Attached files
| file | filename |
|---|---|
| EX-99.4 - EX-99.4 - Horizon Therapeutics Public Ltd Co | d268142dex994.htm |
| EX-99.3 - EX-99.3 - Horizon Therapeutics Public Ltd Co | d268142dex993.htm |
| EX-99.2 - EX-99.2 - Horizon Therapeutics Public Ltd Co | d268142dex992.htm |
| EX-23.1 - EX-23.1 - Horizon Therapeutics Public Ltd Co | d268142dex231.htm |
| 8-K - FORM 8-K - Horizon Therapeutics Public Ltd Co | d268142d8k.htm |
Exhibit 99.1
Overview
We are a biopharmaceutical company focused on improving patients’ lives by identifying, developing, acquiring and commercializing differentiated and accessible medicines that address unmet medical needs. We market nine medicines through our orphan, rheumatology and primary care business units. Our marketed medicines are ACTIMMUNE (interferon gamma-1b), BUPHENYL (sodium phenylbutyrate) Tablets and Powder, DUEXIS (ibuprofen/famotidine), KRYSTEXXA (pegloticase), MIGERGOT (ergotamine tartrate & caffeine suppositories), PENNSAID 2%, RAVICTI (glycerol phenylbutyrate) Oral Liquid, RAYOS (prednisone) delayed-release tablets and VIMOVO (naproxen/esomeprazole magnesium).
We developed DUEXIS and RAYOS, known as LODOTRA outside the United States, acquired the U.S. rights to VIMOVO from AstraZeneca AB (“AstraZeneca”) in November 2013, acquired certain rights to ACTIMMUNE as a result of our merger transaction with Vidara Therapeutics International Public Limited Company (“Vidara” and such transaction, the “Vidara Merger”) in September 2014, acquired the U.S. rights to PENNSAID 2% from Nuvo Research Inc. (“Nuvo”), in October 2014, acquired RAVICTI and BUPHENYL, known as AMMONAPS in certain European countries, as a result of our acquisition of Hyperion Therapeutics, Inc. (“Hyperion”) in May 2015, and acquired KRYSTEXXA and the U.S. rights to MIGERGOT as a result of our acquisition of Crealta Holdings LLC (“Crealta”) in January 2016.
On May 18, 2016, we entered into a definitive agreement with Boehringer Ingelheim International GmbH (“Boehringer Ingelheim International”), to acquire certain rights to interferon gamma-1b (the “IMUKIN Acquisition”), which Boehringer Ingelheim International currently commercializes under the trade names IMUKIN, IMUKINE, IMMUKIN and IMMUKINE in an estimated 30 countries, primarily in Europe and the Middle East. Under the terms of the agreement, we paid Boehringer Ingelheim International €5.0 million ($5.6 million when converted using a Euro-to-Dollar exchange rate of 1.1132) upon signing and will pay €20.0 million upon closing, for certain rights for interferon gamma-1b in all territories outside of the United States, Canada and Japan, as we currently hold marketing rights to interferon gamma-1b in these territories. We currently market interferon gamma-1b as ACTIMMUNE in the United States. The IMUKIN Acquisition is scheduled to close on December 31, 2016 and we are continuing to work with Boehringer Ingelheim International to enable the transfer of applicable marketing authorizations. Under the terms of a separate agreement, we also in-licensed certain U.S., European and Canadian intellectual property rights for interferon gamma-1b for the treatment of FA. Interferon gamma-1b is currently not indicated or approved for the treatment of Friedreich’s ataxia (“FA”).
On September 12, 2016, we, Misneach Corporation, a Delaware corporation and our indirect wholly owned subsidiary (“Merger Sub”), and Raptor Pharmaceutical Corp. (“Raptor”) entered into that certain Agreement and Plan of Merger (the “Merger Agreement”) to acquire Raptor. Raptor’s first commercial medicine, PROCYSBI (cysteamine bitartrate) delayed-release capsules, is an approved therapy for the management of nephropathic cystinosis, a rare, life-threatening metabolic lysosomal storage disorder that causes the rapid, toxic accumulation of cystine in all cells, tissues and organs in the body. Raptor’s second commercial medicine, QUINSAIR (aerosolized form of levofloxacin) a fluroquinolone antibiotic, has received marketing authorization by the European Medicines Agency (“EMA”) for treating chronic lung infection caused by the bacteria Pseudomonas aeruginosa in adults who have cystic fibrosis in March 2015 and by Health Canada in June 2015 for the management of cystic fibrosis in patients aged 18 years or older with chronic pulmonary Pseudomonas aeruginosa infections. QUINSAIR is not approved in the United States and may not be marketed or commercialized in the United States for any indication until it receives U.S. Food and Drug Administration (“FDA”) approval.
Our medicines are dispensed by retail and specialty pharmacies. Part of our commercial strategy for our primary care and rheumatology business units is to offer physicians the opportunity to have their patients fill prescriptions through pharmacies participating in our HorizonCares patient access program. This program does not involve us in the prescribing of medicines. The purpose of this program is solely to assist in ensuring that, when physicians determine that one of our medicines offers a potential clinical benefit to their patients and prescribe the medicine for an eligible patient, financial assistance may be available to reduce a commercial patient’s out-of-pocket costs. In the first six months of 2016, this resulted in 99.8 percent of commercial patients
1
having co-pay amounts of $10 or less when filling prescriptions for our medicines utilizing our patient access program. For commercial patients who are prescribed our primary care medicines or RAYOS, the HorizonCares program offers co-pay assistance when a third-party payer covers a prescription but requires an eligible patient to pay a co-pay or deductible, and offers full subsidization when a third-party payer rejects coverage for an eligible patient. For patients who are prescribed our orphan medicines, our patient access programs provide reimbursement support, a clinical nurse program, co-pay and other patient assistance. The aggregate commercial value of our patient access programs for the six months ended June 30, 2016 was $816.8 million. All pharmacies that dispense prescriptions for our medicines, which we estimate to be about 20,000 in the first half of 2016, are fully independent, including those that participate in HorizonCares. We do not own or possess any option to purchase an ownership stake in any pharmacy that distributes our medicines, and our relationship with each pharmacy is non-exclusive and arm’s length. All of our medicines are dispensed through pharmacies independent of our business.
As an alternative means of ensuring access to our medicines, we have also begun pursuing business arrangements with pharmacy benefit managers (“PBMs”) and other payers to secure formulary status and reimbursement of our medicines, such as our recently announced arrangements with CVS Caremark and Prime Therapeutics LLC (“Prime Therapeutics”). While we believe that, if successful, this strategy would result in broader inclusion of certain of our primary care medicines on healthcare plan formularies, and therefore increase payer reimbursement and lower our cost of providing patient access programs, these arrangements would generally require us to pay administrative and rebate payments to the PBMs and/or other payers.
We have a compliance program in place to address adherence with various laws and regulations relating to the selling, marketing and manufacturing of our medicines, as well as certain third-party relationships, including pharmacies. Specifically with respect to pharmacies, the compliance program utilizes a variety of methods and tools to monitor and audit pharmacies, including those that participate in our access programs, to confirm their activities, adjudication and practices are consistent with our compliance policies and guidance.
We market our medicines in the United States through our field sales force, which numbered approximately 500 representatives as of June 30, 2016. Our strategy is to use the commercial strength and infrastructure we have established in creating a global biopharmaceutical company to continue the successful commercialization of our existing medicine portfolio while also expanding and leveraging these capabilities by identifying, developing, acquiring and commercializing additional differentiated and accessible medicines that address unmet medical needs.
For the twelve months ended June 30, 2016, we generated net sales and adjusted EBITDA of $933 million and $447 million, respectively.
Merger with Raptor
On September 12, 2016, we, Merger Sub and Raptor entered into the Merger Agreement, pursuant to which we, through Merger Sub, commenced a tender offer to acquire all of the outstanding shares of Raptor’s common stock at a price of $9.00 per share in cash on September 26, 2016.
Vidara Merger and Hyperion Acquisition
The Vidara Merger occurred on September 19, 2014 and was accounted for as a reverse acquisition under the acquisition method of accounting for business combinations, with Horizon Pharma, Inc. (“HPI”) treated as the acquiring company for accounting purposes. As part of the Vidara Merger, a wholly owned subsidiary of Vidara merged with and into HPI, with HPI surviving the Vidara Merger as a wholly owned subsidiary of Vidara. Prior to the
2
Vidara Merger, Vidara changed its name to Horizon Pharma plc. Upon the consummation of the Vidara Merger, the historical financial statements of HPI became our historical financial statements.
On May 7, 2015, we completed our acquisition of Hyperion in which we acquired all of the issued and outstanding shares of Hyperion’s common stock for $46.00 per share in cash or approximately $1.1 billion on a fully-diluted basis. Following the completion of our acquisition of Hyperion, Hyperion became our wholly owned subsidiary and was renamed as Horizon Therapeutics, Inc. The consolidated financial statements presented in this report include the results of operations of the acquired business from the date of acquisition.
Competitive Strengths
We believe we have the following strengths:
| • | Diversified business model with increasing shift towards high growth, high margin, orphan medicines. We have successfully diversified our portfolio of medicines from two in 2013 to nine in January 2016, and which will be 11 assuming the completion of our proposed acquisition of Raptor (the “Raptor Acquisition”), across our three business units: orphan, rheumatology and primary care. While we continue to focus on strategies to grow our primary care medicines, we have made a strategic decision to expand our orphan medicine portfolio with the expectation that it will comprise the majority of the business by 2020. This strategy has led to our acquisitions of Crealta, Hyperion and Vidara, as well as the recently announced IMUKIN and Raptor Acquisitions. As a result, on a pro forma basis giving effect to the Raptor Acquisition, for the six months ended June 30, 2016, net sales of our orphan medicines represented approximately 45% of our total net sales. |
| • | Differentiated commercial strategy and significant experience in sales and marketing. Our senior leadership has experience in large pharmaceutical organizations and smaller companies alike with demonstrated capabilities to launch, expand, and grow commercial stage medicines in a variety of orphan, specialty and primary care indications. We have developed our own commercial programs, including HorizonCares, which provides increased patient access to medicines through co-pay assistance, reimbursement support or full subsidization of medicines. |
| • | Strong clinical development capabilities. We have worked diligently to unlock the full therapeutic potential of our medicines by collaborating closely with regulatory agencies, premier academic centers with established study consortiums, healthcare professionals and patient groups to facilitate our clinical development programs and generate data for possible new indications that may help more patients in need. We have a robust clinical development pipeline with a number of separate clinical programs underway for ACTIMMUNE, RAVICTI, KRYSTEXXA and RAYOS. |
| • | Ability to successfully execute business development strategies to drive growth. We completed our initial public offering in 2011 on the NASDAQ Stock Market. In 2013, we acquired VIMOVO, a commercial stage medicine, from AstraZeneca. In 2014, we acquired Vidara and acquired PENNSAID 2% from Nuvo, and in 2015, we acquired Hyperion. In 2016, we acquired Crealta, announced the IMUKIN Acquisition and recently announced the proposed acquisition of Raptor. These completed and proposed acquisitions, coupled with organic growth, have significantly increased, and we expect will continue to increase, our diversification, scale, revenues, profitability and cash flows. We have increased revenues from $75 million in 2013 to $933 million during the twelve months ended June 30, 2016. |
Business Strategy
Our strategy is to utilize the commercial strength and infrastructure we have established in creating a fully-integrated global specialty biopharmaceutical company to continue the successful commercialization of our
3
existing portfolio of medicines while expanding and leveraging these capabilities further through the acquisition of biopharmaceutical medicines and companies. After the completion of the Raptor Acquisition, our strategy will be to continue to enhance growth by driving volume and revenue growth in all three of our business units, integrate PROCYSBI and QUINSAIR into our orphan medicine portfolio, grow our footprint internationally by leveraging the Raptor infrastructure and selectively pursue additional acquisitions in our core areas of focus. We will execute on our strategy by:
| • | Driving volume and revenue growth in all three of our business units. We have seen continued growth in our medicines over the past several years. Net sales of our orphan medicines have grown 304% from the fourth quarter of 2014, the first quarter in which we recognized meaningful net sales from orphan medicines, to the second quarter of 2016, primarily as a result of the addition and growth of RAVICTI and KRYSTEXXA. With our primary care medicines and RAYOS, we have realized prescription volume growth of 173% from the second quarter of 2014 to the second quarter of 2016. During the same period, we have seen our average net realized price for these medicines increase only 8% cumulatively. We expect our recent contracts with pharmacy benefit managers CVS Caremark and Prime Therapeutics will provide increased access to patients as well as sustainability and durability for our primary care business. We continue to be in discussions and negotiations with other PBMs and payers with the goal of further increasing patient access for our clinically relevant primary care medicines. |
| • | Expanding indications across our existing orphan portfolio. As part of our effort to drive growth from our existing portfolio of medicines, we are conducting a number of clinical development programs with the goal of obtaining approval for additional or expanded indications. ACTIMMUNE is in a fully enrolled Phase 3 clinical trial for the treatment of FA, for which we expect to have data by the end of 2016. We estimate that in the United States, ACTIMMUNE in FA represents an annual $500 million to a $1 billion net sales opportunity. For RAVICTI, we have submitted a supplemental new drug application (“sNDA”) to expand its indication to cover patients aged two months to two years in the United States and also expect to launch the medicine in Canada in the second half of 2016 and Europe in 2017. For KRYSTEXXA, we expect to present additional safety and efficacy data at the 2016 American College of Rheumatology Annual Meeting in November 2016. |
| • | Leveraging the Raptor Acquisition to cost efficiently grow our orphan medicine portfolio and expand internationally. Based on our track record of integrating several acquisitions over the past few years, we believe we will be able to efficiently integrate Raptor into our business and that there are meaningful cost savings opportunities that could be realized as the integration is completed. In addition to PROCYSBI’s potential peak annual net sales, which we estimate will be at least $300 million, the Raptor Acquisition will also expand our commercial infrastructure into Europe and other international markets. With both PROCYSBI and QUINSAIR approved in the European Union (“EU”), Raptor has built commercial infrastructure in Europe and has established a number of distribution relationships in other selected international markets. We believe this European infrastructure can be leveraged to support additional international growth opportunities through the expansion of commercial markets for our existing medicines and/or medicines acquired in the future. |
| • | Selectively pursue additional acquisitions in our core areas of focus. We are shifting our business to become a predominantly rare disease focused company by 2020. We focus our business development efforts largely on commercial and late-stage development assets that address unmet medical needs, with orphan, rheumatology and other specialty medicines as priorities. |
4
Credit Highlights
We believe that our credit provides an attractive investment opportunity as we are well-positioned to execute our business strategy based on the following competitive advantages and key strengths:
| • | Increased focus on our long-life, well-protected orphan medicines; |
| • | Well-balanced portfolio of medicines with a diversified patient base; |
| • | Cash-generative and profitable business model; |
| • | Successful and differentiated commercial strategy; |
| • | Proven acquisition and integration capabilities; and |
| • | Highly experienced and capable management team. |
Our Medicines
We believe our medicines address unmet therapeutic needs in orphan diseases, arthritis, pain and/or inflammatory diseases and provide significant advantages over existing therapies.
Our current marketed medicine portfolio consists of the following:
| Medicine |
Disease |
Marketing Rights | ||
| ORPHAN BUSINESS UNIT MEDICINES: |
||||
| ACTIMMUNE |
Chronic granulomatous disease and severe, malignant osteopetrosis | United States and selected foreign countries | ||
| RAVICTI |
Urea cycle disorders | Worldwide(1) | ||
| BUPHENYL/AMMONAPS |
Urea cycle disorders | Worldwide(2) | ||
| RHEUMATOLOGY BUSINESS UNIT MEDICINES: |
||||
| RAYOS/LODOTRA |
Rheumatoid arthritis, polymyalgia rheumatic, systemic lupus erythematosus and multiple other indications | Worldwide(3) | ||
| KRYSTEXXA |
Chronic refractory gout | Worldwide | ||
| PRIMARY CARE BUSINESS UNIT MEDICINES: |
||||
| DUEXIS |
Signs and symptoms of osteoarthritis and rheumatoid arthritis | Worldwide(4) | ||
| VIMOVO |
Signs and symptoms of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis | United States | ||
| PENNSAID 2% |
Pain of osteoarthritis of the knee(s) | United States | ||
| MIGERGOT |
Vascular headache | United States | ||
5
| (1) | RAVICTI distribution rights in the Middle East and North Africa have been licensed to Swedish Orphan Biovitrum AB (“SOBI”). |
| (2) | BUPHENYL/AMMONAPS distribution rights in Europe, certain Asian, Latin American, Middle Eastern, North African and other countries have been licensed to SOBI. |
| (3) | RAYOS/LODOTRA distribution rights in Europe, Australia, certain Asian, Latin American, Middle Eastern, African, and other countries have been licensed to Mundipharma International Corporation Limited (“Mundipharma”). |
| (4) | DUEXIS rights in Latin America have been licensed to Grünenthal S.A. (“Grünenthal”). |
ORPHAN BUSINESS UNIT
Market
Chronic Granulomatous Disease
Chronic granulomatous disease (“CGD”) is a genetic disorder of the immune system. It is described as a primary immunodeficiency disorder, which means it is not caused by another disease or disorder. In people who have CGD, a type of white blood cell, called a phagocyte, is defective. These defective phagocytes cannot generate superoxide, leading to an inability to kill harmful microorganisms such as bacteria and fungi. As a result, the immune system is weakened. People with CGD are more likely to have certain problems such as recurrent severe bacterial and fungal infections and chronic inflammatory conditions. These patients are prone to developing masses called granulomas, which can occur repeatedly in organs throughout the body and cause a variety of problems. CGD is considered to be a condition that patients can live with and manage. Studies suggest overall survival has improved over the last decade with more patients living well into adulthood. Approximately 1 out of every 100,000 to 200,000 babies in the United States is born with CGD.
Severe, Malignant Osteopetrosis
Severe, malignant osteopetrosis (“SMO”) is a form of osteopetrosis and is sometimes referred to as marble bone disease or malignant infantile osteopetrosis because it occurs in very young children. While exact numbers are not known, it has been estimated that 1 out of 250,000 children is born with SMO. During normal bone development, existing bone material is constantly being replaced by new bone. Cells called osteoblasts cause new bone formation while other cells called osteoclasts remove old bone through a process called resorption. In people with osteopetrosis, this balance is not maintained because their osteoclasts do not function properly. As a result, resorption of old bone material decreases while the formation of new bone continues. This leads to an abnormal increase in bone mass, which can make the bones more brittle. Because abnormal bone development affects many different systems in the body, osteopetrosis may cause problems such as blood disorders, decreased ability to fight infection, bone fractures, problems with vision and hearing, and abnormal appearance of the face and head.
Urea Cycle Disorders
Urea cycle disorders (“UCDs”) are inherited metabolic diseases caused by a deficiency of one of the enzymes or transporters that constitute the urea cycle. The urea cycle involves a series of biochemical steps in which ammonia, a potent neurotoxin, is converted to urea, which is excreted in the urine. UCD patients may experience episodes where they get symptoms from the ammonia in their blood being excessively high—called hyperammonemic crises—which may result in irreversible brain damage, coma or death. UCD symptoms may first occur at any age depending on the severity of the disorder, with more severe defects presenting earlier in life.
Our Solutions
ACTIMMUNE
ACTIMMUNE is a biologically manufactured protein called interferon gamma-1b that is similar to a protein the human body makes naturally. In the body, interferon gamma is produced by cells of the immune
6
system and helps to prevent infection in patients with CGD and enhances osteoclast function in patients with SMO. ACTIMMUNE is approved by the FDA to reduce the frequency and severity of serious infections associated with CGD and for delaying time to disease progression in patients with SMO. The precise way that ACTIMMUNE works to help prevent infection in patients with CGD and slow the worsening of SMO is not fully understood, but ACTIMMUNE is believed to work by modifying the cellular function of various cells, including those in the immune system and those that help form bones.
Efficacy in CGD
The International Chronic Granulomatous Disease Cooperative Study Group conducted a controlled clinical trial in 128 patients (ages ranging from 1 to 44 years old) at 13 medical centers across 4 countries. The purpose of this clinical trial was to evaluate the safety and efficacy of ACTIMMUNE in reducing the frequency and severity of serious infections in patients with CGD. Patients enrolled in the trial were randomly selected to receive either ACTIMMUNE or placebo in addition to antibiotics. The number and timing of serious infections were tracked in all patients for up to 1 year. Investigators concluded that ACTIMMUNE is an effective and safe therapy for patients with CGD, because the therapy statistically reduced the frequency of serious infections.
Efficacy in SMO
In a controlled clinical trial, 16 patients were randomized to receive either ACTIMMUNE with calcitriol or calcitriol alone. The age of patients ranged from 1 month to 8 years; with a mean age of 1.5 years. The median time to progression in the ACTIMMUNE plus calcitriol arm was 165 days versus a median of 65 days in the calcitriol only arm. In a separate analysis that combined data from a second trial, 19 of 24 patients on ACTIMMUNE therapy (with or without calcitriol) for at least 6 months had reduced trabecular bone volume compared to baseline.
Commercial Status
ACTIMMUNE is the only drug currently approved by the FDA for the treatment for CGD and SMO. Our licenses allow us to market and sell ACTIMMUNE in the United States, Canada and Japan. We currently commercialize ACTIMMUNE in the United States and also supply ACTIMMUNE to patients in Canada, if so requested by way of a prescription from their treating physicians, through Health Canada’s Special Access Program, which provides access to non-marketed drugs in Canada for practitioners treating patients with serious or life-threatening conditions when conventional therapies have failed, are unsuitable or are unavailable. We have not otherwise registered or sold ACTIMMUNE in any other territories for which we currently hold commercial rights.
Potential for ACTIMMUNE in Friedreich’s ataxia
FA, is a debilitating, life-shortening and degenerative neuro-muscular disorder that affects approximately 3,700 people in the United States. Onset of symptoms can vary from five years old to adulthood, with the childhood onset tending to be associated with a more rapid progression. A progressive loss of coordination and muscle strength leads to motor incapacitation and often the full-time use of a wheelchair. Most young people diagnosed with FA require mobility aids such as a cane, walker or wheelchair by their teens or early twenties. There are currently no approved treatments for FA.
In October 2014, we announced and presented data from the Phase 2 open-label study of ACTIMMUNE treatment in children with FA. The results showed ACTIMMUNE was well tolerated with no serious adverse events, and two subjects reporting severe events and subsequent dose reductions. The safety findings generally reflected the label safety profile for ACTIMMUNE. Changes in frataxin protein levels, the primary study endpoint, were statistically significant in red blood cells, white blood cells and platelets. Mean improvement in the modified Friedreich’s Ataxia Rating Scale (“mFARS”) was statistically significant. The mFARS score is used to measure neurological signs associated with FA, with higher scores reflecting a greater level of disability.
7
In June 2015, we initiated the Safety, Tolerability and Efficacy of ACTIMMUNE Dose Escalation in Friedreich’s Ataxia Study (“STEADFAST”) of ACTIMMUNE for the treatment of people with FA. This Phase 3 trial (NCT02415127) is a randomized, multi-center, double-blind, placebo-controlled study with patients randomized 1:1 to receive subcutaneous doses of either ACTIMMUNE or placebo three times a week for a total of 26 weeks. 92 patients were enrolled at four sites in the United States. The primary endpoint will measure the change in neurological outcome and evaluate the effect of ACTIMMUNE versus placebo as measured by the mFARS score focused on objective neurologic measures such as upper and lower extremity coordination change from baseline. In addition to safety and efficacy, the STEADFAST trial will evaluate the pharmacokinetic characteristics of ACTIMMUNE in people with FA. We completed enrollment in the second quarter of 2016, with top-line data anticipated to become available by the end of 2016. Assuming positive data from the trial, we would plan to submit a supplemental biologics license application in the first quarter of 2017, and given the fast-track designation of ACTIMMUNE for this potential indication, we would request priority review, which, if awarded, would allow us to potentially receive a decision from the FDA within six months of the submission, in the third quarter of 2017.
RAVICTI
RAVICTI is indicated for use as a nitrogen-binding agent for chronic management of adult and pediatric patients 2 years of age and older (2 months of age and older in Europe) with UCDs that cannot be managed by dietary protein restriction and/or amino acid supplementation alone. RAVICTI must be used with dietary protein restriction and, in some cases, dietary supplements (e.g., essential amino acids, arginine, citrulline, or protein-free calorie supplements).
Efficacy in the Treatment of UCDs in Adult Patients
A randomized, double-blind, active-controlled, crossover, noninferiority study compared RAVICTI to sodium phenylbutyrate by evaluating venous ammonia levels in patients with UCDs that had been on sodium phenylbutyrate prior to enrollment for control of their UCD. Patients adhered to a low-protein diet and received amino acid supplements throughout the study. After two weeks of dosing, by which time patients had reached a steady state on each treatment, all patients had 24 hours of ammonia measurements.
Another study was conducted to assess monthly ammonia control and hyperammonemic crisis over a 12-month period. A total of 51 adults were in the study and all but six had been converted from sodium phenylbutyrate to RAVICTI. Venous ammonia levels were monitored monthly. Of 51 adult patients participating in the 12-month, open-label treatment with RAVICTI, seven patients (14 percent) reported a total of 10 hyperammonemic crises.
The efficacy of RAVICTI in pediatric patients two to 17 years of age was evaluated in two fixed-sequence, open-label, sodium phenylbutyrate to RAVICTI switchover studies, seven and 10 days in duration. These studies compared blood ammonia levels of patients on RAVICTI to venous ammonia levels of patients on sodium phenylbutyrate in 26 pediatric UCD patients. Twenty-four hour blood ammonia levels of UCD patients six to 17 years of age (Study 3) and patients two to five years of age (Study 4) were similar between treatments but trended higher with sodium phenylbutyrate.
Long-term (12-month), uncontrolled, open-label studies were conducted to assess monthly ammonia control and hyperammonemic crisis over a 12-month period. Of the 26 pediatric patients six to 17 years of age participating in these two trials, five patients (19 percent) reported a total of five hyperammonemic crises.
Commercial Status
RAVICTI was approved for marketing in the United States in 2013.
On November 30, 2015, we announced the EMA had adopted a binding decision to approve RAVICTI for use as an adjunctive therapy for chronic management of adult and pediatric patients two months of age and
8
older with six subtypes of UCDs. This decision followed the Positive Opinion previously adopted on September 24, 2015 by the Committee for Medicinal Products for Human Use (the “CHMP”) of the EMA. The approval authorizes us to market RAVICTI in all 28 Member States of the EU and the centralized marketing authorization will form the basis for recognition by the Member States of the European Economic Area (the “EEA”), namely Norway, Iceland and Liechtenstein, for the medicine to be placed on the market.
We have worldwide rights to market and distribute RAVICTI. In relation to marketing and distribution rights in the Middle East and North Africa region, we have entered into a distribution agreement with SOBI until 2018. We market and sell RAVICTI in the United States and plan to begin commercializing RAVICTI in Europe in 2017.
We are in the process of seeking approval for label expansions for RAVICTI, with assessments in progress studying the use of RAVICTI in patients both from two months to two years (for which we submitted an sNDA, submission to the FDA in the second quarter of 2016), and from birth to two months (targeted sNDA submission in the first quarter of 2018). Current FDA approval is for patients from two years of age and older only. In patients with UCDs for which RAVICTI is an FDA-approved medicine, there is a variable age of diagnosis (from newborn to adulthood), and the severity of the disease can be associated with the age of onset and enzymatic deficit. However, a prompt diagnosis and careful management of the disease can lead to good clinical outcomes.
BUPHENYL
BUPHENYL tablets for oral administration and BUPHENYL powder for oral, nasogastric, or gastrostomy tube administration are indicated as adjunctive therapy in the chronic management of patients with UCDs involving deficiencies of carbamoyl phosphate synthetase, ornithine transcarbamylase or argininosuccinic acid synthetase.
BUPHENYL is indicated in all patients with neonatal-onset deficiency (complete enzymatic deficiency, presenting within the first 28 days of life). It is also indicated in patients with late-onset disease (partial enzymatic deficiency, presenting after the first month of life) who have a history of hyperammonemic encephalopathy. It is important that the diagnosis be made early and treatment initiated immediately to improve survival. BUPHENYL must be combined with dietary protein restriction and, in some cases, essential amino acid supplementation.
Commercial Status
BUPHENYL was approved by the FDA in the United States in 1996 and by the EMA in Europe in 1999. We commercially market and distribute BUPHENYL in the United States. BUPHENYL is known as AMMONAPS in certain European countries, and the marketing and distribution rights are licensed to SOBI through the end of 2016. We provide BUPHENYL in certain other countries through various Special Access Programs and licensed distributors.
Competition
ACTIMMUNE presently faces limited competition. ACTIMMUNE is the only drug currently approved by the FDA specifically for the treatment for CGD and SMO. While there are additional or alternative approaches used to treat patients with CGD and SMO, including the increasing trend towards the use of bone marrow transplants in patients with CGD, there are currently no medicines on the market that compete directly with ACTIMMUNE.
In the United States, RAVICTI and BUPHENYL compete with generic forms of sodium phenylbutyrate. In Europe and certain other countries, RAVICTI and BUPHENYL compete with Pheburane,
9
which is a sugar-coated version of sodium phenylbutyrate. Pheburane claims a taste advantage over BUPHENYL. However the volume of Pheburane that must be ingested multiple times per day is much greater than BUPHENYL, and significantly greater than RAVICTI, and is a barrier to patient compliance.
RHEUMATOLOGY BUSINESS UNIT
Market
Rheumatoid Arthritis
Rheumatoid arthritis (“RA”) is a chronic disease that causes pain, stiffness and swelling, primarily in the joints. According to a 2006 DataMonitor report, 2.9 million people in the United States suffer from RA, of which 1.8 million are diagnosed and treated with various drugs. RA has no known cause, but unlike osteoarthritis (“OA”), RA is not associated with factors such as aging. RA occurs when the body’s immune system malfunctions, attacking healthy tissue and causing inflammation, which leads to pain and swelling in the joints and may eventually cause permanent joint damage and painful disability. The primary symptoms of RA include progressive immobility and pain, especially in the morning, with long-term sufferers experiencing continual joint destruction for the remainder of their lives. There is no known cure for RA. Once the disease is diagnosed, treatment is prescribed for life to alleviate symptoms and/or to slow or stop disease progression. RA treatments include medications, physical therapy, exercise, education and sometimes surgery. Early, aggressive treatment of RA can delay joint destruction. Treatment of RA usually includes multiple drug therapies taken concurrently. Disease-modifying anti-rheumatic drugs (“DMARDs”) are the current standard of care for the treatment of RA, in addition to rest, exercise and antiinflammatory drugs such as nonsteroidal anti-inflammatory drugs (“NSAIDs”).
Polymyalgia Rheumatica
Polymyalgia Rheumatica (“PMR”) is an inflammatory disorder that causes significant muscle pain and stiffness. The pain and stiffness often occur in the shoulders, neck, upper arms and hip with pronounced morning stiffness lasting at least one hour. Most people who develop PMR are older than 65 years of age. It rarely affects people younger than 50. There are approximately 1.1 million patients with PMR in the United States and it afflicts one in every 133 people over the age of 50. Prednisone is the standard of care for treating PMR and treatment is generally initiated at a relatively high dose (e.g., 10-20 mg per day) and reduced as clinical improvement is seen. Treatment usually lasts 18-24 months. Similar to RA, PMR is associated with circadian patterns of Interleukin 6 (“IL-6”) elevation in early morning hours.
Systemic Lupus Erythematosus
Systemic Lupus Erythematosus (“SLE”) is a chronic autoimmune disease that causes inflammation and pain in the joints and muscles as well as overall fatigue. SLE affects from 161,000 to 322,000 adults in the United States. More than 90 percent of cases of SLE occur in women, frequently starting at childbearing age. In addition to affecting the muscles and joints, it can affect other organs in the body such as the kidneys, tissue lining the lungs (pleura), heart (pericardium), and brain. Most patients feel fatigue and have rashes, arthritis (painful and swollen joints) and fever. SLE flares vary from mild to serious.
In November 2015, we announced our collaboration with the Alliance for Lupus Research (“ALR”) to study the effect of RAYOS on the fatigue experienced by SLE patients. SLE is a chronic autoimmune disease that causes inflammation and pain in the joints and muscles, as well as overall fatigue. RAYOS is currently indicated for patients with SLE. The first study planned as part of the collaboration is an investigator-initiated, randomized, double-blind, active comparator, cross-over study in which patients will be randomized to receive either prednisone for three months or RAYOS at 10 p.m. for three months, and then switched to the alternative medication for an additional three months. Approximately 62 patients across 25 sites will be enrolled in the United States. The primary endpoint will assess fatigue as measured by Functional Assessment of Chronic Illness Therapy-Fatigue, a 13-question survey to be completed by study participants that focuses on the daily fatigue experienced in patients with chronic illnesses.
10
Chronic Refractory Gout
Chronic refractory gout (“CRG”) is a type of arthritis that occurs when uric acid build-up in the blood remains high and inflammation persists even after treatment with conventional therapies. Gout is one of the most common forms of inflammatory arthritis, estimated to affect 8.3 million in the United States, with CRG impacting 40,000 to 50,000 people in the United States. CRG frequently causes crippling disabilities and significant joint damage.
Market Opportunity and Limitations of Existing Treatments
Morning Stiffness, Pain and Immobility
A Medical Marketing Economics May 2008 study of 150 RA patients in the United States, which we sponsored, showed that despite the use of a combination of currently available treatments for RA, more than 90 percent of the patients reported suffering from morning stiffness, pain and immobility, which is linked to peak IL-6 levels in the early morning hours. Patients with RA in general have substantially increased IL-6 levels, with peak IL-6 levels tending to occur in the early morning hours, and low levels typically occurring in the afternoon and evening. Therefore, we believe an optimal treatment would reduce IL-6 levels in the early morning hours.
Side Effects of Current High-Dose Corticosteroid Treatments
According to the 2006 DataMonitor report, approximately 50 percent of RA patients in the United States, Japan, France, Italy, Spain, Germany and the United Kingdom are prescribed combination therapy which often includes corticosteroids, with prednisone being one of the most common. Corticosteroids, including prednisone, are used to suppress various autoimmune, inflammatory and allergic disorders by inhibiting the production of various pro- inflammatory cytokines, such as IL-6 and TNF-alpha. Joint inflammation in RA is driven by excessive production of inflammatory mediators and cytokines such as IL-6 and TNF-alpha. While corticosteroids are potent and effective agents to treat patients with RA, they are often used at high doses to treat RA flares or significant inflammation. High-dose oral corticosteroid treatment is not a viable long-term treatment option due to adverse side effects such as osteoporosis, cardiovascular disease and weight gain. However, clinical studies have shown that the long-term use of low-dose prednisone (<10 mg per day) does not dramatically increase total adverse events. In addition, low doses, typically less than 10 mg daily, of corticosteroids such as prednisone have been shown to treat the symptoms of RA while slowing the overall progression of the disease.
Our Solutions
RAYOS/LODOTRA
The medicine sold and marketed as RAYOS in the United States is known as LODOTRA outside the United States. While the FDA has approved RAYOS for the treatment of RA, ankylosing spondylitis (“AS”), PMR, primary systemic amyloidosis, asthma, chronic obstructive pulmonary disease, SLE and a number of other conditions, we have focused our promotion of RAYOS/LODOTRA on rheumatology indications, including RA and PMR.
The proprietary formulation technology of RAYOS/LODOTRA enables a delayed-release of prednisone approximately four hours after administration. The RAYOS/LODOTRA proprietary delivery system synchronizes the prednisone delivery time with the patient’s elevated cytokine levels, thereby taking effect at a physiologically optimal point to inhibit cytokine production, and thus significantly reduces the signs and symptoms of RA and PMR.
RAYOS/LODOTRA was developed using SkyePharma AG’s (“SkyePharma”) proprietary GeoClock and GeoMatrix technologies, for which we hold an exclusive worldwide license for the delivery of glucocorticoid, a class of corticosteroid. RAYOS/LODOTRA is composed of an active core containing
11
prednisone, which is encapsulated by an inactive porous shell. The inactive shell acts as a barrier between the medicine’s active core and a patient’s gastrointestinal (“GI”) fluids. RAYOS/LODOTRA is intended to be administered at bedtime. At approximately four hours following bedtime administration of RAYOS/LODOTRA, water in the digestive tract diffuses through the shell, causing the active core to expand, which leads to a weakening and breakage of the shell and allows the release of prednisone from the active core. Our pharmacokinetic studies have shown that the blood concentration of prednisone from RAYOS/LODOTRA is similar to immediate release prednisone except for the intended time delay of medicine release after administration.
Commercial Status
We began marketing RAYOS to U.S. rheumatologists in December 2012. LODOTRA received its first approval in Europe in March 2009 and is currently approved for marketing in more than 30 countries outside the United States where Mundipharma holds the commercial rights. Reimbursement has been approved in Germany, Italy and a number of other European countries.
KRYSTEXXA
KRYSTEXXA is an orphan biologic medicine which is the first and only FDA-approved medicine for the treatment of CRG. KRYSTEXXA is a PEGylated uric acid specific enzyme (uricase) indicated for the treatment of CRG in adult patients that are refractory to conventional therapy. Gout refractory to conventional therapy occurs in patients who have failed to normalize serum uric acid and whose signs and symptoms are inadequately controlled with xanthine oxidase inhibitors at the maximum medically appropriate dose or for whom these drugs are contraindicated. KRYSTEXXA has a unique mechanism of action which rapidly reverses disease progression. A PEGylated uric acid specific enzyme catalyzes the conversion of serum uric acid to allantoin, which is then excreted in urine. This PEGylated uric acid specific enzyme is given via an intravenous infusion to patients every two weeks.
Commercial Status
KRYSTEXXA was launched in January 2011. KRYSTEXXA has biologic exclusivity until 2022 and a composition of matter patent until 2026. Orphan drug exclusivity was granted on February 21, 2011, which exclusivity lasts for 7 years and will expire in February 2018.
Competition
RAYOS/LODOTRA competes with a number of medicines on the market to treat RA, including corticosteroids, such as prednisone, traditional DMARDs, such as methotrexate, and biologic agents, such as HUMIRA and Enbrel. The majority of RA patients are treated with DMARDs, which are typically used as initial therapy in patients with RA. Biologic agents are typically added to DMARDs as combination therapy. It is common for an RA patient to take a combination of a DMARD, an oral corticosteroid, an NSAID, and/or a biologic agent. We are not currently aware of any other delayed-release prednisone medicine in development.
As the only FDA approved medication for the treatment of CRG, KRYSTEXXA faces limited direct competition. We believe that the complexity of manufacturing KRYSTEXXA provides a barrier to potential generic competition. However, a number of competitors have medicines in Phase 1 or Phase 2 trials. On December 22, 2015, AstraZeneca secured approval from the FDA for ZURAMPIC (lesinurad) 200mg tablets in combination with a XOI for the treatment of hyperuricemia associated with gout in patients who have not achieved target serum uric acid levels with an xanthine oxidase inhibitor (“XOI”) alone. Although ZURAMPIC is not a direct competitor because it has not been approved for CRG, this therapy could be used prior to use of KRYSTEXXA, and if effective, could reduce the target patient population for KRYSTEXXA.
12
PRIMARY CARE BUSINESS UNIT
Market
Pain is a serious and costly public health concern. In 2010, the U.S. National Center for Health Statistics reported that approximately 30 percent of U.S. adults 18 years of age and over reported recent symptoms of pain, aching or swelling around a joint within the past 30 days.
Some of the most common and debilitating chronic inflammation and pain-related diseases are OA, RA, and acute and chronic pain. According to National Health Interview Survey data analyzed by the U.S. Centers for Disease Control and Prevention, from 2010-2012, 52.5 million U.S. adults 18 years of age and over had reported being diagnosed with some form of arthritis. With the aging of the U.S. population, the prevalence of arthritis is expected to rise by approximately 40 percent by 2030, impacting 67 million people in the United States.
Osteoarthritis
OA is a type of arthritis that is caused by the breakdown and eventual loss of the cartilage of one or more joints. Cartilage is a protein substance that serves as a cushion between the bones of the joints. Among the over 100 different types of arthritis conditions, OA is the most common and occurs more frequently with age. OA commonly affects the hands, feet, spine and large weight-bearing joints, such as the hips and knees. Symptoms of OA manifest in patients as joint pain, tenderness, stiffness, limited joint movement, joint cracking or creaking (crepitation), locking of joints and local inflammation. OA can also lead to joint deformity in later stages of the disease. Many drugs are used to treat the inflammation and pain associated with OA, including aspirin and other NSAIDs, such as ibuprofen, naproxen and diclofenac, that have a rapid analgesic and anti-inflammatory response.
Rheumatoid Arthritis
The market for RA is discussed above under “Rheumatology Business Unit.”
Ankylosing Spondylitis
AS is a type of arthritis that affects the spine. AS symptoms include pain and stiffness from the neck down to the lower back. The spine’s bones (vertebrae) may grow or fuse together, resulting in a rigid spine. These changes may be mild or severe, and may lead to a stooped-over posture. Early diagnosis and treatment helps control pain and stiffness and may reduce or prevent significant deformity.
Market Opportunity and Limitations of Existing Treatments
GI-Associated Adverse Events
NSAIDs are very effective at providing pain relief, including pain associated with OA and RA; however, there are significant upper GI-associated adverse events that can result from the use of NSAIDs. According to a 2004 article published in Alimentary Pharmacology & Therapeutics, significant GI side effects, including serious ulcers, afflict up to approximately 25 percent of all chronic arthritis patients treated with NSAIDs for three months, and OA and RA patients are two to five times more likely than the general population to be hospitalized for NSAID-related GI complications. It is estimated that NSAID-induced GI toxicity causes over 16,500 related deaths in OA and RA patients alone and over 107,000 hospitalizations for serious GI complications each year. In more than 70 percent of patients with these serious GI complications, there are no prior symptoms.
13
Despite the fact that GI ulcers are one of the most prevalent adverse events resulting from the use of NSAIDs in the United States, according to a 2006 article published in BMC Muskoskeletal Disorders, 11 observational studies indicated that physicians do not commonly co-prescribe GI protective agents to high-risk patients. Physicians prescribe concomitant therapy to only 24 percent of NSAID users, and studies show sub-optimal patient compliance with concomitant prophylaxis therapy. According to a 2003 article published in Alimentary Pharmacology & Therapeutics, in a study of 784 patients, 37 percent of patients were non-compliant, a rate increasing to 61 percent in patients treated with three or more drugs. This noncompliance results in a substantial unmet clinical need, which we believe can be appropriately addressed with DUEXIS or VIMOVO, creating smarter solutions for both patients and physicians.
Topical NSAIDs
Within the NSAID market there exists a significant niche for topical NSAIDs, which are prescribed more than 5 million times per year. Topical NSAID treatment may be appropriate for some patients, such as patients who may benefit from the lower systemic exposure in a topical NSAID, patients with OA in just one joint such as the knee, patients who have trouble taking oral medications, or patients who are older. However, applying the correct dosage of the topical NSAID amount can often be a barrier to patient compliance, and there exists a market for a more convenient and more accurate application technique.
