Accordingly, for financial reporting purposes, this transaction was deemed to be a capital transaction in substance and recorded as a reverse recapitalization of Neuro whereby Neuro is deemed to be the continuing, surviving entity for accounting purposes, but through reorganization, has deemed to have adopted the capital structure of the Company. Because the acquisition was considered a reverse recapitalization for accounting purposes, the combined historical financial statements of Neuro became the historical financial statements and from the completion of the acquisition on June 13, 2014, the financial statements have been prepared on a consolidated basis. The assets and liabilities of Neuro have been brought forward at their book value and no goodwill has been recognized in connection with the transaction.
The Company had a wholly-owned subsidiary, 0995162 B.C. Ltd, which was dissolved on October 23, 2014. On December 17, 2014, Neuro incorporated a wholly-owned subsidiary, Helius Medical Technologies (Canada), Inc. (“Helius Canada”). The financial information is presented in United States Dollars.
Going Concern
The Company’s consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”). The Company’s consolidated financial statements have been prepared on the basis of continuity of operations, realization of assets and the satisfaction of liabilities in the ordinary course of business. The Company has incurred a net loss of $6,881,812 for the fiscal year ended March 31, 2016 and, as of March 31, 2016, the Company has an accumulated deficit of $26,305,263. The Company has not generated any product revenues and has not achieved profitable operations. Until the Company generates a level of revenue to support its cost structure, the Company expects to continue to incur substantial operating losses and net cash outflows. There is no assurance that profitable operations will ever be achieved, and, if achieved, will be sustained on a continuing basis.
While the Company had cash and cash equivalents of $2,643,937 as of March 31, 2016, management does not believe these resources will be sufficient to meet the Company’s operating and capital needs for the ensuing fiscal year.
F-10
The Company intends to fund ongoing activities by utilizing current cash and cash equivalents and by raising additional capital through equity or debt financings. There can be no assurance that the Company will be successful in raising additional capital or that such capital, if available, will be on terms that are acceptable to the Company. If the Company is unable to raise sufficient additional capital, the Company may be compelled to reduce the scope of its operations and planned capital expenditure or sell certain assets, including intellectual property assets. This material uncertainty gives rise to substantial doubt about the Company’s ability to continue as a going concern.
| 2. |
SIGNIFICANT ACCOUNTING POLICIES |
Use of Estimates
The preparation of the consolidated financial statements in accordance with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and reported amounts of expenses during the reporting period. Significant estimates include the assumptions used in the fair value pricing model for share-based payment transactions and deferred income tax asset valuation allowances. Financial statements include estimates which, by their nature, are uncertain. Actual outcomes could differ from these estimates.
Principles of Consolidation
The consolidated financial statements include the historic accounts of Neuro and are consolidated with Helius and its subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Cash and Cash Equivalents
Cash and cash equivalents comprise cash at banks and on hand, and short-term highly liquid investments that have an insignificant interest rate risk and an original maturity of 3 months or less.
Concentrations of Credit Risk
The Company is subject to credit risk in respect of its cash. Amounts invested in such instruments are limited by credit rating, maturity, industry group, investment type and issuer. The Company is not currently exposed to any significant concentrations of credit risk from these financial instruments. The Company seeks to maintain safety and preservation of principal and diversification of risk, liquidity of investments sufficient to meet cash flow requirements and a competitive after-tax rate of return.
Accounts Receivable
Accounts receivable are stated at their net realizable value. At March 31, 2016, the accounts receivable balance consisted primarily of GST and QST refunds related to the Company’s Canadian expenditures.
Stock-Based Compensation
The Company accounts for all stock-based payments and awards under the fair value based method. The Company recognizes its stock-based compensation using the straight line method.
Stock-based payments to non-employees are measured at the fair value of the consideration received, or the fair value of the equity instruments issued, or liabilities incurred, whichever is more reliably measurable. The fair value of stock-based payments to non-employees is periodically re-measured until the counterparty performance is complete, and any change therein is recognized over the vesting period of the award and in the same manner as if the Company had paid cash instead of paying with or using equity based instruments. The fair value of the stock-based payments to non-employees that are fully vested and non-forfeitable as at the grant date are measured and recognized at that date.
F-11
The Company accounts for the granting of share purchase options to employees using the fair value method whereby all awards to employees will be measured at fair value on the date of the grant. The fair value of all share purchase options are expensed over their vesting period with a corresponding increase to additional capital surplus. Upon exercise of share purchase options, the consideration paid by the option holder, together with the amount previously recognized in additional paid-in capital is recorded as an increase to share capital. Share purchase options granted to employees are accounted for as liabilities when they contain conditions or other features that are indexed to other than a market, performance or service condition.
The Company uses the Black-Scholes option pricing model to calculate the fair value of share purchase options. The use of the Black-Scholes option pricing model requires management to make assumptions with respect to the expected term of the option, the expected volatility of the common stock consistent with the expected term of the option, risk-free interest rates, the value of the common stock and expected dividend yield of the common stock. Changes in these assumptions can materially affect the fair value estimate.
Foreign Exchange
The functional currency of the Company and Helius Canada is the Canadian dollar (“CAD”) and the functional currency of Neuro is the U.S. dollar (“USD”). The Company’s reporting currency is the U.S. dollar.
Transactions in foreign currencies are remeasured into the functional currency of the relevant subsidiary at the exchange rate in effect at the date of the transaction. Any monetary assets and liabilities arising from these transactions are translated into the functional currency at exchange rates in effect at the balance sheet date or on settlement. Resulting gains and losses are recorded in other foreign exchange gain (loss) within the consolidated statements of operations.
The foreign exchange adjustment in the books of Neuro relating to inter-company advances from Helius that are denominated in Canadian dollars is recorded in the consolidated statements of operations and comprehensive loss. For the years ended March 31, 2016 and 2015, foreign exchange losses of ($18,785) and gains of $865,916 were recognized in the consolidated statements of operations and comprehensive loss.
Income Taxes
The Company accounts for income taxes using the asset and liability method. The asset and liability method provides that deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial reporting and tax bases of assets and liabilities, and for operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using the currently enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company records a valuation allowance to reduce deferred tax assets to the amount that is believed more likely than not to be realized.
The Company has adopted the provisions of Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 740 Income Taxes regarding accounting for uncertainty in income taxes. The Company initially recognizes tax provisions in the financial statements when it is more likely than not the position will be sustained upon examination by the tax authorities. Such tax positions are initially and subsequently measured as the largest amount of the tax benefit that is greater than 50% likely of being realized upon ultimate settlement with the tax authority, assuming full knowledge of the position and all relevant facts. Application requires numerous estimates based on available information. The Company considers many factors when evaluating and estimating its tax positions and tax benefits. These periodic adjustments may have a material impact on the consolidated statements of operations and comprehensive loss. When applicable, the Company classifies penalties and interest associated with uncertain tax positions as a component of income tax expense in its consolidated statements of operations and comprehensive loss.
