Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex23-1.htm |
| EX-32.2 - EXHIBIT 32.2 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex32-2.htm |
| EX-32.1 - EXHIBIT 32.1 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex31-1.htm |
| EX-23.2 - EXHIBIT 23.2 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex23-2.htm |
| EX-21 - EXHIBIT 21 - ELITE PHARMACEUTICALS INC /NV/ | v441688_ex21.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(MARK ONE)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED – MARCH 31, 2016
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM __________ TO __________
Commission File Number: 001 – 15697
ELITE PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Nevada | 22-3542636 |
| (State or other jurisdiction of incorporation) | (IRS Employer Identification No.) |
165 Ludlow Avenue, Northvale, New Jersey 07647
(Address of principal executive offices)
(201) 750 – 2646
(Registrant’s telephone number, including area code)
Securities Registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Exchange on Which Registered | |
| None |
Securities Registered pursuant to Section 12(g) of the Act:
Common Stock, $0.001 par value
| Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act | Yes ¨ | No x | |
| Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act | Yes ¨ | No x | |
| Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that registrant was required to file such reports) and (2) has been subject to such filing requirements for at least the past 90 days. | Yes x | No ¨ | |
| Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). | Yes x | No ¨ | |
| Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. | ¨ |
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a smaller reporting company. See definition of “large accelerated filer”, “accelerated filer” and smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large Accelerated Filer | Accelerated Filer | Non-Accelerated Filer | Smaller Reporting Company |
| ¨ | x | ¨ | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ¨ No x
State the aggregate market value of the voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold as of the last business day of the registrant’s most recently completed second fiscal quarter (for purposes of determining this amount, only directors, executive officers and, based on Schedule 13(d) filings as of September 30, 2015, 10% or greater stockholders, and their respective affiliates, have been deemed affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes).
| Title of Class | Aggregate Market Value | As of Close of Business on | ||||
| Common Stock - $0.001 par value | $ | 156,803,944 | September 30, 2015 | |||
Indicate the number of shares outstanding of each of the registrant’s classes of common stock, as of the latest practical date
| Title of Class | Shares Outstanding | As of Close of Business on | ||||
| Common Stock - $0.001 par value | 726,486,650 | June 7, 2016 | ||||
DOCUMENTS INCORPORATED BY REFERENCE
None.
FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K and the documents incorporated herein contain “forward-looking statements”. Such forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of the Company, or industry results, to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. When used in this report, statements that are not statements of current or historical fact may be deemed to be forward-looking statements. Without limiting the foregoing, the words “plan”, “intend”, “may,” “will,” “expect,” “believe”, “could,” “anticipate,” “estimate,” “forecast”, “contemplate”, “envisage”, or “continue” or similar expressions or other variations or comparable terminology are intended to identify such forward-looking statements. All statements other than statements of historical fact included in this report regarding our financial position, business strategy and plans or objectives for future operations are forward-looking statements. Without limiting the broader description of forward-looking statements above, we specifically note, without limitation, that statements regarding the preliminary nature of the clinical program results and the potential for further product development, that involve known and unknown risks, delays, uncertainties and other factors not under our control, the requirement of substantial future testing, clinical trials, regulatory reviews and approvals by the Food and Drug Administration and other regulatory authorities prior to the commercialization of products under development, and our ability to manufacture and sell any products, gain market acceptance earn a profit from sales or licenses of any drugs or our ability to discover new drugs in the future are all forward-looking in nature. These risks and other factors are discussed in our filings with the Securities and Exchange Commission. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. Except as required by law, the Company undertakes no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise.
| 1 |
Table of Contents
| 2 |
| ITEM 1 | BUSINESS |
General
Elite Pharmaceuticals, Inc., a Nevada corporation (the “Company”, “Elite”, “Elite Pharmaceuticals”, the “registrant”, “we”, “us” or “our”) was incorporated on October 1, 1997 under the laws of the State of Delaware, and its wholly-owned subsidiary, Elite Laboratories, Inc. (“Elite Labs””), was incorporated on August 23, 1990 under the laws of the State of Delaware. On January 5, 2012, Elite Pharmaceuticals was reincorporated under the laws of the State of Nevada.
We are a specialty pharmaceutical company principally engaged in the development and manufacture of oral, controlled-release products, using proprietary know-how and technology, particularly as it relates to abuse resistant products and the manufacture of generic pharmaceuticals. Our strategy includes improving off-patent drug products for life cycle management, developing generic versions of controlled-release drug products with high barriers to entry and the development of branded and generic products that utilize our proprietary and patented abuse resistance technologies.
We own and occupy manufacturing, warehouse, laboratory and office space at 165 Ludlow Avenue and 135 Ludlow Avenue in Northvale, NJ (the “Northvale Facility”). The Northvale Facility operates under Current Good Manufacturing Practice (“cGMP”) and is a United States Drug Enforcement Agency (“DEA”) registered facility for research, development and manufacturing.
Strategy
Elite is focusing its efforts on the following areas: (i) development of Elite’s pain management products; (ii) manufacturing of a line of generic pharmaceutical products with approved ANDAs; (iii) development of additional generic pharmaceutical products; (iv) development of the other products in our pipeline including the products with our partners; (v) commercial exploitation of our products either by license and the collection of royalties, or through the manufacture of our formulations; and (vi) development of new products and the expansion of our licensing agreements with other pharmaceutical companies, including co-development projects, joint ventures and other collaborations.
Elite is focusing on the development of various types of drug products, including branded drug products which require new drug applications (“NDAs”) under Section 505(b)(1) or 505(b)(2) of the Drug Price Competition and Patent Term Restoration Act of 1984 (the “ Drug Price Competition Act ”) as well as generic drug products which require ANDAs.
Elite believes that its business strategy enables it to reduce its risk by having a diverse product portfolio that includes both branded and generic products in various therapeutic categories and to build collaborations and establish licensing agreements with companies with greater resources thereby allowing us to share costs of development and improve cash-flow.
| 3 |
Commercial Products
We own, license, contract manufacture or have certain rights to profits for the following products currently being sold commercially:
| Product |
Branded Product Equivalent |
Therapeutic Category |
Launch Date | |||
|
Phentermine HCl 37.5mg tablets (“Phentermine 37.5mg”) |
Adipex-P® | Bariatric | April 2011 | |||
|
Lodrane D ® Immediate Release capsules (“Lodrane D”) |
n/a | OTC Allergy | September 2011 | |||
|
Methadone HCl 10mg tablets (“Methadone 10mg”) |
Dolophine® | Pain | January 2012 | |||
|
Hydromorphone HCl 8mg tablets (“Hydromorphone 8mg”) |
Dilaudid® | Pain | March 2012 | |||
|
Phendimetrazine Tartrate 35mg tablets (“Phendimetrazine 35mg”) |
Bontril® | Bariatric | November 2012 | |||
|
Phentermine HCl 15mg and 30mg capsules (“Phentermine 15mg” and “Phentermine 30mg”) |
Adipex-P® | Bariatric | April 2013 | |||
|
Naltrexone HCl 50mg tablets (“Naltrexone 50mg”) |
Revia® | Pain | September 2013 | |||
|
Isradipine 2.5mg and 5mg capsules (“Isradipine 2.5mg” and “Isradipine 5mg”) |
n/a | Cardiovascular | January 2015 | |||
|
Hydroxyzine HCl 10mg, 25mg and 50mg tablets (“Hydroxyzine 10mg” and “Hydroxyzine 25mg” and “Hydroxyzine 50mg”) |
Atarax®, Vistaril® | Antihistamine | April 2015 | |||
|
Oxycodone HCl Immediate Release 5mg, 10mg, 15mg, 20mg and 30mg tablets (“OXY IR 5mg”, “Oxy IR 10mg”, “Oxy IR 15mg”, “OXY IR 20mg” and “Oxy IR 30mg”) |
Roxycodone® | Pain | March 2016 |
Note: Phentermine 15mg and Phentermine 30mg are collectively and individually referred to as “Phentermine Capsules”. Isradipine 2.5mg and Isradipine 5mg are collectively and individually referred to as “Isradipine Capsules”. Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg are collectively and individually referred to as “Hydroxyzine”. Oxy IR 5mg, Oxy IR 10mg, Oxy IR 15mg Oxy IR 20mg and Oxy IR 30mg are collectively and individually referred to as “Oxy IR”.
Phentermine 37.5mg tablets
The approved Abbreviated New Drug Application “ANDA” for Phentermine 37.5mg was acquired pursuant to an asset purchase agreement with Epic Pharma LLC (“Epic”) dated September 10, 2010 (the “Phentermine Purchase Agreement”). For further details on the Phentermine Purchase Agreement, please see exhibit 10.7 to the Quarterly Report on Form 10-Q, filed with the SEC on November 15, 2010, with such filing being herein incorporated by reference.
Sales and marketing rights for Phentermine 37.5mg are included in the licensing agreement between the Company and Precision Dose Inc. (“Precision Dose”) dated September 10, 2010 (the “Precision Dose License Agreement”). Please see the section below titled “Precision Dose License Agreement” for further details of this agreement.
The first shipment of Phentermine 37.5mg was made to Precision Dose’s wholly owned subsidiary, TAGI Pharmaceuticals Inc. (“TAGI”), pursuant to the Precision Dose License Agreement, with such initial shipment triggering a milestone payment under this agreement. Phentermine 37.5mg is currently being manufactured by Elite and distributed by TAGI under the Precision Dose License Agreement.
| 4 |
Lodrane D® Immediate Release capsules
On September 27, 2011, the Company, along with ECR Pharmaceuticals (“ECR”), launched Lodrane D®, an immediate release formulation of brompheniramine maleate and pseudoephedrine HCl, an effective, low-sedating antihistamine combined with a decongestant.
ECR products have since been divested so that Lodrane D® is promoted and distributed in the U.S. now by Valeant Pharmaceuticals International Inc. Lodrane D® is available over-the-counter but also has physician promotion. Lodrane D® is one of the only adult brompheniramine containing products available to the consumer at this time.
Lodrane D® is marketed under the Over-the-Counter Monograph (the “OTC Monograph”) and accordingly, under the Code of Federal Regulations can be lawfully marketed in the US without prior approval. Under the Federal Food Drug and Cosmetic Act (“FDCA”), FDA regulations and statements of FDA policy, certain drug products are permitted to be marketed in the U.S. without prior approval. Within the past few years, the FDA has revised its enforcement policies, significantly limiting the circumstances under which these unapproved products may be marketed. If the FDA determines that a company is distributing an unapproved product that requires approval, the FDA may take enforcement action in a variety of ways, including, without limitation, product seizures and seeking a judicial injunction against distribution.
There have been several mergers relating to ECR and successor entities and transfer of brand name ownership since this product was originally launched. Lodrane D® is accordingly currently promoted and distributed in the U.S. by Valeant Pharmaceuticals International Inc. (“Valeant”). Lodrane D® is available over-the-counter but also has physician promotion. Lodrane D® is the one of the only adult brompheniramine containing products available to the consumer at this time.
Elite is manufacturing the product for Valeant and will receive revenues for the manufacturing of Lodrane D®.
Methadone 10mg tablets
Methadone 10mg is contract manufactured by Elite for Ascend Laboratories, LLC (“Ascend”), the owner of the approved ANDA.
On January 17, 2012, Elite commenced shipping Methadone 10mg tablets to Ascend pursuant to a commercial manufacturing and supply agreement dated June 23, 2011, as amended on September 24, 2012, January 19, 2015, and July 20, 2015, between Elite and Ascend (the “Methadone Manufacturing and Supply Agreement”). Under the terms of the Methadone Manufacturing and Supply Agreement, Elite performs manufacturing of Methadone 10mg for Ascend.
Hydromorphone 8mg tablets
The approved ANDA for Hydromorphone 8mg was acquired pursuant to an asset purchase agreement with Mikah Pharma LLC dated May 18, 2010 (the “Hydromorphone Purchase Agreement”). Transfer of the manufacturing process of Hydromorphone 8mg to the Northvale Facility, a prerequisite of the Company’s commercial launch of the product, was approved by the FDA on January 23, 2012.
Sales and marketing rights for Hydromorphone 8mg are included in the Precision Dose License Agreement. Please see the section below titled “Precision Dose License Agreement” for further details of this agreement.
| 5 |
The first shipment of Hydromorphone 8mg was made to TAGI, pursuant to the Precision Dose License Agreement, in March 2012, with such initial shipment triggering a milestone payment under this agreement. Hydromorphone 8mg is currently being manufactured by Elite and distributed by TAGI under the Precision Dose License Agreement.
Phendimetrazine Tartrate 35mg tablets
The ANDA for Phendimetrazine 35mg was acquired by Elite as part of the asset purchase agreement between the Company and Mikah Pharma, dated August 1, 2013 (the “Mikah ANDA Purchase”). Please see “Elite’s Acquisition of 13 Abbreviated New Drug Applications (“ANDAs”)” below for more information on this agreement. The Northvale Facility was already an approved manufacturing site for this product as of the date of the Mikah ANDA Purchase. Prior to the acquisition of this ANDA, Elite had been manufacturing this product on a contract basis pursuant to a manufacturing and supply agreement with Mikah Pharma, dated June 1, 2011.
Phendimetrazine 35mg is currently a commercial product being manufactured by Elite and distributed by Epic Pharma LLC (“Epic”) on a non-exclusive basis, and by Elite.
Phentermine 15mg and 30mg capsules
Phentermine 15mg capsules and Phentermine 30mg capsules were developed by the Company, with Elite receiving approval of the related ANDA in September 2012.
Sales and marketing rights for Phentermine 15mg and Phentermine 30mg are included in the Precision Dose License Agreement. Please see the section below titled “Precision Dose License Agreement” for further details of this agreement.
The first shipments of Phentermine 15mg and Phentermine 30mg were made to TAGI, pursuant to the Precision Dose License Agreement, in April 2013, with such initial shipments triggering a milestone payment under this agreement. Phentermine 15mg and Phentermine 30mg are currently being manufactured by Elite and distributed by TAGI under the Precision Dose License Agreement.
Naltrexone 50mg tablets
The approved ANDA for Naltrexone 50mg was acquired by the Company pursuant to an asset purchase agreement between the Company and Mikah Pharma dated August 27, 2010 (the “Naltrexone Acquisition Agreement”) for aggregate consideration of $200,000.
Sales and marketing rights for Hydromorphone 8mg are included in the Precision Dose License Agreement. Please see the section below titled “Precision Dose License Agreement” for further details of this agreement.
The first shipment of Naltrexone 50mg was made to TAGI, pursuant to the Precision Dose License Agreement, in September 2013, with such initial shipment triggering a milestone payment under this agreement. Naltrexone 50mg is currently being manufactured by Elite and distributed by TAGI under the Precision Dose License Agreement.
Isradipine 2.5mg and Isradipine 5mg capsules
The approved ANDAs for Isradipine 2.5mg and Isradipine 5mg were acquired by Elite as part of the Mikah ANDA Purchase
Sales and marketing rights for Isradipine 2.5mg and Isradipine 5mg are included in the manufacturing and license agreement between the Company and Epic Pharma LLC, dated October 2, 2013 (the “Epic Manufacturing and License Agreement”). Please see the section below titled “Epic Manufacturing and License Agreement” for further details of this agreement.
| 6 |
The first shipment of Isradipine 2.5mg and Isradipine 5mg were made to Epic, pursuant to the Epic Manufacturing and License Agreement, in January 2015. Isradipine 2.5mg and Isradipine 5mg are currently being manufactured by Elite and distributed by Epic under the Epic Manufacturing and License Agreement.
Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg tablets
The approved ANDAs for Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg were acquired by Elite as part of the Mikah ANDA Purchase.
Sales and marketing rights for Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg are included in the Epic Manufacturing and License Agreement.
The first shipment of Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg were made by Epic, pursuant to the Epic Manufacturing and License Agreement, in April 2015. Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg are currently being manufactured and distributed by Epic under the Epic Manufacturing and License Agreement.
Oxycodone 5mg, Oxycodone 10mg, Oxycodone 15mg, Oxycodone 20mg and Oxycodone 30mg tablets
The Company received notification from Epic in October 2015 of the approval by the FDA of Epic’s ANDA for Oxy IR. This product was an Identified IR Product in the Epic Strategic Alliance Agreement Dated March 18, 2009 (the “Epic Strategic Alliance”). Oxy IR was developed at the Northvale Facility pursuant to the Epic Strategic Alliance, with the Company being entitled to a Product Fee of 15% of Profits as defined in the Epic Strategic Alliance.
Epic advised the Company that the first commercial sale of Oxy IR occurred in March 2016 and such sales are ongoing
Approved products not yet commercialized
The Company currently owns six different approved ANDA’s, all of which were acquired as part of the Mikah ANDA Purchase. Each of these approved ANDA’s require manufacturing site transfers as a prerequisite to commencement of commercial manufacturing and distribution. The products are relating to each of these approved ANDA’s are included in the Epic Manufacturing and License Agreement, with Elite granting ANDA specific, exclusive or non-exclusive market rights (depending on the ANDA) to Epic. Commercial manufacturing of these products is expected to be transferred to either Epic or the Northvale Facility, with the required supplements to be filed with FDA in the manner and time frame that is economically beneficial to the Company.
Filed products under FDA review
SequestOx™ - Immediate Release Oxycodone with sequestered Naltrexone
SequestOx™ is Elite’s lead abuse-deterrent candidate for the management of moderate to severe pain where the use of an opioid analgesic is appropriate. SequestOx™ is an immediate-release Oxycodone Hydrochloride containing sequestered Naltrexone which incorporates 5mg, 10mg, 15mg, 20mg and 30mg doses of oxycodone into capsules.
| 7 |
In January 2016, the Company submitted a 505(b)(2) New Drug Application for SequestOx™, after receiving a waiver of the $2.3 million filing fee from the FDA. In March 2016, the Company received notification of the FDA’s acceptance of this filing and that such filing has been granted priority review by the FDA with a target action under the Prescription Drug User Fee Act (“PDUFA”) of July 14, 2016.
This application is currently under review by the FDA.
Please note however that there can be no assurances that this product will receive marketing authorization and achieve commercialization within this time period, or at all. In addition, even if marketing authorization is received, there can be no assurances that there will be future revenues of profits, or that any such future revenues or profits would be in amounts that provide adequate return on the significant investments made to secure this marketing authorization.
Asset Acquisition Agreements
Elite’s Purchase of a Generic Phentermine Product
On September 10, 2010, Elite, together with its subsidiary, Elite Laboratories, Inc., executed a Purchase Agreement (the “Phentermine Purchase Agreement”) with Epic Pharma, LLC (“Epic”) for the purpose of acquiring from Epic an ANDA for a generic phentermine product (the “Phentermine ANDA”), with such being filed with the FDA at the time the Phentermine Purchase Agreement was executed. On February 4, 2011, the FDA approved the Phentermine ANDA. The acquisition of the Phentermine ANDA closed on March 31, 2011 and Elite paid the full acquisition price of $450,000 from the purchase agreement with Epic Pharma.
This product is being marketed and distributed by Precision Dose Inc (“Precision Dose”) and its wholly owned subsidiary, TAGI Pharma Inc. (“TAGI”) pursuant to the Precision Dose License Agreement, a description of which is set forth below.
Elite’s Purchase of a Generic Hydromorphone HCl Product
On May 18, 2010, Elite executed an asset purchase agreement with Mikah Pharma LLC (“Mikah”) (the “Hydromorphone Purchase Agreement”). Pursuant to the Hydromorphone Purchase Agreement, the Company acquired from Mikah an approved ANDA for Hydromorphone 8 mg for aggregate consideration of $225,000, comprised of an initial payment of $150,000, which was made on May 18, 2010. A second payment of $75,000 was due to be paid to Mikah on June 15, 2010, with the Company having the option to make this payment in cash or by issuing to Mikah 937,500 shares of the Company’s Common Stock. The Company elected and did issue 937,500 shares of Common Stock during the quarter ended December 31, 2010, in full payment of the $75,000 due to Mikah pursuant to the asset purchase agreement dated May 18, 2010.
This product is currently being marketed and distributed by Precision Dose and its wholly owned subsidiary, TAGI, pursuant to the Precision Dose License Agreement, a description of which is set forth below.
Elite’s Purchase of a Generic Naltrexone Product
On August 27, 2010, Elite executed an asset purchase with Mikah (the “Naltrexone Acquisition Agreement”). Pursuant to the Naltrexone Acquisition Agreement, Elite acquired from Mikah the ANDA number 75-274 (Naltrexone Hydrochloride Tablets USP, 50 mg), and all amendments thereto, that have to date been filed with the FDA seeking authorization and approval to manufacture, package, ship and sell the products described in this ANDA within the United States and its territories (including Puerto Rico) for aggregate consideration of $200,000. In lieu of cash, Mikah agreed to accept from Elite product development services to be performed by Elite.
| 8 |
This product is currently being marketed and distributed by Precision Dose and its wholly owned subsidiary, TAGI, pursuant to the Precision Dose License Agreement, a description of which is set forth below.
Elite’s Acquisition of 13 Abbreviated New Drug Applications
On August 1, 2013, Elite executed an asset purchase agreement (the “Mikah ANDA Purchase”) with Mikah and acquired from Mikah a total of 13 ANDAs, consisting of 12 ANDAs approved by the FDA and one ANDA under active review with the FDA, and all amendments thereto (the “Mikah 13 ANDA Acquisition”) for aggregate consideration of $10,000,000, payable pursuant to a secured convertible note due in August 2016.
Each of the products referenced in the 12 approved ANDAs require manufacturing site approval with the FDA. Elite believes that the site transfers qualify for CBE 30 review, with one exception, which would allow for the product manufacturing transfer on an expedited basis. However, Elite can give no assurances that all will qualify for CBE 30 review, or on the timing of these transfers of manufacturing site, or on the approval by the FDA of the transfers of manufacturing site.
As of the filing date of this Annual Report on Form 10-K, the following products included in the Mikah Purchase Agreement have successfully achieved manufacturing site transfers:
| · | Phendimetrazine 35mg |
| · | Isradipine 2.5mg and Isradipine 5mg |
| · | Hydroxyzine 10mg, Hydroxyzine 25mg and Hydroxyzine 50mg |
Elite has executed a Manufacturing and License Agreement with Epic Pharma dated October 2, 2013 (the “Epic Pharma Manufacturing and License Agreement”), relating to the manufacturing, marketing and sale of these 12 ANDAs. Please see below for further details on the Epic Pharma Manufacturing and License Agreement.
Licensing, Manufacturing and Development Agreements
Sales and Distribution Licensing Agreement with Epic Pharma LLC for SequestOx™
On June 4, 2015, Elite Pharmaceuticals Inc. and its wholly-owned subsidiary Elite Laboratories, Inc. (collectively, “Elite”) executed an exclusive License Agreement (the “2015 SequestOx™ License Agreement”) with Epic Pharma LLC. (“Epic”), to market and sell in the United States, SequestOx™, an immediate release oxycodone with sequestered naltrexone capsule, owned by Elite. Epic will have the exclusive right to market ELI-200 and its various dosage forms as listed in Schedule A of the Agreement (the “Products”). Epic is responsible for all regulatory and pharmacovigilance matters related to the products. Pursuant to the 2015 SequestOx™ License Agreement, Epic will pay Elite non-refundable payments totaling $15 million, with such amount representing the cost of an exclusive license to SequestOx™, the cost of developing the product, the filing of a New Drug Application (“NDA”) with the U.S. Food and Drug Administration (“FDA”) and the receipt of the approval letter for the NDA from the FDA. As of the date of filing of this annual report on Form 10-K, the Company has received $7.5 million of the $15 million in non-refundable payments due pursuant to the 2015 SequestOx™ License Agreement, with such amount consisting of $5 million being due and owing on the execution date of the 2015 SequestOx™ License Agreement, and $2.5 million being earned as of January 14, 2016, the date of Elite’s filing of an NDA with the FDA for the relevant product. Both of these non-refundable fees (i.e., the $5 million fee and the $2.5 million fee), have been paid by Epic to the Company. The remaining $7.5 million in non-refundable payments due pursuant to the 2015 SequestOx™ License Agreement is due on the FDA’s approval of SequestOx™ for commercial sale in the United States of America. In addition, Elite will receive a license fee computed as a percentage (50%) of net sales of the Products as defined in the Agreement and is entitled to multi million dollar minimum annual license fees Elite will manufacture the product for sale by Epic on a cost plus basis and both parties agree to execute a separate Manufacturing and Supply Agreement. The license fee is payable quarterly for the term of the Agreement. The term of the License Agreement is five years and may be extended for an additional five years upon mutual agreement of the parties. Elite can terminate the Agreement on 90 days’ written notice in the Event that Epic does not pay to Elite certain minimum annual license fees over the initial five year term of the Agreement. Either party may terminate this Agreement upon a material breach and failure to cure that breach by the other party within a specified period.
| 9 |
Manufacturing and License Agreement with Epic Pharma LLC
On October 2, 2013, Elite executed the Epic Pharma Manufacturing and License Agreement (the “Epic Generic Agreement”). This agreement granted Epic Pharma certain rights to manufacture, market and sell in the United States and Puerto Rico the 12 approved ANDAs acquired by Elite pursuant to the Mikah Purchase Agreement. Of the 12 approved ANDAs, Epic Pharma will have the exclusive right to market six products as listed in Schedule A of the Epic Pharma Manufacturing and License Agreement, and a non-exclusive right to market six products as listed in Schedule D of the Epic Pharma Manufacturing and License Agreement. Epic Pharma will manufacture the products and Epic is responsible for all regulatory and pharmacovigilance matters related to the products and for all costs related to the site transfer for all products. Elite has no further obligations or deliverables under the Epic Generic Agreement. Pursuant to the Epic Generic Agreement, Elite will receive a license fee and milestone payments. The license fee will be computed as a percentage of the gross profit, as defined in the Epic Pharma Manufacturing and License Agreement, earned by Epic Pharma a result of sales of the products. The manufacturing cost used for the calculation of the license fee is a predetermined amount per unit plus the cost of the drug substance (API) and the sales cost for the calculation is predetermined based on net sales. If Elite manufactures any product for sale by Epic Pharma, then Epic Pharma shall pay to Elite that same predetermined manufacturing cost per unit plus the cost of the API. The license fee is payable monthly for the term of the Epic Pharma Manufacturing and License Agreement. Epic Pharma shall pay to Elite certain milestone payments as defined by the Epic Pharma Manufacturing and License Agreement. To date, milestones totaling $1,000,000 have been earned and received in relation to the signing of the Epic Pharma Manufacturing and License Agreement and the filing and approval by the FDA of supplements relating to the transfer of manufacturing site for Isradipine 2.5mg and Isradipine 5mg. The term of the Epic Pharma Manufacturing and License Agreement is five years and may be extended for an additional five years upon mutual agreement of the parties. Twelve months following the launch of a product covered by the Epic Pharma Manufacturing and License Agreement, Elite may terminate the marketing rights for any product if the license fee paid by Epic Pharma falls below a designated amount for a six month period of that product. Elite may also terminate the exclusive marketing rights if Epic Pharma is unable to meet the annual unit volume forecast for a designated product group for any year, subject to the ability of Epic Pharma, during the succeeding six month period, to achieve at least one-half of the prior year’s minimum annual unit forecast. The Epic Pharma Manufacturing and License Agreement may be terminated by mutual agreement of Elite and Epic Pharma, as a result of a breach by either party that is not cured within 60 days notice of the breach, or by Elite as a result of Epic Pharma becoming a party to a bankruptcy, reorganization or other insolvency proceeding that continues for a period of 30 days or more.
Methadone Manufacturing and Supply Agreement
On June 23, 2011 and as amended on September 24, 2012, January 19, 2015 and July 20, 2015, Elite entered into an agreement to manufacture and supply Methadone 10mg to ThePharmaNetwork LLC (the “Methadone Manufacturing and Supply Agreement”). ThePharmaNetworkLLC was subsequently acquired by Alkem Laboratories Ltd (“Alkem”) and now goes by the name Ascend Laboratories LLC (“Ascend”) and is a wholly owned subsidiary of Alkem.
| 10 |
Ascend in the owner of the approved ANDA for Methadone 10mg, and the Northvale Facility is an approved manufacturing site for this ANDA. The Methadone Manufacturing and Supply Agreement provides for the manufacture and packaging by the Company of Ascend’s methadone hydrochloride 10mg tablets.
The initial shipment of Methadone 10mg pursuant to the Methadone Manufacturing and Supply Agreement occurred in January 2012.
Licensing Agreement with Precision Dose Inc.
On September 10, 2010, Elite executed a License Agreement with Precision Dose (the “Precision Dose License Agreement”) to market and distribute Phentermine 37.5mg, Phentermine 15mg, Phentermine 30mg, Hydromorphone 8mg, Naltrexone 50mg, and certain additional products that require approval from the FDA, through its wholly-owned subsidiary, TAGI Pharma, Inc. in the United States, Puerto Rico and Canada (the “Precision Dose License Agreement”). Phentermine 37.5mg was launched in April 2011. Hydromorphone 8mg was launched in March 2012. Phentermine 15mg and Phentermine 30mg were launched in April 2013. Naltrexone 50mg was launched in September 2013. Precision Dose will have the exclusive right to market these products in the United States and Puerto Rico and a non-exclusive right to market the products in Canada.
Pursuant to the Precision Dose License Agreement, Elite will receive a license fee and milestone payments. The license fee will be computed as a percentage of the gross profit, as defined in the Precision Dose License Agreement, earned by Precision Dose as a result of sales of the products. The license fee is payable monthly for the term of the Precision Dose License Agreement. The milestone payments will be paid in six installments. The first installment was paid upon execution of the License Agreement. The remaining installments are to be paid upon FDA approval and initial shipment of the products to Precision Dose. The term of the License Agreement is 15 years and may be extended for 3 successive terms, each of 5 years. Please see Part II, Item 1, Legal Proceedings below for details of an arbitration proceeding commenced by Precision Dose related to certain terms and conditions of the Precision Dose License Agreement.
Development agreement with Akorn Pharmaceuticals
On January 10, 2011, Elite and Hi-Tech Pharmacal Co, Inc. (subsequently acquired by Akorn Pharmaceuticals), entered into an agreement for Elite to develop an intermediate product for a generic version of a prescription product for Akorn Pharmaceuticals (“Akorn”). Under the terms of the agreement, Elite will undertake a development program for an intermediate product that Akorn shall then incorporate into a final product. Akorn or its designees, shall be responsible for the filing of the ANDA for the finished product and the ANDA will be filed under the Akorn name. Upon approval of the ANDA, Elite will manufacture the intermediate product. Akorn will manufacture the final product and will be responsible for the marketing and sales of the final product. Akorn will pay Elite milestone payments for the development work. Upon commercialization, Elite will receive payment for the manufacturing of the intermediate product and a percentage of the profits generated from the sale of the product.
As of the date of filing of this annual report on Form 10-K, there has been minimal to no development activity conducted in relation to this agreement for substantially all of the fiscal year ended March 31, 2016. Furthermore, there can be no assurances that development activities will resume or that the resumption of development activities, is such were to occur, would result in the successful development of the product identified.
For further details, please refer to Exhibit 10.51 to this Annual Report on Form 10-K.
| 11 |
Research and Development
Elite’s research and development activities are primarily focused on developing its proprietary abuse deterrent technology and the development of a range of abuse deterrent opioid products that utilize this technology.
Elite’s proprietary abuse-deterrent technology, utilizes the pharmacological approach to abuse deterrence and consists of a multi-particulate capsule which contains an opioid agonist in addition to naltrexone, an opioid antagonist used primarily in the management of alcohol dependence and opioid dependence. When this product is taken as intended, the naltrexone is designed to pass through the body unreleased while the opioid agonist releases over time providing therapeutic pain relief for which it is prescribed. If the multi-particulate beads are crushed or dissolved, the opioid antagonist, naltrexone, is designed to release. The absorption of the naltrexone is intended to block the euphoria by preferentially binding to same receptors in the brain as the opioid agonist and thereby reducing the incentive for abuse or misuse by recreational drug abusers.
We filed an NDA for the first product to utilize our abuse deterrent technology, Immediate Release Oxycodone 5mg, 10mg, 15mg, 20mg and 30mg with sequestered Naltrexone (collectively and individually referred to as “SequestOx™”), on January 14, 2016. On March 17, 2016, the FDA notified Elite that its NDA application was accepted and granted priority review. The Company believes that, subject to the risks described below, approval of this NDA filing could be received during the fiscal year ended March 31, 2017.
The Company believes that the abuse deterrent technology can be applied to and incorporated into a wide range of opioids used today for pain management and has, to date, identified 10 additional products for potential development. All of these products are at early stages of development, with research and development activities mainly consisting of in-house process development and laboratory studies. Extensive efficacy and safety studies, similar to those conducted for SequestOx™ have not yet been conducted for these other products. As a result, costs incurred in relation to the development of these 10 products have not been material.
Research and development costs were $12.4 million, $14.7 million and $4.0 million for the years ended March 31, 2016, 2015 and 2014, respectively, with such costs relating almost entirely to the development of SequestOx™, the first product developed by Elite that incorporates the technology.
On June 4, 2015, the Company entered into a sales and distribution licensing agreement which included a non-refundable payment of $5 million to Elite for prior research and development activities, with such representing the first material net cash inflows being generated by ELI-200. On January 14, 2016, the Company filed an NDA with the FDA for SequestOx™, thereby earning a non-refundable $2.5 million milestone. An additional $7.5 million non-refundable milestone is due upon the FDA’s approval of Elite’s NDA. Please note, as further detailed below, there can be no assurances of the Company receiving marketing authorization for SequestOx™, and accordingly, there can be no assurances that the Company will earn and receive the additional $7.5 million or future license fees. The non receipt by the Company of these payments and or fees may materially and adversely affect our financial condition.
Please note that, while the FDA is required to review applications within certain timeframes, during the review process, the FDA frequently requests that additional information be submitted. The effect of such request and subsequent submission can significantly extend the time for the NDA review process. Until an NDA is actually approved, there can be no assurances that the information requested and submitted will be considered adequate by the FDA to justify approval. The packaging and labeling of our developed products are also subject to FDA regulation. Based on the foregoing, it is impossible to anticipate the amount of time that will be needed to obtain FDA approval to market any product. In addition, there can be no assurances of the Company filing the required application(s) with the FDA or of the FDA approving such application(s) if filed, and the Company’s ability to successfully develop and commercialize products incorporating its abuse deterrent technology is subject to a high level of risk as detailed in “Item 1A-Risk Factors-Risks Related to our Business” of this Annual Report on Form 10-K.
| 12 |
Abuse-Deterrent and Sustained Release Opioids
The abuse-deterrent opioid products utilize our patented abuse-deterrent technology that is based on a pharmacological approach. These products are combinations of a narcotic agonist formulation intended for use in patients with pain, and an antagonist, formulated to deter abuse of the drug. Both, agonist and antagonist, have been on the market for a number of years and sold separately in various dose strengths. Elite has filed INDs for two abuse resistant products under development and has tested products in various pharmacokinetic studies. Elite expects to continue to develop multiple abuse resistant products. Products utilizing the pharmacological approach to deter abuse such as Suboxone®, a product marketed in the United States by Reckitt Benckiser Pharmaceuticals, Inc., and Embeda®, a product marketed in the United States by Pfizer, Inc., have been approved by the FDA and are being marketed in the United States.
Elite has developed, and retains the rights to these abuse resistant and sustained release opioid products. Elite may license these products at a later date to a third party who could provide funding for the remaining clinical studies and who could provide sales and distribution for the product.
Elite also developed controlled release technology for oxycodone under a joint venture with Elan which terminated in 2002. According to the Elan Termination Agreement, Elite acquired all proprietary, development and commercial rights for the worldwide markets for the products developed by the joint venture, including the sustained release opioid products. Upon licensing or commercialization of an oral controlled release formulation of oxycodone for the treatment of pain, Elite will pay a royalty to Elan pursuant to the Termination Agreement. If Elite were to sell the product itself, Elite will pay a 1% royalty to Elan based on the product’s net sales, and if Elite enters into an agreement with another party to sell the product, Elite will pay a 9% royalty to Elan based on Elite’s net revenues from this product. (Elite’s net product revenues would include license fees, royalties, manufacturing profits and milestones) Elite is allowed to recoup all development costs including research, process development, analytical development, clinical development and regulatory costs before payment of any royalties to Elan.
Patents
Since our incorporation, we have secured the following patents, of which two have been assigned for a fee to another pharmaceutical company. Elite’s patents are:
| PATENT | EXPIRATION DATE | |
| U.S. patent 5,837,284 (assigned to Celgene Corporation) | November 2018 | |
| U.S. patent 6,620,439 | October 2020 | |
| U.S. patent 6,635,284 (assigned to Celgene Corporation) | March 2018 | |
| U.S. patent 6,926,909 | April 2023 | |
| U.S. patent 8,182,836 | April 2024 | |
| U.S. patent 8,425,933 | April 2024 | |
| U.S. patent 8,703,186 | April 2024 | |
| Canadian patent 2,521,655 | April 2024 | |
| Canadian patent 2,541,371 | September 2024 | |
| U.S. patent 9,056,054 | June 2030 |
| 13 |
We also have pending applications for two additional U.S. patents and three foreign patents. We intend to apply for patents for other products in the future; however, there can be no assurance that any of the pending applications or other applications which we may file will be granted. We have also filed corresponding foreign applications for key patents.
Prior to the enactment in the United States of new laws adopting certain changes mandated by the General Agreement on Tariffs and Trade (“GATT”), the exclusive rights afforded by a U.S. Patent were for a period of 17 years measured from the date of grant. Under GATT, the term of any U.S. Patent granted on an application filed subsequent to June 8, 1995 terminates 20 years from the date on which the patent application was filed in the United States or the first priority date, whichever occurs first. Future patents granted on an application filed before June 8, 1995, will have a term that terminates 20 years from such date, or 17 years from the date of grant, whichever date is later.
Under the Drug Price Competition Act, a U.S. product patent or use patent may be extended for up to five years under certain circumstances to compensate the patent holder for the time required for FDA regulatory review of the product. Such benefits under the Drug Price Competition Act are available only to the first approved use of the active ingredient in the drug product and may be applied only to one patent per drug product. There can be no assurance that we will be able to take advantage of this law.
Also, different countries have different procedures for obtaining patents, and patents issued by different countries provide different degrees of protection against the use of a patented invention by others. There can be no assurance, therefore, that the issuance to us in one country of a patent covering an invention will be followed by the issuance in other countries of patents covering the same invention, or that any judicial interpretation of the validity, enforceability, or scope of the claims in a patent issued in one country will be similar to the judicial interpretation given to a corresponding patent issued in another country. Furthermore, even if our patents are determined to be valid, enforceable, and broad in scope, there can be no assurance that competitors will not be able to design around such patents and compete with us using the resulting alternative technology.
We also rely upon unpatented proprietary and trade secret technology that we seek to protect, in part, by confidentiality agreements with our collaborative partners, employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. There can be no assurance that these agreements provide meaningful protection or that they will not be breached, that we will have adequate remedies for any such breach, or that our trade secrets, proprietary know-how, and technological advances will not otherwise become known to others. In addition, there can be no assurance that, despite precautions taken by us, others have not and will not obtain access to our proprietary technology.
Trademarks
SequestOx™ is a trademark owned by Elite, which received a Notice of Allowance by the United States Patent and Trademark Office on December 22, 2015.
In addition, we currently plan to license at least some of our products to other entities in the marketing of pharmaceuticals, but may also sell products under our own brand name in which case we may register trademarks for those products.
| 14 |
Discontinued Products, Terminated Agreements, Prior Investments
Discontinued Products - Lodrane 24® and Lodrane 24D®
On March 3, 2011, the FDA announced its intention to remove approximately 500 cough/cold and allergy related products from the U.S. market. The once daily allergy products manufactured by Elite, Lodrane 24® and Lodrane 24D® (the “Lodrane® Extended Release Products”), were included in the FDA list of 500 products. After this announcement by the FDA, the Company’s customer for the Lodrane® Extended Release Products cancelled all outstanding orders and manufacturing of the Lodrane® Extended Release Products has ceased. The shipments made during the quarter ended June 30, 2011 consisted solely of quantities that were in production at the time ECR cancelled all outstanding orders. There were no shipments of the Lodrane Extended Release Products subsequent to those that were made during the quarter ended June 30, 2011.
ECR (the owner and marketer of the Lodrane® Extended Release Products) initiated a formal approval process with the FDA in 2010 regarding the Lodrane® Extended Release Products and issued a press release on March 3, 2011 stating that they will continue to actively pursue approval for the Lodrane® Extended Release Products. In addition, on April 29, 2011, ECR filed a Petition for Review with the United States Court of Appeals for the District of Columbia, petitioning such court to review and set aside the final order of the FDA with relation to the Lodrane® Extended Release Products. The Company has received no further information from ECR with regards to the status of the Petition filed.
The Lodrane® Extended Release Products were co-developed with our partner, ECR, and the Company was receiving revenues from the manufacture of the Lodrane® Products and laboratory stability study services, as well as royalties on in-market sales. Contracts relating to the manufacture and sale of the Lodrane® Extended Release Products were formally terminated on April 26, 2013.
During the three months ended June 30, 2011, Elite made its final shipments of the Lodrane® Extended Release Products. In addition, the Company sold to ECR, at cost without markup, all raw materials related to the manufacture of the Lodrane® Extended Release Products which remained in stock subsequent to the final shipment of the Lodrane® Extended Release Products. As manufacturing of the Lodrane® Extended Release Products has ceased, there will be no further manufacturing revenues derived from the Lodrane® Extended Release Products unless and until such products receive the necessary approvals from the FDA.
Please note that there can be no assurances that such approvals will be granted or that future manufacturing revenues will be earned by the Company from the manufacture of the Lodrane® Extended Release Products, should such approvals be granted by the FDA. Furthermore, the Company has been advised that ECR has decided not to proceed with the development of the extended release formulations marketed under the Lodrane® brand. The Company also has no plans currently to proceed with the development of an extended release brompheniramine/pseudoephedrine product. Notwithstanding the foregoing, Elite may proceed with the development of these formulations and may seek partners in conjunction with such activities, but there can be no assurances that the Company will pursue the development of these formulations, or that such development activities, if pursued, will result in approvals from the FDA. Please also note that the Company does not have ownership of the Lodrane® brand name, and that if any products containing the formulations associated with the Lodrane® brand name are approved and marketed, such would be done under a different brand name.
While Elite’s manufacturing of the Lodrane® Extended Release Products has ceased, the sale of such products in the US market was still permitted by the FDA until August 30, 2011. The Company earned royalties on any in-market sales that occurred up to that date.
| 15 |
Terminated Agreement - Contract Manufacturing of Isradipine and Phendimetrazine
On June 1, 2011, Elite executed a Manufacturing and Supply Agreement (the “Isradipine/ Phendimetrazine Agreement”) with Mikah Pharma, LLC (“Mikah”) to undertake and perform certain services relating to two generic products: Isradipine Capsules USP, 2.5 mg and 5 mg (“Isradipine”) and Phendimetrazine Tartrate Tablets USP, 35 mg (“Phendimetrazine”), including (a) developing and preparing the documentation required for the transfer of the manufacturing process to Elite’s facility and the appropriate regulatory filing for the ANDA, and (b) manufacturing finished dosage forms appropriate for commercial sale, marketing and distribution in the United States, its territories, possessions, and commonwealths in accordance with the requirements of the Isradipine/ Phendimetrazine Agreement; Elite is required to perform, at its sole cost and expense, all Technology Transfer, validation and qualification services (including: equipment, methods and facility qualification), validation and stability services required by Applicable Laws to commence manufacturing Isradipine and Phendimetrazine for commercial sale by Mikah or its designees in accordance with the terms of the Isradipine/ Phendimetrazine Agreement. During the term of the Isradipine/ Phendimetrazine Agreement and subject to the provisions therein, Mikah is required to purchase from Elite and Elite agrees to manufacture and supply solely and exclusively to Mikah, such Isradipine and Phendimetrazine as Mikah may order from time to time pursuant to the Isradipine/ Phendimetrazine Agreement. Mikah will compensate Elite at an agreed upon transfer price for the manufacturing and packaging of Isradipine and Phendimetrazine. For the Isradipine product, Elite will also receive a 10% royalty on net profits of the finished Product. The payment is to be calculated and paid quarterly. Elite will also receive a onetime milestone payment for each Product for the work associated with the Technology transfer. The milestone payment shall be made upon the successful manufacturing and testing of the exhibit batch. The Isradipine/ Phendimetrazine Agreement has a term of five years and automatically renews for additional periods of one year unless Mikah provides written notice of termination to Elite at least six months prior to the expiration of the Term or any Renewal Term.
On November 13, 2012, the Company made the initial shipment of Phendimetrazine tartrate 35mg tablets, the generic equivalent of Bontril PDM® 35mg tablets under a previously announced manufacturing and supply agreement with Mikah Pharma (“Mikah”).
Bontril PDM® and its generic equivalents had total U.S. sales of approximately $3.5 million for the twelve months ended September 2012, based on IMS Health Data. The Company will be compensated at an agreed upon price for the manufacturing and packaging of this product.
On August 1, 2013, Elite executed the Mikah Purchase Agreement in relation to the Mikah 13 ANDA Acquisition, with such transaction including the transfer of ANDAs for Phendimetrazine 35mg and Isradipine 2.5mg and 5mg. In addition, the principal owner of Mikah, Mr. Nasrat Hakim, assumed the position of Elite’s Chief Executive Officer and President on August 2, 2013. Accordingly, the Isradipine/Phendimetrazine Agreement has been terminated by mutual consent of the parties thereto.
Terminated Agreement – Mikah Development Agreement
On January 28, 2015, The Development and License Agreement dated August 27, 2010 and between the Company and Mikah Pharma LLC (the “Mikah Development Agreement”) was terminated by mutual agreement of the Company and Mikah Pharma LLC.
Pursuant to the Mikah Development Agreement, Mikah Pharma LLC (“Mikah”) made advance consideration payments to the Company totaling $200,000 in exchange for product development services to be provided at a future date. Subsequent to the execution of the Mikah Development Agreement, and before any development milestones were achieved, the sole owner of Mikah, Mr. Nasrat Hakim, became the President and Chief Executive Officer of the Company. Mikah has accordingly ceased operating and is in the process of winding down and liquidating its assets.
| 16 |
Any further development of the product related to this agreement will belong to the Company, although there can be no assurances that such development will occur or be successful.
The Mikah Development Agreement requires that the consideration paid in advance to the Company be refunded in the event of no milestones being achieved. Mr. Hakim, as owner of Mikah, has directed that the $200,000 refund due to Mikah not be paid currently, but rather be added to the amounts due under the Hakim Credit Line.
For further details on the Mikah Development Agreement, please see Exhibit 10.6 of the Quarterly Report on Form 10-Q filed with the SEC on November 14, 2010, with such filing being herein incorporated by reference.
For further details on the termination of the Mikah Development Agreement, please see Exhibit 10.84 of the Quarterly Report on Form 10-Q, filed with the SEC on February 17, 2015, with such filing being herein incorporated by reference.
Terminated Agreement - Development and License Agreement with Hong Kong based company
On January 19, 2016, the Development and License Agreement (“D&L Agreement”) between the Company and a private Hong-Kong based company dated March 16, 2016 was terminated. The D&L Agreement was for Elite to develop for the Hong Kong-based Customer a branded prescription pharmaceutical product in the United States. The Hong Kong-based Customer has informed us that it has been in business for more than five years and it has multiple FDA approved manufacturing sites outside of the United States.
Pursuant to the D&L Agreement, the Hong Kong-based Customer engaged Elite to develop and manufacture a prescription pharmaceutical product (the “Prescription Product”), with such development not being successfully completed.
For further details on the D&L Agreement, please refer to Exhibit 10.77 to the Annual Report on Form 10-K filed with the SEC on June 29, 2012.
Novel Labs Investment
At the end of 2006, Elite entered into a joint venture with VGS Pharma, LLC (“VGS”) and created Novel Laboratories, Inc. (“Novel”), a privately-held company specializing in pharmaceutical research, development, manufacturing, licensing, acquisition and marketing of specialty generic pharmaceuticals. Novel's business strategy is to focus on its core strength in identifying and timely executing niche business opportunities in the generic pharmaceutical area. Elite owned less than 10% of the outstanding shares of Class A Voting Common Stock of Novel.
Elite commenced an action against VGS, Novel and related parties (collectively, the “VGS Parties”) related to the Novel transactions. The action was settled and, pursuant to that settlement, in June 2014, Elite received $5,000,000 from the VGS Parties in exchange for 9,800 shares of Novel Class A common stock owned by Elite. This resolved all disputes and claims between the Company and the VGS Parties and ended the Company’s ownership in Novel.
| 17 |
Other Business Factors and Details
Government Regulation and Approval
The design, development and marketing of pharmaceutical compounds, on which our success depends, are intensely regulated by governmental regulatory agencies, in particular the FDA. Non-compliance with applicable requirements can result in fines and other judicially imposed sanctions, including product seizures, injunction actions and criminal prosecution based on products or manufacturing practices that violate statutory requirements. In addition, administrative remedies can involve voluntary withdrawal of products, as well as the refusal of the FDA to approve ANDAs and NDAs. The FDA also has the authority to withdraw approval of drugs in accordance with statutory due process procedures.
Before a drug may be marketed, it must be approved by the FDA either by an NDA or an ANDA, each of which is discussed below.
Please note that, as discussed in “Discontinued Products” above, in March 2011, the FDA announced its intention to remove approximately 500 cough/cold and allergy related products from the U.S. market, with such list of 500 products including the Lodrane Extended Release Products. After this announcement by the FDA, the Company’s customer for the Lodrane Products cancelled all outstanding orders and manufacturing of the Lodrane Products ceased. This cancellation of outstanding orders and the cessation of manufacturing of Lodrane Products had a material adverse effect on revenues for periods beginning subsequent to March 31, 2011.
Lodrane D® which is an immediate release product that is different from the Lodrane Products that were included in the list of products removed from the market by the FDA, is marketed under the Over-the-Counter Monograph (the “OTC Monograph”) and accordingly, under the Code of Federal Regulations can be lawfully marketed in the U.S. without prior approval. Under the Federal Food Drug and Cosmetic Act (“FDCA”), FDA regulations and statements of FDA policy, certain drug products are permitted to be marketed in the U.S. without prior approval. Within the past few years, the FDA has revised its enforcement policies, significantly limiting the circumstances under which these unapproved products may be marketed. If the FDA determines that a company is distributing an unapproved product that requires approval, the FDA may take enforcement action in a variety of ways, including, without limitation, product seizures and seeking a judicial injunction against distribution.
NDAs and NDAs under Section 505(b) of the Drug Price Competition Act
The FDA approval procedure for an NDA is generally a two-step process. During the Initial Product Development stage, an investigational new drug application (“IND”) for each product is filed with the FDA. A 30-day waiting period after the filing of each IND is required by the FDA prior to the commencement of initial clinical testing. If the FDA does not comment on or question the IND within such 30-day period, initial clinical studies may begin. If, however, the FDA has comments or questions, they must be answered to the satisfaction of the FDA before initial clinical testing may begin. In some instances this process could result in substantial delay and expense. Initial clinical studies generally constitute Phase I of the NDA process and are conducted to demonstrate the product tolerance/safety and pharmacokinetic in healthy subjects.
After Phase I testing, extensive efficacy and safety studies in patients must be conducted. After completion of the required clinical testing, an NDA is filed, and its approval, which is required for marketing in the United States, involves an extensive review process by the FDA. The NDA itself is a complicated and detailed application and must include the results of extensive clinical and other testing, the cost of which is substantial. However, the NDA filings contemplated by us, which are already marketed drugs, would be made under Sections 505 (b)(1) or 505 (b)(2) of the Drug Price Competition Act, which do not require certain studies that would otherwise be necessary; accordingly, the development timetable should be shorter. While the FDA is required to review applications within a certain timeframe, during the review process, the FDA frequently requests that additional information be submitted. The effect of such request and subsequent submission can significantly extend the time for the NDA review process. Until an NDA is actually approved, there can be no assurance that the information requested and submitted will be considered adequate by the FDA to justify approval. The packaging and labeling of our developed products are also subject to FDA regulation. It is impossible to anticipate the amount of time that will be needed to obtain FDA approval to market any product.
| 18 |
Whether or not FDA approval has been obtained, approval of the product by comparable regulatory authorities in any foreign country must be obtained prior to the commencement of marketing of the product in that country. We intend to conduct all marketing in territories other than the United States through other pharmaceutical companies based in those countries. The approval procedure varies from country to country, can involve additional testing, and the time required may differ from that required for FDA approval. Although there are some procedures for unified filings for certain European countries, in general each country has its own procedures and requirements, many of which are time consuming and expensive. Thus, there can be substantial delays in obtaining required approvals from both the FDA and foreign regulatory authorities after the relevant applications are filed. After such approvals are obtained, further delays may be encountered before the products become commercially available.
ANDAs
The FDA approval procedure for an ANDA differs from the procedure for a NDA in that the FDA waives the requirement of conducting complete clinical studies, although it normally requires bioavailability and/or bioequivalence studies. “Bioavailability” indicates the rate and extent of absorption and levels of concentration of a drug product in the blood stream needed to produce a therapeutic effect. “Bioequivalence” compares the bioavailability of one drug product with another, and when established, indicates that the rate of absorption and levels of concentration of the active drug substance in the body are equivalent for the generic drug and the previously approved drug. An ANDA may be submitted for a drug on the basis that it is the equivalent of a previously approved drug or, in the case of a new dosage form, is suitable for use for the indications specified.
The timing of final FDA approval of an ANDA depends on a variety of factors, including whether the applicant challenges any listed patents for the drug and whether the brand-name manufacturer is entitled to one or more statutory exclusivity periods, during which the FDA may be prohibited from accepting applications for, or approving, generic products. In certain circumstances, a regulatory exclusivity period can extend beyond the life of a patent, and thus block ANDAs from being approved on the patent expiration date.