Our Solutions
DUEXIS
DUEXIS is a proprietary single-tablet formulation containing a fixed-dose combination of ibuprofen, the most widely prescribed NSAID, and famotidine, a well-established GI agent used to treat dyspepsia, gastroesophageal reflux disease and active ulcers, in one pill. Based on clinical study results, DUEXIS has been proven to reduce the risk of NSAID-induced upper GI ulcers.
Ibuprofen: One of the World’s Most Widely Prescribed NSAIDs
Ibuprofen continues to be one of the most widely prescribed NSAIDs worldwide. According to Intercontinental Marketing Services (“IMS”), in the United States alone, there were over 42 million prescriptions written for ibuprofen in 2015. Ibuprofen’s flexible three times daily dosing allows it to be used for both chronic conditions such as arthritis and chronic back pain, and acute conditions such as sprains and strains.
Famotidine: A Safe and Effective GI Agent
Famotidine is the most potent marketed drug in the class of histamine-2 receptor antagonists (“H2RA”). H2RAs are a class of drugs used to block the action of histamine on the cells in the stomach that secrete gastric acid. Famotidine was chosen as the ideal GI protectant to be combined with ibuprofen as it is a well-studied compound with an estimated 18.8 million patients treated worldwide that provides distinct advantages including:
| • | rapid onset of action; and |
| • | well-tolerated with a low incidence of adverse drug reactions and a demonstrated safety margin of up to eight times the approved prescription dose for an extended period of greater than 12 months. |
Although famotidine as a standalone product is not indicated for risk reduction of GI ulcers, two well-controlled clinical trials of famotidine formulated in DUEXIS found a significant decrease in the risk of developing upper GI ulcers, which in the clinical trials was defined as a gastric and/or duodenal ulcer in patients who are taking ibuprofen for those indications.
14
Benefits of a Fixed-Dose Combination Therapy
Numerous studies have demonstrated that fixed-dose combination therapy provides significant advantages over taking multiple pills. Specifically, fixed-dose combinations can reduce the number of pills, ensure that the correct dosage of each component is taken at the correct time and improve compliance, often associated with better treatment outcomes. DUEXIS has been formulated to provide an optimal dosing regimen of ibuprofen and famotidine together in the convenience of a single pill. Data shows that physicians co-prescribe GI protective agents less than 25 percent of the time when prescribing an NSAID. On occasions where a patient is co-prescribed a GI protective agent, data shows that after three prescriptions, 61 percent of patients no longer take a GI protective agent.
Commercial Status
DUEXIS is indicated for the relief of signs and symptoms of RA and OA and to decrease the risk of developing GI ulcers in patients who are taking ibuprofen for these indications. We began marketing DUEXIS to physicians in December 2011.
In June 2012, we licensed DUEXIS rights in Latin America to Grünenthal, a private company focused on the promotion of pain medicines.
VIMOVO
VIMOVO is a proprietary, fixed-dose, delayed-release tablet. VIMOVO combines enteric-coated naproxen, an NSAID, surrounded by a layer of immediate-release esomeprazole magnesium surrounding the core. Naproxen has proven anti-inflammatory and analgesic properties and esomeprazole magnesium reduces the stomach acid secretions that can cause upper GI ulcers. Both naproxen and esomeprazole magnesium have well-documented and excellent long-term safety profiles and both medicines have been used by millions of patients worldwide. Based on clinical trial results, VIMOVO has been shown to decrease the risk of developing gastric ulcers in patients at risk of developing NSAID associated gastric ulcers.
Naproxen: One of the World’s Most Widely Prescribed NSAIDs
Naproxen is another of the most widely prescribed NSAIDs worldwide. According to IMS, in the United States alone, there were more than 17 million prescriptions written for naproxen in 2015. In addition, naproxen’s twice daily dosing allows it to be used for chronic conditions such as arthritis and AS.
Esomeprazole Magnesium: A Safe and Effective GI Agent
Esomeprazole magnesium, a gastroprotective agent, is a proton pump inhibitor (“PPI”) that works by inhibiting the secretion of gastric acid thus decreasing the amount of acid in the stomach. PPIs are considered to be very potent inhibitors of acid secretion. Esomeprazole magnesium is indicated for reducing the risk of NSAID-induced gastric ulcers.
Benefits of a Fixed-Dose Combination Therapy
VIMOVO is specifically formulated to allow esomeprazole magnesium to achieve its gastroprotective impact before naproxen is released into the system. VIMOVO’s design is intended to produce a sequential delivery of gastroprotective esomeprazole before exposure to naproxen. Data shows that physicians co-prescribe GI protective agents less than 25 percent of the time when prescribing an NSAID. On occasions where a patient is co-prescribed a GI protective agent, data shows that after three prescriptions, 61 percent of patients no longer take a GI protective agent.
15
Commercial Status
Following our acquisition of the U.S. rights to VIMOVO in November 2013, we began marketing VIMOVO in early January 2014.
PENNSAID 2%
PENNSAID 2% is a topical NSAID that is applied directly to the knee and is indicated for the treatment of pain of OA of the knee(s). PENNSAID 2% contains diclofenac sodium, a commonly prescribed NSAID to treat OA pain. PENNSAID 2% also includes dimethyl sulfoxide (“DMSO”), a powerful penetrating agent that helps ensure that diclofenac sodium is absorbed through the skin to the site of inflammation and pain. Topical NSAIDs such as PENNSAID 2% are an alternative to oral NSAID treatment because they reduce systemic exposure to a fraction of that provided by an oral NSAID. PENNSAID 2% is the only topical NSAID offered with the convenience of a metered-dose pump, which ensures that the patient will get the correct amount of PENNSAID 2% solution each time. PENNSAID 2% is easy to apply for patients because PENNSAID 2% is applied in two pumps, twice daily, delivering relief right to the site of OA knee pain.
Commercial Status
On January 16, 2014, the FDA approved PENNSAID 2% for the treatment of the pain of OA of the knee(s). We acquired the U.S. rights to PENNSAID 2% in October 2014, and began marketing PENNSAID 2% with our primary care sales force in early January 2015.
Competition
Our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors in our primary care markets include large pharmaceutical and biotechnology companies, specialty pharmaceutical companies and generic drug companies, although we are not currently aware of any other ibuprofen/famotidine combination medicine or naproxen/esomeprazole magnesium combination medicine in development. We believe that the key competitive factors that will affect the commercial success of our medicines, as well as future drug candidates that we may develop, are their efficacy, safety and tolerability profile, convenience in dosing, price and reimbursement.
DUEXIS and VIMOVO compete with other NSAIDs, including Celebrex which is marketed by Pfizer Inc., and is also a generic medicine known as celecoxib and marketed by other pharmaceutical companies. Celecoxib is an NSAID that selectively inhibits the COX-2 enzyme and is an effective anti-arthritic agent that reduces the risk of ulceration compared to traditional NSAIDs such as ibuprofen.
In general, DUEXIS and VIMOVO also face competition from the separate use of NSAIDs for pain relief and GI medications to address the risk of NSAID-induced ulcers. Use of these therapies separately in generic form may be less expensive than DUEXIS and VIMOVO. We expect to compete with the separate use of NSAIDs and ulcer medications primarily through DUEXIS’ and VIMOVO’s advantages in dosing convenience and patient compliance, and by educating physicians about such advantages. DUEXIS is the only NSAID medicine containing a histamine-2 receptor antagonist with an indication to reduce the risk of NSAID-induced upper GI ulcers and VIMOVO is the only NSAID medicine containing a PPI with an indication to reduce the risk of NSAID-induced ulcers. Data shows that physicians co-prescribe GI protective agents less than 25 percent of the time when prescribing an NSAID. On occasions where a patient is co-prescribed a GI protective agent, data shows that after three prescriptions, 61 percent of patients no longer take a GI protective agent.
PENNSAID 2% faces competition from generic versions of diclofenac sodium topical solutions which are priced significantly lower than the price we charge for PENNSAID 2%. In addition, PENNSAID 2% competes with two other branded topical NSAIDS, including Voltaren Gel, marketed by Endo Pharmaceuticals,
16
which is the market leader in the topical NSAID category. We expect to compete with these other medicines primarily through PENNSAID 2%’s dosing convenience and patient compliance. Unlike the other two medicines that are dosed four times per day and require the patient to measure out the correct dose, only PENNSAID 2% is easy to apply with the convenience of twice-daily dosing and a metered-dose pump, which ensures that the patient will get the correct amount of PENNSAID 2% solution each time.
Distribution
Finished tablets of DUEXIS, VIMOVO, RAYOS, MIGERGOT and BUPHENYL, vials of ACTIMMUNE and KRYSTEXXA, bottles of RAVICTI and PENNSAID 2% and powder of BUPHENYL are shipped to central third-party logistics FDA-compliant warehouses for storage and distribution into the supply chain. Our third-party logistics providers specialize in integrated operations that include warehousing and transportation services that can be scaled and customized to our needs based on market conditions and the demands and delivery service requirements for our medicines and materials. Their services eliminate the need to build dedicated internal infrastructures that would be difficult to scale without significant capital investment. Our third-party logistics providers warehouse all medicines in controlled FDA-registered facilities. Incoming orders are prepared and shipped through an order entry system to ensure just in time delivery of the medicines.
Sales and Marketing
As of June 30, 2016, our sales force was composed of approximately 500 sales representatives consisting of approximately 15 orphan disease sales representatives, 90 rheumatology sales specialists and 395 primary care sales representatives. Our orphan disease representatives focus on marketing our orphan medicines to a limited number of healthcare practitioners who specialize in fields such as pediatric immunology, allergy, infectious diseases, hematology/oncology and metabolic disorders to help them understand the potential benefits of ACTIMMUNE for their patients with CGD and SMO, and the benefits of RAVICTI and BUPHENYL for patients with UCDs. Our rheumatology sales force markets RAYOS, KRYSTEXXA and PENNSAID 2%. With respect to KRYSTEXXA, since the Crealta acquisition, we have increased our sales force from approximately 15 sales representatives to approximately 85 sales representatives and we have added 10 medical scientific liaisons. Our primary care sales force markets DUEXIS, PENNSAID 2% and VIMOVO. We have entered into, and may continue to enter into, agreements with third parties for commercialization of our medicines outside the United States.
Our medicines are dispensed by retail and specialty pharmacies. Part of our commercial strategy for our primary care and rheumatology business units is to offer physicians the opportunity to have their patients fill prescriptions through pharmacies participating in our HorizonCares patient access program. This program does not involve us in the prescribing of medicines. The purpose of this program is solely to assist in ensuring that, when physicians determine that one of our medicines offers a potential clinical benefit to their patients and prescribe the medicine for an eligible patient, financial assistance may be available to reduce a commercial patient’s out-of-pocket costs. In the first six months of 2016, this resulted in 99.8 percent of commercial patients having co-pay amounts of $10 or less when filling prescriptions for our medicines utilizing our patient access program. For commercial patients who are prescribed our primary care medicines or RAYOS, the HorizonCares program offers co-pay assistance when a third-party payer covers a prescription but requires an eligible patient to pay a co-pay or deductible, and offers full subsidization when a third-party payer rejects coverage for an eligible patient. For patients who are prescribed our orphan medicines, our patient access programs provide reimbursement support, a clinical nurse program, co-pay and other patient assistance. The aggregate commercial value of our patient access programs for the six months ended June 30, 2016 was $816.8 million. All pharmacies that dispense prescriptions for our medicines, which we estimate to be about 20,000 in the first half of 2016, are fully independent, including those that participate in HorizonCares. We do not own or possess any option to purchase an ownership stake in any pharmacy that distributes our medicines, and our relationship with each pharmacy is non-exclusive and arm’s length. All of our medicines are dispensed through pharmacies independent of our business. As of December 31, 2015, approximately 25 independent pharmacies participated in the HorizonCares program for our primary care and rheumatology medicines.
17
We have a compliance program in place to address adherence with various laws and regulations relating to our sales, marketing, and manufacturing of our medicines, as well as certain third-party relationships, including pharmacies. Specifically with respect to pharmacies, the compliance program utilizes a variety of methods and tools to monitor and audit pharmacies, including those that participate in our access programs, to confirm their activities, adjudication and practices are consistent with our compliance policies and guidance.
Manufacturing, Commercial and Supply Agreements
We have agreements with third parties for active pharmaceutical ingredients (“APIs”) and product manufacturing, formulation and development services, fill, finish and packaging services, transportation, and distribution and logistics services for certain medicines. In most cases, we retain certain levels of safety stock or maintain alternate supply relationships that we can utilize without undue disruption of our manufacturing processes if a third party fails to perform its contractual obligations.
ACTIMMUNE
ACTIMMUNE is a recombinant protein that is produced by fermentation of a genetically engineered Escherichia coli bacterium containing the DNA which encodes for the human protein. Purification of the active drug substance is achieved by conventional column chromatography. The resulting active drug substance is then formulated as a highly purified sterile solution and filled in a single-use vial for subcutaneous injection, which is the ACTIMMUNE finished drug medicine. In support of its manufacturing process, we and Boehringer Ingelheim International store multiple vials of the Escherichia coli bacterium master cell bank and working cell bank in order to ensure that it will have adequate backup should any cell bank be lost in a catastrophic event.
Boehringer Ingelheim RCV GmbH & Co. KG Supply Agreement
In July 2013, Vidara and Boehringer Ingelheim RCV GmbH & Co. KG (“Boehringer Ingelheim”) entered into an exclusive supply agreement, which we assumed as a result of the Vidara Merger, and which was subsequently amended on June 1, 2015. Pursuant to the agreement, Boehringer Ingelheim manufactures the ACTIMMUNE active drug substance and commercial quantities of the ACTIMMUNE finished drug medicine. Boehringer Ingelheim is our sole source supplier for ACTIMMUNE active drug substance and finished drug medicine. Under the terms of this agreement, we are required to purchase minimum quantities of finished drug medicine of 75,000 vials per annum. Boehringer Ingelheim manufactures our commercial requirements of ACTIMMUNE on an annual basis, and based on our forecasts and the annual contractual minimum purchase quantity. The supply agreement has a term that runs until July 31, 2020 and which can be further renewed by agreement between parties. Under this supply agreement, either we or Boehringer Ingelheim may terminate the agreement for an uncured material breach by the other party or upon the other party’s bankruptcy or insolvency.
Under a development and marketing agreement with Boehringer Ingelheim, we are required to pay royalties on net sales in certain applicable markets in Latin America, Asia, Africa and Eastern Europe if we elect to commercialize ACTIMMUNE in those territories. To date, we have not pursued regulatory or other approvals or commercialized ACTIMMUNE in those territories.
Genentech License Agreement
As a result of the Vidara Merger, we acquired a license agreement, as amended, with Genentech, Inc. (“Genentech”), who was the original developer of ACTIMMUNE. Under such agreement, we are or were obligated to pay royalties to Genentech on our net sales of ACTIMMUNE as follows:
| • | Through November 25, 2014, a royalty of 45 percent of the first $3.7 million in net sales achieved in a calendar year, and 10 percent on all additional net sales in that year; |
18
| • | For the period from November 26, 2014 through May 5, 2018, a royalty in the 20 percent to 30 percent range for the first tier in net sales and in the 1 percent to 9 percent range for the second tier; and |
| • | From May 6, 2018 and for so long as we continue to commercially sell ACTIMMUNE, an annual royalty in the low-single digits as a percentage of annual net sales. |
Either Genentech or we may terminate the agreement if the other party becomes bankrupt or defaults, however, in the case of a default, the defaulting party has 30 days to cure the default before the license agreement may be terminated.
RAVICTI
We have clinical and commercial supplies of glycerol phenylbutyrate API manufactured for us by two alternate suppliers, Helsinn Advanced Synthesis SA (Switzerland) and DSM Fine Chemicals Austria (now known as DPx Fine Chemicals GmbH & Co KG) on a purchase order basis. We have finished RAVICTI drug medicine manufactured by Lyne Laboratories, Inc. under a manufacturing agreement and we have an agreement in place for a fill/finish supplier, Halo Pharmaceuticals, Inc., for European supplies.
Ucyclyd Asset Purchase Agreement
As a result of the Hyperion acquisition, we acquired an asset purchase agreement with Ucyclyd Pharma, Inc. (“Ucyclyd”), pursuant to which we are obligated to pay to Ucyclyd tiered mid- to high- single digit royalties on our global net sales of RAVICTI. The asset purchase agreement cannot be terminated by either party. However, we have a license to certain Ucyclyd manufacturing technology, and Ucyclyd may have a license to certain of our technology, and the party granting a license is permitted to terminate the license if the other party fails to comply with any payment obligations relating to the license and does not cure such failure within a defined time period.
Brusilow License Agreement
As a result of the Hyperion acquisition, we acquired a license agreement with Saul W. Brusilow, M.D. and Brusilow Enterprises, Inc. (“ Brusilow”), pursuant to which we license patented technology related to RAVICTI from Brusilow. Under such agreement, we are obligated to pay low- single digit royalties to Brusilow on net sales of RAVICTI that are covered by a valid claim of a licensed patent. The license agreement may be terminated for any uncured breach as well as bankruptcy. We may terminate also the agreement at any time by giving Brusilow prior written notice, in which case all rights granted to us would revert to Brusilow.
ASD Distribution Services Agreement
As a result of the Hyperion acquisition, we acquired a distribution services agreement, as amended, with ASD Healthcare, a division of ASD Specialty Healthcare, Inc. (“ASD”). Pursuant to the distribution services agreement, ASD is the exclusive reseller of RAVICTI and BUPHENYL in the United States. The distribution services agreement terminates on February 13, 2017, but may be renewed upon mutual written agreement with ASD. Either party may terminate the agreement without cause upon 120 days written notice to the other party, in the case of a material breach that is not cured by the other party, upon 30 days written notice, or in the case of bankruptcy or similar proceeding of the other party, immediately upon written notice.
BUPHENYL
When Hyperion purchased BUPHENYL, Hyperion assumed all of Ucyclyd’s rights and obligations under its manufacturing agreements for the medicine. We assumed these agreements when we acquired Hyperion. We purchase API for BUPHENYL from CU Chemie Uetikon GmbH and final manufacturing, testing and packaging of the medicine is provided by Pharmaceutics International Inc.
19
DUEXIS
The DUEXIS manufacturing process is well-established and we validated the process in accordance with regulatory requirements prior to commercialization in the United States.
The first API in DUEXIS is ibuprofen in a direct compression blend called DC85 and is manufactured for us by BASF Corporation (“BASF”), in Bishop, Texas. The second API in DUEXIS is famotidine, which is available from a number of international suppliers. We currently purchase famotidine manufactured by Dr. Reddy’s in India. We currently receive both APIs in powder form and each is blended with a number of U.S. Pharmacopeia inactive ingredients. We purchase DUEXIS in final, packaged form exclusively from Sanofi-Aventis U.S. LLC (“Sanofi”) for our commercial requirements in North America.
BASF Contract
In July 2010, we entered into a contract with BASF for the purchase of DC85. Pursuant to the agreement, we are obligated to purchase a significant majority of our commercial demand for DC85 from BASF. The contract expires in December 2017. Thereafter, the agreement automatically renews for successive renewal terms of three years each until terminated by either party giving specified prior written notice to the other party. Either party may also terminate the agreement in the event of uncured breach by the other party.
Manufacturing and Supply Agreement with Sanofi
In May 2011, we entered into a manufacturing and supply agreement with Sanofi, which was amended in September 2013. Pursuant to the agreement, Sanofi is obligated to manufacture and supply DUEXIS to us in final, packaged form, and we are obligated to purchase DUEXIS exclusively from Sanofi for our commercial requirements in North America and certain countries and territories in Europe, including the EU member states and Scandinavia, and South America. Sanofi must acquire the components necessary to manufacture DUEXIS, including the APIs, DC85 and famotidine, and is obligated to acquire all DC85 under the terms of our agreements with suppliers, including the current BASF contract. In order to allow Sanofi to perform its obligations under the agreement, we granted Sanofi a non-exclusive license to our related intellectual property. The price for DUEXIS under the agreement varies depending on the volume of DUEXIS we purchase and is subject to annual adjustments to reflect changes in costs as measured by the Producer Price Index published by the U.S. Department of Labor, Bureau of Labor Statistics, and certain other changes and events set forth in the agreement. We have paid for the purchase and installation of equipment necessary to manufacture DUEXIS tablets, and Sanofi is obligated to pay the costs of routine maintenance of the equipment. Upon expiration or termination of the agreement we may also be obligated to reimburse Sanofi for the depreciated net book value of any other equipment purchased by Sanofi in order to fulfill its obligations under the agreement.
The agreement term extends until May 2019, and automatically extends for successive two-year terms unless terminated by either party upon two years prior written notice. Either party may terminate the agreement upon 30 days prior written notice to the other party in the event of breach by the other party that is not cured within 30 days of notice (which notice period may be longer in certain, limited situations) or in the event we lose regulatory approval to market DUEXIS in all countries worldwide, and either party may terminate the agreement without cause upon two years prior written notice to the other party at any time after the third anniversary of the first commercial sale of DUEXIS in any country worldwide.
VIMOVO
AstraZeneca License Agreement
In November 2013, we entered into a license agreement with AstraZeneca (the “AstraZeneca license agreement”), pursuant to which AstraZeneca granted us an exclusive license under certain intellectual property
20
(including patents, know-how, trademarks, copyrights and domain names) of AstraZeneca and its affiliates to develop, manufacture and commercialize VIMOVO in the United States. AstraZeneca also granted us a non-exclusive license under certain intellectual property of AstraZeneca and its affiliates to manufacture, import, export and perform research and development activities with respect to VIMOVO outside the United States but solely for purposes of commercializing VIMOVO in the United States. In addition, AstraZeneca granted us a non-exclusive right of reference and use under certain regulatory documentation controlled by AstraZeneca and its affiliates to develop, manufacture and commercialize VIMOVO in the United States and to manufacture, import, export and perform research and development activities with respect to VIMOVO outside the United States but solely for purposes of commercializing VIMOVO in the United States.
Under the AstraZeneca license agreement, we granted AstraZeneca a non-exclusive sublicense under such licensed intellectual property and a non-exclusive right of reference under certain regulatory documentation controlled by us to manufacture, import, export and perform research and development activities with respect to VIMOVO in the United States but solely for purposes of commercializing VIMOVO outside the United States.
Under the AstraZeneca license agreement, we and our affiliates are subject to certain limitations and restrictions on our ability to develop, commercialize and seek regulatory approval with respect to VIMOVO or other medicines that contain gastroprotective agents in a single fixed combination oral solid dosage form with NSAIDs (excluding DUEXIS). These limitations and restrictions include, among other things, restrictions on indications for which we may commercialize VIMOVO or any such other medicines, restrictions on our ability to develop or seek regulatory approval with respect to such other medicines that contain esomeprazole, restrictions on our ability to develop or seek regulatory approval for VIMOVO for any indications other than the indications for which NSAIDs are indicated, and restrictions on our marketing activities with respect to VIMOVO and any such other medicines.
The AstraZeneca license agreement continues in full force and effect until terminated in accordance with its terms. Under the AstraZeneca license agreement, the parties may terminate upon mutual written agreement by the parties, or either party may terminate rights granted to us with respect to licensed trademarks and licensed domain names under the AstraZeneca license agreement upon uncured material breach by the other party of certain specified provisions of the AstraZeneca license agreement.
Amended and Restated Collaboration and License Agreement with Pozen; Letter Agreement with AstraZeneca and Pozen
We entered into a license agreement with Pozen Inc. (“Pozen”), who subsequently entered into a business combination with Tribute Pharmaceuticals Canada Inc. to become known as Aralez Pharmaceuticals Inc. Under this agreement, we were granted an exclusive, royalty-bearing license under certain of Pozen’s intellectual property in the United States to manufacture, develop and commercialize VIMOVO and other medicines controlled by us that contain gastroprotective agents in a single fixed combination oral solid dosage form with NSAIDs, excluding DUEXIS, in the United States.
Under the Pozen license agreement, we are required to pay Pozen a flat 10 percent royalty based on net sales of VIMOVO and such other medicines sold by us, our affiliates or sublicensees during the royalty term, subject to minimum annual royalty obligations of $7.5 million, which minimum royalty obligations will continue for each year during which one of Pozen’s patents covers such medicines in the United States and there are no competing medicines in the United States. The royalty rate may be reduced to a mid-single digit royalty rate as a result of loss of market share to competing medicines. Our obligation to pay royalties to Pozen will expire upon the later of (a) expiration of the last-to-expire of certain patents covering such medicines in the United States, and (b) 10 years after the first commercial sale of such medicines in the United States. In addition, we will be obligated to reimburse Pozen for costs, including attorneys’ fees, incurred by Pozen in connection with VIMOVO patent litigation moving forward, subject to agreed caps.
21
We are responsible for, and are required to use diligent and reasonable efforts directed to commercializing VIMOVO or another qualified medicine in the United States. We also own and maintain all regulatory filings and marketing approvals in the United States for any such medicines, including all investigational new drugs (“INDs”), and new drug applications (“NDAs”) for VIMOVO. Pozen covenanted that it will not at any time prior to the expiration of the royalty term, and will ensure that its affiliates do not, directly or indirectly, develop or commercialize or license any third party to develop or commercialize certain competing medicines in the United States.
The Pozen license agreement, unless earlier terminated, will expire upon expiration of the royalty term for all such medicines in the United States. Either party has the right to terminate the agreement upon uncured material breach by the other party or upon the bankruptcy or similar proceeding of the other party. We also have the right to terminate the Pozen license agreement for cause upon certain defined medicine failures.
In November 2013, we, AstraZeneca and Pozen entered into a letter agreement in which Pozen consented to AstraZeneca’s assignment of the Pozen license agreement to us and that addresses the rights and responsibilities of the parties in relation to the Pozen license agreement and the amended and restated collaboration and license agreement between Pozen and AstraZeneca for territories outside the United States (the “Pozen-AstraZeneca license agreement”). Under the letter agreement, we and AstraZeneca agreed to pay Pozen milestone payments upon the achievement by us and AstraZeneca, collectively, of certain annual aggregate global net sales thresholds ranging from $550.0 million to $1.25 billion with respect to medicines licensed by Pozen to us under the Pozen license agreement and to AstraZeneca under the Pozen-AstraZeneca license agreement. The aggregate milestone payment amount that may be owed by AstraZeneca and us, collectively, under the letter agreement is $260.0 million, with the amount payable by each of us and AstraZeneca with respect to each milestone to be based upon the proportional sales achieved by each of us and AstraZeneca, respectively, in the applicable year.
The letter agreement will terminate with respect to Pozen and us upon the termination of the Pozen license agreement.
Patheon Agreement
In November 2013, we entered into a master manufacturing services agreement and product agreement (collectively the “Patheon manufacturing agreement”), with Patheon Pharmaceuticals Inc. (“Patheon”), who was AstraZeneca’s contract manufacturer of VIMOVO, for the manufacture and supply of VIMOVO. Under the Patheon manufacturing agreement, we agreed to purchase a specified percentage of our VIMOVO requirements for the United States from Patheon or its affiliates. In addition, under the terms of the Patheon manufacturing agreement, we are able to enter into individual product agreements with Patheon for the manufacture of specific medicines in addition to VIMOVO if agreed by us and Patheon.
Pursuant to the Patheon manufacturing agreement, we are required to supply Patheon with any active materials for VIMOVO. We must pay an agreed price for final, packaged VIMOVO supplied by Patheon subject to adjustments, including certain unilateral adjustments by Patheon, such as annual adjustments for inflation and adjustments to account for certain increases in the cost of components of VIMOVO other than active materials.
The Patheon manufacturing agreement will be effective until December 31, 2019 and will automatically renew for successive terms of three years each if there is any product agreement in effect, unless either party gives written notice to the other party of its intention to terminate the agreement at least 24 months prior to the end of the then current term. Either party may terminate the Patheon manufacturing agreement or any product agreement early for uncured material breach by the other party or upon the other party’s bankruptcy or insolvency. We may terminate any product agreement if any regulatory authority takes any action or raises any objection that prevents us from commercializing the product. Additionally, Patheon may terminate the Patheon manufacturing agreement or any product agreement early if we assign our rights or obligations under the Patheon
22
manufacturing agreement or such product agreement to a competitor of Patheon or to a party that, in the reasonable opinion of Patheon, is not a credit worthy substitute for us, or in certain other circumstances where we assign the Patheon manufacturing agreement or product agreement without Patheon’s consent.
PENNSAID 2%
Nuvo Supply Agreement
In October 2014, in connection with the acquisition of the U.S. rights to PENNSAID 2% from Nuvo, we entered into an exclusive supply agreement with Nuvo, which was amended in February 2016, under which Nuvo will manufacture and supply PENNSAID 2% to us. We have committed to a binding purchase order to Nuvo for delivery of PENNSAID 2%. In addition, at least 90 days prior to the first day of each calendar month during the term of the supply agreement, we are required to submit a binding written purchase order to Nuvo for PENNSAID 2% in minimum batch quantities. The term of our supply agreement is through December 31, 2029, but the agreement may be terminated earlier by either party for any uncured material breach by the other party of its obligations under the supply agreement or upon the bankruptcy or similar proceeding of the other party.
A key excipient used in PENNSAID 2% as a penetration enhancer is DMSO. We and Nuvo rely on a sole proprietary form of DMSO for which we maintain a substantial safety stock. However, should this supply become inadequate, damaged, destroyed or unusable, we and Nuvo may not be able to qualify a second source.
RAYOS/LODOTRA
We rely on well-established third-party manufacturers for the manufacture of RAYOS/LODOTRA. We purchase the primary active ingredients for RAYOS/LODOTRA from Tianjin Tianyao Pharmaceuticals Co., Ltd. in China and from Sanofi Chimie SA in France. We have contracted with Jagotec AG (“Jagotec”) for the production of RAYOS/LODOTRA tablets through its affiliate SkyePharma, and we entered into an agreement with Patheon for the packaging and assembling of RAYOS/LODOTRA.
SkyePharma and Jagotec Agreements
Development and License Agreement
In August 2004, we entered into a development and license agreement with SkyePharma and Jagotec, a wholly owned subsidiary of SkyePharma, regarding certain proprietary technology and know-how owned by SkyePharma for the delayed-release of corticosteroids. Under the agreement, which was amended in August 2007, we received an exclusive, sub-licensable worldwide license to the oral formulation of any glucocorticoid, including prednisone, prednisolone, methylprednisolone and/or cortisone, with delayed-release technology covered by intellectual property rights and know-how owned by SkyePharma. We were also granted an option to acquire a royalty-free, exclusive and sub-licensable right to license and manufacture RAYOS/LODOTRA which we could exercise any time upon specified prior written notice, expiring no earlier than five years after the first launch of RAYOS/LODOTRA. We have exercised the option to acquire the manufacturing license, which became effective in April 2014.
In return for the grant of the license, Jagotec has the right to manufacture, package and supply RAYOS/LODOTRA to us in accordance with terms and conditions of a separate manufacturing and supply agreement we entered into with Jagotec. In addition, Jagotec is entitled to receive a single-digit percentage royalty on net sales of RAYOS/LODOTRA and on any sub-licensing income, which includes any payments not calculated based on the net sales of RAYOS/LODOTRA, such as license fees, and lump sum and milestone payments.
The agreement expires on a country-by-country basis, upon the expiration of the last patent rights for RAYOS/LODOTRA, which will occur between 2024 and 2028. In the event of expiration, the licenses under the agreement will be perpetual, fully paid-up and royalty-free. Either party may also terminate the agreement in the event of a liquidation or bankruptcy of the other party or upon an uncured breach by the other party.
23
Manufacturing and Supply Agreement
In August 2007, we entered into a manufacturing and supply agreement with Jagotec for RAYOS/LODOTRA. Under the agreement, which was amended in March 2011, Jagotec or its affiliates manufacture and supply RAYOS/LODOTRA to us in bulk. Aenova France SAS, a large contract manufacturing organization, is now a subcontractor for Jagotec for the manufacture of RAYOS/LODOTRA, with our consent. As of December 31, 2015, our total remaining minimum purchase commitment was approximately $3.0 million based on tablet pricing under the agreement as of that date, which amount is subject to volume and price adjustments due to, among other things, inflation, order quantities and launch and approval in certain EU countries. We also supply the prednisone API to Jagotec at our expense for use in the manufacture of RAYOS/LODOTRA.
We pay Jagotec, exclusive of any value added tax or similar governmental charges, a price for RAYOS/LODOTRA representing a negotiated mark-up over manufacturing costs. The price is adjusted annually to reflect changes in both manufacturing and materials costs as measured by the Ensemble price index. If Jagotec makes a major capital expenditure during the contract term to fulfill increased orders forecast by us, the price per unit will increase if the actual order falls short of the forecast.
The original agreement term has run such that the agreement now automatically extends on a yearly basis unless terminated by either party upon prior written notice. Either party may also terminate the agreement in the event of insolvency, liquidation or bankruptcy of the other party or upon an uncured breach by the other party. We have the right to receive a continuing supply of RAYOS/LODOTRA from Jagotec for a period of 24 months after termination by Jagotec, regardless of the reason for termination. In April 2015, the agreement automatically renewed for an additional one-year term. Therefore the earliest the right to receive a continuing supply from Jagotec would expire is April 15, 2018, absent any early termination of the agreement.
Pursuant to a letter agreement between Jagotec and us, Jagotec agreed to allow us to give Bayer Pharma AG (“Bayer”) the right to manufacture, test and release quantities of RAYOS/LODOTRA in order to establish and maintain Bayer as a manufacturer of RAYOS/LODOTRA. Under certain circumstances, we may also purchase shortfall quantities of RAYOS/LODOTRA from Bayer to the extent Jagotec is unable to supply us. In March 2013, we entered into an agreement with Bayer to allow us to purchase quantities of RAYOS/LODOTRA for these purposes. We may also purchase quantities of RAYOS/LODOTRA from Bayer pursuant to our agreement with Bayer.
KRYSTEXXA
KRYSTEXXA is produced by fermentation and subsequent purification to produce the urate oxidase enzyme, uricase. Uricase is then PEGylated with a PEGylation agent to produce the bulk medicine, pegloticase. Finally, pegloticase is filled and packaged to produce the final medicine.
NOF Supply Agreement
In August 2015, Crealta and NOF Corporation (“NOF”) entered into an exclusive supply agreement for the PEGylation agent used in the manufacture of KRYSTEXXA. Under the terms of this agreement, we are required to issue NOF forecasts of our requirements for the PEGylation agent, a portion of which are binding. The agreement expires in August 2020, however, either we or NOF may terminate the agreement for any reason upon 24 months’ prior notice. Either we or NOF may also terminate the agreement upon a material breach, if not cured within a specified period of time, or in the event of the other party’s insolvency. While there are no minimum purchase obligations under the agreement, we are required to use NOF as our exclusive supplier for the PEGylation agent, subject to certain exceptions if NOF is unable to supply the PEGylation agent.
Bio-Technology General (Israel) Supply Agreement
In March 2007, Savient Pharmaceuticals, Inc. (as predecessor in interest to Crealta) (“Savient”) entered into a commercial supply agreement with Bio-Technology General (Israel) Ltd. (“BTG Israel”) for the production of the bulk KRYSTEXXA medicine
24
(“bulk medicine”). We assumed this agreement as part of the Crealta acquisition and amended the agreement in September 2016. Under this agreement, we have agreed to purchase certain minimum annual order quantities and are obligated to purchase at least 80 percent of our annual world-wide bulk medicine requirements from BTG Israel. The term of the agreement runs until December 31, 2030, and will automatically renew for successive three year periods unless earlier terminated by either party upon three years prior written notice. The agreement may be terminated earlier by either party in the event of a force majeure, liquidation, dissolution, bankruptcy or insolvency of the other party, uncured material breach by the other party or after January 1, 2024, upon three years prior written notice.
Sigma Tau PharmaSource Supply Agreement
In October 2008, Savient and Sigma Tau PharmaSource, Inc. (as successor in interest to Enzon Pharmaceuticals, Inc.) (“Sigma Tau”) entered into a commercial supply agreement for the packaging and supply of the final drug medicine KRYSTEXXA, which we acquired as part of the Crealta acquisition. This agreement remains in effect until terminated, and either we or Sigma Tau may terminate the agreement with three years notice, given 30 days prior to the agreement anniversary date. Either we or Sigma Tau may also terminate the agreement upon a material default, if not cured within a specified period of time, or in the event of the other party’s insolvency or bankruptcy.
Duke University and Mountain View Pharmaceutical License Agreement
In August 1998, Savient entered into an exclusive, worldwide license agreement with Duke University (“Duke”) and Mountain View Pharmaceuticals, Inc. (“MVP”). Duke developed the recombinant uricase enzyme used in KRYSTEXXA and MVP developed the PEGylation technology used in the manufacture of KRYSTEXXA. Duke and MVP may terminate the agreement if we commit fraud or for our willful misconduct or illegal conduct; upon our material breach of the agreement, if not cured within a specified period of time; upon written notice if we have committed two or more material breaches under the agreement; or in the event of our bankruptcy or insolvency. Under the terms of the agreement, we are obligated to pay Duke a mid-single digit percentage royalty on our global net sales of KRYSTEXXA and a low-double digit percentage royalty on any global sublicense revenue. We are also obligated to pay MVP a mid-single digit percentage royalty on our net sales of KRYSTEXXA outside of the United States and a low-double digit percentage royalty on any sublicense revenue outside of the United States. Royalties terminate upon last to expire of licensed patents on a country-by-country basis, and royalties are reduced by a mid-double digit percentage in countries that never had patents.
Research and Development
We devote significant resources to research and development activities associated with our current branded medicines. For the years ended December 31, 2015, 2014 and 2013, we recorded $41.9 million, $17.5 million and $10.1 million, respectively, in research and development expenses.
25
The following chart depicts our current clinical development pipeline with respect to ACTIMMUNE, RAVICTI, RAYOS and KRYSTEXXA:
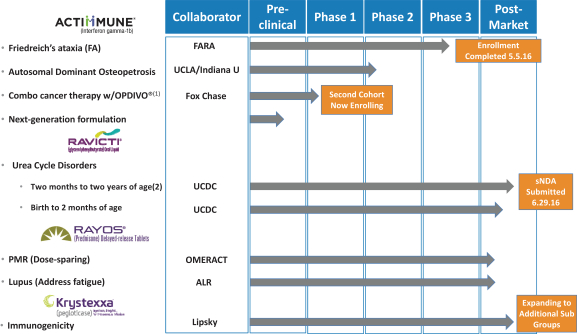
| (1) | Registered trademark of Bristol-Meyers Squibb |
| (2) | Clinical trial was completed and sNDA was submitted in the second quarter of 2016 |
ACTIMMUNE
In February 2015, we submitted an IND application to the FDA for ACTIMMUNE in the treatment of FA, a degenerative neuro-muscular disorder. In June 2015, we commenced the Phase 3 STEADFAST study. This Phase 3 trial (NCT02415127) is a randomized, multi-center, double-blind, placebo-controlled study with patients randomized 1:1 to receive subcutaneous doses of either ACTIMMUNE or placebo three times a week for a total of 26 weeks. 92 patients were enrolled at four sites in the United States. The primary endpoint will measure the change in neurological outcome and evaluate the effect of ACTIMMUNE versus placebo as measured by the mFARS score, focused on objective neurologic measures such as upper and lower extremity coordination improvement from baseline. The mFARS score is used to measure neurological signs associated with FA, with higher scores reflecting a greater level of disability. In addition to safety and efficacy, the STEADFAST trial will evaluate the pharmacokinetic characteristics of ACTIMMUNE in people with FA. We completed enrollment for the STEADFAST trial in the second quarter of 2016, with data anticipated to become available in late 2016. Assuming positive data from the trial, we would plan to submit a supplemental biologics license application in the first quarter of 2017, and given the fast-track designation of ACTIMMUNE for this potential indication, we would request priority review, which, if awarded, would allow us to potentially receive a decision from the FDA within six months of the submission, in the third quarter of 2017.
In July 2015, we announced our collaboration with Fox Chase Cancer Center to study ACTIMMUNE in combination with PD-1/PD-L1 inhibitors in various forms of cancer including advanced urothelial carcinoma (bladder cancer) and renal cell carcinoma. Pre-clinical cell line research has indicated that interferon gamma
26
enhances cellular PD-L1 expression on endothelial cells (inner lining of the blood vessel) and on some tumor cells. By enhancing cellular PD-L1 expression on tumor cells, interferon gamma may promote or enhance the effect of the PD-1 or PD-L1 inhibitors. In December 2015, we announced that an investigator-initiated Phase 1 clinical study had been initiated to evaluate ACTIMMUNE in combination with OPDIVO (nivolumab), a registered trademark of Bristol-Meyers Squibb, in advanced solid tumors. The Phase 1 open label study will evaluate the combination of ACTIMMUNE and nivolumab in patients with advanced solid tumors who have progressed on at least one prior systemic therapy, which may include prior immunotherapy. Patients will be treated with a one week induction phase of ACTIMMUNE (starting dose 50 mcg/m2 subcutaneously), followed by a combination phase with ACTIMMUNE and nivolumab (3 mg/kg intravenously) for three cycles, followed by a single-agent phase of nivolumab alone for up to one year. The study will primarily assess the safety and tolerability of the combination of ACTIMMUNE and nivolumab. Secondary objectives, including overall response rate, progression free survival and overall survival, will also be assessed, as will various correlative analyses. Initial subject enrollment will occur using a modified 6+6 design, and if endpoints for safety (using dose-limiting toxicity criteria) are met, expansion cohorts in renal cell carcinoma (kidney cancer) and urothelial carcinoma (bladder cancer) are planned for up to 15 patients per cohort.
We are collaborating with Indiana University to study ACTIMMUNE in the treatment of type 2 osteopetrosis, autosomal dominant osteopetrosis (“ADO2”). ADO2 is a genetic condition characterized by generalized osteosclerosis predominating in some skeletal sites such as the spine and pelvis. The short-term, open label treatment trial in ADO2 patients aims to determine if administration of ACTIMMUNE increases biochemical markers of bone turnover, and thus determine if the medicine can completely or partially reverse the defective osteoclastic bone resorption in ADO2 patients. The clinical study is expected to run over a period of three years, and commenced in early 2016.
We are also collaborating with several partners to investigate opportunities for next generation formulations of ACTIMMUNE in current and new indications.
RAVICTI
We are in the process of seeking approval for label expansions for RAVICTI, with assessments in progress studying the use of RAVICTI in patients both from two months to two years (for which we submitted a supplemental new drug application sNDA submission to the FDA in the second quarter of 2016), and from birth to two months (targeted sNDA submission in the first quarter of 2018). Current FDA approval is for patients from two years of age and older only. In patients with UCDs for which RAVICTI is an FDA-approved medicine, there is a variable age of diagnosis (from newborn to adulthood), and the severity of the disease can be associated with the age of onset and enzymatic deficit. However, a prompt diagnosis and careful management of the disease can lead to good clinical outcomes.
RAYOS
In November 2015, we announced our collaboration with the ALR to study the effect of RAYOS on the fatigue experienced by SLE patients. SLE is a chronic autoimmune disease that causes inflammation and pain in the joints and muscles, as well as overall fatigue. RAYOS is currently indicated for patients with SLE. The first study planned as part of the collaboration is an investigator-initiated, randomized, double-blind, active comparator, cross-over study in which patients will be randomized to receive either prednisone for three months or RAYOS at 10 p.m. for three months, and then switched to the alternative medication for an additional three months. Approximately 62 patients across 25 sites will be enrolled in the United States. The primary endpoint will assess fatigue as measured by Functional Assessment of Chronic Illness Therapy-Fatigue, a 13-question survey to be completed by study participants that focuses on the daily fatigue experienced in patients with chronic illnesses.