F-12
Research and Development Expenses
Research and development (“R&D”) expenses consist primarily of personnel costs, including salaries, benefits and stock-based compensation, clinical studies performed by contract research organizations and materials and supplies. R&D costs are charged to operations when they are incurred.
Segment Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views their operations and manages their business in one segment.
Derivative Liabilities
The Company evaluates its financial instruments and other contracts to determine if those contracts or embedded components of those contracts qualify as derivatives to be separately accounted for in accordance with ASC 815 Derivatives and Hedging. The result of this accounting treatment is that the fair value of the derivative is marked-to-market at each balance sheet date and recorded as a liability and the change in fair value is recorded in the consolidated statements of operations and comprehensive loss. Upon conversion or exercise of a derivative instrument, the instrument is marked to fair value at the conversion date and then that fair value is reclassified to equity.
The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period. Derivative instruments that become subject to reclassification are reclassified at the fair value of the instrument on the reclassification date. Derivative instrument liabilities will be classified in the balance sheet as current or non-current based on whether or not settlement of the derivative instrument is expected within 12 months of the consolidated balance sheet date.
Fair Value Measurements
The Company’s financial instruments consist primarily of cash and cash equivalents, accounts receivable and the Ximedica project initiation deposit, and accounts payable and accrued liabilities. The book values of these instruments approximate their fair values due to the immediate or short-term nature of those instruments.
ASC 820 establishes a fair value hierarchy based on the level of independent, objective evidence surrounding the inputs used to measure fair value. A financial instrument’s categorization within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. ASC 820 prioritizes the inputs into three levels that may be used to measure fair value:
Level 1 – Quoted prices in active markets for identical assets or liabilities;
Level 2 – Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; and
Level 3 – Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The Company had certain Level 3 derivative liabilities required to be recorded at fair value on a recurring basis in accordance with U.S. GAAP as at March 31, 2016 and 2015. Unobservable inputs used in the valuation of these liabilities includes volatility of the underlying share price and the expected term. See Note 7. for the inputs used in the Black Scholes model at March 31, 2016 and the rollforward of the warrant liability and see Note 8. for the inputs used in the Black Scholes model at March 31, 2016 and 2015 for the rollforward of the derivative liability for non-employee options.
F-13
| Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||
| March 31, 2016 | |||||||||||||
| Liabilities: | |||||||||||||
| Non-employee options | 521,179 | 521,179 | |||||||||||
| Warrants | 1,204,581 | 1,204,581 | |||||||||||
| March 31, 2015 | |||||||||||||
| Assets: | |||||||||||||
| Short-term investments | 378,000 | 378,000 | |||||||||||
| Liabilities: | |||||||||||||
| Non-employee options | 1,581,444 | 1,581,444 | |||||||||||
| Warrants | - | - |
There were no transfers between any of the levels during the years ended March 31, 2016 and 2015.
Basic and Diluted Income (Loss) per Share
Earnings or loss per share (“EPS”) is computed by dividing net income (loss) available to common stockholders by the weighted average number of common shares outstanding for the period. Diluted EPS is computed by dividing net income (loss) by the weighted-average of all potentially dilutive shares of common stock that were outstanding during the periods presented.
The treasury stock method is used in calculating diluted EPS for potentially dilutive stock options and share purchase warrants, which assumes that any proceeds received from the exercise of in-the-money stock options and share purchase warrants, would be used to purchase common shares at the average market price for the period.
EPS for convertible debt is calculated under the “if-converted” method. Under the if-converted method, EPS is calculated as the more dilutive of EPS (i) including all interest (both cash interest and non-cash discount amortization) and excluding all shares underlying the convertible debt or; (ii) excluding all interest and costs directly related to the convertible debt (both cash interest and non-cash discount amortization) and including all shares underlying the convertible debt.
The basic and diluted loss per share for the years ended March 31, 2016 and 2015 were calculated as follows:
| Year ended March 31, 2016 |
Year ended March 31, 2015 |
|||||
| $ | $ | |||||
| Net loss, basic | (6,881,812 | ) | (9,838,317 | ) | ||
| Effect of dilutive securities: Change in fair value of derivative liability | (1,156,002 | ) | - | |||
| Net loss, diluted | (8,037,814 | ) | (9,838,317 | ) | ||
| Denominator Weighted average common shares outstanding |
66,522,564 | 57,048,406 | ||||
| Effective of dilutive
securities Warrants and options |
503,980 | - | ||||
| Diluted weighted average common shares outstanding | 67,026,545 | 57,048,406 | ||||
| Basic net loss per share | $ | (0.10 | ) | $ | (0.17 | ) |
| Diluted net loss per shares | $ | (0.12 | ) | $ | (0.17 | ) |
F-14
The following outstanding securities for the years ended March 31, 2016 and 2015 have been excluded from the computation of diluted weighted shares outstanding, as they would have been anti-dilutive:
| Year ended | Year ended | |||||
| March 31, 2016 | March 31, 2015 | |||||
| Options outstanding | 5,875,360 | 4,920,000 | ||||
| Warrants outstanding | 12,973,009 | 8,444,400 | ||||
| Total | 18,848,369 | 13,364,400 |
Recent Accounting Pronouncements
In March 2016, the FASB issued ASU 2016-09, Compensation—Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. The amendments in this update change existing guidance related to accounting for employee share-based payments affecting the income tax consequences of awards, classification of awards as equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 is effective for annual reporting periods beginning after December 15, 2016, including interim periods within those annual periods, with early adoption permitted. The Company is currently evaluating the potential impact of the adoption of this standard.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). The new standard establishes a right-of-use (“ROU”) model that requires a lessee to record a ROU asset and a lease liability on the consolidated balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the consolidated income statement. ASU 2016-02 is effective for annual periods beginning after December 15, 2018, including interim periods within those annual periods, with early adoption permitted. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. The Company is currently evaluating the potential impact of the adoption of this standard.
In January 2016, the FASB issued ASU 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities. The amendments in this update revise the accounting related to the classification and measurement of investments in equity securities and the presentation of certain fair value changes for financial liabilities measured at fair value. The amendments are effective for annual reporting periods after December 15, 2017, including interim periods within those fiscal years. Early adoption is permitted. The Company is currently evaluating the potential impact of the adoption of this standard.
