Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - OPGEN INC | v434805_ex32-1.htm |
| EX-31.1 - EXHIBIT 31.1 - OPGEN INC | v434805_ex31-1.htm |
| EX-23.1 - EXHIBIT 23.1 - OPGEN INC | v434805_ex23-1.htm |
| EX-31.2 - EXHIBIT 31.2 - OPGEN INC | v434805_ex31-2.htm |
| EX-21.1 - EXHIBIT 21.1 - OPGEN INC | v434805_ex21-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)
| x | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015
| ¨ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______
to _______.
Commission file number 001-31812
OPGEN, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 06-1614015 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 708 Quince Orchard Road, Suite 205 | ||
| Gaithersburg, Maryland | 20878 | |
| (Address of principal executive offices) | (Zip Code) |
(240) 813-1260
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, par value $0.01 per share | The NASDAQ Capital Market | |
| Warrants, exercisable for one share of common stock |
The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of
the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | |
| Non-accelerated filer ¨ (Do not check if a smaller reporting company) |
Smaller reporting company x |
Indicate by check mark whether registrant is a shell company (as defined in Rule 12b-2 of the Act). YES ¨ NO x
The aggregate market value of the voting common stock held by non-affiliates of the registrant as of June 30, 2015, was $39.9 million (based upon the last reported sale price of $3.72 per share on June 30, 2015, on The NASDAQ Capital Market).
As of March 17, 2016, 12,574,303 shares of common stock of the registrant were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the registrant’s 2016 annual meeting of stockholders to be filed within 120 days after the end of the period covered by this Annual Report on Form 10-K are incorporated by reference into Part III of this Annual Report on Form 10-K.
OPGEN, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2015
TABLE OF CONTENTS
| 2 |
INFORMATION REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K for the year ended December 31, 2015 (the “Annual Report”) and certain information incorporated herein by reference contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). In this Annual Report, we refer to OpGen, Inc. as the “Company,” “we,” “our” or “us.” All statements other than statements of historical facts contained herein, including statements regarding our future results of operations and financial position, strategy and plans, and our expectations for future operations, are forward-looking statements. The words “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “design,” “intend,” “expect” or the negative version of these words and similar expressions are intended to identify forward-looking statements.
We have based these forward-looking statements on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, strategy, short- and long-term business operations and objectives, and financial needs. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in Part I Item 1A “Risk Factors.” In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances included herein may not occur, and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
| · | the commercialization of our current products, including our QuickFISH® and PNA FISH diagnostic products for infectious diseases, Acuitas® MDRO test products and completed development and commercialization of our Acuitas Lighthouse™ bioinformatics products and services; |
| · | integration of the operations of AdvanDx, Inc. acquired by merger on July 14, 2015; |
| · | anticipated trends and challenges in our business and the competition that we face; |
| · | the execution of our business plan and our growth strategy; |
| · | our expectations regarding the size of and growth in potential markets; |
| · | changes in laws or regulations applicable to our business, including potential regulation by the FDA; |
| · | our ability to develop and commercialize new products and the timing of commercialization; |
| · | our liquidity and working capital requirements, including our long-term future cash requirements beyond the next 12 months; and |
| · | our expectations regarding future revenue and expenses. |
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. In addition, neither we nor any other person assumes responsibility for the accuracy and completeness of any of these forward-looking statements. Any forward-looking statement made by us in this Annual Report speaks only as of the date on which it is made. We disclaim any duty to update any of these forward looking statements after the date of this Annual Report to confirm these statements to actual results or revised expectations.
These factors should not be construed as exhaustive and should be read in conjunction with our other disclosures, including but not limited to the risk factors described in Part I, Item 1A of this Annual Report. Other risks may be described from time to time in our filings made under the securities laws. New risks emerge from time to time. It is not possible for our management to predict all risks. All forward-looking statements in this Annual Report speak only as of the date made and are based on our current beliefs and expectations. We undertake no obligation to update or revise any forward-looking statement, whether as a result of new information, future events or otherwise.
NOTE REGARDING TRADEMARKS
We own various U.S. federal trademark registrations and applications and unregistered trademarks and servicemarks, including OpGen®, Acuitas®, Acuitas LighthouseTM Argus®, MapIt®, MapSolver™, AdvanDx®, QuickFISH® and PNA FISH®. All other trademarks, servicemarks or trade names referred to in this Annual Report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Annual Report are sometimes referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies, products or services.
| 3 |
Please refer to the Glossary beginning on page 22 of this Business section for definitions or descriptions of scientific, diagnostic, health care and regulatory terms used in this Annual Report.
Overview
We are a precision medicine company using molecular diagnostics and informatics to combat infectious disease. We are developing molecular information solutions to combat infectious disease in global healthcare settings, helping to guide clinicians with more rapid information about life threatening infections, improve patient outcomes, and decrease the spread of infections caused by multidrug-resistant microorganisms. Our proprietary DNA tests and bioinformatics address the rising threat of antibiotic resistance by helping physicians and healthcare providers optimize patient care decisions and protect the hospital biome through customized screening and surveillance solutions.
Our molecular information solution combines Acuitas® DNA tests, Acuitas Lighthouse™ bioinformatics systems, CLIA lab services for MDRO surveillance, and a proprietary data warehouse including genomic data matched with antibiotic susceptibility information for microbes and patient information from healthcare providers. We are working to deliver our molecular information solution to a global network of customers and partners. Our Acuitas DNA tests provide rapid microbial ID, and antibiotic resistance gene information. These products include the QuickFISH® family of FDA-cleared and CE-marked diagnostics used to rapidly detect pathogens in positive blood cultures, the Acuitas MDRO Gene Test to detect, type, track, and trend antibiotic resistant organisms in real-time and our rapid antibiotic resistance test in development. We are working to provide actionable, precise diagnostic information powered by pathogen surveillance data collected through hospital screening programs and a network of hospital and public health laboratories globally. The Acuitas Lighthouse™ data warehouse is a cloud-based HIPAA compliant solution that combines clinical lab test results with patient and hospital information and provides analytics to help manage MDROs in the hospital and patient care environment.
There is rising global concern about the profound health and macroecnomic consequences if the growing threat of antimicrobial resistance is not tackled. Drug resistant infections currently claim at least 50,000 lives each year in the United States and Europe alone, with many hundreds of thousands more dying in other areas of the world. Recognizing this emerging threat, the White House issued a National Action Plan for Combating Antibiotic Resistance Bacteria in March 2015. The National Action Plan aims to achieve major reductions in the incidence of these urgent and serious threats and improvements in antibiotic stewardship during the next five years. The 2016 U.S. government budget includes approximately $1 billion to help combat drug resistant infections. Three key areas have been highlighted for investment: rapid diagnostics, surveillance, and new antibiotics. We are focused in rapid diagnostics where our tests help identify microorganisms and determine their antibiotic resistance genes and susceptibility. Through the use of our Acuitas Lighthouse analytics we are working to provide antibiotic decision support tools to help physicians interpret and act on this information. A second area of our focus is surveillance of microbial infections and colonization with MDROs in the hospital environment. These products and services are designed to help enable effective response to resistant organisms and to help control MDRO transmission and outbreaks in the hospital.
We believe that the diagnostic paradigm for management of drug resistant infections is poised for change. In acute care settings, initial treatment today relies heavily on initial use of broad spectrum antibiotics on an empiric basis. It is common for patients to receive the antibiotic vancomycin for treatment of potential Gram positive infections such as Staphylococcus and the antibiotic cefipime for treatment of potential Gram negative infections from organisms such as Escherichia coli, Klebsiella pneumoniae, or Pseudomonas. These powerful antibiotics are often prescribed without previous knowledge of whether the organism they are intended to treat is present. Current methods require 2-4 days to determine the organism ID and antibiotic susceptibility. During this period in advance of receiving the correct diagnosis, patients are often over treated or treated with the wrong antibiotic leading to potentially undesirable outcomes such as morbidity from expanded infection, drug resistance, and opportunistic C. difficile infections. If the diagnosis is that the initial empiric antibiotic therapy was incorrect, a new therapy must be chosen often resulting in poor clinical outcomes, additional length of stay, and increased health care costs.
| 4 |
Improved diagnostics for detection of resistant bacteria and characterization of resistance patterns will help healthcare providers make optimal treatment decisions and assist public health officials in taking action to prevent and control disease. Improved and more rapid diagnostics will also help decrease unnecessary or inappropriate use of antibiotics. Optimal precision medicine tests for combatting infectious disease will provide diagnostic information in the first hours after presentation of the acutely ill patient in order to impact initial antibiotic selection decisions. Conventional microbiology methods have been largely unchanged, and we believe that it is unlikely that they will be adapted to provide rapid one-hour diagnostic tests for high resolution microbial analysis. DNA analysis technology, such as our Acuitas DNA tests, has the potential to help revolutionize rapid diagnostics for microbiology. DNA tests are highly accurate and can be performed in just 30 minutes to an hour. Our FDA-cleared QuickFISH rapid pathogen ID tests are examples of such rapid detection technology. In addition, DNA sequencing technology now makes it possible to sequence the entire genome of microbes for subsequent analysis, antibiotic selection decision making software, and microbe tracking.
Our suite of DNA-based products and products in development are intended to provide actionable, precise diagnostics powered by microbial surveillance data. The high resolution Acuitas DNA tests use multiplex PCR to help provide reliable and accurate detection of drug resistance. The QuickFISH tests are powered by PNA technology and provide rapid pathogen ID, typically in less than 30 minutes from a positive blood culture result. The Acuitas MDRO Gene Test is used for determining if ICU patients are colonized with MDROs. Positive samples are confirmed using microbiological methods and the Acuitas Resistome™ test for high resolution genotyping. Test results are maintained in the Acuitas Lighthouse™ data warehouse for subsequent interpretation by physicians and healthcare providers.
We are developing a new disruptive one-hour-testing paradigm that we believe could help address many of the current issues with testing for antibiotic resistance. Results from a new high resolution Acuitas DNA test designed to detect the key resistome profiles of Gram negative organisms will be informed by a smart cloud based clinical database that will provide molecular profiles to aid initial antibiotic selection and clinical decision making. Our proprietary Acuitas Lighthouse profiles distill large amounts of data into one actionable profile. We believe our disruptive approach will be globally applicable and could be an important new weapon in the fight against drug-resistant bacteria. The figure below describes the potential workflow and anticipated results from our new testing approach.
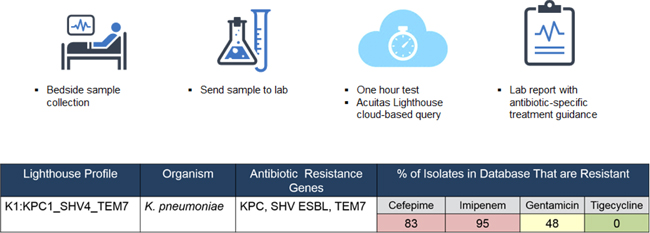
Our Strategy
We are using our current products and products in development to provide precision medicine solutions for combatting infectious diseases with a focus on developing diagnostic tests for rapid pathogen identification and genetic profiling, antibiotic resistance analysis and advanced bioinformatics to store and analyze MDRO and other infectious disease data for hospitals, out-patient settings and other health care providers. We believe more rapid genetic identification methods will reduce morbidity from MDROs, reduce health care costs through reduced length of stay, and assist in the identification of targeted antibiotic therapy. Current conventional microbiology, largely unchanged in 50 years, requires one to two days for growth and phenotypic analysis and often leads to the use of broad spectrum antibiotic therapy in the early stages of infection. Our current QuickFISH, PNA FISH and XpressFISH FDA-cleared, CE-marked diagnostic tests can accelerate accurate pathogen identification by one to three days when compared to conventional methods by providing identification of the pathogen within 20 to 90 minutes of positive blood culture results. We are working to:
| 5 |
| · | Expand our rapid diagnostics product offerings through development of additional Acuitas Gene Tests, with a goal of achieving one hour antibiotic resistance analysis; |
| · | Grow our Acuitas Lighthouse bioinformatics platform to serve as a data warehouse for resistance and susceptibility data in a hospital, hospital system, or broader community; |
| · | Continue development of our Acuitas Lighthouse Portal and decision-making software and work to install Acuitas Lighthouse Portals at all customer sites in the United States and globally who meet minimum test volume license requirements; |
| · | Accelerate the commercialization of our Acuitas Gene Tests and Acuitas Lighthouse informatics; |
| · | Expand our lab service offerings and capabilities through the supply of kits for use on our DNA probe assay platform and commercially available rapid diagnostic testing systems, develop additional MDRO DNA sequencing tests and informatics; |
| · | Partner with reference laboratories, government agencies, diagnostic companies and information technology providers to offer our Acuitas Lighthouse portal on a global basis; and |
| · | Accelerate growth through strategic partnerships, which may include companies developing rapid diagnostic tests for MDROs, sponsored research programs with governments and industry, and strategic acquisitions. |
We believe our products and services can be integrated into a single solution for healthcare providers. By seeking to address institutional needs for informatics, genetic analysis and microbiologic testing, we are working to establish a market leadership position in MDRO analysis. The OpGen solution is intended to help hospitals reduce hospital acquired infection rates by helping to rapidly identify patients colonized with MDROs who should receive contact precautions, and helping to guide antibiotic therapy.
Molecular Information Business
We are working to build a unique and highly proprietary molecular information business. Our approach combines FDA-cleared and CE-marked rapid diagnostics and CLIA lab-based MDRO surveillance tests with our Acuitas Lighthouse bioinformatics systems.
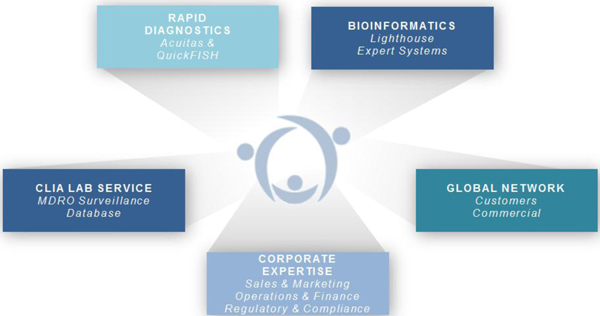
| 6 |
We operate in one segment. Substantially all of our operations are in the United States. Total revenues from customers for the years ended December 31, 2015 and 2014 were $3.2 million and $4.1 million, respectively. Net loss for the years ended December 31, 2015 and 2014 was $17.4 million and $5.7 million, respectively. Total assets at December 31, 2015 and 2014 were $13.8 million and $2.7 million, respectively.
2015 Events
Initial Public Offering
On May 8, 2015, we completed our initial public offering (“IPO”) in which we offered and sold 2,850,000 units, each unit consisting of one share of our common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 units in the IPO. After considering the demand notes, underwriting discounts and commissions and offering expenses, the total net cash proceeds were $12.1 million. On the IPO closing date, the underwriters exercised their over-allotment option to acquire an additional 422,500 stock purchase warrants. In connection with the IPO, all of our then-outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock.
Acquisition of AdvanDx
On July 14, 2015, we completed the strategic acquisition by merger (the “Merger”) of AdvanDx, Inc. and its wholly owned subsidiary AdvanDx A/S, collectively referred to as AdvanDx. Pursuant to the terms of a Merger Agreement, our newly formed subsidiary, Velox Acquisition Corp. merged with and into AdvanDx, Inc. with AdvanDx, Inc. surviving as OpGen’s wholly owned subsidiary.
Merck GHI Investment
In connection with the Merger, on July 14, 2015, the Company entered into a Common Stock and Note Purchase Agreement with Merck Global Health Innovation Fund, LLC (“Merck GHI”) pursuant to which Merck GHI purchased 1,136,364 shares of common stock of the Company at a price of $4.40 per share for gross proceeds of approximately $5,000,000. Pursuant to the Purchase Agreement, the Company also issued to Merck GHI a Senior Secured Promissory Note (the “Note”) in the principal amount of $1,000,000 with a two-year maturity date from the date of issuance. The Company’s obligations under the Note are secured by a lien on all of the Company’s assets.
Market Overview
Antibiotic Resistance – An Urgent Global Issue
We believe that antimicrobial resistance is an urgent global healthcare issue. MDROs have been prioritized as an urgent national and global threat by the CDC, the President of the United States and the WHO. In September 2014, The White House issued a National Strategy for combating antibiotic-resistant bacteria. The strategy calls for the strengthening of surveillance efforts to combat resistance, the development and use of innovative diagnostic tests for identification and characterization of resistant bacteria and antibiotic stewardship and development.
The CDC estimates that in the United States more than two million people are sickened every year with antibiotic-resistant infections, with at least 23,000 dying as a result. Antibiotic-resistant infections add considerable but often avoidable costs to the U.S. healthcare system. In most cases, these infections require prolonged and/or costlier treatments, extended hospital stays, additional doctor visits and healthcare facilities use, and result in greater disability and death compared with infections that are treatable with antibiotics. Estimates for the total economic cost to the U.S. economy range between $20 and $35 billion annually. As described in a December 2014 report issued by the Review on Antimicrobial Resistance commissioned by the U.K. Prime Minister titled “Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations,” 300 million people are expected to die prematurely because of drug resistance over the next 35 years, which could result in $60 to $100 trillion worth of economic output if the problem of antimicrobial drug resistance is not resolved.
In the United States, on August 1, 2014, CMS issued a final rule under the PPACA that, among other things, establishes CMS’ financial incentive program to hospitals that can demonstrate reduction in HAIs. The estimated amount available for these value-based incentive payments in fiscal year 2015 was approximately $1.4 billion. On the other hand, in December 2014, CMS announced its Hospital Acquired Condition Reduction Program, under which CMS will penalize hospitals for excess rates of infections and other patient injuries by reducing Medicare payments. Total penalties are estimated to be approximately $373 million in the first year.
| 7 |
Another emerging global threat are CREs that are either difficult to treat or wholly untreatable. According to CDC Director Dr. Tom Frieden, CREs are a “nightmare bacteria.” The strongest antibiotics do not work and patients are left with potentially untreatable infections with mortality rates ranging between 40% and 80%. CRE strains are transmitted easily in healthcare settings from patients with asymptomatic intestinal colonization, and the CRE strains have the potential to spread antibiotic resistance through plasmid transfer to other bacterial species, including common human flora and potential pathogens such as Escherichia coli. The CDC has called for urgent action to combat the threat of CRE bacteria. Core prevention measures recommended by the CDC for all acute and long-term care facilities include: contact precautions for all patients who are colonized or infected with CRE, single patient room housing or cohorting, laboratory notification procedures, antibiotic stewardship and screening to identify unrecognized CRE colonization in patients admitted to high risk settings such as ICUs, long-term acute care units or facilities, or epidemiologically linked contacts.
Emergence of Superbugs and Lack of Treatment Options
Over the last decade multidrug-resistant Gram negative bacteria, frequently referred to as Superbugs, have been implicated in severe HAIs, and their occurrence has increased steadily. For example, Klebsiella pneumoniae, or K. pneumoniae, is responsible for roughly 15% of Gram-negative infections in hospital intensive care units. Infections caused by KPC strains have few treatment options and are associated with a mortality rate upwards of 50%.
Exacerbating the problems associated with the emergence of these highly resistant KPC strains is their propensity to cause outbreaks in healthcare institutions. These pathogens persist both in the flora of hospitalized patients and in the hospital environment, and they have the capacity to silently colonize patients or hospital personnel by establishing residence in the gastrointestinal tract without causing any signs of infection. Individuals can be silently colonized or become asymptomatic carriers for long periods of time, with detection of these carriers often proving difficult. These silent carriers act as reservoirs for continued transmission, which makes subsequent spread difficult to control and outbreaks difficult to stop. In addition, KPC strains can survive for several hours on the hands of hospital personnel, which likely facilitates spread from patient to patient. Effective control of KPC outbreaks requires a detailed understanding of how transmission occurs, but current technologies do not allow healthcare providers to routinely perform these investigations on a timely basis.
The lack of currently available treatment options and scarcity of new treatment options in development are compounding the emerging Superbug problem. It has been close to 30 years since a new class of antibiotics was developed and successfully introduced. As a result, we believe that rapid, accurate identification of the pathogen and its genetic make-up, screening, infection control and antibiotic stewardship have become the most powerful weapons in the fight to contain this threat.
Based on industry analyses, we believe the global HAI market is a $2 billion dollar market with the molecular diagnostic segment representing a fast growing segment of such market with multiple high acuity patients and significant infectious sites, including urinary tract infections, surgical site infections, pneumonia, bloodstream infections.
The initial focus of our MDRO surveillance business is the U.S. hospital market where there are approximately 5,000 hospitals and a potential market opportunity of six million tests annually for our Acuitas MDRO Gene Test. According to statistics issued by AHA Hospitals Statistics in 2011, there are 1,395 acute care hospitals in the United States with 200 or more beds that are candidates for weekly screening of the approximately 20% of patients who are at high risk for MDRO colonization or infection. There are also 290 long term acute care hospitals where we believe all patients are candidates for bi-weekly screening. We believe the high-risk MDRO testing market opportunity in the United States is approximately $400 million. The trend toward consolidated health systems is resulting in the combination of small and mid-sized hospitals into large health systems that are the initial targets for our test and informatics products. A typical large health system could have more than $4 billion in annual revenue, a central hospital with more than 400 beds and 6-8 smaller hospitals and long-term care facilities. These large health systems have started to centralize their microbiology lab testing, making them an attractive target market for us.
The trend towards forming ACOs is expected to increase the focus on reducing length of stay and the overall cost of hospital procedures. Since HAIs result in increased costs of approximately $24,000 per affected patient, we anticipate ACOs will be particularly receptive to our MDRO management solutions. According to Diagnostic Kit – second edition, March 2014 by Cowen & Co., the MRSA surveillance testing market and C. difficile testing market in the United States are approximately $300 million and $150 million, respectively.
| 8 |
Products
Our current product offerings include our QuickFISH® and PNA FISH® products, which are FDA-cleared, CE-marked in vitro diagnostic tests designed to rapidly identify antimicrobial resistant pathogens significantly earlier than currently available conventional methods, our Acuitas MDRO Gene Test, Acuitas CR Elite Test and Acuitas Resistome Test, each a CLIA lab-based test that provides a profile of MDRO resistant genes for surveillance and response to outbreaks, and our Acuitas Lighthouse bioinformatics platform which is being developed to provide an evergreen database for comprehensive testing and bioinformatics analysis to help guide antibiotic therapy decision making.
FISH Products
We have commercialized 15 QuickFISH, PNA FISH and XpressFISH diagnostic test products in the United States and Europe for the identification of various infectious pathogens. The pathogens identified and differentiated by our FISH products are:
| QuickFISH | PNA FISH | XpressFISH |
| Staphylococcus | Staphylococcus | MRSA |
| Enterococcus | Enterococcus | MSSA |
| Gram-negative bacteria | Gram-negative bacteria | |
| Gram –positive bacteria | Gram-positive bacteria | |
| Candida | Candida |
Our FISH products can provide pathogen identification and differentiation within 20 to 90 minutes of positive blood culture results. Differentiation of the pathogen, such as, for example differentiating a methicillin resistant Staphylococcus aureus (“MRSA”) infection from a methicillin susceptible Staphylococcus aureus (“MSSA”) infection provides actionable information that can be used by the health care provider to determine appropriate antibiotic therapy.
We currently have approximately 100 U.S. hospital customers purchasing our FISH products, and sell our FISH products to hospitals in 10 countries with antibiotic stewardship programs. Our hospital customers include academic medical centers, tertiary care hospitals and community hospitals.
An example of the usefulness of our QuickFISH products at Winter Haven Hospital in Florida was described in a recent publication “The Impact of Implementation of Rapid QuickFISH Testing for Detection of Coagulase Negative Staphylococci at a Community-Based Hospital,” American Journal of Clinical Pathology, January 2016. In such case study our QuickFISH products demonstrated clinical utility and cost effectiveness in the more rapid identification and differentiation of staph-infected patients which resulted in a 90% reduction in pathogen identification (1.4 hours as compared to 17.2 hours from a positive blood culture), decreased utilization of Vancomycin antibiotic therapy, a 30% reduction in length of stay and annual savings of approximately $764,000.
Acuitas Products
Our high resolution DNA tests are marketed under the Acuitas trade name. We have developed Acuitas DNA tests for use in our CLIA lab such as the Acuitas MDRO Gene Test and we are developing a rapid Acuitas DNA test for use in hospital laboratories that will combine rapid pathogen ID and detection of antibiotic resistance genes. In the future we anticipate marketing new FISH products that we may develop under the Acuitas trade name.
| · | Our Acuitas MDRO Gene Test is, to our knowledge, the first CLIA lab-based test able to provide information regarding the presence of ten MDRO resistance genes from one patient specimen. The ten drug-resistant genes identified by our Acuitas MDRO Gene Test are associated with CRE, ESBL and VRE organisms, and are gastrointestinal organisms frequently associated with antibiotic-resistant infections. The test results can be used by healthcare providers to identify patients colonized with organisms expressing the drug-resistant genes or who are actively infected. |
| 9 |
| · | Our Acuitas CR Elite Test adds the ability for the healthcare provider to order a microbiology culture screen to be performed from the same specimen sent for our Acuitas MDRO Gene Test, thereby providing additional information about the organism(s) associated with an active infection, as well as an antibiotic susceptibility profile for such organism(s). |
| · | Our Acuitas Resistome Test, launched in the second quarter of 2015, is a more comprehensive MDRO molecular test which detects 49 genes covering over 900 subtypes associated with antibiotic resistance. The test includes additional resistance genes for carbapenemases, ESBLs and AmpC genes, in replacement of the vancomycin resistant genes found in the Acuitas MDRO Gene Test. We believe the AmpC targets of the Acuitas Resistome Test are more specific for Gram-negative bacteria, thereby strengthening the coverage provided by our Acuitas Resistome Test to detect resistance genes found in Klebsiella pneumoniae, Escherichia coli, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter cloacae, and Citrobacter freundii. We use Acuitas Resistome Test results for Acuitas Lighthouse profiling of specimens collected in hospitals and clinical isolates from infected patients. Information from our Acuitas Resistome Test provides additional gene detection information to supplement our Acuitas MDRO Gene Test. Acuitas Resistome Test results can be used in conjunction with the Acuitas CR Elite Test to provide high resolution Acuitas Lighthouse profiles. Our goal is to provide DNA test-based Acuitas Lighthouse profiles, within 24 hours of sample receipt, and, using the Acuitas CR Elite Test to supplement our Acuitas Lighthouse profiles, with biologically derived, phenotypic antibiotic susceptibility data within 84 hours. We anticipate improving the accuracy, over time, of our Acuitas Resistome Test by performing DNA sequence analysis of microbial isolates within our Acuitas Lighthouse data warehouse. We believe our menu of genotypic and phenotypic tests along with our Acuitas Lighthouse bioinformatics platform profiles, will enable better surveillance and epidemiology, improved infection control practices, improved antibiotic stewardship and individualized patient care, as well as help to facilitate outbreak detection and response in healthcare settings. We also anticipate combining tests for infectious diseases such as C. difficile, MRSA and others to provide enhanced MDRO screening and patient management solutions. |
Acuitas Lighthouse
Our Acuitas Lighthouse bioinformatics platform enables proactive MDRO management to prevent in-hospital transmission events and to help improve patient outcomes. Trend analysis of patient specific data, data specific to individual hospital facilities and health systems can be provided safely and confidentially to healthcare providers. Our Acuitas Lighthouse’s dynamic profiling incorporates identity, phenotype and MDRO gene presence and assigns unique microbe identifiers, or Acuitas Lighthouse profiles, based on MDRO gene composition, and antibiotic susceptibility, or AST, data. We believe our Acuitas Lighthouse profiling will provide a comprehensive diagnostic tracking tool for MDRO infections in the hospital setting. It is based on our CLIA- and HIPAA-compliant LIMS database system. We have developed a web-based portal to allow our customers access to LIMS-based lab reports and Acuitas Lighthouse data reports.
We are also focused on further developing Acuitas Lighthouse as an evergreen database with continual global pathogen data from our CLIA lab and hospital customers, with such data to be used to:
| · | assist in accelerating more rapid diagnosis with improved molecular susceptibility data; |
| · | provide MDRO screening and surveillance capabilities to hospitals to identify pathogen and resistance profiles; and |
| · | potentially accelerate new antibiotic development as the data are used to reveal genetic resistance patterns to direct drug discovery. |
In November 2015, the Company and the District of Columbia Hospital Association (“DCHA”) initiated a comprehensive citywide evaluation, HARP-DC (Healthcare facility Antibiotic Resistance Prevalence-District of Columbia), to be overseen by Washington D.C. public health departments, to gauge the prevalence of the multidrug-resistant CREs in healthcare facilities throughout the District of Columbia. The DC Department of Health led study is being funded by the CDC’s Epidemiology and Laboratory Capacity for Infectious Diseases (“ELC”) Funding program for tracking healthcare associated infections. The DC Department of Health contracted with OpGen to perform the related laboratory services using OpGen’s products, including Acuitas Lighthouse. The HARP-DC study marks the first effort of its kind in the District of Columbia to proactively combat CREs.
| 10 |
Other Products
Prior to our shift in focus to developing and commercializing our MDRO products, OpGen had developed and commercialized the Argus® Whole Genome Mapping System, MapIt® Services and MapSolver™ bioinformatics products and services. Such products and services were sold to academic, public health and corporate customers to allow them to perform Whole Genome Mapping and analysis of microbial, plant, animal and human genomes for life sciences applications. We have more than ten years of experience mapping microbial genomes. Our customers for these products include government and public health agencies such as the CDC, FDA, USDA and biodefense organizations, who use the Argus and MapSolver products in research and development, food safety and public health settings. We continue to provide these products and services to existing customers, however, we anticipate that such revenues will continue to decline as we have shifted our focus to our MDRO, rapid diagnosis and bioinformatics products and services.
In September 2013, we entered into a strategic collaboration with Hitachi High-Technologies Corporation (“Hitachi”) to commercialize our Whole Genome Mapping technology for mapping, assembly and analysis of human DNA. Under that collaboration we developed cloud-based human genome map assembly capabilities. The collaboration agreement ended in December 2015. We have seen declining revenues from our current customers for our Whole Genome Mapping products and services over the past few years, as DNA sequencing techniques and products have grown in popularity. While we continue to provide products and services to our existing customer base, we intend to monetize our Whole Genome Mapping technology by out-licensing or selling our technology to the extent possible.
For the year ended December 31, 2015, revenue earned from Hitachi represented 11% of total revenues. For the year ended December 31, 2014, revenue earned from Hitachi represented 64% of total revenues.
Research and Development
For the years ended December 31, 2015 and 2014, our research and development expenses were $6.0 million and $4.4 million, respectively. We intend to continue to invest in the development of additional Acuitas gene tests, our Acuitas Lighthouse bioinformatics platform, and our QuickFISH rapid identification tests. Our current focus is on completing the development of our product offerings to provide actionable, precise diagnostics powered by surveillance data for rapid diagnostics of pathogens, determination of the correct antibiotics appropriate to treat the infection and accumulation of actionable surveillance data to provide information useful for monitoring and controlling outbreaks and promoting antibiotic stewardship.