We are also collaborating with the University of Alabama at Birmingham School of Medicine in a randomized, open-label, dose-ranging study of RAYOS in patients with untreated PMR. The study aims to
27
determine the relative reduction in the severity of morning stiffness of three night time doses (4mg, 7mg, and 10mg) of RAYOS, compared to the reduction after treatment in the morning with immediate-release 15mg prednisone medicines, in newly diagnosed PMR patients. The selected patients will have had no evidence of other systemic inflammatory diseases and will be known to be responsive to standard treatment in the morning with immediate-release 15mg prednisone medicines.
KRYSTEXXA
In January 2016, following our acquisition of Crealta, we assumed responsibility for a study designed to test the potential reduction of immunogenicity in KRYSTEXXA patients, known as the Tolerization Reduces Intolerance to Pegloticase and Prolongs the Urate Lowering Effect (the “TRIPLE study”). The TRIPLE study is an investigator-initiated, post-market interventional, exploratory open-label, multicenter study of approximately 20 patients to evaluate the effectiveness of a 16-week high zone tolerance regimen of KRYSTEXXA on response to therapy (serum uric acid <6 mg/dL) in adult hyperuricemic patients with gout refractory to conventional therapy. We are also developing a potential registration study to expand the label should the TRIPLE study show positive results. Success in the TRIPLE study and the subsequent registration study would have the potential to significantly expand the patient population and usage of KRYSTEXXA.
As part of the TRIPLE study, initial, more frequent dosing is being examined to determine if this reduces antibody formation by inducing antigen specific non-responsiveness. This would prevent the formation of anti-pegloticase antibodies and prevent the loss of drug response. This involves evaluating the drug’s lowest trough level, which pharmacokinetically occurs between the first and second doses. Increasing this trough level should suppress the high titer antibody formation. Current labelling states that KRYSTEXXA should be given every two weeks. This study adds one extra dose that occurs one week after the initial dose. Active study will last 16 weeks (10 doses) and will compare responder rates with historical control KRYSTEXXA data. The IND for this trial was filed in August 2015, site selection and final investigator meetings were completed in October 2015 and patient enrollment began in November 2015.
An observational study is also being conducted to satisfy certain conditions in the KRYSTEXXA biologics license application (“BLA”) approval letter. A clinical study report was submitted to FDA in February 2016 and the review is ongoing.
Intellectual Property
Our objective is to aggressively patent the technology, inventions and improvements that we consider important to the development of our business. We have a portfolio of patents and applications based on clinical and pharmacokinetic/pharmacodynamic modeling discoveries, and our novel compounds and formulations. We intend to continue filing patent applications seeking intellectual property protection as we generate anticipated formulation refinements, new methods of manufacturing and clinical trial results.
We have multiple patents and patent applications related to DUEXIS. Unless otherwise invalidated, those patents expire in 2026. We continue to prosecute and pursue additional patent coverage on DUEXIS and its uses. However, under the license agreement with Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc. (collectively “Par”), Par may enter the market on January 1, 2023, or earlier under certain circumstances.
We have an exclusive license to U.S. and foreign patents and patent applications from SkyePharma covering RAYOS/LODOTRA. If not otherwise invalidated, those in-licensed patents expire between 2024 and 2028. We continue to prosecute and pursue additional patent coverage on RAYOS/LODOTRA and its uses. However, under the Settlement Agreement with Actavis Laboratories FL, Inc. (formerly known as Watson Laboratories, Inc.—Florida) (“Actavis”), Actavis may enter the market on December 23, 2022, or earlier under certain circumstances.
28
We also have licenses to U.S. patents and patent applications and trademarks covering VIMOVO from Pozen and AstraZeneca. We co-own other U.S. patents and patent applications with Pozen. If not otherwise invalidated, those in-licensed patents expire between 2016 and 2031. We continue to prosecute and pursue patent protection in the United States to obtain additional patent coverage on VIMOVO and its uses.
We also have ownership of U.S. patents and patent applications covering PENNSAID 2% from Nuvo. We also co-own other U.S. patent applications with Mallinckrodt LLC. If not otherwise invalidated, those patents expire between 2027 and 2030. We continue to prosecute and pursue patent protection in the United States to obtain additional patent coverage on PENNSAID 2% and its uses.
We also have ownership of U.S. patents covering ACTIMMUNE. If not otherwise invalidated, those patents expire in 2022. We continue to prosecute and pursue patent protection to obtain additional patent coverage on ACTIMMUNE and its uses.
We also have licenses to U.S. and foreign patents and applications covering KRYSTEXXA. If not otherwise invalidated, those patents expire between 2019 and 2030. We continue to prosecute and pursue patent protection to obtain additional patent coverage on KRYSTEXXA and its uses.
We also have an exclusive license to U.S. and foreign patents from Brusilow Enterprises LLC covering RAVICTI which expire in the United States in 2018 and if extended, in certain countries in Europe in 2021. We also have ownership of U.S. and foreign patents and patent applications covering RAVICTI. If not otherwise invalidated, those patents expire between 2030 and 2032. We continue to prosecute and pursue patent protection to obtain additional patent coverage on RAVICTI and its uses.
We will only be able to protect our technologies and medicines from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them. As such, our commercial success will depend in part on receiving and maintaining patent protection and trade secret protection of our technologies and medicines as well as successfully defending these patents against third-party challenges.
In the United States, ACTIMMUNE has been granted orphan-drug designation for the treatment of FA and we anticipate that ACTIMMUNE will receive seven years of orphan drug exclusivity upon approval for that indication in the United States. In the United States, KRYSTEXXA has received 12 years of biologic exclusivity, expiring in 2022, and seven years of orphan drug exclusivity, expiring in 2017.
In the United States, in addition to patent protections, PENNSAID 2% has been granted three years of marketing exclusivity as a Section 505(b)(2) NDA. This marketing exclusivity period for each medicine began upon marketing approval of such medicine and runs in parallel with any patents that have issued or we expect to be issued protecting such medicine. In the United States, RAVICTI has been granted seven years of orphan drug exclusivity. In the EU, RAVICTI received 10 years of marketing exclusivity protection, beginning with its December 2015 marketing authorization. In the EU, LODOTRA has received 10 years of marketing exclusivity protection, beginning with its March 2009 marketing authorization in Germany.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States. The patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. For example:
| • | we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
29
| • | we or our licensors might not have been the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that none of our pending patent applications or the pending patent applications of our licensors will result in issued patents; |
| • | our issued patents and the issued patents of our licensors may not provide a basis for commercially viable drugs, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; |
| • | we may not be successful in any patent litigation to enforce our patent rights, including our pending patent litigation regarding, PENNSAID 2%, RAVICTI and/or VIMOVO; |
| • | we may not develop additional proprietary technologies or medicine candidates that are patentable; or |
| • | the patents of others may have an adverse effect on our business. |
Third-Party Coverage and Reimbursement
In both U.S. and foreign markets, our ability to commercialize our medicines successfully depends in significant part on the availability of coverage and adequate reimbursement to healthcare providers from third-party payers, including, in the United States, government payers such as the Medicare and Medicaid programs, managed care organizations and private health insurers. Third-party payers are increasingly challenging the prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. This is especially true in markets where over-the-counter and generic options exist. Even if coverage is made available by a third-party payer, the reimbursement rates paid for covered medicines might not be adequate. For example, third-party payers may use tiered coverage and may adversely affect demand for our medicines by not covering our medicines or by placing them in a more expensive formulary tier relative to competitive medicines (where patients have to pay relatively more out of pocket than for medicines in a lower tier). We cannot be certain that our medicines will be covered by third-party payers or that such coverage, where available, will be adequate, or that our medicines will successfully be placed on the list of drugs covered by particular health plan formularies. Many states have also created preferred drug lists and include drugs on those lists only when the manufacturers agree to pay a supplemental rebate. The industry competition to be included on such formularies and preferred drug lists often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug on their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other therapeutic alternative is available. In addition, because each third-party payer individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming and costly process. We may be required to provide scientific and clinical support for the use of any medicine to each third-party payer separately with no assurance that approval would be obtained, and we may need to conduct pharmacoeconomic studies to demonstrate the cost effectiveness of our medicines for formulary coverage and reimbursement. Even with studies, our medicines may be considered less safe, less effective or less cost-effective than competitive medicines, and third-party payers may not provide coverage and adequate reimbursement for our medicines or our medicine candidates. These pricing and reimbursement pressures may create negative perceptions to any medicine price increases, or limit the amount we may be able to increase our medicine prices, which may adversely affect our medicine sales and results of operations. Where coverage and reimbursement are not adequate, physicians may limit how much or under what circumstances they will prescribe or administer such medicines, and patients may decline to purchase them. This, in turn, could affect our ability to successfully commercialize our medicines and impact our profitability, results of operations, financial condition, and future success.
30
The U.S. market has seen a trend in which retail pharmacies have become increasingly involved in determining which prescriptions will be filled with the requested medicine or a substitute medicine, based on a number of factors, including potentially perceived medicine costs and benefits, as well as payer substitution policies. Many states have in place requirements for prescribers to indicate “dispense as written” on their prescriptions if they do not want pharmacies to make substitutions; these requirements are varied and not consistent across states. We may need to increasingly spend time and resources to ensure the prescriptions written for our medicines are filled as written, where appropriate.
Coverage policies, third-party reimbursement rates and medicine pricing regulation may change at any time. Even if favorable coverage and adequate reimbursement status is attained for one or more medicines that receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose extensive requirements upon the clinical development, pre-market approval, manufacture, labeling, marketing, promotion, pricing, import, export, storage and distribution of medicines. These agencies and other regulatory agencies regulate research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, recordkeeping, advertising and promotion of drugs and biologics. Failure to comply with applicable FDA or foreign regulatory agency requirements may result in Warning Letters, fines, civil or criminal penalties, suspension or delays in clinical development, recall or seizure of medicines, partial or total suspension of production or withdrawal of a medicine from the market.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act and its implementing regulations and biologics additionally under the Public Health Service Act. The process required by the FDA before medicine candidates may be marketed in the United States generally involves the following:
| • | submission to the FDA of an IND, which must become effective before human clinical trials may begin and must be updated annually; |
| • | completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s good laboratory practice (“GLP”) regulations; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the medicine candidate for each proposed indication; |
| • | submission to the FDA of an NDA or BLA, as appropriate, after completion of all pivotal clinical trials to demonstrate the safety, purity and potency of the medicine candidate for the indication for use; |
| • | a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities to assess compliance with the FDA’s current good manufacturing practices (“cGMPs”) regulations for pharmaceuticals; and |
| • | FDA review and approval of an NDA or BLA prior to any commercial marketing or sale of the medicine in the United States. |
31
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our medicine candidates will be granted on a timely basis, if at all.
The results of preclinical tests (which include laboratory evaluation as well as GLP studies to evaluate toxicity in animals) for a particular medicine candidate, together with related manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. IND submissions may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during medicine development. Further, an independent IRB for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the institutional review board (“IRB”) or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive good clinical practice regulations and regulations for informed consent and privacy of individually identifiable information. Similar requirements to the U.S. IND are required in the EEA and other jurisdictions in which we may conduct clinical trials.
Clinical Trials. For purposes of NDA or BLA submission and approval, clinical trials are typically conducted in the following sequential phases, which may overlap:
| • | Phase 1. Studies are initially conducted in a limited population to test the medicine candidate for safety, dose tolerance, absorption, distribution, metabolism, and excretion, typically in healthy humans, but in some cases in patients. |
| • | Phase 2. Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, explore the initial efficacy of the medicine for specific targeted indications and to determine dose range or pharmacodynamics. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| • | Phase 3. These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the medicine is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, provide substantial evidence of clinical efficacy and further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial centers. |
| • | Phase 4. The FDA may approve an NDA or BLA for a medicine candidate, but require that the sponsor conduct additional clinical trials to further assess the medicine after approval under a post marketing commitment or post marketing requirement. In addition, a sponsor may decide to conduct additional clinical trials after the FDA has approved a medicine. Post-approval trials are typically referred to as Phase 4 clinical trials. |
The results of drug development, preclinical studies and clinical trials are submitted to the FDA as part of an NDA or BLA, as appropriate. Applications also must contain extensive chemistry, manufacturing and control information. Applications must be accompanied by a significant user fee. Once the submission has been accepted for filing, the FDA’s goal is to review applications within 12 months of submission or, if the application relates to an unmet medical need in a serious or life-threatening indication, eight months from submission. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA will typically conduct a pre-approval inspection of the manufacturer to ensure that the medicine can be reliably produced in compliance with cGMPs and will typically inspect certain clinical trial sites for compliance
32
with good clinical practice (“GCP”). The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it typically follows such recommendations. The FDA may deny approval of an application by issuing a Complete Response Letter if the applicable regulatory criteria are not satisfied. A Complete Response Letter may require additional clinical data and/or trial(s), and/or other significant, expensive and time- consuming requirements related to clinical trials, preclinical studies or manufacturing. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Approval may occur with boxed warnings on medicine labeling or Risk Evaluation and Mitigation Strategies (“REMS”), which limit the labeling, distribution or promotion of a medicine. Once issued, the FDA may withdraw medicine approval if ongoing regulatory requirements are not met or if safety problems occur after the medicine reaches the market. In addition, the FDA may require testing, including Phase 4 clinical trials, and surveillance programs to monitor the safety effects of approved medicines which have been commercialized and the FDA has the power to prevent or limit further marketing of a medicine based on the results of these post-marketing programs or other information.
Expedited Review Programs. A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of NDAs. For example, Fast Track Designation may be granted to a drug intended for treatment of a serious or life-threatening disease or condition that has potential to address unmet medical needs for the disease or condition. The key benefits of fast track designation are the eligibility for priority review, rolling review (submission of portions of an application before the complete marketing application is submitted), and accelerated approval, if relevant criteria are met. Based on results of clinical studies submitted in an NDA, upon the request of an applicant, the FDA may grant the NDA priority review designation, which sets the target date for FDA action on the application at six months after the FDA accepts the application for filing. Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Pediatric Studies and Exclusivity. NDAs must contain data (or a proposal for post-marketing activity) to assess the safety and effectiveness of an investigational new drug product for the claimed indications in all relevant pediatric populations in order to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers if certain criteria are met. The requirements for pediatric data do not apply to any drug for an indication for which orphan designation has been granted.
Pediatric exclusivity is another type of non-patent exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the five-year and three-year non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA or BLA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical study is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of FDA-requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve another application relying on the NDA sponsor’s data.
Orphan Medicines. Under the Orphan Drug Act, the FDA may designate a medicine as an “orphan drug” if it is intended to treat a rare disease or condition, meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and
33
making a medicine available in the United States for treatment of the disease or condition will be recovered from sales of the medicine. A company must request orphan drug designation before submitting an NDA for the drug and rare disease or condition. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan drug designation does not shorten the Prescription Drug User Fee Act (“PDUFA”) goal dates for the regulatory review and approval process, although it does convey certain advantages such as tax benefits and exemption from the PDUFA application fee.
If a medicine with orphan designation receives the first FDA approval for the disease or condition for which it has such designation or for a select indication or use within the rare disease or condition for which it was designated, the medicine generally will receive orphan drug exclusivity. Orphan drug exclusivity means that the FDA may not approve another sponsor’s marketing application for the same drug for the same indication for seven years, except in certain limited circumstances. Orphan exclusivity does not block the approval of a different drug for the same rare disease or condition, nor does it block the approval of the same drug for different indications. If a drug designated as an orphan drug ultimately receives marketing approval for an indication broader than what was designated in its orphan drug application, it may not be entitled to exclusivity. Orphan exclusivity will not bar approval of another medicine under certain circumstances, including if a subsequent medicine with the same drug for the same indication is shown to be clinically superior to the approved medicine on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand.
In the EU, Regulation (EC) No 141/2000 and Regulation (EC) No. 847/2000 provide that a medicine can be designated as an orphan medicinal product by the European Commission (“EC”) if its sponsor can establish: that the medicine is intended for the diagnosis, prevention or treatment of (1) a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons in the EU when the application is made, or (2) a life-threatening, seriously debilitating or serious and chronic condition in the EU and that without incentives the medicinal product is unlikely to be developed. For either of these conditions, the applicant must demonstrate that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the EU or, if such method exists, the medicinal product will be of significant benefit to those affected by that condition. Once authorized, orphan medicinal products are entitled to ten years of market exclusivity in all EU Member States and in addition a range of other benefits during the development and regulatory review process including scientific assistance for study protocols, authorization through the centralized marketing authorization procedure covering all member countries and a reduction or elimination of registration and marketing authorization fees. However, marketing authorization may be granted to a similar medicinal product with the same orphan indication during the ten year period with the consent of the marketing authorization holder for the original orphan medicinal product or if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if this medicine is safer, more effective or otherwise clinically superior to the original orphan medicinal product. The period of market exclusivity may, in addition, be reduced to six years if it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity.
Other Regulatory Requirements. Medicines manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping, annual medicine quality review, payment of medicine and manufacturing establishment fees and reporting requirements. Adverse event experience with the medicine must be reported to the FDA in a timely fashion and pharmacovigilance programs to proactively look for these adverse events are mandated by the FDA. Our medicines may be subject to REMS requirements that affect labeling, distribution or post market reporting. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Following such inspections, the FDA may issue notices on Form 483 and Untitled Letters or Warning Letters that could cause us or our third-party manufacturers to modify certain activities. A
34
Form 483 notice, if issued at the conclusion of an FDA inspection, can list conditions the FDA investigators believe may have violated cGMP or other FDA regulations or guidelines. In addition to Form 483 notices and Untitled Letters or Warning Letters, failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of medicine, injunctive action, import alert or possible civil penalties. The FDA may also require us to recall a drug from distribution or withdraw approval for that medicine.
The FDA closely regulates the post-approval marketing and promotion of pharmaceuticals, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet, including certain social media activities. Medicines may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the medicine, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental application, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in adverse publicity, Warning Letters or “untitled letters”, corrective advertising and potential administrative, civil and criminal penalties, as well as damages, fines, withdrawal of regulatory approval, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to sell our medicines or operate our business and also adversely affect our financial results.
Physicians may, in their independent medical judgment, prescribe legally available pharmaceuticals for uses that are not described in the medicine’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use. Additionally, a significant number of pharmaceutical companies have been the target of inquiries and investigations by various U.S. federal and state regulatory, investigative, prosecutorial and administrative entities in connection with the promotion of medicines for off-label uses and other sales practices. These investigations have alleged violations of various U.S. federal and state laws and regulations, including claims asserting antitrust violations, violations of the Food, Drug and Cosmetic Act, false claims laws, the Prescription Drug Marketing Act, anti-kickback laws, and other alleged violations in connection with the promotion of medicines for unapproved uses, pricing and Medicare and/or Medicaid reimbursement. If our promotional activities, including any promotional activities that a contracted sales force may perform on our behalf, fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to issue warning letters or untitled letters, suspend or withdraw an approved medicine from the market, require corrective advertising or a recall or institute fines or civil fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our business. In addition, the distribution of prescription medicines is subject to the Prescription Drug Marketing Act (the “PDMA”), which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription medicine samples and impose requirements to ensure accountability in distribution, including a drug pedigree which tracks the distribution of prescription drugs. Further, under the Drug Quality and Security Act, drug manufacturers are subject to a number of requirements, including, medicine identification, tracing and verification, among others, that are designed to detect and remove counterfeit, stolen, contaminated or otherwise potentially harmful drugs from the U.S. drug supply chain. These requirements will be phased in over several years and compliance will likely increase the costs of the manufacture and distribution of drug medicines.
35
Outside the United States, the ability of our partners and us to market a medicine is contingent upon obtaining marketing authorization from the appropriate regulatory authorities. The requirements governing marketing authorization, pricing and reimbursement vary widely from country to country and region to region.
The EU and the EEA consist of the 28 Member States of the EU, plus Norway, Iceland and Liechtenstein which are Member States of the EEA. These Member States have all acceded to the single market rules governing the supervision of medicinal products. Under the prevailing rules, medicinal products can only be commercialized after obtaining a Marketing Authorization (“MA”). There are three procedures for a marketing authorization to be obtained:
| • | the Centralized MA, which is issued by the EC through the Centralized Procedure, based on the scientific opinion of the CHMP of the EMA, and which is valid throughout the entire territory of the EU/EEA. When decisions on granting of a Centralized MA are taken by the EU, the EEA Member States will take corresponding decisions on the basis the relevant acts to permit marketing of medicinal products. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EU/EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. |
| • | Decentralized Procedure MAs are available for products not falling within the mandatory scope of the Centralized Procedure. An identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State (the “RMS”), to lead the evaluation of the regulatory submission. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics (“SmPC”), and a draft of the labeling and package leaflet as distilled from the preliminary evaluation, which are sent to the other Member States (referred to as the Concerned Member States (the “CMS”), for their approval. If the CMS raise no objections, based on a potential serious risk to public health, to the assessment, SmPC, labeling, or packaging proposed by the RMS, the RMS records the agreement, closes the procedure and informs the applicant accordingly. Each Member State concerned by the procedure is required to adopt a national decision to grant a national MA in conformity with the approved assessment report, SmPC and the labelling and package leaflet as approved. Where a product has already been authorized for marketing in a Member State of the EEA, the granted national MA can be used for mutual recognition in other Member States through the Mutual Recognition Procedure (the “MRP”) resulting in progressive national approval of the product in the EU/EEA. |
| • | National MAs, which are issued by a single competent authority of the Member States of the EEA and only covers their respective territory, are also available for products not falling within the mandatory scope of the Centralized Procedure. Once a product has been authorized for marketing in a Member State of the EEA through the National Procedure, this National MA can also be recognized in other Member States through the MRP. |
Under the procedures described above, before granting the MA, the EMA or the competent authority(ies) of the Member State(s) of the EEA prepare an assessment of the risk-benefit balance of the product against the scientific criteria concerning its quality, safety and efficacy.
Under Regulation (EC) No 726/2004/EC and Directive 2001/83/EC (each as amended), the EU has adopted a harmonized approach to data and market protection or exclusivity (known as the 8 + 2 + 1 formula). The data exclusivity period begins to run on the date when the first MA is granted in the EU. It confers on the
36
MA holder of the reference medicinal product eight years of data protection and 10 years of market protection. A reference medicinal product is defined to mean a medicinal product authorized based on a full dossier consisting of pharmaceutical and preclinical testing results and clinical trial data, such as a medicinal product containing a new active substance. The 10-year market protection can be extended cumulatively to a maximum period of 11 years if during the first eight years of those ten years of protection period, the MA holder obtains an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies.
The protection period means that an applicant for a generic medicinal product is not permitted to rely on preclinical pharmacological, toxicological, and clinical data contained in the file of the reference medicinal product of the originator until the first eight years of data protection have expired. Thereafter, a generic product application may be submitted and generic companies may rely on the preclinical and clinical data relating to the reference medicinal product to support approval of the generic product. However, a generic cannot market until ten years have elapsed from the initial authorization of the reference medicinal product or eleven years if the protection period is extended, based on the formula of 8+2+1.
The 8 + 2 + 1 exclusivity scheme applies to products that have been authorized in the EU by either the EMA through the Centralized Procedure or the competent authorities of the Member States of the EEA nationally albeit through the Decentralized, or Mutual Recognition procedures.
For a medicinal product which is designated as orphan under Regulation 141/2000, it will benefit from a period of 10 years of orphan market exclusivity which essentially constitutes a period of market monopoly. During this period of orphan market exclusivity, no EU regulatory authority is permitted to accept or approve an application for marketing authorization for a similar medicinal product or an extension application for the same therapeutic indication. This period can be extended cumulatively to a total of 12 years if the marketing authorization holder or applicant complies with the requirements for an agreed pediatric investigation plan pursuant to Regulation 1901/2006.
The holder of a Centralized MA or National MA is subject to various obligations under the applicable EU laws, such as pharmacovigilance obligations, requiring it to, among other things, report and maintain detailed records of adverse reactions, and to submit periodic safety update reports to the competent authorities. The holder must also ensure that the manufacturing and batch release of its product is in compliance with the applicable requirements. The MA holder is further obligated to ensure that the advertising and promotion of its products complies with applicable EU laws and industry code of practice as implemented in the domestic laws of the Member States of the EU/EEA. The advertising and promotional rules are enforced nationally by the EU/EEA Member States.
Healthcare Fraud and Abuse Laws. As a pharmaceutical company, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We may be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. For example, in the United States, there are federal and state anti-kickback laws that prohibit the payment or receipt of kickbacks, bribes or other remuneration intended to induce the purchase or recommendation of healthcare products and services or reward past purchases or recommendations. Violations of these laws can lead to civil and criminal penalties, including fines, imprisonment and exclusion from participation in federal healthcare programs. These laws are potentially applicable to manufacturers of products regulated by the FDA, such as us, and pharmacies, hospitals, physicians and other potential purchasers of such products.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce either the referral of an individual, or the furnishing, recommending, or arranging for a good or service, for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” is not defined in the federal Anti-Kickback Statute and has been broadly interpreted to include anything of value, including for
37
example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of payment, ownership interests and providing anything at less than its fair market value. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute may have been violated, and enforcement will depend on the relevant facts and circumstances. The Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively the “ACA”), among other things, amended the intent requirement of the federal Anti-Kickback Statute to state that a person or entity needs not have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below) or the civil monetary penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent, or to have offered improper inducements to federal health care program beneficiaries to select a particular provider or supplier. The federal Anti-Kickback Statute is broad, and despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs, and do not contain identical safe harbors. In addition, where such activities involve foreign government officials, they may also potentially be subject to the Foreign Corrupt Practices Act. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities, including our activities with physician customers, pharmacies, and patients, as well as our activities pursuant to partnerships with other companies and pursuant to contracts with contract research organizations, could be subject to challenge under one or more of such laws.
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. In addition, the ACA specified that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. The federal False Claims Act has been the basis for numerous enforcement actions and settlements by pharmaceutical and other healthcare companies in connection with various alleged financial relationships with customers. In addition, a number of pharmaceutical manufacturers have reached substantial financial settlements in connection with allegedly causing false claims to be submitted because of the companies’ marketing of products for unapproved, and thus non-reimbursable, uses. Certain marketing practices, including off-label promotion, may also violate false claims laws, as might violations of the federal physician self-referral laws, such as the Stark laws, which prohibit a physician from making a referral to a provider of certain health services with which the physician or the physician’s family member has a financial interest and prohibit submission of a claim for reimbursement pursuant to a prohibited referral. The “qui tam” provisions of the False Claims Act allow a private individual to bring civil actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In addition, various states have enacted similar fraud and abuse statutes or regulations, including, without limitation, false claims laws analogous to the False Claims Act, and laws analogous to the federal Anti-Kickback Statute, that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer, and there are also federal criminal false claims laws.
Separately, there are a number of other fraud and abuse laws that pharmaceutical manufacturers must be mindful of, particularly after a medicine candidate has been approved for marketing in the United States. For example, a federal criminal law enacted as part of, the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payers. The false statements statute prohibits knowingly and willfully
38
falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. There are also federal civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent, as well as federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers.
Healthcare Privacy and Security Laws. We may be subject to, or our marketing activities may be limited by, HIPAA, as amended by the Health Information Technology and Clinical Health Act and their respective implementing regulations, which established uniform standards for certain “covered entities” (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information. Among other things, HIPAA’s privacy and security standards are directly applicable to “business associates”—independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. In addition to possible civil and criminal penalties for violations, state attorneys general are authorized to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. In the EU/EEA, Directive 95/46/EEC (as amended) or its successor applies to identified or identifiable personal data processed by automated means (e.g. a computer database of customers) and data contained in, or intended to be part of, non-automated filing systems (traditional paper files) as well as transfer of such data to a country outside of the EU/EEA.
“Sunshine” and Marketing Disclosure Laws. There are an increasing number of federal and state “sunshine” laws that require pharmaceutical manufacturers to make reports to states on pricing and marketing information. Several states have enacted legislation requiring pharmaceutical companies to, among other things, establish marketing compliance programs, file periodic reports with the state, and make periodic public disclosures on sales and marketing activities, and prohibiting certain other sales and marketing practices. In addition, a similar recently implemented federal requirement requires manufacturers, including pharmaceutical manufacturers, to track and report to the federal government certain payments and other transfers of value made to physicians and other healthcare professionals and teaching hospitals and ownership or investment interests held by physicians and their immediate family members. The federal government began disclosing the reported information on a publicly available website in 2014. These laws may adversely affect our sales, marketing, and other activities with respect to our medicines in the United States by imposing administrative and compliance burdens on us. If we fail to track and report as required by these laws or otherwise comply with these laws, we could be subject to the penalty provisions of the pertinent state and federal authorities. In the EU/EEA, declaration of transfers of value to healthcare professionals is subject to the requirements under the voluntary industry code of practice. France however has a statutory regime similar to the US Sunshine Act.
Government Price Reporting. For those marketed medicines which are covered in the United States by the Medicaid programs, we have various obligations, including government price reporting and rebate requirements, which generally require medicines be offered at substantial rebates/discounts to Medicaid and certain purchasers (including “covered entities” purchasing under the 340B Drug Discount Program). We are also required to discount such medicines to authorized users of the Federal Supply Schedule of the General Services Administration, under which additional laws and requirements apply. These programs require submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations, and the guidance governing such calculations is not always clear. Compliance with such requirements can require significant investment in personnel, systems and resources, but failure to properly calculate our prices, or offer required discounts or rebates could subject us to substantial penalties. One component of the rebate and discount calculations under the Medicaid and 340B programs, respectively, is the “additional rebate”, a complex
39
calculation which is based, in part, on the rate at which a branded drug price increases over time more than the rate of inflation (based on the CPI-U). This comparison is based on the baseline pricing data for the first full quarter of sales associated with a branded drug’s NDA, and baseline data cannot generally be reset, even on transfer of the NDA to another manufacturer. This “additional rebate” calculation can, in some cases where price increase have been relatively high versus the first quarter of sales of the NDA, result in Medicaid rebates up to 100 percent of a drug’s “average manufacturer price” and 340B prices of one penny. Subject to the control of Directive 89/105/EEC, pricing and reimbursement in the EU/EEA is governed by national rules and policy and may vary from Member State to Member State.
In General. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities, in the United States, could be subject to challenge under one or more of such laws. If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including significant civil and criminal penalties, damages, fines, imprisonment, exclusion from participation in U.S. federal or state healthcare programs, and the curtailment or restructuring of our operations. To the extent that any medicine we make is sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals. Any penalties, damages, fines, curtailment or restructuring of our operations could materially adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security, sunshine, government price reporting, and fraud laws may prove costly.
Impact of Healthcare Reform and Recent Public Scrutiny of Specialty Drug Pricing on Coverage, Reimbursement, and Pricing. In the United States and other potentially significant markets for our medicines, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative medicines and therapies, which has resulted in lower average selling prices. Further, the increased scrutiny of prescription drug pricing practices and emphasis on managed healthcare in the United States and on country-specific and regional pricing and reimbursement controls in the EU will put additional pressure on medicine pricing, reimbursement and usage, which may adversely affect our future medicine sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
The U.S. and some foreign jurisdictions are considering or have enacted a number of additional legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our medicines profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs (including a number of proposals pertaining to prescription drugs, specifically), improving quality and/or expanding access. There has been particular and increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices over the course of 2015, particularly with respect to drugs that have been subject to relatively large price increases over relatively short time periods. There have been several recent U.S. Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. In the United States, the pharmaceutical industry has already been significantly affected by major legislative initiatives, including, for example, the ACA. The ACA, among other things, imposes a significant annual fee on companies that manufacture or import branded prescription drug medicines. It also contains substantial provisions intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, and impose additional health policy reforms,
40
any or all of which may affect our business. The ACA, compounded by the intense public scrutiny of drug pricing in the United States, is likely to continue the downward pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Other legislative changes have also been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011 resulted in aggregate reductions in Medicare payments to providers of up to 2 percent per fiscal year, starting in 2013, and the American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Such laws, and others that may affect our business that have been recently enacted or may in the future be enacted, may result in additional reductions in Medicare and other healthcare funding. In the future, there will likely continue to be additional proposals relating to the reform of the U.S. healthcare system, some of which could further limit coverage and reimbursement of medicines, including our medicine candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our medicines.
41
Raptor is a biopharmaceutical company focused on developing and commercializing transformative treatments for people affected by rare and debilitating diseases.
Raptor’s first commercial medicine, PROCYSBI, received marketing approval from the FDA in April 2013 for the management of nephropathic cystinosis in adults and children six years and older with seven years of market exclusivity, through 2020. More recently, PROCYSBI received orphan drug designation from the FDA for the treatment of patients ages two years to six years, through 2022. In Europe, PROCYSBI gastro-resistant hard capsules of cysteamine (as mercaptamine bitartrate), received a marketing authorization in September 2013 from the EC as an orphan medicinal product for the management of proven nephropathic cystinosis in the EU. The EU marketing authorization allows Raptor to commercialize PROCYSBI in the 28 Member States of the EU plus Norway, Liechtenstein and Iceland (which are not EU Member States but are part of the EEA. PROCYSBI received seven and ten years of market exclusivity due to its designation as an orphan drug in the United States and the EU, respectively. Raptor achieved first commercial sales of PROCYSBI in the United States in June 2013 and in the EU, specifically in Germany, in April 2014. A New Drug Submission (“NDS”) for PROCYSBI for the treatment of nephropathic cystinosis is currently under review by Health Canada. Recently, Raptor received notice from Health Canada that it is seeking additional information to complete its review. Raptor intends to submit a response to Health Canada in a timely fashion to preserve Priority Review status and expects that Health Canada’s timeline for review of the NDS will re-commence when it accepts Raptor’s response. As a result, Raptor anticipates a delay in Health Canada’s decision on the potential marketing approval of PROCYSBI.
As of December 31, 2015, insurers of U.S. commercial patients reimburse Raptor for PROCYSBI therapy at a Wholesale Acquisition Cost (“WAC”), price for PROCYSBI of $17,812.50 per bottle of 250 75-mg capsules and $4,275.00 per bottle of 60 25-mg capsules. Prices for PROCYSBI therapy vary among patients because doses are individually based on a patient’s weight. In September 2013, Raptor executed an agreement to participate in the U.S. State Medicare/Medicaid rebate program, which is reflected in Raptor’s net revenues in mandatory rebates on reimbursements for patients receiving state Medicare and Medicaid insurance coverage. As of December 31, 2015, Raptor’s price to German, Swiss and Austrian pharmacies was €5,850.23 per bottle of 250 75-mg capsules and €468.02 per bottle of 60 25-mg capsules.
In October 2015, Raptor acquired various assets and rights related to levofloxacin solution for inhalation, a pharmaceutical product also known as “MP-376” and commercially as QUINSAIR, from Tripex Pharmaceuticals, LLC (“Tripex”). QUINSAIR received marketing authorization by the EMA for treating chronic lung infection caused by the bacteria Pseudomonas aeruginosa in adults who have cystic fibrosis in March 2015 and Health Canada in June 2015 for the management of cystic fibrosis in patients aged 18 years or older with chronic pulmonary Pseudomonas aeruginosa infections. QUINSAIR is the first inhaled fluoroquinolone approved for the management of chronic pulmonary infections due to Pseudomonas aeruginosa in patients with cystic fibrosis 18 years old and older. In April 2016, Raptor launched QUINSAIR in Germany and Denmark, and plans to continue its European launch through 2017, and anticipates launching in Canada in the fourth quarter of 2016. Raptor has discussed the path to potential approval in the same indication in the United States with the FDA and is awaiting additional feedback from the FDA prior to taking any next steps. Raptor also plans to initiate a clinical study for MP-376 in bronchiectasis in 2016. QUINSAIR is not approved in the United States, and Raptor may not market or commercialize QUINSAIR in the United States for any indication unless it receives FDA approval, which it may not be able to obtain.
42
Cysteamine Mechanism of Action
Cysteamine, or 2-aminoethanethiol, the active pharmaceutical ingredient in PROCYSBI, is a molecule generated naturally in human cells during the metabolism of cysteine. Cysteamine is used to construct the key enzymatic cofactor involved in energy produced from sugars and lipids. Cysteamine’s uniquely reactive properties result in a number of physiological effects when given in pharmaceutical doses.
| • | Antioxidation—Cysteamine is known to increase levels of a key cellular antioxidant, glutathione. Glutathione is composed of the amino acids gamma-glutamate, cysteine and glycine. The availability of cysteine is the major rate-limiting factor in glutathione production. Cysteamine may release cysteine in the circulation, or from within the cell. Cysteamine has been shown to activate the NRF2 pathway, which leads to the increased expression of a wide variety of proteins involved in antioxidation which may help to reduce oxidative stress in CNS, hepatic and mitochondrial disorders. |
| • | Proteostasis—Heat shock proteins (“HSPs”) are chaperones that play an important role in protein-protein interactions such as folding and assist in the establishment of proper protein conformation. Proper protein folding may also prevent unwanted protein aggregation. By helping to stabilize partially unfolded proteins, HSPs aid in transporting proteins across membranes within the cell. HSPs are typically produced by cells in response to stress or injury, or other metabolic imbalance. HSPs are part of a cell’s mechanism for protein maintenance. The presence of cysteamine within a cell has been shown to increase transcription of certain HSPs that are key components to the cell’s ability to maintain the integrity of proteins. |
| • | Anti-fibrosis—Cysteamine blocks TGF-ß signaling and thereby inhibits the production and proliferation of myofibroblasts. It also inhibits formation of three cross-links in collagen protein, each of which exacerbate formation of fibrotic tissue: gamma-glutamyl peptide bonds, formed by transglutaminase; oxidized lysyl-lysine conjugates, formed by lysyl oxidase; and inter-chain disulfide bonds. |
| • | Transcription inhibition—Cysteamine inhibits transcription of a variety of collagens and basement membrane-related proteins: |
| • | Metal chelation—In vitro studies have shown that cysteamine chelates metals, including copper, zinc and iron. High doses of cysteamine can lead to copper depletion, implying that chelation effects also occur in vivo. |
| • | Induction of DNA repair mechanisms—Cysteamine has been known for over sixty years to mitigate the effects of radiation by upregulating cell cycle checkpoints and repair mechanisms. |
PROCYSBI
PROCYSBI is an approved therapy for the management of nephropathic cystinosis, a rare, life-threatening metabolic lysosomal storage disorder that causes the rapid, toxic accumulation of cystine in all cells, tissues and organs in the body. PROCYSBI capsules contain cysteamine bitartrate in the form of innovative microspheronized beads that are individually coated to create delayed and extended-release properties, allowing patients to maintain consistent therapeutic systemic drug levels over a 12-hour dosing period. The enteric-coated beads are pH sensitive and bypass the stomach for dissolution and absorption in the more alkaline environment of the proximal small intestine. Randomized controlled clinical trials and extended treatment with PROCYSBI therapy demonstrated consistent cystine depletion as monitored by levels of the biomarker (and surrogate marker), white blood cell cystine.
43
About Nephropathic Cystinosis
Raptor estimates that there are approximately 500 patients diagnosed with cystinosis living in the United States and an estimated 2,000 worldwide. Nephropathic cystinosis comprises 95% of known cases of cystinosis. In these patients, elevated cystine leads to cellular dysfunction and death; without treatment, the disease is usually fatal by the end of the first decade of life. Cystinosis is progressive, eventually causing irreversible tissue damage and multi-organ failure, including kidney failure, blindness, muscle wasting and premature death. Nephropathic cystinosis is usually diagnosed in infancy after children present with symptoms including markedly increased urination, thirst, dehydration, gastrointestinal distress, failure to thrive, rickets, photophobia and kidney symptoms specific to Fanconi syndrome. Management of cystinosis requires lifelong therapy.
Cystine depletion is the only approved treatment strategy for nephropathic cystinosis. Committed adherence and persistence to cystine depletion therapy is critical to achieve optimal clinical outcomes. Failure to adhere to prescribed dosing of cystine depletion therapy results in disease progression, including kidney failure leading to dialysis and kidney transplantation, muscle wasting and in most cases, premature death. Even brief interruptions in daily therapy can permit toxic accumulation of cystine, exposing tissues to renewed, progressive deterioration.
In addition to the population of patients who have already been identified, Raptor believes that a number of patients with atypical phenotypic presentation and end-stage renal disease have their condition as a result of undiagnosed late-onset nephropathic cystinosis, and would benefit from treatment with PROCYSBI. Raptor currently is collaborating with the Marshfield Clinic to develop an algorithm to identify late onset cystinosis patients.
QUINSAIR
QUINSAIR is a formulation of the antibiotic drug levofloxacin, suitable for inhalation via a nebulizer. This route of delivery allows higher concentrations of drug in the lung sputum than can be achieved via systemic (e.g. oral) administration. QUINSAIR, as approved, is administered twice daily in 28-day cycles, using a hand-held nebulizer with a disposable handset known as the Zirela device, manufactured by Raptor’s partner PARI Pharma GmbH (“PARI”), and configured specifically for use with QUINSAIR.
QUINSAIR is the first fluoroquinolone inhaled antibiotic to be approved in Canada and the EU for the treatment of chronic pulmonary infections due to Pseudomonas aeruginosa in cystic fibrosis (“CF”) patients. QUINSAIR was approved in the EU and Canada on the basis of three randomized, controlled studies, one Phase 2 and two Phase 3. In the EU, QUINSAIR is eligible for “new data” regulatory exclusivity of ten years after approval, a period which is concurrent with, and independent from, the period of any applicable patent.
Raptor has discussed filing of a NDA with the FDA for the treatment of Pseudomonas aeruginosa infection in CF based on the studies that were the basis for EU and Canadian marketing approvals. Raptor is awaiting additional feedback from the FDA prior to submitting an NDA based on the FDA’s feedback.
About Pseudomonas aeruginosa infection in Cystic Fibrosis
CF is a rare, life-threatening genetic disease affecting approximately 70,000 people worldwide. CF is caused by a mutation in the cystic fibrosis transmembrane conductance regulator (“CFTR”) gene. Defective or missing CFTR protein causes poor flow of salt and water into or out of the cell in several organs, including the lungs. This leads to the buildup of abnormally thick secretions that can cause chronic lung infections and progressive lung damage in many patients that eventually leads to death.
Patients with CF are highly susceptible to colonization with bacterial infections of the lung, largely because their pulmonary mucous secretions are thicker, stickier, and more difficult to expectorate than those of healthy individuals. This creates an environment in the lung that favors bacterial proliferation. As of 2014,
44
approximately 50% of all patients with CF in the U.S. were colonized with Pseudomonas aeruginosa, a gram-negative bacterial infection. Infection rates climb as patients age, with over 80% of patients colonized by adulthood. In the EU, infection rates vary significantly from country to country, with a median of approximately 35% of patients colonized. These infections are currently treated primarily with inhaled antibiotics, including tobramycin, an aminoglycoside-class antibiotic sold by Novartis Pharmaceuticals Corporation as TOBI or in dry-powder-inhalation format as TOBI Podhaler and sold by others in generic form, and aztreonam, a monobacter-class antibiotic which is marketed in an inhaled formulation by Gilead Sciences, Inc. under the tradename Cayston. Both tobramycin and aztreonam are primarily effective against gram-negative bacteria such as Pseudomonas aeruginosa. However, the prevalence of multi-drug-resistant Pseudomonas aeruginosa is growing. Thus, Raptor believes there is an unmet need that might be addressed with a new class of inhaled antibiotic such as the fluoruquinolone class that levofloxacin represents.