In April 2015, the FASB issued ASU 2015-03, Interest- Imputation of Interest (Subtopic 835-30). This guidance is to simplify the presentation of debt issuance costs by recognizing a debt liability in the balance sheet as a direct deduction from that debt liability consistent with the presentation of a debt discount. The amendments in this update are effective for financial statements issued for fiscal years beginning after December 15, 2015, and interim periods within those fiscal years. The Company has adopted this standard and the adoption did not have a material impact on the Company’s financial position.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements - Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern within one year after the date that the financial statements are issued (or within one year after the date that the financial statements are available to be issued when applicable) and to provide related footnote disclosures. The ASU provides guidance to an organization’s management, with principles and definitions that are intended to reduce diversity in the timing and content of disclosures that are commonly provided by organizations today in the financial statement footnotes. The ASU is effective for annual periods ending after December 15, 2016, and interim periods within annual periods beginning after December 15, 2016, which for the Company is April 1, 2017. Early adoption is permitted. The adoption of this standard will not have a material impact on the Company’s financial position or results of operations.
F-15
The amendments also clarify that the guidance in Topic 275, Risks and Uncertainties, is applicable to entities that have not commenced planned principal operations. The central feature of the guidance on disclosure requirements is that required disclosures are limited to matters significant to a particular entity. The disclosures focus primarily on risks and uncertainties that could significantly affect the amounts reported in the financial statements in the near term or the near-term functioning of the reporting entity.
| 3. |
RECAPITALIZATION |
On June 13, 2014, the Company completed a recapitalization transaction where the Company acquired 100% of the issued and outstanding shares of Neuro. In exchange, the Company issued a total of 35,300,083 shares to the shareholders of Neuro which merged with a wholly-owned subsidiary of the Company, HMT Mergersub, for the purpose of the three-corner amalgamation. As a result, the former Neuro shareholders owned the majority of the outstanding shares of the Company upon completion of the transaction. Prior to the recapitalization transaction, the Company did not meet the definition of a business. Thus, the transaction is considered to be a capital transaction of Neuro accompanied by a recapitalization.
The ongoing Company has adopted the name Helius Medical Technologies, Inc. These consolidated financial statements present the results of Neuro with the exception of common stock which has been retroactively restated to reflect the Recapitalization. In connection with the Recapitalization, the Company advanced Neuro an unsecured loan in the amount of $150,000 (the “Bridge Loan”). The Bridge Loan was for a term of one year commencing on May 30, 2014, and was payable in a lump sum at the end of the term. The Bridge Loan bears interest at a rate of 8% per annum.
F-16
The net assets acquired were as follows,
| Cash and cash equivalents | $ | 23,904 | |
| Receivables | 1,644 | ||
| Bridge loan receivable | 150,000 | ||
| Prepaid expenses | 5,970 | ||
| Accounts payable and accrued liabilities | (18,628 | ) | |
| $ | 162,890 |
The recapitalization transaction reflects a credit to additional paid-in capital of $162,890, the carrying value of the net assets of the Company at the time of the reverse merger.
In connection to the completion of the transaction, the Company completed a private placement of 15,240,000 units at CAD $0.50 per unit for a total of $7,016,002 (CAD $7,620,000) (Note 7). Each unit consisted of one common share of the Company and one-half of a share purchase warrant. Each whole share purchase warrant is exercisable at CAD $1.00 for a period of twenty-four months. In respect of this private placement, the Company paid aggregate finders’ fees of $379,806 (CAD $412,200) and issued 824,400 finders’ warrants. Each finder’s warrant is exercisable at CAD $1.00 per share for a period of two years.
| 4. |
CONVERTIBLE DEBENTURE |
On February 19, 2014, the Company entered into a securities purchase agreement where the Company agreed to sell and issue a note with annual simple interest at 8% (the “Debenture”). A total of $1,000,100 in principal had been received. The Debenture matured on the earliest of (i) February 28, 2015 or such later date as agreed (ii) the closing of a transaction involving a change in control of the Company or (iii) the date of the closing of the Company’s qualified financing being an aggregate amount of at least $2,000,000 (“qualified financing”).
Upon completion of a qualified financing, the Debenture would automatically convert into equity securities of the Company at a price per share equal to 85% of the price per share of the qualified financing. If a qualified financing did not occur on or before the maturity date, at the option of the Company’s board of directors, the outstanding balance of the debenture would be converted into the Company’s equity securities at a conversion price per share determined using a valuation of $8.5 million and the number of shares outstanding at that date.
On June 13, 2014, the Debenture matured on the closing of the Company’s qualified financing and converted into 2,564,705 shares of the Company’s common stock.
The conversion option of the Debenture was accounted for as a contingent beneficial conversion feature valued at $176,488 which was recorded as interest expense in the consolidated statements of operations and comprehensive loss on settlement of the contingency. Upon conversion of the Debenture, the Company issued a total of 2,564,705 common shares. In addition, the Company paid the Debenture holders $11,131 with respect to the accrued and unpaid interest outstanding.
| 5. |
PROMISSORY NOTE |
On September 8, 2015, the Company received $200,000 in exchange for the issuance of a promissory note (the “Promissory Note”). The Promissory Note was to be repaid six months from the date of issuance with interest accruing at the rate of 6% per annum for the first three months and 10% per annum thereafter. In addition, the lender was entitled to receive 30,000 common shares of the Company on the date of the Promissory Note (the “Bonus Shares”) and an additional 30,000 common shares every three months thereafter as long as the principal of the loan remained outstanding. During the year ended March 31, 2016, the Company issued the lender 30,000 Bonus Shares valued at $29,045 based on their quoted market value to the lender. This amount was recorded as a debt discount of the Promissory Note at issuance and was being amortized using the effective interest method over the term of the Promissory Note.
F-17
On October 28, 2015, the Company repaid the Promissory Note in its entirety, along with accrued interest of $1,644. The remaining debt discount was immediately recorded as interest expense on the date of repayment.
| 6. |
CONVERTIBLE NOTE |
On October 9, 2015, in connection with an Asset Purchase Agreement, under which the Company licensed the use of its intellectual property in the People’s Republic of China, Taiwan, Singapore, Hong Kong, and the Macau Special Administrative Region, the Company entered into a US$7.0 million funding commitment with A&B Company Limited (“A&B”) in the form of a convertible promissory note. The funding commitment consisted of (i) an initial $2.0 million under the Note (“$2.0 million note” and (ii) an additional $5.0 million funding commitment, upon which the Company could draw down at any time or from time to time during the six-month period beginning on the issuance date of the promissory note. The $2.0 million note would accrue interest at a rate equal to 6% per annum, payable in cash on the due date of April 9, 2016. The $2.0 million note was unsecured and was convertible at the option of the holder into units of the Company at $0.96 per unit. Each unit would consist of one share of common stock and one half share purchase warrant exercisable at $1.44 for a period of three years from the date of issuance.