Our ongoing research and development efforts include:
| · | Development of a one hour rapid Acuitas DNA test capable of providing genetic resistance information for up to 150 drug resistance genes and a cloud-based Acuitas Lighthouse data warehouse for interpretation of test results and clinical decision making support tools to help select appropriate antibiotic therapies; |
| · | Development of more rapid molecular diagnostic products to achieve actionable pathogen identification and differentiation in the first few hours of presentation or symptoms; |
| · | Automating our QuickFISH products through digital imaging and analysis, new formats requiring less hands on time to process samples, multiplex formats that allow for testing of a broader range of microorganisms; |
| · | Continued investments in our Acuitas Lighthouse bioinformatics platform, focused on (i) data warehouse and portal for MDRO data and (ii) antibiotic analysis; |
| · | Further development of our Acuitas MDRO Gene Test, Acuitas Resistome Test and DNA sequencing; and |
| · | Converting our CLIA lab-based products to in vitro diagnostic kits that can be sold, upon receipt of FDA clearance and other approvals, directly to our customers and to other clinical reference laboratories. |
Our ongoing research and development programs and their stage of development are provided below.
| 11 |
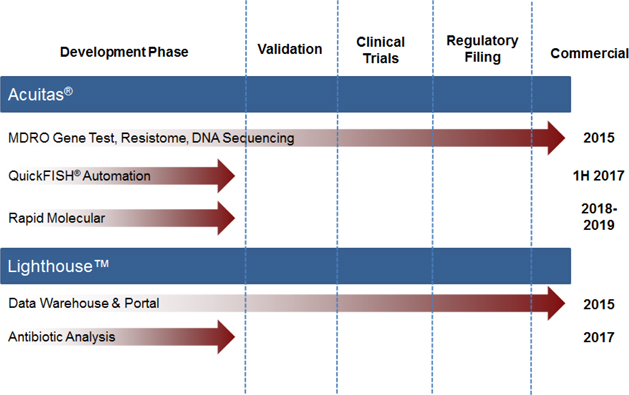
Sales and Marketing
We currently sell and market our products and services in the United States through a 12 person sales and marketing organization including direct sales professionals and a dedicated marketing support organization. Internationally, we sell our products through a network of distributors in 16 countries. We operate a subsidiary in Denmark that provides support for our European customers and to distributors in other parts of the world. We are involved in pilot programs in approximately 10 countries to demonstrate the clinical and cost effectiveness of our FISH products. We are working to expand our market reach by entering into strategic co-marketing relationships with larger diagnostic and pharmaceutical companies and by expanding our network of distributors globally.
Competition
We believe we are currently the only company developing a molecular information business focused on leading a transformation in microbiology and infectious disease through precision medicine solutions combining bioinformatics and clinical diagnostics. Our approach combines proprietary DNA tests developed in our CLIA laboratory, FDA-cleared and CE-marked rapid diagnostics, and our Acuitas Lighthouse bioinformatics and data warehouse offerings. Our competitors include rapid diagnostic testing and traditional microbiology companies, commercial laboratories, information technology companies, and hospital laboratories who may internally develop testing capabilities. Principal competitive factors in our target market include: organizational size, scale, and breadth of product offerings; rapidity of test results; quality and strength of clinical and analytical validation data and confidence in diagnostic results; cost effectiveness; ease of use; and regulatory approval status.
Our principal competition comes from traditional methods used by healthcare providers to diagnose and screen for MDROs and from other molecular diagnostic companies creating screening and diagnostic products such as Cepheid, Becton-Dickinson, bioMerieux, Accelerate Diagnostics, T2 Biosystems and Nanosphere. We believe our focus on identifying antibiotic-resistant genes, rather than organisms, the genes and associated diseases included in our gene tests, and our Acuitas Lighthouse bioinformatics platform products and services to distinguish us from such competitors.
We also face competition from commercial laboratories, such as Bio-Reference Laboratories, Inc., Laboratory Corporation of America Holdings and Quest Diagnostics Incorporated, which have strong infrastructure to support the commercialization of diagnostic services.
| 12 |
Competitors may develop their own versions of our product offerings in countries where we do not have patents or where our intellectual property rights are not recognized.
Many of our potential competitors have widespread brand recognition and substantially greater financial, technical, research and development and selling and marketing capabilities than we do. Others may develop products with prices lower than ours that could be viewed by hospitals, physicians and payers as functionally equivalent to our solution, or offer solutions at prices designed to promote market penetration, which could force us to lower the list prices of our solutions and affect our ability to achieve profitability. If we are unable to change clinical practice in a meaningful way or compete successfully against current and future competitors, we may be unable to increase market acceptance and sales of our products, which could prevent us from increasing our revenue or achieving profitability and could cause our stock price to decline.
Laboratory Operations
Our laboratory operations are headquartered at our CLIA-certified laboratory in Gaithersburg, Maryland, where we perform all Acuitas testing. Samples are transported to the laboratory by FedEx or by courier. Once received, samples are assessed for acceptability, accessioned into our LIMS, prepared for processing and analyzed with traditional microbiology culture methods or using molecular testing instrumentation. Laboratory test data is housed in a proprietary LIMS database that is CLIA laboratory compliant. Customers access CLIA laboratory test results through individual PDF test reports and through our Acuitas Lighthouse portal. Our laboratory also performs testing for research and development purposes and for both the creation and ongoing maintenance of our Acuitas Lighthouse data warehouse.
We believe we have sufficient laboratory capacity to perform Acuitas testing for at least the next 24 months.
Manufacturing
We manufacture our FDA-cleared and CE-marked QuickFISH products in our Woburn, Massachusetts research and manufacturing facility. We are currently operating this facility under a one year lease with an option to extend our lease for subsequent periods. Specialty reagents for our CLIA laboratory are manufactured at our Gaithersburg, Maryland facility.
Manufacturing of our FDA-cleared products is performed under the current Good Manufacturing Practices - Quality System Regulation as required by the FDA for the manufacture of diagnostic products. These regulations carefully control the manufacture, testing and release of diagnostics products as well as raw material receipt and control. Both before and after a product is commercially released, we have ongoing responsibilities under FDA regulations. We are also subject to periodic inspections by the FDA to determine compliance with the FDA’s requirements, including primarily the quality system regulations and medical device reporting regulations. The results of these inspections can include inspectional observations on FDA’s Form 483, warning letters, or other forms of enforcement. Our Woburn, Massachusetts facility was inspected by the FDA in 2015. Following such inspection, the FDA issued a report of its findings and observations, typically referred to as “Form 483 observations," primarily related to our quality systems and testing policies and documentation. We have responded, or intend to respond, to all inspection observations within the required timeframe and are working with the FDA’s Office of Compliance to satisfy the identified deficiencies.
Quality Assurance
Our quality assurance function oversees the quality of our laboratory and our FDA-cleared and CE-marked diagnostic products as well as the quality systems used in research and development, client services, billing operations and sales and marketing. We have established a quality assurance system across our entire business, including implementation and maintenance, document control, supplier qualification, corrective or preventive actions, oversight, and employee training processes. We monitor and seek to improve our quality over time.
| 13 |
Raw Materials and Suppliers
We procure reagents, equipment, chips and other materials we use to perform our Acuitas MDRO Gene Test from sole suppliers such as Fluidigm Corporation. We purchase the PNA probes, glass slides and specialty consumables for our QuickFISH products from third party manufacturers who have long lead times and who manufacture several of these products for us on a sole source basis. We also purchase our collection kits from sole-source suppliers. Some of these items are unique to these suppliers and vendors. While we have developed alternative sourcing strategies for these materials and vendors, we cannot be certain whether these strategies will be effective or whether alternative sources will be available when we need them. If these suppliers can no longer provide us with the materials we need to perform our Acuitas MDRO Gene Test or manufacture our QuickFISH products, if the materials do not meet our quality specifications, or if we cannot obtain acceptable substitute materials, our business would be negatively affected.
Payments and Reimbursement
Our Acuitas MDRO test products, our Acuitas Lighthouse bioinformatics platform and our QuickFISH tests are, and other future products and services will be, sold to hospitals and public health organizations on a fee-for-service basis. When hospital and health system clients purchase our QuickFISH tests we bill them directly for the purchase of test kits and consumables. Hospitals that purchase MDRO services from our CLIA laboratory are billed on a per test basis. Currently we provide access to our Acuitas Lighthouse through portals. The portal capability is provided to our test customers who have sufficient test volume as part of our MDRO test offerings.
In the future we envision selling our Acuitas Lighthouse bioinformatics platform to health systems, hospitals and long-term care facilities under capitated, flat-rate contracts. Health systems and hospitals absorb the costs of extended stay from HAIs and poor treatment outcomes. For healthcare providers to support the use of our tests and services, OpGen needs to demonstrate improved outcomes and reduced costs. Various studies have documented increased hospital stays of six days or more for patients infected with MDROs, resulting in increased costs of $14,000 to $33,000 per infected patient. Determining if an infection is hospital-acquired or was originally obtained from another source is an important issue for hospitals. We believe our tests will help adjudicate payment favorably for hospitals. Isolation procedures are also costly to hospitals, so it is critical that isolation/de-isolation decisions are made accurately. Two recent studies documented a daily extra cost of approximately $101 for contact precaution equipment and approximately $57 for nursing time and contact precaution supplies for each infected patient. In addition to costs to individual hospitals, estimates of the economic costs of antibiotic resistance to the U.S. economy range from $20 billion to $35 billion annually.
Our marketing strategy focuses on the rapid turn-around time of our Acuitas MDRO and QuickFISH test results and the panel of results available from one patient sample. We believe the combination of our Acuitas MDRO test products, including QuickFISH, and our Acuitas Lighthouse bioinformatics platform differentiates us in the marketplace by offering a single sample process for identification and management of MDROs. Our approach can deliver a number of benefits to healthcare organizations including: (1) reduced lengths of stays; (2) cost savings and improved patient outcomes; and (3) avoidance of penalties by third-party payers for HAIs.
We employ diverse marketing programs to inform key stakeholders of the value of our solutions in order to drive adoption. As part of our marketing strategy, we educate hospitals, other health care institutions, and healthcare professionals about our value proposition. We intend to expand our marketing efforts using proceeds from this offering to increase these activities by expanding our sales and marketing efforts to microbiology and infection control professionals and hospital executives. We anticipate supporting efforts to advocate for expanded MDRO hospital surveillance, legislation at the state and federal level to encourage best practices for MDRO surveillance, and clinical practice guidelines. Finally, our website serves as a portal for educational material for hospitals, healthcare professionals and patients.
Third-Party Payers
We do not currently rely on any third-party payers for payment or reimbursement to us for our Acuitas MDRO or QuickFISH test products. Although we do not anticipate seeking direct reimbursement to us, we do believe that federal healthcare programs and other third-party payers may, in the future, reimburse hospitals for implementing institution-wide surveillance, infection control and antibiotic stewardship programs. Our management team has experience seeking reimbursement from federal healthcare programs and other third-party payers, and would work to:
| · | Meet the evidence standards necessary to be consistent with leading clinical guidelines. We believe demonstrating that our solution meets leading clinical practice guidelines plays a critical role in payers’ coverage decisions; |
| 14 |
| · | Engage reimbursement specialists to ensure the payor outreach strategy reacts to and anticipates the changing needs of our customer base. A customer service team would be an integral part of our reimbursement strategy, working with hospitals to navigate the claims process; |
| · | Cultivate a network of key opinion leaders. Key opinion leaders are able to influence clinical practice by publishing research and determining whether new tests should be integrated into practice guidelines. We would collaborate with key opinion leaders early in the development process to ensure our clinical studies are designed and executed in a way that clearly demonstrates the benefits of our tests to physicians and payers; and |
| · | Compile a library of peer-reviewed studies that demonstrate that our Acuitas MDRO test products are effective, accurate and faster than current methods. |
Intellectual Property
In order to remain competitive, we must develop and maintain protection of the proprietary aspects of our technologies. To that end, we rely on a combination of patents, copyrights and trademarks, as well as contracts, such as confidentiality, invention assignment and licensing agreements. We also rely upon trade secret laws to protect unpatented know-how and continuing technological innovation. In addition, we have what we consider to be reasonable security measures in place to maintain confidentiality. Our intellectual property strategy is intended to develop and maintain our competitive position.
As of December 31, 2015, we had total license or ownership rights to 132 patents, including 31 pending United States non-provisional patent applications, and 71 issued United States patents. More specifically, as of December 31, 2015, related to our FISH products, we had license or ownership rights to 84 patents, including 12 pending United States non-provisional patent applications, and 52 issued United States patents. These issued patents began to expire in December 2015 and will be fully expired by January 2029. As of December 31, 2015, related to our Acuitas products, we had license or ownership right to 3 pending United States non-provisional patent applications and no issued United States patents. As of December 31, 2015, related to our other products, we had license or ownership rights to 45 patents, including 16 pending United States non-provisional patent applications, and 19 issued United States patents related to our other products. These issued patents begin to expire in April 2016 and will be fully expired by January 2032.
We intend to file additional patent applications in the United States and abroad to strengthen our intellectual property rights; however, our patent applications (including the patent applications listed above) may not result in issued patents in a timely fashion or at all, and we cannot assure investors that any patents that have issued or might issue will protect our technology.
We require all employees and technical consultants working for us to execute confidentiality agreements, which provide that all confidential information received by them during the course of the employment, consulting or business relationship be kept confidential, except in specified circumstances. Our agreements with our research employees provide that all inventions, discoveries and other types of intellectual property, whether or not patentable or copyrightable, conceived by the individual while he or she is employed by us are assigned to us. We cannot provide any assurance, however, that employees and consultants will abide by the confidentiality or assignment terms of these agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our technology or obtain and use information that we regard as proprietary.
Regulation
The following is a summary of the regulations materially affecting our business and operations.
Clinical Laboratory Improvement Amendments of 1988
As a clinical reference laboratory, we are required to hold certain federal, state and local licenses, certifications and permits to conduct our business. Under CLIA, we are required to hold a certificate applicable to the type of laboratory examinations we perform and to comply with standards covering personnel, facilities administration, quality systems and proficiency testing.
We have a current Certificate of Compliance under CLIA and a Medical Laboratory Permit from the State of Maryland to perform clinical testing at our Gaithersburg, Maryland laboratory. To renew our CLIA certificate, we are subject to survey and inspection every two years to assess compliance with program standards. The regulatory and compliance standards applicable to the testing we perform may change over time, and any such changes could have a material effect on our business. Our current CLIA certificate expires on October 1, 2017, and our Medical Laboratory Permit expires on June 30, 2016.
If our clinical laboratory is out of compliance with CLIA requirements, we may be subject to sanctions such as suspension, limitation or revocation of our CLIA certificate, as well as a directed plan of correction, state on-site monitoring, civil money penalties, civil injunctive suit or criminal penalties. We must maintain CLIA compliance and certification in order to perform clinical laboratory tests and report patient test results. If we were to be found out of compliance with CLIA requirements and subjected to sanction, our business could be harmed.
| 15 |
Federal Oversight of Laboratory Developed Tests and Research-Use-Only Products
Clinical laboratory tests, like our Acuitas MDRO Gene Test, are regulated under CLIA, as well as by applicable state laws. Historically, most laboratory developed tests (“LDTs”), were not subject to FDA regulations applicable to medical devices, although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation. FDA defines the term “laboratory developed test” as an in vitro diagnostic test that is intended for clinical use and designed, manufactured and used within a single laboratory. We believe that our Acuitas MDRO test products are LDTs. Currently, the FDA exercises enforcement discretion with respect to LDTs such that it does not enforce provisions of the Food, Drug and Cosmetic Act applicable to in vitro diagnostic (IVD) devices. In July 2014, due to the increased proliferation of LDTs for complex diagnostic testing, and concerns with several high-risk LDTs related to lack of evidentiary support for claims, erroneous results and falsification of data, the FDA notified Congress that it would issue guidance that, when finalized, would adopt a risk-based framework that would increase FDA oversight of LDTs. As part of this developing framework, the FDA issued draft guidance in October 2014, informing manufacturers of LDTs of its intent to collect information from laboratories regarding their current LDTs and newly developed LDTs through a notification process. The FDA will use this information to classify LDTs and to prioritize enforcement of premarket review requirements for categories of LDTs based on risk, beginning with LDTs that present the highest risk. Specifically, the FDA plans to use advisory panels to provide recommendations to the agency on LDT risks, classification and prioritization of enforcement of applicable regulatory requirements on certain categories of LDTs, as appropriate, for those tests not yet categorized.
Some products are for research use only (“RUO”), or for investigational use only (“IUO”). RUO and IUO products are not intended for human clinical use and must be properly labeled in accordance with FDA guidance. Claims for RUOs and IUOs related to safety, effectiveness, or clinical utility or that are intended for human diagnostic or prognostic use are prohibited. In November 2013, the FDA issued guidance titled “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only - Guidance for Industry and Food and Drug Administration Staff.” This guidance sets forth the requirements to utilize such designations, labeling requirements and acceptable distribution practices, among other requirements. Mere placement of an RUO or IUO label on an in vitro diagnostic product does not render the device exempt from otherwise applicable clearance, approval or other requirements. The FDA may determine that the device is intended for use in clinical diagnosis based on other evidence, including how the device is marketed.
We cannot predict the potential effect the FDA’s current and forthcoming guidance on LDTs and IUOs/RUOs will have on our solutions or materials used to perform our diagnostic services. While we qualify all materials used in our diagnostic services according to CLIA regulations, we cannot be certain that the FDA might not promulgate rules or issue guidance documents that could affect our ability to purchase materials necessary for the performance of our diagnostic services. Should any of the reagents obtained by us from vendors and used in conducting our diagnostic services be affected by future regulatory actions, our business could be adversely affected by those actions, including increasing the cost of service or delaying, limiting or prohibiting the purchase of reagents necessary to perform the service.
We cannot provide any assurance that FDA regulation, including premarket review, will not be required in the future for our surveillance and diagnostic services, whether through additional guidance or regulations issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. On November 17, 2015, the House Committee on Energy and Commerce held one such hearing entitled “Examining the Regulation of Diagnostic Tests and Laboratory Operations.” We expect that new legislative proposals will be introduced from time to time. It is possible that legislation could be enacted into law or regulations or guidance could be issued by the FDA which may result in new or increased regulatory requirements for us to continue to offer our diagnostic services or to develop and introduce new services.
| 16 |
FDA’s Premarket Clearance and Approval Requirements
The FDA classifies medical devices into one of three classes: Class 1, Class 2 or Class 3. Devices deemed to pose lower risk are placed into either Class 1 or Class 2. Class 1 or Class 2 devices that are exempt from the premarket notification process must comply with applicable regulations but can simply list their products with the FDA. Class 1 or Class 2 devices that are non-exempt from the premarket notification process must comply with applicable regulations and submit a 510(k) premarket submission for review to receive clearance to list and market their devices. The 510(k) must establish substantial equivalence to a predicate device. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared device, are placed in Class 3 and require premarket approval (“PMA”) before commercialization. The majority of the medical devices manufactured by OpGen at its Woburn, Massachusetts facility (i.e., the AdvanDx products) are Class 1; one product manufactured in Woburn is a Class 2 device. All of these products are non-exempt and required 510(k) premarket submissions. At this time our CLIA tests are not required to be reviewed by the FDA; however, our LDTs would be considered non-exempt Class 2 devices and would require a 510(k) premarket submission to continue.
All medical device manufacturers must register their establishments with the FDA; such registrations require the payment of user fees. In addition, both 510(k) premarket submissions and PMA applications are subject to the payment of user fees, paid at the time of submission for FDA review. At this time our CLIA lab is not required to register and list with the FDA; however, the Medical Device User Fee Act IV (“MFUFA IV”) negotiations currently taking place between the FDA and medical device manufacturers include discussions regarding user fees for clinical laboratories running LDTs. This new fee would be in addition to the user fees required to operate a clinical laboratory.
The FDA has issued a regulation outlining specific requirements for “specimen transport and storage containers.” “Specimen transport and storage containers” are medical devices if “intended to contain biological specimens, body waste, or body exudate during storage and transport” so that the specimen can be used effectively for diagnostic examination. Since medical devices are subject to registration and listing requirements, the reporting of corrections and removals, and responsible for medical device reporting requirements, if the FDA were to determine that our sample collection container is a medical device, the manufacturer would be required to register and list with the FDA for us to use the container for diagnostic purposes. The specimen collection device would be exempt from premarket review, and from QSR requirements except for recordkeeping and complaint handling requirements, so long as no sterility claims are made, but the manufacturer would still be required to comply with applicable regulations.
510(k) Clearance Pathway
If required to obtain 510(k) clearance for our future products or conversion of our Acuitas MDRO test products to diagnostic kits, such tests would be classified as medical devices and we would have to submit a premarket notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976, for which the FDA has not yet called for the submission of premarket approval applications. FDA’s 510(k) clearance pathway usually takes from three to twelve months. On average the review time is approximately six months, but it can take significantly longer than twelve months in some instances, as the FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, require premarket approval. The FDA requires each manufacturer to determine whether the proposed change requires submission of a new 510(k) notice, or a premarket approval, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or premarket approval is obtained. If the FDA requires us to seek 510(k) clearance or premarket approval for any modifications to a previously cleared product, we may be required to cease marketing or recall the modified device until we obtain this clearance or approval. Also, in these circumstances, we may be subject to significant regulatory fines or penalties. We have made and plan to continue to make additional product enhancements to products that we believe do not require new 510(k) clearances.
Premarket Approval Pathway
A premarket approval application must be submitted if a device cannot be cleared through the 510(k) process. The premarket approval application process is generally more costly and time consuming than the 510(k) process. A premarket approval application must be supported by extensive data including, but not limited to, analytical, preclinical, clinical trials, manufacturing, statutory preapproval inspections, and labeling to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use.
| 17 |
After a premarket approval application is sufficiently complete, the FDA will accept the application and begin an in-depth review of the submitted information. By statute, the FDA has 180 days to review the “accepted application,” although, generally, review of the application can take between one and three years, but it may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The preapproval inspections conducted by the FDA include an evaluation of the manufacturing facility to ensure compliance with the QSR, as well as inspections of the clinical trial sites by the Bioresearch Monitoring group to evaluate compliance with good clinical practice and human subject protections. New premarket approval applications or premarket approval application supplements are required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. Significant changes to an approved PMA require a 180-day supplement, whereas less substantive changes may utilize a 30-day notice, or the 135-day supplement. Premarket approval supplements often require submission of the same type of information as a premarket approval application, except that the supplement is limited to information needed to support any changes from the device covered by the original premarket approval application, and may not require as extensive clinical data or the convening of an advisory panel. None of our products are currently approved under a premarket approval.
Clinical Trials
Clinical trials are almost always required to support a premarket approval application and are usually required to support non-exempt Class 1 and Class 2 510(k) premarket submissions. Clinical trials may also be required to support certain marketing claims. If the device presents a “significant risk,” as defined by the FDA, to human health, the FDA requires the device sponsor to file an investigational device exemption (“IDE”) application with the FDA and obtain IDE approval prior to conducting the human clinical trials. The IDE application must be supported by appropriate data, such as analytical, animal and laboratory testing results, manufacturing information, and an Investigational Review Board (“IRB”) approved protocol showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA prior to initiation of enrollment of human subjects. Clinical trials for a significant risk device may begin once the investigational device exemption application is approved by the FDA. If the clinical trial design is deemed to be “non-significant risk,” the clinical trial may eligible for the “abbreviated” IDE requirements; in some instances IVD clinical trials may be exempt from the more burdensome IDE requirements if certain labeling requirements are met. All clinical trials conducted to support a premarket submission must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent, oversight by an IRB and healthcare privacy requirements. A clinical trial may be suspended by the FDA or the IRB review board at any time for various reasons, including a belief that the risks to the study participants outweigh the benefits of participation in the study. Even if a study is completed, the results of our clinical testing may not demonstrate the safety and efficacy of the device, or may be equivocal or otherwise not be sufficient to obtain approval of our product. Similarly, in Europe the clinical study must be approved by the local ethics committee and in some cases, including studies of high-risk devices, by the Ministry of Health in the applicable country.
Pervasive and Continuing FDA Regulation
Numerous regulatory requirements apply to our products classified as devices would continue to apply. These include:
| · | product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| · | QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the development and manufacturing process; |
| · | labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication; |
| · | clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use of one of our cleared devices; |
| · | approval of product modifications that affect the safety or effectiveness of one of our cleared devices; |
| 18 |
| · | medical device reporting regulations, which require that manufacturers comply with FDA requirements to report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur; |
| · | post-approval restrictions or conditions, including post-approval study commitments; |
| · | post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; |
| · | the FDA’s recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a product that is in violation of governing laws and regulations; |
| · | regulations pertaining to voluntary recalls; and |
| · | notices of corrections or removals. |
OpGen’s Woburn, Massachusetts facility is currently registered as an establishment with the FDA. If the LDTs performed in OpGen’s CLIA lab were deemed medical devices by the FDA, then we and any third-party manufacturers of such devices would need to register with the FDA as medical device manufacturers and obtain all necessary state permits or licenses to operate our business. We and any third-party manufacturers would be subject to announced and unannounced inspections by the FDA to determine our compliance with quality system regulation and other regulations. Our Woburn, Massachusetts facility was inspected by the FDA in 2015. Following such inspection, the FDA issued a report of its findings and observations, typically referred to as “Form 483 observations," primarily related to our quality systems and testing policies and documentation. We have responded, or intend to respond, to all inspection observations within the required time frame and are working with the FDA’s Office of Compliance to satisfy the identified deficiencies.
Failure to comply with applicable regulatory requirements could result in enforcement action by the FDA, which might include any of the following sanctions: (1) untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; (2) unanticipated expenditures to address or defend such actions; (3) customer notifications for repair, replacement and refunds; (4) recall, detention or seizure of our products; (5) operating restrictions or partial suspension or total shutdown of production; (6) refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products; (7) operating restrictions; (8) withdrawing 510(k) clearances or PMA approvals that have already been granted; (9) refusal to grant export approval for our products; or (10) criminal prosecution.
After a medical device is placed on the market, numerous regulatory requirements apply. These include: all of the relevant elements of the Quality System Regulation (“QSR”), labeling regulations, restrictions on promotion and advertising, the medical device reporting (which requires the manufacturer to report to the FDA if its device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur), and the Reports of Corrections and Removals regulations (which requires manufacturers to report certain recalls and field actions to the FDA).
Health Insurance Portability and Accountability Act
Under HIPAA, the Department of Health and Human Services (“HHS”), has issued regulations to protect the privacy and security of protected health information used or disclosed by health care providers, such as us, and by certain vendors of ours, also known as our business associates. The regulations include limitations on the use and disclosure of protected health information and impose notification requirements in the event of a breach of protected health information. HIPAA also regulates standardization of data content, codes and formats used in health care transactions and standardization of identifiers for health plans and providers. Penalties for violations of HIPAA regulations include civil and criminal penalties.
We have developed and implemented policies and procedures designed to comply with these regulations. The requirements under these regulations may change periodically and could have an effect on our business operations if compliance becomes substantially more costly than under current requirements.
In addition to federal privacy regulations, there are a number of state laws governing confidentiality of health information that are applicable to our business. If our business expands internationally, we would be subject to compliance with other laws regarding confidentiality of health information and privacy.
| 19 |
New laws governing privacy may be adopted in the future as well. We have taken steps to comply with health information privacy requirements to which we are aware that we are subject. However, we can provide no assurance that we are or will remain in compliance with diverse privacy requirements in all of the jurisdictions in which we do business. Failure to comply with privacy requirements could result in civil or criminal penalties, which could have a materially adverse effect on our business.
Federal and State Physician Self-referral Prohibitions
As a clinical laboratory, and manufacturer and seller of diagnostic tests, we are subject to the federal physician self-referral prohibitions, commonly known as the Stark Law, and to similar restrictions under the Maryland Physician Self-Referral Law. Together these restrictions generally prohibit us from billing a patient or any governmental or private payor for any clinical laboratory services when the physician ordering the service, or any member of such physician’s immediate family, has an investment interest in or compensation arrangement with us, unless the arrangement meets an exception to the prohibition.
Both the Stark Law and the Maryland Physician Self-Referral Law contain an exception for compensation paid to a physician for personal services rendered by the physician. We have compensation arrangements with a number of physicians for personal services, such as clinical advisory board services, speaking engagements and other consulting activities. We have structured these arrangements with terms intended to comply with the requirements of the personal services exception to Stark and Maryland Physician Self-Referral Law.
However, we cannot be certain that regulators would find these arrangements to be in compliance with Stark, the Maryland Physician Self-Referral Law, or similar state laws. We would be required to refund any payments we receive pursuant to a referral prohibited by these laws to the patient, the payor or the Medicare program, as applicable.
Sanctions for a violation of the Stark Law include the following:
| · | denial of payment for the services provided in violation of the prohibition; |
| · | refunds of amounts collected by an entity in violation of the Stark Law; |
| · | a civil penalty of up to $15,000 for each service arising out of the prohibited referral; |
| · | possible exclusion from federal healthcare programs, including Medicare and Medicaid; and |
| · | a civil penalty of up to $100,000 against parties that enter into a scheme to circumvent the Stark Law’s prohibition. |
These prohibitions apply regardless of the reasons for the financial relationship and the referral. No finding of intent to violate the Stark Law is required for a violation. In addition, knowing violations of the Stark Law may also serve as the basis for liability under the Federal False Claims Act, which prohibits knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to the U.S. Government.
Further, if we submit claims in violation of the Maryland Physician Self-Referral Law, we can be held liable to the payer for any reimbursement received for the services by us. Finally, other states have self-referral restrictions with which we have to comply that differ from those imposed by federal and Maryland law. While we have attempted to comply with the Stark Law and the Maryland Physician Self-Referral Law, it is possible that some of our financial arrangements with physicians could be subject to regulatory scrutiny at some point in the future, and we cannot provide assurance that we will be found to be in compliance with these laws following any such regulatory review.
Federal and State Anti-Kickback Laws
The Federal health care program Anti-Kickback Law makes it a felony for a person or entity, including a laboratory, to knowingly and willfully offer, pay, solicit or receive remuneration, directly or indirectly, in order to induce business that is reimbursable under any federal health care program. A violation of the Anti-Kickback Law may result in imprisonment for up to five years and fines of up to $250,000 in the case of individuals and $500,000 in the case of organizations. Convictions under the Anti-Kickback Law result in mandatory exclusion from federal health care programs for a minimum of five years. In addition, HHS has the authority to impose civil assessments and fines and to exclude health care providers and others engaged in prohibited activities from Medicare, Medicaid and other federal health care programs. Actions which violate the Anti-Kickback Law also incur liability under the Federal False Claims Act.
| 20 |
Although the Anti-Kickback Law applies only to federal health care programs, a number of states, including Maryland, have passed statutes substantially similar to the Anti-Kickback Law pursuant to which similar types of prohibitions are made applicable to all other health plans and third-party payers. Violations of Maryland’s anti-kickback law are punishable by tiered criminal penalties based on the crime with a maximum penalty of life imprisonment and fines of up to $200,000, or both. Civil penalties include three times the amount of any overpayment made in violation of the statute.