CLINICAL DEVELOPMENT
RP103 Clinical Development
Huntington’s Disease
Huntington’s Disease (“HD”) is a rare, inherited neurodegenerative disorder caused by an autosomal dominant mutation in a gene called huntingtin (Htt). The huntingtin gene encodes a protein that is also called “huntingtin.” Expansion of a CAG triplet repeat beyond the normal range within the huntingtin gene results in a mutant form of the protein, which gradually damages cells in the brain. HD causes neuronal degeneration in the cerebral cortex and basal ganglia, which play a key role in movement and behavior control.
RP103, enteric-coated delayed-release cysteamine bitartrate, is currently being evaluated as a potentially neuroprotective treatment for HD. Centre Hospitalier Universitaire d’Angers (“CHU d’Angers”) in France, is conducting a Phase 2/3 clinical trial of RP103, referred to as CYST-HD (Clinicaltrials.gov Identifier:NCT02101957). This trial comprises an 18-month blinded, placebo-controlled phase, followed by an 18-month open-label phase in which all patients transitioned to RP103 and an extension phase for subjects who finished the 36 month trial and wish to continue on RP103. The primary endpoint of the trial was change from the baseline of the Total Motor Score (“TMS”) of the Unified Huntington’s Disease Rating Scale (“UHDRS”) between RP103 and placebo treated patients.
CYST-HD: 18 months results
In February 2014, Raptor announced top line results from the planned 18-month analysis of the study. A total of 96 patients with HD were randomized to treatment with RP103 or placebo. A total of 89 patients completed the initial 18-month phase. A mixed model analysis of all 96 patients enrolled in the trial showed a trend towards slower progression of TMS in patients treated with RP103 versus those patients on placebo after 18 months treatment (4.51 vs. 6.68 respectively, p=0.19). While the results did not reach statistical significance, an overall positive trend was observed.
There were no new or unusual variations from RP103’s clinical safety profile with 48 of 52 patients experiencing at least one adverse event (“AE”) during the 18-month interim evaluation versus 38 of 44 under placebo.
CYST-HD: 36 months results
In December 2015, Raptor announced top line results from the planned 36-month analysis of the study, which included subjects who crossed over from placebo to open-label treatment at the completion of month 18. The study examined the effect of RP103 in Huntington’s disease subjects treated earlier, beginning at Month 0 (RP103/RP103) compared to subjects with a delayed start to treatment, beginning at Month 18 (placebo/RP103). ted 36 months of treatment. The full analyses set included all randomized subjects from Month 0 to Month 36.
45
An evaluation of the change in the progression of the UHDRS-TMS at Month 36 from baseline in the full analyses set in the trial showed a 25% slower progression [10.0 (1.7) vs. 13.3 (1.8), respectively; p=0.18] in patients treated earlier with RP103 relative to those patients on a delayed start. In a completers analysis, these effects were more pronounced with a 35% slower progression [9.2(1.7) vs. 14.1(1.9), respectively p=0.06] in the earlier treatment with RP103 relative to those patients on a delayed start. The 25% treatment effect in TMS favoring subjects treated with RP103 for the full 36 months as compared to the placebo/RP103 arm was not statistically significant.
The safety profile observed for RP103 after completion of the 36 month study was generally consistent with what has been previously reported. Suicides have not been observed in any other RP103 clinical trials or in any patients on clinical or commercial drug.
Regulatory Update
Raptor initiated regulatory discussions with the FDA and the EMA in 2014 based on the 18-month data and most recently received feedback from the EMA in the fourth quarter of 2015 through a Scientific Advice procedure. The outcome of these interactions with both agencies indicated that additional data from a confirmatory study would be required to support an application for marketing authorization.
Under Raptor’s amended collaboration agreement with CHU d’Angers, Raptor supplies RP103 and placebo capsules for the clinical trial and open-label extension study and fund the third-party statistical analysis of clinical trial data in exchange for regulatory and commercial rights to the clinical trial data. Clinical expenses of the study are covered by a grant from the Programme Hospitalier de Recherche Clinique, which is funded by the French government.
In 2008, Raptor received FDA orphan drug designation for cysteamine formulations, including RP103, for the potential treatment of HD. Raptor has received orphan drug designation in the EU for the treatment of HD.
Raptor intends to seek non-dilutive funding or a strategic partnership to advance development of RP103 in HD.
Mitochondrial Disorders including Leigh Syndrome
Leigh syndrome is a severe neurological disorder caused by genetic defects in mitochondrial or nuclear DNA affecting respiratory chain function that typically results in death within the first decade of life. The condition causes increased production of reactive oxygen species that disrupt mitochondrial electron transport and affect cellular function in a variety of tissues. Typically observed during the first year of life, Leigh syndrome is characterized by a failure to thrive, lack of coordination, involuntary and sustained muscle contraction, muscle wasting and multiple organ failure. The incidence of Leigh syndrome and similar mitochondrial disorders in the United States is estimated to be 1 in 40,000 newborns.
RP103 as a Treatment for Mitochondrial Disorders including Leigh Syndrome
In June 2014, Raptor initiated a Phase 2 study in the United States designed to evaluate the safety, tolerability and efficacy of RP103 as a potential treatment for Leigh syndrome and other mitochondrial disorders. RP103 potentially increases mitochondrial glutathione which may act as a scavenging agent of reactive oxygen species and thereby reduce the mitochondrial oxidative stress typically associated with these disorders.
The clinical plan includes an open label, 24 week, Phase 2a study in 24 patients (up to a maximum of 32 patients). Patients with Leigh syndrome are expected to comprise approximately two-thirds of the enrolled population in the study. Employing a statistical plan based on an adaptive design, Raptor conducted interim
46
analyses after four patients and again after 12 patients had completed the study, with the analyses indicating that the trial should continue as planned. The primary endpoint of the study will be the change from baseline in the Newcastle Pediatric Mitochondrial Disease Scale, at 24 weeks. Secondary endpoints will include observations of myopathy, dystonia, seizures, motor development, dyskinesia, quality of life and activities of daily living.
MP-376 Clinical Development
In addition to CF, Raptor believes MP-376 has development potential in two additional orphan diseases with significant unmet need: non-CF bronchiectasis (“BE”) and nontuberculous mycobacteria (“NTM”) lung infections. Currently, few therapeutic options exist for patients with these diseases. BE is characterized by abnormal dilatation and destruction of lung bronchi and bronchioles due to chronic recurring infection and long-term inflammation, which leads to frequent hospitalizations. NTM are a group of microbes that cause severe and recurrent lung infections, often in individuals who are immune-compromised or who have structural lung disease, such as bronchiectasis. Raptor is evaluating the therapeutic potential of MP-376 in these indications and intends to pursue clinical programs in 2016 in non-CF related bronchiectasis and is also planning to do work in preparation to support further clinical development of MP-376 in NTM.
About non-CF Bronchiectasis
BE is a disease in which airways lose elasticity over time, which impairs the lung’s ability to clear out mucus, creating a highly favorable environment for bacteria. These bacterial infections typically result in inflammation that further damages bronchial tissue, creating a negative feedback loop and significant loss of lung function. BE can be due to a variety of causes, including but not limited to chronic obstructive pulmonary disease, smoking history, autoimmune disease, and triggering bacterial or viral infection.
Current standard of care is to reduce the number of exacerbations requiring critical care, with reduction of bacterial load considered to be highly important as a preventative measure. No antibiotic product is approved currently. There is some evidence suggesting that drugs of the macrolide class are effective in treating infections in the context of BE, but these are associated with rapid development of bacterial resistance as well as side effects including hepatotoxicity, hearing loss, and cardiovascular events.
In in vitro studies, levofloxacin has demonstrated the ability to eradicate bacterial colonies of several types that are found with high frequency in the non-CF BE population. In addition, randomized, controlled studies have been conducted with inhaled ciprofloxacin, another drug in the same fluoroquinolone class as levofloxacin, showing increased time to exacerbations and improvements in bacterial load when compared to placebo. Levofloxacin is, in general, as potent as, or more potent in in vitro assays than ciprofloxacin. Thus, Raptor believes it is a favorable candidate for development in BE.
About Non-Tuberculous Mycobacterium Infections
NTM is a bacterial infection of the lung caused by bacteria of the mycobacterium family, but which do not result in tuberculosis. Infections of this type frequently result in progressive loss of lung function, and can be life-threatening if not treated. Symptoms of NTM include fever, weight loss, cough, lack of appetite, night sweats, and loss of energy. Approximately 60,000 patients exist in the United States, with another 30,000 found in the EU.
No treatment is currently approved specifically for use in NTM. Treatment guidelines suggest high doses of systemic (usually oral) antibiotics be used, typically with multiple active agents in a “cocktail” to cover a variety of bacterial strains. These regimens have a high rate of failure to clear the infection, and also cause significant side effects such as gastrointestinal distress, hearing impairment, flu-like reactions, and liver toxicity. Surgical resection of lung tissue is recommended in particularly symptomatic cases.
47
In in vitro studies, levofloxacin (the active agent in MP-376) has demonstrated the ability to inhibit bacterial colonies of several types that are found with high frequency in the NTM population, including m. abcessis and m. kansasii. Based on these results, and the demonstrated ability of MP-376 to reach certain concentrations in lung sputum of patients, as well as on feedback from medical providers and opinion leaders, Raptor believes development of MP-376 for the treatment of this disease is appropriate.
Preclinical Product Candidates
Raptor’s preclinical programs include RP105 and RP106 being developed for a variety of rare diseases.
Intellectual Property
IP Protection for Raptor’s Products and Product Candidates
Raptor seeks to protect its proprietary technology and other intellectual property that it believes is important to its business, including by seeking, maintaining and defending patents. Raptor also relies on trade secrets and know-how to protect its business. Raptor owns certain of these intellectual property rights and has obtained licenses under other of its intellectual property rights.
Raptor’s intellectual property portfolio is directed to the composition of matter (“COM”) the method of use (“MOU”) and the composition for use (“CFU”) of a formulation/pharmaceutical composition for its products, its product candidates, and other proprietary technologies and processes related to its product development candidates. As of February 25, 2016, Raptor’s patent portfolio includes the patents and patent applications described below, which Raptor owns or has exclusively licensed from third parties, along with any patents that may issue from these patents and applications in the future.
With respect to PROCYSBI, Raptor owns, or exclusively licenses from the University of California, San Diego (“UCSD”), five issued patents and three pending patent applications in the United States, and owns, or exclusively licenses from UCSD, three issued foreign patents, and numerous pending foreign patent applications directed to the formulation/composition, the MOU, and the CFU, of PROCYSBI for the treatment of cystinosis.
With respect to RP103 for indications other than nephropathic cystinosis, including HD and mitochondrial disorders, Raptor owns or holds, by exclusive license, six issued US patents and 18 foreign patents, as well as numerous pending applications in the United States and foreign jurisdictions.
IP Protection for QUINSAIR and MP-376
With respect to QUINSAIR, Raptor currently holds seven issued U.S. patents, a Canadian patent and a European patent application that is allowable and issued in August 2016.
General IP Protection
These patents will expire from 2019 to 2031, and additional patents issuing from pending patent applications or others that may be filed, if they issue, will expire after the standard 20-year term, subject to available patent term adjustments.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which Raptor files, the patent term is 20 years from the date of filing the non-provisional application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent.
48
In addition, extensions of the term of a patent that covers an FDA-approved drug are available in the United States, in order to compensate for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent, based on the length of time the drug is under regulatory review, subject to certain limitations. Similar extensions are available in Europe and other foreign jurisdictions for patents that cover an approved drug. Raptor expects to apply for available patent term extensions for patents covering its product candidates.
Despite the measures Raptor takes to protect its intellectual property, any of its patents or other proprietary rights could be challenged, invalidated, infringed or misappropriated or its intellectual property may not prove sufficient to provide Raptor a competitive market advantage.
Trademarks
Raptor’s trademark portfolio consists of several registered U.S. trademarks covering its company and its subsidiaries’ names, the names of its products and services programs (which are additionally registered in additional territories as necessary to protect its rights to the names). Raptor’s trademark RAPTOR is registered in the United States, in the EU and internationally generally and is currently pending registration in several other jurisdictions. Raptor’s trademark PROCYSBI is registered in the United States, the EU and several additional jurisdictions. It is pending registration in Canada. Raptor’s trademark QUINSAIR is registered in the EU and is pending registration in Canada and the United States.
License Agreement with UCSD
In December 2007, by way of a merger with Encode Pharmaceuticals, Inc., (“Encode”) Raptor acquired certain patent rights licensed to Encode by UCSD pursuant to a license agreement dated October 2007, later amended in February 2008, amended and restated in December 2012, and further amended in March 2013 and December 2013. Pursuant to this agreement, Raptor obtained an exclusive, worldwide, sublicenseable license under certain patent rights and know-how controlled by UCSD for the commercial development, use and sale, for human therapeutic purposes, of products covered by such patents or incorporating such know-how, including RP103. This license is exclusive with respect to the licensed patent rights and non-exclusive with respect to the licensed know-how. Under the agreement, UCSD is obligated to diligently prosecute and maintain the licensed patent rights, conditioned upon Raptor’s continued fulfillment of its obligation to reimburse UCSD for related costs incurred.
Pursuant to the license agreement, Raptor is obligated to pay milestone payments (ranging in size for orphan and non-orphan indications), up to an aggregate total of $6,275,000, upon the occurrence of certain specified development-, regulatory- and commercial-related events during the term of the agreement. As of February 2016, Raptor has paid UCSD approximately $2.2 million in total milestone payments. Raptor is also obligated to pay UCSD a royalty on commercial net sales of licensed products, on a country-by-country basis, ranging in the low single-digit to mid-single-digit percentages, based on whether the licensed product sold is covered by the licensed patent rights in such country, as well as a percentage of sublicensing fees and sublicensing royalties Raptor receives under the agreement, if any. In 2013, Raptor began paying UCSD royalties based upon its net sales of PROCYSBI.
Unless earlier terminated, Raptor’s license agreement with UCSD will expire upon the later of (i) on a country-by-country basis, the expiration of the last to expire of the licensed patent rights (in the applicable country), and (ii) ten years from the first commercial sale of any royalty-bearing product. Raptor may terminate the agreement at any time upon a specified period of prior written notice to UCSD. In the event of Raptor’s breach of an obligation under the agreement, which breach is not cured within a specified number of days after receiving notice of such from UCSD, UCSD may terminate the agreement or choose to convert the license into a non-exclusive license with respect to the indication for which Raptor is in breach. Raptor is currently behind in its developmental diligence obligations with respect to HD and non-alcoholic steatohepatitis under this
49
agreement and is in discussions with UCSD to amend the agreement to conform to the product development timeline Raptor expects to achieve. The agreement will immediately terminate if Raptor files a claim asserting that any of the licensed patent rights are invalid or unenforceable.
Asset Purchase Agreement with Tripex
Asset Purchase Agreement
On August 20, 2015, Raptor entered into an Asset Purchase Agreement with Tripex to purchase MP-376 or QUINSAIR and related intellectual property. At the closing of the asset acquisition pursuant to the Amended and Restated Asset Purchase Agreement dated October 2, 2015, which amended agreement provided for the assets to be acquired by Raptor’s subsidiary, Raptor made an upfront payment to Tripex of $35,370,000 in cash consideration, subject to certain deductions, and issued various Tripex stockholders 3,448,001 shares of Raptor’s common stock. As additional consideration, Tripex may become entitled to receive contingent payments from Raptor upon the achievement of certain milestones and variable royalty payments on the net sales of QUINSAIR-related products.
The contingent payments include:
| • | a one-time variable payment which may be as low as $40 million and up to $80 million if the FDA approves QUINSAIR for the treatment of Pseudomonas aeruginosa infections in cystic fibrosis with certain conditions for each level of payment; |
| • | a one-time payment of $20 million if a second-indication registrational trial milestone for QUINSAIR is achieved; |
| • | up to four milestone payments, totaling up to $250 million in the aggregate, payable upon achievement of first commercial sale of QUINSAIR in the United States and/or the European Union for up to two approved label indications in addition to cystic fibrosis; and |
| • | certain royalties would become payable by Raptor to Tripex based on net sales of QUINSAIR-related products by Raptor and its sublicensees. |
In addition, in a change of control, the party acquiring Raptor may be required to prepay portions of certain of Raptor’s contingent payments if, after the change of control event, Raptor has not met certain diligence obligations pertaining to QUINSAIR.
Raptor is obligated to engage in specified levels of effort to undertake activities relevant to the contingent payments, each of which will be subject to various exceptions to performance.
PARI Letter Agreement
Under the purchase agreement with Tripex, Raptor assumed rights and certain obligations under the Development and License Agreement, dated as of February 11, 2006, between PARI, and Mpex Pharmaceuticals, Inc., a prior owner of QUINSAIR. Pursuant to the Development and License Agreement, PARI granted Mpex a worldwide royalty-bearing license to develop, sell and otherwise exploit pharmaceutical preparations formulated for delivery via pulmonary administration, and the parties agreed to perform joint evaluation, research and development of potential formulations of drug compounds for pulmonary delivery with customized PARI nebulizer devices. QUINSAIR was developed pursuant to the Development and License Agreement, and will continue to be developed and commercialized subject to the terms and conditions of the Development and License Agreement, as amended by the PARI Amendment (as defined and described below).
50
On August 20, 2015, Raptor entered into a Letter Agreement with PARI, which provided that PARI and Raptor would enter into Amendment No. 1 to the Development and License Agreement (the “PARI Amendment”) following the closing of Raptor’s acquisition of QUINSAIR from Tripex. The PARI Amendment was entered into on October 4, 2015. Pursuant to the Development and License Agreement, as amended by the PARI Amendment, Raptor will make payments due to PARI upon the achievement of milestones related to regulatory approval and commercialization activities. Raptor is required to comply with diligence milestones related to development and commercialization of QUINSAIR in the United States, to spend a specified minimum amount per year on development activities in the United States until filing of the NDA for QUINSAIR in the United States, and will pay PARI tiered single digit royalties on net sales of QUINSAIR for a specified time period. Raptor will have the right to buy down the royalties under certain conditions by paying an amount determined through a defined net present value calculation. The PARI Amendment further provides that in the event that Raptor decides to cease the development or commercialization of QUINSAIR for exclusive delivery via the PARI nebulizer device in the United States, PARI shall have the right, in its sole discretion, to develop, and/or license its technology for use with other inhaled fluoroquinolones within the United States.
Orphan Drug Designation
Raptor has been granted Orphan Drug Designation from the FDA for use of PROCYSBI to manage cystinosis for patients six years of age and older and separately in the pediatric population for two to six year olds, and the use of RP103 to potentially treat HD, pancreatic cancer and Batten Disease. The Orphan Drug Act of 1983 generally provides incentives, including marketing exclusivity, user fee waivers and tax benefits, to companies that undertake development and marketing of products to treat relatively rare diseases, which are defined as diseases for which there is a patient population of fewer than 200,000 persons in the United States or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. A drug that receives orphan drug designation may receive up to seven years of exclusive marketing in the United States for that indication, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer is unable to assure sufficient product quantity. A drug may be entitled to an additional six months of exclusive marketing if it satisfies the requirements for pediatric exclusivity; Raptor has applied for this additional six-month pediatric extension for PROCYSBI.
PROCYSBI has also been granted Orphan Drug Designation and awarded 10 years of data exclusivity and marketing protection by the EC for treatment of cystinosis, and RP103 has been granted Orphan Drug Designation by the EC for the treatment of HD. QUINSAIR has been awarded 10 years of data exclusivity and marketing protection by the EC for the treatment of Pseudomonas aeruginosa infection in CF patients and has been awarded Orphan Drug Designation by the FDA.
Competition
Cystinosis
Other than PROCYSBI, Raptor is aware of two pharmaceutical products currently approved to treat cystinosis. Cystagon (immediate-release cysteamine bitartrate capsules), is a systemic cystine-depleting therapy for cystinosis in the United States manufactured by Mylan N.V., and by Orphan Europe SARL in markets outside of the United States. Cystagon was approved by the FDA in 1994 and by the EC in 1997. Cystaran (cysteamine ophthalmic solution) was approved by FDA in 2012 for treatment of corneal crystal accumulation in patients with cystinosis and is marketed by Sigma Tau Pharmaceuticals, Inc.
While Raptor believes that PROCYSBI will continue to be well received in the market, Cystagon remains on the market and Raptor expects it will compete with PROCYSBI for the foreseeable future. Raptor is not aware of any pharmaceutical company with an active program to develop an alternative therapy for
51
cystinosis. Academic researchers in the United States and Europe are pursuing gene therapy and stem cell therapy, as well as pro-drug and PEGylated drug approaches as alternatives to cysteamine bitartrate. Raptor believes that the development timeline to an approved product for these approaches is many years with substantial uncertainty.
Huntington’s Disease
Raptor is not aware of any approved available treatments to slow the progression of HD. There is only one approved treatment available for specific symptoms of HD, Xenazine to treat uncontrollable movements (chorea) that result from the disease. There are several pharmaceutical companies pursuing potential cures and disease modifying treatments for HD, as well as numerous academic and foundation sponsored research efforts. Raptor’s product candidate, RP103, is in late-stage clinical development with the goal of slowing motor deterioration with potentially neuroprotective properties through specifically targeting deficient brain derived neurotrophic factor.
Companies with HD product candidates in development include Prana Biotechnology, Omeros, Teva (and formerly Auspex and Neurosearch), Ionis Pharmaceuticals/Roche, Eli Lilly & Co. and Pfizer. Several other companies have drug candidates in preclinical development. Additionally, nutritional supplements including creatine and coenzyme Q10 have been investigated as potential treatments for HD. The Huntington Study Group sponsors numerous studies of potential therapies for HD, including the recently completed trials evaluating coenzyme Q10 and the antibiotic minocycline.
Pseudomonas Aeruginosa Infection in CF Patients
Currently, there are three products approved in the United States for treatment of chronic Pseudomonas aeruginosa lung infections in patients with CF. Tobramycin solution for inhalation is sold by Novartis as TOBI, and is now also available as a generic from multiple sources. TOBI Podhaler, a dry-powder inhalation formulation of tobramycin, is also sold by Novartis. Gilead Sciences sells Cayston, a nebulized formulation of aztreonam. In the EU, an additional competitor, colistimethate, is available in addition to the products currently available in the United States.
Other programs that are in development for treatment of chronic pulmonary infections in CF patients include the inhaled amikacin product developed by Insmed, Inc., an inhaled vancomycin under development by Savara Pharmaceuticals and the tobramycin/fosfomycin treatment under development by CuRx, Inc.
BE and NTM
No products are currently labeled for treatment of either non-CF BE or pulmonary NTM. Systemic antibiotics, labeled for other diseases, are frequently used off-label as first-line treatment. However, several inhaled antibiotic products are currently in development for either BE or NTM, including Insmed’s Arikayce (tm) (liposomal amikacin for inhalation), AG Bayer’s ciprofloxacin dry powder for inhalation, and Aradigm’s two inhaled ciprofloxacin product candidates, one of which has been licensed to Grifols, Inc.
Manufacturing
PROCYSBI drug product and the API, cysteamine bitartrate, are manufactured on a contract basis by third parties. QUINSAIR drug product, and its API, levofloxacin, and the Zirela nebulizer device are all manufactured on a contract basis by third parties. Raptor believes that its third party manufacturers all have sufficient manufacturing capacity to support its commercial and clinical demands for PROCYSBI for the foreseeable future as well as the clinical and commercial requirements for the initial launch of QUINSAIR.
In general Raptor expects to continue to contract with outside providers for manufacturing services, including API and drug product manufacture, encapsulation, vialing and packaging. Third-party manufacturers’
52
facilities are subject to periodic inspections to confirm compliance with applicable law and must be good manufacturing practice certified. Raptor is aware that the EMA plans to inspect the manufacturing facilities of Raptor’s PROCYSBI drug product manufacturer in the first half of 2016. Although Raptor has never experienced a material disruption in supply from its contract manufacturers, Raptor cannot assure that such a disruption will not occur in the future.
Research and Development
Raptor has an active research and development effort. Raptor plans to focus its research and development efforts in the discovery, research, preclinical and clinical development of its drug candidates in order to provide therapies that it believes will be safer, less intrusive and more effective than current approaches in treating a wide variety of disorders. During the years ended December 31, 2015 and 2014, Raptor incurred approximately $58.6 million and $43.5 million, respectively, in research and development expenses.
Regulatory Matters
See “Business—Horizon Pharma—Government Regulation” for a discussion of regulatory matters.
Employees
As of December 31, 2015, Raptor had 158 full time employees (126, 30, and 2 in the United States, EU and Canada, respectively). Of the 158 employees, 49 are sales and marketing, and 44 are general and administrative personnel, 6 are in manufacturing, 15 in quality control and assurance, and 44 are in research and development. None of Raptor’s employees are represented by a collective bargaining unit.
Facilities
Raptor’s primary offices are located at 7 Hamilton Landing, Suite 100, Novato, CA 94949. Raptor’s European headquarters are located at Naritaweg 165, 1043 BW Amsterdam, Netherlands and it has administrative offices in Utrecht, Netherlands.
53
You should consider carefully the risks described below, together with all of the other information included in this report, and in our other filings with the U.S. Securities and Exchange Commission (the “SEC”) before deciding whether to invest in or continue to hold our ordinary shares. The risks described below are all material risks currently known, expected or reasonably foreseeable by us. If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be seriously harmed. This could cause the trading price of our ordinary shares to decline, resulting in a loss of all or part of your investment.
Risks Related to the Acquisition of Raptor Pharmaceutical Corp.
We may fail to realize all of the anticipated benefits of our proposed acquisition of Raptor Pharmaceutical Corp. (“Raptor”), which we refer to as the “Raptor Acquisition”, or those benefits may take longer to realize than expected. We may also encounter significant difficulties in integrating Raptor’s business into our operations.
Our ability to realize the anticipated benefits of the Raptor Acquisition will depend, to a large extent, on our ability to integrate Raptor’s business into our existing operations. The combination of two independent businesses is a complex, costly and time-consuming process that will require significant management attention and resources. The integration process may disrupt the businesses and, if implemented ineffectively, would limit the expected benefits to us of the Raptor Acquisition. The failure to meet the challenges involved in integrating the two businesses and to realize the anticipated benefits of the Raptor Acquisition could cause an interruption of, or a loss of momentum in, the activities of the combined company and could adversely affect the results of operations of the combined company.
In addition, the overall integration of the businesses may result in material unanticipated problems, expenses, liabilities, competitive responses, loss of market share and other business relationships, and diversion of management’s attention. The difficulties of combining the operations of the companies include, among others:
| • | the diversion of management’s attention to integration matters; |
| • | difficulties in achieving anticipated cost savings, synergies, business opportunities and growth prospects from the combination; |
| • | difficulties in the integration of operations and systems; |
| • | conforming standards, controls, procedures and accounting and other policies and compensation structures between the two companies; |
| • | difficulties in the assimilation of employees and corporate cultures; |
| • | potential unknown liabilities, adverse consequences and unforeseen increased expenses associated with the Raptor Acquisition; and |
| • | challenges in retaining key personnel. |
Many of these factors will be outside of our and Raptor’s control and any one of these factors could result in increased costs, decreases in the amount of expected revenues and additional diversion of management’s time and energy, which could materially adversely impact the business, financial condition and results of operations of the combined company. In addition, even if the operations of our business and Raptor’s business are integrated successfully, the full benefits of the Raptor Acquisition may not be realized, including the synergies, cost savings, revenue growth or other benefits that are expected. These benefits may not be achieved
54
within the anticipated time frame, or at all. Further, additional unanticipated costs may be incurred in the integration of our business with Raptor’s business. These costs will be substantial. Non-recurring transaction costs include, but are not limited to, fees paid to legal, financial and accounting advisors, filing fees and printing costs. Additional unanticipated costs may be incurred in the integration of our business with that of Raptor. There can be no assurance that the elimination of certain duplicative costs, as well as the realization of other efficiencies related to the integration of the two businesses, will offset the incremental transaction-related costs over time. As a result, we cannot provide any assurance that the Raptor Acquisition will result in the realization of the full benefits anticipated from the transactions.
The pendency of the Raptor Acquisition could cause disruptions in Raptor’s business, which could have an adverse effect on Raptor’s business and financial results, and consequently on the combined company.
We and Raptor have operated and, until the completion of the Raptor Acquisition, will continue to operate, independently. Uncertainty about the effect of the acquisition on Raptor’s employees, prescribing physicians, customers, distributors and suppliers may have an adverse effect on Raptor and consequently on the combined company. These uncertainties may impair Raptor’s ability to retain and motivate key personnel and could cause disruptions to its existing business relationships or delay or reduce the use of its products, or otherwise impair the operations of Raptor, which may materially and adversely affect Raptor. Due to the limited termination rights agreed to by the parties in the definitive merger agreement, we may be obligated to complete the Raptor Acquisition in spite of any adverse effects resulting from the disruption of Raptor’s ongoing business. Furthermore, this disruption could adversely affect the combined company’s ability to maintain relationships with prescribing physicians, customers, distributors, suppliers and employees after the Raptor Acquisition or to achieve the anticipated benefits of the Raptor Acquisition. Each of these events could adversely affect Raptor in the near term or the combined company thereafter if the Raptor Acquisition is completed.
We and Raptor will incur substantial direct and indirect costs as a result of the Raptor Acquisition.
We and Raptor will incur substantial expenses in connection with and as a result of completing the Raptor Acquisition and, over a period of time following the completion of the Raptor Acquisition, we expect to incur substantial additional expenses in connection with coordinating the businesses, operations, policies and procedures of the combined company. While we have assumed that a certain level of transaction expenses will be incurred, factors beyond our control could affect the total amount or the timing of these expenses. Many of the expenses that will be incurred, by their nature, are difficult to estimate accurately.
We will incur a substantial amount of debt to finance the purchase price and certain other amounts to be paid in connection with the Raptor Acquisition, which could adversely affect our business, including by restricting our ability to engage in additional transactions or incur additional indebtedness.
In connection with that certain Agreement and Plan of Merger (the “Merger Agreement”), dated as of September 12, 2016, by and among Raptor, us and Misneach Corporation, a Delaware corporation and our indirect wholly owned subsidiary (“Merger Sub”), Horizon Pharma, Inc., our indirect wholly owned subsidiary (“HPI”), entered into an amended and restated debt commitment letter (the “Debt Commitment Letter”) with the Bank of America, N.A., JPMorgan Chase Bank, N.A., Jefferies Finance LLC, Citigroup Global Markets, Inc., Cowen and Company, LLC and Cowen Structured Holdings, Inc. (collectively, the “Commitment Parties”) on September 16, 2016, pursuant to which the Commitment Parties have committed to provide up to $675.0 million principal amount of incremental term loans under our existing senior secured credit facility (the “Senior Secured Credit Facility”), the net proceeds of which, together with a portion of our and Raptor’s existing cash on hand, would be available to (i) purchase all of the outstanding shares of Raptor’s common stock, (ii) repay approximately $129.6 million of Raptor’s existing indebtedness (including prepayment premiums), and (iii) pay any other fees and expenses in connection with the foregoing (collectively, the “Transactions”). In lieu of borrowing the entire committed amount pursuant to the Debt Commitment Letter, we expect to borrow up to $375.0 million principal amount of incremental term loans pursuant to the Senior Secured Credit Facility (the “Incremental Term Loans”) and issue $300.0 million principal amount of senior notes due 2024 (the “2024 notes”) pursuant to our proposed offering.
As of June 30, 2016, after giving pro forma effect to the Transactions, the principal amount of our outstanding senior indebtedness would have been approximately $1,946.0 million, of which $771.0 million would have been secured indebtedness. Consequently, following the completion of the Raptor Acquisition, we will have a substantial amount of debt outstanding on a consolidated basis.
55
This substantial amount of debt could have important consequences to our business, including, but not limited to:
| • | reducing the benefits we expect to receive from the Raptor Acquisition; |
| • | making it more difficult for us to satisfy our obligations; |
| • | requiring a substantial portion of our cash flows from operations to be dedicated to the payment of principal and interest on our indebtedness, therefore reducing our ability to use our cash flows to fund acquisitions, capital expenditures, and future business opportunities; |
| • | exposing us to the risk of increased interest rates to the extent of any current or future borrowings, including borrowings under our credit agreement governing the Senior Secured Credit Facility (the “Credit Agreement”), at variable rates of interest; |
| • | limiting our ability to borrow additional funds and increasing the cost of any such borrowing; |
| • | increasing our vulnerability to, and reducing our flexibility to respond to, changes in our business or general adverse economic and industry conditions; |
| • | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate; |
| • | placing us at a disadvantage as compared to our competitors, to the extent that they are not as highly leveraged; and |
| • | restricting us from pursuing certain business opportunities. |
Our credit ratings will impact the cost and availability of future borrowings and our cost of capital. Our ratings at any time will reflect each rating organization’s then-current opinion of our financial strength, operating performance and ability to meet our debt obligations. There can be no assurance that we will achieve a particular rating or maintain a particular rating in the future. Unfavorable ratings or a subsequent reduction in our credit ratings may limit our ability to borrow at acceptable interest rates. If our credit ratings were downgraded or put on watch for a potential downgrade, we may not be able to sell additional debt securities or borrow money in the amounts, at the times or interest rates or upon the more favorable terms and conditions that might otherwise be available. Any impairment of our ability to obtain future financing on favorable terms could have an adverse effect on our ability to refinance any of our then-existing debt and may severely restrict our ability to execute on our business strategy, which includes the continued acquisition of additional products or businesses.
If goodwill or other intangible assets that we record in connection with the Raptor Acquisition become impaired, we could have to take significant charges against earnings.
In connection with the accounting for the Raptor Acquisition, it is expected that we will record a significant amount of intangible assets and may also record goodwill. Under accounting principles generally accepted in the United States (“GAAP”), we must assess, at least annually and potentially more frequently, whether the value of goodwill and other indefinite-lived intangible assets has been impaired. Amortizing intangible assets will be assessed for impairment in the event of an impairment indicator. Any reduction or impairment of the value of goodwill or other intangible assets will result in a charge against earnings, which could materially adversely affect our results of operations and our ability to make payments on our indebtedness.
Our and Raptor’s actual financial positions and results of operations may differ materially from the unaudited pro forma and other financial data included in this report.
The pro forma and other financial information showing changes to our historical financial results contained in this report is presented for illustrative purposes only and may not be an indication
56
of what our financial position or results of operations would have been had the transactions been completed on the dates indicated. This additional financial information has been derived from our audited historical financial statements and the audited and unaudited financial data of Raptor and certain companies previously acquired by us and certain adjustments and assumptions have been made regarding the combined company after giving effect to the indicated transactions. The assets and liabilities of Raptor have been measured at fair value based on various preliminary estimates using assumptions that our management believes are reasonable utilizing information currently available and assumptions about future development plans. The process for estimating the fair value of acquired assets and assumed liabilities requires the use of judgment in determining the appropriate assumptions and estimates. These estimates may be revised as additional information becomes available and as additional analyses are performed. In addition, the pro forma financial information contained in this report is based upon certain assumptions with respect to our financing of the Raptor Acquisition. Whether the assumed financing sources are available and, if available, the terms of our future financings, will be subject to market conditions. The actual sources of financing and the terms on which it is obtained may not be as favorable as those reflected in the pro forma financial information. Moreover, we have presented acquisition-adjusted EBITDA, which combines Horizon’s historical adjusted EBITDA with the historical adjusted EBITDA of Crealta and Raptor, without giving effect to certain adjustments that would ordinarily be required to present combined financial results on a pro forma basis as if the acquisitions had occurred at the beginning of the period presented. Such information may therefore not be indicative of the results that may have been achieved had the acquisitions occurred at the beginning of that period. Differences between preliminary estimates in the pro forma and other supplemental financial information and the final acquisition accounting, as well as between the assumed and actual financing sources and terms, our currently-assumed and future development plans with respect to Raptor’s assets, and our estimated and actual synergies and cost savings from the Raptor Acquisition will occur and could have a material impact on the pro forma and other supplemental financial information and the combined company’s financial position and future results of operations.
Other assumptions used in preparing the pro forma and other supplemental financial information may not prove to be accurate, and other factors may affect our financial condition or results of operations following the closing of the Raptor Acquisition and related transactions. Any potential decline in our financial condition or results of operations may cause significant variations in the price of our ordinary shares.
Litigation has been filed challenging the Raptor Acquisition, and an adverse ruling may prevent the Raptor Acquisition from being completed.
Between October 5 and October 7, 2016, two complaints (captioned Lavrenov v. Raptor Pharmaceutical Corp., et al., Case No. 1:16-cv-00901 and Jordan v. Raptor Pharmaceutical Corp., et al., Case No. 1:16-cv-00913) were filed in the United States District Court for the District of Delaware. Both actions were filed against Raptor and each member of Raptor’s board of directors. We and MergerSub were named as defendants in the Lavrenov action, but not the Jordan action. The actions were brought by purported stockholders of Raptor and seek certification as a class action on behalf of all of Raptor’s public stockholders. The complaints allege, among other things, that the process leading up to the Raptor Acquisition was inadequate and that the public filings related to the Raptor Acquisition omit certain material information or are materially misleading. The complaints seek, among other things, to enjoin the Raptor Acquisition, or in the event the proposed transaction is consummated, to recover money damages. The Parent and the Purchaser intend to vigorously defend against the lawsuit. One of the conditions to the closing of the Raptor Acquisition is that no governmental body, including a court, shall have issued or granted any order, judgment, award, decision, decree, injunction, ruling, writ or assessment, which has the effect of making the Raptor Acquisition illegal or which has the effect of prohibiting or otherwise preventing the consummation of the Raptor Acquisition. Consequently, if plaintiffs secure injunctive or other relief prohibiting, delaying or otherwise adversely affecting our ability to complete the Raptor Acquisition, then such injunctive or other relief may prevent the Raptor Acquisition from becoming effective within the expected time frame or at all.
57
Risks Related to Our Business and Industry
Our ability to generate revenues from our medicines is subject to attaining significant market acceptance among physicians, patients and healthcare payers.
Our current medicines, and other medicines or medicine candidates that we may develop or acquire, may not attain market acceptance among physicians, patients, healthcare payers or the medical community. We have a limited history of commercializing medicines and most of our medicines have not been on the market for an extensive period of time, which subjects us to numerous risks as we attempt to increase our market share. We believe that the degree of market acceptance and our ability to generate revenues from our medicines will depend on a number of factors, including:
| • | timing of market introduction of our medicines as well as competitive medicines; |
| • | efficacy and safety of our medicines; |
| • | continued projected growth of the markets in which our medicines compete; |
| • | prevalence and severity of any side effects; |
| • | if and when we are able to obtain regulatory approvals for additional indications for our medicines; |
| • | acceptance by patients, primary care physicians and key specialists, including rheumatologists, orthopedic surgeons, pain specialists, podiatrists, medical geneticists and specialists in pediatric immunology, allergy, infectious diseases and hematology/oncology; |
| • | availability of coverage and adequate reimbursement and pricing from government and other third-party payers; |
| • | potential or perceived advantages or disadvantages of our medicines over alternative treatments, including cost of treatment and relative convenience and ease of administration; |
| • | strength of sales, marketing and distribution support; |
| • | the price of our medicines, both in absolute terms and relative to alternative treatments; |
| • | impact of past and limitation of future medicine price increases; |
| • | our ability to maintain a continuous supply of medicine for commercial sale; |
| • | the effect of current and future healthcare laws; |
| • | the performance of third-party distribution partners, over which we have limited control; and |
| • | medicine labeling or medicine insert requirements of the U.S. Food and Drug Administration (the “FDA”) or other regulatory authorities. |
With respect to DUEXIS and VIMOVO, studies indicate that physicians do not commonly co-prescribe gastrointestinal (“GI”) protective agents to high-risk patients taking nonsteroidal anti-inflammatory drugs (“NSAIDs”). We believe this is due in part to a lack of awareness among physicians prescribing NSAIDs regarding the risk of NSAID-induced upper GI ulcers, in addition to the inconvenience of prescribing two separate medications and patient compliance issues associated with multiple prescriptions. If physicians remain
58
unaware of, or do not otherwise believe in, the benefits of combining GI protective agents with NSAIDs, our market opportunity for DUEXIS and VIMOVO will be limited. Some physicians may also be reluctant to prescribe DUEXIS or VIMOVO due to the inability to vary the dose of ibuprofen and naproxen, respectively, or if they believe treatment with NSAIDs or GI protective agents other than those contained in DUEXIS and VIMOVO, including those of its competitors, would be more effective for their patients. With respect to each of DUEXIS, PENNSAID 2%, RAYOS/LODOTRA, VIMOVO and BUPHENYL, their higher cost compared to the generic or branded forms of their active ingredients alone may limit adoption by physicians, patients and healthcare payers. With respect to ACTIMMUNE, while it is the only FDA-approved treatment for chronic granulomatous disease (“CGD”) and severe, malignant osteopetrosis (“SMO”), they are very rare conditions and, as a result, our ability to grow ACTIMMUNE sales will depend on our ability to further penetrate this limited market and obtain marketing approval for additional indications. With respect to RAVICTI, which is also approved to treat a very limited patient population, our ability to grow sales will depend in large part on our ability to transition urea cycle disorder (“UCD”) patients from BUPHENYL or generic equivalents, which are comparatively much less expensive, to RAVICTI. With respect to KRYSTEXXA, our ability to grow sales will be affected by the success of our sales and marketing strategies and life cycle management, including studies designed to test reduction of immunogenicity in KRYSTEXXA which could expand the patient population and usage of KRYSTEXXA. With respect to MIGERGOT, our ability to sustain sales will depend on the management of inventory levels and the continued awareness of its benefits among physicians. With respect to PROCYSBI, which is approved to treat a very limited patient population, our ability to grow sales following the Raptor Acquisition will depend in large part on our ability to transition patients from the first-generation therapy to PROCYSBI, to identify additional patients with nephropathic cystinosis, and expand commercialization in Europe. Unless QUINSAIR is approved for marketing in additional countries, our ability to drive growth of this medicine following the Raptor Acquisition will largely depend on expanding its use in Europe and Canada. If our current medicines or any other medicine that we may seek approval for or acquire fail to attain market acceptance, we may not be able to generate significant revenue to achieve or sustain profitability, which would have a material adverse effect on our business, results of operations, financial condition and prospects (including, possibly, the value of our ordinary shares).
Our future prospects are highly dependent on our ability to successfully formulate and execute commercialization strategies for each of our medicines. Failure to do so would adversely impact our financial condition and prospects.
A substantial majority of our resources are focused on the commercialization of our current medicines. Our ability to generate significant medicine revenues and to achieve commercial success in the near-term will initially depend almost entirely on our ability to successfully commercialize these medicines in the United States.