In May 1992, Congress enacted the Generic Drug Enforcement Act of 1992, which allows the FDA to impose debarment and other penalties on individuals and companies that commit certain illegal acts relating to the generic drug approval process. In some situations, the Generic Drug Enforcement Act requires the FDA to not accept or review ANDAs for a period of time from a company or an individual that has committed certain violations. It also provides for temporary denial of approval of applications during the investigation of certain violations that could lead to debarment and also, in more limited circumstances, provides for the suspension of the marketing of approved drugs by the affected company. Lastly, the Generic Drug Enforcement Act allows for civil penalties and withdrawal of previously approved applications. Neither we nor any of our employees have ever been subject to debarment. We do not believe that we receive any services from any debarred person.
Controlled Substances
We are also subject to federal, state, and local laws of general applicability, such as laws relating to working conditions. We are also licensed by, registered with, and subject to periodic inspection and regulation by the Drug Enforcement Agency (“DEA”) and New Jersey state agencies, pursuant to federal and state legislation relating to drugs and narcotics. Certain drugs that we currently develop or may develop in the future may be subject to regulations under the Controlled Substances Act and related statutes. As we manufacture such products, we may become subject to the Prescription Drug Marketing Act, which regulates wholesale distributors of prescription drugs.
| 19 |
cGMP
All facilities and manufacturing techniques used for the manufacture of products for clinical use or for sale must be operated in conformity with cGMP regulations issued by the FDA. We engage in manufacturing on a commercial basis for distribution of products, and operate our facilities in accordance with cGMP regulations. If we hire another company to perform contract manufacturing for us, we must ensure that our contractor’s facilities conform to cGMP regulations.
Compliance with Environmental Laws
We are subject to comprehensive federal, state and local environmental laws and regulations that govern, among other things, air polluting emissions, waste water discharges, solid and hazardous waste disposal, and the remediation of contamination associated with current or past generation handling and disposal activities, including the past practices of corporations as to which we are the legal successor or in possession. We do not expect that compliance with such environmental laws will have a material effect on our capital expenditures, earnings or competitive position in the foreseeable future. There can be no assurance, however, that future changes in environmental laws or regulations, administrative actions or enforcement actions, or remediation obligations arising under environmental laws will not have a material adverse effect on our capital expenditures, earnings or competitive position.
Competition
We have competition with respect to our principal areas of operation. We develop and manufacture generic products, products using controlled-release drug technology, products utilizing abuse deterrent technologies, and we develop and market (either on our own or by license to other companies) generic and proprietary controlled-release and abuse deterrent pharmaceutical products. In both areas, our competition consists of those companies which develop controlled-release, abuse deterrent drugs and alternative drug delivery systems. We do not represent a significant presence in the pharmaceutical industry.
An increasing number of pharmaceutical companies have become interested in the development and commercialization of products incorporating advanced or novel drug delivery systems. Some of the major pharmaceutical companies have invested and are continuing to invest significant resources in the development of their own drug delivery systems and technologies and some have invested funds in such specialized drug delivery companies. Many of these companies have greater financial and other resources as well as more experience than we do in commercializing pharmaceutical products. Certain companies have a track record of success in developing controlled-release drugs. Significant among these are, without limitation, Pfizer, Sandoz (a Novartis company), Durect Corporation, Mylan Laboratories, Inc., Par Pharmaceuticals, Inc., Alkermes, Inc., Teva Pharmaceuticals Industries Ltd., Impax Laboratories, Inc., and Allergen. Each of these companies has developed expertise in certain types of drug delivery systems, although such expertise does not carry over to developing a controlled-release version of all drugs. Such companies may develop new drug formulations and products or may improve existing drug formulations and products more efficiently than we can. In addition, almost all of our competitors have vastly greater resources than we do. While our product development capabilities and, if obtained, patent protection may help us to maintain our market position in the field of advanced drug delivery, there can be no assurance that others will not be able to develop such capabilities or alternative technologies outside the scope of our patents, if any, or that even if patent protection is obtained, such patents will not be successfully challenged in the future.
| 20 |
In addition to competitors that are developing products based on drug delivery technologies, there are also companies that have announced that they are developing opioid abuse-deterrent products that might compete directly or indirectly with Elite’s products. These include, but are not limited to Pfizer Inc., Pain Therapeutics (which has an agreement with Durect Corporation and Pfizer Inc.), Collegium Pharmaceuticals, Inc., Purdue Pharma LP, and Acura Pharmaceuticals, Inc.
We also face competition in the generic pharmaceutical market. The principal competitive factors in the generic pharmaceutical market include: (i) introduction of other generic drug manufacturers’ products in direct competition with our products under development, (ii) introduction of authorized generic products in direct competition with any of our products under development, particularly if such products are approved and sold during exclusivity periods, (iii) consolidation among distribution outlets through mergers and acquisitions and the formation of buying groups, (iv) ability of generic competitors to quickly enter the market after the expiration of patents or exclusivity periods, diminishing the amount and duration of significant profits, (v) the willingness of generic drug customers, including wholesale and retail customers, to switch among pharmaceutical manufacturers, (vi) pricing pressures and product deletions by competitors, (vii) a company’s reputation as a manufacturer and distributor of quality products, (viii) a company’s level of service (including maintaining sufficient inventory levels for timely deliveries), (ix) product appearance and labeling and (x) a company’s breadth of product offerings.
Sources and Availability of Raw Materials; Manufacturing
A significant portion of our raw materials may be available only from foreign sources. Foreign sources can be subject to the special risks of doing business abroad, including:
| · | greater possibility for disruption due to transportation or communication problems; |
| · | the relative instability of some foreign governments and economies; |
| · | interim price volatility based on labor unrest, materials or equipment shortages, export duties, restrictions on the transfer of funds, or fluctuations in currency exchange rates; and |
| · | uncertainty regarding recourse to a dependable legal system for the enforcement of contracts and other rights. |
Please see the Risk Factor in Part I, Item 1A entitled “We are dependent on a small number of suppliers for our raw materials and any delay or unavailability of raw materials can materially adversely affect our ability to produce products”.
While we currently obtain the raw materials that we need from over 20 suppliers, some materials used in our products are currently available from only one supplier or a limited number of suppliers. The FDA requires identification of raw material suppliers in applications for approval of drug products. If raw materials were unavailable from a specified supplier, FDA approval of a new supplier could delay the manufacture of the drug involved.
We have acquired pharmaceutical manufacturing equipment for manufacturing our products. We have registered our facilities with the FDA and the DEA.
| 21 |
Dependence on One or a Few Major Customers
Each year we have had one or a few customers that have accounted for a large percentage of our limited revenues therefore the termination of a contract with a customer may result in the loss of substantially all of our revenues. We are constantly working to develop new relationships with existing or new customers, but despite these efforts we may not, at the time that any of our current contracts expire, have other contracts in place generating similar or material revenue. We have agreements with Epic, Precision Dose and Ascend for the sales and distribution of products that we manufacture. We receive revenues to manufacture these products and also receive a profit split or royalties based on in-market sales of the products. Please see the Risk Factor in Part I, Item 1A entitled “We depend on a limited number of customers and any reduction, delay or cancellation of an order from these customers or the loss of any of these customers could cause our revenue to decline.”
Employees
As of June 7, 2016, we had 35 full time employees. Full-time employees are engaged in operations, administration, research and development. None of our employees is represented by a labor union and we have never experienced a work stoppage. We believe our relationship with our employees to be good. However, our ability to achieve our financial and operational objectives depends in large part upon our continuing ability to attract, integrate, retain and motivate highly qualified personnel, and upon the continued service of our senior management and key personnel.
Available Information
We file our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Exchange Act electronically with the Securities and Exchange Commission, or SEC. The public may read or copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is http://www.sec.gov.
You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports on the day of filing with the SEC on our website on the World Wide Web at http://www.Elitepharma.com under the Investor Relations tab for SEC Filings or by contacting the Investor Relations Department by calling (518) 398-6222 or sending an e-mail message to dianne@elitepharma.com.
| ITEM 1A | RISK FACTORS |
An investment in the Company’s Common Stock involves a high degree of risk. You should carefully consider the risks described below as well as other information provided to you in this report, including information in the section of this document entitled “Forward Looking Statements.” The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our Common Stock could decline, and you may lose all or part of your investment.
In addition to the other information contained in this report, the following risk factors should be considered carefully in evaluating an investment in us and in analyzing our forward-looking statements.
| 22 |
RISKS RELATED TO OUR BUSINESS
Our revenues and operating income/(loss) could fluctuate significantly
Our revenues and operating results may vary significantly from year-to-year and quarter-to-quarter as well as in comparison to the corresponding quarter of the preceding year. Variations my result from one or more factors, including, without limitation:
| · | Timing of approval of applications filed with the FDA; |
| · | Timing of process validation, product launches and market acceptance of products launched; |
| · | Changes in the amounts spent to research, develop, acquire, license or promote new and existing products; |
| · | Results of clinical trial programs; |
| · | Serious or unexpected health or safety concerns with our products, brand products which we have genericized, products currently under development or any other product candidates; |
| · | Introduction of new products by others that render our products obsolete or noncompetitive; |
| · | The ability to maintain selling prices and gross margin on our products; |
| · | The cost and outcome of litigation, in the event that such occurs in relation to, without limitation, intellectual property issues, regulatory or other matters; |
| · | The ability to comply with complex and numerous governmental regulations and regulatory authorities which oversee and regulate many aspects of our business and operations; |
| · | Changes in coverage and reimbursement policies of health plans and other health insurers, including changes to Medicare, Medicaid and similar state programs, especially in relation to those products that are currently manufactured, under development or identified for future development by the Company; |
| · | Increases in the cost of raw materials contained within our products; |
| · | Manufacturing and supply interruptions, including product rejections or recalls due to failure to comply with manufacturing specifications; |
| · | Timing of revenue recognition relating to our licensing and other agreements; |
| · | The ability to protect our intellectual property from being acquired by other entities; |
| · | The ability to avoid infringing the intellectual property of others; |
| · | Our ability to manage growth and integrate acquired products and assets successfully; and |
| · | The addition or loss of customers. |
We have a relatively limited operating history, which makes it difficult to evaluate our future prospects.
Although we have been in operation since 1990, we have a relatively short operating history and limited financial data upon which you may evaluate our business and prospects. In addition, our business model is likely to continue to evolve as we attempt to expand our product offerings and our presence in the generic pharmaceutical market. As a result, our potential for future profitability must be considered in light of the risks, uncertainties, expenses and difficulties frequently encountered by companies that are attempting to move into new markets and continuing to innovate with new and unproven technologies. Some of these risks relate to our potential inability to:
| · | develop new products; |
| · | obtain regulatory approval of our products; |
| · | manage our growth, control expenditures and align costs with revenues; |
| · | attract, retain and motivate qualified personnel; and respond to competitive developments. |
If we do not effectively address the risks we face, our business model may become unworkable and we may not achieve or sustain profitability or successfully develop any products.
| 23 |
We have not been profitable and expect future losses.
To date, we have not been profitable and we may never be profitable or, if we become profitable, we may be unable to sustain profitability. We have sustained losses from operations in each year since our incorporation in 1990. During the past two fiscal years, we incurred net losses from operations of approximately $8.3 million and $16.5 million, respectively. We expect to continue to incur losses until we are able to generate sufficient revenues to support our operations and offset operating costs.
We may require additional financing to meet our business objectives
Although we believe that we have adequate financial resources on hand as of March 31, 2016 to support the anticipated commercial launch of the one abuse resistant opioid product for which an NDA was accepted and granted priority review by the FDA and also ensure operations through March 31, 2017, we cannot assure that we will not need additional funding to accomplish our plans to conduct the clinical development and commercialization of a range of multiple abuse resistant opioids on an accelerated pace.
As of March 31, 2016, we had cash reserves of approximately $11.5 million and a working capital surplus of $12.5 million, and, for the fiscal year ended March 31, 2016, we had losses from operations totaling $8.3 million, net other income totaling $7.1 million and a net loss of $0.7 million.
On April 10, 2014, we executed a purchase agreement with Lincoln Park Capital Fund, LLC (the “LPC Purchase Agreement”), pursuant to which we could raise up to $40 million (see “Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations; Liquidity and Capital Resources; Lincoln Park Capital”). As of June 7, 2016, we have sold approximately 76.7 million shares pursuant to the LPC Purchase Agreement, with proceeds of such sales totaling approximately $21.2 million.
Pursuant to the LPC Purchase Agreement, we may direct Lincoln Park Capital Fund, LLC (“Lincoln Park”) to purchase up to $40,000,000 worth of shares of our common stock under our agreement over a 36 month period generally in amounts up to 500,000 shares on any such business day. However, Lincoln Park shall not be required to purchase more than $760,000 worth of stock on any business day and cannot purchase any shares of our common stock on any business day that the closing sale price of our common stock is less than $0.10 per share, subject to adjustment as set forth in the Purchase Agreement. Assuming a purchase price of $0.33 per share (the closing sale price of the common stock on June 6, 2016) and only approximately 27.5 million shares available for purchase, we would receive $9.1 million in gross proceeds from purchases under the Purchase Agreement by Lincoln Park, inclusive of the $21.2 million already received for sales of shares prior to June 7, 2015.
The extent we rely on Lincoln Park as a source of funding will depend on a number of factors including, the prevailing market price of our common stock and the extent to which we are able to secure working capital from other sources. If obtaining sufficient funding from Lincoln Park were to prove unavailable or prohibitively dilutive, we will need to secure another source of funding in order to satisfy our working capital needs. Even if we sell all remaining shares under the Purchase Agreement to Lincoln Park, we may still need additional capital to fully implement our business, operating and development plans.
We are anticipating that, with the growth of the current generic product line consisting of generic phentermine tablets and capsules, hydromorphone, naltrexone, methadone, phendimetrazine, isradipine, hydroxyzine and immediate release Lodrane D®, combined with the successful transfer of manufacturing site and commercial launch of the 6 remaining approved generic products licensed to Epic Pharma LLC which have not yet been commercialized, profit splits earned from the commercial sale of Oxy-IR by Epic, pursuant to the Epic Strategic Alliance Agreement, and other opportunities in our pipeline, Elite eventually could be profitable. However, there can be no assurances that we will be able to timely raise additional funds, if needed, on acceptable terms through the LPC Purchase Agreement or otherwise, that the sales of the current generic product line will continue, that the 12 approved generic products licensed to Epic Pharma LLC will be successfully commercialization and generate future revenues or that the other opportunities in our pipeline will be successfully commercialized. There can also be no assurances of Elite becoming profitable.
| 24 |
To sustain operations and meet our business objectives we must be able to commercialize our products and other products or pipeline opportunities. If we are unable to timely obtain additional financing, if necessary, and/or we are unable to timely generate greater revenues from our operations, we will be required to reduce and, possibly, cease operations and liquidate our assets. No assurance can be given that we will be able to commercialize the new opportunities, or consummate such other financing or strategic alternative in the time necessary to avoid the cessation of our operations and liquidation of our assets.
Furthermore, the capital and credit markets have experienced extreme volatility. Disruptions in the credit markets make it harder and more expensive to obtain funding. In the event current resources do not satisfy our needs, we may have to seek additional financing. The availability of additional financing will depend on a variety of factors such as market conditions and the general availability of credit. Future debt financing may not be available to us when required or may not be available on acceptable terms, and as a result we may be unable to grow our business, take advantage of business opportunities, or respond to competitive pressures.
We depend on a limited number of customers and any reduction, delay or cancellation of an order from these customers or the loss of any of these customers could cause our revenue to decline.
Each year we have had one or a few customers that have accounted for a large percentage of our limited revenues therefore the termination of a contract with a customer may result in the loss of substantially all of our revenues. We are constantly working to develop new relationships with existing or new customers, but despite these efforts we may not, at the time that any of our current contracts expire, have other contracts in place generating similar or material revenue. We have agreements with Epic, Ascend and Precision Dose for the sales and distribution of products that we manufacture. We receive revenues to manufacture these products and also receive a profit split or royalties based on in-market sales of the products.
In addition, since a significant portion of our revenues is derived from a relatively few customers, any financial difficulties experienced by any one of these customers, or any delay in receiving payments from any one of these customers, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
A notice of default was issued by the New Jersey Economic Development Authority in relation to prior obligations of our tax-exempt bonds. Although we are current in our payments under these bonds, if the principal balances due under these bonds are accelerated pursuant to the notice of default, our ability to operate in the future will be materially and adversely affected.
Although we are current in our payments under the NJEDA Bonds, we previously were in default and a notice of default was issued in March 2009. Should the principal balances due under the NJEDA Bonds be accelerated pursuant to such notice of default, our ability to operate in the future will be materially and adversely affected.
| 25 |
For more information on the NJEDA Bonds, see Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations; Liquidity and Capital Resources; NJEDA Bonds”.
Elite’s pipeline consists of products in various stages of development, including products in early development.
Elite’s product pipeline, including its abuse deterrent opioid products, are in various stages of development. Prior to commercialization, product development must be completed that could include scale-up, clinical studies, regulatory filing, regulatory review, approval by the FDA, and/or other development steps. Additionally, Elite has 6 approved generic products for which a site transfer must be completed prior to product launches. For these generic products, Elite must complete site transfer studies, file a changes being effective in 30 days (CBE 30) and await FDA review and approval. Development is subject to risks. We cannot assure you that development will be successful, or that during development unexpected delays might occur or additional costs might be incurred.
We are subject to significant costs and uncertainties related to compliance with the extensive regulations that govern the manufacturing, labeling, distribution, promotion and sale of pharmaceutical products as well as environmental, safety and health regulations.
The manufacturing, distribution, processing, formulation, packaging, labeling, promotion and sale of our products are subject to extensive regulation by federal agencies, including, without limitation, the FDA, DEA, FTC, Consumer Product Safety Commission and Environmental Protection Agency, among others. We are also subject to state and local laws, regulations and agencies in New Jersey and elsewhere. Such regulations are also subject to change by the relevant federal, state and local agencies. For instance, beginning from January 1, 2015, manufacturers, wholesale distributors, and repackagers of certain prescription drugs are required to provide and capture certain product tracing information under the Drug Quality and Security Act (“DQSA”). Title II of the DQSA, referred to as the Drug Supply Chain Security Act, requires companies in certain prescription drugs’ chain of distribution to build electronic, interoperable systems to identify and trace the products as they are distributed in the United States. Compliance with the DQSA or any future federal or state electronic pedigree requirements may increase the Company's operational expenses and impose significant administrative burdens.
Regulatory agencies such as the FDA regularly inspect our manufacturing facilities and the facilities of our third party suppliers. The failure of the Northvale Facility, or a facility of one of our third party suppliers, to comply with applicable laws and regulations may lead to breach of representations made to our customers or to regulatory or government action against us related to products made in that facility. We have in the past received and successfully resolved Form 483 observations from the FDA regarding certain operations within our manufacturing network. Although we remain committed to continuing to improve our quality control and manufacturing practices, we cannot be assured that the FDA will continue to be satisfied with our quality control and manufacturing systems and standards. If we receive any future FDA observations, we may be subject to regulatory action including, among others, monetary sanctions or penalties, product recalls or seizure, injunctions, total or partial suspension of production and/or distribution, and suspension or withdrawal of regulatory approvals. Further, other federal agencies, our customers and partners in our alliance, development, collaboration and other partnership agreements with respect to our products and services may take any such Form 483 observations into account when considering the award of contracts or the continuation or extension of such partnership agreements. If we receive any future Form 483 observations or warning letters from the FDA, our business, consolidated results of operations and consolidated financial condition could be materially and adversely affected.
| 26 |
With respect to environmental, safety and health laws and regulations, we cannot accurately predict the outcome or timing of future expenditures that we may be required to make in order to comply with such laws as they apply to our operations and facilities. We are also subject to potential liability for the remediation of contamination associated with both present and past hazardous waste generation, handling, and disposal activities. We are subject periodically to environmental compliance reviews by environmental, safety, and health regulatory agencies. Environmental laws are subject to change and we may become subject to stricter environmental standards in the future and face larger capital expenditures in order to comply with environmental laws.
Compliance with federal and state and local law regulations, including compliance with any newly enacted regulations, requires substantial expenditures of time, money and effort to ensure full technical compliance. Failure to comply with the FDA, DEA, EPA and other governmental regulations can result in fines, disgorgement, unanticipated compliance expenditures, recall or seizure of products, exposure to product liability claims, total or partial suspension of production or distribution, suspension of the FDA’s review of NDAs or ANDAs, enforcement actions, injunctions and civil or criminal prosecution, any of which could have a material and adverse effect on our business, results of operations and financial condition.
Legislative or regulatory reform of the healthcare system in the United States may harm our future business.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively commonly referred to as the “Affordable Care Act” may affect the operational results of companies in the pharmaceutical industry such as ours by imposing additional costs. Effective January 1, 2010, the Affordable Care Act, amongst other changes, increased the minimum Medicaid drug rebates for pharmaceutical companies and revised the definition of “average manufacturer price” for reporting purposes, which may affect the amount of Medicaid drug rebates to states related to the sales of our products, whether such sales are made directly by Company or by one of the Company’s licensees. Beginning in 2011, the law also imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
The Affordable Care Act contemplates the promulgation of significant future regulatory action which may also further affect our business. The Affordable Care Act and any further changes to health care laws or regulatory framework that reduce our revenues or increase our costs could also have a material adverse effect on our business, results of operations and financial condition.
If we are unable to satisfy FDA regulatory requirements, we may not be able to commercialize our product candidates.
We need FDA approval prior to marketing our product candidates in the United States of America. If we fail to obtain FDA approval to market our product candidates, we will be unable to sell our product candidates in the United States of America and we will not generate any revenue from the sale of such products.
This regulatory review and approval process, which includes evaluation of preclinical studies and clinical trials of our product candidates, is lengthy, expensive and uncertain. To receive approval, we must, among other things, demonstrate with substantial evidence from well-controlled clinical trials that our product candidates are both safe and effective for each indication where approval is sought. Satisfaction of these requirements typically takes several years and the time needed to satisfy them may vary substantially, based on the type, complexity and novelty of the pharmaceutical product. We cannot predict if or when we might submit for regulatory approval any of our product candidates currently under development. Any approvals we may obtain may not cover all of the clinical indications for which we are seeking approval. Also, an approval might contain significant limitations in the form of narrow indications, warnings, precautions, or contra-indications with respect to conditions of use.
| 27 |
The FDA has substantial discretion in the approval process and may either refuse to accept an application for substantive review or may form the opinion after review of an application that the application is insufficient to allow approval of a product candidate. If the FDA does not accept our application for review or approve our application, it may require that we conduct additional clinical, preclinical or manufacturing validation studies and submit the data before it will reconsider our application. Depending on the extent of these or any other studies that might be required, approval of any applications that we submit may be delayed by several years, or we may be required to expend more resources than we have available. It is also possible that any such additional studies, if performed and completed, may not be considered sufficient by the FDA to make our applications approvable. If any of these outcomes occur, we may be forced to abandon our applications for approval.
We will also be subject to a wide variety of foreign regulations governing the development, manufacture and marketing of our products. Whether or not an FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must still be obtained prior to manufacturing or marketing the product in those countries. The approval process varies from country to country and the time needed to secure approval may be longer or shorter than that required for FDA approval. We cannot assure you that clinical trials conducted in one country will be accepted by other countries or that approval of our product in one country will result in approval in any other country.
Before we can obtain regulatory approval, we need to successfully complete clinical trials, outcomes of which are uncertain.
In order to obtain FDA approval to market a new drug product, we must demonstrate proof of safety and effectiveness in humans. To meet these requirements, we must conduct extensive preclinical testing and “adequate and well-controlled” clinical trials. Conducting clinical trials is a lengthy, time-consuming, and expensive process. Completion of necessary clinical trials may take several years or more. Delays associated with products for which we are directly conducting preclinical or clinical trials may cause us to incur additional operating expenses. The commencement and rate of completion of clinical trials may be delayed by many factors, including, without limitation, for example:
| · | ineffectiveness of our product candidate or perceptions by physicians that the product candidate is not safe or effective for a particular indication; |
| · | inability to manufacture sufficient quantities of the product candidate for use in clinical trials; |
| · | delay or failure in obtaining approval of our clinical trial protocols from the FDA or institutional review boards; |
| · | slower than expected rate of patient recruitment and enrollment; inability to adequately follow and monitor patients after treatment; difficulty in managing multiple clinical sites; |
| · | unforeseen safety issues; |
| · | government or regulatory delays; and |
| · | clinical trial costs that are greater than we currently anticipate. |
| 28 |
Even if we achieve positive interim results in clinical trials, these results do not necessarily predict final results, and positive results in early trials may not be indicative of success in later trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after achieving promising results in earlier trials. Negative or inconclusive results or adverse medical events during a clinical trial could cause us to repeat or terminate a clinical trial or require us to conduct additional trials. We do not know whether our existing or any future clinical trials will demonstrate safety and efficacy sufficiently to result in marketable products. Our clinical trials may be suspended at any time for a variety of reasons, including if the FDA or we believe the patients participating in our trials are exposed to unacceptable health risks or if the FDA finds deficiencies in the conduct of these trials.
Failures or perceived failures in our clinical trials will directly delay our product development and regulatory approval process, damage our business prospects, make it difficult for us to establish collaboration and partnership relationships, and negatively affect our reputation and competitive position in the pharmaceutical community.
Because of these risks, our research and development efforts may not result in any commercially viable products. Any delay in, or termination of, our preclinical or clinical trials will delay the filing of our drug applications with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. If a significant portion of these development efforts are not successfully completed, required regulatory approvals are not obtained, or any approved products are not commercially successful, our business, financial condition, and results of operations may be materially harmed.
If our collaboration or licensing arrangements are unsuccessful, our revenues and product development may be limited.
We have entered into several collaborations and licensing arrangements for the development of products. However, there can be no assurance that any of these agreements will result in FDA approvals, or that we will be able to market any such finished products at a profit. Collaboration and licensing arrangements pose the following risks:
| · | collaborations and licensing arrangements may be terminated, in which case we will experience increased operating expenses and capital requirements if we elect to pursue further development of the related product candidate; |
| · | collaborators and licensees may delay clinical trials and prolong clinical development, under-fund a clinical trial program, stop a clinical trial or abandon a product candidate; |
| · | expected revenue might not be generated because milestones may not be achieved and product candidates may not be developed; |
| · | collaborators and licensees could independently develop, or develop with third parties, products that could compete with our future products; |
| · | the terms of our contracts with current or future collaborators and licensees may not be favorable to us in the future; |
| · | a collaborator or licensee with marketing and distribution rights to one or more of our products may not commit enough resources to the marketing and distribution of our products, limiting our potential revenues from the commercialization of a product; |
| 29 |
| · | disputes may arise delaying or terminating the research, development or commercialization of our product candidates, or result in significant and costly litigation or arbitration; and |
| · | one or more third-party developers could obtain approval for a similar product prior to the collaborator or licensee resulting in unforeseen price competition in connection with the development product. |
If we are unable to protect our intellectual property rights or avoid claims that we infringed on the intellectual property rights of others, our ability to conduct business may be impaired.
Our success depends on our ability to protect our current and future products and to defend our intellectual property rights. If we fail to protect our intellectual property adequately, competitors may manufacture and market products similar to ours.
We currently hold ten patents and we have five patents pending. We intend to file further patent applications in the future. We cannot be certain that our pending patent applications will result in the issuance of patents. If patents are issued, third parties may sue us to challenge our patent protection, and although we know of no reason why they should prevail, it is possible that they could. In addition to modification or revocation of patents in legal proceedings, issued patents may later be modified or revoked by the U.S. Patent and Trademark Office or by analogous foreign offices. It is likewise possible that our patent rights may not prevent or limit our present and future competitors from developing, using or commercializing products that are similar or functionally equivalent to our products.
In addition, we may be required to obtain licenses to patents, or other proprietary rights of third parties, in connection with the development and use of our products and technologies as they relate to other persons’ technologies. At such time as we discover a need to obtain any such license, we will need to establish whether we will be able to obtain such a license on favorable terms, if at all. The failure to obtain the necessary licenses or other rights could preclude the sale, manufacture or distribution of our products.
We rely particularly on trade secrets, unpatented proprietary expertise and continuing innovation that we seek to protect, in part, by entering into confidentiality agreements with licensees, suppliers, employees and consultants. We cannot provide assurance that these agreements will not be breached or circumvented. We also cannot be certain that there will be adequate remedies in the event of a breach. Disputes may arise concerning the ownership of intellectual property or the applicability of confidentiality agreements. We cannot be sure that our trade secrets and proprietary technology will not otherwise be obtained by other entities or become known, obtained or independently developed by our competitors or by other entities. We also cannot be sure that, if patents are not issued with respect to products arising from research, we will be able to maintain the confidentiality of information relating to these products. In addition, efforts to ensure our intellectual property rights can be costly, time-consuming and/or ultimately unsuccessful.
Litigation is common in the pharmaceutical industry, and can be protracted and expensive and could delay and/or prevent entry of our products into the market, which, in turn, could have a material adverse effect on our business.
Litigation concerning patents and proprietary rights can be protracted and expensive. Companies routinely bring litigation against applicants and allege patent infringement or other violations of intellectual property rights as the basis for filing suit against an applicant. Elite develops, owns and/or manufactures generic and branded pharmaceutical products and such drug products may be subject to such litigation. Litigation often involves significant expense and can delay or prevent introduction or sale of our products.
| 30 |
There may also be situations where we use our business judgment and decide to market and sell products, notwithstanding the fact that allegations of patent infringement(s) have not been finally resolved by the courts. The risk involved in doing so can be substantial because the remedies available to the owner of a patent for infringement include, among other things, damages measured by the profits lost by the patent owner and not by the profits earned by the infringer. In the case of a willful infringement, the definition of which is subjective, such damages may be trebled. Moreover, because of the discount pricing typically involved with bioequivalent products, patented brand products generally realize a substantially higher profit margin than bioequivalent products. An adverse decision in a case such as this or in other similar litigation could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our Common Stock to decline.
Please also see “Item 3. Legal Proceedings” below.
The pharmaceutical industry is highly competitive and subject to rapid and significant technological change, which could impair our ability to implement our business model.
The pharmaceutical industry is highly competitive, and we may be unable to compete effectively. In addition, the pharmaceutical industry is undergoing rapid and significant technological change, and we expect competition to intensify as technical advances in each field are made and become more widely known. An increasing number of pharmaceutical companies have been or are becoming interested in the development and commercialization of products incorporating advanced or novel drug delivery systems. We expect that competition in the field of drug delivery will increase in the future as other specialized research and development companies begin to concentrate on this aspect of the business. Some of the major pharmaceutical companies have invested and are continuing to invest significant resources in the development of their own drug delivery systems and technologies and some have invested funds in specialized drug delivery companies. Many of our competitors have longer operating histories and greater financial, research and development, marketing and other resources than we do. Such companies may develop new formulations and products, or may improve existing ones, more efficiently than we can. Our success, if any, will depend in part on our ability to keep pace with the changing technology in the fields in which we operate.
As we expand our presence in the generic pharmaceuticals market our product candidates may face intense competition from brand-name companies that have taken aggressive steps to thwart competition from generic companies. In particular, brand-name companies continue to sell or license their products directly or through licensing arrangements or strategic alliances with generic pharmaceutical companies (so-called “authorized generics”). No significant regulatory approvals are required for a brand-name company to sell directly or through a third party to the generic market, and brand-name companies do not face any other significant barriers to entry into such market. In addition, such companies continually seek to delay generic introductions and to decrease the impact of generic competition, using tactics which include, without limitation:
| · | obtaining new patents on drugs whose original patent protection is about to expire; |
| · | filing patent applications that are more complex and costly to challenge; |
| · | filing suits for patent infringement that automatically delay approval from the FDA; |
| · | filing citizens’ petitions with the FDA contesting approval of the generic versions of products due to alleged health and safety issues; developing controlled-release or other “next-generation” products, which often reduce demand for the generic version of the existing product for which we may be seeking approval; |
| 31 |
| · | changing product claims and product labeling; |
| · | developing and marketing as over-the-counter products those branded products which are about to face generic competition; and |
| · | making arrangements with managed care companies and insurers to reduce the economic incentives to purchase generic pharmaceuticals. |
These strategies may increase the costs and risks associated with our efforts to introduce our generic products under development and may delay or prevent such introduction altogether.
If our product candidates do not achieve market acceptance among physicians, patients, health care payors and the medical community, they will not be commercially successful and our business will be adversely affected.
The degree of market acceptance of any of our approved product candidates among physicians, patients, health care payors and the medical community will depend on a number of factors, including, without limitation:
| · | acceptable evidence of safety and efficacy; |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of any adverse side effects; |
| · | availability of alternative treatments; |
| · | pricing and cost effectiveness; |
| · | effectiveness of sales and marketing strategies; and |
| · | ability to obtain sufficient third-party coverage or reimbursement. |
If we are unable to achieve market acceptance for our product candidates, then such product candidates will not be commercially successful and our business will be adversely affected.
In addition, even if we are able to obtain regulatory approvals for our new products, the success of those products as well as the success of our previously approved products, is dependent upon market acceptance. Levels of market acceptance for our new products could be affected by several factors, including, without limitation:
| · | the availability of alternative products from our competitors; |
| · | the prices of our products relative to those of our competitors; |
| · | the timing of our market entry; |
| · | the ability to market our products effectively at the retail level; |
| · | the perception of patients and the healthcare community, including third-party payers, regarding the safety, efficacy and benefits of our drug products compared to those of competing products; and |
| · | the acceptance of our products by government and private formularies. |
Some of these factors are not within our control, and our products may not achieve expected levels of market acceptance. Additionally, continuing and increasingly sophisticated studies of the proper utilization, safety and efficacy of pharmaceutical products are being conducted by the industry, government agencies and others which can call into question the utilization, safety and efficacy of previously marketed products. In some cases, studies have resulted, and may in the future result, in the discontinuance of product marketing or other risk management programs such as the need for a patient registry.
| 32 |
Legislative or regulatory programs that may influence prices of prescription drugs could have a material adverse effect on our business.
Current or future federal or state laws and regulations may influence the prices of drugs and, therefore, could adversely affect the prices that we receive for our products. Programs in existence in certain states seek to set prices of all drugs sold within those states through the regulation and administration of the sale of prescription drugs. Expansion of these programs, in particular, state Medicaid programs, or changes required in the way in which Medicaid rebates are calculated under such programs, could adversely affect the price we receive for our products and could have a material adverse effect on our business, results of operations and financial condition. Further, prescription drug prices have been the focus of increased scrutiny by the government, including certain state attorneys general, members of congress and the U.S. Department of Justice. Decreases in health care reimbursements or prices of our prescription drugs could limit our ability to sell our products or decrease our revenues, which could have a material adverse effect on our business, results of operations and financial condition.
We are dependent on a small number of suppliers for our raw materials and any delay or unavailability of raw materials can materially adversely affect our ability to produce products.
The FDA requires identification of raw material suppliers in applications for approval of drug products. If raw materials were unavailable from a specified supplier, FDA approval of a new supplier could delay the manufacture of the drug involved.
In addition, some materials used in our products are currently available from only one supplier or a limited number of suppliers and there is a risk of a sole approved supplier significantly raising prices. Please note that such an occurrence has taken place recently, wherein significant price increases from a sole supplier greatly reduced profit margins, sales and delayed product launches. These occurrences were ultimately resolved by the successful FDA approval of an alternate supplier, with such approval process being lengthy and costly.
Further, a significant portion of our raw materials may be available only from foreign sources. Foreign sources can be subject to the special risks of doing business abroad, including, without limitation:
| · | greater possibility for disruption due to transportation or communication problems; |
| · | the relative instability of some foreign governments and economies; |
| · | interim price volatility based on labor unrest, materials or equipment shortages, export duties, restrictions on the transfer of funds, or fluctuations in currency exchange rates; and |
| · | uncertainty regarding recourse to a dependable legal system for the enforcement of contracts and other rights. |
In addition, patent laws in certain foreign jurisdictions (primarily, but not necessarily, in Europe) may make it increasingly difficult to obtain raw materials for research and development prior to expiration of applicable United States or foreign patents. Any delay or inability to obtain raw materials on a timely basis, or any significant price increases that cannot be passed on to customers, can materially adversely affect our ability to produce products. This can materially adversely affect our business and operations.
| 33 |
Even after regulatory approval, we will be subject to ongoing significant regulatory obligations and oversight as evidenced by the FDA’s removal from the market of our Lodrane® extended release product line. In addition, although Lodrane D® is marketed under the Over-the-Counter Monograph and, accordingly, can be lawfully marketed in the US without prior regulatory approval, the FDA has revised its enforcement policies during the past few years, significantly limiting the circumstances under which unapproved products may be marketed.
Even if regulatory approval is obtained for a particular product candidate, the FDA and foreign regulatory authorities may, nevertheless, impose significant restrictions on the indicated uses or marketing of such products, or impose ongoing requirements for post-approval studies. Following any regulatory approval of our product candidates, we will be subject to continuing regulatory obligations, such as safety reporting requirements, and additional post-marketing obligations, including regulatory oversight of the promotion and marketing of our products. If we become aware of previously unknown problems with any of our product candidates here or overseas or at our contract manufacturers’ facilities, a regulatory agency may impose restrictions on our products, our contract manufacturers or on us, including requiring us to reformulate our products, conduct additional clinical trials, make changes in the labeling of our products, implement changes to or obtain re-approvals of our contract manufacturers’ facilities or withdraw the product from the market. In addition, we may experience a significant drop in the sales of the affected products, our reputation in the marketplace may suffer and we may become the target of lawsuits, including class action suits. Moreover, if we fail to comply with applicable regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could harm or prevent sales of the affected products or could substantially increase the costs and expenses of commercializing and marketing these products.
On March 4, 2011, the FDA issued a directive removing from the market approximately 500 cough/cold and allergy products, including our Lodrane® extended release product line. The Lodrane® extended release products constituted approximately 97% of our revenues at the time of FDA’s directive.
Lodrane D® is marketed under the Over-the-Counter Monograph (the “OTC Monograph”) and accordingly, under the Code of Federal Regulations can be lawfully marketed in the US without prior approval. Under the Federal Food Drug and Cosmetic Act (“FDCA”), FDA regulations and statements of FDA policy, certain drug products are permitted to be marketed in the U.S. without prior approval. Within the past few years, the FDA has revised its enforcement policies, significantly limiting the circumstances under which these unapproved products may be marketed. If the FDA determines that a company is distributing an unapproved product that requires approval, the FDA may take enforcement action in a variety of ways, including, without limitation, product seizures and seeking a judicial injunction against distribution.
We depend on qualified scientific and technical employees and are increasingly dependent on our direct sales force, if key personnel were to leave us or if we are unsuccessful in attracting qualified personnel, our ability to develop products and grow our business could be materially harmed.
Because of the specialized scientific nature of our business, we are highly dependent upon our ability to continue to attract and retain qualified scientific and technical personnel. We are not aware of any pending, significant losses of scientific or technical personnel. Loss of the services of, or failure to recruit, key scientific and technical personnel, however, would be significantly detrimental to our product-development programs. As a result of our small size and limited financial and other resources, it may be difficult for us to attract and retain qualified officers and qualified scientific and technical personnel.
In addition, marketing of our branded product, SequestOx™ requires much greater use of a direct sales force compared to marketing of our generic products. Our ability to realize significant revenues from marketing and sales activities depends on our ability or the ability of our partners to attract and retain qualified sales personnel. Competition for qualified sales personnel is intense. Any failure to attract or retain qualified sales personnel could negatively impact our sales revenue and have a material adverse effect on our business, results of operations and financial condition.
| 34 |
We have entered into employment agreements with our executive officers and certain other key employees. We do not maintain “Key Man” life insurance on any executives.
If we were sued on a product liability claim, an award could exceed our insurance coverage and cost us significantly.
The design, development and manufacture of our products involve an inherent risk of product liability claims. We have procured product liability insurance; however, a successful claim against us in excess of the policy limits could be very expensive to us, damaging our financial position. The amount of our insurance coverage, which has been limited due to our limited financial resources, may be materially below the coverage maintained by many of the other companies engaged in similar activities. To the best of our knowledge, no product liability claim has been made against us as of the date hereof.
Our pipeline of products under development include products that would be filed as branded pharmaceuticals and if generic manufacturers use litigation and regulatory means to obtain approval for generic versions of one or more of such branded drugs, our sales may be adversely effected.
Under the Hatch-Waxman Act, the FDA can approve an ANDA for a generic bioequivalent version of a previously approved drug, without undertaking the full clinical testing necessary to obtain approval to market a new drug. In place of such clinical studies, an ANDA applicant usually needs only to submit data demonstrating that its generic product is bioequivalent to the branded product.
Our product development pipeline includes a range of abuse resistant opioid products, with full clinical testing activity being currently planned, in progress or successfully completed. In recent years, various generic manufacturers have filed ANDAs seeking FDA approval for generic versions of opioids and opioids with abuse resistant characteristics. In connection with our filings, these manufacturers may challenge the validity and/or enforceability of one or more of the underlying patents protecting our products. While it is the Company’s intention to vigorously defend and pursue all available legal and regulatory avenues in defense of the intellectual property rights protecting our products, it must also be stressed that litigation is inherently uncertain and we cannot predict the timing or outcome of our efforts. There can also be no assurance that our efforts in defense of the intellectual property rights protecting our products will be successful.
If we are not successful in defending our intellectual property rights, or opt to settle, or if a product’s marketing exclusivity rights expire or become otherwise unenforceable, our competitors could ultimately launch generic versions of one or more of our branded products, after such products have been approved by the FDA, which could significantly decrease our revenues and could have a material adverse effect on our business, financial conditions, results of operations and cash flow. Furthermore such a material adverse effect may result in a material adverse effect on our share price.
| 35 |
Agreements between branded pharmaceutical companies and generic pharmaceutical companies are facing increased government scrutiny in the United States and Internationally.
There are numerous and continuing litigation in which generic companies challenge the validity or enforceability of an innovator products patents and/or the applicability of such patents to a generic applicant’s products. Settlement of such litigation is a common outcome, with review of such agreements by the U.S. Federal Trade Commission (the “FTC”) and the Antitrust Division of the Department of Justice (the “DOJ”) being required by law. The FTC has stated publicly its view that some of these settlement agreements violate antitrust laws and has commenced actions against the branded and generic companies that are parties to these agreements. Accordingly, in the event of the Company being party to a settlement agreement, either as the branded, innovator product owner, or as the generic applicant, we may receive formal or informal requests from the FTC for information about a settlement agreement and there is a risk of the FTC alleging a violation of antitrust laws and commencing an action against us.
In addition, the United States Congress has proposed legislation that would limit the types of settlement agreements generic manufacturers can enter into with brand companies. In 2013, the Supreme Court, in FTC v. Actavis, determined that reverse payment patent settlements between generic and brand companies should be evaluated under the rule of reason, and provided limited guidance beyond the selection of this standard. Due to the court’s non-articulation of a precise rule of lawfulness for such settlements, there may be extensive litigation over what constitutes a reasonable and lawful patent settlement between and brand and generic company.
The impact of such future litigation, if any, legislative proposals and potential future court decisions is uncertain, and there can be no assurances that such impact will not have an adverse effect on the Company’s business, its financial condition, results of operations, cash flows and its stock price.
We may incur significant liability if it is determined that we are promoting or have in the past promoted the “off-label” use of drugs.
In jurisdictions including, without limitation, the United States, a company is not permitted to promote drugs for uses that are not described in the product’s labeling and that differ from those that were approved or cleared by the FDA. Such users are commonly referred to as “off-label uses”. Under what is known as the “practice of medicine”, physicians and other healthcare practitioners may prescribe drug products for off-label or unapproved uses. While the FDA does not regulate a physician’s choice of medications, treatments, or product uses, the Federal Food Drug and Cosmetic Act (“FFDC”) and FDA regulations significantly restrict permissible communications on the subject of off-label uses of drug products by pharmaceutical companies. The FDA, FTC, the Office of the Inspector General of the Department of Health and Human Services (“HHS”), the DOJ and various state Attorneys General actively enforce laws and regulations that prohibit the promotion of off-label uses. A company that is found to have improperly promoted off-label uses may be subject to significant liability, including civil fines, criminal fines and penalties, civil damages, exclusion from federal funded healthcare programs and potential liability under the federal False Claims Act and any applicable state false claims act. Conduct giving rise to such liability could also form the basis for private civil litigation by third-party payers or other persons claiming to be harmed by such conduct.
Notwithstanding the regulatory restrictions on off-label promotion, the FDA’s regulations and judicial case law allows companies to engage in some forms of truthful, non-misleading and non-promotional speech concerning the off-label use of products. Elite believes it and its marketing partners comply with these restrictions.
Nonetheless, the FDA, HHS, DOJ, and/or state Attorneys General, and qui tam relators may take the position that the Company is not in compliance with such requirements, and if such non-compliance is proven, the consequences of such may have an adverse material effect on our business, financial condition, results of operations, cash flows and stock price.
| 36 |
We have significant intangible assets on our balance sheet. Consequently, potential impairment of intangible assets may have an adverse material effect on our profitability.
Intangible assets represent a significant portion of our assets. As of March 31, 2016, intangible assets were approximately $6.4 million, or approximately 20% of our assets.
GAAP requires that intangible assets be subject to regular impairment analysis to determine if changes in circumstances indicate that the value of the asset as recorded may not be recoverable. Such events or changes in circumstances are an inherent risk in the pharmaceutical industry and often cannot be predicted. However, should a change in circumstance occur, requiring the impairment of an intangible asset, the result of such an impairment may have an adverse material effect on our business, financial condition, results of operations, cash flows and stock price.
Our products contain narcotic ingredients. As a result of reports of misuse or abuse of prescription narcotics, the sale of such drugs may be subject to increased litigation risk and new regulation, including the development of REMS, which may prove difficult or expensive to comply with.
Many of our current products and products under development contain narcotics. Misuse or abuse of such drugs can lead to physical or other hard. The FDA and/or the DEA may impose new regulations concerning the manufacture, storage, transportation, distribution and sale of prescription narcotics. Such regulations may include new labeling requirements, the development and implementation of a formal Risk Evaluation and Mitigation Strategy (“REMS”), restrictions on prescription and sale of such products and mandatory reformulation in order to make abuse of such products more difficult. In 2007, Congress passed legislation authorizing the FDA to require companies to undertake post-approval studies in order to assess known or signaled potential serious safety risks and to make any labeling changes necessary to address safety risks. Congress also empowered the FDA to require companies to formulate REMS to confirm a drug’s benefits exceed its risks. In 2011, the FDA issued letters to manufacturers of long-acting and extended-release opioids requiring them to develop and submit to the FDA a post-market REMS plan to require that training is provided to prescribers of these products and that information is provided to prescribers that they can use in counseling patients on the risks and benefits of opioid drug use. Elite does not currently own a product that requires a REMS plan, but some of the products in our pipeline may require a REMS plan. The Obama administration has also released a comprehensive action plan to reduce prescription drug abuse, which may include proposed legislation to amended existing controlled substances laws to require healthcare practitioners who request DEA registration to prescribe controlled substances to receive training on opioid prescribing practices as a condition of registration. In addition, state health departments and boards of pharmacy have authority to regulate distribution and may modify their regulations with respect to prescription narcotics in an attempt to curb abuse.
Such new regulations or requirements may be difficult or cost prohibitive for us to comply with, resulting in delays in the commercialization of new products, and decreased profitability of existing and new products. Such occurrences may have material adverse effects on our business, financial condition, results of operations, cash flows and stock price.
The growth of Elite will depend on developing, commercializing and marketing new products.
Our future revenues and profitability is significantly dependent on our ability to successfully commercialize new branded and generic pharmaceutical products in a timely manner. Accordingly, we must continually develop, test, file, receive marketing authorization and manufacture new products. While we are currently developing products and have plans in place for future products beyond those currently in development, there can be no assurances that any of these products will receive marketing authorization and achieve commercialization. In addition, even if a product receives marketing authorization, there can be no assurances that there will be future revenues or profits, or that any such future revenues or profits would be in amounts that provide adequate return on the significant investments made to secure the marketing authorization and create/support the infrastructure required for the commercial manufacture of such product.
| 37 |
We are engaged in the research and development of pharmaceutical products with the objective of achieving marketing authorizations that enable us to manufacture and sell pharmaceuticals in accordance with specific government regulations. Due to the inherent risk associated with pharmaceutical product research and development, particularly with respect to new/innovative drugs, our research and development expenditures and efforts may not result in a successful regulatory approval and commercialization of new products. Furthermore, after we submit a regulatory application, the relevant government authority may require that we conduct additional studies, resulting in an inability for us to reasonably predict the total research and development costs for a new product.
Circumstances in which the Company is unable to successfully commercialize new products in a timely manner, or circumstances in which the profitability of a new product is not sufficient with respect to the costs and investments required to develop such product may have a material adverse effect on our business, financial condition, results of operations, cash flows and stock price.
If our manufacturing facilities are unable to manufacture our products or the manufacturing process is interrupted due to failure to comply with regulations or for other reasons, it could have a material adverse impact on our business.
If any of our manufacturing facilities, quality and regulatory operations and other business and commercial functions fail to comply with complex and numerous regulatory requirements or encounter other manufacturing difficulties, it could adversely affect our ability to supply products. All facilities and manufacturing processes used for the manufacture of pharmaceutical products must be operated in conformity with cGMP and, in the case of controlled substances, DEA regulations. Compliance with the FDA’s cGMP and DEA requirements applies to both drug products seeking regulatory approval and to approved drug products. In complying with cGMP requirements, pharmaceutical manufacturing facilities must continually expend significant time, money and effort in production, record-keeping and quality assurance and control so that their products meet applicable specifications and other requirements product safety, efficacy and quality. Failure to comply with applicable legal requirements subjects our manufacturing facilities to possible legal or regulatory action, including, without limitation, shutdown, which may adversely affect our ability to manufacture product. Were we not able to manufacture products at our manufacturing facilities because of regulatory, business or any other reason, the manufacture and marketing of these products would be interrupted. This could have a material adverse impact on our business, results of operations, financial condition, cash flows, competitive position and stock price.
The DEA limits the availability of the active ingredients used in many of our current products and products in development, as well as the production and distribution of these products, and, as a result, our procurement, production and distribution quotas may not be sufficient to meet commercial demand or complete clinical trials.
The DEA regulates chemical compounds as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. The active ingredients in some of our current products and products in development, including, without limitation, hydromorphone, methadone, phentermine, phendimetrazine and oxycodone, are listed by the DEA as Scheduled substances under the Controlled Substances Act of 1970. Consequently, their manufacture, shipment, storage, sale and use are subject to a high degree of regulation. Furthermore, the DEA limits the availability of the active ingredients used in many of our current products and products in development and we and/or our contract customers and suppliers, must annually apply to the DEA for procurement quotas in order to obtain and distribute these substances. As a result, our procurement and production quotas may not be sufficient to meet commercial demand or to complete clinical trials. Moreover, the DEA may adjust these quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments. Any delay or refusal by the DEA in establishing our quotas, or modification of our quotas, for controlled substances could delay or result in the stoppage of our clinical trials or product launches, or could cause trade inventory disruptions for those products that already been launched, which could have a material adverse effect on our business, financial position, cash flows and stock price.
| 38 |
Sales of our products may be adversely affected by the continuing consolidation within the retail and wholesale pharmaceutical markets.
Our products, whether sold directly by the Company or through third parties that are licensed to market and distribute our products are sold in large part to a market that is comprised of a relatively few retail drug chains, wholesalers and managed care organizations, with such entities continuing to undergo consolidation. Such consolidation may provide these customers or our products with additional purchasing leverage, and consequently, may increase the pricing pressures faced by us. Additionally, the emergence of large buying groups representing independent retail pharmacies, and the prevalence and influence of managed care organizations and similar institutions, enable those groups to extract price discounts on our products.
In addition, our revenues and quarterly results comparisons may also be affected by fluctuations in the buying patterns of retail chains, major distributors and other trade buyers.
Any delays or unanticipated expenses in connection with the operation of our limited number of facilities could have a material adverse effect on our business.
All of our manufacturing operations are conducted at the Northvale Facility. A significant disruption at this facility, even on a short term basis, whether due to, without limitation, an adverse quality or compliance observation, including a total or partial suspension of production and/or distribution by regulatory authorities, an act of God, civil or political unrest, force majeure situation or other events could impair our ability to produce and ship products on a timely basis, and could, among other consequences, subject us to exposure to claims from customers. Any of these events could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Our business is dependent on market perceptions of us and the safety and efficacy or our products. Negative publicity relating to us or our products could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Market perceptions or our business are important to us, especially market perceptions of the safety and quality of our products. If any of our products or similar products that other companies distribute are subject to market withdrawal, recall, or are proven to be, or are claimed to be, harmful to consumers, then this could have a material adverse effect on our business, results of operations, financial condition and cash flows. Furthermore, due to the importance of market perceptions, negative publicity associated with product quality, illness or other adverse effects resulting from, or perceived to be resulting from, our products, or similar products made by other companies, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
| 39 |
We may discontinue the manufacture and distribution of certain existing products, which may adversely effect our business, results of operations, financial condition and cash flows.
As part of regular evaluations of product performance, we may determine that it is in our best interest to discontinue the manufacture and distribution of certain of our products. We cannot guarantee that we have correctly forecasted, or will correctly forecast in the future, the appropriate products to discontinue or that a decision to discontinue various products is prudent if market conditions change. In addition, there can be no assurances that the discontinuance of products will reduce operating expense or no cause the incurrence of material charges associated with such a decision. Furthermore, the discontinuance of existing products, entails various risks, including, without limitation, the ability to find a purchaser for such products, if there is a decision to sell the product, as well as the risk that the purchase price obtained will not be equal to at least the book value of the net assets relating to such products. Other risks associated with a product discontinuance, include, without limitation, managing the expectations of and maintaining good relations with our customers who previously purchased a discontinued product from us, and the effects such would have on future sales to these customers. We may also incur significant liabilities and costs associated with our product discontinuance. All of the foregoing could have a material adverse effect on our business, results of operations, financial condition and cash flows.
The time necessary to develop generic drugs may adversely affect whether, and the extent to which, we receive a return on our capital.