Federal and state law enforcement authorities scrutinize arrangements between health care providers and potential referral sources to ensure that the arrangements are not designed as a mechanism to induce patient care referrals or induce the purchase or prescribing of particular products or services. The law enforcement authorities, the courts and Congress have also demonstrated a willingness to look behind the formalities of a transaction to determine the underlying purpose of payments between health care providers and actual or potential referral sources. Generally, courts have taken a broad interpretation of the scope of the Anti-Kickback Law, holding that the statute may be violated if merely one purpose of a payment arrangement is to induce referrals or purchases.
In addition to statutory exceptions to the Anti-Kickback Law, regulations provide for a number of safe harbors. If an arrangement meets the provisions of a safe harbor, it is deemed not to violate the Anti-Kickback Law. An arrangement must fully comply with each element of an applicable safe harbor in order to qualify for protection. There are no regulatory safe harbors to the Maryland anti-kickback law.
Among the safe harbors that may be relevant to us is the discount safe harbor. The discount safe harbor potentially applies to discounts provided by providers and suppliers, including laboratories, to physicians or institutions. If the terms of the discount safe harbor are met, the discounts will not be considered prohibited remuneration under the Anti-Kickback Law. Maryland does not have a discount safe harbor.
The personal services safe harbor to the Anti-Kickback Law provides that remuneration paid to a referral source for personal services will not violate the Anti-Kickback Law provided all of the elements of that safe harbor are met. One element is that if the agreement is intended to provide for the services of the physician on a periodic, sporadic or part-time basis, rather than on a full-time basis for the term of the agreement, the agreement specifies exactly the schedule of such intervals, their precise length, and the exact charge for such intervals. Our personal services arrangements with some physicians may not meet the specific requirement of this safe harbor that the agreement specify exactly the schedule of the intervals of time to be spent on the services because the nature of the services, such as speaking engagements, does not lend itself to exact scheduling and therefore meeting this element of the personal services safe harbor is impractical. Failure to meet the terms of the safe harbor does not render an arrangement illegal. Rather, the government may evaluate such arrangements on a case-by-case basis, taking into account all facts and circumstances.
While we believe that we are in compliance with the Anti-Kickback Law and the Maryland anti-kickback law, there can be no assurance that our relationships with physicians, academic institutions and other customers will not be subject to investigation or challenge under such laws. If imposed for any reason, sanctions under the Anti-Kickback Law and the Maryland anti-kickback law could have a negative effect on our business.
Other Federal and State Fraud and Abuse Laws
In addition to the requirements discussed above, several other health care fraud and abuse laws could have an effect on our business. For example, provisions of the Social Security Act permit Medicare and Medicaid to exclude an entity that charges the federal health care programs substantially in excess of its usual charges for its services. The terms “usual charge” and “substantially in excess” are ambiguous and subject to varying interpretations.
Further, the Federal False Claims Act prohibits a person from knowingly submitting a claim, making a false record or statement in order to secure payment or retaining an overpayment by the federal government. In addition to actions initiated by the government itself, the statute authorizes actions to be brought on behalf of the federal government by a private party having knowledge of the alleged fraud, also known as qui tam lawsuits. Because the complaint is initially filed under seal, the action may be pending for some time before the defendant is even aware of the action. If the government is ultimately successful in obtaining redress in the matter or if the plaintiff succeeds in obtaining redress without the government’s involvement, then the plaintiff will receive a percentage of the recovery. It is not uncommon for qui tam lawsuits to be filed by employees, competitors or consultants. Finally, the Social Security Act includes its own provisions that prohibit the filing of false claims or submitting false statements in order to obtain payment. Violation of these provisions may result in fines, imprisonment or both, and possible exclusion from Medicare or Medicaid programs. Maryland has an analogous state false claims act applicable to state health plans and programs, as do many other states.
| 21 |
Maryland Laboratory Licensing
Maryland requires that any site that performs clinical laboratory testing located in the state of Maryland, with limited exceptions, must be licensed by the state, in addition to meeting federal CLIA requirements. As such, our laboratory in Gaithersburg, Maryland holds a current Maryland license and is subject to on site surveys by Maryland’s Office of Health Care Quality. Our license is due to be renewed in June 2016.
Other States’ Laboratory Licensing
In addition to Maryland, other states including California, Florida, New York, Pennsylvania, Rhode Island, and the District of Columbia, require licensing of out-of-state laboratories under certain circumstances. We have obtained a license to receive specimens from Pennsylvania, and we have submitted applications to Florida and New York. We intend to obtain licenses from additional states and jurisdictions where we believe we are required to be licensed, and believe we are in compliance with applicable licensing laws.
From time to time, we may become aware of other states that require out-of-state laboratories to obtain licensure in order to accept specimens from the state, and it is possible that other states do have such requirements or will have such requirements in the future. If we identify any other state with such requirements or if we are contacted by any other state advising us of such requirements, we intend to comply with such requirements.
International Regulation
Sales of diagnostic tests like our Acuitas MDRO test products outside the United States would be subject to foreign government regulations, which vary substantially from country to country. In order to market our products in other countries, we would need to obtain regulatory approvals and comply with extensive safety and quality regulations in other countries. OpGen’s Woburn, Massachusetts facility is currently ISO 13485 certified; the facility passed an inspection by our Notified Body in January 2016. While such certification is not required to distribute products internationally, the ISO 13485 certification implies that we are in compliance with the applicable regulatory requirements to distribute our medical devices internationally. OpGen currently distributes products in the European Union through its Denmark office. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ significantly. If we elect to, or are required to, seek clearance of or approval for any of our products from the FDA, we may be able to commercialize such products with shorter lead time in international markets, but would need to establish international operations in order to do so.
Environmental Matters
Our operations require the use of hazardous materials (including biological materials) which subject us to a variety of federal, state and local environmental and safety laws and regulations. Some of these regulations provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others’, business operations should contamination of the environment or individual exposure to hazardous substances occur. We cannot predict how changes in laws or new regulations will affect our business, operations or the cost of compliance.
Glossary
The following scientific, healthcare, regulatory and OpGen-specific terms are used throughout this Annual Report:
“ACOs” means accountable care organizations, a voluntary combination of doctors, hospitals and other health care providers and other health care system participants, including insurers, formed under the PPACA, to provide coordinated health care to patients.
“Acuitas CR Elite” is a comprehensive test for detection of CRE including our Acuitas MDRO gene test, culture based detection, and Acuitas resistome testing on positive specimens.
| 22 |
“Acuitas Lighthouse” is our bioinformatics product being internally developed to provide real-time information on the MDRO status for patients and hospitals. We combine our molecular test information and microbiology test results from our customized CLIA-based tests to create Acuitas Lighthouse profiles for hospitals, health systems and communities. Acuitas Lighthouse profiling facilitates MDRO tracking and results can be aggregated with hospital data to provide customized reports including alerts, prevalence, trend analysis and transmission information.
“Acuitas MDRO Gene Test” means our internally developed test that detects ten critical MDRO genes, including CRE (7 genes), ESBL (2 genes) and VRE resistant organisms, from one patient swab.
“Acuitas MDRO test products” means our Acuitas MDRO Gene Test, Acuitas CR Elite Test and Acuitas Resistome Test.
“Acuitas Resistome Test” means our rapid, high resolution test that includes additional resistance genes for carbapenems, ESBLs and AmpC.
“Annual Report” means this Annual Report on Form 10-K for the year ended December 31, 2015.
“antibiotic stewardship” has been defined by the CDC to mean hospital-based programs dedicated to improving use of antibiotic therapy with the goal of optimizing the treatment of infections and reducing the adverse events associated with antibiotic use.
“Argus System” means OpGen’s proprietary system used to perform Whole Genome Mapping.
“bioinformatics” refers to methods, algorithms and processes for the collection, classification, storage and analysis of biochemical and biological data and information using computers, especially as applied in molecular genetics and genomics. Our focus is on acquiring such data and information related to MDROs to assist in diagnosis and screening of patients and antibiotic stewardship initiatives by acute care hospitals. When we use the term “advanced bioinformatics,” we mean bioinformatics combined with higher levels of complexity, sophistication and subject matter expertise related to MDROs, diagnostics, antibiotic stewardship, and the development of associated analysis tools, or the novel application of existing bioinformatics in future products or services. In this Annual Report, we also sometimes use the phrase “bioinformatics products and services,” often interchangeably with “bioinformatics platform,” to describe the Company’s focus on the use of bioinformatics and advanced bioinformatics in its current and future product and service offerings.
“bioinformatics platform” means a combination of software tools and analytical processes that streamline the production and analysis of bioinformatics data. When we use the term bioinformatics platform, we are primarily referring to Acuitas Lighthouse.
“CDC” means the U.S. Centers for Disease Control and Prevention.
“C. difficile” means clostridium difficile, an MDRO that causes intestinal tract infections that can lead to sepsis.
“CLIA” means the Clinical Laboratory Improvements Act of 1988, as amended.
“CLIA lab” means a clinical or reference laboratory meeting the requirements of the Clinical Laboratory Improvements Act of 1988, as amended.
“CMS” means the Centers for Medicare and Medicaid Services.
“CRE” means Carbapenem-resistant Enterobacteriaceae, an MDRO.
“DNA probe analysis” is a test where an agent binds directly to a predefined or labeled sequence of nucleotides in a DNA molecule in order to detect unique nucleotide sequences within the molecule.
“DNA sequencing” is the process of determining the precise order of nucleotides within a DNA molecule.
“epidemiologically linked” means situations where it is shown that one person is the source of an infection that spreads through contact to one or more other persons.
| 23 |
“ESBL” means extended spectrum beta lactamase bacteria.
“FDA” means the U.S. Food and Drug Administration.
“HAIs” means hospital acquired infections. Such infections could arise first in the hospital or other healthcare setting, or could result from a patient, colonized with an organism, developing an active infection once admitted to the hospital or other healthcare setting.
“HIPAA” means the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH Act”). HIPAA and HITECH are federal laws mandating security and privacy of protected personal health information of patients.
“hospital biome” is used in this Annual Report to refer to the unique characteristic microbial environment found in a specific hospital or other healthcare setting, which could change from time to time based on the MDRO profile of the institution.
“ICU” means an intensive care unit in a health-care facility.
“KPC” means Klebsiella pneumoniae carbapenemase, an MDRO.
“LIMS” means a laboratory information management system.
“MDRO” means a multidrug-resistant organism.
“microfluidic” means devices or processes that are designed, manufactured or formulated to accommodate applications that require very small volumes of fluid, on the order of nanoliters or picoliters.
“nosocomial” means hospital acquired.
“PPACA” means the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act.
“sensitivity” of a clinical laboratory test reflects the probability that a patient with a specific bacterial organism present will have a positive test result.
“specificity” of a clinical laboratory test reflects the probability that a patient without the specific bacterial organism will have a negative test result.
“USDA” means the United States Department of Agriculture.
“WHO” means the World Health Organization.
“Whole Genome Mapping” means OpGen’s proprietary technology that provides a customer with a high resolution, ordered, whole genome restriction map generated from single DNA molecules extracted from organisms, such as bacteria, yeast or other fungi, plants or animals and humans. Whole Genome Mapping compliments genome assembly and enables scientist to identify highly repetitive regions, tandem repeats and translocations that are difficult to identify and clarify with sequencing alone.
Employees
As of December 31, 2015, we had 48 employees worldwide, with 47 employed in the United States and one employed in Denmark. The 47 employees in the United States primarily work in our Gaithersburg, Maryland and Woburn, Massachusetts locations. None of our employees are the subject of collective bargaining arrangements, and our management considers its relationships with employees to be good.
Corporate Information
OpGen, Inc. was incorporated in Delaware in 2001. On July 14, 2015, it acquired AdvanDx in the Merger. The Company’s headquarters are in Gaithersburg, Maryland, and its principal operations are in Gaithersburg, Maryland and Woburn, Massachusetts. The Company also has operations in Copenhagen, Denmark.
| 24 |
Available Information
The Company maintains a website at www.opgen.com. Our Code of Business Conduct and Ethics is available on our website. We are not incorporating our website into this Annual Report. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and amendments to those reports, filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, are available free of charge on our website as soon as practicable after electronic filing of such material with, or furnishing it to, the U.S. Securities and Exchange Commission (the “SEC”). This information may be read and copied at the Public Reference Room of the SEC at 100 F Street, N.E., Washington D.C. 20549. The SEC also maintains an internet website that contains reports, proxy statements, and other information about issuers, like OpGen who file electronically with the SEC. The address of the site is http://www.sec.gov.
Seasonality of Business
We do not believe our business is subject to seasonality. However, our business can be subject to and affected by the business practices of our business partners. To the extent that the availability of inventory or materials from or development practices of our partners is seasonal, our sales may be subject to fluctuations quarter to quarter or year over year.
| 25 |
The following are significant factors known to us that could materially harm our business, financial condition or operating results or could cause our actual results to differ materially from our anticipated results or other expectations, including those expressed in any forward-looking statement made in this Annual Report. The risks described are not the only risks facing us. Additional risks and uncertainties not currently known to us, or that we currently deem to be immaterial, also may adversely affect our business, financial condition and operating results. If any of these risks actually occur, our business, financial condition, and operating results could suffer significantly.
Risks Related to our Business
We have a history of losses, and we expect to incur losses for the next several years. The report of our independent registered public accounting firm on our financial statements for the years ended December 31, 2015 and 2014 contains explanatory language that substantial doubt exists about our ability to continue as a going concern.
We have incurred substantial losses since our inception, and we expect to continue to incur additional losses for the next several years. For the years ended December 31, 2015 and 2014 we had a net loss of $17.4 million and $5.7 million, respectively. From our inception through December 31, 2015, we had an accumulated deficit of $114.1 million. The report of our independent registered public accounting firm on our financial statements for the years ended December 31, 2015 and 2014 contains explanatory language that substantial doubt exists about our ability to continue as a going concern. In May 2015, the Company completed its initial public offering (“IPO”), pursuant to which the Company offered and sold 2,850,000 units, each unit consisting of one share of common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 Units in the IPO. After considering the demand notes, underwriting discounts and commissions and offering expenses, the total net cash proceeds to the Company were $12.1 million. In connection with the IPO, all of the Company’s outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock. On July 14, 2015, we acquired AdvanDx pursuant to consummation of the Merger under the Merger Agreement. We issued 681,818 shares of our common stock in the AdvanDx Merger transaction. In addition, in July 2015, we received an additional investment of $6.0 million from Merck Global Innovation Fund, LLC (“Merck GHI”), consisting of a $5 million investment in common stock and the issuance of a $1 million senior secured promissory note with a two-year term.
The $1 million senior secured promissory note issued to Merck GHI in July 2015 is secured by a security interest in substantially all of our assets, including our intellectual property assets. The secured promissory note requires interest-only payments at a rate of 8% per annum for two years, with the principal due and payable on July 14, 2017. Such secured creditor rights could negatively impact our ability to raise money in the future. If we default on payments under the promissory note, Merck GHI has the rights of a secured creditor. If those rights are exercised, it could have a material adverse effect on our financial condition.
We expect to continue to incur significant operating expenses and anticipate that our expenses will increase due to costs relating to, among other things:
| · | commercializing our diagnostic test products and Acuitas Lighthouse bioinformatics system and developing rapid molecular diagnostic products and services; |
| · | continued integration of the AdvanDx products into our product offerings; |
| · | developing, presenting and publishing additional clinical and economic utility data intended to increase clinician adoption of our current and future products and services; |
| · | expansion of our operating capabilities; |
| · | maintenance, expansion and protection of our intellectual property portfolio and trade secrets; |
| · | future clinical trials; |
| · | expansion of the size and geographic reach of our sales force and our marketing capabilities to commercialize potential future products and services; |
| · | employment of additional clinical, quality control, scientific, customer service, laboratory, billing and reimbursement and management personnel; and |
| · | employment of operational, financial, accounting and information systems personnel, consistent with expanding our operations and our status as a public company. |
Even if we achieve significant revenues, we may not become profitable, and even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain consistently profitable could adversely affect the market price of our common stock and could significantly impair our ability to raise capital, expand our business or continue to pursue our growth strategy. We anticipate that we will need to raise additional capital to support our operations in 2016. While Merck GHI has the right to participate in future capital raising transactions, there is no assurance that it will invest further in the Company. We have no committed sources of capital and may find it difficult to raise money on terms favorable to us or at all. The failure to obtain sufficient capital to support our operations could have an adverse effect on our business, financial condition and results of operations.
| 26 |
We may fail to realize some or all of the anticipated benefits of the business combination of OpGen and AdvanDx, which may adversely affect the value of our common stock.
The success of the continued integration of AdvanDx will depend, in part, on our ability to realize the anticipated benefits and cost savings from combining the respective business and operations of OpGen and AdvanDx. To realize these anticipated benefits and cost savings, we must successfully combine the acquired business with our legacy operations and integrate our respective operations, technologies and personnel, which is particularly challenging given the geographic and cultural differences between the personnel and facilities based in Maryland and Massachusetts, plus the European operations of AdvanDx, and the lack of experience we have in combining businesses. If we are not able to fully achieve these objectives within the anticipated time frame or at all, the anticipated benefits and cost savings of the acquisition may not be realized fully or at all or may take longer to realize than expected, and the value of our common stock may be adversely affected. In addition, the overall integration of the businesses is a complex, time-consuming and expensive process that, without proper planning and effective and timely implementation, could significantly disrupt our operations.
Risks in integrating AdvanDx into our operations in order to realize the anticipated benefits of the acquisition include, among other factors:
| · | coordinating research and development activities to enhance the introduction of new diagnostic tests and technology of the combined business; |
| · | failure to successfully integrate and harmonize financial reporting and information technology systems of the two companies; |
| · | retaining each company’s relationships with its partners; |
| · | retaining and integrating key employees from OpGen and AdvanDx; |
| · | managing effectively the diversion of management’s attention from business matters to integration issues; |
| · | combining research and development capabilities effectively and quickly; |
| · | integrating partnership efforts so that new partners acquired can easily do business with us; and |
| · | transitioning all facilities to a common information technology environment. |
Actual cost synergies, if achieved at all, may be lower than we expect and may take longer to achieve than anticipated. If we are not able to adequately address these challenges, we may be unable to successfully integrate the operations of the business acquired from AdvanDx into our own, or to realize the anticipated benefits of the integration. The anticipated benefits and synergies assume a successful integration and are based on projections, which are inherently uncertain, and other assumptions. Even if integration is successful, anticipated benefits and synergies may not be achieved. An inability to realize the full extent of, or any of, the anticipated benefits of the acquisition, as well as any delays encountered in the integration process, could have an adverse effect on our business and results of operations, which may affect the value of the shares of our common stock.
We have incurred significant costs related to the Merger. If we are unable to offset the costs of the acquisition through realization of efficiencies, our financial condition, liquidity and results of operations will suffer.
We have incurred, and expect to continue to incur, various non-recurring costs associated with combining the operations of OpGen and AdvanDx, including, but not limited to, legal, accounting and financial advisory fees. The substantial majority of non-recurring expenses have been composed of these costs and expenses related to the execution of the acquisition, facilities and systems consolidation costs and employment-related costs. We have also incurred fees and costs related to formulating and implementing integration plans. Additional unanticipated costs may be incurred in the integration of the businesses. Although we expect that the elimination of duplicative costs, as well as the realization of other efficiencies related to the integration of the businesses, should allow us to offset incremental acquisition and acquisition-related costs over time, this net benefit may not be achieved in the near term, or at all. Furthermore, if we are unable to achieve this net benefit from the acquisition, our financial condition, liquidity and results of operations will suffer.
| 27 |
We expect to make significant additional investment in the future related to our diagnostic products. If we are unable to make such investments our business will suffer.
We anticipate that we will need to make significant investments in the Acuitas MDRO Gene Test and QuickFISH products in order to make the combined business profitable. We have identified potential synergies for future rapid diagnostic test developments based on the combined business’s product offerings, but need to expend significant investments to develop such products. There can be no assurance that we can obtain sufficient resources or capital from operations or future financings to support the combined business operations. In the event the Company is unable to successfully raise additional capital, we will not have sufficient cash flows and liquidity to finance our business operations as currently contemplated. Accordingly, in such circumstances the Company would be compelled to reduce general and administrative expenses and delay research and development projects, including the purchase of scientific equipment and supplies, until it is able to obtain sufficient financing, or pursue other strategic alternatives which may include licensing and/or partnering arrangements or mergers and acquisitions.
We are developing new diagnostic products for the more rapid identification of MDROs and antibiotic therapy selection. If we are unable to successfully develop, receive regulatory clearance or approval for or commercialize such new products, our business will be materially, adversely affected.
We are currently beginning development of a new one-hour rapid array diagnostic product that we believe could help address many of the current issues with the need for more rapid identification of infectious diseases and testing for antibiotic resistance. Development of new diagnostic products is difficult and we cannot assure you that we will be successful in such product development efforts, or, if successful, that we will receive the necessary regulatory clearances to commercialize such products. Our intent is to identify over 100 antibiotic resistance genes to help guide clinician antibiotic therapy decisions when test results are evaluated using the Acuitas Lighthouse. Although we have demonstrated preliminary feasibility, such product development efforts will require us to work collaboratively with other companies, academic and government laboratories, and healthcare providers to access sufficient numbers of microbial isolates, develop the diagnostic tests, identify and license a third party rapid array platform, successfully conduct the necessary clinical trials and apply for and receive regulatory clearances or approvals for the intended use of such diagnostic tests. In addition, we would need to successfully commercialize such products. Such product development, clearance or approval and commercialization activities are time-consuming, expensive and we are not assured that we will have sufficient funds to successfully complete such efforts. We currently estimate that such rapid array diagnostic tests will be commercially available by 2019. Any significant delays or failures in this process could have a material adverse effect on our business and financial condition.
We are an early commercial stage company and may never be profitable.
We rely principally on the commercialization of our QuickFISH and Acuitas MDRO test products and our Acuitas Lighthouse bioinformatics system and services to generate future revenue growth. To date, the Acuitas MDRO test products and Acuitas Lighthouse products and services have delivered only minimal revenue. We believe that our commercialization success is dependent upon our ability to significantly increase the number of hospitals, long-term care facilities and other inpatient healthcare settings that use our products. We have experienced very limited revenue and customer adoption for our Acuitas MDRO products to date. If demand for products does not increase as quickly as we have planned, we may be unable to increase our revenue levels as expected. We are currently not profitable. Even if we succeed in increasing adoption of our products by our target markets, maintaining and creating relationships with our existing and new customers and developing and commercializing additional molecular testing products, we may not be able to generate sufficient revenue to achieve or sustain profitability.
Our products may never achieve significant commercial market acceptance.
Our products and services may never gain significant acceptance in the marketplace and, therefore, may never generate substantial revenue or profits for us. Our ability to achieve commercial market acceptance for our products will depend on several factors, including:
| · | our ability to convince the medical community of the clinical utility of our products and services and their potential advantages over existing tests; |
| · | our ability to successfully develop more rapid diagnostic products and services; |
| · | our ability to convince the medical community of the accuracy and speed of our products and services, as contrasted with the current methods available; |
| · | the willingness of hospitals and physicians to use our products and services; and |
| · | the recognition by inpatient health care facilities of the patient safety, improved outcome and cost-effectiveness benefits of using our products and the willingness to pay for them without reimbursement. |
If any of these actions were to occur it could harm our reputation and cause our product sales and profitability to suffer and may prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
Some of the clearances obtained for the QuickFISH products we currently offer for sale are subject to limitations on the intended uses for which the product may be marketed, which can reduce our potential to successfully commercialize the product and generate revenue from the product. If the FDA determines that our promotional materials, labeling, training or other marketing or educational activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
| 28 |
In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
If we were to lose, or have restrictions imposed on, the FDA clearances received to date, our business, operations, financial condition and results of operations is likely to be significantly adversely affected.
Our future success is dependent upon our ability to expand our customer base.
The current customers we are targeting for our Acuitas MDRO Gene Test are acute care hospitals, particularly those with advanced care units, such as intensive care units, and community-based hospitals. We need to provide a compelling case for the savings, patient safety and recovery, reduced length of stay and reduced costs that come from adopting our MDRO diagnosis and management products and services. If we are not able to successfully increase our customer base, sales of our products and our margins may not meet expectations. Attracting new customers and introducing new products and services requires substantial time and expense. Any failure to expand our existing customer base, or launch new products and services, would adversely affect our ability to improve our operating results.
We have seen declining revenues from our current customers for our QuickFISH products as we work to automate and expand our product offerings. We may not be successful in developing such automated and rapid diagnostic test products, which would materially, adversely affect our business.
Our sales cycle is lengthy and variable, which makes it difficult for us to forecast revenue and other operating results.
The sales cycles for our Acuitas MDRO test products and our Acuitas Lighthouse products are lengthy, which makes it difficult for us to accurately forecast revenues in a given period, and may cause revenue and operating results to vary significantly from period to period. Potential customers for our products typically need to commit significant time and resources to evaluate our products, and their decision to purchase our products may be further limited by budgetary constraints and numerous layers of internal review and approval, which are beyond our control. We spend substantial time and effort assisting potential customers in evaluating our products. Even after initial approval by appropriate decision makers, the negotiation and documentation processes for the actual adoption of our products on a facility-wide basis can be lengthy. As a result of these factors, based on our experience to date, our sales cycle, the time from initial contact with a prospective customer to routine commercial use of our products, has varied and could be 12 months or longer, which has made it difficult for us to accurately project revenues and operating results. In addition, the revenue generated from sales of our products may fluctuate from time to time due to changes in the testing volumes of our customers. As a result, our results may fluctuate on a quarterly basis, which may adversely affect the price of our common stock.
The loss of key members of our senior management team or our inability to attract and retain highly skilled scientists and laboratory and field personnel could adversely affect our business.
Our success depends largely on the skills, experience and performance of key members of our executive management team. The efforts of each of these persons will be critical to us as we continue to develop our products and services and as we attempt to transition to a company with broader product offerings. If we were to lose one or more of these key employees, we may experience difficulties in competing effectively, developing our technologies and implementing our business strategies.
Our research and development programs and commercial laboratory operations depend on our ability to attract and retain highly skilled scientists and technicians, particularly as we seek to further integrate operations of the combined company. We may not be able to attract or retain qualified scientists and technicians in the future due to the intense competition for qualified personnel among life science businesses. We also face competition from universities, public and private research institutions and other organizations in recruiting and retaining highly qualified scientific personnel.
| 29 |
In addition, our success depends on our ability to attract and retain laboratory and field personnel with extensive experience in infection control in inpatient settings. We may have difficulties locating, recruiting or retaining qualified salespeople, which could cause a delay or decline in the rate of adoption of our current and future products and service offerings. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will adversely affect our ability to support our discovery, development, verification and commercialization programs.
We have limited experience in marketing and selling our products, and if we are unable to adequately address our customers’ needs, it could negatively impact sales and market acceptance of our product and we may never generate sufficient revenue to achieve or sustain profitability.
We sell our products through our own direct sales force, which sells both our QuickFISH and our Acuitas MDRO test products. In addition, our product offerings, including our Acuitas Lighthouse bioinformatics product and surveillance product offerings may be offered and sold to different potential customers or involve discussions with multiple personnel in in-patient facilities. Our future sales will depend in large part on our ability to increase our marketing efforts and adequately address our customers’ needs. The inpatient health care facility industry is a large and diverse market. As a result, we believe it is necessary to maintain a sales force that includes sales representatives with specific technical backgrounds that can support our customers’ needs. We will also need to attract and develop sales and marketing personnel with industry expertise. Competition for such employees is intense. We may not be able to attract and retain sufficient personnel to maintain an effective sales and marketing force. If we are unable to successfully market our products and adequately address our customers’ needs, it could negatively impact sales and market acceptance of our products and we may never generate sufficient revenue to achieve or sustain profitability.
We may be unable to manage our future growth effectively, which could make it difficult to execute our business strategy.
We commenced the formal commercial launch of our CLIA lab in late 2013, launched our Acuitas MDRO Gene Test in the second quarter of 2014, launched our Acuitas CR Elite Test in December 2014, our Acuitas Resistome Test in the second quarter of 2015, and we began using Acuitas Lighthouse portal in December 2015. In addition, we integrated the sales of our Acuitas MDRO and QuickFISH products beginning in the third quarter of 2015. We anticipate future growth in our business operations. This future growth could create strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service and sales force management. We may not be able to maintain the quality or expected turn-around times of our diagnostic or screening results, or satisfy customer demand as it grows. Our ability to manage our growth properly will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures. The time and resources required to implement the systems to handle such growth is uncertain, and failure to complete this in a timely and efficient manner could adversely affect our operations.
If the utility of our current products and products in development is not supported by studies published in peer-reviewed medical publications, the rate of adoption of our current and future products and services by clinicians and healthcare facilities may be negatively affected.
The results of our clinical and economic validation studies involving our Acuitas MDRO test products have been presented at major infectious disease and infection control society meetings. We need to maintain and grow a continued presence in peer-reviewed publications to promote clinician adoption of our products. We believe that peer-reviewed journal articles that provide evidence of the utility of our current and future solutions and adoption by key opinion leaders in the infectious disease market are very important to the commercial success of our current and any future products. Clinicians typically take a significant amount of time to adopt new products and testing practices, partly because of perceived liability risks and the uncertainty of a favorable cost/benefit analysis. It is critical to the success of our sales efforts that we educate a sufficient number of clinicians and administrators about our products and demonstrate the clinical benefits of these solutions. Clinicians may not adopt our current and future solutions unless they determine, based on published peer-reviewed journal articles and the experience of other clinicians, that our products provide accurate, reliable, useful and cost-effective information that is useful in MDRO diagnosis, screening and outbreak prevention. If our current and future solutions or the technology underlying our products and services or our future product offerings do not receive sufficient favorable exposure in peer-reviewed publications, the rate of clinician adoption could be negatively affected. The publication of clinical data in peer-reviewed journals is a crucial step in commercializing our products, and our inability to control when, if ever, results are published may delay or limit our ability to derive sufficient revenue from any product that is the subject of a study.
| 30 |
Our products and services are not covered by reimbursement by Medicare, Medicaid and other governmental and third party payors. If we cannot convince our customers that the savings from use of our products and services will increase their overall reimbursement, our business could suffer.
Our products and services do not currently receive reimbursement from Medicare, Medicaid, other governmental payors or commercial third party payors. The recent policy and rule changes in reimbursement announced by CMS, including potential financial incentives for reductions in HAIs, and penalties and decreased Medicare reimbursement for patients with HAIs provide us with an opportunity to establish a business case for the purchase and use of our screening and diagnostic products and services. If we cannot convince our customers that the savings from use of our products and services will increase or stabilize their overall profitability and improve clinical outcomes, our business will suffer.
The performance of clinical and economic utility studies is expensive and demands significant attention from our management team.
The performance of clinical and economic utility studies is expensive and demands significant attention from our management team. Data collected from these studies may not be positive or consistent with our existing data, or may not be statistically significant or compelling to the medical community. If the results obtained from our ongoing or future studies are inconsistent with certain results obtained from our previous studies, adoption of our current and future solutions would suffer and our business would be harmed.