With respect to our orphan medicines, ACTIMMUNE, BUPHENYL and RAVICTI, and following the Raptor Acquisition, PROCYSBI and QUINSAIR, and with respect to our rheumatology medicine KRYSTEXXA, our commercialization strategy includes efforts to increase awareness of the rare conditions that each medicine is designed to treat, enhancing efforts to identify target patients and in certain cases pursue opportunities for label expansion and more effective use through clinical trials. In addition, our strategy with respect to ACTIMMUNE includes pursuing label expansion for additional indications, such as Friedreich’s ataxia (“FA”), and price increases but we cannot be certain that our pricing strategy will not result in downward pressure on sales or that we will be able to successfully complete clinical trials and obtain regulatory approvals in additional indications. With respect to RAVICTI and PROCYSBI, our strategy includes accelerating the transition of patients from first-generation therapies, and increasing the diagnosis of the associated rare conditions through patient and physician outreach. Part of our success in our strategy for RAVICTI will also depend on obtaining approval of RAVICTI for the treatment of UCD in patients less than two years of age. On June 29, 2016, we submitted a supplemental new drug application (“sNDA”) with the FDA for RAVICTI to expand the age range for chronic management of UCDs from two years of age and older to two months of age and older. Subject to positive data from on-going studies, we have targeted an sNDA submission in the first quarter of 2018 in relation to UCD patients during the first two months of life, however, we cannot guarantee that on-going studies will be positive or that we will be
59
able to expand the labeling for RAVICTI on our anticipated timeline or at all. Our strategy with respect to KRYSTEXXA includes the expansion of our sales force, the planned enhancement of the KRYSTEXXA marketing campaign with improved immunogenicity data, continued volume growth and pricing optimization.
Our strategy for RAYOS in the United States is to solely focus on the rheumatology indications approved for RAYOS where our Phase 3 clinical trial data supports our commercial plans.
With respect to our primary care medicines DUEXIS, PENNSAID 2% and VIMOVO, our strategy has included bringing pricing in-line with other branded NSAIDs, thereby significantly increasing the value we realize per prescription, and also increasing sales and marketing support to drive volume growth in prescriptions. We cannot guarantee that this strategy will continue to be effective generally, due to negative reactions to price increases or otherwise. More recently, we have begun entering into rebate agreements with pharmacy benefit managers (“PBMs”) for certain of our primary care medicines where we believe the rebates and costs justify expanded formulary access for patients. However, we cannot guarantee that we will be able to secure additional rebate agreements on commercially reasonable terms. For each of our primary care medicines, we expect that our commercial success will depend on our sales and marketing efforts in the United States.
Our strategy for RAYOS in the United States is to solely focus on the rheumatology indications approved for RAYOS where our Phase 3 clinical trial data supports our commercial plans.
Our overall commercialization strategy also includes plans to expand our sales and market efforts in Europe and other countries outside the United States for certain of our orphan and rheumatology medicines. In November 2015, we received approval of the Committee for Medicinal Products for Human Use of the European Medicines Agency (“EMA”) for RAVICTI for use as an adjunctive therapy for chronic management of adult and pediatric UCD patients greater than two months of age. This authorizes us to market RAVICTI in all 28 Member States of the European Union (“EU”) and will form the basis for recognition by the Member States of the European Economic Area, namely Norway, Iceland and Liechtenstein, for the medicine to be placed on the market. In June 2016, we partnered with Clinigen Group plc’s Idis managed access division to initiate a managed access program in selected European countries. While we expect to commercially launch RAVICTI in Europe in 2017, we cannot guarantee we will be able to successfully implement our commercial plans for RAVICTI in Europe. Although LODOTRA is approved for marketing in countries outside the United States, to date it has only been marketed in a limited number of countries. While we anticipate that LODOTRA will be marketed in additional countries as our distribution partner, Mundipharma International Corporation Limited (“Mundipharma”) formulates its reimbursement strategy, the ability to market LODOTRA in additional countries will depend on Mundipharma’s ability to obtain reimbursement approvals in these countries. With respect to PROCYSBI and QUINSAIR, which are approved for marketing in the EU, we intend to continue evaluating commercial launches in additional EU countries as well as pursuing early access programs. Although LODOTRA is approved for marketing in countries outside the United States, to date it has only been marketed in a limited number of countries. While we anticipate that LODOTRA will be marketed in additional countries as our distribution partner, Mundipharma, formulates its reimbursement strategy, the ability to market LODOTRA in additional countries will depend on Mundipharma’s ability to obtain reimbursement approvals in these countries.
If any of our commercial strategies are unsuccessful or we fail to successfully modify our strategies over time due to changing market conditions, our ability to increase market share for our medicines, grow revenues and sustain profitability will be harmed.
In order to increase adoption and sales of our medicines, we will need to continue developing our commercial organization as well as recruit and retain qualified sales representatives.
Part of our strategy is to continue to build a biopharmaceutical company to successfully execute the commercialization of our medicines in the U.S. market, and in selected markets in Europe where we have commercial rights. We may not be able to successfully commercialize our medicines in the United States or in any other territories where we have commercial rights. Prior to our commercial launch of DUEXIS in the United
60
States in December 2011, we did not have any experience commercializing medicines on our own. In order to commercialize any approved medicines, we must continue to build our sales, marketing, distribution, managerial and other non-technical capabilities. Although we had expanded our sales force to approximately 500 sales representatives as of June 30, 2016, consisting of approximately 15 orphan disease sales representatives, 90 rheumatology sales specialists and 395 primary care sales representatives, we currently have limited resources compared to some of our competitors, and the continued development of our own commercial organization to market our medicines and any additional medicines we may acquire will be expensive and time-consuming. We also cannot be certain that we will be able to continue to successfully develop this capability.
As a result of the evolving role of various constituents in the prescription decision making process, we adjusted the profile of the sales representatives we hire for our primary care and rheumatology business units from those with traditional pharmaceutical sales experience to those with successful business to business experience. For example, we have faced challenges due to pharmacists increasingly switching a patient’s intended prescription from DUEXIS and VIMOVO to a generic or over-the-counter brand of their active ingredients. We have faced similar challenges for RAYOS, BUPHENYL and PENNSAID 2% with respect to generic brands. While we believe the profile of our representatives is better suited for this evolving environment, we cannot be certain that our representatives will be able to successfully protect our market for DUEXIS, PENNSAID 2%, RAYOS, BUPHENYL and VIMOVO or that we will be able to continue attracting and retaining sales representatives with our desired profile and skills. We will also have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train and retain commercial personnel. To the extent we rely on additional third parties to commercialize any approved medicines, we may receive less revenue than if we commercialized these medicines ourselves. In addition, we may have little or no control over the sales efforts of any third parties involved in our commercialization efforts. In the event we are unable to successfully develop and maintain our own commercial organization or collaborate with a third-party sales and marketing organization, we may not be able to commercialize our medicines and medicine candidates and execute on our business plan.
If we are unable to effectively train and equip our sales force, our ability to successfully commercialize our medicines will be harmed.
As we recently acquired additional medicines through acquisition transactions, the members of our sales force may have limited experience promoting these medicines. To the extent we have retained the sales forces promoting recently-acquired medicines, we may not be successful in continuing to retain these employees and we otherwise have limited experience marketing these medicines under our commercial organization. As a result, we are required to expend significant time and resources to train our sales force to be credible and persuasive in convincing physicians to prescribe and pharmacists to dispense our medicines. In addition, we must train our sales force to ensure that a consistent and appropriate message about our medicines is being delivered to our potential customers. Our sales representatives may also experience challenges promoting multiple medicines when we call on physicians and their office staff. We have experienced, and may continue to experience, turnover of the sales representatives that we hired or will hire, requiring us to train new sales representatives. If we are unable to effectively train our sales force and equip them with effective materials, including medical and sales literature to help them inform and educate physicians about the benefits of our medicines and their proper administration and label indication, as well as our access programs, our efforts to successfully commercialize our medicines could be put in jeopardy, which could have a material adverse effect on our financial condition, share price and operations.
If we cannot successfully implement our patient access programs or enter into formulary and reimbursement agreements with pharmacy benefit managers in the face of increasing pressure to reduce the price of medications, the adoption of our medicines by physicians, patients and payers may decline.
There continues to be immense pressure from healthcare payers and PBMs, to use less expensive generics or over-the-counter brands instead of branded medicines. For example, two of the largest PBMs placed
61
DUEXIS and VIMOVO on their formulary exclusion lists beginning in January 2015. Additional healthcare plans, including those that contract with these PBMs but use different formularies, may also choose to exclude our medicines from their formularies or restrict coverage to situations where a generic or over-the-counter medicine has been tried first. Many payers and PBMs also require patients to make co-payments for branded medicines, including many of our medicines, in order to incentivize the use of generic or other lower-priced alternatives instead. Legislation enacted in most states in the United States allows, or in some instances mandates, that a pharmacist dispenses an available generic equivalent when filling a prescription for a branded medicine, in the absence of specific instructions from the prescribing physician. Because our medicines (other than BUPHENYL) do not currently have FDA-approved generic equivalents in the United States, we do not believe our medicines should be subject to mandatory generic substitution laws. However, we understand that some pharmacies may attempt to obtain physician authorization to switch prescriptions for DUEXIS or VIMOVO to prescriptions for multiple generic medicines with similar active pharmaceutical ingredients (“APIs”) to ensure payment for the medicine if the physician’s prescription for the branded medicine is not immediately covered by the payer, despite such substitution being off-label in the case of DUEXIS. If these limitations in coverage and other incentives result in patients refusing to fill prescriptions or being dissatisfied with the out-of-pocket costs of their medications, or if pharmacies otherwise seek and receive physician authorization to switch prescriptions, not only would we lose sales on prescriptions that are ultimately not filled, but physicians may be dissuaded from writing prescriptions for our medicines in the first place in order to avoid potential patient non-compliance or dissatisfaction over medication costs, or to avoid spending the time and effort of responding to pharmacy requests to switch prescriptions.
Part of our commercial strategy to increase adoption and access to our medicines in the face of these incentives to use generic alternatives is to offer physicians the opportunity to have patients fill prescriptions through independent pharmacies participating in our HorizonCares access program. Through HorizonCares, financial assistance may be available to reduce eligible patients’ out-of-pocket costs for prescriptions filled. Because of this assistance, eligible patients’ out-of-pocket cost for our medicines when dispensed through HorizonCares may be significantly lower than such costs when our medicines are dispensed outside of the HorizonCares program. However, to the extent physicians do not direct prescriptions currently filled through traditional pharmacies, including those associated with or controlled by PBMs, to pharmacies participating in our HorizonCares program, we may experience a significant decline in DUEXIS, VIMOVO and PENNSAID 2% prescriptions as a result of formulary exclusions, co-payment requirements or other incentives to use lower-priced alternatives to our medicines. Our ability to increase utilization of our access programs will depend on physician and patient awareness and comfort with the programs, and we have limited ability to influence whether physicians use our access programs to prescribe our medicines or whether patients will agree to receive our medicines through the HorizonCares program. In addition, the HorizonCares program is not available to federal health care program (such as Medicare and Medicaid) beneficiaries. We have also begun to pursue the additional strategy of contracting with PBMs and other payers to secure formulary status and reimbursement for certain of our primary care medicines, which generally require us to pay administrative fees and rebates to the PBMs and other payers for qualifying prescriptions. While we recently announced a business relationship with one of the largest PBMs, CVS Caremark, that has resulted in DUEXIS and VIMOVO being removed from the CVS Caremark 2017 exclusion list, as well as a rebate agreement with another PBM, Prime Therapeutics LLC (“Prime Therapeutics”), and we believe these agreements will secure formulary status for certain of our medicines, we cannot guarantee that we will be able to agree to terms with other PBMs and other payers, or that such terms will be commercially reasonable to us. If we are unable to increase adoption of HorizonCares for filling prescriptions of our medicines or to secure formulary status and reimbursement through arrangements with PBMs and other payers, our ability to maintain or increase prescriptions for our medicines could be impaired.
There has been recent negative publicity regarding the use of specialty pharmacies and drug pricing. Our patient access programs are not involved in the prescribing of medicines and are solely to assist in ensuring that when a physician determines one of our medicines offers a potential clinical benefit to their patients and they prescribe one for an eligible patient, financial assistance may be available to reduce the patient’s out-of-pocket costs. In addition, all pharmacies that fill prescriptions for our medicines are fully independent, including those
62
that participate in HorizonCares. We do not own or possess any option to purchase an ownership stake in any pharmacy that distributes our medicines, and our relationship with each pharmacy is non-exclusive and arm’s length. All of our sales are processed through pharmacies independent of us. Despite this, the recent negative publicity regarding specialty pharmacies may result in physicians being less willing to participate in our patient access programs and thereby limit our ability to increase patient access and adoption of our medicines.
We may also encounter difficulty in forming and maintaining relationships with pharmacies that participate in our patient access programs. We currently depend on a limited number of pharmacies participating in HorizonCares to fulfill patient prescriptions under the HorizonCares program. If these HorizonCares participating pharmacies are unable to process and fulfill the volume of patient prescriptions directed to them under the HorizonCares program, our ability to maintain or increase prescriptions for our medicines will be impaired. The commercialization of our medicines and our operating results could be affected should any of the HorizonCares participating pharmacies choose not to continue participation in our HorizonCares program or by any adverse events at any of those HorizonCares participating pharmacies. For example, pharmacies that dispense our medicines could lose contracts that they currently maintain with managed care organizations (“MCOs”), including PBMs. Pharmacies often enter into agreements with MCOs. They may be required to abide by certain terms and conditions to maintain access to MCO networks, including terms and conditions that could limit their ability to participate in patient access programs like ours. Failure to comply with the terms of their agreements with MCOs could result in a variety of penalties, including termination of their agreement, which could negatively impact the ability of those pharmacies to dispense our medicines and collect reimbursement from MCOs for such medicines.
The HorizonCares program may implicate certain state laws related to, among other things, unlawful schemes to defraud, excessive fees for services, tortious interference with patient contracts and statutory or common law fraud. We have a compliance program in place to address adherence with various laws and regulations relating to the selling, marketing and manufacturing of our medicines, as well as certain third-party relationships, including pharmacies. Specifically with respect to pharmacies, the compliance program utilizes a variety of methods and tools to monitor and audit pharmacies, including those that participate in the HorizonCares program, to confirm their activities, adjudication and practices are consistent with our compliance policies and guidance. Despite our compliance efforts, to the extent the HorizonCares program is found to be inconsistent with applicable laws or the pharmacies that participate in our patient access programs do not comply with applicable laws, we may be required to restructure or discontinue such programs, terminate our relationship with certain pharmacies, or be subject to other significant penalties. In November 2015, we received a subpoena from the U.S. Attorney’s Office for the Southern District of New York requesting documents and information related to our patient access programs and other aspects of our marketing and commercialization activities. We are unable to predict how long this investigation will continue or its outcome, but we anticipate that we may incur significant costs in connection with the investigation, regardless of the outcome. We may also become subject to similar investigations by other governmental agencies. The investigation by the U.S. Attorney’s Office and any additional investigations of our patient access programs and sales and marketing activities may result in damages, fines, penalties or other administrative sanctions against us.
Even if we are successful in increasing the use our patient access programs, these programs may become too costly for us to maintain if we are unable to maintain or enhance payer reimbursement of our medicines. The aggregate commercial value of our patient access programs for the six months ended June 30, 2016 was $816.8 million. If additional formularies place our medicines on their exclusion lists or increase the co-payments applicable to our medicines, our cost of ensuring that patients have low-cost access to our medicines will increase and our profitability could decline. If the cost of maintaining our patient access programs increases relative to our sales revenues, we could be forced to reduce the amount of patient financial assistance that we offer or otherwise scale back or eliminate such programs, which could in turn have a negative impact on physicians’ willingness to prescribe and patients’ willingness to fill prescriptions of our medicines. As an alternative means of ensuring access to our medicines, we have also begun pursuing business arrangements with PBMs and other payers to secure formulary status and reimbursement of our medicines, such as our recently announced arrangements with
63
CVS Caremark and Prime Therapeutics. While we believe that, if successful, this strategy would result in broader inclusion of certain of our primary care medicines on healthcare plan formularies, and therefore increase payer reimbursement and lower our cost of providing patient access programs, these arrangements generally require us to pay administrative and rebate payments to the PBMs and/or other payers. To the extent that we enter into arrangements with PBMs and other payers, we will owe rebate payments not only on prescriptions under plans that currently exclude certain of our medicines from their formulary and for which we provide significant patient assistance to ensure access, but also on prescriptions that are reimbursed by applicable healthcare plans. If our arrangements with PBMs and other payers do not result in increased prescriptions and reductions in our costs to provide our patient access programs that are sufficient to offset the administrative fees and rebate payments to the PBMs and/or other payers, our financial results may be harmed.
If we are unable to successfully implement our commercial plans and facilitate adoption by patients and physicians of any approved medicines through our sales, marketing and commercialization efforts, then we will not be able to generate sustainable revenues from medicine sales which will have a material adverse effect on our business and prospects.
We are solely dependent on third parties to commercialize certain of our medicines outside the United States. Failure of these third parties or any other third parties to successfully commercialize our medicines and medicine candidates in the applicable jurisdictions could have a material adverse effect on our business.
We rely on Mundipharma for commercialization of LODOTRA in various European countries and certain Asian, Latin American, Middle Eastern, African and other countries. We rely on other third-party distributors for commercialization of BUPHENYL (known as AMMONAPS in certain European countries) in certain territories outside the United States for which we currently have rights. We have limited contractual rights to force these third parties to invest significantly in commercialization of LODOTRA or BUPHENYL in our markets. In the event that Mundipharma or our current ex-U.S. distributors for BUPHENYL or any other third-party with any future commercialization rights to any of our medicines or medicine candidates fail to adequately commercialize those medicines or medicine candidates because they lack adequate financial or other resources, decide to focus on other initiatives or otherwise, our ability to successfully commercialize our medicines or medicine candidates in the applicable jurisdictions would be limited, which would adversely affect our business, financial condition, results of operations and prospects. We have had disagreements with Mundipharma under our European agreements and may continue to have disagreements, which could harm commercialization of LODOTRA in Europe or result in the termination of our agreements with Mundipharma. We also rely on Mundipharma’s ability to obtain regulatory approval for LODOTRA in certain Asian, Latin American, Middle Eastern, African and other countries. In addition, our agreements with Mundipharma and our agreements with our current ex-U.S. distributors for BUPHENYL may be terminated by either party in the event of a bankruptcy of the other party or upon an uncured material breach by the other party. If these third parties terminated their agreements, we may not be able to secure an alternative distributor in the applicable territory on a timely basis or at all, in which case our ability to generate revenues from the sale of LODOTRA or BUPHENYL outside the United States would be materially harmed.
Our medicines are subject to extensive regulation, and we may not obtain additional regulatory approvals for our medicines.
The clinical development, manufacturing, labeling, packaging, storage, recordkeeping, advertising, promotion, export, marketing and distribution and other possible activities relating to our medicines and our medicine candidates are, and will be, subject to extensive regulation by the FDA and other regulatory agencies. Failure to comply with FDA and other applicable regulatory requirements may, either before or after medicine approval, subject us to administrative or judicially imposed sanctions.
To market any drugs or biologics outside of the United States, we and current or future collaborators must comply with numerous and varying regulatory and compliance related requirements of other countries.
64
Approval procedures vary among countries and can involve additional medicine testing and additional administrative review periods, including obtaining reimbursement and pricing approval in select markets. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks associated with FDA approval as well as additional, presently unanticipated, risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Applications for regulatory approval, including a marketing authorization application for marketing new drugs in Europe, must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing and controls (“CMC”) to demonstrate the safety and effectiveness of the applicable medicine candidate. The number and types of preclinical studies and clinical trials that will be required for regulatory approval varies depending on the medicine candidate, the disease or the condition that the medicine candidate is designed to target and the regulations applicable to any particular medicine candidate. Despite the time and expense associated with preclinical and clinical studies, failure can occur at any stage, and we could encounter problems that cause us to repeat or perform additional preclinical studies, CMC studies or clinical trials. Regulatory authorities could delay, limit or deny approval of a medicine candidate for many reasons, including because they:
| • | may not deem a medicine candidate to be adequately safe and effective; |
| • | may not find the data from preclinical studies, CMC studies and clinical trials to be sufficient to support a claim of safety and efficacy; |
| • | may interpret data from preclinical studies, CMC studies and clinical trials significantly differently than we do; |
| • | may not approve the manufacturing processes or facilities associated with our medicine candidates; |
| • | may conclude that we have not sufficiently demonstrated long-term stability of the formulation for which we are seeking marketing approval; |
| • | may change approval policies (including with respect to our medicine candidates’ class of drugs) or adopt new regulations; or |
| • | may not accept a submission due to, among other reasons, the content or formatting of the submission. |
Even if we believe that data collected from our preclinical studies, CMC studies and clinical trials of our medicine candidates are promising and that our information and procedures regarding CMC are sufficient, our data may not be sufficient to support marketing approval by regulatory authorities, or regulatory interpretation of these data and procedures may be unfavorable. Even if approved, medicine candidates may not be approved for all indications requested and such approval may be subject to limitations on the indicated uses for which the medicine may be marketed, restricted distribution methods or other limitations. Our business and reputation may be harmed by any failure or significant delay in obtaining regulatory approval for the sale of any of our medicine candidates. We cannot predict when or whether regulatory approval will be obtained for any medicine candidate we develop.
While we anticipate that LODOTRA will be marketed in additional countries as Mundipharma formulates its reimbursement strategy, the ability to market LODOTRA in additional countries will depend on Mundipharma’s ability to obtain regulatory and reimbursement approvals in these countries.
65
Hyperion Therapeutics, Inc. (Hyperion”) submitted a New Drug Submission (“NDS”) to Health Canada (“HC”) for approval to market RAVICTI in Canada. In March 2016, HC issued a Notice of Compliance for RAVICTI for use as an adjunctive therapy for chronic management of adult and pediatric patients two years of age and older with UCDs, and we plan to launch RAVICTI in Canada during 2016. However, if we are unable to obtain any further approvals for RAVICTI outside the United States, Canada and Europe, or determine that commercializing RAVICTI outside the United States, Canada and Europe is not economically viable, the market potential of RAVICTI will be limited.
On July 12, 2016 Raptor received a notice of deficiency (“NOD”) from HC dated July 11, 2016 relating to the NDS Raptor submitted for PROCYSBI in January 2016. The NOD states that information provided in the NDS is insufficient for HC to complete its review. HC’s review of the PROCYSBI NDS will re-commence upon its acceptance of a response to its notice. On September 30, 2016, Raptor was granted a 30-day extension to submit to HC, and Raptor intends to submit the response prior to the revised deadline. If HC accepts a response to the NOD as complete, its decision as to whether to grant marketing approval for PROCYSBI for the treatment of nephropathic cystinosis will be delayed due to the NOD and the time to submit the response.
With respect to QUINSAIR, the FDA has indicated in previous written and verbal communications with Raptor and with the drug’s previous sponsor that it believes the data submitted in connection with EMA’s subsequent approval of QUINSAIR for the management of chronic pulmonary infections due to Pseudomonas aeruginosa in adults with cystic fibrosis does not provide substantial evidence of efficacy and safety to support FDA approval of QUINSAIR for treatment of patients with cystic fibrosis. The FDA identified a number of limitations with the design of the Phase 3 trial (MPEX-209) upon which approval of QUINSAIR in the EU and Canada was based that, in the FDA’s view, impacts its ability to be used as a pivotal efficacy study. The FDA also previously indicated that the pivotal Phase 3 study (MPEX-207) missed its primary endpoint and questioned whether patients in the study achieved any overall benefit. It is possible that additional studies or analyses may be required prior to a submission of an NDA for approval of QUINSAIR for treatment of Pseudomonas aeruginosa in adults with cystic fibrosis. In the second quarter of 2016, Raptor met with the FDA to discuss a planned NDA. The FDA had several questions related to the MPEX-209 study, and asked Raptor to submit information prior to any submission of a new drug application (“NDA”). Raptor submitted data in response to the FDA’s request, and subsequently requested a follow-up meeting to discuss the FDA’s assessment of that information. In preliminary meeting responses, the FDA largely reiterated its view that the prior Phase 3 trial would not be sufficient to file or approve an NDA for QUINSAIR. On August 26, 2016, Raptor had a follow-up in-person meeting with the FDA. The FDA stated in its meeting minutes dated September 22, 2016 that it recognizes that there is an unmet medical need for cystic fibrosis (“CF”) patients. The FDA also stated that although a discussion about the design of a new trial may be helpful, the FDA was willing to reevaluate the existing data and the analysis presented at the meeting to determine if there is enough evidence to support the filing of an NDA. The FDA stated that it will further discuss some of the issues and will provide feedback to Raptor. In addition, the FDA may request additional information. In follow-up verbal communications to Raptor, the FDA has indicated that it anticipates providing feedback by the end of October. Following the acquisition of Raptor, we intend to further evaluate a potential NDA for QUINSAIR and it is possible that we will determine that based on the FDA’s guidance, further pursuit of U.S. approval of QUINSAIR as a treatment of Pseudomonas aeruginosa in adults with cystic fibrosis is not warranted.
Raptor currently plans to pursue a Phase 3 clinical trial of QUINSAIR for use in the indication of bronchiectasis (“BE”), not associated with cystic fibrosis. On September 8, 2016, Raptor met the Medicines and Healthcare Products Regulatory Agency (the “MHRA”) to discuss non-clinical and clinical development aspects of QUINSAIR for the treatment of BE. On September 29, 2016, Raptor received a written response from the MHRA, which included answers to questions on trial design, among other responses. Raptor has also submitted the protocol to the FDA and has not received comments. Raptor is also exploring further clinical development of QUINSAIR for the treatment of pulmonary nontuberculous mycobacteria (“NTM”) infection, based on third-party data generated pertaining to the susceptibility of certain pathogens to treatment with levofloxacin and other fluoroquinolone molecules. No clinical data have been generated with QUINSAIR in patients with BE or with NTM infections, either by Raptor, by us or by other parties. This creates substantial uncertainty as the efficacy of QUINSAIR in these indications.
66
Following the acquisition of Raptor we will evaluate all development opportunities, including all obligations to use commercial reasonable efforts to further develop QUINSAIR. However, we may determine not to pursue such further development.
The ultimate approval and commercial marketing of any of our or Raptor’s medicines in additional indications or geographies is subject to substantial uncertainty. Failure to gain additional regulatory approvals would limit the potential revenues and value of our medicines and could cause our share price to decline.
The amount of Raptor’s product sales in the European Economic Area (the “EEA”) is dependent in part upon the pricing and reimbursement decisions adopted in each of the EEA countries, which may not be at acceptable levels to us.
One or more EEA countries may not support pricing within our target pricing and reimbursement range for our medicines due to budgetary decisions made by regional, national and local health authorities and third-party payers in the EEA, which would negatively affect our revenues. The pricing and reimbursement process in EEA countries can be lengthy, involved and difficult to predict. Failure to timely complete the pricing and reimbursement process in the EEA countries will delay our ability to market PROCYSBI, to bring QUINSAIR to market in the EEA and to derive revenues from those countries.
We may be subject to penalties and litigation and large incremental expenses if we fail to comply with regulatory requirements or experience problems with our medicines.
Even after we achieve regulatory approvals, we are subject to ongoing obligations and continued regulatory review with respect to many operational aspects including our manufacturing processes, labeling, packaging, distribution, storage, adverse event monitoring and reporting, dispensation, advertising, promotion and recordkeeping. These requirements include submissions of safety and other post-marketing information and reports, ongoing maintenance of product registration and continued compliance with current good manufacturing practices (“cGMPs”), good clinical practices (“GCPs”), good pharmacovigilance practice, good distribution practices and good laboratory practices (“GLPs”). We are in the process of implementing corrective and preventive actions that we completed in the first quarter of 2016 related to our pharmacovigilance system to address findings issued in August 2015 following a routine inspection from a European regulatory authority in June 2015 and our own internal reviews of our internal processes.
If we, our medicines or medicine candidates, or the third-party manufacturing facilities for our medicines or medicine candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| • | impose injunctions or restrictions on the marketing, manufacturing or distribution of a product, suspend or withdraw product approvals, revoke necessary licenses or suspend product reimbursement; |
| • | issue warning letters, show cause notices or untitled letters describing alleged violations, which may be publicly available; |
| • | suspend any ongoing clinical trials or delay or prevent the initiation of clinical trials; |
| • | delay or refuse to approve pending applications or supplements to approved applications we have filed; |
| • | refuse to permit drugs or precursor or intermediary chemicals to be imported or exported to or from the United States; |
67
| • | suspend or impose restrictions or additional requirements on operations, including costly new manufacturing quality or pharmacovigilance requirements; |
| • | seize or detain products or require us to initiate a product recall; and/or |
| • | commence criminal investigations and prosecutions. |
Moreover, existing regulatory approvals and any future regulatory approvals that we obtain will be subject to limitations on the approved indicated uses and patient populations for which our medicines may be marketed, the conditions of approval, requirements for potentially costly, post-market testing and requirements for surveillance to monitor the safety and efficacy of the medicines. In the EEA, the advertising and promotion of pharmaceuticals is strictly regulated. The direct-to-consumer promotion of prescription pharmaceuticals is not permitted, and some countries in the EEA require the notification and/or prior authorization of promotional or advertising materials directed at healthcare professionals. The FDA, EMA and other authorities in the EEA countries strictly regulate the promotional claims that may be made about prescription products, and our product labeling, advertising and promotion are subject to continuing regulatory review. Physicians nevertheless may prescribe our medicines to their patients in a manner that is inconsistent with the approved label or that is off-label. Positive clinical trial results in any of our medicine development programs increase the risk that approved pharmaceutical forms of the same active pharmaceutical ingredients may be used off-label in those indications. Our investigational medicine candidate RP103 is comprised of the same API as PROCYSBI. If we are found to have improperly promoted off-label uses of approved medicines, we may be subject to significant sanctions, civil and criminal fines and injunctions prohibiting us from engaging in specified promotional conduct.
In addition, engaging in improper promotion of our medicines for off-label uses in the United States can subject us to false claims litigation under federal and state statutes. These false claims statutes in the United States include the federal False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims or causing to present such false or fraudulent claims for payment by a federal program such as Medicare or Medicaid. Growth in false claims litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay civil money penalties, settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations and be excluded from Medicare, Medicaid and other federal and state healthcare programs.
The regulations, policies or guidance of regulatory agencies may change and new or additional statutes or government regulations may be enacted that could prevent or delay regulatory approval of our medicine candidates or further restrict or regulate post-approval activities. For example, the Food and Drug Administration Safety and Innovation Act requires the FDA to issue new guidance describing its policy regarding internet and social media promotion of regulated medical products, and the FDA may soon specify new restrictions on this type of promotion. In January 2014, the FDA released draft guidance on how drug companies can fulfill their regulatory requirements for post-marketing submission of interactive promotional media, and though the guidance provided insight into how the FDA views a company’s responsibility for certain types of social media promotion, there remains a substantial amount of uncertainty. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from pending or future legislation or administrative action, either in the United States or abroad. If we are unable to achieve and maintain regulatory compliance, we will not be permitted to market our drugs, which would materially adversely affect our business, results of operations and financial condition.
Our limited history of commercial operations makes evaluating our business and future prospects difficult and may increase the risk of any investment in our ordinary shares.
We face considerable risks and difficulties as a company with limited commercial operating history, particularly as a global consolidated entity with operating subsidiaries that also have limited operating histories.
68
If we do not successfully address these risks, our business, prospects, operating results and financial condition will be materially and adversely harmed. Our limited commercial operating history, including our limited history commercializing our current medicines, makes it particularly difficult for us to predict our future operating results and appropriately budget for our expenses. In the event that actual results differ from our estimates or we adjust our estimates in future periods, our operating results and financial position could be materially affected. For example, we may underestimate the resources we will require to successfully integrate recent or future medicine or company acquisitions, or to commercialize our medicines, or not realize the benefits we expect to derive from our recent or future acquisitions. In addition, we have a limited history implementing our commercialization strategy focused on patient access, and we cannot guarantee that we will be able to successfully implement this strategy or that it will represent a viable strategy over the long term.
We have rights to medicines in certain jurisdictions but have no control over third parties that have rights to commercialize those medicines in other jurisdictions, which could adversely affect our commercialization of these medicines.
Boehringer Ingelheim International GmbH (“Boehringer Ingelheim International”) currently has certain rights to commercialize interferon gamma 1b, known as IMUKIN, outside the United States, Canada and Japan. On May 18, 2016, we entered into a definitive agreement with Boehringer Ingelheim International to acquire such rights to IMUKIN, which is scheduled to close on December 31, 2016, and we are continuing to work with Boehringer Ingelheim International to enable the transfer of applicable marketing authorizations. AstraZeneca AB (“Astra Zeneca”) has retained its existing rights to VIMOVO in territories outside of the United States, including the right to use the VIMOVO name and related trademark. While we have the worldwide rights to BUPHENYL, the marketing and distribution rights are licensed to Swedish Orphan Biovitrum AB (“SOBI”) through the end of 2016. Similarly, Nuvo Research Inc. (“Nuvo”) has retained its rights to PENNSAID 2% in territories outside of the United States and has announced its intention to seek commercialization partners outside the United States. We have little or no control over Boehringer Ingelheim International’s activities with respect to IMUKIN outside the United States, Canada and Japan, over AstraZeneca’s activities with respect to VIMOVO outside of the United States, over SOBI’s activities with respect to BUPHENYL in Europe, certain Asian, Latin American, Middle Eastern, North African and other countries or over Nuvo’s or its future commercial partners’ activities with respect to PENNSAID 2% outside of the United States, even though those activities could impact our ability to successfully commercialize ACTIMMUNE, VIMOVO, BUPHENYL and PENNSAID 2%. For example, AstraZeneca or its assignees or Nuvo or its assignees can make statements or use promotional materials with respect to VIMOVO or PENNSAID 2%, respectively, outside of the United States that are inconsistent with our positioning of the medicines in the United States, and could sell VIMOVO or PENNSAID 2%, respectively, in foreign countries, including Canada, at prices that are dramatically lower than the prices we charge in the United States. These activities and decisions, while occurring outside of the United States, could harm our commercialization strategy in the United States, in particular because AstraZeneca is continuing to market VIMOVO outside the United States under the same VIMOVO brand name that we are using in the United States. In addition, medicine recalls or safety issues with ACTIMMUNE, VIMOVO, BUPHENYL or PENNSAID 2% outside the United States, even if not related to the commercial medicine we sell in the United States, could result in serious damage to the brand in the United States and impair our ability to successfully market ACTIMMUNE, VIMOVO, BUPHENYL and PENNSAID 2%. We also rely on Boehringer Ingelheim International, AstraZeneca, SOBI and Nuvo or their assignees to provide us with timely and accurate safety information regarding the use of ACTIMMUNE, VIMOVO, BUPHENYL or PENNSAID 2%, respectively, outside of the United States (and outside of Canada and Japan with regards to Boehringer Ingelheim International), as we have or will have limited access to this information ourselves.
69
We rely on third parties to manufacture commercial supplies of all of our medicines, and we currently intend to rely on third parties to manufacture commercial supplies of any other approved medicines. The commercialization of any of our medicines could be stopped, delayed or made less profitable if those third parties fail to provide us with sufficient quantities of medicine or fail to do so at acceptable quality levels or prices or fail to maintain or achieve satisfactory regulatory compliance.
The facilities used by our third-party manufacturers to manufacture our medicines and medicine candidates must be approved by the applicable regulatory authorities. We do not control the manufacturing processes of third-party manufacturers and are currently completely dependent on our third-party manufacturing partners. In addition, we are required to obtain AstraZeneca’s consent prior to engaging any third-party manufacturers for esomeprazole, one of the APIs in VIMOVO, other than the third-party manufacturer(s) used by AstraZeneca or its affiliates or licensees. To the extent such manufacturers are unwilling or unable to manufacture esomeprazole for us on commercially acceptable terms, we cannot guarantee that AstraZeneca would consent to our use of alternate sources of supply.
We rely on an exclusive supply agreement with Boehringer Ingelheim RCV GmbH & Co. KG (“Boehringer Ingelheim”) for manufacturing and supply of ACTIMMUNE. However, Boehringer Ingelheim also currently manufactures interferon gamma-1b to supply its own commercial needs in its licensed territory, and this may lead to capacity allocation issues and supply constraints to our company. On May 18, 2016, we entered into a definitive agreement with Boehringer Ingelheim International to acquire their rights to interferon gamma-1b, which is scheduled to close on December 31, 2016 and we are continuing to work with Boehringer Ingelheim International to enable the transfer of applicable marketing authorizations. ACTIMMUNE is manufactured by starting with cells from working cell bank samples which are derived from a master cell bank. We and Boehringer Ingelheim separately store multiple vials of the master cell bank. In the event of catastrophic loss at our or Boehringer Ingelheim’s storage facility, it is possible that we could lose multiple cell banks and have the manufacturing capacity of ACTIMMUNE severely impacted by the need to substitute or replace the cell banks. In addition, a key excipient used in PENNSAID 2% as a penetration enhancer is dimethyl sulfoxide (“DMSO”). We and Nuvo, our exclusive supplier of PENNSAID 2%, rely on a sole proprietary form of DMSO for which we maintain a substantial safety stock. However, should this supply become inadequate, damaged, destroyed or unusable, we and Nuvo may not be able to qualify a second source. We rely on NOF Corporation (“NOF”) as our exclusive supplier of the PEGylation agent that is a critical raw material in the manufacture of KRYSTEXXA. If NOF failed to supply such PEGylation agent, it may lead to KRYSTEXXA supply constraints.
If any of our third-party manufacturers cannot successfully manufacture material that conforms to our specifications and the applicable regulatory authorities’ strict regulatory requirements, or pass regulatory inspection, they will not be able to secure or maintain regulatory approval for the manufacturing facilities. In addition, we have no control over the ability of third-party manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or any other applicable regulatory authorities do not approve these facilities for the manufacture of our medicines or if they withdraw any such approval in the future, or if our suppliers or third-party manufacturers decide they no longer want to supply our primary active ingredients or manufacture our medicines, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our medicines. To the extent any third-party manufacturers that we engage with respect to our medicines are different from those currently being used for commercial supply in the United States, the FDA will need to approve the facilities of those third-party manufacturers used in the manufacture of our medicines prior to our sale of any medicine using these facilities.
Although we have entered into supply agreements for the manufacture of our medicines, our manufacturers may not perform as agreed or may terminate their agreements with us. Under our manufacturing and supply agreement with Sanofi-Aventis U.S. LLC (“Sanofi-Aventis U.S.”) either we or Sanofi-Aventis U.S. may terminate the agreement upon an uncured breach by the other party or without cause upon two years prior written notice. Under our master manufacturing services and medicine agreement with Patheon Pharmaceuticals
70
Inc. (“Patheon”) for finished VIMOVO medicine, either we or Patheon may terminate the agreement for uncured material breach by the other party or upon the other party’s bankruptcy or insolvency, we may terminate the agreement if any regulatory authority takes any action or raises any objection that prevents us from commercializing the VIMOVO medicine and Patheon may terminate the agreement if we assign our rights or obligations under the agreement to a competitor of Patheon or to a party that, in the reasonable opinion of Patheon, is not a credit worthy substitute for us, or in certain other circumstances where we assign the agreement without Patheon’s consent. Our manufacturing agreement with Boehringer Ingelheim has a term that runs until July 31, 2020, but the agreement may be terminated earlier by either us or Boehringer Ingelheim for an uncured material breach by the other party or upon the other party’s bankruptcy or insolvency. Under our manufacturing and supply agreement with Jagotec AG (“Jagotec”), either we or Jagotec may terminate the agreement in the event of an insolvency, liquidation or bankruptcy of the other party or upon an uncured breach by the other party. While we have the right to receive a continuing supply of RAYOS/LODOTRA from Jagotec for a period of 24 months after termination, we would need to move our manufacturing to our alternate supplier of RAYOS/ LODOTRA, Bayer Pharma AG, in such an event and we may have to qualify a new back-up manufacturer. The term of our supply agreement with Nuvo for PENNSAID 2% is through December 31, 2029, but the agreement may be terminated earlier by either party for any uncured material breach by the other party of its obligations under the supply agreement or upon the bankruptcy or similar proceeding of the other party. With respect to BUPHENYL, our supply agreement with Pharmaceutics International, Inc. (“PII”) is in place until April 1, 2017, however, the agreement may be terminated earlier by either party. The term of our manufacturing agreement with Halo Pharmaceutical, Inc. for RAVICTI runs until July 4, 2018, however, the agreement may be terminated earlier in the case of breach by either party if the other party is in material breach of any provision of the agreement and the other party fails to remedy such a breach within thirty days, or by us at any time for any reason. Our master services agreement with Lyne Laboratories, Inc. (“Lyne”) for RAVICTI runs until February 1, 2017, with provision for 12 monthly auto renewals thereafter, unless 6 months’ written notice is provided by either party. The agreement may be terminated earlier, on 30 days’ notice, in case of breach by either party. The term of our commercial supply agreement with Bio-Technology General (Israel) Ltd. (“BTG Israel”) for KRYSTEXXA bulk medicine runs until December 31, 2030, and will automatically renew for successive three year periods unless earlier terminated by either party upon three years prior written notice. The agreement may be terminated earlier by either party in the event of a force majeure, liquidation, dissolution, bankruptcy or insolvency of the other party, uncured material breach by the other party or after January 1, 2024, upon three years prior written notice. Raptor currently relies on single source suppliers for cysteamine API and for PROCYSBI drug product. Raptor also relies on single source suppliers for the QUINSAIR API and drug product and the base units, nebulizers and hand-held devices used for the administration of QUINSAIR.
Raptor’s supply agreement with Cambrex Profarmaco Milano (“Cambrex”) for cysteamine API runs until November 3, 2020, and will automatically renew for successive two year periods after such initial term. The agreement may be terminated earlier by either party for any uncured material breach or upon one year advance notice by either party provided such notice is given at least one year prior to the expiration of the then-current term. Following an acquisition of Raptor, we would also be able to terminate the agreement in the event of certain supply failures by Cambrex or if we determined that the products incorporating the cysteamine API will not be marketed or the FDA or EMA withdraws approval or fails to approve such products then in development. Raptor’s supply agreement for QUINSAIR API runs, with respect to the United States, until the seventh anniversary after the first commercial sale in the United States, with respect to Europe, until the fifth anniversary after the first commercial sale in Europe, in each case subject to early termination under specified circumstances. Raptor’s commercial supply agreement with PARI Pharma GmbH (“PARI”) for base units, nebulizers and hand-held devices used for the administration of QUINSAIR may be terminated by either party for any uncured material breach by the other party of its obligations under the agreement, upon the filing of an application for commencement of bankruptcy or similar proceeding against the other party, or following an acquisition of Raptor, if we cease development and commercialization of applicable products for a certain number of years. We rely on safety stock to mitigate the risk of our current suppliers electing to cease producing bulk drug medicine or ceasing to do so at acceptable prices and quality. However, we can provide no assurance that such safety stocks would be sufficient to avoid supply shortfalls in the event we have to identify and qualify new contract manufacturers.