Pursuant to the guidance of ASC 815 Derivatives and Hedging, the Company determined that the conversion feature embedded in the $2.0 Million Note was required to be bifurcated from the Note and accounted for as a derivative liability because it was considered not to be indexed to the Company’s stock due to its exercise price being denominated in a currency other than the Company’s functional currency. Therefore, pursuant to the guidance of ASC 815-15, the Company allocated the proceeds from the issuance of the $2.0 million note first to the fair value of the embedded conversion feature, with a corresponding discount allocated to the Note. This resulted in a debt discount of $425,208. This debt discount would be amortized using the effective interest method over the term of the $2.0 million note. During the year ended March 31, 2016, the Company did not record any accretion in respect of this discount, because the $2.0 million note was immediately converted, as noted below.
On October 9, 2015, the Company received the conversion notice and on November 10th, 2015, the Company issued 2,083,333 shares of common stock at a price of $0.96 per share and 1,041,667 warrants exercisable at $1.44 for a period of three years from the date of issuance. The shares of common stock and the warrants were issued on November 10, 2015. Pursuant to the guidance of ASC 815 Derivatives and Hedging, the Company determined that the warrants are required to be accounted for as liabilities because it they are considered not to be indexed to the Company’s stock due to the exercise price being denominated in a currency other than the Company’s functional currency. The fair value of these warrants was determined to be $206,667 using the Black-Scholes option pricing model. See Note 7. for the derivative liability rollforward.
As a result of the bifurcation of the embedded conversion option, for accounting purposes, two instruments were considered outstanding and, upon exercise of the contractual conversion option, extinguishment accounting has been applied. Consequently, the shares issued pursuant to the conversion are recorded at their fair value on the date of issuance, determined with reference to their quoted market price on the date of conversion. The resulting difference between the fair value of the shares issued, less the fair value of the related conversion feature and the carrying value of the related debt, is recorded as a gain or loss on the consolidated statement of operations. During the year ended March 31, 2016, the Company recorded a gain on extinguishment of debt of $268,334 in connection with the conversion of the Note.
The Company could elect to draw down on the additional $5.0 million funding through the issuance of units of the Company at a price based on the volume weighted average closing price of the Company’s shares of common stock on the date the Company elects to draw down from the commitment (the “Draw Down Price”). Each unit would consist of one shares of common stock of the Company and one half share purchase warrant. The warrant would be exercisable at the price representing a fifty percent (50%) premium to the Draw Down Price.
On December 29, 2015, the Company drew down the remaining $5.0 million commitment through the issuance of 5,555,556 shares of common stock at a price of $0.90 per share and 2,777,778 warrants exercisable at $1.35 for a period of three years from the date of issuance. The shares of common stock and the warrants were issued on January 7, 2016.
F-18
Pursuant to the guidance of ASC 815 Derivatives and Hedging, the Company determined that the warrants are required to be accounted for as liabilities because they are considered not to be indexed to the Company’s stock due to the exercise price being denominated in a currency other than the Company’s functional currency. Consequently, the Company allocated the proceeds from the $5.0 million funding initially to the warrants at their fair value, with the remainder allocated to the common shares. The fair value of these warrants was determined to be $796,945 using the Black Scholes option pricing model. See Note 7. for the derivative liability rollforward relating to these warrants.
| 7. |
COMMON STOCK AND WARRANTS |
As of March 31, 2016, the Company’s certificate of incorporation authorized the Company to issue unlimited Class A common shares without par value. Each Class A common share is entitled to have the right to vote at any shareholder meeting on the basis of one vote per share. Each Class A share held entitles the holder to receive dividends as declared by the directors. No dividends have been declared through March 31, 2016. In the event of the liquidation, dissolution or winding-up of the Company other distribution of assets of the Company among its shareholders for the purposes of winding-up its affairs or upon a reduction of capital the holders of the Class A common shares shall, share equally, share for share, in the remaining assets and property of the Company.
The Company is subject to a stockholders agreement, which places certain restrictions on the Company’s stock and its stockholders. These restrictions include approvals prior to sale or transfer of stock, a right of first refusal to purchase stock held by the Company and a secondary right of refusal to stockholders, right of co-sale whereby certain stockholders may be enabled to participate in a sale of other stockholders to obtain the same price, term and conditions on a pro-rata basis, rights of first offer of new security issuances to current stockholders on a pro-rata basis and certain other restrictions.
Upon completion of the Recapitalization, the Company issued a total of 35,300,083 shares to the shareholders of Neuro (Note 3). In connection with the Recapitalization, the Company also closed a non-brokered private placement (the “Private Placement”) at CAD $0.50 per unit of 15,240,000 units raising $7,016,002 on May 30, 2014 (Note 3). Each unit consists of one common stock of the Company and one half of a warrant of the Company where one full warrant is exercisable for 2 years at CAD $1.00 into one common stock. The fair value of the warrants issued was determined using the Black Scholes option-pricing model and the Company used the relative fair value method to allocate $578,961 of the gross proceeds to Additional Paid-in Capital to account for the warrants issued.
On April 30, 2015, the Company closed a non-brokered private placement (the “First Financing”) raising gross proceeds of $1,825,937 by the issuance of 849,273 units (each a “First Financing Unit”) at a price of $2.15 per First Financing Unit. Each First Financing Unit consists of one (1) common share and one half of one (1/2) common share purchase warrant (each a “First Financing Warrant”). Each whole First Financing Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $3.00 per share for a period of thirty-six (36) months from the closing date of the Financing. The Company paid a cash finder’s fee of $84,074 in connection with this First Financing, as well as 27,396 finder’s warrants (the “First Financing Finder’s Warrants”). Each First Financing Finder’s Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $3.00 per share for a period of thirty-six (36) months from the closing date of the First Financing.
On June 26, 2015, the Company closed a non-brokered private placement (the “Second Financing”) raising gross proceeds of $721,243 by the issuance of 335,463 units (each a “Second Financing Unit”) at a price of $2.15 per Second Financing Unit. Each Second Financing Unit consists of one (1) common share and one half of one (1/2) common share purchase warrant (each a “Second Financing Warrant”). Each whole Second Financing Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $3.00 per share for a period of thirty-six (36) months from the closing date of the Second Financing. The Company paid a cash finder’s fee of $40,803 in connection with this Second Financing, as well as 18,978 finder’s warrants (the “Second Financing Finder’s Warrants”). Each Second Financing Finder’s Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $2.15 per share for a period of sixty (60) months from the closing date of the Second Financing.
F-19
On July 17, 2015, the Company closed a non-brokered private placement (the “Third Financing”) raising gross proceeds of $270,375 by the issuance of 125,756 units (each a “Third Financing Unit”) at a price of $2.15 per Third Financing Unit. Each Third Financing Unit consists of one (1) common share and one half of one (1/2) common share purchase warrant (each a “Third Financing Warrant”). Each whole Third Financing Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $3.00 per share for a period of thirty-six (36) months from the closing date of the Third Financing. The Company paid a cash finder’s fee of $16,223 in connection with this Third Financing, as well as 7,545 finder’s warrants (the “Third Financing Finder’s Warrants”). Each Third Financing Finder’s Warrant entitles the holder thereof to purchase one additional common share of the Company at a price of $2.15 per share for a period of sixty (60) months from the closing date of the Third Financing.