The development process for branded and generic products, including, without limitation, drug formulation, testing, and FDA review and approval, often takes three or more years. This process requires that we expend considerable capital to pursue activities that do not yield an immediate or near-term return. Also, because of the significant time necessary to develop a product, the actual market for a product at the time it is available for sale may be significantly less than the originally projected market for the product. If this were to occur, our potential return on our investment in developing the product, if approved for marketing by the FDA, would be adversely affected and we may never receive a return on our investment in the product. It is also possible for the manufacturer of the brand-name product for which we are developing a generic drug to obtain approvals from the FDA to switch the brand-name drug from the prescription market to the OTC market. If this were to occur, we would be prohibited from marketing our product other than as an OTC drug, in which case revenues could be substantially less than we anticipated.
Research and development efforts invested in our branded pharmaceutical products may not achieve expected results.
The development of branded products requires significant resources from the Company, as well as the potential for resources being acquired through collaborations, in-licensing, or third party product acquisitions. The development proprietary branded drugs involves processes and expertise that is different from that required by the development of generic products, resulting in an increased risk profile for branded development. For example, the time frame from discovery to commercial launch of a branded product can be more than 10 years, involving multiple stages which may consist of intensive preclinical and clinical testing and a highly complex, lengthy and expensive approval process. The longer time frames and increased costs adds increasing risk of achieving product approvals, and if approved, our ability to recover development costs and generate profits.
| 40 |
During each development stage, we may encounter obstacles that delay the process or approval and increase expenses, leading to significant risks that we will not achieve our goals and may be forced to abandon a potential product in which we have invested substantial amounts of time and money. These obstacles may include: preclinical failures; difficulty enrolling patients in clinical trials; delays in completing formulation and other work needed to support an application for approval; adverse reactions or other safety concerns arising during clinical testing; insufficient clinical trial data to support the safety or efficacy of the product candidate; and failure to obtain, or delays in obtaining, the required regulatory approvals for the product candidate or the facilities in which it is manufactured. As a result of the obstacles noted above, our investment in research and development of branded products can involve significant costs with no assurances of future revenues or profits.
Approvals for our new generic drug products may be delayed or become more difficult to obtain if the FDA institutes changes to its approval requirements.
The FDA may institute changes to its ANDA approval requirements, which may make it more difficult or expensive for us to obtain approval for our new generic products. For instance, in July 2012, the Generic Drug Fee User Amendments of 2012 (“GDUFA”) was enacted into law. The GDUFA legislation implemented fees for new ANDAs, Drug Master Files, product and establishment fees and a one-time fee for back-logged ANDAs pending approval as of October 1, 2012. In return, the program is intended to provide faster and more predictable ANDA reviews by the FDA and increased inspections of drug facilities. Under GDUFA, generic product companies face significant penalties for failure to pay the new user fees, including rendering an ANDA not “substantially complete” until the fee is paid. Any failure by us or our suppliers to pay the fees or to comply with the other provisions of GDFUA may impact or delay our ability to file ANDAs, obtain approvals for new generic products, generate revenues and thus may have a material adverse effect on our business, results of operations and financial condition.
In addition to the implementation of new fees and review procedures by the FDA, the FDA may also implement other changes that may directly affect some of our ANDA filings pending approval from the FDA, such as changes to guidance from the FDA regarding bioequivalency requirements for particular drugs. Such changes may cause our development of such generic drugs to be significantly more difficult or result in delays in FDA approval or result in our decision to abandon or terminate certain projects. Any changes in FDA requirements may make it more difficult for us to file ANDAs or obtain approval of our ANDAs and generate revenues and thus have a material adverse effect on our business, results of operations and financial condition.
The risks and uncertainties inherent in conducting clinical trials could delay or prevent the development and commercialization of our own branded products, which could have a material adverse effect on our business, results of operations and financial condition.
With respect to our branded products which do not qualify for the FDA’s abbreviated application procedures, we must demonstrate through clinical trials that these products are safe and effective for use. We have only limited experience in conducting and supervising clinical trials. The process of completing clinical trials and preparing an NDA may take several years and requires substantial resources. Our studies and filings may not result in FDA approval to market our new drug products and, if the FDA grants approval, we cannot predict the timing of any approval. There are substantial filing fees for NDAs, often in excess of $1 million in addition to the cost of product development and clinical trials, that are not refundable if FDA approval is not obtained.
There are a number of risks and uncertainties associated with clinical trials. The results of clinical trials may not be indicative of results that would be obtained from large scale testing. Clinical trials are often conducted with patients having advanced stages of disease and, as a result, during the course of treatment these patients can die or suffer adverse medical effects for reasons that may not be related to the pharmaceutical agents being tested, but which nevertheless affect the clinical trial results. In addition, side effects experienced by the patients may cause delay of approval or limit the profile of an approved product. Moreover, our clinical trials may not demonstrate sufficient safety and efficacy to obtain approval from the FDA or foreign regulatory authorities. The FDA or foreign regulatory authorities may not agree with our assessment of the clinical data or they may interpret it differently. Such regulatory authorities may require additional or expanded clinical trials. Even if the FDA or foreign regulatory authorities approve certain products developed by us, there is no assurance that such regulatory authorities will not subject marketing of such products to certain limits on indicated use.
| 41 |
Failure can occur at any time during the clinical trial process and, in addition, the results from early clinical trials may not be predictive of results obtained in later and larger clinical trials, and product candidates in later clinical trials may fail to show the desired safety or efficacy despite having progressed successfully through earlier clinical testing.
Completion of clinical trials for our product candidates may be delayed or halted for the reasons noted above in addition to many other reasons, including, without limitation:
| · | Delays in patient enrollment, and variability in the number and types of patients available for clinical trials; |
| · | Regulators or institutional review boards may not allow us to commence or continue a clinical trial; |
| · | Our inability, or the inability of our partners, if any, to manufacture or obtain from third parties those materials required to complete clinical trials; |
| · | Delays or failure in reaching agreement on acceptable clinical trial contracts or clinical trial protocols with prospective clinical trial sites; |
| · | Risks associated with trial design, which may result in a failure of the trial to show statistically significant results even if the product candidate is effective; |
| · | Difficulty in maintaining contact with patients after treatment commences, resulting in incomplete data |
| · | Poor effectiveness of product candidates during clinical trials; |
| · | Safety issues, including adverse events associated with product candidates; |
| · | Failure of patients to complete clinical trials due to adverse side effects, dissatisfaction with the product candidate, or other reasons; |
| · | Governmental or regulatory delays or changes in regulatory requirements, policy and guidelines; and |
| · | Varying interpretation of data by the FDA or other relevant regulatory authorities. |
In addition, our product candidates could be subject to competition for clinical study sites and patients from other therapies under development which may delay the enrollment in or initiation of our clinical trials.
The FDA or other relevant regulatory authorities may require us to conduct unanticipated additional clinical trials, which could result in additional expense and delays in bringing our product candidates to market. Any failure or delay in completing clinical trials for our product candidates would prevent or delay the commercialization of our product candidates. We cannot assure that our expenses related to clinical trials will lead to the development of brand-name drugs that will generate revenues in the near future. Delays or failure in the development and commercialization of our own branded products could have a material adverse effect on our business, results of operations and financial condition.
| 42 |
We rely on third parties to conduct clinical trials and testing for our product candidates, and if they do not properly and successfully perform their legal and regulatory obligations, as well as their contractual obligations to us, we may not be able to obtain regulatory approvals for our product candidates.
We design the clinical trials for our product candidates, but rely on contract research organizations and other third parties to assist us in managing, monitoring and otherwise carrying out these trials, including, without limitation, with respect to site selection, contract negotiation, analytical testing and data management. We do not control these third parties and, as a result, delays may occur as a result of the priorities and operations of these third parties differing from those which we may feel would be most optimal to the completion of such activities in the most efficient manner possible.
Although we rely on third parties to conduct our clinical trials and related activities, we are responsible for confirming that each of our clinical trials is conducted in accordance with our general investigational plan and protocol. Moreover, the FDA and other relevant regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices and good laboratory practices, for conducting, recording and reporting the results of clinical trials to ensure that the data and results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. The FDA enforces good clinical practices and good laboratory practices through periodic inspections of trial sponsors, principal investigators and trial sites. If we, our contract research organizations or our study sites fail to comply with applicable good clinical practices and good laboratory practices, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with good clinical practices and good laboratory practices. In addition, our clinical trials must be conducted with product manufactured under the FDA’s current Good Manufacturing Practices, or cGMP, regulations. Our failure or the failure of our contract manufacturers if any are involved in the process, to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
If third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to failure to adhere to our clinical protocols or regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, our clinical trials may not meet regulatory requirements. If our clinical trials do not meet regulatory requirements or if these third parties need to be replaced, our clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates, which could have a material adverse effect on our business, results of operations and financial condition.
The illegal distribution and sale by third parties of counterfeit versions of our products or of stolen products could have a negative impact on our reputation and a material adverse effect on our business, results of operations and financial condition.
Third parties could illegally distribute and sell counterfeit versions of our products, which do not meet the rigorous manufacturing and testing standards that our products undergo. Counterfeit products are frequently unsafe or ineffective, and can be life-threatening. Counterfeit medicines may contain harmful substances, the wrong dose of the active pharmaceutical ingredient or no active pharmaceutical ingredients at all. However, to distributors and users, counterfeit products may be visually indistinguishable from the authentic version.
Reports of adverse reactions to counterfeit drugs or increased levels of counterfeiting could materially affect patient confidence in the authentic product. It is possible that adverse events caused by unsafe counterfeit products will mistakenly be attributed to the authentic product. In addition, thefts of inventory at warehouses, plants or while in-transit, which are not properly stored and which are sold through unauthorized channels could adversely impact patient safety, our reputation and our business.
| 43 |
Public loss of confidence in the integrity of pharmaceutical products as a result of counterfeiting or theft could have a material adverse effect on our business, results of operations and financial condition.
Policies regarding returns, rebates, allowances and chargebacks, and marketing programs adopted by wholesalers may reduce our revenues in future fiscal periods.
Based on industry practice, generic drug manufacturers have liberal return policies and have been willing to give customers post-sale inventory allowances. Such industry practices apply to the current sales of our products by our marketing partners, which in turn effect profit splits and license fees received, and they will also effect prospective future sales made directly by Company.
Under these arrangements, from time to time, customers are given credits on our generic products that are held by them in inventory after there is a decrease in the market prices of the same generic products due to competitive pricing. Therefore, if new competitors enter the marketplace and significantly lower the prices of any of their competing products, the price of our products would also likely be reduced. As a result, we, or are marketing partners, would be obligated to provide credits to our customers who are then holding inventories of such products, which could reduce sales revenue, profit splits, license fees and gross margin for the period the credit is provided. Like most competitors in this market, our marketing partners, or us in the case of prospective direct sales made by the Company, also give credits for chargebacks to wholesalers that have contracts with our marketing partners, or us, prospectively, for their sales to hospitals, group purchasing organizations, pharmacies or other customers. A chargeback is the difference between the price the wholesaler pays and the price that the wholesaler’s end-customer pays for a product. Although, our marketing partners establish, and prospectively we would also establich reserves based on prior experience and best estimates of the impact that these policies may have in subsequent periods, we cannot ensure that such reserves established are adequate or that actual product returns, rebates, allowances and chargebacks will not exceed estimates.
Unstable economic conditions may adversely affect our industry, business, results of operations and financial condition.
The global economy has undergone a period of significant volatility which has led to diminished credit availability, declines in consumer confidence and increases in unemployment rates. There remains caution about the stability of the U.S. economy, and we cannot assure that further deterioration in the financial markets will not occur. These economic conditions have resulted in, and could lead to further, reduced consumer spending related to healthcare in general and pharmaceutical products in particular.
In addition, we have exposure to many different industries and counterparties, including our partners under our alliance and collaboration agreements, suppliers of raw chemical materials, drug wholesalers and other customers that may be affected by an unstable economic environment. Any economic instability may affect these parties’ ability to fulfill their respective contractual obligations to us, cause them to limit or place burdensome conditions upon future transactions with us or drive us and our competitors to decrease prices, each of which could materially and adversely affect our business, results of operations and financial condition.
| 44 |
RISKS RELATED TO OUR COMMON STOCK
Our stock price has been volatile and may fluctuate in the future.
The market price for the publicly traded stock of pharmaceutical companies is generally characterized by high volatility. There has been significant volatility in the market prices for our Common Stock. For the twelve months ended March 31, 2016, the closing sale price on the OTC Bulletin Board (“OTC-BB”) of our Common Stock fluctuated from a high of $0.43 per share to a low of $0.20 per share. The price per share of our Common Stock may not exceed or even remain at current levels in the future. The market price of our Common Stock may be affected by a number of factors, including, without limitation:
| · | Results of our clinical trials; |
| · | Approval or disapproval of our ANDAs or NDAs; |
| · | Announcements of innovations, new products or new patents by us or by our competitors; |
| · | Announcements of other material events; |
| · | Governmental regulation; |
| · | Patent or proprietary rights developments; |
| · | Proxy contests or litigation; |
| · | News regarding the efficacy of, safety of or demand for drugs or drug technologies; |
| · | Economic and market conditions, generally and related to the pharmaceutical industry; |
| · | Healthcare legislation; |
| · | Changes in third-party reimbursement policies for drugs; and |
| · | Fluctuations in our operating results. |
The sale or issuance of our common stock to Lincoln Park or upon conversion of outstanding preferred stock or exercise of outstanding warrants and options may cause dilution and the sale of the shares of common stock acquired by Lincoln Park or the issuance of shares upon conversion or exercise of outstanding preferred stock and warrants, or the perception that such sales and issuances may occur, could cause the price of our common stock to fall.
On April 10, 2014, we entered into the Purchase Agreement with Lincoln Park, pursuant to which Lincoln Park has committed to purchase up to $40,000,000 of our common stock. Concurrently with the execution of the Purchase Agreement, we issued 1,928,641 shares of our common stock to Lincoln Park as a fee for its commitment to purchase shares of our common stock under the Purchase Agreement. The purchase shares that may be sold pursuant to the Purchase Agreement may be sold by us to Lincoln Park at our discretion from time to time over a 36-month period that commenced on May 1, 2014. The purchase price for the shares that we may sell to Lincoln Park under the Purchase Agreement will fluctuate based on the price of our common stock. Depending on market liquidity at the time, sales of such shares may cause the trading price of our common stock to fall.
We generally have the right to control the timing and amount of any sales of our shares to Lincoln Park, except that, pursuant to the terms of our agreements with Lincoln Park, we would be unable to sell shares to Lincoln Park if and when the closing sale price of our common stock is below $0.10 per share, subject to adjustment as set forth in the Purchase Agreement, and in no event would Lincoln Park purchase more than $760,000 worth of our common stock on any single business day, plus an additional “accelerated amount” under certain circumstances. Additional sales of our common stock, if any, to Lincoln Park will depend upon market conditions and other factors to be determined by us. Lincoln Park may ultimately purchase all, some or none of the shares of our common stock that may be sold pursuant to the Purchase Agreement and, after it has acquired shares, Lincoln Park may sell all, some or none of those shares.
| 45 |
In addition, as of June 7, 2016, there were outstanding shares of preferred stock convertible into approximately 142.9 million shares of Common Stock and warrants to purchase an aggregate of approximately 32.2 million shares of Common Stock at exercise prices of $0.625 per share, vested options to purchase an aggregate of approximately 4.5 million shares at an average exercise price of $0.54. Additional shares of Common Stock may be issuable as a result of anti-dilution provisions in the outstanding preferred stock and warrants
As a result of the above discussed potential issuance of securities, such issuances by us could result in substantial dilution to the interests of other holders of our common stock. Additionally, the sale of a substantial number of shares of our common stock to Lincoln Park or pursuant to the conversion or exercise of outstanding shares of preferred stock and warrants, or the anticipation of such issuances, could make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise wish to effect sales.
The issuance of our common stock to Directors, Employees and Consultants in payment of fees and salaries cause dilution and the sale of these shares of common stock so issued, or the perception that sales of these shares so issued may occur, could cause the price of our common stock to fall.
Pursuant to the Company’s policies relating to the compensation of Directors, all director fees are paid via the issuance of shares of Common Stock, with such shares being valued at the simple average of the closing price of the Company’s Common Stock for each day in the period for which the director fees were incurred. In addition, members of the Company’s management, certain employees and consultants receive a portion of their salaries or compensation via the issuance of shares Common Stock, with such shares being valued by the same method as that used for the shares issued in payment of director fees.
The issuance of these shares is dilutive to holders of our Common Stock, and the subsequent sale of these shares, or the perception that the sale of these shares may occur, could cause the price of our common stock to fall.
Raising of additional funding through sales of our securities could cause existing holders of our Common Stock to experience substantial dilution.
Any additional financing that involves the further sale of our securities could cause existing holders of our Common Stock to experience substantial dilution. On the other hand, if we incurred debt, we would be subject to risks associated with indebtedness, including the risk that interest rates might fluctuate and cash flow would be insufficient to pay principal and interest on such indebtedness.
The issuance of additional shares of our Common Stock or our preferred stock could make a change of control more difficult to achieve.
The issuance of additional shares of our Common Stock or the issuance of shares of an additional series of preferred stock could be used to make a change of control of us more difficult and expensive. Under certain circumstances, such shares could be used to create impediments to, or frustrate persons seeking to cause, a takeover or to gain control of us. Such shares could be sold to purchasers who might side with our Board of Directors in opposing a takeover bid that the Board of Directors determines not to be in the best interests of our shareholders. It might also have the effect of discouraging an attempt by another person or entity through the acquisition of a substantial number of shares of our Common Stock to acquire control of us with a view to consummating a merger, sale of all or part of our assets, or a similar transaction, since the issuance of new shares could be used to dilute the stock ownership of such person or entity.
| 46 |
Provisions of our Articles of Incorporation and By-Laws could defer a change of our Management which could discourage or delay offers to acquire us.
Provisions of our Articles of Incorporation and By-Laws law may make it more difficult for someone to acquire control of us or for our shareholders to remove existing management, and might discourage a third party from offering to acquire us, even if a change in control or in Management would be beneficial to our shareholders. For example, as discussed above, our Articles of Incorporation allows us to issue shares of preferred stock without any vote or further action by our shareholders. Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board of Directors also has the authority to issue preferred stock without further shareholder approval. As a result, our Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock. In this regard, on November 15, 2013, we entered into a Shareholder Rights Plan and, under the Rights Plan, our Board of Directors declared a dividend distribution of one Right for each outstanding share of our common stock and one right for each share of Common Stock into which any of our outstanding Preferred Stock is convertible, to shareholders of record at the close of business on that date. Each Right entitles the registered holder to purchase from us one “Unit” consisting of one one-millionth (1/1,000,000) of a share of Series H Junior Participating preferred stock, at a purchase price of $2.10 per Unit, subject to adjustment, and may be redeemed prior to November 15, 2023, the expiration date, at $0.000001 per Right, unless earlier redeemed by the Company. The Rights generally are not transferable apart from the common stock and will not be exercisable unless and until a person or group acquires or commences a tender or exchange offer to acquire, beneficial ownership of 15% or more of our common stock. However, for Mr. Hakim, our Chief Executive Officer, the Rights Plan’s the 15% threshold excludes shares beneficially owned by him as of November 15, 2013 and all shares issuable to him pursuant to his employment agreement and the Mikah Note. Our By-Laws provide for the classification of our Board of Directors into three classes.
There are inherent uncertainties involved in estimates, judgments and assumptions used in the preparation of financial statements in accordance with GAAP. Any future changes in estimates, judgments and assumptions used or necessary revisions to prior estimates, judgments or assumptions could lead to a restatement of our results.
The consolidated financial statements included in this Annual Report on Form 10-K are prepared in accordance with GAAP. This involves making estimates, judgments and assumptions that affect reported amounts of assets (including intangible assets), liabilities, mezzanine equity, stockholders equity, operating revenues, costs of sales, operating expenses, other income and other expenses. Estimates, judgments and assumptions are inherently subject to change in the future and any necessary revisions to prior estimates, judgments or assumptions could lead to a restatement. Any such changes could result in corresponding changes to the amounts of assets (including goodwill and other intangible assets), liabilities, mezzanine equity, stockholders equity, operating revenues, costs of sales, operating expenses, other income and other expenses.
The restatement of our previously issued unaudited quarterly financial statements has been time-consuming and expensive and could expose us to additional risks that could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares to decline.
As discussed herein, we have restated our previously issued unaudited financial statements for the three months ended June 30, 2015 included in the Quarterly Report on Form 10-Q for the quarter ended June 30, 2015 and the unaudited financial statements for the three and six months ended September 30, 2015 included in the Quarterly Report on Form 10-Q for the quarter ended September 30, 2015. In addition, these restated financial statements include corrections of errors in accounting that were made in previously issued audited annual and unaudited interim periods, that we did not consider material pursuant to guidance provided by SEC Staff Accounting Bulletin 99, Materiality (“SAB 99”) and SEC Staff Accounting Bulletin 108, Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements (“SAB 108”), and accordingly reflected on the restated financial statements on a prospective basis.
| 47 |
This restatement (including the review of the errors in accounting that made such restatement necessary) has been time consuming and expensive, requiring the incurrence of substantial and unanticipated expenses and costs, including, without limitation, audit, legal, consulting, research and other professional fees in connection to the identification and correction of errors in accounting, restatement of previously issued financial statements and the ongoing remediation of material weaknesses in our system of internal controls over financial reporting. Certain remediation actions have been recommended, and we are in the process of implementing them (see Item 9A “Controls and Procedures” of this form 10-K for a description of these remediation measures). In an event of and to the extent that these remediation actions are not successful, we could be forced to incur additional time and expense. Furthermore, there is generally an increased risk of shareholder, governmental, or other actions in connection with the restatement of financial statements, with any such proceedings, regardless of outcome, usually consuming a significant amount of management’s time and attention as well as related legal, accounting and other costs. In situations of a company not prevailing in any such proceedings, there is the possibility of substantial damages or settlement costs being required of the company that did not prevail.
We have identified material weaknesses in our internal control over financial reporting which could, if not remediated, adversely affect our ability to report our financial condition, cash flows and results of operations in a timely and accurate manner and/or increase the risk of future misstatements, which could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares and/or debt securities to decline.
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. Based on reviews conducted by management, the Company’s Independent Auditors and specific guidance from subject matter experts engaged by the Company, we have concluded that material weaknesses in the Company’s internal controls over financial report existed that contributed to the errors in accounting that necessitated the restatement of previously issued financial statements. A material weakness is a deficiency, or a combination of deficiencies, in internal controls over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
The Company has identified certain remediation actions and is in the process of implementing them, but such efforts are not complete and remain ongoing. If we do not complete our remediation in a timely manner or if our remedial measures are insufficient to address the material weaknesses, or if additional material weaknesses in our internal controls are discovered or occur in the future, it may materially adversely affect our ability to report our financial condition and results of operations in a timely and accurate manner and there will continue to be an increased risk of future misstatements. Although we regularly review and evaluate internal controls systems to allow management to report on the effectiveness of our internal controls over financial reporting, we may discover additional weaknesses in our internal controls over financial reporting or disclosure controls and procedures. The next time we evaluate our internal controls over financial reporting and disclosure controls and procedures, if we identify one or more new material weaknesses or have been unable to timely remediate our existing material weaknesses, we would be unable to conclude that our internal controls over financial reporting or disclosure controls and procedures are effective. If we are unable to conclude that our internal controls over financial reporting or our disclosure controls and procedures are effective, or if our independent registered public accounting firm expresses an opinion that our internal controls over financial reporting is ineffective, we may not be able to report our financial condition and results of operations in a timely and accurate manner, which could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares to decline. In addition, any potential future restatements could subject us to additional adverse consequences, including sanctions by the SEC, shareholder litigation and other adverse actions. Moreover, we may be the subject of further negative publicity focusing on such financial statement adjustments and resulting restatement and negative reactions from our shareholders, creditors or others with whom we do business. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares to decline.
| 48 |
Our Common Stock is considered a “penny stock”. The application of the “penny stock” rules to our Common Stock could limit the trading and liquidity of our Common Stock, adversely affect the market price of our Common Stock and increase the transaction costs to sell shares of our Common Stock.
Our common stock is a “low-priced” security or “penny stock” under rules promulgated under the Securities Exchange Act of 1934, as amended. In accordance with these rules, broker-dealers participating in transactions in low-priced securities must first deliver a risk disclosure document which describes the risks associated with such stocks, the broker-dealers duties in selling the stock, the customer’s rights and remedies and certain market and other information. Furthermore, the broker-dealer must make a suitability determination approving the customer for low-priced stock transactions based on the customer’s financial situation, investment experience and objectives. Broker-dealers must also disclose these restrictions in writing to the customer, obtain specific written consent from the customer, and provide monthly account statements to the customer. The effect of these restrictions will likely decrease the willingness of broker-dealers to make a market in our Common Stock, will decrease liquidity of our Common Stock and will increase transaction costs for sales and purchases of our Common Stock as compared to other securities.
Our Common Stock is quoted on the Over-the-Counter Bulletin Board. The Over-the-Counter Bulletin Board is a quotation system, not an issuer listing service, market or exchange, therefore, buying and selling stock on the Over-the-Counter Bulletin Board is not as efficient as buying and selling stock through an exchange. As a result, it may be difficult to sell our Common Stock for an optimum trading price or at all.
The Over-the-Counter Bulletin Board (the “OTCBB”) is a regulated quotation service that displays real-time quotes, last sale prices and volume limitations in over-the-counter securities. Because trades and quotations on the OTCBB involve a manual process, the market information for such securities cannot be guaranteed. In addition, quote information, or even firm quotes, may not be available. The manual execution process may delay order processing and intervening price fluctuations may result in the failure of a limit order to execute or the execution of a market order at a significantly different price. Execution of trades, execution reporting and the delivery of legal trade confirmations may be delayed significantly. Consequently, one may not be able to sell shares of our Common Stock at the optimum trading prices.
When fewer shares of a security are being traded on the OTCBB, volatility of prices may increase and price movement may outpace the ability to deliver accurate quote information. Lower trading volumes in a security may result in a lower likelihood of an individual’s orders being executed, and current prices may differ significantly from the price one was quoted by the OTCBB at the time of the order entry. Orders for OTCBB securities may be canceled or edited like orders for other securities. All requests to change or cancel an order must be submitted to, received and processed by the OTCBB. Due to the manual order processing involved in handling OTCBB trades, order processing and reporting may be delayed, and an individual may not be able to cancel or edit his order. Consequently, one may not be able to sell shares of Common Stock at the optimum trading prices.
| 49 |
The dealer’s spread (the difference between the bid and ask prices) may be large and may result in substantial losses to the seller of securities on the OTCBB if the Common Stock or other security must be sold immediately. Further, purchasers of securities may incur an immediate “paper” loss due to the price spread. Moreover, dealers trading on the OTCBB may not have a bid price for securities bought and sold through the OTCBB. Due to the foregoing, demand for securities that are traded through the OTCBB may be decreased or eliminated.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
None.
| ITEM 2. | PROPERTIES. |
We own a facility located at 165 Ludlow Avenue, Northvale, New Jersey (“165 Ludlow”) which contains approximately 15,000 square feet of floor space. This real property and the improvements thereon are encumbered by a mortgage in favor of the New Jersey Economic Development Authority (“NJEDA”) as security for a loan through tax-exempt bonds from the NJEDA to Elite. The mortgage contains certain customary provisions including, without limitation, the right of NJEDA to foreclose upon a default by Elite. The NJEDA has declared the payment of this bond to be in default (For more information on the NJEDA Bonds, see Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations; Liquidity and Capital Resources; NJEDA Bonds”). We are currently using the Facility as a laboratory, manufacturing, storage, distribution and office space.
We entered into a lease for a portion of a one-story warehouse, located at 135 Ludlow Avenue, Northvale, New Jersey (“135 Ludlow”), consisting of approximately 15,000 square feet of floor space. The lease term began on July 1, 2010. On July 14, 2014, this lease was modified, with the material terms of the modification including the Company occupying the entire 35,000 square feet in the building, with such expansion being necessary to support our growing commercial operations.
The lease, as modified, includes an initial term which expires on December 31, 2016 and two tenant renewal options of five years each, with such options being at the sole discretion of the Company. The property related to this lease is used for the manufacture, packaging, storage and distribution of pharmaceutical raw materials, finished goods and related documents and materials. The property requires significant construction and qualification as a prerequisite to achieving suitability for its intended future use. Storage, manufacturing and distribution operations at the initial 15,000 square foot section in January 2013. Such operations continue, currently.
The additional 20,000 square feet for which leasehold rights were secured pursuant to the July 2014 lease modification, require significant leasehold improvements and qualification as a prerequisite for its intended future use. These improvements are currently in progress.
165 Ludlow and 135 Ludlow are hereinafter referred to as the “Facilities” or the “Northvale Facility”.
| 50 |
Properties used in our operation are considered suitable for the purposes for which they are used, at the time they are placed into service, and are believed adequate to meet our needs for the reasonably foreseeable future.
| ITEM 3 | LEGAL PROCEEDINGS |
In the ordinary course of business we may be subject to litigation from time to time. Except as discussed below, there is no current, pending or, to our knowledge, threatened litigation or administrative action to which we are a party or of which our property is the subject (including litigation or actions involving our officers, directors, affiliates, or other key personnel, or holders of record or beneficially of more than 5% of any class of our voting securities, or any associate of any such party) which in our opinion has, or is expected to have, a material adverse effect upon our business, prospects financial condition or operations.
Arbitration with Precision Dose, Inc.
On May 9, 2014, Precision Dose Inc, the parent company of TAGI Pharmaceuticals, Inc., commenced an arbitration against the Company alleging that the Company failed to properly supply, price and satisfy gross profit minimums regarding Phentermine 37.5mg tablets, as required by the parties’ agreements. Elite denied Precision Dose’s allegations and has counterclaimed that Precision Dose is no longer entitled to exclusivity rights with respect to Phentermine 37.5mg tablets, and is responsible for certain costs, expenses, price increases and lost profits relating to Phentermine 37.5mg tablets and the parties’ agreements. The parties have reached agreement in settlement of these issues, with Precision Dose agreeing to pay certain amounts to the Company in exchange for Elite agreeing to restore exclusivity rights with respect to Phentermine 37.5mg tablets, subject to certain defined conditions. Both parties have been complying with the agreed settlement terms and the Company has notified the Arbitrator of this settlement, requesting the issuance of proceeding termination documents.
GAAP requires that a contingency loss may only be recognized if the event is both probable and of an amount that can be reasonably estimated. Due to the agreements reached and adhered to with regards to this issue, the Company has determined that no contingency loss be recorded.
Please see the risk factor in Item 1A titled “We have been dependent on one or a few major customers. If we are unable to develop more customers our business most likely will be adversely affected.”
| ITEM 4 | MINE SAFETY DISCLOSURES. |
Not Applicable.
| ITEM 5 | MARKET FOR COMPANY’S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our Common Stock is quoted on the Over-the-Counter Bulletin Board (OTCBB) under the ticker symbol “ELTP”. The following table shows, for the periods indicated, the high and low bid prices per share of our Common Stock as by OTC Bulletin Board. Over-the-counter market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
| 51 |
| Quarter Ended | High | Low | ||||||
| Fiscal Year Ending March 31, 2016 | ||||||||
| March 31, 2016 | $ | 0.42 | 0.29 | |||||
| December 31, 2015 | $ | 0.44 | 0.21 | |||||
| September 30, 2015 | $ | 0.25 | 0.20 | |||||
| June 30, 2015 | $ | 0.27 | 0.20 | |||||
| Fiscal Year Ending March 31, 2015 | ||||||||
| March 31, 2015 | $ | 0.33 | 0.20 | |||||
| December 31, 2014 | $ | 0.34 | 0.17 | |||||
| September 30, 2014 | $ | 0.45 | 0.28 | |||||
| June 30, 2014 | $ | 0.51 | 0.27 | |||||
As of June 7, 2015, the last reported sale price of our Common Stock, as reported by the OTCBB, was $0.358.
Holders
As of June 7, 2015, there were, respectively, approximately 136 and 1 holders of record of our Common Stock and Series I Preferred Stock.
Dividends
We have never paid cash dividends on our Common Stock. We currently anticipate that we will retain all available funds for use in the operation and expansion of our business.
Stock Performance Graph
The following graph provide a comparison of the cumulative 5 year total shareholder return on the Company’s Common Stock with that of the cumulative total shareholder return on the Russell 3000 Index and a five stock custom composite index, with all cases assuming reinvestment of dividends. The custom composite index, consists of the following companies which were selected as a peer group with comparable market segments and market capitalizations to those of the Company: Durect Corp, Biotime Inc., Biospecifics Technologies Corp, Athersys Inc, Acura Pharmaceuticals Inc.
| 52 |
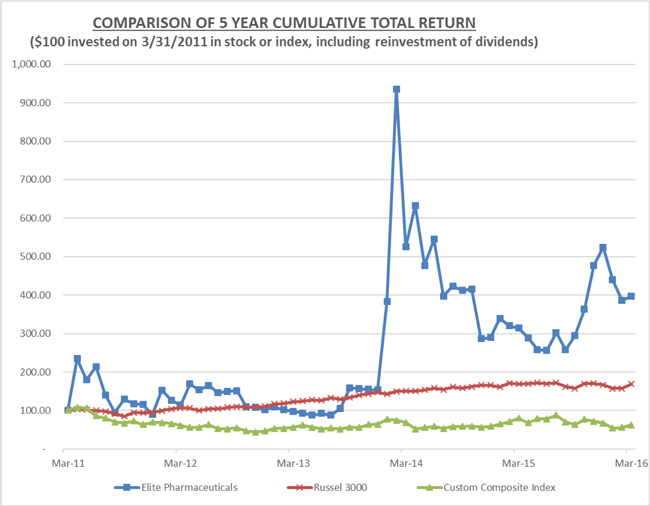
(performance data provided by Bloomberg L.P.)
| Value of $100 Invested on March 31, 2011 | ||||||||||||||||||||||||
| MARCH 31, | ||||||||||||||||||||||||
| 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | |||||||||||||||||||
| Elite Pharmaceuticals Inc. | $ | 100.00 | $ | 114.10 | $ | 97.56 | $ | 525.00 | $ | 314.10 | $ | 397.44 | ||||||||||||
| Russell 3000 Index | $ | 100.00 | $ | 107.18 | $ | 122.80 | $ | 150.56 | $ | 169.18 | $ | 168.60 | ||||||||||||
| Custom Composite Index | $ | 100.00 | $ | 61.51 | $ | 56.33 | $ | 68.61 | $ | 79.94 | $ | 62.58 | ||||||||||||
Recent Sales of Unregistered Securities
During the quarter ended March 31, 2016, the Company issued an aggregate of 13,926,516 shares of Common Stock, with such shares constituting unregistered securities, consisting of 4,554,584 shares of Common Stock issued to Directors and Officers in payment of Directors Fees and Salaries in accordance with the Company’s policy on Director Compensation, or the employment agreements with officers of the Company, as appropriate; and 9,371,932 shares of Common Stock issued pursuant to the exercise of warrants
| 53 |
Securities Authorized for Issuance under Equity Compensation Plans
The following table sets forth certain information regarding Elite’s equity compensation plans as of March 31, 2016.
| Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted- average exercise price per share of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||||
| Equity compensation plans approved by security holders | (1) | — | — | 3,000,000 | ||||||||
| Equity compensation plans not approved by security holders | — | — | 2,595,066 | (2) | ||||||||
| Total | — | — | 5,595,066 | |||||||||
(1) Represents securities reserved and available for grant under the 2014 Equity Incentive Plan
(2) Represents securities reserved and available for grant under the 2009 Equity Incentive Plan
2014 Equity Incentive Plan
Our 2014 Equity Incentive Plan (the “2014 Plan”) was adopted by the Board on March 17, 2014, to attract, motivate and retain officers, employees, consultants, and directors by issuing common stock based incentives to directors, officers, employees and consultants who are selected for participation. By relating incentive compensation to increases in shareholder value, it is hoped that these individuals will both continue in the long-term service of the Company and be motivated to experience a heightened interest and participate in the future success of Company operations. An aggregate of 3,000,000 common shares are reserved for grant and issuance pursuant to the 2014 Plan. The 2014 Plan is administered and interpreted by our Compensation Committee (the “Administrator”). Awards under the 2014 Plan may be granted in any one or all of the following forms: (i) incentive stock options (“ISOs”) intended to qualify under Section 422 of the Internal Revenue Code of 1986, as amended (the “Code”); (ii) non-qualified stock options (“NSOs”); (iii) stock appreciation rights, which may be granted in tandem with options or on a stand-alone basis; (iv) shares of restricted stock; (v) shares of unrestricted stock; (vi) performance shares, and (vii) performance units.
Options may not be granted under the 2014 Plan at an exercise price of less than the fair market value of the common stock on the date of grant and the term of options cannot exceed ten years. ISOs may only be granted to persons who are employees of the Company. The exercise price of an ISO granted to a holder of more than 10% of the common stock must be at least 110% of the fair market value of the common stock on the date of grant, and the term of these options cannot exceed five years.
| 54 |
The Administrator also may grant stock appreciation rights. Stock appreciation rights represent the right to receive upon exercise an amount payable in cash or common stock equal to (A) the number of shares with respect to which the stock appreciation right is being exercised multiplied by (B) the excess of (i) the fair market value of a share of common stock on the date the award is exercised over (ii) the exercise price specified in the award agreement.
Under the performance award component of the 2014 Plan, participants may be granted an award denominated in shares of common stock or in dollars. Achievement of the performance targets, or multiple performance targets established by the Administrator relating to corporate, group, unit or individual performance based upon standards set by the Administrator shall entitle the participant to payment at the full amount or a portion of the amount specified with respect to the award, at the discretion of the Administrator based on its evaluation of the performance of the target goals applicable to such award. Payment may be made in cash, common stock or any combination thereof, as determined by the Administrator, and shall be adjusted in the event the participant ceases to be an employee of the Company before the end of a performance cycle by reason of death, disability or retirement.
Under the stock component of the 2014 Plan, the Administrator may, in selected cases, grant to a plan participant a given number of shares of restricted stock or unrestricted stock. Restricted stock under the 2014 Plan is common stock restricted as to sale pending fulfillment of such vesting schedule and employment requirements as the Administrator shall determine. Prior to the lifting of the restrictions, the participant will nevertheless be entitled to receive distributions in liquidation and dividends on, and to vote the shares of, the restricted stock. The 2014 Plan provides for forfeiture of restricted stock for breach of conditions of grant.
The 2014 Plan also permits the board of directors (and not the Compensation Committee) to grant awards of NSOs, restricted stock or unrestricted stock to non-employee directors. The board may authorize individual grants or adopt one or more formulas for grants of awards to the non-employee directors. All options granted to non-employee directors must have an exercise price equal to the fair market value at the date of grant.
The exercise price of awards may be paid in cash, in shares of common stock (valued at fair market value at the date of exercise), by delivery of a notice of exercise together with irrevocable instructions to a broker to deliver to the Company the proceeds of the sale of common stock or of a loan from the broker sufficient to pay the exercise price, by having the Company withhold from shares being exercised the number of shares having a fair market value equal to the exercise price for all shares being exercised, or by a combination of the foregoing means of payment, as may be determined by the Administrator.
2009 Equity Incentive Plan
Our 2009 Equity Incentive Plan was adopted by the Board on November 24, 2009, to provide incentives to attract, retain and motivate eligible persons whose present and potential contributions are important to the success of Elite and its subsidiaries, by offering them an opportunity to participate in our future performance through awards of Options, the right to purchase Common Stock and Stock Bonuses. An aggregate of 8,000,000 common shares are reserved for grant and issuance pursuant to the 2009 Equity Incentive Plan. The 2009 Equity Incentive Plan is administered and interpreted by our Compensation Committee (the “Compensation Committee”). Under the 2009 Equity Incentive Plan, we are permitted to grant both incentive stock options (“Incentive Stock Options” or “ISOs”) within the meaning of Section 422 of the Internal Revenue Code (the “Code”) to employees, and other options which do not qualify as Incentive Stock Options (the “Non-Qualified Options”) to employees, officers, Directors of and consultants to Elite. The per share purchase price of options granted under the 2009 Equity Incentive Plan may not be less than the fair market value of the shares on the date of the grant, provided that the exercise price of any ISO granted to a ten percent stockholder will not be less than 110% of the fair market value on the date of the grant. Recipients of ISO’s and Non-Qualified Options have no voting, dividend or other rights as stockholders with respect to shares of Common Stock covered by options prior to becoming the holders of record of such shares.
| 55 |
Under the 2009 Equity Incentive Plan, we also are permitted to offer stock awards (“2009 Equity Incentive Plan Stock Awards”) to eligible persons. The 2009 Equity Incentive Plan defines such stock awards as an offer by us to sell to an eligible person shares that may or may not be subject to restrictions. The purchase of price of shares sold pursuant to a 2009 Equity Incentive Plan Stock Award may not be less than the fair market value of the shares on the grant date, provided, however, that the number of shares issued for the payment of employee and officers’ salaries, or directors’ fees will be computed using the average daily closing price, which is defined as the simple average of the closing price of each trading day in the quarter or other applicable period for which payment is due.
We also are permitted to award stock bonuses under the 2009 Equity Incentive Plan, which defines such stock bonuses as an award of shares for extraordinary services rendered to the Company.
Issuer Purchases of Equity Securities
None.
| ITEM 6 | SELECTED FINANCIAL DATA |
The consolidated financial data presented below have been derived from our financial statements. The selected historical consolidated financial data presented below should be read in conjunction with Part II, Item 7. of this report "Management’s Discussion and Analysis of Financial Condition and Results of Operations" and Part II, Item 8. of this report "Financial Statements and Supplementary Data". The selected data in this section is not intended to replace the Consolidated Financial Statements. The information presented below is not necessarily indicative of the results of our future operations. Certain prior period amounts have been restated to reflect corrections to errors in accounting done on a prospective basis. Please see Note2: Restatement of Prior Financial Information for further discussion on prospective restatement of financial information to reflect corrections in accounting error.
| 56 |
| Fiscal Year Ended March 31, | ||||||||||||||||||||
| (dollars in thousands, except per share amounts) | ||||||||||||||||||||
| 2016 | 2015 (restated) | 2014 (restated) | 2013 (restated) | 2012 (restated) | ||||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||||
| Operating revenues | $ | 12,498 | $ | 5,015 | $ | 4,601 | $ | 3,404 | $ | 2,424 | ||||||||||
| Income (Loss) from Operations | (8,317 | ) | (16,507 | ) | (5,284 | ) | (1,563 | ) | (1,966 | ) | ||||||||||
| Other Income (Expense) | 7,113 | 21,724 | (36,270 | ) | 3,259 | (2,348 | ) | |||||||||||||
| Credit for Income Taxes | 520 | 3 | 293 | 354 | 484 | |||||||||||||||
| Net Income (Loss) | (683 | ) | 5,221 | (41,261 | ) | 2,050 | (3,830 | ) | ||||||||||||
| Change in carrying value of convertible preferred share mezzanine equity | (9,286 | ) | 23,709 | (55,314 | ) | (562 | ) | (11,228 | ) | |||||||||||
| Income (Loss) from continuing operations available to common shareholders | (9,969 | ) | 28,930 | (96,575 | ) | 1,488 | (15,058 | ) | ||||||||||||
| Earnings per share: Basic | (0.01 | ) | 0.05 | (0.21 | ) | 0.00 | (0.06 | ) | ||||||||||||
| Earnings per share: Diluted | (0.01 | ) | (0.02 | ) | (0.21 | ) | (0.00 | ) | (0.06 | ) | ||||||||||
| Cash dividends declared per common share | $ | 0.00 | $ | 0.00 | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||||||
| Consolidated Balance Sheet Data: | ||||||||||||||||||||
| Cash | 11,512 | 7,464 | 6,942 | 369 | 668 | |||||||||||||||
| Current Assets | 16,714 | 12.331 | 9,925 | 2,543 | 1,498 | |||||||||||||||
| Total assets | 31,879 | 25,920 | 24,318 | 11,125 | 10,312 | |||||||||||||||
| Current Liabilities | 4,654 | 5,069 | 6,161 | 5,357 | 4,549 | |||||||||||||||
| Working Capital | 12,060 | 7,262 | 3,764 | (2,814 | ) | (3,051 | ) | |||||||||||||
| Long term obligations | 16,061 | 20,583 | 38,373 | 8,107 | 12,240 | |||||||||||||||
| Convertible preferred share mezzanine equity | 44,286 | 35,000 | 60,982 | 6,335 | 8,506 | |||||||||||||||
| Total Equity | (33,122 | ) | (34,731 | ) | (81,198 | ) | (8,673 | ) | (14,984 | ) | ||||||||||
| Other financial data: | ||||||||||||||||||||
| Cash (used in) operating activities | (2,765 | ) | (15,103 | ) | (4,217 | ) | (1,693 | ) | (394 | ) | ||||||||||
| Cash provided by (used in) investing activities | (1,949 | ) | 2,879 | (558 | ) | (192 | ) | (658 | ) | |||||||||||
| Cash provided by (used in) financing activities | 8,762 | 12,746 | 11,347 | 1,585 | (106 | ) | ||||||||||||||
The comparability of the foregoing is impacted by the change in classification of the NJEDA bond liabilities made subsequent to the Company’s repayment of all amounts in arrears during Fiscal 2015. Prior to Fiscal 2015, the entire bond liability was recorded as a current liability as a result of a notice of default being issued pursuant to the Company’s non-payment of scheduled amounts due. As these in arrears amounts were paid in Fiscal 2015, and the Company has remained current on all payments scheduled pursuant to the bond agreement, bond liabilities included in current liabilities consist only of those amounts due within 12 months of the balance sheet date, with all remaining amounts due being classified as non-current liabilities. Please see Note 8: NJEDA Bonds for a further discussion of the bond liability.
The comparison of net income (loss) and long term obligations is significantly impacted by the change in fair value of warrant derivatives, with net income (loss) having a strong inverse correlation to the trading price of the Company’s Common Stock.
| ITEM 7 | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION |
General
The following discussion and analysis should be read with the financial statements and accompanying notes, included elsewhere in this Annual Report on Form 10-K and the information described in Item 1A “Risk Factors” and in “Special Note Regarding Forward Looking Statements” above. The following discussion is intended to assist the reader in understanding and evaluating our financial position.
| 57 |
Critical Accounting Policies and Estimates
Management’s discussion addresses our Consolidated Financial Statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of financial statements and the reported amounts of revenues and expenses during the reporting period. On an ongoing basis, management evaluates its estimates and judgment, including those related to bad debts, intangible assets, income taxes, worker’s compensation, and contingencies and litigation. Management bases its estimates and judgments on historical experience and on various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Management believes the following critical accounting policies, among others, affect its more significant judgments and estimates used in the preparation of its Consolidated Financial Statements. Our most critical accounting policies include the recognition of revenue upon completion of certain phases of projects relating to the licensing of products in research and development. We also assess a need for an allowance to reduce our deferred tax assets to the amount that we believe is more likely than not to be realized. We assess the recoverability of inventory, long-lived assets and intangible assets whenever events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. We assess our exposure to current commitments and contingencies. It should be noted that actual results may differ from these estimates under different assumptions or conditions.
Accounts receivable are comprised of balances due from customers, net of estimated allowances for uncollectible accounts. In determining collectability, historical trends are evaluated and specific customer issues are reviewed on a periodic basis to arrive at appropriate allowances.
The accounting treatment of warrants and preferred share series issued is determined pursuant to the guidance provided by subtopics 470, 480, 815 and 270 of the Accounting Standard Codification (the “ASC”). Each feature of these instruments, including, without limitation, any rights relating to subsequent dilutive issuances, dividend issuances, equity sales, rights offerings, forced conversions, optional redemptions, automatic monthly conversions, dividends and exercise are assessed with determinations made regarding the proper classification on the Company’s statement of financial position, results of operations, cash flow statement and statement of changes in equity.
RECENTLY ISSUED ACCOUNTING PRONOUNCEMENTS
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers. The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The authoritative guidance is effective for annual reporting periods beginning after December 15, 2016. In July 2015, the FASB extended the effective date of the guidance by one year to December 15, 2017. The Company is currently in the process of assessing the impact this guidance will have on the consolidated financial statements.
In July 2015, the FASB issued ASU 2015-11, Inventory — Simplifying the Measurement of Inventory. ASU 2015-11 requires inventory to be subsequently measured using the lower of cost and net realizable value, thereby eliminating the market value approach. Net realizable value is defined as the “estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal and transportation.” ASU 2015-11 is effective for reporting periods beginning after December 15, 2016 and is applied prospectively. Early adoption is permitted. The Company is evaluating the impact, if any, of adopting this new accounting guidance on its financial statements.
| 58 |
In February, 2016, the FASB issued ASU 2016-02, Leases (Topic 842) which provides new guidance on leases. The new guidance will increase transparency and comparability among organizations that lease buildings, equipment, and other assets by recognizing the assets and liabilities that arise from lease transactions. Current off-balance sheet leasing activities will be required to be reflected on balance sheets so that investors and other users of financial statements can more readily and accurately understand the rights and obligations associated with these transactions. Consistent with the current lease standard, the new guidance addresses both finance and operating leases. Finance leases will be accounted for in substantially the same manner as capital leases are accounted for under current GAAP. Operating leases will be accounted for (both in the income statement and statement of cash flows) in a manner consistent with operating leases under existing GAAP. However, as it relates to the balance sheet, lessees will recognize lease liabilities based upon the present value of remaining lease payments and corresponding lease assets for operating leases with limited exceptions. The new guidance will also require lessees and lessors to provide additional qualitative and quantitative disclosures to help investors and other users for financials statements assess the amount, timing and uncertainty of cash flows arising from leases. These disclosures are intended to supplement the amounts recorded in the financial statements so that users can understand more about the nature of an organization’s leasing activities. The new guidance is effective annual reporting periods, including interim reporting periods with those annual periods, beginning after December 15, 2018, with early application being also permitted, but not required. The Company is evaluating the impact of adoption of this guidance on its financial position, results of operations and disclosures.
Product Development Activities
During Fiscal 2016, The Company completed all development activities necessary to file a 505(b)(2) New Drug Application for its lead abuse-deterrent opioid, SequestOx™, Immediate-Release Oxycodone 5mg, 10mg, 15mg, 20mg and 30mg capsules with sequested Naltrexone Hydrochloride, for the treatment of moderate to severe pain. The Company received a waiver from the US-FDA of the $2.3 million filing fee in January 2016 and received notification from the US-FDA in March 2016 that its application was accepted for priority review with a target action date under the Prescription Drug User Fee Act (“PDUFA”) of July 14, 2016. Please note however that there can be no assurances that this product will receive marketing authorization and achieve commercialization within this time period, or at all. In addition, even if marketing authorization is received, there can be no assurances that there will be future revenues of profits, or that any such future revenues or profits would be in amounts that provide adequate return on the significant investments made to secure this marketing authorization.
Liquidity and Capital Resources
Cash and Working Capital
The Company considers cash and working capital balances as several of the factors the Company uses in evaluating its performance, without limitation. As of March 31, 2016, the Company had cash on hand of $11.5 million and a working capital surplus of $12.1 million. The Company believes that such resources, combined with the Company’s access to the remaining balance of the equity line with Lincoln Park Capital, are sufficient to fund operations through the current operating cycle. For the fiscal year ended March 31, 2016 (hereinafter “Fiscal 2016”), we had losses from operations totaling $8.3 million, net other income totaling $7.1 million and a net loss of $0.7 million. In addition, changes in the carrying value of preferred share mezzanine equity for Fiscal 2016 were an increase of $9.3, with such amount being charged to net income available to common shareholders. Please note that the Company’s other income/(expenses) and net income available to common shareholders are significantly influenced by the fluctuations in the fair value of outstanding preferred share and warrant derivatives, and that such fair values bear a strong, inverse correlation to the market share price of the Company’s Common Stock.
| 59 |
The Company does not anticipate being profitable for the fiscal year ending March 31, 2017, due in large part to its plans to conduct clinical development and commercialization activities on a range of abuse deterrent opioid products, on an accelerated and simultaneous basis. Such activities require the investment of significant amounts in clinical trials, safety and efficacy studies, bioequivalence studies, product manufacturing, regulatory expertise and filings, as well as investments in manufacturing and lab equipment and software. In order to finance these significant expenditures, the Company entered into two purchase agreements with Lincoln Park Capital Fund, LLC (“Lincoln Park”), with such agreements providing the company with equity lines totaling $50 million. We believe this amount of financing, if received, is sufficient to fund the commercialization of the abuse deterrent opioid products identified. Please see below for further details on the financing transactions with Lincoln Park.
In addition, the Company had previously received Notices of Default from the Trustee of the NJEDA Bonds as a result of the utilization of the debt service reserve being used to pay interest payments as well as the company’s failure to make scheduled principal payments. All monetary defaults have been cured during Fiscal 2015 and the Company is current on all NJEDA Bond interest and principal payments. See “NJEDA Bonds” below and the Risk Factor in Part I, Item 1A entitled “A notice of default was issued by the New Jersey Economic Development Authority in relation to prior obligations of our tax-exempt bonds. Although we are current in our payments under these bonds, If the principal balances due under these bonds are accelerated pursuant to the notice of default, our ability to operate in the future will be materially and adversely affected”.
Lincoln Park Capital
Pursuant to an April 19, 2013 purchase agreement with Lincoln Park Capital Fund, LLC (“Lincoln Park”) we had the right to sell to and Lincoln Park was obligated to purchase up to $10 million in shares of the Company’s Common Stock, subject to certain limitations, from time to time, over the 36 month period commencing on May 9, 2013. We raised the entire $10 million from the sale of shares to Lincoln Park pursuant to that agreement. That agreement terminated in March 2014 with the sale of all shares covered by that agreement.
On April 10, 2014, we entered into another Purchase Agreement and a Registration Rights Agreement with Lincoln Park. Pursuant to the terms of the Purchase Agreement, Lincoln Park has agreed to purchase from us up to $40 million of our common stock (subject to certain limitations) from time to time over a 36-month period. Pursuant to the terms of the Registration Rights Agreement, we have filed with the SEC a registration statement to register for resale under the Securities Act the shares that have been or may be issued to Lincoln Park under the Purchase Agreement. That registration statement was declared effective by the SEC on May 1, 2014. A post-effective amendment to that Registration Statement was subsequently filed with the SEC and declared effective on July 1, 2014.