If our sole laboratory facility becomes inoperable, we will be unable to perform Acuitas MDRO test products and future solutions, if any, and our business will be harmed.
We perform all of our diagnostic services in our CLIA laboratory located in Gaithersburg, Maryland. We do not have redundant laboratory facilities. Our facility and the equipment we use to perform our diagnostic and screening assays would be costly to replace and could require substantial lead time to repair or replace, if damaged or destroyed. The facility may be harmed or rendered inoperable by natural or man-made disasters, including flooding and power outages, which may render it difficult or impossible for us to perform our tests for some period of time. The inability to perform our tests may result in the loss of customers or harm our reputation, and we may be unable to regain those customers in the future. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
In order to establish a redundant laboratory facility, we would have to spend considerable time and money securing adequate space, constructing the facility, recruiting and training employees, and establishing the additional operational and administrative infrastructure necessary to support a second facility. Additionally, any new clinical laboratory facility opened by us would be required to be certified under CLIA, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. We would also be required to secure and maintain state licenses required by several states, including Maryland, California, Florida, New York and Pennsylvania which can take a significant amount of time and result in delays in our ability to begin operations at that facility. If we failed to secure any such licenses, we would not be able to process samples from recipients in such states. We also expect that it would be difficult, time-consuming and costly to train, equip and use a third-party to perform tests on our behalf. We could only use another facility with the established state licensures and CLIA certification necessary to perform our current or future tests following validation and other required procedures. We cannot assure you that we would be able to find another CLIA-certified facility willing or able to adopt our current or future tests and comply with the required procedures, or that this laboratory would be willing or able to perform the tests for us on commercially reasonable terms.
| 31 |
In order to meet the turn-around time required for our Acuitas MDRO test products, we rely on transport of specimens to our sole laboratory facility; any disruption in such transport could significantly adversely affect our business.
Our current customers for our Acuitas MDRO test products are located near to our sole laboratory facility in Gaithersburg, Maryland. As we expand our customer base, we will need to secure the proper licenses for shipment of specimens and rely on accurate and timely delivery of the specimens by overnight delivery services such as FedEx. Any failure to procure the proper licenses, to comply with the license regulations or to receive undamaged specimens from overnight delivery services could adversely affect our business and reputation.
We rely on a limited number of suppliers or, in some cases, sole suppliers, for some of our laboratory instruments and materials and may not be able to find replacements or immediately transition to alternative suppliers.
We rely on several sole suppliers and manufacturers, including Fluidigm Corporation, for supplying certain laboratory reagents, raw materials, supplies and substances which we use in our laboratory operations and products and to manufacture our products. An interruption in our operations could occur if we encounter delays or difficulties in securing these items or manufacturing our products, and if we cannot, then obtain an acceptable substitute. Any such interruption could significantly affect our business, financial condition, results of operations and reputation.
We believe that there are only a few other equipment manufacturers that are currently capable of supplying and servicing the equipment and other supplies and materials necessary for our laboratory operations. The use of equipment or materials furnished by these replacement suppliers would require us to alter our laboratory operations. Transitioning to a new supplier would be time consuming and expensive, may result in interruptions in our laboratory operations, could affect the performance specifications of our laboratory operations or could require that we revalidate our products. There can be no assurance that we will be able to secure alternative equipment and other materials, and bring such equipment and materials on line and revalidate them without experiencing interruptions in our workflow. If we should encounter delays or difficulties in securing, reconfiguring or revalidating the equipment we require for our products, our business, financial condition, results of operations and reputation could be adversely affected.
If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenue or achieve and sustain profitability.
Our competitors include rapid diagnostic testing and traditional microbiology companies, commercial laboratories, information technology companies, and hospital laboratories who may internally develop testing capabilities. Principal competitive factors in our target market include: organizational size, scale, and breadth of product offerings; rapidity of test results; quality and strength of clinical and analytical validation data and confidence in diagnostic results; cost effectiveness; ease of use; and regulatory approval status.
Our principal competition comes from traditional methods used by healthcare providers to diagnose and screen for MDROs and from other molecular diagnostic companies creating screening and diagnostic products such as Cepheid, Becton-Dickinson, bioMerieux, Accelerate Diagnostics, T2 Biosystems and Nanosphere.
We also face competition from commercial laboratories, such as Bio-Reference Laboratories, Inc., Laboratory Corporation of America Holdings and Quest Diagnostics Incorporated, which have strong infrastructure to support the commercialization of diagnostic services.
Competitors may develop their own versions of our solution in countries where we do not have patents or where our intellectual property rights are not recognized.
Many of our potential competitors have widespread brand recognition and substantially greater financial, technical, research and development and selling and marketing capabilities than we do. Others may develop products with prices lower than ours that could be viewed by hospitals, physicians and payers as functionally equivalent to our solution, or offer solutions at prices designed to promote market penetration, which could force us to lower the list prices of our solutions and affect our ability to achieve profitability. If we are unable to change clinical practice in a meaningful way or compete successfully against current and future competitors, we may be unable to increase market acceptance and sales of our products, which could prevent us from increasing our revenue or achieving profitability and could cause our stock price to decline.
| 32 |
If we are unable to develop products to keep pace with rapid technological, medical and scientific change, our operating results and competitive position could be harmed. New test development involves a lengthy and complex process, and we may not be successful in our efforts to develop and commercialize our diagnostic and screening products and services. The further development and commercialization of additional diagnostic and screening solutions are key to our growth strategy.
A key element of our strategy is to discover, develop, validate and commercialize a portfolio of additional diagnostic and screening products and services to rapidly diagnose and effectively treat MDRO infections and reduce the associated costs to patients, inpatient facilities and the health care industry. We cannot assure you that we will be able to successfully complete development of or commercialize any of our planned future products and services, or that they will be clinically usable. The product development process involves a high degree of risk and may take up to several years or more. Our new product development efforts may fail for many reasons, including:
| · | failure of the test at the research or development stage; |
| · | lack of clinical validation data to support the effectiveness of the test; |
| · | delays resulting from the failure of third-party suppliers or contractors to meet their obligations in a timely and cost-effective manner; |
| · | failure to obtain or maintain necessary certifications, licenses, clearances or approvals to market or perform the test; or |
| · | lack of commercial acceptance by inpatient health care facilities. |
Few research and development projects result in commercial products, and success in early clinical studies often is not replicated in later studies. At any point, we may abandon development of new products, or we may be required to expend considerable resources repeating clinical studies or trials, which would adversely impact the timing for generating potential revenues from those new products. In addition, as we develop new products, we will have to make additional investments in our sales and marketing operations, which may be prematurely or unnecessarily incurred if the commercial launch of a product is abandoned or delayed.
Our insurance policies are expensive and protect us only from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability, employee benefits liability, property, umbrella, business interruption, workers’ compensation, product liability, errors and omissions and directors’ and officers’ insurance. We do not know, however, if we will be able to maintain existing insurance with adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
If we use hazardous materials in a manner that causes injury, we could be liable for damages.
Our activities currently require the use of hazardous materials and the handling of patient samples. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject on an ongoing basis to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. We are, or may be in the future, subject to compliance with additional laws and regulations relating to the protection of the environment and human health and safety, and including those relating to the handling, transportation and disposal of medical specimens, infectious and hazardous waste and Occupational Health and Safety (“OSHA”), requirements.
| 33 |
If we are sued for product liability or errors and omissions liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of our Acuitas MDRO test products could lead to product liability claims if someone were to allege that an Acuitas MDRO test product failed to perform as it was designed. We may also be subject to liability for errors in the results we provide to physicians or for a misunderstanding of, or inappropriate reliance upon, the information we provide. For example, if we diagnosed a patient as having an MDRO but such result was a false positive, the patient could be unnecessarily isolated in an in-patient setting or receive inappropriate treatment. We may also be subject to similar types of claims related to products we may develop in the future. A product liability or errors and omissions liability claim could result in substantial damages and be costly and time consuming for us to defend. Although we maintain product liability and errors and omissions insurance, we cannot assure you that our insurance would fully protect us from the financial impact of defending against these types of claims or any judgments, fines or settlement costs arising out of any such claims. Any product liability or errors and omissions liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could cause injury to our reputation or cause us to suspend sales of our products and solutions. The occurrence of any of these events could have an adverse effect on our business and results of operations.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
We have incurred net losses since inception and do not expect to become profitable in 2016 or for several years thereafter. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until such unused losses expire. We may be unable to use these net operating loss carryforwards (“NOLs”), and certain tax credit carryforwards to offset income before such unused NOLs tax credit carryforwards expire. Under Section 382 of the Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes to offset its post-change income may be further limited. The Merger with AdvanDx resulted in an ownership change for AdvanDx and, accordingly, AdvanDx’s net operating loss carryforwards and certain other tax attributes in U.S. taxing jurisdictions are subject to limitations on their use after the Merger. OpGen’s net operating loss carryforwards may also be subject to limitation as a result of prior shifts in equity ownership and/or the Merger. Additional ownership changes in the future could result in additional limitations on our net operating loss carryforwards. Consequently, even if we achieve profitability, we may not be able to utilize a material portion of our net operating loss carryforwards and other tax attributes, which could have a material adverse effect on cash flow and results of operations. We have not performed an analysis on previous ownership changes. It is possible that we have experienced an ownership change, or that we will experience an ownership change in the future. We had U.S. federal NOL carryforwards of $90.3 million and research and development tax credits of $2.0 million as of December 31, 2015, that may already be or could be limited if we experience an ownership change.
We may be adversely affected by the current economic environment and future adverse economic environments.
Our ability to attract and retain customers, invest in and grow our business and meet our financial obligations depends on our operating and financial performance, which, in turn, is subject to numerous factors, including the prevailing economic conditions and financial, business and other factors beyond our control, such as the rate of unemployment, the number of uninsured persons in the United States and inflationary pressures. We cannot anticipate all the ways in which the current economic climate and financial market conditions, and those in the future, could adversely impact our business.
We are exposed to risks associated with reduced profitability and the potential financial instability of our customers, many of which may be adversely affected by volatile conditions in the financial markets. For example, unemployment and underemployment, and the resultant loss of insurance, may decrease the demand for healthcare services and diagnostic testing. If fewer patients are seeking medical care because they do not have insurance coverage, we may experience reductions in revenues, profitability and/or cash flow. In addition, if economic challenges in the United States result in widespread and prolonged unemployment, either regionally or on a national basis, a substantial number of people may become uninsured or underinsured. To the extent such economic challenges result in less demand for our proprietary tests, our business, results of operations, financial condition and cash flows could be adversely affected.
Risks Related to Regulation of our Business
If we fail to comply with federal, state and foreign laboratory licensing requirements, we could lose the ability to perform our tests or experience disruptions to our business.
We are subject to CLIA for our Acuitas MDRO tests, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA regulations mandate specific standards in the areas of personnel qualifications, administration and participation in proficiency testing, patient test management and quality assurance. CLIA certification is also required in order for us to be eligible to bill state and federal healthcare programs, as well as many private third-party payors. To renew these certifications, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make random inspections of our clinical reference laboratories.
| 34 |
We are also required to maintain state licenses to conduct testing in our laboratories. Maryland law requires that we maintain a state license and establishes standards for the day-to-day operation of our clinical reference laboratory in Gaithersburg, including the training and skills required of personnel and quality control matters. In addition, our clinical reference laboratory is required to be licensed on a test-specific basis by New York State. New York law also mandates proficiency testing for laboratories licensed under New York state law, regardless of whether such laboratories are located in New York. Moreover, several other states including California, Pennsylvania, and Florida require that we hold licenses to test samples from patients in those states. Other states may adopt similar requirements in the future.
If we were to lose, or have restrictions imposed on, our CLIA certificate or Maryland license for our Gaithersburg laboratory, whether as a result of revocation, suspension or limitation, we would no longer be able to perform our test products, which would eliminate our primary source of revenue and harm our business. If we cannot secure a license from New York or from other states where we are required to hold licenses, we will not be able to test specimens from those states.
A number of the AdvanDx products are regulated by the FDA and non-U.S. regulatory authorities. If we or our suppliers fail to comply with ongoing FDA, or other foreign regulatory authority, requirements, or if we experience unanticipated problems with the products, these products could be subject to restrictions or withdrawal from the market.
We do not have significant experience in complying with the rules and regulations of the FDA and foreign regulatory authorities. The AdvanDx products regulated as medical devices, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such products, are subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory bodies. In particular, we and our suppliers are required to comply with FDA’s Quality System Regulations (“QSR”), and International Standards Organization (“ISO”), regulations for the manufacture, labeling, distribution and promotion of the QuickFISH products and other regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval. Regulatory bodies, such as the FDA, enforce the QSR and other regulations through periodic inspections. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA and other regulatory bodies, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in, among other things, any of the following enforcement actions: (1) untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; (2) unanticipated expenditures to address or defend such actions; (3) customer notifications for repair, replacement and refunds; (4) recall, detention or seizure of our products; (5) operating restrictions or partial suspension or total shutdown of production; (6) refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products; (7) operating restrictions; (8) withdrawing 510(k) clearances or PMA approvals that have already been granted; (9) refusal to grant export approval for our products; or (10) criminal prosecution.
If any of these actions were to occur it could harm our reputation and cause our product sales and profitability to suffer and may prevent us from generating revenue. Furthermore, if any of our key component suppliers are not in compliance with all applicable regulatory requirements we may be unable to produce our products on a timely basis and in the required quantities, if at all.
We are also subject to periodic inspections by the FDA to determine compliance with the FDA’s requirements, including primarily the quality system regulations and medical device reporting regulations. The results of these inspections can include inspectional observations on FDA’s Form 483, warning letters, or other forms of enforcement. Since 2009, the FDA has significantly increased its oversight of companies subject to its regulations, by hiring new investigators and stepping up inspections of manufacturing facilities. The FDA has recently also significantly increased the number of warning letters issued to companies. If the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our FDA-cleared products are ineffective or pose an unreasonable health risk, the FDA could take a number of regulatory actions, which could materially adversely affect our business.
| 35 |
Some of the clearances obtained are subject to limitations on the intended uses for which the product may be marketed, which can reduce our potential to successfully commercialize the product and generate revenue from the product. If the FDA determines that our promotional materials, labeling, training or other marketing or educational activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
If we were to lose, or have restrictions imposed on, FDA clearances received to date, our business, operations, financial condition and results of operations would likely be significantly adversely affected.
If the FDA were to begin regulating our laboratory tests, we could incur substantial costs and delays associated with trying to obtain premarket clearance or other approvals.
Clinical laboratory tests, like our Acuitas MDRO Gene Test, are regulated under CLIA, as well as by applicable state laws. Historically, most laboratory developed tests (“LDTs”) were not subject to FDA regulations applicable to medical devices, although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation. The FDA defines the term “laboratory developed test” as an in vitro diagnostic test that is intended for clinical use and designed, manufactured and used within a single laboratory. We believe that our Acuitas MDRO test products are LDTs. Until 2014, the FDA exercised enforcement discretion such that it did not enforce provisions of the Food, Drug, and Cosmetic Act (the “FDA Act”) with respect to LDTs. In July 2014, due to the increased proliferation of LDTs for complex diagnostic testing and concerns with several high-risk LDTs related to lack of evidentiary support for claims, erroneous results and falsification of data, the FDA issued guidance that, when finalized, would adopt a risk-based framework that would increase FDA oversight of LDTs. As part of this developing framework, FDA issued draft guidance in October 2014, informing manufacturers of LDTs of its intent to collect information from laboratories regarding their current LDTs and newly developed LDTs through a notification process. The FDA will use this information to classify LDTs and to prioritize enforcement of premarket review requirements for categories of LDTs based on risk, using a public process. Specifically, the FDA plans to use advisory panels to provide recommendations to the agency on LDT risks, classification and prioritization of enforcement of applicable regulatory requirements on certain categories of LDTs, as appropriate.
We cannot provide any assurance that FDA regulation, including premarket review, will not be required in the future for our tests, whether through additional guidance or regulations issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. It is possible that legislation will be enacted into law, regulations could be promulgated or guidance could be issued by the FDA which may result in increased regulatory burdens for us to continue to offer our tests or to develop and introduce new tests. We cannot predict the timing or content of future legislation enacted, regulations promulgated or guidance issued regarding LDTs, or how it will affect our business.
If FDA premarket review, including clearance or approval, is required for our Acuitas MDRO test products or any of our future tests (either alone or together with sample collection devices), products or services we may develop, or we decide to voluntarily pursue FDA clearance or approval, we may be forced to stop selling our tests while we work to obtain such FDA clearance or approval. Our business would be negatively affected until such review was completed and clearance to market or approval was obtained. The regulatory process may involve, among other things, successfully completing additional clinical studies and submitting premarket notification or filing a premarket approval application with the FDA. If premarket review is required by the FDA or if we decide to voluntarily pursue FDA premarket review of our tests, there can be no assurance that our Acuitas MDRO Gene Test or any tests, products or services we may develop in the future will be cleared or approved on a timely basis, if at all, nor can there be assurance that labeling claims will be consistent with our current claims or adequate to support continued adoption of our tests. If our tests are allowed to remain on the market but there is uncertainty in the marketplace about our tests, if we are required by the FDA to label them investigational, or if labeling claims the FDA allows us to make are limited, orders may decline. Ongoing compliance with FDA regulations would increase the cost of conducting our business, and subject us to heightened regulation by the FDA and penalties for failure to comply with these requirements.
| 36 |
If we are required to but fail to maintain regulatory approvals and clearances, or are unable to obtain, or experience significant delays in obtaining, FDA clearances or approvals for our products or product enhancements, our ability to commercially distribute and market our products could suffer.
If the FDA determines that enforcement discretion is not appropriate or that LDTs are generally subject to FDA regulation and that premarket review, including clearance or approval, is required for our Acuitas MDRO Gene Test or any of our future tests, diagnostic test kits that we may develop, or other products that would be classified as medical devices, the process of obtaining regulatory clearances or approvals to market a medical device can be costly and time consuming, and we may not be able to obtain these clearances or approvals on a timely basis, if at all. In particular, the FDA permits commercial distribution of a new medical device only after the device has received clearance under Section 510(k) of the FDA Act, or is the subject of an approved premarket approval application (“PMA”), unless the device is specifically exempt from those requirements. The FDA will clear marketing of a lower risk medical device through the 510(k) process if the manufacturer demonstrates that the new product is substantially equivalent to other 510(k)-cleared products. High risk devices deemed to pose the greatest risk, such as life-sustaining, life-supporting, or implantable devices, or devices not deemed substantially equivalent to a previously cleared device, require the approval of a PMA. The PMA process is more costly, lengthy and uncertain than the 510(k) clearance process. A PMA application must be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the device for its intended use. Our currently commercialized products have not received FDA clearance or approval, as they are marketed under the FDA’s enforcement discretion for LDTs or are class I medical devices, which are exempt from the requirement for FDA clearance or approval.
Our failure to comply with U.S. federal, state and foreign governmental regulations could lead to the issuance of warning letters or untitled letters, the imposition of injunctions, suspensions or loss of regulatory clearance or approvals, product recalls, termination of distribution, product seizures or civil penalties. In the most extreme cases, criminal sanctions or closure of our manufacturing facility are possible.
Modifications to our marketed products may require new 510(k) clearances or PMA approvals, or may require us to cease marketing or recall the modified products until clearances or approvals are obtained.
If we are required to obtain 510(k) clearance or PMA approval for any of our current or future products, any modification to those products would require additional clearances or approvals. Modifications to a 510(k)-cleared device that could significantly affect its safety or efficacy, or that would constitute a major change in its intended use, requires a new 510(k) clearance or, possibly, a PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review the manufacturer’s decision. The FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. If the FDA requires us to seek 510(k) clearance or a PMA for any modification to a previously cleared product, we may be required to cease marketing and distributing, or to recall the modified product until we obtain such clearance or approval, and we may be subject to significant regulatory fines or penalties. Further, our products could be subject to recall if the FDA determines, for any reason, that our products are not safe or effective. Any recall or FDA requirement that we seek additional approvals or clearances could result in significant delays, fines, increased costs associated with modification of a product, loss of revenue and potential operating restrictions imposed by the FDA.
There is no guarantee that the FDA will grant 510(k) clearance or PMA approval of our future products, and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business.
Some of our future products may require 510(k) clearance from the FDA. Other products, potentially, could require PMA approval. In addition, some of our new products may require clinical trials to support regulatory approval and we may not successfully complete these clinical trials. The FDA may not approve or clear these products for the indications that are necessary or desirable for successful commercialization. Indeed, the FDA may refuse our requests for 510(k) clearance or premarket approval of new products. Failure to receive a required clearance or approval for our new products would have an adverse effect on our ability to expand our business.
| 37 |
Our products may in the future be subject to product recalls that could harm our reputation, business and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of regulated products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. The FDA requires that certain classifications of recalls be reported to FDA within 10 working days after the recall is initiated. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action for failing to report the recalls when they were conducted.
If our products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, medical device and LDT manufacturers are required to report to the FDA information that a device or LDT has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA within the required timeframes, or at all, FDA could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our products for unapproved or "off-label" uses.
We believe that our Acuitas MDRO test products are LDTs, subject to the FDA’s enforcement discretion. To remain within the FDA’s enforcement discretion, we are restricted in the ways we can promote and market our products. Furthermore, certain of our future products, including specimen transport containers we may develop such as Grow on the Go, might be regulated as class I medical devices for which premarket clearance or approval is not required, subject to certain limitations. We believe that our promotional activities for our products fall within the scope of the FDA’s enforcement discretion and applicable premarket exemptions. However, the FDA could disagree and require us to stop promoting our products in certain ways unless and until we obtain FDA clearance or approval for them. In addition, because our products are not currently cleared or approved by the FDA, if the FDA determines that our promotional materials constitute promotion of a use for which premarket clearance or approval is required, it could request that we modify our promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products would be impaired.
| 38 |
We may generate a portion of our future revenue internationally and would then be subject to various risks relating to international activities which could adversely affect our operating results.
We believe that a portion of our future revenue will come from international sources as we implement and expand overseas operations. Engaging in international business involves a number of difficulties and risks, including:
| · | required compliance with existing and changing foreign healthcare and other regulatory requirements and laws, such as those relating to patient privacy; |
| · | required compliance with anti-bribery laws, such as the U.S. Foreign Corrupt Practices Act (“FCPA”), and U.K. Bribery Act, data privacy requirements, labor laws and anti-competition regulations; |
| · | export or import restrictions; |
| · | various reimbursement and insurance regimes; |
| · | laws and business practices favoring local companies; |
| · | longer payment cycles and difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
| · | political and economic instability; |
| · | potentially adverse tax consequences, tariffs, customs charges, bureaucratic requirements and other trade barriers; |
| · | foreign exchange controls; |
| · | difficulties and costs of staffing and managing foreign operations; and |
| · | difficulties protecting or procuring intellectual property rights. |
As we expand internationally, our results of operations and cash flows would become increasingly subject to fluctuations due to changes in foreign currency exchange rates. Our expenses are generally denominated in the currencies in which our operations are located, which is in the United States. If the value of the U.S. dollar increases relative to foreign currencies in the future, in the absence of a corresponding change in local currency prices, our future revenue could be adversely affected as we convert future revenue from local currencies to U.S. dollars. If we dedicate resources to our international operations and are unable to manage these risks effectively, our business, operating results and prospects will suffer.
We face the risk of potential liability under the FCPA for past international distributions of products and to the extent we distribute products or otherwise operate internationally in the future.
In the past, we have distributed certain of our products internationally, and in the future we may distribute our products internationally and possibly engage in additional international operations. The FCPA prohibits companies such as us from engaging, directly or indirectly, in making payments to foreign government and political officials for the purpose of obtaining or retaining business or securing any other improper advantage, including, among other things, the distribution of products and other international business operations. Like other U.S. companies operating abroad, we may face liability under the FCPA if we, or third parties we have used to distribute our products or otherwise advance our international business, have violated the FCPA. Any violations of these laws, or allegations of such violations, could disrupt our operations, involve significant management distraction, involve significant costs and expenses, including legal fees, and could result in a material adverse effect on our business, prospects, financial condition or results of operations. We could also suffer severe penalties, including criminal and civil penalties, disgorgement and other remedial measures.
Risks Related to Compliance with Healthcare and Other Regulations
Changes in healthcare policy, including legislation reforming the U.S. healthcare system, may have a material adverse effect on our financial condition and operations.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, collectively, the PPACA, enacted in March 2010, made changes that significantly affect the pharmaceutical and medical device industries and clinical laboratories. As begun in 2013, each medical device manufacturer must pay a sales tax in an amount equal to 2.3% of the price for which such manufacturer sells its FDA-listed medical devices. The FDA has asserted that clinical laboratory tests such as our Acuitas MDRO Gene Test are medical devices. Our Acuitas MDRO test products are not currently listed as a medical device with the FDA, but we cannot assure you that the tax will not be extended to LDTs such as ours in the future if they were to be regulated as a device.
| 39 |
Other significant measures contained in the PPACA include coordination and promotion of research on comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. The PPACA also includes significant new fraud and abuse measures, including required disclosures of financial arrangements with physician customers, lower thresholds for violations and increasing potential penalties for such violations. In addition, the PPACA establishes an Independent Payment Advisory Board (“IPAB”), to reduce the per capita rate of growth in Medicare spending. The IPAB has broad discretion to propose policies to reduce healthcare expenditures, which may have a negative impact on payment rates for services, including our tests. The IPAB proposals may impact payments for clinical laboratory services for our customers beginning in 2016, and for hospital services beginning in 2020, and may indirectly reduce demand for our product candidates.
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This reduction includes aggregate reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will stay in effect through 2021 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
AdvanDx is an "applicable manufacturer" subject to reporting to CMS under the PPACA Physician Payments Sunshine Act. Sunshine Act reporting requires us to report on certain payments or other transfers of value made to physicians and teaching hospitals. The Sunshine Act reporting obligations have been in effect for two reporting cycles. If we fail to report payments properly or otherwise fail to comply with the Sunshine Act requirements we could be subject to fines and penalties.
The full impact on our business of the PPACA and the other new laws is uncertain. Nor is it clear whether other legislative or regulatory changes will be adopted or how such changes would affect our industry generally or our ability to successfully commercialize our product candidates, if approved. In addition, sales of our tests outside of the United States will subject us to foreign regulatory requirements, which may also change over time.
We cannot predict whether future healthcare initiatives will be implemented at the federal or state level or in countries outside of the United States in which we may do business, or the effect any future legislation or regulation will have on us. The taxes imposed by the new federal legislation and the expansion in government’s effect on the United States healthcare industry may result in decreased profits to us, which may adversely affect our business, financial condition and results of operations.
Failure in our information technology, storage systems or our digital platform technology could significantly disrupt our operations and our research and development efforts, which could adversely impact our revenues, as well as our research, development and commercialization efforts.
Our ability to execute our business strategy depends, in part, on the continued and uninterrupted performance of our information technology (“IT”) systems, which support our operations and our research and development efforts, as well as our storage systems and our analyzers. Due to the sophisticated nature of the technology we use in our products and service offerings, including our Acuitas Lighthouse product, we are substantially dependent on our IT systems. IT systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, sustained or repeated system failures that interrupt our ability to generate and maintain data, and in particular to operate our digital immunoassay platform, could adversely affect our ability to operate our business. Any interruption in the operation of our digital immunoassay platform, due to IT system failures, part failures or potential disruptions in the event we are required to relocate our instruments within our facility or to another facility, could have an adverse effect on our operations.
| 40 |
Security breaches, loss of data and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we collect and store sensitive data, including legally protected health information and personally identifiable information about our customers and their patients. We also store sensitive intellectual property and other proprietary business information, including that of our customers. We manage and maintain our applications and data utilizing a combination of on-site systems and cloud-based data center systems. These applications and data encompass a wide variety of business critical information, including research and development information, commercial information and business and financial information.
We face four primary risks relative to protecting this critical information: loss of access risk, inappropriate disclosure risk, inappropriate modification risk and the risk of our being unable to identify and audit our controls over the first three risks.
We are highly dependent on information technology networks and systems, including the Internet, to securely process, transmit and store this critical information. Security breaches of this infrastructure, including physical or electronic break-ins, computer viruses, attacks by hackers and similar breaches, can create system disruptions, shutdowns or unauthorized disclosure or modification of confidential information. The secure processing, storage, maintenance and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers or viruses or breached due to employee error, malfeasance or other disruptions.
A security breach or privacy violation that leads to disclosure or modification of or prevents access to consumer information (including personally identifiable information or protected health information) could harm our reputation, compel us to comply with disparate state breach notification laws, require us to verify the correctness of database contents and otherwise subject us to liability under laws that protect personal data, resulting in increased costs or loss of revenue. If we are unable to prevent such security breaches or privacy violations or implement satisfactory remedial measures, our operations could be disrupted, and we may suffer loss of reputation, financial loss and other regulatory penalties because of lost or misappropriated information, including sensitive consumer data. In addition, these breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above.
Any such breach or interruption could compromise our networks, and the information stored there could be inaccessible or could be accessed by unauthorized parties, publicly disclosed, lost or stolen. Any such interruption in access, improper access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to perform tests, provide test results, bill facilities or patients, process claims and appeals, provide customer assistance services, conduct research and development activities, collect, process and prepare Company financial information, provide information about our current and future solutions and other patient and clinician education and outreach efforts through our website, and manage the administrative aspects of our business and damage our reputation, any of which could adversely affect our business. Any such breach could also result in the compromise of our trade secrets and other proprietary information, which could adversely affect our competitive position.
In addition, the interpretation and application of consumer, health-related, privacy and data protection laws in the U.S. and elsewhere are often uncertain, contradictory and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. Complying with these various laws could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business.
Payments for our tests and other services could decline because of factors beyond our control.
If hospital patient volumes drop as a result of severe economic conditions, or other unforeseen changes in health care provision or affordability, individual hospitals and health systems may be less willing to invest in our MDRO surveillance and prevention programs. In addition, state and federal funds that are anticipated to be invested in the National Strategy for Combating Antibiotic-Resistant Bacteria could be reduced. If such funds are reduced, the market for our products would be impacted, which may affect our ability to generate revenues.
| 41 |
If we accept payment from federal and state healthcare programs in the future, we will be subject to enforcement actions involving false claims, kickbacks, physician self-referral or other federal or state fraud and abuse laws, and we could incur significant civil and criminal sanctions and loss of reimbursement, which would hurt our business.