On November 10, 2015, upon conversion of the $2.0 million Note, the Company issued 2,083,333 shares of common stock at a price of $0.96 per share and 1,041,667 warrants exercisable at $1.44 for a period of three years from the date of issuance. See Note 6 “Convertible Note”.
On December 29, 2015, the Company drew down the remaining $5.0 million commitment through the issuance of 5,555,556 shares of common stock at a price of $0.90 per share and 2,777,778 warrants exercisable at $1.35 for a period of three years from the date of issuance. The shares of common stock and the warrants were issued on January 7, 2016. See Note 6 “Convertible Note”.
Pursuant to the guidance of ASC 815 Derivatives and Hedging, the Company determined that all of the warrants issued during the year ended March 31, 2016 as described above are required to be accounted for as liabilities because they are considered not to be indexed to the Company’s stock due to the exercise price being denominated in a currency other than the Company’s functional currency. Consequently, the Company determined the fair value of each warrant issuance using the Black-Scholes option pricing model, with the remainder of the proceeds allocated to the common shares.
The warrants having an exercise price denominated in a currency other than the functional currency of the Company that are required to be accounted for as liabilities are summarized as follows for the years ended March 31, 2016 and 2015:
| Year ended March 31, 2016 $ |
Year ended March 31, 2015 $ | |
| Fair value of warrants, beginning of the period | - | - |
| Issuance | 1,536,134 | - |
| Change in fair value of warrants during the period | (331,553) | - |
| Fair value of warrants, end of the period | 1,204,581 | - |
The warrants are required to be re-valued with the change in fair value of the liability recorded as a gain or loss in the change of fair value of derivative liability, included in other income (expense) in the Company’s consolidated statements of operations and comprehensive loss at the end of each reporting period. The fair value of the warrants will continue to be classified as a liability until such time as they are exercised, expire or there is an amendment to the respective agreements that renders these financial instruments to be no longer classified as a liability.
The fair value of warrants as of March 31, 2016 were estimated using the Black-Scholes option pricing model with the following weighted average assumptions:
| March 31, 2016 | |
| Stock price | $0.78 |
| Exercise Price | $1.62 |
| Expected life | 2.65 years |
| Expected volatility | 83.86% |
| Risk – free interest rate | 0.83% |
| Dividend rate | 0.00% |
F-20
The continuity of warrants for the year ended March 31, 2016 is as follows:
| Number of warrants | Weighted Average Exercise Price | |||||||||||
| CAD | US | CAD | US | |||||||||
| Balance, March 31, 2015 | 8,444,400 | - | $ | 1.00 | - | |||||||
| Granted | 4,528,609 | $ | - | 1.62 | ||||||||
| Exercised | (14,400 | ) | - | $ | 1.00 | - | ||||||
| Balance, March 31, 2016 | 8,430,000 | 4,528,609 | $ | 1.00 | 1.62 | |||||||
The warrants outstanding and exercisable at March 31, 2016 are as follows:
| Number of warrants outstanding | Exercise Price | Expiry Date |
| 8,430,000 | CAD $1.00 | May 30, 2016 |
| 452,032 | US $ 3.00 | April 30, 2018 |
| 167,731 | US $ 3.00 | June 26, 2018 |
| 18,978 | US $ 2.15 | June 26, 2020 |
| 62,878 | US $ 3.00 | July 17, 2018 |
| 7,545 | US $ 2.15 | July 17, 2020 |
| 1,041,667 | US $ 1.44 | November 10, 2018 |
| 2,777,778 | US $ 1.35 | December 29, 2018 |
| 8. |
SHARE BASED PAYMENTS |
On June 18, 2014, the Company’s Board of Directors authorized and approved the adoption of the 2014 Plan (“2014 Plan”), under which an aggregate of 12,108,016 shares of common stock may be issued. Pursuant to the terms of the 2014 Plan, the Company is authorized to grant stock options, as well as awards of stock appreciation rights, restricted stock, unrestricted shares, restricted stock units and deferred stock units. These awards may be granted to directors, officers, employees and eligible consultants. Vesting and the term of an option is determined at the discretion of the Board of Directors of the Company. At March 31, 2016, there were 5,432,656 common shares remaining available for grant under the 2014 Plan.
The continuity of stock options for the year ended March 31, 2016is as follows:
| Weighted | |||||||||
| Average | Aggregate | ||||||||
| Exercise Price | Intrinsic Value | ||||||||
| Number | (CAD) | (CAD) | |||||||
| Balance outstanding at March 31, 2015 | 4,920,000 | $ | 1.14 | $ | 10,120,000 | ||||
| Exercised | (94,640 | ) | $ | 0.60 | - | ||||
| Granted | 1,850,000 | $ | 0.88 | - | |||||
| Balance outstanding at March 31, 2016 | 6,675,360 | $ | 1.08 | $ | 1,580,883 | ||||
| Balance exercisable at March 31, 2016 | 4,336,864 | $ | 1.16 | $ | 1,064,963 |
The options outstanding and exercisable at March 31, 2016 are as follows:
F-21
| Options | |||||||||||||||
| outstanding | |||||||||||||||
| remaining | Number of | ||||||||||||||
| Number of | contractual life | Exercise | Grant date fair | options | |||||||||||
| options | Expiry date | (years) | Price (CAD) | value (CAD) | exercisable | ||||||||||
| 3,520,000 | June 18, 2019 | 3.22 | $ | 0.60 | $ | 0.26 | 2,346,669 | ||||||||
| 155,360 | June 20, 2019 | 3.22 | $ | 0.60 | $ | 0.26 | 155,360 | ||||||||
| 100,000 | July 14, 2017 | 1.29 | $ | 2.52 | $ | 1.05 | 100,000 | ||||||||
| 450,000 | December 8, 2019 | 3.69 | $ | 2.92 | $ | 1.65 | 450,000 | ||||||||
| 100,000 | December 8, 2019 | 3.69 | $ | 2.92 | $ | 1.31 | 66,667 | ||||||||
| 400,000 | December 8, 2019 | 3.69 | $ | 2.96 | $ | 1.29 | 300,000 | ||||||||
| 100,000 | March 16, 2020 | 4.96 | $ | 3.20 | $ | 1.42 | 66,667 | ||||||||
| 50,000 | August 15, 2015 | 4.38 | $ | 0.98 | $ | 0.39 | 16,667 | ||||||||
| 750,000 | October 21, 2020 | 4.56 | $ | 0.87 | $ | 0.36 | 187,500 | ||||||||
| 550,000 | October 28, 2020 | 4.58 | $ | 0.84 | $ | 0.44 | 550,000 | ||||||||
| 400,000 | October 28,2020 | 4.58 | $ | 0.84 | $ | 0.36 | 64,000 | ||||||||
| 100,000 | December 31, 2020 | 4.76 | $ | 1.24 | $ | 0.50 | 33,334 | ||||||||
| 6,675,360 | 4,336,864 |
Included in the table above are non-employee awards that are subject to remeasurement each reporting period until vested. As a result, the grant date fair value is not representative of the total expense that will be recorded for these awards. As of March 31, 2016, the unrecognized compensation cost related to non-vested stock options outstanding, was $440,633 to be recognized over a weighted-average remaining vesting period of approximately 0.76 years.