Upon execution of the Purchase Agreement, we have issued 1,928,641 shares of our common stock to Lincoln Park pursuant to the Purchase Agreement as consideration for its commitment to purchase additional shares of our common stock under that agreement and we are obligated to issue up to an additional 1,928,641 commitment shares to Lincoln Park pro rata as up to $40 million of our common stock is purchased by Lincoln Park. Through June 7, 2016, we have sold to Lincoln Park an aggregate of 76.7 million shares under the Purchase Agreement for aggregate gross proceeds of approximately $21.2 million. In addition, we have issued an additional 1.0 million Commitment Shares.
| 60 |
We may, from time to time and at our sole discretion but no more frequently than every other business day, direct Lincoln Park to purchase (a “Regular Purchase”) up to 500,000 shares of our common stock on any such business day, increasing up to 800,000 shares, depending upon the closing sale price of the common stock, provided that in no event shall Lincoln Park purchase more than $760,000 worth of our common stock on any single business day. The purchase price of shares of Common Stock related to the future Regular Purchase funding will be based on the prevailing market prices of such shares at the time of sales (or over a period of up to 10 business days leading up to such time), but in no event will shares be sold to Lincoln Park on a day the Common Stock closing price is less than the floor price of $0.10 per share, subject to adjustment.
In addition to Regular Purchases, on any business day on which we have properly submitted a Regular Purchase notice and the closing sale price is not below $0.15, we may purchase (an “Accelerated Purchase”) an additional “accelerated amount” under certain circumstances. The amount of any Accelerated Purchase cannot exceed the lesser of three times the number of purchase shares purchased pursuant to the corresponding Regular Purchase; and 30% of the aggregate shares of our common stock traded during normal trading hours on the purchase date. The purchase price per share for each such Accelerated Purchase will be equal to the lower of (i) 97% of the volume weighted average price during the purchase date; or (ii) the closing sale price of our common stock on the purchase date.
In the case of both Regular Purchases and Accelerated Purchases, the purchase price per share will be equitably adjusted for any reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar transaction occurring during the business days used to compute the purchase price.
Other than as set forth above, there are no trading volume requirements or restrictions under the Purchase Agreement, and we will control the timing and amount of any sales of our common stock to Lincoln Park.
Our sales of shares of Common Stock to Lincoln Park under the Lincoln Park Purchase Agreement are limited to no more than the number of shares that would result in the beneficial ownership by Lincoln Park and its affiliates, at any single point in time, of more than 9.99% of the then outstanding shares of Common Stock.
The Lincoln Park Purchase Agreement and the Lincoln Park Registration Rights Agreement contain customary representations, warranties, agreements and conditions to completing future sale transactions, indemnification rights and obligations of the parties. The Company has the right to terminate the Lincoln Park Purchase Agreement at any time, at no cost or penalty. Actual sales of shares of Common Stock to Lincoln Park under the Lincoln Park Purchase Agreement will depend on a variety of factors to be determined by the Company from time to time, including, without limitation, market conditions, the trading price of the Common Stock and determinations by the Company as to appropriate sources of funding for the Company and its operations. There are no trading volume requirements or restrictions under the Lincoln Park Purchase Agreement. Lincoln Park has no right to require any sales by the Company, but is obligated to make purchases from the Company as it directs in accordance with the Lincoln Park Purchase Agreement. Lincoln Park has covenanted not to cause or engage in any manner whatsoever, any direct or indirect short selling or hedging of our shares.
The net proceeds under the Purchase Agreement to the Company will depend on the frequency and prices at which the Company sells shares of its stock to Lincoln Park. The Company expects that any proceeds received by the Company from such sales to Lincoln Park under the Lincoln Park Purchase Agreement will be used for general corporate purposes and working capital requirements.
| 61 |
Hakim $1,000,000 Bridge Revolving Credit Line
On October 15, 2013 (the “Hakim Credit Line Effective Date”), and as amended on January 4, 2015, we entered into a bridge loan agreement (the “Hakim Loan Agreement”) with Nasrat Hakim, our President and CEO. Under the terms of the Hakim Loan Agreement, we have the right, in our sole discretion, to a line of credit (“Hakim Credit Line”) in the maximum principal amount of up to $1,000,000 at any one time. Mr. Hakim provided the Credit Line for the purpose of supporting the acceleration of our product development activities. The outstanding amount matured on March 31, 2016. Amounts borrowed under the Hakim Credit Line bear interest at the rate of ten percent (10%) per annum. As of March 31, 2016, the principal balance owed under the Credit Line was $718k with an additional $71k in accrued interest being also owed, in accordance with the terms and conditions of the Credit Line. The entire principal amount due under the Hakim Credit Line, which expired on March 31, 2016, was paid on May 24, 2016. An additional $9k in interest, accrued at an annual rate of 10%, was incurred on the principal balance outstanding during the period beginning on April 1, 2016 and ending on May 24, 2016, the date on which the principal balance was paid. All interest amounts owed in relation to principal balances outstanding on the Hakim Credit Line and consisting of interest amounts due and owing as of March 31, 2016 and interest amounts incurred subsequent to March 31, 2016 and up to the date of principal repayment, were paid on May 24, 2016.
Convertible Note Payable to Mikah Pharma LLC
On August 1, 2013, Elite Laboratories Inc. (“Elite Labs”), a wholly owned subsidiary of the Company, executed an asset purchase agreement (the “Mikah Purchase Agreement”) with Mikah Pharma LLC (“Mikah”), an entity that is wholly owned by Mr. Nasrat Hakim, who, in conjunction with this transaction, was appointed as Elite’s CEO, President and a Director on August 2, 2012, and acquired from Mikah a total of 13 Abbreviated New Drug Applications (“ANDAs”) consisting of 12 ANDAs approved by the FDA and one ANDA under active review with the FDA, and all amendments thereto (the “Acquisition”) for aggregate consideration of $10,000,000, inclusive of imputed interest payable pursuant to a non-interest bearing, secured convertible note due in August 2016 (the “Mikah Note”). Please see “Elite’s Acquisition of a 13 Abbreviated New Drug Applications (“ANDAs”)” in Part I, Item 1 Business, above for more information on the Acquisition. The Mikah Note was amended on February 7, 2014 to make it convertible into shares of the Company’s Series I Convertible Preferred Stock.
The Mikah Note, as amended, was interest free and due and payable on the third anniversary of its issuance. Subject to certain limitations, the principal amount of the Mikah Note was convertible at the option of Mikah into shares of Common Stock at a rate of $0.07 (approximately 14,286 shares per $1,000 in principal amount), the closing market price of the Company’s Common Stock on the date that the asset purchase agreement and Note were executed and/or into shares of the Company’s Series I Convertible Preferred Stock at the rate of 1 share of Series I Preferred Stock for each $100,000 of principal owed on the Mikah Note. The conversion rate was adjustable for customary corporate actions such as stock splits and, subject to certain exclusions, includes weighted average anti-dilution for common stock transactions at prices below the then applicable conversion rate. Pursuant to a security agreement (the “Security Agreement”), repayment of the Mikah Note was secured by the ANDAs acquired in the Acquisition.
On February 7, 2014, Mikah converted the principal amount of $10,000,000, representing the entire principal balance due under the Mikah Note, into 100 shares of the Company’s Series I Preferred Stock.
Please also refer to our audited financial statements and notes to financial statements as and for the fiscal year ended March 31, 2016 for further details.
Despite having entered into the Hakim Credit Line Agreement and the Lincoln Park Purchase Agreement we still may be required to seek additional capital in the future and there can be no assurances that Elite will be able to obtain such additional capital on favorable terms, if at all.
| 62 |
Based upon our current cash position, management has undertaken a review of our operations and implemented cost-cutting measures in an effort to eliminate any expenses which are not deemed critical to our current strategic objectives. We will continue this process without impeding our ability to proceed with our critical strategic goals, which, as noted above, include developing our pain management and other products and manufacturing our current products.
Cash at March 31, 2016 was approximately $11.5 million, an increase of approximately $4 million from the approximately $7.5 million balance of cash at March 31, 2015.
As of March 31, 2016, our principal source of liquidity was approximately $11.5 million of cash. Additionally, we may have access to funds through the exercise of outstanding stock options and warrants and, as mentioned above, from the Lincoln Park Purchase Agreement. There can be no assurance that any of these sources will generate or provide sufficient cash.
NJEDA Bonds
On August 31, 2005, the Company successfully completed a refinancing of a prior 1999 bond issue through the issuance of new tax-exempt bonds (the “Bonds”). The refinancing involved borrowing $4,155,000, evidenced by a 6.5% Series A Note in the principal amount of $3,660,000 maturing on September 1, 2030 and a 9% Series B Note in the principal amount of $495,000 maturing on September 1, 2012. The net proceeds, after payment of issuance costs, were used (i) to redeem the outstanding tax-exempt Bonds originally issued by the Authority on September 2, 1999, (ii) refinance other equipment financing and (iii) for the purchase of certain equipment to be used in the manufacture of pharmaceutical products. As of March 31, 2016, all of the proceeds were utilized by the Company for such stated purposes.
Interest is payable semiannually on March 1 and September 1 of each year. The Bonds are collateralized by a first lien on the Company’s facility and equipment acquired with the proceeds of the original and refinanced Bonds. The related Indenture requires the maintenance of a Debt Service Reserve Fund of $366,000 in relation to the Series A Notes.
Bond issue costs of $354,000 were paid from the bond proceeds and are being amortized over the life of the bonds. Amortization of bond issuance costs amounted to $14,177 for the fiscal year ended March 31, 2016.
The NJEDA Bonds require the Company to make an annual principal payment on September 1st of varying amounts as specified in the loan documents and semi-annual interest payments on March 1st and September 1st, equal to interest due on the outstanding principal at the applicable rate for the semi-annual period just ended.
As of the date of filing of this Annual Report on Form 10-K, there are no interest or principal amounts in arrears. The Series B Notes were retired, at par in July 2014.
Contractual Obligations
The following table lists our enforceable and legally binding noncancellable obligations as of March 31, 2016:
| 63 |
| Total | Less than 1 year | 1-3 years | 3-5 years | More than 5 years | ||||||||||||||||
| Long term debt | $ | 2,760,390 | $ | 426,542 | $ | 516,413 | $ | 347,435 | $ | 1,470,000 | ||||||||||
| Capital lease obligations | 133,701 | 101,722 | 31,979 | — | — | |||||||||||||||
| Operating lease obligations(1) | 155,169 | 155,169 | — | — | — | |||||||||||||||
| Purchase obligations | — | — | — | — | — | |||||||||||||||
| Interest expense | 1,205,859 | 196,408 | 296,930 | 213,645 | 498,876 | |||||||||||||||
| Other Long-Term Liabilities | — | — | — | — | — | |||||||||||||||
| 1 | Consists of lease payments pursuant to the operating lease for 135 Ludlow Ave for the initial lease period, exclusive of taxes and insurance, expiring on December 31, 2016. The lease also includes two five year options, exercised at the sole discretion of the Company and at fixed rates, which are defined in the lease. Due to the relevance to the Company’s operations, of the facility at 135 Ludlow Avenue, the Company expects to exercise the first five year option. If such option were to be exercised, a new contractual obligation would be created, with payments totaling $1.1 million, exclusive of real estate taxes and insurance, over the full five year term of the option period. |
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources that would be considered material to investors.
Effects of Inflation
We are subject to price risks arising from price fluctuations in the market prices of the products that we sell. Management does not believe that inflation risk is material to our business or our consolidated financial position, results of operations, or cash flows.
Cybersecurity
As of March 31, 2016 the Company had no reportable incidents of cybersecurity
Results of Consolidated Operations:
The Company considers operating revenues, research and development costs and income (loss) from operations as several of the factors the Company uses in evaluating its performance, without limitation.
Year Ended March 31, 2016 (“Fiscal 2016”) as compared to the Year Ended March 31, 2015 (“Fiscal 2015”)
Our revenues for Fiscal 2016 were $12.5 million, an increase of $7.5 million or approximately 149% from revenues for the comparable period of the prior year, and consisted of $8.0 million in manufacturing fees and $4.5 million in license fees.
Revenues for Fiscal 2015 consisted of $3.9 million in manufacturing fees, $0.005 million in lab and product development fees, and $1.1 million in license fees. Manufacturing fees increased by approximately 107% as a result of the continued growth in the Company’s generic product sales.
| 64 |
Licensing fees increased by approximately 294% or $3.4 million, from $1.1 million in Fiscal 2015 to $4.5 million in Fiscal 2016. This increase is due to Epic licensing agreements and increase in profit splits from product sales relating to TAGI and Epic.
Research and development costs for Fiscal 2016 were approximately $12.4 million, a decrease of approximately $2.3 million or approximately 16% from $14.7 million of such costs for the comparable period of the prior year. The decrease was primarily due to the timing of ongoing clinical trials related to the development of Elite’s abuse deterrent opioid products. Spending includes equity based payments totaling $0.8 million made pursuant to the Epic strategic Alliance in relation to FDA’s approval of immediate release oxycodone tablets, a product included in the Epic Strategic Alliance. Clinical and lab studies includes (such as category 1, category 2 and category 3 testing as described under the FDA’s draft Guidance for Industry, Abuse-Deterrent Opioids – Evaluation and Labeling, January 2013), process development, analytical development, and regulatory development for products in our pipeline.
General and administrative expenses for Fiscal 2016 were $2.9 million no increase from $2.9 million of general and administrative expenses for the comparable period of the prior year. Please note that these levels of overhead costs are expected to continue or increase in subsequent periods.
Depreciation and amortization for Fiscal 2016 was $0.67 million, an increase of $0.05 million or approximately 8%, from $0.62 million for the comparable period of the prior year. The increase was primarily due to the expansion and upgrading of the Northvale Facility, which has required substantial investments in property, plant and equipment.
Non-cash compensation through the issuance of stock options and warrants for Fiscal 2016 was approximately $0.33 million, an increase of $0.07 million, or approximately 28% from $0.26 million for the comparable period of the prior year. The increase was due to the issuance of options to purchase an aggregate of 4,334,000 shares of Common Stock to various employees during Fiscal 2016, primarily pursuant to employment agreements, and the timing of the amortization schedule established at the time of issuance of the related stock options
As a result of the foregoing, our loss from operations for Fiscal 2016 was $8.3 million, compared to a loss from operations of $16.5 million for Fiscal 2015.
Other income for Fiscal 2016 totaled a net income of $7.1 million, a decrease in net other income of $14.6 million from the net other income of $21.7 million for the comparable period of the prior year. The decrease in other income was due to proceeds from the sale of New Jersey State tax losses totaling $0.5 million and derivative income relating to changes in the fair value of derivative liabilities during Fiscal 2016 totaling $7.4 million, as compared to a net derivative income of $20.3 million and gain on sale of investment totaling $1.7 for the comparable period of the prior year, a $14.6 million overall decrease in other income. Please note that derivative income/(expenses) are most significantly determined by the closing price of the Company’s Common Stock as of the end of each annual or quarterly reporting period, and also as of the date on which shares of the Company’s convertible preferred stock are converted into common stock, with incomes being generated by decreases in such closing prices and expenses being incurred by increases in such closing prices. The closing price of the Company’s Common Stock as of March 31, 2016 was $0.31, as compared to a closing price of $0.25 as of March 31, 2015. These variances in the closing price of the Company’s Common Stock as compared with the closing price at the end of the immediately preceding fiscal year end were significant factors in the derivative income recorded during the year ended March 31, 2016.
As a result of the foregoing, our net loss for Fiscal 2016, including credits for income taxes totaling $0.5 million was $0.7 million, compared to a net income of $5.2 million, inclusive of credit for income taxes totaling $0.003 million for Fiscal 2015.
| 65 |
Changes in maximum redemption value in our convertible preferred mezzanine equity, which is included in the calculation of net loss attributable to common shareholders resulted in the net loss being increased by $9.3 million for fiscal year 2016, as compared to an increase in net income attributable to common shareholders of $23.7 million for the comparable period of the prior year. Accordingly, net income (loss) attributable to common shareholders for fiscal year 2016 was net loss of $10.0 million, compared to net income attributable to common shareholders of $28.9 million for the comparable period of the prior year.
Year Ended March 31, 2015 as compared to the Year Ended March 31, 2014 (“Fiscal 2014”)
Our revenues for Fiscal 2015 were $5.0 million, an increase of $0.4 million or approximately 9% from revenues for the comparable period of the prior year, and consisted of $3.9 million in manufacturing fees, $0.005 million in lab and product development fees and $1.1 million in license fees.
Revenues for Fiscal 2014 consisted of $3.0 million in manufacturing fees, $0.08 million in lab and product development fees, and $1.5 million in license fees. Manufacturing fees increased by approximately 30% as a result of the continued growth in the Company’s generic product sales combine with the launch of Isradipine product line in January 2015.
Licensing fees decreased by approximately 26% or $0.4 million, from $1.5 million in Fiscal 2014 to $1.1 million in Fiscal 2015. This decrease is due to Fiscal 2014 license fee revenues including a one-time milestone of $0.6 million earned pursuant to the Epic Agreement.
Research and development costs for Fiscal 2015 were approximately $14.7 million, an increase of approximately $10.7 million or approximately 275% from $4.0 million of such costs for the comparable period of the prior year. The increase was primarily due to increased activities related to the development of Elite’s abuse deterrent opioid products. Spending included clinical and lab studies (such as category 1, category 2 and category 3 testing as described under the FDA’s draft Guidance for Industry, Abuse-Deterrent Opioids – Evaluation and Labeling, January 2013), process development, analytical development, and regulatory development for products in our pipeline.
General and administrative expenses for Fiscal 2015 were $2.9 million an increase of $0.8 million or approximately 40% from $2.1 million of general and administrative expenses for the comparable period of the prior year. The increase was primarily due to significant increases in regulatory and regulatory compliance costs, including, without limitation, increased fees paid to the US-FDA and the hiring of additional staff to support regulatory compliance activities, additional costs incurred in relation to compliance with the Sarbanes-Oxley Act and significant increases in legal fees, insurance and employee benefits. Please note that these higher levels of overhead costs are expected to continue.
Depreciation and amortization for Fiscal 2015 was $0.6 million, an increase of $0.1 million or approximately 23%, from $0.5 million for the comparable period of the prior year. The increase was primarily due to the expansion and upgrading of the Northvale Facility, which has required substantial investments in property, plant and equipment.
Non-cash compensation through the issuance of stock options and warrants for Fiscal 2015 was approximately $0.3 million, an increase of $0.2 million, or approximately 200% from $0.1 million for the comparable period of the prior year. The increase was due to the issuance of options to purchase an aggregate of 2,590,000 shares of Common Stock to various employees during Fiscal 2015, primarily pursuant to employment agreements, and the timing of the amortization schedule established at the time of issuance of the related stock options
As a result of the foregoing, our loss from operations for Fiscal 2015 was $16.5 million, compared to a loss from operations of $5.3 million for Fiscal 2014.
| 66 |
Other income for Fiscal 2015 totaled a net income of $21.7 million, an increase of net other income of $58.0 million from the net other expense of $36.3 million for the comparable period of the prior year. The increase in other income was due to proceeds from the sale of investment totaling $1.7 and derivative income relating to changes in the fair value of derivative liabilities during Fiscal 2015 totaling $20.3 million, as compared to a net derivative expense of $35.4 million for the comparable period of the prior year. The net other income is offset by interest expense of $0.3 million during Fiscal 2015 as compared to interest expense of $0.9 million for the comparable period of the prior year. Please note that derivative income/(expenses) are most significantly determined by the closing price of the Company’s Common Stock as of the end of each annual or quarterly reporting period, and also as of the date on which shares of the Company’s convertible preferred stock are converted into common stock, with incomes being generated by decreases in such closing prices and expenses being incurred by increases in such closing prices. The closing price of the Company’s Common Stock as of March 31, 2015 was $0.25, as compared to a closing price of $0.41 as of March 31, 2014. These variances in the closing price of the Company’s Common Stock as compared with the closing price at the end of the immediately preceding fiscal year end were significant factors in the derivative income recorded during the year ended March 31, 2015.
As a result of the foregoing, our net income for Fiscal 2015, inclusive of credits for income taxes of $.003 million was $5.2 million, compared to a net loss of $41.3 million, inclusive of credits for income taxes of $0.3 million for Fiscal 2014.
Changes in maximum redemption value in our convertible preferred mezzanine equity, which is included in the calculation of net income (loss) attributable to common shareholders resulted in net income attributable to common shareholders being increased by 23.7 million for fiscal year 2015, as compared to a net loss attributable to common shareholders being increased by $55.3 million for the comparable period of the prior year. Accordingly, net income (loss) attributable to common shareholders for fiscal year 2015 was net income of $28.9 million, compared to net loss of $(96.6) million for the comparable period of the prior year.
Material Changes in Financial Condition
Our working capital (total current assets less total current liabilities) increased by $4.8 million from $7.3 million as of March 31, 2015 to $12.1 million as of March 31, 2016, with such increase being primarily due to the loss from operations sustained during Fiscal 2016 being financed by capital financings that included $6.2 million in proceeds from the sale of Common Stock pursuant to the Purchase Agreement with Lincoln Park and $3.0 million in proceeds from the exercise of cash warrants and options, offset in large part by purchases of fixed assets and leasehold improvements totaling $1.9 million and payment on principal of $0.6 million in NJEDA Bonds and other loans. Please note that capital financings provide cash to the Company without a corresponding current liability and accordingly have an accretive effect on working capital.
Net cash used in operations was $2.8 million for fiscal year ended March 31, 2016, primarily due to our net loss from continuing operations of $0.7 million, offset by non-cash income totaling $4.0 million, which included, without limitation, depreciation and amortization charges of $0.67 million, net income from the change in fair value of derivative liabilities of $7.4 million, non-cash expenses totaling $2.3 million which were paid via the issuance of Common Stock and non-cash compensation accrued of $0.6 million. In addition, net cash provided by operations was effected by changes in the balances of assets and liabilities, including, without limitation, decrease in account receivables and prepaid expenses totaling $0.2 million, and an increase in deferred revenues of $4.2 million, each of which result in a net increase in cash, offset by increases in inventories of $0.3 million and decreases in accounts payables and other current liabilities of $2.2 million, each of which result in a net decrease in cash. In addition to the negative operating cash flow, the Company applied $1.9 million of cash to investing activities, with such amount being used almost entirely for the purchases of property and equipment. These cash outflows were funded by $8.7 million in cash provided by financing, with such amount principally consisting of $6.2 million in proceeds from the sale of Common Stock to Lincoln Park Capital, $3.0 million in proceeds from the exercise of cash warrants and options, offset by $0.6 million in bond and loan principal payments. Overall, as a result of the foregoing, the Company had a net increase in cash of $4.0 million during Fiscal 2016.
| 67 |
Our working capital (total current assets less total current liabilities) increased by $3.5 million from $3.8 million as of March 31, 2014 to $7.3 million as of March 31, 2015, with such increase being primarily due to the loss from operations sustained during Fiscal 2015 being financed by capital financings that included $13.2 million in proceeds from the sale of Common Stock pursuant to the Purchase Agreement with Lincoln Park, $0.8 million in proceeds from the exercise of cash warrants and options and $5.0 million in proceeds from the sale of the Company’s investment in Novel Labs, offset in large part by purchases of fixed assets and leasehold improvements totaling $1.9 million and the retirement of $1.3 million in NJEDA Bonds and other loans. Please note that capital financings provide cash to the Company without a corresponding current liability and accordingly have an accretive effect on working capital.
We experienced negative cash flows from operations of $15.1 million for Fiscal 2015, primarily due to our net income of $28.9 million, offset by non-cash other income items totaling $43.2 million included in the net income, combined with increases in accounts payable and accrued liabilities of $1.2 million (resulting in a positive effect on cash flow), and offset by increases in accounts receivable, inventory and prepaid expenses of $2.1 million (resulting in a negative effective on cash flow). In addition to the negative operating cash flow, the Company realized a net inflow from investments of $2.9 million as a result of the $5 million in proceeds received from the sale of its investment in Novel Laboratories, offset by $2.4 million in investments, which principally consisted of $2.0 million being used for the purchases of property and equipment. Cash outflows were further funded by $12.7 million in cash provided by financing, with such amount principally consisting of $13.2 million in proceeds from the sale of Common Stock to Lincoln Park Capital, $0.9 million in proceeds from the exercise of cash warrants and options, offset by $1.3 million in bond and loan principal payments. Overall, as a result of the foregoing, the Company had a net increase in cash of $0.5 million during Fiscal 2015.
We experienced negative cash flows from operations of $4.2 million for Fiscal 2014, primarily due to our net loss of $41.3 million, offset by net non-cash other expense items totaling $37.1 million included in the net loss, combined with increases in accounts payable and accrued liabilities of $0.7 million (resulting in a positive effect on cash flow), and offset by increases in accounts receivable, inventory and prepaid expenses of $0.8 million (resulting in a negative effective on cash flow). In addition to the negative operating cash flow, the Company applied $0.6 million of cash to investing activities, with such amount being used almost entirely for the purchases of property and equipment. These cash outflows were funded by $11.3 million in cash provided by financing, with such amount principally consisting of $10.0 million in proceeds from the sale of Common Stock to Lincoln Park Capital, $0.9 million in proceeds from the exercise of cash warrants and options and $0.6 million in proceeds from draws on related party lines of credit. Overall, as a result of the foregoing, the Company had a net increase in cash of $6.6 million during Fiscal 2014.
SEGMENT REPORTING
FASB ASC 280-10-50, “Disclosure about Segments of an Enterprise and Related Information” requires use of the “management approach” model for segment reporting. The management approach is based on the way a company’s management organizes segments within the company for making operating decisions and assessing performance. Reportable segments are based on products and services, geography, legal structure, management structure, or any other manner in which management disaggregates a company.
The Company disaggregates its product revenues into the type of marketing authorization relating to each product, specifically the following two reportable segments:
1. ANDA’s for generic products; or
| 68 |
2. NDA’s for branded products.
Asset information is not reviewed or included within the Company’s internal management reporting. Accordingly, the Company does not disclose asset information for each reportable segment.
FAIR VALUE MEASUREMENTS
The Company adopted Accounting Standards Codification (“ASC”) Topic 820, Fair Value Measurements and Disclosures, for financial and non-financial assets and liabilities.
ASC 820 discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow) and the cost approach (cost to replace the service capacity of an asset or replacement cost). The Company utilizes the market approach. The statement utilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
| Level 1: | Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities. |
| Level 2: | Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. |
| Level 3: | Unobservable inputs that reflect the reporting entity’s own assumptions. |
LEASES
Lease agreements are evaluated to determine if they are capital leases meeting any of the following criteria at inception: (a) transfer of ownership; (b) bargain purchase option; (c) the lease term is equal to 75 percent or more of the estimated economic life of the leased property; or (d) the present value at the beginning of the lease term of the minimum lease payments, excluding that portion of the payments representing executory costs such as insurance, maintenance, and taxes to be paid by the lessor, including any profit thereon, equals or exceeds 90 percent of the excess of the fair value of the leased property to the lessor at lease inception over any related investment tax credit retained by the lessor and expected to be realized by the lessor.
If, at its inception, a lease meets any of the four lease criteria above, the lease is classified by the Company as a capital lease; and if none of the four criteria are met, the lease is classified by the Company as an operating lease.
| ITEM 7A | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We believe that our market risk exposures are immaterial as we do not have instruments for trading purposes, and reasonable possible near-term changes in market rates or prices will not result in material near-term losses in earnings, material changes in fair values or cash flows for all instruments.
We maintain all of our cash, cash equivalents and restricted cash in three financial institutions, and we perform periodic evaluations of the relative credit standing of these institutions. However, no assurances can be given that the third party institutions will retain acceptable credit ratings or investment practices.
| 69 |
| ITEM 8 | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Attached hereto and filed as a part of this Annual Report on Form 10-K are our Consolidated Financial Statements, beginning on page F-1.
| ITEM 9 | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
As reported in its Current Report on Form 8-K filed with the Commission on February 4, 2016, effective January 29, 2016, Demetrius Berkower LLC (“Demetrius”) was dismissed as the Company’s independent registered public accounting firm and Buchbinder Tunick & Company was engaged as its new independent registered public accounting firm.
The reports of Demetrius on the Company’s consolidated financial statements for the fiscal year ended March 31, 2015 did not contain an adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
During the fiscal year ended March 31, 2015, and in the subsequent interim period from April 1, 2015 through and including January 29, 2016, there were no disagreements with Demetrius on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which disagreements, if not resolved to Demetrius’s satisfaction, would have caused Demetrius to make reference to the subject matter of the disagreement in connection with its report. During the fiscal year ended March 31, 2015, and in the subsequent interim period from April 1, 2015 through and including January 29, 2016, there were no “reportable events” as that term is described in Item 304(a)(1)(v) of Regulation S-K.
During the fiscal year ended March 31, 2015, and in the subsequent interim period from April 1, 2015 through and including January 29, 2016, the Company did not consult Buchbinder Tunich & Company LLP with respect to the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit option that might be rendered on the Company’s consolidated financial statements.
| ITEM 9A | Evaluation of Disclosure Controls and Procedures |
The Company’s management, with the participation of the Company’s Chief Executive Officer and Chief Financial Officer, have evaluated the effectiveness of the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”)) as of March 31, 2016. Based on that evaluation, the Company’s Chief Executive Officer and the Company’s Chief Financial Officer have concluded that as of the fiscal year ended March 31, 2016, due to the existence of the material weakness in the Company’s internal controls over financial reporting described below, the Company’s disclosure controls and procedures were not effective in providing reasonable assurance that the information required to be disclosed by the Company in the reports that it files or submits under the Exchange Act were recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information was accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosure.
| 70 |
Management’s Annual Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal controls over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. The Company’s internal controls over financial reporting are a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles (“GAAP”).
Because of their inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of management, the Company’s Chief Executive Officer and the Company’s Chief Financial Officer, the Company conducted an evaluation of the effectiveness of its internal control over financial over financial reporting based on the framework described in Internal Control-Integrated Framework issued by the Commission of Sponsoring Organizations of the Treadway Commission, as revised in 2013. Based on that evaluation, management has concluded that the Company did not maintain effective internal control over financial reporting as of the fiscal year ended March 31, 2016 due to the existence of the material weakness in internal control over financial reporting described below.
Material Weakness
A material weakness is a deficiency, or a combination of deficiencies, in internal controls over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
Management has determined that the Company did not maintain effective internal controls over financial reporting as of the fiscal year ended March 31, 2016 due to the existence of the following material weakness identified by management:
Segregation of Duties
Adequate segregation of duties is based on the following principles:
| · | Access to master data is segregated from transaction processing and transaction approval |
| · | The preparation/execution of an activity is segregated from its approval process |
| · | System access rights are restricted and granted on a “need only basis” with user profiles being duly approved by management, properly monitored and documented. |
| · | Mitigating controls being required in situations where proper segregation of duties is not possible. However, it must be noted that mitigating controls are less desirable than proper segregation of duties, because such controls are detective by nature, rather than preventative. Complete reliance on mitigating controls is not ideal due to the increased effort required to investigate and correct errors identified, as opposed to such errors being prevented from occurring under controls which provide adequate segregation of duties. |
| 71 |
The Company did not maintain adequate segregation of duties in our accounting and financial reporting process. The Company has not appropriately restricted access to our accounting applications to appropriate users and does not have processes in place that ensure that appropriate segregation of duties is maintained. Certain personnel have access to financial applications, programs and data beyond that needed to perform their individual job responsibilities and without independent monitoring. This allows for the creation, review and processing of certain financial data without independent review and authorization. There are also certain financial personnel that have incompatible duties, including in the areas of cash disbursements, payroll, and journal entry reviews. The Company has not yet completed the process of assigning different people the responsibilities of authorizing transactions, recording transactions, and maintaining custody of assets to sufficiently reduce the opportunities to allow any person to be in a position to both perpetrate and conceal errors or fraud in the normal course of the person’s duties. Particularly in the areas of purchases, cash disbursements, journal entry review and payroll, certain individuals have incompatible duties that limit our ability to identify and detect errors or fraud that may occur.
Remediation efforts to address the material weakness relating to segregation of duties
The Company recognized that it did not maintain an effective control environment during Fiscal 2016 which contributed to the Company not having effective controls to ensure that potential errors or misstatements may occur, but may not be detected.
During Fiscal 2016, the Company created and staffed a new accounting position, with such position contributing improved segregation of duties in the areas of purchasing, accounts payable processing, and timesheet management. In addition, improved segregation of duties has been achieved in the areas of cash disbursements, banking management, inventory control and manufacturing accounting, through increased delegation of duties to a staff accounting position that was created and staffed in Fiscal 2015.
The Company intends to focus more resources on internal control procedures during Fiscal 2017. The Company has engaged a third party consultant to assist with the enhancement of its control documentation as well as with developing a more robust control environment that once implemented would help remediate the material weakness described above. As part of this process, the Company will develop a Segregation of Duties Matrix and update and enhance business processes, documentation and job roles to fully implement this matrix. The Company will also be evaluating an enhancement in the financial and enterprise resource planning systems for some point in the future but will also focus on effective compensating controls until the financial/ERP software can be upgraded or replaced.
As the Company continues to evaluate and work to improve internal controls over financial reporting, the Company may determine to take additional measures to address the material weakness or determine to modify the remediation efforts described above. Until the remediation efforts discussed above, including any additional remediation efforts that the Company identifies as necessary, are implemented, tested and deemed to be operating effectively, the material weakness described above will continue to exist.
Disclosure controls and procedures and internal controls over financial reporting of complex accounting issues
After receiving a comment letter from the SEC in connection with standard periodic reviews of our Form 10-K for the Fiscal Year Ended March 31, 2015, our Form 10-Q for the Quarterly Period Ended June 30, 2015 and, in the process of review, our Form 10-Q, as amended, for the Quarterly Period Ended September 30, 2015, we identified material weaknesses in internal controls over financial reporting of complex accounting issues. The specific material weaknesses include the fact that the Company experienced difficulty in applying complex accounting and financial reporting and disclosure rules required under GAAP and SEC reporting regulations related to
| · | the recognition of revenue relating to a $5 million non-refundable payment received in relation to a licensing agreement and; |
| · | the accounting treatment of convertible preferred stock and preferred derivative liabilities associated with the share issuances. |
| 72 |
Remediation efforts to address the material weakness relating to financial reporting of complex accounting issues
In response to these material weaknesses, management developed a remediation plan to address the material weaknesses identified. The Company implemented the following remediation actions during Fiscal 2016, with such actions being effective for the quarters ending December 31, 2015 and March 31, 2016:
| · | Additional review and reconciliation steps were added to the revenue recognition process to ensure that review is appropriately recognized in the proper period. |
| · | The Company engaged a third-party subject matter expert to aid in identifying and applying GAAP rules related to complex accounting transactions. |
| · | The Company has improved its in-house technical research resources. |
While Management believes the remediation actions discussed above to be effectively designed, we also believe that there has not yet been a sufficient period of time upon which such actions can demonstrate the successful remediation of the material weaknesses relating to financial reporting and disclosure of complex accounting issues in accordance with GAAP and SEC reporting regulations as of the fiscal year ended March 31, 2016. We will continue to vigorously monitor the operation of these remediation actions to assess their efficacy in remediating the relevant material weakness in internal controls.
The effectiveness of the Company’s internal controls over financial reporting as of March 31, 2016 has been audited by Buchbinder Tunic, the Company’s independent registered public accounting firm, as stated in their report which is included in this Annual Report on Form 10-K.
Changes in internal control over financial reporting
There have been no changes in our internal controls over financial reporting (as such term is defined in Rules 13a-15(f) and 15(d)-15(f) under the Exchange Act) during the fourth quarter of fiscal 2016 that have materially effected, or a reasonably likely to materially effect, our internal controls over financial reporting.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Shareholders of Elite Pharmaceuticals, Inc. and Subsidiary
We have audited Elite Pharmaceuticals, Inc. and Subsidiary’s (“the Company”) internal control over financial reporting as of March 31, 2016, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
| 73 |
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
A material weakness is a control deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weakness has been identified and included in management’s assessment. Material weaknesses in internal control over financials reporting resulted from a lack of segregation of duties in the payroll, accounting and procure to pay cycles, as well as, the lack of oversight in analyzing and recording complex accounting transactions. These material weaknesses were considered in determining the nature, timing, and extent of audit tests applies in our audit of the 2016 financial statements, and this report does not affect our report dated June 15, 2016, on those financial statements.
In our opinion, because of the effect of the material weaknesses described above on the achievement of the objectives of the control criteria, Elite Pharmaceuticals Inc. & Subsidiary has not maintained effective internal control over financial reporting as of March 31, 2016, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets and the related consolidated statements of operations, shareholders’ deficit and cash flows of Elite Pharmaceuticals, Inc. and Subsidiary, and our report dated June 15, 2016, expressed an unqualified opinion.
/s/ Buchbinder Tunick & Company, LLP
Wayne, New Jersey
June 15, 2016
| ITEM 9B | OTHER INFORMATION |
None.
| ITEM 10 | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The following sets forth biographical information about each of our directors and executive officers as of the date of this report:
| Name | Age | Position | Director
/ Officer Since |
Director Tier | ||||
| Nasrat Hakim | 55 | President, Chief Executive Officer and Director | August 1, 2013 | III | ||||
| Barry Dash, Ph. D. | 85 | Director | April 2005 | II | ||||
| Jeffrey Whitnell | 60 | Director | October 2009 | III | ||||
| Eugene Pfeifer | 76 | Director | April 2016 | I | ||||
| Davis Caskey | 68 | Director | April 2016 | I | ||||
| Carter J. Ward | 52 | Chief Financial Officer, Secretary and Treasurer | July 2009 | |||||
| Douglas Plassche | 52 | Executive Vice President of Operations | August 2013 |
The principal occupations and employment of each Director during the past five years is set forth below. In each instance in which dates are not provided in connection with a director’s business experience, such nominee has held the position indicated for at least the past five years.
| 74 |
Each director currently holds office until the expiration of his Tier (each for three years) or until such director’s death, resignation or removal. Pursuant to our recently amended and restated bylaws, our Board of Directors is now classified into three separate tiers of directors, with each respective tier to serve a three-year term and until their successors are duly elected and qualified.
Nasrat Hakim
Nasrat Hakim has served as a Director, President and Chief Executive officer since August 1, 2013. Mr. Hakim has more than 30 years of pharmaceutical and medical industry experience in Quality Assurance, Analytical Research and Development, Technical Services and Regulatory Compliance. He brings with him proven management experience, in-depth knowledge of manufacturing systems, development knowledge in immediate and extended release formulations and extensive regulatory experience of GMP and FDA regulations. From 2004 to 2013, Mr. Hakim was employed by Actavis, Watson and Alpharma in various senior management positions. Most recently, Mr. Hakim served as International Vice President of Quality Assurance at Actavis, overseeing 25 sites with more than 3,000 employees under his leadership. Mr. Hakim also served as Corporate Vice President of Technical Services, Quality and Regulatory Compliance for Actavis U.S., Global Vice President, Quality and Regulatory Compliance for Alpharma, as well as Executive Director of Quality Unit at TheraTech, overseeing manufacturing and research and development. In 2009, Mr. Hakim founded Mikah Pharma, LLC, a virtual, fully functional pharmaceutical company. Mr. Hakim holds a Bachelor in Chemistry/Bio-Chemistry and Masters of Science in Chemistry from California State University at Sacramento, Sacramento, CA; a Masters in Law with Graduate Certification in U.S. and International Taxation from St. Thomas University, School of Law, Miami, FL.; and a Graduate Certification in Regulatory Affairs (RAC) from California State University at San Diego, San Diego, CA. Mr. Hakim’s leadership experience (consisting of extensive experience in senior management positions, responsible for 25 global manufacturing/regulatory sites with more than 3,000 employees under his leadership), industry experience (comprising more than 30 years of pharmaceutical and medical industry experience served in various quality assurance, analytical research and development/technical services and compliance positions) and academic experience (including Bachelor degrees in Chemistry and Bio-Chemistry, Masters degrees in Chemistry and Law, with Graduate Certification in U.S. and International Taxation, and a Graduate Certification in Regulatory Affairs) led to the conclusion that he is qualified to serve as a director.
Barry Dash, Ph.D.
Dr. Barry Dash has served as a Director since April 2005, Member of the Audit Committee since April 2005, Member of the Nominating Committee since April 2005 and Member and Chairman of the Compensation Committee since June 2007. Dr. Dash has been, since 1995, President and Managing Member of Dash Associates, L.L.C., an independent consultant to the pharmaceutical and health industries. From 1983 to 1996 he was employed by Whitehall-Robins Healthcare, a division of American Home Products Corporation (now known as Wyeth), initially as Vice President of Scientific Affairs, then as Senior Vice President of Scientific Affairs and then as Senior Vice President of Advanced Technologies, during which time he personally supervised six separate departments: Medical and Clinical Affairs, Regulatory Affairs, Technical Affairs, Research and Development, Analytical R&D and Quality Management/Q.C. Dr. Dash had been employed by the Whitehall Robins Healthcare from 1960 to 1976, during which time he served as Director of Product Development Research, Assistant Vice President of Product Development and Vice President of Scientific Affairs. Dr. Dash had been employed by J.B. Williams Company (Nabisco Brands, Inc.) from 1978 to 1982. From 1976 to 1978 he was Vice President and Director of Laboratories of the Consumer Products Division of American Can Company. He currently serves on the board of directors of GeoPharma, Inc. (NASDQ: GORX). Dr. Dash holds a Ph.D. from the University of Florida and M.S. and B.S. degrees from Columbia University where he was Assistant Professor at the College of Pharmaceutical Sciences from 1956 to 1960. He is a member of the American Pharmaceutical Association, the American Association for the Advancement of Science and the Society of Cosmetic Chemist, American Association of Pharmaceutical Scientists, Drug Information Association, American Foundation for Pharmaceutical Education, and Diplomate American Board of Forensic Examiners. He is the author of scientific publications and patents in the pharmaceutical field. Dr. Dash’s extensive education in pharmaceutical sciences and his experience in the development of scientific products, including his experience in regulatory affairs, led to the conclusion that he is qualified to serve as a director.
| 75 |
Jeffrey Whitnell
Jeffrey Whitnell has served as a Director since October 23, 2009, Chairman of the Audit Committee since October 23, 2009, member of the Compensation Committee since October 23, 2009 and designated by the Board as an “audit committee financial expert” as defined under applicable rules under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), Since January 2016, Mr. Whitnell has served as the Vice President, Finance & Controller for LifeWatch Services. From June2010 to March 2015, Mr. Whitnell was the Chief Financial Officer for ReliefBand Medical Technologies, a medical device company. From June 2009 to June 2010, Mr. Whitnell provided financial consulting services to various healthcare companies, including ReliefBand Medical Technologies. From June 2004 to June 2009, Mr. Whitnell was Chief Financial Officer and Senior Vice President of Finance at Akorn, Inc. From June 2002 to June 2004, Mr. Whitnell was Vice President of Finance and Treasurer for Ovation Pharmaceuticals. From 1997 to 2001, Mr. Whitnell was Vice President of Finance and Treasurer for MediChem Research. Prior to 1997, Mr. Whitnell held various finance positions at Akzo Nobel and Motorola. Mr. Whitnell began his career as an auditor with Arthur Andersen & Co. He is a certified public accountant and holds an M.B.A. in Finance from the University of Chicago and a B.S. in Accounting from the University of Illinois. Mr. Whitnell’s qualifications as an accounting and audit expert provide specific experience to serve as a director for the Company.
Eugene Pfeifer
Eugene Pfeifer has served as a Director since April 7, 2016. Mr. Pfeifer brings with him more than 45 years of regulatory and trade experience, most recently having served as a law partner at King & Spalding in Washington DC from 1986 to 2009 and prior to that as a law partner at the Burditt, Bowles & Radzius from 1980 to 1985. Since retiring from legal practice in 2009, Mr. Pfeifer has worked as a consultant to companies, including consultation for the Company, by providing his expertise regarding FDA and FTC issues. Among his many accomplishments, he was a major participant in the development of the Drug Price Competition and Patent Term Restoration Act of 1984, and provided strategic counseling to companies affected by that statue. In addition, he has provided regulatory advice and representation on a wide variety of FDA, FTC, and DEA regulated activities, including product approval, advertising, promotion, and compliance issues.
Prior to working at Burditt, Bowles and Radzius, Mr. Pfeifer served from 1974 to 1975 in the General Counsel's office of the Federal Trade Commission, where he represented the FTC in Federal Court to enjoin violations of the Federal Trade Commission Act, and served ten years in the Chief Counsel's Office at the FDA as Associated Chief Counsel for Enforcement, Associate Chief Counsel for Drugs and Deputy Chief Counsel for Regulations and Hearings. During his tenure at the FDA, he was the FDA's lead litigator and Appellate Court advocate, and he briefed the FDA's cases before the Supreme Court. Mr. Pfeifer is a graduate of Brown University and the Georgetown University Law Center. Mr. Pfeifer’s qualifications and extensive experience in the areas of regulatory affairs, legislation and FDA representation, led the Board to conclude that Mr. Pfeifer is qualified to be a member of the Company’s Board of Directors.
| 76 |
Davis Caskey
Davis Caskey has served as a Director since April 28, 2016. He brings more than 40 years of pharmaceutical industry experience to this position. Mr. Caskey is currently President & CEO of Caskey LLC, which he formed in 2013 to serve as an umbrella to manage his pharmaceutical consulting and other business interests. From 1990 to 2013, Davis served as the operating officer of ECR Pharmaceuticals, of which he was a founding member. HiTech Pharmacal acquired the privately held ECR in 2009 and Mr. Caskey continued in his role until retiring in 2013. At ECR, Mr. Caskey was credited with the establishment of the company's sales and marketing structure, its product distribution format, and the development and management of the firm’s internal organization. His responsibilities included the oversight of drug development and regulatory filings, product acquisitions, and acquisition of other companies. A primary focus was to conceive and develop, with the assistance of key strategic partners, unique dosage forms and extended release formulations of products which enhance patient compliance and safety. Prior to ECR, Mr. Caskey was employed by A.H. Robins for 18 years in various field and home office management positions. His experience brings critical insight into the marketing and distribution of pharmaceutical products in a rapid and ever changing competitive marketplace. Mr. Caskey attended the University of Texas (Austin) and Lamar University, and holds bachelor’s and master’s degrees.
Jerry Treppel
Jerry Treppel served as a Director from October 28, 2008 to January 11, 2016, Chairman of the Board from November 6, 2008 to January 11, 2016 and Chief Executive Officer from September 15, 2009 to July 31, 2013. Mr. Treppel is currently a Managing Director of ArcLight Advisors, an investment bank specializing in the health care sector. From October 2008 through March 2013, Mr. Treppel was Managing Director of Ledgemont Capital Group LLC, a boutique merchant bank that provided access to capital and corporate advisory services to public and private companies. Additionally, he served as the managing member of Wheaten Capital Management LLC, a capital management company focusing on investments in the health care sector from 2003 to 2008.Over the past 20 years, Mr. Treppel was an equity research analyst focusing on the specialty pharmaceuticals and generic drug sectors at several investment banking firms including Banc of America Securities, Warburg Dillon Read LLC (now UBS), and Kidder, Peabody & Co. He previously served as a healthcare services analyst at various firms, including Merrill Lynch & Co. He also held administrative positions in the healthcare services industry early in his career. From 2003 to 2009, Mr. Treppel served as a member of the board of directors of Akorn, Incorporated (NASDAQ: AKRX), a specialty pharmaceutical company engaged in the development, manufacturing and marketing of branded and multi-source pharmaceutical products and vaccines. Mr. Treppel also served as the Chair of Akorn’s Nominating and Corporate Governance Committee and as a member of its Audit Committee and Compensation Committee. Mr. Treppel holds a BA in Biology from Rutgers College in New Brunswick, N.J., an MHA in Health Administration from Washington University in St. Louis, Mo., and an MBA in Finance from New York University. Mr. Treppel has been a Chartered Financial Analyst (CFA) since 1988. Mr. Treppel’s knowledge of the pharmaceutical industry as well as his education credentials and his experience as a member of the board of directors of Akorn, Incorporated led to the conclusion that he is qualified to serve as a director.
| 77 |
Jeenarine Narine
Jeenarine Narine served as a Director from June 24, 2009 to April 7, 2016. Mr. Narine was elected as a member of Elite’s Board in June 2009 as one of three directors designated by Epic pursuant to the terms of the Epic Strategic Alliance Agreement (see Item 13: “Certain Relationships And Related Transactions And Director Independence; Certain Related Person Transactions; Strategic Alliance Agreement/Transactions With Epic Pharma LLC And Epic Investments LLC” below). Since December 2010, Mr. Narine has been the President and Chief Operating Officer of Epic Pharma, LLC, a manufacturer of generic pharmaceuticals and Elite’s strategic partner pursuant to the Epic Strategic Alliance Agreement, in which capacity he oversees all manufacturing operations. From July 2008 to December 2010, Mr. Narine served as Epic Pharma’s Executive Vice President of Manufacturing and Operations. Mr. Narine is also the current President of Eniran Manufacturing Inc., a contract manufacturer of dietary and nutritional supplements, and has held such office since 2000. In addition, Mr. Narine has been since 1989 the President of A&J Machine Inc., a company owned by Mr. Narine that is engaged in the sales of new and used pharmaceutical manufacturing equipment. In addition to this professional experience, Mr. Narine graduated from the Guyana Industrial Institute, where he studied Metalology and Welding. Mr. Narine’s experience as President and Chief Operating Officer and, previously, as Executive Vice President of Manufacturing and Operations of Epic Pharma LLC and his knowledge of pharmaceutical manufacturing equipment led to the conclusion that he is qualified to serve as a director.
Ashok G. Nigalaye, Ph.D.
Dr. Ashok G. Nigalaye served as a Director from June 24, 2009 to June 5, 2015, member of the Compensation Committee from October 23, 2009 to June 5, 2015 and Chief Scientific Officer from September 15, 2009 to June 5, 2015. Dr. Nigalaye was elected as a member of Elite’s Board in June 2009 as one of three directors designated by Epic pursuant to the terms of the Epic Strategic Alliance Agreement. Since December 2010, Dr. Nigalaye has been the Chairman and Chief Executive Officer of Epic Pharma, LLC, a manufacturer of generic pharmaceuticals and Elite’s strategic partner pursuant to the Epic Strategic Alliance Agreement. From July 2008 to December 2010, Dr. Nigalaye served as Epic Pharma’s President and Chief Executive Officer. From August 1993 to February 2008, Dr. Nigalaye served as Vice President of Scientific Affairs and Operations of Actavis Totowa LLC, a manufacturer of generic pharmaceuticals, where he was responsible for directing and organizing company activities relating to pharmaceutical drug manufacturing, regulatory affairs and research and development. Dr. Nigalaye currently serves as a director of GTI Inc., a privately held company. Dr. Nigalaye holds a B.S. in Pharmacy from the University of Bombay, an M.S. in Industrial Pharmacy from Long Island University, and a Ph.D. in Industrial Pharmacy from St. John’s University. Dr. Nigalaye is also a licensed pharmacist in the State of New York.
Carter J. Ward
Carter J. Ward has served as Chief Financial Officer, Secretary and Treasurer of the Company since July 1, 2009. Prior to joining the Company, from July 2005 to April 2009, Mr. Ward filled multiple finance and supply chain leadership roles with the Actavis Group and its U.S. subsidiary, Amide Pharmaceuticals. From September 2004 to June 2005, Mr. Ward was a consultant, mainly engaged in improving internal controls and supporting Sarbanes Oxley compliance of Centennial Communications Inc., a NASDAQ listed wireless communications provider. From 1999 to September 2004, Mr. Ward was the Chief Financial Officer for Positive Healthcare/Ceejay Healthcare, a U.S.-Indian joint venture engaged in the manufacture and distribution of generic pharmaceuticals and nutraceuticals in India. Mr. Ward began his career as a certified public accountant in the audit department of KPMG and is a Certified Supply Chain Professional (“CSCP”). Mr. Ward holds a B.S. in Accounting from Long Island University, Brooklyn, NY, from where he graduated summa cum laude Mr. Ward’s experience and expertise in the area of finance and more specifically, as a Certified Supply Chain Professional, provides the qualifications, attributes and skills to serve as an officer for the Company.
| 78 |
Douglas Plassche
Douglas Plassche has served as Executive Vice President of Operations since August 2013.
Prior to joining the Company, from 2009 to 2013, Mr. Plassche served as the Managing Director of the New Jersey Solid Oral Dose Operations of Actavis, overseeing 450 employees and the production of more than 100 products. From 2007 to 2009, Mr. Plassche was the Senior Director of Manufacturing for PAR Pharmaceuticals, overseeing 200 employees and the production of more than 70 products. From 1990 – 2007, Mr. Plassche was employed by Schering-Plough, progressing steadily through multiple disciplines, locations and technical operations sectors with increasing levels of responsibility. Mr. Plassche has a Bachelors Degree in Economics from Rochester University.
There are no family relationships between any of our directors and executive officers.
Compliance with Section 16(a) of the Exchange Act
Section 16(a) of the Exchange Act requires our Officers, Directors, and persons who own more than ten percent of a registered class of equity securities, to file reports with the Securities and Exchange Commission reflecting their initial position of ownership on Form 3 and changes in ownership on Form 4 or Form 5. Based solely on a review of the copies of such Forms received by us, we found that, during the fiscal year ended March 31, 2016, Douglass Plassche filed Form 3 late on Form 5 and reported two transactions late on this Form 5. Jeenarine Narine filed one Form 4 late and reported three transactions late on this Form 4. Jerry Treppel filed two Forms 4 late and reported eight transactions (two and six) late on these Forms 4. Ashok Nigalaye filed two Forms 4 late and reported nine transactions (five and four) late on these Forms 4. Mikah Pharma, LLC filed two Forms 4 late and reported two transactions late on these Forms 4.
Committees of the Board
The Board of Directors has an Audit Committee, a Compensation Committee and a Nominating Committee.