The government has made enforcement of the false claims, anti-kickback, physician self-referral and various other fraud and abuse laws a major priority. In many instances, private whistleblowers also are authorized to enforce these laws even if government authorities choose not to do so. Several clinical diagnostic laboratories and members of their management have been the subject of this enforcement scrutiny, which has resulted in very significant civil and criminal settlement payments. In most of these cases, private whistleblowers brought the allegations to the attention of federal enforcement agencies. The risk of our being found in violation of these laws and regulations is increased by the fact that some of the laws and regulations have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. In the event we begin accepting reimbursement from federal or state healthcare programs for our tests, we would be subject to the following laws:
| · | the federal Anti-Kickback Statute, which constrains certain marketing practices, educational programs, pricing policies and relationships with healthcare providers or other entities by prohibiting, among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| · | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third party payors that are false or fraudulent; |
| · | federal physician self-referral laws, such as the Stark law, which prohibit a physician from making a referral to a provider of certain health services with which the physician or the physician’s family member has a financial interest, and prohibit submission of a claim for reimbursement pursuant to a prohibited referral; and |
| · | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third party payor, including commercial insurers, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
If we or our operations, are found to be in violation of any of these laws and regulations, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in U.S. federal or state healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. We have compliance policies and are in the process of adopting a written compliance plan based on the HHS Office of the Inspector General guidance set forth in its model compliance plan for clinical laboratories, and federal and state fraud and abuse laws. We will monitor changes in government enforcement, particularly in these areas, as we grow and expand our business. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and hurt our reputation. If we were excluded from participation in U.S. federal healthcare programs, we would not be able to receive, or to sell our tests to other parties who receive reimbursement from Medicare, Medicaid and other federal programs, and that could have a material adverse effect on our business.
Risks Related to our Intellectual Property
If we cannot license rights to use technologies on reasonable terms, we may not be able to commercialize new products in the future.
In the future, we may license third-party technology to develop or commercialize new products. In return for the use of a third party’s technology, we may agree to pay the licensor royalties based on sales of our solutions. Royalties are a component of cost of services and affect the margins on our products. We may also need to negotiate licenses to patents and patent applications after introducing a commercial product. Our business may suffer if we are unable to enter into the necessary licenses on acceptable terms, or at all, if any necessary licenses are subsequently terminated, if the licensors fail to abide by the terms of the license or fail to prevent infringement by third parties, or if the licensed patents or other rights are found to be invalid or unenforceable.
| 42 |
If we are unable to protect our intellectual property effectively, our business would be harmed.
We rely on patent protection as well as trademark, copyright, trade secret and other intellectual property rights protection and contractual restrictions to protect our proprietary technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. If we fail to protect our intellectual property, third parties may be able to compete more effectively against us and we may incur substantial litigation costs in our attempts to recover or restrict use of our intellectual property.
In July 2015 we issued a senior secured promissory note, in the principal amount of $1 million, to Merck GHI. Such Note is secured by a lien on our assets, including our intellectual property assets. If we default on our payment obligations under the Note, Merck GHI has the right to control the disposition of our assets, including our intellectual property assets. If such default occurs, and our intellectual property assets are sold or licensed, our business could be materially adversely affected.
We apply for patents covering our products and technologies and uses thereof, as we deem appropriate, however we may fail to apply for patents on important products and technologies in a timely fashion or at all, or we may fail to apply for patents in potentially relevant jurisdictions. It is possible that none of our pending patent applications will result in issued patents in a timely fashion or at all, and even if patents are granted, they may not provide a basis for intellectual property protection of commercially viable products, may not provide us with any competitive advantages, or may be challenged and invalidated by third parties. It is possible that others will design around our current or future patented technologies. We may not be successful in defending any challenges made against our patents or patent applications. Any successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents and increased competition to our business. The outcome of patent litigation can be uncertain and any attempt by us to enforce our patent rights against others may not be successful, or, if successful, may take substantial time and result in substantial cost, and may divert our efforts and attention from other aspects of our business.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States or elsewhere. Courts frequently render opinions in the biotechnology field that may affect the patentability of certain inventions or discoveries, including opinions that may affect the patentability of methods for analyzing or comparing DNA.
In particular, the patent positions of companies engaged in the development and commercialization of genomic diagnostic tests, like ours, are particularly uncertain. Various courts, including the U.S. Supreme Court, have recently rendered decisions that affect the scope of patentability of certain inventions or discoveries relating to certain diagnostic tests and related methods. These decisions state, among other things, that patent claims that recite laws of nature (for example, the relationship between blood levels of certain metabolites and the likelihood that a dosage of a specific drug will be ineffective or cause harm) are not themselves patentable. What constitutes a law of nature is uncertain, and it is possible that certain aspects of genetic diagnostics tests would be considered natural laws. Accordingly, the evolving case law in the United States may adversely affect our ability to obtain patents and may facilitate third-party challenges to any owned and licensed patents. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and we may encounter difficulties protecting and defending such rights in foreign jurisdictions. The legal systems of many other countries do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology, which could make it difficult for us to stop the infringement of our patents in such countries. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Changes in either the patent laws or in interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property. We cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. We may not develop additional proprietary products, methods and technologies that are patentable.
In addition to pursuing patents on our technology, we take steps to protect our intellectual property and proprietary technology by entering into agreements, including confidentiality agreements, non-disclosure agreements and intellectual property assignment agreements, with our employees, consultants, academic institutions, corporate partners and, when needed, our advisors. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure. If we are required to assert our rights against such party, it could result in significant cost and distraction.
Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time consuming, and the outcome would be unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets.
| 43 |
We may also be subject to claims that our employees have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of third parties, or to claims that we have improperly used or obtained such trade secrets. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights and face increased competition to our business. A loss of key research personnel work product could hamper or prevent our ability to commercialize potential products, which could harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Further, competitors could attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. Others may independently develop similar or alternative products and technologies or replicate any of our products and technologies. If our intellectual property does not adequately protect us against competitors’ products and methods, our competitive position could be adversely affected, as could our business.
We have not yet registered certain of our trademarks in all of our potential markets. If we apply to register these trademarks, our applications may not be allowed for registration in a timely fashion or at all, and our registered trademarks may not be maintained or enforced. In addition, opposition or cancellation proceedings may be filed against our trademark applications and registrations, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
To the extent our intellectual property offers inadequate protection, or is found to be invalid or unenforceable, we would be exposed to a greater risk of direct competition. If our intellectual property does not provide adequate coverage of our competitors’ products, our competitive position could be adversely affected, as could our business. Both the patent application process and the process of managing patent disputes can be time consuming and expensive.
We may be involved in litigation related to intellectual property, which could be time-intensive and costly and may adversely affect our business, operating results or financial condition.
We may receive notices of claims of direct or indirect infringement or misappropriation or misuse of other parties’ proprietary rights from time to time. Some of these claims may lead to litigation. We cannot assure you that we will prevail in such actions, or that other actions alleging misappropriation or misuse by us of third-party trade secrets, infringement by us of third-party patents and trademarks or other rights, or the validity of our patents, trademarks or other rights, will not be asserted or prosecuted against us.
We might not have been the first to make the inventions covered by each of our pending patent applications and we might not have been the first to file patent applications for these inventions. To determine the priority of these inventions, we may have to participate in interference proceedings, derivation proceedings, or other post-grant proceedings declared by the United States Patent and Trademark Office that could result in substantial cost to us. No assurance can be given that other patent applications will not have priority over our patent applications. In addition, recent changes to the patent laws of the United States allow for various post-grant opposition proceedings that have not been extensively tested, and their outcome is therefore uncertain. Furthermore, if third parties bring these proceedings against our patents, we could experience significant costs and management distraction.
Litigation may be necessary for us to enforce our patent and proprietary rights or to determine the scope, coverage and validity of the proprietary rights of others. The outcome of any litigation or other proceeding is inherently uncertain and might not be favorable to us, and we might not be able to obtain licenses to technology that we require on acceptable terms or at all. Further, we could encounter delays in product introductions, or interruptions in product sales, as we develop alternative methods or products. In addition, if we resort to legal proceedings to enforce our intellectual property rights or to determine the validity, scope and coverage of the intellectual property or other proprietary rights of others, the proceedings could be burdensome and expensive, even if we were to prevail. Any litigation that may be necessary in the future could result in substantial costs and diversion of resources and could have a material adverse effect on our business, operating results or financial condition.
| 44 |
As we move into new markets and applications for our products, incumbent participants in such markets may assert their patents and other proprietary rights against us as a means of slowing our entry into such markets or as a means to extract substantial license and royalty payments from us. Our competitors and others may now and, in the future, have significantly larger and more mature patent portfolios than we currently have. In addition, future litigation may involve patent holding companies or other adverse patent owners who have no relevant product revenue and against whom our own patents may provide little or no deterrence or protection. Therefore, our commercial success may depend in part on our non-infringement of the patents or proprietary rights of third parties. Numerous significant intellectual property issues have been litigated, and will likely continue to be litigated, between existing and new participants in our existing and targeted markets and competitors may assert that our products infringe their intellectual property rights as part of a business strategy to impede our successful entry into or growth in those markets. Third parties may assert that we are employing their proprietary technology without authorization. In addition, our competitors and others may have patents or may in the future obtain patents and claim that making, having made, using, selling, offering to sell or importing our products infringes these patents. We could incur substantial costs and divert the attention of our management and technical personnel in defending against any of these claims. Parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize and sell products, and could result in the award of substantial damages against us. In the event of a successful claim of infringement against us, we may be required to pay damages and ongoing royalties, and obtain one or more licenses from third parties, or be prohibited from selling certain products. We may not be able to obtain these licenses on acceptable terms, if at all. We could incur substantial costs related to royalty payments for licenses obtained from third parties, which could negatively affect our financial results. In addition, we could encounter delays in product introductions while we attempt to develop alternative methods or products to avoid infringing third-party patents or proprietary rights. Defense of any lawsuit or failure to obtain any of these licenses could prevent us from commercializing products, and the prohibition of sale of any of our products could materially affect our business and our ability to gain market acceptance for our products.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
In addition, our agreements with some of our customers, suppliers or other entities with whom we do business require us to defend or indemnify these parties to the extent they become involved in infringement claims, including the types of claims described above. We could also voluntarily agree to defend or indemnify third parties in instances where we are not obligated to do so if we determine it would be important to our business relationships. If we are required or agree to defend or indemnify third parties in connection with any infringement claims, we could incur significant costs and expenses that could adversely affect our business, operating results, or financial condition.
Risks Related to our Public Company Status
We will incur increased costs and demands on management as a result of compliance with laws and regulations applicable to public companies, which could harm our operating results.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company, including costs associated with public company reporting requirements. In addition, the Sarbanes-Oxley Act of 2002 and the Dodd-Frank Act of 2010, as well as rules implemented by the SEC and The NASDAQ Stock Market, impose a number of requirements on public companies, including with respect to corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance and disclosure obligations. Moreover, these rules and regulations will increase our legal, accounting and financial compliance costs and will make some activities more time-consuming and costly. We also expect that it will be more expensive for us to obtain director and officer liability insurance.
Changes in, or interpretations of, accounting rules and regulations could result in unfavorable accounting changes or require us to change our compensation policies.
Accounting methods and policies for diagnostic companies, including policies governing revenue recognition, research and development and related expenses and accounting for stock-based compensation, are subject to further review, interpretation and guidance from relevant accounting authorities, including the SEC. Changes to, or interpretations of, accounting methods or policies may require us to reclassify, restate or otherwise change or revise our financial statements, including those contained in this Annual Report. Restatement of our financial statements could have a negative impact on our business.
| 45 |
If we are unable to implement and maintain effective internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our reported financial information and the market price of our common stock may be negatively affected.
As a public company, we will be required to maintain internal control over financial reporting and to report any material weaknesses in such internal control. Section 404 of the Sarbanes-Oxley Act of 2002 requires that we evaluate and determine the effectiveness of our internal control over financial reporting and, beginning with our annual report for the year ending December 31, 2016, provide a management report on the internal control over financial reporting. If we have a material weakness in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. We are in the process of compiling the system and processing documentation necessary to perform the evaluation needed to comply with Section 404 of the Sarbanes-Oxley Act of 2002. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion.
During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, our management will be unable to conclude that our internal control over financial reporting is effective. Moreover, when we are no longer an emerging growth company, our independent registered public accounting firm will be required to issue an attestation report on the effectiveness of our internal control over financial reporting. Even if our management concludes that our internal control over financial reporting is effective, our independent registered public accounting firm may conclude that there are material weaknesses with respect to our internal controls or the level at which our internal controls are documented, designed, implemented or reviewed.
If we are unable to conclude that our internal control over financial reporting is effective, or when we are no longer an emerging growth company, if our auditors were to express an adverse opinion on the effectiveness of our internal control over financial reporting because we had one or more material weaknesses, investors could lose confidence in the accuracy and completeness of our financial disclosures, which could cause the price of our common stock to decline. Internal control deficiencies could also result in a restatement of our financial results in the future.
We are an emerging growth company and may elect to comply with reduced public company reporting requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an emerging growth company, as defined under the Securities Act. We will remain an emerging growth company for up to five years, although if our revenue exceeds $1 billion in any fiscal year before that time, we would cease to be an emerging growth company as of the end of that fiscal year. In addition, if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our second fiscal quarter of any fiscal year before the end of that five-year period, we would cease to be an emerging growth company as of December 31 of that year. As an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to certain other public companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced financial statement and financial-related disclosures, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirement of holding a nonbinding advisory vote on executive compensation and obtaining stockholder approval of any golden parachute payments not previously approved by our stockholders. We cannot predict whether investors will find our common stock less attractive if we choose to rely on any of these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure we may make, there may be a less active trading market for our common stock and our stock price may be more volatile.
| 46 |
Trading of our common stock is limited, and trading restrictions imposed on us by applicable regulations may further reduce trading in our common stock, making it difficult for our stockholders to sell their shares; and future sales of common stock could reduce our stock price.
Trading of our common stock is currently conducted on the Nasdaq Capital Market. The liquidity of our common stock is limited, not only in terms of the number of shares that can be bought and sold at a given price, but also as it may be adversely affected by delays in the timing of transactions and reduction in security analysts’ and the media’s coverage of us, if at all. As of December 31, 2015, a significant number of the issued and outstanding shares of our common stock were held by officers, directors and beneficial owners of at least 10% of our outstanding shares, each of whom is subject to certain restrictions with regard to trading our common stock. These factors may result in different prices for our common stock than might otherwise be obtained in a more liquid market and could also result in a larger spread between the bid and asked prices for our common stock. In addition, without a large public float, our common stock is less liquid than the stock of companies with broader public ownership, and, as a result, the trading prices of our common stock may be more volatile. In the absence of an active public trading market, an investor may be unable to liquidate his investment in our common stock. Trading of a relatively small volume of our common stock may have a greater impact on the trading price of our stock than would be the case if our public float were larger. We cannot predict the prices at which our common stock will trade in the future, if at all.
Sales by stockholders of substantial amounts of our shares of common stock, the issuance of new shares of common stock by us or the perception that these sales may occur in the future could materially and adversely affect the market price of our common stock.
As of December 31, 2015, our directors, executive officers, principal stockholders and affiliated entities beneficially owned sufficient shares of our outstanding voting securities that, if some or all of them acted together, they would have the ability to exert substantial influence over the election of our board of directors and the outcome of issues requiring approval by our stockholders. This concentration of ownership may also have the effect of delaying or preventing a change in control of the Company that may be favored by other stockholders. This could prevent transactions in which stockholders might otherwise recover a premium for their shares over current market prices.
Item 1B. Unresolved Staff Comments
None.
The Company leases 20,939 square feet of office and laboratory space at our headquarters in Gaithersburg, Maryland. Pursuant to our lease agreement, as amended, our lease will continue in effect until January 31, 2021 and may be renewed for one additional five-year period at the Company’s election. The Company also leases 12,770 square feet of office space at its facility in Woburn, Massachusetts under an operating lease that expires in January 2017, and provides the Company with options to extend the lease beyond the current expiration date. Additionally, the Company leases 2,967 square feet of office space in Denmark; this lease is currently on a month-to-month basis. Rent expenses under the Company’s facility operating leases for the years ending December 31, 2015 and 2014 were $1.0 million and $0.9 million, respectively.
We believe that our existing facilities are, or any such new facilities will be, adequate to meet our business requirements for at least the next 18 months and that additional space will be available on commercially reasonable terms, if required.
From time to time, we may be party to lawsuits in the ordinary course of business. We are currently not a party to any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
| 47 |
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock and warrants have traded on the NASDAQ Capital Market under the symbols “OPGN” and “OPGNW,” respectively, since May 5, 2015. Prior to such time, there was no public market for our common stock or our warrants. The following table shows the high and low sales price for our common stock and warrants as reported by the NASDAQ Capital Market for the periods indicated:
| High | Low | |||||||
| Common Stock: | ||||||||
| Year Ended December 31, 2015 | ||||||||
| Fourth Quarter | $ | 2.79 | $ | 1.45 | ||||
| Third Quarter | $ | 4.43 | $ | 2.21 | ||||
| Second Quarter (beginning May 5, 2015) | $ | 5.43 | $ | 3.12 | ||||
| Warrants: | ||||||||
| Year Ended December 31, 2015 | ||||||||
| Fourth Quarter | $ | 0.59 | $ | 0.25 | ||||
| Third Quarter | $ | 0.84 | $ | 0.30 | ||||
| Second Quarter (beginning May 5, 2015) | $ | 0.95 | $ | 0.50 | ||||
Stockholder Information
As of March 17, 2016, there were approximately 68 shareholders of record of our common stock, which does not include stockholders that beneficially own shares held in a “nominee” or in “street” name.
Dividends
We have not paid cash dividends in the years ended December 31, 2015 and 2014. We do not anticipate paying cash dividends in the near term.
Sales of Unregistered Securities
The Company issued no unregistered securities during the fourth quarter of 2015.
Use of Proceeds from the Sale of Registered Securities
As of December 31, 2015, we have used approximately $10.7 million of the net cash proceeds from our IPO for sales and marketing, research and development and working capital purposes. There has been no material change in our planned use of the balance of the net proceeds from the IPO as described in our final prospectus dated May 4, 2015 and filed with the SEC pursuant to Rule 424(b) under the Securities Act on May 5, 2015. We have broad discretion in the use of the net proceeds from our IPO. We may find it necessary or advisable to use the net proceeds from our IPO for other purposes than those described in our final prospectus.
Issuer Purchases of Equity Securities
None.
Item 6. Selected Consolidated Financial Data
As a smaller reporting company, we are not required to provide the information required by this item.
| 48 |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read in conjunction with our audited consolidated financial statements and the accompanying notes thereto included elsewhere in this Annual Report. This discussion contains forward-looking statements, based on current expectations and related to future events and our future financial performance, that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many important factors, including those set forth in the section titled “Risk Factors” included under Part I, Item 1A of this Annual Report.
Overview
We are a precision medicine company using molecular diagnostics and informatics to combat infectious disease. We are developing molecular information solutions to combat infectious disease in global healthcare settings, helping to guide clinicians with more rapid information about life threatening infections, improve patient outcomes, and decrease the spread of infections caused by multidrug-resistant microorganisms. Our proprietary DNA tests and bioinformatics address the rising threat of antibiotic resistance by helping physicians and healthcare providers optimize patient care decisions and protect the hospital biome through customized screening and surveillance solutions.
On July 14, 2015, OpGen completed the strategic acquisition (the “Merger”) of AdvanDx, Inc. and its wholly owned subsidiary AdvanDx A/S, collectively referred to as AdvanDx. AdvanDx researches, develops and markets advanced in vitro diagnostic kits for the diagnosis and prevention of infectious diseases, and sells its products principally to hospitals and clinical laboratories in the United States and Europe. The Company acquired AdvanDx principally to use AdvanDx’s diagnostic capabilities with respect to MDROs and leverage AdvanDx’s relationships with hospitals and clinical laboratories to accelerate the sales of OpGen’s products and services.
The Company’s headquarters are in Gaithersburg, Maryland, and its principal operations are in Gaithersburg, Maryland and Woburn, Massachusetts. The Company also has operations in Copenhagen, Denmark. The Company operates in one business segment.
Recent Developments
Since inception, the Company has incurred, and continues to incur, significant losses from operations. The Company has funded its operations primarily through external investor financing arrangements. The Company raised significant funds in 2015, consisting of:
| · | $0.8 million in short-term notes (in the first quarter of 2015, $0.3 million of demand notes held by an entity controlled by our chief executive officer were settled as partial payment for a 2015 convertible note, and in the second quarter of 2015, $0.2 million of notes from a related party were repaid in cash); |
| · | $1.5 million through the issuance of convertible notes; |
| · | $12.1 million in net proceeds from its IPO as discussed further below; and |
| · | $6.0 million in net proceeds from the issuances of common stock and a senior secured promissory note to Merck GHI. |
On May 8, 2015, OpGen completed its IPO pursuant to which it offered and sold 2,850,000 units, each unit consisting of one share of common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 Units in the IPO. After considering the demand notes, underwriting discounts and commissions and offering expenses, the total net cash proceeds were $12.1 million. On the IPO closing date, the underwriters exercised their over-allotment option to acquire an additional 422,500 stock purchase warrants. In connection with the IPO, all of OpGen’s outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock.
On July 14, 2015, the Company completed the strategic acquisition of AdvanDx. Pursuant to the Merger Agreement, a newly formed Merger Sub merged with and into AdvanDx, with AdvanDx surviving as a wholly owned subsidiary of the Company in accordance with the General Corporation Law of the State of Delaware. Under the terms of the Merger Agreement, the Merger Consideration consisted of an aggregate 681,818 shares of the Company's common stock with a value of $2.6 million (based on the closing sales price of our common stock of $3.79 per share on July 13, 2015).
| 49 |
In July 2015, the Company raised $6.0 million by issuing 1,136,364 shares of common stock at $4.40 per share and a $1.0 million senior secured promissory note to Merck GHI. Also in July 2015, the Company entered into a Registration Rights Agreement with Merck GHI and the AdvanDx stockholders who received Merger Consideration in the Merger, which will require the Company to register such shares of Company common stock for resale by such holders in the future. Under the Purchase Agreement, Merck GHI has the right to participate in future securities offerings made by the Company. There is no assurance that Merck GHI will exercise such participation rights in the future.
The Company’s current operating assumptions, which include management’s best estimate of future revenue and operating expenses, indicate that current cash on hand will be sufficient to fund operations into the second quarter of 2016.
In the event the Company is unable to successfully raise additional capital in 2016, the Company will not have sufficient cash flows and liquidity to finance its business operations as currently contemplated. Accordingly, in such circumstances the Company would be compelled to reduce general and administrative expenses and delay research and development projects, including the purchase of scientific equipment and supplies, until it is able to obtain sufficient financing, or pursue other strategic alternatives which may include licensing and/or partnering arrangements or mergers and acquisitions. The consolidated financial statements do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue in existence.
Results of Operations for the Years Ended December 31, 2015 and 2014
Revenues and gross profit
| Year ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Product sales | $ | 2,701,142 | $ | 1,236,349 | ||||
| Laboratory services | 120,476 | 478,909 | ||||||
| Collaboration revenue | 336,102 | 2,411,120 | ||||||
| Total revenue | $ | 3,157,720 | $ | 4,126,378 | ||||
Our total revenue for the year ended December 31, 2015 decreased 23%, to $3.2 million from $4.1 million, when compared to the same period in 2014. This decrease is primarily attributable to:
| · | Product Sales: an increase in revenue of 118% in 2015 as compared to 2014 is attributable to the inclusion of AdvanDx products sales subsequent to the Merger, offset in part by a reduction in the sale of our Argus products, as we transition from our legacy mapping products to the introduction of Acuitas MDRO products; |
| · | Laboratory Services: a decrease in revenue of 75% in 2015 as compared to 2014 as a result of a reduction in sales of mapping products, as we transition from our legacy mapping products to the introduction of Acuitas MDRO products; and |
| · | Collaboration Revenue: a decrease in revenue generated under a collaboration arrangement with Hitachi of 86% in 2015 as compared to 2014 as a result of the completion of our technology development agreement with Hitachi. |
The Company expects an increase in product sales in 2016 due to the inclusion of a full year of QuickFISH and PNA FISH product sales. This increase is expected to be partially offset by a decrease in the sale of Argus products. The Company expects a decline in mapping products and an increase in Acuitas and Lighthouse products.
| 50 |
Operating expenses
| Year ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Costs of products sold | $ | 1,179,771 | $ | 425,541 | ||||
| Costs of services | 367,802 | 526,196 | ||||||
| Research and development | 6,002,941 | 4,368,302 | ||||||
| General and administrative | 5,834,642 | 2,312,935 | ||||||
| Sales and marketing | 4,305,444 | 2,058,085 | ||||||
| Transaction expenses | 526,283 | - | ||||||
| Total operating expenses | $ | 18,216,883 | $ | 9,691,059 | ||||
The Company’s total operating expenses for the year ended December 31, 2015 increased 88%, to $18.2 million from $9.7 million, when compared to the same period in 2014. This increase is primarily attributable to:
| · | Costs of Sales: costs of product sales for the year ended December 31, 2015 increased 177% while costs of services decreased 30%, when compared to the same period in 2014. The change in costs of sales is primarily attributable to the inclusion of costs of QuickFISH products sold along with a decrease in the cost of generating laboratory services revenue; |
| · | Research and Development: an increase in expenses of $1.6 million, primarily due to the inclusion of $1.7 million of expenses related to the development of the automation of our QuickFISH products. The remaining expenses primarily relate to the development of the Lighthouse data warehouse, portal, and antibiotic analysis and the Acuitas rapid molecular diagnostics products; |
| · | General and Administrative: an increase in expenses of $3.5 million primarily due to salaries of $0.8 million, public company costs of $0.8 million, share-based compensation costs of $0.6 million, $0.4 million of legal costs, and $0.2 million of rent, along with $0.7 million related to recruiting, relocation, severance and other support costs; |
| · | Sales and Marketing: an increase in expenses of $2.2 million primarily due to share-based compensation costs of $0.6 million clinical outcome cost benefit studies costs of $0.3 million, salaries of $0.3 million, consulting costs of $0.3 million, and CLIA pilot studies of $0.1 million, along with $0.6 million of recruiting and other support costs; and |
| · | Transaction Expenses: the Company incurred $0.5 million of transaction expenses in connection with the Merger. |
In 2015 and 2014, the Company incurred $370,539 and $138,339 of operating expenses related to agreements with Fluidigm Corporation, a related party. Fluidigm Corporation supplies the Company with its microfluidic test platform for use in manufacturing the Acuitas MDRO Gene Test (see Note 12 to the 2015 audited financial statements included elsewhere herein).
Other income (expense)
| Year ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Interest and other income | $ | 26,657 | $ | 156 | ||||
| Interest expense | (1,801,320 | ) | (111,345 | ) | ||||
| Change in fair value of derivative financial instruments | (647,342 | ) | 4,400 | |||||
| Total other income (expense) | $ | (2,422,005 | ) | $ | (106,789 | ) | ||
Other income (expense) for the year ended December 31, 2015 increased to a net expense of ($2.4 million) from a net expense of ($0.1 million) in 2014. This increase was primarily the result of $1.5 million of non-cash interest expense related to the conversion of our convertible notes in May 2015 and the final mark-to-market adjustment related to our warrant liabilities, which were reclassified to stockholders’ equity on May 8, 2015 when their net cash-settlement features lapsed.
The Company recognized a benefit for income taxes of $0.1 million for the year ended December 31, 2015 (none in 2014) as a result of the net deferred tax liabilities in the U.S. taxing jurisdiction acquired in the Merger.
| 51 |
Liquidity and Capital Resources
At December 31, 2015, the Company had cash and cash equivalents of $7.8 million, compared to $0.7 million at December 31, 2014. The Company raised significant funds in the first nine months of 2015, consisting of:
| · | $0.8 million in short-term notes (in the first quarter of 2015, $0.3 million of demand notes held by an entity controlled by our chief executive officer were settled as partial payment for a 2015 convertible note, and in the second quarter of 2015, $0.2 million of notes from a related party were repaid in cash); |
| · | $1.5 million through the issuance of convertible notes; |
| · | $12.1 million in net proceeds from its IPO, as discussed further below; and |
| · | $6.0 million in net proceeds from the issuances of common stock and senior secured promissory note to Merck GHI. |
On May 8, 2015 the Company completed its IPO pursuant to which the Company offered and sold 2,850,000 units, each unit consisting of one share of common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 Units in the IPO. After considering the demand notes, underwriting discounts and commissions and offering expenses, the total net cash proceeds to the Company were $12.1 million. In connection with the IPO, all of the Company’s outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock.
In July 2015, the Company raised $6.0 million by issuing 1,136,364 shares of common stock at $4.40 per share and a $1.0 million senior secured promissory note to Merck GHI.
The Company’s primary cash requirements are to fund operations as well as research and development programs and collaborations, including those related to the acquisition of AdvanDx in July 2015, and to support general and administrative activities, and to fund acquisitions of products or businesses. The Company’s current operating assumptions, which include management’s best estimate of future revenue and operating expenses, indicate that current cash on hand will be sufficient to fund operations into the second quarter of 2016. The Company does not currently have any bank credit lines.
In the event the Company is unable to successfully raise additional capital in 2016, the Company may not have sufficient cash flows and liquidity to finance its business operations as currently contemplated. Accordingly, in such circumstances the Company would be compelled to reduce general and administrative expenses and delay research and development projects, including the purchase of scientific equipment and supplies, until it is able to obtain sufficient financing, or pursue other strategic alternatives which may include licensing and/or partnering arrangements or mergers and acquisitions.
Sources and uses of cash
The following table summarizes the net cash and cash equivalents provided by (used in) operating activities, investing activities and financing activities for the periods indicated:
| Year ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Net cash used in operating activities | $ | (14,278,004 | ) | $ | (5,385,542 | ) | ||
| Net cash provided by (used in) investing activities | 1,181,915 | (39,537 | ) | |||||
| Net cash provided by financing activities | 20,169,078 | 4,774,251 | ||||||
Net cash used in operating activities
Net cash used in operating activities in 2015 consists primarily of our net loss of ($17.4 million), reduced by certain non-cash items, including non-cash interest expense including that associated with the conversion of our convertible notes in May 2015 of $1.5 million, share-based compensation of $1.4 million, change in the fair value of our warrant liability of $0.6 million, depreciation and amortization expense of $0.6 million, and the net change in operating assets and liabilities of ($1.1 million). Net cash used in operating activities for 2014 consists primarily of our net loss of ($5.7 million), reduced by certain non-cash items, including depreciation and amortization expense of $0.6 million, share-based compensation expense of $0.1 million, and the net change in operating assets and liabilities of ($0.5 million).
| 52 |
Net cash provided by (used in) investing activities
Net cash provided by investing activities in 2015 includes cash on hand at AdvanDx at the date of the Merger of $1.4 million, along with the purchase of property and equipment. Net cash used in investing activities in 2014 includes solely the purchase of property and equipment.
Net cash provided by financing activities
Net cash provided by financing activities in 2015 of $20.2 million consisted primarily of the net proceeds from our IPO of $12.1 million, the net proceeds from the issuance of common stock to Merck GHI of $5.0 million, and the net proceeds from the issuance of debt instruments (including a $1.0 million senior secured promissory note to Merck GHI) of $3.1 million. Net cash provided by financing activities in 2014 of $4.8 million consists primarily of net proceeds from the issuance of preferred stock and convertible notes.
Critical accounting policies and use of estimates
This Management’s Discussion and Analysis of Financial Condition and Results of Operations is based on our audited consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of financial statements in conformity with GAAP requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. In our audited consolidated financial statements, estimates are used for, but not limited to, share-based compensation, allowances for doubtful accounts and inventories, valuation of derivative financial instruments, deferred tax assets and liabilities and related valuation allowance, and depreciation and amortization and estimated useful lives of long-lived assets. Actual results could differ from those estimates.