The fair value of the employee and director stock options granted during the years ended March 31, 2016 and 2015 were estimated using the Black-Scholes option pricing model with the following weighted average assumptions:
| March 31, 2016 | March 31, 2015 | |
| Stock price | $0.82 CAD | $1.85 CAD |
| Exercise Price | $0.87 CAD | $2.20 CAD |
| Expected life | 3.75 years | 3.9 years |
| Expected volatility | 67.85% | 67.85% |
| Risk – free interest rate | 0.63% | 1.32% |
| Dividend rate | 0.00% | 0.00% |
The Company has adopted the simplified method prescribed by the SEC in SAB Topic 14 in respect of estimating the expected term of its stock options as its limited share purchase option history does not provide a reasonable basis to estimate the expected terms. Expected volatility was determined by reference to the average volatility rates of other companies in the same industry due to the Company’s limited trading history. The Company recognizes compensation expense for only the portion of awards that are expected to vest. For the years ended March 31, 2016 and 2015, the Company applied an expected forfeiture rate of 0% based on its historical experience.
Non-Employee Stock Options
In accordance with the guidance of ASC 815-40-15, stock options awarded to non-employees that are performing services for Neuro are required to be accounted for as derivative liabilities once the services have been performed and the options have vested because they are considered not to be indexed to the Company’s stock due to their exercise price being denominated in a currency other than Neuro’s functional currency. Stock options awarded to non-employees that are not vested are re-measured at their respective fair values at each reporting period and accounted for as equity awards until the terms associated with their vesting requirements have been met. The changes in fair of the unvested non-employee awards are reflected in their respective operating expense classification in the Company’s consolidated statements of operations and comprehensive loss.
F-22
The non-employee stock options that are required to be accounted for as liabilities are summarized as follows for the years ended March 31, 2016 and 2015:
| Year ended March 31, 2016 $ |
Year ended March 31, 2015 $ | |
| Fair value of non-employee options, beginning of the period | 1,581,444 | - |
| Issuance | - | 767,879 |
| Reallocation of vested non-employee options | 690,885 | 74,190 |
| Change in fair value of non-employee stock options during the period | (1,751,150) | 739,375 |
| Fair value of non-employee options, end of the period | 521,179 | 1,581,444 |
The non-employee options that have vested are required to be re-valued with the change in fair value of the liability recorded as a gain or loss on the change of fair value of derivative liability and included in other items in the Company’s consolidated statements of operations and comprehensive loss at the end of each reporting period. The fair value of the options will continue to be classified as a liability until such time as they are exercised, expire or there is an amendment to the respective agreements that renders these financial instruments to be no longer classified as a liability.
The fair value of non-employee liability classified awards at March 31, 2016 and 2015 were estimated using the Black-Scholes option pricing model with the following weighted-average assumptions:
| March 31, 2016 | March 31, 2015 | |
| Stock price | $0.97 CAD | $3.20 CAD |
| Exercise Price | $1.52 CAD | $1.90 CAD |
| Expected life | 3.23 years | 4.24 years |
| Expected volatility | 84.62% | 67.85% |
| Risk – free interest rate | 0.53% | 1.31% |
| Dividend rate | 0.00% | 0.00% |
Share-based payments are classified in the Company’s statements of operations and comprehensive loss as follows for the years ended March 31, 2016 and 2015:
| Year ended March 31, 2016 $ |
Year ended March 31, 2015 $ | |
| Consulting fees | 239,064 | 1,167,281 |
| Research and development | 158,396 | 721,601 |
| Wages and salaries | 833,790 | 451,994 |
| 1,231,250 | 2,340,876 |
F-23
| 9. |
INCOME TAXES |
The components of net loss for the years ended March 31, 2016 and 2015 are as follows:
| 2016 | 2015 | |||||
| $ | $ | |||||
| U.S | 5,132,611 | 9,301,988 | ||||
| Non-U.S. | 1,749,201 | 536,329 | ||||
| 6,881,812 | 9,838,317 |
A reconciliation of the income tax provision computed at statutory rates to the reported income tax provision for the years ended March 31, 2016 and 2015 is as follows:
| 2016 | 2015 | |||||
| $ | $ | |||||
| Statutory tax rate | 34.00% | 34.00% | ||||
| Loss before income taxes | (6,882,812 | ) | (9,838,317 | ) | ||
| Expected income tax recovery | (2,340,000 | ) | (3,345,000 | ) | ||
| Increase (decrease) in income tax recovery resulting from: | ||||||
| Derivative liability | (708,000 | ) | 251,000 | |||
| Share based payments | 419,000 | 796,000 | ||||
| Other permanent difference | 49,000 | 12,000 | ||||
| Share issue costs | - | (140,000 | ) | |||
| Effect of change in statutory rate | (147,000 | ) | (41,000 | ) | ||
| Effect of foreign exchange | - | 89,000 | ||||
| Effect of over provision in prior year | (2,164,000 | ) | - | |||
| Foreign income taxed at foreign rate | 100,000 | 14,000 | ||||
| Increase in valuation allowance | 4,791,000 | 2,364,000 | ||||
| Income tax expense | - | - |
The significant components of the Company’s deferred income tax assets and liabilities after applying enacted corporate tax rates at March 31, 2016 and 2015 are as follows:
| 2016 | 2015 | |||||
| $ | $ | |||||
| Deferred income tax assets (liabilities) | ||||||
| Operating losses carried forward | 4,915,000 | 2,074,000 | ||||
| Intangible costs | - | 285,000 | ||||
| Share issuance costs | - | 99,000 | ||||
| Stock compensation | 1,725,000 | - | ||||
| Other | 609,000 | |||||
| Valuation allowance | (7,249,000 | ) | (2,458,000 | ) | ||
| Net deferred income tax asset | - | - |
At March 31, 2016, the Company has accumulated non-capital losses totaling $1,839,000 in Canada and net operating losses of $12,616,000 in the U.S., which may be available to carry forward and offset future years’ taxable income. The losses expire in various amounts starting in 2033.