Audit Committee
During Fiscal 2016, the members of the Audit Committee were Jeffrey Whitnell (Chairman of the Audit Committee), and Dr. Barry Dash. We deem Messrs. Whitnell and Dash to be independent and Mr. Whitnell to be qualified as an audit committee financial expert. The Board of Directors has determined that Messrs. Whitnell and Dash are independent directors as (i) defined in Rule 10A-3(b)(1)(ii) under the Exchange Act and (ii) under Sections 803A(2) and 803B(2)(a) of the NYSE MKT LLC Company Guide (although our securities are not listed on the NYSE MKT LLCE or any other national exchange).
Nominating Committee
During Fiscal 2016, the members of the Nominating Committee were Dr. Barry Dash and Jeenarine Narine. There were no material changes to the procedures by which security holders may recommend nominees to our Board of Directors since the filing of our last Annual Report on Form 10-K. Mr. Narine resigned as a Director on April 7, 2016.
Compensation Committee
During Fiscal 2016, the members of the Compensation Committee were Dr. Barry Dash (Chairman of the Nominating Committee), and Jeffrey Whitnell.
| 79 |
Code of Conduct and Ethics
At the first meeting of the Board of Directors following the annual meeting of stockholders held on June 22, 2004, and as further updated effective July 2009, the Board of Directors adopted a Code of Business Conduct and Ethics that is applicable to the Company’s directors, officers and employees. A copy of the Code of Business Conduct and Ethics is available on our website at www.elitepharma.com, under Investor Relations.
| ITEM 11 | EXECUTIVE COMPENSATION |
Compensation discussion and analysis summary
Our approach to executive compensation, one of the most important and complex aspects of corporate governance, is influenced by our belief in rewarding people for consistently strong execution and performance. We believe that the ability to attract and retain qualified executive officers and other key employees is essential to our long-term success.
Compensation Linked to Attainment of Performance Goals
Our plan to obtain and retain highly skilled employees is to provide significant incentive compensation opportunities and market competitive salaries. The plan was intended to link individual employee objectives with overall company strategies and results, and to reward executive officers and significant employees for their individual contributions to those strategies and results. Furthermore, we believe that equity awards serve to align the interests of our executives with those of our stockholders. As such, equity is a key component of our compensation program.
Role of the Compensation Committee
The Company formed the Compensation Committee in June 2007. Since the formation of the Compensation Committee all elements of the executives’ compensation are determined by the Compensation Committee, which is comprised of a two independent non-employee directors, and one director who is also the Company’s Chief Scientific Officer. However, the Compensation Committee’s decisions concerning the compensation of the Company’s Chief Executive Officer are subject to ratification by the independent directors of the Board of Directors. As of March 31, 2016, the members of the Compensation Committee were Dr. Barry Dash and Jeffrey Whitnell. The Committee operates pursuant to a charter. Under the Compensation Committee charter, the Compensation Committee has authority to retain compensation consultants, outside counsel, and other advisors that the committee deems appropriate, in its sole discretion, to assist it in discharging its duties, and to approve the terms of retention and fees to be paid to such consultants. The Compensation Committee did not engage any advisors.
Named Executive Officers and Key Employees
The named executive officers and key employees for the fiscal year ended March 31, 2016 were:
| · | Nasrat Hakim, Chief Executive Officer and President for the full year. |
| · | Carter J. Ward, Chief Financial Officer, Secretary and Treasurer for the full year. |
| · | Douglas Plassche, Executive Vice President for the full year. |
These individuals are referred to collectively as the “Named Executive Officers”.
| 80 |
Our executive compensation program
Overview
The primary elements of our executive compensation program are base salary, incentive cash and stock bonus opportunities and equity incentives typically in the form of stock option grants or payment of a portion of annual salary as stock. Although we provide other types of compensation, these three elements are the principal means by which we provide the Named Executive Officers with compensation opportunities.
The annual bonus opportunity and equity compensation components of the executive compensation program reflect our belief that a portion of an executive’s compensation should be performance-based. This compensation is performance-based because payment is tied to the achievement of corporate performance goals. To the extent that performance goals are not achieved, executives will receive a lesser amount of total compensation.
Elements of our executive compensation program
Base Salary
We pay a base salary to certain of the Named Executive Officers, with such payments being made in either cash, Common Stock or a combination of cash and Common Stock. In general, base salaries for the Named Executive Officers are determined by evaluating the responsibilities of the executive’s position, the executive’s experience and the competitive marketplace. Base salary adjustments are considered and take into account changes in the executive’s responsibilities, the executive’s performance and changes in the competitive marketplace. We believe that the base salaries of the Named Executive Officers are appropriate within the context of the compensation elements provided to the executives and because they are at a level which remains competitive in the marketplace.
Bonuses
The Board of Directors may authorize us to give discretionary bonuses, payable in cash or shares of Common Stock, to the Named Executive Officers and other key employees. Such bonuses are designed to motivate the Named Executive Officers and other employees to achieve specified corporate, business unit and/or individual, strategic, operational and other performance objectives.
Stock Options
Stock options constitute performance-based compensation because they have value to the recipient only if the price of our Common Stock increases. Stock options for each of the Named Executive Officers generally vest over time, obtainment of a corporate goal or a combination of the two.
The grant of stock options at Elite is designed to motivate our Named Executive Officers to achieve our short-term and long-term corporate goals.
Retirement and Deferred Compensation Benefits
We do not presently provide the Named Executive Officers with a defined benefit pension plan or any supplemental executive retirement plans, nor do we provide the Named Executive Officers with retiree health benefits. We have adopted a deferred compensation plan under Section 401(k) of the Code. The plan provides for employees to defer compensation on a pretax basis subject to certain limits, however, Elite does not provide a matching contribution to its participants.
| 81 |
The retirement and deferred compensation benefits provided to the Named Executive Officers are not material factors considered in making other compensation determinations with respect to Named Executive Officers.
Post-Termination/Change of Control Compensation
Pursuant to his employment agreement, Nasrat Hakim, our Chief Executive Officer, is entitled to a payment in an amount equal to two years base annual salary in effect upon the date of termination, less applicable deductions and withholdings, payable in Common Stock upon a Change of Control (as defined in the Hakim Employment Agreement). For more detailed information, please see “Agreements with Named Executive Officers” below.
We do not presently provide the Named Executive Officers with any plan or arrangement, other than those that may be contained in employment contracts, in connection with any termination, including, without limitation, through retirement, resignation, severance or constructive termination (including a change in responsibilities) of such Named Executive Officer’s employment with the Company.
As part of the Company’s efforts to ensure the retention and continuity of key employees, officers and directors in the event of a change of control of the ownership of the Company, unless otherwise stated in applicable employment contracts, key executives would receive an amount equal to twelve months of such executive’s salary, and certain Directors and managers would receive an amount equal to six months of such Director’s or managers fees or salaries, as applicable. In addition, any outstanding and unvested options would immediately vest, in the event of a change of control.
Perquisites
As described in more detail below, the perquisites provided to certain of the Named Executive Officers consist of car allowances and life insurance premiums. These perquisites represent a small fraction of the total compensation of each such Named Executive Officer. The value of the perquisites we provide are taxable to the Named Executive Officers and the incremental cost to us of providing these perquisites is reflected in the Summary Compensation Table. The Board of Directors believes that the perquisites provided are reasonable and appropriate. For more information on perquisites provided to the Named Executive Officers, please see the “All Other Compensation” column of the Summary Compensation Table and “Agreements with Named Executive Officers,” below.
Agreements with Named Executive Officers
Nasrat Hakim
Pursuant to his August 2013 employment agreement, and as amended on January 12, 2016 (the “Hakim Employment Agreement”), Mr. Hakim receives an annual salary of $500,000 per year. The Salary is paid in shares of the Company’s Common Stock pursuant to the Company’s current procedures for paying Company executives in Stock. He also is entitled to an annual bonus equal to up to 100% of his annual salary, payable in accordance with the Company’s payroll practices, with such being based upon his ability to meet certain Company milestones to be determined by the Company’s Board of Directors. The Board may also award discretionary bonuses in its sole discretion. Mr. Hakim is entitled to employee benefits (e.g., health, vacation, employee benefit plans and programs) consistent with other Company employees of his seniority and a car allowance. The Hakim Employment Agreement contains confidentially, non-competition and other standard restrictive covenants.
| 82 |
Mr. Hakim’s employment is terminable by the Company for cause (as defined in the Hakim Employment Agreement). The Hakim Employment Agreement also may be terminated by the Company upon at least 30 days written notice due to disability (as defined in the Hakim Employment Agreement) or without cause. Mr. Hakim can terminate the Hakim Employment Agreement by resigning, provided he gives notice at least 60 days prior to the effective resignation date. If Mr. Hakim is terminated for cause or he resigns, he only is entitled to accrued and unpaid annual salary, accrued vacation time and any reasonable and necessary business expenses, all through the date of termination and payable in stock (“Basic Termination Benefits”). If Mr. Hakim is terminated because of disability or death, in addition to Basic Termination Benefits, He is entitled his pro rata annual bonus through the date of termination (payable in Stock). If the Company terminates Mr. Hakim without cause, In addition to Basic Termination Benefits, Mr. Hakim is entitled to his pro rata annual bonus through the date of termination and an amount equal to two years’ annual salary (all payable in Stock).
Upon a Change of Control (as defined in the Hakim Employment Agreement), Mr. Hakim is entitled to a payment in an amount equal to two years base annual salary in effect upon the Date of Termination, less applicable deductions and withholdings, payable in Stock computed in the same manner as set forth as the Salary.
Carter J. Ward
On November 12, 2009, the Company entered into an employment agreement with Mr. Carter J. Ward (the “Ward Employment Agreement”). Pursuant to the terms of the Ward Employment Agreement, Mr. Ward continues as an at-will employee of the Company as its Chief Financial Officer. Mr. Ward receives a base salary of $150,000, with $125,000 of such amount being paid in accordance with the Company’s payroll practices and $25,000 of such amount being paid by the issuance of restricted shares of Common Stock, in lieu of cash. The Common Stock component of Mr. Ward’s compensation is to be computed on a quarterly basis, with the number of shares issued equal to the quotient of the quarterly amount due of $6,250 divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
The Board of Directors increased Mr. Ward’s base salary to $155,000 retroactive to January 1, 2013. This $5,000 increase to be paid by the issuance of restricted shares of Common Stock. The Common Stock component of Mr. Ward’s compensation is to be computed on a quarterly basis, with the number of shares issued equal to the quotient of the quarterly amount due of $7,500 divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
On January 1, 2014, Mr. Ward’s compensation was adjusted to include a total compensation of $180,000, consisting of $150,000 being paid in accordance with the Company’s payroll practices and $30,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
On March 1, 2015, Mr. Ward’s compensation was adjusted to include a total compensation of $187,200, consisting of $157,200 being paid in accordance with the Company’s payroll practices and $30,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
On March 1, 2016, Mr. Ward’s compensation was adjusted to include a total compensation of $192,816, consisting of $162,816 being paid in accordance with the Company’s payroll practices and $30,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
| 83 |
The Common Stock component of Mr. Ward’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
Douglas Plassche
On July 20, 2013, the Company entered into an employment agreement with Mr. Douglas Plassche (the “Plassche Employment Agreement”). Pursuant to the Plassche Employment Agreement, Mr. Plassche serves as an at-will employee, in the position of Vice President of Operations, commencing on August 12, 2013. The Plassche Employment Agreement includes a total base compensation of $236,000, consisting of $211,000 being paid in accordance with the Company’s payroll practices and $25,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash. Mr. Plassche is also eligible for an annual bonus in cash and/or equity based awards for up to an equivalent of 30% of base salary, with such annual bonus being granted based upon the achievement of agreed milestones and at the discretion of the Company and its Chief Executive Officer. In addition, pursuant to the Plassche Employment Agreement, he was granted options to purchase 3,000,000 shares of Common Stock, at a price of $ 0.07 per share, (the closing price of the Common Stock on the date of the Plassche Employment Agreement). The options were issued pursuant to the 2004 Employee Stock Option Plan and vest over a period of three years with the vesting period commencing one year from the date of issuance.
Mr. Plassche’s employment is terminable by either party. If the Company terminates Mr. Plassche without cause, Mr. Plassche is entitled to an amount equal to six months of base annual salary in effect upon the date of termination.
On March 1, 2015, Mr. Plassche’s compensation was adjusted to include a total base compensation of $249,800, consisting of $224,800 being paid in accordance with the Company’s payroll practices and $25,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
On March 1, 2016, Mr. Plassche’s compensation was adjusted to include a total base compensation of $253,552, consisting of $228,552 being paid in accordance with the Company’s payroll practices and $25,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
The Common Stock component of Mr. Plassche’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
Barbara Ellison
On March 3, 2014, the Company entered into an employment agreement with Ms. Barbara Ellison (the “Ellison Employment Agreement”). Pursuant to the Ellison Employment Agreement, Ms. Ellison serves as an at-will employee, in the position of Vice President of Quality Operations and Regulatory Affairs, commencing on March 24, 2014. The Ellison Employment Agreement includes a total base compensation of $190,000, consisting of $165,000 being paid in accordance with the Company’s payroll practices and $25,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash. Ms Ellison is also eligible for an annual bonus in cash and/or equity based awards for up to an equivalent of 25% of base salary, with such annual bonus being granted based upon the achievement of agreed milestones in the discretion of the Company and its Chief Executive Officer. In addition, pursuant to the Ellison Employment Agreement, Ms. Ellison was granted options to purchase 600,000 shares of Common Stock, at a price of $ 0.33 per share, (the closing price of the Common Stock on the date of the Ellison Employment Agreement). The options were issued pursuant to the 2009 Employee Stock Option Plan and vest over a period of three years with the vesting period commencing one year from the date of issuance.
| 84 |
Ms. Ellison’s employment is terminable by either party. If the Company terminates Ms. Ellison without cause, Ms. Ellison is entitled to an amount equal to six months of base annual salary in effect upon the date of termination.
On March 1, 2015, Ms. Ellison ‘s compensation was adjusted to include a total base compensation of $193,800, consisting of $168,800 being paid in accordance with the Company’s payroll practices and $25,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
The Common Stock component of Ms. Ellison’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
Ms. Ellison retired from the Company, effective June 1, 2016 and is no longer an employee of the Company. As of the date of Ms. Ellison’s retirement, the options issued to Ms. Ellison were not fully vested, with 400,000 shares being vested and 200,000 shares being non-vested. Pursuant to the 2009 Employee Stock Option Plan, vesting of options require that the employee holder of such options be employed by the Company on each vesting date. Accordingly, future vesting of the non-vested option to purchase 200,000 held by Ms. Ellison at the time of her retirement’s retirement is prohibited. The vested options to purchase up to 400,000 shares at a price of $0.33 per share expire 90 days from the date of termination of Ms. Ellison’s employment with the Company, pursuant to the 2009 Employee Stock Option Plan.
George Kenneth Smith
On October 20, 2014, the Company entered into an employment agreement with Mr. George Kenneth Smith (the “Smith Employment Agreement”). Pursuant to the Smith Employment Agreement, Mr. Smith serves as an at-will employee, in the position of Vice President, Legal, commencing on October 20, 2014. The Smith Employment Agreement includes a total base compensation of $400,000, consisting of $150,000 being paid in accordance with the Company’s payroll practices and $250,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash. Mr Smith is also eligible for an annual bonus and discretionary bonus, with such being at the discretion of the Company and its Chief Executive Officer. In addition, pursuant to the Smith Employment Agreement, Mr. Smith was granted options to purchase 1,500,000 shares of Common Stock, at a price of $ 0.29 per share, (the closing price of the Common Stock on the date of the Smith Employment Agreement). The options were issued pursuant to the 2009 Employee Stock Option Plan and vest over a period of three years with the vesting period commencing one year from the date of issuance.
Mr Smith’s employment is terminable by either party. If the Company terminates Mr. Smith without cause, or if Mr. Smith is terminated upon a change of control event, as defined in the Smith Employment Agreement, Mr. Smith is entitled to an amount equal to one year of base annual salary in effect upon the date of termination.
On March 1, 2016, Mr. Smith’s compensation was adjusted to include a total base compensation of $412,000, consisting of $162,000 being paid in accordance with the Company’s payroll practices and $250,000 being paid by the issuance of restricted shares of Common Stock in lieu of cash.
The Common Stock component of Mr. Smith’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
| 85 |
Jerry Treppel
On December 1, 2008, Elite entered into a compensation agreement with Mr. Treppel (the “First Treppel Agreement”) providing for the terms under which Mr. Treppel will serve as the non-executive Chairman of the Board. Pursuant to the First Treppel Agreement, Mr. Treppel will serve as the non-executive Chairman of the Board until immediately prior to the next annual meeting of the Company’s stockholders; provided, however, that following such annual meeting, and each subsequent annual meeting of the Company’s stockholders, if the Board elects Mr. Treppel as the non-executive Chairman of the Board, the term of the First Treppel Agreement will be extended through the earlier of (a) the date of the next subsequent annual meeting of the Company’s stockholders and (b) the date upon which Mr. Treppel no longer serves as the non-executive Chairman.
During the term of the First Treppel Agreement, including any applicable extensions thereof, Mr. Treppel is entitled to cash compensation of $2,083.33 on a monthly basis in lieu of, and not in addition to, any cash directors’ fees and other compensation paid to other non-employee members of the Board. Mr. Treppel is also entitled to reimbursement of any expenses reasonably incurred in the performance of his duties under the First Treppel Agreement upon presentation of proper written evidence of such expenditures.
In addition, pursuant to the terms of the First Treppel Agreement, Elite granted to Mr. Treppel under its 2004 Stock Option Plan non-qualified stock options to purchase 180,000 shares of Common Stock of Elite, par value $0.001 per share, exercisable for a period of 10 years at an exercise price per share of $0.06, subject to the terms and conditions of the related option agreement.
Under the First Treppel Agreement, Elite has also agreed to indemnify Mr. Treppel to the fullest extent permitted by law in accordance with the By-Laws of Elite against (a) reasonable expenses, including attorneys’ fees, incurred by him in connection with any threatened, pending, or completed civil, criminal, administrative, investigative, or arbitrative action, suit, or proceeding (and any appeal therein) seeking to hold him liable for actions taken in his capacity as Chairman of the Board, and (b) reasonable payments made by him in satisfaction of any judgment, money decree, fine (including assessment of excise tax with respect to an employee benefit plan), penalty or settlement for which he may have become liable in any such action, suit or proceeding, provided that any such expenses or payments are not the result of Mr. Treppel’s gross negligence, willful misconduct or reckless actions.
Either party may terminate the First Treppel Agreement, effective immediately upon the giving of written notice to the other party. If no such written notice is given, then the term of the First Treppel Agreement shall end immediately prior to the next annual meeting of the Company’s stockholders (the “Treppel Term”), provided however, that following such annual meeting, and each subsequent meeting of the Company’s stockholders, if the Board elects Mr. Treppel to continue to serve as the non-executive Chairman of the Board, the Treppel Term shall be extended through the earlier of (a) the date of the next subsequent annual meeting of the Company’s stockholders and (b) the date upon which Mr. Treppel shall no longer serve as the non-executive Chairman of the Board.
On September 15, 2009, Mr. Treppel was appointed Chief Executive Officer of the Company and he served in that capacity until his resignation in August 2013. He continues to also serve as Chairman of the Board and he has agreed to forego any additional compensation related to his activities and Chief Executive Officer. Accordingly, Mr. Treppel’s compensation as Chief Executive Officer and Chairman of the Board remains unchanged from the First Treppel Agreement.
| 86 |
On October 23, 2009, at the meeting of the Board held immediately after the annual stockholders meeting, Mr. Treppel’s compensation as Chairman of the Board was revised to an annual amount of $30,000, payable in common shares of the Company. The amount of common shares to be issued to Mr. Treppel in payment of compensation due to him as Chairman of the Board is calculated on a quarterly basis, and is equal to the quotient of the quarterly amount due of $7,500, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
Mr. Treppel agreed to forego any additional compensation for his services as Chief Executive Officer of the Company.
Mr. Treppel stepped down from his position as Chief Executive Officer and was replaced by Mr. Nasrat Hakim in this position in August 2013. Mr. Treppel resigned from the Board of Directors in January 2016.
Chris C. Dick
On November 13, 2009, the Company entered into an employment agreement with Mr. Dick as our President and Chief Operating Office (the “Dick Employment Agreement”). The Dick Employment Agreement is terminable at the will of either the Company or Mr. Dick, with no notice or cause required. The Dick Employment Agreement provided for a base salary of $200,000, with $175,000 of this amount being paid in accordance with the Company’s payroll practices and $25,000 of this amount being paid via the issuance of restricted shares of Common Stock in lieu of cash. The Common Stock component of Mr. Dick’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
In addition, the Dick Employment Agreement provided for 25 days of paid vacation, the right to participate in all health insurance plans maintained by the Company for its employees, a monthly auto allowance of $700 and term life insurance in the amount of $500,000 payable to Mr. Dick’s estate.
The Dick Employment Agreement also required Mr. Dick’s execution of a Proprietary Rights Agreement.
The Board of Directors of the Company increased Mr. Dick’s base salary to $205,000, effective January 1, 2013, with $175,000 of this amount being paid in accordance with the Company’s payroll practices and $25,000 of this amount being paid via the issuance of restricted shares of Common Stock in lieu of cash. The Common Stock component of Mr. Dick’s compensation is to be computed on a quarterly basis, with the number of shares issued being equal to the quotient of the quarterly amount due, divided by the average daily closing price of the Company’s Common Stock for the quarter just ended.
Mr. Dick stepped down from his employment with the Company on May 24, 2013 and the Dick Employment Agreement was terminated. Mr. Dick continues to provide consulting services to the Company.
Hedging Policy
We do not permit the Named Executive Officers to “hedge” ownership by engaging in short sales or trading in any options contracts involving securities.
| 87 |
Options Exercises and Stock Vested
No options have been exercised by our Named Executive Officers during the 2016 Fiscal Year.
Options to purchase an aggregate of 1,750,000 shares of Common Stock and issued to Named Executive Officers in prior fiscal years vested during Fiscal 2016.
Pension Benefits
We do not provide pension benefits to the Named Executive Officers
Nonqualified Deferred Compensation
We do not have any defined contribution or other plan that provides for the deferral of compensation on a basis that is not tax-qualified.
Potential Payments Upon Termination or Change of Control
We do not presently provide the Named Executive Officers with any plan or arrangement, other than those that may be contained in the employment contracts of Mr. Nasrat Hakim, Mr. Douglass Plassche, and Mr. George Kenneth Smith, as above, in connection with any termination, including, without limitation, through retirement, resignation, severance or constructive termination (including a change in responsibilities) of such Named Executive Officer’s employment with the Company.
As part of the Company’s efforts to ensure the retention and continuity of key employees, officers and directors in the event of a change of control of the ownership of the Company, unless otherwise stated in applicable employment contracts, key executives would receive an amount equal to twelve months of such executive’s salary, and certain Directors and managers would receive an amount equal to six months of such Director’s or manager’s fees or salaries, as applicable. In addition, any outstanding and unvested options would immediately vest, in the event of a change of control.
Compensation of named executive officers
| Summary Compensation Table | ||||||||||||||||||||||||
| Name
and Principal Position | Fiscal Year | Salary(1) ($) | Bonus(1) ($) | Option ($) | All
Other ($) | Total ($) | ||||||||||||||||||
| Nasrat Hakim, President, Chief Executive Officer and Chairman of the Board of Directors | ||||||||||||||||||||||||
| 2016 | (1) | 387,500 | (2) | 387,500 | (3) | — | 18,000 | (4) | 793,000 | |||||||||||||||
| 2015 | (1) | 350,000 | (2) | 350,000 | (3) | — | 18,000 | (4) | 718,000 | |||||||||||||||
| 2014 | (1) | 233,254 | (2) | 233,254 | (3) | — | 12,000 | (4) | 478,666 | |||||||||||||||
| Carter J. Ward, Chief Financial Officer | ||||||||||||||||||||||||
| 2016 | (1) | 187,668 | (5) | 30,000 | (6) | — | — | 217,668 | ||||||||||||||||
| 2015 | (1) | 180,600 | (5) | 36,000 | (6) | — | — | 216,600 | ||||||||||||||||
| 2014 | (1) | 161,250 | (5) | — | — | — | 161,250 | |||||||||||||||||
| Douglas Plassche, Executive Vice President(12) | ||||||||||||||||||||||||
| 2016 | (1) | 244,613 | (7) | 73,140 | (8) | — | 6,000 | (4) | 328,753 | |||||||||||||||
| 2015 | (1) | 231,150 | (7) | 69,000 | (8) | — | 6,000 | (4) | 306,150 | |||||||||||||||
| 2014 | (1) | 146,948 | (7) | 23,926 | (8) | 202,497 | (9) | 3,750 | (4) | 376,770 | ||||||||||||||
| Barbara Ellison, Vice President(13) | ||||||||||||||||||||||||
| 2016 | (1) | 193,800 | (10) | — | — | — | 193,800 | |||||||||||||||||
| 2015 | (1) | 190,317 | (10) | 10,000 | (11) | 188,356 | (12) | 388,673 | ||||||||||||||||
| 2014 | (1) | 4,231 | (10) | — | — | — | 4,231 | |||||||||||||||||
| George Kenneth Smith, Vice President | ||||||||||||||||||||||||
| 2016 | (1) | 401,000 | (14) | — | — | — | 401,000 | |||||||||||||||||
| 2015 | (1) | 178,707 | (14) | — | 412,360 | (15) | — | 591,067 | ||||||||||||||||
| 2014 | (1) | — | — | — | — | — | ||||||||||||||||||
| Jerry Treppel, Chief Executive Officer and Chairman of the Board of Directors(16) | ||||||||||||||||||||||||
| 2016 | (1) | — | — | — | 23,404 | (17) | 23,404 | |||||||||||||||||
| 2015 | (1) | — | — | — | 30,000 | (17) | 30,000 | |||||||||||||||||
| 2014 | (1) | — | — | — | 30,000 | (17) | 30,000 | |||||||||||||||||
| Chris Dick, President and Chief Operating Officer(18) | ||||||||||||||||||||||||
| 2016 | (1) | — | — | — | — | — | ||||||||||||||||||
| 2015 | (1) | — | — | — | — | — | ||||||||||||||||||
| 2014 | (1) | 39,787 | (19) | — | — | 1,400 | (4) | 33,768 | ||||||||||||||||
| 88 |
| (1) | Represents amounts paid or accrued for the fiscal years ended March 31, 2016, 2015 and 2014, respectively. |
| (2) | Represents total salaries paid or accrued to Mr. Hakim pursuant to the Hakim Employment Agreement, with such amounts to be paid via the issuance of Common Shares in lieu of cash. |
A total of 1,061,079 Common Shares have been issued and an additional 384,366 Common Shares are due and owing to Mr. Hakim in relation to salaries earned by Mr. Hakim during Fiscal 2016.
A total of 1,168,806 Common Shares were issued to Mr. Hakim in full payment of salaries earned by Mr. Hakim during Fiscal 2015.
A total of 1,528,822 Common Shares were issued to Mr. Hakim in full payment of salaries earned by Mr. Hakim during Fiscal 2014.
| (3) | Represents bonuses paid or accrued to Mr. Hakim pursuant to the Hakim Employment Agreement, with amounts accrued for periods prior to January 1, 2016 being paid via the issuance of Common Shares in lieu of cash and amounts accrued for periods subsequent to January 1, 2016 to be paid in accordance with the Company’s payroll practices. |
A total of 1,061,079 Common Shares were issued to Mr. Hakim in payment of bonuses totaling $262,500 accrued during Fiscal 2016. The remaining $125,000 in bonuses owed to Mr. Hakim for Fiscal 2016 will be paid in accordance with the Company’s payroll practices.
A total of 1,168,806 Common Shares were issued to Mr. Hakim in full payment of bonuses earned by Mr. Hakim during Fiscal 2015.
A total of 1,528,822 Common Shares were issued to Mr. Hakim in full payment of bonuses earned by Mr. Hakim during Fiscal 2014.
| 89 |
| (4) | Represents amounts paid for auto allowances |
| (5) | Represents salaries earned by Mr. Ward pursuant to the Ward Employment Agreement |
Fiscal 2016 salaries consist of $157,668 being paid in accordance with the Company’s payroll practices and $30,000 being paid via the issuance of 90,950 shares of Common Stock in lieu of cash with an additional 23,062 shares of Common Stock being owed.
Fiscal 2015 salaries consist of $150,600 being paid in accordance with the Company’s payroll practices and $30,000 being paid via the issuance of 100,183 shares of Common Stock in lieu of cash.
Fiscal 2014 salaries consist of $131,750 being paid in accordance with the Company’s payroll practices and $30,000 being paid via the issuance of 262,271 shares of Common Stock in lieu of cash.
| (6) | Discretionary cash bonuses awarded by the Chief Executive Officer |
| (7) | Represents salaries earned by Mr. Plassche pursuant to the Plassche Employment Agreement |
Fiscal 2016 salaries consist of $219,613 being paid in accordance with the Company’s payroll practices and $25,000 being paid via the issuance of 75,791 shares of Common Stock in lieu of cash with an additional 19,218 shares of Common Stock being owed.
Fiscal 2015 salaries consist of $206,150 being paid in accordance with the Company’s payroll practices and $25,000 being paid via the issuance of 83,486 shares of Common Stock in lieu of cash.
Fiscal 2014 salaries consist of $137,222 being paid in accordance with the Company’s payroll practices and $9,726 being paid via the issuance of 101,282 shares of Common Stock in lieu of cash.
| (8) | Cash bonuses paid pursuant to the Plassche Employment Agreement. |
| (9) | Options to purchase 3,000,000 shares of Common Stock granted pursuant to the Plassche Employment Agreement. |
The options include a purchase price equal to the closing price of the Company’s Common Stock as of the date of the Plassche Employment Agreement and vest in 3 equal, annual increments, beginning on the date that is one year after the date of the Plassche Employment Agreement.
Value of the options granted was determined by applying the Black Scholes model for the valuation of options.
| (10) | Represents salaries earned by Ms. Ellison pursuant to the Ellison Employment Agreement |
Fiscal 2016 salaries consist of $168,800 being paid in accordance with the Company’s payroll practices and $25,000 being paid via the issuance of 75,791 shares of Common Stock in lieu of cash with an additional 19,218 shares of Common Stock being owed.
Fiscal 2015 salaries consist of $165,317 being paid in accordance with the Company’s payroll practices and $25,000 being paid via the issuance of 83,541 shares of Common Stock in lieu of cash.
Fiscal 2014 salaries consist of $3,750 being paid in accordance with the Company’s payroll practices and $481 being paid via the issuance of 1,160 shares of Common Stock in lieu of cash.
| (11) | Cash bonuses paid pursuant to the Ellison Employment Agreement. |
| (12) | Options to purchase 600,000 shares of Common Stock granted pursuant to the Ellison Employment Agreement. |
The options include a purchase price equal to the closing price of the Company’s Common Stock as of the date of the Ellison Employment Agreement and vest in 3 equal, annual increments, beginning on the date that is one year after the date of the Ellison Employment Agreement.
Value of the options granted was determined by applying the Black Scholes model for the valuation of options.
| (13) | Ms. Ellison retired on June 1, 2016 and is no longer an employee of the Company |
| 90 |
| (14) | Represents salaries earned by Mr. Smith pursuant to the Smith Employment Agreement |
Fiscal 2016 salaries consist of $151,800 being paid in accordance with the Company’s payroll practices and $250,000 being paid via the issuance of 757,914 shares of Common Stock in lieu of cash with an additional 192,183 shares of Common Stock being owed.
Fiscal 2015 salaries consist of $67,596 being paid in accordance with the Company’s payroll practices and $111,111 being paid via the issuance of 440,512 shares of Common Stock in lieu of cash.
| (15) | Options to purchase 1,500,000 shares of Common Stock granted pursuant to the Smith Employment Agreement. |
The options include a purchase price equal to the closing price of the Company’s Common Stock as of the date of the Smith Employment Agreement and vest in 3 equal, annual increments, beginning on the date that is one year after the date of the Smith Employment Agreement.
Value of the options granted was determined by applying the Black Scholes model for the valuation of options.
| (16) | Mr. Treppel stepped down from his position as Chief Executive Officer in August 2013 and resigned from the Board of Directors in January 2016. |
| (17) | Represents compensation due to Mr. Treppel for his service as Chairman of the Board of Directors. Mr. Treppel received no salary or additional compensation for his service as Chief Executive Officer. Compensation due to Mr. Treppel was paid via the issuance of Common Stock in lieu of cash, pursuant to the Company’s Director compensation policy. |
A total of 93,271 shares of Common Stock were issued to Mr. Treppel for Chairman fees earned during Fiscal 2016.
A total of 100,184 shares of Common Stock were issued to Mr. Treppel for Chairman fees earned during Fiscal 2015.
A total of 349,062 shares of Common Stock were issued to Mr. Treppel for Chairman fees earned during Fiscal 2014.
| (18) | Mr. Dick stepped down from his position as President and Chief Operating Officer in May 2013. |
| (19) | Represents salaries earned by Mr. Dick for the period beginning on April 1, 2013 and ending with Mr. Dick’s resignation on May 24, 2013, and consists of $27,951 in salaries paid in accordance with the Company’s payroll practices and $11,836 being paid via the issuance of 150,390 shares of Common Stock in lieu of cash, pursuant to the Dick Employment Agreement. |
Outstanding Equity Awards at March 31, 2016
| Name | Number of securities underlying unexercised options Exercisable (#) | Number of securities underlying unexercised options Unexercisable (#) | Equity Incentive Plan Awards: Number of securities underlying unexercised unearned options (#) | Options Exercise Price ($) | Option Expiration Date | |||||||||||||
| Carter Ward | 200,000 | — | — | 0.10 | 1/17/2020 | |||||||||||||
| Carter Ward | 150,000 | — | — | 0.12 | 6/19/2022 | |||||||||||||
| Douglas Plassche | 2,000,000 | — | 1,000,000 | (1) | 0.07 | 7/23/2023 | ||||||||||||
| Barbara Ellison | 400,000 | — | 200,000 | (2) | 0.33 | 8/30/2016(3) | ||||||||||||
| George Kenneth Smith | 500,000 | — | 1,000,000 | (4) | 0.29 | 10/20/2024 | ||||||||||||
| 91 |
| (1) | Options vest in July of 2016. |
| (2) | Conditions of vesting include the requirement that the option holder be currently employed by the Company on the vesting date. Due to the retirement of Ms. Ellison, this requirement will not be met and these options will not vest. |
| (3) | Options expire 90 days after Ms. Ellison’s termination of employment with the Company. |
| (4) | Options vest in equal increments in October 2016 and October 2017 |
DIRECTOR COMPENSATION
The following table sets forth information concerning director compensation for the year ended March 31, 2016
| Name | Fees Earned or Paid In Cash ($) | Stock Awards(1) ($) | Option Awards ($) | Non- Equity Incentive Plan Compensation ($) | Non- qualified Deferred Compen- sation ($) | All Other Compen- sation ($) | Total ($) | |||||||||||||||||||||
| Barry Dash | — | 20,000 | (2) | — | — | — | — | 20,000 | ||||||||||||||||||||
| Ashok Nigalaye(5) | — | 4,167 | (3) | — | — | — | — | 4,167 | ||||||||||||||||||||
| Jeenarine Narine(6) | — | 20,000 | (2) | — | — | — | — | 20,000 | ||||||||||||||||||||
| Jeffrey Whitnell | — | 20,000 | (2) | — | — | — | — | 20,000 | ||||||||||||||||||||
| Jerry Treppel(7) | — | 23,404 | (4) | — | — | — | — | 23,404 | ||||||||||||||||||||
| (1) | All director compensation is paid via the issuance of Common Shares in lieu of cash in accordance with the Company’s policy regarding Director Fee Compensation |
| (2) | Represents directors fees earned during the fiscal year ended March 31, 2016, with such fees being paid via the issuance of shares of Common Stock, pursuant to the Company’s policy regarding payment of Director’s fees.
A total of 60,633 shares of Common Stock were issued to, and 15,375 shares of Common Stock are due and owing to each Director in payment of Director’s fees earned during the fiscal year ended March 31, 2016. |
| 92 |
| (3) | Represents director fees earned during the period April 1, 2015 through Mr. Nigalaye’s resignation as a Director on June 15, 2015, with such fees being paid via the issuance of Common Stock, pursuant to the Company’s policy regarding payment of Director’s fees. |
A total of 18,489 shares of Common Stock were issued in full payment of these director fees.
| (4) | Represents director fees earned during the period April 1, 2015 through Mr. Treppel’s resignation as a Director on January 11, 2016, with such fees being paid via the issuance of Common Stock, pursuant to the Company’s policy regarding payment of Director’s fees. |
A total of 93,271 shares of Common Stock were issued in full payment of these director fees.
| (5) | Mr. Nigalaye resigned as a Director on June 15, 2015 |
| (6) | Mr. Narine resigned as a Director on April 7, 2016 |
| (7) | Mr. Treppel resigned as a Director on January 11, 2016 |
Director Fee Compensation
The Company’s policy regarding director fees is as follows: ((i) Directors who are employees or consultants of the Company (and/or any of its subsidiaries), except for Mr. Jerry Treppel, Chief Executive Officer and Dr. Ashok Nigalaye, former Chief Scientific Officer, receive no additional remuneration for serving as directors or members of committees of the Board; (ii) all Directors are entitled to reimbursement for out-of-pocket expenses incurred by them in connection with their attendance at the Board or committee meetings; (iii) Directors who are not employees or consultants of the Company (and/or any of its subsidiaries) receive $30,000 annual retainer fee, with $20,000 of this amount being paid via the issuance of restricted Common Stock of the Company in lieu of cash, as described below, and the remaining $10,000 being paid in cash. (iv) The Chairman of the Board receives a $30,000 annual retainer fee paid via the issuance of restricted shares of Common Stock of the Company in lieu of cash, as described below (v) Directors and the Chairman do not receive any additional compensation for attendance at or chairing of any meetings. (vi) Mr. Jerry Treppel received no additional compensation, above the annual retainer fee due to the Chairman of the Board, for the period that he also served as Chief Executive Officer (vii) Dr. Ashok Nigalaye received no additional compensation, above the annual retainer fee due to Directors, for the period that he also served as Chief Scientific Officer.
Director Equity Compensation
Members of the Board of Directors and the Chairman are paid their annual retainer fees via the issuance of restricted shares of Common Stock of the Company, in lieu of cash. The number of shares to be issued to each Director and the Chairman is equal to the quotient of the quarterly amount due to each Director and the Chairman, respectively, divided by the average daily closing price of the Company’s stock for the quarter just ended.
Members of the Board of Directors during the fiscal years ended March 31, 2016 and March 31, 2015 did not receive any options or equity compensation for serving as directors other than shares of Common Stock earned in lieu of cash in relation to Director and Chairman fees due.
| 93 |
Other
The Company’s Articles of Incorporation provide for the indemnification of each of the Company’s directors to the fullest extent permitted under Nevada General Corporation Law.
| ITEM 12 | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The following table sets forth certain information, as of June 7, 2016 (except as otherwise indicated), regarding beneficial ownership of our Common Stock and our Series I Preferred Stock by (i) each person who is known by us to own beneficially more than 5% of each such class, (ii) each of our directors, (iii) each of our executive officers and (iv) all our directors and executive officers as a group. As of June 7, 2015, we had 726,486,650 shares of Common Stock outstanding (exclusive of 100,000 treasury shares) and 100.000 shares of Series I Preferred Stock outstanding. On any matter presented to the holders of our Common Stock for their action or consideration at any meeting of our Shareholders, each share of Common Stock entitles the holder to one vote and each share of Series I Preferred Stock entitles the holder to the number of votes equal to the number of shares of Common Stock into which such share of Series I Preferred Stock is convertible (1,428,571.4 per whole share).
As used in the table below and elsewhere in this report, the term beneficial ownership with respect to a security consists of sole or shared voting power, including the power to vote or direct the vote, and/or sole or shared investment power, including the power to dispose or direct the disposition, with respect to the security through any contract, arrangement, understanding, relationship, or otherwise, including a right to acquire such power(s) during the 60 days immediately following June 7, 2016. Except as otherwise indicated, the Shareholders listed in the table have sole voting and investment powers with respect to the shares indicated.
| 94 |
Amount and Nature of Beneficial Ownership | Percent (%) of Voting | |||||||||||
| Name and Address Of Beneficial Owner of Common Stock | Common Stock | Series I Preferred Stock | Securities Beneficially Owned | |||||||||
| Nasrat Hakim, President Chief Executive Officer and Director* | 25,125,113 | (1) | 100.000 | (2) | 19.3 | % | ||||||
| Barry Dash, Director* | 1,271,153 | (3) | 0 | ** | ||||||||
| Jeffrey Whitnell, Director* | 1,132,798 | (4) | 0 | ** | ||||||||
| Eugene Pfeifer, Director* | 0 | (5) | 0 | ** | ||||||||
| Davis Caskey, Director* | 0 | (5) | 0 | ** | ||||||||
| Jeenarine Narine, Former Director (10) | 23,605,463 | (6) | 0 | 1.9 | % | |||||||
| Carter J. Ward, Chief Financial Officer * | 3,480,361 | (7) | 0 | ** | ||||||||
| Douglas Plassche, Executive Vice President * | 2,194,248 | (8) | 0 | ** | ||||||||
| Ashok Nigalaye, Former Director(10) | 39,954,695 | (11) | 3.7 | |||||||||
| Jerry Treppel, Former Director(12) | 7,640,561 | (12) | ** | |||||||||
| All Directors and Officers as a group | 33,203,673 | (9) | 100.000 | (2) | 20.2 | % | ||||||
________________________
* The address is c/o Elite Pharmaceuticals Inc., 165 Ludlow Avenue, Northvale, NJ 07647.
** Less than 1%
| (1) | Includes 24,740,747 shares of Common Stock held as per the most recent Form 4 filing, 384,366 shares of Common Stock due and owing to Mr. Hakim as of March 31, 2016 (the latest practicable date) for compensation earned pursuant to Mr. Hakim’s employment agreement with the Company. |
| (2) | Series I Preferred Stock are convertible into Common Stock, with each share of Series I Preferred being convertible into 148,917,143 shares of Common Stock. Series I Preferred Stock also includes voting rights of one vote for each Common Stock equivalent share. |
| (3) | Includes 1,165,778 shares of Common Stock held as per the most recent Form 4 filing and 15,375 shares of Common Stock due and owing to Dr. Dash as of March 31, 2016 (the latest practicable date) for Directors fees accrued as of such date and vested options to purchase 90,000 shares of Common Stock. |
| (4) | Includes 1,117,423 shares of Common Stock held as per the most recent Form 4 filing and 15,375 shares of Common Stock due and owing to Mr. Whitnell as of March 31, 2016 (the latest practicable date) for Directors fees accrued as of such date. |
| (5) | As per most recent Form 3’s filed. Mr. Pfeifer was appointed as a Director on April 7, 2016. Mr. Caskey was appointed as a Director on April 28, 2016. |
| (6) | Mr. Narine resigned from the Board of Directors on April 7, 2016. Includes 16,742,395 shares of Common Stock held as per the most recent Form 4 filing, 16,550 shares of Common Stock due and owing to Mr. Narine for Directors fees accrued prior to Mr. Narine’s resignation, and warrants to purchase 6,846,518 shares of Common Stock. |
| (7) | Includes 3,107,299 shares of Common Stock held as per the most recent Form 4 filing, 23,062 shares of Common Stock due and owing to Mr. Ward as of March 31, 2016 (the latest practicable date) for salaries earned pursuant to Mr. Ward’s employment agreement with the Company, and vested options to purchase 350,000 shares of Common Stock. |
| (8) | Includes 175,030 shares of Common Stock held as per the most recent Form 4 filing, 19,218 shares of Common Stock due and owing to Mr. Plassche as of March 31, 2015 (the latest practicable date) for salaries earned pursuant to Mr. Plassche’s employment agreement with the Company, and vested options to purchase 2,000,000 shares of Common Stock. |
| 95 |
| (9) | Relates only to current directors and officers. Includes 30,306,277 shares of Common Stock held, as per the applicable most recent Form 3 or Form 4 filings, 457,396 shares of Common Stock due and owing as of March 31, 2016 (the latest practicable date) for Chairman fees, directors fees and salaries accrued as of such date, vested options to purchase 2,440,000 shares of Common Stock and 100 Series I Preferred Convertible Shares which are convertible into 142,857,143 shares of Common Stock. Series I Preferred Stock also include voting rights of one vote for each Common Stock equivalent share. |
| (10) | Address is c/o Epic Pharma LLC, 227-15 N. Conduit Ave, Laurelton, NY 11413 |
| (11) | Dr. Nigalaye resigned on June 5, 2015. Includes 31,877,696 shares of Common Stock held as per the most recent Form 4 filing, and warrants to purchase 8,076,999 shares of Common Stock. |
| (12) | Mr. Treppel resigned on January 11, 2016. Address is 282 New Norwalk Road, New Canaan, CT 06840. Includes 7,640,561 shares of Common Stock held as per the most recent Form 4 filing. |
| ITEM 13 | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
Certain Related Person Transactions
Transactions with Nasrat Hakim and Mikah Pharma LLC
On August 1, 2013, Elite Laboratories Inc. (“Elite Labs”), our wholly owned subsidiary, executed an asset purchase agreement (the “Mikah Purchase Agreement”) with Mikah Pharma LLC (“Mikah”), an entity that is wholly owned by Mr. Nasrat Hakim, who, in conjunction with this transaction, was appointed as our Chief Executive Officer, President and a Director on August 2, 2012, and acquired from Mikah a total of 13 Abbreviated New Drug Applications (“ANDAs”) consisting of 12 ANDAs approved by the FDA and one ANDA under active review with the FDA, and all amendments thereto (the “Acquisition”) for aggregate consideration of $10,000,000, inclusive of imputed interest payable pursuant to a non-interest bearing, secured convertible note due in August 2016 (the “Mikah Note”). The Mikah Note was amended on February 7, 2014 to make it convertible into shares of the Company’s Series I Convertible Preferred Stock.
The Mikah Note, as amended, was interest free and due and payable on the third anniversary of its issuance. Subject to certain limitations, the principal amount of the Mikah Note was convertible at the option of Mikah into shares of Common Stock at a rate of $0.07 (approximately 14,286 shares per $1,000 in principal amount), the closing market price of the Company’s Common Stock on the date that the asset purchase agreement and Note were executed and/or into shares of the Company’s Series I Convertible Preferred Stock at the rate of 1 share of Series I Preferred Stock for each $100,000 of principal owed on the Mikah Note. The conversion rate was adjustable for customary corporate actions such as stock splits and, subject to certain exclusions, includes weighted average anti-dilution for common stock transactions at prices below the then applicable conversion rate. Pursuant to a security agreement (the “Security Agreement”), repayment of the Mikah Note was secured by the ANDAs acquired in the Acquisition.
On February 7, 2014, Mikah converted the principal amount of $10,000,000, representing the entire principal balance due under the Mikah Note, into 100 shares of the Company’s Series I Preferred Stock.
On August 27, 2010, Elite executed an asset purchase with Mikah (the “Naltrexone Agreement”). Pursuant to the Naltrexone Agreement, Elite acquired from Mikah the Abbreviated New Drug Application number 75-274 (Naltrexone Hydrochloride Tablets USP, 50 mg), and all amendments thereto (the “ANDA”), that have to date been filed with the FDA seeking authorization and approval to manufacture, package, ship and sell the products described in the ANDA within the United States and its territories (including Puerto Rico) for aggregate consideration of $200,000. In lieu of cash, Mikah agreed to accept from Elite product development services to be performed by Elite, and entered into a Development and License Agreement dated August 27, 2010 between the Company and Mikah (the “Mikah Development Agreement”). A current report on form 8-K was filed on August 27, 2010 in relation to this announcement, such filing being incorporated herein by this reference. Please also refer to exhibit 10.5 of the Quarterly Report on Form 10-Q filed with SEC on November 15, 2010, such filing being incorporated herein by this reference.
| 96 |
The manufacturing of Naltrexone 50mg was successfully transferred to the Company’s Northvale facility, and the first commercial shipment of this product was made in September 2013.
On January 28, 2015, the Mikah Development Agreement was terminated by mutual agreement of the parties thereto. Pursuant to the Mikah Development Agreement, Mikah made advance consideration payments to the Company totaling $200,000 in exchange for product development services to be provided at a future date. Subsequent to the execution of the Mikah Development Agreement, and before any development milestones were achieved, the sole owner of Mikah, Mr. Nasrat Hakim, became the President and Chief Executive Officer of the Company. Mikah has accordingly ceased operating and is in the process of winding down and liquidating its assets.
Any further development of the product related to the Mikah Development Agreement will belong to the Company, although there can be no assurances that such development will occur or be successful.
The Mikah Development Agreement required that the consideration paid in advance to the Company be refunded in the event of no milestones being achieved. Mr. Hakim, as owner of Mikah, has directed that the $200,000 refund due to Mikah not be paid currently, but rather be added to the amounts due under the Hakim Credit Line.
For further details on the Mikah Development Agreement, please see Exhibit 10.85 of the Quarterly Report on Form 10-Q filed with the SEC on February 17, 2015, with such filing being herein incorporated by reference.
On October 15, 2013, we entered into a bridge loan agreement (the “Hakim Loan Agreement”) with Nasrat Hakim, our President and CEO. For details on the Hakim Credit Line, please see “Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations; Liquidity and Capital Resources; Hakim $1,000,000 Bridge Revolving Credit Line” above, the notes to these financial statements, as well as Exhibit 10.16 of the Quarterly Report on Form 10-Q filed with the SEC on November 13, 2013, the Current Report on Form 8-K filed with the SEC on October 16, 2013 and Exhibit 10.84 of the Quarterly Report on Form 10-Q filed with the SEC on February 17, 2015, with all such filings being herein incorporated by reference. The Hakim Loan Agreement expired on March 31, 2016 and all amounts due and owing pursuant to the Hakim Loan Agreement were repaid on May 24, 2016. There are no amounts due and owing on the Hakim Loan Agreement and the Hakim Loan Agreement is expired.
For information about our employment agreement with Mr. Hakim, please see “Executive Compensation-Agreements with Named Executive Officers” above.
| 97 |
Strategic Alliance Agreement/Transactions with Epic Pharma LLC and Epic Investments LLC
On March 18, 2009, the Company entered into the Epic Strategic Alliance Agreement with Epic Pharma, LLC and Epic Investments, LLC, a subsidiary controlled by Epic Pharma LLC. The Strategic Alliance Agreement expired on June 4, 2012. For more information on the Epic Strategic Alliance Agreement please see our Current Reports on Form 8-K, filed with the SEC on March 23, 2009, May 6, 2009 and June 5, 2009, which disclosures are incorporated herein by reference. Ashok G. Nigalaye, Jeenarine Narine and Ram Potti, each were elected as members of our Board of Directors, effective June 24, 2009, as the three directors that Epic was entitled to designate for appointment to the Board pursuant to the terms of the Epic Strategic Alliance Agreement. Mr. Potti resigned from his position as Director of the Company on December 31, 2012, Dr. Nigalaye resigned as a Company Director on June 5, 2015 and Mr. Narine resigned from his position as Director of Company on April 7, 2016. Messrs. Nigalaye, Narine and Potti were also officers of Epic Pharma, LLC, in the following capacities:
• Mr. Nigalaye, Chairman and Chief Executive Officer of Epic Pharma, LLC;
• Mr. Narine, President and Chief Operating Officer of Epic Pharma, LLC;
• Mr. Potti, Vice President of Epic Pharma, LLC.
On March 30, 2016, Humanwell Healthcare Group and PuraCap Pharmaceutical LLC announced that the companies have entered into a definitive agreement to acquire 100% of the membership interests of Epic Pharma, LLC of Laurelton, NY.
The Epic Strategic Alliance Agreement expired on June 4, 2012.
As part of the operation of the strategic alliance, the Company and Epic identified areas of synergy, including, without limitation, raw materials used by both entities, equipment purchases, contract manufacturing/packaging and various regulatory and operational resources existing at Epic that could be utilized by the Company.
With regards to synergies related to raw materials usage, the strategic alliance allowed the Company to purchase such raw materials from Epic, at the Epic acquisition cost, without markup. In all cases, the acquisition cost of Epic was lower than those costs available to the Company, mainly as a result of efficiencies of scale generated by significantly larger volumes purchased by Epic during the course of their normal operations. During Fiscal 2016, 2015 and Fiscal 2014, an aggregate amount of zero, zero and $9,009, respectively, in such materials was purchased from Epic Pharma LLC. All purchases were at Epic Pharma’s acquisition cost, without markup and evidenced by supporting documents of Epic Pharma LLC’s acquisition cost.
With regards to synergies related to regulatory and operational resources, the strategic alliance allowed the Company to utilize Epic’s substantial resources and technical competencies on an “as needed” basis at a cost equal to Epic’s actual cost for only the resources utilized by the Company. Without such access to Epic’s resources, the Company would have to invest significant amounts in human resources and fixed assets as well as incur substantial costs with third party providers to provide the same resources provided by Epic and necessary for the operations of the Company.
During Fiscal 2016, no costs associated with facility maintenance, engineering and regulatory resources from Epic were utilized by the Company. During Fiscal 2015, an aggregate amount of $7,937 was paid to Epic as reimbursement for costs associated with facility maintenance, engineering and regulatory resources utilized by the Company. During Fiscal 2014, an aggregate amount of $30,835 was paid to Epic as reimbursement for costs associated with facility maintenance, engineering and regulatory resources utilized by the Company.
During Fiscal 2016 and Fiscal 2015, the Company did not incur contract manufacturing and/or packaging costs from Epic Pharma. During Fiscal 2014, the Company incurred a total of $29,668 in contract manufacturing and/or packaging costs to Epic Pharma for the Company’s Phentermine, Hydromorphone, Methadone and Immediate Release Lodrane products.
| 98 |
Total purchases from Epic by the Company during the fiscal years ended March 31, 2016, 2015 and 2014 were zero, $7,937 and $69,512, respectively.