A summary of our significant accounting policies is included in Note 3 to the accompanying audited consolidated financial statements. Certain of our accounting policies are considered critical, as these policies require significant, difficult or complex judgments by management, often requiring the use of estimates about the effects of matters that are inherently uncertain.
Revenue Recognition
The Company recognizes revenue primarily from sales of the Argus System, sales of extended warranty service contracts for the Argus System, sales of AdvanDx diagnostic products, providing laboratory services, and from “funded software development” arrangements with collaborative parties. Revenue is recognized when the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred; the selling price is fixed or determinable; and collectability is reasonably assured. At times, the Company sells products and services, or performs software development, under multiple-element arrangements with separate units of accounting; in these situations, total consideration is allocated to the identified units of accounting based on their relative selling prices and revenue is then recognized for each unit based on its specific characteristics.
When an Argus System is sold without the Genome Builder software, total arrangement consideration is recognized as revenue when the system is delivered to the customer. Ancillary performance obligations, including installation, limited customer training and limited consumables, are considered inconsequential and are combined with the Argus System as one unit of accounting. When an Argus System is sold with the Genome Builder software in a multiple-element arrangement, total arrangement consideration is allocated to the Argus System and to the Genome Builder software (considered multiple elements) based on their relative selling prices. Selling prices are determined based on sales of similar systems to similar customers and, where no sales have occurred, on management’s best estimate of the expected selling price relative to similar products. Revenue related to the Argus System is recognized when it is delivered to the customer; revenue for the Genome Builder software is recognized when it is delivered to the customer. Revenue is recognized for Genome Builder software and for consumables, when sold on a stand-alone basis, upon delivery to the customer.
| 53 |
Revenue for the sales of QuickFISH, PNA FISH and XpressFISH diagnostic test products is recognized upon shipment to the customer. Sales are recorded net of accruals for estimated rebates, discounts and other deductions and returns.
The Company recognizes revenue associated with laboratory services contracts when the service has been performed and reports are made available to the customer.
The Company recognizes revenue associated with extended warranty service contracts over the service period in proportion to the costs expected to be incurred over that same period. The Company’s funded software development arrangements generally consist of multiple elements. Total arrangement consideration is allocated to the identified units of accounting based on their relative selling prices and revenue is then recognized for each unit based on its specific characteristics. When funded software development arrangements include substantive research and development milestones, revenue is recognized for each such milestone when the milestone is achieved and is due and collectible. Milestones are considered substantive if all of the following conditions are met: (1) the milestone is nonrefundable; (2) achievement of the milestone was not reasonably assured at the inception of the arrangement; (3) substantive effort is involved to achieve the milestone; and (4) the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with achievement of the milestone.
Impairment of Long-Lived Assets
Property and equipment is reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future undiscounted net cash flows expected to be generated by the asset. Recoverability measurement and estimating of undiscounted cash flows is done at the lowest possible level for which we can identify assets. If such assets are considered to be impaired, impairment is recognized as the amount by which the carrying amount of assets exceeds the fair value of the assets.
Definite-lived intangible assets include trademarks, developed technology and customer relationships. If any indicators were present, the Company would test for recoverability by comparing the carrying amount of the asset to the net undiscounted cash flows expected to be generated from the asset. If those net undiscounted cash flows do not exceed the carrying amount (i.e., the asset is not recoverable), the Company would perform the next step, which is to determine the fair value of the asset and record an impairment loss, if any.
Goodwill represents the excess of the purchase price for AdvanDx over the fair values of the acquired tangible or intangible assets and assumed liabilities. The Company will conduct an impairment test of goodwill on an annual basis as of October 1 of each year, and will also conduct tests if events occur or circumstances change that would, more likely than not, reduce the Company’s fair value below its net equity value.
Share-Based Compensation
Share-based payments to employees, directors and consultants are recognized at fair value. The resulting fair value is recognized ratably over the requisite service period, which is generally the vesting period of the option. The estimated fair value of equity instruments issued to nonemployees is recorded at fair value on the earlier of the performance commitment date or the date the services required are completed.
For all time-vesting awards granted, expense is amortized using the straight-line attribution method. For awards that contain a performance condition, expense is amortized using the accelerated attribution method. Share-based compensation expense recognized is based on the value of the portion of stock-based awards that is ultimately expected to vest during the period. The fair value of share-based payments is estimated, on the date of grant, using the Black-Scholes model. Option valuation models, including the Black-Scholes model, require the input of highly subjective estimates and assumptions, and changes in those estimates and assumptions can materially affect the grant-date fair value of an award. These assumptions include the fair value of the underlying and the expected life of the award.
See additional discussion of the use of estimates relating to share-based compensation, and a discussion of management’s methodology for developing each of the assumptions used in such estimates, in Note 3 to the accompanying unaudited condensed consolidated financial statements.
| 54 |
Recently issued accounting pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued guidance for revenue recognition for contracts, superseding the previous revenue recognition requirements, along with most existing industry-specific guidance. The guidance requires an entity to review contracts in five steps: 1) identify the contract, 2) identify performance obligations, 3) determine the transaction price, 4) allocate the transaction price, and 5) recognize revenue. The new standard will result in enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue arising from contracts with customers. The standard is effective for the Company’s reporting year beginning January 1, 2018 and early adoption is permitted starting January 1, 2017. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In August 2014, the FASB issued guidance requiring management to evaluate on a regular basis whether any conditions or events have arisen that could raise substantial doubt about the entity’s ability to continue as a going concern. The guidance 1) provides a definition for the term ‘‘substantial doubt,’’ 2) requires an evaluation every reporting period, interim periods included, 3) provides principles for considering the mitigating effect of management’s plans to alleviate the substantial doubt, 4) requires certain disclosures if the substantial doubt is alleviated as a result of management’s plans, 5) requires an express statement, as well as other disclosures, if the substantial doubt is not alleviated, and 6) requires an assessment period of one year from the date the financial statements are issued. The standard is effective for the Company’s reporting year beginning January 1, 2017 and early adoption is permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In April 2015, the FASB issued accounting guidance requiring that debt issuance costs related to a recognized liability be presented on the balance sheet as a direct reduction from the carrying amount of that debt liability, consistent with debt discounts. The recognition and measurement guidance for debt issuance costs are not affected. The standard is effective for reporting periods beginning after December 15, 2015. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In July 2015, the FASB issued accounting guidance for inventory. Under the guidance, an entity should measure inventory within the scope of this guidance at the lower of cost and net realizable value, except when inventory is measured using LIFO or the retail inventory method. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. In addition, the FASB has amended some of the other inventory guidance to more clearly articulate the requirements for the measurement and disclosure of inventory. The standard is effective for reporting periods beginning after December 15, 2016. The amendments in this pronouncement should be applied prospectively, with earlier application permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In September 2015, the FASB issued accounting guidance to simplify the accounting for measurement period adjustments resulting from business combinations. Under the guidance, an acquirer will be required to recognize adjustments to provisional amounts identified during the measurement period in the reporting period in which the adjustments are determined. The guidance requires an entity to present separately on the face of the income statement or disclose in the notes to the financial statements the portion of the amount recorded in current-period earnings by line item that would have been recorded in previous reporting periods if the adjustment had been recognized as of the acquisition date. The Company adopted this guidance in the fourth quarter of 2016; see Note 4 to the consolidated financial statements for a discussion of the impact of such adoption.
In February 2016, the FASB issued accounting guidance which will require most leases (with the exception of leases with terms of less than one year) to be recognized on the balance sheet as an asset and a lease liability. Leases will be classified as an operating lease or a financing lease. Operating leases are expensed using the straight-line method whereas financing leases will be treated similarly to a capital lease under the current standard. The new standard will be effective for annual and interim periods, within those fiscal years, beginning after December 15, 2018 but early adoption is permitted. The new standard must be presented using the modified retrospective method beginning with the earliest comparative period presented. The Company is currently evaluating the effect of the new standard on its consolidated financial statements and related disclosures.
| 55 |
Off-Balance Sheet Arrangements
As of each of December 31, 2015 and 2014, we did not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K promulgated by the SEC.
JOBS Act
On April 5, 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including without limitation; (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act of 2002; and (ii) complying with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor’s; discussion and analysis. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) December 31, 2020; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
As a smaller reporting company, we are not required to provide the information required by this item.
Item 8. Consolidated Financial Statements
Our consolidated financial statements and the report of our independent registered public accounting firm are included in this Annual Report as indicated in Part IV, Item 15.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management evaluated, under the supervision and with the participation of our principal executive officer and principal financial officer, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of December 31, 2015. We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow for timely decisions regarding required disclosure. Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of the period covered by this report, our disclosure controls and procedures were effective. In designing and evaluating our disclosure controls and procedures, we recognize that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives.
| 56 |
Management’s Annual Report on Internal Control over Financial Reporting
This Annual Report does not include a report on management’s assessment regarding internal control over financial reporting or an attestation report of the Company’s registered public accounting firm due to a transition period established by the rules of the SEC for newly public companies.
Changes in Internal Control over Financial Reporting
On July 14, 2015, the Company acquired 100% of the capital stock of AdvanDx in the Merger. The Company has not yet completed an assessment of the design and/or operating effectiveness of AdvanDx’s internal control over financial reporting. As of December 31, 2015, AdvanDx had total assets of $4.5 million and AdvanDx generated revenues of $1.8 million for the period subsequent to the Merger. There were no changes in the Company’s internal control over financial reporting during the last quarter that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
None.
| 57 |
Item 10. Directors and Executive Officers and Corporate Governance
Information required by this item is incorporated herein by reference to the similarly named section of our Definitive Proxy Statement for our 2016 Annual Meeting of Stockholders.
Item 11. Executive Compensation
Information required by this item is incorporated herein by reference to the similarly named section of our Definitive Proxy Statement for our 2016 Annual Meeting of Stockholders.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information required by this item is incorporated herein by reference to the similarly named section of our Definitive Proxy Statement for our 2016 Annual Meeting of Stockholders.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information required by this item is incorporated herein by reference to the similarly named section of our Definitive Proxy Statement for our 2016 Annual Meeting of Stockholders.
Item 14. Principal Accountant Fees and Services
Information required by this item is incorporated herein by reference to the similarly named section of our Definitive Proxy Statement for our 2016 Annual Meeting of Stockholders.
| 58 |
Item 15. Exhibits and Financial Statement Schedules
(a)(1) Financial Statements.
The consolidated balance sheets of the Company as of December 31, 2015 and 2014, the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity (deficit) and cash flows for the years then ended, the related notes to the consolidated financial statements and the report of CohnReznick LLP, independent registered public accounting firm, are filed herewith following the signature page.
(a)(2) Financial Statement Schedules.
Not applicable.
(a)(3) Exhibits:
A list of exhibits to this Annual Report is set forth on the Exhibit Index immediately preceding such exhibits and is incorporated herein by reference.
(b) Exhibits
See Exhibit Index.
(c) Not applicable.
| 59 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| OPGEN, INC. | ||
| By: | /s/ Evan Jones | |
| Evan Jones | ||
| Chief Executive Officer | ||
| Date: March 30, 2016 | ||
| By: | /s/ Timothy C. Dec | |
| Timothy C. Dec | ||
| Chief Financial Officer | ||
| Date: March 30, 2016 | ||
POWER OF ATTORNEY
We, the undersigned officers and directors of OpGen, Inc., hereby severally constitute and appoint Evan Jones, Kevin Krenitsky and Timothy C. Dec, our true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution in her or him for her or him and in her or his name, place and stead, and in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as full to all intents and purposes as she or he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or her or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Evan Jones | Chief Executive Officer and Director | March 30, 2016 | ||
| Evan Jones | (principal executive officer) | |||
| /s/ Timothy C. Dec | Chief Financial Officer | March 30, 2016 | ||
| Timothy C. Dec | (principal financial officer and principal accounting officer) | |||
| /s/ Brian G. Atwood | Director | March 30, 2016 | ||
| Brian G. Atwood |
| 60 |
| Signature | Title | Date | ||
| /s/ Timothy J.R. Harris | Director | March 30, 2016 | ||
| Timothy J.R. Harris | ||||
| /s/ Timothy Howe | Director | March 30, 2016 | ||
| Timothy Howe | ||||
| /s/ Laurence R. McCarthy | Director | March 30, 2016 | ||
| Laurence R. McCarthy | ||||
| /s/ David M. Rubin | Director | March 30, 2016 | ||
| David M. Rubin | ||||
| /s/ Misti Ushio | Director | March 30, 2016 | ||
| Misti Ushio |
| 61 |
OPGEN, INC.
Index to Consolidated Financial Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
OpGen, Inc.
We have audited the accompanying consolidated balance sheets of OpGen, Inc. as of December 31, 2015 and 2014, and the related statements of operations and comprehensive loss, stockholders’ equity (deficit) and cash flows for the years then ended. OpGen, Inc.’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of OpGen, Inc. as of December 31, 2015 and 2014 and the results of their operations and their cash flows for the years then ended in conformity with accounting principles generally accepted in the United Stated of America.
The consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in the Note 1 to the consolidated financial statements, the Company has incurred cumulative net losses since inception and will need additional capital to fund future operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/CohnReznick LLP
Vienna, Virginia
March 29, 2016
| F-1 |
Consolidated Balance Sheets
December 31,
| 2015 | 2014 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 7,814,220 | $ | 749,517 | ||||
| Accounts receivable, net | 678,646 | 503,983 | ||||||
| Inventory, net | 826,012 | 369,742 | ||||||
| Prepaid expenses and other current assets | 572,489 | 90,233 | ||||||
| Total current assets | 9,891,367 | 1,713,475 | ||||||
| Property and equipment, net | 1,074,710 | 587,956 | ||||||
| Deferred IPO issuance costs | - | 296,041 | ||||||
| Intangible assets, net | 1,888,814 | - | ||||||
| Goodwill | 637,528 | - | ||||||
| Other noncurrent assets | 270,327 | 57,459 | ||||||
| Total assets | $ | 13,762,746 | $ | 2,654,931 | ||||
| Liabilities, Preferred Stock and Stockholders’ Equity (Deficit) | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 2,285,792 | $ | 1,160,081 | ||||
| Accrued compensation and benefits | 1,081,270 | 423,099 | ||||||
| Deferred rent, current portion | 303,719 | 26,000 | ||||||
| Accrued liabilities | 920,286 | 967,657 | ||||||
| Deferred revenue | 50,925 | 339,171 | ||||||
| Short-term notes payable | - | 1,505,000 | ||||||
| Current maturities of long-term capital lease obligations | 251,800 | 100,499 | ||||||
| Short-term convertible notes, net of discounts | - | 1,500,000 | ||||||
| Total current liabilities | 4,893,792 | 6,021,507 | ||||||
| Long-term capital lease obligations and other liabilities | 377,908 | 134,149 | ||||||
| Notes payable | 1,000,000 | - | ||||||
| Total liabilities | 6,271,700 | 6,155,656 | ||||||
| Commitments and contingencies (Note 10) | ||||||||
| Redeemable convertible preferred stock | ||||||||
| Series A redeemable convertible preferred stock, $.01 par value; 6,000,000 shares authorized and 3,999,864 shares issued and outstanding at December 31, 2014 (none in 2015), respectively; aggregate liquidation preference of $7,999,728 at December 31, 2014 | - | 4,564,899 | ||||||
| Total redeemable convertible preferred stock | - | 4,564,899 | ||||||
| Stockholders' equity (deficit) | ||||||||
| Common stock, $.01 par value; 200,000,000 shares authorized; 12,547,684 and 493,178 shares issued and outstanding at December 31, 2015 and 2014, respectively | 125,477 | 4,932 | ||||||
| Additional paid-in capital | 121,490,994 | 88,701,737 | ||||||
| Accumulated other comprehensive loss | (1,059 | ) | - | |||||
| Accumulated deficit | (114,124,366 | ) | (96,772,293 | ) | ||||
| Total stockholders’ equity (deficit) | 7,491,046 | (8,065,624 | ) | |||||
| Total liabilities, preferred stock and stockholders’ equity (deficit) | $ | 13,762,746 | $ | 2,654,931 | ||||
See accompanying notes to financial statements.
| F-2 |
Consolidated Statements of Operations and Comprehensive Loss
Years Ended December 31,
| 2015 | 2014 | |||||||
| Revenue | ||||||||
| Product sales | $ | 2,701,142 | $ | 1,236,349 | ||||
| Laboratory services | 120,476 | 478,909 | ||||||
| Collaborations revenue | 336,102 | 2,411,120 | ||||||
| Total revenue | 3,157,720 | 4,126,378 | ||||||
| Operating expenses | ||||||||
| Costs of products sold | 1,179,771 | 425,541 | ||||||
| Costs of services | 367,802 | 526,196 | ||||||
| Research and development | 6,002,941 | 4,368,302 | ||||||
| General and administrative | 5,834,642 | 2,312,935 | ||||||
| Sales and marketing | 4,305,444 | 2,058,085 | ||||||
| Transaction expenses | 526,283 | - | ||||||
| Total operating expenses | 18,216,883 | 9,691,059 | ||||||
| Operating loss | (15,059,163 | ) | (5,564,681 | ) | ||||
| Other income (expense) | ||||||||
| Interest and other income | 26,657 | 156 | ||||||
| Interest expense | (1,801,320 | ) | (111,345 | ) | ||||
| Change in fair value of derivative financial instruments and other | (647,342 | ) | 4,400 | |||||
| Total other income (expense) | (2,422,005 | ) | (106,789 | ) | ||||
| Loss before income taxes | (17,481,168 | ) | (5,671,470 | ) | ||||
| Income tax benefit | (129,095 | ) | - | |||||
| Net loss | (17,352,073 | ) | (5,671,470 | ) | ||||
| Accretion of Series A preferred stock | (243,762 | ) | (627,133 | ) | ||||
| Net loss available to common stockholders | $ | (17,595,835 | ) | $ | (6,298,603 | ) | ||
| Net loss per common share - basic and diluted | $ | (2.20 | ) | $ | (16.25 | ) | ||
| Weighted average shares outstanding - basic and diluted | 7,980,995 | 387,590 | ||||||
| Net loss | $ | (17,352,073 | ) | $ | (5,671,470 | ) | ||
| Other comprehensive loss - foreign currency translation | (1,059 | ) | - | |||||
| Comprehensive loss | $ | (17,353,132 | ) | $ | (5,671,470 | ) | ||
See accompanying notes to financial statements.
| F-3 |
Consolidated Statements of Stockholders’ Equity (Deficit)
Years Ended December 31, 2015 and 2014
| Common Stock | ||||||||||||||||||||||||
| Number of Shares | Amount | Additional Paid- in Capital | Accumulated Other Comprehensive Loss | Accumulated Deficit | Total | |||||||||||||||||||
| Balances at December 31, 2013 | 362,537 | $ | 3,625 | $ | 89,265,757 | $ | - | $ | (91,100,823 | ) | $ | (1,831,441 | ) | |||||||||||
| Stock option exercises | 1 | - | 8 | - | - | 8 | ||||||||||||||||||
| Restricted stock unit exercises | 130,640 | 1,307 | (1,307 | ) | - | - | - | |||||||||||||||||
| Stock compensation expense | - | - | 64,412 | - | - | 64,412 | ||||||||||||||||||
| Accretion of Series A preferred stock | - | - | (627,133 | ) | - | - | (627,133 | ) | ||||||||||||||||
| Net loss | - | - | - | - | (5,671,470 | ) | (5,671,470 | ) | ||||||||||||||||
| Balances at December 31, 2014 | 493,178 | 4,932 | 88,701,737 | - | (96,772,293 | ) | (8,065,624 | ) | ||||||||||||||||
| Stock option exercises | 11,472 | 114 | 2,189 | - | - | 2,303 | ||||||||||||||||||
| Beneficial conversion feature | - | - | 1,427,667 | - | - | 1,427,667 | ||||||||||||||||||
| Reclassification of warrant liability to equity | - | - | 719,675 | - | - | 719,675 | ||||||||||||||||||
| Conversion of preferred stock into common shares | 7,374,852 | 73,749 | 7,730,423 | - | - | 7,804,172 | ||||||||||||||||||
| Demand notes tendered for IPO Units | 350,000 | 3,500 | 2,096,500 | - | - | 2,100,000 | ||||||||||||||||||
| Issuance of IPO units, net of offering costs | 2,500,000 | 25,000 | 12,104,133 | - | - | 12,129,133 | ||||||||||||||||||
| Additional IPO issuance costs | (58,566 | ) | - | - | (58,566 | ) | ||||||||||||||||||
| Common shares issued in business combination | 681,818 | 6,818 | 2,577,272 | - | - | 2,584,090 | ||||||||||||||||||
| Common shares issued in financing | 1,136,364 | 11,364 | 4,988,638 | - | - | 5,000,002 | ||||||||||||||||||
| Stock compensation expense | - | - | 1,445,088 | - | - | 1,445,088 | ||||||||||||||||||
| Accretion of Series A preferred stock | - | - | (243,762 | ) | - | - | (243,762 | ) | ||||||||||||||||
| Foreign currency translation | - | - | - | (1,059 | ) | - | (1,059 | ) | ||||||||||||||||
| Net loss | - | - | - | - | (17,352,073 | ) | (17,352,073 | ) | ||||||||||||||||
| Balances at December 31, 2015 | 12,547,684 | $ | 125,477 | $ | 121,490,994 | $ | (1,059 | ) | $ | (114,124,366 | ) | $ | 7,491,046 | |||||||||||
See accompanying notes to financial statements.
| F-4 |
Consolidated Statements of Cash Flows
Years Ended December 31,
| 2015 | 2014 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (17,352,073 | ) | $ | (5,671,470 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 624,653 | 573,918 | ||||||
| Non-cash interest expense including beneficial conversion | 1,598,312 | 84,168 | ||||||
| Bad debt expense | - | 4,000 | ||||||
| Recovery of bad debt | - | (8,400 | ) | |||||
| Deferred tax benefit | (129,095 | ) | - | |||||
| Stock compensation expense | 1,445,088 | 64,412 | ||||||
| Inventory obsolescence expense | - | 21,877 | ||||||
| Change in fair value of derivative financial instruments | 647,342 | - | ||||||
| Other non-cash items | 24,010 | 14,681 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | 359,298 | (257,686 | ) | |||||
| Inventory | 424,505 | (215,906 | ) | |||||
| Other assets | (319,305 | ) | 76,554 | |||||
| Accounts payable | 196,444 | 198,177 | ||||||
| Accrued compensation and other liabilities | (1,508,937 | ) | (99,310 | ) | ||||
| Deferred revenue | (288,246 | ) | (170,557 | ) | ||||
| Net cash used in operating activities | (14,278,004 | ) | (5,385,542 | ) | ||||
| Cash flows from investing activities | ||||||||
| Cash acquired in business combination | 1,367,211 | - | ||||||
| Purchases of property and equipment | (185,296 | ) | (39,537 | ) | ||||
| Net cash provided by (used in) investing activities | 1,181,915 | (39,537 | ) | |||||
| Cash flows from financing activities | ||||||||
| Proceeds from issuance of preferred stock, net of issuance costs | - | 1,937,902 | ||||||
| Proceeds from issuance of common stock, net of issuance costs | 17,366,620 | - | ||||||
| Proceeds from issuance of convertible notes, net of issuance costs | 1,388,815 | 1,472,386 | ||||||
| Proceeds from issuance of promissory notes, net of issuance costs | 1,741,667 | 1,488,229 | ||||||
| Proceeds from exercise of stock options and warrants | 2,293 | 8 | ||||||
| Payments on debt | (155,000 | ) | (5,000 | ) | ||||
| Payments on capital lease obligations | (175,317 | ) | (105,881 | ) | ||||
| Deferred IPO issuance costs | - | (13,393 | ) | |||||
| Net cash provided by financing activities | 20,169,078 | 4,774,251 | ||||||
| Effects of exchange rates on cash | (8,286 | ) | - | |||||
| Net increase (decrease) in cash and cash equivalents | 7,072,989 | (650,828 | ) | |||||
| Cash and cash equivalents at beginning of year | 749,517 | 1,400,345 | ||||||
| Cash and cash equivalents at end of year | $ | 7,814,220 | $ | 749,517 | ||||
| Supplemental disclosure of cash flow information | ||||||||
| Cash paid during the year for interest | $ | 194,288 | $ | 32,296 | ||||
| Supplemental disclosure of noncash investing and financing activities: | ||||||||
| Common stock issued for business combination | $ | 2,584,090 | $ | - | ||||
| Acquisition of equipment purchased through capital leases and leasehold improvement allowances | $ | 580,477 | $ | - | ||||
| Deferred and accrued IPO issuance costs | $ | - | $ | 282,648 | ||||
| Exchange of demand note for convertible debt | $ | 300,000 | $ | - | ||||
| Exchange of demand notes for IPO units | $ | 2,100,000 | $ | - | ||||
| Conversion of convertible notes into Series A preferred stock | $ | 3,000,000 | $ | - | ||||
| Conversion of Series A preferred stock into common shares | $ | 8,183,661 | $ | - | ||||
See accompanying notes to financial statements.
| F-5 |
Notes to Consolidated Financial Statements
Note 1 – Organization
OpGen, Inc. (“OpGen” of the “Company”) was incorporated in Delaware in 2001. On July 14, 2015, OpGen completed the strategic acquisition (the “Merger”) of AdvanDx, Inc. and its wholly owned subsidiary AdvanDx A/S (collectively, “AdvanDx”) (see Note 4). Pursuant to the terms of a merger agreement, Velox Acquisition Corp., OpGen’s wholly owned subsidiary formed for the express purpose of effecting the Merger, merged with and into AdvanDx, Inc. with AdvanDx, Inc. surviving as OpGen’s wholly owned subsidiary (see Note 4). OpGen, AdvanDx, Inc. and AdvanDx A/S are collectively referred to hereinafter as the “Company.” The Company’s headquarters are in Gaithersburg, Maryland, and its principal operations are in Gaithersburg, Maryland and Woburn, Massachusetts. The Company also has operations in Copenhagen, Denmark. The Company operates in one business segment.
OpGen is a precision medicine company using molecular diagnostics and informatics to combat infectious disease. OpGen is developing molecular information solutions to combat infectious disease in global healthcare settings, helping to guide clinicians with more rapid information about life threatening infections, improve patient outcomes, and decrease the spread of infections caused by multidrug-resistant microorganisms. The Company’s proprietary DNA tests and bioinformatics address the rising threat of antibiotic resistance by helping physicians and healthcare providers optimize patient care decisions and protect the hospital biome through customized screening and surveillance solutions. The Company’s molecular information solution combines Acuitas® DNA tests, Acuitas Lighthouse™ bioinformatics, CLIA lab services for MDRO surveillance, and a proprietary data warehouse including genomic data matched with antibiotic susceptibility information for microbes and patient information from healthcare providers. The Company is working to deliver our molecular information solution to a global network of customers and partners. The Acuitas DNA tests provide rapid microbial ID, and antibiotic resistance gene information. These products include the QuickFISH® family of FDA-cleared and CE-marked diagnostics used to rapidly detect pathogens in positive blood cultures, the MDRO Gene Test to detect, type, track, and trend antibiotic resistant organisms in real-time and our rapid antibiotic resistance test in development.
The Company’s operations are subject to certain risks and uncertainties. The risks include rapid technology changes, the need to manage growth, the need to retain key personnel, the need to protect intellectual property and the need to raise additional capital financing on terms acceptable to the Company. The Company’s success depends, in part, on its ability to develop and commercialize its proprietary technology as well as raise additional capital.
Note 2 - Going Concern and Management’s Plans
The accompanying consolidated financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. Since inception, the Company has incurred, and continues to incur, significant losses from operations. The Company has funded its operations primarily through external investor financing arrangements. The Company raised significant funds in 2015, including:
| · | $0.8 million in short-term notes (in the first quarter of 2015, $0.3 million of demand notes held by an entity controlled by our chief executive officer were settled as partial payment for a 2015 convertible note, and in the second quarter of 2015, $0.2 million of notes from a related party were repaid in cash); |
| · | $1.5 million through the issuance of convertible notes; |
| · | $12.1 million in net proceeds from its initial public offering (“IPO”) as discussed further below; and |
| · | $6.0 million in net proceeds from the issuances of common stock and a senior secured promissory note to Merck Global Health Innovation Fund, LLC (“Merck GHI”). |
On May 8, 2015, OpGen completed its IPO pursuant to which it offered and sold 2,850,000 units, each Unit consisting of one share of common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 Units in the IPO. After considering the demand notes, underwriting discounts and commissions and offering expenses, the total net cash proceeds were $12.1 million. On the IPO closing date, the underwriters exercised their over-allotment option to acquire an additional 422,500 stock purchase warrants. In connection with the IPO, all of OpGen’s outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock.
| F-6 |
In July 2015, the Company raised $6.0 million by issuing 1,136,364 shares of common stock at $4.40 per share and a $1.0 million senior secured promissory note to Merck GHI (see Note 5).
The Company’s current operating assumptions, which include management’s best estimate of future revenue and operating expenses, indicate that current cash on hand will be sufficient to fund operations into the second quarter of 2016.
In the event the Company is unable to successfully raise additional capital in 2016, the Company will not have sufficient cash flows and liquidity to finance its business operations as currently contemplated. Accordingly, in such circumstances the Company would be compelled to reduce general and administrative expenses and delay research and development projects, including the purchase of scientific equipment and supplies, until it is able to obtain sufficient financing, or pursue other strategic alternatives which may include licensing and/or partnering arrangements or mergers and acquisitions. The consolidated financial statements do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue in existence.
Note 3 - Summary of Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements are prepared in accordance with generally accepted accounting standards in the United States (“U.S. GAAP”). The consolidated financial statements consolidate the operations of all controlled subsidiaries; all intercompany activity is eliminated. Certain prior period information has been reclassified to conform to the current period presentation.
Foreign Currency
AdvanDx A/S is located in Copenhagen, Denmark and uses the Danish Kroner as its functional currency. As a result, all assets and liabilities are translated into U.S. dollars based on exchange rates at the end of the reporting period. Income and expense items are translated at the average exchange rates prevailing during the reporting period. Translation adjustments are reported in other comprehensive loss, a component of stockholder's equity. Foreign currency translation adjustments are the sole component of accumulated other comprehensive loss at December 31, 2015.
Foreign currency transaction gains and losses, excluding gains and losses on intercompany balances where there is no current intent to settle such amounts in the foreseeable future, are included in the determination of net loss.
Unless otherwise noted, all references to “$” or “dollar” refer to the United States dollar.
Use of Estimates
In preparing financial statements in conformity with GAAP, management is required to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. In the accompanying consolidated financial statements, estimates are used for, but not limited to, share-based compensation, allowances for doubtful accounts and inventory obsolescence, valuation of derivative financial instruments, beneficial conversion features of convertible debt, deferred tax assets and liabilities and related valuation allowance, and depreciation and amortization and estimated useful lives of long-lived assets, and the recoverability of long-lived assets. Actual results could differ from those estimates.
| F-7 |
Fair value of financial instruments
All financial instruments classified as current assets and liabilities are carried at cost, which approximates fair value, because of the short-term maturities of those instruments. Debt is reflective of fair value based on instruments with similar terms available to the Company.