F-24
Under the provisions of the Internal Revenue Code, the net operating loss carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. Net operating loss carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period in excess of 50 percent, as defined under Section 382 of the Internal Revenue Code, as well as similar state provisions. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount of the annual limitation is determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years.
Uncertain Tax Positions
The Company has adopted certain provisions of ASC 740, “Income Taxes”, which prescribes a recognition threshold and measurement attribute for the recognition and measurement of tax positions taken or expected to be taken in income tax returns. The provisions also provide guidance on the de-recognition of income tax assets and liabilities, classification of current and deferred income tax assets and liabilities, and accounting for interest and penalties associated with tax positions.
The Company files income tax returns in the U.S. federal jurisdiction, and in various state and foreign jurisdictions. The Company’s tax returns are subject to tax examinations by U.S. federal and state tax authorities, or examinations by foreign tax authorities until the expiration of the respective statutes of limitation. The Company currently has no tax years under examination.
At March 31, 2016, the Company does not have an accrual relating to uncertain tax positions. It is not anticipated that unrecognized tax benefits would significantly increase or decrease within 12 months of the reporting date.
| 10. |
COMMITMENTS AND CONTINGENCIES |
| (a) |
On January 22, 2013, the Company entered into a license agreement with ANR for an exclusive right on ANR’s patent pending technology, claims and knowhow. In addition to the issuance of 16,035,026 shares, the Company agreed to pay a 4% royalty on net revenue on the sales of devices covered by the patent-pending technology and services related to the therapy or use of devices covered by the patent-pending technology. The Company has not made any royalty payments to date under this agreement. |
| (b) |
On March 7, 2014, the Company entered into a commercial development-to-supply program with Ximedica where Ximedica will design, develop and produce PoNS™ product solution suitable for clinical trial and commercial sale. Under the program, the Company is responsible for ensuring the device is in compliance with relevant laws and regulations. The agreed budget for phase 1B of development is $499,000; phase 2 is $1,065,000; Phase 3 and 4 is $1,389,000 and 2nd software development cycle is $586,000, of which $5,191,368 was expensed as research and development since inception to March 31, 2016. Invoices are to be issued monthly for work in progress. The Company can cancel the project at any time with a written notice at least 30 days prior to the intended date of cancellation. The Company recorded a prepaid expense of $274,000 to Ximedica for the upcoming clinical build of the PoNS™ device. As of March 31, 2016, the Company has expensed $120,448 of the $274,000 prepayment. As of March 31, 2016, the Company also recorded $300,000 project initiation deposit which will be applied once the development-to-supply program has been completed. During the years ended March 31, 2016 and 2015, the Company incurred R&D charges of $1,937,817 and $2,928,289 pursuant to this agreement. |
| (c) |
On January 27, 2015, the Company received a demand letter containing allegations that it had entered into a consulting arrangement with the complainants and breached certain of its terms, and used certain intellectual property in the form of business and marketing plans allegedly prepared by the complainants, and seeking damages. On May 7, 2015, Mr. Rainier Maas and Dr. Jochen Scheld filed a complaint in the U.S. District Court for the Eastern District of Pennsylvania seeking monetary damages in excess of $225,000. On December 2, 2015 the Company entered into a settlement agreement with the plaintiffs for an amount of €57,000 which was subsequently paid on January 12, 2016. The parties have since executed the settlement agreement for the aforementioned amount and the case has been dismissed without prejudice. |
F-25
| (d) |
On January 5, 2015, Wicab Inc. (“Wicab”) filed a complaint against the Company, NHC, its director Mitchell Tyler, and its former director Yuri Danilov, and ANR in the U.S. District Court for the Western District of Wisconsin. The complaint contained various state and common law claims arising from Messrs. Danilov’s and Tyler’s prior employment with Wicab and the Company’s two issued patents for the PoNS™ device. The complaint alleged, among other things, that following their departure from Wicab, Messrs. Danilov and Tyler knowingly filed patent applications for and used ideas and inventions developed at Wicab in violation of various non-competition and confidentiality agreements, and that the Company’s two issued patents are therefore rightfully the property of Wicab. The complaint sought an unspecified amount of monetary damages, an injunction preventing the Company from using the ideas and inventions in the two patents, an order transferring ownership of the patents from the Company to Wicab, and recovery of costs and attorneys’ fees. The complaint was voluntarily dismissed without prejudice on January 14, 2015. |
On October 12, 2015, the Company received a letter from Wicab alleging that the two issued patents were invalid in view of prior art cited in the letter, including scientific publications and patent applications, and that Paul Bach-y-Rita, Wicab’s founder, should have been named as an inventor on these two issued patents. Wicab indicated in the letter that it may file reexamination or inter partes review proceedings with the U.S. Patent Office to attempt to invalidate the claims in the two issued patents. Wicab also stated that it would consider an unspecified “business solution” to resolve this matter. On December 10, 2015, representatives of each of the Company and Wicab met to discuss the parameters of a potential settlement. There can be no guarantee that a settlement will be reached. In the event that a settlement with Wicab is not reached, Wicab may file reexamination or inter partes review proceedings with the U.S. Patent Office to challenge the validity of the two issued patents. If the Company receives an adverse decision from the U.S. Patent Office in connection with these proceedings, some or all of the claims in the two patents may be invalidated or otherwise impaired, which could prevent the Company from bringing an infringement suit against a future competitor for making use of the PoNS™ technology for neurorehabilitation, and could have a material adverse effect on the Company’s business, operating results and financial condition. Wicab may also take other actions against the Company, its assets, intellectual property rights, officers, directors, employees, agents or other persons or entities which may also have a material effect on the Company’s business, operating results and financial condition. The Company believes that the possibility of an economic outlay is remote.
| (e) |
Under the Company’s Asset Purchase Agreement with A&B, if the Company fails to obtain FDA clearance for commercialization of or otherwise fail to ensure that the PoNS™ device is available for purchase by the U.S. Government by December 31, 2017, the Company is subject to a US$2,000,000 contract penalty payable to A&B. |
| (f) |
In November 2014, the Company signed a development and distribution agreement with the Altair company in Russia to apply for registration and distribute the PoNS™ device in the territories of the former Soviet Union. However, there is no assurance that such commercialization will occur. |
| 11. |
RELATED PARTY TRANSACTIONS |
During the years ended March 31, 2016 and 2015, the Company paid $64,210 and $47,100 in consulting fees to directors of the Company. This expense was included in research and development expense. At March 31, 2016, the Company owed $3,450 to a director for consulting services (March 31, 2015: 24,418).