Manufacturing and Licensing Agreement with Epic Pharma LLC
On October 2, 2013, Elite executed the Epic Pharma Manufacturing and License Agreement. This agreement granted Epic Pharma certain rights to manufacture, market and sell in the United States and Puerto Rico the 12 approved ANDAs acquired by Elite pursuant to the Mikah Purchase Agreement. Of the 12 approved ANDAs, Epic Pharma will have the exclusive right to market six products as listed in Schedule A of the Epic Pharma Manufacturing and License Agreement, and a non-exclusive right to market six products as listed in Schedule D of the Epic Pharma Manufacturing and License Agreement. Epic Pharma is responsible for all regulatory and pharmacovigilance matters related to the products and for all costs related to the site transfer for all products. Pursuant to the Epic Pharma Manufacturing and License Agreement, Elite will receive a license fee and milestone payments. The license fee will be computed as a percentage of the gross profit, as defined in the Epic Pharma Manufacturing and License Agreement, earned by Epic Pharma a result of sales of the products. The manufacturing cost used for the calculation of the license fee is a predetermined amount per unit plus the cost of the drug substance (API) and the sales cost for the calculation is predetermined based on net sales. If Elite manufactures any product for sale by Epic Pharma, then Epic Pharma shall pay to Elite that same predetermined manufacturing cost per unit plus the cost of the API. The license fee is payable monthly for the term of the Epic Pharma Manufacturing and License Agreement. Epic Pharma shall pay to Elite certain milestone payments as defined by the Epic Pharma Manufacturing and License Agreement. To date, milestones totaling $1 million have been earned and received in relation to the signing of the Epic Pharma Manufacturing and License Agreement and the filing and approval by the FDA of supplements relating to the transfer of manufacturing site for Isradipine 2.5mg and Isradipine 5mg. The term of the Epic Pharma Manufacturing and License Agreement is five years and may be extended for an additional five years upon mutual agreement of the parties. Twelve months following the launch of a product covered by the Epic Pharma Manufacturing and License Agreement, Elite may terminate the marketing rights for any product if the license fee paid by Epic Pharma falls below a designated amount for a six month period of that product. Elite may also terminate the exclusive marketing rights if Epic Pharma is unable to meet the annual unit volume forecast for a designated product group for any year, subject to the ability of Epic Pharma, during the succeeding six month period, to achieve at least one-half of the prior year’s minimum annual unit forecast. The Epic Pharma Manufacturing and License Agreement may be terminated by mutual agreement of Elite and Epic Pharma, as a result of a breach by either party that is not cured within 60 days notice of the breach, or by Elite as a result of Epic Pharma becoming a party to a bankruptcy, reorganization or other insolvency proceeding that continues for a period of 30 days or more.
Sales and Distribution Licensing Agreement with Epic for Abuse-Deterrent ELI-200
On June 4, 2015, Elite Pharmaceuticals Inc. and its wholly-owned subsidiary Elite Laboratories, Inc. (collectively, “Elite”) executed an exclusive License Agreement (the “Agreement”) with Epic Pharma LLC. (“Epic”), to market and sell in the United States, SequestOx™, an immediate release oxycodone with sequestered naltrexone capsule, owned by Elite. Epic will have the exclusive right to market SequestOx™ and its various dosage forms as listed in Schedule A of the Agreement (the “Products”). Epic is responsible for all regulatory and pharmacovigilance matters related to the products. Pursuant to the Agreement, Elite will receive a license fee and milestone payments. The license fee will be computed as a percentage of net sales of the Products as defined in the Agreement by Epic. Elite will manufacture the product for sale by Epic on a cost plus basis and both parties agree to execute a separate Manufacturing and Supply Agreement. The license fee is payable quarterly for the term of the Agreement. Epic shall pay to Elite certain milestone payments as defined by the Agreement. The first milestone payment was due and was received upon signing the agreement. Subsequent milestone payments are due upon the filing of a New Drug Application (“NDA”) with the U.S. Food and Drug Administration (“FDA”) for the Products and upon receipt of the approval letter for the NDA from the FDA. The term of the License Agreement is five years and may be extended for an additional five years upon mutual agreement of the parties. Elite can terminate the Agreement on 90 days’ written notice in the Event that Epic does not pay to Elite certain minimum annual license fees over the initial five year term of the Agreement. Either party may terminate this Agreement upon a material breach and failure to cure that breach by the other party within a specified period.
| 99 |
Director Independence
All related person transactions are reviewed and, as appropriate, may be approved or ratified by the Board of Directors. If a Director is involved in the transaction, he or she may not participate in any review, approval or ratification of such transaction. Related person transactions are approved by the Board of Directors only if, based on all of the facts and circumstances, they are in, or not inconsistent with, our best interests and the best interests of our stockholders, as the Board of Directors determines in good faith. The Board of Directors takes into account, among other factors it deems appropriate, whether the transaction is on terms generally available to an unaffiliated third-party under the same or similar circumstances and the extent of the related person’s interest in the transaction. The Board of Directors may also impose such conditions as it deems necessary and appropriate on us or the related person in connection with the transaction.
In the case of a transaction presented to the Board of Directors for ratification, the Board of Directors may ratify the transaction or determine whether rescission of the transaction is appropriate.
| ITEM 14 | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The Company’s independent registered public accounting firm is Buchbinder Tunich (“Buchbinder”).
The following table presents fees, including reimbursements for expenses, for professional audit services rendered by Buchbinder and Demetrius Berkower LLC, for the audits of our financial statements and interim reviews of our quarterly financial statements for Fiscal 2016, Fiscal 2015 and Fiscal 2014.
| Fiscal 2016 | Fiscal 2015 | Fiscal 2014 | ||||||||||
| Audit Fees | $ | 110,500 | $ | 103,600 | $ | 81,150 | ||||||
| Audit-Related Fees | 7,000 | 4,000 | 6,400 | |||||||||
| Tax Fees | 12,000 | 12,200 | 7,000 | |||||||||
| All Other Fees | — | — | 1,150 | |||||||||
Audit Fees
Represents fees for professional services provided for the audit of our annual financial statements, services that are performed to comply with generally accepted auditing standards, and review of our financial statements included in our quarterly reports and services in connection with statutory and regulatory filings.
Audit-Related Fees
Represents the fees for assurance and related services that were reasonably related to the performance of the audit or review of our financial statements.
| 100 |
Tax Fees
Represents preparation of Federal, State and Local income tax returns.
The Audit Committee has determined that Buchbinders rendering of these audit-related services was compatible with maintaining auditor’s independence. The Board of Directors considered Buchbinder to be well qualified to serve as our independent public accountants. The Committee also pre-approved the charges for services performed in Fiscal 2016.
The Audit Committee pre-approves all audit related and tax services and the terms thereof (which may include providing comfort letters in connection with securities underwriting) and non-audit services (other than non-audit services prohibited under Section 10A(g) of the Exchange Act or the applicable rules of the SEC or the Public Company Accounting Oversight Board) to be provided to us by the independent auditor; provided, however, the pre-approval requirement is waived with respect to the provisions of non-audit services for us if the “de minimus” provisions of Section 10A (i)(1)(B) of the Exchange Act are satisfied. This authority to pre-approve non-audit services may be delegated to one or more members of the Audit Committee, who shall present all decisions to pre-approve an activity to the full Audit Committee at its first meeting following such decision.
| 101 |
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENTS AND SCHEDULES. |
| (a) | The following are filed as part of this Annual Report on Form 10-K | |
| (1) | The financial statements and schedules required to be filed by Item 8 of this Annual Report on Form 10-K and listed in the Index to Consolidated Financial Statements. | |
| (2) | The Exhibits required by Item 601 of Regulation S-K and listed below in the “Index to Exhibits required by Item 601 of Regulation S-K.” | |
| (b) | The Exhibits are filed with or incorporated by reference in this Annual Report on Form 10-K | |
| (c) | None | |
Index to Exhibits required by Item 601 of Regulation S-K.
Exhibit No. |
Description | |
| 2.1 | Agreement and Plan of Merger between Elite Pharmaceuticals, Inc., a Delaware corporation (“Elite-Delaware”) and Elite Pharmaceuticals, Inc., a Nevada corporation (“Elite-Nevada”), incorporated by reference to Exhibit 2.1 to the Current Report on Form 8-K filed with the SEC on January 9, 2012. | |
| 3.1(a) | Articles of Incorporation of Elite-Nevada, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the SEC on January 9, 2012. | |
| 3.1(b) | Certificate of Incorporation of Elite-Delaware, together with all other amendments thereto, as filed with the Secretary of State of the State of Delaware, incorporated by reference to (a) Exhibit 4.1 to the Registration Statement on Form S-4 (Reg. No. 333-101686), filed with the SEC on December 6, 2002 (the “Form S-4”), (b) Exhibit 3.1 to the Company’s Current Report on Form 8-K dated July 28, 2004 and filed with the SEC on July 29, 2004, (c) Exhibit 3.1 to the Company’s Current Report on Form 8-K dated June 26, 2008 and filed with the SEC on July 2, 2008, and (d) Exhibit 3.1 to the Company’s Current Report on Form 8-K dated December 19, 2008 and filed with the SEC on December 23, 2008.* | |
| 3.1(c) | Certificate of Designations, Preferences and Rights of Series A Preferred Stock, as filed with the Secretary of the State of Delaware, incorporated by reference to Exhibit 4.5 to the Current Report on Form 8-K dated October 6, 2004, and filed with the SEC on October 12, 2004.* | |
| 3.1(d) | Certificate of Retirement with the Secretary of the State of the Delaware to retire 516,558 shares of the Series A Preferred Stock, as filed with the Secretary of State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated March 10, 2006, and filed with the SEC on March 14, 2006.* | |
| 3.1(e) | Certificate of Designations, Preferences and Rights of Series B 8% Convertible Preferred Stock, as filed with the Secretary of the State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated March 15, 2006, and filed with the SEC on March 16, 2006.* | |
| 3.1(f) | Amended Certificate of Designations of Preferences, Rights and Limitations of Series B 8% Convertible Preferred Stock, as filed with the Secretary of State of the State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated April 24, 2007, and filed with the SEC on April 25, 2007.* |
| 102 |
| 3.1(g) | Certificate of Designations, Preferences and Rights of Series C 8% Convertible Preferred Stock, as filed with the Secretary of the State of Delaware, incorporated by reference to Exhibit 3.2 to the Current Report on Form 8-K dated April 24, 2007, and filed with the SEC on April 25, 2007.* | |
| 3.1(h) | Amended Certificate of Designations, Preferences and Rights of Series C 8% Convertible Preferred Stock, as filed with the Secretary of the State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated April 24, 2007, and filed with the SEC on April 25, 2007.* | |
| 3.1(i) | Amended Certificate of Designations of Preferences, Rights and Limitations of Series B 8% Convertible Preferred Stock, as filed with the Secretary of State of the State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated September 15, 2008, and filed with the SEC on September 16, 2008.* | |
| 3.1(j) | Amended Certificate of Designations, Preferences and Rights of Series C 8% Convertible Preferred Stock, as filed with the Secretary of the State of Delaware, incorporated by reference to Exhibit 3.2 to the Current Report on Form 8-K dated September 15, 2008, and filed with the SEC on September 16, 2008.* | |
| 3.1(k) | Amended Certificate of Designations of Preferences, Rights and Limitations of Series D 8% Convertible Preferred Stock, as filed with the Secretary of State of the State of Delaware, incorporated by reference to Exhibit 3.3 to the Current Report on Form 8-K dated September 15, 2008, and filed with the SEC on September 16, 2008.* | |
| 3.1(l) | Certificate of Designation of Preferences, Rights and Limitations of Series E Convertible Preferred Stock, as filed with the Secretary of State of the State of Delaware, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated June 1, 2009, and filed with the SEC on June 5, 2009.* | |
| 3.1(m) | Amended Certificate of Designations of the Series D 8% Convertible Preferred Stock as filed with the Secretary of State of the State of Delaware on June 29, 2010, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K, dated June 24, 2010 and filed with the SEC on July 1, 2010.* | |
| 3.1(n) | Amended Certificate of Designations of the Series E Convertible Preferred Stock as filed with the Secretary of State of the State of Delaware on June 29, 2010, incorporated by reference to Exhibit 3.2 to the Current Report on Form 8-K, dated June 24, 2010 and filed with the SEC on July 1, 2010.* | |
| 3.1(o) | Certificate of Designations of the Series G Convertible Preferred Stock as filed with the Secretary of State of the State of Nevada on April 18, 2013, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated April 18, 2013 and filed with the SEC on April 22, 2013 . | |
| 3.1(p) | Certificate of Designation of the Series H Junior Participating Preferred Stock, incorporated by reference to Exhibit 2 (contained in Exhibit 1) to the Registration Statement on Form 8-A filed with the SEC on November 15, 2013. | |
| 3.1(q) | Certificate of Designations of the Series I Convertible Preferred Stock as filed with the Secretary of State of the State of Nevada on February 6, 2014, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K, dated February 6, 2014 and filed with the SEC on February 7, 2014 | |
| 3.2(a) | Amended and Restated By-Laws of the Company, incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K dated March 17, 2014 and filed with the SEC on March 18, 2014. |
| 103 |
| 3.2(b) | By-Laws of Elite-Delaware, as amended, incorporated by reference to Exhibit 3.2 to the Company’s Registration Statement on Form SB-2 (Reg. No. 333-90633) made effective on February 28, 2000 (the “Form SB-2”).* | |
| 4.1 | Form of specimen certificate for Common Stock of the Company, incorporated by reference to Exhibit 4.1 to the Form SB-2.* | |
| 4.2 | Form of specimen certificate for Series B 8% Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006.* | |
| 4.3 | Form of specimen certificate for Series C 8% Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007.* | |
| 4.4 | Form of Warrant to purchase shares of Common Stock issued to purchasers in the private placement which closed on March 15, 2006 (the “Series B Financing”), incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006.* | |
| 4.5 | Form of Warrant to purchase shares of Common Stock issued to purchasers in the Series B Financing, incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006.* | |
| 4.6 | Form of Warrant to purchase shares of Common Stock issued to the Placement Agent, in connection with the Series B Financing, incorporated by reference to Exhibit 4.4 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006.* | |
| 4.7 | Form of Warrant to purchase 600,000 shares of Common Stock issued to Indigo Ventures, LLC, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated July 12, 2006 and filed with the SEC on July 18, 2006.* | |
| 4.8 | Form of Warrant to purchase up to 478,698 shares of Common Stock issued to VGS PHARMA, LLC, incorporated by reference as Exhibit 3(a) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006.* | |
| 4.9 | Form of Non-Qualified Stock Option Agreement for 1,750,000 shares of Common Stock granted to Veerappan Subramanian, incorporated by reference as Exhibit 3(b) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006.* | |
| 4.10 | Form of Warrant to purchase shares of Common Stock issued to purchasers in the private placement which closed on April 24, 2007 (the “Series C Financing”), incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007.* | |
| 4.11 | Form of Warrant to purchase shares of Common Stock issued to the placement agent in the Series C Financing, incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007.* | |
| 4.12 | Form of specimen certificate for Series D 8% Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated September 15, 2008 and filed with the SEC on September 16, 2008.* | |
| 4.13 | Form of Warrant to purchase shares of Common Stock issued to purchasers in the private placement which closed on September 15, 2008 (the “Series D Financing”), incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated September 15, 2008 and filed with the SEC on September 16, 2008.* |
| 104 |
| 4.14 | Form of Warrant to purchase shares of Common Stock issued to the placement agent in the Series D Financing, incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K, dated September 15, 2008 and filed with the SEC on September 16, 2008.* | |
| 4.15 | Form of specimen certificate for Series E Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K, dated June 1, 2009, and filed with the SEC on June 5, 2009.* | |
| 4.16 | Warrant to purchase shares of Common Stock issued to Epic Investments, LLC in the initial closing of the Strategic Alliance Agreement, dated as of March 18, 2009, by and among the Company, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated June 1, 2009, and filed with the SEC on June 5, 2009.* | |
| 4.17 | Form of specimen certificate for Series G Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated April 18, 2013 and filed with the SEC on April 22, 2013. | |
| 4.18 | Form of specimen certificate for Series I Convertible Preferred Stock of the Company, incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K, dated February 6, 2014 and filed with the SEC on February 7, 2014. | |
| 4.19 | Rights Agreement, dated as of November 15, 2013, between the Company and American Stock Transfer & Trust Company, LLC., incorporated by reference to Exhibit 1 to the Registration Statement on Form 8-A filed with the SEC on November 15, 2013. | |
| 4.20 | Form of Series H Preferred Stock Certificate, incorporated by reference to Exhibit 1 to the Registration Statement on Form 8-A filed with the SEC on November 15, 2013. | |
| 10.1 | Elite Pharmaceuticals, Inc. 2014 Equity Incentive Plan, incorporated by reference to Appendix B to the Company’s Definitive Proxy Statement for its Annual Meeting of Shareholders, filed with the SEC on April 3, 2014. | |
| 10.2 | Form of Confidentiality Agreement (corporate), incorporated by reference to Exhibit 10.7 to the Form SB-2. | |
| 10.3 | Form of Confidentiality Agreement (employee), incorporated by reference to Exhibit 10.8 to the Form SB-2. | |
| 10.4 | Product Development and Commercialization Agreement, dated as of June 21, 2005, between the Company and IntelliPharmaceutics, Corp., incorporated by reference as Exhibit 10.1 to the Current Report on Form 8-K, dated June 21, 2005 and originally filed with the SEC on June 27, 2005, as amended on the Current Report on Form 8-K/A filed September 7, 2005, as further amended by the Current Report on Form 8-K/A filed December 7, 2005 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.5 | Agreement, dated December 12, 2005, by and among the Company, Elite Labs, and IntelliPharmaCeutics Corp., incorporated by reference as Exhibit 10.1 to the Current Report on Form 8-K, dated December 12, 2005, and originally filed with the SEC on December 16, 2005, as amended by the Current Report on Form 8-K/A filed March 7, 2006 (Confidential Treatment granted with respect to portions of the Agreement). |
| 105 |
| 10.6 | Loan Agreement, dated as of August 15, 2005, between New Jersey Economic Development Authority (“NJEDA”) and the Company, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated August 31, 2005 and filed with the SEC on September 6, 2005. | |
| 10.7 | Series A Note in the aggregate principal amount of $3,660,000.00 payable to the order of the NJEDA, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated August 31, 2005 and filed with the SEC on September 6, 2005. | |
| 10.8 | Series B Note in the aggregate principal amount of $495,000.00 payable to the order of the NJEDA, incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K, dated August 31, 2005 and filed with the SEC on September 6, 2005. | |
| 10.9 | Mortgage from the Company to the NJEDA, incorporated by reference to Exhibit 10.4 to the Current Report on Form 8-K, dated August 31, 2005 and filed with the SEC on September 6, 2005. | |
| 10.10 | Indenture between NJEDA and the Bank of New York as Trustee, dated as of August 15, 2005, incorporated by reference to Exhibit 10.5 to the Current Report on Form 8-K, dated August 31, 2005 and filed with the SEC on September 6, 2005. | |
| 10.11 | Form of Securities Purchase Agreement, between the Registrant and the signatories thereto, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006. | |
| 10.12 | Form of Registration Rights Agreement, between the Registrant and signatories thereto, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated March 15, 2006 and filed with the SEC on March 16, 2006. | |
| 10.13 | Form of Placement Agent Agreement, between the Registrant and Indigo Securities, LLC, incorporated by reference as Exhibit 10.3 to the Current Report on Form 8-K, dated March 15, 2006, and filed with the SEC on March 16, 2006. | |
| 10.14 | Financial Advisory Agreement between the Registrant and Indigo Ventures LLC, incorporated by reference as Exhibit 10.1 to the Current Report on Form 8-K dated July 12, 2006 and filed with the SEC on July 18, 2006. | |
| 10.15 | Product Collaboration Agreement between the Registrant and ThePharmaNetwork LLC, incorporated by reference as Exhibit 10.1 to the Current Report on Form 8-K, dated November 10, 2006 and filed with the SEC on November 15, 2006. (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.16 | Strategic Alliance Agreement among the Registrant, VGS Pharma (“VGS”) and Veerappan S. Subramanian (“VS”), incorporated by reference as Exhibit 10(a) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006. | |
| 10.17 | Advisory Agreement, between the Registrant and VS, incorporated by reference as Exhibit 10(b) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006. | |
| 10.18 | Registration Rights Agreement between the Registrant, VGS and VS, incorporated by reference as Exhibit 10(c) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006. | |
| 10.19 | Employment Agreement between Novel Laboratories Inc. (“Novel”) and VS, incorporated by reference as Exhibit 10(d) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006. |
| 106 |
| 10.20 | Stockholders’ Agreement between Registrant, VGS, VS and Novel, incorporated by reference as Exhibit 10(e) to the Current Report on Form 8-K, dated December 6, 2006 and filed with the SEC on December 12, 2006. | |
| 10.21 | Form of Securities Purchase Agreement, between the Registrant and the signatories thereto, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007. | |
| 10.22 | Form of Registration Rights Agreement, between the Registrant and the signatories thereto, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007. | |
| 10.23 | Form of Placement Agent Agreement, between the Company and Oppenheimer & Company, Inc., incorporated by reference as Exhibit 10.3 to the Current Report on Form 8-K, dated April 24, 2007 and filed with the SEC on April 25, 2007. | |
| 10.24 | Form of Securities Purchase Agreement, between the Registrant and the signatories thereto, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated July 17, 2007 and filed with the SEC on July 23, 2007. | |
| 10.25 | Form of Registration Rights Agreement, between the Registrant and the signatories thereto, incorporated by reference as Exhibit 10.2 to the Current Report on Form 8-K, dated July 17, 2007 and filed with the SEC on July 23, 2007. | |
| 10.26 | Consulting Agreement, dated as of July 27, 2007, between the Registrant and Willstar Consultants, Inc., incorporated by reference as Exhibit 10.1 to the Quarterly Report on Form 10-Q for the period ending September 30, 2007 and filed with the SEC on November 14, 2007. | |
| 10.27 | Form of Securities Purchase Agreement, between the Company and the signatories thereto, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated September 15, 2008 and filed with the SEC on September 16, 2008. | |
| 10.28 | Form of Placement Agent Agreement, between the Company, ROTH Capital Partners, LLC and Boenning & Scattergood, Inc., incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K, dated September 15, 2008 and filed with the SEC on September 16, 2008. | |
| 10.29 | Separation Agreement and General Release of Claims, dated as of October 20, 2008, by and between the Company and Stuart Apfel, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated October 15, 2008 and filed with the SEC on October 21, 2008. | |
| 10.30 | Consulting Agreement, dated as of October 20, 2008, by and between the Company and Parallex Clinical Research, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated October 15, 2008 and filed with the SEC on October 21, 2008. | |
| 10.31 | Separation Agreement and General Release of Claims, dated as of November 3, 2008, by and between the Company and Charan Behl, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated October 28, 2008 and filed with the SEC on November 3, 2008. | |
| 10.32 | Consulting Agreement, dated as of November 3, 2008, by and between the Company and Charan Behl, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated October 28, 2008 and filed with the SEC on November 3, 2008. | |
| 10.33 | Separation Agreement and General Release of Claims, dated as of November 5, 2008, by and between the Company and Bernard J. Berk, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated November 6, 2008 and filed with the SEC on November 6, 2008. |
| 107 |
| 10.34 | Compensation Agreement, dated as of December 1, 2008, by and between the Company and Jerry I. Treppel, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated December 1, 2008 and filed with the SEC on December 4, 2008. | |
| 10.35 | Strategic Alliance Agreement, dated as of March 18, 2009, by and among the Company, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated March 18, 2009 and filed with the SEC on March 23, 2009. | |
| 10.36 | Amendment to Strategic Alliance Agreement, dated as of April 30, 2009, by and among the Company, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated April 30, 2009 and filed with the SEC on May 6, 2009. | |
| 10.37 | Second Amendment to Strategic Alliance Agreement, dated as of June 1, 2009, by and among the Company, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated June 1, 2009, and filed with the SEC on June 5, 2009. | |
| 10.38 | Third Amendment to Strategic Alliance Agreement, dated as of Aug 18, 2009, by and among the Company, Epic Pharma LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.3 to the Quarterly Report on Form 10-Q, for the period ending June 30, 2009 and filed with the SEC on August 19, 2009. | |
| 10.39 | Employment Agreement, dated as of November 13, 2009, by and between the Company and Chris Dick, , incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q, for the period ending September 30, 2009 and filed with the SEC on November 16, 2009. | |
| 10.40 | Employment Agreement, dated as of November 13, 2009, by and between the Company and Carter J. Ward, incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q, for the period ending September 30, 2009 and filed with the SEC on November 16, 2009. | |
| 10.41 | Elite Pharmaceuticals Inc. 2009 Equity Incentive Plan, as adopted November 24, 2009, incorporated by reference to Exhibit 10.1 to the Registration Statement Under the Securities Act of 1933 on Form S-8, dated December 18, 2009 and filed with the SEC on December 22, 2009. | |
| 10.42 | Stipulation of Settlement and Release, dated as of June 25, 2010, by and among the Company, Midsummer Investment, Ltd., Bushido Capital Master Fund, LP, BCMF Trustees, LLC, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated June 25, 2010 and filed with the SEC on July 1, 2010 | |
| 10.43 | Amendment Agreement, dated as of June 25, 2010, by and among the Company, and the investors signatory thereto, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated June 25, 2010 and filed with the SEC on July 1, 2010 | |
| 10.44 | Amendment Agreement, dated as of June 2010, by and among the Company, Epic Pharma, LLC and Epic Investments, LLC, incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K, dated June 25, 2010 and filed with the SEC on July 1, 2010 | |
| 10.45 | Asset Purchase Agreement dated as of May 18, 2010, by and among Mikah Pharma LLC and the Company, incorporated by reference to Exhibit 10.4 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010. | |
| 10.46 | Asset Purchase Agreement, dated as of August 27, 2010, by and among Mikah Pharma LLC and the Company, incorporated by reference to Exhibit 10.5 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010 (Confidential Treatment granted with respect to portions of the Agreement). |
| 108 |
| 10.47 | Master Development and License Agreement, dated as of August 27, 2010, by and among Mikah Pharma LLC and the Company incorporated by reference to Exhibit 10.6 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.48 | Purchase Agreement, dated as of September 10, 2010, by and among Epic Pharma LLC and the Company, incorporated by reference to Exhibit 10.7 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.49 | License Agreement, dated as of September 10, 2010, by and among Precision Dose Inc. and the Company, incorporated by reference to Exhibit 10.8 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.50 | Manufacturing and Supply Agreement, dated as of September 10, 2010, by and among Precision Dose Inc. and the Company, incorporated by reference to Exhibit 10.9 to the Quarterly Report on Form 10-Q, for the period ended September 30, 2010 and filed with the SEC on November 15, 2010 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.51 | Product Development Agreement between the Company and Hi-Tech Pharmacal Co., Inc. dated as of January 4, 2011, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated January 4, 2011 and filed with the SEC on January 10, 2011 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.52 | Settlement Agreement between the Company and ThePharmaNetwork, LLC, dated as of March 11, 2011, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated March 11, 2011 and filed with the SEC on March 17, 2011. | |
| 10.53 | Manufacturing & Supply Agreement between the Company and Mikah Pharma LLC, dated as of June 1, 2011, incorporated by reference to Exhibit 10.70 to the Annual Report on Form 10-K, for the period ended March, 31, 2011 and filed with the SEC on June 29, 2011 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.54 | Manufacturing & Supply Agreement between the Company and ThePharmaNetwork, LLC, dated as of June 23, 2011, incorporated by reference to Exhibit 10.71 to the Annual Report on Form 10-K, for the period ended March, 31, 2011 and filed with the SEC on June 29, 2011 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.55 | Amendment, dated as of November 1, 2011, to the Master Development and License Agreement, dated as of August 27, 2010, by and amount Mikah Pharma LLC and the Company (Confidential Treatment granted with respect to portions of the Agreement), incorporated by reference to Exhibit 10.1 to Quarterly Report on Form 10-Q for three and nine months ended December 31, 2011. | |
| 10.56 | Settlement Agreement between the Company and ThePharmaNetwork, LLC, dated as of March 11, 2011, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated March 11, 2011 and filed with the SEC on March 17, 2011. | |
| 10.57 | Securities Purchase Agreement with Socius dated December 30, 2011, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the SEC on January 5, 2012. |
| 109 |
| 10.58 | Amendment to Agreement with Socius dated February 28, 2012, incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K/A filed with the SEC February 29, 2012. | |
| 10.59 | Manufacturing & Supply Agreement between the Company and Mikah Pharma LLC, dated as of June 1, 2011, incorporated by reference to Exhibit 10.70 to the Annual Report on Form 10-K, for the period ended March, 31, 2011 and filed with the SEC on June 29, 2011 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.60 | Amendment, dated as of November 1, 2011, to the Master Development and License Agreement, dated as of August 27, 2010, by and amount Mikah Pharma LLC and the Company (Confidential Treatment granted with respect to portions of the Agreement), incorporated by reference to Exhibit 10.1 to Quarterly Report on Form 10-Q for three and nine months ended December 31, 2011. | |
| 10.61 | Treppel $500,000 Bridge Loan Agreement dated June 12, 2012, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the SEC on June 13, 2012. | |
| 10.62 | December 5, 2012 amendment to the Treppel Bridge Loan Agreement incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the SEC on December 10, 2012. | |
| 10.63 | Development And License Agreement between the Company and a Hong Kong-based client dated March 16, 2012 incorporated by reference to Exhibit 10.77 to the Annual Report on Form 10-K filed with the SEC on June 29,2012 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.64 | Letter Agreement between the Company and ThePharmaNetwork LLC, dated September 21, 2012 incorporated by reference to Exhibit 10.6 to the Quarterly Report on Form 10-Q filed with the SEC on November 14,2012 (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.65 | Purchase Agreement between the Company and Lincoln Park Capital LLC dated April 19, 2013 , incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated April 18, 2013 and filed with the SEC on April 22, 2013. | |
| 10.66 | Registration Rights Agreement between the Company and Lincoln Park Capital LLC dated April 19, 2013 , incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated April 18, 2013 and filed with the SEC on April 22, 2013. | |
| 10.67 | August 1, 2013 Employment Agreement with Nasrat Hakim, incorporated by reference to Exhibit 10.4 to the Current Report on Form 8-K, dated August 1, 2013 and filed with the SEC on August 5, 2013. | |
| 10.68 | August 1, 2013 Mikah LLC Asset Purchase Agreement, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated August 1, 2013 and filed with the SEC on August 5, 2013. (Confidential Treatment granted with respect to portions of the Agreement). | |
| 10.69 | Revised Schedule 1 to the August 1, 2013 Mikah LLC Asset Purchase Agreement (revised to remove confidential treatment with regard to one item set forth thereon) incorporated by reference to Exhibit 10.12 to the Quarterly Report on Form 10-Q for the period ending December 31, 2013, filed with the SEC on February 14, 2014. | |
| 10.70 | August 1, 2013 Secured Convertible Note from the Company to Mikah Pharma LLC., incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated August 1, 2013 and filed with the SEC on August 5, 2013. |
| 110 |
| 10.71 | August 1, 2013 Security Agreement from the Company to Mikah Pharma LLC., incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K, dated August 1, 2013 and filed with the SEC on August 5, 2013. | |
| 10.72 | Termination of June 2011, Manufacturing and Supply Agreement between Mikah Pharma LLC and the Company, incorporated by reference to Exhibit 10.15 of the Quarterly Report on Form 10-Q for the period ending December 31, 2014 and filed with the SEC on February 14, 2014. | |
| 10.73 | October 15, 2013 Hakim Credit Line Agreement, incorporated by reference to Exhibit 10.16 to the Quarterly Report on Form 10-Q for the period ended September 30, 2013. | |
| 10.74 | October 2, 2013 Manufacturing and Licensing Agreement with Epic Pharma LLC, incorporated by reference to Exhibit 10.17 to the Amended Quarterly Report on Form 10-Q/A for the period ended September 30, 2013 and filed with the SEC on April 25, 2014. Confidential Treatment granted with respect to portions of the Agreement. | |
| 10.75 | August 19, 2013, Master Services Agreement with Camargo Pharmaceutical Services, LLC, incorporated by reference to Exhibit 10.18 to the Quarterly Report on Form 10-Q for the period ended September 30, 2013 and filed with the SEC on November 14, 2013 | |
| 10.76 | November 21, 2013 Unsecured Convertible Note from the Company to Jerry Treppel, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated November 26, 2013 and filed with the SEC on November 26, 2013. | |
| 10.77 | February 7,2014 Amendment to Secured Convertible Note from the Company to Mikah, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated February 7, 2014 and filed with the SEC on February 7, 2014. | |
| 10.78 | February 7, 2014 Amendment to Secured Convertible Note from the Company to Jerry Treppel, incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K, dated February 7, 2014 and filed with the SEC on February 7, 2014. | |
| 10.79 | Purchase Agreement between the Company and Lincoln Park Capital LLC dated April 10, 2014 , incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated April 10, 2014 and filed with the SEC on April 14, 2014. | |
| 10.80 | Registration Rights Agreement between the Company and Lincoln Park Capital LLC dated April 10, 2014 , incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K, dated April 10, 2014 and filed with the SEC on April 14, 2014. | |
| 10.81 | Employment Agreement with Dr. G. Kenneth Smith, dated October 20, 2014, incorporated by reference to Exhibit 10.82 to the Quarterly Report on Form 10-Q for the period ended September 30, 2014 and filed with the SEC on November 14, 2014. | |
| 10.82 | January 19, 2015 Second Amendment to TPN-Elite Manufacturing and Supply Agreement dated June 23, 2011 and First Amendment to the TPN-Elite Manufacturing and Supply Agreement dated September 21, 2012, incorporated by reference to Exhibit 10.82 to the Quarterly Report on Form 10-Q for the period ended December 31, 2014, and filed with the SEC on February 17, 2015. Confidential Treatment granted with respect to portions of the Agreement. | |
| 10.83 | January 28, 2015 First Amendment to the Loan Agreement between Nasrat Hakim and Elite Pharmaceuticals dated October 15, 2013, incorporated by reference to Exhibit 10.83 to the Quarterly Report on Form 10-Q for the period ended December 31, 2014 and filed with the SEC on February 17, 2015. |
| 111 |
| 10.84 | January 28, 2015 Termination of Development and License Agreement for Mikah-001 between Elite Pharmaceuticals, Inc. and Mikah Pharma LLC and Transfer of Payment, incorporated by reference to Exhibit 10.84 to the Quarterly Report on Form 10-Q for the period ended December 31, 2014 and filed with the SEC on February 17, 2015 . | |
| 10.85 | June 4, 2015 License Agreement with Epic Pharma LLC. Confidential portions of this exhibit have been redacted and filed separately with the Commission pursuant to a confidential treatment request in accordance with Rule 24b-2 of the Securities Exchange Act of 1934, as amended, incorporated by reference to Exhibit 10.85 to the Annual Report on Form 10-K for the fiscal year ended March 31, 2015 and filed with the SEC on June 15, 2015. | |
| 10.86 | Amendment No. 1 to Hakim Employment Agreement, incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the SEC on January 29, 2016. | |
| 21 | Subsidiaries of the Company.** | |
23.1
23.2 |
Consent of Buchbinder Tunick and Company, Independent Registered Public Accounting Firm**
Consent of Demetrius Berkower LLC, Independent Registered Public Accounting Firm** | |
| 31.1 | Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002** | |
| 31.2 | Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002** | |
| 32.1 | Certification of Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** | |
| 32.2 | Certification of Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** | |
| 101 | The following materials from Elite Pharmaceuticals’ Annual Report on Form 10-K, related to the audited financial statements as and for the fiscal years ended March 31, 2016, 2015 and 2014, formatted in eXtensible Business Reporting Language (“XBRL”); (i) the Consolidated Statements of Income; (ii) the Consolidated Balance Sheets (as of March 31, 2016 and 2015 only); (iii) the Consolidated Statements of Cash Flows; (iv) the Consolidated Statement of Changes in Stockholders Equity (Deficit); and (v) Notes to Consolidated Financial Statements.** |
* On January 5, 2011, the Company changed its domicile from Delaware to Nevada. All corporate documents from Delaware have been superseded by Nevada corporate documents filed or incorporated by reference herein. All outstanding Delaware securities certificates are now outstanding Nevada securities certificates.
** Filed herewith.
| 112 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ELITE PHARMACEUTICALS, INC. | ||
| By: | /s/ Nasrat Hakim | |
| Nasrat Hakim | ||
| Chief Executive Officer | ||
| Dated: June 15, 2016 | ||
| By: | /s/ Carter J. Ward | |
| Carter J. Ward | ||
| Chief Financial Officer | ||
| Dated: June 15, 2016 | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Nasrat Hakim | Chief Executive Officer, President and Chairman of the Board of Directors | June 15, 2016 | ||
| /s/ Carter J. Ward | Chief Financial Officer, Treasurer, Secretary | June 15, 2016 | ||
| /s/ Barry Dash | Director | June 15, 2016 | ||
| /s/ Jeffrey Whitnell | Director | June 15, 2016 | ||
| /s/ Eugene Pfeifer | Director | June 15, 2016 | ||
| /s/ Davis Caskey | Director | June 15, 2016 |
| 113 |
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED MARCH 31, 2016, 2015 AND 2014
CONTENTS
REPORTS OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Shareholders of Elite Pharmaceuticals, Inc. and Subsidiary
We have audited the accompanying consolidated balance sheet of Elite Pharmaceuticals, Inc. and Subsidiary (“the Company”) as of March 31, 2016, and the related consolidated statements of operations, stockholders’ deficit and cash flows for the period ended March 31, 2016. The Company’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Elite Pharmaceuticals, Inc. and Subsidiary as of March 31, 2016 and the results of its operations and their cash flows for the period ended March 31, 2016, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Elite Pharmaceuticals, Inc. and Subsidiary’s internal control over financial reporting as of March 31, 2016, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated June 15, 2016, expressed an adverse opinion.
We have also audited the adjustments described in Note 2 that were applied to restate the March 31, 2015 and 2014 financial statements. In our opinion, such adjustments are appropriate and have been properly applied.
/s/ Buchbinder Tunick & Company LLP
Wayne, New Jersey
June 15, 2016
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To The Board of Directors and Shareholders of
Elite Pharmaceuticals, Inc. and Subsidiary
We have audited, before the effects of the adjustments related to the restatement described in Note 2, the accompanying consolidated balance sheet of Elite Pharmaceuticals, Inc. and Subsidiary (the “Company”), as of March 31, 2015, and the related consolidated statements of operations, changes in stockholders' deficit, and cash flows for each of the years in the two-year period ended March 31, 2015. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, except for the effects of the adjustments related to the restatement described in Note 2, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Elite Pharmaceuticals, Inc. and Subsidiary as of March 31, 2015, and the results of their operations and their cash flows for each of the years in the two-year period ended March 31, 2015, in conformity with accounting principles generally accepted in the United States of America.
We were not engaged to audit, review, or apply any procedures to the adjustments related to the restatement described in Note 2 and, accordingly, we do not express an opinion or any other form of assurance about whether such adjustments are appropriate and have been properly applied. Those adjustments were audited by Buchbinder Tunick & Company LLP.
/s/ Demetrius Berkower LLC
Iselin, New Jersey
June 15, 2015
F-2
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
March 31, 2016 (Audited) and 2015 (Audited and Restated)
| 2016 (Audited) | 2015 (Audited and Restated) | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 11,512,179 | $ | 7,464,180 | ||||
| Accounts receivable (net of allowance for doubtful accounts of $— and $272,620 respectively) | 1,530,296 | 1,446,441 | ||||||
| Inventories | 3,293,729 | 3,032,002 | ||||||
| Prepaid expenses and other current assets | 377,752 | 388,061 | ||||||
| Total Current Assets | 16,713,956 | 12,330,684 | ||||||
| PROPERTY AND EQUIPMENT, net of accumulated depreciation of $6,726,401 and $6,074,117, respectively | 8,110,721 | 6,401,802 | ||||||
| INTANGIBLE ASSETS | 6,411,799 | 6,381,774 | ||||||
| OTHER ASSETS | ||||||||
| Security deposits | 48,714 | 198,481 | ||||||
| Restricted cash – debt service for EDA bonds | 388,959 | 388,959 | ||||||
| EDA bond offering costs, net of accumulated amortization of $150,052 and $135,874, respectively | 204,401 | 218,579 | ||||||
| Total Other Assets | 642,074 | 806,019 | ||||||
| TOTAL ASSETS | $ | 31,878,550 | $ | 25,920,279 | ||||
See accompanying notes
F-3
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
March 31, 2016 (Audited) and 2015 (Audited and Restated)
2016 (Audited) | 2015 (Audited and | |||||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| CURRENT LIABILITIES | ||||||||
| Current portion of EDA bonds payable | $ | 220,000 | $ | 210,000 | ||||
| Short term loans | 34,681 | — | ||||||
| Current portion of long-term debt | 308,263 | 265,165 | ||||||
| Related Party Lines of Credit | 718,309 | 583,071 | ||||||
| Accounts payable | 1,804,429 | 3,383,533 | ||||||
| Accrued expenses | 555,352 | 613,995 | ||||||
| Deferred revenues – current | 1,013,333 | 13,333 | ||||||
| Total Current Liabilities | 4,654,367 | 5,069,097 | ||||||
| LONG TERM LIABILITIES | ||||||||
| EDA Bonds Payable – Non Current | 1,845,000 | 2,065,000 | ||||||
| Deferred revenues | 3,278,887 | 125,557 | ||||||
| Other long term liabilities | 568,251 | 629,138 | ||||||
| Derivative liability – warrants | 10,368,567 | 17,762,573 | ||||||
| Total Long Term Liabilities | 16,060,705 | 20,582,268 | ||||||
| TOTAL LIABILITIES | 20,715,072 | 25,651,365 | ||||||
| MEZZANINE EQUITY | ||||||||
| Convertible preferred shares | 44,285,715 | 35,000,000 | ||||||
| STOCKHOLDERS’ DEFICIT | ||||||||
| Common stock – par value $0.001, Authorized 995,000,000 and 690,000,000 shares, respectively. Issued 711,544,352 shares and 631,160,701 shares, respectively. Outstanding 711,444,352 shares and 631,060,701 shares, respectively. | 711,546 | 631,162 | ||||||
| Additional paid-in-capital | 109,137,805 | 106,926,328 | ||||||
| Accumulated deficit | (142,664,747 | ) | (141,981,735 | ) | ||||
| Treasury stock at cost (100,000 common shares) | (306,841 | ) | (306,841 | ) | ||||
| TOTAL STOCKHOLDERS’ DEFICIT | (33,122,237 | ) | (34,731,086 | ) | ||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT | $ | 31,878,550 | $ | 25,920,279 | ||||
See accompanying notes
F-4
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS
| Years Ended March 31, | ||||||||||||
2016 (Audited) | 2015 (Audited and | 2014 (Audited and | ||||||||||
| REVENUES | ||||||||||||
| Manufacturing Fees | $ | 8,002,866 | $ | 3,870,457 | $ | 2,982,400 | ||||||
| Licensing Fees | 4,495,466 | 1,139,789 | 1,536,039 | |||||||||
| Lab Fee Revenues | — | 5,000 | 82,937 | |||||||||
| Total Revenues | 12,498,332 | 5,015,246 | 4,601,376 | |||||||||
| COSTS OF REVENUES | 4,484,162 | 3,013,592 | 3,236,106 | |||||||||
| Gross Profit | 8,014,170 | 2,001,654 | 1,365,270 | |||||||||
| OPERATING EXPENSES | ||||||||||||
| Research and Development | 12,428,783 | 14,727,472 | 3,959,316 | |||||||||
| General and Administrative | 2,903,178 | 2,904,114 | 2,105,725 | |||||||||
| Non-cash compensation through issuance of stock options | 333,362 | 260,045 | 82,947 | |||||||||
| Depreciation and Amortization | 665,647 | 616,995 | 500,906 | |||||||||
| Total Operating Expenses | 16,330,970 | 18,508,626 | 6,648,894 | |||||||||
| (LOSS) FROM OPERATIONS | (8,316,800 | ) | (16,506,972 | ) | (5,283,624 | ) | ||||||
| OTHER INCOME / (EXPENSES) | ||||||||||||
| Interest expense, net | (280,670 | ) | (287,231 | ) | (859,328 | ) | ||||||
| Change in fair value of derivative liabilities | 7,394,006 | 20,340,874 | (35,389,799 | ) | ||||||||
| Derivative interest expense | —- | — | (40,588 | ) | ||||||||
| Gain on Sale of Investment | — | 1,670,685 | — | |||||||||
| Other Income | — | — | 19,831 | |||||||||
| Total Other Income / (Expense) | 7,113,336 | 21,724,328 | (36,269,884 | ) | ||||||||
| INCOME (LOSS) BEFORE PROVISION FOR INCOME TAXES | (1,203,464 | ) | 5,217,356 | (41,553,508 | ) | |||||||
| CREDIT FOR INCOME TAXES | 520,452 | 3,249 | 292,611 | |||||||||
| NET INCOME (LOSS) | (683,012 | ) | 5,220,605 | (41,260,897 | ) | |||||||
| Change in value of convertible preferred share mezzanine equity | (9,285,715 | ) | 23,709,069 | (55,314,374 | ) | |||||||
| NET INCOME (LOSS) ATTRIBUTABLE TO COMMON SHAREHOLDERS | $ | (9,968,727 | ) | $ | 28,929,674 | $ | (96,575,271 | ) | ||||
| NET INCOME (LOSS) PER SHARE | ||||||||||||
| Basic | $ | (0.01 | ) | $ | 0.05 | $ | (0.21 | ) | ||||
| Diluted | $ | (0.01 | ) | $ | (0.02 | ) | $ | (0.21 | ) | |||
| WEIGHTED AVERAGE NUMBER OF COMMON SHARES OUTSTANDING | ||||||||||||
| Basic | 673,905,485 | 591,214,959 | 463,021,991 | |||||||||
| Diluted | 673,905,485 | 757,579,152 | 463,021,991 | |||||||||
See accompanying notes
F-5
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS (DEFICIT) EQUITY
FOR THE YEARS ENDED MARCH 31, 2016 (Audited) 2015 and 2014(Audited and Restated)
| Common Stock | Treasury Stock | |||||||||||||||||||||||||||
| Shares | Amount | Additional Paid-In Capital | Shares | Amount | Accumulated Deficit | Stockholders’ Deficit | ||||||||||||||||||||||
Balance at March 31, 2013 (restated) | 374,493,959 | $ | 374,495 | $ | 97,200,401 | 100,000 | $ | (306,841 | ) | $ | (105,941,443 | ) | $ | (8,673,388 | ) | |||||||||||||
| Net Income (Loss) | (41,260,897 | ) | (41,260,897 | ) | ||||||||||||||||||||||||
| Change in value of convertible preferred mezzanine equity | (55,314,374 | ) | (55,314,374 | ) | ||||||||||||||||||||||||
| Common shares sold pursuant to the Lincoln Park Capital purchase agreement | 65,143,216 | 65,143 | 9,934,857 | 10,000,000 | ||||||||||||||||||||||||
| Non-cash compensation through the issuance of stock options | 82,947 | 82,947 | ||||||||||||||||||||||||||
| Costs associated with raising capital (net of adjustments) | (47,987 | ) | (47,987 | ) | ||||||||||||||||||||||||
| Common shares issued as commitment shares pursuant to the Lincoln Park purchase agreement | 5,858,230 | 5,858 | (5,858 | ) | — | |||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of warrants | 16,904,038 | 16,904 | 3,584,116 | 3,601,020 | ||||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of options | 308,333 | 308 | 23,992 | 24,300 | ||||||||||||||||||||||||
| Common shares issued in payment of Directors’ Fees | 1,210,583 | 1,211 | 108,789 | 110,000 | ||||||||||||||||||||||||
| Common shares issued in payment of employee salaries | 3,439,467 | 3,439 | 364,794 | 368,233 | ||||||||||||||||||||||||
| Common shares issued in payment of consulting expenses | 210,018 | 210 | 18,626 | 18,836 | ||||||||||||||||||||||||
| Common shares issued in lieu of cash in payment of preferred share derivative interest expense | 878,543 | 879 | 67,210 | 68,089 | ||||||||||||||||||||||||
| Conversion of Series B, Series C and Series E Preferred Shares into Common Shares | 91,796,043 | 91,797 | 9,733,269 | 9,825,066 | ||||||||||||||||||||||||
Balance at March 31, 2014 (restated) | 560,242,430 | $ | 560,244 | $ | 65,750,782 | 100,000 | $ | (306,841 | ) | $ | (147,202,340 | ) | $ | (81,198,155 | ) | |||||||||||||
(continued on next page)
See Accompanying Notes
F-6
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS (DEFICIT) EQUITY
FOR THE YEARS ENDED MARCH 31, 2016 (Audited) 2015, and 2014 (Audited and Restated)
| Common Stock | Treasury Stock | |||||||||||||||||||||||||||
| Shares | Amount | Additional Paid-In Capital | Shares | Amount | Accumulated Deficit | Stockholders’ Deficit | ||||||||||||||||||||||
Balance at March 31, 2014 (restated) | 560,242,430 | $ | 560,244 | $ | 65,750,782 | 100,000 | $ | (306,841 | ) | $ | (147,202,340 | ) | $ | (81,198,155 | ) | |||||||||||||
| Net Income (Loss) | 5,220,605 | 5,220,605 | ||||||||||||||||||||||||||
| Change in value of convertible preferred mezzanine equity | 23,709,069 | 23,709,069 | ||||||||||||||||||||||||||
| Common shares sold pursuant to the Lincoln Park Capital purchase agreement | 47,172,240 | 47,172 | 13,189,452 | 13,236,624 | ||||||||||||||||||||||||
| Non-cash compensation through the issuance of stock options | 260,047 | 260,047 | ||||||||||||||||||||||||||
| Costs associated with raising capital (net of adjustments) | (16,365 | ) | (16,365 | ) | ||||||||||||||||||||||||
| Common shares issued as commitment shares pursuant to the Lincoln Park purchase agreement | 2,566,861 | 2,567 | (2,567 | ) | — | |||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of warrants | 11,985,388 | 11,985 | 762,868 | 774,853 | ||||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of options | 223,334 | 223 | 25,777 | 26,000 | ||||||||||||||||||||||||
| Common shares issued in payment of Directors’ Fees | 321,611 | 322 | 109,678 | 110,000 | ||||||||||||||||||||||||
| Common shares issued in payment of employee salaries | 2,518,668 | 2,519 | 847,218 | 849,737 | ||||||||||||||||||||||||
| Common shares issued in payment of consulting expenses | 70,169 | 70 | 23,929 | 23,999 | ||||||||||||||||||||||||
| Conversion of Series I Preferred Shares into Common Shares | 6,060,000 | 6,060 | 2,266,440 | 2,272,500 | ||||||||||||||||||||||||
Balance at March 31, 2015 (restated) | 631,160,701 | $ | 631,162 | $ | 106,926,328 | 100,000 | $ | (306,841 | ) | $ | (141,981,735 | ) | $ | (34,731,086 | ) | |||||||||||||
(continued on next page)
See Accompanying Notes
F-7
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENT OF CHANGES IN STOCKHOLDERS (DEFICIT) EQUITY
FOR THE YEARS ENDED MARCH 31, 2016 (Audited) 2015, and 2014 (Audited and Restated)
| Common Stock | Treasury Stock | |||||||||||||||||||||||||||
| Shares | Amount | Additional Paid-In Capital | Shares | Amount | Accumulated Deficit | Stockholders’ Deficit | ||||||||||||||||||||||
Balance at March 31, 2015 (restated) | 631,160,701 | $ | 631,162 | $ | 106,926,328 | 100,000 | $ | (306,841 | ) | $ | (141,981,735 | ) | $ | (34,731,086 | ) | |||||||||||||
| Net Income (Loss) | (683,012 | ) | (683,012 | ) | ||||||||||||||||||||||||
| Change in value of convertible preferred mezzanine equity | (9,285,715 | ) | (9,285,715 | ) | ||||||||||||||||||||||||
| Common shares sold pursuant to the Lincoln Park Capital purchase agreement | 23,945,346 | 23,945 | 6,175,698 | 6,199,643 | ||||||||||||||||||||||||
| Non-cash compensation through the issuance of stock options | 333,363 | 333,363 | ||||||||||||||||||||||||||
| Costs associated with raising capital (net of adjustments) | (84,102 | ) | (84,102 | ) | ||||||||||||||||||||||||
| Common shares issued as commitment shares pursuant to the Lincoln Park purchase agreement | 298,923 | 299 | 83,803 | 84,102 | ||||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of warrants | 48,283,968 | 48,284 | 2,969,464 | 3,017,748 | ||||||||||||||||||||||||
| Issuance of Common Shares pursuant to the exercise of options | 112,500 | 113 | 23,638 | 23,751 | ||||||||||||||||||||||||
| Common shares issued in payment of Directors’ Fees | 408,892 | 409 | 99,662 | 100,071 | ||||||||||||||||||||||||
| Common shares issued in payment of employee salaries | 4,236,555 | 4,237 | 1,034,763 | 1,039,000 | ||||||||||||||||||||||||
| Common shares issued in payment of consulting expenses | 97,467 | 97 | 23,903 | 24,000 | ||||||||||||||||||||||||
| Milestone shares issued pursuant to EPIC Strategic Alliance Agreement | 3,000,000 | 3,000 | 837,000 | 840,000 | ||||||||||||||||||||||||
Balance at March 31, 2016 | 711,544,352 | $ | 711,546 | $ | 109,137,805 | 100,000 | $ | (306,841 | ) | $ | (142,664,747 | ) | $ | (33,122,237 | ) | |||||||||||||
See accompanying notes
F-8
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
| YEARS ENDED MARCH 31, | ||||||||||||
2016 (Audited) | 2015 (Audited | 2014 (Audited and | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||||||
| Net Income (Loss) | $ | (683,012 | ) | $ | 5,220,605 | $ | (41,260,897 | ) | ||||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||||||
| Depreciation and amortization | 666,461 | 581,855 | 453,746 | |||||||||
| Change in fair value of derivative liabilities | (7,394,006 | ) | (20,340,874 | ) | 35,389,799 | |||||||
| Preferred share derivative interest satisfied by the issuance of common stock | — | —- | 68,089 | |||||||||
| Non-cash compensation accrued | 573,667 | 679,771 | 95,000 | |||||||||
| Salaries and Directors Fees satisfied by the issuance of common stock | 1,139,071 | 959,737 | 478,233 | |||||||||
| Consulting expenses paid via the issuance of common stock | 24,000 | 23,999 | 18,836 | |||||||||
| Non-cash compensation satisfied by the issuance of common stock and options | 333,363 | 260,047 | 82,947 | |||||||||
| Milestone shares issued pursuant to Epic Strategic Alliance Agreement | 840,000 | — | — | |||||||||
| Non-cash interest expense | — | — | 568,395 | |||||||||
| Non-cash rent expense | (22,996 | ) | 23,703 | (49,439 | ) | |||||||
| Non-cash lease accretion | 1,621 | 1,526 | 1,438 | |||||||||
| Gain on Sale of Investment | — | (1,670,685 | ) | — | ||||||||
| Recovery of Bad Debt | (117,095 | ) | — | — | ||||||||
| Changes in Assets and Liabilities | ||||||||||||
| Accounts receivable | 33,240 | (714,355 | ) | (66,922 | ) | |||||||
| Inventories | (261,727 | ) | (1,099,518 | ) | (574,340 | ) | ||||||
| Prepaid and other current assets | 160,076 | (147,188 | ) | (169,373 | ) | |||||||
| Accounts payable, accrued expenses and other current liabilities | (2,211,414 | ) | 1,131,477 | 788,445 | ||||||||
| Deferred revenues and Customer deposits | 4,153,330 | (13,333 | ) | (13,333 | ) | |||||||
| Derivative interest payable | — | — | (27,500 | ) | ||||||||
| NET CASH USED IN OPERATING ACTIVITIES | (2,765,421 | ) | (15,103,233 | ) | (4,216,876 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||||||
| Purchases of property and equipment | (1,918,804 | ) | (1,965,018 | ) | (502,268 | ) | ||||||
| Costs incurred for intellectual property assets | (30,025 | ) | (31,853 | ) | (58,178 | ) | ||||||
| Withdrawals from restricted cash, net | — | (123,916 | ) | 2,777 | ||||||||
| Proceeds from Sale of Investment in Novel | — | 5,000,000 | — | |||||||||
| NET CASH PROVIDED BY (USED IN) INVESTING ACTIVITIES | (1,948,829 | ) | 2,879,213 | (557,669 | ) | |||||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||||||
| Proceeds from Exercise of Cash Warrants and Options | 3,041,499 | 800,853 | 868,051 | |||||||||
| Proceeds from draws against related party Credit Lines | 135,238 | 54,322 | 528,750 | |||||||||
| Payment of NJEDA Bonds | (210,000 | ) | (1,110,000 | ) | — | |||||||
| Other loan payments | (404,131 | ) | (219,010 | ) | (1,515 | ) | ||||||
| Proceeds from sale of common stock to Lincoln Park Capital | 6,199,643 | 13,236,624 | 10,000,000 | |||||||||
| Costs associated with raising capital | — | (16,365 | ) | (47,987 | ) | |||||||
| NET CASH PROVIDED BY FINANCING ACTIVITIES | 8,762,249 | 12,746,424 | 11,347,299 | |||||||||
| NET CHANGE IN CASH AND CASH EQUIVALENTS | 4,047,999 | 522,404 | 6,572,754 | |||||||||
| CASH AND CASH EQUIVALENTS – beginning of period | 7,464,180 | 6,941,776 | 369,023 | |||||||||
| CASH AND CASH EQUIVALENTS – end of period | $ | 11,512,179 | $ | 7,464,180 | $ | 6,941,777 | ||||||
(See accompanying notes)
Continued on next page
F-9
ELITE PHARMACEUTICALS, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(continued from previous page)
| YEARS ENDED MARCH 31, | ||||||||||||
2016 (Audited) | 2015 (Audited | 2014 (Audited and | ||||||||||
| SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION | ||||||||||||
| Cash paid for interest | $ | 215,878 | $ | 89,336 | $ | 289,494 | ||||||
| Cash paid for taxes | $ | 4,048 | $ | 2,500 | $ | 3,099 | ||||||
| SCHEDULE OF NON-CASH INVESTING AND FINANCING ACTIVITIES | ||||||||||||
| Commitment shares issued to Lincoln Park Capital | $ | 84,102 | $ | 830,521 | $ | 1,112,838 | ||||||
| Conversion of Preferred Shares to Common Shares | — | 2,272,500 | 9,825,066 | |||||||||
| Acquisition of Intellectual Property with convertible note payable | — | — | 5,597,317 | |||||||||
| Issuance of note payable to related party in payment of balance due on line of credit owed to the same related party | — | — | 600,000 | |||||||||
| Issuance of Series I Preferred Shares in satisfaction of amounts due on Notes Payable | — | — | 7,953,591 | |||||||||
| Change in maximum redemption value of convertible preferred mezzanine equity | $ | (9,285,715 | ) | $ | 23,709,069 | $ | 55,314,374 | |||||
| Financing of equipment purchases | $ | 442,399 | $ | 804,861 | $ | 107,960 | ||||||
See accompanying notes
F-10
| NOTE 1 | - | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
BASIS OF PRESENTATION
The accompanying audited financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”).