For additional fair value disclosures, see Note 13.
Cash and cash equivalents and restricted cash
The Company considers all highly liquid instruments with original maturities of three months or less to be cash equivalents. The Company has cash and cash equivalents deposited in financial institutions in which the balances occasionally exceed the federal government agency (FDIC) insured limits of $250,000. The Company has not experienced any losses in such accounts and management believes it is not exposed to any significant credit risk.
At December 31, 2015, the Company has funds totaling $243,380, which are required as collateral for letters of credit benefiting its landlords and for credit card processors. These funds are reflected in other long-term assets on the accompanying consolidated balance sheets.
Accounts Receivable
The Company’s accounts receivable result from revenues earned but not collected from customers. Credit is extended based on an evaluation of a customer’s financial condition and, generally, collateral is not required. Accounts receivable are due within 30 to 60 days and are stated at amounts due from customers. The Company evaluates if an allowance is necessary by considering a number of factors, including the length of time accounts receivable are past due, the Company’s previous loss history and the customer’s current ability to pay its obligation. If amounts become uncollectible, they are charged to operations when that determination is made. The allowance for doubtful accounts was $15,596 and $79,697 as of December 31, 2015 and 2014, respectively.
At December 31, 2015, the Company had accounts receivable from one customer which individually represented 25% of total accounts receivable. At December 31, 2014, the Company had accounts receivable from two customers which individually represent 79% and 15% of total accounts receivable. For the year ended December 31, 2015, revenue earned from Hitachi High-Technologies Corporation (“Hitachi”) represented 11% of total revenues. For the year ended December 31, 2014, revenue earned from Hitachi represented 64% of total revenues. This customer contract was completed as of December 31, 2015.
Inventory
Inventories are valued using the first-in, first-out method and stated at the lower of cost or market and consist of the following:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Raw Materials and supplies | $ | 362,526 | $ | 40,749 | ||||
| Work-in-process | 150,369 | 135,625 | ||||||
| Finished goods | 313,117 | 193,368 | ||||||
| Total inventory | $ | 826,012 | $ | 369,742 | ||||
Inventory includes the Argus Whole Genome Mapping Systems, reagents and supplies used for Argus consumable kits, reagents and components for QuickFISH and PNA FISH kit products, and reagents and supplies used for the Company’s laboratory services. Inventory reserves for obsolescence and expirations were $591,051 and $867,816 at December 31, 2015 and 2014, respectively.
| F-8 |
Intangible assets and goodwill
Intangible assets as of December 31, 2015 were acquired as part of the Merger, and consist of definite-lived intangible assets and goodwill. Intangible assets acquired in prior years were fully amortized as of December 31, 2014.
Definite-lived intangible assets
Definite-lived intangible assets include trademarks, developed technology and customer relationships, and are amortized over their useful lives of 10, 7 and 7 years, respectively, and consisted of the following as of December 31, 2015:
| Cost | Accumulated Amortization | Net balance at December 31, 2015 | ||||||||||
| Trademarks and tradenames | $ | 461,000 | $ | (21,471 | ) | $ | 439,529 | |||||
| Developed technology | 458,000 | (30,474 | ) | 427,526 | ||||||||
| Customer relationships | 1,094,000 | (72,241 | ) | 1,021,759 | ||||||||
| Balance | $ | 2,013,000 | $ | (124,186 | ) | $ | 1,888,814 | |||||
Total amortization expense of intangible assets was $124,186 and $57,594 for the years ended December 31, 2015 and 2014, respectively. Amortization of intangible assets is expected to be approximately $268,000 per year for the next five years.
Definite-lived intangible assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. If any indicators were present, the Company would test for recoverability by comparing the carrying amount of the asset to the net undiscounted cash flows expected to be generated from the asset. If those net undiscounted cash flows do not exceed the carrying amount (i.e., the asset is not recoverable), the Company would perform the next step, which is to determine the fair value of the asset and record an impairment loss, if any. During years ended December 31, 2015 and 2014, the Company determined that its definite-lived intangible assets were not impaired.
Goodwill
Goodwill represents the excess of the purchase price for AdvanDx over the fair values of the acquired tangible or intangible assets and assumed liabilities. Goodwill is not tax deductible in any relevant jurisdictions. As a result of the Merger and subsequent measurement period adjustments (see Note 4), the Company recognized goodwill of $637,528.
The Company conducts an impairment test of goodwill on an annual basis as of October 1 of each year, and will also conduct tests if events occur or circumstances change that would, more likely than not, reduce the Company’s fair value below its net equity value. During year ended December 31, 2015, the Company determined that its goodwill was not impaired.
Property and equipment
Property and equipment is stated at cost and depreciated on a straight-line basis over the estimated useful lives of the related assets. The estimated service lives approximate three to five years. Depreciation expense was $500,467 and $516,324 for the years ended December 31, 2015 and 2014, respectively. Property and equipment consisted of the following at December 31, 2015 and 2014:
| F-9 |
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Laboratory equipment | $ | 3,734,044 | $ | 2,304,615 | ||||
| Office furniture and equipment | 701,557 | 691,032 | ||||||
| Computers | 1,563,177 | 1,169,910 | ||||||
| Leasehold improvements | 659,949 | 245,558 | ||||||
| 6,658,727 | 4,411,115 | |||||||
| Less accumulated depreciation | (5,584,017 | ) | (3,823,159 | ) | ||||
| Property and equipment, net | $ | 1,074,710 | $ | 587,956 | ||||
In 2012, the Company began to provide Argus™ Whole Genome Systems under its Argus Reagent Rental Program to customers, in which the Company retains title without requiring customers to purchase the equipment or enter into an equipment lease or rental contract. The costs associated with these instruments are capitalized and charged to sales and marketing on a straight-line basis over the estimated useful life of the instrument, which is approximately four years. During the years ended December 31, 2015 and 2014, sales and marketing expenses related to these costs were approximately $175,000 and $101,000, respectively. The costs to maintain these systems are charged to operations as incurred.
Property and equipment is reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future undiscounted net cash flows expected to be generated by the asset. Recoverability measurement and estimating of undiscounted cash flows is done at the lowest possible level for which we can identify assets. If such assets are considered to be impaired, impairment is recognized as the amount by which the carrying amount of assets exceeds the fair value of the assets. During the years ended December 31, 2015 and 2014, the Company determined that its property and equipment was not impaired.
Deferred rent
Deferred rent is recorded and amortized to the extent the total minimum rental payments allocated to the current period on a straight-line basis exceed or are less than the cash payments required.
Redeemable convertible preferred stock
All shares of Series A redeemable convertible preferred stock (“Series A Preferred Stock”) (including those shares issued in connection with the conversion of the 2014 and 2015 convertible debt (see Note 6)), were converted into 7,374,852 shares of common stock in connection with the Company’s IPO (see Notes 7 and 8).
Prior to the IPO, the carrying value of the Series A Preferred Stock was increased by the accretion of related discounts, issuance costs and accrued but unpaid dividends so that the carrying amount would equal the redemption amount at the dates the stock becomes redeemable. At December 31, 2014, the Company had 3,999,864 shares of Series A Preferred Stock outstanding. The Series A Preferred Stock was redeemable at the option of the holders of 70% of the outstanding shares of Series A Preferred Stock, subject to certain additional requirements. The Company’s redeemable convertible preferred stock was classified as temporary equity due to redemption provisions outside of the Company’s control.
Revenue recognition
The Company recognizes revenue primarily from sales of its products and services when the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred; the selling price is fixed or determinable; and collectability is reasonably assured. At times, the Company sells products and services, or performs software development, under multiple-element arrangements with separate units of accounting; in these situations, total consideration is allocated to the identified units of accounting based on their relative selling prices and revenue is then recognized for each unit based on its specific characteristics.
| F-10 |
Amounts billed to customers for shipping and handling are included in revenue when the related product or service revenue is recognized. Shipping and handling costs are included in cost of sales.
Revenue from sales of the Argus System
When an Argus System is sold without the Genome Builder software, total arrangement consideration is recognized as revenue when the system is delivered to the customer. Ancillary performance obligations, including installation, limited customer training and limited consumables, are considered inconsequential and are combined with the Argus System as one unit of accounting.
When an Argus System is sold with the Genome Builder software in a multiple-element arrangement, total arrangement consideration is allocated to the Argus System and to the Genome Builder software based on their relative selling prices. Selling prices are determined based on sales of similar systems to similar customers and, where no sales have occurred, on management’s best estimate of the expected selling price relative to similar products. Revenue related to the Argus System is recognized when it is delivered to the customer; revenue for the Genome Builder software is recognized when it is delivered to the customer.
Revenue from sales of QuickFISH, PNA FISH and XpressFISH diagnostic test products
Revenue is recognized upon shipment to the customer. Sales are recorded net of accruals for estimated rebates, discounts and other deductions and returns.
Revenue from sales of Genome Builder Software and consumables (on a stand-alone basis)
Revenue is recognized for Genome Builder Software and for consumables, when sold on a standalone basis, upon delivery to the customer.
Revenue from extended warranty service contracts
The Company recognizes revenue associated with extended warranty service contracts over the service period in proportion to the costs expected to be incurred over that same period.
Revenue from providing laboratory services
The Company recognizes revenue associated with laboratory services contracts when the service has been performed and reports are made available to the customer.
Revenue from funded software development arrangements
The Company’s funded software development arrangements generally consist of multiple elements. Total arrangement consideration is allocated to the identified units of accounting based on their relative selling prices and revenue is then recognized for each unit based on its specific characteristics. When funded software development arrangements include substantive research and development milestones, revenue is recognized for each such milestone when the milestone is achieved and is due and collectible. Milestones are considered substantive if all of the following conditions are met: (1) the milestone is nonrefundable; (2) achievement of the milestone was not reasonably assured at the inception of the arrangement; (3) substantive effort is involved to achieve the milestone; and (4) the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with achievement of the milestone.
Research and development costs
Research and development costs are expensed as incurred. Research and development costs primarily consist of salaries and related expenses for personnel, other resources, laboratory supplies, fees paid to consultants and outside service partners.
Share-based compensation
Share-based payments are recognized at fair value. The fair value of share-based payments to employees and directors is estimated, on the date of grant, using the Black-Scholes model. The resulting fair value is recognized ratably over the requisite service period, which is generally the vesting period of the option. For all time-vesting awards granted, expense is amortized using the straight-line attribution method. Share-based compensation expense recognized is based on the value of the portion of stock-based awards that is ultimately expected to vest during the period.
| F-11 |
Option valuation models, including the Black-Scholes model, require the input of highly subjective assumptions, and changes in the assumptions used can materially affect the grant-date fair value of an award. These assumptions include the risk-free rate of interest, expected dividend yield, expected volatility and the expected life of the award. A discussion of management’s methodology for developing each of the assumptions used in the Black-Scholes model is as follows:
Fair value of common stock
For periods prior to the Company’s IPO, given the lack of an active public market for the common stock, the Company’s board of directors determined the fair value of the common stock. In the absence of a public market, and as an emerging company with no significant revenues, the Company believes that it is appropriate to consider a range of factors to determine the fair market value of the common stock at each grant date. The factors include: (1) the achievement of clinical and operational milestones by the Company; (2) the status of strategic relationships with collaborators; (3) the significant risks associated with the Company’s stage of development; (4) capital market conditions for life science and medical diagnostic companies, particularly similarly situated, privately held, early stage companies; (5) the Company’s available cash, financial condition and results of operations; (6) the most recent sales of the Company’s preferred stock; and (7) the preferential rights of the outstanding preferred stock. Since the IPO, the Company uses the quoted market price of its common stock as its fair value.
Expected volatility
Volatility is a measure of the amount by which a financial variable such as a share price has fluctuated (historical volatility) or is expected to fluctuate (expected volatility) during a period. Until a significant trading history for its common stock develops, the Company has identified several public entities of similar size, complexity and stage of development; accordingly, historical volatility has been calculated using the volatility of this peer group.
Expected dividend yield
The Company has never declared or paid dividends on its common stock and has no plans to do so in the foreseeable future.
Risk-free interest rate
This is the U.S. Treasury rate for the day of each option grant during the year, having a term that most closely resembles the expected term of the option.
Expected term
This is the period of time that the options granted are expected to remain unexercised. Options granted have a maximum term of 10 years. The Company estimates the expected term of the option to be 6.25 years for options with a standard four-year vesting period, using the simplified method. Over time, management will track actual terms of the options and adjust their estimate accordingly so that estimates will approximate actual behavior for similar options.
Expected forfeiture rate
The forfeiture rate is the estimated percentage of options granted that is expected to be forfeited or canceled on an annual basis before becoming fully vested. The Company estimates the forfeiture rate based on turnover data with further consideration given to the class of the employees to whom the options were granted.
The estimated fair value of equity instruments issued to nonemployees are recorded at fair value on the earlier of the performance commitment date or the date the services required are completed.
Income taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the expected future tax consequences attributable to temporary differences between financial statement carrying amounts of existing assets and liabilities and their respective tax basis. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is established when necessary to reduce deferred income tax assets to the amount expected to be realized.
| F-12 |
Tax benefits are initially recognized in the financial statements when it is more likely than not the position will be sustained upon examination by the tax authorities. Such tax positions are initially, and subsequently, measured as the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate settlement with the tax authority, assuming full knowledge of the position and all relevant facts.
The Company had federal net operating loss (“NOL”) carryforwards of $90,297,000 and $76,268,000 at December 31, 2015 and 2014, respectively. Despite the NOL carryforwards, which begin to expire in 2022, the Company may have future tax liability due to alternative minimum tax or state tax requirements. Also, use of the NOL carryforwards may be subject to an annual limitation as provided by Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”). To date, the Company has not performed a formal study to determine if any of its remaining NOL and credit attributes might be further limited due to the ownership change rules of Section 382 or Section 383 of the Code. The Company will continue to monitor this matter going forward. There can be no assurance that the NOL carryforwards will ever be fully utilized.
Loss per share
Basic loss per share is computed by dividing net loss available to common shareholders by the weighted average number of shares of common stock outstanding during the period.
For periods of net income, and when the effects are not anti-dilutive, diluted earnings per share is computed by dividing net income available to common shareholders by the weighted-average number of shares outstanding plus the impact of all potential dilutive common shares, consisting primarily of common stock options and stock purchase warrants using the treasury stock method, and convertible preferred stock and convertible debt using the if-converted method.
For periods of net loss, diluted loss per share is calculated similarly to basic loss per share because the impact of all dilutive potential common shares is anti-dilutive. The number of anti-dilutive shares, consisting of (i) common stock options, (ii) restricted stock units (in 2014), (iii) stock purchase warrants, and (iv) prior to the IPO, convertible preferred stock and convertible debt, exercisable or exchangeable into common stock which have been excluded from the computation of diluted loss per share, was 6.0 million shares and 5.9 million shares for the years ended December 31, 2015 and 2014, respectively. The Company’s convertible preferred stock, prior to its conversion, contained non-forfeitable rights to dividends, and therefore was considered to be a participating security; the calculation of basic and diluted income (loss) per share excludes net income (but not net loss) attributable to the convertible preferred stock from the numerator and excludes the impact of those shares from the denominator in periods prior to the IPO.
Recent accounting pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”), issued guidance for revenue recognition for contracts, superseding the previous revenue recognition requirements, along with most existing industry-specific guidance. The guidance requires an entity to review contracts in five steps: 1) identify the contract, 2) identify performance obligations, 3) determine the transaction price, 4) allocate the transaction price, and 5) recognize revenue. The new standard will result in enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue arising from contracts with customers. The standard is effective for the Company’s reporting year beginning January 1, 2018 and early adoption is permitted starting January 1, 2017. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In August 2014, the FASB issued guidance requiring management to evaluate on a regular basis whether any conditions or events have arisen that could raise substantial doubt about the entity’s ability to continue as a going concern. The guidance 1) provides a definition for the term “substantial doubt,” 2) requires an evaluation every reporting period, interim periods included, 3) provides principles for considering the mitigating effect of management’s plans to alleviate the substantial doubt, 4) requires certain disclosures if the substantial doubt is alleviated as a result of management’s plans, 5) requires an express statement, as well as other disclosures, if the substantial doubt is not alleviated, and 6) requires an assessment period of one year from the date the financial statements are issued. The standard is effective for the Company’s reporting year beginning January 1, 2017 and early adoption is permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
| F-13 |
In April 2015, the FASB issued accounting guidance requiring that debt issuance costs related to a recognized liability be presented on the balance sheet as a direct reduction from the carrying amount of that debt liability, consistent with debt discounts. The recognition and measurement guidance for debt issuance costs are not affected. The standard is effective for reporting periods beginning after December 15, 2015. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In July 2015, the FASB issued accounting guidance for inventory. Under the guidance, an entity should measure inventory within the scope of this guidance at the lower of cost and net realizable value, except when inventory is measured using LIFO or the retail inventory method. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. In addition, the FASB has amended some of the other inventory guidance to more clearly articulate the requirements for the measurement and disclosure of inventory. The standard is effective for reporting periods beginning after December 15, 2016. The amendments in this pronouncement should be applied prospectively, with earlier application permitted. The Company is currently evaluating the impact, if any, that this new accounting pronouncement will have on its consolidated financial statements.
In September 2015, the FASB issued accounting guidance to simplify the accounting for measurement period adjustments resulting from business combinations. Under the guidance, an acquirer will be required to recognize adjustments to provisional amounts identified during the measurement period in the reporting period in which the adjustments are determined. The guidance requires an entity to present separately on the face of the income statement or disclose in the notes to the financial statements the portion of the amount recorded in current-period earnings by line item that would have been recorded in previous reporting periods if the adjustment had been recognized as of the acquisition date. The Company adopted this new guidance in the fourth quarter of 2015; see Note 4 for the impact of adoption.
In February 2016, the FASB issued accounting guidance which will require most leases (with the exception of leases with terms of less than one year) to be recognized on the balance sheet as an asset and a lease liability. Leases will be classified as an operating lease or a financing lease. Operating leases are expensed using the straight-line method whereas financing leases will be treated similarly to a capital lease under the current standard. The new standard will be effective for annual and interim periods, within those fiscal years, beginning after December 15, 2018 but early adoption is permitted. The new standard must be presented using the modified retrospective method beginning with the earliest comparative period presented. The Company is currently evaluating the effect of the new standard on its consolidated financial statements and related disclosures.
The Company has evaluated all other issued and unadopted Accounting Standards Updates and believes the adoption of these standards will not have a material impact on its financial position, results of operations, or cash flows.
Note 4 – Business Combination
On July 14, 2015, the Company acquired 100% of the capital stock of AdvanDx in the Merger in a taxable transaction. AdvanDx researches, develops and markets advanced in vitro diagnostic kits for the diagnosis and prevention of infectious diseases, and sells its products principally to hospitals and clinical laboratories in the United States and Europe. The Company acquired AdvanDx principally to use AdvanDx’s diagnostic capabilities with respect to MDROs and leverage AdvanDx’s relationships with hospitals and clinical laboratories to accelerate the sales of OpGen’s products and services.
Pursuant to an Agreement and Plan of Merger (the “Merger Agreement”), Velox Acquisition Corp. merged with and into AdvanDx, Inc. with AdvanDx, Inc. surviving as a wholly owned subsidiary of the Company in accordance with the General Corporation Law of the State of Delaware. Under the terms of the Merger Agreement, the merger consideration consisted of an aggregate 681,818 shares of the Company’s common stock with a value of $2.6 million (the “Merger Consideration”), which Merger Consideration was distributed in accordance with the liquidation preferences set forth in the AdvanDx, Inc. Restated Certificate of Incorporation, as amended.
| F-14 |
The Company accounted for its acquisition of AdvanDx by recording all tangible and intangible assets acquired and liabilities assumed at their estimated fair values on the acquisition date, with the remaining unallocated purchase price recorded as goodwill. The fair value assigned to identifiable intangible assets acquired was determined using an income approach for trade names and customer relationships, and a cost approach for technology. The fair values are preliminary, are based on the Company’s estimates and may be adjusted from time to time, but no later than July 13, 2016, as better information becomes available. The Company received carryover tax basis in the acquired assets and liabilities and no tax basis in the intangible assets (including goodwill) established on the acquisition date. As a result, the Company recognized deferred tax assets related to foreign taxing jurisdictions of $4.3 million (fully offset by a corresponding valuation allowance) and net deferred tax liabilities of $0.1 million in the U.S. taxing jurisdiction. The net deferred tax liability in the U.S. taxing jurisdiction resulted in an income tax benefit related to a reduction in the Company’s previously established valuation allowance (which reduction is accounted for outside of purchase accounting). The following represents the preliminary allocation of the purchase price:
| Total purchase price - fair value of common stock issued | $ | 2,584,090 | ||
| Fair value of tangible assets acquired: | ||||
| Cash | $ | 1,367,211 | ||
| Receivables | 536,406 | |||
| Inventory | 881,273 | |||
| Property and equipment | 245,479 | |||
| Other assets | 359,587 | |||
| Fair value of identifable intangible assets acquired: | ||||
| Customer relationships | 1,094,000 | |||
| Developed technology | 458,000 | |||
| Trademarks and tradenames | 461,000 | |||
| Fair value of goodwill | 637,528 | |||
| Deferred tax liabilities, net | 129,095 | |||
| Fair value of liabilities assumed | 3,327,299 | |||
| $ | 2,584,090 |
The total consideration paid in the acquisition exceeded the estimated fair value of the tangible and identifiable intangible assets acquired and liabilities assumed, resulting in approximately $0.6 million of goodwill. Goodwill, primarily related to expected synergies gained from combining operations, sales growth from future product offerings and customers, together with certain intangible assets that do not qualify for separate recognition, including assembled workforce, is not tax deductible. The Company expensed acquisition-related costs of approximately $0.5 million related to the Merger in 2015. AdvanDx recognized approximately $1.7 million of revenue and incurred approximately $2.1 million of net losses from the acquisition date to December 31, 2015, which results are included in the Company’s 2015 consolidated financial statements.
Adjustments to Purchase Accounting Estimates
In the fourth quarter of 2015, the Company adopted new accounting guidance with respect to the accounting for measurement period adjustments resulting from business combinations. Under the new guidance, the Company is required to recognize adjustments to provisional amounts identified during the measurement period in the reporting period in which the adjustments are determined and disclose the portion of the amount recorded in current-period losses by line item that would have been recorded in previous reporting periods if the adjustment had been recognized as of the acquisition date.
| F-15 |
In the third quarter of 2015, the Company allocated the total purchase consideration to the assets and liabilities acquired from AdvanDx, based on provisional estimated fair value of those acquired assets and liabilities. During the fourth quarter of 2015, as a result of obtaining new information about facts and circumstances that existed as of the acquisition date, the Company adjusted the provisional estimated fair values, as follows:
| As previously reported (1) | Adjustment | Adjusted amount | ||||||||||
| Accounts receivable | $ | 557,112 | $ | (20,706 | ) | $ | 536,406 | |||||
| Inventory | 1,073,855 | (192,582 | ) | 881,273 | ||||||||
| Property and equipment | 250,636 | (5,157 | ) | 245,479 | ||||||||
| Liabilities | 3,329,058 | (1,759 | ) | 3,327,299 | ||||||||
| Deferred tax liabilities | - | 129,095 | 129,095 | |||||||||
| Goodwill | 291,747 | 345,781 | 637,528 | |||||||||
| Provision (benefit) for income taxes | 1,662 | (130,757 | ) | (129,095 | ) | |||||||
(1) As reported on Form 10-Q for the quarter ended September 30, 2015
In the provisional amounts recorded in the third quarter, the Company had anticipated that it would receive “stepped-up” tax bases in the acquired assets and liabilities as a result of making an election under section 338 of the U.S. Internal Revenue Code. In the fourth quarter of 2015, after further tax analysis, the Company decided that it would not make the 338 election and, as such, the Company will receive “carry-over” tax bases in the assets and liabilities acquired. The change in tax treatment resulted in a reduction of the Company’s deferred tax asset valuation allowance in the U.S. taxing jurisdiction that existed prior to the acquisition.
Pro Forma Disclosures (unaudited)
The following unaudited pro forma financial information summarizes the results of operations for the periods indicated as if the Merger had been completed as of January 1, 2014. Pro forma information primarily reflects adjustments relating to (i) elimination of the interest on AdvanDx’s outstanding debt, and (ii) the amortization of intangibles acquired. The pro forma amounts do not purport to be indicative of the results that would have actually been obtained if the acquisition occurred as of January 1, 2014 or that may be obtained in the future:
| Unaudited pro forma results | Year Ended December 31, | |||||||
| 2015 | 2014 | |||||||
| Revenues | $ | 5,231,844 | $ | 8,904,244 | ||||
| Net loss | $ | (20,751,552 | ) | $ | (14,520,497 | ) | ||
| Net loss per share | $ | (2.52 | ) | $ | (6.87 | ) | ||
Note 5 – Merck GHI Financing
On July 14, 2015, as a condition to the Merger, the Company entered into a Common Stock and Note Purchase Agreement (the “Purchase Agreement”) with Merck GHI, pursuant to which Merck GHI purchased 1,136,364 shares of common stock of the Company at $4.40 per share for gross proceeds of $5.0 million. Pursuant to the Purchase Agreement, the Company also issued to Merck GHI a 8% Senior Secured Promissory Note (the “Note”) in the principal amount of $1.0 million with a two-year maturity date from the date of issuance. The Company’s obligations under the Note are secured by a lien on all of the Company’s assets.
The Company incurred issuance costs of approximately $50,000 related to the financing, of which approximately $8,000 was deferred as debt issuance costs and are being amortized as interest expense over the life of the Note, and $42,000 was charged to additional paid-in capital.
| F-16 |
Note 6 - Redeemable Convertible Preferred Stock
All shares of Series A Preferred Stock (including those shares issued in connection with the conversion of the 2014 and 2015 convertible debt) were converted into 7,374,852 shares of common stock in connection with the Company’s IPO (see Notes 7 and 8). At December 31, 2014, the Company had 3,999,864 shares of Series A Preferred Stock outstanding. The Series A Preferred Stock was redeemable at the option of the holders of 70% of the outstanding shares of Series A Preferred Stock, subject to certain additional requirements. The Company’s redeemable convertible preferred stock was classified as temporary equity due to redemption provisions outside of the Company’s control.
The Company issued 1,999,864 shares of Series A Preferred Stock in December 2013 at $1.00 per share in exchange for $1,999,864 in convertible promissory notes. In February 2014, the Company sold 1,405,096 shares of Series A redeemable convertible preferred stock for gross proceeds of $1,405,096. In April 2014, the Company sold an additional 594,904 shares of Series A Preferred Stock for gross proceeds of $594,904. The Company incurred issuance costs of $62,098 related to the 2014 Series A Preferred Stock sales. As of December 31, 2014, the Company had a total of 3,999,864 shares of Series A Preferred Stock outstanding, convertible into 3,999,864 shares of common stock.
The following table presents the changes in the Series A Preferred Stock during 2014 and 2015:
| Shares | Amount | |||||||
| Balance at December 31, 2013 | 1,999,864 | $ | 1,999,864 | |||||
| February 2014 Issuance | 1,405,096 | 1,361,469 | ||||||
| April 2014 Issuance | 594,904 | 576,433 | ||||||
| 2014 Accretion | - | 627,133 | ||||||
| Balance at December 31, 2014 | 3,999,864 | 4,564,899 | ||||||
| 2015 Accretion | - | 243,762 | ||||||
| 2015 Conversions | (3,999,864 | ) | (4,808,661 | ) | ||||
| Balance at December 31, 2015 | - | $ | - | |||||
The Series A Preferred Stock had the right to receive non-cumulative dividends, at a rate of 8% per annum, when and if declared by the board of directors. The Series A Preferred Stock had preference of payment over all other classes and series of capital stock of the Company with respect to dividends, payment on liquidation and payment on redemption. The liquidation and redemption preferences were at two times the Series A Preferred Stock purchase price. The Series A preferred stockholders were entitled to vote on all matters that come to stockholders on an as-converted basis with holders of the common stock. In addition, the Series A Preferred Stock had broad based anti-dilution rights.
The holders of Series A Preferred Stock had the right to convert such shares, at their option and at any time, into shares of common stock at the then-applicable conversion rate, as defined. The initial conversion rate was one common share for each preferred share, which may be adjusted for specified dilutive transactions. Beginning in December 2019, the Company may have been obligated to redeem shares of Series A Preferred Stock, if requested, by holders of at least 70% of the then-outstanding shares of preferred stock. The redemption, if requested, would have taken place in three equal annual installments. Series A Preferred Stock would have been redeemed at two times the original issue price per share plus all accrued and unpaid dividends. The redemptions were subject to certain equity adjustments for specified anti-dilution transactions, as defined.
| F-17 |
Note 7 – Debt
All the Company’s outstanding demand notes and convertible notes were exchanged for units in the Company’s IPO or otherwise were converted into Series A Preferred Stock and subsequently converted into shares of common stock in connection with the IPO. A short-term 8% promissory note for $150,000 issued in April 2015 was repaid in cash in June 2015. In July 2015, the Company issued a $1.0 million 8% senior secured promissory note to Merck GHI (see Note 5). As of December 31, 2015 and 2014, the Company has a total debt outstanding as follows:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Convertible notes | $ | - | $ | 1,500,000 | ||||
| Demand promissory notes | - | 1,500,000 | ||||||
| Long term promissory notes | 1,000,000 | 5,000 | ||||||
| $ | 1,000,000 | $ | 3,005,000 | |||||
The long-term promissory notes are due in July 2017 (see Note 5).
In 2009, the Company entered into loan agreements with the Department of Business and Economic Development, a principal department of the State of Maryland, and Montgomery County, Maryland. Under the terms of the agreements, the State of Maryland and Montgomery County loaned the Company $100,000 and $10,000, respectively, to assist in the relocation of the Company’s operations from Wisconsin to Gaithersburg, Maryland. Interest on the loans accrued at 3%. The interest was deferred and the loans were forgivable under certain conditions, including the Company maintaining operations in Gaithersburg, Maryland, and attaining a specified level of staffing at that site on or before December 31, 2012. The Company did not attain the required level of staffing at December 31, 2012, and, as a result, these notes and accrued interest became due in 2013. The Company negotiated a settlement with the State of Maryland under which it paid $75,000 in June 2013 in full satisfaction of the $100,000 loan principal balance and accrued interest of $11,811. The Company also negotiated a settlement with Montgomery County under which accrued interest due under the loan provisions was forgiven and the loan would be paid in equal quarterly installments over the eight quarters ending December 31, 2015. The loan was paid in full during 2015 and no amounts remain outstanding.
Demand notes
In the fourth quarter of 2014 and first quarter of 2015, the Company raised a total of $2.3 million through the issuance of short-term demand notes. In the first quarter of 2015, $0.3 million of demand notes, held by an entity controlled by our chief executive officer, were settled as partial payment for a 2015 convertible note. All outstanding demand notes were tendered as payment for 350,000 units in the Company’s IPO (see Note 8). Prior to settlement, the demand notes bore interest at 8% per annum, had a first priority security interest in the assets of the Company, and a term of approximately four months.