71
In addition, we do not have the capability to package any of our medicines for distribution. Under our master manufacturing services agreement with Patheon, we have entered into a medicine agreement for packaging of RAYOS/LODOTRA. Valeant Pharmaceuticals International, Inc. manufactures and supplies DUEXIS to us in final, packaged form for the United States as well as any additional countries as may be agreed to by the parties. Patheon supplies final, packaged VIMOVO medicine pursuant to the master manufacturing services and product agreement we executed in connection with our acquisition of the U.S. rights to VIMOVO. Boehringer Ingelheim supplies final, packaged ACTIMMUNE to us and Nuvo supplies final, packaged PENNSAID 2% to us, in each case under exclusive supply agreements. We have clinical and commercial supplies of BUPHENYL finished medicine manufactured for us by PII on a purchase order basis. We have clinical and commercial supplies of RAVICTI finished drug medicine manufactured by Lyne under a commercial supply agreement and have an agreement in place with Halo Pharmaceutical, Inc. to serve as a finished drug medicine supplier for RAVICTI in the EU. Sigma Tau PharmaSource Inc. supplies final, packaged KRYSTEXXA to us for the United States. G & W Laboratories, Inc. manufactures and supplies MIGERGOT to us in final, packaged form for the United States.
The manufacture of medicines requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of medicines often encounter difficulties in production, particularly in scaling up and validating initial production. These problems include difficulties with production costs and yields, quality control, including stability of the medicine, quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Furthermore, if microbial, viral or other contaminations are discovered in the medicines or in the manufacturing facilities in which our medicines are made, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. We cannot assure you that issues relating to the manufacture of any of our medicines will not occur in the future. Additionally, our manufacturers may experience manufacturing difficulties due to resource constraints or as a result of labor disputes or unstable political environments. If our manufacturers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, our ability to commercialize our medicines in the United States or provide any medicine candidates to patients in clinical trials would be jeopardized.
Any delay or interruption in our ability to meet commercial demand for our medicines will result in the loss of potential revenues and could adversely affect our ability to gain market acceptance for these medicines. In addition, any delay or interruption in the supply of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to commence new clinical trials at additional expense or terminate clinical trials completely.
Failures or difficulties faced at any level of our supply chain could materially adversely affect our business and delay or impede the development and commercialization of any of our medicines or medicine candidates and could have a material adverse effect on our business, results of operations, financial condition and prospects.
We have experienced recent growth and expanded the size of our organization substantially in connection with our recent acquisition transactions, and we may experience difficulties in managing this growth as well as potential additional growth in connection with future medicine or company acquisitions.
As of December 31, 2010 and prior to the commercial launch of DUEXIS, we employed approximately 40 full-time employees as a consolidated entity. As of June 30, 2016, we employed approximately 915 full-time employees, including approximately 500 sales representatives, representing a substantial change to the size of our organization over a relatively short period of time. We have also experienced, and may continue to experience, turnover of the sales representatives that we hired or will hire in connection with the commercialization of our medicines, requiring us to hire and train new sales representatives. Our management, personnel, systems and facilities currently in place may not be adequate to support this recent and anticipated growth, and we may not be able to retain or recruit qualified personnel in the future due to competition for personnel among pharmaceutical businesses.
72
As our commercialization plans and strategies continue to develop, we will need to continue to recruit and train sales and marketing personnel and expect to need to expand the size of our employee base for managerial, operational, financial and other resources as a result of our recent acquisitions. Our ability to manage any future growth effectively may require us to, among other things:
| • | continue to manage and expand the sales and marketing efforts for our existing medicines; |
| • | enhance our operational, financial and management controls, reporting systems and procedures; |
| • | expand our international resources; |
| • | successfully identify, recruit, hire, train, maintain, motivate and integrate additional employees; |
| • | establish and increase our access to commercial supplies of our medicines and medicine candidates; |
| • | expand our facilities and equipment; and |
| • | manage our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors, collaborators, distributors and other third parties. |
In particular, the merger of our business with the business of Vidara Therapeutics International Public Limited Company (“Vidara”) is subject to numerous uncertainties and risks and will continue to require significant efforts and expenditures. For example, we have transitioned from a standalone public Delaware corporation to being part of a combined company organized in Ireland. This combination as well as our other recent acquisitions have resulted in many changes, including significant changes in the corporate business and legal entity structure, the integration of other companies and their personnel with us, and changes in systems. We are currently undertaking numerous complex transition activities associated with our recent acquisitions, and we may encounter unexpected difficulties or incur unexpected costs, including:
| • | difficulties in achieving growth prospects from combining third party businesses with our business; |
| • | difficulties in the integration of operations and systems; |
| • | difficulties in the assimilation of employees and corporate cultures; |
| • | challenges in preparing financial statements and reporting timely results at both a statutory level for multiple entities and jurisdictions and at a consolidated level for public reporting; |
| • | challenges in keeping existing physician prescribers and patients and increasing adoption of acquired medicines; |
| • | difficulties in achieving anticipated cost savings, synergies, business opportunities and growth prospects from the combination; |
| • | potential unknown liabilities, adverse consequences and unforeseen increased expenses associated with the transaction; and |
| • | challenges in attracting and retaining key personnel. |
If any of these factors impair our ability to continue to integrate our operations with those of any companies or businesses we acquire, we may not be able to realize the business opportunities, growth prospects and anticipated tax synergies from combining the businesses. In addition, we may be required to spend additional time or money on integration that otherwise would be spent on the development and expansion of our business.
73
As a result of our anticipated acquisition of Raptor and our plans to launch RAVICTI in Europe, we also expect to continue expanding our operations and to add commercial personnel in Europe. We may not be successful in integrating Raptor’s existing European operations and personnel with our own or in otherwise growing our commercial operations outside the United States, and could encounter other challenges in growing our commercial presence in Europe, including due to risks associated with political and economic instability, operating under different legal requirements and tax complexities. If we are unable to manage our commercial growth outside of the Unites States, our opportunities to expand sales in other countries will be limited or we may experience greater costs with respect to our ex-U.S. commercial operations.
Our management may also have to divert a disproportionate amount of its attention away from day-to-day activities and toward managing these growth and integration activities. Our future financial performance and our ability to execute on our business plan will depend, in part, on our ability to effectively manage any future growth and our failure to effectively manage growth could have a material adverse effect on our business, results of operations, financial condition and prospects.
We face significant competition from other biotechnology and pharmaceutical companies, including those marketing generic medicines and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive. We have competitors both in the United States and international markets, including major multinational pharmaceutical companies, biotechnology companies and universities and other research institutions. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff, experienced marketing and manufacturing organizations and well-established sales forces. Additional consolidations in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors and we will have to find new ways to compete and may have to potentially merge with or acquire other businesses to stay competitive. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or in-licensing on an exclusive basis, medicines that are more effective and/or less costly than our medicines.
DUEXIS and VIMOVO face competition from other NSAIDs, including Celebrex, which is marketed by Pfizer Inc., and is also a generic medicine known as celecoxib and marketed by other pharmaceutical companies. DUEXIS and VIMOVO also face significant competition from the separate use of NSAIDs for pain relief and GI protective medications to reduce the risk of NSAID-induced upper GI ulcers. Both NSAIDs and GI protective medications are available in generic form and may be less expensive to use separately than DUEXIS or VIMOVO. PENNSAID 2% faces competition from generic versions of diclofenac sodium topical solutions that are priced significantly less than the price we charge for PENNSAID 2%, and Voltaren Gel, marketed by Endo Pharmaceuticals Solutions Inc., which is the market leader in the topical NSAID category. Legislation enacted in most states in the United States allows, or in some instances mandates, that a pharmacist dispense an available generic equivalent when filling a prescription for a branded medicine, in the absence of specific instructions from the prescribing physician. Because pharmacists often have economic and other incentives to prescribe lower-cost generics, if physicians prescribe DUEXIS, PENNSAID 2% or VIMOVO, those prescriptions may not result in sales. If physicians do not complete prescriptions through our HorizonCares program or otherwise provide prescribing instructions prohibiting the substitution of generic ibuprofen and famotidine separately as a substitution for DUEXIS or generic naproxen and branded Nexium (esomeprazole) as a substitute for VIMOVO or generic diclofenac sodium topical solutions as a substitute for PENNSAID 2%, sales of DUEXIS, PENNSAID 2% and VIMOVO may suffer despite any success we may have in promoting DUEXIS, PENNSAID 2% or VIMOVO to physicians. In addition, other medicine candidates that contain ibuprofen and famotidine in combination or naproxen and esomeprazole in combination, while not currently known or FDA approved, may be developed and compete with DUEXIS or VIMOVO, respectively, in the future. While KRYSTEXXA faces limited direct competition, a number of competitors have drugs in Phase 1 or Phase 2 trials. On December 22, 2015, AstraZeneca secured approval from the FDA for ZURAMPIC (lesinurad) 200mg tablets
74
in combination with a xanthine oxidase inhibitor (“XOI”) for the treatment of hyperuricemia associated with gout in patients who have not achieved target serum uric acid (sUA) levels with an XOI alone. In April 2016, the U.S. rights to ZURAMPIC were licensed to Ironwood Pharmaceuticals Inc. Although ZURAMPIC is not a direct competitor because it has not been approved for refractory gout, this therapy could be used prior to use of KRYSTEXXA and if effective, could reduce the target patient population for KRYSTEXXA. PROSCYSBI faces competition from Cystagon (immediate-release cysteamine bitartrate capsules) for the treatment of cystinosis and Cystaran (cysteamine ophthalmic solution) for treatment of corneal crystal accumulation in patients with cystinosis. QUINSAIR faces competition from Tobramycin solution, which is available as a generic medicine for treatment of chronic Pseudomonas aeruginosa lung infections in patients with CF, TOBI Podhaler, Cayston and colistimethate.
We have also entered into settlement and license agreements that may allow certain of our competitors to sell generic versions of certain of our medicines in the United States, subject to the terms of such agreements. On August 21, 2013, we entered into a settlement agreement (the “Par settlement agreement”) and license agreement (the “Par license agreement”) with Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc. (collectively “Par”) relating to our patent infringement litigation with Par. Under the Par license agreement, we granted Par a non-exclusive license (that is only royalty-bearing in some circumstances) (the “License”) to manufacture and commercialize Par’s generic version of DUEXIS in the United States after the generic entry date (as defined below) and to take steps necessary to develop inventory of, and obtain regulatory approval for, but not commercialize, Par’s generic version of DUEXIS prior to the generic entry date. Under the Par license agreement, the generic entry date is January 1, 2023; however, Par may be able to enter the market earlier under certain circumstances. Such events relate to the resolution of potential future third-party DUEXIS patent litigation, the entry of other third-party generic versions of DUEXIS or certain specific changes in DUEXIS market conditions. Only in the event that Par enters the DUEXIS market due to the specified changes in DUEXIS market conditions will the License become royalty-bearing, with the royalty obligations ceasing upon the occurrence of one of the other events that would have allowed Par to enter the DUEXIS market.
On October 1, 2015, we, as well as Jagotec, entered into a license and settlement agreement (the “Actavis settlement agreement”) with Actavis Laboratories FL, Inc. (formerly known as Watson Laboratories, Inc.—Florida) (the “Actavis FL”) relating to patent infringement litigation with Actavis FL. Under the Actavis settlement agreement, we and Jagotec granted Actavis FL a non-exclusive license to manufacture and commercialize Actavis FL’s generic version of RAYOS tablets in the United States after the generic entry date (as defined below) and to take steps necessary to develop inventory of, and prepare to commercialize, Actavis FL’s generic version of RAYOS tablets during certain limited periods prior to the generic entry date. We and Jagotec also agreed that during the 180 days after the generic entry date, the license granted to Actavis FL would be exclusive with respect to any third-party generic version of RAYOS tablets. Under the Actavis settlement agreement, the generic entry date is December 23, 2022; however, Actavis FL may be able to enter the market earlier under certain circumstances. Such events relate to the resolution of any other third-party RAYOS patent litigation, the entry of other generic versions of RAYOS tablets or certain substantial reductions in RAYOS prescriptions over specified periods of time. If we or Jagotec enter into any similar agreements with other parties with respect to generic versions of RAYOS tablets, we and Jagotec agreed to amend the Actavis settlement agreement to provide Actavis FL with terms that are no less favorable than those provided to the other parties with respect to the license terms, generic entry date, permitted pre-market activities and notice provisions.
On May 6, 2015, we entered into a settlement and license agreement (the “Perrigo settlement agreement”) with Perrigo Company plc and its subsidiary Paddock Laboratories, LLC (collectively, “Perrigo”) relating to patent infringement litigation with Perrigo. Under the Perrigo settlement agreement, we granted Perrigo a non-exclusive license to manufacture and commercialize Perrigo’s generic version of PENNSAID 2% in the United States after the license effective date (as defined below) and to take steps necessary to develop inventory of, and prepare to commercialize, Perrigo’s generic version of PENNSAID 2% during certain limited periods prior to the license effective date. Under the Perrigo settlement agreement, the license effective date is January 10, 2029; however, Perrigo may be able to enter the market earlier under certain circumstances. Such
75
events relate to the resolution of any other third-party PENNSAID 2% patent litigation, the entry of other third-party generic versions of PENNSAID 2% or certain substantial reductions in our PENNSAID 2% shipments over specified periods of time. In certain circumstances following the entry of other third-party generic versions of PENNSAID 2%, we may be required to supply PENNSAID 2% to Perrigo as our authorized distributor of generic PENNSAID 2%, with us receiving specified percentages of any net sales by Perrigo. We also agreed that if we enter into any similar agreements with other parties with respect to generic versions of PENNSAID 2%, we will amend the Perrigo settlement agreement to provide Perrigo with terms that are no less favorable than those provided to the other parties.
On September 9, 2015, we entered into a settlement and license agreement (the “Taro settlement agreement”) with Taro Pharmaceuticals USA, Inc. and Taro Pharmaceutical Industries, Ltd. (collectively, Taro”) relating to patent infringement litigation with Taro. Under the Taro settlement agreement, we granted Taro a non-exclusive license to manufacture and commercialize Taro’s generic version of PENNSAID 2% in the United States after the license effective date (as defined below) and to take steps necessary to develop inventory of, and prepare to commercialize, Taro’s generic version of PENNSAID 2% during certain limited periods prior to the license effective date. Under the Taro settlement agreement, the license effective date is January 10, 2029; however, Taro may be able to enter the market earlier under certain circumstances. Such events relate to the resolution of any other third-party PENNSAID 2% patent litigation, the entry of other third-party generic versions of PENNSAID 2% or certain substantial reductions in our PENNSAID 2% shipments over specified periods of time. We also agreed that if we enter into any similar agreements with other parties with respect to generic versions of PENNSAID 2%, we will amend the Taro settlement agreement to provide Taro with terms that are no less favorable than those provided to the other parties.
On April 18, 2016, we entered into a settlement and license agreement (the “Amneal settlement agreement”) with Amneal Pharmaceuticals LLC (“Amneal”), relating to patent infringement litigation with Amneal. Under the Amneal settlement agreement, we granted Amneal a non-exclusive license to manufacture and commercialize Amneal’s generic version of PENNSAID 2% in the United States after the license effective date (as defined below) and to take steps necessary to develop inventory of, and prepare to commercialize, Amneal’s generic version of PENNSAID 2% during certain limited periods prior to the license effective date. Under the Amneal settlement agreement, the license effective date is January 10, 2029; however, Amneal may be able to enter the market earlier under certain circumstances. Such events relate to the resolution of any other third-party PENNSAID 2% patent litigation or the entry of other third-party generic versions of PENNSAID 2%. In certain circumstances following the entry of other third-party generic versions of PENNSAID 2%, we may be required to supply PENNSAID 2% to Amneal as our non-exclusive, authorized distributor of generic PENNSAID 2%, with us receiving specified percentages of any net sales by Amneal. We also agreed that if we enter into any similar agreements with other parties with respect to generic versions of PENNSAID 2%, we will amend the Amneal settlement agreement to provide Amneal with terms that are no less favorable than those provided to the other parties.
On May 9, 2016, we entered into a settlement and license agreement (the “Teligent settlement agreement”) with Teligent, Inc. (“Teligent”) relating to patent infringement litigation with Teligent. Under the Teligent settlement agreement, we granted Teligent a non-exclusive license to manufacture and commercialize Taro’s generic version of PENNSAID 2% in the United States after the license effective date (as defined below) and to take steps necessary to develop inventory of, and prepare to commercialize, Teligent’s generic version of PENNSAID 2% during certain limited periods prior to the license effective date. Under the Teligent settlement agreement, the license effective date is January 10, 2029; however, Teligent may be able to enter the market earlier under certain circumstances. Such events relate to the resolution of any other third-party PENNSAID 2% patent litigation, the entry of other third-party generic versions of PENNSAID 2% or certain substantial reductions in our PENNSAID 2% shipments over specified periods of time. We also agreed that if we enter into any similar agreements with other parties with respect to generic versions of PENNSAID 2%, we will amend the Teligent settlement agreement to provide Taro with terms that are no less favorable than those provided to the other parties.
76
Patent litigation is currently pending in the United States District Court for the District of New Jersey against several companies intending to market a generic version of PENNSAID 2% prior to the expiration of certain of our patents listed in the FDA’s Orange Book (the “Orange Book”). These cases are collectively known as the PENNSAID 2% cases, and involve the following sets of defendants: (i) Actavis Laboratories UT, Inc., formerly known as Watson Laboratories, Inc., Actavis, Inc. and Actavis plc (collectively “Actavis”); (ii) Lupin Limited (“Lupin Limited”) and (iii) Amneal, with the Amneal case being settled and awaiting dismissal. These cases arise from Paragraph IV Patent Certification notice letters from each of Actavis, Lupin and Amneal advising each had filed an Abbreviated New Drug Application (“ANDA”) with the FDA seeking approval to market a generic version of PENNSAID 2% before the expiration of the patents-in-suit. No trial date has been set by the court in these actions.
We received from Actavis a Paragraph IV Patent Certification Notice Letter dated July 1, 2016, against Orange Book listed U.S. Patent Nos. 9,339,551 and 9,339,552, advising that Actavis had filed an ANDA with the FDA for a generic version of PENNSAID 2%.
We received from Apotex Inc. (“Apotex”), a Paragraph IV Patent Certification Notice Letter dated April 1, 2016, against Orange Book listed U.S. Patent Nos. 8,217,078, 8,252,838, 8,546,450, 8,563,613, 8,618,164, 8,741,956, 8,871,809, 9,066,913, 9,101,591, 9,132,110, 9,168,304, 9,168,305 and 9,220,784, advising that Apotex had filed an ANDA with the FDA for a generic version of PENNSAID 2%. The Company also received from Apotex a second Paragraph IV Patent Certification Notice Letter dated June 30, 2016, against Orange Book listed U.S. Patent Nos. 9,339,551 and 9,339,552, advising that Apotex had filed an ANDA with the FDA for a generic version of PENNSAID 2%.
Patent litigation is currently pending in the United States District Court for the District of New Jersey against several companies intending to market a generic version of VIMOVO before the expiration of certain of our patents listed in the Orange Book. These cases are collectively known as the VIMOVO cases, and involve the following sets of defendants: (i) Dr. Reddy’s Laboratories Inc. and Dr. Reddy’s Laboratories Ltd. (collectively “Dr. Reddy’s”); (ii) Lupin Limited and Lupin Pharmaceuticals Inc. (collectively “Lupin”); (iii) Mylan Pharmaceuticals Inc., Mylan Laboratories Limited, and Mylan Inc. (collectively “Mylan”); and (iv) Actavis FL and Actavis Pharma, Inc. (collectively “Actavis Pharma”). The cases arise from Paragraph IV Patent Certification notice letters from each of Dr. Reddy’s, Lupin, Mylan and Actavis Pharma advising each had filed an ANDA, with the FDA seeking approval to market generic versions of VIMOVO before the expiration of the patents-in-suit.
Patent litigation is currently pending in the United States District Court for the Eastern District of Texas against Par Pharmaceutical and in the United States District Court for the District of New Jersey against Par Pharmaceutical and against Lupin, who are each intending to market generic versions of RAVICTI prior to the expiration of certain of our patents listed in the Orange Book. These cases are collectively known as the RAVICTI cases and arise from Paragraph IV Patent Certification notice letters from each of Par Pharmaceutical and Lupin advising each had filed an ANDA with the FDA seeking approval to market a generic version of RAVICTI before the expiration of the patents-in-suit.
If we are unsuccessful in any of the VIMOVO cases or PENNSAID 2% cases, we will likely face generic competition with respect to VIMOVO and/or PENNSAID 2% and sales of VIMOVO and/or PENNSAID 2% will be substantially harmed. If we are unsuccessful in any of the RAVICTI cases, RAVICTI would likely face generic competition in the United States when its orphan exclusivity expires (currently scheduled to occur in February 2020), and its sales would likely materially decline.
ACTIMMUNE is the only medicine currently approved by the FDA specifically for the treatment of CGD and SMO. While there are additional or alternative approaches used to treat patients with CGD and SMO, there are currently no medicines on the market that compete directly with ACTIMMUNE. A widely accepted protocol to treat CGD in the United States is the use of concomitant “triple prophylactic therapy” comprising ACTIMMUNE, an oral antibiotic agent and an oral antifungal agent. However, the FDA-approved labeling for
77
ACTIMMUNE does not discuss this “triple prophylactic therapy,” and physicians may choose to prescribe one or both of the other modalities in the absence of ACTIMMUNE. Because of the immediate and life-threatening nature of SMO, the preferred treatment option for SMO is often to have the patient undergo a bone marrow transplant which, if successful, will likely obviate the need for further use of ACTIMMUNE in that patient. Likewise, the use of bone marrow transplants in the treatment of patients with CGD is becoming more prevalent, which could have a material adverse effect on sales of ACTIMMUNE and its profitability. We are aware of a number of research programs investigating the potential of gene therapy as a possible cure for CGD. Additionally, other companies may be pursuing the development of medicines and treatments that target the same diseases and conditions which ACTIMMUNE is currently approved to treat. As a result, it is possible that our competitors may develop new medicines that manage CGD or SMO more effectively, cost less or possibly even cure CGD or SMO. In addition, U.S. healthcare legislation passed in March 2010 authorized the FDA to approve biological products, known as biosimilars, that are similar to or interchangeable with previously approved biological products, like ACTIMMUNE, based upon potentially abbreviated data packages. Biosimilars are likely to be sold at substantially lower prices than branded medicines because the biosimilar manufacturer would not have to recoup the research and development and marketing costs associated with the branded medicine. Though we are not currently aware of any biosimilar under development, the development and commercialization of any competing medicines or the discovery of any new alternative treatment for CGD or SMO could have a material adverse effect on sales of ACTIMMUNE and its profitability.
BUPHENYL’s composition of matter patent protection and orphan drug exclusivity have expired. Because BUPHENYL has no regulatory exclusivity or listed patents, there is nothing to prevent a competitor from submitting an ANDA for a generic version of BUPHENYL and receiving FDA approval. In November 2011, Ampolgen Pharmaceuticals, LLC received FDA approval for a generic version of NaPBA tablets, which may compete with RAVICTI and BUPHENYL in treating UCD. In March 2013, SigmaPharm Laboratories, LLC received FDA approval for a generic version of NaPBA powder, which competes with BUPHENYL and may compete with RAVICTI in treating UCD. In July 2013, Lucane Pharma (“Lucane”), received marketing approval from the EMA for taste-masked NaPBA and has announced a distribution partnership in Canada. In January 2015, Lucane announced it had received marketing approval for its taste-masked NaPBA in Canada. We believe Lucane is also seeking approval via an ANDA in the United States. If this ANDA is approved, this formulation may compete with RAVICTI and BUPHENYL in treating UCD in the United States. Generic versions of BUPHENYL to date have been priced at a discount relative to BUPHENYL or RAVICTI, and physicians, patients, or payers may decide that this less expensive alternative is preferable to BUPHENYL and RAVICTI. If this occurs, sales of BUPHENYL and/or RAVICTI could be materially reduced, but we would nevertheless be required to make royalty payments to Ucyclyd Pharma, Inc. (“Ucyclyd”) and another external party, at the same royalty rates. While Ucyclyd and its affiliates are generally contractually prohibited from developing or commercializing new medicines, anywhere in the world, for the treatment of UCD or hepatic encephalopathy (“HE”), which are chemically similar to RAVICTI, they may still develop and commercialize medicines that compete with RAVICTI. For example, medicines approved for indications other than UCD and HE may still compete with RAVICTI if physicians prescribe such medicines off-label for UCD or HE. We are also aware that Orphan Europe SARL (“Orphan Europe”) is conducting a clinical trial of carglumic acid to treat some of the UCD enzyme deficiencies for which RAVICTI was approved. Promethera Biosciences SA has successfully completed Phase I/II trials of its cell-based therapy for the treatment of UCD and plans to conduct a Phase IIb/III clinical trial. Carglumic acid is approved for maintenance therapy for chronic hyperammonemia and to treat hyperammonenic crises in Nacetylglutamate synthase deficiency, a rare UCD subtype, and is sold under the name Carbaglu. If the results of this trial are successful and Orphan Europe is able to complete development and obtain approval of Carbaglu to treat additional UCD enzyme deficiencies, RAVICTI would face additional competition from this compound.
The availability and price of our competitors’ medicines could limit the demand, and the price we are able to charge, for our medicines. We will not successfully execute on our business objectives if the market acceptance of our medicines is inhibited by price competition, if physicians are reluctant to switch from existing medicines to our medicines, or if physicians switch to other new medicines or choose to reserve our medicines for use in limited patient populations.
78
In addition, established pharmaceutical companies may invest heavily to accelerate discovery and development of novel compounds or to acquire novel compounds that could make our medicines obsolete. Our ability to compete successfully with these companies and other potential competitors will depend largely on our ability to leverage our experience in clinical, regulatory and commercial development to:
| • | develop and acquire medicines that are superior to other medicines in the market; |
| • | attract qualified clinical, regulatory, and sales and marketing personnel; |
| • | obtain patent and/or other proprietary protection for our medicines and technologies; |
| • | obtain required regulatory approvals; and |
| • | successfully collaborate with pharmaceutical companies in the discovery, development and commercialization of new medicine candidates. |
If we are unable to maintain or realize the benefits of orphan drug exclusivity, we may face increased competition with respect to certain of our medicines.
Under the Orphan Drug Act of 1983, the FDA may designate a medicine as an orphan drug if it is a drug intended to treat a rare disease or condition affecting fewer than 200,000 people in the United States. A company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug for a period of seven years from the date of its approval. RAVICTI, KRYSTEXXA, PROSCYSBI have been granted orphan drug exclusivity by the FDA, which we expect will provide orphan drug marketing exclusivity in the United States until May 2020, February 2018 and December 2020, respectively, with exclusivity for PROCYSBI extending to 2022 for patients ages two to six years. However, despite orphan drug exclusivity, the FDA can still approve another drug containing the same active ingredient and used for the same orphan indication if it determines that a subsequent drug is safer, more effective or makes a major contribution to patient care, and orphan exclusivity can be lost if the orphan drug manufacturer is unable to ensure that a sufficient quantity of the orphan drug is available to meet the needs of patients with the rare disease or condition. Orphan drug exclusivity may also be lost if the FDA later determines that the initial request for designation was materially defective. In addition, orphan drug exclusivity does not prevent the FDA from approving competing drugs for the same or similar indication containing a different active ingredient. If orphan drug exclusivity is lost and we were unable to successfully enforce any remaining patents covering RAVICTI, KRYSTEXXA or PROSCYSBI, we could be subject to generic competition and revenues from RAVICTI, KRYSTEXXA or PROCYSBI could decrease materially. In addition, if a subsequent drug is approved for marketing for the same or a similar indication as RAVICTI, KRYSTEXXA or PROCYSBI despite orphan drug exclusivity, we may face increased competition and lose market share with respect to these medicines. KRYSTEXXA does not have orphan drug exclusivity in the EU or other regions of the world. RAVICTI will benefit from a period of 10 years of orphan market exclusivity in the EU, concurrently applied to each of the approved six sub-types of the UCDs. This will run concurrently with its marketing exclusivity status. PROCYSBI received marketing authorization in September 2013 from the European Commission (“EC”), for marketing in the EU as an orphan medicinal product for the management of proven nephropathic cystinosis. PROCYSBI received seven years of market exclusivity, through 2020 for patients six years and older as an orphan drug in the United States and ten years of market exclusivity, through 2023, as an orphan drug in Europe. QUINSAIR received 10 years of market exclusivity in the EU, beginning with its March 2015 marketing authorization. Orphan market exclusivity may be reduced to six years in the EU if the orphan drug designation criteria are no longer met after five years, including where it is shown that the product is sufficiently profitable. As in the United States, loss of orphan marketing exclusivity in the EU may result in early generic competition, which could substantially reduce our revenues from EU sales of these medicines.
79
Our business operations may subject us to numerous commercial disputes, claims and/or lawsuits and such litigation may be costly and time-consuming and could materially and adversely impact our financial position and results of operations.
Operating in the pharmaceutical industry, particularly the commercialization of medicines, involves numerous commercial relationships, complex contractual arrangements, uncertain intellectual property rights, potential product liability and other aspects that create heightened risks of disputes, claims and lawsuits. In particular, we may face claims related to the safety of our medicines, intellectual property matters, employment matters, tax matters, commercial disputes, competition, sales and marketing practices, environmental matters, personal injury, insurance coverage and acquisition or divestiture-related matters. For example, the active ingredient in QUINSAIR, levofloxacin, is currently subject to several product liability claims. Any commercial dispute, claim or lawsuit may divert management’s attention away from our business, we may incur significant expenses in addressing or defending any commercial dispute, claim or lawsuit, and we may be required to pay damage awards or settlements or become subject to equitable remedies that could adversely affect our operations and financial results.
We are currently in litigation with multiple generic drug manufacturers regarding intellectual property infringement. For example, we are currently involved in Hatch Waxman litigation with generic drug manufacturers related to VIMOVO, PENNSAID 2% and RAVICTI.
Similarly, from time to time we are involved in disputes with distributors, PBMs and licensing partners regarding our rights and performance of obligations under contractual arrangements. For example, we were recently in litigation with Express Scripts, Inc. (“Express Scripts”) related to alleged breach of contract claims and in which Express Scripts was seeking payment for rebates relating to DUEXIS, RAYOS and VIMOVO. We counterclaimed against Express Scripts, contesting the amount owed and contending Express Scripts had breached the rebate agreement. In September 2016, we entered into a settlement agreement and mutual release with Express Scripts pursuant to which we and Express Scripts were released from any and all claims relating to the litigation without admitting any fault or wrongdoing and we agreed to pay Express Scripts $65 million.
Litigation related to these disputes may be costly and time-consuming and could materially and adversely impact our financial position and results of operations if resolved against us.
On June 12, 2014, Hyperion acquired Andromeda Biotech Ltd (“Andromeda”), an Israeli company developing DiaPep277 for the treatment of recent onset Type 1 diabetes, from Clal Biotechnology Industries Ltd. (“CBI”). On September 8, 2014, Hyperion announced the termination of further development of DiaPep277 beyond completion of the ongoing clinical trial as a result of evidence Hyperion uncovered that certain employees of Andromeda engaged in serious misconduct that compromised clinical trial results. Hyperion subsequently terminated the Andromeda employees involved in the misconduct and became involved in a legal dispute with CBI related to Andromeda. On February 16, 2015, Hyperion reached a settlement agreement with CBI and Yeda Research and Development Company Ltd. (“Yeda”) the company from which Andromeda licenses the underlying DiaPep277 technology, to resolve DiaPep277-related claims against one another, and Hyperion granted CBI an option to acquire all of the outstanding stock of Andromeda, or if CBI does not exercise the option, CBI agreed to pay Hyperion $400,000. On September 30, 2015, which was the end of the option exercise period, CBI chose not to exercise its option to acquire all of the outstanding stock of Andromeda. In connection with the settlement agreement, the parties appointed a steering committee to oversee the completion of the clinical trial of DiaPep277 until September 30, 2015, with representatives of CBI and Yeda and a non-voting member appointed by Hyperion. Also on February 16, 2015, Hyperion entered into a release with Evotec International GmbH (“Evotec”) pursuant to which Evotec released its previously asserted claims that it was entitled to a milestone payment from Hyperion in connection with Hyperion’s acquisition of Andromeda and that it had suffered harm from recent incidents in relation to DiaPep277 in exchange for a payment of $500,000 from Hyperion. In connection with the settlement agreement, CBI transferred to Hyperion beneficial ownership of 96,612 shares of Hyperion common stock. The voluntary liquidation process of Andromeda was approved by
80
the board of its immediate parent Horizon Pharma Israel Holding Corp. Limited in December 2015. In June 2016, the withholding tax certificate was provided to CBI and in July 2016, CBI completed the transfer of shares to Hyperion and made the $400,000 cash payment.
Although the Andromeda release agreements resolved the disputes among the parties relating to DiaPep277, we cannot be certain that additional legal disputes will not arise with respect to Andromeda, including in connection with the completed Phase 3 clinical trial of DiaPep277, the termination of DiaPep277 development by us and the return of related intellectual property to Yeda following CBI’s decision to not exercise its option. Further, under the terms of the release agreement, Hyperion agreed to retain certain liabilities relating to its ownership of Andromeda, including any liability related to or based on the misconduct of certain former Andromeda employees that led to its decision to terminate further development of DiaPep277. In addition to these potential liabilities, we may incur currently unknown liabilities related to Hyperion’s acquisition of Andromeda. Any such potential legal dispute could lead to costly litigation, divert management’s attention from our core business and harm our business.
A variety of risks associated with operating our business and marketing our medicines internationally could materially adversely affect our business.
In addition to our U.S. operations, we have operations in Ireland, Bermuda, the Grand Duchy of Luxembourg (“Luxembourg”), Switzerland, Germany, Canada and in Israel (through Andromeda). In addition to its U.S. operations, Raptor currently has operations in the Netherlands, France, Germany and the Grand Cayman Islands. Moreover, LODOTRA is currently being marketed in a limited number of countries outside the United States, and Mundipharma is in the process of obtaining pricing and reimbursement approval for, and preparing to market, LODOTRA in other European countries, as well as in certain Asian, Latin American, Middle Eastern and African countries. Also, Grünenthal S.A. is in the registration process for the commercialization of DUEXIS in Latin America. BUPHENYL is currently marketed in various territories outside the United States by third-party distributors. RAVICTI received marketing approval in the EU in November 2015 and we plan to begin commercializing RAVICTI in Europe in 2017. PROCYSBI received marketing authorization from the EMA in September 2013 and is marketed in various countries within the European Economic Area. QUINSAIR received marketing authorization from the EMA in March 2015 and is currently marketed in Germany and Denmark. QUINSAIR received marketing authorization from HC in June 2015 and Raptor anticipates launching in Canada in the fourth quarter of 2016. We face risks associated with our international operations, including possible
81
unfavorable regulatory, pricing and reimbursement, political, tax and labor conditions, which could harm our business. We are subject to numerous risks associated with international business activities, including:
| • | compliance with differing or unexpected regulatory requirements for our medicines; |
| • | compliance with Irish laws and the maintenance of our Irish tax residency with respect to our overall corporate structure and administrative operations, including the need to generally hold meetings of our board of directors and make decisions in Ireland, which may make certain corporate actions more cumbersome, costly and time-consuming; |
| • | difficulties in staffing and managing foreign operations; |
| • | in certain circumstances, including with respect to the commercialization of LODOTRA in Europe and certain Asian, Latin American, Middle Eastern and African countries, commercialization of BUPHENYL in select countries throughout Europe, the Middle East, and the Asia-Pacific region, commercialization of RAVICTI in select countries throughout Europe and commercialization of DUEXIS in Latin America, increased dependence on the commercialization efforts and regulatory compliance of third-party distributors or strategic partners; |
| • | compliance with German laws with respect to our Horizon Pharma GmbH subsidiary through which Horizon Pharma Switzerland GmbH conducts most of its European operations; |
| • | foreign government taxes, regulations and permit requirements; |
| • | U.S. and foreign government tariffs, trade restrictions, price and exchange controls and other regulatory requirements; |
| • | anti-corruption laws, including the Foreign Corrupt Practices Act (the “FCPA”); |
| • | economic weakness, including inflation, natural disasters, war, events of terrorism or political instability in particular foreign countries; |
| • | fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenues, and other obligations related to doing business in another country; |
| • | compliance with tax, employment, immigration and labor laws, regulations and restrictions for employees living or traveling abroad; |
| • | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| • | changes in diplomatic and trade relationships; and |
| • | challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States. |
Our business activities outside of the United States are subject to the FCPA and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate, including the United Kingdom’s Bribery Act 2010 (the “U.K. Bribery Act”). The FCPA and similar anti-corruption laws generally prohibit offering,
82
promising, giving, or authorizing others to give anything of value, either directly or indirectly, to non-U.S. government officials in order to improperly influence any act or decision, secure any other improper advantage, or obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the company and to devise and maintain an adequate system of internal accounting controls. The U.K. Bribery Act prohibits giving, offering, or promising bribes to any person, including non-U.K. government officials and private persons, as well as requesting, agreeing to receive, or accepting bribes from any person. In addition, under the U.K. Bribery Act, companies which carry on a business or part of a business in the United Kingdom (“U.K.”) may be held liable for bribes given, offered or promised to any person, including non-U.K. government officials and private persons, by employees and persons associated with the company in order to obtain or retain business or a business advantage for the company. Liability is strict, with no element of a corrupt state of mind, but a defense of having in place adequate procedures designed to prevent bribery is available. Furthermore, under the U.K. Bribery Act there is no exception for facilitation payments. As described above, our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, any dealings with these prescribers and purchasers may be subject to regulation under the FCPA. Recently the SEC and the U.S. Department of Justice have increased their FCPA enforcement activities with respect to pharmaceutical companies. In addition, under the Dodd-Frank Wall Street Reform and Consumer Protection Act, private individuals who report to the SEC original information that leads to successful enforcement actions may be eligible for a monetary award. We are engaged in ongoing efforts that are designed to ensure our compliance with these laws, including due diligence, training, policies, procedures and internal controls. However, there is no certainty that all employees and third-party business partners (including our distributors, wholesalers, agents, contractors, and other partners) will comply with anti-bribery laws. In particular, we do not control the actions of manufacturers and other third-party agents, although we may be liable for their actions. Violation of these laws may result in civil or criminal sanctions, which could include monetary fines, criminal penalties, and disgorgement of past profits, which could have a material adverse impact on our business and financial condition.
These and other risks associated with our international operations may materially adversely affect our business, financial condition and results of operations.
If we fail to develop or acquire other medicine candidates or medicines, our business and prospects would be limited.
A key element of our strategy is to develop or acquire and commercialize a portfolio of other medicines or medicine candidates in addition to our current medicines, through business or medicine acquisitions. Because we do not engage in proprietary drug discovery, the success of this strategy depends in large part upon the combination of our regulatory, development and commercial capabilities and expertise and our ability to identify, select and acquire approved or clinically enabled medicine candidates for therapeutic indications that complement or augment our current medicines, or that otherwise fit into our development or strategic plans on terms that are acceptable to us. Identifying, selecting and acquiring promising medicines or medicine candidates requires substantial technical, financial and human resources expertise. Efforts to do so may not result in the actual acquisition or license of a particular medicine or medicine candidate, potentially resulting in a diversion of our management’s time and the expenditure of our resources with no resulting benefit. If we are unable to identify, select and acquire suitable medicines or medicine candidates from third parties or acquire businesses at valuations and on other terms acceptable to us, or if we are unable to raise capital required to acquire businesses or new medicines, our business and prospects will be limited.
Moreover, any medicine candidate we acquire may require additional, time-consuming development or regulatory efforts prior to commercial sale or prior to expansion into other indications, including preclinical studies if applicable, and extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities. All medicine candidates are prone to the risk of failure that is inherent in pharmaceutical medicine
83
development, including the possibility that the medicine candidate will not be shown to be sufficiently safe and/or effective for approval by regulatory authorities. In addition, we cannot assure you that any such medicines that are approved will be manufactured or produced economically, successfully commercialized or widely accepted in the marketplace or be more effective or desired than other commercially available alternatives.
In addition, if we fail to successfully commercialize and further develop our medicines, there is a greater likelihood that we will fail to successfully develop a pipeline of other medicine candidates to follow our existing medicines or be able to acquire other medicines to expand our existing portfolio, and our business and prospects would be harmed.
Our recent medicine and company acquisitions and any other strategic transactions that we may pursue in the future could have a variety of negative consequences, and we may not realize the benefits of such transactions or attempts to engage in such transactions.
We have recently completed multiple medicine and company acquisitions and our strategy is to engage in additional strategic transactions with third parties, such as acquisitions of companies or divisions of companies and asset purchases of medicines, medicine candidates or technologies that we believe will complement or augment our existing business, including our proposed acquisition of Raptor. We may also consider a variety of other business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and other investments. Any such transaction may require us to incur non-recurring and other charges, increase our near and long-term expenditures, pose significant integration challenges, create additional tax, legal, accounting and operational complexities in our business, require additional expertise, result in dilution to our existing shareholders and disrupt our management and business, which could harm our operations and financial results. For example, in connection with our acquisition of the U.S. rights to VIMOVO, we assumed primary responsibility for the existing patent infringement litigation with respect to VIMOVO, and have also agreed to reimburse certain legal expenses of Pozen Inc., who subsequently entered into a business combination with Tribute Pharmaceuticals Canada Inc. to become known as Aralez Pharmaceuticals Inc. (“Aralez”) with respect to its continued involvement in such litigation. We also assumed responsibility for the existing patent infringement litigation with respect to RAVICTI upon the closing of the acquisition of Hyperion and have assumed responsibility for completing post-marketing clinical trials of RAVICTI that are required by the FDA and are ongoing. We expect that the RAVICTI litigation will result in substantial on-going expenses and potential distractions to our management team. Following the Raptor Acquisition, we will assume Raptor’s post-marketing clinical study obligations in marketing authorization application (the “MAA”) for QUINSAIR. In addition, we will be subject to the contractual obligations under Raptor’s agreements with Tripex Pharmaceuticals, LLC (“Tripex”) and PARI related to QUINSAIR. Under the agreement with Tripex, we are required to pursue commercially reasonable efforts to initiate, and subsequently to complete, an additional clinical trial of QUINSAIR in a non-cystic-fibrosis patient population within a specified period of time and an obligation to progress toward filing an NDA for approval of QUINSAIR in the United States in all or part of the cystic fibrosis patient population. These obligations are subject to certain exceptions due to, for example, manufacturing delays not under our control, or delays caused by the FDA. If we fail to properly exercise such efforts to initiate and complete an appropriate clinical trial, or fail to file an NDA for U.S. approval in the cystic fibrosis patient population, during the time periods specified in the asset purchase agreement, we may be subject to various claims by Tripex and parties affiliated with Tripex. In addition, if we do not spend a minimum amount on QUINSAIR development in each of the three years following our acquisition of Raptor, we may also be obligated to pre-pay a milestone payment related to initiating a clinical trial for QUINSAIR in a non-cystic-fibrosis indication. Under the license agreement with PARI, we are required to comply with diligence milestones related to development and commercialization of QUINSAIR in the United States, to spend a specified minimum amount per year on development activities in the United States until filing of the NDA for QUINSAIR in the United States. If we do not comply with these obligations, our licenses to certain intellectual property related to QUINSAIR may become non-exclusive in the United States or could be terminated. We will also be subject to contractual obligations under Raptor’s license agreements with the University of California, San Diego (“UCSD”) with respect to PROCYSBI, including diligence obligations to develop PROCYSBI for the treatment
84
of non-alcoholic steatohepatitis (“NASH”) and Huntington’s disease, with which Raptor is currently not in compliance. To the extent that we fail to perform the diligence obligations under the agreement, UCSD may, with respect to such indication, terminate the license or otherwise cause the license to become non-exclusive. If one or more of these licenses was terminated, we would have no further right to use of exploit the related intellectual property, which would limit our ability to develop PROCYSBI or QUINSAIR in other indications, and could impact our ability to continue commercializing PROCYSBI or QUINSAIR in their approved indications.