The consolidated balance sheet as of March 31, 2015 and the consolidated statement of operations for the year ended March 31, 2015, the consolidated statement of cash flows for the year ended March 31, 2015 and the consolidated statement of changes in stockholders equity (deficit) for the year ended March 31, 2015, included in this annual report on form 10-K are restated to correct errors in accounting that were identified in the Company's annual report on Form 10-K for the year ended March 31, 2015. Please refer to Note 2 for further details on the specifics and effects of these corrections of accounting error.
The consolidated balance sheet as of March 31, 2014 and the consolidated statement of operations for the year ended March 31, 2014, the consolidated statement of cash flows for the year ended March 31, 2014 and the consolidated statement of changes in stockholders equity (deficit) for the year ended March 31, 2014, included in this annual report on form 10-K are restated to correct errors in accounting that were identified in the Company's annual report on Form 10-K for the year ended March 31, 2014. Please refer to Note 2 for further details on the specifics and effects of these corrections of accounting error.
PRINCIPLES OF CONSOLIDATION
The consolidated financial statements include the accounts of Elite Pharmaceuticals, Inc. and its wholly-owned subsidiary, Elite Laboratories, Inc. (“Elite Labs”, and collectively, the “Company”). The financial statements of its wholly-owned entity are consolidated and all significant intercompany accounts are eliminated upon consolidation.
NATURE OF BUSINESS
Elite Pharmaceuticals, Inc. was incorporated on October 1, 1997 under the laws of the State of Delaware, and its wholly-owned subsidiary Elite Laboratories, Inc. was incorporated on August 23, 1990 under the laws of the State of Delaware. On January 5, 2012, Elite Pharmaceuticals was reincorporated under the laws of the State of Nevada. Elite Labs engages primarily in researching, developing and licensing proprietary orally administered, controlled-release drug delivery systems and products with abuse deterrent capabilities and the manufacture of generic, oral dose pharmaceuticals. The Company is equipped to manufacture controlled-release products on a contract basis for third parties and itself if and when the products are approved. These products include drugs that cover therapeutic areas for pain, allergy, bariatric and infection. Research and development activities are done so with an objective of developing products that will secure marketing approvals from the United States Food and Drug Administration (“US-FDA”), and thereafter, commercially exploiting such products.
CASH
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents. Cash and cash equivalents consist of cash on deposit with banks and money market instruments. The Company places its cash and cash equivalents with high-quality, U.S. financial institutions and, to date has not experienced losses on any of its balances.
ACCOUNTS RECEIVABLE
Accounts receivable are comprised of balances due from customers, net of estimated allowances for uncollectible accounts. In determining collectability, historical trends are evaluated and specific customer issues are reviewed on a periodic basis to arrive at appropriate allowances.
F-11
INVENTORIES
Inventories are stated at the lower of cost (first-in, first-out basis) or market (net realizable value).
LONG-LIVED ASSETS
The Company periodically evaluates the fair value of long-lived assets, which include property and equipment and intangibles, whenever events or changes in circumstances indicate that its carrying amounts may not be recoverable. Such conditions may include an economic downturn or a change in the assessment of future operations. A charge for impairment is recognized whenever the carrying amount of a long-lived asset exceeds its fair value. Management has determined that no impairment of long-lived assets has occurred.
Property and equipment are stated at cost. Depreciation is provided on the straight-line method based on the estimated useful lives of the respective assets which range from three to forty years. Major repairs or improvements are capitalized. Minor replacements and maintenance and repairs which do not improve or extend asset lives are expensed currently.
Upon retirement or other disposition of assets, the cost and related accumulated depreciation are removed from the accounts and the resulting gain or loss, if any, is recognized in income.
Costs to acquire intangible assets are capitalized and, if such assets are determined to have a finite useful life, amortized to expense on a straight-line method over such finite useful life. Costs to acquire intangible assets that are determined to be indefinitely lived, such as Abbreviated New Drug Applications (“ANDA’s”) are capitalized, but not amortized to expense.
All intangible assets are tested for impairment on at least an annual basis, or sooner, should events or changes in circumstances occur that may indicate a potential impairment of a listed intangible assets.
RESEARCH AND DEVELOPMENT
Research and development expenditures are charged to expense as incurred.
CONCENTRATION OF CREDIT RISK
The Company maintains cash balances, which, at times, may exceed the amounts insured by the Federal Deposit Insurance Corp. Uninsured balances at March 31, 2016 are $11.5 million. Management does not believe that there is any significant risk of losses.
The Company in the normal course of business extends credit to its customers based on contract terms and performs ongoing credit evaluations. An allowance for doubtful accounts due to uncertainty of collection is established based on historical collection experience. Amounts are written off when payment is not received after exhaustive collection efforts. During Fiscal 2016, Fiscal 2015 and Fiscal 2014, the Company generated all its revenues from six, seven and ten companies, respectively. The termination of the contracts with either of such companies will result in the loss of a significant amount of revenues currently being earned.
F-12
USE OF ESTIMATES
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates made by management include, but are not limited to, the recognition of revenue, the amount of the allowance for doubtful accounts receivable and the fair value of intangible assets, stock-based awards and derivatives.
INCOME TAXES
The Company uses the liability method for reporting income taxes, under which current and deferred tax liabilities and assets are recorded in accordance with enacted tax laws and rates. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Under the liability method, the amounts of deferred tax liabilities and assets at the end of each period are determined using the tax rate expected to be in effect when taxes are actually paid or recovered. Further tax benefits are recognized when it is more likely than not, that such benefits will be realized. Valuation allowances are provided to reduce deferred tax assets to the amount considered likely to be realized.
GAAP prescribes a recognition threshold and measurement attribute for how a company should recognize, measure, present, and disclose in its financial statements uncertain tax positions that the company has taken or expects to take on a tax return. GAAP requires that the financial statements reflect expected future tax consequences of such positions presuming the taxing authorities’ full knowledge of the position and all relevant facts, but without considering time values. No adjustments related to uncertain tax positions were recognized during Fiscal 2016 and Fiscal 2015.
The Company recognizes interest and penalties related to uncertain tax positions as a reduction of the income tax benefit. No interest and penalties related to uncertain tax positions were accrued as of March 31, 2016 and March 31, 2015.
The Company operates in multiple tax jurisdictions within the United States of America. Although we do not believe that we are currently under examination in any of our major tax jurisdictions, we remain subject to examination in all of our tax jurisdiction until the applicable statutes of limitation expire. As of March 31, 2016, a summary of the tax years that remain subject to examination in our major tax jurisdictions are: United States – Federal, 2012 and forward, and State, 2008 and forward. The Company did not record unrecognized tax positions for the years ended March 31, 2016, 2015 and 2014.
EARNINGS PER COMMON SHARE
Basic earnings per common share is calculated by dividing net earnings by the weighted average number of shares outstanding during each period presented. Diluted earnings per share are calculated by dividing earnings by the weighted average number of shares and common stock equivalents. The Company’s common stock equivalents consist of options, warrants and convertible securities.
F-13
COLLABORATIVE ARRANGEMENTS
Contracts are considered to be collaborative arrangements when they satisfy the following criteria defined in ASC 808, “Collaborative Arrangements”:
| · | The parties to the contract must actively participate in the joint operating activity; and |
| · | The joint operating activity must expose the parties to the possibility of significant risks and rewards, based on whether or not the activity is successful. |
The Company entered into a sales and distribution licensing agreement with Epic Pharma LLC, dated June 4, 2015 (the “2015 Epic License Agreement”), which has been determined to satisfy the criteria for consideration as a collaborative agreement, and is accounted for accordingly, in accordance with GAAP.
REVENUE RECOGNITION
The Company enters into licensing, manufacturing and development agreements which may include multiple revenue generating activities, including, without limitation, milestones, license fees, product sales and services. These multiple elements are assessed in accordance ASC 605-25 Revenue Recognition for Multiple-Element Arrangements in order to determine whether particular components of the arrangement represent separate units of accounting.
An arrangement component is considered to be a separate unit of accounting if the deliverable relating to the component has value to the customer on a standalone basis, and if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in control of the Company.
The Company recognizes payments received pursuant to a multiple revenue agreement as revenue, only if the related delivered item(s) have stand-alone value, with the arrangement being accordingly accounted for as a separate unit of accounting. If such delivered item(s) are considered to either not have stand-alone value, the arrangement is accounted for as a single unit of accounting, and the payments received are recognized as revenue over the estimated period of when performance obligations relating to the item(s) will be performed.
Whenever the Company determines that an arrangement should be accounted for as a single unit of accounting, it determines the period over which the performance obligations will be performed and revenue will be recognized. If it cannot reasonably estimate the timing and the level of effort to complete its performance obligations under a multiple-element arrangement, revenues are then recognized on a straight-line basis over the period encompassing the expected completion of such obligations, with such period being reassessed at each subsequent reporting period.
Arrangement consideration is allocated at the inception of the arrangement to all deliverables on the basis of their relative selling price (the relative selling price method). When applying the relative selling price method, the selling price of each deliverable is determined using vendor-specific objective evidence of selling price, if such exists; otherwise, third-part evidence of selling price. If neither vendor-specific objective evidence nor third-party evidence of selling price exists for a deliverable, the Company uses its best estimate of the selling price for that deliverable when applying the relative selling price method. In deciding whether we can determine vendor-specific objective evidence or third-party evidence of selling price, the Company does not ignore information that is reasonably available without undue cost and effort.
F-14
When determining the selling price for significant deliverables under a multiple-element revenue arrangement, the Company considers any or all of the following, depending on information available or information that could be reasonably available without undue cost and effort: vendor-specific objective evidence, third party evidence or best estimate of selling price. More specifically, factors considered can include, without limitation and as appropriate, size of market for specific a product, number of suppliers and other competitive market factors, forecast market shares and gross profits, barriers/time frames to market entry/launch, intellectual property rights and protections, exclusive or non-exclusive arrangements, costs of similar/identical deliverables from third parties, contractual terms, including, without limitation, length of contract, renewal rights, commercial terms, profit allocations, and other commercial, financial, tangible and intangible factors that may be relevant in the valuation of a specific deliverable.
Milestone payments are accounted for in accordance with ASC 605-28 “Revenue Recognition-Milestone Method” for any deliverables or units of accounting under which the Company must achieve a defined performance obligation which is contingent upon future events or circumstances that are uncertain as of the inception of the arrangement providing for such future milestone payment. Determination of the substantiveness of a milestone is a matter of subjective assessment performed at the inception of the arrangement, and with consideration earned from the achievement of a milestone meeting all of the following:
· It must be either commensurate with the Company's performance in achieving the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the Company's performance to achieve the milestone; and
· It relates solely to past performance; and
· It is reasonable relative to all of the deliverables and payment terms (including other potential milestone consideration) within the arrangement.
SEGMENT REPORTING
FASB ASC 280-10-50, “Disclosure about Segments of an Enterprise and Related Information” requires use of the “management approach” model for segment reporting. The management approach is based on the way a company’s management organizes segments within the company for making operating decisions and assessing performance. Reportable segments are based on products and services, geography, legal structure, management structure, or any other manner in which management disaggregates a company.
The Company disaggregates its product revenues into the type of marketing authorization relating to each product, specifically the following two reportable segments:
| 1. | ANDA’s for generic products; or |
| 2. | NDA’s for branded products. |
Asset information is not reviewed or included within the Company’s internal management reporting. Accordingly, the Company does not disclose asset information for each reportable segment.
Please see note 3 for further details.
LEASES
Lease agreements are evaluated to determine if they are capital leases meeting any of the following criteria at inception: (a) transfer of ownership; (b) bargain purchase option; (c) the lease term is equal to 75 percent or more of the estimated economic life of the leased property; or (d) the present value at the beginning of the lease term of the minimum lease payments, excluding that portion of the payments representing executory costs such as insurance, maintenance, and taxes to be paid by the lessor, including any profit thereon, equals or exceeds 90 percent of the excess of the fair value of the leased property to the lessor at lease inception over any related investment tax credit retained by the lessor and expected to be realized by the lessor.
If at its inception a lease meets any of the four lease criteria above, the lease is classified by the Company as a capital lease; and if none of the four criteria are met, the lease is classified by the Company as an operating lease.
TREASURY STOCK
The Company records common shares purchased and held in treasury at cost.
FAIR VALUE OF FINANCIAL INSTRUMENTS
The carrying amounts of current assets and liabilities approximate fair value due to the short-term nature of these instruments. The carrying amounts of noncurrent assets are reasonable estimates of their fair values based on management’s evaluation of future cash flows. The long-term liabilities are carried at amounts that approximate fair value based on borrowing rates available to the Company for obligations with similar terms, degrees of risk and remaining maturities.
WARRANTS AND PREFERRED SHARES
The accounting treatment of warrants and preferred share series issued is determined pursuant to the guidance provided by subtopics 470, “Debt”, 480 “Distinguishing liabilities from equity”, and 815, “Derivatives and Hedging” of the Accounting Standard Codification. Each feature of these instruments, including, without limitation, any rights relating to subsequent dilutive issuances, dividend issuances, equity sales, rights offerings, forced conversions, optional redemptions, automatic monthly conversions, dividends and exercise are assessed with determinations made regarding the proper classification on the Company’s statement of financial position, results of operations, cash flow statement and statement of changes in stockholders equity (deficit).
F-15
Please see notes 15 and 16 for further details.
STOCK-BASED COMPENSATION
The Company accounts for all stock-based payments and awards under the fair value based method. Stock-based payments to non-employees are measured at the fair value of the consideration received, or the fair value of the equity instruments issued, or liabilities incurred, whichever is more reliably measurable. The fair value of stock-based payments to non-employees is periodically re-measured until the counterparty performance is complete, and any change therein is recognized over the vesting period of the award and in the same manner as if the Company had paid cash instead of paying with or using equity based instruments on an accelerated basis. The cost of the stock-based payments to nonemployees that are fully vested and non-forfeitable as at the grant date is measured and recognized at that date, unless there is a contractual term for services in which case such compensation would be amortized over the contractual term.
The Company accounts for the granting of share purchase options to employees using the fair value method whereby all awards to employees will be recorded at fair value on the date of the grant. Share based awards granted to employees with a performance condition are measured based on the probable outcome of that performance condition during the requisite service period. Such an award with a performance condition is accrued if it is probable that a performance condition will be achieved. Compensation costs for stock-based payments to employees that do not include performance conditions are recognized on a straight-line basis. The fair value of all share purchase options is expensed over their vesting period with a corresponding increase to additional capital surplus. Upon exercise of share purchase options, the consideration paid by the option holder, together with the amount previously recognized in additional capital surplus, is recorded as an increase to share capital
The Company uses the Black-Scholes option valuation model to calculate the fair value of share purchase options at the date of the grant. Option pricing models require the input of highly subjective assumptions, including the expected price volatility. Changes in these assumptions can materially affect the fair value estimate.
The compensation expense recognized for the years ended March 31, 2016, March 31, 2015 and March 31, 2014 in relation to the granting of share purchase options to employees was $333,362, $260,045 and $82,947 respectively.
FAIR VALUE MEASUREMENTS
The Company adopted Accounting Standards Codification (“ASC”) Topic 820, Fair Value Measurements and Disclosures, for financial and non-financial assets and liabilities.
ASC 820 discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow) and the cost approach (cost to replace the service capacity of an asset or replacement cost). The Company utilizes the market approach. The statement utilizes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
F-16
| Level 1: | Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities. |
| Level 2: | Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. |
| Level 3: | Unobservable inputs that reflect the reporting entity’s own assumptions. |
RECENTLY ISSUED ACCOUNTING PRONOUNCEMENTS
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers. The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The authoritative guidance is effective for annual reporting periods beginning after December 15, 2016. In July 2015, the FASB extended the effective date of the guidance by one year to December 15, 2017. The Company is currently in the process of assessing the impact this guidance will have on the consolidated financial statements.
In July 2015, the FASB issued ASU 2015-11, Inventory — Simplifying the Measurement of Inventory. ASU 2015-11 requires inventory to be subsequently measured using the lower of cost and net realizable value, thereby eliminating the market value approach. Net realizable value is defined as the “estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal and transportation.” ASU 2015-11 is effective for reporting periods beginning after December 15, 2016 and is applied prospectively. Early adoption is permitted. The Company is evaluating the impact, if any, of adopting this new accounting guidance on its financial statements.
In February, 2016, the FASB issued ASU 2016-02, Leases (Topic 842) which provides new guidance on leases. The new guidance will increase transparency and comparability among organizations that lease buildings, equipment, and other assets by recognizing the assets and liabilities that arise from lease transactions. Current off-balance sheet leasing activities will be required to be reflected on balance sheets so that investors and other users of financial statements can more readily and accurately understand the rights and obligations associated with these transactions. Consistent with the current lease standard, the new guidance addresses both finance and operating leases. Finance leases will be accounted for in substantially the same manner as capital leases are accounted for under current GAAP. Operating leases will be accounted for (both in the income statement and statement of cash flows) in a manner consistent with operating leases under existing GAAP. However, as it relates to the balance sheet, lessees will recognize lease liabilities based upon the present value of remaining lease payments and corresponding lease assets for operating leases with limited exceptions. The new guidance will also require lessees and lessors to provide additional qualitative and quantitative disclosures to help investors and other users for financials statements assess the amount, timing and uncertainty of cash flows arising from leases. These disclosures are intended to supplement the amounts recorded in the financial statements so that users can understand more about the nature of an organization’s leasing activities. The new guidance is effective annual reporting periods, including interim reporting periods with those annual periods, beginning after December 15, 2018, with early application being also permitted, but not required. The Company is evaluating the impact of adoption of this guidance on its financial position, results of operations and disclosures.
F-17
| NOTE 2 | - | RESTATEMENT OF PRIOR FINANCIAL INFORMATION |
After receiving a comment letter from the SEC in connection with its standard periodic review of our Form 10-K for the Fiscal Year Ended March 31, 2015, our Form 10-Q for the Quarterly Period Ended June 30, 2015 and, in the process of review, our Form 10-Q, as amended, for the Quarterly Period Ended September 30, 2015, we conducted further reviews of our financial statements. Based on such reviews, the following determinations were made with regards to previously filed annual reports on Form 10-K:
Accounting for convertible preferred shares prior to the fiscal year ended March 31, 2016
The Company determined that the accounting for Convertible Preferred Stock (“Mezzanine Preferred”) for annual periods prior to the fiscal year ended March 31, 2016 was incorrect. Specifically, it has been determined the Mezzanine Preferred which had originally been classified as derivative liabilities on annual reports filed on Form 10-K prior to the fiscal year ended March 31, 2016, should instead be accounted for as quasi equity instruments and recorded as mezzanine equity. In addition, the Mezzanine Preferred which were recorded at fair value each annual reporting period, with changes recorded in net income (loss), will instead be recorded at the maximum redemption amount each annual reporting period with changes to this amount being recorded in additional paid in capital. Accordingly, the change in carrying value of the Mezzanine Preferred, which was originally included in the calculation of net income as well as the calculation of net income attributable to common shareholders for annual periods prior to the fiscal year ended March 31, 2016, should instead be included only in the calculation of net income attributable to common shareholders. Consequently, correction of this error in accounting has no effect on earnings per share.
The correction of this accounting error has no effect on financial statements relating to the fiscal year ended March 31, 2016, which include the correct accounting for the Mezzanine Preferred.
In accordance with the guidance provided by the SEC’s Staff Accounting Bulletin 99, Materiality (“SAB 99”) and Staff Accounting Bulletin 108, Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements (“SAB 108”), the Company has determined that the impact of adjustments relating to the corrections of this accounting error are not material to previously issued annual audited and unaudited consolidated financial statements. Accordingly, these changes are disclosed herein and will be disclosed prospectively.
As a result of the aforementioned correction of accounting errors, the relevant annual financial statements have been restated as follows:
F-18
Effects on financials for the Year Ended March 31, 2015
| As of March 31, 2015 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Balance Sheet | ||||||||||||
| Derivative Liabilities | $ | 52,762,573 | $ | (35,000,000 | )1 | $ | 17,762,573 | |||||
| Convertible preferred shares | — | 35,000,000 | 1 | 35,000,000 | ||||||||
| Additional paid-in capital | 161,0221,568 | (54,095,240 | )1 | 106,926,328 | ||||||||
| Accumulated Deficit | (196,076,975 | ) | 54,095,240 | 1 | (141,981,735 | ) | ||||||
| Year Ended March 31, 2015 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Statement of Operations | ||||||||||||
| Change in Fair Value of derivative Liabilities | $ | 44,049,943 | $ | (23,709,070 | )1 | $ | 20,340,874 | |||||
| Net Income (Loss)2 | 28,929,674 | (23,709,070 | )1 | 5,220,604 | ||||||||
| Change in carrying value of convertible preferred mezzanine equity | — | 23,709,070 | 1 | 23,707,070 | ||||||||
| Net Income (Loss) attributable to common shareholders2 | 28,929,674 | — | 28,929,674 | |||||||||
| Net Income (Loss) per share | ||||||||||||
| Basic | $ | 0.05 | — | $ | 0.05 | |||||||
| Diluted | $ | (0.02 | ) | — | $ | (0.02 | ) | |||||
| Year Ended March 31, 2015 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Statement of Cash Flows | ||||||||||||
| Net Income (Loss)2 | $ | 28,929,674 | $ | (23,709,070 | )1 | $ | 5,220,604 | |||||
| Change in Fair Value of derivative Liabilities | (44,049,943 | ) | 23,709,070 | 1 | (20,340,874 | ) | ||||||
| Net cash used in operating activities | (15,103,233 | ) | — | (15,103,233 | ) | |||||||
| Change in maximum redemption value of convertible preferred mezzanine equity (non-cash financing transaction) | — | 23,709,070 | 1 | 23,709,070 | ||||||||
Effects on financials for the year ended March 31, 2014
| As of March 31, 2014 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Balance Sheet | ||||||||||||
| Derivative Liabilities-Preferred Shares | $ | 60,981,570 | $ | (60,981,570 | )1 | $ | — | |||||
| Convertible Preferred Shares | — | 60,981,570 | 1 | 60,981,570 | ||||||||
| Additional paid-in capital | 143,555,091 | (77,804,310 | )1 | 65,750,781 | ||||||||
| Accumulated Deficit | (255,006,646 | ) | 77,804,310 | 1 | 177,202,336 | |||||||
F-19
| Year Ended March 31, 2014 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Statement of Operations | ||||||||||||
| Change in fair value of derivatives | $ | (90,704,173 | ) | $ | 55,314,374 | 1 | $ | (35,389,799 | ) | |||
| Net Income (Loss)2 | (96,575,271 | ) | 55,314,374 | 1 | (41,260,897 | ) | ||||||
| Change in maximum redemption value of convertible preferred mezzanine equity | — | (55,314,374 | )1 | (55,314,374 | ) | |||||||
| Net Income (Loss) attributable to common shareholders2 | (96,575,271 | ) | — | (96,575,271 | ) | |||||||
| Net Income (Loss) per share | ||||||||||||
| Basic | $ | (0.21 | ) | — | $ | (0.21 | ) | |||||
| Diluted | $ | (0.21 | ) | — | $ | (0.21 | ) | |||||
| Year Ended March 31, 2014 | ||||||||||||
| As Previously Reported | Adjustments | As Restated | ||||||||||
| Condensed Consolidated Statement of Cash Flows | ||||||||||||
| Net Income (Loss)2 | $ | (96,575,271 | ) | $ | 55,314,374 | 1 | $ | (41,260,897 | ) | |||
| Change in Fair Value of derivative Liabilities | 90,704,173 | (55,314,374 | )1 | 35,389,799 | ||||||||
| Net cash used in operating activities | (4,216,875 | ) | — | (4,216,875 | ) | |||||||
| Change in maximum redemption value of convertible preferred mezzanine equity (non-cash financing transaction) | — | (55,314,374 | )1 | (55,314,374 | ) | |||||||
| 1 | Adjustments relate solely to correction of errors in accounting for convertible preferred shares. | |
| 2 | For annual periods prior to the fiscal year ended March 31, 2016, as previously reported Net Income (Loss) and Net Income (Loss) Attributable to Common Shareholders, were the same and, accordingly, both amounts were reported on a single line item identified as Net Income (Loss) Attributable to Common Shareholders |
| NOTE 3 | - | SEGMENT RESULTS |
FASB ASC 280-10-50, “Disclosure about Segments of an Enterprise and Related Information” requires use of the “management approach” model for segment reporting. The management approach is based on the way a company’s management organizes segments within the company for making operating decisions and assessing performance. Reportable segments are based on products and services, geography, legal structure, management structure, or any other manner in which management disaggregates a company.
F-20
The Company disaggregates its product revenues into the type of marketing authorization relating to each product, specifically the following two reportable segments:
| 1. | ANDA’s for generic products; or |
| 2. | NDA’s for branded products. |
The following represents selected information for the Company’s reportable segments for the fiscal years ended March 31, 2016, 2015 and 2014:
| Years Ended March 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Revenues from External Customers | ||||||||||||
| ANDA | $ | 9,164,999 | $ | 5,015,246 | $ | 4,601,376 | ||||||
| NDA | 3,333,333 | — | — | |||||||||
| Total revenues from external customers | $ | 12,498,332 | $ | 5,015,246 | $ | 4,601,376 | ||||||
| Adjusted income from continuing operations before income tax | ||||||||||||
| ANDA | 4,940,515 | 1,650,128 | 1,272,172 | |||||||||
| NDA | (9,305,998 | ) | (14,939,115 | ) | (3,933,203 | ) | ||||||
The table below provides reconciliations of the Company’s segment adjusted income from continuing operations before income tax to our consolidated (loss) income from continuing operations before income taxes, which is determined in accordance with U.S. GAAP, for the fiscal years ended March 31, 2016, 2015 and 2014:
| Years Ended March 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Total segment adjusted (loss) from continuing operations before income tax | $ | (4,365,483 | ) | $ | (13,288,987 | ) | $ | (2,661,031 | ) | |||
| Corporate unallocated costs | (1,772,241 | ) | (1,319,939 | ) | (1,351,921 | ) | ||||||
| Interest revenue | 9,802 | 7 | — | |||||||||
| Interest expense | (290,468 | ) | (287,231 | ) | (859,328 | ) | ||||||
| Depreciation and amortization expense | (665,647 | ) | (616,994 | ) | (500,906 | ) | ||||||
| Significant noncash items other than depreciation and amortization expense | (1,513,433 | ) | (1,281,052 | ) | (749,935 | ) | ||||||
| Change in value of derivatives | 7,394,006 | 20,340,874 | (35,430,386 | ) | ||||||||
| Gain on sale of investment | — | 1,670,678 | — | |||||||||
| Total income (loss) from continuing operations before income tax | $ | (1,203,464 | ) | $ | 5,217,356 | $ | 41,553,507 | |||||
Asset information is not reviewed or included within the Company’s internal management reporting. Accordingly, the Company has not disclosed asset for each reportable segment.
| NOTE 4 | - | INVENTORIES |
Inventories are recorded at the lower of cost or market on a first-in-first-out basis. Inventories at March 31, 2016 and 2015 consist of the following:
F-21
| 2016 (Audited) | 2015 (Audited and | |||||||
| Finished Goods | $ | 225,699 | $ | 122,773 | ||||
| Work-in-Process | 222,784 | 58,770 | ||||||
| Raw Materials | 2,845,246 | 2,850,459 | ||||||
| $ | 3,293,729 | $ | 3,032,002 | |||||
| NOTE 5 | - | PROPERTY AND EQUIPMENT |
Property and equipment at March 31, 2016 and 2015 consists of the following:
| 2016 (Audited) | 2015 (Audited and | |||||||
| Laboratory, manufacturing, and warehouse equipment | $ | 8,255,286 | $ | 7,593,017 | ||||
| Office equipment and Software | 234,634 | 209,551 | ||||||
| Furniture and fixtures | 49,804 | 49,804 | ||||||
| Transportation equipment | 66,855 | 66,855 | ||||||
| Land, building and improvements | 6,230,543 | 4,556,692 | ||||||
| 14,837,122 | 12,475,919 | |||||||
| Less: Accumulated depreciation | (6,726,401 | ) | (6,074,117 | ) | ||||
| $ | 8,110,721 | $ | 6,401,802 | |||||
Depreciation expense amounted to $652,284 and $566,028 for the years ended March 31, 2016 and 2015, respectively.
| NOTE 6 | - | INTANGIBLE ASSETS |
Costs to acquire intangible assets are capitalized and if such assets are determined to have a finite useful life, amortized to expense using a straight line method over this finite useful life. Costs to acquire intangible assets that are determined to be indefinitely lived, such as the costs to acquire Abbreviated New Drug Applications (“ANDA’s”), are capitalized, but not amortized to expense.
Patent application costs capitalized were incurred in relation to the Company’s abuse deterrent opioid technology. Amortization of such patent costs will begin upon the issuance of marketing authorization by the FDA of a product incorporating such patented technology and be calculated on a straight line basis through the expiry of the related patent(s).
All intangible assets are tested for impairment on at least an annual basis, as close to year end as practical, or sooner, should events or changes in circumstances occur that may indicate a potential impairment of a listed intangible asset.
As of March 31, 2016 and 2015, the following costs were recorded as intangible assets on the Company’s balance sheet:
F-22
| 2016 (Audited) | 2015 (Audited and Restated) | |||||||
| Intangible assets at beginning of fiscal year | ||||||||
| Patent application costs | $ | 334,457 | $ | 302,602 | ||||
| ANDA acquisitions | 6,047,317 | 6,047,317 | ||||||
| Less: Accumulated Amortization | — | — | ||||||
| Net Intangible Assets at beginning of fiscal year | $ | 6,381,774 | $ | 6,349,919 | ||||
| Intangible asset costs capitalized during the fiscal year | ||||||||
| Patent application costs | $ | 30,025 | $ | 31,855 | ||||
| ANDA acquisition costs | — | — | ||||||
| Total cost of intangible assets capitalized | $ | 30,025 | $ | 31,855 | ||||
| Intangible assets at end of fiscal year | ||||||||
| Patent application costs | $ | 364,482 | $ | 334,457 | ||||
| ANDA acquisition costs | 6,047,317 | 6,047,317 | ||||||
| Less: Accumulated Amortization | — | — | ||||||
| Net Intangible Assets | $ | 6,411,799 | $ | 6,381,774 | ||||
The costs incurred in patent applications totaling $30,025 and $31,855 for Fiscal 2016 and Fiscal 2015, respectively, were all related to our abuse resistant and extended release opioid product lines. The Company is continuing its efforts to achieve approval of such patents. Additional costs incurred in relation to such patent applications will be capitalized as intangible assets, with amortization of such costs to commence upon approval of the patents and commercialization of products utilizing the patented technologies.
The ANDA acquisition costs of $450,000 recorded as of the beginning of Fiscal 2015 and included as a part of intangible assets as of March 31, 2015 and March 31, 2014, are related to our acquisition of the ANDA for Phentermine 37.5mg tablets.
The ANDA acquisition costs incurred during Fiscal 2014, totaling approximately $5.6 million consist of 12 approved ANDA’s (the “Mikah Approved ANDAs”) and one ANDA that is under active review with the FDA (the “Mikah ANDA Application Product”) which were acquired from Mikah Pharma LLC (“Mikah”) pursuant an asset purchase agreement between the Company and Mikah dated August 1, 2013 (the “Mikah Asset Purchase Agreement”). A Current Report on Form 8-K was filed with the SEC on August 5, 2013 in relation to the Mikah Asset Purchase Agreement, with such filing being herein incorporated by reference.
| NOTE 7 | - | INVESTMENT IN NOVEL LABORATORIES INC. |
At the end of 2006, Elite entered into a joint venture with VGS Pharma, LLC (“VGS”) and created Novel Laboratories, Inc. (“Novel”), a privately-held company specializing in pharmaceutical research, development, manufacturing, licensing, acquisition and marketing of specialty generic pharmaceuticals. Novel's business strategy is to focus on its core strength in identifying and timely executing niche business opportunities in the generic pharmaceutical area. Elite’s ownership interest in Novel consisted of 9,800 shares of Novel’s Class A Voting Common Stock. As of October 1, 2007, Elite deconsolidated its financial statements from Novel and the investment in Novel was accounted for under the cost method of accounting.
F-23
On June 10, 2014, the Company received $5 million in exchange for the 9,800 shares of Novel’s Class A Voting Common Stock owned by the Company. At the time of this transaction, the investment had a book value of $3,329,322, with a gain on sale of investment of $1,670,685 being realized and recorded as an other income item.
| NOTE 8 | - | NJEDA BONDS |
Summary Description and History of NJEDA Bonds
On August 31, 2005, the Company successfully completed a refinancing of a prior 1999 bond issue through the issuance of new tax-exempt bonds (the “Bonds”) via the issuance of the following:
Description | Principal Amount On Issue Date | Interest Rate | Maturity | |||||||
| Series A Note | $ | 3,660,000 | 6.50 | % | September 1, 2030 | |||||
| Series B Note | 495,000 | 9.0 | % | September 1, 2012 | ||||||
The net proceeds, after payment of issuance costs, were used (i) to redeem the outstanding tax-exempt Bonds originally issued by the Authority on September 2, 1999, (ii) refinance other equipment financing and (iii) for the purchase of certain equipment to be used in the manufacture of pharmaceutical products. As of March 31, 2016, all of the proceeds were utilized by the Company for such stated purposes.
On July 23, 2014, the Company retired all outstanding Series B notes, at par, along with all accrued interest due and owing as of such date. As of March 31, 2016, there are no amounts due and owing in relation the Series B notes.
Interest is payable semiannually on March 1 and September 1 of each year. The Bonds are collateralized by a first lien on the Company’s facility and equipment acquired with the proceeds of the original and refinanced Bonds. The related Indenture requires the maintenance of a Debt Service Reserve Fund of $366,000 in relation to the Series A Notes.
The Debt Service Reserve is maintained in restricted cash accounts that are classified in Other Assets.
Bond issue costs were paid from the bond proceeds and are being amortized over the life of the bonds. These costs and amortization activity are summarized as follows:
Description | Balances As of March 31, 2015 | Amortization Expense Current YTD | Balances As of March 31, 2016 | |||||||||
| Bond Issue Costs | $ | 354,453 | $ | 354,453 | ||||||||
| Accumulated Amortization | (135,874 | ) | (14,178 | ) | (150,052 | ) | ||||||
| Unamortized Balance | $ | 218,579 | $ | 204,401 | ||||||||
F-24
The NJEDA Bonds require the Company to make an annual principal payment on September 1st of varying amounts as specified in the loan documents and semi-annual interest payments on March 1st and September 1st, equal to interest due on the outstanding principal at the applicable rate for the semi-annual period just ended.
Balance Sheet Classification of Bond Liability
Bond principal amounts scheduled to mature within 12 months of the balance sheet date were recorded as current liabilities as of such date. Bond principal amounts scheduled to mature on dates later than 12 months from the balance sheet date were recorded as non-current liabilities as of such date.
Bond financing consisting of the following, as of March 31,
| 2016 | 2015 | |||||||
| Refinanced NJEDA Bonds | $ | 2,065,000 | $ | 2,275,000 | ||||
| Current portion | (220,000 | ) | (210,000 | ) | ||||
| Long term portion, net of current maturities | $ | 1,845,000 | $ | 2,065,000 | ||||
Maturities of Bonds for the next five years are as follows:
| YEAR ENDING MARCH 31, | AMOUNT | |||
| 2017 | $ | 220,000 | ||
| 2018 | 85,000 | |||
| 2019 | 90,000 | |||
| 2020 | 95,000 | |||
| 2021 | 105,000 | |||
| Thereafter | 1,470,000 | |||
| $ | 2,065,000 | |||
| NOTE 9 | - | LOANS PAYABLE AND LONG TERM DEBT |
Loans Payable
During the ordinary course of business, the Company has secured loans to support the collateralized financing of fixed asset acquisition, or the renewal of insurance policies. The Company has secured such loans with initial principal amounts totaling $442,399 and $909,477 during Fiscal 2016 and Fiscal 2015, respectively. Payment terms of these loans range from 9 months to 60 months at annual interest rates that range from 6.8% to 12.2%.
Principal amounts scheduled for payment within 12 months of the balance sheet date are classified as current liabilities on the balance sheet.
F-25
Principal amounts scheduled for payment on dates later than 12 months after the balance sheet date are classified as non-current liabilities on the balance sheet.
Loans payable and long term debt consisted of the following:
| March 31, 2016 | March 31, 2015 | |||||||||||||||
| Current | Long- Term | Current | Long- Term | |||||||||||||
| Equipment and Insurance financing loans payable | $ | 342,945 | $ | 520,827 | $ | 265,165 | $ | 560,338 | ||||||||
| Deferred Rent-135 Ludlow Ave Lease (see note 9) | 19,528 | 42,524 | ||||||||||||||
| Lease termination costs – 135 Ludlow Ave lease (see note 9) | — | 27,896 | — | 26,275 | ||||||||||||
| TOTAL | $ | 342,945 | $ | 568,251 | $ | 265,165 | $ | 629,137 | ||||||||
Loan principal payments for the next five years are as follows:
| YEAR ENDING MARCH 31, | AMOUNT | |||
| 2017 | $ | 342,945 | ||
| 2018 | 199,164 | |||
| 2019 | 174,229 | |||
| 2020 | 129,476 | |||
| 2021 | 17,958 | |||
| Thereafter | — | |||
| $ | 863,772 | |||
| NOTE 10 | - | RELATED PARTY LINES OF CREDIT AND NOTES PAYABLE |
Hakim $1,000,000 Bridge Revolving Credit Line
On October 15, 2013 (the “Hakim Credit Line Effective Date”), we entered into a bridge loan agreement (the “Hakim Loan Agreement”) with Mr. Nasrat Hakim, our President and CEO. Under the terms of the Hakim Loan Agreement, we have the right, in our sole discretion, to a line of credit (“Hakim Credit Line”) in the maximum principal amount of up to $1,000,000 at any one time. Mr. Hakim provided the Credit Line for the purpose of supporting the acceleration of our product development activities. The outstanding amount will be evidenced by a promissory note which shall mature on March 31, 2016, as amended., at which time the entire unpaid principal balance plus accrued interest thereon shall be due and payable in full. We may prepay any amounts owed without penalty. Any such prepayments shall first be attributable to interest due and owing and then to principal. Interest only shall be payable quarterly on January 1, April 1, July 1 and October 1 of each year. Prior to maturity or the occurrence of an Event of Default as defined in the Hakim Loan Agreement, we may borrow, repay, and reborrow under the Hakim Credit Line through maturity. Amounts borrowed under the Hakim Credit Line will bear interest at the rate of ten percent (10%) per annum.
F-26
As of March 31, 2016, the principal balance owed under the Hakim Credit Line was $718,309, with an additional $70,784 in accrued interest being also owed, in accordance with the terms and conditions of the Hakim Credit Line. This principal balance was paid in full on May 23, 2016. Accrued interest consisting of $70,784 due and owing on March 31, 2016, plus $9,134 in interest due and owing in principal balances outstanding during the period April 1, 2016 through May 23, 2016 was paid on May 24, 2016. Accordingly, as of May 24, 2016, there are no amounts due and owing under the Hakim Loan Agreement and the Hakim Loan Agreement is expired.
As of March 31, 2015, the principal balance owed under the Hakim Credit Line was $583,071, with an additional $18,105 in accrued interest being also owed, in accordance with the terms and conditions of the Hakim Credit Line.
As of March 31, 2014, the principal balance owed under the Hakim Credit Line was $528,750, with an additional $9,810 in accrued interest being also owed, in accordance with the terms and conditions of the Hakim Credit Line.
Total interest expense accrued pursuant to the Hakim Credit Line was $52,678, $63,947 and $16,674 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively. An additional $9,134 in interest was accrued during the period April 1, 2016 through May 23, 2016, with all principal and interest amounts owed being paid in full on May 24, 2016.
Please note that this transaction is not to be considered as an arms-length transaction.
Convertible Note Payable to Mikah Pharma LLC
On August 1, 2013, Elite Laboratories Inc. (“Elite Labs”), a wholly owned subsidiary of the Company, executed an asset purchase agreement (the “Mikah Purchase Agreement”) with Mikah Pharma LLC (“Mikah”), an entity that is wholly owned by Mr. Nasrat Hakim, who, in conjunction with this transaction, was appointed as Elite’s CEO, President and a Director on August 2, 2012, and acquired from Mikah a total of 13 Abbreviated New Drug Applications (“ANDAs”) consisting of 12 ANDAs approved by the FDA and one ANDA under active review with the FDA, and all amendments thereto (the “Acquisition”) for aggregate consideration of $10,000,000, inclusive of imputed interest payable pursuant to a non-interest bearing, secured convertible note due in August 2016 (the “Mikah Note”). The Mikah Note was amended on February 7, 2014 to make it convertible into shares of the Company’s Series I Convertible Preferred Stock.
The Mikah Note, as amended, was interest free and due and payable on the third anniversary of its issuance. Subject to certain limitations, the principal amount of the Mikah Note was convertible at the option of Mikah into shares of Common Stock at a rate of $0.07 (approximately 14,286 shares per $1,000 in principal amount), the closing market price of the Company’s Common Stock on the date that the asset purchase agreement and Note were executed and/or into shares of the Company’s Series I Convertible Preferred Stock at the rate of 1 share of Series I Preferred Stock for each $100,000 of principal owed on the Mikah Note. The conversion rate was adjustable for customary corporate actions such as stock splits and, subject to certain exclusions, includes weighted average anti-dilution for common stock transactions at prices below the then applicable conversion rate. Pursuant to a security agreement (the “Security Agreement”), repayment of the Mikah Note was secured by the ANDAs acquired in the Acquisition.
On February 7, 2014, Mikah converted the principal amount of $10,000,000, representing the entire principal balance due under the Mikah Note, into 100 shares of the Company’s Series I Preferred Stock, with the Mikah Note being retired.
Please note that this transaction is not to be considered as an arms-length transaction.
| NOTE 11 | - | LEASES OF RENTAL PROPERTIES |
The following leases for rental properties were operative during the year ended March 31, 2016:
F-27
135 Ludlow Ave (see notes 10 and 11) | ||||
| Effective Date | July 1, 2010(3) | |||
| Termination Date | December 31, 2016 | |||
| Lease term | 6 years with 2 tenant renewal options for 5 years each | |||
| Rent expense for the 2016 Fiscal Year | $ 180,854 | |||
| Rent expense for the 2015 Fiscal Year | $ 153,430 | |||
| Minimum 5 Year Lease Payments(1) | ||||
| Fiscal year ended March 31, 2017(2) | 155,169 | |||
| Fiscal year ended March 31, 2018(2) | — | |||
| Fiscal year ended March 31, 2019(2) | — | |||
| Fiscal year ended March 31, 2020(2) | — | |||
| Fiscal year ended March 31, 2021(2) | — | |||
| $ 155,169 | ||||
| (1) | Minimum lease payments are exclusive of additional expenses related to certain expenses incurred in the operation and maintenance of the premises, including, without limitation, real estate taxes and common area charges which may be due under the terms and conditions of the lease. |
| (2) | Minimum lease payments calculated for the initial term of the lease only, with such initial term expiring on December 31, 2016. |
| (3) | Inclusive of a modification of lease agreement dated July 29, 2014 |
Rent expense related to the operating lease at 135 Ludlow was recorded using the straight line method and summarized as follows:
Summary of Rent Expense – 135 Ludlow Avenue
Fiscal Year Ended March 31, 2016 | Fiscal Year Ended March 31, 2015 | |||||||
| Rent Expense | $ | 180,854 | $ | 153,430 | ||||
| Actual lease payments | 203,850 | 114,321 | ||||||
| Increase (Decrease) in deferred rent liability | (22,996 | ) | 39,109 | |||||
| Adjustments to deferred rent liability | — | (15,409 | ) | |||||
| Balance of deferred rent liability | 19,528 | 42,524 | ||||||
| NOTE 12 | - | LEASE OF 135 LUDLOW AVENUE |
The Company entered into a lease for a portion of a one-story warehouse, located at 135 Ludlow Avenue, Northvale, New Jersey, consisting of approximately 15,000 square feet of floor space. The lease term began on July 1, 2010 and is classified as an operating lease.
F-28
On July 29, 2014, the Company modified this operating lease, with the material terms of the modification including the Company being permitted to occupy the entire 35,000 square feet in the building, with this expansion being necessary to support the Company’s growing and projected commercial operations.
The lease, as modified, includes an initial term which expires on December 31, 2016 and two tenant renewal options of five years each, with such options being at the sole discretion of the Company. The property related to this lease will be used for the storage of pharmaceutical finished goods, raw materials, equipment and documents as well as pharmaceutical manufacturing, packaging, distribution activities and related regulatory activities.
The additional 20,000 square feet for which the Company secured occupancy rights pursuant to the July 2014 modification agreement requires significant leasehold improvements and qualification as a prerequisite for its intended future use. These improvements are currently in progress.
Please refer to Note 9 of these financial statements for details on minimum lease payments, rent expense and deferred rent liabilities.
| NOTE 13 | - | LEASE TERMINATION COSTS - 135 LUDLOW AVENUE |
The lease for the property located at 135 Ludlow Avenue, Northvale NJ, includes a requirement that, at termination, the Company return the property to its condition at the inception of the lease, with normal wear and tear excepted. Such requirement accordingly represents an unconditional obligation associated with the retirement of a long-lived asset and subject to ASC 410 of the Codification. The Company estimates such costs would amount to $50,000, at lease termination, and pursuant to ASC 410 has recorded a liability and offsetting asset equal to the present value, at lease inception, of such obligation. This liability is accreted over the term of the lease (including extensions), using the interest method and principally included within interest expense.
| NOTE 14 | - | DEFERRED REVENUES |
Deferred revenues in the aggregate amount of $4,292,220, consisting of a current component of $1,013,333 and a long term component of $3,278,887. These line items represent the unamortized amounts of a $200,000 advance payment received for a TAGI licensing agreement with a fifteen year term beginning in September 2010 and ending in August 2025 and the $5,000,000 advance payment Epic Collaborative Agreement with a five year term beginning in June 2015 and ending in May 2020. The advance payment was recorded as deferred revenue when received and is earned, on a straight line basis over the life of the license. The current component is equal to the amount of revenue to be earned during the 12 month period immediately subsequent to the balance date and the long term component is equal to the amount of revenue to be earned thereafter.
F-29
| NOTE 15 | - | MEZZANINE EQUITY – CONVERTIBLE PREFERRED SHARES |
On February 6, 2014, the Company created the Series I Convertible Preferred Stock (“Series I Preferred”). A total of 500 shares of Series I Preferred are authorized and as of the current Balance Sheet Date, 100 shares are issued and outstanding, with a stated value of $100,000 and a par value of $0.01. The Certificate of Designations (“COD”) for the Series I Preferred contain the following features:
| · | Conversion feature - the Series I Preferred Shares may be converted, at the option of the Holder, into the Company’s Common Stock at a stated conversion price of $0.07. |
| · | Subsequent dilutive issuances - if the Company issues options at a price below the Conversion Price, then the Conversion Price will be reduced. |
| · | Subsequent dividend issuances - if the Company issues Common Stock in lieu of cash in satisfaction of its dividend obligation on its Series C Certificate, the applicable Conversion Price of the Series I Preferred is adjusted. |
Management has determined that the Series I Preferred host instrument is more akin to equity than debt and also that the above financial instruments are clearly and closely related to the host instrument, with bifurcation and classification as a derivative liability being not required.
Based on Management’s review of the COD, the host instrument, the Series I Preferred Shares, will be classified as mezzanine equity. The above identified embedded financial instruments: Conversion Feature, Subsequent Dilutive Issuances and Subsequent Dividend Issuances will not be bifurcated from the host and are therefore classified as mezzanine equity with the Series I Preferred. The Series I Preferred will be carried at the maximum redemption value, with changes in this value charged to retained earnings or to additional paid-in capital in the absence of retained earnings.
Changes in carrying value are also subtracted from net income (loss), (in a manner similar to the treatment of dividends paid on preferred stock), in arriving at net income (loss) available to common stockholders used in the calculation of earnings per share.
CONVERTIBLE PREFERRED MEZZANINE EQUITY
| March 31, 2016 | March 31, 2015 | |||||||
| Shares authorized | 500 | 500 | ||||||
| Shares outstanding | 100 | 100 | ||||||
| Par value | $ | 0.01 | $ | 0.01 | ||||
| Stated value | $ | 100,000 | $ | 100,000 | ||||
| Conversion Price | $ | 0.07 | $ | 0.07 | ||||
| Common shares to be issued upon redemption | 142,857,143 | 142,857,143 | ||||||
| Closing price on valuation date | $ | 0.31 | $ | 0.2450 | ||||
| Carrying value of convertible preferred mezzanine equity | $ | 44,285,714 | $ | 35,000,000 | ||||
INCREASE / (DECREASE) IN VALUE OF CONVERTIBLE PREFERRED MEZZANINE EQUITY
| FISCAL YEAR ENDED | ||||||||
| March 31, 2016 | March 31, 2015 | |||||||
| Series I Preferred | 9,285,715 | (23,709,069 | ) | |||||
F-30
| NOTE 16 | - | DERIVATIVE LIABILITIES - WARRANTS |
To date, the Company has authorized the issuance of Common Stock Purchase Warrants, with terms of five to seven years, to various corporations and individuals, in connection with the sale of securities, loan agreements and consulting agreements. Exercise prices on those warrants outstanding during Fiscal 2016 and Fiscal 2015 range from $0.0625 to $0.25 per warrant. The warrants expire at various times through April 25, 2018.