2014 convertible debt
In July, August and September 2014, the Company raised $1.5 million through the issuance of convertible debt. All outstanding 2014 convertible debt was converted into Series A Preferred Stock and then into 1,500,000 shares of common stock in connection with the Company’s IPO (see Note 8). Prior to its conversion, the debt was convertible, at the option of the holders or in certain cases at the Company’s option, into shares of Series A Preferred Stock or other potential equity securities, bore interest at 8% and was due in full on July 11, 2015.
2015 convertible debt
In February and March 2015, the Company raised $1.5 million in capital through the issuance of 8% secured convertible notes with detachable stock purchase warrants. All outstanding 2015 convertible debt was converted into Series A Preferred Stock and then into 1,875,000 shares of common stock in connection with the Company’s IPO (see Note 8). Prior to its conversion, the 2015 convertible notes were prepayable by the Company without penalty at any time following the three-month anniversary of the closing of the IPO (provided that before the six-month anniversary of the closing of an IPO, the 2015 convertible notes could only be prepaid out of newly issued capital subsequent to the IPO), and were puttable by the holder to the Company in the event of a defined default. The 2015 convertible notes were each convertible, at the election of the holder, into (i) shares of Series A Preferred Stock, at a conversion rate of 1.25 shares of Series A Preferred Stock for each $1.00 converted if the conversion occurs prior to closing of an IPO, or (ii) shares of common stock at a conversion rate of one share of common stock for each $1.00 converted if the conversion occurs after the closing of an IPO.
| F-18 |
The conversion option embedded in the convertible notes was determined to contain beneficial conversion features, resulting in the bifurcation of those features as an equity instrument (resulting in an additional debt discount) at issuance. After allocation of the gross proceeds to the detachable stock purchase warrants (discussed below) and beneficial conversion feature, the total debt discount recognized was equal to the face value of the 2015 convertible notes. Upon conversion in May 2015, the remaining unamortized beneficial conversion feature of approximately $1.5 million was charged to interest expense in the accompanying consolidated statement of operations. Remaining unamortized deferred financing costs of $71,421 were also charged to interest expense upon conversion.
The 2015 convertible note holders also received detachable stock purchase warrants exercisable for 225,011 shares of common stock at 110% of the IPO price and exercisable only if the IPO occurred, and then exercisable beginning on the six-month anniversary of the closing of the IPO. Prior to the IPO, as a result of net settlement features, the stock purchase warrants were considered derivative liabilities, were initially recorded at fair value (resulting in a debt discount) and were marked-to-market at each balance sheet date through earnings. As a result of the elimination of the net settlement features in the IPO, the stock purchase warrants were marked to fair value of $0.7 million on May 8, 2015 and then reclassified to equity.
Total interest expense on all debt instruments was $1.8 million and $0.1 million in 2015 and 2014, respectively.
Note 8 - Stockholders’ Equity
As of December 31, 2015, the Company has 200,000,000 shares of authorized common shares and 12,547,684 issued and outstanding, and 10,000,000 of authorized preferred shares, none of which were issued or outstanding.
On May 8, 2015, the Company completed its IPO pursuant to which the Company offered and sold 2,850,000 units, each Unit consisting of one share of common stock and a detachable stock purchase warrant to purchase an additional share of common stock, at an initial offering price of $6.00 per unit. Of the total gross proceeds of $17.1 million, approximately $2.1 million was used to satisfy outstanding demand notes by exchanging such notes for 350,000 Units in the IPO. After considering the demand notes, and underwriting discounts, commissions and offering expenses of $2.9 million (which were charged to additional paid-in capital), the total net cash proceeds to the Company was $12.1 million. On the IPO closing date, the underwriters exercised a portion of their over-allotment option to acquire an additional 422,500 stock purchase warrants for cash of $4,225. In connection with the IPO, all of the Company’s outstanding Series A Preferred Stock, 2014 convertible notes and 2015 convertible notes were converted into 7,374,852 shares of common stock.
The stock purchase warrants issued as part of the units (including over-allotment option) are exercisable for 3,272,500 shares of common stock at $6.60 per share beginning six months after the closing of the IPO for five years, expiring on May 8, 2020. Additionally, the Company issued additional warrants to its investment bankers to purchase 185,250 shares of common stock, on the same terms as the warrants issued with the units. The warrants were valued using the Black-Scholes option pricing model and are classified as equity.
In July 2015, the Company issued 1,136,364 shares of common stock to Merck GHI for cash consideration of $5.0 million (see Note 5).
Stock options
In 2002, the Company adopted the 2002 Stock Option and Restricted Stock Plan (the “2002 Plan”), pursuant to which the Company’s Board of Directors could grant either incentive stock options or non-qualified stock options, shares of restricted stock, shares of unrestricted common stock, and other share-based awards to officers and employees. In 2008, the Company adopted the 2008 Stock Option and Restricted Stock Plan (the “2008 Plan”), pursuant to which the Company’s Board of Directors may grant either incentive or non-qualified stock options or shares of restricted stock to directors, key employees, consultants and advisors.
In April 2015, the Company adopted, and the Company’s stockholders approved, the 2015 Equity Incentive Plan (the “2015 Plan”); the 2015 Plan became effective upon the execution and delivery of the underwriting agreement for the Company’s IPO. Following the effectiveness of the 2015 Plan, no further grants will be made under the 2002 Plan or 2008 Plan. The 2015 Plan provides for the granting of incentive stock options within the meaning of Section 422 of the Internal Revenue Code to employees and the granting of non-qualified stock options to employees, non-employee directors and consultants. The 2015 Plan also provides for the grants of restricted stock, restricted stock units, stock appreciation rights, dividend equivalents and stock payments to employees, non-employee directors and consultants.
| F-19 |
Under the 2015 Plan, the aggregate number of shares of the common stock authorized for issuance may not exceed (1) 1,355,000 plus (2) the sum of the number of shares subject to outstanding awards under the 2008 Plan as of the 2015 Plan’s effective date, that are subsequently forfeited or terminated for any reason before being exercised or settled, plus (3) the number of shares subject to vesting restrictions under the 2008 Plan as of the 2015 Plan’s effective date that are subsequently forfeited. In addition, the number of shares that have been authorized for issuance under the 2015 Plan will be automatically increased on the first day of each fiscal year beginning on January 1, 2016 and ending on (and including) January 1, 2025, in an amount equal to the lesser of (1) 4% of the outstanding shares of common stock on the last day of the immediately preceding fiscal year, or (2) another lesser amount determined by the Company’s Board of Directors. Shares subject to awards granted under the 2015 Plan that are forfeited or terminated before being exercised or settled, or are not delivered to the participant because such award is settled in cash, will again become available for issuance under the 2015 Plan. However, shares that have actually been issued shall not again become available unless forfeited. As of December 31, 2015, 233,562 shares remain available for issuance under the 2015 Plan.
For the years ended December 31, 2015 and 2014, the Company recorded $1.4 million and $0.1 million, respectively, of stock compensation expense. No income tax benefit for stock-based compensation arrangements was recognized in the consolidated statements of operations due to the Company’s net loss position. The allocation of share-based compensation expense by operating expenses is as follows:
| Year Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Research and development | $ | 240,739 | $ | 5,234 | ||||
| General and administrative | 619,899 | 55,802 | ||||||
| Sales and marketing | 584,450 | 3,376 | ||||||
| $ | 1,445,088 | $ | 64,412 | |||||
During 2015 and 2014, the Company granted stock options to acquire 1,961,637 and 401,053 shares of common stock, respectively, at average exercise prices of $2.68 and $0.05 per share and with a weighted average grant date fair values of $2.80 and $0.03, respectively. At December 31, 2015, the Company had unrecognized expense related to its stock options of $3.4 million which will be recognized over a weighted-average period of 1.57 years.
A summary of the status of options granted under the plan is presented below as of and for the years ended December 31, 2015 and 2014:
| F-20 |
| Number of Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life (in years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at January 1, 2014 | 20,956 | 8.1 | $ | - | ||||||||||||
| Granted | 401,053 | $ | 0.05 | |||||||||||||
| Exercised | (1 | ) | $ | 7.91 | $ | - | ||||||||||
| Forfeited | (17,736 | ) | $ | 40.34 | ||||||||||||
| Outstanding at December 31, 2014 | 404,272 | $ | 1.13 | 9.3 | $ | - | ||||||||||
| Granted | 1,961,637 | $ | 2.68 | |||||||||||||
| Exercised | (11,472 | ) | $ | 0.20 | $ | 19,519 | ||||||||||
| Forfeited | (193,657 | ) | $ | 0.55 | ||||||||||||
| Outstanding at December 31, 2015 | 2,160,780 | $ | 2.60 | 9.1 | $ | 1,575,646 | ||||||||||
| Exercisable at December 31, 2015 | 432,340 | $ | 1.36 | 8.6 | $ | 632,052 | ||||||||||
| Vested and expected to vest | 2,138,606 | $ | 2.60 | 9.1 | $ | 1,555,185 | ||||||||||
The weighted-average grant-date fair value for the option awards granted during the years ended December 31, 2015 and 2014 was $2.80 and $0.03, respectively. The total fair value of options vested in the years ended December 31, 2015 and 2014, was $1,140,079 and $47,331, respectively. The fair value of each option grant was estimated at the date of grant using the Black -Scholes option pricing model based on the assumptions below:
| Year Ended December 31, | ||||||||
| 2015 | 2014 | |||||||
| Annual dividend | - | - | ||||||
| Expected life (in years) | 5.5-6.25 | 6.25 | ||||||
| Risk free interest rate | 1.46-1.9 | % | 1.84-2.02 | % | ||||
| Expected volatility | 47.7-65.0 | % | 60.0 | % | ||||
On October 23, 2014, the Company’s Board of Directors approved grants of stock options to acquire approximately 825,000 shares of common stock under the 2002 Plan, contingent upon obtaining and approving an independent valuation of the fair value of the Company's common stock. These options were approved by the Board of Directors in February 2015, at which time the Company began recognizing stock-based compensation expense.
Restricted stock units
In March 2014, the Company awarded restricted stock units to acquire 130,640 shares of common stock to its Chief Executive Officer (“CEO”). The restricted stock units were compensation for his service as CEO from October 2013 through June 2014 and were subject to forfeiture if he did not continue to perform management services through October 24, 2014. The restricted stock units vested on October 24, 2014 and 130,640 shares of common stock were issued to the CEO. In the fourth quarter of 2015, the Company granted additional restricted stock units to acquire 75,000 shares of common stock, with a weighted average grant date fair value of $1.70 per share, all of which remain outstanding as of December 31, 2015.
Stock purchase warrants
At December 31, 2015 and 2014, the following warrants to purchase shares of common stock were outstanding:
| F-21 |
| Outstanding at December 31, | ||||||||||||||
| Issuance | Exercise Price | Expiration | 2015 | 2014 | ||||||||||
| August 2007 | $ | 7.91 | August 2017 | 8,921 | 8,921 | |||||||||
| March 2008 | $ | 790.54 | March 2018 | 46 | 46 | |||||||||
| November 2009 | $ | 7.91 | November 2019 | 6,674 | 6,674 | |||||||||
| January 2010 | $ | 7.91 | January 2020 | 6,674 | 6,674 | |||||||||
| March 2010 | $ | 7.91 | March 2020 | 1,277 | 1,277 | |||||||||
| November 2011 | $ | 7.91 | November 2021 | 5,213 | 5,213 | |||||||||
| December 2011 | $ | 7.91 | December 2021 | 664 | 664 | |||||||||
| March 2012 | $ | 109.90 | March 2019 | 4,125 | 4,125 | |||||||||
| February 2015 | $ | 6.60 | February 2025 | 225,011 | - | |||||||||
| May 2015 | $ | 6.60 | May 2020 | 3,457,750 | - | |||||||||
| 3,716,355 | 33,594 | |||||||||||||
The warrants listed above were issued in connection with various debt, preferred stock or development contract agreements. The warrants issued in February 2015 were initially classified as a liability since the exercise price was variable. The exercise price became fixed as a result of the Company’s IPO and, as such, the warrant liability was marked to fair value at that time and reclassified to equity (see Note 13).
Note 9 - Income Taxes
At December 31, 2015 and 2014, the Company had net deferred tax assets of $41,554,045 and $31,505,287, respectively, primarily consisting of NOL carry forwards, research and experimental (“R&E”) credits, and differences between depreciation and amortization recorded for financial statement and tax purposes. The Company’s net deferred tax assets at December 31, 2015 and 2014 have been offset by a valuation allowance of $41,545,045 and $31,505,287, respectively. The valuation allowance has been recorded due to the uncertainty of realization of the deferred tax assets. The Company’s deferred tax assets and liabilities as of December 31, 2015 and 2014 are as follows:
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| Deferred tax assets: | ||||||||
| NOL carryforward | $ | 38,797,762 | $ | 28,704,237 | ||||
| R&E credit carryforward | 1,994,478 | 1,894,478 | ||||||
| Share-based compensation | 383,153 | 144,742 | ||||||
| Inventory reserve | 226,299 | 334,578 | ||||||
| Depreciation | 313,714 | 246,233 | ||||||
| Accruals and other | 495,640 | 185,702 | ||||||
| Total deferred tax assets | 42,211,046 | 31,509,970 | ||||||
| Valuation allowance | (41,554,045 | ) | (31,505,287 | ) | ||||
| Deferred tax liabilities: | ||||||||
| Intangible assets | (657,001 | ) | - | |||||
| Fixed assets | - | (4,683 | ) | |||||
| Net deferred tax liability | $ | - | $ | - | ||||
| F-22 |
The difference between the Company’s expected income tax provision (benefit) from applying federal statutory tax rates to the pre-tax loss and actual income tax provision (benefit) relates to the effect of the following:
| 2015 | 2014 | |||||||
| Federal income tax benefit at statutory rates | 35.0 | % | 34.0 | % | ||||
| State income tax benefit, net of Federal benefit | 3.3 | % | 3.6 | % | ||||
| Change in valuation allowance | (33.1) | % | (51.1) | % | ||||
| Change in state tax rates and other | (4.5) | % | 13.5 | % | ||||
| 0.7 | % | 0.0 | % | |||||
Additionally, despite the NOL carryforwards, the Company may have future tax liability due to alternative minimum tax or state tax requirements. The Company has federal NOL carryforwards of $90,297,225 and $76,267,809 at December 31, 2015 and 2014, respectively. The NOL carry forwards begin to expire in 2022. Utilization of the NOL carryforward may be subject to an annual limitation as provided by Section 382 of the Internal Revenue Code. There can be no assurance that the NOL carryforward will ever be fully utilized. To date, the Company has not performed a formal study to determine if any of its remaining NOL and credit attributes might be further limited due to the ownership change rules of Section 382 or Section 383 of the Internal Revenue Code of 1986, as amended. The Company will continue to monitor this matter going forward. There can be no assurance that the NOL carryforwards will ever be fully utilized.
Note 10 - Commitments
Operating leases
During the second quarter 2015, the Company extended the term of its Gaithersburg, Maryland office lease, effective May 7, 2015, through January 31, 2021, with one additional five-year renewal at the Company’s election. The Company is responsible for all utilities, repairs, insurance, and taxes under this operating lease. Effective July 1, 2015, the Company further modified its lease agreement to add additional leased space to its headquarters. The Company also leases a facility in Woburn, Massachusetts under an operating lease that expires in January 2017, and provides the Company with options to extend the lease beyond the current expiration date. Additionally, the Company leases office space in Denmark; this lease is currently on a month-to-month basis. Rent expense under the Company’s facility operating leases for the years ended December 31, 2015 and 2014, was $1,027,174 and $883,155, respectively.
Capital leases
The Company leases computer equipment, office furniture, and equipment under various capital leases. The leases expire at various dates through 2020. The leases require monthly principal and interest payments. Following is a schedule by year of the estimated future minimum payments under all operating and capital leases as of December 31, 2015:
| F-23 |
| Year ending December 31, | Capital Leases | Operating Leases | Total | |||||||||
| 2016 | $ | 294,141 | $ | 870,484 | $ | 1,164,625 | ||||||
| 2017 | 204,354 | 633,480 | 837,834 | |||||||||
| 2018 | 109,272 | 568,410 | 677,682 | |||||||||
| 2019 | 21,266 | - | 21,266 | |||||||||
| 2020 and thereafter | 19,494 | - | 19,494 | |||||||||
| Total | $ | 648,527 | $ | 2,072,374 | $ | 2,720,901 | ||||||
| Less: amount representing interest | (70,255 | ) | ||||||||||
| Net present value of future minimum lease payments | $ | 578,272 | ||||||||||
| Current maturities | (251,800 | ) | ||||||||||
| Long-term maturities | $ | 326,472 | ||||||||||
Assets under capital leases were included in the following balance sheet categories as of December 31:
| 2015 | 2014 | |||||||
| Laboratory equipment | $ | 803,500 | $ | 364,471 | ||||
| Office furniture | 89,140 | - | ||||||
| Computers | 153,693 | 153,693 | ||||||
| Less accumulated amortization | (402,066 | ) | (245,030 | ) | ||||
| Capital lease assets, net | $ | 644,267 | $ | 273,134 | ||||
Amortization expense associated with equipment under capital leases for the years ended December 31, 2015 and 2014 was $157,036 and $122,411, respectively, and is included within depreciation and amortization expense in the consolidated statements of operations.
Registration and other shareholder rights
In connection with the Merger and the investment transactions (see Notes 4 and 5), the Company also entered into a Registration Rights Agreement with the AdvanDx stockholders receiving Merger Consideration and with Merck GHI, pursuant to which the investors were granted certain demand registration rights and piggyback registration rights in connection with subsequent registered offerings of the Company’s common stock. Merck GHI also received rights to participate on a pro-rata basis in future securities offerings by the Company.
On December 18, 2013, the Company entered into the Third Amended and Restated Investors’ Rights Agreement (the “Investors’ Rights Agreement”) with investors acquiring promissory notes convertible into shares of the Company's Series A Preferred Stock. Following the IPO, the holders of 20% or more of such shares of common stock subject to the Investors’ Rights Agreement have demand registration rights and piggyback registration rights in connection with subsequent registered offerings of the Company’s common stock.
Note 11 - License Agreements, Research Collaborations and Development Agreements
The Company is a party to three license agreements to acquire certain patent rights and technologies. Royalties are incurred upon the sale of a product or service which utilizes the licensed technology. Certain of the agreements require the Company to pay minimum royalties or license maintenance fees. The Company recognized $205,147 and $97,134 of net royalty expense for the years ended December 31, 2015 and 2014, respectively. In 2016, future minimum royalty fees are $270,000 under these agreements.
| F-24 |
In September 2013, the Company entered into a technology development agreement in which the Company would receive fixed milestone payments for meeting development milestones under the agreement. Since the milestones are substantive, the Company recognizes revenue in the periods in which the substantive milestones are achieved. In addition, the Company received an upfront payment of $250,000, which is recognized on a straight-line basis over the term of the technology development agreement. The Company recognized total revenue of $336,102 and $2,411,120 during the years ended December 31, 2015 and 2014, respectively, relating to this arrangement.
Note 12 – Related Party Transactions
In March 2014, the Company entered into a supply agreement with Fluidigm Corporation, or Fluidigm, under which Fluidigm supplies the Company with its microfluidic test platform for use in manufacturing the Acuitas MDRO Gene Test. The Company’s CEO and Chairman of the Board of Directors of the Company, is a director of Fluidigm. On July 12, 2015, the Company entered into a letter agreement (the “Fluidigm Agreement”) with Fluidigm to expand the companies’ existing relationship to include collaborating on the development of test kits and custom analytic instruments for identification, screening and surveillance testing of MDROs. The Fluidigm Agreement also expands the existing Supply Agreement between the Company and Fluidigm, and provides for expansion of the gene targets and organisms to be tested on the Company’s existing CLIA lab-based tests, the Acuitas MDRO Gene Test and the Acuitas Resistome Test, using Fluidigm technologies and products. Additionally, Fluidigm has agreed not to develop or directly collaborate with any third party to develop an FDA approved or CE-marked diagnostic test for the purpose of detecting resistome genes for identified MDROs if the Company meets certain minimum purchase commitments and other requirements. The initial term of the Fluidigm Agreement is five years. Both parties have the ability to extend the term for an additional five years. Under the expanded Supply Agreement, the term is extended until March 17, 2018, and the Company has the right to extend the term of the Supply Agreement for up to two additional three-year terms. The Company incurred and paid $370,539 and $138,339 related to these agreements in 2015 and 2014, respectively.
In addition, the Company has several capital lease arrangements for laboratory equipment manufactured by Fluidigm. The Company paid $119,919 and $59,412 related to the leased equipment in 2015 and 2014, respectively.
Note 13 – Fair Value Measurements
The Company classifies its financial instruments using a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include:
| · | Level 1 - defined as observable inputs such as quoted prices in active markets; |
| · | Level 2 - defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and |
| · | Level 3 - defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions such as expected revenue growth and discount factors applied to cash flow projections. |
Financial assets and liabilities measured at fair value on a recurring basis
The Company evaluates financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level at which to classify them each reporting period. This determination requires the Company to make subjective judgments as to the significance of inputs used in determining fair value and where such inputs lie within the hierarchy.
Prior to its IPO, certain stock purchase warrants contained cash settlement features and, accordingly, the Company considered them to be derivative financial instruments and accounted for them at fair value using level 3 inputs. As a result of the Company’s IPO and elimination of the cash settlement features pursuant to their terms, those stock purchase warrants were reclassified to equity. For periods prior to the IPO, the Company determined the fair value of these derivative liabilities using a hybrid valuation method that consisted of a probability weighted expected return method that values the Company’s equity securities assuming various possible future economic outcomes while using an option pricing method (that treated all equity linked instruments as call options on the Company’s equity value with exercise prices based on the liquidation preference of the Series A Preferred Stock ) to estimate the allocation of value within one or more of the scenarios. Using this hybrid method, unobservable inputs included the Company’s equity value, the exercise price for each option value, expected timing of possible economic outcomes such as initial public offering, risk free interest rates and stock price volatility. The following tables set forth a summary of changes in the fair value of Level 3 liabilities measured at fair value on a recurring basis for the year ended December 31, 2015:
| F-25 |
| Description | Balance at December 31, 2014 | Established in 2015 | Change in Fair Value | Reclassified to Equity | Balance at December 31, 2015 | |||||||||||||||
| Derivative warrant liability | $ | - | $ | 72,333 | $ | 647,342 | $ | (719,675 | ) | $ | - | |||||||||
Financial assets and liabilities carried at fair value on a non-recurring basis
The Company does not have any financial assets and liabilities measured at fair value on a non-recurring basis.
Non-financial assets and liabilities carried at fair value on a recurring basis
The Company does not have any non-financial assets and liabilities measured at fair value on a recurring basis.
Non-financial assets and liabilities carried at fair value on a non-recurring basis
The Company measures its long-lived assets, including property and equipment and intangible assets (including goodwill), at fair value on a non-recurring basis when they are deemed to be impaired. No such fair value impairment was recognized in 2015 and 2014.
See Note 4 for a discussion of the fair value of assets acquired and liabilities assumed in the Merger.
| F-26 |
EXHIBIT INDEX
|
Exhibit |
Description | |
| 2.1 | Agreement and Plan of Merger, dated as of July 14, 2015, among the Registrant, Velox Acquisition Corp, AdvanDx, Inc., Stockholder Parties and Representatives (incorporated by reference to Exhibit 2.1 of Current Report on Form 8-K, File No. 001-37367, filed on July 16, 2015) | |
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 of Current Report on Form 8-K, File No. 001-37367, filed on May 13, 2015) | |
| 3.2 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 of Form S-1, File No. 333-202478, filed on March 3, 2015) | |
| 4.1 | Form of Common Stock Certificate of the Registrant (incorporated by reference to Exhibit 4.1 of Form S-1/A, File No. 333-202478, filed on April 28, 2015) | |
| 4.2 | Second Stockholders’ Agreements Amendment, dated as of February 7, 2015, among the Registrant and certain investors (incorporated by reference to Exhibit 4.4 of Form S-1, File No. 333-202478, filed on March 3, 2015) | |
| 4.3 | Form of 2015 Warrant to Purchase Common Stock of the Registrant (incorporated by reference to Exhibit 4.6 of Form S-1/A, File No. 333-202478, filed on March 20, 2015) | |
| 4.4 | Form of Underwriters’ Warrant to Purchase Common Stock of the Registrant (incorporated by reference to Exhibit 4.2 of Current Report on Form 8-K, File No. 001-37367, filed on May 13, 2015) | |
| 4.5 | Form of Offered Warrant to Purchase Common Stock of the Registrant (incorporated by reference to Exhibit 4.8 of Form S-1/A, File No. 333-202478, filed on April 23, 2015) | |
| 10.1 | Lease Agreement, dated as of June 30, 2008, between the Registrant and ARE-708 Quince Orchard, LLC (the "Landlord") (incorporated by reference to Exhibit 10.1 of Form S-1/A, file No. 333-202478, filed March 3, 2015) | |
| 10.1.1 | First Amendment to Lease, dated as of April 4, 2011, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1.1 of Form S-1, File No. 333-202478, filed March 3, 2015) | |
| 10.1.2 | Second Amendment to Lease, dated as of August 15, 2012, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1.2 of Form S-1, File No. 333-202478, filed March 3, 2015) | |
| 10.1.3 | Third Amendment to Lease, dated as of December 30, 2013, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1.3 of Form S-1, File No. 333-202478, filed March 3, 2015) | |
| 10.1.4 | Fourth Amendment to Lease, dated as of March 21, 2014, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.4 of Form S-1, File No. 333-202478, filed March 3, 2015) | |
| 10.1.5 | Fifth Amendment to Lease Agreement, dated as of March 20, 2015, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1.5 of Form S-1/A, File No. 333-202478, filed on March 20, 2015) | |
| 10.1.6 | Sixth Amendment to Lease Agreement, dated as of April 30, 2015, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1.6 of Form S-1/A, File No. 333-202478, filed on May 1, 2015) | |
| 10.1.7 | Seventh Amendment to Lease Agreement, dated as of June 30, 2015, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.1 of Current Report on Form 8-K, filed on July 7, 2015) | |
| 10.1.8 | Eighth Amendment to Lease Agreement, dated September 8, 2015, between the Registrant and the Landlord (incorporated by reference to Exhibit 10.6 of Quarterly Report on Form 10-Q, filed on November 13, 2015) | |
| 10.2 | Form of Indemnification Agreement between the Registrant and each of its directors and executive officers (incorporated by reference to Exhibit 10.2 of Form S-1, File No. 333-202478, filed on March 3, 2015) | |
| 10.3 | Notes Purchase Agreement, dated February 17, 2015, by and among the Registrant and the investors party thereto (including as Exhibit B the form of convertible note) (incorporated by reference to Exhibit 10.9 of Form S-1, File No. 333-202478, filed on March 3, 2015) | |
| 10.4 | Form of Amended and Restated Secured Convertible Promissory Note (incorporated by reference to Exhibit 10.10 of Form S-1/A, File No. 333-202478, filed on March 20, 2015) | |
| 10.5 | Amended and Restated Intercreditor Agreement, dated as of February 17, 2015, by and among, the Registrant, Harris & Harris Group, Inc., as collateral agent, and each of the Secured Parties party thereto (incorporated by reference to Exhibit 10.11 of Form S-1/A, File No. 333-202478, filed on March 20, 2015) |
| 1 |
| Exhibit Number |
Description | |
| 10.6 | Form of Security Agreement, by and among the Registrant, the Secured Parties party thereto and Harris & Harris Group, Inc., as collateral agent (incorporated by reference to Exhibit 10.12 of Form S-1/A, File No. 333-202478, filed on March 20, 2015) | |
| 10.7# | 2015 Equity Incentive Plan (incorporated by reference to Exhibit 10.13 of Form S-1/A, File No. 333-202478, filed on April 6, 2015) | |
| 10.8# | Executive Employment, Change in Control and Severance Benefit Agreement, dated April 17, 2015, by and between the Registrant and Kevin Krenitsky, M.D. (incorporated by reference to Exhibit 10.17 of Form S-1/A, File No. 333-202478, filed on April 17, 2015) | |
| 10.9# | Employment Agreement, dated April 17, 2015, by and between the Registrant and Timothy C. Dec (incorporated by reference to Exhibit 10.18 of Form S-1/A, File No. 333-202478, filed on April 17, 2015) | |
| 10.10 | Consulting Agreement, effective May 4, 2015, by and between the Registrant and C. Eric Winzer (incorporated by reference to Exhibit 10.14 of Form S-1/A, File No. 333-202478, filed on April 6, 2015) | |
| 10.11 | Form of Secured Demand Note (incorporated by reference to Exhibit 10.15 of Form S-1/A, File No. 333-202478, filed on April 6, 2015) | |
| 10.12 | Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.16 of Form S-1/A, File No. 333-202478, filed on April 6, 2015) | |
| 10.13 | Warrant Agreement, dated as of May 8, 2015, between the Registrant and Philadelphia Stock Transfer, Inc., as warrant agent (incorporated by reference to Exhibit 10.1 of Current Report on Form 8-K, filed on May 13, 2015) | |
| 10.14# | Form of Stock Option Agreement under the 2015 Stock Incentive Plan (incorporated by reference to Exhibit 10.14 of Quarterly Report on Form 10-Q, filed on June 18, 2015) | |
| 10.15 | Common Stock and Note Purchase Agreement, dated as of July 14, 2015, between the Registrant and Merck Global Health Innovation Fund, LLC (incorporated by reference to Exhibit 10.1 of Current Report on Form 8-K, filed on July 16, 2015) | |
| 10.16 | Senior Secured Promissory Note, dated as of July 14, 2015, between the Registrant and Merck Global Health Innovation Fund, LLC (incorporated by reference to Exhibit 10.1 of Current Report on Form 8-K, filed on July 16, 2015) | |
| 10.17 | Registration Rights Agreement, dated as of July 14, 2015, among the Registrant, Merck Global Health Innovation Fund, LLC, SLS Invest AB and LD Pensions (incorporated by reference to Exhibit 10.1 of Current Report on Form 8-K, filed on July 16, 2015) | |
| 10.18± | Letter Agreement, dated July 12, 2015, between the Registrant and Fluidigm Corporation (incorporated by reference to Exhibit 10.5 of Quarterly Report on Form 10-Q, filed on August 14, 2015) | |
| 21.1* | Subsidiaries of the Registrant | |
| 23.1* | Consent of CohnReznick LLP | |
| 31.1* | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 31.2* | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 32.1* |
Certification of Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 2 |
| Exhibit Number |
Description | |
| 101* | Interactive data files pursuant to Rule 405 of Regulation S-T; (i) the Balance Sheets, (ii) the Statements of Operations, (iii) the Statements of Stockholders’ Equity (Deficit), (iv) Statements of Cash Flows and (v) the Notes to the Financial Statements | |
| * | Filed herewith | |
| # | Management contract or compensatory arrangement. | |
| ± | Confidential treatment has been granted for certain provisions of this agreement pursuant to an application for confidential treatment filed with the Securities and Exchange Commission on August 15, 2015. Such provisions have been separately filed with the Commission. |
| 3 |