We face significant competition in seeking appropriate strategic transaction opportunities and the negotiation process for any strategic transaction can be time-consuming and complex. In addition, we may not be successful in our efforts to engage in certain strategic transactions because our financial resources may be insufficient and/or third parties may not view our commercial and development capabilities as being adequate. We may not be able to expand our business or realize our strategic goals if we do not have sufficient funding or cannot borrow or raise additional capital. There is no assurance that following any of our recent acquisition transactions or any other strategic transaction, we will achieve the anticipated revenues, net income, tax or other benefits that we believe justify such transactions. In addition, any failures or delays in entering into strategic transactions anticipated by analysts or the investment community could seriously harm our consolidated business, financial condition, results of operations or cash flow.
Our parent company may not be able to successfully maintain its current advantageous tax status and resulting tax rates, which could adversely affect our business and financial condition, results of operations and growth prospects.
Our parent company is incorporated in Ireland and maintains subsidiaries in multiple jurisdictions, including Ireland, the U.K, the United States, Switzerland, Luxembourg, Germany, Canada and Bermuda. Prior to our merger transaction with Vidara (the “Vidara Merger”), Vidara was able to achieve a favorable tax rate through the performance of certain functions and ownership of certain assets in tax-efficient jurisdictions, including Ireland and Bermuda, together with intra-group service and transfer pricing agreements, each on an arm’s length basis. We are continuing a substantially similar structure and arrangements. Taxing authorities, such as the U.S. Internal Revenue Service (the “IRS”), actively audit and otherwise challenge these types of arrangements, and have done so in the pharmaceutical industry. We expect that these challenges will continue as a result of the recent increase in scrutiny and political attention on corporate tax structures. The IRS may challenge our structure and transfer pricing arrangements through an audit or lawsuit. Responding to or defending such a challenge could be expensive and consume time and other resources, and divert management’s time and focus from operating our business. We cannot predict whether taxing authorities will conduct an audit or file a lawsuit challenging this structure, the cost involved in responding to any such audit or lawsuit, or the outcome. If we are unsuccessful in defending such a challenge, we may be required to pay taxes for prior periods, interest, fines or penalties, and may be obligated to pay increased taxes in the future, any of which could require us to reduce our operating expenses, decrease efforts in support of our medicines or seek to raise additional funds, all of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
The IRS may not agree with our conclusion that our parent company should be treated as a foreign corporation for U.S. federal income tax purposes following the combination of the businesses of Horizon Pharma, Inc. and Vidara Therapeutics International plc.
Although our parent company is incorporated in Ireland, the IRS may assert that it should be treated as a U.S. corporation (and, therefore, a U.S. tax resident) for U.S. federal income tax purposes pursuant to Section 7874 of the Internal Revenue Code of 1986, as amended (the “Code”). A corporation is generally considered a tax resident in the jurisdiction of its organization or incorporation for U.S. federal income tax purposes. Because our parent company is an Irish incorporated entity, it would generally be classified as a foreign corporation (and, therefore, a non-U.S. tax resident) under these rules. Section 7874 provides an exception pursuant to which a foreign incorporated entity may, in certain circumstances, be treated as a U.S. corporation for U.S. federal income tax purposes.
85
Under Section 7874, a foreign corporation will be treated as a U.S. corporation for U.S. federal tax purposes if, due to an acquisition of a U.S. corporation, at least 80 percent of its stock (by vote or value) is held by former stockholders of the acquired U.S. corporation. We believe that we should be treated as a foreign corporation because the former stockholders of HPI owned (within the meaning of Section 7874) less than 80 percent (by both vote and value) of the combined entity’s stock immediately after the Vidara Merger. However, there can be no assurance that there will not exist in the future a subsequent change in the facts or in law which might cause our parent company to be treated as a domestic corporation for U.S. federal income tax purposes, including with retroactive effect.
Further, there can be no assurance that the IRS will agree with the position that the ownership test was satisfied. There is limited guidance regarding the application of Section 7874 of the Code, including with respect to the provisions regarding the application of the ownership test. If our parent company were unable to be treated as a foreign corporation for U.S. federal income tax purposes, one of our significant strategic reasons for completing the Vidara Merger would be nullified and we may not be able to recoup the significant investment in completing the transaction.
Future changes to U.S. and non-U.S. tax laws could materially adversely affect our company.
Under current law, we expect our parent company to be treated as a foreign corporation for U.S. federal income tax purposes. However, changes to the rules in Section 7874 of the Code or regulations promulgated thereunder or other guidance issued by the U.S. Department of the Treasury (the “U.S. Treasury”) or the IRS could adversely affect our parent company’s status as a foreign corporation for U.S. federal income tax purposes, and any such changes could have prospective or retroactive application. If our parent company is treated as a domestic corporation, more of our income will be taxed by the United States which may substantially increase our effective tax rate.
On April 4, 2016, the U.S. Treasury and the IRS issued temporary regulations that expand the scope of transactions subject to the rules designed to eliminate the U.S. tax benefits of inversions. Under the temporary regulations, the former stockholders of U.S. corporations acquired by a foreign corporation within 36 months of the signing date of the last such acquisition are aggregated for the purpose of determining whether the foreign corporation will be treated as a domestic corporation for U.S. federal tax purposes because at least 80 percent of the stock of the foreign corporation is held by former stockholders of a U.S. corporation. The requirement to aggregate the stockholders in such acquisitions for the purpose of determining whether the 80 percent threshold is met may limit our ability to use our stock to acquire U.S. corporations or their assets in the future.
The U.S. Treasury and the IRS also issued proposed regulations on April 4, 2016 that address whether an interest in a related corporation is debt or equity. The proposed regulations would treat certain inter-company debt issued on or after that date as equity including, subject to certain exceptions, inter-company debt issued in certain distributions, acquisitions of related party stock and asset reorganizations. As drafted, the proposed regulations would limit the ability of our U.S. group to deduct interest on such new inter-company debt. The proposed regulations could also result in recharacterization of inter-company debt to equity for inter-company debt incurred to provide funding for an acquisition by the U.S. group if, and to the extent of, certain cash or property transfers by our U.S. group to the foreign affiliates within 36 months before or after these inter-company borrowings. These limitations could result in more of our future income being taxed by the United States and thereby increase our effective tax rate.
In July 2015, the International Tax Bipartisan Tax Working Group of the United States Senate Committee on Finance (the “Finance Committee”) issued its report on international tax reform. The Finance Committee’s co-chairs concluded that it will be necessary to limit earnings stripping by foreign multinationals through interest deductions on inter-company debt in order to eliminate a competitive advantage that foreign multinationals would otherwise have over domestic multinational companies. This and other international tax reforms proposed by the Finance Committee could result in more of our income being taxed by the United States and thereby increase our effective tax rate.
86
In addition, the Organization for Economic Co-operation and Development released its Base Erosion and Profit Shifting project final report on October 5, 2015. This report provides the basis for international standards for corporate taxation that are designed to prevent, among other things, the artificial shifting of income to tax havens and low-tax jurisdictions, the erosion of the tax base through interest deductions on inter-company debt and the artificial avoidance of permanent establishments (i.e., tax nexus with a jurisdiction). Legislation to adopt these standards has been enacted or is currently under consideration in a number of jurisdictions. As a result, our income may be taxed in jurisdictions where it is not currently taxed and at higher rates of tax than it is currently taxed, which may substantially increase our effective tax rate.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceuticals industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management, sales and marketing and scientific and medical personnel, including our executive committee composed of our Chairman, President and Chief Executive Officer, Timothy P. Walbert; our Executive Vice President, Chief Business Officer, Robert F. Carey; our Executive Vice President, Chief Financial Officer, Paul W. Hoelscher; our Executive Vice President, Company Secretary and Managing Director, Ireland, David G. Kelly; our Executive Vice President, Chief Operating Officer, Barry J. Moze; our Executive Vice President, Research and Development and Chief Medical Officer, Jeffrey W. Sherman, M.D., FACP; our Executive Vice President, General Counsel, Brian K. Beeler; our Executive Vice President, Strategy and Investor Relations, John B. Thomas; our Executive Vice President, Global Orphan Business Unit, Primary Care and International Operations, George Hampton; our Senior Vice President, Commercial Operations, Timothy J. Ackerman; and our Senior Vice President, Corporate Communications, Geoffrey M. Curtis. In order to retain valuable employees at our company, in addition to salary and cash incentives, we provide performance stock units (“PSUs”), and stock options and restricted stock units that vest over time. The value to employees of PSUs, stock options and restricted stock units will be significantly affected by movements in our share price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies.
Despite our efforts to retain valuable employees, members of our management, sales and marketing, regulatory affairs, clinical development, medical affairs and development teams may terminate their employment with us on short notice. Although we have written employment arrangements with all of our employees, these employment arrangements generally provide for at-will employment, which means that our employees can leave our employment at any time, with or without notice. The loss of the services of any of our executive officers or other key employees and our inability to find suitable replacements could potentially harm our business, financial condition and prospects. We do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level, and senior managers as well as junior, mid-level, and senior sales and marketing and scientific and medical personnel.
Many of the other biotechnology and pharmaceutical companies with whom we compete for qualified personnel have greater financial and other resources, different risk profiles and longer histories in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than that which we have to offer. If we are unable to continue to attract and retain high quality personnel, the rate and success at which we can develop and commercialize medicines and medicine candidates will be limited.
87
We are, with respect to our current medicines, and will be, with respect to any other medicine or medicine candidate for which we obtain FDA or EMA approval or which we acquire, subject to ongoing FDA or the EMA obligations and continued regulatory review, which may result in significant additional expense. Additionally, any other medicine candidate, if approved by the FDA or the EMA, could be subject to labeling and other restrictions and market withdrawal, and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our medicines.
Any regulatory approvals that we obtain for our medicine candidates may also be subject to limitations on the approved indicated uses for which the medicine may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials and surveillance to monitor the safety and efficacy of the medicine candidate. In addition, with respect to our current FDA-approved medicines (and with respect to our medicine candidates, if approved), the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the medicine are subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs, GCPs, international conference on harmonization regulations (“ICH regulations”), and GLPs, which are regulations and guidelines enforced by the FDA for all of our medicines in clinical development, for any clinical trials that we conduct post-approval. In connection with our November 2013 acquisition of the U.S. rights to VIMOVO, we assumed responsibility for completing an ongoing Pediatric Research Equity Act post-marketing requirement study in children 12 years to 16 years and 11 months of age with juvenile rheumatoid arthritis. This report was submitted to the FDA in December 2015. With respect to RAVICTI, the FDA imposed several post-marketing requirements and a post-marketing commitment, which include remaining obligations to conduct studies in UCD patients during the first two months of life and from two months to two years of age, including a study of the pharmacokinetics in both age groups, and a randomized study to determine the safety and efficacy in UCD patients who are treatment naïve to phenylbutyrate treatment. Although we are committed to carrying out these commitments, there are challenges in conducting studies in pediatric patients including availability of study sites, patients, and obtaining parental informed consent. On June 29, 2016, we submitted an sNDA with the FDA for RAVICTI to expand the age range for chronic management of UCDs from two years of age and older to two months of age and older. Subject to positive data from on-going studies, we have targeted an sNDA submission in the first quarter of 2018 in relation to UCD patients during the first two months of life. In connection with our acquisition of Crealta Holdings LLC (“Crealta”) in January 2016, we assumed responsibility for an observational study related to KRYSTEXXA. Thus far in this study there have been no new safety signals and the reported safety results parallel those in the KRYSTEXXA product label. We are continuing to screen and enroll patients in the near term. With respect to QUINSAIR following the Raptor Acquisition, we will be required to conduct post-marketing clinical studies in cystic fibrosis patients pursuant to obligations in the MAA for QUINSAIR and submit data to the EMA regularly regarding observed clinical product profile and safety assessment.
In addition, the FDA closely regulates the marketing and promotion of drugs and biologics. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ promotional communications. A significant number of pharmaceutical companies have been the target of inquiries and investigations by various U.S. federal and state regulatory, investigative, prosecutorial and administrative entities in connection with the promotion of medicines for off-label uses and other sales practices. These investigations have alleged violations of various U.S. federal and state laws and regulations, including claims asserting antitrust violations, violations of the Food, Drug and Cosmetic Act, false claims laws, the Prescription Drug Marketing Act, anti-kickback laws, and other alleged violations in connection with the promotion of medicines for unapproved uses, pricing and Medicare and/or Medicaid reimbursement.
88
Later discovery of previously unknown problems with a medicine, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • | restrictions on the marketing or manufacturing of the medicine, withdrawal of the medicine from the market, or voluntary or mandatory medicine recalls; |
| • | fines, warning letters or holds on clinical trials; |
| • | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our strategic partners, or suspension or revocation of medicine license approvals; |
| • | medicine seizure or detention, or refusal to permit the import or export of medicines; and |
| • | injunctions, the imposition of civil or criminal penalties, or exclusion, debarment or suspension from government healthcare programs. |
If we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would have a material adverse effect on our business, results of operations, financial condition and prospects.
Coverage and reimbursement may not be available, or reimbursement may be available at only limited levels, for our medicines, which could make it difficult for us to sell our medicines profitably or to successfully execute planned medicine price increases.
Market acceptance and sales of our medicines will depend in large part on global coverage and reimbursement policies and may be affected by future healthcare reform measures, both in the United States and other key international markets. Successful commercialization of our medicines will depend in part on the availability of governmental and third-party payer reimbursement for the cost of our medicines. Government health administration authorities, private health insurers and other organizations generally provide reimbursement for healthcare. In particular, in the United States, private health insurers and other third-party payers often provide reimbursement for medicines and services based on the level at which the government (through the Medicare or Medicaid programs) provides reimbursement for such treatments. In the United States, the EU and other significant or potentially significant markets for our medicines and medicine candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medicines and services, particularly for new and innovative medicines and therapies, which has resulted in lower average selling prices. Further, the increased scrutiny of prescription drug pricing practices and emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the EU will put additional pressure on medicine pricing, reimbursement and usage, which may adversely affect our medicine sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general. These pressures may create negative reactions to any medicine price increases, or limit the amount by which we may be able to increase our medicine prices, which may adversely affect our medicine sales and results of operations.
Patients are unlikely to use our medicines unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our medicines. Third-party payers may limit coverage to specific medicines on an approved list, also known as a formulary, which might not include all of the FDA-approved medicines for a particular indication. Moreover, a third-party payer’s decision to provide coverage for a medicine does not imply that an adequate reimbursement rate will be approved. Additionally, one third-party payer’s decision to cover a particular medicine does not ensure that other payers will also provide coverage for the medicine, or will provide coverage at an adequate reimbursement rate. Even though we have contracts with some
89
PBMs in the United States, that does not guarantee that they will perform in accordance with the contracts, nor does that preclude them from taking adverse actions against us, which could materially adversely affect our operating results. In addition, the existence of such PBM contracts does not guarantee coverage by such PBM’s contracted health plans or adequate reimbursement to their respective providers for our medicines. For example, two significant PBMs placed DUEXIS and VIMOVO on their exclusion lists beginning in 2015, which has resulted in a loss of coverage for patients whose healthcare plans have adopted these PBM lists. While DUEXIS and VIMOVO were removed from the CVS Caremark 2017 exclusion list, we cannot guarantee that CVS Caremark will not later add these medicines back to its exclusion list or that we will be able to otherwise expand formulary access for DUEXIS and VIMOVO under health plans that contract with CVS Caremark. Also, as noted above, we were recently in a contract and rebate dispute with Express Scripts involving DUEXIS, RAYOS and VIMOVO. In September 2016, we entered into a settlement agreement and mutual release with Express Scripts pursuant to which we and Express Scripts were released from any and all claims relating to the litigation without admitting any fault or wrongdoing and we agreed to pay Express Scripts $65 million. Additional healthcare plan formularies may also exclude our medicines from coverage due to the actions of certain PBMs, future price increases we may implement, our use of the HorizonCares program or any other co-pay programs, or other reasons. If our strategies to mitigate formulary exclusions are not effective, these events may reduce the likelihood that physicians prescribe our medicines and increase the likelihood that prescriptions for our medicines are not filled.
Outside of the United States, the success of our medicines, including BUPHENYL, LODOTRA, and RAVICTI and, following the Raptor Acquisition, PROCYSBI and QUINSAIR, and following the IMUKIN Acquisition, interferon gamma-1b (currently commercialized under the trade names IMUKIN, IMUKINE, IMMUKIN and IMMUKINE), will depend largely on obtaining and maintaining government coverage, because in many countries patients are unlikely to use prescription drugs that are not covered by their government healthcare programs. To date, reimbursement for LODOTRA has been obtained in Germany, Italy, Sweden and Switzerland. Mundipharma is seeking coverage for LODOTRA in a number of countries and currently sells LODOTRA without coverage in a limited number of countries. BUPHENYL is marketed in select countries throughout Europe, the Middle East and the Asia-Pacific region. With respect to RAVICTI, we expect to begin commercializing the medicine in Europe in 2017. PROCYSBI is marketed in select countries in Europe and QUINSAIR was recently launched in certain countries in Europe, but we cannot be certain that existing reimbursement in EU countries will be maintained or that we will be able to secure reimbursement in additional countries. Negotiating coverage and reimbursement with governmental authorities can delay commercialization by 12 months or more. Coverage and reimbursement policies may adversely affect our ability to sell our medicines on a profitable basis. In many international markets, governments control the prices of prescription pharmaceuticals, including through the implementation of reference pricing, price cuts, rebates, revenue-related taxes and profit control, and we expect prices of prescription pharmaceuticals to decline over the life of the medicine or as volumes increase. Many countries in the EU have increased the amount of discounts required on medicines, which we believe has impacted the reimbursement rates and timing to launch for LODOTRA to date, and we expect these discounts to continue as countries attempt to manage healthcare expenditures, especially in light of current economic conditions. As a result of these pricing practices, it may become difficult to achieve or sustain profitability or expected rates of growth in revenue or results of operations. Any shortfalls in revenue could adversely affect our business, financial condition and results of operations.
In light of such policies and the uncertainty surrounding proposed regulations and changes in the coverage and reimbursement policies of governments and third-party payers, we cannot be sure that coverage and reimbursement will be available for any of our medicines in any additional markets or for any other medicine candidates that we may develop. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, our medicines. If coverage and reimbursement are not available or are available only at limited levels, we may not be able to successfully commercialize our medicines.
We expect to experience pricing pressures in connection with the sale of our medicines due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional
90
legislative proposals relating to outcomes and quality. For example, in March 2016, the Centers for Medicare & Medicaid Services (“CMS”) announced a Proposed Rule that would test new payment models for Medicare Part B prescription drugs, and provider services incident to, or otherwise related to, such drugs. Generally, the Proposed Rule includes payment models that are designed on quality and value propositions and include incentives to drive utilization of efficient therapies and make payments based on clinical outcomes. The Proposed Rule greatly differs from the current reimbursement methodology for Medicare Part B drugs. The comment period for the Proposed Rule closed on May 9, 2016. Since the closing of the comment period, the Proposed Rule has been subject to significant and ongoing discussion among stakeholders including industry, payers, healthcare providers and other interested organizations. The Proposed Rule would not take effect until CMS considers the comments received and issues a Final Rule.
There may be additional pressure by payers, healthcare providers, and members of Congress, to use generic drugs that contain the active ingredients found in our medicines or any other medicine candidates that we may develop or acquire. If we fail to successfully secure and maintain coverage and adequate reimbursement for our medicines or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our medicines and expected revenue and profitability which would have a material adverse effect on our business, results of operations, financial condition and prospects.
We may also experience pressure from payers concerning certain promotional approaches that we may implement such as our HorizonCares program or any other co-pay or free medicine programs whereby we assist qualified patients with certain out-of-pocket expenditures for our medicine. If we are unsuccessful with our HorizonCares program or any other co-pay initiatives or free medicine programs, or we alternatively are unable to secure expanded formulary access through additional arrangements with PBMs or other payers, we would be at a competitive disadvantage in terms of pricing versus preferred branded and generic competitors. We may also experience financial pressure in the future which would make it difficult to support investment levels in areas such as managed care contract rebates, HorizonCares and other access tools.
We are subject to federal, state and foreign healthcare laws and regulations and implementation or changes to such healthcare laws and regulations could adversely affect our business and results of operations.
The U.S. and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to regulate and to change the healthcare system in ways that could affect our ability to sell our medicines profitably. In the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs (including a number of proposals pertaining to prescription drugs, specifically), improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
If we are found to be in violation of any of these laws or any other federal or state regulations, we may be subject to civil and/or criminal penalties, damages, fines, exclusion from federal health care programs and the restructuring of our operations. Any of these could have a material adverse effect on our business and financial results. Since many of these laws have not been fully interpreted by the courts, there is an increased risk that we may be found in violation of one or more of their provisions. Any action against us for violation of these laws, even if we ultimately are successful in our defense, will cause us to incur significant legal expenses and divert our management’s attention away from the operation of our business.
We expect that the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively the “ACA”), as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we may receive for any approved medicine. An expansion in the government’s role in the U.S. healthcare industry may cause general downward pressure on the prices of prescription medicines, lower reimbursements for providers using our medicines, reduce medicine utilization and adversely affect our business
91
and results of operations. It is unclear whether and to what extent, if at all, other potential developments resulting from the ACA, such as an increase in the number of people with health insurance and an increased focus on preventive medicine, may provide us with additional revenue to offset the annual excise tax (on certain medicine sales) enacted under the ACA, subject to limited exceptions. It is possible that the tax burden, if ours is not excepted, would adversely affect our financial performance, which in turn could cause the price of our ordinary shares to decline. The ACA, among other things, also established a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50 percent point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Further, certain hospitals and other providers, or covered entities, may access the 340B Drug Pricing Program, administered by the Health Resources and Services Administration (“HRSA”), to obtain discounted prices on “covered outpatient drugs” (prescription drugs and biologics other than vaccines) from drug manufacturers. Generally, manufacturers must offer 340B discounts to these covered entities as a prerequisite to Medicaid reimbursement for the drugs. The discount amount to “patients” (as defined by HRSA) of covered entities is significant. The number of covered entities that may access the 340B prices has been growing and includes certain free-standing cancer hospitals.
Moreover, certain politicians, including presidential candidates, have announced plans to regulate the prices of medicines. The majority of our medicines are purchased by private payers, and we do not believe that any such legislation, if enacted, would have a material effect on us or our business. However, we cannot know what form any such legislation may take or the market’s perception of how such legislation would affect us. Any reduction in reimbursement from government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our current medicines and/or those for which we may receive regulatory approval in the future.
We are subject, directly or indirectly, to federal and state healthcare fraud and abuse and false claims laws and regulations. Prosecutions under such laws have increased in recent years and we may become subject to such litigation. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
In the United States, we are subject directly, or indirectly through our customers, to various state and federal fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act, civil monetary penalty statutes prohibiting beneficiary inducements, and similar state laws, federal and state privacy and security laws, sunshine laws, government price reporting laws, and other fraud laws. These laws may impact, among other things, our current and proposed sales, marketing and educational programs, as well as other possible relationships with customers, pharmacies, physicians, payers, and patients.
Compliance with these laws, including the development of a comprehensive compliance program, is difficult, costly and time consuming. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. These risks may be increased where there are evolving interpretations of applicable regulatory requirements, such as those applicable to manufacturer co-pay initiatives. Pharmaceutical manufacturer co-pay initiatives and free medicine programs are the subject of ongoing litigation (involving other manufacturers and to which we are not a party) and evolving interpretations of applicable regulatory requirements and certain state laws, and any change in the regulatory or enforcement environment regarding such programs could impact our ability to offer such programs. If we are unsuccessful with our HorizonCares programs, any other co-pay initiatives or free medicine programs, we would be at a competitive disadvantage in terms of pricing versus preferred branded and generic competitors, or be subject to significant penalties. We are engaged in various business arrangements with current and potential customers, and we can give no assurance that such arrangements would not be subject to scrutiny under such laws, despite our efforts to properly structure such arrangements. Even if we structure our programs with the intent of compliance with such laws, there can be no certainty that we would not need to defend our business activities against enforcement or
92
litigation. Further, we cannot give any assurances that prior business activities or arrangements of other companies that we acquire will not be scrutinized or subject to enforcement or litigation.
There has also been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The ACA, among other things, imposed new reporting requirements on drug manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in significant civil monetary penalties.
We are unable to predict whether we could be subject to actions under any of these or other healthcare laws, or the impact of such actions. If we are found to be in violation of, or to encourage or assist the violation by third parties of any of the laws described above or other applicable state and federal fraud and abuse laws, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, withdrawal of regulatory approval, imprisonment, exclusion from government healthcare reimbursement programs, contractual damages, reputational harm, diminished profits and future earnings, injunctions and other associated remedies, or private “qui tam” actions brought by individual whistleblowers in the name of the government, and the curtailment or restructuring of our operations, all of which could have a material adverse effect on our business and results of operations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Our medicines or any other medicine candidate that we develop may cause undesirable side effects or have other properties that could delay or prevent regulatory approval or commercialization, result in medicine re-labeling or withdrawal from the market or have a significant impact on customer demand.
Undesirable side effects caused by any medicine candidate that we develop could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, or cause us to evaluate the future of our development programs. In our two Phase 3 clinical trials with DUEXIS, the most commonly reported treatment-emergent adverse events were nausea, dyspepsia, diarrhea, constipation and upper respiratory tract infection. In Phase 3 endoscopic registration clinical trials with VIMOVO, the most commonly reported treatment-emergent adverse events were erosive gastritis, dyspepsia, gastritis, diarrhea, gastric ulcer, upper abdominal pain, nausea and upper respiratory tract infection. The most common side effects observed in pivotal trials for ACTIMMUNE were “flu-like” or constitutional symptoms such as fever, headache, chills, myalgia and fatigue. The most commonly reported treatment-emergent adverse events in the Phase 3 clinical trials with RAYOS/LODOTRA included flare in rheumatoid arthritis related symptoms, abdominal pain, nasopharyngitis, headache, flushing, upper respiratory tract infection, back pain and weight gain. The most common adverse events reported in a Phase 2 clinical trial of PENNSAID 2% were application site reactions, such as dryness, exfoliation, erythema, pruritus, pain, induration, rash and scabbing. With respect to BUPHENYL, the most common side effects are change in the frequency of breathing, lack of or irregular menstruation, lower back, side, or stomach pain, mood or mental changes, muscle pain or twitching, nausea or vomiting, nervousness or restlessness, swelling of the feet or lower legs, unpleasant taste and unusual tiredness or weakness. With respect to RAVICTI, the most common side effects are diarrhea, nausea, decreased appetite, gas, vomiting, high blood levels of ammonia, headache, tiredness and dizziness. With respect to KRYSTEXXA, the most commonly reported serious adverse reactions in the pivotal trial were gout flares, infusion reactions, nausea, contusion or ecchymosis, nasopharyngitis, constipation, chest pain, anaphylaxis, exacerbation of pre-existing congestive heart failure and vomiting. With respect to MIGERGOT, the most commonly reported adverse reactions are ischemia, cyanosis, absence of pulse, cold extremities, gangrene, precordial distress and pain, electrocardiogram change, muscle pain, nausea and vomiting, rectal or anal ulcer, parathesias, numbness weakness, vertigo, localized edemas and itching. With respect to PROCYSBI, the most common side effects include vomiting, nausea, abdominal pain, breath odor, diarrhea, skin odor, fatigue, rash and headache. With respect to QUINSAIR, the most common side effects include itching, wheezing, hives, rash, swelling, pale skin color, fast heartbeat, and faintness. The FDA or other regulatory authorities may also require, or we may undertake, additional clinical trials to support the safety profile of our medicines or medicine candidates.
93
In addition, if we or others identify undesirable side effects caused by our medicines or any other medicine candidate that we may develop that receives marketing approval, or if there is a perception that the medicine is associated with undesirable side effects:
| • | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| • | regulatory authorities may withdraw their approval of the medicine or place restrictions on the way it is prescribed; |
| • | we may be required to change the way the medicine is administered, conduct additional clinical trials or change the labeling of the medicine or implement a risk evaluation and mitigation strategy; and |
| • | we may be subject to increased exposure to product liability and/or personal injury claims. |
If any of these events occurred with respect to our medicines, our ability to generate significant revenues from the sale of these medicines would be significantly harmed.
We rely on third parties to conduct our preclinical and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines or if they experience regulatory compliance issues, we may not be able to obtain regulatory approval for or commercialize our medicine candidates and our business could be substantially harmed.
We have agreements with third-party contract research organizations (“CROs”) to conduct our clinical programs, including those required for post-marketing commitments, and we expect to continue to rely on CROs for the completion of on-going and planned clinical trials. We may also have the need to enter into other such agreements in the future if we were to develop other medicine candidates or conduct clinical trials in additional indications for our existing medicines. In connection with our on-going Phase 3 study to evaluate ACTIMMUNE for the treatment of FA, we are working with the Clinical Trials Coordination Center, an academic research organization (“ARO”) that is part of the Center for Human Experimental Therapeutics at the University of Rochester as well as collaborating with the Friedreich’s Ataxia Research Alliance (“FARA”) and select investigators of FARA’s Collaborative Clinical Research Network in FA. In connection with the investigator-initiated study to evaluate ACTIMMUNE in combination with PD-1/PD-L1 inhibitors in various forms of cancer including advanced urothelial carcinoma (bladder cancer) and renal cell carcinoma, we are collaborating with Fox Chase Cancer Center. In connection with our ongoing study to evaluate RAYOS/LODOTRA on the fatigue experienced by systemic lupus erythematosus patients, we are collaborating with the Alliance for Lupus Research. We rely heavily on these parties for the execution of our clinical studies and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol. We, our CROs and our ARO are required to comply with current GCP or ICH regulations. The FDA enforces these GCP or ICH regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or our CROs or collaborators fail to comply with applicable GCP or ICH regulations, the data generated in our clinical trials may be deemed unreliable and our submission of marketing applications may be delayed or the FDA may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply or complied with GCP or ICH regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations, and may require a large number of test subjects. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, our business may be implicated if any of our CROs or collaborators violates federal or state fraud and abuse or false claims laws and regulations or healthcare privacy and security laws. We must also obtain certain third-party institutional review board (“IRB”) and ethics committee approvals in order to conduct our clinical trials. Delays by IRBs and ethics committees in providing such approvals may delay our clinical trials.
94
If any of our relationships with these third-party CROs or collaborators terminate, we may not be able to enter into similar arrangements on commercially reasonable terms, or at all. If CROs or collaborators do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our medicines and medicine candidates. As a result, our results of operations and the commercial prospects for our medicines and medicine candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Switching or adding additional CROs or collaborators can involve substantial cost and require extensive management time and focus. In addition, there is a natural transition period when a new CRO or collaborator commences work. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs and collaborators, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition or prospects.
Clinical development of drugs and biologics involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing is expensive and can take many years to complete, and its outcome is uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of potential medicine candidates may not be predictive of the results of later-stage clinical trials. Medicine candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical testing. For example, Raptor announced on in September 2015, based on information then available, that it would not advance its program for the treatment of pediatric NASH with PROCYSBI after a Phase 2b trial failed achieve its primary endpoints.
With respect to our on-going Phase 3 clinical trial to evaluate ACTIMMUNE for the treatment of FA, and the investigator-initiated study to evaluate ACTIMMUNE in combination with OPDIVO nivolumab in advanced solid tumors and to the extent that we are required to conduct additional clinical development of any of our existing or later acquired medicines or we conduct clinical development of earlier stage medicine candidates or for other additional indications for RAYOS/LODOTRA, we may experience delays in these clinical trials or investigator-initiated studies. We do not know whether any additional clinical trials will be initiated in the future, begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| • | obtaining regulatory approval to commence a trial; |
| • | reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| • | obtaining IRB or ethics committee approval at each site; |
| • | recruiting suitable patients to participate in a trial; |
| • | having patients complete a trial or return for post-treatment follow-up; |
| • | clinical sites dropping out of a trial; |
95
| • | adding new sites; or |
| • | manufacturing sufficient quantities of medicine candidates for use in clinical trials. |
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the medicine candidate being studied in relation to other available therapies, including any new drugs or biologics that may be approved for the indications we are investigating. Furthermore, we rely and expect to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our future clinical trials and while we have and intend to have agreements governing their committed activities, we will have limited influence over their actual performance.
We could encounter delays if prescribing physicians encounter unresolved ethical issues associated with enrolling patients in clinical trials of our medicine candidates in lieu of prescribing existing treatments that have established safety and efficacy profiles. Further, a clinical trial may be suspended or terminated by us, our collaborators, the FDA or other regulatory authorities due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a medicine candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. If we experience delays in the completion of, or if we terminate, any clinical trial of our medicine candidates, the commercial prospects of our medicine candidates will be harmed, and our ability to generate medicine revenues from any of these medicine candidates will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our medicine development and approval process and jeopardize our ability to commence medicine sales and generate revenues.
Moreover, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of marketing approval of one or more of our medicine candidates.
Any of these occurrences may harm our business, financial condition, results of operations and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our medicine candidates.
Business interruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions. While we carry insurance for certain of these events and have implemented disaster management plans and contingencies, the occurrence of any of these business interruptions could seriously harm our business and financial condition and increase our costs and expenses. We conduct significant management operations at both our global headquarters located in Dublin, Ireland and our U.S. office located in Lake Forest, Illinois. If our Dublin or Lake Forest offices were affected by a natural or man-made disaster or other business interruption, our ability to manage our domestic and foreign operations could be
96
impaired, which could materially and adversely affect our results of operations and financial condition. We currently rely, and intend to rely in the future, on third-party manufacturers and suppliers to produce our medicines and third-party logistics partners to ship our medicines. Our ability to obtain commercial supplies of our medicines could be disrupted and our results of operations and financial condition could be materially and adversely affected if the operations of these third-party suppliers or logistics partners were affected by a man-made or natural disaster or other business interruption. The ultimate impact of such events on us, our significant suppliers and our general infrastructure is unknown.
We are dependent on information technology systems, infrastructure and data, which exposes us to data security risks.
We are dependent upon information technology systems, infrastructure and data, including mobile technologies, to operate our business. The multitude and complexity of our computer systems make them inherently vulnerable to service interruption or destruction, malicious intrusion and random attack. Likewise, data privacy or security breaches by employees or others may pose a risk that sensitive data, including our intellectual property, trade secrets or personal information of our employees, patients, customers or other business partners may be exposed to unauthorized persons or to the public. Cyber-attacks are increasing in their frequency, sophistication and intensity. Cyber-attacks could include the deployment of harmful malware, denial-of-service, social engineering and other means to affect service reliability and threaten data confidentiality, integrity and availability. Our business partners face similar risks and any security breach of their systems could adversely affect our security posture. A security breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could harm our reputation, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to liability under laws and regulations that protect personal data, any of which could disrupt our business and/or result in increased costs or loss of revenue. Moreover, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information, trade secrets or other intellectual property. While we have invested, and continue to invest, in the protection of our data and information technology infrastructure, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our medicines.
We face an inherent risk of product liability claims as a result of the commercial sales of our medicines and the clinical testing of our medicine candidates. For example, we may be sued if any of our medicines or medicine candidates allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the medicine, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our medicines and medicine candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| • | decreased demand for our medicines or medicine candidates that we may develop; |
| • | injury to our reputation; |
97
| • | withdrawal of clinical trial participants; |
| • | initiation of investigations by regulators; |
| • | costs to defend the related litigation; |
| • | a diversion of management’s time and resources; |
| • | substantial monetary awards to trial participants or patients; |
| • | medicine recalls, withdrawals or labeling, marketing or promotional restrictions; |
| • | loss of revenue; |
| • | exhaustion of any available insurance and our capital resources; and |
| • | the inability to commercialize our medicines or medicine candidates. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of medicines we develop. We currently carry product liability insurance covering our clinical studies and commercial medicine sales in the amount of $75 million in the aggregate. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. If we determine that it is prudent to increase our product liability coverage due to the on-going commercialization of our current medicines in the United States, and/or the potential commercial launches of any of our medicines in additional markets or for additional indications, we may be unable to obtain such increased coverage on acceptable terms or at all. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
Our business involves the use of hazardous materials, and we and our third-party manufacturers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our third-party manufacturers’ activities involve the controlled storage, use and disposal of hazardous materials owned by us, including the components of our medicine candidates and other hazardous compounds. We and our manufacturers are subject to federal, state and local as well as foreign laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In the event of an accident, state, federal or foreign authorities may curtail the use of these materials and interrupt our business operations. We do not currently maintain hazardous materials insurance coverage. If we are subject to any liability as a result of our third-party manufacturers’ activities involving hazardous materials, our business and financial condition may be adversely affected. In the future we may seek to establish longer-term third-party manufacturing arrangements, pursuant to which we would seek to obtain contractual indemnification protection from such third-party manufacturers potentially limiting this liability exposure.
98
Our employees, independent contractors, principal investigators, consultants, vendors, distributors and CROs may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, consultants, vendors, distributors and CROs may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or unauthorized activities that violate FDA regulations, including those laws that require the reporting of true, complete and accurate information to the FDA, manufacturing standards, federal and state healthcare fraud and abuse laws and regulations, and laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by our employees and other third parties may also include the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a Code of Business Conduct and Ethics, but it is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment.
Risks Related to our Financial Position and Capital Requirements
In the past we have incurred significant operating losses, and we recently achieved operating profitability.
We have a limited operating history and even less history operating as a combined organization following the acquisitions of Vidara, Hyperion and Crealta. We have financed our operations primarily through equity and debt financings and have incurred significant operating losses in the past. We had operating income of $4.3 million and $55.4 million for the six months ended June 30, 2016 and the year ended December 31, 2015, respectively, and operating losses of $8.5 million and $42.9 million for the years ended December 31, 2014 and 2013, respectively. We had a net loss of $30.4 million and a net income of $39.5 million for the six months ended June 30, 2016 and the year ended December 31, 2015, respectively, and net losses of $263.6 million and $149.0 million for the years ended December 31, 2014 and 2013, respectively. As of June 30, 2016, we had an accumulated deficit of $711.6 million. Our prior losses have resulted principally from costs incurred in our development activities for our medicines and medicine candidates, commercialization activities related to our medicines, costs associated with our acquisition transactions and costs associated with derivative liability accounting. Our prior losses, combined with possible future losses, have had and will continue to have an adverse effect on our shareholders’ deficit and working capital. While we anticipate that we will continue to generate operating profits in the future, whether we can sustain this will depend on the revenues we generate from the sale of our medicines being sufficient to cover our operating expenses.
We have limited sources of revenues and significant expenses. We cannot be certain that we will sustain profitability, which would depress the market price of our ordinary shares and could cause our investors to lose all or a part of their investment.
Our ability to sustain profitability depends upon our ability to generate sales of our medicines. We have a limited history of commercializing our medicines as a company, and commercialization has been primarily in
99
the United States. We may never be able to successfully commercialize our medicines or develop or commercialize other medicines in the United States or in the EU, which we believe represents our most significant commercial opportunity. Our ability to generate future revenues depends heavily on our success in:
| • | continued commercialization of our existing medicines and any other medicine candidates for which we obtain approval; |
| • | obtaining FDA approvals for additional indications for ACTIMMUNE and RAVICTI; |
| • | securing additional foreign regulatory approvals for our medicines in territories where we have commercial rights; and |
| • | developing, acquiring and commercializing a portfolio of other medicines or medicine candidates in addition to our current medicines. |
Even if we do generate additional medicine sales, we may not be able to sustain profitability on a quarterly or annual basis. Our failure to remain profitable would depress the market price of our ordinary shares and could impair our ability to raise capital, expand our business, diversify our medicine offerings or continue our operations.
We may need to obtain additional financing to fund additional acquisitions.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to:
| • | commercialize our existing medicines in the United States, including the substantial expansion of our sales force in recent years, and our planned commercial launch of RAVICTI in Europe in 2017; |
| • | complete the regulatory approval process, and any future required clinical development related thereto, for our medicines and medicine candidates; |
| • | potentially acquire other businesses or additional complementary medicines or medicines that augment our current medicine portfolio, including costs associated with refinancing debt of acquired companies; and |
| • | conduct clinical trials with respect to potential additional indications, as well as conduct post-marketing requirements and commitments, with respect to our medicines and medicines we acquire. |
While we believe that our existing cash and cash equivalents will be sufficient to fund our operations based on our current expectations of continued revenue growth, we may need to raise additional funds if we choose to expand our commercialization or development efforts more rapidly than presently anticipated, if we develop or acquire additional medicines or acquire companies, or if our revenue does not meet expectations.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our medicines or medicine candidates or one or more of our other research and development initiatives, or delay, cut back or abandon our plans to grow the business through acquisition. We also could be required to:
| • | seek collaborators for one or more of our current or future medicine candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; or |
100
| • | relinquish or license on unfavorable terms our rights to technologies or medicine candidates that we would otherwise seek to develop or commercialize ourselves. |
In addition, if we are unable to secure financing to support future acquisitions, our ability to execute on a key aspect of our overall growth strategy would be impaired.
Any of the above events could significantly harm our business, financial condition and prospects.
We have incurred a substantial amount of debt, which could adversely affect our business, including by restricting our ability to engage in additional transactions or incur additional indebtedness, and prevent us from meeting our debt obligations.
After giving pro forma effect to the Transactions, we will be highly leveraged. As of June 30, 2016, after giving pro forma effect to the Transactions, the principal amount of our outstanding senior indebtedness would have been approximately $1,946.0 million, of which $771.0 million would have been secured indebtedness.