A summary of warrant activity for the fiscal years indicated below is as follows:
| Fiscal Year 2016 | Fiscal Year 2015 | |||||||||||||||
| Warrant Shares | Weighted Average Exercise Price | Warrant Shares | Weighted Average Exercise Price | |||||||||||||
| Balance at beginning of year | 89,870,034 | $ | 0.06 | 102,143,091 | $ | 0.06 | ||||||||||
| Warrant exercises, forfeited or expired | 48,283,968 | $ | 0.06 | 12,273,057 | $ | 0.07 | ||||||||||
| Ending Balance | 41,586,066 | $ | 0.06 | 89,870,034 | $ | 0.06 | ||||||||||
Accounting Standard Codification “ASC” 815 – Derivatives and Hedging, which provides guidance on determining what types of instruments or embedded features in an instrument issued by a reporting entity can be considered indexed to its own stock for the purpose of evaluating the first criteria of the scope exception in the pronouncement on accounting for derivatives. These requirements can affect the accounting for warrants and convertible preferred instruments issued by the Company. As the conversion features within, and the detachable warrants, if any, issued with the Company’s Series E and Series I Preferred Stock, do not have fixed settlement provisions because their conversion and exercise prices may be lowered if the Company issues securities at lower prices in the future, we have concluded that the instruments are not indexed to the Company’s stock and are to be treated as derivative liabilities.
The Warrant Derivative Liabilities are measured at fair market value, using the market approach and a level 3 fair value hierarchy, on a recurring basis as of March 31, 2016 and March 31, 2015, in accordance with the valuation techniques discussed in ASC 820.
The portion of derivative liabilities related to outstanding warrants was valued using the Black-Scholes option valuation model, a level 3 fair value hierarchy using the following assumptions:
March 31 2016 | March 31 2015 | March 31 2014 | ||||
| Risk-Free interest rate | 0.18% - 0.73% | 0.05% - 0.89% | 0.05% - 1.32% | |||
| Expected volatility | 52% - 81% | 93% - 113% | 111% - 207% | |||
| Expected life (in years) | 0.2 – 2.1 | 1.2 – 3.1 | 0.3 - 4.1 | |||
| Expected dividend yield | 0% | 0% | 0% | |||
| Number of warrants | 41,586,066 | 89,870,034 | 102,143,093 | |||
| Fair value – Warrant Derivative Liability | $10,368,567 | $17,762,573 | $38,103,446 | |||
| Change in warrant derivative liability for the twelve months ended | $7,394,006 | $(18,447,573) | $32,997,869 |
F-31
The risk free interest rate was based on rates established by the US Treasury Department. The expected volatility was based on the historical volatility of the Company’s share price for periods equal to the expected life of the outstanding warrants at each valuation date. The expected dividend rate was based on the fact that the Company has not historically paid dividends on common stock and does not expect to pay dividends on common stock in the future.
The changes of $7,394,006, $(18,447,573) and $32,997,869 in value of the warrant derivative liability occurring during the years ended March 31, 2016, 2015 and 2014, respectively, are included in the amounts reported in the “Other Income/(Expense)” section of the statement of operations. Increases in value are reported as other expenses and decreases in value are reported as other income.
The following table summarizes, as of March 31, 2016, 2015 and 2014, the warrant activity subject to Level 3 inputs which are measured on a recurring basis:
Fair value measurements of warrants using significant unobservable inputs (Level 3)
| March 31 | March 31 | March 31 | ||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Balance at Beginning of Fiscal Year | $ | 17,762,573 | $ | 38,103,446 | $ | 7,862,848 | ||||||
| Warrants Exercised | (14,788,012 | ) | (2,578,300 | ) | (2,757,271 | ) | ||||||
| Change in fair value of warrant liability | 7,394,006 | (18,447,573 | ) | 32,997,869 | ||||||||
| Balance at End of Fiscal Year | $ | 10,368,567 | $ | 17,762,573 | $ | 38,103,446 | ||||||
| NOTE 17 | - | COMMON STOCK |
Lincoln Park Capital
Pursuant to an April 19, 2013 purchase agreement with Lincoln Park Capital Fund, LLC (“Lincoln Park”) we had the right to sell to and Lincoln Park was obligated to purchase up to $10 million in shares of the Company’s Common Stock, subject to certain limitations, from time to time, over the 36 month period commencing on May 9, 2013. We raised the entire $10 million from the sale of shares to Lincoln Park pursuant to that agreement. That agreement terminated in March 2014 with the sale of all shares covered by that agreement.
On April 10, 2014, we entered into another Purchase Agreement and a Registration Rights Agreement with Lincoln Park. Pursuant to the terms of the Purchase Agreement, Lincoln Park has agreed to purchase from us up to $40 million of our common stock (subject to certain limitations) from time to time over a 36-month period. Pursuant to the terms of the Registration Rights Agreement, we have filed with the SEC a registration statement to register for resale under the Securities Act the shares that have been or may be issued to Lincoln Park under the Purchase Agreement. That registration statement was declared effective by the SEC on May 1, 2014. A post-effective amendment to that Registration Statement was subsequently filed with the SEC and declared effective on July 1, 2014.
F-32
Upon execution of the Purchase Agreement, we have issued 1,928,641 shares of our common stock to Lincoln Park pursuant to the Purchase Agreement as consideration for its commitment to purchase additional shares of our common stock under that agreement and we are obligated to issue up to an additional 1,928,641 commitment shares to Lincoln Park pro rata as up to $40 million of our common stock is purchased by Lincoln Park. Through June 7, 2016, we have sold to Lincoln Park an aggregate of 76.7 million shares under the Purchase Agreement for aggregate gross proceeds of approximately $21.2 million. In addition, we have issued an additional 1.0 million Commitment Shares.
We may, from time to time and at our sole discretion but no more frequently than every other business day, direct Lincoln Park to purchase (a “Regular Purchase”) up to 500,000 shares of our common stock on any such business day, increasing up to 800,000 shares, depending upon the closing sale price of the common stock, provided that in no event shall Lincoln Park purchase more than $760,000 worth of our common stock on any single business day. The purchase price of shares of Common Stock related to the future Regular Purchase funding will be based on the prevailing market prices of such shares at the time of sales (or over a period of up to 10 business days leading up to such time), but in no event will shares be sold to Lincoln Park on a day the Common Stock closing price is less than the floor price of $0.10 per share, subject to adjustment.
In addition to Regular Purchases, on any business day on which we have properly submitted a Regular Purchase notice and the closing sale price is not below $0.15, we may purchase (an “Accelerated Purchase”) an additional “accelerated amount” under certain circumstances. The amount of any Accelerated Purchase cannot exceed the lesser of three times the number of purchase shares purchased pursuant to the corresponding Regular Purchase; and 30% of the aggregate shares of our common stock traded during normal trading hours on the purchase date. The purchase price per share for each such Accelerated Purchase will be equal to the lower of (i) 97% of the volume weighted average price during the purchase date; or (ii) the closing sale price of our common stock on the purchase date.
In the case of both Regular Purchases and Accelerated Purchases, the purchase price per share will be equitably adjusted for any reorganization, recapitalization, non-cash dividend, stock split, reverse stock split or other similar transaction occurring during the business days used to compute the purchase price.
Other than as set forth above, there are no trading volume requirements or restrictions under the Purchase Agreement, and we will control the timing and amount of any sales of our common stock to Lincoln Park.
Our sales of shares of Common Stock to Lincoln Park under the Lincoln Park Purchase Agreement are limited to no more than the number of shares that would result in the beneficial ownership by Lincoln Park and its affiliates, at any single point in time, of more than 9.99% of the then outstanding shares of Common Stock.
The Lincoln Park Purchase Agreement and the Lincoln Park Registration Rights Agreement contain customary representations, warranties, agreements and conditions to completing future sale transactions, indemnification rights and obligations of the parties. The Company has the right to terminate the Lincoln Park Purchase Agreement at any time, at no cost or penalty. Actual sales of shares of Common Stock to Lincoln Park under the Lincoln Park Purchase Agreement will depend on a variety of factors to be determined by the Company from time to time, including, without limitation, market conditions, the trading price of the Common Stock and determinations by the Company as to appropriate sources of funding for the Company and its operations. There are no trading volume requirements or restrictions under the Lincoln Park Purchase Agreement. Lincoln Park has no right to require any sales by the Company, but is obligated to make purchases from the Company as it directs in accordance with the Lincoln Park Purchase Agreement. Lincoln Park has covenanted not to cause or engage in any manner whatsoever, any direct or indirect short selling or hedging of our shares.
F-33
The net proceeds under the Purchase Agreement to the Company will depend on the frequency and prices at which the Company sells shares of its stock to Lincoln Park. The Company expects that any proceeds received by the Company from such sales to Lincoln Park under the Lincoln Park Purchase Agreement will be used for general corporate purposes and working capital requirements.
Summary of Common Stock Activity
During Fiscal Years 2016, 2015 and 2014 the Company issued a total of 80,386,651, 70,918,271 and 185,748,471 shares of Common Stock, respectively, with such issuances of Common Stock being summarized as follows:
| Description | Fiscal Year 2016 | Fiscal Year 2015 | Fiscal Year 2014 | |||||||||
| Common shares sold pursuant to the Lincoln Park Capital Purchase Agreements, with net proceeds of such shares totaling $6,199,643, $13,236,624 and $10,000,000 in Fiscal 2016, Fiscal 2015, and Fiscal 2014, respectively. | 23,945,346 | 47,172,240 | 65,143,216 | |||||||||
| Common shares issued as commitment shares pursuant to the Lincoln Park Capital Purchase Agreements | 298,923 | 2,566,861 | 5,858,230 | |||||||||
| Common Shares issued in lieu of cash payment in payment of preferred share derivative interest expenses totaling zero, zero and $68,089 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively | --- | --- | 878,543 | |||||||||
| Common Shares issued pursuant to the conversion of Series B, Series C, Series E and Series I Convertible Preferred Share derivatives, with such derivative liabilities totaling zero, $2,272,500, and 9,825,066 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively, at the time of their conversion. | --- | 6,060,000 | 91,796,043 | |||||||||
| Common Shares issued in payment of Director’s fees totaling $100,071, $110,000 and $110,000 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively | 408,892 | 321,611 | 1,210,583 | |||||||||
| Common shares issued in payment of employee salaries totaling $1,039,000, $849,737 and $368,233 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively. | 4,236,555 | 2,518,668 | 3,439,467 | |||||||||
| Common shares issued in payment of consulting expenses totaling $24,000, $23,999 and $18,836 for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively | 97,467 | 70,169 | 210,018 | |||||||||
| Common shares issued pursuant to warrants exercised | 48,283,968 | 11,985,388 | 16,904,038 | |||||||||
| Common shares issued pursuant to options exercised | 112,500 | 223,334 | 308,333 | |||||||||
| Milestone shares issued pursuant to EPIC Strategic Alliance Agreement totaling $840,000, zero and zero for Fiscal 2016, Fiscal 2015 and Fiscal 2014, respectively | 3,000,000 | --- | --- | |||||||||
| Total Common Shares issued during Fiscal 2016, Fiscal 2015 and Fiscal 2014 | 80,383,651 | 70,918,271 | 185,748,471 | |||||||||
| Common Shares issued at March 31, | 711,544,352 | 631,160,701 | 560,242,430 | |||||||||
F-34
| NOTE 18 | - | PER SHARE INFORMATION |
Basic earnings per share of common stock (“Basic EPS”) is computed by dividing the net income(loss) by the weighted-average number of shares of common stock outstanding. Diluted earnings per share of common stock (“Diluted EPS”) is computed by dividing the net income(loss) by the weighted-average number of shares of common stock and dilutive common stock equivalents and convertible securities then outstanding. GAAP requires the presentation of both Basic EPS and Diluted EPS, if such Diluted EPS is not anti-dilutive, on the face of the Company’s Consolidated Statements of Operations. As the Company had a net loss for Fiscal Year 2016 and 2014, Diluted EPS is not presented as the effect of the Company’s common stock equivalents and convertible securities is anti-dilutive.
Basic EPS is calculated as follows:
Fiscal Year 2016 | Fiscal Year 2015 | Fiscal Year 2014 | ||||||||||
| Numerator | ||||||||||||
| Net Income (Loss) attributable to common shareholders | $ | (9,968,727 | ) | $ | 28,929,674 | $ | (96,575,271 | ) | ||||
| Denominator | ||||||||||||
| Weighted average shares of common stock outstanding | 673,905,485 | 591,214,959 | 463,021,991 | |||||||||
| Net Earnings (Loss) per Share – Basic | $ | (0.01 | ) | $ | 0.05 | $ | (0.21 | ) | ||||
| Potentially dilutive securities excluded from the calculation of diluted loss per share for Fiscal 2016 and 2014 | ||||||||||||
| Stock Options | 581,020 | n/a | 174,359 | |||||||||
| Convertible Preferred Mezzanine Equity | 142,857,143 | n/a | 148,917,143 | |||||||||
| Warrants | 22,926,029 | n/a | 15,782,718 | |||||||||
Diluted Earnings (Loss) per share (for Fiscal 2015) is calculated as follows:
Fiscal Year 2015 | ||||
| Net Income attributable to common shareholders | $ | 28,929,674 | ||
| Adjustments to Net Income | ||||
| Reversal of Change in Value of Warrant Derivatives | (18,447,573 | ) | ||
| Reversal of Change in Value of Convertible Preferred Share Mezzanine Equity | (25,602,370 | ) | ||
| Net loss attributable to common shareholders on a diluted basis | $ | (15,120,269 | ) | |
| Denominator | ||||
| Weighted average shares of common stock outstanding | 591,214,959 | |||
| Dilutive effects of convertible preferred mezzanine equity and warrants | ||||
| Convertible preferred mezzanine equity | 142,857,143 | |||
| Warrants | 22,926,029 | |||
| Stock Options | 581,020 | |||
| Weighted average shares outstanding - diluted | 757,579,152 | |||
| Fully Diluted Earnings (Loss) per Share | $ | (0.02 | ) | |
F-35
| NOTE 19 | - | STOCK-BASED COMPENSATION |
Part or all of the compensation paid by the Company to its Directors and employees consists of the issuance of Common Stock or via the granting of options to purchase Common Stock
Stock-based Director Compensation
The Company’s Director compensation policy instituted in October 2009 includes provisions that Director’s fees are to be paid via the issuance of shares of the Company’s Common Stock, in lieu of cash, with the valuation of such shares being calculated on a quarterly basis and equal to the average closing price of the Company’s common stock for the quarter just ended.
During Fiscal 2016, the Company issued 408,892 shares of Common Stock to its Directors in payment of Director’s fees in the aggregate amount of $100,071 and related to the calendar year ending on December 31, 2015. Please note that the shares issued during Fiscal 2016, include those shares owed and not yet issued at the end of Fiscal Year 2015.
During Fiscal 2015, the Company issued 321,611 shares of Common Stock to its Directors in payment of Director’s fees in the aggregate amount of $110,000 and related to the calendar year ending on December 31, 2014. Please note that the shares issued during Fiscal 2015, include those shares owed and not yet issued at the end of Fiscal Year 2014.
During Fiscal 2014, the Company issued 1,210,583 shares of Common Stock to its Directors in payment of Director’s fees in the aggregate amount of $110,000 and related to the calendar year ending on December 31, 2013. Please note that the shares issued during Fiscal 2014, include those shares owed and not yet issued at the end of Fiscal Year 2013.
As of March 31, 2016, the Company owes its Directors a total of 46,125 shares of Common Stock in payment of Directors Fees totaling $15,000 for the three months ended March 31, 2016. The Company anticipates that these shares of Common Stock will be issued during the fiscal year ended March 31, 2017.
Stock-based Employee Compensation
Employment contracts with the Company’s President and Chief Executive Officer, Chief Financial Officer and certain other employees includes provisions for a portion of each employee’s salaries to be paid via the issuance of shares of the Company’s Common, in lieu of cash, with the valuation of such shares being calculated on a quarterly basis and equal to the average closing price of the Company’s common stock for the quarter just ended.
During Fiscal Year 2016, the Company issued a total of 4,236,555 shares of Common Stock to its President and Chief Executive Officer, Chief Financial Officer and certain other employees in payment of salaries in the aggregate amount of $1,039,000 and related to the calendar year ended December 31, 2015. Please note that the shares issued during Fiscal 2016, include those shares owed and not yet issued at the end of Fiscal 2015.
During Fiscal Year 2015, the Company issued a total of 2,518,668 shares of Common Stock to its President and Chief Executive Officer, Chief Financial Officer and certain other employees in payment of salaries in the aggregate amount of $849,737 and related to the calendar year ended December 31, 2014. Please note that the shares issued during Fiscal 2015, include those shares owed and not yet issued at the end of Fiscal 2014.
F-36
During Fiscal Year 2014, the Company issued a total of 3,439,467 shares of Common Stock to its President and Chief Executive Officer, Chief Financial Officer and certain other employees in payment of salaries in the aggregate amount of $368,233 and related to the period calendar year ended December 31, 2013. Please note that the shares issued during Fiscal 2014, include those shares owed and not yet issued at the end of Fiscal 2013.
As of March 31, 2016, the Company owes its President and Chief Executive Officer, Chief Financial Officer and certain other employees a total of 638,407 shares of Common Stock in payment of salaries totaling $207,500 for the three months ended March 31, 2016, with such amount being recorded in accrued expenses. The Company anticipates that these shares of Common Stock will be issued during the fiscal year ended March 31, 2017.
Stock option based Employee Compensation
During Fiscal 2016, the Company issued, to various employees, options to purchase a total of 360,000 shares Common Stock, in aggregate (the “2016 Options”). The 2016 Options have exercise prices equal to the closing price of the Company’s common stock on the grant date of each such option, such prices ranging from $0.23 per share to $0.415 per share. The 2016 Options vest in equal increments over a three year period which commences one year from the date of grant. The 2016 Options expire ten years from the date of grant. The fair value of the 2016 Options was $129,913 computed using the Black-Scholes options pricing model on the grant date. Such fair value is being amortized by the Company, on a straight line basis, over the vesting period and recorded on the Company’s Statement of Income as “Non-cash compensation through the issuance of stock options”.
During Fiscal 2015, the Company issued, to various employees, options to purchase a total of 2,590,000 shares Common Stock, in aggregate (the “2015 Options”). The 2015 Options have exercise prices equal to the closing price of the Company’s common stock on the grant date of each such option, such prices ranging from $0.26 per share to $0.46 per share. The 2015 Options vest in equal increments over a three year period which commences one year from the date of grant. The 2015 Options expire ten years from the date of grant. The fair value of the 2015 Options was $769,400, computed using the Black-Scholes options pricing model on the grant date. Such fair value is being amortized by the Company, on a straight line basis, over the vesting period and recorded on the Company’s Statement of Income as “Non-cash compensation through the issuance of stock options”.
F-37
During Fiscal 2014, the Company issued, to various employees, options to purchase a total of 3,000,000 shares Common Stock, in aggregate (the “2014 Options”). The 2014 Options have exercise prices equal to the closing price of the Company’s common stock on the grant date of each such option, such prices being $0.07 per share. The 2014 Options vest in equal increments over a three year period which commences one year from the date of grant. The 2014 Options expire ten years from the date of grant. The fair value of the 2015 Options was $202,497, computed using the Black-Scholes options pricing model on the grant date. Such fair value is being amortized by the Company, on a straight line basis, over the vesting period and recorded on the Company’s Statement of Income as “Non-cash compensation through the issuance of stock options”.
The Company measures stock-based compensation cost for options using the Black-Scholes option pricing model. The following table presents a summary of the assumptions used to estimate fair values of the stock options granted during Fiscal 2016, Fiscal 2015 and Fiscal 2014:
| Fiscal Year 2016 | Fiscal Year 2015 | Fiscal Year 2014 | ||||||||||
| Exercise prices | $0.23 - $0.42 | $0.27 - $0.46 | $ | 0.07 | ||||||||
| Options Granted (shares) | 360,000 | 2,590,000 | 3,000,000 | |||||||||
| Risk-free interest rate | 2.1% - 2.2% | 2.2% - 2.8% | 2.5 | % | ||||||||
| Expected volatility | 119% - 120% | 120% - 121% | 130 | % | ||||||||
| Expected dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Forfeiture rate | 2.7 | % | 0.0 | % | 0.0 | % | ||||||
| Expected term (in years) | 10 | 10 | 10 | |||||||||
| Fair value of options granted | $ | 129,913 | $ | 769,421 | $ | 202,497 | ||||||
| Non-cash compensation through issuance of stock options | $ | 333,363 | $ | 260,045 | $ | 82,947 | ||||||
As of March 31, 2016, the total remaining unrecognized non-cash compensation costs related to stock options granted was $462,308. This amount will be recognized as non-cash compensation over the course of the next three fiscal years.
| NOTE 20 | - | STOCK OPTION PLANS |
Under its 2014 Stock Option Plan and prior options plans, the Company may grant stock options to officers, selected employees, as well as members of the Board of Directors and advisory board members. All options have generally been granted at a price equal to or greater than the fair market value of the Company’s Common Stock at the date of the grant. Generally, options are granted with a vesting period of up to three years and expire ten years from the date of grant.
Transactions under the plans for the years indicated were as follows:
| Fiscal Year 2016 | Fiscal Year 2015 | Fiscal Year 2014 | ||||||||||||||||||||||
| Options | Weighted Average Exercise Price | Options | Weighted Average Exercise Price | Options | Weighted Average Exercise Price | |||||||||||||||||||
| Outstanding at beginning of year | 7,642,167 | $ | 0.48 | 5,435,667 | $ | 0.54 | 3,939,000 | $ | 1.18 | |||||||||||||||
| Options Granted | 360,000 | $ | 0.38 | 2,590,000 | $ | 0.31 | 3,000,000 | $ | 0.07 | |||||||||||||||
| Options Exercised | 112,500 | $ | 0.21 | 223,334 | $ | 0.12 | 308,333 | $ | 0.08 | |||||||||||||||
| Options Expired/Forfeited | 280,000 | $ | 0.60 | 160,166 | $ | 0.18 | 1,195,000 | $ | 1.60 | |||||||||||||||
| Outstanding at end of year | 7,609,667 | $ | 0.48 | 7,642,167 | $ | 0.48 | 5,435,667 | $ | 0.54 | |||||||||||||||
The following table summarizes information about stock options outstanding at March 31, 2016:
| Range | Options Outstanding | Weighted Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | Options Exercisable | Weighted Average Exercise Price | |||||||||||||||
| $ 0.01 – 0.25 | 3,766,667 | 6.9 | $ | 0.08 | 2,766,668 | $ | 0.09 | |||||||||||||
| $ 0.26 – 0.50 | 2,650,000 | 8.5 | $ | 0.32 | 783,333 | $ | 1.08 | |||||||||||||
| $ 0.51 – 1.00 | — | — | — | — | ||||||||||||||||
| $ 1.01 – 2.00 | 96,000 | 1.8 | 1.08 | 96,000 | ||||||||||||||||
| $ 2.01 – 3.00 | 1,097,000 | 0.7 | $ | 2.16 | 847,000 | $ | 2.18 | |||||||||||||
| $ 0.01 – 3.00 | 7,609,667 | 6.5 | $ | 0.48 | 4,493,001 | $ | 0.54 | |||||||||||||
F-38
As of March 31, 2016, there were 5,595,066 options available for future grant under our Stock Option Plans.
The aggregate intrinsic value of options outstanding as of March 31, 2016, calculated as the difference between the exercise price and the closing price of the Company’s Common Stock on March 31, 2016 was $904,409. The aggregate intrinsic value of vested options outstanding as of March 31, 2016 was $642,981.
The total intrinsic value of options exercised during the Fiscal 2016, 2015 and 2014 was $22,173, $31,513 and $101,962, respectively.
| NOTE 21 | - | INCOME TAXES |
The components of the credit for income taxes are as follows:
| Year Ended March 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Federal: | ||||||||||||
| Current | $ | — | $ | — | $ | — | ||||||
| Deferred | — | — | — | |||||||||
| State | ||||||||||||
| Current | (4,048 | ) | $ | (3,248 | ) | $ | (3,099 | ) | ||||
| Deferred | — | — | — | |||||||||
| Sale of New Jersey Net Operating Losses | 524,500 | — | 295,710 | |||||||||
| Net Credit for Income Taxes | $ | 520,452 | $ | (3,248 | ) | $ | 292,611 | |||||
The Major components of deferred tax assets and liabilities at March 31, 2016 and 2015 are as follows (amounts in thousands of dollars):
| March 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Federal | ||||||||||||
| Net Operating Loss Carry forward | $ | 27,033 | $ | 24,547 | $ | 19,813 | ||||||
| Valuation Allowance | (27,033 | ) | (24,547 | ) | (19,813 | ) | ||||||
| $ | — | $ | — | $ | — | |||||||
| State | ||||||||||||
| Net Operating Loss Carryforwards | $ | 2,722 | $ | 2,602 | $ | 1,318 | ||||||
| Valuation Allowance | (2,722 | ) | (2,602 | ) | (1,318 | ) | ||||||
| $ | — | $ | — | $ | — | |||||||
At March 31, 2016 and 2015, a 100% valuation allowance is provided, as it is uncertain if the deferred tax assets will provide any future benefits because of the uncertainty about the Company’s ability to generate the future taxable income necessary to use the net operating loss carryforwards.
The company believes that temporary timing differences between accrual and payment of income taxes are not material to the financial position of the Company.
F-39
| NOTE 22 | - | CONCENTRATIONS |
Revenue Concentrations
Five customers accounted for substantially all of the Company’s revenues for Fiscal 2016. Included in these customers are three customers that accounted for approximately 37%, 28% and 27% of revenues each, respectively.
Seven customers accounted for substantially all of the Company’s revenues for Fiscal 2015. Included in these customers are three customers that accounted for approximately 55%, 24% and 11% of revenues each, respectively.
Ten customers accounted for substantially all of the Company’s revenues for Fiscal 2014. Included in these customers are three customers that accounted for approximately 88 percent of revenues for Fiscal 2014.
Accounts Receivable Concentrations
Four customers accounted for substantially all of the Company’s accounts receivable as of March 31, 2016. Included in these customers are three customers that accounted for approximately 54%, 30% and 8% of revenues each, respectively.
Five customers accounted for substantially all of the Company’s accounts receivable as of March 31, 2015. Included in these customers are four customers that accounted for approximately 46%, 26%, 17% and 11% of revenues each, respectively.
Five customers accounted for substantially all of the Company’s accounts receivable as of March 31, 2014. Included in these customers are three customers that accounted for approximately 83% of accounts receivable as of March 31, 2014.
Purchasing Concentrations
Seven suppliers accounted for more than 80% of the Company’s purchases of raw materials for Fiscal 2016. Included in these seven suppliers are three suppliers that accounted for approximately 44%, 20% and 10% of revenues each, respectively.
Seven suppliers accounted for more than 80% of the Company’s purchases of raw materials for Fiscal 2015. Included in these seven suppliers are four suppliers that accounted for approximately 38%, 11%, 10% and 10% of revenues each, respectively.
Seven suppliers accounted for more than 80% of the Company’s purchases of raw materials for Fiscal 2014. Included in these seven suppliers are two suppliers that accounted for approximately 52% of raw material purchases for Fiscal 2014.
F-40
| NOTE 23 | - | COLLABORATIVE AGREEMENT WITH EPIC PHARMA LLC |
On June 4, 2015, the Company entered into the 2015 Epic License Agreement, which provides for the exclusive right to market, sell and distribute, by Epic Pharma LLC (“Epic”) of SequestOx™, an abuse deterrent opioid which employs the Company’s proprietary pharmacological abuse-deterrent technology. Epic will be responsible for payment of product development and pharmacovigilance costs, sales and marketing of SequestOx™, and Elite will be responsible for the manufacture of the product. Under the 2015 Epic License Agreement, Epic will pay Elite non-refundable payments totaling $15 million, with such amount representing the cost of an exclusive license to ELI-200, the cost of developing the product and certain filings and a royalty based on an amount equal to 50% of profits derived from net product sales as defined in the 2015 Epic License Agreement. The initial term of the exclusive right to product development sales and distribution is five years (“Epic Exclusivity Period”); the license is renewable upon mutual agreement at the end of the initial term.
In June 2015, Elite received non-refundable payments totaling $5 million from Epic for the exclusive right to product development sales and distribution of SequestOx™ pursuant to the Epic Collaborative Agreement, under which it agreed to not permit marketing or selling of SequestOx™ within the United States of America to any other party. Such exclusive rights are considered a significant deliverable element of the Epic Collaborative Agreement pursuant to ASC 605-25, Revenue Recognition – Multiple Element Arrangements. These nonrefundable payments represent consideration for certain exclusive rights to ELI-200 and will be recognized ratably over the Epic Exclusivity Period.
In addition, in January 2016, a New Drug Application (“NDA”) for SequestOx™ was filed, thereby earning the Company a non-refundable $2.5 million milestone, pursuant to the 2015 Epic License Agreement. The filing of this NDA represents a significant deliverable element as defined within the Epic Collaborative pursuant to ASC 605-25, Revenue Recognition – Multiple Element Arrangements. Accordingly, the Company has recognized the $2.5 million milestone, which was paid by Epic and related to this deliverable as income during the year ended March 31, 2016.
In total, during Fiscal 2016, the Company received payments totaling $7.5 million pursuant to the 2015 Epic License Agreement, with all amounts being non-refundable. An additional $7.5 million is due upon approval by the FDA of the NDA filed for SequestOx™, and license fees based on commercial sales of SequestOx™. Revenues relating to these additional amounts due under the 2015 Epic License Agreement will be recognized as the defined elements are completed and collectability is reasonably assured.
| NOTE 24 | - | RELATED PARTY TRANSACTION AGREEMENTS WITH EPIC PHARMA LLC |
The Company has entered into two agreements with Epic which constitute agreements with a related party due to the management of Epic including a member on our Board of Directors.
On June 4, 2015, the Company entered into the 2015 Epic License Agreement (please see Note 23 above)
The 2015 Epic License Agreement includes milestone payments totaling $10 million upon the filing with and approval of a New Drug Application (“NDA”) with the FDA. The Company has determined these milestones to be substantive, with such assessment being made at the inception of the 2015 Epic License Agreement, and based on the following:
| · | The Company’s performance is required to achieve each milestone; and |
| · | The milestones will relate to past performance, when achieved; and |
| · | The milestones are reasonable relative to all of the deliverables and payment terms within the 2015 Epic License Agreement. |
F-41
After marketing authorization is received from the FDA, Elite will receive a license fee which is based on profits achieved from the commercial sales of ELI-200. On January 14, 2016, the Company filed an NDA with the FDA for SequestOx™, thereby earning a $2.5 million milestone pursuant to the 2015 Epic License Agreement. The Company has received payment of this amount from Epic. There can be no assurances of the Company receiving marketing authorization for SequestOx™, and accordingly, there can be no assurances that the Company will earn and receive the additional $7.5 million or future license fees. If the Company does not receive these payments or fees, it will materially and adversely affect our financial condition.
On October 2, 2013, Elite executed the Epic Pharma Manufacturing and License Agreement (the “Epic Generic Agreement”), which granted rights to Epic to manufacture twelve generic products whose ANDA’s are owned by Elite, and to market, in the United States and Puerto Rico, six of these products on an exclusive basis, and the remaining six products on a non-exclusive basis. These products will be manufactured at Epic, with Epic being responsible for the manufacturing site transfer supplements that are a prerequisite to each product being approved for commercial sale. In addition, Epic is responsible for all regulatory and pharmacovigilance matters, as well as all marketing and distribution activities. Elite has no further obligations or deliverables under the Epic Generic Agreement.
Pursuant to the Epic Generic Agreement, Elite will receive $1.8 million, payable in increments that require the commercialization of all six exclusive products if the full amount is to be received, plus license fees equal to a percentage that is not less than 50% and not greater than 60% of profits achieved from commercial sales of the products, as defined in the Epic Generic Agreement. While Epic has launched four of the six exclusive products and Elite has collected $1.0 million of the $1.8 million total fee, collection of the remaining $800k is contingent upon Epic filing the required supplements with and receiving approval from the FDA for the remaining exclusive generic products. There can be no assurances of Epic filing these supplements, or getting approval of any supplements filed. Accordingly, there can be no assurances of Elite receiving the remaining $800k due under the Epic Generic Agreement, or future license fees related thereto. Please also note that all commercialization, regulatory, manufacturing, marketing and distribution activities are being conducted solely by Epic, without Elite’s participation.
Both the 2015 Epic License Agreement and the Epic Generic Agreement contain license fees that will be earned and payable to the Company, after the FDA has issued marketing authorization(s) for the related product(s). License fees are based on commercial sales of the products achieved by Epic and calculated as a percentage of net sales dollars realized from such commercial sales. Net sales dollars consist of gross invoiced sales less those costs and deductions directly attributable to each invoiced sale, including, without limitation, cost of goods sold, cash discounts, Medicaid rebates, state program rebates, price adjustments, returns, short date adjustments, charge backs, promotions and marketing costs. The rate applied to the net sales dollars to determine license fees due to the Company is equal to an amount negotiated and agreed to by the parties to each agreement, with the following significant factors, inputs, assumptions and methods, without limitation, being considered by either or both parties:
F-42
| · | Assessment of the opportunity for each product in the market, including consideration of the following, without limitation: market size, number of competitors, the current and estimated future regulatory, legislative and social environment for abuse deterrent opioids and the other generic products to which the underlying contracts are relevant; |
| · | Assessment of various avenues for monetizing SequestOx™ and the twelve ANDA’s owned by the Company, including the various combinations of sites of manufacture and marketing options; |
| · | Elite’s resources and capabilities with regards to the concurrent development of abuse deterrent opioids and expansion of its generic business segment, including financial and operational resources required to achieve manufacturing site transfers for twelve approved ANDA’s; |
| · | Capabilities of each party with regards to various factors, including, one or more of the following: manufacturing, marketing, regulatory and financial resources, distribution capabilities, ownership structure, personnel, assessments of operational efficiencies and entity stability, company culture and image; |
| · | Stage of development of SequestOx™ and manufacturing site transfer and regulatory requirements relating to the commercialization of the generic products at the time of the discussions/negotiations, and an assessment of the risks, probability and time frames for achieving marketing authorizations from the FDA for each product. |
| · | Assessment of consideration offered; and |
| · | Comparison of the above factors among the various entities with whom the Company was engaged in discussions relating to the commercialization of SequestOx™ and the manufacture/marketing of the twelve generics related to the Epic Generic Agreement. |
Please note that this transaction is not to be considered as an arms-length transaction.
| NOTE 25 | - | TRANSACTIONS WITH RELATED PARTIES – NASRAT HAKIM AND MIKAH PHARMA LLC |
On August 1, 2013, Elite Laboratories Inc. (“Elite Labs”), our wholly owned subsidiary, executed an asset purchase agreement (the “Mikah Purchase Agreement”) with Mikah Pharma LLC (“Mikah”), an entity that is wholly owned by Mr. Nasrat Hakim, who, in conjunction with this transaction, was appointed as our Chief Executive Officer, President and a Director on August 2, 2012, and acquired from Mikah a total of 13 Abbreviated New Drug Applications (“ANDAs”) consisting of 12 ANDAs approved by the FDA and one ANDA under active review with the FDA, and all amendments thereto (the “Acquisition”) for aggregate consideration of $10,000,000, inclusive of imputed interest payable pursuant to a non-interest bearing, secured convertible note due in August 2016 (the “Mikah Note”). The Mikah Note was amended on February 7, 2014 to make it convertible into shares of the Company’s Series I Convertible Preferred Stock.
The Mikah Note, as amended, was interest free and due and payable on the third anniversary of its issuance. Subject to certain limitations, the principal amount of the Mikah Note was convertible at the option of Mikah into shares of Common Stock at a rate of $0.07 (approximately 14,286 shares per $1,000 in principal amount), the closing market price of the Company’s Common Stock on the date that the asset purchase agreement and Note were executed and/or into shares of the Company’s Series I Convertible Preferred Stock at the rate of 1 share of Series I Preferred Stock for each $100,000 of principal owed on the Mikah Note. The conversion rate was adjustable for customary corporate actions such as stock splits and, subject to certain exclusions, includes weighted average anti-dilution for common stock transactions at prices below the then applicable conversion rate. Pursuant to a security agreement (the “Security Agreement”), repayment of the Mikah Note was secured by the ANDAs acquired in the Acquisition.
On February 7, 2014, Mikah converted the principal amount of $10,000,000, representing the entire principal balance due under the Mikah Note, into 100 shares of the Company’s Series I Preferred Stock.
F-43
On August 27, 2010, Elite executed an asset purchase with Mikah (the “Naltrexone Agreement”). Pursuant to the Naltrexone Agreement, Elite acquired from Mikah the Abbreviated New Drug Application number 75-274 (Naltrexone Hydrochloride Tablets USP, 50 mg), and all amendments thereto (the “ANDA”), that have to date been filed with the FDA seeking authorization and approval to manufacture, package, ship and sell the products described in the ANDA within the United States and its territories (including Puerto Rico) for aggregate consideration of $200,000. In lieu of cash, Mikah agreed to accept from Elite product development services to be performed by Elite, and entered into a Development and License Agreement dated August 27, 2010 between the Company and Mikah (the “Mikah Development Agreement”).
The manufacturing of Naltrexone 50mg was successfully transferred to the Company’s Northvale facility, and the first commercial shipment of this product was made in September 2013.
On January 28, 2015, the Mikah Development Agreement was terminated by mutual agreement of the parties thereto. Pursuant to the Mikah Development Agreement, Mikah made advance consideration payments to the Company totaling $200,000 in exchange for product development services to be provided at a future date. Subsequent to the execution of the Mikah Development Agreement, and before any development milestones were achieved, the sole owner of Mikah, Mr. Nasrat Hakim, became the President and Chief Executive Officer of the Company. Mikah has accordingly ceased operating and is in the process of winding down and liquidating its assets.
Any further development of the product related to the Mikah Development Agreement will belong to the Company, although there can be no assurances that such development will occur or be successful.
The Mikah Development Agreement requires that the consideration paid in advance to the Company be refunded in the event of no milestones being achieved. Mr. Hakim, as owner of Mikah, has directed that the $200,000 refund due to Mikah not be paid currently, but rather be added to the amounts due under the Hakim Credit Line.
Please note that this transaction is not to be considered as an arms-length transaction.
| NOTE 26 | - | MANUFACTURING, LICENSE AND DEVELOPMENT AGREEMENTS |
The Company has entered into the following active agreements:
| · | License agreement with Precision Dose, dated September 10, 2010 (the “Precision Dose License Agreement”) |
| · | Manufacturing and Supply Agreement with Ascend Laboratories Inc., dated June 23, 2011 and as amended on September 24, 2012 and January 19, 2015 (the “Ascend Manufacturing Agreement”) and |
| · | Development agreement with Akorn Pharmaceuticals, dated January 10, 2011 (the “Akorn Agreement”). |
F-44
The Precision Dose Agreement provides for the marketing and distribution, by Precision Dose and its wholly owned subsidiary, TAGI Pharma, of Phentermine 37.5mg tablets (launched in April 2011), Phentermine 15mg capsules (launched in April 2013), Phentermine 30mg capsules (launched in April 2013), Hydromorphone 8mg tablets (launched in March 2012), Naltrexone 50mg tablets (launched in September 2013) and certain additional products that require approval from the FDA which has not been received. Precision Dose will have the exclusive right to market these products in the United States and Puerto Rico and a non-exclusive right to market the products in Canada. Pursuant to the Precision Dose License Agreement, Elite received $200k at signing, and is receiving milestone payments and a license fee which is based on profits achieved from the commercial sale of the products included in the agreement.
Revenue from the $200k payment made upon signing of the Precision Dose Agreement is being recognized over the life of the Precision Dose Agreement.
The milestones, totaling $500k, consist of amounts due upon the first shipment of each identified product, as follows: Phentermine 37.5mg tablets ($145k), Phentermine 15 & 30mg capsules ($45k), Hydromorphone 8mg ($125k), Naltrexone 50mg ($95k) and the balance of $95k due in relation to the first shipment of generic products which still require marketing authorizations from the FDA, and to which there can be no assurances of such marketing authorizations being granted and accordingly there can be no assurances that the Company will earn and receive these milestone amounts. These milestones have been determined to be substantive, with such determination being made by the Company after assessments based on the following:
| · | The Company’s performance is required to achieve each milestone; and |
| · | The milestones will relate to past performance, when achieved; and |
| · | The milestones are reasonable relative to all of the deliverables and payment terms within the Precision Dose License Agreement. |
The license fees provided for in the Precision Dose Agreement are calculated as a percentage of net sales dollars realized from commercial sales of the related products. Net sales dollars consist of gross invoiced sales less those costs and deductions directly attributable to each invoiced sale, including, without limitation, cost of goods sold, cash discounts, Medicaid rebates, state program rebates, price adjustments, returns, short date adjustments, charge backs, promotions and marketing costs. The rate applied to the net sales dollars to determine license fees due to the Company is equal to an amount negotiated and agreed to by the parties to the Precision Dose License Agreement, with the following significant factors, inputs, assumptions and methods, without limitation, being considered by either or both parties:
| · | Assessment of the opportunity for each generic product in the market, including consideration of the following, without limitation: market size, number of competitors, the current and estimated future regulatory, legislative and social environment for each generic product, and the maturity of the market; |
| · | Assessment of various avenues for monetizing the generic products, including the various combinations of sites of manufacture and marketing options; |
| · | Capabilities of each party with regards to various factors, including, one or more of the following: manufacturing resources, marketing resources, financial resources, distribution capabilities, ownership structure, personnel, assessment of operational efficiencies and stability, company culture and image; |
| · | Stage of development of each generic products, all of which did not have FDA approval at the time of the discussions/negotiations and an assessment of the risks, probability and time frame for achieving marketing authorizations from the FDA for the products; |
| · | Assessment of consideration offered by Precision and other entities with whom discussions were conducted; and |
F-45
| · | Comparison of the above factors among the various entities with whom the Company was engaged in discussions relating to the commercialization of the generic products. |
The Ascend Manufacturing Agreement provides for the manufacturing by Elite of Methadone 10mg for supply to Ascend Laboratories LLC (“Ascend”). Ascend is the owner of the approved ANDA for Methadone 10mg, and the Northvale Facility is an approved manufacturing site for this ANDA. There are no license fees or milestones relating to this agreement. All revenues earned are recognized as manufacturing revenues on the date of shipment of the product, when title for the goods is transferred, and for which the price is agreed to and it has been determined that collectability is reasonably assured. The initial shipment of Methadone 10mg pursuant to the Ascend Manufacturing Agreement occurred in January 2012.
The Akorn Agreement was executed on January 10, 2011 between Hi-Tech Pharmacal Inc. (subsequently acquired by Akorn Pharmaceuticals) and provides for Elite to develop an intermediate product which will be incorporated into the finished formulation of a generic version of a prescription product for Akorn Pharmaceuticals (“Akorn”). There is currently no development activity being conducted pursuant to this agreement and there was no activity during the last fiscal year as well. There can be no assurances that development activities will resume or that a resumption of development activities will result in the successful development of the relevant product.
| NOTE 27 | - | CONVERSIONS OF PREFERRED STOCK MEZZANINE EQUITY TO COMMON STOCK |
The Certificate of Designations of the Series I Convertible Preferred Stock Mezzanine Equity (the “Series I Mezzanine Equity”), includes provisions entitling its rights to convert shares of the Series I Mezzanine Equity into shares of Common Stock.
Conversions of Series I Mezzanine Equity to Common Stock during Fiscal 2016 and Fiscal 2015 are summarized as follows:
| Fiscal 2016 | Fiscal 2015 | |||||||
| Series I Preferred Derivatives | ||||||||
| Number of Derivative Shares Converted | — | 4.242 | ||||||
| Number of Common Shares issued pursuant to conversion | — | 6,060,000 | ||||||
| Value of Preferred Derivative shares at time of conversion (represents decrease in derivative liability resulting from conversions) | — | 2,272,500 | ||||||
| Change in value of preferred share derivative liability recorded at time of conversion | — | (303,000 | ) | |||||
| Par value of Common Shares issued | — | 6,060 | ||||||
| Additional paid in capital recorded as a result of the conversions | — | 2,266,440 | ||||||
F-46
Please also see Note 15 for further details on the Series I Mezzanine Equity.
| NOTE 28 | - | CONTINGENCIES |
As part of the Company’s efforts to ensure the retention and continuity of key employees, officers and directors in the event of a change of control of the ownership of the Company, the Board of Directors passed a resolution whereby, in the event of a change in control of the ownership of the Company, key executives would receive an amount equal to twelve months of such executive’s salary, and certain Directors and managers would receive an amount equal to six months of such Director’s or managers fees or salaries, as applicable. In addition, the resolution passed provided for the immediate vesting of outstanding options, in the event of a change of control.
| NOTE 29 | - | RIGHTS PLAN |
On November 15, 2013, our board of directors declared a dividend distribution of one right for each outstanding share of our common stock and one right for each share of Common Stock into which any of our outstanding Preferred Stock is convertible, to stockholders of record at the close of business on that date. Each Right entitles the registered holder to purchase from us one “Unit” consisting of one one-millionth (1/1,000,000) of a share of Series H Junior Participating preferred stock, par value $0.01 per share (the “H Preferred Stock”), at a purchase price of $2.10 per Unit, subject to adjustment, and may be redeemed prior to November 15, 2023, the expiration date, at $0.000001 per Right, unless earlier redeemed by the Company. The Rights generally are not transferable apart from the common stock and will not be exercisable unless and until a person or group acquires or commences a tender or exchange offer to acquire, beneficial ownership of 15% or more of our common stock. However, for Mr. Hakim, our Chief Executive Officer, the Rights Plan's the 15% threshold excludes shares beneficially owned by him as of November 15, 2013 and all shares issuable to him pursuant to his employment agreement and the Mikah Note. The description and terms of the Rights are set forth in a Rights Agreement (“Rights Agreement”) between the Company and American Stock Transfer & Trust Company, LLC, as Rights Agent.
| NOTE 30 | - | LEGAL PROCEEDINGS |
In the ordinary course of business, we may be subject to litigation from time to time. Except as discussed below, there is no current, pending or, to our knowledge, threatened litigation or administrative action to which we are a party or of which our property is the subject (including litigation or actions involving our officers, directors, affiliates, or other key personnel, or holders of record or beneficially of more than 5% of any class of our voting securities, or any associate of such party) which in our opinion has, or is expected to have, a material adverse effect upon our business, prospects, financial condition or operations.
Arbitration with Precision Dose Inc.
On May 9, 2014, Precision Dose Inc, the parent company of TAGI Pharmaceuticals, Inc., commenced an arbitration against the Company alleging that the Company failed to properly supply, price and satisfy gross profit minimums regarding Phentermine 37.5mg tablets, as required by the parties' agreements. Elite denied Precision Dose's allegations and has counterclaimed that Precision Dose is no longer entitled to exclusivity rights with respect to Phentermine 37.5mg tablets, and is responsible for certain costs, expenses, price increases and lost profits relating to Phentermine 37.5mg tablets and the parties' agreements. The parties have reached agreement in settlement of these issues, with Precision Dose agreeing to pay certain amounts to the Company in exchange for Elite agreeing to restore exclusivity rights with respect to Phentermine 37.5mg tablets, subject to certain defined conditions. Both parties have been complying with the agreed settlement terms and the Company has notified the Arbitrator of this settlement, requesting the issuance of proceeding termination documents.
F-47
Generally Accepted Accounting Principles require that a contingency loss may only be recognized if the event is (i) probable and (ii) the amount of the loss can be reasonably estimated. There were no liabilities meeting this criteria at March 31, 2016.
| NOTE 31 | - | QUARTERLY FINANCIAL INFORMATION (UNADITED) |
The Company’s consolidated results of operations are shown below:
| (In thousands, except per share data) | Fourth Quarter | Third Quarter | Second (restated) | First (restated) | ||||||||||||
| Fiscal year ended March 31, 2016 | ||||||||||||||||
| Total revenues | $ | 5,195 | $ | 2,194 | $ | 2,947 | $ | 2,163 | ||||||||
| Costs of revenues | 1,036 | 836 | 1,415 | 1,197 | ||||||||||||
| Gross Profit | 4,159 | 1,358 | 1,532 | 966 | ||||||||||||
| Operating Expenses | 3,588 | 4,071 | 5,299 | 3,373 | ||||||||||||
| Income (Loss) from Operations | 571 | (2,713 | ) | (3,767 | ) | (2,407 | ) | |||||||||
| Other income (expense) | 7,408 | (9,520 | ) | 2,086 | 7,139 | |||||||||||
| Income tax (credit) expense | (520 | ) | — | — | — | |||||||||||
| Net Income | 8,499 | (12,233 | ) | (1,681 | ) | 4,732 | ||||||||||
| Change in carrying value of convertible preferred mezzanine equity | 14,142 | (24,786 | ) | (5,071 | ) | 6,429 | ||||||||||
| Net income attributable to common shareholders | 22,641 | (37,019 | ) | (6,753 | ) | 11,161 | ||||||||||
| Earnings per share – basic | $ | 0.03 | $ | (0.05 | ) | $ | (0.01 | ) | $ | 0.02 | ||||||
| Earnings per share - Diluted | $ | 0.00 | $ | (0.05 | ) | $ | (0.01 | ) | $ | (0.00 | ) | |||||
| (In thousands, except per share data) | Fourth (restated) | Third (restated) | Second (restated) | First (restated) | ||||||||||||
| Fiscal year ended March 31, 2015 | ||||||||||||||||
| Total revenues | 1,234 | 1,363 | 1,256 | 1,162 | ||||||||||||
| Costs of revenues | 904 | 700 | 682 | 729 | ||||||||||||
| Gross Profit | 331 | 663 | 575 | 433 | ||||||||||||
| Operating Expenses | 5,788 | 3,221 | 4,636 | 4,864 | ||||||||||||
| Income (Loss) from Operations | (5,457 | ) | (2,557 | ) | (4,061 | ) | (4,431 | ) | ||||||||
| Other income (expense) | (898 | ) | 9,974 | 10,310 | 2,338 | |||||||||||
| Income tax (credit) expense | (3 | ) | — | — | — | |||||||||||
| Net Income | (6,350 | ) | 7,417 | 6,248 | (2,094 | ) | ||||||||||
| Change in carrying value of convertible preferred mezzanine equity | (2,715 | ) | 13,600 | 15,132 | (2,308 | ) | ||||||||||
| Net income attributable to common shareholders | (9,065 | ) | 21,017 | 21,380 | (4,402 | ) | ||||||||||
| Earnings (Loss) per share – basic | $ | (0.01 | ) | $ | 0.03 | $ | 0.04 | $ | (0.01 | ) | ||||||
| Earnings (Loss) per share - diluted | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.01 | ) | ||||
F-48
| (In thousands, except per share data) | Fourth (restated) | Third (restated) | Second (restated) | First (restated) | ||||||||||||
| Fiscal year ended March 31, 2014 | ||||||||||||||||
| Total revenues | 1,028 | 1,693 | 1,159 | 722 | ||||||||||||
| Costs of revenues | 1,046 | 995 | 617 | 579 | ||||||||||||
| Gross Profit | (18 | ) | 699 | 542 | 143 | |||||||||||
| Operating Expenses | 2,365 | 1,936 | 1,229 | 1,118 | ||||||||||||
| Income (Loss) from Operations | (2,384 | ) | (1,237 | ) | (687 | ) | (976 | ) | ||||||||
| Other income (expense) | (31,652 | ) | 175 | (6,887 | ) | 2,095 | ||||||||||
| Income tax (credit) expense | (295 | ) | — | 2 | — | |||||||||||
| Net Income | (33,741 | ) | (1,061 | ) | (7,576 | ) | 1,119 | |||||||||
| Change in carrying value of convertible preferred mezzanine equity | (53,056 | ) | 1 | (2,061 | ) | (197 | ) | |||||||||
| Net income attributable to common shareholders | (86,798 | ) | (1,062 | ) | (9,638 | ) | 922 | |||||||||
| Earnings (Loss) per share – basic | $ | (0.16 | ) | $ | (0.00 | ) | $ | (0.02 | ) | $ | 0.00 | |||||
| Earnings (Loss) per share - diluted | $ | (0.16 | ) | $ | (0.00 | ) | $ | (0.02 | ) | $ | (0.00 | ) | ||||
| NOTE 32 | - | ADVERTISING COSTS |
The Company's advertising costs for Fiscal 2016, Fiscal 2015 and Fiscal 2014 were immaterial.
| NOTE 33 | - | SUBSEQUENT EVENTS |
The Company has evaluated subsequent events from the balance sheet date through June 15, 2016, the date the accompanying financial statements were issued. The following are material subsequent events:
Common Stock sold pursuant to the LPC-40 Purchase Agreement
Subsequent to March 31, 2016 and up to June 7, 2016 (the latest practicable date), a total of 5.5 million shares of Common Stock were sold and 0.1 million additional commitment shares were issued, pursuant to the LPC-40 Purchase Agreement. Proceeds received from such transactions totaled $1.7 million.
Common Stock issued pursuant to the exercise of cash warrants
Subsequent to March 31, 2016 and up to June 7, 2016 (the latest practicable date), a total of 9.3 million shares of Common Stock were issued pursuant to the exercise of cash warrants. Proceeds received from such warrant exercises totaled $0.6 million.
Repayment of Hakim Credit Line
On May 24, 2016 all amounts due and owing pursuant to the Hakim Credit Line, which expired on March 31, 2016, were paid in full. These payments, which totaled $798,227 consisted of principal due of $718,309 plus accrued interest totaling $79,918. As of May 24, 2016 there are no amounts due and owing under the Hakim Loan Agreement and the Hakim Loan Agreement is expired. Please also see Note 10 above.
F-49