During the years ended March 31, 2016 and 2015, a benefit of $195,709 and an expense of $1,040,854 was included in research & development expense as the fair value of stock-based compensation attributed to the options granted to two directors and a consultant for consulting services rendered with respect to the design and development of the PoNS™ device.
| 12. |
SOLE-SOURCE COST-SHARING AGREEMENT |
During the fiscal year ended March 31, 2016, the Company entered into a sole source cost sharing contract executed with the USAMRMC. Under the terms of the contract, the USAMRMC will reimburse the Company up to a maximum of $2,996,244 representing approximately 62% of the Company’s estimated costs for the registrational trial (“the trial”) investigating the safety and effectiveness of the portable neuromodulation stimulator for mild to moderate traumatic brain injury. The trial expires on December 31, 2016 however the Company is working with the USAMRMC to extend the contract into 2017 based on the current trial forecast timelines. As of March 31, 2016, the Company has received a total of $1,458,374 in respect of expenses reimbursed. All reimbursement amounts received are credited directly to the accounts in which the original expense is recorded, including research and development, wages and salaries, and legal expenses.
Under the terms of the agreement, the USAMRMC may terminate their obligation at any time with 30 days written notice.
F-26
| 13. |
SUPPLEMENTAL CASH FLOW INFORMATION |
Investing and financing activities that do not have a direct impact on current cash flows are excluded from the consolidated statements of cash flows.
During the year ended March 31, 2016, the Company had the following non-cash financing transactions:
|
|
i) |
Fair value of warrants issued in conjunction with private placements in April, June and July 2015 was $532,522. |
|
|
|
|
|
|
ii) |
Fair value of warrants issued in conjunction with A&B credit facility (including upon conversion of $2.0 million convertible note and draw down of $5.0 million) was $1,003,612. |
|
|
|
|
|
iii) |
The Company issued 30,000 shares of common stock having a fair value of $29,045 based on the quoted market price as a bonus in connection with the issuance of a promissory note; | |
| iv) | A gain on extinguishment of debt of $268,334 based on the difference between the fair value of the shares issued to settle the A&B credit facility and the fair value of the related conversion feature and the carrying value of the related debt. |
During the year ended March 31, 2015:
|
|
i) |
the Company issued 2,564,705 common shares valued at $1,000,100 based on the carrying value of the convertible debenture upon its conversion; |
|
|
|
|
|
|
ii) |
the Company recorded a beneficial conversion feature of $176,488 in respect of a qualifying transaction recorded in connection with the convertible debenture; |
|
|
|
|
|
|
iii) |
the Company recorded a credit to additional paid-in capital of $162,890 representing the carrying values of the net assets acquired in a reverse merger recapitalization transaction. |
| 14. |
SUBSEQUENT EVENTS |
On April 18, 2016, the Company closed its short form prospectus offering in Canada and a concurrent U.S. private placement (the "Offering") of units (the "Units") with gross proceeds to the Company of CAD $9,215,000 through the issuance of Units at a price of CAD $1.00 per Unit. Each Unit consists of one Class A common share in the capital of the Company (a “Common Share”) and one half of one Common Share purchase warrant (each whole warrant, a “Warrant”). Each Warrant entitles the holder thereof to acquire one additional Common Share at an exercise price of CAD $1.50 on or before April 18, 2019. Mackie Research Capital Corporation (the "Agent") acted as agent and sole bookrunner in connection with the Offering. The Company paid the Agent a cash commission of CAD $436,050 and has granted to the Agent compensation options exercisable to purchase 436,050 Units at an exercise price of CAD $1.00 per Unit for a period of 24 months from the closing of the Offering.
As at March 231, 2016, the Company had received $150,000 in respect of the Offering; these funds were reflected as a current liability on Company's consolidated balance sheet as the issuance of the Units in connection with these proceeds was contingent on the Company achieving a minimum financing threshold. Subsequent to March 31, 2016, the Company issued the Units in connection with these proceeds.
In addition, the Company has recorded other current assets of $495,415 which represent legal fees pertaining to the Offering invoiced to the Company prior to March 31, 2016. The amount will be recorded as a share issuance cost upon closing of the Offering.
F-27
On May 2, 2016, the Company closed the sale of the additional units issued pursuant to the exercise of the over-allotment option (“Over-Allotment Option”) granted to the Agent in connection with the Offering. The Offering was made pursuant to a short form prospectus filed with the securities regulatory authorities in each of the provinces of Canada, except Québec. Pursuant to the exercise of the Over-Allotment Option, the Company issued an additional 1,090,125 Units (the "Over-Allotment Units") at a price of CAD $1.00 per Over-Allotment Unit for additional gross proceeds to the Company of CAD $1,090,125, bringing the total aggregate gross proceeds to the Company under the Offering to CAD $10,305,125. Each Over-Allotment Unit consists of one Class A common share in the capital of the Company (an “Over-Allotment Common Share”) and one half of one Common Share purchase warrant (each whole warrant, an “Over-Allotment Warrant”). Each Over-Allotment Warrant entitles the holder thereof to acquire one additional Over-Allotment Common Share at an exercise price of CAD $1.50 on or before April 18, 2019. In connection with the closing of the Over-Allotment Option, the Company paid the Agent a cash commission of CAD $65,408 and granted to the Agent compensation options exercisable to purchase 65,407 Over-Allotment Units at an exercise price of CAD $1.00 per Over-Allotment Unit for a period of 24 months from the closing of the Offering.
On June 6th, 2016, the Company announced that it received proceeds of CAD $1,825,600 from the exercise of outstanding warrants which were issued in connection with the Company’s private placement of subscription receipts that closed on May 30, 2014.
F-28
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| HELIUS MEDICAL TECHNOLOGIES, INC. | ||
| Dated: October 31, 2016 | By: | /s/ Philippe Deschamps |
| Philippe Deschamps | ||
| President, Chief Executive Officer and a Director | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| By | /s/ Philippe Deschamps | Date: October 31, 2016 | |
| Philippe Deschamps | |||
| President, Chief Executive Officer and a | |||
| Director | |||
| By | /s/ Joyce LaViscount | Date: October 31, 2016 | |
| Joyce LaViscount | |||
| Chief Financial Officer (Principal Accounting | |||
| Officer), and Corporate Secretary | |||
| By | /s/ Savio Chiu | Date: October 31, 2016 | |
| Savio Chiu | |||
| Director | |||
| By | /s/ Blane Walter | Date: October 31, 2016 | |
| Blane Walter | |||
| Director | |||
| By | /s/ Mitchell E. Tyler | Date: October 31, 2016 | |
| Mitchell E. Tyler | |||
| Director | |||
| By | /s/ Edward M. Straw | Date: October 31, 2016 | |
| Edward M. Straw | |||
| Director | |||
| By | /s/ Huaizheng Peng | Date: October 31, 2016 | |
| Huaizheng Peng | |||
| Director |