This substantial level of debt could have important consequences to our business, including, but not limited to:
| • | reducing the benefits we expect to receive from our recent and any future acquisition transactions; |
| • | making it more difficult for us to satisfy our obligations; |
| • | requiring a substantial portion of our cash flows from operations to be dedicated to the payment of principal and interest on our indebtedness, therefore reducing our ability to use our cash flows to fund acquisitions, capital expenditures, and future business opportunities; |
| • | exposing us to the risk of increased interest rates to the extent of any future borrowings, including borrowings under our Credit Agreement, at variable rates of interest; |
| • | making it more difficult for us to satisfy our obligations with respect to our indebtedness, including our outstanding notes, our Credit Agreement, and any failure to comply with the obligations of any of our debt instruments, including restrictive covenants and borrowing conditions, could result in an event of default under the agreements governing such indebtedness; |
| • | increasing our vulnerability to, and reducing our flexibility to respond to, changes in our business or general adverse economic and industry conditions; |
| • | limiting our ability to obtain additional financing for working capital, capital expenditures, debt service requirements, acquisitions, and general corporate or other purposes and increasing the cost of any such financing; |
| • | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate; and placing us at a competitive disadvantage as compared to our competitors, to the extent they are not as highly leveraged and who, therefore, may be able to take advantage of opportunities that our leverage may prevent us from exploiting; and |
| • | restricting us from pursuing certain business opportunities. |
The Credit Agreement, the indenture governing our 6.625% senior notes due 2023 (the “2023 notes”) and the indenture that will govern the 2024 notes impose, and the terms of any future indebtedness may impose, various covenants that limit our ability and/
101
or the ability of our restricted subsidiaries (as designated under such agreements) to, among other things, pay dividends or distributions, repurchase equity, prepay junior debt and make certain investments, incur additional debt and issue certain preferred stock, incur liens on assets, engage in certain asset sales, consolidate with or merge or sell all or substantially all of our assets, enter into transactions with affiliates, designate subsidiaries as unrestricted subsidiaries, and allow to exist certain restrictions on the ability of restricted subsidiaries to pay dividends or make other payments to us.
Our ability to obtain future financing and engage in other transactions may be restricted by these covenants. In addition, any credit ratings will impact the cost and availability of future borrowings and our cost of capital. Our ratings at any time will reflect each rating organization’s then opinion of our financial strength, operating performance and ability to meet our debt obligations. There can be no assurance that we will achieve a particular rating or maintain a particular rating in the future. A reduction in our credit ratings may limit our ability to borrow at acceptable interest rates. If our credit ratings were downgraded or put on watch for a potential downgrade, we may not be able to sell additional debt securities or borrow money in the amounts, at the times or interest rates or upon the more favorable terms and conditions that might otherwise be available. Any impairment of our ability to obtain future financing on favorable terms could have an adverse effect on our ability to refinance any of our then-existing debt and may severely restrict our ability to execute on our business strategy, which includes the continued acquisition of additional medicines or businesses.
We may not be able to generate sufficient cash to service all of our indebtedness and may be forced to take other actions to satisfy our obligations under our indebtedness, which may not be successful.
Our ability to make scheduled payments under or to refinance our debt obligations depends on our financial condition and operating performance, which is subject to prevailing economic, industry and competitive conditions and to certain financial, business and other factors beyond our control. Our ability to generate cash flow to meet our payment obligations under our debt may also depend on the successful implementation of our operating and growth strategies, including the Raptor Acquisition. Any refinancing of our debt could be at higher interest rates and may require us to comply with more onerous covenants, which could further restrict our business operations. We cannot assure you that we will maintain a level of cash flows from operating activities sufficient to pay the principal, premium, if any, and interest on our indebtedness.
If our cash flows and capital resources are insufficient to fund our debt service obligations, we may be forced to reduce or delay capital expenditures, sell assets or business operations, seek additional capital or restructure or refinance our indebtedness. We cannot ensure that we would be able to take any of these actions, that these actions would be successful and permit us to meet our scheduled debt service obligations or that these actions would be permitted under the terms of existing or future debt agreements, including the indenture that governs the 2023 notes, the indenture that will govern the 2024 notes and the Credit Agreement. In addition, any failure to make payments of interest and principal on our outstanding indebtedness on a timely basis would likely result in a reduction of our credit rating, which could harm our ability to incur additional indebtedness.
If we cannot make scheduled payments on our debt, we will be in default and, as a result:
| • | our debt holders could declare all outstanding principal and interest to be due and payable; |
| • | the administrative agent and/or the lenders under the Credit Agreement could foreclose against the assets securing the borrowings then outstanding; and |
| • | we could be forced into bankruptcy or liquidation, which could result in you losing your investment. |
102
We generally have broad discretion in the use of our cash and may not use it effectively.
Our management has broad discretion in the application of our cash, and investors will be relying on the judgment of our management regarding the use of our cash. Our management may not apply our cash in ways that ultimately increase the value of any investment in our securities. We expect to use our existing cash to fund commercialization activities for our medicines, to potentially fund additional medicine or business acquisitions, to potentially fund additional regulatory approvals of certain of our medicines, to potentially fund development, life cycle management or manufacturing activities of our medicines for other indications, to potentially fund share repurchases, and for working capital, capital expenditures and general corporate purposes. We may also invest our cash in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our shareholders. If we do not invest or apply our cash in ways that enhance shareholder value, we may fail to achieve expected financial results, which could cause the price of our ordinary shares to decline.
Our ability to use net operating loss carryforwards and certain other tax attributes may be limited.
Under Sections 382 and 383 of the Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50 percent change (by value) in its equity ownership over a three year period), the corporation’s ability to use pre-change net operating loss carryforwards and other pre-change tax attributes to offset post-change income may be limited. In September 2014, the Vidara Merger triggered an “ownership change” limitation and, as a result, we are subject to annual limits on our ability to use the net operating loss carryforwards of Horizon Pharma Inc. and its subsidiaries. We estimate this will result in annual limits of approximately $90 million in the years from 2016 through to 2031. Furthermore, we continue to carry forward our annual limitation resulting from an ownership change date of August 2, 2012. The limitation on pre-change net operating losses incurred prior to the August 2, 2012 change date is approximately $20 million for 2016, $15 million for 2017 and $8 million in the years from 2018 through to 2028. During the second quarter of 2015, we also recognized additional net operating losses and federal and state tax credits as a result of our acquisition of Hyperion on May 7, 2015 in the amount of approximately $31 million of federal net operating losses, state operating losses of approximately $68 million (net of federal effect) and approximately $30 million of federal and state tax credits. We continue to carry forward the annual limitation related to Hyperion of $50 million resulting from the last ownership change date in 2014. Further, as a result of our acquisition of Crealta in the first quarter of 2016, we have recognized an additional estimated net operating loss carryforward of approximately $4 million. The net operating loss carryforward limitation is cumulative such that any use of the carryforwards below the limitations in one year will result in a corresponding increase in the limitations for the subsequent tax year.
Following certain acquisitions of a U.S. corporation by a foreign corporation, Section 7874 of the Code limits the ability of the acquired U.S. corporation and its U.S. affiliates to utilize U.S. tax attributes such as net operating losses to offset U.S. taxable income resulting from certain transactions. Based on the limited guidance available, we expect this limitation is applicable following the Vidara Merger. As a result, it is not currently expected that we or our other U.S. affiliates will be able to utilize their U.S. tax attributes to offset their U.S. taxable income, if any, resulting from certain taxable transactions following the Vidara Merger. Notwithstanding this limitation, we expect that we will be able to fully use our U.S. net operating losses prior to their expiration. As a result of this limitation, however, it may take HPI longer to use its net operating losses. Moreover, contrary to these expectations, it is possible that the limitation under Section 7874 of the Code on the utilization of U.S. tax attributes could prevent us from fully utilizing our U.S. tax attributes prior to their expiration if we do not generate sufficient taxable income.
Any limitation on our ability to use our net operating loss and tax credit carryforwards, including the carryforwards of companies that we acquire, will likely increase the taxes we would otherwise pay in future years if we were not subject to such limitations.
103
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and share price.
As widely reported, global credit and financial markets have experienced extreme disruptions in the past several years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, and uncertainty about economic stability. While there has been some recent improvement in some of these financial metrics, there can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment and continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate again, or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and share price and could require us to delay or abandon commercialization or development plans. There is a risk that one or more of our current service providers, manufacturers and other partners may not survive these difficult economic times, which could directly affect our ability to attain our operating goals on schedule and on budget. Additionally, the SEC has proposed new rules on U.S. domiciled money market funds due to come into effect in the fourth quarter of 2016, which may temporarily suspend redemptions or impose liquidity fees on investors withdrawing assets during volatile periods, and this may adversely affect our investment strategy and/or liquidity.
The U.K.’s referendum to leave the EU or “Brexit,” has and may continue to cause disruptions to capital and currency markets worldwide. The full impact of the Brexit decision, however, remains uncertain. A process of negotiation will determine the future terms of the U.K.’s relationship with the EU. During this period of negotiation, our results of operations and access to capital may be negatively affected by interest rate, exchange rate and other market and economic volatility, as well as regulatory and political uncertainty. Brexit may also have a detrimental effect on our customers, distributors and suppliers, which would, in turn, adversely affect our revenues and financial condition.
At June 30, 2016, we had $424.5 million of cash and cash equivalents consisting of cash and money market funds. While we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents since June 30, 2016, no assurance can be given that further deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents or our ability to meet our financing objectives. Further dislocations in the credit market may adversely impact the value and/or liquidity of marketable securities owned by us.
Changes in accounting rules or policies may affect our financial position and results of operations.
GAAP and related implementation guidelines and interpretations can be highly complex and involve subjective judgments. Changes in these rules or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations. In addition, our operation as an Irish company with multiple subsidiaries in different jurisdictions adds additional complexity to the application of GAAP and this complexity will be exacerbated further if we complete additional strategic transactions. Changes in the application of existing rules or guidance applicable to us or our wholly owned subsidiaries could significantly affect our consolidated financial position and results of operations.
104
Covenants under the indenture governing the 2023 notes, the indenture that will govern the 2024 notes and the Credit Agreement may restrict our business and operations in many ways, and if we do not effectively manage our covenants, our financial conditions and results of operations could be adversely affected.
The indenture governing the 2023 notes, the indenture that will govern the 2024 notes and the Credit Agreement impose various covenants that limit our ability and/or our restricted subsidiaries’ ability to, among other things:
| • | pay dividends or distributions, repurchase equity, prepay, redeem or repurchase certain debt and make certain investments; |
| • | incur additional debt and issue certain preferred stock; |
| • | provide guarantees in respect of obligations of other persons |
| • | incur liens on assets; |
| • | engage in certain asset sales; |
| • | merge, consolidate with or sell all or substantially all of our assets to another person; |
| • | enter into transactions with affiliates; |
| • | sell assets and capital stock of our subsidiaries; |
| • | enter into agreements that restrict distributions from our subsidiaries |
| • | designate subsidiaries as unrestricted subsidiaries; and |
| • | allow to exist certain restrictions on the ability of restricted subsidiaries to pay dividends or make other payments to us. |
These covenants may:
| • | limit our ability to borrow additional funds for working capital, capital expenditures, acquisitions or other general business purposes; |
| • | limit our ability to use our cash flow or obtain additional financing for future working capital, capital expenditures, acquisitions or other general business purposes; |
| • | require us to use a substantial portion of our cash flow from operations to make debt service payments; |
| • | limit our flexibility to plan for, or react to, changes in our business and industry; |
| • | place us at a competitive disadvantage compared to less leveraged competitors; and |
| • | increase our vulnerability to the impact of adverse economic and industry conditions. |
If we are unable to successfully manage the limitations and decreased flexibility on our business due to our significant debt obligations, we may not be able to capitalize on strategic opportunities or grow our business to the extent we would be able to without these limitations.
105
Our failure to comply with any of the covenants could result in a default under the Credit Agreement, the indenture governing the 2023 notes or the indenture that will govern the 2024 notes, which could permit the administrative agent or the trustee, as applicable, or permit the lenders or the holders of the 2023 notes or the 2024 notes to cause the administrative agent or the trustee, as applicable, to declare all or part of any outstanding senior secured term loans the 2023 notes, or the 2024 notes to be immediately due and payable or to exercise any remedies provided to the administrative agent or the trustee, including, in the case of the Credit Agreement proceeding against the collateral granted to secure our obligations under the Credit Agreement. An event of default under the Credit Agreement, the indenture governing the 2023 notes or the indenture that will govern the 2024 notes could also lead to an event of default under the terms of the other agreement and the indenture governing our 2.50% exchangeable senior notes due 2022 (the “2022 notes”). Any such event of default or any exercise of rights and remedies by our creditors could seriously harm our business.
If intangible assets that we have recorded in connection with our acquisition transactions become impaired, we could have to take significant charges against earnings.
In connection with the accounting for our various acquisition transactions, we have recorded significant amounts of intangible assets. Under GAAP, we must assess, at least annually and potentially more frequently, whether the value of goodwill and other indefinite-lived intangible assets has been impaired. Amortizing intangible assets will be assessed for impairment in the event of an impairment indicator. Any reduction or impairment of the value of goodwill or other intangible assets will result in a charge against earnings, which could materially adversely affect our results of operations and shareholders’ equity in future periods.
Risks Related to Our Intellectual Property
If we are unable to obtain or protect intellectual property rights related to our medicines and medicine candidates, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our medicines and medicine candidates. The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications that we own may fail to result in issued patents with claims that cover our medicines in the United States or in other foreign countries. If this were to occur, early generic competition could be expected against our current medicines and other medicine candidates in development. There is no assurance that all potentially relevant prior art relating to our patents and patent applications has been found, which prior art can invalidate a patent or prevent a patent from issuing based on a pending patent application. In particular, because the APIs in DUEXIS, VIMOVO and RAYOS/LODOTRA have been on the market as separate medicines for many years, it is possible that these medicines have previously been used off-label in such a manner that such prior usage would affect the validity of our patents or our ability to obtain patents based on our patent applications. In addition, claims directed to dosing and dose adjustment may be substantially less likely to issue in light of the Supreme Court decision in Mayo Collaborative Services v. Prometheus Laboratories, Inc., where the court held that claims directed to methods of determining whether to adjust drug dosing levels based on drug metabolite levels in the red blood cells were not patent eligible because they were directed to a law of nature. This decision may have wide-ranging implications on the validity and scope of pharmaceutical method claims.
Even if patents do successfully issue, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed or invalidated.
Patent litigation is currently pending in the United States District Court for the District of New Jersey against several companies intending to market a generic version of VIMOVO before the expiration of certain of our patents listed in the Orange Book. These cases are collectively known as the VIMOVO cases, and involve the following sets of defendants: (i) Dr. Reddy’s; (ii) Lupin; (iii) Mylan; and (iv) Actavis Pharma. The cases arise from Paragraph IV Patent Certification notice letters from each of Dr. Reddy’s, Lupin, Mylan and Actavis Pharma advising each had filed an ANDA with the FDA seeking approval to market generic versions of VIMOVO before the expiration of the patents-in-suit.
106
On February 24, 2015, Dr. Reddy’s Laboratories, Inc. filed a Petition for inter partes review (“IPR”) of U.S. Patent No. 8,557,285, one of the patents in litigation in the above referenced VIMOVO cases. On October 9, 2015, the United States Patent and Trademark Office (the “U.S. PTO”) denied such Petition for IPR.
On May 21, 2015, the Coalition for Affordable Drugs VII LLC (the “Coalition for Affordable Drugs”) filed a Petition for IPR of U.S. Patent No. 6,926,907, one of the patents in litigation in the above referenced VIMOVO cases. On December 8, 2015, the U.S. PTO denied such Petition for IPR.
On June 5, 2015, the Coalition for Affordable Drugs filed another Petition for IPR of U.S. Patent No. 8,858,996, one of the patents in litigation in the above referenced VIMOVO cases. On December 17, 2015, the U.S. PTO denied such Petition for IPR.
On August 7, 2015, the Coalition for Affordable Drugs filed another Petition for IPR of U.S. Patent No. 8,852,636, one of the patents in litigation in the above referenced VIMOVO cases. On February 11, 2016, the U.S. PTO denied such Petition for IPR.
On August 12, 2015, the Coalition for Affordable Drugs filed another Petition for IPR of U.S. Patent No. 8,945,621, one of the patents in litigation in the above referenced VIMOVO cases. On February 22, 2016, the Patent Trial and Appeal Board (the “PTAB”) issued a decision to institute the IPR.
On August 19, 2015, Lupin filed Petitions for IPR of U.S. Patent Nos. 8,858,996, 8,852,636, and 8,865,190, all patents in litigation in the above referenced VIMOVO cases. On March 1, 2016, the PTAB issued decisions to institute the IPRs for U.S. Patent Nos. 8,858,996 and 8,865,190. Also, on March 1, 2016, the PTAB denied the Petition for IPR for U.S. Patent No. 8,852,636.
Patent litigation is currently pending in the United States District Court for the District of New Jersey against several companies intending to market a generic version of PENNSAID 2% prior to the expiration of certain of our patents listed in the Orange Book. These cases are collectively known as the PENNSAID 2% cases, and involve the following sets of defendants: (i) Actavis; (ii) Lupin Limited; and (iii) Amneal (settled, awaiting dismissal). These cases arise from Paragraph IV Patent Certification notice letters from each of Actavis, Lupin Limited and Amneal advising each had filed an ANDA with the FDA seeking approval to market a generic version of PENNSAID 2% before the expiration of the patents-in-suit. The status of these cases is as set forth above.
The Company received from Actavis a Paragraph IV Patent Certification Notice Letter dated July 1, 2016, against Orange Book listed U.S. Patent Nos. 9,339,551 and 9,339,552, advising that Actavis had filed an ANDA with the FDA for a generic version of PENNSAID 2%.
The Company has received from Apotex two Paragraph IV Patent Certification Notice Letters dated April 1, 2016, and June 30, 2016, against Orange Book listed U.S. Patent Nos. 8,217,078, 8,252,838, 8,546,450, 8,563,613, 8,618,164, 8,741,956, 8,871,809, 9,066,913, 9,101,591, 9,132,110, 9,168,304, 9,168,305, 9,220,784, 9,339,551 and 9,339,552, advising that Apotex had filed an ANDA with the FDA for a generic version of PENNSAID 2%.
Patent litigation is currently pending in the United States District Court for the Eastern District of Texas against Par Pharmaceutical and in the United States District Court for the District of New Jersey against Lupin and against Par Pharmaceutical, who are each intending to market generic versions of RAVICTI prior to the expiration of certain of our patents listed in the Orange Book. These cases are collectively known as the RAVICTI cases, and arise from Paragraph IV Patent Certification notice letters from each of Par Pharmaceutical and Lupin advising each had filed an ANDA with the FDA seeking approval to market a generic version of RAVICTI before the expiration of the patents-in-suit.
107
On April 29, 2015, Par Pharmaceutical filed Petitions for IPR of U.S. Patent No. 8,404,215 and U.S. Patent No. 8,642,012, two of the patents involved in the above mentioned RAVICTI cases. On November 4, 2015, the PTAB issued decisions instituting such IPRs and on December 14, 2015, the District Court Judge Roy Payne issued a stay pending a final written decision from the PTAB with respect to such IPRs. On September 29, 2016, the PTAB found all of the claims in U.S. Patent No. 8,404,215 to be unpatentable. The PTAB’s decision may be appealed to the Federal Circuit. The PTAB must issue a final written decision on the IPR of U.S. Patent No. 8,642,012 by no later than November 4, 2016.
On April 1, 2016, Lupin filed a Petition for IPR of U.S. Patent No. 9,095,559, a patent currently at issue in the Lupin RAVICTI case. On September 30, 2016, the PTAB issued a decision instituting the IPR. The PTAB must issue a final written decision on the IPR no later than September 30, 2017.
We intend to vigorously defend our intellectual property rights relating to our medicines, but we cannot predict the outcome of the VIMOVO cases, the PENNSAID 2% cases, the RAVICTI cases or the IPRs. Any adverse outcome in these matters or any new generic challenges that may arise could result in one or more generic versions of our medicines being launched before the expiration of the listed patents, which could adversely affect our ability to successfully execute our business strategy to increase sales of our medicines, and would negatively impact our financial condition and results of operations, including causing a significant decrease in our revenues and cash flows.
Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the patent applications we hold with respect to our medicines fail to issue or if their breadth or strength of protection is threatened, it could dissuade companies from collaborating with us to develop them and threaten our ability to commercialize our medicines. We cannot offer any assurances about which, if any, patents will issue or whether any issued patents will be found not invalid and not unenforceable or will go unthreatened by third parties. Since patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we were the first to file any patent application related to our medicines or any other medicine candidates. Furthermore, if third parties have filed such patent applications, an interference proceeding in the United States can be provoked by a third-party or instituted by us to determine who was the first to invent any of the subject matter covered by the patent claims of our applications.
With respect to RAVICTI, the composition of matter patent we hold would have expired in the United States in February 2015 without term extension. However, Hyperion applied for a term extension of approximately four years for this patent under the Drug Price Competition and Patent Term Restoration Act. Hyperion recently received notice that the U.S. PTO has determined that the length of the extension is 1,267 days. We cannot guarantee that pending patent applications related to RAVICTI will result in additional patents or that other existing and future patents related to RAVICTI will be held valid and enforceable or will be sufficient to deter generic competition in the United States. Therefore, it is possible that upon expiration of the RAVICTI composition of matter patent, we would need to rely on forms of regulatory exclusivity, to the extent available, to protect against generic competition.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our drug discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we expect all of our employees to assign their inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques.
108
Our ability to obtain patents is highly uncertain because, to date, some legal principles remain unresolved, there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States and the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. For example, on September 16, 2011, the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. The U.S. PTO has developed new and untested regulations and procedures to govern the full implementation of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective in March 2013. The Leahy-Smith Act has also introduced procedures making it easier for third-parties to challenge issued patents, as well as to intervene in the prosecution of patent applications. Finally, the Leahy-Smith Act contains new statutory provisions that still require the U.S. PTO to issue new regulations for their implementation and it may take the courts years to interpret the provisions of the new statute. Accordingly, it is too early to tell what, if any, impact the Leahy-Smith Act will have on the operation of our business and the protection and enforcement of our intellectual property. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. In addition, the ACA allows applicants seeking approval of biosimilar or interchangeable versions of biological products such as ACTIMMUNE to initiate a process for challenging some or all of the patents covering the innovator biological product used as the reference product. This process is complicated and could result in the limitation or loss of certain patent rights. An inability to obtain, enforce and defend patents covering our proprietary technologies would materially and adversely affect our business prospects and financial condition.
Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. For example, if the issuance, in a given country, of a patent to us, covering an invention, is not followed by the issuance, in other countries, of patents covering the same invention, or if any judicial interpretation of the validity, enforceability, or scope of the claims in, or the written description or enablement in, a patent issued in one country is not similar to the interpretation given to the corresponding patent issued in another country, our ability to protect our intellectual property in those countries may be limited. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may materially diminish the value of our intellectual property or narrow the scope of our patent protection. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on us avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions and inter party reexamination proceedings before the U.S. PTO. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which our collaborators are developing medicine candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our medicine candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of
109
manufacture or methods for treatment related to the use or manufacture of our medicines and/or any other medicine candidates. Because patent applications can take many years to issue, there may be currently pending patent applications, which may later result in issued patents that our medicine candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our medicine candidates, any molecules formed during the manufacturing process or any final medicine itself, the holders of any such patents may be able to block our ability to commercialize such medicine candidate unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patent were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy, the holders of any such patent may be able to block our ability to develop and commercialize the applicable medicine candidate unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our medicine candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing medicines, which may be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our medicine candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize one or more of our medicine candidates, which could harm our business significantly. We cannot provide any assurances that third-party patents do not exist which might be enforced against our medicines, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties.
If we fail to comply with our obligations in the agreements under which we license rights to technology from third parties, we could lose license rights that are important to our business.
We are party to a number of technology licenses that are important to our business and expect to enter into additional licenses in the future. For example, we hold an exclusive license to SkyePharma AG’s (“SkyePharma”) proprietary technology and know-how covering the delayed-release of corticosteroids relating to RAYOS/LODOTRA. If we fail to comply with our obligations under our agreement with SkyePharma or our other license agreements, or if we are subject to a bankruptcy, the licensor may have the right to terminate the license, in which event we would not be able to market medicines covered by the license, including RAYOS/ LODOTRA.
In connection with our November 2013 acquisition of the U.S. rights to VIMOVO, we (i) received the benefit of a covenant not to sue under AstraZeneca’s patent portfolio with respect to Nexium (which shall automatically become a license under such patent portfolio if and when AstraZeneca reacquires control of such patent portfolio from Merck Sharp & Dohme Corp. and certain of its affiliates), (ii) were assigned AstraZeneca’s amended and restated collaboration and license agreement for the United States with Aralez, under which AstraZeneca has in-licensed exclusive rights under certain of Aralez’s patents with respect to VIMOVO, and (iii) acquired AstraZeneca’s co-ownership rights with Aralez with respect to certain joint patents covering VIMOVO, all for the commercialization of VIMOVO in the United States. If we fail to comply with our obligations under our agreements with AstraZeneca or if we fail to comply with our obligations under our agreements with Aralez, our rights to commercialize VIMOVO in the United States may be adversely affected or terminated by AstraZeneca or Aralez.
110
We also license rights to patents, know-how and trademarks for ACTIMMUNE from Genentech Inc. (“Genentech”), under an agreement that remains in effect for so long as we continue to commercialize and sell ACTIMMUNE. However, Genentech may terminate the agreement upon our material default, if not cured within a specified period of time. Genentech may also terminate the agreement in the event of our bankruptcy or insolvency. Upon such a termination of the agreement, all intellectual property rights conveyed to us under the agreement, including the rights to the ACTIMMUNE trademark, revert to Genentech. If we fail to comply with our obligations under this agreement, we could lose the ability to market and distribute ACTIMMUNE, which would have a material adverse effect on our business, financial condition and results of operations.
We rely on a license from Ucyclyd with respect to technology developed by Ucyclyd in connection with the manufacturing of RAVICTI. The purchase agreement under which Hyperion purchased the worldwide rights to RAVICTI contains obligations to pay Ucyclyd regulatory and sales milestone payments relating to RAVICTI, as well as royalties on the net sales of RAVICTI. On May 31, 2013, when Hyperion acquired BUPHENYL under a restated collaboration agreement with Ucyclyd, Hyperion received a license to use some of the manufacturing technology developed by Ucyclyd in connection with the manufacturing of BUPHENYL. The restated collaboration agreement also contains obligations to pay Ucyclyd regulatory and sales milestone payments, as well as royalties on net sales of BUPHENYL. If we fail to make a required payment to Ucyclyd and do not cure the failure within the required time period, Ucyclyd may be able to terminate the license to use its manufacturing technology for RAVICTI and BUPHENYL. If we lose access to the Ucyclyd manufacturing technology, we cannot guarantee that an acceptable alternative method of manufacture could be developed or acquired. Even if alternative technology could be developed or acquired, the loss of the Ucyclyd technology could still result in substantial costs and potential periods where we would not be able to market and sell RAVICTI and/or BUPHENYL. We also license intellectual property necessary for commercialization of RAVICTI from an external party. This party may be entitled to terminate the license if we breach the agreement, including failure to pay required royalties on net sales of RAVICTI, or we do not meet specified diligence obligations in our development and commercialization of RAVICTI, and we do not cure the failure within the required time period. If the license is terminated, it may be difficult or impossible for us to continue to commercialize RAVICTI, which would have a material adverse effect on our business, financial condition and results of operations.
We also hold an exclusive license to patents and technology from Duke University (“Duke”) and Mountain View Pharmaceuticals, Inc. (“MVP”) covering KRYSTEXXA. Duke and MVP may terminate the license if we commit fraud or for our willful misconduct or illegal conduct. Duke and MVP may also terminate the license upon our material breach of the agreement, if not cured within a specified period of time, or upon written notice if we have committed two or more material breaches under the agreement. Duke and MVP may also terminate the license in the event of our bankruptcy or insolvency. If the license is terminated, it may be impossible for us to continue to commercialize KRYSTEXXA, which would have a material adverse effect on our business, financial condition and results of operations.
In addition, we will be subject to the contractual obligations under Raptor’s agreements with Tripex and PARI related to QUINSAIR. Under the agreement with Tripex, we are required to pursue commercially reasonable efforts to initiate, and subsequently to complete, an additional clinical trial of QUINSAIR in a non-cystic-fibrosis patient population within a specified period of time and an obligation to progress toward filing an NDA for approval of QUINSAIR in the United States in all or part of the cystic fibrosis patient population. These obligations are subject to certain exceptions due to, for example, manufacturing delays not under our control, or delays caused by the FDA. If we fail to properly exercise such efforts to initiate and complete an appropriate clinical trial, or fail to file an NDA for U.S. approval in the cystic fibrosis patient population, during the time periods specified in the asset purchase agreement, we may be subject to various claims by Tripex and parties affiliated with Tripex. In addition, if we do not spend a minimum amount on QUINSAIR development in each of the three years following our acquisition of Raptor, we may also be obligated to pre-pay a milestone payment related to initiating a clinical trial for QUINSAIR in a non-cystic-fibrosis indication. Under the license agreement with PARI, we are required to comply with diligence milestones related to development and commercialization of QUINSAIR in the United States, to spend a specified minimum amount per year on development activities in
111
the United States until filing of the NDA for QUINSAIR in the United States. If we do not comply with these obligations, our licenses to certain intellectual property related to QUINSAIR may become non-exclusive in the United States or could be terminated. We will also be subject to contractual obligations under Raptor’s license agreements with the UCSD with respect to PROCYSBI, including diligence obligations to develop PROCYSBI for the treatment of NASH and Huntington’s disease, with which Raptor is currently not in compliance. To the extent that we fail to perform the diligence obligations under the agreement, UCSD may, with respect to such indication, terminate the license or otherwise cause the license to become non-exclusive. If one or more of these licenses was terminated, we would have no further right to use of exploit the related intellectual property, which would limit our ability to develop PROCYSBI or QUINSAIR in other indications, and could impact our ability to continue commercializing PROCYSBI or QUINSAIR in their approved indications.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one of our patents, or a patent of one of our licensors, is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
There are numerous post grant review proceedings available at the U.S. PTO (including IPR, post-grant review and ex-parte reexamination) and similar proceedings in other countries of the world that could be initiated by a third-party that could potentially negatively impact our issued patents.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our ordinary shares.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the U.S. PTO and foreign patent agencies in several stages over the lifetime of the patent. The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in
112
abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or licensors that control the prosecution and maintenance of our licensed patents fail to maintain the patents and patent applications covering our medicine candidates, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of our employees’ former employers or other third parties. We may also be subject to claims that former employers or other third parties have an ownership interest in our patents. Litigation may be necessary to defend against these claims. There is no guarantee of success in defending these claims, and even if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
Risks Related to Ownership of Our Ordinary Shares
The market price of our ordinary shares historically has been volatile and is likely to continue to be volatile, and you could lose all or part of any investment in our ordinary shares.
The trading price of our ordinary shares has been volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this report, these factors include:
| • | our failure to successfully execute our commercialization strategy with respect to our approved medicines, particularly our commercialization of our medicines in the United States; |
| • | actions or announcements by third-party or government payors with respect to coverage and reimbursement of our medicines; |
| • | disputes or other developments relating to intellectual property and other proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our medicines and medicine candidates; |
| • | unanticipated serious safety concerns related to the use of our medicines; |
| • | adverse regulatory decisions; |
| • | changes in laws or regulations applicable to our business, medicines or medicine candidates, including but not limited to clinical trial requirements for approvals or tax laws; |
| • | inability to comply with our debt covenants and to make payments as they become due; |
| • | inability to obtain adequate commercial supply for any approved medicine or inability to do so at acceptable prices; |
| • | developments concerning our commercial partners, including but not limited to those with our sources of manufacturing supply; |
| • | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| • | adverse results or delays in clinical trials; |
| • | our failure to successfully develop and/or acquire additional medicine candidates or obtain approvals for additional indications for our existing medicine candidates; |
| • | introduction of new medicines or services offered by us or our competitors; |
| • | overall performance of the equity markets, including the pharmaceutical sector, and general political and economic conditions; |
| • | failure to meet or exceed revenue and financial projections that we may provide to the public; |
| • | actual or anticipated variations in quarterly operating results; |
| • | failure to meet or exceed the estimates and projections of the investment community; |
113
| • | inaccurate or significant adverse media coverage; |
| • | publication of research reports about us or our industry or positive or negative recommendations or withdrawal of research coverage by securities analysts; |
| • | our inability to successfully enter new markets; |
| • | the termination of a collaboration or the inability to establish additional collaborations; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| • | our inability to maintain an adequate rate of growth; |
| • | ineffectiveness of our internal controls or our inability to otherwise comply with financial reporting requirements; |
| • | adverse U.S. and foreign tax exposure; |
| • | additions or departures of key management, commercial or regulatory personnel; |
| • | issuances of debt or equity securities; |
| • | significant lawsuits, including patent or shareholder litigation; |
| • | changes in the market valuations of similar companies to us; |
| • | sales of our ordinary shares by us or our shareholders in the future; |
| • | trading volume of our ordinary shares; |
| • | effects of natural or man-made catastrophic events or other business interruptions; and |
| • | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and The NASDAQ Global Select Market and the stock of biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may adversely affect the market price of our ordinary shares, regardless of our actual operating performance.
We have never declared or paid dividends on our share capital and we do not anticipate paying dividends in the foreseeable future.
We have never declared or paid any cash dividends on our ordinary shares. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future, including due to limitations that are currently imposed by the Senior Secured Credit Facility. Any return to shareholders will therefore be limited to the increase, if any, of our ordinary share price.
We have incurred and will continue to incur significant increased costs as a result of operating as a public company and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we have incurred and will continue to incur significant legal, accounting and other expenses that we did not incur as a private company. In particular, the Sarbanes-Oxley Act of 2000 (the “Sarbanes-Oxley Act”)
114
as well as rules subsequently implemented by the SEC and the NASDAQ Stock Market, Inc. (“NASDAQ”) impose significant requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. These rules and regulations have substantially increased our legal and financial compliance costs and have made some activities more time-consuming and costly. These effects are exacerbated by our transition to an Irish company and the integration of numerous acquired businesses and operations into our historical business and operating structure. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations. The increased costs will continue to decrease our net income or increase our net loss, and may require us to reduce costs in other areas of our business or increase the prices of our medicines or services. For example, these rules and regulations make it more difficult and more expensive for us to obtain and maintain director and officer liability insurance. We cannot predict or estimate the amount or timing of additional costs that we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers. If we fail to comply with the continued listing requirements of NASDAQ, our ordinary shares could be delisted from The NASDAQ Global Select Market, which would adversely affect the liquidity of our ordinary shares and our ability to obtain future financing.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. In particular, we are required to perform annual system and process evaluation and testing of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act, or Section 404. Our independent registered public accounting firm is also required to deliver a report on the effectiveness of our internal control over financial reporting. Our testing, or the testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses. Our compliance with Section 404 requires that we incur substantial expense and expend significant management efforts, particularly because of our Irish parent company structure and international operations. In particular, prior to the acquisition of Crealta, Crealta and its affiliated entities were not subject to the requirements of the Sarbanes-Oxley Act. We are taking measures to establish or implement an internal control environment at these entities aimed at successfully adopting the requirements of Section 404. However, it is possible that we may experience delays in implementing or be unable to implement the required internal controls over financial reporting and other disclosure controls and procedures. If we are not able to comply with the requirements of Section 404 or if we or our independent registered public accounting firm identify deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses, the market price of our ordinary shares could decline and we could be subject to sanctions or investigations by NASDAQ, the SEC or other regulatory authorities, which would require additional financial and management resources.
New laws and regulations as well as changes to existing laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act and rules adopted by the SEC and by NASDAQ, would likely result in increased costs as we respond to their requirements.
Sales of a substantial number of our ordinary shares in the public market could cause our share price to decline.
If our existing shareholders sell, or indicate an intention to sell, substantial amounts of our ordinary shares in the public market, the trading price of such ordinary shares could decline. In addition, our ordinary shares that are either subject to outstanding options or reserved for future issuance under our employee benefit plans are or may become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act of 1933, as amended (the “Securities Act”). If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of our ordinary shares could decline.
Certain holders of our ordinary shares are entitled to rights with respect to the registration of their shares under the Securities Act. Registration of these shares under the Securities Act would result in the shares
115
becoming freely tradable without restriction under the Securities Act, except for shares purchased by our affiliates. For example, we are subject to a registration rights agreement with certain former Vidara shareholders that acquired our ordinary shares in connection with our acquisition of Vidara. Pursuant to this agreement, we filed and are required to maintain a registration statement covering the resale of ordinary shares held by these shareholders and in certain circumstances, these holders can require us to participate in an underwritten public offering of their ordinary shares. Any sales of securities by these shareholders or a public announcement of such sales could have a material adverse effect on the trading price of our ordinary shares.
In addition, any conversion or exchange of our 2022 notes, whether pursuant to their terms or pursuant to privately negotiated transactions between the issuer and/or us and a holder of such securities, could depress the market price for our ordinary shares.
Future sales and issuances of our ordinary shares, securities convertible into our ordinary shares or rights to purchase ordinary shares or convertible securities could result in additional dilution of the percentage ownership of our shareholders and could cause our share price to decline.
Additional capital may be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities or securities convertible into or exchangeable for ordinary shares, our shareholders may experience substantial dilution. We may sell ordinary shares, and we may sell convertible or exchangeable securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell such ordinary shares, convertible or exchangeable securities or other equity securities in subsequent transactions, existing shareholders may be materially diluted. New investors in such subsequent transactions could gain rights, preferences and privileges senior to those of holders of ordinary shares. We also maintain equity incentive plans, including our Amended and Restated 2014 Equity Incentive Plan, 2014 Non-Employee Equity Plan and 2014 Employee Share Purchase Plan, and intend to grant additional ordinary share awards under these and future plans, which will result in additional dilution to our existing shareholders.
Irish law differs from the laws in effect in the United States and may afford less protection to holders of our securities.
It may not be possible to enforce court judgments obtained in the United States against us in Ireland based on the civil liability provisions of the U.S. federal or state securities laws. In addition, there is some uncertainty as to whether the courts of Ireland would recognize or enforce judgments of U.S. courts obtained against us or our directors or officers based on the civil liabilities provisions of the U.S. federal or state securities laws or hear actions against us or those persons based on those laws. We have been advised that the U.S. currently does not have a treaty with Ireland providing for the reciprocal recognition and enforcement of judgments in civil and commercial matters. Therefore, a final judgment for the payment of money rendered by any U.S. federal or state court based on civil liability, whether or not based solely on U.S. federal or state securities laws, would not automatically be enforceable in Ireland.
As an Irish company, we are governed by the Irish Companies Acts, which differ in some material respects from laws generally applicable to U.S. corporations and shareholders, including, among others, differences relating to interested director and officer transactions and shareholder lawsuits. Likewise, the duties of directors and officers of an Irish company generally are owed to the company only. Shareholders of Irish companies generally do not have a personal right of action against directors or officers of the company and may exercise such rights of action on behalf of the company only in limited circumstances. Accordingly, holders of our securities may have more difficulty protecting their interests than would holders of securities of a corporation incorporated in a jurisdiction of the United States.
116
Provisions of our articles of association could delay or prevent a takeover of us by a third-party.
Our articles of association could delay, defer or prevent a third-party from acquiring us, despite the possible benefit to our shareholders, or otherwise adversely affect the price of our ordinary shares. For example, our articles of association:
| • | permit our board of directors to issue one or more series of preferred shares with rights and preferences designated by our board of directors; |
| • | impose advance notice requirements for shareholder proposals and nominations of directors to be considered at shareholder meetings; |
| • | stagger the terms of our board of directors into three classes; and |
| • | require the approval of a supermajority of the voting power of the shares of our share capital entitled to vote generally at a meeting of shareholders to amend or repeal our articles of association. |
In addition, several mandatory provisions of Irish law could prevent or delay an acquisition of us. For example, Irish law does not permit shareholders of an Irish public limited company to take action by written consent with less than unanimous consent. We are also subject to various provisions of Irish law relating to mandatory bids, voluntary bids, requirements to make a cash offer and minimum price requirements, as well as substantial acquisition rules and rules requiring the disclosure of interests in our ordinary shares in certain circumstances.
These provisions may discourage potential takeover attempts, discourage bids for our ordinary shares at a premium over the market price or adversely affect the market price of, and the voting and other rights of the holders of, our ordinary shares. These provisions could also discourage proxy contests and make it more difficult for you and our other shareholders to elect directors other than the candidates nominated by our board of directors, and could depress the market price of our ordinary shares.
A transfer of our ordinary shares may be subject to Irish stamp duty.
In certain circumstances, the transfer of shares in an Irish incorporated company will be subject to Irish stamp duty, which is a legal obligation of the buyer. This duty is currently charged at the rate of 1.0 percent of the price paid or the market value of the shares acquired, if higher. Because our ordinary shares are traded on a recognized stock exchange in the United States, an exemption from this stamp duty is available to transfers by shareholders who hold ordinary shares beneficially through brokers which in turn hold those shares through the Depositary Trust Company (“DTC”) to holders who also hold through DTC. However, a transfer by or to a record holder who holds ordinary shares directly in his, her or its own name could be subject to this stamp duty. We, in our absolute discretion and insofar as the Companies Acts or any other applicable law permit, may, or may provide that one of our subsidiaries will pay Irish stamp duty arising on a transfer of our ordinary shares on behalf of the transferee of such ordinary shares. If stamp duty resulting from the transfer of ordinary shares which would otherwise be payable by the transferee is paid by us or any of our subsidiaries on behalf of the transferee, then in those circumstances, we will, on our behalf or on behalf of such subsidiary (as the case may be), be entitled to (i) seek reimbursement of the stamp duty from the transferee, (ii) set-off the stamp duty against any dividends payable to the transferee of those ordinary shares and (iii) claim a first and permanent lien on the ordinary shares on which stamp duty has been paid by us or such subsidiary for the amount of stamp duty paid. Our lien shall extend to all dividends paid on those ordinary shares.
Dividends paid by us may be subject to Irish dividend withholding tax.
In certain circumstances, as an Irish tax resident company, we will be required to deduct Irish dividend withholding tax (currently at the rate of 20%) from dividends paid to our shareholders. Shareholders that are resident in the United States, EU countries (other than Ireland) or other countries with which Ireland has signed a tax treaty (whether the treaty has been ratified or not) generally should not be subject to Irish withholding tax so long as the shareholder has provided its broker, for onward transmission to our qualifying intermediary or other designated agent (in the case of shares held beneficially), or our or its transfer agent (in the case of shares held directly), with all the necessary documentation by the appropriate due date prior to payment of the dividend. However, some shareholders may be subject to withholding tax, which could adversely affect the price of our ordinary shares.
117
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our ordinary shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our rating or publish inaccurate or unfavorable research about our business, our share price could decline. If one or more of these analysts cease coverage of our company or fail to publish reports on our company regularly, demand for our ordinary shares could decrease, which might cause our share price and trading volume to decline.
Securities class action litigation could divert our management’s attention and harm our business and could subject us to significant liabilities.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the equity securities of pharmaceutical companies. These broad market fluctuations may cause the market price of our ordinary shares to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. For example, following declines in our stock price, two federal securities class action lawsuits were filed in March 2016 against us and certain of our current and former officers alleging violations of the Securities Exchange Act of 1934, as amended. Subsequently, the two actions were consolidated, and plaintiff added claims under the Securities Act and named additional defendants. This consolidated class action (captioned Schaffer v. Horizon Pharma plc, et al., Case No. 1:16-cv-01763) is currently pending in the United States District Court for the Southern District of New York. Even if we are successful in defending against this or any similar claims that may be brought in the future, litigation could result in substantial costs and may be a distraction to our management, and may lead to an unfavorable outcome that could adversely impact our financial condition and prospects.
118